
ストロンチウム、ビスマス、タンタル系前駆物質及びその製造法
【課題】Sr,Bi,Ta系酸化物を構成するストロンチム、ビスマス、タンタル、それぞれの金属を含み、かつ高い均質性、有機溶媒に対する高い溶解性、縮合安定性及び保存安定性に優れた高分子量体として、鉛系金属酸化物の前駆物質、及びその製造法を提供することである。
【解決手段】Sr,Bi及びTaの錯体化合物とアルカノールアミンを反応させて、錯体分子間でアルカノールアミンの水酸基を介した架橋反応を起こすことで得られる高分子量体の酸化物前駆物質、及びその製造法。
【解決手段】Sr,Bi及びTaの錯体化合物とアルカノールアミンを反応させて、錯体分子間でアルカノールアミンの水酸基を介した架橋反応を起こすことで得られる高分子量体の酸化物前駆物質、及びその製造法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ストロンチム、ビスマス及びタンタルからなるSr,Bi,Ta系酸化物の製造方法に関する。さらに詳しくは、Sr,Bi,Ta系酸化物を形成する際の原料となる前駆物質の調製、Sr,Bi,Ta系酸化物の形成方法等に関する。
【背景技術】
【0002】
結晶性のタンタル酸ストロンチウムビスマス(以下SBTと称す。)薄膜は高い誘電率、優れた強誘電特性から、種々のキャパシタや不揮発性メモリなどのデバイスへの応用が期待されている。
現在、実用化に至っている強誘電体である鉛系金属酸化物は、鉛が環境問題に対応できず、今後使用ができなく可能性があり、鉛系金属酸化物に代わる非鉛系金属酸化物強誘電体の開発が待たれている。そこで、非鉛系金属酸化物強誘電体として最も有望であるもののひとつがSBTである。
ストロンチム、ビスマス及びタンタルからなるSr,Bi,Ta系酸化物を700℃から1000℃で焼成することで、強誘電特性を示すタンタル酸ストロンチウムビスマスが得られることが分かっている(非特許文献1または非特許文献2参照)。
代表的なSBT薄膜の作製方法として、スパッタリング法、気相蒸着法、レーザーアブレーション法などがあるが、特に液相法には、金属有機化合物堆積法(Metal Organic Deposition:MOD)法やゾル・ゲル法などがある。
MOD法では、原料として有機金属化合物である、酢酸塩、クエン酸塩、シュウ酸塩、カルボン酸塩、金属アルコキシド、キレート試薬などを用いて簡潔なプロセスで前駆体溶液が調製されている。前駆体溶液は、混合物なので分子単体で存在すること、安定化することによってそれぞれの分子間の反応が阻害されていることなどから、前駆体溶液内の均質性、縮合安定性に欠けることが懸念される。
【0003】
ゾル・ゲル法では一般的に、金属アルコキシド化合物の加水分解・縮重合を経て、前駆体溶液が調製される。金属アルコキシド化合物は反応性が高いため、大気中の水分で容易に加水分解と縮重合が進むため、取扱いに困難が伴う。また、加水分解・縮重合を経た前駆体溶液も、大気中の水分で、さらに加水分解と縮重合が進み、保存安定性がしばしば問題となり、その後の成膜時に不良を生じる原因となる。そのため、工業的な応用を図るために、β−ジケトン類、β−ケトエステル類、カルボン酸類、エタノールアミン類、ジオール類、エステル類を前駆体の安定化剤として各々のPb系金属酸化物モル数に対して0.2倍〜5倍モル添加することで高い安定性を有する前駆体が調製されている。例えば、βージケトン、βーケトエステル、オキシ酸、ジオール、トリオール、高級カルボン酸、アルカノールアミン及び多価アミンよりなる群から選ばれた1種又は2種以上の安定化剤を組成物中の金属合計量1モルに対して、0.2〜3モルの割合で含有することを特徴としたSBT系強誘電体薄膜形成用組成物(特許文献1参照)などの発明が知られている。しかし、安定化剤を使用しても、依然として前駆体溶液の安定性の更なる改善が望まれている。
【特許文献1】特開2003−2650号公報
【非特許文献1】「ソリッドステイトマテリアルズ フォー アドバンス テクノロジー(solid−state materials for advanced technology)」,B97号,2003年,p.275−282
【非特許文献2】「ジャーナル オブ ヨーロピアン セラミックス ソサイエティー(Journal of the European Ceramic Society)」,24号,2004年,p.3043−3048
【発明の開示】
【発明が解決しようとする課題】
【0004】
そこで、本発明の目的は、Sr,Bi,Ta系酸化物を構成するストロンチム、ビスマス、タンタル、それぞれの金属を含み、かつ高い均質性、有機溶媒に対する高い溶解性、縮合安定性及び保存安定性に優れた高分子量体として、鉛系金属酸化物の前駆物質、及びその製造法を提供することである。
【課題を解決するための手段】
【0005】
本発明者は、以上の課題を解決するために鋭意検討した結果、本発明を完成するに至った。即ち、本発明は、
1.Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られることを特徴とするSr,Bi,Ta系酸化物前駆物質。
2.アルカノールアミンがジエタノールアミンまたはトリエタノールアミンである上記1記載のSr,Bi,Ta系酸化物前駆物質。
3.Sr、Bi及びTaの錯体化合物がアセト酢酸エチル錯体である上記1又は2に記載のSr,Bi,Ta系酸化物前駆物質。
4.Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させることを特徴とするSr,Bi,Ta系酸化物前駆物質の製造法。
5.アルカノールアミンがジエタノールアミンまたはトリエタノールアミンである上記4記載のSr,Bi,Ta系酸化物前駆物質の製造法。
6.Sr、Bi及びTaの錯体化合物がアセト酢酸エチル錯体である上記4又は5に記載のSr,Bi,Ta系酸化物前駆物質の製造法。
7.上記1〜3のいずれかに記載のSr,Bi,Ta系酸化物前駆物質を含有するSr,Bi,Ta系酸化物前駆体。
8.上記7に記載のSr,Bi,Ta系酸化物前駆体を用いて得られるSr,Bi,Ta系酸化物。
9.Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られるSr,Bi,Ta系酸化物前駆物質を用いることを特徴とするSr,Bi,Ta系酸化物前駆体の製造法。
10.Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られるSr,Bi,Ta系酸化物前駆物質を含有するSr,Bi,Ta系酸化物前駆体を用いるSr,Bi,Ta系酸化物膜の製造方法。
に関する。
【発明の効果】
【0006】
本発明のSr,Bi,Ta系酸化物前駆物質の製造方法では、Sr,Bi,Ta系酸化物を構成するそれぞれの金属を含み、かつ高い均質性、有機溶媒に対する高い溶解性、縮合安定性及び保存安定性に優れた高分子量体として、Sr,Bi,Ta系酸化物前駆物質を製造することができる。そして、得られた前駆物質を用いて調製した前駆体溶液を塗布液とした薄膜の製造により、Sr,Bi,Ta系酸化物を作製することができる。
【発明を実施するための最良の形態】
【0007】
本発明について詳細に述べる。本発明のSr,Bi,Ta系酸化物前駆物質は、ストロンチム、ビスマス及びタンタルの錯体化合物とアルカノールアミン(トリエタノールアミンまたはジエタノールアミンなどとを有機溶媒中で反応させた後、濃縮により溶媒および反応により生成した錯体化合物の配位子を留去して得られ、高い均質性、有機溶媒に対する高い溶解性、縮合安定性及び保存安定性に優れる高分子量体のSr,Bi,Ta系酸化物前駆物質である。また、この前駆物質は高粘度の液体または有機溶媒可溶性の粉末として得られ、これらを用いて前駆体溶液を調製し、前駆物質、前駆体溶液及び前駆体溶液を塗布液として製造した薄膜を減圧下熱分解ですることで高均質Sr,Bi,Ta系酸化物を製造することができる。
本発明はストロンチム、ビスマス及びタンタルの錯体化合物、特にアセト酢酸エチル錯体の性質に着目してなされたものである。即ち、これらの錯体は、それぞれの金属アルコキシドやカルボン酸塩に比べ、分子会合性が低いため一般の有機溶媒に対する溶解性が極めて高く取扱いが容易である。
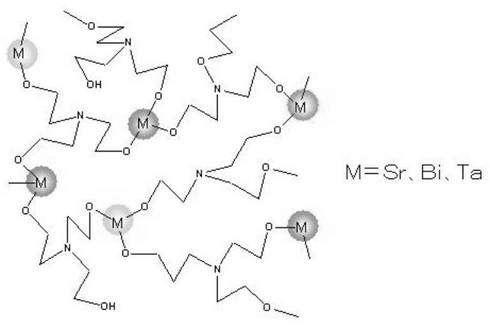
一方、ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンとの反応については明確なことは不明であるが、これらの錯体分子間でトリエタノールアミンまたはジエタノールアミンの水酸基を介した架橋反応が容易に起こり、これらの錯体化合物の高分子量化が起こっていると推定される(図1)。
【0008】
従って、ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンとを有機溶媒中で反応させることにより、錯体化合物の高分子量化に伴うと推定される高粘度化または乾固が起こり、有機溶媒に対して良好な溶解性を示す、高粘性前駆物質液体または有機溶媒可溶性前駆物質粉末が得られる。さらに、上記前駆物質を用いて調製した前駆体より、Sr,Bi,Ta系酸化物薄膜、高純度Sr,Bi,Ta系酸化物粉末が容易に得られる。
ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンの反応においては、錯体化合物の配位子が外れることによって、錯体分子間でトリエタノールアミンまたはジエタノールアミンの水酸基を介した架橋反応が進行する。そのため、反応後の溶液に配位子の存在を確認することによって、高分子量化が進んでいることが確かめられる。
【0009】
本発明の前駆物質の製造に用いられるストロンチム、ビスマス及びタンタルの錯体化合物は、β−ジケトン類、β−ケトエステル類、カルボン酸類、ジオール類(エチレングリコール、プロピレングリコール、1,2−ブタンジオール、1,3−ブタンジオール、1,4−ブタンジオール、2,3−ブタンジオール、ヘキシレングリコールなど)、エステル類などの錯体化合物であり、これらが1種類以上用いられる。
好ましくはアセト酢酸エチル錯体及びトリエタノールアミン錯体を用いることによって、有機溶媒に対する溶解性が極めて高く、また加水分解に対する安定性が高い、さらに製造時の取扱い及び貯蔵安定性が特に優れた金属酸化物の前駆体を製造することができる。
本発明のSr,Bi,Ta系酸化物前駆物質の製造方法は、[1]ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンの調製→[2]ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンとの反応→[3]生成した前駆物質溶液の濃縮・乾固よりなる。以下に詳細を示す。
【0010】
[1]ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンの調製
金属酸化物の前駆物質の原料であるストロンチム、ビスマス及びタンタルの錯体化合物はモル比でSr:Bi:Ta=0.6〜1.1:2.0〜2.6:1.0の組成よりなるように調製される。上記組成範囲の各錯体化合物を有機溶媒に混合溶解した後、トリエタノールアミンまたはジエタノールアミンを、Biの錯体化合物に対して0.5〜5.0倍モルの範囲で添加する。
添加するトリエタノールアミンまたはジエタノールアミンが0.5〜1.0未満の範囲では高分子量化に必要なトリエタノールアミンまたはジエタノールアミンの量が不十分なため、原料と高分子量体の進行した前駆物質の混合物として得られる。1.0〜2.0倍モルの範囲では、前駆物質は粉末として得られ、3.0〜5.0倍モルの範囲では前駆物質は固体と液体の混合物または液体として得られる。高分子量化に関わるトリエタノールアミンまたはジエタノールアミンの量は前駆物質が粉末で得られる範囲であり、この範囲ではトリエタノールアミンまたはジエタノールアミンが増えるにしたがって、高分子量化が進行する。これ以上の範囲では、トリエタノールアミンまたはジエタノールアミンが過剰に存在し、前駆物質がトリエタノールアミンまたはジエタノールアミンに一部溶解した固体と液体の混合物またはすべて溶解した液体となる。よって、5.0倍モルより大きくても濃縮に時間がかかり経済的でないため、より好ましくはトリエタノールアミンまたはジエタノールアミンは1.0〜2.0倍モルの範囲で添加される。
ストロンチム、ビスマス及びタンタルの錯体化合物は各金属を酸化物に換算して1〜10wt%含まれるように調製される。酸化物含量が1wt%以下の場合、濃縮するために多量の溶媒を留去しなければならず経済的でない。一方、10wt%を超えると鉛、ストロンチム、ビスマス及びタンタルの錯体化合物が溶解しづらくなり、且つ均一な反応を行わせることが困難になる。
【0011】
[2]ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンとの反応
反応温度は通常60〜100℃であるが、用いる有機溶媒の還流温度が簡便である。また、反応時間は反応温度によって異なり、即ち低温では長時間を要し、高温では短時間で済む。
[3]生成した前駆物質溶液の濃縮・乾固
反応終了後、温度40〜80℃、圧力20〜60mmHgの減圧下で2〜3時間かけて反応に用いた有機溶媒及び反応で生成した配位子を留去し、酸化物濃度25〜30wt%となるまで濃縮することにより、粘度50mPa・s〜1000mPa・sの高粘性溶液が得られる。
さらに、得られた高粘性溶液を温度40〜80℃、圧力10mmHg以下の減圧下で高分子量化に生じる配位子の留去し、重量減少がなくなるまで濃縮、乾固することにより、有機溶媒可溶性前駆物質粉末、高粘性前駆物質液体またはそれらの混合物を得ることができる。減圧度10mmHg以上では、トリエタノールアミンまたはジエタノールアミンや高分子量化の反応中に生じたアセト酢酸エチルなどの錯体化合物の配位子の一部が充分に揮散しない。
反応終了後、溶液を10mmHg以下において40〜80℃で処理することにより、有機溶媒の留去による濃縮及び高分子量化に生じる配位子の留去による濃縮、乾固を同時に行い、有機溶媒可溶性前駆物質粉末、高粘性前駆物質液体またはそれらの混合物を得ることもできる。
【0012】
本発明方法に用いる有機溶媒としては、ストロンチム、ビスマス及びタンタルの錯体化合物を溶解する物が用いられる。一例を挙げるならば、アルコール類(メタノール、エタノール、1−プロパノール、2−プロパノール、1−ブタノール、2−ブタノール、ヘキサノールなど)、テトラヒドロフラン、ジオキサンなどのエーテル類、セロソルブ類(メチルセロソルブ、エチルセロソルブ、プロピルセロソルブ、ブチルセロソルブなど)、グリコール類(エチレングリコール、プロピレングリコール、1,2−ブタンジオール、1,3−ブタンジオール、1,4−ブタンジオール、2,3−ブタンジオール、ヘキシレングリコールなど)、アセトン、クロロホルム、四塩化炭素、ベンゼン、トルエンなどであり、これらの一種以上を用いることができる。
本発明のSr,Bi,Ta系酸化物前駆体は、このようにして得られた有機溶媒可溶性前駆物質粉末、高粘性前駆物質液体またはそれらの混合物を有機溶媒に溶解したものを用いる。用いる有機溶媒としては、前駆物質を溶解するものであれば特に限定されない。その具体例としては、メタノール、エタノール、テトラヒドロフラン、クロロホルムなどが挙げられる。
【0013】
このようにして得られるSr,Bi,Ta系酸化物前駆体は、通常用いられる塗布法、例えばディッピング法、スピンコート法、フレキソ印刷法などを用いて、ガラス、金属、セラミックスなどの基板に塗布することができる。優れた成膜性を得るためには、酸化物換算濃度として1〜20wt%とすることが好ましい。酸化物濃度が1%以下では粘度が低く成膜性が低く、一方20wt%以上では成形加工性が悪くなり、成膜が困難である。従って、あらかじめ成形された薄膜などを上記条件下で処理することにより、薄膜などの形状を保持したまま分解し、不融化することができる。また、有機溶媒可溶性前駆物質粉末、高粘性前駆物質液体またはそれらの混合物に同様の処理を行うことにより高純度のSr,Bi,Ta系酸化物を得ることができる。
本発明のSr,Bi,Ta系酸化物前駆体である有機溶媒可溶性前駆物質粉末、高粘性前駆物質液体またはそれらの混合物は、クロロホルム、テトラヒドロフラン、ジオキサン、アセトン、プロパノール、エタノール、メタノールなどの極性溶媒に対して優れた溶解性を示し、ベンゼン、四塩化炭素などの非極性溶媒には溶解性を示さない。従って、上記前駆物質を再溶解し、塗布液として用いる場合には、上記極性溶媒を用いる必要がある。ただし、塗工性の向上を目的として、前駆物質の溶解性を損なわない範囲で、他の非極性溶媒を一部加えることは差し支えない。
以下に実施例を挙げ、本発明を更に具体的に説明するが、本発明はこれらに限定されるものではない。
【実施例】
【0014】
実施例で用いた分析法は以下の通りである。
[1] 熱重量示差熱分析(TG−DTA)
TG−DTA装置:TG−DTA2020S((株)マック・サイエンス製)で測定した。測定条件:空気気流中、昇温速度:10℃/min。
[2] ガスクロマトグラフィー分析(GC)
シリカゲル(GLサイエンス社製SE−30)を充填したステンレス製カラム(2m×3mmφ)を装着した島津製作所製(株)GC−14B型(インジェクション温度:290℃、カラム温度:180℃、熱電対型検出器温度:290℃)を用い、キャリアーガスとしてヘリウムガスを流量5.5ml/minで流通させた。鉛、チタン及びジルコニウムの錯体化合物、鉛のトリエタノールアミンまたはジエタノールアミン錯体、トリエタノールアミンまたはジエタノールアミン、それらの反応後、濃縮・乾固で留去した溶媒の上記条件でGC分析を行い、アセト酢酸エチルのピークの有無を確認した。
[3] 融点、沸点、分解点の測定
密閉したガラス細管に試料を充填し、ヤナコ製Micro Meltingpoint Apparatus MP−S3型融点測定器を用いて測定した。同一試料について試料が液化した温度前後で数回測定を繰り返し、毎回同一温度で液化した場合を融点とし、試料に形状や色の変化が見られ、その温度前後で数回測定を繰り返し、毎回その温度が変化する場合を分解点とした。
[4] 溶解性の評価方法
試料0.01gを計り取り、ヘキサン、ベンゼン、トルエン、四塩化炭素、クロロホルム、テトラヒドロフラン(THF)、メタノール(MeOH)、エタノール(EtOH)及び水を1mL,5mL,10mL加えて沸点まで加熱し,溶液に残存する粉末の有無を目視によって確認した。サンプルが溶解したときの溶媒の量が1ml以下で++:Easily soluble、1〜5mlで+:soluble、5〜10mlで−:slightly soluble、10ml以上で−−:insolubleとし、分解したものはD:decompositionとした。加えた溶媒にすべての試料が溶解したと目視できるときに溶解とし、溶媒を加えても試料が溶解せずに残存していることを目視したときに不溶解とした。また、化学変化により試料が変質した場合を分解とした。
【0015】
〔実施例1〕 前駆物質Aの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに溶解し、モル比がSr:Bi:Ta=1:2:2の溶液を調製した。この溶液にトリエタノールアミン0.063gを加え60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで0.71gの前駆物質Aを得た。この前駆物質Aは茶色の粘性液体であり、留去した溶媒をGC分析した結果、アセト酢酸エチルのピークが確認できた。
【0016】
〔実施例2〕前駆物質Bの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに溶解し、モル比がPb:Zr:Ti=2:1:1の溶液を調製した。この溶液にトリエタノールアミン0.125gを加え60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで0.780gの前駆物質Bを得た。留去した溶媒をGC分析した結果、アセト酢酸エチルのピークが確認できた。得られた前駆物質Bは茶色の粉末であり、濃縮段階で単一成分の析出が全く見られず、均質なものであった。ベンゼン、クロロホルム、テトラヒドロフラン、メタノール、エタノールに非常に高い溶解性を示し、また、TG−DTA測定の結果(図2)から、800℃でのセラミック収率は47.5%であり、得られた前駆物質Bは450℃程度で完全にセラミック化したことが示された。
【0017】
〔実施例3〕前駆物質Cの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに、溶解し、モル比がPb:Zr:Ti=2:1:1の溶液を調製した。この溶液にトリエタノールアミン0.250gを加え60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで1.01gの前駆物質Cを得た。留去した溶媒をGC分析した結果、アセト酢酸エチルのピークが確認できた。得られた前駆物質Cは、濃縮段階で単一成分の析出が全く見ず、均質なものであった。この前駆物質Cは茶色の粉末であり、クロロホルム、テトラヒドロフラン、メタノール、エタノールに高い溶解性を示した。また、TG−DTA測定の結果から、800℃でのセラミック収率は50.6%であり、得られた前駆物質は450℃程度で完全にセラミック化したことが示された。
【0018】
〔実施例4〕前駆物質Dの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに溶解し、モル比がPb:Zr:Ti=2:1:1の溶液を調製した。この溶液にトリエタノールアミン0.376gを加え60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで1.08gの前駆物質Dを得た。この前駆物質Dは粉末と茶色の液体の混合物であった。留去した溶媒をGC分析した結果、アセト酢酸エチルのピークが確認できた。
【0019】
〔実施例5〕前駆物質Eの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに溶解し、モル比がPb:Zr:Ti=2:1:1の溶液を調製した。この溶液にトリエタノールアミン0.501gを加え60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで1.13gの前駆物質Eを得た。この前駆物質Eは粉末と茶色の液体の混合物であった。留去した溶媒をGC分析した結果、アセト酢酸エチルのピークが確認できた。
【0020】
〔比較例1〕前駆物質Fの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに溶解し、モル比がPb:Zr:Ti=2:1:1の溶液を調製した。この溶液を60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで1.05gの前駆物質を得た。前駆物質は白色粉末が分散したの黄色の液体であった。留去した溶媒をGC分析した結果、アセト酢酸エチルのピークは確認できなかった。
【0021】
〔実施例6〕 原料、前駆物質の融点、沸点または分解点の測定
ストロンチウムのアセト酢酸エチル錯体及びタンタルのアセト酢酸エチル錯体の沸点、ビスマスのアセト酢酸エチル錯体、前駆物質B及びCの分解点を測定した。結果を表1に示す。
【0022】
【表1】

【0023】
前駆物質A、B、C、D及びEは、反応後に留去した溶媒のGC分析の結果から、前駆物質の原料の配位子であるアセト酢酸エチルのピークが確認できた。一方、トリエタノールアミンを添加していない前駆物質Fでは、反応後に留去した溶媒のGC分析でのアセト酢酸エチルのピークが確認できなかった。また、前駆物質B及びCの分解点が110.1℃〜及び122.8℃〜であること、ストロンチウムのアセト酢酸エチル錯体及びタンタルのアセト酢酸エチル錯体の沸点がいずれも150℃以上であること及びビスマスのアセト酢酸エチル錯体の分解点は前駆物質B及びCの分解点とは異なることから、前駆物質B及びCは原料の混合物ではない。
これらのことから、本発明の製造法によって、前駆物質の原料が高分子量化し、そのため、得られた前駆物質は高分子量体であることが示された。
【0024】
〔実施例7〕 前駆物質の溶解性の評価
実施例B及びCで得られた前駆物質B及びCについて、各溶媒について溶解性の評価を行ったところ、以下の結果が得られ、クロロホルム、テトラヒドロフラン、メタノール、エタノールに非常に高い溶解性を示した。結果を表2に示す。前駆物質B及びCは水を加えると加水分解により白色固体が生成した。
【0025】
【表2】
【産業上の利用可能性】
【0026】
本発明によって得られたSr,Bi,Ta系酸化物の前駆物質を適当な条件で焼成することにより、Sr,Bi,Ta系酸化物強誘電体を得ることができ、強誘電体キャパシタや強誘電メモリなどのデバイスに好適に用いることができる。
【図面の簡単な説明】
【0027】
【図1】本発明で得られたと推定される高分子量化した強誘電前駆物質の構造。
【図2】〔実施例2〕で得られた生成物のTG−DTA測定結果である。
【技術分野】
【0001】
本発明は、ストロンチム、ビスマス及びタンタルからなるSr,Bi,Ta系酸化物の製造方法に関する。さらに詳しくは、Sr,Bi,Ta系酸化物を形成する際の原料となる前駆物質の調製、Sr,Bi,Ta系酸化物の形成方法等に関する。
【背景技術】
【0002】
結晶性のタンタル酸ストロンチウムビスマス(以下SBTと称す。)薄膜は高い誘電率、優れた強誘電特性から、種々のキャパシタや不揮発性メモリなどのデバイスへの応用が期待されている。
現在、実用化に至っている強誘電体である鉛系金属酸化物は、鉛が環境問題に対応できず、今後使用ができなく可能性があり、鉛系金属酸化物に代わる非鉛系金属酸化物強誘電体の開発が待たれている。そこで、非鉛系金属酸化物強誘電体として最も有望であるもののひとつがSBTである。
ストロンチム、ビスマス及びタンタルからなるSr,Bi,Ta系酸化物を700℃から1000℃で焼成することで、強誘電特性を示すタンタル酸ストロンチウムビスマスが得られることが分かっている(非特許文献1または非特許文献2参照)。
代表的なSBT薄膜の作製方法として、スパッタリング法、気相蒸着法、レーザーアブレーション法などがあるが、特に液相法には、金属有機化合物堆積法(Metal Organic Deposition:MOD)法やゾル・ゲル法などがある。
MOD法では、原料として有機金属化合物である、酢酸塩、クエン酸塩、シュウ酸塩、カルボン酸塩、金属アルコキシド、キレート試薬などを用いて簡潔なプロセスで前駆体溶液が調製されている。前駆体溶液は、混合物なので分子単体で存在すること、安定化することによってそれぞれの分子間の反応が阻害されていることなどから、前駆体溶液内の均質性、縮合安定性に欠けることが懸念される。
【0003】
ゾル・ゲル法では一般的に、金属アルコキシド化合物の加水分解・縮重合を経て、前駆体溶液が調製される。金属アルコキシド化合物は反応性が高いため、大気中の水分で容易に加水分解と縮重合が進むため、取扱いに困難が伴う。また、加水分解・縮重合を経た前駆体溶液も、大気中の水分で、さらに加水分解と縮重合が進み、保存安定性がしばしば問題となり、その後の成膜時に不良を生じる原因となる。そのため、工業的な応用を図るために、β−ジケトン類、β−ケトエステル類、カルボン酸類、エタノールアミン類、ジオール類、エステル類を前駆体の安定化剤として各々のPb系金属酸化物モル数に対して0.2倍〜5倍モル添加することで高い安定性を有する前駆体が調製されている。例えば、βージケトン、βーケトエステル、オキシ酸、ジオール、トリオール、高級カルボン酸、アルカノールアミン及び多価アミンよりなる群から選ばれた1種又は2種以上の安定化剤を組成物中の金属合計量1モルに対して、0.2〜3モルの割合で含有することを特徴としたSBT系強誘電体薄膜形成用組成物(特許文献1参照)などの発明が知られている。しかし、安定化剤を使用しても、依然として前駆体溶液の安定性の更なる改善が望まれている。
【特許文献1】特開2003−2650号公報
【非特許文献1】「ソリッドステイトマテリアルズ フォー アドバンス テクノロジー(solid−state materials for advanced technology)」,B97号,2003年,p.275−282
【非特許文献2】「ジャーナル オブ ヨーロピアン セラミックス ソサイエティー(Journal of the European Ceramic Society)」,24号,2004年,p.3043−3048
【発明の開示】
【発明が解決しようとする課題】
【0004】
そこで、本発明の目的は、Sr,Bi,Ta系酸化物を構成するストロンチム、ビスマス、タンタル、それぞれの金属を含み、かつ高い均質性、有機溶媒に対する高い溶解性、縮合安定性及び保存安定性に優れた高分子量体として、鉛系金属酸化物の前駆物質、及びその製造法を提供することである。
【課題を解決するための手段】
【0005】
本発明者は、以上の課題を解決するために鋭意検討した結果、本発明を完成するに至った。即ち、本発明は、
1.Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られることを特徴とするSr,Bi,Ta系酸化物前駆物質。
2.アルカノールアミンがジエタノールアミンまたはトリエタノールアミンである上記1記載のSr,Bi,Ta系酸化物前駆物質。
3.Sr、Bi及びTaの錯体化合物がアセト酢酸エチル錯体である上記1又は2に記載のSr,Bi,Ta系酸化物前駆物質。
4.Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させることを特徴とするSr,Bi,Ta系酸化物前駆物質の製造法。
5.アルカノールアミンがジエタノールアミンまたはトリエタノールアミンである上記4記載のSr,Bi,Ta系酸化物前駆物質の製造法。
6.Sr、Bi及びTaの錯体化合物がアセト酢酸エチル錯体である上記4又は5に記載のSr,Bi,Ta系酸化物前駆物質の製造法。
7.上記1〜3のいずれかに記載のSr,Bi,Ta系酸化物前駆物質を含有するSr,Bi,Ta系酸化物前駆体。
8.上記7に記載のSr,Bi,Ta系酸化物前駆体を用いて得られるSr,Bi,Ta系酸化物。
9.Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られるSr,Bi,Ta系酸化物前駆物質を用いることを特徴とするSr,Bi,Ta系酸化物前駆体の製造法。
10.Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られるSr,Bi,Ta系酸化物前駆物質を含有するSr,Bi,Ta系酸化物前駆体を用いるSr,Bi,Ta系酸化物膜の製造方法。
に関する。
【発明の効果】
【0006】
本発明のSr,Bi,Ta系酸化物前駆物質の製造方法では、Sr,Bi,Ta系酸化物を構成するそれぞれの金属を含み、かつ高い均質性、有機溶媒に対する高い溶解性、縮合安定性及び保存安定性に優れた高分子量体として、Sr,Bi,Ta系酸化物前駆物質を製造することができる。そして、得られた前駆物質を用いて調製した前駆体溶液を塗布液とした薄膜の製造により、Sr,Bi,Ta系酸化物を作製することができる。
【発明を実施するための最良の形態】
【0007】
本発明について詳細に述べる。本発明のSr,Bi,Ta系酸化物前駆物質は、ストロンチム、ビスマス及びタンタルの錯体化合物とアルカノールアミン(トリエタノールアミンまたはジエタノールアミンなどとを有機溶媒中で反応させた後、濃縮により溶媒および反応により生成した錯体化合物の配位子を留去して得られ、高い均質性、有機溶媒に対する高い溶解性、縮合安定性及び保存安定性に優れる高分子量体のSr,Bi,Ta系酸化物前駆物質である。また、この前駆物質は高粘度の液体または有機溶媒可溶性の粉末として得られ、これらを用いて前駆体溶液を調製し、前駆物質、前駆体溶液及び前駆体溶液を塗布液として製造した薄膜を減圧下熱分解ですることで高均質Sr,Bi,Ta系酸化物を製造することができる。
本発明はストロンチム、ビスマス及びタンタルの錯体化合物、特にアセト酢酸エチル錯体の性質に着目してなされたものである。即ち、これらの錯体は、それぞれの金属アルコキシドやカルボン酸塩に比べ、分子会合性が低いため一般の有機溶媒に対する溶解性が極めて高く取扱いが容易である。
一方、ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンとの反応については明確なことは不明であるが、これらの錯体分子間でトリエタノールアミンまたはジエタノールアミンの水酸基を介した架橋反応が容易に起こり、これらの錯体化合物の高分子量化が起こっていると推定される(図1)。
【0008】
従って、ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンとを有機溶媒中で反応させることにより、錯体化合物の高分子量化に伴うと推定される高粘度化または乾固が起こり、有機溶媒に対して良好な溶解性を示す、高粘性前駆物質液体または有機溶媒可溶性前駆物質粉末が得られる。さらに、上記前駆物質を用いて調製した前駆体より、Sr,Bi,Ta系酸化物薄膜、高純度Sr,Bi,Ta系酸化物粉末が容易に得られる。
ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンの反応においては、錯体化合物の配位子が外れることによって、錯体分子間でトリエタノールアミンまたはジエタノールアミンの水酸基を介した架橋反応が進行する。そのため、反応後の溶液に配位子の存在を確認することによって、高分子量化が進んでいることが確かめられる。
【0009】
本発明の前駆物質の製造に用いられるストロンチム、ビスマス及びタンタルの錯体化合物は、β−ジケトン類、β−ケトエステル類、カルボン酸類、ジオール類(エチレングリコール、プロピレングリコール、1,2−ブタンジオール、1,3−ブタンジオール、1,4−ブタンジオール、2,3−ブタンジオール、ヘキシレングリコールなど)、エステル類などの錯体化合物であり、これらが1種類以上用いられる。
好ましくはアセト酢酸エチル錯体及びトリエタノールアミン錯体を用いることによって、有機溶媒に対する溶解性が極めて高く、また加水分解に対する安定性が高い、さらに製造時の取扱い及び貯蔵安定性が特に優れた金属酸化物の前駆体を製造することができる。
本発明のSr,Bi,Ta系酸化物前駆物質の製造方法は、[1]ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンの調製→[2]ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンとの反応→[3]生成した前駆物質溶液の濃縮・乾固よりなる。以下に詳細を示す。
【0010】
[1]ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンの調製
金属酸化物の前駆物質の原料であるストロンチム、ビスマス及びタンタルの錯体化合物はモル比でSr:Bi:Ta=0.6〜1.1:2.0〜2.6:1.0の組成よりなるように調製される。上記組成範囲の各錯体化合物を有機溶媒に混合溶解した後、トリエタノールアミンまたはジエタノールアミンを、Biの錯体化合物に対して0.5〜5.0倍モルの範囲で添加する。
添加するトリエタノールアミンまたはジエタノールアミンが0.5〜1.0未満の範囲では高分子量化に必要なトリエタノールアミンまたはジエタノールアミンの量が不十分なため、原料と高分子量体の進行した前駆物質の混合物として得られる。1.0〜2.0倍モルの範囲では、前駆物質は粉末として得られ、3.0〜5.0倍モルの範囲では前駆物質は固体と液体の混合物または液体として得られる。高分子量化に関わるトリエタノールアミンまたはジエタノールアミンの量は前駆物質が粉末で得られる範囲であり、この範囲ではトリエタノールアミンまたはジエタノールアミンが増えるにしたがって、高分子量化が進行する。これ以上の範囲では、トリエタノールアミンまたはジエタノールアミンが過剰に存在し、前駆物質がトリエタノールアミンまたはジエタノールアミンに一部溶解した固体と液体の混合物またはすべて溶解した液体となる。よって、5.0倍モルより大きくても濃縮に時間がかかり経済的でないため、より好ましくはトリエタノールアミンまたはジエタノールアミンは1.0〜2.0倍モルの範囲で添加される。
ストロンチム、ビスマス及びタンタルの錯体化合物は各金属を酸化物に換算して1〜10wt%含まれるように調製される。酸化物含量が1wt%以下の場合、濃縮するために多量の溶媒を留去しなければならず経済的でない。一方、10wt%を超えると鉛、ストロンチム、ビスマス及びタンタルの錯体化合物が溶解しづらくなり、且つ均一な反応を行わせることが困難になる。
【0011】
[2]ストロンチム、ビスマス及びタンタルの錯体化合物とトリエタノールアミンまたはジエタノールアミンとの反応
反応温度は通常60〜100℃であるが、用いる有機溶媒の還流温度が簡便である。また、反応時間は反応温度によって異なり、即ち低温では長時間を要し、高温では短時間で済む。
[3]生成した前駆物質溶液の濃縮・乾固
反応終了後、温度40〜80℃、圧力20〜60mmHgの減圧下で2〜3時間かけて反応に用いた有機溶媒及び反応で生成した配位子を留去し、酸化物濃度25〜30wt%となるまで濃縮することにより、粘度50mPa・s〜1000mPa・sの高粘性溶液が得られる。
さらに、得られた高粘性溶液を温度40〜80℃、圧力10mmHg以下の減圧下で高分子量化に生じる配位子の留去し、重量減少がなくなるまで濃縮、乾固することにより、有機溶媒可溶性前駆物質粉末、高粘性前駆物質液体またはそれらの混合物を得ることができる。減圧度10mmHg以上では、トリエタノールアミンまたはジエタノールアミンや高分子量化の反応中に生じたアセト酢酸エチルなどの錯体化合物の配位子の一部が充分に揮散しない。
反応終了後、溶液を10mmHg以下において40〜80℃で処理することにより、有機溶媒の留去による濃縮及び高分子量化に生じる配位子の留去による濃縮、乾固を同時に行い、有機溶媒可溶性前駆物質粉末、高粘性前駆物質液体またはそれらの混合物を得ることもできる。
【0012】
本発明方法に用いる有機溶媒としては、ストロンチム、ビスマス及びタンタルの錯体化合物を溶解する物が用いられる。一例を挙げるならば、アルコール類(メタノール、エタノール、1−プロパノール、2−プロパノール、1−ブタノール、2−ブタノール、ヘキサノールなど)、テトラヒドロフラン、ジオキサンなどのエーテル類、セロソルブ類(メチルセロソルブ、エチルセロソルブ、プロピルセロソルブ、ブチルセロソルブなど)、グリコール類(エチレングリコール、プロピレングリコール、1,2−ブタンジオール、1,3−ブタンジオール、1,4−ブタンジオール、2,3−ブタンジオール、ヘキシレングリコールなど)、アセトン、クロロホルム、四塩化炭素、ベンゼン、トルエンなどであり、これらの一種以上を用いることができる。
本発明のSr,Bi,Ta系酸化物前駆体は、このようにして得られた有機溶媒可溶性前駆物質粉末、高粘性前駆物質液体またはそれらの混合物を有機溶媒に溶解したものを用いる。用いる有機溶媒としては、前駆物質を溶解するものであれば特に限定されない。その具体例としては、メタノール、エタノール、テトラヒドロフラン、クロロホルムなどが挙げられる。
【0013】
このようにして得られるSr,Bi,Ta系酸化物前駆体は、通常用いられる塗布法、例えばディッピング法、スピンコート法、フレキソ印刷法などを用いて、ガラス、金属、セラミックスなどの基板に塗布することができる。優れた成膜性を得るためには、酸化物換算濃度として1〜20wt%とすることが好ましい。酸化物濃度が1%以下では粘度が低く成膜性が低く、一方20wt%以上では成形加工性が悪くなり、成膜が困難である。従って、あらかじめ成形された薄膜などを上記条件下で処理することにより、薄膜などの形状を保持したまま分解し、不融化することができる。また、有機溶媒可溶性前駆物質粉末、高粘性前駆物質液体またはそれらの混合物に同様の処理を行うことにより高純度のSr,Bi,Ta系酸化物を得ることができる。
本発明のSr,Bi,Ta系酸化物前駆体である有機溶媒可溶性前駆物質粉末、高粘性前駆物質液体またはそれらの混合物は、クロロホルム、テトラヒドロフラン、ジオキサン、アセトン、プロパノール、エタノール、メタノールなどの極性溶媒に対して優れた溶解性を示し、ベンゼン、四塩化炭素などの非極性溶媒には溶解性を示さない。従って、上記前駆物質を再溶解し、塗布液として用いる場合には、上記極性溶媒を用いる必要がある。ただし、塗工性の向上を目的として、前駆物質の溶解性を損なわない範囲で、他の非極性溶媒を一部加えることは差し支えない。
以下に実施例を挙げ、本発明を更に具体的に説明するが、本発明はこれらに限定されるものではない。
【実施例】
【0014】
実施例で用いた分析法は以下の通りである。
[1] 熱重量示差熱分析(TG−DTA)
TG−DTA装置:TG−DTA2020S((株)マック・サイエンス製)で測定した。測定条件:空気気流中、昇温速度:10℃/min。
[2] ガスクロマトグラフィー分析(GC)
シリカゲル(GLサイエンス社製SE−30)を充填したステンレス製カラム(2m×3mmφ)を装着した島津製作所製(株)GC−14B型(インジェクション温度:290℃、カラム温度:180℃、熱電対型検出器温度:290℃)を用い、キャリアーガスとしてヘリウムガスを流量5.5ml/minで流通させた。鉛、チタン及びジルコニウムの錯体化合物、鉛のトリエタノールアミンまたはジエタノールアミン錯体、トリエタノールアミンまたはジエタノールアミン、それらの反応後、濃縮・乾固で留去した溶媒の上記条件でGC分析を行い、アセト酢酸エチルのピークの有無を確認した。
[3] 融点、沸点、分解点の測定
密閉したガラス細管に試料を充填し、ヤナコ製Micro Meltingpoint Apparatus MP−S3型融点測定器を用いて測定した。同一試料について試料が液化した温度前後で数回測定を繰り返し、毎回同一温度で液化した場合を融点とし、試料に形状や色の変化が見られ、その温度前後で数回測定を繰り返し、毎回その温度が変化する場合を分解点とした。
[4] 溶解性の評価方法
試料0.01gを計り取り、ヘキサン、ベンゼン、トルエン、四塩化炭素、クロロホルム、テトラヒドロフラン(THF)、メタノール(MeOH)、エタノール(EtOH)及び水を1mL,5mL,10mL加えて沸点まで加熱し,溶液に残存する粉末の有無を目視によって確認した。サンプルが溶解したときの溶媒の量が1ml以下で++:Easily soluble、1〜5mlで+:soluble、5〜10mlで−:slightly soluble、10ml以上で−−:insolubleとし、分解したものはD:decompositionとした。加えた溶媒にすべての試料が溶解したと目視できるときに溶解とし、溶媒を加えても試料が溶解せずに残存していることを目視したときに不溶解とした。また、化学変化により試料が変質した場合を分解とした。
【0015】
〔実施例1〕 前駆物質Aの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに溶解し、モル比がSr:Bi:Ta=1:2:2の溶液を調製した。この溶液にトリエタノールアミン0.063gを加え60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで0.71gの前駆物質Aを得た。この前駆物質Aは茶色の粘性液体であり、留去した溶媒をGC分析した結果、アセト酢酸エチルのピークが確認できた。
【0016】
〔実施例2〕前駆物質Bの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに溶解し、モル比がPb:Zr:Ti=2:1:1の溶液を調製した。この溶液にトリエタノールアミン0.125gを加え60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで0.780gの前駆物質Bを得た。留去した溶媒をGC分析した結果、アセト酢酸エチルのピークが確認できた。得られた前駆物質Bは茶色の粉末であり、濃縮段階で単一成分の析出が全く見られず、均質なものであった。ベンゼン、クロロホルム、テトラヒドロフラン、メタノール、エタノールに非常に高い溶解性を示し、また、TG−DTA測定の結果(図2)から、800℃でのセラミック収率は47.5%であり、得られた前駆物質Bは450℃程度で完全にセラミック化したことが示された。
【0017】
〔実施例3〕前駆物質Cの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに、溶解し、モル比がPb:Zr:Ti=2:1:1の溶液を調製した。この溶液にトリエタノールアミン0.250gを加え60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで1.01gの前駆物質Cを得た。留去した溶媒をGC分析した結果、アセト酢酸エチルのピークが確認できた。得られた前駆物質Cは、濃縮段階で単一成分の析出が全く見ず、均質なものであった。この前駆物質Cは茶色の粉末であり、クロロホルム、テトラヒドロフラン、メタノール、エタノールに高い溶解性を示した。また、TG−DTA測定の結果から、800℃でのセラミック収率は50.6%であり、得られた前駆物質は450℃程度で完全にセラミック化したことが示された。
【0018】
〔実施例4〕前駆物質Dの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに溶解し、モル比がPb:Zr:Ti=2:1:1の溶液を調製した。この溶液にトリエタノールアミン0.376gを加え60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで1.08gの前駆物質Dを得た。この前駆物質Dは粉末と茶色の液体の混合物であった。留去した溶媒をGC分析した結果、アセト酢酸エチルのピークが確認できた。
【0019】
〔実施例5〕前駆物質Eの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに溶解し、モル比がPb:Zr:Ti=2:1:1の溶液を調製した。この溶液にトリエタノールアミン0.501gを加え60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで1.13gの前駆物質Eを得た。この前駆物質Eは粉末と茶色の液体の混合物であった。留去した溶媒をGC分析した結果、アセト酢酸エチルのピークが確認できた。
【0020】
〔比較例1〕前駆物質Fの調製
ビスアセト酢酸エチルストロンチウム0.145g、トリスアセト酢酸エチルビスマス0.500g、モノアセト酢酸エチルタンタルテトラエトキシド0.411gを100mLのエタノールに溶解し、モル比がPb:Zr:Ti=2:1:1の溶液を調製した。この溶液を60℃で1時間加熱後、温度40℃、圧力2mmHgで2時間濃縮することで1.05gの前駆物質を得た。前駆物質は白色粉末が分散したの黄色の液体であった。留去した溶媒をGC分析した結果、アセト酢酸エチルのピークは確認できなかった。
【0021】
〔実施例6〕 原料、前駆物質の融点、沸点または分解点の測定
ストロンチウムのアセト酢酸エチル錯体及びタンタルのアセト酢酸エチル錯体の沸点、ビスマスのアセト酢酸エチル錯体、前駆物質B及びCの分解点を測定した。結果を表1に示す。
【0022】
【表1】
【0023】
前駆物質A、B、C、D及びEは、反応後に留去した溶媒のGC分析の結果から、前駆物質の原料の配位子であるアセト酢酸エチルのピークが確認できた。一方、トリエタノールアミンを添加していない前駆物質Fでは、反応後に留去した溶媒のGC分析でのアセト酢酸エチルのピークが確認できなかった。また、前駆物質B及びCの分解点が110.1℃〜及び122.8℃〜であること、ストロンチウムのアセト酢酸エチル錯体及びタンタルのアセト酢酸エチル錯体の沸点がいずれも150℃以上であること及びビスマスのアセト酢酸エチル錯体の分解点は前駆物質B及びCの分解点とは異なることから、前駆物質B及びCは原料の混合物ではない。
これらのことから、本発明の製造法によって、前駆物質の原料が高分子量化し、そのため、得られた前駆物質は高分子量体であることが示された。
【0024】
〔実施例7〕 前駆物質の溶解性の評価
実施例B及びCで得られた前駆物質B及びCについて、各溶媒について溶解性の評価を行ったところ、以下の結果が得られ、クロロホルム、テトラヒドロフラン、メタノール、エタノールに非常に高い溶解性を示した。結果を表2に示す。前駆物質B及びCは水を加えると加水分解により白色固体が生成した。
【0025】
【表2】
【産業上の利用可能性】
【0026】
本発明によって得られたSr,Bi,Ta系酸化物の前駆物質を適当な条件で焼成することにより、Sr,Bi,Ta系酸化物強誘電体を得ることができ、強誘電体キャパシタや強誘電メモリなどのデバイスに好適に用いることができる。
【図面の簡単な説明】
【0027】
【図1】本発明で得られたと推定される高分子量化した強誘電前駆物質の構造。
【図2】〔実施例2〕で得られた生成物のTG−DTA測定結果である。
【特許請求の範囲】
【請求項1】
Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られることを特徴とするSr,Bi,Ta系酸化物前駆物質。
【請求項2】
アルカノールアミンがジエタノールアミンまたはトリエタノールアミンである請求項1記載のSr,Bi,Ta系酸化物前駆物質。
【請求項3】
Sr、Bi及びTaの錯体化合物がアセト酢酸エチル錯体である請求項1又は2に記載のSr,Bi,Ta系酸化物前駆物質。
【請求項4】
Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させることを特徴とするSr,Bi,Ta系酸化物前駆物質の製造法。
【請求項5】
アルカノールアミンがジエタノールアミンまたはトリエタノールアミンである請求項4記載のSr,Bi,Ta系酸化物前駆物質の製造法。
【請求項6】
Sr、Bi及びTaの錯体化合物がアセト酢酸エチル錯体である請求項4又は5に記載のSr,Bi,Ta系酸化物前駆物質の製造法。
【請求項7】
請求項1〜3のいずれかに記載のSr,Bi,Ta系酸化物前駆物質を含有するSr,Bi,Ta系酸化物前駆体。
【請求項8】
請求項7に記載のSr,Bi,Ta系酸化物前駆体を用いて得られるSr,Bi,Ta系酸化物。
【請求項9】
Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られるSr,Bi,Ta系酸化物前駆物質を用いることを特徴とするSr,Bi,Ta系酸化物前駆体の製造法。
【請求項10】
Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られるSr,Bi,Ta系酸化物前駆物質を含有するSr,Bi,Ta系酸化物前駆体を用いるSr,Bi,Ta系酸化物膜の製造方法。
【請求項1】
Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られることを特徴とするSr,Bi,Ta系酸化物前駆物質。
【請求項2】
アルカノールアミンがジエタノールアミンまたはトリエタノールアミンである請求項1記載のSr,Bi,Ta系酸化物前駆物質。
【請求項3】
Sr、Bi及びTaの錯体化合物がアセト酢酸エチル錯体である請求項1又は2に記載のSr,Bi,Ta系酸化物前駆物質。
【請求項4】
Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させることを特徴とするSr,Bi,Ta系酸化物前駆物質の製造法。
【請求項5】
アルカノールアミンがジエタノールアミンまたはトリエタノールアミンである請求項4記載のSr,Bi,Ta系酸化物前駆物質の製造法。
【請求項6】
Sr、Bi及びTaの錯体化合物がアセト酢酸エチル錯体である請求項4又は5に記載のSr,Bi,Ta系酸化物前駆物質の製造法。
【請求項7】
請求項1〜3のいずれかに記載のSr,Bi,Ta系酸化物前駆物質を含有するSr,Bi,Ta系酸化物前駆体。
【請求項8】
請求項7に記載のSr,Bi,Ta系酸化物前駆体を用いて得られるSr,Bi,Ta系酸化物。
【請求項9】
Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られるSr,Bi,Ta系酸化物前駆物質を用いることを特徴とするSr,Bi,Ta系酸化物前駆体の製造法。
【請求項10】
Sr、Bi及びTaの錯体化合物とアルカノールアミンを反応させて得られるSr,Bi,Ta系酸化物前駆物質を含有するSr,Bi,Ta系酸化物前駆体を用いるSr,Bi,Ta系酸化物膜の製造方法。
【図1】

【図2】

【図2】
【公開番号】特開2006−76843(P2006−76843A)
【公開日】平成18年3月23日(2006.3.23)
【国際特許分類】
【出願番号】特願2004−263682(P2004−263682)
【出願日】平成16年9月10日(2004.9.10)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 2004年3月1日 社団法人日本化学会発行の「化学と工業 2004年3月号 第57巻第3号」に発表
【出願人】(000003986)日産化学工業株式会社 (510)
【Fターム(参考)】
【公開日】平成18年3月23日(2006.3.23)
【国際特許分類】
【出願日】平成16年9月10日(2004.9.10)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 2004年3月1日 社団法人日本化学会発行の「化学と工業 2004年3月号 第57巻第3号」に発表
【出願人】(000003986)日産化学工業株式会社 (510)
【Fターム(参考)】
[ Back to top ]