
スクロース代謝能を有する遺伝子組み換え微生物
本発明はスクロース代謝能を有する遺伝子組み換え微生物に関し、より具体的には、スクロースホスホトランスフェラーゼ及び/またはスクロース−6−リン酸ヒドロラーゼをコードする遺伝子が導入されているスクロース代謝能を有する遺伝子組み換え微生物、またはβ−フルクトフラノシダーゼをコードする遺伝子が導入されたスクロース代謝能を有する遺伝子組み換え微生物に関する。本発明によると、高価なグルコースの代わりに低価格のスクロースを炭素源として使用できる遺伝子組み換え微生物を提供する効果がある。更に、スクロースを炭素源として利用できなかった微生物の培養においてもグルコースをはじめとする他の炭素源をスクロースに代えることができる。

【発明の詳細な説明】
【技術分野】
【0001】
本発明はスクロース代謝能を有する遺伝子組み換え微生物に関し、より具体的には、スクロースホスホトランスフェラーゼ及び/またはスクロース−6−リン酸ヒドロラーゼをコードする遺伝子が導入されているスクロース代謝能を有する遺伝子組み換え微生物またはβ−フルクトフラノシダーゼをコードする遺伝子が導入されたスクロース代謝能を有する遺伝子組み換え微生物に関する。
【背景技術】
【0002】
人類の持続可能な開発(sustainable development)のために再生可能な生物資源から有用な物質を生産するための産業生命工学への高い関心と共にこれを発展させる研究が活発に行われている。現在までの微生物発酵を介した化学物質生産は、グルコースを主原料物質に使用して成されているが、グルコースの高価格によって、原油を主原料物質として使う化学合成法を介する生産価格より発酵による生産価格が高く、微生物発酵を介する化学物質生産の商用化には困難がある。従って、高価なグルコースを炭素源として利用する方法から脱皮して、豊富な生物資源から容易に得られる多種の安価な炭素源を探し出すための研究が活発に行われている。例えば、木質系加水生成物、グリセロール、ホエー(whey)、コーンスティープリカー(corn steep liquor)などの比較的安価な原料物質を炭素源として使って、多様な一次代謝産物を生産するための研究が本発明者等を含み、多くの研究者によって行われてきた(Samuelov et al., Appl. Environ. Microbiol., 65:2260, 1999; Lee et al., Appl. Microbiol.Biotechnol., 54:23, 2000; Lee et al., Biotechnol.Bioeng., 72:41, 2001; Lee et al., Biotechnol. Lett., 25:111, 2003; Lee et al., Bioproc. Biosystems Eng., 26:63, 2003)。しかし、現在まで報告された研究結果によると、グルコースの代りに前記原料物質を炭素源として使った場合には、目的代謝産物の生産性あるいは生産収率がグルコースを炭素源として使った場合より顕著に低く、これを克服するための研究開発が切に求められている。
【0003】
砂糖として広く知られるスクロースはグルコースとフラクトースで形成された二糖類の一つであり、光合成能を有する全ての植物から生産されて、自然界に非常に豊富に存在する炭素源である。特に、サトウキビとサトウダイコンは過量のスクロースを含有しており、現在全世界のスクロースの60%以上がサトウキビから生産されている。何よりもスクロースはサトウキビの機械的な圧搾で得られた抽出液の単純な蒸発/濃縮工程を介して産業的に生産が可能で、生産価格が非常に安い。Koutinasらは2004年に微生物を利用した化学物質生産に使用可能な多様な原料物質の価格をグルコース含有量を基準に計算し、グルコース含有量1キログラムを基準にスクロースの価格は26.1セントとして、これは小麦、糖蜜、及びグルコースの価格に対して、77%、50%、28.9%に過ぎない非常に安い価格である(Koutinas et al., Ind. Crops and Products, 20:75, 2004)。
【0004】
International Sugar Agreement(ISA) daily price報告書は1994〜1995年を頂点にスクロースの価格が供給過剰によって下降安定傾向を維持しており、このような下落傾向は持続すると展望している。従って、スクロースは微生物発酵によって多様な化学物質生産に現在使われている高価なグルコースを代えられる最も有力な炭素源として注目されている。特に、主にトウモロコシ澱粉から生産されているグルコースの場合には持続的なトウモロコシ価格の上昇と共に、トウモロコシからの澱粉抽出、熱的/化学的澱粉前処理、酵素反応によるグルコースへの転換、及びこれの精製など、非常に複雑な工程を経て生産されるためグルコース生産単価をスクロースレベルに下げるのは非常に困難であることを当業界の従事者は周知している。このような理由から最近米国におけるトウモロコシ基盤バイオエタノール生産は次第に減少しているが(Mae-Wan Ho, Science in Society, 33:40, 2007)、ブラジルのサトウキビ、即ちスクロース基盤バイオエタノール生産工程は急激な成長を示している。
【0005】
現在までのスクロースを炭素源として利用した有用な物質生産に関する研究は高濃度細胞培養を介する生分解性高分子(Lee et al., Biotechnol. Lett., 15:971, 1993; Lee et al., Biotechnol. Techniques, 1:59, 1997)、クエン酸(Forster et al., App. Microbiol.Biotechnol., 75:1409, 2007)、アセトン、ブタノール、エタノール、イソプロパノール(George et al., Appl. Environ. Microbiol., 45:1160, 1983; Durre, Appl. Microbiol.Biotechnol., 49:639, 1998)、イタコン酸(Kautola et al., Biotechnol. Lett., 11:313, 1989)、キサンタンガム(Letisse et al., Appl. Microbiol.Biotechnol., 55:417, 2001)等を中心に進められてきた。特に、国際バイオエネルギー及びバイオ燃料市場を分析する国際エネルギーエージェンシー(International Energy Agency)の2006年のバイオエネルギータスク40(Bioenergy Task 40)報告書は、ブラジルのスクロースを含むサトウキビからバイオエタノール生産工程を環境に優しく持続可能なバイオ燃料生産の優れたモデルとして高く評価した。
【0006】
微生物発酵による化学物質生産のための原料物質としてスクロースは価格が安いだけでなく、スクロース自体が過量の目的代謝産物を含有する外部環境から細胞膜を完全に維持させる役割を果たすことによって、高濃度の目的代謝産物を生産できる優秀な機能を有している。Kilimann等(Biochimica et Biophysica Acta, 1764, 2006)はスクロースを含んだ培養培地とこれを含まない培養培地に、致死的な温度で微生物を露出させた後、それらの生存率と蛋白質の2次構造を調べた。その結果、スクロースを含んだ培養培地に露出した微生物の細胞内に存在する蛋白質の2次構造は保存状態が非常に良好であったが、スクロースを含まない培養培地に露出した微生物の細胞内の蛋白質構造は非常に変性されていたことを確認した。特に、スクロースを含んでいる培養培地に露出した微生物の生存率はスクロースを含まない培養培地に露出した微生物の生存率と比較して、非常に高かった。これは有害な外部環境に対するスクロースの微生物保護機能を直接的に証明するものである。
【0007】
前記において記述したスクロースの安い価格と外部環境に対する微生物保護機能という原料物質として優秀な長所を有しているにも拘らず、実際にスクロースを炭素源として使って、微生物発酵を介する目的化学物質生産に成功し、実際に商業化した例は、未だに報告されていない。これは多数の微生物がスクロースを細胞内に輸送した後、これを分解して、解糖過程につなげるメカニズムを完全に有しないか、あるいは不完全に有していることでスクロースを炭素源として利用できないためである。たとえスクロースを利用できるメカニズムを有している微生物の場合にも炭素源としてこれを摂取して分解する速度が非常に遅いため、目的代謝産物を効率よく生産できない。
【0008】
微生物発酵を介する多様な化学物質生産が産業的に経済性を有するためには前記に記述したとおりスクロースのように価格が安い原料物質の使用が求められ、さらにスクロースを炭素源として効率よく迅速に摂取してこれを分解できる酵素の同定及びこれを利用できる研究開発が必要である。特に、微生物発酵を介する化学物質生産において原料物質の価格が最終生産価格の約50%を占める事実を考慮した場合、安い原料物質であるスクロースを効率よく利用させるようにする酵素の同定及びこれの応用開発が非常に急がれている。
【0009】
現在まで高濃度細胞培養を通した生分解性高分子、クエン酸、イタコン酸、アセトン、ブタノール、エタノール、イソプロパノール、及びキサンタンガムなどの多様なバイオ製品の生産にスクロースを活用することができる例が報告されている。しかし、実際にスクロースを炭素源として使って、微生物発酵を介する目的化学物質生産に成功して、実際に商業化した例は多くない。
【0010】
従って、環境に優しいグリーン成長の核心技術として微生物発酵を介する多様な化学物質生産が産業化に成功するためには、スクロースのような安価でかつ豊富に存在する炭素源を利用して、これを効果的に摂取して分解できる微生物の開発が必要である。
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の主な目的は、スクロースを炭素源として使えるようにする新規スクロース代謝関連遺伝子、及びこれによってコードされる酵素を提供することである。
【0012】
本発明の他の目的は、前記スクロース代謝関連遺伝子が導入されているスクロース代謝能を有する遺伝子組み換え微生物、及びこれを利用した代謝産物、生分解性高分子または遺伝子組み換え蛋白質の製造方法を提供することである。
【課題を解決するための手段】
【0013】
前記目的を達成するために、本発明は配列番号1のアミノ酸配列を有するスクロースホスホトランスフェラーゼ、これをコードする遺伝子(ptsG)、並びに前記遺伝子(ptsG)及びスクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)を含有する遺伝子組み換えベクターを提供する。
【0014】
本発明はまた、前記遺伝子が細菌、酵母及びカビから構成された群より選択された宿主細胞に導入されて、スクロース代謝能を有する遺伝子組み換え微生物、及びこれをスクロースを炭素源として含有する培地で培養することを特徴とする代謝産物、生分解性高分子、または遺伝子組み換え蛋白質の製造方法を提供する。
【0015】
本発明はまた、β−D−フルクトフラノシド結合を加水分解して、フラクトースを遊離する活性を有するβ−フルクトフラノシダーゼ及びこれをコードする遺伝子を提供する。
【0016】
本発明はまた、前記遺伝子が細菌、酵母及びカビから構成された群より選択された宿主細胞に導入されて、スクロース代謝能を有する遺伝子組み換え微生物及びこれをスクロースを炭素源として含有する培地で培養することを特徴とする代謝産物、生分解性高分子、または遺伝子組み換え蛋白質の製造方法を提供する。
【0017】
本発明の他の特徴及び具現例は次の詳細な説明及び添付された特許請求範囲からより一層明白になるであろう。
【図面の簡単な説明】
【0018】
【図1】スクロースを代謝可能にするM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来の新規スクロース代謝関連酵素、スクロースホスホトランスフェラーゼ、スクロース−6−リン酸ヒドロラーゼ及びフラクトキナーゼをスクロースを代謝できない微生物に導入した時、スクロースが摂取されて分解され、解糖過程を介して、代謝される経路を示した模式図である。太い矢印(→):導入したM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来の新規スクロース代謝関連酵素によって制御される代謝経路を示し、細い矢印(→):遺伝子組み換え微生物が本来有している解糖過程の代謝経路を示す。
【図2】スクロースを代謝可能にするM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来の新規β−フルクトフラノシダーゼを、スクロースを代謝できない微生物に導入した時、この酵素がスクロースの代謝にどのように係わるのかを示した4種の可能な経路を示した模式図である(太い矢印(→):導入したM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来の新規β−フルクトフラノシダーゼが係わる可能性がある4種の可能な経路を示し、細い矢印(→):遺伝子組み換え微生物が本来有している糖類分解過程の代謝経路を示す)。
【図3】スクロースホスホトランスフェラーゼ、スクロース−6−リン酸ヒドロラーゼ及びフラクトキナーゼをコードする遺伝子(ptsG、sacC、rbsK)を含有する遺伝子組み換えベクターpMSscrIIAの地図である。
【図4】母菌株MBEL55E、遺伝子組み換えMptsG、遺伝子組み換えMsacC菌株のスクロース培地上における成長を示したグラフである(●:MBEL55E、▲:MptsG、及び△:MsacC)。
【図5】遺伝子組み換えベクターpTac15Kの切断地図である。
【図6】スクロース−6−リン酸ヒドロラーゼをコードする遺伝子を含有する遺伝子組み換えベクターpTac15KsacCの切断地図である。
【図7】遺伝子組み換えE. coli W3110 pTac15Kのスクロース含有M9培地における成長を示すグラフである(●を含む実線:スクロース濃度;◆を含む実線:OD600)。
【図8】本発明によるスクロース代謝能を有する遺伝子組み換えE. coli W3110 pTac15KsacCのスクロース含有M9培地における生長グラフである(●を含む実線:スクロース濃度;◆を含む実線:OD600;●を含む点線:グルコース濃度;○を含む実線:フラクトース濃度;▼を含む点線:酢酸濃度)。
【図9】E. coli W3110 pTac15KEWcscA及びE. coli W3110 pTac15KBSsacAのスクロース含有M9培地における成長を示すグラフである(●を含む実線:E. coli W3110 pTac15KEWcscA;▲を含む実線:E. coli W3110 pTac15KBSsacA)
【図10】本発明によるスクロース代謝能を有する遺伝子組み換えE. coli W3110 pTac15KsacCを嫌気条件で発酵した成長、及び代謝産物生産を示すグラフである(●を含む実線:スクロース濃度;△を含む実線: OD600;▼を含む実線:グルコース濃度;■を含む実線:フラクトース濃度;●を含む点線:コハク酸濃度;▽を含む点線:乳酸濃度;■を含む点線:ギ酸濃度;◇を含む点線:酢酸濃度;▲を含む点線:エタノール濃度)
【発明を実施するための形態】
【0019】
本発明者は安価でかつ豊富に存在する炭素源の一つとして砂糖として広く知られたスクロースを活用したバイオ製品の生産、正確にはスクロースを炭素源として活用できる微生物、または活用できない微生物の場合においてもこれを活用できるようにする酵素を探し出して、これを活用して、多様なバイオ製品の生産に応用しようとする。
【0020】
スクロースはグルコースとフラクトースで形成された二糖類として地球上に豊富に存在する炭素源の一つである。光合成能力を有する多くの植物から生産され、特に、熱帯及び亜熱帯性作物のサトウキビとサトウダイコンは過量のスクロースを含有している。スクロースはサトウキビの機械的な圧搾で得られた抽出液の単純な蒸発/濃縮工程を介して産業的に生産可能であり、現在全世界のスクロースの60%以上がサトウキビから非常に安く生産されている。グルコース含有量を基準に、微生物の発酵の原料として使用可能な多様な原料物質の価格を計算したKoutinas等の2004年度の論文によると、グルコース含有量1kgを基準にスクロースの価格は26.1セントとしてグルコース価格の約1/4で、これは小麦、糖蜜の価格と比較しても1/2〜2/3の価格に相当する(Koutinas et al., Ind. Crops and Products, 20:75, 2004)。さらに、過去15年間International Sugar Organizationに公示されたスクロース価格の推移を調べてみると、1995年の13.28cents/1bを頂点に、1999年6.27cents/lbまで暴落し、2008年3月頃14.20cents/lbを経て、2008年5月7日の価格が12.44cents/lbに維持されており、供給過剰によって、下降安定傾向を維持すると予想している。このような価格的なメリット以外にもスクロースは穀物から得られるのではないため、最近の穀物価格の高騰のような食糧危機状況下においても安定的に供給可能な長所がある。
【0021】
現在まで高濃度細胞培養による生分解性高分子、クエン酸、イタコン酸、アセトン、ブタノール、エタノール、イソプロパノール、及びキサンタンガム(Lee et al., Biotechnol. Lett., 15:971, 1993; Lee et al., Biotechnol. Techniques, 1:59, 1997; Forster et al., App. Microbiol.Biotechnol., 75:1409, 2007; George et al., Appl. Environ. MicroBiol., 45:1160, 1983; Durre, Appl. Microbiol.Biotechnol., 49:639, 1998; Kautola et al., Biotechnol. Lett., 11:313, 1989; Letisse et al., Appl. Microbiol.Biotechnol., 55:417, 2001)等の多くのバイオ製品の生産にスクロースを活用した例が報告され、これはスクロース活用能力の向上が正に効果的なバイオ製品の生産に直接影響を与えられると予想される。
【0022】
しかし、原料としての長所とスクロース自体が有する蛋白質変性に対する保護能、外部環境に対する細胞の保護能などの種々のメリットにも係わらず(Kilimann et al.,Biochimica et Biophysica Acta, 1764, 2006)、実際にスクロースを炭素源として使って、微生物発酵による目的化学物質生産に成功して、実際商業化した例は多くない。これは多数の微生物がスクロースを代謝できるメカニズムを有していないか、有している場合にも代謝速度が遅く、目的とするバイオ製品を効果的に生産できないのがその原因の一つである。従って、環境に優しいグリーン成長の重要技術として微生物発酵による多様な化学物質生産が産業化に成功するためには、スクロースのように安価でかつ豊富に存在する炭素源を利用して、これを効果的に摂取して、分解できる微生物の開発が必要である。そのためにはスクロースを速く分解して代謝できる酵素の同定及び活用が伴わなければならない。
【0023】
現在まで幾人の研究者によって、スクロースを活用可能にする酵素群の開発がなされて来た。代表的なものが、大腸菌由来のPTS(phosphoenol pyruvate-dependent phosphotransferase system)及びnon−PTSのスクロース輸送システム(sucrose transport system)を導入して、アミノ酸生産に活用した味の素(Ajinomoto Co.)の技術が挙げられ、これはPTSまたはnon−PTS関連全体遺伝子群を導入して発明を完成したことにその特徴がある。
【0024】
しかし、本発明においては、マンヘミア・サクシニシプロデュセンスMBEL55E(Mannheimia succiniciproducens MBEL55E:KCTC 0769BP)の遺伝情報を基にスクロースを細胞内に輸送して、これを分解して解糖過程へつなげるのに関与する酵素[スクロースホスホトランスフェラーゼ(pstG、MS0784)、スクロース−6−リン酸ヒドロラーゼ(SacC、MS1233)及びフラクトキナーゼ(RbsK、MS1233)]をコードする新規遺伝子(ptsG、sacC、及びrbsK)を探し出し、これらの配列と機能を解明した。
【0025】
従って、本発明は一観点において、スクロースを細胞内に輸送し、これを分解して、解糖過程へつなげるのに関与する酵素の配列番号1のアミノ酸配列を有するスクロースホスホトランスフェラーゼ、配列番号3のアミノ酸配列を有するスクロース−6−リン酸ヒドロラーゼ及び配列番号5のアミノ酸配列を有するフラクトキナーゼに関する。本発明はまた、前記スクロースホスホトランスフェラーゼをコードする遺伝子(ptsG)、前記スクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)、及び前記フラクトキナーゼをコードする遺伝子(rbsK)に関する。
【0026】
本発明において、前記スクロースホスホトランスフェラーゼ(pstG)はスクロースをスクロース−6−リン酸に転換させながら、細胞中に輸送する役割を果たし、前記スクロース−6−リン酸ヒドロラーゼ(SacC)はスクロース−6−リン酸をグルコース−6−リン酸及びフラクトースに転換させる活性を有し、前記フラクトキナーゼ(RbsK)はフラクトースをフラクトース−6−リン酸に転換させる活性を有する。
【0027】
本発明において、前記ptsGは配列番号2の塩基配列で表されることを特徴とし、前記sacCは配列番号4の塩基配列で表されることを特徴とし、前記rbsKは配列番号6の塩基配列で表されることを特徴とする。
【0028】
具体的には、本発明においては前記ptsG及びsacC遺伝子を含有する遺伝子組み換えベクターを作製した後、これをスクロースを炭素源として使えない大腸菌に導入して、遺伝子組み換え大腸菌を作製して、前記作製された遺伝子組み換え大腸菌を単一炭素源でスクロースを含有する培地で培養した結果、スクロース代謝能を有するということを確認した。
【0029】
これは図1に示したように、スクロースが前記スクロースホスホトランスフェラーゼ(pstG)によりスクロース−6−リン酸に転換されながら、細胞中に移送されて、細胞内に移送されたスクロース−6−リン酸は前記スクロース−6−リン酸ヒドロラーゼ(SacC)によりグルコース−6−リン酸及びフラクトースに転換された後、フラクトースはフラクトキナーゼ(RbsK)によりフラクトース−6−リン酸に転換されて、解糖過程につながることと推定することができる。
【0030】
一方、本発明者等は、マンヘミア・サクシニシプロデュセンスMBEL55E(Mannheimia succiniciproducens MBEL55E:KCTC 0769BP)由来のβ−フルクトフラノシダーゼ(β-fructofuranosidase)であるスクロース−6−リン酸ヒドロラーゼ(SacC、MS0909)をはじめとして、他のβ−フルクトフラノシダーゼ(β-fructofuranosidase)をコードする新規遺伝子(cscA、sacA)を導入することによっても、スクロースを代謝できない微生物を、スクロースを代謝可能にできることを示し、本菌株を活用した多様な代謝産物の生産する例を示すことで本発明を完成した。換言すると、マンヘミア・サクシニシプロデュセンスMBEL55E(Mannheimia succiniciproducens MBEL55E:KCTC 0769BP)、E. coli W及びバシラス・サブチリス(Bacillus subtilis)の遺伝情報を基にスクロースを分解して、解糖過程につなげるのに関与する酵素β−フルクトフラノシダーゼ(β-fructofuranosidase)をコードする新規遺伝子(sacC、cscA、sacA)を探し出し、これらの配列と機能を解明したものである。
【0031】
IUBMB(International Union of Biochemistry and Molecular Biology、国際生化学・分子生物学連合(http://www.iubmb.org/)の機能に応じる酵素分類法によって付与されたEC番号(Enzyme Comission number)は酵素を区分するために最も広く活用される番号である。
【0032】
スクロース−6−リン酸ヒドロラーゼの正式名称はβ−フルクトフラノシダーゼ(β-fructofuranosidase、(EC 3.2.1.26))であり、β-D-fructofuranoside fructohydrolase、invertase、saccharase、glucosucrase、β-h-fructosidase、β-fructosidase、sucrase、maxinvert L 1000、fructosylinvertase、alkaline invertase、acid invertaseなどの他の名称が存在する(http://www.chem.qmul.ac.uk/iubmb/enzyme/EC3/2/1/26.html)。即ち、スクロース−6−リン酸ヒドロラーゼ及びスクラーゼは、EC 3.2.1.26に該当するβ−フルクトフラノシダーゼ(β-fructofuranosidase)という正式名称を有している。
【0033】
このようなβ−フルクトフラノシダーゼは、β−D−フルクトフラノシド(β-D-fructofuranoside)において、ターミナル(terminal)非還元β-D-fructofuranoside残基を加水分解する反応に係わり、その係わる反応の基質によって、スクロースを加水分解すると、スクラーゼまたはインバターゼ、スクロース−6−リン酸を基質とするとスクロース−6−リン酸ヒドロラーゼという慣用名で使われる。
【0034】
そこで、本実施例においては、マンヘミア(Mannheimia)由来のスクロース−6−リン酸ヒドロラーゼ(EC 3.2.1.26、SacC)、即ち、β−フルクトフラノシダーゼ(β-fructofuranosidase)の導入によるスクロース代謝能の証明に加えて、一般的なβ−フルクトフラノシダーゼ(β-fructofuranosidase)の導入がスクロース代謝能を持たせるようにすることを示そうと努力した結果、例えば、E. coli W菌株由来のインバターゼ(EC 3.2.1.26、CscA)とバシラス・サブチリス(Bacillus subtilis)由来のβ−フルクトフラノシダーゼ(EC 3.2.1.26、SacA)の各々を導入することで、スクロースを代謝できない微生物がβ−フルクトフラノシダーゼ(EC 3.2.1.26)を導入することでスクロースを代謝して、成長することを示す。
【0035】
従って、本発明は他の観点において、スクロースを加水分解して、グルコースとフルクトースに転換する活性、及びスクロース−6−リン酸を加水分解して、グルコース−6−リン酸及びフラクトースに転換する活性などを含む、β−D−フルクトフラノシド結合を加水分解して、フラクトースを遊離する活性を有するβ−フルクトフラノシダーゼに関する。また、前記β−フルクトフラノシダーゼは、配列番号3、配列番号7及び配列番号9のアミノ酸配列で構成された群から選択されたアミノ酸配列を有してもよいが、これに制限されるものではない。
【0036】
具体的には、本発明において、前記β−フルクトフラノシダーゼの一例であるスクロース−6−リン酸ヒドロラーゼ(sacC)は、スクロース−6−リン酸をグルコース−6−リン酸及びフラクトースに転換させる活性、またはスクロースをグルコース及びフラクトースに転換させる活性を有する。また、実施例においてはβ−フルクトフラノシダーゼはマンヘミア由来であってもよいが、その他の異なる微生物から由来したもの、また本発明の範囲に含まれ、配列番号3のアミノ酸配列を有してもよく、これをコードする遺伝子(sacC)は配列番号4の塩基配列で表されることを特徴とする。
【0037】
本発明において、前記β−フルクトフラノシダーゼの一例である、インバターゼ、スクラーゼ、スクロースヒドロラーゼ(CscA)はスクロースを分解して、解糖過程につなげるのに関与する酵素で、スクロース−6−リン酸をグルコース−6−リン酸及びフラクトースに転換させる活性、またはスクロースをグルコース及びフラクトースに転換させる活性を有する。また、実施例においては前記β−フルクトフラノシダーゼは大腸菌(E. coli W)由来を例示しているが、その他の異なる微生物から由来したものも本発明の範囲に含まれ、配列番号7のアミノ酸配列を有してもよく、これをコードする遺伝子(cscA)は配列番号8の塩基配列で表されることを特徴とする。
【0038】
本発明において、前記β−フルクトフラノシダーゼの一例であるスクロース−6−リン酸ヒドロラーゼ(sacA)は、スクロースを分解して、解糖過程につなげるのに関与する酵素で、スクロース−6−リン酸をグルコース−6−リン酸及びフラクトースに転換させる活性、またはスクロースをグルコース及びフラクトースに転換させる活性を有する。また、実施例においては前記β−フルクトフラノシダーゼはバシラス・サブチリス(Bacillus subtilis)由来を例示しているが、その他の異なる微生物から由来したものも本発明の範囲に含まれ、配列番号9のアミノ酸配列を有してもよく、これをコードする遺伝子(sacA)は配列番号10の塩基配列で表されることを特徴とする(sacA、BSU38040、sucrose-6-phosphate hydrolase)。
【0039】
また、マンヘミア由来のsacCによって、コードされるβ−フルクトフラノシダーゼとprotein BLAST(http://blast.ncbi.nlm.nih.gov/Blast.cgi)検索を介する酵素のアミノ酸シーケンス間の相同性比較を実施してみると、E. coli W菌株由来のcscAによって、コードされるインバターゼの場合は28%の相同性を有し、バシラス・サブチリス(Bacillus subtilis)由来のsacAによってコードされるスクロース−6−リン酸ヒドロラーゼ(β−フルクトフラノシダーゼ)の場合は35%の相同性を有する。そして、二つの菌株由来のβ−フルクトフラノシダーゼは共に、「SacC」と称されるβ−フルクトシダーゼ(COG1621)といわれる保存されたドメイン(conserved domain)を有する。これは相同性において、たとえある程度差があっても、「SacC」と称されるβ−フルクトシダーゼ(COG1621)といわれる保存されたドメインが存在すると、下記の通り、これを微生物内に導入した時、スクロースを代謝して成長できることを示している。従って、前記β−フルクトフラノシダーゼに関して本明細書においては配列番号3、7または9のアミノ酸配列を例示したが、これに制限されることなく、β−D−フルクトフラノシド結合を加水分解して、フラクトースを遊離する活性を有するβ−フルクトフラノシダーゼであれば、全て本発明に含まれる。例えば、配列番号3、7または9のアミノ酸配列と配列同一性が少なくとも70%、80%、90%以上のアミノ酸配列も本発明に含まれる。
【0040】
同様に、前記β−フルクトフラノシダーゼをコードする遺伝子は、例えば、配列番号4、8または10の塩基配列を有しながら、この配列中いずれか一つの塩基配列が置換、欠失、挿入及び付加されて、突然変異が起きることによって前記本発明による塩基配列と少なくとも70%、80%、望ましくは90%、より望ましくは95%の配列同一性を有するDNAも本発明に含まれる。
【0041】
前記「配列同一性」とは、2個のポリヌクレオチド間、またはアミノ酸間残基の配列類似性をいう。前記「配列同一性」は比較対象の塩基配列、またはアミノ酸配列の領域に跨って、最適状態でアラインメントされた2個の塩基配列またはアミノ酸配列を比較することによって決定される。ここで、比較対象のポリヌクレオチドまたはアミノ酸配列は、2個の配列の最適なアラインメントのための参考配列(例えば、コンセンサス配列など)と比較して、付加または欠失(例えば、ギャップ、オーバーハング等)を有してもよい。配列同一性の数値は両方の配列に存在する同じ核酸塩基またはアミノ酸を決めて、適合部位の数を決めて、続いて、比較対象の配列領域内の塩基またはアミノ酸の合計数で前記適合部位の数を割り算して得られた数値に100を乗算することによって算出される。核酸またはアミノ酸の間の配列同一性は、例えば、配列解釈ソフト、具体的にはBLASTN、FASTAなどを使って測定される。前記BLASTは、http://www.ncbi.nlm.nih.gov/Blast/から普通に利用することができる。
【0042】
本発明において、スクロース(sucrose)は先に説明したように、D−グルコースとD−フラクトースで形成される二糖類で、それ自体が細胞内の蛋白質の変性を防いで安定化させたり、外部環境の変化から細胞の溶解を極力抑制させる役割を果たすと知られており、高濃度の基礎化学物質生産や高濃度細胞培養時にさらに有効活用可能である。
【0043】
図2に示したように、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来の新規β−フルクトフラノシダーゼルスクロース−6−リン酸ヒドロラーゼを、スクロースを代謝できない微生物に導入した時、前記導入された酵素が4種類の可能な経路を介して代謝されると予想される。
【0044】
最初の可能な経路(反応I)は、導入した前記スクロース−6−リン酸ヒドロラーゼが細胞外空間(Extracellular space)でスクロースをグルコースとフラクトースに分解した後、各々の特定のホスホトランスフェラーゼなどの輸送に関与する酵素を介して細胞内部に導入される場合である。
【0045】
二番目の可能な経路(反応II)は、導入されたスクロース−6−リン酸ヒドロラーゼが周辺細胞質(periplasm)内でスクロースをグルコースとフラクトースに分解した後、各々の特定のホスホトランスフェラーゼなどの輸送に関与する酵素を介して細胞内部に導入される場合である。
【0046】
三番目の可能な経路(反応III)は、スクロースがホスホトランスフェラーゼ系列でないパーミアーゼ(permease)酵素によってスクロースのまま細胞内に入ってきて、前記導入されたスクロース−6−リン酸ヒドロラーゼによって、グルコースとフラクトースに分解されて利用できる場合である。
【0047】
四番目の可能な経路(反応IV)は、スクロースがホスホトランスフェラーゼによって、細胞内部に入って来ると共に、スクロース−6−リン酸に転換され、導入されたスクロース−6−リン酸ヒドロラーゼによって、フラクトースとグルコース−6−リン酸に転換されて利用できる場合である。
【0048】
また、前記sacC遺伝子は、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC 0769BP)由来であり、前記菌株はアクチノバシラス・サクシノゲネス130Z(Actinobacillus succinogenes 130Z:ATCC 55618)と共にパスツレラ科(Pasteurellaceae)(family)に属する。特に、前記M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC 0769BP)とA.サクシノゲネス130Z(A. succinogenes 130Z:ATCC 55618)のゲノム配列及び細胞生理(cell physiology)が非常に類似している。前記二つの菌株のゲノム配列は今まで解読された全てのゲノム配列の中で最も類似していると知られている(Mckinlay et al., Appl. Microbiol.Biotechnol., 76:727, 2007)。また、前記sacC遺伝子に対してA.サクシノゲネス130Z(A. succinogenes 130Z)由来であるものが、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来のものと非常に高い相同性を有し、最も類似する酵素と知られているため、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来のスクロース−6−リン酸ヒドロラーゼ(SacC、MS0909)酵素及びこれをコードする遺伝子とA.サクシノゲネス130Z(A. succinogenes 130Z)のスクロース−6−リン酸ヒドロラーゼ(Asuc_1829)酵素及びこれをコードする遺伝子も同様に適用されるのは、当業界の通常の知識を有するた者には明らかに理解できる。
【0049】
従って、本発明は他の観点において、スクロースホスホトランスフェラーゼ及び/またはスクロース−6−リン酸ヒドロラーゼをコードする遺伝子が導入されたスクロース代謝能を有する遺伝子組み換え微生物及び前記遺伝子組み換え微生物をスクロースを炭素源として含有する培地で培養するのを特徴とする代謝産物、生分解性高分子または遺伝子組み換え蛋白質の製造方法に関する。
【0050】
本発明において、遺伝子組み換えベクターは、適切な宿主細胞で蛋白質を発現できるベクターであり、蛋白質の発現を調節できる調節配列(control sequences)に作動可能に連結されたDNA配列及びその他遺伝子操作を容易にしたり蛋白質の発現を最適化したり、またはベクターの複製に必要な配列を含有するDNA作製(DNA construct)をいう。前記調節配列には転写を調節するためのプロモーター(promoter)、転写を調節するために選択的に付加されたオペレーター(operator)、適切なmRNAリボソーム結合部位及び転写/翻訳の終了を調節する配列が含まれる。例えば、前記遺伝子組み換えベクターは下記の実施例において使われたpTrc99Aのような遺伝子組み換えベクターであってもよいが、このベクター以外に他の公知のベクターも適用可能であり、pKK223−3、pTac99A、pET series、pMAL seriesのような発現ベクターだけでなく、pACYC、pBluescript SK−、pBR322、pGEM series、pMB1等のクローニングベクターにもsacC遺伝子を含む発現カセットを挿入することで適用可能であり、その他にも本発明が属する技術分野の公知の遺伝子組み換えベクター(Sambrook J, Russell D, Molecular cloning:a laboratory manual, 3rd ed., Cold Spring Harbor Lab (CSHL) Press, New York, 2001)を利用することも可能である。また、アンピシリン抵抗遺伝子以外にも公知の他の多くの抵抗遺伝子を含むものも本発明に適用可能である。
【0051】
また、前記遺伝子はM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC 0769BP)由来のものであり、前記菌株はアクチノバシラス・サクシノゲネス130Z(Actinobacillus succinogenes 130Z:ATCC 55618)と共にパスツレラ科(Pasteurellaceae)(family)に属する。特に、前記二つの菌株、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC 0769BP)とA.サクシノゲネス130Z(A. succinogenes 130Z:ATCC 55618)のゲノム配列及び細胞生理が非常に類似する。McKinlay等の2007年度の報告によると、前記二つの菌株のゲノム配列は今まで解読された全てのゲノム配列の中で最も類似すると知られている(Appl. Microbiol.Biotechnol., 76:727, 2007)。従って、前記遺伝子はM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC 0769BP)と共にA.サクシノゲネス130Z(A. succinogenes 130Z:ATCC 55618)から由来した遺伝子にも適用できることは、当業界の通常の知識を有するた者には明らかに理解できる。
【0052】
本発明の実施例においては、前記ptsG、sacC及びrbsK遺伝子を含む遺伝子組み換えベクターを利用して、スクロースを炭素源として使えない微生物を形質転換させてスクロース代謝能を有する遺伝子組み換え微生物を作製したが、前記遺伝子を、既に知らされた公知の方法で、スクロースを炭素源として使えない他の微生物の染色体に挿入させて、染色体上で操作された(genome-engineered)スクロース代謝能を有する遺伝子組み換え微生物を作製してもよい。
【0053】
また、本発明は他の観点において、前記sacC遺伝子をはじめとするβ−フルクトフラノシダーゼをコードする遺伝子を含有する遺伝子組み換えベクターを作製した後、これをスクロースを炭素源として使えない大腸菌に導入することによって、前記遺伝子組み換えベクター、前記遺伝子組み換えベクターによって形質転換されたスクロース代謝能を有する遺伝子組み換え微生物、前記作製された遺伝子組み換え微生物を単一炭素源としてスクロースを含有する培地において培養した結果、スクロース代謝能を有することを確認して、これを利用した代謝産物、生分解性高分子、又は遺伝子組み換え蛋白質の製造方法に関する。
【0054】
前記遺伝子組み換えベクターは図5の切断地図を有する遺伝子組み換えベクターであってもよいが、図5に示されたpTac15Kベクター以外に他の公知のベクターも適用可能であり、pTrc99A、pTac99A、pMAL seriesのような発現ベクターだけでなく、pACYC、pBluescript SK−、pBR322、pGEM series、pMB1等のクローニングベクターにもsacC遺伝子をはじめとするβ−フルクトフラノシダーゼをコードする遺伝子を含む発現カセットを挿入することで適用可能であり、その他にも本発明が属する技術分野の公知の遺伝子組み換えベクター(Sambrook J, Russell D, Molecular cloning: a laboratory manual, 3rd ed., Cold Spring Harbor Lab (CSHL) Press, New York, 2001)も利用可能である。また、カナマイシン抵抗遺伝子以外にも公知の他の多くの抵抗遺伝子を含むものも本発明に適用可能である。
【0055】
本発明の下記実施例において、前記sacC、cscA、sacAを含む遺伝子組み換えベクターを利用して、スクロースを炭素源として使えない微生物を形質転換させてスクロース代謝能を有する遺伝子組み換え微生物を作製したが、スクロースを炭素源として使えない他の微生物の染色体に前記遺伝子を公知の方法で挿入させてスクロース代謝能を有する遺伝子組み換え微生物を作製することができる。それと共に、本発明の実施例においてはスクロースを炭素源として使えない宿主微生物として特定大腸菌のみを示したが、その他の異なる大腸菌だけでなく、細菌、酵母及びカビなどのスクロースを炭素源として使えない宿主微生物に導入しても同様の結果を得られることは当業者には明らかに理解できる。
【0056】
また、本発明において、代謝産物とは、代謝過程を介して生成される中間産物及び生成物を総称するものであって、一次代謝産物と二次代謝産物に区分される。例えば、本発明はバイオ燃料、一次代謝産物、二次代謝産物、生分解性高分子、遺伝子組み換え蛋白質生産菌株作製時に遺伝子組み換えまたは染色体上における操作(genome-engineered)等の手法でスクロースを活用できない微生物に多様に適用可能である。例えば、ブタノール(Atsumi et al., Nature., 451:7174, 2008)、エタノール(Lindsay et al., Appl. Microbiol.Biotechnol, 43:70, 1995)、乳酸(Zhou, Appl. Environ. Microbiol, 69:399 2003)、コハク酸及びリンゴ酸(Jantama et al.,Biotechnol.Bioeng., 99:1140, 2008)、アミノ酸(Park et al., PNAS, 104:7797, 2006; Lee et al., Molecular Systems Biology, 3:1, 2007)、生分解性高分子(Ahn et al., Appl. Environl. MicroBiol., 66:3624, 2000; Park et al.,Biomacromolecules, 2:248, 2001; Park et al.,Biotechnol.Bioeng., 74:81, 2001)、遺伝子組み換え蛋白質(Jeong et al., Appl. Environl. MicroBiol., 65:3027, 1999; Han et al., Appl. Environl. MicroBiol., 69:5772, 2003)などの生産時に今まではグルコースを主炭素源として活用して、その目的バイオ製品を生産したが、本発明を前記公知の菌株に導入すると、スクロースを炭素源として活用して、目的する有用バイオ製品を生産可能である。
【0057】
他にも生分解性高分子、遺伝子組み換え蛋白質の生産にも本発明を当業者は容易に適用可能である。下記実施例においては代謝産物として、例えば、トレオニンなどを挙げて代謝産物を生産する場合を示したが、それ以外に生分解性高分子、遺伝子組み換え蛋白質の生産にも本発明を当業者は容易に適用可能である。
【実施例】
【0058】
以下、実施例によって、本発明をより一層詳細に説明する。これらの実施例は単に本発明を例示するものであり、本発明の範囲がこれらの実施例によって制限されると解釈されないことは、当業界において通常の知識を有するた者には明らかに理解できる。
特に、下記実施例においては本発明による遺伝子を発現させるためにスクロース代謝を行えない宿主菌株として特定大腸菌を示したが、他の大腸菌や他属の微生物を使っても、本発明によるスクロース代謝能に関与する遺伝子を導入してこれを利用してスクロースをはじめとする代謝産物を製造することも当業界において通常の知識を有するた者には明らに理解できる。
【0059】
[実施例1]ptsG、sacC及びrbsK遺伝子のスクロース代謝能確認
1.1:ptsG、sacC及びrbsK遺伝子の分離
本発明による遺伝子(ptsG、sacC、及びrbsK)が共にスクロース代謝に関与するかを調べてみるため、先ず前記遺伝子をM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC0769BP)から分離した。
【0060】
先ず、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC0769BP)のゲノムDNAを鋳型にして、配列番号:11及び12のプライマーを使ったPCRを行ってptsG(MS0784)のDNAを増幅した。配列番号:13及び14と配列番号:15及び16のプライマーを使ったPCRを同様に行って、各々sacC(MS0909)とrbsK(MS1233)のDNAを増幅し、これらを混合したものを鋳型にして配列番号:13と16のプライマーを使ってオーバーラッピングPCR(overlapping PCR)を行った。前記pstG(MS0784)、sacC(MS0909)とrbsK(MS1233)は、順にスクロースホスホトランスフェラーゼ、スクロース−6−リン酸ヒドロラーゼ、及びフラクトキナーゼにコードする遺伝子である。
【0061】
配列番号:11:5'−GGAATTCATGCTCGTTTTAGCTAGAATTGG
配列番号:12:5'−TCCGAGCTCTTACTATTCTTTTGCGTTAGCTCTTG
配列番号:13:5'−ACCTGCGAGCTCTTTCACACAGGAAACAATTTTCATGCGGTCGTTTTTACCG
配列番号:14:5'−CAAATTTTGTTTGTCATATGCATGAAATCTGTTTCCTGTGTGAAATTACTATTTATATTCAATTTCTTTCGGATA
配列番号:15:5'−TATCCGAAAGAAATTGAATATAAATAGTAATTTCACACAGGAAACAGATTTCATGCATATGACAAACAAAATTTG
配列番号:16:5'−ACCTGCGGGTACCCTATTAGTTTGCTAAAAATTCCGCT
【0062】
1.2:遺伝子組み換えベクターpMSscrIIAの作製
マンヘミア由来のスクロースホスホトランスフェラーゼ、スクロース−6−リン酸ヒドロラーゼ、及びフラクトキナーゼを各々コードする遺伝子、pstG(MS0784)、sacC(MS0909)及びrbsK(MS1233)を大腸菌から発現させるために発現ベクターを次のように作製した。
【0063】
実施例1.1において増幅されたsacC(MS0909)及びrbsK(MS1233)遺伝子を含む一つのDNA片を切断酵素SacIとKpnIで切断して、これを同じ切断酵素で切断したpTrc99A(Pharmacia Biotech., Uppsala, Sweden)と結合させてpTrc99AsacCrbsKを作製した。
【0064】
次に、実施例1.1において取得されたptsG(MS0784)DNA片をEcoRIとSacIで切断し、これを同じ切断酵素で切断した発現ベクターpTrc99AsacCrbsKと結合させてpTrc99AptsGsacCrbsKを作製して、これをpMSscrIIAと命名した(図3)。
【0065】
1.3:Escherichia coli W3110 pMSscrIIA及びW3110 pTrc99A菌株の作製
スクロースを代謝できないと知られるE. coli K-12菌株の亜株(substrain)のE. coli W3110をモデル微生物として下記の実験を行った。
【0066】
前記E. coli W3110をLB固体培地に塗抹して、8時間37℃で培養した後、コロニーをLB液体培地10mLに接種して、8時間培養した。前記培養液をLB液体培地100mLに1%(v/v)接種して、37℃の振盪培養器で培養した。
【0067】
約2時間後、OD600が0.30〜0.35程度になると、氷に20分間放置して、細胞の成長を停止させる。4℃、3,000rpm、15分の条件で遠心分離して細胞を取得した後、4℃状態のRFI溶液32mLで細胞を懸濁した。そして、前記懸濁液を4℃、3,000rpm、15分の条件で遠心分離して細胞を取得した。これをRFII溶液8mLで細胞を再懸濁した後、氷に15分間放置した。最終的に前記再懸濁液を100μLずつ分株して−70℃で保管した。RFIの組成は100mM RbCl、50mM MnCl2−4H2O、0.1M CH3COOK、10mM CaCl2及び15%(w/v)グリセロールで構成され、0.2M 酢酸を添加してpHを5.8に合わせた。RFIIは10mM MOPS、10mM RbCl、100mM CaCl2、及び15%(w/v)グリセロールで構成され、NaOHを添加することによってpHを6.8に合わせた。
【0068】
このように準備されたE. coli W3110菌株に実施例1.2において作製された発現ベクターpMSscrIIAまたは対照群でpTrc99A(Pharmacia Biotech., Uppsala, Sweden)を混合した後、42℃で90秒間ヒートショックを与えることによって形質転換を行った。ヒートショック後、LB液体培地0.8mLを加えて、37℃の振盪培養器で1時間培養した。
【0069】
培養液を抗生剤アンピシリン(最終濃度50μg/L)を含有したLB固体培地に塗抹して、37℃で12時間以上培養した。形成されたE.coli W3110 pMSscrIIA及びE.coli W3110 pTrc99Aコロニーを、LB液体培地に接種をして、37℃で8時間以上培養した。ベクターが上手く導入されたのか確認するために前記培養された菌株から、GeneAll(商標) Plasmid SV(GeneAll biotechnology, Korea)を利用してベクターを分離し、電気泳動によって確認した。E. coli W3110 pMSscrIIA菌株の場合、下記配列番号:17〜24までのプライマーを利用してソルジェント(Solgent, Korea)にシーケンシング(sequencing)を依頼して、塩基配列の一致の有無を確認した。
【0070】
配列番号:17:5'−GGAAACAGACCATGGAATT
配列番号:18:5'−CCGCAAAAGATTTATTCGAAGAAG
配列番号:19:5'−CCTGGTTATATGATACTTTAGG
配列番号:20:5'−TAGTGCTGGGCGCAAGAGCTAA
配列番号:21:5'−ACCAGTGGGCGATAAAATCG
配列番号:22:5'−TGATCAAGGTTTCGATTTCT
配列番号:23:5'−TTTTCCTGAATGACGGCGAA
配列番号:24:5'−CGATCTGCCGCAATTTCAAG
【0071】
1.4:遺伝子組み換え大腸菌のスクロース代謝能確認
実施例1.3において作製された固体培地上のE. coli W3110 pMSscrIIA菌株とE. coli W3110 pTrc99Aのコロニーを5g/Lスクロースを単一炭素源として含んだ10mLのM9最小培地に接種して、37℃で16時間培養した後、これを再びスクロースを含んだ100mLのM9最小培地に3%(v/v)接種した後37℃で培養した。この時、抗生剤として最終濃度50μg/Lのアンピシリンを添加した。M9最小培地は33.9g/LのNa2HPO4、15g/LのKH2PO4、2.5g/LのNaCl、5g/LのNH4Cl、及び0.36g/LのMgSO4で構成された。培養液内の細胞濃度は分光光度計を利用してOD600の値として推定した。培養中、周期的にサンプルを採取し、採取された試料は13,000rpmで5分間遠心分離した後、上澄み液のスクロースの濃度を液体クロマトグラフィー(HPLC)で分析した。
【0072】
その結果、表1に示したように、E. coli W3110 pTrc99A菌株の場合、スクロースを単一炭素源として含んだM9最小培地において全く生長できないが、E. coli W3110 pMSscrIIA菌株はスクロースの優れた代謝能を示す。E. coli W3110 pMSscrIIA菌株は19時間約2.2g/Lのスクロースを代謝して、OD600基準に3.12のバイオマスの増加を示し、0.67g/Lの硝酸を付加生成物として生産した。従って、前記ptsG、sacC及びrbsK遺伝子を全て含有する場合、優れたスクロース代謝能を示すことが確認された。
【0073】
【表1】
【0074】
前記に示したように、本発明のスクロースホスホトランスフェラーゼ、またはこれとスクロース−6−リン酸ヒドロラーゼ遺伝子を導入することだけでも、スクロースの代謝能を備えると共に、スクロースを炭素源として活用して代謝産物の一つとして酢酸を生産することを示した。
【0075】
従って、酢酸以外にもギ酸、乳酸、コハク酸、エタノールなどの他にも、バイオブタノールを含むバイオ燃料及びエネルギー、そして生分解性高分子、遺伝子組み換え蛋白質の生産にも本発明を当業者は容易に適用可能である。
【0076】
[実施例2]pstG遺伝子及びsacC遺伝子各々のスクロース代謝能確認
2.1:pstG遺伝子及びsacC遺伝子のスクロース代謝能確認のため遺伝子組み換えベクター作製
本発明によるptsG及びsacCが単独でもスクロース代謝能に係わるのかを確認するため、ptsG(MS0784)欠失のためのベクター(pSacHR06ptsG)とsacC(MS0909)欠失のためのベクター(pSacHR06sacC)を作製してノックアウト(knock-out)実験を行った。
【0077】
先ず、スクロースホスホトランスフェラーゼ遺伝子(ptsG)を相同性遺伝子組み換え方法で破壊するため遺伝子交換ベクターを次のように作製した。マンヘミア・サクシニシプロデュセンスMBEL55E(Mannheimia succiniciproducens MBEL55E:KCTC 0769BP)のゲノムDNAを鋳型にして、下記配列番号:25と26のプライマーを利用して、左側ホモロガスアーム(homologous arm)部分を増幅し、配列番号:27と28のプライマーを利用して、同じ鋳型で右側ホモロガスアーム(homologous arm)部分を増幅し、配列番号:29と30のプライマーを利用してpECmuloxベクター(Kim et al., FEMS Microbiol. Lett., 278:78, 2008)を鋳型にして抗生剤マーカーと変異lox siteを含んだ部分のDNA断片を増幅し、これら三つのDNA断片を配列番号:25と28プライマーを利用してオーバーラッピングPCR(overlapping PCR)を行った。
【0078】
配列番号:25:5'−ATATCTGCAGCCGGCATTAAATATTAGTCAAC
配列番号:26:5'−CGTTCTAACGGAGGTTGAAAACTGCCCTTT
配列番号:27:5'−GTCTCCCTATCACGCCGTTATTTTCATTATT
配列番号:28:5'−ATTAGTCGACACCATCCCCACGGAATACAT
配列番号:29:5'−TTTCAACCTCCGTTAGAACGCGGCTACAAT
配列番号:30:5'−TAACGGCGTGATAGGGAGACCGGCAGATCC
【0079】
このように増幅された最終DNA断片は制限酵素PstIとsalIで切断し、同じ酵素で切断されたpSacHR06ベクター(Park et al., Proc. Nat. Acad. Sci. (PNAS), 104:7797, 2007)にクローニングしてpSacHR06ptsGを作製した。また、前記のような方法で配列番号:31から36までのプライマーを利用して、pSacHR06sacCを作製した。
【0080】
配列番号:31:5'−ATACACTGCAGTTATGCAATTTATCGCACCC
配列番号:32:5'−AATCTGCTCTGATGCGGTCGTGAAATGCTTCCA
配列番号:33:5'−CACAGAATCAGGACAAATGGCATTCAATGCTG
配列番号:34:5'−ATACTGTCGACTCAATGGCATATGCAGCG
配列番号:35:5'−AAGCATTTCACGACCGCATCAGAGCAGATTGTACTGAGAG
配列番号:36:5'−TGAATGCCATTTGTCCTGATTCTGTGGATAACCGTATTAC
【0081】
2.2:M.サクシニシプロデュセンスMptsG(M. succiniciproducens MptsG)及びM.サクシニシプロデュセンスMsacC(M. succiniciproducens MsacC)菌株の作製
実施例2.1において作製されたptsG遺伝子除去用交換ベクターpSacHR06ptsGとsacC遺伝子除去用交換ベクターpSacHR06sacC各々を利用して、Kimら(FEMS Microbiol. Lett., 278:78, 2008)により報告された方法でM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC0769BP)のゲノムで前記遺伝子を欠失させて、変異菌株であるM.サクシニシプロデュセンスMptsG(M. succiniciproducens MptsG)及びM.サクシニシプロデュセンスMsacC(M. succiniciproducens MsacC)菌株を作製した。
【0082】
即ち、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC0769BP)を10g/Lのグルコースを含有したLB−グルコース固体培地に塗抹して、36時間37℃で培養した後、コロニーをLB−グルコース液体培地10mLに接種して、12時間培養した。十分に培養された培養液をLB−グルコース液体培地100mLに1%接種して、200rpm、37℃の振盪培養器で培養した。
【0083】
約4〜5時間後、OD600が0.3〜0.4程度になったら4℃、4,500rpm、20分の条件で遠心分離して、細胞を取得した後、4℃の状態の10%グリセロール溶液200mLで細胞を再懸濁した。そして、4℃、5,500rpm、20分の条件で遠心分離して細胞を取得した。再懸濁時に使われるグリセロール溶液を半分に減らしながら、これを2回繰り返し実行した後、細胞とグリセロール溶液の体積比が1:1になるべく再懸濁して細胞濃縮液を前記取得された細胞濃縮液と前記実施例2.1において作製された遺伝子交換ベクターpSacHR06ptsGまたはpSacHR06sacC各々を混合した後、2.5kV、25μF、400ohmsの条件でelectroporationを行うことで、前記遺伝子交換ベクターを培養されたM.サクシニシプロデュセンスMsacC(M. succiniciproducens)に各々導入させた。電気衝撃後、LB−グルコース液体培地1mLを加えて、200rpm、37℃の振盪培養器で1時間培養した。培養液を、抗生剤クロラムフェニコール(最終濃度6.8μg/L)を含有したLB−グルコース固体培地に塗抹して、37℃で48時間以上培養した。形成されたコロニーの中から、ダブルクロスオーバー(double crossover)だけが起きたものを選び出すため、これをクロラムフェニコール(最終濃度6.8μg/L)を含有したLB−スクロース(100g/Lのスクロースを含有したLB培地)培地にストリーキング(streaking)した後、24時間後に形成されたコロニーを同じプレートに再びストリーキング(streaking)した。
【0084】
前記プレートで形成されたコロニー(変異菌株)を抗生剤が含まれたLB−グルコース液体培地で培養して、培養された菌株からRochelle等による方法(Rchelle et al., FEMS Microbiol. Lett., 100:59, 1992)を応用して、ゲノムDNAを分離した。前記分離した変異株のゲノムDNAを鋳型にしてPCRを行った後、取得されたPCR産物を電気泳動することによってptsG及びsacCの欠失の有無を各々の変異菌株において確認した。
【0085】
MptsG菌株からptsGの欠失の有無を確認するため、下記のように配列番号:37と38のプライマーを利用したPCRと、配列番号:39と40のプライマーを利用したPCRによって確認し、MsacC菌株においてsacCの欠失の有無を確認するため、配列番号:39と40のプライマーと、配列番号:41と42のプライマーを利用したPCRによって確認した。
【0086】
配列番号:37:5'−CGGGGCGAAAGTGATTGAGA
配列番号:38:5'−AATTGCCGCCTGGGTATTGG
配列番号:39:5'−ACCTTTACTACCGCACTGCTGG
配列番号:40:5'−GCGGGAGTCAGTGAACAGGTAC
配列番号:41:5'−GATCTTGAGTCCGTAAAACAGGCTT
配列番号:42:5'−TTCCGCTCAAGCCATTGTAGTG
【0087】
2.3:MptsG菌株、MsacC菌株、及び母菌株(MBEL55E)における生長比較
前記実施例2.2において作られた遺伝子組み換えMptsG、MsacC菌株をBHI(Bacto(商標)Brain Heart Infusion; Becton, Dickinson and Company, Sparks, MD)培養培地で8時間ほど培養した後、10mLを300mL MH5S培養培地(liter当たり:2.5g 酵母エキス、2.5g ポリペプトン、1g NaCl、8.7gK2HPO4、10gNaHCO3、0.02gCaCl22H2O、0.2gMgCl26H2O 及び10gスクロース)に接種した後、同様の条件で培養したこれらの母菌株であるMBEL55Eの生長曲線と比較した。図3はOD600で測定したMBEL55E(●)、MptsG(▲)、及びMsacC(△)菌株の生長曲線である。図3に示したように、母菌株(MBEL55E)を有していたスクロース培地上における成長能力が各々の遺伝子を除去することで、殆ど消えることが示され、これは各々の遺伝子がスクロース培地上において生長するのに必須であることを意味する。
【0088】
一方、フラクトキナーゼ(RbsK、MS1233)をコードするrbsK遺伝子の場合には前記同様の実験を行った、増殖が遅くなるなどの成長の変化は観察されなかった。また、酵素活性測定結果においても母菌株とrbsK除去菌株との間に大きい差はなく、両方とも一般的なフラクトキナーゼの活性に比べて、非常に低いレベルであり、即ち、値を無視できるほどの活性が測定された。このような根拠として、rbsK遺伝子はフラクトキナーゼをコードしなかったか、フラクトキナーゼをコードしたとしても非常に弱い活性を有すると推定される。
【0089】
2.4:MptsG及びMsacC菌株と母菌株の該当酵素活性比較
スクロースPTSの活性測定のためにJacobsonら(J. BIOL. CHEM., 254:249, 1979)とLodgeら(Infect. Immun., 56:2594, 1988)による方法を導入した。
【0090】
先ず、細胞を透過(permeabilize)させるために、OD600値が1.2近傍の細胞培養液1mLをTDMバッファー(50mM Tris/HCl、10mM MgCl2、1mM DTT;pH7.5)で洗浄した後、同じバッファー1mLに再懸濁した後、0.01mLトルエンを加えて45秒間強く攪拌(agitation)。12,000×gで遠心分離して集めた細胞をTDMバッファーで2回洗浄した。このような過程をもう一度繰り返して、最終的にTDMバッファー50μLに再懸濁することで、透過性細胞(permeabilized cell)を準備した。PEP−依存的スクロース燐酸化は、全反応体積100μLに25mM Tris/HCl(pH8.0)、1mM DTT、5mM MgCl2、10mM KF、1μCi[U−14C]スクロースに5μLの透過させた前記細胞懸濁液を加えた状態で、1mM PEPを含めるものと含まないものの2種類の反応の差を測定して成された。
【0091】
前記混合液は37℃で10分間反応し、1mLの冷たい水を加えることで反応を停止させた。最終反応物はろ過装置上のDEAEフィルターディスク(Whatman DE81)を通過させて、10倍体積の冷たい水を加えて洗浄した後、公知の方法でBeckman LS6500 liquid scintilation counter(Beckman, Ramsey, MN)を利用して、放射活性を測定した。スクロース−6−リン酸ヒドロラーゼの活性はMartinら(Appl. Environ. Microbiol., 53:2388, 1987)による方法に若干修正を加えて測定した。OD600値が1近傍の細胞培養液20mLを前記スクロースPTS活性測定時のような方法で透過(Permeabilize)させて、最終的に1mLのTDMバッファーに再懸濁して準備した。全反応物の体積は300μLであり、50mM Sodium-Potassium Phosphateバッファー(pH7.2)、5mM MgCl2、4mMスクロース、0.8mM NADP、6.4Uのグルコース−6−ホスフェート デヒドロレース(glucose-6-phosphate dehydrolase)、そして、30μLの透過性細胞(permeabilized cell)を加えた状態で、10mM PEPを含めたものと含めないもの2種類の反応の差を測定した。
【0092】
各々のptsG及びsacC遺伝子が除去された変異菌株とその母菌株との活性を測定した結果を表2に示す。この結果から、MS0784(ptsG)がコードするスクロースPTS酵素がPTS活性を有することを意味し、MS0909(sacC)がコードするスクロース−6−リン酸ヒドロラーゼ酵素が各々細胞内において該当機能を有することを意味する。
【0093】
【表2】
a BHI, Bacto(商標) Brain Heart Infusion(Becton, Dickinson and Company, Spark, MD)、MH5S培地組成はリッター当り:2.5g酵母エキス、2.5gポリペプトン、1gNaCl、8.7gK2HPO4、10gNaHCO3、0.02gCaCl2・H2O and 0.2gMaCl2・6H2O及び10gスクロースを含む。
b スクロースホスホトランスフェラーゼの活性は液体シンチレーションカウンターによって測定されたcpm(count per minute)値を、mU/mg unit単位に転換して表記した。
c 相対的活性度は、*表記されたものを100として、残りの値を計算した。
【0094】
[実施例3]スクロース代謝に関与する酵素をコードする新しい遺伝子の分離
前記実施例2の結果を基に、スクロース代謝能があると確認されたsacCをはじめとして、β−フルクトフラノシダーゼをコードする遺伝子を下記のような方法で各々マンヘミア、大腸菌、及びバシラス・サチリスから分離した。
【0095】
3.1:マンヘミア由来スクロース−6−リン酸ヒドロラーゼをコードする遺伝子の分離
先ず、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC0769BP)のゲノムDNAを鋳型にして、配列番号43及び44のプライマーを使ってPCRを行うことによってsacC(MS0909)遺伝子のDNAを増幅した。前記sacC(MS0909)はβ−フルクトフラノシダーゼ(スクロース−6−リン酸ヒドロラーゼ)をコードする遺伝子(配列番号4)である。
【0096】
配列番号43:5'−ACTGAGCCATGGCGAAAATCAATAAAGTAGATC−3'
配列番号44:5'−TGATCCGAGCTCCTATTATTCCAGTGTTCCCGCC−3'
【0097】
3.2:大腸菌由来インバターゼをコードする遺伝子の分離
E. coli WのゲノムDNAを鋳型にして、配列番号45及び46のプライマーを使ってPCRを行うことでcscA遺伝子のDNAを増幅した。前記cscAはβ−フルクトフラノシダーゼ(インバターゼ)をコードする遺伝子(配列番号8)である。
【0098】
配列番号45:ACTCCGGAATTCATGACGCAATCTCGATTGCA
配列番号46:ACCTGCGAGCTCCCGTTGTTCCACCTGATATTATG
【0099】
3.3:バシラス・サチリス由来スクロース−6−リン酸ヒドロラーゼをコードする遺伝子の分離
バシラス・サブチリス(Bacillus subtilis)のゲノムDNAを鋳型にして、配列番号47及び48のプライマーを使ってPCRを行うことによってsacA遺伝子のDNAを増幅した。前記sacAはβ−フルクトフラノシダーゼ(スクロース−6−リン酸ヒドロラーゼ)をコードする遺伝子(配列番号10)である。
【0100】
配列番号47:GCATAGAATTCATGACAGCACATGACCAGGAGCT
配列番号48:GCATAGAGCTCCTACATAAGTGTCCAAATTCCGACATTC
【0101】
[実施例4]遺伝子組み換えベクターの作製
4.1:pTac15Kの準備
pKK223−3(Pharmacia Biotech., Uppsala, Sweden)のtrcプロモーターとトランスクリプションターミネーター(trascription terminator)部分をpACYC177(NEB, Beverly, MA, USA)に挿入して作製した。pTac15Kは構成的(constitutive)発現のための発現ベクターであり、図5の切断地図に示された構成を有している。
【0102】
4.2:遺伝子組み換えベクターpTac15KsacCの作製
マンヘミア由来のβ−フルクトフラノシダーゼ(スクロース−6−リン酸ヒドロラーゼ)をコードする遺伝子sacC(MS0909)を大腸菌に発現させるため、発現ベクターを次のように作製した。
【0103】
実施例3.1において増幅されたsacC(MS0909)遺伝子を含むPCR断片をEcoRIとSacIで切断して、これを同じ切断酵素で切断したpTac15Kと結合させる公知の分子工学的手法に従って、pTac15KsacCを作製した(図6)。そして、下記配列番号49乃至52のプライマーを利用してソルジェント(Solgent, Korea)にシーケンシングを依頼して、塩基配列の一致の有無を確認した。
【0104】
配列番号49:5'−CCCGTTCTGGATAATGTTTT−3'
配列番号50:5'−AAAGTCACGGTTGTTATTCC−3'
配列番号51:5'−CATTTAATGCCGCTCATATT−3'
配列番号52:5'−ACCGCTCAATTATTGAGATT−3'
【0105】
4.3:遺伝子組み換えベクターpTac15KEWcscAの作製
実施例3.2において得られたDNA断片をEcoRIとSacIで処理して、同じ酵素で処理したpTac15Kと結合させる公知の分子工学的手法に従って、pTac15KEWcscAを作製した。
【0106】
4.4遺伝子組み換えベクターpTac15KBSsacAの作製
実施例3.3において得られたDNA断片をEcoRI及びSacIで処理して、同じ酵素で処理したpTac15Kと結合させる公知の分子工学的手法に従って、pTac15KBSsacAを作製した。
【0107】
[実施例5]遺伝子組み換え菌株の作製
5.1:Escherichia coli W3110 pTac15KsacC菌株の作製
スクロースを代謝できないと知られたE. coli K-12菌株の亜株(substrain)であるE.coli W3110をモデル微生物として下記の実験を行った。前記E.coli W3110をLB固体培地に塗抹して、8時間37℃で培養した後、コロニーをLB液体培地10mLに接種して、8時間培養した。前記培養液をLB液体培地100mLに1%(v/v)接種して、37℃の振盪培養器で培養した。
【0108】
約2時間後、OD600が0.30〜0.35程度になると、氷に20分間放置して、細胞の成長を停止させた。4℃、3,000rpm、15分の条件で遠心分離して、細胞を取得した後、4℃状態のRFI溶液32mLで細胞を懸濁した。そして、前記懸濁液を4℃、3,000rpm、15分の条件で遠心分離して細胞を取得した。これをRFII溶液8mLで細胞を再懸濁した後、氷に15分間放置した。最終的に前記再懸濁液を100μLずつ分株して−70℃で保管した。RFIの組成は100mM RbCl、50mM MnCl2−4H2O、0.1M CH3COOK、10mM CaCl2及び15%(w/v)グリセロールで構成され、0.2M酢酸を添加してpHを5.8に合わせた。RFIIは10mM MOPS、10mM RbCl、100mM CaCl2、及び15%(w/v)グリセロールで構成され、NaOHを添加することによってpHを6.8に合わせた。
【0109】
このように準備されたE. coli W3110菌株に実施例2.2において作製された発現ベクターpTac15KsacC、または対照群でpTac15K(Pharmacia Biotech., Uppsala, Sweden)を混合した後、42℃で90秒間ヒートショックを与えることによって形質転換を行った。ヒートショック後、LB液体培地0.8mLを加えて、37℃の振盪培養器で1時間培養した。
【0110】
培養液を抗生剤カナマイシン最終濃度50μg/L)を含有したLB固体培地に塗抹して、37℃で12時間以上培養した。形成されたE. coli W3110 pTac15K及びE. coli W3110 pTac15KsacCコロニーを、LB液体培地に接種して、37℃で8時間以上培養した。ベクターが上手く導入されたかを確認するために前記培養された菌株から、GeneAll(商標) Plasmid SV(GeneAll Biotechnology, Korea)を利用して、ベクターを分離して、電気泳動によって確認した。
【0111】
5.2:E. coli W3110 pTac15KEWcscAとE. coli W3110 pTac15KBSsacA菌株の作製
実施例5.1に記載された方法と同じ方法で、スクロースを代謝できないと知られたE. coli K-12菌株の亜株(substrain)のE. coli W3110を実施例4.3及び4.4において作製されたpTac15KEWcscA及びpTac15KBSsacAを利用して形質転換させて、ベクターが上手く導入されたのか電気泳動によって確認した。
【0112】
[実施例6]遺伝子組み換え大腸菌のスクロース代謝能及び成長確認
前記実施例5.1において作製された固体培地上のE. coli W3110 pTac15KsacC菌株とE. coli W3110 pTac15Kのコロニーを10mLのLB培地に接種して、37℃で8時間培養した後、さらにこれを10g/Lのスクロースを含んだ100mLのM9最小培地に5%(v/v)接種した後、37℃で培養した。この時、抗生剤として最終濃度50μg/Lのカナマイシンを添加した。前記LB培地は10g/Lのトリプトン、10g/LのNaCl、及び5g/Lの酵母エキスで構成され、M9最小培地は33.9g/LのNa2HPO4、15g/LのKH2PO4、2.5g/LのNaCl、5g/LのNH4Cl、及び0.36g/LのMgSO4で構成された。培養液内の細胞濃度は分光光度計を利用してOD600の値で測定した。培養中、周期的にサンプルを採取し、採取された試料は13,000rpmで5分間遠心分離した後、上澄み液のスクロース及び代謝産物の濃度を液体クロマトグラフィー(HPLC)で分析した。
【0113】
その結果、図7、図8及び表3に示したように、E. coli W3110 pTac15K菌株の場合、スクロースを単一炭素源として含んだM9最小培地で全く生長できないが、E. coli W3110 pTac15KsacC菌株はスクロースの優れた代謝力を示した。E. coli W3110 pTac15KsacC菌株は17時間11.08g/Lのスクロースを代謝して、OD600基準に3.71のバイオマスの増加を示した。これは通常の大腸菌のOD600と乾燥細胞重量(dry cell weight:DCW、g/L)の換算係数(conversion factor:1 OD600 = 0.37g/L DCW)を考慮すると、約1.37g/Lのバイオマスに該当する量である。1.42g/Lの酢酸が生成され、2.01g/Lのグルコース及び4.51g/Lのフルクトースが残って、これらの二つの糖類は時間と伴って次第に消耗されることが分かる。
【0114】
【表3】
【0115】
W3110 pTac15K菌株の成長グラフにおいて(図7)、接種後8時間目にODが若干増加したことは、複合培地である接種培地(5%(v/v)接種する)に入っていた成分によるものと考えられ、LC分析結果、スクロースを分解することも、またスクロースを単独炭素源として成長することもできないことを示す。
【0116】
また、前記実施例5.2において形質転換された遺伝子組み換え菌株E. coli W3110 pTac15KEWcscAとE. coli W3110 pTac15KBSsacAをスクロースを単独炭素源として、前記E. coli W3110 pTac15KsacC菌株と同じ方法で培養した結果、図9と同様に、優れた成長能を示し、E. coli W3110 pTac15KEWcscAは初期スクロース7.77g/Lから47時間後1.82 g/Lに減少することを確認し、E. coli W3110 pTac15KBSsacAは初期8.49g/Lから47時間後7.98 g/Lに減少することを確認し、これらがスクロースを代謝して成長する可能性があることを確認した。
【0117】
[実施例7]遺伝子組み換え大腸菌を利用してスクロースを炭素源とする代謝産物生産
先に述べたように、M.サクシニシプロデュセンス(M. succiniciproducens)由来のスクロース−6−リン酸ヒドロラーゼの導入はスクロースを炭素源として成長できない微生物をスクロース単独炭素源として成長可能にした。即ち、スクロースを炭素源として活用できない微生物を、スクロースを単独炭素源として最小培地上で培養可能にすることで、既にグルコースなどを炭素源とする多くの一次、二次代謝産物、遺伝子組み換え蛋白質、生分解性高分子などの多くのバイオ製品生産システムなどをそのまま本発明に適用することができる。
【0118】
そこで、本実施例においては嫌気条件でスクロースを活用して、多様な代謝産物を生産する例を示す。
【0119】
前記実施例5.1において作製されたE. coli W3110 pTac15KsacC菌株を10mLのLB培地に接種して、37℃で8時間培養した後、さらにこれを200mLのLB培地に接種して、37℃で8時間培養し、これを容積2.5Lの発酵槽(New Brunswick System, BioFlo 3000)に接種した。発酵槽の培養培地は20g/Lスクロースを含んだR/2最小培地で構成され、操業条件はpH6.8、37℃、200rpmに100% CO2を0.5vvmの速度で注入した。抗生剤として最終濃度50μg/Lのカナマイシンを添加した。R/2培地は6.75g/LのKH2PO4、2g/Lの(NH4)2HPO4、0.85g/LのC6H8O7H2O、及び0.7g/LのMgSO47H2Oと5mL/Lの微量金属溶液(trace metal solution:10g/L FeSO47H20、2g/L CaCl2、2.2g/L ZnSO47H2O、0.54g/L MnSO45H2O、1g/L CuSO45H2O、0.1g/L NH4Mo7O247H20、0.02g/L Na2B4O710H2O、及び5mL HCl)で構成された。培養液内の細胞濃度は分光光度計を利用してOD600の値で測定し、周期的に採取された試料は13,000rpmで5分間遠心分離した後、上澄み液のスクロース及び代謝産物の濃度を液体クロマトグラフィー(HPLC)で分析した。
【0120】
その結果、図10及び表4に示したように、酢酸、ギ酸、乳酸、コハク酸、及びエタノール(acetic, formic, lactic, and succinic acid and ethanol)を成功的に生産することができた。前記のような嫌気条件で52.5時間22.13g/Lのスクロース及びこれから由来した糖類をすべて消耗し、その結果4.49g/Lの酢酸、3.74g/Lのギ酸、4.19g/Lの乳酸、5.10g/Lのコハク酸、2.66g/Lのエタノールをスクロース基準に基質当たり0.20、0.17、0.19、0.23、及び0.12 g/gスクロースの収率で生産し、これらの収率の合計は0.91g/gスクロースとして理論値に近接した相当高い収率を示した。また、培養開始後52.5時間のOD600 = 2.7であるため、これは前記に言及した換算係数を考慮すると、gDCW = 0.999に該当するため、菌体重さ当たり収率である比収率(specific yield)は表2のようになる。これは、前記のスクロース−6−リン酸ヒドロラーゼの単独導入により、スクロースを利用して、多様な代謝産物を高収率と高比収率を示しており、成功的に生産できることを示す。
【0121】
【表4】
【0122】
[実施例8]トレオニン(Threonine)生産菌株への適用による代謝工学的に改良された有用バイオ製品生産への応用
代謝工学的に改良された、遺伝子設計された(genome-engineered)または遺伝子組み換え大腸菌菌株にも本発明の遺伝子を導入することで、グルコースあるいは既に使われていた他の炭素源の代わりにスクロースを活用して、生産可能である例として下記のようなトレオニン生産菌株を利用して実験を行った。具体的に、大腸菌(Escherichia coli)W3110を基盤に代謝工学的に改良された遺伝子設計された(genome-engineered)及び遺伝子組み換え菌株のTH28C pBRThrABCR3菌株(Lee et al., Molecular. Systems. Biology., 3:149, 2007)をモデルにして、下記の実験を行った。
【0123】
実施例5において説明した方法と同じ方法で前記TH28C pBRThrABCR3菌株を、実施例4において作製したスクロース代謝能を持たせるプラスミドpTac15KsacCで形質転換させて、TH28C pBRThrABCR3 pTac15KsacCを作製した。
【0124】
前記作製されたTH28C pBRTHrABCR3 pTac15KsacC菌株を10mLのLB培地に接種して、31℃で12時間培養した後、さらにこれを50mLのLB培地に接種して、31℃で12時間培養したし、これを容積2.5Lの発酵槽(New Brunswick System, BioFlo 3000)に接種した。発酵槽の培養培地は20g/Lスクロースを含んだTPM2培地で構成され、操業条件はpH6.5、31℃、溶存酸素濃度は1vvmのAirの供給と自動的にrpmを1000まで上げられるように調節することによって40%以上の空気が飽和された状態になるよう維持した。抗生剤としては最終濃度30μg/Lのクロラムフェニコール、40μg/Lのカナマイシン、50μg/Lのアンピシリンを添加した。TPM2培地は2g/Lの酵母エキス、2g/LのMgSO47H2O、2g/LのKH2PO4、10g/L(NH4)2SO4及び0.2g/L L−メチオニンと0.2g/L−リジンと0.05g/LのL−イソロイシンと、10mL/Lの微量金属溶液(trace metal solution:10g/L FeSO47H2O、2g/L CaCl2、2.2g/L ZnSO47H2O、0.54g/L MnSO45H2O、1g/L CuSO45H2O、0.1g/L NH4Mo7O247H2O、0.02g/L Na2B4O710H2O、及び5mL HCl)で構成された。培養液内の細胞濃度は分光光度計を利用してOD600の値で測定し、周期的に採取された試料は13,000rpmで5分間遠心分離した後、上澄み液のスクロース及び代謝産物の濃度を液体クロマトグラフィー(HPLC)で分析した。アミノ酸の場合、5mLの培養液を遠心分離とろ過過程を経た後、Science Lab Center Co.(Daejeon, Korea)に依頼して、陽イオン分離カラム(caution seperation column, LCA K06/Na1.6150mm; SykamGmbH, Eresing, Germany)によって分離した後、Sykam S433アミノ酸分析機を活用して分析した。
【0125】
その結果、表5に示したように、スクロースを活用して、4.7g/Lのトレオニンを成功的に生産することができた。
【0126】
【表5】
【0127】
前記に示したように本発明のβ−フルクトフラノシダーゼ遺伝子を導入することだけでも、スクロースを代謝する能力を備えることと共にスクロースを炭素源として活用して、酢酸、ギ酸、乳酸、コハク酸、エタノールなどの多様な代謝産物の生産を可能にした。また、代謝工学的に改良された菌株にも同様の方法で導入して、目的代謝産物、即ち、本例ではトレオニンを成功裡に生産可能だった。それ以外にも生分解性高分子、遺伝子組み換え蛋白質の生産にも本発明を当業者は容易に適用可能である。
【産業上利用の可能性】
【0128】
以上、詳細に説明したように、高価なグルコースの代わりに低価格のスクロースを炭素源として使用できる遺伝子組み換え微生物を提供する効果がある。従来スクロースを炭素源として利用できなかった微生物の培養においてもグルコースをはじめとする他の炭素源をスクロースに代えることは可能である。本発明においては安価であり、自然界に豊富に存在するスクロースを炭素源として効率よく使用可能にする新しい酵素群を同定して開発することによって今後微生物発酵を介する有用な化学物質の製造や医療用遺伝子組み換え蛋白質生産において、スクロースをその炭素源としてより効率的かつ経済的に生産するのに利用するであろう。特に、スクロースはそれ自体で細胞内の蛋白質を安定化させたり、細胞の溶解を極力抑制させる役割を果たすと知られているため、高濃度の基礎化学物質生産や高濃度細胞培養時にさらに有効活用される。
【0129】
以上で本発明内容の特定部分を詳細に記述したが、当業界の通常の知識を有する者において、このような具体的技術は単に望ましい実施様態に過ぎなく、これによって、本発明の範囲が制限されるのではない点は明らかであろう。従って、本発明の実質的な範囲は添付された請求項とその等価物によって定義される。
【技術分野】
【0001】
本発明はスクロース代謝能を有する遺伝子組み換え微生物に関し、より具体的には、スクロースホスホトランスフェラーゼ及び/またはスクロース−6−リン酸ヒドロラーゼをコードする遺伝子が導入されているスクロース代謝能を有する遺伝子組み換え微生物またはβ−フルクトフラノシダーゼをコードする遺伝子が導入されたスクロース代謝能を有する遺伝子組み換え微生物に関する。
【背景技術】
【0002】
人類の持続可能な開発(sustainable development)のために再生可能な生物資源から有用な物質を生産するための産業生命工学への高い関心と共にこれを発展させる研究が活発に行われている。現在までの微生物発酵を介した化学物質生産は、グルコースを主原料物質に使用して成されているが、グルコースの高価格によって、原油を主原料物質として使う化学合成法を介する生産価格より発酵による生産価格が高く、微生物発酵を介する化学物質生産の商用化には困難がある。従って、高価なグルコースを炭素源として利用する方法から脱皮して、豊富な生物資源から容易に得られる多種の安価な炭素源を探し出すための研究が活発に行われている。例えば、木質系加水生成物、グリセロール、ホエー(whey)、コーンスティープリカー(corn steep liquor)などの比較的安価な原料物質を炭素源として使って、多様な一次代謝産物を生産するための研究が本発明者等を含み、多くの研究者によって行われてきた(Samuelov et al., Appl. Environ. Microbiol., 65:2260, 1999; Lee et al., Appl. Microbiol.Biotechnol., 54:23, 2000; Lee et al., Biotechnol.Bioeng., 72:41, 2001; Lee et al., Biotechnol. Lett., 25:111, 2003; Lee et al., Bioproc. Biosystems Eng., 26:63, 2003)。しかし、現在まで報告された研究結果によると、グルコースの代りに前記原料物質を炭素源として使った場合には、目的代謝産物の生産性あるいは生産収率がグルコースを炭素源として使った場合より顕著に低く、これを克服するための研究開発が切に求められている。
【0003】
砂糖として広く知られるスクロースはグルコースとフラクトースで形成された二糖類の一つであり、光合成能を有する全ての植物から生産されて、自然界に非常に豊富に存在する炭素源である。特に、サトウキビとサトウダイコンは過量のスクロースを含有しており、現在全世界のスクロースの60%以上がサトウキビから生産されている。何よりもスクロースはサトウキビの機械的な圧搾で得られた抽出液の単純な蒸発/濃縮工程を介して産業的に生産が可能で、生産価格が非常に安い。Koutinasらは2004年に微生物を利用した化学物質生産に使用可能な多様な原料物質の価格をグルコース含有量を基準に計算し、グルコース含有量1キログラムを基準にスクロースの価格は26.1セントとして、これは小麦、糖蜜、及びグルコースの価格に対して、77%、50%、28.9%に過ぎない非常に安い価格である(Koutinas et al., Ind. Crops and Products, 20:75, 2004)。
【0004】
International Sugar Agreement(ISA) daily price報告書は1994〜1995年を頂点にスクロースの価格が供給過剰によって下降安定傾向を維持しており、このような下落傾向は持続すると展望している。従って、スクロースは微生物発酵によって多様な化学物質生産に現在使われている高価なグルコースを代えられる最も有力な炭素源として注目されている。特に、主にトウモロコシ澱粉から生産されているグルコースの場合には持続的なトウモロコシ価格の上昇と共に、トウモロコシからの澱粉抽出、熱的/化学的澱粉前処理、酵素反応によるグルコースへの転換、及びこれの精製など、非常に複雑な工程を経て生産されるためグルコース生産単価をスクロースレベルに下げるのは非常に困難であることを当業界の従事者は周知している。このような理由から最近米国におけるトウモロコシ基盤バイオエタノール生産は次第に減少しているが(Mae-Wan Ho, Science in Society, 33:40, 2007)、ブラジルのサトウキビ、即ちスクロース基盤バイオエタノール生産工程は急激な成長を示している。
【0005】
現在までのスクロースを炭素源として利用した有用な物質生産に関する研究は高濃度細胞培養を介する生分解性高分子(Lee et al., Biotechnol. Lett., 15:971, 1993; Lee et al., Biotechnol. Techniques, 1:59, 1997)、クエン酸(Forster et al., App. Microbiol.Biotechnol., 75:1409, 2007)、アセトン、ブタノール、エタノール、イソプロパノール(George et al., Appl. Environ. Microbiol., 45:1160, 1983; Durre, Appl. Microbiol.Biotechnol., 49:639, 1998)、イタコン酸(Kautola et al., Biotechnol. Lett., 11:313, 1989)、キサンタンガム(Letisse et al., Appl. Microbiol.Biotechnol., 55:417, 2001)等を中心に進められてきた。特に、国際バイオエネルギー及びバイオ燃料市場を分析する国際エネルギーエージェンシー(International Energy Agency)の2006年のバイオエネルギータスク40(Bioenergy Task 40)報告書は、ブラジルのスクロースを含むサトウキビからバイオエタノール生産工程を環境に優しく持続可能なバイオ燃料生産の優れたモデルとして高く評価した。
【0006】
微生物発酵による化学物質生産のための原料物質としてスクロースは価格が安いだけでなく、スクロース自体が過量の目的代謝産物を含有する外部環境から細胞膜を完全に維持させる役割を果たすことによって、高濃度の目的代謝産物を生産できる優秀な機能を有している。Kilimann等(Biochimica et Biophysica Acta, 1764, 2006)はスクロースを含んだ培養培地とこれを含まない培養培地に、致死的な温度で微生物を露出させた後、それらの生存率と蛋白質の2次構造を調べた。その結果、スクロースを含んだ培養培地に露出した微生物の細胞内に存在する蛋白質の2次構造は保存状態が非常に良好であったが、スクロースを含まない培養培地に露出した微生物の細胞内の蛋白質構造は非常に変性されていたことを確認した。特に、スクロースを含んでいる培養培地に露出した微生物の生存率はスクロースを含まない培養培地に露出した微生物の生存率と比較して、非常に高かった。これは有害な外部環境に対するスクロースの微生物保護機能を直接的に証明するものである。
【0007】
前記において記述したスクロースの安い価格と外部環境に対する微生物保護機能という原料物質として優秀な長所を有しているにも拘らず、実際にスクロースを炭素源として使って、微生物発酵を介する目的化学物質生産に成功し、実際に商業化した例は、未だに報告されていない。これは多数の微生物がスクロースを細胞内に輸送した後、これを分解して、解糖過程につなげるメカニズムを完全に有しないか、あるいは不完全に有していることでスクロースを炭素源として利用できないためである。たとえスクロースを利用できるメカニズムを有している微生物の場合にも炭素源としてこれを摂取して分解する速度が非常に遅いため、目的代謝産物を効率よく生産できない。
【0008】
微生物発酵を介する多様な化学物質生産が産業的に経済性を有するためには前記に記述したとおりスクロースのように価格が安い原料物質の使用が求められ、さらにスクロースを炭素源として効率よく迅速に摂取してこれを分解できる酵素の同定及びこれを利用できる研究開発が必要である。特に、微生物発酵を介する化学物質生産において原料物質の価格が最終生産価格の約50%を占める事実を考慮した場合、安い原料物質であるスクロースを効率よく利用させるようにする酵素の同定及びこれの応用開発が非常に急がれている。
【0009】
現在まで高濃度細胞培養を通した生分解性高分子、クエン酸、イタコン酸、アセトン、ブタノール、エタノール、イソプロパノール、及びキサンタンガムなどの多様なバイオ製品の生産にスクロースを活用することができる例が報告されている。しかし、実際にスクロースを炭素源として使って、微生物発酵を介する目的化学物質生産に成功して、実際に商業化した例は多くない。
【0010】
従って、環境に優しいグリーン成長の核心技術として微生物発酵を介する多様な化学物質生産が産業化に成功するためには、スクロースのような安価でかつ豊富に存在する炭素源を利用して、これを効果的に摂取して分解できる微生物の開発が必要である。
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の主な目的は、スクロースを炭素源として使えるようにする新規スクロース代謝関連遺伝子、及びこれによってコードされる酵素を提供することである。
【0012】
本発明の他の目的は、前記スクロース代謝関連遺伝子が導入されているスクロース代謝能を有する遺伝子組み換え微生物、及びこれを利用した代謝産物、生分解性高分子または遺伝子組み換え蛋白質の製造方法を提供することである。
【課題を解決するための手段】
【0013】
前記目的を達成するために、本発明は配列番号1のアミノ酸配列を有するスクロースホスホトランスフェラーゼ、これをコードする遺伝子(ptsG)、並びに前記遺伝子(ptsG)及びスクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)を含有する遺伝子組み換えベクターを提供する。
【0014】
本発明はまた、前記遺伝子が細菌、酵母及びカビから構成された群より選択された宿主細胞に導入されて、スクロース代謝能を有する遺伝子組み換え微生物、及びこれをスクロースを炭素源として含有する培地で培養することを特徴とする代謝産物、生分解性高分子、または遺伝子組み換え蛋白質の製造方法を提供する。
【0015】
本発明はまた、β−D−フルクトフラノシド結合を加水分解して、フラクトースを遊離する活性を有するβ−フルクトフラノシダーゼ及びこれをコードする遺伝子を提供する。
【0016】
本発明はまた、前記遺伝子が細菌、酵母及びカビから構成された群より選択された宿主細胞に導入されて、スクロース代謝能を有する遺伝子組み換え微生物及びこれをスクロースを炭素源として含有する培地で培養することを特徴とする代謝産物、生分解性高分子、または遺伝子組み換え蛋白質の製造方法を提供する。
【0017】
本発明の他の特徴及び具現例は次の詳細な説明及び添付された特許請求範囲からより一層明白になるであろう。
【図面の簡単な説明】
【0018】
【図1】スクロースを代謝可能にするM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来の新規スクロース代謝関連酵素、スクロースホスホトランスフェラーゼ、スクロース−6−リン酸ヒドロラーゼ及びフラクトキナーゼをスクロースを代謝できない微生物に導入した時、スクロースが摂取されて分解され、解糖過程を介して、代謝される経路を示した模式図である。太い矢印(→):導入したM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来の新規スクロース代謝関連酵素によって制御される代謝経路を示し、細い矢印(→):遺伝子組み換え微生物が本来有している解糖過程の代謝経路を示す。
【図2】スクロースを代謝可能にするM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来の新規β−フルクトフラノシダーゼを、スクロースを代謝できない微生物に導入した時、この酵素がスクロースの代謝にどのように係わるのかを示した4種の可能な経路を示した模式図である(太い矢印(→):導入したM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来の新規β−フルクトフラノシダーゼが係わる可能性がある4種の可能な経路を示し、細い矢印(→):遺伝子組み換え微生物が本来有している糖類分解過程の代謝経路を示す)。
【図3】スクロースホスホトランスフェラーゼ、スクロース−6−リン酸ヒドロラーゼ及びフラクトキナーゼをコードする遺伝子(ptsG、sacC、rbsK)を含有する遺伝子組み換えベクターpMSscrIIAの地図である。
【図4】母菌株MBEL55E、遺伝子組み換えMptsG、遺伝子組み換えMsacC菌株のスクロース培地上における成長を示したグラフである(●:MBEL55E、▲:MptsG、及び△:MsacC)。
【図5】遺伝子組み換えベクターpTac15Kの切断地図である。
【図6】スクロース−6−リン酸ヒドロラーゼをコードする遺伝子を含有する遺伝子組み換えベクターpTac15KsacCの切断地図である。
【図7】遺伝子組み換えE. coli W3110 pTac15Kのスクロース含有M9培地における成長を示すグラフである(●を含む実線:スクロース濃度;◆を含む実線:OD600)。
【図8】本発明によるスクロース代謝能を有する遺伝子組み換えE. coli W3110 pTac15KsacCのスクロース含有M9培地における生長グラフである(●を含む実線:スクロース濃度;◆を含む実線:OD600;●を含む点線:グルコース濃度;○を含む実線:フラクトース濃度;▼を含む点線:酢酸濃度)。
【図9】E. coli W3110 pTac15KEWcscA及びE. coli W3110 pTac15KBSsacAのスクロース含有M9培地における成長を示すグラフである(●を含む実線:E. coli W3110 pTac15KEWcscA;▲を含む実線:E. coli W3110 pTac15KBSsacA)
【図10】本発明によるスクロース代謝能を有する遺伝子組み換えE. coli W3110 pTac15KsacCを嫌気条件で発酵した成長、及び代謝産物生産を示すグラフである(●を含む実線:スクロース濃度;△を含む実線: OD600;▼を含む実線:グルコース濃度;■を含む実線:フラクトース濃度;●を含む点線:コハク酸濃度;▽を含む点線:乳酸濃度;■を含む点線:ギ酸濃度;◇を含む点線:酢酸濃度;▲を含む点線:エタノール濃度)
【発明を実施するための形態】
【0019】
本発明者は安価でかつ豊富に存在する炭素源の一つとして砂糖として広く知られたスクロースを活用したバイオ製品の生産、正確にはスクロースを炭素源として活用できる微生物、または活用できない微生物の場合においてもこれを活用できるようにする酵素を探し出して、これを活用して、多様なバイオ製品の生産に応用しようとする。
【0020】
スクロースはグルコースとフラクトースで形成された二糖類として地球上に豊富に存在する炭素源の一つである。光合成能力を有する多くの植物から生産され、特に、熱帯及び亜熱帯性作物のサトウキビとサトウダイコンは過量のスクロースを含有している。スクロースはサトウキビの機械的な圧搾で得られた抽出液の単純な蒸発/濃縮工程を介して産業的に生産可能であり、現在全世界のスクロースの60%以上がサトウキビから非常に安く生産されている。グルコース含有量を基準に、微生物の発酵の原料として使用可能な多様な原料物質の価格を計算したKoutinas等の2004年度の論文によると、グルコース含有量1kgを基準にスクロースの価格は26.1セントとしてグルコース価格の約1/4で、これは小麦、糖蜜の価格と比較しても1/2〜2/3の価格に相当する(Koutinas et al., Ind. Crops and Products, 20:75, 2004)。さらに、過去15年間International Sugar Organizationに公示されたスクロース価格の推移を調べてみると、1995年の13.28cents/1bを頂点に、1999年6.27cents/lbまで暴落し、2008年3月頃14.20cents/lbを経て、2008年5月7日の価格が12.44cents/lbに維持されており、供給過剰によって、下降安定傾向を維持すると予想している。このような価格的なメリット以外にもスクロースは穀物から得られるのではないため、最近の穀物価格の高騰のような食糧危機状況下においても安定的に供給可能な長所がある。
【0021】
現在まで高濃度細胞培養による生分解性高分子、クエン酸、イタコン酸、アセトン、ブタノール、エタノール、イソプロパノール、及びキサンタンガム(Lee et al., Biotechnol. Lett., 15:971, 1993; Lee et al., Biotechnol. Techniques, 1:59, 1997; Forster et al., App. Microbiol.Biotechnol., 75:1409, 2007; George et al., Appl. Environ. MicroBiol., 45:1160, 1983; Durre, Appl. Microbiol.Biotechnol., 49:639, 1998; Kautola et al., Biotechnol. Lett., 11:313, 1989; Letisse et al., Appl. Microbiol.Biotechnol., 55:417, 2001)等の多くのバイオ製品の生産にスクロースを活用した例が報告され、これはスクロース活用能力の向上が正に効果的なバイオ製品の生産に直接影響を与えられると予想される。
【0022】
しかし、原料としての長所とスクロース自体が有する蛋白質変性に対する保護能、外部環境に対する細胞の保護能などの種々のメリットにも係わらず(Kilimann et al.,Biochimica et Biophysica Acta, 1764, 2006)、実際にスクロースを炭素源として使って、微生物発酵による目的化学物質生産に成功して、実際商業化した例は多くない。これは多数の微生物がスクロースを代謝できるメカニズムを有していないか、有している場合にも代謝速度が遅く、目的とするバイオ製品を効果的に生産できないのがその原因の一つである。従って、環境に優しいグリーン成長の重要技術として微生物発酵による多様な化学物質生産が産業化に成功するためには、スクロースのように安価でかつ豊富に存在する炭素源を利用して、これを効果的に摂取して、分解できる微生物の開発が必要である。そのためにはスクロースを速く分解して代謝できる酵素の同定及び活用が伴わなければならない。
【0023】
現在まで幾人の研究者によって、スクロースを活用可能にする酵素群の開発がなされて来た。代表的なものが、大腸菌由来のPTS(phosphoenol pyruvate-dependent phosphotransferase system)及びnon−PTSのスクロース輸送システム(sucrose transport system)を導入して、アミノ酸生産に活用した味の素(Ajinomoto Co.)の技術が挙げられ、これはPTSまたはnon−PTS関連全体遺伝子群を導入して発明を完成したことにその特徴がある。
【0024】
しかし、本発明においては、マンヘミア・サクシニシプロデュセンスMBEL55E(Mannheimia succiniciproducens MBEL55E:KCTC 0769BP)の遺伝情報を基にスクロースを細胞内に輸送して、これを分解して解糖過程へつなげるのに関与する酵素[スクロースホスホトランスフェラーゼ(pstG、MS0784)、スクロース−6−リン酸ヒドロラーゼ(SacC、MS1233)及びフラクトキナーゼ(RbsK、MS1233)]をコードする新規遺伝子(ptsG、sacC、及びrbsK)を探し出し、これらの配列と機能を解明した。
【0025】
従って、本発明は一観点において、スクロースを細胞内に輸送し、これを分解して、解糖過程へつなげるのに関与する酵素の配列番号1のアミノ酸配列を有するスクロースホスホトランスフェラーゼ、配列番号3のアミノ酸配列を有するスクロース−6−リン酸ヒドロラーゼ及び配列番号5のアミノ酸配列を有するフラクトキナーゼに関する。本発明はまた、前記スクロースホスホトランスフェラーゼをコードする遺伝子(ptsG)、前記スクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)、及び前記フラクトキナーゼをコードする遺伝子(rbsK)に関する。
【0026】
本発明において、前記スクロースホスホトランスフェラーゼ(pstG)はスクロースをスクロース−6−リン酸に転換させながら、細胞中に輸送する役割を果たし、前記スクロース−6−リン酸ヒドロラーゼ(SacC)はスクロース−6−リン酸をグルコース−6−リン酸及びフラクトースに転換させる活性を有し、前記フラクトキナーゼ(RbsK)はフラクトースをフラクトース−6−リン酸に転換させる活性を有する。
【0027】
本発明において、前記ptsGは配列番号2の塩基配列で表されることを特徴とし、前記sacCは配列番号4の塩基配列で表されることを特徴とし、前記rbsKは配列番号6の塩基配列で表されることを特徴とする。
【0028】
具体的には、本発明においては前記ptsG及びsacC遺伝子を含有する遺伝子組み換えベクターを作製した後、これをスクロースを炭素源として使えない大腸菌に導入して、遺伝子組み換え大腸菌を作製して、前記作製された遺伝子組み換え大腸菌を単一炭素源でスクロースを含有する培地で培養した結果、スクロース代謝能を有するということを確認した。
【0029】
これは図1に示したように、スクロースが前記スクロースホスホトランスフェラーゼ(pstG)によりスクロース−6−リン酸に転換されながら、細胞中に移送されて、細胞内に移送されたスクロース−6−リン酸は前記スクロース−6−リン酸ヒドロラーゼ(SacC)によりグルコース−6−リン酸及びフラクトースに転換された後、フラクトースはフラクトキナーゼ(RbsK)によりフラクトース−6−リン酸に転換されて、解糖過程につながることと推定することができる。
【0030】
一方、本発明者等は、マンヘミア・サクシニシプロデュセンスMBEL55E(Mannheimia succiniciproducens MBEL55E:KCTC 0769BP)由来のβ−フルクトフラノシダーゼ(β-fructofuranosidase)であるスクロース−6−リン酸ヒドロラーゼ(SacC、MS0909)をはじめとして、他のβ−フルクトフラノシダーゼ(β-fructofuranosidase)をコードする新規遺伝子(cscA、sacA)を導入することによっても、スクロースを代謝できない微生物を、スクロースを代謝可能にできることを示し、本菌株を活用した多様な代謝産物の生産する例を示すことで本発明を完成した。換言すると、マンヘミア・サクシニシプロデュセンスMBEL55E(Mannheimia succiniciproducens MBEL55E:KCTC 0769BP)、E. coli W及びバシラス・サブチリス(Bacillus subtilis)の遺伝情報を基にスクロースを分解して、解糖過程につなげるのに関与する酵素β−フルクトフラノシダーゼ(β-fructofuranosidase)をコードする新規遺伝子(sacC、cscA、sacA)を探し出し、これらの配列と機能を解明したものである。
【0031】
IUBMB(International Union of Biochemistry and Molecular Biology、国際生化学・分子生物学連合(http://www.iubmb.org/)の機能に応じる酵素分類法によって付与されたEC番号(Enzyme Comission number)は酵素を区分するために最も広く活用される番号である。
【0032】
スクロース−6−リン酸ヒドロラーゼの正式名称はβ−フルクトフラノシダーゼ(β-fructofuranosidase、(EC 3.2.1.26))であり、β-D-fructofuranoside fructohydrolase、invertase、saccharase、glucosucrase、β-h-fructosidase、β-fructosidase、sucrase、maxinvert L 1000、fructosylinvertase、alkaline invertase、acid invertaseなどの他の名称が存在する(http://www.chem.qmul.ac.uk/iubmb/enzyme/EC3/2/1/26.html)。即ち、スクロース−6−リン酸ヒドロラーゼ及びスクラーゼは、EC 3.2.1.26に該当するβ−フルクトフラノシダーゼ(β-fructofuranosidase)という正式名称を有している。
【0033】
このようなβ−フルクトフラノシダーゼは、β−D−フルクトフラノシド(β-D-fructofuranoside)において、ターミナル(terminal)非還元β-D-fructofuranoside残基を加水分解する反応に係わり、その係わる反応の基質によって、スクロースを加水分解すると、スクラーゼまたはインバターゼ、スクロース−6−リン酸を基質とするとスクロース−6−リン酸ヒドロラーゼという慣用名で使われる。
【0034】
そこで、本実施例においては、マンヘミア(Mannheimia)由来のスクロース−6−リン酸ヒドロラーゼ(EC 3.2.1.26、SacC)、即ち、β−フルクトフラノシダーゼ(β-fructofuranosidase)の導入によるスクロース代謝能の証明に加えて、一般的なβ−フルクトフラノシダーゼ(β-fructofuranosidase)の導入がスクロース代謝能を持たせるようにすることを示そうと努力した結果、例えば、E. coli W菌株由来のインバターゼ(EC 3.2.1.26、CscA)とバシラス・サブチリス(Bacillus subtilis)由来のβ−フルクトフラノシダーゼ(EC 3.2.1.26、SacA)の各々を導入することで、スクロースを代謝できない微生物がβ−フルクトフラノシダーゼ(EC 3.2.1.26)を導入することでスクロースを代謝して、成長することを示す。
【0035】
従って、本発明は他の観点において、スクロースを加水分解して、グルコースとフルクトースに転換する活性、及びスクロース−6−リン酸を加水分解して、グルコース−6−リン酸及びフラクトースに転換する活性などを含む、β−D−フルクトフラノシド結合を加水分解して、フラクトースを遊離する活性を有するβ−フルクトフラノシダーゼに関する。また、前記β−フルクトフラノシダーゼは、配列番号3、配列番号7及び配列番号9のアミノ酸配列で構成された群から選択されたアミノ酸配列を有してもよいが、これに制限されるものではない。
【0036】
具体的には、本発明において、前記β−フルクトフラノシダーゼの一例であるスクロース−6−リン酸ヒドロラーゼ(sacC)は、スクロース−6−リン酸をグルコース−6−リン酸及びフラクトースに転換させる活性、またはスクロースをグルコース及びフラクトースに転換させる活性を有する。また、実施例においてはβ−フルクトフラノシダーゼはマンヘミア由来であってもよいが、その他の異なる微生物から由来したもの、また本発明の範囲に含まれ、配列番号3のアミノ酸配列を有してもよく、これをコードする遺伝子(sacC)は配列番号4の塩基配列で表されることを特徴とする。
【0037】
本発明において、前記β−フルクトフラノシダーゼの一例である、インバターゼ、スクラーゼ、スクロースヒドロラーゼ(CscA)はスクロースを分解して、解糖過程につなげるのに関与する酵素で、スクロース−6−リン酸をグルコース−6−リン酸及びフラクトースに転換させる活性、またはスクロースをグルコース及びフラクトースに転換させる活性を有する。また、実施例においては前記β−フルクトフラノシダーゼは大腸菌(E. coli W)由来を例示しているが、その他の異なる微生物から由来したものも本発明の範囲に含まれ、配列番号7のアミノ酸配列を有してもよく、これをコードする遺伝子(cscA)は配列番号8の塩基配列で表されることを特徴とする。
【0038】
本発明において、前記β−フルクトフラノシダーゼの一例であるスクロース−6−リン酸ヒドロラーゼ(sacA)は、スクロースを分解して、解糖過程につなげるのに関与する酵素で、スクロース−6−リン酸をグルコース−6−リン酸及びフラクトースに転換させる活性、またはスクロースをグルコース及びフラクトースに転換させる活性を有する。また、実施例においては前記β−フルクトフラノシダーゼはバシラス・サブチリス(Bacillus subtilis)由来を例示しているが、その他の異なる微生物から由来したものも本発明の範囲に含まれ、配列番号9のアミノ酸配列を有してもよく、これをコードする遺伝子(sacA)は配列番号10の塩基配列で表されることを特徴とする(sacA、BSU38040、sucrose-6-phosphate hydrolase)。
【0039】
また、マンヘミア由来のsacCによって、コードされるβ−フルクトフラノシダーゼとprotein BLAST(http://blast.ncbi.nlm.nih.gov/Blast.cgi)検索を介する酵素のアミノ酸シーケンス間の相同性比較を実施してみると、E. coli W菌株由来のcscAによって、コードされるインバターゼの場合は28%の相同性を有し、バシラス・サブチリス(Bacillus subtilis)由来のsacAによってコードされるスクロース−6−リン酸ヒドロラーゼ(β−フルクトフラノシダーゼ)の場合は35%の相同性を有する。そして、二つの菌株由来のβ−フルクトフラノシダーゼは共に、「SacC」と称されるβ−フルクトシダーゼ(COG1621)といわれる保存されたドメイン(conserved domain)を有する。これは相同性において、たとえある程度差があっても、「SacC」と称されるβ−フルクトシダーゼ(COG1621)といわれる保存されたドメインが存在すると、下記の通り、これを微生物内に導入した時、スクロースを代謝して成長できることを示している。従って、前記β−フルクトフラノシダーゼに関して本明細書においては配列番号3、7または9のアミノ酸配列を例示したが、これに制限されることなく、β−D−フルクトフラノシド結合を加水分解して、フラクトースを遊離する活性を有するβ−フルクトフラノシダーゼであれば、全て本発明に含まれる。例えば、配列番号3、7または9のアミノ酸配列と配列同一性が少なくとも70%、80%、90%以上のアミノ酸配列も本発明に含まれる。
【0040】
同様に、前記β−フルクトフラノシダーゼをコードする遺伝子は、例えば、配列番号4、8または10の塩基配列を有しながら、この配列中いずれか一つの塩基配列が置換、欠失、挿入及び付加されて、突然変異が起きることによって前記本発明による塩基配列と少なくとも70%、80%、望ましくは90%、より望ましくは95%の配列同一性を有するDNAも本発明に含まれる。
【0041】
前記「配列同一性」とは、2個のポリヌクレオチド間、またはアミノ酸間残基の配列類似性をいう。前記「配列同一性」は比較対象の塩基配列、またはアミノ酸配列の領域に跨って、最適状態でアラインメントされた2個の塩基配列またはアミノ酸配列を比較することによって決定される。ここで、比較対象のポリヌクレオチドまたはアミノ酸配列は、2個の配列の最適なアラインメントのための参考配列(例えば、コンセンサス配列など)と比較して、付加または欠失(例えば、ギャップ、オーバーハング等)を有してもよい。配列同一性の数値は両方の配列に存在する同じ核酸塩基またはアミノ酸を決めて、適合部位の数を決めて、続いて、比較対象の配列領域内の塩基またはアミノ酸の合計数で前記適合部位の数を割り算して得られた数値に100を乗算することによって算出される。核酸またはアミノ酸の間の配列同一性は、例えば、配列解釈ソフト、具体的にはBLASTN、FASTAなどを使って測定される。前記BLASTは、http://www.ncbi.nlm.nih.gov/Blast/から普通に利用することができる。
【0042】
本発明において、スクロース(sucrose)は先に説明したように、D−グルコースとD−フラクトースで形成される二糖類で、それ自体が細胞内の蛋白質の変性を防いで安定化させたり、外部環境の変化から細胞の溶解を極力抑制させる役割を果たすと知られており、高濃度の基礎化学物質生産や高濃度細胞培養時にさらに有効活用可能である。
【0043】
図2に示したように、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来の新規β−フルクトフラノシダーゼルスクロース−6−リン酸ヒドロラーゼを、スクロースを代謝できない微生物に導入した時、前記導入された酵素が4種類の可能な経路を介して代謝されると予想される。
【0044】
最初の可能な経路(反応I)は、導入した前記スクロース−6−リン酸ヒドロラーゼが細胞外空間(Extracellular space)でスクロースをグルコースとフラクトースに分解した後、各々の特定のホスホトランスフェラーゼなどの輸送に関与する酵素を介して細胞内部に導入される場合である。
【0045】
二番目の可能な経路(反応II)は、導入されたスクロース−6−リン酸ヒドロラーゼが周辺細胞質(periplasm)内でスクロースをグルコースとフラクトースに分解した後、各々の特定のホスホトランスフェラーゼなどの輸送に関与する酵素を介して細胞内部に導入される場合である。
【0046】
三番目の可能な経路(反応III)は、スクロースがホスホトランスフェラーゼ系列でないパーミアーゼ(permease)酵素によってスクロースのまま細胞内に入ってきて、前記導入されたスクロース−6−リン酸ヒドロラーゼによって、グルコースとフラクトースに分解されて利用できる場合である。
【0047】
四番目の可能な経路(反応IV)は、スクロースがホスホトランスフェラーゼによって、細胞内部に入って来ると共に、スクロース−6−リン酸に転換され、導入されたスクロース−6−リン酸ヒドロラーゼによって、フラクトースとグルコース−6−リン酸に転換されて利用できる場合である。
【0048】
また、前記sacC遺伝子は、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC 0769BP)由来であり、前記菌株はアクチノバシラス・サクシノゲネス130Z(Actinobacillus succinogenes 130Z:ATCC 55618)と共にパスツレラ科(Pasteurellaceae)(family)に属する。特に、前記M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC 0769BP)とA.サクシノゲネス130Z(A. succinogenes 130Z:ATCC 55618)のゲノム配列及び細胞生理(cell physiology)が非常に類似している。前記二つの菌株のゲノム配列は今まで解読された全てのゲノム配列の中で最も類似していると知られている(Mckinlay et al., Appl. Microbiol.Biotechnol., 76:727, 2007)。また、前記sacC遺伝子に対してA.サクシノゲネス130Z(A. succinogenes 130Z)由来であるものが、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来のものと非常に高い相同性を有し、最も類似する酵素と知られているため、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E)由来のスクロース−6−リン酸ヒドロラーゼ(SacC、MS0909)酵素及びこれをコードする遺伝子とA.サクシノゲネス130Z(A. succinogenes 130Z)のスクロース−6−リン酸ヒドロラーゼ(Asuc_1829)酵素及びこれをコードする遺伝子も同様に適用されるのは、当業界の通常の知識を有するた者には明らかに理解できる。
【0049】
従って、本発明は他の観点において、スクロースホスホトランスフェラーゼ及び/またはスクロース−6−リン酸ヒドロラーゼをコードする遺伝子が導入されたスクロース代謝能を有する遺伝子組み換え微生物及び前記遺伝子組み換え微生物をスクロースを炭素源として含有する培地で培養するのを特徴とする代謝産物、生分解性高分子または遺伝子組み換え蛋白質の製造方法に関する。
【0050】
本発明において、遺伝子組み換えベクターは、適切な宿主細胞で蛋白質を発現できるベクターであり、蛋白質の発現を調節できる調節配列(control sequences)に作動可能に連結されたDNA配列及びその他遺伝子操作を容易にしたり蛋白質の発現を最適化したり、またはベクターの複製に必要な配列を含有するDNA作製(DNA construct)をいう。前記調節配列には転写を調節するためのプロモーター(promoter)、転写を調節するために選択的に付加されたオペレーター(operator)、適切なmRNAリボソーム結合部位及び転写/翻訳の終了を調節する配列が含まれる。例えば、前記遺伝子組み換えベクターは下記の実施例において使われたpTrc99Aのような遺伝子組み換えベクターであってもよいが、このベクター以外に他の公知のベクターも適用可能であり、pKK223−3、pTac99A、pET series、pMAL seriesのような発現ベクターだけでなく、pACYC、pBluescript SK−、pBR322、pGEM series、pMB1等のクローニングベクターにもsacC遺伝子を含む発現カセットを挿入することで適用可能であり、その他にも本発明が属する技術分野の公知の遺伝子組み換えベクター(Sambrook J, Russell D, Molecular cloning:a laboratory manual, 3rd ed., Cold Spring Harbor Lab (CSHL) Press, New York, 2001)を利用することも可能である。また、アンピシリン抵抗遺伝子以外にも公知の他の多くの抵抗遺伝子を含むものも本発明に適用可能である。
【0051】
また、前記遺伝子はM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC 0769BP)由来のものであり、前記菌株はアクチノバシラス・サクシノゲネス130Z(Actinobacillus succinogenes 130Z:ATCC 55618)と共にパスツレラ科(Pasteurellaceae)(family)に属する。特に、前記二つの菌株、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC 0769BP)とA.サクシノゲネス130Z(A. succinogenes 130Z:ATCC 55618)のゲノム配列及び細胞生理が非常に類似する。McKinlay等の2007年度の報告によると、前記二つの菌株のゲノム配列は今まで解読された全てのゲノム配列の中で最も類似すると知られている(Appl. Microbiol.Biotechnol., 76:727, 2007)。従って、前記遺伝子はM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC 0769BP)と共にA.サクシノゲネス130Z(A. succinogenes 130Z:ATCC 55618)から由来した遺伝子にも適用できることは、当業界の通常の知識を有するた者には明らかに理解できる。
【0052】
本発明の実施例においては、前記ptsG、sacC及びrbsK遺伝子を含む遺伝子組み換えベクターを利用して、スクロースを炭素源として使えない微生物を形質転換させてスクロース代謝能を有する遺伝子組み換え微生物を作製したが、前記遺伝子を、既に知らされた公知の方法で、スクロースを炭素源として使えない他の微生物の染色体に挿入させて、染色体上で操作された(genome-engineered)スクロース代謝能を有する遺伝子組み換え微生物を作製してもよい。
【0053】
また、本発明は他の観点において、前記sacC遺伝子をはじめとするβ−フルクトフラノシダーゼをコードする遺伝子を含有する遺伝子組み換えベクターを作製した後、これをスクロースを炭素源として使えない大腸菌に導入することによって、前記遺伝子組み換えベクター、前記遺伝子組み換えベクターによって形質転換されたスクロース代謝能を有する遺伝子組み換え微生物、前記作製された遺伝子組み換え微生物を単一炭素源としてスクロースを含有する培地において培養した結果、スクロース代謝能を有することを確認して、これを利用した代謝産物、生分解性高分子、又は遺伝子組み換え蛋白質の製造方法に関する。
【0054】
前記遺伝子組み換えベクターは図5の切断地図を有する遺伝子組み換えベクターであってもよいが、図5に示されたpTac15Kベクター以外に他の公知のベクターも適用可能であり、pTrc99A、pTac99A、pMAL seriesのような発現ベクターだけでなく、pACYC、pBluescript SK−、pBR322、pGEM series、pMB1等のクローニングベクターにもsacC遺伝子をはじめとするβ−フルクトフラノシダーゼをコードする遺伝子を含む発現カセットを挿入することで適用可能であり、その他にも本発明が属する技術分野の公知の遺伝子組み換えベクター(Sambrook J, Russell D, Molecular cloning: a laboratory manual, 3rd ed., Cold Spring Harbor Lab (CSHL) Press, New York, 2001)も利用可能である。また、カナマイシン抵抗遺伝子以外にも公知の他の多くの抵抗遺伝子を含むものも本発明に適用可能である。
【0055】
本発明の下記実施例において、前記sacC、cscA、sacAを含む遺伝子組み換えベクターを利用して、スクロースを炭素源として使えない微生物を形質転換させてスクロース代謝能を有する遺伝子組み換え微生物を作製したが、スクロースを炭素源として使えない他の微生物の染色体に前記遺伝子を公知の方法で挿入させてスクロース代謝能を有する遺伝子組み換え微生物を作製することができる。それと共に、本発明の実施例においてはスクロースを炭素源として使えない宿主微生物として特定大腸菌のみを示したが、その他の異なる大腸菌だけでなく、細菌、酵母及びカビなどのスクロースを炭素源として使えない宿主微生物に導入しても同様の結果を得られることは当業者には明らかに理解できる。
【0056】
また、本発明において、代謝産物とは、代謝過程を介して生成される中間産物及び生成物を総称するものであって、一次代謝産物と二次代謝産物に区分される。例えば、本発明はバイオ燃料、一次代謝産物、二次代謝産物、生分解性高分子、遺伝子組み換え蛋白質生産菌株作製時に遺伝子組み換えまたは染色体上における操作(genome-engineered)等の手法でスクロースを活用できない微生物に多様に適用可能である。例えば、ブタノール(Atsumi et al., Nature., 451:7174, 2008)、エタノール(Lindsay et al., Appl. Microbiol.Biotechnol, 43:70, 1995)、乳酸(Zhou, Appl. Environ. Microbiol, 69:399 2003)、コハク酸及びリンゴ酸(Jantama et al.,Biotechnol.Bioeng., 99:1140, 2008)、アミノ酸(Park et al., PNAS, 104:7797, 2006; Lee et al., Molecular Systems Biology, 3:1, 2007)、生分解性高分子(Ahn et al., Appl. Environl. MicroBiol., 66:3624, 2000; Park et al.,Biomacromolecules, 2:248, 2001; Park et al.,Biotechnol.Bioeng., 74:81, 2001)、遺伝子組み換え蛋白質(Jeong et al., Appl. Environl. MicroBiol., 65:3027, 1999; Han et al., Appl. Environl. MicroBiol., 69:5772, 2003)などの生産時に今まではグルコースを主炭素源として活用して、その目的バイオ製品を生産したが、本発明を前記公知の菌株に導入すると、スクロースを炭素源として活用して、目的する有用バイオ製品を生産可能である。
【0057】
他にも生分解性高分子、遺伝子組み換え蛋白質の生産にも本発明を当業者は容易に適用可能である。下記実施例においては代謝産物として、例えば、トレオニンなどを挙げて代謝産物を生産する場合を示したが、それ以外に生分解性高分子、遺伝子組み換え蛋白質の生産にも本発明を当業者は容易に適用可能である。
【実施例】
【0058】
以下、実施例によって、本発明をより一層詳細に説明する。これらの実施例は単に本発明を例示するものであり、本発明の範囲がこれらの実施例によって制限されると解釈されないことは、当業界において通常の知識を有するた者には明らかに理解できる。
特に、下記実施例においては本発明による遺伝子を発現させるためにスクロース代謝を行えない宿主菌株として特定大腸菌を示したが、他の大腸菌や他属の微生物を使っても、本発明によるスクロース代謝能に関与する遺伝子を導入してこれを利用してスクロースをはじめとする代謝産物を製造することも当業界において通常の知識を有するた者には明らに理解できる。
【0059】
[実施例1]ptsG、sacC及びrbsK遺伝子のスクロース代謝能確認
1.1:ptsG、sacC及びrbsK遺伝子の分離
本発明による遺伝子(ptsG、sacC、及びrbsK)が共にスクロース代謝に関与するかを調べてみるため、先ず前記遺伝子をM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC0769BP)から分離した。
【0060】
先ず、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC0769BP)のゲノムDNAを鋳型にして、配列番号:11及び12のプライマーを使ったPCRを行ってptsG(MS0784)のDNAを増幅した。配列番号:13及び14と配列番号:15及び16のプライマーを使ったPCRを同様に行って、各々sacC(MS0909)とrbsK(MS1233)のDNAを増幅し、これらを混合したものを鋳型にして配列番号:13と16のプライマーを使ってオーバーラッピングPCR(overlapping PCR)を行った。前記pstG(MS0784)、sacC(MS0909)とrbsK(MS1233)は、順にスクロースホスホトランスフェラーゼ、スクロース−6−リン酸ヒドロラーゼ、及びフラクトキナーゼにコードする遺伝子である。
【0061】
配列番号:11:5'−GGAATTCATGCTCGTTTTAGCTAGAATTGG
配列番号:12:5'−TCCGAGCTCTTACTATTCTTTTGCGTTAGCTCTTG
配列番号:13:5'−ACCTGCGAGCTCTTTCACACAGGAAACAATTTTCATGCGGTCGTTTTTACCG
配列番号:14:5'−CAAATTTTGTTTGTCATATGCATGAAATCTGTTTCCTGTGTGAAATTACTATTTATATTCAATTTCTTTCGGATA
配列番号:15:5'−TATCCGAAAGAAATTGAATATAAATAGTAATTTCACACAGGAAACAGATTTCATGCATATGACAAACAAAATTTG
配列番号:16:5'−ACCTGCGGGTACCCTATTAGTTTGCTAAAAATTCCGCT
【0062】
1.2:遺伝子組み換えベクターpMSscrIIAの作製
マンヘミア由来のスクロースホスホトランスフェラーゼ、スクロース−6−リン酸ヒドロラーゼ、及びフラクトキナーゼを各々コードする遺伝子、pstG(MS0784)、sacC(MS0909)及びrbsK(MS1233)を大腸菌から発現させるために発現ベクターを次のように作製した。
【0063】
実施例1.1において増幅されたsacC(MS0909)及びrbsK(MS1233)遺伝子を含む一つのDNA片を切断酵素SacIとKpnIで切断して、これを同じ切断酵素で切断したpTrc99A(Pharmacia Biotech., Uppsala, Sweden)と結合させてpTrc99AsacCrbsKを作製した。
【0064】
次に、実施例1.1において取得されたptsG(MS0784)DNA片をEcoRIとSacIで切断し、これを同じ切断酵素で切断した発現ベクターpTrc99AsacCrbsKと結合させてpTrc99AptsGsacCrbsKを作製して、これをpMSscrIIAと命名した(図3)。
【0065】
1.3:Escherichia coli W3110 pMSscrIIA及びW3110 pTrc99A菌株の作製
スクロースを代謝できないと知られるE. coli K-12菌株の亜株(substrain)のE. coli W3110をモデル微生物として下記の実験を行った。
【0066】
前記E. coli W3110をLB固体培地に塗抹して、8時間37℃で培養した後、コロニーをLB液体培地10mLに接種して、8時間培養した。前記培養液をLB液体培地100mLに1%(v/v)接種して、37℃の振盪培養器で培養した。
【0067】
約2時間後、OD600が0.30〜0.35程度になると、氷に20分間放置して、細胞の成長を停止させる。4℃、3,000rpm、15分の条件で遠心分離して細胞を取得した後、4℃状態のRFI溶液32mLで細胞を懸濁した。そして、前記懸濁液を4℃、3,000rpm、15分の条件で遠心分離して細胞を取得した。これをRFII溶液8mLで細胞を再懸濁した後、氷に15分間放置した。最終的に前記再懸濁液を100μLずつ分株して−70℃で保管した。RFIの組成は100mM RbCl、50mM MnCl2−4H2O、0.1M CH3COOK、10mM CaCl2及び15%(w/v)グリセロールで構成され、0.2M 酢酸を添加してpHを5.8に合わせた。RFIIは10mM MOPS、10mM RbCl、100mM CaCl2、及び15%(w/v)グリセロールで構成され、NaOHを添加することによってpHを6.8に合わせた。
【0068】
このように準備されたE. coli W3110菌株に実施例1.2において作製された発現ベクターpMSscrIIAまたは対照群でpTrc99A(Pharmacia Biotech., Uppsala, Sweden)を混合した後、42℃で90秒間ヒートショックを与えることによって形質転換を行った。ヒートショック後、LB液体培地0.8mLを加えて、37℃の振盪培養器で1時間培養した。
【0069】
培養液を抗生剤アンピシリン(最終濃度50μg/L)を含有したLB固体培地に塗抹して、37℃で12時間以上培養した。形成されたE.coli W3110 pMSscrIIA及びE.coli W3110 pTrc99Aコロニーを、LB液体培地に接種をして、37℃で8時間以上培養した。ベクターが上手く導入されたのか確認するために前記培養された菌株から、GeneAll(商標) Plasmid SV(GeneAll biotechnology, Korea)を利用してベクターを分離し、電気泳動によって確認した。E. coli W3110 pMSscrIIA菌株の場合、下記配列番号:17〜24までのプライマーを利用してソルジェント(Solgent, Korea)にシーケンシング(sequencing)を依頼して、塩基配列の一致の有無を確認した。
【0070】
配列番号:17:5'−GGAAACAGACCATGGAATT
配列番号:18:5'−CCGCAAAAGATTTATTCGAAGAAG
配列番号:19:5'−CCTGGTTATATGATACTTTAGG
配列番号:20:5'−TAGTGCTGGGCGCAAGAGCTAA
配列番号:21:5'−ACCAGTGGGCGATAAAATCG
配列番号:22:5'−TGATCAAGGTTTCGATTTCT
配列番号:23:5'−TTTTCCTGAATGACGGCGAA
配列番号:24:5'−CGATCTGCCGCAATTTCAAG
【0071】
1.4:遺伝子組み換え大腸菌のスクロース代謝能確認
実施例1.3において作製された固体培地上のE. coli W3110 pMSscrIIA菌株とE. coli W3110 pTrc99Aのコロニーを5g/Lスクロースを単一炭素源として含んだ10mLのM9最小培地に接種して、37℃で16時間培養した後、これを再びスクロースを含んだ100mLのM9最小培地に3%(v/v)接種した後37℃で培養した。この時、抗生剤として最終濃度50μg/Lのアンピシリンを添加した。M9最小培地は33.9g/LのNa2HPO4、15g/LのKH2PO4、2.5g/LのNaCl、5g/LのNH4Cl、及び0.36g/LのMgSO4で構成された。培養液内の細胞濃度は分光光度計を利用してOD600の値として推定した。培養中、周期的にサンプルを採取し、採取された試料は13,000rpmで5分間遠心分離した後、上澄み液のスクロースの濃度を液体クロマトグラフィー(HPLC)で分析した。
【0072】
その結果、表1に示したように、E. coli W3110 pTrc99A菌株の場合、スクロースを単一炭素源として含んだM9最小培地において全く生長できないが、E. coli W3110 pMSscrIIA菌株はスクロースの優れた代謝能を示す。E. coli W3110 pMSscrIIA菌株は19時間約2.2g/Lのスクロースを代謝して、OD600基準に3.12のバイオマスの増加を示し、0.67g/Lの硝酸を付加生成物として生産した。従って、前記ptsG、sacC及びrbsK遺伝子を全て含有する場合、優れたスクロース代謝能を示すことが確認された。
【0073】
【表1】
【0074】
前記に示したように、本発明のスクロースホスホトランスフェラーゼ、またはこれとスクロース−6−リン酸ヒドロラーゼ遺伝子を導入することだけでも、スクロースの代謝能を備えると共に、スクロースを炭素源として活用して代謝産物の一つとして酢酸を生産することを示した。
【0075】
従って、酢酸以外にもギ酸、乳酸、コハク酸、エタノールなどの他にも、バイオブタノールを含むバイオ燃料及びエネルギー、そして生分解性高分子、遺伝子組み換え蛋白質の生産にも本発明を当業者は容易に適用可能である。
【0076】
[実施例2]pstG遺伝子及びsacC遺伝子各々のスクロース代謝能確認
2.1:pstG遺伝子及びsacC遺伝子のスクロース代謝能確認のため遺伝子組み換えベクター作製
本発明によるptsG及びsacCが単独でもスクロース代謝能に係わるのかを確認するため、ptsG(MS0784)欠失のためのベクター(pSacHR06ptsG)とsacC(MS0909)欠失のためのベクター(pSacHR06sacC)を作製してノックアウト(knock-out)実験を行った。
【0077】
先ず、スクロースホスホトランスフェラーゼ遺伝子(ptsG)を相同性遺伝子組み換え方法で破壊するため遺伝子交換ベクターを次のように作製した。マンヘミア・サクシニシプロデュセンスMBEL55E(Mannheimia succiniciproducens MBEL55E:KCTC 0769BP)のゲノムDNAを鋳型にして、下記配列番号:25と26のプライマーを利用して、左側ホモロガスアーム(homologous arm)部分を増幅し、配列番号:27と28のプライマーを利用して、同じ鋳型で右側ホモロガスアーム(homologous arm)部分を増幅し、配列番号:29と30のプライマーを利用してpECmuloxベクター(Kim et al., FEMS Microbiol. Lett., 278:78, 2008)を鋳型にして抗生剤マーカーと変異lox siteを含んだ部分のDNA断片を増幅し、これら三つのDNA断片を配列番号:25と28プライマーを利用してオーバーラッピングPCR(overlapping PCR)を行った。
【0078】
配列番号:25:5'−ATATCTGCAGCCGGCATTAAATATTAGTCAAC
配列番号:26:5'−CGTTCTAACGGAGGTTGAAAACTGCCCTTT
配列番号:27:5'−GTCTCCCTATCACGCCGTTATTTTCATTATT
配列番号:28:5'−ATTAGTCGACACCATCCCCACGGAATACAT
配列番号:29:5'−TTTCAACCTCCGTTAGAACGCGGCTACAAT
配列番号:30:5'−TAACGGCGTGATAGGGAGACCGGCAGATCC
【0079】
このように増幅された最終DNA断片は制限酵素PstIとsalIで切断し、同じ酵素で切断されたpSacHR06ベクター(Park et al., Proc. Nat. Acad. Sci. (PNAS), 104:7797, 2007)にクローニングしてpSacHR06ptsGを作製した。また、前記のような方法で配列番号:31から36までのプライマーを利用して、pSacHR06sacCを作製した。
【0080】
配列番号:31:5'−ATACACTGCAGTTATGCAATTTATCGCACCC
配列番号:32:5'−AATCTGCTCTGATGCGGTCGTGAAATGCTTCCA
配列番号:33:5'−CACAGAATCAGGACAAATGGCATTCAATGCTG
配列番号:34:5'−ATACTGTCGACTCAATGGCATATGCAGCG
配列番号:35:5'−AAGCATTTCACGACCGCATCAGAGCAGATTGTACTGAGAG
配列番号:36:5'−TGAATGCCATTTGTCCTGATTCTGTGGATAACCGTATTAC
【0081】
2.2:M.サクシニシプロデュセンスMptsG(M. succiniciproducens MptsG)及びM.サクシニシプロデュセンスMsacC(M. succiniciproducens MsacC)菌株の作製
実施例2.1において作製されたptsG遺伝子除去用交換ベクターpSacHR06ptsGとsacC遺伝子除去用交換ベクターpSacHR06sacC各々を利用して、Kimら(FEMS Microbiol. Lett., 278:78, 2008)により報告された方法でM.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC0769BP)のゲノムで前記遺伝子を欠失させて、変異菌株であるM.サクシニシプロデュセンスMptsG(M. succiniciproducens MptsG)及びM.サクシニシプロデュセンスMsacC(M. succiniciproducens MsacC)菌株を作製した。
【0082】
即ち、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC0769BP)を10g/Lのグルコースを含有したLB−グルコース固体培地に塗抹して、36時間37℃で培養した後、コロニーをLB−グルコース液体培地10mLに接種して、12時間培養した。十分に培養された培養液をLB−グルコース液体培地100mLに1%接種して、200rpm、37℃の振盪培養器で培養した。
【0083】
約4〜5時間後、OD600が0.3〜0.4程度になったら4℃、4,500rpm、20分の条件で遠心分離して、細胞を取得した後、4℃の状態の10%グリセロール溶液200mLで細胞を再懸濁した。そして、4℃、5,500rpm、20分の条件で遠心分離して細胞を取得した。再懸濁時に使われるグリセロール溶液を半分に減らしながら、これを2回繰り返し実行した後、細胞とグリセロール溶液の体積比が1:1になるべく再懸濁して細胞濃縮液を前記取得された細胞濃縮液と前記実施例2.1において作製された遺伝子交換ベクターpSacHR06ptsGまたはpSacHR06sacC各々を混合した後、2.5kV、25μF、400ohmsの条件でelectroporationを行うことで、前記遺伝子交換ベクターを培養されたM.サクシニシプロデュセンスMsacC(M. succiniciproducens)に各々導入させた。電気衝撃後、LB−グルコース液体培地1mLを加えて、200rpm、37℃の振盪培養器で1時間培養した。培養液を、抗生剤クロラムフェニコール(最終濃度6.8μg/L)を含有したLB−グルコース固体培地に塗抹して、37℃で48時間以上培養した。形成されたコロニーの中から、ダブルクロスオーバー(double crossover)だけが起きたものを選び出すため、これをクロラムフェニコール(最終濃度6.8μg/L)を含有したLB−スクロース(100g/Lのスクロースを含有したLB培地)培地にストリーキング(streaking)した後、24時間後に形成されたコロニーを同じプレートに再びストリーキング(streaking)した。
【0084】
前記プレートで形成されたコロニー(変異菌株)を抗生剤が含まれたLB−グルコース液体培地で培養して、培養された菌株からRochelle等による方法(Rchelle et al., FEMS Microbiol. Lett., 100:59, 1992)を応用して、ゲノムDNAを分離した。前記分離した変異株のゲノムDNAを鋳型にしてPCRを行った後、取得されたPCR産物を電気泳動することによってptsG及びsacCの欠失の有無を各々の変異菌株において確認した。
【0085】
MptsG菌株からptsGの欠失の有無を確認するため、下記のように配列番号:37と38のプライマーを利用したPCRと、配列番号:39と40のプライマーを利用したPCRによって確認し、MsacC菌株においてsacCの欠失の有無を確認するため、配列番号:39と40のプライマーと、配列番号:41と42のプライマーを利用したPCRによって確認した。
【0086】
配列番号:37:5'−CGGGGCGAAAGTGATTGAGA
配列番号:38:5'−AATTGCCGCCTGGGTATTGG
配列番号:39:5'−ACCTTTACTACCGCACTGCTGG
配列番号:40:5'−GCGGGAGTCAGTGAACAGGTAC
配列番号:41:5'−GATCTTGAGTCCGTAAAACAGGCTT
配列番号:42:5'−TTCCGCTCAAGCCATTGTAGTG
【0087】
2.3:MptsG菌株、MsacC菌株、及び母菌株(MBEL55E)における生長比較
前記実施例2.2において作られた遺伝子組み換えMptsG、MsacC菌株をBHI(Bacto(商標)Brain Heart Infusion; Becton, Dickinson and Company, Sparks, MD)培養培地で8時間ほど培養した後、10mLを300mL MH5S培養培地(liter当たり:2.5g 酵母エキス、2.5g ポリペプトン、1g NaCl、8.7gK2HPO4、10gNaHCO3、0.02gCaCl22H2O、0.2gMgCl26H2O 及び10gスクロース)に接種した後、同様の条件で培養したこれらの母菌株であるMBEL55Eの生長曲線と比較した。図3はOD600で測定したMBEL55E(●)、MptsG(▲)、及びMsacC(△)菌株の生長曲線である。図3に示したように、母菌株(MBEL55E)を有していたスクロース培地上における成長能力が各々の遺伝子を除去することで、殆ど消えることが示され、これは各々の遺伝子がスクロース培地上において生長するのに必須であることを意味する。
【0088】
一方、フラクトキナーゼ(RbsK、MS1233)をコードするrbsK遺伝子の場合には前記同様の実験を行った、増殖が遅くなるなどの成長の変化は観察されなかった。また、酵素活性測定結果においても母菌株とrbsK除去菌株との間に大きい差はなく、両方とも一般的なフラクトキナーゼの活性に比べて、非常に低いレベルであり、即ち、値を無視できるほどの活性が測定された。このような根拠として、rbsK遺伝子はフラクトキナーゼをコードしなかったか、フラクトキナーゼをコードしたとしても非常に弱い活性を有すると推定される。
【0089】
2.4:MptsG及びMsacC菌株と母菌株の該当酵素活性比較
スクロースPTSの活性測定のためにJacobsonら(J. BIOL. CHEM., 254:249, 1979)とLodgeら(Infect. Immun., 56:2594, 1988)による方法を導入した。
【0090】
先ず、細胞を透過(permeabilize)させるために、OD600値が1.2近傍の細胞培養液1mLをTDMバッファー(50mM Tris/HCl、10mM MgCl2、1mM DTT;pH7.5)で洗浄した後、同じバッファー1mLに再懸濁した後、0.01mLトルエンを加えて45秒間強く攪拌(agitation)。12,000×gで遠心分離して集めた細胞をTDMバッファーで2回洗浄した。このような過程をもう一度繰り返して、最終的にTDMバッファー50μLに再懸濁することで、透過性細胞(permeabilized cell)を準備した。PEP−依存的スクロース燐酸化は、全反応体積100μLに25mM Tris/HCl(pH8.0)、1mM DTT、5mM MgCl2、10mM KF、1μCi[U−14C]スクロースに5μLの透過させた前記細胞懸濁液を加えた状態で、1mM PEPを含めるものと含まないものの2種類の反応の差を測定して成された。
【0091】
前記混合液は37℃で10分間反応し、1mLの冷たい水を加えることで反応を停止させた。最終反応物はろ過装置上のDEAEフィルターディスク(Whatman DE81)を通過させて、10倍体積の冷たい水を加えて洗浄した後、公知の方法でBeckman LS6500 liquid scintilation counter(Beckman, Ramsey, MN)を利用して、放射活性を測定した。スクロース−6−リン酸ヒドロラーゼの活性はMartinら(Appl. Environ. Microbiol., 53:2388, 1987)による方法に若干修正を加えて測定した。OD600値が1近傍の細胞培養液20mLを前記スクロースPTS活性測定時のような方法で透過(Permeabilize)させて、最終的に1mLのTDMバッファーに再懸濁して準備した。全反応物の体積は300μLであり、50mM Sodium-Potassium Phosphateバッファー(pH7.2)、5mM MgCl2、4mMスクロース、0.8mM NADP、6.4Uのグルコース−6−ホスフェート デヒドロレース(glucose-6-phosphate dehydrolase)、そして、30μLの透過性細胞(permeabilized cell)を加えた状態で、10mM PEPを含めたものと含めないもの2種類の反応の差を測定した。
【0092】
各々のptsG及びsacC遺伝子が除去された変異菌株とその母菌株との活性を測定した結果を表2に示す。この結果から、MS0784(ptsG)がコードするスクロースPTS酵素がPTS活性を有することを意味し、MS0909(sacC)がコードするスクロース−6−リン酸ヒドロラーゼ酵素が各々細胞内において該当機能を有することを意味する。
【0093】
【表2】
a BHI, Bacto(商標) Brain Heart Infusion(Becton, Dickinson and Company, Spark, MD)、MH5S培地組成はリッター当り:2.5g酵母エキス、2.5gポリペプトン、1gNaCl、8.7gK2HPO4、10gNaHCO3、0.02gCaCl2・H2O and 0.2gMaCl2・6H2O及び10gスクロースを含む。
b スクロースホスホトランスフェラーゼの活性は液体シンチレーションカウンターによって測定されたcpm(count per minute)値を、mU/mg unit単位に転換して表記した。
c 相対的活性度は、*表記されたものを100として、残りの値を計算した。
【0094】
[実施例3]スクロース代謝に関与する酵素をコードする新しい遺伝子の分離
前記実施例2の結果を基に、スクロース代謝能があると確認されたsacCをはじめとして、β−フルクトフラノシダーゼをコードする遺伝子を下記のような方法で各々マンヘミア、大腸菌、及びバシラス・サチリスから分離した。
【0095】
3.1:マンヘミア由来スクロース−6−リン酸ヒドロラーゼをコードする遺伝子の分離
先ず、M.サクシニシプロデュセンスMBEL55E(M. succiniciproducens MBEL55E:KCTC0769BP)のゲノムDNAを鋳型にして、配列番号43及び44のプライマーを使ってPCRを行うことによってsacC(MS0909)遺伝子のDNAを増幅した。前記sacC(MS0909)はβ−フルクトフラノシダーゼ(スクロース−6−リン酸ヒドロラーゼ)をコードする遺伝子(配列番号4)である。
【0096】
配列番号43:5'−ACTGAGCCATGGCGAAAATCAATAAAGTAGATC−3'
配列番号44:5'−TGATCCGAGCTCCTATTATTCCAGTGTTCCCGCC−3'
【0097】
3.2:大腸菌由来インバターゼをコードする遺伝子の分離
E. coli WのゲノムDNAを鋳型にして、配列番号45及び46のプライマーを使ってPCRを行うことでcscA遺伝子のDNAを増幅した。前記cscAはβ−フルクトフラノシダーゼ(インバターゼ)をコードする遺伝子(配列番号8)である。
【0098】
配列番号45:ACTCCGGAATTCATGACGCAATCTCGATTGCA
配列番号46:ACCTGCGAGCTCCCGTTGTTCCACCTGATATTATG
【0099】
3.3:バシラス・サチリス由来スクロース−6−リン酸ヒドロラーゼをコードする遺伝子の分離
バシラス・サブチリス(Bacillus subtilis)のゲノムDNAを鋳型にして、配列番号47及び48のプライマーを使ってPCRを行うことによってsacA遺伝子のDNAを増幅した。前記sacAはβ−フルクトフラノシダーゼ(スクロース−6−リン酸ヒドロラーゼ)をコードする遺伝子(配列番号10)である。
【0100】
配列番号47:GCATAGAATTCATGACAGCACATGACCAGGAGCT
配列番号48:GCATAGAGCTCCTACATAAGTGTCCAAATTCCGACATTC
【0101】
[実施例4]遺伝子組み換えベクターの作製
4.1:pTac15Kの準備
pKK223−3(Pharmacia Biotech., Uppsala, Sweden)のtrcプロモーターとトランスクリプションターミネーター(trascription terminator)部分をpACYC177(NEB, Beverly, MA, USA)に挿入して作製した。pTac15Kは構成的(constitutive)発現のための発現ベクターであり、図5の切断地図に示された構成を有している。
【0102】
4.2:遺伝子組み換えベクターpTac15KsacCの作製
マンヘミア由来のβ−フルクトフラノシダーゼ(スクロース−6−リン酸ヒドロラーゼ)をコードする遺伝子sacC(MS0909)を大腸菌に発現させるため、発現ベクターを次のように作製した。
【0103】
実施例3.1において増幅されたsacC(MS0909)遺伝子を含むPCR断片をEcoRIとSacIで切断して、これを同じ切断酵素で切断したpTac15Kと結合させる公知の分子工学的手法に従って、pTac15KsacCを作製した(図6)。そして、下記配列番号49乃至52のプライマーを利用してソルジェント(Solgent, Korea)にシーケンシングを依頼して、塩基配列の一致の有無を確認した。
【0104】
配列番号49:5'−CCCGTTCTGGATAATGTTTT−3'
配列番号50:5'−AAAGTCACGGTTGTTATTCC−3'
配列番号51:5'−CATTTAATGCCGCTCATATT−3'
配列番号52:5'−ACCGCTCAATTATTGAGATT−3'
【0105】
4.3:遺伝子組み換えベクターpTac15KEWcscAの作製
実施例3.2において得られたDNA断片をEcoRIとSacIで処理して、同じ酵素で処理したpTac15Kと結合させる公知の分子工学的手法に従って、pTac15KEWcscAを作製した。
【0106】
4.4遺伝子組み換えベクターpTac15KBSsacAの作製
実施例3.3において得られたDNA断片をEcoRI及びSacIで処理して、同じ酵素で処理したpTac15Kと結合させる公知の分子工学的手法に従って、pTac15KBSsacAを作製した。
【0107】
[実施例5]遺伝子組み換え菌株の作製
5.1:Escherichia coli W3110 pTac15KsacC菌株の作製
スクロースを代謝できないと知られたE. coli K-12菌株の亜株(substrain)であるE.coli W3110をモデル微生物として下記の実験を行った。前記E.coli W3110をLB固体培地に塗抹して、8時間37℃で培養した後、コロニーをLB液体培地10mLに接種して、8時間培養した。前記培養液をLB液体培地100mLに1%(v/v)接種して、37℃の振盪培養器で培養した。
【0108】
約2時間後、OD600が0.30〜0.35程度になると、氷に20分間放置して、細胞の成長を停止させた。4℃、3,000rpm、15分の条件で遠心分離して、細胞を取得した後、4℃状態のRFI溶液32mLで細胞を懸濁した。そして、前記懸濁液を4℃、3,000rpm、15分の条件で遠心分離して細胞を取得した。これをRFII溶液8mLで細胞を再懸濁した後、氷に15分間放置した。最終的に前記再懸濁液を100μLずつ分株して−70℃で保管した。RFIの組成は100mM RbCl、50mM MnCl2−4H2O、0.1M CH3COOK、10mM CaCl2及び15%(w/v)グリセロールで構成され、0.2M酢酸を添加してpHを5.8に合わせた。RFIIは10mM MOPS、10mM RbCl、100mM CaCl2、及び15%(w/v)グリセロールで構成され、NaOHを添加することによってpHを6.8に合わせた。
【0109】
このように準備されたE. coli W3110菌株に実施例2.2において作製された発現ベクターpTac15KsacC、または対照群でpTac15K(Pharmacia Biotech., Uppsala, Sweden)を混合した後、42℃で90秒間ヒートショックを与えることによって形質転換を行った。ヒートショック後、LB液体培地0.8mLを加えて、37℃の振盪培養器で1時間培養した。
【0110】
培養液を抗生剤カナマイシン最終濃度50μg/L)を含有したLB固体培地に塗抹して、37℃で12時間以上培養した。形成されたE. coli W3110 pTac15K及びE. coli W3110 pTac15KsacCコロニーを、LB液体培地に接種して、37℃で8時間以上培養した。ベクターが上手く導入されたかを確認するために前記培養された菌株から、GeneAll(商標) Plasmid SV(GeneAll Biotechnology, Korea)を利用して、ベクターを分離して、電気泳動によって確認した。
【0111】
5.2:E. coli W3110 pTac15KEWcscAとE. coli W3110 pTac15KBSsacA菌株の作製
実施例5.1に記載された方法と同じ方法で、スクロースを代謝できないと知られたE. coli K-12菌株の亜株(substrain)のE. coli W3110を実施例4.3及び4.4において作製されたpTac15KEWcscA及びpTac15KBSsacAを利用して形質転換させて、ベクターが上手く導入されたのか電気泳動によって確認した。
【0112】
[実施例6]遺伝子組み換え大腸菌のスクロース代謝能及び成長確認
前記実施例5.1において作製された固体培地上のE. coli W3110 pTac15KsacC菌株とE. coli W3110 pTac15Kのコロニーを10mLのLB培地に接種して、37℃で8時間培養した後、さらにこれを10g/Lのスクロースを含んだ100mLのM9最小培地に5%(v/v)接種した後、37℃で培養した。この時、抗生剤として最終濃度50μg/Lのカナマイシンを添加した。前記LB培地は10g/Lのトリプトン、10g/LのNaCl、及び5g/Lの酵母エキスで構成され、M9最小培地は33.9g/LのNa2HPO4、15g/LのKH2PO4、2.5g/LのNaCl、5g/LのNH4Cl、及び0.36g/LのMgSO4で構成された。培養液内の細胞濃度は分光光度計を利用してOD600の値で測定した。培養中、周期的にサンプルを採取し、採取された試料は13,000rpmで5分間遠心分離した後、上澄み液のスクロース及び代謝産物の濃度を液体クロマトグラフィー(HPLC)で分析した。
【0113】
その結果、図7、図8及び表3に示したように、E. coli W3110 pTac15K菌株の場合、スクロースを単一炭素源として含んだM9最小培地で全く生長できないが、E. coli W3110 pTac15KsacC菌株はスクロースの優れた代謝力を示した。E. coli W3110 pTac15KsacC菌株は17時間11.08g/Lのスクロースを代謝して、OD600基準に3.71のバイオマスの増加を示した。これは通常の大腸菌のOD600と乾燥細胞重量(dry cell weight:DCW、g/L)の換算係数(conversion factor:1 OD600 = 0.37g/L DCW)を考慮すると、約1.37g/Lのバイオマスに該当する量である。1.42g/Lの酢酸が生成され、2.01g/Lのグルコース及び4.51g/Lのフルクトースが残って、これらの二つの糖類は時間と伴って次第に消耗されることが分かる。
【0114】
【表3】
【0115】
W3110 pTac15K菌株の成長グラフにおいて(図7)、接種後8時間目にODが若干増加したことは、複合培地である接種培地(5%(v/v)接種する)に入っていた成分によるものと考えられ、LC分析結果、スクロースを分解することも、またスクロースを単独炭素源として成長することもできないことを示す。
【0116】
また、前記実施例5.2において形質転換された遺伝子組み換え菌株E. coli W3110 pTac15KEWcscAとE. coli W3110 pTac15KBSsacAをスクロースを単独炭素源として、前記E. coli W3110 pTac15KsacC菌株と同じ方法で培養した結果、図9と同様に、優れた成長能を示し、E. coli W3110 pTac15KEWcscAは初期スクロース7.77g/Lから47時間後1.82 g/Lに減少することを確認し、E. coli W3110 pTac15KBSsacAは初期8.49g/Lから47時間後7.98 g/Lに減少することを確認し、これらがスクロースを代謝して成長する可能性があることを確認した。
【0117】
[実施例7]遺伝子組み換え大腸菌を利用してスクロースを炭素源とする代謝産物生産
先に述べたように、M.サクシニシプロデュセンス(M. succiniciproducens)由来のスクロース−6−リン酸ヒドロラーゼの導入はスクロースを炭素源として成長できない微生物をスクロース単独炭素源として成長可能にした。即ち、スクロースを炭素源として活用できない微生物を、スクロースを単独炭素源として最小培地上で培養可能にすることで、既にグルコースなどを炭素源とする多くの一次、二次代謝産物、遺伝子組み換え蛋白質、生分解性高分子などの多くのバイオ製品生産システムなどをそのまま本発明に適用することができる。
【0118】
そこで、本実施例においては嫌気条件でスクロースを活用して、多様な代謝産物を生産する例を示す。
【0119】
前記実施例5.1において作製されたE. coli W3110 pTac15KsacC菌株を10mLのLB培地に接種して、37℃で8時間培養した後、さらにこれを200mLのLB培地に接種して、37℃で8時間培養し、これを容積2.5Lの発酵槽(New Brunswick System, BioFlo 3000)に接種した。発酵槽の培養培地は20g/Lスクロースを含んだR/2最小培地で構成され、操業条件はpH6.8、37℃、200rpmに100% CO2を0.5vvmの速度で注入した。抗生剤として最終濃度50μg/Lのカナマイシンを添加した。R/2培地は6.75g/LのKH2PO4、2g/Lの(NH4)2HPO4、0.85g/LのC6H8O7H2O、及び0.7g/LのMgSO47H2Oと5mL/Lの微量金属溶液(trace metal solution:10g/L FeSO47H20、2g/L CaCl2、2.2g/L ZnSO47H2O、0.54g/L MnSO45H2O、1g/L CuSO45H2O、0.1g/L NH4Mo7O247H20、0.02g/L Na2B4O710H2O、及び5mL HCl)で構成された。培養液内の細胞濃度は分光光度計を利用してOD600の値で測定し、周期的に採取された試料は13,000rpmで5分間遠心分離した後、上澄み液のスクロース及び代謝産物の濃度を液体クロマトグラフィー(HPLC)で分析した。
【0120】
その結果、図10及び表4に示したように、酢酸、ギ酸、乳酸、コハク酸、及びエタノール(acetic, formic, lactic, and succinic acid and ethanol)を成功的に生産することができた。前記のような嫌気条件で52.5時間22.13g/Lのスクロース及びこれから由来した糖類をすべて消耗し、その結果4.49g/Lの酢酸、3.74g/Lのギ酸、4.19g/Lの乳酸、5.10g/Lのコハク酸、2.66g/Lのエタノールをスクロース基準に基質当たり0.20、0.17、0.19、0.23、及び0.12 g/gスクロースの収率で生産し、これらの収率の合計は0.91g/gスクロースとして理論値に近接した相当高い収率を示した。また、培養開始後52.5時間のOD600 = 2.7であるため、これは前記に言及した換算係数を考慮すると、gDCW = 0.999に該当するため、菌体重さ当たり収率である比収率(specific yield)は表2のようになる。これは、前記のスクロース−6−リン酸ヒドロラーゼの単独導入により、スクロースを利用して、多様な代謝産物を高収率と高比収率を示しており、成功的に生産できることを示す。
【0121】
【表4】
【0122】
[実施例8]トレオニン(Threonine)生産菌株への適用による代謝工学的に改良された有用バイオ製品生産への応用
代謝工学的に改良された、遺伝子設計された(genome-engineered)または遺伝子組み換え大腸菌菌株にも本発明の遺伝子を導入することで、グルコースあるいは既に使われていた他の炭素源の代わりにスクロースを活用して、生産可能である例として下記のようなトレオニン生産菌株を利用して実験を行った。具体的に、大腸菌(Escherichia coli)W3110を基盤に代謝工学的に改良された遺伝子設計された(genome-engineered)及び遺伝子組み換え菌株のTH28C pBRThrABCR3菌株(Lee et al., Molecular. Systems. Biology., 3:149, 2007)をモデルにして、下記の実験を行った。
【0123】
実施例5において説明した方法と同じ方法で前記TH28C pBRThrABCR3菌株を、実施例4において作製したスクロース代謝能を持たせるプラスミドpTac15KsacCで形質転換させて、TH28C pBRThrABCR3 pTac15KsacCを作製した。
【0124】
前記作製されたTH28C pBRTHrABCR3 pTac15KsacC菌株を10mLのLB培地に接種して、31℃で12時間培養した後、さらにこれを50mLのLB培地に接種して、31℃で12時間培養したし、これを容積2.5Lの発酵槽(New Brunswick System, BioFlo 3000)に接種した。発酵槽の培養培地は20g/Lスクロースを含んだTPM2培地で構成され、操業条件はpH6.5、31℃、溶存酸素濃度は1vvmのAirの供給と自動的にrpmを1000まで上げられるように調節することによって40%以上の空気が飽和された状態になるよう維持した。抗生剤としては最終濃度30μg/Lのクロラムフェニコール、40μg/Lのカナマイシン、50μg/Lのアンピシリンを添加した。TPM2培地は2g/Lの酵母エキス、2g/LのMgSO47H2O、2g/LのKH2PO4、10g/L(NH4)2SO4及び0.2g/L L−メチオニンと0.2g/L−リジンと0.05g/LのL−イソロイシンと、10mL/Lの微量金属溶液(trace metal solution:10g/L FeSO47H2O、2g/L CaCl2、2.2g/L ZnSO47H2O、0.54g/L MnSO45H2O、1g/L CuSO45H2O、0.1g/L NH4Mo7O247H2O、0.02g/L Na2B4O710H2O、及び5mL HCl)で構成された。培養液内の細胞濃度は分光光度計を利用してOD600の値で測定し、周期的に採取された試料は13,000rpmで5分間遠心分離した後、上澄み液のスクロース及び代謝産物の濃度を液体クロマトグラフィー(HPLC)で分析した。アミノ酸の場合、5mLの培養液を遠心分離とろ過過程を経た後、Science Lab Center Co.(Daejeon, Korea)に依頼して、陽イオン分離カラム(caution seperation column, LCA K06/Na1.6150mm; SykamGmbH, Eresing, Germany)によって分離した後、Sykam S433アミノ酸分析機を活用して分析した。
【0125】
その結果、表5に示したように、スクロースを活用して、4.7g/Lのトレオニンを成功的に生産することができた。
【0126】
【表5】
【0127】
前記に示したように本発明のβ−フルクトフラノシダーゼ遺伝子を導入することだけでも、スクロースを代謝する能力を備えることと共にスクロースを炭素源として活用して、酢酸、ギ酸、乳酸、コハク酸、エタノールなどの多様な代謝産物の生産を可能にした。また、代謝工学的に改良された菌株にも同様の方法で導入して、目的代謝産物、即ち、本例ではトレオニンを成功裡に生産可能だった。それ以外にも生分解性高分子、遺伝子組み換え蛋白質の生産にも本発明を当業者は容易に適用可能である。
【産業上利用の可能性】
【0128】
以上、詳細に説明したように、高価なグルコースの代わりに低価格のスクロースを炭素源として使用できる遺伝子組み換え微生物を提供する効果がある。従来スクロースを炭素源として利用できなかった微生物の培養においてもグルコースをはじめとする他の炭素源をスクロースに代えることは可能である。本発明においては安価であり、自然界に豊富に存在するスクロースを炭素源として効率よく使用可能にする新しい酵素群を同定して開発することによって今後微生物発酵を介する有用な化学物質の製造や医療用遺伝子組み換え蛋白質生産において、スクロースをその炭素源としてより効率的かつ経済的に生産するのに利用するであろう。特に、スクロースはそれ自体で細胞内の蛋白質を安定化させたり、細胞の溶解を極力抑制させる役割を果たすと知られているため、高濃度の基礎化学物質生産や高濃度細胞培養時にさらに有効活用される。
【0129】
以上で本発明内容の特定部分を詳細に記述したが、当業界の通常の知識を有する者において、このような具体的技術は単に望ましい実施様態に過ぎなく、これによって、本発明の範囲が制限されるのではない点は明らかであろう。従って、本発明の実質的な範囲は添付された請求項とその等価物によって定義される。
【特許請求の範囲】
【請求項1】
配列番号1のアミノ酸配列を有するスクロースホスホトランスフェラーゼ。
【請求項2】
スクロースをスクロース−6−リン酸に転換させ、細胞中に輸送する機能を有する請求項1に記載のスクロースホスホトランスフェラーゼ。
【請求項3】
請求項1に記載のスクロースホスホトランスフェラーゼをコードする遺伝子(ptsG)。
【請求項4】
配列番号2の塩基配列で表されることを特徴とする請求項3に記載の遺伝子(ptsG)。
【請求項5】
β−D−フルクトフラノシド結合を加水分解して、フラクトースを遊離する活性によりスクロース代謝能を有するβ−フルクトフラノシダーゼ。
【請求項6】
配列番号3、配列番号7及び配列番号9のアミノ酸配列からなる群より選択されたアミノ酸配列を有することを特徴とする請求項5に記載のβ−フルクトフラノシダーゼ。
【請求項7】
請求項5に記載のβ−フルクトフラノシダーゼをコードする遺伝子。
【請求項8】
配列番号4、配列番号8及び配列番号10の塩基配列からなる群より選択された塩基配列を有することを特徴とする請求項7に記載の遺伝子。
【請求項9】
請求項3に記載の遺伝子(ptsG)、及びスクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)を含有する組み換えベクター。
【請求項10】
前記スクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)は配列番号4の塩基配列で表されることを特徴とする請求項9に記載の組み換えベクター。
【請求項11】
請求項7に記載の遺伝子を含有する組み換えベクター。
【請求項12】
前記遺伝子は配列番号4、配列番号8及び配列番号10の塩基配列からなる群より選択された塩基配列を有する遺伝子であることを特徴とする請求項11に記載の組み換えベクター。
【請求項13】
細菌、酵母及びカビから構成された群より選択された宿主細胞に請求項9または請求項11に記載の組み換えベクターが導入されていることを特徴とするスクロース代謝能を有する組み換え微生物。
【請求項14】
大腸菌であることを特徴とする請求項13に記載の組み換え微生物。
【請求項15】
細菌、酵母及びカビから構成された群より選択された宿主細胞の染色体DNAに請求項3に記載の遺伝子(ptsG)及びスクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)が導入されていることを特徴とするスクロース代謝能を有する組み換え微生物。
【請求項16】
前記スクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)は配列番号4の塩基配列で表されることを特徴とする請求項15に記載の組み換え微生物。
【請求項17】
大腸菌であることを特徴とする請求項15に記載の組み換え微生物。
【請求項18】
細菌、酵母及びカビから構成された群より選択された宿主細胞の染色体DNAに請求項7に記載の遺伝子が導入されていることを特徴とするスクロース代謝能を有する組み換え微生物。
【請求項19】
前記遺伝子は配列番号4、配列番号8及び配列番号10の塩基配列からなる群より選択された塩基配列を有することを特徴とする請求項18に記載の組み換え微生物。
【請求項20】
大腸菌であることを特徴とする請求項18に記載の組み換え微生物。
【請求項21】
請求項13に記載の組み換え微生物を、スクロースを炭素源として含有する培地で培養することを特徴とする代謝産物、生分解性高分子、または遺伝子組み換え蛋白質の製造方法。
【請求項22】
請求項15または請求項18に記載の組み換え微生物を、スクロースを炭素源として含有する培地で培養することを特徴とする代謝産物、生分解性高分子、または遺伝子組み換え蛋白質の製造方法。
【請求項1】
配列番号1のアミノ酸配列を有するスクロースホスホトランスフェラーゼ。
【請求項2】
スクロースをスクロース−6−リン酸に転換させ、細胞中に輸送する機能を有する請求項1に記載のスクロースホスホトランスフェラーゼ。
【請求項3】
請求項1に記載のスクロースホスホトランスフェラーゼをコードする遺伝子(ptsG)。
【請求項4】
配列番号2の塩基配列で表されることを特徴とする請求項3に記載の遺伝子(ptsG)。
【請求項5】
β−D−フルクトフラノシド結合を加水分解して、フラクトースを遊離する活性によりスクロース代謝能を有するβ−フルクトフラノシダーゼ。
【請求項6】
配列番号3、配列番号7及び配列番号9のアミノ酸配列からなる群より選択されたアミノ酸配列を有することを特徴とする請求項5に記載のβ−フルクトフラノシダーゼ。
【請求項7】
請求項5に記載のβ−フルクトフラノシダーゼをコードする遺伝子。
【請求項8】
配列番号4、配列番号8及び配列番号10の塩基配列からなる群より選択された塩基配列を有することを特徴とする請求項7に記載の遺伝子。
【請求項9】
請求項3に記載の遺伝子(ptsG)、及びスクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)を含有する組み換えベクター。
【請求項10】
前記スクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)は配列番号4の塩基配列で表されることを特徴とする請求項9に記載の組み換えベクター。
【請求項11】
請求項7に記載の遺伝子を含有する組み換えベクター。
【請求項12】
前記遺伝子は配列番号4、配列番号8及び配列番号10の塩基配列からなる群より選択された塩基配列を有する遺伝子であることを特徴とする請求項11に記載の組み換えベクター。
【請求項13】
細菌、酵母及びカビから構成された群より選択された宿主細胞に請求項9または請求項11に記載の組み換えベクターが導入されていることを特徴とするスクロース代謝能を有する組み換え微生物。
【請求項14】
大腸菌であることを特徴とする請求項13に記載の組み換え微生物。
【請求項15】
細菌、酵母及びカビから構成された群より選択された宿主細胞の染色体DNAに請求項3に記載の遺伝子(ptsG)及びスクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)が導入されていることを特徴とするスクロース代謝能を有する組み換え微生物。
【請求項16】
前記スクロース−6−リン酸ヒドロラーゼをコードする遺伝子(sacC)は配列番号4の塩基配列で表されることを特徴とする請求項15に記載の組み換え微生物。
【請求項17】
大腸菌であることを特徴とする請求項15に記載の組み換え微生物。
【請求項18】
細菌、酵母及びカビから構成された群より選択された宿主細胞の染色体DNAに請求項7に記載の遺伝子が導入されていることを特徴とするスクロース代謝能を有する組み換え微生物。
【請求項19】
前記遺伝子は配列番号4、配列番号8及び配列番号10の塩基配列からなる群より選択された塩基配列を有することを特徴とする請求項18に記載の組み換え微生物。
【請求項20】
大腸菌であることを特徴とする請求項18に記載の組み換え微生物。
【請求項21】
請求項13に記載の組み換え微生物を、スクロースを炭素源として含有する培地で培養することを特徴とする代謝産物、生分解性高分子、または遺伝子組み換え蛋白質の製造方法。
【請求項22】
請求項15または請求項18に記載の組み換え微生物を、スクロースを炭素源として含有する培地で培養することを特徴とする代謝産物、生分解性高分子、または遺伝子組み換え蛋白質の製造方法。
【図1】

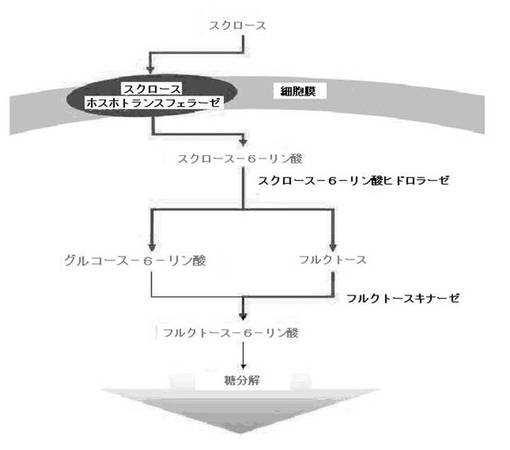
【図2】
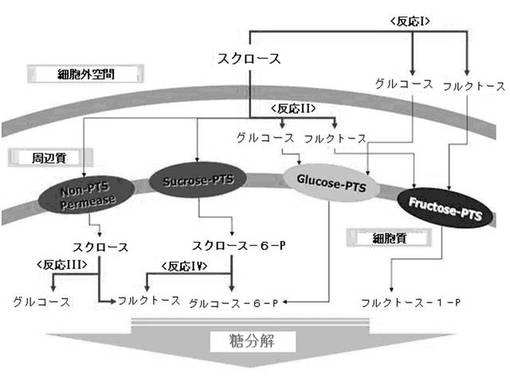
【図3】
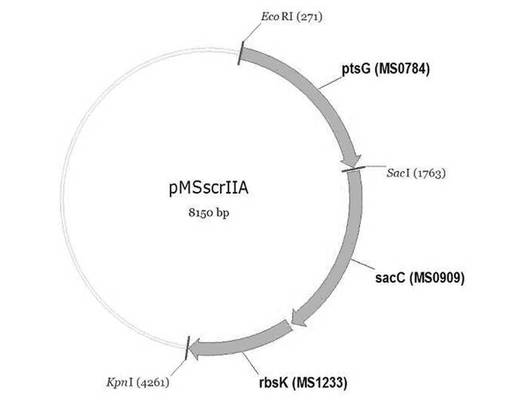
【図4】
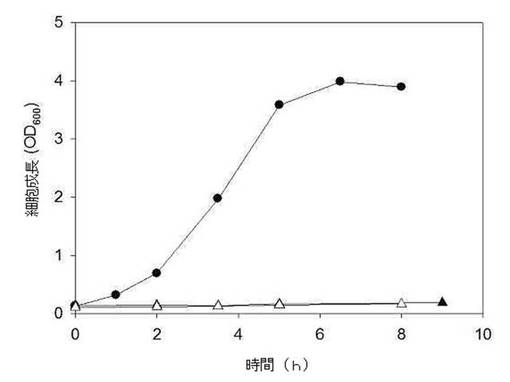
【図5】
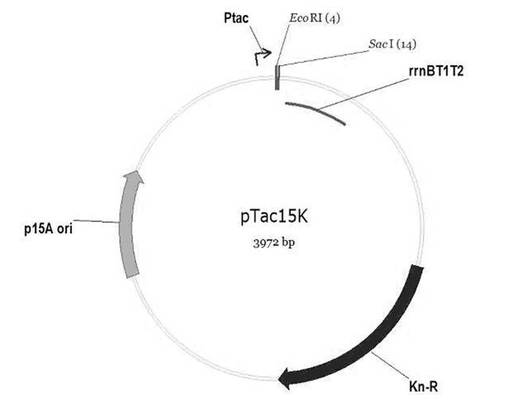
【図6】
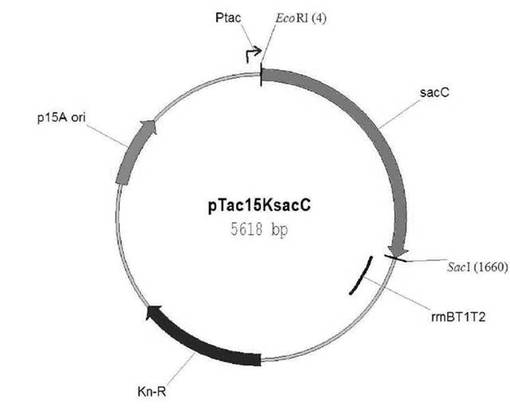
【図7】
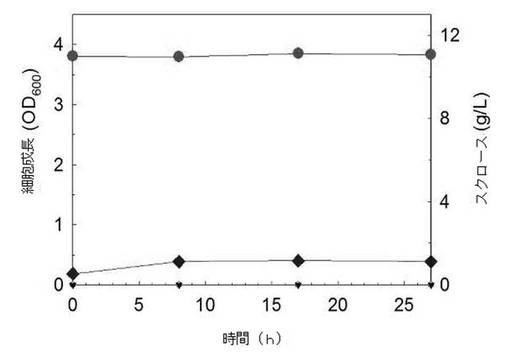
【図8】
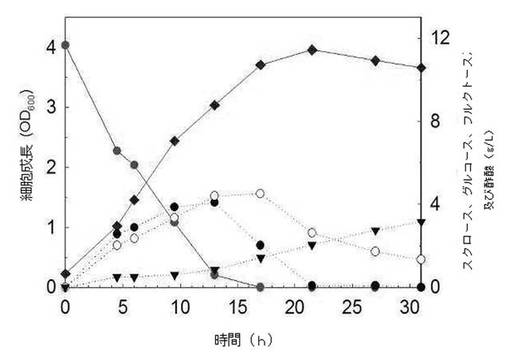
【図9】
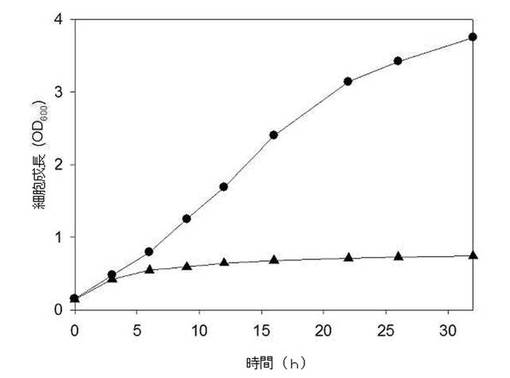
【図10】
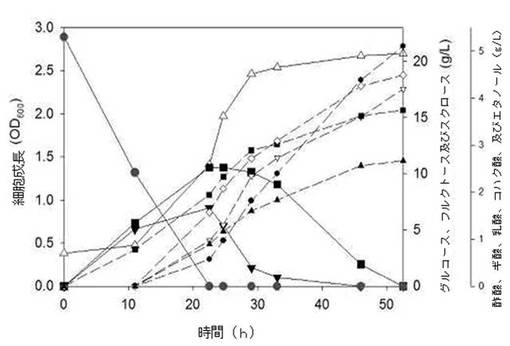
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2011−505869(P2011−505869A)
【公表日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願番号】特願2010−539302(P2010−539302)
【出願日】平成20年12月18日(2008.12.18)
【国際出願番号】PCT/KR2008/007533
【国際公開番号】WO2009/078687
【国際公開日】平成21年6月25日(2009.6.25)
【出願人】(502318478)コリア アドバンスド インスティチュート オブ サイエンス アンド テクノロジィ (27)
【Fターム(参考)】
【公表日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願日】平成20年12月18日(2008.12.18)
【国際出願番号】PCT/KR2008/007533
【国際公開番号】WO2009/078687
【国際公開日】平成21年6月25日(2009.6.25)
【出願人】(502318478)コリア アドバンスド インスティチュート オブ サイエンス アンド テクノロジィ (27)
【Fターム(参考)】
[ Back to top ]