
ジカルボン酸化合物の製造方法

【課題】ポリエステル樹脂、ポリアミド樹脂、ポリアリレート樹脂、ポリベンゾオキサゾール樹脂及びポリベンゾチアゾール樹脂などの原料として有用な、ジカルボン酸化合物を、短い反応工程かつ、高収率で提供する。
【解決手段】一般式(3)で表されるジカルボン酸化合物を製造する方法であって、エステル化合物と、酸ジクロライド化合物とを反応させることを特徴とする、ジカルボン酸化合物の製造方法。

[式中、R1及びR3は、置換もしくは無置換の芳香環を有する有機基、あるいは置換もしくは無置換の脂肪族基を表す。]
【解決手段】一般式(3)で表されるジカルボン酸化合物を製造する方法であって、エステル化合物と、酸ジクロライド化合物とを反応させることを特徴とする、ジカルボン酸化合物の製造方法。
[式中、R1及びR3は、置換もしくは無置換の芳香環を有する有機基、あるいは置換もしくは無置換の脂肪族基を表す。]
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ジカルボン酸化合物の製造方法に関するものである。
【背景技術】
【0002】
一分子に2つのカルボキシル基を有するジカルボン酸化合物及びその酸塩化物誘導体は、ポリエステル樹脂、ポリアミド樹脂、ポリアリレート樹脂、ポリベンゾオキサゾール樹脂及びポリベンゾチアゾール樹脂などの原料として用いられている。これらの樹脂は、その用途に応じて、様々な構造の樹脂が合成されており、ジカルボン酸化合物及びその酸塩化物も樹脂構造に対応する様々な構造が選択され使用されている。
【0003】
これらの樹脂の主鎖に、エステル基などのメソゲン基を有する場合、液晶性を示すポリマーになり得るため、特に、高弾性、高強度、高耐熱樹脂へと広く検討されている。また、エステル基を有するジカルボン酸及びその酸塩化物誘導体は、液晶性を示すポリマーの原料として広く知られている(例えば、特許文献1参照。)。
【0004】
これらのエステル基を有するジカルボン酸の製造方法は、一般的に、下記反応式で示されるように、酸ジクロライドと、保護されたカルボキシル基を有する2当量の水酸基化合物とを反応させた後、脱保護する方法が知られており、1分子内に2つのエステル結合を有する対称なジカルボン酸化合物が得られる。
【0005】
【化6】
[式中、R、R’及びR’’は、それぞれ、置換もしくは無置換の芳香環を有する有機基、あるいは置換もしくは無置換の脂肪族基を表す。また、R’は脱離可能なカルボキシル基の保護基を表す。]
【0006】
一方で、分子内にエステル結合を1つ有する非対称なジカルボン酸の製造方法は、カルボキシル基の保護・脱保護を繰り返す多段階に及ぶ反応のため、収率及びコストの観点から、十分でないという問題があった。(例えば、非特許文献1参照。)
【特許文献1】特表平8−509020号公報
【非特許文献1】Macromolecules 1983,16,1034
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明は、ジカルボン酸化合物を、高収率で、しかも容易な工程で製造することができる製造方法を提供することである。
【課題を解決するための手段】
【0008】
本発明者は、前述したような問題点を解決すべく、鋭意検討を重ねた結果、次のような事項を見出し、本発明を完成するに至った。
【0009】
即ち、下記第(1)項〜第(6)項の本発明により達成される。
【0010】
(1) 一般式(3)で表されるジカルボン酸化合物を製造する方法であって、一般式(1)で表されるエステル化合物と、一般式(2)で表される酸ジクロライド化合物とを反応させることを特徴とする、ジカルボン酸化合物の製造方法。
【0011】
【化1】
【化2】
【化3】
[式中、R1、R2及びR3は、それぞれ、置換もしくは無置換の芳香族基、又は置換もしくは無置換の脂肪族基を示す。]
【0012】
(2) 一般式(1)で表されるエステル化合物が、前記一般式(1)におけるR1として、芳香族基を有するものである、上記第(1)項に記載のジカルボン酸化合物の製造方法。
(3) 一般式(2)で表される酸ジクロライド化合物が、一般式(2)におけるR3として、芳香族基を有するものである、上記第(1)項又は第(2)項に記載のジカルボン酸化合物の製造方法。
(4) 一般式(1)で表されるエステル化合物が、一般式(4)で表されるエステル化合物である、上記第(1)項〜第(3)項のいずれか1項に記載のジカルボン酸化合物の製造方法。
【0013】
【化4】
[式中、R4は、保護基を示す。]
【0014】
(5) 一般式(1)で表されるエステル化合物が、一般式(5)で表されるエステル化合物である、上記第(1)項〜第(4)項のいずれか1項に記載のジカルボン酸化合物の製造方法。
【0015】
【化5】
【0016】
(6) 前記一般式(1)で表されるエステル化合物と、前記一般式(2)で表される酸ジクロライド化合物との反応は、−60℃以上20℃以下の温度領域で行うものである上記第(1)項〜第(5)項のいずれか1項に記載のジカルボン酸化合物の製造方法。
【発明の効果】
【0017】
本発明のジカルボン酸の製造方法によれば、簡単な反応工程のもと高収率で、ジカルボン酸化合物が製造できる。
【発明を実施するための最良の形態】
【0018】
以下、本発明のジカルボン酸化合物の製造方法の好適実施形態について説明する。
【0019】
本発明により得られるジカルボン酸化合物としては、カルボキシル基を有する2つの基が、エステル結合により結合した構造を有するものであり、前記一般式(3)で表される構造を有するものである。
【0020】
本発明の一般式(3)で表されるジカルボン酸化合物は、一般式(3)中のR1及びR3として、それぞれ、置換もしくは無置換の芳香族基、又は置換もしくは無置換の脂肪族基を有するものである。
【0021】
前記一般式(3)におけるR1としては、例えば、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基、メチル基、エチル基及びn−ブチル基などの鎖状脂肪族基、またシクロヘキシル基などの脂環式脂肪族基などの置換もしくは無置換の脂肪族基が挙げられる。前記芳香族基における置換基としては、炭素数1〜6の脂肪族基やメトキシ基及びエトキシ基などのアルコキシ基などが挙げられる。前記脂肪族における置換基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、t−ブチル基及びヘキシル基などが挙げられる。これらの中でも、R1としては、熱安定性に優れるジカルボン酸化合物を得る上では、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基がより好ましい。
【0022】
また、前記一般式(3)におけるR3としては、例えば、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基、メチル基、エチル基及びn−ブチル基などの鎖状脂肪族基、またシクロヘキシル基などの脂環式脂肪族基などの置換もしくは無置換の脂肪族基が挙げられる。前記芳香族基における置換基としては、炭素数1〜6の脂肪族基やメトキシ基及びエトキシ基などのアルコキシ基などが挙げられる。前記脂肪族における置換基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、t−ブチル基及びヘキシル基などが挙げられる。これらの中でも、R3としては、熱安定性に優れるジカンルボン酸化合物を得る上では、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基がより好ましい。
【0023】
上記一般式(3)で表されるジカルボン酸化合物としては、例えば、2−カルボキシフェニルハイドロジェンテレフタレート、3−カルボキシフェニルハイドロジェンテレフタレート、4−カルボキシフェニルハイドロジェンテレフタレート、2−カルボキシフェニルハイドロジェンイソフタレート、3−カルボキシフェニルハイドロジェンイソフタレート、4−カルボキシフェニルハイドロジェンイソフタレート、2−カルボキシフェニルハイドロジェンフタレート、3−カルボキシフェニルハイドロジェンフタレート、4−カルボキシフェニルハイドロジェンフタレート、2,2’−(6−カルボキシナフチル)ハイドロジェンナフタレンジカルボキシレート、(2−カルボキシフェニルハイドロジェン)−2,2’−ビフェニルジカルボキシレート、(3−カルボキシフェニルハイドロジェン)−2,2’−ビフェニルジカルボキシレート、(4−カルボキシフェニルハイドロジェン)−2,2’−ビフェニルジカルボキシレート、2−カルボキシシクロヘキシルハイドロジェンシクロヘキシルジカルボキシレート、3−カルボキシシクロヘキシルハイドロジェンシクロヘキシルジカルボキシレート及び4−カルボキシシクロヘキシルハイドロジェンシクロヘキシルジカルボキシレートなどを挙げることができるが、これらに限定されるものではない。
【0024】
本発明に用いる、一般式(1)で表されるエステル化合物は、一般式(1)中のR1及びR2として、それぞれ、置換若しくは無置換の芳香族基、又は置換若しくは無置換の脂肪族基を有するものであり、それぞれ、同一であっても異なっていても良い。また、R2は、脱離可能なカルボキシル基の保護基である。
【0025】
前記一般式(1)におけるR1としては、一般式(3)中のR1における置換もしくは無置換の芳香族基、又は置換もしくは無置換の脂肪族基と同様のものを挙げることができ、熱安定性に優れるジカルボン酸を得る上では、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基がより好ましい。
【0026】
前記一般式(1)におけるカルボキシル基の保護基R2としては、加溶媒分解反応や水素添加反応などで脱離する有機基が挙げられ、例えば、フェニル基、ナフチル基及びベンジル基などの置換もしくは無置換の芳香族基、メチル基、メトキシメチル基、エチル基及びt−ブチル基などの炭素数1〜4の脂肪族基、トリメチルシリル基、トリエチルシリル基、トリイソプロピルシリル基、t−ブチルジメチルシリル基などのトリアルキルシリル基などの置換もしくは無置換の脂肪族基が挙げられる。前記芳香族基における置換基としては、炭素数1〜6の脂肪族基やメトキシ基及びエトキシ基などのアルコキシ基などが挙げられる。前記脂肪族における置換基としては、炭素数1〜6の脂肪族基やメトキシ基及びエトキシ基などのアルコキシ基などが挙げられる。これらの中でも、R2としては、カルボキシル基への脱保護反応における脱離性の点から、ベンジル基などの置換芳香族基、メチル基、エチル基及びt−ブチル基などの無置換脂肪族基、メトキシメチル基、トリメチルシリル基、トリエチルシリル基、トリイソプロピルシリル基及びt−ブチルジメチルシリル基などの置換脂肪族基がより好ましい。
【0027】
また、これらの一般式(1)で表されるエステル化合物のうち、より好ましいエステル化合物としては、前記一般式(4)で表される、エステル化合物を挙げることができる。
【0028】
前記一般式(4)における保護基R4としては、前記一般式(1)におけるカルボキシル基の保護基R2としての加溶媒分解反応や水素添加反応などで脱離する有機基と同様のものを挙げることができ、これらの中でも、R2としては、カルボキシル基への脱保護反応における脱離性の点から、ベンジル基、メチル基、メトキシメチル基、エチル基、t−ブチル基、トリメチルシリル基、トリエチルシリル基、トリイソプロピルシリル基、t−ブチルジメチルシリル基などの置換芳香族基または無置換芳香族基または無置換脂肪族基がより好ましい。
【0029】
また、これらのエステル化合物の中でのうち、さらにより好ましいエステル化合物としては、前記一般式(5)で表される、エステル化合物を挙げることができる。
【0030】
本発明に用いる、一般式(2)で表される酸ジクロライド化合物は、2つのカルボニルクロライド基を有するものである。
【0031】
前記一般式(2)におけるR3としては、一般式(3)中のR3における置換もしくは無置換の芳香族基、又は置換もしくは無置換の脂肪族基と同様のものを挙げることができ、これらの中でも、R3としては、熱安定性の点から、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基がより好ましい。
【0032】
ここで、本発明のジカルボン酸化合物の製造方法について、詳細に説明する。
【0033】
本発明のジカルボン酸化合物の製造方法としては、前記一般式(1)で表されるエステル化合物と、前記一般式(2)で表される酸ジクロライド化合物とを反応させて、前記一般式(3)で表されるジカルボン酸化合物を製造するものであるが、その具体例としては、前記一般式(1)で表されるエステル化合物を反応溶媒に溶解した溶液を、前記一般式(2)で表される酸ジクロライド化合物を反応溶媒に溶解した溶液中に、滴下して、混合し、エステル結合形成反応を行った後、前記一般式(1)で表されるエステル化合物由来のカルボキシル基の保護基を、脱保護反応により除去する製造方法を挙げることができ、かかる製造方法により、容易かつ高収率で合成することが可能である。
【0034】
上記一般式(1)で表されるエステル化合物の具体例としては、例えば、2−ヒドロキシ安息香酸メチル、3−ヒドロキシ安息香酸メチル、4−ヒドロキシ安息香酸メチル、2−ヒドロキシ安息香酸エチル、3−ヒドロキシ安息香酸エチル、4−ヒドロキシ安息香酸エチル、2−ヒドロキシ安息香酸メトキシメチル、3−ヒドロキシ安息香酸メトキシメチル、4−ヒドロキシ安息香酸メトキシメチル、2−ヒドロキシ安息香酸−t−ブチル、3−ヒドロキシ安息香酸−t−ブチル、4−ヒドロキシ安息香酸−t−ブチル、2−ヒドロキシ安息香酸ベンジル、3−ヒドロキシ安息香酸ベンジル、4−ヒドロキシ安息香酸ベンジル、2−ヒドロキシ安息香酸ベンジロキシメチル、3−ヒドロキシ安息香酸ベンジロキシメチル、4−ヒドロキシ安息香酸ベンジロキシメチル、2−ヒドロキシ安息香酸トリメチルシリル、3−ヒドロキシ安息香酸トリメチルシリル、4−ヒドロキシ安息香酸トリメチルシリル、2−ヒドロキシ安息香酸トリエチルシリル、3−ヒドロキシ安息香酸トリエチルシリル、4−ヒドロキシ安息香酸トリエチルシリル、2−ヒドロキシ安息香酸トリイソプロピルシリル、3−ヒドロキシ安息香酸トリイソプロピルシリル、4−ヒドロキシ安息香酸トリイソプロピルシリル、2−ヒドロキシ安息香酸−t−ブチルジメチルシリル、3−ヒドロキシ安息香酸−t−ブチルジメチルシリル、4−ヒドロキシ安息香酸−t−ブチルジメチルシリル、6−ヒドロキシ−2−ナフトエ酸メチル、6−ヒドロキシ−2−ナフトエ酸エチル、6−ヒドロキシ−2−ナフトエ酸メトキシメチル、6−ヒドロキシ−2−ナフトエ酸−t−ブチル、6−ヒドロキシ−2−ナフトエ酸ベンジル、6−ヒドロキシ−2−ナフトエ酸ベンジロキシメチル、6−ヒドロキシ−2−ナフトエ酸トリメチルシリル、6−ヒドロキシ−2−ナフトエ酸トリエチルシリル、6−ヒドロキシ−2−ナフトエ酸トリイソプロピルシリル、6−ヒドロキシ−2−ナフトエ酸−t−ブチルジメチルシリル、メチル−4−ヒドロキシシクロヘキサンカルボキシレート、エチル−4−ヒドロキシシクロヘキサンカルボキシレート、メトキシメチル−4−ヒドロキシシクロヘキサンカルボキシレート、t−ブチル−4−ヒドロキシシクロヘキサンカルボキシレート、ベンジル−4−ヒドロキシシクロヘキサンカルボキシレート、ベンジロキシメチル−4−ヒドロキシシクロヘキサンカルボキシレート、トリメチルシリル−4−ヒドロキシシクロヘキサンカルボキシレート、トリエチルシリル−4−ヒドロキシシクロヘキサンカルボキシレート、トリイソプロピルシリル−4−ヒドロキシシクロヘキサンカルボキシレート及びt−ブチルジメチルシリル−4−ヒドロキシシクロヘキサンカルボキシレートなどを挙げることができる。これらのなかでも、上記一般式(4)で表されるエステル化合物である、2−ヒドロキシ安息香酸メチル、3−ヒドロキシ安息香酸メチル、4−ヒドロキシ安息香酸メチル、2−ヒドロキシ安息香酸エチル、3−ヒドロキシ安息香酸エチル、4−ヒドロキシ安息香酸エチル、2−ヒドロキシ安息香酸メトキシメチル、3−ヒドロキシ安息香酸メトキシメチル、4−ヒドロキシ安息香酸メトキシメチル、2−ヒドロキシ安息香酸−t−ブチル、3−ヒドロキシ安息香酸−t−ブチル、4−ヒドロキシ安息香酸−t−ブチル、2−ヒドロキシ安息香酸ベンジル、3−ヒドロキシ安息香酸ベンジル、4−ヒドロキシ安息香酸ベンジル、2−ヒドロキシ安息香酸ベンジロキシメチル、3−ヒドロキシ安息香酸ベンジロキシメチル、4−ヒドロキシ安息香酸ベンジロキシメチル、2−ヒドロキシ安息香酸トリメチルシリル、3−ヒドロキシ安息香酸トリメチルシリル、4−ヒドロキシ安息香酸トリメチルシリル、2−ヒドロキシ安息香酸トリエチルシリル、3−ヒドロキシ安息香酸トリエチルシリル、4−ヒドロキシ安息香酸トリエチルシリル、2−ヒドロキシ安息香酸トリイソプロピルシリル、3−ヒドロキシ安息香酸トリイソプロピルシリル、4−ヒドロキシ安息香酸トリイソプロピルシリル、2−ヒドロキシ安息香酸−t−ブチルジメチルシリル、3−ヒドロキシ安息香酸−t−ブチルジメチルシリル及び4−ヒドロキシ安息香酸−t−ブチルジメチルシリルが好ましく、更には、上記一般式(5)で表されるエステル化合物である、2−ヒドロキシ安息香酸ベンジル、3−ヒドロキシ安息香酸ベンジル、4−ヒドロキシ安息香酸ベンジルがより好ましい。
【0035】
上記一般式(2)で表される酸ジクロライド化合物の具体例としては、例えば、フタロイルクロライド、イソフタロイルクロライド、テレフタロイルクロライド、2,3−ナフタレンジカルボニルジクロライド、2,6−ナフタレンジカルボニルジクロライド、2,2’−ビフェニルジカルボニルジクロライド、4,4’−ビフェニルジカルボニルジクロライド、1,3−シクロヘキシルジカルボニルジクロライド及び1,4−シクロヘキシルジカルボニルジクロライドなどを挙げることができる。これらのなかでも、得られるジカルボン酸化合物の熱安定性の点から、芳香族酸ジクロライド化合物が好ましく、具体例としては、フタロイルクロライド、イソフタロイルクロライド、テレフタロイルクロライド、2,3−ナフタレンジカルボニルジクロライド、2,6−ナフタレンジカルボニルジクロライド、2,2’−ビフェニルジカルボニルジクロライド及び4,4’−ビフェニルジカルボニルジクロライドがより好ましい。
【0036】
上記のエステル結合形成反応においては、反応溶媒として、例えば、テトラヒドロフラン、N,N−ジメチルホルムアミド、ジメチルスルホキシド、γ−ブチロラクトン及びN−メチルピロリドン等の有機溶媒を用いることができる。
【0037】
上記のエステル結合形成反応において、前記一般式(1)で表されるエステル化合物(A)と、前記一般式(2)で表される酸ジクロライド化合物(B)との反応モル比(A/B)としては、コスト、純度及び収率などの観点から、0.6〜1.2の間であることが好ましく、0.8〜1.0の間であることがより好ましい。
【0038】
上記のエステル結合形成反応における反応温度は、反応により得られるジカルボン酸化合物の純度及び収率の観点から、−60℃以上20℃以下の温度領域で行うのが好ましく、−20℃以上10℃以下の温度領域がより好ましい。
【0039】
上記の脱保護反応は、例えば、酸による加溶媒分解反応、アルカリによる加溶媒分解反応、水素添加反応などを挙げることができる。
【0040】
上記酸による加溶媒分解反応としては、塩酸、硫酸及び硝酸などの無機酸、酢酸、トリフルオロ酢酸及びp−トルエンスルホン酸などの有機酸、塩化リチウム、塩化アルミニウム、塩化鉄、臭化リチウム、臭化アルミニウム及び臭化鉄などのルイス酸と、メタノール、エタノール、プロパノール、イソプロパノール及びブタノールなどのアルコール溶媒や水との混合溶媒中で、上記エステル形成反応により得られた反応生成物を攪拌する方法を挙げることができる。
【0041】
上記アルカリによる加溶媒分解反応としては、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化カルシウム、水酸化バリウム、炭酸カリウム、炭酸カルシウム及び炭酸セシウムなどの無機塩基と、メタノール、エタノール、プロパノール、イソプロパノール及びブタノールなどのアルコール溶媒や水との混合溶媒中で、上記エステル形成反応により得られた反応生成物を攪拌する方法を挙げることができる。
【0042】
上記水素添加反応としては、水素雰囲気下にて、パラジウム活性炭素触媒やラネーニッケル触媒などの遷移金属触媒を、メタノール、エタノール、テトラヒドロフラン、N,N’−ジメチルホルムアミド、ジメチルスルホキシド及びN−メチルピロリドンなどの溶媒中で、上記エステル形成反応により得られた反応生成物を攪拌する方法が挙げられる。
【0043】
上記の脱保護反応により得られる反応物は、メタノール及びエタノールなどのアルコール溶媒、N,N−ジメチルホルムアミド及びジメチルスルホキシドなどの両親媒性溶媒、酢酸及びトリフルオロ酢酸などの酸、水、これら単独又は複数の混合溶媒で再結晶することにより、精製して純度を上げることも可能である。
【0044】
なお、本発明のジカルボン酸化合物の製造方法は、上記の合成反応ルートが一般的であるが、これらに何ら限定されるものではない。
【実施例】
【0045】
次に、本発明の具体的実施例について説明する。
【0046】
(実施例1)
冷却管及び撹拌装置付きのセパラブルフラスコ(容量:200mL)に、4−ヒドロキシ安息香酸13.8g(0.10mol)、臭化ベンジル17.1g(0.10mol)、炭酸カリウム6.9g(0.05mol)及びN,N−ジメチルホルムアミド100mLを仕込み、攪拌下120℃で4時間反応させ、析出した塩を含む反応溶液を得た。析出した塩を吸引濾過で除いた後、反応溶液を1Lビーカーに移し、これに、水300mLと酢酸エチル200mLを加え、分液ロートで有機層を回収した。回収した溶液に、硫酸マグネシウムを加えて、水分を除去した後、溶媒を減圧下で留去し、4−ヒドロキシ安息香酸ベンジルを得た。
次に、窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、テレフタロイルクロライド12.2g(0.06mol)を仕込み、上記で得た4−ヒドロキシ安息香酸ベンジルを11.4g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて、攪拌下0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃で、さらに5時間攪拌を続けた後、生成物を300mLの水に添加し、析出した固体を吸引濾過で回収した。撹拌装置付きのナスフラスコ(容量:500mL)に、上記で得られた固体、N,N−ジメチルホルムアミド200mL及び10%パラジウム活性炭素1.06g(0.001mol)を仕込み、水素雰囲気下で、24時間攪拌した。パラジウム活性炭素を吸引濾過で除いた後、1Lビーカーに移した反応溶液に水500mLを加え、析出した固体を吸引濾過で回収した真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶10.3gを得た。
【0047】
この生成物をC1とした。生成物C1を、1H−NMR、マススペクトル、元素分析で分析した結果は次の通りであった。
生成物の元素分析:
(実験値) C:63.3%,H:3.3%,O:33.4%,
(理論値) C:63.0%,H:3.5%,O:33.5%
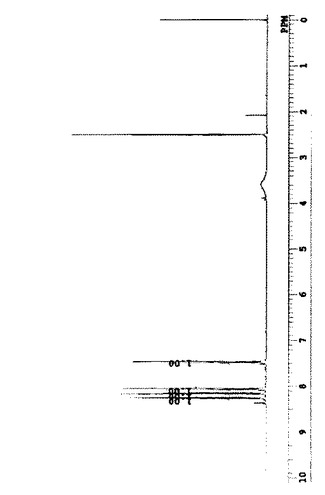
生成物C1の1H−NMRスペクトルを図1に示す。
分析結果より、得られた生成物C1は下記式(7)で表される目的のジカルボン酸化合物であることが確認された。得られたC1の収率は、72%であった。
【0048】
【化7】
【0049】
(実施例2)
冷却管及び撹拌装置付きのセパラブルフラスコ(容量:200mL)に、3−ヒドロキシ安息香酸13.8g(0.10mol)、ブロモメトキシメタン12.5g(0.10mol)、炭酸カリウム6.9g(0.05mol)及びN,N−ジメチルホルムアミド100mLを仕込み、攪拌下120℃で4時間反応させ、塩が析出した反応溶液を得た。析出した塩を吸引濾過で除いた後、反応溶液を1Lビーカーに移し、これに水300mLと酢酸エチル200mLを加え、分液ロートで有機層を回収した。回収した溶液に硫酸マグネシウムを加えて水分を除去した後、溶媒を減圧下で留去し、3−ヒドロキシ安息香酸メトキシメチルを得た。
次に、窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、イソフタロイルクロライド12.2g(0.06mol)を仕込み、上記で得た3−ヒドロキシ安息香酸メトキシメチルを9.11g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて、攪拌下、0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃で、さらに5時間攪拌を続けた後、生成物を、300mLの水に添加し、析出した固体を吸引濾過で回収した。
撹拌装置付きのセパラブルフラスコ(容量:500mL)に、上記で得られた固体、メタノール100mL及び水100mLを仕込み、60℃で一晩攪拌した。これにより析出した固体を吸引濾過で回収した。上記で得た固体を真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶10.0gを得た。分析結果より、得られた生成物C2は下記式(8)で表される目的のジカルボン酸化合物であることが確認された。
【0050】
【化8】
【0051】
(実施例3)
撹拌装置付きのセパラブルフラスコ(容量:200mL)に、6−ヒドロキシナフトイックアシッド18.8g(0.10mol)、t−ブチルジメチルシリルクロライド15.1g(0.10mol)、トリエチルアミン10.1g(0.10mol)及びピリジン100mLを仕込み、攪拌下30℃で8時間反応した。析出した塩を吸引濾過で除いた後、溶媒を減圧下で留去し、6−ヒドロキシ−2−ナフトエ酸−t−ブチルジメチルシリルを得た。
窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、2,6−ナフタレンジカルボニルジクロライド15.2g(0.06mol)を仕込み、上記で得た6−ヒドロキシ−2−ナフトエ酸−t−ブチルジメチルシリルを15.1g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて攪拌下0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃で、さらに5時間攪拌を続けた後、生成物を300mLの水に添加し、析出した固体を吸引濾過で回収した。
撹拌装置付きのセパラブルフラスコ(容量:500mL)に、上記で得られた固体、酢酸150mL、テトラヒドロフラン50mL,及び水50mLを仕込み、60℃で一晩攪拌した。これにより、析出した固体を吸引濾過で回収した。上記で得た固体を、真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶14.7gを得た。分析結果より、得られた生成物C3は下記式(9)で表される目的のジカルボン酸化合物であることが確認された。
【0052】
【化9】
【0053】
(実施例4)
撹拌装置付きのセパラブルフラスコ(容量:200mL)に、4−ヒドロキシベンゾイックアシッドナトリウム塩16.1g(0.10mol)、ベンジルクロロメチルエーテル15.7g(0.10mol)及びヘキサメチルリン酸トリアミド100mLを仕込み、攪拌下30℃で8時間反応させ、塩が析出した反応溶液を得た。析出した塩を吸引濾過で除いた後、反応溶液を1Lビーカーに移し、水300mLと酢酸エチル200mLを加え、分液ロートで有機層を回収した。回収した溶液に、硫酸マグネシウムを加えて水分を除去した後、溶媒を減圧下で留去し、4−ヒドロキシ安息香酸ベンジロキシメチルを得た。
窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、2,2’−ビフェニルジカルボニルジクロライド16.8g(0.06mol)を仕込み、4−ヒドロキシ安息香酸ベンジロキシメチルを12.9g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて攪拌下0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃で、さらに5時間攪拌を続けた後、生成物を300mLの水に添加し、析出した固体を吸引濾過で回収した。
撹拌装置付きのセパラブルフラスコ(容量:500mL)に、上記で得られた固体、0.2mol/L塩酸水溶液50mL及びテトラヒドロフラン250mLを仕込み、60℃で一晩攪拌した。これにより、析出した固体を吸引濾過で回収した。上記で得た固体を真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶12.1gを得た。分析結果より、得られた生成物C4は下記式(10)で表される目的のジカルボン酸化合物であることが確認された。
【0054】
【化10】
【0055】
(実施例5)
窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、1,4−シクロヘキシルジカルボニルジクロライド12.6g(0.06mol)を仕込み、エチル−4−ヒドロキシシクロヘキサンカルボキシレートを8.60g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて攪拌下0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃でさらに5時間攪拌を続けた。得られた反応溶液を1Lビーカーに移し、水300mLと酢酸エチル200mLを加え、分液ロートで有機層を回収した。回収した有機層に、硫酸マグネシウムを加えて水分を除去した後、溶媒を減圧下で留去し、反応物を回収した。
撹拌装置付きのセパラブルフラスコ(容量:500mL)に、上記で回収した反応物、0.2mol/L水酸化ナトリウム水溶液50mL及びメタノール250mLを仕込み、30℃で一晩攪拌した。これにより析出した固体を吸引濾過で回収した。上記で得た固体を真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶8.94gを得た。分析結果より、得られた生成物C5は下記式(11)で表される目的のジカルボン酸化合物であることが確認された。
【0056】
【化11】
【0057】
実施例1〜5の合成結果と、得られた生成物のマススペクトル(MSスペクトル)及び元素分析による分析結果を表1にまとめた。
【0058】
【表1】
【0059】
(比較例1)
非特許文献、Macromolecules 1983,16,1034に記載の方法に従い、実施例1と同様の構造を有するジカルボン酸C1の合成を行った。
テレフタル酸と臭化ベンジルとを反応させ、テレフタル酸ジベンジルを合成した後、アルカリ加水分解によりテレフタル酸モノベンジルとし、塩化チオニルと反応させることで、テレフタル酸モノベンジルクロライドを合成した。次いで、p−ヒドロキシ安息香酸ベンジルとテレフタル酸モノベンジルクロライドとにより、エステル結合形成反応を行った。次に、得られたベンジル−4−(カルボベンゾキシ)フェニルテレフタレートを、トリフルオロ酢酸中、臭化水素による加溶媒分解により、脱保護反応を行った。析出した固体を吸引濾過で回収した。真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶5.00gを得た。得られた結晶を1H−NMR、マススペクトル、元素分析で分析した結果、目的の化合物C1が得られた。(収率35%)
【0060】
(比較例2)
窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、テレフタロイルクロライド12.2g(0.06mol)を仕込み、p−ヒドロキシ安息香酸を6.90g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて攪拌下0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃でさらに5時間攪拌を続けた後、生成物を300mLの水に添加し、析出固体を吸引濾過で回収した。真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶を得た。得られた結晶を1H−NMR、マススペクトル、元素分析で分析した結果、実施例1と同様の構造を有する化合物C1、テレフタル酸、2分子のp−ヒドロキシ安息香酸がエステル結合形成反応を起こした生成物である、ビス(カルボキシフェニル)テレフタレートの混合物が得られた。(純度85%、収率70%)
【0061】
比較例1では、多段階に及ぶ保護・脱保護反応を繰り返すため、本発明の製造方法よりも反応工程がかかるうえ、コストや収率の観点からも好ましくない。
比較例2では、カルボキシル基の保護がなされていないため、本発明の製造方法よりも反応の選択性が低下するため、純度の観点から好ましくない。
【産業上の利用可能性】
【0062】
本発明によれば、短い反応工程かつ、高収率でジカルボン酸化合物を得ることができ、本発明のジカルボン酸化合物は、ポリエステル樹脂、ポリアミド樹脂、ポリアリレート樹脂、ポリベンゾオキサゾール樹脂及びポリベンゾチアゾール樹脂などの原料として有用である。
【図面の簡単な説明】
【0063】
【図1】生成物C1の1H−NMR(400MHz, DMSO−d6)スペクトル
【技術分野】
【0001】
本発明は、ジカルボン酸化合物の製造方法に関するものである。
【背景技術】
【0002】
一分子に2つのカルボキシル基を有するジカルボン酸化合物及びその酸塩化物誘導体は、ポリエステル樹脂、ポリアミド樹脂、ポリアリレート樹脂、ポリベンゾオキサゾール樹脂及びポリベンゾチアゾール樹脂などの原料として用いられている。これらの樹脂は、その用途に応じて、様々な構造の樹脂が合成されており、ジカルボン酸化合物及びその酸塩化物も樹脂構造に対応する様々な構造が選択され使用されている。
【0003】
これらの樹脂の主鎖に、エステル基などのメソゲン基を有する場合、液晶性を示すポリマーになり得るため、特に、高弾性、高強度、高耐熱樹脂へと広く検討されている。また、エステル基を有するジカルボン酸及びその酸塩化物誘導体は、液晶性を示すポリマーの原料として広く知られている(例えば、特許文献1参照。)。
【0004】
これらのエステル基を有するジカルボン酸の製造方法は、一般的に、下記反応式で示されるように、酸ジクロライドと、保護されたカルボキシル基を有する2当量の水酸基化合物とを反応させた後、脱保護する方法が知られており、1分子内に2つのエステル結合を有する対称なジカルボン酸化合物が得られる。
【0005】
【化6】
[式中、R、R’及びR’’は、それぞれ、置換もしくは無置換の芳香環を有する有機基、あるいは置換もしくは無置換の脂肪族基を表す。また、R’は脱離可能なカルボキシル基の保護基を表す。]
【0006】
一方で、分子内にエステル結合を1つ有する非対称なジカルボン酸の製造方法は、カルボキシル基の保護・脱保護を繰り返す多段階に及ぶ反応のため、収率及びコストの観点から、十分でないという問題があった。(例えば、非特許文献1参照。)
【特許文献1】特表平8−509020号公報
【非特許文献1】Macromolecules 1983,16,1034
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明は、ジカルボン酸化合物を、高収率で、しかも容易な工程で製造することができる製造方法を提供することである。
【課題を解決するための手段】
【0008】
本発明者は、前述したような問題点を解決すべく、鋭意検討を重ねた結果、次のような事項を見出し、本発明を完成するに至った。
【0009】
即ち、下記第(1)項〜第(6)項の本発明により達成される。
【0010】
(1) 一般式(3)で表されるジカルボン酸化合物を製造する方法であって、一般式(1)で表されるエステル化合物と、一般式(2)で表される酸ジクロライド化合物とを反応させることを特徴とする、ジカルボン酸化合物の製造方法。
【0011】
【化1】
【化2】
【化3】
[式中、R1、R2及びR3は、それぞれ、置換もしくは無置換の芳香族基、又は置換もしくは無置換の脂肪族基を示す。]
【0012】
(2) 一般式(1)で表されるエステル化合物が、前記一般式(1)におけるR1として、芳香族基を有するものである、上記第(1)項に記載のジカルボン酸化合物の製造方法。
(3) 一般式(2)で表される酸ジクロライド化合物が、一般式(2)におけるR3として、芳香族基を有するものである、上記第(1)項又は第(2)項に記載のジカルボン酸化合物の製造方法。
(4) 一般式(1)で表されるエステル化合物が、一般式(4)で表されるエステル化合物である、上記第(1)項〜第(3)項のいずれか1項に記載のジカルボン酸化合物の製造方法。
【0013】
【化4】
[式中、R4は、保護基を示す。]
【0014】
(5) 一般式(1)で表されるエステル化合物が、一般式(5)で表されるエステル化合物である、上記第(1)項〜第(4)項のいずれか1項に記載のジカルボン酸化合物の製造方法。
【0015】
【化5】
【0016】
(6) 前記一般式(1)で表されるエステル化合物と、前記一般式(2)で表される酸ジクロライド化合物との反応は、−60℃以上20℃以下の温度領域で行うものである上記第(1)項〜第(5)項のいずれか1項に記載のジカルボン酸化合物の製造方法。
【発明の効果】
【0017】
本発明のジカルボン酸の製造方法によれば、簡単な反応工程のもと高収率で、ジカルボン酸化合物が製造できる。
【発明を実施するための最良の形態】
【0018】
以下、本発明のジカルボン酸化合物の製造方法の好適実施形態について説明する。
【0019】
本発明により得られるジカルボン酸化合物としては、カルボキシル基を有する2つの基が、エステル結合により結合した構造を有するものであり、前記一般式(3)で表される構造を有するものである。
【0020】
本発明の一般式(3)で表されるジカルボン酸化合物は、一般式(3)中のR1及びR3として、それぞれ、置換もしくは無置換の芳香族基、又は置換もしくは無置換の脂肪族基を有するものである。
【0021】
前記一般式(3)におけるR1としては、例えば、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基、メチル基、エチル基及びn−ブチル基などの鎖状脂肪族基、またシクロヘキシル基などの脂環式脂肪族基などの置換もしくは無置換の脂肪族基が挙げられる。前記芳香族基における置換基としては、炭素数1〜6の脂肪族基やメトキシ基及びエトキシ基などのアルコキシ基などが挙げられる。前記脂肪族における置換基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、t−ブチル基及びヘキシル基などが挙げられる。これらの中でも、R1としては、熱安定性に優れるジカルボン酸化合物を得る上では、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基がより好ましい。
【0022】
また、前記一般式(3)におけるR3としては、例えば、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基、メチル基、エチル基及びn−ブチル基などの鎖状脂肪族基、またシクロヘキシル基などの脂環式脂肪族基などの置換もしくは無置換の脂肪族基が挙げられる。前記芳香族基における置換基としては、炭素数1〜6の脂肪族基やメトキシ基及びエトキシ基などのアルコキシ基などが挙げられる。前記脂肪族における置換基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、t−ブチル基及びヘキシル基などが挙げられる。これらの中でも、R3としては、熱安定性に優れるジカンルボン酸化合物を得る上では、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基がより好ましい。
【0023】
上記一般式(3)で表されるジカルボン酸化合物としては、例えば、2−カルボキシフェニルハイドロジェンテレフタレート、3−カルボキシフェニルハイドロジェンテレフタレート、4−カルボキシフェニルハイドロジェンテレフタレート、2−カルボキシフェニルハイドロジェンイソフタレート、3−カルボキシフェニルハイドロジェンイソフタレート、4−カルボキシフェニルハイドロジェンイソフタレート、2−カルボキシフェニルハイドロジェンフタレート、3−カルボキシフェニルハイドロジェンフタレート、4−カルボキシフェニルハイドロジェンフタレート、2,2’−(6−カルボキシナフチル)ハイドロジェンナフタレンジカルボキシレート、(2−カルボキシフェニルハイドロジェン)−2,2’−ビフェニルジカルボキシレート、(3−カルボキシフェニルハイドロジェン)−2,2’−ビフェニルジカルボキシレート、(4−カルボキシフェニルハイドロジェン)−2,2’−ビフェニルジカルボキシレート、2−カルボキシシクロヘキシルハイドロジェンシクロヘキシルジカルボキシレート、3−カルボキシシクロヘキシルハイドロジェンシクロヘキシルジカルボキシレート及び4−カルボキシシクロヘキシルハイドロジェンシクロヘキシルジカルボキシレートなどを挙げることができるが、これらに限定されるものではない。
【0024】
本発明に用いる、一般式(1)で表されるエステル化合物は、一般式(1)中のR1及びR2として、それぞれ、置換若しくは無置換の芳香族基、又は置換若しくは無置換の脂肪族基を有するものであり、それぞれ、同一であっても異なっていても良い。また、R2は、脱離可能なカルボキシル基の保護基である。
【0025】
前記一般式(1)におけるR1としては、一般式(3)中のR1における置換もしくは無置換の芳香族基、又は置換もしくは無置換の脂肪族基と同様のものを挙げることができ、熱安定性に優れるジカルボン酸を得る上では、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基がより好ましい。
【0026】
前記一般式(1)におけるカルボキシル基の保護基R2としては、加溶媒分解反応や水素添加反応などで脱離する有機基が挙げられ、例えば、フェニル基、ナフチル基及びベンジル基などの置換もしくは無置換の芳香族基、メチル基、メトキシメチル基、エチル基及びt−ブチル基などの炭素数1〜4の脂肪族基、トリメチルシリル基、トリエチルシリル基、トリイソプロピルシリル基、t−ブチルジメチルシリル基などのトリアルキルシリル基などの置換もしくは無置換の脂肪族基が挙げられる。前記芳香族基における置換基としては、炭素数1〜6の脂肪族基やメトキシ基及びエトキシ基などのアルコキシ基などが挙げられる。前記脂肪族における置換基としては、炭素数1〜6の脂肪族基やメトキシ基及びエトキシ基などのアルコキシ基などが挙げられる。これらの中でも、R2としては、カルボキシル基への脱保護反応における脱離性の点から、ベンジル基などの置換芳香族基、メチル基、エチル基及びt−ブチル基などの無置換脂肪族基、メトキシメチル基、トリメチルシリル基、トリエチルシリル基、トリイソプロピルシリル基及びt−ブチルジメチルシリル基などの置換脂肪族基がより好ましい。
【0027】
また、これらの一般式(1)で表されるエステル化合物のうち、より好ましいエステル化合物としては、前記一般式(4)で表される、エステル化合物を挙げることができる。
【0028】
前記一般式(4)における保護基R4としては、前記一般式(1)におけるカルボキシル基の保護基R2としての加溶媒分解反応や水素添加反応などで脱離する有機基と同様のものを挙げることができ、これらの中でも、R2としては、カルボキシル基への脱保護反応における脱離性の点から、ベンジル基、メチル基、メトキシメチル基、エチル基、t−ブチル基、トリメチルシリル基、トリエチルシリル基、トリイソプロピルシリル基、t−ブチルジメチルシリル基などの置換芳香族基または無置換芳香族基または無置換脂肪族基がより好ましい。
【0029】
また、これらのエステル化合物の中でのうち、さらにより好ましいエステル化合物としては、前記一般式(5)で表される、エステル化合物を挙げることができる。
【0030】
本発明に用いる、一般式(2)で表される酸ジクロライド化合物は、2つのカルボニルクロライド基を有するものである。
【0031】
前記一般式(2)におけるR3としては、一般式(3)中のR3における置換もしくは無置換の芳香族基、又は置換もしくは無置換の脂肪族基と同様のものを挙げることができ、これらの中でも、R3としては、熱安定性の点から、フェニル基、メチルフェニル基、メトキシフェニル基、ナフチル基及びビフェニル基などの置換もしくは無置換の芳香族基がより好ましい。
【0032】
ここで、本発明のジカルボン酸化合物の製造方法について、詳細に説明する。
【0033】
本発明のジカルボン酸化合物の製造方法としては、前記一般式(1)で表されるエステル化合物と、前記一般式(2)で表される酸ジクロライド化合物とを反応させて、前記一般式(3)で表されるジカルボン酸化合物を製造するものであるが、その具体例としては、前記一般式(1)で表されるエステル化合物を反応溶媒に溶解した溶液を、前記一般式(2)で表される酸ジクロライド化合物を反応溶媒に溶解した溶液中に、滴下して、混合し、エステル結合形成反応を行った後、前記一般式(1)で表されるエステル化合物由来のカルボキシル基の保護基を、脱保護反応により除去する製造方法を挙げることができ、かかる製造方法により、容易かつ高収率で合成することが可能である。
【0034】
上記一般式(1)で表されるエステル化合物の具体例としては、例えば、2−ヒドロキシ安息香酸メチル、3−ヒドロキシ安息香酸メチル、4−ヒドロキシ安息香酸メチル、2−ヒドロキシ安息香酸エチル、3−ヒドロキシ安息香酸エチル、4−ヒドロキシ安息香酸エチル、2−ヒドロキシ安息香酸メトキシメチル、3−ヒドロキシ安息香酸メトキシメチル、4−ヒドロキシ安息香酸メトキシメチル、2−ヒドロキシ安息香酸−t−ブチル、3−ヒドロキシ安息香酸−t−ブチル、4−ヒドロキシ安息香酸−t−ブチル、2−ヒドロキシ安息香酸ベンジル、3−ヒドロキシ安息香酸ベンジル、4−ヒドロキシ安息香酸ベンジル、2−ヒドロキシ安息香酸ベンジロキシメチル、3−ヒドロキシ安息香酸ベンジロキシメチル、4−ヒドロキシ安息香酸ベンジロキシメチル、2−ヒドロキシ安息香酸トリメチルシリル、3−ヒドロキシ安息香酸トリメチルシリル、4−ヒドロキシ安息香酸トリメチルシリル、2−ヒドロキシ安息香酸トリエチルシリル、3−ヒドロキシ安息香酸トリエチルシリル、4−ヒドロキシ安息香酸トリエチルシリル、2−ヒドロキシ安息香酸トリイソプロピルシリル、3−ヒドロキシ安息香酸トリイソプロピルシリル、4−ヒドロキシ安息香酸トリイソプロピルシリル、2−ヒドロキシ安息香酸−t−ブチルジメチルシリル、3−ヒドロキシ安息香酸−t−ブチルジメチルシリル、4−ヒドロキシ安息香酸−t−ブチルジメチルシリル、6−ヒドロキシ−2−ナフトエ酸メチル、6−ヒドロキシ−2−ナフトエ酸エチル、6−ヒドロキシ−2−ナフトエ酸メトキシメチル、6−ヒドロキシ−2−ナフトエ酸−t−ブチル、6−ヒドロキシ−2−ナフトエ酸ベンジル、6−ヒドロキシ−2−ナフトエ酸ベンジロキシメチル、6−ヒドロキシ−2−ナフトエ酸トリメチルシリル、6−ヒドロキシ−2−ナフトエ酸トリエチルシリル、6−ヒドロキシ−2−ナフトエ酸トリイソプロピルシリル、6−ヒドロキシ−2−ナフトエ酸−t−ブチルジメチルシリル、メチル−4−ヒドロキシシクロヘキサンカルボキシレート、エチル−4−ヒドロキシシクロヘキサンカルボキシレート、メトキシメチル−4−ヒドロキシシクロヘキサンカルボキシレート、t−ブチル−4−ヒドロキシシクロヘキサンカルボキシレート、ベンジル−4−ヒドロキシシクロヘキサンカルボキシレート、ベンジロキシメチル−4−ヒドロキシシクロヘキサンカルボキシレート、トリメチルシリル−4−ヒドロキシシクロヘキサンカルボキシレート、トリエチルシリル−4−ヒドロキシシクロヘキサンカルボキシレート、トリイソプロピルシリル−4−ヒドロキシシクロヘキサンカルボキシレート及びt−ブチルジメチルシリル−4−ヒドロキシシクロヘキサンカルボキシレートなどを挙げることができる。これらのなかでも、上記一般式(4)で表されるエステル化合物である、2−ヒドロキシ安息香酸メチル、3−ヒドロキシ安息香酸メチル、4−ヒドロキシ安息香酸メチル、2−ヒドロキシ安息香酸エチル、3−ヒドロキシ安息香酸エチル、4−ヒドロキシ安息香酸エチル、2−ヒドロキシ安息香酸メトキシメチル、3−ヒドロキシ安息香酸メトキシメチル、4−ヒドロキシ安息香酸メトキシメチル、2−ヒドロキシ安息香酸−t−ブチル、3−ヒドロキシ安息香酸−t−ブチル、4−ヒドロキシ安息香酸−t−ブチル、2−ヒドロキシ安息香酸ベンジル、3−ヒドロキシ安息香酸ベンジル、4−ヒドロキシ安息香酸ベンジル、2−ヒドロキシ安息香酸ベンジロキシメチル、3−ヒドロキシ安息香酸ベンジロキシメチル、4−ヒドロキシ安息香酸ベンジロキシメチル、2−ヒドロキシ安息香酸トリメチルシリル、3−ヒドロキシ安息香酸トリメチルシリル、4−ヒドロキシ安息香酸トリメチルシリル、2−ヒドロキシ安息香酸トリエチルシリル、3−ヒドロキシ安息香酸トリエチルシリル、4−ヒドロキシ安息香酸トリエチルシリル、2−ヒドロキシ安息香酸トリイソプロピルシリル、3−ヒドロキシ安息香酸トリイソプロピルシリル、4−ヒドロキシ安息香酸トリイソプロピルシリル、2−ヒドロキシ安息香酸−t−ブチルジメチルシリル、3−ヒドロキシ安息香酸−t−ブチルジメチルシリル及び4−ヒドロキシ安息香酸−t−ブチルジメチルシリルが好ましく、更には、上記一般式(5)で表されるエステル化合物である、2−ヒドロキシ安息香酸ベンジル、3−ヒドロキシ安息香酸ベンジル、4−ヒドロキシ安息香酸ベンジルがより好ましい。
【0035】
上記一般式(2)で表される酸ジクロライド化合物の具体例としては、例えば、フタロイルクロライド、イソフタロイルクロライド、テレフタロイルクロライド、2,3−ナフタレンジカルボニルジクロライド、2,6−ナフタレンジカルボニルジクロライド、2,2’−ビフェニルジカルボニルジクロライド、4,4’−ビフェニルジカルボニルジクロライド、1,3−シクロヘキシルジカルボニルジクロライド及び1,4−シクロヘキシルジカルボニルジクロライドなどを挙げることができる。これらのなかでも、得られるジカルボン酸化合物の熱安定性の点から、芳香族酸ジクロライド化合物が好ましく、具体例としては、フタロイルクロライド、イソフタロイルクロライド、テレフタロイルクロライド、2,3−ナフタレンジカルボニルジクロライド、2,6−ナフタレンジカルボニルジクロライド、2,2’−ビフェニルジカルボニルジクロライド及び4,4’−ビフェニルジカルボニルジクロライドがより好ましい。
【0036】
上記のエステル結合形成反応においては、反応溶媒として、例えば、テトラヒドロフラン、N,N−ジメチルホルムアミド、ジメチルスルホキシド、γ−ブチロラクトン及びN−メチルピロリドン等の有機溶媒を用いることができる。
【0037】
上記のエステル結合形成反応において、前記一般式(1)で表されるエステル化合物(A)と、前記一般式(2)で表される酸ジクロライド化合物(B)との反応モル比(A/B)としては、コスト、純度及び収率などの観点から、0.6〜1.2の間であることが好ましく、0.8〜1.0の間であることがより好ましい。
【0038】
上記のエステル結合形成反応における反応温度は、反応により得られるジカルボン酸化合物の純度及び収率の観点から、−60℃以上20℃以下の温度領域で行うのが好ましく、−20℃以上10℃以下の温度領域がより好ましい。
【0039】
上記の脱保護反応は、例えば、酸による加溶媒分解反応、アルカリによる加溶媒分解反応、水素添加反応などを挙げることができる。
【0040】
上記酸による加溶媒分解反応としては、塩酸、硫酸及び硝酸などの無機酸、酢酸、トリフルオロ酢酸及びp−トルエンスルホン酸などの有機酸、塩化リチウム、塩化アルミニウム、塩化鉄、臭化リチウム、臭化アルミニウム及び臭化鉄などのルイス酸と、メタノール、エタノール、プロパノール、イソプロパノール及びブタノールなどのアルコール溶媒や水との混合溶媒中で、上記エステル形成反応により得られた反応生成物を攪拌する方法を挙げることができる。
【0041】
上記アルカリによる加溶媒分解反応としては、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化カルシウム、水酸化バリウム、炭酸カリウム、炭酸カルシウム及び炭酸セシウムなどの無機塩基と、メタノール、エタノール、プロパノール、イソプロパノール及びブタノールなどのアルコール溶媒や水との混合溶媒中で、上記エステル形成反応により得られた反応生成物を攪拌する方法を挙げることができる。
【0042】
上記水素添加反応としては、水素雰囲気下にて、パラジウム活性炭素触媒やラネーニッケル触媒などの遷移金属触媒を、メタノール、エタノール、テトラヒドロフラン、N,N’−ジメチルホルムアミド、ジメチルスルホキシド及びN−メチルピロリドンなどの溶媒中で、上記エステル形成反応により得られた反応生成物を攪拌する方法が挙げられる。
【0043】
上記の脱保護反応により得られる反応物は、メタノール及びエタノールなどのアルコール溶媒、N,N−ジメチルホルムアミド及びジメチルスルホキシドなどの両親媒性溶媒、酢酸及びトリフルオロ酢酸などの酸、水、これら単独又は複数の混合溶媒で再結晶することにより、精製して純度を上げることも可能である。
【0044】
なお、本発明のジカルボン酸化合物の製造方法は、上記の合成反応ルートが一般的であるが、これらに何ら限定されるものではない。
【実施例】
【0045】
次に、本発明の具体的実施例について説明する。
【0046】
(実施例1)
冷却管及び撹拌装置付きのセパラブルフラスコ(容量:200mL)に、4−ヒドロキシ安息香酸13.8g(0.10mol)、臭化ベンジル17.1g(0.10mol)、炭酸カリウム6.9g(0.05mol)及びN,N−ジメチルホルムアミド100mLを仕込み、攪拌下120℃で4時間反応させ、析出した塩を含む反応溶液を得た。析出した塩を吸引濾過で除いた後、反応溶液を1Lビーカーに移し、これに、水300mLと酢酸エチル200mLを加え、分液ロートで有機層を回収した。回収した溶液に、硫酸マグネシウムを加えて、水分を除去した後、溶媒を減圧下で留去し、4−ヒドロキシ安息香酸ベンジルを得た。
次に、窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、テレフタロイルクロライド12.2g(0.06mol)を仕込み、上記で得た4−ヒドロキシ安息香酸ベンジルを11.4g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて、攪拌下0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃で、さらに5時間攪拌を続けた後、生成物を300mLの水に添加し、析出した固体を吸引濾過で回収した。撹拌装置付きのナスフラスコ(容量:500mL)に、上記で得られた固体、N,N−ジメチルホルムアミド200mL及び10%パラジウム活性炭素1.06g(0.001mol)を仕込み、水素雰囲気下で、24時間攪拌した。パラジウム活性炭素を吸引濾過で除いた後、1Lビーカーに移した反応溶液に水500mLを加え、析出した固体を吸引濾過で回収した真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶10.3gを得た。
【0047】
この生成物をC1とした。生成物C1を、1H−NMR、マススペクトル、元素分析で分析した結果は次の通りであった。
生成物の元素分析:
(実験値) C:63.3%,H:3.3%,O:33.4%,
(理論値) C:63.0%,H:3.5%,O:33.5%
生成物C1の1H−NMRスペクトルを図1に示す。
分析結果より、得られた生成物C1は下記式(7)で表される目的のジカルボン酸化合物であることが確認された。得られたC1の収率は、72%であった。
【0048】
【化7】
【0049】
(実施例2)
冷却管及び撹拌装置付きのセパラブルフラスコ(容量:200mL)に、3−ヒドロキシ安息香酸13.8g(0.10mol)、ブロモメトキシメタン12.5g(0.10mol)、炭酸カリウム6.9g(0.05mol)及びN,N−ジメチルホルムアミド100mLを仕込み、攪拌下120℃で4時間反応させ、塩が析出した反応溶液を得た。析出した塩を吸引濾過で除いた後、反応溶液を1Lビーカーに移し、これに水300mLと酢酸エチル200mLを加え、分液ロートで有機層を回収した。回収した溶液に硫酸マグネシウムを加えて水分を除去した後、溶媒を減圧下で留去し、3−ヒドロキシ安息香酸メトキシメチルを得た。
次に、窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、イソフタロイルクロライド12.2g(0.06mol)を仕込み、上記で得た3−ヒドロキシ安息香酸メトキシメチルを9.11g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて、攪拌下、0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃で、さらに5時間攪拌を続けた後、生成物を、300mLの水に添加し、析出した固体を吸引濾過で回収した。
撹拌装置付きのセパラブルフラスコ(容量:500mL)に、上記で得られた固体、メタノール100mL及び水100mLを仕込み、60℃で一晩攪拌した。これにより析出した固体を吸引濾過で回収した。上記で得た固体を真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶10.0gを得た。分析結果より、得られた生成物C2は下記式(8)で表される目的のジカルボン酸化合物であることが確認された。
【0050】
【化8】
【0051】
(実施例3)
撹拌装置付きのセパラブルフラスコ(容量:200mL)に、6−ヒドロキシナフトイックアシッド18.8g(0.10mol)、t−ブチルジメチルシリルクロライド15.1g(0.10mol)、トリエチルアミン10.1g(0.10mol)及びピリジン100mLを仕込み、攪拌下30℃で8時間反応した。析出した塩を吸引濾過で除いた後、溶媒を減圧下で留去し、6−ヒドロキシ−2−ナフトエ酸−t−ブチルジメチルシリルを得た。
窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、2,6−ナフタレンジカルボニルジクロライド15.2g(0.06mol)を仕込み、上記で得た6−ヒドロキシ−2−ナフトエ酸−t−ブチルジメチルシリルを15.1g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて攪拌下0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃で、さらに5時間攪拌を続けた後、生成物を300mLの水に添加し、析出した固体を吸引濾過で回収した。
撹拌装置付きのセパラブルフラスコ(容量:500mL)に、上記で得られた固体、酢酸150mL、テトラヒドロフラン50mL,及び水50mLを仕込み、60℃で一晩攪拌した。これにより、析出した固体を吸引濾過で回収した。上記で得た固体を、真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶14.7gを得た。分析結果より、得られた生成物C3は下記式(9)で表される目的のジカルボン酸化合物であることが確認された。
【0052】
【化9】
【0053】
(実施例4)
撹拌装置付きのセパラブルフラスコ(容量:200mL)に、4−ヒドロキシベンゾイックアシッドナトリウム塩16.1g(0.10mol)、ベンジルクロロメチルエーテル15.7g(0.10mol)及びヘキサメチルリン酸トリアミド100mLを仕込み、攪拌下30℃で8時間反応させ、塩が析出した反応溶液を得た。析出した塩を吸引濾過で除いた後、反応溶液を1Lビーカーに移し、水300mLと酢酸エチル200mLを加え、分液ロートで有機層を回収した。回収した溶液に、硫酸マグネシウムを加えて水分を除去した後、溶媒を減圧下で留去し、4−ヒドロキシ安息香酸ベンジロキシメチルを得た。
窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、2,2’−ビフェニルジカルボニルジクロライド16.8g(0.06mol)を仕込み、4−ヒドロキシ安息香酸ベンジロキシメチルを12.9g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて攪拌下0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃で、さらに5時間攪拌を続けた後、生成物を300mLの水に添加し、析出した固体を吸引濾過で回収した。
撹拌装置付きのセパラブルフラスコ(容量:500mL)に、上記で得られた固体、0.2mol/L塩酸水溶液50mL及びテトラヒドロフラン250mLを仕込み、60℃で一晩攪拌した。これにより、析出した固体を吸引濾過で回収した。上記で得た固体を真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶12.1gを得た。分析結果より、得られた生成物C4は下記式(10)で表される目的のジカルボン酸化合物であることが確認された。
【0054】
【化10】
【0055】
(実施例5)
窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、1,4−シクロヘキシルジカルボニルジクロライド12.6g(0.06mol)を仕込み、エチル−4−ヒドロキシシクロヘキサンカルボキシレートを8.60g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて攪拌下0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃でさらに5時間攪拌を続けた。得られた反応溶液を1Lビーカーに移し、水300mLと酢酸エチル200mLを加え、分液ロートで有機層を回収した。回収した有機層に、硫酸マグネシウムを加えて水分を除去した後、溶媒を減圧下で留去し、反応物を回収した。
撹拌装置付きのセパラブルフラスコ(容量:500mL)に、上記で回収した反応物、0.2mol/L水酸化ナトリウム水溶液50mL及びメタノール250mLを仕込み、30℃で一晩攪拌した。これにより析出した固体を吸引濾過で回収した。上記で得た固体を真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶8.94gを得た。分析結果より、得られた生成物C5は下記式(11)で表される目的のジカルボン酸化合物であることが確認された。
【0056】
【化11】
【0057】
実施例1〜5の合成結果と、得られた生成物のマススペクトル(MSスペクトル)及び元素分析による分析結果を表1にまとめた。
【0058】
【表1】
【0059】
(比較例1)
非特許文献、Macromolecules 1983,16,1034に記載の方法に従い、実施例1と同様の構造を有するジカルボン酸C1の合成を行った。
テレフタル酸と臭化ベンジルとを反応させ、テレフタル酸ジベンジルを合成した後、アルカリ加水分解によりテレフタル酸モノベンジルとし、塩化チオニルと反応させることで、テレフタル酸モノベンジルクロライドを合成した。次いで、p−ヒドロキシ安息香酸ベンジルとテレフタル酸モノベンジルクロライドとにより、エステル結合形成反応を行った。次に、得られたベンジル−4−(カルボベンゾキシ)フェニルテレフタレートを、トリフルオロ酢酸中、臭化水素による加溶媒分解により、脱保護反応を行った。析出した固体を吸引濾過で回収した。真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶5.00gを得た。得られた結晶を1H−NMR、マススペクトル、元素分析で分析した結果、目的の化合物C1が得られた。(収率35%)
【0060】
(比較例2)
窒素置換した撹拌装置付きのセパラブルフラスコ(容量:200mL)に、N−メチル−2−ピロリドン80mL、テレフタロイルクロライド12.2g(0.06mol)を仕込み、p−ヒドロキシ安息香酸を6.90g(0.05mol)、ピリジン9.49g(0.12mol)及びN−メチル−2−ピロリドン50mLを予め溶解させ、滴下ロートを用いて攪拌下0〜10℃で1時間かけてゆっくりセパラブルフラスコに滴下した。滴下終了後、0〜10℃でさらに5時間攪拌を続けた後、生成物を300mLの水に添加し、析出固体を吸引濾過で回収した。真空乾燥後、酢酸/水混合溶液で再結晶を行い、白色結晶を得た。得られた結晶を1H−NMR、マススペクトル、元素分析で分析した結果、実施例1と同様の構造を有する化合物C1、テレフタル酸、2分子のp−ヒドロキシ安息香酸がエステル結合形成反応を起こした生成物である、ビス(カルボキシフェニル)テレフタレートの混合物が得られた。(純度85%、収率70%)
【0061】
比較例1では、多段階に及ぶ保護・脱保護反応を繰り返すため、本発明の製造方法よりも反応工程がかかるうえ、コストや収率の観点からも好ましくない。
比較例2では、カルボキシル基の保護がなされていないため、本発明の製造方法よりも反応の選択性が低下するため、純度の観点から好ましくない。
【産業上の利用可能性】
【0062】
本発明によれば、短い反応工程かつ、高収率でジカルボン酸化合物を得ることができ、本発明のジカルボン酸化合物は、ポリエステル樹脂、ポリアミド樹脂、ポリアリレート樹脂、ポリベンゾオキサゾール樹脂及びポリベンゾチアゾール樹脂などの原料として有用である。
【図面の簡単な説明】
【0063】
【図1】生成物C1の1H−NMR(400MHz, DMSO−d6)スペクトル
【特許請求の範囲】
【請求項1】
一般式(3)で表されるジカルボン酸化合物を製造する方法であって、一般式(1)で表されるエステル化合物と、一般式(2)で表される酸ジクロライド化合物とを反応させることを特徴とする、ジカルボン酸化合物の製造方法。
【化1】
【化2】
【化3】
[式中、R1及びR3は、それぞれ、置換もしくは無置換の芳香族基、又は置換もしくは無置換の脂肪族基を示し、R2は、保護基を示す。]
【請求項2】
一般式(1)で表されるエステル化合物が、前記一般式(1)におけるR1として芳香族基を有するももである、請求項1に記載のジカルボン酸化合物の製造方法。
【請求項3】
一般式(2)で表される酸ジクロライド化合物が、前記一般式(2)におけるR3として、芳香族基を有するものである、請求項1又は2に記載のジカルボン酸化合物の製造方法。
【請求項4】
一般式(1)で表されるエステル化合物が、一般式(4)で表されるエステル化合物である、請求項1〜3のいずれか1項に記載のジカルボン酸化合物の製造方法。
【化4】
[式中、R4は保護基を示す。]
【請求項5】
一般式(1)で表されるエステル化合物が、一般式(5)で表されるエステル化合物である、請求項1〜4のいずれか1項に記載のジカルボン酸化合物の製造方法。
【化5】
【請求項6】
前記一般式(1)で表されるエステル化合物と、前記一般式(2)で表される酸ジクロライド化合物との反応は、−60℃以上20℃以下の温度領域で行うものである、請求項1〜5のいずれか1項に記載のジカルボン酸化合物の製造方法。
【請求項1】
一般式(3)で表されるジカルボン酸化合物を製造する方法であって、一般式(1)で表されるエステル化合物と、一般式(2)で表される酸ジクロライド化合物とを反応させることを特徴とする、ジカルボン酸化合物の製造方法。
【化1】
【化2】
【化3】
[式中、R1及びR3は、それぞれ、置換もしくは無置換の芳香族基、又は置換もしくは無置換の脂肪族基を示し、R2は、保護基を示す。]
【請求項2】
一般式(1)で表されるエステル化合物が、前記一般式(1)におけるR1として芳香族基を有するももである、請求項1に記載のジカルボン酸化合物の製造方法。
【請求項3】
一般式(2)で表される酸ジクロライド化合物が、前記一般式(2)におけるR3として、芳香族基を有するものである、請求項1又は2に記載のジカルボン酸化合物の製造方法。
【請求項4】
一般式(1)で表されるエステル化合物が、一般式(4)で表されるエステル化合物である、請求項1〜3のいずれか1項に記載のジカルボン酸化合物の製造方法。
【化4】
[式中、R4は保護基を示す。]
【請求項5】
一般式(1)で表されるエステル化合物が、一般式(5)で表されるエステル化合物である、請求項1〜4のいずれか1項に記載のジカルボン酸化合物の製造方法。
【化5】
【請求項6】
前記一般式(1)で表されるエステル化合物と、前記一般式(2)で表される酸ジクロライド化合物との反応は、−60℃以上20℃以下の温度領域で行うものである、請求項1〜5のいずれか1項に記載のジカルボン酸化合物の製造方法。
【図1】

【公開番号】特開2007−277137(P2007−277137A)
【公開日】平成19年10月25日(2007.10.25)
【国際特許分類】
【出願番号】特願2006−104210(P2006−104210)
【出願日】平成18年4月5日(2006.4.5)
【出願人】(000002141)住友ベークライト株式会社 (2,927)
【Fターム(参考)】
【公開日】平成19年10月25日(2007.10.25)
【国際特許分類】
【出願日】平成18年4月5日(2006.4.5)
【出願人】(000002141)住友ベークライト株式会社 (2,927)
【Fターム(参考)】
[ Back to top ]