
ジスルフィド結合により安定化された機能的可溶性MHCクラスIIヘテロ二量体
本発明は、ジスルフィド結合により安定化された組換えMHCクラスII分子に関する。具体的には、本発明は、(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分とを含む組換えMHCクラスII分子であって、(i)および(ii)が、機能的ペプチド結合ドメインを提供し、(i)および(ii)が、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基が天然MHCクラスIIのα2およびβ2ドメインに存在しない、組換えMHCクラスII分子を提供する。原核系でこれらの分子を作製する方法およびこれらの分子の各種の使用がさらなる態様を形成する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ジスルフィド結合により安定化された組換えMHCクラスII分子に関する。
【背景技術】
【0002】
主要組織適合複合体(MHC)分子は、脊椎動物免疫系の中心的な構成要素であり、すべての有核細胞表面に存在する。MHCは2つの主要な型、すなわちMHCクラスIおよびクラスIIとして存在する。両型とも、所与のMHCを発現する細胞自体の中で、T細胞エピトープと呼ばれるタンパク質分解処理されたペプチドとともに機能的複合体を形成するということは重要である。生じたペプチド/MHC(pMHC)複合体は、次いで細胞表面の膜貫通型の複合体となり、この現象は抗原提示と呼ばれる。次いで、細胞表面に結合したpMHCは、その同族パートナー、すなわちTリンパ球表面に存在するT細胞受容体(TCR)と相互作用し得る。
【0003】
pMHC−TCR相互作用は適応免疫において極めて重要な役割を果たしているため、基礎および応用科学の分野では、細胞および分子の両レベルで理解し、組換え型の両分子を入手することに大きな注目が集められている。さらに、その系の生態を研究し理解することを可能にするためにも、また新規な治療剤および診断剤を開発するためにも、こうした組換え分子を利用できることが極めて重要であるということも理解されつつある。
【0004】
多くの重要な医学的状態では、患者の免疫系の活性を調節する治療的介入が必要である。例えば自己免疫疾患およびアレルギーでは、免疫系の亢進および慢性炎症を抑える必要がある。これに対し、免疫促進は、免疫細胞を癌性細胞に対して活性化して標的化する、感染症および癌に適切なアプローチである。さらに、移植レシピエントは通常、免疫抑制を必要とする。同時にこれにより、膨大な量の免疫調節剤が開発されるようになり、現在では数十億ドル規模の産業となっている。
【0005】
これらの疾患の免疫的構成要素を理解し、新たな治療法をスクリーニングする上で鍵となるのは、抗原提示細胞とT細胞の間の相互作用、つまり、より具体的にはMHCクラスIおよびII分子とTCRの間の相互作用である。クラスIIMHCは外来性のペプチドと特異的に結合し、それをCD4+Tヘルパー細胞(TH細胞)に提示する。次いでTH細胞が活性化され、各種サイトカインを分泌するエフェクター細胞となる。これらのサイトカインは、脅威への対処に関与する他の様々な免疫細胞を活性化する。このシステムが機能不全に陥ると、例えば、自己免疫疾患を引き起こしたり、癌細胞が生き残って分裂したりする可能性がある。このように、自己免疫疾患はMHCとの強い関連性および標的器官へのT細胞浸潤を特徴とする。
【0006】
免疫調節剤スクリーニングのためのプラットフォームには、安定で完全に機能的な可溶性MHCクラスII分子が必要である。重要なのは、可溶性MHCクラスII分子の作製は現在、分子安定性の欠如という深刻な問題に阻まれているということである。
【0007】
ここ数年の間に、可溶性MHCクラスI分子を四量体として作製する能力(テトラマー技術)により、基礎および応用免疫学が大きな変化を遂げた(Constantin et al.,2002,Biological Research for Nursing,4:115−127)。テトラマー技術により、抗原およびT細胞応答の両方に関して特定の方法で、主としてフローサイトメトリーにより評価される免疫応答の過程を追跡する能力が大幅に向上したというのがその理由である。またこの能力は、免疫系のより一層深い理解にもつながり、新規な診断ツールを実際に生み出す可能性もある。
【0008】
組換えMHCクラスIの作製に関する技術的問題点の多くが解決されてきたのと同様に、四量体試薬はこれまで、ほとんどがMHCクラスI分子に限られてきた。実際、MHCクラスII分子に関しては、この課題が極めて困難であることが証明されている。したがって、独立した例が文献中にも、また市販の試薬としても見られるが、利用できるMHCクラスII四量体作製の一般的なプロトコルは現在のところない(Vollers,S.およびStern,L.,2008,Immunology 123:305−313)。MHCクラスII四量体が利用可能な少数例では、MHCクラスII四量体が広範囲で使用され、疾患発症の理解に大きな影響を与えた。したがって、組換えMHCクラスI作製の成功により既に示されている影響を考えれば、新規なMHCクラスII作製手段にさらに力を注ぐ強く明確な学術的・商業上的動機が存在することがわかる。しかし、現在直面している問題を考えれば、成功するか否かは定かではない。
【0009】
部分的に不明である理由により、MHCクラスII分子は可溶型の安定な組換え分子として作製することが特に難しいことが証明されている。天然の分子はα鎖とβ鎖を含む非共有結合性の膜貫通型ヘテロ二量体であり、αおよびβの両鎖は膜貫通領域を有し、免疫グロブリン(Ig)スーパーファミリーに属する。各鎖の細胞外部分は、それぞれ約90個のアミノ酸残基からなる2つのドメインから構成され、そのうちの2つの膜遠位ドメインであるα1ドメインとβ1ドメインが、T細胞エピトープのペプチド結合性に不可欠な、互いに格子状になったα/β構造を形成している。2つの膜近位ドメインであるα2ドメインとβ2ドメインは、ともに分離したIgドメインを形成している。αおよびβの両鎖では、約20個のアミノ酸残基の配列が細胞膜を横断し、膜の細胞質側にはごく短いペプチドセグメントがある。
【0010】
α鎖とβ鎖の二量体化は、(i)膜貫通セグメント、(ii)ペプチド結合および(iii)膜にある推定上の付属構成要素により生じると考えられている。したがって、MHCクラスII分子を一度その天然の状態から切り離し、可溶性分子として作製すると、本来の低い安定性と非常に低い生産レベルに悩まされることが多い。さらには大規模で供給源を要する、場合に応じた最適化を行わなければならない。
【0011】
また、上述の二量体化のための必要事項を考慮すれば、任意の非天然の状態、すなわち、細胞が天然でMHCクラスII分子を発現するような細胞膜と関連した産生以外の任意の状態でMHCクラスII分子を作製するための一般的方法、例えば、他の生物学的存在、細胞または粒子の表面に示される不可溶性分子として、例えば、ウイルスキャプシドタンパク質との融合によりファージ表面で発現される不溶性分子として作製することは、容易とは言い難い。
【0012】
したがって、安定化されたMHCクラスIIヘテロ二量体を作製することができる一般的な戦略は、現在のところ存在しない。しかし、すべて各場合に特有の成功例を示す各種の研究が行われており、これらの例は、むしろこの課題の複雑さを反映している。そのような例は、(i)脂質テザー(GPIアンカー)の使用による真核細胞表面での膜結合MHCクラスIIヘテロ二量体の異所性発現(Wettstein et al.,1991,J.of Exp.Medicine,174:219−228を参照されたい);(ii)昆虫細胞でのMHCクラスII外部ドメイン発現(Wallny et al.,1995,Eur.J.Immunology,25:1262−1266を参照されたい);(iii)昆虫細胞での産生と組み合わせた、MHCクラスII分子C−末端へのロイシンジッパーのような異種二量体化モチーフの導入(Quarsten et al.,2001,J.Immunol.,167:4861−4868およびCrawford et al.,2006,Immunological Reviews,210:156−170を参照されたい);(iv)昆虫細胞での産生と組み合わせた、抗体/MHCクラスIIキメラの産生(Casares et al.,1997,Protein Engineering,10:1295−1301を参照されたい);(v)封入体からのリフォールディング法により機能的pMHC三量体の形成が可能な細菌発現系の使用(Arimilli et al.,1995,J.Biol.Chem.,270:971−977を参照されたい);または(vi)封入体からのリフォールディング法により機能的pMHCの形成が可能な、α1ドメインとβ1ドメインのみからなる短縮された細菌産生一本鎖MHCクラスII形式の使用(Burrows et al.,1999,Protein Engineering,12:771−778を参照されたい)である。さらに、Landais et al.,2009,J.Immunol.,183:7949−7957 には、安定化されたマウスI−AdOVA MHCクラスII四量体作製のための、内部の人工的なジスルフィド架橋を外来性ロイシンジッパーとともに用いる昆虫細胞発現系が記載されている。重要なのは、改変による発現レベルの増加にもかかわらず、安定性が見かけ上増加した上記分子は、いずれも特異的なT細胞染色を示さなかったということである。
【発明の概要】
【0013】
本明細書に記載の戦略は、あらゆるMHCクラスII分子に適用可能な一般的戦略を提供するだけでなく、原核発現系でも実施可能であり、かつロイシンジッパーのような異種性/外来性の二量体化モチーフを使用する必要がないため、かかる先行技術の方法よりも優れた重要な利点を提供する。さらに、本明細書に記載の戦略は、導入された改変に直接起因する、特異的T細胞染色として示される機能性の増加につながる。
【0014】
しかし驚くべきことに、本発明者らは、組換えMHCクラスII分子に関連した不安定性の問題を克服する、または大幅に軽減することができる一般的方法を特定した。この戦略では、αおよびβ鎖の細胞外部分が存在し、かつα2−β2ドメイン接合点を用いて2つの鎖を結合する操作された/人工的なジスルフィド架橋により、α鎖とβ鎖のヘテロ二量体が安定化されている、組換えMHCクラスII分子を作製する。ジスルフィド結合は、分子が完全に機能する安定なコンホメーションを固定する。MHCクラスIIヘテロ二量体を安定化させるこの戦略は、汎用性があり、また単一の組換え形式に限定されないため、上で議論したような広範で供給源を要する各場合に応じた最適化の必要性が回避されるという点が重要であり、また有利である。さらにこの戦略は、数多く既存の方法とは異なり、原核発現系/宿主で実施することができる。原核宿主には、正確なジスルフィド架橋形成に一般に必要とされる真核細胞の複雑な仕組みがなく、したがって機能的分子が形成されないということを考えれば、このこと自体が驚くべきことである。本発明は、免疫調節剤の探索および開発の分野の妨げとなっている現在の問題点を解決するだけでなく、T細胞を開発および改変し、それを治療法として使用するための重要なツールとなるであろう。
【0015】
したがって、本発明の一態様では、
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含む組換えMHCクラスII分子が提供され、
(i)および(ii)は、機能的ペプチド結合ドメインを提供し、(i)および(ii)は、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基は、天然MHCクラスIIのα2およびβ2ドメイン中には存在しない。
【0016】
上で述べたように、2つ以上のポリペプチドから構成され、膜貫通ドメインを有するMHCクラスII分子のようなタンパク質は、特にその膜貫通領域およびあるいは膜に付属する構成要素により安定化される場合が多いため、天然の膜から外れた状態で、かつ特に可溶型において作製するのが困難なことがある。このことはMHCクラスII分子の場合に当てはまり、文献にもそれが反映されており、MHCクラスII分子が不安定であり、十分な収率で作製できない、またはペプチドを認識してそれと結合することができないことが文献で報告されている。したがって、本発明の分子は安定しており、かつペプチドを認識してそれと結合することもでき、またT細胞の染色も可能であるため、先行技術を上回る大きな進歩を示すものである。
【0017】
上で議論したように、天然のMHCクラスII分子はα鎖とβ鎖を含み、両鎖とも膜貫通領域を有し、免疫グロブリン(Ig)スーパーファミリーに属する。各鎖の細胞外部分は、それぞれ約90個のアミノ酸残基からなる2つのドメインから構成され、そのうちの2つの膜遠位ドメインであるα1ドメインとβ1ドメインが、T細胞エピトープのペプチド結合性に不可欠である、互いに格子状になったα/β構造を形成している。2つの膜近位ドメインであるα2ドメインとβ2ドメインは、ともに分離したIgドメインを形成している。αおよびβの両鎖では、約20個のアミノ酸残基の配列が細胞膜を横断し、膜の細胞質側にはごく短いペプチドセグメントがある。
【0018】
本発明の分子は、MHCクラスIIα鎖の細胞外部分の全部または一部分と、MHCクラスIIβ鎖の細胞外部分の全部または一部分とを含む。MHCクラスIIα鎖の細胞外部分は、シグナル配列と、膜遠位α1ドメインと、膜近位α2ドメイン(分離したIgドメインを形成している)とを含む。膜貫通ドメインとα2ドメインの間にスペーサー領域も存在する。同様に、MHCクラスIIβ鎖の細胞外部分は、シグナル配列と、膜遠位β1ドメインと、膜近位β2ドメイン(分離したIgドメインを形成している)と、スペーサー領域とを含む。
【0019】
したがって、本明細書で使用される「MHCクラスIIα鎖の細胞外部分」という用語は、α鎖の膜貫通ドメインまたは細胞質ドメインを包含しない。実際、本発明の好ましい実施形態では、組換えMHCクラスII分子は、前記膜貫通ドメインのアミノ酸残基(すなわち、専用の膜貫通エクソンによりコードされるアミノ酸)または前記細胞質ドメインのアミノ酸残基のいずれも含まない。
【0020】
したがって同様に、本明細書で使用される「MHCクラスIIβ鎖の細胞外部分」という用語は、β鎖の膜貫通ドメインまたは細胞質ドメインを包含しない。実際、本発明の好ましい実施形態では、組換えMHCクラスII分子は、前記膜貫通ドメインのアミノ酸残基(すなわち、専用の膜貫通エクソンによりコードされるアミノ酸)または前記細胞質ドメインのアミノ酸残基のいずれも含まない。
【0021】
αおよび/またはβ鎖のすべての前記細胞外部分が本発明の組換え分子(すなわち、シグナルペプチド、α1/β1ドメイン、α2/β2ドメインおよびスペーサー領域)中に存在し得る。しかし、組換え分子が、適当なペプチド、例えばT細胞エフェクターペプチドと結合する能力に関して依然として機能的である限り、また前記分子が、人工的な(非天然の)システイン残基を、組換えMHCクラスII分子の安定化に機能するジスルフィド結合の形成に使用可能な形態または立体配置で含む限り、代わりにαおよび/またはβ鎖の細胞外部分の一部分だけが本発明の分子中に存在していればよい。
【0022】
シグナルペプチドは、特にMHCクラスII分子を原核細胞で発現させる場合、本発明の組換えMHC分子のαおよび/またはβ鎖中に欠けていてもよい。適当な数のN末端アミノ酸残基を欠くこのような構築物は、当業者が容易に設計し作製することができる。またかかる欠如が機能に対して、例えば、ペプチドと結合するまたはT細胞を活性化もしくは染色する能力、あるいは分子の安定性に対して影響を及ぼすか否かを容易に試験することもできる。
【0023】
またスペーサー領域も、例えば、α鎖スペーサーおよびβ鎖スペーサーの9個のアミノ酸がいずれも含まれていない実験例に示されるように、その全体が欠けていても、または短縮されていてもよい。ここでも、適当な数のスペーサーアミノ酸残基を欠くこのような構築物は、当業者が容易に設計し作製することができる。またかかる欠如が機能に対して、例えば、ペプチドと結合するまたはT細胞を活性化もしくは染色する能力、あるいは分子の安定性に対して影響を及ぼすか否かを容易に試験することもできる。
【0024】
本発明の好ましい分子は、α1ドメインの少なくとも一部分とβ1ドメインの少なくとも一部分とを含む。ただし、これらのドメインがペプチドと結合する機能、または本明細書に記載の他の機能、例えば、かかるペプチドをT細胞受容体(TCR)に提示する機能が影響を受けない。さらに組換えMHCクラスII分子が全体として、ペプチドをT細胞受容体(TCR)に提示するために、かかるペプチドと結合する能力または本明細書に記載の他の機能に関して、依然として機能的でなければならない。好ましくは、完全または完全長のα1および/またはβ1ドメインが存在するか、または実質的に完全または実質的に完全長のα1および/またはβ1ドメインが存在し、前記実質的に完全または実質的に完全長のドメインは、例えば、ペプチドをT細胞受容体(TCR)に提示するために、かかるドメインがかかるペプチドと結合する機能、または本明細書に記載の他の機能に影響を与えない、天然配列からの変化、例えば、アミノ酸の付加、欠失または置換を含む。かかる機能に影響を与えずに変異させる、修飾するまたは削除することができるアミノ酸の決定は、当業者の技能の範囲内であろう。MHCクラスII分子が保持するべき他の好ましい機能は、T細胞を活性化する能力、より好ましくは、T細胞を染色する能力である。
【0025】
したがって、本発明におけるこれらの好ましい分子は、例えば、ペプチドをTCRに提示する、T細胞を活性化する、またはT細胞の染色を可能にするために、かかるペプチドと結合することができるように、十分なα1およびβ1ドメイン残基を含む。
【0026】
本発明の他の好ましい分子は、α2ドメインの少なくとも一部分とβ2ドメインの少なくとも一部分とを含む。ただし、ジスルフィド結合を形成するために使用される非天然のシステイン残基が存在する。かかるシステイン残基は、システイン残基間にジスルフィド架橋を形成することができるように、また組換えMHCクラスII分子の安定化に作用することができるように、互いに適切な方向および距離で存在している必要がある。さらに組換えMHCクラスII分子が全体として、ペプチドをT細胞受容体(TCR)に提示するために、かかるペプチドと結合する能力または本明細書に記載の他の機能に関して、依然として機能的でなければならない。好ましくは、完全または完全長のα2および/またはβ2ドメインが存在するか、または実質的に完全または実質的に完全長のα2および/またはβ2ドメインが存在し、前記実質的に完全または実質的に完全長のドメインは、α2ドメインとβ2ドメイン間のジスルフィド結合の形成およびその結果としての組換えMHCクラスII分子の安定化に影響を与えず、かつα2またはβ2ドメインのフォールディングに悪影響を及ぼさない、天然配列からの変化、例えば、アミノ酸の付加、欠失または置換を含む。かかる機能に影響を与えずに変異させる、修飾するまたは削除することができるアミノ酸の決定は、当業者の技能の範囲内であろう。
【0027】
各種の構造的および機能的ドメインアミノ酸位置ならびにαおよびβMHCクラスII鎖の領域は、当該技術分野において公知であり記載されている(例えば、Burrows et al.,1999,上記)。また各種ドメインの配置も図3に示されており、このような情報を用いて、本明細書に記載の本発明の組換え分子(例えば、図7を参照されたい)を容易に設計することができる。
【0028】
本発明の組換え分子がペプチドと結合する、T細胞を活性化する、またはT細胞を染色する能力を評価するための適当な機能試験も当業者に公知であり、例えば、Biacoreのような表面プラズモン共鳴(SPR)技術およびFACSのようなフローサイトメトリー技術がこれに含まれるであろう。フローサイトメトリーは生細胞と組み合わせて使用できるため、これを使用することが特に好ましい。
【0029】
本明細書で使用される「ジスルフィド結合」という用語は、MHCクラスIIヘテロ二量体のα鎖のα2ドメイン中に位置するシステイン残基とβ鎖のβ2ドメイン中に位置するシステイン残基との間で形成される(すなわち、鎖内ではなく、鎖間のジスルフィド結合である)、任意のジスルフィド架橋、例えば、操作されたまたは人工的なジスルフィド架橋を表す。前記ジスルフィド結合はα鎖とβ鎖間で共有結合を形成し、MHCクラスIIヘテロ二量体の安定化に作用する。したがって、本発明の組換え分子は安定であるが、完全な機能性を保持している。例えば、本発明の組換え分子は、その同族T細胞受容体リガンドに対する本来の特異性を保持し、好ましくは、T細胞を活性化する、またはT細胞を染色する能力を保持している。
【0030】
安定であるまたは安定化されているという本発明のMHCクラスII分子の特性は、温度変性に対する耐性の増加(例えば、ELISA法、SPR法または円二色法により測定される)のような公知の方法により評価することができる。さらに好ましい試験は、ヘテロ二量体の保存をSDS PAGEゲル上で評価することである。インタクトで安定なジスルフィド結合ヘテロ二量体は、非還元条件下で泳動したSDS PAGEゲル上で容易に確認することができ、この場合、インタクトのヘテロ二量体に対応する適当な分子量の位置にあるバンドを可視化することができる。適当なアッセイを実施例で示す(図5および10を参照されたい)。
【0031】
かかるジスルフィド結合は、天然のMHCクラスIIα2およびβ2ドメインには通常は存在しないシステイン残基間で形成される。したがって、例えば、部位特異的突然変異により天然分子中の適当な非システイン残基からシステイン残基に変異させることにより、システイン残基を操作するかまたは人工的に導入して、α2およびβ2ドメイン内に新たに導入されたシステイン残基間でジスルフィド結合を形成させる。このような結合は内部ジスルフィド結合とも呼ばれる。
【0032】
システインへの変異に適した残基は、天然のMHCクラスIIヘテロ二量体において、約6Å(0.6nm)、7Å(0.7nm)またはそれ未満だけ離れた、例えば、4Å(0.4nm)または5Å(0.5nm)〜6.5Å(0.65nm)または7Å(0.7nm)の範囲、好ましくは、5Å(0.5nm)〜6.5Å(0.65nm)の範囲、最も好ましくは、4Å(0.4nm)または5Å(0.5nm)〜5.6Å(0.56nm)または6Å(0.6nm)の範囲だけ離れた別々のβ炭素を有することが好ましい。変異に好ましい部位は、種間ならびに特定種内のアイソフォーム間およびアイソタイプ間で保存されている。具体的には変異に好ましい部位は、マウスとヒトのMHCクラスII配列間で保存されているか、またはヒトMHCクラスIIアイソタイプ(例えばDP、DQおよびDRなど)間またはマウスアイソタイプ(例えばI−EおよびI−Aなど)間で保存されている。
【0033】
代わりにまたは加えて、結晶構造の3D重ね合わせに基づく構造評価により、変異に好ましい部位を特定する。このようにして、α2ドメインとβ2ドメイン間の接合点を形成している残基を調べることができ、またさらなる解析またはシステイン残基への変異のために、7Å以下(または実際は、上記の他の任意の距離もしくは範囲)の距離だけ離れたβ炭素を有する側鎖を選択することができる。かかる構造評価を行うための方法は当業者に公知であろう。例えば、この解析を行うために、MHCクラスII分子の結晶構造を、例えばRCSB PDBタンパク質データバンクから自由に入手することができる。かかる結晶構造の3D重ね合わせ行うための適当なソフトウェアまたはその他の手段も当該技術分野において、例えば、PyMOL、MOLMOL、DeepViewのような自由に入手できるソフトウェア、またはiSuperposeのような専用のウェブサイトを用いて利用することができる。
【0034】
特に好ましい部位の対は、ジスルフィド結合を形成するようにシステインが導入された、以下に挙げる対のうちの1つ以上、すなわち、Pro96α2−Ser119β2(1位)、Ser95α2−Ser121β2(2位)、Arg94α2−Asn151β2(3位)、Phe148α2−Gly152β2(4位)、Pro96α2−Thr101β2(5位)、Pro96α2−Ser121β2(6位)、Ile106α2−Asn151β2(7位)およびSer95α2−Asp122β2(8位)のうちの1つ以上の対である。これらの対はβ炭素の近接度の順に並べられており、どの対を用いてもよいが、1位〜4位または1位〜3位の対が好ましい。特に1位または2位の対が好ましい。
【0035】
α2およびβ2ドメイン中にあるおよび上記の残基の対におけるアミノ酸位置およびその位置の天然アミノ酸の性質は、マウスI−EアイソタイプのMHCクラスII分子に適している。アミノ酸番号付けは、成熟ペプチドのアミノ酸に関するものである(すなわち、番号付けにはシグナルペプチドは含まれていない)。改変システイン残基の位置を特定するために使用することができる参照配列の例は、IMGTデータベースで得られるI−E配列であり、これを図3に示す(α鎖ではH−2EA*02(配列番号1)およびβ鎖ではH−2EB*01(配列番号2)で示される)。実際、図3では1位、2位および3位の残基の位置に黒い影で印が付してあり、その他の順位の残基は図3から容易にわかる。
【0036】
したがって、別途記述がない限り、本明細書に記載のMHCクラスIIアミノ酸残基の番号付けおよび性質は、Lefranc,M−P.ら,2009(Nuc.Acids Res.,37:D1006−D1012,データベース記事)に記載されているIMGT方式および以下のウェブサイト参考文献:http://imgt.cines.fr;http://www.imgt.org.にあるIMGTデータベースに従っている。関連するGenBankアクセッション番号も実施例に記載されている。例えば、H−2E(マウスI−E)で関連するアクセッション番号はK00971(α鎖)およびAF050157(β鎖)である。
【0037】
本発明の好ましい実施形態では、ジスルフィド結合は、マウスI−Eアイソタイプの成熟ポリペプチドのPro96α2−Ser119β2(1位)、Ser95α2−Ser121β2(2位)もしくはArg94α2−Asn151β2(3位)に対応する残基、または別のMHCクラスIIアイソタイプのこれに相当する位置に位置するシステイン残基間にある。かかるシステイン残基の位置を決定するための参照配列が本明細書に記載されている。
【0038】
上で議論した好ましい部位の対は、1FNG番号付け、すなわち、Pro96α2−Ser118β2(1位)、Ser95α2−Ser120β2(2位)、Arg94α2−Asn150β2(3位)、Phe148α2−Gly151β2(4位)、Pro96α2−Thr100β2(5位)、Pro96α2−Ser120β2(6位)、Ile106α2−Asn150β2(7位)およびSer95α2−Asp121β2(8位)のうちの1つ以上、ならびにIMGT番号付けの両方を用いて、表2にも示されている。1FNG番号付けに関して、表2のアミノ酸番号付けは、生体高分子の三次元構造情報のタンパク質データバンク(PDB)データベース(登録番号PDB ID:1FNG)で番号付けに対応しており、このデータベース登録のβ鎖中の適当な残基の位置が、IMGTデータベース配列のこれに相当する残基よりも1アミノ酸小さいということがわかる。したがって、この命名法は少し異なっている。
【0039】
表2および図3に示されている残基に相当する残基は、他のマウスアイソタイプ、例えばI−Aアイソタイプ、またはヒトアイソタイプにおいて、例えば、Clustalソフトウェアのような適当なソフトウェアを用いたアライメントにより容易に特定することができる。実際にマウスI−Aアイソタイプとのアライメントが図3に示されている。さらに、ヒトMHCクラスIIアロタイプであるHLA−DP、−DQおよび−DRとのアライメントの例も図7に示されている。表2および図3に示されている1位、2位および3位の残基に相当する残基の位置は、α鎖に関しては図7Aおよびβ鎖に関しては図7Bの中で黒い影で印が付してあり、その他の順位の残基の位置は図7から容易にわかる。表2および図3に示されているα2およびβ2の位置は、ヒトHLAレパートリー全体で完全に保存されていることがわかる。
【0040】
したがって、所望の順位のジスルフィド結合を形成するために、図7に示される配列アライメントを上の情報とともに用いて、任意のヒトMHCクラスII対立遺伝子のα2およびβ2ドメインにおいて、システインに変異させるのに適当な残基の位置を容易に特定することができる。同様のアライメント法を用いて、他の任意の種またはアイソタイプにおいて相当する残基を特定することができる。
【0041】
本明細書で議論されるジスルフィド結合による安定化結合は、先行技術で述べられているような、MHCクラスII分子を安定化する他の方法および手段と適合性がある。例えば、ジスルフィド結合を、ロイシンジッパーモチーフのような各種の二量体化モチーフ(例えば、Quarsten et al.,2001およびCrawford et al.,2006(上記)に記載されているような)とともに、またはIg融合(すなわち、例えばCasares et al.,1997(上記)に記載されているような免疫グロブリンのFc部分との融合)とともに用いることができ、また本発明の分子を、それをコードするベクターとともに適当に設計することができる。
【0042】
本明細書の他の箇所に記載されているように、原核宿主、例えば細菌宿主で、本発明の分子の発現または産生させることが好ましく、このような実施形態、具体的には分子を封入体から単離する実施形態では、ロイシンジッパーモチーフまたはその他の二量体化モチーフを使用しないことが好ましい。
【0043】
したがって、本発明の好ましい態様では、
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含み、細菌宿主で発現可能な組換えMHCクラスII分子が提供され、
(i)および(ii)は、機能的ペプチド結合ドメインを提供し、(i)および(ii)は、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基は、天然MHCクラスIIのα2およびβ2ドメイン中には存在しない。
【0044】
別の好ましい態様では、
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含む組換えMHCクラスII分子が提供され、
(i)および(ii)は、機能的ペプチド結合ドメインを提供し、(i)および(ii)は、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基は、天然MHCクラスIIのα2およびβ2ドメイン中には存在せず、さらに前記組換え分子はロイシンジッパーモチーフを含まない。他の実施形態では、二量体化モチーフが全く含まれない。これらの実施形態では、組換えMHCクラスII分子が原核宿主、例えば細菌宿主で発現させることができることが好ましい。
【0045】
本発明のジスルフィド結合による結合を、MHCクラスII分子を安定化させる他の方法および手段とともに使用することができるが、前記ジスルフィド結合は、MHCクラスII分子を安定化させる単独の手段を提供する。実際、このような実施形態が好ましい。本明細書で使用される「MHCクラスII分子を安定化させる単独の手段」という用語は、特定のMHCクラスII分子に天然にまたは本来的に存在するあらゆる安定化以外の、分子を安定化させるための単独または唯一の手段を提供するジスルフィド結合を表す。例えば、本発明のジスルフィド結合は、MHCクラスII分子を安定化させるための単独または唯一の、人工的または操作されたまたは非天然の手段を提供する。したがって、このような実施形態には、当該技術分野において記載されている、ロイシンジッパーまたはその他の二量体化モチーフのような他の安定化の手段の使用は含まれていない。
【0046】
かかるジスルフィド結合が、二量体化モチーフのような非天然の安定化の手段をさらに使用することなくMHCクラスII分子を十分に安定化させるという驚くべき発見は、本明細書に記載のデータに明確に示されており、本明細書では、MHCクラスII分子が繊維状ファージの表面に安定かつ機能的な形態で示される。この発見は、Igフォールドトポロジーで作製される操作された分子が原核宿主で発現されたということを考慮すると、特に驚くべきことであった。天然では生存している真核ドメインでのみ生じるIgフォールドは、その機能的なトポロジーに達するために、天然のシステイン間で形成される保存されたドメイン内S−S架橋を必要とする(Halaby,D.M.,ら,1999,Protein Eng.,12(7):563)。したがって、原核宿主には正確なS−S架橋形成に必要な真核細胞の複雑なシャペロン機構が必然的に欠けているため、MHCクラスII分子がそこで発現された場合、異常なS−S架橋形成が顕著であるため、機能的な発現が行われないということが一般に認められている。したがってこれらの系における、明らかに人工的なS−S架橋形成とまでは言わないが、システイン数の増加を含む操作戦略は、一般に発展不可能であると考えられている。ここで本発明者らは、共有結合による二量体形成(図5および10)により示されるようにファージ上に機能的分子が示され、それらが同族リガンドとの特異的結合(図6および9)を示しているため、このことが当てはまらないという証拠を明確に記載する。
【0047】
一実施形態では、本発明のMHCクラスII分子のαおよびβ鎖は、鎖内ジスルフィド結合、例えば、天然のシステイン残基間に存在する天然の鎖内ジスルフィド結合も含む。このような天然の鎖内ジスルフィド結合は、例えば、α2およびβ2ドメインのIgフォールドトポロジーを形成するために、これらのドメイン中に存在していてもよい。さらに、MHCクラスII分子の大部分はβ1ドメイン内に、βシート底面とαへリックス部分とを結び付けるドメイン内ジスルフィド架橋を有する。さらに重要なのは、多くのMHCクラスII分子が、どのジスルフィド架橋形成にも関与しないが、新たに導入された非天然のシステイン残基と誤ったジスルフィド結合を形成し得る遊離システインを、β1ドメインのβシート底面にさらに含むということである。したがって、本発明の好ましい実施形態、特に原核発現を用いる実施形態では、例えば、全体の構造を保つセリンまたはアラニンのような別の残基への変異により、この残基を除去する。当業者は、任意のMHCクラスII対立遺伝子において、このシステイン残基の位置を容易に特定することができるであろう。例えば、このシステインは、図3Bに示される完全長のIMGT参照配列H−2EB*01(配列番号2)中のβ鎖残基38または成熟IMGT参照配列(すなわち、シグナルペプチドを含まない)中の残基12に対応する。
【0048】
新たに導入された非天然のシステイン残基との誤ったジスルフィド結合の形成にも関与し得るその他のβ1ドメインシステイン(架橋を形成する)も、当業者により容易に位置が特定され得る。例えば、これらは、図3Bに示される完全長のIMGT参照配列H−2EB*01(配列番号2)残基42および106または成熟IMGT参照配列(すなわち、シグナルペプチドを含まない)のそれぞれ残基16および80の位置にある。本発明の別の好ましい実施形態では、1つ以上のこのようなシステイン残基が存在しない。結合形成を防ぐために、1つ以上の適当な天然のシステイン残基をジスルフィド結合形成に関与しない別のアミノ酸残基へ変異させることにより、これを達成することができる。システインと置き換わる残基の例は、分子の全体構造を保つ残基、例えば水素結合を保つ残基であろう。上で議論したSerまたはAlaのいずれかが好ましい選択であろう。
【0049】
したがって、上で議論した1つ以上の天然システイン残基の除去は、天然のシステイン残基と本発明の新たに操作された非天然のシステイン残基との間での誤ったジスルフィド結合形成を防ぐのに役立つであろう。したがって、本発明の好ましい実施形態では、β1ドメイン中の1つ以上の天然システイン残基を除去する。本発明の特に好ましい実施形態では、完全長の参照配列H−2EB*01(配列番号2)の38、42もしくは106の位置に対応する1つ以上のシステイン残基(または成熟参照配列中のそれに相当する残基)、または別のMHCクラスIIアイソタイプのそれに相当する位置にある1つ以上のシステイン残基を除去する。
【0050】
本明細書で使用される「機能的ペプチド結合ドメイン」という用語は、ペプチド、例えばT細胞エフェクターペプチドまたは抗原ペプチドと結合することができる、本発明の組換えMHCクラスII分子中のドメインを表す。ペプチドとのかかる結合は検出可能なレベルであるべきであり、また結合を検出するための適当な方法は当業者に公知であろう(例えば、表面プラズモン共鳴(SPR)技術またはフローサイトメトリー技術)。いくつかの実施形態では、前記ペプチドとは、前記ペプチドのTCRへの提示が可能なように、または少なくとも前記ペプチドのTCRへの提示が可能か否かを試験できるように結合する。
【0051】
好ましくは、かかるペプチドは、TCRとpMHC複合体の結合が可能となるようにMHCクラスII分子と結合または会合する。より好ましくは、TCRとのかかる相互作用により、pMHC複合体を認識するT細胞が染色されるか、または他の方法で可視化される。好ましくは、かかるペプチド結合ドメインは、ペプチドと結合し、次いでTCRを介してT細胞の活性化を引き起こすことができる。例えば、かかるペプチド結合ドメインは、Tヘルパー細胞の、例えばサイトカイン(IL−2など)の分泌または増殖の誘導(例えば、BrdU取込みによりcpmとして測定される)を誘発することができる。かかるペプチド結合ドメインは一般に、MHCクラスII分子のα1およびβ1ドメイン由来の残基により形成さる。
【0052】
本発明のMHCクラスII分子を、空の形態または未負荷の形態で、すなわち、ペプチドが上記ペプチド結合ドメインと結合していない状態(すなわち、無ペプチド)で提供し得る。この場合、後でMHCクラスII分子に適当なペプチドをインビトロで負荷することができる。このようなインビトロでの負荷は、クラスIIヘテロ二量体の安定性を補助するために、例えば、短いリンカーを介してMHCβ鎖と共有結合した融合タンパク質としてペプチドを作製することにより、ペプチドが付加した状態で作製しなければならない多くの先行技術のMHCクラスII分子よりも有利である。インビトロ負荷により、異なるMHC−ペプチド複合体それぞれに対する異なる発現ベクターを得る必要なしに、真に汎用性のあるMHCクラスII分子を作製することができる。
【0053】
本発明のいくつかの実施形態では、組換えMHCクラスII分子は、上記ペプチド結合ドメインと結合または会合するペプチドを有する。MHCクラスII分子のα1およびβ1ドメイン由来の残基により形成されたペプチド結合ドメインとの結合または会合に適した任意のペプチドを使用することができる。一般にこのようなペプチドは、12〜25マーであり、通常、α1およびβ1ドメインにより形成される溝の両端からはみ出ている。
【0054】
天然のMHCクラスII分子では、かかるペプチドは外来抗原に由来する。本発明では、任意のこのようなペプチドを使用し得る。例えば、当該技術分野において、MHCクラスII分子により提示されてTCR結合およびT細胞活性化を生じる特定のT細胞エフェクターペプチドがいくつか同定され記載されており、そのいずれも本発明とともに使用し得る。特に、MHCクラスII分子上で提示される特定のペプチドが、特定の疾患と関連するものとして同定されており、組換えMHCクラスII分子がペプチドと会合する本発明の実施形態では、このような疾患特異的なペプチドが好ましい。このようなペプチドは当該技術分野においていくつか記載されており、将来的には他のペプチドも同定されるであろうが、上記ペプチドはいずれも、本明細書に記載のMHCクラスII分子との使用に適するであろう。
【0055】
MHCクラスII分子のペプチド結合ドメインと会合または結合するペプチドの代表例には、HLA−DQ2 MHCクラスII分子上で提示されることがわかっており、セリアック病と関連するヒトα−II−グリアジン(N−PQPELPYPOPE−C);HLA−DR4 MHCクラスII分子上で提示されることがわかっており、関節リウマチと関連するヒトhCkappaaa40-48(N−WKIDGSERQ−C);HLA−DP1 MHCクラスII分子上で提示されることがわかっており、破傷風と関連するヒトTTaa947-967(N−FNNFTVSFWLRVPKVSASHLE−C);およびマウスI−EdクラスII分子上で提示されることがわかっている、H.インフルエンザのヘマグルチニン(HA)に由来するペプチド(aa110−120:N−SFERFEIFPKE−C)がある。
【0056】
あるいは他のペプチドを使用して、例えば、そのペプチドがペプチド結合ドメインと結合することができるか否か、またそのペプチドにより、TCRを介したT細胞結合および好ましくはT細胞染色が可能であるか否か、またはT細胞活性化が可能であるか否かを確認してもよい。このように、本発明の分子を用いて、T細胞エピトープとして働く、これまで未知であった新規なペプチド(例えば、抗原ペプチド)を同定することができる。
【0057】
あるいは、組換えMHCクラスII分子を、無関係な、すなわちT細胞エフェクターでないペプチド(いわゆる「スタッファー」ペプチド)(このペプチドをMHC分子と結合させているリンカーの切断によりMHCクラスII分子から解離し得る)と会合させ、次いで、インビトロペプチド交換反応で目的とするT細胞エフェクターペプチドと置き換えることができる。
【0058】
任意の適当な方法で、かかるペプチドとMHCクラスII分子との結合または会合を促進することができる。例えば、かかるペプチドがMHCクラスII分子とともに産生されるように本発明の構築物を操作する(例えば、これらを同じ構築物にコードさせる、例えば、これらを適当なリンカー配列によりβMHCクラスII鎖またはαMHCクラスII鎖と共有結合させてコードさせることにより、または同一宿主細胞内の異なる構築物にコードさせることにより)ことにより、適当なTヘルパー細胞による認識に適した前記ペプチドを提示する、またはそれと結合もしくは会合する組換えMHCクラスII分子を産生させることが可能である。このようなペプチド関連分子を作製するための適当な方法は、Kozono et al.(1994)により記載されている(Nature,369:151−154)。Kozonoの方法では18アミノ酸残基のリンカーを使用するが、本発明の好ましい実施形態では、これよりも短いリンカー、例えば15アミノ酸リンカー(例えば、(G4S)3のようなGly−Serリンカー)または6、7もしくは8アミノ酸残基のリンカー(例えば、Gly−Serリンカー、例えばGSGSGS、GGSGSGS、SGSGSGSまたはSGGSGSGSなど)など、最も好ましくは、6アミノ酸残基のリンカーを使用する。
【0059】
本発明は、任意のタイプのMHCクラスII分子に対して一般的に適用可能である。例えば本発明は、任意の種由来のMHCクラスII分子およびその種内のMHCクラスII分子の任意のサブタイプに対して適用可能である。特に、本明細書で特定された、非天然のシステイン残基に変異させて1つ以上の鎖間ジスルフィド結合を形成させることが可能な残基が種間で保存されていることにより、このアプローチの一般性が支えられている。したがって、本発明をあらゆる哺乳動物のMHCクラスII分子、例えば、ヒト、マウス、ラット、ブタ、ヤギおよびヒツジ、特にヒトおよびマウスの分子に適用することができる。例えば、本発明は、DP、DQおよびDRヒトMHCクラスII分子(すなわち、ヒトにおいて同定されているMHCクラスII分子の3つの機能的タイプ)に対して、またマウスI−AおよびI−E分子(例えば、I−EdおよびI−Ek分子、好ましくはI−Ed)に対しても適用可能である。I−AおよびI−E分子の他の例を下の表に示す。
【0060】
【表1】

【0061】
最も好ましいMHCクラスII分子はヒトである。
【0062】
システインへの変異のために特定された残基は種間で保存されているため、当業者であれば、任意の種由来の任意のMHCクラスII分子において、変異のための対応する、同等の適当な残基を容易に特定し、1つ以上の適当な鎖間ジスルフィド結合により安定化された組換えMHCクラスII分子を作製することができるであろう。
【0063】
本発明の一実施形態では、組換えMHCクラスII分子を、細胞または別の生物学的存在もしくはパッケージの表面、例えば、繊維状ファージの表面で発現させる。この型は、本明細書では「不溶性」型または「不溶性」分子と呼ばれることがある。このような実施形態では、一般に本発明のMHCクラスII分子のα鎖またはβ鎖が、当該存在物の表面で通常発現される、または当該存在物の表面に組み込まれているかそれに付随しているタンパク質との融合タンパク質として操作される。
【0064】
ファージ表面タンパク質との融合体としての発現が好ましく、このような実施形態では、本発明のMHCクラスII分子のα鎖またはβ鎖と任意の適当なファージ表面タンパク質との融合が考えられる。好ましい例は、gpIII、gpVIII、gpVIIまたはgpIXとの融合、最も好ましくはgpIIIとの融合である。ファージ粒子表面での本発明のMHCクラスII分子発現の方法論は、当業者の技術の範囲内であり、技術および方法の例が実施例に記載されている。好ましい方法論は、例えば国際公開第09/024591号に記載されている。
【0065】
本発明の別の実施形態では、組換えMHC分子は可溶性分子、例えば、ウイルスキャプシドタンパク質との融合体または生物学的存在の表面との会合または複合体形成を生じる他のタンパク質との融合体などとして細胞または生物学的存在の表面に付随していないまたはそこで発現されない、本発明のMHCクラスII分子である。したがって、可溶性分子の例としては、本発明の分子の(i)および(ii)の細胞外部分のみを含む分子が挙げられ、また分子の溶解性に影響を与えない他の短い構成要素、例えば親和性タグおよび/または二量体化モチーフのような短いC末端伸長などを含めた外部ドメインも挙げられる。作製方法に応じて、かかる可溶性分子は、宿主細胞から分泌されるか、または他の任意の適当な方法により宿主細胞から入手され得る。かかる可溶性分子を、実質的に純粋な形態で、または精製もしくは単離された調製物として得てもよい。例えば、かかる可溶性分子を、実質的に他のタンパク質を含まない形態で作製し得る。
【0066】
本発明の好ましい実施形態では、組換えMHCクラスII分子を、例えば多価性の多量体型で得てもよい。一般に単一のpMHC複合体とT細胞受容体との間の親和性は非常に低いため、かかる多量体型は、MHCクラスII分子とT細胞受容体の結合を可能にするのに有利である場合が多い。かかる多量体型は、複数(2つ以上)の本発明の組換えMHCクラスII分子を含む。複数のMHCクラスII分子はそれぞれ同一であることが好ましい。
【0067】
多量体型のMHCクラスII分子を調製する任意の適当な方法を使用してよく、そのうちのいくつかは、当該技術分野において記載されている(例えば、Vollers et al.,2008(上記)を参照されたい)。多量体型の本発明のMHCクラスII分子を調製するのに特に有利な方法は、繊維状ファージ表面での提示を利用するものである。このような方法では、MHCクラスII分子が融合し付随する適当なファージ構造タンパク質を選択することにより、多量体を作製するためにファージ分子の天然の構造を利用することができる。例えば、gpIII、gpVIIまたはgpIXとの融合、好ましくはgpIIIとの融合を用いて、各ファージ粒子の表面に3〜5コピーのMHCクラスII分子を得ることができる。gpVIIIとの融合を用いて、これよりも多くのコピーを得ることができる(野生型ファージには、約2700コピーのgpVIIIが存在する)。したがって重要なのは、pVIII提示法により、MHCの価数がgpIII提示法および古典的なテトラマー技術に比べて少なくとも一桁分増加するため、感度が大幅に増加するということである。これにより新たな興味深い適用が開拓されるであろう。
【0068】
既知のファージ提示法および構築物設計技術、例えば、ファージミド構築物および改変型のヘルパーファージを用いて、ファージ表面のMHCクラスII分子のコピー数を変化させることができる。ファージ表面に多量体MHCクラスII分子を生成する能力を用いれば、1回の迅速な費用効率の高い工程で多量体MHCクラスII分子の製造または作製が可能となるという点で有利である。Vollers et al.(2008)に記載されているような従来のテトラマー技術では、各構成要素を別々に作製し、混合して複合体を形成し、精製した後に試薬が下流での適用に適した状態になるが、これとは極めて対照的である。
【0069】
したがって、好ましい多量体は、互いに会合した2つ、3つ、4つ、5つまたはそれを上回る組換えMHCクラスII分子を含む。このような会合は、当該技術分野において既知であり記載されている方法を用いて行われ得るが、一般的には、リンカー分子のような別の結合により仲介される。適当なリンカー分子は当該技術分野において公知で、記載されており、特に適切なリンカー分子は、組換え分子が結合することができる複数の結合部位を有する。例えば、それぞれが複数のビオチン結合部位を有する、アビジンおよびストレプトアビジン(またはビオチンと多価的に結合する他の任意の分子)のような複数の結合分子を使用し得る。したがって、当該技術分野で公知の方法(例えば、AviTagまたはその他のBirA基質を用いて酵素的ビオチン化を可能にする)により組換えMHC分子へビオチンを組み込むことにより、多量体、例えば四量体のMHC分子を形成することが可能である。また、例えば、蛍光ストレプトアビジンを用いて、または標識とファージコートタンパク質(例えば、MHCクラスII分子の提示用に選択したものとは異なるタイプのコートタンパク質)との融合により、標識を多量体型に好都合に組み込むこともできる。こうした標識を組み込むことにより、既知の各種技術で四量体を迅速に検出することが可能となる。例えば、蛍光標識により、特に有利な、FACS解析のようなフローサイトメトリー技術を用いることが可能となる。
【0070】
また、本発明のMHCクラスII分子またはその多量体を、平面状または粒子状の固体支持体のような固体支持体、例えば膜、プレートまたはビーズの上にコーティングし得る。このための技術は当該技術分野において公知であり、記載されている。
【0071】
本発明のMHCクラスII分子、および前記分子を安定化させるために、本明細書に記載のようにMHCクラスII分子のα2ドメインとβ2ドメイン間の1つ以上の操作されたジスルフィド結合を用いることは、MHCクラスII分子作製のためのあらゆる先行技術の方法と適合性がある。したがって、本発明は、当該技術分野において記載されている従来の型のMHCクラスII分子のいずれとも適合性があり、作製効率および生成物の安定性を向上させるであろう。特に、本発明により組換えMHCクラスIIの適用性および汎用性が広がるため、分子は、例えば、(i)細菌ペリプラズム内の機能的可溶性分子、(ii)好ましくは、現在の標準的なプロトコルにより得られるもの(最大で出発物質の30%、例えば、Arimilli et al.,1995(上記)を参照されたい)よりも実質的に高い収率で精製した後に、可溶化し機能的な存在にリフォールディングすることができる、細菌封入体の非機能的構成要素、(iii)繊維状ファージ系でファージ提示される構成要素、(iv)真核細胞内のIg融合体、および(v)真核細胞内の可溶性MHCクラスII分子として、ジスルフィド結合により安定化された形態で作製され得る。
【0072】
アプローチ(ii)(封入体アプローチ)およびアプローチ(iii)(ファージ提示アプローチ)は、組換えMHCクラスII分子の効率的な使用で非常に重要な2つの適用を直ちに行うことが可能な点で有利であり、このうちアプローチ(iii)は全く新規なものである。この点に関して、繊維状ファージビリオン上でのpMHCクラスIIの良好な機能的提示法はこれまで達成されたことがなく、ビリオンをわずかなコストで迅速に作製し得ることを除けば、一連の新たなまたは向上した適用、例えば(a)MHCクラスIIに関連したファージ提示ペプチドライブラリー;(b)所与のpMHCクラスIIの組合せの極めて迅速かつ簡便な作製;および(c)四量体のような従来のpMHCクラスII多量体に代わるMHCクラスII分子の多価の繊維状ファージ提示などを開拓するものである。
【0073】
本発明のMHCクラスII分子には、研究試薬として特定の価値および用途がある。このことは、本明細書に記載の多価型およびファージ提示型(MHCクラスII分子が提示されるファージ粒子の固有の構造により、多価になるよう容易に設計することができる)について特に当てはまる。したがって、四量体型およびファージ提示型のMHCクラスII分子、特に多価ファージ提示型がこの用途には好適である。本発明の四量体型または多価ファージ提示型のMHCクラスII分子を、従来の四量体が使用されている任意の適用に好ましい代替試薬として使用することができる。本発明の分子を従来の四量体の代わりに使用することができる好ましい適用は、一般に二量体およびより高次のオリゴマーのようなpMHCオリゴマーを用いたフローサイトメトリーによるT細胞の検出を含む、T細胞のMHC染色におけるものである。本明細書に示されているデータは、本発明のMHC分子をフローサイトメトリー解析で使用しT細胞を染色することができるということを示している。先行技術の多くのMHCクラスII四量体は、T細胞を活性化することができても、抗原特異的TH細胞を染色することができないため、このことは非常に有利かつ重要な特性である。したがって、本発明の好ましいMHCクラスII分子は、例えばフローサイトメトリーを用いて検出できるように、T細胞を染色する能力を有する。単量体型を用いてT細胞の染色を観察することは可能であるが、この特性は、特に多量体または多価型の分子で観察される。多量体ファージ提示型のさらなる利点は、一般に、比較的力価の低いファージを用いて同じ結果、例えばT細胞染色を達成することができるということである。
【0074】
かかるMHCクラスII分子を上述のように試薬として提供する場合、分子を関連するペプチドとともにまたはそれなしで提供することができる。かかる試薬を、タンパク質として、またはかかるMHCクラスII分子をコードする核酸として、例えば1つ以上の発現ベクターの形態で提供することができる。
【0075】
かかる試薬を、特に上述のようにT細胞染色に使用する場合、特定のペプチドに対するT細胞応答を研究するために、例えば、T細胞応答のレパートリーおよび動力学を研究するために、ならびに例えば末梢血中での抗原特異的CD4+T細胞の直接的なエクスビボ解析を可能にするために使用することができる。末梢血の解析はこれまで、組換えMHCクラスII分子が関係する課題であった。またこれらを用いて、抗原特異的T細胞をインビボ、インビトロおよびエクスビボで分離し同定することもできる。このようにして同定されたT細胞は、さらなる研究、拡張または活性化に供することができ、また治療で使用される可能性がある。
【0076】
また本発明の組換えMHCクラスII分子を、免疫調節剤をスクリーニングするためのプラットフォームとして使用することもできる。免疫系が関与する疾患において重要な段階は、抗原提示細胞とT細胞間の相互作用である。特に抗原提示細胞上のMHCクラスII複合体は、外部に由来するペプチドと結合して、それをTヘルパー細胞に提示するため重要である。次いで、Tヘルパー細胞が活性化されて、広範なエフェクター細胞を活性化する各種サイトカインを分泌する。
【0077】
本明細書に記載されているような安定で完全に機能的なMHCクラスII分子および特にこの分子の可溶型を提供することは、Tヘルパー細胞とMHCクラスII分子間の相互作用ならびにそれに続くTヘルパー細胞およびエフェクター細胞の活性化を調節する、例えば上方または下方調節することができる免疫調節剤のスクリーニングのためのプラットフォームを提供するのに重要である。本発明までは、安定なMHCクラスII分子を高収率かつ低コストで作製することの難しさが、こうしたスクリーニングの妨げとなっていた。したがって、本発明の組換えMHCクラスII分子を、免疫調節剤のスクリーニングプラットフォームとして用いることができる。
【0078】
かかる方法に使用するのに好ましいMHCクラスII分子は、本明細書の他の箇所に記載されている。好ましくは、前記MHCクラスII分子は疾患特異的ペプチドと会合する。このようなペプチドの例は、本明細書および先行技術に記載されており、セリアック病特異的またはI型糖尿病特異的ペプチドのような疾患特異的ペプチドと会合するヒトHLA−DQ2分子、関節リウマチの疾患特異的ペプチドと会合するヒトHLA−DR4 MHCクラスII分子、および特定の破傷風ペプチドと会合するヒトHLA−DP1 MHCクラスII分子がこれに含まれる。本発明のMHCクラスII分子と会合する他のペプチドも同様に使用することができる。
【0079】
次いで、かかる疾患関連ペプチドをMHCクラスIIとの関連で特異的に認識することができるT細胞を、例えば、本発明のMHCクラスII分子を上記のように用いて特定するか、または他の方法で入手(例えば、こうした細胞系は科学者により既にいくつか開発されている)した後、1つ以上の化合物がT細胞と抗原提示細胞間の相互作用またはその下流の事象(例えばサイトカイン分泌およびエフェクター細胞機能など)を調節する能力を評価することができる。
【0080】
本発明の組換えMHCクラスII分子および特にこの分子のファージ提示型を、エピトープ発見、例えば、T細胞により認識される抗原ペプチドエピトープ(T細胞エピトープ)の同定および特徴付けに使用することができる。したがって、本発明のさらなる実施形態では、T細胞により認識され得る抗原ペプチドエピトープを同定する方法が提供され、本発明の組換えMHCクラスII分子をT細胞受容体と接触させることと、前記組換えMHCクラスII分子と前記T細胞受容体との結合を検出することとを含む。T細胞受容体の結合は、MHCクラスII分子と会合した抗原ペプチドエピトープの存在を示すものであり、その後、このエピトープをさらに解析し特徴付けることができる。
【0081】
例えば、固定されたMHCとの関連で提示される抗原ペプチドに多様性を導入することにより、エピトープのライブラリーを作出することができる。次いで、例えば、組換え可溶性TCRもしくはファージ提示TCR、またはその他のT細胞集団(例えば、患者由来のT細胞)を用いて、このライブラリーのスクリーニングを行うことができる。低スループットのバキュロウイルスライブラリーに基づくものであり、生細胞で選択を行うことができないというさらなる不利な点を抱える現在の方法(Crawford et al.,2004,PLos Biology,2:0523−0533)と比較すれば、これは大きな進歩であろう。
【0082】
本発明の組換えMHCクラスII分子を診断試薬として使用することができる。例えば、本明細書の他の箇所に記載されているように、疾患特異的ペプチドを本発明のMHCクラスII分子と複合体形成または会合させ、これを用いて試料中、例えば、その疾患を有する可能性のある患者から採取した血液試料中の疾患特異的T細胞の有無を検出することができる。疾患特異的T細胞、特に、疾患を有さない患者で見られるレベルと比較して有意な数の疾患特異的T細胞の存在は、陽性の診断を示すことになる。
【0083】
したがって、本発明のさらなる態様では、試料中の抗原特異的T細胞を検出する方法が提供され、前記方法は、本発明の組換えMHCクラスII分子を前記試料と接触させることと、前記組換えMHCクラスII分子と前記T細胞との結合を検出することとを含む。前記組換えMHCクラスII分子とT細胞との結合は、抗原特異的T細胞の存在を示すものである。
【0084】
かかる方法を用いて、試料中の疾患特異的T細胞の存在を検出し、疾患の有無を診断することができる。この方法を用いて診断することができる適当な疾患は、MHCクラスII分子が疾患特異的ペプチドと結合する疾患である。疾患の例は本明細書の他の箇所に記載されており、セリアック病、関節リウマチ、破傷風およびインフルエンザがこれに含まれる。
【0085】
特定の疾患の背景にある機構をより詳細に理解するために、捕捉された疾患特異的T細胞およびこのT細胞と本発明のMHCクラスII分子間の相互作用を特徴付けることもできる。したがって、これも、例えば特定のT細胞応答をモニターし特徴付けるために、本発明のMHCクラスII分子を研究試薬として使用することができる方法もう1つの例である。
【0086】
本発明の組換えMHCクラスII分子がペプチドと会合する場合、それをTCR親和性成熟の標的としても使用することができる。
【0087】
当該技術分野において公知であり記載されているいくつかの方法のいずれでも、本発明の組換えMHCクラスII分子を調製し得るが、組換え法を用いて調製するのが最も好ましいということを当業者は理解するであろう。
【0088】
したがって、遺伝子クローニング技術を用いて本発明のMHCクラスII分子を作製してもよく、例えば、J.Sambrook et al.,Molecular Cloning,第2版,Cold Spring Harbor Laboratory Press,1989年、に適当な技術が開示されている。したがって、本発明のMHCクラスII分子の鎖(例えば、αおよび/またはβ鎖)をコードする配列またはこれに相補的な配列を含む核酸分子は、本発明のさらなる態様を形成する。
【0089】
本明細書に記載のMHCクラスII分子のα鎖(すなわち、鎖(i))またはβ鎖(すなわち、鎖(ii))をコードする核酸分子を、任意の適当な方法により、例えばクローニングまたは合成により、入手または作製することができる。例えば、適当な源由来(例えば、標準的なバフィーコートから分離された白血球のような細胞由来)の適当な配列をクローニングすることによりこのような配列を調製することができ、関係する特定のMHCクラスII対立遺伝子の配列を用いて設計された適当なプライマーを用いた標準的RT−PCRのようなPCR技術を用いて、適当な配列をクローニングすることができる。特定のMHCクラスII対立遺伝子の配列はGenBankで入手することができるが、IMGTデータベースを介した方が容易にアクセスすることができる。また全遺伝子合成法により、収率を最大化するのに重要となり得るコドン最適化が可能となるため、特に原核発現でこれを行うこともできる。
【0090】
最初の配列が一度クローニングまたは合成されれば、当該技術分野において公知であり記載されている方法を用いて、例えば部位特異的変異誘発により、本発明の変異システイン残基をコードする核酸分子を得るために配列に対して任意の付属の改変を行うことができる。
【0091】
さらに必要に応じて、本発明の分子を作製するために、MHCクラスII分子のαおよび/またはβ鎖の他の部分を操作してもよい。したがって、例えば、膜貫通ドメインおよび細胞質ドメインをコードするヌクレオチドを除去してもよく、同様に、本発明の分子が機能的であるために必要でないと考えられる他の任意の領域も除去してもよい。
【0092】
本発明のMHCクラスII分子の2つの鎖をコードする核酸フラグメントが得られれば、例えば、本明細書の他の箇所に記載されているように、調節配列などの他の所望の構成要素を含ませる、または非天然のシステイン残基を組み込む、または天然のシステイン残基を除去するために、そのフラグメントを標準的な組換えDNA技術でさらに操作してもよい。通常は、またはこのさらなる操作手順の一部として、本発明のMHCクラスII分子の作製を容易にするために、本発明のMHCクラスII分子をコードする核酸フラグメントを1つ以上の適当な発現ベクター内に組み込み、このベクター(1つまたは複数)を宿主細胞内に組み込む。かかる発現ベクターおよびかかる発現ベクターを含む宿主細胞は、本発明のさらなる態様を形成する。
【0093】
したがって、さらなる態様では、1つ以上の本発明の核酸分子を含む発現構築物または発現ベクターが提供される。好ましくは、発現構築物またはベクターは組換えによるものである。好ましくは、前記構築物またはベクターは、本発明の核酸分子によりコードされるタンパク質配列の転写および翻訳に必要な調節配列をさらに含む。
【0094】
さらなる態様では、1つ以上の本発明の発現構築物または発現ベクターを含む宿主細胞が提供される。また、1つ以上の本発明の核酸分子を含む宿主細胞も提供される。本発明のMHCクラスII分子を発現する宿主細胞は、さらなる態様を形成する。
【0095】
本発明のさらなる態様では、例えば本発明の宿主細胞を培養することを含む、本発明のMHCクラスII分子の作製方法が提供される。好ましい方法は、(i)コードされているMHCクラスII分子のαおよびβ鎖の発現に適した条件下で本発明の1つ以上の組換え発現ベクターまたは1つ以上の核酸配列を含む宿主細胞を培養すること;ならびに任意に(ii)宿主細胞由来または増殖培地/上清由来のタンパク質を単離または入手すること、を含む。この作製方法はタンパク質産物を精製することも含み得る。この作製方法は原核宿主細胞、例えば、本明細書の他の箇所に記載されている細菌宿主細胞で行うことが好ましい。
【0096】
したがって、本発明のさらなる態様では、
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含む組換えMHCクラスII分子を作製する方法が提供され、
(i)および(ii)は、機能的ペプチド結合ドメインを提供し、(i)および(ii)は、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基は、天然MHCクラスIIのα2およびβ2ドメイン中には存在せず、
前記方法は、前記組換え分子を原核宿主、好ましくは細菌宿主で発現させることを含む。
【0097】
上で議論したように、原核宿主細胞を用いて、ジスルフィド結合を組み込んで本発明の機能的MHCクラスII分子を作製することができるということは、驚くべきことであり、また有利である。
【0098】
本発明のMHCクラスII分子は2つ以上のポリペプチド鎖から構成される(すなわち、ヘテロ二量体である)ため、いくつかの実施形態では、例えば、ファージ提示法、細菌ペリプラズムでの発現および真核発現を含む実施形態では、すべてのポリペプチドを同一の宿主細胞内で、同じまたは異なる発現ベクターから発現させて、その宿主細胞内で完全な二量体タンパク質が組み立てられ、そこから単離または精製され得るようにすることが好ましい。他の実施形態、例えば意図的な封入体産生による細菌での大量生産などでは、各鎖を別々の宿主細胞(同一のタイプ/菌株の宿主細胞であることに留意)で発現させなければならない。
【0099】
任意の作製系を使用することができ(例えば、原核系も真核系もともに使用することができる)、発現ベクターを選択および設計することにより、使用する宿主細胞と確実に適合させる。したがって、発現ベクターが「宿主細胞の形質転換に適している」ということは、その発現ベクターが1つ以上の本発明の核酸分子と、発現に使用する宿主細胞に基づき選択され、核酸分子と作動可能に連結された調節配列とを含むことを意味する。作動可能に連結されたという表現は、核酸が発現されるように調節配列と連結されているという意味であることが意図される。
【0100】
MHCクラスII分子が不溶型である、例えば、別の生物粒子または生物膜の表面に提示される本発明の実施形態では、当該技術分野において記載されている方法により、本発明のMHC分子の提示を達成することができる。例えば、MHCクラスII分子が繊維状ファージ表面に提示される本発明の好ましい実施形態では、当該技術分野で記載されている任意の適当な方法を用いることができる。例えば、標準的なテキストとして、例えば「Phage Display in Biotechnology and Drug Discovery」(Sachdev S.Sidhu,1995)または「Phage Display:A Laboratory Manual」(Barbas et al.,1994)などを参照することができる。方法および系の例は、例えば、国際公開第09/024591号に記載されており、また実例にも示されている。
【0101】
一般に、本発明のファージ提示法の態様にはファージミドベクターの使用が好ましく、これには、当業者により容易に選択され得る適当なヘルパー株の使用が必要である。例えば、VCSM13ヘルパーファージ(Stratagene)およびHyperPhage(商標)(Progen Biotechnik GmbH)のような適当なヘルパーファージ株が多数市販されており、そのいずれも好都合に使用し得る。MHCクラスIIヘテロ二量体の一方の鎖、すなわちα鎖またはβ鎖のどちらかを、選択されたコートタンパク質、例えば、本明細書の他の箇所で議論されているgpIIIまたはその他のコートタンパク質の1つとの融合体としてベクターに組み込み、またもう一方の鎖を可溶性分子として、すなわち、コートタンパク質との融合体としてではなく産生させるのが好都合である。コートタンパク質との融合は一般に、前記コートタンパク質のN末端で行う。付属の実施例では、β鎖がコートタンパク質との融合体として産生され、α鎖が可溶型で産生される。
【0102】
他の適当な構成要素も構築物/ファージミドベクター中に存在していてよく、その多くはこのようなベクターでは普通のことである。例えば、タンパク質を細胞周辺腔へ方向付けるpelBのようなシグナル配列が、例えば、適当なプロモーター/オペレーター配列、リボソーム結合部位、複製起点、転写ターミネーターなどとともに一般に組み込まれる。ファージミドベクターおよび構成要素の例を図2に示すが、これらの構成要素のうちの1つ以上を本発明の発現ベクター内に組み込むことができる。
【0103】
組換えMHCクラスII分子のペプチド結合ドメインとの結合のためのペプチドを得ることが好都合な本発明の実施形態では、これらも構築物/発現ベクターの適当な位置に組み込んでよい。一般に、これらはリンカーペプチドによりαまたはβ鎖、例えばβ鎖のN−末端との共有結合による融合体として産生され、その例が本明細書の他の箇所に記載されている。
【0104】
MHCクラスII分子が可溶型である本発明の実施形態では、この分子を大腸菌(E.coli)のような細菌宿主細胞内の封入体として発現させ、次いでインビトロでリフォールディングさせることにより入手し得る。かかるインビトロでのリフォールディングは、標準的なプロトコル(例えば、Qoronfleh et al.,2007,Protein Expression and Purification 55:209−224に記載されている)およびそれを適当に修正したものを用いた、適切なリフォールディング条件下で生じ得る。
【0105】
あるいは、本発明の可溶性MHCクラスII分子を大腸菌(E.coli)のような細菌宿主細胞での発現により入手してもよく、ここでは、前記分子は宿主のペリプラズムで産生される。ペリプラズムでの産生は、適当な発現ベクターの使用により、例えば、タンパク質を細胞周辺腔へ方向付けるpelBのようなシグナルペプチドを組み込んで達成され得る。
【0106】
原核宿主、特に細菌宿主での本発明のMHCクラスII分子の作製は、本発明の好ましい実施形態である。この作製方法は、本明細書に記載のファージ提示法および可溶型のMHCクラスII分子の両方と適合性があり、この方法を用いて、高度に純化したタンパク質を大量に得ることができる。高度に純化したタンパク質を大量に得るためには、封入体から可溶性分子として産生されることが特に好ましい。操作されたジスルフィド結合を含むことにより安定性が増加した本発明の組換えMHCクラスII分子は、特に高収率での封入体産生に適しており、好ましくは、現在達成可能な収率(現在、出発物質の10%未満の範囲の収率はごく普通である)よりも高い収率で封入体から機能的MHCクラスII分子を産生させることを可能にする。
【0107】
任意の適当な原核宿主つまり細菌宿主をかかる産生に使用することができる。好ましい宿主細胞は、グラム陰性宿主細胞、より好ましくは大腸菌(E.coli)であろう。ファージ提示型に適した大腸菌(E.coli)宿主は当業者に公知であり、例えばXL1−blueがある。特に封入体からの精製が関係する場合に、可溶型の産生に好ましい大腸菌(E.coli)宿主は、大腸菌(E.coli)K12の派生物、最も好ましくはBL21表現型を有するものおよびその派生物である(Terpe,K.,Appl Microbiol Biotechnol.2006 Sep;72(2):211−22)。
【0108】
可溶性のペリプラズム発現に適した細菌宿主は当業者に公知であろう(例えば、Terpe,K.,Appl Microbiol Biotechnol.2006 Sep;72(2):211−22による総説を参照されたい)。
【0109】
あるいは、本発明の可溶性MHCクラスII分子を、酵母(pichiaを含む)、哺乳動物細胞または昆虫細胞のような真核細胞系で発現させることにより入手し得る。このような作製技術に適した宿主細胞は当業者に公知であろう。真核宿主細胞を用いた本発明の好ましい実施形態は、本発明の組換えMHCクラスII分子を免疫グロブリン(Ig)のFc部分との融合体、すなわち、例えばCasares et al.(1997,同上)に記載されているようなIg融合体として作製することである。このような実施形態では、免疫グロブリン分子のFc部分、例えばIgG2a分子のFc部分をαまたはβMHCクラスII鎖(通常はβ鎖のC−末端)と融合させて、本発明の組換えMHCクラスII分子(すなわち、本発明の人工的なジスルフィド結合を含む分子)のα鎖とβ鎖の二量体化を達成するために使用することができる。かかる分子を昆虫細胞系(または別の適当な系)で、例えば、昆虫細胞をバキュロウイルスに感染させて発現させることができ、これによりジスルフィド結合により安定化された分泌型のMHCクラスII分子が産生され得る。
【0110】
本発明の発現ベクターでの使用に適した調節配列は、細菌、真菌、ウイルス、哺乳動物または昆虫の遺伝子を含めた各種の源に由来し得る(例えば、Goeddel,1990,Gene Expression Technology:Methods in Enzymology 185,Academic Press,San Diego,CA,1990に記載されている調節配列を参照されたい)。適当な調節配列の選択は選択される宿主細胞によって決まり、当業者はこれを容易に行い得る。かかる調節配列の例としては、転写プロモーター/エンハンサーまたはRNAポリメラーゼ結合配列、翻訳開始シグナルを含むリボソーム結合配列が挙げられる。さらに、選択する宿主細胞および使用するベクターによっては、他の配列、例えば複製起点、追加のDNA制限部位、エンハンサーおよび転写誘導能を付与する配列などを発現ベクター内に組み込んでもよい。
【0111】
また本発明の発現ベクターは、本発明の組換え分子で形質転換された、またはそれが形質移入された宿主細胞の選択を容易にする選択マーカー遺伝子も含み得る。選択マーカー遺伝子の例は、特定の薬剤に対する耐性を付与するネオマイシンおよびヒグロマイシンのようなタンパク質、β−ガラクトシダーゼ、クロラムフェニコールアセチルトランスフェラーゼ、ホタルルシフェラーゼ、または免疫グロブリンもしくはその一部分(例えば免疫グロブリン、好ましくはIgGのFc部分など)をコードする遺伝子である。目的とする核酸とは別のベクターに選択マーカーを導入し得るということが理解されるであろう。
【0112】
また組換え発現ベクターは、組換えタンパク質の発現増加;組換えタンパク質の溶解性増加をもたらす;およびアフィニティー精製時のリガンド(例えば、精製および/または同定を可能にする適当な「タグ」、例えばHisタグ、HAタグ、FLAGタグまたはmycタグが存在し得る)として働くことにより標的組換えタンパク質の精製に役立つ融合部分をコードする遺伝子も含み得る。例えば、タンパク質分解性切断部位を標的組換えタンパク質に加えて、融合タンパク質の精製後に組換えタンパク質を融合部分から分離させ得る。典型的な融合発現ベクターにはpGEX(Amrad Corp.、Melbourne、Australia)、pMal(New England Biolabs、Beverly、MA)およびpRIT5(Pharmacia、Piscataway、NJ)があり、それぞれグルタチオンS−トランスフェラーゼ、マルトースE結合タンパク質またはプロテインAを組換えタンパク質と融合させる。
【0113】
発現ベクターのその他の任意選択の構成要素としては、AviTagまたは別のBirA基質のようなビオチン化配列(特に多量体型に関係する実施形態において)、検出のための標識、例えば、フローサイトメトリー技術の実施を可能にする蛍光標識が挙げられる。
【0114】
発現ベクターを宿主細胞内に導入して、形質転換宿主細胞を作製することができる。「〜で形質転換された」、「〜が形質移入された」、「形質転換」および「形質移入」という用語は、当該技術分野で公知の数多くの可能な技術のいずれか1つによる、細胞への核酸(例えば、ベクター)の導入を包含することが意図される。本明細書で使用される「形質転換宿主細胞」という用語は、本発明の組換え発現ベクターにより形質転換された、グリコシル化が可能な細胞も包含することが意図される。例えば、エレクトロポレーションまたは塩化カルシウムによる形質転換を用いて、原核細胞を核酸で形質転換することができる。例えば、従来技術、例えばリン酸カルシウムもしくは塩化カルシウム共沈殿、DEAE−デキストランによる形質移入、リポフェクション、エレクトロポレーションまたはマイクロインジェクションなどにより、核酸を哺乳動物細胞内に導入することができる。宿主細胞の形質転換および形質移入に適した方法は、Sambrook et al.(1989)およびその他の実験用教科書に記載されている。ファージ提示法が関係する本発明の態様では、関連する技術に関して、「Phage Display in Biotechnology and Drug Discovery」(Sachdev S.Sidhu,1995)または「Phage Display:A Laboratory Manual」(Barbas et al.,1994)のような汎用的なファージ提示法の教科書を参照することができる。
【0115】
他の分子とコンジュゲートされた本発明のタンパク質を含むN末端またはC末端融合タンパク質を、組換え技術による融合を用いて調製し得る。得られた融合タンパク質は、さらなる選択されたタンパク質(例えば、本明細書に記載のマーカータンパク質またはタグタンパク質)と融合した本発明のタンパク質を含む。また本発明のタンパク質を、他の既知の技術を用いて他のタンパク質とコンジュゲートさせてもよい。例えば、国際公開第90/10457号に記載されているヘテロ二官能性チオール含有リンカー、N−スクシンイミジル−3−(2−ピリジルジチオ−プロプリオナート(proprionate))またはN−スクシンイミジル−5チオアセタートを用いてタンパク質を結合させてもよい。融合タンパク質またはコンジュゲートを調製するために使用し得るタンパク質の例としては、免疫グロブリンのような細胞結合タンパク質、ホルモン、増殖因子、レクチン、インスリン、低密度リポタンパク質、グルカゴン、エンドルフィン、トランスフェリン、ボンベシン、アシアロ糖タンパク質、グルタチオン−S−トランスフェラーゼ(GST)、赤血球凝集素(HA)および短縮mycなどが挙げられる。
【0116】
本発明の第一のMHCクラスII分子をコードする核酸分子の調製方法に関係なく、さらなる適切なバリアント核酸分子を標準的な分子生物学的技術により容易に調製し得る。本発明のMHCクラスII分子をコードする任意のバリアント、変異体または第二世代の核酸分子が本発明での使用に適することを確認するために、本明細書の他の箇所に記載されているように、核酸分子に関して機能性を確認するための試験を行う。またバリアント、変異体または第二世代の核酸セグメントに関しても、標準的な条件下、より好ましくは標準的なストリンジェントなハイブリダイゼーション条件下でのハイブリダイゼーションを確認するための試験を行う。適切なハイブリダイゼーション条件の例は、約50℃で、約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、約1mMのEDTA中でのハイブリダイゼーション;および約42℃で約1%のSDSによる洗浄を含む。
【0117】
本明細書および添付の特許請求の範囲で使用される、単数形「a」、「an」および「the」は、文脈上そうでないことが明らかでない限り、複数形の指示対象を包含する。
【0118】
以下の非限定的な実施例に関連して、以下の図面を参照しながら本発明をさらに説明する。
【図面の簡単な説明】
【0119】
【図1】マウスMHCクラスII分子I−Ek(PDB:1FNG)のα2−β2ドメインを結び付ける推定ジスルフィド架橋のコンピュータモデリングを示す図である。wt(上段)および対応する変異体(下段)の両方について、表2の上位3位のアミノ酸対の残基:1位(A)、2位(B)および3位(C)を関連する原子間距離とともに示している。
【図2】pFABDFNファージミドのベクター骨格がpIII提示ファージミドpSEX81(GenBankアクセション番号:Y14584)に基づくことを示す図である。ファージミドは、インフレームの外来配列のセグメント(E1およびE2と呼ぶ)を、それぞれNcoI/SpeIおよびMluI/SfiI部分のカセット交換により収めることができる。I−Edα鎖(aa26〜204)およびβ鎖(aa27〜216)をそれぞれE1およびE2に挿入した。略語:lacPO、lacプロモーター;sd、Shine−Dalgarno配列;pelB、細菌ペクチン酸リアーゼのシグナル配列;t、T7転写ターミネーター;xPO、fkpAの天然プロモーター;FkpA、fkpAのオープンリーディングフレーム;f1 IG、ファージf1の遺伝子間領域;AmpR、β−ラクタマーゼをコードする領域;ColE1、複製起点。
【図3】マウスMHCクラスII分子I−EおよびI−AのIMGT参照α鎖(A)およびβ鎖(B)配列を、クローニングされたI−Ed(_ELの記号)およびI−Ek(1FNGの記号)のものと比較した、ClustalW2.02多重配列アライメントを示す図である。システイン置換のために選択された位置が黒い影で強調してある(番号付けは表2/成熟IMGT I−E配列に従った):1位(α96−β118/α96−β119)、2位(α95−β120/α95−β121)および3位(α94−β150/α94−β151)。α鎖の完全長IMGT配列が示されているが(H−2EA*02)、これは本明細書中で配列番号1と呼ばれる場合がある。β鎖の完全長IMGT配列が示されているが(H−2EB1*01)、これは本明細書中で配列番号2と呼ばれる場合がある。α鎖の成熟IMGT配列、すなわち、シグナルペプチドを含まない完全長配列は、本明細書中で配列番号3と呼ばれる場合がある(シグナルペプチドを含まないH−2EA*02)。β鎖のIMGT配列、すなわち、シグナルペプチドを含まない完全長配列は、本明細書中で配列番号4と呼ばれる場合がある(シグナルペプチドを含まないH−2EB1*01)。上記各配列の注釈は、PDB ID:1FNGおよびBurrows et al.,1999,Protein Engineering,12:771−778を参照して行った。
【図4】ファージELISAによるI−Ed−pIIIの整合性の解析を示す図である。対象とする抗体を固定化し、正規量のビリオンを反応させた。捕捉されたビリオンを抗M13HRPコンジュゲートで検出し、ELISAをTMBで発色させてA450nmで読み取った。phOx−BSA特異的Fabにより提示される抗体が対照として含まれていた。
【図5】抗pIIIで展開したSDS PAGE/ウエスタンブロット法による多価I−Ed−pIII提示レベルの解析を示す図である。残基の番号付けはPDB ID:1FNGに従った(g表2を参照されたい)。
【図6】図6aは、P96α2−S119Cβ2(1位、表2)により安定化されたI−Edを提示するファージが、LD1 T細胞ハイブリドーマ細胞を特異的に染色することを示す図である。LD1細胞をブロックし、氷上で2時間、I−Ed−pIII提示ファージミドにより染色した後、ビオチン化したウサギ抗fdビオチンコンジュゲートおよびストレプトアビジンPEとともにインキュベートした。実線はHAaa110-120/I−Ed−pIII提示を表し、破線はI−Ed−pIII提示を表す。非常に低い力価(5×109ファージ/ml)でも特異的染色が達成され、また達成可能な最大力価は約1013ファージ/mlであることに注目されたい。図6bは、R94Cα2−N151Cβ2(3位、表2)、S95Cα2−S121Cβ2(2位、表2)またはP96Cα2−S119Cβ2(1位、表2)のいずれかにより安定化されたI−Edを提示するファージが、LD1 T細胞ハイブリドーマ細胞を特異的に染色することを示す図である。LD1細胞を室温で30分間、I−Ed−pIII提示ファージミドにより染色した後、ビオチン化したウサギ抗fdビオチンコンジュゲートおよびストレプトアビジンAPCとともにインキュベートした。各ファージ調製物の平均蛍光強度(MFI)を棒グラフで表す。非常に低い力価(2×109ファージ/ml)でも特異的染色が達成され、また達成可能な最大力価は約1013ファージ/mlであることに注目されたい。
【図7】代表的なマウスおよびヒトMHCクラスIIアロタイプであるI−E、I−A、HLA−DP、−DQおよび−DR配列のIMGTのClustalW2.02多重配列アライメントを示す図である。α鎖のアライメントをAに、β鎖のアライメントをBにそれぞれ示す。表2、図1および図3に示されるシステイン置換のために特定された位置が黒い影で強調してある。特定された位置はすべて、保存されたα2/β2ドメイン中にある。
【図8】重ね合った8つの異なるヒトHLAクラスIIの構造を表す図であり、この構造は、分子の四次構造が広範な遺伝的多型にもかかわらず高度に保存されており、したがって図7のアライメントを構造レベル(図8A)で検証するものであることを示している。例えば、Aの全体的に重ね合った構造のR94α2−N150β2残基の対(3位、表2)に着目すれば、全体的にRMSDが0.29〜1.18Åで変化する高度に保存された配置が明らかとなる(図8B)。したがって、観察される差が、単一分子の無関係な構造に対して予想される実験変動内にあるに過ぎないため、これらの構造はトポロジーレベルで実質的に同一であると見なすことができる。
【図9】S95Cα2−S121Cβ2(2位、表2)により安定化されたHLA−DRまたはHLA−DQ2.5を提示するファージが、ヒトT細胞クローンを特異的に染色することを示す図である。T18、TCC820.26およびTCC820.88細胞をブロックし、室温で1時間、HLAクラスII−pIII提示ファージミドにより染色した後、ビオチン化したウサギ抗fdビオチンコンジュゲートおよびストレプトアビジンPEとともに氷上でインキュベートした。各ファージ調製物の幾何平均強度(GMI)を棒グラフで表す。
【図10】非還元または還元SDS PAGE、次いで抗pIIIで検出したウエスタンブロッティングによる、一価(M13K07)および多価(HyperPhage)のI−Ed−pIII提示レベルの解析を示す図である。残基の番号付けはIMGTに従った(表2を参照されたい)。
【発明を実施するための形態】
【0120】
実施例
【0121】
実施例1
材料および方法
構造評価、分子モデリングおよび配列解析
最初のジスルフィド結合予測にヘモグロビン(Hb)由来ペプチド(Hbaa65-76)1と複合したマウスMHCクラスII分子I−Ekの1.9Å分解能の結晶構造(PDB ID:1FNG)を用いた。α2−β2定常ドメイン間の接合点を形成している残基を調べ、さらなる解析のためにβ炭素が7Å未満の距離にある側鎖を選択した。最初に、選択された側鎖をコンピュータ上での突然変異によりシステインに変化させて、回転異性体ライブラリーのスキャンにより最適な回転異性体を選択した。選択された残基対のチオール基がジスルフィド結合形成に最適でない位置にある場合、好ましい角度になるまで側鎖のχ1二面角を互いに回転させた。次いでGromos96力場を用いて、制約付きエネルギー最小化により局所の幾何学的位置を最適化した。
【0122】
H−2E(GenBankアクセション番号:K00971およびAF050157)およびH−2A(GenBankアクセション番号:V00832およびM13538)の翻訳したマウスIMG参照配列、本発明者らがクローニングしたH−2Ed(下を参照されたい)ならびにPDB:1FNG H−2E配列を、ClustalW2.02(http://www.ebi.ac.uk/)を用いて整列させ、手動で注釈を付けた。代表的なマウスH−2AおよびE、ならびにヒトHLA−DP、DQおよびDRをIMGT(http://imgt.cines.fr/textes/IMGTrepertoireMHC/LocusGenes/RepresentativeMHCgc.html)からダウンロードして翻訳し、ClustalW2.02(http://www.ebi.ac.uk/)を用いて整列させた。使用したIMGT参照配列(GenBankアクセションコード(α鎖/β鎖))は、I−A(V00832/M13538)、I−E(K00971/AF050157)、HLA−DP(X03100/M23907)、HLA−DQ(M23907/U92032)およびHLA−DR(J00204/AJ297583)であった。H−2E、およびPDB ID:1FNGの構造解析に従って、アライメントに手動で影をつけた。この配列アライメントを、代表的なPDB登録物:1FNG(I−Ek)、1S9V(HLA−DQ2)、1JK8(HLA−DQ8)、1DLH(HLA−DR1)、1BX2(HLA−DR2)、1HQR(HLA−DR2a)、1A6A(HLA−DR3)、1D6E(HLA−DR4)および2Q6W(HLA−DR52a)の構造アライメントおよび同時の構造重ね合わせにより検証した。分子の可視化および操作作業では、すべてspdbビューア3.7SP5ソフトウェア2を用いた。
【0123】
細菌株、ヘルパーファージおよびプラスミド
大腸菌(E.coli)株XL1−BlueおよびVCSM13ヘルパーファージはStratagene(LaJolla、CA、USA)から購入した。HyperPhage(商標)ヘルパーファージはProgen Biotechnik GmbH(Heidelberg、Germany)から購入した。phOx−BSAに対して特異的な一本鎖Fv(scFv)を有するpIII提示ファージミドpSEX813は、Affitech AS(Oslo、Norway)の厚意により提供されたが、PROGEN Biotechnik GmbH(Heidelberg、Germany)から入手することもできる。pSEX81系pFAB提示およびphOx−BSA特異性を有するpFABDFNファージミドは既に記載されている4。
【0124】
細胞系
T細胞ハイブリドーマLD15は、マウスMHCクラスII分子I−Ed上に提示されたインフルエンザ赤血球凝集素由来ペプチド(アミノ酸110〜120;N−SFERFEIFPKE−C)に対して特異的である。ヒトCD4+T細胞クローンT18はマウスIgCカッパ(アミノ酸40〜48;N−WKIDGSERQ−C)に対して特異的であり、HLA−DR4(DRA1、B1*0401)(PMID:12456590)拘束性である。ヒトT細胞クローンであるTCC820.26およびTCC820.88は、それぞれDQ2−αI−エピトープ(アミノ酸57〜68;N−QLQPFPQPELPY−C)およびDQ2−αII−エピトープ(アミノ酸62〜72;N−PQPELPYPQPE−C)を認識する。各細胞はS.−W.Qiao博士(オスロ大学免疫学研究所、ノルウェー)の厚意により提供された。細胞はすべて、10%のFCS、0,1mMの非必須アミノ酸、1mMのピルビン酸ナトリウム、50μMのモノチオグリセロールおよび12μg/mlの硫酸ゲンタマイシンを添加したRPMI1640で維持した。
【0125】
抗体および追加の試薬
すべての培地および緩衝液は、実質的にはSambrook et al.6に記載されている通りに調製した。マウス抗pIII、ウサギ抗fd、ウサギ抗fdビオチンコンジュゲート、ヒツジ抗M13−HRPおよびヒツジ抗マウス−HRP抗体は、それぞれMoBiTec(Goettingen、Germany)、Sigma−Aldrich(Oslo、Norway)およびAmersham Biosciences(Uppsala、Sweden)から購入した。抗I−E/I−A抗体2G9、ならびにストレプトアビジン−PEおよびストレプトアビジン−APCコンジュゲートはBD Pharmingen(San Jose、CA、USA)から購入した。ラット抗マウスCD32抗体2.4G2(ATCC(登録商標)番号HB−197(商標))は自家製であった。ハプテンである、ウシ血清アルブミン(BSA)とコンジュゲートされた2−フェニルオキサゾール−5−オン(phOx)は、実質的に記載されている通りに調製した7。制限酵素(RE)は、Stratagene(LaJolla、CA、USA)から入手したDpnI以外は、New England Biolabs(Ipswich、MA、USA)から購入した。DNAオリゴは、MWG Biotech AG(Ebersberg、Germany)、DNA Technology(Aarhus、Denmark)またはSigma−Aldrichから購入した。BSAおよびTween20はSigma−Aldrich(Oslo、Norway)から購入した。Pfu Turbo DNAポリメラーゼはStratagene(LaJolla、CA、USA)から購入した。トリプシン/EDTAはBioWhittaker(Lonza Group Ltd.、Visp、Switzerland)から購入した。
【0126】
I−Ed−pIII提示ファージミドの構築
実質的にはCasares et al.8により記載されている通りに、A20 BALB/c Bリンパ腫細胞由来のH−2E遺伝子をRT−PCRにより回収し、別々に真核pLNOH2ベクター中にクローニングして、I−Ed−Ig融合体を作出した。I−EdαおよびI−Edβをコードする遺伝子を、以下のプライマー対を用いてこれらのベクターから増幅した:I−Edαには、pFAB I−Eda fw(5’−TATACCATGGCCATCAAAGAGGAACACACCATCATCCAGG−3’)およびpFAB I−Eda rv(5’−TATAACTAGTCATTACTCCCAGTGCTTCCGCAGAG−3’);I−Edβには、pFAB I−Edb fw(5’−TATAACGCGTCAGAGACACCAGACCACGGTTTTTG−3’)およびpFAB I−Edb rv(5’−TATAGGCCGCAGCGGCCCCTTTCCACTCGACCGTGACAGGGT−3’)。I−Edβ遺伝子がM13pIII表面タンパク質と融合し、I−Edα鎖が可溶性の物質として産生されるように、I−Ed遺伝子をpFABDFNファージミド中にクローニングした(図2)。インフルエンザ赤血球凝集素由来ペプチド(アミノ酸110〜120)および15アミノ酸リンカー(G4S)3をコードするDNAを、記載されている重複伸長による遺伝子スプライシング9および以下のプライマーを用いてI−Edβ鎖のN末端に挿入した:pMHCHA SOEing fw(5’−AAAGGAAGGAGGTGGTGGCTCCGGTGGAGGGGGAAGTGGAGGTGGAGGGTCTGTCAGAGACACCAGACCACGGTT−3’)およびpMHCHA SOEing rv(5’−GGAGCCACCACCTCCTTCCTTTGGGAAGATCTCGAACCTTTCGAATGATACGCGTGCCATCGCCG−3’)。クローニングのために、製造者のプロトコル(Stratagene、LaJolla、CA、USA)に従い、プライマー:QCIEdbPstI fw(5’−TGGACACGTACTGTAGACACAACTATGAGAT−3’)およびQCIEdbPstI rv(5’−ATCTCATAGTTGTGTCTACAGTACGTGTCCA−3’)を用いたQuikChange(商標)での部位特異的変異誘発により、I−Edβ遺伝子内のPstI制限部位を除去した。製造者のプロトコル(Stratagene、LaJolla、CA、USA)に従い、QuikChange(商標)での部位特異的変異誘発により、ジスルフィド結合を導入するための点突然変異を導入した。使用したプライマーは、I−EdαのR94C変異には、Mut R94C I−Edアルファfw(5’−GACTGTACTCTCCTGTAGCCCTGTGAACC−3’)およびMut R94C I−Edアルファrv(5’−GGTTCACAGGGCTACAGGAGAGTACAGTC−3’);I−EdβのN150C変異には、Mut N151C I−Edベータfw(5’−CCTGGTCCGATGTGGAGACTGGACCTTC−3’)およびMut N151C I−Edベータrv(5’−GAAGGTCCAGTCTCCACATCGGACCAGG−3’);I−EdαのS95C変異には、QCIEdaS95C fw(5’−ACTCTCCAGATGCCCTGTGAAC−3’)およびQCIEdaS95C rv(5’−GTTCACAGGGCATCTGGAGAGT−3’);I−EdβのS120C変異には、QCIEdbS120C fw(5’−GTCTGCTCTGTGTGTGACTTCTAC−3’)およびQCIEdbS120C rv(5’−GTAGAAGTCACACACAGAGCAGAC−3’);I−EdαのP96C変異には、QCIEdaP96C fw(5’−CAGAAGCTGTGTGAACCTGGGA−3’)およびQCIEdaP96C rv(5’−TCCCAGGTTCACACAGCTTCTG−3’);I−EdβのS118C変異には、QCIEdbS118C fw(5’−CCTGGTCTGCTGTGTGAGTGAC−3’)およびQCIEdbS118C rv(5’−GTCACTCACACAGCAGACCAGG−3’)であった。
【0127】
MHCII提示バクテリオファージの調製
VCSM13またはHyperPhage(商標)ヘルパーファージおよびビリオン会合を用いた大腸菌(E.coli)XL1−Blueからのファージミドレスキューを、記載されているスポット力価測定法10によりモニターした。
【0128】
ファージ捕捉ELISA
捕捉抗体およびphOx−BSAを、pH7.4のPBS中2.5〜5μg/mlの濃度で、4℃で一晩、MaxiSorp(商標)マイクロタイタープレートウェル(Nunc、Roskilde、Denmark)に吸着させた。ウェルをPBSTM(0.05%v/vのTween20および4%w/vの脱脂乳を添加したPBS)により、室温で1時間ブロックした。次いで、正規量のビリオン調製物(VCSM13レスキュー試料:1×1010cfuampR/ウェル;HyperPhageレスキュー試料:1×108cfuampR/ウェル)を加え、室温で1〜2時間反応させた後、捕捉されたビリオンを室温で1時間、抗M13−HRP(1:5,000)により検出した。TMB可溶性基質(Merck KGaA、Darmstadt、Germany)でウェルを発色させ、30分後に1MのHClで停止させ、A450nmで吸光度を読み取った。
【0129】
SDS−PAGEおよびウエスタンブロット法
ビリオン(108cfuampR/レーン)を非還元および還元4〜12%ビス/トリスXT CriterionプレキャストSDS−PAGE(Bio−Rad、Hercules、CA、USA)により分離し、セミドライブロッティング装置(Bio−Rad、Hercules、CA、USA)を用いて、トリス/グリシン緩衝液(25mMのトリス、192mMのグリシンおよび20%のメタノール、pH8.3)中、25Vで30分間、ポリフッ化ビニリデン膜(Millipore、Madison、USA)にブロットした。膜をPBSTM中でブロックした後、マウス抗pIIIMAb(1:4,000)、次いでヒツジ抗マウス−HRP(1:10,000)でpIII融合体を検出した。膜を洗浄し、SuperSignal(商標)West Femto基質(Pierce、Rockford、IL、USA)で発色させ、BioMax(商標)MRフィルム(Kodak、Fernwald、Germany)に感光させた。
【0130】
人工的S−S架橋を含むHLA−DP、−DRおよび−DQの構築
IMGT(http://imgt.cines.fr/)により定義される代表的なHLA−DR(DRA*0101/DRB1*1402)および−DQ(DQA1*0501/DQB1*0301)をコードする遺伝子を化学的に合成した(Genscript、Piscataway、NJ)。大腸菌(E.coli)コドンバイアスを修正してDNA配列を最適化し、2位のα2−β2システイン対を導入するための点突然変異を組み込んだ(表2)。DNAフラグメントを上記のようにpFABDFNファージミド中にクローニングした。
【0131】
あるいは、IMGT(http://imgt.cines.fr/)により定義される代表的なHLA−DP(DPA1*0103/DPB1*0401)、−DR(DRA*0101/DRB1*1402)および−DQ(DQA1*0501/DQB1*0301)をコードする遺伝子を、新たに単離したヒトPBMCからRT−PCRにより増幅し、上記のようにpFABDFNファージミド中にクローニングする。2位(表2)のα2−β2システイン対を導入するための点突然変異を、製造者のプロトコル(Stratagene、LaJolla、CA、USA)に従いQuikChange(商標)での部位特異的変異誘発により組み込む。使用したプライマー(表1に記載)は、HLA−DPA α2−ドメインのS95C変異には、DPA_S95C_センスおよびDPA_S95C_アンチセンス;HLA−DPB β2−ドメインのS121C変異には、DPB_S121C_センスおよびDPB_S121C_アンチセンス;HLA−DRA α2−ドメインのS95C変異には、DRA_S95C_センスおよびDRA_S95C_アンチセンス;HLA−DRB β2−ドメインのS121C変異には、DRB_S121C_センスおよびDRB_S121C_アンチセンス;HLA−DQA α2−ドメインのS95C変異には、DQA_S95C_センスおよびDQA_S95C_アンチセンス;HLA−DQB β2−ドメインのS121C変異には、DQB_S121C_センスおよびDQB_S121C_アンチセンスである。
【0132】
【表2】
【0133】
フローサイトメトリー
LD1細胞の単細胞懸濁液を、氷上で15分間、60%で熱不活性化したラット血清および1×PBS中200μg/mlの抗CD32mAb(HB197)によりブロックするか(図6a)、またはブロックしなかった(図6b)。HyperPhageにパッケージングされたI−Ed−pIII提示の染色を、氷上で2時間、染色緩衝液(0.5%のBSAを含む1×PBS)中で(図6a)、または室温で30分間、1%のBSAを含む1×PBS中で(図6b)行った。図6aで使用したファージ投入量は、HAaa110-120−I−Ed提示には5×109cfuampR、I−Ed提示には1.6×1010cfuampRであった。図6bで使用したファージ投入量は、VCSM13ヘルパーファージ(ネガティブ対照、1×1011cfukanR/ml)を除くすべての調製物で2×109cfuampRであった。次いで試料を、10μg/mlのビオチン化ウサギ抗fdとともに氷上で1時間(図6a)または30分間(図6b)インキュベートし、次いで2μg/mlのストレプトアビジンPEとともに氷上で15分間インキュベートした。
【0134】
3つのT細胞クローン、すなわちT18、TCC820.26またはTCC820.88のそれぞれの単細胞懸濁液を染色緩衝液(0.5%のBSAを含む1×PBS)で調製した。HLAクラスII−pIII融合体(図9)を多価(HyperPhageにパッケージング)または一価(VCSM13にパッケージング)で提示するファージとともに室温で1時間、細胞を染色した。使用したファージ投入量は、それぞれ約1×109cfuampR(図9a)、約2×109cfuampR(図9b)および約2×1012cfuampR(図9cおよび9d)であった。次いで試料を、10μg/mlのビオチン化ウサギ抗fdとともに氷上で1時間インキュベートし、次いで2μg/mlのストレプトアビジン−PEとともに氷上で30分間インキュベートした。FACSCaliburフローサイトメータ(BD Biosciences)で事象を捕捉し、CellQuest PROソフトウェア(BD Biosciences)を用いてそれを解析した。
【0135】
結果
分子モデリング
表2(1FNG番号付け)は、I−Edα/I−Ekβ構造(PDB ID:1FNG)内のα2/β2定常ドメイン接合点中の選択された残基対のβ炭素側鎖の距離を示している。実際、1位〜3位の残基対の、システインへのコンピュータ上での突然変異誘発および側鎖の最適化により、タンパク質の三次構造を不安定にすることなく2.03〜2.07Åの間のジスルフィド結合距離が得られた(図1)。この結果は、変異体ポリペプチドにおいてこれらのアミノ酸置換が可能になり、ジスルフィド架橋を形成された可能性があり、したがって、さらなる解析に選択されたということを強く示唆するものであった。PDB ID 1FNGで受託されているアミノ酸番号付けは、β鎖のIMGT配列番号付け(図3Bを参照されたい)からずれていることに留意されたい。
【0136】
【表3】
【0137】
MHC−pIII融合体の設計
I−Ed遺伝子セグメントを、αおよびβ鎖が2つの別々のポリペプチドとして翻訳され、そのうちβ鎖がウイルスキャプシドタンパク質pIIIのN末端にインフレームで直接融合されるようにpFABDFNファージミド内に挿入した(図2)。両鎖とも、ペリプラズムへの標的化のためのN末端シグナル配列(pelB)を含んでいた。挿入されたα鎖部分(図3A、H−2EA_EL)は、図3Aに示されるIMGT H−2EA*02参照配列(GenBankアクセションコード:K00971、配列番号1)のアミノ酸26〜204に対応する。同様に、図3Bに示されるIMGT H−2EB1*01参照配列(GenBankアクセションコード:AF050157、配列番号2)のアミノ酸27〜216に対応するβ鎖部分を用いた(図3B、H−2EB_EL)。したがって、I−E IMGT参照配列に基づけば、表2の1位〜3位のシステイン置換の位置は、それぞれ成熟ペプチド(すなわち、シグナルペプチドを含まないペプチド)のα鎖の位置94〜96(図3A)およびβ鎖の位置119、121および151(図3B)に対応する。このβ鎖残基の番号付けは、1FNGテンプレートファイルに受託されている番号付けからずれていることに留意されたい。
【0138】
ファージミド由来のMHCII−pIII提示の解析
pMHC部分の提示の傾向および整合性を比較するために、I−E特異的モノクローナル抗体(MAb)である2G9で正規量のビリオンを捕捉することにより、ファージELISAを行った。結果は、多価および低価数の提示の両方で、すべての試料がMAb 2G9により認識されることを示していたが(図4)、このことは、構造上の整合性が全体的に保持されたことを示唆している。MAb 2G9は、pIIIと直接融合させたI−Eβ鎖と結合する。ファージ上に共有結合のα鎖ヘテロ二量体が存在するか否かを評価するために、非還元および還元SDS PAGE、次いで抗pIIIでのウエスタンブロット検出により正規量のビリオンを解析した(図5)。結果は、wt(図5、レーン5)およびHAペプチドを有するα95/β120変異体(図5、レーン3)の2つを除く全試料中に、有意な割合の共有結合のヘテロ二量体が存在することを示していた。しかし、α95/β120変異体の試料は、試料負荷量が非常に少ないため適切な分離が妨げられたことを示していたのは意外であった。すべての共有結合複合体形成が試料の還元の際に打ち消された(図5、レーン11〜20)。さらに抗体Fab対照(図5、レーン10および20)に関して、pMHC部分の改変に対して1つの共有結合ヘテロ二量体種のみが予想されたが、すべての関連試料においてかかる種が複数観察され、予想とは明らかに異なっていた。おそらくこの現象は、導入された変異のほかにも形成された、異所での天然システイン残基間のジスルフィド架橋により説明されるであろう。したがって、実際に提示されたタンパク質のごく一部だけが、人工的に導入された架橋のほかに天然のジスルフィド架橋をα2−β2ドメイン中に有する鎖である。
【0139】
I−Ed−pIII構築物の一価および多価の提示を同時に解する同様の実験を行った。図10にその結果を示す。低レベルのファージミド提示(一価)では、約1〜10%のビリオンが融合タンパク質を有する(BradburyおよびMarks,J Immunol Methods.2004,290(1−2):29.)。したがって、はるかに感度の高いELISAアッセイでは融合体が容易に検出されるのにもかかわらず、これらの試料(M13K07試料)では融合タンパク質がはっきりとは見られない(図4)。
【0140】
細胞表面染色試薬としてのpMHC−pIII
pIIIとの融合体としてバクテリオファージ上に提示されたペプチド−MHCクラスIIを従来のpMHCII四量体と同様に染色試薬として使用できるか否かを調べるために、本発明者らは、HAaa110-120/I−Ed−pIIIまたは融合ペプチドを含まないI−Ed−pIIIでLD1細胞を染色した。図6aからわかるように、関連ペプチドなしで産生されたバクテリオファージ上のI−Ed−pIII融合体では染色が観察されなかったが、HAaa110−120/I−Ed−pIIIは、約0.8logだけ明るい強度で細胞を染色した。図6bからわかるように、調べたすべてのジスルフィド架橋でペプチド特異的染色が観察される。陽性染色は、I−Ed−pIIIのジスルフィド安定化型および抗原ペプチド有する型(HAaa110-120)でのみ観察された。3つの操作されたジスルフィド架橋間で明らかな差は見られなかった。したがって、これらは同様の効率で作用すると思われる。
【0141】
pMHC−pIIIの機能性にさらに取り組むために、VCSM13またはHyperPhage(商標)を用いたファージミドレスキューにより、mCκaa40-48/HLA−DR4−pIII、αIaa57-68/HLA−DQ2.5またはαIIaa62-72/HLA−DQ2.5を提示するビリオンを調製した。また対照として、すべてのpMHC−pIII融合体を抗原ペプチドなしでも作製した。次いでこれらのビリオンを、記載されているようにフローサイトメトリー実験で試験した。図9aからわかるように、mCκaa40-48/HLA−DR4−pIII染色細胞でペプチド特異的染色が観察された。図9bおよび9cでは、αIaa57-68/HLA−DQ2.5の多価および一価の提示がペプチド特異的染色を示している。ファージ投入量は、VCSM13にパッケージングされたαIaa57-68/HLA−DQ2.5提示において1012cfuampRであったということに留意されたい。αIIaa62-72−HLA−DQ2.5のペプチド特異的染色を図9dに示す。
【0142】
考察
可溶性の組換えMHCクラスII分子の効果的な使用の主な障害は、このような操作された分子に固有の安定性の問題が原因である。この制限を克服するために様々な立案されたアプローチが採用されてきたが、汎用性のあるアプローチは現在のところ存在しない11。また、現在用いられている手段はすべて、コストと労働力がかかる方法であり、事実上、高スループットなアプローチの妨げとなっている。本発明者らは、組換えMHCクラスII作製とファージ提示技術12を合わせることにより、この問題に対する魅力的な解決方法を見出した。これは非常に迅速かつ安価な技術プラットフォームであるばかりでなく、極めて多様なコンビナトリアルスクリーニングのアプローチを可能にするという固有の特性も有している。MHC分子によりもたらされる広大な抗原プロテオーム空間は、例えばエピトープ発見において、このコンビナトリアル性から大きな恩恵を受けるであろう。しかし現在のところ、こうした発見はまだバキュロウイルスに基づく煩雑で低スループットな技術13に限定されている。
【0143】
ファージ提示を使用すれば、十分に記述された簡便な形式で12、より大きな桁数のレパートリーサイズに容易に手が届くであろう。このような提示によりMHCクラスI分子が探求されてきたが14〜16、MHCクラスIファージ提示のアプローチはいずれも、細胞特異的染色がまだ得られていないことから、現在のところ可溶性四量体の代替物として機能しないことがわかっている16。MHCクラスI分子は単一のポリペプチドとして効率的に作製することができる18のとは対照的に、MHCクラスIIは、等しく関与する別々の鎖のヘテロ二量体であることから、その作製は複雑である。MHCクラスIおよびIIの機能的なフォールドはともに、Igフォールドトポロジーで構築されるため、ドメイン内のジスルフィド架橋の形成も必要とする19。
【0144】
MHCクラスIIとその同族リガンド間の天然の相互作用の鍵となる特徴は、固有の弱い親和性である20。組換えMHCクラスII分子を検出試薬として用いる場合にこの感度における制限を克服するために、分子を四量体または五量体に重合することが必要であった11。こうしたポリマーの作製には、実質的な操作を伴う多段階のプロトコルが含まれている。ここでファージ提示を使用すれば、ファージ上での多量体提示を可能にする既に利用可能な技術の使用により、かかる重合が非常に容易に行われる21。
【0145】
ここで本発明者らは、pIIIファージミド提示により機能的に提示されるMHCクラスII分子を得るための戦略を提供する。これを達成するために、αおよびβの2つの鎖を大腸菌(E.coli)内で2つの別々のポリペプチド鎖として発現させ、そのうちの1つの鎖はpIIIキャプシドと融合され、ペリプラズムへ標的化されている。酸化ペリプラズムでは、2つの鎖が、α2ドメインとβ2ドメインを結合させる新規な人工的ジスルフィド架橋を利用して、安定なヘテロ二量体を形成する。マウスMHCクラスII分子I−Edの保存されたα2−β2接合点中の複数の位置を、人工的な鎖間ジスルフィド架橋を可能にするアミノ酸置換の標的とすることができる(図1および3)。このような人工のMHC分子を、標準的なファージミドによるファージ提示を用いてpIII上に提示させることにより、共有結合のヘテロ二量体が実際にビリオン上に提示される(図5および10)。wtおよび変異体両方のすべての分子が、マウスMHCクラスII I−E分子に特異的なモノクローナル抗体(MAB 2G9)と反応することから、分子全体の整合性は保持されると思われる(図4)。I−Ed分子を、β鎖との共有結合テザーとして十分に特徴付けられているHAaa110-120抗原ペプチド22を提示するように、実質的には記載されている通りに設計した23。ジスルフィド架橋により安定化されたこれらの組換え分子を提示するファージをフローサイトメトリーで用いて、I−Ed/HAaa110-120複合体に対して特異的なT細胞受容体を有するLD1 T細胞ハイブリドーマ5の特異的染色が得られた(図6aおよび6b)。重要なのは、特異的染色が抗原ペプチドを有するジスルフィド安定化I−Ed−pIII融合体でのみ見られ、このことは真にこのアプローチの機能性を示しているということである。さらに、一価の提示では同様の染色が見られなかったことから(データ不掲載)、MHC部分の多価の提示が染色に不可欠であると思われ、このことは、機能的な親和性を天然の相互作用の親和性よりも高める必要性を強調している。これは現在のところ、ファージ提示されたMHC分子を特定のT細胞の直接的な可視化に使用した最初の例であり、古典的なMHCテトラマー技術を補完するものである。
【0146】
成功の鍵は、MHCクラスII分子のα2−β2接合点を安定化させる人工的なジスルフィド架橋の導入である。特定された位置は、HLAを含めたMHCクラスII分子間で高度に保存されているため(図7および8)、これは汎用性のあるアプローチであると思われる。この汎用性は、試験した他の3つのMHCクラスII複合体、すなわちmCκaa40-48/HLA−DR4、αIaa57-68/HLA−DQ2.5およびαIIaa62-72/HLA−DQ2.5はすべて、pIII融合体として、サイトメトリーにおいて抗原ペプチド特異的に特異的細胞染色を示したということにより強く支持される(図9)。予想されたように、特定のpMHCII複合体およびそれと反応するT細胞クローンの中で染色能および多価相互作用への依存に違いが見られた。したがって、多価相互作用への依存は全か無かの事象ではなく、このことは一価型のαIaa57-68/HLA−DQ2.5およびαIIaa62-72/HLA−DQ2.5の両方による特異的細胞染色により示された。フローサイトメトリーにおいて特異的に陽性に染色する能力は、特定のT細胞受容体のpMHCに対する本来の親和性、T細胞上での受容体密度および膜での受容体のクラスター形成能のようなパラメーターに依存するであろう。αIaa57-68/HLA−DQ2.5による多価(図9b)と一価(図9c)の染色では、多価型の方がファージ投入量が1000倍少なかったにもかかわらず、ともにほぼ同じであったということは注目すべきことである。
【0147】
参考文献
1. Kersh,G.J.et al.Structural and Functional Consequences of Altering a Peptide MHC Anchor Residue.Journal of Immunology 166,3345−3354(2001).
2. Guex,N.& Peitsch,M.C.SWISS−MODEL and the Swiss−PdbViewer:an environment for comparative protein modeling.Electrophoresis 18,2714−2723(1997).
3. Welschof,M.et al.The antigen−binding domain of a human IgG−anti−F(ab’)2autoantibody.PNAS 94,1902−1907(1997).
4. Loset,G.A.,Lunde,E.,Bogen,B.,Brekke,O.H.& Sandlie,I.Functional phage display of two murine α/β T−cell receptors is strongly dependent on fusion format,mode and periplasmic folding assistance.Protein Eng Des Sel.20,461−472(2007).
5. Taylor,A.H.,Haberman,A.M.,Gerhard,W.& Caton,A.J.Structure−function relationships among highly diverse T cells that recognize a determinant from influenza virus hemagglutinin.J Exp Med 172,1643−1651(1990).
6. Sambrook,J.& Russell,D.W.Molecular Cloning:A Laboratory Manual,Edn.3.(Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY;2001).
7. Makela,O.,Kaartinen,M.,Pelkonen,J.L.& Karjalainen,K.Inheritance of antibody specificity V.Anti−2−phenyloxazolone in the mouse.J Exp Med 148,1644−1660(1978).
8. Casares,S.,Bona,C.A.& Brumeanu,T.D.Engineering and characterization of a murine MHC class II−immunoglobulin chimera expressing an immunodominant CD4 T viral epitope.Protein Eng 10,1295−1301(1997).
9. Horton,R.M.,Cai,Z.L.,Ho,S.N.& Pease,L.R.Gene splicing by overlap extension:tailor−made genes using the polymerase chain reaction.Biotechniques 8,528−535(1990).
10. Loset,G.A.,Kristinsson,S.G.& Sandlie,I.Reliable titration of filamentous bacteriophages independent of pIII fusion moiety and genome size by using trypsin to restore wild−type pIII phenotype.BioTechniques 44,551−554(2008).
11. Vollers,S.S.& Stern,L.J.Class II major histocompatibility complex tetramer staining:progress,problems,and prospects.Immunology 123,305−313(2008).
12. Bratkovic,T.Progress in phage display:evolution of the technique and its applications.Cell Mol Life Sci(2009).
13. Crawford,F.et al.Use of baculovirus MHC/peptide display libraries to characterize T−cell receptor ligands.Immunol Rev 210,156−170(2006).
14. Le Doussal,J.,Piqueras,B.,Dogan,I.,Debre,P.& Gorochov,G.Phage display of peptide/major histocompatibility complex.J Immunol Methods 241,147−158.(2000).
15. Kurokawa,M.S.et al.Expression of MHC class I molecules together with antigenic peptides on filamentous phages.Immunol Lett 80,163−168(2002).
16. Vest Hansen,N.,Ostergaard Pedersen,L.,Stryhn,A.& Buus,S.Phage display of peptide/major histocompatibility class I complexes.Eur J Immunol 31,32−38(2001).
17. Constantin,C.M.,Bonney,E.E.,Altman,J.D.& Strickland,O.L.Major histocompatibility complex(MHC)tetramer technology:an evaluation.Biol Res Nurs 4,115−127(2002).
18. Leisner,C.et al.One−Pot,Mix−and−Read Peptide−MHC Tetramers.PLoS ONE 3,e1678(2008).
19. Halaby,D.M.,Poupon,A.& Mornon,J.The immunoglobulin fold family:sequence analysis and 3D structure comparisons.Protein Eng 12,563−571(1999).
20. Rudolph,M.G.,Stanfield,R.L.& Wilson,I.A.How TCRs bind MHCs,peptides,and co−receptors.Annu Rev Immunol 24,419−466(2006).
21. Soltes,G.et al.On the influence of vector design on antibody phage display.Journal of Biotechnology 127,626−637(2007).
22. Schild,H.et al.Natural ligand motifs of H−2E molecules are allele specific and illustrate homology to HLA−DR molecules.Int Immunol 7,1957−1965(1995).
23. Kozono,H.,White,J.,Clements,J.,Marrack,P.& Kappler,J.Production of soluble MHC class II p
【技術分野】
【0001】
本発明は、ジスルフィド結合により安定化された組換えMHCクラスII分子に関する。
【背景技術】
【0002】
主要組織適合複合体(MHC)分子は、脊椎動物免疫系の中心的な構成要素であり、すべての有核細胞表面に存在する。MHCは2つの主要な型、すなわちMHCクラスIおよびクラスIIとして存在する。両型とも、所与のMHCを発現する細胞自体の中で、T細胞エピトープと呼ばれるタンパク質分解処理されたペプチドとともに機能的複合体を形成するということは重要である。生じたペプチド/MHC(pMHC)複合体は、次いで細胞表面の膜貫通型の複合体となり、この現象は抗原提示と呼ばれる。次いで、細胞表面に結合したpMHCは、その同族パートナー、すなわちTリンパ球表面に存在するT細胞受容体(TCR)と相互作用し得る。
【0003】
pMHC−TCR相互作用は適応免疫において極めて重要な役割を果たしているため、基礎および応用科学の分野では、細胞および分子の両レベルで理解し、組換え型の両分子を入手することに大きな注目が集められている。さらに、その系の生態を研究し理解することを可能にするためにも、また新規な治療剤および診断剤を開発するためにも、こうした組換え分子を利用できることが極めて重要であるということも理解されつつある。
【0004】
多くの重要な医学的状態では、患者の免疫系の活性を調節する治療的介入が必要である。例えば自己免疫疾患およびアレルギーでは、免疫系の亢進および慢性炎症を抑える必要がある。これに対し、免疫促進は、免疫細胞を癌性細胞に対して活性化して標的化する、感染症および癌に適切なアプローチである。さらに、移植レシピエントは通常、免疫抑制を必要とする。同時にこれにより、膨大な量の免疫調節剤が開発されるようになり、現在では数十億ドル規模の産業となっている。
【0005】
これらの疾患の免疫的構成要素を理解し、新たな治療法をスクリーニングする上で鍵となるのは、抗原提示細胞とT細胞の間の相互作用、つまり、より具体的にはMHCクラスIおよびII分子とTCRの間の相互作用である。クラスIIMHCは外来性のペプチドと特異的に結合し、それをCD4+Tヘルパー細胞(TH細胞)に提示する。次いでTH細胞が活性化され、各種サイトカインを分泌するエフェクター細胞となる。これらのサイトカインは、脅威への対処に関与する他の様々な免疫細胞を活性化する。このシステムが機能不全に陥ると、例えば、自己免疫疾患を引き起こしたり、癌細胞が生き残って分裂したりする可能性がある。このように、自己免疫疾患はMHCとの強い関連性および標的器官へのT細胞浸潤を特徴とする。
【0006】
免疫調節剤スクリーニングのためのプラットフォームには、安定で完全に機能的な可溶性MHCクラスII分子が必要である。重要なのは、可溶性MHCクラスII分子の作製は現在、分子安定性の欠如という深刻な問題に阻まれているということである。
【0007】
ここ数年の間に、可溶性MHCクラスI分子を四量体として作製する能力(テトラマー技術)により、基礎および応用免疫学が大きな変化を遂げた(Constantin et al.,2002,Biological Research for Nursing,4:115−127)。テトラマー技術により、抗原およびT細胞応答の両方に関して特定の方法で、主としてフローサイトメトリーにより評価される免疫応答の過程を追跡する能力が大幅に向上したというのがその理由である。またこの能力は、免疫系のより一層深い理解にもつながり、新規な診断ツールを実際に生み出す可能性もある。
【0008】
組換えMHCクラスIの作製に関する技術的問題点の多くが解決されてきたのと同様に、四量体試薬はこれまで、ほとんどがMHCクラスI分子に限られてきた。実際、MHCクラスII分子に関しては、この課題が極めて困難であることが証明されている。したがって、独立した例が文献中にも、また市販の試薬としても見られるが、利用できるMHCクラスII四量体作製の一般的なプロトコルは現在のところない(Vollers,S.およびStern,L.,2008,Immunology 123:305−313)。MHCクラスII四量体が利用可能な少数例では、MHCクラスII四量体が広範囲で使用され、疾患発症の理解に大きな影響を与えた。したがって、組換えMHCクラスI作製の成功により既に示されている影響を考えれば、新規なMHCクラスII作製手段にさらに力を注ぐ強く明確な学術的・商業上的動機が存在することがわかる。しかし、現在直面している問題を考えれば、成功するか否かは定かではない。
【0009】
部分的に不明である理由により、MHCクラスII分子は可溶型の安定な組換え分子として作製することが特に難しいことが証明されている。天然の分子はα鎖とβ鎖を含む非共有結合性の膜貫通型ヘテロ二量体であり、αおよびβの両鎖は膜貫通領域を有し、免疫グロブリン(Ig)スーパーファミリーに属する。各鎖の細胞外部分は、それぞれ約90個のアミノ酸残基からなる2つのドメインから構成され、そのうちの2つの膜遠位ドメインであるα1ドメインとβ1ドメインが、T細胞エピトープのペプチド結合性に不可欠な、互いに格子状になったα/β構造を形成している。2つの膜近位ドメインであるα2ドメインとβ2ドメインは、ともに分離したIgドメインを形成している。αおよびβの両鎖では、約20個のアミノ酸残基の配列が細胞膜を横断し、膜の細胞質側にはごく短いペプチドセグメントがある。
【0010】
α鎖とβ鎖の二量体化は、(i)膜貫通セグメント、(ii)ペプチド結合および(iii)膜にある推定上の付属構成要素により生じると考えられている。したがって、MHCクラスII分子を一度その天然の状態から切り離し、可溶性分子として作製すると、本来の低い安定性と非常に低い生産レベルに悩まされることが多い。さらには大規模で供給源を要する、場合に応じた最適化を行わなければならない。
【0011】
また、上述の二量体化のための必要事項を考慮すれば、任意の非天然の状態、すなわち、細胞が天然でMHCクラスII分子を発現するような細胞膜と関連した産生以外の任意の状態でMHCクラスII分子を作製するための一般的方法、例えば、他の生物学的存在、細胞または粒子の表面に示される不可溶性分子として、例えば、ウイルスキャプシドタンパク質との融合によりファージ表面で発現される不溶性分子として作製することは、容易とは言い難い。
【0012】
したがって、安定化されたMHCクラスIIヘテロ二量体を作製することができる一般的な戦略は、現在のところ存在しない。しかし、すべて各場合に特有の成功例を示す各種の研究が行われており、これらの例は、むしろこの課題の複雑さを反映している。そのような例は、(i)脂質テザー(GPIアンカー)の使用による真核細胞表面での膜結合MHCクラスIIヘテロ二量体の異所性発現(Wettstein et al.,1991,J.of Exp.Medicine,174:219−228を参照されたい);(ii)昆虫細胞でのMHCクラスII外部ドメイン発現(Wallny et al.,1995,Eur.J.Immunology,25:1262−1266を参照されたい);(iii)昆虫細胞での産生と組み合わせた、MHCクラスII分子C−末端へのロイシンジッパーのような異種二量体化モチーフの導入(Quarsten et al.,2001,J.Immunol.,167:4861−4868およびCrawford et al.,2006,Immunological Reviews,210:156−170を参照されたい);(iv)昆虫細胞での産生と組み合わせた、抗体/MHCクラスIIキメラの産生(Casares et al.,1997,Protein Engineering,10:1295−1301を参照されたい);(v)封入体からのリフォールディング法により機能的pMHC三量体の形成が可能な細菌発現系の使用(Arimilli et al.,1995,J.Biol.Chem.,270:971−977を参照されたい);または(vi)封入体からのリフォールディング法により機能的pMHCの形成が可能な、α1ドメインとβ1ドメインのみからなる短縮された細菌産生一本鎖MHCクラスII形式の使用(Burrows et al.,1999,Protein Engineering,12:771−778を参照されたい)である。さらに、Landais et al.,2009,J.Immunol.,183:7949−7957 には、安定化されたマウスI−AdOVA MHCクラスII四量体作製のための、内部の人工的なジスルフィド架橋を外来性ロイシンジッパーとともに用いる昆虫細胞発現系が記載されている。重要なのは、改変による発現レベルの増加にもかかわらず、安定性が見かけ上増加した上記分子は、いずれも特異的なT細胞染色を示さなかったということである。
【発明の概要】
【0013】
本明細書に記載の戦略は、あらゆるMHCクラスII分子に適用可能な一般的戦略を提供するだけでなく、原核発現系でも実施可能であり、かつロイシンジッパーのような異種性/外来性の二量体化モチーフを使用する必要がないため、かかる先行技術の方法よりも優れた重要な利点を提供する。さらに、本明細書に記載の戦略は、導入された改変に直接起因する、特異的T細胞染色として示される機能性の増加につながる。
【0014】
しかし驚くべきことに、本発明者らは、組換えMHCクラスII分子に関連した不安定性の問題を克服する、または大幅に軽減することができる一般的方法を特定した。この戦略では、αおよびβ鎖の細胞外部分が存在し、かつα2−β2ドメイン接合点を用いて2つの鎖を結合する操作された/人工的なジスルフィド架橋により、α鎖とβ鎖のヘテロ二量体が安定化されている、組換えMHCクラスII分子を作製する。ジスルフィド結合は、分子が完全に機能する安定なコンホメーションを固定する。MHCクラスIIヘテロ二量体を安定化させるこの戦略は、汎用性があり、また単一の組換え形式に限定されないため、上で議論したような広範で供給源を要する各場合に応じた最適化の必要性が回避されるという点が重要であり、また有利である。さらにこの戦略は、数多く既存の方法とは異なり、原核発現系/宿主で実施することができる。原核宿主には、正確なジスルフィド架橋形成に一般に必要とされる真核細胞の複雑な仕組みがなく、したがって機能的分子が形成されないということを考えれば、このこと自体が驚くべきことである。本発明は、免疫調節剤の探索および開発の分野の妨げとなっている現在の問題点を解決するだけでなく、T細胞を開発および改変し、それを治療法として使用するための重要なツールとなるであろう。
【0015】
したがって、本発明の一態様では、
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含む組換えMHCクラスII分子が提供され、
(i)および(ii)は、機能的ペプチド結合ドメインを提供し、(i)および(ii)は、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基は、天然MHCクラスIIのα2およびβ2ドメイン中には存在しない。
【0016】
上で述べたように、2つ以上のポリペプチドから構成され、膜貫通ドメインを有するMHCクラスII分子のようなタンパク質は、特にその膜貫通領域およびあるいは膜に付属する構成要素により安定化される場合が多いため、天然の膜から外れた状態で、かつ特に可溶型において作製するのが困難なことがある。このことはMHCクラスII分子の場合に当てはまり、文献にもそれが反映されており、MHCクラスII分子が不安定であり、十分な収率で作製できない、またはペプチドを認識してそれと結合することができないことが文献で報告されている。したがって、本発明の分子は安定しており、かつペプチドを認識してそれと結合することもでき、またT細胞の染色も可能であるため、先行技術を上回る大きな進歩を示すものである。
【0017】
上で議論したように、天然のMHCクラスII分子はα鎖とβ鎖を含み、両鎖とも膜貫通領域を有し、免疫グロブリン(Ig)スーパーファミリーに属する。各鎖の細胞外部分は、それぞれ約90個のアミノ酸残基からなる2つのドメインから構成され、そのうちの2つの膜遠位ドメインであるα1ドメインとβ1ドメインが、T細胞エピトープのペプチド結合性に不可欠である、互いに格子状になったα/β構造を形成している。2つの膜近位ドメインであるα2ドメインとβ2ドメインは、ともに分離したIgドメインを形成している。αおよびβの両鎖では、約20個のアミノ酸残基の配列が細胞膜を横断し、膜の細胞質側にはごく短いペプチドセグメントがある。
【0018】
本発明の分子は、MHCクラスIIα鎖の細胞外部分の全部または一部分と、MHCクラスIIβ鎖の細胞外部分の全部または一部分とを含む。MHCクラスIIα鎖の細胞外部分は、シグナル配列と、膜遠位α1ドメインと、膜近位α2ドメイン(分離したIgドメインを形成している)とを含む。膜貫通ドメインとα2ドメインの間にスペーサー領域も存在する。同様に、MHCクラスIIβ鎖の細胞外部分は、シグナル配列と、膜遠位β1ドメインと、膜近位β2ドメイン(分離したIgドメインを形成している)と、スペーサー領域とを含む。
【0019】
したがって、本明細書で使用される「MHCクラスIIα鎖の細胞外部分」という用語は、α鎖の膜貫通ドメインまたは細胞質ドメインを包含しない。実際、本発明の好ましい実施形態では、組換えMHCクラスII分子は、前記膜貫通ドメインのアミノ酸残基(すなわち、専用の膜貫通エクソンによりコードされるアミノ酸)または前記細胞質ドメインのアミノ酸残基のいずれも含まない。
【0020】
したがって同様に、本明細書で使用される「MHCクラスIIβ鎖の細胞外部分」という用語は、β鎖の膜貫通ドメインまたは細胞質ドメインを包含しない。実際、本発明の好ましい実施形態では、組換えMHCクラスII分子は、前記膜貫通ドメインのアミノ酸残基(すなわち、専用の膜貫通エクソンによりコードされるアミノ酸)または前記細胞質ドメインのアミノ酸残基のいずれも含まない。
【0021】
αおよび/またはβ鎖のすべての前記細胞外部分が本発明の組換え分子(すなわち、シグナルペプチド、α1/β1ドメイン、α2/β2ドメインおよびスペーサー領域)中に存在し得る。しかし、組換え分子が、適当なペプチド、例えばT細胞エフェクターペプチドと結合する能力に関して依然として機能的である限り、また前記分子が、人工的な(非天然の)システイン残基を、組換えMHCクラスII分子の安定化に機能するジスルフィド結合の形成に使用可能な形態または立体配置で含む限り、代わりにαおよび/またはβ鎖の細胞外部分の一部分だけが本発明の分子中に存在していればよい。
【0022】
シグナルペプチドは、特にMHCクラスII分子を原核細胞で発現させる場合、本発明の組換えMHC分子のαおよび/またはβ鎖中に欠けていてもよい。適当な数のN末端アミノ酸残基を欠くこのような構築物は、当業者が容易に設計し作製することができる。またかかる欠如が機能に対して、例えば、ペプチドと結合するまたはT細胞を活性化もしくは染色する能力、あるいは分子の安定性に対して影響を及ぼすか否かを容易に試験することもできる。
【0023】
またスペーサー領域も、例えば、α鎖スペーサーおよびβ鎖スペーサーの9個のアミノ酸がいずれも含まれていない実験例に示されるように、その全体が欠けていても、または短縮されていてもよい。ここでも、適当な数のスペーサーアミノ酸残基を欠くこのような構築物は、当業者が容易に設計し作製することができる。またかかる欠如が機能に対して、例えば、ペプチドと結合するまたはT細胞を活性化もしくは染色する能力、あるいは分子の安定性に対して影響を及ぼすか否かを容易に試験することもできる。
【0024】
本発明の好ましい分子は、α1ドメインの少なくとも一部分とβ1ドメインの少なくとも一部分とを含む。ただし、これらのドメインがペプチドと結合する機能、または本明細書に記載の他の機能、例えば、かかるペプチドをT細胞受容体(TCR)に提示する機能が影響を受けない。さらに組換えMHCクラスII分子が全体として、ペプチドをT細胞受容体(TCR)に提示するために、かかるペプチドと結合する能力または本明細書に記載の他の機能に関して、依然として機能的でなければならない。好ましくは、完全または完全長のα1および/またはβ1ドメインが存在するか、または実質的に完全または実質的に完全長のα1および/またはβ1ドメインが存在し、前記実質的に完全または実質的に完全長のドメインは、例えば、ペプチドをT細胞受容体(TCR)に提示するために、かかるドメインがかかるペプチドと結合する機能、または本明細書に記載の他の機能に影響を与えない、天然配列からの変化、例えば、アミノ酸の付加、欠失または置換を含む。かかる機能に影響を与えずに変異させる、修飾するまたは削除することができるアミノ酸の決定は、当業者の技能の範囲内であろう。MHCクラスII分子が保持するべき他の好ましい機能は、T細胞を活性化する能力、より好ましくは、T細胞を染色する能力である。
【0025】
したがって、本発明におけるこれらの好ましい分子は、例えば、ペプチドをTCRに提示する、T細胞を活性化する、またはT細胞の染色を可能にするために、かかるペプチドと結合することができるように、十分なα1およびβ1ドメイン残基を含む。
【0026】
本発明の他の好ましい分子は、α2ドメインの少なくとも一部分とβ2ドメインの少なくとも一部分とを含む。ただし、ジスルフィド結合を形成するために使用される非天然のシステイン残基が存在する。かかるシステイン残基は、システイン残基間にジスルフィド架橋を形成することができるように、また組換えMHCクラスII分子の安定化に作用することができるように、互いに適切な方向および距離で存在している必要がある。さらに組換えMHCクラスII分子が全体として、ペプチドをT細胞受容体(TCR)に提示するために、かかるペプチドと結合する能力または本明細書に記載の他の機能に関して、依然として機能的でなければならない。好ましくは、完全または完全長のα2および/またはβ2ドメインが存在するか、または実質的に完全または実質的に完全長のα2および/またはβ2ドメインが存在し、前記実質的に完全または実質的に完全長のドメインは、α2ドメインとβ2ドメイン間のジスルフィド結合の形成およびその結果としての組換えMHCクラスII分子の安定化に影響を与えず、かつα2またはβ2ドメインのフォールディングに悪影響を及ぼさない、天然配列からの変化、例えば、アミノ酸の付加、欠失または置換を含む。かかる機能に影響を与えずに変異させる、修飾するまたは削除することができるアミノ酸の決定は、当業者の技能の範囲内であろう。
【0027】
各種の構造的および機能的ドメインアミノ酸位置ならびにαおよびβMHCクラスII鎖の領域は、当該技術分野において公知であり記載されている(例えば、Burrows et al.,1999,上記)。また各種ドメインの配置も図3に示されており、このような情報を用いて、本明細書に記載の本発明の組換え分子(例えば、図7を参照されたい)を容易に設計することができる。
【0028】
本発明の組換え分子がペプチドと結合する、T細胞を活性化する、またはT細胞を染色する能力を評価するための適当な機能試験も当業者に公知であり、例えば、Biacoreのような表面プラズモン共鳴(SPR)技術およびFACSのようなフローサイトメトリー技術がこれに含まれるであろう。フローサイトメトリーは生細胞と組み合わせて使用できるため、これを使用することが特に好ましい。
【0029】
本明細書で使用される「ジスルフィド結合」という用語は、MHCクラスIIヘテロ二量体のα鎖のα2ドメイン中に位置するシステイン残基とβ鎖のβ2ドメイン中に位置するシステイン残基との間で形成される(すなわち、鎖内ではなく、鎖間のジスルフィド結合である)、任意のジスルフィド架橋、例えば、操作されたまたは人工的なジスルフィド架橋を表す。前記ジスルフィド結合はα鎖とβ鎖間で共有結合を形成し、MHCクラスIIヘテロ二量体の安定化に作用する。したがって、本発明の組換え分子は安定であるが、完全な機能性を保持している。例えば、本発明の組換え分子は、その同族T細胞受容体リガンドに対する本来の特異性を保持し、好ましくは、T細胞を活性化する、またはT細胞を染色する能力を保持している。
【0030】
安定であるまたは安定化されているという本発明のMHCクラスII分子の特性は、温度変性に対する耐性の増加(例えば、ELISA法、SPR法または円二色法により測定される)のような公知の方法により評価することができる。さらに好ましい試験は、ヘテロ二量体の保存をSDS PAGEゲル上で評価することである。インタクトで安定なジスルフィド結合ヘテロ二量体は、非還元条件下で泳動したSDS PAGEゲル上で容易に確認することができ、この場合、インタクトのヘテロ二量体に対応する適当な分子量の位置にあるバンドを可視化することができる。適当なアッセイを実施例で示す(図5および10を参照されたい)。
【0031】
かかるジスルフィド結合は、天然のMHCクラスIIα2およびβ2ドメインには通常は存在しないシステイン残基間で形成される。したがって、例えば、部位特異的突然変異により天然分子中の適当な非システイン残基からシステイン残基に変異させることにより、システイン残基を操作するかまたは人工的に導入して、α2およびβ2ドメイン内に新たに導入されたシステイン残基間でジスルフィド結合を形成させる。このような結合は内部ジスルフィド結合とも呼ばれる。
【0032】
システインへの変異に適した残基は、天然のMHCクラスIIヘテロ二量体において、約6Å(0.6nm)、7Å(0.7nm)またはそれ未満だけ離れた、例えば、4Å(0.4nm)または5Å(0.5nm)〜6.5Å(0.65nm)または7Å(0.7nm)の範囲、好ましくは、5Å(0.5nm)〜6.5Å(0.65nm)の範囲、最も好ましくは、4Å(0.4nm)または5Å(0.5nm)〜5.6Å(0.56nm)または6Å(0.6nm)の範囲だけ離れた別々のβ炭素を有することが好ましい。変異に好ましい部位は、種間ならびに特定種内のアイソフォーム間およびアイソタイプ間で保存されている。具体的には変異に好ましい部位は、マウスとヒトのMHCクラスII配列間で保存されているか、またはヒトMHCクラスIIアイソタイプ(例えばDP、DQおよびDRなど)間またはマウスアイソタイプ(例えばI−EおよびI−Aなど)間で保存されている。
【0033】
代わりにまたは加えて、結晶構造の3D重ね合わせに基づく構造評価により、変異に好ましい部位を特定する。このようにして、α2ドメインとβ2ドメイン間の接合点を形成している残基を調べることができ、またさらなる解析またはシステイン残基への変異のために、7Å以下(または実際は、上記の他の任意の距離もしくは範囲)の距離だけ離れたβ炭素を有する側鎖を選択することができる。かかる構造評価を行うための方法は当業者に公知であろう。例えば、この解析を行うために、MHCクラスII分子の結晶構造を、例えばRCSB PDBタンパク質データバンクから自由に入手することができる。かかる結晶構造の3D重ね合わせ行うための適当なソフトウェアまたはその他の手段も当該技術分野において、例えば、PyMOL、MOLMOL、DeepViewのような自由に入手できるソフトウェア、またはiSuperposeのような専用のウェブサイトを用いて利用することができる。
【0034】
特に好ましい部位の対は、ジスルフィド結合を形成するようにシステインが導入された、以下に挙げる対のうちの1つ以上、すなわち、Pro96α2−Ser119β2(1位)、Ser95α2−Ser121β2(2位)、Arg94α2−Asn151β2(3位)、Phe148α2−Gly152β2(4位)、Pro96α2−Thr101β2(5位)、Pro96α2−Ser121β2(6位)、Ile106α2−Asn151β2(7位)およびSer95α2−Asp122β2(8位)のうちの1つ以上の対である。これらの対はβ炭素の近接度の順に並べられており、どの対を用いてもよいが、1位〜4位または1位〜3位の対が好ましい。特に1位または2位の対が好ましい。
【0035】
α2およびβ2ドメイン中にあるおよび上記の残基の対におけるアミノ酸位置およびその位置の天然アミノ酸の性質は、マウスI−EアイソタイプのMHCクラスII分子に適している。アミノ酸番号付けは、成熟ペプチドのアミノ酸に関するものである(すなわち、番号付けにはシグナルペプチドは含まれていない)。改変システイン残基の位置を特定するために使用することができる参照配列の例は、IMGTデータベースで得られるI−E配列であり、これを図3に示す(α鎖ではH−2EA*02(配列番号1)およびβ鎖ではH−2EB*01(配列番号2)で示される)。実際、図3では1位、2位および3位の残基の位置に黒い影で印が付してあり、その他の順位の残基は図3から容易にわかる。
【0036】
したがって、別途記述がない限り、本明細書に記載のMHCクラスIIアミノ酸残基の番号付けおよび性質は、Lefranc,M−P.ら,2009(Nuc.Acids Res.,37:D1006−D1012,データベース記事)に記載されているIMGT方式および以下のウェブサイト参考文献:http://imgt.cines.fr;http://www.imgt.org.にあるIMGTデータベースに従っている。関連するGenBankアクセッション番号も実施例に記載されている。例えば、H−2E(マウスI−E)で関連するアクセッション番号はK00971(α鎖)およびAF050157(β鎖)である。
【0037】
本発明の好ましい実施形態では、ジスルフィド結合は、マウスI−Eアイソタイプの成熟ポリペプチドのPro96α2−Ser119β2(1位)、Ser95α2−Ser121β2(2位)もしくはArg94α2−Asn151β2(3位)に対応する残基、または別のMHCクラスIIアイソタイプのこれに相当する位置に位置するシステイン残基間にある。かかるシステイン残基の位置を決定するための参照配列が本明細書に記載されている。
【0038】
上で議論した好ましい部位の対は、1FNG番号付け、すなわち、Pro96α2−Ser118β2(1位)、Ser95α2−Ser120β2(2位)、Arg94α2−Asn150β2(3位)、Phe148α2−Gly151β2(4位)、Pro96α2−Thr100β2(5位)、Pro96α2−Ser120β2(6位)、Ile106α2−Asn150β2(7位)およびSer95α2−Asp121β2(8位)のうちの1つ以上、ならびにIMGT番号付けの両方を用いて、表2にも示されている。1FNG番号付けに関して、表2のアミノ酸番号付けは、生体高分子の三次元構造情報のタンパク質データバンク(PDB)データベース(登録番号PDB ID:1FNG)で番号付けに対応しており、このデータベース登録のβ鎖中の適当な残基の位置が、IMGTデータベース配列のこれに相当する残基よりも1アミノ酸小さいということがわかる。したがって、この命名法は少し異なっている。
【0039】
表2および図3に示されている残基に相当する残基は、他のマウスアイソタイプ、例えばI−Aアイソタイプ、またはヒトアイソタイプにおいて、例えば、Clustalソフトウェアのような適当なソフトウェアを用いたアライメントにより容易に特定することができる。実際にマウスI−Aアイソタイプとのアライメントが図3に示されている。さらに、ヒトMHCクラスIIアロタイプであるHLA−DP、−DQおよび−DRとのアライメントの例も図7に示されている。表2および図3に示されている1位、2位および3位の残基に相当する残基の位置は、α鎖に関しては図7Aおよびβ鎖に関しては図7Bの中で黒い影で印が付してあり、その他の順位の残基の位置は図7から容易にわかる。表2および図3に示されているα2およびβ2の位置は、ヒトHLAレパートリー全体で完全に保存されていることがわかる。
【0040】
したがって、所望の順位のジスルフィド結合を形成するために、図7に示される配列アライメントを上の情報とともに用いて、任意のヒトMHCクラスII対立遺伝子のα2およびβ2ドメインにおいて、システインに変異させるのに適当な残基の位置を容易に特定することができる。同様のアライメント法を用いて、他の任意の種またはアイソタイプにおいて相当する残基を特定することができる。
【0041】
本明細書で議論されるジスルフィド結合による安定化結合は、先行技術で述べられているような、MHCクラスII分子を安定化する他の方法および手段と適合性がある。例えば、ジスルフィド結合を、ロイシンジッパーモチーフのような各種の二量体化モチーフ(例えば、Quarsten et al.,2001およびCrawford et al.,2006(上記)に記載されているような)とともに、またはIg融合(すなわち、例えばCasares et al.,1997(上記)に記載されているような免疫グロブリンのFc部分との融合)とともに用いることができ、また本発明の分子を、それをコードするベクターとともに適当に設計することができる。
【0042】
本明細書の他の箇所に記載されているように、原核宿主、例えば細菌宿主で、本発明の分子の発現または産生させることが好ましく、このような実施形態、具体的には分子を封入体から単離する実施形態では、ロイシンジッパーモチーフまたはその他の二量体化モチーフを使用しないことが好ましい。
【0043】
したがって、本発明の好ましい態様では、
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含み、細菌宿主で発現可能な組換えMHCクラスII分子が提供され、
(i)および(ii)は、機能的ペプチド結合ドメインを提供し、(i)および(ii)は、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基は、天然MHCクラスIIのα2およびβ2ドメイン中には存在しない。
【0044】
別の好ましい態様では、
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含む組換えMHCクラスII分子が提供され、
(i)および(ii)は、機能的ペプチド結合ドメインを提供し、(i)および(ii)は、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基は、天然MHCクラスIIのα2およびβ2ドメイン中には存在せず、さらに前記組換え分子はロイシンジッパーモチーフを含まない。他の実施形態では、二量体化モチーフが全く含まれない。これらの実施形態では、組換えMHCクラスII分子が原核宿主、例えば細菌宿主で発現させることができることが好ましい。
【0045】
本発明のジスルフィド結合による結合を、MHCクラスII分子を安定化させる他の方法および手段とともに使用することができるが、前記ジスルフィド結合は、MHCクラスII分子を安定化させる単独の手段を提供する。実際、このような実施形態が好ましい。本明細書で使用される「MHCクラスII分子を安定化させる単独の手段」という用語は、特定のMHCクラスII分子に天然にまたは本来的に存在するあらゆる安定化以外の、分子を安定化させるための単独または唯一の手段を提供するジスルフィド結合を表す。例えば、本発明のジスルフィド結合は、MHCクラスII分子を安定化させるための単独または唯一の、人工的または操作されたまたは非天然の手段を提供する。したがって、このような実施形態には、当該技術分野において記載されている、ロイシンジッパーまたはその他の二量体化モチーフのような他の安定化の手段の使用は含まれていない。
【0046】
かかるジスルフィド結合が、二量体化モチーフのような非天然の安定化の手段をさらに使用することなくMHCクラスII分子を十分に安定化させるという驚くべき発見は、本明細書に記載のデータに明確に示されており、本明細書では、MHCクラスII分子が繊維状ファージの表面に安定かつ機能的な形態で示される。この発見は、Igフォールドトポロジーで作製される操作された分子が原核宿主で発現されたということを考慮すると、特に驚くべきことであった。天然では生存している真核ドメインでのみ生じるIgフォールドは、その機能的なトポロジーに達するために、天然のシステイン間で形成される保存されたドメイン内S−S架橋を必要とする(Halaby,D.M.,ら,1999,Protein Eng.,12(7):563)。したがって、原核宿主には正確なS−S架橋形成に必要な真核細胞の複雑なシャペロン機構が必然的に欠けているため、MHCクラスII分子がそこで発現された場合、異常なS−S架橋形成が顕著であるため、機能的な発現が行われないということが一般に認められている。したがってこれらの系における、明らかに人工的なS−S架橋形成とまでは言わないが、システイン数の増加を含む操作戦略は、一般に発展不可能であると考えられている。ここで本発明者らは、共有結合による二量体形成(図5および10)により示されるようにファージ上に機能的分子が示され、それらが同族リガンドとの特異的結合(図6および9)を示しているため、このことが当てはまらないという証拠を明確に記載する。
【0047】
一実施形態では、本発明のMHCクラスII分子のαおよびβ鎖は、鎖内ジスルフィド結合、例えば、天然のシステイン残基間に存在する天然の鎖内ジスルフィド結合も含む。このような天然の鎖内ジスルフィド結合は、例えば、α2およびβ2ドメインのIgフォールドトポロジーを形成するために、これらのドメイン中に存在していてもよい。さらに、MHCクラスII分子の大部分はβ1ドメイン内に、βシート底面とαへリックス部分とを結び付けるドメイン内ジスルフィド架橋を有する。さらに重要なのは、多くのMHCクラスII分子が、どのジスルフィド架橋形成にも関与しないが、新たに導入された非天然のシステイン残基と誤ったジスルフィド結合を形成し得る遊離システインを、β1ドメインのβシート底面にさらに含むということである。したがって、本発明の好ましい実施形態、特に原核発現を用いる実施形態では、例えば、全体の構造を保つセリンまたはアラニンのような別の残基への変異により、この残基を除去する。当業者は、任意のMHCクラスII対立遺伝子において、このシステイン残基の位置を容易に特定することができるであろう。例えば、このシステインは、図3Bに示される完全長のIMGT参照配列H−2EB*01(配列番号2)中のβ鎖残基38または成熟IMGT参照配列(すなわち、シグナルペプチドを含まない)中の残基12に対応する。
【0048】
新たに導入された非天然のシステイン残基との誤ったジスルフィド結合の形成にも関与し得るその他のβ1ドメインシステイン(架橋を形成する)も、当業者により容易に位置が特定され得る。例えば、これらは、図3Bに示される完全長のIMGT参照配列H−2EB*01(配列番号2)残基42および106または成熟IMGT参照配列(すなわち、シグナルペプチドを含まない)のそれぞれ残基16および80の位置にある。本発明の別の好ましい実施形態では、1つ以上のこのようなシステイン残基が存在しない。結合形成を防ぐために、1つ以上の適当な天然のシステイン残基をジスルフィド結合形成に関与しない別のアミノ酸残基へ変異させることにより、これを達成することができる。システインと置き換わる残基の例は、分子の全体構造を保つ残基、例えば水素結合を保つ残基であろう。上で議論したSerまたはAlaのいずれかが好ましい選択であろう。
【0049】
したがって、上で議論した1つ以上の天然システイン残基の除去は、天然のシステイン残基と本発明の新たに操作された非天然のシステイン残基との間での誤ったジスルフィド結合形成を防ぐのに役立つであろう。したがって、本発明の好ましい実施形態では、β1ドメイン中の1つ以上の天然システイン残基を除去する。本発明の特に好ましい実施形態では、完全長の参照配列H−2EB*01(配列番号2)の38、42もしくは106の位置に対応する1つ以上のシステイン残基(または成熟参照配列中のそれに相当する残基)、または別のMHCクラスIIアイソタイプのそれに相当する位置にある1つ以上のシステイン残基を除去する。
【0050】
本明細書で使用される「機能的ペプチド結合ドメイン」という用語は、ペプチド、例えばT細胞エフェクターペプチドまたは抗原ペプチドと結合することができる、本発明の組換えMHCクラスII分子中のドメインを表す。ペプチドとのかかる結合は検出可能なレベルであるべきであり、また結合を検出するための適当な方法は当業者に公知であろう(例えば、表面プラズモン共鳴(SPR)技術またはフローサイトメトリー技術)。いくつかの実施形態では、前記ペプチドとは、前記ペプチドのTCRへの提示が可能なように、または少なくとも前記ペプチドのTCRへの提示が可能か否かを試験できるように結合する。
【0051】
好ましくは、かかるペプチドは、TCRとpMHC複合体の結合が可能となるようにMHCクラスII分子と結合または会合する。より好ましくは、TCRとのかかる相互作用により、pMHC複合体を認識するT細胞が染色されるか、または他の方法で可視化される。好ましくは、かかるペプチド結合ドメインは、ペプチドと結合し、次いでTCRを介してT細胞の活性化を引き起こすことができる。例えば、かかるペプチド結合ドメインは、Tヘルパー細胞の、例えばサイトカイン(IL−2など)の分泌または増殖の誘導(例えば、BrdU取込みによりcpmとして測定される)を誘発することができる。かかるペプチド結合ドメインは一般に、MHCクラスII分子のα1およびβ1ドメイン由来の残基により形成さる。
【0052】
本発明のMHCクラスII分子を、空の形態または未負荷の形態で、すなわち、ペプチドが上記ペプチド結合ドメインと結合していない状態(すなわち、無ペプチド)で提供し得る。この場合、後でMHCクラスII分子に適当なペプチドをインビトロで負荷することができる。このようなインビトロでの負荷は、クラスIIヘテロ二量体の安定性を補助するために、例えば、短いリンカーを介してMHCβ鎖と共有結合した融合タンパク質としてペプチドを作製することにより、ペプチドが付加した状態で作製しなければならない多くの先行技術のMHCクラスII分子よりも有利である。インビトロ負荷により、異なるMHC−ペプチド複合体それぞれに対する異なる発現ベクターを得る必要なしに、真に汎用性のあるMHCクラスII分子を作製することができる。
【0053】
本発明のいくつかの実施形態では、組換えMHCクラスII分子は、上記ペプチド結合ドメインと結合または会合するペプチドを有する。MHCクラスII分子のα1およびβ1ドメイン由来の残基により形成されたペプチド結合ドメインとの結合または会合に適した任意のペプチドを使用することができる。一般にこのようなペプチドは、12〜25マーであり、通常、α1およびβ1ドメインにより形成される溝の両端からはみ出ている。
【0054】
天然のMHCクラスII分子では、かかるペプチドは外来抗原に由来する。本発明では、任意のこのようなペプチドを使用し得る。例えば、当該技術分野において、MHCクラスII分子により提示されてTCR結合およびT細胞活性化を生じる特定のT細胞エフェクターペプチドがいくつか同定され記載されており、そのいずれも本発明とともに使用し得る。特に、MHCクラスII分子上で提示される特定のペプチドが、特定の疾患と関連するものとして同定されており、組換えMHCクラスII分子がペプチドと会合する本発明の実施形態では、このような疾患特異的なペプチドが好ましい。このようなペプチドは当該技術分野においていくつか記載されており、将来的には他のペプチドも同定されるであろうが、上記ペプチドはいずれも、本明細書に記載のMHCクラスII分子との使用に適するであろう。
【0055】
MHCクラスII分子のペプチド結合ドメインと会合または結合するペプチドの代表例には、HLA−DQ2 MHCクラスII分子上で提示されることがわかっており、セリアック病と関連するヒトα−II−グリアジン(N−PQPELPYPOPE−C);HLA−DR4 MHCクラスII分子上で提示されることがわかっており、関節リウマチと関連するヒトhCkappaaa40-48(N−WKIDGSERQ−C);HLA−DP1 MHCクラスII分子上で提示されることがわかっており、破傷風と関連するヒトTTaa947-967(N−FNNFTVSFWLRVPKVSASHLE−C);およびマウスI−EdクラスII分子上で提示されることがわかっている、H.インフルエンザのヘマグルチニン(HA)に由来するペプチド(aa110−120:N−SFERFEIFPKE−C)がある。
【0056】
あるいは他のペプチドを使用して、例えば、そのペプチドがペプチド結合ドメインと結合することができるか否か、またそのペプチドにより、TCRを介したT細胞結合および好ましくはT細胞染色が可能であるか否か、またはT細胞活性化が可能であるか否かを確認してもよい。このように、本発明の分子を用いて、T細胞エピトープとして働く、これまで未知であった新規なペプチド(例えば、抗原ペプチド)を同定することができる。
【0057】
あるいは、組換えMHCクラスII分子を、無関係な、すなわちT細胞エフェクターでないペプチド(いわゆる「スタッファー」ペプチド)(このペプチドをMHC分子と結合させているリンカーの切断によりMHCクラスII分子から解離し得る)と会合させ、次いで、インビトロペプチド交換反応で目的とするT細胞エフェクターペプチドと置き換えることができる。
【0058】
任意の適当な方法で、かかるペプチドとMHCクラスII分子との結合または会合を促進することができる。例えば、かかるペプチドがMHCクラスII分子とともに産生されるように本発明の構築物を操作する(例えば、これらを同じ構築物にコードさせる、例えば、これらを適当なリンカー配列によりβMHCクラスII鎖またはαMHCクラスII鎖と共有結合させてコードさせることにより、または同一宿主細胞内の異なる構築物にコードさせることにより)ことにより、適当なTヘルパー細胞による認識に適した前記ペプチドを提示する、またはそれと結合もしくは会合する組換えMHCクラスII分子を産生させることが可能である。このようなペプチド関連分子を作製するための適当な方法は、Kozono et al.(1994)により記載されている(Nature,369:151−154)。Kozonoの方法では18アミノ酸残基のリンカーを使用するが、本発明の好ましい実施形態では、これよりも短いリンカー、例えば15アミノ酸リンカー(例えば、(G4S)3のようなGly−Serリンカー)または6、7もしくは8アミノ酸残基のリンカー(例えば、Gly−Serリンカー、例えばGSGSGS、GGSGSGS、SGSGSGSまたはSGGSGSGSなど)など、最も好ましくは、6アミノ酸残基のリンカーを使用する。
【0059】
本発明は、任意のタイプのMHCクラスII分子に対して一般的に適用可能である。例えば本発明は、任意の種由来のMHCクラスII分子およびその種内のMHCクラスII分子の任意のサブタイプに対して適用可能である。特に、本明細書で特定された、非天然のシステイン残基に変異させて1つ以上の鎖間ジスルフィド結合を形成させることが可能な残基が種間で保存されていることにより、このアプローチの一般性が支えられている。したがって、本発明をあらゆる哺乳動物のMHCクラスII分子、例えば、ヒト、マウス、ラット、ブタ、ヤギおよびヒツジ、特にヒトおよびマウスの分子に適用することができる。例えば、本発明は、DP、DQおよびDRヒトMHCクラスII分子(すなわち、ヒトにおいて同定されているMHCクラスII分子の3つの機能的タイプ)に対して、またマウスI−AおよびI−E分子(例えば、I−EdおよびI−Ek分子、好ましくはI−Ed)に対しても適用可能である。I−AおよびI−E分子の他の例を下の表に示す。
【0060】
【表1】
【0061】
最も好ましいMHCクラスII分子はヒトである。
【0062】
システインへの変異のために特定された残基は種間で保存されているため、当業者であれば、任意の種由来の任意のMHCクラスII分子において、変異のための対応する、同等の適当な残基を容易に特定し、1つ以上の適当な鎖間ジスルフィド結合により安定化された組換えMHCクラスII分子を作製することができるであろう。
【0063】
本発明の一実施形態では、組換えMHCクラスII分子を、細胞または別の生物学的存在もしくはパッケージの表面、例えば、繊維状ファージの表面で発現させる。この型は、本明細書では「不溶性」型または「不溶性」分子と呼ばれることがある。このような実施形態では、一般に本発明のMHCクラスII分子のα鎖またはβ鎖が、当該存在物の表面で通常発現される、または当該存在物の表面に組み込まれているかそれに付随しているタンパク質との融合タンパク質として操作される。
【0064】
ファージ表面タンパク質との融合体としての発現が好ましく、このような実施形態では、本発明のMHCクラスII分子のα鎖またはβ鎖と任意の適当なファージ表面タンパク質との融合が考えられる。好ましい例は、gpIII、gpVIII、gpVIIまたはgpIXとの融合、最も好ましくはgpIIIとの融合である。ファージ粒子表面での本発明のMHCクラスII分子発現の方法論は、当業者の技術の範囲内であり、技術および方法の例が実施例に記載されている。好ましい方法論は、例えば国際公開第09/024591号に記載されている。
【0065】
本発明の別の実施形態では、組換えMHC分子は可溶性分子、例えば、ウイルスキャプシドタンパク質との融合体または生物学的存在の表面との会合または複合体形成を生じる他のタンパク質との融合体などとして細胞または生物学的存在の表面に付随していないまたはそこで発現されない、本発明のMHCクラスII分子である。したがって、可溶性分子の例としては、本発明の分子の(i)および(ii)の細胞外部分のみを含む分子が挙げられ、また分子の溶解性に影響を与えない他の短い構成要素、例えば親和性タグおよび/または二量体化モチーフのような短いC末端伸長などを含めた外部ドメインも挙げられる。作製方法に応じて、かかる可溶性分子は、宿主細胞から分泌されるか、または他の任意の適当な方法により宿主細胞から入手され得る。かかる可溶性分子を、実質的に純粋な形態で、または精製もしくは単離された調製物として得てもよい。例えば、かかる可溶性分子を、実質的に他のタンパク質を含まない形態で作製し得る。
【0066】
本発明の好ましい実施形態では、組換えMHCクラスII分子を、例えば多価性の多量体型で得てもよい。一般に単一のpMHC複合体とT細胞受容体との間の親和性は非常に低いため、かかる多量体型は、MHCクラスII分子とT細胞受容体の結合を可能にするのに有利である場合が多い。かかる多量体型は、複数(2つ以上)の本発明の組換えMHCクラスII分子を含む。複数のMHCクラスII分子はそれぞれ同一であることが好ましい。
【0067】
多量体型のMHCクラスII分子を調製する任意の適当な方法を使用してよく、そのうちのいくつかは、当該技術分野において記載されている(例えば、Vollers et al.,2008(上記)を参照されたい)。多量体型の本発明のMHCクラスII分子を調製するのに特に有利な方法は、繊維状ファージ表面での提示を利用するものである。このような方法では、MHCクラスII分子が融合し付随する適当なファージ構造タンパク質を選択することにより、多量体を作製するためにファージ分子の天然の構造を利用することができる。例えば、gpIII、gpVIIまたはgpIXとの融合、好ましくはgpIIIとの融合を用いて、各ファージ粒子の表面に3〜5コピーのMHCクラスII分子を得ることができる。gpVIIIとの融合を用いて、これよりも多くのコピーを得ることができる(野生型ファージには、約2700コピーのgpVIIIが存在する)。したがって重要なのは、pVIII提示法により、MHCの価数がgpIII提示法および古典的なテトラマー技術に比べて少なくとも一桁分増加するため、感度が大幅に増加するということである。これにより新たな興味深い適用が開拓されるであろう。
【0068】
既知のファージ提示法および構築物設計技術、例えば、ファージミド構築物および改変型のヘルパーファージを用いて、ファージ表面のMHCクラスII分子のコピー数を変化させることができる。ファージ表面に多量体MHCクラスII分子を生成する能力を用いれば、1回の迅速な費用効率の高い工程で多量体MHCクラスII分子の製造または作製が可能となるという点で有利である。Vollers et al.(2008)に記載されているような従来のテトラマー技術では、各構成要素を別々に作製し、混合して複合体を形成し、精製した後に試薬が下流での適用に適した状態になるが、これとは極めて対照的である。
【0069】
したがって、好ましい多量体は、互いに会合した2つ、3つ、4つ、5つまたはそれを上回る組換えMHCクラスII分子を含む。このような会合は、当該技術分野において既知であり記載されている方法を用いて行われ得るが、一般的には、リンカー分子のような別の結合により仲介される。適当なリンカー分子は当該技術分野において公知で、記載されており、特に適切なリンカー分子は、組換え分子が結合することができる複数の結合部位を有する。例えば、それぞれが複数のビオチン結合部位を有する、アビジンおよびストレプトアビジン(またはビオチンと多価的に結合する他の任意の分子)のような複数の結合分子を使用し得る。したがって、当該技術分野で公知の方法(例えば、AviTagまたはその他のBirA基質を用いて酵素的ビオチン化を可能にする)により組換えMHC分子へビオチンを組み込むことにより、多量体、例えば四量体のMHC分子を形成することが可能である。また、例えば、蛍光ストレプトアビジンを用いて、または標識とファージコートタンパク質(例えば、MHCクラスII分子の提示用に選択したものとは異なるタイプのコートタンパク質)との融合により、標識を多量体型に好都合に組み込むこともできる。こうした標識を組み込むことにより、既知の各種技術で四量体を迅速に検出することが可能となる。例えば、蛍光標識により、特に有利な、FACS解析のようなフローサイトメトリー技術を用いることが可能となる。
【0070】
また、本発明のMHCクラスII分子またはその多量体を、平面状または粒子状の固体支持体のような固体支持体、例えば膜、プレートまたはビーズの上にコーティングし得る。このための技術は当該技術分野において公知であり、記載されている。
【0071】
本発明のMHCクラスII分子、および前記分子を安定化させるために、本明細書に記載のようにMHCクラスII分子のα2ドメインとβ2ドメイン間の1つ以上の操作されたジスルフィド結合を用いることは、MHCクラスII分子作製のためのあらゆる先行技術の方法と適合性がある。したがって、本発明は、当該技術分野において記載されている従来の型のMHCクラスII分子のいずれとも適合性があり、作製効率および生成物の安定性を向上させるであろう。特に、本発明により組換えMHCクラスIIの適用性および汎用性が広がるため、分子は、例えば、(i)細菌ペリプラズム内の機能的可溶性分子、(ii)好ましくは、現在の標準的なプロトコルにより得られるもの(最大で出発物質の30%、例えば、Arimilli et al.,1995(上記)を参照されたい)よりも実質的に高い収率で精製した後に、可溶化し機能的な存在にリフォールディングすることができる、細菌封入体の非機能的構成要素、(iii)繊維状ファージ系でファージ提示される構成要素、(iv)真核細胞内のIg融合体、および(v)真核細胞内の可溶性MHCクラスII分子として、ジスルフィド結合により安定化された形態で作製され得る。
【0072】
アプローチ(ii)(封入体アプローチ)およびアプローチ(iii)(ファージ提示アプローチ)は、組換えMHCクラスII分子の効率的な使用で非常に重要な2つの適用を直ちに行うことが可能な点で有利であり、このうちアプローチ(iii)は全く新規なものである。この点に関して、繊維状ファージビリオン上でのpMHCクラスIIの良好な機能的提示法はこれまで達成されたことがなく、ビリオンをわずかなコストで迅速に作製し得ることを除けば、一連の新たなまたは向上した適用、例えば(a)MHCクラスIIに関連したファージ提示ペプチドライブラリー;(b)所与のpMHCクラスIIの組合せの極めて迅速かつ簡便な作製;および(c)四量体のような従来のpMHCクラスII多量体に代わるMHCクラスII分子の多価の繊維状ファージ提示などを開拓するものである。
【0073】
本発明のMHCクラスII分子には、研究試薬として特定の価値および用途がある。このことは、本明細書に記載の多価型およびファージ提示型(MHCクラスII分子が提示されるファージ粒子の固有の構造により、多価になるよう容易に設計することができる)について特に当てはまる。したがって、四量体型およびファージ提示型のMHCクラスII分子、特に多価ファージ提示型がこの用途には好適である。本発明の四量体型または多価ファージ提示型のMHCクラスII分子を、従来の四量体が使用されている任意の適用に好ましい代替試薬として使用することができる。本発明の分子を従来の四量体の代わりに使用することができる好ましい適用は、一般に二量体およびより高次のオリゴマーのようなpMHCオリゴマーを用いたフローサイトメトリーによるT細胞の検出を含む、T細胞のMHC染色におけるものである。本明細書に示されているデータは、本発明のMHC分子をフローサイトメトリー解析で使用しT細胞を染色することができるということを示している。先行技術の多くのMHCクラスII四量体は、T細胞を活性化することができても、抗原特異的TH細胞を染色することができないため、このことは非常に有利かつ重要な特性である。したがって、本発明の好ましいMHCクラスII分子は、例えばフローサイトメトリーを用いて検出できるように、T細胞を染色する能力を有する。単量体型を用いてT細胞の染色を観察することは可能であるが、この特性は、特に多量体または多価型の分子で観察される。多量体ファージ提示型のさらなる利点は、一般に、比較的力価の低いファージを用いて同じ結果、例えばT細胞染色を達成することができるということである。
【0074】
かかるMHCクラスII分子を上述のように試薬として提供する場合、分子を関連するペプチドとともにまたはそれなしで提供することができる。かかる試薬を、タンパク質として、またはかかるMHCクラスII分子をコードする核酸として、例えば1つ以上の発現ベクターの形態で提供することができる。
【0075】
かかる試薬を、特に上述のようにT細胞染色に使用する場合、特定のペプチドに対するT細胞応答を研究するために、例えば、T細胞応答のレパートリーおよび動力学を研究するために、ならびに例えば末梢血中での抗原特異的CD4+T細胞の直接的なエクスビボ解析を可能にするために使用することができる。末梢血の解析はこれまで、組換えMHCクラスII分子が関係する課題であった。またこれらを用いて、抗原特異的T細胞をインビボ、インビトロおよびエクスビボで分離し同定することもできる。このようにして同定されたT細胞は、さらなる研究、拡張または活性化に供することができ、また治療で使用される可能性がある。
【0076】
また本発明の組換えMHCクラスII分子を、免疫調節剤をスクリーニングするためのプラットフォームとして使用することもできる。免疫系が関与する疾患において重要な段階は、抗原提示細胞とT細胞間の相互作用である。特に抗原提示細胞上のMHCクラスII複合体は、外部に由来するペプチドと結合して、それをTヘルパー細胞に提示するため重要である。次いで、Tヘルパー細胞が活性化されて、広範なエフェクター細胞を活性化する各種サイトカインを分泌する。
【0077】
本明細書に記載されているような安定で完全に機能的なMHCクラスII分子および特にこの分子の可溶型を提供することは、Tヘルパー細胞とMHCクラスII分子間の相互作用ならびにそれに続くTヘルパー細胞およびエフェクター細胞の活性化を調節する、例えば上方または下方調節することができる免疫調節剤のスクリーニングのためのプラットフォームを提供するのに重要である。本発明までは、安定なMHCクラスII分子を高収率かつ低コストで作製することの難しさが、こうしたスクリーニングの妨げとなっていた。したがって、本発明の組換えMHCクラスII分子を、免疫調節剤のスクリーニングプラットフォームとして用いることができる。
【0078】
かかる方法に使用するのに好ましいMHCクラスII分子は、本明細書の他の箇所に記載されている。好ましくは、前記MHCクラスII分子は疾患特異的ペプチドと会合する。このようなペプチドの例は、本明細書および先行技術に記載されており、セリアック病特異的またはI型糖尿病特異的ペプチドのような疾患特異的ペプチドと会合するヒトHLA−DQ2分子、関節リウマチの疾患特異的ペプチドと会合するヒトHLA−DR4 MHCクラスII分子、および特定の破傷風ペプチドと会合するヒトHLA−DP1 MHCクラスII分子がこれに含まれる。本発明のMHCクラスII分子と会合する他のペプチドも同様に使用することができる。
【0079】
次いで、かかる疾患関連ペプチドをMHCクラスIIとの関連で特異的に認識することができるT細胞を、例えば、本発明のMHCクラスII分子を上記のように用いて特定するか、または他の方法で入手(例えば、こうした細胞系は科学者により既にいくつか開発されている)した後、1つ以上の化合物がT細胞と抗原提示細胞間の相互作用またはその下流の事象(例えばサイトカイン分泌およびエフェクター細胞機能など)を調節する能力を評価することができる。
【0080】
本発明の組換えMHCクラスII分子および特にこの分子のファージ提示型を、エピトープ発見、例えば、T細胞により認識される抗原ペプチドエピトープ(T細胞エピトープ)の同定および特徴付けに使用することができる。したがって、本発明のさらなる実施形態では、T細胞により認識され得る抗原ペプチドエピトープを同定する方法が提供され、本発明の組換えMHCクラスII分子をT細胞受容体と接触させることと、前記組換えMHCクラスII分子と前記T細胞受容体との結合を検出することとを含む。T細胞受容体の結合は、MHCクラスII分子と会合した抗原ペプチドエピトープの存在を示すものであり、その後、このエピトープをさらに解析し特徴付けることができる。
【0081】
例えば、固定されたMHCとの関連で提示される抗原ペプチドに多様性を導入することにより、エピトープのライブラリーを作出することができる。次いで、例えば、組換え可溶性TCRもしくはファージ提示TCR、またはその他のT細胞集団(例えば、患者由来のT細胞)を用いて、このライブラリーのスクリーニングを行うことができる。低スループットのバキュロウイルスライブラリーに基づくものであり、生細胞で選択を行うことができないというさらなる不利な点を抱える現在の方法(Crawford et al.,2004,PLos Biology,2:0523−0533)と比較すれば、これは大きな進歩であろう。
【0082】
本発明の組換えMHCクラスII分子を診断試薬として使用することができる。例えば、本明細書の他の箇所に記載されているように、疾患特異的ペプチドを本発明のMHCクラスII分子と複合体形成または会合させ、これを用いて試料中、例えば、その疾患を有する可能性のある患者から採取した血液試料中の疾患特異的T細胞の有無を検出することができる。疾患特異的T細胞、特に、疾患を有さない患者で見られるレベルと比較して有意な数の疾患特異的T細胞の存在は、陽性の診断を示すことになる。
【0083】
したがって、本発明のさらなる態様では、試料中の抗原特異的T細胞を検出する方法が提供され、前記方法は、本発明の組換えMHCクラスII分子を前記試料と接触させることと、前記組換えMHCクラスII分子と前記T細胞との結合を検出することとを含む。前記組換えMHCクラスII分子とT細胞との結合は、抗原特異的T細胞の存在を示すものである。
【0084】
かかる方法を用いて、試料中の疾患特異的T細胞の存在を検出し、疾患の有無を診断することができる。この方法を用いて診断することができる適当な疾患は、MHCクラスII分子が疾患特異的ペプチドと結合する疾患である。疾患の例は本明細書の他の箇所に記載されており、セリアック病、関節リウマチ、破傷風およびインフルエンザがこれに含まれる。
【0085】
特定の疾患の背景にある機構をより詳細に理解するために、捕捉された疾患特異的T細胞およびこのT細胞と本発明のMHCクラスII分子間の相互作用を特徴付けることもできる。したがって、これも、例えば特定のT細胞応答をモニターし特徴付けるために、本発明のMHCクラスII分子を研究試薬として使用することができる方法もう1つの例である。
【0086】
本発明の組換えMHCクラスII分子がペプチドと会合する場合、それをTCR親和性成熟の標的としても使用することができる。
【0087】
当該技術分野において公知であり記載されているいくつかの方法のいずれでも、本発明の組換えMHCクラスII分子を調製し得るが、組換え法を用いて調製するのが最も好ましいということを当業者は理解するであろう。
【0088】
したがって、遺伝子クローニング技術を用いて本発明のMHCクラスII分子を作製してもよく、例えば、J.Sambrook et al.,Molecular Cloning,第2版,Cold Spring Harbor Laboratory Press,1989年、に適当な技術が開示されている。したがって、本発明のMHCクラスII分子の鎖(例えば、αおよび/またはβ鎖)をコードする配列またはこれに相補的な配列を含む核酸分子は、本発明のさらなる態様を形成する。
【0089】
本明細書に記載のMHCクラスII分子のα鎖(すなわち、鎖(i))またはβ鎖(すなわち、鎖(ii))をコードする核酸分子を、任意の適当な方法により、例えばクローニングまたは合成により、入手または作製することができる。例えば、適当な源由来(例えば、標準的なバフィーコートから分離された白血球のような細胞由来)の適当な配列をクローニングすることによりこのような配列を調製することができ、関係する特定のMHCクラスII対立遺伝子の配列を用いて設計された適当なプライマーを用いた標準的RT−PCRのようなPCR技術を用いて、適当な配列をクローニングすることができる。特定のMHCクラスII対立遺伝子の配列はGenBankで入手することができるが、IMGTデータベースを介した方が容易にアクセスすることができる。また全遺伝子合成法により、収率を最大化するのに重要となり得るコドン最適化が可能となるため、特に原核発現でこれを行うこともできる。
【0090】
最初の配列が一度クローニングまたは合成されれば、当該技術分野において公知であり記載されている方法を用いて、例えば部位特異的変異誘発により、本発明の変異システイン残基をコードする核酸分子を得るために配列に対して任意の付属の改変を行うことができる。
【0091】
さらに必要に応じて、本発明の分子を作製するために、MHCクラスII分子のαおよび/またはβ鎖の他の部分を操作してもよい。したがって、例えば、膜貫通ドメインおよび細胞質ドメインをコードするヌクレオチドを除去してもよく、同様に、本発明の分子が機能的であるために必要でないと考えられる他の任意の領域も除去してもよい。
【0092】
本発明のMHCクラスII分子の2つの鎖をコードする核酸フラグメントが得られれば、例えば、本明細書の他の箇所に記載されているように、調節配列などの他の所望の構成要素を含ませる、または非天然のシステイン残基を組み込む、または天然のシステイン残基を除去するために、そのフラグメントを標準的な組換えDNA技術でさらに操作してもよい。通常は、またはこのさらなる操作手順の一部として、本発明のMHCクラスII分子の作製を容易にするために、本発明のMHCクラスII分子をコードする核酸フラグメントを1つ以上の適当な発現ベクター内に組み込み、このベクター(1つまたは複数)を宿主細胞内に組み込む。かかる発現ベクターおよびかかる発現ベクターを含む宿主細胞は、本発明のさらなる態様を形成する。
【0093】
したがって、さらなる態様では、1つ以上の本発明の核酸分子を含む発現構築物または発現ベクターが提供される。好ましくは、発現構築物またはベクターは組換えによるものである。好ましくは、前記構築物またはベクターは、本発明の核酸分子によりコードされるタンパク質配列の転写および翻訳に必要な調節配列をさらに含む。
【0094】
さらなる態様では、1つ以上の本発明の発現構築物または発現ベクターを含む宿主細胞が提供される。また、1つ以上の本発明の核酸分子を含む宿主細胞も提供される。本発明のMHCクラスII分子を発現する宿主細胞は、さらなる態様を形成する。
【0095】
本発明のさらなる態様では、例えば本発明の宿主細胞を培養することを含む、本発明のMHCクラスII分子の作製方法が提供される。好ましい方法は、(i)コードされているMHCクラスII分子のαおよびβ鎖の発現に適した条件下で本発明の1つ以上の組換え発現ベクターまたは1つ以上の核酸配列を含む宿主細胞を培養すること;ならびに任意に(ii)宿主細胞由来または増殖培地/上清由来のタンパク質を単離または入手すること、を含む。この作製方法はタンパク質産物を精製することも含み得る。この作製方法は原核宿主細胞、例えば、本明細書の他の箇所に記載されている細菌宿主細胞で行うことが好ましい。
【0096】
したがって、本発明のさらなる態様では、
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含む組換えMHCクラスII分子を作製する方法が提供され、
(i)および(ii)は、機能的ペプチド結合ドメインを提供し、(i)および(ii)は、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基は、天然MHCクラスIIのα2およびβ2ドメイン中には存在せず、
前記方法は、前記組換え分子を原核宿主、好ましくは細菌宿主で発現させることを含む。
【0097】
上で議論したように、原核宿主細胞を用いて、ジスルフィド結合を組み込んで本発明の機能的MHCクラスII分子を作製することができるということは、驚くべきことであり、また有利である。
【0098】
本発明のMHCクラスII分子は2つ以上のポリペプチド鎖から構成される(すなわち、ヘテロ二量体である)ため、いくつかの実施形態では、例えば、ファージ提示法、細菌ペリプラズムでの発現および真核発現を含む実施形態では、すべてのポリペプチドを同一の宿主細胞内で、同じまたは異なる発現ベクターから発現させて、その宿主細胞内で完全な二量体タンパク質が組み立てられ、そこから単離または精製され得るようにすることが好ましい。他の実施形態、例えば意図的な封入体産生による細菌での大量生産などでは、各鎖を別々の宿主細胞(同一のタイプ/菌株の宿主細胞であることに留意)で発現させなければならない。
【0099】
任意の作製系を使用することができ(例えば、原核系も真核系もともに使用することができる)、発現ベクターを選択および設計することにより、使用する宿主細胞と確実に適合させる。したがって、発現ベクターが「宿主細胞の形質転換に適している」ということは、その発現ベクターが1つ以上の本発明の核酸分子と、発現に使用する宿主細胞に基づき選択され、核酸分子と作動可能に連結された調節配列とを含むことを意味する。作動可能に連結されたという表現は、核酸が発現されるように調節配列と連結されているという意味であることが意図される。
【0100】
MHCクラスII分子が不溶型である、例えば、別の生物粒子または生物膜の表面に提示される本発明の実施形態では、当該技術分野において記載されている方法により、本発明のMHC分子の提示を達成することができる。例えば、MHCクラスII分子が繊維状ファージ表面に提示される本発明の好ましい実施形態では、当該技術分野で記載されている任意の適当な方法を用いることができる。例えば、標準的なテキストとして、例えば「Phage Display in Biotechnology and Drug Discovery」(Sachdev S.Sidhu,1995)または「Phage Display:A Laboratory Manual」(Barbas et al.,1994)などを参照することができる。方法および系の例は、例えば、国際公開第09/024591号に記載されており、また実例にも示されている。
【0101】
一般に、本発明のファージ提示法の態様にはファージミドベクターの使用が好ましく、これには、当業者により容易に選択され得る適当なヘルパー株の使用が必要である。例えば、VCSM13ヘルパーファージ(Stratagene)およびHyperPhage(商標)(Progen Biotechnik GmbH)のような適当なヘルパーファージ株が多数市販されており、そのいずれも好都合に使用し得る。MHCクラスIIヘテロ二量体の一方の鎖、すなわちα鎖またはβ鎖のどちらかを、選択されたコートタンパク質、例えば、本明細書の他の箇所で議論されているgpIIIまたはその他のコートタンパク質の1つとの融合体としてベクターに組み込み、またもう一方の鎖を可溶性分子として、すなわち、コートタンパク質との融合体としてではなく産生させるのが好都合である。コートタンパク質との融合は一般に、前記コートタンパク質のN末端で行う。付属の実施例では、β鎖がコートタンパク質との融合体として産生され、α鎖が可溶型で産生される。
【0102】
他の適当な構成要素も構築物/ファージミドベクター中に存在していてよく、その多くはこのようなベクターでは普通のことである。例えば、タンパク質を細胞周辺腔へ方向付けるpelBのようなシグナル配列が、例えば、適当なプロモーター/オペレーター配列、リボソーム結合部位、複製起点、転写ターミネーターなどとともに一般に組み込まれる。ファージミドベクターおよび構成要素の例を図2に示すが、これらの構成要素のうちの1つ以上を本発明の発現ベクター内に組み込むことができる。
【0103】
組換えMHCクラスII分子のペプチド結合ドメインとの結合のためのペプチドを得ることが好都合な本発明の実施形態では、これらも構築物/発現ベクターの適当な位置に組み込んでよい。一般に、これらはリンカーペプチドによりαまたはβ鎖、例えばβ鎖のN−末端との共有結合による融合体として産生され、その例が本明細書の他の箇所に記載されている。
【0104】
MHCクラスII分子が可溶型である本発明の実施形態では、この分子を大腸菌(E.coli)のような細菌宿主細胞内の封入体として発現させ、次いでインビトロでリフォールディングさせることにより入手し得る。かかるインビトロでのリフォールディングは、標準的なプロトコル(例えば、Qoronfleh et al.,2007,Protein Expression and Purification 55:209−224に記載されている)およびそれを適当に修正したものを用いた、適切なリフォールディング条件下で生じ得る。
【0105】
あるいは、本発明の可溶性MHCクラスII分子を大腸菌(E.coli)のような細菌宿主細胞での発現により入手してもよく、ここでは、前記分子は宿主のペリプラズムで産生される。ペリプラズムでの産生は、適当な発現ベクターの使用により、例えば、タンパク質を細胞周辺腔へ方向付けるpelBのようなシグナルペプチドを組み込んで達成され得る。
【0106】
原核宿主、特に細菌宿主での本発明のMHCクラスII分子の作製は、本発明の好ましい実施形態である。この作製方法は、本明細書に記載のファージ提示法および可溶型のMHCクラスII分子の両方と適合性があり、この方法を用いて、高度に純化したタンパク質を大量に得ることができる。高度に純化したタンパク質を大量に得るためには、封入体から可溶性分子として産生されることが特に好ましい。操作されたジスルフィド結合を含むことにより安定性が増加した本発明の組換えMHCクラスII分子は、特に高収率での封入体産生に適しており、好ましくは、現在達成可能な収率(現在、出発物質の10%未満の範囲の収率はごく普通である)よりも高い収率で封入体から機能的MHCクラスII分子を産生させることを可能にする。
【0107】
任意の適当な原核宿主つまり細菌宿主をかかる産生に使用することができる。好ましい宿主細胞は、グラム陰性宿主細胞、より好ましくは大腸菌(E.coli)であろう。ファージ提示型に適した大腸菌(E.coli)宿主は当業者に公知であり、例えばXL1−blueがある。特に封入体からの精製が関係する場合に、可溶型の産生に好ましい大腸菌(E.coli)宿主は、大腸菌(E.coli)K12の派生物、最も好ましくはBL21表現型を有するものおよびその派生物である(Terpe,K.,Appl Microbiol Biotechnol.2006 Sep;72(2):211−22)。
【0108】
可溶性のペリプラズム発現に適した細菌宿主は当業者に公知であろう(例えば、Terpe,K.,Appl Microbiol Biotechnol.2006 Sep;72(2):211−22による総説を参照されたい)。
【0109】
あるいは、本発明の可溶性MHCクラスII分子を、酵母(pichiaを含む)、哺乳動物細胞または昆虫細胞のような真核細胞系で発現させることにより入手し得る。このような作製技術に適した宿主細胞は当業者に公知であろう。真核宿主細胞を用いた本発明の好ましい実施形態は、本発明の組換えMHCクラスII分子を免疫グロブリン(Ig)のFc部分との融合体、すなわち、例えばCasares et al.(1997,同上)に記載されているようなIg融合体として作製することである。このような実施形態では、免疫グロブリン分子のFc部分、例えばIgG2a分子のFc部分をαまたはβMHCクラスII鎖(通常はβ鎖のC−末端)と融合させて、本発明の組換えMHCクラスII分子(すなわち、本発明の人工的なジスルフィド結合を含む分子)のα鎖とβ鎖の二量体化を達成するために使用することができる。かかる分子を昆虫細胞系(または別の適当な系)で、例えば、昆虫細胞をバキュロウイルスに感染させて発現させることができ、これによりジスルフィド結合により安定化された分泌型のMHCクラスII分子が産生され得る。
【0110】
本発明の発現ベクターでの使用に適した調節配列は、細菌、真菌、ウイルス、哺乳動物または昆虫の遺伝子を含めた各種の源に由来し得る(例えば、Goeddel,1990,Gene Expression Technology:Methods in Enzymology 185,Academic Press,San Diego,CA,1990に記載されている調節配列を参照されたい)。適当な調節配列の選択は選択される宿主細胞によって決まり、当業者はこれを容易に行い得る。かかる調節配列の例としては、転写プロモーター/エンハンサーまたはRNAポリメラーゼ結合配列、翻訳開始シグナルを含むリボソーム結合配列が挙げられる。さらに、選択する宿主細胞および使用するベクターによっては、他の配列、例えば複製起点、追加のDNA制限部位、エンハンサーおよび転写誘導能を付与する配列などを発現ベクター内に組み込んでもよい。
【0111】
また本発明の発現ベクターは、本発明の組換え分子で形質転換された、またはそれが形質移入された宿主細胞の選択を容易にする選択マーカー遺伝子も含み得る。選択マーカー遺伝子の例は、特定の薬剤に対する耐性を付与するネオマイシンおよびヒグロマイシンのようなタンパク質、β−ガラクトシダーゼ、クロラムフェニコールアセチルトランスフェラーゼ、ホタルルシフェラーゼ、または免疫グロブリンもしくはその一部分(例えば免疫グロブリン、好ましくはIgGのFc部分など)をコードする遺伝子である。目的とする核酸とは別のベクターに選択マーカーを導入し得るということが理解されるであろう。
【0112】
また組換え発現ベクターは、組換えタンパク質の発現増加;組換えタンパク質の溶解性増加をもたらす;およびアフィニティー精製時のリガンド(例えば、精製および/または同定を可能にする適当な「タグ」、例えばHisタグ、HAタグ、FLAGタグまたはmycタグが存在し得る)として働くことにより標的組換えタンパク質の精製に役立つ融合部分をコードする遺伝子も含み得る。例えば、タンパク質分解性切断部位を標的組換えタンパク質に加えて、融合タンパク質の精製後に組換えタンパク質を融合部分から分離させ得る。典型的な融合発現ベクターにはpGEX(Amrad Corp.、Melbourne、Australia)、pMal(New England Biolabs、Beverly、MA)およびpRIT5(Pharmacia、Piscataway、NJ)があり、それぞれグルタチオンS−トランスフェラーゼ、マルトースE結合タンパク質またはプロテインAを組換えタンパク質と融合させる。
【0113】
発現ベクターのその他の任意選択の構成要素としては、AviTagまたは別のBirA基質のようなビオチン化配列(特に多量体型に関係する実施形態において)、検出のための標識、例えば、フローサイトメトリー技術の実施を可能にする蛍光標識が挙げられる。
【0114】
発現ベクターを宿主細胞内に導入して、形質転換宿主細胞を作製することができる。「〜で形質転換された」、「〜が形質移入された」、「形質転換」および「形質移入」という用語は、当該技術分野で公知の数多くの可能な技術のいずれか1つによる、細胞への核酸(例えば、ベクター)の導入を包含することが意図される。本明細書で使用される「形質転換宿主細胞」という用語は、本発明の組換え発現ベクターにより形質転換された、グリコシル化が可能な細胞も包含することが意図される。例えば、エレクトロポレーションまたは塩化カルシウムによる形質転換を用いて、原核細胞を核酸で形質転換することができる。例えば、従来技術、例えばリン酸カルシウムもしくは塩化カルシウム共沈殿、DEAE−デキストランによる形質移入、リポフェクション、エレクトロポレーションまたはマイクロインジェクションなどにより、核酸を哺乳動物細胞内に導入することができる。宿主細胞の形質転換および形質移入に適した方法は、Sambrook et al.(1989)およびその他の実験用教科書に記載されている。ファージ提示法が関係する本発明の態様では、関連する技術に関して、「Phage Display in Biotechnology and Drug Discovery」(Sachdev S.Sidhu,1995)または「Phage Display:A Laboratory Manual」(Barbas et al.,1994)のような汎用的なファージ提示法の教科書を参照することができる。
【0115】
他の分子とコンジュゲートされた本発明のタンパク質を含むN末端またはC末端融合タンパク質を、組換え技術による融合を用いて調製し得る。得られた融合タンパク質は、さらなる選択されたタンパク質(例えば、本明細書に記載のマーカータンパク質またはタグタンパク質)と融合した本発明のタンパク質を含む。また本発明のタンパク質を、他の既知の技術を用いて他のタンパク質とコンジュゲートさせてもよい。例えば、国際公開第90/10457号に記載されているヘテロ二官能性チオール含有リンカー、N−スクシンイミジル−3−(2−ピリジルジチオ−プロプリオナート(proprionate))またはN−スクシンイミジル−5チオアセタートを用いてタンパク質を結合させてもよい。融合タンパク質またはコンジュゲートを調製するために使用し得るタンパク質の例としては、免疫グロブリンのような細胞結合タンパク質、ホルモン、増殖因子、レクチン、インスリン、低密度リポタンパク質、グルカゴン、エンドルフィン、トランスフェリン、ボンベシン、アシアロ糖タンパク質、グルタチオン−S−トランスフェラーゼ(GST)、赤血球凝集素(HA)および短縮mycなどが挙げられる。
【0116】
本発明の第一のMHCクラスII分子をコードする核酸分子の調製方法に関係なく、さらなる適切なバリアント核酸分子を標準的な分子生物学的技術により容易に調製し得る。本発明のMHCクラスII分子をコードする任意のバリアント、変異体または第二世代の核酸分子が本発明での使用に適することを確認するために、本明細書の他の箇所に記載されているように、核酸分子に関して機能性を確認するための試験を行う。またバリアント、変異体または第二世代の核酸セグメントに関しても、標準的な条件下、より好ましくは標準的なストリンジェントなハイブリダイゼーション条件下でのハイブリダイゼーションを確認するための試験を行う。適切なハイブリダイゼーション条件の例は、約50℃で、約7%のドデシル硫酸ナトリウム(SDS)、約0.5MのNaPO4、約1mMのEDTA中でのハイブリダイゼーション;および約42℃で約1%のSDSによる洗浄を含む。
【0117】
本明細書および添付の特許請求の範囲で使用される、単数形「a」、「an」および「the」は、文脈上そうでないことが明らかでない限り、複数形の指示対象を包含する。
【0118】
以下の非限定的な実施例に関連して、以下の図面を参照しながら本発明をさらに説明する。
【図面の簡単な説明】
【0119】
【図1】マウスMHCクラスII分子I−Ek(PDB:1FNG)のα2−β2ドメインを結び付ける推定ジスルフィド架橋のコンピュータモデリングを示す図である。wt(上段)および対応する変異体(下段)の両方について、表2の上位3位のアミノ酸対の残基:1位(A)、2位(B)および3位(C)を関連する原子間距離とともに示している。
【図2】pFABDFNファージミドのベクター骨格がpIII提示ファージミドpSEX81(GenBankアクセション番号:Y14584)に基づくことを示す図である。ファージミドは、インフレームの外来配列のセグメント(E1およびE2と呼ぶ)を、それぞれNcoI/SpeIおよびMluI/SfiI部分のカセット交換により収めることができる。I−Edα鎖(aa26〜204)およびβ鎖(aa27〜216)をそれぞれE1およびE2に挿入した。略語:lacPO、lacプロモーター;sd、Shine−Dalgarno配列;pelB、細菌ペクチン酸リアーゼのシグナル配列;t、T7転写ターミネーター;xPO、fkpAの天然プロモーター;FkpA、fkpAのオープンリーディングフレーム;f1 IG、ファージf1の遺伝子間領域;AmpR、β−ラクタマーゼをコードする領域;ColE1、複製起点。
【図3】マウスMHCクラスII分子I−EおよびI−AのIMGT参照α鎖(A)およびβ鎖(B)配列を、クローニングされたI−Ed(_ELの記号)およびI−Ek(1FNGの記号)のものと比較した、ClustalW2.02多重配列アライメントを示す図である。システイン置換のために選択された位置が黒い影で強調してある(番号付けは表2/成熟IMGT I−E配列に従った):1位(α96−β118/α96−β119)、2位(α95−β120/α95−β121)および3位(α94−β150/α94−β151)。α鎖の完全長IMGT配列が示されているが(H−2EA*02)、これは本明細書中で配列番号1と呼ばれる場合がある。β鎖の完全長IMGT配列が示されているが(H−2EB1*01)、これは本明細書中で配列番号2と呼ばれる場合がある。α鎖の成熟IMGT配列、すなわち、シグナルペプチドを含まない完全長配列は、本明細書中で配列番号3と呼ばれる場合がある(シグナルペプチドを含まないH−2EA*02)。β鎖のIMGT配列、すなわち、シグナルペプチドを含まない完全長配列は、本明細書中で配列番号4と呼ばれる場合がある(シグナルペプチドを含まないH−2EB1*01)。上記各配列の注釈は、PDB ID:1FNGおよびBurrows et al.,1999,Protein Engineering,12:771−778を参照して行った。
【図4】ファージELISAによるI−Ed−pIIIの整合性の解析を示す図である。対象とする抗体を固定化し、正規量のビリオンを反応させた。捕捉されたビリオンを抗M13HRPコンジュゲートで検出し、ELISAをTMBで発色させてA450nmで読み取った。phOx−BSA特異的Fabにより提示される抗体が対照として含まれていた。
【図5】抗pIIIで展開したSDS PAGE/ウエスタンブロット法による多価I−Ed−pIII提示レベルの解析を示す図である。残基の番号付けはPDB ID:1FNGに従った(g表2を参照されたい)。
【図6】図6aは、P96α2−S119Cβ2(1位、表2)により安定化されたI−Edを提示するファージが、LD1 T細胞ハイブリドーマ細胞を特異的に染色することを示す図である。LD1細胞をブロックし、氷上で2時間、I−Ed−pIII提示ファージミドにより染色した後、ビオチン化したウサギ抗fdビオチンコンジュゲートおよびストレプトアビジンPEとともにインキュベートした。実線はHAaa110-120/I−Ed−pIII提示を表し、破線はI−Ed−pIII提示を表す。非常に低い力価(5×109ファージ/ml)でも特異的染色が達成され、また達成可能な最大力価は約1013ファージ/mlであることに注目されたい。図6bは、R94Cα2−N151Cβ2(3位、表2)、S95Cα2−S121Cβ2(2位、表2)またはP96Cα2−S119Cβ2(1位、表2)のいずれかにより安定化されたI−Edを提示するファージが、LD1 T細胞ハイブリドーマ細胞を特異的に染色することを示す図である。LD1細胞を室温で30分間、I−Ed−pIII提示ファージミドにより染色した後、ビオチン化したウサギ抗fdビオチンコンジュゲートおよびストレプトアビジンAPCとともにインキュベートした。各ファージ調製物の平均蛍光強度(MFI)を棒グラフで表す。非常に低い力価(2×109ファージ/ml)でも特異的染色が達成され、また達成可能な最大力価は約1013ファージ/mlであることに注目されたい。
【図7】代表的なマウスおよびヒトMHCクラスIIアロタイプであるI−E、I−A、HLA−DP、−DQおよび−DR配列のIMGTのClustalW2.02多重配列アライメントを示す図である。α鎖のアライメントをAに、β鎖のアライメントをBにそれぞれ示す。表2、図1および図3に示されるシステイン置換のために特定された位置が黒い影で強調してある。特定された位置はすべて、保存されたα2/β2ドメイン中にある。
【図8】重ね合った8つの異なるヒトHLAクラスIIの構造を表す図であり、この構造は、分子の四次構造が広範な遺伝的多型にもかかわらず高度に保存されており、したがって図7のアライメントを構造レベル(図8A)で検証するものであることを示している。例えば、Aの全体的に重ね合った構造のR94α2−N150β2残基の対(3位、表2)に着目すれば、全体的にRMSDが0.29〜1.18Åで変化する高度に保存された配置が明らかとなる(図8B)。したがって、観察される差が、単一分子の無関係な構造に対して予想される実験変動内にあるに過ぎないため、これらの構造はトポロジーレベルで実質的に同一であると見なすことができる。
【図9】S95Cα2−S121Cβ2(2位、表2)により安定化されたHLA−DRまたはHLA−DQ2.5を提示するファージが、ヒトT細胞クローンを特異的に染色することを示す図である。T18、TCC820.26およびTCC820.88細胞をブロックし、室温で1時間、HLAクラスII−pIII提示ファージミドにより染色した後、ビオチン化したウサギ抗fdビオチンコンジュゲートおよびストレプトアビジンPEとともに氷上でインキュベートした。各ファージ調製物の幾何平均強度(GMI)を棒グラフで表す。
【図10】非還元または還元SDS PAGE、次いで抗pIIIで検出したウエスタンブロッティングによる、一価(M13K07)および多価(HyperPhage)のI−Ed−pIII提示レベルの解析を示す図である。残基の番号付けはIMGTに従った(表2を参照されたい)。
【発明を実施するための形態】
【0120】
実施例
【0121】
実施例1
材料および方法
構造評価、分子モデリングおよび配列解析
最初のジスルフィド結合予測にヘモグロビン(Hb)由来ペプチド(Hbaa65-76)1と複合したマウスMHCクラスII分子I−Ekの1.9Å分解能の結晶構造(PDB ID:1FNG)を用いた。α2−β2定常ドメイン間の接合点を形成している残基を調べ、さらなる解析のためにβ炭素が7Å未満の距離にある側鎖を選択した。最初に、選択された側鎖をコンピュータ上での突然変異によりシステインに変化させて、回転異性体ライブラリーのスキャンにより最適な回転異性体を選択した。選択された残基対のチオール基がジスルフィド結合形成に最適でない位置にある場合、好ましい角度になるまで側鎖のχ1二面角を互いに回転させた。次いでGromos96力場を用いて、制約付きエネルギー最小化により局所の幾何学的位置を最適化した。
【0122】
H−2E(GenBankアクセション番号:K00971およびAF050157)およびH−2A(GenBankアクセション番号:V00832およびM13538)の翻訳したマウスIMG参照配列、本発明者らがクローニングしたH−2Ed(下を参照されたい)ならびにPDB:1FNG H−2E配列を、ClustalW2.02(http://www.ebi.ac.uk/)を用いて整列させ、手動で注釈を付けた。代表的なマウスH−2AおよびE、ならびにヒトHLA−DP、DQおよびDRをIMGT(http://imgt.cines.fr/textes/IMGTrepertoireMHC/LocusGenes/RepresentativeMHCgc.html)からダウンロードして翻訳し、ClustalW2.02(http://www.ebi.ac.uk/)を用いて整列させた。使用したIMGT参照配列(GenBankアクセションコード(α鎖/β鎖))は、I−A(V00832/M13538)、I−E(K00971/AF050157)、HLA−DP(X03100/M23907)、HLA−DQ(M23907/U92032)およびHLA−DR(J00204/AJ297583)であった。H−2E、およびPDB ID:1FNGの構造解析に従って、アライメントに手動で影をつけた。この配列アライメントを、代表的なPDB登録物:1FNG(I−Ek)、1S9V(HLA−DQ2)、1JK8(HLA−DQ8)、1DLH(HLA−DR1)、1BX2(HLA−DR2)、1HQR(HLA−DR2a)、1A6A(HLA−DR3)、1D6E(HLA−DR4)および2Q6W(HLA−DR52a)の構造アライメントおよび同時の構造重ね合わせにより検証した。分子の可視化および操作作業では、すべてspdbビューア3.7SP5ソフトウェア2を用いた。
【0123】
細菌株、ヘルパーファージおよびプラスミド
大腸菌(E.coli)株XL1−BlueおよびVCSM13ヘルパーファージはStratagene(LaJolla、CA、USA)から購入した。HyperPhage(商標)ヘルパーファージはProgen Biotechnik GmbH(Heidelberg、Germany)から購入した。phOx−BSAに対して特異的な一本鎖Fv(scFv)を有するpIII提示ファージミドpSEX813は、Affitech AS(Oslo、Norway)の厚意により提供されたが、PROGEN Biotechnik GmbH(Heidelberg、Germany)から入手することもできる。pSEX81系pFAB提示およびphOx−BSA特異性を有するpFABDFNファージミドは既に記載されている4。
【0124】
細胞系
T細胞ハイブリドーマLD15は、マウスMHCクラスII分子I−Ed上に提示されたインフルエンザ赤血球凝集素由来ペプチド(アミノ酸110〜120;N−SFERFEIFPKE−C)に対して特異的である。ヒトCD4+T細胞クローンT18はマウスIgCカッパ(アミノ酸40〜48;N−WKIDGSERQ−C)に対して特異的であり、HLA−DR4(DRA1、B1*0401)(PMID:12456590)拘束性である。ヒトT細胞クローンであるTCC820.26およびTCC820.88は、それぞれDQ2−αI−エピトープ(アミノ酸57〜68;N−QLQPFPQPELPY−C)およびDQ2−αII−エピトープ(アミノ酸62〜72;N−PQPELPYPQPE−C)を認識する。各細胞はS.−W.Qiao博士(オスロ大学免疫学研究所、ノルウェー)の厚意により提供された。細胞はすべて、10%のFCS、0,1mMの非必須アミノ酸、1mMのピルビン酸ナトリウム、50μMのモノチオグリセロールおよび12μg/mlの硫酸ゲンタマイシンを添加したRPMI1640で維持した。
【0125】
抗体および追加の試薬
すべての培地および緩衝液は、実質的にはSambrook et al.6に記載されている通りに調製した。マウス抗pIII、ウサギ抗fd、ウサギ抗fdビオチンコンジュゲート、ヒツジ抗M13−HRPおよびヒツジ抗マウス−HRP抗体は、それぞれMoBiTec(Goettingen、Germany)、Sigma−Aldrich(Oslo、Norway)およびAmersham Biosciences(Uppsala、Sweden)から購入した。抗I−E/I−A抗体2G9、ならびにストレプトアビジン−PEおよびストレプトアビジン−APCコンジュゲートはBD Pharmingen(San Jose、CA、USA)から購入した。ラット抗マウスCD32抗体2.4G2(ATCC(登録商標)番号HB−197(商標))は自家製であった。ハプテンである、ウシ血清アルブミン(BSA)とコンジュゲートされた2−フェニルオキサゾール−5−オン(phOx)は、実質的に記載されている通りに調製した7。制限酵素(RE)は、Stratagene(LaJolla、CA、USA)から入手したDpnI以外は、New England Biolabs(Ipswich、MA、USA)から購入した。DNAオリゴは、MWG Biotech AG(Ebersberg、Germany)、DNA Technology(Aarhus、Denmark)またはSigma−Aldrichから購入した。BSAおよびTween20はSigma−Aldrich(Oslo、Norway)から購入した。Pfu Turbo DNAポリメラーゼはStratagene(LaJolla、CA、USA)から購入した。トリプシン/EDTAはBioWhittaker(Lonza Group Ltd.、Visp、Switzerland)から購入した。
【0126】
I−Ed−pIII提示ファージミドの構築
実質的にはCasares et al.8により記載されている通りに、A20 BALB/c Bリンパ腫細胞由来のH−2E遺伝子をRT−PCRにより回収し、別々に真核pLNOH2ベクター中にクローニングして、I−Ed−Ig融合体を作出した。I−EdαおよびI−Edβをコードする遺伝子を、以下のプライマー対を用いてこれらのベクターから増幅した:I−Edαには、pFAB I−Eda fw(5’−TATACCATGGCCATCAAAGAGGAACACACCATCATCCAGG−3’)およびpFAB I−Eda rv(5’−TATAACTAGTCATTACTCCCAGTGCTTCCGCAGAG−3’);I−Edβには、pFAB I−Edb fw(5’−TATAACGCGTCAGAGACACCAGACCACGGTTTTTG−3’)およびpFAB I−Edb rv(5’−TATAGGCCGCAGCGGCCCCTTTCCACTCGACCGTGACAGGGT−3’)。I−Edβ遺伝子がM13pIII表面タンパク質と融合し、I−Edα鎖が可溶性の物質として産生されるように、I−Ed遺伝子をpFABDFNファージミド中にクローニングした(図2)。インフルエンザ赤血球凝集素由来ペプチド(アミノ酸110〜120)および15アミノ酸リンカー(G4S)3をコードするDNAを、記載されている重複伸長による遺伝子スプライシング9および以下のプライマーを用いてI−Edβ鎖のN末端に挿入した:pMHCHA SOEing fw(5’−AAAGGAAGGAGGTGGTGGCTCCGGTGGAGGGGGAAGTGGAGGTGGAGGGTCTGTCAGAGACACCAGACCACGGTT−3’)およびpMHCHA SOEing rv(5’−GGAGCCACCACCTCCTTCCTTTGGGAAGATCTCGAACCTTTCGAATGATACGCGTGCCATCGCCG−3’)。クローニングのために、製造者のプロトコル(Stratagene、LaJolla、CA、USA)に従い、プライマー:QCIEdbPstI fw(5’−TGGACACGTACTGTAGACACAACTATGAGAT−3’)およびQCIEdbPstI rv(5’−ATCTCATAGTTGTGTCTACAGTACGTGTCCA−3’)を用いたQuikChange(商標)での部位特異的変異誘発により、I−Edβ遺伝子内のPstI制限部位を除去した。製造者のプロトコル(Stratagene、LaJolla、CA、USA)に従い、QuikChange(商標)での部位特異的変異誘発により、ジスルフィド結合を導入するための点突然変異を導入した。使用したプライマーは、I−EdαのR94C変異には、Mut R94C I−Edアルファfw(5’−GACTGTACTCTCCTGTAGCCCTGTGAACC−3’)およびMut R94C I−Edアルファrv(5’−GGTTCACAGGGCTACAGGAGAGTACAGTC−3’);I−EdβのN150C変異には、Mut N151C I−Edベータfw(5’−CCTGGTCCGATGTGGAGACTGGACCTTC−3’)およびMut N151C I−Edベータrv(5’−GAAGGTCCAGTCTCCACATCGGACCAGG−3’);I−EdαのS95C変異には、QCIEdaS95C fw(5’−ACTCTCCAGATGCCCTGTGAAC−3’)およびQCIEdaS95C rv(5’−GTTCACAGGGCATCTGGAGAGT−3’);I−EdβのS120C変異には、QCIEdbS120C fw(5’−GTCTGCTCTGTGTGTGACTTCTAC−3’)およびQCIEdbS120C rv(5’−GTAGAAGTCACACACAGAGCAGAC−3’);I−EdαのP96C変異には、QCIEdaP96C fw(5’−CAGAAGCTGTGTGAACCTGGGA−3’)およびQCIEdaP96C rv(5’−TCCCAGGTTCACACAGCTTCTG−3’);I−EdβのS118C変異には、QCIEdbS118C fw(5’−CCTGGTCTGCTGTGTGAGTGAC−3’)およびQCIEdbS118C rv(5’−GTCACTCACACAGCAGACCAGG−3’)であった。
【0127】
MHCII提示バクテリオファージの調製
VCSM13またはHyperPhage(商標)ヘルパーファージおよびビリオン会合を用いた大腸菌(E.coli)XL1−Blueからのファージミドレスキューを、記載されているスポット力価測定法10によりモニターした。
【0128】
ファージ捕捉ELISA
捕捉抗体およびphOx−BSAを、pH7.4のPBS中2.5〜5μg/mlの濃度で、4℃で一晩、MaxiSorp(商標)マイクロタイタープレートウェル(Nunc、Roskilde、Denmark)に吸着させた。ウェルをPBSTM(0.05%v/vのTween20および4%w/vの脱脂乳を添加したPBS)により、室温で1時間ブロックした。次いで、正規量のビリオン調製物(VCSM13レスキュー試料:1×1010cfuampR/ウェル;HyperPhageレスキュー試料:1×108cfuampR/ウェル)を加え、室温で1〜2時間反応させた後、捕捉されたビリオンを室温で1時間、抗M13−HRP(1:5,000)により検出した。TMB可溶性基質(Merck KGaA、Darmstadt、Germany)でウェルを発色させ、30分後に1MのHClで停止させ、A450nmで吸光度を読み取った。
【0129】
SDS−PAGEおよびウエスタンブロット法
ビリオン(108cfuampR/レーン)を非還元および還元4〜12%ビス/トリスXT CriterionプレキャストSDS−PAGE(Bio−Rad、Hercules、CA、USA)により分離し、セミドライブロッティング装置(Bio−Rad、Hercules、CA、USA)を用いて、トリス/グリシン緩衝液(25mMのトリス、192mMのグリシンおよび20%のメタノール、pH8.3)中、25Vで30分間、ポリフッ化ビニリデン膜(Millipore、Madison、USA)にブロットした。膜をPBSTM中でブロックした後、マウス抗pIIIMAb(1:4,000)、次いでヒツジ抗マウス−HRP(1:10,000)でpIII融合体を検出した。膜を洗浄し、SuperSignal(商標)West Femto基質(Pierce、Rockford、IL、USA)で発色させ、BioMax(商標)MRフィルム(Kodak、Fernwald、Germany)に感光させた。
【0130】
人工的S−S架橋を含むHLA−DP、−DRおよび−DQの構築
IMGT(http://imgt.cines.fr/)により定義される代表的なHLA−DR(DRA*0101/DRB1*1402)および−DQ(DQA1*0501/DQB1*0301)をコードする遺伝子を化学的に合成した(Genscript、Piscataway、NJ)。大腸菌(E.coli)コドンバイアスを修正してDNA配列を最適化し、2位のα2−β2システイン対を導入するための点突然変異を組み込んだ(表2)。DNAフラグメントを上記のようにpFABDFNファージミド中にクローニングした。
【0131】
あるいは、IMGT(http://imgt.cines.fr/)により定義される代表的なHLA−DP(DPA1*0103/DPB1*0401)、−DR(DRA*0101/DRB1*1402)および−DQ(DQA1*0501/DQB1*0301)をコードする遺伝子を、新たに単離したヒトPBMCからRT−PCRにより増幅し、上記のようにpFABDFNファージミド中にクローニングする。2位(表2)のα2−β2システイン対を導入するための点突然変異を、製造者のプロトコル(Stratagene、LaJolla、CA、USA)に従いQuikChange(商標)での部位特異的変異誘発により組み込む。使用したプライマー(表1に記載)は、HLA−DPA α2−ドメインのS95C変異には、DPA_S95C_センスおよびDPA_S95C_アンチセンス;HLA−DPB β2−ドメインのS121C変異には、DPB_S121C_センスおよびDPB_S121C_アンチセンス;HLA−DRA α2−ドメインのS95C変異には、DRA_S95C_センスおよびDRA_S95C_アンチセンス;HLA−DRB β2−ドメインのS121C変異には、DRB_S121C_センスおよびDRB_S121C_アンチセンス;HLA−DQA α2−ドメインのS95C変異には、DQA_S95C_センスおよびDQA_S95C_アンチセンス;HLA−DQB β2−ドメインのS121C変異には、DQB_S121C_センスおよびDQB_S121C_アンチセンスである。
【0132】
【表2】
【0133】
フローサイトメトリー
LD1細胞の単細胞懸濁液を、氷上で15分間、60%で熱不活性化したラット血清および1×PBS中200μg/mlの抗CD32mAb(HB197)によりブロックするか(図6a)、またはブロックしなかった(図6b)。HyperPhageにパッケージングされたI−Ed−pIII提示の染色を、氷上で2時間、染色緩衝液(0.5%のBSAを含む1×PBS)中で(図6a)、または室温で30分間、1%のBSAを含む1×PBS中で(図6b)行った。図6aで使用したファージ投入量は、HAaa110-120−I−Ed提示には5×109cfuampR、I−Ed提示には1.6×1010cfuampRであった。図6bで使用したファージ投入量は、VCSM13ヘルパーファージ(ネガティブ対照、1×1011cfukanR/ml)を除くすべての調製物で2×109cfuampRであった。次いで試料を、10μg/mlのビオチン化ウサギ抗fdとともに氷上で1時間(図6a)または30分間(図6b)インキュベートし、次いで2μg/mlのストレプトアビジンPEとともに氷上で15分間インキュベートした。
【0134】
3つのT細胞クローン、すなわちT18、TCC820.26またはTCC820.88のそれぞれの単細胞懸濁液を染色緩衝液(0.5%のBSAを含む1×PBS)で調製した。HLAクラスII−pIII融合体(図9)を多価(HyperPhageにパッケージング)または一価(VCSM13にパッケージング)で提示するファージとともに室温で1時間、細胞を染色した。使用したファージ投入量は、それぞれ約1×109cfuampR(図9a)、約2×109cfuampR(図9b)および約2×1012cfuampR(図9cおよび9d)であった。次いで試料を、10μg/mlのビオチン化ウサギ抗fdとともに氷上で1時間インキュベートし、次いで2μg/mlのストレプトアビジン−PEとともに氷上で30分間インキュベートした。FACSCaliburフローサイトメータ(BD Biosciences)で事象を捕捉し、CellQuest PROソフトウェア(BD Biosciences)を用いてそれを解析した。
【0135】
結果
分子モデリング
表2(1FNG番号付け)は、I−Edα/I−Ekβ構造(PDB ID:1FNG)内のα2/β2定常ドメイン接合点中の選択された残基対のβ炭素側鎖の距離を示している。実際、1位〜3位の残基対の、システインへのコンピュータ上での突然変異誘発および側鎖の最適化により、タンパク質の三次構造を不安定にすることなく2.03〜2.07Åの間のジスルフィド結合距離が得られた(図1)。この結果は、変異体ポリペプチドにおいてこれらのアミノ酸置換が可能になり、ジスルフィド架橋を形成された可能性があり、したがって、さらなる解析に選択されたということを強く示唆するものであった。PDB ID 1FNGで受託されているアミノ酸番号付けは、β鎖のIMGT配列番号付け(図3Bを参照されたい)からずれていることに留意されたい。
【0136】
【表3】
【0137】
MHC−pIII融合体の設計
I−Ed遺伝子セグメントを、αおよびβ鎖が2つの別々のポリペプチドとして翻訳され、そのうちβ鎖がウイルスキャプシドタンパク質pIIIのN末端にインフレームで直接融合されるようにpFABDFNファージミド内に挿入した(図2)。両鎖とも、ペリプラズムへの標的化のためのN末端シグナル配列(pelB)を含んでいた。挿入されたα鎖部分(図3A、H−2EA_EL)は、図3Aに示されるIMGT H−2EA*02参照配列(GenBankアクセションコード:K00971、配列番号1)のアミノ酸26〜204に対応する。同様に、図3Bに示されるIMGT H−2EB1*01参照配列(GenBankアクセションコード:AF050157、配列番号2)のアミノ酸27〜216に対応するβ鎖部分を用いた(図3B、H−2EB_EL)。したがって、I−E IMGT参照配列に基づけば、表2の1位〜3位のシステイン置換の位置は、それぞれ成熟ペプチド(すなわち、シグナルペプチドを含まないペプチド)のα鎖の位置94〜96(図3A)およびβ鎖の位置119、121および151(図3B)に対応する。このβ鎖残基の番号付けは、1FNGテンプレートファイルに受託されている番号付けからずれていることに留意されたい。
【0138】
ファージミド由来のMHCII−pIII提示の解析
pMHC部分の提示の傾向および整合性を比較するために、I−E特異的モノクローナル抗体(MAb)である2G9で正規量のビリオンを捕捉することにより、ファージELISAを行った。結果は、多価および低価数の提示の両方で、すべての試料がMAb 2G9により認識されることを示していたが(図4)、このことは、構造上の整合性が全体的に保持されたことを示唆している。MAb 2G9は、pIIIと直接融合させたI−Eβ鎖と結合する。ファージ上に共有結合のα鎖ヘテロ二量体が存在するか否かを評価するために、非還元および還元SDS PAGE、次いで抗pIIIでのウエスタンブロット検出により正規量のビリオンを解析した(図5)。結果は、wt(図5、レーン5)およびHAペプチドを有するα95/β120変異体(図5、レーン3)の2つを除く全試料中に、有意な割合の共有結合のヘテロ二量体が存在することを示していた。しかし、α95/β120変異体の試料は、試料負荷量が非常に少ないため適切な分離が妨げられたことを示していたのは意外であった。すべての共有結合複合体形成が試料の還元の際に打ち消された(図5、レーン11〜20)。さらに抗体Fab対照(図5、レーン10および20)に関して、pMHC部分の改変に対して1つの共有結合ヘテロ二量体種のみが予想されたが、すべての関連試料においてかかる種が複数観察され、予想とは明らかに異なっていた。おそらくこの現象は、導入された変異のほかにも形成された、異所での天然システイン残基間のジスルフィド架橋により説明されるであろう。したがって、実際に提示されたタンパク質のごく一部だけが、人工的に導入された架橋のほかに天然のジスルフィド架橋をα2−β2ドメイン中に有する鎖である。
【0139】
I−Ed−pIII構築物の一価および多価の提示を同時に解する同様の実験を行った。図10にその結果を示す。低レベルのファージミド提示(一価)では、約1〜10%のビリオンが融合タンパク質を有する(BradburyおよびMarks,J Immunol Methods.2004,290(1−2):29.)。したがって、はるかに感度の高いELISAアッセイでは融合体が容易に検出されるのにもかかわらず、これらの試料(M13K07試料)では融合タンパク質がはっきりとは見られない(図4)。
【0140】
細胞表面染色試薬としてのpMHC−pIII
pIIIとの融合体としてバクテリオファージ上に提示されたペプチド−MHCクラスIIを従来のpMHCII四量体と同様に染色試薬として使用できるか否かを調べるために、本発明者らは、HAaa110-120/I−Ed−pIIIまたは融合ペプチドを含まないI−Ed−pIIIでLD1細胞を染色した。図6aからわかるように、関連ペプチドなしで産生されたバクテリオファージ上のI−Ed−pIII融合体では染色が観察されなかったが、HAaa110−120/I−Ed−pIIIは、約0.8logだけ明るい強度で細胞を染色した。図6bからわかるように、調べたすべてのジスルフィド架橋でペプチド特異的染色が観察される。陽性染色は、I−Ed−pIIIのジスルフィド安定化型および抗原ペプチド有する型(HAaa110-120)でのみ観察された。3つの操作されたジスルフィド架橋間で明らかな差は見られなかった。したがって、これらは同様の効率で作用すると思われる。
【0141】
pMHC−pIIIの機能性にさらに取り組むために、VCSM13またはHyperPhage(商標)を用いたファージミドレスキューにより、mCκaa40-48/HLA−DR4−pIII、αIaa57-68/HLA−DQ2.5またはαIIaa62-72/HLA−DQ2.5を提示するビリオンを調製した。また対照として、すべてのpMHC−pIII融合体を抗原ペプチドなしでも作製した。次いでこれらのビリオンを、記載されているようにフローサイトメトリー実験で試験した。図9aからわかるように、mCκaa40-48/HLA−DR4−pIII染色細胞でペプチド特異的染色が観察された。図9bおよび9cでは、αIaa57-68/HLA−DQ2.5の多価および一価の提示がペプチド特異的染色を示している。ファージ投入量は、VCSM13にパッケージングされたαIaa57-68/HLA−DQ2.5提示において1012cfuampRであったということに留意されたい。αIIaa62-72−HLA−DQ2.5のペプチド特異的染色を図9dに示す。
【0142】
考察
可溶性の組換えMHCクラスII分子の効果的な使用の主な障害は、このような操作された分子に固有の安定性の問題が原因である。この制限を克服するために様々な立案されたアプローチが採用されてきたが、汎用性のあるアプローチは現在のところ存在しない11。また、現在用いられている手段はすべて、コストと労働力がかかる方法であり、事実上、高スループットなアプローチの妨げとなっている。本発明者らは、組換えMHCクラスII作製とファージ提示技術12を合わせることにより、この問題に対する魅力的な解決方法を見出した。これは非常に迅速かつ安価な技術プラットフォームであるばかりでなく、極めて多様なコンビナトリアルスクリーニングのアプローチを可能にするという固有の特性も有している。MHC分子によりもたらされる広大な抗原プロテオーム空間は、例えばエピトープ発見において、このコンビナトリアル性から大きな恩恵を受けるであろう。しかし現在のところ、こうした発見はまだバキュロウイルスに基づく煩雑で低スループットな技術13に限定されている。
【0143】
ファージ提示を使用すれば、十分に記述された簡便な形式で12、より大きな桁数のレパートリーサイズに容易に手が届くであろう。このような提示によりMHCクラスI分子が探求されてきたが14〜16、MHCクラスIファージ提示のアプローチはいずれも、細胞特異的染色がまだ得られていないことから、現在のところ可溶性四量体の代替物として機能しないことがわかっている16。MHCクラスI分子は単一のポリペプチドとして効率的に作製することができる18のとは対照的に、MHCクラスIIは、等しく関与する別々の鎖のヘテロ二量体であることから、その作製は複雑である。MHCクラスIおよびIIの機能的なフォールドはともに、Igフォールドトポロジーで構築されるため、ドメイン内のジスルフィド架橋の形成も必要とする19。
【0144】
MHCクラスIIとその同族リガンド間の天然の相互作用の鍵となる特徴は、固有の弱い親和性である20。組換えMHCクラスII分子を検出試薬として用いる場合にこの感度における制限を克服するために、分子を四量体または五量体に重合することが必要であった11。こうしたポリマーの作製には、実質的な操作を伴う多段階のプロトコルが含まれている。ここでファージ提示を使用すれば、ファージ上での多量体提示を可能にする既に利用可能な技術の使用により、かかる重合が非常に容易に行われる21。
【0145】
ここで本発明者らは、pIIIファージミド提示により機能的に提示されるMHCクラスII分子を得るための戦略を提供する。これを達成するために、αおよびβの2つの鎖を大腸菌(E.coli)内で2つの別々のポリペプチド鎖として発現させ、そのうちの1つの鎖はpIIIキャプシドと融合され、ペリプラズムへ標的化されている。酸化ペリプラズムでは、2つの鎖が、α2ドメインとβ2ドメインを結合させる新規な人工的ジスルフィド架橋を利用して、安定なヘテロ二量体を形成する。マウスMHCクラスII分子I−Edの保存されたα2−β2接合点中の複数の位置を、人工的な鎖間ジスルフィド架橋を可能にするアミノ酸置換の標的とすることができる(図1および3)。このような人工のMHC分子を、標準的なファージミドによるファージ提示を用いてpIII上に提示させることにより、共有結合のヘテロ二量体が実際にビリオン上に提示される(図5および10)。wtおよび変異体両方のすべての分子が、マウスMHCクラスII I−E分子に特異的なモノクローナル抗体(MAB 2G9)と反応することから、分子全体の整合性は保持されると思われる(図4)。I−Ed分子を、β鎖との共有結合テザーとして十分に特徴付けられているHAaa110-120抗原ペプチド22を提示するように、実質的には記載されている通りに設計した23。ジスルフィド架橋により安定化されたこれらの組換え分子を提示するファージをフローサイトメトリーで用いて、I−Ed/HAaa110-120複合体に対して特異的なT細胞受容体を有するLD1 T細胞ハイブリドーマ5の特異的染色が得られた(図6aおよび6b)。重要なのは、特異的染色が抗原ペプチドを有するジスルフィド安定化I−Ed−pIII融合体でのみ見られ、このことは真にこのアプローチの機能性を示しているということである。さらに、一価の提示では同様の染色が見られなかったことから(データ不掲載)、MHC部分の多価の提示が染色に不可欠であると思われ、このことは、機能的な親和性を天然の相互作用の親和性よりも高める必要性を強調している。これは現在のところ、ファージ提示されたMHC分子を特定のT細胞の直接的な可視化に使用した最初の例であり、古典的なMHCテトラマー技術を補完するものである。
【0146】
成功の鍵は、MHCクラスII分子のα2−β2接合点を安定化させる人工的なジスルフィド架橋の導入である。特定された位置は、HLAを含めたMHCクラスII分子間で高度に保存されているため(図7および8)、これは汎用性のあるアプローチであると思われる。この汎用性は、試験した他の3つのMHCクラスII複合体、すなわちmCκaa40-48/HLA−DR4、αIaa57-68/HLA−DQ2.5およびαIIaa62-72/HLA−DQ2.5はすべて、pIII融合体として、サイトメトリーにおいて抗原ペプチド特異的に特異的細胞染色を示したということにより強く支持される(図9)。予想されたように、特定のpMHCII複合体およびそれと反応するT細胞クローンの中で染色能および多価相互作用への依存に違いが見られた。したがって、多価相互作用への依存は全か無かの事象ではなく、このことは一価型のαIaa57-68/HLA−DQ2.5およびαIIaa62-72/HLA−DQ2.5の両方による特異的細胞染色により示された。フローサイトメトリーにおいて特異的に陽性に染色する能力は、特定のT細胞受容体のpMHCに対する本来の親和性、T細胞上での受容体密度および膜での受容体のクラスター形成能のようなパラメーターに依存するであろう。αIaa57-68/HLA−DQ2.5による多価(図9b)と一価(図9c)の染色では、多価型の方がファージ投入量が1000倍少なかったにもかかわらず、ともにほぼ同じであったということは注目すべきことである。
【0147】
参考文献
1. Kersh,G.J.et al.Structural and Functional Consequences of Altering a Peptide MHC Anchor Residue.Journal of Immunology 166,3345−3354(2001).
2. Guex,N.& Peitsch,M.C.SWISS−MODEL and the Swiss−PdbViewer:an environment for comparative protein modeling.Electrophoresis 18,2714−2723(1997).
3. Welschof,M.et al.The antigen−binding domain of a human IgG−anti−F(ab’)2autoantibody.PNAS 94,1902−1907(1997).
4. Loset,G.A.,Lunde,E.,Bogen,B.,Brekke,O.H.& Sandlie,I.Functional phage display of two murine α/β T−cell receptors is strongly dependent on fusion format,mode and periplasmic folding assistance.Protein Eng Des Sel.20,461−472(2007).
5. Taylor,A.H.,Haberman,A.M.,Gerhard,W.& Caton,A.J.Structure−function relationships among highly diverse T cells that recognize a determinant from influenza virus hemagglutinin.J Exp Med 172,1643−1651(1990).
6. Sambrook,J.& Russell,D.W.Molecular Cloning:A Laboratory Manual,Edn.3.(Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY;2001).
7. Makela,O.,Kaartinen,M.,Pelkonen,J.L.& Karjalainen,K.Inheritance of antibody specificity V.Anti−2−phenyloxazolone in the mouse.J Exp Med 148,1644−1660(1978).
8. Casares,S.,Bona,C.A.& Brumeanu,T.D.Engineering and characterization of a murine MHC class II−immunoglobulin chimera expressing an immunodominant CD4 T viral epitope.Protein Eng 10,1295−1301(1997).
9. Horton,R.M.,Cai,Z.L.,Ho,S.N.& Pease,L.R.Gene splicing by overlap extension:tailor−made genes using the polymerase chain reaction.Biotechniques 8,528−535(1990).
10. Loset,G.A.,Kristinsson,S.G.& Sandlie,I.Reliable titration of filamentous bacteriophages independent of pIII fusion moiety and genome size by using trypsin to restore wild−type pIII phenotype.BioTechniques 44,551−554(2008).
11. Vollers,S.S.& Stern,L.J.Class II major histocompatibility complex tetramer staining:progress,problems,and prospects.Immunology 123,305−313(2008).
12. Bratkovic,T.Progress in phage display:evolution of the technique and its applications.Cell Mol Life Sci(2009).
13. Crawford,F.et al.Use of baculovirus MHC/peptide display libraries to characterize T−cell receptor ligands.Immunol Rev 210,156−170(2006).
14. Le Doussal,J.,Piqueras,B.,Dogan,I.,Debre,P.& Gorochov,G.Phage display of peptide/major histocompatibility complex.J Immunol Methods 241,147−158.(2000).
15. Kurokawa,M.S.et al.Expression of MHC class I molecules together with antigenic peptides on filamentous phages.Immunol Lett 80,163−168(2002).
16. Vest Hansen,N.,Ostergaard Pedersen,L.,Stryhn,A.& Buus,S.Phage display of peptide/major histocompatibility class I complexes.Eur J Immunol 31,32−38(2001).
17. Constantin,C.M.,Bonney,E.E.,Altman,J.D.& Strickland,O.L.Major histocompatibility complex(MHC)tetramer technology:an evaluation.Biol Res Nurs 4,115−127(2002).
18. Leisner,C.et al.One−Pot,Mix−and−Read Peptide−MHC Tetramers.PLoS ONE 3,e1678(2008).
19. Halaby,D.M.,Poupon,A.& Mornon,J.The immunoglobulin fold family:sequence analysis and 3D structure comparisons.Protein Eng 12,563−571(1999).
20. Rudolph,M.G.,Stanfield,R.L.& Wilson,I.A.How TCRs bind MHCs,peptides,and co−receptors.Annu Rev Immunol 24,419−466(2006).
21. Soltes,G.et al.On the influence of vector design on antibody phage display.Journal of Biotechnology 127,626−637(2007).
22. Schild,H.et al.Natural ligand motifs of H−2E molecules are allele specific and illustrate homology to HLA−DR molecules.Int Immunol 7,1957−1965(1995).
23. Kozono,H.,White,J.,Clements,J.,Marrack,P.& Kappler,J.Production of soluble MHC class II p
【特許請求の範囲】
【請求項1】
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含む組換えMHCクラスII分子であって、
(i)および(ii)が、機能的ペプチド結合ドメインを提供し、かつ(i)および(ii)が、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基が、天然MHCクラスIIのα2およびβ2ドメイン中には存在しない、組換えMHCクラスII分子。
【請求項2】
前記組換え分子が、さらにロイシンジッパーモチーフを含まない、請求項1に記載の組換えMHCクラスII分子。
【請求項3】
前記ジスルフィド結合が、マウスI−EアイソタイプのPro96α2−Ser119β2(1位)、Ser95α2−Ser121β2(2位)もしくはArg94α2−Asn151β2(3位)または別のMHCクラスIIアイソタイプにおいて相当する位置のシステイン残基間にある、請求項1または2に記載の組換えMHCクラスII分子。
【請求項4】
前記ジスルフィド結合が、マウスI−EアイソタイプのSer95α2−Ser121β2(2位)または別のMHCクラスIIアイソタイプにおいて相当する位置のシステイン残基間にある、請求項3に記載の組換えMHCクラスII分子。
【請求項5】
さらに参照配列H−2EB*01(配列番号2)の38、42もしくは106の位置に対応する1つ以上のシステイン残基または別のMHCクラスIIアイソタイプの相当する位置の1つ以上のシステイン残基が除去されている、請求項1〜4のいずれか1項に記載の組換えMHCクラスII分子。
【請求項6】
前記組換え分子が、繊維状ファージ表面に発現されている、請求項1〜5のいずれか1項に記載の組換えMHCクラスII分子。
【請求項7】
前記組換え分子が、ファージ表面タンパク質gpIII、gpVII、gpVIIIまたはgpIXとの融合体として発現されている、請求項6に記載の組換えMHCクラスII分子。
【請求項8】
前記ファージ表面タンパク質がgpIXである、請求項7に記載の組換えMHCクラスII分子。
【請求項9】
前記分子が可溶性分子である、請求項1〜5のいずれか1項に記載の組換えMHCクラスII分子。
【請求項10】
前記分子の(i)または(ii)が免疫グロブリンのFc部分と融合している、請求項9に記載の組換えMHCクラスII分子。
【請求項11】
前記分子が多量体である、請求項1〜10のいずれか1項に記載の組換えMHCクラスII分子。
【請求項12】
前記分子の(i)および(ii)が、マウスMHCクラスII分子またはヒトMHCクラスII分子、好ましくはヒトMHCクラスII分子に由来する、請求項1〜11のいずれか1項に記載の組換えMHCクラスII分子。
【請求項13】
前記分子が細菌宿主により産生されている、請求項1〜12のいずれか1項に記載の組換えMHCクラスII分子。
【請求項14】
前記細菌宿主が、大腸菌(E.coli)、好ましくは大腸菌(E.coli)K12派生物、より好ましくはBL21表現型を有する大腸菌(E.coli)またはその派生物である、請求項13に記載の組換えMHCクラスII分子。
【請求項15】
前記ジスルフィド結合が、MHCクラスII分子安定化の唯一の手段を提供する、請求項1〜14のいずれか1項に記載の組換えMHCクラスII分子。
【請求項16】
前記分子がT細胞を染色することができる、請求項1〜15のいずれか1項に記載の組換えMHCクラスII分子。
【請求項17】
前記分子が前記ペプチド結合ドメインと結合したペプチドをさらに含む、請求項1〜16のいずれか1項に記載の組換えMHCクラスII分子。
【請求項18】
原核宿主で発現させることができる組換えMHCクラスII分子であって、
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含み、
(i)および(ii)が、機能的ペプチド結合ドメインを提供し、かつ(i)および(ii)が、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基が、天然MHCクラスIIのα2およびβ2ドメイン中には存在しない組換えMHCクラスII分子。
【請求項19】
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含む組換えMHCクラスII分子の作製方法であって、
(i)および(ii)が、機能的ペプチド結合ドメインを提供し、かつ(i)および(ii)が、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基が、天然MHCクラスIIのα2およびβ2ドメイン中には存在せず、
前記組換え分子を原核宿主で発現させることを含む、方法。
【請求項20】
T細胞により認識され得る抗原ペプチドエピトープを同定する方法であって、
前記方法が、請求項17に記載の組換えMHCクラスII分子をT細胞受容体と接触させることと、
前記組換えMHCクラスII分子と前記T細胞受容体との結合を検出することと、
を含む方法。
【請求項21】
試料中の抗原特異的T細胞を検出する方法であって、
前記方法が、請求項17に記載の組換えMHCクラスII分子を前記試料と接触させることと、
前記組換えMHCクラスII分子と前記T細胞との結合を検出することと、
を含む方法。
【請求項22】
前記方法が、試料中の疾患特異的T細胞の存在を検出して疾患の有無を診断するために使用される、請求項21に記載の方法。
【請求項1】
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含む組換えMHCクラスII分子であって、
(i)および(ii)が、機能的ペプチド結合ドメインを提供し、かつ(i)および(ii)が、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基が、天然MHCクラスIIのα2およびβ2ドメイン中には存在しない、組換えMHCクラスII分子。
【請求項2】
前記組換え分子が、さらにロイシンジッパーモチーフを含まない、請求項1に記載の組換えMHCクラスII分子。
【請求項3】
前記ジスルフィド結合が、マウスI−EアイソタイプのPro96α2−Ser119β2(1位)、Ser95α2−Ser121β2(2位)もしくはArg94α2−Asn151β2(3位)または別のMHCクラスIIアイソタイプにおいて相当する位置のシステイン残基間にある、請求項1または2に記載の組換えMHCクラスII分子。
【請求項4】
前記ジスルフィド結合が、マウスI−EアイソタイプのSer95α2−Ser121β2(2位)または別のMHCクラスIIアイソタイプにおいて相当する位置のシステイン残基間にある、請求項3に記載の組換えMHCクラスII分子。
【請求項5】
さらに参照配列H−2EB*01(配列番号2)の38、42もしくは106の位置に対応する1つ以上のシステイン残基または別のMHCクラスIIアイソタイプの相当する位置の1つ以上のシステイン残基が除去されている、請求項1〜4のいずれか1項に記載の組換えMHCクラスII分子。
【請求項6】
前記組換え分子が、繊維状ファージ表面に発現されている、請求項1〜5のいずれか1項に記載の組換えMHCクラスII分子。
【請求項7】
前記組換え分子が、ファージ表面タンパク質gpIII、gpVII、gpVIIIまたはgpIXとの融合体として発現されている、請求項6に記載の組換えMHCクラスII分子。
【請求項8】
前記ファージ表面タンパク質がgpIXである、請求項7に記載の組換えMHCクラスII分子。
【請求項9】
前記分子が可溶性分子である、請求項1〜5のいずれか1項に記載の組換えMHCクラスII分子。
【請求項10】
前記分子の(i)または(ii)が免疫グロブリンのFc部分と融合している、請求項9に記載の組換えMHCクラスII分子。
【請求項11】
前記分子が多量体である、請求項1〜10のいずれか1項に記載の組換えMHCクラスII分子。
【請求項12】
前記分子の(i)および(ii)が、マウスMHCクラスII分子またはヒトMHCクラスII分子、好ましくはヒトMHCクラスII分子に由来する、請求項1〜11のいずれか1項に記載の組換えMHCクラスII分子。
【請求項13】
前記分子が細菌宿主により産生されている、請求項1〜12のいずれか1項に記載の組換えMHCクラスII分子。
【請求項14】
前記細菌宿主が、大腸菌(E.coli)、好ましくは大腸菌(E.coli)K12派生物、より好ましくはBL21表現型を有する大腸菌(E.coli)またはその派生物である、請求項13に記載の組換えMHCクラスII分子。
【請求項15】
前記ジスルフィド結合が、MHCクラスII分子安定化の唯一の手段を提供する、請求項1〜14のいずれか1項に記載の組換えMHCクラスII分子。
【請求項16】
前記分子がT細胞を染色することができる、請求項1〜15のいずれか1項に記載の組換えMHCクラスII分子。
【請求項17】
前記分子が前記ペプチド結合ドメインと結合したペプチドをさらに含む、請求項1〜16のいずれか1項に記載の組換えMHCクラスII分子。
【請求項18】
原核宿主で発現させることができる組換えMHCクラスII分子であって、
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含み、
(i)および(ii)が、機能的ペプチド結合ドメインを提供し、かつ(i)および(ii)が、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基が、天然MHCクラスIIのα2およびβ2ドメイン中には存在しない組換えMHCクラスII分子。
【請求項19】
(i)MHCクラスIIα鎖の細胞外部分の全部または一部分と、
(ii)MHCクラスIIβ鎖の細胞外部分の全部または一部分と
を含む組換えMHCクラスII分子の作製方法であって、
(i)および(ii)が、機能的ペプチド結合ドメインを提供し、かつ(i)および(ii)が、前記α鎖のα2ドメイン中に位置するシステイン残基と前記β鎖のβ2ドメイン中に位置するシステイン残基との間のジスルフィド結合により結合しており、前記システイン残基が、天然MHCクラスIIのα2およびβ2ドメイン中には存在せず、
前記組換え分子を原核宿主で発現させることを含む、方法。
【請求項20】
T細胞により認識され得る抗原ペプチドエピトープを同定する方法であって、
前記方法が、請求項17に記載の組換えMHCクラスII分子をT細胞受容体と接触させることと、
前記組換えMHCクラスII分子と前記T細胞受容体との結合を検出することと、
を含む方法。
【請求項21】
試料中の抗原特異的T細胞を検出する方法であって、
前記方法が、請求項17に記載の組換えMHCクラスII分子を前記試料と接触させることと、
前記組換えMHCクラスII分子と前記T細胞との結合を検出することと、
を含む方法。
【請求項22】
前記方法が、試料中の疾患特異的T細胞の存在を検出して疾患の有無を診断するために使用される、請求項21に記載の方法。
【図1】


【図2】

【図4】

【図5】

【図6a】

【図6b】

【図7A】

【図7B】

【図8】
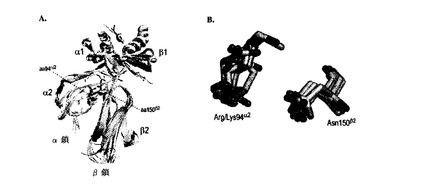
【図9】
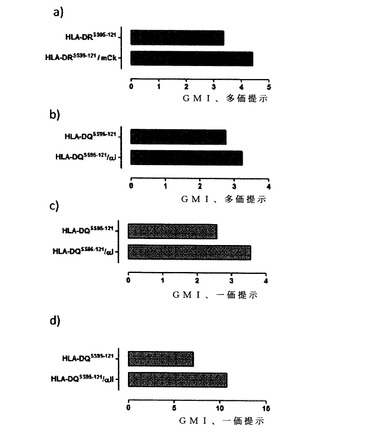
【図10】
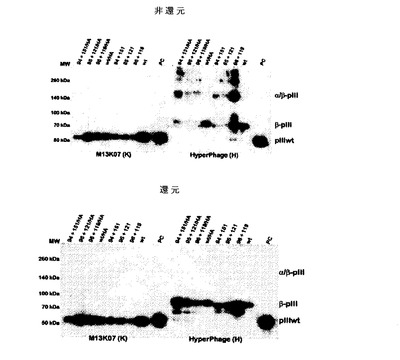
【図3】

【図2】
【図4】
【図5】
【図6a】
【図6b】
【図7A】
【図7B】
【図8】
【図9】
【図10】
【図3】
【公表番号】特表2013−519721(P2013−519721A)
【公表日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願番号】特願2012−553409(P2012−553409)
【出願日】平成23年2月18日(2011.2.18)
【国際出願番号】PCT/GB2011/050325
【国際公開番号】WO2011/101681
【国際公開日】平成23年8月25日(2011.8.25)
【出願人】(510179537)
【Fターム(参考)】
【公表日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願日】平成23年2月18日(2011.2.18)
【国際出願番号】PCT/GB2011/050325
【国際公開番号】WO2011/101681
【国際公開日】平成23年8月25日(2011.8.25)
【出願人】(510179537)
【Fターム(参考)】
[ Back to top ]