
スネイル−p53間の結合を阻害する化合物及びそれを有効成分として含有する癌疾患治療剤
スネイル−p53間の結合を阻害する化合物及びそれを有効成分として含有する癌疾患治療剤を提供する。スネイル−p53間の結合阻害剤は、K−Ras突然変異細胞株でp53の発現を誘導することによって、膵膓癌、肺癌、胆管癌及び大膓癌のように診断や治療が容易ではないK−Ras突然変異性癌疾患を効果的に治療するか、予防することができる。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、スネイル−p53間の結合を阻害してp53の発現を誘導することによって、膵膓癌、肺癌、胆管癌及び大膓癌のようなK−Ras突然変異性癌疾患治療に有用に使われるスネイル−p53間の結合を阻害する化合物及びそれを有効成分として含有する癌疾患治療剤に関する。
【背景技術】
【0002】
抗癌剤及び診断技術の改善によって全体癌患者の5年生存率が50%増加した。しかし、いくつかの癌疾患、例えば、肺癌と膵膓癌は、相変らず10%以下の低い生存率を表わしている。したがって、このような患者の生存率を増加させるために、このような疾患の初期診断方法の開発が至急である。また、このような類型の癌は、K−Rasでよく変化を示している。特に、発癌性K−Rasは、5%以下の生存率を表わす膵膓癌で圧倒的に優勢な傾向を表わしている。
【0003】
発癌性Rasは、p53活性化を通じる老化及び連続した細胞死を誘導すると知られており、発癌性Ras媒介腫瘍形成は、p53欠乏条件下で起こると推定されており、特に、H−Ras誘導肝癌細胞種は、素早く活性化されたp53によって抑制された。
【0004】
現在使われている肺癌、膵膓癌などの治療剤は、薬物効果の持続時間が短い一方、多様な副作用が誘発されるので、このような疾患を効果的に治療するか、初期診断することができる薬物の開発が至急である。
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明者は、発癌性K−Rasがスネイルの誘導を通じてp53を抑制するということを明らかにし、p53及びスネイル間の相互作用を遮断することができる化合物を同定し、このような化合物が、K−Ras突然変異細胞株でp53発現を誘導することを発見して、本発明を完成した。
【0006】
これにより、本発明の目的は、スネイル−p53の結合を阻害する候補薬物を選別する段階を含むK−Ras突然変異性癌疾患治療剤のスクリーニング方法を提供することである。
【0007】
また、本発明の他の目的は、スネイル−p53間の結合を阻害する化合物及びそれを有効成分として含有する癌疾患治療剤を提供することである。
【0008】
また、本発明者は、p53の特定領域、例えば、DNA結合ドメインは、K−Ras突然変異細胞での透過性が増加するので、それを担体として用いてK−Ras突然変異細胞に特異的に薬物を伝達することができるという点を明らかにし、またスネイルの自動抗体の発現有無を測定してK−Ras突然変異性癌疾患、例えば、膵膓癌、肺癌、胆管癌、大膓癌などを初期に診断することができるという点を明らかにすることによって、本発明を完成した。
【0009】
これにより、本発明のまた他の目的は、p53のDNA結合ドメインのエンドサイトーシスを用いてK−Ras突然変異細胞に特異的に薬物を伝達することができる薬物伝達方法を提供することである。
【0010】
また、本発明のさらに他の目的は、スネイル自動抗体の発現有無を測定してK−Ras突然変異性癌疾患を診断することができる初期診断方法を提供することである。
【課題を解決するための手段】
【0011】
前記目的を果たすために、本発明は、p53を固定させたプレートにスネイル及び候補薬物を培養する段階と、ELISAリーダーでスネイル−p53の結合を阻害する候補薬物を選別する段階と、を含んでなることを特徴とするK−Ras突然変異性癌疾患治療剤のスクリーニング方法を提供する。
【0012】
また、本発明は、下記化学式1で表される化合物、またはその塩を提供する:
【化1】
前記化学式1で、
mは、0ないし10の整数、n及びpは、それぞれ0または1の整数であり、
Zは、−NH(CH2)qCH3、−OH、4−フェニルピペリジン基、4−フェニルピペラジン基、イソブチルアミノ基及びイソブチルオキシ基からなる群から選択され、
qは、0ないし9の整数である。
【0013】
より望ましくは、前記化合物は、2−ノニルアミノ−5、8−ジメトキシ−1、4−ナフトキノン;2−デシルアミノ−5、8−ジメトキシ−1、4−ナフトキノン;3−(5,8−ジメトキシ−1、4−ジオキソ−ナフタレン−2−イルチオ)プロパン酸;11−(5,8−ジメトキシ−1、4−ジオキソ−ナフタレン−2−イルチオ)ウンデカン酸;イソブチル−11−(5,8−ジメトキシ−1、4−ジオキソ−1、4−ジヒドロナフタレン−2−イルチオ)−ウンデカノアート;11−(5,8−ジメトキシ−1、4−ジオキソ−1、4−ジヒドロナフタレン−2−イルチオ)−N−イソブチルウンデカンアミド;及びイソブチル−11−(5,8−ジメトキシ−1、4−ジオキソ−1、4−ジヒドロナフタレン−2−イルスルフィニル)ウンデカノアートからなる群から選択された化合物、またはその塩であり得る。
【0014】
前記化学式1の化合物は、前記K−Ras突然変異性癌疾患治療剤のスクリーニング方法を利用すれば、スネイル−p53の結合を阻害する薬物を特異的に選別することができて、膵膓癌、肺癌及び大膓癌のように診断や治療が容易ではないK−Ras突然変異性癌疾患を効果的に治療するか、予防することができる。
【発明の効果】
【0015】
本発明によるK−Ras突然変異性癌疾患治療剤のスクリーニング方法を利用すれば、スネイル−p53間の結合を阻害する薬物を特異的に選別することができて、膵膓癌、肺癌、胆管癌及び大膓癌のように診断や治療が容易ではないK−Ras突然変異性癌疾患を効果的に治療するか、予防することができる。
【0016】
また、p53のDNA結合ドメインを担体として用いてK−Ras突然変異細胞に特異的に薬物を伝達することができて、K−Ras突然変異性癌疾患の治療に非常に有用であり、スネイルの自動抗体の発現有無を測定してK−Ras突然変異によって誘発される癌疾患を初期に診断することができて、診断が困難な膵膓癌などの早期診断が可能であって、癌患者の生存率を延長させるか、治療に効率的に対処することができる。
【図面の簡単な説明】
【0017】
【図1】スネイル媒介p53発現抑制に関する分子的機転の考察を示す図である。
【図2】スネイル媒介p53発現抑制に関する分子的機転の考察を示す図である。
【図3】スネイル媒介p53発現抑制に関する分子的機転の考察を示す図である。
【図4】発癌性K−Ras媒介p53抑制が化学的阻害剤によって遮断されないものを示す図である。
【図5】スネイルが発癌性K−Ras媒介p53抑制のために重要な媒介体であるものを示す図である。
【図6】スネイルとp53との間の直接的な相互作用を示す図である。
【図7】K−Ras誘導性スネイル安定化において、ATR活性の必要条件を示す図である。
【図8】スネイル及びp53間の直接的な結合を示す図である。
【図9】スネイル及びp53間の結合阻害剤の同定に関する図である。
【図10】スネイル及びp53間の結合遮断がK−Ras突然変異細胞でp53機能を誘導するものを示す図である。
【図11】スネイル及びp53間の結合遮断がK−Ras突然変異細胞でp53機能を誘導するものを示す図である。
【図12】スネイル媒介p53発現抑制に関する分子的機転の考察を示す図である。
【図13】p53の核から細胞質への移動機転に関する結果である。
【図14】p53が小嚢輸送を通じて分泌するものを示す図である。
【図15】p53がプロテアーゼ及びエンドサイトーシスによって除去される一方、スネイルは、これらに抵抗性を表わした結果である。
【図16】K−Ras突然変異細胞でのHis−p53の再吸収を示す図である。
【図17】癌組織でのp53及びスネイル分泌を示す図である。
【図18】肺癌血清で抗−スネイルAbを分析した図である。
【図19】発癌性K−Rasでp53及びスネイルの分泌態様を模式化した図である。
【図20】実施例1〜5で合成された化合物のp53及びp21の誘導能を確認するウェスタンブロット結果を示す図である。
【図21】Nutlin−3と前記化合物とのうちから5oと7aとを選択してスネイル−p53結合阻害効果を確認するGSTプルダウン分析結果を示す図である。
【図22】K−Ras突然変異癌細胞株とK−Ras野生型癌細胞株とに化合物5o、7a及びNutlin−3を処理して表われた細胞死滅率を示す図である。
【図23】p53突然変異細胞株に化合物5oとNutlin−3とを処理してp21の活性が誘導されるものを表わすウェスタンブロット結果を数値化して示す図である。
【図24】腹腔注射でA549細胞を非胸腺マウスに注入後、化合物5o処理によるマウスの生存率を示す図である。
【図25】腹腔注射後、発生した腫瘍の組職写真を示す図である。
【図26】A549細胞で化合物5o処理による総体的な解剖学的異常所見を観察した図である。
【発明を実施するための形態】
【0018】
一つの具体的様態として、前記化学式1の化合物は、塩の形態で存在することができる。前記塩は、塩酸または硫酸などのような無機酸、またはp−トルエンスルホン酸などのような有機酸の薬剤学的に許容可能な塩であり得る。
【0019】
前記化学式1の化合物は、下記反応式1ないし5のような方法で製造可能である。
【化2】
【0020】
【化3】
【0021】
【化4】
【0022】
【化5】
【0023】
【化6】
【0024】
反応式2の工程を具体的に説明すれば、次の通りである。
1,5−ジヒドロキシナフタレン(2)を出発物質として用いて、既に知られた3段階の反応を経て、1,4,5,8−テトラメトキシナフタレン(3)を合成し、再び脱メチル化して合成中間体である5,8−ジメトキシ−1,4−ナフトキノン(4)を合成した。詳しい合成方法は、合成スキームにある参考文献で見られるが、このとき使われた溶媒は、反応に悪影響を及ぼさない溶媒である水酸化ナトリウム、アセトニトリル、無水メタノール、N,N−ジメチルホルムアミド、クロロホルムなどを使った。初めのメチル化反応は、窒素ガス存在下で水酸化ナトリウムに溶かした1,5−ジヒドロキシナフタレンに硫酸ジメチルを1時間滴加して2時間反応させて得た。後でベンゼンで再結晶して1,5−ジメトキシナフタレンを得た。
【0025】
1,4,5,8−テトラメトキシナフタレン(3)を合成する時は、無水条件でナトリウムメトキシドとヨウ化銅とをジメチルホルムアミドとメタノールとを入れて30時間加熱還流して得た。反応に対する温度は、80℃以上にして還流させ続けた。中間体5,8−ジメトキシ−1,4−ナフトキノンを合成する時は、硝酸二アンモニウムセリウムを使ったが、室温で30分間滴加して30分間さらに反応させて得た。化合物5aないし5pは、中間体5,8−ジメトキシ−1,4−ナフトキノン(4)をメタノールに溶かして、得ようとするアミンやメルカプタンまたは末端にカルボキシル基やヒドロキシル基が付いたメルカプタンを入れて室温で4時間から一晩撹拌してTLCで反応進行状況を確認し、硫酸と重クロム酸ナトリウム水溶液とでウォークアップさせてシリカゲルカラムクロマトグラフィーで分離して得た。
【0026】
次の段階で、化合物5oの2番位置カルボキシル基に4−フェニルピペリジンまたは4−フェニルピペラジン誘導体を付ける時は、1,3−ジシクロヘキシルカルボジイミド(DCC)とN,N−ジメチルアミノピリジン(DMAP)よりは、N−(3−ジメチルアミノプロピル)−N´−エチルカルボジイミド塩酸塩(EDC)を入れて反応させることによって、化合物6aと6bとを合成することができ、前記反応は、収率も高くて分離過程にウレアがなしにきれいに分離することができる。次の段階で、化合物5pを用いてイソブチルアルコールとイソブチルアミンとを付ける時にも、同様にN−(3−ジメチルアミノプロピル)−N´−エチルカルボジイミド塩酸塩(EDC)を使って、得ようとした化合物7a、7bを容易に得られる。スルホキシド化合物8を合成する時には、化合物7aを用いてMCPBAを使い、TLCで反応状況を確認しながら合成することができる。最後に、重炭酸ナトリウムで反応を中止させてシリカゲルカラムをかけて精製された化合物7aが得られる。
【0027】
しかし、前記反応式2ないし5による方法は、化学式1の化合物の製造方法の一例に該当し、例えば、反応溶媒、塩基、反応物質の使用量のような反応条件が、前記から説明されたことのみで限定されるものではなく、前記反応式2ないし5の方法の以外にも、当業者に知られた公知の多様な合成方法を用いて、前記化学式1の化合物を製造することができる。
【0028】
また、本発明は、スネイル−p53間の結合を阻害する化合物を有効成分として含有する癌疾患治療剤を提供する。
【0029】
前記化合物は、前記化学式1で表される化合物、またはその塩であり、より望ましくは、2−ノニルアミノ−5,8−ジメトキシ−1,4−ナフトキノン;2−デシルアミノ−5,8−ジメトキシ−1,4−ナフトキノン;3−(5,8−ジメトキシ−1,4−ジオキソナフタレン−2−イルチオ)プロパン酸;11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)ウンデカン酸;イソブチル−11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロキシナフタレン−2−イルチオ)−ウンデカノアート;11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)−N−イソブチルウンデカンアミド;及びイソブチル−11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルスルフィニル)ウンデカノアートからなる群から選択された化合物、またはその塩であり得る。
【0030】
前記癌疾患は、K−Ras突然変異性癌疾患であり、望ましくは、膵膓癌、肺癌、胆管癌及び大膓癌からなる群から選択された癌疾患であり得る。
【0031】
本発明による癌疾患治療剤は、薬学組成物の製造に通常使う適切な担体、賦形剤または希釈剤をさらに含みうる。
【0032】
本発明で使用可能な担体、賦形剤または希釈剤としては、ラクトース、デキストロース、スクロース、ソルビトール、マンニトール、キシリトール、エリスリトール、マルチトール、澱粉、アカシアゴム、アルギン酸、ゼラチン、燐酸カルシウム、ケイ酸カルシウム、セルロース、メチルセルロース、微晶質セルロース、ポリビニルピロリドン、水、ヒドロキシ安息香酸メチル、ヒドロキシ安息香酸プロピル、ステアリン酸マグネシウムまたは鉱物類などが挙げられる。
【0033】
前記癌疾患治療剤は、それぞれ通常の方法によって、散剤、顆粒剤、錠剤、カプセル剤、懸濁液、エマルジョン、シロップ、エアゾールなどの経口型剤型、外用剤、座剤及び滅菌注射溶液の形態で剤型化して使われる。
【0034】
製剤化する場合には、普通使う充填剤、増量剤、結合剤、湿潤剤、崩解剤、界面活性剤などの希釈剤または賦形剤を使って調剤される。経口投与のための固型製剤には、錠剤、丸剤、散剤、顆粒剤、カプセル剤などが含まれ、このような固型製剤は、前記化合物は少なくとも一つ以上の賦形剤、例えば、澱粉、炭酸カルシウム(calcium carbonate)、スクロース(sucrose)またはラクトース(lactose)、ゼラチンなどを混ぜて調剤する。
【0035】
また、単純な賦形剤の以外に、ステアリン酸マグネシウム、タルクのような潤滑剤も使われる。経口のための液状製剤としては、懸濁剤、内用液剤、乳剤、シロップ剤などが該当するが、よく使われる単純希釈剤である水、リキッドパラフィンの以外に、さまざまな賦形剤、例えば、湿潤剤、甘味剤、芳香剤、保存剤などが含まれうる。
【0036】
非経口投与のための製剤には、滅菌された水溶液、非水性溶剤、懸濁剤、乳剤、凍結乾燥製剤、座剤が含まれる。非水性溶剤、懸濁剤としては、プロピレングリコール(propylene glycol)、ポリエチレングリコール、オリーブオイルのような植物性油、オレイン酸エチルのような注射可能なエステルなどが使われる。座剤の基剤としては、ハードファット(witepsol)、マクロゴ−ル、トウイーン(tween)61、カカオ脂、ラウリン脂、グリセロゼラチンなどが使われる。
【0037】
前記癌疾患治療剤の使用量は、患者の年齢、性別、体重によって変わりうるが、0.1ないし100mg/kgの量を一日1回ないし数回投与することができる。
【0038】
また、このような癌疾患治療剤の投与量は、投与経路、疾病の程度、性別、体重、年齢などによって増減される。したがって、前記投与量は、如何なる面でも、本発明の範囲を限定するものではない。
【0039】
前記癌疾患治療剤は、モルモット、マウス、家畜、人間などの哺乳動物に多様な経路で投与されることがある。投与のあらゆる方式は予想されるが、例えば、経口、直腸または静脈、筋肉、皮下、子宮内硬膜または脳血管内(intracerebroventricular)注射によって投与されることがある。
【0040】
また、本発明は、p53のDNA結合ドメインのエンドサイトーシス(endocytosis)を用いて伝達しようとする薬物をK−Ras突然変異細胞に特異的に伝達することを特徴とするK−Ras突然変異細胞の特異的薬物伝達方法を提供する。
【0041】
前記DNA結合ドメインは、人間p53(Genbank Accession No.P04637)の90番目から280番目までのアミノ酸配列を含む。
【0042】
望ましくは、前記薬物伝達方法は、p53のDNA結合ドメイン及び伝達しようとする薬物を共に細胞に処理する段階と、前記p53のDNA結合ドメインによるエンドサイトーシスによって隣合うK−Ras突然変異細胞に薬物が伝達される段階と、を含んでなりうる。
【0043】
また、本発明は、スネイル抗体の発現有無を測定することを特徴とするK−Ras突然変異性癌疾患の初期診断方法を提供する。
【0044】
前記スネイル抗体の発現は、K−Ras突然変異性癌疾患を有した患者の血清から測定することができ、K−Ras突然変異性癌疾患は、膵膓癌、肺癌、胆管癌及び大膓癌からなる群から選択された癌疾患であり得る。
【0045】
本発明の理解を助けるために、望ましい実施例を下記に提示する。しかし、このような実施例は、本発明をより容易に理解するために提供されるものであって、本発明が、下記の実施例に限定されるものではない。
[スネイル媒介p53抑制に関する分子的機転の考察]
<実施例1>
【0046】
1.マウス線維芽細胞の分離及び無限分裂
6ヶ月齢の雄マウスを犠牲させて肺線維芽細胞(fibroblast)を集めた。肺を分離した後、組職メスを用いて組職を切って溶出した。20% FBSを含有したDMEM培地で3日間培養した後、付着された細胞を培養皿に分周し、jetPEIを用いて製造者の指針によって突然変異H−Ras、N−Ras及びK−Rasで形質感染させた。72時間後、DMEM含有G418 400μg/mlを用いて形質感染された細胞を選別した。
2.細胞培養及び試薬準備
【0047】
本発明で使った細胞株は、ATCCから購入し、10% FBSを含有したRPMI−1640またはDMEMで維持された。また、使われた抗体は、Santa CruzまたはCell Signaling(p53−R、P−Erk)から購買した。Ras発現ベクター及びSnailベクターは、それぞれDr.Chi SG及びHung M−Cから提供された。p53 S46D及び46Aは、Mayo LDから提供された。また、本発明で使われた化学物質は、Calbiochemから購買し、組替えp53は、Assay designsから購買した。
【0048】
また、細胞分画分析は、Subcell fraction Kit(Merck)を用いて製造者の指針によって行った。培地分析のために、細胞培養された培地を集めてCentricon(Millipore)またはEtOH沈降を用いて濃縮した。
3.免疫染色及びウェスタンブロット
【0049】
細胞染色のために、先行培養された細胞を洗浄して100%メタノールで固定し、抗体(第1次抗体;1:200、4℃で一晩中、第2次抗体;1:1000、室温で2時間)で培養した。分泌されたp53及びSnailを測定するために、HCT116 p53−/−細胞を1mL RPMI 1640培地で24時間ベクターで形質感染させ、洗浄なしに2% PFA 1mLを加えて固定させた。固定後、細胞をPBSで2回洗浄してブロッキング緩衝液[PBS+抗人間Ab(1:500)]で培養して、非特異的結合を除去した。PBS洗浄後、細胞を抗−p53 Ab及び抗−Snail Abと培養して、第2次Abとマッチングした。タンパク質分析のために、RIPA緩衝液でタンパク質を抽出してSDS−PAGEに試料を適用し、一般的なウェスタンブロット(WB)プロトコルによって行った。
【0050】
免疫沈降分析は、一般的なプロトコルによって行われた。すなわち、細胞溶出物を4時間Abと先に培養し、タンパク質−A/G−アガロースで2時間培養した。遠心分離後、3回洗浄した。沈降された複合体を用いてSDS−PAGE/WB分析を行った。
【0051】
4.形質感染及びsi−RNA効果の検討
細胞形質感染のために、製造者のプロトコルによってjetPEIを利用した。細胞を完全培地で24時間DNA/jetPEI混合物で培養した。生体外遺伝子ノックアウトのために、Snail及びMDM2に対するsi−RNAを作った。jetPEIを用いてsi−RNAを形質感染させて24時間後、その効果を検討した。
5.実験結果
【0052】
図1Aに示したように、N−RasまたはH−Rasの形質感染がアポトーシスまたは老化を誘導したが、K−Rasで形質感染された細胞は、6ヶ月が経過されても引き続き成長して維持された。
【0053】
また、発癌性K−Rasの強制発現が、野生型p53−含有細胞株でp53発現を抑制し(図2A参照)、K−Rasは、N−RasまたはH−Rasと異なってp53抑制を引き起こし、これは、si−MDM2によって遮断されなかった(図2B参照)。しかし、野生型K−Rasは、p53発現を抑制しなかった(図2C及び図2D参照)。
【0054】
図1Bに示したように、このようなp53は、K−Ras濃度依存的に抑制された。DN−RaseによるRas活性の遮断が、K−Ras突然変異A549でp53発現を増加させたが、HepG2では増加させなかった(図2E)。
【0055】
前記結果を総合すれば、内因性発癌性K−Rasが、p53発現を抑制するということを示唆する。しかし、図1Cのように、DN−RaseがDNA損傷−媒介p53抑制に関して明確であり、相加的な効果を観察することができず、これは、強い遺伝毒性ストレスが発癌性K−Ras媒介p53抑制を克服できるということを示唆する。
【0056】
図2F及び図2Gに示したように、K−Ras媒介p53抑制が点突然変異p53で確認されたが、p53活性型であるp53 S46Dは、K−Ras媒介p53抑制に抵抗性を表わした。このような結果は、遺伝毒性誘導性p53活性化がK−Ras媒介抑制を克服できるという図1Cの結果とも一致する。
【0057】
22/23突然変異がMDM2と関連しないために、K−Ras媒介p53抑制は、MDM2と独立した経路で行なわれるということを確認することができ、プロテアソーム−阻害剤がK−Ras媒介p53を遮断することができなかったために、MDM2またはp53ユビキチンシステムと関係のないということが分った(図1A及び図2H参照)。
【0058】
また、K−Ras媒介p53抑制に対するMAPK信号阻害剤の効果を検討した結果、MAPK経路遮断が、K−Ras媒介p53抑制に関する効果を無くすことができなかった。このような結果からK−Ras媒介p53抑制は、他の新規経路で行われると判断される。
【0059】
また、不溶性分画でスネイルによるp53の分布を検討した結果、図12Aのように、p53は、スネイルまたは発癌性K−Rasによって減少し、細胞を4つの分画、すなわち、核、細胞質、膜/器官及び不溶性分画に分けて検討した結果、図12Bのように、スネイルによって減少したp53は、いかなる細胞分画でも回復されなかった。
【0060】
また、K−Ras突然変異細胞でスネイルの除去がsi−MDM2より圧倒的にp53を誘導した(図12C)。そして、図12D及び図12Eのように、スネイルによるp53の減少は、MDM2−媒介抑制とは異なってプロテアソーム阻害剤によって回復されなかった。
<実施例2>
1.ウェスタンブロット及び生体外のキナーゼまたはバインディング分析
【0061】
Snail及びp53間の直接結合を検討するために、典型的なSDS−PAGE及びゲル移動方式を通じて組替えp53またはSnailまたはp53形質感染された細胞溶出物でローディングされたメンブレンを準備した。5%非脂肪乾燥油でブロッキングした後、メンブレンをp53またはSnail形質感染されたp53−/−HCT116細胞溶出物で4℃で4時間培養した。洗浄後、メンブレンをp53 AbまたはSnail Abを用いて典型的なWB手続きを進行した。
【0062】
インビトロ結合のために、再結合p53及びGST−Snailを交互に4℃で1時間培養し、p53 AbまたはGST Abを有したIP及びGSTまたはp53 Abを有したWBを行った。Snailの変形を調査するために、293細胞を形質感染のために使った。フラクションまたはリシス後、溶出物をGSTまたはGST−Snailで25℃で1時間培養し、SDS−PAGE及びWB分析を行った。p−MAPK及びp−ATM/ATR基質に対する抗体をcell signalingから購入した。
2.実験結果
【0063】
図3Aに示したように、スネイル発現に対するK−Rasの効果を検討した結果、スネイルがK−Rasによって誘導されうるということを観察した。また、細胞株でp53発現に対するスネイルの効果を検討した結果、図3Bのように、スネイルの過発現がA549及びHepG2細胞株でp53を抑制することができる一方、スネイルノックダウンがA549(発癌性K−Ras含有細胞株)でのみp53を誘導するだけで、HepG2では誘導することができなかった。
【0064】
また、si−スネイルがDNA損傷薬剤に対して敏感性を増加させ(図4F)、したがって、スネイルの過発現が細胞増殖を増進させてDNA損傷誘導性細胞死に抵抗性を表わした(図4G)。
【0065】
図3Cのように、スネイルは、K−Rasと同様にendo−p53だけではなく、exo−p53を抑制し、図3Dないし図3Fのように、スネイルとp53とを共に形質感染させた時、突然変異p53と関係なくスネイル及びp53いずれも減少した。しかし、スネイル及びp53のmRNAは減少しなかった(図3C及び図3D)。また、p53転写物に対するスネイルの効果を検討した結果、スネイルは、p53 mRNAを減少させなかった(図4H)。
【0066】
したがって、p53及びスネイルは、たとえよく知られた転写調節剤であるとしても、これらの減少は、転写調節と無関係であるいうことを示唆する。また、図3G及び図3Hのように、スネイルの除去がK−Ras媒介p53抑制を遮断することができる。また、図5A及び図5Bのように、exo−p53でも類似した結果を得た。このような結果からK−Ras媒介p53抑制は、スネイル誘導を通じて得られるということが分かる。
【0067】
また、図5Cのように、発癌性K−Rasは、4時間以内にp53を誘導する一方、p53は6時間後に減少し、これは、p53抑制が形質感染の人為的結果ではなく、形質感染されたタンパク質の効果であるということを表わす。
【0068】
また、図5Eのように、K−Ras/スネイル形質感染された細胞でアポトーシス及び細胞周期を検討し、アポトーシス及び細胞周期阻害は、K−Ras/スネイルによって明らかに誘導されなかった。
【0069】
また、Aphidicolin処理細胞でK−Ras/スネイルによってp53の減少が観察されて、p53の減少は細胞周期とは関連しないものと判断される。
【0070】
また、CHX−pulse chaseを通じてp53の半減期を対するスネイルの効果を検討し、図5Fのように、スネイルは、p53半減期を短縮させなかった。そして、p53 S46Dの発現に対するスネイルの効果を検討し、図6Aのように、スネイルがK−Rasと異なってp53 S46D発現を抑制した。
【0071】
また、スネイル発現に対するp53 S46Dの効果を検討した結果、図6Bのように、S46Dが転写及び翻訳レベルでスネイル発現を抑制した。したがって、si−スネイルがp53抑制を回復させることができる野生型p53とは異なって、図6Cのように、S46Dで形質感染される場合には、si−スネイルがp53発現を誘導することができなかった。
【0072】
このような結果から或るストレス条件下でセリン46残基で変形によって活性化されたp53は、スネイル転写物の抑制を通じてK−Ras媒介抑制機転を克服することができる。
【0073】
また、スネイル誘導がATRを通じてなされた。
K−Rasが、スネイルを如何に誘導するかを検証するために、AKTの関連を検討した。Rasは、AKTを活性化してGSK−3β媒介スネイル不安定性を抑制できると知られている。
【0074】
しかし、図7Aのように、AKT−KDは、スネイルまたはK−Ras誘導p53抑制を遮断することができなかった。一方、図7Bのように、si−RNAを通じるATRの抑制がp53抑制を遮断した。また、図7C及び図7Dのように、スネイルは、ATR処理によって増加するが、ATM及びノコダゾール処理によっては増加しなかった。図6Dのように、インビトロキナーゼ分析でスネイルは、ATRによってリン酸化された。ATRを活性化させるものと知られたK−Rasは、図7Eのように、p−スネイルをATR−依存性方式と同様に増加させ、図7Fのように、スネイルの半減期を延長させた。
<実施例3>
1.再組合タンパク質及びGST−pull down分析
【0075】
3つの人間Snail断片(残基1−90、91−112及び113−264)及びp53断片(1−93及び93−292)をGST−融合タンパク質として大膓菌で発現した。各断片をGSH−アガロースにローディングし、洗浄した後、20mM還元グルタチオンを含有した緩衝液を用いて溶出した。溶出分画を陰イオン−交換クロマトグラフィー(HitrapQ)を用いて錠剤した。再組合人間p53タンパク質(残基94−292)をベクターpET28Aを用いて大膓菌で発現させた。前記ベクターpET28Aは、C末端にヘキサ−ヒスチジンタグを含有した。
【0076】
Ni−NTA親和力及び大きさ排除クロマトグラフィー(Superdex200)を用いてp53タンパク質を錠剤した。p53及びSnail間の直接結合を検討するために、アガロース−ビーズ接合GSTまたはGST−Snailを4℃で45分間RIPAで細胞溶出物、またはHis−p53と培養した。PBS及びRIPAで洗浄した後、沈澱されたタンパク質をSDS−PAGE及びWBを行った。
【0077】
2.実験結果
スネイルは、核タンパク質であるために、スネイル及びp53を共に形質感染させた時、スネイル及びp53がいずれも消えた(図3Dないし図3F参照)。図8Aのように、endo−IPからこれらタンパク質が互いに関連するということが分かった。図8B及び図8Cのように、Far−western blot分析及びGST−pull down分析を通じてスネイル及びp53が互いに直接的に相互作用するというが分かった。また、p53のDNA結合ドメイン及びスネイルの中間領域が、結合ドメインとして役割を行っ
た(図8Dないし図8F、図6E、図6F参照)。
<実施例4>
1.化学的スクリーニングのためのELISAシステムの製造
【0078】
Snail−p53結合阻害剤を分離するために、ELISAシステムを製造した。0.5% PFAを用いて96ウェルプレートにHis−p53(93−292)を固定させた。乾燥して洗浄した後、0.1μM化学物質を有したGST−Snailで培養した。1時間培養後、96ウェルプレートをTBSTで洗浄して抗−GST−Ab(1:10000、45分)及び抗−マウス−IgG−HRP(1:50000、30分)で培養した。2回洗浄後、プレートをTMB溶液及び停止液で培養した。ELISAリーダーを用いて測定した。
2.実験結果
【0079】
図9A及び図9Bのように、p53及びスネイル間の結合抑制をELISAシステムで検討した。図9Cのように、150余個の化学物質からスネイル及びp53の結合阻害剤で3つの化学物質を同定した。図10A及び図11Aのように、このような化学物質は、濃度依存的にスネイル及びp53間の結合を抑制した。
【0080】
GST−pull down分析を通じて、このような化合物処理後、p53及びその標的の発現を検討し、3つの化学物質がいずれもp53及びスネイルの相互作用発現を抑制し、p53発現を誘導した(図8B及び図11B)。
【0081】
また、図10Cのように、このような化学物質処理によってPUMA及びp21の誘導を観察することができた。特に、p53の誘導がK−Ras突然変異細胞でのみ検出され、野生型K−Ras細胞では検出されなかった(図10D)。ケルセチン及びモリンの類似した構造が、本発明のスクリーニングシステムの信頼性を高めた(図10E)。
【0082】
また、スネイル媒介p53抑制に対する化学物質の効果を検討し、図10E及び図10Fのように、スネイルの共形質感染によるp53の減少が、#3及び#9による化学物質処理によって遮断された。このような結果からp53−スネイルの相互作用の遮断が、p53発現を回復することができる。
【0083】
また、トリパンブルー染色を用いて細胞増殖に対するこれら化学物質の効果を検討した。A549で、これら化学物質が細胞増殖を明らかに抑制した一方、MKN45(K−Ras野生型細胞)に対する抗増殖効果を示していない(図11C)。
【0084】
そして、フェルラ酸は、K−Ras突然変異細胞で細胞死を誘発した(図11D)。また、ケルセチン(#2)は、スネイル−p53相互作用抑制剤として確認された。
[p53のDNA結合ドメインのエンドサイトーシスを利用したK−Ras突然変異細胞の特異的に薬物伝達方法の考察]
<実施例5>
【0085】
1.p53の核から細胞質への移動機転の検討
GST−pull down分析のために、先ず人間スネイル及びp53再組合タンパク質を知られた方法によって製造した(Neoplasia 11:1−10,2009)。培地及び前細胞ライセートでp53及びスネイル間の直接的な結合を検討するために、アガロース−ビーズ接合GSTまたはGST−スネイルを細胞ライセートまたは培養培地と4℃で2時間培養した。PBS及びRIPAで洗浄後、沈降されたタンパク質を以前方法と同様にSDS−PAGE及びWBに適用した。
2.実験結果
【0086】
p53の核から細胞質への移動配列で変異を起こすp53 NESに対するスネイルの効果を検討した結果、スネイルは、p53 NES発現を減少させ(図13A)、レプトマイシンB(LMB;核から細胞質への移動遮断剤)は、スネイル媒介−p53減少を遮断することができなかった(図13B及び図13C)。スネイルによるp53減少がリアルタイムで確認された(図13D)。培養培地でp53の発現を検討した結果、2時間以内に培地でp53の発現を検出することができた(図13E)。そして、p53は、スネイルまたはK−Ras形質感染細胞で小嚢のような構造でサイトゾルから検出され、最終的に、細胞外の領域から検出された(図13F)。一方、スネイルは、p53と共にサイトゾルの小嚢に存在した(図17A)。
【0087】
K−Ras突然変異細胞でp53の分泌を検討するために、培養培地及び細胞ライセートでスネイル−GSTを利用したGST−Pull down分析を行った。スネイル関連p53が、あらゆる細胞ライセートから検出されても、K−Ras突然変異膵膓癌細胞株でのみ培地p53を観察することができた(図13G)。また、K−Ras突然変異細胞の培養培地でスネイル−pull downなしに培地p53を観察することができた(図13H)。
【0088】
p53は、小嚢のような輸送を通じて減少するために、ノコダゾール(nocodazole;Noc)による細胞骨格ネットワークを妨害してp53の発現を検討した結果、Noc処理は、スネイルまたはK−Ras媒介p53減少を遮断することができた(図14A)。Noc−処理細胞でp53のサイトゾル小嚢性染色にもかかわらず、細胞形象は対照群と区別されるように変化された(図14B)。Nocは、p53減少を遮断することができた(図14C)。Noc−処理によって培地p53が消えた一方、p53は小嚢として細胞質で蓄積された(図14D及び図14E)。Aph/Nocは、p53の減少を遮断することができ、これは、LMBによって得られることができなかった(図14F及び図14G)。
<実施例6>
1.細胞外p53及びスネイルの態様検討
【0089】
1)組職分析
正常及び腫瘍が一対を成す胆管癌及び肝組職を順天郷医薬センターから得た。組職をdeep freezerで素早く凍結し、該凍結された組職を切って0.5mgの組職を0.25mL血清フリー培地で37℃で30分間培養して組職液を遊離した。培養後、培養培地を集めて0.5mL 100% EtOHで沈降させた。該沈降された物質をRIPAを用いて溶解させ、以前方法と同様にSDS−PAGE及びWB分析を行った。また、同じ方法で同一培養培地を得てp53 Abを検出した。
【0090】
2)p53 ELISA分析
p53の調査のために、製造者の指針(Assay Design)によってELISAを行った。すなわち、0.2mL組職培養培地を各ウェルに加えて検出Abと培養した。洗浄緩衝液で洗浄した後、0.2mL基質ゾル及び0.05mL停止液を加えた。
【0091】
3)血液試料でスネイルAb検出
人間血液試料を順天郷大学(膵膓癌及び胆石患者)から得て、釜山大学の医薬センター(肺癌)から得た。正常血液試料を志願者または癌ではない患者から集めた。血清は遠心分離して集め、使用前まで−70℃で保管された。3μlの血清をGST−タンパク質と事前承認後、アガロース−接合GST−スネイル−Nと培養した。沈降されたGST−スネイル−Ab複合体は、RIPA及びSDS試料緩衝液で溶解し、以前方法と同様にSDS−PAGEに適用した。PVDF膜に移した後、タンパク質を抗−人間Ab及び抗−GST Abと培養した。
2.実験結果
【0092】
先ず、A549及びMKN45細胞に組替えp53を処理し、これらの位置を調査した。培地から回収された対照群タンパク質(His−lamin A)と比較して、His−p53は、A549の前細胞ライセートで発現された(図15A)。そして、His−p53は、MKN45細胞及びその培養培地で完全に除去された(図15A)。
【0093】
より具体的な検討のために、本発明では、新鮮な培地、A549−培養培地、PC3及びHCT116を用いて組替えp53を培養した。培地で回収されたHis−laminAと比較して、p53−Hisは、HCT116の前細胞ライセートから検出された。そして、p53はPC3だけではなく、A549培養培地で回収されなかった。このような結果からp53は、培養された細胞から分泌されるプロテアーゼによって消化され、K−Ras突然変異細胞によって再吸収されるということが分かる。
【0094】
また、プロテアーゼ阻害剤(PMSF)及びエンドサイトーシス阻害剤(Brefeldin A;BFA)をA549及びCapan−1に処理した。細胞内p53発現に対する未反応にもかかわらず、これら化合物は、培地p53の発現を増加させた(図15B)。BFAは、A549前細胞ライセートで組替えp53の局在を遮断した(図15C)。
【0095】
分泌されたスネイルの態様を確認するために、再組合スネイルで処理してp53の目的地を比較した。p53中間領域は、A549のWCLから回収され、これは、BFAによって抑制された。しかし、MKN45に処理された組替えp53は、培地及びWCLで消えた。このような結果から分泌されたp53は、セリンプロテアーゼだけではなく、MMPのような他種のプロテアーゼによって消化されると判断された。一方、スネイルは、培地から回収され、プロテアーゼ阻害剤なしに再組合スネイルを回収した。このような結果からスネイルは、プロテアーゼ媒介消化だけではなく、エンドサイトーシスに抵抗性があると判断された。
【0096】
p53−エンドサイトーシスに対するK−Rasの効果を検討した結果、組替えp53は、K−Ras形質感染細胞によって選択的に除去された。p53中間領域が、K−Ras突然変異細胞によって選択的に再び入ることができるならば、このような特徴は、K−Ras突然変異細胞に対する化合物の伝達のために非常に有用である。
【0097】
それを確認するために、PI(propidium iodine;Red dye、50ug/mL)及びHis−p53(2ug/mL)をA549及びMKN45に処理した結果、PIのみ処理した場合には、二つの細胞株で細胞として蓄積されなかったが、PI及びp53を共に処理すれば、A549の細胞内にPIの蓄積を観察することができた(図15E、図16Aないし図16B)。しかし、MKN45では、このような観察ができなかった。このような結果からp53は、K−Ras特異的薬物伝達システムとして有用であると判断された。
【0098】
スネイル媒介分泌されたp53によってp53の自動Abが生成されるか否かを検討するために、組職液でのp53及びスネイルの発現態様を調査した結果、胆管癌では、p53及びスネイルが検出されたが、肝癌細胞種及び非癌組職液では検出されなかった(図17A及び図17B)。図17Cのように、ELISAを用いて組職液でのp53の存在を確認した結果、抗−p53 Ab及び抗−スネイルAbは胆管癌では検出されたが、肝癌細胞種では検出されなかった(図17D及び図17E)。また、膵膓癌または胆管癌患者の血清でp53及びスネイルに対する自動Abを調査した結果、抗−p53 Abは、癌と関連性を示していない。このような結果からp53自動Abは、癌の状態と関連しないものと判断され、分泌されたp53は、迅速にプロテアーゼ及びエンドサイトーシスによって除去された(図15A)。
【0099】
【表1】
【0100】
一方、スネイルは、プロテアーゼ及びエンドサイトーシスに対する抵抗性を表わした(図15D)。したがって、血清でスネイル自動Abを測定した。スネイルAbの発現は、膵膓癌患者の血清及び胆石患者の血清から検出された(図15F)。一方、スネイルAbは、正常健康人では検出されなかった(図15G)。また、肺癌患者の血清でも、スネイルAbが観察された(図18)。したがって、スネイルAbの存在が癌診断マーカーとして非常に有用であると判断された。
【0101】
【表2】
【0102】
[スネイル−p53間の結合阻害剤の同定]
<実施例1>
1−1.2−メチルチオ−1,4−ナフトキノン(2−Methylthio−1,4−naphthoquinone、1a)の合成
【0103】
1口100mlのラウンドフラスコに、1,4−ナフトキノン(1,4−naphthoquinone)0.617mMをメタノール30mlに溶解させた後、ナトリウムチオメトキシド(sodium thiomethoxide)1.54mMを入れて一晩中撹拌させた。反応混液に飽和塩化ナトリウム溶液50mlを入れてクロロホルム50mlで3回抽出した後、有機層を無水芒硝で脱水させた後、濾過した。該濾過した濾液を減圧濃縮して得た残渣をメタノールで再結晶して黄色結晶の表題化合物を得た。
【0104】
収率:14.0%、融点:185−186℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.07(m、2H)、7.78−7.70(m、2H)、6.58(s、1H)、2.40(s、3H)、m/z 205.1(M+H)+。
1−2.2−エチルチオ−1,4−ナフトキノン(2−Ethylthio−1,4−naphthoquinone、1b)の合成
【0105】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、エチルメルカプタン(Ethylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0106】
収率:40.7%、融点:135−136℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.08(m、2H)、7.78−7.69(m、2H)、6.62(s、1H)、2.87(q、J=7.2Hz、2H)、1.44(t、J=7.2Hz、3H)、m/z 219.1(M+H)+。
1−3.2−プロピルチオ−1,4−ナフトキノン(2−Propylthylthio−1,4−naphthoquinone、1c)の合成
【0107】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、プロピルメルカプタン(Propylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0108】
収率:33.9%、融点:118−119℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.07(m、2H)、7.77−7.68(m、2H)、6.61(s、1H)、2.82(t、J=7.6Hz、2H)、1.86−1.77(m、2H)、1.11(t、J=7.2Hz、3H)、m/z 233.0(M+H)+。
1−4.2−ブチルチオ−1,4−ナフトキノン(2−Butylthio−1,4−naphthoquinone、1d)の合成
【0109】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、ブチルメルカプタン(Butylmercaptan)を使って黄色結晶2−ブチルチオ−1,4−ナフトキノン(1d)を得た。その収率及び物性は、下記の通りであった。
【0110】
収率:33.9%、融点:97−98℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.07(m、2H)、7.77−7.68(m、2H)、6.61(s、1H)、2.84(t、J=7.2Hz、2H)、1.80−1.72(m、2H)、1.57−1.48(m、2H)、0.98(t、J=7.6Hz、3H)、m/z 247.1(M+H)+。
1−5.2−ペンチルチオ−1,4−ナフトキノン(2−Pentylthio−1,4−naphthoquinone、1e)の合成
【0111】
前記合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、ペンチルメルカプタン(pentylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0112】
収率:15.3%、融点:111−112℃、1H−NMR(CDCl3、400MHz):δ 8.11−8.06(m、2H)、7.77−7.68(m、2H)、6.60(s、1H)、2.83(t、J=7.6Hz、2H)、2.17−1.74(m、2H)、1.51−1.33(m、4H)、0.93(t、J=7.2Hz、3H)、m/z 261.2(M+H)+。
1−6.2−ヘキシルチオ−1,4−ナフトキノン(2−Hexylthio−1,4−naphthoquinone、1e)の合成
【0113】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、ヘキシルメルカプタン(hexylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0114】
収率:15.0%、融点:101−102℃、1H−NMR(CDCl3、400MHz):δ 8.13−8.08(m、2H)、7.77−7.69(m、2H)、6.61(s、1H)、2.83(t、J=7.6Hz、2H)、1.81−1.73(m、2H)、1.56−1.46(m、2H)、1.35−1.31(m、4H)、0.91(t、J=6.8Hz、3H)、m/z 275.3(M+H)+。
1−7.2−ヘプチルチオ−1,4−ナフトキノン(2−Heptylthio−1,4−naphthoquinone、1g)の合成
【0115】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、ヘブチルメルカプタン(heptylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0116】
収率:46.4%、融点:114−115℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.07(m、2H)、7.77−7.68(m、2H)、6.61(s、1H)、2.83(t、J=14.8Hz、2H)、1.77(quint、J=7.6Hz、2H)、1.52−1.45(m、2H)、1.35−1.29(m、6H)、0.90(t、J=6.8Hz、3H)、m/z 289.2(M+H)+。
1−8.2−オクチルチオ−1,4−ナフトキノン(2−Octylthio−1,4−naphthoquinone、1h)の合成
【0117】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、オクチルメルカプタン(octylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0118】
収率:76.8%、融点:114−115℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.10(m、2H)、7.78−7.70(m、2H)、6.61(s、1H)、2.84(t、J=7.6Hz、2H)、1.77(quint、J=7.6Hz、2H)、1.49−1.47(m、2H)、1.35−1.29(m、8H)、0.89(t、J=6.8Hz、3H)、m/z 304.5(M+H)+。
1−9.2−ノニルチオ−1,4−ナフトキノン(2−Nonylthio−1,4−naphthoquinone、1i)の合成
【0119】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、ノニルメルカプタン(nonylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0120】
収率:87.4%、融点:105−106℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.07(m、2H)、7.77−7.68(m、2H)、6.61(s、1H)、2.83(t、J=7.6Hz、2H)、1.77(quint、J=7.6Hz、2H)、1.51−1.45(m、2H)、1.33−1.29(m、10H)、0.89(t、J=6.4Hz、3H)、m/z 317.5(M+H)+。
1−10.2−デシルチオ−1,4−ナフトキノン(2−Decylthio−1,4−naphthoquinone、1j)の合成
【0121】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、デシルメルカプタン(Decylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0122】
収率:87.4%、融点:101−102℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.08(m、2H)、7.77−7.68(m、2H)、6.61(s、1H)、2.83(t、J=7.2Hz、2H)、1.77(quint、J=7.6Hz、2H)、1.50−1.42(m、2H)、1.33−1.29(m、12H)、0.88(t、J=6.4Hz、3H)、m/z 331.1(M+H)+。
<実施例2>
2−1.2−メチルアミノ−5,8−ジメトキシ−1.4−ナフトキノン(2−Methylamino−5,8− dimethoxy−1,4 −naphthoquinone、5a)の合成
【0123】
1口100mlのラウンドフラスコに、前から製造された5,8−ジメトキシ−1,4−ナフトキノン(5,8−dimethoxy−1,4−naphthoquinone、4)を0.45mMをメタノール30mlに溶解させた後、メチルアミン(Methylamine)0.687mmolを入れて室温で3時間撹拌させた。反応混液に重クロム酸ナトリウム(Sodim dichromate)0.64mMと硫酸(sulfuric acid)0.18mMとを水に溶解させてゆっくり滴加した後、室温で3分間撹拌した。反応混液に飽和塩化ナトリウム溶液50mlを入れてクロロホルム50mlで3回抽出した後、有機層を合わせて無水芒硝で脱水させた後、濾過した。該濾過した濾液を減圧濃縮して得た残渣をシリカゲルカラム分離を通じてさび色の表題化合物を得た。表題化合物の収率及び物性は、下記の通りであった。
【0124】
収率:56.7%、融点:203−204℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6MHz)、7.19(d、J=9.2MHz、1H)、5.75(BR、1H)、5.60(s、1H)、3.96(s、3H)、3.94(s、3H)、2.87(d、J=5.2MHz、3H)、m/z 248(M+H)+。
2−2.2−エチルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Ethylamino−5,8−dimethoxy−1,4−naphthoquinone、5b)の合成
【0125】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、エチルアミン(ethylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5b)を製造した。その収率及び物性は、下記の通りであった。
【0126】
収率:23.6%、融点:172−173℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.2Hz、1H)、7.19(d、J=9.6Hz、1H)、5.63(BR、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.09(q、2H)、1.29(t、J=7.2Hz、3H)、m/z 262.1(M+H)+。
2−3.2−プロピルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Propylamino−5,8−dimethoxy−1,4−naphthoquinone、5c)の合成
【0127】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、プロピルアミン(propylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5c)を製造した。その収率及び物性は、下記の通りであった。
【0128】
収率:46.5%、融点:175−176℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.2Hz、1H)、7.19(d、J=9.2Hz、1H)、5.72(BR、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.09(q、2H)、1.68(J=6.8Hz、2H)、0.99(t、J=7.6Hz、3H)、m/z 276(M+H)+。
2−4.2−ブチルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Butylamino−5,8−dimethoxy−1,4−naphthoquinone、5d)の合成
【0129】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、ブチルアミン(butylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5d)を製造した。その収率及び物性は、下記の通りであった。
【0130】
収率:46.2%、融点:104−105℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.19(d、J=9.6Hz、1H)、5.70(BR、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.64(p、2H)、1.46−1.38(m、2H)、0.95(t、J=7.2Hz、3H)、m/z 290(M+H)+。
2−5.2−ペンチルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Pentylamino−5,8−dimethoxy−1,4−naphthoquinone、5e)の合成
【0131】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、ペンチルアミン(pentylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5e)を製造した。その収率及び物性は、下記の通りであった。
【0132】
収率:55.9%、融点:102−103℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.2MHz、1H)、7.19(d、J=9.2Hz、1H)、5.70(BR、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.29(t、J=7.2Hz、3H)、m/z 303.6(M+H)+。
2−6.2−ヘキシルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Hexylamino−5,8−dimethoxy−1,4−naphthoquinone、5f)の合成
【0133】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、ヘキシルアミン(Hexylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5f)を製造した。その収率及び物性は、下記の通りであった。
【0134】
収率:47.3%、融点:83−84℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.18(d、J=9.6Hz、1H)、5.69(BR、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.66−1.58(m、4H)、1.32−1.30(m、4H)、0.89(t、J=6.8Hz、3H)、m/z 318(M+H)+。
2−7.2−ヘブチルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Heptylamino−5,8− dimethoxy−1,4−naphthoquinone、5g)の合成
【0135】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、ヘブチルアミン(Heptylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5g)を製造した。その収率及び物性は、下記の通りであった。
【0136】
収率:41.8%、融点:74−75℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.33(d、J=9.6Hz、1H)、7.18(d、J=9.6Hz、1H)、5.69(BR、1H)、5.60(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.66−1.61(m、2H)、1.35−1.29(m、8H)、0.89(t、J=6.4Hz、3H)、m/z332(M+H)+。
2−8.2−オクチルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Octylamino−5,8−dimethoxy−1,4−naphthoquinone、5h)の合成
【0137】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、オクチルアミン(octylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5h)を製造した。その収率及び物性は、下記の通りであった。
【0138】
収率:45.1%、融点:81−82℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.18(d、J=9.6Hz、1H)、5.69(br、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.68−1.61(m、2H)、1.41−1.20(m、10H)、0.88(t、J=6.4Hz、3H)、m/z 346(M+H)+。
2−9.2−ノニルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Nonylamino−5,8−dimethoxy−1,4−naphthoquinone、5i)の合成
【0139】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、ノニルアミン(nonylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5i)を製造した。その収率及び物性は、下記の通りであった。
【0140】
収率:44.0%、融点:85−86℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.18(d、J=9.6Hz、1H)、5.69(br、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(m、2H)、1.66−1.61(m、2H)、1.41−1.20(m、12H)、0.88(t、J=6.4Hz、3H)、m/z 360(M+H)+。
2−10.2−デシルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Decylamino−5,8−dimethoxy−1,4−naphthoquinone、5j)の合成
【0141】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、デシルアミン(decylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5j)を製造した。その収率及び物性は、下記の通りであった。
【0142】
収率:17.6%、融点:86−87℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.2Hz、1H)、7.18(d、J=9.2Hz、1H)、5.69(br、1H)、5.60(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.66−1.61(m、2H)、1.40−1.20(m、14H)、0.88(t、J=6.4Hz、3H)、m/z 374(M+H)+。
2−11.2−(2−ヒドロキシエチルチオ)−5,8−ジメトキシ−1,4−ナフトキノン[2−(2−Hydroxyethylthio)−5,8−dimethoxy−1,4−naphthoquinone、5k]の合成
【0143】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、2−メルカプトエタノール(2−mercaptoethanol)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5k)を製造した。その収率及び物性は、下記の通りであった。
【0144】
収率:59.5%、融点:117−118℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.28(d、J=9.6Hz、1H)、6.61(s、1H)、3.96(s、6H)、3.93(t、J=6.4Hz、2H)、3.05(t、J=6.4Hz、2H)、m/z 316.9(M+Na)+。
2−12.2−(3−ヒドロキシプロピルチオ)−5,8−ジメトキシ−1,4−ナフトキノン[2−(3−Hydroxypropylthio)−5,8−dimethoxy−1,4−naphthoquinone、5l]の合成
【0145】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、3−メルカプト−1−プロパノール(3−mercaptopropanol)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5l)を製造した。その収率及び物性は、下記の通りであった。
【0146】
収率:69.9%、融点:125−126℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.6Hz、1H)、7.27(d、J=9.6Hz、1H)、6.51(s、1H)、3.96(s、6H)、3.81(t、J=6.4Hz、2H)、2.91(t、J=7.2Hz、2H)、1.99(m、2H)、m/z 331.1(M+Na)+。
2−13.2−(4−ヒドロキシブチルチオ)−5,8−ジメトキシ−1,4−ナフトキノン[2−(4−Hydroxybutylthio)−5,8−dimethoxy−1,4−naphthoquinone、5m]の合成
【0147】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、4−メルカプトブタノール(4−mercaptobutanol)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5m)を製造した。その収率及び物性は、下記の通りであった。
【0148】
収率:64.0%、融点:122−123℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.6Hz、1H)、7.29(d、J=10.0Hz、1H)、6.46(s、1H)、3.96(s、3H)、3.95(s、3H)、3.71(t、J=6.4Hz、2H)、1.87−1.81(m、2H)、1.78−1.50(m、2H)、m/z 345.1(M+Na)+。
2−14.2−(6−ヒドロキシヘキシルチオ)−5,8−ジメトキシ−1,4−ナフトキノン[2−(6−Hydroxyhexylthio)−5,8−dimethoxy−1,4−naphthoquinone、5n]の合成
【0149】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、6−メルカプトヘキサノール(6−mercaptohexanol)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5n)を製造した。その収率及び物性は、下記の通りであった。
【0150】
収率:38.2%、融点:87−88℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.6Hz、1H)、7.27(d、J=9.6Hz、1H)、6.45(s、1H)、3.96(s、6H)、3.66(t、J=6.4Hz、2H)、2.76(t、J=7.6Hz、2H)、1.78−1.12(m、2H)、1.61−1.25(m、6H)、m/z 372.9(M+Na)+。
2−15.3−(5,8−ジメトキシ−1,4−ジオキソナフタレン−2−イルチオ)プロパン酸[3−(5,8−dimethoxy−1,4−dioxo−naphthalen−2−ylthio)propanoic acid、5o]の合成
【0151】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、3−メルカプトプロピオン酸(3−mercaptopropionic acid)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5o)を製造した。その収率及び物性は、下記の通りであった。
【0152】
収率:80.6%、融点:208−209℃、1H−NMR(CDCl3、400MHz):δ 7.35(d、J=9.2Hz、1H)、7.28(d、J=13.6Hz、1H)、6.51(s、1H)、3.97(s、3H)、3.96(s、3H)、3.07(t、J=7.2Hz、2H)、2.81(t、J=7.2Hz、2H)、m/z 348.4(M+Na)+。
2−16.11−(5,8−ジメトキシ−1,4−ジオキソ−ナフタレン−2−イルチオ)ウンデカン酸[11−(5,8−dimethoxy−1,4−dioxo−naphthalen−2−ylthio)undecanoic acid、5p]の合成
【0153】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、11−メルカプトウンデカン酸(11−mercaptoundecanoic acid)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5p)を製造した。
【0154】
収率:77.9%、融点:146−147℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.6Hz、1H)、7.27(d、J=9.6Hz、1H)、6.46(s、1H)、3.96(s、3H)、3.95(s、3H)、3.34(t、J=7.2Hz、2H)、2.75(t、J=2.7Hz、2H)、2.41−2.32(m、5H)、2.06−2.00(m、2H)、1.76−1.56(m、5H)、1.48−1.39(m、2H)、0.97−0.88(m、2H)、m/z 435(M+H)+。
<実施例3>
3−1.5,8−ジメトキシ−2−[3−オキソ−3−(4−フェニルピペリジン−4−イル)プロピルチオ]−1,4−ナフトキノン[5,8−dimethoxy−2−(3−oxo−3−(4−phenylpiperazin−1−yl)propylthio)naphthalene−1,4−dione、6a]の合成
【0155】
1口100mlのラウンドフラスコに、前から製造された3−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)プロパン酸、0.163mMをクロロホルム40mlに溶解させた後、N−(3−ジメチルアミノプロピル)−N´−エチルカルボジイミド塩酸塩(EDC)0.26mMと4−フェニルピペリジン(4−phenylpiperidine)0.26mMとを入れて一晩撹拌させた。反応混液に1N塩酸溶液を入れた後、室温で3分間撹拌した。再び飽和塩化ナトリウム溶液50mlを入れてクロロホルム50mlで3回抽出した後、有機層を合わせて無水芒硝で脱水させた後、濾過した。該濾過した濾液を減圧濃縮して得た残渣をシリカゲルカラム分離を通じてさび色の表題化合物(6a)を得た。表題化合物の収率及び物性は、下記の通りであった。
【0156】
収率:73.5%、融点:94−95℃、1H−NMR(CDCl3、400MHz):δ 7.34−7.28(m、5H)、7.19(d、J=7.6Hz)、6.26(s、1H)、3.95(s、3H)、3.94(s、3H)、3.14(t、4H)、2.79−2.74(q、4H)、2.67(t、J=12Hz、1H)、1.89(t、J=12.4Hz、2H)、1.65(t、J=12.4Hz、2H)、m/z 466.3(M+H)+。
<実施例4>
4−1.イソブチル−11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)−ウンデカノアート[Isobutyl−11−(5,8−dimethoxy−1,4−dioxo−1,4−dihydronaphthalene−2−ylthio)undecanoate、7a]の合成
【0157】
1口100mlのラウンドフラスコに、前から製造された11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)ウンデカン酸(5p)1.15mMをクロロホルム60mlに溶解させた後、N−(3−ジメチルアミノプロピル)−N´−エチルカルボジイミド塩酸塩(EDC)1.38mMとイソブチルアルコール(isobutylalcohol)1.38mMとを入れて一晩撹拌させた。反応混液に1N塩酸溶液を入れた後、室温で3分間撹拌した。再び飽和塩化ナトリウム溶液50mlを入れてクロロホルム50mlで3回抽出した後、有機層を合わせて無水芒硝で脱水させた後、濾過した。該濾過した濾液を減圧濃縮して得た残渣をシリカゲルカラム分離を通じてさび色の表題化合物(7a)を得た。表題化合物の収率及び物性は、下記の通りであった。
【0158】
収率:52.4%、融点:58−59℃、1H−NMR(CDCl3、400MHz):δ 7.32(d、J=9.6Hz、1H)、7.26(d、J=9.6Hz、1H)、6.44(s、1H)、3.96(s、3H)、3.95(s、3H)、3.85(d、J=6.8Hz、2H)、2.75(t、J=7.6Hz、2H)、2.31(t、7.6Hz、2H)、1.96−1.89(m、1H)、1.72−1.68(m、4H)、1.29−1.25(m、12H)、0.94(s、3H)、0.92(s、3H)、m/z 491(M+H)+。
4−2.11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)−N−イソブチルウンデカンアミド[11−(5,8−dimethoxy−1,4−dioxo−1,4−dihydronaphthalene−2−ylthio)−N−isobutyl undecanamide、7b]の合成
【0159】
前記実施例4−1のラウンドフラスコにイソブチルアルコールの代りに、イソブチルアミンを使用する点を除いては、実施例4−1の反応と同じ工程を行って下記物性値を有する表題化合物(7b)を得た。収率及び物性は、下記の通りであった。
【0160】
収率:77.9%、融点:74−75℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.6Hz、1H)、7.27(d、J=9.2Hz)、6.44(s、1H)、5.55(s、1H)、3.96(s、3H)、3.95(s、3H)、3.10(t、J=6.6Hz、2H)、2.75(t、J=7.6Hz、2H)、2.17(t、J=7.4Hz、2H)、1.78−1.7(m、2H)、1.63(m、2H)、1.45(m、1H)、0.92(s、3H)、0.90(s、3H)、m/z 490.0(M+H)+。
<実施例5>
5−1.イソブチル11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルスルフィニル)ウンデカノアート[isobutyl11−(5,8−dimethoxyl−1,4−dioxo−1,4−dihydronaphthalene−2−yl sulfinyl)undecanoate、8]の合成
【0161】
1口100mlのラウンドフラスコに、前から製造されたイソブチル11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロキシナフタレン−2−イルチオ)−ウンデカノアート0.163mM(7a)をジクロロメタン30mlに溶解させた後、3−クロロ過安息香酸(3−chloroperoxybbenzoic acid)0.196mMを入れて2時間撹拌させた。反応混液に重炭酸ナトリウム溶液を入れた後、室温で3分間撹拌した。再び飽和塩化ナトリウム溶液50mlを入れてクロロホルム50mlで3回抽出した後、有機層を合わせて無水芒硝で脱水させた後、濾過した。該濾過した濾液を減圧濃縮して得た残渣をシリカゲルカラム分離を通じて赤色の表題化合物(8)を得た。表題化合物の収率及び物性は、下記の通りであった。
【0162】
収率:45.8%、融点:92−93℃、1H−NMR(CDCl3、400MHz):δ 7.41(d、J=9.6Hz、1H)、7.36(d、J=9.6Hz、1H)、7.31(s、1H)、3.99(s、3H)、3.98(s、3H)、3.85(d、J=6.8Hz、1H)、3.28−3.21(m、1H)、2.96−1.89(m、2H)、1.69−1.59(m、4H)、1.41−1.23(m、12H)、0.93(d、J=6.8MHz、6H)、m/z 507(M+H)+。
【0163】
前記方法で合成した化合物群を用いてスネイル−p53の結合抑制効果を次の実験を通じて確認した。
<実験例1>p53活性回復及びターゲット遺伝子誘導確認
【0164】
前記実施例1〜5で合成された化合物が、スネイル−p53結合阻害効果があるか、細胞レベルで効果的であるか否かを立証するために、前記化合物をK−Ras突然変異型癌細胞株HCT116に処理し、ウェスタンブロットを用いて、この結果を確認した。
【0165】
K−Rasは、スネイルの活性を抑制してp53を誘導する作用をする遺伝子であって、前記細胞株HCT116は、K−Rasの突然変異によってスネイルが常に発現してp53と結合した後、p53が正常に作動しないようにする役割を果たす。前記K−Ras突然変異型癌細胞株HCT116にスネイル−p53結合を抑制する化合物を処理すれば、p53が正常作用することによって、発現量が増加してp53のターゲット遺伝子が誘導される。
【0166】
優先的に、p53とターゲット遺伝子であるp21との発現を確認するために、ウェスタンブロットを行った。本発明で使ったあらゆる細胞株は、ATCCから購入し、10%FBSを含有したRPMI−1640またはDMEMで維持された。RIPA緩衝液でタンパク質を抽出し、典型的なSDS−PAGE及びゲル移動方式を通じて細胞溶出物でローディングされたメンブレンを準備した。5%非脂肪乾燥油でブロッキングした後、各遺伝子に該当する抗体を用いて典型的なウェスタンブロット手続きを進行した。本発明で使われた抗体は、Cell signaling、SantaCruzから購買した。p53の活性は、p53のターゲット遺伝子であるp21の発現量を通じて確認することができた。図1は、実施例1〜5で合成した化合物の一部を用いてK−Ras突然変異型癌細胞株であるHCT116細胞でウェスタンブロットを行った結果を表わす図及びそれを数値化して表わしたグラフであり、表3は、この数値を表わした図表である。
【0167】
【表3】
【0168】
前記化合物をK−Ras突然変異型HCT116癌細胞株に処理した時、対照群(DMSO control)と比較して、前記化合物を10μM濃度で処理した所でp53を2〜5倍程度で、p21は、2〜8倍程度で発現量が大きく増加する化合物があるということを確認することができた(図20及び表3参考)。前記化合物が、p53とp53とのターゲット遺伝子であるp21の発現量を増幅させたことは、スネイルが常に活性化されている状態のK−Ras突然変異細胞株であるHCT116細胞でスネイルの役割が、前記化合物を通じて抑制されてスネイル−p53の結合阻害状態が維持されるということを表わす。
<実験例2>スネイル−p53結合阻害効果の確認
【0169】
次いで、p53とp21とを同時に誘導しながら発現量が多かった前記化合物のうちから二つの化合物5oまたは7aを選択して、K−Ras、スネイル及びp53の関係を確認するために使う。スネイル−p53の結合が阻害されるということを立証するために、前記二つの化合物5o及び7aを用いてGSTプルダウン分析(GST pull down assay)を行って確認した。
【0170】
GSTプルダウン分析は、二つのタンパク質の結合程度を確認することができる方法でGSTが融合されたスネイルタンパク質とp53再組合タンパク質とを製造して、前記化合物5oあるいは7aを共に処理してGST−スネイルタンパク質とp53タンパク質との結合程度を確認した。前記化合物の比較群としては、Nutlin−3を使った。Nutlin−3は、p53の陰性調節因子であるMDM2とp53との結合を遮断する物質で多くの腫瘍細胞で過発現されるタンパク質として知られている。MDM2が過発現されれば、p53のタンパク分解が誘導されてp53のターゲット遺伝子を通じて起こるアポトーシス(apoptosis)などの反応を抑制し、細胞の抗増殖効果などを誘導する。代表的なp53の調節因子であるが、K−Ras突然変異性疾患や突然変異p53を有した疾患には作用しない。
【0171】
GSTプルダウン分析のために、3つの人間スネイル断片(残基1−90、91−112及び113−264)及びp53断片(1−93及び93−292)をGST−融合タンパク質として大膓菌で発現し、各断片をGSH−アガロースにローディングし、洗浄した後、20mM還元グルタチオンを含有した緩衝液を用いて溶出した。溶出分画を陰イオン−交換クロマトグラフィー(HitrapQ)を用いて錠剤した。再組合人間p53タンパク質(残基94−292)をベクターpET28Aを用いて大膓菌で発現させた。前記ベクターpET28Aは、C末端にヘキサ−ヒスチジンタグを含有した。Ni−NTA親和力及び大きさ排除クロマトグラフィー(Superdex 200)を用いてヒスチジンが付いたp53タンパク質を錠剤した。p53及びスネイル間の直接結合を検討するために、アガロース−ビーズ接合GSTまたはGST−スネイルを4℃で45分間PBSでHis−p539(ヒスチジン−p53)と培養した。PBSで洗浄した後、沈澱されたタンパク質をSDS−PAGE及びウェスタンブロットを行った。
【0172】
図21に示したように、GSTプルダウン分析結果を確認して見れば、対照群(c)の場合、GST−スネイルとp53とが多量に結合されていることを確認することができる。Nutlin−3もMDM2を調節してp53活性を誘導する物質であるために、スネイル−p53の結合を阻害できず、対照群と類似している結合程度を表わしている。一方、前記二つの化合物5oと7aとを使った分画では、GST−スネイルタンパク質にp53が結合される程度が確実に減ることを確認することができ、前記化合物が、スネイル−p53の結合を抑制したということを立証することができた。
<実験例3>K−Ras依存的なp53誘導能の確認
【0173】
次いで、前記化合物が、正常細胞には作用せず、K−Ras突然変異癌細胞にのみ選択的に作用して死滅させる能力があるか否かを確認するために、細胞生存率を通じて前記二つの化合物5oと7aとに対する細胞毒性及び死滅効果をトリパンブルー溶液を用いて細胞数を測定して確認した。前記化合物のp53誘導能が、K−Rasと関係があるかを検討するために、K−Ras突然変異癌細胞株であるA549とHCT116、K−Ras野生型癌細胞株であるMKN45とを利用した。
【0174】
表4は、それぞれの細胞株に対する死滅率を数値化して表わした図表であり、図22は、死滅率に対する数値を表わしたグラフである。
【0175】
【表4】
【0176】
細胞死滅率を通じて表われた結果は、前記化合物5oが、K−Ras突然変異癌細胞株であるA549とHCT116とから10μM程度でそれぞれ51%と57%程度の死滅率を示し、7aも同一濃度10μMでほぼ34%と44%程度の死滅率を示した(表4及び図22参照)。一方、K−Ras野生型癌細胞株であるMKN45では、同一濃度10μMで前記化合物5oは、15%程度の死滅率を、前記化合物7aは、ほとんど1%の死滅率を示してK−Ras突然変異型癌細胞株と確実に異なる結果を表わした。これにより、前記化合物が、K−Rasが正常に作用しない癌細胞株でのみ選択的に作用して高い死滅率を表わすことを確認することができた。これは、前記化合物が、K−Rasが損傷されている癌細胞にのみ選択的に作用することができ、K−Rasが損傷されて癌にかかった患者に有用であるということを暗示する。一方、Nutlin−3は、K−Ras突然変異癌細胞株であるA549とHCT116細胞株で同一濃度10μMでそれぞれ19%と32%死滅率を表わし、K−Ras野生型細胞株であるMKN45では、同一濃度10μMで細胞死滅率が38%程度でさらに高くなるなどの一括的ではない類型を表わすことを確認することができた。このように、Nutlin−3は、K−Rasが正常に作用をするか、突然変異状態であるものに関わらずに或る有意な反応を表わさず、K−Rasに選択的な反応を表わさないということを確認することができた。これで、前記化合物5oと7aとがNutlin−3と異なってK−Rasに選択的作用効果に優れるということを立証した。
<実験例4>p53のターゲット遺伝子の活性誘導
【0177】
次いで、突然変異p53がある時の前記化合物5oの効果を確認した。
前記化合物5oを突然変異p53類型であるMT/WT−p53遺伝子を有するMDA−MB 468という人間乳房癌細胞株に処理してウェスタンブロットを実施した。図23は、MDA−MBに5oを処理して実施したウェスタンブロットの結果を表わすグラフであり、表5は、このグラフ値を数値で表わした図表である。
【0178】
【表5】
【0179】
前記化合物5oとNutlin−3とをp53突然変異型癌細胞株MDA−MB 468に処理した時、5oが処理された所でのみ多量のp21が発現されることを確認することができた。(図23及び表5参照)。この結果からNutlin−3は、突然変異p53がある時、ターゲット遺伝子であるp21に何らの影響を及ぼすことができない一方、前記化合物5oは、p21の活性を誘導してNutlin−3と差別的な影響力があるということを表わす。
<実験例5>生体内異種移植(xenograft)の考察
【0180】
1.実験方法
非胸腺マウスをDaehan Biolink Co.Ltdから購入し、温度及び光の調節条件(20−23℃、12時間光/12時間暗周期)下で生育させ、滅菌食餌と水とを自在に供給した。2週後、非胸腺マウス(n=21)に腹腔注射を通じてA549細胞を1X107細胞で接種した。2週後、各グループを3つのサブグループに分けてPBS、10mg/kgまたは20mg/kgの5oを一週間に一回ずつ10週間腹腔内に投与し、生存率を観察した。動物試験は、釜山大学校動物保護委員会の承認及び指針によって行われた。
【0181】
2.実験結果
図24のように、10mg/kgと20mg/kgとの5oの処理によって腫瘍による死亡率が遮断された一方、PBS処理群では、10週後に生存率が50%以下になり、図25に示したように、5o処理群での腫瘍は、ほとんど死滅する形状が観察された。また、図26のように、体重減少や化合物注射による総体的な解剖学的異常所見を観察することができなかった。下記表6は、腫瘍の発生位置及び形態学的特徴を要約したものである。
【0182】
【表6】
【0183】
[配列目録フリーテキスト]
配列番号1は、人間p53細胞性腫瘍抗原のアミノ酸配列を表わしたものである。
【産業上の利用可能性】
【0184】
本発明は、スネイル−p53間の結合を阻害する化合物及びそれを有効成分として含有する癌疾患治療剤関連の分野に適用可能である。
【技術分野】
【0001】
本発明は、スネイル−p53間の結合を阻害してp53の発現を誘導することによって、膵膓癌、肺癌、胆管癌及び大膓癌のようなK−Ras突然変異性癌疾患治療に有用に使われるスネイル−p53間の結合を阻害する化合物及びそれを有効成分として含有する癌疾患治療剤に関する。
【背景技術】
【0002】
抗癌剤及び診断技術の改善によって全体癌患者の5年生存率が50%増加した。しかし、いくつかの癌疾患、例えば、肺癌と膵膓癌は、相変らず10%以下の低い生存率を表わしている。したがって、このような患者の生存率を増加させるために、このような疾患の初期診断方法の開発が至急である。また、このような類型の癌は、K−Rasでよく変化を示している。特に、発癌性K−Rasは、5%以下の生存率を表わす膵膓癌で圧倒的に優勢な傾向を表わしている。
【0003】
発癌性Rasは、p53活性化を通じる老化及び連続した細胞死を誘導すると知られており、発癌性Ras媒介腫瘍形成は、p53欠乏条件下で起こると推定されており、特に、H−Ras誘導肝癌細胞種は、素早く活性化されたp53によって抑制された。
【0004】
現在使われている肺癌、膵膓癌などの治療剤は、薬物効果の持続時間が短い一方、多様な副作用が誘発されるので、このような疾患を効果的に治療するか、初期診断することができる薬物の開発が至急である。
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明者は、発癌性K−Rasがスネイルの誘導を通じてp53を抑制するということを明らかにし、p53及びスネイル間の相互作用を遮断することができる化合物を同定し、このような化合物が、K−Ras突然変異細胞株でp53発現を誘導することを発見して、本発明を完成した。
【0006】
これにより、本発明の目的は、スネイル−p53の結合を阻害する候補薬物を選別する段階を含むK−Ras突然変異性癌疾患治療剤のスクリーニング方法を提供することである。
【0007】
また、本発明の他の目的は、スネイル−p53間の結合を阻害する化合物及びそれを有効成分として含有する癌疾患治療剤を提供することである。
【0008】
また、本発明者は、p53の特定領域、例えば、DNA結合ドメインは、K−Ras突然変異細胞での透過性が増加するので、それを担体として用いてK−Ras突然変異細胞に特異的に薬物を伝達することができるという点を明らかにし、またスネイルの自動抗体の発現有無を測定してK−Ras突然変異性癌疾患、例えば、膵膓癌、肺癌、胆管癌、大膓癌などを初期に診断することができるという点を明らかにすることによって、本発明を完成した。
【0009】
これにより、本発明のまた他の目的は、p53のDNA結合ドメインのエンドサイトーシスを用いてK−Ras突然変異細胞に特異的に薬物を伝達することができる薬物伝達方法を提供することである。
【0010】
また、本発明のさらに他の目的は、スネイル自動抗体の発現有無を測定してK−Ras突然変異性癌疾患を診断することができる初期診断方法を提供することである。
【課題を解決するための手段】
【0011】
前記目的を果たすために、本発明は、p53を固定させたプレートにスネイル及び候補薬物を培養する段階と、ELISAリーダーでスネイル−p53の結合を阻害する候補薬物を選別する段階と、を含んでなることを特徴とするK−Ras突然変異性癌疾患治療剤のスクリーニング方法を提供する。
【0012】
また、本発明は、下記化学式1で表される化合物、またはその塩を提供する:
【化1】
前記化学式1で、
mは、0ないし10の整数、n及びpは、それぞれ0または1の整数であり、
Zは、−NH(CH2)qCH3、−OH、4−フェニルピペリジン基、4−フェニルピペラジン基、イソブチルアミノ基及びイソブチルオキシ基からなる群から選択され、
qは、0ないし9の整数である。
【0013】
より望ましくは、前記化合物は、2−ノニルアミノ−5、8−ジメトキシ−1、4−ナフトキノン;2−デシルアミノ−5、8−ジメトキシ−1、4−ナフトキノン;3−(5,8−ジメトキシ−1、4−ジオキソ−ナフタレン−2−イルチオ)プロパン酸;11−(5,8−ジメトキシ−1、4−ジオキソ−ナフタレン−2−イルチオ)ウンデカン酸;イソブチル−11−(5,8−ジメトキシ−1、4−ジオキソ−1、4−ジヒドロナフタレン−2−イルチオ)−ウンデカノアート;11−(5,8−ジメトキシ−1、4−ジオキソ−1、4−ジヒドロナフタレン−2−イルチオ)−N−イソブチルウンデカンアミド;及びイソブチル−11−(5,8−ジメトキシ−1、4−ジオキソ−1、4−ジヒドロナフタレン−2−イルスルフィニル)ウンデカノアートからなる群から選択された化合物、またはその塩であり得る。
【0014】
前記化学式1の化合物は、前記K−Ras突然変異性癌疾患治療剤のスクリーニング方法を利用すれば、スネイル−p53の結合を阻害する薬物を特異的に選別することができて、膵膓癌、肺癌及び大膓癌のように診断や治療が容易ではないK−Ras突然変異性癌疾患を効果的に治療するか、予防することができる。
【発明の効果】
【0015】
本発明によるK−Ras突然変異性癌疾患治療剤のスクリーニング方法を利用すれば、スネイル−p53間の結合を阻害する薬物を特異的に選別することができて、膵膓癌、肺癌、胆管癌及び大膓癌のように診断や治療が容易ではないK−Ras突然変異性癌疾患を効果的に治療するか、予防することができる。
【0016】
また、p53のDNA結合ドメインを担体として用いてK−Ras突然変異細胞に特異的に薬物を伝達することができて、K−Ras突然変異性癌疾患の治療に非常に有用であり、スネイルの自動抗体の発現有無を測定してK−Ras突然変異によって誘発される癌疾患を初期に診断することができて、診断が困難な膵膓癌などの早期診断が可能であって、癌患者の生存率を延長させるか、治療に効率的に対処することができる。
【図面の簡単な説明】
【0017】
【図1】スネイル媒介p53発現抑制に関する分子的機転の考察を示す図である。
【図2】スネイル媒介p53発現抑制に関する分子的機転の考察を示す図である。
【図3】スネイル媒介p53発現抑制に関する分子的機転の考察を示す図である。
【図4】発癌性K−Ras媒介p53抑制が化学的阻害剤によって遮断されないものを示す図である。
【図5】スネイルが発癌性K−Ras媒介p53抑制のために重要な媒介体であるものを示す図である。
【図6】スネイルとp53との間の直接的な相互作用を示す図である。
【図7】K−Ras誘導性スネイル安定化において、ATR活性の必要条件を示す図である。
【図8】スネイル及びp53間の直接的な結合を示す図である。
【図9】スネイル及びp53間の結合阻害剤の同定に関する図である。
【図10】スネイル及びp53間の結合遮断がK−Ras突然変異細胞でp53機能を誘導するものを示す図である。
【図11】スネイル及びp53間の結合遮断がK−Ras突然変異細胞でp53機能を誘導するものを示す図である。
【図12】スネイル媒介p53発現抑制に関する分子的機転の考察を示す図である。
【図13】p53の核から細胞質への移動機転に関する結果である。
【図14】p53が小嚢輸送を通じて分泌するものを示す図である。
【図15】p53がプロテアーゼ及びエンドサイトーシスによって除去される一方、スネイルは、これらに抵抗性を表わした結果である。
【図16】K−Ras突然変異細胞でのHis−p53の再吸収を示す図である。
【図17】癌組織でのp53及びスネイル分泌を示す図である。
【図18】肺癌血清で抗−スネイルAbを分析した図である。
【図19】発癌性K−Rasでp53及びスネイルの分泌態様を模式化した図である。
【図20】実施例1〜5で合成された化合物のp53及びp21の誘導能を確認するウェスタンブロット結果を示す図である。
【図21】Nutlin−3と前記化合物とのうちから5oと7aとを選択してスネイル−p53結合阻害効果を確認するGSTプルダウン分析結果を示す図である。
【図22】K−Ras突然変異癌細胞株とK−Ras野生型癌細胞株とに化合物5o、7a及びNutlin−3を処理して表われた細胞死滅率を示す図である。
【図23】p53突然変異細胞株に化合物5oとNutlin−3とを処理してp21の活性が誘導されるものを表わすウェスタンブロット結果を数値化して示す図である。
【図24】腹腔注射でA549細胞を非胸腺マウスに注入後、化合物5o処理によるマウスの生存率を示す図である。
【図25】腹腔注射後、発生した腫瘍の組職写真を示す図である。
【図26】A549細胞で化合物5o処理による総体的な解剖学的異常所見を観察した図である。
【発明を実施するための形態】
【0018】
一つの具体的様態として、前記化学式1の化合物は、塩の形態で存在することができる。前記塩は、塩酸または硫酸などのような無機酸、またはp−トルエンスルホン酸などのような有機酸の薬剤学的に許容可能な塩であり得る。
【0019】
前記化学式1の化合物は、下記反応式1ないし5のような方法で製造可能である。
【化2】
【0020】
【化3】
【0021】
【化4】
【0022】
【化5】
【0023】
【化6】
【0024】
反応式2の工程を具体的に説明すれば、次の通りである。
1,5−ジヒドロキシナフタレン(2)を出発物質として用いて、既に知られた3段階の反応を経て、1,4,5,8−テトラメトキシナフタレン(3)を合成し、再び脱メチル化して合成中間体である5,8−ジメトキシ−1,4−ナフトキノン(4)を合成した。詳しい合成方法は、合成スキームにある参考文献で見られるが、このとき使われた溶媒は、反応に悪影響を及ぼさない溶媒である水酸化ナトリウム、アセトニトリル、無水メタノール、N,N−ジメチルホルムアミド、クロロホルムなどを使った。初めのメチル化反応は、窒素ガス存在下で水酸化ナトリウムに溶かした1,5−ジヒドロキシナフタレンに硫酸ジメチルを1時間滴加して2時間反応させて得た。後でベンゼンで再結晶して1,5−ジメトキシナフタレンを得た。
【0025】
1,4,5,8−テトラメトキシナフタレン(3)を合成する時は、無水条件でナトリウムメトキシドとヨウ化銅とをジメチルホルムアミドとメタノールとを入れて30時間加熱還流して得た。反応に対する温度は、80℃以上にして還流させ続けた。中間体5,8−ジメトキシ−1,4−ナフトキノンを合成する時は、硝酸二アンモニウムセリウムを使ったが、室温で30分間滴加して30分間さらに反応させて得た。化合物5aないし5pは、中間体5,8−ジメトキシ−1,4−ナフトキノン(4)をメタノールに溶かして、得ようとするアミンやメルカプタンまたは末端にカルボキシル基やヒドロキシル基が付いたメルカプタンを入れて室温で4時間から一晩撹拌してTLCで反応進行状況を確認し、硫酸と重クロム酸ナトリウム水溶液とでウォークアップさせてシリカゲルカラムクロマトグラフィーで分離して得た。
【0026】
次の段階で、化合物5oの2番位置カルボキシル基に4−フェニルピペリジンまたは4−フェニルピペラジン誘導体を付ける時は、1,3−ジシクロヘキシルカルボジイミド(DCC)とN,N−ジメチルアミノピリジン(DMAP)よりは、N−(3−ジメチルアミノプロピル)−N´−エチルカルボジイミド塩酸塩(EDC)を入れて反応させることによって、化合物6aと6bとを合成することができ、前記反応は、収率も高くて分離過程にウレアがなしにきれいに分離することができる。次の段階で、化合物5pを用いてイソブチルアルコールとイソブチルアミンとを付ける時にも、同様にN−(3−ジメチルアミノプロピル)−N´−エチルカルボジイミド塩酸塩(EDC)を使って、得ようとした化合物7a、7bを容易に得られる。スルホキシド化合物8を合成する時には、化合物7aを用いてMCPBAを使い、TLCで反応状況を確認しながら合成することができる。最後に、重炭酸ナトリウムで反応を中止させてシリカゲルカラムをかけて精製された化合物7aが得られる。
【0027】
しかし、前記反応式2ないし5による方法は、化学式1の化合物の製造方法の一例に該当し、例えば、反応溶媒、塩基、反応物質の使用量のような反応条件が、前記から説明されたことのみで限定されるものではなく、前記反応式2ないし5の方法の以外にも、当業者に知られた公知の多様な合成方法を用いて、前記化学式1の化合物を製造することができる。
【0028】
また、本発明は、スネイル−p53間の結合を阻害する化合物を有効成分として含有する癌疾患治療剤を提供する。
【0029】
前記化合物は、前記化学式1で表される化合物、またはその塩であり、より望ましくは、2−ノニルアミノ−5,8−ジメトキシ−1,4−ナフトキノン;2−デシルアミノ−5,8−ジメトキシ−1,4−ナフトキノン;3−(5,8−ジメトキシ−1,4−ジオキソナフタレン−2−イルチオ)プロパン酸;11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)ウンデカン酸;イソブチル−11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロキシナフタレン−2−イルチオ)−ウンデカノアート;11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)−N−イソブチルウンデカンアミド;及びイソブチル−11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルスルフィニル)ウンデカノアートからなる群から選択された化合物、またはその塩であり得る。
【0030】
前記癌疾患は、K−Ras突然変異性癌疾患であり、望ましくは、膵膓癌、肺癌、胆管癌及び大膓癌からなる群から選択された癌疾患であり得る。
【0031】
本発明による癌疾患治療剤は、薬学組成物の製造に通常使う適切な担体、賦形剤または希釈剤をさらに含みうる。
【0032】
本発明で使用可能な担体、賦形剤または希釈剤としては、ラクトース、デキストロース、スクロース、ソルビトール、マンニトール、キシリトール、エリスリトール、マルチトール、澱粉、アカシアゴム、アルギン酸、ゼラチン、燐酸カルシウム、ケイ酸カルシウム、セルロース、メチルセルロース、微晶質セルロース、ポリビニルピロリドン、水、ヒドロキシ安息香酸メチル、ヒドロキシ安息香酸プロピル、ステアリン酸マグネシウムまたは鉱物類などが挙げられる。
【0033】
前記癌疾患治療剤は、それぞれ通常の方法によって、散剤、顆粒剤、錠剤、カプセル剤、懸濁液、エマルジョン、シロップ、エアゾールなどの経口型剤型、外用剤、座剤及び滅菌注射溶液の形態で剤型化して使われる。
【0034】
製剤化する場合には、普通使う充填剤、増量剤、結合剤、湿潤剤、崩解剤、界面活性剤などの希釈剤または賦形剤を使って調剤される。経口投与のための固型製剤には、錠剤、丸剤、散剤、顆粒剤、カプセル剤などが含まれ、このような固型製剤は、前記化合物は少なくとも一つ以上の賦形剤、例えば、澱粉、炭酸カルシウム(calcium carbonate)、スクロース(sucrose)またはラクトース(lactose)、ゼラチンなどを混ぜて調剤する。
【0035】
また、単純な賦形剤の以外に、ステアリン酸マグネシウム、タルクのような潤滑剤も使われる。経口のための液状製剤としては、懸濁剤、内用液剤、乳剤、シロップ剤などが該当するが、よく使われる単純希釈剤である水、リキッドパラフィンの以外に、さまざまな賦形剤、例えば、湿潤剤、甘味剤、芳香剤、保存剤などが含まれうる。
【0036】
非経口投与のための製剤には、滅菌された水溶液、非水性溶剤、懸濁剤、乳剤、凍結乾燥製剤、座剤が含まれる。非水性溶剤、懸濁剤としては、プロピレングリコール(propylene glycol)、ポリエチレングリコール、オリーブオイルのような植物性油、オレイン酸エチルのような注射可能なエステルなどが使われる。座剤の基剤としては、ハードファット(witepsol)、マクロゴ−ル、トウイーン(tween)61、カカオ脂、ラウリン脂、グリセロゼラチンなどが使われる。
【0037】
前記癌疾患治療剤の使用量は、患者の年齢、性別、体重によって変わりうるが、0.1ないし100mg/kgの量を一日1回ないし数回投与することができる。
【0038】
また、このような癌疾患治療剤の投与量は、投与経路、疾病の程度、性別、体重、年齢などによって増減される。したがって、前記投与量は、如何なる面でも、本発明の範囲を限定するものではない。
【0039】
前記癌疾患治療剤は、モルモット、マウス、家畜、人間などの哺乳動物に多様な経路で投与されることがある。投与のあらゆる方式は予想されるが、例えば、経口、直腸または静脈、筋肉、皮下、子宮内硬膜または脳血管内(intracerebroventricular)注射によって投与されることがある。
【0040】
また、本発明は、p53のDNA結合ドメインのエンドサイトーシス(endocytosis)を用いて伝達しようとする薬物をK−Ras突然変異細胞に特異的に伝達することを特徴とするK−Ras突然変異細胞の特異的薬物伝達方法を提供する。
【0041】
前記DNA結合ドメインは、人間p53(Genbank Accession No.P04637)の90番目から280番目までのアミノ酸配列を含む。
【0042】
望ましくは、前記薬物伝達方法は、p53のDNA結合ドメイン及び伝達しようとする薬物を共に細胞に処理する段階と、前記p53のDNA結合ドメインによるエンドサイトーシスによって隣合うK−Ras突然変異細胞に薬物が伝達される段階と、を含んでなりうる。
【0043】
また、本発明は、スネイル抗体の発現有無を測定することを特徴とするK−Ras突然変異性癌疾患の初期診断方法を提供する。
【0044】
前記スネイル抗体の発現は、K−Ras突然変異性癌疾患を有した患者の血清から測定することができ、K−Ras突然変異性癌疾患は、膵膓癌、肺癌、胆管癌及び大膓癌からなる群から選択された癌疾患であり得る。
【0045】
本発明の理解を助けるために、望ましい実施例を下記に提示する。しかし、このような実施例は、本発明をより容易に理解するために提供されるものであって、本発明が、下記の実施例に限定されるものではない。
[スネイル媒介p53抑制に関する分子的機転の考察]
<実施例1>
【0046】
1.マウス線維芽細胞の分離及び無限分裂
6ヶ月齢の雄マウスを犠牲させて肺線維芽細胞(fibroblast)を集めた。肺を分離した後、組職メスを用いて組職を切って溶出した。20% FBSを含有したDMEM培地で3日間培養した後、付着された細胞を培養皿に分周し、jetPEIを用いて製造者の指針によって突然変異H−Ras、N−Ras及びK−Rasで形質感染させた。72時間後、DMEM含有G418 400μg/mlを用いて形質感染された細胞を選別した。
2.細胞培養及び試薬準備
【0047】
本発明で使った細胞株は、ATCCから購入し、10% FBSを含有したRPMI−1640またはDMEMで維持された。また、使われた抗体は、Santa CruzまたはCell Signaling(p53−R、P−Erk)から購買した。Ras発現ベクター及びSnailベクターは、それぞれDr.Chi SG及びHung M−Cから提供された。p53 S46D及び46Aは、Mayo LDから提供された。また、本発明で使われた化学物質は、Calbiochemから購買し、組替えp53は、Assay designsから購買した。
【0048】
また、細胞分画分析は、Subcell fraction Kit(Merck)を用いて製造者の指針によって行った。培地分析のために、細胞培養された培地を集めてCentricon(Millipore)またはEtOH沈降を用いて濃縮した。
3.免疫染色及びウェスタンブロット
【0049】
細胞染色のために、先行培養された細胞を洗浄して100%メタノールで固定し、抗体(第1次抗体;1:200、4℃で一晩中、第2次抗体;1:1000、室温で2時間)で培養した。分泌されたp53及びSnailを測定するために、HCT116 p53−/−細胞を1mL RPMI 1640培地で24時間ベクターで形質感染させ、洗浄なしに2% PFA 1mLを加えて固定させた。固定後、細胞をPBSで2回洗浄してブロッキング緩衝液[PBS+抗人間Ab(1:500)]で培養して、非特異的結合を除去した。PBS洗浄後、細胞を抗−p53 Ab及び抗−Snail Abと培養して、第2次Abとマッチングした。タンパク質分析のために、RIPA緩衝液でタンパク質を抽出してSDS−PAGEに試料を適用し、一般的なウェスタンブロット(WB)プロトコルによって行った。
【0050】
免疫沈降分析は、一般的なプロトコルによって行われた。すなわち、細胞溶出物を4時間Abと先に培養し、タンパク質−A/G−アガロースで2時間培養した。遠心分離後、3回洗浄した。沈降された複合体を用いてSDS−PAGE/WB分析を行った。
【0051】
4.形質感染及びsi−RNA効果の検討
細胞形質感染のために、製造者のプロトコルによってjetPEIを利用した。細胞を完全培地で24時間DNA/jetPEI混合物で培養した。生体外遺伝子ノックアウトのために、Snail及びMDM2に対するsi−RNAを作った。jetPEIを用いてsi−RNAを形質感染させて24時間後、その効果を検討した。
5.実験結果
【0052】
図1Aに示したように、N−RasまたはH−Rasの形質感染がアポトーシスまたは老化を誘導したが、K−Rasで形質感染された細胞は、6ヶ月が経過されても引き続き成長して維持された。
【0053】
また、発癌性K−Rasの強制発現が、野生型p53−含有細胞株でp53発現を抑制し(図2A参照)、K−Rasは、N−RasまたはH−Rasと異なってp53抑制を引き起こし、これは、si−MDM2によって遮断されなかった(図2B参照)。しかし、野生型K−Rasは、p53発現を抑制しなかった(図2C及び図2D参照)。
【0054】
図1Bに示したように、このようなp53は、K−Ras濃度依存的に抑制された。DN−RaseによるRas活性の遮断が、K−Ras突然変異A549でp53発現を増加させたが、HepG2では増加させなかった(図2E)。
【0055】
前記結果を総合すれば、内因性発癌性K−Rasが、p53発現を抑制するということを示唆する。しかし、図1Cのように、DN−RaseがDNA損傷−媒介p53抑制に関して明確であり、相加的な効果を観察することができず、これは、強い遺伝毒性ストレスが発癌性K−Ras媒介p53抑制を克服できるということを示唆する。
【0056】
図2F及び図2Gに示したように、K−Ras媒介p53抑制が点突然変異p53で確認されたが、p53活性型であるp53 S46Dは、K−Ras媒介p53抑制に抵抗性を表わした。このような結果は、遺伝毒性誘導性p53活性化がK−Ras媒介抑制を克服できるという図1Cの結果とも一致する。
【0057】
22/23突然変異がMDM2と関連しないために、K−Ras媒介p53抑制は、MDM2と独立した経路で行なわれるということを確認することができ、プロテアソーム−阻害剤がK−Ras媒介p53を遮断することができなかったために、MDM2またはp53ユビキチンシステムと関係のないということが分った(図1A及び図2H参照)。
【0058】
また、K−Ras媒介p53抑制に対するMAPK信号阻害剤の効果を検討した結果、MAPK経路遮断が、K−Ras媒介p53抑制に関する効果を無くすことができなかった。このような結果からK−Ras媒介p53抑制は、他の新規経路で行われると判断される。
【0059】
また、不溶性分画でスネイルによるp53の分布を検討した結果、図12Aのように、p53は、スネイルまたは発癌性K−Rasによって減少し、細胞を4つの分画、すなわち、核、細胞質、膜/器官及び不溶性分画に分けて検討した結果、図12Bのように、スネイルによって減少したp53は、いかなる細胞分画でも回復されなかった。
【0060】
また、K−Ras突然変異細胞でスネイルの除去がsi−MDM2より圧倒的にp53を誘導した(図12C)。そして、図12D及び図12Eのように、スネイルによるp53の減少は、MDM2−媒介抑制とは異なってプロテアソーム阻害剤によって回復されなかった。
<実施例2>
1.ウェスタンブロット及び生体外のキナーゼまたはバインディング分析
【0061】
Snail及びp53間の直接結合を検討するために、典型的なSDS−PAGE及びゲル移動方式を通じて組替えp53またはSnailまたはp53形質感染された細胞溶出物でローディングされたメンブレンを準備した。5%非脂肪乾燥油でブロッキングした後、メンブレンをp53またはSnail形質感染されたp53−/−HCT116細胞溶出物で4℃で4時間培養した。洗浄後、メンブレンをp53 AbまたはSnail Abを用いて典型的なWB手続きを進行した。
【0062】
インビトロ結合のために、再結合p53及びGST−Snailを交互に4℃で1時間培養し、p53 AbまたはGST Abを有したIP及びGSTまたはp53 Abを有したWBを行った。Snailの変形を調査するために、293細胞を形質感染のために使った。フラクションまたはリシス後、溶出物をGSTまたはGST−Snailで25℃で1時間培養し、SDS−PAGE及びWB分析を行った。p−MAPK及びp−ATM/ATR基質に対する抗体をcell signalingから購入した。
2.実験結果
【0063】
図3Aに示したように、スネイル発現に対するK−Rasの効果を検討した結果、スネイルがK−Rasによって誘導されうるということを観察した。また、細胞株でp53発現に対するスネイルの効果を検討した結果、図3Bのように、スネイルの過発現がA549及びHepG2細胞株でp53を抑制することができる一方、スネイルノックダウンがA549(発癌性K−Ras含有細胞株)でのみp53を誘導するだけで、HepG2では誘導することができなかった。
【0064】
また、si−スネイルがDNA損傷薬剤に対して敏感性を増加させ(図4F)、したがって、スネイルの過発現が細胞増殖を増進させてDNA損傷誘導性細胞死に抵抗性を表わした(図4G)。
【0065】
図3Cのように、スネイルは、K−Rasと同様にendo−p53だけではなく、exo−p53を抑制し、図3Dないし図3Fのように、スネイルとp53とを共に形質感染させた時、突然変異p53と関係なくスネイル及びp53いずれも減少した。しかし、スネイル及びp53のmRNAは減少しなかった(図3C及び図3D)。また、p53転写物に対するスネイルの効果を検討した結果、スネイルは、p53 mRNAを減少させなかった(図4H)。
【0066】
したがって、p53及びスネイルは、たとえよく知られた転写調節剤であるとしても、これらの減少は、転写調節と無関係であるいうことを示唆する。また、図3G及び図3Hのように、スネイルの除去がK−Ras媒介p53抑制を遮断することができる。また、図5A及び図5Bのように、exo−p53でも類似した結果を得た。このような結果からK−Ras媒介p53抑制は、スネイル誘導を通じて得られるということが分かる。
【0067】
また、図5Cのように、発癌性K−Rasは、4時間以内にp53を誘導する一方、p53は6時間後に減少し、これは、p53抑制が形質感染の人為的結果ではなく、形質感染されたタンパク質の効果であるということを表わす。
【0068】
また、図5Eのように、K−Ras/スネイル形質感染された細胞でアポトーシス及び細胞周期を検討し、アポトーシス及び細胞周期阻害は、K−Ras/スネイルによって明らかに誘導されなかった。
【0069】
また、Aphidicolin処理細胞でK−Ras/スネイルによってp53の減少が観察されて、p53の減少は細胞周期とは関連しないものと判断される。
【0070】
また、CHX−pulse chaseを通じてp53の半減期を対するスネイルの効果を検討し、図5Fのように、スネイルは、p53半減期を短縮させなかった。そして、p53 S46Dの発現に対するスネイルの効果を検討し、図6Aのように、スネイルがK−Rasと異なってp53 S46D発現を抑制した。
【0071】
また、スネイル発現に対するp53 S46Dの効果を検討した結果、図6Bのように、S46Dが転写及び翻訳レベルでスネイル発現を抑制した。したがって、si−スネイルがp53抑制を回復させることができる野生型p53とは異なって、図6Cのように、S46Dで形質感染される場合には、si−スネイルがp53発現を誘導することができなかった。
【0072】
このような結果から或るストレス条件下でセリン46残基で変形によって活性化されたp53は、スネイル転写物の抑制を通じてK−Ras媒介抑制機転を克服することができる。
【0073】
また、スネイル誘導がATRを通じてなされた。
K−Rasが、スネイルを如何に誘導するかを検証するために、AKTの関連を検討した。Rasは、AKTを活性化してGSK−3β媒介スネイル不安定性を抑制できると知られている。
【0074】
しかし、図7Aのように、AKT−KDは、スネイルまたはK−Ras誘導p53抑制を遮断することができなかった。一方、図7Bのように、si−RNAを通じるATRの抑制がp53抑制を遮断した。また、図7C及び図7Dのように、スネイルは、ATR処理によって増加するが、ATM及びノコダゾール処理によっては増加しなかった。図6Dのように、インビトロキナーゼ分析でスネイルは、ATRによってリン酸化された。ATRを活性化させるものと知られたK−Rasは、図7Eのように、p−スネイルをATR−依存性方式と同様に増加させ、図7Fのように、スネイルの半減期を延長させた。
<実施例3>
1.再組合タンパク質及びGST−pull down分析
【0075】
3つの人間Snail断片(残基1−90、91−112及び113−264)及びp53断片(1−93及び93−292)をGST−融合タンパク質として大膓菌で発現した。各断片をGSH−アガロースにローディングし、洗浄した後、20mM還元グルタチオンを含有した緩衝液を用いて溶出した。溶出分画を陰イオン−交換クロマトグラフィー(HitrapQ)を用いて錠剤した。再組合人間p53タンパク質(残基94−292)をベクターpET28Aを用いて大膓菌で発現させた。前記ベクターpET28Aは、C末端にヘキサ−ヒスチジンタグを含有した。
【0076】
Ni−NTA親和力及び大きさ排除クロマトグラフィー(Superdex200)を用いてp53タンパク質を錠剤した。p53及びSnail間の直接結合を検討するために、アガロース−ビーズ接合GSTまたはGST−Snailを4℃で45分間RIPAで細胞溶出物、またはHis−p53と培養した。PBS及びRIPAで洗浄した後、沈澱されたタンパク質をSDS−PAGE及びWBを行った。
【0077】
2.実験結果
スネイルは、核タンパク質であるために、スネイル及びp53を共に形質感染させた時、スネイル及びp53がいずれも消えた(図3Dないし図3F参照)。図8Aのように、endo−IPからこれらタンパク質が互いに関連するということが分かった。図8B及び図8Cのように、Far−western blot分析及びGST−pull down分析を通じてスネイル及びp53が互いに直接的に相互作用するというが分かった。また、p53のDNA結合ドメイン及びスネイルの中間領域が、結合ドメインとして役割を行っ
た(図8Dないし図8F、図6E、図6F参照)。
<実施例4>
1.化学的スクリーニングのためのELISAシステムの製造
【0078】
Snail−p53結合阻害剤を分離するために、ELISAシステムを製造した。0.5% PFAを用いて96ウェルプレートにHis−p53(93−292)を固定させた。乾燥して洗浄した後、0.1μM化学物質を有したGST−Snailで培養した。1時間培養後、96ウェルプレートをTBSTで洗浄して抗−GST−Ab(1:10000、45分)及び抗−マウス−IgG−HRP(1:50000、30分)で培養した。2回洗浄後、プレートをTMB溶液及び停止液で培養した。ELISAリーダーを用いて測定した。
2.実験結果
【0079】
図9A及び図9Bのように、p53及びスネイル間の結合抑制をELISAシステムで検討した。図9Cのように、150余個の化学物質からスネイル及びp53の結合阻害剤で3つの化学物質を同定した。図10A及び図11Aのように、このような化学物質は、濃度依存的にスネイル及びp53間の結合を抑制した。
【0080】
GST−pull down分析を通じて、このような化合物処理後、p53及びその標的の発現を検討し、3つの化学物質がいずれもp53及びスネイルの相互作用発現を抑制し、p53発現を誘導した(図8B及び図11B)。
【0081】
また、図10Cのように、このような化学物質処理によってPUMA及びp21の誘導を観察することができた。特に、p53の誘導がK−Ras突然変異細胞でのみ検出され、野生型K−Ras細胞では検出されなかった(図10D)。ケルセチン及びモリンの類似した構造が、本発明のスクリーニングシステムの信頼性を高めた(図10E)。
【0082】
また、スネイル媒介p53抑制に対する化学物質の効果を検討し、図10E及び図10Fのように、スネイルの共形質感染によるp53の減少が、#3及び#9による化学物質処理によって遮断された。このような結果からp53−スネイルの相互作用の遮断が、p53発現を回復することができる。
【0083】
また、トリパンブルー染色を用いて細胞増殖に対するこれら化学物質の効果を検討した。A549で、これら化学物質が細胞増殖を明らかに抑制した一方、MKN45(K−Ras野生型細胞)に対する抗増殖効果を示していない(図11C)。
【0084】
そして、フェルラ酸は、K−Ras突然変異細胞で細胞死を誘発した(図11D)。また、ケルセチン(#2)は、スネイル−p53相互作用抑制剤として確認された。
[p53のDNA結合ドメインのエンドサイトーシスを利用したK−Ras突然変異細胞の特異的に薬物伝達方法の考察]
<実施例5>
【0085】
1.p53の核から細胞質への移動機転の検討
GST−pull down分析のために、先ず人間スネイル及びp53再組合タンパク質を知られた方法によって製造した(Neoplasia 11:1−10,2009)。培地及び前細胞ライセートでp53及びスネイル間の直接的な結合を検討するために、アガロース−ビーズ接合GSTまたはGST−スネイルを細胞ライセートまたは培養培地と4℃で2時間培養した。PBS及びRIPAで洗浄後、沈降されたタンパク質を以前方法と同様にSDS−PAGE及びWBに適用した。
2.実験結果
【0086】
p53の核から細胞質への移動配列で変異を起こすp53 NESに対するスネイルの効果を検討した結果、スネイルは、p53 NES発現を減少させ(図13A)、レプトマイシンB(LMB;核から細胞質への移動遮断剤)は、スネイル媒介−p53減少を遮断することができなかった(図13B及び図13C)。スネイルによるp53減少がリアルタイムで確認された(図13D)。培養培地でp53の発現を検討した結果、2時間以内に培地でp53の発現を検出することができた(図13E)。そして、p53は、スネイルまたはK−Ras形質感染細胞で小嚢のような構造でサイトゾルから検出され、最終的に、細胞外の領域から検出された(図13F)。一方、スネイルは、p53と共にサイトゾルの小嚢に存在した(図17A)。
【0087】
K−Ras突然変異細胞でp53の分泌を検討するために、培養培地及び細胞ライセートでスネイル−GSTを利用したGST−Pull down分析を行った。スネイル関連p53が、あらゆる細胞ライセートから検出されても、K−Ras突然変異膵膓癌細胞株でのみ培地p53を観察することができた(図13G)。また、K−Ras突然変異細胞の培養培地でスネイル−pull downなしに培地p53を観察することができた(図13H)。
【0088】
p53は、小嚢のような輸送を通じて減少するために、ノコダゾール(nocodazole;Noc)による細胞骨格ネットワークを妨害してp53の発現を検討した結果、Noc処理は、スネイルまたはK−Ras媒介p53減少を遮断することができた(図14A)。Noc−処理細胞でp53のサイトゾル小嚢性染色にもかかわらず、細胞形象は対照群と区別されるように変化された(図14B)。Nocは、p53減少を遮断することができた(図14C)。Noc−処理によって培地p53が消えた一方、p53は小嚢として細胞質で蓄積された(図14D及び図14E)。Aph/Nocは、p53の減少を遮断することができ、これは、LMBによって得られることができなかった(図14F及び図14G)。
<実施例6>
1.細胞外p53及びスネイルの態様検討
【0089】
1)組職分析
正常及び腫瘍が一対を成す胆管癌及び肝組職を順天郷医薬センターから得た。組職をdeep freezerで素早く凍結し、該凍結された組職を切って0.5mgの組職を0.25mL血清フリー培地で37℃で30分間培養して組職液を遊離した。培養後、培養培地を集めて0.5mL 100% EtOHで沈降させた。該沈降された物質をRIPAを用いて溶解させ、以前方法と同様にSDS−PAGE及びWB分析を行った。また、同じ方法で同一培養培地を得てp53 Abを検出した。
【0090】
2)p53 ELISA分析
p53の調査のために、製造者の指針(Assay Design)によってELISAを行った。すなわち、0.2mL組職培養培地を各ウェルに加えて検出Abと培養した。洗浄緩衝液で洗浄した後、0.2mL基質ゾル及び0.05mL停止液を加えた。
【0091】
3)血液試料でスネイルAb検出
人間血液試料を順天郷大学(膵膓癌及び胆石患者)から得て、釜山大学の医薬センター(肺癌)から得た。正常血液試料を志願者または癌ではない患者から集めた。血清は遠心分離して集め、使用前まで−70℃で保管された。3μlの血清をGST−タンパク質と事前承認後、アガロース−接合GST−スネイル−Nと培養した。沈降されたGST−スネイル−Ab複合体は、RIPA及びSDS試料緩衝液で溶解し、以前方法と同様にSDS−PAGEに適用した。PVDF膜に移した後、タンパク質を抗−人間Ab及び抗−GST Abと培養した。
2.実験結果
【0092】
先ず、A549及びMKN45細胞に組替えp53を処理し、これらの位置を調査した。培地から回収された対照群タンパク質(His−lamin A)と比較して、His−p53は、A549の前細胞ライセートで発現された(図15A)。そして、His−p53は、MKN45細胞及びその培養培地で完全に除去された(図15A)。
【0093】
より具体的な検討のために、本発明では、新鮮な培地、A549−培養培地、PC3及びHCT116を用いて組替えp53を培養した。培地で回収されたHis−laminAと比較して、p53−Hisは、HCT116の前細胞ライセートから検出された。そして、p53はPC3だけではなく、A549培養培地で回収されなかった。このような結果からp53は、培養された細胞から分泌されるプロテアーゼによって消化され、K−Ras突然変異細胞によって再吸収されるということが分かる。
【0094】
また、プロテアーゼ阻害剤(PMSF)及びエンドサイトーシス阻害剤(Brefeldin A;BFA)をA549及びCapan−1に処理した。細胞内p53発現に対する未反応にもかかわらず、これら化合物は、培地p53の発現を増加させた(図15B)。BFAは、A549前細胞ライセートで組替えp53の局在を遮断した(図15C)。
【0095】
分泌されたスネイルの態様を確認するために、再組合スネイルで処理してp53の目的地を比較した。p53中間領域は、A549のWCLから回収され、これは、BFAによって抑制された。しかし、MKN45に処理された組替えp53は、培地及びWCLで消えた。このような結果から分泌されたp53は、セリンプロテアーゼだけではなく、MMPのような他種のプロテアーゼによって消化されると判断された。一方、スネイルは、培地から回収され、プロテアーゼ阻害剤なしに再組合スネイルを回収した。このような結果からスネイルは、プロテアーゼ媒介消化だけではなく、エンドサイトーシスに抵抗性があると判断された。
【0096】
p53−エンドサイトーシスに対するK−Rasの効果を検討した結果、組替えp53は、K−Ras形質感染細胞によって選択的に除去された。p53中間領域が、K−Ras突然変異細胞によって選択的に再び入ることができるならば、このような特徴は、K−Ras突然変異細胞に対する化合物の伝達のために非常に有用である。
【0097】
それを確認するために、PI(propidium iodine;Red dye、50ug/mL)及びHis−p53(2ug/mL)をA549及びMKN45に処理した結果、PIのみ処理した場合には、二つの細胞株で細胞として蓄積されなかったが、PI及びp53を共に処理すれば、A549の細胞内にPIの蓄積を観察することができた(図15E、図16Aないし図16B)。しかし、MKN45では、このような観察ができなかった。このような結果からp53は、K−Ras特異的薬物伝達システムとして有用であると判断された。
【0098】
スネイル媒介分泌されたp53によってp53の自動Abが生成されるか否かを検討するために、組職液でのp53及びスネイルの発現態様を調査した結果、胆管癌では、p53及びスネイルが検出されたが、肝癌細胞種及び非癌組職液では検出されなかった(図17A及び図17B)。図17Cのように、ELISAを用いて組職液でのp53の存在を確認した結果、抗−p53 Ab及び抗−スネイルAbは胆管癌では検出されたが、肝癌細胞種では検出されなかった(図17D及び図17E)。また、膵膓癌または胆管癌患者の血清でp53及びスネイルに対する自動Abを調査した結果、抗−p53 Abは、癌と関連性を示していない。このような結果からp53自動Abは、癌の状態と関連しないものと判断され、分泌されたp53は、迅速にプロテアーゼ及びエンドサイトーシスによって除去された(図15A)。
【0099】
【表1】
【0100】
一方、スネイルは、プロテアーゼ及びエンドサイトーシスに対する抵抗性を表わした(図15D)。したがって、血清でスネイル自動Abを測定した。スネイルAbの発現は、膵膓癌患者の血清及び胆石患者の血清から検出された(図15F)。一方、スネイルAbは、正常健康人では検出されなかった(図15G)。また、肺癌患者の血清でも、スネイルAbが観察された(図18)。したがって、スネイルAbの存在が癌診断マーカーとして非常に有用であると判断された。
【0101】
【表2】
【0102】
[スネイル−p53間の結合阻害剤の同定]
<実施例1>
1−1.2−メチルチオ−1,4−ナフトキノン(2−Methylthio−1,4−naphthoquinone、1a)の合成
【0103】
1口100mlのラウンドフラスコに、1,4−ナフトキノン(1,4−naphthoquinone)0.617mMをメタノール30mlに溶解させた後、ナトリウムチオメトキシド(sodium thiomethoxide)1.54mMを入れて一晩中撹拌させた。反応混液に飽和塩化ナトリウム溶液50mlを入れてクロロホルム50mlで3回抽出した後、有機層を無水芒硝で脱水させた後、濾過した。該濾過した濾液を減圧濃縮して得た残渣をメタノールで再結晶して黄色結晶の表題化合物を得た。
【0104】
収率:14.0%、融点:185−186℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.07(m、2H)、7.78−7.70(m、2H)、6.58(s、1H)、2.40(s、3H)、m/z 205.1(M+H)+。
1−2.2−エチルチオ−1,4−ナフトキノン(2−Ethylthio−1,4−naphthoquinone、1b)の合成
【0105】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、エチルメルカプタン(Ethylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0106】
収率:40.7%、融点:135−136℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.08(m、2H)、7.78−7.69(m、2H)、6.62(s、1H)、2.87(q、J=7.2Hz、2H)、1.44(t、J=7.2Hz、3H)、m/z 219.1(M+H)+。
1−3.2−プロピルチオ−1,4−ナフトキノン(2−Propylthylthio−1,4−naphthoquinone、1c)の合成
【0107】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、プロピルメルカプタン(Propylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0108】
収率:33.9%、融点:118−119℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.07(m、2H)、7.77−7.68(m、2H)、6.61(s、1H)、2.82(t、J=7.6Hz、2H)、1.86−1.77(m、2H)、1.11(t、J=7.2Hz、3H)、m/z 233.0(M+H)+。
1−4.2−ブチルチオ−1,4−ナフトキノン(2−Butylthio−1,4−naphthoquinone、1d)の合成
【0109】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、ブチルメルカプタン(Butylmercaptan)を使って黄色結晶2−ブチルチオ−1,4−ナフトキノン(1d)を得た。その収率及び物性は、下記の通りであった。
【0110】
収率:33.9%、融点:97−98℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.07(m、2H)、7.77−7.68(m、2H)、6.61(s、1H)、2.84(t、J=7.2Hz、2H)、1.80−1.72(m、2H)、1.57−1.48(m、2H)、0.98(t、J=7.6Hz、3H)、m/z 247.1(M+H)+。
1−5.2−ペンチルチオ−1,4−ナフトキノン(2−Pentylthio−1,4−naphthoquinone、1e)の合成
【0111】
前記合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、ペンチルメルカプタン(pentylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0112】
収率:15.3%、融点:111−112℃、1H−NMR(CDCl3、400MHz):δ 8.11−8.06(m、2H)、7.77−7.68(m、2H)、6.60(s、1H)、2.83(t、J=7.6Hz、2H)、2.17−1.74(m、2H)、1.51−1.33(m、4H)、0.93(t、J=7.2Hz、3H)、m/z 261.2(M+H)+。
1−6.2−ヘキシルチオ−1,4−ナフトキノン(2−Hexylthio−1,4−naphthoquinone、1e)の合成
【0113】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、ヘキシルメルカプタン(hexylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0114】
収率:15.0%、融点:101−102℃、1H−NMR(CDCl3、400MHz):δ 8.13−8.08(m、2H)、7.77−7.69(m、2H)、6.61(s、1H)、2.83(t、J=7.6Hz、2H)、1.81−1.73(m、2H)、1.56−1.46(m、2H)、1.35−1.31(m、4H)、0.91(t、J=6.8Hz、3H)、m/z 275.3(M+H)+。
1−7.2−ヘプチルチオ−1,4−ナフトキノン(2−Heptylthio−1,4−naphthoquinone、1g)の合成
【0115】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、ヘブチルメルカプタン(heptylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0116】
収率:46.4%、融点:114−115℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.07(m、2H)、7.77−7.68(m、2H)、6.61(s、1H)、2.83(t、J=14.8Hz、2H)、1.77(quint、J=7.6Hz、2H)、1.52−1.45(m、2H)、1.35−1.29(m、6H)、0.90(t、J=6.8Hz、3H)、m/z 289.2(M+H)+。
1−8.2−オクチルチオ−1,4−ナフトキノン(2−Octylthio−1,4−naphthoquinone、1h)の合成
【0117】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、オクチルメルカプタン(octylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0118】
収率:76.8%、融点:114−115℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.10(m、2H)、7.78−7.70(m、2H)、6.61(s、1H)、2.84(t、J=7.6Hz、2H)、1.77(quint、J=7.6Hz、2H)、1.49−1.47(m、2H)、1.35−1.29(m、8H)、0.89(t、J=6.8Hz、3H)、m/z 304.5(M+H)+。
1−9.2−ノニルチオ−1,4−ナフトキノン(2−Nonylthio−1,4−naphthoquinone、1i)の合成
【0119】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、ノニルメルカプタン(nonylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0120】
収率:87.4%、融点:105−106℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.07(m、2H)、7.77−7.68(m、2H)、6.61(s、1H)、2.83(t、J=7.6Hz、2H)、1.77(quint、J=7.6Hz、2H)、1.51−1.45(m、2H)、1.33−1.29(m、10H)、0.89(t、J=6.4Hz、3H)、m/z 317.5(M+H)+。
1−10.2−デシルチオ−1,4−ナフトキノン(2−Decylthio−1,4−naphthoquinone、1j)の合成
【0121】
前記実施例1−1の合成方法と同じ方法で製造するが、単にナトリウムチオメトキシドの代りに、デシルメルカプタン(Decylmercaptan)を使って黄色結晶の表題化合物を得た。その収率及び物性は、下記の通りであった。
【0122】
収率:87.4%、融点:101−102℃、1H−NMR(CDCl3、400MHz):δ 8.12−8.08(m、2H)、7.77−7.68(m、2H)、6.61(s、1H)、2.83(t、J=7.2Hz、2H)、1.77(quint、J=7.6Hz、2H)、1.50−1.42(m、2H)、1.33−1.29(m、12H)、0.88(t、J=6.4Hz、3H)、m/z 331.1(M+H)+。
<実施例2>
2−1.2−メチルアミノ−5,8−ジメトキシ−1.4−ナフトキノン(2−Methylamino−5,8− dimethoxy−1,4 −naphthoquinone、5a)の合成
【0123】
1口100mlのラウンドフラスコに、前から製造された5,8−ジメトキシ−1,4−ナフトキノン(5,8−dimethoxy−1,4−naphthoquinone、4)を0.45mMをメタノール30mlに溶解させた後、メチルアミン(Methylamine)0.687mmolを入れて室温で3時間撹拌させた。反応混液に重クロム酸ナトリウム(Sodim dichromate)0.64mMと硫酸(sulfuric acid)0.18mMとを水に溶解させてゆっくり滴加した後、室温で3分間撹拌した。反応混液に飽和塩化ナトリウム溶液50mlを入れてクロロホルム50mlで3回抽出した後、有機層を合わせて無水芒硝で脱水させた後、濾過した。該濾過した濾液を減圧濃縮して得た残渣をシリカゲルカラム分離を通じてさび色の表題化合物を得た。表題化合物の収率及び物性は、下記の通りであった。
【0124】
収率:56.7%、融点:203−204℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6MHz)、7.19(d、J=9.2MHz、1H)、5.75(BR、1H)、5.60(s、1H)、3.96(s、3H)、3.94(s、3H)、2.87(d、J=5.2MHz、3H)、m/z 248(M+H)+。
2−2.2−エチルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Ethylamino−5,8−dimethoxy−1,4−naphthoquinone、5b)の合成
【0125】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、エチルアミン(ethylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5b)を製造した。その収率及び物性は、下記の通りであった。
【0126】
収率:23.6%、融点:172−173℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.2Hz、1H)、7.19(d、J=9.6Hz、1H)、5.63(BR、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.09(q、2H)、1.29(t、J=7.2Hz、3H)、m/z 262.1(M+H)+。
2−3.2−プロピルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Propylamino−5,8−dimethoxy−1,4−naphthoquinone、5c)の合成
【0127】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、プロピルアミン(propylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5c)を製造した。その収率及び物性は、下記の通りであった。
【0128】
収率:46.5%、融点:175−176℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.2Hz、1H)、7.19(d、J=9.2Hz、1H)、5.72(BR、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.09(q、2H)、1.68(J=6.8Hz、2H)、0.99(t、J=7.6Hz、3H)、m/z 276(M+H)+。
2−4.2−ブチルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Butylamino−5,8−dimethoxy−1,4−naphthoquinone、5d)の合成
【0129】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、ブチルアミン(butylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5d)を製造した。その収率及び物性は、下記の通りであった。
【0130】
収率:46.2%、融点:104−105℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.19(d、J=9.6Hz、1H)、5.70(BR、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.64(p、2H)、1.46−1.38(m、2H)、0.95(t、J=7.2Hz、3H)、m/z 290(M+H)+。
2−5.2−ペンチルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Pentylamino−5,8−dimethoxy−1,4−naphthoquinone、5e)の合成
【0131】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、ペンチルアミン(pentylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5e)を製造した。その収率及び物性は、下記の通りであった。
【0132】
収率:55.9%、融点:102−103℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.2MHz、1H)、7.19(d、J=9.2Hz、1H)、5.70(BR、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.29(t、J=7.2Hz、3H)、m/z 303.6(M+H)+。
2−6.2−ヘキシルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Hexylamino−5,8−dimethoxy−1,4−naphthoquinone、5f)の合成
【0133】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、ヘキシルアミン(Hexylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5f)を製造した。その収率及び物性は、下記の通りであった。
【0134】
収率:47.3%、融点:83−84℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.18(d、J=9.6Hz、1H)、5.69(BR、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.66−1.58(m、4H)、1.32−1.30(m、4H)、0.89(t、J=6.8Hz、3H)、m/z 318(M+H)+。
2−7.2−ヘブチルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Heptylamino−5,8− dimethoxy−1,4−naphthoquinone、5g)の合成
【0135】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、ヘブチルアミン(Heptylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5g)を製造した。その収率及び物性は、下記の通りであった。
【0136】
収率:41.8%、融点:74−75℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.33(d、J=9.6Hz、1H)、7.18(d、J=9.6Hz、1H)、5.69(BR、1H)、5.60(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.66−1.61(m、2H)、1.35−1.29(m、8H)、0.89(t、J=6.4Hz、3H)、m/z332(M+H)+。
2−8.2−オクチルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Octylamino−5,8−dimethoxy−1,4−naphthoquinone、5h)の合成
【0137】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、オクチルアミン(octylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5h)を製造した。その収率及び物性は、下記の通りであった。
【0138】
収率:45.1%、融点:81−82℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.18(d、J=9.6Hz、1H)、5.69(br、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.68−1.61(m、2H)、1.41−1.20(m、10H)、0.88(t、J=6.4Hz、3H)、m/z 346(M+H)+。
2−9.2−ノニルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Nonylamino−5,8−dimethoxy−1,4−naphthoquinone、5i)の合成
【0139】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、ノニルアミン(nonylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5i)を製造した。その収率及び物性は、下記の通りであった。
【0140】
収率:44.0%、融点:85−86℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.18(d、J=9.6Hz、1H)、5.69(br、1H)、5.61(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(m、2H)、1.66−1.61(m、2H)、1.41−1.20(m、12H)、0.88(t、J=6.4Hz、3H)、m/z 360(M+H)+。
2−10.2−デシルアミノ−5,8−ジメトキシ−1,4−ナフトキノン(2−Decylamino−5,8−dimethoxy−1,4−naphthoquinone、5j)の合成
【0141】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、デシルアミン(decylamine)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5j)を製造した。その収率及び物性は、下記の通りであった。
【0142】
収率:17.6%、融点:86−87℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.2Hz、1H)、7.18(d、J=9.2Hz、1H)、5.69(br、1H)、5.60(s、1H)、3.96(s、3H)、3.94(s、3H)、3.11(q、2H)、1.66−1.61(m、2H)、1.40−1.20(m、14H)、0.88(t、J=6.4Hz、3H)、m/z 374(M+H)+。
2−11.2−(2−ヒドロキシエチルチオ)−5,8−ジメトキシ−1,4−ナフトキノン[2−(2−Hydroxyethylthio)−5,8−dimethoxy−1,4−naphthoquinone、5k]の合成
【0143】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、2−メルカプトエタノール(2−mercaptoethanol)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5k)を製造した。その収率及び物性は、下記の通りであった。
【0144】
収率:59.5%、融点:117−118℃、1H−NMR(CDCl3、400MHz):δ 7.34(d、J=9.6Hz、1H)、7.28(d、J=9.6Hz、1H)、6.61(s、1H)、3.96(s、6H)、3.93(t、J=6.4Hz、2H)、3.05(t、J=6.4Hz、2H)、m/z 316.9(M+Na)+。
2−12.2−(3−ヒドロキシプロピルチオ)−5,8−ジメトキシ−1,4−ナフトキノン[2−(3−Hydroxypropylthio)−5,8−dimethoxy−1,4−naphthoquinone、5l]の合成
【0145】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、3−メルカプト−1−プロパノール(3−mercaptopropanol)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5l)を製造した。その収率及び物性は、下記の通りであった。
【0146】
収率:69.9%、融点:125−126℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.6Hz、1H)、7.27(d、J=9.6Hz、1H)、6.51(s、1H)、3.96(s、6H)、3.81(t、J=6.4Hz、2H)、2.91(t、J=7.2Hz、2H)、1.99(m、2H)、m/z 331.1(M+Na)+。
2−13.2−(4−ヒドロキシブチルチオ)−5,8−ジメトキシ−1,4−ナフトキノン[2−(4−Hydroxybutylthio)−5,8−dimethoxy−1,4−naphthoquinone、5m]の合成
【0147】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、4−メルカプトブタノール(4−mercaptobutanol)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5m)を製造した。その収率及び物性は、下記の通りであった。
【0148】
収率:64.0%、融点:122−123℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.6Hz、1H)、7.29(d、J=10.0Hz、1H)、6.46(s、1H)、3.96(s、3H)、3.95(s、3H)、3.71(t、J=6.4Hz、2H)、1.87−1.81(m、2H)、1.78−1.50(m、2H)、m/z 345.1(M+Na)+。
2−14.2−(6−ヒドロキシヘキシルチオ)−5,8−ジメトキシ−1,4−ナフトキノン[2−(6−Hydroxyhexylthio)−5,8−dimethoxy−1,4−naphthoquinone、5n]の合成
【0149】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、6−メルカプトヘキサノール(6−mercaptohexanol)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5n)を製造した。その収率及び物性は、下記の通りであった。
【0150】
収率:38.2%、融点:87−88℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.6Hz、1H)、7.27(d、J=9.6Hz、1H)、6.45(s、1H)、3.96(s、6H)、3.66(t、J=6.4Hz、2H)、2.76(t、J=7.6Hz、2H)、1.78−1.12(m、2H)、1.61−1.25(m、6H)、m/z 372.9(M+Na)+。
2−15.3−(5,8−ジメトキシ−1,4−ジオキソナフタレン−2−イルチオ)プロパン酸[3−(5,8−dimethoxy−1,4−dioxo−naphthalen−2−ylthio)propanoic acid、5o]の合成
【0151】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、3−メルカプトプロピオン酸(3−mercaptopropionic acid)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5o)を製造した。その収率及び物性は、下記の通りであった。
【0152】
収率:80.6%、融点:208−209℃、1H−NMR(CDCl3、400MHz):δ 7.35(d、J=9.2Hz、1H)、7.28(d、J=13.6Hz、1H)、6.51(s、1H)、3.97(s、3H)、3.96(s、3H)、3.07(t、J=7.2Hz、2H)、2.81(t、J=7.2Hz、2H)、m/z 348.4(M+Na)+。
2−16.11−(5,8−ジメトキシ−1,4−ジオキソ−ナフタレン−2−イルチオ)ウンデカン酸[11−(5,8−dimethoxy−1,4−dioxo−naphthalen−2−ylthio)undecanoic acid、5p]の合成
【0153】
前記実施例2−1のラウンドフラスコにメチルアミンの代りに、11−メルカプトウンデカン酸(11−mercaptoundecanoic acid)を使用する点を除いては、実施例2−1の反応と同じ工程を行って表題化合物(5p)を製造した。
【0154】
収率:77.9%、融点:146−147℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.6Hz、1H)、7.27(d、J=9.6Hz、1H)、6.46(s、1H)、3.96(s、3H)、3.95(s、3H)、3.34(t、J=7.2Hz、2H)、2.75(t、J=2.7Hz、2H)、2.41−2.32(m、5H)、2.06−2.00(m、2H)、1.76−1.56(m、5H)、1.48−1.39(m、2H)、0.97−0.88(m、2H)、m/z 435(M+H)+。
<実施例3>
3−1.5,8−ジメトキシ−2−[3−オキソ−3−(4−フェニルピペリジン−4−イル)プロピルチオ]−1,4−ナフトキノン[5,8−dimethoxy−2−(3−oxo−3−(4−phenylpiperazin−1−yl)propylthio)naphthalene−1,4−dione、6a]の合成
【0155】
1口100mlのラウンドフラスコに、前から製造された3−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)プロパン酸、0.163mMをクロロホルム40mlに溶解させた後、N−(3−ジメチルアミノプロピル)−N´−エチルカルボジイミド塩酸塩(EDC)0.26mMと4−フェニルピペリジン(4−phenylpiperidine)0.26mMとを入れて一晩撹拌させた。反応混液に1N塩酸溶液を入れた後、室温で3分間撹拌した。再び飽和塩化ナトリウム溶液50mlを入れてクロロホルム50mlで3回抽出した後、有機層を合わせて無水芒硝で脱水させた後、濾過した。該濾過した濾液を減圧濃縮して得た残渣をシリカゲルカラム分離を通じてさび色の表題化合物(6a)を得た。表題化合物の収率及び物性は、下記の通りであった。
【0156】
収率:73.5%、融点:94−95℃、1H−NMR(CDCl3、400MHz):δ 7.34−7.28(m、5H)、7.19(d、J=7.6Hz)、6.26(s、1H)、3.95(s、3H)、3.94(s、3H)、3.14(t、4H)、2.79−2.74(q、4H)、2.67(t、J=12Hz、1H)、1.89(t、J=12.4Hz、2H)、1.65(t、J=12.4Hz、2H)、m/z 466.3(M+H)+。
<実施例4>
4−1.イソブチル−11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)−ウンデカノアート[Isobutyl−11−(5,8−dimethoxy−1,4−dioxo−1,4−dihydronaphthalene−2−ylthio)undecanoate、7a]の合成
【0157】
1口100mlのラウンドフラスコに、前から製造された11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)ウンデカン酸(5p)1.15mMをクロロホルム60mlに溶解させた後、N−(3−ジメチルアミノプロピル)−N´−エチルカルボジイミド塩酸塩(EDC)1.38mMとイソブチルアルコール(isobutylalcohol)1.38mMとを入れて一晩撹拌させた。反応混液に1N塩酸溶液を入れた後、室温で3分間撹拌した。再び飽和塩化ナトリウム溶液50mlを入れてクロロホルム50mlで3回抽出した後、有機層を合わせて無水芒硝で脱水させた後、濾過した。該濾過した濾液を減圧濃縮して得た残渣をシリカゲルカラム分離を通じてさび色の表題化合物(7a)を得た。表題化合物の収率及び物性は、下記の通りであった。
【0158】
収率:52.4%、融点:58−59℃、1H−NMR(CDCl3、400MHz):δ 7.32(d、J=9.6Hz、1H)、7.26(d、J=9.6Hz、1H)、6.44(s、1H)、3.96(s、3H)、3.95(s、3H)、3.85(d、J=6.8Hz、2H)、2.75(t、J=7.6Hz、2H)、2.31(t、7.6Hz、2H)、1.96−1.89(m、1H)、1.72−1.68(m、4H)、1.29−1.25(m、12H)、0.94(s、3H)、0.92(s、3H)、m/z 491(M+H)+。
4−2.11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)−N−イソブチルウンデカンアミド[11−(5,8−dimethoxy−1,4−dioxo−1,4−dihydronaphthalene−2−ylthio)−N−isobutyl undecanamide、7b]の合成
【0159】
前記実施例4−1のラウンドフラスコにイソブチルアルコールの代りに、イソブチルアミンを使用する点を除いては、実施例4−1の反応と同じ工程を行って下記物性値を有する表題化合物(7b)を得た。収率及び物性は、下記の通りであった。
【0160】
収率:77.9%、融点:74−75℃、1H−NMR(CDCl3、400MHz):δ 7.33(d、J=9.6Hz、1H)、7.27(d、J=9.2Hz)、6.44(s、1H)、5.55(s、1H)、3.96(s、3H)、3.95(s、3H)、3.10(t、J=6.6Hz、2H)、2.75(t、J=7.6Hz、2H)、2.17(t、J=7.4Hz、2H)、1.78−1.7(m、2H)、1.63(m、2H)、1.45(m、1H)、0.92(s、3H)、0.90(s、3H)、m/z 490.0(M+H)+。
<実施例5>
5−1.イソブチル11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルスルフィニル)ウンデカノアート[isobutyl11−(5,8−dimethoxyl−1,4−dioxo−1,4−dihydronaphthalene−2−yl sulfinyl)undecanoate、8]の合成
【0161】
1口100mlのラウンドフラスコに、前から製造されたイソブチル11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロキシナフタレン−2−イルチオ)−ウンデカノアート0.163mM(7a)をジクロロメタン30mlに溶解させた後、3−クロロ過安息香酸(3−chloroperoxybbenzoic acid)0.196mMを入れて2時間撹拌させた。反応混液に重炭酸ナトリウム溶液を入れた後、室温で3分間撹拌した。再び飽和塩化ナトリウム溶液50mlを入れてクロロホルム50mlで3回抽出した後、有機層を合わせて無水芒硝で脱水させた後、濾過した。該濾過した濾液を減圧濃縮して得た残渣をシリカゲルカラム分離を通じて赤色の表題化合物(8)を得た。表題化合物の収率及び物性は、下記の通りであった。
【0162】
収率:45.8%、融点:92−93℃、1H−NMR(CDCl3、400MHz):δ 7.41(d、J=9.6Hz、1H)、7.36(d、J=9.6Hz、1H)、7.31(s、1H)、3.99(s、3H)、3.98(s、3H)、3.85(d、J=6.8Hz、1H)、3.28−3.21(m、1H)、2.96−1.89(m、2H)、1.69−1.59(m、4H)、1.41−1.23(m、12H)、0.93(d、J=6.8MHz、6H)、m/z 507(M+H)+。
【0163】
前記方法で合成した化合物群を用いてスネイル−p53の結合抑制効果を次の実験を通じて確認した。
<実験例1>p53活性回復及びターゲット遺伝子誘導確認
【0164】
前記実施例1〜5で合成された化合物が、スネイル−p53結合阻害効果があるか、細胞レベルで効果的であるか否かを立証するために、前記化合物をK−Ras突然変異型癌細胞株HCT116に処理し、ウェスタンブロットを用いて、この結果を確認した。
【0165】
K−Rasは、スネイルの活性を抑制してp53を誘導する作用をする遺伝子であって、前記細胞株HCT116は、K−Rasの突然変異によってスネイルが常に発現してp53と結合した後、p53が正常に作動しないようにする役割を果たす。前記K−Ras突然変異型癌細胞株HCT116にスネイル−p53結合を抑制する化合物を処理すれば、p53が正常作用することによって、発現量が増加してp53のターゲット遺伝子が誘導される。
【0166】
優先的に、p53とターゲット遺伝子であるp21との発現を確認するために、ウェスタンブロットを行った。本発明で使ったあらゆる細胞株は、ATCCから購入し、10%FBSを含有したRPMI−1640またはDMEMで維持された。RIPA緩衝液でタンパク質を抽出し、典型的なSDS−PAGE及びゲル移動方式を通じて細胞溶出物でローディングされたメンブレンを準備した。5%非脂肪乾燥油でブロッキングした後、各遺伝子に該当する抗体を用いて典型的なウェスタンブロット手続きを進行した。本発明で使われた抗体は、Cell signaling、SantaCruzから購買した。p53の活性は、p53のターゲット遺伝子であるp21の発現量を通じて確認することができた。図1は、実施例1〜5で合成した化合物の一部を用いてK−Ras突然変異型癌細胞株であるHCT116細胞でウェスタンブロットを行った結果を表わす図及びそれを数値化して表わしたグラフであり、表3は、この数値を表わした図表である。
【0167】
【表3】
【0168】
前記化合物をK−Ras突然変異型HCT116癌細胞株に処理した時、対照群(DMSO control)と比較して、前記化合物を10μM濃度で処理した所でp53を2〜5倍程度で、p21は、2〜8倍程度で発現量が大きく増加する化合物があるということを確認することができた(図20及び表3参考)。前記化合物が、p53とp53とのターゲット遺伝子であるp21の発現量を増幅させたことは、スネイルが常に活性化されている状態のK−Ras突然変異細胞株であるHCT116細胞でスネイルの役割が、前記化合物を通じて抑制されてスネイル−p53の結合阻害状態が維持されるということを表わす。
<実験例2>スネイル−p53結合阻害効果の確認
【0169】
次いで、p53とp21とを同時に誘導しながら発現量が多かった前記化合物のうちから二つの化合物5oまたは7aを選択して、K−Ras、スネイル及びp53の関係を確認するために使う。スネイル−p53の結合が阻害されるということを立証するために、前記二つの化合物5o及び7aを用いてGSTプルダウン分析(GST pull down assay)を行って確認した。
【0170】
GSTプルダウン分析は、二つのタンパク質の結合程度を確認することができる方法でGSTが融合されたスネイルタンパク質とp53再組合タンパク質とを製造して、前記化合物5oあるいは7aを共に処理してGST−スネイルタンパク質とp53タンパク質との結合程度を確認した。前記化合物の比較群としては、Nutlin−3を使った。Nutlin−3は、p53の陰性調節因子であるMDM2とp53との結合を遮断する物質で多くの腫瘍細胞で過発現されるタンパク質として知られている。MDM2が過発現されれば、p53のタンパク分解が誘導されてp53のターゲット遺伝子を通じて起こるアポトーシス(apoptosis)などの反応を抑制し、細胞の抗増殖効果などを誘導する。代表的なp53の調節因子であるが、K−Ras突然変異性疾患や突然変異p53を有した疾患には作用しない。
【0171】
GSTプルダウン分析のために、3つの人間スネイル断片(残基1−90、91−112及び113−264)及びp53断片(1−93及び93−292)をGST−融合タンパク質として大膓菌で発現し、各断片をGSH−アガロースにローディングし、洗浄した後、20mM還元グルタチオンを含有した緩衝液を用いて溶出した。溶出分画を陰イオン−交換クロマトグラフィー(HitrapQ)を用いて錠剤した。再組合人間p53タンパク質(残基94−292)をベクターpET28Aを用いて大膓菌で発現させた。前記ベクターpET28Aは、C末端にヘキサ−ヒスチジンタグを含有した。Ni−NTA親和力及び大きさ排除クロマトグラフィー(Superdex 200)を用いてヒスチジンが付いたp53タンパク質を錠剤した。p53及びスネイル間の直接結合を検討するために、アガロース−ビーズ接合GSTまたはGST−スネイルを4℃で45分間PBSでHis−p539(ヒスチジン−p53)と培養した。PBSで洗浄した後、沈澱されたタンパク質をSDS−PAGE及びウェスタンブロットを行った。
【0172】
図21に示したように、GSTプルダウン分析結果を確認して見れば、対照群(c)の場合、GST−スネイルとp53とが多量に結合されていることを確認することができる。Nutlin−3もMDM2を調節してp53活性を誘導する物質であるために、スネイル−p53の結合を阻害できず、対照群と類似している結合程度を表わしている。一方、前記二つの化合物5oと7aとを使った分画では、GST−スネイルタンパク質にp53が結合される程度が確実に減ることを確認することができ、前記化合物が、スネイル−p53の結合を抑制したということを立証することができた。
<実験例3>K−Ras依存的なp53誘導能の確認
【0173】
次いで、前記化合物が、正常細胞には作用せず、K−Ras突然変異癌細胞にのみ選択的に作用して死滅させる能力があるか否かを確認するために、細胞生存率を通じて前記二つの化合物5oと7aとに対する細胞毒性及び死滅効果をトリパンブルー溶液を用いて細胞数を測定して確認した。前記化合物のp53誘導能が、K−Rasと関係があるかを検討するために、K−Ras突然変異癌細胞株であるA549とHCT116、K−Ras野生型癌細胞株であるMKN45とを利用した。
【0174】
表4は、それぞれの細胞株に対する死滅率を数値化して表わした図表であり、図22は、死滅率に対する数値を表わしたグラフである。
【0175】
【表4】
【0176】
細胞死滅率を通じて表われた結果は、前記化合物5oが、K−Ras突然変異癌細胞株であるA549とHCT116とから10μM程度でそれぞれ51%と57%程度の死滅率を示し、7aも同一濃度10μMでほぼ34%と44%程度の死滅率を示した(表4及び図22参照)。一方、K−Ras野生型癌細胞株であるMKN45では、同一濃度10μMで前記化合物5oは、15%程度の死滅率を、前記化合物7aは、ほとんど1%の死滅率を示してK−Ras突然変異型癌細胞株と確実に異なる結果を表わした。これにより、前記化合物が、K−Rasが正常に作用しない癌細胞株でのみ選択的に作用して高い死滅率を表わすことを確認することができた。これは、前記化合物が、K−Rasが損傷されている癌細胞にのみ選択的に作用することができ、K−Rasが損傷されて癌にかかった患者に有用であるということを暗示する。一方、Nutlin−3は、K−Ras突然変異癌細胞株であるA549とHCT116細胞株で同一濃度10μMでそれぞれ19%と32%死滅率を表わし、K−Ras野生型細胞株であるMKN45では、同一濃度10μMで細胞死滅率が38%程度でさらに高くなるなどの一括的ではない類型を表わすことを確認することができた。このように、Nutlin−3は、K−Rasが正常に作用をするか、突然変異状態であるものに関わらずに或る有意な反応を表わさず、K−Rasに選択的な反応を表わさないということを確認することができた。これで、前記化合物5oと7aとがNutlin−3と異なってK−Rasに選択的作用効果に優れるということを立証した。
<実験例4>p53のターゲット遺伝子の活性誘導
【0177】
次いで、突然変異p53がある時の前記化合物5oの効果を確認した。
前記化合物5oを突然変異p53類型であるMT/WT−p53遺伝子を有するMDA−MB 468という人間乳房癌細胞株に処理してウェスタンブロットを実施した。図23は、MDA−MBに5oを処理して実施したウェスタンブロットの結果を表わすグラフであり、表5は、このグラフ値を数値で表わした図表である。
【0178】
【表5】
【0179】
前記化合物5oとNutlin−3とをp53突然変異型癌細胞株MDA−MB 468に処理した時、5oが処理された所でのみ多量のp21が発現されることを確認することができた。(図23及び表5参照)。この結果からNutlin−3は、突然変異p53がある時、ターゲット遺伝子であるp21に何らの影響を及ぼすことができない一方、前記化合物5oは、p21の活性を誘導してNutlin−3と差別的な影響力があるということを表わす。
<実験例5>生体内異種移植(xenograft)の考察
【0180】
1.実験方法
非胸腺マウスをDaehan Biolink Co.Ltdから購入し、温度及び光の調節条件(20−23℃、12時間光/12時間暗周期)下で生育させ、滅菌食餌と水とを自在に供給した。2週後、非胸腺マウス(n=21)に腹腔注射を通じてA549細胞を1X107細胞で接種した。2週後、各グループを3つのサブグループに分けてPBS、10mg/kgまたは20mg/kgの5oを一週間に一回ずつ10週間腹腔内に投与し、生存率を観察した。動物試験は、釜山大学校動物保護委員会の承認及び指針によって行われた。
【0181】
2.実験結果
図24のように、10mg/kgと20mg/kgとの5oの処理によって腫瘍による死亡率が遮断された一方、PBS処理群では、10週後に生存率が50%以下になり、図25に示したように、5o処理群での腫瘍は、ほとんど死滅する形状が観察された。また、図26のように、体重減少や化合物注射による総体的な解剖学的異常所見を観察することができなかった。下記表6は、腫瘍の発生位置及び形態学的特徴を要約したものである。
【0182】
【表6】
【0183】
[配列目録フリーテキスト]
配列番号1は、人間p53細胞性腫瘍抗原のアミノ酸配列を表わしたものである。
【産業上の利用可能性】
【0184】
本発明は、スネイル−p53間の結合を阻害する化合物及びそれを有効成分として含有する癌疾患治療剤関連の分野に適用可能である。
【特許請求の範囲】
【請求項1】
下記化学式1で表されることを特徴とする化合物、またはその塩:
【化1】
前記化学式1で、
mは、0ないし10の整数、n及びpは、それぞれ0または1の整数であり、
Zは、−NH(CH2)qCH3、−OH、4−フェニルピペリジン基、4−フェニルピペラジン基、イソブチルアミノ基及びイソブチルオキシ基からなる群から選択され、
qは、0ないし9の整数である。
【請求項2】
前記化合物は、2−ノニルアミノ−5,8−ジメトキシ−1,4−ナフトキノン;2−デシルアミノ−5,8−ジメトキシ−1,4−ナフトキノン;3−(5,8−ジメトキシ−1,4−ジオキソ−ナフタレン−2−イルチオ)プロパン酸;11−(5,8−ジメトキシ−1,4−ジオキソ−ナフタレン−2−イルチオ)ウンデカン酸;イソブチル−11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)−ウンデカノアート;11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)−N−イソブチルウンデカンアミド;及びイソブチル−11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルスルフィニル)ウンデカノアートからなる群から選択されたことを特徴とする請求項1に記載の化合物、またはその塩。
【請求項3】
スネイル−p53間の結合を阻害する化合物を有効成分として含有することを特徴とする癌疾患治療剤。
【請求項4】
前記化合物は、下記化学式1で表される化合物、またはその塩であることを特徴とする請求項3に記載の癌疾患治療剤:
【化2】
前記化学式1で、
mは、0ないし10の整数、n及びpは、それぞれ0または1の整数であり、
Zは、−NH(CH2)qCH3、−OH、4−フェニルピペリジン基、4−フェニルピペラジン基、イソブチルアミノ基及びイソブチルオキシ基からなる群から選択され、
qは、0ないし9の整数である。
【請求項5】
前記癌疾患は、K−Ras突然変異性癌疾患であることを特徴とする請求項3または4に記載の癌疾患治療剤。
【請求項6】
前記K−Ras突然変異性癌疾患は、膵膓癌、肺癌、胆管癌及び大膓癌からなる群から選択された何れか一つであること特徴とする請求項5に記載の癌疾患治療剤。
【請求項7】
p53を固定させたプレートにスネイル及び候補薬物を培養する段階と、
ELISAリーダーでスネイル−p53の結合を阻害する候補薬物を選別する段階と、
を含んでなることを特徴とするK−Ras突然変異性癌疾患治療剤のスクリーニング方法。
【請求項8】
p53のDNA結合ドメインのエンドサイトーシス(endocytosis)を用いて伝達しようとする薬物をK−Ras突然変異細胞に特異的に伝達することを特徴とするK−Ras突然変異細胞の特異的薬物伝達方法。
【請求項9】
前記DNA結合ドメインは、人間p53の90番目から280番目までのアミノ酸配列を含むこと特徴とする請求項8に記載のK−Ras突然変異細胞の特異的薬物伝達方法。
【請求項10】
前記薬物伝達方法は、p53のDNA結合ドメイン及び伝達しようとする薬物を共に細胞に処理する段階と、
前記p53のDNA結合ドメインによるエンドサイトーシスによって隣合うK−Ras突然変異細胞に薬物が伝達される段階と、
を含んでなること特徴とする請求項8または9に記載のK−Ras突然変異細胞の特異的薬物伝達方法。
【請求項11】
スネイル抗体の発現有無を測定することを特徴とするK−Ras突然変異性癌疾患の初期診断方法。
【請求項12】
前記スネイル抗体の発現は、K−Ras突然変異性癌疾患を有した患者の血清から測定すること特徴とする請求項11に記載のK−Ras突然変異性癌疾患の初期診断方法。
【請求項13】
前記K−Ras突然変異性癌疾患は、膵膓癌、肺癌、胆管癌及び大膓癌からなる群から選択された何れか一つであること特徴とする請求項11または12に記載のK−Ras突然変異性癌疾患の初期診断方法。
【請求項1】
下記化学式1で表されることを特徴とする化合物、またはその塩:
【化1】
前記化学式1で、
mは、0ないし10の整数、n及びpは、それぞれ0または1の整数であり、
Zは、−NH(CH2)qCH3、−OH、4−フェニルピペリジン基、4−フェニルピペラジン基、イソブチルアミノ基及びイソブチルオキシ基からなる群から選択され、
qは、0ないし9の整数である。
【請求項2】
前記化合物は、2−ノニルアミノ−5,8−ジメトキシ−1,4−ナフトキノン;2−デシルアミノ−5,8−ジメトキシ−1,4−ナフトキノン;3−(5,8−ジメトキシ−1,4−ジオキソ−ナフタレン−2−イルチオ)プロパン酸;11−(5,8−ジメトキシ−1,4−ジオキソ−ナフタレン−2−イルチオ)ウンデカン酸;イソブチル−11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)−ウンデカノアート;11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルチオ)−N−イソブチルウンデカンアミド;及びイソブチル−11−(5,8−ジメトキシ−1,4−ジオキソ−1,4−ジヒドロナフタレン−2−イルスルフィニル)ウンデカノアートからなる群から選択されたことを特徴とする請求項1に記載の化合物、またはその塩。
【請求項3】
スネイル−p53間の結合を阻害する化合物を有効成分として含有することを特徴とする癌疾患治療剤。
【請求項4】
前記化合物は、下記化学式1で表される化合物、またはその塩であることを特徴とする請求項3に記載の癌疾患治療剤:
【化2】
前記化学式1で、
mは、0ないし10の整数、n及びpは、それぞれ0または1の整数であり、
Zは、−NH(CH2)qCH3、−OH、4−フェニルピペリジン基、4−フェニルピペラジン基、イソブチルアミノ基及びイソブチルオキシ基からなる群から選択され、
qは、0ないし9の整数である。
【請求項5】
前記癌疾患は、K−Ras突然変異性癌疾患であることを特徴とする請求項3または4に記載の癌疾患治療剤。
【請求項6】
前記K−Ras突然変異性癌疾患は、膵膓癌、肺癌、胆管癌及び大膓癌からなる群から選択された何れか一つであること特徴とする請求項5に記載の癌疾患治療剤。
【請求項7】
p53を固定させたプレートにスネイル及び候補薬物を培養する段階と、
ELISAリーダーでスネイル−p53の結合を阻害する候補薬物を選別する段階と、
を含んでなることを特徴とするK−Ras突然変異性癌疾患治療剤のスクリーニング方法。
【請求項8】
p53のDNA結合ドメインのエンドサイトーシス(endocytosis)を用いて伝達しようとする薬物をK−Ras突然変異細胞に特異的に伝達することを特徴とするK−Ras突然変異細胞の特異的薬物伝達方法。
【請求項9】
前記DNA結合ドメインは、人間p53の90番目から280番目までのアミノ酸配列を含むこと特徴とする請求項8に記載のK−Ras突然変異細胞の特異的薬物伝達方法。
【請求項10】
前記薬物伝達方法は、p53のDNA結合ドメイン及び伝達しようとする薬物を共に細胞に処理する段階と、
前記p53のDNA結合ドメインによるエンドサイトーシスによって隣合うK−Ras突然変異細胞に薬物が伝達される段階と、
を含んでなること特徴とする請求項8または9に記載のK−Ras突然変異細胞の特異的薬物伝達方法。
【請求項11】
スネイル抗体の発現有無を測定することを特徴とするK−Ras突然変異性癌疾患の初期診断方法。
【請求項12】
前記スネイル抗体の発現は、K−Ras突然変異性癌疾患を有した患者の血清から測定すること特徴とする請求項11に記載のK−Ras突然変異性癌疾患の初期診断方法。
【請求項13】
前記K−Ras突然変異性癌疾患は、膵膓癌、肺癌、胆管癌及び大膓癌からなる群から選択された何れか一つであること特徴とする請求項11または12に記載のK−Ras突然変異性癌疾患の初期診断方法。
【図1】

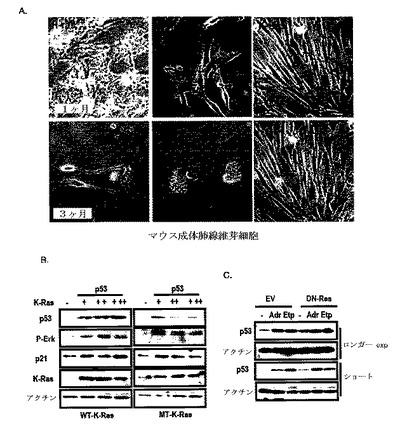
【図2】
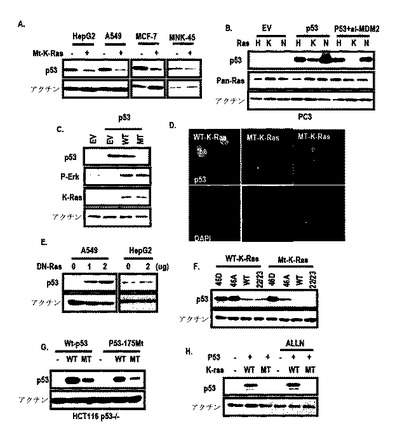
【図3】
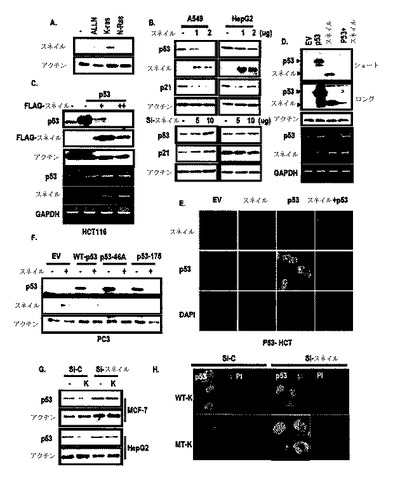
【図4】
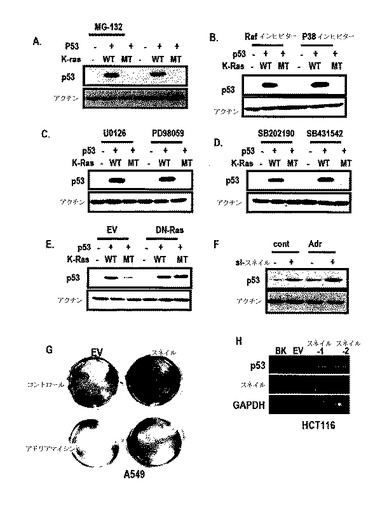
【図5】
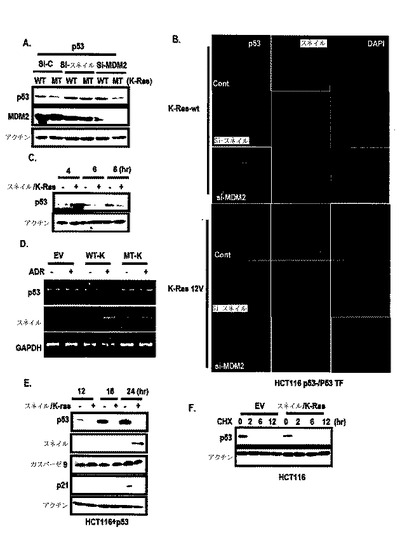
【図6】
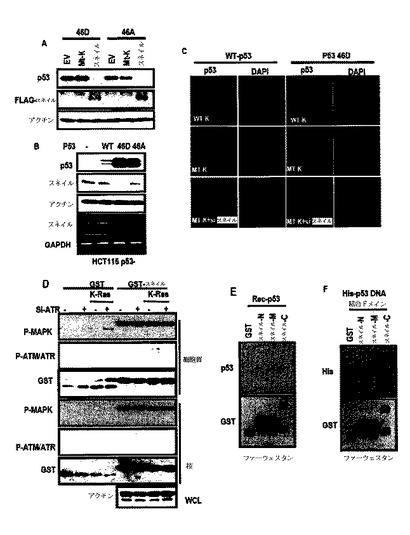
【図7】
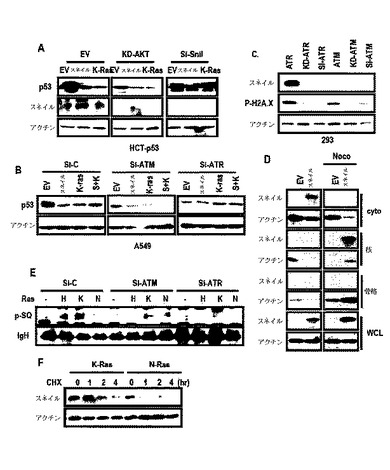
【図8】
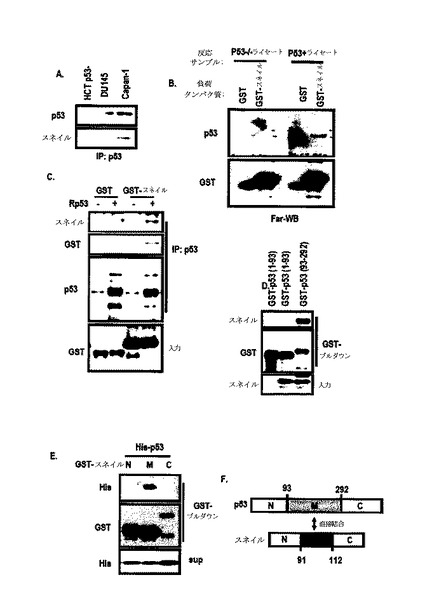
【図9】
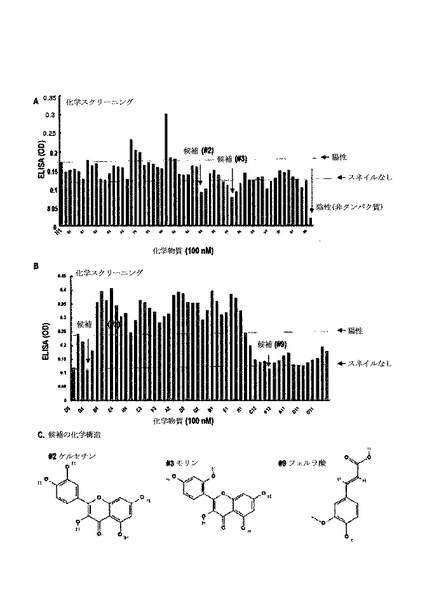
【図10】
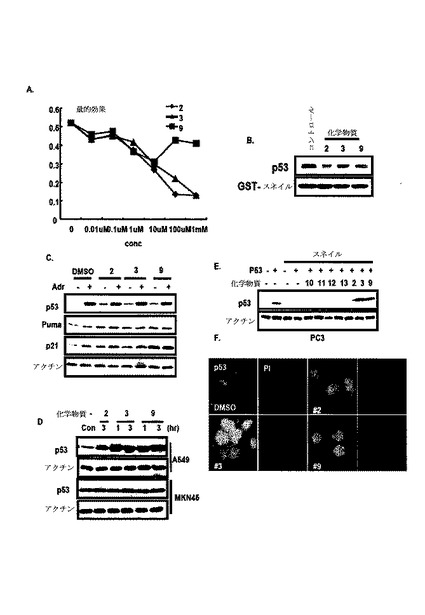
【図11】
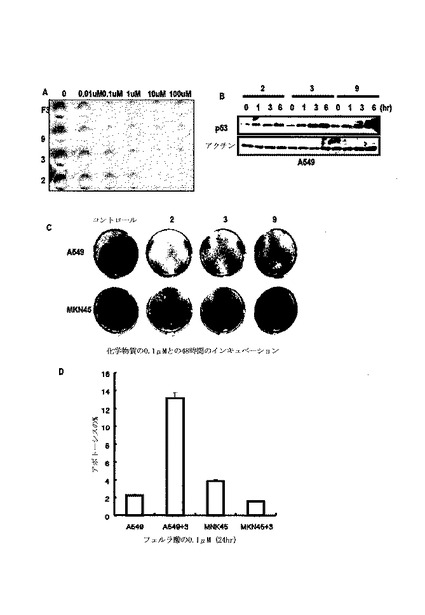
【図12】
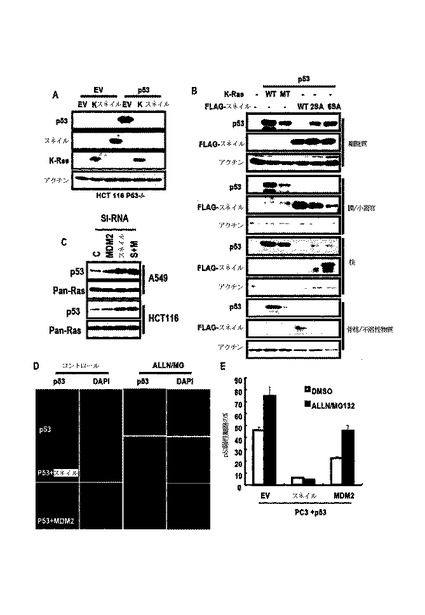
【図13】
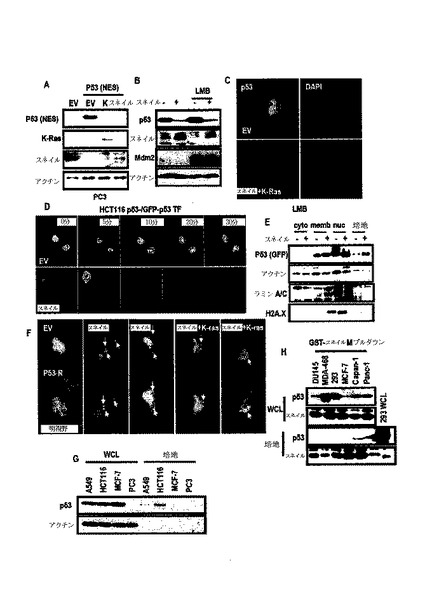
【図14】
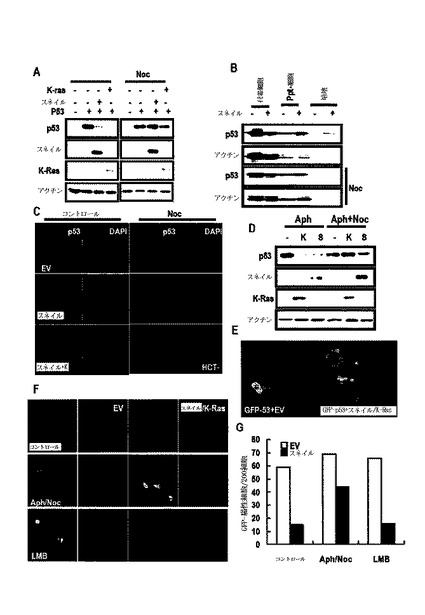
【図15】
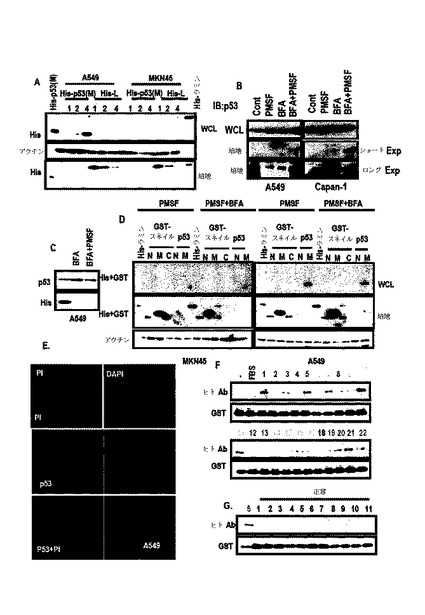
【図16】

【図17】
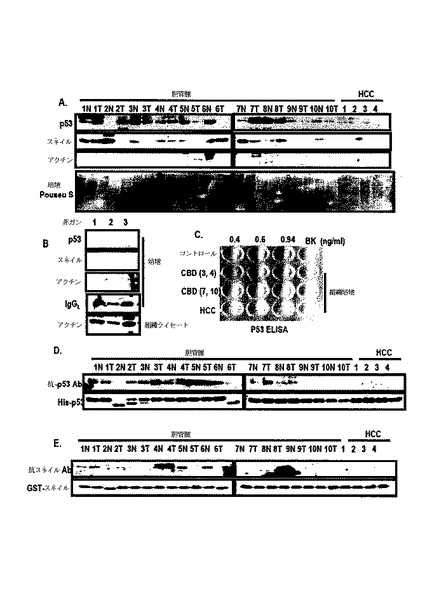
【図18】
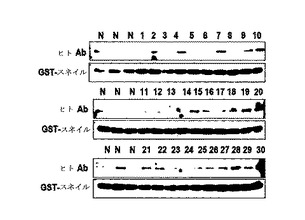
【図19】
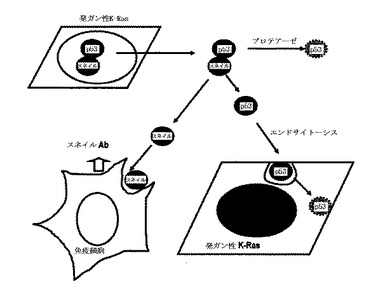
【図20】
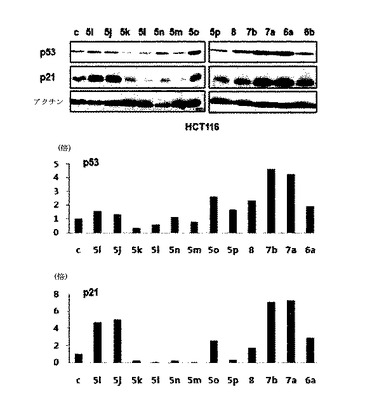
【図21】
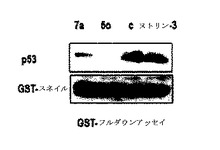
【図22】
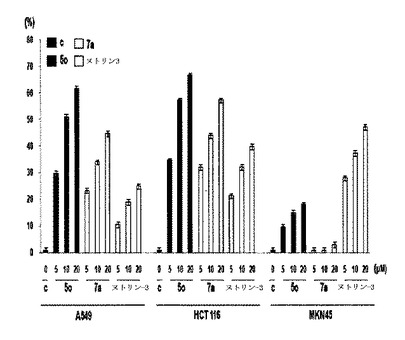
【図23】
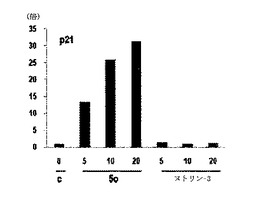
【図24】
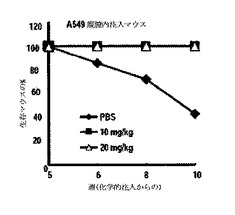
【図25】
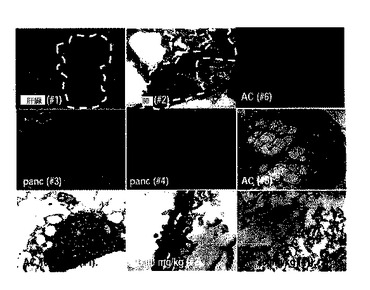
【図26】
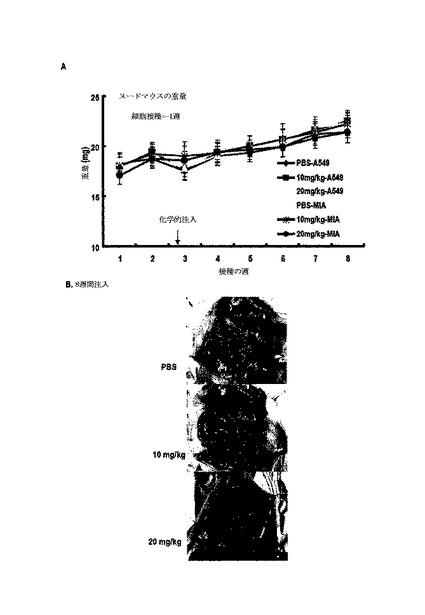
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【公表番号】特表2012−509319(P2012−509319A)
【公表日】平成24年4月19日(2012.4.19)
【国際特許分類】
【出願番号】特願2011−537370(P2011−537370)
【出願日】平成21年11月23日(2009.11.23)
【国際出願番号】PCT/KR2009/006896
【国際公開番号】WO2010/059004
【国際公開日】平成22年5月27日(2010.5.27)
【出願人】(511123902)プサン ナショナル ユニヴァーシティ インダストリー ユニヴァーシティ (1)
【氏名又は名称原語表記】Pusan National University Industry−University
【出願人】(509020516)ザ インダストリー アンド アカデミック クーパレイション イン チュンナン ナショナル ユニバーシティー (2)
【氏名又は名称原語表記】The Industry & Academic Cooperation in Chungnam National University
【住所又は居所原語表記】Department of Nanomaterials, Engineering, Chungnam National University, 220 Gung−dong Yuseong−gu, Daejeon 305−764 Korea
【Fターム(参考)】
【公表日】平成24年4月19日(2012.4.19)
【国際特許分類】
【出願日】平成21年11月23日(2009.11.23)
【国際出願番号】PCT/KR2009/006896
【国際公開番号】WO2010/059004
【国際公開日】平成22年5月27日(2010.5.27)
【出願人】(511123902)プサン ナショナル ユニヴァーシティ インダストリー ユニヴァーシティ (1)
【氏名又は名称原語表記】Pusan National University Industry−University
【出願人】(509020516)ザ インダストリー アンド アカデミック クーパレイション イン チュンナン ナショナル ユニバーシティー (2)
【氏名又は名称原語表記】The Industry & Academic Cooperation in Chungnam National University
【住所又は居所原語表記】Department of Nanomaterials, Engineering, Chungnam National University, 220 Gung−dong Yuseong−gu, Daejeon 305−764 Korea
【Fターム(参考)】
[ Back to top ]