
スピロ型シクロトリシロキサン誘導体、その製造方法、それを用いた製膜法及び膜
【課題】本発明の課題は、低い誘電率を有する機械強度に優れた電気絶縁膜を作製するためのプレカーサーとして有用なスピロ型シクロトリシロキサン誘導体を提供することにある。
【解決手段】一般式(1)
【化1】

(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されるシクロトリシロキサン化合物を用いて形成させた膜は、所望の低い誘電率を有し、機械強度に優れた電気絶縁膜となる。
【解決手段】一般式(1)
【化1】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されるシクロトリシロキサン化合物を用いて形成させた膜は、所望の低い誘電率を有し、機械強度に優れた電気絶縁膜となる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、特にプラズマ促進化学気相蒸着(PECVD)法による製膜プロセスに有用なスピロ型シクロトリシロキサン誘導体及びその製造方法に関する。さらに本発明は、スピロ型シクロトリシロキサン誘導体を用いて製膜して得られる、機械的強度に優れた膜に関する。
【背景技術】
【0002】
有機ケイ素化合物をPECVD法などで基板上に堆積させた絶縁膜は、一般に化学構造が明確でないが、その構成元素から、SiOCH膜と呼ぶことがある。シクロトリシロキサン誘導体を原料に用いてPECVD法によってSiOCH膜を作製した例としては、特許文献1および2をあげることができる。更にこれらSiOCH膜は比較的低い誘電率を有する絶縁膜として利用できることが示されている。
【0003】
スピロ型シクロトリシロキサン誘導体の報告例として非特許文献1および2を挙げることができる。非特許文献1には2,2,4,4,6,6−トリ(1,4−ブタンジイル)シクロトリシロキサン及び2,2,4,4,6,6−トリ(1,5−ペンタンジイル)シクロトリシロキサンの合成について記載がある。また非特許文献2には2,2,4,4,6,6−トリ(2−ブテン−1,4−ジイル)シクロトリシロキサンが反応混合物中に存在していることを示されているものの、このものの単離精製は行われていない。これら非特許文献中では、いずれの化合物についても製膜材料としての提案および材料物性についての記載は無い。さらに先に挙げた特許文献2ではスピロ型シクロトリシロキサン誘導体として2,2−(ブタン−1,4−ジイル)−4,4,6,6−テトラメチルシクロトリシロキサン及び2,2−(ブタン−1,4−ジイル)−4,6−ジメチルシクロトリシロキサンの合成についての記載がある。しかしながら、いずれのスピロ型シクロトリシロキサン誘導体も、同一ケイ素原子上に結合しているアルキレン基またはアルケニレン基の炭素原子上に水素のみが結合しており、このアルキレン基またはアルケニレン基の炭素原子上に炭化水素基が結合したスピロ型シクロトリシロキサン誘導体の合成例は無い。
【0004】
然るにシクロトリシロキサンのすべてのケイ素原子上に、一つ以上のアルキル基で置換された1,4−ブタンジイル基または2−ブテン−1,4−ジイル基を導入したスピロ型シクロトリシロキサン誘導体が合成された例は無く、ましてやこれを用いて電気絶縁膜を作製した例も一切無い。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2007−96237号公報
【特許文献2】米国特許第6572923号明細書
【非特許文献】
【0006】
【非特許文献1】Francis J. Bajerら、Journal of Organic Chemistry,28巻(7号),1941ー1942頁(1963年)。
【0007】
【非特許文献2】A.N.Polivanovら、Zhurnal Obshchei Khimii,48巻(7号),1662頁(1978年)。
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明の課題は、所望の低い誘電率を有し、機械強度に優れた電気絶縁膜を形成可能なプレカーサーとしてのスピロ型シクロトリシロキサン誘導体及びその製造方法を提供することにある。さらにスピロ型シクロトリシロキサン誘導体を用いて製造される膜及びその製造方法を提供することにある。
【課題を解決するための手段】
【0009】
本発明者等は上記の課題を解決すべく鋭意検討した結果、シクロトリシロキサンのすべてのケイ素原子上に、アルキル基が置換されている1,4−ブタンジイル基または2−ブテン−1,4−ジイル基を導入したスピロ型シクロトリシロキサン誘導体が、上記課題を解決し得る性能を有する化合物であることを見いだし、本発明を完成させるに至った。
【0010】
すなわち本発明は、一般式(1)
【0011】
【化1】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されることを特徴とする、スピロ型シクロトリシロキサン誘導体である。
【0012】
また本発明は、一般式(2)
【0013】
【化2】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表し、Xはそれぞれ独立に塩素原子、メトキシ基又はエトキシ基を表す。)で表されるシラン誘導体を環化剤の存在下に反応させることを特徴とする、一般式(1)で表されるスピロ型シクロトリシロキサン誘導体の製造方法である。
【0014】
さらに本発明は、一般式(1)で表されるスピロ型シクロトリシロキサン誘導体を原料として用いて製膜することを特徴とする、膜の製造法である。また本発明は、上述の製造法により製造されることを特徴とする膜である。さらに本発明は、上述の膜から成ることを特徴とする、電気絶縁膜である。また本発明は、上述の電気絶縁膜を配してなることを特徴とする、半導体電子デバイスである。
【0015】
以下に本発明をさらに詳細に説明する。一般式(1)及び(2)並びに後述の一般式(2a)で表される化合物の置換基R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表すが、R1、R2、R3及びR4は同時に水素原子ではない。また、炭素数が1乃至3のアルキル基としてはメチル基、エチル基、プロピル基、イソプロピル基、シクロプロピル基を挙げることができる。PECVD法による製膜プロセスに一般式(1)で表されるスピロ型シクロトリシロキサン誘導体を用いる場合、蒸気圧の観点からR1、R2、R3又はR4のうち一つもしくは二つがメチル基で残りが水素原子、またはR1、R2、R3又はR4のうち一つがエチル基で三つが水素原子であることが望ましく、原料の入手の容易さなどからR2、R3及びR4が水素原子、R1がメチル基であり、破線を伴う実線が単結合であるか、R1、R3及びR4が水素原子、R2がメチル基であり、破線を伴う実線が単結合であるか、又は、R1、R3及びR4が水素原子、R2がメチル基であり、破線を伴う実線が二重結合であることがさらに望ましい。
【0016】
一般式(2)で表されるシラン誘導体を環化剤存在下に反応させることによって一般式(1)で表されるスピロ型シクロトリシロキサン誘導体の製造が可能である。このとき用いることのできる環化剤としては、一般式(2)で表されるシラン誘導体を環化させることができるものであれば特に限定はないが、例えばスルホキシド化合物、金属酸化物、水、または酸性水溶液等を挙げることができる。
【0017】
Xが塩素原子であるシラン誘導体(2)を用いて対応するスピロ型シクロトリシロキサン誘導体(1)を製造する際、環化剤としてスルホキシド化合物が好適に用いられる。用いることのできるスルホキシド化合物として、反応が進行すれば特に制限は無いが、ジメチルスルホキシド、テトラメチレンスルホキシド、ジオクチルスルホキシド、ジベンジルスルホキシド、ジフェニルスルホキシド、ジ−p−トリルスルホキシド、ジ−p−クロロフェニルスルホキシドなどを例示できる。反応が速やかに進行する点でジメチルスルホキシド、テトラメチレンスルホキシド、ジオクチルスルホキシドが好ましく、安価である点でジメチルスルホキシドが更に好ましい。スルホキシド化合物の添加量は特に制限は無いが、収率、効率の点でジクロロシラン化合物に対して2〜4当量加えることが望ましい。
【0018】
反応は有機溶媒中で行なうことができる。用いることのできる有機溶媒としてはシラン誘導体(2)やスルホキシド化合物と反応したり、溶解性が低かったりして所望の環化反応が阻害されなければ特に制限は無く、ヘキサン、シクロヘキサン、ヘプタン、ベンゼン、トルエン、キシレンなどの炭化水素系有機溶媒、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサンなどのエーテル系有機溶媒、酢酸エチル、アセトン、アセトニトリル、メチルエチルケトン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミドなどの非プロトン性の極性有機溶媒、四塩化炭素、クロロホルム、ジクロロメタン、1,2−ジクロロエタンなどのハロゲン系有機溶媒などを例示できるが、収率の点で炭化水素系有機溶媒、エーテル系有機溶媒およびハロゲン系有機溶媒が好適に用いられる。
【0019】
またシラン誘導体(2)とスルホキシド化合物の添加の方法は、スルホキシド化合物にシラン誘導体(2)を加えても、シラン誘導体(2)にスルホキシド化合物を加えても良い。反応温度はシラン誘導体(2)とスルホキシド化合物を混合できれば特に制限は無いが、−50〜100℃の範囲に、好ましくは−20℃〜50℃の範囲に制御して、反応を行なうことが好ましい。反応が30分間から1日程度で終了するように有機溶媒の種類や量、撹拌効率を制御すると良い。反応混合物から目的のスピロ構造を有するシクロトリシロキサン化合物は、水洗浄、カラムクロマトグラフィー、蒸留などを組み合わせて精製することができるが、反応で副生するスルフィド化合物由来の臭いがこれらの精製工程において除くことができない場合は、適宜活性炭処理、酸化剤の添加処理などを行なうと良い。
【0020】
Xが塩素原子であるシラン誘導体(2)を用いて対応するスピロ型シクロトリシロキサン誘導体(1)を製造する際、環化剤として金属酸化物も好適に用いられる。用いることのできる金属酸化物として、反応が進行すれば特に制限は無いが、副生物が少なく、所望の形状の金属酸化物の入手の容易さ、収率の点で、酸化亜鉛、酸化銅(II)及び酸化マグネシウムが好適に用いられる。環化剤として用いることのできる金属酸化物の形状として、反応が進行すれば特に制限は無いが、反応が速やかに進行する点で粉末状が望ましく、粒径1mm程度より細かい粉末が好適に用いられる。金属酸化物の添加量は特に制限は無いが、収率や効率の点でシラン誘導体(2)に対して1〜2当量加えることが望ましい。
【0021】
反応は有機溶媒中で行なうことができる。用いることのできる有機溶媒としてはシラン誘導体(2)や金属酸化物と反応したり、溶解性が低かったりして所望の環化反応が阻害されなければ特に制限は無く、ヘキサン、シクロヘキサン、ヘプタン、ベンゼン、トルエン、キシレンなどの炭化水素系有機溶媒、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサンなどのエーテル系有機溶媒、酢酸エチル、アセトン、アセトニトリル、メチルエチルケトン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミドなどの非プロトン性の極性有機溶媒、四塩化炭素、クロロホルム、ジクロロメタン、1,2−ジクロロエタンなどのハロゲン系有機溶媒などを例示できる。反応温度はシラン誘導体(2)と金属酸化物を混合できれば特に制限は無いが、−80〜150℃の範囲で行なうことができる。生産性や、反応制御のしやすさを考慮すると、例えば酸化亜鉛を用いた場合は、−50℃〜50℃の範囲で反応を行なうことが好ましく、酸化銅(II)又は酸化マグネシウムを用いた場合は、室温〜150℃の範囲で行なうことが好ましい。反応が30分間から1日程度で終了するように有機溶媒の種類や量、撹拌効率を制御すると良い。反応混合物から目的のシクロトリシロキサン化合物は、ろ過、水洗浄、カラムクロマトグラフィー、蒸留などを組み合わせて精製することができる。
【0022】
さらに本発明のスピロ型シクロトリシロキサン誘導体(1)は、シラン誘導体(2)に水または酸性水溶液を環化剤として作用させることによっても製造することができる。使用可能な酸としては塩酸、硫酸などの無機酸、p−トルエンスルホン酸、トリフルオロ酢酸、トリフルオロメタンスルホン酸などの有機酸などを例示できる。シラン誘導体(2)のXが塩素原子の場合は加水分解によって塩酸が発生するので、水を環化剤として加えることにより、スピロ型シクロトリシロキサン誘導体(1)を得ることもできる。この際、水の量は特に制限は無いがシラン誘導体(2)に対し1当量以上あれば良い。Xがメトキシ基あるいはエトキシ基の場合は、酸性水溶液を環化剤として作用させることにより、本発明のスピロ型シクロトリシロキサン誘導体(1)を得ることができる。使用可能な酸としては塩酸、硫酸などの無機酸、p−トルエンスルホン酸、トリフルオロ酢酸、トリフルオロメタンスルホン酸などの有機酸などを例示でき、これらの水溶液を環化剤として用いることができる。収率、取扱の容易さ、経済性などの観点から硫酸が好適に用いられる。加える酸の量に制限はなく、シラン誘導体(2)に対し0.01当量程度の触媒量でも大過剰量でも収率よくスピロ型シクロトリシロキサン誘導体(1)を得ることができる。酸性水溶液の使用量に特に制限はないが、反応液中の水がシラン誘導体(2)に対し1当量以上になるように使用することが好ましい。
【0023】
これらいずれの反応も有機溶媒を用いて行なうことができ、例えばヘキサン、シクロヘキサン、ヘプタン、ベンゼン、トルエン、キシレンなどの炭化水素系有機溶媒、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサンなどのエーテル系有機溶媒、四塩化炭素、クロロホルム、ジクロロメタン、1,2−ジクロロエタンなどのハロゲン系有機溶媒などを例示できる。反応温度は0℃以上で、有機溶媒の沸点および原料のシラン化合物の沸点や反応速度を考慮して決めれば良い。反応混合物から目的のスピロ型シクロトリシロキサン誘導体(1)は、水洗浄、カラムクロマトグラフィー、蒸留などを組み合わせて精製することができる。
【0024】
一般式(2a)
【0025】
【化3】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されるジクロロシラン誘導体に、メタノール又はエタノールを反応させてXがメトキシ基又はエトキシ基であるシラン誘導体(2)を得た(工程1)後、前述の水や酸性水溶液を環化剤として作用させる(工程2)ことにより、スピロ型シクロトリシロキサン誘導体(1)を得ることができる。工程1において、ジクロロシラン誘導体(2a)に、メタノール又はエタノールを反応させる際、トリエチルアミンやピリジンなどの有機塩基で副生する塩化水素を中和することができる。加えるメタノール又はエタノールの量はジクロロシラン誘導体(2a)に対して任意の量で良く、好ましくは2当量以上の過剰量を用いることにより収率良くXがメトキシ基又はエトキシ基であるシラン誘導体(2)を得ることができるが、必ずしもすべてのXをメトキシ基又はエトキシ基に変換しなくてもよい。工程1において反応終了後、蒸留精製によって2つのXが共にメトキシ基又はエトキシ基であるシラン誘導体(2)を分離することができるが、あえて分離する必要はなく、ジクロロシラン誘導体(2a)や、1つのXが塩素原子、もう1つのXがメトキシ基又はエトキシ基であるシラン誘導体(2)が混在していてもそのまま工程2に用いることができる。工程1と工程2は連続して行なっても良い。工程1で有機塩基を用いた場合は、工程2において液性が酸性になるように酸性水溶液を加えることによって、収率良くスピロ型シクロトリシロキサン誘導体(1)を得ることができる。
【0026】
本発明のスピロ型シクロトリシロキサン誘導体(1)を原料として用いて製膜することにより、膜を製造することができる。この膜は電気絶縁膜として使用することができ、半導体電子デバイスに配して用いることができる。膜は、スピンキャスト法、化学気相蒸着(CVD)法などにより製造することができるが、本発明の所望の低い誘電率を示し機械的強度に優れた絶縁膜を製造するためにPECVD法が好適に用いられる。その後、紫外線照射処理、オゾン処理などの硬化工程を行うことができる。
【0027】
スピロ型シクロトリシロキサン誘導体(1)は室温で固体または液体である。固体の場合、減圧下で昇華させてガスとしてPECVDプロセスに供することができる。また、融点以上に加熱することによって液体としたのち、気化させて原料ガスの供給系に導入することができる。スピロ型シクロトリシロキサン誘導体(1)は、例えば特許文献1で開示されている環状シロキサン化合物、2,4,6,8−テトラメチルシクロテトラシロキサン、テトラエトキシシランなどの、PECVD法で絶縁膜を製造可能な含ケイ素化合物と混合して原料ガスの供給系に導入することもできる。混合の割合は均一に混合できれば特に制限は無く、例えば、スピロ型シクロトリシロキサン誘導体(1)と特許文献1で開示されている環状シロキサン化合物やPECVD法で絶縁膜を製造可能な含ケイ素化合物とを重量パーセントで99対1〜10対90の割合で混合することが可能である。スピロ型シクロトリシロキサン誘導体(1)が室温で液体である場合、またはスピロ型シクロトリシロキサン誘導体(1)とPECVD法で絶縁膜を製造可能な含ケイ素化合物との混合物が液体である場合、ガスインジェクション法、リキッドインジェクション法のいずれを用いてPECVDプロセスに供しても良い。
【発明の効果】
【0028】
本発明によるスピロ型シクロトリシロキサン誘導体(1)を原料として用いて製膜することができる。得られた膜は、比誘電率2.5以下の低い値を示し、且つ弾性率が概ね10GPa以上、硬度が概ね1GPa以上である機械的強度に優れた電気絶縁膜であり、半導体電子デバイスに用いることができる。
【図面の簡単な説明】
【0029】
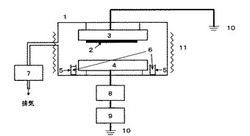
【図1】実施例ー16〜27、比較例ー1〜4で用いたPECVD製膜装置の概略図である。
【実施例】
【0030】
以下、参考例、実施例及び試験例を挙げて本発明をさらに詳細に説明するが、本発明はこれらに限定されるものではない。
【0031】
参考例−1
ジムロート冷却管、磁気撹拌子および滴下ロート(200mL)を備えた3口フラスコ(1000mL)をアルゴンで置換し、マグネシウム24.98g(1.028mol)および脱水ジエチルエーテル580mLを収め、滴下ロートから1,4−ジブロモペンタン100.7g(437.9mmol)をゆっくりと加えながら室温で12時間撹拌し、ジグリニャール試薬を調製した。別に、ジムロート冷却管、磁気撹拌子および滴下ロート(300mL)を備えた3口フラスコ(1000mL)を用意し、アルゴン雰囲気下、四塩化ケイ素74.55g(438.8mmol)のジエチルエーテル50mL溶液を収め、−78℃に冷却した。ここに、先に調製したジグリニャール試薬のエーテル溶液を滴下ロートからゆっくりと滴下し、12時間撹拌した。この混合物を更に室温で一日撹拌した。反応混合物をろ過後、ろ液を濃縮し、得られた粗生成物を減圧蒸留(沸点:45℃/1.6kPa)することにより、1,1−ジクロロ−2−メチル−1−シラシクロペンタンを無色液体として40.66g(収率:54.90%、GC純度:98%)得た。EI−MS(70eV),m/z(相対強度):168([M]+,27),140(100),127(53)。1H−NMR,δ(250MHz,CDCl3,ppm):1.117(d,3H,J=6.3Hz),1.20〜1.41(m,4H),1.41〜1.70(m,2H),1.83〜2.03(m,1H)。
【0032】
参考例−2
メチルコハク酸無水物はJournal of Chemical Society,Perkin Transactions 1、1980年、2029頁を参考に合成した。ジムロート冷却管、磁気撹拌子および滴下ロート(300mL)を備えた3口フラスコ(1000mL)に、メチルコハク酸101.2g(766.0mmol)を収め、滴下ロートから塩化アセチル400mLをゆっくり滴下した。この混合溶液を2時間撹拌し、濃縮後減圧蒸留(沸点:82℃/100Pa)することにより、メチルコハク酸無水物を無色液体として83.6g(収率:95.7%)得た。IR,ν(neat,cm−1):2989,2943,1770,1228,1063,991,903,725。
【0033】
参考例−3
2−メチル−1,4−ブタンジオールはThe Journal of Organic Chemistry、54巻(10号)、2471頁(1989年)を参考に合成した。ジムロート冷却管、磁気撹拌子および滴下ロート(300mL)を備えた3口フラスコ(2000mL)をアルゴンで置換し、水素化リチウムアルミニウム26.3g(705mmol)および脱水テトラヒドロフラン800mLを収めた。これに、メチルコハク酸無水物41.0g(359mmol)の脱水テトラヒドロフラン200mL溶液を滴下ロートより1時間かけて滴下し、2日間撹拌した。滴下ロートからゆっくりと水100mLとテトラヒドロフラン100mLの混合物を滴下し、得られたスラリーにさらにテトラヒドロフラン500mLを加えた。これをろ過し、ろ液をエバポレーターで濃縮後クロロホルム500mLを加えた。これを無水硫酸ナトリウムで乾燥し、ろ過後再度濃縮した。得られた粗生成物を蒸留(沸点:76℃/70Pa)することにより、2−メチル−1,4−ブタンジオールを無色粘稠液体として28.5g(収率:76.2%)得た。IR,ν(neat,cm−1):3280,2924,2873,1458,1381,1059,1005。
【0034】
参考例−4
1,4−ジブロモ−2−メチルブタンはJournal of The American Chemical Society、73巻(8号)、3632頁(1951年)を参考に合成した。リービッヒ冷却管、磁気撹拌子および滴下ロートを備えた3口フラスコ(200mL)に2−メチル−1,4−ブタンジオール28.0g(269mmol)を収め、別のフラスコ中でテトラリンと臭素の反応により発生させた臭化水素ガスを、70℃で3時間吹き込んだ。これを分液ロートに移し、ジエチルエーテルで希釈後に水で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、さらに水素化カルシウムで乾燥した。これを濃縮後減圧蒸留(沸点:82℃/2.4kPa)することにより、1,4−ジブロモ−2−メチルブタンを無色液体として31.6g(収率:51.1%)得た。IR,ν(neat,cm−1):2964,2931,1458,1438,1379,1261,1234,746。
【0035】
参考例−5
ジムロート冷却管、磁気撹拌子および滴下ロート(200mL)を備えた3口フラスコ(300mL)に削り状マグネシウム8.53g(0.351mol)を収め、アルゴンで置換した。これに脱水ジエチルエーテル50mLを加え、少量の1,2−ジブロモエタンでマグネシウムを活性化させた。これに1時間かけて1,4−ジブロモ−2−メチルブタン25.0g(109mmol)の脱水ジエチルエーテル90mL溶液をゆっくりと滴下し、12時間撹拌してジグリニャール試薬を調製した。別に、ジムロート冷却管、磁気撹拌子および滴下ロート(300mL)を備えた3口フラスコ(500mL)をアルゴンで置換し、四塩化ケイ素20.4g(120mmol)および脱水ジエチルエーテルを収め、先に調製したジグリニャール試薬を滴下した。これを3日間撹拌後、生じた塩をろ過して除いた。ろ液を濃縮後減圧蒸留(沸点:89℃/12kPa)することにより、1,1−ジクロロ−3−メチル−1−シラシクロペンタンを無色液体として6.0g(収率:33%)得た。EI−MS(70eV),m/z(相対強度):168(M+,37),153(3),140(100),127(74),125(60),112(28)。1H−NMR,δ(500MHz,CDCl3,ppm):0.72(dd,1H,J=10.8,15.4),0.80〜0.90(m,1H),1.10(d,3H,J=6.5Hz),1.20〜1.30(m,1H),1.36(ddd,1H,J=2.9,7.2,15.1Hz),1.43(ddd,1H,J=2.0,6.5,15.4Hz),1.88〜1.96(m,1H),1.96〜2.03(m,1H)。13C−NMR,δ(126MHz,CDCl3,ppm):18.6,22.8,27.1,33.0,33.6。29Si−NMR,δ(99MHz,CDCl3,ppm):44.6。IR,ν(neat,cm−1):2954,2924,2870,1456,1396,1076,820,789,750,735,675。
【0036】
参考例−6
1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エンはJournal of Organometallic Chemistry、391巻(1号)、7頁(1990年)を参考に合成した。アルゴン置換したステンレス製オートクレーブ(200mL)に、トリクロロシラン60.1g(444mmol)、イソプレン13.8g(203mmol)およびテトラブチルホスホニウムクロリド2.30g(7.80mmol)を収め、密封後150℃で13時間反応させた。反応終了後大気圧に戻し、得られた反応混合物中の低沸点分を減圧下で留去した。残さを減圧蒸留(沸点:70℃/5.5kPa)し、1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エンを無色透明液体として21.1g(収率:62.2%、GC純度:96%)得た。EI−MS(70eV),m/z(相対強度):166(M+,44),151(10),138(15),130(100)。IR,ν(neat,cm−1):3016,2964,2914,1598,1390,1159,1024,729。
【0037】
参考例−7
ジムロート冷却管および撹拌子を備えた2口フラスコ(100mL)に、10%パラジウム担持炭素粉末730mg(パラジウム:0.686mmol)を収め、減圧下で1時間乾燥した。容器をアルゴンで置換し、1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エン7.35g(44.0mmol)を加えた。反応容器を1気圧の水素ガスで置換し、12時間撹拌した。反応混合物を室温、減圧下(0.1kPa)で留去し、留去成分を回収した。これを蒸留精製(沸点:159℃)することにより、1,1−ジクロロ−3−メチル−1−シラシクロペンタンを無色液体として7.33g(収率:98.5%,GC純度:95%)得た。
【0038】
参考例−8
1,1−ジクロロ−3,4−ジメチル−1−シラシクロペンタ−3−エンはOrganometallics、22巻(13号)、2551頁(2003年)を参考に合成した。撹拌子を備えたステンレス製オートクレーブ(200mL)をアルゴンで置換し、2,3−ジメチル−1,3−ブタジエン16.4g(200mmol)、トリクロロシラン80.5g(594mmol)および無水テトラブチルホスホニウムクロリド2.70g(9.16mmol)を収め、密閉した。これを激しく撹拌しながら150℃で48時間加熱した。混合物をナスフラスコ(100mL)に移し、減圧下で過剰のトリクロロシランを除いた。残さを減圧蒸留(沸点:78℃/2.8kPa)することにより、1,1−ジクロロ−3,4−ジメチル−1−シラシクロペンタ−3−エン29.6g(収率:81.8%)を無色液体として得た。EI−MS(70eV),m/z(相対強度):180(M+,100),165(73),144(59),138(40),129(62)。1H−NMR,δ(250MHz,CDCl3,ppm):1.751(t,6H,J=1Hz),1.882(m,4H)。IR,ν(neat,cm−1):2983,2914,1441,1392,1173,982,769,752,694。
【0039】
実施例−1
アルゴン置換した2口フラスコ(100mL)に脱水トルエン50mLおよび脱水ジメチルスルホキシド8.42g(108mmol)を収め、シリンジより1,1−ジクロロ−2−メチル−1−シラシクロペンタン5.13g(30.3mmol)を1時間かけてゆっくりと滴下した。反応混合物を2時間撹拌し、分液ロートに移して水(50mL)を加えて有機層を抽出した。有機層を濃縮後、クーゲルロールを用いて蒸留(蒸留温度:108℃/5Pa)することにより、2,2,4,4,6,6−トリ(1,4−ペンタンジイル)シクロトリシロキサン(化合物1)の異性体混合物0.97g(収率:28%、異性体混合物のトータルGC純度:99%)を得た。融点:88.3℃.EI−MS(70eV),m/z(相対強度):262(M+,16),247(33),234(27),219(27),193(100)。1H−NMR,δ(250MHz,CDCl3,ppm):0.45〜0.60(m,3H),0.60〜0.80(m,6H),0.95〜1.05(m,9H),1.10〜1.25(m,3H),1.30〜1.50(m,3H),1.70〜1.90(m,3H)。
【0040】
実施例−2
リービッヒ冷却管および撹拌子を備えた2口ナスフラスコ(200mL)に酸化亜鉛10.09g(124.0mmol)を収め、減圧乾燥後にアルゴンで置換した。反応容器に脱水酢酸エチル140mLを収め、1,1−ジクロロ−2−メチル−1−シラシクロペンタン15.84g(93.66mmol)をシリンジより1時間かけて滴下したのち、混合物を12時間撹拌した。水150mLを加えて、有機層を抽出し、硫酸ナトリウムで乾燥させた。ろ液を濃縮後クーゲルロール蒸留(蒸留温度:110℃/0.5Pa)することにより3.30g(収率:30.8%、異性体混合物のトータルGC純度:99%)の化合物1を無色固体として得た。
【0041】
実施例−3
撹拌子を備えたシュレンク管(100mL)に酸化銅(II)800mg(10.1mmol)、1,1−ジクロロ−2−メチル−1−シラシクロペンタン1.69g(10.0mmol)および脱水テトラヒドロフラン20mLを収めた。これを室温で80時間撹拌した。反応混合物にヘキサン50mLを加え、ろ過し、ロータリーエバポレーターで濃縮した。得られた混合物をクーゲルロール蒸留(蒸留温度145℃/60Pa)することにより、183mg(収率:16.0%、異性体混合物のトータルGC純度:94%)の化合物1を無色結晶性固体として得た。
【0042】
実施例−4
ジムロート冷却管、撹拌子および滴下ロート(50mL)を備えた3口フラスコ(300mL)にジエチルエーテル250mL、トリエチルアミン6.10g(60.2mmol)および1,1−ジクロロ−2−メチル−1−シラシクロペンタン4.99g(29.5mmol)を収めた。混合物を0℃に冷却し、滴下ロートから蒸留水540mg(30.0mmol)のテトラヒドロフラン50mL溶液をゆっくり加えた。これを室温に戻し、14時間撹拌した。反応混合物を分液ロートに移し、水300mLを加えて、有機層を抽出した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。得られた混合物をクーゲルロール蒸留(蒸留温度:140℃/50Pa)することにより、543mg(収率:16.1%、異性体混合物のトータルGC純度:93%)の化合物1を無色結晶性固体として得た。
【0043】
実施例−5
ジムロート冷却管および撹拌子を備えた2口フラスコ(100mL)をアルゴンで置換し、脱水ベンゼン24mLおよび脱水ジメチルスルホキシド2.77g(35.5mmol)を収めた。シリンジより1,1−ジクロロ−3−メチル−1−シラシクロペンタン2.00g(11.8mmol)を滴下し、室温で13時間撹拌した。反応混合物を分液ロートに移し、水(30mL)を加えて有機層を抽出した。有機層を無水硫酸ナトリウムで乾燥し、ろ過後濃縮した。これをクーゲルロールを用いて減圧蒸留(蒸留温度:130℃/50Pa)することにより、2,2,4,4,6,6−トリ(2−メチルブタン−1,4−ジイル)シクロトリシロキサン(化合物2)の異性体混合物311mg(収率:23%、異性体混合物のトータルGC純度:94%)を無色固体として得た。EI−MS(70eV),m/z(相対強度):342(M+,25),314(100),300(41),286(35),272(53),244(62)。1H−NMR,δ(250MHz,CDCl3,ppm):0.05〜0.19(m,3H),0.42〜0.59(m,3H),0.50〜0.90(m,6H),1.00(d,9H,J=6.3Hz),0.92〜1.10(m,3H),1.60〜1.88(m,6H)。IR,ν(neat,cm−1):2947,2922,2865,1454,1255,1074,1002,972,829,757。
【0044】
実施例−6
ジムロート冷却管および撹拌子を備えた3口フラスコ(200mL)に酸化マグネシウム480mg(11.9mmol)、1,1−ジクロロ−3−メチル−1−シラシクロペンタン2.00g(11.8mmol)および脱水テトラヒドロフラン30mLを収めた。混合物を70℃で16時間加熱撹拌した。得られた反応混合物をロータリーエバポレーターで濃縮し、残さにヘキサン50mLを加え、分液ロートに移して水50mLで洗浄した。有機層を無水硫酸マグネシウムで乾燥し、ロータリーエバポレーターで濃縮した。得られた混合物をクーゲルロール蒸留(蒸留温度135℃/50Pa)することにより、215mg(収率:15.9%、異性体混合物のトータルGC純度:99%)の化合物2を無色結晶性固体として得た。
【0045】
実施例−7
ジムロート冷却管、撹拌子および滴下ロート(100mL)を備えた2口フラスコ(200mL)にジエチルエーテル150mL、トリエチルアミン4.59g(45.4mmol)および1,1−ジクロロ−3−メチル−1−シラシクロペンタン3.64g(21.6mmol)を収めた。混合物を0℃に冷却し、滴下ロートから蒸留水400mg(22.2mmol)のテトラヒドロフラン65mL溶液をゆっくり加えた。滴下後にゆっくりと室温に戻し、4時間撹拌した。反応混合物を分液ロートに移し、水100mLで有機層を2回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。これをクーゲルロール蒸留(蒸留温度:135℃/50Pa)することにより、391mg(収率:15.9%、異性体混合物のトータルGC純度:96%)の化合物2を無色結晶性固体として得た。
【0046】
実施例−8
ベンゼン200mLおよび脱水ジメチルスルホキシド23.4g(300mmol)の混合物中に、1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エン16.7g(99.9mmol)の20mLベンゼン溶液をゆっくり滴下した。滴下後混合物を4時間撹拌した。反応混合物を分液ロートに移し、水200mLを加えて有機層を抽出した。有機層を無水塩化カルシウムで乾燥後濃縮し、次いでクーゲルロールを用いて蒸留(蒸留温度:140℃/50Pa)することにより、2,2,4,4,6,6−トリ(2−メチル−2−ブテン−1,4−ジイル)シクロトリシロキサン(化合物3)の異性体混合物5.13g(収率:45.8%、異性体混合物のトータルGC純度:95%)を無色液体として得た。これをシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン)で精製し、さらにクーゲルロールを用いて蒸留し、化合物3の異性体混合物1.59g(収率:14.2%、異性体混合物のトータルGC純度:99%)を得た。EI−MS(70eV),m/z(相対強度):336(M+,100),308(79),268(32),133(32)。1H−NMR,δ(250MHz,CDCl3,ppm):1.23(m,6H),1.33(m,6H),1.73(m,9H),5.52(m,3H)。13C−NMR,δ(126MHz,C6D6,ppm):17.27(Si−CH2),20.21(Si−CH2),23.55(−CH3),123.73(−CH=C(Me)−),123.79(−CH=C(Me)−),123.85(−CH=C(Me)−),138.76(−CH=C(Me)−),138.82(−CH=C(Me)−),138.88(−CH=C(Me)−)。29Si−NMR,δ(99MHz,C6D6,ppm):−6.64。IR,ν(neat,cm−1):2962,2914,1437,1377,1159,1030,1012,787,764。
【0047】
実施例−9
ジムロート冷却管および撹拌子を備えた2口フラスコ(100mL)に酸化亜鉛1.95g(24.0mmol)を収め、真空下で乾燥させた。このものに、脱水ジエチルエーテル50mLおよび1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エン3.97g(23.9mmol)をアルゴン雰囲気下で加えた。この混合物を室温で14時間撹拌した。反応混合物を分液ロートに移し、水50mLを加えて有機層を抽出した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。残さをクーゲルロール蒸留(蒸留温度:145℃/60Pa)することにより、261mg(収率:9.8%、異性体混合物のトータルGC純度:98%)の化合物3を無色液体として得た。
【0048】
実施例−10
ジムロート冷却管、撹拌子および滴下ロート(50mL)を備えた3口フラスコ(200mL)にジエチルエーテル200mL、トリエチルアミン4.99g(49.3mmol)および1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エン4.00g(23.9mmol)を収めた。混合物を0℃に冷却し、滴下ロートから蒸留水490mg(27.2mmol)のテトラヒドロフラン40mL溶液をゆっくり加え、徐々に室温に戻し、14時間撹拌した。反応混合物を分液ロートに移し、水200mLを加えて有機層を抽出した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。これをクーゲルロール蒸留(蒸留温度:135℃/50Pa)することにより、62mg(収率:2.3%、異性体混合物のトータルGC純度:95%)の化合物3を無色液体として得た。
【0049】
実施例−11
ジムロート冷却管、撹拌子および滴下ロート(50mL)を備えた3口フラスコ(200mL)をアルゴンで置換し、脱水ベンゼン75mLおよび脱水ジメチルスルホキシド11.7g(150mmol)を収めた。この混合物に、滴下ロートから1,1−ジクロロ−3,4−ジメチル−1−シラシクロペンタ−3−エン9.05g(50.0mmol)のベンゼン25mL溶液を30分かけて滴下し、室温で4時間撹拌した。反応混合物を分液ロートに移し、水100mLを加えて有機層を抽出した。有機層を無水塩化カルシウムで乾燥し、ろ過後濃縮した。残さをクーゲルロールを用いて減圧蒸留(蒸留温度:150℃/45Pa)することにより、2,2,4,4,6,6−トリ(2,3−ジメチル−2−ブテン−1,4−ジイル)シクロトリシロキサン2.09g(収率:33.1%、GC純度:99%)を無色結晶として得た。1H−NMR,δ(250MHz,CDCl3,ppm):1.342(m,12H),1.702(s,18H)。IR,ν(neat,cm−1):2978,2906,2879,1439,1171,1130,1009。
【0050】
参考例−9
ジムロート冷却管、撹拌子および滴下ロート(200mL)を備えた3口フラスコ(1000mL)に、アルゴン雰囲気下で脱水ジエチルエーテル900mL、1,1−ジクロロ−2−メチル−1−シラシクロペンタン20.3g(0.120mol)および脱水トリエチルアミン24.3g(0.240mol)を収めた。この混合物に滴下ロートから脱水エタノール11.1g(0.240mol)の脱水ジエチルエーテル100mL溶液を2時間かけて滴下した後、室温で16時間撹拌し、生じたアミン塩酸塩をろ過により除き、ろ液を減圧濃縮した。残さを減圧蒸留(沸点:70℃/1.7kPa)することにより、1,1−ジエトキシ−2−メチル−1−シラシクロペンタン10.8g(収率:48.0%、GC純度95%)を無色液体として得た。EI−MS(70eV),m/z(相対強度):188(M+,38),160(82),145(51),132(29),131(28),118(100),103(60)。1H−NMR,δ(500MHz,CDCl3,ppm):0.45〜0.53(m,1H),0.58〜0.65(m,1H),0.76〜0.85(m,1H),1.01(d,3H,J=7.5Hz),1.12〜1.18(m,1H),1.20(t,6H,J=7.0Hz),1.36〜1.42(m,1H),1.33〜1.44(m,1H),1.71〜1.84(m,2H),3.76(q,3H,J=7.0Hz),3.80(q,3H,J=7.0Hz)。13C−NMR,δ(126MHz,CDCl3,ppm):8.5,14.2,17.0,18.3,18.4,22.5,35.2,58.7,58.8。29Si−NMR,δ(99MHz,CDCl3,ppm):7.3。IR,ν(neat,cm−1):2972,2925,2866,1389,1101,1076,949,789,760。
【0051】
参考例−10
ジムロート冷却管、撹拌子および滴下ロート(100mL)を備えた3口フラスコ(200mL)をアルゴン置換し、脱水ジエチルエーテル100mL、1,1−ジクロロ−2−メチル−1−シラシクロペンタン5.89g(34.8mmol)および脱水トリエチルアミン7.50g(74.1mmol)を収めた。この混合物に、滴下ロートから脱水メタノール2.27g(70.8mmol)の脱水テトラヒドロフラン80mL溶液を1時間かけて滴下し、室温で12時間撹拌し、生じたアミン塩酸塩をろ過により除き、ろ液を濃縮した。濃縮物を減圧蒸留(沸点:81℃/5.8kPa)することにより、1,1−ジメトキシ−2−メチル−1−シラシクロペンタン4.93g(収率:88.3%、GC純度:94%)を無色液体として得た。EI−MS(70eV),m/z(相対強度):160(M+,33),132(100),118(62),117(54),104(45)。1H−NMR,δ(500MHz,CDCl3,ppm):0.52(ddd,1H,J=8.4,9.5,15.3Hz),0.62〜0.69(m,1H),0.82〜0.90(m,1H),1.06(d,3H,J=7.5Hz),1.15〜1.24(m,1H),1.38〜1.47(m,1H),1.75〜1.89(m,2H),3.55(s,3H),3.58(s,3H)。13C−NMR,δ(126MHz,CDCl3,ppm):7.6,14.1,16.7,22.5,35.1,50.7,50.9。29Si−NMR,δ(99MHz,CDCl3,ppm):10.9。IR,ν(neat,cm−1):2937,2835,1454,1188,1076,800,773,731。
【0052】
参考例−11
ジムロート冷却管、撹拌子および滴下ロート(200mL)を備えた3口フラスコ(500mL)に、アルゴン雰囲気下で脱水ジエチルエーテル300mL、1,1−ジクロロ−3−メチル−1−シラシクロペンタン17.0g(0.101mol)および脱水トリエチルアミン20.7g(0.204mol)を収めた。この混合物に、滴下ロートから脱水エタノール9.50g(0.206mol)の脱水テトラヒドロフラン200mL溶液を1時間かけて滴下した後、室温で3時間撹拌した。生じたアミン塩酸塩をろ過により除き、ろ液を濃縮した。濃縮物を減圧蒸留(沸点:74℃/2.5kPa)することにより、1,1−ジエトキシ−3−メチル−1−シラシクロペンタン15.4g(収率:81.5%、GC純度97%)を無色液体として得た。EI−MS(70eV),m/z(相対強度):188(M+,17),160(63),146(32),145(28),131(20),118(100),103(51)。1H−NMR,δ(500MHz,CDCl3,ppm):0.12(dd,1H,J=11.0,15.0Hz),0.49〜0.56(m,1H),0.75〜0.81(m,1H),0.83〜0.89(m,2H),1.03(d,3H,J=6.5Hz),1.23(t,3H,J=7.0Hz),1.24(t,3H,J=7.0Hz),1.65〜1.73(m,1H),1.82〜1.89(m,1H),3.79(q,3H,J=7.0Hz).3.80(q,3H,J=7.0Hz)。13C−NMR,δ(126MHz,CDCl3,ppm)9.5,18.3,18.4,23.8,33.2,33.3,58.7,58.8。29Si−NMR,δ(99MHz,CDCl3,ppm)11.4。IR,ν(neat,cm−1)2949,2924,1389,1101,1074,949,822,771。
【0053】
参考例−12
ジムロート冷却管、撹拌子および滴下ロート(200mL)を備えた3口フラスコ(500mL)に、アルゴン雰囲気下で脱水ジエチルエーテル300mL、1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エン18.3g(0.110mol)および脱水トリエチルアミン32.1g(0.317mol)を収めた。混合物に滴下ロートから脱水エタノール13.5g(0.293mol)の脱水テトラヒドロフラン200mL溶液を1時間かけて滴下した後、室温で3時間撹拌した。生じたアミン塩酸塩をろ過により除き、ろ液を濃縮した。濃縮物を減圧蒸留(沸点:73℃/1.4kPa)することにより、1,1−ジエトキシ−3−メチル−1−シラシクロペンタ−3−エン15.8g(収率:77.1%、GC純度:97%)を無色液体として得た。EI−MS(70eV),m/z(相対強度):186(M+,57),157(18),140(100),113(47),103(36)。1H−NMR,δ(500MHz,CDCl3,ppm):1.20(s,br,1H),1.24(t,6H,J=7.0Hz),1.30(s,br,2H),1.76(s,br,3H),3.82(q,4H,J=7.0Hz),5.56〜5.57(m,1H)。13C−NMR,δ(126MHz,CDCl3,ppm):14.3,17.7,18.3,23.5,58.9,123.7,139.0。29Si−NMR,δ(99MHz,CDCl3,ppm):8.4。IR,ν(neat,cm−1):2972,2885,1390,1157,1074,949,773,750。
【0054】
実施例−12
ジムロート冷却管および撹拌子を備えた2口フラスコ(50mL)にジエチルエーテル10mL、蒸留水2.30g(128mmol)および硫酸10.5g(107mmol)を収めた。これを0℃に冷却し、1,1−ジエトキシ−2−メチル−1−シラシクロペンタン3.49g(18.5mmol)をゆっくり加えた。混合物をゆっくりと室温に戻し、24時間撹拌した。ジムロート冷却管をト字管に付け替え、80℃まで昇温して低沸点溶媒を留去した。残さを分液ロートに移し、ヘキサン50mLを加え、水50mLで有機層を3回洗浄した。有機層を無水硫酸マグネシウムで乾燥後、ロータリーエバポレーターで濃縮した。残さをクーゲルロール蒸留(蒸留温度:130℃/50Pa)することにより、1.11g(収率:52.3%、異性体混合物のトータルGC純度:97%)の化合物1を無色結晶性固体として得た。
【0055】
実施例−13
ジムロート冷却管および撹拌子を備えた2口フラスコ(50mL)に蒸留水4.13g(229mmol)、硫酸7.14g(72.8mmol)およびジエチルエーテル25mLを収めた。これを0℃に冷却し、1,1−ジメトキシ−2−メチル−1−シラシクロペンタン1.71g(10.6mmol)をゆっくり加えた。混合物を室温に戻し、12時間撹拌した。反応混合物を分液ロートに移し、ヘキサン50mLを加え、水50mLで有機層を3回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。残さをクーゲルロール蒸留(蒸留温度125℃/40Pa)することにより、564mg(収率:46.4%、異性体混合物のトータルGC純度:99%)の化合物1を無色結晶性固体として得た。
【0056】
実施例−14
ジムロート冷却管および撹拌子を備えた2口フラスコ(50mL)にジエチルエーテル25mL、蒸留水3.09g(172mmol)および硫酸2.51g(25.6mmol)を収めた。この混合物をを0℃に冷却し、1,1−ジエトキシ−3−メチル−1−シラシクロペンタン4.74g(25.1mmol)をゆっくり加え、徐々に室温に戻した後、さらに40時間撹拌した。ジムロート冷却管をト字管に付け替え、80℃まで昇温して低沸点成分を溜去した。残査を分液ロートに移し、ヘキサン50mLを加え、水50mLで有機層を3回洗浄した。有機層を無水硫酸マグネシウムで乾燥後、ロータリーエバポレーターで濃縮した。残査をクーゲルロール蒸留(蒸留温度:140℃/50Pa)することにより、384mg(収率:13.4%、異性体混合物のトータルGC純度:93%)の化合物2を無色結晶性固体として得た。
【0057】
実施例−15
ジムロート冷却管および撹拌子を備えた2口フラスコ(50mL)にジエチルエーテル25mL、蒸留水3.04g(169mmol)および硫酸2.52g(25.7mmol)を収めた。この混合物を0℃に冷却し、1,1−ジエトキシ−3−メチル−1−シラシクロペンタ−3−エン3.70g(19.9mmol)をゆっくり加え、徐々に室温に戻した後、さらに18時間撹拌した。反応混合物を分液ロートに移し、ヘキサン50mLを加え、水50mLで有機層を3回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。残査をクーゲルロール蒸留(蒸留温度:140℃/50Pa)することにより、120mg(収率:5.4%、異性体混合物のトータルGC純度:94%)の化合物3を無色液体として得た。
【0058】
参考例−13
ステンレス製オートクレーブに撹拌子を入れ、アルゴン置換した。この中に酸化白金100mg、化合物3を396mg(1.18mmol)、および脱水テトラヒドロフラン5mLを収め、水素ガスで内部を置換した。これを90℃で5日間撹拌した。反応混合物をセライトでろ過し、ろ液をロータリーエバポレーターで濃縮した。残査をクーゲルロールで蒸留(蒸留温度:110〜140℃/50Pa)し、化合物2の異性体混合物149mg(収率:36.9%、異性体混合物のトータルGC純度:79%)を無色半固体として得た。
【0059】
実施例−16〜27、比較例1〜4
実施例で製造した本発明のスピロ型シクロトリシロキサン誘導体を、試験例−1に示す方法により、初期内圧及び製膜時間を変えて製膜し、得られた膜の膜厚、弾性率、硬度及び比誘電率を試験例−2及び3に示す方法により測定した(実施例−16〜27)。結果を表1に纏めて示した。比較例として市販のヘキサメチルシクロトリシロキサン(HMCTS)およびヘキサエチルシクロトリシロキサン(HECTS)を用いて、試験例1に従い製膜し、得られた膜の膜厚、弾性率、硬度及び比誘電率を試験例−2及び3に示す方法により測定し(比較例−1〜4)、結果を合わせて表1に示した。なお特に温度の表示の無いものはチャンバー内温55℃で製膜を行なった。
【0060】
試験例−1(製膜)
図1に示す、50mmの間隔で対向電極を上下に配した平行平板容量結合型PECVD装置を用い、片方の電極に基板として用いたシリコンウェーハを張付け、チャンバー内の所定の位置に本発明のスピロ型シクロトリシロキサン誘導体(約1g)を設置した。チャンバー内を真空ポンプで減圧し、ヒーター加熱により内温を所定温度に保持した。電源周波数13.56MHzのRF電源出力を所定の値にセットし、所定時間製膜を行った。
試験例−2(膜厚および比誘電率測定)
製膜された膜について日本分光株式会社製のエリプソメーター(型式:MEL−30S)を用いて膜厚と屈折率を測定し、屈折率を二乗することによって比誘電率とした。
試験例−3(弾性率および硬度測定)
製膜された膜について、Hysitron社製のTRIBOSCOPEを用いてナノインデンテーション法により弾性率および硬度を測定した。検量線はBerkovich型の圧子を用い、溶融石英を参照サンプルとして作成した。測定は膜の膜厚に対して押し込み深さを10%として測定を行った。
【0061】
【表1】
【符号の説明】
【0062】
1.PECVDチャンバー
2.基板
3.上部電極
4.下部電極
5.原料ガラス容器
6.原料
7.真空ポンプ
8.マッチング回路
9.RF電源
10.アース
11.ヒーター
【技術分野】
【0001】
本発明は、特にプラズマ促進化学気相蒸着(PECVD)法による製膜プロセスに有用なスピロ型シクロトリシロキサン誘導体及びその製造方法に関する。さらに本発明は、スピロ型シクロトリシロキサン誘導体を用いて製膜して得られる、機械的強度に優れた膜に関する。
【背景技術】
【0002】
有機ケイ素化合物をPECVD法などで基板上に堆積させた絶縁膜は、一般に化学構造が明確でないが、その構成元素から、SiOCH膜と呼ぶことがある。シクロトリシロキサン誘導体を原料に用いてPECVD法によってSiOCH膜を作製した例としては、特許文献1および2をあげることができる。更にこれらSiOCH膜は比較的低い誘電率を有する絶縁膜として利用できることが示されている。
【0003】
スピロ型シクロトリシロキサン誘導体の報告例として非特許文献1および2を挙げることができる。非特許文献1には2,2,4,4,6,6−トリ(1,4−ブタンジイル)シクロトリシロキサン及び2,2,4,4,6,6−トリ(1,5−ペンタンジイル)シクロトリシロキサンの合成について記載がある。また非特許文献2には2,2,4,4,6,6−トリ(2−ブテン−1,4−ジイル)シクロトリシロキサンが反応混合物中に存在していることを示されているものの、このものの単離精製は行われていない。これら非特許文献中では、いずれの化合物についても製膜材料としての提案および材料物性についての記載は無い。さらに先に挙げた特許文献2ではスピロ型シクロトリシロキサン誘導体として2,2−(ブタン−1,4−ジイル)−4,4,6,6−テトラメチルシクロトリシロキサン及び2,2−(ブタン−1,4−ジイル)−4,6−ジメチルシクロトリシロキサンの合成についての記載がある。しかしながら、いずれのスピロ型シクロトリシロキサン誘導体も、同一ケイ素原子上に結合しているアルキレン基またはアルケニレン基の炭素原子上に水素のみが結合しており、このアルキレン基またはアルケニレン基の炭素原子上に炭化水素基が結合したスピロ型シクロトリシロキサン誘導体の合成例は無い。
【0004】
然るにシクロトリシロキサンのすべてのケイ素原子上に、一つ以上のアルキル基で置換された1,4−ブタンジイル基または2−ブテン−1,4−ジイル基を導入したスピロ型シクロトリシロキサン誘導体が合成された例は無く、ましてやこれを用いて電気絶縁膜を作製した例も一切無い。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2007−96237号公報
【特許文献2】米国特許第6572923号明細書
【非特許文献】
【0006】
【非特許文献1】Francis J. Bajerら、Journal of Organic Chemistry,28巻(7号),1941ー1942頁(1963年)。
【0007】
【非特許文献2】A.N.Polivanovら、Zhurnal Obshchei Khimii,48巻(7号),1662頁(1978年)。
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明の課題は、所望の低い誘電率を有し、機械強度に優れた電気絶縁膜を形成可能なプレカーサーとしてのスピロ型シクロトリシロキサン誘導体及びその製造方法を提供することにある。さらにスピロ型シクロトリシロキサン誘導体を用いて製造される膜及びその製造方法を提供することにある。
【課題を解決するための手段】
【0009】
本発明者等は上記の課題を解決すべく鋭意検討した結果、シクロトリシロキサンのすべてのケイ素原子上に、アルキル基が置換されている1,4−ブタンジイル基または2−ブテン−1,4−ジイル基を導入したスピロ型シクロトリシロキサン誘導体が、上記課題を解決し得る性能を有する化合物であることを見いだし、本発明を完成させるに至った。
【0010】
すなわち本発明は、一般式(1)
【0011】
【化1】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されることを特徴とする、スピロ型シクロトリシロキサン誘導体である。
【0012】
また本発明は、一般式(2)
【0013】
【化2】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表し、Xはそれぞれ独立に塩素原子、メトキシ基又はエトキシ基を表す。)で表されるシラン誘導体を環化剤の存在下に反応させることを特徴とする、一般式(1)で表されるスピロ型シクロトリシロキサン誘導体の製造方法である。
【0014】
さらに本発明は、一般式(1)で表されるスピロ型シクロトリシロキサン誘導体を原料として用いて製膜することを特徴とする、膜の製造法である。また本発明は、上述の製造法により製造されることを特徴とする膜である。さらに本発明は、上述の膜から成ることを特徴とする、電気絶縁膜である。また本発明は、上述の電気絶縁膜を配してなることを特徴とする、半導体電子デバイスである。
【0015】
以下に本発明をさらに詳細に説明する。一般式(1)及び(2)並びに後述の一般式(2a)で表される化合物の置換基R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表すが、R1、R2、R3及びR4は同時に水素原子ではない。また、炭素数が1乃至3のアルキル基としてはメチル基、エチル基、プロピル基、イソプロピル基、シクロプロピル基を挙げることができる。PECVD法による製膜プロセスに一般式(1)で表されるスピロ型シクロトリシロキサン誘導体を用いる場合、蒸気圧の観点からR1、R2、R3又はR4のうち一つもしくは二つがメチル基で残りが水素原子、またはR1、R2、R3又はR4のうち一つがエチル基で三つが水素原子であることが望ましく、原料の入手の容易さなどからR2、R3及びR4が水素原子、R1がメチル基であり、破線を伴う実線が単結合であるか、R1、R3及びR4が水素原子、R2がメチル基であり、破線を伴う実線が単結合であるか、又は、R1、R3及びR4が水素原子、R2がメチル基であり、破線を伴う実線が二重結合であることがさらに望ましい。
【0016】
一般式(2)で表されるシラン誘導体を環化剤存在下に反応させることによって一般式(1)で表されるスピロ型シクロトリシロキサン誘導体の製造が可能である。このとき用いることのできる環化剤としては、一般式(2)で表されるシラン誘導体を環化させることができるものであれば特に限定はないが、例えばスルホキシド化合物、金属酸化物、水、または酸性水溶液等を挙げることができる。
【0017】
Xが塩素原子であるシラン誘導体(2)を用いて対応するスピロ型シクロトリシロキサン誘導体(1)を製造する際、環化剤としてスルホキシド化合物が好適に用いられる。用いることのできるスルホキシド化合物として、反応が進行すれば特に制限は無いが、ジメチルスルホキシド、テトラメチレンスルホキシド、ジオクチルスルホキシド、ジベンジルスルホキシド、ジフェニルスルホキシド、ジ−p−トリルスルホキシド、ジ−p−クロロフェニルスルホキシドなどを例示できる。反応が速やかに進行する点でジメチルスルホキシド、テトラメチレンスルホキシド、ジオクチルスルホキシドが好ましく、安価である点でジメチルスルホキシドが更に好ましい。スルホキシド化合物の添加量は特に制限は無いが、収率、効率の点でジクロロシラン化合物に対して2〜4当量加えることが望ましい。
【0018】
反応は有機溶媒中で行なうことができる。用いることのできる有機溶媒としてはシラン誘導体(2)やスルホキシド化合物と反応したり、溶解性が低かったりして所望の環化反応が阻害されなければ特に制限は無く、ヘキサン、シクロヘキサン、ヘプタン、ベンゼン、トルエン、キシレンなどの炭化水素系有機溶媒、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサンなどのエーテル系有機溶媒、酢酸エチル、アセトン、アセトニトリル、メチルエチルケトン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミドなどの非プロトン性の極性有機溶媒、四塩化炭素、クロロホルム、ジクロロメタン、1,2−ジクロロエタンなどのハロゲン系有機溶媒などを例示できるが、収率の点で炭化水素系有機溶媒、エーテル系有機溶媒およびハロゲン系有機溶媒が好適に用いられる。
【0019】
またシラン誘導体(2)とスルホキシド化合物の添加の方法は、スルホキシド化合物にシラン誘導体(2)を加えても、シラン誘導体(2)にスルホキシド化合物を加えても良い。反応温度はシラン誘導体(2)とスルホキシド化合物を混合できれば特に制限は無いが、−50〜100℃の範囲に、好ましくは−20℃〜50℃の範囲に制御して、反応を行なうことが好ましい。反応が30分間から1日程度で終了するように有機溶媒の種類や量、撹拌効率を制御すると良い。反応混合物から目的のスピロ構造を有するシクロトリシロキサン化合物は、水洗浄、カラムクロマトグラフィー、蒸留などを組み合わせて精製することができるが、反応で副生するスルフィド化合物由来の臭いがこれらの精製工程において除くことができない場合は、適宜活性炭処理、酸化剤の添加処理などを行なうと良い。
【0020】
Xが塩素原子であるシラン誘導体(2)を用いて対応するスピロ型シクロトリシロキサン誘導体(1)を製造する際、環化剤として金属酸化物も好適に用いられる。用いることのできる金属酸化物として、反応が進行すれば特に制限は無いが、副生物が少なく、所望の形状の金属酸化物の入手の容易さ、収率の点で、酸化亜鉛、酸化銅(II)及び酸化マグネシウムが好適に用いられる。環化剤として用いることのできる金属酸化物の形状として、反応が進行すれば特に制限は無いが、反応が速やかに進行する点で粉末状が望ましく、粒径1mm程度より細かい粉末が好適に用いられる。金属酸化物の添加量は特に制限は無いが、収率や効率の点でシラン誘導体(2)に対して1〜2当量加えることが望ましい。
【0021】
反応は有機溶媒中で行なうことができる。用いることのできる有機溶媒としてはシラン誘導体(2)や金属酸化物と反応したり、溶解性が低かったりして所望の環化反応が阻害されなければ特に制限は無く、ヘキサン、シクロヘキサン、ヘプタン、ベンゼン、トルエン、キシレンなどの炭化水素系有機溶媒、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサンなどのエーテル系有機溶媒、酢酸エチル、アセトン、アセトニトリル、メチルエチルケトン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミドなどの非プロトン性の極性有機溶媒、四塩化炭素、クロロホルム、ジクロロメタン、1,2−ジクロロエタンなどのハロゲン系有機溶媒などを例示できる。反応温度はシラン誘導体(2)と金属酸化物を混合できれば特に制限は無いが、−80〜150℃の範囲で行なうことができる。生産性や、反応制御のしやすさを考慮すると、例えば酸化亜鉛を用いた場合は、−50℃〜50℃の範囲で反応を行なうことが好ましく、酸化銅(II)又は酸化マグネシウムを用いた場合は、室温〜150℃の範囲で行なうことが好ましい。反応が30分間から1日程度で終了するように有機溶媒の種類や量、撹拌効率を制御すると良い。反応混合物から目的のシクロトリシロキサン化合物は、ろ過、水洗浄、カラムクロマトグラフィー、蒸留などを組み合わせて精製することができる。
【0022】
さらに本発明のスピロ型シクロトリシロキサン誘導体(1)は、シラン誘導体(2)に水または酸性水溶液を環化剤として作用させることによっても製造することができる。使用可能な酸としては塩酸、硫酸などの無機酸、p−トルエンスルホン酸、トリフルオロ酢酸、トリフルオロメタンスルホン酸などの有機酸などを例示できる。シラン誘導体(2)のXが塩素原子の場合は加水分解によって塩酸が発生するので、水を環化剤として加えることにより、スピロ型シクロトリシロキサン誘導体(1)を得ることもできる。この際、水の量は特に制限は無いがシラン誘導体(2)に対し1当量以上あれば良い。Xがメトキシ基あるいはエトキシ基の場合は、酸性水溶液を環化剤として作用させることにより、本発明のスピロ型シクロトリシロキサン誘導体(1)を得ることができる。使用可能な酸としては塩酸、硫酸などの無機酸、p−トルエンスルホン酸、トリフルオロ酢酸、トリフルオロメタンスルホン酸などの有機酸などを例示でき、これらの水溶液を環化剤として用いることができる。収率、取扱の容易さ、経済性などの観点から硫酸が好適に用いられる。加える酸の量に制限はなく、シラン誘導体(2)に対し0.01当量程度の触媒量でも大過剰量でも収率よくスピロ型シクロトリシロキサン誘導体(1)を得ることができる。酸性水溶液の使用量に特に制限はないが、反応液中の水がシラン誘導体(2)に対し1当量以上になるように使用することが好ましい。
【0023】
これらいずれの反応も有機溶媒を用いて行なうことができ、例えばヘキサン、シクロヘキサン、ヘプタン、ベンゼン、トルエン、キシレンなどの炭化水素系有機溶媒、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサンなどのエーテル系有機溶媒、四塩化炭素、クロロホルム、ジクロロメタン、1,2−ジクロロエタンなどのハロゲン系有機溶媒などを例示できる。反応温度は0℃以上で、有機溶媒の沸点および原料のシラン化合物の沸点や反応速度を考慮して決めれば良い。反応混合物から目的のスピロ型シクロトリシロキサン誘導体(1)は、水洗浄、カラムクロマトグラフィー、蒸留などを組み合わせて精製することができる。
【0024】
一般式(2a)
【0025】
【化3】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されるジクロロシラン誘導体に、メタノール又はエタノールを反応させてXがメトキシ基又はエトキシ基であるシラン誘導体(2)を得た(工程1)後、前述の水や酸性水溶液を環化剤として作用させる(工程2)ことにより、スピロ型シクロトリシロキサン誘導体(1)を得ることができる。工程1において、ジクロロシラン誘導体(2a)に、メタノール又はエタノールを反応させる際、トリエチルアミンやピリジンなどの有機塩基で副生する塩化水素を中和することができる。加えるメタノール又はエタノールの量はジクロロシラン誘導体(2a)に対して任意の量で良く、好ましくは2当量以上の過剰量を用いることにより収率良くXがメトキシ基又はエトキシ基であるシラン誘導体(2)を得ることができるが、必ずしもすべてのXをメトキシ基又はエトキシ基に変換しなくてもよい。工程1において反応終了後、蒸留精製によって2つのXが共にメトキシ基又はエトキシ基であるシラン誘導体(2)を分離することができるが、あえて分離する必要はなく、ジクロロシラン誘導体(2a)や、1つのXが塩素原子、もう1つのXがメトキシ基又はエトキシ基であるシラン誘導体(2)が混在していてもそのまま工程2に用いることができる。工程1と工程2は連続して行なっても良い。工程1で有機塩基を用いた場合は、工程2において液性が酸性になるように酸性水溶液を加えることによって、収率良くスピロ型シクロトリシロキサン誘導体(1)を得ることができる。
【0026】
本発明のスピロ型シクロトリシロキサン誘導体(1)を原料として用いて製膜することにより、膜を製造することができる。この膜は電気絶縁膜として使用することができ、半導体電子デバイスに配して用いることができる。膜は、スピンキャスト法、化学気相蒸着(CVD)法などにより製造することができるが、本発明の所望の低い誘電率を示し機械的強度に優れた絶縁膜を製造するためにPECVD法が好適に用いられる。その後、紫外線照射処理、オゾン処理などの硬化工程を行うことができる。
【0027】
スピロ型シクロトリシロキサン誘導体(1)は室温で固体または液体である。固体の場合、減圧下で昇華させてガスとしてPECVDプロセスに供することができる。また、融点以上に加熱することによって液体としたのち、気化させて原料ガスの供給系に導入することができる。スピロ型シクロトリシロキサン誘導体(1)は、例えば特許文献1で開示されている環状シロキサン化合物、2,4,6,8−テトラメチルシクロテトラシロキサン、テトラエトキシシランなどの、PECVD法で絶縁膜を製造可能な含ケイ素化合物と混合して原料ガスの供給系に導入することもできる。混合の割合は均一に混合できれば特に制限は無く、例えば、スピロ型シクロトリシロキサン誘導体(1)と特許文献1で開示されている環状シロキサン化合物やPECVD法で絶縁膜を製造可能な含ケイ素化合物とを重量パーセントで99対1〜10対90の割合で混合することが可能である。スピロ型シクロトリシロキサン誘導体(1)が室温で液体である場合、またはスピロ型シクロトリシロキサン誘導体(1)とPECVD法で絶縁膜を製造可能な含ケイ素化合物との混合物が液体である場合、ガスインジェクション法、リキッドインジェクション法のいずれを用いてPECVDプロセスに供しても良い。
【発明の効果】
【0028】
本発明によるスピロ型シクロトリシロキサン誘導体(1)を原料として用いて製膜することができる。得られた膜は、比誘電率2.5以下の低い値を示し、且つ弾性率が概ね10GPa以上、硬度が概ね1GPa以上である機械的強度に優れた電気絶縁膜であり、半導体電子デバイスに用いることができる。
【図面の簡単な説明】
【0029】
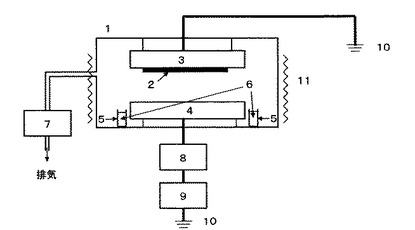
【図1】実施例ー16〜27、比較例ー1〜4で用いたPECVD製膜装置の概略図である。
【実施例】
【0030】
以下、参考例、実施例及び試験例を挙げて本発明をさらに詳細に説明するが、本発明はこれらに限定されるものではない。
【0031】
参考例−1
ジムロート冷却管、磁気撹拌子および滴下ロート(200mL)を備えた3口フラスコ(1000mL)をアルゴンで置換し、マグネシウム24.98g(1.028mol)および脱水ジエチルエーテル580mLを収め、滴下ロートから1,4−ジブロモペンタン100.7g(437.9mmol)をゆっくりと加えながら室温で12時間撹拌し、ジグリニャール試薬を調製した。別に、ジムロート冷却管、磁気撹拌子および滴下ロート(300mL)を備えた3口フラスコ(1000mL)を用意し、アルゴン雰囲気下、四塩化ケイ素74.55g(438.8mmol)のジエチルエーテル50mL溶液を収め、−78℃に冷却した。ここに、先に調製したジグリニャール試薬のエーテル溶液を滴下ロートからゆっくりと滴下し、12時間撹拌した。この混合物を更に室温で一日撹拌した。反応混合物をろ過後、ろ液を濃縮し、得られた粗生成物を減圧蒸留(沸点:45℃/1.6kPa)することにより、1,1−ジクロロ−2−メチル−1−シラシクロペンタンを無色液体として40.66g(収率:54.90%、GC純度:98%)得た。EI−MS(70eV),m/z(相対強度):168([M]+,27),140(100),127(53)。1H−NMR,δ(250MHz,CDCl3,ppm):1.117(d,3H,J=6.3Hz),1.20〜1.41(m,4H),1.41〜1.70(m,2H),1.83〜2.03(m,1H)。
【0032】
参考例−2
メチルコハク酸無水物はJournal of Chemical Society,Perkin Transactions 1、1980年、2029頁を参考に合成した。ジムロート冷却管、磁気撹拌子および滴下ロート(300mL)を備えた3口フラスコ(1000mL)に、メチルコハク酸101.2g(766.0mmol)を収め、滴下ロートから塩化アセチル400mLをゆっくり滴下した。この混合溶液を2時間撹拌し、濃縮後減圧蒸留(沸点:82℃/100Pa)することにより、メチルコハク酸無水物を無色液体として83.6g(収率:95.7%)得た。IR,ν(neat,cm−1):2989,2943,1770,1228,1063,991,903,725。
【0033】
参考例−3
2−メチル−1,4−ブタンジオールはThe Journal of Organic Chemistry、54巻(10号)、2471頁(1989年)を参考に合成した。ジムロート冷却管、磁気撹拌子および滴下ロート(300mL)を備えた3口フラスコ(2000mL)をアルゴンで置換し、水素化リチウムアルミニウム26.3g(705mmol)および脱水テトラヒドロフラン800mLを収めた。これに、メチルコハク酸無水物41.0g(359mmol)の脱水テトラヒドロフラン200mL溶液を滴下ロートより1時間かけて滴下し、2日間撹拌した。滴下ロートからゆっくりと水100mLとテトラヒドロフラン100mLの混合物を滴下し、得られたスラリーにさらにテトラヒドロフラン500mLを加えた。これをろ過し、ろ液をエバポレーターで濃縮後クロロホルム500mLを加えた。これを無水硫酸ナトリウムで乾燥し、ろ過後再度濃縮した。得られた粗生成物を蒸留(沸点:76℃/70Pa)することにより、2−メチル−1,4−ブタンジオールを無色粘稠液体として28.5g(収率:76.2%)得た。IR,ν(neat,cm−1):3280,2924,2873,1458,1381,1059,1005。
【0034】
参考例−4
1,4−ジブロモ−2−メチルブタンはJournal of The American Chemical Society、73巻(8号)、3632頁(1951年)を参考に合成した。リービッヒ冷却管、磁気撹拌子および滴下ロートを備えた3口フラスコ(200mL)に2−メチル−1,4−ブタンジオール28.0g(269mmol)を収め、別のフラスコ中でテトラリンと臭素の反応により発生させた臭化水素ガスを、70℃で3時間吹き込んだ。これを分液ロートに移し、ジエチルエーテルで希釈後に水で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、さらに水素化カルシウムで乾燥した。これを濃縮後減圧蒸留(沸点:82℃/2.4kPa)することにより、1,4−ジブロモ−2−メチルブタンを無色液体として31.6g(収率:51.1%)得た。IR,ν(neat,cm−1):2964,2931,1458,1438,1379,1261,1234,746。
【0035】
参考例−5
ジムロート冷却管、磁気撹拌子および滴下ロート(200mL)を備えた3口フラスコ(300mL)に削り状マグネシウム8.53g(0.351mol)を収め、アルゴンで置換した。これに脱水ジエチルエーテル50mLを加え、少量の1,2−ジブロモエタンでマグネシウムを活性化させた。これに1時間かけて1,4−ジブロモ−2−メチルブタン25.0g(109mmol)の脱水ジエチルエーテル90mL溶液をゆっくりと滴下し、12時間撹拌してジグリニャール試薬を調製した。別に、ジムロート冷却管、磁気撹拌子および滴下ロート(300mL)を備えた3口フラスコ(500mL)をアルゴンで置換し、四塩化ケイ素20.4g(120mmol)および脱水ジエチルエーテルを収め、先に調製したジグリニャール試薬を滴下した。これを3日間撹拌後、生じた塩をろ過して除いた。ろ液を濃縮後減圧蒸留(沸点:89℃/12kPa)することにより、1,1−ジクロロ−3−メチル−1−シラシクロペンタンを無色液体として6.0g(収率:33%)得た。EI−MS(70eV),m/z(相対強度):168(M+,37),153(3),140(100),127(74),125(60),112(28)。1H−NMR,δ(500MHz,CDCl3,ppm):0.72(dd,1H,J=10.8,15.4),0.80〜0.90(m,1H),1.10(d,3H,J=6.5Hz),1.20〜1.30(m,1H),1.36(ddd,1H,J=2.9,7.2,15.1Hz),1.43(ddd,1H,J=2.0,6.5,15.4Hz),1.88〜1.96(m,1H),1.96〜2.03(m,1H)。13C−NMR,δ(126MHz,CDCl3,ppm):18.6,22.8,27.1,33.0,33.6。29Si−NMR,δ(99MHz,CDCl3,ppm):44.6。IR,ν(neat,cm−1):2954,2924,2870,1456,1396,1076,820,789,750,735,675。
【0036】
参考例−6
1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エンはJournal of Organometallic Chemistry、391巻(1号)、7頁(1990年)を参考に合成した。アルゴン置換したステンレス製オートクレーブ(200mL)に、トリクロロシラン60.1g(444mmol)、イソプレン13.8g(203mmol)およびテトラブチルホスホニウムクロリド2.30g(7.80mmol)を収め、密封後150℃で13時間反応させた。反応終了後大気圧に戻し、得られた反応混合物中の低沸点分を減圧下で留去した。残さを減圧蒸留(沸点:70℃/5.5kPa)し、1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エンを無色透明液体として21.1g(収率:62.2%、GC純度:96%)得た。EI−MS(70eV),m/z(相対強度):166(M+,44),151(10),138(15),130(100)。IR,ν(neat,cm−1):3016,2964,2914,1598,1390,1159,1024,729。
【0037】
参考例−7
ジムロート冷却管および撹拌子を備えた2口フラスコ(100mL)に、10%パラジウム担持炭素粉末730mg(パラジウム:0.686mmol)を収め、減圧下で1時間乾燥した。容器をアルゴンで置換し、1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エン7.35g(44.0mmol)を加えた。反応容器を1気圧の水素ガスで置換し、12時間撹拌した。反応混合物を室温、減圧下(0.1kPa)で留去し、留去成分を回収した。これを蒸留精製(沸点:159℃)することにより、1,1−ジクロロ−3−メチル−1−シラシクロペンタンを無色液体として7.33g(収率:98.5%,GC純度:95%)得た。
【0038】
参考例−8
1,1−ジクロロ−3,4−ジメチル−1−シラシクロペンタ−3−エンはOrganometallics、22巻(13号)、2551頁(2003年)を参考に合成した。撹拌子を備えたステンレス製オートクレーブ(200mL)をアルゴンで置換し、2,3−ジメチル−1,3−ブタジエン16.4g(200mmol)、トリクロロシラン80.5g(594mmol)および無水テトラブチルホスホニウムクロリド2.70g(9.16mmol)を収め、密閉した。これを激しく撹拌しながら150℃で48時間加熱した。混合物をナスフラスコ(100mL)に移し、減圧下で過剰のトリクロロシランを除いた。残さを減圧蒸留(沸点:78℃/2.8kPa)することにより、1,1−ジクロロ−3,4−ジメチル−1−シラシクロペンタ−3−エン29.6g(収率:81.8%)を無色液体として得た。EI−MS(70eV),m/z(相対強度):180(M+,100),165(73),144(59),138(40),129(62)。1H−NMR,δ(250MHz,CDCl3,ppm):1.751(t,6H,J=1Hz),1.882(m,4H)。IR,ν(neat,cm−1):2983,2914,1441,1392,1173,982,769,752,694。
【0039】
実施例−1
アルゴン置換した2口フラスコ(100mL)に脱水トルエン50mLおよび脱水ジメチルスルホキシド8.42g(108mmol)を収め、シリンジより1,1−ジクロロ−2−メチル−1−シラシクロペンタン5.13g(30.3mmol)を1時間かけてゆっくりと滴下した。反応混合物を2時間撹拌し、分液ロートに移して水(50mL)を加えて有機層を抽出した。有機層を濃縮後、クーゲルロールを用いて蒸留(蒸留温度:108℃/5Pa)することにより、2,2,4,4,6,6−トリ(1,4−ペンタンジイル)シクロトリシロキサン(化合物1)の異性体混合物0.97g(収率:28%、異性体混合物のトータルGC純度:99%)を得た。融点:88.3℃.EI−MS(70eV),m/z(相対強度):262(M+,16),247(33),234(27),219(27),193(100)。1H−NMR,δ(250MHz,CDCl3,ppm):0.45〜0.60(m,3H),0.60〜0.80(m,6H),0.95〜1.05(m,9H),1.10〜1.25(m,3H),1.30〜1.50(m,3H),1.70〜1.90(m,3H)。
【0040】
実施例−2
リービッヒ冷却管および撹拌子を備えた2口ナスフラスコ(200mL)に酸化亜鉛10.09g(124.0mmol)を収め、減圧乾燥後にアルゴンで置換した。反応容器に脱水酢酸エチル140mLを収め、1,1−ジクロロ−2−メチル−1−シラシクロペンタン15.84g(93.66mmol)をシリンジより1時間かけて滴下したのち、混合物を12時間撹拌した。水150mLを加えて、有機層を抽出し、硫酸ナトリウムで乾燥させた。ろ液を濃縮後クーゲルロール蒸留(蒸留温度:110℃/0.5Pa)することにより3.30g(収率:30.8%、異性体混合物のトータルGC純度:99%)の化合物1を無色固体として得た。
【0041】
実施例−3
撹拌子を備えたシュレンク管(100mL)に酸化銅(II)800mg(10.1mmol)、1,1−ジクロロ−2−メチル−1−シラシクロペンタン1.69g(10.0mmol)および脱水テトラヒドロフラン20mLを収めた。これを室温で80時間撹拌した。反応混合物にヘキサン50mLを加え、ろ過し、ロータリーエバポレーターで濃縮した。得られた混合物をクーゲルロール蒸留(蒸留温度145℃/60Pa)することにより、183mg(収率:16.0%、異性体混合物のトータルGC純度:94%)の化合物1を無色結晶性固体として得た。
【0042】
実施例−4
ジムロート冷却管、撹拌子および滴下ロート(50mL)を備えた3口フラスコ(300mL)にジエチルエーテル250mL、トリエチルアミン6.10g(60.2mmol)および1,1−ジクロロ−2−メチル−1−シラシクロペンタン4.99g(29.5mmol)を収めた。混合物を0℃に冷却し、滴下ロートから蒸留水540mg(30.0mmol)のテトラヒドロフラン50mL溶液をゆっくり加えた。これを室温に戻し、14時間撹拌した。反応混合物を分液ロートに移し、水300mLを加えて、有機層を抽出した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。得られた混合物をクーゲルロール蒸留(蒸留温度:140℃/50Pa)することにより、543mg(収率:16.1%、異性体混合物のトータルGC純度:93%)の化合物1を無色結晶性固体として得た。
【0043】
実施例−5
ジムロート冷却管および撹拌子を備えた2口フラスコ(100mL)をアルゴンで置換し、脱水ベンゼン24mLおよび脱水ジメチルスルホキシド2.77g(35.5mmol)を収めた。シリンジより1,1−ジクロロ−3−メチル−1−シラシクロペンタン2.00g(11.8mmol)を滴下し、室温で13時間撹拌した。反応混合物を分液ロートに移し、水(30mL)を加えて有機層を抽出した。有機層を無水硫酸ナトリウムで乾燥し、ろ過後濃縮した。これをクーゲルロールを用いて減圧蒸留(蒸留温度:130℃/50Pa)することにより、2,2,4,4,6,6−トリ(2−メチルブタン−1,4−ジイル)シクロトリシロキサン(化合物2)の異性体混合物311mg(収率:23%、異性体混合物のトータルGC純度:94%)を無色固体として得た。EI−MS(70eV),m/z(相対強度):342(M+,25),314(100),300(41),286(35),272(53),244(62)。1H−NMR,δ(250MHz,CDCl3,ppm):0.05〜0.19(m,3H),0.42〜0.59(m,3H),0.50〜0.90(m,6H),1.00(d,9H,J=6.3Hz),0.92〜1.10(m,3H),1.60〜1.88(m,6H)。IR,ν(neat,cm−1):2947,2922,2865,1454,1255,1074,1002,972,829,757。
【0044】
実施例−6
ジムロート冷却管および撹拌子を備えた3口フラスコ(200mL)に酸化マグネシウム480mg(11.9mmol)、1,1−ジクロロ−3−メチル−1−シラシクロペンタン2.00g(11.8mmol)および脱水テトラヒドロフラン30mLを収めた。混合物を70℃で16時間加熱撹拌した。得られた反応混合物をロータリーエバポレーターで濃縮し、残さにヘキサン50mLを加え、分液ロートに移して水50mLで洗浄した。有機層を無水硫酸マグネシウムで乾燥し、ロータリーエバポレーターで濃縮した。得られた混合物をクーゲルロール蒸留(蒸留温度135℃/50Pa)することにより、215mg(収率:15.9%、異性体混合物のトータルGC純度:99%)の化合物2を無色結晶性固体として得た。
【0045】
実施例−7
ジムロート冷却管、撹拌子および滴下ロート(100mL)を備えた2口フラスコ(200mL)にジエチルエーテル150mL、トリエチルアミン4.59g(45.4mmol)および1,1−ジクロロ−3−メチル−1−シラシクロペンタン3.64g(21.6mmol)を収めた。混合物を0℃に冷却し、滴下ロートから蒸留水400mg(22.2mmol)のテトラヒドロフラン65mL溶液をゆっくり加えた。滴下後にゆっくりと室温に戻し、4時間撹拌した。反応混合物を分液ロートに移し、水100mLで有機層を2回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。これをクーゲルロール蒸留(蒸留温度:135℃/50Pa)することにより、391mg(収率:15.9%、異性体混合物のトータルGC純度:96%)の化合物2を無色結晶性固体として得た。
【0046】
実施例−8
ベンゼン200mLおよび脱水ジメチルスルホキシド23.4g(300mmol)の混合物中に、1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エン16.7g(99.9mmol)の20mLベンゼン溶液をゆっくり滴下した。滴下後混合物を4時間撹拌した。反応混合物を分液ロートに移し、水200mLを加えて有機層を抽出した。有機層を無水塩化カルシウムで乾燥後濃縮し、次いでクーゲルロールを用いて蒸留(蒸留温度:140℃/50Pa)することにより、2,2,4,4,6,6−トリ(2−メチル−2−ブテン−1,4−ジイル)シクロトリシロキサン(化合物3)の異性体混合物5.13g(収率:45.8%、異性体混合物のトータルGC純度:95%)を無色液体として得た。これをシリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン)で精製し、さらにクーゲルロールを用いて蒸留し、化合物3の異性体混合物1.59g(収率:14.2%、異性体混合物のトータルGC純度:99%)を得た。EI−MS(70eV),m/z(相対強度):336(M+,100),308(79),268(32),133(32)。1H−NMR,δ(250MHz,CDCl3,ppm):1.23(m,6H),1.33(m,6H),1.73(m,9H),5.52(m,3H)。13C−NMR,δ(126MHz,C6D6,ppm):17.27(Si−CH2),20.21(Si−CH2),23.55(−CH3),123.73(−CH=C(Me)−),123.79(−CH=C(Me)−),123.85(−CH=C(Me)−),138.76(−CH=C(Me)−),138.82(−CH=C(Me)−),138.88(−CH=C(Me)−)。29Si−NMR,δ(99MHz,C6D6,ppm):−6.64。IR,ν(neat,cm−1):2962,2914,1437,1377,1159,1030,1012,787,764。
【0047】
実施例−9
ジムロート冷却管および撹拌子を備えた2口フラスコ(100mL)に酸化亜鉛1.95g(24.0mmol)を収め、真空下で乾燥させた。このものに、脱水ジエチルエーテル50mLおよび1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エン3.97g(23.9mmol)をアルゴン雰囲気下で加えた。この混合物を室温で14時間撹拌した。反応混合物を分液ロートに移し、水50mLを加えて有機層を抽出した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。残さをクーゲルロール蒸留(蒸留温度:145℃/60Pa)することにより、261mg(収率:9.8%、異性体混合物のトータルGC純度:98%)の化合物3を無色液体として得た。
【0048】
実施例−10
ジムロート冷却管、撹拌子および滴下ロート(50mL)を備えた3口フラスコ(200mL)にジエチルエーテル200mL、トリエチルアミン4.99g(49.3mmol)および1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エン4.00g(23.9mmol)を収めた。混合物を0℃に冷却し、滴下ロートから蒸留水490mg(27.2mmol)のテトラヒドロフラン40mL溶液をゆっくり加え、徐々に室温に戻し、14時間撹拌した。反応混合物を分液ロートに移し、水200mLを加えて有機層を抽出した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。これをクーゲルロール蒸留(蒸留温度:135℃/50Pa)することにより、62mg(収率:2.3%、異性体混合物のトータルGC純度:95%)の化合物3を無色液体として得た。
【0049】
実施例−11
ジムロート冷却管、撹拌子および滴下ロート(50mL)を備えた3口フラスコ(200mL)をアルゴンで置換し、脱水ベンゼン75mLおよび脱水ジメチルスルホキシド11.7g(150mmol)を収めた。この混合物に、滴下ロートから1,1−ジクロロ−3,4−ジメチル−1−シラシクロペンタ−3−エン9.05g(50.0mmol)のベンゼン25mL溶液を30分かけて滴下し、室温で4時間撹拌した。反応混合物を分液ロートに移し、水100mLを加えて有機層を抽出した。有機層を無水塩化カルシウムで乾燥し、ろ過後濃縮した。残さをクーゲルロールを用いて減圧蒸留(蒸留温度:150℃/45Pa)することにより、2,2,4,4,6,6−トリ(2,3−ジメチル−2−ブテン−1,4−ジイル)シクロトリシロキサン2.09g(収率:33.1%、GC純度:99%)を無色結晶として得た。1H−NMR,δ(250MHz,CDCl3,ppm):1.342(m,12H),1.702(s,18H)。IR,ν(neat,cm−1):2978,2906,2879,1439,1171,1130,1009。
【0050】
参考例−9
ジムロート冷却管、撹拌子および滴下ロート(200mL)を備えた3口フラスコ(1000mL)に、アルゴン雰囲気下で脱水ジエチルエーテル900mL、1,1−ジクロロ−2−メチル−1−シラシクロペンタン20.3g(0.120mol)および脱水トリエチルアミン24.3g(0.240mol)を収めた。この混合物に滴下ロートから脱水エタノール11.1g(0.240mol)の脱水ジエチルエーテル100mL溶液を2時間かけて滴下した後、室温で16時間撹拌し、生じたアミン塩酸塩をろ過により除き、ろ液を減圧濃縮した。残さを減圧蒸留(沸点:70℃/1.7kPa)することにより、1,1−ジエトキシ−2−メチル−1−シラシクロペンタン10.8g(収率:48.0%、GC純度95%)を無色液体として得た。EI−MS(70eV),m/z(相対強度):188(M+,38),160(82),145(51),132(29),131(28),118(100),103(60)。1H−NMR,δ(500MHz,CDCl3,ppm):0.45〜0.53(m,1H),0.58〜0.65(m,1H),0.76〜0.85(m,1H),1.01(d,3H,J=7.5Hz),1.12〜1.18(m,1H),1.20(t,6H,J=7.0Hz),1.36〜1.42(m,1H),1.33〜1.44(m,1H),1.71〜1.84(m,2H),3.76(q,3H,J=7.0Hz),3.80(q,3H,J=7.0Hz)。13C−NMR,δ(126MHz,CDCl3,ppm):8.5,14.2,17.0,18.3,18.4,22.5,35.2,58.7,58.8。29Si−NMR,δ(99MHz,CDCl3,ppm):7.3。IR,ν(neat,cm−1):2972,2925,2866,1389,1101,1076,949,789,760。
【0051】
参考例−10
ジムロート冷却管、撹拌子および滴下ロート(100mL)を備えた3口フラスコ(200mL)をアルゴン置換し、脱水ジエチルエーテル100mL、1,1−ジクロロ−2−メチル−1−シラシクロペンタン5.89g(34.8mmol)および脱水トリエチルアミン7.50g(74.1mmol)を収めた。この混合物に、滴下ロートから脱水メタノール2.27g(70.8mmol)の脱水テトラヒドロフラン80mL溶液を1時間かけて滴下し、室温で12時間撹拌し、生じたアミン塩酸塩をろ過により除き、ろ液を濃縮した。濃縮物を減圧蒸留(沸点:81℃/5.8kPa)することにより、1,1−ジメトキシ−2−メチル−1−シラシクロペンタン4.93g(収率:88.3%、GC純度:94%)を無色液体として得た。EI−MS(70eV),m/z(相対強度):160(M+,33),132(100),118(62),117(54),104(45)。1H−NMR,δ(500MHz,CDCl3,ppm):0.52(ddd,1H,J=8.4,9.5,15.3Hz),0.62〜0.69(m,1H),0.82〜0.90(m,1H),1.06(d,3H,J=7.5Hz),1.15〜1.24(m,1H),1.38〜1.47(m,1H),1.75〜1.89(m,2H),3.55(s,3H),3.58(s,3H)。13C−NMR,δ(126MHz,CDCl3,ppm):7.6,14.1,16.7,22.5,35.1,50.7,50.9。29Si−NMR,δ(99MHz,CDCl3,ppm):10.9。IR,ν(neat,cm−1):2937,2835,1454,1188,1076,800,773,731。
【0052】
参考例−11
ジムロート冷却管、撹拌子および滴下ロート(200mL)を備えた3口フラスコ(500mL)に、アルゴン雰囲気下で脱水ジエチルエーテル300mL、1,1−ジクロロ−3−メチル−1−シラシクロペンタン17.0g(0.101mol)および脱水トリエチルアミン20.7g(0.204mol)を収めた。この混合物に、滴下ロートから脱水エタノール9.50g(0.206mol)の脱水テトラヒドロフラン200mL溶液を1時間かけて滴下した後、室温で3時間撹拌した。生じたアミン塩酸塩をろ過により除き、ろ液を濃縮した。濃縮物を減圧蒸留(沸点:74℃/2.5kPa)することにより、1,1−ジエトキシ−3−メチル−1−シラシクロペンタン15.4g(収率:81.5%、GC純度97%)を無色液体として得た。EI−MS(70eV),m/z(相対強度):188(M+,17),160(63),146(32),145(28),131(20),118(100),103(51)。1H−NMR,δ(500MHz,CDCl3,ppm):0.12(dd,1H,J=11.0,15.0Hz),0.49〜0.56(m,1H),0.75〜0.81(m,1H),0.83〜0.89(m,2H),1.03(d,3H,J=6.5Hz),1.23(t,3H,J=7.0Hz),1.24(t,3H,J=7.0Hz),1.65〜1.73(m,1H),1.82〜1.89(m,1H),3.79(q,3H,J=7.0Hz).3.80(q,3H,J=7.0Hz)。13C−NMR,δ(126MHz,CDCl3,ppm)9.5,18.3,18.4,23.8,33.2,33.3,58.7,58.8。29Si−NMR,δ(99MHz,CDCl3,ppm)11.4。IR,ν(neat,cm−1)2949,2924,1389,1101,1074,949,822,771。
【0053】
参考例−12
ジムロート冷却管、撹拌子および滴下ロート(200mL)を備えた3口フラスコ(500mL)に、アルゴン雰囲気下で脱水ジエチルエーテル300mL、1,1−ジクロロ−3−メチル−1−シラシクロペンタ−3−エン18.3g(0.110mol)および脱水トリエチルアミン32.1g(0.317mol)を収めた。混合物に滴下ロートから脱水エタノール13.5g(0.293mol)の脱水テトラヒドロフラン200mL溶液を1時間かけて滴下した後、室温で3時間撹拌した。生じたアミン塩酸塩をろ過により除き、ろ液を濃縮した。濃縮物を減圧蒸留(沸点:73℃/1.4kPa)することにより、1,1−ジエトキシ−3−メチル−1−シラシクロペンタ−3−エン15.8g(収率:77.1%、GC純度:97%)を無色液体として得た。EI−MS(70eV),m/z(相対強度):186(M+,57),157(18),140(100),113(47),103(36)。1H−NMR,δ(500MHz,CDCl3,ppm):1.20(s,br,1H),1.24(t,6H,J=7.0Hz),1.30(s,br,2H),1.76(s,br,3H),3.82(q,4H,J=7.0Hz),5.56〜5.57(m,1H)。13C−NMR,δ(126MHz,CDCl3,ppm):14.3,17.7,18.3,23.5,58.9,123.7,139.0。29Si−NMR,δ(99MHz,CDCl3,ppm):8.4。IR,ν(neat,cm−1):2972,2885,1390,1157,1074,949,773,750。
【0054】
実施例−12
ジムロート冷却管および撹拌子を備えた2口フラスコ(50mL)にジエチルエーテル10mL、蒸留水2.30g(128mmol)および硫酸10.5g(107mmol)を収めた。これを0℃に冷却し、1,1−ジエトキシ−2−メチル−1−シラシクロペンタン3.49g(18.5mmol)をゆっくり加えた。混合物をゆっくりと室温に戻し、24時間撹拌した。ジムロート冷却管をト字管に付け替え、80℃まで昇温して低沸点溶媒を留去した。残さを分液ロートに移し、ヘキサン50mLを加え、水50mLで有機層を3回洗浄した。有機層を無水硫酸マグネシウムで乾燥後、ロータリーエバポレーターで濃縮した。残さをクーゲルロール蒸留(蒸留温度:130℃/50Pa)することにより、1.11g(収率:52.3%、異性体混合物のトータルGC純度:97%)の化合物1を無色結晶性固体として得た。
【0055】
実施例−13
ジムロート冷却管および撹拌子を備えた2口フラスコ(50mL)に蒸留水4.13g(229mmol)、硫酸7.14g(72.8mmol)およびジエチルエーテル25mLを収めた。これを0℃に冷却し、1,1−ジメトキシ−2−メチル−1−シラシクロペンタン1.71g(10.6mmol)をゆっくり加えた。混合物を室温に戻し、12時間撹拌した。反応混合物を分液ロートに移し、ヘキサン50mLを加え、水50mLで有機層を3回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。残さをクーゲルロール蒸留(蒸留温度125℃/40Pa)することにより、564mg(収率:46.4%、異性体混合物のトータルGC純度:99%)の化合物1を無色結晶性固体として得た。
【0056】
実施例−14
ジムロート冷却管および撹拌子を備えた2口フラスコ(50mL)にジエチルエーテル25mL、蒸留水3.09g(172mmol)および硫酸2.51g(25.6mmol)を収めた。この混合物をを0℃に冷却し、1,1−ジエトキシ−3−メチル−1−シラシクロペンタン4.74g(25.1mmol)をゆっくり加え、徐々に室温に戻した後、さらに40時間撹拌した。ジムロート冷却管をト字管に付け替え、80℃まで昇温して低沸点成分を溜去した。残査を分液ロートに移し、ヘキサン50mLを加え、水50mLで有機層を3回洗浄した。有機層を無水硫酸マグネシウムで乾燥後、ロータリーエバポレーターで濃縮した。残査をクーゲルロール蒸留(蒸留温度:140℃/50Pa)することにより、384mg(収率:13.4%、異性体混合物のトータルGC純度:93%)の化合物2を無色結晶性固体として得た。
【0057】
実施例−15
ジムロート冷却管および撹拌子を備えた2口フラスコ(50mL)にジエチルエーテル25mL、蒸留水3.04g(169mmol)および硫酸2.52g(25.7mmol)を収めた。この混合物を0℃に冷却し、1,1−ジエトキシ−3−メチル−1−シラシクロペンタ−3−エン3.70g(19.9mmol)をゆっくり加え、徐々に室温に戻した後、さらに18時間撹拌した。反応混合物を分液ロートに移し、ヘキサン50mLを加え、水50mLで有機層を3回洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ロータリーエバポレーターで濃縮した。残査をクーゲルロール蒸留(蒸留温度:140℃/50Pa)することにより、120mg(収率:5.4%、異性体混合物のトータルGC純度:94%)の化合物3を無色液体として得た。
【0058】
参考例−13
ステンレス製オートクレーブに撹拌子を入れ、アルゴン置換した。この中に酸化白金100mg、化合物3を396mg(1.18mmol)、および脱水テトラヒドロフラン5mLを収め、水素ガスで内部を置換した。これを90℃で5日間撹拌した。反応混合物をセライトでろ過し、ろ液をロータリーエバポレーターで濃縮した。残査をクーゲルロールで蒸留(蒸留温度:110〜140℃/50Pa)し、化合物2の異性体混合物149mg(収率:36.9%、異性体混合物のトータルGC純度:79%)を無色半固体として得た。
【0059】
実施例−16〜27、比較例1〜4
実施例で製造した本発明のスピロ型シクロトリシロキサン誘導体を、試験例−1に示す方法により、初期内圧及び製膜時間を変えて製膜し、得られた膜の膜厚、弾性率、硬度及び比誘電率を試験例−2及び3に示す方法により測定した(実施例−16〜27)。結果を表1に纏めて示した。比較例として市販のヘキサメチルシクロトリシロキサン(HMCTS)およびヘキサエチルシクロトリシロキサン(HECTS)を用いて、試験例1に従い製膜し、得られた膜の膜厚、弾性率、硬度及び比誘電率を試験例−2及び3に示す方法により測定し(比較例−1〜4)、結果を合わせて表1に示した。なお特に温度の表示の無いものはチャンバー内温55℃で製膜を行なった。
【0060】
試験例−1(製膜)
図1に示す、50mmの間隔で対向電極を上下に配した平行平板容量結合型PECVD装置を用い、片方の電極に基板として用いたシリコンウェーハを張付け、チャンバー内の所定の位置に本発明のスピロ型シクロトリシロキサン誘導体(約1g)を設置した。チャンバー内を真空ポンプで減圧し、ヒーター加熱により内温を所定温度に保持した。電源周波数13.56MHzのRF電源出力を所定の値にセットし、所定時間製膜を行った。
試験例−2(膜厚および比誘電率測定)
製膜された膜について日本分光株式会社製のエリプソメーター(型式:MEL−30S)を用いて膜厚と屈折率を測定し、屈折率を二乗することによって比誘電率とした。
試験例−3(弾性率および硬度測定)
製膜された膜について、Hysitron社製のTRIBOSCOPEを用いてナノインデンテーション法により弾性率および硬度を測定した。検量線はBerkovich型の圧子を用い、溶融石英を参照サンプルとして作成した。測定は膜の膜厚に対して押し込み深さを10%として測定を行った。
【0061】
【表1】
【符号の説明】
【0062】
1.PECVDチャンバー
2.基板
3.上部電極
4.下部電極
5.原料ガラス容器
6.原料
7.真空ポンプ
8.マッチング回路
9.RF電源
10.アース
11.ヒーター
【特許請求の範囲】
【請求項1】
一般式(1)
【化1】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されることを特徴とする、スピロ型シクロトリシロキサン誘導体。
【請求項2】
R2、R3及びR4が水素原子、R1がメチル基であり、破線を伴う実線が単結合であるか、R1、R3及びR4が水素原子、R2がメチル基であり、破線を伴う実線が単結合であるか、又は、R1、R3及びR4が水素原子、R2がメチル基であり、破線を伴う実線が二重結合である、請求項1に記載のスピロ型シクロトリシロキサン誘導体。
【請求項3】
一般式(2)
【化2】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表し、Xはそれぞれ独立に塩素原子、メトキシ基又はエトキシ基を表す。)で表されるシラン誘導体を環化剤の存在下に反応させることを特徴とする、一般式(1)
【化3】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されるスピロ型シクロトリシロキサン誘導体の製造方法。
【請求項4】
環化剤がスルホキシド化合物、金属酸化物、水又は酸性水溶液である、請求項3に記載の製造方法。
【請求項5】
Xが塩素原子である一般式(2)で表されるシラン誘導体に、環化剤としてジメチルスルホキシド、酸化亜鉛、酸化銅(II)、酸化マグネシウム又は水を作用させる、請求項3又は4に記載の製造方法。
【請求項6】
Xがメトキシ基又はエトキシ基である一般式(2)で表されるシラン誘導体に、環化剤として硫酸水溶液を作用させる、請求項3又は4に記載の製造方法。
【請求項7】
一般式(2a)
【化4】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。破線を伴う実線は単結合又は二重結合を表す。)で表されるジクロロシラン誘導体に、メタノール又はエタノールを反応させて得られた、Xがメトキシ基又はエトキシ基である一般式(2)で表されるシラン誘導体を用いる、請求項3、4又は6に記載の製造方法。
【請求項8】
一般式(1)
【化5】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されるスピロ型シクロトリシロキサン誘導体を原料として用いて製膜することを特徴とする、膜の製造法。
【請求項9】
製膜をプラズマ促進化学気相蒸着法により行う、請求項8に記載の製造法。
【請求項10】
請求項8または9に記載の製造法により製造されることを特徴とする膜。
【請求項11】
請求項10に記載の膜から成ることを特徴とする、電気絶縁膜。
【請求項12】
請求項11に記載の電気絶縁膜を配してなることを特徴とする、半導体電子デバイス。
【請求項1】
一般式(1)
【化1】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されることを特徴とする、スピロ型シクロトリシロキサン誘導体。
【請求項2】
R2、R3及びR4が水素原子、R1がメチル基であり、破線を伴う実線が単結合であるか、R1、R3及びR4が水素原子、R2がメチル基であり、破線を伴う実線が単結合であるか、又は、R1、R3及びR4が水素原子、R2がメチル基であり、破線を伴う実線が二重結合である、請求項1に記載のスピロ型シクロトリシロキサン誘導体。
【請求項3】
一般式(2)
【化2】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表し、Xはそれぞれ独立に塩素原子、メトキシ基又はエトキシ基を表す。)で表されるシラン誘導体を環化剤の存在下に反応させることを特徴とする、一般式(1)
【化3】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されるスピロ型シクロトリシロキサン誘導体の製造方法。
【請求項4】
環化剤がスルホキシド化合物、金属酸化物、水又は酸性水溶液である、請求項3に記載の製造方法。
【請求項5】
Xが塩素原子である一般式(2)で表されるシラン誘導体に、環化剤としてジメチルスルホキシド、酸化亜鉛、酸化銅(II)、酸化マグネシウム又は水を作用させる、請求項3又は4に記載の製造方法。
【請求項6】
Xがメトキシ基又はエトキシ基である一般式(2)で表されるシラン誘導体に、環化剤として硫酸水溶液を作用させる、請求項3又は4に記載の製造方法。
【請求項7】
一般式(2a)
【化4】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。破線を伴う実線は単結合又は二重結合を表す。)で表されるジクロロシラン誘導体に、メタノール又はエタノールを反応させて得られた、Xがメトキシ基又はエトキシ基である一般式(2)で表されるシラン誘導体を用いる、請求項3、4又は6に記載の製造方法。
【請求項8】
一般式(1)
【化5】
(式中、R1、R2、R3及びR4はそれぞれ独立に水素原子又は炭素数が1乃至3のアルキル基を表す。但し、R1、R2、R3及びR4は同時に水素原子ではない。また破線を伴う実線は単結合又は二重結合を表す。)で表されるスピロ型シクロトリシロキサン誘導体を原料として用いて製膜することを特徴とする、膜の製造法。
【請求項9】
製膜をプラズマ促進化学気相蒸着法により行う、請求項8に記載の製造法。
【請求項10】
請求項8または9に記載の製造法により製造されることを特徴とする膜。
【請求項11】
請求項10に記載の膜から成ることを特徴とする、電気絶縁膜。
【請求項12】
請求項11に記載の電気絶縁膜を配してなることを特徴とする、半導体電子デバイス。
【図1】

【公開番号】特開2011−111399(P2011−111399A)
【公開日】平成23年6月9日(2011.6.9)
【国際特許分類】
【出願番号】特願2009−267358(P2009−267358)
【出願日】平成21年11月25日(2009.11.25)
【出願人】(000003300)東ソー株式会社 (1,901)
【出願人】(000173762)公益財団法人相模中央化学研究所 (151)
【Fターム(参考)】
【公開日】平成23年6月9日(2011.6.9)
【国際特許分類】
【出願日】平成21年11月25日(2009.11.25)
【出願人】(000003300)東ソー株式会社 (1,901)
【出願人】(000173762)公益財団法人相模中央化学研究所 (151)
【Fターム(参考)】
[ Back to top ]