
ジンゲロール配糖体、その製造方法およびその用途
【課題】新規ジンゲロール配糖体、その製法およびその用途を提供すること。
【解決手段】ジンゲロールのOH基にグルコース、マルトースまたはマルトオリゴ糖が脱水縮合していることを特徴とするジンゲロール配糖体。
【解決手段】ジンゲロールのOH基にグルコース、マルトースまたはマルトオリゴ糖が脱水縮合していることを特徴とするジンゲロール配糖体。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規な化合物およびその製造法ならびに用途に関し、詳しくはショウガ成分であるジンゲロールの配糖体、当該化合物の製造方法、当該化合物の用途に関する。
【背景技術】
【0002】
ジンゲロールは、ショウガ(Zingiber officinale)などZingiberaceae属植物の根茎等に含まれる精油成分であり、アディポネクチン産生増強効果(特許文献1)、脂肪蓄積抑制効果(非特許文献1)などの効能が知られ、肥満予防、糖尿病治療、冷え改善などの効能を有する飲食品および医薬品等への応用が期待されている。
【0003】
上記精油成分は、例えばショウガから公知の手法により抽出することで得ることができる。
【0004】
ジンゲロールを含有するショウガは薬用として利用される他、香辛料として食用に供されるなど、歴史的に安全な食品であることが証明されている。医薬品としては、消化薬、緩下剤、鎮咳剤、制酸薬の原料として使用される。食品としては調味料、清涼飲料、アルコール飲料、菓子類などに、また化粧品の香料としても使用されている。
【0005】
しかしながら、有効成分のジンゲロールは、水に不溶であり、また、強い辛味を呈し、刺激がある。また、加熱あるいは酸性条件下においてより辛みの強く脂溶性の高いショウガオールまたはジンゲロンに変換されやすいことが知られている。このことから、ジンゲロールを安定した品質で、飲食品あるいは医薬品等へ利用することが困難であった。
【0006】
そのような問題から、ジンゲロールの水溶性、安定性改善を目的として、特許文献2においては、ジンゲロールまたはジンゲロール含有物と糖類の熱処理物を組み合わせることにより、安定性および水溶性の高いジンゲロール含有水溶性組成物を得たことが開示されている。しかしながら、得られる組成物はカラメル状であることから汎用性が低く、また、エタノールが残存しているために直接飲食品へ利用しにくい等の問題点があった。
【0007】
したがって、ジンゲロールの水溶性、安定性および味質などの物性が改善され、飲食品、医薬部外品および医薬品工業等において取扱が容易な汎用性の高いジンゲロール誘導体があれば、その産業利用価値が高くなることが期待された。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2011−1386号公報
【特許文献2】特開2008−56572号公報
【非特許文献】
【0009】
【非特許文献1】J.Med.Chem.2011、54:6295−6304
【発明の概要】
【発明が解決しようとする課題】
【0010】
そこで本発明の目的は、水に可溶かつ安定性の高い新規ジンゲロール配糖体、その製法およびその用途を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、従来のジンゲロールの欠点を改善するために、とりわけ、糖転移反応を利用した新しいジンゲロール誘導体の開発を目指し、研究を重ねてきた。
【0012】
その結果、水に可溶で、室温において固形粉末として存在可能であり、安定性の高いジンゲロール配糖体を、少なくともジンゲロールと、食品に利用可能なマルトース等の糖供与体を含む混合溶液に、糖転移酵素を作用させることにより合成可能であることを見出した。また、ある特定の糖転移酵素を使用することによって、ジンゲロールの側鎖に存在する、従来ジンゲロールの不安定さの原因とされていたβ−ヒドロキシケトンのOH基を選択的に配糖化可能であることを見出し、さらに、その製造法並びに飲食物、医薬部外品および医薬品ならびに化粧品等への用途を開発して本発明を完成した。
【0013】
すなわち、本発明は以下の通りである。
【0014】
<1> ジンゲロールのOH基にグルコース、マルトースまたはマルトオリゴ糖が脱水縮合していることを特徴とするジンゲロール配糖体。
<2> 下記一般式(I)で示されるジンゲロール配糖体である、<1>に記載のジンゲロール配糖体。
【0015】
【化1】

【0016】
(式中、
Rはグルコシル基、マルトシル基またはマルトオリゴ糖のグルコシル基を示し、
nは2,4,6または8のいずれかの整数を示す。)
<3> ジンゲロール配糖体が、下記式(II)で示される、6−ジンゲロールの側鎖のOH基にグルコースがα−1位で結合したジンゲロール配糖体である、<1>または<2>に記載のジンゲロール配糖体。
【0017】
【化2】
【0018】
<4> ジンゲロール配糖体が、下記式(III)で示される、6−ジンゲロールの側鎖のOH基にマルトースがα−1位で結合したジンゲロール配糖体である、<1>または<2>に記載のジンゲロール配糖体。
【0019】
【化3】
【0020】
<5> <1>〜<4>のいずれかに記載のジンゲロール配糖体を含有する組成物。
<6> 飲食品である、<5>に記載の組成物。
<7> 化粧品である、<5>に記載の組成物。
<8> 医薬部外品または医薬品である、<5>に記載の組成物。
【0021】
<9> ジンゲロールまたはジンゲロールを含む植物抽出物に糖転移酵素を糖供与体の存在下で作用させる工程を含むことを特徴とする、<1>〜<4>のいずれかに記載のジンゲロール配糖体の製造方法。
<10> 糖転移酵素が、Halomonas属細菌由来である、<9>に記載のジンゲロール配糖体の製造方法。
<11> 植物が、ショウガである<9>または<10>に記載のジンゲロール配糖体の製造方法。
【発明の効果】
【0022】
本発明により、有用な生理作用を有し、かつジンゲロールに比べ水溶性、安定性が向上したジンゲロール配糖体を提供可能である。当該配糖体は、水に溶解しやすく、室温において固形粉末として存在可能であり、さらに品質が安定であることから、飲食品、医薬部外品および医薬品ならびに化粧品等へ広く利用することができる。さらに、ジンゲロールを配糖化することでジンゲロール自身の辛みが低減されることに加え、ショウガオール、ジンゲロンへの変換を抑制できることから、ショウガオールまたはジンゲロン由来の辛味を低減することが可能となり、味質を改善することが可能である。
【図面の簡単な説明】
【0023】
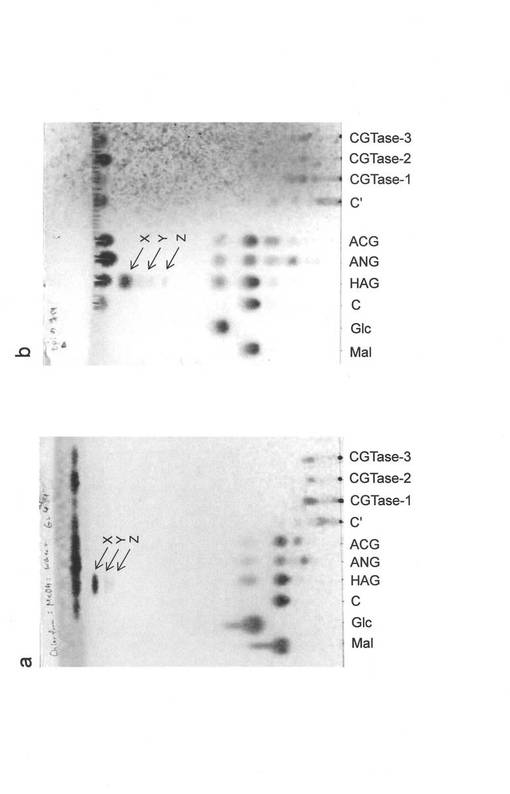
【図1】各種糖転移酵素による6−ジンゲロールの配糖化を検討した薄層クロマトグラフィーの図(写真)である。図中のMalはマルトース標準品、Glcはグルコース標準品、Cはα−グルコシダーゼによる反応のコントロール、C´はシクロデキストリングルカノトランスフェラーゼによる反応のコントロールを表す。その他の表記は、使用した酵素名を示している。図1aは展開溶媒クロロホルム:メタノール:水=6:4:1(v/v)の場合、図1bは展開溶媒2−プロパノール: 1−ブタノ―ル: 水=2:2:1(v/v)の場合である。
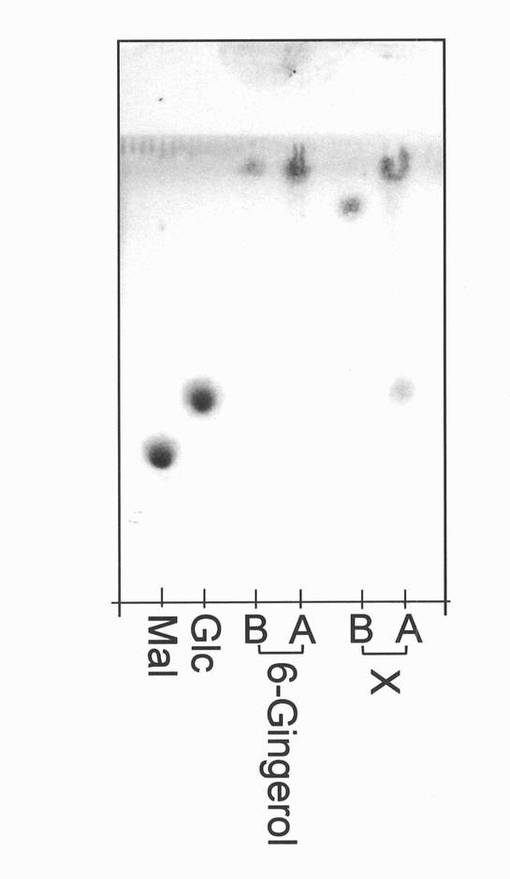
【図2】反応生成物Xのトリフルオロ酢酸(TFA)による分解試験の結果を示す薄層クロマトグラフィーの図(写真)である。図中のMalはマルトース標準品、Glcはグルコース標準品を表す。BおよびAは、それぞれトリフルオロ酢酸処理前および処理後を意味する。
【図3】反応生成物XのMSスペクトルを表す図である。
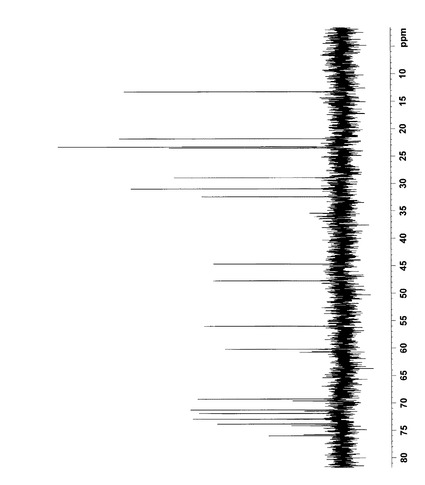
【図4】反応生成物Xの1H NMRスペクトルを表す図である。(全体図)
【図5】反応生成物Xの1H NMRスペクトルを表す図である。(4〜9ppm拡大図)
【図6】反応生成物Xの1H NMRスペクトルを表す図である。(2〜5ppm拡大図)
【図7】反応生成物Xの1H NMRスペクトルを表す図である。(0.6〜3.0ppm拡大図)
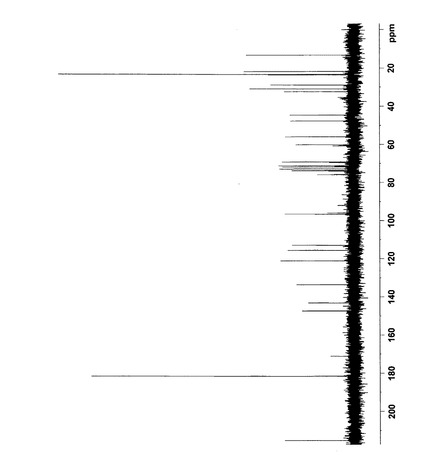
【図8】反応生成物Xの13C NMRスペクトルを表す図である。(全体図)
【図9】反応生成物Xの13C NMRスペクトルを表す図である。(5〜80ppm拡大図)
【図10】反応生成物YのMSスペクトルを表す図である。
【図11】反応生成物ZのMSスペクトルを表す図である。
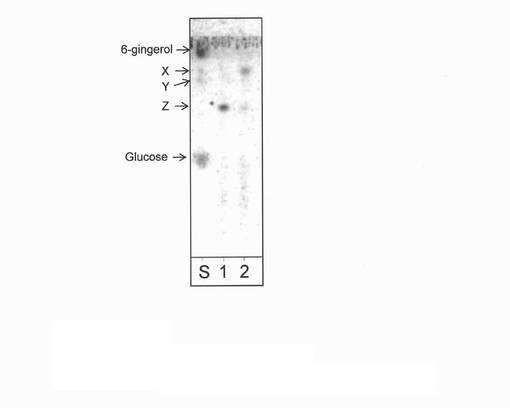
【図12】反応生成物Zのグルコアミラーゼ処理による分解結果を表す薄層クロマトグラフィーの図(写真)である。図中のSはグルコース、反応生成物XおよびY、および6−ジンゲロールを混合した標準物質である。1は反応生成物Z、2は反応生成物Zのグルコアミラーゼ処理物を示す。
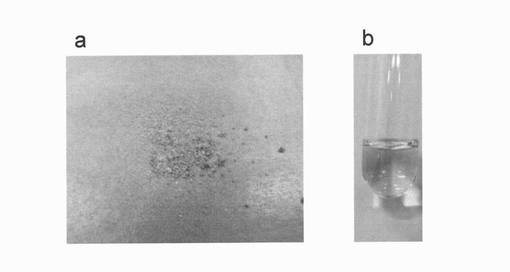
【図13】反応生成物X(ジンゲロール6−グルコシド)の外観図(写真)である。aは凍結乾燥粉体、bは0.2w/v%水溶液の様子。
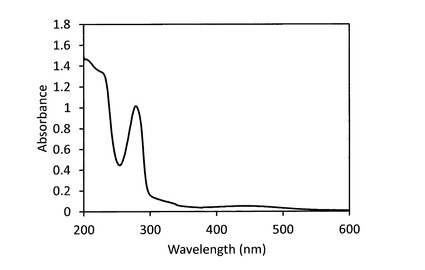
【図14】反応生成物X(ジンゲロール6−グルコシド)の0.2w/v%水溶液の吸収スペクトルである。
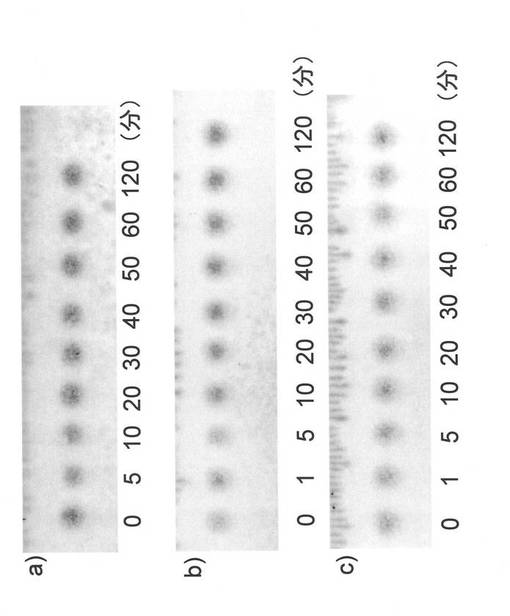
【図15】反応生成物X(ジンゲロール6−グルコシド)の消化性試験結果を表すTLCの図(写真)である。図中の数字は反応時間(分)を意味する。aはラット小腸アセトン抽出物、bはヒト人工胃液、cはヒト人工腸液による試験結果を示す。
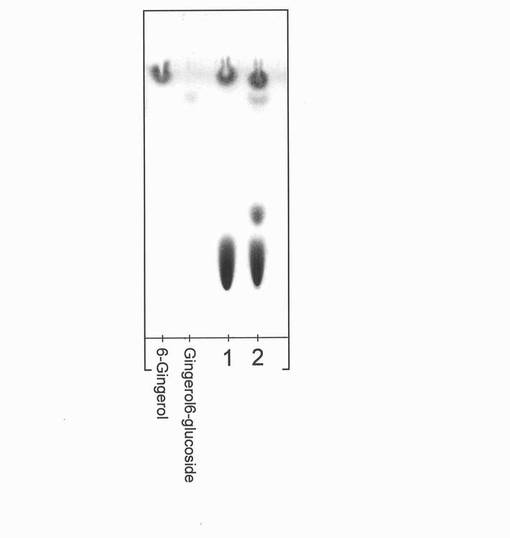
【図16】ジンジャーオイルを基質とした酵素反応による反応生成物X(ジンゲロール6−グルコシド)の生成を表すTLCの図(写真)である。図中の6−ジンゲロールおよびジンゲロール6−グルコシドは、それぞれの標準物質を示し、1および2は、それぞれコントロールおよび酵素反応溶液である。
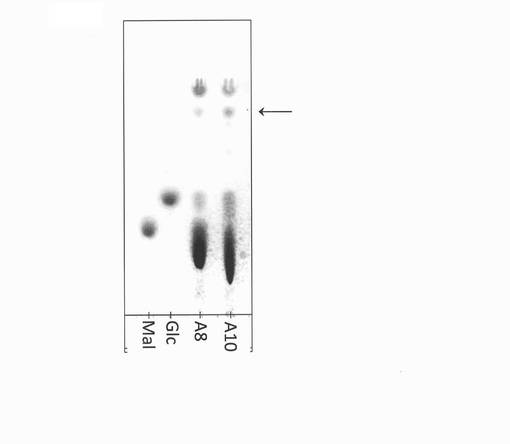
【図17】Halomonas sp.A8株およびA10株の粗酵素液によるジンゲロール6−グルコシドの生成を表すTLCの図(写真)である。図中のMalはマルトース標準品、Glcはグルコース標準品、A8はHalomonas sp.A8株の粗酵素による反応溶液、A10はHalomonas sp.A10株の粗酵素による反応溶液を示す。図中矢印はジンゲロール6−グルコシドを示す。
【発明を実施するための形態】
【0024】
〈1〉ジンゲロール配糖体
本発明のジンゲロール配糖体は、ジンゲロールのOH基にグルコース、マルトースまたはマルトオリゴ糖が結合したものである。
【0025】
ここで、マルトオリゴ糖のグルコース重合度は特に限定されないが、通常グルコース重合度3〜8であり、好ましくは3〜6である。
【0026】
本発明により、上記ジンゲロール配糖体の構造、物性等が明らかにされたため、本明細書の記載に基づいて、上記ジンゲロール配糖体を合成及び使用することができる。本発明のジンゲロール配糖体としては、上記ジンゲロール配糖体であれば、その製法は問わず、生化学的手法による製法であっても、通常の有機化学的手法を用いた有機化学的合成法であってもよい。
【0027】
しかし、安全性、経済性の観点から、ジンゲロールまたはジンゲロールを含むショウガ抽出物と糖供与体を含有する溶液に糖転移酵素を作用させる生化学的手法により合成することが望ましい。
【0028】
本明細書でいうジンゲロールとは、6−ジンゲロール、4−ジンゲロール、8−ジンゲロール、10−ジンゲロール、及び3S,5S−6−ジンゲジオール、3R,5S−6−ジンゲジオール等のジンゲジオールから選ばれる1種または2種以上の混合物をいう。すなわち、本発明のジンゲロール配糖体は、6−ジンゲロール、4−ジンゲロール、8−ジンゲロール、10−ジンゲロール、3S,5S−6−ジンゲジオール、3R,5S−6−ジンゲジオールから選ばれる1種の配糖体、または2種以上の配糖体の混合物であってよい。
【0029】
一つの形態では、本発明のジンゲロール配糖体は、下記一般式(I)で示されるジンゲロール配糖体である。
【0030】
【化4】
【0031】
(式中、
Rはグルコシル基、マルトシル基またはマルトオリゴ糖のグルコシル基を示し、
nは2,4,6または8のいずれかの整数を示す。)
【0032】
別の形態では、本発明のジンゲロール配糖体は、下記一般式(II)で示される、6−ジンゲロールの側鎖のOH基にグルコースがα−1位で結合したジンゲロール配糖体である。
【0033】
【化5】
【0034】
別の形態では、本発明のジンゲロール配糖体は、ジンゲロール配糖体が、下記式(III)で示される、6−ジンゲロールの側鎖のOH基にマルトースがα−1位で結合したジンゲ
ロール配糖体である。
【0035】
【化6】
【0036】
別の形態では、本発明のジンゲロール配糖体は、下記式(IIa)で示されるジンゲロール配糖体である。
【0037】
【化7】
【0038】
別の形態では、本発明のジンゲロール配糖体は、下記式(IIIa)で示されるジンゲロール配糖体である。
【0039】
【化8】
【0040】
〈2〉ジンゲロール配糖体の製造方法
本発明のジンゲロール配糖体の製造方法は、ジンゲロールまたはジンゲロールを含む植物抽出物に糖転移酵素を糖供与体の存在下で作用させる工程を含むことを特徴とする、ジンゲロール配糖体の製造方法である。
【0041】
(糖転移反応に用いるジンゲロール)
本発明の製造方法の原料、すなわち、糖転移反応に用いるジンゲロール原料としては、通常、6−ジンゲロール、4−ジンゲロール、8−ジンゲロール、10−ジンゲロール、3S,5S−6−ジンゲジオール、3R,5S−6−ジンゲジオール等のジンゲロール類だけでなく、ショウガ抽出物等の6−ジンゲロール、4−ジンゲロール、8−ジンゲロール、10−ジンゲロール、3S,5S−6−ジンゲジオール、3R,5S−6−ジンゲジオール等のジンゲロール類を含む植物抽出物等の混合物、または、その他のジンゲロール類を含む混合物を適宜用いることができる。
本発明におけるショウガ抽出物等の植物抽出物とは、その成分としてジンゲロールを含有している限りその原料および製法(抽出法)に特段制限は無く、公知の製法により得ることができる。例えば、ショウガ(Zingiber officinale)などZingiberaceae属植物等の植物全体、あるいは葉部、茎部、花部、果肉部、根部等を破砕、搾汁、粉末化、ペースト化等の加工を行うこと、更には必要に応じて熱水抽出、エタノール抽出、超臨界抽出等の抽出処理を行うことで得られる。市販のショウガ抽出物としてショウガオイル等を利用することも可能である。
【0042】
(糖供与体)
生化学的手法により本発明のジンゲロール配糖体を製造する場合、糖転移酵素の作用によりジンゲロール配糖体が生成するものであれば、糖供与体に特に制限は無く、使用する糖転移酵素の特性に応じて糖供与体を適宜選択することができる。糖転移酵素としてα−グルコシダーゼまたは/およびシクロデキストリングルカノトランスフェラーゼを用いる場合であれば、グルコース、マルトース、マルトオリゴ糖残基を含有するもの、例えば、マルトース、マルトトリオース、マルトテトラオース、マルトペンタオース、マルトヘキサオース、マルトヘプタオース、マルトオクタオースなどのマルトオリゴ糖、デキストリン、シクロデキストリン、アミロース等のα−グルカンと称される澱粉加水分解物、または澱粉およびそれらの混合物等を用いることができる。
糖転移酵素を用いてジンゲロール配糖体を製造する際は、澱粉や澱粉分解物にα−アミラーゼ、β−アミラーゼ、イソアミラーゼ等の加水分解酵素を作用させて糖供与体を生成
させた後、糖転移酵素を作用させても、上記澱粉や澱粉分解物を糖供与前駆体として酵素反応溶液に添加し、加水分解酵素と糖転移酵素を共に作用させてもよい。
【0043】
(糖転移酵素)
本発明の糖転移酵素は、ジンゲロールと糖供与体とを含む溶液から、ジンゲロール配糖体を生成可能な酵素であれば種類を問わないが、α−グルコシダーゼ(酵素番号:EC.3.2.1.20)、シクロデキストリングルカノトランスフェラーゼ(酵素番号:EC.2.4.1.19)、スクロースホスホリラーゼ(酵素番号:EC.2.4.1.7)などが挙げられる。
本発明でいうα−グルコシダーゼとはα−グルカンの非還元末端のα−グルコシド結合の加水分解反応を触媒し、α−グルコースを生じさせる酵素全般を意味する。また、α−グルコシダーゼには、加水分解反応とともに糖転移反応を触媒するものが存在し、本発明においては、加水分解反応とともに糖転移反応を触媒するα−グルコシダーゼが用いられる。
上記糖転移酵素のうち、好ましくはα−グルコシダーゼが挙げられ、より好ましくはHalomonas属微生物由来のα−グルコシダーゼが挙げられる。
上記糖転移酵素、特にα−グルコシダーゼの由来には、ジンゲロール配糖体を生成可能な酵素であれば特に制限は無く、動物、植物由来およびMucor属、Penicillium属、Aspergillus属、Saccharomyces属およびHalomonas属等の微生物由来酵素を適宜選択すればよい。これらの内、好ましくはジンゲロールの複数あるOH基のうち、側鎖のOH基を選択的に配糖化する酵素、例えばHalomonas属微生物由来のα−グルコシダーゼが挙げられ、例えば、Halomonas sp. A8株(平成23年5月2日付、受託番号NITE P−1096、独立行政法人製品評価技術基盤機構 特許微生物寄託センターに寄託)、Halomonas sp. A10株(平成23年5月2日付、受託番号NITE P−1097、独立行政法人製品評価技術基盤機構 特許微生物寄託センターに寄託)、Halomonas sp. H11株(平成23年5月2日付、受託番号NITE P−1098、独立行政法人製品評価技術基盤機構 特許微生物寄託センターに寄託)由来のα−グルコシダーゼが挙げられる。
【0044】
また、Halomonas属由来α−グルコシダーゼを用いれば、ジンゲロールの複数あるOH基のうち、側鎖のOH基を選択的に配糖化することが可能である。
【0045】
これらの糖転移酵素は、ジンゲロール配糖体を作製可能である範囲であれば、必ずしも精製して使用する必要はなく、通常ならば粗酵素であってよい。また、組換え・非組換えを問わず、必要に応じて宿主を選択すればよく、公知の方法を駆使して精製して
使用してもよい。また、糖転移酵素源として、糖転移酵素を産生する微生物を用いてもよい。
【0046】
本発明の製造方法において、本発明の効果を妨げない限り、使用する糖転移酵素に応じて、酵素の活性化剤等を用いてもよい。Halomonas属由来のα−グルコシダーゼを使用する場合には、必要に応じて、酵素の活性化剤として塩化アンモニウム、硫酸アンモニウム、塩化カリウム、塩化ルビジウム等のアルカリ金属塩またはアンモニウム塩等を添加することにより、酵素添加量または酵素反応時間を削減することができる。
【0047】
活性化剤の濃度は、酵素が活性化される濃度であれば特に制限されず、通常カチオン濃度として1μmol/L〜1mol/Lが適当である。経済性およびイオン負荷の点から、1μmol/L〜100mmol/L程度とすることが望ましい。
【0048】
使用酵素と反応時間には、密接な関係があることから、都合に応じて酵素使用量および
反応時間を選択すれば良いが、通常は、経済性の点から、約1〜100時間で反応が終了できるような酵素添加量を選択する。このような濃度としては、通常、糖供与体1gあたり0.1〜100U、好ましくは、糖供与体1gあたり0.2〜50Uであればよい。
本発明の製造方法における、酵素反応溶液におけるジンゲロールの濃度は、通常、1w/v%以上、好ましくは、2〜30w/v%であればよく、糖供与体濃度(w/v)は、ジンゲロールに対して、0.5〜30倍の範囲が好ましい。
【0049】
(反応条件)
本発明の製造方法における反応様式は、酵素反応が進む条件であれば、反応様式は特に限定されず、固定化された糖転移酵素をバッチ式で繰り返し、または連続式で反応に使用することも可能である。糖転移酵素を産生する微生物の増殖中の培地にジンゲロールと糖供与体を共存させ、目的物質を生成させることも有利に実施できる。
【0050】
本発明におけるジンゲロール配糖体調製のための反応方法は、通常、前述のジンゲロールと糖供与体とを含有する溶液に糖転移酵素を加え、該酵素が十分に作用する条件、通常、pH5〜10、温度15〜53℃の範囲から選ばれる条件に維持して行う。
【0051】
また、通常、ジンゲロールは水相と分離するため、アジテーターまたはスターラー等で攪拌しながら、またはローテーター等で反応容器自体を回転または振動させながら反応を行うことが好適である。必要であれば、乳化剤を混合させておいてもよい。
【0052】
このように生じた生成物および然る糖供与体を共存させた状態で、さらにα−グルコシダーゼまたはシクロデキストリングルカノトランスフェラーゼ、またはその他の糖転移酵素を作用させることにより、グルコース重合度を2以上とすること、またはグルコース以外の糖を、グルコシル基に転移することも当然可能であり、また、そのようにして生じた物質はモノグルコシドよりも水溶性が高いと予想される。そのようにして生じたジンゲロール配糖体及びその派生物質も本発明の範囲に包含される。
【0053】
酵素反応によりジンゲロール配糖体が生成した反応溶液には、通常、目的物であるジンゲロール配糖体の他、未反応物であるジンゲロール、糖供与体、場合によっては酵素、およびグルコース等の反応副生成物が混合している。
【0054】
このような混合物には、必要に応じて、α−アミラーゼ(EC.3.2.1.1)、β−アミラーゼ(EC.3.2.1.2)、グルコアミラーゼ(EC3.1.1.3)などによって加水分解処理を行い、残存した糖供与体を低分子化させることができる。
【0055】
上記混合物および混合物加水分解処理物をそのまま、ジンゲロール配糖体を含む、飲食品用、化粧品用、医薬部外品用または医薬品用組成物として、飲食品、化粧品、医薬部外品または医薬品に使用することができる。
【0056】
また、反応溶液を加熱する、またはpHを下げる等して酵素を失活させ、ろ過・濃縮して溶液状の、更には、乾燥、粉末化してジンゲロール配糖体含有組成物(製品)にし、飲食品、化粧品、医薬部外品または医薬品に使用することができる。
【0057】
ジンゲロールとジンゲロール配糖体の水溶性の違いを利用して、然る抽出操作を行うことにより、それぞれを、油相および水相に分離させ、双方を回収することも有利に実施できる。通常は、ジンゲロール配糖体はより親水性の高い溶媒へ抽出されることから、これを更にイオン交換カラムクロマトグラフィー、ゲルろ過クロマトグラフィー、疎水クロマトグラフィーなどの方法で分離精製すれば、糖供与体、酵素、およびグルコース等の反応副生成物とも分離が可能であり、ジンゲロール配糖体の高純度品を容易に得ることができ
る。
【0058】
この際、分離されるジンゲロールおよび糖供与体を、それぞれ回収し、糖転移反応の原料として再利用することも可能である。
【0059】
また、糖転移反応終了後、抽出およびクロマトグラフィーなどの分離手段にかけるまでに、必要に応じてろ過、活性炭処理による脱色、酸性および塩基性イオン交換樹脂による脱ミネラル処理等をすることも有利に実施できる。
【0060】
このようにして得られるジンゲロール配糖体は、以下の特徴を有する。
(1)下記に示す化合物
【0061】
【化9】
【0062】
(2)下記に示す化合物
【0063】
【化10】
【0064】
〈3〉ジンゲロール配糖体を含む組成物
上記のとおり、本発明のジンゲロール配糖体は、本発明の効果を妨げない限り、ジンゲロール配糖体以外に、植物抽出物成分、未反応物、酵素、反応副生成物等を含む組成物の形態でも使用することができる。
本発明のジンゲロール配糖体を含む組成物は、本発明のジンゲロール配糖体、及び通常飲食品、化粧品、医薬部外品または医薬品用成分として使用される成分を含むことができ、飲食品、化粧品、医薬部外品または医薬品等として使用することができる。
ジンゲロール配糖体の含有量は、各製品におけるジンゲロールの含有量を参考にして規定することができ、限定されないが、飲食品であれば0.01〜10質量%程度、化粧品であれば0.01〜5質量%程度、医薬部外品または医薬品であれば0.01〜10質量%程度である。
本発明の組成物は、本発明のジンゲロール配糖体を含有する以外は、上記の様な通常の成分を用いて、通常の方法により、製造できる。
本発明のジンゲロール配糖体を添加することで、ジンゲロールの作用を利用した、アディポネクチン産生増強効果や脂肪蓄積抑制効果といった有用な生理作用を有した飲食品、化粧品、医薬部外品または医薬品を製造することができる。さらに、後述の実施例5および図13に示した通り、本発明のジンゲロール配糖体は室温にて赤オレンジ色の粉末状であるため、着色料として飲食品、化粧品、医薬部外品または医薬品に添加することもできる。
【0065】
上記飲食品としては、醤油、粉末醤油、味噌、粉末味噌、もろみ、ひしお、フリカケ、マヨネーズ、ドレッシング、食酢、三杯酢、粉末すし酢、中華の素、天つゆ、麺つゆ、ソース、ケチャップ、焼き肉のタレ、カレールウ、シチューの素、スープの素、ダシの素、複合調味料、みりん、新みりん、テーブルシュガー、コーヒーシュガーなどの各種調味料、せんべい、あられ、おこし、求肥、餅類、まんじゅう、ういろう、餡類、羊羹、水羊羹、錦玉、ゼリー、カステラ、飴玉などの各種和菓子、パン、ビスケット、クラッカー、クッキー、パイ、プリン、バタークリーム、カスタードクリーム、シュークリーム、ワッフル、スポンジケーキ、ドーナツ、チョコレート、チューインガム、キャラメル、ヌガー、キャンディーなどの各種洋菓子、アイスクリーム、シャーベットなどの氷菓、果実のシロップ漬、氷蜜などのシロップ類、フラワーペースト、ピーナッツペースト、フルーツペーストなどのペースト類、ジャム、マーマレード、シロップ漬、糖果などの果実、野菜の加工食品類、福神漬け、べったら漬、千枚漬などの漬物類、たくわん漬の素、白菜漬の素などの漬物の素、ハム、ソーセージなどの畜肉製品類、魚肉ハム、魚肉ソーセージ、カマボコ、チクワ、天ぷらなどの魚肉製品類、ウニ、イカの塩辛、酢コンブ、さきするめ、タラ、タイ、エビなどの田麩などの各種珍味類、海苔、山菜、するめ、小魚、貝などで製造される佃煮類、煮豆、煮魚、ポテトサラダ、コンブ巻などの惣菜食品、乳製品、魚肉、畜肉、果実、野菜の瓶詰、缶詰類、プリンミックス、ホットケーキミックス、即席ジュース、即席コーヒー、即席汁粉、即席スープなどの即席食品、冷凍食品、果汁含有飲料、果汁ジュース、野菜ジュースなどの果実・野菜飲料、サイダー、ジンジャーエールなどの炭酸飲料、アイソトニック飲料、アミノ酸飲料などのスポーツ飲料、コーヒー飲料、緑茶などの茶系飲料、乳酸飲料、ココアなどの乳系飲料、チューハイ、清酒、果実酒などのアルコール飲料、栄養ドリンク等が挙げられるが、これらに限定されるものではない。
【0066】
上記化粧品とは、薬事法により定められた化粧品を指し、例えば、日焼け防止剤、ローション、エッセンス、乳液、クリーム、ハップ剤、ペースト剤、ゲル剤、パウダー、ファンデーション、化粧水、パック剤などの基礎化粧品およびメイクアップ化粧品、石鹸、洗顔料、ボディソープなどの皮膚洗浄剤、シャンプー、リンス、ヘアートニック、ヘアートリートメント剤、養毛剤、育毛剤、ヘアスプレーなどのヘアケア製品、浴用剤等が挙げられるが、これらに限定されるものではない。
【0067】
上記医薬部外品および医薬品とは、薬事法に定められた医薬部外品および医薬品を指し、例えば経口投与用製剤としてエキス剤、エリキシル剤、シロップ剤、チンキ剤、リモナーデ剤等の液剤とカプセル剤、顆粒剤、細粒剤、丸剤、散剤、錠剤等の固形剤等が挙げられるが、これらに限定されるものではない。
【0068】
〈4〉試験及び評価方法
(酵素力価の測定)
本発明において、α−グルコシダーゼの酵素力価の測定は、例えば、0.2w/v%マルトースを基質として、pH7、30℃、100μLの系で、特定量の酵素量から生じたグルコース量を測定することにより行うことができる。具体的には、例えば、上記系にて10分間の酵素反応後、2mol/LのTris−HCl(pH7)を200μl添加することにより酵素反応を終了させ、そこに、グルコーステストワコーC II(和光純薬製)を100μL添加し37℃で30分間インキュベートする。波長490nmの吸光度を測定することにより、生じたグルコース量を算出する。上記条件にて1分間に1μmolのマルトースを加水分解する酵素量を1Uと定義する。
【0069】
シクロデキストリングルカノトランスフェラーゼの酵素力価の測定は、例えば、0.9w/v%の可溶性でんぷんを基質として、pH6、40℃、1mLの系で、特定の酵素量から生じたβ−シクロデキストリンを測定することにより以下の方法により行うことができる。具体的には、例えば、上記系にて10分間の酵素反応後、40mMの水酸化ナトリウム水溶液を2.5mL添加し酵素反応を終了させ、そこに250μMの炭酸ナトリウムおよび0.1mg/mLのフェノールフタレインを含む溶液を0.3mL添加し、550nmの吸光度を測定することにより、生じたβ−シクロデキストリン量を算出する。上記条件にて、1分間に1mgのβ−シクロデキストリンを生成する酵素量を1Uとする。
【実施例】
【0070】
以下に実施例を挙げて本発明の詳細を説明するが、本発明は以下の実施例に限定されるものではない。
<実施例1>各種酵素による配糖体の調製検討
各種糖転移酵素により、ジンゲロールへの配糖化能を検討した。
【0071】
まず、α−グルコシダーゼとして、Aspergillus niger由来、Acremonium sp.由来およびHalomonas sp.H11株(受託番号:NITE P−1098)由来のものを検討した。以後、Aspergillus niger由来の酵素を「ANG」、Acremonium sp.由来の酵素を「ACG」およびHalomonas sp.H11株由来の酵素を「HAG」と表記する。なお、ANGおよびACGはそれぞれ「製品名:トランスグルコシダーゼアマノ、アマノエンザイム製」および「製品名:テイスターゼ、キリン協和フーズ製」である。HAGは、下記の参考例1の方法にて調製し、SDS−ポリアクリルアミド電気泳動的に単一バンドとして得られた精製酵素を使用した。
【0072】
10w/v%マルトース(日本食品化工製)、2.5w/v% 6−ジンゲロール(ナカライテスク製)、50mM緩衝液、各種α−グルコシダーゼをマルトース1gあたり1.5Uとなるよう反応溶液を調製し、10μLの系にて40℃にて反応させた。HAGおよびACGの反応にはHEPES−NaOH緩衝液(pH7)、ANGには酢酸ナトリウム緩衝液(pH5)を使用した。また、HAGにおける反応系には、硫酸アンモニウムを5mMとなるよう添加した。酵素の代わりに水を添加したpH7における溶液をコントロールとした。
【0073】
シクロデキストリングルカノトランスフェラーゼによる反応においては、Bacillus stearothermophillus由来、Bacillus sp. No.38−2由来およびBacillus coagulans由来の酵素を使用した。以後、これらの酵素をそれぞれCGTase−1、CGTase−2およびCGTase−3と表記することがある。なお、CGTase−1はノボザイムズ製、CGTase−2およびCGTase−3は日本食品化工製である。
酵素反応条件は以下の通りである。
すなわち、10w/v%パインデックス#100(松谷化学工業製)、2.5 w/v%6−ジンゲロール、50mM 酢酸ナトリウム緩衝液(pH6)、12mM Ca2+ 、パインデックス1gあたりシクロデキストリングルカノトランスフェラーゼ5Uとなるよう反応溶液を調製し、10μLの系にて反応させた。酵素の代わりに水を添加した溶液をコントロールとした。40℃にて24時間反応させ、反応溶液をメタノールにより10倍希釈し、そのうち1μLを薄層クロマトグラフィー(TLC)による分析に供した。
【0074】
TLCには、TLCアルミプレート(シリカゲル60 F254、 Merck製)を使用し、展開溶媒として、クロロホルム:メタノール:水=6:4:1(v/v)または2−プロパノール: 1−ブタノ―ル: 水=2:2:1(v/v)を使用し、10 v/v%硫酸/メタノール溶液を噴霧しオーブンで加熱することにより呈色させた。以後、上記TLC条件をそれぞれ、TLC条件1およびTLC条件2と記載する。標品としてグルコース(関東化学製)、マルトース(日本食品化工製)の1w/v %水溶液を1μL使用した。
【0075】
その結果、HAGによる反応溶液にのみ図1中矢印で示した特異的な反応産物が観察された。本分析で使用したいずれの展開溶媒においても主要な反応生成物スポットが1つ確認された。また、若干ではあるものの、その下に6−ジンゲロール由来と推察されるスポットが2つ確認された。そこで、生成物をTLCにおいて上から、反応生成物X、Y、およびZとし、これらを単離精製した。
【0076】
<参考例1>Halomonas sp. H11由来α−グルコシダーゼの組換え発現および精製
可溶性澱粉(関東化学製)10g/L、ポリペプトン(和光純薬製)5g/L、酵母抽出物(商品名『イーストエキストラクト』、ベクトン・ディッキンソン製)5g/L、塩化ナトリウム(和光純薬製)35g/L、リン酸二水素カリウム(関東化学製)1g/L、硫酸マグネシウム七水和物(関東化学製)0.2g/L、および水からなるpH7の液体培地10mLを200mLのバッフル付きフラスコに調製し、オートクレーブで121℃、20分間滅菌し冷却した。
【0077】
上記培地10mLに、Halomonas sp.H11株のグリセロールストックを接種し、37℃、150rpmで16時間培養した。培養液を1.5mL容エッペンドルフチューブに1mLずつ分注し遠心分離(4℃、14,000rpm、2分間)し上清を取り除いた。5mg/mLのリゾチーム(生化学工業製)、TE溶液(pH8)をチューブ1本当たり350μL添加し、37℃にて1時間保持した。10%ドデシル硫酸ナトリウム溶液を50μL添加し転倒混和し37℃にて30分間保持した。TE飽和フェノールを400μL添加し穏やかに転倒混和した後遠心分離(室温、14,000rpm、5分間)した。上層を新たなエッペンドルフチューブに移した後、フェノールクロロホルム溶液を400μL添加し穏やかに転倒混和し、上記と同様に遠心分離した。それぞれの上層を一つのエッペンドルフチューブに移し、3mol/L酢酸ナトリウム水溶液および95(v/v%)冷エタノールを、それぞれ溶液の1/10容量および2倍容量添加し遠心分離(4℃、14,000rpm、15分間)した。得られた沈殿物を70%エタノールにてリンス洗浄し風乾した。0.2mg/mlのRNase/TE溶液を400μL添加し、35℃にて30分間保持した。フェノールクロロホルム抽出およびエタノール沈殿法を上記と同様に行い、乾燥させた沈殿物をTE溶液100μLに溶解し、ゲノム溶液とした。
【0078】
クローニング用オリゴヌクレオチド、HAG−F−NdeI(5´−AAACATAT
GCAAGACAACATGATGTGGTG−3´)(配列番号1)およびHAG−R−XhoI(5´−AAACTCGAGTTAGGCAACCTGCATAAAGG−3´)(配列番号2)を用いてHalomonas sp. H11株のα−グルコシダーゼをクローニングした。すなわち、ゲノム100ng、10×KOD ver.2 buffer、 5μL、2mmol/L dNTPs 5μL、25mmol/L MgSO4 3μL、20mmol/Lのプライマー溶液各1μL、KOD plus polymerase 1μLを混合し、水で50μLにメスアップした。なお、上記PCR用の試薬は全て東洋紡製である。PCR条件は以下の通りである。94℃に2分間保持した後、94℃に30秒間、55℃に30秒間、72℃に2分間からなるサイクルを30回繰り返し、最後に72℃に3分間保持した。このうち4μLを電気泳動に供し、約1.5kbの増幅断片を確認した。当該断片を市販のキット(GEヘルスケア製、商品名『illustra GFXTM PCR DNA and Gel Band Purification Kit』、以後、「GFX」と表記する)にて精製した。これを、NdeIおよびXhoI(いずれもタカラバイオ製)で酵素処理し、GFXにて精製した。本DNA断片を、上記と同様にNdeIおよびXhoIで制限酵素処理したpET22b(ノバジェン製)に、通常のライゲーション反応にて連結させ発現用プラスミドを作製した。本プラスミドを『pET22b−HAG−H11』と命名した。
【0079】
上記pET22b−HAG−H11で、大腸菌BL21(DE3) Codon Plus RIL(ストラタジーン製)を形質転換した。
すなわち、50μg/mlアンピシリンを含むLB培地3mLに、上記形質転換体を接種し、37℃で一晩前培養した。このうち1mLを600mLの同培地に接種し、37℃で3時間培養した。それを氷冷し、0.1mol/Lのイソプロピル−β−チオガラクトピラノシド(和光純薬製)を600μL添加した。これを16℃で24時間回転振盪培養した。得られた培養物を、定法に従い、遠心分離して菌体を回収した。菌体を20mmol/L HEPES−NaOH緩衝液(pH7)約40mLに懸濁して、超音波破砕機(microsom(商標) ultrasonic cell disruptor)にて細胞を破砕した(出力:4ワット、全出力時間: 20分間)。これを遠心分離した上清を粗酵素液とした。
【0080】
粗酵素液には89.1 mgのタンパク質、83.3 Uのα−グルコシダーゼ活性が含まれていた。比活性は0.93U/mgであった。なお、タンパク質の定量は、DC protein assay kit(Bio Rad社製)を使用して、添付のプロトコルに従って行った。タンパク質の標準曲線は0〜1mg/mlのbovine serum albumin(Bio Rad社製)を用いて作成した。以下の実施例においても、特に示さない限り、同様である。
【0081】
粗酵素液を20mmol/L HEPES−NaOH緩衝液(pH7)で平衡化されたイオン交換カラムに、以下の条件で供し、塩化ナトリウム濃度0.1mol/L付近に溶出された画分(部分精製酵素1)を回収した。
【0082】
(条件)
カラム : TOYOPEARL(登録商標) DEAE−650M(東ソー株式会社製)
カラムサイズ: φ26mm×200mm
ゲル量 : 106mL
溶出液 : 20mmol/L HEPES−NaOH緩衝液(pH 7)
塩化ナトリウム濃度0mol/L〜0.5mol/Lのリニアグラジエント
流速 : 2mL/分
溶出液量 : 120mL
【0083】
部分精製酵素1には、54mgのタンパク質、374Uの酵素活性が含まれていた。比活性は6.93U/mgであった。なお、本α―グルコシダーゼは、アルカリ金属塩に活性化される特徴を有することから、粗酵素液と比較して総活性が上昇した原因は、溶出液中に含まれるNaClの影響であると考えられた。
【0084】
前記部分精製酵素1に、硫酸アンモニウムを少量ずつ加え、終濃度1.5mol/Lとした。これを1.5mol/L硫酸アンモニウムを含む20mmol/L HEPES緩衝液(pH7)で平衡化した疎水カラムに、以下の条件で供した。そして、硫酸アンモニウム濃度1.0mol/L付近で溶出された画分、約50mLを回収した。これを、5mmol/LのHEPES−NaOH緩衝液(pH7)5Lで2回透析し、精製酵素とした。
【0085】
(条件)
カラム : TOYOPEARL(登録商標) BUTHYL−650M(東ソー株式会社製)
カラムサイズ : φ16mm×150mm
ゲル量 : 30mL
溶出液 : 20mmol/L HEPES緩衝液(pH7)
硫酸アンモニウム濃度1.5mol/L〜0mol/Lのリニアグラジエント
流速 : 1mL/分
溶出液量 : 120mL
【0086】
精製酵素には、17.8mgのタンパク質と、67.5Uの酵素活性が含まれており、比活性は3.8U/mgであった。粗酵素液からの回収率は、81.4%であり、精製度は4.1倍であった。この画分を1μg、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動に供した結果、1本のバンドのみが検出されたことから、酵素は単一に精製されていることが分かった。得られた精製酵素を実施例に用いた。
【0087】
上記記載において、比活性はタンパク質1mgあたりの酵素活性(U/mg)を示し、回収率は、粗酵素液の総活性に対する酵素活性の残存率(%)を示し、精製度は、粗酵素液の比活性に対する倍率(倍)を示す。
【0088】
<実施例2>生成物の精製
ジンゲロール配糖体の単離精製法は以下の通りである。まず、30w/v%マルトース、5w/v%6−ジンゲロール、5mM硫酸アンモニウム、30mM HEPES−NaOH緩衝液(pH7)およびマルトース1gあたり3UのHAGとなるよう、計3mLの溶液を調製しよく攪拌した後、40℃で48時間インキュベートした。反応溶液をPLCガラスプレート(シリカゲル60F254、2mm、Merck製)に500μL/枚となるようにスポッティングし、クロロホルム:メタノール:水=6:4:1(v/v)またはクロロホルム:メタノール:酢酸=80:20:5で展開した。展開プレートの方端を前述の方法と同様に呈色させ、各反応生成物のスポットと同Rf値の展開をスパーテルで掻き取り、50v/v%メタノール水溶液で抽出した。抽出液をろ紙(No.5、アドバンテック製)でろ過し、減圧濃縮した。これを水に溶解し、0.45μmフィルター(ミリポア製)にてろ過し凍結乾燥させた。これを以後の実験に用いた。
【0089】
<実施例3>反応生成物Xの構造解析
(トリフルオロ酢酸による分解)
精製した生成物Xおよび6−ジンゲロール標品をそれぞれ0.5 mgずつ量り取り、それに純水50μLを添加した。これに4Mトリフルオロ酢酸(関東化学製、以下TFAと表記することがある)を400μL添加し、100℃に1時間保持した。減圧下にて濃縮・乾固させたものを50v/v%メタノール水溶液40μLに溶解し、実施例1で上述したTLC条件2で分析した。なお、TFA添加前のサンプルも同様に分析した。その結果、生成物Xは、図2に示すように、TFA処理により6−ジンゲロール(もしくはその分解物)とグルコースの位置にスポットを生じた。このことから、反応生成物Xは6−ジンゲロールとグルコースから構成される化合物である可能性が示唆された。
【0090】
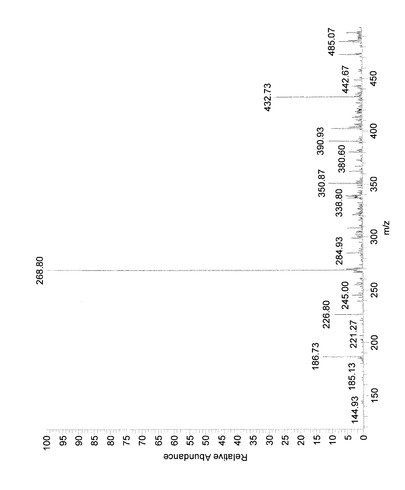
(MS(質量分析、Mass Spectrometry)解析)
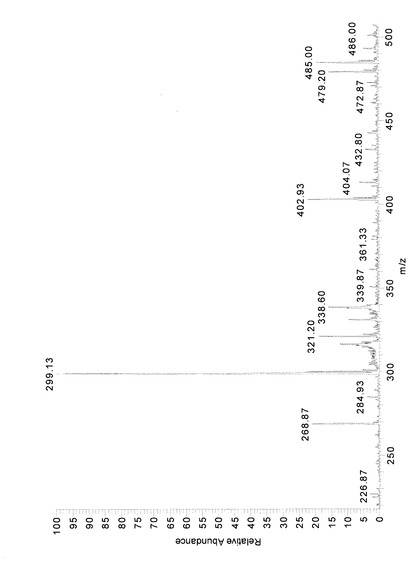
反応生成物Xを0.1mg/mLとなるようメタノールに溶解し、質量分析に供した。分析には、LCQ(Thermo electron製)を用い、エレクトロスプレーイオン化法(ESI)により実施した。測定はポジティブモードで行った。その結果、図3に示したようなMSスペクトルが得られ、そのピークおよび対応する分子量(M)は、表1の通りであった。
【0091】
【表1】
【0092】
すなわち、反応生成物Xにおいては、6−ジンゲロール(分子量294)にグルコースが一つ付加した配糖体(分子量456)およびESIによってその配糖体のグリコシド結合が切断され、さらに、それにより生じた6−ジンゲロールが脱水し生成したと考えられるショウガオール(分子量276)に相当するピークが検出された。
【0093】
(NMR解析)
BRUKER社製 AV400MdigitalNMR(400MHz)を使用しH−H COSY、HMQCおよびHMBCの二次元分析を行った。詳細には、精製し凍結乾燥した反応生成物X10mgを0.5mLのD2O(ナカライテスク、99.90%D)に溶解し、標準物質として0.1mg/mLの3−(トリメチルシリル)プロピオン酸ナトリウム−2,2,3,3−d4(TSP、和光純薬)を2μL滴下したものを分析に供した。分析時の温度を303ケルビンに設定した。
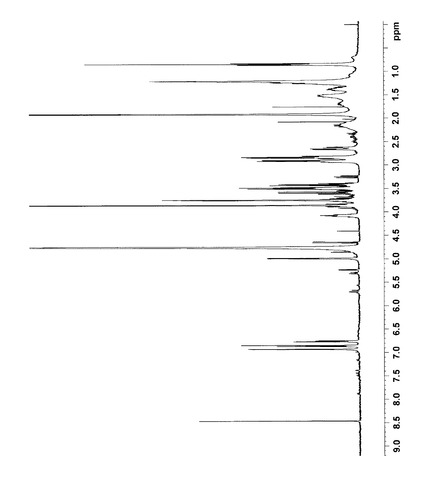
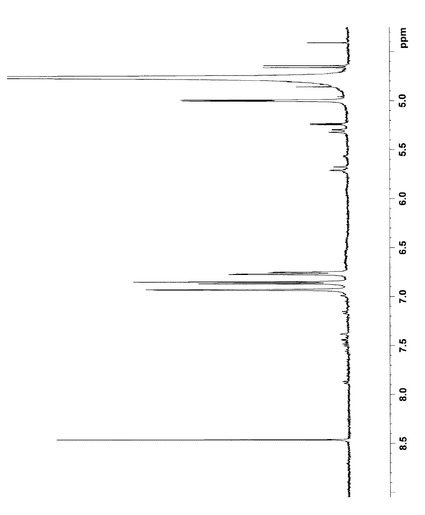
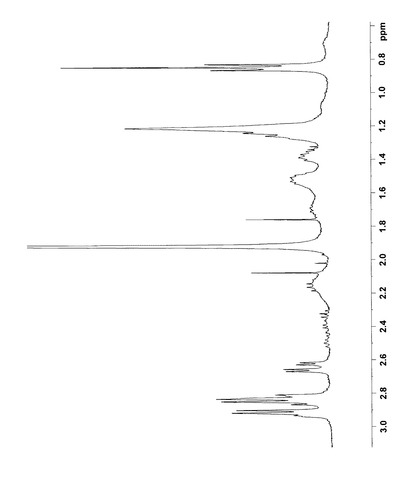
NMR解析の結果から、各シグナルを以下のように帰属した。なお、1H NMRおよび13C NMRの解析結果は図4〜9に示した。
【0094】
1H NMR (400MHz,D2O)
δ6.93(s,1H,16),6.88(d,1H,13), 6.75(d,1H,12),5.00(d,1H,1´),4.05(m,1H,6),3.90(dd,1H,6´a),3.85(s,3H,17),3.70(dd,1H,6´b), 3.60(t,1H,3´),3.50(t,1H,2´),3.49(t,1H,5´),3.42(t,1H,4´),2.95(d,2H,9),2.88(dd,1H,7a),2.85(d,2H,10),2.52(dd,1H,7b),1.53(m,1H,5a), 1.38(m,1H,5b),1.25(m,2H,2),1.22(t,2H,3), 1.22(t,2H,4),0.85(t,3H,1)
【0095】
13C NMR (400 MHz, D2O)
δ147.36(15),143.10(14),133.56(11),121.08(12),115.56(13),112.88(16),96.57(1´),75.97(5´),74.16(6),73.84(3´),72.95(8),71.28(2´),69.29(4´),60.19(6´),56.01(17),47.43(7),44.66(9),32.41(5),30.97(4),28.92(10),23.55(3),21.83(2),13.26(1)
【0096】
HMBCにおいてH1´−C6およびH6−C1´の相関が確認されたため、配糖化部位をC6のOH基と決定した。
以上、TFA処理、MS解析およびNMR解析の結果より、HAGによる6−ジンゲロールの配糖化反応により生じた主生成物Xは、下記式(IV)で示される、6−ジンゲロールの側鎖のOH基にグルコースがα−1位で結合したジンゲロール配糖体であることが明らかとなった。以降、反応生成物Xを簡易的に、ジンゲロール6−グルコシドと呼ぶことがある。
【0097】
【化11】
【0098】
<実施例4>反応生成物YおよびZの構造解析
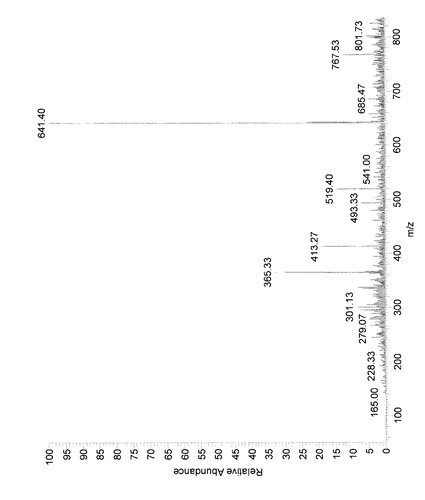
(反応生成物YおよびZのMS解析)
各精製物を実験3と同様の方法にてMS解析した。その結果、図10および11に示したようなMSスペクトルが得られ、その主要ピークおよび対応する分子量(M)は表2の通りであった。
【0099】
【表2】
【0100】
すなわち、反応生成物YのMS分析において検出された分子量は、6−ジンゲロール(分子量294)が脱メトキシされた物質(分子量246)および当該物質のモノグルコシド(410)に相当していた。本生成物は、生成量が微量でありNMR解析に不十分であったことから、MS分析結果より以下の構造であると推定した。
反応生成物Y:脱メトキシされた6−ジンゲロールのいずれかのOH基にグルコースが脱水結合した化合物。
反応生成物Zの、641m/zは、6−ジンゲロールにグルコース (分子量180)が2分子縮合し、ナトリウムイオンが付加した分子量 (294+180+180−18−18+23=641)と一致した。
【0101】
(反応生成物Zのグルコアミラーゼ処理)
反応生成物Zは、Yと同様に生成量がごく微量であったことから、NMR解析に不十分であった。そこで、反応生成物Zにグルコアミラーゼを作用させ、その反応生成物をTLCにて分析することにより、反応生成物Zの構造を推定した。すなわち、0.1g/mL反応生成物Z、4U/mLグルコアミラーゼ(生化学工業製)、4mM酢酸ナトリウム緩衝液(pH5)となるよう混合し、40℃にて24時間インキュベートした。コントロールとして、グルコアミラーゼ無添加のものを同様にインキュベートした。2μLをTLCに供し、実施例1で上述のTLC条件2により分析した。なお、6−ジンゲロール、反応生成物XおよびY、およびグルコースを混合し50%メタノール水溶液に溶解させた溶液を標準物質としてスポッティングした。その結果、図12に示すように、Zをグルコアミラーゼ処理すると反応生成物X(ジンゲロール6−グルコシド)を生成することが分かった。これは、反応生成物Zは、反応生成物Xのグルコース残基にさらにもう一分子のグルコースがα−1,4グルコシド結合した構造を取っていることを意味していた。さらに、本実施例の酵素反応において使用したHalomonas属由来α−グルコシダーゼは、α1,4型の転移酵素であることから、その結合様式はα−1,4であると考えられた。以上、MSおよび本結果より、反応生成物Zは、以下の構造であると推定した。
反応生成物Z:6−ジンゲロールの側鎖OH基にマルトースがα−1位で結合した化合物であることが明らかとなった。
【0102】
<実施例5>ジンゲロール6−グルコシドの外観、溶解性および吸収極大
精製したジンゲロール6−グルコシドの凍結乾燥品は、図13に示すように、室温にて赤オレンジ色の粉末状であり、水に可溶であった。また、0.2w/v%の水溶液を調製し、幅10mmの石英セルにて、波長600〜200nmにおける吸収スペクトルを測定したところ、ジンゲロール6−グルコシドは、図14に示すように、波長278nmに吸収極大を有していることがわかった。
【0103】
<実施例6>反応生成物の消化性
ジンゲロール6−グルコシドの、ラット小腸アセトンパウダー、ヒト人工胃液およびヒト人工腸液による、消化性試験を行った。
(ラット小腸アセトンパウダーによる消化性)
ラット小腸アセトンパウダー(Sigma製)50mgを純水1mLに溶解し、ボルテックスにてよく混合した後氷冷した。これを遠心分離(15,000rpm、10分、4℃)し、上清を酵素液として用いた。ジンゲロール6−グルコシド1mgに0.5Mリン酸ナトリウム緩衝液(pH7)10μL、および酵素液90μLを添加しよく混合し37℃でインキュベートした。これを経時的にサンプリングし、1μLをTLCプレートにスポッティングしドライヤーで即座に乾燥させることにより反応を停止した。実施例1で上述したTLC条件2にて分析し、スポットの消失を観察した。
【0104】
(ヒト人工胃液による消化性)
Pepsin 1:100 (和光純薬製)3.2mg、NaCl2.0mgおよびH
Cl 7μLを混合し、純水にて0.8mLにメスアップしたものを人工胃液とした。ジンゲロール6−グルコシドを0.5mg量り取り、純水10μLに溶解した。これに人工胃液を40μL添加し、37℃にてインキュベートした。これを経時的にサンプリングした。上記と同様の方法にて反応を停止し、TLC分析した。
【0105】
(ヒト人工腸液による消化性)
Pancreatin(和光純薬製)10 mg、0.2M KH2PO4 250μLおよび0.2M NaOH 118μLを混合し、純水にて0.8mLにメスアップしたものを人工腸液とした。ジンゲロール6−グルコシドを0.5mg量り取り、純水10μLに溶解した。これに人工腸液を40μL添加し、37℃にてインキュベートした。以後の操作は上記と同様である。
【0106】
各種消化酵素による、ジンゲロール6−グルコシドの消化性試験の結果、図15に示すように、ラット小腸アセトン抽出物、ヒト人工胃液およびヒト人工腸液にいずれにおいても、120分間の処理においてジンゲロール6−グルコシドは減少しなかった。すなわち、ジンゲロール6−グルコシドは、これらの消化酵素には分解されず、このような消化酵素に対して安定であることがわかった。
【0107】
<実施例7>ジンゲロール6−グルコシドの安定性
ジンゲロール6−グルコシドを1mg量り取り、50%エタノール、0.1M HClの水溶液1mLに溶解した。これを、密栓可能な容器で、60℃にて18時間インキュベートしたときの残存率を測定することにより、ジンゲロール6−グルコシドの安定性を評価した。すなわち、サンプルを20μLずつ採取し、これをアセトニトリル:水=80:20(v/v)の溶液180μLと混合した。これをHPLC分析に供した。インキュベート0時間におけるジンゲロール6−グルコシドのUV吸収ピーク面積を100(%)としたときの、18時間後における相対ピーク面積を残存率(%)とした。なお、比較対象として、6−ジンゲロールの安定性を同様に評価した。
【0108】
測定条件は下記の通りである。
カラム : YMC Pack Pro C18(AS−303、φ4.6mm×250mm)
溶媒 : アセトニトリル:水=7:3(v/v)
流速 : 1mL/分
温度 : 30℃
検出 : UV280nm
注入量 : 20μL
【0109】
なお、本条件における各物質の溶出時間は以下の通りである。
ジンゲロール6−グルコシド : 3.0分
6−ジンゲロール : 4.7分
6−ショウガオール : 7.8分
本試験の結果、処理18時間後において、6−ジンゲロールは、約半量が6−ショウガオールへ変換しており、残存率は49%であった。一方で、ジンゲロール6−グルコシドの残存率は94%であった。このことから、6−ジンゲロールの構造が変化しやすい0.1MのHCl溶液においても、ジンゲロール6−グルコシドは安定であることがわかった。
【0110】
<実施例8>ジンジャーオイルを基質とした反応
10%v/vジンジャーオイル(稲葉香料製)、20%w/vマルトース、5mM硫酸アンモニウム、50mM HEPES−NaOH緩衝液(pH7)および0.3U/mL
のHAGを含む1mLの水溶液を調製し、40℃にて93時間酵素反応させた。なお、酵素無添加のものをコントロールとした。反応溶液を熱失活し、1μLを、実施例1で上記に示したTLC条件2にて分析した。なお、標品として、6−ジンゲロールおよび単離精製したジンゲロール6−グルコシドを分析に供した。
その結果、図16に示すように、反応後の主生成物としてジンゲロール6−グルコシドと同Rf値にスポットが現れた。このことから、6−ジンゲロール以外の成分を含むジンジャーオイルを基質とした場合においても、HAGによりジンゲロール6−グルコシドを生成可能であることがわかった。
【0111】
<実施例9>Halomonas sp.A8株およびA10株由来酵素による反応
参考例1に示したHalomonas sp.H11株の培養と同条件において、Halomonas sp.A8株およびA10株を100mLの培地にて好気的に培養し、遠心分離により集菌した。菌体を20mM HEPES−NaOH緩衝液(pH7)30mLに懸濁し、それぞれ超音波破砕機(microsom(商標) ultrasonic cell disruptor)にて細胞を破砕した(出力:4ワット、全出力時間:20分間)。これを遠心分離した上清を粗酵素液とした。Halomonas sp.A8株およびA10株の粗酵素液のα−グルコシダーゼ活性は、それぞれ0.032U/mLおよび0.023U/mLであった。
【0112】
10w/v%マルトース(日本食品化工製)、2.5w/v% 6−ジンゲロール(ナカライテスク製)、5mM硫酸アンモニウム、50mM HEPES−NaOH緩衝液(pH7)、上記粗酵素液を、α−グルコシダーゼ活性としてマルトース1gあたり0.15Uとなるよう反応溶液を調製し、10μLの系にて37℃にて17時間反応させた。反応溶液1μLを実施例1で上記に示したTLC条件2にて分析した。その結果、図17に示すように、ジンゲロール6−グルコシドが生成していた。このことから、Halomonas sp.A8およびA10株の未精製酵素においてもジンゲロール6−グルコシドを生成可能であることがわかった。
【技術分野】
【0001】
本発明は、新規な化合物およびその製造法ならびに用途に関し、詳しくはショウガ成分であるジンゲロールの配糖体、当該化合物の製造方法、当該化合物の用途に関する。
【背景技術】
【0002】
ジンゲロールは、ショウガ(Zingiber officinale)などZingiberaceae属植物の根茎等に含まれる精油成分であり、アディポネクチン産生増強効果(特許文献1)、脂肪蓄積抑制効果(非特許文献1)などの効能が知られ、肥満予防、糖尿病治療、冷え改善などの効能を有する飲食品および医薬品等への応用が期待されている。
【0003】
上記精油成分は、例えばショウガから公知の手法により抽出することで得ることができる。
【0004】
ジンゲロールを含有するショウガは薬用として利用される他、香辛料として食用に供されるなど、歴史的に安全な食品であることが証明されている。医薬品としては、消化薬、緩下剤、鎮咳剤、制酸薬の原料として使用される。食品としては調味料、清涼飲料、アルコール飲料、菓子類などに、また化粧品の香料としても使用されている。
【0005】
しかしながら、有効成分のジンゲロールは、水に不溶であり、また、強い辛味を呈し、刺激がある。また、加熱あるいは酸性条件下においてより辛みの強く脂溶性の高いショウガオールまたはジンゲロンに変換されやすいことが知られている。このことから、ジンゲロールを安定した品質で、飲食品あるいは医薬品等へ利用することが困難であった。
【0006】
そのような問題から、ジンゲロールの水溶性、安定性改善を目的として、特許文献2においては、ジンゲロールまたはジンゲロール含有物と糖類の熱処理物を組み合わせることにより、安定性および水溶性の高いジンゲロール含有水溶性組成物を得たことが開示されている。しかしながら、得られる組成物はカラメル状であることから汎用性が低く、また、エタノールが残存しているために直接飲食品へ利用しにくい等の問題点があった。
【0007】
したがって、ジンゲロールの水溶性、安定性および味質などの物性が改善され、飲食品、医薬部外品および医薬品工業等において取扱が容易な汎用性の高いジンゲロール誘導体があれば、その産業利用価値が高くなることが期待された。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2011−1386号公報
【特許文献2】特開2008−56572号公報
【非特許文献】
【0009】
【非特許文献1】J.Med.Chem.2011、54:6295−6304
【発明の概要】
【発明が解決しようとする課題】
【0010】
そこで本発明の目的は、水に可溶かつ安定性の高い新規ジンゲロール配糖体、その製法およびその用途を提供することにある。
【課題を解決するための手段】
【0011】
本発明者らは、従来のジンゲロールの欠点を改善するために、とりわけ、糖転移反応を利用した新しいジンゲロール誘導体の開発を目指し、研究を重ねてきた。
【0012】
その結果、水に可溶で、室温において固形粉末として存在可能であり、安定性の高いジンゲロール配糖体を、少なくともジンゲロールと、食品に利用可能なマルトース等の糖供与体を含む混合溶液に、糖転移酵素を作用させることにより合成可能であることを見出した。また、ある特定の糖転移酵素を使用することによって、ジンゲロールの側鎖に存在する、従来ジンゲロールの不安定さの原因とされていたβ−ヒドロキシケトンのOH基を選択的に配糖化可能であることを見出し、さらに、その製造法並びに飲食物、医薬部外品および医薬品ならびに化粧品等への用途を開発して本発明を完成した。
【0013】
すなわち、本発明は以下の通りである。
【0014】
<1> ジンゲロールのOH基にグルコース、マルトースまたはマルトオリゴ糖が脱水縮合していることを特徴とするジンゲロール配糖体。
<2> 下記一般式(I)で示されるジンゲロール配糖体である、<1>に記載のジンゲロール配糖体。
【0015】
【化1】
【0016】
(式中、
Rはグルコシル基、マルトシル基またはマルトオリゴ糖のグルコシル基を示し、
nは2,4,6または8のいずれかの整数を示す。)
<3> ジンゲロール配糖体が、下記式(II)で示される、6−ジンゲロールの側鎖のOH基にグルコースがα−1位で結合したジンゲロール配糖体である、<1>または<2>に記載のジンゲロール配糖体。
【0017】
【化2】
【0018】
<4> ジンゲロール配糖体が、下記式(III)で示される、6−ジンゲロールの側鎖のOH基にマルトースがα−1位で結合したジンゲロール配糖体である、<1>または<2>に記載のジンゲロール配糖体。
【0019】
【化3】
【0020】
<5> <1>〜<4>のいずれかに記載のジンゲロール配糖体を含有する組成物。
<6> 飲食品である、<5>に記載の組成物。
<7> 化粧品である、<5>に記載の組成物。
<8> 医薬部外品または医薬品である、<5>に記載の組成物。
【0021】
<9> ジンゲロールまたはジンゲロールを含む植物抽出物に糖転移酵素を糖供与体の存在下で作用させる工程を含むことを特徴とする、<1>〜<4>のいずれかに記載のジンゲロール配糖体の製造方法。
<10> 糖転移酵素が、Halomonas属細菌由来である、<9>に記載のジンゲロール配糖体の製造方法。
<11> 植物が、ショウガである<9>または<10>に記載のジンゲロール配糖体の製造方法。
【発明の効果】
【0022】
本発明により、有用な生理作用を有し、かつジンゲロールに比べ水溶性、安定性が向上したジンゲロール配糖体を提供可能である。当該配糖体は、水に溶解しやすく、室温において固形粉末として存在可能であり、さらに品質が安定であることから、飲食品、医薬部外品および医薬品ならびに化粧品等へ広く利用することができる。さらに、ジンゲロールを配糖化することでジンゲロール自身の辛みが低減されることに加え、ショウガオール、ジンゲロンへの変換を抑制できることから、ショウガオールまたはジンゲロン由来の辛味を低減することが可能となり、味質を改善することが可能である。
【図面の簡単な説明】
【0023】
【図1】各種糖転移酵素による6−ジンゲロールの配糖化を検討した薄層クロマトグラフィーの図(写真)である。図中のMalはマルトース標準品、Glcはグルコース標準品、Cはα−グルコシダーゼによる反応のコントロール、C´はシクロデキストリングルカノトランスフェラーゼによる反応のコントロールを表す。その他の表記は、使用した酵素名を示している。図1aは展開溶媒クロロホルム:メタノール:水=6:4:1(v/v)の場合、図1bは展開溶媒2−プロパノール: 1−ブタノ―ル: 水=2:2:1(v/v)の場合である。
【図2】反応生成物Xのトリフルオロ酢酸(TFA)による分解試験の結果を示す薄層クロマトグラフィーの図(写真)である。図中のMalはマルトース標準品、Glcはグルコース標準品を表す。BおよびAは、それぞれトリフルオロ酢酸処理前および処理後を意味する。
【図3】反応生成物XのMSスペクトルを表す図である。
【図4】反応生成物Xの1H NMRスペクトルを表す図である。(全体図)
【図5】反応生成物Xの1H NMRスペクトルを表す図である。(4〜9ppm拡大図)
【図6】反応生成物Xの1H NMRスペクトルを表す図である。(2〜5ppm拡大図)
【図7】反応生成物Xの1H NMRスペクトルを表す図である。(0.6〜3.0ppm拡大図)
【図8】反応生成物Xの13C NMRスペクトルを表す図である。(全体図)
【図9】反応生成物Xの13C NMRスペクトルを表す図である。(5〜80ppm拡大図)
【図10】反応生成物YのMSスペクトルを表す図である。
【図11】反応生成物ZのMSスペクトルを表す図である。
【図12】反応生成物Zのグルコアミラーゼ処理による分解結果を表す薄層クロマトグラフィーの図(写真)である。図中のSはグルコース、反応生成物XおよびY、および6−ジンゲロールを混合した標準物質である。1は反応生成物Z、2は反応生成物Zのグルコアミラーゼ処理物を示す。
【図13】反応生成物X(ジンゲロール6−グルコシド)の外観図(写真)である。aは凍結乾燥粉体、bは0.2w/v%水溶液の様子。
【図14】反応生成物X(ジンゲロール6−グルコシド)の0.2w/v%水溶液の吸収スペクトルである。
【図15】反応生成物X(ジンゲロール6−グルコシド)の消化性試験結果を表すTLCの図(写真)である。図中の数字は反応時間(分)を意味する。aはラット小腸アセトン抽出物、bはヒト人工胃液、cはヒト人工腸液による試験結果を示す。
【図16】ジンジャーオイルを基質とした酵素反応による反応生成物X(ジンゲロール6−グルコシド)の生成を表すTLCの図(写真)である。図中の6−ジンゲロールおよびジンゲロール6−グルコシドは、それぞれの標準物質を示し、1および2は、それぞれコントロールおよび酵素反応溶液である。
【図17】Halomonas sp.A8株およびA10株の粗酵素液によるジンゲロール6−グルコシドの生成を表すTLCの図(写真)である。図中のMalはマルトース標準品、Glcはグルコース標準品、A8はHalomonas sp.A8株の粗酵素による反応溶液、A10はHalomonas sp.A10株の粗酵素による反応溶液を示す。図中矢印はジンゲロール6−グルコシドを示す。
【発明を実施するための形態】
【0024】
〈1〉ジンゲロール配糖体
本発明のジンゲロール配糖体は、ジンゲロールのOH基にグルコース、マルトースまたはマルトオリゴ糖が結合したものである。
【0025】
ここで、マルトオリゴ糖のグルコース重合度は特に限定されないが、通常グルコース重合度3〜8であり、好ましくは3〜6である。
【0026】
本発明により、上記ジンゲロール配糖体の構造、物性等が明らかにされたため、本明細書の記載に基づいて、上記ジンゲロール配糖体を合成及び使用することができる。本発明のジンゲロール配糖体としては、上記ジンゲロール配糖体であれば、その製法は問わず、生化学的手法による製法であっても、通常の有機化学的手法を用いた有機化学的合成法であってもよい。
【0027】
しかし、安全性、経済性の観点から、ジンゲロールまたはジンゲロールを含むショウガ抽出物と糖供与体を含有する溶液に糖転移酵素を作用させる生化学的手法により合成することが望ましい。
【0028】
本明細書でいうジンゲロールとは、6−ジンゲロール、4−ジンゲロール、8−ジンゲロール、10−ジンゲロール、及び3S,5S−6−ジンゲジオール、3R,5S−6−ジンゲジオール等のジンゲジオールから選ばれる1種または2種以上の混合物をいう。すなわち、本発明のジンゲロール配糖体は、6−ジンゲロール、4−ジンゲロール、8−ジンゲロール、10−ジンゲロール、3S,5S−6−ジンゲジオール、3R,5S−6−ジンゲジオールから選ばれる1種の配糖体、または2種以上の配糖体の混合物であってよい。
【0029】
一つの形態では、本発明のジンゲロール配糖体は、下記一般式(I)で示されるジンゲロール配糖体である。
【0030】
【化4】
【0031】
(式中、
Rはグルコシル基、マルトシル基またはマルトオリゴ糖のグルコシル基を示し、
nは2,4,6または8のいずれかの整数を示す。)
【0032】
別の形態では、本発明のジンゲロール配糖体は、下記一般式(II)で示される、6−ジンゲロールの側鎖のOH基にグルコースがα−1位で結合したジンゲロール配糖体である。
【0033】
【化5】
【0034】
別の形態では、本発明のジンゲロール配糖体は、ジンゲロール配糖体が、下記式(III)で示される、6−ジンゲロールの側鎖のOH基にマルトースがα−1位で結合したジンゲ
ロール配糖体である。
【0035】
【化6】
【0036】
別の形態では、本発明のジンゲロール配糖体は、下記式(IIa)で示されるジンゲロール配糖体である。
【0037】
【化7】
【0038】
別の形態では、本発明のジンゲロール配糖体は、下記式(IIIa)で示されるジンゲロール配糖体である。
【0039】
【化8】
【0040】
〈2〉ジンゲロール配糖体の製造方法
本発明のジンゲロール配糖体の製造方法は、ジンゲロールまたはジンゲロールを含む植物抽出物に糖転移酵素を糖供与体の存在下で作用させる工程を含むことを特徴とする、ジンゲロール配糖体の製造方法である。
【0041】
(糖転移反応に用いるジンゲロール)
本発明の製造方法の原料、すなわち、糖転移反応に用いるジンゲロール原料としては、通常、6−ジンゲロール、4−ジンゲロール、8−ジンゲロール、10−ジンゲロール、3S,5S−6−ジンゲジオール、3R,5S−6−ジンゲジオール等のジンゲロール類だけでなく、ショウガ抽出物等の6−ジンゲロール、4−ジンゲロール、8−ジンゲロール、10−ジンゲロール、3S,5S−6−ジンゲジオール、3R,5S−6−ジンゲジオール等のジンゲロール類を含む植物抽出物等の混合物、または、その他のジンゲロール類を含む混合物を適宜用いることができる。
本発明におけるショウガ抽出物等の植物抽出物とは、その成分としてジンゲロールを含有している限りその原料および製法(抽出法)に特段制限は無く、公知の製法により得ることができる。例えば、ショウガ(Zingiber officinale)などZingiberaceae属植物等の植物全体、あるいは葉部、茎部、花部、果肉部、根部等を破砕、搾汁、粉末化、ペースト化等の加工を行うこと、更には必要に応じて熱水抽出、エタノール抽出、超臨界抽出等の抽出処理を行うことで得られる。市販のショウガ抽出物としてショウガオイル等を利用することも可能である。
【0042】
(糖供与体)
生化学的手法により本発明のジンゲロール配糖体を製造する場合、糖転移酵素の作用によりジンゲロール配糖体が生成するものであれば、糖供与体に特に制限は無く、使用する糖転移酵素の特性に応じて糖供与体を適宜選択することができる。糖転移酵素としてα−グルコシダーゼまたは/およびシクロデキストリングルカノトランスフェラーゼを用いる場合であれば、グルコース、マルトース、マルトオリゴ糖残基を含有するもの、例えば、マルトース、マルトトリオース、マルトテトラオース、マルトペンタオース、マルトヘキサオース、マルトヘプタオース、マルトオクタオースなどのマルトオリゴ糖、デキストリン、シクロデキストリン、アミロース等のα−グルカンと称される澱粉加水分解物、または澱粉およびそれらの混合物等を用いることができる。
糖転移酵素を用いてジンゲロール配糖体を製造する際は、澱粉や澱粉分解物にα−アミラーゼ、β−アミラーゼ、イソアミラーゼ等の加水分解酵素を作用させて糖供与体を生成
させた後、糖転移酵素を作用させても、上記澱粉や澱粉分解物を糖供与前駆体として酵素反応溶液に添加し、加水分解酵素と糖転移酵素を共に作用させてもよい。
【0043】
(糖転移酵素)
本発明の糖転移酵素は、ジンゲロールと糖供与体とを含む溶液から、ジンゲロール配糖体を生成可能な酵素であれば種類を問わないが、α−グルコシダーゼ(酵素番号:EC.3.2.1.20)、シクロデキストリングルカノトランスフェラーゼ(酵素番号:EC.2.4.1.19)、スクロースホスホリラーゼ(酵素番号:EC.2.4.1.7)などが挙げられる。
本発明でいうα−グルコシダーゼとはα−グルカンの非還元末端のα−グルコシド結合の加水分解反応を触媒し、α−グルコースを生じさせる酵素全般を意味する。また、α−グルコシダーゼには、加水分解反応とともに糖転移反応を触媒するものが存在し、本発明においては、加水分解反応とともに糖転移反応を触媒するα−グルコシダーゼが用いられる。
上記糖転移酵素のうち、好ましくはα−グルコシダーゼが挙げられ、より好ましくはHalomonas属微生物由来のα−グルコシダーゼが挙げられる。
上記糖転移酵素、特にα−グルコシダーゼの由来には、ジンゲロール配糖体を生成可能な酵素であれば特に制限は無く、動物、植物由来およびMucor属、Penicillium属、Aspergillus属、Saccharomyces属およびHalomonas属等の微生物由来酵素を適宜選択すればよい。これらの内、好ましくはジンゲロールの複数あるOH基のうち、側鎖のOH基を選択的に配糖化する酵素、例えばHalomonas属微生物由来のα−グルコシダーゼが挙げられ、例えば、Halomonas sp. A8株(平成23年5月2日付、受託番号NITE P−1096、独立行政法人製品評価技術基盤機構 特許微生物寄託センターに寄託)、Halomonas sp. A10株(平成23年5月2日付、受託番号NITE P−1097、独立行政法人製品評価技術基盤機構 特許微生物寄託センターに寄託)、Halomonas sp. H11株(平成23年5月2日付、受託番号NITE P−1098、独立行政法人製品評価技術基盤機構 特許微生物寄託センターに寄託)由来のα−グルコシダーゼが挙げられる。
【0044】
また、Halomonas属由来α−グルコシダーゼを用いれば、ジンゲロールの複数あるOH基のうち、側鎖のOH基を選択的に配糖化することが可能である。
【0045】
これらの糖転移酵素は、ジンゲロール配糖体を作製可能である範囲であれば、必ずしも精製して使用する必要はなく、通常ならば粗酵素であってよい。また、組換え・非組換えを問わず、必要に応じて宿主を選択すればよく、公知の方法を駆使して精製して
使用してもよい。また、糖転移酵素源として、糖転移酵素を産生する微生物を用いてもよい。
【0046】
本発明の製造方法において、本発明の効果を妨げない限り、使用する糖転移酵素に応じて、酵素の活性化剤等を用いてもよい。Halomonas属由来のα−グルコシダーゼを使用する場合には、必要に応じて、酵素の活性化剤として塩化アンモニウム、硫酸アンモニウム、塩化カリウム、塩化ルビジウム等のアルカリ金属塩またはアンモニウム塩等を添加することにより、酵素添加量または酵素反応時間を削減することができる。
【0047】
活性化剤の濃度は、酵素が活性化される濃度であれば特に制限されず、通常カチオン濃度として1μmol/L〜1mol/Lが適当である。経済性およびイオン負荷の点から、1μmol/L〜100mmol/L程度とすることが望ましい。
【0048】
使用酵素と反応時間には、密接な関係があることから、都合に応じて酵素使用量および
反応時間を選択すれば良いが、通常は、経済性の点から、約1〜100時間で反応が終了できるような酵素添加量を選択する。このような濃度としては、通常、糖供与体1gあたり0.1〜100U、好ましくは、糖供与体1gあたり0.2〜50Uであればよい。
本発明の製造方法における、酵素反応溶液におけるジンゲロールの濃度は、通常、1w/v%以上、好ましくは、2〜30w/v%であればよく、糖供与体濃度(w/v)は、ジンゲロールに対して、0.5〜30倍の範囲が好ましい。
【0049】
(反応条件)
本発明の製造方法における反応様式は、酵素反応が進む条件であれば、反応様式は特に限定されず、固定化された糖転移酵素をバッチ式で繰り返し、または連続式で反応に使用することも可能である。糖転移酵素を産生する微生物の増殖中の培地にジンゲロールと糖供与体を共存させ、目的物質を生成させることも有利に実施できる。
【0050】
本発明におけるジンゲロール配糖体調製のための反応方法は、通常、前述のジンゲロールと糖供与体とを含有する溶液に糖転移酵素を加え、該酵素が十分に作用する条件、通常、pH5〜10、温度15〜53℃の範囲から選ばれる条件に維持して行う。
【0051】
また、通常、ジンゲロールは水相と分離するため、アジテーターまたはスターラー等で攪拌しながら、またはローテーター等で反応容器自体を回転または振動させながら反応を行うことが好適である。必要であれば、乳化剤を混合させておいてもよい。
【0052】
このように生じた生成物および然る糖供与体を共存させた状態で、さらにα−グルコシダーゼまたはシクロデキストリングルカノトランスフェラーゼ、またはその他の糖転移酵素を作用させることにより、グルコース重合度を2以上とすること、またはグルコース以外の糖を、グルコシル基に転移することも当然可能であり、また、そのようにして生じた物質はモノグルコシドよりも水溶性が高いと予想される。そのようにして生じたジンゲロール配糖体及びその派生物質も本発明の範囲に包含される。
【0053】
酵素反応によりジンゲロール配糖体が生成した反応溶液には、通常、目的物であるジンゲロール配糖体の他、未反応物であるジンゲロール、糖供与体、場合によっては酵素、およびグルコース等の反応副生成物が混合している。
【0054】
このような混合物には、必要に応じて、α−アミラーゼ(EC.3.2.1.1)、β−アミラーゼ(EC.3.2.1.2)、グルコアミラーゼ(EC3.1.1.3)などによって加水分解処理を行い、残存した糖供与体を低分子化させることができる。
【0055】
上記混合物および混合物加水分解処理物をそのまま、ジンゲロール配糖体を含む、飲食品用、化粧品用、医薬部外品用または医薬品用組成物として、飲食品、化粧品、医薬部外品または医薬品に使用することができる。
【0056】
また、反応溶液を加熱する、またはpHを下げる等して酵素を失活させ、ろ過・濃縮して溶液状の、更には、乾燥、粉末化してジンゲロール配糖体含有組成物(製品)にし、飲食品、化粧品、医薬部外品または医薬品に使用することができる。
【0057】
ジンゲロールとジンゲロール配糖体の水溶性の違いを利用して、然る抽出操作を行うことにより、それぞれを、油相および水相に分離させ、双方を回収することも有利に実施できる。通常は、ジンゲロール配糖体はより親水性の高い溶媒へ抽出されることから、これを更にイオン交換カラムクロマトグラフィー、ゲルろ過クロマトグラフィー、疎水クロマトグラフィーなどの方法で分離精製すれば、糖供与体、酵素、およびグルコース等の反応副生成物とも分離が可能であり、ジンゲロール配糖体の高純度品を容易に得ることができ
る。
【0058】
この際、分離されるジンゲロールおよび糖供与体を、それぞれ回収し、糖転移反応の原料として再利用することも可能である。
【0059】
また、糖転移反応終了後、抽出およびクロマトグラフィーなどの分離手段にかけるまでに、必要に応じてろ過、活性炭処理による脱色、酸性および塩基性イオン交換樹脂による脱ミネラル処理等をすることも有利に実施できる。
【0060】
このようにして得られるジンゲロール配糖体は、以下の特徴を有する。
(1)下記に示す化合物
【0061】
【化9】
【0062】
(2)下記に示す化合物
【0063】
【化10】
【0064】
〈3〉ジンゲロール配糖体を含む組成物
上記のとおり、本発明のジンゲロール配糖体は、本発明の効果を妨げない限り、ジンゲロール配糖体以外に、植物抽出物成分、未反応物、酵素、反応副生成物等を含む組成物の形態でも使用することができる。
本発明のジンゲロール配糖体を含む組成物は、本発明のジンゲロール配糖体、及び通常飲食品、化粧品、医薬部外品または医薬品用成分として使用される成分を含むことができ、飲食品、化粧品、医薬部外品または医薬品等として使用することができる。
ジンゲロール配糖体の含有量は、各製品におけるジンゲロールの含有量を参考にして規定することができ、限定されないが、飲食品であれば0.01〜10質量%程度、化粧品であれば0.01〜5質量%程度、医薬部外品または医薬品であれば0.01〜10質量%程度である。
本発明の組成物は、本発明のジンゲロール配糖体を含有する以外は、上記の様な通常の成分を用いて、通常の方法により、製造できる。
本発明のジンゲロール配糖体を添加することで、ジンゲロールの作用を利用した、アディポネクチン産生増強効果や脂肪蓄積抑制効果といった有用な生理作用を有した飲食品、化粧品、医薬部外品または医薬品を製造することができる。さらに、後述の実施例5および図13に示した通り、本発明のジンゲロール配糖体は室温にて赤オレンジ色の粉末状であるため、着色料として飲食品、化粧品、医薬部外品または医薬品に添加することもできる。
【0065】
上記飲食品としては、醤油、粉末醤油、味噌、粉末味噌、もろみ、ひしお、フリカケ、マヨネーズ、ドレッシング、食酢、三杯酢、粉末すし酢、中華の素、天つゆ、麺つゆ、ソース、ケチャップ、焼き肉のタレ、カレールウ、シチューの素、スープの素、ダシの素、複合調味料、みりん、新みりん、テーブルシュガー、コーヒーシュガーなどの各種調味料、せんべい、あられ、おこし、求肥、餅類、まんじゅう、ういろう、餡類、羊羹、水羊羹、錦玉、ゼリー、カステラ、飴玉などの各種和菓子、パン、ビスケット、クラッカー、クッキー、パイ、プリン、バタークリーム、カスタードクリーム、シュークリーム、ワッフル、スポンジケーキ、ドーナツ、チョコレート、チューインガム、キャラメル、ヌガー、キャンディーなどの各種洋菓子、アイスクリーム、シャーベットなどの氷菓、果実のシロップ漬、氷蜜などのシロップ類、フラワーペースト、ピーナッツペースト、フルーツペーストなどのペースト類、ジャム、マーマレード、シロップ漬、糖果などの果実、野菜の加工食品類、福神漬け、べったら漬、千枚漬などの漬物類、たくわん漬の素、白菜漬の素などの漬物の素、ハム、ソーセージなどの畜肉製品類、魚肉ハム、魚肉ソーセージ、カマボコ、チクワ、天ぷらなどの魚肉製品類、ウニ、イカの塩辛、酢コンブ、さきするめ、タラ、タイ、エビなどの田麩などの各種珍味類、海苔、山菜、するめ、小魚、貝などで製造される佃煮類、煮豆、煮魚、ポテトサラダ、コンブ巻などの惣菜食品、乳製品、魚肉、畜肉、果実、野菜の瓶詰、缶詰類、プリンミックス、ホットケーキミックス、即席ジュース、即席コーヒー、即席汁粉、即席スープなどの即席食品、冷凍食品、果汁含有飲料、果汁ジュース、野菜ジュースなどの果実・野菜飲料、サイダー、ジンジャーエールなどの炭酸飲料、アイソトニック飲料、アミノ酸飲料などのスポーツ飲料、コーヒー飲料、緑茶などの茶系飲料、乳酸飲料、ココアなどの乳系飲料、チューハイ、清酒、果実酒などのアルコール飲料、栄養ドリンク等が挙げられるが、これらに限定されるものではない。
【0066】
上記化粧品とは、薬事法により定められた化粧品を指し、例えば、日焼け防止剤、ローション、エッセンス、乳液、クリーム、ハップ剤、ペースト剤、ゲル剤、パウダー、ファンデーション、化粧水、パック剤などの基礎化粧品およびメイクアップ化粧品、石鹸、洗顔料、ボディソープなどの皮膚洗浄剤、シャンプー、リンス、ヘアートニック、ヘアートリートメント剤、養毛剤、育毛剤、ヘアスプレーなどのヘアケア製品、浴用剤等が挙げられるが、これらに限定されるものではない。
【0067】
上記医薬部外品および医薬品とは、薬事法に定められた医薬部外品および医薬品を指し、例えば経口投与用製剤としてエキス剤、エリキシル剤、シロップ剤、チンキ剤、リモナーデ剤等の液剤とカプセル剤、顆粒剤、細粒剤、丸剤、散剤、錠剤等の固形剤等が挙げられるが、これらに限定されるものではない。
【0068】
〈4〉試験及び評価方法
(酵素力価の測定)
本発明において、α−グルコシダーゼの酵素力価の測定は、例えば、0.2w/v%マルトースを基質として、pH7、30℃、100μLの系で、特定量の酵素量から生じたグルコース量を測定することにより行うことができる。具体的には、例えば、上記系にて10分間の酵素反応後、2mol/LのTris−HCl(pH7)を200μl添加することにより酵素反応を終了させ、そこに、グルコーステストワコーC II(和光純薬製)を100μL添加し37℃で30分間インキュベートする。波長490nmの吸光度を測定することにより、生じたグルコース量を算出する。上記条件にて1分間に1μmolのマルトースを加水分解する酵素量を1Uと定義する。
【0069】
シクロデキストリングルカノトランスフェラーゼの酵素力価の測定は、例えば、0.9w/v%の可溶性でんぷんを基質として、pH6、40℃、1mLの系で、特定の酵素量から生じたβ−シクロデキストリンを測定することにより以下の方法により行うことができる。具体的には、例えば、上記系にて10分間の酵素反応後、40mMの水酸化ナトリウム水溶液を2.5mL添加し酵素反応を終了させ、そこに250μMの炭酸ナトリウムおよび0.1mg/mLのフェノールフタレインを含む溶液を0.3mL添加し、550nmの吸光度を測定することにより、生じたβ−シクロデキストリン量を算出する。上記条件にて、1分間に1mgのβ−シクロデキストリンを生成する酵素量を1Uとする。
【実施例】
【0070】
以下に実施例を挙げて本発明の詳細を説明するが、本発明は以下の実施例に限定されるものではない。
<実施例1>各種酵素による配糖体の調製検討
各種糖転移酵素により、ジンゲロールへの配糖化能を検討した。
【0071】
まず、α−グルコシダーゼとして、Aspergillus niger由来、Acremonium sp.由来およびHalomonas sp.H11株(受託番号:NITE P−1098)由来のものを検討した。以後、Aspergillus niger由来の酵素を「ANG」、Acremonium sp.由来の酵素を「ACG」およびHalomonas sp.H11株由来の酵素を「HAG」と表記する。なお、ANGおよびACGはそれぞれ「製品名:トランスグルコシダーゼアマノ、アマノエンザイム製」および「製品名:テイスターゼ、キリン協和フーズ製」である。HAGは、下記の参考例1の方法にて調製し、SDS−ポリアクリルアミド電気泳動的に単一バンドとして得られた精製酵素を使用した。
【0072】
10w/v%マルトース(日本食品化工製)、2.5w/v% 6−ジンゲロール(ナカライテスク製)、50mM緩衝液、各種α−グルコシダーゼをマルトース1gあたり1.5Uとなるよう反応溶液を調製し、10μLの系にて40℃にて反応させた。HAGおよびACGの反応にはHEPES−NaOH緩衝液(pH7)、ANGには酢酸ナトリウム緩衝液(pH5)を使用した。また、HAGにおける反応系には、硫酸アンモニウムを5mMとなるよう添加した。酵素の代わりに水を添加したpH7における溶液をコントロールとした。
【0073】
シクロデキストリングルカノトランスフェラーゼによる反応においては、Bacillus stearothermophillus由来、Bacillus sp. No.38−2由来およびBacillus coagulans由来の酵素を使用した。以後、これらの酵素をそれぞれCGTase−1、CGTase−2およびCGTase−3と表記することがある。なお、CGTase−1はノボザイムズ製、CGTase−2およびCGTase−3は日本食品化工製である。
酵素反応条件は以下の通りである。
すなわち、10w/v%パインデックス#100(松谷化学工業製)、2.5 w/v%6−ジンゲロール、50mM 酢酸ナトリウム緩衝液(pH6)、12mM Ca2+ 、パインデックス1gあたりシクロデキストリングルカノトランスフェラーゼ5Uとなるよう反応溶液を調製し、10μLの系にて反応させた。酵素の代わりに水を添加した溶液をコントロールとした。40℃にて24時間反応させ、反応溶液をメタノールにより10倍希釈し、そのうち1μLを薄層クロマトグラフィー(TLC)による分析に供した。
【0074】
TLCには、TLCアルミプレート(シリカゲル60 F254、 Merck製)を使用し、展開溶媒として、クロロホルム:メタノール:水=6:4:1(v/v)または2−プロパノール: 1−ブタノ―ル: 水=2:2:1(v/v)を使用し、10 v/v%硫酸/メタノール溶液を噴霧しオーブンで加熱することにより呈色させた。以後、上記TLC条件をそれぞれ、TLC条件1およびTLC条件2と記載する。標品としてグルコース(関東化学製)、マルトース(日本食品化工製)の1w/v %水溶液を1μL使用した。
【0075】
その結果、HAGによる反応溶液にのみ図1中矢印で示した特異的な反応産物が観察された。本分析で使用したいずれの展開溶媒においても主要な反応生成物スポットが1つ確認された。また、若干ではあるものの、その下に6−ジンゲロール由来と推察されるスポットが2つ確認された。そこで、生成物をTLCにおいて上から、反応生成物X、Y、およびZとし、これらを単離精製した。
【0076】
<参考例1>Halomonas sp. H11由来α−グルコシダーゼの組換え発現および精製
可溶性澱粉(関東化学製)10g/L、ポリペプトン(和光純薬製)5g/L、酵母抽出物(商品名『イーストエキストラクト』、ベクトン・ディッキンソン製)5g/L、塩化ナトリウム(和光純薬製)35g/L、リン酸二水素カリウム(関東化学製)1g/L、硫酸マグネシウム七水和物(関東化学製)0.2g/L、および水からなるpH7の液体培地10mLを200mLのバッフル付きフラスコに調製し、オートクレーブで121℃、20分間滅菌し冷却した。
【0077】
上記培地10mLに、Halomonas sp.H11株のグリセロールストックを接種し、37℃、150rpmで16時間培養した。培養液を1.5mL容エッペンドルフチューブに1mLずつ分注し遠心分離(4℃、14,000rpm、2分間)し上清を取り除いた。5mg/mLのリゾチーム(生化学工業製)、TE溶液(pH8)をチューブ1本当たり350μL添加し、37℃にて1時間保持した。10%ドデシル硫酸ナトリウム溶液を50μL添加し転倒混和し37℃にて30分間保持した。TE飽和フェノールを400μL添加し穏やかに転倒混和した後遠心分離(室温、14,000rpm、5分間)した。上層を新たなエッペンドルフチューブに移した後、フェノールクロロホルム溶液を400μL添加し穏やかに転倒混和し、上記と同様に遠心分離した。それぞれの上層を一つのエッペンドルフチューブに移し、3mol/L酢酸ナトリウム水溶液および95(v/v%)冷エタノールを、それぞれ溶液の1/10容量および2倍容量添加し遠心分離(4℃、14,000rpm、15分間)した。得られた沈殿物を70%エタノールにてリンス洗浄し風乾した。0.2mg/mlのRNase/TE溶液を400μL添加し、35℃にて30分間保持した。フェノールクロロホルム抽出およびエタノール沈殿法を上記と同様に行い、乾燥させた沈殿物をTE溶液100μLに溶解し、ゲノム溶液とした。
【0078】
クローニング用オリゴヌクレオチド、HAG−F−NdeI(5´−AAACATAT
GCAAGACAACATGATGTGGTG−3´)(配列番号1)およびHAG−R−XhoI(5´−AAACTCGAGTTAGGCAACCTGCATAAAGG−3´)(配列番号2)を用いてHalomonas sp. H11株のα−グルコシダーゼをクローニングした。すなわち、ゲノム100ng、10×KOD ver.2 buffer、 5μL、2mmol/L dNTPs 5μL、25mmol/L MgSO4 3μL、20mmol/Lのプライマー溶液各1μL、KOD plus polymerase 1μLを混合し、水で50μLにメスアップした。なお、上記PCR用の試薬は全て東洋紡製である。PCR条件は以下の通りである。94℃に2分間保持した後、94℃に30秒間、55℃に30秒間、72℃に2分間からなるサイクルを30回繰り返し、最後に72℃に3分間保持した。このうち4μLを電気泳動に供し、約1.5kbの増幅断片を確認した。当該断片を市販のキット(GEヘルスケア製、商品名『illustra GFXTM PCR DNA and Gel Band Purification Kit』、以後、「GFX」と表記する)にて精製した。これを、NdeIおよびXhoI(いずれもタカラバイオ製)で酵素処理し、GFXにて精製した。本DNA断片を、上記と同様にNdeIおよびXhoIで制限酵素処理したpET22b(ノバジェン製)に、通常のライゲーション反応にて連結させ発現用プラスミドを作製した。本プラスミドを『pET22b−HAG−H11』と命名した。
【0079】
上記pET22b−HAG−H11で、大腸菌BL21(DE3) Codon Plus RIL(ストラタジーン製)を形質転換した。
すなわち、50μg/mlアンピシリンを含むLB培地3mLに、上記形質転換体を接種し、37℃で一晩前培養した。このうち1mLを600mLの同培地に接種し、37℃で3時間培養した。それを氷冷し、0.1mol/Lのイソプロピル−β−チオガラクトピラノシド(和光純薬製)を600μL添加した。これを16℃で24時間回転振盪培養した。得られた培養物を、定法に従い、遠心分離して菌体を回収した。菌体を20mmol/L HEPES−NaOH緩衝液(pH7)約40mLに懸濁して、超音波破砕機(microsom(商標) ultrasonic cell disruptor)にて細胞を破砕した(出力:4ワット、全出力時間: 20分間)。これを遠心分離した上清を粗酵素液とした。
【0080】
粗酵素液には89.1 mgのタンパク質、83.3 Uのα−グルコシダーゼ活性が含まれていた。比活性は0.93U/mgであった。なお、タンパク質の定量は、DC protein assay kit(Bio Rad社製)を使用して、添付のプロトコルに従って行った。タンパク質の標準曲線は0〜1mg/mlのbovine serum albumin(Bio Rad社製)を用いて作成した。以下の実施例においても、特に示さない限り、同様である。
【0081】
粗酵素液を20mmol/L HEPES−NaOH緩衝液(pH7)で平衡化されたイオン交換カラムに、以下の条件で供し、塩化ナトリウム濃度0.1mol/L付近に溶出された画分(部分精製酵素1)を回収した。
【0082】
(条件)
カラム : TOYOPEARL(登録商標) DEAE−650M(東ソー株式会社製)
カラムサイズ: φ26mm×200mm
ゲル量 : 106mL
溶出液 : 20mmol/L HEPES−NaOH緩衝液(pH 7)
塩化ナトリウム濃度0mol/L〜0.5mol/Lのリニアグラジエント
流速 : 2mL/分
溶出液量 : 120mL
【0083】
部分精製酵素1には、54mgのタンパク質、374Uの酵素活性が含まれていた。比活性は6.93U/mgであった。なお、本α―グルコシダーゼは、アルカリ金属塩に活性化される特徴を有することから、粗酵素液と比較して総活性が上昇した原因は、溶出液中に含まれるNaClの影響であると考えられた。
【0084】
前記部分精製酵素1に、硫酸アンモニウムを少量ずつ加え、終濃度1.5mol/Lとした。これを1.5mol/L硫酸アンモニウムを含む20mmol/L HEPES緩衝液(pH7)で平衡化した疎水カラムに、以下の条件で供した。そして、硫酸アンモニウム濃度1.0mol/L付近で溶出された画分、約50mLを回収した。これを、5mmol/LのHEPES−NaOH緩衝液(pH7)5Lで2回透析し、精製酵素とした。
【0085】
(条件)
カラム : TOYOPEARL(登録商標) BUTHYL−650M(東ソー株式会社製)
カラムサイズ : φ16mm×150mm
ゲル量 : 30mL
溶出液 : 20mmol/L HEPES緩衝液(pH7)
硫酸アンモニウム濃度1.5mol/L〜0mol/Lのリニアグラジエント
流速 : 1mL/分
溶出液量 : 120mL
【0086】
精製酵素には、17.8mgのタンパク質と、67.5Uの酵素活性が含まれており、比活性は3.8U/mgであった。粗酵素液からの回収率は、81.4%であり、精製度は4.1倍であった。この画分を1μg、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動に供した結果、1本のバンドのみが検出されたことから、酵素は単一に精製されていることが分かった。得られた精製酵素を実施例に用いた。
【0087】
上記記載において、比活性はタンパク質1mgあたりの酵素活性(U/mg)を示し、回収率は、粗酵素液の総活性に対する酵素活性の残存率(%)を示し、精製度は、粗酵素液の比活性に対する倍率(倍)を示す。
【0088】
<実施例2>生成物の精製
ジンゲロール配糖体の単離精製法は以下の通りである。まず、30w/v%マルトース、5w/v%6−ジンゲロール、5mM硫酸アンモニウム、30mM HEPES−NaOH緩衝液(pH7)およびマルトース1gあたり3UのHAGとなるよう、計3mLの溶液を調製しよく攪拌した後、40℃で48時間インキュベートした。反応溶液をPLCガラスプレート(シリカゲル60F254、2mm、Merck製)に500μL/枚となるようにスポッティングし、クロロホルム:メタノール:水=6:4:1(v/v)またはクロロホルム:メタノール:酢酸=80:20:5で展開した。展開プレートの方端を前述の方法と同様に呈色させ、各反応生成物のスポットと同Rf値の展開をスパーテルで掻き取り、50v/v%メタノール水溶液で抽出した。抽出液をろ紙(No.5、アドバンテック製)でろ過し、減圧濃縮した。これを水に溶解し、0.45μmフィルター(ミリポア製)にてろ過し凍結乾燥させた。これを以後の実験に用いた。
【0089】
<実施例3>反応生成物Xの構造解析
(トリフルオロ酢酸による分解)
精製した生成物Xおよび6−ジンゲロール標品をそれぞれ0.5 mgずつ量り取り、それに純水50μLを添加した。これに4Mトリフルオロ酢酸(関東化学製、以下TFAと表記することがある)を400μL添加し、100℃に1時間保持した。減圧下にて濃縮・乾固させたものを50v/v%メタノール水溶液40μLに溶解し、実施例1で上述したTLC条件2で分析した。なお、TFA添加前のサンプルも同様に分析した。その結果、生成物Xは、図2に示すように、TFA処理により6−ジンゲロール(もしくはその分解物)とグルコースの位置にスポットを生じた。このことから、反応生成物Xは6−ジンゲロールとグルコースから構成される化合物である可能性が示唆された。
【0090】
(MS(質量分析、Mass Spectrometry)解析)
反応生成物Xを0.1mg/mLとなるようメタノールに溶解し、質量分析に供した。分析には、LCQ(Thermo electron製)を用い、エレクトロスプレーイオン化法(ESI)により実施した。測定はポジティブモードで行った。その結果、図3に示したようなMSスペクトルが得られ、そのピークおよび対応する分子量(M)は、表1の通りであった。
【0091】
【表1】
【0092】
すなわち、反応生成物Xにおいては、6−ジンゲロール(分子量294)にグルコースが一つ付加した配糖体(分子量456)およびESIによってその配糖体のグリコシド結合が切断され、さらに、それにより生じた6−ジンゲロールが脱水し生成したと考えられるショウガオール(分子量276)に相当するピークが検出された。
【0093】
(NMR解析)
BRUKER社製 AV400MdigitalNMR(400MHz)を使用しH−H COSY、HMQCおよびHMBCの二次元分析を行った。詳細には、精製し凍結乾燥した反応生成物X10mgを0.5mLのD2O(ナカライテスク、99.90%D)に溶解し、標準物質として0.1mg/mLの3−(トリメチルシリル)プロピオン酸ナトリウム−2,2,3,3−d4(TSP、和光純薬)を2μL滴下したものを分析に供した。分析時の温度を303ケルビンに設定した。
NMR解析の結果から、各シグナルを以下のように帰属した。なお、1H NMRおよび13C NMRの解析結果は図4〜9に示した。
【0094】
1H NMR (400MHz,D2O)
δ6.93(s,1H,16),6.88(d,1H,13), 6.75(d,1H,12),5.00(d,1H,1´),4.05(m,1H,6),3.90(dd,1H,6´a),3.85(s,3H,17),3.70(dd,1H,6´b), 3.60(t,1H,3´),3.50(t,1H,2´),3.49(t,1H,5´),3.42(t,1H,4´),2.95(d,2H,9),2.88(dd,1H,7a),2.85(d,2H,10),2.52(dd,1H,7b),1.53(m,1H,5a), 1.38(m,1H,5b),1.25(m,2H,2),1.22(t,2H,3), 1.22(t,2H,4),0.85(t,3H,1)
【0095】
13C NMR (400 MHz, D2O)
δ147.36(15),143.10(14),133.56(11),121.08(12),115.56(13),112.88(16),96.57(1´),75.97(5´),74.16(6),73.84(3´),72.95(8),71.28(2´),69.29(4´),60.19(6´),56.01(17),47.43(7),44.66(9),32.41(5),30.97(4),28.92(10),23.55(3),21.83(2),13.26(1)
【0096】
HMBCにおいてH1´−C6およびH6−C1´の相関が確認されたため、配糖化部位をC6のOH基と決定した。
以上、TFA処理、MS解析およびNMR解析の結果より、HAGによる6−ジンゲロールの配糖化反応により生じた主生成物Xは、下記式(IV)で示される、6−ジンゲロールの側鎖のOH基にグルコースがα−1位で結合したジンゲロール配糖体であることが明らかとなった。以降、反応生成物Xを簡易的に、ジンゲロール6−グルコシドと呼ぶことがある。
【0097】
【化11】
【0098】
<実施例4>反応生成物YおよびZの構造解析
(反応生成物YおよびZのMS解析)
各精製物を実験3と同様の方法にてMS解析した。その結果、図10および11に示したようなMSスペクトルが得られ、その主要ピークおよび対応する分子量(M)は表2の通りであった。
【0099】
【表2】
【0100】
すなわち、反応生成物YのMS分析において検出された分子量は、6−ジンゲロール(分子量294)が脱メトキシされた物質(分子量246)および当該物質のモノグルコシド(410)に相当していた。本生成物は、生成量が微量でありNMR解析に不十分であったことから、MS分析結果より以下の構造であると推定した。
反応生成物Y:脱メトキシされた6−ジンゲロールのいずれかのOH基にグルコースが脱水結合した化合物。
反応生成物Zの、641m/zは、6−ジンゲロールにグルコース (分子量180)が2分子縮合し、ナトリウムイオンが付加した分子量 (294+180+180−18−18+23=641)と一致した。
【0101】
(反応生成物Zのグルコアミラーゼ処理)
反応生成物Zは、Yと同様に生成量がごく微量であったことから、NMR解析に不十分であった。そこで、反応生成物Zにグルコアミラーゼを作用させ、その反応生成物をTLCにて分析することにより、反応生成物Zの構造を推定した。すなわち、0.1g/mL反応生成物Z、4U/mLグルコアミラーゼ(生化学工業製)、4mM酢酸ナトリウム緩衝液(pH5)となるよう混合し、40℃にて24時間インキュベートした。コントロールとして、グルコアミラーゼ無添加のものを同様にインキュベートした。2μLをTLCに供し、実施例1で上述のTLC条件2により分析した。なお、6−ジンゲロール、反応生成物XおよびY、およびグルコースを混合し50%メタノール水溶液に溶解させた溶液を標準物質としてスポッティングした。その結果、図12に示すように、Zをグルコアミラーゼ処理すると反応生成物X(ジンゲロール6−グルコシド)を生成することが分かった。これは、反応生成物Zは、反応生成物Xのグルコース残基にさらにもう一分子のグルコースがα−1,4グルコシド結合した構造を取っていることを意味していた。さらに、本実施例の酵素反応において使用したHalomonas属由来α−グルコシダーゼは、α1,4型の転移酵素であることから、その結合様式はα−1,4であると考えられた。以上、MSおよび本結果より、反応生成物Zは、以下の構造であると推定した。
反応生成物Z:6−ジンゲロールの側鎖OH基にマルトースがα−1位で結合した化合物であることが明らかとなった。
【0102】
<実施例5>ジンゲロール6−グルコシドの外観、溶解性および吸収極大
精製したジンゲロール6−グルコシドの凍結乾燥品は、図13に示すように、室温にて赤オレンジ色の粉末状であり、水に可溶であった。また、0.2w/v%の水溶液を調製し、幅10mmの石英セルにて、波長600〜200nmにおける吸収スペクトルを測定したところ、ジンゲロール6−グルコシドは、図14に示すように、波長278nmに吸収極大を有していることがわかった。
【0103】
<実施例6>反応生成物の消化性
ジンゲロール6−グルコシドの、ラット小腸アセトンパウダー、ヒト人工胃液およびヒト人工腸液による、消化性試験を行った。
(ラット小腸アセトンパウダーによる消化性)
ラット小腸アセトンパウダー(Sigma製)50mgを純水1mLに溶解し、ボルテックスにてよく混合した後氷冷した。これを遠心分離(15,000rpm、10分、4℃)し、上清を酵素液として用いた。ジンゲロール6−グルコシド1mgに0.5Mリン酸ナトリウム緩衝液(pH7)10μL、および酵素液90μLを添加しよく混合し37℃でインキュベートした。これを経時的にサンプリングし、1μLをTLCプレートにスポッティングしドライヤーで即座に乾燥させることにより反応を停止した。実施例1で上述したTLC条件2にて分析し、スポットの消失を観察した。
【0104】
(ヒト人工胃液による消化性)
Pepsin 1:100 (和光純薬製)3.2mg、NaCl2.0mgおよびH
Cl 7μLを混合し、純水にて0.8mLにメスアップしたものを人工胃液とした。ジンゲロール6−グルコシドを0.5mg量り取り、純水10μLに溶解した。これに人工胃液を40μL添加し、37℃にてインキュベートした。これを経時的にサンプリングした。上記と同様の方法にて反応を停止し、TLC分析した。
【0105】
(ヒト人工腸液による消化性)
Pancreatin(和光純薬製)10 mg、0.2M KH2PO4 250μLおよび0.2M NaOH 118μLを混合し、純水にて0.8mLにメスアップしたものを人工腸液とした。ジンゲロール6−グルコシドを0.5mg量り取り、純水10μLに溶解した。これに人工腸液を40μL添加し、37℃にてインキュベートした。以後の操作は上記と同様である。
【0106】
各種消化酵素による、ジンゲロール6−グルコシドの消化性試験の結果、図15に示すように、ラット小腸アセトン抽出物、ヒト人工胃液およびヒト人工腸液にいずれにおいても、120分間の処理においてジンゲロール6−グルコシドは減少しなかった。すなわち、ジンゲロール6−グルコシドは、これらの消化酵素には分解されず、このような消化酵素に対して安定であることがわかった。
【0107】
<実施例7>ジンゲロール6−グルコシドの安定性
ジンゲロール6−グルコシドを1mg量り取り、50%エタノール、0.1M HClの水溶液1mLに溶解した。これを、密栓可能な容器で、60℃にて18時間インキュベートしたときの残存率を測定することにより、ジンゲロール6−グルコシドの安定性を評価した。すなわち、サンプルを20μLずつ採取し、これをアセトニトリル:水=80:20(v/v)の溶液180μLと混合した。これをHPLC分析に供した。インキュベート0時間におけるジンゲロール6−グルコシドのUV吸収ピーク面積を100(%)としたときの、18時間後における相対ピーク面積を残存率(%)とした。なお、比較対象として、6−ジンゲロールの安定性を同様に評価した。
【0108】
測定条件は下記の通りである。
カラム : YMC Pack Pro C18(AS−303、φ4.6mm×250mm)
溶媒 : アセトニトリル:水=7:3(v/v)
流速 : 1mL/分
温度 : 30℃
検出 : UV280nm
注入量 : 20μL
【0109】
なお、本条件における各物質の溶出時間は以下の通りである。
ジンゲロール6−グルコシド : 3.0分
6−ジンゲロール : 4.7分
6−ショウガオール : 7.8分
本試験の結果、処理18時間後において、6−ジンゲロールは、約半量が6−ショウガオールへ変換しており、残存率は49%であった。一方で、ジンゲロール6−グルコシドの残存率は94%であった。このことから、6−ジンゲロールの構造が変化しやすい0.1MのHCl溶液においても、ジンゲロール6−グルコシドは安定であることがわかった。
【0110】
<実施例8>ジンジャーオイルを基質とした反応
10%v/vジンジャーオイル(稲葉香料製)、20%w/vマルトース、5mM硫酸アンモニウム、50mM HEPES−NaOH緩衝液(pH7)および0.3U/mL
のHAGを含む1mLの水溶液を調製し、40℃にて93時間酵素反応させた。なお、酵素無添加のものをコントロールとした。反応溶液を熱失活し、1μLを、実施例1で上記に示したTLC条件2にて分析した。なお、標品として、6−ジンゲロールおよび単離精製したジンゲロール6−グルコシドを分析に供した。
その結果、図16に示すように、反応後の主生成物としてジンゲロール6−グルコシドと同Rf値にスポットが現れた。このことから、6−ジンゲロール以外の成分を含むジンジャーオイルを基質とした場合においても、HAGによりジンゲロール6−グルコシドを生成可能であることがわかった。
【0111】
<実施例9>Halomonas sp.A8株およびA10株由来酵素による反応
参考例1に示したHalomonas sp.H11株の培養と同条件において、Halomonas sp.A8株およびA10株を100mLの培地にて好気的に培養し、遠心分離により集菌した。菌体を20mM HEPES−NaOH緩衝液(pH7)30mLに懸濁し、それぞれ超音波破砕機(microsom(商標) ultrasonic cell disruptor)にて細胞を破砕した(出力:4ワット、全出力時間:20分間)。これを遠心分離した上清を粗酵素液とした。Halomonas sp.A8株およびA10株の粗酵素液のα−グルコシダーゼ活性は、それぞれ0.032U/mLおよび0.023U/mLであった。
【0112】
10w/v%マルトース(日本食品化工製)、2.5w/v% 6−ジンゲロール(ナカライテスク製)、5mM硫酸アンモニウム、50mM HEPES−NaOH緩衝液(pH7)、上記粗酵素液を、α−グルコシダーゼ活性としてマルトース1gあたり0.15Uとなるよう反応溶液を調製し、10μLの系にて37℃にて17時間反応させた。反応溶液1μLを実施例1で上記に示したTLC条件2にて分析した。その結果、図17に示すように、ジンゲロール6−グルコシドが生成していた。このことから、Halomonas sp.A8およびA10株の未精製酵素においてもジンゲロール6−グルコシドを生成可能であることがわかった。
【特許請求の範囲】
【請求項1】
ジンゲロールのOH基にグルコース、マルトースまたはマルトオリゴ糖が脱水縮合していることを特徴とするジンゲロール配糖体。
【請求項2】
ジンゲロール配糖体が、下記一般式(I)で示されるジンゲロール配糖体である、請求項1に記載のジンゲロール配糖体。
【化1】
(式中、
Rはグルコシル基、マルトシル基またはマルトオリゴ糖のグルコシル基を示し、
nは2,4,6または8のいずれかの整数を示す。)
【請求項3】
ジンゲロール配糖体が、下記式(II)で示される、6−ジンゲロールの側鎖のOH基にグルコースがα−1位で結合したジンゲロール配糖体である、請求項1または2に記載のジンゲロール配糖体。
【化2】
【請求項4】
ジンゲロール配糖体が、下記式(III)で示される、6−ジンゲロールの側鎖のOH基にマルトースがα−1位で結合したジンゲロール配糖体である、請求項1または2に記載のジンゲロール配糖体。
【化3】
【請求項5】
請求項1〜4のいずれか1項に記載のジンゲロール配糖体を含有する組成物。
【請求項6】
飲食品である、請求項5に記載の組成物。
【請求項7】
化粧品である、請求項5に記載の組成物。
【請求項8】
医薬部外品または医薬品である、請求項5に記載の組成物。
【請求項9】
ジンゲロールまたはジンゲロールを含む植物抽出物に糖転移酵素を糖供与体の存在下で作用させる工程を含むことを特徴とする、請求項1〜4のいずれか1項に記載のジンゲロール配糖体の製造方法。
【請求項10】
糖転移酵素が、Halomonas属細菌由来である、請求項9に記載のジンゲロール配糖体の製造方法。
【請求項11】
植物が、ショウガである請求項9または10に記載のジンゲロール配糖体の製造方法。
【請求項1】
ジンゲロールのOH基にグルコース、マルトースまたはマルトオリゴ糖が脱水縮合していることを特徴とするジンゲロール配糖体。
【請求項2】
ジンゲロール配糖体が、下記一般式(I)で示されるジンゲロール配糖体である、請求項1に記載のジンゲロール配糖体。
【化1】
(式中、
Rはグルコシル基、マルトシル基またはマルトオリゴ糖のグルコシル基を示し、
nは2,4,6または8のいずれかの整数を示す。)
【請求項3】
ジンゲロール配糖体が、下記式(II)で示される、6−ジンゲロールの側鎖のOH基にグルコースがα−1位で結合したジンゲロール配糖体である、請求項1または2に記載のジンゲロール配糖体。
【化2】
【請求項4】
ジンゲロール配糖体が、下記式(III)で示される、6−ジンゲロールの側鎖のOH基にマルトースがα−1位で結合したジンゲロール配糖体である、請求項1または2に記載のジンゲロール配糖体。
【化3】
【請求項5】
請求項1〜4のいずれか1項に記載のジンゲロール配糖体を含有する組成物。
【請求項6】
飲食品である、請求項5に記載の組成物。
【請求項7】
化粧品である、請求項5に記載の組成物。
【請求項8】
医薬部外品または医薬品である、請求項5に記載の組成物。
【請求項9】
ジンゲロールまたはジンゲロールを含む植物抽出物に糖転移酵素を糖供与体の存在下で作用させる工程を含むことを特徴とする、請求項1〜4のいずれか1項に記載のジンゲロール配糖体の製造方法。
【請求項10】
糖転移酵素が、Halomonas属細菌由来である、請求項9に記載のジンゲロール配糖体の製造方法。
【請求項11】
植物が、ショウガである請求項9または10に記載のジンゲロール配糖体の製造方法。
【図3】

【図4】
【図5】
【図6】

【図7】
【図8】
【図9】
【図10】
【図11】
【図14】
【図1】
【図2】
【図12】
【図13】
【図15】
【図16】
【図17】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図14】
【図1】
【図2】
【図12】
【図13】
【図15】
【図16】
【図17】
【公開番号】特開2013−112623(P2013−112623A)
【公開日】平成25年6月10日(2013.6.10)
【国際特許分類】
【出願番号】特願2011−258152(P2011−258152)
【出願日】平成23年11月25日(2011.11.25)
【出願人】(000231453)日本食品化工株式会社 (68)
【Fターム(参考)】
【公開日】平成25年6月10日(2013.6.10)
【国際特許分類】
【出願日】平成23年11月25日(2011.11.25)
【出願人】(000231453)日本食品化工株式会社 (68)
【Fターム(参考)】
[ Back to top ]