
ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール誘導体
式(I)の化合物、ならびにナトリウム−グルコース輸送体阻害剤(特に、SGLT2阻害剤)によって媒介される疾患、状態および/または障害の治療のためのその使用が、本明細書に記載されている。本明細書に開示されている化合物は、肥満症およびその関連する共存疾患、特にII型(2型)糖尿病の予防および治療に有用である。
【図1】

【図1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ナトリウム−グルコース共輸送体(SGLT)阻害剤としてのジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール誘導体、その結晶構造、医薬組成物および使用に関する。
【背景技術】
【0002】
肥満症は、高血圧症、インスリン抵抗性、糖尿病、冠動脈疾患および心不全(代謝症候群と総称されている)などの共存疾患が含まれるその深刻な合併症のために、重大な健康問題である。肥満症およびその関連する共存疾患は、先進国世界において健康上の問題を拡大させ続けており、発展途上世界にもまた影響を及ぼし始めている。肥満症は健康への悪影響によって、米国において予防可能な死の第2の主要原因となっており、社会に重大な経済的および心理社会的影響をもたらしている。McGinnis M、Foege WH.、「Actual Causes of Death in the United States」、JAMA、270、2207〜12(1993)を参照されたい。肥満症およびそれに伴う共存疾患、特に、II型(2型)糖尿病を治療および/または予防する新規な薬剤を同定および開発することが求められている。
【0003】
より最近では、ナトリウム−グルコース共輸送(SGLT)阻害剤、特にSGLT2阻害剤は、糸球体における腎臓濾過液からのグルコースの再吸収を遮断し、それによって尿中のグルコース排泄を誘発することが示された。過剰なグルコースが排泄されると、血糖値の減少、グルコースの肝臓貯蔵の減少、インスリン分泌の減少、それらに続き炭水化物の脂肪への変換の減少、最終的には蓄積された脂肪の減少が起こる。SGLT2の選択的阻害は、グルコース排泄を増強することによって血漿グルコースを正常化することが期待されている。結果的に、SGLT2阻害剤は、体重増加または低血糖の危険性を伴わずに糖尿病状態を改善するための魅力的な手段を提供する。Isaji,M.、Current Opinion Investigational Drugs、8(4)、285〜292(2007)を参照されたい。治療標的としてのSGLTの一般的な概説については、Asano,T.ら、Drugs of the Future、29(5)、461〜466(2004)をまた参照されたい。
【0004】
NIDDMおよび肥満症の治療に有用であることが示されているグリコシドの代表例は、下記の開示に見出すことができる:米国特許第6,515,117号、同第6,414,126号、同第7,101,856号、同第7,169,761号、および同第7,202,350号、米国特許出願公開第US2002/0111315号、同第US2002/0137903号、同第US2004/0138439号、同第US2005/0233988号、同第US2006/0025349号、同第US2006/0035841号、および同第US2006/0632722号、およびPCT国際公開第WO01/027128号、同第WO02/044192号、同第WO02/088157号、同第WO03/099836号、同第WO04/087727号、同第WO05/021566号、同第WO05/085267号、同第WO06/008038号、同第WO06/002912号、同第WO06/062224号、同第WO07/000445号、同第WO07/093610号、および同第WO08/002824号。
【0005】
特定のグリコシドは遺伝毒性であり、潜在的に変異原性または発癌性であるように細胞の遺伝物質に影響を及ぼす。遺伝毒性物質は、In Vitro Mammalian Cell Micronuleus Test(MNvit)、Organization for Economic Co−Operation and Development(OECD)Draft Test Guideline(Draft TG)487(2007);In vitro Mammalian Chromosomal Aberration Test、OECD TG 473(1997);Bacterial Reverse Mutation Test、OECD TG 471(1997);Mammalian Erythrocyte Micronucleus Test、OECD TG 474(1997)または同様のものなどの標準的アッセイを使用して検出することができる。結果的に、肥満症ならびにそれに伴う共存疾患、特に、2型糖尿病および関連する障害のより有効で安全な治療的処置および/または予防が依然として求められている。
【発明の概要】
【発明が解決しようとする課題】
【0006】
【課題を解決するための手段】
【0007】
式(A)および式(B)の化合物は、ナトリウム−グルコース共輸送(SGLT)阻害剤、特に、SGLT2阻害剤として作用し、したがって、このような阻害によって媒介される疾患(例えば、肥満症に関連する疾患、2型糖尿病、ならびに肥満症に関連する共存疾患および糖尿病に関連する共存疾患)の治療において使用し得ることが見出されている。これらの化合物は、下記に示されているような式(A)および(B)によって表すことができる
【0008】
【化1】
[式中、R1は、H、(C1〜C4)アルキル、(C1〜C4)アルコキシ、Cl、F、シアノ、フルオロ置換(C1〜C2)アルキル、(C1〜C4)アルキル−SO2−、または(C3〜C6)シクロアルキルであり、
R2は、(C1〜C4)アルキル、(C1〜C4)アルコキシ、(C2〜C4)アルキニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、Cl、F、シアノ、フルオロ置換(C1〜C2)アルキル、(C1〜C4)アルキル−SO2−、(C3〜C6)シクロアルキル、または各々独立にN、O、もしくはSから選択される1個もしくは2個のヘテロ原子を有する(C5〜C6)複素環である]。
【0009】
選択された置換基(複数可)が化合物の薬理学的特性に悪影響を与えず、または医薬の使用を不利に妨げない限り、様々な置換基を式(A)または式(B)の化合物に付加してもよいことを一般に当業者は理解する。
【0010】
式(A)の特定の化合物には、(1S,2S,3S,4R,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;2−(4−メトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)ベンゾニトリル;2−(4−エトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)ベンゾニトリル;(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;および(1S,2S,3S,4R,5S)−5−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオールが含まれる。
【0011】
式(B)の特定の化合物には、(1S,2S,3S,4S,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4S,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;および(1S,2S,3S,4S,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオールが含まれる。
【0012】
本発明のさらなる態様は、式(4A)を有する化合物を含む結晶である。
【0013】
【化2】
【0014】
本発明の別の態様は、(1)本発明の化合物と、(2)薬学的に許容できる添加剤、賦形剤、または担体とを含む医薬組成物である。好ましくは、組成物は、治療有効量の本発明の化合物を含む。組成物はまた、少なくとも1種のさらなる医薬品(本明細書に記載されている)を含有し得る。好ましい薬剤には、抗肥満剤および/または抗糖尿病剤(本明細書において下記に記載されている)が含まれる。
【0015】
本発明のまた別の態様において、動物においてSGLT2阻害によって調節される疾患、障害、または状態を治療するための方法であって、このような治療を必要としている動物(好ましくは、ヒト)に、治療有効量の本発明の化合物(またはその医薬組成物)を投与するステップを含む方法が提供される。SGLT2阻害によって調節される疾患、状態、および/または障害には、例えば、II型糖尿病、糖尿病性腎症、インスリン抵抗性症候群、高血糖症、高インスリン血症、高脂血症、耐糖能異常、肥満症(体重管理または体重維持を含めた)、高血圧症、および血糖値の減少が含まれる。
【0016】
本発明の化合物は、他の医薬品(特に、本明細書において下記に記載されている抗肥満剤および抗糖尿病剤)と組み合わせて投与されてもよい。併用療法は、(a)本発明の化合物、本明細書に記載されている少なくとも1種のさらなる医薬品、および薬学的に許容できる添加剤、賦形剤、もしくは担体を含む単一の医薬組成物、または(b)(i)本発明の化合物と、薬学的に許容できる添加剤、賦形剤、または担体とを含む第1の組成物、および(ii)本明細書に記載されている少なくとも1種のさらなる医薬品と、薬学的に許容できる添加剤、賦形剤、または担体とを含む第2の組成物を含む2つの別々の医薬組成物として投与することができる。医薬組成物は、同時または順次に、および任意の順序で投与することができる。
【0017】
上記の概要および下記の詳細な説明は両方とも例示的および説明的なものにすぎず、特許請求している本発明を制限するものではないことを理解すべきである。
【図面の簡単な説明】
【0018】
【技術分野】
【0001】
本発明は、ナトリウム−グルコース共輸送体(SGLT)阻害剤としてのジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール誘導体、その結晶構造、医薬組成物および使用に関する。
【背景技術】
【0002】
肥満症は、高血圧症、インスリン抵抗性、糖尿病、冠動脈疾患および心不全(代謝症候群と総称されている)などの共存疾患が含まれるその深刻な合併症のために、重大な健康問題である。肥満症およびその関連する共存疾患は、先進国世界において健康上の問題を拡大させ続けており、発展途上世界にもまた影響を及ぼし始めている。肥満症は健康への悪影響によって、米国において予防可能な死の第2の主要原因となっており、社会に重大な経済的および心理社会的影響をもたらしている。McGinnis M、Foege WH.、「Actual Causes of Death in the United States」、JAMA、270、2207〜12(1993)を参照されたい。肥満症およびそれに伴う共存疾患、特に、II型(2型)糖尿病を治療および/または予防する新規な薬剤を同定および開発することが求められている。
【0003】
より最近では、ナトリウム−グルコース共輸送(SGLT)阻害剤、特にSGLT2阻害剤は、糸球体における腎臓濾過液からのグルコースの再吸収を遮断し、それによって尿中のグルコース排泄を誘発することが示された。過剰なグルコースが排泄されると、血糖値の減少、グルコースの肝臓貯蔵の減少、インスリン分泌の減少、それらに続き炭水化物の脂肪への変換の減少、最終的には蓄積された脂肪の減少が起こる。SGLT2の選択的阻害は、グルコース排泄を増強することによって血漿グルコースを正常化することが期待されている。結果的に、SGLT2阻害剤は、体重増加または低血糖の危険性を伴わずに糖尿病状態を改善するための魅力的な手段を提供する。Isaji,M.、Current Opinion Investigational Drugs、8(4)、285〜292(2007)を参照されたい。治療標的としてのSGLTの一般的な概説については、Asano,T.ら、Drugs of the Future、29(5)、461〜466(2004)をまた参照されたい。
【0004】
NIDDMおよび肥満症の治療に有用であることが示されているグリコシドの代表例は、下記の開示に見出すことができる:米国特許第6,515,117号、同第6,414,126号、同第7,101,856号、同第7,169,761号、および同第7,202,350号、米国特許出願公開第US2002/0111315号、同第US2002/0137903号、同第US2004/0138439号、同第US2005/0233988号、同第US2006/0025349号、同第US2006/0035841号、および同第US2006/0632722号、およびPCT国際公開第WO01/027128号、同第WO02/044192号、同第WO02/088157号、同第WO03/099836号、同第WO04/087727号、同第WO05/021566号、同第WO05/085267号、同第WO06/008038号、同第WO06/002912号、同第WO06/062224号、同第WO07/000445号、同第WO07/093610号、および同第WO08/002824号。
【0005】
特定のグリコシドは遺伝毒性であり、潜在的に変異原性または発癌性であるように細胞の遺伝物質に影響を及ぼす。遺伝毒性物質は、In Vitro Mammalian Cell Micronuleus Test(MNvit)、Organization for Economic Co−Operation and Development(OECD)Draft Test Guideline(Draft TG)487(2007);In vitro Mammalian Chromosomal Aberration Test、OECD TG 473(1997);Bacterial Reverse Mutation Test、OECD TG 471(1997);Mammalian Erythrocyte Micronucleus Test、OECD TG 474(1997)または同様のものなどの標準的アッセイを使用して検出することができる。結果的に、肥満症ならびにそれに伴う共存疾患、特に、2型糖尿病および関連する障害のより有効で安全な治療的処置および/または予防が依然として求められている。
【発明の概要】
【発明が解決しようとする課題】
【0006】
【課題を解決するための手段】
【0007】
式(A)および式(B)の化合物は、ナトリウム−グルコース共輸送(SGLT)阻害剤、特に、SGLT2阻害剤として作用し、したがって、このような阻害によって媒介される疾患(例えば、肥満症に関連する疾患、2型糖尿病、ならびに肥満症に関連する共存疾患および糖尿病に関連する共存疾患)の治療において使用し得ることが見出されている。これらの化合物は、下記に示されているような式(A)および(B)によって表すことができる
【0008】
【化1】
[式中、R1は、H、(C1〜C4)アルキル、(C1〜C4)アルコキシ、Cl、F、シアノ、フルオロ置換(C1〜C2)アルキル、(C1〜C4)アルキル−SO2−、または(C3〜C6)シクロアルキルであり、
R2は、(C1〜C4)アルキル、(C1〜C4)アルコキシ、(C2〜C4)アルキニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、Cl、F、シアノ、フルオロ置換(C1〜C2)アルキル、(C1〜C4)アルキル−SO2−、(C3〜C6)シクロアルキル、または各々独立にN、O、もしくはSから選択される1個もしくは2個のヘテロ原子を有する(C5〜C6)複素環である]。
【0009】
選択された置換基(複数可)が化合物の薬理学的特性に悪影響を与えず、または医薬の使用を不利に妨げない限り、様々な置換基を式(A)または式(B)の化合物に付加してもよいことを一般に当業者は理解する。
【0010】
式(A)の特定の化合物には、(1S,2S,3S,4R,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;2−(4−メトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)ベンゾニトリル;2−(4−エトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)ベンゾニトリル;(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;および(1S,2S,3S,4R,5S)−5−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオールが含まれる。
【0011】
式(B)の特定の化合物には、(1S,2S,3S,4S,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4S,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;および(1S,2S,3S,4S,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオールが含まれる。
【0012】
本発明のさらなる態様は、式(4A)を有する化合物を含む結晶である。
【0013】
【化2】
【0014】
本発明の別の態様は、(1)本発明の化合物と、(2)薬学的に許容できる添加剤、賦形剤、または担体とを含む医薬組成物である。好ましくは、組成物は、治療有効量の本発明の化合物を含む。組成物はまた、少なくとも1種のさらなる医薬品(本明細書に記載されている)を含有し得る。好ましい薬剤には、抗肥満剤および/または抗糖尿病剤(本明細書において下記に記載されている)が含まれる。
【0015】
本発明のまた別の態様において、動物においてSGLT2阻害によって調節される疾患、障害、または状態を治療するための方法であって、このような治療を必要としている動物(好ましくは、ヒト)に、治療有効量の本発明の化合物(またはその医薬組成物)を投与するステップを含む方法が提供される。SGLT2阻害によって調節される疾患、状態、および/または障害には、例えば、II型糖尿病、糖尿病性腎症、インスリン抵抗性症候群、高血糖症、高インスリン血症、高脂血症、耐糖能異常、肥満症(体重管理または体重維持を含めた)、高血圧症、および血糖値の減少が含まれる。
【0016】
本発明の化合物は、他の医薬品(特に、本明細書において下記に記載されている抗肥満剤および抗糖尿病剤)と組み合わせて投与されてもよい。併用療法は、(a)本発明の化合物、本明細書に記載されている少なくとも1種のさらなる医薬品、および薬学的に許容できる添加剤、賦形剤、もしくは担体を含む単一の医薬組成物、または(b)(i)本発明の化合物と、薬学的に許容できる添加剤、賦形剤、または担体とを含む第1の組成物、および(ii)本明細書に記載されている少なくとも1種のさらなる医薬品と、薬学的に許容できる添加剤、賦形剤、または担体とを含む第2の組成物を含む2つの別々の医薬組成物として投与することができる。医薬組成物は、同時または順次に、および任意の順序で投与することができる。
【0017】
上記の概要および下記の詳細な説明は両方とも例示的および説明的なものにすぎず、特許請求している本発明を制限するものではないことを理解すべきである。
【図面の簡単な説明】
【0018】
【特許請求の範囲】
【請求項1】
式(A)または式(B)の化合物
【化1】
[式中、
R1は、H、(C1〜C4)アルキル、(C1〜C4)アルコキシ、Cl、F、シアノ、フルオロ置換(C1〜C2)アルキル、(C1〜C4)アルキル−SO2−、または(C3〜C6)シクロアルキルであり、
R2は、(C1〜C4)アルキル、(C1〜C4)アルコキシ、(C2〜C4)アルキニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、Cl、F、シアノ、フルオロ置換(C1〜C2)アルキル、(C1〜C4)アルキル−SO2−、(C3〜C6)シクロアルキル、または各々独立にN、O、もしくはSから選択される1個もしくは2個のヘテロ原子を有する(C5〜C6)複素環である]。
【請求項2】
式(A)の化合物である、請求項1に記載の化合物。
【請求項3】
R1が、H、メチル、エチル、プロピル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、シクロプロピル、またはシクロブチルであり、
R2が、メチル、エチル、プロピル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである、請求項1または2に記載の化合物。
【請求項4】
R1が、H、メチル、エチル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、またはシクロプロピルであり、
R2が、メチル、エチル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである、請求項3に記載の化合物。
【請求項5】
R1が、H、メチル、エチル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、またはシクロプロピルであり、
R2が、メチル、エチル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである、請求項4に記載の化合物。
【請求項6】
R1が、メチル、エチル、F、Cl、シアノ、CF3、またはシクロプロピルであり、
R2が、メトキシ、またはエトキシである、請求項5に記載の化合物。
【請求項7】
(1S,2S,3S,4R,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
2−(4−メトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)ベンゾニトリル;
2−(4−エトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)ベンゾニトリル;
(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;および
(1S,2S,3S,4R,5S)−5−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオールからなる群から選択される化合物。
【請求項8】
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオールである化合物。
【請求項9】
(1S,2S,3S,4S,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4S,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;および
(1S,2S,3S,4S,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオールからなる群から選択される化合物。
【請求項10】
式(4A)を有する化合物を含む結晶。
【化2】
【請求項11】
L−プロリンまたはL−ピログルタミン酸をさらに含む、請求項10に記載の結晶。
【請求項12】
L−ピログルタミン酸をさらに含み、
a)P2(1)2(1)2(1)の空間群および実質的に下記と等しい単位格子パラメータ
a=7.4907(10)Å α=90°
b=12.8626(15)Å β=90°
c=28.029(4)Å γ=90°、
b)2θ値(CuKα放射線、1.54056Åの波長)6.4±0.2、16.7±0.2、17.4±0.2および21.1±0.2を含む粉末X線回折パターン、または
c)500MHz分光計で決定すると、29.5ppmの結晶性アダマンチンに対して、16.5±0.2、131.1±0.2、158.7±0.2、および181.5±0.2ppmにおいてピーク位置を含む固体13C NMRスペクトル
の1つまたは複数を有する、請求項10に記載の結晶。
【請求項13】
L−ピログルタミン酸をさらに含み、式(4A)の化合物およびL−ピログルタミン酸を1:1の化学量論比で含む共結晶である、請求項10に記載の結晶。
【請求項14】
L−ピログルタミン酸をさらに含み、
b)2θ値(CuKα放射線、1.54056Åの波長)6.4±0.2を含む粉末X線回折パターン、および
d)500MHz分光計で決定すると、29.5ppmの結晶性アダマンチンに対して、16.5±0.2、158.7±0.2、および181.5±0.2ppmにおいてピーク位置を含む固体13C NMRスペクトル
を有する、請求項10に記載の結晶。
【請求項15】
L−プロリンをさらに含み、
a)C2の空間群および実質的に下記と等しい単位格子パラメータ
a=32.8399(16)Å α=90°
b=7.2457(4)Å β=101.268(5)°
c=11.8023(6)Å γ=90°、または
b)2θ値(CuKα放射線、1.54056Åの波長)7.6±0.2、12.1±0.2、20.3±0.2および28.8±0.2を含む粉末X線回折パターン
の1つまたは複数を有する、請求項10に記載の結晶。
【請求項16】
(i)請求項1から9のいずれかに記載の化合物、または請求項10から15のいずれかに記載の結晶と、(ii)薬学的に許容できる添加剤、賦形剤、または担体とを含む医薬組成物。
【請求項17】
動物において肥満症および肥満症に関連する障害を治療する方法であって、このような治療を必要としている動物に、治療有効量の請求項1から9のいずれか一項に記載の化合物、または治療有効量の請求項10から15のいずれか一項に記載の結晶を投与するステップを含む、方法。
【請求項18】
動物において2型糖尿病および糖尿病に関連する障害を治療、または進行もしくは発症を遅延させる方法であって、このような治療を必要としている動物に、治療有効量の請求項1から9のいずれか一項に記載の化合物、または治療有効量の請求項10から15のいずれか一項に記載の結晶を投与するステップを含む、方法。
【請求項19】
動物において肥満症および肥満症に関連する障害を治療する方法であって、このような治療を必要としている動物に、請求項16に記載の医薬組成物を投与するステップを含む、方法。
【請求項20】
動物において2型糖尿病および糖尿病に関連する障害を治療、または進行もしくは発症を遅延させる方法であって、このような治療を必要としている動物に、請求項16に記載の医薬組成物を投与するステップを含む、方法。
【請求項1】
式(A)または式(B)の化合物
【化1】
[式中、
R1は、H、(C1〜C4)アルキル、(C1〜C4)アルコキシ、Cl、F、シアノ、フルオロ置換(C1〜C2)アルキル、(C1〜C4)アルキル−SO2−、または(C3〜C6)シクロアルキルであり、
R2は、(C1〜C4)アルキル、(C1〜C4)アルコキシ、(C2〜C4)アルキニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、Cl、F、シアノ、フルオロ置換(C1〜C2)アルキル、(C1〜C4)アルキル−SO2−、(C3〜C6)シクロアルキル、または各々独立にN、O、もしくはSから選択される1個もしくは2個のヘテロ原子を有する(C5〜C6)複素環である]。
【請求項2】
式(A)の化合物である、請求項1に記載の化合物。
【請求項3】
R1が、H、メチル、エチル、プロピル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、シクロプロピル、またはシクロブチルであり、
R2が、メチル、エチル、プロピル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである、請求項1または2に記載の化合物。
【請求項4】
R1が、H、メチル、エチル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、またはシクロプロピルであり、
R2が、メチル、エチル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである、請求項3に記載の化合物。
【請求項5】
R1が、H、メチル、エチル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、またはシクロプロピルであり、
R2が、メチル、エチル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである、請求項4に記載の化合物。
【請求項6】
R1が、メチル、エチル、F、Cl、シアノ、CF3、またはシクロプロピルであり、
R2が、メトキシ、またはエトキシである、請求項5に記載の化合物。
【請求項7】
(1S,2S,3S,4R,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
2−(4−メトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)ベンゾニトリル;
2−(4−エトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)ベンゾニトリル;
(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;および
(1S,2S,3S,4R,5S)−5−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオールからなる群から選択される化合物。
【請求項8】
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオールである化合物。
【請求項9】
(1S,2S,3S,4S,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4S,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;
(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール;および
(1S,2S,3S,4S,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオールからなる群から選択される化合物。
【請求項10】
式(4A)を有する化合物を含む結晶。
【化2】
【請求項11】
L−プロリンまたはL−ピログルタミン酸をさらに含む、請求項10に記載の結晶。
【請求項12】
L−ピログルタミン酸をさらに含み、
a)P2(1)2(1)2(1)の空間群および実質的に下記と等しい単位格子パラメータ
a=7.4907(10)Å α=90°
b=12.8626(15)Å β=90°
c=28.029(4)Å γ=90°、
b)2θ値(CuKα放射線、1.54056Åの波長)6.4±0.2、16.7±0.2、17.4±0.2および21.1±0.2を含む粉末X線回折パターン、または
c)500MHz分光計で決定すると、29.5ppmの結晶性アダマンチンに対して、16.5±0.2、131.1±0.2、158.7±0.2、および181.5±0.2ppmにおいてピーク位置を含む固体13C NMRスペクトル
の1つまたは複数を有する、請求項10に記載の結晶。
【請求項13】
L−ピログルタミン酸をさらに含み、式(4A)の化合物およびL−ピログルタミン酸を1:1の化学量論比で含む共結晶である、請求項10に記載の結晶。
【請求項14】
L−ピログルタミン酸をさらに含み、
b)2θ値(CuKα放射線、1.54056Åの波長)6.4±0.2を含む粉末X線回折パターン、および
d)500MHz分光計で決定すると、29.5ppmの結晶性アダマンチンに対して、16.5±0.2、158.7±0.2、および181.5±0.2ppmにおいてピーク位置を含む固体13C NMRスペクトル
を有する、請求項10に記載の結晶。
【請求項15】
L−プロリンをさらに含み、
a)C2の空間群および実質的に下記と等しい単位格子パラメータ
a=32.8399(16)Å α=90°
b=7.2457(4)Å β=101.268(5)°
c=11.8023(6)Å γ=90°、または
b)2θ値(CuKα放射線、1.54056Åの波長)7.6±0.2、12.1±0.2、20.3±0.2および28.8±0.2を含む粉末X線回折パターン
の1つまたは複数を有する、請求項10に記載の結晶。
【請求項16】
(i)請求項1から9のいずれかに記載の化合物、または請求項10から15のいずれかに記載の結晶と、(ii)薬学的に許容できる添加剤、賦形剤、または担体とを含む医薬組成物。
【請求項17】
動物において肥満症および肥満症に関連する障害を治療する方法であって、このような治療を必要としている動物に、治療有効量の請求項1から9のいずれか一項に記載の化合物、または治療有効量の請求項10から15のいずれか一項に記載の結晶を投与するステップを含む、方法。
【請求項18】
動物において2型糖尿病および糖尿病に関連する障害を治療、または進行もしくは発症を遅延させる方法であって、このような治療を必要としている動物に、治療有効量の請求項1から9のいずれか一項に記載の化合物、または治療有効量の請求項10から15のいずれか一項に記載の結晶を投与するステップを含む、方法。
【請求項19】
動物において肥満症および肥満症に関連する障害を治療する方法であって、このような治療を必要としている動物に、請求項16に記載の医薬組成物を投与するステップを含む、方法。
【請求項20】
動物において2型糖尿病および糖尿病に関連する障害を治療、または進行もしくは発症を遅延させる方法であって、このような治療を必要としている動物に、請求項16に記載の医薬組成物を投与するステップを含む、方法。
【図1】SHELXTLプロッティングパッケージを使用してプロットした実施例8Aの化合物についての精密な結晶構造を表す図である。
【図2】SHELXTLプロッティングパッケージを使用してプロットした実施例9Aの化合物についての精密な結晶構造を表す図である。
【図3】実施例22:実施例4Aの化合物とL−プロリンとの実施例18の共結晶について観察された粉末X線回折パターンを表す図である。
【図4】実施例22:実施例4Aの化合物とL−ピログルタミン酸との実施例20の共結晶について観察された粉末X線回折パターンを表す図である。
【図5】実施例23:実施例4Aの化合物とL−プロリンとの実施例18の共結晶について観察された示差走査熱量測定サーモグラムを表す図である。
【図6】実施例23:実施例4Aの化合物とL−ピログルタミン酸との実施例20の共結晶について観察された示差走査熱量測定サーモグラムを表す図である。
【図7】SHELXTLプロッティングパッケージを使用してプロットした実施例24:実施例4Aの化合物とL−プロリンとの共結晶についての精密な結晶構造を表す図である。
【図8】SHELXTLプロッティングパッケージを使用してプロットした実施例25:実施例4Aの化合物とL−ピログルタミン酸との共結晶についての精密な結晶構造を表す図である。
【図9】実施例26:実施例4Aの化合物とL−ピログルタミン酸との共結晶についての観察される13C固体核磁気共鳴スペクトルを表す図である。アスタリスクでマークされているピークは、スピニングサイドバンドである。
【発明を実施するための形態】
【0019】
下記の本発明の例示的実施形態の詳細な説明およびその中に含まれている実施例を参照することによって、本発明はさらにより容易に理解し得る。
【0020】
本発明の化合物、組成物および方法を開示および記載する前に、本発明は、当然ながら変化し得る作製の特定の合成法に限定されないことを理解すべきである。本明細書において使用される用語は、特定の実施形態を記載する目的のためにすぎず、限定的なものではないこともまた理解すべきである。複数形および単数形は、数が示されている場合以外は、交換可能なものとして扱うべきである。
【0021】
本明細書において使用する場合、「アルキル」という用語は、一般式CnH2n+1の炭化水素基を意味する。アルカン基は、直鎖状または分岐状であってよい。例えば、「(C1〜C6)アルキル」という用語は、1〜6個の炭素原子を含有する一価の直鎖状または分岐状の脂肪族基(例えば、メチル、エチル、n−プロピル、i−プロピル、n−ブチル、i−ブチル、s−ブチル、t−ブチル、n−ペンチル、1−メチルブチル、2−メチルブチル、3−メチルブチル、ネオペンチル、3,3−ジメチルプロピル、ヘキシル、2−メチルペンチルなど)を意味する。同様に、アルコキシ、アシル(例えば、アルカノイル)、アルキルアミノ、ジアルキルアミノ、アルキルスルホニル、およびアルキルチオ基のアルキル部位(すなわち、アルキル部分)は、上記と同じ定義を有する。「置換されていてもよい」と示されているとき、アルカン基またはアルキル部分は、非置換であるか、または「置換されている」についての定義において下記で一覧表示した置換基の群から独立に選択される1個または複数の置換基(ペルクロロまたはペルフルオロアルキルなどのハロゲン置換基の場合を除いて、一般に1〜3個の置換基)で置換されていてもよい。「ハロ置換アルキル」とは、1個または複数のハロゲン原子で置換されているアルキル基(例えば、フルオロメチル、ジフルオロメチル、トリフルオロメチル、ペルフルオロエチル、1,1−ジフルオロエチルなど)を意味する。
【0022】
「シクロアルキル」という用語は、完全に水素化され、かつ単環式環、二環式環またはスピロ環として存在し得る非芳香環を意味する。他に特定しない限り、炭素環は、一般に3〜8員環である。例えば、シクロアルキルには、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘキセニル、ノルボルニル(ビシクロ[2.2.1]ヘプチル)、ビシクロ[2.2.2]オクチルなどなどの基が含まれる。
【0023】
「複素環」という用語は、完全に水素化され、かつ単環式環、二環式環またはらせん環として存在し得る非芳香環を意味する。他に特定しない限り、複素環は一般に、硫黄、酸素および/または窒素から独立に選択される1〜3個のヘテロ原子(好ましくは、1個または2個のヘテロ原子)を含有する3〜6員環である。複素環には、エポキシ、アジリジニル、テトラヒドロフラニル、ピロリジニル、N−メチルピロリジニル、ピペリジニル、ピペラジニル、ピラゾリジニル、4H−ピラニル、モルホリノ、チオモルホリノ、テトラヒドロチエニル、テトラヒドロチエニル1,1−ジオキシドなどの基が含まれる。
【0024】
「治療有効量」という語句は、(i)特定の疾患、状態、または障害を治療し、(ii)特定の疾患、状態、または障害の1つまたは複数の症状を軽減、緩和、もしくは解消し、または(iii)本明細書に記載されている特定の疾患、状態、または障害の1つまたは複数の症状の発症を予防もしくは遅延させる本発明の化合物の量を意味する。
【0025】
「動物」という用語は、ヒト(男性または女性)、伴侶動物(例えば、イヌ、ネコおよびウマ)、食料源動物、動物園動物、海生動物、鳥および他の類似の動物種を意味する。「食用動物」とは、ウシ、ブタ、ヒツジおよび家禽などの食料源動物を意味する。
【0026】
「薬学的に許容できる」という語句は、物質または組成物が、製剤を構成する他の成分、および/またはそれによって治療を受ける哺乳動物と、化学的および/または毒物学的に適合性でなくてはならないことを示す。
【0027】
「治療すること」、「治療する」、または「治療」という用語は、防止的、すなわち、予防的および姑息的治療の両方を包含する。
【0028】
「調節される」または「調節すること」、または「調節する」という用語は、本明細書において使用する場合、他に示さない限り、本発明の化合物によってナトリウム−グルコース輸送体(特に、SGLT2)を阻害し、それによって輸送体によるグルコース輸送を部分的または完全に防止することを意味する。
【0029】
「本発明の化合物」という用語は、(特に別に特定されなければ)式(A)、式(B)の化合物、ならびに全ての純粋および混合立体異性体(ジアステレオマーおよびエナンチオマーを含めて)、互変異性体および同位体標識化合物を意味する。本発明の化合物の水和物および溶媒和物は、化合物が各々水または溶媒と会合している本発明の組成物と考えられる。化合物はまた、1種または複数の結晶状態で、すなわち、共結晶、多形として存在することもあり、またはアモルファス固体として存在することがある。全てのこのような形態は、特許請求の範囲に包含されている。
【0030】
一実施形態において、R1は、H、メチル、エチル、プロピル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、シクロプロピル、またはシクロブチルである。別の実施形態において、R1は、H、メチル、エチル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、またはシクロプロピルである。さらなる実施形態において、R1は、H、メチル、エチル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、またはシクロプロピルである。さらなる実施形態において、R1は、メチル、エチル、F、Cl、シアノ、CF3、またはシクロプロピルである。
【0031】
一実施形態において、R2は、メチル、エチル、プロピル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである。別の実施形態において、R2は、メチル、エチル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである。さらなる実施形態において、R2は、メチル、エチル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである。さらなる実施形態において、R2は、メトキシまたはエトキシである。
【0032】
一実施形態において、結晶は、化合物4Aと、L−プロリンまたはL−ピログルタミン酸とを含む。
【0033】
さらなる実施形態において、結晶は、
a)P2(1)2(1)2(1)の空間群および実質的に下記と等しい単位格子パラメータ
a=7.4907(10)Å α=90°
b=12.8626(15)Å β=90°
c=28.029(4)Å γ=90°、
b)2θ値(CuKα放射線、1.54056Åの波長)6.4±0.2、16.7±0.2、17.4±0.2および21.1±0.2を含む粉末X線回折パターン、
c)500MHz分光計で決定すると、29.5ppmの結晶性アダマンチンに対して、16.5±0.2、131.1±0.2、158.7±0.2、および181.5±0.2ppmにおいてピーク位置を有する固体13C NMRスペクトル、または
d)約142.5±2℃の吸熱を有する示差走査熱量測定サーモグラム
の1つまたは複数を有する。
【0034】
さらなる実施形態において、結晶は、式(4A)の化合物およびL−ピログルタミン酸を1:1の化学量論比で含む共結晶である。
【0035】
本発明の化合物は、特に本明細書に含有されている記載に照らして、化学技術分野において周知のものと類似のプロセスを含む合成経路によって合成することができる。出発物質は一般に、Aldrich Chemicals(Milwaukee、WI)などの商業的供給源から入手可能であり、または当業者には周知の方法(例えば、Louis F.FieserおよびMary Fieser、Reagents for Organic Synthesis、第1〜19巻、Wiley、New York(1967〜1999編)、もしくはBeilsteins Handbuch der organischen Chemie、4版、Aufl.編、Springer−Verlag、Berlin(追補を含めて)(Beilsteinオンラインデータベースによっても入手可能である)に一般に記載されている方法によって調製する)を使用して容易に調製される。
【0036】
例示の目的のために、下記に示す反応スキームは、本発明の化合物および重要中間体を合成するための可能な経路を提供する。個々の反応段階のより詳細な記載については、下記の実施例の項を参照されたい。他の合成経路を使用して、本発明の化合物を合成し得ることを当業者であれば理解するであろう。特定の出発物質および試薬をスキームにおいて示し、下記で議論するが、他の出発物質および試薬を容易に代用して、種々の誘導体および/または反応条件を提供することができる。さらに、下記の方法によって調製した化合物の多くは、当業者には周知の従来の化学作用を使用してこの開示に照らしさらに修飾することができる。
【0037】
本発明の化合物の調製において、中間体の遠隔官能基の保護が必要なことがある。このような保護の必要性は、遠隔官能基の性質および調製法の条件によって変化するであろう。「ヒドロキシ保護基」は、ヒドロキシ官能基を遮断または保護するヒドロキシ基の置換基を意味する。適切なヒドロキシル保護基(O−Pg)には、例えば、アリル、アセチル(Ac)、シリル(トリメチルシリ(TMS)またはtert−ブチルジメチルシリル(TBS)など)、ベンジル(Bn)、パラ−メトキシベンジル(PMB)、トリチル(Tr)、パラ−ブロモベンゾイル、パラ−ニトロベンゾイルなど(1,3−ジオールの保護のためにはベンジリデン)が含まれる。このような保護の必要性は、当業者に容易に決定される。保護基およびそれらの使用の概要については、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley & Sons、New York、1991を参照されたい。
【0038】
スキーム1は、本発明の化合物を提供するために使用することができる一般手順を略述する。
【0039】
【化3】

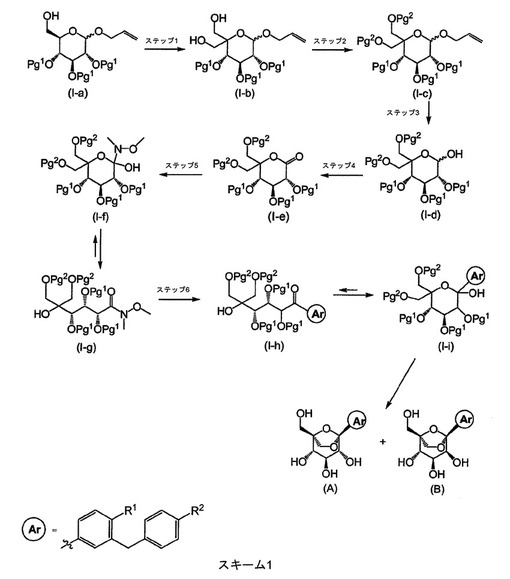
【0040】
アリル2,3,4−トリ−O−ベンジル−D−グルコピラノシド(I−a、Pg1はベンジル基である)は、Shinya Hanashimaら、Bioorganic & Medicinal Chemistry、9、367(2001);Patricia A.Gentら、Journal of the Chemical Society、Perkin1、1835(1974);Hans Peter Wessel、Journal of Carbohydrate Chemistry、7、263、(1988);またはYoko Yuasaら、Organic Process Research & Development、8、405〜407(2004)によって記載された手順によって調製することができる。スキーム1のステップ1において、ヒドロキシメチレン基は、スワーン酸化、続いてアルカリ金属水酸化物(例えば、水酸化ナトリウム)の存在下でホルムアルデヒドによる処理によって、グリコシド上に導入することができる。これはアルドール−カニッツァーロ反応と称される。スワーン酸化は、Kanji OmuraおよびDaniel SwernによってTetrahedron、34、1651(1978)に記載されている。当業者には既知のこの方法の変更形態もまた使用し得る。例えば、Ozanne,A.ら、Organic Letters、5、2903(2003)によって記載された安定化した2−ヨードキシ安息香酸などの他のオキシダント、および当業者が既知の他のオキシダントもまた使用することができる。アルドールカニッツァーロの連続した処理は、Robert Schaffer、Journal of The American Chemical Society、81、5452(1959)およびAmigues,E.J.ら、Tetrahedron、63、10042(2007)によって記載されている。
【0041】
スキーム1のステップ2において、保護基(Pg2)は、中間体(I−b)を所望の特定の保護基のための適当な試薬および手順で処理することによって付加することができる。例えば、p−メトキシベンジル(PMB)基は、水素化ナトリウム、水素化カリウム、カリウムtert−ブトキシドの存在下で、テトラヒドロフラン、1,2−ジメトキシエタンまたはN,N−ジメチルホルムアミド(DMF)などの溶媒中、中間体(I−b)をp−メトキシベンジルブロミドまたはp−メトキシベンジルクロリドで処理することによって導入することができる。触媒量の酸(例えば、トリフルオロメタンスルホン酸、メタンスルホン酸、またはカンファースルホン酸)の存在下で、ジクロロメタン、ヘプタンまたはヘキサンなどの溶媒中、パラ−メトキシベンジルトリクロロアセトイミデートを伴う条件もまた使用することができる。ベンジル(Bn)基は、水素化ナトリウム、水素化カリウム、カリウムtert−ブトキシドの存在下で、テトラヒドロフラン、1,2−ジメトキシエタンまたはN,N−ジメチルホルムアミドなどの溶媒中、中間体(I−b)を臭化ベンジルまたは塩化ベンジルで処理することによって導入することができる。触媒量の酸(例えば、トリフルオロメタンスルホン酸、メタンスルホン酸、またはカンファースルホン酸)の存在下で、ジクロロメタン、ヘプタンまたはヘキサンなどの溶媒中、ベンジルトリクロロアセトイミデートを伴う条件もまた使用することができる。
【0042】
スキーム1のステップ3において、アリル保護基を除去して(例えば、メタノール中の塩化パラジウムによる処理によって、ジクロロメタンなどの共溶媒もまた使用し得る、当業者には既知の他の条件もまた使用することができる、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley & Sons、New York、1991を参照されたい)、ラクトール(I−d)が形成される。
【0043】
スキーム1のステップ4において、次いで保護されていないヒドロキシル基のオキソ基への酸化(例えば、スワーン酸化)によって、ラクトン(I−e)が形成される。
【0044】
スキーム1のステップ5において、ラクトン(I−e)は、N,O−ジメチルヒドロキシルアミン塩酸塩と反応して、相当するワインレブアミドが形成され、これは閉環/開環形態(I−f/I−g)の平衡状態で存在し得る。「ワインレブアミド」(I−g)は、当業者には周知の手順を使用して作製することができる。Nahm,S.およびS.M.Weinreb、Tetrahedron Letters、22(39)、3815〜1818(1981)を参照されたい。例えば、中間体(I−f/I−g)は、市販のN,O−ジメチルヒドロキシルアミン塩酸塩および活性化剤(例えば、トリメチルアルミニウム)から調製することができる。
【0045】
スキーム1のステップ6において、アリールベンジル基(Ar)は、約−78℃〜約20℃の範囲の温度でテトラヒドロフラン(THF)中、所望の有機金属反応剤(例えば、有機リチウム化合物(ArLi)または有機マグネシウム化合物(ArMgX))を使用して導入され、続いて(プロトン性条件に静置すると)相当するラクトール(I−i)に加水分解され、これは相当するケトン(I−h)と平衡状態で存在し得る。(A)および(B)に見出される架橋ケタールモチーフは、用いる保護基に適当な試薬を使用して保護基(Pg2)を除去することによって調製することができる。例えば、PMB保護基は、アニソールおよびジクロロメタン(DCM)の存在下で約0℃〜約23℃(室温)にてトリフルオロ酢酸で処理することによって除去することができる。次いで、残った保護基(Pg1)は、特定の保護基に適当な化学作用を使用して除去することができる。例えば、ベンジル保護基は、パラジウム(Pd黒)の存在下で、プロトン性溶媒(例えば、エタノール/THF)中、約室温にてギ酸で処理することによって除去され、最終生成物(A)および(B)を生成し得る。R1がCNであるとき、約−78℃〜約室温の範囲の温度で、ジクロロメタンまたは1,2−ジクロロエタンなどの溶媒中、三塩化ホウ素のようなルイス酸をまた使用して、ベンジル保護基および/またはパラ−メトキシベンジル保護基を除去し得る。
【0046】
中間体(I−i)または生成物(A)もしくは(B)において、R1がCNであり、R2が(C1〜C4)アルコキシであるとき、三塩化ホウ素または三臭化ホウ素などのルイス酸による処理によって、相当するフェノールへの部分的から完全な脱アルキル化が起こり、相当する化合物(A)または(B)(R1はCNであり、R2はOHである)をもたらし得る。これが起こった場合、(C1〜C4)アルコキシ基を、穏やかな塩基性条件(例えば、アセトン中の炭酸カリウム)下で、約室温〜摂氏約56度の範囲の温度で、(C1〜C4)ヨウ化アルキルを使用した選択的アルキル化によって再導入し得る。
【0047】
R1および/またはR2が(C1〜C4)アルキル−SO2−であるとき、有機金属の添加ステップ6(スキーム1)は、相当する(C1〜C4)アルキル−S−含有有機金属反応剤で行われることを当業者は理解する。次いで、チオ−アルキルを、当業者に既知の従来の方法を使用して後の段階で相当するスルホンに酸化させる。
【0048】
本発明の化合物は、任意の適切な方法を使用して共結晶として調製し得る。このような共結晶を調製するための代表的なスキームを、スキーム2に記載する。
【0049】
【化4】
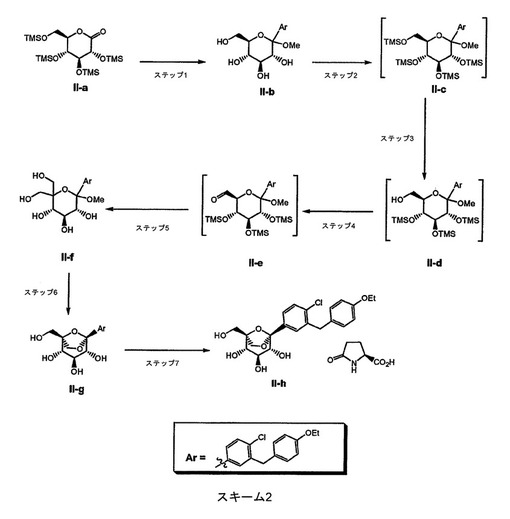
【0050】
スキーム2(Meはメチルであり、Etはエチルである)のステップ1において、1−(5−ブロモ−2−クロロベンジル)−4−エトキシベンゼンを3:1のトルエン:テトラヒドロフランに溶解し、続いて得られた溶液を−70℃未満に冷却する。この溶液に、ヘキシルリチウムを添加し、その間に反応物を−65℃以下に維持し、続いて1時間撹拌する。(3R,4S,5R,6R)−3,4,5−トリス(トリメチルシリルオキシ)−6−((トリメチルシリルオキシ)メチル)−テトラヒドロピラン−2−オン(II−a)をトルエンに溶解し、得られた溶液を−15℃に冷却する。次いで、この溶液を−70℃のアリールリチウム溶液に添加し、続いて1時間撹拌する。次いで、メタンスルホン酸のメタノール溶液を添加し、続いて室温に温め、16〜24時間撹拌する。α−アノマーのレベルが3%以下であるとき、反応は完了していると見なす。次いで、5Mの水酸化ナトリウム水溶液を添加することによって反応物を塩基性化する。得られた塩を濾過して除去し、続いて粗生成物溶液を濃縮する。2−メチルテトラヒドロフランを共溶媒として添加し、有機相を水で2度抽出する。次いで、有機相をトルエン中4容量に濃縮する。次いで、この濃縮物を5:1のヘプタン:トルエン溶液に添加し、沈殿物を形成させる。固体を集め、真空下で乾燥させ、固体を得る。
【0051】
スキーム2のステップ2において、塩化メチレン中の(II−b)にイミダゾールを添加し、続いて0℃に冷却し、次いでトリメチルシリルクロリドを添加し、ペルシリル化生成物を得る。反応物を室温に温め、水を添加することによってクエンチし、有機相を水で洗浄する。(II−c)のこの粗塩化メチレン溶液を硫酸ナトリウム上で乾燥させ、次いで粗製物を次のステップにかける。
【0052】
スキーム2のステップ3において、塩化メチレン中の(II−c)の粗溶液を低容量に濃縮し、次いで溶媒をメタノールに交換する。(II−c)のメタノール溶液を0℃に冷却し、次いで1mol%の炭酸カリウムをメタノール中の溶液として添加し、続いて5時間撹拌する。次いで、メタノール中の1mol%酢酸を添加することによって反応物をクエンチし、続いて室温に温め、溶媒を酢酸エチルに交換し、次いで少量の無機固体の濾過を行う。(II−d)の粗酢酸エチル溶液を、次のステップに直接かける。
【0053】
スキーム2のステップ4において、(II−d)の粗溶液を低容量に濃縮し、次いで塩化メチレンおよびジメチルスルホキシドで希釈する。トリエチルアミンを添加し、続いて10℃に冷却し、次いで三酸化硫黄ピリジン錯体を10分間隔にて3分割で固体として添加する。反応物を10℃でさらに3時間撹拌し、その後水でクエンチし、室温に温める。相を分離し、続いて塩化メチレン層を塩化アンモニウム水溶液で洗浄する。(II−e)の粗塩化メチレン溶液を、次のステップに直接かける。
【0054】
スキーム2のステップ5において、(II−e)の粗溶液を低容量に濃縮し、次いで溶媒をエタノールに交換する。30当量のホルムアルデヒド水溶液を添加し、続いて55℃に温める。2当量の三塩基性リン酸カリウムの水溶液を添加し、続いて55℃で24時間撹拌する。次いで、反応温度を70℃にさらに12時間上げる。反応物を室温に冷却し、tert−ブチルメチルエーテルおよびブラインで希釈する。相を分離し、続いて有機相の溶媒を酢酸エチルへ交換する。酢酸エチル相をブラインで洗浄し、低容量に濃縮する。次いで、粗濃縮物を、5%メタノール、95%トルエンで溶出するシリカゲルフラッシュクロマトグラフィーによって精製する。生成物含有画分を合わせ、低容量に濃縮する。メタノールを添加し、続いて沈殿が起こるまで撹拌する。懸濁液を冷却し、固体を集め、ヘプタンですすぎ、続いて乾燥させる。生成物(II−f)を固体として単離する。
【0055】
スキーム2のステップ6において、化合物(II−f)を5容量の塩化メチレンに溶解し、続いて1mol%SiliaBond(登録商標)トシル酸を添加し、18時間室温で撹拌する。酸触媒を濾過して除去し、(II−g)の塩化メチレン溶液を、次のステップである共結晶化手順に直接かける。
【0056】
スキーム2のステップ7において、(II−g)の塩化メチレン溶液を濃縮し、次いで溶媒を2−プロパノールに交換する。水を添加し、続いて55℃に温める。L−ピログルタミン酸の水溶液を添加し、続いて得られた溶液を室温に冷却する。次いで、溶液に種晶添加し、18時間造粒する。冷却後、固体を集め、ヘプタンですすぎ、続いて乾燥させる。生成物(II−h)を固体として単離する。
【0057】
本発明の化合物(A)のための代替合成経路をスキーム3において示し、下記に記載する。
【0058】
【化5】

【0059】
(III−a)(R3は、アルキルまたはフルオロ置換アルキルである(酸素原子に隣接した炭素を除いて))の合成は、スキーム2のステップ1に記載されているのと同様の方法で調製することができる。スキーム3のステップ1において、第一級ヒドロキシル基は、適当な保護基によって選択的に保護される。例えば、トリチル基(Pg3=Tr)は、ピリジンなどの塩基の存在下で、トルエン、テトラヒドロフランまたはジクロロメタンなどの溶媒中、摂氏約0度〜約室温の範囲の温度で、中間体(III−a)をクロロトリフェニルメタンで処理することによって導入することができる。このような保護基および実験条件のさらなる例は当業者に既知であり、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley & Sons、New York、1991に見出すことができる。
【0060】
スキーム3のステップ2において、第二級ヒドロキシル基は、適当な保護基によって保護することができる。例えば、ベンジル基(Pg4はBnである)は、水素化ナトリウム、水素化カリウム、カリウムtert−ブトキシドの存在下で、テトラヒドロフラン、1,2−ジメトキシエタンまたはN,N−ジメチルホルムアミドなどの溶媒中、摂氏約0度〜摂氏約80度の範囲の温度にて、中間体(III−b)を臭化ベンジルまたは塩化ベンジルで処理することによって導入することができる。アセチルまたはベンゾイル基(Pg4=AcまたはBzである)は、トリエチルアミン、N,N−ジイソプロピルエチルアミンまたは4−(ジメチルアミノ)ピリジンなどの塩基の存在下で、テトラヒドロフラン、1,2−ジメトキシエタンまたはジクロロメタンなどの溶媒中、摂氏約0度〜摂氏約80度の範囲の温度にて、中間体(III−b)を塩化アセチル、臭化アセチルまたは無水酢酸または塩化ベンゾイルまたは無水安息香酸で処理することによって導入し得る。
【0061】
スキーム3のステップ3において、第一級ヒドロキシル基が脱保護され、中間体(III−d)がもたらされる。Pg3がTrであるとき、摂氏約−20度〜約室温の範囲の温度にて、パラ−トルエンスルホン酸など酸の存在下、メタノールなどのアルコール性溶媒中で中間体(III−c)を処理し、中間体(III−d)が提供される。クロロホルムのような共溶媒を使用し得る。
【0062】
スキーム3のステップ4において、ヒドロキシメチレン基は、スキーム1(ステップ1)およびスキーム2(ステップ4および5)に既に記載されているものと同様の方法によって導入される。エタノールなどの溶媒中、約室温〜摂氏約70度の範囲の温度にて、アルカリ金属アルコキシドの存在下で、パラホルムアルデヒドのような他のホルムアルデヒド源をまた、このステップにおいて使用することができる。Pg4がBnであるとき、このステップは、中間体(III−e)を提供し、Pg4がAcまたはBzであるとき、このステップは、中間体(III−f)を提供する。
【0063】
スキーム3のステップ5において、中間体(III−e)を、ジクロロメタンなどの溶媒中、摂氏約−10度〜約室温の範囲の温度にて、トリフルオロ酢酸または酸性樹脂などの酸で処理して、中間体(III−g)が生成される。
【0064】
スキーム3のステップ6において、次いで、残った保護基(Pg4)を、特定の保護基に適当な化学作用を使用して除去することができる。例えば、ベンジル保護基は、パラジウム(Pd黒)の存在下で、プロトン性溶媒(例えば、エタノール/THF)中、約室温にてギ酸で処理することによって除去され、最終生成物(A)を生成し得る。
【0065】
スキーム3のステップ7において、中間体(III−f)を、ジクロロメタンなどの溶媒中、摂氏約−10度〜約室温の範囲の温度にて、トリフルオロ酢酸または酸性樹脂などの酸で処理して、最終生成物(A)が生成される。
【0066】
生成物(A)を合成するための別の代替スキームを、スキーム4に示し、下記に記載する。
【0067】
【化6】
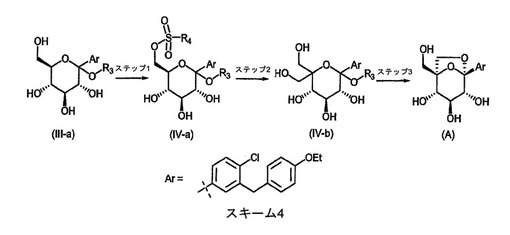
【0068】
スキーム4のステップ1において、中間体(III−a)を、ピリジン、トリエチルアミン、N,N−ジイソプロピルエチルアミンなどの塩基の存在下で、テトラヒドロフラン、2−メチルテトラヒドロフランなどの溶媒中、摂氏約−20度〜約室温の範囲の温度にて、適当なアリールスルホニルクロリドR4SO2Clまたはアリールスルホン酸無水物R4S(O)2OS(O)2R4(式中、R4は、アリールスルホニルクロリド、4−メチル−ベンゼンスルホニルクロリド、4−ニトロ−ベンゼンスルホニルクロリド、4−フルオロ−ベンゼンスルホニルクロリド、2,6−ジクロロ−ベンゼンスルホニルクロリド、4−フルオロ−2−メチル−ベンゼンスルホニルクロリド、および2,4,6−トリクロロ−ベンゼンスルホニルクロリドにおいて、およびアリールスルホン酸無水物、p−トルエンスルホン酸無水物において見出されるものなどの、置換されていてもよいアリール基である)で処理する。臭化亜鉛(II)などのいくつかのルイス酸を、添加物として使用してもよい。
【0069】
スキーム4のステップ2において、中間体(IV−a)をコーンブルム型酸化(Kornblum,N.ら、Journal of The American Chemical Society、81、4113(1959)を参照されたい)に供して、相当する水和物および/またはヘミアセタール形態と平衡状態で存在し得る相当するアルデヒドを生成する。例えば、ピリジン、2,6−ルチジン、2,4,6−コリジン、N,N−ジイソプロピルエチルアミン、4−(ジメチルアミノ)ピリジンなどの塩基の存在下で、ジメチルスルホキシドなどの溶媒中、約室温〜摂氏約150度の範囲の温度で、中間体(IV−a)を処理する。次いで、生成されたアルデヒド中間体を、ステップ1(スキーム1)およびステップ5(スキーム2)について記載したアルドール/カニッツァーロ条件に供して、中間体(IV−b)を生成する。
【0070】
スキーム4のステップ3において、中間体(IV−b)を、ジクロロメタンなどの溶媒中、摂氏約−10度〜約室温の範囲の温度にて、トリフルオロ酢酸または酸性樹脂などの酸で処理して、最終生成物(A)が生成される。
【0071】
R2が(C2〜C4)アルキニルであるとき、このプロセスは、スキーム5(R6は、Hまたは(C1〜C2)アルキルである)を使用して行うことができる。
【0072】
【化7】
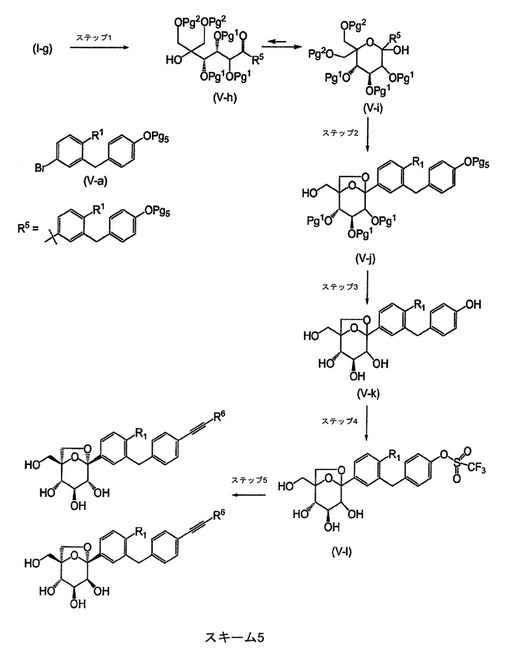
【0073】
中間体(V−i)を提供するスキーム5のステップ1において、有機金属の添加ステップを、(V−a)(Pg5は、ヒドロキシル基のための適切な保護基である)に由来する有機金属反応剤を使用して、スキーム1、ステップ6に記載したものと同様の方法で行う。例えば、Pg5は、tert−ブチルジメチルシリル基(TBS)とすることができる(例えば{4−[(5−ブロモ−2−クロロ−フェニル)−メチル]−フェノキシ}−tert−ブチル−ジメチル−シランの調製については、US2007/0054867を参照されたい)。
【0074】
スキーム5のステップ2において、Pg2=PMBのとき、中間体(V−i)を、アニソールの存在下で、ジクロロメタンなどの溶媒中、摂氏約−10度〜約室温の範囲の温度で、トリフルオロ酢酸、メタンスルホン酸または酸性樹脂などの酸で処理して、中間体(V−j)を生成する。
【0075】
スキーム5のステップ3において、保護基(Pg5)および(Pg1)を除去して、(V−k)を提供することができる。典型的には、(Pg5)はTBSであり、Pg1はBnである。この状況において、(V−j)を、1)テトラヒドロフランまたは2−メチルテトラヒドロフランのような溶媒中、摂氏0度〜摂氏約40度の範囲の温度にて、フッ化テトラブチルアンモニウム、および2)パラジウム(Pd黒)の存在下で、プロトン性溶媒(例えば、エタノール/THF)中、約室温にてギ酸によって連続的に処理することによって、保護基を除去する。この連続した処理において、2つの反応の順序は交換可能である。
【0076】
スキーム5のステップ4において、中間体(V−k)を、トリエチルアミンまたは4−ジメチアミノピリジンなどの塩基の存在下で、ジクロロメタンまたは1,2−ジクロロエタンなどの溶媒中、摂氏0度〜摂氏約40度の範囲の温度にて、N,N−ビス−(トリフルオロメタンスルホニル)−アニリンで処理して、中間体(V−l)を生成する。
【0077】
スキーム5のステップ5において、中間体(V−l)を、薗頭型反応に供する(Sonogashira,K.、Coupling Reactions Between sp2 and sp Carbon Centers、Comprehensive Organic Synthesis(Trost,B.M.、Fleming,I.編)、3、521〜549、(Pergamon、Oxford、1991)を参照されたい)。例えば、(V−l)は、ヨウ化銅(I)、ビス−(トリフェニルホスフィン)−パラジウムジクロリドまたはテトラキス(トリフェニルホスフィン)パラジウム(0)などの触媒の存在下で、トリエチルアミンまたはN,N−ジイソプロピルエチルアミンなどの塩基の存在下で、N,N−ジメチルホルムアミドなどの溶媒中、約室温〜摂氏約120度の範囲の温度にて、適当な末端アルキンHCCR6で処理して、所望の生成物(A)および(B)を生成する。R6がHであるとき、トリメチルシリルアセチレンを使用することがより好都合である。この場合、上記の反応から得られた粗材料を、MeOHなどのアルコール性溶媒中、約室温にて炭酸カリウムなどの塩基で処理して、当業者には既知の古典的な後処理の後、所望の生成物(A)および(B)(R2は、−CCHである)が生成される。
【0078】
スキーム1〜5において上記で記載した化学的手法は、中間体(V−k)を得る様々な方法を表すことを当業者であれば理解するであろう。そしてまた、特にR1がClであるとき、(V−k)は、古典的な条件下で一般に好まれるアルキル化剤で処理して、フェノール基を選択的にアルキル化して、(A)(ならびにスキーム1および5において(B))(R2は(C1〜C4)アルコキシである)を生成することができる。
【0079】
本発明の化合物は、不斉中心またはキラル中心を含有し、したがって、異なる立体異性体の形態で存在する。他に特定しない限り、本発明の化合物の全ての立体異性体の形態、およびラセミ混合物を含めたその混合物は、本発明の一部を形成することが意図される。さらに、本発明は、全ての幾何異性体および位置異性体を包含する。例えば、本発明の化合物が二重結合または縮合環を組み込む場合、cis−およびtrans−形態の両方、ならびに混合物は、本発明の範囲内に包含される。
【0080】
ジアステレオマー混合物は、当業者には周知の方法(クロマトグラフィーおよび/または分別結晶、蒸留、昇華など)によるそれらの物理的化学的差異に基づいて、それらの個々のジアステレオマーに分離することができる。エナンチオマーは、適当な光学活性な化合物(例えば、キラルアルコールまたはモーシェル酸クロリドなどのキラル補助基)との反応によってエナンチオマー混合物をジアステレオマー混合物に変換し、ジアステレオマーを分離し、個々のジアステレオマーを相当する純粋なエナンチオマーに変換(例えば、加水分解)することによって分離することができる。また、本発明の化合物のいくつかは、アトロプ異性体(例えば、置換ビアリール)である場合があり、本発明の一部と考えられる。エナンチオマーはまた、キラルHPLC(高圧液体クロマトグラフィー)カラムを使用することによって分離することができる。
【0081】
本発明の中間体および化合物は異なる互変異性型で存在する場合があることもまた考えられ、全てのこのような形態は本発明の範囲内に包含される。「互変異性体」または「互変異性型」という用語は、低いエネルギー障壁によって相互変換可能な異なるエネルギーの構造異性体を意味する。例えば、プロトン互変異性体(プロトン性互変異性体としてもまた既知である)には、プロトンの移動による相互変換(ケト−エノールおよびイミン−エナミン異性化など)が含まれる。プロトン互変異性体の具体例は、プロトンが2個の環窒素の間を移動するイミダゾール部分である。原子価互変異性体には、結合電子のいくつかの再編成による相互変換が含まれる。いくつかの中間体(および/または中間体の混合物)の閉環形態と開環形態との間の平衡状態は、当業者に既知のアルドースが関与する変旋光の過程を連想させる。
【0082】
本発明はまた、1個または複数の原子が、天然に通常見出される原子質量または質量数と異なる原子質量または質量数を有する原子によって置き換えられているという事実を別にすれば、本明細書において記載したものと同一の同位体標識された本発明の化合物を包含する。本発明の化合物中に組み込むことができる同位体の例には、水素、炭素、窒素、酸素、リン、硫黄、フッ素、ヨウ素、および塩素の同位体(各々、2H、3H、11C、13C、14C、13N、15N、15O、17O、18O、31P、32P、35S、18F、123I、125Iおよび36Clなど)が含まれる。
【0083】
特定の同位体標識された本発明の化合物(例えば、3Hおよび14Cで標識されたもの)は、化合物および/または基質組織分布アッセイにおいて有用である。それらの調製の容易さおよび検出能のために、トリチウム標識(すなわち、3H)および炭素−14(すなわち、14C)同位体が特に好ましい。さらに、重水素(すなわち、2H)などのより重い同位体による置換によって、より高い代謝安定性からもたらされる特定の治療上の利点(例えば、インビボ半減期の増加または投与必要量の減少)が可能となることがあり、したがって、ある状況において好ましいことがある。15O、13N、11C、および18Fなどのポジトロン放出同位体は、基質占有率を調査するためのポジトロン放出断層撮影(PET)研究のために有用である。同位体標識された本発明の化合物は一般に、同位体標識されていない試薬を同位体標識された試薬で置換することによって、スキームおよび/または本明細書における下記の実施例において開示されているものと類似の手順に従うことによって調製することができる。
【0084】
本発明の化合物は、ナトリウム−グルコース輸送体(特に、SGLT2)の阻害によって調節される疾患、状態および/または障害を治療するのに有用である。したがって、本発明の別の実施形態は、治療有効量の本発明の化合物、および薬学的に許容できる添加剤、賦形剤または担体を含む医薬組成物である。本発明の化合物(組成物およびそこで使用されている方法を含めて)はまた、本明細書に記載されている治療用途のための医薬の製造において使用することができる。
【0085】
典型的な製剤は、本発明の化合物および担体、賦形剤または添加剤を混合することによって調製する。適切な担体、賦形剤および添加剤は当業者には周知であり、炭水化物、ワックス、水溶性および/または膨潤性ポリマー、親水性または疎水性材料、ゼラチン、油、溶媒、水などの材料が含まれる。使用される特定の担体、賦形剤または添加剤は、本発明の化合物を適用する手段および目的によって決まる。溶媒は一般に、哺乳動物に投与されても安全であると当業者に認められる(GRAS)溶媒に基づいて選択される。一般に、安全な溶媒は、水、および水中で可溶性または混和性の他の無毒性溶媒などの無毒性水性溶媒である。適切な水性溶媒には、水、エタノール、プロピレングリコール、ポリエチレングリコール(例えば、PEG400、PEG300)など、およびこれらの混合物が含まれる。製剤にはまた、1種または複数の緩衝液、安定化剤、界面活性剤、湿潤剤、滑沢剤、乳化剤、懸濁化剤、保存剤、抗酸化剤、不透明化剤、流動促進剤、加工助剤、着色剤、甘味剤、香料剤、香味剤、および薬物(すなわち、本発明の化合物またはその医薬組成物)の洗練された外観をもたらし、または医薬品(すなわち、医薬)の製造に役立つ他の既知の添加物が含まれることがある。
【0086】
製剤は、従来の溶解および混合手順を使用して調製し得る。例えば、原薬(すなわち、本発明の化合物、または化合物の安定化した形態(例えば、シクロデキストリン誘導体または他の既知の複合体形成剤との複合体))を、上記の添加剤の1種または複数の存在下で適切な溶媒に溶解する。本発明の化合物は典型的には、医薬品剤形に製剤されて、容易に管理可能な薬物の投与量を提供し、洗練された容易に取り扱うことができる製品を患者に与える。
【0087】
医薬組成物にはまた、式(I)の化合物の溶媒和物および水和物が含まれる。「溶媒和物」という用語は、1個または複数の溶媒分子を有する式(I)によって表される化合物の分子複合体(薬学的に許容できるその塩を含めて)を意味する。このような溶媒分子は、製薬技術において通常使用されるものであり、レシピエントにとって無害であることが知られている(例えば、水、エタノール、エチレングリコールなど)。「水和物」という用語は、溶媒分子が水である複合体を意味する。溶媒和物および/または水和物は好ましくは、結晶形態で存在する。他の溶媒を、より望ましい溶媒和物の調製における中間体溶媒和物として使用してもよい(メタノール、メチルt−ブチルエーテル、酢酸エチル、酢酸メチル、(S)−プロピレングリコール、(R)−プロピレングリコール、1,4−ブチン−ジオールなど)。結晶形態はまた、他の無害な小分子(L−フェニルアラニン、L−プロリン、L−ピログルタミン酸など)との複合体として、共結晶、または溶媒和物もしくは水和物の共結晶性材料としても存在し得る。溶媒和物、水和物および共結晶化合物は、参照により本明細書中に組み込まれているPCT国際公開第WO08/002824号に記載されている手順、または当業者には周知の他の手順を使用して調製することができる。
【0088】
適用するための医薬組成物(または製剤)は、薬物を投与するために使用される方法によって種々の方法でパッケージし得る。一般に、分配するための物品には、その中に適当な形態の医薬製剤を入れた容器が含まれる。適切な容器は当業者には周知であり、ボトル(プラスチックおよびガラス)、小袋、アンプル、ビニール袋、金属製円筒などの材料が含まれる。容器にはまた、パッケージの内容物への不注意による接触を防止するための誤使用防止アセンブリが含まれることがある。さらに、容器には、容器の内容について記載したラベルがその上に貼ってある。ラベルにはまた、適当な警告が含まれることがある。
【0089】
本発明は、動物においてナトリウム−グルコース輸送体の阻害によって調節される疾患、状態および/または障害を治療する方法であって、このような治療を必要としている動物に、治療有効量の本発明の化合物、または有効量の本発明の化合物を含む医薬組成物、および薬学的に許容できる添加剤、賦形剤、または担体を投与するステップを含む、方法をさらに提供する。この方法は、SGLT2の阻害が有益である疾患、状態および/または障害を治療するのに特に有用である。
【0090】
本発明の一態様は、肥満症、および肥満症に関連する障害(例えば、過体重、体重増加、または体重維持)の治療である。
【0091】
肥満症および過体重は一般に肥満度指数(BMI)によって定義され、これは総体脂肪と相関し、疾患の相対危険度を推定する。BMIは、体重(キログラム)を身長(メートル)の2乗で割ることによって計算する(kg/m2)。過体重は典型的には、25〜29.9kg/m2のBMIとして定義され、肥満症は典型的には、30kg/m2のBMIとして定義される。例えば、National Heart,Lung,and Blood Institute、Clinical Guidelines on the Identification,Evaluation,and Treatment of Overweight and Obesity in Adults、The Evidence Report、Washington,DC:U.S. Department of Health and Human Services、NIH出版番号98−4083(1998)を参照されたい。
【0092】
本発明の別の態様は、糖尿病、または糖尿病に関連する障害(1型(インスリン依存性糖尿病、「IDDM」とも称される)および2型(非インスリン依存性糖尿病、「NIDDM」とも称される)糖尿病、耐糖能異常、創傷治癒の遅延、高インスリン血症、脂肪酸の血中濃度の上昇、高脂血症、高トリグリセリド血症、X症候群、高密度リポタンパク質レベルの上昇、インスリン抵抗性、高血糖症、ならびに糖尿病性合併症(アテローム性動脈硬化症、冠動脈心疾患、脳卒中、末梢血管疾患、腎症、高血圧症、ニューロパシー、および網膜症など)を含めた)の治療、または進行もしくは発症を遅延させるためである。
【0093】
本発明のまた別の態様は、代謝症候群などの肥満症の共存疾患の治療である。代謝症候群には、異脂肪血症、高血圧症、インスリン抵抗性、糖尿病(例えば、2型糖尿病)、冠動脈疾患および心不全などの疾患、状態または障害が含まれる。代謝症候群についてのさらなる詳細な情報については、例えば、Zimmet,P.Z.ら、「The Metabolic Syndrome:Perhaps an Etiologic Mystery but Far From a Myth−Where Does the International Diabetes Federation Stand?」、Diabetes & Endocrinology、7(2)、(2005)、およびAlberti,K.G.ら、「The Metabolic Syndrome−A New Worldwide Definition」、Lancet、366、1059〜62(2005)を参照されたい。
【0094】
好ましくは、本発明の化合物の投与は、薬物を含有しないビヒクル対照と比較して、血漿レプチン、C反応性タンパク質(CRP)および/またはコレステロールの減少などの、少なくとも1つの心血管疾患の危険因子の統計的に有意な(p<0.05)低下をもたらす。本発明の化合物の投与はまた、血清グルコースレベルの統計的に有意な(p<0.05)低下をもたらし得る。
【0095】
約100kgの体重を有する正常な成人のヒトについて、体重1キログラム当たり約0.001mg〜約10mg、好ましくは約0.01mg/kg〜約5.0mg/kg、さらに好ましくは約0.01mg/kg〜約1mg/kgの範囲の投与量が典型的には十分である。しかし、一般の投与量範囲におけるいくらかの変動は、治療を受ける対象の年齢および体重、意図される投与経路、投与される特定の化合物などに応じて必要とされることがある。特定の患者についての投与量範囲および最適な投与量を決定することは、本開示の利益を有する当業者の能力の十分に範囲内である。本発明の化合物は、それらの形態もまた当業者に周知である持続放出、制御放出、および遅延放出製剤において使用できることもまた留意される。
【0096】
本発明の化合物はまた、本明細書に記載されている疾患、状態および/または障害の治療のために他の医薬品と併せて使用し得る。したがって、他の医薬品と組み合わせて本発明の化合物を投与することを含む治療方法もまた提供される。本発明の化合物と組み合わせて使用し得る適切な医薬品には、抗肥満剤(食欲抑制剤を含めて)、抗糖尿病剤、抗高血糖剤、脂質低下剤、抗炎症剤および降圧剤が含まれる。
【0097】
適切な抗肥満剤には、カンナビノイド−1(CB−1)アンタゴニスト(リモナバンなど)、11β−ヒドロキシステロイドデヒドロゲナーゼ−1(11β−HSD1型)阻害剤、ステアロイル−CoAデサチュラーゼ−1(SCD−1)阻害剤、MCR−4アゴニスト、コレシストキニン−A(CCK−A)アゴニスト、モノアミン再取込み阻害剤(シブトラミンなど)、交感神経様作用剤、β3アドレナリン作動性アゴニスト、ドーパミンアゴニスト(ブロモクリプチンなど)、メラニン細胞刺激ホルモン類似体、5HT2cアゴニスト、メラニン凝集ホルモンアンタゴニスト、レプチン(OBタンパク質)、レプチン類似体、レプチンアゴニスト、ガラニンアンタゴニスト、リパーゼ阻害剤(テトラヒドロリプスタチン、すなわち、オルリスタットなど)、食欲抑制剤(ボンベシンアゴニストなど)、ニューロペプチド−Yアンタゴニスト(例えば、NPY Y5アンタゴニスト)、PYY3−36(その類似体を含めて)、甲状腺模倣剤、デヒドロエピアンドロステロンまたはその類似体、グルココルチコイドアゴニストまたはアンタゴニスト、オレキシンアンタゴニスト、グルカゴン様ペプチド−1アゴニスト、繊毛様神経栄養因子(Regeneron Pharmaceuticals、Inc.、Tarrytown、NYおよびProcter & Gamble Company、Cincinnati、OHから入手可能なAxokine(商標)など)、ヒトアグーチ関連タンパク質(AGRP)阻害剤、グレリンアンタゴニスト、ヒスタミン3アンタゴニストまたはインバースアゴニスト、ニューロメジンUアゴニスト、MTP/ApoB阻害剤(例えば、ジルロタピドなどの消化管選択的MTP阻害剤)、オピオイドアンタゴニスト、オレキシンアンタゴニストなどが含まれる。
【0098】
本発明の組合せの態様において使用するための好ましい抗肥満剤には、CB−1アンタゴニスト(例えば、リモナバン、タラナバン、スリナバン、オテナバント、SLV319(CAS番号464213−10−3)およびAVE1625(CAS番号358970−97−5))、消化管選択的MTP阻害剤(例えば、ジルロタピド、ミトラタピドおよびインプリタピド、R56918(CAS番号403987)およびCAS番号913541−47−6)、CCKaアゴニスト(例えば、PCT国際公開第WO2005/116034号または米国特許出願公開第2005−0267100A1号に記載されているN−ベンジル−2−[4−(1H−インドール−3−イルメチル)−5−オキソ−1−フェニル−4,5−ジヒドロ−2,3,6,10b−テトラアザ−ベンゾ[e]アズレン−6−イル]−N−イソプロピル−アセトアミド)、5HT2cアゴニスト(例えば、ロルカセリン)、MCR4アゴニスト(例えば、US6,818,658に記載されている化合物)、リパーゼ阻害剤(例えば、セチリスタット)、PYY3−36(本明細書において使用する場合、「PYY3−36」には、ペグ化PYY3−36、例えば、米国特許出願公開第2006/0178501号に記載されているものなどの類似体が含まれる)、オピオイドアンタゴニスト(例えば、ナルトレキソン)、オレオイル−エストロン(CAS番号180003−17−2)、オビネピチド(TM30338)、プラムリンチド(Symlin(登録商標))、テソフェンシン(NS2330)、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エキセナチド(Byetta(登録商標))、AOD−9604(CAS番号221231−10−3)およびシブトラミンが含まれる。好ましくは、本発明の化合物および併用療法は、運動および分別のある食事と併せて投与する。
【0099】
適切な抗糖尿病剤には、アセチル−CoAカルボキシラーゼ−2(ACC−2)阻害剤、ホスホジエステラーゼ(PDE)−10阻害剤、ジアシルグリセロールアシルトランスフェラーゼ(DGAT)1または2阻害剤、スルホニル尿素(例えば、アセトヘキサミド、クロルプロパミド、ジアビネス、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド、グリキドン、グリソラミド、トラザミド、およびトルブタミド)、メグリチニド、α−アミラーゼ阻害剤(例えば、テンダミスタット、トレスタチンおよびAL−3688)、α−グルコシドヒドロラーゼ阻害剤(例えば、アカルボース)、α−グルコシダーゼ阻害剤(例えば、アジポシン、カミグリボース、エミグリタート、ミグリトール、ボグリボース、プラジミシン−Q、およびサルボスタチン)、PPARγアゴニスト(例えば、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン、ピオグリタゾン、ロシグリタゾンおよびトログリタゾン)、PPARα/γアゴニスト(例えば、CLX−0940、GW−1536、GW−1929、GW−2433、KRP−297、L−796449、LR−90、MK−0767およびSB−219994)、ビグアニド(例えば、メトホルミン)、グルカゴン様ペプチド1(GLP−1)アゴニスト(例えば、エキセンディン−3およびエキセンディン−4)、タンパク質チロシンホスファターゼ−1B(PTP−1B)阻害剤(例えば、トロズスクエミン、ヒルチオサール抽出物、およびZhang,S.ら、Drug Discovery Today、12(9/10)、373〜381(2007)によって開示されている化合物)、SIRT−1阻害剤(例えば、レセルバトロール)、ジペプチジルペプチダーゼIV(DPP−IV)阻害剤(例えば、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチン)、インスリン分泌促進物質、脂肪酸酸化阻害剤、A2アンタゴニスト、c−junアミノ末端キナーゼ(JNK)阻害剤、インスリン、インスリン模倣物、グリコーゲンホスホリラーゼ阻害剤、VPAC2受容体アゴニストおよびグルコキナーゼ活性化剤が含まれる。好ましい抗糖尿病剤は、メトホルミンおよびDPP−IV阻害剤(例えば、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチン)である。
【0100】
適切な抗炎症剤には、生殖管/尿路感染症の予防薬および治療薬が含まれる。例示的な薬剤には、クランベリー(すなわち、オオミノツルコケモモ(Vaccinium macrocarpon))およびクランベリー誘導体(クランベリージュース、クランベリー抽出物またはクランベリーのフラボノールなど)が含まれる。クランベリー抽出物には、1種もしくは複数のフラボノール(すなわち、アントシアニンおよびプロアントシアニジン)、または精製されたクランベリーフラボノール化合物(ミリセチン−3−β−キシロピラノシド、ケルセチン−3−β−グルコシド、ケルセチン−3−α−アラビノピラノシド、3’−メトキシケルセチン−3−α−キシロピラノシド、ケルセチン−3−O−(6’’−p−クマロイル)−β−ガラクトシド、ケルセチン−3−O−(6’’−ベンゾイル)−β−ガラクトシド、および/もしくはケルセチン−3−α−アラビノフラノシドを含めた)が含まれ得る。
【0101】
本発明の実施形態を、下記の実施例によって例示する。しかし、他のその変形形態が当業者には既知であり、または本開示に照らし合わせて明らかであるため、本発明の実施形態は、これらの実施例の特定の詳細に限定されないことが理解されるべきである。
【実施例】
【0102】
他に特定しない限り、出発物質は一般に、Aldrich Chemicals Co.(Milwaukee、WI)、Lancaster Synthesis、Inc.(Windham、NH)、Acros Organics(Fairlawn、NJ)、Maybridge Chemical Company、Ltd.(Cornwall、英国)、Tyger Scientific(Princeton、NJ)、AstraZeneca Pharmaceuticals(London、英国)、およびAccela ChemBio(San Diego、CA)などの商業的供給源から入手可能である。
【0103】
全般的な実験手順
NMRスペクトルは、Varian Unity(商標)400(Varian Inc.、Palo Alto、CAから入手可能)で室温にてプロトンについて400MHzで記録した。化学シフトは、内部参照として残留溶媒に対する百万分率(δ)で表す。ピーク形状は下記のように表す。s、一重線;d、二重線;dd、二重線の二重線;t、三重線;q、四重線;m、多重線;bsまたはbr.s.、幅広い一重線;2s、2本の一重線;br.d.、幅広い二重線。エレクトロスプレーイオン化質量スペクトル(ES)は、Waters(商標)ZMD機器(キャリアガス:窒素;溶媒A:水/0.01%ギ酸、溶媒B:アセトニトリル/0.005%ギ酸;Waters Corp.、Milford、MAから入手可能)で得た。高分解能質量スペクトル(HRMS)は、Agilent(商標)Model6210(飛行時間型)で得た。単一の塩素または単一の臭素を含有するイオンの強度が記載されている場合、予想される強度比が観察された(35Cl/37Cl含有イオンについて概ね3:1、および79Br/81Br含有イオンについて1:1)。より低い質量のイオンのみの強度を示す。場合によっては、代表的な1H NMRピークのみを示す。
【0104】
カラムクロマトグラフィーは、ガラス製カラムまたはFlash40Biotage(商標)カラム(ISC、Inc.、Shelton、CT)中の、Baker(商標)シリカゲル(40μm;J.T.Baker、Phillipsburg、NJ)またはSilica Gel50(EM Sciences(商標)、Gibbstown、NJ)で行った。MPLC(中圧液体クロマトグラフィー)は、Biotage(商標)SP精製システムまたはTeledyne(商標)Isco(商標)からのCombiflash(登録商標)Companion(登録商標)を使用して行った。低窒素圧力下でBiotage(商標)SNAPカートリッジKPsilまたはRedisep Rfシリカ(Teledyne(商標)Isco(商標)から)を使用した。
【0105】
HPLC(高圧液体クロマトグラフィー)は、Shimadzu(商標)10A LC−UVまたはAgilent(商標)1100分取HPLCを使用して行った。
【0106】
特に断りのない限り、全ての反応は、無水溶媒中窒素ガスの不活性雰囲気下で行った。また、特に断りのない限り、全ての反応は、室温で(約23℃)行った。
【0107】
TLC(薄層クロマトグラフィー)を行うとき、Rfは、化合物が移動する距離を溶離液が移動する距離で割った比として定義する。
Rt(保持時間)。
【0108】
出発物質
一般に、下記の出発物質のいずれかは、米国特許出願公開第2008/0132563号のスキーム7もしくは8、または代わりに、米国特許出願公開第2007/0259821号のスキーム2、3もしくは8に記載されている手順を使用して調製することができる。さらに具体的には、下記の実施例において使用される下記の出発物質は、相当する参考文献に記載されている手順を使用して調製することができ、または相当する販売業者から購入することができる。
【0109】
4−ブロモ−2−(4−メトキシ−ベンジル)−1−メチル−ベンゼンは、PCT国際公開第WO01/027128号の実施例8に記載されている手順によって調製することができる。
【0110】
4−ブロモ−2−(4−エトキシ−ベンジル)−1−メチル−ベンゼンは、US2008/0132563の実施例17の調製に記載されている手順によって調製することができる。
【0111】
4−ブロモ−1−クロロ−2−(4−メトキシ−ベンジル)−ベンゼンは、US2008/0132563の実施例19またはUS2007/0259821の実施例Vの調製に記載されている手順によって調製することができる。
【0112】
4−ブロモ−1−クロロ−2−(4−エトキシ−ベンジル)−ベンゼンは、Shanghai Haoyuan Chemexpress Co.、Ltd.、Shanghai、中華人民共和国から購入し得る。
【0113】
4−ブロモ−2−(4−メトキシ−ベンジル)−ベンゾニトリルは、US2007/0259821の実施例XXIIに記載されている手順によって調製することができる。
【0114】
下記の出発物質は、下記のように調製した。
【0115】
4−ブロモ−1−フルオロ−2−(4−メトキシ−ベンジル)−ベンゼンの調製:
塩化オキサリル(11.0mL、126mmol)を、よく撹拌した5−ブロモ−2−フルオロ−安息香酸(25.0g、114mmol)のジクロロメタン(150mL)およびN,N−ジメチルホルムアミド(1.5mL)懸濁液に0℃にて滴下で添加した。得られた混合物を、室温に徐々に温めた。18時間後、固体が溶液となった。得られた薄オレンジ色の溶液を減圧下で濃縮し、ジエチルエーテルで2度抽出し、5−ブロモ−2−フルオロ−ベンゾイルクロリド(27.0g、定量的収率)を淡オレンジ色の油として得た。
【0116】
5−ブロモ−2−フルオロ−ベンゾイルクロリド(27.0g、114mmol)およびアニソール(12.9g、13.0mL、119mmol)のジクロロメタン(150mL)溶液に、内部温度を10℃未満に維持するように、0℃で三塩化アルミニウム(16.2g、119mmol)を少しずつ添加した。0℃で4時間撹拌した後、溶液を砕いた氷の上に注ぎ、得られた混合物を撹拌した。30分後、有機相を除去し、水相をジクロロメタンで2度抽出した。合わせた有機相を1Mの塩酸水溶液で1度洗浄し、1Mの水酸化ナトリウム水溶液で1度洗浄し、ブラインで1度洗浄した。有機相を硫酸ナトリウム上で乾燥させ、濾過し、減圧下で濃縮した。得られた残渣をエタノールから再結晶させ、(5−ブロモ−2−フルオロ−フェニル)−(4−メトキシ−フェニル)−メタノン(22.5g、64%)を白色の固体として得た。
【0117】
よく撹拌した(5−ブロモ−2−フルオロ−フェニル)−(4−メトキシ−フェニル)−メタノン(22.5g、72.80mmol)およびトリエチルシラン(27.9mL、20.3g、175.0mmol)のジクロロメタン(20mL)およびアセトニトリル(60mL)溶液に、0℃で三フッ化ホウ素エーテラート(32.0mL、36.2g、255.0mmol)を滴下で添加した。内部温度が20℃を超えないような速度で三フッ化ホウ素エーテラートを添加した。反応溶液を室温に温め、一晩撹拌した。全部で18時間後、水酸化カリウム(5.0g)の水(15.0mL)溶液を添加し、得られた混合物を2時間撹拌した。有機相を分離し、水相をジエチルエーテルで2度抽出した。合わせた有機相を1Mの水酸化ナトリウム水溶液で1度洗浄し、ブラインで1度洗浄した。有機相を硫酸ナトリウム上で乾燥させ、濾過し、減圧下で濃縮した。エタノールを得られた残渣に添加すると、白色の固体が形成された。固体を集め、高真空下にて乾燥させ、4−ブロモ−1−フルオロ−2−(4−メトキシ−ベンジル)−ベンゼン(20.1g、93%収率)を白色の固体として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 3.79
(s, 3 H), 3.89 (s, 2 H), 6.85 (d, J=8.6 Hz, 2 H), 6.91 (t, 1 H), 7.12 (d, J=8.8
Hz, 2 H), 7.21 - 7.31 (m, 2 H).
【0118】
出発物質4−ブロモ−2−(4−エトキシ−ベンジル)−ベンゾニトリルの調製:
エチル(4−エトキシ−フェニル)アセテート(2.68g、12.87mmol)、4−ブロモ−2−フルオロ−ベンゾニトリル(2.74g、13.70mmol)のN−メチルピロリドン(4mL)溶液を、カリウムtert−ブトキシド(3.14g、27.98mmol)のN−メチルピロリドン(13mL)懸濁液に0℃でゆっくりと添加した。添加すると、溶液は暗赤色となった。暗赤色の混合物を0℃で30分間、次いで室温で1時間撹拌した。メタノール(10mL)および1Mの水酸化ナトリウム水溶液(13.7mL)を添加し、混合物を室温で一晩撹拌した。pHを塩酸(1Mの水溶液)で約4に調節し、混合物を酢酸エチル(50mL×4)で抽出した。合わせた有機層をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、蒸発乾固させた。N,N−ジメチルホルムアミド(5mL)および炭酸カリウム(7g)を添加し、混合物を100℃に1時間加熱し、室温に冷却した。水を添加し、混合物を酢酸エチル(60mL×3)で抽出した。合わせた有機層をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、蒸発乾固させた。粗製物を、シリカゲル(ヘプタン中の0〜14%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、2.26gの粗生成物(所望の生成物および別の生成物を含有)を得た。粗生成物をメタノールで沈殿させ、4−ブロモ−2−(4−エトキシ−ベンジル)−ベンゾニトリル(1.2g、4.15ppmの四重線、および1.5ppmの三重線でNMRピークを有する5%の別の化合物を含有)を得た。
1H NMR
(400 MHz, クロロホルム-d) δ 7.48-7.38
(m, 3 H), 7.13 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 4.08 (s, 2H),
4.03 (q, J = 7.2 Hz, 2H), 1.41 (t, J = 7.2 Hz, 3H).
【0119】
出発物質4−ブロモ−2−(4−エトキシ−ベンジル)−1−フルオロ−ベンゼンの調製
4−ブロモ−1−フルオロ−2−(4−メトキシ−ベンジル)−ベンゼン(4.2g、14.2mmol)のジクロロメタン(20mL)溶液に、0℃で三臭化ホウ素のジクロロメタン(15.7mL、16.0mmol)溶液(1M)をゆっくりと10分に亘り滴下で添加した。三臭化ホウ素の添加が完了すると、反応混合物を室温に徐々に温めた。4時間後、反応混合物を0℃に冷却し、1Nの塩酸水溶液(20mL)をゆっくりと添加することによってクエンチした。反応混合物を30分間撹拌し、ジクロロメタンで2度抽出した。合わせた有機層を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮し、薄ピンク色の固体(3.83g、96%)を得た。粗生成物4−(5−ブロモ−2−フルオロ−ベンジル)−フェノールを、それ以上精製することなく次のステップで使用した。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 3.88
(s, 2 H), 4.76 (br. s., 1 H), 6.77 (d, J=8.2 Hz, 2 H), 6.91 (t, J=9.1 Hz, 1 H),
7.07 (d, J=8.6 Hz, 2 H), 7.23 (dd, J=6.8, 2.3 Hz, 1 H), 7.26 - 7.31 (m, 1 H).
【0120】
0℃に冷却した4−(5−ブロモ−2−フルオロ−ベンジル)−フェノール(6.0g、21.0mmol)の無水N,N−ジメチルホルムアミド(20mL)溶液に、水素化ナトリウム(鉱油中60%分散物、1.02g、25.6mmol)を添加した。0℃にて45分間撹拌した後、ヨードエタン(2.08mL、25.6mmol)を滴下で添加し、得られた混合物を室温に温めた。18時間後、反応混合物を水でクエンチし、酢酸エチルで2度抽出した。合わせた有機層を水で2度洗浄し、ブラインで1度洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、ヘプタン中の0〜10%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、4.6g(58%収率)の所望の生成物を黄色の油として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 1.40
(t, J=7.0 Hz, 3 H), 3.89 (s, 2 H), 4.01 (q, J=6.9 Hz, 2 H), 6.83 (d, J=8.4 Hz,
2 H), 6.91 (t, J=9.0 Hz, 1 H), 7.10 (d, J=8.8 Hz, 2 H), 7.20 - 7.30 (m, 2 H).
【0121】
トルエン−4−スルホン酸テトラヒドロ−フラン−3−イルエステルの調製
3−ヒドロキシテトラヒドロフラン(2.5g、28.0mmol)の無水ピリジン(60mL)溶液に、室温で4−トルエンスルホニルクロリド(6.49g、34.0mmol)を添加した。室温で反応混合物を18時間撹拌した後、反応混合物を減圧下で濃縮した。得られた残渣は、ヘプタン中の0〜30%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、3.5g(51%収率)の所望の生成物を無色の油として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 2.05
- 2.12 (m, 2 H), 2.45 (s, 3 H), 3.77 - 3.92 (m, 4 H), 5.09 - 5.14 (m, 1 H),
7.35 (d, J=8.00 Hz, 2 H), 7.79 (d, 2 H).
【0122】
トルエン−4−スルホン酸オキセタン−3−イルエステルの調製
オキセタン−3−オール(1.0g、13.0mmol)の無水ピリジン(25mL)溶液に、室温で4−トルエンスルホニルクロリド(3.09g、16.2mmol)を添加した。室温で反応混合物を18時間撹拌した後、反応混合物を減圧下で濃縮した。得られた残渣は、ヘプタン中の0〜30%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、1.9g(62%収率)の所望の生成物を白色の固体として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 2.46
(s, 3 H), 4.63 - 4.75 (m, 4 H), 5.26 - 5.34 (m, 1 H), 7.36 (d, J=8.00 Hz, 2 H),
7.78 (d, J=8.40 Hz, 2 H).
【0123】
出発物質3−[4−(5−ブロモ−2−フルオロ−ベンジル)−フェノキシ]−テトラヒドロ−フランの調製
4−(5−ブロモ−2−フルオロ−ベンジル)−フェノール(1.5g、5.3mmol)および炭酸セシウム(2.61g、8.0mmol)のN,N−ジメチルホルムアミド(15.0mL)溶液に、室温でトルエン−4−スルホン酸テトラヒドロ−フラン−3−イルエステル(1.94g、8.0mmol)のN,N−ジメチホルムアミド(10.0mL)溶液を添加した。次いで、反応混合物を一晩50℃で撹拌した。全部で18時間後、反応混合物を室温に冷却し、ブラインで希釈し、酢酸エチルで3度抽出した。合わせた有機層を水で2度洗浄し、ブラインで1度洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下で濃縮した。得られた粗残渣は、ヘプタン中の0〜30%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、1.66g(89%収率)の所望の生成物を無色の油として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 2.09
- 2.24 (m, 2 H), 3.86 - 4.01 (m, 6 H), 4.86 - 4.91 (m, 1 H), 6.80 (d, J=8.6 Hz,
2 H), 6.91 (t, J=9 Hz, 1 H), 7.10 (d, J=8.6 Hz, 2 H), 7.23 (dd, J=6.8, 2.5 Hz,
1 H), 7.26 - 7.31 (m, 1 H).
【0124】
出発物質3−[4−(5−ブロモ−2−フルオロ−ベンジル)−フェノキシ]−オキセタンの調製
4−(5−ブロモ−2−フルオロ−ベンジル)−フェノール(1.1g、3.9mmol)および炭酸セシウム(1.91g、5.87mmol)のN.N−ジメチルホルムアミド(15.0mL)溶液に、室温でトルエン−4−スルホン酸オキセタン−3−イルエステル(1.34g、8.0mmol)のN,N−ジメチルホルムアミド(10.0mL)溶液を添加した。次いで、反応混合物を一晩65℃で撹拌した。全部で18時間後、反応混合物を室温に冷却し、ブラインで希釈し、酢酸エチルで3度抽出した。合わせた有機層を水で2度洗浄し、ブラインで1度洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、ヘプタン中の0〜30%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、0.948g(72%収率)の所望の生成物を白色の固体として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 3.88
(s, 2 H), 4.76 (dd, J=7.22, 5.3 Hz, 2 H), 4.95 (t, J=6.6 Hz, 2 H), 5.14 - 5.21
(m, 1 H), 6.63 (d, J=8.4 Hz, 2 H), 6.92 (dd, 1 H), 7.10 (d, J=8.6 Hz, 2 H),
7.23 (dd, J=6.6, 2.15 Hz, 1 H), 7.26 - 7.31 (m, 1 H).
【0125】
3−(4−(5−ブロモ−2−クロロベンジル)フェノキシ)オキセタンの調製:
4−ブロモ−1−クロロ−2−(4−メトキシベンジル)−ベンゼン(10g、32mmol)をジクロロメタン(32mL)に溶解し、窒素下で0℃に冷却した。三臭化ホウ素のジクロロメタン(35.3mL、34.3mmol)溶液(1.0M)を、10分に亘り滴下で添加した。添加の後、氷浴を取り除き、溶液を室温で1時間撹拌した。反応混合物を0℃に冷却し、1Nの塩酸水溶液(45mL)を添加することによってクエンチした。混合物を30分間撹拌し、分液漏斗に移し、有機層を集め、水層をジクロロメタン(45mL)で抽出した。合わせた有機抽出物を硫酸マグネシウム上で乾燥させ、濾過し、真空中で濃縮し、4−(5−ブロモ−2−クロロベンジル)フェノール(9.5g、99%収率)を白色の固体として得た。
【0126】
粗4−(5−ブロモ−2−クロロベンジル)フェノール(3.0g、10mmol)および炭酸セシウム(4.9g、15mmol)のN,N−ジメチルホルムアミド(77.5mL)溶液に、室温でトルエン−4−スルホン酸オキセタン−3−イルエステル(3.5g、15mmol)のN,N−ジメチルホルムアミド(8mL)溶液を添加した。混合物を65℃に22時間加熱し、そこでさらなる一定分量の炭酸セシウム(3.3g、10mmol)を添加した。反応混合物を120℃でさらに12時間撹拌し、室温に冷却し、そこで水および酢酸エチルを添加し、混合物を1Nの塩酸水溶液で注意深く酸性化した。有機層を分離し、ブラインで洗浄し(3度)、真空中で濃縮した。Biotage MPLC(シリカゲル、ヘプタン中の0〜25%酢酸エチルの勾配で溶出)による精製によって、3−(4−(5−ブロモ−2−クロロベンジル)フェノキシ)オキセタン(2.5g、70%収率)を白色の固体として得た。
1H NMR
(400 MHz, ジクロロメタン-d2) δ ppm
7.34 - 7.28 (m, 2 H), 7.26 (d, J=8.4 Hz, 1 H), 7.14 - 7.09 (m, 2 H), 6.69 -
6.35 (m, 2 H), 5.22 - 5.16 (m, 1 H), 4.96 - 4.91 (m, 2H), 4.72 - 4.68 (m, 2H),
4.01 (s, 2H).
【0127】
4−ブロモ−2−(4−クロロ−ベンジル)−1−フルオロ−ベンゼンの調製
5−ブロモ−2−フルオロベンズアルデヒド(10.2g、50mmol)の無水テトラヒドロフラン(200mL)溶液を、−78℃に冷却した。4−クロロフェニル−マグネシウムブロミドの溶液(ジエチルエーテル中1M、60mL、60mmol)を、シリンジによって8分に亘り添加した。低温で5分間撹拌を続け、反応物を室温に温め、1時間この温度で撹拌した。溶液を氷水浴中で冷却し、飽和塩化アンモニウム水溶液(40mL)を添加することによってクエンチした。有機相をデカントし、水性残渣を減圧下で濃縮し、残った有機溶媒を除去した。水相を酢酸エチル(200mL×2)で抽出し、抽出物をデカントしたテトラヒドロフラン溶液と合わせた。この溶液をブライン(25mL)で洗浄し、乾燥させ(硫酸ナトリウム)、濾過し、減圧下で濃縮し、粗(5−ブロモ−2−フルオロフェニル)−(4−クロロフェニル)−メタノール(15.2g、96%収率)を黄色の固体として得た。
【0128】
上記の(5−ブロモ−2−フルオロフェニル)−(4−クロロフェニル)−メタノール(15.0g、48mmol)およびトリエチルシラン(18.5mL、116mmol)のジクロロメタン(40mL)およびアセトニトリル(20mL)溶液に、0℃にて窒素下で三フッ化ホウ素ジエチルエーテラート(22.7mL、181mmol)をゆっくりと添加した。得られた溶液を18時間撹拌し、その間に室温にゆっくりと温めた。反応物を氷水浴中で冷却し、7Mの水酸化カリウム水溶液(30mL)をゆっくりと添加することによってクエンチし、メチルtert−ブチルエーテル(200mL×2)で抽出した。合わせた有機溶液を水(25mL×2)、ブライン(25mL×2)で洗浄し、乾燥させ(硫酸ナトリウム)、濾過し、減圧下で濃縮した。ヘプタン中の酢酸エチルの勾配で溶出するシリカゲル上のフラッシュカラムクロマトグラフィーによる精製によって、2−(4−クロロベンジル)−4−ブロモ−1−フルオロベンゼン(5.0g、35%収率)を無色の油として得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 7.33-7.22 (m, 4H), 7.13 (d, J =
8.4 Hz, 2H), 6.93 (dd, J = 9.2, 9.2 Hz, 1H), 3.92 (s, 2H).
【0129】
中間体の調製
中間体((2R,3R,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−テトラヒドロ−ピラン−2−イル)−メタノール(I−1a)の調製:
【0130】
【化8】
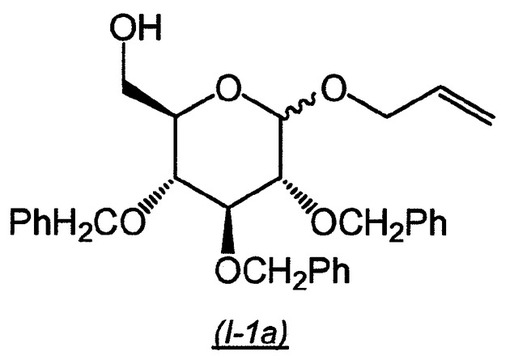
D−グルコース(1.2kg、6.6mol)、トリフルオロメタンスルホン酸(12mL)およびアリルアルコール(5L)の懸濁液を、80℃で3日間加熱した。混合物を室温に冷却し、揮発性物質を真空中で除去し、残渣をN,N−ジメチルホルムアミド(8L)に溶解した。これを4つの等しい反応物に分割し、各々に塩化トリチル(463g、1.67mol)およびトリエチルアミン(231mL、1.67mol)を添加した。トリエチルアミンを添加する間に、僅かな発熱量が観察された。反応混合物を30℃で2日間撹拌し、次いで各反応物を半分に分け、8つの等しい反応物を得た。これらの反応物の各々に、塩化ベンジル(300mL、2.60mol)を添加し、続いて水素化ナトリウム(102.5g、2.60mol)を少しずつ添加し、反応温度を40〜50℃に維持した。完全に添加した後、反応混合物を室温で20時間撹拌した。次いで、各反応物を氷/水(2L)上に注ぎ、酢酸エチル(2.5L)で抽出した。各々の有機相を飽和ブライン/水(1:1、2×2L)で洗浄し、合わせ、硫酸マグネシウム上で乾燥させた(3:1のヘキサン/酢酸エチル中の生成物Rf0.85)。濾過および蒸発後、残渣をジクロロメタン(16L)およびメタノール(4L)の混合物に溶解した。混合物を5つの等しい部分に分割し、各々に硫酸(32mL)を添加した。反応物を3時間撹拌し、ブライン/2Mの水酸化ナトリウム水溶液(1:1、2×2L)で洗浄し、合わせ、硫酸マグネシウム上で乾燥させた。濾過および真空濃縮後、残渣をトルエン中の30%酢酸エチルで溶出するシリカゲル上でさらに精製し、中間化合物(I−1a)をアノマーの混合物として得た(1.77kg、D−グルコースから54%収率)。3:1のヘキサン/酢酸エチル中Rf0.15。
【0131】
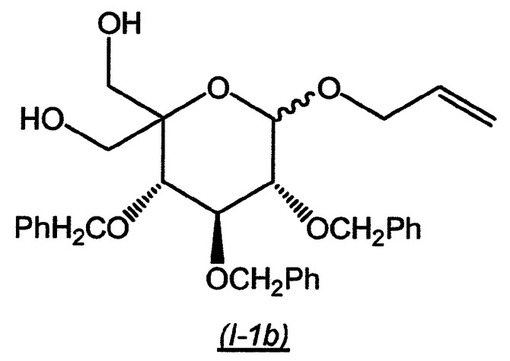
中間体((3S,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−2−ヒドロキシメチル−テトラヒドロ−ピラン−2−イル)−メタノール(I−1b)の調製:
【0132】
【化9】
ジメチルスルホキシド(87mL、1.22mol)のジクロロメタン(160mL)溶液を、塩化オキサリル(64.7mL、0.76mol)のジクロロメタン(2.5L)溶液に−78℃にて滴下で添加した。完全に添加した後、中間体(I−1a)(287g、0.59mol)のジクロロメタン(500mL)溶液を、−78℃にて滴下で添加した。完全に添加した後、反応混合物を30分間撹拌し、トリエチルアミン(417mL、2.9mol)を滴下で添加した。完全に添加した後、反応混合物は、室温にそれ自体で温まった。次いで、反応物を1Mの塩酸水溶液(2L)および水(2L)で洗浄し、次いで硫酸マグネシウム上で乾燥させた。この反応手順を6つの等しい反応物で繰返し、乾燥させた後それらを合わせ、蒸発させ、アルデヒドを黄色の油(1.71kg)として得た。この油をイソプロパノール(2.57L)に溶解し、7つの等しい反応物に分割した。これらの各々に、37%ホルムアルデヒド水溶液(0.79L、10mol)を添加し、続いて水酸化ナトリウム(32g、0.8mol)の水(130mL)溶液を滴下で添加した。完全に添加した後、反応混合物を室温で2日間撹拌した。反応混合物をブライン(2L)で希釈し、酢酸エチル(2L)で抽出した。有機相を飽和炭酸水素ナトリウム水溶液(2L)、ブライン(2L)でさらに洗浄し、次いで硫酸マグネシウム上で乾燥させた。7回の反応からの有機相を合わせ、蒸発させ、残渣をシリカゲル(4対1から1対1までの酢酸エチル中のヘキサンで溶出する)上で精製し、中間化合物(I−1b)をアノマーの混合物(980g、2ステップに亘り53%収率)として得た。1:1のヘキサン/酢酸エチル中Rf0.57および0.60。
【0133】
(3S,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−2,2−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン(I−1c):
【0134】
【化10】
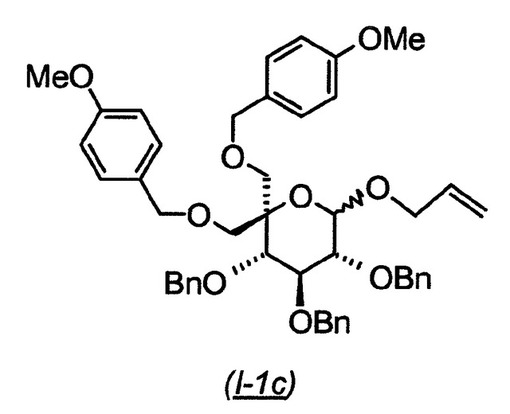
出発物質のジオール[((3S,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−2−ヒドロキシメチル−テトラヒドロ−ピラン−2−イル)−メタノール(I−1b:10g、19.208mmol)をN,N−ジメチルホルムアミド(70mL)に溶解し、0℃に冷却した。水素化ナトリウム(鉱油中60%分散物、1.69g、42.3mmol)を添加し、反応物を0℃で1時間撹拌し、その後1−ブロモメチル−4−メトキシ−ベンゼン(5.96mL、40.3mmol)を添加した。次いで、反応物を60℃に一晩加熱した。混合物を室温に冷却し、反応物を水でクエンチし、酢酸エチル(2度)で抽出した。合わせた有機層を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下濃縮した。次いで、反応物をシリカゲル(ヘプタン中の0〜80%酢酸エチルの勾配で溶出)上でクロマトグラフィーにかけ、7.55g(52%収率)の生成物(I−1c)を得た。MS 778.8 (M+NH4+; ポジティブモード).
【0135】
(3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−1d):
【0136】
【化11】
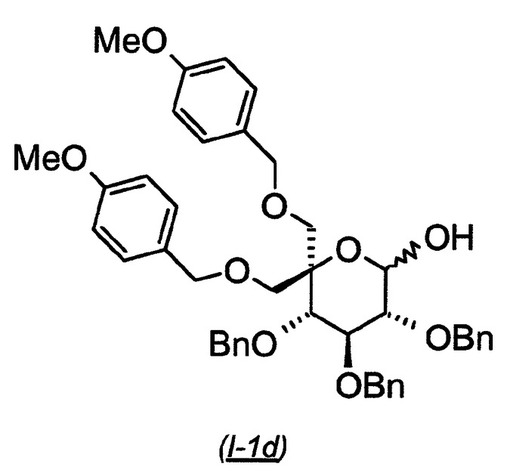
出発物質((3S,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−2,2−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン(I−1c:7.55g、9.92mmol)のメタノール(60mL)およびジクロロメタン(20mL)溶液に、室温で塩化パラジウム(II)(528mg、2.98mmol)を添加し、得られた混合物をこの温度で4時間撹拌した。TLCは、より極性の生成物の明らかな形成を示した。反応物をCelite(登録商標)を通して濾過し、減圧下で濃縮した。粗材料を、ヘプタン中の0〜80%酢酸エチルの勾配で溶出するシリカゲル上のクロマトグラフィーにかけ、5.6g(78%収率)の生成物(I−1d)を得た。MS 738.8 (M+NH4+; ポジティブモード).
【0137】
(3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オン(I−1e):
【0138】
【化12】

二塩化オキサリル(1.9mL、23mmol)のジクロロメタン(65mL)溶液に、−78℃でジメチルスルホキシド(3.3mL、47mmol)のジクロロメタン(5mL)溶液を添加し、得られた溶液をこの温度で30分間撹拌した。出発物質((3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−1d、5.6g、7.7mmol)のジクロロメタン(15.0mL)溶液を、次いで滴下で添加し、得られた混合物を30分間撹拌し、温度を−60℃に上げた。トリエチルアミン(9.7mL、69.5mmol)を滴下で添加し、混合物を1時間に亘り0℃に温めた。反応物を飽和塩化アンモニウム水溶液の添加によってクエンチし、有機相を硫酸マグネシウム上で乾燥させ、濾過し、減圧下濃縮した。粗材料は、ヘプタン中の0〜60%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、生成物(I−1e)(4g、72%収率)を生成した。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 3.24
(d, J=10 Hz, 1 H), 3.40 - 3.47 (m, 2 H), 3.74 (s, 3 H), 3.77 (s, 3 H), 3.86 (d,
J=10 Hz, 1 H), 4.07 (d, J=8.6 Hz, 1 H), 4.15 (d, J=9.6 Hz, 1 H), 4.35 - 4.55
(m, 6 H), 4.65 - 4.72 (m, 2 H), 4.82 (d, J=11 Hz,1 H), 4.87 (d, J=11.2 Hz, 1
H), 5.10 (d, J=11.1 Hz, 1 H), 6.74 - 6.79 (m, 2 H), 6.81 - 6.85 (m, 2 H), 7.11
(dd, J=7.0, 2.5 Hz, 2 H), 7.17 - 7.41 (m, 17 H).
【0139】
(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)および/または(3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−2−(メトキシ−メチル−アミノ)−テトラヒドロ−ピラン−2−オール(I−1f):
【0140】
【化13】
ラクトン((3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オン(I−1e:10.4g、14.5mmol)およびN,O−ジメチル−ヒドロキシルアミン塩酸塩(1.77g、29.0mmol)のジクロロメタン(100mL)溶液に、0℃でトリメチルアルミニウムのヘキサン(14.5mL、29.0mmol)溶液(2.0M)を滴下で添加し、得られた溶液を室温で16時間撹拌した。反応混合物を0℃に冷却し、1Nの塩酸水溶液をゆっくり添加することによってクエンチした。得られた混合物を1時間撹拌した。有機相を分離し、1Nの塩酸水溶液で洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下で濃縮した。粗材料を中圧クロマトグラフィー(ヘプタン中の5〜40%酢酸エチルの勾配)によって精製し、6.5g(58%)の生成物を得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 2.62
(br. s, 1 H), 2.94 (br. s., 3 H), 3.23 (br. s., 3 H), 3.42 (d, J=9.4 Hz, 1 H),
3.50 - 3.60 (m, 3 H), 3.75 (s, 3 H), 3.77 (s, 3 H), 4.03 (d, J=6.9 Hz, 1 H),
4.20 (dd, J=6.9, 3.3 Hz, 1 H), 4.31 - 4.44 (m, 5 H), 4.46 - 4.51 (m , 2H), 4.53
(d, J=12 Hz, 1 H), 4.66 (d, J=12 Hz, 1 H), 4.80 (br. d, J=11.5 Hz, 1 H), 4.87
(d, J=11.4 Hz, 1 H), 6.77 - 6.83 (m, 4 H), 7.15 - 7.35 (m, 19 H). ([M+H+]
780.8, ポジティブモード; [M+HCO2-] 824.7,
ネガティブモード). HRMS C46H54NO10の計算値 (M+H+) 780.3742, 実測値 780.3708.
【0141】
(2R,3S,4S)−2,3,4,6−テトラキス−ベンジルオキシ−5−ベンジルオキシメチル−5−ヒドロキシ−ヘキサン酸メトキシ−メチル−アミド(I−6g)および/または(3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−ベンジルオキシメチル−2−(メトキシ−メチル−アミノ)−テトラヒドロ−ピラン−2−オール(I−6f):
【0142】
【化14】
この化合物は、(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)および/または(3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−2−(メトキシ−メチル−アミノ)−テトラヒドロ−ピラン−2−オール(I−1f)の合成について記載したものと同様の手順((I−1b)から(I−1c)への変換を記載した実験の部において使用したアルキル化剤が、パラ−メトキシベンジルブロミドの代わりに臭化ベンジルであったこと以外)を使用して、[((3S,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−2−ヒドロキシメチル−テトラヒドロ−ピラン−2−イル)−メタノール(I−1b)から出発して調製した。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 2.66
(br. s, 1 H), 2.94 (br. s., 3 H), 3.23 (br. s., 3 H), 3.48 (d, J=9.4 Hz, 1 H),
3.55 - 3.66 (m, 3 H), 4.05 (d, J=6.9 Hz, 1 H), 4.21 (dd, J=6.9, 3.3 Hz, 1 H),
4.36 (d, 1H, J = 11.7 Hz), 4.41 - 4.58 (m, 7 H), 4.68 (d, J=11.9 Hz, 1 H), 4.81
(br. d, J=11.5 Hz, 1 H), 4.89 (d, J=11.5 Hz, 1 H), 7.15 - 7.35 (m, 25 H). MS
[M+H+] 720.7, ポジティブモード; [M+HCO2-]
764.7, ネガティブモード).
【0143】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−1i):
【0144】
【化15】
n−ブチルリチウム(0.97mL、2.5M/ヘキサン、3.15当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−2−(4−メトキシ−ベンジル)−1−メチル−ベンゼン(690mg、3当量)の無水テトラヒドロフラン(2.7mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度でさらに1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(608mg)の無水テトラヒドロフラン(1.35mL)溶液を、次いでシリンジポンプを使用して1.5時間に亘り滴下で添加し、得られた混合物を−78℃で1時間撹拌し、その後−20℃に14時間に亘り温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。ジエチルエーテルを添加し、1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮した。ヘプタン中の20〜50%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーによって、生成物を異性体の混合物(440mg、61%収率)として得た。
HRMS C59H62O10Naの計算値
(M+Na+) 953.4235, 実測値953.4236.
【0145】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−1k):
【0146】
【化16】
中間体I−1i(150mg)のジクロロメタン(3mL)溶液に、アニソール(90μL、5当量)、続いて20%トリフルオロ酢酸のジクロロメタン溶液(3mL)を添加し、得られた混合物を、室温で約1時間撹拌した。混合物を濃縮し、粗製物をシリカゲル(ヘプタン中の10〜30%酢酸エチルの勾配を使用)上のクロマトグラフィーにかけ、所望の生成物を異性体の混合物として得た(66mg、61%収率)。MS (LCMS) 673.9 (M+H+, ポジティブモード).
【0147】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−2i):
【0148】
【化17】
n−ブチルリチウム(0.312mL、2.5M/ヘキサン、3.05当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−2−(4−エトキシ−ベンジル)−1−メチル−ベンゼン(238mg、3.05当量)の無水テトラヒドロフラン(0.9mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度でさらに1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(200mg)の無水テトラヒドロフラン(0.6mL)溶液を、次いでシリンジポンプを使用して1.5時間に亘り滴下で添加し、得られた混合物を−78℃で1時間撹拌し、その後16時間に亘り室温に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。ジエチルエーテルを添加し、1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮した。Biotage(商標)自動クロマトグラフィーユニット(2つの積重ねた10gシリカゲルカラム、ヘプタン中の0〜60%酢酸エチルの勾配で溶出)を使用して粗材料をクロマトグラフィーにかけ、生成物を異性体の混合物(136mg、56%収率)として得た。MS (LCMS) 968 (M+Na+, ポジティブモード).
【0149】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−2k):
【0150】
【化18】
中間体I−2i(136mg、0.145mmol)のジクロロメタン(4mL)溶液に、アニソール(310μL、約5当量)、続いて20%トリフルオロ酢酸のジクロロメタン溶液(4mL)を添加し、得られた混合物を室温で1.5時間撹拌した。混合物を濃縮し、粗製物は、ISCO(商標)combiflash(登録商標)companion(登録商標)自動クロマトグラフィーユニット(4gのシリカゲルカラム)を使用して、ヘプタン中の0〜70%酢酸エチルの勾配で溶出するクロマトグラフィーにかけ、所望の生成物を異性体の混合物として得た(85mg、85%収率)。MS (LCMS) 687.7 (M+H+, ポジティブモード).
【0151】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−3i):
【0152】
【化19】
n−ブチルリチウム(0.97mL、2.5M/ヘキサン、3.15当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−1−クロロ−2−(4−メトキシ−ベンジル)−ベンゼン(725mg、2.95当量)の無水テトラヒドロフラン(2.7mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度でさらに1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(616mg)の無水テトラヒドロフラン(1.35mL)溶液を、次いでシリンジポンプを使用して1.5時間に亘り滴下で添加し、得られた混合物を−78℃で1時間撹拌し、その後14時間に亘り−20℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。ジエチルエーテルを添加し、1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮した。ヘプタン中の10〜40%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーによって、生成物を異性体の混合物(530mg、71%収率)として得た。
【0153】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−3k):
【0154】
【化20】
中間体I−3i(530mg)のジクロロメタン(11mL)溶液に、アニソール(300μL、5当量)、続いて20%トリフルオロ酢酸のジクロロメタン溶液(11mL)を添加し、得られた混合物を室温で1時間撹拌した。混合物を濃縮し、粗製物を、ヘプタン中の10〜40%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーにかけ、生成物を異性体の混合物として得た(229mg、59%収率)。
MS (LCMS) 693.6 (M+H+, ポジティブモード).
【0155】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−4i):
【0156】
【化21】

n−ブチルリチウム(1.0mL、2.5M/ヘキサン、3.25当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−1−クロロ−2−(4−エトキシ−ベンジル)−ベンゼン(815mg、3.25当量)の無水テトラヒドロフラン(2.9mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度でさらに1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(600mg)の無水テトラヒドロフラン(1.45mL)溶液を、次いでシリンジポンプを使用して1.3時間に亘り滴下で添加し、得られた混合物を−78℃で1時間撹拌し、その後14時間に亘り−25℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。ジエチルエーテルを添加し、1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮した。ヘプタン中の10〜40%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーによって、生成物を異性体の混合物(280mg、38%収率)として得た。
HRMS C59H61O10ClNaの計算値 (M+Na+) 987.3845, 実測値987.3840.
【0157】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−4k):
【0158】
【化22】
中間体I−4i(1.46g)のジクロロメタン(31mL)溶液に、アニソール(900μL、約5当量)、続いて20%トリフルオロ酢酸のジクロロメタン溶液(31mL)を添加し、得られた混合物を室温で1時間撹拌した。混合物を濃縮し、粗製物を、ヘプタン中の10〜30%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーにかけ、生成物を異性体の混合物として得た(670mg、63%収率)。
HRMS C43H44O7Clの計算値 (M+H+) 707.2770, 実測値707.2765.
【0159】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−6,6−ビス−(4メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−5i):
【0160】
【化23】

n−ブチルリチウム(462μL、2.5M/ヘキサン、3.0当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−1−フルオロ−2−(4−メトキシ−ベンジル)−ベンゼン(341mg、3当量)の無水テトラヒドロフラン(1.4mL)溶液に−78℃にて窒素下にて滴下で添加した(5秒毎に1滴)。得られた溶液をこの温度で1時間撹拌した。次いで、(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(300mg、0.385mmol)の無水テトラヒドロフラン(0.70mL)溶液を、非常にゆっくりと滴下で添加し(5秒毎に1滴)、得られた混合物を−78℃でさらに1時間撹拌し、その後12時間に亘り10℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。反応物をジエチルエーテルで希釈し、1Nの塩酸水溶液を滴下で添加することによってクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜40%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(199mg、55%収率)として得た。
【0161】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−5k):
【0162】
【化24】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−6,6−ビス−(4メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−5i;191mg、0.204mmol)のジクロロメタン(3.75mL)溶液に、アニソール(0.178mL、1.63mmol)、続いてトリフルオロ酢酸のジクロロメタン(3.75mL)溶液(20%)を室温で窒素下にて添加した。室温で1時間撹拌した後、反応混合物を減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜30%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(115mg、83%収率)として得た。MS (LCMS) 677.7 (M+H+, ポジティブモード).
【0163】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−10i)
【0164】
【化25】
n−ブチルリチウム(508μL、2.5M/ヘキサン、3.0当量)を、酸素脱気した4−ブロモ−2−(4−エトキシ−ベンジル)−1−フルオロ−ベンゼン(392.0mg、1.27mmol)の無水テトラヒドロフラン(1.5mL)溶液に−78℃にて窒素下にて滴下で添加した(5秒毎に1滴)。得られた溶液をこの温度で1時間撹拌した。次いで、(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミドI−1g(330.0mg、0.423mmol)の無水テトラヒドロフラン(0.75mL)溶液を非常にゆっくりと滴下で添加し(5秒毎に1滴)、得られた混合物を−78℃でさらに1時間撹拌し、その後12時間に亘り10℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する)。反応物をジエチルエーテルで希釈し、1Nの塩酸水溶液を滴下で添加することによってクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜40%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(180mg、44%収率)として得た。
【0165】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−10k)
【0166】
【化26】
中間体I−10i(180.0mg、0.19mmol)のジクロロメタン(2.0mL)溶液に、アニソール(0.175mL、1.60mmol)、続いてトリフルオロ酢酸のジクロロメタン(2.0mL)溶液(20%)を室温にて窒素下で添加した。1時間撹拌した後、反応混合物を減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜30%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物として得た(85.0mg、64%収率)。
【0167】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−11i)
【0168】
【化27】
n−ブチルリチウム(1.0mL、2.5M/ヘキサン、3.0当量)を、酸素脱気した3−[4−(5−ブロモ−2−フルオロ−ベンジル)−フェノキシ]−テトラヒドロ−フラン(878mg、2.50mmol)の無水テトラヒドロフラン(3.0mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度で1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミドI−1g(650mg、0.833mmol)の無水テトラヒドロフラン(1.5mL)溶液を、次いで非常にゆっくりと滴下で添加し(0.9mL/時間)、得られた混合物を−78℃でさらに1時間撹拌し、その後12時間に亘り10℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する)。反応物をジエチルエーテルで希釈し、1Nの塩酸水溶液を滴下で添加することによってクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜40%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(287mg、34%収率)として得た。
【0169】
((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノール(I−11k)
【0170】
【化28】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オールI−11i(275mg、0.28mmol)のジクロロメタン(2.0mL)溶液に、アニソール(0.250mL、2.29mmol)、続いてトリフルオロ酢酸のジクロロメタン(8.0mL)溶液(20%)を室温にて窒素下で添加した。1時間撹拌した後、反応混合物を減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜30%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(168mg、83%収率)として得た。
【0171】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[3−(4−クロロ−ベンジル)−4−フルオロ−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−12i):
【0172】
【化29】

n−ブチルリチウム(1.0mL、2.5M/ヘキサン、3.1当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−2−(4−クロロ−ベンジル)−1−フルオロ−ベンゼン(702mg、2.9当量)の無水テトラヒドロフラン(3.0mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度で25分間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(621mg)の無水テトラヒドロフラン(1.5mL)溶液を、次いでシリンジポンプを使用して滴下で添加し(0.9mL/時間)、得られた混合物を低温でさらに17時間撹拌した(アルミ箔で覆われた深いデュワー中に置いて、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。1Mの塩酸水溶液(1.5mL)を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で30分間撹拌した。混合物を飽和塩化アンモニウム水溶液(15mL)で希釈し、酢酸エチル(15mL×3)で抽出した。合わせた有機溶液をブライン(30mL)で洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮した。ヘプタン中の10〜40%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーによって、生成物を異性体の混合物(477mg、64%収率)として得た。
【0173】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−クロロ−ベンジル)−4−フルオロ−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−12k):
【0174】
【化30】
中間体I−12i(243mg)のジクロロメタン(9mL)溶液に、アニソール(0.15mL、5.3当量)、続いてトリフルオロ酢酸(1.0mL、50当量)を添加し、得られた混合物を室温で2時間撹拌した。混合物を濃縮し、粗製物を、ヘプタン中の10〜30%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーにかけ、生成物を異性体の混合物として得た(102mg、58%収率)。
【0175】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−13i)
【0176】
【化31】
n−ブチルリチウム(1.12mL、2.5M/ヘキサン、3.0当量)を、酸素脱気した3−[4−(5−ブロモ−2−フルオロ−ベンジル)−フェノキシ]−オキセタン(942.0mg、2.79mmol)の無水テトラヒドロフラン(3.0mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度で1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミドI−1g(725.0mg、0.930mmol)の無水テトラヒドロフラン(1.5mL)溶液を、次いで非常にゆっくりと滴下で添加し(0.9ml/時間)、得られた混合物を−78℃でさらに1時間撹拌し、その後12時間に亘り10℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する)。反応物をジエチルエーテルで希釈し、1Nの塩酸水溶液を滴下で添加することによってクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜40%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(535mg、59%収率)として得た。
【0177】
((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノール(I−13k)
【0178】
【化32】
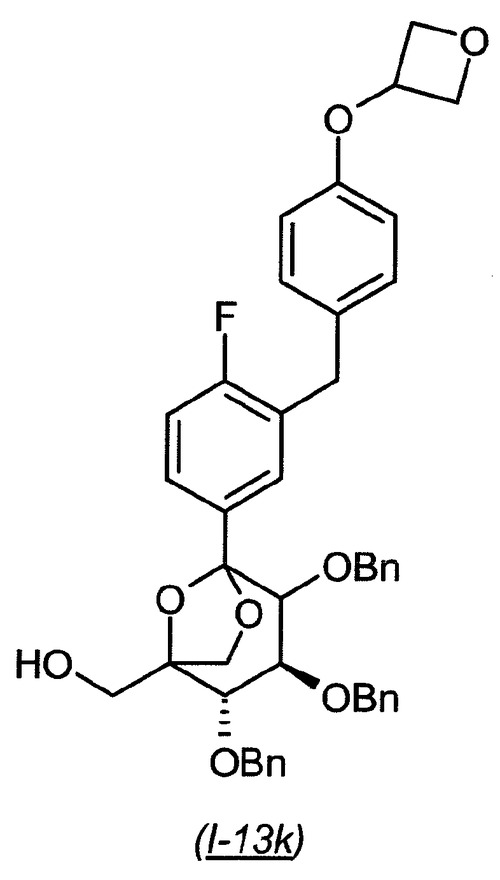
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オールI−13i(535mg、0.548mmol)のジクロロメタン(2.0mL)溶液に、アニソール(0.480mL、4.38mmol)、続いてトリフルオロ酢酸のジクロロメタン(8.0mL)溶液(20%)を室温にて窒素下で添加した。1時間撹拌した後、反応混合物を減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜30%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(300mg、76%収率)として得た。
【0179】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−14i):
【0180】
【化33】
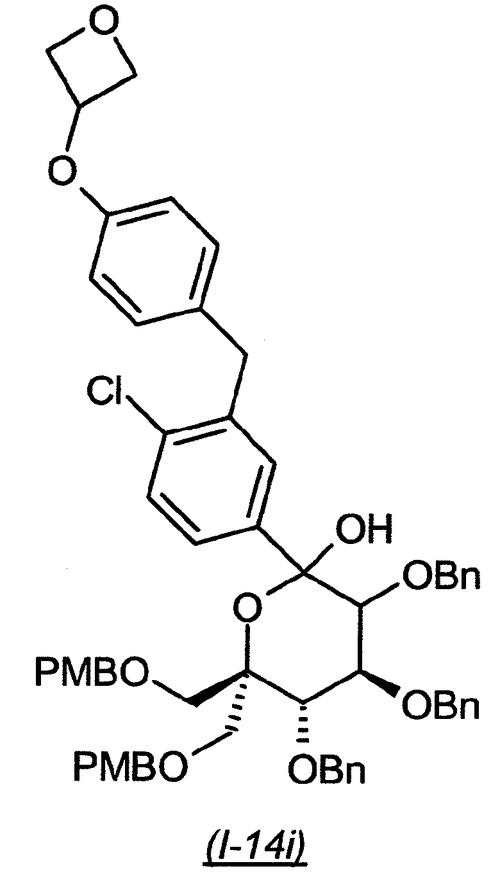
n−ブチルリチウム(0.97mL、2.5M/ヘキサン、3.15当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)3−(4−(5−ブロモ−2−クロロベンジル)フェノキシ)オキセタン(824mg、2.95当量)の無水テトラヒドロフラン(2.7mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度でさらに1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(616mg)の無水テトラヒドロフラン(1.35mL)溶液を、次いでシリンジポンプを使用して1.5時間に亘り滴下で添加し、得られた混合物を−78℃で1時間撹拌し、その後14時間に亘り−20℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。ジエチルエーテルを添加し、1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。ヘプタン中の0〜50%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーによって、生成物を異性体の混合物(563mg、72%収率)として得た。
【0181】
((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノール(I−14k):
【0182】
【化34】
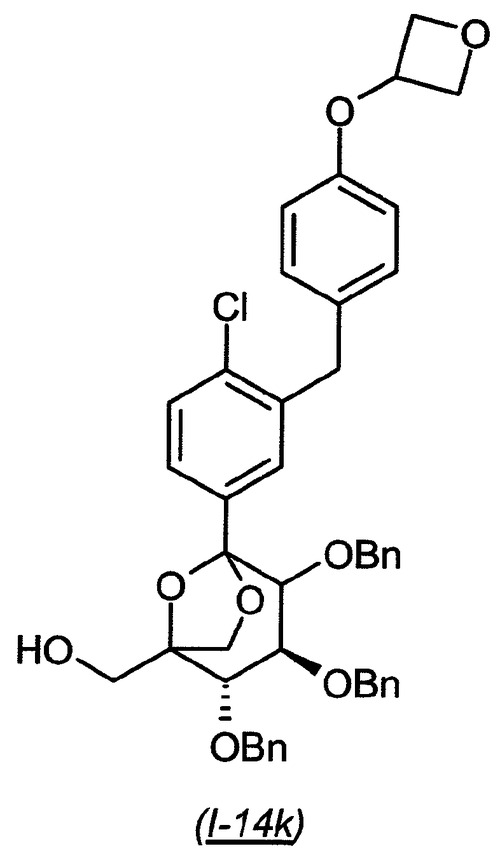
中間体(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オールI−14i(282mg)のジクロロメタン(2.84mL)溶液に、アニソール(200μL、約7当量)、続いて20%トリフルオロ酢酸のジクロロメタン溶液(3.07mL)を添加し、得られた混合物を室温で1.5時間撹拌した。混合物を濃縮し、粗製物を、ヘプタン中の10〜50%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーにかけ、生成物を異性体の混合物として得た(186mg、89%収率)。
【0183】
(実施例1)
(1S,2S,3S,4R,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(1A)および(1S,2S,3S,4S,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(1B):
【0184】
【化35】
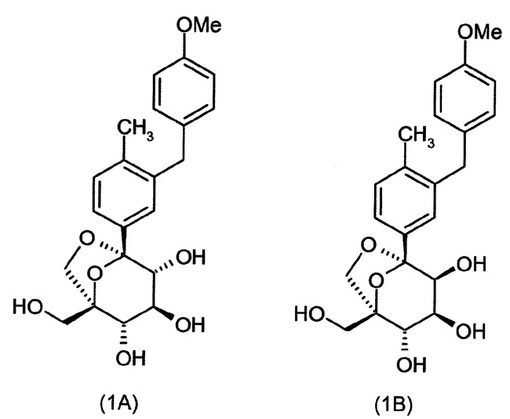
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−1k:236mg)のエタノール/テトラヒドロフラン(7mL、4/1容)溶液に、ギ酸(270μL、19当量)およびパラジウム黒(150mg、4当量)を連続的に添加し、得られた混合物を室温で3時間撹拌した。パラジウムを濾過し、溶媒の蒸発の後に得られた粗混合物を、ヘプタン中の85〜100%酢酸エチルの勾配で溶出するシリカゲル上のクロマトグラフィーによって精製した。得られた生成物の混合物を分取HPLCによって精製した。
【0185】
HPLC分取法:逆相C18phenomenexカラムLuna、5マイクロメーター、150×21.20mm、20mL/分、20分に亘りアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;20〜60%のアセトニトリル/0.1%ギ酸の勾配。UV検出:254nm。HPLCは、3:1(1A:1B)のジアステレオマー比を示した。
【0186】
1A:(55mg、39%収率);Rt=10.9分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。MS (LCMS) 403.3 (M+H+; ポジティブモード)
447.3 (M+HCO2-, ネガティブモード).
1H NMR
(400 MHz, メタノール-d4) δ 7.33 (d, 1H, J = 1.6 Hz), 7.30 (dd, 1H, J = 7.6および1.6 Hz), 7.10 (d, 1H, J = 7.6 Hz), 7.02-6.98 (m, 2H), 6.79-6.75 (m,
2H), 4.13 (d, 1H, J = 7.4 Hz), 3.90 (s, 2H), 3.82 (d, 1H, J = 12.5 Hz), 3.77
(dd, 1H, J = 8.2および1.2 Hz), 3.72 (s, 3H), 3.66 (d, 1H,
J = 12.5 Hz), 3.65 (t, 1H, J = 8.0 Hz), 3.59 (d, 1H, J = 7.8 Hz), 3.58 (dd, 1H,
J = 7.5および1.5 Hz), 2.16 (s, 3H). HRMS C22H27O7の計算値 (M+H+) 403.1751, 実測値 403.1737.
【0187】
1B:(20mg、14%収率);Rt=11.5分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。MS (LCMS) 403 (M+H+; ポジティブモード)
447 (M+HCO2-, ネガティブモード).
1H NMR
(400 MHz, メタノール-d4) δ 7.38 (d, 1H, J = 1.8 Hz) 7.33 (dd, 1H, J = 7.9および1.8 Hz), 7.10 (d, 1H, J = 7.9 Hz), 7.02-6.97 (m, 2H), 6.79-6.74 (m,
2H), 4.02 (d, 1H, J = 7.4 Hz), 3.93 (t, 1H, J = 2.2 Hz), 3.91 (br. s, 2H), 3.88
(d, 1H, J = 12.5 Hz), 3.84 (d, 2H, J = 2.4 Hz), 3.75 (d, 1H, J = 12.5 Hz), 3.71
(s, 3H), 3.49 (d, 1H, J = 7.4 Hz), 2.16 (s, 3H).
【0188】
(実施例2)
(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(2A)および(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(2B)
【0189】
【化36】
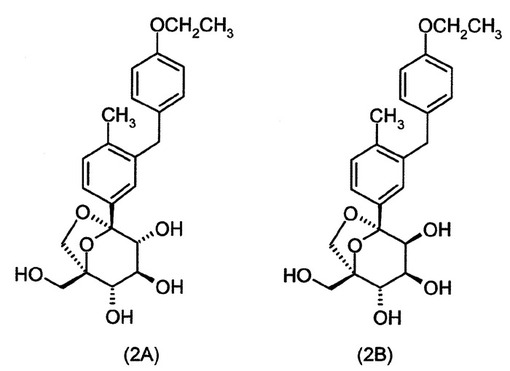
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−2k:85mg、0.12mmol)のエタノール/テトラヒドロフラン(7mL、約4/1容)溶液に、ギ酸(95μL、19当量)およびパラジウム黒(53mg、4当量)を連続的に添加し、得られた混合物を室温で3時間撹拌した。パラジウムを濾過し、溶媒の蒸発の後に得られた粗混合物を分取HPLCによって精製した。
【0190】
HPLC分取法:逆相C18phenomenexカラムLuna、5マイクロメーター、150×21.20mm、20mL/分、20分に亘りアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;20〜60%のアセトニトリル/0.1%ギ酸の勾配。UV検出:254nm。HPLCは、4:1(2A:2B)のジアステレオマー比を示した。
【0191】
2A:(20mg;38%収率)Rt=12.7分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
MS (LCMS) 417.3 (M+H+; ポジティブモード); 461.4 (M+HCO2-; ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ ppm 1.34 (t, J=6.9 Hz, 3 H), 2.18 (s, 3 H), 3.60 (d,
J=8 Hz, 2 H), 3.66 (t, J=8 Hz, 1 H), 3.68 (d, J=12.5 Hz, 1 H), 3.78 (d, 1H, J=
8.8 Hz), 3.84 (d, J=12.4 Hz, 1 H), 3.92 (s, 2 H), 3.97 (q, J=7 Hz, 2 H), 4.15
(d, J=7.5 Hz, 1 H), 6.77 (m, 2 H), 7.00 (m, 2 H), 7.12 (d, J=7.7 Hz, 1 H), 7.31
(dd, J=7.9および1.4 Hz, 1 H), 7.34 (s, 1 H).
【0192】
2B:(5mg;9%収率)Rt=13.2分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
MS (LCMS) 417.3 (M+H+; ポジティブモード); 461.4 (M+HCO2-; ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ ppm 1.34 (t, J=6.9 Hz, 3 H), 2.18 (s, 3 H), 3.52 (d,
1H, J= 7.4 Hz), 3.77 (d, J=12.5 Hz, 1 H), 4.00-3.84 (m, 8 H), 4.04 (d, J=7.4
Hz, 1 H), 6.79-6.75 (m, 2 H), 7.03-6.98 (m, 2 H), 7.12 (d, J=7.9 Hz, 1 H), 7.35
(dd, J=7.7および1.9 Hz, 1 H), 7.39 (d, J = 1.9 Hz, 1 H).
【0193】
(実施例3)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(3A)および(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(3B):
【0194】
【化37】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−3k:229mg)のエタノール/テトラヒドロフラン(7mL、4/1容)溶液に、ギ酸(270μL、20当量)およびパラジウム黒(140mg、4当量)を連続的に添加し、得られた混合物を室温で撹拌した。1時間後、さらなるギ酸(270μL、20当量)およびパラジウム黒(140mg、4当量)を添加し、混合物をさらに1時間室温で撹拌した。パラジウムを濾過し、溶媒の蒸発の後に得られた粗混合物を分取HPLCによって精製した。
【0195】
HPLC分取法:逆相C18phenomenexカラムLuna、5マイクロメーター、150×21.20mm、20mL/分、20分に亘るアセトニトリル/0.1%ギ酸:水/0,1%ギ酸;20〜60%のアセトニトリル/0.1%ギ酸の勾配。UV検出:254nm。HPLCは、1.4:1(3A:3B)のジアステレオマー比を示した。
【0196】
3A:(50mg;36%収率)Rt=12.1分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で濃縮した。
MS (LCMS) 423.3 (M+H+; ポジティブモード); 467.3 (M+HCO2-; ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ 7.43 (s, 1H), 7.38-7.30 (m, 2H), 7.08 (d, 2H), 6.79
(d, 2H), 4.12 (d, 1H, J = 7.5 Hz), 4.01 (s, 2H), 3.81 (d, 1H, J = 12.5 Hz),
3.75 (d, 1H, J = 8.4 Hz), 3.73 (s, 3H), 3.66 (d, 1H, J = 11.7 Hz), 3.63 (t, 1H,
J = 8.2 Hz), 3.57 (d, 1H, J = 7.4 Hz), 3.52 (d, 1H, J = 7.8 Hz). HRMS C21H24O7Clの計算値 (M+H+) 423.1205, 実測値 423.1192.
【0197】
3B:(37mg;27%収率)Rt=12.8分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で濃縮した。
MS (LCMS) 423.3 (M+H+; ポジティブモード) 467.3 (M+HCO2-, ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ 7.50 (d, 1H, J = 1.9 Hz) 7.42 (dd, 1H, J = 8.3および1.9 Hz), 7.35 (d, 1H, J = 8.3 Hz), 7.12-7.07 (m, 2H), 6.83-6.78 (m,
2H), 4.06-4.01 (m, 3H), 3.91-3.83 (m, 4H), 3.78-3.72 (m, 4H), 3.51 (d, 1H, J =
7.5 Hz).
【0198】
(実施例4)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(4A)および(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(4B):
【0199】
【化38】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−4k:335mg)のエタノール/テトラヒドロフラン(10mL、4/1容)溶液に、ギ酸(420μL、22当量)およびパラジウム黒(208mg、4当量)を連続的に添加し、得られた混合物を室温で撹拌した。1時間後、さらなるギ酸(420μL、22当量)およびパラジウム黒(208mg、4当量)を添加し、混合物をさらに1時間室温で撹拌した。パラジウムを濾過し、溶媒の蒸発の後に得られた粗混合物を分取HPLCによって精製した。
【0200】
分取HPLC:逆相C18Geminiカラム、5マイクロメーター、30×100mm、40mL/分、18分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;25〜50%のアセトニトリル/0.1%ギ酸の勾配;UV検出:220nm。HPLCは、1.1:1(4A:4B)のジアステレオマー比を示した。
【0201】
4A:(60mg、29%収率);Rt=12.4分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
MS (LCMS) 437.3 (M+H+; ポジティブモード); 481.3 (M+HCO2-; ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ 7.43 (d, 1H, J = 1.9 Hz), 7.36 (dd, 1H, J = 8.3および2 Hz), 7.32 (d, 1H, J = 8.3 Hz), 7.08-7.04 (m, 2H), 6.79-6.75 (m,
2H), 4.12 (d, 1H, J = 7.5 Hz), 4.00 (s, 2H), 3.96 (q, 2H, J = 7.0 Hz), 3.81 (d,
1H, J = 12.5 Hz), 3.75 (dd, 1H, J = 8.3および1.3 Hz), 3.65
(d, 1H, J = 12.5 Hz), 3.63 (t, 1H, J = 8.2 Hz), 3.57 (dd, 1H, J = 7.5および1.3 Hz), 3.52 (d, 1H, J = 8.0 Hz), 1.33 (t, 3H, J = 6.9 Hz). HRMS C22H26O7Clの計算値 (M+H+) 437.1361, 実測値 437.1360.
【0202】
4B:(30mg、15%収率);Rt=13.2分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
MS (LCMS) 437.3 (M+H+; ポジティブモード) 481.3 (M+HCO2-, ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ 7.48 (d, 1H, J = 1.9 Hz) 7.40 (dd, 1H, J = 8.1および1.9 Hz), 7.32 (d, 1H, J = 8.3 Hz), 7.08-7.03 (m, 2H), 6.80-6.74 (m,
2H), 4.04-3.99 (m, 3H), 3.95 (q, 2H, J = 7 Hz), 3.89-3.81 (m, 4H), 3.73 (d, 1H,
J = 12.5 Hz), 3.49 (d, 1H, J = 7.3 Hz), 1.32 (t, 3H, J = 7 Hz). HRMS C22H26O7Clの計算値 (M+H+) 437.1361, 実測値 437.1358.
【0203】
(実施例5)
(1S,2S,3S,4R,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(5A)および(1S,2S,3S,4S,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(5B):
【0204】
【化39】
エタノール/テトラヒドロフランの4:1溶液(10mL)中の{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(115mg、0.170mmol)の溶液に、ギ酸(137μL、3.42mmol)およびパラジウム黒(73mg、0.687mmol)を連続的に添加した。得られた混合物を室温で撹拌した。3時間後、さらなるギ酸(137μL、3.42mmol)およびパラジウム黒(73mg、0.687mmol)を添加した。18時間後、反応混合物を濾過し、濾液を減圧下で濃縮した。得られた粗残渣は、シリカゲル(ジクロロメタン中の0〜15%メタノールの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、64mgの白色の固体を得た。異性体の混合物を分取HPLCによって精製した。
【0205】
HPLC分取法:逆相C18phenomenexカラムLuna、5マイクロメーター、150×21.20mm、20mL/分、20分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;20〜80%のアセトニトリル/0.1%ギ酸の勾配)。UV検出:254nm。HPLCは、1:1(5A:5B)のジアステレオマー比を示した。
【0206】
5A:(6mg;9%収率)Rt=8.5分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
1H NMR
(400 MHz, メタノール-d4) δ ppm 3.55 (d, J=7.8 Hz, 1 H), 3.58 (dd, J=7.5, 1.2 Hz, 1 H), 3.64
(t, J=8.2 Hz, 1 H), 3.67 (d, J=12.5 Hz, 1 H), 3.74 (s, 3 H), 3.77 (dd, J=8.3,
1.2 Hz, 1 H), 3.83 (d, J=12.5 Hz, 1 H), 3.91 (s, 2 H), 4.14 (d, J=7.4 Hz, 1 H),
6.76 - 6.84 (m, 2 H), 7.02 (dd, J=9.9, 8.3 Hz, 1 H), 7.09 - 7.13 (m, 2 H), 7.37
- 7.44 (m, 2 H); MS: 407.4 (M+ H+; ポジティブモード);
451.3 (M+HCO2-; ネガティブモード)
【0207】
5B:(12mg;17%収率)Rt=9分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
1H NMR
(400 MHz, メタノール-d4) δ ppm 3.51 (d, J=7.4 Hz, 1 H), 3.74 (s, 3 H), 3.75 (d, 1H, J = 13
Hz), 3.83 - 3.93 (m, 6 H), 4.03 (d, J=7.4 Hz, 1 H), 6.78 - 6.82 (m, 2 H), 7.02
(dd, J=9.9, 8.5 Hz, 1 H), 7.09 - 7.13 (m, 2 H), 7.42 - 7.49 (m, 2 H); MS: 407.4
(M+ H+; ポジティブモード); 451.3 (M+HCO2-;
ネガティブモード)
【0208】
(実施例6)
2−(4−メトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)−ベンゾニトリル(6A):
【0209】
【化40】
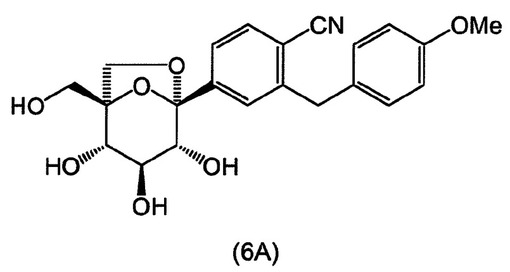
n−ブチルリチウム(1.04mL、2.6mmol、ヘキサン中2.5M)を、臭化イソプロピルマグネシウムの溶液(1.27mL、1.27mmol、テトラヒドロフラン中1M)に0℃で添加した。30分間撹拌した後、得られた混合物を−78℃に冷却し、4−ブロモ−2−(4−エトキシ−ベンジル)−ベンゾニトリル(380mg、1.20mmol)の無水テトラヒドロフラン(1mL)溶液を添加した。緑がかった混合物を−78℃で1時間撹拌し、(2R,3S,4S)−2,3,4,6−テトラキス−ベンジルオキシ−5−ベンジルオキシメチル−5−ヒドロキシ−ヘキサン酸メトキシ−メチル−アミド(I−6g)(700mg、0.972mmol)の無水テトラヒドロフラン(2mL)溶液を、非常にゆっくりと添加した(20分に亘る、5秒毎に1滴)。溶液を−78℃で1時間撹拌し、室温に3時間に亘ってゆっくり温めた。1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチし、次いで酢酸エチルで希釈した。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮し、粗生成物を得た。粗生成物は、ヘプタン中の0〜20%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、所望の中間体2−(4−エトキシ−ベンジル)−4−((4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−ベンジルオキシメチル−2−ヒドロキシ−テトラヒドロ−ピラン−2−イル)−ベンゾニトリル(300mg;34%収率)を得た。MS 918.8 (M+Na+, ポジティブモード).
【0210】
三塩化ホウ素(4.18mL、4.18mmol、ヘキサン中の1M溶液)を、上記の中間体(250mg、0.279mmol)のCH2Cl2(2mL)溶液に−78℃で添加した。混合物を−78℃で10分間撹拌し、次いで、室温に一晩温めた。混合物を水(10mL)でクエンチし、酢酸エチル(50mL)で抽出した。有機層を硫酸ナトリウム上で乾燥させ、蒸発乾固させた。シリカゲル(ジクロロメタン中のメタノールで溶出:1〜9容)上のフラッシュクロマトグラフィーによる精製によって、所望の中間体2−(4−ヒドロキシ−ベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクト−5−イル)−ベンゾニトリル(35mg、30%収率)を得た。
【0211】
炭酸カリウム(28mg、0.2mmol)を、上記の中間体(34mg、0.077mmol)のアセトン(0.4mL)溶液に添加し、続いてヨードメタン(7μL、0.11mmol)を室温で添加した。混合物を45℃で一晩撹拌した。混合物を酢酸エチル(60mL)で希釈し、水で洗浄した。有機層を硫酸ナトリウム上で乾燥させ、蒸発乾固させた。シリカゲル(ジクロロメタン中のメタノールで溶出:1〜9容)上の分取薄層クロマトグラフィーによる精製によって、所望の生成物6A(18mg;57%収率)の単離が可能となった。
1H NMR
(400 MHz, メタノール-d4) δ 7.69 (d, J = 8 Hz, 1H), 7.61 (s, 1H), 7.56 (d, J = 8 Hz, 1H),
7.19-7.14 (m, 2H), 6.87-6.82 (m, 2H), 4.18 (d, J = 7.6 Hz, 1H), 4.14 (s, 2H),
3.86 (d, J = 12.7 Hz, 1H); 3.81 (d, J = 8.3 Hz, 1H), 3.76 (s, 3H), 3.69
(d, J = 12.5 Hz, 1H), 3.67 (t, J = 8.1 Hz, 1H), 3.61 (d, J = 7.6 Hz, 1H), 3.54
(d, J = 8 Hz, 1H); MS 458.4 (M+HCO2-; ネガティブモード).
【0212】
(実施例7)
2−(4−エトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)−ベンゾニトリル(7A):
【0213】
【化41】
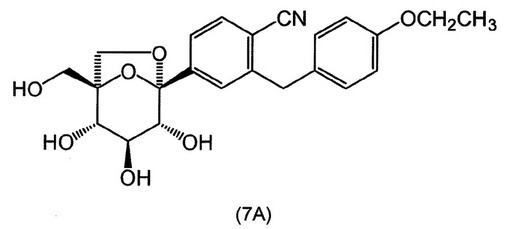
炭酸カリウム(8mg、0.058mmol)を、中間体2−(4−ヒドロキシ−ベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクト−5−イル)−ベンゾニトリル(実施例6を参照されたい;8.9mg、0.022mmol)のアセトン(0.4mL)溶液に添加し、続いてヨードエタン(4μL、0.044mmol)を室温で添加した。混合物を45℃で一晩撹拌した。混合物を酢酸エチル(60mL)で希釈し、水で洗浄した。有機層を硫酸ナトリウム上で乾燥させ、蒸発乾固させた。シリカゲル(ジクロロメタン中のメタノールで溶出:1〜9容)上の分取薄層クロマトグラフィーによる精製によって、所望の生成物7A(2.4mg;26%収率)の単離が可能となった。
1H NMR
(メタノール-d4) δ 7.69
(d, J = 8.0 Hz, 1H), 7.61 (d, J = 1.5 Hz, 1H), 7.56 (dd, J = 8.0, 1.5 Hz, 1H),
7.17-7.13 (m, 2H), 6.86-6.81 (m, 2H), 4.18 (d, J = 7.5 Hz, 1H), 4.14 (s, 2H),
4.01 (q, J = 7.0 Hz, 2H); 3.86 (d, J = 12.5 Hz, 1H); 3.80 (dd, J = 8.0および1.2 Hz, 1H), 3.70 (d, J = 11.7 Hz, 1H), 3.67 (t, J = 8.0 Hz, 1H),
3.61 (dd, J = 7.5および1.2 Hz, 1H), 3.54 (d, J = 7.8 Hz,
1H), 1.37 (t, J = 7.0 Hz, 3H); MS 472.1 (M+HCO2-; ネガティブモード).
【0214】
実施例8は、実施例3Bの構造および立体配置を確認するための、実施例3Bの化合物の結晶性誘導体の調製を例示する。
【0215】
(実施例8)
(8A)を得るための(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(3B)のペル4−ブロモベンゾイル化:
【0216】
【化42】
(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(3B)(11mg、0.026mmol)の無水テトラヒドロフラン(600μL)溶液に、室温でN,N−ジイソプロピルエチルアミン(32μL、7当量)および4−ジメチルアミノピリジン(3mg、0.9当量)、続いてパラ−ブロモベンゾイルクロリド(35mg、6当量)を添加し、得られた混合物を室温で62時間撹拌した。酢酸エチルおよび水を添加し、有機相を0.5Mの塩酸水溶液およびブラインで連続的に洗浄した。有機相を硫酸マグネシウム上で乾燥させ、濾過し、濃縮し、粗製物を、ヘプタン中の15〜30%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、27mgの生成物(90%収率)を得た。
1H NMR
(400 MHz, クロロホルム-d) δ 7.82 (m,
2H), 7.74-7.64 (m, 4H), 7.58-7.46 (m, 8H), 7.42-7.34 (m, 4H), 7.29 (d, 1H, J =
8.3 Hz), 6.89 (m, 2H), 6.63 (m, 2H), 6.04 (dd, 1H, J = 9.6および1 Hz), 5.98 (dd, 1H, J = 9.6および4.4 Hz), 5.89
(d, 1H, J = 4.4 Hz), 4.70 (d, 1H, J = 12.4 Hz), 4.65 (d, 1H, J = 12.4 Hz), 4.60
(d, 1H, J = 8 Hz), 3.98-3.88 (m, 3H), 3.73 (s, 3H).
【0217】
単結晶は、ヘプタンおよび酢酸エチルを溶媒として使用して蒸気拡散技術によって得た。融点=191℃。単結晶X線分析。代表的な結晶を調査し、1Åデータセット(最大sinΘ/λ=0.5)をBruker APEX II/R回折計で集めた。絶対配置の決定を容易にするために、フリーデル対を集めた。原子散乱因子は、International Tables for Crystallographyから取った。International Tables for Crystallography、第C巻、219頁、500、Kluwer Academic Publishers、1992を参照されたい。全ての結晶学的計算を、SHELXTLシステムによって容易にした。SHELXTL、バージョン5.1、Bruker AXS、(1997)を参照されたい。全ての回折計データは室温で集めた。関連する結晶、データ収集、および精密化を、下記の表1に要約する。
【0218】
【表1】

【0219】
試行構造は、直接法によって得た。この試行構造は常に精密化した。水素の位置を可能な限り計算した。メチル水素は、差フーリエ技術によって位置決めし、次いで理想化した。水素パラメータを構造因子計算に加えたが、精密化しなかった。最小二乗精密化の最終サイクルにおいて計算されたシフトは全て、0.1未満の相当する標準偏差であった。最終R指数は、3.71%であった。最終差フーリエは、電子密度の欠損または誤った配置を示さなかった。精密な構造は、SHELXTLプロッティングパッケージを使用してプロットした(図1)。絶対配置は、Flackの方法によって決定した。Flack,H.D.、Acta Crystallogr、A39、876、(1983)を参照されたい。
【0220】
実施例9は、実施例4Aの構造および立体配置を確認するための、実施例4Aの化合物の結晶性誘導体の調製を例示する。
【0221】
(実施例9)
(9A)を得るための(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(4A)のペル4−ニトロベンゾイル化:
【0222】
【化43】

0℃に冷却した(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(4A:10.6mg、0.024mmol)の無水テトラヒドロフラン(300μL)溶液に、N,N−ジイソプロピルエチルアミン(30μL、7当量)および4−ジメチルアミノピリジン(3mg、1当量)、続いてパラ−ニトロベンゾイルクロリド(27mg、6当量)を添加し、得られた混合物を60℃で6時間撹拌した。混合物を室温に冷却し、酢酸エチルおよび水を添加し、有機相を0.5Mの塩酸水溶液およびブラインで連続的に洗浄した。有機相を硫酸マグネシウム上で乾燥させ、濾過し、濃縮し、粗製物を、ヘプタン中の10〜50%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、18mgの生成物(73%収率)を得た。
1H NMR
(400 MHz, クロロホルム-d) δ 8.33 (m,
2H), 8.28-8.12 (m, 8H), 8.07 (m, 2H), 8.00 (m, 2H), 7.91 (m, 2H), 7.45-7.40 (m,
2H), 7.34 (d, 1H, J = 8.2 Hz), 6.87 (m, 2H), 6.64 (m, 2H), 6.13 (d, 1H, J = 8.6
Hz), 6.06 (t, 1H, J = 8.3 Hz), 5.86 (d, 1H, J = 8.1 Hz), 4.81 (d, 1H, J = 8.3
Hz), 4.75 (d, 1H, J = 12.7 Hz), 4.60 (d, 1H, J = 12.8 Hz), 4.06 (d, 1H, J = 8.5
Hz), 3.98-3.90 (m, 4H), 1.39 (t, 3H, J = 7 Hz).
【0223】
単結晶は、溶媒としてアセトニトリル/イソプロパノールからのゆっくりとした再結晶によって得た。融点=211℃。代表的な結晶を調査し、0.88Åデータセット(最大sinΘ/λ=0.57)をBruker APEX II/R回折計上で集めた。絶対配置の決定を容易にするために、フリーデル対を集めた。原子散乱因子は、International Tables for Crystallographyから取った。International Tables for Crystallography、第C巻、219頁、500、Kluwer Academic Publishers、1992を参照されたい。全ての結晶学的計算を、SHELXTLシステムによって容易にした。SHELXTL、バージョン5.1、Bruker AXS、(1997)を参照されたい。全ての回折計データは室温で集めた。関連する結晶、データ収集、および精密化を、下記の表2に要約する。
【0224】
【表2】

【0225】
試行構造は、直接法によって得た。この試行構造は常に精密化した。水素の位置を可能な限り計算した。メチル水素を差フーリエ技術によって位置決めし、次いで理想化した。水素パラメータを構造因子計算に加えたが、精密化しなかった。最小二乗精密化の最終サイクルにおいて計算されたシフトは全て、0.1未満の相当する標準偏差であった。最終R指数は、4.36%であった。最終差フーリエは、電子密度の欠損または誤った配置を示さなかった。
【0226】
精密な構造を、SHELXTLプロッティングパッケージ(図2)を使用してプロットした。絶対配置はFlackの方法によって決定した。Flack,H.D.、Acta Crystallogr.、A39、876、(1983)を参照されたい。
【0227】
(実施例10)
(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(10A)および(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(10B)
【0228】
【化44】
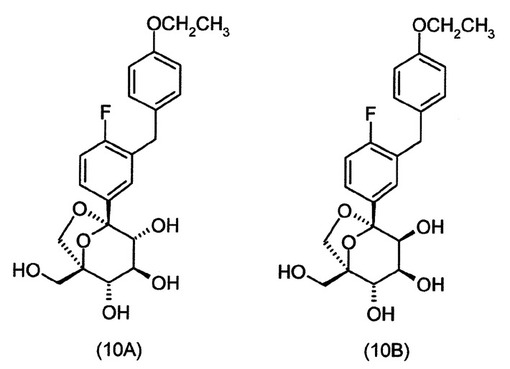
エタノール/テトラヒドロフランの4:1溶液(10mL)中の{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノールI−10k(80.0mg、0.120mmol)の溶液に、ギ酸(93μL、2.32mmol)およびパラジウム黒(62mg、0.580mmol)を連続的に添加した。得られた混合物を室温で撹拌した。3時間後、さらなるギ酸(93μL、2.32mmol)およびパラジウム黒(62mg、0.580mmol)を添加した。5時間後、反応混合物を濾過し、濾液を減圧下で濃縮した。得られた粗残渣は、シリカゲル(ジクロロメタン中の0〜15%メタノールの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、35.0mgの白色の固体(異性体の混合物)を得た。異性体の混合物を分取HPLCによって精製した。
【0229】
HPLC分取法:逆相C18Geminiカラム、5マイクロメーター、30×100mm、40mL/分の流量、18分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;25〜50%アセトニトリル/0.1%ギ酸の勾配;UV検出:220nm。
【0230】
HPLC分析法:逆相C18Geminiカラム、5μm、4.6×150mm、1mL/分の流量、12分に亘るアセトニトリル/0.1%トリフルオロ酢酸:水/0.1%トリフルオロ酢酸;5〜100%アセトニトリル/0.1%トリフルオロ酢酸の勾配;UV検出:220nm。
【0231】
10A:(2.2mg、4.5%収率)Rt=7分(分析法)。生成物含有画分を減圧下で濃縮した。
MS (LCMS) 421.4 (M+H+; ポジティブモード) 465.3 (M+HCO2-, ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ ppm 1.33 (t, J=7.0 Hz, 3 H), 3.53 (d, J=8.0 Hz, 1 H),
3.57 (dd, J=7.5, 1.5 Hz, 1 H), 3.60 - 3.67 (m, 2 H), 3.75 (dd, J=8.3, 1.3 Hz, 1
H), 3.81 (d, J=12.5 Hz, 1 H), 3.89 (s, 2 H), 3.96 (q, J=6.9 Hz, 2 H), 4.12 (d,
J=7.4 Hz, 1 H), 6.77 (m, 2 H), 7.00 (dd, J=9.4, 8.2 Hz, 1 H), 7.08 (m, 2 H),
7.36 - 7.41 (m, 2 H).
【0232】
10B:(1.8mg、3.7%収率)Rt=7.13分(分析法)。生成物含有画分を減圧下で濃縮した。
MS (LCMS) 421.4 (M+H+; ポジティブモード) 465.3 (M+HCO2-, ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ ppm 1.34 (t, J=7.0 Hz, 3 H), 3.51 (d, J=7.4 Hz, 1 H),
3.75 (d, 1 H, J = 12.5 Hz), 3.82 - 4.01 (m, 8 H), 4.03 (d, J=7.4 Hz, 1 H), 6.79
(m, 2 H), 7.02 (dd, J=9.8, 8.4 Hz, 1 H), 7.10 (m, 2 H), 7.41 - 7.49 (m, 2 H).
注記:分取HPLC後、これらの生成物を含有する画分を濃縮し、シリカゲル(ジクロロメタン中の0〜10%メタノールの勾配で溶出)上のフラッシュクロマトグラフィーによって再精製した。
【0233】
(実施例11)
(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(11A)
【0234】
【化45】
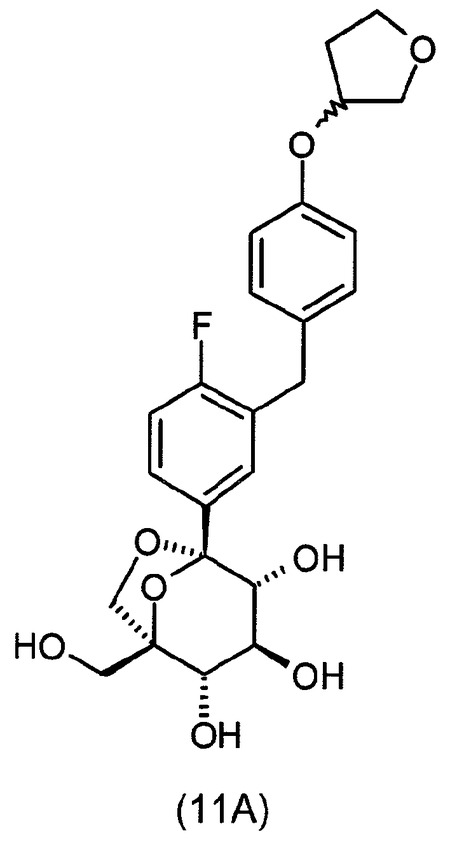
エタノール/テトラヒドロフランの4:1溶液(10mL)中の((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノールI−11k(160.0mg、0.218mmol)の溶液に、ギ酸(185μL、4.64mmol)およびパラジウム黒(148mg、1.39mmol)を連続的に添加した。得られた混合物を室温で撹拌した。3時間後、さらなるギ酸(185μL、4.64mmol)およびパラジウム黒(148mg、1.39mmol)を添加した。5時間後、反応混合物を濾過し、濾液を減圧下で濃縮した。得られた粗残渣は、シリカゲル(ジクロロメタン中の0〜15%メタノールの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、100mgの白色の固体(異性体の混合物)を得た。異性体の混合物を分取HPLCによって精製した。
【0235】
HPLC分取法:逆相C18Geminiカラム、5マイクロメーター、30×100mm、40mL/分の流量、18分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;25〜50%アセトニトリル/0.1%ギ酸の勾配;UV検出:220nm。
【0236】
HPLC分析法:逆相C18Geminiカラム、5マイクロメーター、4.6×150mm、1mL/分の流量、12分に亘るアセトニトリル/0.1%トリフルオロ酢酸:水/0.1%トリフルオロ酢酸;5〜100%アセトニトリル/0.1%トリフルオロ酢酸の勾配;UV検出:220nm。
【0237】
11A:(19mg、19%収率)Rt=6.43分(分析法)。生成物含有画分を減圧下で濃縮した。
1H NMR
(400 MHz, メタノール-d4) δ ppm 2.03 - 2.11 (m, 1 H), 2.15 - 2.25 (m, 1 H), 3.55 (d, 1 H, J = 8
Hz), 3.59 (dd, 1 H, J = 7.4および1 Hz), 3.61 - 3.69 (m, 2
H), 3.77 (dd, J=8.2および1 Hz, 1 H), 3.81 - 3.96 (m, 7 H),
4.14 (d, J=7.4 Hz, 1 H), 4.94 - 4.98 (m, 1 H), 6.79 (m, 2 H), 7.02 (dd, J=9.9,
8.5 Hz, 1 H), 7.12 (m, 2 H), 7.37 - 7.45 (m, 2 H).
【0238】
(実施例12)
(1S,2S,3S,4R,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオール(12A)および(1S,2S,3S,4S,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオール(12B)
【0239】
【化46】
エタノール/テトラヒドロフラン(2mL、4/1容)中の中間体I−12k(102mg)およびパラジウム黒(98mg、6.1当量)の混合物に、ギ酸(0.9mL)を添加し、得られた混合物を室温で撹拌した。1時間後、さらなるパラジウム黒(67mg、4.2当量)を添加し、混合物をさらに1時間室温で撹拌した。パラジウムを、Celite(登録商標)を通した濾過によって除去し、濾液を濃縮し、生成物の混合物を得た。この材料を、分取HPLCによる精製のために、粗材料の第2のバッチ(上記の手順に従って中間体I−12k(80mg)から調製)と合わせた。
【0240】
分取HPLC条件:逆相C18Geminiカラム、5マイクロメーター、30×100mm、流量40mL/分、18分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;25〜50%のアセトニトリル/0.1%ギ酸の勾配)、UV検出:220nm。
【0241】
HPLC分析法:逆相C18Geminiカラム、5μm、4.6×150mm、1mL/分の流量、12分に亘るアセトニトリル/0.1%トリフルオロ酢酸:水/0.1%トリフルオロ酢酸;5〜100%アセトニトリル/0.1%トリフルオロ酢酸の勾配;UV検出:220nm。
【0242】
12A:(18mg、16%収率)Rt=7.11分(分析法); MS
(LCMS) 411.3 (M+H+; ポジティブモード); 409.2 (M-H+;
ネガティブモード). 1H NMR (400 MHz, メタノール-d4) δ ppm 7.45-7.42 (m, 2H),
7.25 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H), 7.05 (dd, J = 9.6, 9.2 Hz,
1H), 4.15 (d, J = 7.6 Hz, 1H), 3.98 (s, 2H), 3.84 (d, J = 12.4 Hz, 1H), 3.78
(dd, J = 8.4, 1.2 Hz, 1H), 3.68 (d, J = 12.8 Hz, 1H), 3.66 (t, J = 8.2 Hz, 1H),
3.60 (dd, J = 7.4, 1.4 Hz, 1H), 3.56 (d, J = 7.6 Hz, 1H).
12B:(12mg、11%収率)Rt=7.25分(分析法); MS
(LCMS) 411.3 (M+H+; ポジティブモード); 409.1 (M-H+;
ネガティブモード). 1H NMR (400 MHz, メタノール-d4) δ ppm 7.52-7.45 (m, 2H),
7.25 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H), 7.05 (dd, J = 9.8, 8.6 Hz,
1H), 4.05 (d, J = 7.2 Hz, 1H), 3.98 (s, 2H), 3.91-3.84 (m, 4H), 3.76 (d, J =
12.4 Hz, 1H), 3.52 (d, J = 7.6 Hz, 1H).
【0243】
(実施例13)
(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(13A)
【0244】
【化47】

エタノール/テトラヒドロフランの4:1溶液(10mL)中の((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノールI−13k(300mg、0.417mmol)の溶液に、ギ酸(333μL、8.34mmol)およびパラジウム黒(266mg、2.50mmol)を連続的に添加した。得られた混合物を室温で撹拌した。3時間後、さらなるギ酸(333μL、8.34mmol)およびパラジウム黒(266mg、2.50mmol)を添加した。5時間後、反応混合物を濾過し、濾液を減圧下で濃縮した。得られた粗残渣は、シリカゲル(ジクロロメタン中の0〜15%メタノールの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、153.0mgの白色の固体(異性体の混合物)を得た。異性体の混合物を分取HPLCによって精製した。
【0245】
HPLC分取法:逆相C18Geminiカラム、5マイクロメーター、30×100mm、40mL/分の流量、18分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;25〜50%アセトニトリル/0.1%ギ酸の勾配;UV検出:220nm。
【0246】
13A:(23mg、12%収率)Rt=7.9分。生成物含有画分を減圧下で濃縮した。
1H NMR
(400 MHz, メタノール-d4) δ ppm 3.52 (d, J=7.8 Hz, 1 H), 3.57 (d, J=7.2 Hz, 1 H), 3.60 - 3.68
(m, 2 H), 3.75 (d, J=8.2 Hz, 1 H), 3.81 (d, J=12.5 Hz, 1 H), 3.89 (s, 2 H),
4.12 (d, J=7.4 Hz, 1 H), 4.63 (dd, J=7.3, 4.8 Hz, 2 H), 4.95 (t, J=6.5 Hz, 2
H), 5.16 - 5.23 (m, 1 H), 6.63 (m, 2 H), 7.00 (dd, J=9.7, 8.5 Hz, 1 H), 7.10
(m, 2 H), 7.36 - 7.42 (m, 2 H).
【0247】
(実施例14)
(1S,2S,3S,4R,5S)−5−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(14A)
【0248】
【化48】
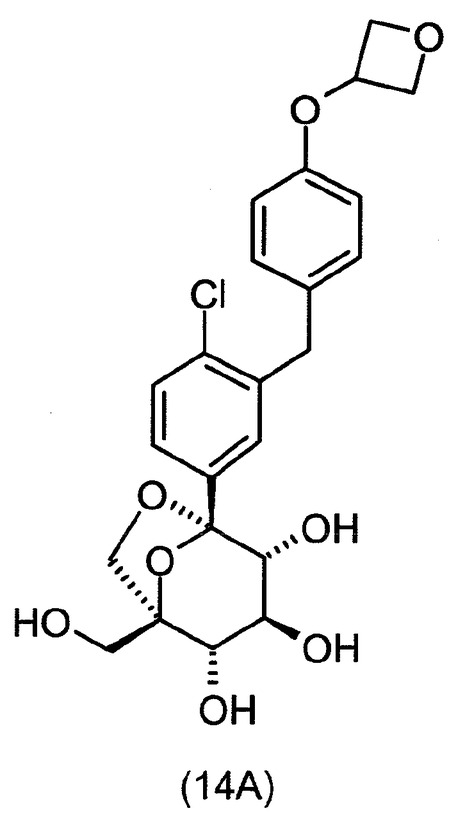
中間体((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノールI−14k(182mg)のエタノール/テトラヒドロフラン(14mL、4/1容)溶液に、ギ酸(190μL、20当量)およびパラジウム黒(106mg、4当量)を連続的に添加し、得られた混合物を室温で撹拌した。2時間後さらに1mLのテトラヒドロフランを添加し、得られた混合物を室温でさらに1時間撹拌した。この時点で、さらなるギ酸(190μL、20当量)およびパラジウム黒(106mg、4当量)を添加し、混合物をさらに1時間室温で撹拌した。パラジウムを濾過し、溶媒(異性体の混合物を含有)の蒸発の後に得られた粗混合物を分取HPLCによって精製した。
【0249】
HPLC分取法:逆相C18Xbridgeカラム、5マイクロメーター、100×30mm、流量40mL/分、11分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;30〜55%のアセトニトリル/0.1%ギ酸の勾配;UV検出:220nm。
【0250】
14A:(20mg、17%収率);Rt=4.43分。生成物含有画分を減圧下で濃縮し、白色の固体を得た。
MS (LCMS) 465.3 (M+H+; ポジティブモード); 509.2 (M+HCO2-; ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ ppm 3.53 (d, J=8.0 Hz, 1 H), 3.58 (dd, J=7.4, 1.4 Hz,
1 H), 3.64 (t, J=8.2 Hz, 1 H), 3.67 (d, J=12.4 Hz, 1 H), 3.77 (dd, J=8.4, 1.4
Hz, 1 H), 3.83 (d, J=12.6 Hz, 1 H), 4.03 (s, 2 H), 4.14 (d, J=7.4 Hz, 1 H),
4.65 (m, 2 H), 4.97 (t, J=6.6 Hz, 2 H), 5.22 (m, 1 H), 6.65 (m, 2 H), 7.11 (m,
2 H), 7.34 (d, J=8.4 Hz, 1 H), 7.38 (dd, J=8.4, 2.2 Hz, 1 H), 7.45 (d, J=2.0
Hz, 1 H).
【0251】
(実施例15)
(15)を得るためのL−プロリンとの(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
水(概ね480mg/mL)に溶解したL−プロリンを、実施例4Aの化合物に添加した(1モル(実施例4Aの化合物)毎に、概ね80モルのL−プロリン)。エタノールによって容量を2倍にし、溶液をキャップし、概ね12時間撹拌した。ベンチ上での蒸発によって容量を半分に減少させた。エタノールを使用して容量を2倍にし、蒸発を使用して溶液の容量をまた半分に減少させた。遠心濾過を使用して固体を回収した。
【0252】
(実施例16)
(16)を得るためのL−プロリンとの(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
水に溶解したL−プロリン(概ね480mg/mL)を、実施例4Aの化合物に添加した(実施例4Aの化合物1モル当たり概ね59モルのL−プロリン)。メタノールによって容量を2倍にし、溶液は透明であった。アセトンを使用して容量を25%増加させた。溶液をキャップし、概ね12時間撹拌した。ベンチ上での蒸発によって容量を概ね60%減少させた。メタノールを使用して容量を2倍にし、残りの溶媒を蒸発させ、固体の白色の沈殿物が残った。
【0253】
(実施例17)
(17)を得るためのL−プロリンとの(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
L−プロリンで飽和したエタノールの溶液を、ガラス製バイアル中の実施例4Aの化合物に添加した(実施例4Aの化合物1モル当たり概ね2.2モルのL−プロリン)。透明な溶液をキャップし、概ね72時間撹拌した。室温で蒸発させることによって容量を半分に減少させた。沈殿物が認められ、バイアルをキャップし、概ね12時間撹拌した。遠心濾過を使用して白色の固体を集めた。
【0254】
(実施例18)
(18)を得るためのL−プロリンとの(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
溶液が濁るまで、水に溶解したL−プロリン(330mg/mL)を、イソプロパノールに溶解した概ね2mLの実施例4Aの化合物に滴下した(98mg/mL)。15〜20分後、沈殿が観察され、懸濁液は濃厚となった。概ね8mLの水を添加し、溶液をキャップし、一晩撹拌した。真空濾過を使用して白色の固体を集め、50℃の真空オーブン中で概ね2時間乾燥させた。
【0255】
(実施例19)
(19)を得るためのL−ピログルタミン酸との(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
イソプロピルアルコール中の化合物(4A)(97.97mg/mL)(153μL)を、水中のL−ピログルタミン酸(213.0mg/mL)(500μL)中にピペットで移した。溶液をキャップし、一晩撹拌した。概ね5〜10mgのさらなる固体L−ピログルタミン酸を添加した。100μLのエタノールを添加した。溶液をキャップし、一晩撹拌した。総容量が概ね2mLに調整されるまでエタノールを添加した。溶液のキャップを取り、フード中に一晩置いた。概ね10〜30mgのさらなる実施例4Aの化合物を添加した。溶液をキャップし、概ね2日間撹拌した。白色の沈殿物が見出された。懸濁液を、0.45μmのナイロンフィルター膜インサートを備えたCo−star微量遠心分離管にピペットで移した。固体が溶液から分離されるまで溶液を遠心分離した。共結晶(19)を回収した。
【0256】
(実施例20)
(20)を得るためのL−ピログルタミン酸との(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
4〜5mLの1:1エタノール/水溶液をL−ピログルタミン酸(412.1mg/mL)で飽和させた。730mgの実施例4Aの化合物を、3.2mLのL−ピログルタミン酸溶液に添加した。概ね1分後、沈殿が見出された。溶液は濃厚すぎて撹拌することができず、したがって2mLの1:1エタノール/水溶液を添加した。溶液を一晩撹拌した。0.45μmのナイロンフィルター膜上の真空濾過を使用して固体を集めた。固体を50℃の真空オーブン中で概ね2時間乾燥させた。概ね960mgの共結晶複合体(20)を回収した。L−ピログルタミン酸に対する実施例4Aの化合物の化学量論比は、定量的NMRを使用して1:1.63であると決定した。材料をエタノール中で懸濁させることによって過剰なL−ピログルタミン酸を除去し、1:1の共結晶(20)を得た。
【0257】
(実施例21)
(21)を得るためのL−ピログルタミン酸との(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
494mgの実施例4Aの化合物を、1.5mLのイソプロパノールおよびエタノールの溶液(各々、4:1)に溶解した。917.2mgのL−ピログルタミン酸を3mLの水に溶解した。両方の溶液を40℃に加熱した。200μLのL−ピログルタミン酸溶液を、全ての溶液が移されるまでずっと実施例4Aの化合物溶液に添加した(溶液が移されない限り、両方の溶液をキャップする)。L−ピログルタミン酸溶液を有するバイアルを200μLのエタノールで洗浄し、溶液を実施例4Aの化合物溶液に移した。溶液を5分間撹拌し、次いで熱を止めた(溶液は3分毎に概ね摂氏1度冷却した)。30℃で、溶液を周囲温度の撹拌機に入れ、20℃で20分間撹拌した。溶液は透明であった。概ね2mLの乾燥種晶を添加した。次の2時間に亘り懸濁液は濃厚となった。溶液を一晩撹拌した。Pyrex2mL10〜15M焼結ガラス漏斗フィルター上の真空濾過を使用して固体を回収した。固体を50℃の真空オーブン中で24時間乾燥した。
【0258】
(実施例22)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−プロリンとの共結晶、および(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−ピログルタミン酸との共結晶:
粉末X線回折分析:実施例4Aの化合物とL−プロリンとの共結晶、および実施例4Aの化合物とL−ピログルタミン酸との共結晶の粉末X線回折パターンは、銅放射線(波長:1.54056Å)を使用したBruker D5000回折計で行った。管電圧およびアンペア数を、各々、40kVおよび40mAに設定した。発散スリットおよび散乱スリットは1mmに設定し、受光スリットは0.6mmに設定した。回折放射線は、Kevex PSI検出器によって検出した。3.0から40°2θの毎分2.4°(0.04°ステップ毎に1秒)でのθ−2θ連続スキャンを使用した。アルミナ標準物質を分析し、機器の整合性を点検した。データを集め、Bruker axisソフトウェアバージョン7.0を使用して分析した。試料は、それらを石英ホルダー中に置くことによって調製した。Bruker InstrumentsはSiemensを買収したことに留意すべきである。したがって、Bruker D5000機器は、Siemens D5000と本質的に同じである。Eva Application13.0.0.3ソフトウェアを使用して、PXRDスペクトルを可視化および評価した。PXRDデータファイル(未加工)は、ピーク探索の前に処理しなかった。一般に、2の閾値および0.3の幅値を使用して、予備的ピーク割当を作製した。自動化割当のアウトプットを視覚的に検査し、妥当性を確実にし、必要に応じて調節を手動で行った。
【0259】
本明細書において報告した測定のために使用するBrukerシステムなどのBragg−Brentano機器上でX線回折測定を行うために、試料を典型的には、空洞を有するホルダー中に置く。試料粉末をスライドガラスまたは相当するものによって押し、ランダム面および妥当な試料高さを確実にする。次いで、試料保持器を機器中に置く。入射X線ビームを試料に向け、最初にホルダーの面に対して低角度で、次いで入射ビームおよびホルダーの面の間の角度を連続的に増加させる弧に沿って動かす。このようなX線粉末分析に関連する測定の差異は、(a)試料調製におけるエラー(例えば、試料高さ)、(b)機器エラー(例えば、平板試料エラー)、(c)較正誤差、(d)操作者のエラー(ピーク位置を決定するときに生じるそれらのエラーを含めて)、および(e)材料の性質(例えば、選択方位および透明性のエラー)を含めた種々の要因からもたらされる。較正誤差および試料高さのエラーは、同じ方向への全てのピークのシフトをもたらすことが多い。フラットホルダーを使用するときの試料高さの小さな差異は、XRPDピーク位置の大きな変位をもたらす。典型的なブラッグ−ブレンターノ配置においてShimadzu XRD−6000を使用して、1mmの試料高さの差異は、1°2θもの高いピークシフトをもたらすことが系統的研究によって示された(Chenら;J Pharmaceutical and Biomedical Analysis、2001;26,63)。これらのシフトは、X線ディフラクトグラムから同定することができ、シフトを相殺する(全てのピーク位置値に系統的補正因子を適用する)ことによって、または機器を再較正することによって除去することができる。上記のように、様々な機械からの測定を、系統的補正因子を適用することによって修正し、ピーク位置と一致させることが可能である。一般に、この補正因子は、Brukerから測定したピーク位置を予想されるピーク位置と一致させるが、0〜0.2°2θの範囲のことがある。
【0260】
粉末X線回折値は一般に、機器および試験条件の僅かな変動によって±0.2の2θ度以内で正確である。
【0261】
実施例18からの実施例4Aの化合物とL−プロリンとの共結晶は、図3において提供する下記の粉末X線回折パターンによって特徴付けられる(2θ度および相対強度に関して表す、CuKα放射線によるBruker D5000回折計で測定して≧2.7%の相対強度を有する)。
【0262】
【表3】
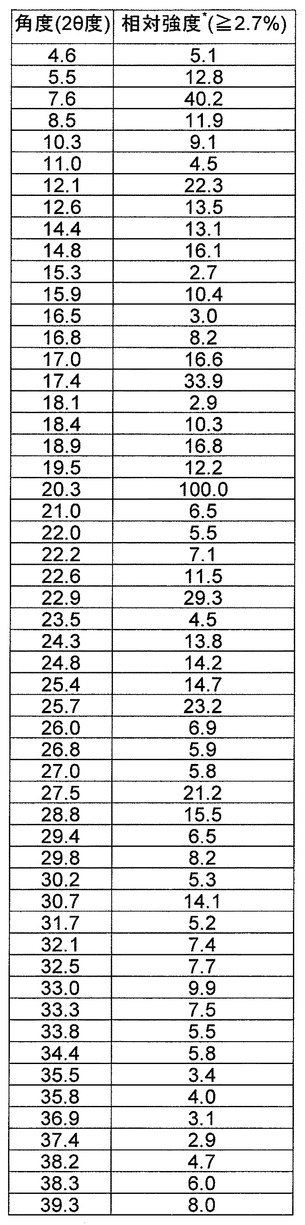
*相対強度は、結晶サイズおよび形態によって変化し得る。
【0263】
実施例4Aの化合物とL−プロリンとの共結晶の特徴的な2θピークまたは組合せ。
【0264】
【表4】
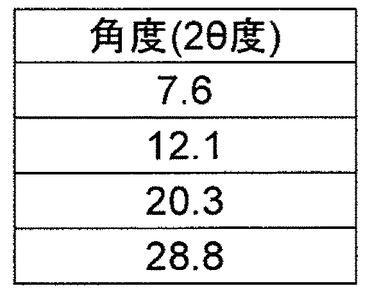
【0265】
実施例20からの実施例4Aの化合物とL−ピログルタミン酸との共結晶は、図4において提供する下記の粉末X線回折パターンによって特徴付けられる(2θ度および相対強度に関して表す、CuKα放射線によるBruker D5000回折計で測定して≧2.7%の相対強度を有する)。
【0266】
【表5】
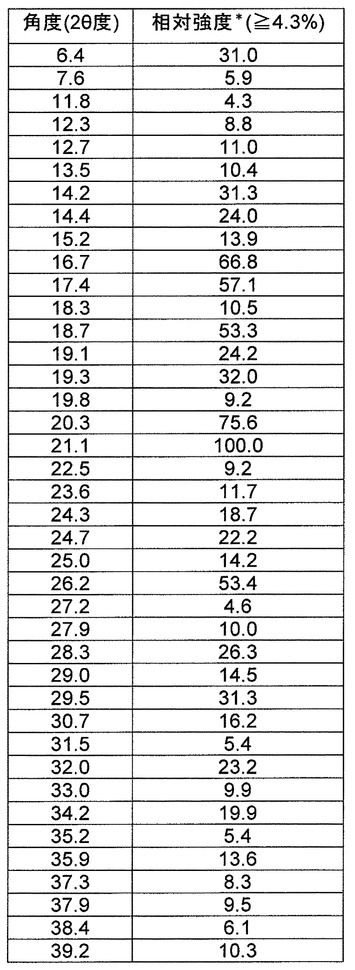
*相対強度は、結晶サイズおよび形態によって変化し得る。
【0267】
実施例4Aの化合物とL−ピログルタミン酸との共結晶の特徴的な2θピークまたは組合せ。
【0268】
【表6】
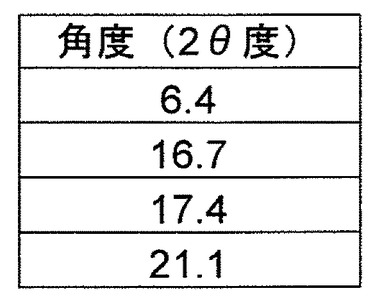
【0269】
(実施例23)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−プロリンとの共結晶、および(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−ピログルタミン酸との共結晶:
示差走査熱量測定サーモグラム分析:
サーモグラムは、TA Instruments Q1000示差走査熱量計(DSC)で得た。1〜2mgの試料をアルミニウム試料パン中に置き、次いでせん孔された蓋で覆った。温度が25℃から200〜300℃に毎分10℃で上がるにつれ、空のパンに対してエネルギーを測定した。融解吸熱の開始温度は、融解温度として報告した。融解吸熱の開始温度は、他の要因の中でも加熱速度、試料の純度、結晶および試料のサイズによって決まる。典型的には、DSC結果は、約±2℃以内、好ましくは±1.5℃以内で正確である。
【0270】
実施例4Aの化合物とL−プロリンとの実施例18の共結晶のDSCの結果を図5に示す。
【0271】
実施例4Aの化合物とL−ピログルタミン酸との実施例20の共結晶のDSCの結果を図6に示す。
【0272】
(実施例24)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−プロリンとの共結晶
単結晶X線分析。実施例17からの濾液を使用し、ゆっくりとした蒸発による濃縮を使用した代表的な結晶を調査し、0.85Åデータセット(最大sinΘ/λ=0.60)をBruker APEX回折計で集めた。絶対配置の決定を容易にするために、フリーデル対を集めた。原子散乱因子は、International Tables for Crystallography、第C巻、219頁、500、Kluwer Academic Publishers、1992から取った。全ての結晶学的計算を、SHELXTLシステム、バージョン5.1、Bruker AXS、1997によって容易にした。全ての回折計データは室温で集めた。関連する結晶、データ収集、および精密化を、表24−1に要約する。
【0273】
試行構造は直接法によって得た。この試行構造は、予想外の水分子およびL−プロリンによる配座障害以外は常に精密化した。L−プロリンは、「半いす形」および「封筒型」配座において約60/40占有率でモデル化した。非常に同様の障害が、H.D.Flack、Acta Crystallogr.、A39、876、1983において観察された。
【0274】
N1、O6およびO7に結合している水素原子は差フーリエ技術によって位置決めし、距離を制限して精密化した。O5に結合した関連する水素原子をフーリエ技術から位置決めしたが、削除し、理想化した位置(HFIX83)に配置した。O4に結合した関連する水素原子は、フーリエ技術で見出すことができず、理想化した位置(HFIX83)に配置した。水分子上の水素原子は、位置決めすることができず、溶液から除外した。水素パラメータを構造因子計算に加えたが、精密化しなかった。最小二乗精密化の最終サイクルにおいて計算されたシフトは全て、0.1未満の相当する標準偏差であった。最終R指数は5.15%であった。最終差フーリエは、電子密度の欠損または誤った配置を示さなかった。
【0275】
精密な構造は、SHELXTLプロッティングパッケージを使用してプロットした(図7)。絶対配置は、Flackの方法によって決定した4。座標、異方性温度因子、距離および角度は、補助的材料として入手可能である(表24−2〜24−5)。
【0276】
【表7】

【0277】
【表8】
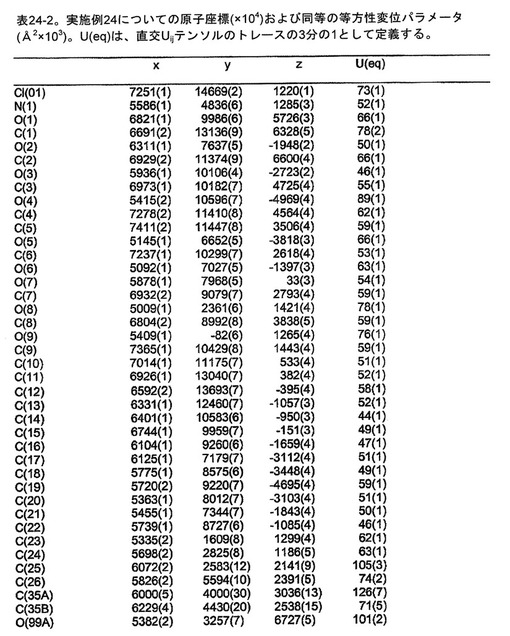
【0278】
【表9−1】

【0279】
【表9−2】

【0280】
【表9−3】

【0281】
【表9−4】

【0282】
【表9−5】

【0283】
【表9−6】

【0284】
【表10】

【0285】
【表11】
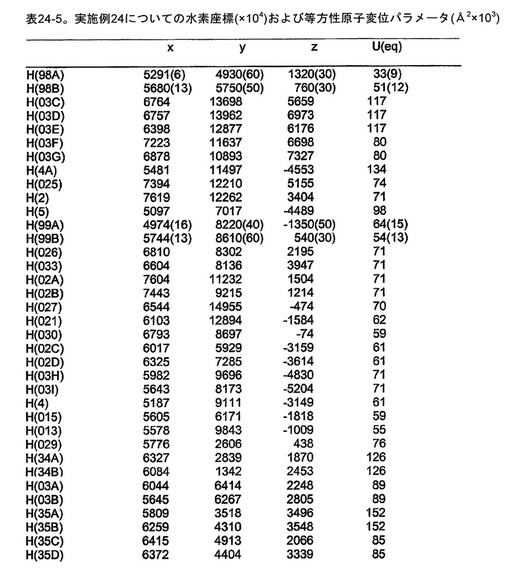
【0286】
(実施例25)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−ピログルタミン酸との実施例20からの共結晶:
単結晶X線分析。実施例20からの試料の代表的な結晶を調査し、0.90Åデータセット(最大sinΘ/λ=0.56)をBruker APEX回折計で集めた。絶対配置の決定を容易にするために、フリーデル対を集めた。立体配置は、flackパラメータから、およびまた共形成物(L−ピログルタミン酸)の既知のキラリティーから決定した。原子散乱因子は、International Tables for Crystallography、第C巻、219頁、500、Kluwer Academic Publishers、1992から取った。全ての結晶学的計算を、SHELXTLバージョン5.1、Bruker AXS、1997システムによって容易にした。全ての回折計データは室温で集めた。関連する結晶、データ収集、および精密化を、表25−1に要約する。
【0287】
試行構造は直接法によって得た。この試行構造は、0.1化学量論的水として精密化された低い残差ピーク以外は常に精密化した。水の化学量論は、分子上のヒドロキシル基を最初に削除し、精密化し、得られたqピークを測定し、次いでこのピークを水分子からの残差ピークと比較することによって見出した。この方法を使用して、1〜0.1の比(水に対する分子)を推定した。さらに、溶液から水分子を除去し、Material Studio、Platon、およびMercuryによって結晶における空隙空間を捜索することによって、水分子について33立法オングストロームという可能性が高い容量が明らかになった(水は典型的には約40立法オングストロームの空間を有する)。窒素および酸素上の水素原子は、差フーリエ技術によって位置決めし、制約なしに自由に精密化した。ヘテロ原子に結合しているプロトンのいくつか(H97a、H97b、H97c、およびH97c)は、いくらか短い結合距離(予測値約0.96に対して実測値約0.8オングストローム)を示したが、距離は規制しなかった。O99上の水素原子(水)は差異マップからは見出せず、構造解明から除外した。水素パラメータを構造因子計算に加えたが、精密化しなかった。最小二乗精密化の最終サイクルにおいて計算されたシフトは全て、0.2未満の相当する標準偏差であった。最終R指数は、3.58%であった。最終差フーリエは、電子密度の欠損または誤った配置を示さなかった。残った残差のうち、1つはO39に結合したプロトン(カルボン酸)について合理的な位置にある。この残差は、H98aプロトン(O39に結合したプロトン)についてのさらなる占有位置のことがあるが、このように精密化しなかった。
【0288】
精密な構造は、SHELXTLプロッティングパッケージを使用してプロットした(図8)。絶対配置は、Flack(H.D.Flack、Acta Crystallogr.、A39、876、1983)の方法によって決定した。座標、異方性温度因子、距離および角度は、補助的材料として入手可能である(表25−2から25−5)。
【0289】
【表12】

【0290】
【表13】

【0291】
【表14−1】

【0292】
【表14−2】

【0293】
【表14−3】

【0294】
【表15】

【0295】
【表16】
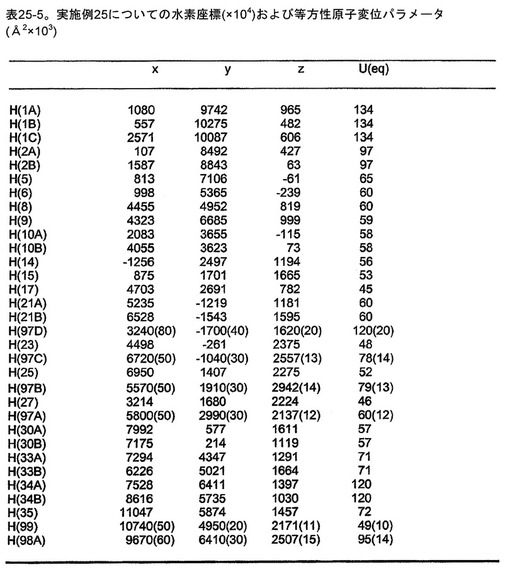
【0296】
(実施例26)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−ピログルタミン酸との共結晶:
固体NMR:
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とスキーム2に記載されている方法を使用して調製されたL−ピログルタミン酸との概ね80mgの共結晶を、4mmのZrO2ローター中にしっかりと詰めた。スペクトルは、大口径Bruker−Biospin DSX500MHz(1H周波数)NMR分光計中に位置するBruker−Biospin4mm BL CPMASプローブで室温および室内圧力にて集めた。詰めたローターはマジック角で方向付け、15.0kHzで回転した。13C固体スペクトルは、プロトンデカップル交差分極マジック角回転実験(CPMAS)を使用して集めた。交差分極接触時間は、2.0msに設定した。概ね85kHzのプロトンデカップリング領域を適用した。14秒の待ち時間で1448スキャンを集めた。炭素スペクトルは結晶性アダマンタンの外部標準を使用して参照し、その高磁場共鳴を29.5ppmに設定した。化学シフトデータは、他の要因の中でも、試験条件(すなわち、スピニング速度および試料保持器)、参照材料、およびデータプロセシングパラメータによって決まる。典型的には、ss−NMR結果は、約±0.2ppm以内で正確である。
【0297】
【表17】
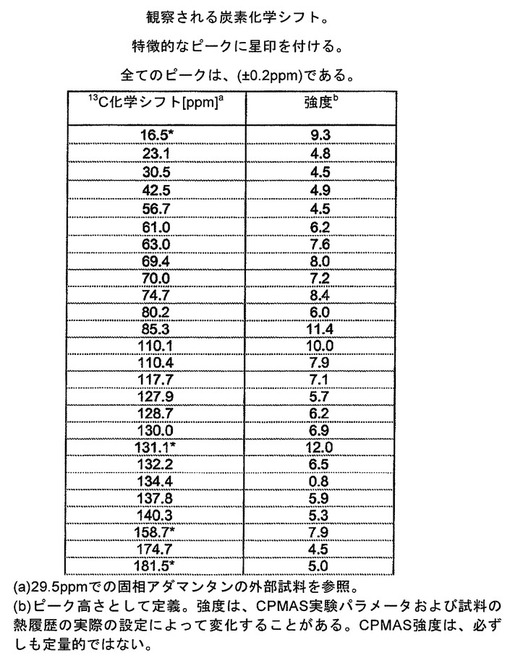
【0298】
実施例26のSSNMR13C CPMASスペクトルを図9に示す。図9においてアスタリスクによってマークされているピークは、スピニングサイドバンドである。
【0299】
薬理試験
SGLT2の阻害によって調節される疾患の治療のための本発明の実施は、本明細書において下記に記載されているプロトコルの少なくとも1つにおける活性によって証明することができる。
【0300】
生物学的アッセイ
インビトロアッセイ
SGLT2機能アッセイは、SGLT2輸送体によるメチル−α−Dグルコピラノシド(AMG−グルコースの代謝可能でない形態)取込みの阻害を検出するために設計した。SGLT2輸送体は、腎臓の近位尿細管からのグルコースを回収する。その阻害によって、糖が尿中に廃棄されることをもたらす。陽性対照化合物であるフロリジンは、SGLT2についてのグルコース取込みの既知の阻害剤である。それを試験化合物のSGLT2阻害の高率の作用を比較するために使用した。
【0301】
ヒトSGLT2(pcDNA5/FRT)を安定的に発現しているCHO−FlpIn(Invitrogen、Carlsbad、CA)細胞を、100μLの増殖培地(1:1のF−12/DMEM培地(Gibco、Carlsbad、CA)、10%FBS(Sigma、St.Louis MO)、1×Pen/Strep(Gibco、Carlsbad、CA)、600μg/mLのハイグロマイシン(Invitrogen、Carlsbad、CA))中で、100,000個の細胞/ウェルの密度で、Iso−TC96ウェルプレート(Perkin Elmer、Waltham、MA)に蒔いた。試験化合物で処理する前に、コンフルエントな細胞を、1:1のF−12/DMEM培地(1時間後、新鮮なF−12/DMEM培地と交換)中で37℃にて2時間、血清飢餓培養を行った。ジメチルスルホキシド(Sigma、St.Louis、MO)中の試験化合物は、取込み緩衝液で予めすすいだ細胞プレートに、取込み緩衝液(140mMのNaCl(Promega、Madison、WI)、2mMのKCl(Teknova、Hollister、CA)、1mMのCaCl2(Teknova、Hollister、CA)、1mMのMgCl2(Teknova、Hollister、CA)、および10mMのHEPES(Gibco、Carlsbad、CA))中で100倍に希釈した。ウェル毎に50μLのAMG(非標識AMG(Aldrich、St.Louis、MO)中の40nCi AMG[U−14C](Perkin Elmer、Waltham、MA))を添加(11.33μMのAMGの最終濃度を生じさせた)する前に、細胞を試験化合物と共に15分間プレインキュベートした。次いで、細胞プレートを、AMG取込みのために37℃にて3時間インキュベートした。インキュベーション後、細胞を氷冷の洗浄緩衝液(200μMのフロリジン(Sigma)を含有する取込み緩衝液)で2度洗浄し、空気乾燥させ、オービタルシェーカーで30μLのNaOH(200mM)および1%SDS緩衝液に溶解させた。Microscint40(Perkin Elmer、Waltham、MA)を溶解した細胞(200μLの最終容量とする)に添加し、30分間オービタルシェーカーにかけることによって混合した。プレートを暗所で一晩保管し、14C検出のための正規化されたプロトコルを使用して1540Microbeta Trilux(Wallac、Waltham、MA)中で定量化した。AMG取込みを阻害する試験化合物の作用割合は、下記の計算を使用して計算した。
[作用%=((ZPE−T)/(ZPE−HPE))×100%]
式中、「ZPE」は、0.5%DMSOを含有する対照ウェル中の毎分補正計数(CCPM)であり、Tは、様々な濃度の標準曲線における試験化合物を含有するウェル中のCCPMであり、HPEは、10μMのフロリジンを含有する対照ウェル中のCCPMに関する高率作用である。IC50値は用量反応式を使用して計算した。表3において試験した化合物について要約する。
【0302】
インビトロ試験の記載において使用された略語には、以下が含まれる。
SGLT2 ナトリウム/グルコース共輸送体2型
AMG メチル−α−Dグルコピラノシド
DMEM ダルベッコ改変イーグル培地
IC50 50%阻害濃度
FBS ウシ胎児血清
DMSO ジメチルスルホキシド
SDS ドデシル硫酸ナトリウム
CHO−FlpIn FRT部位を含有するチャイニーズハムスター卵巣細胞
【0303】
【表18−1】
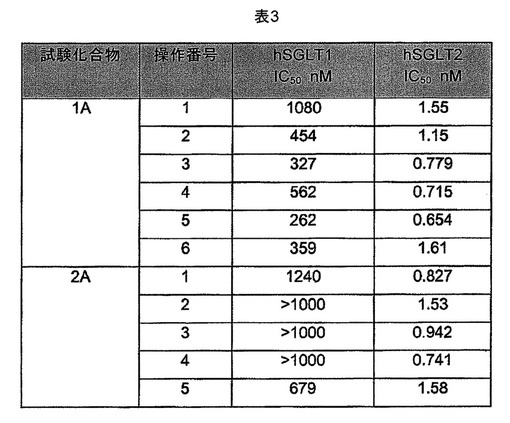
【0304】
【表18−2】
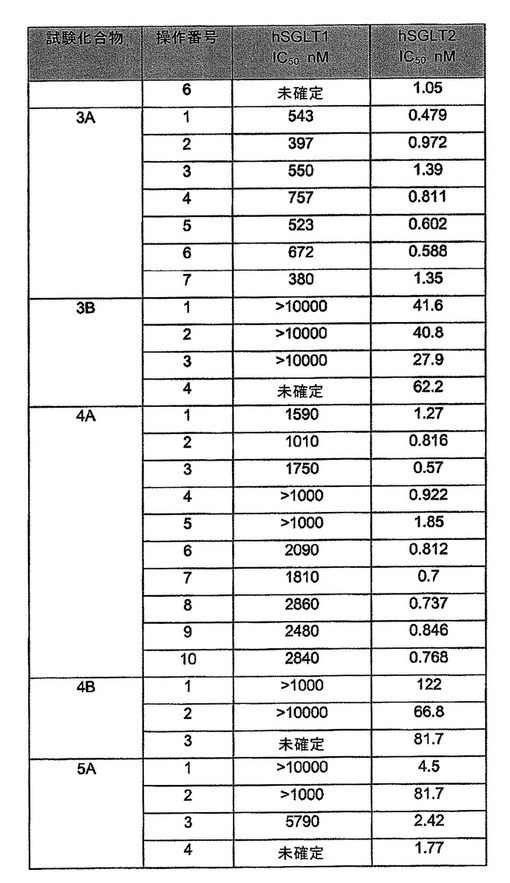
【0305】
【表18−3】

【0306】
【表18−4】
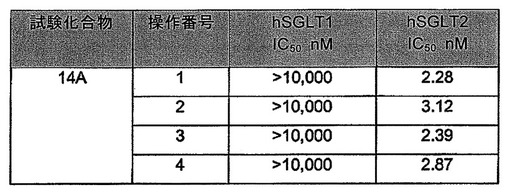
【0307】
インビボアッセイ
実施例1Aおよび4Aをラットにおいて試験し、尿中グルコース排泄によるグルコース輸送の阻害を評価した。雄性Sprague−Dawleyラット(約300g)は、尿採取のための代謝ケージに単独で収容した。ラットは、標準的飼料および水に自由にアクセスできた。ラット(n=2〜5/群)に、経口胃管栄養法によってビヒクルまたは化合物を投与した。投与溶液は、各々、0.1mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgおよび60mg/kg用量について、0.03mg/mL、0.3mg/mL、0.9mg/mL、3mg/mL、9mg/mLおよび18mg/mLであった。投与容量は、全ての用量について1mL/300g(体重)であった。1つの群は、10mg/kg用量の実施例1Aを投与し、他の群は、0.1、1、3、10、30または60mg/kg用量の実施例4Aを投与した。ビヒクルは、20%v/vのPEG400および24%v/vのヒドロキシプロピルβシクロデキストリン(HPBCD)であった。経口投与に続いて、尿を24時間集めた。Roche Hitachi917分光光度計(Diamond Diagnostics、Holliston、MA)を使用した340nmでのUV吸光度分光測定によって、グルコース濃度を尿中で測定した。尿中に排泄されるグルコースの総量を、下記の式を使用して尿中濃度および尿量の積として計算した。
排泄される尿中グルコース(mg)/200g(体重)=尿中グルコース濃度(mg/dL)×尿量(dL)×200/ラット体重(g)。排泄される尿中グルコース(UGE)の量は、上記の方法によって実施例1Aおよび実施例4Aについてラットから得た。それを表4に示す。血液(0.1mL)をPKサテライト群動物から投与後1、2、4、7、24時間に集め、血漿を得て、LC−MS/MSによって分析した。試験した様々な用量における平均PKパラメータを、表4に示す。
【0308】
【表19】
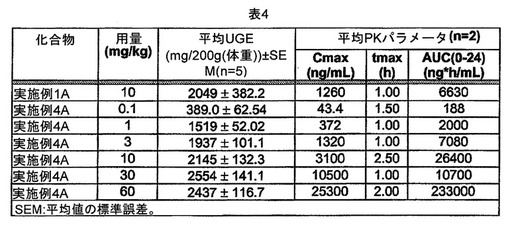
【0309】
ラットにおける薬物動態試験
実施例1A、2A、4A、12A、および14Aをラットで試験し、最高濃度(Cmax)、血漿濃度時間曲線下面積(AUC)、クリアランス(CL)、定常状態分布容積(Vss)、半減期(t1/2)、およびバイオアベイラビリティー(F)を含めた薬物動態パラメータを評価した。雄性Sprague−Dawleyラット(約300g)を使用した。ラットに静脈内(IV)または経口胃管栄養法(PO)投与によって化合物を投与した。投与溶液を製剤するための、ビヒクルを含めた試験した用量を、表5に一覧表示する。
【0310】
IVまたはPO投与に続いて、0.2mLの血液を様々な時点(表5)で頸静脈から試料採取した。20μL分量の血漿試料および標準物質を、内部標準を含有するアセトニトリルによるタンパク質沈殿に供した。試料をボルテックスし、遠心分離し、上清を得て、それをLC−MS/MSによって分析した。Analyst(バージョン1.4.1)を使用してピーク面積を測定し、分析物と内部標準とのピーク面積比を計算した。LC−MS/MS条件は、下記の通りである。質量分析計+ソースタイプは、Sciex API4000−Turbo Sprayであった。HPLCポンプは、島津製作所であった。オートサンプラーは、CTC PALオートサンプラーであった。注入量は、3.0〜10μLであった。勾配は、移動相A:水中の10mMの酢酸アンモニウムおよび1%イソプロピルアルコール;B:アセトニトリルで使用した。流量、毎分0.300mL(カラム2.0×30mm、5μm、LUNA C18カラム(phenomenex))。検出モードはネガティブであった。
【0311】
較正曲線は、重み付き線形(1/xまたは1/x2)回帰を適用することによって、標準物質の内部標準に対するピーク面積比から構築した。標準曲線の測定範囲は、5.00ng/mL〜5000ng/mLであった。
【0312】
薬物動態パラメータは、Watson(バージョン7.2)でノンコンパートメント分析を使用して個々の動物データから決定した。定量下限未満(BLQ)の濃度は、計算において使用するために0ng/mLとして記録した。
【0313】
下記の計算を使用した。
AUC(0−τ)=線形台形法を使用して決定
AUC(0−∞)=AUC(0−τ)、およびτにおける血漿濃度を最終対数線形相の勾配で割ることによって決定した外挿領域
CL=用量/AUC(0−∞)
Vdss=CL×MRT
Cmax=血漿濃度時間曲線から直接記録
Tmax=血漿濃度時間曲線から直接記録
t1/2=ln(0.5)/最終対数線形相の勾配
F%=用量毎のAUC(0−∞)PO/用量毎のAUC(0−∞)IV
C(0)=IV投与後の明らかな分布相からの線形回帰によって外挿する
MRT=AUMC(AUC(0−∞)/AUC(0−∞)
【0314】
【表20】
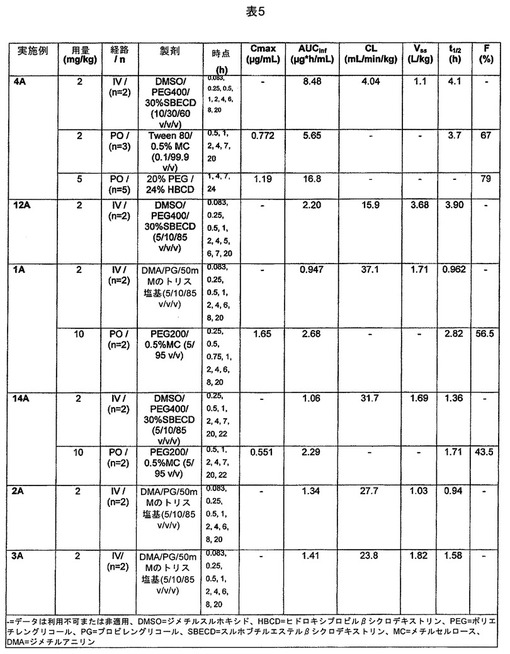
【0315】
本出願に亘って、様々な公開資料を参照する。これらの公開資料の開示はその全体が、全ての目的のために参照により本出願に組み込まれている。
【0316】
本発明の範囲または趣旨を逸脱することなく、本発明において様々な改変および変形を行えることは当業者であれば明らかであろう。本発明の他の実施形態は、ここにおいて開示されている本発明の明細書および実施を考慮すると当業者であれば明らかであろう。実施例を含めた明細書は例示のみとして考えられ、本発明の本当の範囲および精神は下記の特許請求の範囲によって示されることを意図する。
【図1】
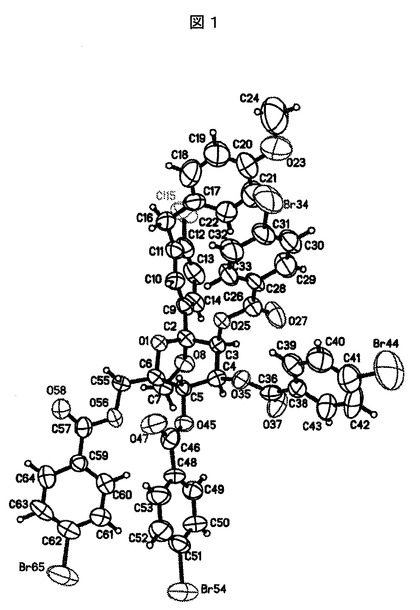
【図2】
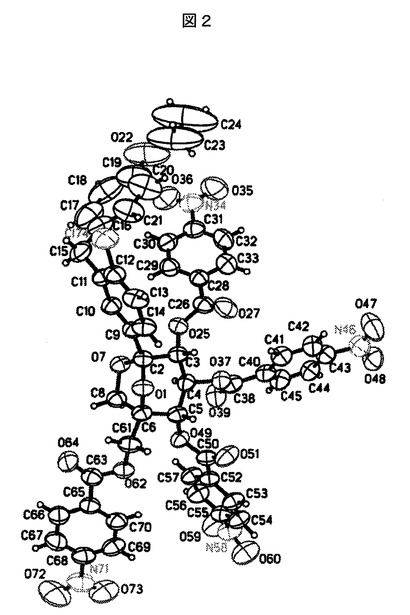
【図3】
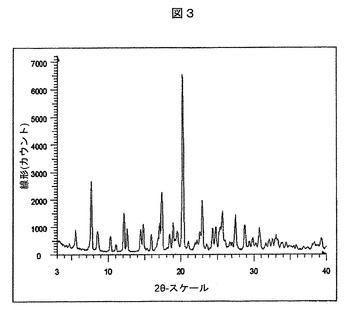
【図4】
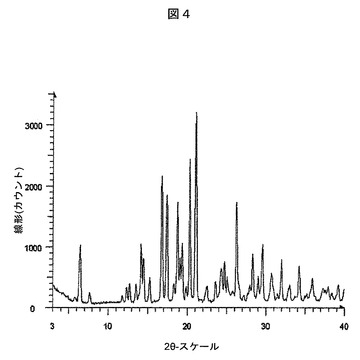
【図5】
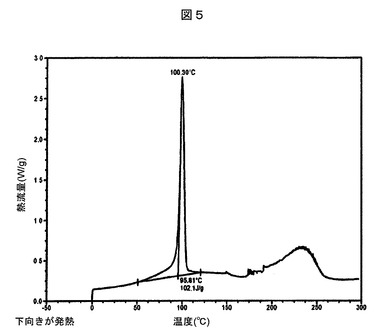
【図6】

【図7】
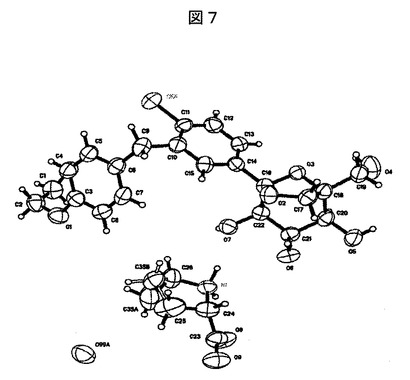
【図8】

【図9】
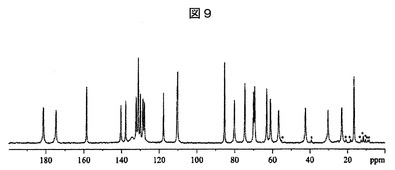
【図2】SHELXTLプロッティングパッケージを使用してプロットした実施例9Aの化合物についての精密な結晶構造を表す図である。
【図3】実施例22:実施例4Aの化合物とL−プロリンとの実施例18の共結晶について観察された粉末X線回折パターンを表す図である。
【図4】実施例22:実施例4Aの化合物とL−ピログルタミン酸との実施例20の共結晶について観察された粉末X線回折パターンを表す図である。
【図5】実施例23:実施例4Aの化合物とL−プロリンとの実施例18の共結晶について観察された示差走査熱量測定サーモグラムを表す図である。
【図6】実施例23:実施例4Aの化合物とL−ピログルタミン酸との実施例20の共結晶について観察された示差走査熱量測定サーモグラムを表す図である。
【図7】SHELXTLプロッティングパッケージを使用してプロットした実施例24:実施例4Aの化合物とL−プロリンとの共結晶についての精密な結晶構造を表す図である。
【図8】SHELXTLプロッティングパッケージを使用してプロットした実施例25:実施例4Aの化合物とL−ピログルタミン酸との共結晶についての精密な結晶構造を表す図である。
【図9】実施例26:実施例4Aの化合物とL−ピログルタミン酸との共結晶についての観察される13C固体核磁気共鳴スペクトルを表す図である。アスタリスクでマークされているピークは、スピニングサイドバンドである。
【発明を実施するための形態】
【0019】
下記の本発明の例示的実施形態の詳細な説明およびその中に含まれている実施例を参照することによって、本発明はさらにより容易に理解し得る。
【0020】
本発明の化合物、組成物および方法を開示および記載する前に、本発明は、当然ながら変化し得る作製の特定の合成法に限定されないことを理解すべきである。本明細書において使用される用語は、特定の実施形態を記載する目的のためにすぎず、限定的なものではないこともまた理解すべきである。複数形および単数形は、数が示されている場合以外は、交換可能なものとして扱うべきである。
【0021】
本明細書において使用する場合、「アルキル」という用語は、一般式CnH2n+1の炭化水素基を意味する。アルカン基は、直鎖状または分岐状であってよい。例えば、「(C1〜C6)アルキル」という用語は、1〜6個の炭素原子を含有する一価の直鎖状または分岐状の脂肪族基(例えば、メチル、エチル、n−プロピル、i−プロピル、n−ブチル、i−ブチル、s−ブチル、t−ブチル、n−ペンチル、1−メチルブチル、2−メチルブチル、3−メチルブチル、ネオペンチル、3,3−ジメチルプロピル、ヘキシル、2−メチルペンチルなど)を意味する。同様に、アルコキシ、アシル(例えば、アルカノイル)、アルキルアミノ、ジアルキルアミノ、アルキルスルホニル、およびアルキルチオ基のアルキル部位(すなわち、アルキル部分)は、上記と同じ定義を有する。「置換されていてもよい」と示されているとき、アルカン基またはアルキル部分は、非置換であるか、または「置換されている」についての定義において下記で一覧表示した置換基の群から独立に選択される1個または複数の置換基(ペルクロロまたはペルフルオロアルキルなどのハロゲン置換基の場合を除いて、一般に1〜3個の置換基)で置換されていてもよい。「ハロ置換アルキル」とは、1個または複数のハロゲン原子で置換されているアルキル基(例えば、フルオロメチル、ジフルオロメチル、トリフルオロメチル、ペルフルオロエチル、1,1−ジフルオロエチルなど)を意味する。
【0022】
「シクロアルキル」という用語は、完全に水素化され、かつ単環式環、二環式環またはスピロ環として存在し得る非芳香環を意味する。他に特定しない限り、炭素環は、一般に3〜8員環である。例えば、シクロアルキルには、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘキセニル、ノルボルニル(ビシクロ[2.2.1]ヘプチル)、ビシクロ[2.2.2]オクチルなどなどの基が含まれる。
【0023】
「複素環」という用語は、完全に水素化され、かつ単環式環、二環式環またはらせん環として存在し得る非芳香環を意味する。他に特定しない限り、複素環は一般に、硫黄、酸素および/または窒素から独立に選択される1〜3個のヘテロ原子(好ましくは、1個または2個のヘテロ原子)を含有する3〜6員環である。複素環には、エポキシ、アジリジニル、テトラヒドロフラニル、ピロリジニル、N−メチルピロリジニル、ピペリジニル、ピペラジニル、ピラゾリジニル、4H−ピラニル、モルホリノ、チオモルホリノ、テトラヒドロチエニル、テトラヒドロチエニル1,1−ジオキシドなどの基が含まれる。
【0024】
「治療有効量」という語句は、(i)特定の疾患、状態、または障害を治療し、(ii)特定の疾患、状態、または障害の1つまたは複数の症状を軽減、緩和、もしくは解消し、または(iii)本明細書に記載されている特定の疾患、状態、または障害の1つまたは複数の症状の発症を予防もしくは遅延させる本発明の化合物の量を意味する。
【0025】
「動物」という用語は、ヒト(男性または女性)、伴侶動物(例えば、イヌ、ネコおよびウマ)、食料源動物、動物園動物、海生動物、鳥および他の類似の動物種を意味する。「食用動物」とは、ウシ、ブタ、ヒツジおよび家禽などの食料源動物を意味する。
【0026】
「薬学的に許容できる」という語句は、物質または組成物が、製剤を構成する他の成分、および/またはそれによって治療を受ける哺乳動物と、化学的および/または毒物学的に適合性でなくてはならないことを示す。
【0027】
「治療すること」、「治療する」、または「治療」という用語は、防止的、すなわち、予防的および姑息的治療の両方を包含する。
【0028】
「調節される」または「調節すること」、または「調節する」という用語は、本明細書において使用する場合、他に示さない限り、本発明の化合物によってナトリウム−グルコース輸送体(特に、SGLT2)を阻害し、それによって輸送体によるグルコース輸送を部分的または完全に防止することを意味する。
【0029】
「本発明の化合物」という用語は、(特に別に特定されなければ)式(A)、式(B)の化合物、ならびに全ての純粋および混合立体異性体(ジアステレオマーおよびエナンチオマーを含めて)、互変異性体および同位体標識化合物を意味する。本発明の化合物の水和物および溶媒和物は、化合物が各々水または溶媒と会合している本発明の組成物と考えられる。化合物はまた、1種または複数の結晶状態で、すなわち、共結晶、多形として存在することもあり、またはアモルファス固体として存在することがある。全てのこのような形態は、特許請求の範囲に包含されている。
【0030】
一実施形態において、R1は、H、メチル、エチル、プロピル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、シクロプロピル、またはシクロブチルである。別の実施形態において、R1は、H、メチル、エチル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、またはシクロプロピルである。さらなる実施形態において、R1は、H、メチル、エチル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、またはシクロプロピルである。さらなる実施形態において、R1は、メチル、エチル、F、Cl、シアノ、CF3、またはシクロプロピルである。
【0031】
一実施形態において、R2は、メチル、エチル、プロピル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである。別の実施形態において、R2は、メチル、エチル、イソプロピル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである。さらなる実施形態において、R2は、メチル、エチル、メトキシ、エトキシ、F、Cl、シアノ、−CF3、−CF2CH3、エチニル、3−オキセタニルオキシ、3−テトラヒドロフラニルオキシ、またはシクロプロピルである。さらなる実施形態において、R2は、メトキシまたはエトキシである。
【0032】
一実施形態において、結晶は、化合物4Aと、L−プロリンまたはL−ピログルタミン酸とを含む。
【0033】
さらなる実施形態において、結晶は、
a)P2(1)2(1)2(1)の空間群および実質的に下記と等しい単位格子パラメータ
a=7.4907(10)Å α=90°
b=12.8626(15)Å β=90°
c=28.029(4)Å γ=90°、
b)2θ値(CuKα放射線、1.54056Åの波長)6.4±0.2、16.7±0.2、17.4±0.2および21.1±0.2を含む粉末X線回折パターン、
c)500MHz分光計で決定すると、29.5ppmの結晶性アダマンチンに対して、16.5±0.2、131.1±0.2、158.7±0.2、および181.5±0.2ppmにおいてピーク位置を有する固体13C NMRスペクトル、または
d)約142.5±2℃の吸熱を有する示差走査熱量測定サーモグラム
の1つまたは複数を有する。
【0034】
さらなる実施形態において、結晶は、式(4A)の化合物およびL−ピログルタミン酸を1:1の化学量論比で含む共結晶である。
【0035】
本発明の化合物は、特に本明細書に含有されている記載に照らして、化学技術分野において周知のものと類似のプロセスを含む合成経路によって合成することができる。出発物質は一般に、Aldrich Chemicals(Milwaukee、WI)などの商業的供給源から入手可能であり、または当業者には周知の方法(例えば、Louis F.FieserおよびMary Fieser、Reagents for Organic Synthesis、第1〜19巻、Wiley、New York(1967〜1999編)、もしくはBeilsteins Handbuch der organischen Chemie、4版、Aufl.編、Springer−Verlag、Berlin(追補を含めて)(Beilsteinオンラインデータベースによっても入手可能である)に一般に記載されている方法によって調製する)を使用して容易に調製される。
【0036】
例示の目的のために、下記に示す反応スキームは、本発明の化合物および重要中間体を合成するための可能な経路を提供する。個々の反応段階のより詳細な記載については、下記の実施例の項を参照されたい。他の合成経路を使用して、本発明の化合物を合成し得ることを当業者であれば理解するであろう。特定の出発物質および試薬をスキームにおいて示し、下記で議論するが、他の出発物質および試薬を容易に代用して、種々の誘導体および/または反応条件を提供することができる。さらに、下記の方法によって調製した化合物の多くは、当業者には周知の従来の化学作用を使用してこの開示に照らしさらに修飾することができる。
【0037】
本発明の化合物の調製において、中間体の遠隔官能基の保護が必要なことがある。このような保護の必要性は、遠隔官能基の性質および調製法の条件によって変化するであろう。「ヒドロキシ保護基」は、ヒドロキシ官能基を遮断または保護するヒドロキシ基の置換基を意味する。適切なヒドロキシル保護基(O−Pg)には、例えば、アリル、アセチル(Ac)、シリル(トリメチルシリ(TMS)またはtert−ブチルジメチルシリル(TBS)など)、ベンジル(Bn)、パラ−メトキシベンジル(PMB)、トリチル(Tr)、パラ−ブロモベンゾイル、パラ−ニトロベンゾイルなど(1,3−ジオールの保護のためにはベンジリデン)が含まれる。このような保護の必要性は、当業者に容易に決定される。保護基およびそれらの使用の概要については、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley & Sons、New York、1991を参照されたい。
【0038】
スキーム1は、本発明の化合物を提供するために使用することができる一般手順を略述する。
【0039】
【化3】
【0040】
アリル2,3,4−トリ−O−ベンジル−D−グルコピラノシド(I−a、Pg1はベンジル基である)は、Shinya Hanashimaら、Bioorganic & Medicinal Chemistry、9、367(2001);Patricia A.Gentら、Journal of the Chemical Society、Perkin1、1835(1974);Hans Peter Wessel、Journal of Carbohydrate Chemistry、7、263、(1988);またはYoko Yuasaら、Organic Process Research & Development、8、405〜407(2004)によって記載された手順によって調製することができる。スキーム1のステップ1において、ヒドロキシメチレン基は、スワーン酸化、続いてアルカリ金属水酸化物(例えば、水酸化ナトリウム)の存在下でホルムアルデヒドによる処理によって、グリコシド上に導入することができる。これはアルドール−カニッツァーロ反応と称される。スワーン酸化は、Kanji OmuraおよびDaniel SwernによってTetrahedron、34、1651(1978)に記載されている。当業者には既知のこの方法の変更形態もまた使用し得る。例えば、Ozanne,A.ら、Organic Letters、5、2903(2003)によって記載された安定化した2−ヨードキシ安息香酸などの他のオキシダント、および当業者が既知の他のオキシダントもまた使用することができる。アルドールカニッツァーロの連続した処理は、Robert Schaffer、Journal of The American Chemical Society、81、5452(1959)およびAmigues,E.J.ら、Tetrahedron、63、10042(2007)によって記載されている。
【0041】
スキーム1のステップ2において、保護基(Pg2)は、中間体(I−b)を所望の特定の保護基のための適当な試薬および手順で処理することによって付加することができる。例えば、p−メトキシベンジル(PMB)基は、水素化ナトリウム、水素化カリウム、カリウムtert−ブトキシドの存在下で、テトラヒドロフラン、1,2−ジメトキシエタンまたはN,N−ジメチルホルムアミド(DMF)などの溶媒中、中間体(I−b)をp−メトキシベンジルブロミドまたはp−メトキシベンジルクロリドで処理することによって導入することができる。触媒量の酸(例えば、トリフルオロメタンスルホン酸、メタンスルホン酸、またはカンファースルホン酸)の存在下で、ジクロロメタン、ヘプタンまたはヘキサンなどの溶媒中、パラ−メトキシベンジルトリクロロアセトイミデートを伴う条件もまた使用することができる。ベンジル(Bn)基は、水素化ナトリウム、水素化カリウム、カリウムtert−ブトキシドの存在下で、テトラヒドロフラン、1,2−ジメトキシエタンまたはN,N−ジメチルホルムアミドなどの溶媒中、中間体(I−b)を臭化ベンジルまたは塩化ベンジルで処理することによって導入することができる。触媒量の酸(例えば、トリフルオロメタンスルホン酸、メタンスルホン酸、またはカンファースルホン酸)の存在下で、ジクロロメタン、ヘプタンまたはヘキサンなどの溶媒中、ベンジルトリクロロアセトイミデートを伴う条件もまた使用することができる。
【0042】
スキーム1のステップ3において、アリル保護基を除去して(例えば、メタノール中の塩化パラジウムによる処理によって、ジクロロメタンなどの共溶媒もまた使用し得る、当業者には既知の他の条件もまた使用することができる、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley & Sons、New York、1991を参照されたい)、ラクトール(I−d)が形成される。
【0043】
スキーム1のステップ4において、次いで保護されていないヒドロキシル基のオキソ基への酸化(例えば、スワーン酸化)によって、ラクトン(I−e)が形成される。
【0044】
スキーム1のステップ5において、ラクトン(I−e)は、N,O−ジメチルヒドロキシルアミン塩酸塩と反応して、相当するワインレブアミドが形成され、これは閉環/開環形態(I−f/I−g)の平衡状態で存在し得る。「ワインレブアミド」(I−g)は、当業者には周知の手順を使用して作製することができる。Nahm,S.およびS.M.Weinreb、Tetrahedron Letters、22(39)、3815〜1818(1981)を参照されたい。例えば、中間体(I−f/I−g)は、市販のN,O−ジメチルヒドロキシルアミン塩酸塩および活性化剤(例えば、トリメチルアルミニウム)から調製することができる。
【0045】
スキーム1のステップ6において、アリールベンジル基(Ar)は、約−78℃〜約20℃の範囲の温度でテトラヒドロフラン(THF)中、所望の有機金属反応剤(例えば、有機リチウム化合物(ArLi)または有機マグネシウム化合物(ArMgX))を使用して導入され、続いて(プロトン性条件に静置すると)相当するラクトール(I−i)に加水分解され、これは相当するケトン(I−h)と平衡状態で存在し得る。(A)および(B)に見出される架橋ケタールモチーフは、用いる保護基に適当な試薬を使用して保護基(Pg2)を除去することによって調製することができる。例えば、PMB保護基は、アニソールおよびジクロロメタン(DCM)の存在下で約0℃〜約23℃(室温)にてトリフルオロ酢酸で処理することによって除去することができる。次いで、残った保護基(Pg1)は、特定の保護基に適当な化学作用を使用して除去することができる。例えば、ベンジル保護基は、パラジウム(Pd黒)の存在下で、プロトン性溶媒(例えば、エタノール/THF)中、約室温にてギ酸で処理することによって除去され、最終生成物(A)および(B)を生成し得る。R1がCNであるとき、約−78℃〜約室温の範囲の温度で、ジクロロメタンまたは1,2−ジクロロエタンなどの溶媒中、三塩化ホウ素のようなルイス酸をまた使用して、ベンジル保護基および/またはパラ−メトキシベンジル保護基を除去し得る。
【0046】
中間体(I−i)または生成物(A)もしくは(B)において、R1がCNであり、R2が(C1〜C4)アルコキシであるとき、三塩化ホウ素または三臭化ホウ素などのルイス酸による処理によって、相当するフェノールへの部分的から完全な脱アルキル化が起こり、相当する化合物(A)または(B)(R1はCNであり、R2はOHである)をもたらし得る。これが起こった場合、(C1〜C4)アルコキシ基を、穏やかな塩基性条件(例えば、アセトン中の炭酸カリウム)下で、約室温〜摂氏約56度の範囲の温度で、(C1〜C4)ヨウ化アルキルを使用した選択的アルキル化によって再導入し得る。
【0047】
R1および/またはR2が(C1〜C4)アルキル−SO2−であるとき、有機金属の添加ステップ6(スキーム1)は、相当する(C1〜C4)アルキル−S−含有有機金属反応剤で行われることを当業者は理解する。次いで、チオ−アルキルを、当業者に既知の従来の方法を使用して後の段階で相当するスルホンに酸化させる。
【0048】
本発明の化合物は、任意の適切な方法を使用して共結晶として調製し得る。このような共結晶を調製するための代表的なスキームを、スキーム2に記載する。
【0049】
【化4】
【0050】
スキーム2(Meはメチルであり、Etはエチルである)のステップ1において、1−(5−ブロモ−2−クロロベンジル)−4−エトキシベンゼンを3:1のトルエン:テトラヒドロフランに溶解し、続いて得られた溶液を−70℃未満に冷却する。この溶液に、ヘキシルリチウムを添加し、その間に反応物を−65℃以下に維持し、続いて1時間撹拌する。(3R,4S,5R,6R)−3,4,5−トリス(トリメチルシリルオキシ)−6−((トリメチルシリルオキシ)メチル)−テトラヒドロピラン−2−オン(II−a)をトルエンに溶解し、得られた溶液を−15℃に冷却する。次いで、この溶液を−70℃のアリールリチウム溶液に添加し、続いて1時間撹拌する。次いで、メタンスルホン酸のメタノール溶液を添加し、続いて室温に温め、16〜24時間撹拌する。α−アノマーのレベルが3%以下であるとき、反応は完了していると見なす。次いで、5Mの水酸化ナトリウム水溶液を添加することによって反応物を塩基性化する。得られた塩を濾過して除去し、続いて粗生成物溶液を濃縮する。2−メチルテトラヒドロフランを共溶媒として添加し、有機相を水で2度抽出する。次いで、有機相をトルエン中4容量に濃縮する。次いで、この濃縮物を5:1のヘプタン:トルエン溶液に添加し、沈殿物を形成させる。固体を集め、真空下で乾燥させ、固体を得る。
【0051】
スキーム2のステップ2において、塩化メチレン中の(II−b)にイミダゾールを添加し、続いて0℃に冷却し、次いでトリメチルシリルクロリドを添加し、ペルシリル化生成物を得る。反応物を室温に温め、水を添加することによってクエンチし、有機相を水で洗浄する。(II−c)のこの粗塩化メチレン溶液を硫酸ナトリウム上で乾燥させ、次いで粗製物を次のステップにかける。
【0052】
スキーム2のステップ3において、塩化メチレン中の(II−c)の粗溶液を低容量に濃縮し、次いで溶媒をメタノールに交換する。(II−c)のメタノール溶液を0℃に冷却し、次いで1mol%の炭酸カリウムをメタノール中の溶液として添加し、続いて5時間撹拌する。次いで、メタノール中の1mol%酢酸を添加することによって反応物をクエンチし、続いて室温に温め、溶媒を酢酸エチルに交換し、次いで少量の無機固体の濾過を行う。(II−d)の粗酢酸エチル溶液を、次のステップに直接かける。
【0053】
スキーム2のステップ4において、(II−d)の粗溶液を低容量に濃縮し、次いで塩化メチレンおよびジメチルスルホキシドで希釈する。トリエチルアミンを添加し、続いて10℃に冷却し、次いで三酸化硫黄ピリジン錯体を10分間隔にて3分割で固体として添加する。反応物を10℃でさらに3時間撹拌し、その後水でクエンチし、室温に温める。相を分離し、続いて塩化メチレン層を塩化アンモニウム水溶液で洗浄する。(II−e)の粗塩化メチレン溶液を、次のステップに直接かける。
【0054】
スキーム2のステップ5において、(II−e)の粗溶液を低容量に濃縮し、次いで溶媒をエタノールに交換する。30当量のホルムアルデヒド水溶液を添加し、続いて55℃に温める。2当量の三塩基性リン酸カリウムの水溶液を添加し、続いて55℃で24時間撹拌する。次いで、反応温度を70℃にさらに12時間上げる。反応物を室温に冷却し、tert−ブチルメチルエーテルおよびブラインで希釈する。相を分離し、続いて有機相の溶媒を酢酸エチルへ交換する。酢酸エチル相をブラインで洗浄し、低容量に濃縮する。次いで、粗濃縮物を、5%メタノール、95%トルエンで溶出するシリカゲルフラッシュクロマトグラフィーによって精製する。生成物含有画分を合わせ、低容量に濃縮する。メタノールを添加し、続いて沈殿が起こるまで撹拌する。懸濁液を冷却し、固体を集め、ヘプタンですすぎ、続いて乾燥させる。生成物(II−f)を固体として単離する。
【0055】
スキーム2のステップ6において、化合物(II−f)を5容量の塩化メチレンに溶解し、続いて1mol%SiliaBond(登録商標)トシル酸を添加し、18時間室温で撹拌する。酸触媒を濾過して除去し、(II−g)の塩化メチレン溶液を、次のステップである共結晶化手順に直接かける。
【0056】
スキーム2のステップ7において、(II−g)の塩化メチレン溶液を濃縮し、次いで溶媒を2−プロパノールに交換する。水を添加し、続いて55℃に温める。L−ピログルタミン酸の水溶液を添加し、続いて得られた溶液を室温に冷却する。次いで、溶液に種晶添加し、18時間造粒する。冷却後、固体を集め、ヘプタンですすぎ、続いて乾燥させる。生成物(II−h)を固体として単離する。
【0057】
本発明の化合物(A)のための代替合成経路をスキーム3において示し、下記に記載する。
【0058】
【化5】
【0059】
(III−a)(R3は、アルキルまたはフルオロ置換アルキルである(酸素原子に隣接した炭素を除いて))の合成は、スキーム2のステップ1に記載されているのと同様の方法で調製することができる。スキーム3のステップ1において、第一級ヒドロキシル基は、適当な保護基によって選択的に保護される。例えば、トリチル基(Pg3=Tr)は、ピリジンなどの塩基の存在下で、トルエン、テトラヒドロフランまたはジクロロメタンなどの溶媒中、摂氏約0度〜約室温の範囲の温度で、中間体(III−a)をクロロトリフェニルメタンで処理することによって導入することができる。このような保護基および実験条件のさらなる例は当業者に既知であり、T.W.Greene、Protective Groups in Organic Synthesis、John Wiley & Sons、New York、1991に見出すことができる。
【0060】
スキーム3のステップ2において、第二級ヒドロキシル基は、適当な保護基によって保護することができる。例えば、ベンジル基(Pg4はBnである)は、水素化ナトリウム、水素化カリウム、カリウムtert−ブトキシドの存在下で、テトラヒドロフラン、1,2−ジメトキシエタンまたはN,N−ジメチルホルムアミドなどの溶媒中、摂氏約0度〜摂氏約80度の範囲の温度にて、中間体(III−b)を臭化ベンジルまたは塩化ベンジルで処理することによって導入することができる。アセチルまたはベンゾイル基(Pg4=AcまたはBzである)は、トリエチルアミン、N,N−ジイソプロピルエチルアミンまたは4−(ジメチルアミノ)ピリジンなどの塩基の存在下で、テトラヒドロフラン、1,2−ジメトキシエタンまたはジクロロメタンなどの溶媒中、摂氏約0度〜摂氏約80度の範囲の温度にて、中間体(III−b)を塩化アセチル、臭化アセチルまたは無水酢酸または塩化ベンゾイルまたは無水安息香酸で処理することによって導入し得る。
【0061】
スキーム3のステップ3において、第一級ヒドロキシル基が脱保護され、中間体(III−d)がもたらされる。Pg3がTrであるとき、摂氏約−20度〜約室温の範囲の温度にて、パラ−トルエンスルホン酸など酸の存在下、メタノールなどのアルコール性溶媒中で中間体(III−c)を処理し、中間体(III−d)が提供される。クロロホルムのような共溶媒を使用し得る。
【0062】
スキーム3のステップ4において、ヒドロキシメチレン基は、スキーム1(ステップ1)およびスキーム2(ステップ4および5)に既に記載されているものと同様の方法によって導入される。エタノールなどの溶媒中、約室温〜摂氏約70度の範囲の温度にて、アルカリ金属アルコキシドの存在下で、パラホルムアルデヒドのような他のホルムアルデヒド源をまた、このステップにおいて使用することができる。Pg4がBnであるとき、このステップは、中間体(III−e)を提供し、Pg4がAcまたはBzであるとき、このステップは、中間体(III−f)を提供する。
【0063】
スキーム3のステップ5において、中間体(III−e)を、ジクロロメタンなどの溶媒中、摂氏約−10度〜約室温の範囲の温度にて、トリフルオロ酢酸または酸性樹脂などの酸で処理して、中間体(III−g)が生成される。
【0064】
スキーム3のステップ6において、次いで、残った保護基(Pg4)を、特定の保護基に適当な化学作用を使用して除去することができる。例えば、ベンジル保護基は、パラジウム(Pd黒)の存在下で、プロトン性溶媒(例えば、エタノール/THF)中、約室温にてギ酸で処理することによって除去され、最終生成物(A)を生成し得る。
【0065】
スキーム3のステップ7において、中間体(III−f)を、ジクロロメタンなどの溶媒中、摂氏約−10度〜約室温の範囲の温度にて、トリフルオロ酢酸または酸性樹脂などの酸で処理して、最終生成物(A)が生成される。
【0066】
生成物(A)を合成するための別の代替スキームを、スキーム4に示し、下記に記載する。
【0067】
【化6】
【0068】
スキーム4のステップ1において、中間体(III−a)を、ピリジン、トリエチルアミン、N,N−ジイソプロピルエチルアミンなどの塩基の存在下で、テトラヒドロフラン、2−メチルテトラヒドロフランなどの溶媒中、摂氏約−20度〜約室温の範囲の温度にて、適当なアリールスルホニルクロリドR4SO2Clまたはアリールスルホン酸無水物R4S(O)2OS(O)2R4(式中、R4は、アリールスルホニルクロリド、4−メチル−ベンゼンスルホニルクロリド、4−ニトロ−ベンゼンスルホニルクロリド、4−フルオロ−ベンゼンスルホニルクロリド、2,6−ジクロロ−ベンゼンスルホニルクロリド、4−フルオロ−2−メチル−ベンゼンスルホニルクロリド、および2,4,6−トリクロロ−ベンゼンスルホニルクロリドにおいて、およびアリールスルホン酸無水物、p−トルエンスルホン酸無水物において見出されるものなどの、置換されていてもよいアリール基である)で処理する。臭化亜鉛(II)などのいくつかのルイス酸を、添加物として使用してもよい。
【0069】
スキーム4のステップ2において、中間体(IV−a)をコーンブルム型酸化(Kornblum,N.ら、Journal of The American Chemical Society、81、4113(1959)を参照されたい)に供して、相当する水和物および/またはヘミアセタール形態と平衡状態で存在し得る相当するアルデヒドを生成する。例えば、ピリジン、2,6−ルチジン、2,4,6−コリジン、N,N−ジイソプロピルエチルアミン、4−(ジメチルアミノ)ピリジンなどの塩基の存在下で、ジメチルスルホキシドなどの溶媒中、約室温〜摂氏約150度の範囲の温度で、中間体(IV−a)を処理する。次いで、生成されたアルデヒド中間体を、ステップ1(スキーム1)およびステップ5(スキーム2)について記載したアルドール/カニッツァーロ条件に供して、中間体(IV−b)を生成する。
【0070】
スキーム4のステップ3において、中間体(IV−b)を、ジクロロメタンなどの溶媒中、摂氏約−10度〜約室温の範囲の温度にて、トリフルオロ酢酸または酸性樹脂などの酸で処理して、最終生成物(A)が生成される。
【0071】
R2が(C2〜C4)アルキニルであるとき、このプロセスは、スキーム5(R6は、Hまたは(C1〜C2)アルキルである)を使用して行うことができる。
【0072】
【化7】
【0073】
中間体(V−i)を提供するスキーム5のステップ1において、有機金属の添加ステップを、(V−a)(Pg5は、ヒドロキシル基のための適切な保護基である)に由来する有機金属反応剤を使用して、スキーム1、ステップ6に記載したものと同様の方法で行う。例えば、Pg5は、tert−ブチルジメチルシリル基(TBS)とすることができる(例えば{4−[(5−ブロモ−2−クロロ−フェニル)−メチル]−フェノキシ}−tert−ブチル−ジメチル−シランの調製については、US2007/0054867を参照されたい)。
【0074】
スキーム5のステップ2において、Pg2=PMBのとき、中間体(V−i)を、アニソールの存在下で、ジクロロメタンなどの溶媒中、摂氏約−10度〜約室温の範囲の温度で、トリフルオロ酢酸、メタンスルホン酸または酸性樹脂などの酸で処理して、中間体(V−j)を生成する。
【0075】
スキーム5のステップ3において、保護基(Pg5)および(Pg1)を除去して、(V−k)を提供することができる。典型的には、(Pg5)はTBSであり、Pg1はBnである。この状況において、(V−j)を、1)テトラヒドロフランまたは2−メチルテトラヒドロフランのような溶媒中、摂氏0度〜摂氏約40度の範囲の温度にて、フッ化テトラブチルアンモニウム、および2)パラジウム(Pd黒)の存在下で、プロトン性溶媒(例えば、エタノール/THF)中、約室温にてギ酸によって連続的に処理することによって、保護基を除去する。この連続した処理において、2つの反応の順序は交換可能である。
【0076】
スキーム5のステップ4において、中間体(V−k)を、トリエチルアミンまたは4−ジメチアミノピリジンなどの塩基の存在下で、ジクロロメタンまたは1,2−ジクロロエタンなどの溶媒中、摂氏0度〜摂氏約40度の範囲の温度にて、N,N−ビス−(トリフルオロメタンスルホニル)−アニリンで処理して、中間体(V−l)を生成する。
【0077】
スキーム5のステップ5において、中間体(V−l)を、薗頭型反応に供する(Sonogashira,K.、Coupling Reactions Between sp2 and sp Carbon Centers、Comprehensive Organic Synthesis(Trost,B.M.、Fleming,I.編)、3、521〜549、(Pergamon、Oxford、1991)を参照されたい)。例えば、(V−l)は、ヨウ化銅(I)、ビス−(トリフェニルホスフィン)−パラジウムジクロリドまたはテトラキス(トリフェニルホスフィン)パラジウム(0)などの触媒の存在下で、トリエチルアミンまたはN,N−ジイソプロピルエチルアミンなどの塩基の存在下で、N,N−ジメチルホルムアミドなどの溶媒中、約室温〜摂氏約120度の範囲の温度にて、適当な末端アルキンHCCR6で処理して、所望の生成物(A)および(B)を生成する。R6がHであるとき、トリメチルシリルアセチレンを使用することがより好都合である。この場合、上記の反応から得られた粗材料を、MeOHなどのアルコール性溶媒中、約室温にて炭酸カリウムなどの塩基で処理して、当業者には既知の古典的な後処理の後、所望の生成物(A)および(B)(R2は、−CCHである)が生成される。
【0078】
スキーム1〜5において上記で記載した化学的手法は、中間体(V−k)を得る様々な方法を表すことを当業者であれば理解するであろう。そしてまた、特にR1がClであるとき、(V−k)は、古典的な条件下で一般に好まれるアルキル化剤で処理して、フェノール基を選択的にアルキル化して、(A)(ならびにスキーム1および5において(B))(R2は(C1〜C4)アルコキシである)を生成することができる。
【0079】
本発明の化合物は、不斉中心またはキラル中心を含有し、したがって、異なる立体異性体の形態で存在する。他に特定しない限り、本発明の化合物の全ての立体異性体の形態、およびラセミ混合物を含めたその混合物は、本発明の一部を形成することが意図される。さらに、本発明は、全ての幾何異性体および位置異性体を包含する。例えば、本発明の化合物が二重結合または縮合環を組み込む場合、cis−およびtrans−形態の両方、ならびに混合物は、本発明の範囲内に包含される。
【0080】
ジアステレオマー混合物は、当業者には周知の方法(クロマトグラフィーおよび/または分別結晶、蒸留、昇華など)によるそれらの物理的化学的差異に基づいて、それらの個々のジアステレオマーに分離することができる。エナンチオマーは、適当な光学活性な化合物(例えば、キラルアルコールまたはモーシェル酸クロリドなどのキラル補助基)との反応によってエナンチオマー混合物をジアステレオマー混合物に変換し、ジアステレオマーを分離し、個々のジアステレオマーを相当する純粋なエナンチオマーに変換(例えば、加水分解)することによって分離することができる。また、本発明の化合物のいくつかは、アトロプ異性体(例えば、置換ビアリール)である場合があり、本発明の一部と考えられる。エナンチオマーはまた、キラルHPLC(高圧液体クロマトグラフィー)カラムを使用することによって分離することができる。
【0081】
本発明の中間体および化合物は異なる互変異性型で存在する場合があることもまた考えられ、全てのこのような形態は本発明の範囲内に包含される。「互変異性体」または「互変異性型」という用語は、低いエネルギー障壁によって相互変換可能な異なるエネルギーの構造異性体を意味する。例えば、プロトン互変異性体(プロトン性互変異性体としてもまた既知である)には、プロトンの移動による相互変換(ケト−エノールおよびイミン−エナミン異性化など)が含まれる。プロトン互変異性体の具体例は、プロトンが2個の環窒素の間を移動するイミダゾール部分である。原子価互変異性体には、結合電子のいくつかの再編成による相互変換が含まれる。いくつかの中間体(および/または中間体の混合物)の閉環形態と開環形態との間の平衡状態は、当業者に既知のアルドースが関与する変旋光の過程を連想させる。
【0082】
本発明はまた、1個または複数の原子が、天然に通常見出される原子質量または質量数と異なる原子質量または質量数を有する原子によって置き換えられているという事実を別にすれば、本明細書において記載したものと同一の同位体標識された本発明の化合物を包含する。本発明の化合物中に組み込むことができる同位体の例には、水素、炭素、窒素、酸素、リン、硫黄、フッ素、ヨウ素、および塩素の同位体(各々、2H、3H、11C、13C、14C、13N、15N、15O、17O、18O、31P、32P、35S、18F、123I、125Iおよび36Clなど)が含まれる。
【0083】
特定の同位体標識された本発明の化合物(例えば、3Hおよび14Cで標識されたもの)は、化合物および/または基質組織分布アッセイにおいて有用である。それらの調製の容易さおよび検出能のために、トリチウム標識(すなわち、3H)および炭素−14(すなわち、14C)同位体が特に好ましい。さらに、重水素(すなわち、2H)などのより重い同位体による置換によって、より高い代謝安定性からもたらされる特定の治療上の利点(例えば、インビボ半減期の増加または投与必要量の減少)が可能となることがあり、したがって、ある状況において好ましいことがある。15O、13N、11C、および18Fなどのポジトロン放出同位体は、基質占有率を調査するためのポジトロン放出断層撮影(PET)研究のために有用である。同位体標識された本発明の化合物は一般に、同位体標識されていない試薬を同位体標識された試薬で置換することによって、スキームおよび/または本明細書における下記の実施例において開示されているものと類似の手順に従うことによって調製することができる。
【0084】
本発明の化合物は、ナトリウム−グルコース輸送体(特に、SGLT2)の阻害によって調節される疾患、状態および/または障害を治療するのに有用である。したがって、本発明の別の実施形態は、治療有効量の本発明の化合物、および薬学的に許容できる添加剤、賦形剤または担体を含む医薬組成物である。本発明の化合物(組成物およびそこで使用されている方法を含めて)はまた、本明細書に記載されている治療用途のための医薬の製造において使用することができる。
【0085】
典型的な製剤は、本発明の化合物および担体、賦形剤または添加剤を混合することによって調製する。適切な担体、賦形剤および添加剤は当業者には周知であり、炭水化物、ワックス、水溶性および/または膨潤性ポリマー、親水性または疎水性材料、ゼラチン、油、溶媒、水などの材料が含まれる。使用される特定の担体、賦形剤または添加剤は、本発明の化合物を適用する手段および目的によって決まる。溶媒は一般に、哺乳動物に投与されても安全であると当業者に認められる(GRAS)溶媒に基づいて選択される。一般に、安全な溶媒は、水、および水中で可溶性または混和性の他の無毒性溶媒などの無毒性水性溶媒である。適切な水性溶媒には、水、エタノール、プロピレングリコール、ポリエチレングリコール(例えば、PEG400、PEG300)など、およびこれらの混合物が含まれる。製剤にはまた、1種または複数の緩衝液、安定化剤、界面活性剤、湿潤剤、滑沢剤、乳化剤、懸濁化剤、保存剤、抗酸化剤、不透明化剤、流動促進剤、加工助剤、着色剤、甘味剤、香料剤、香味剤、および薬物(すなわち、本発明の化合物またはその医薬組成物)の洗練された外観をもたらし、または医薬品(すなわち、医薬)の製造に役立つ他の既知の添加物が含まれることがある。
【0086】
製剤は、従来の溶解および混合手順を使用して調製し得る。例えば、原薬(すなわち、本発明の化合物、または化合物の安定化した形態(例えば、シクロデキストリン誘導体または他の既知の複合体形成剤との複合体))を、上記の添加剤の1種または複数の存在下で適切な溶媒に溶解する。本発明の化合物は典型的には、医薬品剤形に製剤されて、容易に管理可能な薬物の投与量を提供し、洗練された容易に取り扱うことができる製品を患者に与える。
【0087】
医薬組成物にはまた、式(I)の化合物の溶媒和物および水和物が含まれる。「溶媒和物」という用語は、1個または複数の溶媒分子を有する式(I)によって表される化合物の分子複合体(薬学的に許容できるその塩を含めて)を意味する。このような溶媒分子は、製薬技術において通常使用されるものであり、レシピエントにとって無害であることが知られている(例えば、水、エタノール、エチレングリコールなど)。「水和物」という用語は、溶媒分子が水である複合体を意味する。溶媒和物および/または水和物は好ましくは、結晶形態で存在する。他の溶媒を、より望ましい溶媒和物の調製における中間体溶媒和物として使用してもよい(メタノール、メチルt−ブチルエーテル、酢酸エチル、酢酸メチル、(S)−プロピレングリコール、(R)−プロピレングリコール、1,4−ブチン−ジオールなど)。結晶形態はまた、他の無害な小分子(L−フェニルアラニン、L−プロリン、L−ピログルタミン酸など)との複合体として、共結晶、または溶媒和物もしくは水和物の共結晶性材料としても存在し得る。溶媒和物、水和物および共結晶化合物は、参照により本明細書中に組み込まれているPCT国際公開第WO08/002824号に記載されている手順、または当業者には周知の他の手順を使用して調製することができる。
【0088】
適用するための医薬組成物(または製剤)は、薬物を投与するために使用される方法によって種々の方法でパッケージし得る。一般に、分配するための物品には、その中に適当な形態の医薬製剤を入れた容器が含まれる。適切な容器は当業者には周知であり、ボトル(プラスチックおよびガラス)、小袋、アンプル、ビニール袋、金属製円筒などの材料が含まれる。容器にはまた、パッケージの内容物への不注意による接触を防止するための誤使用防止アセンブリが含まれることがある。さらに、容器には、容器の内容について記載したラベルがその上に貼ってある。ラベルにはまた、適当な警告が含まれることがある。
【0089】
本発明は、動物においてナトリウム−グルコース輸送体の阻害によって調節される疾患、状態および/または障害を治療する方法であって、このような治療を必要としている動物に、治療有効量の本発明の化合物、または有効量の本発明の化合物を含む医薬組成物、および薬学的に許容できる添加剤、賦形剤、または担体を投与するステップを含む、方法をさらに提供する。この方法は、SGLT2の阻害が有益である疾患、状態および/または障害を治療するのに特に有用である。
【0090】
本発明の一態様は、肥満症、および肥満症に関連する障害(例えば、過体重、体重増加、または体重維持)の治療である。
【0091】
肥満症および過体重は一般に肥満度指数(BMI)によって定義され、これは総体脂肪と相関し、疾患の相対危険度を推定する。BMIは、体重(キログラム)を身長(メートル)の2乗で割ることによって計算する(kg/m2)。過体重は典型的には、25〜29.9kg/m2のBMIとして定義され、肥満症は典型的には、30kg/m2のBMIとして定義される。例えば、National Heart,Lung,and Blood Institute、Clinical Guidelines on the Identification,Evaluation,and Treatment of Overweight and Obesity in Adults、The Evidence Report、Washington,DC:U.S. Department of Health and Human Services、NIH出版番号98−4083(1998)を参照されたい。
【0092】
本発明の別の態様は、糖尿病、または糖尿病に関連する障害(1型(インスリン依存性糖尿病、「IDDM」とも称される)および2型(非インスリン依存性糖尿病、「NIDDM」とも称される)糖尿病、耐糖能異常、創傷治癒の遅延、高インスリン血症、脂肪酸の血中濃度の上昇、高脂血症、高トリグリセリド血症、X症候群、高密度リポタンパク質レベルの上昇、インスリン抵抗性、高血糖症、ならびに糖尿病性合併症(アテローム性動脈硬化症、冠動脈心疾患、脳卒中、末梢血管疾患、腎症、高血圧症、ニューロパシー、および網膜症など)を含めた)の治療、または進行もしくは発症を遅延させるためである。
【0093】
本発明のまた別の態様は、代謝症候群などの肥満症の共存疾患の治療である。代謝症候群には、異脂肪血症、高血圧症、インスリン抵抗性、糖尿病(例えば、2型糖尿病)、冠動脈疾患および心不全などの疾患、状態または障害が含まれる。代謝症候群についてのさらなる詳細な情報については、例えば、Zimmet,P.Z.ら、「The Metabolic Syndrome:Perhaps an Etiologic Mystery but Far From a Myth−Where Does the International Diabetes Federation Stand?」、Diabetes & Endocrinology、7(2)、(2005)、およびAlberti,K.G.ら、「The Metabolic Syndrome−A New Worldwide Definition」、Lancet、366、1059〜62(2005)を参照されたい。
【0094】
好ましくは、本発明の化合物の投与は、薬物を含有しないビヒクル対照と比較して、血漿レプチン、C反応性タンパク質(CRP)および/またはコレステロールの減少などの、少なくとも1つの心血管疾患の危険因子の統計的に有意な(p<0.05)低下をもたらす。本発明の化合物の投与はまた、血清グルコースレベルの統計的に有意な(p<0.05)低下をもたらし得る。
【0095】
約100kgの体重を有する正常な成人のヒトについて、体重1キログラム当たり約0.001mg〜約10mg、好ましくは約0.01mg/kg〜約5.0mg/kg、さらに好ましくは約0.01mg/kg〜約1mg/kgの範囲の投与量が典型的には十分である。しかし、一般の投与量範囲におけるいくらかの変動は、治療を受ける対象の年齢および体重、意図される投与経路、投与される特定の化合物などに応じて必要とされることがある。特定の患者についての投与量範囲および最適な投与量を決定することは、本開示の利益を有する当業者の能力の十分に範囲内である。本発明の化合物は、それらの形態もまた当業者に周知である持続放出、制御放出、および遅延放出製剤において使用できることもまた留意される。
【0096】
本発明の化合物はまた、本明細書に記載されている疾患、状態および/または障害の治療のために他の医薬品と併せて使用し得る。したがって、他の医薬品と組み合わせて本発明の化合物を投与することを含む治療方法もまた提供される。本発明の化合物と組み合わせて使用し得る適切な医薬品には、抗肥満剤(食欲抑制剤を含めて)、抗糖尿病剤、抗高血糖剤、脂質低下剤、抗炎症剤および降圧剤が含まれる。
【0097】
適切な抗肥満剤には、カンナビノイド−1(CB−1)アンタゴニスト(リモナバンなど)、11β−ヒドロキシステロイドデヒドロゲナーゼ−1(11β−HSD1型)阻害剤、ステアロイル−CoAデサチュラーゼ−1(SCD−1)阻害剤、MCR−4アゴニスト、コレシストキニン−A(CCK−A)アゴニスト、モノアミン再取込み阻害剤(シブトラミンなど)、交感神経様作用剤、β3アドレナリン作動性アゴニスト、ドーパミンアゴニスト(ブロモクリプチンなど)、メラニン細胞刺激ホルモン類似体、5HT2cアゴニスト、メラニン凝集ホルモンアンタゴニスト、レプチン(OBタンパク質)、レプチン類似体、レプチンアゴニスト、ガラニンアンタゴニスト、リパーゼ阻害剤(テトラヒドロリプスタチン、すなわち、オルリスタットなど)、食欲抑制剤(ボンベシンアゴニストなど)、ニューロペプチド−Yアンタゴニスト(例えば、NPY Y5アンタゴニスト)、PYY3−36(その類似体を含めて)、甲状腺模倣剤、デヒドロエピアンドロステロンまたはその類似体、グルココルチコイドアゴニストまたはアンタゴニスト、オレキシンアンタゴニスト、グルカゴン様ペプチド−1アゴニスト、繊毛様神経栄養因子(Regeneron Pharmaceuticals、Inc.、Tarrytown、NYおよびProcter & Gamble Company、Cincinnati、OHから入手可能なAxokine(商標)など)、ヒトアグーチ関連タンパク質(AGRP)阻害剤、グレリンアンタゴニスト、ヒスタミン3アンタゴニストまたはインバースアゴニスト、ニューロメジンUアゴニスト、MTP/ApoB阻害剤(例えば、ジルロタピドなどの消化管選択的MTP阻害剤)、オピオイドアンタゴニスト、オレキシンアンタゴニストなどが含まれる。
【0098】
本発明の組合せの態様において使用するための好ましい抗肥満剤には、CB−1アンタゴニスト(例えば、リモナバン、タラナバン、スリナバン、オテナバント、SLV319(CAS番号464213−10−3)およびAVE1625(CAS番号358970−97−5))、消化管選択的MTP阻害剤(例えば、ジルロタピド、ミトラタピドおよびインプリタピド、R56918(CAS番号403987)およびCAS番号913541−47−6)、CCKaアゴニスト(例えば、PCT国際公開第WO2005/116034号または米国特許出願公開第2005−0267100A1号に記載されているN−ベンジル−2−[4−(1H−インドール−3−イルメチル)−5−オキソ−1−フェニル−4,5−ジヒドロ−2,3,6,10b−テトラアザ−ベンゾ[e]アズレン−6−イル]−N−イソプロピル−アセトアミド)、5HT2cアゴニスト(例えば、ロルカセリン)、MCR4アゴニスト(例えば、US6,818,658に記載されている化合物)、リパーゼ阻害剤(例えば、セチリスタット)、PYY3−36(本明細書において使用する場合、「PYY3−36」には、ペグ化PYY3−36、例えば、米国特許出願公開第2006/0178501号に記載されているものなどの類似体が含まれる)、オピオイドアンタゴニスト(例えば、ナルトレキソン)、オレオイル−エストロン(CAS番号180003−17−2)、オビネピチド(TM30338)、プラムリンチド(Symlin(登録商標))、テソフェンシン(NS2330)、レプチン、リラグルチド、ブロモクリプチン、オルリスタット、エキセナチド(Byetta(登録商標))、AOD−9604(CAS番号221231−10−3)およびシブトラミンが含まれる。好ましくは、本発明の化合物および併用療法は、運動および分別のある食事と併せて投与する。
【0099】
適切な抗糖尿病剤には、アセチル−CoAカルボキシラーゼ−2(ACC−2)阻害剤、ホスホジエステラーゼ(PDE)−10阻害剤、ジアシルグリセロールアシルトランスフェラーゼ(DGAT)1または2阻害剤、スルホニル尿素(例えば、アセトヘキサミド、クロルプロパミド、ジアビネス、グリベンクラミド、グリピジド、グリブリド、グリメピリド、グリクラジド、グリペンチド、グリキドン、グリソラミド、トラザミド、およびトルブタミド)、メグリチニド、α−アミラーゼ阻害剤(例えば、テンダミスタット、トレスタチンおよびAL−3688)、α−グルコシドヒドロラーゼ阻害剤(例えば、アカルボース)、α−グルコシダーゼ阻害剤(例えば、アジポシン、カミグリボース、エミグリタート、ミグリトール、ボグリボース、プラジミシン−Q、およびサルボスタチン)、PPARγアゴニスト(例えば、バラグリタゾン、シグリタゾン、ダルグリタゾン、エングリタゾン、イサグリタゾン、ピオグリタゾン、ロシグリタゾンおよびトログリタゾン)、PPARα/γアゴニスト(例えば、CLX−0940、GW−1536、GW−1929、GW−2433、KRP−297、L−796449、LR−90、MK−0767およびSB−219994)、ビグアニド(例えば、メトホルミン)、グルカゴン様ペプチド1(GLP−1)アゴニスト(例えば、エキセンディン−3およびエキセンディン−4)、タンパク質チロシンホスファターゼ−1B(PTP−1B)阻害剤(例えば、トロズスクエミン、ヒルチオサール抽出物、およびZhang,S.ら、Drug Discovery Today、12(9/10)、373〜381(2007)によって開示されている化合物)、SIRT−1阻害剤(例えば、レセルバトロール)、ジペプチジルペプチダーゼIV(DPP−IV)阻害剤(例えば、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチン)、インスリン分泌促進物質、脂肪酸酸化阻害剤、A2アンタゴニスト、c−junアミノ末端キナーゼ(JNK)阻害剤、インスリン、インスリン模倣物、グリコーゲンホスホリラーゼ阻害剤、VPAC2受容体アゴニストおよびグルコキナーゼ活性化剤が含まれる。好ましい抗糖尿病剤は、メトホルミンおよびDPP−IV阻害剤(例えば、シタグリプチン、ビルダグリプチン、アログリプチンおよびサクサグリプチン)である。
【0100】
適切な抗炎症剤には、生殖管/尿路感染症の予防薬および治療薬が含まれる。例示的な薬剤には、クランベリー(すなわち、オオミノツルコケモモ(Vaccinium macrocarpon))およびクランベリー誘導体(クランベリージュース、クランベリー抽出物またはクランベリーのフラボノールなど)が含まれる。クランベリー抽出物には、1種もしくは複数のフラボノール(すなわち、アントシアニンおよびプロアントシアニジン)、または精製されたクランベリーフラボノール化合物(ミリセチン−3−β−キシロピラノシド、ケルセチン−3−β−グルコシド、ケルセチン−3−α−アラビノピラノシド、3’−メトキシケルセチン−3−α−キシロピラノシド、ケルセチン−3−O−(6’’−p−クマロイル)−β−ガラクトシド、ケルセチン−3−O−(6’’−ベンゾイル)−β−ガラクトシド、および/もしくはケルセチン−3−α−アラビノフラノシドを含めた)が含まれ得る。
【0101】
本発明の実施形態を、下記の実施例によって例示する。しかし、他のその変形形態が当業者には既知であり、または本開示に照らし合わせて明らかであるため、本発明の実施形態は、これらの実施例の特定の詳細に限定されないことが理解されるべきである。
【実施例】
【0102】
他に特定しない限り、出発物質は一般に、Aldrich Chemicals Co.(Milwaukee、WI)、Lancaster Synthesis、Inc.(Windham、NH)、Acros Organics(Fairlawn、NJ)、Maybridge Chemical Company、Ltd.(Cornwall、英国)、Tyger Scientific(Princeton、NJ)、AstraZeneca Pharmaceuticals(London、英国)、およびAccela ChemBio(San Diego、CA)などの商業的供給源から入手可能である。
【0103】
全般的な実験手順
NMRスペクトルは、Varian Unity(商標)400(Varian Inc.、Palo Alto、CAから入手可能)で室温にてプロトンについて400MHzで記録した。化学シフトは、内部参照として残留溶媒に対する百万分率(δ)で表す。ピーク形状は下記のように表す。s、一重線;d、二重線;dd、二重線の二重線;t、三重線;q、四重線;m、多重線;bsまたはbr.s.、幅広い一重線;2s、2本の一重線;br.d.、幅広い二重線。エレクトロスプレーイオン化質量スペクトル(ES)は、Waters(商標)ZMD機器(キャリアガス:窒素;溶媒A:水/0.01%ギ酸、溶媒B:アセトニトリル/0.005%ギ酸;Waters Corp.、Milford、MAから入手可能)で得た。高分解能質量スペクトル(HRMS)は、Agilent(商標)Model6210(飛行時間型)で得た。単一の塩素または単一の臭素を含有するイオンの強度が記載されている場合、予想される強度比が観察された(35Cl/37Cl含有イオンについて概ね3:1、および79Br/81Br含有イオンについて1:1)。より低い質量のイオンのみの強度を示す。場合によっては、代表的な1H NMRピークのみを示す。
【0104】
カラムクロマトグラフィーは、ガラス製カラムまたはFlash40Biotage(商標)カラム(ISC、Inc.、Shelton、CT)中の、Baker(商標)シリカゲル(40μm;J.T.Baker、Phillipsburg、NJ)またはSilica Gel50(EM Sciences(商標)、Gibbstown、NJ)で行った。MPLC(中圧液体クロマトグラフィー)は、Biotage(商標)SP精製システムまたはTeledyne(商標)Isco(商標)からのCombiflash(登録商標)Companion(登録商標)を使用して行った。低窒素圧力下でBiotage(商標)SNAPカートリッジKPsilまたはRedisep Rfシリカ(Teledyne(商標)Isco(商標)から)を使用した。
【0105】
HPLC(高圧液体クロマトグラフィー)は、Shimadzu(商標)10A LC−UVまたはAgilent(商標)1100分取HPLCを使用して行った。
【0106】
特に断りのない限り、全ての反応は、無水溶媒中窒素ガスの不活性雰囲気下で行った。また、特に断りのない限り、全ての反応は、室温で(約23℃)行った。
【0107】
TLC(薄層クロマトグラフィー)を行うとき、Rfは、化合物が移動する距離を溶離液が移動する距離で割った比として定義する。
Rt(保持時間)。
【0108】
出発物質
一般に、下記の出発物質のいずれかは、米国特許出願公開第2008/0132563号のスキーム7もしくは8、または代わりに、米国特許出願公開第2007/0259821号のスキーム2、3もしくは8に記載されている手順を使用して調製することができる。さらに具体的には、下記の実施例において使用される下記の出発物質は、相当する参考文献に記載されている手順を使用して調製することができ、または相当する販売業者から購入することができる。
【0109】
4−ブロモ−2−(4−メトキシ−ベンジル)−1−メチル−ベンゼンは、PCT国際公開第WO01/027128号の実施例8に記載されている手順によって調製することができる。
【0110】
4−ブロモ−2−(4−エトキシ−ベンジル)−1−メチル−ベンゼンは、US2008/0132563の実施例17の調製に記載されている手順によって調製することができる。
【0111】
4−ブロモ−1−クロロ−2−(4−メトキシ−ベンジル)−ベンゼンは、US2008/0132563の実施例19またはUS2007/0259821の実施例Vの調製に記載されている手順によって調製することができる。
【0112】
4−ブロモ−1−クロロ−2−(4−エトキシ−ベンジル)−ベンゼンは、Shanghai Haoyuan Chemexpress Co.、Ltd.、Shanghai、中華人民共和国から購入し得る。
【0113】
4−ブロモ−2−(4−メトキシ−ベンジル)−ベンゾニトリルは、US2007/0259821の実施例XXIIに記載されている手順によって調製することができる。
【0114】
下記の出発物質は、下記のように調製した。
【0115】
4−ブロモ−1−フルオロ−2−(4−メトキシ−ベンジル)−ベンゼンの調製:
塩化オキサリル(11.0mL、126mmol)を、よく撹拌した5−ブロモ−2−フルオロ−安息香酸(25.0g、114mmol)のジクロロメタン(150mL)およびN,N−ジメチルホルムアミド(1.5mL)懸濁液に0℃にて滴下で添加した。得られた混合物を、室温に徐々に温めた。18時間後、固体が溶液となった。得られた薄オレンジ色の溶液を減圧下で濃縮し、ジエチルエーテルで2度抽出し、5−ブロモ−2−フルオロ−ベンゾイルクロリド(27.0g、定量的収率)を淡オレンジ色の油として得た。
【0116】
5−ブロモ−2−フルオロ−ベンゾイルクロリド(27.0g、114mmol)およびアニソール(12.9g、13.0mL、119mmol)のジクロロメタン(150mL)溶液に、内部温度を10℃未満に維持するように、0℃で三塩化アルミニウム(16.2g、119mmol)を少しずつ添加した。0℃で4時間撹拌した後、溶液を砕いた氷の上に注ぎ、得られた混合物を撹拌した。30分後、有機相を除去し、水相をジクロロメタンで2度抽出した。合わせた有機相を1Mの塩酸水溶液で1度洗浄し、1Mの水酸化ナトリウム水溶液で1度洗浄し、ブラインで1度洗浄した。有機相を硫酸ナトリウム上で乾燥させ、濾過し、減圧下で濃縮した。得られた残渣をエタノールから再結晶させ、(5−ブロモ−2−フルオロ−フェニル)−(4−メトキシ−フェニル)−メタノン(22.5g、64%)を白色の固体として得た。
【0117】
よく撹拌した(5−ブロモ−2−フルオロ−フェニル)−(4−メトキシ−フェニル)−メタノン(22.5g、72.80mmol)およびトリエチルシラン(27.9mL、20.3g、175.0mmol)のジクロロメタン(20mL)およびアセトニトリル(60mL)溶液に、0℃で三フッ化ホウ素エーテラート(32.0mL、36.2g、255.0mmol)を滴下で添加した。内部温度が20℃を超えないような速度で三フッ化ホウ素エーテラートを添加した。反応溶液を室温に温め、一晩撹拌した。全部で18時間後、水酸化カリウム(5.0g)の水(15.0mL)溶液を添加し、得られた混合物を2時間撹拌した。有機相を分離し、水相をジエチルエーテルで2度抽出した。合わせた有機相を1Mの水酸化ナトリウム水溶液で1度洗浄し、ブラインで1度洗浄した。有機相を硫酸ナトリウム上で乾燥させ、濾過し、減圧下で濃縮した。エタノールを得られた残渣に添加すると、白色の固体が形成された。固体を集め、高真空下にて乾燥させ、4−ブロモ−1−フルオロ−2−(4−メトキシ−ベンジル)−ベンゼン(20.1g、93%収率)を白色の固体として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 3.79
(s, 3 H), 3.89 (s, 2 H), 6.85 (d, J=8.6 Hz, 2 H), 6.91 (t, 1 H), 7.12 (d, J=8.8
Hz, 2 H), 7.21 - 7.31 (m, 2 H).
【0118】
出発物質4−ブロモ−2−(4−エトキシ−ベンジル)−ベンゾニトリルの調製:
エチル(4−エトキシ−フェニル)アセテート(2.68g、12.87mmol)、4−ブロモ−2−フルオロ−ベンゾニトリル(2.74g、13.70mmol)のN−メチルピロリドン(4mL)溶液を、カリウムtert−ブトキシド(3.14g、27.98mmol)のN−メチルピロリドン(13mL)懸濁液に0℃でゆっくりと添加した。添加すると、溶液は暗赤色となった。暗赤色の混合物を0℃で30分間、次いで室温で1時間撹拌した。メタノール(10mL)および1Mの水酸化ナトリウム水溶液(13.7mL)を添加し、混合物を室温で一晩撹拌した。pHを塩酸(1Mの水溶液)で約4に調節し、混合物を酢酸エチル(50mL×4)で抽出した。合わせた有機層をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、蒸発乾固させた。N,N−ジメチルホルムアミド(5mL)および炭酸カリウム(7g)を添加し、混合物を100℃に1時間加熱し、室温に冷却した。水を添加し、混合物を酢酸エチル(60mL×3)で抽出した。合わせた有機層をブラインで洗浄し、硫酸ナトリウム上で乾燥させ、蒸発乾固させた。粗製物を、シリカゲル(ヘプタン中の0〜14%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、2.26gの粗生成物(所望の生成物および別の生成物を含有)を得た。粗生成物をメタノールで沈殿させ、4−ブロモ−2−(4−エトキシ−ベンジル)−ベンゾニトリル(1.2g、4.15ppmの四重線、および1.5ppmの三重線でNMRピークを有する5%の別の化合物を含有)を得た。
1H NMR
(400 MHz, クロロホルム-d) δ 7.48-7.38
(m, 3 H), 7.13 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 4.08 (s, 2H),
4.03 (q, J = 7.2 Hz, 2H), 1.41 (t, J = 7.2 Hz, 3H).
【0119】
出発物質4−ブロモ−2−(4−エトキシ−ベンジル)−1−フルオロ−ベンゼンの調製
4−ブロモ−1−フルオロ−2−(4−メトキシ−ベンジル)−ベンゼン(4.2g、14.2mmol)のジクロロメタン(20mL)溶液に、0℃で三臭化ホウ素のジクロロメタン(15.7mL、16.0mmol)溶液(1M)をゆっくりと10分に亘り滴下で添加した。三臭化ホウ素の添加が完了すると、反応混合物を室温に徐々に温めた。4時間後、反応混合物を0℃に冷却し、1Nの塩酸水溶液(20mL)をゆっくりと添加することによってクエンチした。反応混合物を30分間撹拌し、ジクロロメタンで2度抽出した。合わせた有機層を硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮し、薄ピンク色の固体(3.83g、96%)を得た。粗生成物4−(5−ブロモ−2−フルオロ−ベンジル)−フェノールを、それ以上精製することなく次のステップで使用した。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 3.88
(s, 2 H), 4.76 (br. s., 1 H), 6.77 (d, J=8.2 Hz, 2 H), 6.91 (t, J=9.1 Hz, 1 H),
7.07 (d, J=8.6 Hz, 2 H), 7.23 (dd, J=6.8, 2.3 Hz, 1 H), 7.26 - 7.31 (m, 1 H).
【0120】
0℃に冷却した4−(5−ブロモ−2−フルオロ−ベンジル)−フェノール(6.0g、21.0mmol)の無水N,N−ジメチルホルムアミド(20mL)溶液に、水素化ナトリウム(鉱油中60%分散物、1.02g、25.6mmol)を添加した。0℃にて45分間撹拌した後、ヨードエタン(2.08mL、25.6mmol)を滴下で添加し、得られた混合物を室温に温めた。18時間後、反応混合物を水でクエンチし、酢酸エチルで2度抽出した。合わせた有機層を水で2度洗浄し、ブラインで1度洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、ヘプタン中の0〜10%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、4.6g(58%収率)の所望の生成物を黄色の油として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 1.40
(t, J=7.0 Hz, 3 H), 3.89 (s, 2 H), 4.01 (q, J=6.9 Hz, 2 H), 6.83 (d, J=8.4 Hz,
2 H), 6.91 (t, J=9.0 Hz, 1 H), 7.10 (d, J=8.8 Hz, 2 H), 7.20 - 7.30 (m, 2 H).
【0121】
トルエン−4−スルホン酸テトラヒドロ−フラン−3−イルエステルの調製
3−ヒドロキシテトラヒドロフラン(2.5g、28.0mmol)の無水ピリジン(60mL)溶液に、室温で4−トルエンスルホニルクロリド(6.49g、34.0mmol)を添加した。室温で反応混合物を18時間撹拌した後、反応混合物を減圧下で濃縮した。得られた残渣は、ヘプタン中の0〜30%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、3.5g(51%収率)の所望の生成物を無色の油として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 2.05
- 2.12 (m, 2 H), 2.45 (s, 3 H), 3.77 - 3.92 (m, 4 H), 5.09 - 5.14 (m, 1 H),
7.35 (d, J=8.00 Hz, 2 H), 7.79 (d, 2 H).
【0122】
トルエン−4−スルホン酸オキセタン−3−イルエステルの調製
オキセタン−3−オール(1.0g、13.0mmol)の無水ピリジン(25mL)溶液に、室温で4−トルエンスルホニルクロリド(3.09g、16.2mmol)を添加した。室温で反応混合物を18時間撹拌した後、反応混合物を減圧下で濃縮した。得られた残渣は、ヘプタン中の0〜30%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、1.9g(62%収率)の所望の生成物を白色の固体として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 2.46
(s, 3 H), 4.63 - 4.75 (m, 4 H), 5.26 - 5.34 (m, 1 H), 7.36 (d, J=8.00 Hz, 2 H),
7.78 (d, J=8.40 Hz, 2 H).
【0123】
出発物質3−[4−(5−ブロモ−2−フルオロ−ベンジル)−フェノキシ]−テトラヒドロ−フランの調製
4−(5−ブロモ−2−フルオロ−ベンジル)−フェノール(1.5g、5.3mmol)および炭酸セシウム(2.61g、8.0mmol)のN,N−ジメチルホルムアミド(15.0mL)溶液に、室温でトルエン−4−スルホン酸テトラヒドロ−フラン−3−イルエステル(1.94g、8.0mmol)のN,N−ジメチホルムアミド(10.0mL)溶液を添加した。次いで、反応混合物を一晩50℃で撹拌した。全部で18時間後、反応混合物を室温に冷却し、ブラインで希釈し、酢酸エチルで3度抽出した。合わせた有機層を水で2度洗浄し、ブラインで1度洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下で濃縮した。得られた粗残渣は、ヘプタン中の0〜30%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、1.66g(89%収率)の所望の生成物を無色の油として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 2.09
- 2.24 (m, 2 H), 3.86 - 4.01 (m, 6 H), 4.86 - 4.91 (m, 1 H), 6.80 (d, J=8.6 Hz,
2 H), 6.91 (t, J=9 Hz, 1 H), 7.10 (d, J=8.6 Hz, 2 H), 7.23 (dd, J=6.8, 2.5 Hz,
1 H), 7.26 - 7.31 (m, 1 H).
【0124】
出発物質3−[4−(5−ブロモ−2−フルオロ−ベンジル)−フェノキシ]−オキセタンの調製
4−(5−ブロモ−2−フルオロ−ベンジル)−フェノール(1.1g、3.9mmol)および炭酸セシウム(1.91g、5.87mmol)のN.N−ジメチルホルムアミド(15.0mL)溶液に、室温でトルエン−4−スルホン酸オキセタン−3−イルエステル(1.34g、8.0mmol)のN,N−ジメチルホルムアミド(10.0mL)溶液を添加した。次いで、反応混合物を一晩65℃で撹拌した。全部で18時間後、反応混合物を室温に冷却し、ブラインで希釈し、酢酸エチルで3度抽出した。合わせた有機層を水で2度洗浄し、ブラインで1度洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、ヘプタン中の0〜30%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、0.948g(72%収率)の所望の生成物を白色の固体として得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 3.88
(s, 2 H), 4.76 (dd, J=7.22, 5.3 Hz, 2 H), 4.95 (t, J=6.6 Hz, 2 H), 5.14 - 5.21
(m, 1 H), 6.63 (d, J=8.4 Hz, 2 H), 6.92 (dd, 1 H), 7.10 (d, J=8.6 Hz, 2 H),
7.23 (dd, J=6.6, 2.15 Hz, 1 H), 7.26 - 7.31 (m, 1 H).
【0125】
3−(4−(5−ブロモ−2−クロロベンジル)フェノキシ)オキセタンの調製:
4−ブロモ−1−クロロ−2−(4−メトキシベンジル)−ベンゼン(10g、32mmol)をジクロロメタン(32mL)に溶解し、窒素下で0℃に冷却した。三臭化ホウ素のジクロロメタン(35.3mL、34.3mmol)溶液(1.0M)を、10分に亘り滴下で添加した。添加の後、氷浴を取り除き、溶液を室温で1時間撹拌した。反応混合物を0℃に冷却し、1Nの塩酸水溶液(45mL)を添加することによってクエンチした。混合物を30分間撹拌し、分液漏斗に移し、有機層を集め、水層をジクロロメタン(45mL)で抽出した。合わせた有機抽出物を硫酸マグネシウム上で乾燥させ、濾過し、真空中で濃縮し、4−(5−ブロモ−2−クロロベンジル)フェノール(9.5g、99%収率)を白色の固体として得た。
【0126】
粗4−(5−ブロモ−2−クロロベンジル)フェノール(3.0g、10mmol)および炭酸セシウム(4.9g、15mmol)のN,N−ジメチルホルムアミド(77.5mL)溶液に、室温でトルエン−4−スルホン酸オキセタン−3−イルエステル(3.5g、15mmol)のN,N−ジメチルホルムアミド(8mL)溶液を添加した。混合物を65℃に22時間加熱し、そこでさらなる一定分量の炭酸セシウム(3.3g、10mmol)を添加した。反応混合物を120℃でさらに12時間撹拌し、室温に冷却し、そこで水および酢酸エチルを添加し、混合物を1Nの塩酸水溶液で注意深く酸性化した。有機層を分離し、ブラインで洗浄し(3度)、真空中で濃縮した。Biotage MPLC(シリカゲル、ヘプタン中の0〜25%酢酸エチルの勾配で溶出)による精製によって、3−(4−(5−ブロモ−2−クロロベンジル)フェノキシ)オキセタン(2.5g、70%収率)を白色の固体として得た。
1H NMR
(400 MHz, ジクロロメタン-d2) δ ppm
7.34 - 7.28 (m, 2 H), 7.26 (d, J=8.4 Hz, 1 H), 7.14 - 7.09 (m, 2 H), 6.69 -
6.35 (m, 2 H), 5.22 - 5.16 (m, 1 H), 4.96 - 4.91 (m, 2H), 4.72 - 4.68 (m, 2H),
4.01 (s, 2H).
【0127】
4−ブロモ−2−(4−クロロ−ベンジル)−1−フルオロ−ベンゼンの調製
5−ブロモ−2−フルオロベンズアルデヒド(10.2g、50mmol)の無水テトラヒドロフラン(200mL)溶液を、−78℃に冷却した。4−クロロフェニル−マグネシウムブロミドの溶液(ジエチルエーテル中1M、60mL、60mmol)を、シリンジによって8分に亘り添加した。低温で5分間撹拌を続け、反応物を室温に温め、1時間この温度で撹拌した。溶液を氷水浴中で冷却し、飽和塩化アンモニウム水溶液(40mL)を添加することによってクエンチした。有機相をデカントし、水性残渣を減圧下で濃縮し、残った有機溶媒を除去した。水相を酢酸エチル(200mL×2)で抽出し、抽出物をデカントしたテトラヒドロフラン溶液と合わせた。この溶液をブライン(25mL)で洗浄し、乾燥させ(硫酸ナトリウム)、濾過し、減圧下で濃縮し、粗(5−ブロモ−2−フルオロフェニル)−(4−クロロフェニル)−メタノール(15.2g、96%収率)を黄色の固体として得た。
【0128】
上記の(5−ブロモ−2−フルオロフェニル)−(4−クロロフェニル)−メタノール(15.0g、48mmol)およびトリエチルシラン(18.5mL、116mmol)のジクロロメタン(40mL)およびアセトニトリル(20mL)溶液に、0℃にて窒素下で三フッ化ホウ素ジエチルエーテラート(22.7mL、181mmol)をゆっくりと添加した。得られた溶液を18時間撹拌し、その間に室温にゆっくりと温めた。反応物を氷水浴中で冷却し、7Mの水酸化カリウム水溶液(30mL)をゆっくりと添加することによってクエンチし、メチルtert−ブチルエーテル(200mL×2)で抽出した。合わせた有機溶液を水(25mL×2)、ブライン(25mL×2)で洗浄し、乾燥させ(硫酸ナトリウム)、濾過し、減圧下で濃縮した。ヘプタン中の酢酸エチルの勾配で溶出するシリカゲル上のフラッシュカラムクロマトグラフィーによる精製によって、2−(4−クロロベンジル)−4−ブロモ−1−フルオロベンゼン(5.0g、35%収率)を無色の油として得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 7.33-7.22 (m, 4H), 7.13 (d, J =
8.4 Hz, 2H), 6.93 (dd, J = 9.2, 9.2 Hz, 1H), 3.92 (s, 2H).
【0129】
中間体の調製
中間体((2R,3R,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−テトラヒドロ−ピラン−2−イル)−メタノール(I−1a)の調製:
【0130】
【化8】
D−グルコース(1.2kg、6.6mol)、トリフルオロメタンスルホン酸(12mL)およびアリルアルコール(5L)の懸濁液を、80℃で3日間加熱した。混合物を室温に冷却し、揮発性物質を真空中で除去し、残渣をN,N−ジメチルホルムアミド(8L)に溶解した。これを4つの等しい反応物に分割し、各々に塩化トリチル(463g、1.67mol)およびトリエチルアミン(231mL、1.67mol)を添加した。トリエチルアミンを添加する間に、僅かな発熱量が観察された。反応混合物を30℃で2日間撹拌し、次いで各反応物を半分に分け、8つの等しい反応物を得た。これらの反応物の各々に、塩化ベンジル(300mL、2.60mol)を添加し、続いて水素化ナトリウム(102.5g、2.60mol)を少しずつ添加し、反応温度を40〜50℃に維持した。完全に添加した後、反応混合物を室温で20時間撹拌した。次いで、各反応物を氷/水(2L)上に注ぎ、酢酸エチル(2.5L)で抽出した。各々の有機相を飽和ブライン/水(1:1、2×2L)で洗浄し、合わせ、硫酸マグネシウム上で乾燥させた(3:1のヘキサン/酢酸エチル中の生成物Rf0.85)。濾過および蒸発後、残渣をジクロロメタン(16L)およびメタノール(4L)の混合物に溶解した。混合物を5つの等しい部分に分割し、各々に硫酸(32mL)を添加した。反応物を3時間撹拌し、ブライン/2Mの水酸化ナトリウム水溶液(1:1、2×2L)で洗浄し、合わせ、硫酸マグネシウム上で乾燥させた。濾過および真空濃縮後、残渣をトルエン中の30%酢酸エチルで溶出するシリカゲル上でさらに精製し、中間化合物(I−1a)をアノマーの混合物として得た(1.77kg、D−グルコースから54%収率)。3:1のヘキサン/酢酸エチル中Rf0.15。
【0131】
中間体((3S,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−2−ヒドロキシメチル−テトラヒドロ−ピラン−2−イル)−メタノール(I−1b)の調製:
【0132】
【化9】
ジメチルスルホキシド(87mL、1.22mol)のジクロロメタン(160mL)溶液を、塩化オキサリル(64.7mL、0.76mol)のジクロロメタン(2.5L)溶液に−78℃にて滴下で添加した。完全に添加した後、中間体(I−1a)(287g、0.59mol)のジクロロメタン(500mL)溶液を、−78℃にて滴下で添加した。完全に添加した後、反応混合物を30分間撹拌し、トリエチルアミン(417mL、2.9mol)を滴下で添加した。完全に添加した後、反応混合物は、室温にそれ自体で温まった。次いで、反応物を1Mの塩酸水溶液(2L)および水(2L)で洗浄し、次いで硫酸マグネシウム上で乾燥させた。この反応手順を6つの等しい反応物で繰返し、乾燥させた後それらを合わせ、蒸発させ、アルデヒドを黄色の油(1.71kg)として得た。この油をイソプロパノール(2.57L)に溶解し、7つの等しい反応物に分割した。これらの各々に、37%ホルムアルデヒド水溶液(0.79L、10mol)を添加し、続いて水酸化ナトリウム(32g、0.8mol)の水(130mL)溶液を滴下で添加した。完全に添加した後、反応混合物を室温で2日間撹拌した。反応混合物をブライン(2L)で希釈し、酢酸エチル(2L)で抽出した。有機相を飽和炭酸水素ナトリウム水溶液(2L)、ブライン(2L)でさらに洗浄し、次いで硫酸マグネシウム上で乾燥させた。7回の反応からの有機相を合わせ、蒸発させ、残渣をシリカゲル(4対1から1対1までの酢酸エチル中のヘキサンで溶出する)上で精製し、中間化合物(I−1b)をアノマーの混合物(980g、2ステップに亘り53%収率)として得た。1:1のヘキサン/酢酸エチル中Rf0.57および0.60。
【0133】
(3S,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−2,2−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン(I−1c):
【0134】
【化10】
出発物質のジオール[((3S,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−2−ヒドロキシメチル−テトラヒドロ−ピラン−2−イル)−メタノール(I−1b:10g、19.208mmol)をN,N−ジメチルホルムアミド(70mL)に溶解し、0℃に冷却した。水素化ナトリウム(鉱油中60%分散物、1.69g、42.3mmol)を添加し、反応物を0℃で1時間撹拌し、その後1−ブロモメチル−4−メトキシ−ベンゼン(5.96mL、40.3mmol)を添加した。次いで、反応物を60℃に一晩加熱した。混合物を室温に冷却し、反応物を水でクエンチし、酢酸エチル(2度)で抽出した。合わせた有機層を水、ブラインで洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下濃縮した。次いで、反応物をシリカゲル(ヘプタン中の0〜80%酢酸エチルの勾配で溶出)上でクロマトグラフィーにかけ、7.55g(52%収率)の生成物(I−1c)を得た。MS 778.8 (M+NH4+; ポジティブモード).
【0135】
(3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−1d):
【0136】
【化11】
出発物質((3S,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−2,2−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン(I−1c:7.55g、9.92mmol)のメタノール(60mL)およびジクロロメタン(20mL)溶液に、室温で塩化パラジウム(II)(528mg、2.98mmol)を添加し、得られた混合物をこの温度で4時間撹拌した。TLCは、より極性の生成物の明らかな形成を示した。反応物をCelite(登録商標)を通して濾過し、減圧下で濃縮した。粗材料を、ヘプタン中の0〜80%酢酸エチルの勾配で溶出するシリカゲル上のクロマトグラフィーにかけ、5.6g(78%収率)の生成物(I−1d)を得た。MS 738.8 (M+NH4+; ポジティブモード).
【0137】
(3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オン(I−1e):
【0138】
【化12】
二塩化オキサリル(1.9mL、23mmol)のジクロロメタン(65mL)溶液に、−78℃でジメチルスルホキシド(3.3mL、47mmol)のジクロロメタン(5mL)溶液を添加し、得られた溶液をこの温度で30分間撹拌した。出発物質((3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−1d、5.6g、7.7mmol)のジクロロメタン(15.0mL)溶液を、次いで滴下で添加し、得られた混合物を30分間撹拌し、温度を−60℃に上げた。トリエチルアミン(9.7mL、69.5mmol)を滴下で添加し、混合物を1時間に亘り0℃に温めた。反応物を飽和塩化アンモニウム水溶液の添加によってクエンチし、有機相を硫酸マグネシウム上で乾燥させ、濾過し、減圧下濃縮した。粗材料は、ヘプタン中の0〜60%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、生成物(I−1e)(4g、72%収率)を生成した。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 3.24
(d, J=10 Hz, 1 H), 3.40 - 3.47 (m, 2 H), 3.74 (s, 3 H), 3.77 (s, 3 H), 3.86 (d,
J=10 Hz, 1 H), 4.07 (d, J=8.6 Hz, 1 H), 4.15 (d, J=9.6 Hz, 1 H), 4.35 - 4.55
(m, 6 H), 4.65 - 4.72 (m, 2 H), 4.82 (d, J=11 Hz,1 H), 4.87 (d, J=11.2 Hz, 1
H), 5.10 (d, J=11.1 Hz, 1 H), 6.74 - 6.79 (m, 2 H), 6.81 - 6.85 (m, 2 H), 7.11
(dd, J=7.0, 2.5 Hz, 2 H), 7.17 - 7.41 (m, 17 H).
【0139】
(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)および/または(3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−2−(メトキシ−メチル−アミノ)−テトラヒドロ−ピラン−2−オール(I−1f):
【0140】
【化13】
ラクトン((3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オン(I−1e:10.4g、14.5mmol)およびN,O−ジメチル−ヒドロキシルアミン塩酸塩(1.77g、29.0mmol)のジクロロメタン(100mL)溶液に、0℃でトリメチルアルミニウムのヘキサン(14.5mL、29.0mmol)溶液(2.0M)を滴下で添加し、得られた溶液を室温で16時間撹拌した。反応混合物を0℃に冷却し、1Nの塩酸水溶液をゆっくり添加することによってクエンチした。得られた混合物を1時間撹拌した。有機相を分離し、1Nの塩酸水溶液で洗浄し、硫酸ナトリウム上で乾燥させ、濾過し、減圧下で濃縮した。粗材料を中圧クロマトグラフィー(ヘプタン中の5〜40%酢酸エチルの勾配)によって精製し、6.5g(58%)の生成物を得た。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 2.62
(br. s, 1 H), 2.94 (br. s., 3 H), 3.23 (br. s., 3 H), 3.42 (d, J=9.4 Hz, 1 H),
3.50 - 3.60 (m, 3 H), 3.75 (s, 3 H), 3.77 (s, 3 H), 4.03 (d, J=6.9 Hz, 1 H),
4.20 (dd, J=6.9, 3.3 Hz, 1 H), 4.31 - 4.44 (m, 5 H), 4.46 - 4.51 (m , 2H), 4.53
(d, J=12 Hz, 1 H), 4.66 (d, J=12 Hz, 1 H), 4.80 (br. d, J=11.5 Hz, 1 H), 4.87
(d, J=11.4 Hz, 1 H), 6.77 - 6.83 (m, 4 H), 7.15 - 7.35 (m, 19 H). ([M+H+]
780.8, ポジティブモード; [M+HCO2-] 824.7,
ネガティブモード). HRMS C46H54NO10の計算値 (M+H+) 780.3742, 実測値 780.3708.
【0141】
(2R,3S,4S)−2,3,4,6−テトラキス−ベンジルオキシ−5−ベンジルオキシメチル−5−ヒドロキシ−ヘキサン酸メトキシ−メチル−アミド(I−6g)および/または(3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−ベンジルオキシメチル−2−(メトキシ−メチル−アミノ)−テトラヒドロ−ピラン−2−オール(I−6f):
【0142】
【化14】
この化合物は、(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)および/または(3R,4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−2−(メトキシ−メチル−アミノ)−テトラヒドロ−ピラン−2−オール(I−1f)の合成について記載したものと同様の手順((I−1b)から(I−1c)への変換を記載した実験の部において使用したアルキル化剤が、パラ−メトキシベンジルブロミドの代わりに臭化ベンジルであったこと以外)を使用して、[((3S,4S,5R)−6−アリルオキシ−3,4,5−トリス−ベンジルオキシ−2−ヒドロキシメチル−テトラヒドロ−ピラン−2−イル)−メタノール(I−1b)から出発して調製した。
1H NMR
(400 MHz, クロロホルム-d) δ ppm 2.66
(br. s, 1 H), 2.94 (br. s., 3 H), 3.23 (br. s., 3 H), 3.48 (d, J=9.4 Hz, 1 H),
3.55 - 3.66 (m, 3 H), 4.05 (d, J=6.9 Hz, 1 H), 4.21 (dd, J=6.9, 3.3 Hz, 1 H),
4.36 (d, 1H, J = 11.7 Hz), 4.41 - 4.58 (m, 7 H), 4.68 (d, J=11.9 Hz, 1 H), 4.81
(br. d, J=11.5 Hz, 1 H), 4.89 (d, J=11.5 Hz, 1 H), 7.15 - 7.35 (m, 25 H). MS
[M+H+] 720.7, ポジティブモード; [M+HCO2-]
764.7, ネガティブモード).
【0143】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−1i):
【0144】
【化15】
n−ブチルリチウム(0.97mL、2.5M/ヘキサン、3.15当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−2−(4−メトキシ−ベンジル)−1−メチル−ベンゼン(690mg、3当量)の無水テトラヒドロフラン(2.7mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度でさらに1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(608mg)の無水テトラヒドロフラン(1.35mL)溶液を、次いでシリンジポンプを使用して1.5時間に亘り滴下で添加し、得られた混合物を−78℃で1時間撹拌し、その後−20℃に14時間に亘り温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。ジエチルエーテルを添加し、1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮した。ヘプタン中の20〜50%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーによって、生成物を異性体の混合物(440mg、61%収率)として得た。
HRMS C59H62O10Naの計算値
(M+Na+) 953.4235, 実測値953.4236.
【0145】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−1k):
【0146】
【化16】
中間体I−1i(150mg)のジクロロメタン(3mL)溶液に、アニソール(90μL、5当量)、続いて20%トリフルオロ酢酸のジクロロメタン溶液(3mL)を添加し、得られた混合物を、室温で約1時間撹拌した。混合物を濃縮し、粗製物をシリカゲル(ヘプタン中の10〜30%酢酸エチルの勾配を使用)上のクロマトグラフィーにかけ、所望の生成物を異性体の混合物として得た(66mg、61%収率)。MS (LCMS) 673.9 (M+H+, ポジティブモード).
【0147】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−2i):
【0148】
【化17】
n−ブチルリチウム(0.312mL、2.5M/ヘキサン、3.05当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−2−(4−エトキシ−ベンジル)−1−メチル−ベンゼン(238mg、3.05当量)の無水テトラヒドロフラン(0.9mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度でさらに1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(200mg)の無水テトラヒドロフラン(0.6mL)溶液を、次いでシリンジポンプを使用して1.5時間に亘り滴下で添加し、得られた混合物を−78℃で1時間撹拌し、その後16時間に亘り室温に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。ジエチルエーテルを添加し、1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮した。Biotage(商標)自動クロマトグラフィーユニット(2つの積重ねた10gシリカゲルカラム、ヘプタン中の0〜60%酢酸エチルの勾配で溶出)を使用して粗材料をクロマトグラフィーにかけ、生成物を異性体の混合物(136mg、56%収率)として得た。MS (LCMS) 968 (M+Na+, ポジティブモード).
【0149】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−2k):
【0150】
【化18】
中間体I−2i(136mg、0.145mmol)のジクロロメタン(4mL)溶液に、アニソール(310μL、約5当量)、続いて20%トリフルオロ酢酸のジクロロメタン溶液(4mL)を添加し、得られた混合物を室温で1.5時間撹拌した。混合物を濃縮し、粗製物は、ISCO(商標)combiflash(登録商標)companion(登録商標)自動クロマトグラフィーユニット(4gのシリカゲルカラム)を使用して、ヘプタン中の0〜70%酢酸エチルの勾配で溶出するクロマトグラフィーにかけ、所望の生成物を異性体の混合物として得た(85mg、85%収率)。MS (LCMS) 687.7 (M+H+, ポジティブモード).
【0151】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−3i):
【0152】
【化19】
n−ブチルリチウム(0.97mL、2.5M/ヘキサン、3.15当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−1−クロロ−2−(4−メトキシ−ベンジル)−ベンゼン(725mg、2.95当量)の無水テトラヒドロフラン(2.7mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度でさらに1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(616mg)の無水テトラヒドロフラン(1.35mL)溶液を、次いでシリンジポンプを使用して1.5時間に亘り滴下で添加し、得られた混合物を−78℃で1時間撹拌し、その後14時間に亘り−20℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。ジエチルエーテルを添加し、1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮した。ヘプタン中の10〜40%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーによって、生成物を異性体の混合物(530mg、71%収率)として得た。
【0153】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−3k):
【0154】
【化20】
中間体I−3i(530mg)のジクロロメタン(11mL)溶液に、アニソール(300μL、5当量)、続いて20%トリフルオロ酢酸のジクロロメタン溶液(11mL)を添加し、得られた混合物を室温で1時間撹拌した。混合物を濃縮し、粗製物を、ヘプタン中の10〜40%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーにかけ、生成物を異性体の混合物として得た(229mg、59%収率)。
MS (LCMS) 693.6 (M+H+, ポジティブモード).
【0155】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−4i):
【0156】
【化21】
n−ブチルリチウム(1.0mL、2.5M/ヘキサン、3.25当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−1−クロロ−2−(4−エトキシ−ベンジル)−ベンゼン(815mg、3.25当量)の無水テトラヒドロフラン(2.9mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度でさらに1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(600mg)の無水テトラヒドロフラン(1.45mL)溶液を、次いでシリンジポンプを使用して1.3時間に亘り滴下で添加し、得られた混合物を−78℃で1時間撹拌し、その後14時間に亘り−25℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。ジエチルエーテルを添加し、1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮した。ヘプタン中の10〜40%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーによって、生成物を異性体の混合物(280mg、38%収率)として得た。
HRMS C59H61O10ClNaの計算値 (M+Na+) 987.3845, 実測値987.3840.
【0157】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−4k):
【0158】
【化22】
中間体I−4i(1.46g)のジクロロメタン(31mL)溶液に、アニソール(900μL、約5当量)、続いて20%トリフルオロ酢酸のジクロロメタン溶液(31mL)を添加し、得られた混合物を室温で1時間撹拌した。混合物を濃縮し、粗製物を、ヘプタン中の10〜30%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーにかけ、生成物を異性体の混合物として得た(670mg、63%収率)。
HRMS C43H44O7Clの計算値 (M+H+) 707.2770, 実測値707.2765.
【0159】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−6,6−ビス−(4メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−5i):
【0160】
【化23】
n−ブチルリチウム(462μL、2.5M/ヘキサン、3.0当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−1−フルオロ−2−(4−メトキシ−ベンジル)−ベンゼン(341mg、3当量)の無水テトラヒドロフラン(1.4mL)溶液に−78℃にて窒素下にて滴下で添加した(5秒毎に1滴)。得られた溶液をこの温度で1時間撹拌した。次いで、(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(300mg、0.385mmol)の無水テトラヒドロフラン(0.70mL)溶液を、非常にゆっくりと滴下で添加し(5秒毎に1滴)、得られた混合物を−78℃でさらに1時間撹拌し、その後12時間に亘り10℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。反応物をジエチルエーテルで希釈し、1Nの塩酸水溶液を滴下で添加することによってクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜40%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(199mg、55%収率)として得た。
【0161】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−5k):
【0162】
【化24】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−6,6−ビス−(4メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−5i;191mg、0.204mmol)のジクロロメタン(3.75mL)溶液に、アニソール(0.178mL、1.63mmol)、続いてトリフルオロ酢酸のジクロロメタン(3.75mL)溶液(20%)を室温で窒素下にて添加した。室温で1時間撹拌した後、反応混合物を減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜30%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(115mg、83%収率)として得た。MS (LCMS) 677.7 (M+H+, ポジティブモード).
【0163】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−10i)
【0164】
【化25】
n−ブチルリチウム(508μL、2.5M/ヘキサン、3.0当量)を、酸素脱気した4−ブロモ−2−(4−エトキシ−ベンジル)−1−フルオロ−ベンゼン(392.0mg、1.27mmol)の無水テトラヒドロフラン(1.5mL)溶液に−78℃にて窒素下にて滴下で添加した(5秒毎に1滴)。得られた溶液をこの温度で1時間撹拌した。次いで、(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミドI−1g(330.0mg、0.423mmol)の無水テトラヒドロフラン(0.75mL)溶液を非常にゆっくりと滴下で添加し(5秒毎に1滴)、得られた混合物を−78℃でさらに1時間撹拌し、その後12時間に亘り10℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する)。反応物をジエチルエーテルで希釈し、1Nの塩酸水溶液を滴下で添加することによってクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜40%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(180mg、44%収率)として得た。
【0165】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−10k)
【0166】
【化26】
中間体I−10i(180.0mg、0.19mmol)のジクロロメタン(2.0mL)溶液に、アニソール(0.175mL、1.60mmol)、続いてトリフルオロ酢酸のジクロロメタン(2.0mL)溶液(20%)を室温にて窒素下で添加した。1時間撹拌した後、反応混合物を減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜30%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物として得た(85.0mg、64%収率)。
【0167】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−11i)
【0168】
【化27】
n−ブチルリチウム(1.0mL、2.5M/ヘキサン、3.0当量)を、酸素脱気した3−[4−(5−ブロモ−2−フルオロ−ベンジル)−フェノキシ]−テトラヒドロ−フラン(878mg、2.50mmol)の無水テトラヒドロフラン(3.0mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度で1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミドI−1g(650mg、0.833mmol)の無水テトラヒドロフラン(1.5mL)溶液を、次いで非常にゆっくりと滴下で添加し(0.9mL/時間)、得られた混合物を−78℃でさらに1時間撹拌し、その後12時間に亘り10℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する)。反応物をジエチルエーテルで希釈し、1Nの塩酸水溶液を滴下で添加することによってクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜40%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(287mg、34%収率)として得た。
【0169】
((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノール(I−11k)
【0170】
【化28】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オールI−11i(275mg、0.28mmol)のジクロロメタン(2.0mL)溶液に、アニソール(0.250mL、2.29mmol)、続いてトリフルオロ酢酸のジクロロメタン(8.0mL)溶液(20%)を室温にて窒素下で添加した。1時間撹拌した後、反応混合物を減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜30%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(168mg、83%収率)として得た。
【0171】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−[3−(4−クロロ−ベンジル)−4−フルオロ−フェニル]−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−12i):
【0172】
【化29】
n−ブチルリチウム(1.0mL、2.5M/ヘキサン、3.1当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)4−ブロモ−2−(4−クロロ−ベンジル)−1−フルオロ−ベンゼン(702mg、2.9当量)の無水テトラヒドロフラン(3.0mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度で25分間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(621mg)の無水テトラヒドロフラン(1.5mL)溶液を、次いでシリンジポンプを使用して滴下で添加し(0.9mL/時間)、得られた混合物を低温でさらに17時間撹拌した(アルミ箔で覆われた深いデュワー中に置いて、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。1Mの塩酸水溶液(1.5mL)を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で30分間撹拌した。混合物を飽和塩化アンモニウム水溶液(15mL)で希釈し、酢酸エチル(15mL×3)で抽出した。合わせた有機溶液をブライン(30mL)で洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮した。ヘプタン中の10〜40%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーによって、生成物を異性体の混合物(477mg、64%収率)として得た。
【0173】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−クロロ−ベンジル)−4−フルオロ−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−12k):
【0174】
【化30】
中間体I−12i(243mg)のジクロロメタン(9mL)溶液に、アニソール(0.15mL、5.3当量)、続いてトリフルオロ酢酸(1.0mL、50当量)を添加し、得られた混合物を室温で2時間撹拌した。混合物を濃縮し、粗製物を、ヘプタン中の10〜30%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーにかけ、生成物を異性体の混合物として得た(102mg、58%収率)。
【0175】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−13i)
【0176】
【化31】
n−ブチルリチウム(1.12mL、2.5M/ヘキサン、3.0当量)を、酸素脱気した3−[4−(5−ブロモ−2−フルオロ−ベンジル)−フェノキシ]−オキセタン(942.0mg、2.79mmol)の無水テトラヒドロフラン(3.0mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度で1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミドI−1g(725.0mg、0.930mmol)の無水テトラヒドロフラン(1.5mL)溶液を、次いで非常にゆっくりと滴下で添加し(0.9ml/時間)、得られた混合物を−78℃でさらに1時間撹拌し、その後12時間に亘り10℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する)。反応物をジエチルエーテルで希釈し、1Nの塩酸水溶液を滴下で添加することによってクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜40%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(535mg、59%収率)として得た。
【0177】
((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノール(I−13k)
【0178】
【化32】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オールI−13i(535mg、0.548mmol)のジクロロメタン(2.0mL)溶液に、アニソール(0.480mL、4.38mmol)、続いてトリフルオロ酢酸のジクロロメタン(8.0mL)溶液(20%)を室温にて窒素下で添加した。1時間撹拌した後、反応混合物を減圧下で濃縮した。粗残渣は、シリカゲル(ヘプタン中の10〜30%酢酸エチルの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、生成物を異性体の混合物(300mg、76%収率)として得た。
【0179】
(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オール(I−14i):
【0180】
【化33】
n−ブチルリチウム(0.97mL、2.5M/ヘキサン、3.15当量)を、酸素脱気した(そのキャップで密封した事前に乾燥させたBiotage(商標)マイクロ波バイアル(10〜20mL)中に入れ、窒素ガスの正流下に置く)3−(4−(5−ブロモ−2−クロロベンジル)フェノキシ)オキセタン(824mg、2.95当量)の無水テトラヒドロフラン(2.7mL)溶液に−78℃にて滴下で添加し(5秒毎に1滴)、得られた溶液をこの温度でさらに1時間撹拌した。(2R,3S,4S)−2,3,4−トリス−ベンジルオキシ−5−ヒドロキシ−6−(4−メトキシ−ベンジルオキシ)−5−(4−メトキシ−ベンジルオキシメチル)−ヘキサン酸メトキシ−メチル−アミド(I−1g)(616mg)の無水テトラヒドロフラン(1.35mL)溶液を、次いでシリンジポンプを使用して1.5時間に亘り滴下で添加し、得られた混合物を−78℃で1時間撹拌し、その後14時間に亘り−20℃に温めた(アルミ箔で覆われた深いデュワー中に入れ、低温を維持する。デュワーのサイズ:外径10cm、内径8cm、高さ9cm)。ジエチルエーテルを添加し、1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチした。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。ヘプタン中の0〜50%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーによって、生成物を異性体の混合物(563mg、72%収率)として得た。
【0181】
((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノール(I−14k):
【0182】
【化34】
中間体(4S,5S)−3,4,5−トリス−ベンジルオキシ−2−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,6−ビス−(4−メトキシ−ベンジルオキシメチル)−テトラヒドロ−ピラン−2−オールI−14i(282mg)のジクロロメタン(2.84mL)溶液に、アニソール(200μL、約7当量)、続いて20%トリフルオロ酢酸のジクロロメタン溶液(3.07mL)を添加し、得られた混合物を室温で1.5時間撹拌した。混合物を濃縮し、粗製物を、ヘプタン中の10〜50%酢酸エチルの勾配を使用したシリカゲル上のクロマトグラフィーにかけ、生成物を異性体の混合物として得た(186mg、89%収率)。
【0183】
(実施例1)
(1S,2S,3S,4R,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(1A)および(1S,2S,3S,4S,5S)−1−ヒドロキシメチル−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(1B):
【0184】
【化35】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−メトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−1k:236mg)のエタノール/テトラヒドロフラン(7mL、4/1容)溶液に、ギ酸(270μL、19当量)およびパラジウム黒(150mg、4当量)を連続的に添加し、得られた混合物を室温で3時間撹拌した。パラジウムを濾過し、溶媒の蒸発の後に得られた粗混合物を、ヘプタン中の85〜100%酢酸エチルの勾配で溶出するシリカゲル上のクロマトグラフィーによって精製した。得られた生成物の混合物を分取HPLCによって精製した。
【0185】
HPLC分取法:逆相C18phenomenexカラムLuna、5マイクロメーター、150×21.20mm、20mL/分、20分に亘りアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;20〜60%のアセトニトリル/0.1%ギ酸の勾配。UV検出:254nm。HPLCは、3:1(1A:1B)のジアステレオマー比を示した。
【0186】
1A:(55mg、39%収率);Rt=10.9分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。MS (LCMS) 403.3 (M+H+; ポジティブモード)
447.3 (M+HCO2-, ネガティブモード).
1H NMR
(400 MHz, メタノール-d4) δ 7.33 (d, 1H, J = 1.6 Hz), 7.30 (dd, 1H, J = 7.6および1.6 Hz), 7.10 (d, 1H, J = 7.6 Hz), 7.02-6.98 (m, 2H), 6.79-6.75 (m,
2H), 4.13 (d, 1H, J = 7.4 Hz), 3.90 (s, 2H), 3.82 (d, 1H, J = 12.5 Hz), 3.77
(dd, 1H, J = 8.2および1.2 Hz), 3.72 (s, 3H), 3.66 (d, 1H,
J = 12.5 Hz), 3.65 (t, 1H, J = 8.0 Hz), 3.59 (d, 1H, J = 7.8 Hz), 3.58 (dd, 1H,
J = 7.5および1.5 Hz), 2.16 (s, 3H). HRMS C22H27O7の計算値 (M+H+) 403.1751, 実測値 403.1737.
【0187】
1B:(20mg、14%収率);Rt=11.5分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。MS (LCMS) 403 (M+H+; ポジティブモード)
447 (M+HCO2-, ネガティブモード).
1H NMR
(400 MHz, メタノール-d4) δ 7.38 (d, 1H, J = 1.8 Hz) 7.33 (dd, 1H, J = 7.9および1.8 Hz), 7.10 (d, 1H, J = 7.9 Hz), 7.02-6.97 (m, 2H), 6.79-6.74 (m,
2H), 4.02 (d, 1H, J = 7.4 Hz), 3.93 (t, 1H, J = 2.2 Hz), 3.91 (br. s, 2H), 3.88
(d, 1H, J = 12.5 Hz), 3.84 (d, 2H, J = 2.4 Hz), 3.75 (d, 1H, J = 12.5 Hz), 3.71
(s, 3H), 3.49 (d, 1H, J = 7.4 Hz), 2.16 (s, 3H).
【0188】
(実施例2)
(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(2A)および(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(2B)
【0189】
【化36】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−エトキシ−ベンジル)−4−メチル−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−2k:85mg、0.12mmol)のエタノール/テトラヒドロフラン(7mL、約4/1容)溶液に、ギ酸(95μL、19当量)およびパラジウム黒(53mg、4当量)を連続的に添加し、得られた混合物を室温で3時間撹拌した。パラジウムを濾過し、溶媒の蒸発の後に得られた粗混合物を分取HPLCによって精製した。
【0190】
HPLC分取法:逆相C18phenomenexカラムLuna、5マイクロメーター、150×21.20mm、20mL/分、20分に亘りアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;20〜60%のアセトニトリル/0.1%ギ酸の勾配。UV検出:254nm。HPLCは、4:1(2A:2B)のジアステレオマー比を示した。
【0191】
2A:(20mg;38%収率)Rt=12.7分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
MS (LCMS) 417.3 (M+H+; ポジティブモード); 461.4 (M+HCO2-; ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ ppm 1.34 (t, J=6.9 Hz, 3 H), 2.18 (s, 3 H), 3.60 (d,
J=8 Hz, 2 H), 3.66 (t, J=8 Hz, 1 H), 3.68 (d, J=12.5 Hz, 1 H), 3.78 (d, 1H, J=
8.8 Hz), 3.84 (d, J=12.4 Hz, 1 H), 3.92 (s, 2 H), 3.97 (q, J=7 Hz, 2 H), 4.15
(d, J=7.5 Hz, 1 H), 6.77 (m, 2 H), 7.00 (m, 2 H), 7.12 (d, J=7.7 Hz, 1 H), 7.31
(dd, J=7.9および1.4 Hz, 1 H), 7.34 (s, 1 H).
【0192】
2B:(5mg;9%収率)Rt=13.2分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
MS (LCMS) 417.3 (M+H+; ポジティブモード); 461.4 (M+HCO2-; ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ ppm 1.34 (t, J=6.9 Hz, 3 H), 2.18 (s, 3 H), 3.52 (d,
1H, J= 7.4 Hz), 3.77 (d, J=12.5 Hz, 1 H), 4.00-3.84 (m, 8 H), 4.04 (d, J=7.4
Hz, 1 H), 6.79-6.75 (m, 2 H), 7.03-6.98 (m, 2 H), 7.12 (d, J=7.9 Hz, 1 H), 7.35
(dd, J=7.7および1.9 Hz, 1 H), 7.39 (d, J = 1.9 Hz, 1 H).
【0193】
(実施例3)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(3A)および(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(3B):
【0194】
【化37】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−3k:229mg)のエタノール/テトラヒドロフラン(7mL、4/1容)溶液に、ギ酸(270μL、20当量)およびパラジウム黒(140mg、4当量)を連続的に添加し、得られた混合物を室温で撹拌した。1時間後、さらなるギ酸(270μL、20当量)およびパラジウム黒(140mg、4当量)を添加し、混合物をさらに1時間室温で撹拌した。パラジウムを濾過し、溶媒の蒸発の後に得られた粗混合物を分取HPLCによって精製した。
【0195】
HPLC分取法:逆相C18phenomenexカラムLuna、5マイクロメーター、150×21.20mm、20mL/分、20分に亘るアセトニトリル/0.1%ギ酸:水/0,1%ギ酸;20〜60%のアセトニトリル/0.1%ギ酸の勾配。UV検出:254nm。HPLCは、1.4:1(3A:3B)のジアステレオマー比を示した。
【0196】
3A:(50mg;36%収率)Rt=12.1分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で濃縮した。
MS (LCMS) 423.3 (M+H+; ポジティブモード); 467.3 (M+HCO2-; ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ 7.43 (s, 1H), 7.38-7.30 (m, 2H), 7.08 (d, 2H), 6.79
(d, 2H), 4.12 (d, 1H, J = 7.5 Hz), 4.01 (s, 2H), 3.81 (d, 1H, J = 12.5 Hz),
3.75 (d, 1H, J = 8.4 Hz), 3.73 (s, 3H), 3.66 (d, 1H, J = 11.7 Hz), 3.63 (t, 1H,
J = 8.2 Hz), 3.57 (d, 1H, J = 7.4 Hz), 3.52 (d, 1H, J = 7.8 Hz). HRMS C21H24O7Clの計算値 (M+H+) 423.1205, 実測値 423.1192.
【0197】
3B:(37mg;27%収率)Rt=12.8分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で濃縮した。
MS (LCMS) 423.3 (M+H+; ポジティブモード) 467.3 (M+HCO2-, ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ 7.50 (d, 1H, J = 1.9 Hz) 7.42 (dd, 1H, J = 8.3および1.9 Hz), 7.35 (d, 1H, J = 8.3 Hz), 7.12-7.07 (m, 2H), 6.83-6.78 (m,
2H), 4.06-4.01 (m, 3H), 3.91-3.83 (m, 4H), 3.78-3.72 (m, 4H), 3.51 (d, 1H, J =
7.5 Hz).
【0198】
(実施例4)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(4A)および(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(4B):
【0199】
【化38】
{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(I−4k:335mg)のエタノール/テトラヒドロフラン(10mL、4/1容)溶液に、ギ酸(420μL、22当量)およびパラジウム黒(208mg、4当量)を連続的に添加し、得られた混合物を室温で撹拌した。1時間後、さらなるギ酸(420μL、22当量)およびパラジウム黒(208mg、4当量)を添加し、混合物をさらに1時間室温で撹拌した。パラジウムを濾過し、溶媒の蒸発の後に得られた粗混合物を分取HPLCによって精製した。
【0200】
分取HPLC:逆相C18Geminiカラム、5マイクロメーター、30×100mm、40mL/分、18分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;25〜50%のアセトニトリル/0.1%ギ酸の勾配;UV検出:220nm。HPLCは、1.1:1(4A:4B)のジアステレオマー比を示した。
【0201】
4A:(60mg、29%収率);Rt=12.4分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
MS (LCMS) 437.3 (M+H+; ポジティブモード); 481.3 (M+HCO2-; ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ 7.43 (d, 1H, J = 1.9 Hz), 7.36 (dd, 1H, J = 8.3および2 Hz), 7.32 (d, 1H, J = 8.3 Hz), 7.08-7.04 (m, 2H), 6.79-6.75 (m,
2H), 4.12 (d, 1H, J = 7.5 Hz), 4.00 (s, 2H), 3.96 (q, 2H, J = 7.0 Hz), 3.81 (d,
1H, J = 12.5 Hz), 3.75 (dd, 1H, J = 8.3および1.3 Hz), 3.65
(d, 1H, J = 12.5 Hz), 3.63 (t, 1H, J = 8.2 Hz), 3.57 (dd, 1H, J = 7.5および1.3 Hz), 3.52 (d, 1H, J = 8.0 Hz), 1.33 (t, 3H, J = 6.9 Hz). HRMS C22H26O7Clの計算値 (M+H+) 437.1361, 実測値 437.1360.
【0202】
4B:(30mg、15%収率);Rt=13.2分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
MS (LCMS) 437.3 (M+H+; ポジティブモード) 481.3 (M+HCO2-, ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ 7.48 (d, 1H, J = 1.9 Hz) 7.40 (dd, 1H, J = 8.1および1.9 Hz), 7.32 (d, 1H, J = 8.3 Hz), 7.08-7.03 (m, 2H), 6.80-6.74 (m,
2H), 4.04-3.99 (m, 3H), 3.95 (q, 2H, J = 7 Hz), 3.89-3.81 (m, 4H), 3.73 (d, 1H,
J = 12.5 Hz), 3.49 (d, 1H, J = 7.3 Hz), 1.32 (t, 3H, J = 7 Hz). HRMS C22H26O7Clの計算値 (M+H+) 437.1361, 実測値 437.1358.
【0203】
(実施例5)
(1S,2S,3S,4R,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(5A)および(1S,2S,3S,4S,5S)−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(5B):
【0204】
【化39】
エタノール/テトラヒドロフランの4:1溶液(10mL)中の{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[4−フルオロ−3−(4−メトキシ−ベンジル)−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノール(115mg、0.170mmol)の溶液に、ギ酸(137μL、3.42mmol)およびパラジウム黒(73mg、0.687mmol)を連続的に添加した。得られた混合物を室温で撹拌した。3時間後、さらなるギ酸(137μL、3.42mmol)およびパラジウム黒(73mg、0.687mmol)を添加した。18時間後、反応混合物を濾過し、濾液を減圧下で濃縮した。得られた粗残渣は、シリカゲル(ジクロロメタン中の0〜15%メタノールの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、64mgの白色の固体を得た。異性体の混合物を分取HPLCによって精製した。
【0205】
HPLC分取法:逆相C18phenomenexカラムLuna、5マイクロメーター、150×21.20mm、20mL/分、20分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;20〜80%のアセトニトリル/0.1%ギ酸の勾配)。UV検出:254nm。HPLCは、1:1(5A:5B)のジアステレオマー比を示した。
【0206】
5A:(6mg;9%収率)Rt=8.5分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
1H NMR
(400 MHz, メタノール-d4) δ ppm 3.55 (d, J=7.8 Hz, 1 H), 3.58 (dd, J=7.5, 1.2 Hz, 1 H), 3.64
(t, J=8.2 Hz, 1 H), 3.67 (d, J=12.5 Hz, 1 H), 3.74 (s, 3 H), 3.77 (dd, J=8.3,
1.2 Hz, 1 H), 3.83 (d, J=12.5 Hz, 1 H), 3.91 (s, 2 H), 4.14 (d, J=7.4 Hz, 1 H),
6.76 - 6.84 (m, 2 H), 7.02 (dd, J=9.9, 8.3 Hz, 1 H), 7.09 - 7.13 (m, 2 H), 7.37
- 7.44 (m, 2 H); MS: 407.4 (M+ H+; ポジティブモード);
451.3 (M+HCO2-; ネガティブモード)
【0207】
5B:(12mg;17%収率)Rt=9分。生成物含有画分を減圧下で濃縮した。粗材料を酢酸エチルおよびヘプタンから沈殿させた。得られた白色の固体をヘプタンで2度洗浄し、減圧下で乾燥させた。
1H NMR
(400 MHz, メタノール-d4) δ ppm 3.51 (d, J=7.4 Hz, 1 H), 3.74 (s, 3 H), 3.75 (d, 1H, J = 13
Hz), 3.83 - 3.93 (m, 6 H), 4.03 (d, J=7.4 Hz, 1 H), 6.78 - 6.82 (m, 2 H), 7.02
(dd, J=9.9, 8.5 Hz, 1 H), 7.09 - 7.13 (m, 2 H), 7.42 - 7.49 (m, 2 H); MS: 407.4
(M+ H+; ポジティブモード); 451.3 (M+HCO2-;
ネガティブモード)
【0208】
(実施例6)
2−(4−メトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)−ベンゾニトリル(6A):
【0209】
【化40】
n−ブチルリチウム(1.04mL、2.6mmol、ヘキサン中2.5M)を、臭化イソプロピルマグネシウムの溶液(1.27mL、1.27mmol、テトラヒドロフラン中1M)に0℃で添加した。30分間撹拌した後、得られた混合物を−78℃に冷却し、4−ブロモ−2−(4−エトキシ−ベンジル)−ベンゾニトリル(380mg、1.20mmol)の無水テトラヒドロフラン(1mL)溶液を添加した。緑がかった混合物を−78℃で1時間撹拌し、(2R,3S,4S)−2,3,4,6−テトラキス−ベンジルオキシ−5−ベンジルオキシメチル−5−ヒドロキシ−ヘキサン酸メトキシ−メチル−アミド(I−6g)(700mg、0.972mmol)の無水テトラヒドロフラン(2mL)溶液を、非常にゆっくりと添加した(20分に亘る、5秒毎に1滴)。溶液を−78℃で1時間撹拌し、室温に3時間に亘ってゆっくり温めた。1Mの塩酸水溶液を滴下で添加することによって反応物をクエンチし、次いで酢酸エチルで希釈した。得られた二相性混合物を室温で15分間撹拌した。有機相を分離し、ブラインで洗浄し、硫酸マグネシウム上で乾燥させ、濾過し、濃縮し、粗生成物を得た。粗生成物は、ヘプタン中の0〜20%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、所望の中間体2−(4−エトキシ−ベンジル)−4−((4S,5S)−3,4,5−トリス−ベンジルオキシ−6,6−ビス−ベンジルオキシメチル−2−ヒドロキシ−テトラヒドロ−ピラン−2−イル)−ベンゾニトリル(300mg;34%収率)を得た。MS 918.8 (M+Na+, ポジティブモード).
【0210】
三塩化ホウ素(4.18mL、4.18mmol、ヘキサン中の1M溶液)を、上記の中間体(250mg、0.279mmol)のCH2Cl2(2mL)溶液に−78℃で添加した。混合物を−78℃で10分間撹拌し、次いで、室温に一晩温めた。混合物を水(10mL)でクエンチし、酢酸エチル(50mL)で抽出した。有機層を硫酸ナトリウム上で乾燥させ、蒸発乾固させた。シリカゲル(ジクロロメタン中のメタノールで溶出:1〜9容)上のフラッシュクロマトグラフィーによる精製によって、所望の中間体2−(4−ヒドロキシ−ベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクト−5−イル)−ベンゾニトリル(35mg、30%収率)を得た。
【0211】
炭酸カリウム(28mg、0.2mmol)を、上記の中間体(34mg、0.077mmol)のアセトン(0.4mL)溶液に添加し、続いてヨードメタン(7μL、0.11mmol)を室温で添加した。混合物を45℃で一晩撹拌した。混合物を酢酸エチル(60mL)で希釈し、水で洗浄した。有機層を硫酸ナトリウム上で乾燥させ、蒸発乾固させた。シリカゲル(ジクロロメタン中のメタノールで溶出:1〜9容)上の分取薄層クロマトグラフィーによる精製によって、所望の生成物6A(18mg;57%収率)の単離が可能となった。
1H NMR
(400 MHz, メタノール-d4) δ 7.69 (d, J = 8 Hz, 1H), 7.61 (s, 1H), 7.56 (d, J = 8 Hz, 1H),
7.19-7.14 (m, 2H), 6.87-6.82 (m, 2H), 4.18 (d, J = 7.6 Hz, 1H), 4.14 (s, 2H),
3.86 (d, J = 12.7 Hz, 1H); 3.81 (d, J = 8.3 Hz, 1H), 3.76 (s, 3H), 3.69
(d, J = 12.5 Hz, 1H), 3.67 (t, J = 8.1 Hz, 1H), 3.61 (d, J = 7.6 Hz, 1H), 3.54
(d, J = 8 Hz, 1H); MS 458.4 (M+HCO2-; ネガティブモード).
【0212】
(実施例7)
2−(4−エトキシベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−(ヒドロキシメチル)−6,8−ジオキサ−ビシクロ[3,2,1]オクト−5−イル)−ベンゾニトリル(7A):
【0213】
【化41】
炭酸カリウム(8mg、0.058mmol)を、中間体2−(4−ヒドロキシ−ベンジル)−4−((1S,2S,3S,4R,5S)−2,3,4−トリヒドロキシ−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクト−5−イル)−ベンゾニトリル(実施例6を参照されたい;8.9mg、0.022mmol)のアセトン(0.4mL)溶液に添加し、続いてヨードエタン(4μL、0.044mmol)を室温で添加した。混合物を45℃で一晩撹拌した。混合物を酢酸エチル(60mL)で希釈し、水で洗浄した。有機層を硫酸ナトリウム上で乾燥させ、蒸発乾固させた。シリカゲル(ジクロロメタン中のメタノールで溶出:1〜9容)上の分取薄層クロマトグラフィーによる精製によって、所望の生成物7A(2.4mg;26%収率)の単離が可能となった。
1H NMR
(メタノール-d4) δ 7.69
(d, J = 8.0 Hz, 1H), 7.61 (d, J = 1.5 Hz, 1H), 7.56 (dd, J = 8.0, 1.5 Hz, 1H),
7.17-7.13 (m, 2H), 6.86-6.81 (m, 2H), 4.18 (d, J = 7.5 Hz, 1H), 4.14 (s, 2H),
4.01 (q, J = 7.0 Hz, 2H); 3.86 (d, J = 12.5 Hz, 1H); 3.80 (dd, J = 8.0および1.2 Hz, 1H), 3.70 (d, J = 11.7 Hz, 1H), 3.67 (t, J = 8.0 Hz, 1H),
3.61 (dd, J = 7.5および1.2 Hz, 1H), 3.54 (d, J = 7.8 Hz,
1H), 1.37 (t, J = 7.0 Hz, 3H); MS 472.1 (M+HCO2-; ネガティブモード).
【0214】
実施例8は、実施例3Bの構造および立体配置を確認するための、実施例3Bの化合物の結晶性誘導体の調製を例示する。
【0215】
(実施例8)
(8A)を得るための(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(3B)のペル4−ブロモベンゾイル化:
【0216】
【化42】
(1S,2S,3S,4S,5S)−5−[4−クロロ−3−(4−メトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(3B)(11mg、0.026mmol)の無水テトラヒドロフラン(600μL)溶液に、室温でN,N−ジイソプロピルエチルアミン(32μL、7当量)および4−ジメチルアミノピリジン(3mg、0.9当量)、続いてパラ−ブロモベンゾイルクロリド(35mg、6当量)を添加し、得られた混合物を室温で62時間撹拌した。酢酸エチルおよび水を添加し、有機相を0.5Mの塩酸水溶液およびブラインで連続的に洗浄した。有機相を硫酸マグネシウム上で乾燥させ、濾過し、濃縮し、粗製物を、ヘプタン中の15〜30%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、27mgの生成物(90%収率)を得た。
1H NMR
(400 MHz, クロロホルム-d) δ 7.82 (m,
2H), 7.74-7.64 (m, 4H), 7.58-7.46 (m, 8H), 7.42-7.34 (m, 4H), 7.29 (d, 1H, J =
8.3 Hz), 6.89 (m, 2H), 6.63 (m, 2H), 6.04 (dd, 1H, J = 9.6および1 Hz), 5.98 (dd, 1H, J = 9.6および4.4 Hz), 5.89
(d, 1H, J = 4.4 Hz), 4.70 (d, 1H, J = 12.4 Hz), 4.65 (d, 1H, J = 12.4 Hz), 4.60
(d, 1H, J = 8 Hz), 3.98-3.88 (m, 3H), 3.73 (s, 3H).
【0217】
単結晶は、ヘプタンおよび酢酸エチルを溶媒として使用して蒸気拡散技術によって得た。融点=191℃。単結晶X線分析。代表的な結晶を調査し、1Åデータセット(最大sinΘ/λ=0.5)をBruker APEX II/R回折計で集めた。絶対配置の決定を容易にするために、フリーデル対を集めた。原子散乱因子は、International Tables for Crystallographyから取った。International Tables for Crystallography、第C巻、219頁、500、Kluwer Academic Publishers、1992を参照されたい。全ての結晶学的計算を、SHELXTLシステムによって容易にした。SHELXTL、バージョン5.1、Bruker AXS、(1997)を参照されたい。全ての回折計データは室温で集めた。関連する結晶、データ収集、および精密化を、下記の表1に要約する。
【0218】
【表1】
【0219】
試行構造は、直接法によって得た。この試行構造は常に精密化した。水素の位置を可能な限り計算した。メチル水素は、差フーリエ技術によって位置決めし、次いで理想化した。水素パラメータを構造因子計算に加えたが、精密化しなかった。最小二乗精密化の最終サイクルにおいて計算されたシフトは全て、0.1未満の相当する標準偏差であった。最終R指数は、3.71%であった。最終差フーリエは、電子密度の欠損または誤った配置を示さなかった。精密な構造は、SHELXTLプロッティングパッケージを使用してプロットした(図1)。絶対配置は、Flackの方法によって決定した。Flack,H.D.、Acta Crystallogr、A39、876、(1983)を参照されたい。
【0220】
実施例9は、実施例4Aの構造および立体配置を確認するための、実施例4Aの化合物の結晶性誘導体の調製を例示する。
【0221】
(実施例9)
(9A)を得るための(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(4A)のペル4−ニトロベンゾイル化:
【0222】
【化43】
0℃に冷却した(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(4A:10.6mg、0.024mmol)の無水テトラヒドロフラン(300μL)溶液に、N,N−ジイソプロピルエチルアミン(30μL、7当量)および4−ジメチルアミノピリジン(3mg、1当量)、続いてパラ−ニトロベンゾイルクロリド(27mg、6当量)を添加し、得られた混合物を60℃で6時間撹拌した。混合物を室温に冷却し、酢酸エチルおよび水を添加し、有機相を0.5Mの塩酸水溶液およびブラインで連続的に洗浄した。有機相を硫酸マグネシウム上で乾燥させ、濾過し、濃縮し、粗製物を、ヘプタン中の10〜50%酢酸エチルの勾配で溶出するシリカゲル上のフラッシュクロマトグラフィーによって精製し、18mgの生成物(73%収率)を得た。
1H NMR
(400 MHz, クロロホルム-d) δ 8.33 (m,
2H), 8.28-8.12 (m, 8H), 8.07 (m, 2H), 8.00 (m, 2H), 7.91 (m, 2H), 7.45-7.40 (m,
2H), 7.34 (d, 1H, J = 8.2 Hz), 6.87 (m, 2H), 6.64 (m, 2H), 6.13 (d, 1H, J = 8.6
Hz), 6.06 (t, 1H, J = 8.3 Hz), 5.86 (d, 1H, J = 8.1 Hz), 4.81 (d, 1H, J = 8.3
Hz), 4.75 (d, 1H, J = 12.7 Hz), 4.60 (d, 1H, J = 12.8 Hz), 4.06 (d, 1H, J = 8.5
Hz), 3.98-3.90 (m, 4H), 1.39 (t, 3H, J = 7 Hz).
【0223】
単結晶は、溶媒としてアセトニトリル/イソプロパノールからのゆっくりとした再結晶によって得た。融点=211℃。代表的な結晶を調査し、0.88Åデータセット(最大sinΘ/λ=0.57)をBruker APEX II/R回折計上で集めた。絶対配置の決定を容易にするために、フリーデル対を集めた。原子散乱因子は、International Tables for Crystallographyから取った。International Tables for Crystallography、第C巻、219頁、500、Kluwer Academic Publishers、1992を参照されたい。全ての結晶学的計算を、SHELXTLシステムによって容易にした。SHELXTL、バージョン5.1、Bruker AXS、(1997)を参照されたい。全ての回折計データは室温で集めた。関連する結晶、データ収集、および精密化を、下記の表2に要約する。
【0224】
【表2】
【0225】
試行構造は、直接法によって得た。この試行構造は常に精密化した。水素の位置を可能な限り計算した。メチル水素を差フーリエ技術によって位置決めし、次いで理想化した。水素パラメータを構造因子計算に加えたが、精密化しなかった。最小二乗精密化の最終サイクルにおいて計算されたシフトは全て、0.1未満の相当する標準偏差であった。最終R指数は、4.36%であった。最終差フーリエは、電子密度の欠損または誤った配置を示さなかった。
【0226】
精密な構造を、SHELXTLプロッティングパッケージ(図2)を使用してプロットした。絶対配置はFlackの方法によって決定した。Flack,H.D.、Acta Crystallogr.、A39、876、(1983)を参照されたい。
【0227】
(実施例10)
(1S,2S,3S,4R,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(10A)および(1S,2S,3S,4S,5S)−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(10B)
【0228】
【化44】
エタノール/テトラヒドロフランの4:1溶液(10mL)中の{(2S,3S)−2,3,4−トリス−ベンジルオキシ−5−[3−(4−エトキシ−ベンジル)−4−フルオロ−フェニル]−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル}−メタノールI−10k(80.0mg、0.120mmol)の溶液に、ギ酸(93μL、2.32mmol)およびパラジウム黒(62mg、0.580mmol)を連続的に添加した。得られた混合物を室温で撹拌した。3時間後、さらなるギ酸(93μL、2.32mmol)およびパラジウム黒(62mg、0.580mmol)を添加した。5時間後、反応混合物を濾過し、濾液を減圧下で濃縮した。得られた粗残渣は、シリカゲル(ジクロロメタン中の0〜15%メタノールの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、35.0mgの白色の固体(異性体の混合物)を得た。異性体の混合物を分取HPLCによって精製した。
【0229】
HPLC分取法:逆相C18Geminiカラム、5マイクロメーター、30×100mm、40mL/分の流量、18分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;25〜50%アセトニトリル/0.1%ギ酸の勾配;UV検出:220nm。
【0230】
HPLC分析法:逆相C18Geminiカラム、5μm、4.6×150mm、1mL/分の流量、12分に亘るアセトニトリル/0.1%トリフルオロ酢酸:水/0.1%トリフルオロ酢酸;5〜100%アセトニトリル/0.1%トリフルオロ酢酸の勾配;UV検出:220nm。
【0231】
10A:(2.2mg、4.5%収率)Rt=7分(分析法)。生成物含有画分を減圧下で濃縮した。
MS (LCMS) 421.4 (M+H+; ポジティブモード) 465.3 (M+HCO2-, ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ ppm 1.33 (t, J=7.0 Hz, 3 H), 3.53 (d, J=8.0 Hz, 1 H),
3.57 (dd, J=7.5, 1.5 Hz, 1 H), 3.60 - 3.67 (m, 2 H), 3.75 (dd, J=8.3, 1.3 Hz, 1
H), 3.81 (d, J=12.5 Hz, 1 H), 3.89 (s, 2 H), 3.96 (q, J=6.9 Hz, 2 H), 4.12 (d,
J=7.4 Hz, 1 H), 6.77 (m, 2 H), 7.00 (dd, J=9.4, 8.2 Hz, 1 H), 7.08 (m, 2 H),
7.36 - 7.41 (m, 2 H).
【0232】
10B:(1.8mg、3.7%収率)Rt=7.13分(分析法)。生成物含有画分を減圧下で濃縮した。
MS (LCMS) 421.4 (M+H+; ポジティブモード) 465.3 (M+HCO2-, ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ ppm 1.34 (t, J=7.0 Hz, 3 H), 3.51 (d, J=7.4 Hz, 1 H),
3.75 (d, 1 H, J = 12.5 Hz), 3.82 - 4.01 (m, 8 H), 4.03 (d, J=7.4 Hz, 1 H), 6.79
(m, 2 H), 7.02 (dd, J=9.8, 8.4 Hz, 1 H), 7.10 (m, 2 H), 7.41 - 7.49 (m, 2 H).
注記:分取HPLC後、これらの生成物を含有する画分を濃縮し、シリカゲル(ジクロロメタン中の0〜10%メタノールの勾配で溶出)上のフラッシュクロマトグラフィーによって再精製した。
【0233】
(実施例11)
(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(11A)
【0234】
【化45】
エタノール/テトラヒドロフランの4:1溶液(10mL)中の((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−フルオロ−3−[4−(テトラヒドロ−フラン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノールI−11k(160.0mg、0.218mmol)の溶液に、ギ酸(185μL、4.64mmol)およびパラジウム黒(148mg、1.39mmol)を連続的に添加した。得られた混合物を室温で撹拌した。3時間後、さらなるギ酸(185μL、4.64mmol)およびパラジウム黒(148mg、1.39mmol)を添加した。5時間後、反応混合物を濾過し、濾液を減圧下で濃縮した。得られた粗残渣は、シリカゲル(ジクロロメタン中の0〜15%メタノールの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、100mgの白色の固体(異性体の混合物)を得た。異性体の混合物を分取HPLCによって精製した。
【0235】
HPLC分取法:逆相C18Geminiカラム、5マイクロメーター、30×100mm、40mL/分の流量、18分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;25〜50%アセトニトリル/0.1%ギ酸の勾配;UV検出:220nm。
【0236】
HPLC分析法:逆相C18Geminiカラム、5マイクロメーター、4.6×150mm、1mL/分の流量、12分に亘るアセトニトリル/0.1%トリフルオロ酢酸:水/0.1%トリフルオロ酢酸;5〜100%アセトニトリル/0.1%トリフルオロ酢酸の勾配;UV検出:220nm。
【0237】
11A:(19mg、19%収率)Rt=6.43分(分析法)。生成物含有画分を減圧下で濃縮した。
1H NMR
(400 MHz, メタノール-d4) δ ppm 2.03 - 2.11 (m, 1 H), 2.15 - 2.25 (m, 1 H), 3.55 (d, 1 H, J = 8
Hz), 3.59 (dd, 1 H, J = 7.4および1 Hz), 3.61 - 3.69 (m, 2
H), 3.77 (dd, J=8.2および1 Hz, 1 H), 3.81 - 3.96 (m, 7 H),
4.14 (d, J=7.4 Hz, 1 H), 4.94 - 4.98 (m, 1 H), 6.79 (m, 2 H), 7.02 (dd, J=9.9,
8.5 Hz, 1 H), 7.12 (m, 2 H), 7.37 - 7.45 (m, 2 H).
【0238】
(実施例12)
(1S,2S,3S,4R,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオール(12A)および(1S,2S,3S,4S,5S)−5−[3−(4−クロロベンジル)−4−フルオロフェニル]−1−ヒドロキシメチル−6,8−ジオキサビシクロ[3.2.1]オクタン−2,3,4−トリオール(12B)
【0239】
【化46】
エタノール/テトラヒドロフラン(2mL、4/1容)中の中間体I−12k(102mg)およびパラジウム黒(98mg、6.1当量)の混合物に、ギ酸(0.9mL)を添加し、得られた混合物を室温で撹拌した。1時間後、さらなるパラジウム黒(67mg、4.2当量)を添加し、混合物をさらに1時間室温で撹拌した。パラジウムを、Celite(登録商標)を通した濾過によって除去し、濾液を濃縮し、生成物の混合物を得た。この材料を、分取HPLCによる精製のために、粗材料の第2のバッチ(上記の手順に従って中間体I−12k(80mg)から調製)と合わせた。
【0240】
分取HPLC条件:逆相C18Geminiカラム、5マイクロメーター、30×100mm、流量40mL/分、18分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;25〜50%のアセトニトリル/0.1%ギ酸の勾配)、UV検出:220nm。
【0241】
HPLC分析法:逆相C18Geminiカラム、5μm、4.6×150mm、1mL/分の流量、12分に亘るアセトニトリル/0.1%トリフルオロ酢酸:水/0.1%トリフルオロ酢酸;5〜100%アセトニトリル/0.1%トリフルオロ酢酸の勾配;UV検出:220nm。
【0242】
12A:(18mg、16%収率)Rt=7.11分(分析法); MS
(LCMS) 411.3 (M+H+; ポジティブモード); 409.2 (M-H+;
ネガティブモード). 1H NMR (400 MHz, メタノール-d4) δ ppm 7.45-7.42 (m, 2H),
7.25 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H), 7.05 (dd, J = 9.6, 9.2 Hz,
1H), 4.15 (d, J = 7.6 Hz, 1H), 3.98 (s, 2H), 3.84 (d, J = 12.4 Hz, 1H), 3.78
(dd, J = 8.4, 1.2 Hz, 1H), 3.68 (d, J = 12.8 Hz, 1H), 3.66 (t, J = 8.2 Hz, 1H),
3.60 (dd, J = 7.4, 1.4 Hz, 1H), 3.56 (d, J = 7.6 Hz, 1H).
12B:(12mg、11%収率)Rt=7.25分(分析法); MS
(LCMS) 411.3 (M+H+; ポジティブモード); 409.1 (M-H+;
ネガティブモード). 1H NMR (400 MHz, メタノール-d4) δ ppm 7.52-7.45 (m, 2H),
7.25 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H), 7.05 (dd, J = 9.8, 8.6 Hz,
1H), 4.05 (d, J = 7.2 Hz, 1H), 3.98 (s, 2H), 3.91-3.84 (m, 4H), 3.76 (d, J =
12.4 Hz, 1H), 3.52 (d, J = 7.6 Hz, 1H).
【0243】
(実施例13)
(1S,2S,3S,4R,5S)−5−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(13A)
【0244】
【化47】
エタノール/テトラヒドロフランの4:1溶液(10mL)中の((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−フルオロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノールI−13k(300mg、0.417mmol)の溶液に、ギ酸(333μL、8.34mmol)およびパラジウム黒(266mg、2.50mmol)を連続的に添加した。得られた混合物を室温で撹拌した。3時間後、さらなるギ酸(333μL、8.34mmol)およびパラジウム黒(266mg、2.50mmol)を添加した。5時間後、反応混合物を濾過し、濾液を減圧下で濃縮した。得られた粗残渣は、シリカゲル(ジクロロメタン中の0〜15%メタノールの勾配で溶出)上のフラッシュクロマトグラフィーによって精製し、153.0mgの白色の固体(異性体の混合物)を得た。異性体の混合物を分取HPLCによって精製した。
【0245】
HPLC分取法:逆相C18Geminiカラム、5マイクロメーター、30×100mm、40mL/分の流量、18分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;25〜50%アセトニトリル/0.1%ギ酸の勾配;UV検出:220nm。
【0246】
13A:(23mg、12%収率)Rt=7.9分。生成物含有画分を減圧下で濃縮した。
1H NMR
(400 MHz, メタノール-d4) δ ppm 3.52 (d, J=7.8 Hz, 1 H), 3.57 (d, J=7.2 Hz, 1 H), 3.60 - 3.68
(m, 2 H), 3.75 (d, J=8.2 Hz, 1 H), 3.81 (d, J=12.5 Hz, 1 H), 3.89 (s, 2 H),
4.12 (d, J=7.4 Hz, 1 H), 4.63 (dd, J=7.3, 4.8 Hz, 2 H), 4.95 (t, J=6.5 Hz, 2
H), 5.16 - 5.23 (m, 1 H), 6.63 (m, 2 H), 7.00 (dd, J=9.7, 8.5 Hz, 1 H), 7.10
(m, 2 H), 7.36 - 7.42 (m, 2 H).
【0247】
(実施例14)
(1S,2S,3S,4R,5S)−5−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(14A)
【0248】
【化48】
中間体((2S,3S)−2,3,4−トリス−ベンジルオキシ−5−{4−クロロ−3−[4−(オキセタン−3−イルオキシ)−ベンジル]−フェニル}−6,8−ジオキサ−ビシクロ[3.2.1]オクト−1−イル)−メタノールI−14k(182mg)のエタノール/テトラヒドロフラン(14mL、4/1容)溶液に、ギ酸(190μL、20当量)およびパラジウム黒(106mg、4当量)を連続的に添加し、得られた混合物を室温で撹拌した。2時間後さらに1mLのテトラヒドロフランを添加し、得られた混合物を室温でさらに1時間撹拌した。この時点で、さらなるギ酸(190μL、20当量)およびパラジウム黒(106mg、4当量)を添加し、混合物をさらに1時間室温で撹拌した。パラジウムを濾過し、溶媒(異性体の混合物を含有)の蒸発の後に得られた粗混合物を分取HPLCによって精製した。
【0249】
HPLC分取法:逆相C18Xbridgeカラム、5マイクロメーター、100×30mm、流量40mL/分、11分に亘るアセトニトリル/0.1%ギ酸:水/0.1%ギ酸;30〜55%のアセトニトリル/0.1%ギ酸の勾配;UV検出:220nm。
【0250】
14A:(20mg、17%収率);Rt=4.43分。生成物含有画分を減圧下で濃縮し、白色の固体を得た。
MS (LCMS) 465.3 (M+H+; ポジティブモード); 509.2 (M+HCO2-; ネガティブモード). 1H NMR (400 MHz, メタノール-d4)
δ ppm 3.53 (d, J=8.0 Hz, 1 H), 3.58 (dd, J=7.4, 1.4 Hz,
1 H), 3.64 (t, J=8.2 Hz, 1 H), 3.67 (d, J=12.4 Hz, 1 H), 3.77 (dd, J=8.4, 1.4
Hz, 1 H), 3.83 (d, J=12.6 Hz, 1 H), 4.03 (s, 2 H), 4.14 (d, J=7.4 Hz, 1 H),
4.65 (m, 2 H), 4.97 (t, J=6.6 Hz, 2 H), 5.22 (m, 1 H), 6.65 (m, 2 H), 7.11 (m,
2 H), 7.34 (d, J=8.4 Hz, 1 H), 7.38 (dd, J=8.4, 2.2 Hz, 1 H), 7.45 (d, J=2.0
Hz, 1 H).
【0251】
(実施例15)
(15)を得るためのL−プロリンとの(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
水(概ね480mg/mL)に溶解したL−プロリンを、実施例4Aの化合物に添加した(1モル(実施例4Aの化合物)毎に、概ね80モルのL−プロリン)。エタノールによって容量を2倍にし、溶液をキャップし、概ね12時間撹拌した。ベンチ上での蒸発によって容量を半分に減少させた。エタノールを使用して容量を2倍にし、蒸発を使用して溶液の容量をまた半分に減少させた。遠心濾過を使用して固体を回収した。
【0252】
(実施例16)
(16)を得るためのL−プロリンとの(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
水に溶解したL−プロリン(概ね480mg/mL)を、実施例4Aの化合物に添加した(実施例4Aの化合物1モル当たり概ね59モルのL−プロリン)。メタノールによって容量を2倍にし、溶液は透明であった。アセトンを使用して容量を25%増加させた。溶液をキャップし、概ね12時間撹拌した。ベンチ上での蒸発によって容量を概ね60%減少させた。メタノールを使用して容量を2倍にし、残りの溶媒を蒸発させ、固体の白色の沈殿物が残った。
【0253】
(実施例17)
(17)を得るためのL−プロリンとの(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
L−プロリンで飽和したエタノールの溶液を、ガラス製バイアル中の実施例4Aの化合物に添加した(実施例4Aの化合物1モル当たり概ね2.2モルのL−プロリン)。透明な溶液をキャップし、概ね72時間撹拌した。室温で蒸発させることによって容量を半分に減少させた。沈殿物が認められ、バイアルをキャップし、概ね12時間撹拌した。遠心濾過を使用して白色の固体を集めた。
【0254】
(実施例18)
(18)を得るためのL−プロリンとの(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
溶液が濁るまで、水に溶解したL−プロリン(330mg/mL)を、イソプロパノールに溶解した概ね2mLの実施例4Aの化合物に滴下した(98mg/mL)。15〜20分後、沈殿が観察され、懸濁液は濃厚となった。概ね8mLの水を添加し、溶液をキャップし、一晩撹拌した。真空濾過を使用して白色の固体を集め、50℃の真空オーブン中で概ね2時間乾燥させた。
【0255】
(実施例19)
(19)を得るためのL−ピログルタミン酸との(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
イソプロピルアルコール中の化合物(4A)(97.97mg/mL)(153μL)を、水中のL−ピログルタミン酸(213.0mg/mL)(500μL)中にピペットで移した。溶液をキャップし、一晩撹拌した。概ね5〜10mgのさらなる固体L−ピログルタミン酸を添加した。100μLのエタノールを添加した。溶液をキャップし、一晩撹拌した。総容量が概ね2mLに調整されるまでエタノールを添加した。溶液のキャップを取り、フード中に一晩置いた。概ね10〜30mgのさらなる実施例4Aの化合物を添加した。溶液をキャップし、概ね2日間撹拌した。白色の沈殿物が見出された。懸濁液を、0.45μmのナイロンフィルター膜インサートを備えたCo−star微量遠心分離管にピペットで移した。固体が溶液から分離されるまで溶液を遠心分離した。共結晶(19)を回収した。
【0256】
(実施例20)
(20)を得るためのL−ピログルタミン酸との(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
4〜5mLの1:1エタノール/水溶液をL−ピログルタミン酸(412.1mg/mL)で飽和させた。730mgの実施例4Aの化合物を、3.2mLのL−ピログルタミン酸溶液に添加した。概ね1分後、沈殿が見出された。溶液は濃厚すぎて撹拌することができず、したがって2mLの1:1エタノール/水溶液を添加した。溶液を一晩撹拌した。0.45μmのナイロンフィルター膜上の真空濾過を使用して固体を集めた。固体を50℃の真空オーブン中で概ね2時間乾燥させた。概ね960mgの共結晶複合体(20)を回収した。L−ピログルタミン酸に対する実施例4Aの化合物の化学量論比は、定量的NMRを使用して1:1.63であると決定した。材料をエタノール中で懸濁させることによって過剰なL−ピログルタミン酸を除去し、1:1の共結晶(20)を得た。
【0257】
(実施例21)
(21)を得るためのL−ピログルタミン酸との(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)の共結晶化:
494mgの実施例4Aの化合物を、1.5mLのイソプロパノールおよびエタノールの溶液(各々、4:1)に溶解した。917.2mgのL−ピログルタミン酸を3mLの水に溶解した。両方の溶液を40℃に加熱した。200μLのL−ピログルタミン酸溶液を、全ての溶液が移されるまでずっと実施例4Aの化合物溶液に添加した(溶液が移されない限り、両方の溶液をキャップする)。L−ピログルタミン酸溶液を有するバイアルを200μLのエタノールで洗浄し、溶液を実施例4Aの化合物溶液に移した。溶液を5分間撹拌し、次いで熱を止めた(溶液は3分毎に概ね摂氏1度冷却した)。30℃で、溶液を周囲温度の撹拌機に入れ、20℃で20分間撹拌した。溶液は透明であった。概ね2mLの乾燥種晶を添加した。次の2時間に亘り懸濁液は濃厚となった。溶液を一晩撹拌した。Pyrex2mL10〜15M焼結ガラス漏斗フィルター上の真空濾過を使用して固体を回収した。固体を50℃の真空オーブン中で24時間乾燥した。
【0258】
(実施例22)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−プロリンとの共結晶、および(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−ピログルタミン酸との共結晶:
粉末X線回折分析:実施例4Aの化合物とL−プロリンとの共結晶、および実施例4Aの化合物とL−ピログルタミン酸との共結晶の粉末X線回折パターンは、銅放射線(波長:1.54056Å)を使用したBruker D5000回折計で行った。管電圧およびアンペア数を、各々、40kVおよび40mAに設定した。発散スリットおよび散乱スリットは1mmに設定し、受光スリットは0.6mmに設定した。回折放射線は、Kevex PSI検出器によって検出した。3.0から40°2θの毎分2.4°(0.04°ステップ毎に1秒)でのθ−2θ連続スキャンを使用した。アルミナ標準物質を分析し、機器の整合性を点検した。データを集め、Bruker axisソフトウェアバージョン7.0を使用して分析した。試料は、それらを石英ホルダー中に置くことによって調製した。Bruker InstrumentsはSiemensを買収したことに留意すべきである。したがって、Bruker D5000機器は、Siemens D5000と本質的に同じである。Eva Application13.0.0.3ソフトウェアを使用して、PXRDスペクトルを可視化および評価した。PXRDデータファイル(未加工)は、ピーク探索の前に処理しなかった。一般に、2の閾値および0.3の幅値を使用して、予備的ピーク割当を作製した。自動化割当のアウトプットを視覚的に検査し、妥当性を確実にし、必要に応じて調節を手動で行った。
【0259】
本明細書において報告した測定のために使用するBrukerシステムなどのBragg−Brentano機器上でX線回折測定を行うために、試料を典型的には、空洞を有するホルダー中に置く。試料粉末をスライドガラスまたは相当するものによって押し、ランダム面および妥当な試料高さを確実にする。次いで、試料保持器を機器中に置く。入射X線ビームを試料に向け、最初にホルダーの面に対して低角度で、次いで入射ビームおよびホルダーの面の間の角度を連続的に増加させる弧に沿って動かす。このようなX線粉末分析に関連する測定の差異は、(a)試料調製におけるエラー(例えば、試料高さ)、(b)機器エラー(例えば、平板試料エラー)、(c)較正誤差、(d)操作者のエラー(ピーク位置を決定するときに生じるそれらのエラーを含めて)、および(e)材料の性質(例えば、選択方位および透明性のエラー)を含めた種々の要因からもたらされる。較正誤差および試料高さのエラーは、同じ方向への全てのピークのシフトをもたらすことが多い。フラットホルダーを使用するときの試料高さの小さな差異は、XRPDピーク位置の大きな変位をもたらす。典型的なブラッグ−ブレンターノ配置においてShimadzu XRD−6000を使用して、1mmの試料高さの差異は、1°2θもの高いピークシフトをもたらすことが系統的研究によって示された(Chenら;J Pharmaceutical and Biomedical Analysis、2001;26,63)。これらのシフトは、X線ディフラクトグラムから同定することができ、シフトを相殺する(全てのピーク位置値に系統的補正因子を適用する)ことによって、または機器を再較正することによって除去することができる。上記のように、様々な機械からの測定を、系統的補正因子を適用することによって修正し、ピーク位置と一致させることが可能である。一般に、この補正因子は、Brukerから測定したピーク位置を予想されるピーク位置と一致させるが、0〜0.2°2θの範囲のことがある。
【0260】
粉末X線回折値は一般に、機器および試験条件の僅かな変動によって±0.2の2θ度以内で正確である。
【0261】
実施例18からの実施例4Aの化合物とL−プロリンとの共結晶は、図3において提供する下記の粉末X線回折パターンによって特徴付けられる(2θ度および相対強度に関して表す、CuKα放射線によるBruker D5000回折計で測定して≧2.7%の相対強度を有する)。
【0262】
【表3】
*相対強度は、結晶サイズおよび形態によって変化し得る。
【0263】
実施例4Aの化合物とL−プロリンとの共結晶の特徴的な2θピークまたは組合せ。
【0264】
【表4】
【0265】
実施例20からの実施例4Aの化合物とL−ピログルタミン酸との共結晶は、図4において提供する下記の粉末X線回折パターンによって特徴付けられる(2θ度および相対強度に関して表す、CuKα放射線によるBruker D5000回折計で測定して≧2.7%の相対強度を有する)。
【0266】
【表5】
*相対強度は、結晶サイズおよび形態によって変化し得る。
【0267】
実施例4Aの化合物とL−ピログルタミン酸との共結晶の特徴的な2θピークまたは組合せ。
【0268】
【表6】
【0269】
(実施例23)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−プロリンとの共結晶、および(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−ピログルタミン酸との共結晶:
示差走査熱量測定サーモグラム分析:
サーモグラムは、TA Instruments Q1000示差走査熱量計(DSC)で得た。1〜2mgの試料をアルミニウム試料パン中に置き、次いでせん孔された蓋で覆った。温度が25℃から200〜300℃に毎分10℃で上がるにつれ、空のパンに対してエネルギーを測定した。融解吸熱の開始温度は、融解温度として報告した。融解吸熱の開始温度は、他の要因の中でも加熱速度、試料の純度、結晶および試料のサイズによって決まる。典型的には、DSC結果は、約±2℃以内、好ましくは±1.5℃以内で正確である。
【0270】
実施例4Aの化合物とL−プロリンとの実施例18の共結晶のDSCの結果を図5に示す。
【0271】
実施例4Aの化合物とL−ピログルタミン酸との実施例20の共結晶のDSCの結果を図6に示す。
【0272】
(実施例24)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−プロリンとの共結晶
単結晶X線分析。実施例17からの濾液を使用し、ゆっくりとした蒸発による濃縮を使用した代表的な結晶を調査し、0.85Åデータセット(最大sinΘ/λ=0.60)をBruker APEX回折計で集めた。絶対配置の決定を容易にするために、フリーデル対を集めた。原子散乱因子は、International Tables for Crystallography、第C巻、219頁、500、Kluwer Academic Publishers、1992から取った。全ての結晶学的計算を、SHELXTLシステム、バージョン5.1、Bruker AXS、1997によって容易にした。全ての回折計データは室温で集めた。関連する結晶、データ収集、および精密化を、表24−1に要約する。
【0273】
試行構造は直接法によって得た。この試行構造は、予想外の水分子およびL−プロリンによる配座障害以外は常に精密化した。L−プロリンは、「半いす形」および「封筒型」配座において約60/40占有率でモデル化した。非常に同様の障害が、H.D.Flack、Acta Crystallogr.、A39、876、1983において観察された。
【0274】
N1、O6およびO7に結合している水素原子は差フーリエ技術によって位置決めし、距離を制限して精密化した。O5に結合した関連する水素原子をフーリエ技術から位置決めしたが、削除し、理想化した位置(HFIX83)に配置した。O4に結合した関連する水素原子は、フーリエ技術で見出すことができず、理想化した位置(HFIX83)に配置した。水分子上の水素原子は、位置決めすることができず、溶液から除外した。水素パラメータを構造因子計算に加えたが、精密化しなかった。最小二乗精密化の最終サイクルにおいて計算されたシフトは全て、0.1未満の相当する標準偏差であった。最終R指数は5.15%であった。最終差フーリエは、電子密度の欠損または誤った配置を示さなかった。
【0275】
精密な構造は、SHELXTLプロッティングパッケージを使用してプロットした(図7)。絶対配置は、Flackの方法によって決定した4。座標、異方性温度因子、距離および角度は、補助的材料として入手可能である(表24−2〜24−5)。
【0276】
【表7】
【0277】
【表8】
【0278】
【表9−1】
【0279】
【表9−2】
【0280】
【表9−3】
【0281】
【表9−4】
【0282】
【表9−5】
【0283】
【表9−6】
【0284】
【表10】
【0285】
【表11】
【0286】
(実施例25)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−ピログルタミン酸との実施例20からの共結晶:
単結晶X線分析。実施例20からの試料の代表的な結晶を調査し、0.90Åデータセット(最大sinΘ/λ=0.56)をBruker APEX回折計で集めた。絶対配置の決定を容易にするために、フリーデル対を集めた。立体配置は、flackパラメータから、およびまた共形成物(L−ピログルタミン酸)の既知のキラリティーから決定した。原子散乱因子は、International Tables for Crystallography、第C巻、219頁、500、Kluwer Academic Publishers、1992から取った。全ての結晶学的計算を、SHELXTLバージョン5.1、Bruker AXS、1997システムによって容易にした。全ての回折計データは室温で集めた。関連する結晶、データ収集、および精密化を、表25−1に要約する。
【0287】
試行構造は直接法によって得た。この試行構造は、0.1化学量論的水として精密化された低い残差ピーク以外は常に精密化した。水の化学量論は、分子上のヒドロキシル基を最初に削除し、精密化し、得られたqピークを測定し、次いでこのピークを水分子からの残差ピークと比較することによって見出した。この方法を使用して、1〜0.1の比(水に対する分子)を推定した。さらに、溶液から水分子を除去し、Material Studio、Platon、およびMercuryによって結晶における空隙空間を捜索することによって、水分子について33立法オングストロームという可能性が高い容量が明らかになった(水は典型的には約40立法オングストロームの空間を有する)。窒素および酸素上の水素原子は、差フーリエ技術によって位置決めし、制約なしに自由に精密化した。ヘテロ原子に結合しているプロトンのいくつか(H97a、H97b、H97c、およびH97c)は、いくらか短い結合距離(予測値約0.96に対して実測値約0.8オングストローム)を示したが、距離は規制しなかった。O99上の水素原子(水)は差異マップからは見出せず、構造解明から除外した。水素パラメータを構造因子計算に加えたが、精密化しなかった。最小二乗精密化の最終サイクルにおいて計算されたシフトは全て、0.2未満の相当する標準偏差であった。最終R指数は、3.58%であった。最終差フーリエは、電子密度の欠損または誤った配置を示さなかった。残った残差のうち、1つはO39に結合したプロトン(カルボン酸)について合理的な位置にある。この残差は、H98aプロトン(O39に結合したプロトン)についてのさらなる占有位置のことがあるが、このように精密化しなかった。
【0288】
精密な構造は、SHELXTLプロッティングパッケージを使用してプロットした(図8)。絶対配置は、Flack(H.D.Flack、Acta Crystallogr.、A39、876、1983)の方法によって決定した。座標、異方性温度因子、距離および角度は、補助的材料として入手可能である(表25−2から25−5)。
【0289】
【表12】
【0290】
【表13】
【0291】
【表14−1】
【0292】
【表14−2】
【0293】
【表14−3】
【0294】
【表15】
【0295】
【表16】
【0296】
(実施例26)
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とL−ピログルタミン酸との共結晶:
固体NMR:
(1S,2S,3S,4R,5S)−5−[4−クロロ−3−(4−エトキシ−ベンジル)−フェニル]−1−ヒドロキシメチル−6,8−ジオキサ−ビシクロ[3.2.1]オクタン−2,3,4−トリオール(実施例4Aの化合物)とスキーム2に記載されている方法を使用して調製されたL−ピログルタミン酸との概ね80mgの共結晶を、4mmのZrO2ローター中にしっかりと詰めた。スペクトルは、大口径Bruker−Biospin DSX500MHz(1H周波数)NMR分光計中に位置するBruker−Biospin4mm BL CPMASプローブで室温および室内圧力にて集めた。詰めたローターはマジック角で方向付け、15.0kHzで回転した。13C固体スペクトルは、プロトンデカップル交差分極マジック角回転実験(CPMAS)を使用して集めた。交差分極接触時間は、2.0msに設定した。概ね85kHzのプロトンデカップリング領域を適用した。14秒の待ち時間で1448スキャンを集めた。炭素スペクトルは結晶性アダマンタンの外部標準を使用して参照し、その高磁場共鳴を29.5ppmに設定した。化学シフトデータは、他の要因の中でも、試験条件(すなわち、スピニング速度および試料保持器)、参照材料、およびデータプロセシングパラメータによって決まる。典型的には、ss−NMR結果は、約±0.2ppm以内で正確である。
【0297】
【表17】
【0298】
実施例26のSSNMR13C CPMASスペクトルを図9に示す。図9においてアスタリスクによってマークされているピークは、スピニングサイドバンドである。
【0299】
薬理試験
SGLT2の阻害によって調節される疾患の治療のための本発明の実施は、本明細書において下記に記載されているプロトコルの少なくとも1つにおける活性によって証明することができる。
【0300】
生物学的アッセイ
インビトロアッセイ
SGLT2機能アッセイは、SGLT2輸送体によるメチル−α−Dグルコピラノシド(AMG−グルコースの代謝可能でない形態)取込みの阻害を検出するために設計した。SGLT2輸送体は、腎臓の近位尿細管からのグルコースを回収する。その阻害によって、糖が尿中に廃棄されることをもたらす。陽性対照化合物であるフロリジンは、SGLT2についてのグルコース取込みの既知の阻害剤である。それを試験化合物のSGLT2阻害の高率の作用を比較するために使用した。
【0301】
ヒトSGLT2(pcDNA5/FRT)を安定的に発現しているCHO−FlpIn(Invitrogen、Carlsbad、CA)細胞を、100μLの増殖培地(1:1のF−12/DMEM培地(Gibco、Carlsbad、CA)、10%FBS(Sigma、St.Louis MO)、1×Pen/Strep(Gibco、Carlsbad、CA)、600μg/mLのハイグロマイシン(Invitrogen、Carlsbad、CA))中で、100,000個の細胞/ウェルの密度で、Iso−TC96ウェルプレート(Perkin Elmer、Waltham、MA)に蒔いた。試験化合物で処理する前に、コンフルエントな細胞を、1:1のF−12/DMEM培地(1時間後、新鮮なF−12/DMEM培地と交換)中で37℃にて2時間、血清飢餓培養を行った。ジメチルスルホキシド(Sigma、St.Louis、MO)中の試験化合物は、取込み緩衝液で予めすすいだ細胞プレートに、取込み緩衝液(140mMのNaCl(Promega、Madison、WI)、2mMのKCl(Teknova、Hollister、CA)、1mMのCaCl2(Teknova、Hollister、CA)、1mMのMgCl2(Teknova、Hollister、CA)、および10mMのHEPES(Gibco、Carlsbad、CA))中で100倍に希釈した。ウェル毎に50μLのAMG(非標識AMG(Aldrich、St.Louis、MO)中の40nCi AMG[U−14C](Perkin Elmer、Waltham、MA))を添加(11.33μMのAMGの最終濃度を生じさせた)する前に、細胞を試験化合物と共に15分間プレインキュベートした。次いで、細胞プレートを、AMG取込みのために37℃にて3時間インキュベートした。インキュベーション後、細胞を氷冷の洗浄緩衝液(200μMのフロリジン(Sigma)を含有する取込み緩衝液)で2度洗浄し、空気乾燥させ、オービタルシェーカーで30μLのNaOH(200mM)および1%SDS緩衝液に溶解させた。Microscint40(Perkin Elmer、Waltham、MA)を溶解した細胞(200μLの最終容量とする)に添加し、30分間オービタルシェーカーにかけることによって混合した。プレートを暗所で一晩保管し、14C検出のための正規化されたプロトコルを使用して1540Microbeta Trilux(Wallac、Waltham、MA)中で定量化した。AMG取込みを阻害する試験化合物の作用割合は、下記の計算を使用して計算した。
[作用%=((ZPE−T)/(ZPE−HPE))×100%]
式中、「ZPE」は、0.5%DMSOを含有する対照ウェル中の毎分補正計数(CCPM)であり、Tは、様々な濃度の標準曲線における試験化合物を含有するウェル中のCCPMであり、HPEは、10μMのフロリジンを含有する対照ウェル中のCCPMに関する高率作用である。IC50値は用量反応式を使用して計算した。表3において試験した化合物について要約する。
【0302】
インビトロ試験の記載において使用された略語には、以下が含まれる。
SGLT2 ナトリウム/グルコース共輸送体2型
AMG メチル−α−Dグルコピラノシド
DMEM ダルベッコ改変イーグル培地
IC50 50%阻害濃度
FBS ウシ胎児血清
DMSO ジメチルスルホキシド
SDS ドデシル硫酸ナトリウム
CHO−FlpIn FRT部位を含有するチャイニーズハムスター卵巣細胞
【0303】
【表18−1】
【0304】
【表18−2】
【0305】
【表18−3】
【0306】
【表18−4】
【0307】
インビボアッセイ
実施例1Aおよび4Aをラットにおいて試験し、尿中グルコース排泄によるグルコース輸送の阻害を評価した。雄性Sprague−Dawleyラット(約300g)は、尿採取のための代謝ケージに単独で収容した。ラットは、標準的飼料および水に自由にアクセスできた。ラット(n=2〜5/群)に、経口胃管栄養法によってビヒクルまたは化合物を投与した。投与溶液は、各々、0.1mg/kg、1mg/kg、3mg/kg、10mg/kg、30mg/kgおよび60mg/kg用量について、0.03mg/mL、0.3mg/mL、0.9mg/mL、3mg/mL、9mg/mLおよび18mg/mLであった。投与容量は、全ての用量について1mL/300g(体重)であった。1つの群は、10mg/kg用量の実施例1Aを投与し、他の群は、0.1、1、3、10、30または60mg/kg用量の実施例4Aを投与した。ビヒクルは、20%v/vのPEG400および24%v/vのヒドロキシプロピルβシクロデキストリン(HPBCD)であった。経口投与に続いて、尿を24時間集めた。Roche Hitachi917分光光度計(Diamond Diagnostics、Holliston、MA)を使用した340nmでのUV吸光度分光測定によって、グルコース濃度を尿中で測定した。尿中に排泄されるグルコースの総量を、下記の式を使用して尿中濃度および尿量の積として計算した。
排泄される尿中グルコース(mg)/200g(体重)=尿中グルコース濃度(mg/dL)×尿量(dL)×200/ラット体重(g)。排泄される尿中グルコース(UGE)の量は、上記の方法によって実施例1Aおよび実施例4Aについてラットから得た。それを表4に示す。血液(0.1mL)をPKサテライト群動物から投与後1、2、4、7、24時間に集め、血漿を得て、LC−MS/MSによって分析した。試験した様々な用量における平均PKパラメータを、表4に示す。
【0308】
【表19】
【0309】
ラットにおける薬物動態試験
実施例1A、2A、4A、12A、および14Aをラットで試験し、最高濃度(Cmax)、血漿濃度時間曲線下面積(AUC)、クリアランス(CL)、定常状態分布容積(Vss)、半減期(t1/2)、およびバイオアベイラビリティー(F)を含めた薬物動態パラメータを評価した。雄性Sprague−Dawleyラット(約300g)を使用した。ラットに静脈内(IV)または経口胃管栄養法(PO)投与によって化合物を投与した。投与溶液を製剤するための、ビヒクルを含めた試験した用量を、表5に一覧表示する。
【0310】
IVまたはPO投与に続いて、0.2mLの血液を様々な時点(表5)で頸静脈から試料採取した。20μL分量の血漿試料および標準物質を、内部標準を含有するアセトニトリルによるタンパク質沈殿に供した。試料をボルテックスし、遠心分離し、上清を得て、それをLC−MS/MSによって分析した。Analyst(バージョン1.4.1)を使用してピーク面積を測定し、分析物と内部標準とのピーク面積比を計算した。LC−MS/MS条件は、下記の通りである。質量分析計+ソースタイプは、Sciex API4000−Turbo Sprayであった。HPLCポンプは、島津製作所であった。オートサンプラーは、CTC PALオートサンプラーであった。注入量は、3.0〜10μLであった。勾配は、移動相A:水中の10mMの酢酸アンモニウムおよび1%イソプロピルアルコール;B:アセトニトリルで使用した。流量、毎分0.300mL(カラム2.0×30mm、5μm、LUNA C18カラム(phenomenex))。検出モードはネガティブであった。
【0311】
較正曲線は、重み付き線形(1/xまたは1/x2)回帰を適用することによって、標準物質の内部標準に対するピーク面積比から構築した。標準曲線の測定範囲は、5.00ng/mL〜5000ng/mLであった。
【0312】
薬物動態パラメータは、Watson(バージョン7.2)でノンコンパートメント分析を使用して個々の動物データから決定した。定量下限未満(BLQ)の濃度は、計算において使用するために0ng/mLとして記録した。
【0313】
下記の計算を使用した。
AUC(0−τ)=線形台形法を使用して決定
AUC(0−∞)=AUC(0−τ)、およびτにおける血漿濃度を最終対数線形相の勾配で割ることによって決定した外挿領域
CL=用量/AUC(0−∞)
Vdss=CL×MRT
Cmax=血漿濃度時間曲線から直接記録
Tmax=血漿濃度時間曲線から直接記録
t1/2=ln(0.5)/最終対数線形相の勾配
F%=用量毎のAUC(0−∞)PO/用量毎のAUC(0−∞)IV
C(0)=IV投与後の明らかな分布相からの線形回帰によって外挿する
MRT=AUMC(AUC(0−∞)/AUC(0−∞)
【0314】
【表20】
【0315】
本出願に亘って、様々な公開資料を参照する。これらの公開資料の開示はその全体が、全ての目的のために参照により本出願に組み込まれている。
【0316】
本発明の範囲または趣旨を逸脱することなく、本発明において様々な改変および変形を行えることは当業者であれば明らかであろう。本発明の他の実施形態は、ここにおいて開示されている本発明の明細書および実施を考慮すると当業者であれば明らかであろう。実施例を含めた明細書は例示のみとして考えられ、本発明の本当の範囲および精神は下記の特許請求の範囲によって示されることを意図する。
【図1】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2012−500842(P2012−500842A)
【公表日】平成24年1月12日(2012.1.12)
【国際特許分類】
【出願番号】特願2011−524489(P2011−524489)
【出願日】平成21年8月17日(2009.8.17)
【特許番号】特許第4825322号(P4825322)
【特許公報発行日】平成23年11月30日(2011.11.30)
【国際出願番号】PCT/IB2009/053626
【国際公開番号】WO2010/023594
【国際公開日】平成22年3月4日(2010.3.4)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
【公表日】平成24年1月12日(2012.1.12)
【国際特許分類】
【出願日】平成21年8月17日(2009.8.17)
【特許番号】特許第4825322号(P4825322)
【特許公報発行日】平成23年11月30日(2011.11.30)
【国際出願番号】PCT/IB2009/053626
【国際公開番号】WO2010/023594
【国際公開日】平成22年3月4日(2010.3.4)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
[ Back to top ]