
スルホンアミド含有インドール化合物の結晶およびその製造方法
粉末X線回折において、回折角度(2θ±0.2°)19.1°に回折ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、血管新生阻害作用を有する抗腫瘍剤として有用なスルホンアミド含有インドール化合物の結晶およびその製造方法に関する。
【背景技術】
【0002】
スルホンアミド含有インドール化合物は血管新生阻害作用を有する抗腫瘍剤として有用であり、中でもN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(以下、化合物(5b)と示す。)は顕著な抗腫瘍効果を示す(特許文献1参照)。また、化合物(5b)の製造方法は特許文献1の実施例1に開示されている。
【0003】
【特許文献1】国際公開第00/50395号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0004】
本発明者らが特許文献1の実施例1を数回追試したところ、必ずしも一定の結晶が得られないことが分かった。医薬の有効成分としては一定品質の製品を安定供給する必要がある。そのため、医薬の有効成分が結晶性物質として得られる場合は単一の結晶形からなり、光などに対し安定である等、良好な物性を持つことが望ましい。また、そのような結晶を安定して工業的規模で製造できる方法の開発も望まれている。そこで、本発明は、化合物(5b)の単一の結晶形からなる結晶およびその製造方法を提供することを目的とする。
【課題を解決するための手段】
【0005】
本発明者らは、精力的に研究を重ねた結果、化合物(5b)の晶析に際して特定の溶媒を用いることにより、単一の結晶形の化合物(5b)が得られることを見出して本発明を完成した。
【0006】
すなわち、本発明は、以下の[1]〜[30]を提供する。
[1]粉末X線回折において、回折角度(2θ±0.2°)11.4°に回折ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
[2]粉末X線回折において、更に、回折角度(2θ±0.2°)19.1°に回折ピークを有する、[1]記載の結晶(C晶)。
[3]赤外吸収スペクトル(KBr法)において、波数1410±1cm−1に吸収ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
[4]赤外吸収スペクトル(KBr法)において、更に、波数1443±1cm−1に吸収ピークを有する、[3]記載の結晶(C晶)。
[4−2]赤外吸収スペクトル(KBr法)において、波数1410±1cm−1に吸収ピークを有する、[1]または[2]に記載の結晶(C晶)。
[4−3]赤外吸収スペクトル(KBr法)において、更に、波数1443±1cm−1に吸収ピークを有する、[4−2]に記載の結晶(C晶)。
[5]13C固体NMRスペクトルにおいて、化学シフト約143.4ppmにピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
[6]13C固体NMRスペクトルにおいて、更に、化学シフト約131.1ppmにピークを有する、[5]記載の結晶(C晶)。
[6−2]13C固体NMRスペクトルにおいて、化学シフト約143.4ppmにピークを有する、[1]〜[4−3]いずれか1に記載の結晶(C晶)。
[6−3]13C固体NMRスペクトルにおいて、更に、化学シフト約131.1ppmにピークを有する、[6−2]に記載の結晶(C晶)。
[7]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドをn−プロピルアルコール、イソプロピルアルコール、n−ブチルアルコール、s−ブチルアルコール、t−ブチルアルコールおよび水からなる群から選ばれる単一溶媒またはこれらの混合溶媒を晶析溶媒として用いて晶析させることを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[8]晶析溶媒が、イソプロピルアルコールもしくはs−ブチルアルコールの単一溶媒、s−ブチルアルコールと水との混合溶媒またはイソプロピルアルコールと水との混合溶媒である[7]記載の製造方法。
[9]晶析溶媒が、s−ブチルアルコールと水との混合溶媒(容量比3:1〜5:1)またはイソプロピルアルコールと水との混合溶媒(容量比9:1〜10:1)である[7]記載の製造方法。
[10]晶析溶媒が、s−ブチルアルコールと水との混合溶媒(容量比3.9:1〜4.1:1)である[7]記載の製造方法。
[11]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを溶媒に加熱溶解して晶析させることを特徴とする[7]に記載の製造方法。
[12]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを溶媒に加熱溶解後に徐冷して晶析させることを特徴とする[7]に記載の製造方法。
[13]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを80〜130℃で加熱することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[14]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを水中で60〜90℃にて加熱撹拌することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[15]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を80〜130℃で加熱することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[16]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を水中で60〜90℃にて加熱撹拌することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[17]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの無水物の結晶およびN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を含む混合物を80〜130℃で加熱することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[18]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの無水物の結晶およびN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を含む混合物を水中で60〜90℃にて加熱撹拌することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[19]粉末X線回折において、回折角度(2θ±0.2°)8.5°に回折ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
[20]粉末X線回折において、更に、回折角度(2θ±0.2°)25.8°に回折ピークを有する、[19]記載の結晶(A晶)。
[21]赤外吸収スペクトル(KBr法)において、波数616±1cm−1に吸収ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
[22]赤外吸収スペクトル(KBr法)において、更に、波数802±1cm−1に吸収ピークを有する、[21]記載の結晶(A晶)。
[22−2]赤外吸収スペクトル(KBr法)において、波数616±1cm−1に吸収ピークを有する、前記[19]または[20]に記載の結晶(A晶)。
[22−3]赤外吸収スペクトル(KBr法)において、更に、波数802±1cm−1に吸収ピークを有する、前記[22−2]に記載の結晶(A晶)。
[23]13C固体NMRスペクトルにおいて、化学シフト約134.7ppmにピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
[24]13C固体NMRスペクトルにおいて、更に、化学シフト約126.3ppmにピークを有する、請求項23記載の結晶(A晶)。
[24−2]13C固体NMRスペクトルにおいて、化学シフト約134.7ppmにピークを有する、前記[19]〜[22−3]に記載の結晶(A晶)。
[24−3]13C固体NMRスペクトルにおいて、更に、化学シフト約126.3ppmにピークを有する、前記[24−2]に記載の結晶(A晶)。
[25][1]〜[6−3]いずれか1に記載の結晶を含有する医薬組成物。
[26][1]〜[6−3]いずれか1に記載の結晶を含有する血管新生抑制剤。
[27][1]〜[6−3]いずれか1に記載の結晶を含有する抗腫瘍剤、すい臓癌治療剤、大腸癌治療剤、胃癌治療剤、乳癌治療剤、前立腺癌治療剤、肺癌治療剤、卵巣癌治療剤、癌転移抑制剤、糖尿病性網膜症治療剤、リューマチ性関節炎治療剤または血管腫治療剤。
[28][1]〜[6−3]いずれか1に記載の結晶の薬理学上有効量を患者に投与する、血管新生阻害が有効な疾患の治療または予防方法。
[29][1]〜[6−3]いずれか1に記載の結晶の薬理学上有効量を患者に投与する、抗腫瘍、すい臓癌、大腸癌、胃癌、乳癌、前立腺癌、肺癌、卵巣癌、癌転移、糖尿病性網膜症、リューマチ性関節炎または血管腫の治療または予防方法。
[30]抗腫瘍剤、すい臓癌治療剤、大腸癌治療剤、胃癌治療剤、乳癌治療剤、前立腺癌治療剤、肺癌治療剤、卵巣癌治療剤、癌転移抑制剤、糖尿病性網膜症治療剤、リューマチ性関節炎治療剤または血管腫治療剤の製造のための、[1]〜[6−3]いずれか1に記載の結晶の使用。
【発明の効果】
【0007】
本発明の製造方法によれば、化合物(5b)の単一の結晶形からなる結晶(C晶)を容易に工業的規模で製造することが可能である。本発明の各結晶(A晶、C晶)は、晶析などにより単一の結晶態様として製造することができ、また、光に対して安定である等良好な物性を有し、抗腫瘍剤の有効成分として使用するのに適している。また、A晶は、熱転移法によってC晶を製造する際の中間体としても有用である。
【図面の簡単な説明】
【0008】
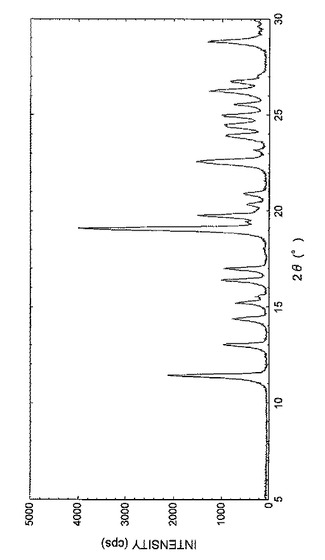
【図1】図1は、実施例3Aで得られた結晶の粉末X線回析パターンを表す図である。
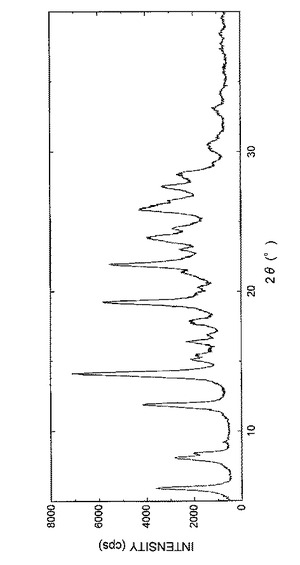
【図2】図2は、実施例1Bで得られた結晶の粉末X線回析パターンを表す図である。
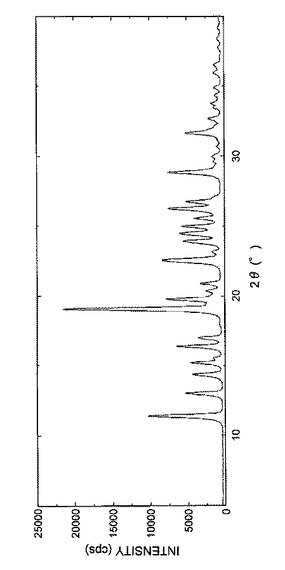
【図3】図3は、実施例1Cで得られた結晶の粉末X線回析パターンを表す図である。
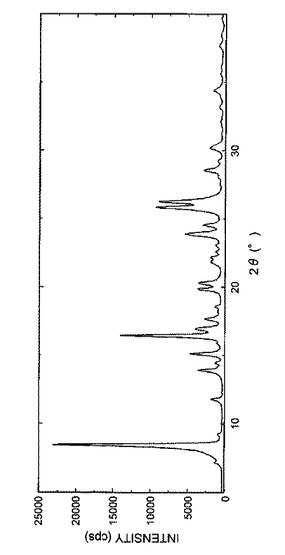
【図4】図4は、実施例1Fで得られた結晶の粉末X線回析パターンを表す図である。
【図5】図5は、実施例1Cで得られた結晶の13C固体NMRスペクトルを表す図である。
【図6】図6は、実施例1Fで得られた結晶の13C固体NMRスペクトルを表す図である。
【図7】図7は、実施例1Cで得られた結晶の赤外線吸収スペクトル(KBr法)を表す図である。
【図8】図8は、実施例1Fで得られた結晶の赤外線吸収スペクトル(KBr法)を表す図である。
【図9】図9は、実施例1Eで得られた結晶の粉末X線回析パターンを表す図である。
【発明を実施するための最良の形態】
【0009】
以下、本発明の内容について詳細に説明する。本発明の結晶は、以下の特徴を有する化合物(5b)の結晶(A晶、C晶)である。粉末X線回折、赤外吸収スペクトル(KBr法)、13C固体NMRスペクトルの各測定条件は特に限定されないが、本明細書に記載の測定条件で測定することが好ましい。
【0010】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの無水結晶(C晶)
本発明の結晶(C晶)は、化合物(5b)の単一の結晶形からなる無水物の結晶であって、粉末X線回折において回折角度(2θ±0.2°)11.4°に回折ピークを有することを特徴とする結晶、または回折角度(2θ±0.2°)11.4°および19.1°に回折ピークを有することを特徴とする結晶である。粉末X線回折におけるこれらの特徴的ピークは特許文献1に開示の製造方法によって得られた結晶(後述の実施例1B、表6および図2を参照)では観察されない。また、本発明の結晶(C晶)は、赤外吸収スペクトル(KBr法)において波数1410±1cm−1に吸収ピークを有することを特徴とする結晶、または波数1410±1cm−1および1443±1cm−1に吸収ピークを有することを特徴とする結晶である。さらに、本発明の結晶(C晶)は、13C固体NMRスペクトルにおいて化学シフト約143.4ppmにピークを有することを特徴とする結晶、または化学シフト約143.4ppmおよび約131.1ppmにピークを有することを特徴とする結晶である。
【0011】
一般に、粉末X線回折における回折角度(2θ)は回折角±0.2°の範囲内で誤差が生じ得ることから、上記の回折角度の値は±0.2°程度の範囲内の数値も含むものとして理解される必要がある。したがって、粉末X線回折におけるピークの回折角度が完全に一致する結晶だけでなく、ピークの回折角度が±0.2°程度の誤差で一致する結晶も本発明に含まれる。
【0012】
具体的には、本明細書において「回折角度(2θ±0.2°)11.4°に回折ピークを有する」とは、「回折角度(2θ)11.2°〜11.6°の範囲内に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)19.1°に回折ピークを有する」とは、「回折角度(2θ)18.9°〜19.3°の範囲内に回折ピークを有する」ということを意味する。
【0013】
同様に、本明細書において「波数1410±1cm−1に吸収ピークを有する」とは、「波数1409〜1411cm−1の範囲内に吸収ピークを有する」ということを意味する。また、本明細書において「波数1443±1cm−1の範囲内に吸収ピークを有する」とは、「波数1442〜1443cm−1に吸収ピークを有する」ということを意味する。
【0014】
本明細書において「化学シフト約143.4ppmにピークを有する」とは、「通常の測定条件にて13C固体NMRスペクトル測定を行い、化学シフト143.4ppmと同等と考えられるピークを有する」ことを意味する。また、本明細書において「化学シフト約131.1ppmにピークを有する」とは、「通常の測定条件にて13C固体NMRスペクトル測定を行い、化学シフト131.1ppmと同等と考えられるピークを有する」ことを意味する。
【0015】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)
本発明の結晶(A晶)は、化合物(5b)の単一の結晶形からなる水和物の結晶であって、粉末X線回折において回折角度(2θ±0.2°)8.5°に回折ピークを有することを特徴とする結晶、または回折角度(2θ±0.2°)8.5°及び25.8°に回折ピークを有することを特徴とする結晶である。また、本発明の結晶(A晶)は、赤外吸収スペクトル(KBr法)において波数616±1cm−1に吸収ピークを有することを特徴とする結晶、または波数616±1cm−1および波数802±1cm−1に吸収ピークを有することを特徴とする結晶である。さらに、本発明の結晶(A晶)は、13C固体NMRスペクトルにおいて化学シフト約134.7ppmにピークを有することを特徴とする結晶、または化学シフト約134.7ppmおよび約126.3ppmにピークを有することを特徴とする結晶である。
【0016】
一般に、粉末X線回折における回折角度(2θ)は回折角±0.2°の範囲内で誤差が生じ得るから、上記の回折角度の値は±0.2°程度の範囲内の数値も含むものとして理解される必要がある。したがって、粉末X線回折におけるピークの回折角度が完全に一致する結晶だけでなく、ピークの回折角度が±0.2°程度の誤差で一致する結晶も本発明に含まれる。
【0017】
本明細書において「回折角度(2θ±0.2°)8.5°に回折ピークを有する」とは、「回折角度(2θ)8.3°〜8.7°の範囲内に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)25.8°に回折ピークを有する」とは、「回折角度(2θ)25.6°〜26.0°の範囲内に回折ピークを有する」ということを意味する。
【0018】
同様に、本明細書において「波数616±1cm−1に吸収ピークを有する」とは、「波数615〜617cm−1の範囲内に吸収ピークを有する」ということを意味する。また、本明細書において「波数802±1cm−1に吸収ピークを有する」とは、「波数801〜803cm−1の範囲内に吸収ピークを有する」ということを意味する。
【0019】
本明細書において「化学シフト約134.7ppmにピークを有する」とは、「通常の測定条件にて13C固体NMRスペクトル測定を行い、化学シフト134.7ppmと同等と考えられるピークを有する」ことを意味する。また、本明細書において「化学シフト約126.3ppmにピークを有する」とは、「通常の測定条件にて13C固体NMRスペクトル測定を行い、化学シフト126.3ppmと同等と考えられるピークを有する」ことを意味する。
【0020】
本発明の結晶(A晶)は、例えば、本発明の結晶(C晶)をエタノールと水との混合溶媒から再結晶することにより得られる。
【0021】
C晶の製造方法(晶析法)
本発明の結晶(C晶)は、特許文献1の実施例1または本明細書の製造例3Aにしたがって化合物(5b)を製造し、化合物(5b)を特定の溶媒から晶析させることにより、工業的規模で安定して製造することができる。晶析に使用する化合物(5b)は、どのような形態であってもよい。すなわち、水和物でも無水物でもよく、非晶質でも結晶質(複数の結晶多形からなるものを含む)でもよく、これらの混合物であってもよい。
【0022】
晶析に使用する溶媒は、n−プロピルアルコール、イソプロピルアルコール、n−ブチルアルコール、s−ブチルアルコール、t−ブチルアルコールおよび水からなる群より選ばれる単一溶媒またはこれらの混合溶媒である。混合溶媒は前記群より選ばれる2種類の溶媒の混合溶媒であることが好ましい。好ましい溶媒は、イソプロピルアルコールもしくはs−ブチルアルコールの単一溶媒、s−ブチルアルコールと水との混合溶媒またはイソプロピルアルコールと水との混合溶媒であり、より好ましくはs−ブチルアルコールと水との混合溶媒またはイソプロピルアルコールと水との混合溶媒であり、さらに好ましくはs−ブチルアルコールと水の混合溶媒である。
【0023】
s−ブチルアルコールと水との混合溶媒を使用する場合の混合比(容量比)は、好ましくは3:1〜5:1であり、より好ましくは3.9:1〜4.1:1であり、さらに好ましくは4:1である。
【0024】
イソプロピルアルコールと水との混合溶媒を使用する場合の混合比(容量比)は、好ましくは5:1〜100:1であり、より好ましくは9:1〜100:1であり、さらに好ましくは9.9:1〜10.1:1であり、もっとも好ましくは10:1である。
【0025】
また、溶媒の使用量は、化合物(5b)が加熱により溶解する量を下限とし、結晶の収量が著しく低下しない量を上限として適宜選択することができるが、好ましくは化合物(5b)の重量に対する容量比で3〜40倍量(v/w)であり、より好ましくは10〜20倍量(v/w)であり、さらに好ましくは15〜17倍量(v/w)であり、最も好ましくは15.7〜16.3倍量(v/w)である。
【0026】
化合物(5b)を溶解する温度は、溶媒に応じて化合物(5b)が溶解する温度を適宜選択すればよいが、好ましくは75℃〜加熱還流温度である。晶析時の冷却は、結晶の品質や粒度等への影響を考慮して適宜冷却速度を調整して実施することが望ましく、好ましくは徐冷(40℃/時間以下の速度での冷却)である。より好ましい冷却速度は5〜20℃/時間で、さらに好ましくは約10℃/時間である。また、最終的な晶析温度は、結晶の収量と品質等から適宜選択することができるが、好ましくは室温〜0℃であり、より好ましくは9〜5℃であり、さらに好ましくは6.5〜7.5℃である。
【0027】
晶析した結晶を通常の濾過操作で分離し、必要に応じて適切な溶媒で洗浄し、さらに乾燥して目的の結晶を得ることができる。結晶の洗浄に使用する溶媒は、晶析溶媒と共通であるが、好ましくはs−ブチルアルコールである。
【0028】
濾過操作で分離した結晶が(主として)無水物結晶(C晶)である場合の乾燥は、大気下に放置することでも可能であるが、大量に製造する場合には効率的でなく、加熱によって乾燥することが好ましい。乾燥温度としては、製造量に応じて適宜選択することができるが、好ましくは40℃〜130℃であり、より好ましくは65〜75℃であり、さらに好ましくは70℃である。乾燥時間は、残留溶媒が所定の量を下回るまでの時間を製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよい。また、乾燥は通風下でも減圧下でも行うことができるが、減圧下で行うことが好ましい。減圧度は、製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよい。
【0029】
C晶の製造方法(熱転移法)
化合物(5b)を熱転移させることによっても、C晶を製造することが可能である。熱転移に使用する化合物(5b)は、どのような形態であってもよい。すなわち、水和物でも無水物でもよく、非晶質でも結晶質(複数の結晶多形からなるものも含む)でもよく、これらの混合物であってもよい。特に、好ましい形態は、化合物(5b)の水和物の結晶、または化合物(5b)の無水物結晶および化合物(5b)の水和物の結晶の混合物である。かかる混合物としては、例えば、化合物(5b)の再結晶の際に急冷等によって得られる混合物(本明細書の実施例2Bおよび1D)等を用いることが可能である。
【0030】
化合物(5b)を加熱乾燥装置にて加熱乾燥することにより、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド無水物結晶(C晶)を得ることができる。
【0031】
加熱温度としては、製造量に応じて適宜選択することができるが、好ましくは80〜130℃であり、より好ましくは119〜121℃であり、さらに好ましくは120℃である。乾燥時間は、残留溶媒が所定の量を下回るまでの時間を製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよいが、好ましくは10分〜12時間であり、より好ましくは30分〜3時間である。また、乾燥は通風下でも減圧下でも行うことができるが、減圧下で行うことが好ましい。減圧度は、製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよい。
【0032】
また、化合物(5b)を水に懸濁させ、加熱撹拌した後、濾取することにより、C晶を得ることができる。用いる水の量は特に制限されないが、好ましくは懸濁させる水和物を含む結晶の5〜30倍量(v/w)、より好ましくは18〜22倍量(v/w)、さらに好ましくは20倍量(v/w)を用いる。加熱撹拌を行う温度は60〜90℃であり、好ましくは75〜85℃で、より好ましくは80℃である。加熱撹拌を行う時間は1時間〜24時間であるが、より好ましくは3〜18時間で、さらに好ましくは16〜18時間である。
【0033】
得られた無水物結晶は、晶析法に記載の乾燥の方法・条件と同様の方法・条件にて、さらに乾燥を行うことができる。
【0034】
上記の方法によって得られたC晶は単一の結晶形からなり、この結晶形は安定であって、容易に他の結晶形や非晶質に転移することがなく、また吸湿性もない等の良好な物性を有しており、製剤化にも適している。
【0035】
本発明の結晶を含有する医薬組成物
化合物(5b)の抗腫瘍剤としての使用に関しては特許文献1に詳細に開示されており、それと同様に本発明の結晶は抗腫瘍剤の有効成分として使用することができる。特許文献1の開示のすべてを参照として本明細書の開示に含める。また、本発明のC晶は、その良好な安定性と物性とから、抗腫瘍剤の有効成分としての化合物(5b)の使用に最も適した形態である。
【0036】
本発明にかかる結晶は、慣用されている方法により錠剤、散剤、細粒剤、顆粒剤、被覆錠剤、カプセル剤、シロップ剤、トローチ剤、吸入剤、坐剤、注射剤、軟膏剤、眼軟膏剤、点眼剤、点鼻剤、点耳剤、パップ剤、ローション剤等として製剤化することができる。製剤化には通常用いられる賦形剤、結合剤、滑沢剤、着色剤、矯味矯臭剤や、および必要により安定化剤、乳化剤、吸収促進剤、界面活性剤、pH調整剤、防腐剤、抗酸化剤などを使用することができ、一般に医薬品製剤の原料として用いられる成分を配合して常法により製剤化される。
【0037】
これらの成分としては例えば、大豆油、牛脂、合成グリセライド等の動植物油;流動パラフィン、スクワラン、固形パラフィン等の炭化水素;ミリスチン酸オクチルドデシル、ミリスチン酸イソプロピル等のエステル油;セトステアリルアルコール、ベヘニルアルコール等の高級アルコール;シリコン樹脂;シリコン油;ポリオキシエチレン脂肪酸エステル、ソルビタン脂肪酸エステル、グリセリン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレン硬化ひまし油、ポリオキシエチレンポリオキシプロピレンブロックコポリマー等の界面活性剤;ヒドロキシエチルセルロース、ポリアクリル酸、カルボキシビニルポリマー、ポリエチレングリコール、ポリビニルピロリドン、メチルセルロースなどの水溶性高分子;エタノール、イソプロピルアルコールなどの低級アルコール;グリセリン、プロピレングリコール、ジプロピレングリコール、ソルビトールなどの多価アルコール;グルコース、ショ糖などの糖;無水ケイ酸、ケイ酸アルミニウムマグネシウム、ケイ酸アルミニウムなどの無機粉体、精製水などが挙げられる。
【0038】
賦形剤としては、例えば乳糖、コーンスターチ、白糖、ブドウ糖、マンニトール、ソルビット、結晶セルロース、二酸化ケイ素などが、結合剤としては、例えばポリビニルアルコール、ポリビニルエーテル、メチルセルロース、エチルセルロース、アラビアゴム、トラガント、ゼラチン、シェラック、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ポリビニルピロリドン、ポリプロピレングリコール・ポリオキシエチレン・ブロックポリマー、メグルミンなどが、崩壊剤としては、例えば澱粉、寒天、ゼラチン末、結晶セルロース、炭酸カルシウム、炭酸水素ナトリウム、クエン酸カルシウム、デキストリン、ペクチン、カルボキシメチルセルロース・カルシウム等が、滑沢剤としては、例えばステアリン酸マグネシウム、タルク、ポリエチレングリコール、シリカ、硬化植物油等が、着色剤としては医薬品に添加することが許可されているものが、矯味矯臭剤としては、ココア末、ハッカ脳、芳香散、ハッカ油、竜脳、桂皮末等が用いられる。
【0039】
経口製剤を製造するには、本発明にかかる化合物またはその薬理学的に許容される塩と賦形剤、さらに必要に応じて結合剤、崩壊剤、滑沢剤、着色剤、矯味矯臭剤などを加えた後、常法により散剤、細粒剤、顆粒剤、錠剤、被覆錠剤、カプセル剤等とする。
【0040】
これらの錠剤・顆粒剤には糖衣、その他必要により適宜コーティングすることはもちろん差支えない。
【0041】
また、シロップ剤や注射用製剤等の液剤を製造する際には、本発明にかかる化合物またはその薬理学的に許容される塩にpH調整剤、溶解剤、等張化剤などと、必要に応じて溶解補助剤、安定化剤などを加えて、常法により製剤化する。
【0042】
外用剤を製造する際の方法は限定されず、常法により製造することができる。すなわち製剤化にあたり使用する基剤原料としては、医薬品、医薬部外品、化粧品等に通常使用される各種原料を用いることが可能である。使用する基剤原料として具体的には、例えば動植物油、鉱物油、エステル油、ワックス類、高級アルコール類、脂肪酸類、シリコン油、界面活性剤、リン脂質類、アルコール類、多価アルコール類、水溶性高分子類、粘土鉱物類、精製水などの原料が挙げられ、さらに必要に応じ、pH調整剤、抗酸化剤、キレート剤、防腐防黴剤、着色料、香料などを添加することができるが、本発明にかかる外用剤の基剤原料はこれらに限定されない。また必要に応じて血流促進剤、殺菌剤、消炎剤、細胞賦活剤、ビタミン類、アミノ酸、保湿剤、角質溶解剤等の成分を配合することもできる。なお上記基剤原料の添加量は、通常外用剤の製造にあたり設定される濃度になる量である。
【0043】
本発明にかかる結晶を投与する場合、その形態は特に限定されず、通常用いられる方法により経口投与でも非経口投与でもよい。例えば錠剤、散剤、顆粒剤、カプセル剤、シロップ剤、トローチ剤、吸入剤、坐剤、注射剤、軟膏剤、眼軟膏剤、点眼剤、点鼻剤、点耳剤、パップ剤、ローション剤などの剤として製剤化し、投与することができる。本発明にかかる医薬の投与量は、患者の年齢、性別、体重、症状の程度、疾患の具体的な種類、投与形態・塩の種類等に応じて適宜選ぶことができる。
【0044】
以下に、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの無水物結晶(C晶)(化合物(5b)C晶)を含有する、人への治療または予防などに用いるための製剤処方の例を示す。
【0045】
100mg製剤全処方(1錠あたりの含量)
【0046】
【表1】

【0047】
50mg製剤全処方(1錠あたりの含量)
【0048】
【表2】
【0049】
10mg製剤全処方(1錠あたりの含量)
【0050】
【表3】
【0051】
2mg製剤全処方(1錠あたりの含量)
【0052】
【表4】
【0053】
なお、上記処方の製剤は、製剤学的に一般的に用いられている方法によって得ることができる。
【実施例】
【0054】
以下の実施例により本発明を詳細に且つ具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
【0055】
実施例1A:3−シアノ−4−メチル−7−ニトロ−1H−インドールの製造
【0056】
【化1】
【0057】
ジメチルホルムアミド740mLに0℃でオキシ塩化リン235mL(2.52mol)を加え、その後、0℃で0.5時間攪拌した。次いでこの反応溶液中に4−メチル−7−ニトロ−1H−インドール370g(2.10mol)(WO00/50395号)のジメチルホルムアミド溶液(1110mL)を0℃で加え、60℃で2時間加熱攪拌した。
【0058】
次に、この反応液中にヒドロキシルアミン塩酸塩292g(4.20mol)のジメチルホルムアミド溶液(1850mL)を内温80℃以上にならないように滴下し、60℃で40分間加熱攪拌した。反応混合液に氷冷下で氷水11.1Lを加え、さらに終夜撹拌した。析出した結晶を濾取し、水洗した。結晶を水11.1Lに懸濁し、この懸濁液に1N水酸化ナトリウム溶液を加えてpH7に調整した後、結晶を濾取、水洗し、標記化合物412gを得た(収率:97.6%)。
【0059】
HPLC分析により、WO00/50395号記載の3−シアノ−4−メチル−7−ニトロ−1H−インドールと同じ化合物であることを確認した。
(HPLC条件)
移動相:CH3CN/H2O/70%HClO4=500/500/1(v/v/v)
流速:1.0mL/分
検出:UV(254nm)
カラム:YMC−Pack Pro C18 250×4.6mm
【0060】
実施例2A:7−アミノ−3−シアノ−4−メチル−1H−インドールの製造
【0061】
【化2】
【0062】
実施例1Aで得た3−シアノ−4−メチル−7−ニトロ−1H−インドール400g(1.99mol)を酢酸エチル6Lとメタノール6Lの混合液に懸濁し、10%パラジウム−炭素40gの存在下、常温4気圧で水素添加した。触媒を濾別した後、濾液を活性炭処理し、濃縮し、粗結晶を得た。外温60℃にて粗結晶を1,2−ジメトキシエタン6Lに溶解した後、水12Lを滴下した。結晶の析出を確認後、氷冷下1.5時間攪拌し、濾過し、結晶を水(1L)で2回洗浄した。この結晶を50℃で16時間通風乾燥することにより標記化合物289gを得た(収率:84.8%)。
【0063】
HPLC分析により、WO00/50395号記載の7−アミノ−3−シアノ−4−メチル−1H−インドールと同じ化合物であることを確認した。
(HPLC条件)
移動相:CH3CN/H2O/70%HClO4=400/600/1(v/v/v)
流速:1.0mL/分
検出:UV(282nm)
カラム:YMC−Pack Pro C18 250×4.6mm
【0064】
実施例3A:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶))の製造
【0065】
【化3】
【0066】
実施例2Aで得た7−アミノ−3−シアノ−4−メチル−1H−インドール5.0g(29mmol)および3−シアノベンゼンスルホニルクロリド6.48g(32mmol)[CAS No.56542−67−7]を酢酸メチル150mLに懸濁し、次いで水75mL、ピリジン2.83mL(35mmol)を加えて2時間40分攪拌した。反応液に濃塩酸0.73mL(9mmol)を加えた後、分液し、有機層を水75mL、エタノール17.5mLの混液で洗浄した。有機層に活性炭を加えて45〜50℃で30分攪拌した後、濾過し濃縮した。こうして得られた粗結晶に2−ブタノール96mLおよび水24mLを加えて、75℃で溶解させた後、約10℃/時間で7℃まで徐冷し、終夜攪拌した。析出した結晶を濾取し、2−ブタノール10mLずつで2回洗浄し、標記化合物の結晶8.17g(乾燥前の重量)を得た。さらに、この結晶を70℃で2時間減圧乾燥することにより7.54gの標記化合物の結晶を得た。
【0067】
HPLC分析により、WO00/50395号記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドと同じ化合物であることを確認した。
(HPLC条件)
移動相:CH3CN/H2O/70%HClO4=500/500/1(v/v/v)
流速:1.0mL/分
検出:UV(282nm)
カラム:YMC−Pack Pro C18 250×4.6mm
【0068】
また、得られた結晶の粉末X線回折パターンを図1に示し、回折角(2θ)のピークおよびピーク強度を表5に示した。
【0069】
【表5】
【0070】
実施例1B:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(B晶)を主に含む混合物)の製造
WO00/50359号記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの反応条件、再結晶条件に基づいて標記化合物の結晶を合成した。
【0071】
7−アミノ−3−シアノ−4−メチル−1H−インドール(10g、58.4mmol)のテトラヒドロフラン(200ml)溶液に、ピリジン(20ml)と3−シアノベンゼンスルホニルクロライド(12.5g)を加え、室温で3.5時間攪拌した。2N塩酸(100ml)を加え、酢酸エチルで抽出した。有機層を水(2回)、飽和食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1〜3:2)で精製した。これにエタノール−ヘキサン混合溶媒(1:2)を加え、ソニケーション後、沈殿を濾取し、これをエタノール−ヘキサン混合溶媒(1:3)で洗浄した。一晩減圧乾燥し、標記化合物(9.33g、27.7mmol、収率47%)を得た。
【0072】
1H−NMRスペクトル(DMSO−d6)δ(ppm):2.58(3H,s),6.52(1H,d,J=7.6Hz),6.80(1H,d,J=7.6Hz),7.74(1H,m),7.92(1H,d,J=8.0Hz),8.12(2H,m),8.19(1H,d,J=3.2Hz),10.13(1H,s),12.03(1H,s)。
【0073】
得られた結晶の粉末X線回折パターンを図2に示し、回折角(2θ)のピークおよびピーク強度を表6に示した。
【0074】
【表6】
【0075】
実施例1Bと同様の方法により同一の結晶の製造を試みたが、粉末X線回折パターンは一致しなかった。すなわち、実施例1Bで得られた結晶は単一結晶ではなく、複数の結晶の混合物であると考えられる。また、実施例1Bの方法では、単一結晶を製造することはできないと考えられる。
【0076】
実施例2B:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶))の製造(別法1)
実施例1Bと同様の方法で得た、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(水和物の結晶を主に含む混合物)(1.00g)をイソプロピルアルコール(5.0ml)に懸濁し、加熱還流した。ここに、イソプロピルアルコール(16.0ml)を徐々に加え、結晶を完全に溶解した。この溶液をさらに30分間加熱還流した後、油浴の加熱を停止し、そのまま12時間攪拌した。析出した結晶を濾取し、結晶をイソプロピルアルコール(2ml×3)で洗浄後、室温で10分間吸引乾燥した。こうして得られた結晶を50℃で13.5時間乾燥した後、乳鉢ですりつぶした。これをさらに50℃で13時間乾燥し、淡黄色〜淡褐色結晶として、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物結晶と無水物結晶(C晶)の混合物(744mg)を得た。
【0077】
このうち200mgを120℃で30分間乾燥することにより、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(194mg)を得た。
【0078】
得られた結晶の粉末X線回折パターンを測定したところ、実施例1Cで得られた結晶の回折パターンと一致しており、得られた結晶は実施例1Cで得られた結晶と同一の結晶(無水物結晶(C晶))であることを確認した。
【0079】
実施例1C:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶))の製造(別法2)
7−アミノ−3−シアノ−4−メチル−1H−インドール2.50kg(14.6mol)および3−シアノベンゼンスルホニルクロリド3.24kg(16.06mol)[CAS No.56542−67−7]を酢酸メチル25Lに懸濁し、次いで酢酸メチル87.5Lおよび水37.5Lを加えた。次にピリジン1.39kg(17.52mol)を滴下し、2時間攪拌した。
【0080】
反応液に濃塩酸0.36L(4.38mol)を加えた後、分液し、有機層を水37.5Lおよびエタノール8.8Lの混液で洗浄した。有機層に活性炭を加えて50℃で30分攪拌した後、ろ過し濃縮した。これにイソプロピルアルコール30Lを加え再濃縮した後、イソプロピルアルコール91Lと水9.1Lを加え70℃に加熱した。2時間後溶解を確認し清澄濾過後、イソプロピルアルコール11.4Lと水1.1Lを加えた。この溶液を10℃/時間で7℃まで徐冷し(64℃で種結晶を投入)、7℃で終夜攪拌後、結晶を濾取した。結晶を減圧下70℃で乾燥することにより標記化合物3.6kgの白色結晶粉末を得た(収率:73%)。
【0081】
得られた白色結晶粉末の水分含量をカールフィシャー法により測定したところ0.1%であり、得られた結晶は無水物の結晶であることを確認した。また、HPLC分析により、得られた結晶がN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドであることを確認した。
(HPLC条件)
移動相:CH3CN/H2O/70%HClO4=500/500/1(v/v/v)
流速:1.0ml/分
検出:UV(282nm)
カラム:YMC−Pack Pro C18 250×4.6mm
カラム温度:25℃
保持時間:8.3分
【0082】
得られた結晶の粉末X線回折パターンを図3に示し、回折角(2θ)のピークおよびピーク強度を表7に示した。
【0083】
【表7】
【0084】
また、得られた結晶の13C固体NMRスペクトルを図5に示し、化学シフトを表8にまとめた。
【0085】
【表8】
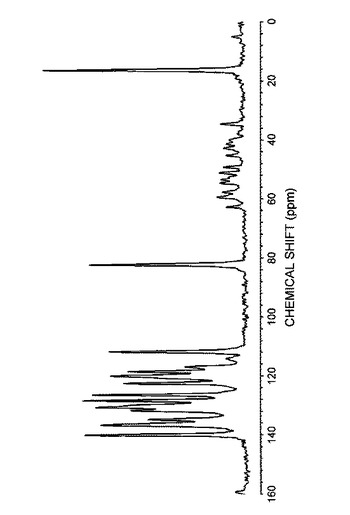
【0086】
さらに、得られた結晶の赤外スペクトル(KBr法)を図7に示し、吸収ピークの波数(cm−1)および透過率(%T)を表9に示した。
【0087】
【表9】
【0088】
実施例1D:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(水和物結晶 A晶)の製造
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(31.8g)をイソプロピルアルコール(954ml)および水(9.5ml)中、外温80℃で溶解した。無水物の種結晶(95.4mg)を加え、一気に氷冷した。30分間攪拌後、結晶を濾取し、イソプロピルアルコール(60ml)で2回洗浄し、19℃で3.5時間減圧乾燥し、標記化合物の白色結晶(28.1g)を得た。得られた結晶の粉末X線回折パターンを測定したところ、実施例1Fで得られた結晶の粉末X線回折パターンと一致し、本実施例で得られた結晶は実施例1Fで得られた結晶と同じ態様であることを確認した。
【0089】
実施例2D:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶))の製造(別法3)
実施例1Dで得たN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(5g)を水(100ml)に懸濁し、80℃で17時間加熱撹拌した。室温まで放冷後、結晶を濾取し、水(20ml)で洗浄し、さらに70℃で22時間減圧乾燥し、標記化合物の結晶(4.20g)を得た(収率:97.7%)。
【0090】
得られた結晶の粉末X線回折パターンを測定したところ、実施例1Cで得られた結晶の回折パターンと一致しており、得られた結晶は実施例1Cで得られた結晶と同一の結晶(無水物結晶(C晶))であることを確認した。
【0091】
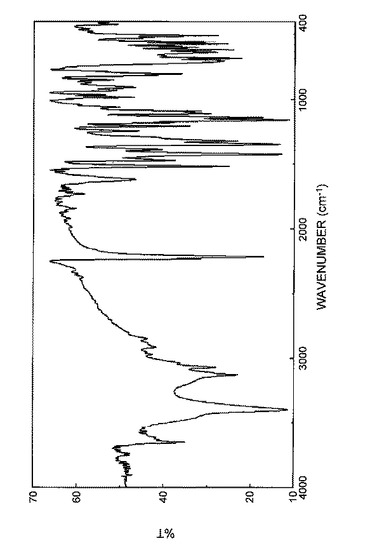
実施例1E:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(B晶))の製造
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶)、1.0g)を無水エタノール(36mL)と水(6mL)の混液で70℃の水浴中で溶解させ、次いで、その溶液を氷水中に静置させた。析出した結晶を濾過し、得られた結晶を200℃で乾燥させ、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(B晶))を得た。
【0092】
得られた結晶の粉末X線回折パターンを図9に示した。
【0093】
実施例1F:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(水和物結晶(A晶))の製造
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶)、1.0g)を無水エタノール(36mL)と水(6mL)の混液で70℃の水浴中で溶解させ、次いで、その溶液を氷水浴中に静置させた。析出した結晶を濾過し、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(水和物結晶(A晶))を得た。
【0094】
得られた結晶の粉末X線回折パターンを図4に示し、回折角(2θ)のピーク及びピーク強度を表10に示した。
【0095】
【表10】
【0096】
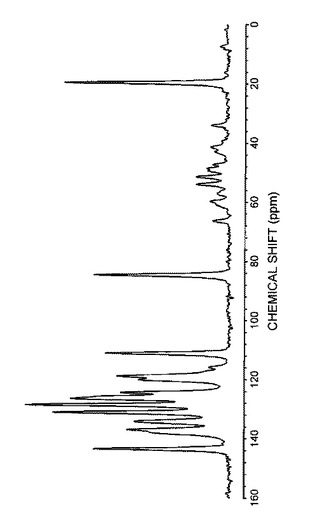
また、得られた結晶の13C固体NMRスペクトルを図6に示し、化学シフトを表11に示した。
【0097】
【表11】
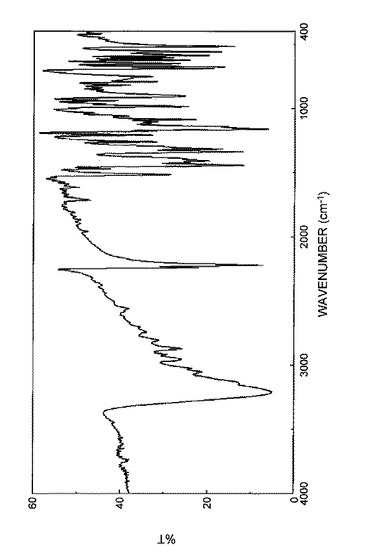
【0098】
さらに、得られた結晶の赤外スペクトル(KBr法)を図8に示し、吸収ピークの波数(cm−1)及び透過率(%T)を表12に示した。
【0099】
【表12】
【0100】
(粉末X線回折パターンの測定)
各実施例で得られた結晶の粉末X線回折測定は、以下の測定条件で行った。
[測定条件A]
使用X線:CuKα線
管電圧:40kV
管電流:20mA
発散スリット:1°
受光スリット:0.15mm
散乱スリット:1°
走査速度:2°/分
[測定条件B]
使用X線:CuKα線
管電圧:40kV
管電流:200mA
発散スリット:1/2°
受光スリット:0.3mm
散乱スリット:1/2°
走査速度:2°/分
【0101】
実施例3Aで得られた結晶は上記測定条件Aにて測定し、実施例1B、1C、1Eおよび1Fで得られた結晶は上記測定条件Bにて測定した。
【0102】
(13C固体NMRスペクトルの測定)
実施例1Cおよび1Fで得られた結晶の13C固体NMRスペクトル測定を以下の条件で行った。
測定温度:室温(〜22℃)
基準物質:シリコーンゴム(内部基準:1.56ppm)
測定核:13C(75.188829MHz)
パルス繰り返し時間:70秒(A晶:実施例1F)150秒(C晶:実施例1C)
パルスモード:CP/MAS測定(VACPX−pm)
【0103】
(赤外吸収スペクトル(KBr法)の測定)
実施例1Cおよび1Fで得られた結晶の赤外スペクトル測定は、臭化カリウム錠剤法で行った。
【0104】
試験例1:実施例1Bで得られる結晶の純度
HPLC法により、実施例1Bで得られる結晶中に含まれる不純物量を測定した。
【0105】
(HPLC条件)
カラム:ODSカラム(内径4.6mm,カラム長250mm,粒子径5μm)
カラム温度:30℃
検出波長:282nm
流速:1.0mL/分
移動相:
A液 CH3CN/H2O/70%HClO4=100/900/1(v/v/v)
B液 CH3CN/H2O/70%HClO4=900/100/1(v/v/v)
グラジエントプログラムを表13に示した。
【0106】
【表13】
【0107】
(不純物量の計算方法)
クロマトグラムから、全てのピークのピーク面積を算出し、以下の式により各ピーク(N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドのピークを除く)について不純物量を算出した。
個々の不純物量(%)=(個々の不純物ピーク面積)/(全ピーク面積の総和)×100
不純物量が0.05%以上のピークを不純物ピークとし、それらの総和を結晶に含まれる不純物量とした。
不純物量(%)=個々の不純物量(%)の総和
【0108】
HPLC分析の結果、実施例1Bで得られた結晶は、不純物を2.17%含んでいることが分かった。
【0109】
試験例2:光による固体安定性
実施例1C、実施例1Eおよび実施例1Fで得られる各結晶を、25℃/1000Lx(光安定性試験装置,LT−120D 3J,ナガノ科学機械製作所,日本)で1ヶ月間及び3ヶ月保存したあと,HPLC法により不純物量を測定した。
【0110】
(HPLC条件)
表14に示したグラジエントプログラムにより溶出させた以外は、試験例1に示した条件と同一の条件でHPLC法を行った。
【0111】
【表14】
【0112】
試験例1に示した計算方法と同様の方法により、結晶に含まれる不純物量を求めた。保存の前後で、各実施例で得られた結晶に含まれる不純物量を表15に示した。表15から明らかなように、実施例1Fおよび実施例1Cでは保存の前後で不純物量に変化が認められなかったが、実施例1Eは保存により不純物量が増加した。すなわち、実施例1Fおよび1Cで得られた結晶(それぞれ、A晶およびC晶)は、光に対する安定性が高いことが明らかとなった。
【0113】
【表15】
【0114】
以上の結果から、本発明のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法によれば、純度の高い結晶を得られることが明らかとなり、また得られた結晶(C晶)は光に対する安定性が高く、製剤化に適した物性を備えていることが明らかとなった。
【産業上の利用可能性】
【0115】
本発明は、単一の結晶形からなり、光に対する安定に優れた、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶およびその製造方法を提供する。かかる結晶は、医薬組成物の有効成分として用いることができ、特に、血管新生抑制剤、抗腫瘍剤、すい臓癌治療剤、大腸癌治療剤、胃癌治療剤、乳癌治療剤、前立腺癌治療剤、肺癌治療剤、卵巣癌治療剤、癌転移抑制剤、糖尿病性網膜症治療剤、リューマチ性関節炎治療剤および血管腫治療剤の有効成分として用いるのに適している。
【技術分野】
【0001】
本発明は、血管新生阻害作用を有する抗腫瘍剤として有用なスルホンアミド含有インドール化合物の結晶およびその製造方法に関する。
【背景技術】
【0002】
スルホンアミド含有インドール化合物は血管新生阻害作用を有する抗腫瘍剤として有用であり、中でもN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(以下、化合物(5b)と示す。)は顕著な抗腫瘍効果を示す(特許文献1参照)。また、化合物(5b)の製造方法は特許文献1の実施例1に開示されている。
【0003】
【特許文献1】国際公開第00/50395号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0004】
本発明者らが特許文献1の実施例1を数回追試したところ、必ずしも一定の結晶が得られないことが分かった。医薬の有効成分としては一定品質の製品を安定供給する必要がある。そのため、医薬の有効成分が結晶性物質として得られる場合は単一の結晶形からなり、光などに対し安定である等、良好な物性を持つことが望ましい。また、そのような結晶を安定して工業的規模で製造できる方法の開発も望まれている。そこで、本発明は、化合物(5b)の単一の結晶形からなる結晶およびその製造方法を提供することを目的とする。
【課題を解決するための手段】
【0005】
本発明者らは、精力的に研究を重ねた結果、化合物(5b)の晶析に際して特定の溶媒を用いることにより、単一の結晶形の化合物(5b)が得られることを見出して本発明を完成した。
【0006】
すなわち、本発明は、以下の[1]〜[30]を提供する。
[1]粉末X線回折において、回折角度(2θ±0.2°)11.4°に回折ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
[2]粉末X線回折において、更に、回折角度(2θ±0.2°)19.1°に回折ピークを有する、[1]記載の結晶(C晶)。
[3]赤外吸収スペクトル(KBr法)において、波数1410±1cm−1に吸収ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
[4]赤外吸収スペクトル(KBr法)において、更に、波数1443±1cm−1に吸収ピークを有する、[3]記載の結晶(C晶)。
[4−2]赤外吸収スペクトル(KBr法)において、波数1410±1cm−1に吸収ピークを有する、[1]または[2]に記載の結晶(C晶)。
[4−3]赤外吸収スペクトル(KBr法)において、更に、波数1443±1cm−1に吸収ピークを有する、[4−2]に記載の結晶(C晶)。
[5]13C固体NMRスペクトルにおいて、化学シフト約143.4ppmにピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
[6]13C固体NMRスペクトルにおいて、更に、化学シフト約131.1ppmにピークを有する、[5]記載の結晶(C晶)。
[6−2]13C固体NMRスペクトルにおいて、化学シフト約143.4ppmにピークを有する、[1]〜[4−3]いずれか1に記載の結晶(C晶)。
[6−3]13C固体NMRスペクトルにおいて、更に、化学シフト約131.1ppmにピークを有する、[6−2]に記載の結晶(C晶)。
[7]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドをn−プロピルアルコール、イソプロピルアルコール、n−ブチルアルコール、s−ブチルアルコール、t−ブチルアルコールおよび水からなる群から選ばれる単一溶媒またはこれらの混合溶媒を晶析溶媒として用いて晶析させることを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[8]晶析溶媒が、イソプロピルアルコールもしくはs−ブチルアルコールの単一溶媒、s−ブチルアルコールと水との混合溶媒またはイソプロピルアルコールと水との混合溶媒である[7]記載の製造方法。
[9]晶析溶媒が、s−ブチルアルコールと水との混合溶媒(容量比3:1〜5:1)またはイソプロピルアルコールと水との混合溶媒(容量比9:1〜10:1)である[7]記載の製造方法。
[10]晶析溶媒が、s−ブチルアルコールと水との混合溶媒(容量比3.9:1〜4.1:1)である[7]記載の製造方法。
[11]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを溶媒に加熱溶解して晶析させることを特徴とする[7]に記載の製造方法。
[12]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを溶媒に加熱溶解後に徐冷して晶析させることを特徴とする[7]に記載の製造方法。
[13]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを80〜130℃で加熱することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[14]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを水中で60〜90℃にて加熱撹拌することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[15]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を80〜130℃で加熱することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[16]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を水中で60〜90℃にて加熱撹拌することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[17]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの無水物の結晶およびN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を含む混合物を80〜130℃で加熱することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[18]N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの無水物の結晶およびN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を含む混合物を水中で60〜90℃にて加熱撹拌することを特徴とする[1]〜[6−3]いずれか1に記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
[19]粉末X線回折において、回折角度(2θ±0.2°)8.5°に回折ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
[20]粉末X線回折において、更に、回折角度(2θ±0.2°)25.8°に回折ピークを有する、[19]記載の結晶(A晶)。
[21]赤外吸収スペクトル(KBr法)において、波数616±1cm−1に吸収ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
[22]赤外吸収スペクトル(KBr法)において、更に、波数802±1cm−1に吸収ピークを有する、[21]記載の結晶(A晶)。
[22−2]赤外吸収スペクトル(KBr法)において、波数616±1cm−1に吸収ピークを有する、前記[19]または[20]に記載の結晶(A晶)。
[22−3]赤外吸収スペクトル(KBr法)において、更に、波数802±1cm−1に吸収ピークを有する、前記[22−2]に記載の結晶(A晶)。
[23]13C固体NMRスペクトルにおいて、化学シフト約134.7ppmにピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
[24]13C固体NMRスペクトルにおいて、更に、化学シフト約126.3ppmにピークを有する、請求項23記載の結晶(A晶)。
[24−2]13C固体NMRスペクトルにおいて、化学シフト約134.7ppmにピークを有する、前記[19]〜[22−3]に記載の結晶(A晶)。
[24−3]13C固体NMRスペクトルにおいて、更に、化学シフト約126.3ppmにピークを有する、前記[24−2]に記載の結晶(A晶)。
[25][1]〜[6−3]いずれか1に記載の結晶を含有する医薬組成物。
[26][1]〜[6−3]いずれか1に記載の結晶を含有する血管新生抑制剤。
[27][1]〜[6−3]いずれか1に記載の結晶を含有する抗腫瘍剤、すい臓癌治療剤、大腸癌治療剤、胃癌治療剤、乳癌治療剤、前立腺癌治療剤、肺癌治療剤、卵巣癌治療剤、癌転移抑制剤、糖尿病性網膜症治療剤、リューマチ性関節炎治療剤または血管腫治療剤。
[28][1]〜[6−3]いずれか1に記載の結晶の薬理学上有効量を患者に投与する、血管新生阻害が有効な疾患の治療または予防方法。
[29][1]〜[6−3]いずれか1に記載の結晶の薬理学上有効量を患者に投与する、抗腫瘍、すい臓癌、大腸癌、胃癌、乳癌、前立腺癌、肺癌、卵巣癌、癌転移、糖尿病性網膜症、リューマチ性関節炎または血管腫の治療または予防方法。
[30]抗腫瘍剤、すい臓癌治療剤、大腸癌治療剤、胃癌治療剤、乳癌治療剤、前立腺癌治療剤、肺癌治療剤、卵巣癌治療剤、癌転移抑制剤、糖尿病性網膜症治療剤、リューマチ性関節炎治療剤または血管腫治療剤の製造のための、[1]〜[6−3]いずれか1に記載の結晶の使用。
【発明の効果】
【0007】
本発明の製造方法によれば、化合物(5b)の単一の結晶形からなる結晶(C晶)を容易に工業的規模で製造することが可能である。本発明の各結晶(A晶、C晶)は、晶析などにより単一の結晶態様として製造することができ、また、光に対して安定である等良好な物性を有し、抗腫瘍剤の有効成分として使用するのに適している。また、A晶は、熱転移法によってC晶を製造する際の中間体としても有用である。
【図面の簡単な説明】
【0008】
【図1】図1は、実施例3Aで得られた結晶の粉末X線回析パターンを表す図である。
【図2】図2は、実施例1Bで得られた結晶の粉末X線回析パターンを表す図である。
【図3】図3は、実施例1Cで得られた結晶の粉末X線回析パターンを表す図である。
【図4】図4は、実施例1Fで得られた結晶の粉末X線回析パターンを表す図である。
【図5】図5は、実施例1Cで得られた結晶の13C固体NMRスペクトルを表す図である。
【図6】図6は、実施例1Fで得られた結晶の13C固体NMRスペクトルを表す図である。
【図7】図7は、実施例1Cで得られた結晶の赤外線吸収スペクトル(KBr法)を表す図である。
【図8】図8は、実施例1Fで得られた結晶の赤外線吸収スペクトル(KBr法)を表す図である。
【図9】図9は、実施例1Eで得られた結晶の粉末X線回析パターンを表す図である。
【発明を実施するための最良の形態】
【0009】
以下、本発明の内容について詳細に説明する。本発明の結晶は、以下の特徴を有する化合物(5b)の結晶(A晶、C晶)である。粉末X線回折、赤外吸収スペクトル(KBr法)、13C固体NMRスペクトルの各測定条件は特に限定されないが、本明細書に記載の測定条件で測定することが好ましい。
【0010】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの無水結晶(C晶)
本発明の結晶(C晶)は、化合物(5b)の単一の結晶形からなる無水物の結晶であって、粉末X線回折において回折角度(2θ±0.2°)11.4°に回折ピークを有することを特徴とする結晶、または回折角度(2θ±0.2°)11.4°および19.1°に回折ピークを有することを特徴とする結晶である。粉末X線回折におけるこれらの特徴的ピークは特許文献1に開示の製造方法によって得られた結晶(後述の実施例1B、表6および図2を参照)では観察されない。また、本発明の結晶(C晶)は、赤外吸収スペクトル(KBr法)において波数1410±1cm−1に吸収ピークを有することを特徴とする結晶、または波数1410±1cm−1および1443±1cm−1に吸収ピークを有することを特徴とする結晶である。さらに、本発明の結晶(C晶)は、13C固体NMRスペクトルにおいて化学シフト約143.4ppmにピークを有することを特徴とする結晶、または化学シフト約143.4ppmおよび約131.1ppmにピークを有することを特徴とする結晶である。
【0011】
一般に、粉末X線回折における回折角度(2θ)は回折角±0.2°の範囲内で誤差が生じ得ることから、上記の回折角度の値は±0.2°程度の範囲内の数値も含むものとして理解される必要がある。したがって、粉末X線回折におけるピークの回折角度が完全に一致する結晶だけでなく、ピークの回折角度が±0.2°程度の誤差で一致する結晶も本発明に含まれる。
【0012】
具体的には、本明細書において「回折角度(2θ±0.2°)11.4°に回折ピークを有する」とは、「回折角度(2θ)11.2°〜11.6°の範囲内に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)19.1°に回折ピークを有する」とは、「回折角度(2θ)18.9°〜19.3°の範囲内に回折ピークを有する」ということを意味する。
【0013】
同様に、本明細書において「波数1410±1cm−1に吸収ピークを有する」とは、「波数1409〜1411cm−1の範囲内に吸収ピークを有する」ということを意味する。また、本明細書において「波数1443±1cm−1の範囲内に吸収ピークを有する」とは、「波数1442〜1443cm−1に吸収ピークを有する」ということを意味する。
【0014】
本明細書において「化学シフト約143.4ppmにピークを有する」とは、「通常の測定条件にて13C固体NMRスペクトル測定を行い、化学シフト143.4ppmと同等と考えられるピークを有する」ことを意味する。また、本明細書において「化学シフト約131.1ppmにピークを有する」とは、「通常の測定条件にて13C固体NMRスペクトル測定を行い、化学シフト131.1ppmと同等と考えられるピークを有する」ことを意味する。
【0015】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)
本発明の結晶(A晶)は、化合物(5b)の単一の結晶形からなる水和物の結晶であって、粉末X線回折において回折角度(2θ±0.2°)8.5°に回折ピークを有することを特徴とする結晶、または回折角度(2θ±0.2°)8.5°及び25.8°に回折ピークを有することを特徴とする結晶である。また、本発明の結晶(A晶)は、赤外吸収スペクトル(KBr法)において波数616±1cm−1に吸収ピークを有することを特徴とする結晶、または波数616±1cm−1および波数802±1cm−1に吸収ピークを有することを特徴とする結晶である。さらに、本発明の結晶(A晶)は、13C固体NMRスペクトルにおいて化学シフト約134.7ppmにピークを有することを特徴とする結晶、または化学シフト約134.7ppmおよび約126.3ppmにピークを有することを特徴とする結晶である。
【0016】
一般に、粉末X線回折における回折角度(2θ)は回折角±0.2°の範囲内で誤差が生じ得るから、上記の回折角度の値は±0.2°程度の範囲内の数値も含むものとして理解される必要がある。したがって、粉末X線回折におけるピークの回折角度が完全に一致する結晶だけでなく、ピークの回折角度が±0.2°程度の誤差で一致する結晶も本発明に含まれる。
【0017】
本明細書において「回折角度(2θ±0.2°)8.5°に回折ピークを有する」とは、「回折角度(2θ)8.3°〜8.7°の範囲内に回折ピークを有する」ということを意味し、「回折角度(2θ±0.2°)25.8°に回折ピークを有する」とは、「回折角度(2θ)25.6°〜26.0°の範囲内に回折ピークを有する」ということを意味する。
【0018】
同様に、本明細書において「波数616±1cm−1に吸収ピークを有する」とは、「波数615〜617cm−1の範囲内に吸収ピークを有する」ということを意味する。また、本明細書において「波数802±1cm−1に吸収ピークを有する」とは、「波数801〜803cm−1の範囲内に吸収ピークを有する」ということを意味する。
【0019】
本明細書において「化学シフト約134.7ppmにピークを有する」とは、「通常の測定条件にて13C固体NMRスペクトル測定を行い、化学シフト134.7ppmと同等と考えられるピークを有する」ことを意味する。また、本明細書において「化学シフト約126.3ppmにピークを有する」とは、「通常の測定条件にて13C固体NMRスペクトル測定を行い、化学シフト126.3ppmと同等と考えられるピークを有する」ことを意味する。
【0020】
本発明の結晶(A晶)は、例えば、本発明の結晶(C晶)をエタノールと水との混合溶媒から再結晶することにより得られる。
【0021】
C晶の製造方法(晶析法)
本発明の結晶(C晶)は、特許文献1の実施例1または本明細書の製造例3Aにしたがって化合物(5b)を製造し、化合物(5b)を特定の溶媒から晶析させることにより、工業的規模で安定して製造することができる。晶析に使用する化合物(5b)は、どのような形態であってもよい。すなわち、水和物でも無水物でもよく、非晶質でも結晶質(複数の結晶多形からなるものを含む)でもよく、これらの混合物であってもよい。
【0022】
晶析に使用する溶媒は、n−プロピルアルコール、イソプロピルアルコール、n−ブチルアルコール、s−ブチルアルコール、t−ブチルアルコールおよび水からなる群より選ばれる単一溶媒またはこれらの混合溶媒である。混合溶媒は前記群より選ばれる2種類の溶媒の混合溶媒であることが好ましい。好ましい溶媒は、イソプロピルアルコールもしくはs−ブチルアルコールの単一溶媒、s−ブチルアルコールと水との混合溶媒またはイソプロピルアルコールと水との混合溶媒であり、より好ましくはs−ブチルアルコールと水との混合溶媒またはイソプロピルアルコールと水との混合溶媒であり、さらに好ましくはs−ブチルアルコールと水の混合溶媒である。
【0023】
s−ブチルアルコールと水との混合溶媒を使用する場合の混合比(容量比)は、好ましくは3:1〜5:1であり、より好ましくは3.9:1〜4.1:1であり、さらに好ましくは4:1である。
【0024】
イソプロピルアルコールと水との混合溶媒を使用する場合の混合比(容量比)は、好ましくは5:1〜100:1であり、より好ましくは9:1〜100:1であり、さらに好ましくは9.9:1〜10.1:1であり、もっとも好ましくは10:1である。
【0025】
また、溶媒の使用量は、化合物(5b)が加熱により溶解する量を下限とし、結晶の収量が著しく低下しない量を上限として適宜選択することができるが、好ましくは化合物(5b)の重量に対する容量比で3〜40倍量(v/w)であり、より好ましくは10〜20倍量(v/w)であり、さらに好ましくは15〜17倍量(v/w)であり、最も好ましくは15.7〜16.3倍量(v/w)である。
【0026】
化合物(5b)を溶解する温度は、溶媒に応じて化合物(5b)が溶解する温度を適宜選択すればよいが、好ましくは75℃〜加熱還流温度である。晶析時の冷却は、結晶の品質や粒度等への影響を考慮して適宜冷却速度を調整して実施することが望ましく、好ましくは徐冷(40℃/時間以下の速度での冷却)である。より好ましい冷却速度は5〜20℃/時間で、さらに好ましくは約10℃/時間である。また、最終的な晶析温度は、結晶の収量と品質等から適宜選択することができるが、好ましくは室温〜0℃であり、より好ましくは9〜5℃であり、さらに好ましくは6.5〜7.5℃である。
【0027】
晶析した結晶を通常の濾過操作で分離し、必要に応じて適切な溶媒で洗浄し、さらに乾燥して目的の結晶を得ることができる。結晶の洗浄に使用する溶媒は、晶析溶媒と共通であるが、好ましくはs−ブチルアルコールである。
【0028】
濾過操作で分離した結晶が(主として)無水物結晶(C晶)である場合の乾燥は、大気下に放置することでも可能であるが、大量に製造する場合には効率的でなく、加熱によって乾燥することが好ましい。乾燥温度としては、製造量に応じて適宜選択することができるが、好ましくは40℃〜130℃であり、より好ましくは65〜75℃であり、さらに好ましくは70℃である。乾燥時間は、残留溶媒が所定の量を下回るまでの時間を製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよい。また、乾燥は通風下でも減圧下でも行うことができるが、減圧下で行うことが好ましい。減圧度は、製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよい。
【0029】
C晶の製造方法(熱転移法)
化合物(5b)を熱転移させることによっても、C晶を製造することが可能である。熱転移に使用する化合物(5b)は、どのような形態であってもよい。すなわち、水和物でも無水物でもよく、非晶質でも結晶質(複数の結晶多形からなるものも含む)でもよく、これらの混合物であってもよい。特に、好ましい形態は、化合物(5b)の水和物の結晶、または化合物(5b)の無水物結晶および化合物(5b)の水和物の結晶の混合物である。かかる混合物としては、例えば、化合物(5b)の再結晶の際に急冷等によって得られる混合物(本明細書の実施例2Bおよび1D)等を用いることが可能である。
【0030】
化合物(5b)を加熱乾燥装置にて加熱乾燥することにより、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド無水物結晶(C晶)を得ることができる。
【0031】
加熱温度としては、製造量に応じて適宜選択することができるが、好ましくは80〜130℃であり、より好ましくは119〜121℃であり、さらに好ましくは120℃である。乾燥時間は、残留溶媒が所定の量を下回るまでの時間を製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよいが、好ましくは10分〜12時間であり、より好ましくは30分〜3時間である。また、乾燥は通風下でも減圧下でも行うことができるが、減圧下で行うことが好ましい。減圧度は、製造量、乾燥装置、乾燥温度等に応じて適宜選択すればよい。
【0032】
また、化合物(5b)を水に懸濁させ、加熱撹拌した後、濾取することにより、C晶を得ることができる。用いる水の量は特に制限されないが、好ましくは懸濁させる水和物を含む結晶の5〜30倍量(v/w)、より好ましくは18〜22倍量(v/w)、さらに好ましくは20倍量(v/w)を用いる。加熱撹拌を行う温度は60〜90℃であり、好ましくは75〜85℃で、より好ましくは80℃である。加熱撹拌を行う時間は1時間〜24時間であるが、より好ましくは3〜18時間で、さらに好ましくは16〜18時間である。
【0033】
得られた無水物結晶は、晶析法に記載の乾燥の方法・条件と同様の方法・条件にて、さらに乾燥を行うことができる。
【0034】
上記の方法によって得られたC晶は単一の結晶形からなり、この結晶形は安定であって、容易に他の結晶形や非晶質に転移することがなく、また吸湿性もない等の良好な物性を有しており、製剤化にも適している。
【0035】
本発明の結晶を含有する医薬組成物
化合物(5b)の抗腫瘍剤としての使用に関しては特許文献1に詳細に開示されており、それと同様に本発明の結晶は抗腫瘍剤の有効成分として使用することができる。特許文献1の開示のすべてを参照として本明細書の開示に含める。また、本発明のC晶は、その良好な安定性と物性とから、抗腫瘍剤の有効成分としての化合物(5b)の使用に最も適した形態である。
【0036】
本発明にかかる結晶は、慣用されている方法により錠剤、散剤、細粒剤、顆粒剤、被覆錠剤、カプセル剤、シロップ剤、トローチ剤、吸入剤、坐剤、注射剤、軟膏剤、眼軟膏剤、点眼剤、点鼻剤、点耳剤、パップ剤、ローション剤等として製剤化することができる。製剤化には通常用いられる賦形剤、結合剤、滑沢剤、着色剤、矯味矯臭剤や、および必要により安定化剤、乳化剤、吸収促進剤、界面活性剤、pH調整剤、防腐剤、抗酸化剤などを使用することができ、一般に医薬品製剤の原料として用いられる成分を配合して常法により製剤化される。
【0037】
これらの成分としては例えば、大豆油、牛脂、合成グリセライド等の動植物油;流動パラフィン、スクワラン、固形パラフィン等の炭化水素;ミリスチン酸オクチルドデシル、ミリスチン酸イソプロピル等のエステル油;セトステアリルアルコール、ベヘニルアルコール等の高級アルコール;シリコン樹脂;シリコン油;ポリオキシエチレン脂肪酸エステル、ソルビタン脂肪酸エステル、グリセリン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリオキシエチレン硬化ひまし油、ポリオキシエチレンポリオキシプロピレンブロックコポリマー等の界面活性剤;ヒドロキシエチルセルロース、ポリアクリル酸、カルボキシビニルポリマー、ポリエチレングリコール、ポリビニルピロリドン、メチルセルロースなどの水溶性高分子;エタノール、イソプロピルアルコールなどの低級アルコール;グリセリン、プロピレングリコール、ジプロピレングリコール、ソルビトールなどの多価アルコール;グルコース、ショ糖などの糖;無水ケイ酸、ケイ酸アルミニウムマグネシウム、ケイ酸アルミニウムなどの無機粉体、精製水などが挙げられる。
【0038】
賦形剤としては、例えば乳糖、コーンスターチ、白糖、ブドウ糖、マンニトール、ソルビット、結晶セルロース、二酸化ケイ素などが、結合剤としては、例えばポリビニルアルコール、ポリビニルエーテル、メチルセルロース、エチルセルロース、アラビアゴム、トラガント、ゼラチン、シェラック、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ポリビニルピロリドン、ポリプロピレングリコール・ポリオキシエチレン・ブロックポリマー、メグルミンなどが、崩壊剤としては、例えば澱粉、寒天、ゼラチン末、結晶セルロース、炭酸カルシウム、炭酸水素ナトリウム、クエン酸カルシウム、デキストリン、ペクチン、カルボキシメチルセルロース・カルシウム等が、滑沢剤としては、例えばステアリン酸マグネシウム、タルク、ポリエチレングリコール、シリカ、硬化植物油等が、着色剤としては医薬品に添加することが許可されているものが、矯味矯臭剤としては、ココア末、ハッカ脳、芳香散、ハッカ油、竜脳、桂皮末等が用いられる。
【0039】
経口製剤を製造するには、本発明にかかる化合物またはその薬理学的に許容される塩と賦形剤、さらに必要に応じて結合剤、崩壊剤、滑沢剤、着色剤、矯味矯臭剤などを加えた後、常法により散剤、細粒剤、顆粒剤、錠剤、被覆錠剤、カプセル剤等とする。
【0040】
これらの錠剤・顆粒剤には糖衣、その他必要により適宜コーティングすることはもちろん差支えない。
【0041】
また、シロップ剤や注射用製剤等の液剤を製造する際には、本発明にかかる化合物またはその薬理学的に許容される塩にpH調整剤、溶解剤、等張化剤などと、必要に応じて溶解補助剤、安定化剤などを加えて、常法により製剤化する。
【0042】
外用剤を製造する際の方法は限定されず、常法により製造することができる。すなわち製剤化にあたり使用する基剤原料としては、医薬品、医薬部外品、化粧品等に通常使用される各種原料を用いることが可能である。使用する基剤原料として具体的には、例えば動植物油、鉱物油、エステル油、ワックス類、高級アルコール類、脂肪酸類、シリコン油、界面活性剤、リン脂質類、アルコール類、多価アルコール類、水溶性高分子類、粘土鉱物類、精製水などの原料が挙げられ、さらに必要に応じ、pH調整剤、抗酸化剤、キレート剤、防腐防黴剤、着色料、香料などを添加することができるが、本発明にかかる外用剤の基剤原料はこれらに限定されない。また必要に応じて血流促進剤、殺菌剤、消炎剤、細胞賦活剤、ビタミン類、アミノ酸、保湿剤、角質溶解剤等の成分を配合することもできる。なお上記基剤原料の添加量は、通常外用剤の製造にあたり設定される濃度になる量である。
【0043】
本発明にかかる結晶を投与する場合、その形態は特に限定されず、通常用いられる方法により経口投与でも非経口投与でもよい。例えば錠剤、散剤、顆粒剤、カプセル剤、シロップ剤、トローチ剤、吸入剤、坐剤、注射剤、軟膏剤、眼軟膏剤、点眼剤、点鼻剤、点耳剤、パップ剤、ローション剤などの剤として製剤化し、投与することができる。本発明にかかる医薬の投与量は、患者の年齢、性別、体重、症状の程度、疾患の具体的な種類、投与形態・塩の種類等に応じて適宜選ぶことができる。
【0044】
以下に、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの無水物結晶(C晶)(化合物(5b)C晶)を含有する、人への治療または予防などに用いるための製剤処方の例を示す。
【0045】
100mg製剤全処方(1錠あたりの含量)
【0046】
【表1】
【0047】
50mg製剤全処方(1錠あたりの含量)
【0048】
【表2】
【0049】
10mg製剤全処方(1錠あたりの含量)
【0050】
【表3】
【0051】
2mg製剤全処方(1錠あたりの含量)
【0052】
【表4】
【0053】
なお、上記処方の製剤は、製剤学的に一般的に用いられている方法によって得ることができる。
【実施例】
【0054】
以下の実施例により本発明を詳細に且つ具体的に説明するが、本発明はこれらの実施例に限定されるものではない。
【0055】
実施例1A:3−シアノ−4−メチル−7−ニトロ−1H−インドールの製造
【0056】
【化1】
【0057】
ジメチルホルムアミド740mLに0℃でオキシ塩化リン235mL(2.52mol)を加え、その後、0℃で0.5時間攪拌した。次いでこの反応溶液中に4−メチル−7−ニトロ−1H−インドール370g(2.10mol)(WO00/50395号)のジメチルホルムアミド溶液(1110mL)を0℃で加え、60℃で2時間加熱攪拌した。
【0058】
次に、この反応液中にヒドロキシルアミン塩酸塩292g(4.20mol)のジメチルホルムアミド溶液(1850mL)を内温80℃以上にならないように滴下し、60℃で40分間加熱攪拌した。反応混合液に氷冷下で氷水11.1Lを加え、さらに終夜撹拌した。析出した結晶を濾取し、水洗した。結晶を水11.1Lに懸濁し、この懸濁液に1N水酸化ナトリウム溶液を加えてpH7に調整した後、結晶を濾取、水洗し、標記化合物412gを得た(収率:97.6%)。
【0059】
HPLC分析により、WO00/50395号記載の3−シアノ−4−メチル−7−ニトロ−1H−インドールと同じ化合物であることを確認した。
(HPLC条件)
移動相:CH3CN/H2O/70%HClO4=500/500/1(v/v/v)
流速:1.0mL/分
検出:UV(254nm)
カラム:YMC−Pack Pro C18 250×4.6mm
【0060】
実施例2A:7−アミノ−3−シアノ−4−メチル−1H−インドールの製造
【0061】
【化2】
【0062】
実施例1Aで得た3−シアノ−4−メチル−7−ニトロ−1H−インドール400g(1.99mol)を酢酸エチル6Lとメタノール6Lの混合液に懸濁し、10%パラジウム−炭素40gの存在下、常温4気圧で水素添加した。触媒を濾別した後、濾液を活性炭処理し、濃縮し、粗結晶を得た。外温60℃にて粗結晶を1,2−ジメトキシエタン6Lに溶解した後、水12Lを滴下した。結晶の析出を確認後、氷冷下1.5時間攪拌し、濾過し、結晶を水(1L)で2回洗浄した。この結晶を50℃で16時間通風乾燥することにより標記化合物289gを得た(収率:84.8%)。
【0063】
HPLC分析により、WO00/50395号記載の7−アミノ−3−シアノ−4−メチル−1H−インドールと同じ化合物であることを確認した。
(HPLC条件)
移動相:CH3CN/H2O/70%HClO4=400/600/1(v/v/v)
流速:1.0mL/分
検出:UV(282nm)
カラム:YMC−Pack Pro C18 250×4.6mm
【0064】
実施例3A:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶))の製造
【0065】
【化3】
【0066】
実施例2Aで得た7−アミノ−3−シアノ−4−メチル−1H−インドール5.0g(29mmol)および3−シアノベンゼンスルホニルクロリド6.48g(32mmol)[CAS No.56542−67−7]を酢酸メチル150mLに懸濁し、次いで水75mL、ピリジン2.83mL(35mmol)を加えて2時間40分攪拌した。反応液に濃塩酸0.73mL(9mmol)を加えた後、分液し、有機層を水75mL、エタノール17.5mLの混液で洗浄した。有機層に活性炭を加えて45〜50℃で30分攪拌した後、濾過し濃縮した。こうして得られた粗結晶に2−ブタノール96mLおよび水24mLを加えて、75℃で溶解させた後、約10℃/時間で7℃まで徐冷し、終夜攪拌した。析出した結晶を濾取し、2−ブタノール10mLずつで2回洗浄し、標記化合物の結晶8.17g(乾燥前の重量)を得た。さらに、この結晶を70℃で2時間減圧乾燥することにより7.54gの標記化合物の結晶を得た。
【0067】
HPLC分析により、WO00/50395号記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドと同じ化合物であることを確認した。
(HPLC条件)
移動相:CH3CN/H2O/70%HClO4=500/500/1(v/v/v)
流速:1.0mL/分
検出:UV(282nm)
カラム:YMC−Pack Pro C18 250×4.6mm
【0068】
また、得られた結晶の粉末X線回折パターンを図1に示し、回折角(2θ)のピークおよびピーク強度を表5に示した。
【0069】
【表5】
【0070】
実施例1B:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(B晶)を主に含む混合物)の製造
WO00/50359号記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの反応条件、再結晶条件に基づいて標記化合物の結晶を合成した。
【0071】
7−アミノ−3−シアノ−4−メチル−1H−インドール(10g、58.4mmol)のテトラヒドロフラン(200ml)溶液に、ピリジン(20ml)と3−シアノベンゼンスルホニルクロライド(12.5g)を加え、室温で3.5時間攪拌した。2N塩酸(100ml)を加え、酢酸エチルで抽出した。有機層を水(2回)、飽和食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥後、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=2:1〜3:2)で精製した。これにエタノール−ヘキサン混合溶媒(1:2)を加え、ソニケーション後、沈殿を濾取し、これをエタノール−ヘキサン混合溶媒(1:3)で洗浄した。一晩減圧乾燥し、標記化合物(9.33g、27.7mmol、収率47%)を得た。
【0072】
1H−NMRスペクトル(DMSO−d6)δ(ppm):2.58(3H,s),6.52(1H,d,J=7.6Hz),6.80(1H,d,J=7.6Hz),7.74(1H,m),7.92(1H,d,J=8.0Hz),8.12(2H,m),8.19(1H,d,J=3.2Hz),10.13(1H,s),12.03(1H,s)。
【0073】
得られた結晶の粉末X線回折パターンを図2に示し、回折角(2θ)のピークおよびピーク強度を表6に示した。
【0074】
【表6】
【0075】
実施例1Bと同様の方法により同一の結晶の製造を試みたが、粉末X線回折パターンは一致しなかった。すなわち、実施例1Bで得られた結晶は単一結晶ではなく、複数の結晶の混合物であると考えられる。また、実施例1Bの方法では、単一結晶を製造することはできないと考えられる。
【0076】
実施例2B:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶))の製造(別法1)
実施例1Bと同様の方法で得た、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(水和物の結晶を主に含む混合物)(1.00g)をイソプロピルアルコール(5.0ml)に懸濁し、加熱還流した。ここに、イソプロピルアルコール(16.0ml)を徐々に加え、結晶を完全に溶解した。この溶液をさらに30分間加熱還流した後、油浴の加熱を停止し、そのまま12時間攪拌した。析出した結晶を濾取し、結晶をイソプロピルアルコール(2ml×3)で洗浄後、室温で10分間吸引乾燥した。こうして得られた結晶を50℃で13.5時間乾燥した後、乳鉢ですりつぶした。これをさらに50℃で13時間乾燥し、淡黄色〜淡褐色結晶として、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物結晶と無水物結晶(C晶)の混合物(744mg)を得た。
【0077】
このうち200mgを120℃で30分間乾燥することにより、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(194mg)を得た。
【0078】
得られた結晶の粉末X線回折パターンを測定したところ、実施例1Cで得られた結晶の回折パターンと一致しており、得られた結晶は実施例1Cで得られた結晶と同一の結晶(無水物結晶(C晶))であることを確認した。
【0079】
実施例1C:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶))の製造(別法2)
7−アミノ−3−シアノ−4−メチル−1H−インドール2.50kg(14.6mol)および3−シアノベンゼンスルホニルクロリド3.24kg(16.06mol)[CAS No.56542−67−7]を酢酸メチル25Lに懸濁し、次いで酢酸メチル87.5Lおよび水37.5Lを加えた。次にピリジン1.39kg(17.52mol)を滴下し、2時間攪拌した。
【0080】
反応液に濃塩酸0.36L(4.38mol)を加えた後、分液し、有機層を水37.5Lおよびエタノール8.8Lの混液で洗浄した。有機層に活性炭を加えて50℃で30分攪拌した後、ろ過し濃縮した。これにイソプロピルアルコール30Lを加え再濃縮した後、イソプロピルアルコール91Lと水9.1Lを加え70℃に加熱した。2時間後溶解を確認し清澄濾過後、イソプロピルアルコール11.4Lと水1.1Lを加えた。この溶液を10℃/時間で7℃まで徐冷し(64℃で種結晶を投入)、7℃で終夜攪拌後、結晶を濾取した。結晶を減圧下70℃で乾燥することにより標記化合物3.6kgの白色結晶粉末を得た(収率:73%)。
【0081】
得られた白色結晶粉末の水分含量をカールフィシャー法により測定したところ0.1%であり、得られた結晶は無水物の結晶であることを確認した。また、HPLC分析により、得られた結晶がN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドであることを確認した。
(HPLC条件)
移動相:CH3CN/H2O/70%HClO4=500/500/1(v/v/v)
流速:1.0ml/分
検出:UV(282nm)
カラム:YMC−Pack Pro C18 250×4.6mm
カラム温度:25℃
保持時間:8.3分
【0082】
得られた結晶の粉末X線回折パターンを図3に示し、回折角(2θ)のピークおよびピーク強度を表7に示した。
【0083】
【表7】
【0084】
また、得られた結晶の13C固体NMRスペクトルを図5に示し、化学シフトを表8にまとめた。
【0085】
【表8】
【0086】
さらに、得られた結晶の赤外スペクトル(KBr法)を図7に示し、吸収ピークの波数(cm−1)および透過率(%T)を表9に示した。
【0087】
【表9】
【0088】
実施例1D:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(水和物結晶 A晶)の製造
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(31.8g)をイソプロピルアルコール(954ml)および水(9.5ml)中、外温80℃で溶解した。無水物の種結晶(95.4mg)を加え、一気に氷冷した。30分間攪拌後、結晶を濾取し、イソプロピルアルコール(60ml)で2回洗浄し、19℃で3.5時間減圧乾燥し、標記化合物の白色結晶(28.1g)を得た。得られた結晶の粉末X線回折パターンを測定したところ、実施例1Fで得られた結晶の粉末X線回折パターンと一致し、本実施例で得られた結晶は実施例1Fで得られた結晶と同じ態様であることを確認した。
【0089】
実施例2D:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶))の製造(別法3)
実施例1Dで得たN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(5g)を水(100ml)に懸濁し、80℃で17時間加熱撹拌した。室温まで放冷後、結晶を濾取し、水(20ml)で洗浄し、さらに70℃で22時間減圧乾燥し、標記化合物の結晶(4.20g)を得た(収率:97.7%)。
【0090】
得られた結晶の粉末X線回折パターンを測定したところ、実施例1Cで得られた結晶の回折パターンと一致しており、得られた結晶は実施例1Cで得られた結晶と同一の結晶(無水物結晶(C晶))であることを確認した。
【0091】
実施例1E:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(B晶))の製造
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶)、1.0g)を無水エタノール(36mL)と水(6mL)の混液で70℃の水浴中で溶解させ、次いで、その溶液を氷水中に静置させた。析出した結晶を濾過し、得られた結晶を200℃で乾燥させ、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(B晶))を得た。
【0092】
得られた結晶の粉末X線回折パターンを図9に示した。
【0093】
実施例1F:N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(水和物結晶(A晶))の製造
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(無水物結晶(C晶)、1.0g)を無水エタノール(36mL)と水(6mL)の混液で70℃の水浴中で溶解させ、次いで、その溶液を氷水浴中に静置させた。析出した結晶を濾過し、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミド(水和物結晶(A晶))を得た。
【0094】
得られた結晶の粉末X線回折パターンを図4に示し、回折角(2θ)のピーク及びピーク強度を表10に示した。
【0095】
【表10】
【0096】
また、得られた結晶の13C固体NMRスペクトルを図6に示し、化学シフトを表11に示した。
【0097】
【表11】
【0098】
さらに、得られた結晶の赤外スペクトル(KBr法)を図8に示し、吸収ピークの波数(cm−1)及び透過率(%T)を表12に示した。
【0099】
【表12】
【0100】
(粉末X線回折パターンの測定)
各実施例で得られた結晶の粉末X線回折測定は、以下の測定条件で行った。
[測定条件A]
使用X線:CuKα線
管電圧:40kV
管電流:20mA
発散スリット:1°
受光スリット:0.15mm
散乱スリット:1°
走査速度:2°/分
[測定条件B]
使用X線:CuKα線
管電圧:40kV
管電流:200mA
発散スリット:1/2°
受光スリット:0.3mm
散乱スリット:1/2°
走査速度:2°/分
【0101】
実施例3Aで得られた結晶は上記測定条件Aにて測定し、実施例1B、1C、1Eおよび1Fで得られた結晶は上記測定条件Bにて測定した。
【0102】
(13C固体NMRスペクトルの測定)
実施例1Cおよび1Fで得られた結晶の13C固体NMRスペクトル測定を以下の条件で行った。
測定温度:室温(〜22℃)
基準物質:シリコーンゴム(内部基準:1.56ppm)
測定核:13C(75.188829MHz)
パルス繰り返し時間:70秒(A晶:実施例1F)150秒(C晶:実施例1C)
パルスモード:CP/MAS測定(VACPX−pm)
【0103】
(赤外吸収スペクトル(KBr法)の測定)
実施例1Cおよび1Fで得られた結晶の赤外スペクトル測定は、臭化カリウム錠剤法で行った。
【0104】
試験例1:実施例1Bで得られる結晶の純度
HPLC法により、実施例1Bで得られる結晶中に含まれる不純物量を測定した。
【0105】
(HPLC条件)
カラム:ODSカラム(内径4.6mm,カラム長250mm,粒子径5μm)
カラム温度:30℃
検出波長:282nm
流速:1.0mL/分
移動相:
A液 CH3CN/H2O/70%HClO4=100/900/1(v/v/v)
B液 CH3CN/H2O/70%HClO4=900/100/1(v/v/v)
グラジエントプログラムを表13に示した。
【0106】
【表13】
【0107】
(不純物量の計算方法)
クロマトグラムから、全てのピークのピーク面積を算出し、以下の式により各ピーク(N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドのピークを除く)について不純物量を算出した。
個々の不純物量(%)=(個々の不純物ピーク面積)/(全ピーク面積の総和)×100
不純物量が0.05%以上のピークを不純物ピークとし、それらの総和を結晶に含まれる不純物量とした。
不純物量(%)=個々の不純物量(%)の総和
【0108】
HPLC分析の結果、実施例1Bで得られた結晶は、不純物を2.17%含んでいることが分かった。
【0109】
試験例2:光による固体安定性
実施例1C、実施例1Eおよび実施例1Fで得られる各結晶を、25℃/1000Lx(光安定性試験装置,LT−120D 3J,ナガノ科学機械製作所,日本)で1ヶ月間及び3ヶ月保存したあと,HPLC法により不純物量を測定した。
【0110】
(HPLC条件)
表14に示したグラジエントプログラムにより溶出させた以外は、試験例1に示した条件と同一の条件でHPLC法を行った。
【0111】
【表14】
【0112】
試験例1に示した計算方法と同様の方法により、結晶に含まれる不純物量を求めた。保存の前後で、各実施例で得られた結晶に含まれる不純物量を表15に示した。表15から明らかなように、実施例1Fおよび実施例1Cでは保存の前後で不純物量に変化が認められなかったが、実施例1Eは保存により不純物量が増加した。すなわち、実施例1Fおよび1Cで得られた結晶(それぞれ、A晶およびC晶)は、光に対する安定性が高いことが明らかとなった。
【0113】
【表15】
【0114】
以上の結果から、本発明のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法によれば、純度の高い結晶を得られることが明らかとなり、また得られた結晶(C晶)は光に対する安定性が高く、製剤化に適した物性を備えていることが明らかとなった。
【産業上の利用可能性】
【0115】
本発明は、単一の結晶形からなり、光に対する安定に優れた、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶およびその製造方法を提供する。かかる結晶は、医薬組成物の有効成分として用いることができ、特に、血管新生抑制剤、抗腫瘍剤、すい臓癌治療剤、大腸癌治療剤、胃癌治療剤、乳癌治療剤、前立腺癌治療剤、肺癌治療剤、卵巣癌治療剤、癌転移抑制剤、糖尿病性網膜症治療剤、リューマチ性関節炎治療剤および血管腫治療剤の有効成分として用いるのに適している。
【特許請求の範囲】
【請求項1】
粉末X線回折において、回折角度(2θ±0.2°)11.4°に回折ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
【請求項2】
粉末X線回折において、更に、回折角度(2θ±0.2°)19.1°に回折ピークを有する、請求項1記載の結晶(C晶)。
【請求項3】
赤外吸収スペクトル(KBr法)において、波数1410±1cm−1に吸収ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
【請求項4】
赤外吸収スペクトル(KBr法)において、更に、波数1443±1cm−1に吸収ピークを有する、請求項3記載の結晶(C晶)。
【請求項5】
13C固体NMRスペクトルにおいて、化学シフト約143.4ppmにピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
【請求項6】
13C固体NMRスペクトルにおいて、更に、化学シフト約131.1ppmにピークを有する、請求項5記載の結晶(C晶)。
【請求項7】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドをn−プロピルアルコール、イソプロピルアルコール、n−ブチルアルコール、s−ブチルアルコール、t−ブチルアルコールおよび水からなる群から選ばれる単一溶媒またはこれらの混合溶媒を晶析溶媒として用いて晶析させることを特徴とする請求項1〜6いずれか1項記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
【請求項8】
晶析溶媒が、イソプロピルアルコールもしくはs−ブチルアルコールの単一溶媒、s−ブチルアルコールと水との混合溶媒またはイソプロピルアルコールと水との混合溶媒である請求項7記載の製造方法。
【請求項9】
晶析溶媒が、s−ブチルアルコールと水との混合溶媒(容量比3:1〜5:1)またはイソプロピルアルコールと水との混合溶媒(容量比9:1〜10:1)である請求項7記載の製造方法。
【請求項10】
晶析溶媒が、s−ブチルアルコールと水との混合溶媒(容量比3.9:1〜4.1:1)である請求項7記載の製造方法。
【請求項11】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを溶媒に加熱溶解して晶析させることを特徴とする請求項7に記載の製造方法。
【請求項12】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを溶媒に加熱溶解後に徐冷して晶析させることを特徴とする請求項7に記載の製造方法。
【請求項13】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを80〜130℃で加熱することを特徴とする請求項1〜6いずれか1項記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
【請求項14】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを水中で60〜90℃にて加熱撹拌することを特徴とする請求項1〜6いずれか1項記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
【請求項15】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を80〜130℃で加熱することを特徴とする請求項1〜6いずれか1項記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
【請求項16】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を水中で60〜90℃にて加熱撹拌することを特徴とする請求項1〜6いずれか1項記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
【請求項17】
粉末X線回折において、回折角度(2θ±0.2°)8.5°に回折ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
【請求項18】
粉末X線回折において、更に、回折角度(2θ±0.2°)25.8°に回折ピークを有する、請求項17記載の結晶(A晶)。
【請求項19】
赤外吸収スペクトル(KBr法)において、波数616±1cm−1に吸収ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
【請求項20】
赤外吸収スペクトル(KBr法)において、更に、波数802±1cm−1に吸収ピークを有する、請求項19記載の結晶(A晶)。
【請求項21】
13C固体NMRスペクトルにおいて、化学シフト約134.7ppmにピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
【請求項22】
13C固体NMRスペクトルにおいて、更に、化学シフト約126.3ppmにピークを有する、請求項21記載の結晶(A晶)。
【請求項1】
粉末X線回折において、回折角度(2θ±0.2°)11.4°に回折ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
【請求項2】
粉末X線回折において、更に、回折角度(2θ±0.2°)19.1°に回折ピークを有する、請求項1記載の結晶(C晶)。
【請求項3】
赤外吸収スペクトル(KBr法)において、波数1410±1cm−1に吸収ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
【請求項4】
赤外吸収スペクトル(KBr法)において、更に、波数1443±1cm−1に吸収ピークを有する、請求項3記載の結晶(C晶)。
【請求項5】
13C固体NMRスペクトルにおいて、化学シフト約143.4ppmにピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)。
【請求項6】
13C固体NMRスペクトルにおいて、更に、化学シフト約131.1ppmにピークを有する、請求項5記載の結晶(C晶)。
【請求項7】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドをn−プロピルアルコール、イソプロピルアルコール、n−ブチルアルコール、s−ブチルアルコール、t−ブチルアルコールおよび水からなる群から選ばれる単一溶媒またはこれらの混合溶媒を晶析溶媒として用いて晶析させることを特徴とする請求項1〜6いずれか1項記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
【請求項8】
晶析溶媒が、イソプロピルアルコールもしくはs−ブチルアルコールの単一溶媒、s−ブチルアルコールと水との混合溶媒またはイソプロピルアルコールと水との混合溶媒である請求項7記載の製造方法。
【請求項9】
晶析溶媒が、s−ブチルアルコールと水との混合溶媒(容量比3:1〜5:1)またはイソプロピルアルコールと水との混合溶媒(容量比9:1〜10:1)である請求項7記載の製造方法。
【請求項10】
晶析溶媒が、s−ブチルアルコールと水との混合溶媒(容量比3.9:1〜4.1:1)である請求項7記載の製造方法。
【請求項11】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを溶媒に加熱溶解して晶析させることを特徴とする請求項7に記載の製造方法。
【請求項12】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを溶媒に加熱溶解後に徐冷して晶析させることを特徴とする請求項7に記載の製造方法。
【請求項13】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを80〜130℃で加熱することを特徴とする請求項1〜6いずれか1項記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
【請求項14】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドを水中で60〜90℃にて加熱撹拌することを特徴とする請求項1〜6いずれか1項記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
【請求項15】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を80〜130℃で加熱することを特徴とする請求項1〜6いずれか1項記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
【請求項16】
N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶を水中で60〜90℃にて加熱撹拌することを特徴とする請求項1〜6いずれか1項記載のN−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの結晶(C晶)の製造方法。
【請求項17】
粉末X線回折において、回折角度(2θ±0.2°)8.5°に回折ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
【請求項18】
粉末X線回折において、更に、回折角度(2θ±0.2°)25.8°に回折ピークを有する、請求項17記載の結晶(A晶)。
【請求項19】
赤外吸収スペクトル(KBr法)において、波数616±1cm−1に吸収ピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
【請求項20】
赤外吸収スペクトル(KBr法)において、更に、波数802±1cm−1に吸収ピークを有する、請求項19記載の結晶(A晶)。
【請求項21】
13C固体NMRスペクトルにおいて、化学シフト約134.7ppmにピークを有する、N−(3−シアノ−4−メチル−1H−インドール−7−イル)−3−シアノベンゼンスルホンアミドの水和物の結晶(A晶)。
【請求項22】
13C固体NMRスペクトルにおいて、更に、化学シフト約126.3ppmにピークを有する、請求項21記載の結晶(A晶)。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
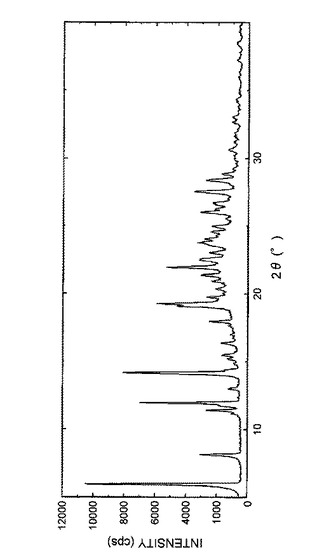
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【国際公開番号】WO2005/026118
【国際公開日】平成17年3月24日(2005.3.24)
【発行日】平成19年11月8日(2007.11.8)
【国際特許分類】
【出願番号】特願2005−513843(P2005−513843)
【国際出願番号】PCT/JP2004/012649
【国際出願日】平成16年9月1日(2004.9.1)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
【国際公開日】平成17年3月24日(2005.3.24)
【発行日】平成19年11月8日(2007.11.8)
【国際特許分類】
【国際出願番号】PCT/JP2004/012649
【国際出願日】平成16年9月1日(2004.9.1)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
[ Back to top ]