
スフィンゴシンキナーゼタイプ1阻害物質、上記阻害物質を使用するための合成物と方法
【課題】 スフィンゴシンキナーゼタイプ1(Sphk1)を独自に阻害する、更にはがん細胞を殺すかあるいは損傷すること、アポトーシスを誘発すること、生長を阻害すること、がん細胞、白血病の増殖、転移や化学療法抵抗性の発達を阻害すること、抗がん剤の効果の増強、免疫反応性を弱めること、がん細胞における生存信号伝達の阻害および多発性硬化症の症状の軽減を含む多くの応用において有用である新規な合成物を提供する。
【解決手段】 合成物は下記の化1の構造を有する。
【解決手段】 合成物は下記の化1の構造を有する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、全般的に言えば、スフィンゴ脂質媒介、スフィンゴ脂質媒体および特にタイプ1を含むスフィンゴキナーゼの阻害物質、及びがん、ぜんそく、アナフィラキシー、自食作用、中枢神経系統、その他の分野に関する。
【背景技術】
【0002】
この出願において引用されるか又は明らかにされているすべての特許、特許出願、特許刊行物、科学論文等は、本発明が属する技術分野の状況をより完全に記述するために、ここにそっくりそのまま引用文献によって取り入れられている。
この出願は2008年4月29日に出願された暫定的な出願No.61/048,638に関連するものである。
【0003】
スフィンゴシン−1−ホスフェイト(S1P)、即ちスフィンゴシンキナーゼ(SphKs)によってスフィンゴシンから製造される効力のある脂質の媒介物質は、がんの進行にとって重要な細胞の成長及び生存を含む多くの過程を調節する(非特許文献1)。
S1Pとは対照的に、その前駆体、スフィンゴシン、及びセラミドは、アポトーシスの成長抑止や誘導に関連している(非特許文献2)。かくしてこれら相互転換出来るスフィンゴ脂質代謝産物は、細胞の壽命を測定する細胞質可変抵抗器として見られてきた(非特許文献3)。
今までの多くの研究が、S1P/セラミド制御点中の摂動が、造血起点に対する化学療法や放射線療法を含む腫瘍性細胞の化学療法や放射線療法に対する抵抗性調節に含まれることを示している(非特許文献4及び非特許文献5)。
【0004】
二つのSphkイソ酵素、SphK1およびSphK2が多くの特徴を共有するとは言え(非特許文献6及び非特許文献7)、異なった機能を示すことが記述されている。
SphK1は細胞成長と細胞生存を促進する(非特許文献8〜非特許文献11)。
一方SphK2は過剰発現されるとき、逆の効果を有する(非特許文献12及び非特許文献13)。
SphK1はS1P/セラミド制御点を調節するキー酵素である(非特許文献14及び非特許文献15)。
【0005】
実際以前からS1PとSphK1とが一次白血病性細胞と白血病性細胞系の両方が、一般的に使用される細胞毒性薬剤によって導かれるアポトーシスに対する抵抗に関与しているとされて来た(非特許文献16〜18)。
レートレオージヒドロスフィンゴジン(safingol)およびN,N−デメチルスフィンゴジン(DMS)のような、SphKsの非イソ酵素特定阻害物質は、白血病細胞に対して有毒である(非特許文献19及び非特許文献20)。興味深いことには、多数の薬剤に対する抵抗性を有するHL-60骨髄性白血病性細胞は親細胞よりもDMSに対してより感受性が強かった(非特許文献20)。更にSphK1の活性は、MDR1-又はMRP1-陽性HL-60細胞中よりもドキソルビシン/またはエトポサイドに対して感受性のあるHL-60細胞中の方が低かった。
感受性のあるHL-60細胞中のSphK1の強調された発現はアポトーシスをブロックしたが、一方SphK1の反応抑制作用は、ミトコンドリア依存のアポトーシスを誘導することによって化学薬剤抵抗性を克服した(非特許文献21)。これらの観察は、MDR発現が骨髄性白血病(AML)における強力な徴候の指標である証拠(非特許文献22)、そして一般的にアントラサイクリン又は植物ベースのアルカロイドによるAMLの治療の後に続くのであるが、MDR表現形は化学療法の成功を妨げる役割をすると考えられている証拠に照らして見れば、更なる重要性をもたらすものである。
【0006】
加えて、K562ヒト慢性骨髄性白血病性細胞のイマチニブに対する抵抗性、Bcr-Abl-チロシンキナーゼの阻害はSphK1の発現とS1Pの生成に相互に関連し、一方SphK1の反応抑制は抵抗細胞中のイマチニブ−誘導−アポトーシスに対する感受性を増強する(非特許文献23)。かくしてSphK1の効果的な特効のある阻害物質の開発は不活性型生存S1Pのレベルを減少させることのみならず、ケラミド生成の助長に、少なくとも部分的に一定の細胞毒薬剤の不活性型アポトーシス作用を仲介する過程において有用であることがわかる(非特許文献24〜26)。
シンゴシンキナーゼ阻害物質は以前から記述されている(非特許文献27〜31)。これらの刊行物は、しかしながら本発明の新規なスフィンゴシンキナーゼタイプ1阻害物質については記述していない。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Spiegel et al., Nature Rev Mol Cell Biol. 4:397-407, 2003
【非特許文献2】Ogretman & Hannun, Nature Rev Cancer 4:604-616, 2004
【非特許文献3】Cuvillier et ao., Nature 381:800-803, 1996
【非特許文献4】Ogretmann et al., supra.; Hait et al., Biochem Biophys Acta 1758:2016-2026. 2006
【非特許文献5】Milstien & Spiegel, Cancer Cell 9:148-150, 2006
【非特許文献6】Kohama et al., J. Biol Chem 273:23722-23728, 1998
【非特許文献7】Liu et al., J. Biol Chem 275: 19513-19520, 2000
【非特許文献8】Olivera et al., J Cell Biol 147:545-558, 1999
【非特許文献9】Xia et al., J. Biol Chem 277:7996-8003, 2002
【非特許文献10】Bonhoure et al., Leukemia 20:95-102, 2006
【非特許文献11】Sukocheva et al., J Cell Biol 173:301-310, 2006
【非特許文献12】Maceyka et al., J Biol Chem 280:37118-37129,2005
【非特許文献13】Okada et al., J Biol Chem 280:36318-36325, 2005
【非特許文献14】Maceyka et al., supra.; Berdyshev et al., Cell Signal 18:1779-1792, 2006
【非特許文献15】Taha et al., FASEB J 20:482-484, 2006
【非特許文献16】Cuvillier et al., Nature, 2004 supra.; Cuvillier et al., J Biol Chem 273:2910-2916, 1998
【非特許文献17】Cuvillier et al., Blood 98:2828-2836, 2001
【非特許文献18】Jendiroba et al., Leuk Res 26:301-310, 2002
【非特許文献19】Jarvis et al., Mol Pharmacol 54:844-856, 1998
【非特許文献20】Jendiroba et al., 2002, supra.
【非特許文献21】Bonhoure et al., 2006, supra.
【非特許文献22】Filipits et al., Leukemia 14:68-76, 2000
【非特許文献23】Baran et al., J Biol Chem 282:10922-10934, 2007
【非特許文献24】Maggio et al., Cancer Res 64:2590-2600, 2004
【非特許文献25】Rahmani et al., Cancer Res 65:2422-2432, 2005
【非特許文献26】Rosato et al., Mol Pharmacol 69:216-225, 2006
【非特許文献27】Kim et al., Bioorg & Med Chem 13:3475-3485, 2005
【非特許文献28】Kono et al., J. Antibiotics 53:459-466, 2000
【非特許文献29】Kono et al., J. Antibiotics 53:753-758, 2000
【非特許文献30】Marsolais & Rosen, Nature Reviews/Drug Discovery 8:297-307, 2009
【非特許文献31】US 2008/0167352 A1 (Smith et al., published July 10, 2008)
【発明の概要】
【発明が解決しようとする課題】
【0008】
ここに我々は種々の信号発信および生存関連蛋白質の活性化における多重摂動を引き起こすSphK1(SK1-l)の効力のある水溶性阻害物質を説明する。
SK1-lはAMLによって患者から得られた芽細胞のみならずヒト白血病性細胞系におけるアポトーシスを明らかに誘発し、そしてAML異種移植腫瘍の成長を阻害した。
SK1-lは以下に記載する他の関連合成物に対するモデルとして役立つ。
【課題を解決するための手段】
【0009】
この発明はまた、スフィンゴシンキナーゼ活性を測定する試験管内測定法でスフィンゴシンキナーゼ1(SphK1)をスフィンゴシンキナーゼ2(SphK2)よりも少なくとも5倍以上強く阻害する合成物を提供する。
本発明は下記の化1の構造からなる合成物を提供する。
【化1】

この図のXはC(炭素)あるいはN(窒素)であり、R1、R2、R3、R4はそれぞれ水素、直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、あるいはエーテルグループで構成され、そしてそのなかで、R1、R2、R3、R4は独立してお互いに融合して一つあるいはそれ以上の環をつくる、あるいは上述のもののあらゆる組み合わせでもあり得る。
【発明の効果】
【0010】
本発明の合成物は、がん細胞を殺したり損傷させたりすること、アポトーシス(プログラムされた細胞死)を誘導すること、がん細胞、白血病で増殖、転移、薬剤耐性の発現を阻害すること、抗がん剤の効果を増強すること、免疫反応性を弱めること、がん細胞の生存信号を阻害すること、多発性硬化症の症状を和らげること、など多くの応用あるいは基礎づけに有用である。
【図面の簡単な説明】
【0011】
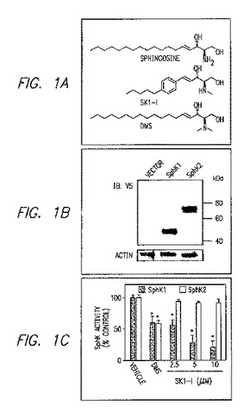
【図1A】SK1-l (BML-258)の構造をしめす。
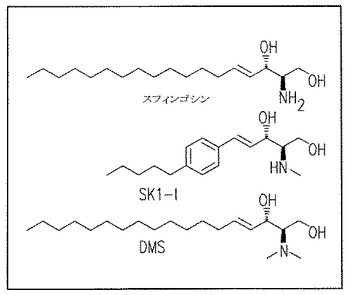
【図1B】組み替えSphK1およびSphK2へのSK1-lの影響を示す結果である。
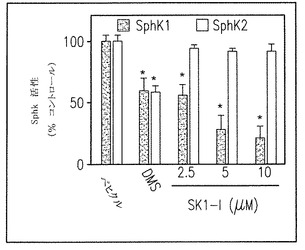
【図1C】組み替えSphK1およびSphK2へのSK1-lの影響を示す結果である。
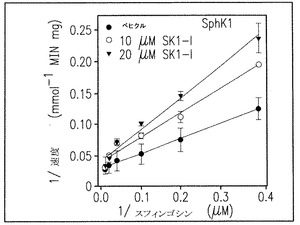
【図1D】組み替えSphK1およびSphK2へのSK1-lの影響を示す結果である。
【図1E】組み替えSphK1およびSphK2へのSK1-lの影響を示す結果である。
【図1F】SK1-lの種々のタンパク質キナーゼ活性への影響を示す結果である。
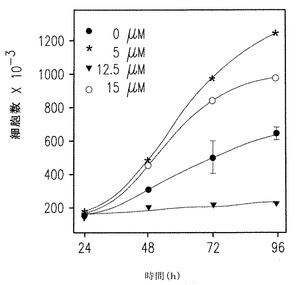
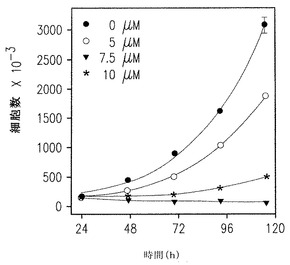
【図2A】U937細胞の増殖へのSK1-Iの濃度効果を示す。
【図2B】Tリンパ球性Jurkat細胞の増殖へのSK1-lの濃度効果を示す。
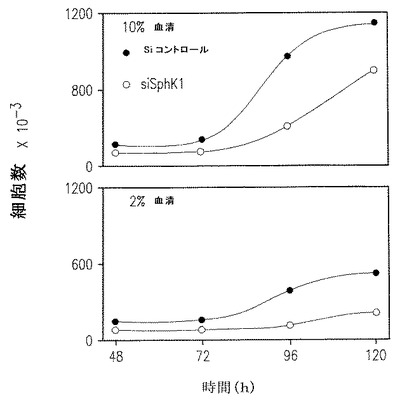
【図2C】siRNAを標的としてSphK1発現を低下させたU937細胞の増殖速度の低下を示す。
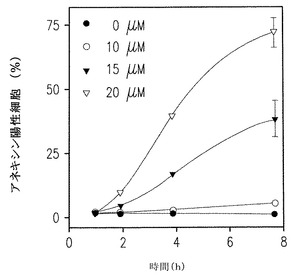
【図2D】SK1-lにさらすことによるU937細胞のアポトーシスの時間および濃度依存的な増加を示す。
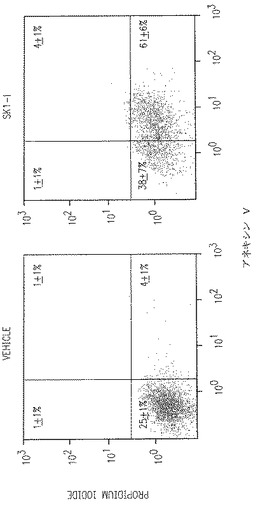
【図2E】大多数の細胞が初期アポトーシスの状態で少数の細胞が壊死状態(Pl-陽性)にあるU937細胞の様子を図示する。
【図2F】TUNEL測定法で決められるDNA鎖断裂と結果が密接に関連していることを示している。
【図2G】U937細胞への血清濃度の効果を示す。
【図3A】U937細胞のSK1-l処理がカスパーゼ3とカスパーゼ9活性化を増強し、アポトーシス誘導を伴うポリADPリボースポリメラーゼ(PARP)の開裂を引き起こすことを示す。
【図3B】U937細胞のSK1-l処理がカスパーゼ3とカスパーゼ9活性化を増強し、アポトーシス誘導を伴うポリADPリボースポリメラーゼ(PARP)の開裂を引き起こすことを示す。
【図3C】汎カスパーゼ阻害剤であるZVADとBOCでのU937細胞の前処理のSK1-lで誘導されるアポトーシスとDNA損傷剤エトポシドによる誘導への結果を示す。
【図3D】SK1-Iで誘導される致死性に対するBcl-2の異所性発現の効果を示す。
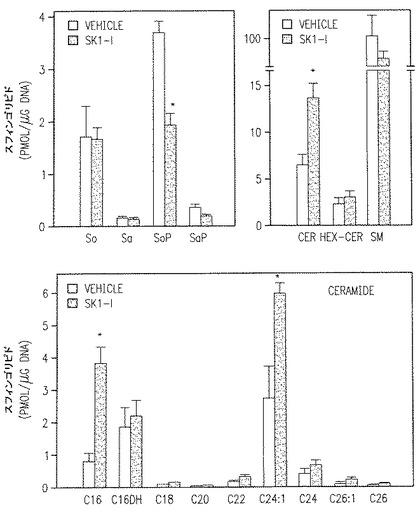
【図4A】HPLC ESI-MS/MSにより決定したスフィンゴ脂質代謝物のレベルに対するSK1-lの影響を示す。
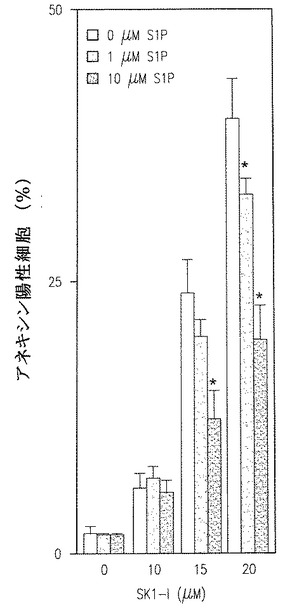
【図4B】量依存的に外から加えたS1PによるSK1-l誘導アポトーシスの結果を示す。
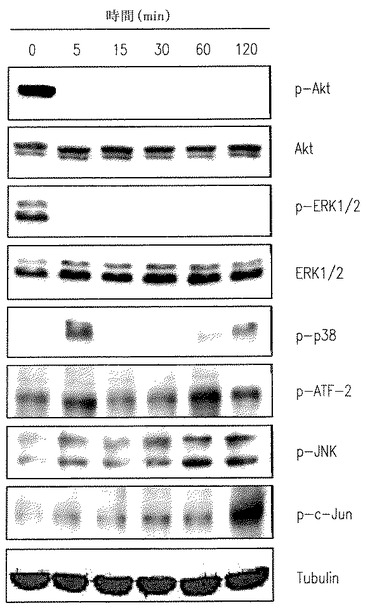
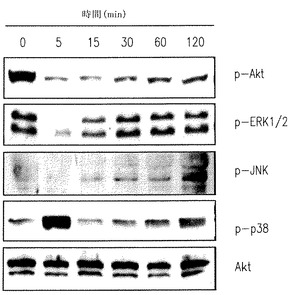
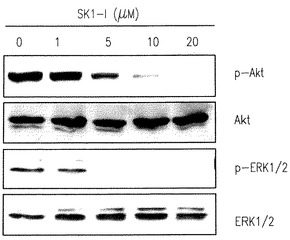
【図5A】U937細胞がSK1-lで処理されたときERK1/2とAktのリン酸化の減少を示す。
【図5B】U937細胞がSK1-lで処理されたときERK1/2とAktのリン酸化の減少を示す。
【図5C】U937細胞がSK1-lで処理されたときERK1/2とAktのリン酸化の減少を示す。
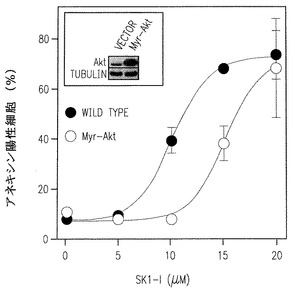
【図5D】U937細胞中に構成的に活性化されたミリスチン酸化Aktの過剰発現の15μM以下の濃度でのSK1-lに誘導されるアポトーシスに対する影響を示す。
【図6A】SK1-lにさらされたときの二人の患者試料でのアポトーシスの増加を示す。
【図6B】SK1-lにさらされたときの二人の患者試料でのアポトーシスの増加を示す。
【図7A】免疫能の欠乏したマウスへ異所移植した腫瘍増殖のSK1-l投与による減少を示す。
【図7B】マウスでの腫瘍重量へのSK1-l処理の効果を示す。
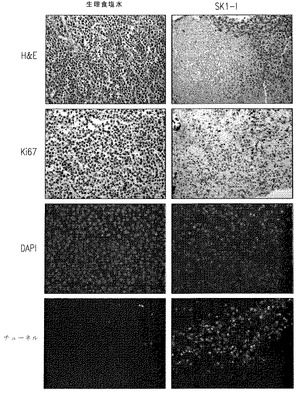
【図7C】SK1-I処理マウスからの腫瘍の免疫組織化学的分析を示す。
【発明を実施するための形態】
【0012】
本発明は化1の構造からなる合成物を提供する。
【化1】
化1のXはC(炭素)あるいはN(窒素)であり、R1、R2、R3、R4は独立に水素、直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、エーテルグループで構成される。そしてそのなかで、R1、R2、R3、R4は独立してお互いに融合して一つあるいはそれ以上の環を作り、あるいは上述のもののあらゆる組み合わせでもあり得る。
【0013】
本発明はまた化2の構造からなる合成物を提供する。
【化2】
【0014】
本発明は化3の構造からなる合成物を提供する。
【化3】
化3のR5とR6は独立に直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、あるいはエーテルグループあるいは上述のもののあらゆる組み合わせで構成される。
【0015】
また本発明では、R5とR6がお互いに結合し環を作って為るさらなる合成物として、以下の化4の構造を持つ合成物を提供する。
【化4】
化4のR7は水素、直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、エーテルグループあるいは上述のもののあらゆる組み合わせで構成される。
【0016】
ほかの例では、R1がH(水素)でR2がCH3である合成物が提供される。さらにまたほかの合成物では、R3あるいはR4がサルファイド、SR5であり、R5が水素、直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、エーテルグループあるいは上述のもののあらゆる組み合わせで構成される。
【0017】
SR5が以下の化5の構造を持つもう一つの合成物が提供される。
【化5】
【0018】
さらに他の合成物では、XがC、R1とR3がH、そしてR4がSR5であり、合成物は以下の化6の構造を持つ。
【化6】
【0019】
R2がCH3、XがCで有り得るものであり、R1とR4がHであり、そしてR3がSR5である合成物は以下の化7の構造を持つ。
【化7】
上記の合成物では、R2はCH3、R3はHであり得る。
【0020】
他の例では、XがC、R1がH、R2がCH3そしてR4が(CH2)4CH3で、合成物は以下の化8の構造を持つ。
【化8】
R4の末端の炭素は一つあるいはそれ以上のハロゲン化物で置換でき、ハロゲン化物は臭化物、塩化物、フッ化物である。
【0021】
他の例では合成物は以下の化9の構造を持つ。
【化9】
R4はHであり得、R2はメチルグループ、R1は水素あるいはメチルグループである。
また、R1とR2は融合し、置換基の入ったあるいは入らない環をつくることができる。
【0022】
他の例では、本発明は以下の化10の構造を持つ合成物を提供する。
【化10】
XはCであり得る。
【0023】
もう一つの合成物は以下の化11の構造を持つ。
【化11】
ここでは、XはCである。
【0024】
また、R4はエーテルであり得、合成物は以下の化12の構造を持つ。
【化12】
ここでは、R5は直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、エーテルグループあるいは上述のもののあらゆる組み合わせを含んでいる。
さらなる例では、XはCであり得、R1はHそしてR2はCH3である。
【0025】
多くの有用な過程が上述の合成物で提供されている。
これらは当該がん細胞を殺すあるいは損傷するに十分な量の合成物にがん細胞をさらす段階を含み、がん細胞を殺すあるいは損傷させる過程を含んでいる。このようながん細胞としては、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫あるいは腎がん細胞およびこれらの組み合わせを含んでいる。
【0026】
この発明で提供されるもう一つの過程は、がん細胞にアポトーシスを起こさせ、また、当該がん細胞にアポトーシスを起こさせるに十分量の合成物に対してがん細胞をさらすステップを含んでいる。もう一度言うと、そのようながん細胞は白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫あるいは腎がん細胞およびこれらの組み合わせを含んでいる。
この発明はまた、当該がん細胞の増殖、転移、薬剤耐性の展開を阻害するのに十分量の合成物へがん細胞をさらすステップを含む、がん細胞の増殖、転移、薬剤耐性の展開を阻害する過程を提供する。もう一度言うと、がん細胞には白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫あるいは腎がん細胞およびこれらの組み合わせが含まれる。
【0027】
もう一つの過程は、当該患者で白血病の症状を処置し、和らげるに十分量の合成物を当該患者に導入するステップを含んで、必要な患者で白血病の症状を処置し、和らげる過程である。
さらにもう一つの過程は、抗がん剤を患者に投与するステップを含んで、必要とする患者内のがん細胞を殺すための抗がん剤の能力を増加させる過程である。そして該合成物、該合成物は患者中のがん細胞を殺すための抗がん剤の能力を増加するために十分な量を投与される。がん細胞には白血病細胞、胸部がん細胞、前立腺がん、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞あるいは腎がん細胞およびこれらの組み合わせが含まれうる。
また、患者の免疫反応性を減ずるに十分量の合成物を患者に投与するステップを含んで、必要な患者の免疫反応性を減弱する過程が提供される。さらなる具体例では、肥満細胞機能の減弱により免疫反応性の減弱が引き起こされている。また、免疫反応性の減弱が必要とされる患者のぜんそくの症状を和らげるようにしむけられことができる、免疫反応性の減弱はアナフィラキシーショック症状を和らげあるいは自己免疫病の症状を和らげるのにしむけられることができる。
【0028】
提供されるさらなるもう一つの過程は、請求項1の合成物を、生存信号を阻害するのに十分量癌様細胞に投与するステップを含んで、癌様細胞の生存信号を阻害するためのものである。もう一つの具体例では、生存信号の阻害はAktあるいはERK1/2あるいはこの両者のリン酸化の減少で達成されている。
さらに多発性硬化症患者の症状を和らげるのに十分量の合成物を患者に投与することを含んで、必要とされる多発性硬化症患者の症状を和らげるもう一つの過程が提供されている。
【0029】
この発明はまた、スフィンゴシンキナーゼを測定する試験管内測定法で、スフィンゴシンキナーゼ1(SphK1)をスフィンゴシンキナーゼ2(SphK2)を阻害するよりもよりもすくなくとも5倍以上強く阻害する合成物を提供する。SphK1はまた当該SphK2の阻害よりすくなくとも10倍以上阻害される。試験管内測定での阻害は10μM濃度で測定される。試験管内測定法での阻害はまたSphK1を50%阻害する濃度で測定することができる。
上記の合成物はまた多くの応用と基礎づけに有用である。これらは、がん細胞を殺すか損傷させるに十分量の上記合成物にがん細胞をさらすステップを含み、がん細胞を殺し、または損傷させる過程を含む。がん細胞には白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞あるいは腎がん細胞およびこれらの組み合わせが含まれうる。
【0030】
もう一つの具体例では、上記合成物は、がん細胞をアポトーシスに向かわせるのを引き起こすのに十分量の請求項44の合成物にがん細胞をさらすステップを含んで、がん細胞をアポトーシス向かわせるのを引き起こす過程に有用である。ここでも、がん細胞には白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞あるいは腎がん細胞およびこれらの組み合わせが含まれうる。
【0031】
上記合成物を含むもう一つの過程では、この発明は、がん細胞の増殖、転移、薬剤耐性の発現の阻害に十分量の合成物にがん細胞をさらす過程を含んで、がん細胞の増殖、転移、薬剤耐性の発現を阻害する過程を提供する。がん細胞には白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞あるいは腎がん細胞およびこれらの組み合わせが含まれうる。
患者の有する白血病の症状を治療または軽減するための方法は該患者の白血病の症状を治療または軽減するために十分な量の最後に記載した例の合成物を該患者に投与するステップからなる。
【0032】
もう一つの具体例では、抗がん剤を患者に投与するステップを含んで、必要とされる患者内のがん細胞を殺す抗がん剤の能力を増強する過程を提供する。そして最後に記載した例の合成物は抗がん剤の患者のがん細胞を殺すための能力を増加するために十分な量を投与される。がん細胞には白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞あるいは腎がん細胞およびこれらの組み合わせが含まれうる。
また、患者の免疫反応性を弱めるのに十分量の上記合成物を患者に投与するステップを含み、必要とされる患者の免疫反応性を弱める過程が提供される。他の具体例では、免疫反応性の減弱は肥満細胞機能の減弱によりもたらされる。免疫反応性の減弱は、必要とされる患者の喘息の症状を和らげ、アナフィラキシーショックの症状を和らげ、自己免疫病の症状を和らげることに導かれる。
【0033】
もう一つの具体例では、この発明は、生存信号を阻害するに十分量の上記合成物を癌様細胞に投与するステップを含んで、癌様細胞の生存信号を阻害する過程を提供する。生存信号の阻害はAktあるいはERK1/2あるいは両者のリン酸化の減弱により達成されうる。
この発明で提供されるもう一つの過程は、患者の多発性硬化症の症状を和らげるに十分量の上記合成物を患者に投与するステップを含んで、必要とされる患者の多発性硬化症の症状を和らげるために提供される。
【0034】
ここに述べられる本発明の合成物のすべてが、二量体あるいは多量体の合成物で明瞭に示されることが認識されるべきである。
また、この合成物は、従来よく知られている過程を用いて通常の材料と結合されてきた薬用合成物に明示することができる。これらの合成物は患者が摂取するために、タブレット、ピル、カプセル、液体、ゲル、シロップ、水混合物、懸濁液やこれに似たものとして明示されうる。
【実施例】
【0035】
以下の例は例証としてこの発明の提示されるものであり、発明を限界づけるものではない。
〔例1〕
記載され以下に使われている合成物、BML-258、は以下の案と手順に従って合成された。
【化13】
窒素中、-20℃で65 ml乾燥THF中の4-n-phenylphenylacetylene 1(3.343g, 0.01776 mol) にn-BuLi (1.6 M hexane中に10.2 ml、0.01628 mol)を滴下して加えた。反応混液を-20℃で2時間攪拌した。25ml 乾燥THF中のmethyl(R)-(+)-3-(t-butoxycarbonyl)-2,2-dimethyl-4- oxazolidinecarboxylate 2 (3.393g, 0.01480 mol)をカニューレを通して加えた。反応液を-20℃で一晩攪拌した。TLC(薄層クロマトグラフィー)(20% ethylacetate/hexanes)では反応が完了していた。
反応液をET2O で希釈し、水と塩水で注意深く洗った。フラッシュカラムクロマトグラフィー(12% ethyl acetate/hexanes, silica gel)で、収量はerythroとthreo の混合物として4.50g (73 %)であった。
調達用のHPLC(高速液体クロマトグラフィー)(Dynamax Si, 15% ethyl acetate/hexanes, 260 nm)で収量は3.71gのerythro 3と0.49g threoであった。
1H NMR(CDCl3) erythro:7.34-7.32(d, 2H), 7.12-709(d,2H), 5.19-5.16(d, 1H) 4.73-4.70(d,1H), 4.26-3.96(m, 3H), 2.61-2.56(t, 2H), 1.62(s, 3H), 1.60-1.50(m,2H),1.54(s, 3H), 1.50(s, 9H), 1.34-1.27(m, 4H), 0.91-0.86(t, 3H)。
【0036】
【化14】
100 ml MeOH中のoxazolidine 3 (3.48g, 0.00814 mol)にAmberlyst-15 (200 mg)を加えた。反応液は室温で一夜攪拌した。TLC(30% Ethyl acetate/hexanes)では反応は完全であった。混合物は濾過され、迅速クロマトグラフィー(5% MeOH/methylene chloride, silica gel) で2.44g (79%)のaminoalcohol 4 がとれた。
1H NMR(CDCl3): 7.34-7.32(d, 2H), 7.12-709(d,2H), 5.45-5.38(d, 1H) 4.88-4.82(m,1H), 4.25-4.19(m, 1H), 3.91-3.80(m, 2H), 3.26-3.23(d, 1H), 2.61-2.56(t,2H), 1.63-1.54(m, 2H), 1.49(s, 9H), 1.35-1.26(m, 4H), 0.91-0.86(t, 3H)。
【0037】
【化15】
窒素中、0℃で125 ml 乾燥Et2Oのalkyne 4 (2.44g, 0.00646 mol)にRed-Al (9.85 ml, toluene中に65 wt%, 0.03232 mol)を滴下しながら加えた。滴下後、反応液を室温にし、36時間攪拌した。TLC (40% Ethyl acetate/hexenes)では反応は完結していた。反応液を0℃に冷やし、注意深く15% NaOH液で急冷した。この混合物を、両方の層が透明になるまで強く攪拌した(45 分)。両層を分離し、水層をchloroformで抽出した(3回)。有機層一緒にして15% NaOH、水、塩水で洗浄した。フラッシュクロマトグラフィー(5% MeOH/methylene chlorideから20% MeOH/methylene chloride + 1% NH4OH, silica gelの濃度勾配)で1.76g (72%)のtrans alkene 5 を得た。
1H NMR(CDCl3): 7.31-7.29(d, 2H), 7.15-712(d,2H), 6.70-6.65(d, 1H, J=16Hz) 6.26-6.18(dd,1H, J=16Hz), 5.35-5.32(d, 1H), 4.55-4.49(m, 1H), 4.03-3.96(m, 1H), 3.80-3.68(m,2H), 2.83-2.79(d, 1H), 2.61-2.56(t, 2H), 1.65-1.55(m, 2H), 1.44(s, 9H).1.34-1.25(m, 4H), 0.91-0.86 (1,3H)。
【0038】
【化16】
窒素中で20ml 乾燥THFに溶かしたBOC-alkene 5 (0.350g, 0.00092 mol)に室温でDIBAL(THF中に1Mを9.22ml, 0.00922 mol)を注意深く加えた。加えた後、反応液を還流した。24時間還流後、混合物を室温に冷却し、さらに5.0 mlのDIBAL液(0.00500 mol)を加えた。還流をさらに24時間続けた。反応液を0℃に冷やし、水(0.60 ml)、15% NaOH(0.60 ml)そしてもう一度水(1.50 ml)を加えた。THF(50 ml)を加え、混合物を15分間強く攪拌した。それからNa2SO4(2 g)、celite (2 g)を加え、室温まで暖めながら、攪拌を30分続けた。混合物を濾過し、濾過した固まりを大量のTHFで抽出した。フラッシュクロマトグラフィー(2% MeOH/methylene chlorideから10% MeOH/methylene chloride + 0.75% NH4OHの濃度勾配)で0.187g (73%)のamine 6 を得た。
1H NMR(CDCl3): 7.31-7.29(d, 2H), 7.15-712(d,2H), 6.68-6.63(d, 1H, J=16Hz) 6.22-6.14(dd, 1H, J=16Hz), 4.51-4.47(m, 1H), 3.80-3.74(m, 3H), 2.61-2.56(t, 2H), 2.50(s,3H), 2.40-2.10(broad, 2H), 1.65-1.55(m, 2H), 1.34-1.25(m, 4H), 0.91-0.86(t, 3H). HRMS(MH+): Calc. - 278.2120, Found - 278.2199.
【0039】
【化17】
15 ml の乾燥Et2O中のamine 6 (0.335g, 0.00121 mol))に3 ml の1M HCl/Et2Oを加えた。すぐに白い沈殿ができた。室温で15分攪拌後沈殿を濾過し、Et2Oで洗い、0.352g(89%)のBML-258を得た。
1H NMR(DMSO): 8.75-8.50(bd, 2H), 7.38-734(d,2H), 7.19-7.15(d, 2H,), 6.65-6.60(dd, 1H, J=16Hz), 6.30-6.22(dd, 1H, J=16Hz), 5.84-5.82(m, 1H), 5.30-5.25(m, 1H), 4.60-4.54(m,1H), 3.76-3.72(m, 2H), 3.18-3.10(m, 1H), 2.64(s, 3H), 2.56-2.50(t, 2H)1.60-1.50(m,2H), 1.34-1.23(m, 4H), 0.90-0.85(t, 3H).
【0040】
SK1-I、(2R,3S,4E)-N-methyl-5-(4'-pentylphenyl)-2-aminopent-4-ene-1,3-diol (BML-258)は例1に述べたようにBIOMOL International (Plymouth Meeting, PA)により合成された。SphingosineとN,N-dimethylsphingosineはMIOMOLから得た。[y-32P]ATP(3000 Ci/mmol)はPerkin Elmer (Boston, MA)から購入した。Boc-D-FMK(BOC), Z-VAD-FMK(ZVAD)、etoposideはEMD Biosciences (San Diego, CA)からのものである。流動細胞計測法(フローサイトメトリー)のためのTerminal deoxynucleotidyl trensferase Br-dUTP nick end labeling (TUNEL) kitはSigma Aldrich (St. Louis, Mo)からのものである。免疫組織化学用のTUNELキットはRoche Applied Science (Indianapolis, IN)からのものである。アポトーシス用のFITC4標識annexin V/propidium iodide染色キットはBD Biosciences (San Jose, CA)からのものである。
【0041】
〔細胞と細胞培養〕
U937 ヒト組織細胞型白血病およびJurkat急性T細胞白血病細胞はAmerican Type Culture Collectionから入手した。特に指示しない限り、細胞はL-glutamate, penicillin, streptomycin, 10% 仔牛血清(Invitrogen, Carlsbad, CA)を加えたRPMI 1640培地に培養し、対数増殖期に維持された(Dai et al., Cancer Res 61: 5106-5115,2001)。Bcl-2、Bcl-xL、構成的に活性なAkt (Mycがついたミリオスチン酸化Akt)およびそれらの対応する空ベクターを安定的に過剰発現しているU937をえて、記載されたと全く同じように適当な選択抗生剤の存在下で培養した(Rahmani et al., 2005, 上記)。
Institutional Review Board of Virginia Commonwealth Universityからの認可のもとに、通常の特徴的な吸引をしている二人のAML患者から、インフォームドコンセントのもとに白血病芽球を得た。インフォームドコンセントはヘルシンキ宣言にしたがって行った。二人の患者試料の特徴は次の通りである。
患者1:FABサブタイプはM2、知られている融合あるいは変異タンパク質はない、知られている染色体異常はない。
患者2:FABサブタイプはM4、知られている融合あるいは変異タンパク質はない、16番染色体に逆位がある。
それぞれ85%の芽細胞を含むサンプルは室温でFicoll/Hypaque(比重1.077-1.081; Sigma, St. Louis, MO, USA)上で400 x g の遠心分離で分けた。芽細胞を含む境界層を滅菌したパスツールピペットでとり、10% FBSを含む培養液に再懸濁した。細胞はトリパンブルー排除法で95%以上の生存率を示し、上記のように培養した。末梢血の単核白血球は同じ方法で健常人から分離した。
【0042】
〔RNA干渉〕
Qiagen (Valencia, CA)から入手したSphK1 を標的とした100 pmol のRNAi オリゴヌクレオチド(標的配列:GGGCAAGGCCTTGCAGCTC (SEQ ID NO: 1)) と標的としない対照siRNA (非特異的なランダムな配列)をU937細胞に導入した。導入はCell Line Nucleofector Kit V (Amaxa GmbH, Cologne, Germany)のAmaxa Nucleofecter (program V-001)により製造者の指示に従って行った
【0043】
〔スフィンゴシンキナーゼの発現と活性〕
HEK 293細胞を10%子牛血清を含むDMEMで培養し、以前に述べたようにLipofectamine PLUS (Invitrogen)を用いて、V5-His-pcDNA3.1 ベクター(Invitrogen)、V5-His標識ヒトSphK1あるいはV5-His標識ヒトSphK2を導入した(Paugh et al., FEBS Lett 554: 189-193, 2003)。それから細胞を2日間培養し、凍結融解で溶解し、[y-32P]ATP (10pCi, 1 mM, 10 mM MgCl2中)とスフィンゴシンを用いて、SphK2を阻害する0.25% Triton X-100中でSphK1活性を測定した (Hait et al., J Biol Chem280: 29462-29469, 2005)。SphK2活性は1 M KClの存在下で、4 mg/ml BSA との複合体で加えたスフィンゴシンを用いて測定された。この条件はSphK2活性に至適であり、SphK1は強く阻害される (Hait et al., J Bio Chem, 2005,上記)。標識されたS1Pは抽出され、クロロフォルム/アセトン/メタノール/酢酸/水(10:4:3:2:1. v/v)を溶剤するシリカゲルG60上でのTLCで分離した。S1Pに相当する放射能を持つバンドはFX Molecular Imager (Bio-Rad, Hercules, CA)で定量化した。SphKの比活性はmgタンパク質当たり生成するpmolのS1Pとして表した。
【0044】
〔ウエスタンブロット分析〕
細胞は細胞融解緩衝液(50 mM Tris pH 7.5, 150 mM NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, 1mM PMSF, 5μg/ml leupeptin, 5μg/ml aprotinin, 1 mM DTT)に再懸濁された。同じ量のタンパク質(60 pg)が10% SDS-PAGEで分離され、ニトロセルロースに転写された。ブロットが、5%の無脂肪ドライミルクと0.1% Tween 20を含むトリス緩衝液(TBS)中で一次抗体(1:1000) と一晩保温し、続いて抗ウサギHRP標識IgG(1:10,000, Jackson Immunoresearch Laboratories)で保温した。免疫複合体はKodakまたはPhenix Research ProductsのX腺フィルムでの高感度化学発光(Pierce)で可視化した。ウエスタン物はAlfa Innotech Corporation (San Leandro) からのAlphaEaseFC 4.0.0ソフトを用いて定量化された。
次のものが一次抗体として使われた:phospho-p44/42 MAP キナーゼ(Thr202/Tyr204)抗体、phospho-p38 MAPキナーゼ(Thr180/Tyr182)抗体(Cell Signaling, Beverly, MA, USA)、phospho-JNK (Thr183/Tyr185)抗体、Bcl-xS/L抗体(S-18, Santa Cruz, Santa Cruz, CA)、抗ヒトBcl-2 (Dako, Carpinteria, CA)、Mcl-1抗体、抗caspase-3と抗caspase-9 (Pharmingen)および抗PARP (Biomol)。
【0045】
〔タンパク質キナーゼの概観〕
SK1-lの種々のタンパク質キナーゼ活性への影響はSelectScreenTM Kinase Profiling (Invitrogen Drug Discovery Solution, Madison, WI)で評価された。簡単に述べると、5 pM SK1-lの存在下あるいは非存在下で、それぞれのタンパク質キナーゼ対してのKmappのATP濃度で、FRETを構成する二つの蛍光をもたらすものを含むペプチド基質を用いて、蛍光共鳴エネルギー転移(FRET)に基づくキナーゼ測定系により384穴プレート中で測定が行われた。発展液はリン酸化されていないペプチドを特異的に消化するタンパク質分解酵素を含み蛍光信号をつくりだす。Coumarin蛍光と蛍光のFRET信号はそれぞれ445 nmと520 nmで測定された。coumarin放射は開裂していない(リン酸化された)基質ペプチドのみでFRETによる蛍光を引き起こす。
ATPの非存在下でリン酸化されていないペプチドとキナーゼ含む反応と化学量論的にリン酸化されたペプチドを含む反応がそれぞれ0%および100%リン酸化の対照に供された。生の蛍光値は背景レベルに対して調整された。反応の終わりは蛍光FRET信号でcoumarin蛍光を割った放射比率として計算された。これらの比率はその後100%リン酸化対照で得られた比率で標準化された。
【0046】
〔アポトーシスのAnnexin V/PI測定〕
細胞はannexin V-フルオレッセインイソチオシアネートとプロピジウムイオダイド (PI)で染色し、それから製造者(BD PharMingen, San Diego,CA)の指示に従ってフローサイトメトリーによりアポトーシスを評価した。簡単に言うと、106個の細胞をリン酸緩衝食塩水(PBS)で洗い、10 mM HEPES, pH 7.4、140 mM NaOHおよび2.5 mM CaCl2を含んだ緩衝液中の5μl annexin V-フルオレッセインイソチオシアネートと5μlヨウ素化プロピジュウム(50μg/ml)で室温、暗所で15分間染色した。アポトーシス細胞はEXPO32 フローサイトメトリー分析プログラムのついたCoulter Epics-XL-MCL細胞蛍光光度計を使い決定した。下右1/4のパーセントは初期のアポトーシス細胞(annexin陽性)に一致し、上右1/4のパーセントは後期のアポトーシス細胞(annexin VとPI陽性)に一致する。
【0047】
〔TUNEL測定によるDNA鎖開裂検出〕
細胞(106)は氷上で1% (w/v)パラフォルムアルデヒドで15分間固定され、PBSで2回洗い、氷上で30分70% エタノール中で透過性をあげる処理をした。細胞を洗い、ターミナルデオキシヌクレオチドトランスフェラーゼとブロモデオキシウリヂン(BrdU)を含んだDNA標識液に再懸濁し、37℃で1時間製造者(Sigma)の指示に従って保温した。それから、細胞は抗BrdU蛍光抗体と暗所で室温30分間保温し、EXPO32 Flow Cytometry分析プログラム(Beckman Coulter)のついたCoulter Epics-XL-MCL細胞蛍光光度計を使い分析した。
【0048】
〔スフィンゴシンと代謝物の質量分析計による分析〕
細胞は冷やしたPBSでよく洗い、2000 xg 10分間の遠心分離で沈殿とした。細胞の一部をDNAとタンパク質の測定のためにとった。残りに内部標準を加え(それぞれ0.5 nmolのC12-SM, C12-Cer, C12-GlcCer, C12-LacCer, C17-sphingosine, C17-sphingosine 1-phosphate, C17-sphinganine-1-phosphateとC12-Cer-phosphate, Avanti Polar Lipids, Alabaster, AL)、脂質を抽出し、個々のセラミドアシル鎖種を液体クロマトグラフィーとelectrospray ionization-tandem質量分析計(ESI-MS/MS, 4000 QTRAP, Applied Biosystems)により前に述べたように定量化した(Sullards et al., Science STKE 2001: L1, 2001)。
【0049】
〔異所性移植の腫瘍モデル〕
動物を含む全ての実験はVCU IACUCにより認可されている。U937細胞(滅菌PBS 100μl に懸濁した2×106個)が6週齢のCB17 SCID/beigeマウス (Taconic Farms, Germantown, NY)両脇腹の2カ所に注入され、7日間、触診できる腫瘍に成長させた。腫瘍が50-100 mm3の量に達したときに、動物を任意の2グループに分け、引き続いて7日間200μlの食塩水またはSK1-I (20 mg/kg)を腹腔内に注入した。腫瘍測定はカリパスで毎日行い、腫瘍量は計算式 (πx[ミリで計った長さ]×[ミリで計った幅]2)/6 を使って計算した。実験の終わりに、動物を殺し、腫瘍を取り、ホルマリンで固定しパラフィン包埋するか液体窒素で凍結した。ホルマリン固定切片はヘマトキシリン−エオジンで染色されるかKi-67に対する抗体で染色された(Novocastra, Newcastle, UK)。抗体の結合は免疫組織化学およびパーオキシダーゼ標識種特異的2次抗体で検出され、3,3-ジアミノベンチジンで可視化された。パラフィン切片はロウを落とし、再水和し、浸透化の前にproteinase Kで処理された。凍結切片は蛍光TUNEL標識キットで染色し、DAPIで対抗染色した。スライドは蛍光顕微鏡で分析された。
【0050】
〔統計分析〕
実験は矛盾しない結果で少なくとも3回繰り返された。それぞれの実験に対して3つのサンプルからのデータが計算され、平均±S.D.として表された。実験条件の間の有意差は相手のない観察に対するStudent t試験を使って決めた。
【0051】
〔結果〕
(SK1-lはSphK2ではなくSphK1の強力な選択的な阻害剤である)
現在は、阻害剤の合理的な設計のためにcomputational docking methodsが使えるようにするための構造的な情報がSphKについてはない。従って、他のやり方は、もっと強力で選択的な阻害剤を設計するための阻害剤研究から得られる情報を用いることである。いろいろな化学合成された短鎖スフィンゴシンやジヒドロスフィンゴシン類似物がSphKの阻害剤として以前から研究されてきた(Edsall et al.,Biochemistry 37:12892-12898, 1998; De Jonghe et al., Bioorg Med Chem Lett 9:3175-3180, 1999; Johnson et al., J. Pharmacol Exp309:452-461, 2004: そしてNiiro et al., Bioorg Med Chem12:45-51, 2004)。アルキル鎖をフェニール環に置き換えるとか3-ヒドロキシグループをフッ素に置換すると強力なSphK阻害剤を生ずることは知られていた。
さらに、4,5-トランス型の二重結合を持つ同族体は一般により上位の阻害剤であった。(Johnson et al., J Pharmacol Exp, 2004, 上記)。
これらの以前の観察に基づき、我々は(2R,3S,4E)-N-methyl-5-(4'-pentylphenyl)-2-aminopent-4-ene-1,3-diol(図1A)を合成し、組み替えSphK1とSphK2へのその効果を調べた(図1B-E)。この水溶性のスフィンゴシン類似物は、投与量依存的に強力にsphK1活性を阻害し(図1C)、5μMで60-70% 阻害する。以前に報告されたように(Edsall et al., Biochemistry, 1998上記、およびKohama et al., J Biol Chem, 1998上記)、N,N-dimethylsphingosine (DMS)もまた強くはないがSphK1活性を阻害する。重要なことは、SphK2 (Liu et al., J Biol Chem, 2000, 上記)やセラミドキナーゼ (Sugiura et al., J Biol Chem, 277:23294-23300, 2002)を阻害するDMSとは対照的に、我々の合成物は組み替えSphK2(図1C, E)やセラミドキナーゼ(データは示していない)を阻害しないことである。このように、SphK1への特異的な阻害の理由で、この合成物をSK1-lと称する。
Lineweaver-Burk分析でSphKl活性は、スフィンゴシンに対するKmとほぼ同じKi値約10μMでSK1-lにより完全に阻害されることが明らかとなった(図1D)。SK1-lはSphK1あるいはSphK2のどちらによってもリン酸化されなかった(データは示していない)。DMSといくつかの他の一般的なSphK阻害剤はタンパク質キナーゼC(Igarashi et al., Biochemistry28:6796-6800, 1989)と潜在的に他のキナーゼ(De Luca et al., Biofactors25:43-60, 2005とGamble et al., Int J Cancer 118: 2412-2420, 2006)をもまた阻害するのでSK1-lの他のタンパク質キナーゼへの効果を調べることは重要であった。いくつかの組み替えタンパク質キナーゼ、蛍光標識したポリペプチド基質、それぞれのキナーゼに対してKmapp でのATPを含むタンパク質キナーゼ活性が選別された。SK1-lは、PKC類, PKCα, PKCδ, PKA, Akt1, ERKl, EGFR, CDK2, 1KKβあるいはCamKllβの二つの異なったメンバーを含む全てのタンパク質キナーゼを大きくは阻害しない(図1F)。従って、SK1-lは、イソ酵素選択性、水への溶解性、タンパク質キナーゼへの影響がないことの観点から、知られているSphK阻害剤のうちで特徴的である。
【0052】
(SK1-lはヒト白血病細胞の増殖を強く阻害する)
以前の研究は一般のSphK阻害剤DMSはU937やJurkat T細胞のアポトーシスを強く引き起こすことを示していた(Cuvillier et al., Nature, 1996, 上記;Jarvis et al., Mol Pharmacol 52:935-947; Edsall et al., 上記; Hamada et al., Biochem Biophys Res Commun 244:745-750, 1998およびCuvillier et al., Blood, 2001, 上記 )。図2Aに示したように、SK1-lは、72時間培養後に明らかになるように5μMの低濃度で10% 血清の存在下で培養したU937細胞の増殖を十分に低下させた。5μMで50%、10μMで完全に増殖が阻害されるように、T-リンパ芽球性Jurkat細胞はSK1-lに対してより敏感であった(図2B)。同様に、10μM SK1-lは、promyelocytic HL60、Molt-4 T細胞白血病およびK-562 CML細胞をそれぞれ50%、70%、90%阻害することを含めて、処理後48時間以内に他の白血病細胞の増殖を減少させた。
SK1-l処理の影響に対すると同様にまた他の白血病細胞での研究と一致して(Bonhoure et al., 上記とBaran et al., 上記)、SphK1タンパク質とmRNAレベルを60%以上(図2C) 減ずる特定の配列を標的としたSphK1発現のダウンレギュレーションが、2%あるいは10%血清の存在下で培養したU937細胞の増殖速度を著しく減少させた(図2C)。総合すると、これらの発見は、薬学的あるいは遺伝的方法によるSphK1の特異的阻害が骨髄性およびリンパ性白血病細胞の増殖を阻害するという見解と一致する。
【0053】
(SK1-lはヒト白血病細胞のアポトーシスを引き起こす)
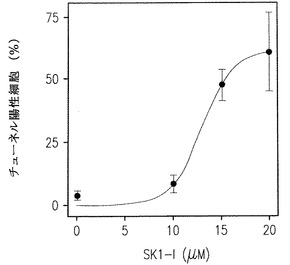
DMSによるSphKの阻害あるいはSphK1発現のダウンレギュレーションはヒト白血病細胞を含め多くの細胞型でアポトーシスの誘導と関連づけられてきた(Cuvillier et al., Nature, 1996, 上記;Jarvis et al., 1998, 上記、Jendiroba et al., 2002, 上記そしてBonhoure et al., 上記)。このように、われわれは次に膜の透過性の基準としてannexin V染色とPlによる細胞膜外側にホスファチジルセリンを発現している細胞をモニターする流動細胞計を用いてU937細胞のアポトーシスに対するSK1-lの影響を調べた。SK1-IにさらしたU937細胞のアポトーシスには時間および濃度への依存が増加した(図2D)。図2Eに示されるように、大部分の細胞が初期のアポトーシス状態にあり、ほんのわずかのパーセントのものが壊死的(Plにのみ陽性)であった。これらの結果はTUNEL測定で決められたDNA鎖開裂の存在と密接に関連していた(図2F)。さらに、細胞が低濃度の血清下で培養されるときにより効果的に細胞増殖を阻害するSphKlのダウンレギュレーションと同様に(図2C)、U937細胞は、血清濃度が減少したときSK1-lで引き起こされるアポトーシスに対して、より敏感になった(図2G)。
【0054】
(SK1-lで誘導される細胞死におけるカスパーゼ活性化とBcl-2開裂の機能的役割)
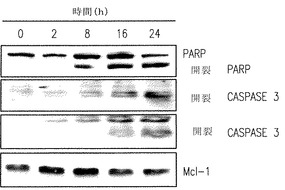
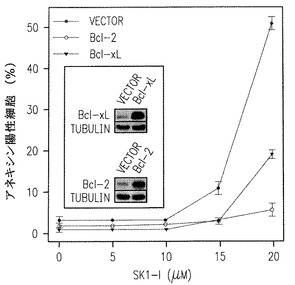
siRNAでのHL60細胞におけるSphK1のダウンレギュレーション(Bonhoure et al., 上記) あるいはJurkat細胞におけるDMSでのSphKの阻害(Cuvillier et al., J Biol Chem 275:15691-15700, 2000)はアポトーシスの証明であるPARPの開裂と同様にexecutionerカスパーゼ3の活性化を起こさせるのに十分である。同様に、アポトーシスの惹起に付随して、SK1-lでのU937細胞の処理はカスパーゼ-3と-9の活性化を増加させ、ポリADPリボースポリメラーゼ(PARP)の開裂を引き起こす(図3A,B)。さらに、16-24時間SK1-lにさらすとミトコンドリアの機能障害を起こさないようにする抗アポトーシスタンパク質であるBcl-2の開裂(図3B)が生じる。他方、悪性造血細胞の生存に鍵となる役割をする抗アポトーシスタンパク質(Moulding et al., Blood 96:1756-1763, 2000)はそれほど多くは変えられなかった(図3A,B)。次に我々はSK1-lで引き起こされる致死でのカスパーゼ活性化とBcl-2開裂の機能的役割を研究した。汎カスパーゼ阻害剤ZVADとBOCでのU937細胞の前処理は、DNA損傷剤エトポシドにより引き起こされるアポトーシスと同様にSK1-lでもたらされるアポトーシスを十分に弱めた(図3C)。さらに、Bcl-2の異所性の発現はSK1-lの引き起こす致死から守り、Bcl-xLの発現は細胞死を60%減弱した(図3D)。総合すると、これらの発見はBcl-2によるのとは反対にSK1-lの致死性は固有のミトコンドリア過程を通して基本的に達成されていることを示している。
SphK1はアポトーシスに向かわせるセラミドと抗アポトーシス的S1Pの間のバランスの決定的な調節剤であるから(Olivere et al., 上記とBonhoure et al., 上記)、これらのスフィンゴシン代謝物のレベルへのSK1-lの影響は高性能液体クロマトグラフィーESI-MS/MSで決められた(Sullards et al., 上記)。SK1-l処理は、スフィンゴシンあるいはジヒドロスフィンゴシン(sphinganine)のレベルを変えることなく、全細胞セラミドの増加とスフィンゴミエリンの減少を伴って全細胞S1Pの50%の減少を引き起こした(図4A)。U937細胞にもっとも多量にあるセラミド種は24:1であった(図4A)。SK1-l処理はC16:0とC24:1セラミド種のレベルをそれぞれ3倍、2倍増加させるが、他のセラミド種には大きな影響をもたらさない(図4A)。
SK1-lのアポトーシス効果がそのSphK1阻害能力によることを確かめるために、S1P添加背景実験を行った。SK1-lによるS1Pレベルの減少に伴い、SK1-lによるアポトーシスは外からのS1Pの添加により添加依存性で減少した(図4B)。総体的に、この発見はSK1-lがSphK1を阻害することによるヒト白血病細胞のアポトーシスとセラミド増加を伴うS1Pの産生を引き起こすことを示している。
【0055】
(SKl-iにより引き起こされるアポトーシスはERKl/2とAkt生存信号の不活性化と関係する)
分裂促進因子で活性化されたタンパクキナーゼ(ERK1/2、JNKおよびp38 MAPK)とAktは白血病細胞の運命に重要な役割を果たすことは豊富な証拠で示されている(Steelman et al., Leukemia 18:189-218, 2004)。U937細胞をSK1-lで処理するとERK1/2とAktのリン酸化の急で大きな減少を引き起こす(図5A-C)。生存信号のこのような不活性化は低濃度血清下で2時間まで続くが(図5A)、一方10%血清の存在下ではp-ERK1/2 とp-Aktレベルの減少は徐々に克服され(図5B)、SK1-lの濃度に依存した(図5C)。さらに、p38リン酸化の一時的な増加が5分間観察され、後の強くない活性化が続いて起きた(図5A,B)。加うるに、JNKの活性化とc-Junリン酸化もまた後の時間に検出された(図A,B)。SK1-lはAktを強く不活性化するので、SK1-lの致死効果でのその役割を決めることは興味深い。構成的に活性のあるミリストイル化されたAktのU937細胞内での過剰発現は15μM以下の濃度のSK1-lで引き起こされるアポトーシスを大きく減少させ(図5D)、Aktの不活性化がSK1-lのアポトーシス効果に役立つ要素の一つであるかもしれないことを示唆している。
【0056】
(原発性ヒトAML芽球はSK1-lで引き起こされるアポトーシスに非常に影響されやすい)
原発AML試料へのSK1-lの影響を調べるために、二人のAML患者(FABサブタイプM2)からの骨髄から得た白血病芽球で平行した実験を行った。SK1-lの濃度を増していった芽球の処理はU937とJurkat細胞と比較して、アポトーシスの誘導に対する感受性が増加していることを明らかにした。両患者のサンプルはSK1-lに24時間さらしたときアポトーシスの顕著な増加を示し、annexin V/PI分析で明らかになったように7.5μMで40-50%のアポトーシスが観察された(図6A)。前の結果と一致して(Rosato et al., Mol Cancer Ther 6:692-702, 2007)、処理しない場合には正常末梢血単核球細胞のアポトーシスと非常に似たアポトーシスを10%以下の芽球が示した。注目すべきことに、SK1-lは正常の末梢血単核細胞の生存に重要な影響をもたらさなかった(図6B)。これらの結果はSphK1を過剰発現していると報告された原発性ヒトAML細胞 (Sobue et al., Leukemia20:2042-2046, 2006) は長期培養された白血病細胞よりSK1-lに対し感受性があるが、SK1-lは正常の末梢血単核白血球には比較的害がないことを示唆している。
【0057】
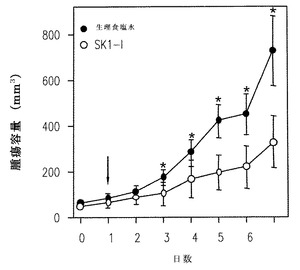
(生体内でのSK1-lの抗白血病活性)
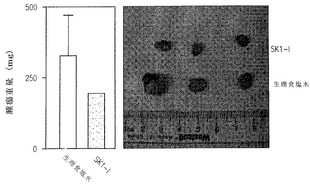
我々は次に、いくつかの新しい処理法の発展を助けるのに非常によく使われてきた免疫不全マウスへの異種移植で、SK1-lの白血病細胞の腫瘍増殖を阻害する能力を評価した (McCormack et al., Leukemia 19:687-706,2005)。SCID/beigeマウスの脇腹に皮下注射したU937細胞は急激に増殖する腫瘍を急速に生じた。腫瘍が50-100 mm3の量に達したときマウスに食塩水あるいはSK1-l (20 mg/kg)を毎日腹腔内注射した。図7Aに見られるように、SK1-lは腫瘍増殖を著しく減少させた。7日後、SK1-lで処理したマウスのU937腫瘍の平均の大きさは食塩水処理マウスの腫瘍より50%以上小さかった(対照群の平均=747 mm3、SK1-l群平均=332 mm3, p<0.001)。SK1-l処理マウスの腫瘍の死後の重量もまた著しく低かった(図7B)。SK1-l処理マウスは消耗した様子は示さず、7日後の体重は対照標準と著しくは違わなかった。
期待されたように、食塩水処理マウスからの腫瘍はKi67で強く染色され、TUNELで染色されるわずかのアポトーシス細胞とともに高い増殖性細胞の腫瘍組成を示していた(図7C)。対照的に、SK1-l処理マウスからの腫瘍細胞の免疫組織化学分析で核の断片化(TUNEL染色)と凝縮した核で決められる多くのアポトーシス様細胞が示された(図7C)。SK1-lはまた腫瘍中の分裂している細胞を劇的に減らした(図7C)。これらの結果はSK1-lが生体内で強い抗白血病活性を持つことを示している。
【0058】
〔討議〕
がんの多くの型で、S1Pの産生が制御低下を起こし、異常な細胞増殖や生存信号にいたることを多くの証拠が示している (Milstien et al., 上記とRA Sabbadini, Br J Cancer 95:1131-1135, 2006)。SphK1は種々の固形癌(French et al., Cancer Res63:5962-5969, 2003)またAML(Sobue et al., 上記)で過剰発現している。さらに、慢性骨髄性白血病の共通の遺伝的異常と急性白血病の患者の少なくとも20%で予後不良の尺度であるbcr/ablがSphK1発現の上昇制御していることが示されている(Li et al., Oncogene 26:7904-7908,2007)。したがって、SphK1はいまでは、そのレベルが薬剤耐性や放射線耐性と関連している白血病細胞では特に、薬学的な干渉の潜在的な標的である考えられている(Bonhoure et al., 上記とBaran et al., 上記)。白血病細胞でのSphK1の役割の以前の研究は、特異的siRNAによるそのダウンレギュレーションあるいはSphK1およびSphK2、潜在的にタンパク質キナーゼを阻害する薬剤の使用に焦点が当てられてきた。本研究はもっとも強力で水溶性のSphK1イソ酵素特異的阻害剤、SK1-Iの開発について述べる。さらに、SK1-lはPKCあるいは大多数の他のタンパク質キナーゼを阻害しない。よく保存されているATP結合ポケットでATPと競合しまた潜在的に交差反応するほとんどの低分子タンパク質キナーゼ阻害剤とは対照的に、SK1-Iは液体基質と競合的である。
【0059】
フォスファチジルセリンの外部化、DNA鎖切断の増加、カスパーゼ3と9の活性化、およびPARPとBcl-2の開裂を反映して、SK1-lはいくつかの白血病細胞系とAML白血病芽球でアポトーシスを強く引き起こす。どのような機構でSK1-lがこれらの致死的な効果を非常に強く引き起こすのかについて考察すると、いくつかのお互いに排除しあわない関連した作用によるものであろうと予測される。SK1-lは、その細胞内標的が解明されてはいないが、細胞内で増殖と生存を増強するように働く生存に有利なS1Pの産生を阻害する(Kohno, et al., Mol Cell Biol26:7211-7223,2006)。細胞内で産生されたS1Pが細胞から放出されることができ(Mitra et al., Proc Natl Acad Sci USA 103:16394-16399,2006)、またERK1/2とAktを含む生存経路につながる細胞表面受容体を通して作用する(Spiegel et al., 上記)ことはまた周知されている。この点について、白血病細胞中の活性化されたERK1/2とAktを減少させるSK1-lの能力は、Raf/MEK/ERKおよびPl3K/AktがしばしばAML中で構成的に活性化されているものと関連づけられうる(Steelman et al., 上記およびNyakern et al., Leukemia 20:230-238, 2006)。ERK1/2はSphK1をリン酸化し活性化し(Pitson et al., J Exp Med 201:49-54, 2005)、そのあとERK1/2を刺激することができるS1Pの増加に導くので、SK1-Iは、SphK1を阻害すること、増殖と生存のS1Pを減少させ一方で同時に前駆体である前アポトーシス状のセラミドを増加させることにより、この正のフィードバック環を断ち切ることができる。それゆえ、SK1-Iは白血病での多様な分子治療標的を構成する。
【0060】
セラミド産生はヒト白血病細胞にアポトーシス引き起こすと長い間関連づけて考えられてきた(Jarvis et al., Proc Natl Acad Sci USA 91:73-77,1994)、そして最近、いくつかの異なったシグナル伝達阻害剤のアポトーシスへの相乗効果がセラミド産生の増加と関連づけられてきた。例えば、ヒト白血病細胞にペリフォシンとヒストンデアセチラーゼ同時に投与すると、AktとERKの崩壊、セラミドと反応性の酸素種の産生の著しい増加、ミトコンドリア傷害とアポトーシスの驚くべき増加を引き起こす(Rahmani et al., 上記とRosato et al., Mol Pharmacol 69:216-225, 2006)。セラミドはそのアポトーシス作用を多くの経路で変換できる(Ogretmen et al., 上記)。重要な同定されたセラミド標的は、Bcl-2の別のスプライシングの調節役であるSRタンパク質と同様にAktを脱リン酸化するセリン/スレオニンタンパク質ホスファターゼであるPP1とPP2Aを含んでいる(Ogretmen et al., 上記)。我々はSK1-lにさらすと、ミトコンドリア依存性のアポトーシス関連づけられてきた反応であるBcl-2の開裂が起きることを見いだした(Cheng et al., Science 278:1966-1968,1977)。さらに、Bcl-2の過剰発現は、セラミド生成を妨げ、急性リンパ芽球性白血病やAMLを含む多くの細胞でのセラミドにより誘起されるアポトーシスから守ることは立証されている (Zhang et al., Proc Nat Acad Sci USA 93:5325-5328, 1996、Amarante-Mendes et al., Blood 91:1700-1705, 1998とOgretmen et al., 上記)。これと一致して、我々はBcl-2の過剰発現が、内在するミトコンドリアの死経路の重要性を強調するSK1-l誘発の致死性をもまた妨げることを発見した。この見解と一致して最近、S1Pが、MEK/ERK1/2依存的な方法でミトコンドリアへのBaxの移転をブロックすることでJurkat細胞のアポトーシス間のミトコンドリア内の事象に細胞防御的な効果を与えることが示されている (Betio et al., Biochem Biophys Res Commun340:1273-1277.2006)。最近の研究では、小胞体でのセラミドの持続的な増加は同時にER stress responseを活性化し、抗アポトーシス性のAktを不活性化しアポトーシスに向かわせることが示唆されている(Swanton et al., Cancer Cell 11:498-512, 2007)。したがって、SK1-lで引き起こされるAktの不活性化はS1P生成の減少によるばかりでなくセラミドの増加によってももたらされることが可能である。SK1-lと構造的に似ており、SphKsの拮抗阻害剤であるよりは拮抗基質である免疫抑制剤ETY720もまたAktリン酸化を減少させることが示されている (Ng et al., Int J Oncol 30:375-380, 2007)。多発性硬化症の臨床試験で比較的毒性が少ないFTY720 (Brinkman et al., Pharmacol Ther 115:84-105, 2007)は芽球急変期の慢性骨髄性白血病とフィラデルフィア染色体陽性の急性リンパ性白血病を治療する別の方法であることが最近提唱されている (Neviani et al., J Clin Invest 117:2408-2421, 2007)。
【0061】
SK1-lがAML患者から分離した白血病芽球のアポトーシスを強く引き起こすが正常末梢血単核白血球には比較的細胞毒性がないことは、白血病細胞への選択性を強調する。さらに、異種移植AML中で、試験管内での作用と同様に、SK1-lが腫瘍増殖を抑え、腫瘍のアポトーシスを誘導し、増殖を減少させる明瞭な信号発信剤の活性を持っていた。肝臓、腎臓、膵臓への毒性の予備的分析では、目立った効果は全くなかった。このように、特異的SphK1阻害剤は、単独であるいは通常のまたは他の知られた標的剤の付加物として使用される、白血病の強力な薬学的な介在物として考慮に値する。
【0062】
上記の詳細な記載と本発明の例にてらして、多くの明らかな差違が疑いもなくその分野の当業者のそれらに対して示唆される。同じ目的を達成するために計算されたいかなる取り決めも、示された特殊な具体化物で置き換えられ得ることが当業者により評価されるであろう。この応用と発明は、本発明のいかなる応用物あるいは変化物をも包含するように意図されている。以下に続く請求範囲により特別に決められているように、全てのこのような変化物は発明の範囲と意図に完全に含まれている。
【技術分野】
【0001】
本発明は、全般的に言えば、スフィンゴ脂質媒介、スフィンゴ脂質媒体および特にタイプ1を含むスフィンゴキナーゼの阻害物質、及びがん、ぜんそく、アナフィラキシー、自食作用、中枢神経系統、その他の分野に関する。
【背景技術】
【0002】
この出願において引用されるか又は明らかにされているすべての特許、特許出願、特許刊行物、科学論文等は、本発明が属する技術分野の状況をより完全に記述するために、ここにそっくりそのまま引用文献によって取り入れられている。
この出願は2008年4月29日に出願された暫定的な出願No.61/048,638に関連するものである。
【0003】
スフィンゴシン−1−ホスフェイト(S1P)、即ちスフィンゴシンキナーゼ(SphKs)によってスフィンゴシンから製造される効力のある脂質の媒介物質は、がんの進行にとって重要な細胞の成長及び生存を含む多くの過程を調節する(非特許文献1)。
S1Pとは対照的に、その前駆体、スフィンゴシン、及びセラミドは、アポトーシスの成長抑止や誘導に関連している(非特許文献2)。かくしてこれら相互転換出来るスフィンゴ脂質代謝産物は、細胞の壽命を測定する細胞質可変抵抗器として見られてきた(非特許文献3)。
今までの多くの研究が、S1P/セラミド制御点中の摂動が、造血起点に対する化学療法や放射線療法を含む腫瘍性細胞の化学療法や放射線療法に対する抵抗性調節に含まれることを示している(非特許文献4及び非特許文献5)。
【0004】
二つのSphkイソ酵素、SphK1およびSphK2が多くの特徴を共有するとは言え(非特許文献6及び非特許文献7)、異なった機能を示すことが記述されている。
SphK1は細胞成長と細胞生存を促進する(非特許文献8〜非特許文献11)。
一方SphK2は過剰発現されるとき、逆の効果を有する(非特許文献12及び非特許文献13)。
SphK1はS1P/セラミド制御点を調節するキー酵素である(非特許文献14及び非特許文献15)。
【0005】
実際以前からS1PとSphK1とが一次白血病性細胞と白血病性細胞系の両方が、一般的に使用される細胞毒性薬剤によって導かれるアポトーシスに対する抵抗に関与しているとされて来た(非特許文献16〜18)。
レートレオージヒドロスフィンゴジン(safingol)およびN,N−デメチルスフィンゴジン(DMS)のような、SphKsの非イソ酵素特定阻害物質は、白血病細胞に対して有毒である(非特許文献19及び非特許文献20)。興味深いことには、多数の薬剤に対する抵抗性を有するHL-60骨髄性白血病性細胞は親細胞よりもDMSに対してより感受性が強かった(非特許文献20)。更にSphK1の活性は、MDR1-又はMRP1-陽性HL-60細胞中よりもドキソルビシン/またはエトポサイドに対して感受性のあるHL-60細胞中の方が低かった。
感受性のあるHL-60細胞中のSphK1の強調された発現はアポトーシスをブロックしたが、一方SphK1の反応抑制作用は、ミトコンドリア依存のアポトーシスを誘導することによって化学薬剤抵抗性を克服した(非特許文献21)。これらの観察は、MDR発現が骨髄性白血病(AML)における強力な徴候の指標である証拠(非特許文献22)、そして一般的にアントラサイクリン又は植物ベースのアルカロイドによるAMLの治療の後に続くのであるが、MDR表現形は化学療法の成功を妨げる役割をすると考えられている証拠に照らして見れば、更なる重要性をもたらすものである。
【0006】
加えて、K562ヒト慢性骨髄性白血病性細胞のイマチニブに対する抵抗性、Bcr-Abl-チロシンキナーゼの阻害はSphK1の発現とS1Pの生成に相互に関連し、一方SphK1の反応抑制は抵抗細胞中のイマチニブ−誘導−アポトーシスに対する感受性を増強する(非特許文献23)。かくしてSphK1の効果的な特効のある阻害物質の開発は不活性型生存S1Pのレベルを減少させることのみならず、ケラミド生成の助長に、少なくとも部分的に一定の細胞毒薬剤の不活性型アポトーシス作用を仲介する過程において有用であることがわかる(非特許文献24〜26)。
シンゴシンキナーゼ阻害物質は以前から記述されている(非特許文献27〜31)。これらの刊行物は、しかしながら本発明の新規なスフィンゴシンキナーゼタイプ1阻害物質については記述していない。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Spiegel et al., Nature Rev Mol Cell Biol. 4:397-407, 2003
【非特許文献2】Ogretman & Hannun, Nature Rev Cancer 4:604-616, 2004
【非特許文献3】Cuvillier et ao., Nature 381:800-803, 1996
【非特許文献4】Ogretmann et al., supra.; Hait et al., Biochem Biophys Acta 1758:2016-2026. 2006
【非特許文献5】Milstien & Spiegel, Cancer Cell 9:148-150, 2006
【非特許文献6】Kohama et al., J. Biol Chem 273:23722-23728, 1998
【非特許文献7】Liu et al., J. Biol Chem 275: 19513-19520, 2000
【非特許文献8】Olivera et al., J Cell Biol 147:545-558, 1999
【非特許文献9】Xia et al., J. Biol Chem 277:7996-8003, 2002
【非特許文献10】Bonhoure et al., Leukemia 20:95-102, 2006
【非特許文献11】Sukocheva et al., J Cell Biol 173:301-310, 2006
【非特許文献12】Maceyka et al., J Biol Chem 280:37118-37129,2005
【非特許文献13】Okada et al., J Biol Chem 280:36318-36325, 2005
【非特許文献14】Maceyka et al., supra.; Berdyshev et al., Cell Signal 18:1779-1792, 2006
【非特許文献15】Taha et al., FASEB J 20:482-484, 2006
【非特許文献16】Cuvillier et al., Nature, 2004 supra.; Cuvillier et al., J Biol Chem 273:2910-2916, 1998
【非特許文献17】Cuvillier et al., Blood 98:2828-2836, 2001
【非特許文献18】Jendiroba et al., Leuk Res 26:301-310, 2002
【非特許文献19】Jarvis et al., Mol Pharmacol 54:844-856, 1998
【非特許文献20】Jendiroba et al., 2002, supra.
【非特許文献21】Bonhoure et al., 2006, supra.
【非特許文献22】Filipits et al., Leukemia 14:68-76, 2000
【非特許文献23】Baran et al., J Biol Chem 282:10922-10934, 2007
【非特許文献24】Maggio et al., Cancer Res 64:2590-2600, 2004
【非特許文献25】Rahmani et al., Cancer Res 65:2422-2432, 2005
【非特許文献26】Rosato et al., Mol Pharmacol 69:216-225, 2006
【非特許文献27】Kim et al., Bioorg & Med Chem 13:3475-3485, 2005
【非特許文献28】Kono et al., J. Antibiotics 53:459-466, 2000
【非特許文献29】Kono et al., J. Antibiotics 53:753-758, 2000
【非特許文献30】Marsolais & Rosen, Nature Reviews/Drug Discovery 8:297-307, 2009
【非特許文献31】US 2008/0167352 A1 (Smith et al., published July 10, 2008)
【発明の概要】
【発明が解決しようとする課題】
【0008】
ここに我々は種々の信号発信および生存関連蛋白質の活性化における多重摂動を引き起こすSphK1(SK1-l)の効力のある水溶性阻害物質を説明する。
SK1-lはAMLによって患者から得られた芽細胞のみならずヒト白血病性細胞系におけるアポトーシスを明らかに誘発し、そしてAML異種移植腫瘍の成長を阻害した。
SK1-lは以下に記載する他の関連合成物に対するモデルとして役立つ。
【課題を解決するための手段】
【0009】
この発明はまた、スフィンゴシンキナーゼ活性を測定する試験管内測定法でスフィンゴシンキナーゼ1(SphK1)をスフィンゴシンキナーゼ2(SphK2)よりも少なくとも5倍以上強く阻害する合成物を提供する。
本発明は下記の化1の構造からなる合成物を提供する。
【化1】
この図のXはC(炭素)あるいはN(窒素)であり、R1、R2、R3、R4はそれぞれ水素、直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、あるいはエーテルグループで構成され、そしてそのなかで、R1、R2、R3、R4は独立してお互いに融合して一つあるいはそれ以上の環をつくる、あるいは上述のもののあらゆる組み合わせでもあり得る。
【発明の効果】
【0010】
本発明の合成物は、がん細胞を殺したり損傷させたりすること、アポトーシス(プログラムされた細胞死)を誘導すること、がん細胞、白血病で増殖、転移、薬剤耐性の発現を阻害すること、抗がん剤の効果を増強すること、免疫反応性を弱めること、がん細胞の生存信号を阻害すること、多発性硬化症の症状を和らげること、など多くの応用あるいは基礎づけに有用である。
【図面の簡単な説明】
【0011】
【図1A】SK1-l (BML-258)の構造をしめす。
【図1B】組み替えSphK1およびSphK2へのSK1-lの影響を示す結果である。
【図1C】組み替えSphK1およびSphK2へのSK1-lの影響を示す結果である。
【図1D】組み替えSphK1およびSphK2へのSK1-lの影響を示す結果である。
【図1E】組み替えSphK1およびSphK2へのSK1-lの影響を示す結果である。
【図1F】SK1-lの種々のタンパク質キナーゼ活性への影響を示す結果である。
【図2A】U937細胞の増殖へのSK1-Iの濃度効果を示す。
【図2B】Tリンパ球性Jurkat細胞の増殖へのSK1-lの濃度効果を示す。
【図2C】siRNAを標的としてSphK1発現を低下させたU937細胞の増殖速度の低下を示す。
【図2D】SK1-lにさらすことによるU937細胞のアポトーシスの時間および濃度依存的な増加を示す。
【図2E】大多数の細胞が初期アポトーシスの状態で少数の細胞が壊死状態(Pl-陽性)にあるU937細胞の様子を図示する。
【図2F】TUNEL測定法で決められるDNA鎖断裂と結果が密接に関連していることを示している。
【図2G】U937細胞への血清濃度の効果を示す。
【図3A】U937細胞のSK1-l処理がカスパーゼ3とカスパーゼ9活性化を増強し、アポトーシス誘導を伴うポリADPリボースポリメラーゼ(PARP)の開裂を引き起こすことを示す。
【図3B】U937細胞のSK1-l処理がカスパーゼ3とカスパーゼ9活性化を増強し、アポトーシス誘導を伴うポリADPリボースポリメラーゼ(PARP)の開裂を引き起こすことを示す。
【図3C】汎カスパーゼ阻害剤であるZVADとBOCでのU937細胞の前処理のSK1-lで誘導されるアポトーシスとDNA損傷剤エトポシドによる誘導への結果を示す。
【図3D】SK1-Iで誘導される致死性に対するBcl-2の異所性発現の効果を示す。
【図4A】HPLC ESI-MS/MSにより決定したスフィンゴ脂質代謝物のレベルに対するSK1-lの影響を示す。
【図4B】量依存的に外から加えたS1PによるSK1-l誘導アポトーシスの結果を示す。
【図5A】U937細胞がSK1-lで処理されたときERK1/2とAktのリン酸化の減少を示す。
【図5B】U937細胞がSK1-lで処理されたときERK1/2とAktのリン酸化の減少を示す。
【図5C】U937細胞がSK1-lで処理されたときERK1/2とAktのリン酸化の減少を示す。
【図5D】U937細胞中に構成的に活性化されたミリスチン酸化Aktの過剰発現の15μM以下の濃度でのSK1-lに誘導されるアポトーシスに対する影響を示す。
【図6A】SK1-lにさらされたときの二人の患者試料でのアポトーシスの増加を示す。
【図6B】SK1-lにさらされたときの二人の患者試料でのアポトーシスの増加を示す。
【図7A】免疫能の欠乏したマウスへ異所移植した腫瘍増殖のSK1-l投与による減少を示す。
【図7B】マウスでの腫瘍重量へのSK1-l処理の効果を示す。
【図7C】SK1-I処理マウスからの腫瘍の免疫組織化学的分析を示す。
【発明を実施するための形態】
【0012】
本発明は化1の構造からなる合成物を提供する。
【化1】
化1のXはC(炭素)あるいはN(窒素)であり、R1、R2、R3、R4は独立に水素、直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、エーテルグループで構成される。そしてそのなかで、R1、R2、R3、R4は独立してお互いに融合して一つあるいはそれ以上の環を作り、あるいは上述のもののあらゆる組み合わせでもあり得る。
【0013】
本発明はまた化2の構造からなる合成物を提供する。
【化2】
【0014】
本発明は化3の構造からなる合成物を提供する。
【化3】
化3のR5とR6は独立に直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、あるいはエーテルグループあるいは上述のもののあらゆる組み合わせで構成される。
【0015】
また本発明では、R5とR6がお互いに結合し環を作って為るさらなる合成物として、以下の化4の構造を持つ合成物を提供する。
【化4】
化4のR7は水素、直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、エーテルグループあるいは上述のもののあらゆる組み合わせで構成される。
【0016】
ほかの例では、R1がH(水素)でR2がCH3である合成物が提供される。さらにまたほかの合成物では、R3あるいはR4がサルファイド、SR5であり、R5が水素、直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、エーテルグループあるいは上述のもののあらゆる組み合わせで構成される。
【0017】
SR5が以下の化5の構造を持つもう一つの合成物が提供される。
【化5】
【0018】
さらに他の合成物では、XがC、R1とR3がH、そしてR4がSR5であり、合成物は以下の化6の構造を持つ。
【化6】
【0019】
R2がCH3、XがCで有り得るものであり、R1とR4がHであり、そしてR3がSR5である合成物は以下の化7の構造を持つ。
【化7】
上記の合成物では、R2はCH3、R3はHであり得る。
【0020】
他の例では、XがC、R1がH、R2がCH3そしてR4が(CH2)4CH3で、合成物は以下の化8の構造を持つ。
【化8】
R4の末端の炭素は一つあるいはそれ以上のハロゲン化物で置換でき、ハロゲン化物は臭化物、塩化物、フッ化物である。
【0021】
他の例では合成物は以下の化9の構造を持つ。
【化9】
R4はHであり得、R2はメチルグループ、R1は水素あるいはメチルグループである。
また、R1とR2は融合し、置換基の入ったあるいは入らない環をつくることができる。
【0022】
他の例では、本発明は以下の化10の構造を持つ合成物を提供する。
【化10】
XはCであり得る。
【0023】
もう一つの合成物は以下の化11の構造を持つ。
【化11】
ここでは、XはCである。
【0024】
また、R4はエーテルであり得、合成物は以下の化12の構造を持つ。
【化12】
ここでは、R5は直鎖あるいは枝分かれした(C1−C18)アルキルグループ、置換基の入った直鎖あるいは枝分かれした(C1−C16)アルキルグループ、シクロアルキルグループ、置換基の入ったシクロアルキルグループ、ヘテロサイクリックグループ、置換基の入ったヘテロサイクリックグループ、アリルアルキルグループ、置換基の入ったアリルアルキルグループ、ヘテロアリルアルキルグループ、置換基の入ったヘテロアリルアルキルグループ、アルコキシグループ、置換基の入ったアルコキシグループ、アルケングループ、置換基の入ったアルケングループ、アルキングループ、アシルグループ、サルファイド、エーテルグループあるいは上述のもののあらゆる組み合わせを含んでいる。
さらなる例では、XはCであり得、R1はHそしてR2はCH3である。
【0025】
多くの有用な過程が上述の合成物で提供されている。
これらは当該がん細胞を殺すあるいは損傷するに十分な量の合成物にがん細胞をさらす段階を含み、がん細胞を殺すあるいは損傷させる過程を含んでいる。このようながん細胞としては、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫あるいは腎がん細胞およびこれらの組み合わせを含んでいる。
【0026】
この発明で提供されるもう一つの過程は、がん細胞にアポトーシスを起こさせ、また、当該がん細胞にアポトーシスを起こさせるに十分量の合成物に対してがん細胞をさらすステップを含んでいる。もう一度言うと、そのようながん細胞は白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫あるいは腎がん細胞およびこれらの組み合わせを含んでいる。
この発明はまた、当該がん細胞の増殖、転移、薬剤耐性の展開を阻害するのに十分量の合成物へがん細胞をさらすステップを含む、がん細胞の増殖、転移、薬剤耐性の展開を阻害する過程を提供する。もう一度言うと、がん細胞には白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫あるいは腎がん細胞およびこれらの組み合わせが含まれる。
【0027】
もう一つの過程は、当該患者で白血病の症状を処置し、和らげるに十分量の合成物を当該患者に導入するステップを含んで、必要な患者で白血病の症状を処置し、和らげる過程である。
さらにもう一つの過程は、抗がん剤を患者に投与するステップを含んで、必要とする患者内のがん細胞を殺すための抗がん剤の能力を増加させる過程である。そして該合成物、該合成物は患者中のがん細胞を殺すための抗がん剤の能力を増加するために十分な量を投与される。がん細胞には白血病細胞、胸部がん細胞、前立腺がん、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞あるいは腎がん細胞およびこれらの組み合わせが含まれうる。
また、患者の免疫反応性を減ずるに十分量の合成物を患者に投与するステップを含んで、必要な患者の免疫反応性を減弱する過程が提供される。さらなる具体例では、肥満細胞機能の減弱により免疫反応性の減弱が引き起こされている。また、免疫反応性の減弱が必要とされる患者のぜんそくの症状を和らげるようにしむけられことができる、免疫反応性の減弱はアナフィラキシーショック症状を和らげあるいは自己免疫病の症状を和らげるのにしむけられることができる。
【0028】
提供されるさらなるもう一つの過程は、請求項1の合成物を、生存信号を阻害するのに十分量癌様細胞に投与するステップを含んで、癌様細胞の生存信号を阻害するためのものである。もう一つの具体例では、生存信号の阻害はAktあるいはERK1/2あるいはこの両者のリン酸化の減少で達成されている。
さらに多発性硬化症患者の症状を和らげるのに十分量の合成物を患者に投与することを含んで、必要とされる多発性硬化症患者の症状を和らげるもう一つの過程が提供されている。
【0029】
この発明はまた、スフィンゴシンキナーゼを測定する試験管内測定法で、スフィンゴシンキナーゼ1(SphK1)をスフィンゴシンキナーゼ2(SphK2)を阻害するよりもよりもすくなくとも5倍以上強く阻害する合成物を提供する。SphK1はまた当該SphK2の阻害よりすくなくとも10倍以上阻害される。試験管内測定での阻害は10μM濃度で測定される。試験管内測定法での阻害はまたSphK1を50%阻害する濃度で測定することができる。
上記の合成物はまた多くの応用と基礎づけに有用である。これらは、がん細胞を殺すか損傷させるに十分量の上記合成物にがん細胞をさらすステップを含み、がん細胞を殺し、または損傷させる過程を含む。がん細胞には白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞あるいは腎がん細胞およびこれらの組み合わせが含まれうる。
【0030】
もう一つの具体例では、上記合成物は、がん細胞をアポトーシスに向かわせるのを引き起こすのに十分量の請求項44の合成物にがん細胞をさらすステップを含んで、がん細胞をアポトーシス向かわせるのを引き起こす過程に有用である。ここでも、がん細胞には白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞あるいは腎がん細胞およびこれらの組み合わせが含まれうる。
【0031】
上記合成物を含むもう一つの過程では、この発明は、がん細胞の増殖、転移、薬剤耐性の発現の阻害に十分量の合成物にがん細胞をさらす過程を含んで、がん細胞の増殖、転移、薬剤耐性の発現を阻害する過程を提供する。がん細胞には白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞あるいは腎がん細胞およびこれらの組み合わせが含まれうる。
患者の有する白血病の症状を治療または軽減するための方法は該患者の白血病の症状を治療または軽減するために十分な量の最後に記載した例の合成物を該患者に投与するステップからなる。
【0032】
もう一つの具体例では、抗がん剤を患者に投与するステップを含んで、必要とされる患者内のがん細胞を殺す抗がん剤の能力を増強する過程を提供する。そして最後に記載した例の合成物は抗がん剤の患者のがん細胞を殺すための能力を増加するために十分な量を投与される。がん細胞には白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠腫がん細胞、大腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞あるいは腎がん細胞およびこれらの組み合わせが含まれうる。
また、患者の免疫反応性を弱めるのに十分量の上記合成物を患者に投与するステップを含み、必要とされる患者の免疫反応性を弱める過程が提供される。他の具体例では、免疫反応性の減弱は肥満細胞機能の減弱によりもたらされる。免疫反応性の減弱は、必要とされる患者の喘息の症状を和らげ、アナフィラキシーショックの症状を和らげ、自己免疫病の症状を和らげることに導かれる。
【0033】
もう一つの具体例では、この発明は、生存信号を阻害するに十分量の上記合成物を癌様細胞に投与するステップを含んで、癌様細胞の生存信号を阻害する過程を提供する。生存信号の阻害はAktあるいはERK1/2あるいは両者のリン酸化の減弱により達成されうる。
この発明で提供されるもう一つの過程は、患者の多発性硬化症の症状を和らげるに十分量の上記合成物を患者に投与するステップを含んで、必要とされる患者の多発性硬化症の症状を和らげるために提供される。
【0034】
ここに述べられる本発明の合成物のすべてが、二量体あるいは多量体の合成物で明瞭に示されることが認識されるべきである。
また、この合成物は、従来よく知られている過程を用いて通常の材料と結合されてきた薬用合成物に明示することができる。これらの合成物は患者が摂取するために、タブレット、ピル、カプセル、液体、ゲル、シロップ、水混合物、懸濁液やこれに似たものとして明示されうる。
【実施例】
【0035】
以下の例は例証としてこの発明の提示されるものであり、発明を限界づけるものではない。
〔例1〕
記載され以下に使われている合成物、BML-258、は以下の案と手順に従って合成された。
【化13】
窒素中、-20℃で65 ml乾燥THF中の4-n-phenylphenylacetylene 1(3.343g, 0.01776 mol) にn-BuLi (1.6 M hexane中に10.2 ml、0.01628 mol)を滴下して加えた。反応混液を-20℃で2時間攪拌した。25ml 乾燥THF中のmethyl(R)-(+)-3-(t-butoxycarbonyl)-2,2-dimethyl-4- oxazolidinecarboxylate 2 (3.393g, 0.01480 mol)をカニューレを通して加えた。反応液を-20℃で一晩攪拌した。TLC(薄層クロマトグラフィー)(20% ethylacetate/hexanes)では反応が完了していた。
反応液をET2O で希釈し、水と塩水で注意深く洗った。フラッシュカラムクロマトグラフィー(12% ethyl acetate/hexanes, silica gel)で、収量はerythroとthreo の混合物として4.50g (73 %)であった。
調達用のHPLC(高速液体クロマトグラフィー)(Dynamax Si, 15% ethyl acetate/hexanes, 260 nm)で収量は3.71gのerythro 3と0.49g threoであった。
1H NMR(CDCl3) erythro:7.34-7.32(d, 2H), 7.12-709(d,2H), 5.19-5.16(d, 1H) 4.73-4.70(d,1H), 4.26-3.96(m, 3H), 2.61-2.56(t, 2H), 1.62(s, 3H), 1.60-1.50(m,2H),1.54(s, 3H), 1.50(s, 9H), 1.34-1.27(m, 4H), 0.91-0.86(t, 3H)。
【0036】
【化14】
100 ml MeOH中のoxazolidine 3 (3.48g, 0.00814 mol)にAmberlyst-15 (200 mg)を加えた。反応液は室温で一夜攪拌した。TLC(30% Ethyl acetate/hexanes)では反応は完全であった。混合物は濾過され、迅速クロマトグラフィー(5% MeOH/methylene chloride, silica gel) で2.44g (79%)のaminoalcohol 4 がとれた。
1H NMR(CDCl3): 7.34-7.32(d, 2H), 7.12-709(d,2H), 5.45-5.38(d, 1H) 4.88-4.82(m,1H), 4.25-4.19(m, 1H), 3.91-3.80(m, 2H), 3.26-3.23(d, 1H), 2.61-2.56(t,2H), 1.63-1.54(m, 2H), 1.49(s, 9H), 1.35-1.26(m, 4H), 0.91-0.86(t, 3H)。
【0037】
【化15】
窒素中、0℃で125 ml 乾燥Et2Oのalkyne 4 (2.44g, 0.00646 mol)にRed-Al (9.85 ml, toluene中に65 wt%, 0.03232 mol)を滴下しながら加えた。滴下後、反応液を室温にし、36時間攪拌した。TLC (40% Ethyl acetate/hexenes)では反応は完結していた。反応液を0℃に冷やし、注意深く15% NaOH液で急冷した。この混合物を、両方の層が透明になるまで強く攪拌した(45 分)。両層を分離し、水層をchloroformで抽出した(3回)。有機層一緒にして15% NaOH、水、塩水で洗浄した。フラッシュクロマトグラフィー(5% MeOH/methylene chlorideから20% MeOH/methylene chloride + 1% NH4OH, silica gelの濃度勾配)で1.76g (72%)のtrans alkene 5 を得た。
1H NMR(CDCl3): 7.31-7.29(d, 2H), 7.15-712(d,2H), 6.70-6.65(d, 1H, J=16Hz) 6.26-6.18(dd,1H, J=16Hz), 5.35-5.32(d, 1H), 4.55-4.49(m, 1H), 4.03-3.96(m, 1H), 3.80-3.68(m,2H), 2.83-2.79(d, 1H), 2.61-2.56(t, 2H), 1.65-1.55(m, 2H), 1.44(s, 9H).1.34-1.25(m, 4H), 0.91-0.86 (1,3H)。
【0038】
【化16】
窒素中で20ml 乾燥THFに溶かしたBOC-alkene 5 (0.350g, 0.00092 mol)に室温でDIBAL(THF中に1Mを9.22ml, 0.00922 mol)を注意深く加えた。加えた後、反応液を還流した。24時間還流後、混合物を室温に冷却し、さらに5.0 mlのDIBAL液(0.00500 mol)を加えた。還流をさらに24時間続けた。反応液を0℃に冷やし、水(0.60 ml)、15% NaOH(0.60 ml)そしてもう一度水(1.50 ml)を加えた。THF(50 ml)を加え、混合物を15分間強く攪拌した。それからNa2SO4(2 g)、celite (2 g)を加え、室温まで暖めながら、攪拌を30分続けた。混合物を濾過し、濾過した固まりを大量のTHFで抽出した。フラッシュクロマトグラフィー(2% MeOH/methylene chlorideから10% MeOH/methylene chloride + 0.75% NH4OHの濃度勾配)で0.187g (73%)のamine 6 を得た。
1H NMR(CDCl3): 7.31-7.29(d, 2H), 7.15-712(d,2H), 6.68-6.63(d, 1H, J=16Hz) 6.22-6.14(dd, 1H, J=16Hz), 4.51-4.47(m, 1H), 3.80-3.74(m, 3H), 2.61-2.56(t, 2H), 2.50(s,3H), 2.40-2.10(broad, 2H), 1.65-1.55(m, 2H), 1.34-1.25(m, 4H), 0.91-0.86(t, 3H). HRMS(MH+): Calc. - 278.2120, Found - 278.2199.
【0039】
【化17】
15 ml の乾燥Et2O中のamine 6 (0.335g, 0.00121 mol))に3 ml の1M HCl/Et2Oを加えた。すぐに白い沈殿ができた。室温で15分攪拌後沈殿を濾過し、Et2Oで洗い、0.352g(89%)のBML-258を得た。
1H NMR(DMSO): 8.75-8.50(bd, 2H), 7.38-734(d,2H), 7.19-7.15(d, 2H,), 6.65-6.60(dd, 1H, J=16Hz), 6.30-6.22(dd, 1H, J=16Hz), 5.84-5.82(m, 1H), 5.30-5.25(m, 1H), 4.60-4.54(m,1H), 3.76-3.72(m, 2H), 3.18-3.10(m, 1H), 2.64(s, 3H), 2.56-2.50(t, 2H)1.60-1.50(m,2H), 1.34-1.23(m, 4H), 0.90-0.85(t, 3H).
【0040】
SK1-I、(2R,3S,4E)-N-methyl-5-(4'-pentylphenyl)-2-aminopent-4-ene-1,3-diol (BML-258)は例1に述べたようにBIOMOL International (Plymouth Meeting, PA)により合成された。SphingosineとN,N-dimethylsphingosineはMIOMOLから得た。[y-32P]ATP(3000 Ci/mmol)はPerkin Elmer (Boston, MA)から購入した。Boc-D-FMK(BOC), Z-VAD-FMK(ZVAD)、etoposideはEMD Biosciences (San Diego, CA)からのものである。流動細胞計測法(フローサイトメトリー)のためのTerminal deoxynucleotidyl trensferase Br-dUTP nick end labeling (TUNEL) kitはSigma Aldrich (St. Louis, Mo)からのものである。免疫組織化学用のTUNELキットはRoche Applied Science (Indianapolis, IN)からのものである。アポトーシス用のFITC4標識annexin V/propidium iodide染色キットはBD Biosciences (San Jose, CA)からのものである。
【0041】
〔細胞と細胞培養〕
U937 ヒト組織細胞型白血病およびJurkat急性T細胞白血病細胞はAmerican Type Culture Collectionから入手した。特に指示しない限り、細胞はL-glutamate, penicillin, streptomycin, 10% 仔牛血清(Invitrogen, Carlsbad, CA)を加えたRPMI 1640培地に培養し、対数増殖期に維持された(Dai et al., Cancer Res 61: 5106-5115,2001)。Bcl-2、Bcl-xL、構成的に活性なAkt (Mycがついたミリオスチン酸化Akt)およびそれらの対応する空ベクターを安定的に過剰発現しているU937をえて、記載されたと全く同じように適当な選択抗生剤の存在下で培養した(Rahmani et al., 2005, 上記)。
Institutional Review Board of Virginia Commonwealth Universityからの認可のもとに、通常の特徴的な吸引をしている二人のAML患者から、インフォームドコンセントのもとに白血病芽球を得た。インフォームドコンセントはヘルシンキ宣言にしたがって行った。二人の患者試料の特徴は次の通りである。
患者1:FABサブタイプはM2、知られている融合あるいは変異タンパク質はない、知られている染色体異常はない。
患者2:FABサブタイプはM4、知られている融合あるいは変異タンパク質はない、16番染色体に逆位がある。
それぞれ85%の芽細胞を含むサンプルは室温でFicoll/Hypaque(比重1.077-1.081; Sigma, St. Louis, MO, USA)上で400 x g の遠心分離で分けた。芽細胞を含む境界層を滅菌したパスツールピペットでとり、10% FBSを含む培養液に再懸濁した。細胞はトリパンブルー排除法で95%以上の生存率を示し、上記のように培養した。末梢血の単核白血球は同じ方法で健常人から分離した。
【0042】
〔RNA干渉〕
Qiagen (Valencia, CA)から入手したSphK1 を標的とした100 pmol のRNAi オリゴヌクレオチド(標的配列:GGGCAAGGCCTTGCAGCTC (SEQ ID NO: 1)) と標的としない対照siRNA (非特異的なランダムな配列)をU937細胞に導入した。導入はCell Line Nucleofector Kit V (Amaxa GmbH, Cologne, Germany)のAmaxa Nucleofecter (program V-001)により製造者の指示に従って行った
【0043】
〔スフィンゴシンキナーゼの発現と活性〕
HEK 293細胞を10%子牛血清を含むDMEMで培養し、以前に述べたようにLipofectamine PLUS (Invitrogen)を用いて、V5-His-pcDNA3.1 ベクター(Invitrogen)、V5-His標識ヒトSphK1あるいはV5-His標識ヒトSphK2を導入した(Paugh et al., FEBS Lett 554: 189-193, 2003)。それから細胞を2日間培養し、凍結融解で溶解し、[y-32P]ATP (10pCi, 1 mM, 10 mM MgCl2中)とスフィンゴシンを用いて、SphK2を阻害する0.25% Triton X-100中でSphK1活性を測定した (Hait et al., J Biol Chem280: 29462-29469, 2005)。SphK2活性は1 M KClの存在下で、4 mg/ml BSA との複合体で加えたスフィンゴシンを用いて測定された。この条件はSphK2活性に至適であり、SphK1は強く阻害される (Hait et al., J Bio Chem, 2005,上記)。標識されたS1Pは抽出され、クロロフォルム/アセトン/メタノール/酢酸/水(10:4:3:2:1. v/v)を溶剤するシリカゲルG60上でのTLCで分離した。S1Pに相当する放射能を持つバンドはFX Molecular Imager (Bio-Rad, Hercules, CA)で定量化した。SphKの比活性はmgタンパク質当たり生成するpmolのS1Pとして表した。
【0044】
〔ウエスタンブロット分析〕
細胞は細胞融解緩衝液(50 mM Tris pH 7.5, 150 mM NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, 1mM PMSF, 5μg/ml leupeptin, 5μg/ml aprotinin, 1 mM DTT)に再懸濁された。同じ量のタンパク質(60 pg)が10% SDS-PAGEで分離され、ニトロセルロースに転写された。ブロットが、5%の無脂肪ドライミルクと0.1% Tween 20を含むトリス緩衝液(TBS)中で一次抗体(1:1000) と一晩保温し、続いて抗ウサギHRP標識IgG(1:10,000, Jackson Immunoresearch Laboratories)で保温した。免疫複合体はKodakまたはPhenix Research ProductsのX腺フィルムでの高感度化学発光(Pierce)で可視化した。ウエスタン物はAlfa Innotech Corporation (San Leandro) からのAlphaEaseFC 4.0.0ソフトを用いて定量化された。
次のものが一次抗体として使われた:phospho-p44/42 MAP キナーゼ(Thr202/Tyr204)抗体、phospho-p38 MAPキナーゼ(Thr180/Tyr182)抗体(Cell Signaling, Beverly, MA, USA)、phospho-JNK (Thr183/Tyr185)抗体、Bcl-xS/L抗体(S-18, Santa Cruz, Santa Cruz, CA)、抗ヒトBcl-2 (Dako, Carpinteria, CA)、Mcl-1抗体、抗caspase-3と抗caspase-9 (Pharmingen)および抗PARP (Biomol)。
【0045】
〔タンパク質キナーゼの概観〕
SK1-lの種々のタンパク質キナーゼ活性への影響はSelectScreenTM Kinase Profiling (Invitrogen Drug Discovery Solution, Madison, WI)で評価された。簡単に述べると、5 pM SK1-lの存在下あるいは非存在下で、それぞれのタンパク質キナーゼ対してのKmappのATP濃度で、FRETを構成する二つの蛍光をもたらすものを含むペプチド基質を用いて、蛍光共鳴エネルギー転移(FRET)に基づくキナーゼ測定系により384穴プレート中で測定が行われた。発展液はリン酸化されていないペプチドを特異的に消化するタンパク質分解酵素を含み蛍光信号をつくりだす。Coumarin蛍光と蛍光のFRET信号はそれぞれ445 nmと520 nmで測定された。coumarin放射は開裂していない(リン酸化された)基質ペプチドのみでFRETによる蛍光を引き起こす。
ATPの非存在下でリン酸化されていないペプチドとキナーゼ含む反応と化学量論的にリン酸化されたペプチドを含む反応がそれぞれ0%および100%リン酸化の対照に供された。生の蛍光値は背景レベルに対して調整された。反応の終わりは蛍光FRET信号でcoumarin蛍光を割った放射比率として計算された。これらの比率はその後100%リン酸化対照で得られた比率で標準化された。
【0046】
〔アポトーシスのAnnexin V/PI測定〕
細胞はannexin V-フルオレッセインイソチオシアネートとプロピジウムイオダイド (PI)で染色し、それから製造者(BD PharMingen, San Diego,CA)の指示に従ってフローサイトメトリーによりアポトーシスを評価した。簡単に言うと、106個の細胞をリン酸緩衝食塩水(PBS)で洗い、10 mM HEPES, pH 7.4、140 mM NaOHおよび2.5 mM CaCl2を含んだ緩衝液中の5μl annexin V-フルオレッセインイソチオシアネートと5μlヨウ素化プロピジュウム(50μg/ml)で室温、暗所で15分間染色した。アポトーシス細胞はEXPO32 フローサイトメトリー分析プログラムのついたCoulter Epics-XL-MCL細胞蛍光光度計を使い決定した。下右1/4のパーセントは初期のアポトーシス細胞(annexin陽性)に一致し、上右1/4のパーセントは後期のアポトーシス細胞(annexin VとPI陽性)に一致する。
【0047】
〔TUNEL測定によるDNA鎖開裂検出〕
細胞(106)は氷上で1% (w/v)パラフォルムアルデヒドで15分間固定され、PBSで2回洗い、氷上で30分70% エタノール中で透過性をあげる処理をした。細胞を洗い、ターミナルデオキシヌクレオチドトランスフェラーゼとブロモデオキシウリヂン(BrdU)を含んだDNA標識液に再懸濁し、37℃で1時間製造者(Sigma)の指示に従って保温した。それから、細胞は抗BrdU蛍光抗体と暗所で室温30分間保温し、EXPO32 Flow Cytometry分析プログラム(Beckman Coulter)のついたCoulter Epics-XL-MCL細胞蛍光光度計を使い分析した。
【0048】
〔スフィンゴシンと代謝物の質量分析計による分析〕
細胞は冷やしたPBSでよく洗い、2000 xg 10分間の遠心分離で沈殿とした。細胞の一部をDNAとタンパク質の測定のためにとった。残りに内部標準を加え(それぞれ0.5 nmolのC12-SM, C12-Cer, C12-GlcCer, C12-LacCer, C17-sphingosine, C17-sphingosine 1-phosphate, C17-sphinganine-1-phosphateとC12-Cer-phosphate, Avanti Polar Lipids, Alabaster, AL)、脂質を抽出し、個々のセラミドアシル鎖種を液体クロマトグラフィーとelectrospray ionization-tandem質量分析計(ESI-MS/MS, 4000 QTRAP, Applied Biosystems)により前に述べたように定量化した(Sullards et al., Science STKE 2001: L1, 2001)。
【0049】
〔異所性移植の腫瘍モデル〕
動物を含む全ての実験はVCU IACUCにより認可されている。U937細胞(滅菌PBS 100μl に懸濁した2×106個)が6週齢のCB17 SCID/beigeマウス (Taconic Farms, Germantown, NY)両脇腹の2カ所に注入され、7日間、触診できる腫瘍に成長させた。腫瘍が50-100 mm3の量に達したときに、動物を任意の2グループに分け、引き続いて7日間200μlの食塩水またはSK1-I (20 mg/kg)を腹腔内に注入した。腫瘍測定はカリパスで毎日行い、腫瘍量は計算式 (πx[ミリで計った長さ]×[ミリで計った幅]2)/6 を使って計算した。実験の終わりに、動物を殺し、腫瘍を取り、ホルマリンで固定しパラフィン包埋するか液体窒素で凍結した。ホルマリン固定切片はヘマトキシリン−エオジンで染色されるかKi-67に対する抗体で染色された(Novocastra, Newcastle, UK)。抗体の結合は免疫組織化学およびパーオキシダーゼ標識種特異的2次抗体で検出され、3,3-ジアミノベンチジンで可視化された。パラフィン切片はロウを落とし、再水和し、浸透化の前にproteinase Kで処理された。凍結切片は蛍光TUNEL標識キットで染色し、DAPIで対抗染色した。スライドは蛍光顕微鏡で分析された。
【0050】
〔統計分析〕
実験は矛盾しない結果で少なくとも3回繰り返された。それぞれの実験に対して3つのサンプルからのデータが計算され、平均±S.D.として表された。実験条件の間の有意差は相手のない観察に対するStudent t試験を使って決めた。
【0051】
〔結果〕
(SK1-lはSphK2ではなくSphK1の強力な選択的な阻害剤である)
現在は、阻害剤の合理的な設計のためにcomputational docking methodsが使えるようにするための構造的な情報がSphKについてはない。従って、他のやり方は、もっと強力で選択的な阻害剤を設計するための阻害剤研究から得られる情報を用いることである。いろいろな化学合成された短鎖スフィンゴシンやジヒドロスフィンゴシン類似物がSphKの阻害剤として以前から研究されてきた(Edsall et al.,Biochemistry 37:12892-12898, 1998; De Jonghe et al., Bioorg Med Chem Lett 9:3175-3180, 1999; Johnson et al., J. Pharmacol Exp309:452-461, 2004: そしてNiiro et al., Bioorg Med Chem12:45-51, 2004)。アルキル鎖をフェニール環に置き換えるとか3-ヒドロキシグループをフッ素に置換すると強力なSphK阻害剤を生ずることは知られていた。
さらに、4,5-トランス型の二重結合を持つ同族体は一般により上位の阻害剤であった。(Johnson et al., J Pharmacol Exp, 2004, 上記)。
これらの以前の観察に基づき、我々は(2R,3S,4E)-N-methyl-5-(4'-pentylphenyl)-2-aminopent-4-ene-1,3-diol(図1A)を合成し、組み替えSphK1とSphK2へのその効果を調べた(図1B-E)。この水溶性のスフィンゴシン類似物は、投与量依存的に強力にsphK1活性を阻害し(図1C)、5μMで60-70% 阻害する。以前に報告されたように(Edsall et al., Biochemistry, 1998上記、およびKohama et al., J Biol Chem, 1998上記)、N,N-dimethylsphingosine (DMS)もまた強くはないがSphK1活性を阻害する。重要なことは、SphK2 (Liu et al., J Biol Chem, 2000, 上記)やセラミドキナーゼ (Sugiura et al., J Biol Chem, 277:23294-23300, 2002)を阻害するDMSとは対照的に、我々の合成物は組み替えSphK2(図1C, E)やセラミドキナーゼ(データは示していない)を阻害しないことである。このように、SphK1への特異的な阻害の理由で、この合成物をSK1-lと称する。
Lineweaver-Burk分析でSphKl活性は、スフィンゴシンに対するKmとほぼ同じKi値約10μMでSK1-lにより完全に阻害されることが明らかとなった(図1D)。SK1-lはSphK1あるいはSphK2のどちらによってもリン酸化されなかった(データは示していない)。DMSといくつかの他の一般的なSphK阻害剤はタンパク質キナーゼC(Igarashi et al., Biochemistry28:6796-6800, 1989)と潜在的に他のキナーゼ(De Luca et al., Biofactors25:43-60, 2005とGamble et al., Int J Cancer 118: 2412-2420, 2006)をもまた阻害するのでSK1-lの他のタンパク質キナーゼへの効果を調べることは重要であった。いくつかの組み替えタンパク質キナーゼ、蛍光標識したポリペプチド基質、それぞれのキナーゼに対してKmapp でのATPを含むタンパク質キナーゼ活性が選別された。SK1-lは、PKC類, PKCα, PKCδ, PKA, Akt1, ERKl, EGFR, CDK2, 1KKβあるいはCamKllβの二つの異なったメンバーを含む全てのタンパク質キナーゼを大きくは阻害しない(図1F)。従って、SK1-lは、イソ酵素選択性、水への溶解性、タンパク質キナーゼへの影響がないことの観点から、知られているSphK阻害剤のうちで特徴的である。
【0052】
(SK1-lはヒト白血病細胞の増殖を強く阻害する)
以前の研究は一般のSphK阻害剤DMSはU937やJurkat T細胞のアポトーシスを強く引き起こすことを示していた(Cuvillier et al., Nature, 1996, 上記;Jarvis et al., Mol Pharmacol 52:935-947; Edsall et al., 上記; Hamada et al., Biochem Biophys Res Commun 244:745-750, 1998およびCuvillier et al., Blood, 2001, 上記 )。図2Aに示したように、SK1-lは、72時間培養後に明らかになるように5μMの低濃度で10% 血清の存在下で培養したU937細胞の増殖を十分に低下させた。5μMで50%、10μMで完全に増殖が阻害されるように、T-リンパ芽球性Jurkat細胞はSK1-lに対してより敏感であった(図2B)。同様に、10μM SK1-lは、promyelocytic HL60、Molt-4 T細胞白血病およびK-562 CML細胞をそれぞれ50%、70%、90%阻害することを含めて、処理後48時間以内に他の白血病細胞の増殖を減少させた。
SK1-l処理の影響に対すると同様にまた他の白血病細胞での研究と一致して(Bonhoure et al., 上記とBaran et al., 上記)、SphK1タンパク質とmRNAレベルを60%以上(図2C) 減ずる特定の配列を標的としたSphK1発現のダウンレギュレーションが、2%あるいは10%血清の存在下で培養したU937細胞の増殖速度を著しく減少させた(図2C)。総合すると、これらの発見は、薬学的あるいは遺伝的方法によるSphK1の特異的阻害が骨髄性およびリンパ性白血病細胞の増殖を阻害するという見解と一致する。
【0053】
(SK1-lはヒト白血病細胞のアポトーシスを引き起こす)
DMSによるSphKの阻害あるいはSphK1発現のダウンレギュレーションはヒト白血病細胞を含め多くの細胞型でアポトーシスの誘導と関連づけられてきた(Cuvillier et al., Nature, 1996, 上記;Jarvis et al., 1998, 上記、Jendiroba et al., 2002, 上記そしてBonhoure et al., 上記)。このように、われわれは次に膜の透過性の基準としてannexin V染色とPlによる細胞膜外側にホスファチジルセリンを発現している細胞をモニターする流動細胞計を用いてU937細胞のアポトーシスに対するSK1-lの影響を調べた。SK1-IにさらしたU937細胞のアポトーシスには時間および濃度への依存が増加した(図2D)。図2Eに示されるように、大部分の細胞が初期のアポトーシス状態にあり、ほんのわずかのパーセントのものが壊死的(Plにのみ陽性)であった。これらの結果はTUNEL測定で決められたDNA鎖開裂の存在と密接に関連していた(図2F)。さらに、細胞が低濃度の血清下で培養されるときにより効果的に細胞増殖を阻害するSphKlのダウンレギュレーションと同様に(図2C)、U937細胞は、血清濃度が減少したときSK1-lで引き起こされるアポトーシスに対して、より敏感になった(図2G)。
【0054】
(SK1-lで誘導される細胞死におけるカスパーゼ活性化とBcl-2開裂の機能的役割)
siRNAでのHL60細胞におけるSphK1のダウンレギュレーション(Bonhoure et al., 上記) あるいはJurkat細胞におけるDMSでのSphKの阻害(Cuvillier et al., J Biol Chem 275:15691-15700, 2000)はアポトーシスの証明であるPARPの開裂と同様にexecutionerカスパーゼ3の活性化を起こさせるのに十分である。同様に、アポトーシスの惹起に付随して、SK1-lでのU937細胞の処理はカスパーゼ-3と-9の活性化を増加させ、ポリADPリボースポリメラーゼ(PARP)の開裂を引き起こす(図3A,B)。さらに、16-24時間SK1-lにさらすとミトコンドリアの機能障害を起こさないようにする抗アポトーシスタンパク質であるBcl-2の開裂(図3B)が生じる。他方、悪性造血細胞の生存に鍵となる役割をする抗アポトーシスタンパク質(Moulding et al., Blood 96:1756-1763, 2000)はそれほど多くは変えられなかった(図3A,B)。次に我々はSK1-lで引き起こされる致死でのカスパーゼ活性化とBcl-2開裂の機能的役割を研究した。汎カスパーゼ阻害剤ZVADとBOCでのU937細胞の前処理は、DNA損傷剤エトポシドにより引き起こされるアポトーシスと同様にSK1-lでもたらされるアポトーシスを十分に弱めた(図3C)。さらに、Bcl-2の異所性の発現はSK1-lの引き起こす致死から守り、Bcl-xLの発現は細胞死を60%減弱した(図3D)。総合すると、これらの発見はBcl-2によるのとは反対にSK1-lの致死性は固有のミトコンドリア過程を通して基本的に達成されていることを示している。
SphK1はアポトーシスに向かわせるセラミドと抗アポトーシス的S1Pの間のバランスの決定的な調節剤であるから(Olivere et al., 上記とBonhoure et al., 上記)、これらのスフィンゴシン代謝物のレベルへのSK1-lの影響は高性能液体クロマトグラフィーESI-MS/MSで決められた(Sullards et al., 上記)。SK1-l処理は、スフィンゴシンあるいはジヒドロスフィンゴシン(sphinganine)のレベルを変えることなく、全細胞セラミドの増加とスフィンゴミエリンの減少を伴って全細胞S1Pの50%の減少を引き起こした(図4A)。U937細胞にもっとも多量にあるセラミド種は24:1であった(図4A)。SK1-l処理はC16:0とC24:1セラミド種のレベルをそれぞれ3倍、2倍増加させるが、他のセラミド種には大きな影響をもたらさない(図4A)。
SK1-lのアポトーシス効果がそのSphK1阻害能力によることを確かめるために、S1P添加背景実験を行った。SK1-lによるS1Pレベルの減少に伴い、SK1-lによるアポトーシスは外からのS1Pの添加により添加依存性で減少した(図4B)。総体的に、この発見はSK1-lがSphK1を阻害することによるヒト白血病細胞のアポトーシスとセラミド増加を伴うS1Pの産生を引き起こすことを示している。
【0055】
(SKl-iにより引き起こされるアポトーシスはERKl/2とAkt生存信号の不活性化と関係する)
分裂促進因子で活性化されたタンパクキナーゼ(ERK1/2、JNKおよびp38 MAPK)とAktは白血病細胞の運命に重要な役割を果たすことは豊富な証拠で示されている(Steelman et al., Leukemia 18:189-218, 2004)。U937細胞をSK1-lで処理するとERK1/2とAktのリン酸化の急で大きな減少を引き起こす(図5A-C)。生存信号のこのような不活性化は低濃度血清下で2時間まで続くが(図5A)、一方10%血清の存在下ではp-ERK1/2 とp-Aktレベルの減少は徐々に克服され(図5B)、SK1-lの濃度に依存した(図5C)。さらに、p38リン酸化の一時的な増加が5分間観察され、後の強くない活性化が続いて起きた(図5A,B)。加うるに、JNKの活性化とc-Junリン酸化もまた後の時間に検出された(図A,B)。SK1-lはAktを強く不活性化するので、SK1-lの致死効果でのその役割を決めることは興味深い。構成的に活性のあるミリストイル化されたAktのU937細胞内での過剰発現は15μM以下の濃度のSK1-lで引き起こされるアポトーシスを大きく減少させ(図5D)、Aktの不活性化がSK1-lのアポトーシス効果に役立つ要素の一つであるかもしれないことを示唆している。
【0056】
(原発性ヒトAML芽球はSK1-lで引き起こされるアポトーシスに非常に影響されやすい)
原発AML試料へのSK1-lの影響を調べるために、二人のAML患者(FABサブタイプM2)からの骨髄から得た白血病芽球で平行した実験を行った。SK1-lの濃度を増していった芽球の処理はU937とJurkat細胞と比較して、アポトーシスの誘導に対する感受性が増加していることを明らかにした。両患者のサンプルはSK1-lに24時間さらしたときアポトーシスの顕著な増加を示し、annexin V/PI分析で明らかになったように7.5μMで40-50%のアポトーシスが観察された(図6A)。前の結果と一致して(Rosato et al., Mol Cancer Ther 6:692-702, 2007)、処理しない場合には正常末梢血単核球細胞のアポトーシスと非常に似たアポトーシスを10%以下の芽球が示した。注目すべきことに、SK1-lは正常の末梢血単核細胞の生存に重要な影響をもたらさなかった(図6B)。これらの結果はSphK1を過剰発現していると報告された原発性ヒトAML細胞 (Sobue et al., Leukemia20:2042-2046, 2006) は長期培養された白血病細胞よりSK1-lに対し感受性があるが、SK1-lは正常の末梢血単核白血球には比較的害がないことを示唆している。
【0057】
(生体内でのSK1-lの抗白血病活性)
我々は次に、いくつかの新しい処理法の発展を助けるのに非常によく使われてきた免疫不全マウスへの異種移植で、SK1-lの白血病細胞の腫瘍増殖を阻害する能力を評価した (McCormack et al., Leukemia 19:687-706,2005)。SCID/beigeマウスの脇腹に皮下注射したU937細胞は急激に増殖する腫瘍を急速に生じた。腫瘍が50-100 mm3の量に達したときマウスに食塩水あるいはSK1-l (20 mg/kg)を毎日腹腔内注射した。図7Aに見られるように、SK1-lは腫瘍増殖を著しく減少させた。7日後、SK1-lで処理したマウスのU937腫瘍の平均の大きさは食塩水処理マウスの腫瘍より50%以上小さかった(対照群の平均=747 mm3、SK1-l群平均=332 mm3, p<0.001)。SK1-l処理マウスの腫瘍の死後の重量もまた著しく低かった(図7B)。SK1-l処理マウスは消耗した様子は示さず、7日後の体重は対照標準と著しくは違わなかった。
期待されたように、食塩水処理マウスからの腫瘍はKi67で強く染色され、TUNELで染色されるわずかのアポトーシス細胞とともに高い増殖性細胞の腫瘍組成を示していた(図7C)。対照的に、SK1-l処理マウスからの腫瘍細胞の免疫組織化学分析で核の断片化(TUNEL染色)と凝縮した核で決められる多くのアポトーシス様細胞が示された(図7C)。SK1-lはまた腫瘍中の分裂している細胞を劇的に減らした(図7C)。これらの結果はSK1-lが生体内で強い抗白血病活性を持つことを示している。
【0058】
〔討議〕
がんの多くの型で、S1Pの産生が制御低下を起こし、異常な細胞増殖や生存信号にいたることを多くの証拠が示している (Milstien et al., 上記とRA Sabbadini, Br J Cancer 95:1131-1135, 2006)。SphK1は種々の固形癌(French et al., Cancer Res63:5962-5969, 2003)またAML(Sobue et al., 上記)で過剰発現している。さらに、慢性骨髄性白血病の共通の遺伝的異常と急性白血病の患者の少なくとも20%で予後不良の尺度であるbcr/ablがSphK1発現の上昇制御していることが示されている(Li et al., Oncogene 26:7904-7908,2007)。したがって、SphK1はいまでは、そのレベルが薬剤耐性や放射線耐性と関連している白血病細胞では特に、薬学的な干渉の潜在的な標的である考えられている(Bonhoure et al., 上記とBaran et al., 上記)。白血病細胞でのSphK1の役割の以前の研究は、特異的siRNAによるそのダウンレギュレーションあるいはSphK1およびSphK2、潜在的にタンパク質キナーゼを阻害する薬剤の使用に焦点が当てられてきた。本研究はもっとも強力で水溶性のSphK1イソ酵素特異的阻害剤、SK1-Iの開発について述べる。さらに、SK1-lはPKCあるいは大多数の他のタンパク質キナーゼを阻害しない。よく保存されているATP結合ポケットでATPと競合しまた潜在的に交差反応するほとんどの低分子タンパク質キナーゼ阻害剤とは対照的に、SK1-Iは液体基質と競合的である。
【0059】
フォスファチジルセリンの外部化、DNA鎖切断の増加、カスパーゼ3と9の活性化、およびPARPとBcl-2の開裂を反映して、SK1-lはいくつかの白血病細胞系とAML白血病芽球でアポトーシスを強く引き起こす。どのような機構でSK1-lがこれらの致死的な効果を非常に強く引き起こすのかについて考察すると、いくつかのお互いに排除しあわない関連した作用によるものであろうと予測される。SK1-lは、その細胞内標的が解明されてはいないが、細胞内で増殖と生存を増強するように働く生存に有利なS1Pの産生を阻害する(Kohno, et al., Mol Cell Biol26:7211-7223,2006)。細胞内で産生されたS1Pが細胞から放出されることができ(Mitra et al., Proc Natl Acad Sci USA 103:16394-16399,2006)、またERK1/2とAktを含む生存経路につながる細胞表面受容体を通して作用する(Spiegel et al., 上記)ことはまた周知されている。この点について、白血病細胞中の活性化されたERK1/2とAktを減少させるSK1-lの能力は、Raf/MEK/ERKおよびPl3K/AktがしばしばAML中で構成的に活性化されているものと関連づけられうる(Steelman et al., 上記およびNyakern et al., Leukemia 20:230-238, 2006)。ERK1/2はSphK1をリン酸化し活性化し(Pitson et al., J Exp Med 201:49-54, 2005)、そのあとERK1/2を刺激することができるS1Pの増加に導くので、SK1-Iは、SphK1を阻害すること、増殖と生存のS1Pを減少させ一方で同時に前駆体である前アポトーシス状のセラミドを増加させることにより、この正のフィードバック環を断ち切ることができる。それゆえ、SK1-Iは白血病での多様な分子治療標的を構成する。
【0060】
セラミド産生はヒト白血病細胞にアポトーシス引き起こすと長い間関連づけて考えられてきた(Jarvis et al., Proc Natl Acad Sci USA 91:73-77,1994)、そして最近、いくつかの異なったシグナル伝達阻害剤のアポトーシスへの相乗効果がセラミド産生の増加と関連づけられてきた。例えば、ヒト白血病細胞にペリフォシンとヒストンデアセチラーゼ同時に投与すると、AktとERKの崩壊、セラミドと反応性の酸素種の産生の著しい増加、ミトコンドリア傷害とアポトーシスの驚くべき増加を引き起こす(Rahmani et al., 上記とRosato et al., Mol Pharmacol 69:216-225, 2006)。セラミドはそのアポトーシス作用を多くの経路で変換できる(Ogretmen et al., 上記)。重要な同定されたセラミド標的は、Bcl-2の別のスプライシングの調節役であるSRタンパク質と同様にAktを脱リン酸化するセリン/スレオニンタンパク質ホスファターゼであるPP1とPP2Aを含んでいる(Ogretmen et al., 上記)。我々はSK1-lにさらすと、ミトコンドリア依存性のアポトーシス関連づけられてきた反応であるBcl-2の開裂が起きることを見いだした(Cheng et al., Science 278:1966-1968,1977)。さらに、Bcl-2の過剰発現は、セラミド生成を妨げ、急性リンパ芽球性白血病やAMLを含む多くの細胞でのセラミドにより誘起されるアポトーシスから守ることは立証されている (Zhang et al., Proc Nat Acad Sci USA 93:5325-5328, 1996、Amarante-Mendes et al., Blood 91:1700-1705, 1998とOgretmen et al., 上記)。これと一致して、我々はBcl-2の過剰発現が、内在するミトコンドリアの死経路の重要性を強調するSK1-l誘発の致死性をもまた妨げることを発見した。この見解と一致して最近、S1Pが、MEK/ERK1/2依存的な方法でミトコンドリアへのBaxの移転をブロックすることでJurkat細胞のアポトーシス間のミトコンドリア内の事象に細胞防御的な効果を与えることが示されている (Betio et al., Biochem Biophys Res Commun340:1273-1277.2006)。最近の研究では、小胞体でのセラミドの持続的な増加は同時にER stress responseを活性化し、抗アポトーシス性のAktを不活性化しアポトーシスに向かわせることが示唆されている(Swanton et al., Cancer Cell 11:498-512, 2007)。したがって、SK1-lで引き起こされるAktの不活性化はS1P生成の減少によるばかりでなくセラミドの増加によってももたらされることが可能である。SK1-lと構造的に似ており、SphKsの拮抗阻害剤であるよりは拮抗基質である免疫抑制剤ETY720もまたAktリン酸化を減少させることが示されている (Ng et al., Int J Oncol 30:375-380, 2007)。多発性硬化症の臨床試験で比較的毒性が少ないFTY720 (Brinkman et al., Pharmacol Ther 115:84-105, 2007)は芽球急変期の慢性骨髄性白血病とフィラデルフィア染色体陽性の急性リンパ性白血病を治療する別の方法であることが最近提唱されている (Neviani et al., J Clin Invest 117:2408-2421, 2007)。
【0061】
SK1-lがAML患者から分離した白血病芽球のアポトーシスを強く引き起こすが正常末梢血単核白血球には比較的細胞毒性がないことは、白血病細胞への選択性を強調する。さらに、異種移植AML中で、試験管内での作用と同様に、SK1-lが腫瘍増殖を抑え、腫瘍のアポトーシスを誘導し、増殖を減少させる明瞭な信号発信剤の活性を持っていた。肝臓、腎臓、膵臓への毒性の予備的分析では、目立った効果は全くなかった。このように、特異的SphK1阻害剤は、単独であるいは通常のまたは他の知られた標的剤の付加物として使用される、白血病の強力な薬学的な介在物として考慮に値する。
【0062】
上記の詳細な記載と本発明の例にてらして、多くの明らかな差違が疑いもなくその分野の当業者のそれらに対して示唆される。同じ目的を達成するために計算されたいかなる取り決めも、示された特殊な具体化物で置き換えられ得ることが当業者により評価されるであろう。この応用と発明は、本発明のいかなる応用物あるいは変化物をも包含するように意図されている。以下に続く請求範囲により特別に決められているように、全てのこのような変化物は発明の範囲と意図に完全に含まれている。
【特許請求の範囲】
【請求項1】
下記の化1の構造を有することを特徴とする合成物。
【化1】
ここにXはCまたはN、R1、R2、R3、およびR4は、それぞれ独立して水素、C1〜C18の直鎖または分岐アルキル基、C1〜C16の置換直鎖または分岐アルキル基、シクロアルキル基、置換シクロアルキル基、複素環基、置換複素環基、アリルアルキル基、置換アリルアルキル基、複素環式アリルアルキル基、置換複素環式アリルアルキル基、アルコキシ基、置換アルコキシ基、アルケン基、置換アルケン基、アルキン基、アシル基、スルファイド、またはエーテル基からなり、上記R1とR2、R3、R4とは相互に独立して一体化することによって1個またはそれ以上の環を形成するか、または前記のいずれかの2個またはそれ以上の結合体を形成する。
【請求項2】
下記の化2の構造からなる請求項1に記載の合成物。
【化2】
【請求項3】
請求項2に記載の合成物であって、R3およびR4は下記の化3の構造を有する。
【化3】
ここにR5、R6は、それぞれ独立して水素、C1〜C18の直鎖または分岐アルキル基、C1〜C16の置換直鎖または分岐アルキル基、シクロアルキル基、置換シクロアルキル基、複素環基、置換複素環基、アリルアルキル基、置換アリルアルキル基、複素環式アリルアルキル基、置換複素環式アリルアルキル基、アルコキシ基、置換アルコキシ基、アルケン基、置換アルケン基、アルキン基、アシル基、スルファイド、エーテル基、または上記のいずれかの2個またはそれ以上の結合体からなる。
【請求項4】
請求項3に記載の合成物であって、R5、R6とは相互に結合して環を形成し、上記合成物は下記の化4の構造からなる。
【化4】
ここにR7は、水素、C1〜C18の直鎖または分岐アルキル基、C1〜C16の置換直鎖または分岐アルキル基、シクロアルキル基、置換シクロアルキル基、複素環基、置換複素環基、アリルアルキル基、置換アリルアルキル基、複素環式アリルアルキル基、置換複素環式アリルアルキル基、アルコキシ基、置換アルコキシ基、アルケン基、置換アルケン基、アルキン基、アシル基、スルファイド、エーテル基、または上記のいずれかの2個またはそれ以上の結合体からなる。
【請求項5】
請求項3または4に記載の合成物であって、R1は水素、そしてR2はCH3である。
【請求項6】
請求項2に記載の合成物であって、R3またはR4はスルファイド、SR5であり、R5は、水素、C1〜C18の直鎖または分岐アルキル基、C1〜C16の置換直鎖または分岐アルキル基、シクロアルキル基、置換シクロアルキル基、複素環基、置換複素環基、アリルアルキル基、置換アリルアルキル基、複素環式アリルアルキル基、置換複素環式アリルアルキル基、アルコキシ基、置換アルコキシ基、アルケン基、置換アルケン基、アルキン基、アシル基、スルファイドである。
【請求項7】
請求項6に記載の合成物であって、SR5は下記の化5の構造を有する。
【化5】
【請求項8】
請求項7に記載の合成物であって、XはC、R1とR3はH、R4はSR5であり、上記合成物は下記の化6の構造を有する。
【化6】
【請求項9】
請求項8に記載の合成物であって、R2はCH3である。
【請求項10】
請求項7に記載の合成物であって、XはC、R1およびR4はH、そしてR3はSR5であり、該合成物は下記の化7の構造を有する。
【化7】
【請求項11】
請求項10に記載の合成物であって、R2はCH3である。
【請求項12】
請求項2に記載の合成物であって、R3はHである。
【請求項13】
請求項12に記載の合成物であって、XはC、R1はH、R2はCH3そしてR4は(CH2)4CH3であり、該合成物は下記の化8の構造を有する。
【化8】
【請求項14】
請求項13に記載の合成物であって、R4の末端炭素は1個または2個以上のハロゲン化物によって置換されている。
【請求項15】
請求項14に記載の合成物であって、該ハロゲン化物は臭化物、塩化物、フッ化物である。
【請求項16】
請求項14に記載の合成物であって、該合成物は下記の化9の構造を有する。
【化9】
【請求項17】
請求項2に記載の合成物であって、R4はHである。
【請求項18】
請求項12又は17に記載の合成物であって、R2はメチル基である。
【請求項19】
請求項18に記載の合成物であって、R1は水素である。
【請求項20】
請求項18に記載の合成物であって、R1はメチル基である。
【請求項21】
請求項12または17に記載の合成物であって、R1とR2とは相互に一体化することによって置換または非置換の環を形成する。
【請求項22】
請求項21に記載の合成物は下記の化10の構造を有する。
【化10】
【請求項23】
請求項22に記載の合成物であって、XはCである。
【請求項24】
請求項21に記載の合成物であって、合成物は下記の化11の構造を有する。
【化11】
【請求項25】
請求項24に記載の合成物であって、XはCである。
【請求項26】
請求項12に記載の合成物であって、R4はエーテルであり、該合成物は下記の化12の構造を有する。
【化12】
ここにR5はC1〜C18の直鎖または分岐アルキル基、C1〜C16の置換直鎖または分岐アルキル基、シクロアルキル基、置換シクロアルキル基、複素環基、置換複素環基、アリルアルキル基、置換アリルアルキル基、複素環式アリルアルキル基、置換複素環式アリルアルキル基、アルコキシ基、置換アルコキシ基、アルケン基、置換アルケン基、アルキン基、アシル基、スルファイド、エーテル基、または上記のいずれかの2個またはそれ以上の結合体からなる。
【請求項27】
請求項26に記載の合成物であって、XはC、R1はH、そしてR2はCH3である。
【請求項28】
がん細胞を請求項1に記載の合成物の該がん細胞を殺すかまたは損傷を与えるのに充分な量に暴露するステップからなることを特徴とするがん細胞を殺すかまたは損傷を与える方法。
【請求項29】
請求項28の方法において、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項30】
がん細胞に対して該がん細胞にアポトーシスを引き起こすための充分な量の請求項1に記載の合成物を暴露するステップからなることを特徴とするがん細胞にアポトーシスを引き起こす方法。
【請求項31】
請求項30に記載の方法であって、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項32】
がん細胞を該がん細胞の増殖、転移、および該がん細胞における化学療法抵抗性の増大を抑制するために充分な量の請求項1に記載の合成物に暴露するステップからなることを特徴とするがん細胞の増殖、転移、および化学療法抵抗性の増大を抑制するための方法。
【請求項33】
請求項32に記載の方法であって、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項34】
白血病患者の症状を治療または軽減するために充分な量の請求項1に記載の合成物を該患者に投与するステップからなることを特徴とする対象患者の白血病の症状を治療または軽減するための方法。
【請求項35】
がん患者中のがん細胞を殺すための抗がん剤の効力を増強するために充分な量の請求項1に記載の合成物を該がん患者に投与するステップからなることを特徴とする対象患者のがん細胞を殺すための抗がん剤の効力を増強する方法。
【請求項36】
請求項35に記載の方法であって、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項37】
対象とする患者の免疫反応性を弱めるに充分な量の請求項1に記載の合成物を該患者に投与するステップからなることを特徴とする対象患者における免疫反応性を弱める方法。
【請求項38】
上記免疫反応性を弱めることは肥満細胞機能を弱めることによって行われる請求項37に記載の方法。
【請求項39】
請求項37に記載の方法において、免疫反応性を弱めることは、すなわち対象患者のぜんそく症状を弱めることを指向する。
【請求項40】
請求項37に記載の方法であって、免疫反応性を弱めることは、すなわちアナフィラキシーの症状を弱めることを指向する。
【請求項41】
請求項37に記載の方法であって、免疫反応性を弱めることは、すなわち自己免疫病の症状を軽減することを指向する。
【請求項42】
請求項1に記載の合成物の生存信号を阻害するために充分な量を上記がんにかかった細胞に投与するステップからなることを特徴とするがんにかかった細胞における生存信号を阻害するための方法。
【請求項43】
請求項42に記載の方法において、上記生存信号の阻害はAktまたはERKあるいはその両方のリン酸化を弱めることによって行われる。
【請求項44】
請求項1に記載の合成物の薬剤における多発性硬化症の症状を軽減するために充分な量を該患者に投与するステップからなることを特徴とする該患者における多発性硬化症の症状を軽減する方法。
【請求項45】
スフィンゴシンキナーゼ活性を測定する試験管内の検定においてスフィンゴシンキナーゼ2(Sphk2)を阻害するよりも5倍大きくスフィンゴシンキナーゼ1(Sphk1)を阻害することを特徴とする合成物。
【請求項46】
請求項45に記載の合成物であって、上記Sphk1はSphk2の阻害よりも少なくとも10倍大きく阻害される。
【請求項47】
請求項45に記載の合成物であって、上記試験管内の検定における阻害は10μM濃度で測定される。
【請求項48】
請求項45に記載の合成物であって、上記試験管内の検定における阻害はSphk1の50%阻害を与える濃度で測定される。
【請求項49】
がん細胞を殺すかまたは損傷するに充分な量の請求項45に記載の合成物に暴露するステップからなるがん細胞を殺すかまたは損傷を与えることを特徴とする方法。
【請求項50】
請求項45に記載の方法において、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項51】
がん細胞を、該がん細胞にアポトーシスを引き起こすために充分な量の請求項45に記載の合成物に暴露するステップからなることを特徴とするがん細胞にアポトーシスを引き起こす方法。
【請求項52】
請求項51に記載の方法において、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項53】
がん細胞を、該がん細胞の成長、転移、および化学薬剤耐性の進化を阻害するに充分な量の請求項45に記載の合成物に暴露するステップからなることを特徴とするがん細胞の成長、転移、および化学薬剤耐性の進化を阻害する方法。
【請求項54】
請求項53に記載の方法であって、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項55】
患者の白血病の症状を治療または軽減するために充分な量の請求項45に記載の合成物を該患者に投与するステップからなることを特徴とするそれを必要とする患者に対して白血病の症状を治療または軽減することを特徴とする方法。
【請求項56】
患者に対して抗がん剤と、該抗がん剤が該患者のがん細胞を殺す能力を増強するために充分な量の請求項45に記載の合成物とを投与するステップからなることを特徴とするそれらを必要とする患者のがん細胞を殺すため抗がん剤の能力を増強することを特徴とする方法。
【請求項57】
請求項56に記載の方法であって、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項58】
それを必要とする患者における免疫反応性を弱めるために充分な量の請求項45に記載の合成物を該患者に投与することを特徴とする該患者の免疫反応性を弱めるための方法。
【請求項59】
上記免疫反応性を弱めることは肥満細胞機能を弱めることによって行われる請求項58に記載の方法。
【請求項60】
請求項58に記載の方法において、免疫反応性を弱めることは、それを必要とする患者のぜんそくの症状を弱めることを指向する。
【請求項61】
請求項58に記載の方法において、免疫反応性を弱めることは、すなわちアナフィラキシーの症状を弱めることを指向する。
【請求項62】
請求項58に記載の方法において、免疫反応性を弱めることは、すなわち自己免疫病の症状を軽減することを指向する。
【請求項63】
がんにかかった細胞における生存信号を阻害するための方法であって、該方法は請求項45記載の合成物の生存信号を阻害するために充分な量を上記がんにかかった細胞に投与するステップからなる。
【請求項64】
請求項63に記載の方法において、上記生存信号の阻害はAktまたはERKあるいはその両方のリン酸化を弱めることによって行われる。
【請求項65】
請求項45の合成物を必要とする患者の多発性硬化症の症状を軽減するために充分な量を該患者に投与するステップからなることを特徴とする患者の多発性硬化症の症状を軽減することを特徴とする方法。
【請求項66】
オリゴマーまたはポリマーである請求項1に記載の合成物からなることを特徴とする合成物。
【請求項1】
下記の化1の構造を有することを特徴とする合成物。
【化1】
ここにXはCまたはN、R1、R2、R3、およびR4は、それぞれ独立して水素、C1〜C18の直鎖または分岐アルキル基、C1〜C16の置換直鎖または分岐アルキル基、シクロアルキル基、置換シクロアルキル基、複素環基、置換複素環基、アリルアルキル基、置換アリルアルキル基、複素環式アリルアルキル基、置換複素環式アリルアルキル基、アルコキシ基、置換アルコキシ基、アルケン基、置換アルケン基、アルキン基、アシル基、スルファイド、またはエーテル基からなり、上記R1とR2、R3、R4とは相互に独立して一体化することによって1個またはそれ以上の環を形成するか、または前記のいずれかの2個またはそれ以上の結合体を形成する。
【請求項2】
下記の化2の構造からなる請求項1に記載の合成物。
【化2】
【請求項3】
請求項2に記載の合成物であって、R3およびR4は下記の化3の構造を有する。
【化3】
ここにR5、R6は、それぞれ独立して水素、C1〜C18の直鎖または分岐アルキル基、C1〜C16の置換直鎖または分岐アルキル基、シクロアルキル基、置換シクロアルキル基、複素環基、置換複素環基、アリルアルキル基、置換アリルアルキル基、複素環式アリルアルキル基、置換複素環式アリルアルキル基、アルコキシ基、置換アルコキシ基、アルケン基、置換アルケン基、アルキン基、アシル基、スルファイド、エーテル基、または上記のいずれかの2個またはそれ以上の結合体からなる。
【請求項4】
請求項3に記載の合成物であって、R5、R6とは相互に結合して環を形成し、上記合成物は下記の化4の構造からなる。
【化4】
ここにR7は、水素、C1〜C18の直鎖または分岐アルキル基、C1〜C16の置換直鎖または分岐アルキル基、シクロアルキル基、置換シクロアルキル基、複素環基、置換複素環基、アリルアルキル基、置換アリルアルキル基、複素環式アリルアルキル基、置換複素環式アリルアルキル基、アルコキシ基、置換アルコキシ基、アルケン基、置換アルケン基、アルキン基、アシル基、スルファイド、エーテル基、または上記のいずれかの2個またはそれ以上の結合体からなる。
【請求項5】
請求項3または4に記載の合成物であって、R1は水素、そしてR2はCH3である。
【請求項6】
請求項2に記載の合成物であって、R3またはR4はスルファイド、SR5であり、R5は、水素、C1〜C18の直鎖または分岐アルキル基、C1〜C16の置換直鎖または分岐アルキル基、シクロアルキル基、置換シクロアルキル基、複素環基、置換複素環基、アリルアルキル基、置換アリルアルキル基、複素環式アリルアルキル基、置換複素環式アリルアルキル基、アルコキシ基、置換アルコキシ基、アルケン基、置換アルケン基、アルキン基、アシル基、スルファイドである。
【請求項7】
請求項6に記載の合成物であって、SR5は下記の化5の構造を有する。
【化5】
【請求項8】
請求項7に記載の合成物であって、XはC、R1とR3はH、R4はSR5であり、上記合成物は下記の化6の構造を有する。
【化6】
【請求項9】
請求項8に記載の合成物であって、R2はCH3である。
【請求項10】
請求項7に記載の合成物であって、XはC、R1およびR4はH、そしてR3はSR5であり、該合成物は下記の化7の構造を有する。
【化7】
【請求項11】
請求項10に記載の合成物であって、R2はCH3である。
【請求項12】
請求項2に記載の合成物であって、R3はHである。
【請求項13】
請求項12に記載の合成物であって、XはC、R1はH、R2はCH3そしてR4は(CH2)4CH3であり、該合成物は下記の化8の構造を有する。
【化8】
【請求項14】
請求項13に記載の合成物であって、R4の末端炭素は1個または2個以上のハロゲン化物によって置換されている。
【請求項15】
請求項14に記載の合成物であって、該ハロゲン化物は臭化物、塩化物、フッ化物である。
【請求項16】
請求項14に記載の合成物であって、該合成物は下記の化9の構造を有する。
【化9】
【請求項17】
請求項2に記載の合成物であって、R4はHである。
【請求項18】
請求項12又は17に記載の合成物であって、R2はメチル基である。
【請求項19】
請求項18に記載の合成物であって、R1は水素である。
【請求項20】
請求項18に記載の合成物であって、R1はメチル基である。
【請求項21】
請求項12または17に記載の合成物であって、R1とR2とは相互に一体化することによって置換または非置換の環を形成する。
【請求項22】
請求項21に記載の合成物は下記の化10の構造を有する。
【化10】
【請求項23】
請求項22に記載の合成物であって、XはCである。
【請求項24】
請求項21に記載の合成物であって、合成物は下記の化11の構造を有する。
【化11】
【請求項25】
請求項24に記載の合成物であって、XはCである。
【請求項26】
請求項12に記載の合成物であって、R4はエーテルであり、該合成物は下記の化12の構造を有する。
【化12】
ここにR5はC1〜C18の直鎖または分岐アルキル基、C1〜C16の置換直鎖または分岐アルキル基、シクロアルキル基、置換シクロアルキル基、複素環基、置換複素環基、アリルアルキル基、置換アリルアルキル基、複素環式アリルアルキル基、置換複素環式アリルアルキル基、アルコキシ基、置換アルコキシ基、アルケン基、置換アルケン基、アルキン基、アシル基、スルファイド、エーテル基、または上記のいずれかの2個またはそれ以上の結合体からなる。
【請求項27】
請求項26に記載の合成物であって、XはC、R1はH、そしてR2はCH3である。
【請求項28】
がん細胞を請求項1に記載の合成物の該がん細胞を殺すかまたは損傷を与えるのに充分な量に暴露するステップからなることを特徴とするがん細胞を殺すかまたは損傷を与える方法。
【請求項29】
請求項28の方法において、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項30】
がん細胞に対して該がん細胞にアポトーシスを引き起こすための充分な量の請求項1に記載の合成物を暴露するステップからなることを特徴とするがん細胞にアポトーシスを引き起こす方法。
【請求項31】
請求項30に記載の方法であって、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項32】
がん細胞を該がん細胞の増殖、転移、および該がん細胞における化学療法抵抗性の増大を抑制するために充分な量の請求項1に記載の合成物に暴露するステップからなることを特徴とするがん細胞の増殖、転移、および化学療法抵抗性の増大を抑制するための方法。
【請求項33】
請求項32に記載の方法であって、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項34】
白血病患者の症状を治療または軽減するために充分な量の請求項1に記載の合成物を該患者に投与するステップからなることを特徴とする対象患者の白血病の症状を治療または軽減するための方法。
【請求項35】
がん患者中のがん細胞を殺すための抗がん剤の効力を増強するために充分な量の請求項1に記載の合成物を該がん患者に投与するステップからなることを特徴とする対象患者のがん細胞を殺すための抗がん剤の効力を増強する方法。
【請求項36】
請求項35に記載の方法であって、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項37】
対象とする患者の免疫反応性を弱めるに充分な量の請求項1に記載の合成物を該患者に投与するステップからなることを特徴とする対象患者における免疫反応性を弱める方法。
【請求項38】
上記免疫反応性を弱めることは肥満細胞機能を弱めることによって行われる請求項37に記載の方法。
【請求項39】
請求項37に記載の方法において、免疫反応性を弱めることは、すなわち対象患者のぜんそく症状を弱めることを指向する。
【請求項40】
請求項37に記載の方法であって、免疫反応性を弱めることは、すなわちアナフィラキシーの症状を弱めることを指向する。
【請求項41】
請求項37に記載の方法であって、免疫反応性を弱めることは、すなわち自己免疫病の症状を軽減することを指向する。
【請求項42】
請求項1に記載の合成物の生存信号を阻害するために充分な量を上記がんにかかった細胞に投与するステップからなることを特徴とするがんにかかった細胞における生存信号を阻害するための方法。
【請求項43】
請求項42に記載の方法において、上記生存信号の阻害はAktまたはERKあるいはその両方のリン酸化を弱めることによって行われる。
【請求項44】
請求項1に記載の合成物の薬剤における多発性硬化症の症状を軽減するために充分な量を該患者に投与するステップからなることを特徴とする該患者における多発性硬化症の症状を軽減する方法。
【請求項45】
スフィンゴシンキナーゼ活性を測定する試験管内の検定においてスフィンゴシンキナーゼ2(Sphk2)を阻害するよりも5倍大きくスフィンゴシンキナーゼ1(Sphk1)を阻害することを特徴とする合成物。
【請求項46】
請求項45に記載の合成物であって、上記Sphk1はSphk2の阻害よりも少なくとも10倍大きく阻害される。
【請求項47】
請求項45に記載の合成物であって、上記試験管内の検定における阻害は10μM濃度で測定される。
【請求項48】
請求項45に記載の合成物であって、上記試験管内の検定における阻害はSphk1の50%阻害を与える濃度で測定される。
【請求項49】
がん細胞を殺すかまたは損傷するに充分な量の請求項45に記載の合成物に暴露するステップからなるがん細胞を殺すかまたは損傷を与えることを特徴とする方法。
【請求項50】
請求項45に記載の方法において、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項51】
がん細胞を、該がん細胞にアポトーシスを引き起こすために充分な量の請求項45に記載の合成物に暴露するステップからなることを特徴とするがん細胞にアポトーシスを引き起こす方法。
【請求項52】
請求項51に記載の方法において、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項53】
がん細胞を、該がん細胞の成長、転移、および化学薬剤耐性の進化を阻害するに充分な量の請求項45に記載の合成物に暴露するステップからなることを特徴とするがん細胞の成長、転移、および化学薬剤耐性の進化を阻害する方法。
【請求項54】
請求項53に記載の方法であって、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項55】
患者の白血病の症状を治療または軽減するために充分な量の請求項45に記載の合成物を該患者に投与するステップからなることを特徴とするそれを必要とする患者に対して白血病の症状を治療または軽減することを特徴とする方法。
【請求項56】
患者に対して抗がん剤と、該抗がん剤が該患者のがん細胞を殺す能力を増強するために充分な量の請求項45に記載の合成物とを投与するステップからなることを特徴とするそれらを必要とする患者のがん細胞を殺すため抗がん剤の能力を増強することを特徴とする方法。
【請求項57】
請求項56に記載の方法であって、上記がん細胞は、白血病細胞、胸部がん細胞、前立腺がん細胞、膵臓がん細胞、神経膠がん細胞、結腸がん細胞、肺がん細胞、卵巣がん細胞、黒色腫細胞、または腎臓がん細胞およびそれらの複合である。
【請求項58】
それを必要とする患者における免疫反応性を弱めるために充分な量の請求項45に記載の合成物を該患者に投与することを特徴とする該患者の免疫反応性を弱めるための方法。
【請求項59】
上記免疫反応性を弱めることは肥満細胞機能を弱めることによって行われる請求項58に記載の方法。
【請求項60】
請求項58に記載の方法において、免疫反応性を弱めることは、それを必要とする患者のぜんそくの症状を弱めることを指向する。
【請求項61】
請求項58に記載の方法において、免疫反応性を弱めることは、すなわちアナフィラキシーの症状を弱めることを指向する。
【請求項62】
請求項58に記載の方法において、免疫反応性を弱めることは、すなわち自己免疫病の症状を軽減することを指向する。
【請求項63】
がんにかかった細胞における生存信号を阻害するための方法であって、該方法は請求項45記載の合成物の生存信号を阻害するために充分な量を上記がんにかかった細胞に投与するステップからなる。
【請求項64】
請求項63に記載の方法において、上記生存信号の阻害はAktまたはERKあるいはその両方のリン酸化を弱めることによって行われる。
【請求項65】
請求項45の合成物を必要とする患者の多発性硬化症の症状を軽減するために充分な量を該患者に投与するステップからなることを特徴とする患者の多発性硬化症の症状を軽減することを特徴とする方法。
【請求項66】
オリゴマーまたはポリマーである請求項1に記載の合成物からなることを特徴とする合成物。
【図1A】

【図1B】

【図1C】
【図1D】
【図1E】

【図1F】

【図2A】
【図2B】
【図2C】

【図2D】
【図2E】
【図2F】
【図2G】

【図3A】
【図3B】

【図3C】

【図3D】
【図4A】
【図4B】
【図5A】
【図5B】
【図5C】
【図5D】
【図6A】

【図6B】

【図7A】
【図7B】
【図7C】
【図1B】
【図1C】
【図1D】
【図1E】
【図1F】
【図2A】
【図2B】
【図2C】
【図2D】
【図2E】
【図2F】
【図2G】
【図3A】
【図3B】
【図3C】
【図3D】
【図4A】
【図4B】
【図5A】
【図5B】
【図5C】
【図5D】
【図6A】
【図6B】
【図7A】
【図7B】
【図7C】
【公表番号】特表2012−525421(P2012−525421A)
【公表日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願番号】特願2012−508719(P2012−508719)
【出願日】平成22年4月29日(2010.4.29)
【国際出願番号】PCT/US2010/032939
【国際公開番号】WO2010/127093
【国際公開日】平成22年11月4日(2010.11.4)
【出願人】(597020845)エンゾー セラピューティクス インコーポレイティド (4)
【氏名又は名称原語表記】ENZO THERAPEUTICS INC.
【住所又は居所原語表記】10 Executive Boulevard, Farmingdale, New York 11735 (US).
【出願人】(511264102)バージニア コモンウェルス ユニバーシティ (1)
【氏名又は名称原語表記】VIRGINIA COMMONWEALTH UNIVERSITY
【Fターム(参考)】
【公表日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願日】平成22年4月29日(2010.4.29)
【国際出願番号】PCT/US2010/032939
【国際公開番号】WO2010/127093
【国際公開日】平成22年11月4日(2010.11.4)
【出願人】(597020845)エンゾー セラピューティクス インコーポレイティド (4)
【氏名又は名称原語表記】ENZO THERAPEUTICS INC.
【住所又は居所原語表記】10 Executive Boulevard, Farmingdale, New York 11735 (US).
【出願人】(511264102)バージニア コモンウェルス ユニバーシティ (1)
【氏名又は名称原語表記】VIRGINIA COMMONWEALTH UNIVERSITY
【Fターム(参考)】
[ Back to top ]