
スルホネート界面活性剤及び製造方法及び使用
新しい界面活性剤、及び当該界面活性剤の製造方法及び用途を提供する。この界面活性剤は、式Iの化合物である。

(上記式中、R、R1、R2、R3、R4、R5及びR6は明細書中に規定される通りである)
(上記式中、R、R1、R2、R3、R4、R5及びR6は明細書中に規定される通りである)
【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本出願は2009年7月16日付けで出願された米国仮出願番号61/226,109号明細書の優先権を主張する。前記明細書は、これを全体的に参照することにより本明細書中に組み入れられる。
【0002】
本発明は、新しいスルホネート界面活性剤、新しいスルホネート界面活性剤の組成物、及び当該界面活性剤の製造方法及び使用に関する。
【背景技術】
【0003】
アニオン性界面活性剤はよく知られており、例えばクリーナー及び洗剤を含む種々の用途において使用されている。しかし、一般的なアニオン性界面活性剤、例えば線状アルキルベンゼンスルホネート(LAS)、及びアルコールスルフェート(例えばナトリウムラウリルスルフェート)は、冷水又は硬水中、及び苛性物質中での溶解度が低い。例えばエピクロロヒドリン、線状アルキルアルコール、及び亜硫酸ナトリウムから調製されたアルキルグリセロールスルホネート(AGS)界面活性剤も同様である。このような溶解性不足は、これらの界面活性剤の配合選択肢及び性能を制限する。
【0004】
高い界面活性を維持しつつ、改善された一連の特性、例えば冷水、硬水、苛性水、及びイオン水中での高い溶解度を有するアニオン性界面活性剤を提供することは、アニオン性界面活性剤にとって依然として難題である。
【発明の概要】
【課題を解決するための手段】
【0005】
1つの態様において、本発明は、新規のアニオン性スルホネート界面活性剤を提供する。これらのアニオン性界面活性剤は、式I:
【0006】
【化1】
【0007】
(上記式中、R、R1、R2、R3、R4、R5及びR6は下記で規定される通りである)
の化合物、又はこれらのうちの2種もしくは3種以上の混合物である。
【0008】
別の態様では、本発明は、式Iの1種又は2種以上のアニオン性界面活性剤を含有する配合物を提供する。
【0009】
さらなる別の態様では、本発明は、式Iの界面活性剤を含むクリーニング組成物を提供する。
【0010】
さらに別の態様では、本発明は、式Iのアニオン性界面活性剤又はこれらのうちの2種又は3種以上の混合物を製造する方法を提供する。
【図面の簡単な説明】
【0011】
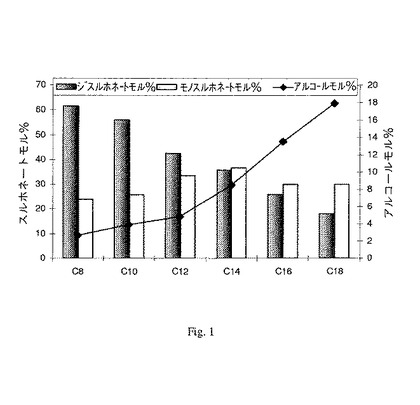
【図1】図1は、本発明の方法によって得ることができる界面活性剤組成物の例を示すグラフである。
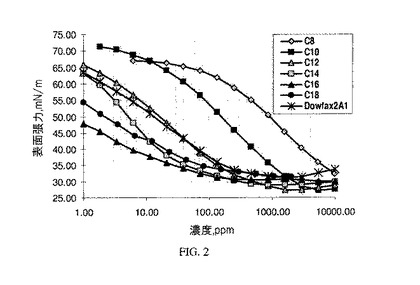
【図2】図2は、本発明のものではない界面活性剤と比較した本発明のアニオン性界面活性剤の表面張力の結果を示すグラフである。
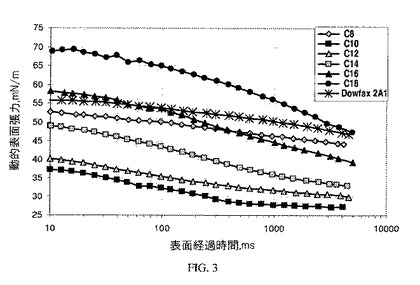
【図3】図3は、本発明のものではない界面活性剤と比較した本発明のアニオン性界面活性剤の動的表面張力の結果を示すグラフである。
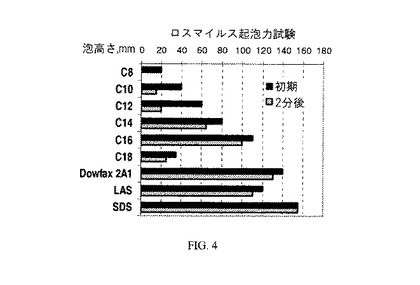
【図4】図4は、本発明のものではない界面活性剤と比較した本発明のアニオン性界面活性剤のロスマイルス(Ross−Miles)起泡力試験結果を示すグラフである。
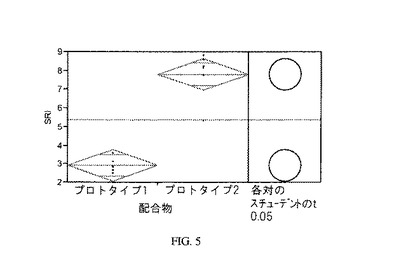
【図5】図5は、本発明の配合物のクリーニング効率を、比較配合物と比較するグラフである。
【発明を実施するための形態】
【0012】
上記のように、1つの態様において、本発明は新規のアニオン性界面活性剤を提供する。本発明のアニオン性界面活性剤は、2つのスルホネート基、又は1つのスルホネート基と1つのヒドロキシ基とを、分枝状アルキルエーテル主鎖上に含有する。アニオン性界面活性剤は、他の既知のジスルホネート界面活性剤及びヒドロキシスルホネート(例えばAGS)界面活性剤と比較して改善された一連の特性を呈し、界面活性剤が種々様々な用途に使用されるのを可能にする。例えば、本発明の界面活性剤は高い水溶性を有し、硬水、電解質、及び苛性溶液に対して高い耐容性を有し、そしてクリーニング組成物中で使用されると、油汚れの除去の際に優れた性能を発揮する。加えて、以下の例によって実証されるように、本発明の界面活性剤は、極めて好ましい水中毒性を示す。
【0013】
本発明のアニオン性界面活性剤は、
式I:
【0014】
【化2】
【0015】
(上記式中、Rは線状又は分枝状C2−C22アルキルであり;
R1、R2、及びR3は独立して、H又は線状もしくは分枝状C1−C18アルキルであり;
R4はH、CH2SO3-M+、又はCH2OHであり;
R5はOH、SO3-M+、又は式:
【0016】
【化3】
【0017】
の基であり;
R6はH、CH2SO3-M+、又はCH2OHであり;そして
M+はH+、又は一価又は二価カチオン、例えばナトリウム、カリウム、アンモニウム、カルシウム、マグネシウム、又はアルキル化アンモニウムであり、
R4及びR6のうちの一方はHであり、そしてR4、R5、又はR6のうちの1つ又は2つはSO3-M+を含有する)
の化合物である。
【0018】
本発明の界面活性剤を製造するための下記方法は結果として、式I化合物の混合物を形成することができる。個々の式I化合物は混合物から単離することができるものの、この工程は必要なものではなく、実際には界面活性剤を混合物の形態で使用することが好ましいことがある。こうして、式I化合物の混合物である界面活性剤が考えられ、これも本発明の範囲に含まれる。
【0019】
式Iの好ましい化合物は、Rが線状C2−C22アルキルである式Iの化合物である、式I−1の化合物(又はこれらの混合物)を含む。さらに好ましくは、Rは、線状又は分枝状、より好ましくは線状のC4−C16アルキルである。
【0020】
式I、及び式I−1の好ましい化合物は、R1がHである式I、又は式I−1の化合物である、式I−2の化合物を含む。
【0021】
式I、式I−1、及び式I−2の好ましい化合物は、R2がHである式I、式I−1、又は式I−2の化合物である、式I−3の化合物を含む。
【0022】
式I、式I−1、式I−2、及び式I−3の好ましい化合物は、R3がHである式I、式I−1、式I−2、又は式I−3の化合物である、式I−4の化合物を含む。
【0023】
式I、式I−1、式I−2、式I−3、及び式I−4の好ましい化合物は、R4がCH2SO3-M+、又はCH2OHである式I、式I−1、式I−2、式I−3、又は式I−4の化合物である、式I−5の化合物を含む。1つの態様では、R4は好ましくはCH2SO3-M+である。別の態様では、R4は好ましくはCH2OHである。
【0024】
式I、式I−1、式I−2、式I−3、式I−4、及び式I−5の好ましい化合物は、R5がOH、又はSO3-M+である式I、式I−1、式I−2、式I−3、式I−4、又は式I−5の化合物である、式I−6の化合物を含む。1つの態様では、R5は好ましくはSO3-M+である。別の態様では、R5は好ましくはOHである。
【0025】
式I、式I−1、式I−2、式I−3、式I−4、式I−5、及び式I−6の好ましい化合物は、R6がHである式I、式I−1、式I−2、式I−3、式I−4、式I−5、又は式I−6の化合物である、式I−7の化合物を含む。
【0026】
式I、式I−1、式I−2、式I−3、式I−4、式I−5、式I−6、及び式I−7の好ましい化合物は、M+がH+、又は一価カチオンである式I、式I−1、式I−2、式I−3、式I−4、式I−5、式I−6、又は式I−7の化合物である、式I−8の化合物を含む。さらに好ましくは、M+はH+、Na+、K+、アンモニウム、又はアルキル化アンモニウムである。特に好ましいのはNa+である。
【0027】
式Iの好ましい化合物はさらに、式II:
【0028】
【化4】
【0029】
(上記式中、Rは線状又は分枝状C2−C22アルキルであり;
R1、R2、及びR3は独立して、H又は線状もしくは分枝状C1−C18アルキルであり;
R4はCH2SO3-M+、又はCH2OHであり;
R5はOH、又はSO3-M+であり;そして
M+はH+、又は一価又は二価カチオンであり、R4及びR5のうちの一方又は両方がSO3-M+を含有する)
の化合物を含む。
【0030】
式IIの好ましい化合物は、Rが線状C2−C22アルキルである式IIの化合物である、式II−1の化合物を含む。さらに好ましくは、Rは、線状又は分枝状、より好ましくは線状のC4−C16アルキルである。
【0031】
式II、及びII−1の好ましい化合物は、R1、R2、及びR3がそれぞれHである式II、又はII−1の化合物である、式II−2の化合物を含む。
【0032】
式II、式II−1、及び式II−2の好ましい化合物は、R4がCH2SO3-M+であり、そしてR5がOHである式II、式II−1、又は式II−2の化合物である、式II−3の化合物を含む。
【0033】
式II、式II−1、及び式II−2の好ましい化合物は、R4がCH2SO3-M+であり、そしてR5がSO3-M+である式II、式II−1、又は式II−2の化合物である、式II−4の化合物を含む。
【0034】
式II、式II−1、式II−2、式II−3、及び式II−4の好ましい化合物は、M+がH+、又は一価カチオンである、式II、式II−1、式II−2、式II−3、又は式II−4である式II−5の化合物を含む。さらに好ましくは、M+はH+、Na+、K+、アンモニウム、又はアルキル化アンモニウムである。特に好ましいのはNa+である。
【0035】
本発明のいくつかの態様の場合、式Iの化合物は式III:
【0036】
【化5】
【0037】
(上記式中、Rは線状又は分枝状C2−C22アルキルであり;
R1、R2、及びR3は独立して、H又は線状もしくは分枝状C1−C18アルキルであり;
R5はOH、又はSO3-M+であり;
R6はCH2SO3-M+、又はCH2OHであり;そして
M+はH+、又は一価又は二価カチオンであり、R5及びR6のうちの一方又は両方がSO3-M+を含有する)
を有する。
【0038】
式IIIの好ましい化合物は、Rが線状C2−C22アルキルである式IIIの化合物である、式III−1の化合物を含む。さらに好ましくは、Rは、線状又は分枝状、より好ましくは線状のC4−C16アルキルである。
【0039】
式III、及びIII−1の好ましい化合物は、R1、R2、及びR3がそれぞれHである式III、又はIII−1の化合物である、式III−2の化合物を含む。
【0040】
式III、式III−1、及び式III−2の好ましい化合物は、R5がOHであり、そしてR6がCH2SO3-M+である式III、式III−1、又は式III−2の化合物である、式III−3の化合物を含む。
【0041】
式III、式III−1、及び式III−2の好ましい化合物は、R5がSO3-M+であり、そしてR6がCH2OHである式III、式III−1、又は式III−2の化合物である、式III−4の化合物を含む。
【0042】
式III、式III−1、及び式III−2の好ましい化合物は、R5がSO3-M+であり、そしてR6がCH2SO3-M+である式III、式III−1、又は式III−2の化合物である、式III−5の化合物を含む。
【0043】
式III、式III−1、式III−2、式III−3、式III−4、及び式III−5の好ましい化合物は、M+がH+、又は一価カチオンである、式III、式III−1、式III−2、式III−3、式III−4、又は式III−5の化合物である、式III−6の化合物を含む。さらに好ましくは、M+はH+、Na+、K+、アンモニウム、又はアルキル化アンモニウムである。特に好ましいのはNa+である。
【0044】
本発明の好ましいアニオン性界面活性剤は、表1に示された化合物を含む:
【0045】
【表1−1】
【0046】
【表1−2】
【0047】
【表1−3】
【0048】
表1に示された2−位置におけるアルキル鎖の置換に加えて、アルキル鎖の他の第2級炭素のうちのいずれかで置換が生じている構造も好ましい。さらに好ましいのは、このような化合物の異性体混合物である。
【0049】
上述のように、本発明の界面活性剤の製造方法は、結果として、個々の化合物に分離する必要なしに、直接に界面活性剤として任意に使用できる式I化合物の混合物を形成することができる。
【0050】
例えば、1つの好ましい混合物は、1つのスルホネート基及び1つのヒドロキシ基を含有する式I化合物;及び2つのスルホネート基を含有する式I化合物を含む。さらなる例は、式II−3の化合物、及び式II−4の化合物を含む組成物である。図1は、さらなる非制限的例を示している。
【0051】
別の好ましい組成物は、アルキル主鎖(R、R1、R2、及びR3と、これらが結合される炭素とによって形成される)が、少なくとも2つの異なる第2級炭素においてエーテルによって置換されている2種又は3種以上の式I化合物を含む異性体混合物を含む。
【0052】
別の態様において、本発明は、式Iのアニオン性界面活性剤を製造する方法を提供する。1つの態様の場合、この方法は、
(a)式A:
【0053】
【化6】
【0054】
(上記式中、R、R1、R2、及びR3は上記で規定された通りであり;そして
R7はH、又はCH2Xであり、R8はXであり、そしてR9はH、又はCH2Xであり、R7又はR9の一方はHであり;そして
XはF、Cl、Br、又はI(好ましくはCl)である)
のエーテル化合物を用意する工程;そして
(b)式I化合物を提供するために、スルホン化条件下で式Aのエーテル化合物をスルホン化する工程を含む。
【0055】
式Aのエーテル化合物は、2009年4月27日付けで出願された、同一出願人による米国特許出願番号12/430,171号明細書(全体的に参照することにより本明細書中に組み込まれる)に記載されているように調製されてよい。一般に、合成は、酸性エーテル化触媒の存在においてアルコール化合物とオレフィンとを反応させることを含む。典型的には、等モル又は僅かに過剰のオレフィンを使用する。溶媒は使用してよいが、必要というわけではない。反応は高温、例えば50〜150℃で行ってよい。一旦所望の量のエーテル化合生成物が形成されたら(例えばガスクロマトグラフィによって検出する)、反応混合物を冷却し、そしてこれにコンベンショナルなワークアップを施す。例えば、均一酸触媒を除去するために、冷却された混合物を、重炭酸塩及び/又は塩化物塩を含有する水に添加し、そしてエーテル化合物を含有する混合物の有機液層を取り出す。エーテル化合物は、周知の技術、例えば蒸留によってさらに精製してよい。
【0056】
上記合成のアルコールは一般に下記式:
【0057】
【化7】
【0058】
(上記式中、R7、R8、及びR9は上記で規定した通りである)
を有する。
【0059】
合成のための好ましいアルコールは、1,3−ジハロ−2−プロパノール、及び2,3−ジハロプロパノール、及びこれらの混合物を含む。特に好ましいのは、1,3−ジクロロ−2−プロパノール、及び2,3−ジクロロプロパノール、及びこれらの混合物である。
【0060】
上記合成において使用するためのオレフィンは好ましくは、炭素原子数4〜22の線状又は分枝状アルファ−オレフィン(すなわち1−アルケン)、又は内部及び/又は第三オレフィン異性体と一緒に4〜22の炭素原子を含有する線状又は分枝状1−アルケンの異性体の混合物である。好ましくは、アルケンは線状であり、炭素原子数が6〜18である。特に好ましいアルファ−オレフィンの非限定的な例は:1−ブテン、1−ペンテン、1−ヘキセン、4−メチル−1−ペンテン、1−ヘプテン、1−オクテン、1−ノネン、1−デセン、1−ウンデセン、1−ドデセン、1−トリデセン、1−テトラデセン、1−ペンタデセン、1−ヘキサデセン、1−ヘプタデセン、1−オクタデセン、1−ノナデセン、1−エイコセン、又はこれらのうちの2種又は3種以上の混合物を含む。
【0061】
酸性エーテル化触媒と接触させるとオレフィンを異性体化することができるので、アルファ−オレフィンを使用することは必要でなく、そして、炭素原子数4〜22の内部オレフィン、又は線状又は分枝状アルケンの異性体の混合物も使用に適している。好適な内部オレフィンの非限定的な例は、2−ブテン、2−ペンテン、2−ヘキセン、3−ヘキセン、2−ヘプテン、3−ヘプテン、2−オクテン、3−オクテン、4−オクテン、2−ノネン、3−ノネン、4−ノネン、2−デセン、3−デセン、4−デセン、5−デセン、など、又はこれらのうちの2種又は3種以上の混合物を含む。
【0062】
エーテル化合物の合成の際に使用するのに適した酸性エーテル化触媒の一例としては、酸性イオン交換樹脂、例えばThe Dow Chemical Companyから入手可能なDOWEX DR−2030、粘土、ゼオライト、スルホン化ポリスチレンビーズ、及び不均一表面上に固定化された酸、例えばシリカビーズ上のテトラフルオロエタンスルホン酸、ブレンステッド酸、例えばtriflic(トリフルオロメタンスルホン)酸、メタンスルホン酸、又は硫酸、ルイス酸、例えばBF3及びその誘導体(例えば二水和物又はエーテル)、及びトリメチルシリルトリフレートが挙げられる。触媒と反応物質との比はさほど重要ではなく、一般に、所望の反応速度を得るように調節される。好ましくは、触媒は、エーテル化反応を容易にするために、プロセス中、約50〜150℃の温度にある。
【0063】
プロセスの工程(b)は、式I化合物を提供するために、スルホン化条件下でエーテル化合物をスルホン化することである。典型的には、エーテル化合物を、スルホン化剤、例えば亜硫酸ナトリウム、又は亜硫酸ナトリウムと炭酸ナトリウムとの組み合わせと接触させる。反応は水中で行われてよく、典型的には高い温度及び圧力、例えば150〜220℃で、100〜350psigで実施される。反応が生じるのに十分な時間(例えば24時間)に続いて、反応混合物を冷却し、周囲条件まで減圧し、そして次いでこれにコンベンショナルなワークアップを施す。式I化合物を必要に応じてさらに精製する。コンベンショナルな技術、例えば抽出、濾過、クロマトグラフィ、及び/又は結晶化を用いて精製を行うことができる。所望の場合には、例えば過酸化水素を添加することにより、過剰の亜硫酸塩を酸化して硫酸塩にすることができる。
【0064】
スルホン酸塩官能基を導入する他の典型的な方法、例えばエーテル化合物を硫化物、又はポリ硫化物と反応させ、次いで酸化させるという方法を、式I化合物を発生させるために用いてもよい。
【0065】
本発明のアニオン性界面活性剤は、上記二官能性ハロゲン化エーテル出発材料(式A)から調製することができる。両ハロゲンをスルホン化剤によって置換する場合、ジスルホネート化合物が形成される。置換及び加水分解の両方が行われる場合には、ヒドロキシスルホネートが形成される。なお、ヒドロキシスルホネートは、直接置換によって生じることができ、或いは、スルホン化条件下で容易に加水分解され、従って反応混合物中では検出されないスルホン中間体を介して生じることもできる。
【0066】
本発明のアニオン性界面活性剤が式I化合物の混合物を含む場合、混合物の組成は、アルキル鎖長、反応温度、塩基度、及び試薬ローディング量を含むスルホン化反応条件を変えることによって制御することができる。こうして、界面活性剤生成物の特徴及び特性は、所望の用途のニーズに合致するように調整することができる。本発明のプロセスによって得ることができる種々様々な界面活性剤組成物の非限定的な例が図1に示されている。また加水分解によって形成される副生成物アルキルアルコール(すなわちC12界面活性剤例の場合にはドデカノール)のレベルも図1に示されている。
【0067】
式Aのエーテル化合物から調製するのに加えて、アルキルグリセリルエーテルスルホネート(AGS)界面活性剤を形成するために一般に用いられるのと同様の方法によって、本発明のアニオン性界面活性剤のいくつか(具体的には1つのスルホネート基と1つのヒドロキシ基とを含有する界面活性剤)を調製することもできる。典型的には、このような方法は;a)エポキシ化合物を形成するためにアルコールとエピクロロヒドリンとを反応させ;そしてb)ヒドロキシモノスルホネートを形成するためにスルホン化することを含む。
【0068】
式Iのアニオン性界面活性剤は、界面活性剤の存在が所望されるか又は必要とされる種々様々な組成物及び用途において使用されてよい。非限定的な例を挙げるならば、界面活性剤は洗濯洗剤、ペイント及び塗料配合物、エマルジョン重合剤又は配合物、家庭用及び工業用クリーナー、農業用配合物、ラテックス配合物、環境復旧剤、油田用化学薬品、石油増進回収剤、ガス処理配合物、布地処理剤及び仕上げ剤、紙パルプ処理剤、香料可溶化剤配合物、金属作業流体、例えば切削流体、及びパーソナルケア製品(スキンケア及びヘアケア製品、例えばシャンプー)、などとして、又はこれらの中に使用されてよい。これらの用途に使用されるべき式Iの化合物の量及び組成は、用途及び所望の結果に応じて変化し、過度の実験なしに当業者によって決定することができる。一般に、界面活性剤として式I化合物を含む組成物は、組成物の総重量を基準として、少なくとも約0.01重量パーセントの界面活性剤を含有することになる。
【0069】
好ましい態様の場合、式Iの界面活性剤は、クリーニング組成物、例えば洗濯洗剤中に使用される。産業界及び消費者がクリーニング組成物に関して目下直面する問題点の1つは、油汚れを除去する効率が不足していることである。この効率不足は、クリーニングが室温で、且つ/又は硬水の存在において行われる場合に一層顕著である。別の問題点は、金属イオン、例えばカルシウム及びマグネシウムの存在におけるアニオン性界面活性剤の不安定性である。
【0070】
式Iのアニオン界面活性剤は前述の問題点に対処する。具体的には、界面活性剤は、油汚れの除去に優れた性能を発揮し、カルシウム・イオン及びマグネシウム・イオンの存在において可溶性である。油汚れに加えて、本発明の界面活性剤を含有するクリーニング組成物は、例えば粒子状汚れ、酸化性汚れ、有機及び無機汚れを含む他のタイプの汚染物を除去するために使用することもできる。
【0071】
クリーニング組成物中に使用されるべき式Iの界面活性剤量は、当業者によって容易に決定することができる。例えば、その量は、クリーニング組成物の総重量を基準として、典型的には約0.01重量%〜30重量%、好ましくは1重量%〜20重量%である。実際の使用時、例えば洗濯用途では、クリーニング組成物を洗浄時間前又は洗浄時間中に水で希釈することにより、式I界面活性剤の0.01重量%〜5重量%、好ましくは0.01重量%〜1重量%、及びより好ましくは0.01重量%〜0.5重量%の濃度を提供することが一般に好ましい。
【0072】
クリーニング組成物は、例えば他のアニオン性界面活性剤、非イオン性界面活性剤、カチオン性界面活性剤、両性界面活性剤、酵素、溶剤、ヒドロトロープ、ビルダー、増粘剤、キレート剤、香料、色素、乳白剤、蛍光増白剤、漂白剤、及びpH緩衝剤から選択された1種又は2種以上の追加の添加剤を含む、このような組成物中に一般に使用される他の添加剤を任意選択的に含有してよい。組成物中の好ましいpH4〜14を維持するために、緩衝剤を使用するのが典型的である。
【0073】
このような任意選択の添加剤が存在する場合、その量は次の通りであるのが好ましい;0.01%〜50%、好ましくは0.01%〜25%、より好ましくは1%〜20%のアニオン性界面活性剤;0.01%〜20%、好ましくは0.01%〜15%、より好ましくは0.5%〜10%の非イオン性界面活性剤;0.01%〜20%、好ましくは0.01%〜15%、より好ましくは0.5%〜10%のカチオン性界面活性剤;0.01%〜20%、好ましくは0.01%〜15%、より好ましくは0.5%〜10%の両性界面活性剤;0.0001%〜6%の酵素;0.01%〜20%、好ましくは0.01%〜15%、より好ましくは0.5%〜10%の溶剤;1%〜60%のビルダー;0.1%〜20%のキレート剤;及び0.1%〜15%、好ましくは0.5%〜10%のヒドロトロープ。
【0074】
上記のように、本発明は、改善された一連の特性を呈する新しいアニオン性界面活性剤を提供し、これらの界面活性剤が種々様々な用途に使用されるのを可能にする。このような特性は、低い水中毒性、高い水溶性、硬水、電解質、及び苛性溶液に対する高い耐容性を含み、そしてクリーニング組成物中で使用されると、油汚れの除去の際に優れた性能を発揮する。
【0075】
下記例は本発明の一例であって、本発明の範囲を限定しようとするものではない。特に断りのない場合には、本明細書中に使用される比、パーセンテージ、部などは重量で表される。
【実施例】
【0076】
例1
1,3−ジクロロ−2−プロパノールによるアルファ−オレフィンのエーテル化
式I化合物の前駆体である模範的なエーテル化合物を、下記プロトコルによって調製することができる。
樹脂ビーズを保持するためのグラスウール栓を有する底部ドレン・ディーン・スターク・トラップに、16.2gのDOWEX DR−2030樹脂を装入する。樹脂を11.5gの1,3−ジクロロ−2−プロパノールで湿潤し、この装置を1L丸底フラスコに取り付ける。フラスコに1.1モルのアルファ−オレフィン、及び139.7gの1,3−ジクロロ−2−プロパノールを装入する(全部で1.17モル)。真空を施し、そして1Lフラスコからの蒸留物を、加熱された樹脂を含有するディーン・スターク・トラップ内に凝縮し、そして1Lフラスコに戻すように、丸底フラスコを加熱する。1Lフラスコ内の温度は、蒸留を続けるにつれて上昇する。反応混合物を蒸留によって精製することにより、アルキル1,3−ジクロロプロピルエーテルをもたらす。
【0077】
例2
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたスルホネートの調製
本発明の模範的な界面活性剤を次のプロトコルによって形成することができる。2L Parr反応器に0.456モルのアルキル1,3−ジクロロプロピルエーテル(例1から)、0.783モルの亜硫酸ナトリウム、0.180モルのメタ−亜硫酸水素ナトリウム、0.289モルの炭酸ナトリウム、及び590gの水を装入する。窒素フラッシュ及び圧力チェックに続いて、システムを20時間にわたって200℃まで加熱する。この温度に達したあとの圧力は250psigである。溶液を周囲温度まで冷却し、そして取り出すことにより反応生成物をもたらす。
【0078】
例3
1,3−ジクロロ−2−プロパノールを用いた1−ドデセンのエーテル化
実質的に例1に記載されたプロトコルに従って形成されるエーテルを次のように製造した。2L反応蒸留装置をここに記載するように組み立てた。磁気攪拌器を備えた2L丸底フラスコを加熱マントル内に装着し、そして蒸留物受容体に接続した。磁気攪拌器及び温度プローブを含有するサイドアーム蒸留物受容体内に蒸留物を凝縮した。蒸留物受容体と2Lフラスコとの間の弁付きラインが、蒸留物受容体内に公称容積約100mLを与えた。触媒床を提供するためにそれぞれの端部に90μmのスクリーンフィルタを備えた長さ21インチ及び直径3/4インチのステンレス鋼管へ、蒸留物受容体の底部から液体をポンプ供給した。触媒含有管を覆うジャケット・システムを、再循環熱油浴を使用して加熱した。触媒床の出口は、液体を蒸留物受容体に戻した。反応蒸留が10〜300torrの圧力で行われ得るように、システムを真空ポンプに接続した。2L反応蒸留装置の触媒床には、60gのDOWEX DR−2030を装入した。2L容器に684.72g(5.304モル)の1,3−ジクロロ−2−プロパノール及び843.05g(5.009モル)の1−ドデセンを装入した。真空を22torrに調節し、そして初期温度79℃で蒸留をもたらすように、蒸気温度70℃で2L容器を加熱した。触媒床を出る反応生成物中の温度を80〜88℃にするために、触媒床油浴を110℃に設定した。凝縮器温度は約−11℃〜−5℃であった。蒸留物受容体温度は63〜69℃であった。さらに加熱するにつれて、底部温度は192℃に達し、オーバーヘッド温度は80℃であった。混合物を冷却し、そして取り出した。蒸留物受容体内及び触媒床内の溶液(96.30g、ローディングされた質量の6.3%)を廃棄した。2L容器内の溶液(1302.33g、ローディングされた質量の85.2%)をGC分析によって分析した(1.803面積%、1.10重量%のドデセン、0.708面積%、0.48重量%のドデカノール、0.01面積%、0.03重量%の1,3−ジクロロ−2−プロパノール、89.843面積%、88.71重量%のC12 1,3−ジクロロプロピルエーテル)。一部(1298.01)を2L丸底フラスコにローディングし、そして、還流スプリッタを載置された14インチ真空ジャケット付きVigreuxカラムを使用して0.2〜0.6torrで蒸留することによりこれを精製した。オーバーヘッド温度25〜105℃、底部温度146〜189℃で15:1の還流比を用いて、最初の留分(30.36g)を捕集した。オーバーヘッド温度104〜118℃、底部温度190〜220℃で15:1の還流比を用いて生成物留分を捕集することにより、1217.88g(4.09モル)の、ドデカンの1,3−ジクロロプロピルエーテルをもたらした(1,3−ジクロロプロパン−2−イルオキシドデカン、94.8面積%のC12 DCPエーテル、位置異性体の混合物、93.8%蒸留収率)。42.10gの残留物が蒸留ボトムとして残った。
【0079】
例4
含水亜硫酸ナトリウムを用いたC6スルホネート反応生成物の調製
出発材料の適切な置換を用いて事実上例3に記載されているように調製された44.48g(0.209モル)の1,3−ジクロロプロパン−2−イルオキシヘキサン、68.7g(0.545モル)の亜硫酸ナトリウム、3.06g(0.029モル)の炭酸ナトリウム、及び251.25gの脱イオン水を2L Parr反応器に装入した。窒素フラッシュ及び圧力チェックに続いて、システムを24時間にわたって170℃に加熱した。結果として生じた圧力は130psigであった。溶液を周囲温度まで冷却することにより、338.69gの溶液を提供した。溶液を酢酸エチル(173.7g、次いで45.0g)で抽出し、そして合体された上側の酢酸エチル相を蒸発させて1.59gの残留物を形成した。残留物のGC分析では、これが18重量%のC6 1,3−ジクロロ−2−プロパノールエーテルを含有することが判った。水溶液を濾過し、そして部分的に濃縮することにより、267.95gの溶液をもたらした。NMR分析では、17重量%(0.135モル、理論値の65%)のナトリウム2−(ヘキシルオキシ)プロパン−1,3−ジスルホネート(C6ジスルホネート)及び2.6重量%(0.026モル、理論値の12%)のナトリウム2−(ヘキシルオキシ)−3−ヒドロキシプロパン−1−スルホネート(C6 モノスルホネート)が見いだされた。
【0080】
例5
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC8スルホネートの調製
実質的に例2に記載されたプロトコルに従って形成される本発明の界面活性剤を、次のように生成した。2L Parr反応器に、出発材料の適切な置換を用いて実質的に例3に記載されているように調製された156.0g(0.646モル)の1,3−ジクロロプロパン−2−イルオキシオクタン、137.1g(1.09モル)の亜硫酸ナトリウム、48.1(0.253モル)のメタ−亜硫酸水素ナトリウム、53.88g(0.508モル)の炭酸ナトリウム、及び600.0gの水を装入した。窒素フラッシュ及び圧力チェックに続いて、システムを20時間にわたって180℃まで加熱した。この温度で圧力は150psigであり、そして一晩で190psigまで上昇させた。溶液を周囲温度まで冷却し、そして取り出すことにより、上側に褐色油層を有する淡褐色反応生成物944.18gをもたらした。pHは7.14であった。水性溶液のHPLC分析では、15.3重量%(0.381モル、理論値の59.0%)のC8ジスルホネート(ナトリウム2−(オクチルオキシ)プロパン−1,3−ジスルホネート)及び4.1重量%(0.206モル、理論値の21%)のC8モノスルホネート(ナトリウム2−(オクチルオキシ)−3−ヒドロキシプロパン−1−スルホネート)が見いだされた。全反応生成物のうちの24.70gの部分を取り出して12.73gの酢酸エチルで抽出した。11.55gの有機相のGC分析では、0.418重量%のオクタノール(944.18gの溶液中1.88g、0.0119モル、装入されたC8 1,3−ジクロロ−2−プロパノールエーテルの1.8モル%)、及び3.75重量%のC8 1,3−ジクロロ−2−プロパノールエーテル及びC8オレフィンの二量体生成物(ヘキサデセン異性体)(944.18gの溶液中16.9g、装入されたC8 1,3−ジクロロ−2−プロパノールエーテルの10.8重量%)が見いだされた。
【0081】
例6
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC10スルホネートの調製
出発材料(138.6gの1,3−ジクロロ−2−プロパン−2−イルオキシデカン、112.5gの亜硫酸ナトリウム、39.0gのメタ−亜硫酸水素ナトリウム、34.52gの炭酸ナトリウム、及び550.43gの水、190℃、20時間)の適切な置換を用いて、実質的に上述のプロトコルに従って化合物を形成した。反応に続いて、反応混合物を周囲温度まで冷却し、そして取り出すことにより、淡褐色反応生成物826.5gをもたらした。pHは7.11であった。水性溶液のHPLC分析では、14.3重量%(0.292モル、理論値の56.6%)のC10ジスルホネート(ナトリウム2−(デシルオキシ)プロパン−1,3−ジスルホネート)及び5.4重量%(0.139モル、理論値の27.0%)のC10モノスルホネート(ナトリウム2−(デシルオキシ)−3−ヒドロキシプロパン−1−スルホネート)が見いだされた。18.44gの部分を取り出して6.45gの酢酸エチルで抽出した。5.06gの有機相のGC分析では、0.662重量%のデセン(826.5gの溶液中1.50g、装入されたC10 1,3−ジクロロ−2−プロパノールエーテルの1.1%)、及び1.44重量%のデカノール(826.5gの溶液中3.26g、0.0206モル、装入されたC10 1,3−ジクロロ−2−プロパノールエーテルの4.0モル%)、1.15重量%のC10 1,3−ジクロロ−2−プロパノールエーテル(826.5gの溶液中2.60g、装入されたC10 1,3−ジクロロ−2−プロパノールエーテルの1.9%)、及び0.615重量%のC10オレフィン二量体生成物エイコセン(826.5gの溶液中1.39g、装入されたC10 1,3−ジクロロ−2−プロパノールエーテルの1重量%)が見いだされた。
【0082】
例7
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC12スルホネートの調製
出発材料(135.5gの1,3−ジクロロ−2−プロパン−2−イルオキシデカン、98.66gの亜硫酸ナトリウム、34.18gのメタ−亜硫酸水素ナトリウム、30.66gの炭酸ナトリウム、及び590gの水、200℃、20時間)の適切な置換を用いて、実質的に上述のプロトコルに従って化合物を形成した。反応に続いて、反応混合物を周囲温度まで冷却し、そして取り出すことにより、淡褐色反応生成物865.3gをもたらした。pHは7.75であった。水性溶液のHPLC分析では、9.14重量%(0.183モル、理論値の40%)のナトリウム2−(ドデカニルオキシ)プロパン−1,3−ジスルホネート)(C12ジスルホネート)及び5.69重量%(0.142モル、理論値の31%)のナトリウム2−(ドデカニルオキシ)−3−ヒドロキシプロパン−1−スルホネート(C12モノスルホネート)が見いだされた。10.4gの部分を取り出して4.00gの酢酸エチルで抽出した。2.77gの有機相のGC分析では、0.2重量%のドデセン(865.3gの溶液中0.46g、装入されたC12 1,3−ジクロロ−2−プロパノールエーテルの0.34%)、1.98重量%のデカノール(865.3gの溶液中4.56g、0.0245モル、装入されたC12 1,3−ジクロロ−2−プロパノールエーテルの5.4モル%)、及び1.35重量%のC12 1,3−ジクロロ−2−プロパノールエーテル(865.3gの溶液中3.11g、装入されたC12 1,3−ジクロロ−2−プロパノールエーテルの2.3%)が見いだされた。
【0083】
例8
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC14スルホネートの調製
出発材料(160.8gの1,3−ジクロロ−2−プロパン−2−イルオキシテトラデカン、108.53gの亜硫酸ナトリウム、37.46gのメタ−亜硫酸水素ナトリウム、35.14gの炭酸ナトリウム、及び828.5gの水、203℃、20時間)の適切な置換を用いて、実質的に上述のプロトコルに従って化合物を形成した。反応に続いて、反応混合物を周囲温度まで冷却し、そして取り出すことにより、黄色反応生成物1156.0gをもたらした。pHは6.67であった。水性溶液のHPLC分析では、7.19重量%(0.181モル、理論値の37%)のC14ジスルホネート(ナトリウム2−(テトラデシルオキシ)プロパン−1,3−ジスルホネート)及び5.70重量%(0.176モル、理論値の36%)のC14モノスルホネート(ナトリウム2−(テトラデシルオキシ)−3−ヒドロキシプロパン−1−スルホネート)が見いだされた。19.80gの部分を取り出して8.76gの酢酸エチルで抽出した。7.06gの有機相のGC分析では、0.29重量%のテトラドデセン(1156gの溶液中1.18g、装入されたC14 1,3−ジクロロ−2−プロパノールエーテルの0.73%)、1.99重量%のテトラデカノール(1156gの溶液中8.2g、0.0383モル、装入されたC14 1,3−ジクロロ−2−プロパノールエーテルの7.7モル%)、及び1.53重量%のC14 1,3−ジクロロ−2−プロパノールエーテル(1156gの溶液中6.3g、装入されたC14 1,3−ジクロロ−2−プロパノールエーテルの3.9%)が見いだされた。
【0084】
例9
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC16スルホネートの調製
出発材料(87.10gの1,3−ジクロロ−2−プロパン−2−イルオキシヘキサデカン、53.57gの亜硫酸ナトリウム、19.83gのメタ−亜硫酸水素ナトリウム、19.11gの炭酸ナトリウム、及び558.44gの水、207℃、31時間)の適切な置換を用いて、実質的に上述のプロトコルに従って化合物を形成した。反応に続いて、反応混合物を周囲温度まで冷却し、そして取り出すことにより、乳化反応混合物677g(ローディングされた質量の91.7%)をもたらした。エマルジョンの一部が反応器の壁に残った。9.5891gの部分を86.30gの水で希釈することによって提供される透明溶液のHPLC分析では、3.39重量%のC16ジスルホネート(ナトリウム2−(ヘキサデシルオキシ)プロパン−1,3−ジスルホネート)(738gの生成物溶液中0.051モルに相当、又は20.8モル%)、及び4.74重量%のC16モノスルホネート(ナトリウム2−(ヘキサデシルオキシ)−3−ヒドロキシプロパン−1−スルホネート)(738gの生成物溶液中0.087モルに相当、又は35.3モル%)、が見いだされた。生成混合物の38.41gの部分を19.04gの酢酸エチルで希釈すると、2つの透明な相がもたらされた。上側の16.80gの有機相のGC分析では、0.619重量%のヘキサドデセン(738gの反応生成物溶液中2.0g、装入されたC16 1,3−ジクロロ−2−プロパノールエーテルの2.3%)、2.28重量%のヘキサデカノール(738gの反応生成物溶液中7.35g、0.0303モル、装入されたC16 1,3−ジクロロ−2−プロパノールエーテルの12.2モル%)、及び0.57重量%のC16 1,3−ジクロロ−2−プロパノールエーテル(738gの反応生成物溶液中1.85g、装入されたC16 1,3−ジクロロ−2−プロパノールエーテルの2.1モル%)が見いだされた。
【0085】
例10
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC18スルホネートの調製
出発材料(107.36gの1,3−ジクロロ−2−プロパン−2−イルオキシオクタデカン、62.2gの亜硫酸ナトリウム、21.74gのメタ−亜硫酸水素ナトリウム、21.08gの炭酸ナトリウム、及び584.7gの水、212℃、24時間)の適切な置換を用いて、実質的に上述のプロトコルに従って化合物を形成した。反応に続いて、反応混合物を周囲温度まで冷却し、そして取り出すことにより、極めて高粘度の白色エマルジョン607.54g(ローディングされた質量の76.2%)をもたらした。エマルジョンの一部が反応器の壁に残った。生成混合物の281.55gの部分を307.14gの酢酸エチル及び254gの水で希釈すると、2つの透明な相がもたらされた。上側の有機相(288.25g)を蒸発させて13.36gの残留物を形成した。残留物のGC分析では、9.97重量%のオクタデセン(797gの反応生成物中3.79g、装入されたC18 1,3−ジクロロ−2−プロパノールエーテルの3.5%)、36.32重量%のオクタデカノール(797gの反応生成物中13.81g、0.0504モル、装入されたC18 1,3−ジクロロ−2−プロパノールエーテルの17.9モル%)、及び47.52重量%のC18 1,3−ジクロロ−2−プロパノールエーテル(797gの反応生成物中18.1g、装入されたC18 1,3−ジクロロ−2−プロパノールエーテルの16.8%)が見いだされた。反応生成物の5.50gの部分を44.16gの水、及び5.34gの2−プロパノールで希釈することにより、濁った溶液(1:10希釈)を提供した。溶液のHPLC分析では、反応生成物が3.25重量%のC18ジスルホネート(ナトリウム2−(オクタデシルオキシ)プロパン−1,3−ジスルホネート)(797gの生成物溶液中0.0501モルに相当、又は17.8モル%)、及び4.47重量%のC18モノスルホネート(ナトリウム2−(オクタデシルオキシ)−3−ヒドロキシプロパン−1−スルホネート)(797gの生成物溶液中0.0828モルに相当、又は29.5モル%)を含有することを見いだした。
【0086】
例11
個々の界面活性剤成分の単離
個々の界面活性剤成分は本発明のスルホネート組成物から単離されてよい。例えば例7のC12スルホネート組成物を、C18逆相クロマトグラフィ樹脂を含有するカラムに装入し、そしてアセトニトリルの水溶液で溶離する。所望の成分を含有する画分を捕集して蒸発させることにより、ナトリウム2−(ドデカニルオキシ)プロパン−1,3−ジスルホネート(C12ジスルホネート)、及びナトリウム2−(ドデカニルオキシ)−3−ヒドロキシプロパン−1−スルホネート(C12モノスルホネート)の単離された固形試料を提供する。
【0087】
例12
表面張力評価
Kruss K100張力計を使用してウィルヘルミー板によって、表面張力を測定する。逆CMC拡張法(reverse CMC extended method)を利用する。この方法では、界面活性剤溶液を試料容器内に先ず入れ、次いで脱イオン水で段階的に希釈する。それぞれの水添加後、試料を攪拌し、次いで、同量の溶液を取り出し、一定の体積を維持する。0.1M塩化ナトリウム中の、例5〜10で生成された本発明のC8〜C18スルホネート組成物の表面張力結果を、アルキルジフェニルオキシドジスルホネート(Dow Chemical CompanyからDOWFAX(登録商標)2A1として入手可能)と比較して、図2に示す。結果は、本発明のこれらのスルホン化材料が良好な界面活性剤特性を呈することを示している。それというのも、これらの材料は表面張力が低く(30mN/m未満)、そしてC14及びC16スルホネートの場合、DOWFAX 2A1よりも低い濃度で、最小表面張力に達することを示しており、この場合、CMCは、DOWFAX 2A1の232mN/mに対して、それぞれ68mN/m及び74mN/mである。
【0088】
例13
動的表面張力評価
最大気泡圧力法を用いて液体の動的表面張力を測定するKruss気泡圧力張力計BP2を利用して、1重量%の界面活性剤の動的表面張力を測定する。動的表面張力は、いかに素早く界面活性剤分子が界面へ移動するか、ひいてはいかに効果的に界面活性剤が適用時間スケール内で表面張力を低減するかを示すので、重要な界面活性剤特性である。
【0089】
例5〜10で生成された本発明のC8〜C18スルホネート組成物の動的表面張力は、これらの界面活性剤が、DOWFAX(登録商標)2A1よりも速く界面へ移動することを示している(図3)。このことは、例えば動的プロセス、例えばインクジェット印刷にとって極めて重要な利点であり得る。
【0090】
例14
起泡力評価
0.1重量%界面活性剤溶液を使用してASTM法D1173に従って、ロスマイルス起泡力試験を行う。例5〜10で生成された本発明のC8〜C18スルホネート組成物に対するロスマイルス起泡力試験結果を、DOWFAX(登録商標)2A1と比較して、図4に示す。結果は、初期注入時点、及び2分後の泡の高さとして表す。生成した泡の高さが40mm未満の界面活性剤、又は2分後に40mmまで崩壊する界面活性剤が、低起泡力界面活性剤と考えられるのに対して、120mmを上回る高さの泡を生成する界面活性剤は高起泡力界面活性剤と考えられる。界面活性剤の用途はしばしば特定の起泡力のニーズ及び/又は制限を有する。起泡力の結果は、低起泡力要件又は高起泡力要件の両方が生じる可能性のある用途に、疎水性物質の組成に応じて、本発明の界面活性剤を合わせ得ることを示している。
【0091】
例15
溶解度評価
水性苛性物質中の溶解度評価のために、2mlの1%界面活性剤溶液を8mlの脱イオン水と合体し、バイアルを手で振ることによりよく混ぜる。次いで10mlの20% NaOH溶液を添加し、そして試料バイアルを30秒間にわたって振る。最終濃度は界面活性剤が0.1重量%、NaOHが10重量%である。
【0092】
塩化カルシウムとの溶解度評価のために、2mlの1%界面活性剤溶液を8mlの脱イオン水と合体し、バイアルを手で振ることによりよく混ぜる。次いで10mlの2MCaCl2溶液を添加し、そして試料バイアルを30秒間にわたって振る。最終濃度は界面活性剤が0.1重量%、CaCl2が1Mである。
【0093】
10%水酸化ナトリウム、1M塩化カルシウム、及び7℃における、例5、7、及び8で生成された、本発明のC8、C12、及びC14スルホネート組成物の0.1重量%溶液の視覚的評価を、LAS及びDOWFAX(登録商標)2A1と比較して下記表2に示し、そして評価は、これらの新しい材料が望ましい特性を有することを示す。
【0094】
【表2】
【0095】
例16
水中毒性
研究手順及び試験法は、下記ガイドラインの推奨事項に基づく:
経済協力開発機構(OECD):化学薬品の試験のためのOECDガイドライン、「淡水性藻類及びシアノバクテリア、成長阻害試験」手順201,2006年3月23日付け採択;欧州経済共同体(EEC):1992年7月31日付けの委員会指示92/69/EEC、環境毒性の検出法、C.3,「藻類阻害試験」。
【0096】
例11の分離・精製されたC12モノ及びジスルホネートの急性毒性試験のデータを表3に示す。
【0097】
【表3】
【0098】
US EPAによって使用される分類システムに従って、毒性を報告する:極めて高い毒性(EC50</=0.1mg/l);高い毒性(EC50>0.1且つ</=1mg/l);多少の毒性(EC50>1且つ</=10mg/l);僅かな毒性(EC50>10且つ</=10mg/l);事実上非毒性(EC50>100mg/l)。データは、本発明の界面活性剤が極めて好ましい毒性プロフィールを有することを示す。
【0099】
例17
クリーニング効率及び溶解度の評価
この例では、表4に示された配合物を試験する。
【0100】
【表4】
【0101】
調理用油脂及びスーダン・レッドLot#1276、産業界における標準汚れで染色されたScientific Services S/D, Inc.の「Cotton 400」見本を、表4に示す2種の洗濯用配合物の水溶液で独立して洗う。
【0102】
評価のために用いられる手順は次の通りである:
タージトメータ(Tergitometer)を使用して洗浄を行う(タージトメータは、ラボラトリー規模で洗濯物を回すための産業用標準機器である)。タージトメータは100rpmで回るように設定する。各洗濯ポットは1リットルの総溶液(水+配合物)と6つの見本とを含む。洗浄の直前に、配合物を硬水中に2500ppmで希釈する。洗浄液のpHをNaOHの希釈溶液で10に調節する。4リットルの脱イオン水中に1.37gのCaCl2・2H2O及び0.43gのMgCl2・6H2Oを希釈することにより、硬水を調製する。洗浄を23℃で行う。洗浄時間は25分間である。濯ぎ時間は脱イオン水中で2分間である。
【0103】
下記式に従って、見本のL、a*、b*色パラメータの測定値に基づいて、クリーニング効率(染み除去指数:Stain Removal Index)を計算する:
【0104】
【数1】
【0105】
(上記式中、X1=(L,a*,b*)洗濯が行われる前のパラメータ、
上記式中、X2=(L,a*,b*)洗濯が行われた後のパラメータ)
【0106】
図5は、配合物1及び2のSRIの比較を示すグラフである。データは、本発明の配合物2が比較配合物1よりも、優れた油脂除去性能を呈することを示している。
【0107】
クリーニング効率に加えて、カルシウム・イオンの存在において、2種の配合物を溶解度の点に関しても観察する。視覚検査は、比較配合物1がモノスルフェート・アニオン性界面活性剤のカルシウム塩の不溶性によって引き起こされる予期される濁りを呈し、これに対して、本発明のモノスルホネート化合物及びジスルホネート化合物を含有する本発明の配合物2は透明であり、不溶性の塩は形成されないことを示す。
【0108】
視覚検査に加えて、画像のグレー値を測定するために、imageJソフトウェアを使用して試験溶液の画像上でデジタル分析を行う。比較配合物1はグレー値19を示すのに対して、本発明の配合物2はグレー値9を示す。この結果は、不溶性塩が配合物2よりも著しく大きな規模で配合物1に形成されることを裏付ける。
【0109】
例18
スチレン−ブタジエンコポリマーのエマルジョン重合
例7で調製された(本発明の)C12スルホネート組成物を、スチレン−ブタジエンコポリマーのエマルジョン重合において試験し、DOWFAX(登録商標)2A1を界面活性剤として利用する重合と比較する。次のスチレン−ブタジエン・プロトコルを使用する。0.454部のシード・ラテックス、79.2部の水、36.5部のブタジエン、28.3部のスチレン、1部のt−ドデシルメルカプタンを反応器に装入する。反応器内にさらに30.2部のスチレン、5部のアクリル酸、15部の水、1部の過硫酸ナトリウム、0.3部の本発明のスルホネート組成物(対0.5部のDOWFAX(登録商標)2A1)、及び0.1部の水酸化ナトリウムを添加する。水酸化ナトリウムを用いてpH6.2に中和された本発明のC12スルホネート界面活性剤を用いて形成されたポリマーの分析結果は下記の通りである:
100及び325メッシュ篩を介して反応器を濾過した後に観察される残留物レベル:26mg/L、固形分:50.9%、粒度:130nm。重合及び水蒸気ストリッピング中の安定性;優。ポリマーエマルジョン表面張力(1%):25℃で48.6dyn/cm。Ca2+イオン安定性(凝集するまでラテックスに10%塩化カルシウム溶液を添加することによって見極める):16ml/100g。
【0110】
本発明の界面活性剤は、100及び325メッシュ篩を介して反応器を濾過した後で観察される低い残留物レベルによって示されるように、優れた安定性を提供する。界面活性剤は粒度制御を妨げない。基準ラテックス(DOWFAX(登録商標)2A1で形成される)と同様の粒度が観察される。このことは、重合・水蒸気ストリッピング過程中の良好な安定性を示す。粒度分布の著しい相違は観察されない。界面活性剤は、反応器試料中の残留スチレン量によって示されるように、モノマー変換に影響を及ぼすことはない。本発明の界面活性剤で形成されたポリマーと、比較DOWFAX(登録商標)2A1で形成されたポリマーとの間には、同様のガラス転移温度及びゲル含量が観察される。カルボン酸モノマーは、セーラム相、ラテックス粒子表面の間に分布されているか、又はポリマー粒子内部に埋め込まれている。界面活性剤のタイプを変更した場合に、酸分布に差は生じない。
【0111】
C12スルホネート界面活性剤が反応速度特性及び粒度制御に不都合な影響を与えることはない。この処方における界面活性剤の量が、商業的なベンチマークと比較して本発明のスルホネート組成物において40%だけ低下した場合に、同様のラテックス及びポリマーの特性が達成される。界面活性剤は単独で、又は他の界面活性剤(非イオン性又は硫酸化アルキルエトキシレートを含む)との組み合わせで使用することができる。
【0112】
例19
界面活性剤溶解度のさらなる比較
この例において、本発明のアニオン性界面活性剤を、本発明のものではないアルキルグリセリルエーテルスルホネート(AGS)界面活性剤と比較する。比較を目的として、本発明の界面活性剤及び比較界面活性剤の両方を、AGS界面活性剤を形成するために一般に用いられる方法によって調製する(例えば米国特許第2989547号明細書、同第4976953号明細書、同第5246613号明細書、国際公開第1997/040131号パンフレット、同第4917823号明細書、同第5062973号明細書、欧州特許出願公開第717032号明細書、米国特許第4954281号明細書参照)。この方法は、a)アルコールとエピクロロヒドリンとを反応させることにより、式Bのエポキシ化合物を形成し、次いでb)式Cのヒドロキシモノスルホネートを形成するためにスルホン化することを伴う。
【0113】
【化8】
【0114】
本発明の界面活性剤及び比較(本発明のものではない)界面活性剤の水溶性を表5に示す。本発明のものではない界面活性剤は驚くべきことに、式CのR=C12及びR=C16に関して<0.1及び<0.025重量%という低い水溶性を示す。低い溶解度は、これらの界面活性剤としての有用性を制限する。比較すると、本発明のC8〜C12スルホネート界面活性剤の混合分枝状アルキル鎖の場合、9重量%を上回る、そして17重量%もの高さの界面活性剤濃度が観察される。
【0115】
【表5】
【0116】
本発明を好ましい態様に従って説明してきたが、本発明はこの開示内容の思想及び範囲の中で改変することができる。従って本出願は、本明細書中に開示された一般原理を用いた、本発明のいかなる変更形、使用、又は適応形にも範囲が及ぶものとする。さらに、本出願は、本発明が関連する技術分野において周知の又は習慣的な実施の圏内に入るような、そして下記請求項の範囲内に含まれる、本開示内容からの逸脱にも範囲が及ぶものとする。
【技術分野】
【0001】
関連出願の相互参照
本出願は2009年7月16日付けで出願された米国仮出願番号61/226,109号明細書の優先権を主張する。前記明細書は、これを全体的に参照することにより本明細書中に組み入れられる。
【0002】
本発明は、新しいスルホネート界面活性剤、新しいスルホネート界面活性剤の組成物、及び当該界面活性剤の製造方法及び使用に関する。
【背景技術】
【0003】
アニオン性界面活性剤はよく知られており、例えばクリーナー及び洗剤を含む種々の用途において使用されている。しかし、一般的なアニオン性界面活性剤、例えば線状アルキルベンゼンスルホネート(LAS)、及びアルコールスルフェート(例えばナトリウムラウリルスルフェート)は、冷水又は硬水中、及び苛性物質中での溶解度が低い。例えばエピクロロヒドリン、線状アルキルアルコール、及び亜硫酸ナトリウムから調製されたアルキルグリセロールスルホネート(AGS)界面活性剤も同様である。このような溶解性不足は、これらの界面活性剤の配合選択肢及び性能を制限する。
【0004】
高い界面活性を維持しつつ、改善された一連の特性、例えば冷水、硬水、苛性水、及びイオン水中での高い溶解度を有するアニオン性界面活性剤を提供することは、アニオン性界面活性剤にとって依然として難題である。
【発明の概要】
【課題を解決するための手段】
【0005】
1つの態様において、本発明は、新規のアニオン性スルホネート界面活性剤を提供する。これらのアニオン性界面活性剤は、式I:
【0006】
【化1】
【0007】
(上記式中、R、R1、R2、R3、R4、R5及びR6は下記で規定される通りである)
の化合物、又はこれらのうちの2種もしくは3種以上の混合物である。
【0008】
別の態様では、本発明は、式Iの1種又は2種以上のアニオン性界面活性剤を含有する配合物を提供する。
【0009】
さらなる別の態様では、本発明は、式Iの界面活性剤を含むクリーニング組成物を提供する。
【0010】
さらに別の態様では、本発明は、式Iのアニオン性界面活性剤又はこれらのうちの2種又は3種以上の混合物を製造する方法を提供する。
【図面の簡単な説明】
【0011】
【図1】図1は、本発明の方法によって得ることができる界面活性剤組成物の例を示すグラフである。
【図2】図2は、本発明のものではない界面活性剤と比較した本発明のアニオン性界面活性剤の表面張力の結果を示すグラフである。
【図3】図3は、本発明のものではない界面活性剤と比較した本発明のアニオン性界面活性剤の動的表面張力の結果を示すグラフである。
【図4】図4は、本発明のものではない界面活性剤と比較した本発明のアニオン性界面活性剤のロスマイルス(Ross−Miles)起泡力試験結果を示すグラフである。
【図5】図5は、本発明の配合物のクリーニング効率を、比較配合物と比較するグラフである。
【発明を実施するための形態】
【0012】
上記のように、1つの態様において、本発明は新規のアニオン性界面活性剤を提供する。本発明のアニオン性界面活性剤は、2つのスルホネート基、又は1つのスルホネート基と1つのヒドロキシ基とを、分枝状アルキルエーテル主鎖上に含有する。アニオン性界面活性剤は、他の既知のジスルホネート界面活性剤及びヒドロキシスルホネート(例えばAGS)界面活性剤と比較して改善された一連の特性を呈し、界面活性剤が種々様々な用途に使用されるのを可能にする。例えば、本発明の界面活性剤は高い水溶性を有し、硬水、電解質、及び苛性溶液に対して高い耐容性を有し、そしてクリーニング組成物中で使用されると、油汚れの除去の際に優れた性能を発揮する。加えて、以下の例によって実証されるように、本発明の界面活性剤は、極めて好ましい水中毒性を示す。
【0013】
本発明のアニオン性界面活性剤は、
式I:
【0014】
【化2】
【0015】
(上記式中、Rは線状又は分枝状C2−C22アルキルであり;
R1、R2、及びR3は独立して、H又は線状もしくは分枝状C1−C18アルキルであり;
R4はH、CH2SO3-M+、又はCH2OHであり;
R5はOH、SO3-M+、又は式:
【0016】
【化3】
【0017】
の基であり;
R6はH、CH2SO3-M+、又はCH2OHであり;そして
M+はH+、又は一価又は二価カチオン、例えばナトリウム、カリウム、アンモニウム、カルシウム、マグネシウム、又はアルキル化アンモニウムであり、
R4及びR6のうちの一方はHであり、そしてR4、R5、又はR6のうちの1つ又は2つはSO3-M+を含有する)
の化合物である。
【0018】
本発明の界面活性剤を製造するための下記方法は結果として、式I化合物の混合物を形成することができる。個々の式I化合物は混合物から単離することができるものの、この工程は必要なものではなく、実際には界面活性剤を混合物の形態で使用することが好ましいことがある。こうして、式I化合物の混合物である界面活性剤が考えられ、これも本発明の範囲に含まれる。
【0019】
式Iの好ましい化合物は、Rが線状C2−C22アルキルである式Iの化合物である、式I−1の化合物(又はこれらの混合物)を含む。さらに好ましくは、Rは、線状又は分枝状、より好ましくは線状のC4−C16アルキルである。
【0020】
式I、及び式I−1の好ましい化合物は、R1がHである式I、又は式I−1の化合物である、式I−2の化合物を含む。
【0021】
式I、式I−1、及び式I−2の好ましい化合物は、R2がHである式I、式I−1、又は式I−2の化合物である、式I−3の化合物を含む。
【0022】
式I、式I−1、式I−2、及び式I−3の好ましい化合物は、R3がHである式I、式I−1、式I−2、又は式I−3の化合物である、式I−4の化合物を含む。
【0023】
式I、式I−1、式I−2、式I−3、及び式I−4の好ましい化合物は、R4がCH2SO3-M+、又はCH2OHである式I、式I−1、式I−2、式I−3、又は式I−4の化合物である、式I−5の化合物を含む。1つの態様では、R4は好ましくはCH2SO3-M+である。別の態様では、R4は好ましくはCH2OHである。
【0024】
式I、式I−1、式I−2、式I−3、式I−4、及び式I−5の好ましい化合物は、R5がOH、又はSO3-M+である式I、式I−1、式I−2、式I−3、式I−4、又は式I−5の化合物である、式I−6の化合物を含む。1つの態様では、R5は好ましくはSO3-M+である。別の態様では、R5は好ましくはOHである。
【0025】
式I、式I−1、式I−2、式I−3、式I−4、式I−5、及び式I−6の好ましい化合物は、R6がHである式I、式I−1、式I−2、式I−3、式I−4、式I−5、又は式I−6の化合物である、式I−7の化合物を含む。
【0026】
式I、式I−1、式I−2、式I−3、式I−4、式I−5、式I−6、及び式I−7の好ましい化合物は、M+がH+、又は一価カチオンである式I、式I−1、式I−2、式I−3、式I−4、式I−5、式I−6、又は式I−7の化合物である、式I−8の化合物を含む。さらに好ましくは、M+はH+、Na+、K+、アンモニウム、又はアルキル化アンモニウムである。特に好ましいのはNa+である。
【0027】
式Iの好ましい化合物はさらに、式II:
【0028】
【化4】
【0029】
(上記式中、Rは線状又は分枝状C2−C22アルキルであり;
R1、R2、及びR3は独立して、H又は線状もしくは分枝状C1−C18アルキルであり;
R4はCH2SO3-M+、又はCH2OHであり;
R5はOH、又はSO3-M+であり;そして
M+はH+、又は一価又は二価カチオンであり、R4及びR5のうちの一方又は両方がSO3-M+を含有する)
の化合物を含む。
【0030】
式IIの好ましい化合物は、Rが線状C2−C22アルキルである式IIの化合物である、式II−1の化合物を含む。さらに好ましくは、Rは、線状又は分枝状、より好ましくは線状のC4−C16アルキルである。
【0031】
式II、及びII−1の好ましい化合物は、R1、R2、及びR3がそれぞれHである式II、又はII−1の化合物である、式II−2の化合物を含む。
【0032】
式II、式II−1、及び式II−2の好ましい化合物は、R4がCH2SO3-M+であり、そしてR5がOHである式II、式II−1、又は式II−2の化合物である、式II−3の化合物を含む。
【0033】
式II、式II−1、及び式II−2の好ましい化合物は、R4がCH2SO3-M+であり、そしてR5がSO3-M+である式II、式II−1、又は式II−2の化合物である、式II−4の化合物を含む。
【0034】
式II、式II−1、式II−2、式II−3、及び式II−4の好ましい化合物は、M+がH+、又は一価カチオンである、式II、式II−1、式II−2、式II−3、又は式II−4である式II−5の化合物を含む。さらに好ましくは、M+はH+、Na+、K+、アンモニウム、又はアルキル化アンモニウムである。特に好ましいのはNa+である。
【0035】
本発明のいくつかの態様の場合、式Iの化合物は式III:
【0036】
【化5】
【0037】
(上記式中、Rは線状又は分枝状C2−C22アルキルであり;
R1、R2、及びR3は独立して、H又は線状もしくは分枝状C1−C18アルキルであり;
R5はOH、又はSO3-M+であり;
R6はCH2SO3-M+、又はCH2OHであり;そして
M+はH+、又は一価又は二価カチオンであり、R5及びR6のうちの一方又は両方がSO3-M+を含有する)
を有する。
【0038】
式IIIの好ましい化合物は、Rが線状C2−C22アルキルである式IIIの化合物である、式III−1の化合物を含む。さらに好ましくは、Rは、線状又は分枝状、より好ましくは線状のC4−C16アルキルである。
【0039】
式III、及びIII−1の好ましい化合物は、R1、R2、及びR3がそれぞれHである式III、又はIII−1の化合物である、式III−2の化合物を含む。
【0040】
式III、式III−1、及び式III−2の好ましい化合物は、R5がOHであり、そしてR6がCH2SO3-M+である式III、式III−1、又は式III−2の化合物である、式III−3の化合物を含む。
【0041】
式III、式III−1、及び式III−2の好ましい化合物は、R5がSO3-M+であり、そしてR6がCH2OHである式III、式III−1、又は式III−2の化合物である、式III−4の化合物を含む。
【0042】
式III、式III−1、及び式III−2の好ましい化合物は、R5がSO3-M+であり、そしてR6がCH2SO3-M+である式III、式III−1、又は式III−2の化合物である、式III−5の化合物を含む。
【0043】
式III、式III−1、式III−2、式III−3、式III−4、及び式III−5の好ましい化合物は、M+がH+、又は一価カチオンである、式III、式III−1、式III−2、式III−3、式III−4、又は式III−5の化合物である、式III−6の化合物を含む。さらに好ましくは、M+はH+、Na+、K+、アンモニウム、又はアルキル化アンモニウムである。特に好ましいのはNa+である。
【0044】
本発明の好ましいアニオン性界面活性剤は、表1に示された化合物を含む:
【0045】
【表1−1】
【0046】
【表1−2】
【0047】
【表1−3】
【0048】
表1に示された2−位置におけるアルキル鎖の置換に加えて、アルキル鎖の他の第2級炭素のうちのいずれかで置換が生じている構造も好ましい。さらに好ましいのは、このような化合物の異性体混合物である。
【0049】
上述のように、本発明の界面活性剤の製造方法は、結果として、個々の化合物に分離する必要なしに、直接に界面活性剤として任意に使用できる式I化合物の混合物を形成することができる。
【0050】
例えば、1つの好ましい混合物は、1つのスルホネート基及び1つのヒドロキシ基を含有する式I化合物;及び2つのスルホネート基を含有する式I化合物を含む。さらなる例は、式II−3の化合物、及び式II−4の化合物を含む組成物である。図1は、さらなる非制限的例を示している。
【0051】
別の好ましい組成物は、アルキル主鎖(R、R1、R2、及びR3と、これらが結合される炭素とによって形成される)が、少なくとも2つの異なる第2級炭素においてエーテルによって置換されている2種又は3種以上の式I化合物を含む異性体混合物を含む。
【0052】
別の態様において、本発明は、式Iのアニオン性界面活性剤を製造する方法を提供する。1つの態様の場合、この方法は、
(a)式A:
【0053】
【化6】
【0054】
(上記式中、R、R1、R2、及びR3は上記で規定された通りであり;そして
R7はH、又はCH2Xであり、R8はXであり、そしてR9はH、又はCH2Xであり、R7又はR9の一方はHであり;そして
XはF、Cl、Br、又はI(好ましくはCl)である)
のエーテル化合物を用意する工程;そして
(b)式I化合物を提供するために、スルホン化条件下で式Aのエーテル化合物をスルホン化する工程を含む。
【0055】
式Aのエーテル化合物は、2009年4月27日付けで出願された、同一出願人による米国特許出願番号12/430,171号明細書(全体的に参照することにより本明細書中に組み込まれる)に記載されているように調製されてよい。一般に、合成は、酸性エーテル化触媒の存在においてアルコール化合物とオレフィンとを反応させることを含む。典型的には、等モル又は僅かに過剰のオレフィンを使用する。溶媒は使用してよいが、必要というわけではない。反応は高温、例えば50〜150℃で行ってよい。一旦所望の量のエーテル化合生成物が形成されたら(例えばガスクロマトグラフィによって検出する)、反応混合物を冷却し、そしてこれにコンベンショナルなワークアップを施す。例えば、均一酸触媒を除去するために、冷却された混合物を、重炭酸塩及び/又は塩化物塩を含有する水に添加し、そしてエーテル化合物を含有する混合物の有機液層を取り出す。エーテル化合物は、周知の技術、例えば蒸留によってさらに精製してよい。
【0056】
上記合成のアルコールは一般に下記式:
【0057】
【化7】
【0058】
(上記式中、R7、R8、及びR9は上記で規定した通りである)
を有する。
【0059】
合成のための好ましいアルコールは、1,3−ジハロ−2−プロパノール、及び2,3−ジハロプロパノール、及びこれらの混合物を含む。特に好ましいのは、1,3−ジクロロ−2−プロパノール、及び2,3−ジクロロプロパノール、及びこれらの混合物である。
【0060】
上記合成において使用するためのオレフィンは好ましくは、炭素原子数4〜22の線状又は分枝状アルファ−オレフィン(すなわち1−アルケン)、又は内部及び/又は第三オレフィン異性体と一緒に4〜22の炭素原子を含有する線状又は分枝状1−アルケンの異性体の混合物である。好ましくは、アルケンは線状であり、炭素原子数が6〜18である。特に好ましいアルファ−オレフィンの非限定的な例は:1−ブテン、1−ペンテン、1−ヘキセン、4−メチル−1−ペンテン、1−ヘプテン、1−オクテン、1−ノネン、1−デセン、1−ウンデセン、1−ドデセン、1−トリデセン、1−テトラデセン、1−ペンタデセン、1−ヘキサデセン、1−ヘプタデセン、1−オクタデセン、1−ノナデセン、1−エイコセン、又はこれらのうちの2種又は3種以上の混合物を含む。
【0061】
酸性エーテル化触媒と接触させるとオレフィンを異性体化することができるので、アルファ−オレフィンを使用することは必要でなく、そして、炭素原子数4〜22の内部オレフィン、又は線状又は分枝状アルケンの異性体の混合物も使用に適している。好適な内部オレフィンの非限定的な例は、2−ブテン、2−ペンテン、2−ヘキセン、3−ヘキセン、2−ヘプテン、3−ヘプテン、2−オクテン、3−オクテン、4−オクテン、2−ノネン、3−ノネン、4−ノネン、2−デセン、3−デセン、4−デセン、5−デセン、など、又はこれらのうちの2種又は3種以上の混合物を含む。
【0062】
エーテル化合物の合成の際に使用するのに適した酸性エーテル化触媒の一例としては、酸性イオン交換樹脂、例えばThe Dow Chemical Companyから入手可能なDOWEX DR−2030、粘土、ゼオライト、スルホン化ポリスチレンビーズ、及び不均一表面上に固定化された酸、例えばシリカビーズ上のテトラフルオロエタンスルホン酸、ブレンステッド酸、例えばtriflic(トリフルオロメタンスルホン)酸、メタンスルホン酸、又は硫酸、ルイス酸、例えばBF3及びその誘導体(例えば二水和物又はエーテル)、及びトリメチルシリルトリフレートが挙げられる。触媒と反応物質との比はさほど重要ではなく、一般に、所望の反応速度を得るように調節される。好ましくは、触媒は、エーテル化反応を容易にするために、プロセス中、約50〜150℃の温度にある。
【0063】
プロセスの工程(b)は、式I化合物を提供するために、スルホン化条件下でエーテル化合物をスルホン化することである。典型的には、エーテル化合物を、スルホン化剤、例えば亜硫酸ナトリウム、又は亜硫酸ナトリウムと炭酸ナトリウムとの組み合わせと接触させる。反応は水中で行われてよく、典型的には高い温度及び圧力、例えば150〜220℃で、100〜350psigで実施される。反応が生じるのに十分な時間(例えば24時間)に続いて、反応混合物を冷却し、周囲条件まで減圧し、そして次いでこれにコンベンショナルなワークアップを施す。式I化合物を必要に応じてさらに精製する。コンベンショナルな技術、例えば抽出、濾過、クロマトグラフィ、及び/又は結晶化を用いて精製を行うことができる。所望の場合には、例えば過酸化水素を添加することにより、過剰の亜硫酸塩を酸化して硫酸塩にすることができる。
【0064】
スルホン酸塩官能基を導入する他の典型的な方法、例えばエーテル化合物を硫化物、又はポリ硫化物と反応させ、次いで酸化させるという方法を、式I化合物を発生させるために用いてもよい。
【0065】
本発明のアニオン性界面活性剤は、上記二官能性ハロゲン化エーテル出発材料(式A)から調製することができる。両ハロゲンをスルホン化剤によって置換する場合、ジスルホネート化合物が形成される。置換及び加水分解の両方が行われる場合には、ヒドロキシスルホネートが形成される。なお、ヒドロキシスルホネートは、直接置換によって生じることができ、或いは、スルホン化条件下で容易に加水分解され、従って反応混合物中では検出されないスルホン中間体を介して生じることもできる。
【0066】
本発明のアニオン性界面活性剤が式I化合物の混合物を含む場合、混合物の組成は、アルキル鎖長、反応温度、塩基度、及び試薬ローディング量を含むスルホン化反応条件を変えることによって制御することができる。こうして、界面活性剤生成物の特徴及び特性は、所望の用途のニーズに合致するように調整することができる。本発明のプロセスによって得ることができる種々様々な界面活性剤組成物の非限定的な例が図1に示されている。また加水分解によって形成される副生成物アルキルアルコール(すなわちC12界面活性剤例の場合にはドデカノール)のレベルも図1に示されている。
【0067】
式Aのエーテル化合物から調製するのに加えて、アルキルグリセリルエーテルスルホネート(AGS)界面活性剤を形成するために一般に用いられるのと同様の方法によって、本発明のアニオン性界面活性剤のいくつか(具体的には1つのスルホネート基と1つのヒドロキシ基とを含有する界面活性剤)を調製することもできる。典型的には、このような方法は;a)エポキシ化合物を形成するためにアルコールとエピクロロヒドリンとを反応させ;そしてb)ヒドロキシモノスルホネートを形成するためにスルホン化することを含む。
【0068】
式Iのアニオン性界面活性剤は、界面活性剤の存在が所望されるか又は必要とされる種々様々な組成物及び用途において使用されてよい。非限定的な例を挙げるならば、界面活性剤は洗濯洗剤、ペイント及び塗料配合物、エマルジョン重合剤又は配合物、家庭用及び工業用クリーナー、農業用配合物、ラテックス配合物、環境復旧剤、油田用化学薬品、石油増進回収剤、ガス処理配合物、布地処理剤及び仕上げ剤、紙パルプ処理剤、香料可溶化剤配合物、金属作業流体、例えば切削流体、及びパーソナルケア製品(スキンケア及びヘアケア製品、例えばシャンプー)、などとして、又はこれらの中に使用されてよい。これらの用途に使用されるべき式Iの化合物の量及び組成は、用途及び所望の結果に応じて変化し、過度の実験なしに当業者によって決定することができる。一般に、界面活性剤として式I化合物を含む組成物は、組成物の総重量を基準として、少なくとも約0.01重量パーセントの界面活性剤を含有することになる。
【0069】
好ましい態様の場合、式Iの界面活性剤は、クリーニング組成物、例えば洗濯洗剤中に使用される。産業界及び消費者がクリーニング組成物に関して目下直面する問題点の1つは、油汚れを除去する効率が不足していることである。この効率不足は、クリーニングが室温で、且つ/又は硬水の存在において行われる場合に一層顕著である。別の問題点は、金属イオン、例えばカルシウム及びマグネシウムの存在におけるアニオン性界面活性剤の不安定性である。
【0070】
式Iのアニオン界面活性剤は前述の問題点に対処する。具体的には、界面活性剤は、油汚れの除去に優れた性能を発揮し、カルシウム・イオン及びマグネシウム・イオンの存在において可溶性である。油汚れに加えて、本発明の界面活性剤を含有するクリーニング組成物は、例えば粒子状汚れ、酸化性汚れ、有機及び無機汚れを含む他のタイプの汚染物を除去するために使用することもできる。
【0071】
クリーニング組成物中に使用されるべき式Iの界面活性剤量は、当業者によって容易に決定することができる。例えば、その量は、クリーニング組成物の総重量を基準として、典型的には約0.01重量%〜30重量%、好ましくは1重量%〜20重量%である。実際の使用時、例えば洗濯用途では、クリーニング組成物を洗浄時間前又は洗浄時間中に水で希釈することにより、式I界面活性剤の0.01重量%〜5重量%、好ましくは0.01重量%〜1重量%、及びより好ましくは0.01重量%〜0.5重量%の濃度を提供することが一般に好ましい。
【0072】
クリーニング組成物は、例えば他のアニオン性界面活性剤、非イオン性界面活性剤、カチオン性界面活性剤、両性界面活性剤、酵素、溶剤、ヒドロトロープ、ビルダー、増粘剤、キレート剤、香料、色素、乳白剤、蛍光増白剤、漂白剤、及びpH緩衝剤から選択された1種又は2種以上の追加の添加剤を含む、このような組成物中に一般に使用される他の添加剤を任意選択的に含有してよい。組成物中の好ましいpH4〜14を維持するために、緩衝剤を使用するのが典型的である。
【0073】
このような任意選択の添加剤が存在する場合、その量は次の通りであるのが好ましい;0.01%〜50%、好ましくは0.01%〜25%、より好ましくは1%〜20%のアニオン性界面活性剤;0.01%〜20%、好ましくは0.01%〜15%、より好ましくは0.5%〜10%の非イオン性界面活性剤;0.01%〜20%、好ましくは0.01%〜15%、より好ましくは0.5%〜10%のカチオン性界面活性剤;0.01%〜20%、好ましくは0.01%〜15%、より好ましくは0.5%〜10%の両性界面活性剤;0.0001%〜6%の酵素;0.01%〜20%、好ましくは0.01%〜15%、より好ましくは0.5%〜10%の溶剤;1%〜60%のビルダー;0.1%〜20%のキレート剤;及び0.1%〜15%、好ましくは0.5%〜10%のヒドロトロープ。
【0074】
上記のように、本発明は、改善された一連の特性を呈する新しいアニオン性界面活性剤を提供し、これらの界面活性剤が種々様々な用途に使用されるのを可能にする。このような特性は、低い水中毒性、高い水溶性、硬水、電解質、及び苛性溶液に対する高い耐容性を含み、そしてクリーニング組成物中で使用されると、油汚れの除去の際に優れた性能を発揮する。
【0075】
下記例は本発明の一例であって、本発明の範囲を限定しようとするものではない。特に断りのない場合には、本明細書中に使用される比、パーセンテージ、部などは重量で表される。
【実施例】
【0076】
例1
1,3−ジクロロ−2−プロパノールによるアルファ−オレフィンのエーテル化
式I化合物の前駆体である模範的なエーテル化合物を、下記プロトコルによって調製することができる。
樹脂ビーズを保持するためのグラスウール栓を有する底部ドレン・ディーン・スターク・トラップに、16.2gのDOWEX DR−2030樹脂を装入する。樹脂を11.5gの1,3−ジクロロ−2−プロパノールで湿潤し、この装置を1L丸底フラスコに取り付ける。フラスコに1.1モルのアルファ−オレフィン、及び139.7gの1,3−ジクロロ−2−プロパノールを装入する(全部で1.17モル)。真空を施し、そして1Lフラスコからの蒸留物を、加熱された樹脂を含有するディーン・スターク・トラップ内に凝縮し、そして1Lフラスコに戻すように、丸底フラスコを加熱する。1Lフラスコ内の温度は、蒸留を続けるにつれて上昇する。反応混合物を蒸留によって精製することにより、アルキル1,3−ジクロロプロピルエーテルをもたらす。
【0077】
例2
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたスルホネートの調製
本発明の模範的な界面活性剤を次のプロトコルによって形成することができる。2L Parr反応器に0.456モルのアルキル1,3−ジクロロプロピルエーテル(例1から)、0.783モルの亜硫酸ナトリウム、0.180モルのメタ−亜硫酸水素ナトリウム、0.289モルの炭酸ナトリウム、及び590gの水を装入する。窒素フラッシュ及び圧力チェックに続いて、システムを20時間にわたって200℃まで加熱する。この温度に達したあとの圧力は250psigである。溶液を周囲温度まで冷却し、そして取り出すことにより反応生成物をもたらす。
【0078】
例3
1,3−ジクロロ−2−プロパノールを用いた1−ドデセンのエーテル化
実質的に例1に記載されたプロトコルに従って形成されるエーテルを次のように製造した。2L反応蒸留装置をここに記載するように組み立てた。磁気攪拌器を備えた2L丸底フラスコを加熱マントル内に装着し、そして蒸留物受容体に接続した。磁気攪拌器及び温度プローブを含有するサイドアーム蒸留物受容体内に蒸留物を凝縮した。蒸留物受容体と2Lフラスコとの間の弁付きラインが、蒸留物受容体内に公称容積約100mLを与えた。触媒床を提供するためにそれぞれの端部に90μmのスクリーンフィルタを備えた長さ21インチ及び直径3/4インチのステンレス鋼管へ、蒸留物受容体の底部から液体をポンプ供給した。触媒含有管を覆うジャケット・システムを、再循環熱油浴を使用して加熱した。触媒床の出口は、液体を蒸留物受容体に戻した。反応蒸留が10〜300torrの圧力で行われ得るように、システムを真空ポンプに接続した。2L反応蒸留装置の触媒床には、60gのDOWEX DR−2030を装入した。2L容器に684.72g(5.304モル)の1,3−ジクロロ−2−プロパノール及び843.05g(5.009モル)の1−ドデセンを装入した。真空を22torrに調節し、そして初期温度79℃で蒸留をもたらすように、蒸気温度70℃で2L容器を加熱した。触媒床を出る反応生成物中の温度を80〜88℃にするために、触媒床油浴を110℃に設定した。凝縮器温度は約−11℃〜−5℃であった。蒸留物受容体温度は63〜69℃であった。さらに加熱するにつれて、底部温度は192℃に達し、オーバーヘッド温度は80℃であった。混合物を冷却し、そして取り出した。蒸留物受容体内及び触媒床内の溶液(96.30g、ローディングされた質量の6.3%)を廃棄した。2L容器内の溶液(1302.33g、ローディングされた質量の85.2%)をGC分析によって分析した(1.803面積%、1.10重量%のドデセン、0.708面積%、0.48重量%のドデカノール、0.01面積%、0.03重量%の1,3−ジクロロ−2−プロパノール、89.843面積%、88.71重量%のC12 1,3−ジクロロプロピルエーテル)。一部(1298.01)を2L丸底フラスコにローディングし、そして、還流スプリッタを載置された14インチ真空ジャケット付きVigreuxカラムを使用して0.2〜0.6torrで蒸留することによりこれを精製した。オーバーヘッド温度25〜105℃、底部温度146〜189℃で15:1の還流比を用いて、最初の留分(30.36g)を捕集した。オーバーヘッド温度104〜118℃、底部温度190〜220℃で15:1の還流比を用いて生成物留分を捕集することにより、1217.88g(4.09モル)の、ドデカンの1,3−ジクロロプロピルエーテルをもたらした(1,3−ジクロロプロパン−2−イルオキシドデカン、94.8面積%のC12 DCPエーテル、位置異性体の混合物、93.8%蒸留収率)。42.10gの残留物が蒸留ボトムとして残った。
【0079】
例4
含水亜硫酸ナトリウムを用いたC6スルホネート反応生成物の調製
出発材料の適切な置換を用いて事実上例3に記載されているように調製された44.48g(0.209モル)の1,3−ジクロロプロパン−2−イルオキシヘキサン、68.7g(0.545モル)の亜硫酸ナトリウム、3.06g(0.029モル)の炭酸ナトリウム、及び251.25gの脱イオン水を2L Parr反応器に装入した。窒素フラッシュ及び圧力チェックに続いて、システムを24時間にわたって170℃に加熱した。結果として生じた圧力は130psigであった。溶液を周囲温度まで冷却することにより、338.69gの溶液を提供した。溶液を酢酸エチル(173.7g、次いで45.0g)で抽出し、そして合体された上側の酢酸エチル相を蒸発させて1.59gの残留物を形成した。残留物のGC分析では、これが18重量%のC6 1,3−ジクロロ−2−プロパノールエーテルを含有することが判った。水溶液を濾過し、そして部分的に濃縮することにより、267.95gの溶液をもたらした。NMR分析では、17重量%(0.135モル、理論値の65%)のナトリウム2−(ヘキシルオキシ)プロパン−1,3−ジスルホネート(C6ジスルホネート)及び2.6重量%(0.026モル、理論値の12%)のナトリウム2−(ヘキシルオキシ)−3−ヒドロキシプロパン−1−スルホネート(C6 モノスルホネート)が見いだされた。
【0080】
例5
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC8スルホネートの調製
実質的に例2に記載されたプロトコルに従って形成される本発明の界面活性剤を、次のように生成した。2L Parr反応器に、出発材料の適切な置換を用いて実質的に例3に記載されているように調製された156.0g(0.646モル)の1,3−ジクロロプロパン−2−イルオキシオクタン、137.1g(1.09モル)の亜硫酸ナトリウム、48.1(0.253モル)のメタ−亜硫酸水素ナトリウム、53.88g(0.508モル)の炭酸ナトリウム、及び600.0gの水を装入した。窒素フラッシュ及び圧力チェックに続いて、システムを20時間にわたって180℃まで加熱した。この温度で圧力は150psigであり、そして一晩で190psigまで上昇させた。溶液を周囲温度まで冷却し、そして取り出すことにより、上側に褐色油層を有する淡褐色反応生成物944.18gをもたらした。pHは7.14であった。水性溶液のHPLC分析では、15.3重量%(0.381モル、理論値の59.0%)のC8ジスルホネート(ナトリウム2−(オクチルオキシ)プロパン−1,3−ジスルホネート)及び4.1重量%(0.206モル、理論値の21%)のC8モノスルホネート(ナトリウム2−(オクチルオキシ)−3−ヒドロキシプロパン−1−スルホネート)が見いだされた。全反応生成物のうちの24.70gの部分を取り出して12.73gの酢酸エチルで抽出した。11.55gの有機相のGC分析では、0.418重量%のオクタノール(944.18gの溶液中1.88g、0.0119モル、装入されたC8 1,3−ジクロロ−2−プロパノールエーテルの1.8モル%)、及び3.75重量%のC8 1,3−ジクロロ−2−プロパノールエーテル及びC8オレフィンの二量体生成物(ヘキサデセン異性体)(944.18gの溶液中16.9g、装入されたC8 1,3−ジクロロ−2−プロパノールエーテルの10.8重量%)が見いだされた。
【0081】
例6
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC10スルホネートの調製
出発材料(138.6gの1,3−ジクロロ−2−プロパン−2−イルオキシデカン、112.5gの亜硫酸ナトリウム、39.0gのメタ−亜硫酸水素ナトリウム、34.52gの炭酸ナトリウム、及び550.43gの水、190℃、20時間)の適切な置換を用いて、実質的に上述のプロトコルに従って化合物を形成した。反応に続いて、反応混合物を周囲温度まで冷却し、そして取り出すことにより、淡褐色反応生成物826.5gをもたらした。pHは7.11であった。水性溶液のHPLC分析では、14.3重量%(0.292モル、理論値の56.6%)のC10ジスルホネート(ナトリウム2−(デシルオキシ)プロパン−1,3−ジスルホネート)及び5.4重量%(0.139モル、理論値の27.0%)のC10モノスルホネート(ナトリウム2−(デシルオキシ)−3−ヒドロキシプロパン−1−スルホネート)が見いだされた。18.44gの部分を取り出して6.45gの酢酸エチルで抽出した。5.06gの有機相のGC分析では、0.662重量%のデセン(826.5gの溶液中1.50g、装入されたC10 1,3−ジクロロ−2−プロパノールエーテルの1.1%)、及び1.44重量%のデカノール(826.5gの溶液中3.26g、0.0206モル、装入されたC10 1,3−ジクロロ−2−プロパノールエーテルの4.0モル%)、1.15重量%のC10 1,3−ジクロロ−2−プロパノールエーテル(826.5gの溶液中2.60g、装入されたC10 1,3−ジクロロ−2−プロパノールエーテルの1.9%)、及び0.615重量%のC10オレフィン二量体生成物エイコセン(826.5gの溶液中1.39g、装入されたC10 1,3−ジクロロ−2−プロパノールエーテルの1重量%)が見いだされた。
【0082】
例7
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC12スルホネートの調製
出発材料(135.5gの1,3−ジクロロ−2−プロパン−2−イルオキシデカン、98.66gの亜硫酸ナトリウム、34.18gのメタ−亜硫酸水素ナトリウム、30.66gの炭酸ナトリウム、及び590gの水、200℃、20時間)の適切な置換を用いて、実質的に上述のプロトコルに従って化合物を形成した。反応に続いて、反応混合物を周囲温度まで冷却し、そして取り出すことにより、淡褐色反応生成物865.3gをもたらした。pHは7.75であった。水性溶液のHPLC分析では、9.14重量%(0.183モル、理論値の40%)のナトリウム2−(ドデカニルオキシ)プロパン−1,3−ジスルホネート)(C12ジスルホネート)及び5.69重量%(0.142モル、理論値の31%)のナトリウム2−(ドデカニルオキシ)−3−ヒドロキシプロパン−1−スルホネート(C12モノスルホネート)が見いだされた。10.4gの部分を取り出して4.00gの酢酸エチルで抽出した。2.77gの有機相のGC分析では、0.2重量%のドデセン(865.3gの溶液中0.46g、装入されたC12 1,3−ジクロロ−2−プロパノールエーテルの0.34%)、1.98重量%のデカノール(865.3gの溶液中4.56g、0.0245モル、装入されたC12 1,3−ジクロロ−2−プロパノールエーテルの5.4モル%)、及び1.35重量%のC12 1,3−ジクロロ−2−プロパノールエーテル(865.3gの溶液中3.11g、装入されたC12 1,3−ジクロロ−2−プロパノールエーテルの2.3%)が見いだされた。
【0083】
例8
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC14スルホネートの調製
出発材料(160.8gの1,3−ジクロロ−2−プロパン−2−イルオキシテトラデカン、108.53gの亜硫酸ナトリウム、37.46gのメタ−亜硫酸水素ナトリウム、35.14gの炭酸ナトリウム、及び828.5gの水、203℃、20時間)の適切な置換を用いて、実質的に上述のプロトコルに従って化合物を形成した。反応に続いて、反応混合物を周囲温度まで冷却し、そして取り出すことにより、黄色反応生成物1156.0gをもたらした。pHは6.67であった。水性溶液のHPLC分析では、7.19重量%(0.181モル、理論値の37%)のC14ジスルホネート(ナトリウム2−(テトラデシルオキシ)プロパン−1,3−ジスルホネート)及び5.70重量%(0.176モル、理論値の36%)のC14モノスルホネート(ナトリウム2−(テトラデシルオキシ)−3−ヒドロキシプロパン−1−スルホネート)が見いだされた。19.80gの部分を取り出して8.76gの酢酸エチルで抽出した。7.06gの有機相のGC分析では、0.29重量%のテトラドデセン(1156gの溶液中1.18g、装入されたC14 1,3−ジクロロ−2−プロパノールエーテルの0.73%)、1.99重量%のテトラデカノール(1156gの溶液中8.2g、0.0383モル、装入されたC14 1,3−ジクロロ−2−プロパノールエーテルの7.7モル%)、及び1.53重量%のC14 1,3−ジクロロ−2−プロパノールエーテル(1156gの溶液中6.3g、装入されたC14 1,3−ジクロロ−2−プロパノールエーテルの3.9%)が見いだされた。
【0084】
例9
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC16スルホネートの調製
出発材料(87.10gの1,3−ジクロロ−2−プロパン−2−イルオキシヘキサデカン、53.57gの亜硫酸ナトリウム、19.83gのメタ−亜硫酸水素ナトリウム、19.11gの炭酸ナトリウム、及び558.44gの水、207℃、31時間)の適切な置換を用いて、実質的に上述のプロトコルに従って化合物を形成した。反応に続いて、反応混合物を周囲温度まで冷却し、そして取り出すことにより、乳化反応混合物677g(ローディングされた質量の91.7%)をもたらした。エマルジョンの一部が反応器の壁に残った。9.5891gの部分を86.30gの水で希釈することによって提供される透明溶液のHPLC分析では、3.39重量%のC16ジスルホネート(ナトリウム2−(ヘキサデシルオキシ)プロパン−1,3−ジスルホネート)(738gの生成物溶液中0.051モルに相当、又は20.8モル%)、及び4.74重量%のC16モノスルホネート(ナトリウム2−(ヘキサデシルオキシ)−3−ヒドロキシプロパン−1−スルホネート)(738gの生成物溶液中0.087モルに相当、又は35.3モル%)、が見いだされた。生成混合物の38.41gの部分を19.04gの酢酸エチルで希釈すると、2つの透明な相がもたらされた。上側の16.80gの有機相のGC分析では、0.619重量%のヘキサドデセン(738gの反応生成物溶液中2.0g、装入されたC16 1,3−ジクロロ−2−プロパノールエーテルの2.3%)、2.28重量%のヘキサデカノール(738gの反応生成物溶液中7.35g、0.0303モル、装入されたC16 1,3−ジクロロ−2−プロパノールエーテルの12.2モル%)、及び0.57重量%のC16 1,3−ジクロロ−2−プロパノールエーテル(738gの反応生成物溶液中1.85g、装入されたC16 1,3−ジクロロ−2−プロパノールエーテルの2.1モル%)が見いだされた。
【0085】
例10
亜硫酸ナトリウム/メタ−亜硫酸水素ナトリウムを用いたC18スルホネートの調製
出発材料(107.36gの1,3−ジクロロ−2−プロパン−2−イルオキシオクタデカン、62.2gの亜硫酸ナトリウム、21.74gのメタ−亜硫酸水素ナトリウム、21.08gの炭酸ナトリウム、及び584.7gの水、212℃、24時間)の適切な置換を用いて、実質的に上述のプロトコルに従って化合物を形成した。反応に続いて、反応混合物を周囲温度まで冷却し、そして取り出すことにより、極めて高粘度の白色エマルジョン607.54g(ローディングされた質量の76.2%)をもたらした。エマルジョンの一部が反応器の壁に残った。生成混合物の281.55gの部分を307.14gの酢酸エチル及び254gの水で希釈すると、2つの透明な相がもたらされた。上側の有機相(288.25g)を蒸発させて13.36gの残留物を形成した。残留物のGC分析では、9.97重量%のオクタデセン(797gの反応生成物中3.79g、装入されたC18 1,3−ジクロロ−2−プロパノールエーテルの3.5%)、36.32重量%のオクタデカノール(797gの反応生成物中13.81g、0.0504モル、装入されたC18 1,3−ジクロロ−2−プロパノールエーテルの17.9モル%)、及び47.52重量%のC18 1,3−ジクロロ−2−プロパノールエーテル(797gの反応生成物中18.1g、装入されたC18 1,3−ジクロロ−2−プロパノールエーテルの16.8%)が見いだされた。反応生成物の5.50gの部分を44.16gの水、及び5.34gの2−プロパノールで希釈することにより、濁った溶液(1:10希釈)を提供した。溶液のHPLC分析では、反応生成物が3.25重量%のC18ジスルホネート(ナトリウム2−(オクタデシルオキシ)プロパン−1,3−ジスルホネート)(797gの生成物溶液中0.0501モルに相当、又は17.8モル%)、及び4.47重量%のC18モノスルホネート(ナトリウム2−(オクタデシルオキシ)−3−ヒドロキシプロパン−1−スルホネート)(797gの生成物溶液中0.0828モルに相当、又は29.5モル%)を含有することを見いだした。
【0086】
例11
個々の界面活性剤成分の単離
個々の界面活性剤成分は本発明のスルホネート組成物から単離されてよい。例えば例7のC12スルホネート組成物を、C18逆相クロマトグラフィ樹脂を含有するカラムに装入し、そしてアセトニトリルの水溶液で溶離する。所望の成分を含有する画分を捕集して蒸発させることにより、ナトリウム2−(ドデカニルオキシ)プロパン−1,3−ジスルホネート(C12ジスルホネート)、及びナトリウム2−(ドデカニルオキシ)−3−ヒドロキシプロパン−1−スルホネート(C12モノスルホネート)の単離された固形試料を提供する。
【0087】
例12
表面張力評価
Kruss K100張力計を使用してウィルヘルミー板によって、表面張力を測定する。逆CMC拡張法(reverse CMC extended method)を利用する。この方法では、界面活性剤溶液を試料容器内に先ず入れ、次いで脱イオン水で段階的に希釈する。それぞれの水添加後、試料を攪拌し、次いで、同量の溶液を取り出し、一定の体積を維持する。0.1M塩化ナトリウム中の、例5〜10で生成された本発明のC8〜C18スルホネート組成物の表面張力結果を、アルキルジフェニルオキシドジスルホネート(Dow Chemical CompanyからDOWFAX(登録商標)2A1として入手可能)と比較して、図2に示す。結果は、本発明のこれらのスルホン化材料が良好な界面活性剤特性を呈することを示している。それというのも、これらの材料は表面張力が低く(30mN/m未満)、そしてC14及びC16スルホネートの場合、DOWFAX 2A1よりも低い濃度で、最小表面張力に達することを示しており、この場合、CMCは、DOWFAX 2A1の232mN/mに対して、それぞれ68mN/m及び74mN/mである。
【0088】
例13
動的表面張力評価
最大気泡圧力法を用いて液体の動的表面張力を測定するKruss気泡圧力張力計BP2を利用して、1重量%の界面活性剤の動的表面張力を測定する。動的表面張力は、いかに素早く界面活性剤分子が界面へ移動するか、ひいてはいかに効果的に界面活性剤が適用時間スケール内で表面張力を低減するかを示すので、重要な界面活性剤特性である。
【0089】
例5〜10で生成された本発明のC8〜C18スルホネート組成物の動的表面張力は、これらの界面活性剤が、DOWFAX(登録商標)2A1よりも速く界面へ移動することを示している(図3)。このことは、例えば動的プロセス、例えばインクジェット印刷にとって極めて重要な利点であり得る。
【0090】
例14
起泡力評価
0.1重量%界面活性剤溶液を使用してASTM法D1173に従って、ロスマイルス起泡力試験を行う。例5〜10で生成された本発明のC8〜C18スルホネート組成物に対するロスマイルス起泡力試験結果を、DOWFAX(登録商標)2A1と比較して、図4に示す。結果は、初期注入時点、及び2分後の泡の高さとして表す。生成した泡の高さが40mm未満の界面活性剤、又は2分後に40mmまで崩壊する界面活性剤が、低起泡力界面活性剤と考えられるのに対して、120mmを上回る高さの泡を生成する界面活性剤は高起泡力界面活性剤と考えられる。界面活性剤の用途はしばしば特定の起泡力のニーズ及び/又は制限を有する。起泡力の結果は、低起泡力要件又は高起泡力要件の両方が生じる可能性のある用途に、疎水性物質の組成に応じて、本発明の界面活性剤を合わせ得ることを示している。
【0091】
例15
溶解度評価
水性苛性物質中の溶解度評価のために、2mlの1%界面活性剤溶液を8mlの脱イオン水と合体し、バイアルを手で振ることによりよく混ぜる。次いで10mlの20% NaOH溶液を添加し、そして試料バイアルを30秒間にわたって振る。最終濃度は界面活性剤が0.1重量%、NaOHが10重量%である。
【0092】
塩化カルシウムとの溶解度評価のために、2mlの1%界面活性剤溶液を8mlの脱イオン水と合体し、バイアルを手で振ることによりよく混ぜる。次いで10mlの2MCaCl2溶液を添加し、そして試料バイアルを30秒間にわたって振る。最終濃度は界面活性剤が0.1重量%、CaCl2が1Mである。
【0093】
10%水酸化ナトリウム、1M塩化カルシウム、及び7℃における、例5、7、及び8で生成された、本発明のC8、C12、及びC14スルホネート組成物の0.1重量%溶液の視覚的評価を、LAS及びDOWFAX(登録商標)2A1と比較して下記表2に示し、そして評価は、これらの新しい材料が望ましい特性を有することを示す。
【0094】
【表2】
【0095】
例16
水中毒性
研究手順及び試験法は、下記ガイドラインの推奨事項に基づく:
経済協力開発機構(OECD):化学薬品の試験のためのOECDガイドライン、「淡水性藻類及びシアノバクテリア、成長阻害試験」手順201,2006年3月23日付け採択;欧州経済共同体(EEC):1992年7月31日付けの委員会指示92/69/EEC、環境毒性の検出法、C.3,「藻類阻害試験」。
【0096】
例11の分離・精製されたC12モノ及びジスルホネートの急性毒性試験のデータを表3に示す。
【0097】
【表3】
【0098】
US EPAによって使用される分類システムに従って、毒性を報告する:極めて高い毒性(EC50</=0.1mg/l);高い毒性(EC50>0.1且つ</=1mg/l);多少の毒性(EC50>1且つ</=10mg/l);僅かな毒性(EC50>10且つ</=10mg/l);事実上非毒性(EC50>100mg/l)。データは、本発明の界面活性剤が極めて好ましい毒性プロフィールを有することを示す。
【0099】
例17
クリーニング効率及び溶解度の評価
この例では、表4に示された配合物を試験する。
【0100】
【表4】
【0101】
調理用油脂及びスーダン・レッドLot#1276、産業界における標準汚れで染色されたScientific Services S/D, Inc.の「Cotton 400」見本を、表4に示す2種の洗濯用配合物の水溶液で独立して洗う。
【0102】
評価のために用いられる手順は次の通りである:
タージトメータ(Tergitometer)を使用して洗浄を行う(タージトメータは、ラボラトリー規模で洗濯物を回すための産業用標準機器である)。タージトメータは100rpmで回るように設定する。各洗濯ポットは1リットルの総溶液(水+配合物)と6つの見本とを含む。洗浄の直前に、配合物を硬水中に2500ppmで希釈する。洗浄液のpHをNaOHの希釈溶液で10に調節する。4リットルの脱イオン水中に1.37gのCaCl2・2H2O及び0.43gのMgCl2・6H2Oを希釈することにより、硬水を調製する。洗浄を23℃で行う。洗浄時間は25分間である。濯ぎ時間は脱イオン水中で2分間である。
【0103】
下記式に従って、見本のL、a*、b*色パラメータの測定値に基づいて、クリーニング効率(染み除去指数:Stain Removal Index)を計算する:
【0104】
【数1】
【0105】
(上記式中、X1=(L,a*,b*)洗濯が行われる前のパラメータ、
上記式中、X2=(L,a*,b*)洗濯が行われた後のパラメータ)
【0106】
図5は、配合物1及び2のSRIの比較を示すグラフである。データは、本発明の配合物2が比較配合物1よりも、優れた油脂除去性能を呈することを示している。
【0107】
クリーニング効率に加えて、カルシウム・イオンの存在において、2種の配合物を溶解度の点に関しても観察する。視覚検査は、比較配合物1がモノスルフェート・アニオン性界面活性剤のカルシウム塩の不溶性によって引き起こされる予期される濁りを呈し、これに対して、本発明のモノスルホネート化合物及びジスルホネート化合物を含有する本発明の配合物2は透明であり、不溶性の塩は形成されないことを示す。
【0108】
視覚検査に加えて、画像のグレー値を測定するために、imageJソフトウェアを使用して試験溶液の画像上でデジタル分析を行う。比較配合物1はグレー値19を示すのに対して、本発明の配合物2はグレー値9を示す。この結果は、不溶性塩が配合物2よりも著しく大きな規模で配合物1に形成されることを裏付ける。
【0109】
例18
スチレン−ブタジエンコポリマーのエマルジョン重合
例7で調製された(本発明の)C12スルホネート組成物を、スチレン−ブタジエンコポリマーのエマルジョン重合において試験し、DOWFAX(登録商標)2A1を界面活性剤として利用する重合と比較する。次のスチレン−ブタジエン・プロトコルを使用する。0.454部のシード・ラテックス、79.2部の水、36.5部のブタジエン、28.3部のスチレン、1部のt−ドデシルメルカプタンを反応器に装入する。反応器内にさらに30.2部のスチレン、5部のアクリル酸、15部の水、1部の過硫酸ナトリウム、0.3部の本発明のスルホネート組成物(対0.5部のDOWFAX(登録商標)2A1)、及び0.1部の水酸化ナトリウムを添加する。水酸化ナトリウムを用いてpH6.2に中和された本発明のC12スルホネート界面活性剤を用いて形成されたポリマーの分析結果は下記の通りである:
100及び325メッシュ篩を介して反応器を濾過した後に観察される残留物レベル:26mg/L、固形分:50.9%、粒度:130nm。重合及び水蒸気ストリッピング中の安定性;優。ポリマーエマルジョン表面張力(1%):25℃で48.6dyn/cm。Ca2+イオン安定性(凝集するまでラテックスに10%塩化カルシウム溶液を添加することによって見極める):16ml/100g。
【0110】
本発明の界面活性剤は、100及び325メッシュ篩を介して反応器を濾過した後で観察される低い残留物レベルによって示されるように、優れた安定性を提供する。界面活性剤は粒度制御を妨げない。基準ラテックス(DOWFAX(登録商標)2A1で形成される)と同様の粒度が観察される。このことは、重合・水蒸気ストリッピング過程中の良好な安定性を示す。粒度分布の著しい相違は観察されない。界面活性剤は、反応器試料中の残留スチレン量によって示されるように、モノマー変換に影響を及ぼすことはない。本発明の界面活性剤で形成されたポリマーと、比較DOWFAX(登録商標)2A1で形成されたポリマーとの間には、同様のガラス転移温度及びゲル含量が観察される。カルボン酸モノマーは、セーラム相、ラテックス粒子表面の間に分布されているか、又はポリマー粒子内部に埋め込まれている。界面活性剤のタイプを変更した場合に、酸分布に差は生じない。
【0111】
C12スルホネート界面活性剤が反応速度特性及び粒度制御に不都合な影響を与えることはない。この処方における界面活性剤の量が、商業的なベンチマークと比較して本発明のスルホネート組成物において40%だけ低下した場合に、同様のラテックス及びポリマーの特性が達成される。界面活性剤は単独で、又は他の界面活性剤(非イオン性又は硫酸化アルキルエトキシレートを含む)との組み合わせで使用することができる。
【0112】
例19
界面活性剤溶解度のさらなる比較
この例において、本発明のアニオン性界面活性剤を、本発明のものではないアルキルグリセリルエーテルスルホネート(AGS)界面活性剤と比較する。比較を目的として、本発明の界面活性剤及び比較界面活性剤の両方を、AGS界面活性剤を形成するために一般に用いられる方法によって調製する(例えば米国特許第2989547号明細書、同第4976953号明細書、同第5246613号明細書、国際公開第1997/040131号パンフレット、同第4917823号明細書、同第5062973号明細書、欧州特許出願公開第717032号明細書、米国特許第4954281号明細書参照)。この方法は、a)アルコールとエピクロロヒドリンとを反応させることにより、式Bのエポキシ化合物を形成し、次いでb)式Cのヒドロキシモノスルホネートを形成するためにスルホン化することを伴う。
【0113】
【化8】
【0114】
本発明の界面活性剤及び比較(本発明のものではない)界面活性剤の水溶性を表5に示す。本発明のものではない界面活性剤は驚くべきことに、式CのR=C12及びR=C16に関して<0.1及び<0.025重量%という低い水溶性を示す。低い溶解度は、これらの界面活性剤としての有用性を制限する。比較すると、本発明のC8〜C12スルホネート界面活性剤の混合分枝状アルキル鎖の場合、9重量%を上回る、そして17重量%もの高さの界面活性剤濃度が観察される。
【0115】
【表5】
【0116】
本発明を好ましい態様に従って説明してきたが、本発明はこの開示内容の思想及び範囲の中で改変することができる。従って本出願は、本明細書中に開示された一般原理を用いた、本発明のいかなる変更形、使用、又は適応形にも範囲が及ぶものとする。さらに、本出願は、本発明が関連する技術分野において周知の又は習慣的な実施の圏内に入るような、そして下記請求項の範囲内に含まれる、本開示内容からの逸脱にも範囲が及ぶものとする。
【特許請求の範囲】
【請求項1】
式Iの化合物:
【化1】
(上記式中、Rは線状又は分枝状C2−C22アルキルであり;
R1、R2、及びR3は独立して、H又は線状もしくは分枝状C1−C18アルキルであり;
R4はH、CH2SO3-M+、又はCH2OHであり;
R5はOH、SO3-M+、又は式:
【化2】
の基であり;
R6はH、CH2SO3-M+、又はCH2OHであり;そして
M+はH+、又は一価又は二価カチオンであり、
R4及びR6のうちの一方はHであり、そしてR4、R5、及びR6のうちの1つ又は2つはSO3-M+を含有する)。
【請求項2】
Rが線状C4−C16アルキルである、請求項1に記載の化合物。
【請求項3】
R1、R2、及びR3がそれぞれHである、請求項1又は2に記載の化合物。
【請求項4】
R4がCH2SO3-M+又はCH2OHであり、R5がSO3-M+であり、そしてR6がHである、請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
ナトリウム2−ヘキサン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−ヘキサン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−オクタン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−オクタン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−デカン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−デカン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−ドデカン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−ドデカン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−テトラデカン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−テトラデカン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−ヘキサデカン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−ヘキサデカン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−オクタデカン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−オクタデカン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;又はこれらのうちの2種又は3種以上の混合物である、請求項1〜4のいずれか1項に記載の化合物。
【請求項6】
請求項1〜5のいずれか1項に記載の化合物又はこれらのうちの2種又は3種以上の混合物を含む、洗濯洗剤、ペイント及び塗料配合物、エマルジョン重合剤又は配合物、家庭用及び工業用クリーナー、農業用配合物、ラテックス配合物、環境復旧剤、油田用化学薬品、石油増進回収剤、ガス処理配合物、布地処理剤及び仕上げ剤、紙パルプ処理剤、香料可溶化剤配合物、金属作業流体、例えば切削流体、及びパーソナルケア製品から選択される配合物。
【請求項7】
請求項1〜5のいずれか1項に記載の化合物又はこれらのうちの2種又は3種以上の混合物を含む洗濯用組成物。
【請求項8】
他のアニオン性界面活性剤、非イオン性界面活性剤、カチオン性界面活性剤、両性界面活性剤、酵素、溶剤、ヒドロトロープ、ビルダー、増粘剤、キレート剤、香料、色素、乳白剤、蛍光増白剤、漂白剤、及びpH緩衝剤から選択される1種又は2種以上の追加の添加剤をさらに含む、請求項7に記載の洗濯用組成物。
【請求項9】
請求項1〜5のいずれか1項に記載の化合物を製造する方法であって、
(a)式B:
【化3】
(上記式中、R、R1、R2、及びR3は請求項1に規定された通りであり;そして
R7はH又はCH2Xであり、R8はXであり、そしてR9はH又はCH2Xであり、R7又はR9の一方はHであり;そして
XはF、Cl、Br、又はIである)のエーテル化合物を用意する工程;そして
(b)請求項1に記載の化合物を提供するために、スルホン化条件下で式Bのエーテル化合物をスルホン化する工程
ことを含む方法。
【請求項10】
請求項1〜5のいずれか1項に記載の2種又は3種以上の化合物を含む組成物。
【請求項1】
式Iの化合物:
【化1】
(上記式中、Rは線状又は分枝状C2−C22アルキルであり;
R1、R2、及びR3は独立して、H又は線状もしくは分枝状C1−C18アルキルであり;
R4はH、CH2SO3-M+、又はCH2OHであり;
R5はOH、SO3-M+、又は式:
【化2】
の基であり;
R6はH、CH2SO3-M+、又はCH2OHであり;そして
M+はH+、又は一価又は二価カチオンであり、
R4及びR6のうちの一方はHであり、そしてR4、R5、及びR6のうちの1つ又は2つはSO3-M+を含有する)。
【請求項2】
Rが線状C4−C16アルキルである、請求項1に記載の化合物。
【請求項3】
R1、R2、及びR3がそれぞれHである、請求項1又は2に記載の化合物。
【請求項4】
R4がCH2SO3-M+又はCH2OHであり、R5がSO3-M+であり、そしてR6がHである、請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
ナトリウム2−ヘキサン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−ヘキサン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−オクタン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−オクタン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−デカン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−デカン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−ドデカン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−ドデカン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−テトラデカン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−テトラデカン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−ヘキサデカン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−ヘキサデカン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;ナトリウム2−オクタデカン−2−イルオキシプロパン−1,3−ジスルホネート;ナトリウム2−オクタデカン−2−イルオキシ−3−ヒドロキシプロパン−1−ジスルホネート;又はこれらのうちの2種又は3種以上の混合物である、請求項1〜4のいずれか1項に記載の化合物。
【請求項6】
請求項1〜5のいずれか1項に記載の化合物又はこれらのうちの2種又は3種以上の混合物を含む、洗濯洗剤、ペイント及び塗料配合物、エマルジョン重合剤又は配合物、家庭用及び工業用クリーナー、農業用配合物、ラテックス配合物、環境復旧剤、油田用化学薬品、石油増進回収剤、ガス処理配合物、布地処理剤及び仕上げ剤、紙パルプ処理剤、香料可溶化剤配合物、金属作業流体、例えば切削流体、及びパーソナルケア製品から選択される配合物。
【請求項7】
請求項1〜5のいずれか1項に記載の化合物又はこれらのうちの2種又は3種以上の混合物を含む洗濯用組成物。
【請求項8】
他のアニオン性界面活性剤、非イオン性界面活性剤、カチオン性界面活性剤、両性界面活性剤、酵素、溶剤、ヒドロトロープ、ビルダー、増粘剤、キレート剤、香料、色素、乳白剤、蛍光増白剤、漂白剤、及びpH緩衝剤から選択される1種又は2種以上の追加の添加剤をさらに含む、請求項7に記載の洗濯用組成物。
【請求項9】
請求項1〜5のいずれか1項に記載の化合物を製造する方法であって、
(a)式B:
【化3】
(上記式中、R、R1、R2、及びR3は請求項1に規定された通りであり;そして
R7はH又はCH2Xであり、R8はXであり、そしてR9はH又はCH2Xであり、R7又はR9の一方はHであり;そして
XはF、Cl、Br、又はIである)のエーテル化合物を用意する工程;そして
(b)請求項1に記載の化合物を提供するために、スルホン化条件下で式Bのエーテル化合物をスルホン化する工程
ことを含む方法。
【請求項10】
請求項1〜5のいずれか1項に記載の2種又は3種以上の化合物を含む組成物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2012−533543(P2012−533543A)
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−520657(P2012−520657)
【出願日】平成22年6月30日(2010.6.30)
【国際出願番号】PCT/US2010/040570
【国際公開番号】WO2011/008570
【国際公開日】平成23年1月20日(2011.1.20)
【出願人】(502141050)ダウ グローバル テクノロジーズ エルエルシー (1,383)
【Fターム(参考)】
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願日】平成22年6月30日(2010.6.30)
【国際出願番号】PCT/US2010/040570
【国際公開番号】WO2011/008570
【国際公開日】平成23年1月20日(2011.1.20)
【出願人】(502141050)ダウ グローバル テクノロジーズ エルエルシー (1,383)
【Fターム(参考)】
[ Back to top ]