
スチレン改質ポリオレフィン系樹脂粒子、発泡性樹脂粒子、予備発泡粒子及び発泡成形体
【課題】融着性と耐熱性を両方とも満足する発泡成型体を製造可能なスチレン改質ポリオレフィン系樹脂粒子を提供することを課題とする。
【解決手段】(A)密度[d(kg/m3)]が910以上950以下、(B)190℃、2.16kg荷重で測定したメルトフローレート[MFR(g/10分)]が0.1以上20以下、(C)末端ビニル数が1000炭素原子当たり0.2個以下、(D)160℃で測定した溶融張力[MS160(mN)]とMFRの関係が、MS160>90−130×log(MFR)を満足、(E)190℃で測定した溶融張力[MS190(mN)]とMS160の関係が、MS160/MS190<1.8を満足、(F)連続昇温溶出分別法による溶出温度−溶出量曲線にピークが2個以上存在、の要件を満足するポリオレフィン系樹脂を含むスチレン改質ポリオレフィン系樹脂粒子により上記課題を解決する。
【解決手段】(A)密度[d(kg/m3)]が910以上950以下、(B)190℃、2.16kg荷重で測定したメルトフローレート[MFR(g/10分)]が0.1以上20以下、(C)末端ビニル数が1000炭素原子当たり0.2個以下、(D)160℃で測定した溶融張力[MS160(mN)]とMFRの関係が、MS160>90−130×log(MFR)を満足、(E)190℃で測定した溶融張力[MS190(mN)]とMS160の関係が、MS160/MS190<1.8を満足、(F)連続昇温溶出分別法による溶出温度−溶出量曲線にピークが2個以上存在、の要件を満足するポリオレフィン系樹脂を含むスチレン改質ポリオレフィン系樹脂粒子により上記課題を解決する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、スチレン改質ポリオレフィン系樹脂粒子、発泡性樹脂粒子、予備発泡粒子及び発泡成形体に関する。更に詳しくは、本発明は、発泡成形体に優れた耐熱性や加熱寸法安定性を与えうるスチレン改質ポリオレフィン系樹脂粒子、発泡性樹脂粒子及び予備発泡粒子、それら粒子から得られた発泡成形体に関する。
【背景技術】
【0002】
従来から、ポリスチレン系樹脂からなる予備発泡粒子を型内に充填し、次いで加熱することで発泡させて得られる発泡成形体は、剛性、断熱性、軽量性、耐水性及び発泡成形性に優れていることが知られている。そのため、この発泡成形体は、緩衝材や建材用断熱材として広く用いられている。しかし、ポリスチレン系樹脂からなる発泡成形体は、耐薬品性及び耐衝撃性が劣るという課題があった。
【0003】
一方、ポリエチレンやポリプロピレン等のポリオレフィン系樹脂からなる発泡成形体は、耐薬品性及び耐衝撃性に優れていることが知られている。そのため、この発泡成形体は、自動車関連部品に使用されている。しかし、ポリオレフィン系樹脂は、発泡剤の保持性が劣ることから、発泡成形条件を精密に制御する必要がある。そのため製造コストが高くつくという課題があった。加えて、この発泡成形体は、ポリスチレン系樹脂からなる発泡成形体に比べて、剛性が劣るという課題もあった。
【0004】
上記ポリスチレン系樹脂及びポリオレフィン系樹脂からなる発泡成形体の課題を解決するために、ポリスチレン系樹脂とポリオレフィン系樹脂とを混合した粒子から得られた発泡成形体が報告されている。この発泡成形体は、ポリスチレン系樹脂の優れた剛性及び発泡成形性と、ポリオレフィン系樹脂の優れた耐薬品性及び耐衝撃性とを兼ね備えている。
ところで、自動車関連部品は、ガソリン、灯油、ブレーキオイル、塩化ビニルに使用される可塑剤等の薬品に接触したり、強い衝撃を受けたりすることがある。そのためこれら用途に使用される発泡成形体には、高い耐薬品性及び耐衝撃性が要求されているが、上記発泡成形体では不十分であった。
【0005】
そこで、上記要求を満たす方法として、ポリオレフィン系樹脂としてのエチレン−酢酸ビニル共重合体(EVA)、高圧法低密度ポリエチレン(LDPE)又は直鎖状低密度ポリエチレン(LLDPE)の粒子を、ポリスチレン系樹脂で改質し、改質粒子から発泡成形体を得る方法が提案されている(特開2005−97555号公報:特許文献1)が、上記公報に記載の方法では、高い耐薬品性及び耐衝撃性を有する発泡成形体が得られるものの、例えば、高温に晒されるようなより厳しい条件下での用途では、耐熱性や加熱寸法安定性が不足する場合があり、更に改善した発泡成形体の提供が望まれていた。
また、上記要求に答えうる技術として、特開2007−270116号公報(特許文献2)に記載された技術が提案されている。この公報に記載された技術では、より厳しい条件下での用途における、耐熱性や加熱寸法安定性がある程度確保されている。
【0006】
【特許文献1】特開2005−97555号公報
【特許文献2】特開2007−270116号公報
【発明の開示】
【発明が解決しようとする課題】
【0007】
特許文献2に記載されたスチレン改質ポリオレフィン系樹脂粒子は、発泡性の優れた発泡性樹脂粒子を提供できる。しかし、発泡成形体の曲げ強度、曲げ破断変位等の物性をより向上させる観点から、加熱寸法安定性に寄与する発泡粒子同士の融着性を更に改善可能なスチレン改質ポリオレフィン系樹脂粒子が望まれている。
発泡粒子同士の融着性を改善させるには、ポリオレフィン系樹脂の結晶化度を下げることが考えられる。しかし、結晶化度を単純に下げると、発泡成型体の耐熱性が低下することになる。
そのため、融着性と耐熱性を両方とも満足する発泡成型体を製造可能なスチレン改質ポリオレフィン系樹脂粒子の提供が望まれていた。
【課題を解決するための手段】
【0008】
本発明の発明者等は、耐熱性や加熱寸法安定性を更に改善するために、発泡成形体の原料を見直した結果、特定の性質を有するポリオレフィン系樹脂を使用することで、耐熱性や加熱寸法安定性を更に改善できることを見出し、本発明に至った。
かくして本発明によれば、エチレンから導かれる繰り返し単位、又はエチレンから導かれる繰り返し単位及び炭素数3〜8のα−オレフィンから導かれる繰り返し単位から構成され、かつ下記要件(A)〜(H)
(A)密度[d(kg/m3)]が910以上950以下
(B)190℃、2.16kg荷重で測定したメルトフローレート[MFR(g/10分)]が0.1以上20以下
(C)末端ビニル数が1000炭素原子当たり0.2個以下
(D)160℃で測定した溶融張力[MS160(mN)]とMFRの関係が、下記式(1)を満足
MS160>90−130×log(MFR) (1)
(E)190℃で測定した溶融張力[MS190(mN)]とMS160の関係が、下記式(2)を満足
MS160/MS190<1.8 (2)
(F)連続昇温溶出分別法(TREF)による溶出温度−溶出量曲線にピークが2個以上存在
の要件を満足するポリオレフィン系樹脂と、前記ポリオレフィン系樹脂を改質するスチレン系樹脂とを含み、前記スチレン系樹脂が、前記ポリオレフィン系樹脂100重量部に対して、20〜600重量部使用されることを特徴とするスチレン改質ポリオレフィン系樹脂粒子が提供される。
【0009】
更に、本発明によれば、上記スチレン改質ポリオレフィン系樹脂粒子100重量部と、揮発性発泡剤5〜25重量部とを含むことを特徴とする発泡性樹脂粒子が提供される。
また、本発明によれば、上記発泡性樹脂粒子を予備発泡させて得られた嵩密度0.01〜0.2g/cm3の予備発泡粒子が提供される。
更にまた、上記予備発泡粒子を型内発泡成形させて得られた密度0.01〜0.2g/cm3の発泡成形体が提供される。
【発明の効果】
【0010】
本発明によれば、特定の性質を有するポリオレフィン系樹脂を使用したスチレン改質ポリオレフィン系樹脂粒子により、耐熱性や加熱寸法安定性が更に改善された発泡成形体を提供できる。
【発明を実施するための最良の形態】
【0011】
(スチレン改質ポリオレフィン系樹脂粒子)
本発明のスチレン改質ポリオレフィン系樹脂粒子は、ポリオレフィン系樹脂粒子をスチレン系樹脂で改質した粒子(以下、改質樹脂粒子と称する)である。ここで、本発明は、改質樹脂粒子を構成するポリオレフィン系樹脂の量よりスチレン系樹脂の量が多い場合を含むが、本明細書では、その場合でもポリオレフィン系樹脂粒子がスチレン系樹脂で改質されているとする。
ポリオレフィン系樹脂粒子のスチレン系樹脂での改質方法は、特に限定されず、公知の方法を種々使用できるが、例えば、ポリオレフィン系樹脂粒子にスチレン系モノマーを含浸させ、次いでスチレン系モノマーを重合させる方法が挙げられる。
【0012】
(1)ポリオレフィン系樹脂
ポリオレフィン系樹脂は、エチレン単独重合体又はエチレンと炭素数3〜8のα−オレフィンとの共重合体からなる。上記炭素数3〜8のα−オレフィンとしては、プロピレン、1−ブテン、1−ヘキセン、1−オクテン、4−メチル−1−ペンテン、3−メチル−1−ブテン、ビニルシクロアルカン(例えば、ビニルシクロペンタン、ビニルシクロヘキサン)、環状オレフィン(例えば、ノルボルネン、ノルボルナジエン)、ジエン(例えば、ブタジエン、1,4−ヘキサジエン)等が挙げられる。炭素数3〜8のオレフィン由来の成分が、ポリオレフィン系樹脂に占める割合は、特に限定されないが、50重量%以下であることが好ましく、20重量%以下であることがより好ましい。なお、発明を阻害しない範囲で、スチレンをエチレンと共重合させてもよい。
【0013】
更に、ポリオレフィン系樹脂は、以下の要件を満足する樹脂である。
(A)密度[d(kg/m3)]が910以上950以下
(B)190℃、2.16kg荷重で測定したメルトフローレート[MFR(g/10分)]が0.1以上20以下
(C)末端ビニル数が1000炭素原子当たり0.2個以下
(D)160℃で測定した溶融張力[MS160(mN)]とMFRの関係が、下記式(1)を満足
MS160>90−130×log(MFR) (1)
(E)190℃で測定した溶融張力[MS190(mN)]とMS160の関係が、下記式(2)を満足
MS160/MS190<1.8 (2)
(F)連続昇温溶出分別法(TREF)による溶出温度−溶出量曲線にピークが2個以上存在。
【0014】
要件(A)において、密度[d(kg/m3)]は910kg/m3以上950kg/m3以下であり、特に925〜945kg/m3であることが好ましい。ここで、密度が910kg/m3未満のポリオレフィン系樹脂は、融解温度が低いため、得られる発泡成形体は耐熱性に劣るものとなる。一方、950kg/m3を超えるポリオレフィン系樹脂は、融解温度が高いため発泡成形温度が高くなり生産性が低下することに加えて、得られる発泡成形体は耐衝撃性に劣るものとなる。なお、密度の測定法は、実施例の欄に記載する。
【0015】
要件(B)において、190℃、2.16kg荷重で測定したメルトフローレート[MFR(g/10分)](以下、MFRと記す。)は0.1g/10分以上20g/10分以下である。ここで、MFRが0.1g/10分未満のポリオレフィン系樹脂は、発泡倍率が低下するため好ましくない。また、20g/10分を超えると溶融張力が小さくなり発泡倍率が低下することに加え、発泡成形体の強度も低下するため好ましくない。より好ましいMFRは2〜10g/10分である。MFRの測定法は、実施例の欄に記載する。
【0016】
要件(C)において、末端ビニル数は1000炭素原子当たり0.2個以下であり、特に0.05個以下であることが好ましい。ここで、末端ビニル数が1000炭素原子当たり0.2個を越えるポリオレフィン系樹脂は、熱劣化、とくに黄変が生じることがある。末端ビニル数の測定法は、実施例の欄に記載する。
【0017】
要件(D)において、160℃で測定した溶融張力[MS160(mN)](以下、MS160と記す。)と、190℃で、2.16kg荷重で測定したMFRとの関係が、下記式(1)を満足するものである。
MS160>90−130×log(MFR) (1)
特に、MS160は、MS160>110−130×log(MFR)を満足することが好ましい。
ここで、MS160が[90−130×log(MFR)]以下の範囲にあるオレフィン系樹脂は、ガスの保持力が不足し、発泡性が劣る場合がある。
【0018】
要件(E)において、190℃で測定した溶融張力[MS190(mN)](以下、MS190と記す。)とMS160の関係が、下記式(2)を満足するものである。
MS160/MS190<1.8 (2)
ここで、MS160/MS190が1.8以上のポリオレフィン系樹脂である場合、発泡温度による溶融張力が大きく変化することがある。そのため、成形温度の厳密な調節が必要となり、ひいては成形可能範囲が狭くなり、発泡成形性に劣るポリオレフィン系樹脂となる。より好ましいMS160/MS190は1.6より小さい値である。
なお、MS160とMS190の測定法は、実施例の欄に記載する。
【0019】
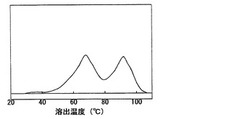
要件(F)において、連続昇温溶出分別法(以下、TREFと記す。)により求めた溶出温度−溶出量曲線においてピークが2個以上存在するポリオレフィン系樹脂である。更に、ポリオレフィン系樹脂は、2個のピークを有することが好ましい。特に2個のピークの内、結晶化度が上昇することから発泡成形体とした際の耐熱性及び剛性を向上できる観点から、高温側のピークは85〜100℃の間に存在することが好ましい。また、発泡粒子同士の融着特性に優れることから低温側のピークは65〜80℃の間に存在することが好ましい。
【0020】
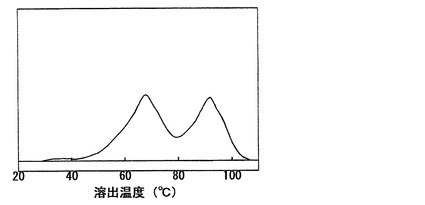
なお、図1に本発明に使用可能なポリオレフィン系樹脂の代表的なTREF溶出温度−溶出量曲線を示す。また、図2には代表的なメタロセン触媒により得られたエチレン・α−オレフィン共重合体のTREF溶出温度−溶出量曲線を示す。本発明に使用可能なポリオレフィン系樹脂は、組成分布が広いために溶出温度−溶出量曲線が2つのピークを有しており、1つのピークの従来のメタロセン触媒により得られるエチレン・α−オレフィン共重合体とは異なった樹脂である。図1及び2において、縦軸は溶出量を意味する。
なお、上記曲線の測定法は、実施例の欄に記載する。
【0021】
ポリオレフィン系樹脂は、更に、下記(G)及び(H)
(G)流動の活性化エネルギー[Ea(kJ/mol)]と密度の関係が、下記式(3)を満足
127−0.107d<Ea<88−0.060d (3)
(H)50℃におけるn−ヘプタン抽出量が0.2重量%以下
の要件を満足することが好ましい。
【0022】
要件(G)において、流動の活性化エネルギー[Ea(kJ/mol)](以下、Eaと記す。)と密度dとの関係が、Ea≦(127−0.107d)の場合、発泡成形性に問題が生じることがある。一方、Ea≧(88−0.060d)の場合、溶融粘度の温度依存性が大きく、成形温度の厳密な調節が必要となり、ひいては発泡成形可能範囲が狭くなるという問題が生じることがある。特に、Eaは、127−0.107d<Ea<87−0.060dの要件を満足することが好ましい。
なお、Eaの測定法は、実施例の欄に記載する。
【0023】
要件(H)において、50℃におけるn−ヘプタン抽出量は0.2重量%以下である。ここで、抽出量が0.2重量%を超えるポリオレフィン系樹脂である場合、融着性に劣るものとなる。より好ましい抽出量は、0.1重量%以下である。
更に、ポリオレフィン系樹脂は、特に機械強度に優れる発泡成形体を得ることが可能となることから、重量平均分子量(以下、Mwと記す。)と数平均分子量(以下、Mnと記す。)の比(Mw/Mn)が4.5以上7.5以下であることが好ましく、特に5.0以上7.0以下であることが好ましい(要件(I))。なお、Mw及びMnの測定法は、実施例の欄に記載する。
【0024】
本発明のエチレン系重合体の製造方法としては、上記要件(A)〜(F)(特に、要件(A)〜(I))を満足するポリオレフィン系樹脂の製造が可能であれば如何なる製造方法を用いることも可能である。例えば重合触媒及び/又は重合条件を多段階で変更する多段重合法、複数の重合触媒を混合した触媒による重合法、同一又は異なる重合触媒で調製した複数のポリオレフィン系樹脂をブレンドする方法等を挙げることができる。
【0025】
上記要件(A)〜(F)を満たし、好ましくは前記要件(A)〜(I)を満足するポリオレフィン系樹脂は、後述する実施例の製造条件そのもの、あるいは条件因子のマイナー変動によって任意に作り分けることが可能である。条件因子のマイナー変動とは、例えば、単独の重合触媒成分を複数の重合触媒成分の組み合わせに替えること(例えば、複数の重合触媒成分を用いる場合、各々を成分(a)及び成分(b)等とする)や重合条件を変更すること等が該当する。具体的には成分(a)及び成分(b)として重合触媒のメタロセン化合物の種類、担体種類あるいは調製方法の異なる重合触媒を用いることや、それらを組み合わせて用いる場合の成分(a)に対する成分(b)の重量比を変更することや、重合触媒と組み合わせて用いることのできる助触媒成分の種類及び量を変更する等重合触媒成分に関して操作することや、重合反応を行う際の重合温度、エチレン分圧、共存させる水素等の分子量調整剤の量、添加するコモノマー量等で示されるいわゆる重合条件を制御すること、によって作り分けることも可能である。また更に多段重合との組み合わせで、物性の範囲を拡大することも可能である。
【0026】
より具体的には、例えばエチレン分圧を低下させること、コモノマー添加量を減少させること、成分(a)の構造を変えること等によって、末端ビニル数を変化させることが可能である。また、溶融張力は、成分(a)の構造を変えること、末端ビニル数を増加させること、成分(b)の構造を変えること、エチレン分圧を低下させること、長鎖分岐数を増加させること、長鎖分岐長さを増加させること、成分(a)に対する成分(b)の量を変えること、Mw/Mnを増加させること等により増加させることが可能である。更に流動の活性化エネルギー(Ea)は、成分(a)の構造、末端ビニル数、成分(b)の構造、エチレン分圧、長鎖分岐数、長鎖分岐長さ、成分(a)に対する成分(b)の量により制御が可能である。
【0027】
ポリオレフィン系樹脂の製造に用いる重合触媒としては、例えば、特開2004−346304号公報、特開2005−248013号公報、特開2006−321991号公報、特開2007−169341号公報及び2008−050278号公報に記載の重合触媒及びそれらを公知の方法により組み合わせた重合触媒が挙げられる。
ポリオレフィン系樹脂の製造において、重合温度は−100〜120℃が好ましく、特に生産性を考慮すると20〜120℃、更には50〜120℃の範囲であることが好ましい。重合時間は10秒〜20時間の範囲が好ましく、重合圧力は常圧〜300MPaの範囲であることが好ましい。
【0028】
エチレンと炭素数3〜8のα−オレフィンからなるポリオレフィン系樹脂を使用する場合、エチレン/炭素数3〜8のα−オレフィン(モル比)は、1〜200であることが好ましく、より好ましくは3〜100、更に好ましくは5〜50である。また、重合時に水素等を用いて分子量の調節を行うことも可能である。
【0029】
更に、ポリオレフィン系樹脂は、マクロモノマーの存在下に、オレフィンを重合させることで得られた樹脂であることが好ましい。
具体的には、末端にビニル基を有するエチレンの単独重合体又はエチレンと炭素数3以上のオレフィンとの共重合体からなるマクロモノマーと、炭素数2以上のオレフィンとを共重合させることにより得られるポリオレフィン系樹脂である。
【0030】
ここで、マクロモノマーは、2000以上のMnと、2〜5のMw/Mnとを有していることが好ましい。炭素数3以上のオレフィンとしては、プロピレン、1−ブテン、1−ヘキセン、1−オクテン、4−メチル−1−ペンテン、3−メチル−1−ブテン、ビニルシクロアルカン(例えば、ビニルシクロペンタン、ビニルシクロヘキサン)等が挙げられる。これらオレフィンは、単独でも、2種以上の組み合わせでもよい。なお、炭素数2以上のオレフィンは、上記エチレンと炭素数3〜8のα−オレフィンから選択される。
マクロモノマーのMnは、5000以上であることがより好ましく、10000以上であることが更に好ましい。上限は、100000であることが好ましい。また、Mw/Mnは、2〜4であることがより好ましく、2〜3.5であることが更に好ましい。
【0031】
更に、マクロモノマーの主鎖であるメチレン炭素1000個当たりのビニル末端数をX、マクロモノマーの主鎖であるメチレン炭素1000個当たりの飽和末端数をYとした場合、式Z=X/[(X+Y)×2]で表されるZが0.25〜1であることが好ましい。Zは0.5〜1であることがより好ましい。なお、ビニル末端及び飽和末端は、1H−NMR、13C−NMR又はFT−IRによりその数を測定できることは、当業者によく知られている。例えば、13C−NMRの場合、ビニル末端は114ppmと139ppmに、飽和末端は32.3ppm、22.9ppm及び14.1ppmにピークを有し、このピークからその数を測定できる。
【0032】
上記マクロモノマーとオレフィンとを共重合させることで、本発明の使用に好適なポリオレフィン系樹脂が得られる。ここで、マクロモノマー以外の炭素数2以上のオレフィンに由来する樹脂(樹脂A)の全樹脂に対する割合は、1〜99重量%が好ましく、5〜90重量%がより好ましく、30〜80重量%が更に好ましい。樹脂Aの割合の測定は、樹脂のGPCチャートを、マクロモノマーのGPCチャートと比較することにより行うことができる。具体的には、両チャートの比較により樹脂Aに由来するピークを決定し、そのピークの面積の全ピークの面積に対する割合が、樹脂Aの割合に相当する。
重合はバッチ式、半連続式、連続式のいずれの方法でも行うことが可能であり、重合条件を変えて2段階以上に分けて行うことも可能である。また、ポリオレフィン系樹脂は、重合終了後に従来既知の方法により重合溶媒から分離回収され、乾燥して単離できる。
【0033】
重合はスラリー状態、溶液状態又は気相状態で実施することができ、特に、重合をスラリー状態で行う場合には粒子形状の整ったポリオレフィン系樹脂を効率よく、安定的に生産することができる。また、重合に用いる溶媒は一般に用いられる有機溶媒であればいずれでもよく、具体的には例えばベンゼン、トルエン、キシレン、プロパン、イソブタン、ペンタン、ヘキサン、ヘプタン、シクロヘキサン、ガソリン等が挙げられる。更には、プロピレン、1−ブテン、1−ヘキセン、1−オクテン等のオレフィン自身を溶媒として用いることもできる。
【0034】
ポリオレフィン系樹脂は、本発明の目的を逸脱しない限りにおいて、他の樹脂を含んでいてもよい。他の樹脂としては、上記(A)〜(F)の性質を少なくとも1つ備えていない他のポリオレフィン系樹脂が好ましい。他のポリオレフィン系樹脂としては、炭素数2〜20のα−オレフィン単独重合体及び共重合体が挙げられる。具体的には、高密度ポリエチレン、低密度ポリエチレン、直鎖状低密度ポリエチレン、ポリプロピレン、ポリ1−ブテン、ポリ(4−メチル−1−ペンテン)、ポリ1−ペンテン、エチレン/ブロピレン共重合体、エチレン/1−ブテン共重合体、プロピレン/1−ブテン共重合体、エチレン/プロピレン/1−ブテン共重合体、4−メチル−1−ペンテン/エチレン共重合体、エチレン/プロピレン/ポリエン共重合体、種々のプロピレン系ブロック共重合体やプロピレン系ランダム共重合体等が挙げられる。これら他の樹脂の配合割合は、全ポリオレフィン系樹脂量に対して、50重量%以下が好ましく、5〜30重量%がより好ましい。
【0035】
ポリオレフィン系樹脂には、必要に応じて、着色剤、安定剤、充填材(補強材)、高級脂肪酸金属塩、難燃剤、帯電防止剤、滑剤、天然又は合成油、ワックス、紫外線吸収剤、耐候安定剤、防曇剤、坑ブロッキング剤、スリップ剤、被覆剤、中性子遮蔽剤等の添加剤が含まれていてもよい。この内、着色剤としては、無機及び有機の着色剤(顔料又は染料)をいずれも使用できる。特に、酸化鉄、カーボンブラック等の無機着色剤が好ましい。
【0036】
酸化鉄としては、黄色系統のものとしてα−FeOOH(含水結晶)、赤色系統のものとしてα−Fe2O3、黒色系統のものとして(FeO)x(Fe2O3)y等が挙げられる。これら酸化鉄は、Feの一部が、Zn、Mg等の他の金属で置き換えられていてもよい。更に、これら酸化鉄は、所望の色を得るために、混合して用いてもよい。この内、黒色系統の(FeO)x(Fe2O3)yに含まれるFe3O4であることが好ましい。
【0037】
酸化鉄は、0.1〜1μmの平均粒径を有していることが好ましく、0.2〜0.8μmがより好ましい。平均粒径は、レーザー回折式粒度分布計(日本電子社製ロドス)により測定できる。酸化鉄は、ポリオレフィン系樹脂中、1.5〜70重量%の範囲で含まれていることが好ましく、5〜40重量%の範囲がより好ましく、10〜30重量%の範囲が更に好ましい。1.5重量%未満であれば、ポリオレフィン系樹脂が十分着色されない場合があるため好ましくない。70重量%より多い場合、ポリオレフィン系樹脂中に混合することが困難となり易い。加えて、酸化鉄の比重がポリオレフィン樹脂より大きいため、ポリオレフィン系樹脂粒子が重くなり、スチレン系モノマーを均一に含浸させることが困難となり易い。
【0038】
カーボンブラックとしては、ファーネスブラック、チャンネルブラック、サーマルブラック、アセチレンブラック、黒鉛、炭素繊維等が挙げられる。
カーボンブラックは、ポリオレフィン系樹脂中、1〜50重量%の範囲で含まれていることが好ましく、2〜30重量%の範囲がより好ましい。1重量%未満であれば、ポリオレフィン系樹脂が十分着色されない場合がある。50重量%より多い場合、ポリオレフィン系樹脂中に混合することが困難となり易い。
【0039】
安定剤は、酸化劣化や熱劣化等を防止する役割を果たし、公知物をいずれも使用できる。例えば、フェノール系安定剤、有機ホスファイト系安定剤、チオエーテル系安定剤、ヒンダードアミン系安定剤等が挙げられる。
充填材としては、タルク、ガラス等が挙げられ、その形状は球状、板状、繊維状等特に限定されない。
高級脂肪族金属塩としては、ステアリン酸、オレイン酸、ラウリン酸等の高級脂肪酸と、アルカリ土類金属(マグネシウム、カルシウム、バリウム等)やアルカリ金属(ナトリウム、カリウム、リチウム等)との塩が挙げられる。
【0040】
(2)スチレン系樹脂
スチレン系樹脂としては、例えば、スチレン、α−メチルスチレン、p−メチルスチレン、t−ブチルスチレン等のスチレン系モノマーに由来する樹脂が挙げられる。更に、スチレン系樹脂は、スチレン系モノマーと、スチレン系モノマーと共重合可能な他のモノマーとの共重合体からなる成分であってもよい。他のモノマーとしては、ジビニルベンゼンのような多官能性モノマーや、(メタ)アクリル酸ブチルのような構造中にベンゼン環を含まない(メタ)アクリル酸アルキルエステル等が例示される。これら他のモノマーを、スチレン系樹脂に対して5重量%を超えない範囲で使用してもよい。
【0041】
スチレン系樹脂の量は、ポリオレフィン系樹脂100重量部に対して20〜600重量部、好ましくは100〜550重量部、より好ましくは130〜500重量部である。また、600重量部を超える場合、発泡成形体の耐薬品性及び耐衝撃性が低下することがある。20重量部より少ない場合、発泡成形体の剛性が低下することがある。
【0042】
(3)改質樹脂粒子の形状
改質樹脂粒子は、粒子の長さをL、平均径をDとした場合のL/Dが0.6〜1.6である円筒状、略球状ないしは球状であり、平均粒径が0.3〜3.0mmであることが好ましい。L/Dが0.6より小さくないしは1.6より大きく扁平度が大きい場合は、改質樹脂粒子から得られる予備発泡粒子を、金型に充填して発泡成形体を得る際に、金型への充填性が悪くなることがある。また形状は、充填性をよくするには略球状ないしは球状がより好ましい。
平均粒径は0.3mm未満の場合、発泡性樹脂粒子に使用する場合、発泡剤の保持性が低くなり、低密度化が困難となり易い。3.0mmを超える場合、充填性が悪くなり易く、発泡成形体の薄肉化も困難となり易い。
【0043】
(発泡性樹脂粒子)
発泡性樹脂粒子は、上記改質樹脂粒子に揮発性発泡剤を含浸させた粒子である。揮発性発泡剤としては、例えば、プロパン、n−ブタン、イソブタン、ペンタン、イソペンタン、シクロペンタン、ヘキサン、ジメチルエーテル等が挙げられる。これら揮発性発泡剤は、単独もしくは2種以上混合して用いることができる。揮発性発泡剤の含有量は、改質樹脂粒子100重量部に対して、5〜25重量部であることが好ましい。
発泡性樹脂粒子のL/D及び平均粒径は、上記改質樹脂粒子と同程度とすることができる。また形状は、充填性をよくするには略球状ないしは球状がより好ましい。
【0044】
(予備発泡粒子)
予備発泡粒子は、上記発泡性樹脂粒子を予備発泡させて得られた粒子である。
予備発泡粒子は、0.01〜0.2g/cm3の嵩密度を有する。好ましい嵩密度は、0.014〜0.15g/cm3である。嵩密度が0.01g/cm3より小さい場合、発泡させたときに独立気泡率が低下して、予備発泡粒子から得られる発泡成形体の強度が低下することがある。一方、0.2g/cm3より大きい場合、得られる発泡成形体の重量が増加することがある。嵩密度の測定法は、実施例の欄に記載する。
【0045】
(発泡成形体)
発泡成形体は、上記予備発泡粒子を型内発泡成形させて得られた成形体である。
発泡成形体は、0.01〜0.2g/cm3の密度を有する。好ましい密度は、0.014〜0.15g/cm3である。嵩密度が0.01g/cm3より小さい場合、独立気泡率が多くなるため、強度が低下することがある。一方、0.2g/cm3より大きい場合、重量が増加することがある。密度の測定法は、実施例の欄に記載する。
【0046】
本発明の発泡成形体は、耐薬品性、衝撃強度及び剛性が優れていることに加えて、耐熱性や加熱寸法安定性が更に改善されている。
本発明の発泡成形体は、種々の用途に使用できるが、バンパーの芯材、ドア内装緩衝材等の車両用緩衝材、電子部品、各種工業資材、食品の搬送容器等の各種用途に使用できる。特に、車両用緩衝材に好適に使用できる。
【0047】
(改質樹脂粒子、発泡性樹脂粒子、予備発泡粒子及び発泡成形体の製造方法)
まず、改質樹脂粒子は、ポリオレフィン系樹脂とスチレン系樹脂とを溶融混練し、粒子状に切断することによって形成できるが、例えば、以下のように製造することが好ましい。すなわち、水性懸濁液中に、上記ポリオレフィン系樹脂の粒子100重量部と、スチレン系モノマー20〜600重量部と、重合開始剤とを分散させる。なお、スチレン系モノマーと重合開始剤とを予め混合して用いてもよい。
【0048】
ポリオレフィン系樹脂の粒子は、公知の方法により得ることができる。例えば、ポリオレフィン系樹脂を、必要に応じて無機核剤と添加剤と共に、押出機中で溶融混練して押出すことでストランドを得、得られたストランドを、空気中でカット、水中でカット、加熱しつつカットすることで、造粒する方法が挙げられる。
【0049】
ポリオレフィン系樹脂の粒子は、粒子の長さをL、平均径をDとした場合のL/Dが0.6〜1.6である円筒状、略球状ないしは球状であり、平均粒径が0.2〜1.5mmであることが好ましい。L/Dが0.6より小さくないしは1.6より大きく扁平度が大きい場合は、発泡性樹脂粒子として予備発泡させ、金型に充填して発泡成形体を得る際に、金型への充填性が悪くなり易い。また形状は、充填性をよくするには略球状ないしは球状がより好ましい。平均粒径は0.2mm未満の場合、発泡剤の保持性が低くなり、低密度化が困難となり易いので好ましくない。1.5mmを超える場合、充填性が悪くなるだけでなく発泡成形体の薄肉化も困難となり易い。
【0050】
無機核剤としては、例えば、タルク、二酸化珪素、マイカ、クレー、ゼオライト、炭酸カルシウム等が挙げられる。無機核剤の使用量は、ポリオレフィン系樹脂100重量部に対して、2重量部以下が好ましく、0.2〜1.5重量部がより好ましい。
水性懸濁液を構成する水性媒体としては、水、水と水溶性溶媒(例えば、低級アルコール)との混合媒体が挙げられる。
【0051】
重合開始剤としては、一般にスチレン系モノマーの懸濁重合用の開始剤として用いられているものが使用できる。例えば、ベンゾイルパーオキサイド、ジt−ブチルパーオキサイド、t−ブチルパーオキシベンゾエート、ジクミルパーオキサイド、2,5−ジメチル−2,5−ジ−t−ブチルパーオキシヘキサン、t−ブチルパーオキシ−3,5,5−トリメチルヘキサノエート、t−ブチル−パーオキシ−2−エチルヘキシルカーボネート等の有機化過酸化物である。これらの重合開始剤は単独もしくは2種以上を併用してもよい。
【0052】
重合開始剤の使用量は、スチレン系モノマー100重量部に対して、0.1〜0.9重量部が好ましく、0.2〜0.5重量部がより好ましい。0.1重量部未満ではスチレン系モノマーの重合に時間がかかり過ぎることがある。0.9重量部を超える重合開始剤の使用は、スチレン系樹脂の分子量が低くなることがある。
【0053】
水系懸濁液には、必要に応じて分散剤を添加してもよい。分散剤としては、特に限定されず、公知のものをいずれも使用することができる。具体的には、リン酸カルシウム、ピロリン酸マグネシウム、ピロリン酸ナトリウム、酸化マグネシウム等の難溶性無機物が挙げられる。更に、ドデシルベンゼンスルホン酸ソーダのような界面活性剤を使用してもよい。
【0054】
次に、得られた分散液をスチレン系モノマーが実質的に重合しない温度に加熱してスチレン系モノマーをポリオレフィン系樹脂粒子に含浸させる。
ポリオレフィン系樹脂粒子内部にスチレン系モノマーを含浸させる時間は、30分〜2時間が適当である。十分に含浸させる前に重合が進行するとスチレン系樹脂の重合体粉末を生成してしまうからである。前記モノマーが実質的に重合しない温度とは、高い方が含浸速度を速めるには有利であるが、重合開始剤の分解温度を考慮して決定する必要がある。
【0055】
次いで、スチレン系モノマーの重合を行う。重合は、特に限定されないが、105〜140℃で、1.5〜5時間行うことが好ましい。重合は、通常、加圧可能な密閉容器中で行われる。
なお、スチレン系モノマーの含浸と重合を複数回に分けて行ってもよい。複数回に分けることで、ポリスチレンの重合体粉末の発生を極力少なくできる。
上記工程により改質樹脂粒子を得ることができる。得られた改質樹脂粒子は、内部がスチレン系樹脂粒子がリッチであり、外殻部がポリオレフィン樹脂がリッチであるため、発泡成形体の物性に好影響を与えると発明者等は考えている。
【0056】
次に、発泡性樹脂粒子は、上記重合中もしくは重合終了後の改質樹脂粒子に発泡剤を含浸することで得ることができる。この含浸は、それ自体公知の方法により行うことができる。例えば、重合中での含浸は、重合反応を密閉式の容器中で行い、容器中に発泡剤を圧入することにより行うことができる。重合終了後の含浸は、密閉式の容器中で、発泡剤を圧入することにより行われる。
更に、予備発泡粒子は、上記発泡性樹脂粒子を、公知の方法で所定の嵩密度に予備発泡させることで得ることができる。
更に、発泡成形体は、予備発泡粒子を発泡成形機の金型内に充填し、再度加熱して予備発泡粒子を発泡させながら、発泡粒同士を熱融着させることで得ることができる。加熱用の媒体は水蒸気が好適に使用できる。
【実施例】
【0057】
以下、実施例により本発明を具体的に説明するが、本発明はこれに限定されるものではない。なお、断りのない限り用いた試薬等は市販品を用いた。有機化合物で処理された粘土鉱物の調製、エチレン系重合体製造用触媒の調製、エチレン系重合体の製造及び溶媒精製は全て不活性ガス雰囲気下で行った。また、溶媒は全て予め公知の方法で精製、乾燥、脱酸素を行ったものを用いた。トリイソブチルアルミニウムのヘキサン溶液(0.714M)は東ソーファインケム社製を用いた。
【0058】
以下の実施例におけるポリオレフィン系樹脂の密度、MFR、末端ビニル数、溶融張力(MS160、MS190)、溶出温度−溶出量曲線、活性化エネルギーEa、n−ヘプタン抽出量、分子量(Mn、Mw)、融点及び融着強度、予備発泡粒子の嵩密度、発泡成形体の密度、融着率、加熱寸法変化率及び耐薬品性の測定法を下記する。
【0059】
<密度(d)の測定>
JIS K6760(1995)に準拠して密度勾配管法で測定する。
<MFR>
MFRは、JIS K6922−1:1998に準拠して、190℃、2.16kg荷重で測定する。
【0060】
<末端ビニル数の測定>
フーリエ変換赤外分光光度計(FT−IR)(Perkin Elmer社製、商品名SPECTRUM ONE)を用いて、エチレン系重合体を熱プレスした後、氷冷して調製したフィルムを4000cm-1〜400cm-1の範囲で吸光度を測定し、下式を用い算出する。
1000炭素原子当たりの末端ビニル数(個/1000C)=a×A/L/d
式中、aは吸光光度係数、Aは末端ビニルに帰属する909cm-1の吸光度、Lはフィルムの厚み、dは密度を示す。なお、aは、1H−NMR測定より、1000炭素原子当たりの末端ビニル数を確認したサンプルを用いて作成した検量線から求める。1H−NMR測定は、核磁気共鳴測定装置(日本電子社製、商品名GSX400)を用い、重水素化ベンゼンとo−ジクロロベンゼンの混合溶媒中、130℃において実施する。1000炭素原子当たりの末端ビニル数は、メチレンに帰属するピークと末端ビニルに帰属するピークの積分比から算出する。テトラメチルシランを基準(0ppm)として、化学シフトが1.3ppmのピークをメチレン、4.8−5.0ppmのピークを末端ビニルに帰属するピークとする。)
【0061】
<溶融張力(MS160、MS190)の測定>
ポリオレフィン系樹脂に、耐熱安定剤(チバスペシャリティケミカルズ社製、イルガノックス1010TM;1500ppm、イルガフォス168TM;1500ppm)を添加したものを、インターナルミキサー(東洋精機製作所製、商品名ラボプラストミル)を用いて、窒素気流下、190℃、回転数30rpmで30分間混練したもの測定用試料とする。
溶融張力は、バレル直径9.55mmの毛管粘度計(東洋精機製作所、商品名キャピログラフ)に、長さが8mm、直径が2.095mmのダイス状の試料を流入角が90°になるように装着して溶融張力を測定する。
【0062】
MS160は、温度を160℃に設定し、ピストン降下速度を10mm/分、延伸比を47に設定し、引き取りに必要な荷重(mN)である。なお、最大延伸比が47未満の場合、破断しない最高の延伸比での引き取りに必要な荷重(mN)をMS160とする。また、温度を190℃に設定すること以外は、160℃と同様の方法で測定した荷重(mN)をMS190とする。
【0063】
<TREFにより溶出温度−溶出量曲線の測定>
ポリオレフィン系樹脂に、耐熱安定剤(チバスペシャリティケミカルズ社製、イルガノックス1010TM;1500ppm、イルガフォス168TM;1500ppm)を添加したものを、インターナルミキサー(東洋精機製作所製、商品名ラボプラストミル)を用いて、窒素気流下、190℃、回転数30rpmで30分間混練する。混練物を、ODCBに、その濃度が0.05重量%となるように135℃で加熱溶解する。この加熱溶液5ミリリットルを、ガラスビーズを充填したカラムに注入した後、0.1℃/分の冷却速度で25℃まで冷却し、試料をガラスビーズ表面に沈着する。次に、このカラムにODCBを一定流量で流しながら、カラム温度を50℃/hrの一定速度で昇温し、各温度において溶液に溶解可能な試料を準備溶出させる。
【0064】
この際、溶剤中の試料濃度はメチレンの非対象伸縮振動の波数2925cm-1に対する吸収を赤外検出器で連続的に検出することで得られる。連続的に検出された濃度から、溶出温度−溶出量曲線を得ることができる。TREF分析は極少量の試料で、温度変化に対する溶出速度の変化を連続的に分析できるため、分別法では検出できない比較的細かいピークの検出が可能である。
【0065】
<流動の活性化エネルギー(Ea)の測定>
ポリオレフィン系樹脂に、耐熱安定剤(チバスペシャリティケミカルズ社製、イルガノックス1010TM;1500ppm、イルガフォス168TM;1500ppm)を添加したものを、インターナルミキサー(東洋精機製作所製、商品名ラボプラストミル)を用いて、窒素気流下、190℃、回転数30rpmで30分間混練したもの測定用試料とする。
【0066】
Eaは、円板−円板レオメーター(アントンパール社製、商品名MCR−300)を用い、150℃、170℃、190℃の各温度で角速度0.1〜100rad/sの範囲のせん断貯蔵弾性率G’、せん断損失弾性率G”を求める。両弾性率から、基準温度150℃での横軸のシフトファクターを求め、以下のアレニウス型の式により計算した。
粘度(ηo)=Aexp(Ea/RT)
式中、Aは温度に無関係な定数(頻度因子)、Eaは流動の活性化エネルギー(kJ/mol)、Rは気体定数、Tは絶対温度(単位:K)である。なお、縦軸の移動は行っていない。
【0067】
具体的には、流動の活性化エネルギー(Ea)は、温度−時間重ね合わせ原理に基づいて、150℃での剪断貯蔵弾性率G'、せん断損失弾性率G"(単位:Pa)の角周波数(単位:rad/sec)依存性を示すマスターカーブを作成する際のシフトファクター(aT)からアレニウス型方程式により算出される数値であって、以下に示す方法で求められる値である。すなわち、150℃、170℃及び190℃夫々の温度(t、単位:℃)におけるオレフィン系樹脂の弾性率−角周波数曲線を、温度−時間重ね合わせ原理に基づいて、各温度(t)での溶融複素粘度−角周波数曲線毎に、150℃での弾性率−角周波数曲線に重ね合わせた際に得られる各温度(t)でのシフトファクター(aT)を求め、夫々の温度(t)と、各温度(t)でのシフトファクター(aT)とから、最小自乗法により[ln(aT)]と[1/(t+273.16)]との一次近似式(下記(I)式)を算出する。次に、該一次近似式(I)の傾きmを用いて下記式(II)より流動の活性化エネルギー(Ea)を求める。
ln(aT)=m(1/(t+273.16))+n (I)
(I)式中、aTはシフトファクター、tは温度(単位:℃)、mは一次近似式(I)の傾き、nは一次近似式(I)の切片である。なお、温度t(単位:℃)を絶対温度T(単位:K)に換算するには、T=t+273.16で行う。
Ea=|0.008314×m| (II)
(II)式中、Eaは流動の活性化エネルギー(単位:kJ/mol)、mは一次近似式(I)の傾きである。
【0068】
なお、シフトファクター(aT)は、夫々の温度(t)における弾性率−角周波数の両対数曲線を、log(Y)=−log(X)軸方向に移動させて(但し、Y軸を弾性率、X軸を角周波数とする。)、150℃での弾性率−角周波数曲線に重ね合わせた際の移動量であり、該重ね合わせでは、夫々の温度(t)における弾性率−角周波数の両対数曲線は、各曲線ごとに、角周波数をaT倍に、弾性率を1/aT倍に移動させる。また、150℃、170℃及び190℃の3点の値から(I)式を最小自乗法で求めるときの相関係数は、通常、0.99以上である。
【0069】
<50℃におけるn−ヘプタン抽出量>
50℃におけるn−ヘプタン抽出量の測定方法は以下の通りである。
200メッシュパスの粉砕試料10gを秤量し、400ミリリットルのn−ヘプタンを加えて、50℃で2時間抽出を行う。抽出液からn−ヘプタンを蒸発させて、乾燥固化させて得た抽出物の重量の初期重量(10g)に対する割合を求めることにより算出する。
【0070】
<重量平均分子量(Mw)、数平均分子量(Mn)及び重量平均分子量と数平均分子量の比(Mw/Mn)の測定>
ゲル・パーミエーション・クロマトグラフィー(以下、GPC)によって上記値を次のように測定する。GPC装置(東ソー社製 商品名HLC−8121GPC/HT、カラム(東ソー社製 商品名TSKgel GMHhr−H(20)HTを装着)、溶離液1,2,4−トリクロロベンゼンを使用し、カラム温度を140℃とする。測定試料は1.0mg/ミリリットルの濃度で調製し、カラムに0.3ミリリットル注入する。GPCにより得られた溶出曲線から検量線を用いて分子量を求める。分子量の検量線は、分子量既知のポリスチレン試料を用いて校正したものを用いる。なお、Mw及びMnは直鎖状ポリエチレン換算の値として求める。
【0071】
<融点の測定>
DSC(パーキンエルマー社製、商品名:DSC−7)を用いて測定を行なう。具体的には、5〜10mgのサンプルをアルミニウムパンに挿填し、DSCに設置した後、80℃/分の昇温速度で230℃まで昇温し、230℃で3分間放置する。その後、10℃/分の降温速度で−10℃まで冷却し、再度10℃/分の昇温速度で−10℃から150℃まで昇温する手順で昇温/降温操作を行い、2回目の昇温時に観測される吸熱曲線のピーク温度を融点とする。
【0072】
<ポリオレフィン系樹脂の融着強度の評価>
ポリオレフィン系樹脂のペレットを、圧縮成形機(180℃、圧力0.2MPa)を使用して厚さ1mmのシートに成形する。得られたシートを、東洋精機社製熱版式ヒートシーラー(シール温度:融点+5℃、シール圧力:0.07MPa、シール時間:35秒)を使用して融着させる。融着強度は引張試験機テンシロン(オリエンテック社製)を用い、引張速度50mm/分で測定する。
【0073】
<嵩密度>
予備発泡粒子の嵩密度は、下記の要領で測定する。
まず、予備発泡粒子をメスシリンダに500cm3の目盛りまで充填する。但し、メスシリンダを水平方向から目視し、予備発泡粒子が一粒でも500cm3の目盛りに達していれば、充填を終了する。次に、メスシリンダ内に充填した予備発泡粒子の重量を小数点以下2位の有効数字で秤量し、その重量をW(g)とする。次式により予備発泡粒子の嵩密度を算出する。
嵩密度(g/cm3)=W/500
<発泡成形体の密度>
発泡成形体の密度は、JIS A 9511:1995「発泡プラスチック保温板」記載の方法で測定する。
【0074】
<融着率>
縦400mm×横300mmの上面を有し、厚み30mmの直方体形状の発泡成形体の上面に、カッターで横方向に沿って長さ300mm、深さ約5mmの切り込み線を入れ、この切り込み線に沿って発泡成形体を二分割する。そして、二分割された発泡成形体の破断面の発泡粒子について、発泡粒子内で破断している発泡粒子数(a)と、発泡粒子間の界面で破断している発泡粒子数(b)を測定し、下記式に基づいて融着率を算出する。
融着率(%)=100×(a)/〔(a)+(b)〕
【0075】
<加熱寸法変化率>
加熱寸法変化率は、JIS K6767:1999「発泡プラスチック−ポリエチレン−試験方法」記載のB法にて測定する。発泡成形体から縦150mm×横150mmの上面を有し、厚み50mmの試験片を切り出す。この試験片の上面の中央部に縦及び横方向に沿ってそれぞれ互いに平行に3本の直線を50mm間隔になるように記入する。そして、試験片を90℃の熱風循環式乾燥機の中に22時間置いた後に取り出し、標準状態(20±2℃、湿度65±5%)の場所に1時間放置後、縦及び横線の寸法を測定する。加熱前の直線の長さの平均値をL1、加熱後の直線の長さの平均値をL0とし、下記式に基づいて加熱寸法変化率を算出する。
加熱寸法変化率S=100×(L1−L0)/L0
加熱寸法変化率の評価は以下の通り
○:0≦S<3;寸法変化率が低く、寸法の安定性が良好である。
△:3≦S<7;寸法の変化が見られるものの、実用上使用可能である。
×:S≧7;寸法の変化が著しく見られ、実用上使用不可能である。
【0076】
<耐薬品性>
発泡成形体から縦100mm×横100mm×厚み20mmの平面長方形状の板状試験片を切り出し、23℃、湿度50%の条件で24時間放置する。なお、試験片の上面全面が発泡成形体の表面から形成されるように試験片を発泡成形体から切り出す。
次に、試験片の上面にガソリン1gを均一に塗布し、23℃、湿度50%の条件で60分放置する。その後、試験片の上面から薬品を拭き取り、試験片の上面を目視観察して下記基準に基づいて判断する。
○:良好 変化なし
△:やや悪い 表面軟化
×:悪い 表面陥没(収縮)
【0077】
製造例1
[変性ヘクトライトの調製]
水3リットルにエタノール3リットルと37%濃塩酸100ミリリットルを加えた後、得られた溶液にN,N−ジメチル−オクタデシルアミン330g(1.1mol)を添加し、60℃に加熱することによって、塩酸塩溶液を調製した。この溶液にヘクトライト1kgを懸濁させた。この懸濁液を60℃で、3時間撹拌し、上澄液を除去した後、60℃の水50Lで洗浄した。その後、60℃、10-3torrで24時間乾燥し、ジェットミルで粉砕することによって、平均粒径5.2μmの変性ヘクトライトを得た。
【0078】
[重合触媒(p)の調製]
上記変性ヘクトライト500gをヘキサン1.7リットルに懸濁させ、1,1,3,3−テトラメチルジシロキサン−1,3−ジイル−ビス(シクロペンタジエニル)ジルコニウムジクロライド8.45g(20.0mmol)とトリイソブチルアルミニウムのヘキサン溶液(0.714M)2.8リットル(2mol)の混合液を添加し、60℃で3時間攪拌した。この後、1,1,3,3−テトラメチルジシロキサン−1,3−ジイル−ビス(シクロペンタジエニル)ジルコニウムジクロライドに対して15mol%のジフェニル(1−シクロペンタジエニル)(2,7−ジ−tert−ブチル−9−フルオレニル)ジルコニウムジクロライド2.36g(3.53mmol)を添加して室温で6時間撹拌した。静置して上澄み液を除去、更にトリイソブチルアルミニウムのヘキサン溶液(0.15M)を添加して最終的に100g/Lの触媒スラリーを得た。
【0079】
[重合触媒(q)の調製]
前記変性ヘクトライト500gをヘキサン1.7リットルに懸濁させ、プロパン−1,3−ジイルビス(シクロペンタジエニル)ジルコニウムジクロリド6.63g(20.0mmol)とトリイソブチルアルミニウムのヘキサン溶液(0.714M)2.8リットル(2mol)の混合液を添加し、60℃で3時間攪拌した後、プロパン−1,3−ジイルビス(シクロペンタジエニル)ジルコニウムジクロリドに対して5mol%のジフェニルメチレン(1−シクロペンタジエニル)(9−フルオレニル)ジルコニウムジクロリド0.58g(1.05mmol)を添加して室温で6時間撹拌した。静置して上澄み液を除去、更にトリイソブチルアルミニウムのヘキサン溶液(0.15M)を添加して最終的に100g/Lの触媒スラリーを得た。
【0080】
[ポリオレフィン系樹脂の製造]
内容積540リットルの重合器に、ヘキサン300リットル及び1−ブテン1.6リットルを導入し、オートクレーブの内温を80℃に昇温した。このオートクレーブに前記重合触媒(p)74ミリリットル及び前記重合触媒(q)125ミリリットルを添加し、エチレン/水素混合ガス(水素:1500ppm含)を分圧が0.9MPaになるまで導入して重合を開始した。重合中、分圧が0.9MPaに保たれるようにエチレン/水素混合ガスを連続的に導入した。また、重合温度を80℃に制御した。重合開始90分後に重合器の内圧を脱圧した後、内容物をろ過し、乾燥して54kgのポリオレフィン系樹脂粉末を得た。ポリオレフィン系樹脂粉末を200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでポリオレフィン系樹脂ペレットを得た。得られたポリオレフィン系樹脂ペレットの密度は937kg/m3、MFRは8g/10分であった。
【0081】
製造例2
[重合触媒(r)の調製]
上記変性ヘクトライト500gをヘキサン1.7リットルに懸濁させ、ジメチルシランジイルビス(シクロペンタジエニル)ジルコニウムジクロリド6.97g(20.0mmol)とトリイソブチルアルミニウムのヘキサン溶液(0.714M)2.8リットル(2mol)の混合液を添加し、60℃で3時間攪拌した。この後、ジメチルシランジイルビス(シクロペンタジエニル)ジルコニウムジクロリドに対して15mol%のジフェニル(1−シクロペンタジエニル)(2,7−ジ−tert−ブチル−9−フルオレニル)ジルコニウムジクロライド2.36g(3.53mmol)を添加して室温で6時間撹拌した。静置して上澄み液を除去、更にトリイソブチルアルミニウムのヘキサン溶液(0.15M)を添加して最終的に100g/Lの触媒スラリーを得た。
【0082】
[ポリオレフィン系樹脂(R)の製造]
内容積540リットルの重合器に、ヘキサン300リットル及び1−ブテン1.6リットルを導入し、オートクレーブの内温を80℃に昇温した。このオートクレーブに[重合触媒(r)の調製]で調製した重合触媒(r)147ミリリットルを添加し、エチレン/水素混合ガス(水素:1000ppm含)を分圧が0.9MPaになるまで導入して重合を開始した。重合中、分圧が0.9MPaに保たれるようにエチレン/水素混合ガスを連続的に導入した。また、重合温度を80℃に制御した。重合開始90分後に重合器の内圧を脱圧した後、内容物をろ過し、乾燥して50kgのポリオレフィン系樹脂(R)粉末を得た。ポリオレフィン系樹脂粉末を200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでポリオレフィン系樹脂(R)ペレットを得た。
得られたポリオレフィン系樹脂(R)ペレットの密度は945kg/m3、MFRは10g/10分であった。
【0083】
[ポリオレフィン系樹脂(Q)の製造]
内容積540リットルの重合器に、ヘキサン300リットル及び1−ブテン6.1リットルを導入し、オートクレーブの内温を80℃に昇温した。このオートクレーブに合成例1で得られた重合触媒(q)250ミリリットルを添加し、エチレン/水素混合ガス(水素:2000ppm含)を分圧が0.9MPaになるまで導入して重合を開始した。重合中、分圧が0.9MPaに保たれるようにエチレン/水素混合ガスを連続的に導入した。また、重合温度を80℃に制御した。重合開始90分後に重合器の内圧を脱圧した後、内容物をろ過し、乾燥して53kgのポリオレフィン系樹脂(Q)粉末を得た。この粉末を200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでポリオレフィン系樹脂(Q)ペレットを得た。
得られたポリオレフィン系樹脂(Q)ペレット)の密度は930kg/m3、MFRは4g/10分であった。
【0084】
[ポリオレフィン系樹脂の調製]
前記ポリオレフィン系樹脂(R)と前記ポリオレフィン系樹脂(Q)を50対50の割合でブレンドし、200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでポリオレフィン系樹脂ペレットを得た。混合物の密度は938kg/m3、MFRは7.9g/10分であった。
【0085】
製造例3
[ポリオレフィン系樹脂の製造方法]
内容積540リットルのオートクレーブに、ヘキサン300リットル及び1−ブテン10.8リットルを導入し、オートクレーブの内温を60℃に昇温した。このオートクレーブに製造例1で調製した重合触媒(p)74ミリリットル及び重合触媒(q)125ミリリットルを添加し、エチレンを分圧が0.9MPaに保たれるように連続的に導入した。また、重合温度を60℃に制御した。重合開始30分後に1−ブテン9.1リットルを添加し、重合開始90分後に重合器(オートクレーブ)の内圧を脱圧した後、内容物をデカンテーションし、真空乾燥することで36kgのポリオレフィン系樹脂を得た。ポリオレフィン系樹脂を200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでエチレン系共重合体ペレットを得た。得られたポリオレフィン系樹脂のペレットの密度は915kg/m3、MFRは6.1g/10分であった。
【0086】
製造例4
[ポリオレフィン系樹脂の製造方法]
製造例1のポリオレフィン系樹脂の製造において、エチレン/水素混合ガスの代わりにエチレンを単独に用い、オートクレーブの内温を70℃に保つ以外、同様な方法で重合反応を行った。内容物をろ過し、乾燥して35kgのポリオレフィン系樹脂の粉末を得た。製造例1と同様にペレタイズすることで得られたポリオレフィン系樹脂のペレットの密度は935kg/m3、MFRは0.15g/10分であった。
【0087】
製造例5
[ポリオレフィン系樹脂の製造方法]
製造例1のポリオレフィン系樹脂の製造において、エチレン/水素混合ガスの濃度を3200ppmとした以外、同様な方法で重合反応を行った。内容物をろ過し、乾燥して57kgのポリオレフィン系樹脂の粉末を得た。製造例1と同様にペレタイズすることで得られたポリオレフィン系樹脂のペレットの密度は939kg/m3、MFRは18.8g/10分であった。
【0088】
製造例6
[ポリオレフィン系樹脂の製造方法]
製造例1のポリオレフィン系樹脂の製造において、エチレン/水素混合ガスの代わりにエチレンを単独に用い、オートクレーブの内温を60℃に保つ以外、同様な方法で重合反応を行った。内容物をろ過し、乾燥して21kgのポリオレフィン系樹脂の粉末を得た。製造例1と同様にペレタイズすることで得られたポリオレフィン系樹脂のペレットの密度は935kg/m3、MFRは0.06g/10分であった。
【0089】
製造例7
[ポリオレフィン系樹脂の製造方法]
製造例1のポリオレフィン系樹脂の製造において、エチレン/水素混合ガスの濃度を4800ppmとした以外、同様な方法で重合反応を行った。内容物をろ過し、乾燥して51kgのポリオレフィン系樹脂の粉末を得た。製造例1と同様にペレタイズすることで得られたポリオレフィン系樹脂のペレットの密度は939kg/m3、MFRは22.5g/10分であった。
【0090】
製造例8
[ポリオレフィン系樹脂の製造方法]
製造例2のポリオレフィン系樹脂の製造において、1−ブテンの量を最初18.4リットルとし、重合開始の30分後に9.5リットル添加した以外、同様な方法で重合反応を行った。重合開始30分後に1−ブテンを9.5リットル添加した時点で重合温度の制御が不能となり反応を停止した。内容物を乾燥させ12kgのポリオレフィン系樹脂を得た。ポリオレフィン系樹脂の密度は908kg/m3、MFRは2.0g/10分であった。
【0091】
製造例9
[ポリオレフィン系樹脂の製造方法]
製造例1のポリオレフィン系樹脂の製造において、エチレン/水素混合ガスの濃度を1000ppmとし、1−ブテンの量を0.9リットルとした以外、同様な方法で重合反応を行った。内容物をろ過し、乾燥して58kgのポリオレフィン系樹脂の粉末を得た。製造例1と同様にペレタイズすることで得られたポリオレフィン系樹脂のペレットの密度は955kg/m3、MFRは4.5g/10分であった。
【0092】
製造例10
[ポリオレフィン系樹脂の製造方法]
内容積540リットルの重合器(オートクレーブ)に、ヘキサン300リットル及び1−ブテン9.5リットルを導入し、オートクレーブの内温を80℃に昇温した。このオートクレーブに製造例2で調製した重合触媒(r)200ミリリットルを添加し、エチレンを分圧が0.9MPaに保たれるように連続的に導入した。また、重合温度を80℃に制御した。重合開始90分後に重合器(オートクレーブ)の内圧を脱圧した後、内容物をろ過し、乾燥することで57kgのポリオレフィン系樹脂を得た。得られたポリオレフィン系樹脂のペレットの密度は937kg/m3、MFRは8.0g/10分であった。
【0093】
実施例1
製造例1で得られたポリオレフィン系樹脂ペレット100重量部を押出機に供給して溶融混練して水中カット方式により造粒して楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を得た。この樹脂粒子の平均重量は0.6mgであった。
次に、攪拌機付の5リットルのオートクレーブに、ピロリン酸マグネシウム50g、ドデシルベンゼンスルホン酸ソーダ3.5gを純水2kgに分散させて分散用媒体を得た。
【0094】
分散用媒体に30℃で上記高密度ポリオレフィン系樹脂粒子600gを分散させて10分間保持し、次いで60℃に昇温して懸濁液を得た。
更に、この懸濁液に、重合開始剤としてジクミルパーオキサイドを0.6g溶解させたスチレンモノマー0.30kgを30分かけて滴下した。滴下後、30分間保持することで、高密度ポリオレフィン系樹脂粒子中にスチレン系モノマーを含浸させた。含浸後、130℃に昇温し、この温度で2時間重合(第1重合)させた。
【0095】
次に、120℃に下げた懸濁液中に、ジクミルパーオキサイドを4.2g溶解させたスチレンモノマー1.10kgを4時間30分かけて滴下した。滴下後、120℃で1時間保持することで、高密度ポリオレフィン系樹脂粒子中にスチレン系モノマーを含浸させた。含浸後、140℃に昇温し、この温度で2時間30分間保持して重合(第2重合)させた。この重合の結果、改質樹脂粒子を得ることができた。
【0096】
次いで、常温(約23℃)まで冷却し、オートクレーブから改質樹脂粒子を取り出した。改質樹脂粒子2kgと水2リットルとを、5リットルの攪拌機付オートクレーブに入れた。更に、発泡剤としてブタン520ミリリットル(300g)をオートクレーブに入れた。この後、70℃に昇温し、4時間攪拌を続けることで発泡性樹脂粒子を得ることができた。
その後、常温まで冷却して、発泡性樹脂粒子をオートクレーブから取り出し、脱水乾燥させた。
【0097】
次いで、得られた発泡性樹脂粒子を嵩密度0.033g/cm3に予備発泡させることで、予備発泡粒子を得た。得られた予備発泡粒子を1日間室温(23℃)に放置した後、400mm×300mm×30mmの大きさの成形用金型に入れた。その後、0.12MPaの水蒸気を50秒間導入して加熱し、次いで、発泡成形体の最高面圧が0.01MPaに低下するまで冷却することで、密度0.033g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。結果を表3に示す。
【0098】
実施例2
製造例1で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を760g使用し、第1重合において、ジクミルパーオキサイドを0.8g、スチレンモノマーを0.38kg使用し、第2重合において、ジクミルパーオキサイドを3.7g、スチレンモノマーを0.86kg使用し、滴下時間を4時間15分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.040g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.040g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0099】
実施例3
製造例2で得られたポリオレフィン系樹脂ペレット100重量部を押出機に供給して溶融混練して水中カット方式により造粒して楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を得た。この樹脂粒子の平均重量は0.6mgであった。
上記で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を600g使用し、第1重合において、ジクミルパーオキサイドを0.6g、スチレンモノマーを0.30kg使用し、第2重合において、ジクミルパーオキサイドを4.2g、スチレンモノマーを1.10kg使用し、滴下時間を4時間30分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
【0100】
次いで、得られた発泡性樹脂粒子を嵩密度0.033g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.033g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0101】
実施例4
製造例2で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を760g使用し、第1重合において、ジクミルパーオキサイドを0.8g、スチレンモノマーを0.38kg使用し、第2重合において、ジクミルパーオキサイドを3.7g、スチレンモノマーを0.86kg使用し、滴下時間を4時間15分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.045g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.045g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0102】
実施例5
製造例3で得られたポリオレフィン系樹脂ペレット100重量部を押出機に供給して溶融混練して水中カット方式により造粒して楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を得た。この樹脂粒子の平均重量は0.6mgであった。
【0103】
上記で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を600g使用し、第1重合において、ジクミルパーオキサイドを0.6g、スチレンモノマーを0.30kg使用し、第2重合において、ジクミルパーオキサイドを4.2g、スチレンモノマーを1.10kg使用し、滴下時間を4時間30分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
【0104】
次いで、得られた発泡性樹脂粒子を嵩密度0.033g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.033g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0105】
実施例6
製造例3で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を760g使用し、第1重合において、ジクミルパーオキサイドを0.8g、スチレンモノマーを0.38kg使用し、第2重合において、ジクミルパーオキサイドを3.7g、スチレンモノマーを0.86kg使用し、滴下時間を4時間15分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.050g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.050g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0106】
実施例7
製造例4で得られたポリオレフィン系樹脂ペレット100重量部を押出機に供給して溶融混練して水中カット方式により造粒して楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を得た。この樹脂粒子の平均重量は0.6mgであった。
上記で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を600g使用し、第1重合において、ジクミルパーオキサイドを0.6g、スチレンモノマーを0.30kg使用し、第2重合において、ジクミルパーオキサイドを4.2g、スチレンモノマーを1.10kg使用し、滴下時間を4時間30分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
【0107】
次いで、得られた発泡性樹脂粒子を嵩密度0.050g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.050g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0108】
実施例8
製造例4で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を760g使用し、第1重合において、ジクミルパーオキサイドを0.8g、スチレンモノマーを0.38kg使用し、第2重合において、ジクミルパーオキサイドを3.7g、スチレンモノマーを0.86kg使用し、滴下時間を4時間30分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.055g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.055g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0109】
比較例1
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例5で得られたポリオレフィン系樹脂ペレット(MFR=18.8g/10分、密度=939kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.040g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.040g/cm3の発泡成形体を得た。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0110】
比較例2
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例6で得られたポリオレフィン系樹脂ペレット(MFR=0.06g/10分、密度=935kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
比較例3
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例7で得られたポリオレフィン系樹脂ペレット(MFR=22.5g/10分、密度=939kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
【0111】
比較例4
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例8で得られたポリオレフィン系樹脂ペレット(MFR=2.0g/10分、密度=908kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.033g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.033g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0112】
比較例5
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例9で得られたポリオレフィン系樹脂ペレット(MFR=4.5g/10分、密度=955kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.065g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.065g/cm3の発泡成形体を得た。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0113】
比較例6
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例10で得られたポリオレフィン系樹脂ペレット(MFR=8.0g/10分、密度=937kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.033g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.033g/cm3の発泡成形体を得た。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0114】
比較例7
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、示差走査型熱量計による昇温測定において得られる吸熱曲線のピークが1つである、市販の直鎖状低密度ポリエチレン(ユメリット4540F、宇部興産社製、MFR=3.9g/10分、密度=944kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
【0115】
比較例8
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、示差走査型熱量計による昇温測定において得られる吸熱曲線のピークが1つである、市販の高密度ポリエチレン(ニポロンハード2500、東ソー社製、MFR=8.0g/10分、密度=961kg/m3)とした以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
【0116】
比較例9
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、示差走査型熱量計による昇温測定において得られる吸熱曲線のピークが1つである、市販の直鎖状低密度ポリエチレン(ニポロンZZF260、東ソー社製、MFR=2.0g/10分、密度=936kg/m3)を用いた以外は実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
【0117】
比較例10
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、示差走査型熱量計による昇温測定において得られる吸熱曲線のピークが1つである、市販の低密度ポリエチレン(ペトロセン 203、東ソー(株)製、MFR=8.0g/10分、密度=919kg/m3)を用いた以外は実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子、予備発泡粒子(嵩密度0.033g/cm3)、発泡成形体(密度0.033g/cm3)を得た。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0118】
比較例11
[重合触媒(r)の調製]
上記変性ヘクトライト500gをヘキサン1.7リットルに懸濁させ、ジメチルシランジイルビス(シクロペンタジエニル)ジルコニウムジクロリド6.97g(20.0mmol)とトリイソブチルアルミニウムのヘキサン溶液(0.714M)2.8リットル(2mol)の混合液を添加し、60℃で3時間攪拌した後、ジメチルシランジイルビス(シクロペンタジエニル)ジルコニウムジクロリドに対して15mol%のジフェニル(1−シクロペンタジエニル)(2,7−ジ−tert−ブチル−9−フルオレニル)ジルコニウムジクロライド2.36g(3.53mmol)を添加して室温で6時間撹拌した。静置して上澄み液を除去、更にトリイソブチルアルミニウムのヘキサン溶液(0.15M)を添加して最終的に100g/Lの触媒スラリーを得た。
【0119】
[ポリオレフィン系樹脂(S)の製造]
内容積540リットルの重合器に、ヘキサン300リットル及び1−ブテン1.3リットルを導入し、オートクレーブの内温を80℃に昇温した。このオートクレーブに[重合触媒(r)の調製]で調製した重合触媒(r)147ミリリットルを添加し、エチレン/水素混合ガス(水素:1000ppm含)を分圧が0.9MPaになるまで導入して重合を開始した。重合中、分圧が0.9MPaに保たれるようにエチレン/水素混合ガスを連続的に導入した。また、重合温度を80℃に制御した。重合開始90分後に重合器の内圧を脱圧した後、内容物をろ過し、乾燥して50kgのポリオレフィン系樹脂(S)粉末を得た。ポリオレフィン系樹脂粉末を200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでポリオレフィン系樹脂(S)ペレットを得た。
得られたポリオレフィン系樹脂(S)ペレットの密度は950kg/m3、MFRは8g/10分であった。
【0120】
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、上記で得られたポリオレフィン系樹脂(S)ペレット(MFR=8.0g/10分、密度=950kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子、予備発泡粒子(嵩密度0.033g/cm3)、発泡成形体(密度0.033g/cm3)を得た。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0121】
比較例12
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、示差走査型熱量計による昇温測定において得られる吸熱曲線のピークが複数個ある、市販のメタロセンポリエチレン(ハーモレックス NH745S、日本ポリエチレン社製、MFR=9.2g/10分、密度=912kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
【0122】
以下、表1及び2に実施例及び比較例で使用した触媒の各種物性値を示す。
【0123】
【表1】

【0124】
【表2】
【0125】
更に、表3及び4に実施例及び比較例の製造条件、予備発泡粒子の嵩密度、発泡成形体の融着率、加熱寸法変化率及び耐薬品性を示す。
【0126】
【表3】
【0127】
【表4】
【0128】
表3及び4から以下のことが分かる。
実施例と、比較例4、5及び8とから、オレフィン系樹脂の密度(kg・cm3)が910〜950の範囲内であることで、融着率、加熱寸法変化率及び耐薬品性の全てが優れた発泡成形体が得られることが分かる。
実施例と、比較例2及び3とから、MFR(g/10分)が0.1〜20の範囲内であることで、融着率、加熱寸法変化率及び耐薬品性の全てが優れた発泡成形体が得られることが分かる。
実施例と比較例7とから、末端ビニル数が1000炭素原子当たり0.2以下であることで、融着率、加熱寸法変化率及び耐薬品性の全てが優れた発泡成形体が得られることが分かる。
【0129】
実施例と、比較例7及び9とから、MS160>90−130×log(MFR)の関係を有することで、融着率、加熱寸法変化率及び耐薬品性の全てが優れた発泡成形体が得られることが分かる。
実施例と比較例1とから、MS160/MS190>1.8の関係を有することで、融着率が優れた発泡成形体が得られることが分かる。
実施例と、比較例6〜11とから、TREFによる溶出温度−溶出量曲線にピークが2個以上存在することで、融着率、加熱寸法変化率及び耐薬品性の全てが優れた発泡成形体が得られることが分かる。
【図面の簡単な説明】
【0130】
【図1】本発明に使用可能なポリオレフィン系樹脂の代表的なTREF溶出温度−溶出量曲線である。
【図2】代表的なメタロセン触媒により得られたエチレン・α−オレフィン共重合体のTREF溶出温度−溶出量曲線である。
【技術分野】
【0001】
本発明は、スチレン改質ポリオレフィン系樹脂粒子、発泡性樹脂粒子、予備発泡粒子及び発泡成形体に関する。更に詳しくは、本発明は、発泡成形体に優れた耐熱性や加熱寸法安定性を与えうるスチレン改質ポリオレフィン系樹脂粒子、発泡性樹脂粒子及び予備発泡粒子、それら粒子から得られた発泡成形体に関する。
【背景技術】
【0002】
従来から、ポリスチレン系樹脂からなる予備発泡粒子を型内に充填し、次いで加熱することで発泡させて得られる発泡成形体は、剛性、断熱性、軽量性、耐水性及び発泡成形性に優れていることが知られている。そのため、この発泡成形体は、緩衝材や建材用断熱材として広く用いられている。しかし、ポリスチレン系樹脂からなる発泡成形体は、耐薬品性及び耐衝撃性が劣るという課題があった。
【0003】
一方、ポリエチレンやポリプロピレン等のポリオレフィン系樹脂からなる発泡成形体は、耐薬品性及び耐衝撃性に優れていることが知られている。そのため、この発泡成形体は、自動車関連部品に使用されている。しかし、ポリオレフィン系樹脂は、発泡剤の保持性が劣ることから、発泡成形条件を精密に制御する必要がある。そのため製造コストが高くつくという課題があった。加えて、この発泡成形体は、ポリスチレン系樹脂からなる発泡成形体に比べて、剛性が劣るという課題もあった。
【0004】
上記ポリスチレン系樹脂及びポリオレフィン系樹脂からなる発泡成形体の課題を解決するために、ポリスチレン系樹脂とポリオレフィン系樹脂とを混合した粒子から得られた発泡成形体が報告されている。この発泡成形体は、ポリスチレン系樹脂の優れた剛性及び発泡成形性と、ポリオレフィン系樹脂の優れた耐薬品性及び耐衝撃性とを兼ね備えている。
ところで、自動車関連部品は、ガソリン、灯油、ブレーキオイル、塩化ビニルに使用される可塑剤等の薬品に接触したり、強い衝撃を受けたりすることがある。そのためこれら用途に使用される発泡成形体には、高い耐薬品性及び耐衝撃性が要求されているが、上記発泡成形体では不十分であった。
【0005】
そこで、上記要求を満たす方法として、ポリオレフィン系樹脂としてのエチレン−酢酸ビニル共重合体(EVA)、高圧法低密度ポリエチレン(LDPE)又は直鎖状低密度ポリエチレン(LLDPE)の粒子を、ポリスチレン系樹脂で改質し、改質粒子から発泡成形体を得る方法が提案されている(特開2005−97555号公報:特許文献1)が、上記公報に記載の方法では、高い耐薬品性及び耐衝撃性を有する発泡成形体が得られるものの、例えば、高温に晒されるようなより厳しい条件下での用途では、耐熱性や加熱寸法安定性が不足する場合があり、更に改善した発泡成形体の提供が望まれていた。
また、上記要求に答えうる技術として、特開2007−270116号公報(特許文献2)に記載された技術が提案されている。この公報に記載された技術では、より厳しい条件下での用途における、耐熱性や加熱寸法安定性がある程度確保されている。
【0006】
【特許文献1】特開2005−97555号公報
【特許文献2】特開2007−270116号公報
【発明の開示】
【発明が解決しようとする課題】
【0007】
特許文献2に記載されたスチレン改質ポリオレフィン系樹脂粒子は、発泡性の優れた発泡性樹脂粒子を提供できる。しかし、発泡成形体の曲げ強度、曲げ破断変位等の物性をより向上させる観点から、加熱寸法安定性に寄与する発泡粒子同士の融着性を更に改善可能なスチレン改質ポリオレフィン系樹脂粒子が望まれている。
発泡粒子同士の融着性を改善させるには、ポリオレフィン系樹脂の結晶化度を下げることが考えられる。しかし、結晶化度を単純に下げると、発泡成型体の耐熱性が低下することになる。
そのため、融着性と耐熱性を両方とも満足する発泡成型体を製造可能なスチレン改質ポリオレフィン系樹脂粒子の提供が望まれていた。
【課題を解決するための手段】
【0008】
本発明の発明者等は、耐熱性や加熱寸法安定性を更に改善するために、発泡成形体の原料を見直した結果、特定の性質を有するポリオレフィン系樹脂を使用することで、耐熱性や加熱寸法安定性を更に改善できることを見出し、本発明に至った。
かくして本発明によれば、エチレンから導かれる繰り返し単位、又はエチレンから導かれる繰り返し単位及び炭素数3〜8のα−オレフィンから導かれる繰り返し単位から構成され、かつ下記要件(A)〜(H)
(A)密度[d(kg/m3)]が910以上950以下
(B)190℃、2.16kg荷重で測定したメルトフローレート[MFR(g/10分)]が0.1以上20以下
(C)末端ビニル数が1000炭素原子当たり0.2個以下
(D)160℃で測定した溶融張力[MS160(mN)]とMFRの関係が、下記式(1)を満足
MS160>90−130×log(MFR) (1)
(E)190℃で測定した溶融張力[MS190(mN)]とMS160の関係が、下記式(2)を満足
MS160/MS190<1.8 (2)
(F)連続昇温溶出分別法(TREF)による溶出温度−溶出量曲線にピークが2個以上存在
の要件を満足するポリオレフィン系樹脂と、前記ポリオレフィン系樹脂を改質するスチレン系樹脂とを含み、前記スチレン系樹脂が、前記ポリオレフィン系樹脂100重量部に対して、20〜600重量部使用されることを特徴とするスチレン改質ポリオレフィン系樹脂粒子が提供される。
【0009】
更に、本発明によれば、上記スチレン改質ポリオレフィン系樹脂粒子100重量部と、揮発性発泡剤5〜25重量部とを含むことを特徴とする発泡性樹脂粒子が提供される。
また、本発明によれば、上記発泡性樹脂粒子を予備発泡させて得られた嵩密度0.01〜0.2g/cm3の予備発泡粒子が提供される。
更にまた、上記予備発泡粒子を型内発泡成形させて得られた密度0.01〜0.2g/cm3の発泡成形体が提供される。
【発明の効果】
【0010】
本発明によれば、特定の性質を有するポリオレフィン系樹脂を使用したスチレン改質ポリオレフィン系樹脂粒子により、耐熱性や加熱寸法安定性が更に改善された発泡成形体を提供できる。
【発明を実施するための最良の形態】
【0011】
(スチレン改質ポリオレフィン系樹脂粒子)
本発明のスチレン改質ポリオレフィン系樹脂粒子は、ポリオレフィン系樹脂粒子をスチレン系樹脂で改質した粒子(以下、改質樹脂粒子と称する)である。ここで、本発明は、改質樹脂粒子を構成するポリオレフィン系樹脂の量よりスチレン系樹脂の量が多い場合を含むが、本明細書では、その場合でもポリオレフィン系樹脂粒子がスチレン系樹脂で改質されているとする。
ポリオレフィン系樹脂粒子のスチレン系樹脂での改質方法は、特に限定されず、公知の方法を種々使用できるが、例えば、ポリオレフィン系樹脂粒子にスチレン系モノマーを含浸させ、次いでスチレン系モノマーを重合させる方法が挙げられる。
【0012】
(1)ポリオレフィン系樹脂
ポリオレフィン系樹脂は、エチレン単独重合体又はエチレンと炭素数3〜8のα−オレフィンとの共重合体からなる。上記炭素数3〜8のα−オレフィンとしては、プロピレン、1−ブテン、1−ヘキセン、1−オクテン、4−メチル−1−ペンテン、3−メチル−1−ブテン、ビニルシクロアルカン(例えば、ビニルシクロペンタン、ビニルシクロヘキサン)、環状オレフィン(例えば、ノルボルネン、ノルボルナジエン)、ジエン(例えば、ブタジエン、1,4−ヘキサジエン)等が挙げられる。炭素数3〜8のオレフィン由来の成分が、ポリオレフィン系樹脂に占める割合は、特に限定されないが、50重量%以下であることが好ましく、20重量%以下であることがより好ましい。なお、発明を阻害しない範囲で、スチレンをエチレンと共重合させてもよい。
【0013】
更に、ポリオレフィン系樹脂は、以下の要件を満足する樹脂である。
(A)密度[d(kg/m3)]が910以上950以下
(B)190℃、2.16kg荷重で測定したメルトフローレート[MFR(g/10分)]が0.1以上20以下
(C)末端ビニル数が1000炭素原子当たり0.2個以下
(D)160℃で測定した溶融張力[MS160(mN)]とMFRの関係が、下記式(1)を満足
MS160>90−130×log(MFR) (1)
(E)190℃で測定した溶融張力[MS190(mN)]とMS160の関係が、下記式(2)を満足
MS160/MS190<1.8 (2)
(F)連続昇温溶出分別法(TREF)による溶出温度−溶出量曲線にピークが2個以上存在。
【0014】
要件(A)において、密度[d(kg/m3)]は910kg/m3以上950kg/m3以下であり、特に925〜945kg/m3であることが好ましい。ここで、密度が910kg/m3未満のポリオレフィン系樹脂は、融解温度が低いため、得られる発泡成形体は耐熱性に劣るものとなる。一方、950kg/m3を超えるポリオレフィン系樹脂は、融解温度が高いため発泡成形温度が高くなり生産性が低下することに加えて、得られる発泡成形体は耐衝撃性に劣るものとなる。なお、密度の測定法は、実施例の欄に記載する。
【0015】
要件(B)において、190℃、2.16kg荷重で測定したメルトフローレート[MFR(g/10分)](以下、MFRと記す。)は0.1g/10分以上20g/10分以下である。ここで、MFRが0.1g/10分未満のポリオレフィン系樹脂は、発泡倍率が低下するため好ましくない。また、20g/10分を超えると溶融張力が小さくなり発泡倍率が低下することに加え、発泡成形体の強度も低下するため好ましくない。より好ましいMFRは2〜10g/10分である。MFRの測定法は、実施例の欄に記載する。
【0016】
要件(C)において、末端ビニル数は1000炭素原子当たり0.2個以下であり、特に0.05個以下であることが好ましい。ここで、末端ビニル数が1000炭素原子当たり0.2個を越えるポリオレフィン系樹脂は、熱劣化、とくに黄変が生じることがある。末端ビニル数の測定法は、実施例の欄に記載する。
【0017】
要件(D)において、160℃で測定した溶融張力[MS160(mN)](以下、MS160と記す。)と、190℃で、2.16kg荷重で測定したMFRとの関係が、下記式(1)を満足するものである。
MS160>90−130×log(MFR) (1)
特に、MS160は、MS160>110−130×log(MFR)を満足することが好ましい。
ここで、MS160が[90−130×log(MFR)]以下の範囲にあるオレフィン系樹脂は、ガスの保持力が不足し、発泡性が劣る場合がある。
【0018】
要件(E)において、190℃で測定した溶融張力[MS190(mN)](以下、MS190と記す。)とMS160の関係が、下記式(2)を満足するものである。
MS160/MS190<1.8 (2)
ここで、MS160/MS190が1.8以上のポリオレフィン系樹脂である場合、発泡温度による溶融張力が大きく変化することがある。そのため、成形温度の厳密な調節が必要となり、ひいては成形可能範囲が狭くなり、発泡成形性に劣るポリオレフィン系樹脂となる。より好ましいMS160/MS190は1.6より小さい値である。
なお、MS160とMS190の測定法は、実施例の欄に記載する。
【0019】
要件(F)において、連続昇温溶出分別法(以下、TREFと記す。)により求めた溶出温度−溶出量曲線においてピークが2個以上存在するポリオレフィン系樹脂である。更に、ポリオレフィン系樹脂は、2個のピークを有することが好ましい。特に2個のピークの内、結晶化度が上昇することから発泡成形体とした際の耐熱性及び剛性を向上できる観点から、高温側のピークは85〜100℃の間に存在することが好ましい。また、発泡粒子同士の融着特性に優れることから低温側のピークは65〜80℃の間に存在することが好ましい。
【0020】
なお、図1に本発明に使用可能なポリオレフィン系樹脂の代表的なTREF溶出温度−溶出量曲線を示す。また、図2には代表的なメタロセン触媒により得られたエチレン・α−オレフィン共重合体のTREF溶出温度−溶出量曲線を示す。本発明に使用可能なポリオレフィン系樹脂は、組成分布が広いために溶出温度−溶出量曲線が2つのピークを有しており、1つのピークの従来のメタロセン触媒により得られるエチレン・α−オレフィン共重合体とは異なった樹脂である。図1及び2において、縦軸は溶出量を意味する。
なお、上記曲線の測定法は、実施例の欄に記載する。
【0021】
ポリオレフィン系樹脂は、更に、下記(G)及び(H)
(G)流動の活性化エネルギー[Ea(kJ/mol)]と密度の関係が、下記式(3)を満足
127−0.107d<Ea<88−0.060d (3)
(H)50℃におけるn−ヘプタン抽出量が0.2重量%以下
の要件を満足することが好ましい。
【0022】
要件(G)において、流動の活性化エネルギー[Ea(kJ/mol)](以下、Eaと記す。)と密度dとの関係が、Ea≦(127−0.107d)の場合、発泡成形性に問題が生じることがある。一方、Ea≧(88−0.060d)の場合、溶融粘度の温度依存性が大きく、成形温度の厳密な調節が必要となり、ひいては発泡成形可能範囲が狭くなるという問題が生じることがある。特に、Eaは、127−0.107d<Ea<87−0.060dの要件を満足することが好ましい。
なお、Eaの測定法は、実施例の欄に記載する。
【0023】
要件(H)において、50℃におけるn−ヘプタン抽出量は0.2重量%以下である。ここで、抽出量が0.2重量%を超えるポリオレフィン系樹脂である場合、融着性に劣るものとなる。より好ましい抽出量は、0.1重量%以下である。
更に、ポリオレフィン系樹脂は、特に機械強度に優れる発泡成形体を得ることが可能となることから、重量平均分子量(以下、Mwと記す。)と数平均分子量(以下、Mnと記す。)の比(Mw/Mn)が4.5以上7.5以下であることが好ましく、特に5.0以上7.0以下であることが好ましい(要件(I))。なお、Mw及びMnの測定法は、実施例の欄に記載する。
【0024】
本発明のエチレン系重合体の製造方法としては、上記要件(A)〜(F)(特に、要件(A)〜(I))を満足するポリオレフィン系樹脂の製造が可能であれば如何なる製造方法を用いることも可能である。例えば重合触媒及び/又は重合条件を多段階で変更する多段重合法、複数の重合触媒を混合した触媒による重合法、同一又は異なる重合触媒で調製した複数のポリオレフィン系樹脂をブレンドする方法等を挙げることができる。
【0025】
上記要件(A)〜(F)を満たし、好ましくは前記要件(A)〜(I)を満足するポリオレフィン系樹脂は、後述する実施例の製造条件そのもの、あるいは条件因子のマイナー変動によって任意に作り分けることが可能である。条件因子のマイナー変動とは、例えば、単独の重合触媒成分を複数の重合触媒成分の組み合わせに替えること(例えば、複数の重合触媒成分を用いる場合、各々を成分(a)及び成分(b)等とする)や重合条件を変更すること等が該当する。具体的には成分(a)及び成分(b)として重合触媒のメタロセン化合物の種類、担体種類あるいは調製方法の異なる重合触媒を用いることや、それらを組み合わせて用いる場合の成分(a)に対する成分(b)の重量比を変更することや、重合触媒と組み合わせて用いることのできる助触媒成分の種類及び量を変更する等重合触媒成分に関して操作することや、重合反応を行う際の重合温度、エチレン分圧、共存させる水素等の分子量調整剤の量、添加するコモノマー量等で示されるいわゆる重合条件を制御すること、によって作り分けることも可能である。また更に多段重合との組み合わせで、物性の範囲を拡大することも可能である。
【0026】
より具体的には、例えばエチレン分圧を低下させること、コモノマー添加量を減少させること、成分(a)の構造を変えること等によって、末端ビニル数を変化させることが可能である。また、溶融張力は、成分(a)の構造を変えること、末端ビニル数を増加させること、成分(b)の構造を変えること、エチレン分圧を低下させること、長鎖分岐数を増加させること、長鎖分岐長さを増加させること、成分(a)に対する成分(b)の量を変えること、Mw/Mnを増加させること等により増加させることが可能である。更に流動の活性化エネルギー(Ea)は、成分(a)の構造、末端ビニル数、成分(b)の構造、エチレン分圧、長鎖分岐数、長鎖分岐長さ、成分(a)に対する成分(b)の量により制御が可能である。
【0027】
ポリオレフィン系樹脂の製造に用いる重合触媒としては、例えば、特開2004−346304号公報、特開2005−248013号公報、特開2006−321991号公報、特開2007−169341号公報及び2008−050278号公報に記載の重合触媒及びそれらを公知の方法により組み合わせた重合触媒が挙げられる。
ポリオレフィン系樹脂の製造において、重合温度は−100〜120℃が好ましく、特に生産性を考慮すると20〜120℃、更には50〜120℃の範囲であることが好ましい。重合時間は10秒〜20時間の範囲が好ましく、重合圧力は常圧〜300MPaの範囲であることが好ましい。
【0028】
エチレンと炭素数3〜8のα−オレフィンからなるポリオレフィン系樹脂を使用する場合、エチレン/炭素数3〜8のα−オレフィン(モル比)は、1〜200であることが好ましく、より好ましくは3〜100、更に好ましくは5〜50である。また、重合時に水素等を用いて分子量の調節を行うことも可能である。
【0029】
更に、ポリオレフィン系樹脂は、マクロモノマーの存在下に、オレフィンを重合させることで得られた樹脂であることが好ましい。
具体的には、末端にビニル基を有するエチレンの単独重合体又はエチレンと炭素数3以上のオレフィンとの共重合体からなるマクロモノマーと、炭素数2以上のオレフィンとを共重合させることにより得られるポリオレフィン系樹脂である。
【0030】
ここで、マクロモノマーは、2000以上のMnと、2〜5のMw/Mnとを有していることが好ましい。炭素数3以上のオレフィンとしては、プロピレン、1−ブテン、1−ヘキセン、1−オクテン、4−メチル−1−ペンテン、3−メチル−1−ブテン、ビニルシクロアルカン(例えば、ビニルシクロペンタン、ビニルシクロヘキサン)等が挙げられる。これらオレフィンは、単独でも、2種以上の組み合わせでもよい。なお、炭素数2以上のオレフィンは、上記エチレンと炭素数3〜8のα−オレフィンから選択される。
マクロモノマーのMnは、5000以上であることがより好ましく、10000以上であることが更に好ましい。上限は、100000であることが好ましい。また、Mw/Mnは、2〜4であることがより好ましく、2〜3.5であることが更に好ましい。
【0031】
更に、マクロモノマーの主鎖であるメチレン炭素1000個当たりのビニル末端数をX、マクロモノマーの主鎖であるメチレン炭素1000個当たりの飽和末端数をYとした場合、式Z=X/[(X+Y)×2]で表されるZが0.25〜1であることが好ましい。Zは0.5〜1であることがより好ましい。なお、ビニル末端及び飽和末端は、1H−NMR、13C−NMR又はFT−IRによりその数を測定できることは、当業者によく知られている。例えば、13C−NMRの場合、ビニル末端は114ppmと139ppmに、飽和末端は32.3ppm、22.9ppm及び14.1ppmにピークを有し、このピークからその数を測定できる。
【0032】
上記マクロモノマーとオレフィンとを共重合させることで、本発明の使用に好適なポリオレフィン系樹脂が得られる。ここで、マクロモノマー以外の炭素数2以上のオレフィンに由来する樹脂(樹脂A)の全樹脂に対する割合は、1〜99重量%が好ましく、5〜90重量%がより好ましく、30〜80重量%が更に好ましい。樹脂Aの割合の測定は、樹脂のGPCチャートを、マクロモノマーのGPCチャートと比較することにより行うことができる。具体的には、両チャートの比較により樹脂Aに由来するピークを決定し、そのピークの面積の全ピークの面積に対する割合が、樹脂Aの割合に相当する。
重合はバッチ式、半連続式、連続式のいずれの方法でも行うことが可能であり、重合条件を変えて2段階以上に分けて行うことも可能である。また、ポリオレフィン系樹脂は、重合終了後に従来既知の方法により重合溶媒から分離回収され、乾燥して単離できる。
【0033】
重合はスラリー状態、溶液状態又は気相状態で実施することができ、特に、重合をスラリー状態で行う場合には粒子形状の整ったポリオレフィン系樹脂を効率よく、安定的に生産することができる。また、重合に用いる溶媒は一般に用いられる有機溶媒であればいずれでもよく、具体的には例えばベンゼン、トルエン、キシレン、プロパン、イソブタン、ペンタン、ヘキサン、ヘプタン、シクロヘキサン、ガソリン等が挙げられる。更には、プロピレン、1−ブテン、1−ヘキセン、1−オクテン等のオレフィン自身を溶媒として用いることもできる。
【0034】
ポリオレフィン系樹脂は、本発明の目的を逸脱しない限りにおいて、他の樹脂を含んでいてもよい。他の樹脂としては、上記(A)〜(F)の性質を少なくとも1つ備えていない他のポリオレフィン系樹脂が好ましい。他のポリオレフィン系樹脂としては、炭素数2〜20のα−オレフィン単独重合体及び共重合体が挙げられる。具体的には、高密度ポリエチレン、低密度ポリエチレン、直鎖状低密度ポリエチレン、ポリプロピレン、ポリ1−ブテン、ポリ(4−メチル−1−ペンテン)、ポリ1−ペンテン、エチレン/ブロピレン共重合体、エチレン/1−ブテン共重合体、プロピレン/1−ブテン共重合体、エチレン/プロピレン/1−ブテン共重合体、4−メチル−1−ペンテン/エチレン共重合体、エチレン/プロピレン/ポリエン共重合体、種々のプロピレン系ブロック共重合体やプロピレン系ランダム共重合体等が挙げられる。これら他の樹脂の配合割合は、全ポリオレフィン系樹脂量に対して、50重量%以下が好ましく、5〜30重量%がより好ましい。
【0035】
ポリオレフィン系樹脂には、必要に応じて、着色剤、安定剤、充填材(補強材)、高級脂肪酸金属塩、難燃剤、帯電防止剤、滑剤、天然又は合成油、ワックス、紫外線吸収剤、耐候安定剤、防曇剤、坑ブロッキング剤、スリップ剤、被覆剤、中性子遮蔽剤等の添加剤が含まれていてもよい。この内、着色剤としては、無機及び有機の着色剤(顔料又は染料)をいずれも使用できる。特に、酸化鉄、カーボンブラック等の無機着色剤が好ましい。
【0036】
酸化鉄としては、黄色系統のものとしてα−FeOOH(含水結晶)、赤色系統のものとしてα−Fe2O3、黒色系統のものとして(FeO)x(Fe2O3)y等が挙げられる。これら酸化鉄は、Feの一部が、Zn、Mg等の他の金属で置き換えられていてもよい。更に、これら酸化鉄は、所望の色を得るために、混合して用いてもよい。この内、黒色系統の(FeO)x(Fe2O3)yに含まれるFe3O4であることが好ましい。
【0037】
酸化鉄は、0.1〜1μmの平均粒径を有していることが好ましく、0.2〜0.8μmがより好ましい。平均粒径は、レーザー回折式粒度分布計(日本電子社製ロドス)により測定できる。酸化鉄は、ポリオレフィン系樹脂中、1.5〜70重量%の範囲で含まれていることが好ましく、5〜40重量%の範囲がより好ましく、10〜30重量%の範囲が更に好ましい。1.5重量%未満であれば、ポリオレフィン系樹脂が十分着色されない場合があるため好ましくない。70重量%より多い場合、ポリオレフィン系樹脂中に混合することが困難となり易い。加えて、酸化鉄の比重がポリオレフィン樹脂より大きいため、ポリオレフィン系樹脂粒子が重くなり、スチレン系モノマーを均一に含浸させることが困難となり易い。
【0038】
カーボンブラックとしては、ファーネスブラック、チャンネルブラック、サーマルブラック、アセチレンブラック、黒鉛、炭素繊維等が挙げられる。
カーボンブラックは、ポリオレフィン系樹脂中、1〜50重量%の範囲で含まれていることが好ましく、2〜30重量%の範囲がより好ましい。1重量%未満であれば、ポリオレフィン系樹脂が十分着色されない場合がある。50重量%より多い場合、ポリオレフィン系樹脂中に混合することが困難となり易い。
【0039】
安定剤は、酸化劣化や熱劣化等を防止する役割を果たし、公知物をいずれも使用できる。例えば、フェノール系安定剤、有機ホスファイト系安定剤、チオエーテル系安定剤、ヒンダードアミン系安定剤等が挙げられる。
充填材としては、タルク、ガラス等が挙げられ、その形状は球状、板状、繊維状等特に限定されない。
高級脂肪族金属塩としては、ステアリン酸、オレイン酸、ラウリン酸等の高級脂肪酸と、アルカリ土類金属(マグネシウム、カルシウム、バリウム等)やアルカリ金属(ナトリウム、カリウム、リチウム等)との塩が挙げられる。
【0040】
(2)スチレン系樹脂
スチレン系樹脂としては、例えば、スチレン、α−メチルスチレン、p−メチルスチレン、t−ブチルスチレン等のスチレン系モノマーに由来する樹脂が挙げられる。更に、スチレン系樹脂は、スチレン系モノマーと、スチレン系モノマーと共重合可能な他のモノマーとの共重合体からなる成分であってもよい。他のモノマーとしては、ジビニルベンゼンのような多官能性モノマーや、(メタ)アクリル酸ブチルのような構造中にベンゼン環を含まない(メタ)アクリル酸アルキルエステル等が例示される。これら他のモノマーを、スチレン系樹脂に対して5重量%を超えない範囲で使用してもよい。
【0041】
スチレン系樹脂の量は、ポリオレフィン系樹脂100重量部に対して20〜600重量部、好ましくは100〜550重量部、より好ましくは130〜500重量部である。また、600重量部を超える場合、発泡成形体の耐薬品性及び耐衝撃性が低下することがある。20重量部より少ない場合、発泡成形体の剛性が低下することがある。
【0042】
(3)改質樹脂粒子の形状
改質樹脂粒子は、粒子の長さをL、平均径をDとした場合のL/Dが0.6〜1.6である円筒状、略球状ないしは球状であり、平均粒径が0.3〜3.0mmであることが好ましい。L/Dが0.6より小さくないしは1.6より大きく扁平度が大きい場合は、改質樹脂粒子から得られる予備発泡粒子を、金型に充填して発泡成形体を得る際に、金型への充填性が悪くなることがある。また形状は、充填性をよくするには略球状ないしは球状がより好ましい。
平均粒径は0.3mm未満の場合、発泡性樹脂粒子に使用する場合、発泡剤の保持性が低くなり、低密度化が困難となり易い。3.0mmを超える場合、充填性が悪くなり易く、発泡成形体の薄肉化も困難となり易い。
【0043】
(発泡性樹脂粒子)
発泡性樹脂粒子は、上記改質樹脂粒子に揮発性発泡剤を含浸させた粒子である。揮発性発泡剤としては、例えば、プロパン、n−ブタン、イソブタン、ペンタン、イソペンタン、シクロペンタン、ヘキサン、ジメチルエーテル等が挙げられる。これら揮発性発泡剤は、単独もしくは2種以上混合して用いることができる。揮発性発泡剤の含有量は、改質樹脂粒子100重量部に対して、5〜25重量部であることが好ましい。
発泡性樹脂粒子のL/D及び平均粒径は、上記改質樹脂粒子と同程度とすることができる。また形状は、充填性をよくするには略球状ないしは球状がより好ましい。
【0044】
(予備発泡粒子)
予備発泡粒子は、上記発泡性樹脂粒子を予備発泡させて得られた粒子である。
予備発泡粒子は、0.01〜0.2g/cm3の嵩密度を有する。好ましい嵩密度は、0.014〜0.15g/cm3である。嵩密度が0.01g/cm3より小さい場合、発泡させたときに独立気泡率が低下して、予備発泡粒子から得られる発泡成形体の強度が低下することがある。一方、0.2g/cm3より大きい場合、得られる発泡成形体の重量が増加することがある。嵩密度の測定法は、実施例の欄に記載する。
【0045】
(発泡成形体)
発泡成形体は、上記予備発泡粒子を型内発泡成形させて得られた成形体である。
発泡成形体は、0.01〜0.2g/cm3の密度を有する。好ましい密度は、0.014〜0.15g/cm3である。嵩密度が0.01g/cm3より小さい場合、独立気泡率が多くなるため、強度が低下することがある。一方、0.2g/cm3より大きい場合、重量が増加することがある。密度の測定法は、実施例の欄に記載する。
【0046】
本発明の発泡成形体は、耐薬品性、衝撃強度及び剛性が優れていることに加えて、耐熱性や加熱寸法安定性が更に改善されている。
本発明の発泡成形体は、種々の用途に使用できるが、バンパーの芯材、ドア内装緩衝材等の車両用緩衝材、電子部品、各種工業資材、食品の搬送容器等の各種用途に使用できる。特に、車両用緩衝材に好適に使用できる。
【0047】
(改質樹脂粒子、発泡性樹脂粒子、予備発泡粒子及び発泡成形体の製造方法)
まず、改質樹脂粒子は、ポリオレフィン系樹脂とスチレン系樹脂とを溶融混練し、粒子状に切断することによって形成できるが、例えば、以下のように製造することが好ましい。すなわち、水性懸濁液中に、上記ポリオレフィン系樹脂の粒子100重量部と、スチレン系モノマー20〜600重量部と、重合開始剤とを分散させる。なお、スチレン系モノマーと重合開始剤とを予め混合して用いてもよい。
【0048】
ポリオレフィン系樹脂の粒子は、公知の方法により得ることができる。例えば、ポリオレフィン系樹脂を、必要に応じて無機核剤と添加剤と共に、押出機中で溶融混練して押出すことでストランドを得、得られたストランドを、空気中でカット、水中でカット、加熱しつつカットすることで、造粒する方法が挙げられる。
【0049】
ポリオレフィン系樹脂の粒子は、粒子の長さをL、平均径をDとした場合のL/Dが0.6〜1.6である円筒状、略球状ないしは球状であり、平均粒径が0.2〜1.5mmであることが好ましい。L/Dが0.6より小さくないしは1.6より大きく扁平度が大きい場合は、発泡性樹脂粒子として予備発泡させ、金型に充填して発泡成形体を得る際に、金型への充填性が悪くなり易い。また形状は、充填性をよくするには略球状ないしは球状がより好ましい。平均粒径は0.2mm未満の場合、発泡剤の保持性が低くなり、低密度化が困難となり易いので好ましくない。1.5mmを超える場合、充填性が悪くなるだけでなく発泡成形体の薄肉化も困難となり易い。
【0050】
無機核剤としては、例えば、タルク、二酸化珪素、マイカ、クレー、ゼオライト、炭酸カルシウム等が挙げられる。無機核剤の使用量は、ポリオレフィン系樹脂100重量部に対して、2重量部以下が好ましく、0.2〜1.5重量部がより好ましい。
水性懸濁液を構成する水性媒体としては、水、水と水溶性溶媒(例えば、低級アルコール)との混合媒体が挙げられる。
【0051】
重合開始剤としては、一般にスチレン系モノマーの懸濁重合用の開始剤として用いられているものが使用できる。例えば、ベンゾイルパーオキサイド、ジt−ブチルパーオキサイド、t−ブチルパーオキシベンゾエート、ジクミルパーオキサイド、2,5−ジメチル−2,5−ジ−t−ブチルパーオキシヘキサン、t−ブチルパーオキシ−3,5,5−トリメチルヘキサノエート、t−ブチル−パーオキシ−2−エチルヘキシルカーボネート等の有機化過酸化物である。これらの重合開始剤は単独もしくは2種以上を併用してもよい。
【0052】
重合開始剤の使用量は、スチレン系モノマー100重量部に対して、0.1〜0.9重量部が好ましく、0.2〜0.5重量部がより好ましい。0.1重量部未満ではスチレン系モノマーの重合に時間がかかり過ぎることがある。0.9重量部を超える重合開始剤の使用は、スチレン系樹脂の分子量が低くなることがある。
【0053】
水系懸濁液には、必要に応じて分散剤を添加してもよい。分散剤としては、特に限定されず、公知のものをいずれも使用することができる。具体的には、リン酸カルシウム、ピロリン酸マグネシウム、ピロリン酸ナトリウム、酸化マグネシウム等の難溶性無機物が挙げられる。更に、ドデシルベンゼンスルホン酸ソーダのような界面活性剤を使用してもよい。
【0054】
次に、得られた分散液をスチレン系モノマーが実質的に重合しない温度に加熱してスチレン系モノマーをポリオレフィン系樹脂粒子に含浸させる。
ポリオレフィン系樹脂粒子内部にスチレン系モノマーを含浸させる時間は、30分〜2時間が適当である。十分に含浸させる前に重合が進行するとスチレン系樹脂の重合体粉末を生成してしまうからである。前記モノマーが実質的に重合しない温度とは、高い方が含浸速度を速めるには有利であるが、重合開始剤の分解温度を考慮して決定する必要がある。
【0055】
次いで、スチレン系モノマーの重合を行う。重合は、特に限定されないが、105〜140℃で、1.5〜5時間行うことが好ましい。重合は、通常、加圧可能な密閉容器中で行われる。
なお、スチレン系モノマーの含浸と重合を複数回に分けて行ってもよい。複数回に分けることで、ポリスチレンの重合体粉末の発生を極力少なくできる。
上記工程により改質樹脂粒子を得ることができる。得られた改質樹脂粒子は、内部がスチレン系樹脂粒子がリッチであり、外殻部がポリオレフィン樹脂がリッチであるため、発泡成形体の物性に好影響を与えると発明者等は考えている。
【0056】
次に、発泡性樹脂粒子は、上記重合中もしくは重合終了後の改質樹脂粒子に発泡剤を含浸することで得ることができる。この含浸は、それ自体公知の方法により行うことができる。例えば、重合中での含浸は、重合反応を密閉式の容器中で行い、容器中に発泡剤を圧入することにより行うことができる。重合終了後の含浸は、密閉式の容器中で、発泡剤を圧入することにより行われる。
更に、予備発泡粒子は、上記発泡性樹脂粒子を、公知の方法で所定の嵩密度に予備発泡させることで得ることができる。
更に、発泡成形体は、予備発泡粒子を発泡成形機の金型内に充填し、再度加熱して予備発泡粒子を発泡させながら、発泡粒同士を熱融着させることで得ることができる。加熱用の媒体は水蒸気が好適に使用できる。
【実施例】
【0057】
以下、実施例により本発明を具体的に説明するが、本発明はこれに限定されるものではない。なお、断りのない限り用いた試薬等は市販品を用いた。有機化合物で処理された粘土鉱物の調製、エチレン系重合体製造用触媒の調製、エチレン系重合体の製造及び溶媒精製は全て不活性ガス雰囲気下で行った。また、溶媒は全て予め公知の方法で精製、乾燥、脱酸素を行ったものを用いた。トリイソブチルアルミニウムのヘキサン溶液(0.714M)は東ソーファインケム社製を用いた。
【0058】
以下の実施例におけるポリオレフィン系樹脂の密度、MFR、末端ビニル数、溶融張力(MS160、MS190)、溶出温度−溶出量曲線、活性化エネルギーEa、n−ヘプタン抽出量、分子量(Mn、Mw)、融点及び融着強度、予備発泡粒子の嵩密度、発泡成形体の密度、融着率、加熱寸法変化率及び耐薬品性の測定法を下記する。
【0059】
<密度(d)の測定>
JIS K6760(1995)に準拠して密度勾配管法で測定する。
<MFR>
MFRは、JIS K6922−1:1998に準拠して、190℃、2.16kg荷重で測定する。
【0060】
<末端ビニル数の測定>
フーリエ変換赤外分光光度計(FT−IR)(Perkin Elmer社製、商品名SPECTRUM ONE)を用いて、エチレン系重合体を熱プレスした後、氷冷して調製したフィルムを4000cm-1〜400cm-1の範囲で吸光度を測定し、下式を用い算出する。
1000炭素原子当たりの末端ビニル数(個/1000C)=a×A/L/d
式中、aは吸光光度係数、Aは末端ビニルに帰属する909cm-1の吸光度、Lはフィルムの厚み、dは密度を示す。なお、aは、1H−NMR測定より、1000炭素原子当たりの末端ビニル数を確認したサンプルを用いて作成した検量線から求める。1H−NMR測定は、核磁気共鳴測定装置(日本電子社製、商品名GSX400)を用い、重水素化ベンゼンとo−ジクロロベンゼンの混合溶媒中、130℃において実施する。1000炭素原子当たりの末端ビニル数は、メチレンに帰属するピークと末端ビニルに帰属するピークの積分比から算出する。テトラメチルシランを基準(0ppm)として、化学シフトが1.3ppmのピークをメチレン、4.8−5.0ppmのピークを末端ビニルに帰属するピークとする。)
【0061】
<溶融張力(MS160、MS190)の測定>
ポリオレフィン系樹脂に、耐熱安定剤(チバスペシャリティケミカルズ社製、イルガノックス1010TM;1500ppm、イルガフォス168TM;1500ppm)を添加したものを、インターナルミキサー(東洋精機製作所製、商品名ラボプラストミル)を用いて、窒素気流下、190℃、回転数30rpmで30分間混練したもの測定用試料とする。
溶融張力は、バレル直径9.55mmの毛管粘度計(東洋精機製作所、商品名キャピログラフ)に、長さが8mm、直径が2.095mmのダイス状の試料を流入角が90°になるように装着して溶融張力を測定する。
【0062】
MS160は、温度を160℃に設定し、ピストン降下速度を10mm/分、延伸比を47に設定し、引き取りに必要な荷重(mN)である。なお、最大延伸比が47未満の場合、破断しない最高の延伸比での引き取りに必要な荷重(mN)をMS160とする。また、温度を190℃に設定すること以外は、160℃と同様の方法で測定した荷重(mN)をMS190とする。
【0063】
<TREFにより溶出温度−溶出量曲線の測定>
ポリオレフィン系樹脂に、耐熱安定剤(チバスペシャリティケミカルズ社製、イルガノックス1010TM;1500ppm、イルガフォス168TM;1500ppm)を添加したものを、インターナルミキサー(東洋精機製作所製、商品名ラボプラストミル)を用いて、窒素気流下、190℃、回転数30rpmで30分間混練する。混練物を、ODCBに、その濃度が0.05重量%となるように135℃で加熱溶解する。この加熱溶液5ミリリットルを、ガラスビーズを充填したカラムに注入した後、0.1℃/分の冷却速度で25℃まで冷却し、試料をガラスビーズ表面に沈着する。次に、このカラムにODCBを一定流量で流しながら、カラム温度を50℃/hrの一定速度で昇温し、各温度において溶液に溶解可能な試料を準備溶出させる。
【0064】
この際、溶剤中の試料濃度はメチレンの非対象伸縮振動の波数2925cm-1に対する吸収を赤外検出器で連続的に検出することで得られる。連続的に検出された濃度から、溶出温度−溶出量曲線を得ることができる。TREF分析は極少量の試料で、温度変化に対する溶出速度の変化を連続的に分析できるため、分別法では検出できない比較的細かいピークの検出が可能である。
【0065】
<流動の活性化エネルギー(Ea)の測定>
ポリオレフィン系樹脂に、耐熱安定剤(チバスペシャリティケミカルズ社製、イルガノックス1010TM;1500ppm、イルガフォス168TM;1500ppm)を添加したものを、インターナルミキサー(東洋精機製作所製、商品名ラボプラストミル)を用いて、窒素気流下、190℃、回転数30rpmで30分間混練したもの測定用試料とする。
【0066】
Eaは、円板−円板レオメーター(アントンパール社製、商品名MCR−300)を用い、150℃、170℃、190℃の各温度で角速度0.1〜100rad/sの範囲のせん断貯蔵弾性率G’、せん断損失弾性率G”を求める。両弾性率から、基準温度150℃での横軸のシフトファクターを求め、以下のアレニウス型の式により計算した。
粘度(ηo)=Aexp(Ea/RT)
式中、Aは温度に無関係な定数(頻度因子)、Eaは流動の活性化エネルギー(kJ/mol)、Rは気体定数、Tは絶対温度(単位:K)である。なお、縦軸の移動は行っていない。
【0067】
具体的には、流動の活性化エネルギー(Ea)は、温度−時間重ね合わせ原理に基づいて、150℃での剪断貯蔵弾性率G'、せん断損失弾性率G"(単位:Pa)の角周波数(単位:rad/sec)依存性を示すマスターカーブを作成する際のシフトファクター(aT)からアレニウス型方程式により算出される数値であって、以下に示す方法で求められる値である。すなわち、150℃、170℃及び190℃夫々の温度(t、単位:℃)におけるオレフィン系樹脂の弾性率−角周波数曲線を、温度−時間重ね合わせ原理に基づいて、各温度(t)での溶融複素粘度−角周波数曲線毎に、150℃での弾性率−角周波数曲線に重ね合わせた際に得られる各温度(t)でのシフトファクター(aT)を求め、夫々の温度(t)と、各温度(t)でのシフトファクター(aT)とから、最小自乗法により[ln(aT)]と[1/(t+273.16)]との一次近似式(下記(I)式)を算出する。次に、該一次近似式(I)の傾きmを用いて下記式(II)より流動の活性化エネルギー(Ea)を求める。
ln(aT)=m(1/(t+273.16))+n (I)
(I)式中、aTはシフトファクター、tは温度(単位:℃)、mは一次近似式(I)の傾き、nは一次近似式(I)の切片である。なお、温度t(単位:℃)を絶対温度T(単位:K)に換算するには、T=t+273.16で行う。
Ea=|0.008314×m| (II)
(II)式中、Eaは流動の活性化エネルギー(単位:kJ/mol)、mは一次近似式(I)の傾きである。
【0068】
なお、シフトファクター(aT)は、夫々の温度(t)における弾性率−角周波数の両対数曲線を、log(Y)=−log(X)軸方向に移動させて(但し、Y軸を弾性率、X軸を角周波数とする。)、150℃での弾性率−角周波数曲線に重ね合わせた際の移動量であり、該重ね合わせでは、夫々の温度(t)における弾性率−角周波数の両対数曲線は、各曲線ごとに、角周波数をaT倍に、弾性率を1/aT倍に移動させる。また、150℃、170℃及び190℃の3点の値から(I)式を最小自乗法で求めるときの相関係数は、通常、0.99以上である。
【0069】
<50℃におけるn−ヘプタン抽出量>
50℃におけるn−ヘプタン抽出量の測定方法は以下の通りである。
200メッシュパスの粉砕試料10gを秤量し、400ミリリットルのn−ヘプタンを加えて、50℃で2時間抽出を行う。抽出液からn−ヘプタンを蒸発させて、乾燥固化させて得た抽出物の重量の初期重量(10g)に対する割合を求めることにより算出する。
【0070】
<重量平均分子量(Mw)、数平均分子量(Mn)及び重量平均分子量と数平均分子量の比(Mw/Mn)の測定>
ゲル・パーミエーション・クロマトグラフィー(以下、GPC)によって上記値を次のように測定する。GPC装置(東ソー社製 商品名HLC−8121GPC/HT、カラム(東ソー社製 商品名TSKgel GMHhr−H(20)HTを装着)、溶離液1,2,4−トリクロロベンゼンを使用し、カラム温度を140℃とする。測定試料は1.0mg/ミリリットルの濃度で調製し、カラムに0.3ミリリットル注入する。GPCにより得られた溶出曲線から検量線を用いて分子量を求める。分子量の検量線は、分子量既知のポリスチレン試料を用いて校正したものを用いる。なお、Mw及びMnは直鎖状ポリエチレン換算の値として求める。
【0071】
<融点の測定>
DSC(パーキンエルマー社製、商品名:DSC−7)を用いて測定を行なう。具体的には、5〜10mgのサンプルをアルミニウムパンに挿填し、DSCに設置した後、80℃/分の昇温速度で230℃まで昇温し、230℃で3分間放置する。その後、10℃/分の降温速度で−10℃まで冷却し、再度10℃/分の昇温速度で−10℃から150℃まで昇温する手順で昇温/降温操作を行い、2回目の昇温時に観測される吸熱曲線のピーク温度を融点とする。
【0072】
<ポリオレフィン系樹脂の融着強度の評価>
ポリオレフィン系樹脂のペレットを、圧縮成形機(180℃、圧力0.2MPa)を使用して厚さ1mmのシートに成形する。得られたシートを、東洋精機社製熱版式ヒートシーラー(シール温度:融点+5℃、シール圧力:0.07MPa、シール時間:35秒)を使用して融着させる。融着強度は引張試験機テンシロン(オリエンテック社製)を用い、引張速度50mm/分で測定する。
【0073】
<嵩密度>
予備発泡粒子の嵩密度は、下記の要領で測定する。
まず、予備発泡粒子をメスシリンダに500cm3の目盛りまで充填する。但し、メスシリンダを水平方向から目視し、予備発泡粒子が一粒でも500cm3の目盛りに達していれば、充填を終了する。次に、メスシリンダ内に充填した予備発泡粒子の重量を小数点以下2位の有効数字で秤量し、その重量をW(g)とする。次式により予備発泡粒子の嵩密度を算出する。
嵩密度(g/cm3)=W/500
<発泡成形体の密度>
発泡成形体の密度は、JIS A 9511:1995「発泡プラスチック保温板」記載の方法で測定する。
【0074】
<融着率>
縦400mm×横300mmの上面を有し、厚み30mmの直方体形状の発泡成形体の上面に、カッターで横方向に沿って長さ300mm、深さ約5mmの切り込み線を入れ、この切り込み線に沿って発泡成形体を二分割する。そして、二分割された発泡成形体の破断面の発泡粒子について、発泡粒子内で破断している発泡粒子数(a)と、発泡粒子間の界面で破断している発泡粒子数(b)を測定し、下記式に基づいて融着率を算出する。
融着率(%)=100×(a)/〔(a)+(b)〕
【0075】
<加熱寸法変化率>
加熱寸法変化率は、JIS K6767:1999「発泡プラスチック−ポリエチレン−試験方法」記載のB法にて測定する。発泡成形体から縦150mm×横150mmの上面を有し、厚み50mmの試験片を切り出す。この試験片の上面の中央部に縦及び横方向に沿ってそれぞれ互いに平行に3本の直線を50mm間隔になるように記入する。そして、試験片を90℃の熱風循環式乾燥機の中に22時間置いた後に取り出し、標準状態(20±2℃、湿度65±5%)の場所に1時間放置後、縦及び横線の寸法を測定する。加熱前の直線の長さの平均値をL1、加熱後の直線の長さの平均値をL0とし、下記式に基づいて加熱寸法変化率を算出する。
加熱寸法変化率S=100×(L1−L0)/L0
加熱寸法変化率の評価は以下の通り
○:0≦S<3;寸法変化率が低く、寸法の安定性が良好である。
△:3≦S<7;寸法の変化が見られるものの、実用上使用可能である。
×:S≧7;寸法の変化が著しく見られ、実用上使用不可能である。
【0076】
<耐薬品性>
発泡成形体から縦100mm×横100mm×厚み20mmの平面長方形状の板状試験片を切り出し、23℃、湿度50%の条件で24時間放置する。なお、試験片の上面全面が発泡成形体の表面から形成されるように試験片を発泡成形体から切り出す。
次に、試験片の上面にガソリン1gを均一に塗布し、23℃、湿度50%の条件で60分放置する。その後、試験片の上面から薬品を拭き取り、試験片の上面を目視観察して下記基準に基づいて判断する。
○:良好 変化なし
△:やや悪い 表面軟化
×:悪い 表面陥没(収縮)
【0077】
製造例1
[変性ヘクトライトの調製]
水3リットルにエタノール3リットルと37%濃塩酸100ミリリットルを加えた後、得られた溶液にN,N−ジメチル−オクタデシルアミン330g(1.1mol)を添加し、60℃に加熱することによって、塩酸塩溶液を調製した。この溶液にヘクトライト1kgを懸濁させた。この懸濁液を60℃で、3時間撹拌し、上澄液を除去した後、60℃の水50Lで洗浄した。その後、60℃、10-3torrで24時間乾燥し、ジェットミルで粉砕することによって、平均粒径5.2μmの変性ヘクトライトを得た。
【0078】
[重合触媒(p)の調製]
上記変性ヘクトライト500gをヘキサン1.7リットルに懸濁させ、1,1,3,3−テトラメチルジシロキサン−1,3−ジイル−ビス(シクロペンタジエニル)ジルコニウムジクロライド8.45g(20.0mmol)とトリイソブチルアルミニウムのヘキサン溶液(0.714M)2.8リットル(2mol)の混合液を添加し、60℃で3時間攪拌した。この後、1,1,3,3−テトラメチルジシロキサン−1,3−ジイル−ビス(シクロペンタジエニル)ジルコニウムジクロライドに対して15mol%のジフェニル(1−シクロペンタジエニル)(2,7−ジ−tert−ブチル−9−フルオレニル)ジルコニウムジクロライド2.36g(3.53mmol)を添加して室温で6時間撹拌した。静置して上澄み液を除去、更にトリイソブチルアルミニウムのヘキサン溶液(0.15M)を添加して最終的に100g/Lの触媒スラリーを得た。
【0079】
[重合触媒(q)の調製]
前記変性ヘクトライト500gをヘキサン1.7リットルに懸濁させ、プロパン−1,3−ジイルビス(シクロペンタジエニル)ジルコニウムジクロリド6.63g(20.0mmol)とトリイソブチルアルミニウムのヘキサン溶液(0.714M)2.8リットル(2mol)の混合液を添加し、60℃で3時間攪拌した後、プロパン−1,3−ジイルビス(シクロペンタジエニル)ジルコニウムジクロリドに対して5mol%のジフェニルメチレン(1−シクロペンタジエニル)(9−フルオレニル)ジルコニウムジクロリド0.58g(1.05mmol)を添加して室温で6時間撹拌した。静置して上澄み液を除去、更にトリイソブチルアルミニウムのヘキサン溶液(0.15M)を添加して最終的に100g/Lの触媒スラリーを得た。
【0080】
[ポリオレフィン系樹脂の製造]
内容積540リットルの重合器に、ヘキサン300リットル及び1−ブテン1.6リットルを導入し、オートクレーブの内温を80℃に昇温した。このオートクレーブに前記重合触媒(p)74ミリリットル及び前記重合触媒(q)125ミリリットルを添加し、エチレン/水素混合ガス(水素:1500ppm含)を分圧が0.9MPaになるまで導入して重合を開始した。重合中、分圧が0.9MPaに保たれるようにエチレン/水素混合ガスを連続的に導入した。また、重合温度を80℃に制御した。重合開始90分後に重合器の内圧を脱圧した後、内容物をろ過し、乾燥して54kgのポリオレフィン系樹脂粉末を得た。ポリオレフィン系樹脂粉末を200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでポリオレフィン系樹脂ペレットを得た。得られたポリオレフィン系樹脂ペレットの密度は937kg/m3、MFRは8g/10分であった。
【0081】
製造例2
[重合触媒(r)の調製]
上記変性ヘクトライト500gをヘキサン1.7リットルに懸濁させ、ジメチルシランジイルビス(シクロペンタジエニル)ジルコニウムジクロリド6.97g(20.0mmol)とトリイソブチルアルミニウムのヘキサン溶液(0.714M)2.8リットル(2mol)の混合液を添加し、60℃で3時間攪拌した。この後、ジメチルシランジイルビス(シクロペンタジエニル)ジルコニウムジクロリドに対して15mol%のジフェニル(1−シクロペンタジエニル)(2,7−ジ−tert−ブチル−9−フルオレニル)ジルコニウムジクロライド2.36g(3.53mmol)を添加して室温で6時間撹拌した。静置して上澄み液を除去、更にトリイソブチルアルミニウムのヘキサン溶液(0.15M)を添加して最終的に100g/Lの触媒スラリーを得た。
【0082】
[ポリオレフィン系樹脂(R)の製造]
内容積540リットルの重合器に、ヘキサン300リットル及び1−ブテン1.6リットルを導入し、オートクレーブの内温を80℃に昇温した。このオートクレーブに[重合触媒(r)の調製]で調製した重合触媒(r)147ミリリットルを添加し、エチレン/水素混合ガス(水素:1000ppm含)を分圧が0.9MPaになるまで導入して重合を開始した。重合中、分圧が0.9MPaに保たれるようにエチレン/水素混合ガスを連続的に導入した。また、重合温度を80℃に制御した。重合開始90分後に重合器の内圧を脱圧した後、内容物をろ過し、乾燥して50kgのポリオレフィン系樹脂(R)粉末を得た。ポリオレフィン系樹脂粉末を200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでポリオレフィン系樹脂(R)ペレットを得た。
得られたポリオレフィン系樹脂(R)ペレットの密度は945kg/m3、MFRは10g/10分であった。
【0083】
[ポリオレフィン系樹脂(Q)の製造]
内容積540リットルの重合器に、ヘキサン300リットル及び1−ブテン6.1リットルを導入し、オートクレーブの内温を80℃に昇温した。このオートクレーブに合成例1で得られた重合触媒(q)250ミリリットルを添加し、エチレン/水素混合ガス(水素:2000ppm含)を分圧が0.9MPaになるまで導入して重合を開始した。重合中、分圧が0.9MPaに保たれるようにエチレン/水素混合ガスを連続的に導入した。また、重合温度を80℃に制御した。重合開始90分後に重合器の内圧を脱圧した後、内容物をろ過し、乾燥して53kgのポリオレフィン系樹脂(Q)粉末を得た。この粉末を200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでポリオレフィン系樹脂(Q)ペレットを得た。
得られたポリオレフィン系樹脂(Q)ペレット)の密度は930kg/m3、MFRは4g/10分であった。
【0084】
[ポリオレフィン系樹脂の調製]
前記ポリオレフィン系樹脂(R)と前記ポリオレフィン系樹脂(Q)を50対50の割合でブレンドし、200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでポリオレフィン系樹脂ペレットを得た。混合物の密度は938kg/m3、MFRは7.9g/10分であった。
【0085】
製造例3
[ポリオレフィン系樹脂の製造方法]
内容積540リットルのオートクレーブに、ヘキサン300リットル及び1−ブテン10.8リットルを導入し、オートクレーブの内温を60℃に昇温した。このオートクレーブに製造例1で調製した重合触媒(p)74ミリリットル及び重合触媒(q)125ミリリットルを添加し、エチレンを分圧が0.9MPaに保たれるように連続的に導入した。また、重合温度を60℃に制御した。重合開始30分後に1−ブテン9.1リットルを添加し、重合開始90分後に重合器(オートクレーブ)の内圧を脱圧した後、内容物をデカンテーションし、真空乾燥することで36kgのポリオレフィン系樹脂を得た。ポリオレフィン系樹脂を200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでエチレン系共重合体ペレットを得た。得られたポリオレフィン系樹脂のペレットの密度は915kg/m3、MFRは6.1g/10分であった。
【0086】
製造例4
[ポリオレフィン系樹脂の製造方法]
製造例1のポリオレフィン系樹脂の製造において、エチレン/水素混合ガスの代わりにエチレンを単独に用い、オートクレーブの内温を70℃に保つ以外、同様な方法で重合反応を行った。内容物をろ過し、乾燥して35kgのポリオレフィン系樹脂の粉末を得た。製造例1と同様にペレタイズすることで得られたポリオレフィン系樹脂のペレットの密度は935kg/m3、MFRは0.15g/10分であった。
【0087】
製造例5
[ポリオレフィン系樹脂の製造方法]
製造例1のポリオレフィン系樹脂の製造において、エチレン/水素混合ガスの濃度を3200ppmとした以外、同様な方法で重合反応を行った。内容物をろ過し、乾燥して57kgのポリオレフィン系樹脂の粉末を得た。製造例1と同様にペレタイズすることで得られたポリオレフィン系樹脂のペレットの密度は939kg/m3、MFRは18.8g/10分であった。
【0088】
製造例6
[ポリオレフィン系樹脂の製造方法]
製造例1のポリオレフィン系樹脂の製造において、エチレン/水素混合ガスの代わりにエチレンを単独に用い、オートクレーブの内温を60℃に保つ以外、同様な方法で重合反応を行った。内容物をろ過し、乾燥して21kgのポリオレフィン系樹脂の粉末を得た。製造例1と同様にペレタイズすることで得られたポリオレフィン系樹脂のペレットの密度は935kg/m3、MFRは0.06g/10分であった。
【0089】
製造例7
[ポリオレフィン系樹脂の製造方法]
製造例1のポリオレフィン系樹脂の製造において、エチレン/水素混合ガスの濃度を4800ppmとした以外、同様な方法で重合反応を行った。内容物をろ過し、乾燥して51kgのポリオレフィン系樹脂の粉末を得た。製造例1と同様にペレタイズすることで得られたポリオレフィン系樹脂のペレットの密度は939kg/m3、MFRは22.5g/10分であった。
【0090】
製造例8
[ポリオレフィン系樹脂の製造方法]
製造例2のポリオレフィン系樹脂の製造において、1−ブテンの量を最初18.4リットルとし、重合開始の30分後に9.5リットル添加した以外、同様な方法で重合反応を行った。重合開始30分後に1−ブテンを9.5リットル添加した時点で重合温度の制御が不能となり反応を停止した。内容物を乾燥させ12kgのポリオレフィン系樹脂を得た。ポリオレフィン系樹脂の密度は908kg/m3、MFRは2.0g/10分であった。
【0091】
製造例9
[ポリオレフィン系樹脂の製造方法]
製造例1のポリオレフィン系樹脂の製造において、エチレン/水素混合ガスの濃度を1000ppmとし、1−ブテンの量を0.9リットルとした以外、同様な方法で重合反応を行った。内容物をろ過し、乾燥して58kgのポリオレフィン系樹脂の粉末を得た。製造例1と同様にペレタイズすることで得られたポリオレフィン系樹脂のペレットの密度は955kg/m3、MFRは4.5g/10分であった。
【0092】
製造例10
[ポリオレフィン系樹脂の製造方法]
内容積540リットルの重合器(オートクレーブ)に、ヘキサン300リットル及び1−ブテン9.5リットルを導入し、オートクレーブの内温を80℃に昇温した。このオートクレーブに製造例2で調製した重合触媒(r)200ミリリットルを添加し、エチレンを分圧が0.9MPaに保たれるように連続的に導入した。また、重合温度を80℃に制御した。重合開始90分後に重合器(オートクレーブ)の内圧を脱圧した後、内容物をろ過し、乾燥することで57kgのポリオレフィン系樹脂を得た。得られたポリオレフィン系樹脂のペレットの密度は937kg/m3、MFRは8.0g/10分であった。
【0093】
実施例1
製造例1で得られたポリオレフィン系樹脂ペレット100重量部を押出機に供給して溶融混練して水中カット方式により造粒して楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を得た。この樹脂粒子の平均重量は0.6mgであった。
次に、攪拌機付の5リットルのオートクレーブに、ピロリン酸マグネシウム50g、ドデシルベンゼンスルホン酸ソーダ3.5gを純水2kgに分散させて分散用媒体を得た。
【0094】
分散用媒体に30℃で上記高密度ポリオレフィン系樹脂粒子600gを分散させて10分間保持し、次いで60℃に昇温して懸濁液を得た。
更に、この懸濁液に、重合開始剤としてジクミルパーオキサイドを0.6g溶解させたスチレンモノマー0.30kgを30分かけて滴下した。滴下後、30分間保持することで、高密度ポリオレフィン系樹脂粒子中にスチレン系モノマーを含浸させた。含浸後、130℃に昇温し、この温度で2時間重合(第1重合)させた。
【0095】
次に、120℃に下げた懸濁液中に、ジクミルパーオキサイドを4.2g溶解させたスチレンモノマー1.10kgを4時間30分かけて滴下した。滴下後、120℃で1時間保持することで、高密度ポリオレフィン系樹脂粒子中にスチレン系モノマーを含浸させた。含浸後、140℃に昇温し、この温度で2時間30分間保持して重合(第2重合)させた。この重合の結果、改質樹脂粒子を得ることができた。
【0096】
次いで、常温(約23℃)まで冷却し、オートクレーブから改質樹脂粒子を取り出した。改質樹脂粒子2kgと水2リットルとを、5リットルの攪拌機付オートクレーブに入れた。更に、発泡剤としてブタン520ミリリットル(300g)をオートクレーブに入れた。この後、70℃に昇温し、4時間攪拌を続けることで発泡性樹脂粒子を得ることができた。
その後、常温まで冷却して、発泡性樹脂粒子をオートクレーブから取り出し、脱水乾燥させた。
【0097】
次いで、得られた発泡性樹脂粒子を嵩密度0.033g/cm3に予備発泡させることで、予備発泡粒子を得た。得られた予備発泡粒子を1日間室温(23℃)に放置した後、400mm×300mm×30mmの大きさの成形用金型に入れた。その後、0.12MPaの水蒸気を50秒間導入して加熱し、次いで、発泡成形体の最高面圧が0.01MPaに低下するまで冷却することで、密度0.033g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。結果を表3に示す。
【0098】
実施例2
製造例1で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を760g使用し、第1重合において、ジクミルパーオキサイドを0.8g、スチレンモノマーを0.38kg使用し、第2重合において、ジクミルパーオキサイドを3.7g、スチレンモノマーを0.86kg使用し、滴下時間を4時間15分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.040g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.040g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0099】
実施例3
製造例2で得られたポリオレフィン系樹脂ペレット100重量部を押出機に供給して溶融混練して水中カット方式により造粒して楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を得た。この樹脂粒子の平均重量は0.6mgであった。
上記で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を600g使用し、第1重合において、ジクミルパーオキサイドを0.6g、スチレンモノマーを0.30kg使用し、第2重合において、ジクミルパーオキサイドを4.2g、スチレンモノマーを1.10kg使用し、滴下時間を4時間30分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
【0100】
次いで、得られた発泡性樹脂粒子を嵩密度0.033g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.033g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0101】
実施例4
製造例2で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を760g使用し、第1重合において、ジクミルパーオキサイドを0.8g、スチレンモノマーを0.38kg使用し、第2重合において、ジクミルパーオキサイドを3.7g、スチレンモノマーを0.86kg使用し、滴下時間を4時間15分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.045g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.045g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0102】
実施例5
製造例3で得られたポリオレフィン系樹脂ペレット100重量部を押出機に供給して溶融混練して水中カット方式により造粒して楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を得た。この樹脂粒子の平均重量は0.6mgであった。
【0103】
上記で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を600g使用し、第1重合において、ジクミルパーオキサイドを0.6g、スチレンモノマーを0.30kg使用し、第2重合において、ジクミルパーオキサイドを4.2g、スチレンモノマーを1.10kg使用し、滴下時間を4時間30分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
【0104】
次いで、得られた発泡性樹脂粒子を嵩密度0.033g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.033g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0105】
実施例6
製造例3で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を760g使用し、第1重合において、ジクミルパーオキサイドを0.8g、スチレンモノマーを0.38kg使用し、第2重合において、ジクミルパーオキサイドを3.7g、スチレンモノマーを0.86kg使用し、滴下時間を4時間15分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.050g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.050g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0106】
実施例7
製造例4で得られたポリオレフィン系樹脂ペレット100重量部を押出機に供給して溶融混練して水中カット方式により造粒して楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を得た。この樹脂粒子の平均重量は0.6mgであった。
上記で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を600g使用し、第1重合において、ジクミルパーオキサイドを0.6g、スチレンモノマーを0.30kg使用し、第2重合において、ジクミルパーオキサイドを4.2g、スチレンモノマーを1.10kg使用し、滴下時間を4時間30分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
【0107】
次いで、得られた発泡性樹脂粒子を嵩密度0.050g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.050g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0108】
実施例8
製造例4で得られた楕円球状(卵状)の高密度ポリオレフィン系樹脂粒子を760g使用し、第1重合において、ジクミルパーオキサイドを0.8g、スチレンモノマーを0.38kg使用し、第2重合において、ジクミルパーオキサイドを3.7g、スチレンモノマーを0.86kg使用し、滴下時間を4時間30分とすること以外は、実施例1と同様にして、改質樹脂粒子及び発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.055g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.055g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表3に示す。
【0109】
比較例1
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例5で得られたポリオレフィン系樹脂ペレット(MFR=18.8g/10分、密度=939kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.040g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.040g/cm3の発泡成形体を得た。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0110】
比較例2
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例6で得られたポリオレフィン系樹脂ペレット(MFR=0.06g/10分、密度=935kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
比較例3
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例7で得られたポリオレフィン系樹脂ペレット(MFR=22.5g/10分、密度=939kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
【0111】
比較例4
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例8で得られたポリオレフィン系樹脂ペレット(MFR=2.0g/10分、密度=908kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.033g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.033g/cm3の発泡成形体を得た。得られた発泡成形体の外観及び融着は共に良好であった。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0112】
比較例5
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例9で得られたポリオレフィン系樹脂ペレット(MFR=4.5g/10分、密度=955kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.065g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.065g/cm3の発泡成形体を得た。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0113】
比較例6
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、製造例10で得られたポリオレフィン系樹脂ペレット(MFR=8.0g/10分、密度=937kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。
次いで、得られた発泡性樹脂粒子を嵩密度0.033g/cm3に予備発泡させることで、予備発泡粒子を得た。また、実施例1と同様にして密度0.033g/cm3の発泡成形体を得た。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0114】
比較例7
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、示差走査型熱量計による昇温測定において得られる吸熱曲線のピークが1つである、市販の直鎖状低密度ポリエチレン(ユメリット4540F、宇部興産社製、MFR=3.9g/10分、密度=944kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
【0115】
比較例8
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、示差走査型熱量計による昇温測定において得られる吸熱曲線のピークが1つである、市販の高密度ポリエチレン(ニポロンハード2500、東ソー社製、MFR=8.0g/10分、密度=961kg/m3)とした以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
【0116】
比較例9
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、示差走査型熱量計による昇温測定において得られる吸熱曲線のピークが1つである、市販の直鎖状低密度ポリエチレン(ニポロンZZF260、東ソー社製、MFR=2.0g/10分、密度=936kg/m3)を用いた以外は実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
【0117】
比較例10
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、示差走査型熱量計による昇温測定において得られる吸熱曲線のピークが1つである、市販の低密度ポリエチレン(ペトロセン 203、東ソー(株)製、MFR=8.0g/10分、密度=919kg/m3)を用いた以外は実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子、予備発泡粒子(嵩密度0.033g/cm3)、発泡成形体(密度0.033g/cm3)を得た。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0118】
比較例11
[重合触媒(r)の調製]
上記変性ヘクトライト500gをヘキサン1.7リットルに懸濁させ、ジメチルシランジイルビス(シクロペンタジエニル)ジルコニウムジクロリド6.97g(20.0mmol)とトリイソブチルアルミニウムのヘキサン溶液(0.714M)2.8リットル(2mol)の混合液を添加し、60℃で3時間攪拌した後、ジメチルシランジイルビス(シクロペンタジエニル)ジルコニウムジクロリドに対して15mol%のジフェニル(1−シクロペンタジエニル)(2,7−ジ−tert−ブチル−9−フルオレニル)ジルコニウムジクロライド2.36g(3.53mmol)を添加して室温で6時間撹拌した。静置して上澄み液を除去、更にトリイソブチルアルミニウムのヘキサン溶液(0.15M)を添加して最終的に100g/Lの触媒スラリーを得た。
【0119】
[ポリオレフィン系樹脂(S)の製造]
内容積540リットルの重合器に、ヘキサン300リットル及び1−ブテン1.3リットルを導入し、オートクレーブの内温を80℃に昇温した。このオートクレーブに[重合触媒(r)の調製]で調製した重合触媒(r)147ミリリットルを添加し、エチレン/水素混合ガス(水素:1000ppm含)を分圧が0.9MPaになるまで導入して重合を開始した。重合中、分圧が0.9MPaに保たれるようにエチレン/水素混合ガスを連続的に導入した。また、重合温度を80℃に制御した。重合開始90分後に重合器の内圧を脱圧した後、内容物をろ過し、乾燥して50kgのポリオレフィン系樹脂(S)粉末を得た。ポリオレフィン系樹脂粉末を200℃に設定した50mm径の単軸押出機を使用して溶融混練、ペレタイズすることでポリオレフィン系樹脂(S)ペレットを得た。
得られたポリオレフィン系樹脂(S)ペレットの密度は950kg/m3、MFRは8g/10分であった。
【0120】
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、上記で得られたポリオレフィン系樹脂(S)ペレット(MFR=8.0g/10分、密度=950kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子、予備発泡粒子(嵩密度0.033g/cm3)、発泡成形体(密度0.033g/cm3)を得た。得られた発泡成形体の融着率、加熱寸法変化率、耐薬品性を測定した。その結果を表4に示す。
【0121】
比較例12
製造例1で得られたポリオレフィン系樹脂ペレットに代えて、示差走査型熱量計による昇温測定において得られる吸熱曲線のピークが複数個ある、市販のメタロセンポリエチレン(ハーモレックス NH745S、日本ポリエチレン社製、MFR=9.2g/10分、密度=912kg/m3)を用いた以外は、実施例1と同様に、改質樹脂粒子、発泡性樹脂粒子を得た。得られた発泡性樹脂粒子を予備発泡させたが、ほとんど発泡せず、発泡可能な予備発泡粒子は得られなかった。
【0122】
以下、表1及び2に実施例及び比較例で使用した触媒の各種物性値を示す。
【0123】
【表1】
【0124】
【表2】
【0125】
更に、表3及び4に実施例及び比較例の製造条件、予備発泡粒子の嵩密度、発泡成形体の融着率、加熱寸法変化率及び耐薬品性を示す。
【0126】
【表3】
【0127】
【表4】
【0128】
表3及び4から以下のことが分かる。
実施例と、比較例4、5及び8とから、オレフィン系樹脂の密度(kg・cm3)が910〜950の範囲内であることで、融着率、加熱寸法変化率及び耐薬品性の全てが優れた発泡成形体が得られることが分かる。
実施例と、比較例2及び3とから、MFR(g/10分)が0.1〜20の範囲内であることで、融着率、加熱寸法変化率及び耐薬品性の全てが優れた発泡成形体が得られることが分かる。
実施例と比較例7とから、末端ビニル数が1000炭素原子当たり0.2以下であることで、融着率、加熱寸法変化率及び耐薬品性の全てが優れた発泡成形体が得られることが分かる。
【0129】
実施例と、比較例7及び9とから、MS160>90−130×log(MFR)の関係を有することで、融着率、加熱寸法変化率及び耐薬品性の全てが優れた発泡成形体が得られることが分かる。
実施例と比較例1とから、MS160/MS190>1.8の関係を有することで、融着率が優れた発泡成形体が得られることが分かる。
実施例と、比較例6〜11とから、TREFによる溶出温度−溶出量曲線にピークが2個以上存在することで、融着率、加熱寸法変化率及び耐薬品性の全てが優れた発泡成形体が得られることが分かる。
【図面の簡単な説明】
【0130】
【図1】本発明に使用可能なポリオレフィン系樹脂の代表的なTREF溶出温度−溶出量曲線である。
【図2】代表的なメタロセン触媒により得られたエチレン・α−オレフィン共重合体のTREF溶出温度−溶出量曲線である。
【特許請求の範囲】
【請求項1】
エチレンから導かれる繰り返し単位、又はエチレンから導かれる繰り返し単位及び炭素数3〜8のα−オレフィンから導かれる繰り返し単位から構成され、かつ下記要件(A)〜(H)
(A)密度[d(kg/m3)]が910以上950以下
(B)190℃、2.16kg荷重で測定したメルトフローレート[MFR(g/10分)]が0.1以上20以下
(C)末端ビニル数が1000炭素原子当たり0.2個以下
(D)160℃で測定した溶融張力[MS160(mN)]とMFRの関係が、下記式(1)を満足
MS160>90−130×log(MFR) (1)
(E)190℃で測定した溶融張力[MS190(mN)]とMS160の関係が、下記式(2)を満足
MS160/MS190<1.8 (2)
(F)連続昇温溶出分別法(TREF)による溶出温度−溶出量曲線にピークが2個以上存在
の要件を満足するポリオレフィン系樹脂と、前記ポリオレフィン系樹脂を改質するスチレン系樹脂とを含み、前記スチレン系樹脂が、前記ポリオレフィン系樹脂100重量部に対して、20〜600重量部使用されることを特徴とするスチレン改質ポリオレフィン系樹脂粒子。
【請求項2】
前記ポリオレフィン系樹脂が、更に、下記(G)及び(H)
(G)流動の活性化エネルギー[Ea(kJ/mol)]と密度の関係が、下記式(3)を満足
127−0.107d<Ea<88−0.060d (3)
(H)50℃におけるn−ヘプタン抽出量が0.2重量%以下
の要件を満足する請求項1に記載のスチレン改質ポリオレフィン系樹脂粒子。
【請求項3】
前記要件(F)のピークが2個存在し、一方のピークが85〜100℃の範囲に、他方のピークが65〜80℃の範囲に存在する請求項1又は2に記載のスチレン改質ポリオレフィン系樹脂粒子。
【請求項4】
前記要件(A)の密度が925〜945の範囲、
前記要件(B)のメルトフローレートが2〜10の範囲、
前記要件(C)の末端ビニル数が、0.05個以下、
前記要件(E)の式(2)がMS160/MS190<1.6
を満足する請求項1〜3のいずれか1つに記載のスチレン改質ポリオレフィン系樹脂粒子。
【請求項5】
前記スチレン改質ポリオレフィン系樹脂粒子が、前記ポリオレフィン系樹脂を与えるポリオレフィン系樹脂粒子100重量部に、水性媒体中、重合開始剤の存在下で、スチレン系モノマー20〜600重量部を含浸させつつ重合させることで得られる請求項1〜4のいずれか1つに記載のスチレン改質ポリオレフィン系樹脂粒子。
【請求項6】
前記ポリオレフィン系樹脂が、末端にビニル基を有するエチレンの単独重合体又はエチレンと炭素数3以上のオレフィンとの共重合体からなるマクロモノマーと、炭素数2以上のオレフィンとを共重合させることにより得られる請求項1〜5のいずれか1つに記載のスチレン改質ポリオレフィン系樹脂粒子。
【請求項7】
請求項1〜6いずれか1つに記載のスチレン改質ポリオレフィン系樹脂粒子100重量部と、揮発性発泡剤5〜25重量部とを含むことを特徴とする発泡性樹脂粒子。
【請求項8】
請求項7に記載の発泡性樹脂粒子を予備発泡させて得られた嵩密度0.01〜0.2g/cm3の予備発泡粒子。
【請求項9】
請求項8に記載の予備発泡粒子を型内発泡成形させて得られた密度0.01〜0.2g/cm3の発泡成形体。
【請求項1】
エチレンから導かれる繰り返し単位、又はエチレンから導かれる繰り返し単位及び炭素数3〜8のα−オレフィンから導かれる繰り返し単位から構成され、かつ下記要件(A)〜(H)
(A)密度[d(kg/m3)]が910以上950以下
(B)190℃、2.16kg荷重で測定したメルトフローレート[MFR(g/10分)]が0.1以上20以下
(C)末端ビニル数が1000炭素原子当たり0.2個以下
(D)160℃で測定した溶融張力[MS160(mN)]とMFRの関係が、下記式(1)を満足
MS160>90−130×log(MFR) (1)
(E)190℃で測定した溶融張力[MS190(mN)]とMS160の関係が、下記式(2)を満足
MS160/MS190<1.8 (2)
(F)連続昇温溶出分別法(TREF)による溶出温度−溶出量曲線にピークが2個以上存在
の要件を満足するポリオレフィン系樹脂と、前記ポリオレフィン系樹脂を改質するスチレン系樹脂とを含み、前記スチレン系樹脂が、前記ポリオレフィン系樹脂100重量部に対して、20〜600重量部使用されることを特徴とするスチレン改質ポリオレフィン系樹脂粒子。
【請求項2】
前記ポリオレフィン系樹脂が、更に、下記(G)及び(H)
(G)流動の活性化エネルギー[Ea(kJ/mol)]と密度の関係が、下記式(3)を満足
127−0.107d<Ea<88−0.060d (3)
(H)50℃におけるn−ヘプタン抽出量が0.2重量%以下
の要件を満足する請求項1に記載のスチレン改質ポリオレフィン系樹脂粒子。
【請求項3】
前記要件(F)のピークが2個存在し、一方のピークが85〜100℃の範囲に、他方のピークが65〜80℃の範囲に存在する請求項1又は2に記載のスチレン改質ポリオレフィン系樹脂粒子。
【請求項4】
前記要件(A)の密度が925〜945の範囲、
前記要件(B)のメルトフローレートが2〜10の範囲、
前記要件(C)の末端ビニル数が、0.05個以下、
前記要件(E)の式(2)がMS160/MS190<1.6
を満足する請求項1〜3のいずれか1つに記載のスチレン改質ポリオレフィン系樹脂粒子。
【請求項5】
前記スチレン改質ポリオレフィン系樹脂粒子が、前記ポリオレフィン系樹脂を与えるポリオレフィン系樹脂粒子100重量部に、水性媒体中、重合開始剤の存在下で、スチレン系モノマー20〜600重量部を含浸させつつ重合させることで得られる請求項1〜4のいずれか1つに記載のスチレン改質ポリオレフィン系樹脂粒子。
【請求項6】
前記ポリオレフィン系樹脂が、末端にビニル基を有するエチレンの単独重合体又はエチレンと炭素数3以上のオレフィンとの共重合体からなるマクロモノマーと、炭素数2以上のオレフィンとを共重合させることにより得られる請求項1〜5のいずれか1つに記載のスチレン改質ポリオレフィン系樹脂粒子。
【請求項7】
請求項1〜6いずれか1つに記載のスチレン改質ポリオレフィン系樹脂粒子100重量部と、揮発性発泡剤5〜25重量部とを含むことを特徴とする発泡性樹脂粒子。
【請求項8】
請求項7に記載の発泡性樹脂粒子を予備発泡させて得られた嵩密度0.01〜0.2g/cm3の予備発泡粒子。
【請求項9】
請求項8に記載の予備発泡粒子を型内発泡成形させて得られた密度0.01〜0.2g/cm3の発泡成形体。
【図1】

【図2】
【図2】
【公開番号】特開2010−24353(P2010−24353A)
【公開日】平成22年2月4日(2010.2.4)
【国際特許分類】
【出願番号】特願2008−187515(P2008−187515)
【出願日】平成20年7月18日(2008.7.18)
【出願人】(000002440)積水化成品工業株式会社 (1,335)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
【公開日】平成22年2月4日(2010.2.4)
【国際特許分類】
【出願日】平成20年7月18日(2008.7.18)
【出願人】(000002440)積水化成品工業株式会社 (1,335)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
[ Back to top ]