
スフィンゴミエリン合成酵素の活性測定法
【課題】簡便かつ高速にSMS活性を測定するための新規な測定方法、およびそれを利用したSMS活性の阻害薬または促進薬の高速スクリーニング方法の提供。
【解決手段】SMSによって生成したスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させ、該蛍光物質の蛍光強度を測定することを特徴とするSMS活性の新規測定方法、およびそれを利用したSMS活性の阻害薬または促進薬の高速スクリーニング方法を提供する。
【解決手段】SMSによって生成したスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させ、該蛍光物質の蛍光強度を測定することを特徴とするSMS活性の新規測定方法、およびそれを利用したSMS活性の阻害薬または促進薬の高速スクリーニング方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、スフィンゴミエリン合成酵素の活性測定方法、およびその方法を用いたスフィンゴミエリン合成酵素活性の阻害薬または促進薬のスクリーニング方法に関する。
【背景技術】
【0002】
スフィンゴミエリン合成酵素(以下、SMSと略記することがある。)は、ホスファチジルコリンのリン酸コリン部分をセラミドに転移してスフィンゴミエリンを生成するとともに、ジアシルグリセロールを生成する酵素であり、SMS1とSMS2の2種類の存在が知られている。SMSは、セラミドとスフィンゴミエリンの代謝に関与するため、SMS活性阻害薬は、細胞増殖抑制を必要とする疾患、例えば、白血病、癌、自己免疫疾患等の治療に、SMS活性促進薬は、細胞増殖を必要とする疾患、例えば、骨髄異形成症候群、骨髄移植後の造血抑制等の治療に有用であることが報告されている(特許文献1参照)。
【0003】
これまで、SMSの活性測定には、放射性または蛍光標識された基質(セラミドまたはホスファチジルコリン)を用いてSMSの酵素反応を行った後、脂質を抽出し、薄層クロマトグラフィー等で分離後、生成したスフィンゴミエリンの放射活性または蛍光量を測定する方法が利用されている(非特許文献1参照)。しかしながら、この方法では、操作が煩雑なため多くの時間を要し、SMS活性の阻害薬または促進薬の高速スクリーニングに利用することはできなかった。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2005−192537号公報
【非特許文献】
【0005】
【非特許文献1】メソッズ・イン・エンザイモロジー(Methods in Enzymology)311巻、31〜42頁(2000年)
【発明の概要】
【発明が解決しようとする課題】
【0006】
スフィンゴミエリン合成酵素(SMS)活性の阻害薬または促進薬の高速スクリーニングに利用できる、SMS活性を簡便かつ高速に測定する方法の構築が望まれていた。
【0007】
従って、本発明の課題は、簡便かつ高速にSMS活性を測定するための新規な測定方法、およびそれを利用したSMS活性の阻害薬または促進薬の高速スクリーニング方法を提供することにある。
【課題を解決するための手段】
【0008】
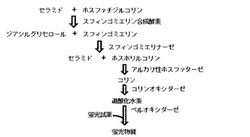
本発明者らは、上記課題を解決すべく鋭意検討した結果、スフィンゴミエリン合成酵素の活性測定方法として、標識したセラミドまたは標識したホスファチジルコリンを使用しなくても良く、かつスフィンゴミエリンを分離することなく、スフィンゴミエリン合成酵素の活性を測定できる新規の方法を見出した。すなわち、本発明は、図1に示すように、SMSによって生成したスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させ、次いで該蛍光物質の蛍光強度を測定することを特徴とするSMS活性の測定方法(以下、本発明方法と略記することがある。)に関する。本発明方法では、基質(セラミドまたはホスファチジルコリン)を標識する手間が不要であり、さらに従来の測定方法で操作を煩雑にしていた、脂質の抽出や薄層クロマトグラフィーを用いたスフィンゴミエリンの分離など多くの時間を要するステップを必要としないため、SMS活性を簡便かつ高速に測定することができる。また、本発明方法は、高感度であり、SMS活性の定量を行うことも可能である。また、驚くべきことに、本発明方法では、昆虫細胞の膜画分中におけるSMS活性を、哺乳類細胞の膜画分中におけるSMS活性よりも、高感度に測定できることを見出した。これまで、昆虫細胞の発現系は、発現効率や安定発現の向上を目的に利用されてきたが、本発明においては、活性測定時のバックグランドが低下するという新たな効果が昆虫細胞発現系の利用によって得られた。さらに、本発明者らは、本発明方法を利用した、SMS活性の阻害薬または促進薬の高速スクリーニング方法(以下、本発明のスクリーニング方法と略記することがある。)を見出し、本発明を完成させた。
【0009】
すなわち、本発明は、
(1)スフィンゴミエリン合成酵素の活性測定方法であって、標識したセラミドまたは標識したホスファチジルコリンを使用しなくてもよく、かつスフィンゴミエリンを分離することなく、スフィンゴミエリン合成酵素の活性を測定する方法、
(2)下記工程(i)〜(iv):
(i)スフィンゴミエリン合成酵素を含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、
(ii)工程(i)の後、スフィンゴミエリン合成酵素を失活させる工程、
(iii)工程(ii)の後、工程(i)で生じるスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、および
(iv)工程(iii)で生じる蛍光物質の蛍光強度を測定する工程
を含む、前記(1)記載の方法、
(3)工程(i)が、スフィンゴミエリン合成酵素を含有する検体を用いて、非標識のセラミドおよび非標識のホスファチジルコリンからスフィンゴミエリンを生成させる工程である前記(1)または前記(2)記載の方法、
(4)スフィンゴミエリン合成酵素を含有する検体が、スフィンゴミエリン合成酵素発現昆虫細胞の膜画分である前記(1)〜(3)記載の方法、
(5)スフィンゴミエリン合成酵素が、スフィンゴミエリン合成酵素SMS1および/またはSMS2である前記(1)〜(4)記載の方法、
(6)スフィンゴミエリン合成酵素の活性を阻害または促進する被検物質のスクリーニング方法であって、下記工程(a)〜(e):
(a)被検物質の存在下または非存在下、スフィンゴミエリン合成酵素を含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、
(b)工程(a)の後、スフィンゴミエリン合成酵素を失活させる工程、
(c)工程(b)の後、工程(a)で生じるスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、
(d)工程(c)で生じる蛍光物質の蛍光強度を測定する工程、および
(e)該被検物質の存在下における該蛍光強度と、該被検物質の非存在下における該蛍光強度を比較して、該被検物質がスフィンゴミエリン合成酵素活性を阻害する物質であるか、あるいは、促進する物質であるかを判断する工程
を含むスクリーニング方法、
(7)工程(a)が、被検物質の存在下または非存在下、スフィンゴミエリン合成酵素を含有する検体を用いて、非標識のセラミドおよび非標識のホスファチジルコリンからスフィンゴミエリンを生成させる工程である前記(6)記載の方法、
(8)スフィンゴミエリン合成酵素を含有する検体が、スフィンゴミエリン合成酵素発現昆虫細胞の膜画分である前記(6)または前記(7)記載の方法、
(9)スフィンゴミエリン合成酵素が、スフィンゴミエリン合成酵素SMS1および/またはSMS2である前記(6)〜(8)記載の方法に関する。
【発明の効果】
【0010】
本発明により、SMS活性を簡便かつ高速に測定することが可能となる。また、本発明方法を利用することで、SMS活性の阻害薬または促進薬の高速スクリーニングが可能となり、該SMS活性を標的とした薬剤の新薬開発に大きく貢献できる。本発明は、SMSのサブタイプであるSMS1およびSMS2のいずれに対しても適用可能であり、いずれのサブタイプでも使用することができる。
【図面の簡単な説明】
【0011】
【図1】本発明のスキームを示す。
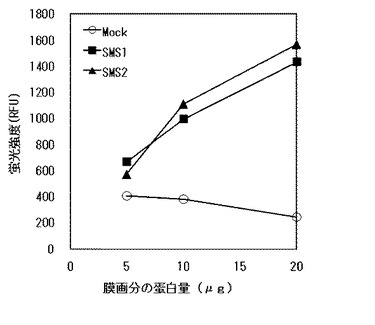
【図2】SMS1またはSMS2発現Sf9細胞の膜画分中のSMS活性を示す。縦軸は蛍光強度(RFU)を示し、横軸は膜画分の蛋白量(μg)を示す。黒四角印はSMS1発現Sf9細胞の膜画分を用いた結果を、黒三角印はSMS2発現Sf9細胞の膜画分を用いた結果を、白丸印はmockの膜画分についての結果をそれぞれ示す。
【発明を実施するための形態】
【0012】
本発明において、スフィンゴミエリン合成酵素(SMS)(EC 2.7.8.27)としては、SMS1とSMS2のいずれのサブタイプであってもよい。
【0013】
本発明において、SMSを含有する検体としては、SMSを含むものなら特に制限されないが、例えば、単離されたSMS、SMS発現細胞、SMS発現細胞の膜画分等であり、好ましくはSMS発現細胞の膜画分である。
【0014】
本発明において、SMS発現細胞とは、SMSを発現している限り、特に制限されないが、例えば、SMSを元々発現する細胞やSMS発現遺伝子を導入した細胞等が挙げられる。好ましくは、SMS発現遺伝子を導入した細胞であり、より好ましくはSMS発現遺伝子を導入した昆虫細胞(以下、SMS発現昆虫細胞と略記することがある。)である。
【0015】
SMSを含有する検体として、特に好ましくはSMS発現昆虫細胞の膜画分である。
【0016】
本発明に利用される昆虫細胞としては、例えばヨトウガの幼虫由来株化細胞(Spodoptera frugiperda cell;Sf細胞)、Trichoplusia niの中腸由来のMG1細胞、Trichoplusia niの卵由来のHigh FiveTM(商標)細胞、Mamestrabrassicae由来の細胞、Estigmena acrea由来の細胞、蚕由来株化細胞(Bombyx mori N;BmN細胞)等の他、カイコ幼虫個体が挙げられる。昆虫細胞として好ましくは、Sf細胞(例えば、Sf9細胞、Sf21細胞)であり、より好ましくはSf9細胞である。
【0017】
本発明において、SMS発現昆虫細胞とは、SMSをコードするポリヌクレオチドを導入した昆虫細胞を意味する。
【0018】
SMSをコードするポリヌクレオチドは、SMSまたはそれと実質的に同等のSMS活性を有するポリペプチドをコードするポリヌクレオチドである限り、特に限定されるものではない。
【0019】
本発明において、SMS1をコードするポリヌクレオチドとしては、具体的には、下記(1)または(2)のポリヌクレオチドが挙げられる。
(1)NCBI GENEBANKアクセッション番号:NM_147156.3塩基番号955〜2196で表される塩基配列(以下、塩基配列1と略記することがある。)、またはそれに相補的な塩基配列からなるポリヌクレオチド。
(2)塩基配列1またはそれに相補的な塩基配列のポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつ、スフィンゴミエリン合成酵素活性を有するポリペプチドをコードするポリヌクレオチド。
【0020】
本発明において、SMS2をコードするポリヌクレオチドとしては、具体的、下記(3)または(4)のポリヌクレオチドが挙げられる。
(3)NCBI GENEBANKアクセッション番号: NM_152621.5塩基番号640〜1737で表される塩基配列(以下、塩基配列2と略記することがある。)、またはそれに相補的な塩基配列からなるポリヌクレオチド。
(4)塩基配列2またはそれに相補的な塩基配列のポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつ、スフィンゴミエリン合成酵素活性を有するポリペプチドをコードするポリヌクレオチド。
なお、本発明において、ポリヌクレオチドには、特に言及しない限り、DNAおよびRNAの双方が含まれる。また、DNAには、特に言及しない限り、cDNA、ゲノムDNAおよび合成DNAが含まれる。RNAには、特に言及しない限り、totalRNA、mRNA、rRNA、tRNAおよび合成RNAが含まれる。
【0021】
(1)または(3)のポリヌクレオチドは、例えば、それぞれ塩基配列1または塩基配列2に基づき合成したオリゴヌクレオチドをプローブとして用いてヒトcDNAライブラリーをハイブリダイゼーションによりスクリーニングする方法により得ることができる。また、ヒトcDNAライブラリーをPCRの鋳型として用い、例えば塩基配列1または塩基配列2を基に設計したプライマーを用いて、常法に従いPCRを行うことによっても得ることができる。また、化学合成によっても得ることができる。
【0022】
(2)または(4)のポリヌクレオチドは、塩基配列1または塩基配列2またはそれらに相補的な塩基配列のポリヌクレオチドとストリンジェントな条件下でハイブリダイズする。「あるポリヌクレオチドにストリンジェントな条件下でハイブリダイズするポリヌクレオチド」は、例えばMolecular Cloning: A Laboratory Manual (Sambrookら編、コールド・スプリング・ハーバー・ラボラトリー・プレス、コールド・スプリング・ハーバー、ニューヨーク、1989年)等に記載の方法によって得ることができる。
【0023】
本発明において、「ストリンジェントな条件」としては、例えば、6xSSC、0.5%SDSおよび50%ホルムアミドの溶液中で42℃で加温した後、0.1xSSC、0.5%SDSの溶液中で68℃で洗浄した場合に、陽性のハイブリタイズのシグナルが観察される条件等が挙げられる。
【0024】
(2)または(4)のポリヌクレオチドには、例えば、それぞれ塩基配列1または塩基配列2のヒトのポリヌクレオチドに対応するマウスやラットのような他生物種のポリヌクレオチドや、天然または非天然のアレル変異体等が含まれる。
【0025】
(1)または(3)のポリヌクレオチドのヌクレオチドを置換して、(2)または(4)のポリヌクレオチドを定めるに当たっては、翻訳後に得られるアミノ酸が、極性、電荷、可溶性、親水性/疎水性、極性等の点で、置換前のアミノ酸と類似した性質を有するような塩基の置換を行うことができる。また、一つのアミノ酸が数個の翻訳コドンを有する場合、この翻訳コドン内での塩基の置換も勿論可能である。
【0026】
さらに、これら(1)〜(4)のポリヌクレオチドに、精製、検出用のタグ、例えば従来公知のヒスチジンタグや、GSTタグをコードする塩基配列等を付加させてもよい。
【0027】
昆虫細胞にSMSをコードするポリヌクレオチドを導入し、SMS発現昆虫細胞を製造する方法としては、バイオ/テクノロジー(Bio/Technology),6,47(1988)等に記載の当該分野で周知慣用の方法を用いることができる。例えば、SMSのポリヌクレオチドを挿入したトランスファーベクターとバキュロウイルスを昆虫細胞にコトランスフェクションして昆虫細胞培養上清中に組換えウイルスを得た後、さらに組換えウイルスを昆虫細胞に感染させ、SMSのポリペプチドを発現させる。
【0028】
昆虫細胞へのコトランスフェクションの方法としては、リン酸カルシウム法(Cell,11,223(1983))、リポフェクション法(Proceedings of the National Academy of Sciences USA,84,7413(1987))等を挙げることができる。また、動物細胞にDNAを導入する方法と同様の方法を用いて、昆虫細胞にDNAを導入することもでき、例えば、エレクトロポレーション法(Cytotechnology,3,133(1990))等を挙げることができる。
【0029】
バキュロウイルスとしては、核多角体病ウイルス(nuclear polyhedrosis virus:NPV)等が挙げられる。好ましくは、Autographa californica NPV(AcNPV)、カイコガBombyx mori NPV(BmNPV)であり、特に好ましくは、AcNPVである。
【0030】
バキュロウイルスの昆虫細胞への感染方法としては、MOI(感染多重度)が約0.1〜100、好ましくは1〜10、特に好ましくは1になるようにバキュロウイルスを昆虫細胞の培養液に添加する方法が用いられる。
【0031】
トランスファーベクターの種類は特に限定されないが、例えばpPSC8、pFastBac等であり、好ましくはpPSC8である。これらのトランスファーベクターは市販のものを用いることもできる。
【0032】
発現したSMSの量と質の確認は、それ自体公知の方法、例えば、ザ・ジャーナル・オブ・バイオロジカル・ケミストリー(J. Biol. Chem.),267,19555-19559(1992)に記載の方法に従って行うことができる。
【0033】
本発明のSMS発現昆虫細胞の膜画分としては、前記SMS発現昆虫細胞を破砕した後、それ自体公知の方法で得られる細胞膜が多く含まれる画分のことを言う。細胞の破砕方法としては、例えばホモジナイザー(例えば、Potter−Elvehiem型ホモジナイザー)で細胞を押し潰す方法、ワーリングブレンダーまたはポリトロン(Kinematica社)による破砕、超音波による破砕、あるいはフレンチプレス等で加圧しながら細いノズルから細胞を噴出させることによる破砕等が挙げられる。また、細胞膜の分画方法としては、例えば、分画遠心分離法や密度勾配遠心分離法等の遠心力による分画法が挙げられる。
[SMS活性の測定方法]
本発明のSMS活性測定方法は、(1)SMSを含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、(2)該スフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、(3)該蛍光物質の蛍光強度を測定する工程の3段階の操作からなる。
【0034】
工程(1)のSMSによる、セラミドとホスファチジルコリンからスフィンゴミエリンを生成する反応は、公知の反応であり、その条件等は公知の方法を考慮して適宜決定できる。例えば、適当なアッセイバッファー(好ましくは、50mmol/L Tris−HCl pH 7.4、25mmol/L KCl、0.5mmol/L EDTA)中で、SMSを含有する検体とセラミド(好ましくはC6セラミド)とホスファチジルコリンを含有する基質溶液を混合し、37度付近で約30分〜約4時間、好ましくは1時間反応させることにより行うことができる。
【0035】
工程(1)のセラミドとホスファチジルコリンは、標識されたものを使用しなくても良いため、非標識なものでも標識されたものでも構わないが、好ましくは非標識なものである。
【0036】
工程(1)として好ましくは、SMSを含有する検体を用いて、非標識のセラミドと非標識のホスファチジルコリンからスフィンゴミエリンを生成させる工程である。
【0037】
本発明において、セラミドとホスファチジルコリンは市販のものを使用でき、その混合比は、SMS酵素反応が効率よく進むものならどのような比率でも構わないが、例えば、ホスファチジルコリンを重量比でセラミド1に対し、約0.1〜20、好ましくは1〜10、より好ましくは10の割合で用いる。
【0038】
工程(1)のSMSの酵素反応を行った後、工程(2)の酵素反応を行う前に、SMSを失活させる工程を含むことが適当である。SMSは、例えば、工程(1)の反応終了後の混合物に塩酸(好ましくは、0.125N塩酸)を加えることで失活させることができる。
【0039】
SMSを失活させた反応混合物は、次いで工程(2)に用いられる。
【0040】
工程(2)の反応は公知の反応であり、条件等は公知の方法を考慮して適宜決定できる。
工程(2)の反応は、例えば、適当なアッセイバッファー(好ましくは、50mmol/L Tris−HCl pH 7.4、25mmol/L KCl、0.5mmol/L EDTA)中で、前記SMSを失活させた反応混合物、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を混合し、37度付近で約30分〜約4時間、好ましくは1時間反応させることにより行うことができる。
【0041】
工程(2)の蛍光試薬として好ましくは、Amplex(登録商標)Redである。ペルオキダーゼの存在下で、過酸化水素とAmplex(登録商標)Redを反応させると、強い蛍光性の化合物(蛍光物質)であるレゾルフィンが生じる。
【0042】
工程(3)の蛍光強度の測定は、公知の各種の方法で行うことができる。レゾルフィンを測定する場合には、例えば、モルキュラーデバイス社製Flex Station3により励起波長530〜560nm(好ましくは555nm)、検出波長590〜600nm(好ましくは595nm)における蛍光強度を測定することができる。
【0043】
上記蛍光強度は、SMSによって生成するスフィンゴミエリン量に依存するため、SMS活性は、上記蛍光強度を指標に測定できる。
【0044】
本発明において、SMSを含有する検体が、SMS発現遺伝子を導入した細胞またはその膜画分である場合には、偽形質転換した細胞(mock)またはその膜画分を陰性対照として置き、陰性対照との差を算出することで、より正確にSMS活性を測定することができる。
【0045】
本発明において、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよびAmplex(登録商標)Redは、市販のものを使用できる。
[SMS活性の阻害薬または促進薬のスクリーニング方法]
本発明のSMS活性の阻害薬または促進薬のスクリーニング方法は、(1)被検物質の存在下または非存在下、SMSを含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、(2)該スフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、(3)該蛍光物質の蛍光強度を測定する工程、(4)該被検物質の存在下における該蛍光強度と、該被検物質の非存在下における該蛍光強度を比較して、該被検物質がSMS活性を阻害する物質であるか、あるいは、促進する物質であるかを判断する工程の4段階の操作からなる。
【0046】
(1)の被検物質の共存下でのSMSの酵素反応は、被検物質を共存させること以外は、前記SMS活性の測定方法の(1)の工程と同様に行うことができる。また、必要により、被検物質とSMSを含有する検体のプレインキュベーションを行った後、セラミドとホスファチジルコリンを加えて、酵素反応を開始してもよい。プレインキュベーションを行う時間として、例えば約10分〜60分が挙げられる。
【0047】
本発明において、被検物質は、低分子化合物でも、オリゴマーまたはポリマーで有っても良く、また単独でも複数の組み合わせでもよい。
【0048】
本発明において、SMSを含有する検体としては、測定感度の観点から、SMS発現昆虫細胞の膜画分が最適である。
【0049】
(2)および(3)の工程は、前記SMS活性の測定方法の(2)および(3)の工程と同様に行うことができる。
【0050】
(4)の工程では、被検物質存在下における蛍光強度と、被検物質非存在下における蛍光強度を比較し、前者のほうの蛍光強度が低い場合(例えば、被検物質非存在下の蛍光強度と比較して、5%以上あるいは10%以上低値である場合)には、その被検物質は、SMS活性を阻害する物質であると決定することができる。また、被検物質存在下における蛍光強度と、被検物質非存在下における蛍光強度を比較し、前者のほうの蛍光強度が高い場合(例えば、被検物質非存在下の蛍光強度と比較して、5%以上あるいは10%以上高値である場合)には、その被検物質は、SMS活性を促進する物質であると決定することができる。
【実施例】
【0051】
本発明を下記実施例により具体的に説明するが、本発明はこれら実施例により限定されるものではない。
実施例1:SMS1発現昆虫細胞の膜画分の調整
(1)SMS1発現ベクターの構築
SMS1をコードするDNA(塩基配列1)をトランスファーベクター(pPSC8)にクローニングし、大腸菌DH5αで形質転換し、形質転換体を得た。形質転換体から目的配列を有するクローンを選択した。
(2)コトランスフェクション
次に、25cm2フラスコに播いた1.0x106個のSf9細胞(ATCC、CRL1711)に、上記SMS1発現ベクター約2μg、Linear AcNPV DNA85ng、及びInsect GeneJuice(メルク社)5μLを含むSF900II(インビトロジェン社)200μLを加えた。28℃で5日間培養後、培養上清を回収し、コトランスフェクション溶液とした。
(3)組換えウイルスの純化
組換えウイルスの純化はプラークアッセイ法で行った。上記コトランスフェクション液をSF900IIで102、103、104、105、106及び107倍に希釈し、直径60mmのシャーレに播いた2.0x106個のSf9細胞に1mLずつ加え、室温で1時間穏やかに振盪した。シャーレのウイルス液を取り除き、0.5% SeaKemGTG agarose(タカラバイオ社)を含むSF900II4mLをシャーレに流し込み、28℃で8日間培養した。多角体の無いプラーク3個を培地ごと採取し、各々1mLのSF900IIに懸濁させ、組換えウイルス原液とした。
(4)組換えウイルス液の作成
25cm2フラスコに播いた2.0x106個のSf9細胞に、上記組換えウイルス原液を0.5mL加え、28℃で3日間培養した(第1代ウイルス液)。75cm2フラスコに播いた6.0x106個のSf9細胞に、上記第1代ウイルス液を全量加え、28℃で3日間培養した。培養後培養上清を回収し、3,000xg、4℃で15分間遠心分離し、上清と沈殿に分画した。この上清をウイルス液として回収した(第2代ウイルス液)。1.5x106個/mLになるようにSF900IIで希釈したexpresSF+(登録商標)細胞を125mL三角フラスコ1本に50mL用意した。これに第2代ウイルス液を0.5mL添加し、130rpm、28℃で3日間振盪培養した。培養後、10,000xg、4℃で20分間遠心分離し、上清と沈殿に分画した。この培養上清をウイルス液として回収し、第3代ウイルス液とした。
(5)SMS1発現昆虫細胞の生産
1.5x106個/mLになるようにSf900IIで希釈したexpresSF+(登録商標)細胞1L相当を用意した。これに上記第3代ウイルス液をMOI(感染多重度)=1となるよう添加し、130rpm、28℃で振盪培養した。48時間後に細胞培養液を回収し、3,000xg、4℃で20分間遠心分離を行い、細胞を回収した。
(6)SMS1発現昆虫細胞の膜画分の調整
回収した細胞をホモジナイズ用緩衝液(20mM Tris−HCl pH 7.4、50mM NaCl、1mM EDTA、1mM EGTA、10% Glycerol)に懸濁後、PHYSCOTRON(マイクロテック・ニチオン株式会社)でoutput 8、10秒間の破砕を20回行った。その後、4℃、500xgで30分間遠心分離し、上清を回収した。回収した上清を次に4℃、100,000xgで60分間遠心し、得られた沈殿をホモジナイズ用緩衝液6mLに再懸濁し、これを細胞膜画分とした。調製した膜画分は、適当量に分注して使用時まで−80℃で保存した。
実施例2:SMS2発現昆虫細胞の膜画分の調整
(1)SMS2発現ベクターの構築
SMS1をコードするDNA(塩基配列1)の代わりに、SMS2をコードするDNA(塩基配列2)を用いて実施例1の(1)と同様の方法によりSMS2発現ベクターを構築した。
(2)SMS2発現昆虫細胞の膜画分の調整
SMS1発現ベクターの代わりに、SMS2発現ベクターを用いて、実施例1の(2)〜(6)と同様の方法を用いて、SMS2発現Sf9細胞の膜画分を調整した。
実施例3:SMS発現昆虫細胞の膜画分中のSMS活性測定
(1)基質溶液(C6セラミドとホスファチジルコリンのコンプレックス(1:10))の調整
10mgのC6セラミド(Avanti Polar Lipids)を適量のクロロホルムに溶かし、100mg相当のホスファチジルコリン(Avanti Polar Lipids)のクロロホルム溶液を加え、ボルテックスした。窒素ガスを静かに吹き付けつつ37℃でインキュベートし、クロロホルムを蒸発させた。1%オクチルβ−グルコシド溶液33.3ml加え、ボルテックスとソニケーションを行い、溶液が均一で透明であることを確認した。適当量に分注して、使用時まで−20℃で保存した。
(2)活性測定
96ウェルプレートに、実施例1または実施例2で調整したSMS1またはSMS2発現Sf9細胞の膜画分、あるいは偽形質転換したSf9細胞(mock)の膜画分20μL、実施例3(1)で調整した基質溶液10μL、アッセイバッファー(50mM Tris−HCl pH 7.4、25mM KCl、0.5mM EDTA)70μLを加え、37℃で1時間反応を行った。反応後、0.125N塩酸を100μL添加し、酵素反応を停止した。次いで、この反応液20μL、1U/mLスフィンゴミエリナーゼ2.5μL、25U/mLアルカリ性ホスファターゼ0.2μL、300U/mLコリンオキシダーゼ0.1μL、1000U/mLペルオキシダーゼ0.2μL、10mM Amplex(登録商標)Red0.75μL、アッセイバッファー86.25μLを混合し、37℃で1時間反応を行った。一方、膜画分のみ、または基質のみを含有するウェルをブランクとした。
【0052】
反応後、モルキュラーデバイス社製Flex Station3により、励起波長555nm、蛍光波長595nmにおける蛍光強度を測定し、得られた結果から、ブランクを差し引いた値を、SMS活性の指標とした。
【0053】
結果を図2に示す。なお、図中、黒四角印はSMS1発現Sf9細胞の膜画分を用いた結果を、黒三角印はSMS2発現Sf9細胞の膜画分を用いた結果を、白丸印はmockの膜画分についての結果をそれぞれ示す。
【0054】
図2から、SMS1およびSMS2発現Sf9細胞の膜画分中のSMS活性はともに、膜画分の蛋白量に応じた上昇を示し、定量的な測定結果が得られた。また、mockとの差を算出することにより、より正確にSMS活性を測定できることがわかった。
【0055】
以上の結果から、本発明方法を用いることで、SMS活性を簡便、かつ高速に測定できることが明らかとなった。また、本発明方法は、SMS活性の阻害薬または促進薬の高速スクリーニングに利用できる。
【産業上の利用可能性】
【0056】
本発明は、細胞増殖抑制を必要とする疾患、例えば、白血病、癌、自己免疫疾患等の治療に極めて有用であると考えられているSMS活性阻害薬や、細胞増殖を必要とする疾患、例えば、骨髄異形成症候群、骨髄移植後の造血抑制等の治療に極めて有用であると考えられているSMS活性促進薬を、簡便かつ高速にスクリーニングすることができる新規なSMS活性測定方法を提供するものであり、新規の医薬品を創出するのに有用である。
【技術分野】
【0001】
本発明は、スフィンゴミエリン合成酵素の活性測定方法、およびその方法を用いたスフィンゴミエリン合成酵素活性の阻害薬または促進薬のスクリーニング方法に関する。
【背景技術】
【0002】
スフィンゴミエリン合成酵素(以下、SMSと略記することがある。)は、ホスファチジルコリンのリン酸コリン部分をセラミドに転移してスフィンゴミエリンを生成するとともに、ジアシルグリセロールを生成する酵素であり、SMS1とSMS2の2種類の存在が知られている。SMSは、セラミドとスフィンゴミエリンの代謝に関与するため、SMS活性阻害薬は、細胞増殖抑制を必要とする疾患、例えば、白血病、癌、自己免疫疾患等の治療に、SMS活性促進薬は、細胞増殖を必要とする疾患、例えば、骨髄異形成症候群、骨髄移植後の造血抑制等の治療に有用であることが報告されている(特許文献1参照)。
【0003】
これまで、SMSの活性測定には、放射性または蛍光標識された基質(セラミドまたはホスファチジルコリン)を用いてSMSの酵素反応を行った後、脂質を抽出し、薄層クロマトグラフィー等で分離後、生成したスフィンゴミエリンの放射活性または蛍光量を測定する方法が利用されている(非特許文献1参照)。しかしながら、この方法では、操作が煩雑なため多くの時間を要し、SMS活性の阻害薬または促進薬の高速スクリーニングに利用することはできなかった。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2005−192537号公報
【非特許文献】
【0005】
【非特許文献1】メソッズ・イン・エンザイモロジー(Methods in Enzymology)311巻、31〜42頁(2000年)
【発明の概要】
【発明が解決しようとする課題】
【0006】
スフィンゴミエリン合成酵素(SMS)活性の阻害薬または促進薬の高速スクリーニングに利用できる、SMS活性を簡便かつ高速に測定する方法の構築が望まれていた。
【0007】
従って、本発明の課題は、簡便かつ高速にSMS活性を測定するための新規な測定方法、およびそれを利用したSMS活性の阻害薬または促進薬の高速スクリーニング方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明者らは、上記課題を解決すべく鋭意検討した結果、スフィンゴミエリン合成酵素の活性測定方法として、標識したセラミドまたは標識したホスファチジルコリンを使用しなくても良く、かつスフィンゴミエリンを分離することなく、スフィンゴミエリン合成酵素の活性を測定できる新規の方法を見出した。すなわち、本発明は、図1に示すように、SMSによって生成したスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させ、次いで該蛍光物質の蛍光強度を測定することを特徴とするSMS活性の測定方法(以下、本発明方法と略記することがある。)に関する。本発明方法では、基質(セラミドまたはホスファチジルコリン)を標識する手間が不要であり、さらに従来の測定方法で操作を煩雑にしていた、脂質の抽出や薄層クロマトグラフィーを用いたスフィンゴミエリンの分離など多くの時間を要するステップを必要としないため、SMS活性を簡便かつ高速に測定することができる。また、本発明方法は、高感度であり、SMS活性の定量を行うことも可能である。また、驚くべきことに、本発明方法では、昆虫細胞の膜画分中におけるSMS活性を、哺乳類細胞の膜画分中におけるSMS活性よりも、高感度に測定できることを見出した。これまで、昆虫細胞の発現系は、発現効率や安定発現の向上を目的に利用されてきたが、本発明においては、活性測定時のバックグランドが低下するという新たな効果が昆虫細胞発現系の利用によって得られた。さらに、本発明者らは、本発明方法を利用した、SMS活性の阻害薬または促進薬の高速スクリーニング方法(以下、本発明のスクリーニング方法と略記することがある。)を見出し、本発明を完成させた。
【0009】
すなわち、本発明は、
(1)スフィンゴミエリン合成酵素の活性測定方法であって、標識したセラミドまたは標識したホスファチジルコリンを使用しなくてもよく、かつスフィンゴミエリンを分離することなく、スフィンゴミエリン合成酵素の活性を測定する方法、
(2)下記工程(i)〜(iv):
(i)スフィンゴミエリン合成酵素を含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、
(ii)工程(i)の後、スフィンゴミエリン合成酵素を失活させる工程、
(iii)工程(ii)の後、工程(i)で生じるスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、および
(iv)工程(iii)で生じる蛍光物質の蛍光強度を測定する工程
を含む、前記(1)記載の方法、
(3)工程(i)が、スフィンゴミエリン合成酵素を含有する検体を用いて、非標識のセラミドおよび非標識のホスファチジルコリンからスフィンゴミエリンを生成させる工程である前記(1)または前記(2)記載の方法、
(4)スフィンゴミエリン合成酵素を含有する検体が、スフィンゴミエリン合成酵素発現昆虫細胞の膜画分である前記(1)〜(3)記載の方法、
(5)スフィンゴミエリン合成酵素が、スフィンゴミエリン合成酵素SMS1および/またはSMS2である前記(1)〜(4)記載の方法、
(6)スフィンゴミエリン合成酵素の活性を阻害または促進する被検物質のスクリーニング方法であって、下記工程(a)〜(e):
(a)被検物質の存在下または非存在下、スフィンゴミエリン合成酵素を含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、
(b)工程(a)の後、スフィンゴミエリン合成酵素を失活させる工程、
(c)工程(b)の後、工程(a)で生じるスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、
(d)工程(c)で生じる蛍光物質の蛍光強度を測定する工程、および
(e)該被検物質の存在下における該蛍光強度と、該被検物質の非存在下における該蛍光強度を比較して、該被検物質がスフィンゴミエリン合成酵素活性を阻害する物質であるか、あるいは、促進する物質であるかを判断する工程
を含むスクリーニング方法、
(7)工程(a)が、被検物質の存在下または非存在下、スフィンゴミエリン合成酵素を含有する検体を用いて、非標識のセラミドおよび非標識のホスファチジルコリンからスフィンゴミエリンを生成させる工程である前記(6)記載の方法、
(8)スフィンゴミエリン合成酵素を含有する検体が、スフィンゴミエリン合成酵素発現昆虫細胞の膜画分である前記(6)または前記(7)記載の方法、
(9)スフィンゴミエリン合成酵素が、スフィンゴミエリン合成酵素SMS1および/またはSMS2である前記(6)〜(8)記載の方法に関する。
【発明の効果】
【0010】
本発明により、SMS活性を簡便かつ高速に測定することが可能となる。また、本発明方法を利用することで、SMS活性の阻害薬または促進薬の高速スクリーニングが可能となり、該SMS活性を標的とした薬剤の新薬開発に大きく貢献できる。本発明は、SMSのサブタイプであるSMS1およびSMS2のいずれに対しても適用可能であり、いずれのサブタイプでも使用することができる。
【図面の簡単な説明】
【0011】
【図1】本発明のスキームを示す。
【図2】SMS1またはSMS2発現Sf9細胞の膜画分中のSMS活性を示す。縦軸は蛍光強度(RFU)を示し、横軸は膜画分の蛋白量(μg)を示す。黒四角印はSMS1発現Sf9細胞の膜画分を用いた結果を、黒三角印はSMS2発現Sf9細胞の膜画分を用いた結果を、白丸印はmockの膜画分についての結果をそれぞれ示す。
【発明を実施するための形態】
【0012】
本発明において、スフィンゴミエリン合成酵素(SMS)(EC 2.7.8.27)としては、SMS1とSMS2のいずれのサブタイプであってもよい。
【0013】
本発明において、SMSを含有する検体としては、SMSを含むものなら特に制限されないが、例えば、単離されたSMS、SMS発現細胞、SMS発現細胞の膜画分等であり、好ましくはSMS発現細胞の膜画分である。
【0014】
本発明において、SMS発現細胞とは、SMSを発現している限り、特に制限されないが、例えば、SMSを元々発現する細胞やSMS発現遺伝子を導入した細胞等が挙げられる。好ましくは、SMS発現遺伝子を導入した細胞であり、より好ましくはSMS発現遺伝子を導入した昆虫細胞(以下、SMS発現昆虫細胞と略記することがある。)である。
【0015】
SMSを含有する検体として、特に好ましくはSMS発現昆虫細胞の膜画分である。
【0016】
本発明に利用される昆虫細胞としては、例えばヨトウガの幼虫由来株化細胞(Spodoptera frugiperda cell;Sf細胞)、Trichoplusia niの中腸由来のMG1細胞、Trichoplusia niの卵由来のHigh FiveTM(商標)細胞、Mamestrabrassicae由来の細胞、Estigmena acrea由来の細胞、蚕由来株化細胞(Bombyx mori N;BmN細胞)等の他、カイコ幼虫個体が挙げられる。昆虫細胞として好ましくは、Sf細胞(例えば、Sf9細胞、Sf21細胞)であり、より好ましくはSf9細胞である。
【0017】
本発明において、SMS発現昆虫細胞とは、SMSをコードするポリヌクレオチドを導入した昆虫細胞を意味する。
【0018】
SMSをコードするポリヌクレオチドは、SMSまたはそれと実質的に同等のSMS活性を有するポリペプチドをコードするポリヌクレオチドである限り、特に限定されるものではない。
【0019】
本発明において、SMS1をコードするポリヌクレオチドとしては、具体的には、下記(1)または(2)のポリヌクレオチドが挙げられる。
(1)NCBI GENEBANKアクセッション番号:NM_147156.3塩基番号955〜2196で表される塩基配列(以下、塩基配列1と略記することがある。)、またはそれに相補的な塩基配列からなるポリヌクレオチド。
(2)塩基配列1またはそれに相補的な塩基配列のポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつ、スフィンゴミエリン合成酵素活性を有するポリペプチドをコードするポリヌクレオチド。
【0020】
本発明において、SMS2をコードするポリヌクレオチドとしては、具体的、下記(3)または(4)のポリヌクレオチドが挙げられる。
(3)NCBI GENEBANKアクセッション番号: NM_152621.5塩基番号640〜1737で表される塩基配列(以下、塩基配列2と略記することがある。)、またはそれに相補的な塩基配列からなるポリヌクレオチド。
(4)塩基配列2またはそれに相補的な塩基配列のポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつ、スフィンゴミエリン合成酵素活性を有するポリペプチドをコードするポリヌクレオチド。
なお、本発明において、ポリヌクレオチドには、特に言及しない限り、DNAおよびRNAの双方が含まれる。また、DNAには、特に言及しない限り、cDNA、ゲノムDNAおよび合成DNAが含まれる。RNAには、特に言及しない限り、totalRNA、mRNA、rRNA、tRNAおよび合成RNAが含まれる。
【0021】
(1)または(3)のポリヌクレオチドは、例えば、それぞれ塩基配列1または塩基配列2に基づき合成したオリゴヌクレオチドをプローブとして用いてヒトcDNAライブラリーをハイブリダイゼーションによりスクリーニングする方法により得ることができる。また、ヒトcDNAライブラリーをPCRの鋳型として用い、例えば塩基配列1または塩基配列2を基に設計したプライマーを用いて、常法に従いPCRを行うことによっても得ることができる。また、化学合成によっても得ることができる。
【0022】
(2)または(4)のポリヌクレオチドは、塩基配列1または塩基配列2またはそれらに相補的な塩基配列のポリヌクレオチドとストリンジェントな条件下でハイブリダイズする。「あるポリヌクレオチドにストリンジェントな条件下でハイブリダイズするポリヌクレオチド」は、例えばMolecular Cloning: A Laboratory Manual (Sambrookら編、コールド・スプリング・ハーバー・ラボラトリー・プレス、コールド・スプリング・ハーバー、ニューヨーク、1989年)等に記載の方法によって得ることができる。
【0023】
本発明において、「ストリンジェントな条件」としては、例えば、6xSSC、0.5%SDSおよび50%ホルムアミドの溶液中で42℃で加温した後、0.1xSSC、0.5%SDSの溶液中で68℃で洗浄した場合に、陽性のハイブリタイズのシグナルが観察される条件等が挙げられる。
【0024】
(2)または(4)のポリヌクレオチドには、例えば、それぞれ塩基配列1または塩基配列2のヒトのポリヌクレオチドに対応するマウスやラットのような他生物種のポリヌクレオチドや、天然または非天然のアレル変異体等が含まれる。
【0025】
(1)または(3)のポリヌクレオチドのヌクレオチドを置換して、(2)または(4)のポリヌクレオチドを定めるに当たっては、翻訳後に得られるアミノ酸が、極性、電荷、可溶性、親水性/疎水性、極性等の点で、置換前のアミノ酸と類似した性質を有するような塩基の置換を行うことができる。また、一つのアミノ酸が数個の翻訳コドンを有する場合、この翻訳コドン内での塩基の置換も勿論可能である。
【0026】
さらに、これら(1)〜(4)のポリヌクレオチドに、精製、検出用のタグ、例えば従来公知のヒスチジンタグや、GSTタグをコードする塩基配列等を付加させてもよい。
【0027】
昆虫細胞にSMSをコードするポリヌクレオチドを導入し、SMS発現昆虫細胞を製造する方法としては、バイオ/テクノロジー(Bio/Technology),6,47(1988)等に記載の当該分野で周知慣用の方法を用いることができる。例えば、SMSのポリヌクレオチドを挿入したトランスファーベクターとバキュロウイルスを昆虫細胞にコトランスフェクションして昆虫細胞培養上清中に組換えウイルスを得た後、さらに組換えウイルスを昆虫細胞に感染させ、SMSのポリペプチドを発現させる。
【0028】
昆虫細胞へのコトランスフェクションの方法としては、リン酸カルシウム法(Cell,11,223(1983))、リポフェクション法(Proceedings of the National Academy of Sciences USA,84,7413(1987))等を挙げることができる。また、動物細胞にDNAを導入する方法と同様の方法を用いて、昆虫細胞にDNAを導入することもでき、例えば、エレクトロポレーション法(Cytotechnology,3,133(1990))等を挙げることができる。
【0029】
バキュロウイルスとしては、核多角体病ウイルス(nuclear polyhedrosis virus:NPV)等が挙げられる。好ましくは、Autographa californica NPV(AcNPV)、カイコガBombyx mori NPV(BmNPV)であり、特に好ましくは、AcNPVである。
【0030】
バキュロウイルスの昆虫細胞への感染方法としては、MOI(感染多重度)が約0.1〜100、好ましくは1〜10、特に好ましくは1になるようにバキュロウイルスを昆虫細胞の培養液に添加する方法が用いられる。
【0031】
トランスファーベクターの種類は特に限定されないが、例えばpPSC8、pFastBac等であり、好ましくはpPSC8である。これらのトランスファーベクターは市販のものを用いることもできる。
【0032】
発現したSMSの量と質の確認は、それ自体公知の方法、例えば、ザ・ジャーナル・オブ・バイオロジカル・ケミストリー(J. Biol. Chem.),267,19555-19559(1992)に記載の方法に従って行うことができる。
【0033】
本発明のSMS発現昆虫細胞の膜画分としては、前記SMS発現昆虫細胞を破砕した後、それ自体公知の方法で得られる細胞膜が多く含まれる画分のことを言う。細胞の破砕方法としては、例えばホモジナイザー(例えば、Potter−Elvehiem型ホモジナイザー)で細胞を押し潰す方法、ワーリングブレンダーまたはポリトロン(Kinematica社)による破砕、超音波による破砕、あるいはフレンチプレス等で加圧しながら細いノズルから細胞を噴出させることによる破砕等が挙げられる。また、細胞膜の分画方法としては、例えば、分画遠心分離法や密度勾配遠心分離法等の遠心力による分画法が挙げられる。
[SMS活性の測定方法]
本発明のSMS活性測定方法は、(1)SMSを含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、(2)該スフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、(3)該蛍光物質の蛍光強度を測定する工程の3段階の操作からなる。
【0034】
工程(1)のSMSによる、セラミドとホスファチジルコリンからスフィンゴミエリンを生成する反応は、公知の反応であり、その条件等は公知の方法を考慮して適宜決定できる。例えば、適当なアッセイバッファー(好ましくは、50mmol/L Tris−HCl pH 7.4、25mmol/L KCl、0.5mmol/L EDTA)中で、SMSを含有する検体とセラミド(好ましくはC6セラミド)とホスファチジルコリンを含有する基質溶液を混合し、37度付近で約30分〜約4時間、好ましくは1時間反応させることにより行うことができる。
【0035】
工程(1)のセラミドとホスファチジルコリンは、標識されたものを使用しなくても良いため、非標識なものでも標識されたものでも構わないが、好ましくは非標識なものである。
【0036】
工程(1)として好ましくは、SMSを含有する検体を用いて、非標識のセラミドと非標識のホスファチジルコリンからスフィンゴミエリンを生成させる工程である。
【0037】
本発明において、セラミドとホスファチジルコリンは市販のものを使用でき、その混合比は、SMS酵素反応が効率よく進むものならどのような比率でも構わないが、例えば、ホスファチジルコリンを重量比でセラミド1に対し、約0.1〜20、好ましくは1〜10、より好ましくは10の割合で用いる。
【0038】
工程(1)のSMSの酵素反応を行った後、工程(2)の酵素反応を行う前に、SMSを失活させる工程を含むことが適当である。SMSは、例えば、工程(1)の反応終了後の混合物に塩酸(好ましくは、0.125N塩酸)を加えることで失活させることができる。
【0039】
SMSを失活させた反応混合物は、次いで工程(2)に用いられる。
【0040】
工程(2)の反応は公知の反応であり、条件等は公知の方法を考慮して適宜決定できる。
工程(2)の反応は、例えば、適当なアッセイバッファー(好ましくは、50mmol/L Tris−HCl pH 7.4、25mmol/L KCl、0.5mmol/L EDTA)中で、前記SMSを失活させた反応混合物、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を混合し、37度付近で約30分〜約4時間、好ましくは1時間反応させることにより行うことができる。
【0041】
工程(2)の蛍光試薬として好ましくは、Amplex(登録商標)Redである。ペルオキダーゼの存在下で、過酸化水素とAmplex(登録商標)Redを反応させると、強い蛍光性の化合物(蛍光物質)であるレゾルフィンが生じる。
【0042】
工程(3)の蛍光強度の測定は、公知の各種の方法で行うことができる。レゾルフィンを測定する場合には、例えば、モルキュラーデバイス社製Flex Station3により励起波長530〜560nm(好ましくは555nm)、検出波長590〜600nm(好ましくは595nm)における蛍光強度を測定することができる。
【0043】
上記蛍光強度は、SMSによって生成するスフィンゴミエリン量に依存するため、SMS活性は、上記蛍光強度を指標に測定できる。
【0044】
本発明において、SMSを含有する検体が、SMS発現遺伝子を導入した細胞またはその膜画分である場合には、偽形質転換した細胞(mock)またはその膜画分を陰性対照として置き、陰性対照との差を算出することで、より正確にSMS活性を測定することができる。
【0045】
本発明において、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよびAmplex(登録商標)Redは、市販のものを使用できる。
[SMS活性の阻害薬または促進薬のスクリーニング方法]
本発明のSMS活性の阻害薬または促進薬のスクリーニング方法は、(1)被検物質の存在下または非存在下、SMSを含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、(2)該スフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、(3)該蛍光物質の蛍光強度を測定する工程、(4)該被検物質の存在下における該蛍光強度と、該被検物質の非存在下における該蛍光強度を比較して、該被検物質がSMS活性を阻害する物質であるか、あるいは、促進する物質であるかを判断する工程の4段階の操作からなる。
【0046】
(1)の被検物質の共存下でのSMSの酵素反応は、被検物質を共存させること以外は、前記SMS活性の測定方法の(1)の工程と同様に行うことができる。また、必要により、被検物質とSMSを含有する検体のプレインキュベーションを行った後、セラミドとホスファチジルコリンを加えて、酵素反応を開始してもよい。プレインキュベーションを行う時間として、例えば約10分〜60分が挙げられる。
【0047】
本発明において、被検物質は、低分子化合物でも、オリゴマーまたはポリマーで有っても良く、また単独でも複数の組み合わせでもよい。
【0048】
本発明において、SMSを含有する検体としては、測定感度の観点から、SMS発現昆虫細胞の膜画分が最適である。
【0049】
(2)および(3)の工程は、前記SMS活性の測定方法の(2)および(3)の工程と同様に行うことができる。
【0050】
(4)の工程では、被検物質存在下における蛍光強度と、被検物質非存在下における蛍光強度を比較し、前者のほうの蛍光強度が低い場合(例えば、被検物質非存在下の蛍光強度と比較して、5%以上あるいは10%以上低値である場合)には、その被検物質は、SMS活性を阻害する物質であると決定することができる。また、被検物質存在下における蛍光強度と、被検物質非存在下における蛍光強度を比較し、前者のほうの蛍光強度が高い場合(例えば、被検物質非存在下の蛍光強度と比較して、5%以上あるいは10%以上高値である場合)には、その被検物質は、SMS活性を促進する物質であると決定することができる。
【実施例】
【0051】
本発明を下記実施例により具体的に説明するが、本発明はこれら実施例により限定されるものではない。
実施例1:SMS1発現昆虫細胞の膜画分の調整
(1)SMS1発現ベクターの構築
SMS1をコードするDNA(塩基配列1)をトランスファーベクター(pPSC8)にクローニングし、大腸菌DH5αで形質転換し、形質転換体を得た。形質転換体から目的配列を有するクローンを選択した。
(2)コトランスフェクション
次に、25cm2フラスコに播いた1.0x106個のSf9細胞(ATCC、CRL1711)に、上記SMS1発現ベクター約2μg、Linear AcNPV DNA85ng、及びInsect GeneJuice(メルク社)5μLを含むSF900II(インビトロジェン社)200μLを加えた。28℃で5日間培養後、培養上清を回収し、コトランスフェクション溶液とした。
(3)組換えウイルスの純化
組換えウイルスの純化はプラークアッセイ法で行った。上記コトランスフェクション液をSF900IIで102、103、104、105、106及び107倍に希釈し、直径60mmのシャーレに播いた2.0x106個のSf9細胞に1mLずつ加え、室温で1時間穏やかに振盪した。シャーレのウイルス液を取り除き、0.5% SeaKemGTG agarose(タカラバイオ社)を含むSF900II4mLをシャーレに流し込み、28℃で8日間培養した。多角体の無いプラーク3個を培地ごと採取し、各々1mLのSF900IIに懸濁させ、組換えウイルス原液とした。
(4)組換えウイルス液の作成
25cm2フラスコに播いた2.0x106個のSf9細胞に、上記組換えウイルス原液を0.5mL加え、28℃で3日間培養した(第1代ウイルス液)。75cm2フラスコに播いた6.0x106個のSf9細胞に、上記第1代ウイルス液を全量加え、28℃で3日間培養した。培養後培養上清を回収し、3,000xg、4℃で15分間遠心分離し、上清と沈殿に分画した。この上清をウイルス液として回収した(第2代ウイルス液)。1.5x106個/mLになるようにSF900IIで希釈したexpresSF+(登録商標)細胞を125mL三角フラスコ1本に50mL用意した。これに第2代ウイルス液を0.5mL添加し、130rpm、28℃で3日間振盪培養した。培養後、10,000xg、4℃で20分間遠心分離し、上清と沈殿に分画した。この培養上清をウイルス液として回収し、第3代ウイルス液とした。
(5)SMS1発現昆虫細胞の生産
1.5x106個/mLになるようにSf900IIで希釈したexpresSF+(登録商標)細胞1L相当を用意した。これに上記第3代ウイルス液をMOI(感染多重度)=1となるよう添加し、130rpm、28℃で振盪培養した。48時間後に細胞培養液を回収し、3,000xg、4℃で20分間遠心分離を行い、細胞を回収した。
(6)SMS1発現昆虫細胞の膜画分の調整
回収した細胞をホモジナイズ用緩衝液(20mM Tris−HCl pH 7.4、50mM NaCl、1mM EDTA、1mM EGTA、10% Glycerol)に懸濁後、PHYSCOTRON(マイクロテック・ニチオン株式会社)でoutput 8、10秒間の破砕を20回行った。その後、4℃、500xgで30分間遠心分離し、上清を回収した。回収した上清を次に4℃、100,000xgで60分間遠心し、得られた沈殿をホモジナイズ用緩衝液6mLに再懸濁し、これを細胞膜画分とした。調製した膜画分は、適当量に分注して使用時まで−80℃で保存した。
実施例2:SMS2発現昆虫細胞の膜画分の調整
(1)SMS2発現ベクターの構築
SMS1をコードするDNA(塩基配列1)の代わりに、SMS2をコードするDNA(塩基配列2)を用いて実施例1の(1)と同様の方法によりSMS2発現ベクターを構築した。
(2)SMS2発現昆虫細胞の膜画分の調整
SMS1発現ベクターの代わりに、SMS2発現ベクターを用いて、実施例1の(2)〜(6)と同様の方法を用いて、SMS2発現Sf9細胞の膜画分を調整した。
実施例3:SMS発現昆虫細胞の膜画分中のSMS活性測定
(1)基質溶液(C6セラミドとホスファチジルコリンのコンプレックス(1:10))の調整
10mgのC6セラミド(Avanti Polar Lipids)を適量のクロロホルムに溶かし、100mg相当のホスファチジルコリン(Avanti Polar Lipids)のクロロホルム溶液を加え、ボルテックスした。窒素ガスを静かに吹き付けつつ37℃でインキュベートし、クロロホルムを蒸発させた。1%オクチルβ−グルコシド溶液33.3ml加え、ボルテックスとソニケーションを行い、溶液が均一で透明であることを確認した。適当量に分注して、使用時まで−20℃で保存した。
(2)活性測定
96ウェルプレートに、実施例1または実施例2で調整したSMS1またはSMS2発現Sf9細胞の膜画分、あるいは偽形質転換したSf9細胞(mock)の膜画分20μL、実施例3(1)で調整した基質溶液10μL、アッセイバッファー(50mM Tris−HCl pH 7.4、25mM KCl、0.5mM EDTA)70μLを加え、37℃で1時間反応を行った。反応後、0.125N塩酸を100μL添加し、酵素反応を停止した。次いで、この反応液20μL、1U/mLスフィンゴミエリナーゼ2.5μL、25U/mLアルカリ性ホスファターゼ0.2μL、300U/mLコリンオキシダーゼ0.1μL、1000U/mLペルオキシダーゼ0.2μL、10mM Amplex(登録商標)Red0.75μL、アッセイバッファー86.25μLを混合し、37℃で1時間反応を行った。一方、膜画分のみ、または基質のみを含有するウェルをブランクとした。
【0052】
反応後、モルキュラーデバイス社製Flex Station3により、励起波長555nm、蛍光波長595nmにおける蛍光強度を測定し、得られた結果から、ブランクを差し引いた値を、SMS活性の指標とした。
【0053】
結果を図2に示す。なお、図中、黒四角印はSMS1発現Sf9細胞の膜画分を用いた結果を、黒三角印はSMS2発現Sf9細胞の膜画分を用いた結果を、白丸印はmockの膜画分についての結果をそれぞれ示す。
【0054】
図2から、SMS1およびSMS2発現Sf9細胞の膜画分中のSMS活性はともに、膜画分の蛋白量に応じた上昇を示し、定量的な測定結果が得られた。また、mockとの差を算出することにより、より正確にSMS活性を測定できることがわかった。
【0055】
以上の結果から、本発明方法を用いることで、SMS活性を簡便、かつ高速に測定できることが明らかとなった。また、本発明方法は、SMS活性の阻害薬または促進薬の高速スクリーニングに利用できる。
【産業上の利用可能性】
【0056】
本発明は、細胞増殖抑制を必要とする疾患、例えば、白血病、癌、自己免疫疾患等の治療に極めて有用であると考えられているSMS活性阻害薬や、細胞増殖を必要とする疾患、例えば、骨髄異形成症候群、骨髄移植後の造血抑制等の治療に極めて有用であると考えられているSMS活性促進薬を、簡便かつ高速にスクリーニングすることができる新規なSMS活性測定方法を提供するものであり、新規の医薬品を創出するのに有用である。
【特許請求の範囲】
【請求項1】
スフィンゴミエリン合成酵素の活性測定方法であって、標識したセラミドまたは標識したホスファチジルコリンを使用しなくてもよく、かつスフィンゴミエリンを分離することなく、スフィンゴミエリン合成酵素の活性を測定する方法。
【請求項2】
下記工程(i)〜(iv):
(i)スフィンゴミエリン合成酵素を含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、
(ii)工程(i)の後、スフィンゴミエリン合成酵素を失活させる工程、
(iii)工程(ii)の後、工程(i)で生じるスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、および
(iv)工程(iii)で生じる蛍光物質の蛍光強度を測定する工程
を含む、請求項1記載の方法。
【請求項3】
工程(i)が、スフィンゴミエリン合成酵素を含有する検体を用いて、非標識のセラミドおよび非標識のホスファチジルコリンからスフィンゴミエリンを生成させる工程である請求項1または請求項2記載の方法。
【請求項4】
スフィンゴミエリン合成酵素を含有する検体が、スフィンゴミエリン合成酵素発現昆虫細胞の膜画分である請求項1〜3記載の方法。
【請求項5】
スフィンゴミエリン合成酵素が、スフィンゴミエリン合成酵素SMS1および/またはSMS2である請求項1〜4記載の方法。
【請求項6】
スフィンゴミエリン合成酵素の活性を阻害または促進する被検物質のスクリーニング方法であって、下記工程(a)〜(e):
(a)被検物質の存在下または非存在下、スフィンゴミエリン合成酵素を含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、
(b)工程(a)の後、スフィンゴミエリン合成酵素を失活させる工程、
(c)工程(b)の後、工程(a)で生じるスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、
(d)工程(c)で生じる蛍光物質の蛍光強度を測定する工程、および
(e)該被検物質の存在下における該蛍光強度と、該被検物質の非存在下における該蛍光強度を比較して、該被検物質がスフィンゴミエリン合成酵素活性を阻害する物質であるか、あるいは、促進する物質であるかを判断する工程
を含むスクリーニング方法。
【請求項7】
工程(a)が、被検物質の存在下または非存在下、スフィンゴミエリン合成酵素を含有する検体を用いて、非標識のセラミドおよび非標識のホスファチジルコリンからスフィンゴミエリンを生成させる工程である請求項6記載の方法。
【請求項8】
スフィンゴミエリン合成酵素を含有する検体が、スフィンゴミエリン合成酵素発現昆虫細胞の膜画分である請求項6または請求項7記載の方法。
【請求項9】
スフィンゴミエリン合成酵素が、スフィンゴミエリン合成酵素SMS1および/またはSMS2である請求項6〜8記載の方法。
【請求項1】
スフィンゴミエリン合成酵素の活性測定方法であって、標識したセラミドまたは標識したホスファチジルコリンを使用しなくてもよく、かつスフィンゴミエリンを分離することなく、スフィンゴミエリン合成酵素の活性を測定する方法。
【請求項2】
下記工程(i)〜(iv):
(i)スフィンゴミエリン合成酵素を含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、
(ii)工程(i)の後、スフィンゴミエリン合成酵素を失活させる工程、
(iii)工程(ii)の後、工程(i)で生じるスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、および
(iv)工程(iii)で生じる蛍光物質の蛍光強度を測定する工程
を含む、請求項1記載の方法。
【請求項3】
工程(i)が、スフィンゴミエリン合成酵素を含有する検体を用いて、非標識のセラミドおよび非標識のホスファチジルコリンからスフィンゴミエリンを生成させる工程である請求項1または請求項2記載の方法。
【請求項4】
スフィンゴミエリン合成酵素を含有する検体が、スフィンゴミエリン合成酵素発現昆虫細胞の膜画分である請求項1〜3記載の方法。
【請求項5】
スフィンゴミエリン合成酵素が、スフィンゴミエリン合成酵素SMS1および/またはSMS2である請求項1〜4記載の方法。
【請求項6】
スフィンゴミエリン合成酵素の活性を阻害または促進する被検物質のスクリーニング方法であって、下記工程(a)〜(e):
(a)被検物質の存在下または非存在下、スフィンゴミエリン合成酵素を含有する検体を用いて、セラミドとホスファチジルコリンからスフィンゴミエリンを生成させる工程、
(b)工程(a)の後、スフィンゴミエリン合成酵素を失活させる工程、
(c)工程(b)の後、工程(a)で生じるスフィンゴミエリンを分離することなく、スフィンゴミエリナーゼ、アルカリ性ホスファターゼ、コリンオキシダーゼ、ペルオキシダーゼおよび蛍光試薬を含む組成物と混合して蛍光物質を発生させる工程、
(d)工程(c)で生じる蛍光物質の蛍光強度を測定する工程、および
(e)該被検物質の存在下における該蛍光強度と、該被検物質の非存在下における該蛍光強度を比較して、該被検物質がスフィンゴミエリン合成酵素活性を阻害する物質であるか、あるいは、促進する物質であるかを判断する工程
を含むスクリーニング方法。
【請求項7】
工程(a)が、被検物質の存在下または非存在下、スフィンゴミエリン合成酵素を含有する検体を用いて、非標識のセラミドおよび非標識のホスファチジルコリンからスフィンゴミエリンを生成させる工程である請求項6記載の方法。
【請求項8】
スフィンゴミエリン合成酵素を含有する検体が、スフィンゴミエリン合成酵素発現昆虫細胞の膜画分である請求項6または請求項7記載の方法。
【請求項9】
スフィンゴミエリン合成酵素が、スフィンゴミエリン合成酵素SMS1および/またはSMS2である請求項6〜8記載の方法。
【図1】


【図2】
【図2】
【公開番号】特開2013−17394(P2013−17394A)
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願番号】特願2011−150762(P2011−150762)
【出願日】平成23年7月7日(2011.7.7)
【出願人】(000185983)小野薬品工業株式会社 (180)
【Fターム(参考)】
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願日】平成23年7月7日(2011.7.7)
【出願人】(000185983)小野薬品工業株式会社 (180)
【Fターム(参考)】
[ Back to top ]