
スフィンゴ脂質の酵素的合成
【課題】リゾスフィンゴ脂質を酵素的にアミド化する方法に関する。
【解決手段】本発明は、スフィンゴ脂質の酵素的合成、およびリゾスフィンゴ脂質とカルボニックエステル由来のスフィンゴ脂質を含む組成物、ならびに、前記スフィンゴ脂質または組成物を含む、化粧用製剤、皮膚用製剤または医薬製剤に関する。
【解決手段】本発明は、スフィンゴ脂質の酵素的合成、およびリゾスフィンゴ脂質とカルボニックエステル由来のスフィンゴ脂質を含む組成物、ならびに、前記スフィンゴ脂質または組成物を含む、化粧用製剤、皮膚用製剤または医薬製剤に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、スフィンゴ脂質の酵素的合成ならびに、リゾスフィンゴ脂質とカルボキシルエステル由来のスフィンゴ脂質を含む組成物に関し、また、こうしたスフィンゴ脂質または組成物を含む化粧製剤、皮膚用製剤または医薬製剤に関する。
【背景技術】
【0002】
本発明は、一般式I
【化1】

で表されるスフィンゴ脂質の生体触媒的調製方法であって、
一般式II
【化2】
で表されるリゾスフィンゴ脂質を、一般式III
【化3】
(ここで、R1、R2、R3およびXは以下に定義の通りである)
で表されるカルボキシルエステルと反応させることによる調製方法に関する。
【0003】
本スフィンゴ脂質は、化粧用および/または皮膚用有効成分として使用される。
【0004】
R2=Hである一般式Iのスフィンゴ脂質は、セラミドと言われ、皮膚(角質層)内に天然に存在する極性脂質である。外側の角質細胞では、セラミドは、細胞膜の主成分(脂質含量の40〜65%)に相当し、たとえば、透水性を調節することにより、その保護機能において中心的役割を果たす。加齢とともに、ケラチノサイトは、セラミド合成能の大部分を失い、それ故皮膚はその保護作用の一部を失い、たとえば、もはや表皮の水分喪失を完全に抑制することはできない。これは、セラミドの局所適用によって、少なくとも部分的に補うことができる(たとえば、Farwick, M. et al., Int. J. Cosm. Sci., 2007, 29(4), 319-329またはKlee, S.K. et al. Basic Clinic. Dermatol., 2007, 40, 155-165)。
【0005】
加えて、アトピー性皮膚炎の治療における、セラミドの好ましい作用が報告されている(Kerscher, M. et al., Eur. J. Dermatol., 1991, 1, 39-43; Imokawa, G. et al., J. Soc. Cosmet. Chem. 1989, 40, 500-507)。
【0006】
多くの先進工業諸国、特にドイツにおける人口統計推移を考慮すると、セラミドの需要はさらに増大すると予想される。
【0007】
先行技術では、セラミドは、活性化カルボン酸誘導体を使用して、R2=Hである一般式IIのリゾスフィンゴ脂質、たとえば、フィトスフィンゴシン(R2=HかつX=CH2−HCOHである、一般式II)等の、ショッテン・バウマン(Schotten Baumann)類似N−アシル化によって調製される。
【0008】
フィトスフィンゴシンは、一般に発酵起源である。
【0009】
活性化カルボン酸は一般に、それ自体として使用されるか、またはカルボン酸からイン・シチュで調製されるかのいずれかである、塩化カルボニルである。米国特許第6420604号(Cosmoferm, NL)は、スフィンゴシン塩基を好適なカルボニルハロゲン化物と反応させることによる、幾つかのセラミドの合成について記述している。この方法の際立った不都合は、有毒な有機塩素化合物を使用する必要性がある一方で、結果として生じる最終生成物が高塩負荷であることである。さらに、極めて反応しやすいカルボニルハロゲン化物を使用する際には、かなりの量の不要な副産物の生成を予想しなければならないことは、当業者に明白である。
【0010】
代替合成経路は、無水物によって進行する。したがって、たとえば、国際公開第93/20038号パンフレット(Gist-Brocades, NL)は、フィトスフィンゴシンのN−アシル化用の反応性カルボン酸誘導体を得るために、カルボン酸とアルキルフェニルスルホニルクロリドから、混合無水物を塩基触媒合成することを教示している。
【0011】
これらの経路全部に共通しているのは、反応性カルボン酸誘導体の使用に依存していることである。これに関連した不都合は、こうした物質の高い腐蝕性およびそれらの健康に対する危険性の両者であり、これにより特別の反応器システムおよび注意が必要となり、したがって、調製の複雑さが増大する。加えて、本生成物を局所適用できるためには、反応しやすく有毒な前駆体および副産物が本生成物から除去されていることが保証されなければならない。高塩負荷、ならびにそれに関連した生成物精製および廃棄物処理のための付加的な複雑さもさらに予想される。先行技術の方法のさらなる不都合は、低い前駆体濃度および生成物濃度しか得られないことであり;たとえば、国際公開第93/20038号パンフレット(Gist-Brocades, NL)は、反応性かつ有毒な共試薬である、塩化p−トルエンスルホニルおよびトリエチルアミンの同時使用と共に、有毒な溶剤塩化メチレン中の最大15%(w/v)の前駆体溶液を記している。加えて、本生成物の純度または収量(いずれの場合にも、80%)はいずれも満足のいくものではない。
【0012】
したがって、本発明の目的は、無害かつ産業上利用することができる様式で、先行技術の不都合の少なくとも1つを克服することができる、セラミドを得る代替方法を提供することである。
【0013】
生体触媒的に、すなわち、酵素を触媒として使用して、アミド化反応を得ることもできる。酵素の産業上の利用の概説は、たとえばLieseら(Industrial Biotransformations; Second, Completely Revised Edition, Wiley-VCH, Weinheim, 2006)に見られる。カルボン酸誘導体を合成するのに好ましい生体触媒は、加水分解酵素(E.C. 3.1.x.x)、特にリパーゼ(トリアシルグリセロール加水分解酵素、E.C. 3.1.1.3)およびエステラーゼ(E.C. 3.1.1.1)である。
【0014】
リパーゼは、細胞の代謝における自然機能に従って、エステルの加水開裂を優先的に触媒する。しかし、エステルの縮合的生成も、文献に多数記述されている。それらの代表例は、Drauz and Waldmann (Enzyme Catalysis in Organic Synthesis, A Comprehensive Handbook; Second, Completely Revised and Enlarged Edition, Vol. II, Wiley-VCH, Weinheim, 2002)、Aehle (Enzymes in Industry, Production and Applications; Second, Completely Revised Edition, Wiley-VCH, Weinheim, 2004)またはBornscheuer (Enzymes in Lipid Modification, Wiley-VCH, Weinheim, 2000)に見られる。
【0015】
リパーゼ−触媒縮合反応における求核剤としてのアミンの使用例も、文献にある。
【0016】
Y.-M. Xia et al., J Mol Catal B, 2004, 31,111-115は、たとえば、カンジダ・アンタークティカ(Candida antarctica (CALB))由来のリパーゼにより触媒される反応性一級アミンを用いたラウリン酸メチルのアミド化によるN−ラウロイル−β−アミノプロプリオニトリルの合成について記述している。記載の方法の不都合は、基質溶液の希釈(50mM未満)の制限に加え、僅か94.3%までの変換という、比較的低い変換である。さらに、示された反応には、位置選択性または化学選択性は全く必要ない。
【0017】
しかし、前駆体分子、たとえば、フィトスフィンゴシン等は、複数の反応性官能基を有する可能性があるため、本発明による反応には、このような選択性が絶対的に必要であろう。この選択性の問題は、たとえばP.Tufvesson et al.: Biotechnol.Bioeng., 2007, 97(3), 447-453により示され:エタノールアミンとカルボン酸とを用いたCALB−触媒による反応において、酵素触媒によるエステル化を回避する単純な選択的アミド化は不可能であった。前駆体の多回添加および蒸留による反応水の除去によってのみ、所望のアミドの濃縮を達成することが可能であった。
【0018】
酵素的アミド化のさらなる例で、M.Nechab et al.: JOC, 2007, 72, 6918-6923は、CALBを使用した、(R)立体配置二級アミンの立体選択的反応について記述している。生成物の高い光学純度(>99% ee)が確認されたことから、CALBの(R)立体配置アミンに対する強い優先傾向が示唆され、したがって、たとえばフィトスフィンゴシンは、S−立体配置であるため、この触媒で、アミン炭素原子に反応するかどうか疑わしい。加えて、やはり、非常に希釈された基質溶液(100mM)が、高濃度(27%w(CALB)/w(基質))の生体触媒と同時に使用され、記載の方法の産業上の魅力を著しく制限している。
【0019】
国際公開第94/26919号パンフレット(Gist-Brocades, NL)は、細菌リパーゼまたは哺乳リパーゼの使用とともに、カルボキシルエステルを使用するフィトスフィンゴシンの酵素的N−アシル化について記述している。細菌リパーゼおよび哺乳リパーゼ、特に、シュードモナス・アルカリゲネス(Pseudomonas alcaligenes)由来のリパーゼは、記載の方法で好ましく使用される。この方法の不都合は、特に、中程度の変換(最高78%)を達成するだけのために、大量の生体触媒(30〜95%w/w)を必要とすることである。加えて、かなりの量の不要なN,O−ジアシル化生成物(最高17%)が確認された。さらに、基質の希釈溶液のみが使用され(59〜93mM)、その結果として空時収量が低くなる。さらなる不都合は、基質としてフィトスフィンゴシン塩基に制限されることであり、フィトスフィンゴシンの酸付加生成物、たとえばフィトスフィンゴシンサルフェートの使用と比較して、基質価格の激増を伴う。さらに、酵母および真菌に由来するリパーゼは、触媒として不安定であることが明確に指摘されている。したがって、国際公開第94/26919号パンフレットに記述されている方法は、既存の化学的方法に代わる経済的に価値ある代替法としては除外される。
【発明の概要】
【発明が解決しようとする課題】
【0020】
このように、従来実現できなかった、アルキルカルボキシレートからのスフィンゴ脂質の生体触媒的合成の実施を可能にするために、先行技術の不都合を克服する、リゾスフィンゴ脂質を酵素的にアミド化する方法が引き続き必要である。
【0021】
したがって、本発明の目的は、先行技術の方法の不都合の1つまたは複数がない方法を提供することであった。特に、その意図は、容易に入手できるリゾスフィンゴ脂質を前駆体として、さらには高濃度で、使用することができる方法を開発することであった。
【0022】
明確に言及されていないさらなる目的は、以下の説明、実施例および特許請求の範囲の文脈から明白である。
【課題を解決するための手段】
【0023】
驚くべきことに、先行技術とは対照的に、真菌リパーゼがカルボキシルエステルとリゾスフィンゴ脂質を選択的に反応させ、高純度のスフィンゴ脂質を形成可能なことが見出された。さらに、この反応は、経済的に理にかなった様式で実施可能なことが見出された。
【0024】
本発明による方法および/または本発明で使用される生体触媒は、比較的低い酵素濃度、たとえば使用する前駆体を基準にして10重量%未満を実施に使用する必要とするため、および/または当該生体触媒を何度も、たとえば少なくとも10回、再使用できるため、経済的に価値のある方法でアミド化反応を実施できるという利点を有する。したがって、本発明による方法のさらなる利点は、クロマトグラフィまたは分別結晶等の精巧な完成反応なしで、高純度、たとえば、有効含量>90%およびN,O−ジアシル化生成物の比率5%未満の生成物を調製することができることである。本発明の方法の特有の利点は、カルボン酸成分として、容易に入手可能なアルキルカルボキシレート(例えば、メチルエステル)であって任意の望ましくない二次反応を引き起こさないものを使用することを含む。
【0025】
以下の図面は実施例の一部である。
【図面の簡単な説明】
【0026】
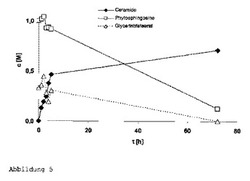
【図1】フィトスフィンゴシンと3.4倍過剰のアシルドナーとの酵素反応の経過。
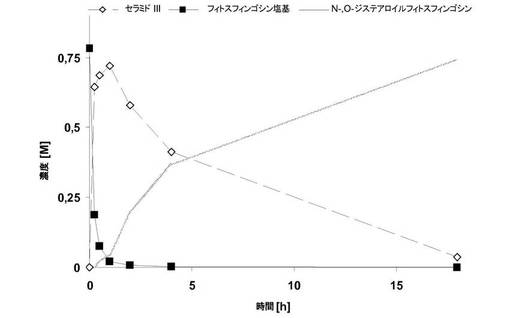
【図2】フィトスフィンゴシンとステアリン酸メチルとの酵素反応の経過の比較。三角:N−ステアロイルフィトスフィンゴシン追加あり、四角:N−ステアロイルフィトスフィンゴシン追加なし。
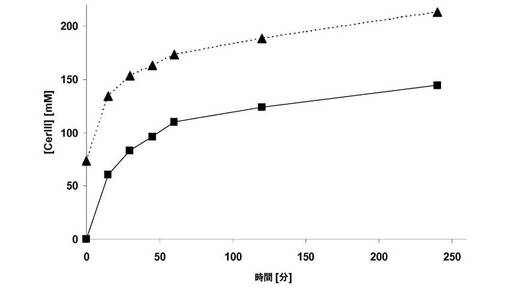
【図3】生体触媒のリサイクル:関連サイクルの初期活性は黒色で再現され、24時間後の収率は斜線で再現されている。
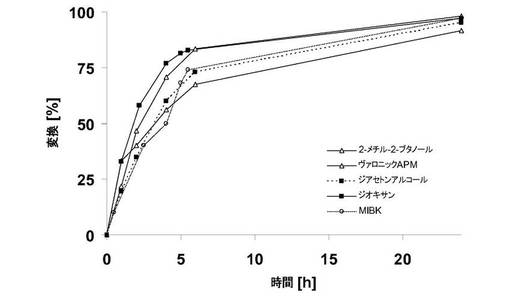
【図4】様々な溶剤中での、フィトスフィンゴシンとステアリン酸メチルとの酵素反応の時間変換曲線。
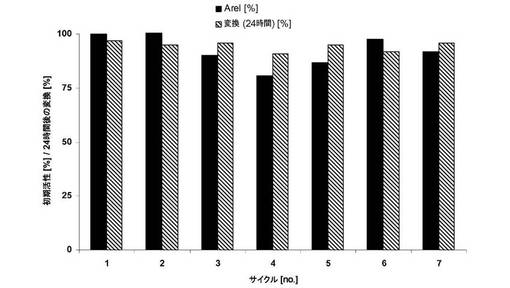
【図5】トリステアリンをアシルドナーとして使用する、酵素的セラミドIII合成の経過。
【発明を実施するための形態】
【0027】
本発明はしたがって、一般式I
【化4】
で示されるスフィンゴ脂質の生体触媒的調製方法であって、
一般式II
【化5】
で表されるリゾスフィンゴ脂質と、
一般式III
【化6】
で表されるカルボキシルエステル
[式中、R1は、場合により1つもしくは複数の多重結合および/または芳香環もしくは芳香族複素環を含んでもよく、場合により酸素原子またはエステル官能基もしくはアミド官能基が割り込んでいてもよく、また場合によりアルキル基、ヒドロキシ基、ケト基またはアミン基から選択される少なくとも1つのさらなる基で置換されていてもよい、2〜55個の炭素原子を有する直鎖または分岐したアルキル鎖を表し、好ましくは、場合により1つもしくは複数の多重結合および/または芳香環もしくは芳香族複素環を含んでもよく、場合により酸素原子またはエステル官能基もしくはアミド官能基が割り込んでいてもよく、また場合によりアルキル基、ヒドロキシ基、ケト基またはアミン基から選択される少なくとも1つのさらなる基で置換されていてもよい−CH2−Y−CH3(式中、Yは炭素−炭素結合、あるいは、1〜53個の炭素原子を有する直鎖または分岐したアルキレン鎖である)を表し、
R2は、H、ホスホコリン、セリン、エタノールアミンまたは糖を表し、好ましくは糖またはHを表し、最も好ましくはHを表し、
Xは、CH=CH、CH2−CH2またはCH2−HCOHを表し、好ましくはCH2−HCOHを表し、
R3は、少なくとも1つの基−OR4(式中、R4は、独立して、
H
および
−C(O)R1
からなる群から選択される、同一または非同一の基である)で置換されていてもよい、1〜12個の炭素原子を有する直鎖または分岐したアルキル基を表す。]
を反応させることによる生体触媒的調製方法に関し、使用される生体触媒が以下の:
菌界の生物から単離することができる酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素、および
菌界の生物から単離することができる酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素と、アミノ酸レベルで少なくとも60%、好ましくは少なくとも80%、より好ましくは少なくとも90%、特に好ましくは少なくとも95%、98%または99%相同である、酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素
を含む群から選択される酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素を少なくとも1つ含むことを特徴とする。
【0028】
「アミノ酸レベルでの相同性」は、本発明の文脈で、以下、既知の方法を用いて決定することができる「アミノ酸同一性」を意味する。一般に、特定の要求を考慮に入れたアルゴリズムを用いた特別のコンピュータープログラムを使用する。同一性を決定する好ましい方法は、まず第一に比較すべき配列間の最大一致を生じさせる。同一性を決定するためのコンピュータープログラムとしては、
−GAP (Deveroy, J. et al., Nucleic Acid Research 12 (1984), page 387, Genetics Computer Group University of Wisconsin, Medicine (WI)、および
−BLASTP、BLASTNおよびFASTA (Altschul, S. et al., Journal of Molecular Biology 215 (1990), pages 403-410を含むGCGプログラムパッケージなどがあるが、この限りではない。このBLASTプログラムは、National Centre For Biotechnology Information(NCBI)およびさらなるソース(BLAST handbook, Altschul S. et al., NCBI NLM NIH Bethesda ND 22894; Altschul S. et al.、上記)から入手できる。
【0029】
当業者は、2つのヌクレオチド間またはアミノ酸配列間の類似性または同一性を計算するために、様々なコンピュータープログラムを利用できることを承知している。したがって、2つのアミノ酸配列間のパーセンテージ同一性は、たとえばGCGソフトウェアパッケージ(http://www.gcg.comを介して得られる)のGAPプログラムに組み込まれているNeedleman and Wunsch (J. Mol. Biol. (48): 444-453 (1970))アルゴリズムによって、具体的には、ブロッサム(Blossom)62マトリックスまたはPAM250マトリックスのいずれか、ギャップウェイト16、14、12、10、8、6、または4および長さウェイト1、2、3、4、5、または6を使用して、決定することができる。異なるパラメータの使用は、僅かに異なる結果につながるであろうが、2つのアミノ酸配列間のパーセンテージ同一性は、全体として著しく異ならないであろうことを、当業者は理解するであろう。通常は、プリセット(ギャップウェイト:12、長さウェイト:1)を適用し、ブロッサム62マトリックスが使用される。
【0030】
上述のアルゴリズムによる60%の同一性は、本発明との関連で、60%相同性を意味する。同じことが、より高い同一性にも当てはまる。
【0031】
本発明による方法において、一般式IIIのカルボキシルエステルは、特に好ましくは、カルボン酸と、アルコールR3OHの−R3基とのエステルであり、ここで、カルボン酸は、6〜30個の炭素原子、特に8〜22個の炭素原子を有する、天然の植物油または動物油をベースとする天然の脂肪酸の群から選択される。天然の脂肪酸は、分岐しておらず、また偶数の炭素原子から成る。どの二重結合も、シス立体配置である。例は:カプロン酸、カプリル酸、カプリン酸、ラウリン酸、ミリスチン酸、パルミチン酸、パルミトレイン酸、イソステアリン酸、ステアリン酸、12−ヒドロキシステアリン酸、ジヒドロキシステアリン酸、オレイン酸、リノール酸、リノレン酸、ペトロセリン酸、エライジン酸、アラキン酸、ベヘン酸、エルカ酸、ガドレイン酸、エイコサペンタエン酸、ドコサヘキサエン酸、アラキドン酸であり、これらのエステル生成物が最も特に好ましい。
【0032】
さらに、本発明による方法において、一般式IIIのカルボキシルエステルは、好ましくは、カルボン酸と、アルコールR3OHの−R3基とのエステルであり、ここで、カルボン酸は、多価不飽和脂肪酸のヒドロキシル化誘導体の群から選択される。そのような脂肪酸は、例えば、9−または13−ヒドロキシオクタデカジエン酸、15−ヒドロキシエイコサテトラエン酸および一連のいわゆるα−ヒドロキシ酸である。同様にこの関連で、カルボン酸は、特に好ましくは、ヒドロキシ−官能化酸の重縮合生成物(たとえばポリ−12−ヒドロキシステアリン酸またはポリリシノール酸)である。
【0033】
同様にこの関連で、カルボン酸は、特に好ましくは、芳香族置換基を含む、合成または天然に存在するカルボン酸であり、たとえば、安息香酸、桂皮酸、フェルラ酸、プロトカテク酸、没食子酸、バニリン酸、シリンガ酸、イソフェルラ酸、シナピン酸、カフェイン酸、ゲニチン酸(genitic acid)、サリチル酸、サリチル酸(salicyuric acid)またはニコチン酸である。
【0034】
本発明による方法において、R3は、好ましくは、1〜4個の炭素原子を有する非置換アルキル基であり、好ましくは、R3は、メチル、エチル、ビニル、プロピル、イソプロピル、n−ブチル、sec−ブチルおよびtert−ブチル基を含む群から選択される。
【0035】
本発明による方法のさらなる態様において、一般式IIIのカルボキシルエステルは、ポリオールと、少なくとも1つの酸R1COOHとの完全または部分エステルである。ポリオールの完全または部分エステルとしての使用に好ましいものは、グリコール、グリセリル、1,2−ペンタンジイル、1,3−ペンタンジイル、1,2−ブタジイル、1,3−ブタジイル、1,4−ブタジイルエステルならびにそれらの異性体および不飽和類似体である。
【0036】
R1COOHのエステル化を介してR3基を決定する上述の出発物質は、純物質としてまたは混合物で存在することができ、したがって、一般式IIIの構造は、場合によっては混合物を示すことがある。
【0037】
本発明の方法は、異なるR1を有する一般式IIIの混合物の使用によって、一般式Iのスフィンゴ脂質の混合物の生体触媒的調製に使用することができる。
【0038】
同様に、本発明の方法は、異なるR3を有する一般式IIIのカルボキシルエステルの混合物を使用することができる。
【0039】
本発明によれば、たとえばP. Lersch, U. Schick, Spec. Chem. Mag., 2003, 23(6), 30-31に記載の既報のフィトスフィンゴシンの場合に確立された発酵方法の結果として得られるもののような、リゾスフィンゴ脂質の酸付加生成物(酸付加塩ともよばれる)を使用することも可能である。好ましく使用されるリゾスフィンゴ脂質の酸付加生成物は、カルボン酸カルボキシレート、硫酸塩、リン酸塩、硝酸塩、炭酸塩、水酸化物またはハロゲン化物、特に好ましくは、リゾスフィンゴ脂質の塩化物および硫酸塩である。
【0040】
本発明による方法でリゾスフィンゴ脂質の酸付加生成物を使用するとき、リゾスフィンゴ脂質は、酵素反応の前に、リゾスフィンゴ脂質の酸付加生成物の脱プロトン化によって調製されることが好ましい。
【0041】
こうして得られるリゾスフィンゴ脂質を、本明細書において以降、リゾスフィンゴ脂質塩基とも呼ぶ。
【0042】
脱プロトン処理は、従来の有機溶剤中、リゾスフィンゴ脂質の酸付加生成物の溶液または懸濁液で行うことができる。使用できる溶剤の例は:パラフィン、一価または多価アルコール(たとえば:メタノール、エタノール、イソプロパノール、プロパノール、ブタノール、ペンタノール、ヘキサノール、シクロヘキサノール、メチルシクロヘキサノール、2−ブチル−1−オクタノールおよびそれらの異性体、エチレングリコール、グリセロール、ジアセトンアルコール、イソブタノール、等々)、エーテル(ジエチルエーテル、tert−ブチルメチルエーテル、テトラヒドロフラン、ジオキサン、ポリエトキシレート、ポリプロポキシレートおよびコポリマー等々)、ケトン(アセトン、メチルイソブチルケトン、メチルエチルケトン、等々)、エステル(クエン酸トリエチル、クエン酸トリブチル、イソ酪酸イソブチル、酢酸イソブチル、イソノナン酸イソノニル、酢酸2−エチルヘキシル、酢酸シクロヘキシル、酢酸4−tert−ブチルシクロヘキシル)である。メチルイソブチルケトンまたは2−メチル−2−ブタノール等の、毒物学的に許容できる溶剤が好ましい。
【0043】
脱プロトン化ステップは、溶剤が液体状態である反応温度で実施することができる。好ましくは、−80℃〜+150℃の間、特に好ましくは0℃〜120℃、極めて特に好ましくは+25℃〜100℃の間の反応温度で実施される。
【0044】
このようにして得られるリゾスフィンゴ脂質塩基の溶液は、それ自身、酵素反応に使用することもでき、予め濾過することもできる。
【0045】
本発明のさらなる実施態様では、当業者によく知られている方法(たとえば蒸留、またはリゾスフィンゴ脂質塩基の沈殿/結晶化とその後の濾過による除去)で、脱プロトン化ステップの溶剤を除去することができる。このリゾスフィンゴ脂質塩基は場合により、たとえば沈殿または結晶化により、あるいは蒸留で溶剤を除去することにより、酵素反応の前に、単離してもよい。脱プロトン処理につながる塩が除去される濾過ステップは、リゾスフィンゴ脂質の酸付加生成物の脱プロトン化と生体触媒的調製との間に行われることが好ましい。
【0046】
代替法として、生体触媒作用中に、リゾスフィンゴ脂質の酸付加生成物の脱プロトン化によって、リゾスフィンゴ脂質塩基を調製することができる。
【0047】
活性化と関連した脱プロトン化は、塩基を使用することにより、好ましくは有機塩基または無機塩基を用いて、行うことができる。無機塩基として好ましく使用されるものは、水酸化物、炭酸塩、金属水素化物(たとえば:水素化リチウムアルミニウム、水素化カルシウム、水素化ナトリウム等々)、イオン交換体(たとえば、陽イオン交換体または陰イオン交換体等)であり、有機塩基としては、金属オルガニル(たとえば、ブチルリチウム等)、アルコラート、アミドおよびそれらの金属塩、たとえば、リチウムジイソプロピルアミド等である。
【0048】
驚くべきことに、酵素反応で特に容易に反応させることができる特に高品質のリゾスフィンゴ脂質塩基は、特に、酸付加塩との反応で正式に水を少しも生成しない塩基によって提供されることが分かった。したがって、特に好ましい塩基は、その反応で、水を全く遊離しないものである。それらは、プロトン化後に水を少しも遊離しないものである。アルカリ金属アルコラートは、好ましくは塩基として使用される。これは、たとえばアルコール性溶液中に存在してもよい。ナトリウムメトキシドおよびカリウムメトキシドは特に好ましい。これらは同様に、有機溶剤中の溶液として使用することができる。
【0049】
反応水を回避するための、したがって同様に本発明による方法で好ましい、さらなる代替法は、アルカリ金属水酸化物および/またはアルカリ土金属水酸化物を塩基として使用する際に、その反応で生成される水を結合するために、水結合性の塩、好ましくは水結合性無機塩、たとえば、Na2SO4等を使用することである。好ましくは、Na2SO4が使用される。
【0050】
本発明によれば、本方法におけるリゾスフィンゴ脂質の酸付加生成物と塩基との間のモル比は、10:1〜0.05:1、好ましくは3:1〜0.2:1、特に好ましくは1.4:1〜0.6:1の範囲内であり、極めて特に好ましくは、リゾスフィンゴ脂質の酸付加生成物と塩基との間のモル比は、等モルである。
【0051】
本発明の特定の実施態様では、リゾスフィンゴ脂質塩基を調製するために、存在する任意の水が脱プロトン化混合物から除去される。このために使用される除水方法は、たとえば、物理的方法(たとえば蒸留、膜方法、抽出または吸着(たとえば分子篩))あるいは化学的方法(たとえば:金属水素化物および金属酸化物との反応、または、無水物、エステル等の他の反応物質との反応)等である。
【0052】
冒頭に既述の通り、国際公開第94/26919号パンフレット(Gist-Brocades)に記載の先行技術を考慮すると、真菌リパーゼを、本発明による方法で効率よい生体触媒として使用できることは、全く驚きである。
【0053】
本発明による方法における生体触媒としては、とくに、菌界の生物から単離することができる酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素、および/またはアミノ酸レベルで、菌界の生物から単離できるものと少なくとも60%、好ましくは少なくとも80%、より好ましくは少なくとも90%、特に好ましくは少なくとも95%、98%または99%相同である、カルボキシルエステル加水分解酵素があり、その生物は、Aspergillus属、Bipolaris属、Candida属、Fusarium属、Geotrichum属、Humicola属、Microsporum属、Mucor属、Pichia属、Thermomyces属、Penicillium属、Rhizopus属、Rhizomucor属、Microsporum属、Mucor属、Nocardia属、Saccharomyces属、Streptomyces属、Thermomyces属、Trichosporon属、Zygosaccharomyces属の群から選択される。
【0054】
典型的な代表は、たとえば:
Aspergillus caesiellus、A. candidus、A. carbonarius、A. carneus、A. chevalieri、A. clavatus、A. costaricaensis、A. cretensis、A. deflectus、A. flaviceps、A. flavus、A. flocculatosus、A. fumigatus、A. glaucus、A. ibericus、A. lacticoffeatus、A. lentulus、A. neobridgeri、A. nidulans、A. niger、A. niveus、A. ochraceus、A. oryzae、A. parasiticus、A. penicilloides、A. piperis、A. pseudoelegans、A. pseudofisheri、A. restrictus、A. roseoglobulus、A. sclerotioniger、A. sojae、A. steynii、A. sydowi、A. terreus、A. udagawae、A. ustus、A. versicolor、A. westerdijkiae、Bipolaris australiensis、B. hawaiiensis、B. picifera、Candida antarctica、C. cylindracea、C. lipolytica、C. rugosa、C. utilis、C. robusta、C. kefyr、C. albicans、C. glabrata、C. krusei、C. lusitaniae、C. parapsilosis、C. tropicalis、Fusarium chlamydosporum、F. coeruleum、F. dimerum、F. incarnatum、F. moniliforme F. oxysporum、F. proliferatum、F. sacchari、F. semitectum、F. solani、F. sporotrichoides、F. sub glutinans、F. tabacinum、F. verticillioides、Geotrichum candidum、G.klebahnii、Humicola fuscoatra var. Fuscoatra、H.a grisea var. Grisea、H. grisea var. Thermoidea、H. insolens、Microsporum amazonicum、M. audouinii、M. audouinii var. Langeronii、M. audouinii var. Rivalierii、M. boullardii、M. canis、M. canis var. distortum、M. cookei、M. equinum、M. ferrugineum、M. fulvum、M. gallinae、M. gypseum、M. nanum、M. persicolor、M. praecox、M. racemosum、M. vanbreuseghemii Mucor javanicus、M. pusillus、M. plumbeus、M. racemosus、M. hiemalis、Nocardia asteroides、N. brasiliensis、N. otitidiscaviarum、Penicillium aurantiogriseum、P. brevicompactum、P. chrysogenum、P. camemberti、P. digitatum、P. citrinum、P. commune、P. corylophilum、P. crustosum、P. cyclopium、P. expansum、P. funiculosum、P. glabrum、P. glaucum、P. griseofulvum、P. italicum、P. marneffei、P. nalgiovense、P. nordicum、P. notatum、P. palitans、P. purpurrescens、P. purpurogenum、P. olsonii、P. oryzae、P. roqueforti、P. variabile、P. viridicatum、P. verrucosum、Pichia acaciae、P. amylophilia、P. ciferrii、P. pastoris、P. alni、P. americana、P. amethionina、P. angophorae、P. angusta、P. anomala、P. antillensis、P. barkeri、P. besseyi、P. bimundalis、P. bispora、P. bovis、P. cactophila、P. canadensis、P. capsulata、P. caribaea、P. castillae、P. chambardii、P. delftensis、P. deserticola、P. dryadoides、P. euphorbiae、P. euphorbiiphila、P. fabianii、P. faecalis、P. farinosa、P. fermentans、P. finlandica、P. fluxuum、P. galaeiformis、P. glucozyma、P. guilliermondii、P. hampshirensis、P. haplophila、P. heedii、P. heimii、P. henricii、P. holstii、P. inositovora、P. jadinii、P. japonica、P. kluyveri、P. kodamae、P. lynferdii、P. maganishii、P. media、P. membranifaciens、P. methanolica、P. methylivoria、P. mexicana、P. meyerae、P. minuta、P. mississippiensis、P. nakasei、P. nakazawae、P. norvegensis、P. oezlemii、P. ofunaensis、P. ohmeri、P. onychis、P. opuntiae、P. petersonii、P. philodendri、P. philogaea、P. pijperi、P. pini、P. populi、P. pseudocactophila、P. quercuum、P. rabaulensis、P. rhodanensis、P. salicaria、P. scolyti、P. segobiensis、P. silvicola、P. spartinae、P. stipitis、P. strasburgensis、P. subpelliculosa、P. sydowiorum、P. tannicola、P. thermotolerans、P. toletana、P. trehalophila、P. triangularis、P. veronae、P. wickerhamii、P. xylosa、Thermomyces lanuginosa、Rhizopus arrhizus、R. delemar、R. niveus、R. oryzae、R. azygosporus、R. microsporus、R. schipperae、R. stolonifer、Rhizomucor miehei、Saccharomyces barnettii、S. bayanus、S. castellii、S. cerevisiae、S. dairenensis、S. exiguus、S. paradoxus、S. pastorianus、S. rosinii、S. servazii、S. spencoerorum、S. transvaalensis、S. unisporus、S. bailii、Streptomyces spp.、Trichosporon asahii、T. asteroides、T. beigelii、T. coremiiforme、T. cutaneum、T. faecale、T. gracile、T. inkin、T. mucoides、T. ovoides、T. pullulans、Zygosaccharomyces bisporus、Z. cidri、Z. fermantati、Z. florentinus、Z. mellis、Z. microellipsoides、Z. mrakii、Z. rouxiiである。
【0055】
本発明による方法で特に好ましく使用されるカルボキシルエステル加水分解酵素は、Thermomyces lanuginosus(受入番号O59952)由来のリパーゼ、Candida antarctica(受入番号P41365)由来のリパーゼAおよびBならびにMucor miehei(受入番号P19515)由来のリパーゼ、Rhizomucor javanicus(受入番号S32492)由来のリパーゼ、Rhizopus oryzae(受入番号P61872)由来のリパーゼ、Candida rugosa(受入番号P20261、P32946、P32947、P3294およびP32949)由来のリパーゼ、Rhizopus niveus(受入番号P61871)由来のリパーゼ、Penicillium camemberti(受入番号P25234)由来のリパーゼ、Aspergillus niger(ABG73613、ABG73614およびABG37906)由来のリパーゼおよびPenicillium cyclopium(受入番号P61869)由来のリパーゼの群から選択される酵素、ならびに、各場合において、アミノ酸レベルで、少なくとも60%、好ましくは少なくとも80%、より好ましくは少なくとも90%、特に好ましくは少なくとも95%、98%または99%相同のものである。相同性に関しては、前述の定義を参照することが可能である。
【0056】
市販例および同様に本発明による方法で好ましく使用されるカルボキシルエステル加水分解酵素は、商品Lipozyme TL IM、Novozym 435、Lipozyme IM 20、Lipase SP382、Lipase SP525、Lipase SP523、(全てNovozymes A/S, Bagsvaerd, Denmarkの商品)、Chirazyme L2、Chirazyme L5、Chirazyme L8、Chirazyme L9(全てRoche Molecular Biochemicals, Mannheim, Germanyの商品)、およびLipase M “Amano”、Lipase F-AP 15 “Amano”、Lipase AY “Amano”、Lipase N “Amano”、Lipase R “Amano”、Lipase A “Amano”、Lipase D “Amano”、Lipase G “Amano”(全てAmano, Japanの商品)である。
【0057】
本生体触媒は、無水物または部分的水和物の形で好ましく使用される。本生体触媒は、固定化されるかまたは凍結乾燥物として使用することができる。固定化のために使用することができる担体は、不活性な有機担体または無機担体である。固定化型酵素に好ましく使用されるかまたは存在する不活性な担体は、粒子の少なくとも90%が10〜5000μm、好ましくは50〜2000μmの粒径を有する粒径分布を有する粒状担体である。特に使用することができる有機担体は、ポリアクリレート、ポリメタクリレート、ポリビニルスチレン、スチレン−ジビニルベンゼンコポリマー、ポリプロピレン、ポリエチレン、ポリエチレンテレフタレート、PTFEおよび/または他のポリマーを含むか、またはこれらから成るものである。固定化すべき酵素に応じて、特に酸性イオン交換樹脂または塩基性イオン交換樹脂、たとえばDuolite A568、Duolite XAD 761、Duolite XAD 1180、Duolite XAD 7HP、Amberlite IR 120、Amberlite IR 400、Amberlite CG 50、Amberlyst 15(全てRohm and Haasの製品)またはLewatit CNP 105およびLewatit VP OC 1600(Lanxess, Leverkusen, Germanyの製品)を、担体材料として使用することができる。使用することができる無機担体は、最新技術で周知の酸化物(oxidic)担体および/またはセラミック担体である。特に使用できる無機担体は、たとえばセライト、沸石、シリカ、細孔制御ガラス(CPG)またはたとえば、L. Cao, “Carrier-bound Immobilized Enzymes: Principles, Application and Design”, Wiley-VCH: 2005, Weinheim, Germanyに記載されているもののような他の担体である。固定化型酵素に存在する不活性な担体または固定化型酵素の製造に使用される不活性な担体は、特に好ましくはポリビニルスチレン、ポリメタクリレートまたはポリアクリレートから成る。
【0058】
本発明によれば、共有結合的または非共有結合的に、好ましくは非共有結合的に、粒子上に固定化することが可能である。非共有結合的固定化の場合、たとえば酵素水溶液(たとえば無機塩または洗浄剤等のさらなる成分を含んでもよい)を用いて、担体をインキュベートするかまたは含浸させる。このインキュベーション/含浸は、たとえば0℃〜50℃、好ましくは0℃〜40℃の温度で、実施することができる。このインキュベーション/含浸は、数分から数時間までの期間にわたって好ましく行われる。インキュベーションの進行は、従来の蛋白質測定方法で溶液中の酵素濃度を測定することにより追跡することができる。所望の程度の固定化に到達した後、好ましくは担体を水洗し、必要に応じて乾燥させる。
【0059】
好適な生体触媒を含む全細胞を、静止細胞としてまたは乾燥型としてのいずれかで、好ましくは先行技術の方法で透過処理して、使用することも可能である。
【0060】
本発明によれば、使用される基質に対して、0.01〜300%(w/w)の量比で、生体触媒を使用することができる。それぞれの反応条件で(特に、基質、それらの濃度、溶剤および反応温度に応じて)、好ましい質量比は、生体触媒の比活性から得られる。こうしたパラメータに応じて、生体触媒の好ましい量は、1〜96時間、好ましくは8〜24時間の期間内に、反応物質の完全変換を可能にする量である。生体触媒の量は、前駆体の全質量を基準にして、1〜100%(w/w)および極めて特に好ましくは1〜50%(w/w)、特に1〜20%(w/w)の範囲で、好ましく使用される。
【0061】
ある特定の実施態様では、この生体触媒は回収される(recovered)。
【0062】
本発明によれば、酵素反応の開始時に、反応物質は、1:10〜10:1というリゾスフィンゴ脂質とカルボキシルエステルとのモル比で存在することが可能である。この関連で、用語「モル比」は、リゾスフィンゴ脂質アミンとアシルドナーカルボキシレート基とのモル比に関する。1:3〜3:1の量比が、好ましく使用される。1:1.4〜1.4:1の量比、とりわけ等モル量比が、特に好ましく使用される。
【0063】
反応物質は、さらなる溶解剤を含まずに溶融状態で存在してもよく、有機溶剤中に溶解または懸濁されていてもよい。反応は、好ましくは有機溶剤中で実施される。特に適した溶剤は、20〜130℃の範囲内の温度で、反応物質を高濃度で溶解することができるものである。特に適していると証明されている例は、たとえば、メチルイソブチルケトンまたはシクロヘキサノン等のケトン、2−ブチル−1−オクタノール、メチルシクロヘキサノール、1−メトキシ−2−プロパノール、2,3−ブタンジオール2−オクタノール、ジアセトンアルコール、2−メチル−2−ブタノール等の立体障害性二級アルコール、酢酸4−tert−ブチルシクロヘキシル、酢酸2−エチルヘキシル、酢酸シクロヘキシル等の立体障害性エステルならびに1,4−ジオキサン、テトラヒドロフランおよびVaronic APM(Evonik Goldschmidt GmbH、Essen、Germany)等のエーテルであるが、こうした溶剤に限定されない。不適当であると証明されている溶剤は、とりわけ生体触媒によるかなり大きい活性で変換されるエステルである。フィトスフィンゴシンを前駆体として用いて、酪酸エチルを使用した反応で、たとえば所望の生成物に加えて、かなりの量の、N−ブチリルフィトスフィンゴシンと使用したアシルドナー(たとえばステアリン酸メチル)とのエステル交換生成物を検出することができた。同じことが、酢酸ブチルの使用にも当てはまる(N−アセチルフィトスフィンゴシンとアシルドナーブチルエステルの形成)。
【0064】
本反応は、カールフィッシャー(Karl Fischer)法で検出される含水量が0.100M以下、0.010M以下および特に好ましくは最高でも0.005Mとして定義された無水条件で好ましく実施される。
【0065】
高基質濃度は、反応速度に特に有利である。変換率が高いと、生成物が高濃度になるが、驚くべきことに、変換率が高いとき、酵素活性に対して悪影響はない。酵素反応では生成物阻害が通常観察されるため、このことは当業者には驚くべきことである。加えて、驚くべきことに、非常に高い変換乃至定量的変換を達成するために、反応生成物(セラミドまたはアルコール)の1つを反応混合物から除去することが、絶対的に必要というわけではないことが分かった。当業者によく知られているLe Chatelierの原理に従えば、このことが本平衡反応に関して予想されたであろう。僅か67〜78%という先行技術(国際公開第94/26919号パンフレット、Gist−Brocades)の反応率が、その必要を予想させることになった。しかし、本発明によれば、より低い基質濃度でも、効率よく反応することが可能である。
【0066】
本発明の文脈で好ましい反応開始時の反応物質濃度は、各反応物質について、0.01M〜3Mの範囲内、特に0.2M〜2Mの範囲内、特に好ましくは0.5M〜1.0Mの範囲内である。
【0067】
反応温度に関して、特に適した温度は、基質が溶剤に均質に溶解する温度であることは分かっていた。これに関連して、20〜130℃の範囲内の反応温度が有利であった。とりわけ高温では、変色のため、反応混合物の注意深い脱気が必要である。しかし、驚くべきことに100℃以下の反応温度では、この種の用心は無用であることが分かった。したがって、反応温度は、好ましくは40〜90℃の範囲内であり、極めて特に好ましくは、70〜85℃の範囲内である。
【0068】
反応は好ましくは、1bar未満、好ましくは0.5bar未満、特に好ましくは0.05bar未満の圧力で実施される。
【0069】
この反応は、バッチ方法(撹拌容器)で実施することもでき、または固定床反応器で連続的または半連続的に実施することもできる。高速の生体触媒失活は、とりわけバッチ方法で推定されるものであった。撹拌によって生じる機械的ストレスによる生体触媒の機械的破壊、ならびに反応物質および溶剤に起因する失活作用と対をなす生体触媒の熱失活について留意すべきである。このような作用は当業者に周知であり、また好適な救済策が文献に開示されている。したがって、たとえば生体触媒の機械的疲弊が撹拌方法で起きるのであれば、固定床を使用することにより、これを回避することが可能である。さらなる反応器は、当業者に周知であり、同様に、機械的酵素失活を回避するのに好適である。
【0070】
さらに、エステルをアシルドナーとして使用するとき、遊離したアルコールによって生体触媒が化学的に失活されることが予想される(たとえば:Y. Yoshida et al., J. Biosci. Bioeng., 2006, 102(1), 66-68)。このような失活を回避する方法は当業者に周知であり、たとえば、蒸留による反応混合物からのアルコール除去や膜方法の使用などがある。しかし、驚くべきことに、上述の方法を使用しなくても、活性を著しく損失せずに生体触媒を何度も再使用できることが分かった。
【0071】
結果として得られる生成物が、スフィンゴ脂質および高比率(全生成物の最高19%)の対応するN,O−ジアシル化生成物の組成物であることは、先行技術に記載されている全ての方法に共通である。こうした組成物は、たとえば、N,O−ジアシル化生成物の比率が5%以上であるため、化粧用組成物に全く使用できない。さらに、本発明による方法では、驚くべきことに、二級アミン官能基のアミド化は、アルコール官能基の1つのエステル化より紛れもなく好ましいことが分かった。反対の挙動、特に、一級、すなわち立体的に容易に近づける、アルコール官能基の選好が予想されたであろう。先行技術(国際公開第94/26919号パンフレット、Gist−Brocades)は、類似した状況について記している:細菌リパーゼを使用し、アシルドナーがフィトスフィンゴシンより60%モル過剰であっても、その状況で得られた反応生成物は、かなりの比率のN,O−ジアシル化生成物(モノジアシル化生成物とジアシル化生成物の比率>1:5)を含んでいた。
【0072】
それとは反対に、リゾスフィンゴ脂質より3.6倍過剰のアシルドナーを用いても、リゾスフィンゴ脂質が反応混合物中に存在する限り、顕著なジアシル化がないことを、本発明による方法で示すことが可能であった。リゾスフィンゴ脂質が完全にスフィンゴ脂質に変換されていたときに限って、緩徐なエステル化活性を検出することが可能である。リゾスフィンゴ脂質およびアシルドナーを化学量論的に使用したとき、上述の不要な副反応はほとんど認められなかった。
【0073】
したがって本発明は、酵素反応による粗生成物としてでさえ、特に純粋なN−アシルフィトスフィンゴシンが得られる方法に関する。式Iで表される標的化合物に関連した変換は、理論的に予想される変換の好ましくは80%超、特に好ましくは85%超、および極めて特に好ましくは90%超である。
【0074】
本発明による方法はさらに、式Iおよび0.001〜19質量%、好ましくは0.005〜19質量%、特に好ましくは0.01〜5質量%の、対応するN,O−ジアシル化生成物を含む組成物を調製するのに好適である。
【0075】
したがって、本発明は同様に、本発明による方法を使用して調製される、式Iおよび0.001〜19質量%、好ましくは0.005〜19質量%、特に好ましくは0.01〜5質量%の、対応するN,O−ジアシル化生成物を含む組成物に関する。
【0076】
本発明は同様に、本発明による方法を使用して調製されるスフィンゴ脂質に関する。
【0077】
本発明はさらに、スフィンゴ脂質を反応混合物から分離する方法に関する。スフィンゴ脂質は好都合な物理的性質を有するため、スフィンゴ脂質は結晶化/沈殿により反応溶液から容易に除去することができ、したがって、既に高い粗生成物の純度を、簡単な方法でさらに高めることができる。
【0078】
スフィンゴ脂質および本発明による組成物は、化粧用製剤および医薬製剤の製造に特に適する。したがって本発明は同様に、本発明による少なくとも1つのスフィンゴ脂質および/または本発明による1つの組成物を含む、化粧用製剤または医薬製剤に関する。
【0079】
本発明を、以下に詳述する実施例において例として説明するが、本発明は、実施例で言及されている実施態様に限定されることは意図されず、その適用範囲は完全な記述および特許請求の範囲から明白である。
【実施例】
【0080】
実施例1:PSサルフェートの脱プロトン化
フィトスフィンゴシンサルフェート(Cosmoferm,Delft,NL)100gを、55℃のメタノール600g中に懸濁させ、ナトリウムメトキシド46gと混合し、55℃で150分間撹拌する。次いで、この懸濁液を熱濾過するとき、その濾液は高濃度である。淡いベージュ色の固体(78g)をさらなる実験に使用する。
【0081】
実施例2:アミド化反応性に関する、市販のリパーゼのスクリーニング
ジオキサン(11mL)中の、フィトスフィンゴシン(実施例1から)およびステアリン酸メチル(各1M)の等モル溶液に、表1に記載の真菌リパーゼ各0.35g(5% w/w)を懸濁させ、80℃で72時間、リパーゼで振盪する。次いで、生体触媒を熱濾去し、その濾液をガスクロマトグラフィで分析する。
【0082】
【表1】
【0083】
カンジダ・アンタークチカ(Candida antarctica)由来のリパーゼBを、さらなる実験に使用した。
【0084】
実施例4:陰性対照
フィトスフィンゴシンとアシルドナーとの、可能性のあるあらゆる無触媒反応を除外するために、0.2M強度、80℃で、フィトスフィンゴシンをステアリン酸メチルに溶解し、4時間インキュベートした。ガスクロマトグラフィによるその後の分析は、有意な(定量化可能な)変換を示さなかった。
【0085】
実施例5:フィトスフィンゴシンサルフェートとの反応
フィトスフィンゴシンサルフェートを脱プロトン化する必要性を証明するために、以下の比較実験を実施した:フィトスフィンゴシンサルフェート8.18gおよびステアリン酸メチル6.72gを、80℃のジオキサン15mLに懸濁させ、Novozym 435 0.16gと混合した。撹拌しながら4時間インキュベーションした後、この反応混合物をガスクロマトグラフィで分析した。僅か31.7mMのN−ステアロイルフィトスフィンゴシンが確認されただけであった。炭酸ナトリウム2.4gを加えた後であること以外は同一条件での比較実験で、N−ステアロイルフィトスフィンゴシン368mMが確認された。
【0086】
実施例6:酵素的アシル化の位置選択性
フィトスフィンゴシンに比して超過剰のアシルドナーを得るために、フィトスフィンゴシン4.43gを120℃のステアリン酸メチル15.09gに溶解した。この溶液をNovozym 435 0.98gに溶解し(5% w/w)、標準気圧で撹拌した。定期的に試料を採取して、ガスクロマトグラフィで分析した。
【0087】
反応経過(図1)から分かる通り、不要なN,O−ジステアリルフィトスフィンゴシンの顕著な蓄積は、使用したフィトスフィンゴシンが完全に変換された時にのみ起こる。1時間後、19mM(変換>99.5%)の残余フィトスフィンゴシン濃度で、N,O−ジアシル化生成物はまだ検出されず、N,O−ジアシル化生成物の形成は、約5mM未満のフィトスフィンゴシン濃度でのみ確認される。加えて、二次反応の反応速度は、25を超える倍数(factor)で所望の反応速度を下回る。
【0088】
実施例7:生成物阻害に関する実験
あらゆる生成物阻害を除外するために、2つの並行実験を実施した。この目的のため、各ケースにおいて、フィトスフィンゴシン1.59gを、ステアリン酸メチル7.45gと共に、80℃のジオキサン25mLに溶解し、さらに、各ケースにおいてNovozyme 435 0.49gと混合した。2つの反応混合物の1つを、N−ステアロイルフィトスフィンゴシン(Cosmoferm, Delft, NL)1.3gとさらに混合した。2つの時間−変換曲線の比較が示す通り(図2)、その反応軌跡は全く同じであり、したがって任意の顕著な生成物阻害を除外することが可能である。
【0089】
実施例8:生体触媒の再使用可能性
生体触媒の再使用可能性を評価するために、以下の通りに一連の実験を実施した:フィトスフィンゴシン3.3gをステアリン酸メチル14.9gと一緒に、80℃のジオキサン50mL中に溶解し、Novo 435 0.98gを加えることによって反応を開始した。試料を定期的に採取し、ガスクロマトグラフィで分析し、初期速度および変換率をそれから算出した。24時間後、その反応バッチを熱濾過し、後に残っている酵素を、毎回、熱い(50℃)ジオキサン50mlで3度洗浄する。次いで、このようにしてリサイクルした生体触媒を、上述と同じ条件で再使用する。この手順を全6回繰り返す。
【0090】
図3から明白なように、観測可能な活性低下なしに少なくとも7度、この生体触媒を反応に使用することができる。
【0091】
実施例9:生体触媒の速度論的パラメータの測定
酵素的N−アシル化の速度論的パラメータを、以下の通りに測定した:一方の反応物濃度は変えるが、他方の濃度は一定に保った。反応は、いずれの場合にも同量の酵素を使用して、80℃のジオキサン中で実施した。試料を定期的に採取してガスクロマトグラフィで分析した;それから、それぞれの初期速度を決定し、Lineweaver-Burkに準拠して解析した。決定された結果は、フィトスフィンゴシンに関してはKM値400mMであり、またステアリン酸メチルに関してはKM値196mMであった。
【0092】
実施例10:温度が反応速度に及ぼす影響
酵素反応を様々な温度で実施し、基質は、すべての場合に等モル量で使用した。使用した酵素量は、いずれの場合にも1.4%(w/w)であった。初期速度のガスクロマトグラフ評価の結果および4時間後の変換の結果も、表2に報告する。
【0093】
【表2】
【0094】
実施例11:様々な溶媒の使用
フィトスフィンゴシンおよびステアリン酸メチルを、80℃のそれぞれの溶剤中に1Mで各々溶解し、5%(w/w)のNovozym 435と混合して撹拌した。定期的に試料を採取してガスクロマトグラフィで分析した。図4から明白なように、変換時間曲線および個々の反応の24時間後の変換も、ほぼ一致した。
【0095】
実施例12:フィトスフィンゴシンと様々な脂肪酸エステルとの酵素反応
フィトスフィンゴシンおよび表3に報告したアシルドナーを、各々1Mで80℃の溶剤に溶解し、5%(w/w)のNovozym 435と混合して撹拌した。定期的に試料を採取して、ガスクロマトグラフィで分析した。
【0096】
【表3】
【0097】
実施例13:生成物の選択的結晶化
ステアリン酸メチル(734mM)およびフィトスフィンゴシン(146mM)を、80℃の2−メチル−2−ブタノールに溶解し、5%(w/w)のNovozym 435と混合し、80℃で20時間撹拌した。次いで、酵素を熱濾去し、透き通った出発溶液を一晩、45℃に温度調節した。沈殿した固体を濾去し、濾液および濾過残渣を、ガスクロマトグラフで分析した。表4から明らかなように、基質が超過剰に存在する場合でも、分別沈殿によって、所望の生成物を選択的に濃縮することが可能になる。
【0098】
【表4】
【0099】
実施例14:アシルドナーとしてのトリステアリン
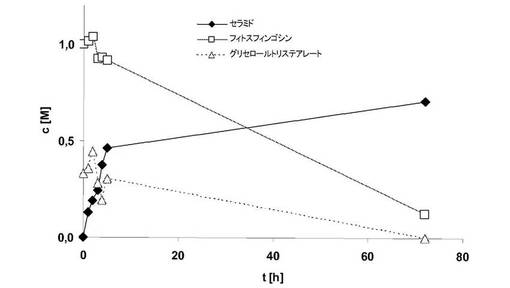
トリグリセリド(トリステアリン)をアシルドナーとして使用して、酵素反応を実施した。80℃で、ジオキサン17g中にフィトスフィンゴシン5.4gおよびトリステアリン5gを含む溶液に、Novozym 435 0.5gを加え、大気圧下で撹拌した。試料を定期的に採取し、ガスクロマトグラフィで分析した。
【0100】
図5から明らかなように、アシルドナーの完全変換は可能である。驚くべきことに、中間体部分グリセリド(グリセロールジステアレートまたはグリセロールモノステアレート)はほとんど確認されないことが、ガスクロマトグラフィによる分析から明らかになった。
【0101】
実施例15:フィトスフィンゴシンサルフェートからのN−ステアロイルフィトスフィンゴシンのワンポット合成
フィトスフィンゴシンサルフェート(Cosmoferm, Delft,NL)100gを、55℃のMIBK(メチルイソブチルケトン)100mlに懸濁させ、ナトリウムメトキシド46gと混合し、55℃で180分間、撹拌する。次いで、ステアリン酸メチル73.3gおよびNovo435 5gを加える。このようにして得た懸濁液を、80℃で36時間撹拌し、熱濾去する。透き通った濾液を、室温で4時間静置し、沈殿した透き通ったベージュ色の固体(N−ステアロイルフィトスフィンゴシン122.3g、純度>97%)を濾去する。
【0102】
実施例16:固定床反応器でのパイロットバッチ
フィトスフィンゴシンサルフェート207.8gを、NaOCH3溶液(メタノール中25%濃度)108.04gと共に55℃のMIBK 500mlに懸濁させ、続いてフィルタープレスを介して固定床反応器に熱いうちに移し、ステアリン酸メチル149.3gと混合する。この固定床は、Novozyme 435 17.8gを含む。反応器全体を、80℃に温度調節する。約25ml分−1のポンプ流量で反応混合物を固定床に流す。0.8barの圧力下、過剰のメタノールを除去する。24時間後、この反応器に窒素を通気し、反応器を空にして、この反応溶液を室温で一晩静置する。沈殿した固体を濾去する(N−ステアロイルフィトスフィンゴシン287.1g、純度98.7%)。
【技術分野】
【0001】
本発明は、スフィンゴ脂質の酵素的合成ならびに、リゾスフィンゴ脂質とカルボキシルエステル由来のスフィンゴ脂質を含む組成物に関し、また、こうしたスフィンゴ脂質または組成物を含む化粧製剤、皮膚用製剤または医薬製剤に関する。
【背景技術】
【0002】
本発明は、一般式I
【化1】
で表されるスフィンゴ脂質の生体触媒的調製方法であって、
一般式II
【化2】
で表されるリゾスフィンゴ脂質を、一般式III
【化3】
(ここで、R1、R2、R3およびXは以下に定義の通りである)
で表されるカルボキシルエステルと反応させることによる調製方法に関する。
【0003】
本スフィンゴ脂質は、化粧用および/または皮膚用有効成分として使用される。
【0004】
R2=Hである一般式Iのスフィンゴ脂質は、セラミドと言われ、皮膚(角質層)内に天然に存在する極性脂質である。外側の角質細胞では、セラミドは、細胞膜の主成分(脂質含量の40〜65%)に相当し、たとえば、透水性を調節することにより、その保護機能において中心的役割を果たす。加齢とともに、ケラチノサイトは、セラミド合成能の大部分を失い、それ故皮膚はその保護作用の一部を失い、たとえば、もはや表皮の水分喪失を完全に抑制することはできない。これは、セラミドの局所適用によって、少なくとも部分的に補うことができる(たとえば、Farwick, M. et al., Int. J. Cosm. Sci., 2007, 29(4), 319-329またはKlee, S.K. et al. Basic Clinic. Dermatol., 2007, 40, 155-165)。
【0005】
加えて、アトピー性皮膚炎の治療における、セラミドの好ましい作用が報告されている(Kerscher, M. et al., Eur. J. Dermatol., 1991, 1, 39-43; Imokawa, G. et al., J. Soc. Cosmet. Chem. 1989, 40, 500-507)。
【0006】
多くの先進工業諸国、特にドイツにおける人口統計推移を考慮すると、セラミドの需要はさらに増大すると予想される。
【0007】
先行技術では、セラミドは、活性化カルボン酸誘導体を使用して、R2=Hである一般式IIのリゾスフィンゴ脂質、たとえば、フィトスフィンゴシン(R2=HかつX=CH2−HCOHである、一般式II)等の、ショッテン・バウマン(Schotten Baumann)類似N−アシル化によって調製される。
【0008】
フィトスフィンゴシンは、一般に発酵起源である。
【0009】
活性化カルボン酸は一般に、それ自体として使用されるか、またはカルボン酸からイン・シチュで調製されるかのいずれかである、塩化カルボニルである。米国特許第6420604号(Cosmoferm, NL)は、スフィンゴシン塩基を好適なカルボニルハロゲン化物と反応させることによる、幾つかのセラミドの合成について記述している。この方法の際立った不都合は、有毒な有機塩素化合物を使用する必要性がある一方で、結果として生じる最終生成物が高塩負荷であることである。さらに、極めて反応しやすいカルボニルハロゲン化物を使用する際には、かなりの量の不要な副産物の生成を予想しなければならないことは、当業者に明白である。
【0010】
代替合成経路は、無水物によって進行する。したがって、たとえば、国際公開第93/20038号パンフレット(Gist-Brocades, NL)は、フィトスフィンゴシンのN−アシル化用の反応性カルボン酸誘導体を得るために、カルボン酸とアルキルフェニルスルホニルクロリドから、混合無水物を塩基触媒合成することを教示している。
【0011】
これらの経路全部に共通しているのは、反応性カルボン酸誘導体の使用に依存していることである。これに関連した不都合は、こうした物質の高い腐蝕性およびそれらの健康に対する危険性の両者であり、これにより特別の反応器システムおよび注意が必要となり、したがって、調製の複雑さが増大する。加えて、本生成物を局所適用できるためには、反応しやすく有毒な前駆体および副産物が本生成物から除去されていることが保証されなければならない。高塩負荷、ならびにそれに関連した生成物精製および廃棄物処理のための付加的な複雑さもさらに予想される。先行技術の方法のさらなる不都合は、低い前駆体濃度および生成物濃度しか得られないことであり;たとえば、国際公開第93/20038号パンフレット(Gist-Brocades, NL)は、反応性かつ有毒な共試薬である、塩化p−トルエンスルホニルおよびトリエチルアミンの同時使用と共に、有毒な溶剤塩化メチレン中の最大15%(w/v)の前駆体溶液を記している。加えて、本生成物の純度または収量(いずれの場合にも、80%)はいずれも満足のいくものではない。
【0012】
したがって、本発明の目的は、無害かつ産業上利用することができる様式で、先行技術の不都合の少なくとも1つを克服することができる、セラミドを得る代替方法を提供することである。
【0013】
生体触媒的に、すなわち、酵素を触媒として使用して、アミド化反応を得ることもできる。酵素の産業上の利用の概説は、たとえばLieseら(Industrial Biotransformations; Second, Completely Revised Edition, Wiley-VCH, Weinheim, 2006)に見られる。カルボン酸誘導体を合成するのに好ましい生体触媒は、加水分解酵素(E.C. 3.1.x.x)、特にリパーゼ(トリアシルグリセロール加水分解酵素、E.C. 3.1.1.3)およびエステラーゼ(E.C. 3.1.1.1)である。
【0014】
リパーゼは、細胞の代謝における自然機能に従って、エステルの加水開裂を優先的に触媒する。しかし、エステルの縮合的生成も、文献に多数記述されている。それらの代表例は、Drauz and Waldmann (Enzyme Catalysis in Organic Synthesis, A Comprehensive Handbook; Second, Completely Revised and Enlarged Edition, Vol. II, Wiley-VCH, Weinheim, 2002)、Aehle (Enzymes in Industry, Production and Applications; Second, Completely Revised Edition, Wiley-VCH, Weinheim, 2004)またはBornscheuer (Enzymes in Lipid Modification, Wiley-VCH, Weinheim, 2000)に見られる。
【0015】
リパーゼ−触媒縮合反応における求核剤としてのアミンの使用例も、文献にある。
【0016】
Y.-M. Xia et al., J Mol Catal B, 2004, 31,111-115は、たとえば、カンジダ・アンタークティカ(Candida antarctica (CALB))由来のリパーゼにより触媒される反応性一級アミンを用いたラウリン酸メチルのアミド化によるN−ラウロイル−β−アミノプロプリオニトリルの合成について記述している。記載の方法の不都合は、基質溶液の希釈(50mM未満)の制限に加え、僅か94.3%までの変換という、比較的低い変換である。さらに、示された反応には、位置選択性または化学選択性は全く必要ない。
【0017】
しかし、前駆体分子、たとえば、フィトスフィンゴシン等は、複数の反応性官能基を有する可能性があるため、本発明による反応には、このような選択性が絶対的に必要であろう。この選択性の問題は、たとえばP.Tufvesson et al.: Biotechnol.Bioeng., 2007, 97(3), 447-453により示され:エタノールアミンとカルボン酸とを用いたCALB−触媒による反応において、酵素触媒によるエステル化を回避する単純な選択的アミド化は不可能であった。前駆体の多回添加および蒸留による反応水の除去によってのみ、所望のアミドの濃縮を達成することが可能であった。
【0018】
酵素的アミド化のさらなる例で、M.Nechab et al.: JOC, 2007, 72, 6918-6923は、CALBを使用した、(R)立体配置二級アミンの立体選択的反応について記述している。生成物の高い光学純度(>99% ee)が確認されたことから、CALBの(R)立体配置アミンに対する強い優先傾向が示唆され、したがって、たとえばフィトスフィンゴシンは、S−立体配置であるため、この触媒で、アミン炭素原子に反応するかどうか疑わしい。加えて、やはり、非常に希釈された基質溶液(100mM)が、高濃度(27%w(CALB)/w(基質))の生体触媒と同時に使用され、記載の方法の産業上の魅力を著しく制限している。
【0019】
国際公開第94/26919号パンフレット(Gist-Brocades, NL)は、細菌リパーゼまたは哺乳リパーゼの使用とともに、カルボキシルエステルを使用するフィトスフィンゴシンの酵素的N−アシル化について記述している。細菌リパーゼおよび哺乳リパーゼ、特に、シュードモナス・アルカリゲネス(Pseudomonas alcaligenes)由来のリパーゼは、記載の方法で好ましく使用される。この方法の不都合は、特に、中程度の変換(最高78%)を達成するだけのために、大量の生体触媒(30〜95%w/w)を必要とすることである。加えて、かなりの量の不要なN,O−ジアシル化生成物(最高17%)が確認された。さらに、基質の希釈溶液のみが使用され(59〜93mM)、その結果として空時収量が低くなる。さらなる不都合は、基質としてフィトスフィンゴシン塩基に制限されることであり、フィトスフィンゴシンの酸付加生成物、たとえばフィトスフィンゴシンサルフェートの使用と比較して、基質価格の激増を伴う。さらに、酵母および真菌に由来するリパーゼは、触媒として不安定であることが明確に指摘されている。したがって、国際公開第94/26919号パンフレットに記述されている方法は、既存の化学的方法に代わる経済的に価値ある代替法としては除外される。
【発明の概要】
【発明が解決しようとする課題】
【0020】
このように、従来実現できなかった、アルキルカルボキシレートからのスフィンゴ脂質の生体触媒的合成の実施を可能にするために、先行技術の不都合を克服する、リゾスフィンゴ脂質を酵素的にアミド化する方法が引き続き必要である。
【0021】
したがって、本発明の目的は、先行技術の方法の不都合の1つまたは複数がない方法を提供することであった。特に、その意図は、容易に入手できるリゾスフィンゴ脂質を前駆体として、さらには高濃度で、使用することができる方法を開発することであった。
【0022】
明確に言及されていないさらなる目的は、以下の説明、実施例および特許請求の範囲の文脈から明白である。
【課題を解決するための手段】
【0023】
驚くべきことに、先行技術とは対照的に、真菌リパーゼがカルボキシルエステルとリゾスフィンゴ脂質を選択的に反応させ、高純度のスフィンゴ脂質を形成可能なことが見出された。さらに、この反応は、経済的に理にかなった様式で実施可能なことが見出された。
【0024】
本発明による方法および/または本発明で使用される生体触媒は、比較的低い酵素濃度、たとえば使用する前駆体を基準にして10重量%未満を実施に使用する必要とするため、および/または当該生体触媒を何度も、たとえば少なくとも10回、再使用できるため、経済的に価値のある方法でアミド化反応を実施できるという利点を有する。したがって、本発明による方法のさらなる利点は、クロマトグラフィまたは分別結晶等の精巧な完成反応なしで、高純度、たとえば、有効含量>90%およびN,O−ジアシル化生成物の比率5%未満の生成物を調製することができることである。本発明の方法の特有の利点は、カルボン酸成分として、容易に入手可能なアルキルカルボキシレート(例えば、メチルエステル)であって任意の望ましくない二次反応を引き起こさないものを使用することを含む。
【0025】
以下の図面は実施例の一部である。
【図面の簡単な説明】
【0026】
【図1】フィトスフィンゴシンと3.4倍過剰のアシルドナーとの酵素反応の経過。
【図2】フィトスフィンゴシンとステアリン酸メチルとの酵素反応の経過の比較。三角:N−ステアロイルフィトスフィンゴシン追加あり、四角:N−ステアロイルフィトスフィンゴシン追加なし。
【図3】生体触媒のリサイクル:関連サイクルの初期活性は黒色で再現され、24時間後の収率は斜線で再現されている。
【図4】様々な溶剤中での、フィトスフィンゴシンとステアリン酸メチルとの酵素反応の時間変換曲線。
【図5】トリステアリンをアシルドナーとして使用する、酵素的セラミドIII合成の経過。
【発明を実施するための形態】
【0027】
本発明はしたがって、一般式I
【化4】
で示されるスフィンゴ脂質の生体触媒的調製方法であって、
一般式II
【化5】
で表されるリゾスフィンゴ脂質と、
一般式III
【化6】
で表されるカルボキシルエステル
[式中、R1は、場合により1つもしくは複数の多重結合および/または芳香環もしくは芳香族複素環を含んでもよく、場合により酸素原子またはエステル官能基もしくはアミド官能基が割り込んでいてもよく、また場合によりアルキル基、ヒドロキシ基、ケト基またはアミン基から選択される少なくとも1つのさらなる基で置換されていてもよい、2〜55個の炭素原子を有する直鎖または分岐したアルキル鎖を表し、好ましくは、場合により1つもしくは複数の多重結合および/または芳香環もしくは芳香族複素環を含んでもよく、場合により酸素原子またはエステル官能基もしくはアミド官能基が割り込んでいてもよく、また場合によりアルキル基、ヒドロキシ基、ケト基またはアミン基から選択される少なくとも1つのさらなる基で置換されていてもよい−CH2−Y−CH3(式中、Yは炭素−炭素結合、あるいは、1〜53個の炭素原子を有する直鎖または分岐したアルキレン鎖である)を表し、
R2は、H、ホスホコリン、セリン、エタノールアミンまたは糖を表し、好ましくは糖またはHを表し、最も好ましくはHを表し、
Xは、CH=CH、CH2−CH2またはCH2−HCOHを表し、好ましくはCH2−HCOHを表し、
R3は、少なくとも1つの基−OR4(式中、R4は、独立して、
H
および
−C(O)R1
からなる群から選択される、同一または非同一の基である)で置換されていてもよい、1〜12個の炭素原子を有する直鎖または分岐したアルキル基を表す。]
を反応させることによる生体触媒的調製方法に関し、使用される生体触媒が以下の:
菌界の生物から単離することができる酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素、および
菌界の生物から単離することができる酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素と、アミノ酸レベルで少なくとも60%、好ましくは少なくとも80%、より好ましくは少なくとも90%、特に好ましくは少なくとも95%、98%または99%相同である、酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素
を含む群から選択される酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素を少なくとも1つ含むことを特徴とする。
【0028】
「アミノ酸レベルでの相同性」は、本発明の文脈で、以下、既知の方法を用いて決定することができる「アミノ酸同一性」を意味する。一般に、特定の要求を考慮に入れたアルゴリズムを用いた特別のコンピュータープログラムを使用する。同一性を決定する好ましい方法は、まず第一に比較すべき配列間の最大一致を生じさせる。同一性を決定するためのコンピュータープログラムとしては、
−GAP (Deveroy, J. et al., Nucleic Acid Research 12 (1984), page 387, Genetics Computer Group University of Wisconsin, Medicine (WI)、および
−BLASTP、BLASTNおよびFASTA (Altschul, S. et al., Journal of Molecular Biology 215 (1990), pages 403-410を含むGCGプログラムパッケージなどがあるが、この限りではない。このBLASTプログラムは、National Centre For Biotechnology Information(NCBI)およびさらなるソース(BLAST handbook, Altschul S. et al., NCBI NLM NIH Bethesda ND 22894; Altschul S. et al.、上記)から入手できる。
【0029】
当業者は、2つのヌクレオチド間またはアミノ酸配列間の類似性または同一性を計算するために、様々なコンピュータープログラムを利用できることを承知している。したがって、2つのアミノ酸配列間のパーセンテージ同一性は、たとえばGCGソフトウェアパッケージ(http://www.gcg.comを介して得られる)のGAPプログラムに組み込まれているNeedleman and Wunsch (J. Mol. Biol. (48): 444-453 (1970))アルゴリズムによって、具体的には、ブロッサム(Blossom)62マトリックスまたはPAM250マトリックスのいずれか、ギャップウェイト16、14、12、10、8、6、または4および長さウェイト1、2、3、4、5、または6を使用して、決定することができる。異なるパラメータの使用は、僅かに異なる結果につながるであろうが、2つのアミノ酸配列間のパーセンテージ同一性は、全体として著しく異ならないであろうことを、当業者は理解するであろう。通常は、プリセット(ギャップウェイト:12、長さウェイト:1)を適用し、ブロッサム62マトリックスが使用される。
【0030】
上述のアルゴリズムによる60%の同一性は、本発明との関連で、60%相同性を意味する。同じことが、より高い同一性にも当てはまる。
【0031】
本発明による方法において、一般式IIIのカルボキシルエステルは、特に好ましくは、カルボン酸と、アルコールR3OHの−R3基とのエステルであり、ここで、カルボン酸は、6〜30個の炭素原子、特に8〜22個の炭素原子を有する、天然の植物油または動物油をベースとする天然の脂肪酸の群から選択される。天然の脂肪酸は、分岐しておらず、また偶数の炭素原子から成る。どの二重結合も、シス立体配置である。例は:カプロン酸、カプリル酸、カプリン酸、ラウリン酸、ミリスチン酸、パルミチン酸、パルミトレイン酸、イソステアリン酸、ステアリン酸、12−ヒドロキシステアリン酸、ジヒドロキシステアリン酸、オレイン酸、リノール酸、リノレン酸、ペトロセリン酸、エライジン酸、アラキン酸、ベヘン酸、エルカ酸、ガドレイン酸、エイコサペンタエン酸、ドコサヘキサエン酸、アラキドン酸であり、これらのエステル生成物が最も特に好ましい。
【0032】
さらに、本発明による方法において、一般式IIIのカルボキシルエステルは、好ましくは、カルボン酸と、アルコールR3OHの−R3基とのエステルであり、ここで、カルボン酸は、多価不飽和脂肪酸のヒドロキシル化誘導体の群から選択される。そのような脂肪酸は、例えば、9−または13−ヒドロキシオクタデカジエン酸、15−ヒドロキシエイコサテトラエン酸および一連のいわゆるα−ヒドロキシ酸である。同様にこの関連で、カルボン酸は、特に好ましくは、ヒドロキシ−官能化酸の重縮合生成物(たとえばポリ−12−ヒドロキシステアリン酸またはポリリシノール酸)である。
【0033】
同様にこの関連で、カルボン酸は、特に好ましくは、芳香族置換基を含む、合成または天然に存在するカルボン酸であり、たとえば、安息香酸、桂皮酸、フェルラ酸、プロトカテク酸、没食子酸、バニリン酸、シリンガ酸、イソフェルラ酸、シナピン酸、カフェイン酸、ゲニチン酸(genitic acid)、サリチル酸、サリチル酸(salicyuric acid)またはニコチン酸である。
【0034】
本発明による方法において、R3は、好ましくは、1〜4個の炭素原子を有する非置換アルキル基であり、好ましくは、R3は、メチル、エチル、ビニル、プロピル、イソプロピル、n−ブチル、sec−ブチルおよびtert−ブチル基を含む群から選択される。
【0035】
本発明による方法のさらなる態様において、一般式IIIのカルボキシルエステルは、ポリオールと、少なくとも1つの酸R1COOHとの完全または部分エステルである。ポリオールの完全または部分エステルとしての使用に好ましいものは、グリコール、グリセリル、1,2−ペンタンジイル、1,3−ペンタンジイル、1,2−ブタジイル、1,3−ブタジイル、1,4−ブタジイルエステルならびにそれらの異性体および不飽和類似体である。
【0036】
R1COOHのエステル化を介してR3基を決定する上述の出発物質は、純物質としてまたは混合物で存在することができ、したがって、一般式IIIの構造は、場合によっては混合物を示すことがある。
【0037】
本発明の方法は、異なるR1を有する一般式IIIの混合物の使用によって、一般式Iのスフィンゴ脂質の混合物の生体触媒的調製に使用することができる。
【0038】
同様に、本発明の方法は、異なるR3を有する一般式IIIのカルボキシルエステルの混合物を使用することができる。
【0039】
本発明によれば、たとえばP. Lersch, U. Schick, Spec. Chem. Mag., 2003, 23(6), 30-31に記載の既報のフィトスフィンゴシンの場合に確立された発酵方法の結果として得られるもののような、リゾスフィンゴ脂質の酸付加生成物(酸付加塩ともよばれる)を使用することも可能である。好ましく使用されるリゾスフィンゴ脂質の酸付加生成物は、カルボン酸カルボキシレート、硫酸塩、リン酸塩、硝酸塩、炭酸塩、水酸化物またはハロゲン化物、特に好ましくは、リゾスフィンゴ脂質の塩化物および硫酸塩である。
【0040】
本発明による方法でリゾスフィンゴ脂質の酸付加生成物を使用するとき、リゾスフィンゴ脂質は、酵素反応の前に、リゾスフィンゴ脂質の酸付加生成物の脱プロトン化によって調製されることが好ましい。
【0041】
こうして得られるリゾスフィンゴ脂質を、本明細書において以降、リゾスフィンゴ脂質塩基とも呼ぶ。
【0042】
脱プロトン処理は、従来の有機溶剤中、リゾスフィンゴ脂質の酸付加生成物の溶液または懸濁液で行うことができる。使用できる溶剤の例は:パラフィン、一価または多価アルコール(たとえば:メタノール、エタノール、イソプロパノール、プロパノール、ブタノール、ペンタノール、ヘキサノール、シクロヘキサノール、メチルシクロヘキサノール、2−ブチル−1−オクタノールおよびそれらの異性体、エチレングリコール、グリセロール、ジアセトンアルコール、イソブタノール、等々)、エーテル(ジエチルエーテル、tert−ブチルメチルエーテル、テトラヒドロフラン、ジオキサン、ポリエトキシレート、ポリプロポキシレートおよびコポリマー等々)、ケトン(アセトン、メチルイソブチルケトン、メチルエチルケトン、等々)、エステル(クエン酸トリエチル、クエン酸トリブチル、イソ酪酸イソブチル、酢酸イソブチル、イソノナン酸イソノニル、酢酸2−エチルヘキシル、酢酸シクロヘキシル、酢酸4−tert−ブチルシクロヘキシル)である。メチルイソブチルケトンまたは2−メチル−2−ブタノール等の、毒物学的に許容できる溶剤が好ましい。
【0043】
脱プロトン化ステップは、溶剤が液体状態である反応温度で実施することができる。好ましくは、−80℃〜+150℃の間、特に好ましくは0℃〜120℃、極めて特に好ましくは+25℃〜100℃の間の反応温度で実施される。
【0044】
このようにして得られるリゾスフィンゴ脂質塩基の溶液は、それ自身、酵素反応に使用することもでき、予め濾過することもできる。
【0045】
本発明のさらなる実施態様では、当業者によく知られている方法(たとえば蒸留、またはリゾスフィンゴ脂質塩基の沈殿/結晶化とその後の濾過による除去)で、脱プロトン化ステップの溶剤を除去することができる。このリゾスフィンゴ脂質塩基は場合により、たとえば沈殿または結晶化により、あるいは蒸留で溶剤を除去することにより、酵素反応の前に、単離してもよい。脱プロトン処理につながる塩が除去される濾過ステップは、リゾスフィンゴ脂質の酸付加生成物の脱プロトン化と生体触媒的調製との間に行われることが好ましい。
【0046】
代替法として、生体触媒作用中に、リゾスフィンゴ脂質の酸付加生成物の脱プロトン化によって、リゾスフィンゴ脂質塩基を調製することができる。
【0047】
活性化と関連した脱プロトン化は、塩基を使用することにより、好ましくは有機塩基または無機塩基を用いて、行うことができる。無機塩基として好ましく使用されるものは、水酸化物、炭酸塩、金属水素化物(たとえば:水素化リチウムアルミニウム、水素化カルシウム、水素化ナトリウム等々)、イオン交換体(たとえば、陽イオン交換体または陰イオン交換体等)であり、有機塩基としては、金属オルガニル(たとえば、ブチルリチウム等)、アルコラート、アミドおよびそれらの金属塩、たとえば、リチウムジイソプロピルアミド等である。
【0048】
驚くべきことに、酵素反応で特に容易に反応させることができる特に高品質のリゾスフィンゴ脂質塩基は、特に、酸付加塩との反応で正式に水を少しも生成しない塩基によって提供されることが分かった。したがって、特に好ましい塩基は、その反応で、水を全く遊離しないものである。それらは、プロトン化後に水を少しも遊離しないものである。アルカリ金属アルコラートは、好ましくは塩基として使用される。これは、たとえばアルコール性溶液中に存在してもよい。ナトリウムメトキシドおよびカリウムメトキシドは特に好ましい。これらは同様に、有機溶剤中の溶液として使用することができる。
【0049】
反応水を回避するための、したがって同様に本発明による方法で好ましい、さらなる代替法は、アルカリ金属水酸化物および/またはアルカリ土金属水酸化物を塩基として使用する際に、その反応で生成される水を結合するために、水結合性の塩、好ましくは水結合性無機塩、たとえば、Na2SO4等を使用することである。好ましくは、Na2SO4が使用される。
【0050】
本発明によれば、本方法におけるリゾスフィンゴ脂質の酸付加生成物と塩基との間のモル比は、10:1〜0.05:1、好ましくは3:1〜0.2:1、特に好ましくは1.4:1〜0.6:1の範囲内であり、極めて特に好ましくは、リゾスフィンゴ脂質の酸付加生成物と塩基との間のモル比は、等モルである。
【0051】
本発明の特定の実施態様では、リゾスフィンゴ脂質塩基を調製するために、存在する任意の水が脱プロトン化混合物から除去される。このために使用される除水方法は、たとえば、物理的方法(たとえば蒸留、膜方法、抽出または吸着(たとえば分子篩))あるいは化学的方法(たとえば:金属水素化物および金属酸化物との反応、または、無水物、エステル等の他の反応物質との反応)等である。
【0052】
冒頭に既述の通り、国際公開第94/26919号パンフレット(Gist-Brocades)に記載の先行技術を考慮すると、真菌リパーゼを、本発明による方法で効率よい生体触媒として使用できることは、全く驚きである。
【0053】
本発明による方法における生体触媒としては、とくに、菌界の生物から単離することができる酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素、および/またはアミノ酸レベルで、菌界の生物から単離できるものと少なくとも60%、好ましくは少なくとも80%、より好ましくは少なくとも90%、特に好ましくは少なくとも95%、98%または99%相同である、カルボキシルエステル加水分解酵素があり、その生物は、Aspergillus属、Bipolaris属、Candida属、Fusarium属、Geotrichum属、Humicola属、Microsporum属、Mucor属、Pichia属、Thermomyces属、Penicillium属、Rhizopus属、Rhizomucor属、Microsporum属、Mucor属、Nocardia属、Saccharomyces属、Streptomyces属、Thermomyces属、Trichosporon属、Zygosaccharomyces属の群から選択される。
【0054】
典型的な代表は、たとえば:
Aspergillus caesiellus、A. candidus、A. carbonarius、A. carneus、A. chevalieri、A. clavatus、A. costaricaensis、A. cretensis、A. deflectus、A. flaviceps、A. flavus、A. flocculatosus、A. fumigatus、A. glaucus、A. ibericus、A. lacticoffeatus、A. lentulus、A. neobridgeri、A. nidulans、A. niger、A. niveus、A. ochraceus、A. oryzae、A. parasiticus、A. penicilloides、A. piperis、A. pseudoelegans、A. pseudofisheri、A. restrictus、A. roseoglobulus、A. sclerotioniger、A. sojae、A. steynii、A. sydowi、A. terreus、A. udagawae、A. ustus、A. versicolor、A. westerdijkiae、Bipolaris australiensis、B. hawaiiensis、B. picifera、Candida antarctica、C. cylindracea、C. lipolytica、C. rugosa、C. utilis、C. robusta、C. kefyr、C. albicans、C. glabrata、C. krusei、C. lusitaniae、C. parapsilosis、C. tropicalis、Fusarium chlamydosporum、F. coeruleum、F. dimerum、F. incarnatum、F. moniliforme F. oxysporum、F. proliferatum、F. sacchari、F. semitectum、F. solani、F. sporotrichoides、F. sub glutinans、F. tabacinum、F. verticillioides、Geotrichum candidum、G.klebahnii、Humicola fuscoatra var. Fuscoatra、H.a grisea var. Grisea、H. grisea var. Thermoidea、H. insolens、Microsporum amazonicum、M. audouinii、M. audouinii var. Langeronii、M. audouinii var. Rivalierii、M. boullardii、M. canis、M. canis var. distortum、M. cookei、M. equinum、M. ferrugineum、M. fulvum、M. gallinae、M. gypseum、M. nanum、M. persicolor、M. praecox、M. racemosum、M. vanbreuseghemii Mucor javanicus、M. pusillus、M. plumbeus、M. racemosus、M. hiemalis、Nocardia asteroides、N. brasiliensis、N. otitidiscaviarum、Penicillium aurantiogriseum、P. brevicompactum、P. chrysogenum、P. camemberti、P. digitatum、P. citrinum、P. commune、P. corylophilum、P. crustosum、P. cyclopium、P. expansum、P. funiculosum、P. glabrum、P. glaucum、P. griseofulvum、P. italicum、P. marneffei、P. nalgiovense、P. nordicum、P. notatum、P. palitans、P. purpurrescens、P. purpurogenum、P. olsonii、P. oryzae、P. roqueforti、P. variabile、P. viridicatum、P. verrucosum、Pichia acaciae、P. amylophilia、P. ciferrii、P. pastoris、P. alni、P. americana、P. amethionina、P. angophorae、P. angusta、P. anomala、P. antillensis、P. barkeri、P. besseyi、P. bimundalis、P. bispora、P. bovis、P. cactophila、P. canadensis、P. capsulata、P. caribaea、P. castillae、P. chambardii、P. delftensis、P. deserticola、P. dryadoides、P. euphorbiae、P. euphorbiiphila、P. fabianii、P. faecalis、P. farinosa、P. fermentans、P. finlandica、P. fluxuum、P. galaeiformis、P. glucozyma、P. guilliermondii、P. hampshirensis、P. haplophila、P. heedii、P. heimii、P. henricii、P. holstii、P. inositovora、P. jadinii、P. japonica、P. kluyveri、P. kodamae、P. lynferdii、P. maganishii、P. media、P. membranifaciens、P. methanolica、P. methylivoria、P. mexicana、P. meyerae、P. minuta、P. mississippiensis、P. nakasei、P. nakazawae、P. norvegensis、P. oezlemii、P. ofunaensis、P. ohmeri、P. onychis、P. opuntiae、P. petersonii、P. philodendri、P. philogaea、P. pijperi、P. pini、P. populi、P. pseudocactophila、P. quercuum、P. rabaulensis、P. rhodanensis、P. salicaria、P. scolyti、P. segobiensis、P. silvicola、P. spartinae、P. stipitis、P. strasburgensis、P. subpelliculosa、P. sydowiorum、P. tannicola、P. thermotolerans、P. toletana、P. trehalophila、P. triangularis、P. veronae、P. wickerhamii、P. xylosa、Thermomyces lanuginosa、Rhizopus arrhizus、R. delemar、R. niveus、R. oryzae、R. azygosporus、R. microsporus、R. schipperae、R. stolonifer、Rhizomucor miehei、Saccharomyces barnettii、S. bayanus、S. castellii、S. cerevisiae、S. dairenensis、S. exiguus、S. paradoxus、S. pastorianus、S. rosinii、S. servazii、S. spencoerorum、S. transvaalensis、S. unisporus、S. bailii、Streptomyces spp.、Trichosporon asahii、T. asteroides、T. beigelii、T. coremiiforme、T. cutaneum、T. faecale、T. gracile、T. inkin、T. mucoides、T. ovoides、T. pullulans、Zygosaccharomyces bisporus、Z. cidri、Z. fermantati、Z. florentinus、Z. mellis、Z. microellipsoides、Z. mrakii、Z. rouxiiである。
【0055】
本発明による方法で特に好ましく使用されるカルボキシルエステル加水分解酵素は、Thermomyces lanuginosus(受入番号O59952)由来のリパーゼ、Candida antarctica(受入番号P41365)由来のリパーゼAおよびBならびにMucor miehei(受入番号P19515)由来のリパーゼ、Rhizomucor javanicus(受入番号S32492)由来のリパーゼ、Rhizopus oryzae(受入番号P61872)由来のリパーゼ、Candida rugosa(受入番号P20261、P32946、P32947、P3294およびP32949)由来のリパーゼ、Rhizopus niveus(受入番号P61871)由来のリパーゼ、Penicillium camemberti(受入番号P25234)由来のリパーゼ、Aspergillus niger(ABG73613、ABG73614およびABG37906)由来のリパーゼおよびPenicillium cyclopium(受入番号P61869)由来のリパーゼの群から選択される酵素、ならびに、各場合において、アミノ酸レベルで、少なくとも60%、好ましくは少なくとも80%、より好ましくは少なくとも90%、特に好ましくは少なくとも95%、98%または99%相同のものである。相同性に関しては、前述の定義を参照することが可能である。
【0056】
市販例および同様に本発明による方法で好ましく使用されるカルボキシルエステル加水分解酵素は、商品Lipozyme TL IM、Novozym 435、Lipozyme IM 20、Lipase SP382、Lipase SP525、Lipase SP523、(全てNovozymes A/S, Bagsvaerd, Denmarkの商品)、Chirazyme L2、Chirazyme L5、Chirazyme L8、Chirazyme L9(全てRoche Molecular Biochemicals, Mannheim, Germanyの商品)、およびLipase M “Amano”、Lipase F-AP 15 “Amano”、Lipase AY “Amano”、Lipase N “Amano”、Lipase R “Amano”、Lipase A “Amano”、Lipase D “Amano”、Lipase G “Amano”(全てAmano, Japanの商品)である。
【0057】
本生体触媒は、無水物または部分的水和物の形で好ましく使用される。本生体触媒は、固定化されるかまたは凍結乾燥物として使用することができる。固定化のために使用することができる担体は、不活性な有機担体または無機担体である。固定化型酵素に好ましく使用されるかまたは存在する不活性な担体は、粒子の少なくとも90%が10〜5000μm、好ましくは50〜2000μmの粒径を有する粒径分布を有する粒状担体である。特に使用することができる有機担体は、ポリアクリレート、ポリメタクリレート、ポリビニルスチレン、スチレン−ジビニルベンゼンコポリマー、ポリプロピレン、ポリエチレン、ポリエチレンテレフタレート、PTFEおよび/または他のポリマーを含むか、またはこれらから成るものである。固定化すべき酵素に応じて、特に酸性イオン交換樹脂または塩基性イオン交換樹脂、たとえばDuolite A568、Duolite XAD 761、Duolite XAD 1180、Duolite XAD 7HP、Amberlite IR 120、Amberlite IR 400、Amberlite CG 50、Amberlyst 15(全てRohm and Haasの製品)またはLewatit CNP 105およびLewatit VP OC 1600(Lanxess, Leverkusen, Germanyの製品)を、担体材料として使用することができる。使用することができる無機担体は、最新技術で周知の酸化物(oxidic)担体および/またはセラミック担体である。特に使用できる無機担体は、たとえばセライト、沸石、シリカ、細孔制御ガラス(CPG)またはたとえば、L. Cao, “Carrier-bound Immobilized Enzymes: Principles, Application and Design”, Wiley-VCH: 2005, Weinheim, Germanyに記載されているもののような他の担体である。固定化型酵素に存在する不活性な担体または固定化型酵素の製造に使用される不活性な担体は、特に好ましくはポリビニルスチレン、ポリメタクリレートまたはポリアクリレートから成る。
【0058】
本発明によれば、共有結合的または非共有結合的に、好ましくは非共有結合的に、粒子上に固定化することが可能である。非共有結合的固定化の場合、たとえば酵素水溶液(たとえば無機塩または洗浄剤等のさらなる成分を含んでもよい)を用いて、担体をインキュベートするかまたは含浸させる。このインキュベーション/含浸は、たとえば0℃〜50℃、好ましくは0℃〜40℃の温度で、実施することができる。このインキュベーション/含浸は、数分から数時間までの期間にわたって好ましく行われる。インキュベーションの進行は、従来の蛋白質測定方法で溶液中の酵素濃度を測定することにより追跡することができる。所望の程度の固定化に到達した後、好ましくは担体を水洗し、必要に応じて乾燥させる。
【0059】
好適な生体触媒を含む全細胞を、静止細胞としてまたは乾燥型としてのいずれかで、好ましくは先行技術の方法で透過処理して、使用することも可能である。
【0060】
本発明によれば、使用される基質に対して、0.01〜300%(w/w)の量比で、生体触媒を使用することができる。それぞれの反応条件で(特に、基質、それらの濃度、溶剤および反応温度に応じて)、好ましい質量比は、生体触媒の比活性から得られる。こうしたパラメータに応じて、生体触媒の好ましい量は、1〜96時間、好ましくは8〜24時間の期間内に、反応物質の完全変換を可能にする量である。生体触媒の量は、前駆体の全質量を基準にして、1〜100%(w/w)および極めて特に好ましくは1〜50%(w/w)、特に1〜20%(w/w)の範囲で、好ましく使用される。
【0061】
ある特定の実施態様では、この生体触媒は回収される(recovered)。
【0062】
本発明によれば、酵素反応の開始時に、反応物質は、1:10〜10:1というリゾスフィンゴ脂質とカルボキシルエステルとのモル比で存在することが可能である。この関連で、用語「モル比」は、リゾスフィンゴ脂質アミンとアシルドナーカルボキシレート基とのモル比に関する。1:3〜3:1の量比が、好ましく使用される。1:1.4〜1.4:1の量比、とりわけ等モル量比が、特に好ましく使用される。
【0063】
反応物質は、さらなる溶解剤を含まずに溶融状態で存在してもよく、有機溶剤中に溶解または懸濁されていてもよい。反応は、好ましくは有機溶剤中で実施される。特に適した溶剤は、20〜130℃の範囲内の温度で、反応物質を高濃度で溶解することができるものである。特に適していると証明されている例は、たとえば、メチルイソブチルケトンまたはシクロヘキサノン等のケトン、2−ブチル−1−オクタノール、メチルシクロヘキサノール、1−メトキシ−2−プロパノール、2,3−ブタンジオール2−オクタノール、ジアセトンアルコール、2−メチル−2−ブタノール等の立体障害性二級アルコール、酢酸4−tert−ブチルシクロヘキシル、酢酸2−エチルヘキシル、酢酸シクロヘキシル等の立体障害性エステルならびに1,4−ジオキサン、テトラヒドロフランおよびVaronic APM(Evonik Goldschmidt GmbH、Essen、Germany)等のエーテルであるが、こうした溶剤に限定されない。不適当であると証明されている溶剤は、とりわけ生体触媒によるかなり大きい活性で変換されるエステルである。フィトスフィンゴシンを前駆体として用いて、酪酸エチルを使用した反応で、たとえば所望の生成物に加えて、かなりの量の、N−ブチリルフィトスフィンゴシンと使用したアシルドナー(たとえばステアリン酸メチル)とのエステル交換生成物を検出することができた。同じことが、酢酸ブチルの使用にも当てはまる(N−アセチルフィトスフィンゴシンとアシルドナーブチルエステルの形成)。
【0064】
本反応は、カールフィッシャー(Karl Fischer)法で検出される含水量が0.100M以下、0.010M以下および特に好ましくは最高でも0.005Mとして定義された無水条件で好ましく実施される。
【0065】
高基質濃度は、反応速度に特に有利である。変換率が高いと、生成物が高濃度になるが、驚くべきことに、変換率が高いとき、酵素活性に対して悪影響はない。酵素反応では生成物阻害が通常観察されるため、このことは当業者には驚くべきことである。加えて、驚くべきことに、非常に高い変換乃至定量的変換を達成するために、反応生成物(セラミドまたはアルコール)の1つを反応混合物から除去することが、絶対的に必要というわけではないことが分かった。当業者によく知られているLe Chatelierの原理に従えば、このことが本平衡反応に関して予想されたであろう。僅か67〜78%という先行技術(国際公開第94/26919号パンフレット、Gist−Brocades)の反応率が、その必要を予想させることになった。しかし、本発明によれば、より低い基質濃度でも、効率よく反応することが可能である。
【0066】
本発明の文脈で好ましい反応開始時の反応物質濃度は、各反応物質について、0.01M〜3Mの範囲内、特に0.2M〜2Mの範囲内、特に好ましくは0.5M〜1.0Mの範囲内である。
【0067】
反応温度に関して、特に適した温度は、基質が溶剤に均質に溶解する温度であることは分かっていた。これに関連して、20〜130℃の範囲内の反応温度が有利であった。とりわけ高温では、変色のため、反応混合物の注意深い脱気が必要である。しかし、驚くべきことに100℃以下の反応温度では、この種の用心は無用であることが分かった。したがって、反応温度は、好ましくは40〜90℃の範囲内であり、極めて特に好ましくは、70〜85℃の範囲内である。
【0068】
反応は好ましくは、1bar未満、好ましくは0.5bar未満、特に好ましくは0.05bar未満の圧力で実施される。
【0069】
この反応は、バッチ方法(撹拌容器)で実施することもでき、または固定床反応器で連続的または半連続的に実施することもできる。高速の生体触媒失活は、とりわけバッチ方法で推定されるものであった。撹拌によって生じる機械的ストレスによる生体触媒の機械的破壊、ならびに反応物質および溶剤に起因する失活作用と対をなす生体触媒の熱失活について留意すべきである。このような作用は当業者に周知であり、また好適な救済策が文献に開示されている。したがって、たとえば生体触媒の機械的疲弊が撹拌方法で起きるのであれば、固定床を使用することにより、これを回避することが可能である。さらなる反応器は、当業者に周知であり、同様に、機械的酵素失活を回避するのに好適である。
【0070】
さらに、エステルをアシルドナーとして使用するとき、遊離したアルコールによって生体触媒が化学的に失活されることが予想される(たとえば:Y. Yoshida et al., J. Biosci. Bioeng., 2006, 102(1), 66-68)。このような失活を回避する方法は当業者に周知であり、たとえば、蒸留による反応混合物からのアルコール除去や膜方法の使用などがある。しかし、驚くべきことに、上述の方法を使用しなくても、活性を著しく損失せずに生体触媒を何度も再使用できることが分かった。
【0071】
結果として得られる生成物が、スフィンゴ脂質および高比率(全生成物の最高19%)の対応するN,O−ジアシル化生成物の組成物であることは、先行技術に記載されている全ての方法に共通である。こうした組成物は、たとえば、N,O−ジアシル化生成物の比率が5%以上であるため、化粧用組成物に全く使用できない。さらに、本発明による方法では、驚くべきことに、二級アミン官能基のアミド化は、アルコール官能基の1つのエステル化より紛れもなく好ましいことが分かった。反対の挙動、特に、一級、すなわち立体的に容易に近づける、アルコール官能基の選好が予想されたであろう。先行技術(国際公開第94/26919号パンフレット、Gist−Brocades)は、類似した状況について記している:細菌リパーゼを使用し、アシルドナーがフィトスフィンゴシンより60%モル過剰であっても、その状況で得られた反応生成物は、かなりの比率のN,O−ジアシル化生成物(モノジアシル化生成物とジアシル化生成物の比率>1:5)を含んでいた。
【0072】
それとは反対に、リゾスフィンゴ脂質より3.6倍過剰のアシルドナーを用いても、リゾスフィンゴ脂質が反応混合物中に存在する限り、顕著なジアシル化がないことを、本発明による方法で示すことが可能であった。リゾスフィンゴ脂質が完全にスフィンゴ脂質に変換されていたときに限って、緩徐なエステル化活性を検出することが可能である。リゾスフィンゴ脂質およびアシルドナーを化学量論的に使用したとき、上述の不要な副反応はほとんど認められなかった。
【0073】
したがって本発明は、酵素反応による粗生成物としてでさえ、特に純粋なN−アシルフィトスフィンゴシンが得られる方法に関する。式Iで表される標的化合物に関連した変換は、理論的に予想される変換の好ましくは80%超、特に好ましくは85%超、および極めて特に好ましくは90%超である。
【0074】
本発明による方法はさらに、式Iおよび0.001〜19質量%、好ましくは0.005〜19質量%、特に好ましくは0.01〜5質量%の、対応するN,O−ジアシル化生成物を含む組成物を調製するのに好適である。
【0075】
したがって、本発明は同様に、本発明による方法を使用して調製される、式Iおよび0.001〜19質量%、好ましくは0.005〜19質量%、特に好ましくは0.01〜5質量%の、対応するN,O−ジアシル化生成物を含む組成物に関する。
【0076】
本発明は同様に、本発明による方法を使用して調製されるスフィンゴ脂質に関する。
【0077】
本発明はさらに、スフィンゴ脂質を反応混合物から分離する方法に関する。スフィンゴ脂質は好都合な物理的性質を有するため、スフィンゴ脂質は結晶化/沈殿により反応溶液から容易に除去することができ、したがって、既に高い粗生成物の純度を、簡単な方法でさらに高めることができる。
【0078】
スフィンゴ脂質および本発明による組成物は、化粧用製剤および医薬製剤の製造に特に適する。したがって本発明は同様に、本発明による少なくとも1つのスフィンゴ脂質および/または本発明による1つの組成物を含む、化粧用製剤または医薬製剤に関する。
【0079】
本発明を、以下に詳述する実施例において例として説明するが、本発明は、実施例で言及されている実施態様に限定されることは意図されず、その適用範囲は完全な記述および特許請求の範囲から明白である。
【実施例】
【0080】
実施例1:PSサルフェートの脱プロトン化
フィトスフィンゴシンサルフェート(Cosmoferm,Delft,NL)100gを、55℃のメタノール600g中に懸濁させ、ナトリウムメトキシド46gと混合し、55℃で150分間撹拌する。次いで、この懸濁液を熱濾過するとき、その濾液は高濃度である。淡いベージュ色の固体(78g)をさらなる実験に使用する。
【0081】
実施例2:アミド化反応性に関する、市販のリパーゼのスクリーニング
ジオキサン(11mL)中の、フィトスフィンゴシン(実施例1から)およびステアリン酸メチル(各1M)の等モル溶液に、表1に記載の真菌リパーゼ各0.35g(5% w/w)を懸濁させ、80℃で72時間、リパーゼで振盪する。次いで、生体触媒を熱濾去し、その濾液をガスクロマトグラフィで分析する。
【0082】
【表1】
【0083】
カンジダ・アンタークチカ(Candida antarctica)由来のリパーゼBを、さらなる実験に使用した。
【0084】
実施例4:陰性対照
フィトスフィンゴシンとアシルドナーとの、可能性のあるあらゆる無触媒反応を除外するために、0.2M強度、80℃で、フィトスフィンゴシンをステアリン酸メチルに溶解し、4時間インキュベートした。ガスクロマトグラフィによるその後の分析は、有意な(定量化可能な)変換を示さなかった。
【0085】
実施例5:フィトスフィンゴシンサルフェートとの反応
フィトスフィンゴシンサルフェートを脱プロトン化する必要性を証明するために、以下の比較実験を実施した:フィトスフィンゴシンサルフェート8.18gおよびステアリン酸メチル6.72gを、80℃のジオキサン15mLに懸濁させ、Novozym 435 0.16gと混合した。撹拌しながら4時間インキュベーションした後、この反応混合物をガスクロマトグラフィで分析した。僅か31.7mMのN−ステアロイルフィトスフィンゴシンが確認されただけであった。炭酸ナトリウム2.4gを加えた後であること以外は同一条件での比較実験で、N−ステアロイルフィトスフィンゴシン368mMが確認された。
【0086】
実施例6:酵素的アシル化の位置選択性
フィトスフィンゴシンに比して超過剰のアシルドナーを得るために、フィトスフィンゴシン4.43gを120℃のステアリン酸メチル15.09gに溶解した。この溶液をNovozym 435 0.98gに溶解し(5% w/w)、標準気圧で撹拌した。定期的に試料を採取して、ガスクロマトグラフィで分析した。
【0087】
反応経過(図1)から分かる通り、不要なN,O−ジステアリルフィトスフィンゴシンの顕著な蓄積は、使用したフィトスフィンゴシンが完全に変換された時にのみ起こる。1時間後、19mM(変換>99.5%)の残余フィトスフィンゴシン濃度で、N,O−ジアシル化生成物はまだ検出されず、N,O−ジアシル化生成物の形成は、約5mM未満のフィトスフィンゴシン濃度でのみ確認される。加えて、二次反応の反応速度は、25を超える倍数(factor)で所望の反応速度を下回る。
【0088】
実施例7:生成物阻害に関する実験
あらゆる生成物阻害を除外するために、2つの並行実験を実施した。この目的のため、各ケースにおいて、フィトスフィンゴシン1.59gを、ステアリン酸メチル7.45gと共に、80℃のジオキサン25mLに溶解し、さらに、各ケースにおいてNovozyme 435 0.49gと混合した。2つの反応混合物の1つを、N−ステアロイルフィトスフィンゴシン(Cosmoferm, Delft, NL)1.3gとさらに混合した。2つの時間−変換曲線の比較が示す通り(図2)、その反応軌跡は全く同じであり、したがって任意の顕著な生成物阻害を除外することが可能である。
【0089】
実施例8:生体触媒の再使用可能性
生体触媒の再使用可能性を評価するために、以下の通りに一連の実験を実施した:フィトスフィンゴシン3.3gをステアリン酸メチル14.9gと一緒に、80℃のジオキサン50mL中に溶解し、Novo 435 0.98gを加えることによって反応を開始した。試料を定期的に採取し、ガスクロマトグラフィで分析し、初期速度および変換率をそれから算出した。24時間後、その反応バッチを熱濾過し、後に残っている酵素を、毎回、熱い(50℃)ジオキサン50mlで3度洗浄する。次いで、このようにしてリサイクルした生体触媒を、上述と同じ条件で再使用する。この手順を全6回繰り返す。
【0090】
図3から明白なように、観測可能な活性低下なしに少なくとも7度、この生体触媒を反応に使用することができる。
【0091】
実施例9:生体触媒の速度論的パラメータの測定
酵素的N−アシル化の速度論的パラメータを、以下の通りに測定した:一方の反応物濃度は変えるが、他方の濃度は一定に保った。反応は、いずれの場合にも同量の酵素を使用して、80℃のジオキサン中で実施した。試料を定期的に採取してガスクロマトグラフィで分析した;それから、それぞれの初期速度を決定し、Lineweaver-Burkに準拠して解析した。決定された結果は、フィトスフィンゴシンに関してはKM値400mMであり、またステアリン酸メチルに関してはKM値196mMであった。
【0092】
実施例10:温度が反応速度に及ぼす影響
酵素反応を様々な温度で実施し、基質は、すべての場合に等モル量で使用した。使用した酵素量は、いずれの場合にも1.4%(w/w)であった。初期速度のガスクロマトグラフ評価の結果および4時間後の変換の結果も、表2に報告する。
【0093】
【表2】
【0094】
実施例11:様々な溶媒の使用
フィトスフィンゴシンおよびステアリン酸メチルを、80℃のそれぞれの溶剤中に1Mで各々溶解し、5%(w/w)のNovozym 435と混合して撹拌した。定期的に試料を採取してガスクロマトグラフィで分析した。図4から明白なように、変換時間曲線および個々の反応の24時間後の変換も、ほぼ一致した。
【0095】
実施例12:フィトスフィンゴシンと様々な脂肪酸エステルとの酵素反応
フィトスフィンゴシンおよび表3に報告したアシルドナーを、各々1Mで80℃の溶剤に溶解し、5%(w/w)のNovozym 435と混合して撹拌した。定期的に試料を採取して、ガスクロマトグラフィで分析した。
【0096】
【表3】
【0097】
実施例13:生成物の選択的結晶化
ステアリン酸メチル(734mM)およびフィトスフィンゴシン(146mM)を、80℃の2−メチル−2−ブタノールに溶解し、5%(w/w)のNovozym 435と混合し、80℃で20時間撹拌した。次いで、酵素を熱濾去し、透き通った出発溶液を一晩、45℃に温度調節した。沈殿した固体を濾去し、濾液および濾過残渣を、ガスクロマトグラフで分析した。表4から明らかなように、基質が超過剰に存在する場合でも、分別沈殿によって、所望の生成物を選択的に濃縮することが可能になる。
【0098】
【表4】
【0099】
実施例14:アシルドナーとしてのトリステアリン
トリグリセリド(トリステアリン)をアシルドナーとして使用して、酵素反応を実施した。80℃で、ジオキサン17g中にフィトスフィンゴシン5.4gおよびトリステアリン5gを含む溶液に、Novozym 435 0.5gを加え、大気圧下で撹拌した。試料を定期的に採取し、ガスクロマトグラフィで分析した。
【0100】
図5から明らかなように、アシルドナーの完全変換は可能である。驚くべきことに、中間体部分グリセリド(グリセロールジステアレートまたはグリセロールモノステアレート)はほとんど確認されないことが、ガスクロマトグラフィによる分析から明らかになった。
【0101】
実施例15:フィトスフィンゴシンサルフェートからのN−ステアロイルフィトスフィンゴシンのワンポット合成
フィトスフィンゴシンサルフェート(Cosmoferm, Delft,NL)100gを、55℃のMIBK(メチルイソブチルケトン)100mlに懸濁させ、ナトリウムメトキシド46gと混合し、55℃で180分間、撹拌する。次いで、ステアリン酸メチル73.3gおよびNovo435 5gを加える。このようにして得た懸濁液を、80℃で36時間撹拌し、熱濾去する。透き通った濾液を、室温で4時間静置し、沈殿した透き通ったベージュ色の固体(N−ステアロイルフィトスフィンゴシン122.3g、純度>97%)を濾去する。
【0102】
実施例16:固定床反応器でのパイロットバッチ
フィトスフィンゴシンサルフェート207.8gを、NaOCH3溶液(メタノール中25%濃度)108.04gと共に55℃のMIBK 500mlに懸濁させ、続いてフィルタープレスを介して固定床反応器に熱いうちに移し、ステアリン酸メチル149.3gと混合する。この固定床は、Novozyme 435 17.8gを含む。反応器全体を、80℃に温度調節する。約25ml分−1のポンプ流量で反応混合物を固定床に流す。0.8barの圧力下、過剰のメタノールを除去する。24時間後、この反応器に窒素を通気し、反応器を空にして、この反応溶液を室温で一晩静置する。沈殿した固体を濾去する(N−ステアロイルフィトスフィンゴシン287.1g、純度98.7%)。
【特許請求の範囲】
【請求項1】
一般式I
【化1】
で表されるスフィンゴ脂質の生体触媒的調製方法であって、
一般式II
【化2】
で表されるリゾスフィンゴ脂質と、一般式III
【化3】
で表されるカルボキシルエステル
[式中、R1は、場合により1つもしくは複数の多重結合および/または芳香環もしくは芳香族複素環を含んでもよく、場合により酸素原子またはエステル官能基もしくはアミド官能基が割り込んでいてもよく、また場合によりアルキル基、ヒドロキシ基、ケト基またはアミン基から選択される少なくとも1つのさらなる基で置換されていてもよい、2〜55個の炭素原子を有する直鎖または分岐したアルキル鎖を表し、
R2は、H、ホスホコリン、エタノールアミン、セリンまたは糖を表し、
Xは、CH=CH、CH2−CH2またはCH2−HCOHを表し、
R3は、少なくとも1つの基−OR4(式中、R4は、独立して、
H
および
−C(O)R1(式中、R1は上記定義の通りである)
からなる群から選択される、同一または非同一の基である)で置換されていてもよい、1〜12個の炭素原子を有する直鎖または分岐したアルキル基を表す。]
とを、反応させることによる生体触媒的調製方法であり、使用する生体触媒が、
菌界の生物から単離することができる酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素、および
菌界の生物から単離することができる酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素と、アミノ酸レベルで少なくとも80%相同である、酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素
からなる群から選択される酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素を少なくとも1つ含むことを特徴とする、生体触媒的調製方法。
【請求項2】
R3が、1〜4個の炭素原子を有する非置換アルキル基であることを特徴とする、請求項1に記載の方法。
【請求項3】
一般式IIIのカルボキシルエステルが、ポリオールと少なくとも1つの酸R1COOHとの完全または部分エステルであることを特徴とする、請求項1に記載の方法。
【請求項4】
前記リゾスフィンゴ脂質が、前記酵素反応の前に、前記リゾスフィンゴ脂質の酸付加生成物の脱プロトン化によって調製されることを特徴とする、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記リゾスフィンゴ脂質の酸付加生成物の脱プロトン化と、生体触媒的調製との間に濾過ステップが行われることを特徴とする、請求項4に記載の方法。
【請求項6】
前記リゾスフィンゴ脂質が、生体触媒作用中に、前記リゾスフィンゴ脂質の酸付加生成物の脱プロトン化によって調製されることを特徴とする、請求項1〜3のいずれか1項に記載の方法。
【請求項7】
前記リゾスフィンゴ脂質の、カルボン酸カルボキシレート、硫酸塩、リン酸塩、硝酸塩、炭酸塩、水酸化物またはハロゲン化物が、前記リゾスフィンゴ脂質の酸付加生成物として使用されることを特徴とする、請求項4〜6のいずれか1項に記載の方法。
【請求項8】
有機塩基または無機塩基が前記脱プロトン化に使用されることを特徴とする、請求項4〜7のいずれか1項に記載の方法。
【請求項9】
プロトン化後に水を少しも遊離しない塩基が使用されることを特徴とする、請求項8に記載の方法。
【請求項10】
アルカリ金属アルコラートが塩基として使用されることを特徴とする、請求項8または9に記載の方法。
【請求項11】
前記リゾスフィンゴ脂質の酸付加生成物と塩基との間のモル比が、10:1〜0.05:1の範囲内であることを特徴とする、請求項8〜10のいずれか1項に記載の方法。
【請求項12】
前記菌界の生物が、Aspergillus属、Bipolaris属、Candida属、Fusarium属、Geotrichum属、Humicola属、Microsporum属、Mucor属、Pichia属、Thermomyces属、Penicillium属、Rhizopus属、Rhizomucor属、Microsporum属、Mucor属、Nocardia属、Saccharomyces属、Streptomyces属、Thermomyces属、Trichosporon属、Zygosaccharomyces属の群から選択されることを特徴とする、請求項1〜11のいずれか1項に記載の方法。
【請求項13】
前記生体触媒の量が、前駆体の全質量を基準にして、1%(w/w)〜100%(w/w)の範囲内で使用されることを特徴とする、請求項1〜12のいずれか1項に記載の方法。
【請求項14】
前記生体触媒が回収されることを特徴とする、請求項1〜13のいずれか1項に記載の方法。
【請求項15】
前記酵素反応の開始時に、リゾスフィンゴ脂質とカルボキシルエステルとのモル比1:10〜10:1で反応物質が存在することを特徴とする、請求項1〜14のいずれか1項に記載の方法。
【請求項16】
前記反応が、有機溶剤中で実施されることを特徴とする、請求項1〜15のいずれか1項に記載の方法。
【請求項17】
カールフィッシャー法で検出される水含量0.1M以下として定義された無水条件下で前記反応が実施されることを特徴とする、請求項1〜16のいずれか1項に記載の方法。
【請求項18】
前記反応開始時の前記反応物質濃度が、各反応物質について、0.01M〜3Mの範囲内であることを特徴とする、請求項1〜17のいずれか1項に記載の方法。
【請求項19】
前記反応温度が、20℃〜130℃の範囲内であることを特徴とする、請求項1〜18のいずれか1項に記載の方法。
【請求項20】
前記反応が、1bar未満の圧力下で実施されることを特徴とする、請求項1〜19のいずれか1項に記載の方法。
【請求項21】
式Iの標的化合物に関連した変換が、理論的に予想される変換の80%超であることを特徴とする、請求項1〜20のいずれか1項に記載の方法。
【請求項22】
式Iおよび0.001〜19質量%の対応するN,O−ジアセチル化生成物を含む組成物が得られることを特徴とする、請求項1〜21のいずれか1項に記載の方法。
【請求項23】
請求項1〜21のいずれか1項に記載の方法により調製されるスフィンゴ脂質。
【請求項24】
請求項22に記載の方法を使用して調製される、式Iおよび0.001〜19質量%の対応するN,O−ジアセチル化生成物を含む組成物。
【請求項25】
少なくとも1つの請求項23に記載のスフィンゴ脂質および/または1つの請求項24に記載の組成物を含む、化粧用製剤または医薬製剤。
【請求項1】
一般式I
【化1】
で表されるスフィンゴ脂質の生体触媒的調製方法であって、
一般式II
【化2】
で表されるリゾスフィンゴ脂質と、一般式III
【化3】
で表されるカルボキシルエステル
[式中、R1は、場合により1つもしくは複数の多重結合および/または芳香環もしくは芳香族複素環を含んでもよく、場合により酸素原子またはエステル官能基もしくはアミド官能基が割り込んでいてもよく、また場合によりアルキル基、ヒドロキシ基、ケト基またはアミン基から選択される少なくとも1つのさらなる基で置換されていてもよい、2〜55個の炭素原子を有する直鎖または分岐したアルキル鎖を表し、
R2は、H、ホスホコリン、エタノールアミン、セリンまたは糖を表し、
Xは、CH=CH、CH2−CH2またはCH2−HCOHを表し、
R3は、少なくとも1つの基−OR4(式中、R4は、独立して、
H
および
−C(O)R1(式中、R1は上記定義の通りである)
からなる群から選択される、同一または非同一の基である)で置換されていてもよい、1〜12個の炭素原子を有する直鎖または分岐したアルキル基を表す。]
とを、反応させることによる生体触媒的調製方法であり、使用する生体触媒が、
菌界の生物から単離することができる酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素、および
菌界の生物から単離することができる酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素と、アミノ酸レベルで少なくとも80%相同である、酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素
からなる群から選択される酵素クラスE.C.3.1.1のカルボキシルエステル加水分解酵素を少なくとも1つ含むことを特徴とする、生体触媒的調製方法。
【請求項2】
R3が、1〜4個の炭素原子を有する非置換アルキル基であることを特徴とする、請求項1に記載の方法。
【請求項3】
一般式IIIのカルボキシルエステルが、ポリオールと少なくとも1つの酸R1COOHとの完全または部分エステルであることを特徴とする、請求項1に記載の方法。
【請求項4】
前記リゾスフィンゴ脂質が、前記酵素反応の前に、前記リゾスフィンゴ脂質の酸付加生成物の脱プロトン化によって調製されることを特徴とする、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記リゾスフィンゴ脂質の酸付加生成物の脱プロトン化と、生体触媒的調製との間に濾過ステップが行われることを特徴とする、請求項4に記載の方法。
【請求項6】
前記リゾスフィンゴ脂質が、生体触媒作用中に、前記リゾスフィンゴ脂質の酸付加生成物の脱プロトン化によって調製されることを特徴とする、請求項1〜3のいずれか1項に記載の方法。
【請求項7】
前記リゾスフィンゴ脂質の、カルボン酸カルボキシレート、硫酸塩、リン酸塩、硝酸塩、炭酸塩、水酸化物またはハロゲン化物が、前記リゾスフィンゴ脂質の酸付加生成物として使用されることを特徴とする、請求項4〜6のいずれか1項に記載の方法。
【請求項8】
有機塩基または無機塩基が前記脱プロトン化に使用されることを特徴とする、請求項4〜7のいずれか1項に記載の方法。
【請求項9】
プロトン化後に水を少しも遊離しない塩基が使用されることを特徴とする、請求項8に記載の方法。
【請求項10】
アルカリ金属アルコラートが塩基として使用されることを特徴とする、請求項8または9に記載の方法。
【請求項11】
前記リゾスフィンゴ脂質の酸付加生成物と塩基との間のモル比が、10:1〜0.05:1の範囲内であることを特徴とする、請求項8〜10のいずれか1項に記載の方法。
【請求項12】
前記菌界の生物が、Aspergillus属、Bipolaris属、Candida属、Fusarium属、Geotrichum属、Humicola属、Microsporum属、Mucor属、Pichia属、Thermomyces属、Penicillium属、Rhizopus属、Rhizomucor属、Microsporum属、Mucor属、Nocardia属、Saccharomyces属、Streptomyces属、Thermomyces属、Trichosporon属、Zygosaccharomyces属の群から選択されることを特徴とする、請求項1〜11のいずれか1項に記載の方法。
【請求項13】
前記生体触媒の量が、前駆体の全質量を基準にして、1%(w/w)〜100%(w/w)の範囲内で使用されることを特徴とする、請求項1〜12のいずれか1項に記載の方法。
【請求項14】
前記生体触媒が回収されることを特徴とする、請求項1〜13のいずれか1項に記載の方法。
【請求項15】
前記酵素反応の開始時に、リゾスフィンゴ脂質とカルボキシルエステルとのモル比1:10〜10:1で反応物質が存在することを特徴とする、請求項1〜14のいずれか1項に記載の方法。
【請求項16】
前記反応が、有機溶剤中で実施されることを特徴とする、請求項1〜15のいずれか1項に記載の方法。
【請求項17】
カールフィッシャー法で検出される水含量0.1M以下として定義された無水条件下で前記反応が実施されることを特徴とする、請求項1〜16のいずれか1項に記載の方法。
【請求項18】
前記反応開始時の前記反応物質濃度が、各反応物質について、0.01M〜3Mの範囲内であることを特徴とする、請求項1〜17のいずれか1項に記載の方法。
【請求項19】
前記反応温度が、20℃〜130℃の範囲内であることを特徴とする、請求項1〜18のいずれか1項に記載の方法。
【請求項20】
前記反応が、1bar未満の圧力下で実施されることを特徴とする、請求項1〜19のいずれか1項に記載の方法。
【請求項21】
式Iの標的化合物に関連した変換が、理論的に予想される変換の80%超であることを特徴とする、請求項1〜20のいずれか1項に記載の方法。
【請求項22】
式Iおよび0.001〜19質量%の対応するN,O−ジアセチル化生成物を含む組成物が得られることを特徴とする、請求項1〜21のいずれか1項に記載の方法。
【請求項23】
請求項1〜21のいずれか1項に記載の方法により調製されるスフィンゴ脂質。
【請求項24】
請求項22に記載の方法を使用して調製される、式Iおよび0.001〜19質量%の対応するN,O−ジアセチル化生成物を含む組成物。
【請求項25】
少なくとも1つの請求項23に記載のスフィンゴ脂質および/または1つの請求項24に記載の組成物を含む、化粧用製剤または医薬製剤。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2011−522550(P2011−522550A)
【公表日】平成23年8月4日(2011.8.4)
【国際特許分類】
【出願番号】特願2011−512917(P2011−512917)
【出願日】平成21年5月22日(2009.5.22)
【国際出願番号】PCT/EP2009/056206
【国際公開番号】WO2009/150022
【国際公開日】平成21年12月17日(2009.12.17)
【出願人】(507375465)エヴォニク ゴールドシュミット ゲーエムベーハー (100)
【Fターム(参考)】
【公表日】平成23年8月4日(2011.8.4)
【国際特許分類】
【出願日】平成21年5月22日(2009.5.22)
【国際出願番号】PCT/EP2009/056206
【国際公開番号】WO2009/150022
【国際公開日】平成21年12月17日(2009.12.17)
【出願人】(507375465)エヴォニク ゴールドシュミット ゲーエムベーハー (100)
【Fターム(参考)】
[ Back to top ]