
スルホン酸基を有する置換ポリアセチレン膜の製造方法及びそれによって得られる膜とその用途
【課題】 均一にスルホン酸基を導入した固体電解質膜で、その電極接合体を電気化学デバイス及び燃料電池として用いる置換ポリアセチレン電解質膜の製造方法と電解質膜を提供する。
【解決手段】 式(1)で表される繰り返し単位を有する置換ポリアセチレンを膜状に成形し、スルホン化剤に接触させてスルホン化するスルホン酸基を有する置換ポリアセチレン膜の製造方法、及び、該製造方法によって製造され、スルホン酸基の分布が膜厚方向に均一である置換ポリアセチレン膜としたものである。

【解決手段】 式(1)で表される繰り返し単位を有する置換ポリアセチレンを膜状に成形し、スルホン化剤に接触させてスルホン化するスルホン酸基を有する置換ポリアセチレン膜の製造方法、及び、該製造方法によって製造され、スルホン酸基の分布が膜厚方向に均一である置換ポリアセチレン膜としたものである。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は燃料電池、二次電池、湿度センサー、イオンセンサー、ガスセンサー、デシカント剤等、種々の電気化学デバイスにおいて好適に用いられる電解質及び電解質膜としてのスルホン酸基を用する置換ポリアセチレン膜の製造方法、並びにこれらを用いた電気化学デバイス及び燃料電池に関する。
【背景技術】
【0002】
電解質及び電解質膜は、燃料電池、二次電池、湿度センサー、イオンセンサー、ガスセンサー、デシカント剤等の電気化学デバイスにおいて用いられ、それらのデバイスの性能に最も大きな影響を及ぼす部材である。こうした部材を構成する電解質材料として、酸解離性官能基を有するフッ素系高分子は、電解質特性、機械的特性、化学安定性などにおいて優れた性能を発揮するため、広範な用途に開発されている。
フッ素系電解質以外では、主に芳香族系高分子電解質の開発が盛んである。耐熱性や機械的特性、化学的安定性に優れた芳香族系高分子の主鎖骨格としては、例えば、ポリベンズイミダゾール、ポリスルホン、ポリエーテルエーテルケトン、ポリアミド、ポリイミドなど様々な骨格が利用されている。一方、最近、機能性材料として注目されているフラーレンに酸解離性の官能基を導入して、さらにポリマーバインダーで成形したものや、共役系高分子電解質など新しいタイプの電解質膜が開発されている。
【0003】
一方、ポリアセチレンは、アセチレンを遷移金属を用いて配位重合させることによって、主鎖骨格に二重結合と単結合が交互につながった構造を有する。この二重結合においてトランス配座で結合したものは、主鎖のπ電子が共役するため、半導体的性質を示す。また、化学ドーピングなどを施すことによって金属光沢を示し、金属と同等の導電性を発現することが知られている(非特許文献1)。
さらに、一置換アセチレン誘導体を重合させたものは、ポリアセチレンの側鎖に様々な機能性置換基を導入できることから、液晶性や光機能性を付与した導電性ポリアセチレンや、スルホン酸やホスホン酸などの極性基を導入したポリアセチレン電解質など、新しい機能性材料として注目されている(非特許文献2〜3)。
また、二置換のアセチレン誘導体を重合させたものについても報告されている。例えば、1−トリメチルシリル−1−プロピンなど嵩高い置換基を導入したポリアセチレン膜は、酸素富化膜などへの応用が期待されている。ジフェニルアセチレンの誘導体を配位重合させることによって、高分子量のシス配座リッチな重合体が得られ、これを用いた膜の気体透過性なども報告されている(特許文献1、非特許文献4、非特許文献5)。
【0004】
また、置換ポリアセチレンにイオン解離基を導入して、固体高分子電解質膜を作製する方法が記載されている(特許文献2)。記載されたスルホン化方法は、主に二通りであり、重合したポリアセチレンをクロロスルホン酸、濃硫酸などのスルホン化剤と接触した後に製膜する方法と、スルホン酸基を持つモノマーを重合した後に製膜する方法である。しかしながら、我々の研究においては、ポリマー溶液にスルホン化剤を加え、スルホン化した場合には、得られたポリマーは、N,N−ジメチルスルホキシド、N,N−ジメチルアセトアミド、水、メタノール、アセトン、酢酸エチルなどの一般的な溶媒には不溶となり、製膜は困難であった。さらに、スルホン酸基の導入量を増加させると水溶性となり、製膜は可能であっても、固体電解質特に燃料電池用固体電解質膜としては適用困難であると予想される。さらに、スルホン酸基を持つモノマーを重合又は共重合した場合にも、得られる構造は同様であり、上記のような溶媒に不溶であることが予想される。また、スルホン酸基をアミンなどで保護してスルホンアミドとした後に重合し、加水分解する方法も例示されているが、一般にスルホンアミドは、臭化水素や過塩素酸などの強酸や、ソジウム ナフタレニド、ソジウム アントラセニドといった強塩基でのみ脱離することが知られている。これらの反応を用いる場合には、主鎖の切断などの副反応を生じる可能性があり、好ましい方法とはいえない(非特許文献6)。
【0005】
先に挙げた酸解離性官能基を有するフッ素系高分子電解質は、電解質特性、加工性、機械的強度、化学的安定性に優れるものの、耐熱性が十分でなく、原料及び製造コストが高いという問題があった。また、構造中にフッ素原子を含むため、製造工程あるいは製造物が廃棄される際の環境へのフッ素イオン又はフッ化物の放出が、生体への影響又は環境負荷などの点で危惧されている。そうした社会的側面もうけて、フッ素系に代わる炭化水素系高分子電解質の開発が活発に行われているが、芳香族系高分子電解質、フラーレン含有電解質、共役系高分子電解質は、いずれにおいても、機械的強度が低い、加工性が悪いなどの問題があった。
【0006】
一方、ポリアセチレン誘導体については、電子伝導性、イオン伝導性などの報告があり、また、上記のようにイオン交換基を導入した置換ポリアセチレン電解質も知られているが、導入するイオン交換基がスルホン酸基である場合のスルホン化方法と製膜方法については、上記のように十分な技術であるとはいえなかった。
【0007】
本発明者らは、基材となる置換ポリアセチレンを製膜した後にスルホン化することによってスルホン酸基を導入すれば、上記の不溶化の問題を克服できると考えた。しかしながら、ポリジフェニルアセチレン膜を、濃硫酸等のスルホン化剤に浸漬してスルホン化を行うと、スルホン酸基は、膜の内部には導入されず、膜厚方向に均一にスルホン酸基を導入できないことが明らかとなった。
【特許文献1】特開2002−322293号公報
【特許文献2】特開2004−296141号公報
【非特許文献1】H. Shirakawa, T. Masuda and K. Takeda, The chemistry of triple-bonded functional groups, Chapter 17, pp 945-1016, Ed. By S. Patai, John Wiley & Sons, Chichester, 2004
【非特許文献2】K. Akagi, T. Kadokura and H. Shirakawa, Polymer, 1992, 33, 4058.
【非特許文献3】H. Onouchi, D. Kashiwagi, K. Hayashi, K. Maeda and E. Yashima, Macromolecules, 2004, 37, 5495-5503.
【非特許文献4】K. Nagai, T. Masuda, T. Nakagawa, B. D. Freeman and I. Pinnau, Prog. Polym. Sci., 2001, 26, 721-798.
【非特許文献5】T. Masuda, M. Teraguchi and R. Nomura, Am. Chem. Soc. Sym. Ser., 1999 , 733, 28-37.
【非特許文献6】T. W. Greene, P. G. M. Wuts, PROTECTIVE GROUPS IN ORGANIC SYNTHESIS 3rd ed., pp 605, JOHN WILY & SONS, New York, 1999.
【非特許文献7】T. Masuda, H. Tachimori, Pure Appl. Chem., 1994, A31, 1675-1690.
【非特許文献8】Peter G. M. Wuts, Katherun E. Wilson, Synthesis, 1998, 1593-1595.
【非特許文献9】R. W. Bott, C. Eaborn, Tadashi Hashimoto, J. Organometallic. Chem., 1965, 3, 442-447.
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、上記既知事実に鑑み、電気化学デバイスで使用するのに充分な電解質特性を示し、かつ用途に応じて充分な耐熱性、機械的強度を有し、環境負荷の大きなフッ素などのハロゲン元素を含まず、製膜性など加工性に優れた固体電解質膜、電極接合体及びそれらを用いた電気化学デバイス及び燃料電池を提供するために、均一にスルホン酸基を導入した固体電解質膜、好ましくは、そのイオン交換容量が従来より大きいスルホン酸基を有する置換ポリアセチレン電解質膜の製造方法とそれによって得られる電解質膜を提供することを課題とする。
【課題を解決するための手段】
【0009】
上記課題を解決するために、本発明では、下記式(1)で表される繰り返し単位を有する置換ポリアセチレンを膜状に成形し、該成形物をスルホン化剤に接触させてスルホン化することを特徴とするスルホン酸基を有する置換ポリアセチレン膜の製造方法としたものである。
【化1】
〔ただし、式(1)中、R1,R2は、いずれか一方又はいずれもが下記式(2)で表されるシリル基であり、残りの一方は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、t-ブチルジメチルシリルオキシ基、アセチルオキシ基、又は下記式(3)で表される。〕
【化2】
〔ただし、式(2)中、X1,X2,X3は、独立に炭素数1〜6の直鎖又は分岐のアルキル基を表す。〕
【化3】
〔ただし、式(3)中、R3は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、トリメチルシリル基、t-ブチルジメチルシリルオキシ基又はアセチルオキシ基又は式(2)を表す。〕
【0010】
このとき、モノマー合成の際に用いる出発原料の入手の容易さ、置換ポリアセチレンの重合度又は有機溶媒への溶解性を考慮すると、好ましくは、式(2)において、X1,X2,X3が独立に炭素数1〜4のアルキル基であり、さらに好ましくは、式(2)はトリメチルシリル基である。また、このときに用いる前記スルホン化剤としては、濃硫酸、濃硫酸と溶媒の混合溶液、発煙硫酸、三酸化硫黄−ジオキサン、三酸化硫黄-ピリジン、クロロスルホン酸、亜硫酸から選択されるいずれか1つ又は複数の組み合わせからなるスルホン化剤が特に好ましい。
【0011】
また、本発明では、上記のいずれかに記載の製造方法によって製造され、スルホン酸基の分布が膜厚芳香に均一であるスルホン酸基を有する置換ポリアセチレン膜としたものであり、スルホン酸基が膜内部まで導入され、スルホン酸基の分布が、膜厚方向に均一であるスルホン酸基を有する置換ポリアセチレン膜である。このとき、スルホン酸基の分布が膜厚方向に均一であることの指標として、例えば、SEM−EDSによって測定されるスルホン酸基を構成する硫黄原子由来の特性X線(SKa)強度比を用いることができ、膜中心部の強度が、測定範囲のSKaの最大値の好ましくは、70%以上、より好ましくは80%以上、もっとも好ましくは90%以上である。またさらに、上記のいずれかに記載の製造方法によって製造されるスルホン酸基を有する置換ポリアセチレン膜においては、イオン交換容量が2.0−3.5meq/gであることが特に好ましい。
【0012】
本発明によって得られる上記のスルホン基を有する置換ポリアセチレン膜の好ましい用途としては、このスルホン基を有する置換ポリアセチレン膜に電極を付与した、置換ポリアセチレン膜/電極接合体があり、また、この置換ポリアセチレン膜/電極接合体は、電気化学デバイスに好適に用いることができる。電気化学デバイスとしては、例えば、燃料電池、二次電池、湿度センサー、イオンセンサー、ガスセンサー、デシカント剤等、種々の電気デバイスに好適に用いることができ、特に燃料電池として最も好適に用いることができる。
【0013】
前記本発明は、公知のスルホン化方法である濃硫酸との接触においては、濃硫酸の粘度が比較的大きいために膜内部まで濃硫酸が浸透せず、スルホン化が進行しないものと推測し、その結果に基づき、硫酸(スルホン化剤)が内部に浸透しやすい膜であれば、容易に膜内部までスルホン化できると着想し、そのような構造としてポリマー分子鎖の間隙が大きい膜を用いることを試みた。
例えば、トリメチルシリル基を含有するポリアセチレン膜は、嵩高いトリメチルシリル基の影響でポリマー分子鎖の間隙が大きい構造を有していることが知られている(T. Masuda, H. Tachimori, Pure Appl. Chem., 1994, A31, 1675-1690.)。さらに、トリメチルシリル基を含有する芳香環を、三酸化硫黄-ジオキサン又は三酸化硫黄を用いて、トリメチルシリル基との置換反応によりスルホン化する方法がそれぞれ知られている(Peter G. M. Wuts, Katherun E. Wilson, Synthesis, 1998, 1593-1595. R. W. Bott, C. Eaborn, Tadashi Hashimoto, J. Organometallic. Chem., 1965, 3, 442-447)
【0014】
本発明者らは、これらの知見を応用することにより、本発明の課題を解決するための手段を発案し、本発明を完成するに至った。しかしながら、このような知見を高分子電解質の合成、さらには置換ポリアセチレンのスルホン化に用いた例は存在せず、さらには、膜状基材のスルホン化に関するスルホン酸基導入の均一性については、上記文献には一切例示も示唆もされていない。
すなわち、本発明は、嵩高い炭素数1〜6の直鎖又は分岐の含ケイ素置換基(シリル基)を有するアセチレンモノマーを配位重合すること等により得られる、シリル基を有する置換ポリアセチレンを用い、これを製膜した後に、濃硫酸などの強酸、濃硫酸と溶媒との混合溶液、三酸化硫黄又は三酸化硫黄―ジオキサンなどのスルホン化剤と接触させることにより、内部までスルホン化剤を浸透させ、さらには、シリル基の脱離を伴ってスルホン酸基を膜厚方向に均一に導入する技術である。
【0015】
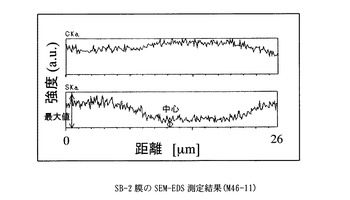
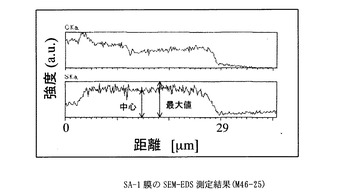
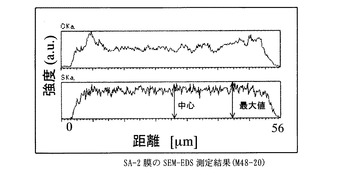
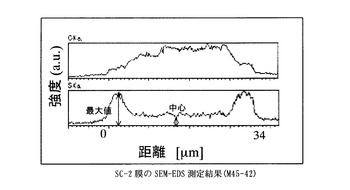
なお、本発明において、導入されたスルホン酸基が内部まで導入され均一であるとは、SEM-EDS(Scanning Electron Microscope - Energy Dispersive X-ray Spectrometer)によって膜厚方向のスルホン酸基(硫黄原子Sに基づく)分布を調べた場合に、膜中心部の硫黄の特性X線(SKa)の強度が、測定範囲中のSKaの最大値の70%以上、より好ましくは80%以上、もっとも好ましくは90%以上であることを指す。
【0016】
さらに、本発明の方法によって得られたスルホン酸基を有する置換ポリアセチレン膜は、膜厚方向に均一にスルホン酸基を有するため、従来技術で作製された置換ポリアセチレン電解質膜とは異なり、条件によってはイオン交換容量が2.0meq/g以上であっても、十分な膜強度を持ち、且つ水やメタノール水溶液に溶解しないスルホン酸基を有する置換ポリアセチレン膜である。ただし、イオン交換容量が3.5meq/gを越えると、スルホン酸基を有する置換ポリアセチレン膜が水又はメタノール水溶液に溶解する虞がある。これら置換ポリアセチレン膜は、スルホン酸基が均一に導入され、さらには条件によっては大きなイオン交換容量を有するため、プロトン(水素イオン)、リチウムイオンなどのイオン伝導性に優れた固体高分子電解質である。加えて、先述のように大きなイオン交換容量を有する場合には、低湿度状態であっても高いプロトン伝導性を有することが期待できる。勿論、構成する化学構造中に、ハロゲン元素は共有結合で導入されていないため、ハロゲン元素に関連する環境負荷は小さいことが期待される。
【発明の効果】
【0017】
本発明により、膜厚方向にスルホン酸基を均一に導入し、条件によっては従来では合成できなかった、イオン交換容量が2.0−3.5meq/gスルホン酸基を有する置換ポリアセチレン膜を合成することができる。これらスルホン酸基を有する置換アセチレン膜は、プロトン伝導性、イオン伝導性に優れた固体電解質として用いることができる。これらは、電気化学デバイスに用いるのに十分な機械的強度を有し、耐熱性に優れ、かつ化学構造中にハロゲン元素を共有結合で導入していないことから、製造及び廃棄時の環境負荷が小さいことが期待されるため、燃料電池やイオンセンサーなどの電気デバイス又は燃料電池に用いるのに好適である。
【発明を実施するための最良の形態】
【0018】
以下に、本発明をさらに詳細に説明する。本発明に使用される置換ポリアセチレンは、その分子構造中に式(1)の構造を含んでいれば特に制限はなく、一種類のアセチレン誘導体を重合してなる単独重合体でも2種以上のアセチレン誘導体を重合してなる共重合体でもよい。アセチレン誘導体は、所望のシリル基を有するハロゲン化アリーレン化合物とフェニルアセチレンなどのアセチレン化合物から、薗頭-萩原カップリング法などの公知の方法を用いて合成することができる。置換ポリアセチレンは、当該アセチレン誘導体をNb,Ta,Mo,Wなどの遷移金属触媒、又は、これら触媒とテトラブチル錫などの助触媒を用いて、脱水溶媒中で加熱することにより得られるが、公知すべての重合方法を用いることができる。置換ポリアセチレンの分子量は、電解質膜の耐熱性及び機械的強度に大きな影響を与え、分子量が小さすぎると、耐熱性及び機械的強度の低下を招き、分子量が大きすぎると、溶解性の低下、製膜時の溶媒量の増加などを招くので、おおむね1万から1000万の範囲、より望ましくは5万から500万の範囲の分子量の置換ポリアセチレンを用いるとよい。
【0019】
【化4】
〔ただし、式(1)中、R1,R2は、いずれか一方又はいずれもが下記式(2)で表されるシリル基であり、残りの一方は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、t-ブチルジメチルシリルオキシ基、アセチルオキシ基、又は下記式(3)で表される。〕
【化5】
〔ただし、式(2)中、X1,X2,X3は、独立に炭素数1〜6の直鎖又は分岐のアルキル基を表す。〕
【化6】
〔ただし、式(3)中、R3は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、トリメチルシリル基、t-ブチルジメチルシリルオキシ基、アセチルオキシ基又は一般式(2)を表す。〕
【0020】
芳香環上の置換基R1、R2及びR3がアルキル基又はアルコキシ基の場合、具体的には、メチル基、エチル基、n-プロピル基、i-プロピル基、シクロプロピル基、n-ブチル基、t-ブチル基、1-メチルプロピル基、2-メチルプロピル基、シクロブチル基、シクロプロピルメチル基、n-ペンチル基、1-メチルブチル基、2-メチルブチル基、3-メチルブチル基、シクロペンチル基、シクロブチルメチル基、n-ヘキシル基、4-メチルペンチル基、2-エチルブチル基、1-エチル-1-メチルプロピル基、シクロヘキシル基、n-ヘプチル基、1-メチルヘキシル基、シクロヘキシルメチル基、4-メチルシクロヘキシル基、シクロヘプチル基、n-オクチル基、2-エチルヘキシル基、メトキシ基、エトキシ基、n-プロポキシ基、i-プロポキシ基、n-ブトキシ基、t-ブトキシ基、1-メチルプロポキシ基、2-メチルプロポキシ基、シクロプロピルメトキシ基、シクロブトキシ基、n-ペンチルオキシ基、1-メチルブトキシ基、2-メチルブトキシ基、3-メチルブトキシ基、1-エチルプロポキシ基、1,1-ジメチルプロポキシ基、1,2-ジメチルプロポキシ基、2,2-ジメチルプロポキシ基、シクロペンチルオキシ基、1-メチルシクロプロピルメトキシ基、2-メチルシクロプロピルメトキシ基、n-ヘキシルオキシ基、1-メチルペンチルオキシ基、2-メチルペンチルオキシ基、3-メチルペンチルオキシ基、4-メチルペンチルオキシ基、1-エチルブトキシ基、2-エチルブトキシ基、1,1-ジメチルブトキシ基、3,3-ジメチルブトキシ基、1,2-ジメチルブトキシ基、1,3-ジメチルブトキシ基、1,1,2-トリメチルプロポキシ基、1,2,2-トリメチルプロポキシ基、1-メチル-1-エチルプロポキシ基、2-メチル-1-エチルプロポキシ基、シクロヘキシルオキシ基、シクロペンチルメトキシ基、1-メチルシクロペンチルオキシ基、2-メチルシクロペンチルオキシ基、3-メチルシクロペンチルオキシ基、n-ヘプチルオキシ基、1-メチルヘキシルオキシ基、1-エチルペンチルオキシ基、5-エチルペンチルオキシ基、1,1-ジメチルペンチルオキシ基、1,4-ジメチルペンチルオキシ基、1-(1-メチルエチル)ブトキシ基、1,3,3-トリメチルブトキシ基、1-エチル-2,2-ジメチルプロポキシ基、1-エチル-1,2-ジメチルプロポキシ基、1,1-ジエチルプロポキシ基、ジイソプロピルメトキシ基、シクロヘプチルオキシ基、シクロヘキシルメトキシ基、1-シクロペンチルエトキシ基、1-メチルシクロヘキシルオキシ基、2-メチルシクロヘキシルオキシ基、3-メチルシクロヘキシルオキシ基、4-メチルシクロヘキシルオキシ基、n-オクチルオキシ基、1-メチルヘプチルオキシ基、2-エチルヘキシルオキシ基、1,5-ジメチルヘキシルオキシ基、2-プロピルペンチルオキシ基、2-メチル-1-エチルペンチルオキシ基、2,4,4-トリメチルペンチルオキシ基、シクロオクチルオキシ基、1-シクロヘキシルエトキシ基、2-シクロヘキシルエトキシ基、2-エチルシクロヘキシルオキシ基、4-エチルシクロヘキシルオキシ基、2,3-ジメチルシクロヘキシルオキシ基、2,6-ジメチルシクロヘキシルオキシ基、3,5-ジメチルシクロヘキシルオキシ基、3-シクロペンチルプロポキシ基などを例示できる。
【0021】
さらに、式(2)のX1、X2、X3は、具体的には、メチル基、エチル基、n-プロピル基、i-プロピル基、n-ブチル基、1-メチルプロピル基、2-メチルプロピル基、t-ブチル基、n-ペンチル基、1-メチルブチル基、2-メチルブチル基、3-メチルブチル基、1,1-ジメチルプロピル基、1,2-ジメチルプロピル基、2,2-ジメチルプロピル基、3,3-ジメチルプロピル基、1-エチルプロピル基、n-ヘキシル基、1-メチルペンチル基、2-メチルペンチル基、3-メチルペンチル基、4-メチルペンチル基、1,1-ジメチルブチル基、1,2-ジメチルブチル基、1,3-ジメチルブチル基、1,1,2-トリメチルプロピル基、1,2,2-トリメチルプロピル基、1,1,2,2-テトラメチルエチル基、1-エチルブチル基、2-エチルブチル基を挙げることができる。さらに出発原料の入手の容易さ、置換ポリアセチレンの重合度、有機溶媒に対する溶解性を考慮すると、式(2)のX1、X2、X3は、独立に炭素数1〜4の直鎖又は分岐のアルキル基であることが好ましく、具体的にはメチル基、エチル基、n-プロピル基、i-プロピル基、n-ブチル基、1-メチルプロピル基、2-メチルプロピル基、t-ブチル基である。さらに好ましくは式(2)はトリメチルシリル基である。これら置換基を持つ置換ポリアセチレンを公知の方法により製膜し、置換ポリアセチレン膜を得ることができる。
【0022】
公知の製膜方法としては、溶媒キャスト法、スピンコート法、転写法、印刷法等による製膜方法の他、必要に応じて加熱処理や、圧延・延伸などの機械的処理を組み合わせてもよく、膜状に成形できれば特に制限はない。
【0023】
スルホン化方法としては、濃硫酸、濃硫酸と溶媒の混合溶液、発煙硫酸、三酸化硫黄-ジオキサン、三酸化硫黄-ピリジン、クロロスルホン酸又は亜硫酸などのスルホン化剤を用いることが出来る。液相中でスルホン化する場合には、必要に応じて溶媒や界面活性剤を用いることができる。この溶媒及び界面活性剤としては、膜の性状、スルホン化の制御において悪影響を及ぼさなければ特に制限はなく、例えば、溶媒としては、水、炭素数1〜8のアルコール、酢酸エチル、酢酸ブチル、クロロホルム、ジクロロメタン、1,2-ジクロロエタン、ギ酸、酢酸、酪酸、無水酢酸、クロロ酢酸、トリフルオロ酢酸、トリフルオロ酢酸無水物、ニトロベンゼンなどを単独、あるいは2種以上混合して用いることができる。界面活性剤としては、テトラメチルアンモニウム、テトラエチルアンモニウム、テトラプロピルアンモニウム、テトラブチルアンモニウムなどの4級アンモニウムイオンと塩化物イオン、臭化物イオン、ヨウ化物イオン、硫酸水素イオンなどとの塩、又はノナニルベンゼンスルホン酸、ラウリル硫酸、安息香酸などのナトリウム塩やアンモニウム塩といったイオン性界面活性剤、プロピレングリコールやポリオキシエチレングリコールモノラウリルエーテルなどの非イオン性界面活性剤などを、単独又は2種以上混合して用いることができる。気相中で反応させる場合に、例えば亜硫酸ガスに直接膜を曝しても良いが、スルホン化雰囲気の制御又は移動相として、窒素ガス、空気、又は先にあげた溶媒の蒸気などを単独又は2種以上混合して用いることができる。スルホン化試薬の量、溶媒や界面活性剤の量、ガス量、スルホン化処理に要す時間、温度などは、膜の性状への影響、目的の電気化学デバイスで必要な電気化学特性に応じたスルホン化量をもとに決めればよく、生産効率を考慮して、処理時間が数分から数時間になるように決めればよい。
【0024】
スルホン化剤の中では、工業上安価でかつ取り扱いが比較的容易であり、さらには再利用が可能な、濃硫酸又は濃硫酸と溶媒の混合溶液が好ましい。濃硫酸と混合する溶媒としては、濃硫酸と反応しない溶媒であれば特に制限はなく、上記の溶媒を用いることが出来る。濃硫酸と溶媒の濃度は、濃硫酸100〜20重量%、好ましくは100〜50重量%、さらに好ましくは100〜80重量%である。
置換ポリアセチレン膜をこれらスルホン化剤に浸漬させる場合には、予め膜を溶媒に浸漬して膨潤させておいても良い。膨潤させる溶媒は、膜が溶解しなければ特に制限はなく、酢酸エチル、ジエチルエーテルなどが例示できる。さらには、置換ポリアセチレン膜を浸漬させる温度については、用いた溶媒の沸点以下であれば特に制限はないが、好ましくは-30〜200℃、より好ましくは0〜100℃である。
【実施例】
【0025】
以下実施例に基づいて本発明をさらに詳しく説明する。なお、実施例及び比較例において、使用した膜の作製方法をモノマー合成、ポリマー合成、製膜方法、脱シリル化方法の順に記載した。さらに、比較例として、ポリマー溶液にスルホン化剤を加えてポリマーをスルホン化した結果、及び当該シリル基を有しない置換ポリアセチレン膜のスルホン化結果を記載した。なお、1H−NMRスペクトル、FT−IRスペクトル、分子量、膜厚、イオン交換容量、含水率、膨潤度、イオン交換容量及びスルホン酸基分布は以下のようにして求めた。
【0026】
1.1H−NMRスペクトル
核磁気共鳴装置(BurkerBiospin製, 商品名AVNCE DRX 400)を用いて測定した。
2.FT−IRスペクトル
FT-IR測定装置(PerkinElmer製、商品名PARAGON FT-IR)を用いてKBrディスク法により測定した。
3.分子量
分子量は得られたポリマーをテトラヒドロフラン(THF)に溶解し、ゲルパーミエーションクロマトグラフィー(GPC)(東ソー製、商品名HLC-802A)により、数平均分子量及び重量平均分子量を測定した。溶離液にはTHFを用い、標準試料としてポリスチレンを用いた。
4.膜厚
所定の膜を110℃にて16時間真空乾燥した後、膜厚計(Mitutoyo製、商品名クイックマイクロ)を用いて膜の周囲及び中心部5点を測定し、その平均値を算出した。
【0027】
5.イオン交換容量
所定量の膜を110℃にて16時間真空乾燥し、重量測定を行った。その後、膜を0.1mol/l塩化ナトリウム水溶液50mlに浸漬し、16時間緩やかに撹拌した。その後膜を取り出し、1/50N水酸化ナトリウム水溶液にて滴定した。滴定には自動滴定装置(東亜電波工業製、商品名AUT−501)を用い、滴的曲線の変曲点を中和点(終点)として、(式1)に従いイオン交換容量を算出した。
イオン交換容量(meq/g)=〔0.02×ファクター×1/50N水酸化ナトリウム水溶液の消費量(mL)〕/〔膜の重量(g)〕 (式1)
6.含水率
所定量の膜を1.0mol/l硫酸水溶液にて1時間煮沸し、さらに純水にて1時間煮沸したのち、膜の重量を測定した。その後、膜を110℃にて16時間真空乾燥し重量測定を行い、(式2)に従って含水率を算出した。
含水率(%)=〔含水膜の重量(g)−乾燥時の重量(g)〕/〔乾燥時の重量(g)〕×100 (式2)
【0028】
7.膨潤度
所定量の膜を1.0mol/l硫酸水溶液にて1時間煮沸し、さらに純水にて1時間煮沸したのち、膜の大きさ(縦×横×厚さ)を測定した。その後、膜を110℃にて16時間真空乾燥し膜の大きさを測定し、(式3)に従って膨潤度を算出した。
膨潤度(%)=〔膨潤時の体積(mm3)〕/〔乾燥時の体積(mm3)〕× 100 (式3)
8.イオン伝導度
膜を2cm ´ 5cmの大きさに切り、1mol/lの硫酸水溶液で1時間煮沸処理した。続いて蒸留水で1時間煮沸した後、0.5cm離して平行に配置した長さ4cmの金電極上に密着して、恒温恒湿槽内で90 ℃、相対湿度90%に制御しながらインピーダンスアナライザ(東陽テクニカ製、商品名Solartron1260)を用いて周波数0.5 Hz 〜10 MHzの範囲でインピーダンス測定を行った。得られたNyquist Plotよりインピーダンスを求め、(式4)に従いイオン伝導度を算出した。
イオン伝導度(S/cm)=〔0.5 (cm)〕/〔インピーダンス(Ω)´(4 (cm) ´ 膜厚(cm)〕 (式4)
【0029】
9.膜厚方向のスルホン酸基分布測定
膜を小片状に切り出し、サンプルホルダーに固定したのち、PT−Pd蒸着を行い、サンプルの膜厚方向の炭素と硫黄の分布をSEM-EDS(Scanning Electron Microscope - Energy Dispersive X-ray Spectrometer)、(日本電子製、商品名JSM-5800LV)により分析した。図中、CKaは、炭素の特性X線の強度、SKaは、硫黄の特性X線の強度を表す。CKaは、置換ポリアセチレン膜の、SKaは、スルホン酸基の存在位置及び存在量の相対値にそれぞれ相当する。スルホン酸基が均一に導入されていることを示す指標として、SKaの最大値に対する膜中心部のSKaの強度比(a)を用いた。
【0030】
【化7】
(モノマーM-1の合成)
アルゴン雰囲気下、200024ml三口フラスコにビス(トリフェニルホスフィン)パラジウム(II)ジクロライド17 mg (0.024mmol)とヨウ化銅23 mg (0.12mmol)及びトリフェニルホスフィン32mg(0.12mmol)を量り取った。その後、予め水素化カルシウムにて脱水したトリエチルアミン 70ml (0.50mmol)を加えた。さらに、1−ブロモ−4−(トリメチルシリル)ベンゼン 1.6ml (8.0mmol)とフェニルアセチレン 0.90ml(8.0mmol)を加え、90℃にて16時間撹拌した。
【0031】
その後、トリエチルアミンを留去し、ジエチルエーテルを加えろ過した。ろ液を濃縮し、シリカゲルカラムクロマトグラフィー(溶媒:ヘキサン)にて精製した。その後、アルミナカラムクロマトグラフィー(溶媒:ヘキサン)にて精製を行ったところ、透明で粘ちょうな液体1.2 g(収率:61 %)を得た。1H-NMR, IR測定により、M−1であることを確認した。
1H-NMR, d (ppm, CDCl3, 400 MHz): 0.28 (9H, s, CH3´3), 7.33 (2H, m, Ph), 7.35 (1H, m, Ph), 7.50 (4H, s, Ph), 7.53 (2H, m, Ph).
IR, n (KBr disk, cm-1): 3065 (w), 2956 (m, C-H), 2219 (vw, CoC), 1601 (m, arC-C), 1249 (s), 1101 (w), 855 (s, Si-C), 839 (s), 820 (s), 755 (m), 690 (s), 627 (w), 633 (m).
【0032】
(ポリマーP−1の合成)
50mlナスフラスコに、グローブボックス中にて、五塩化タンタル (V)55mg(0.15mmol)及びテトラブチル錫(IV) 0.10ml(0.31mmol)を加えた。さらに、脱水トルエン4.0mlを加え、80℃にて20分間撹拌し、触媒溶液を熟成した。また、アルゴン雰囲気下50 mlナスフラスコにM−11.0g(4.0mmol)を量り取り、脱水トルエン4.0mlを加えた。その後、キャヌラによりモノマー溶液を触媒溶液に加え、80℃にて2時間撹拌した。反応液を希釈し、メタノール中に析出させたところ、黄色繊維 0.70g(収率:73%)を得た。IR測定により、P−1であることを確認した。また、GPC測定により、平均分子量を測定した。
IR, n (KBr disk, cm-1): 3055 (w, arC-H), 3017 (w), 2957 (s, C-H), 1646 (vw, >C=C<), 1597 (w, arC-C), 1494 (w), 1248 (s), 1118 (m), 855 (s, Si-C), 835 (s), 814 (s), 755 (s), 689 (s), 630 (w), 554 (s).
GPC測定結果:数平均分子量= 6.0×105、重量平均分子量=6.1×105
【0033】
(A膜の作成)
300mlナスフラスコにP−1を0.2g量り取り、トルエン50mlを加え100℃にて16時間溶解させた。その後、恒温槽内に水平に設置した10cm×10cmのガラス板に、幅1cmのテフロン(登録商標)枠を取り付け、P−1のトルエン溶液を溜延した。60℃にて3日間静置したところ、膜厚29μmの丈夫な黄色のA−1膜を得た。さらに、P−1の量を変化させることによりその他は同様の手順により、膜厚の異なるA−2膜(膜厚56μm)を合成した。
【0034】
(B膜の作製)(A膜の脱シリル化処理)
【化8】
10mlナスフラスコに、ヘキサン:トリフルオロ酢酸=1:1溶液50mlを加え、A−1膜を含浸させ、室温にて24時間撹拌した。その後膜をヘキサン溶液50mlに含浸させ16時間撹拌した。その後110℃にて16時間膜を乾燥したところ、膜厚28μmの丈夫な黄色のB−1膜を得た。IR測定により脱シリル化されたことを確認した。同様にして、A−2膜を脱シリル化することにより、B−2膜を得た。
IR, n (cm-1, KBr disk): 3085 (w), 3054 (s, arC-H), 3019 (m), 2959 (vw C-H), 2923 (vw, C-H), 1661 (w, >C=C<), 1599 (w, arC-C), 1576 (w), 1494 (s, arC-C), 1442 (s, arC-C), 1250 (w), 1156 (w), 1076 (w), 1030 (w), 902 (m), 830 (w), 769 (s), 691 (s), 553 (s).
【0035】
【化9】
(モノマーM−2の合成)
アルゴン雰囲気下、200ml三口フラスコにビス(トリフェニルホスフィン)パラジウム(II)ジクロライド 62mg (0.089mmlo), ヨウ化銅 85mg (0.44mmol)及びトリフェニルホスフィン0.12g(0.44mmol)を量り取った。その後、予め水素化カルシウムにて脱水したトリエチルアミン30mlを加え、さらにフェニルアセチレン3.3ml(30mmol)及び4−ブロモジフェニルエーテル5.2ml (30mmol)を加え、90℃にて4時間撹拌した。トリエチルアミンを留去した後、ジエチルエーテルを加えて抽出し、ろ過を行った。ろ液を水洗し、さらにエバポレートした後、シリカゲルカラムクロマトグラフィー(溶媒:ヘキサン)にて精製したところ、白色固体3.1g(収率:39 %)を得た。1H-NMR, IR測定により、M-2であることを確認した。
1H-NMR, d (ppm, CDCl3, 400 Mz): 6.97 (2H, d, J=8.8Hz, Ph), 7.20 (2H, d, J=8.8Hz,Ph), 7.15 (1H, t, J=8Hz, Ph), 7.32-7.39 (5H, m, Ph), 7.50-7.54 (4H, m, Ph).
IR, n (cm-1, KBr disk): 3050 (m, arC-H), 2360 (w, C≡C), 1591 (s, arC-C), 1490 (s, arC-C), 1286 (s), 1258 (s, arC-O-arC), 1105 (s), 1071 (s), 838 (s), 751 (s), 691 (s).
【0036】
(ポリマーP-2の合成)
アルゴン雰囲気下、100ml二口フラスコにグローブボックス中にて五塩化タンタル 0.26g(0.69mmol)及びテトラブチル錫(IV) 0.45ml(1.4mmol)を加えた。さらに、脱水トルエン15mlを加え、80℃にて20分間撹拌し、触媒溶液を熟成した。また、アルゴン雰囲気下100mlナスフラスコにM-21.0g(2.9mmol)を量り取り、脱水トルエン15mlを加えた。その後、キャヌラによりモノマー溶液を触媒溶液に加え24時間撹拌した。その後、メタノール中に析出させたところ黄褐色繊維0.62g(収率:62%)を得た。IR測定により、P−2であることを確認した。またGPC測定により、平均分子量を測定した。
IR, ν (cm-1, KBr disk): 3052 (w, arC-H), 1588 (m, arC-C), 1489 (s, arC-C), 1237 (s,ar C-O-arC), 890 (s), 750 (m).
GPC測定結果:数平均分子量=1.4×106、重量平均分子量=1.5×106
【0037】
(C膜の作成)
500mlナスフラスコにP−20.20gを量り取り、トルエン50mlを加え、90℃にて16時間撹拌した。その後、恒温槽内に水平に設置した10cm×10cmのガラス板に、幅1cmのテフロン(登録商標)枠を取り付け、P−2のトルエン溶液を溜延した。その後、50℃にて6時間静置したところ、膜厚29μmの黄色のC−1膜を得た。さらに、P−2の量を変化させることによりその他は同様の手順で、膜厚の異なるC−2膜(膜厚35μm)、C−3膜(膜厚55μm)を合成した。
【0038】
【化10】
(モノマーM−3の合成)
アルゴン雰囲気下、200ml三口フラスコにビス(トリフェニルホスフィン)パラジウム(II)ジクロライド 25mg (0.035mmlo),ヨウ化銅 6.7 mg (0.035mmol),トリフェニルホスフィン 0.047 g (0.18mmol)及び4−エチニルジフェニルエーテル2.3g(12mmol)を量り取った。その後、予め水素化カルシウムにて脱水したトリエチルアミン 5.9mlを加え、さらに1−ブロモ−4−(トリメチルシリル)ベンゼン 2.5ml(12mmol)を加え、90℃にて4時間撹拌した。トリエチルアミンを留去した後、ジエチルエーテルを加えて抽出し、ろ過を行った。ろ液を水洗し、さらにエバポレートした後、シリカゲルカラムクロマトグラフィー(溶媒:ヘキサン)にて精製したところ、白色固体3.0 g(収率:75 %)を得た。1H-NMR, IR測定により、M−3であることを確認した。
1H-NMR, d (ppm, CDCl3, 400 Mz): 0.28 (9H, s, CH3´3), 6.97 (2H, m, Ph), 7.05 (2H, m, Ph), 7.15 (1H, m, Ph), 7.37 (2H, m, Ph), 7.50 (6H, m, Ph).
IR, n (cm-1, KBr disk): 3067 (m, arC-H), 2954 (w, C-H st), 2213 (w, C≡C), 1587 (m, arC-C), 1508 (m), 1487 (s, arC-C), 1244 (s, arC-O-arC), 1163 (m), 1099 (s), 838 (s, Si-C),819 (s), 751 (s), 690 (m), 518 (m).
【0039】
(ポリマーP−3の合成)
アルゴン雰囲気下、100ml二口フラスコにグローブボックス中にて五塩化タンタル 0.45g(1.17mmol)及びテトラブチル錫(IV)0.81ml(2.3mmol)を加えた。さらに、脱水トルエン38mlを加え、80℃にて20分間撹拌し、触媒溶液を熟成した。またアルゴン雰囲気下100mlナスフラスコにM−3を2.0g(5.9mmol)量り取り、脱水トルエン20mlを加えた。その後、キャヌラによりモノマー溶液を触媒溶液に加え24時間撹拌した。その後、メタノール中に析出させたところ黄褐色繊維1.0g(収率:50 %)を得た。IR測定によりP−3であることを確認した。またGPC測定により、平均分子量を測定した。
IR, ν (cm-1, KBr disk): 3064 (w, arC-H), 1590 (s, arC-C), 1488 (s, arC-C), 1241 (s,ar C-O-arC), 836 (s, Si-C), 750 (s), 690 (s).
GPC測定結果:数平均分子量=3.8×106、重量平均分子量=5.8×106
【0040】
(D膜の作成)
500mlナスフラスコにP−3を0.20g量り取り、テトラヒドロフラン60mlを加え、70℃にて16時間撹拌した。その後、恒温槽内に水平に設置した10cm×10cmのガラス板に、幅1cmのテフロン(登録商標)枠を取り付け、P−3のテトラヒドロフラン溶液を溜延した。その後、50℃にて6時間静置したところ、膜厚50μmの黄色のD−1膜を得た。さらに、P−3の量を変えることにより、その他は同様の手順で、膜厚の異なる膜D−2(膜厚:40μm)を合成した。
比較例1 P−1溶液にスルホン化剤を加えたスルホン化
【0041】
P−1の20mgをアルゴン雰囲気下、50mlナスフラスコに量り取り、ジクロロメタン(脱水)7.0mlを加え、室温にて16時間撹拌して、P−1のジクロロメタン溶液を調整した。このポリマー溶液に、クロロスルホン酸 : ジクロロメタン=1:99(容積比)0.5mlを滴下したところ、繊維状の沈殿物が生じた。2時間撹拌した後、反応溶液をエーテル中に加え、沈殿物をろ別し、60℃、16時間真空乾燥した。その後、IR測定を行ったところ、トリメチルシリル基の脱離とスルホン酸基の導入が確認された。一方、沈殿物はN,N-ジメチルスルホキシド、N,N-ジメチルアセトアミド、m-クレゾール、メタノール、アセトン、酢酸エチル、水に不溶であった。
IR, ν (cm-1, KBr disk): 3443 (s), 1637 (m, arC-C), 1216 (m), 1178 (m), 1128 (m, SO3H), 1036 (m, SO3H), 1009 (s), 759 (w), 689 (m), 578 (w)
【0042】
比較例2 P−2溶液にスルホン化剤を加えたP−2のスルホン化
P−2の20mgをアルゴン雰囲気下、50mlナスフラスコに量り取り、ジクロロメタン(脱水)3.5mlを加え、室温にて16時間撹拌して、P−2のジクロロメタン溶液を調整した。このポリマー溶液にクロロスルホン酸:ジクロロメタン=1:99(容積比)0.25mlを滴下したところ繊維状の沈殿物が生じた。2時間撹拌した後、反応溶液をエーテル中に加え、沈殿物をろ別した後、60℃、16時間真空乾燥した。その後IR測定を行ったところ、トリメチルシリル基の脱離とスルホン酸の導入が確認された。一方、沈殿物はN,N−ジメチルスルホキシド、N,N−ジメチルアセトアミド、m−クレゾール、メタノール、アセトン、酢酸エチル、水に不溶であった。
IR, ν (cm-1, KBr disk): 3444 (s), 1637 (m, arC-C), 1490 (m, arC-C), 1241 (m, arC-O-arC), 1169 (m), 1125 (w, SO3H), 1033 (m, SO3H), 1007 (w), 694 (m), 607 (w), 552 (w).
【0043】
比較例3 B−1膜のスルホン化(SB−1膜の合成)
50mlナスフラスコに、濃硫酸(97%)10mlを量り取り、B−1膜の1.8mgを浸漬して室温にて3時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、SB−1膜を得た。IR測定を行ったが、スルホン酸の導入は確認されなかった。
IR, n (cm-1, KBr disk): 3055 (s, arC-H), 1599 (w, arC-C), 1491 (s, arC-C), 1440 (m), 1246 (m), 1162 (w), 903 (m), 832 (w), 754 (m), 687 (s), 548 (s).
得た膜は、膜厚28μm、イオン交換容量は検出限界以下であった。
【0044】
比較例4 B-2膜のスルホン化(SB−2膜の合成)
100mlナスフラスコに、濃硫酸(97%)50mlを量り取り、B−2膜41mgを浸漬して室温にて16時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色のSB−2膜を得た。IR測定により、スルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1632 (m, arC-C), 1490 (s, arC-C), 1440 (s), 1254 (s), 1168 (m), 1128 (w, SO3H), 1032 (w, SO3H), 1003 (w), 906 (w), 829 (w), 755 (s), 690 (s), 567 (m).
得た膜は、膜厚26μm、イオン交換容量1.4meq/g、含水率21%、膨潤度156%、イオン伝導度は5.6 x 10-3S/cmであった(90℃,RH90%)。
【0045】
実施例1 A−1膜のスルホン化(SA−1膜の合成)
100mlナスフラスコに、濃硫酸(97%)50mlを量り取り、A−1膜69mgを浸漬して室温にて3時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色のSA−1膜を得た。IR測定により、脱シリル化反応及びスルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1642 (m, arC-C), 1492 (s, arC-C), 1442 (w), 1218 (s), 1154 (s), 1128 (w, SO3H), 1033 (w, SO3H), 1005 (w), 907 (w), 825 (w), 755 (s), 691 (s), 572 (m).
得た膜は、膜厚29μm、イオン交換容量2.3meq/g、含水率80 %、膨潤度282%、イオン伝導度は3.7x10-1S/cmであった(90℃,RH90%)。
【0046】
実施例2 A−2膜のスルホン化 (SA−2膜の合成)
100mlナスフラスコに、濃硫酸(97%)と酢酸エチルの混合溶液(濃硫酸:酢酸エチル=801:20)50mlを量り取り、A−2膜52mgを浸漬して室温にて16時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色のSA−2膜を得た。IR測定により、脱シリル化反応及びスルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1634 (m, arC-C), 1490 (s, arC-C), 1441 (w), 1215 (s), 1159 (s), 1128 (s, SO3H), 1032 (w, SO3H), 1003 (w), 910 (w), 827 (w), 756 (s), 693 (s), 572 (m).
得た膜は、膜厚56μm、イオン交換容量2.1meq/g、含水率78 %、膨潤度375%、イオン伝導度は2.4x10-1S/cmであった(90℃, RH90%)。
【0047】
比較例5 C−1膜のスルホン化 (SC−1膜の合成) (M48−11)
100mlナスフラスコに、濃硫酸(97%)30mlを量り取り、C−1膜56mgを浸漬して室温にて16分間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色のSC−1膜を得た。IR測定を行ったがスルホン酸の導入は確認されなかった。
IR, n (cm-1, KBr disk): 3053 (m, arC-H), 1588 (m, arC-C), 1487 (s, arC-C), 1238 (s, arC-O-arC), 1164 (s), 869(w), 750 (s), 689 (s).
得た膜は、膜厚28μm、イオン交換容量は検出限界以下であった。
【0048】
比較例6 C−2膜のスルホン化 (SC−2膜の合成)(M45−42)
100mlナスフラスコに、濃硫酸(97%)50mlを量り取り、C−2膜69mgを浸漬して室温にて1時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色のSC−2膜を得た。IR測定により、スルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1588 (m, arC-C), 1489 (s, arC-C), 1240 (s, arC-O-arC), 1166 (s), 1123(s, SO3H), 1030 (s, SO3H), 1003 (s), 831(w), 748 (s), 690 (s).
得た膜は、膜厚34μm、イオン交換容量1.1meq/g、含水率15%、膨潤度106%、イオン伝導度は1.0x10-1S/cmであった(90℃,RH90%)。
【0049】
比較例7 C−3膜のスルホン化 (SC−3膜の合成)(M48−5)
100mlナスフラスコに、濃硫酸(97%)と酢酸エチルの混合溶液(濃硫酸:酢酸エチル=80:20)50mlを量り取り、C−3膜65mgを浸漬して室温にて3時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色の膜SC−3を得た。IR測定により、スルホン化反応が進行していることを確認した。
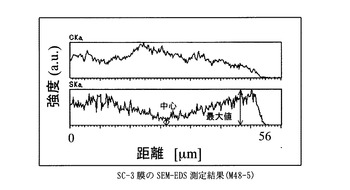
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1588 (m, arC-C), 1489 (s, arC-C), 1239 (s, arC-O-alC), 1164 (s), 1123(s, SO3H), 1029 (s, SO3H), 1004 (s), 832(w), 751 (s), 691 (s).
得た膜は、膜厚56μm、イオン交換容量1.0meq/g、含水率22%、膨潤度127%、イオン伝導度は3.8x10-2S/cmであった(90℃,RH90%)
【0050】
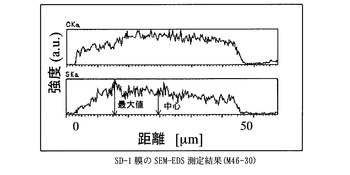
実施例3 D−1膜のスルホン化 (SD−1膜の合成)
100mlナスフラスコに、濃硫酸(97%)50mlを量り取り、D−1膜49mgを浸漬して室温にて16分間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色の膜SD−1を得た。IR測定により、脱シリル化反応及びスルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1588 (m, arC-C), 1489 (s, arC-C), 1238 (s, arC-O-arC), 1164 (s), 1122(s, SO3H), 1028 (s, SO3H), 1002 (s), 830(w), 753 (s), 691 (s).
得た膜は、膜厚50μm、イオン交換容量1.9meq/g、含水率65%、膨潤度151%、イオン伝導度は8.0x 10-2S/cmであった(90℃,RH90%)
【0051】
実施例4 D−2膜のスルホン化(SD−2)膜の合成
100mlナスフラスコに、濃硫酸(97%)と酢酸エチルの混合溶媒(濃硫酸:酢酸エチル=80:20)50mlを量り取り、D−2膜30mgを浸漬して室温にて3時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色の膜SD−2を得た。IR測定により、脱シリル化反応及びスルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1587 (m, arC-C), 1492 (s, arC-C), 1239 (s, arC-O-arC), 1163 (s), 1122(s, SO3H), 1028 (s, SO3H), 1001 (s), 830(w), 752 (s), 692 (s).
得た膜は、膜厚40μm、イオン交換容量1.5meq/g、含水率63%、膨潤度262%、イオン伝導度は4.7x10-1S/cmであった(90℃,RH90%)
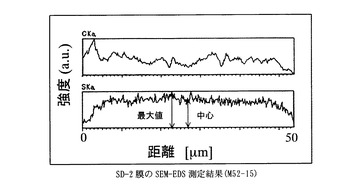
図1〜図7に比較例及び実施例の膜厚方向のSEM−EDS分析結果を示す。さらに、表1に各サンプルのaを示した。比較例に対して実施例の膜中心部の強度は大きく、均一にスルホン酸基が導入されていることが示された。
【0052】
【表1】
【図面の簡単な説明】
【0053】
【図1】SB−2膜のSEM−EDS測定結果に示すグラフ。
【図2】SA−1膜のSEM−EDS測定結果に示すグラフ。
【図3】SA−2膜のSEM−EDS測定結果に示すグラフ。
【図4】SC−2膜のSEM−EDS測定結果に示すグラフ。
【図5】SC−3膜のSEM−EDS測定結果に示すグラフ。
【図6】SD−1膜のSEM−EDS測定結果に示すグラフ。
【図7】SD−2膜のSEM−EDS測定結果に示すグラフ。
【技術分野】
【0001】
本発明は燃料電池、二次電池、湿度センサー、イオンセンサー、ガスセンサー、デシカント剤等、種々の電気化学デバイスにおいて好適に用いられる電解質及び電解質膜としてのスルホン酸基を用する置換ポリアセチレン膜の製造方法、並びにこれらを用いた電気化学デバイス及び燃料電池に関する。
【背景技術】
【0002】
電解質及び電解質膜は、燃料電池、二次電池、湿度センサー、イオンセンサー、ガスセンサー、デシカント剤等の電気化学デバイスにおいて用いられ、それらのデバイスの性能に最も大きな影響を及ぼす部材である。こうした部材を構成する電解質材料として、酸解離性官能基を有するフッ素系高分子は、電解質特性、機械的特性、化学安定性などにおいて優れた性能を発揮するため、広範な用途に開発されている。
フッ素系電解質以外では、主に芳香族系高分子電解質の開発が盛んである。耐熱性や機械的特性、化学的安定性に優れた芳香族系高分子の主鎖骨格としては、例えば、ポリベンズイミダゾール、ポリスルホン、ポリエーテルエーテルケトン、ポリアミド、ポリイミドなど様々な骨格が利用されている。一方、最近、機能性材料として注目されているフラーレンに酸解離性の官能基を導入して、さらにポリマーバインダーで成形したものや、共役系高分子電解質など新しいタイプの電解質膜が開発されている。
【0003】
一方、ポリアセチレンは、アセチレンを遷移金属を用いて配位重合させることによって、主鎖骨格に二重結合と単結合が交互につながった構造を有する。この二重結合においてトランス配座で結合したものは、主鎖のπ電子が共役するため、半導体的性質を示す。また、化学ドーピングなどを施すことによって金属光沢を示し、金属と同等の導電性を発現することが知られている(非特許文献1)。
さらに、一置換アセチレン誘導体を重合させたものは、ポリアセチレンの側鎖に様々な機能性置換基を導入できることから、液晶性や光機能性を付与した導電性ポリアセチレンや、スルホン酸やホスホン酸などの極性基を導入したポリアセチレン電解質など、新しい機能性材料として注目されている(非特許文献2〜3)。
また、二置換のアセチレン誘導体を重合させたものについても報告されている。例えば、1−トリメチルシリル−1−プロピンなど嵩高い置換基を導入したポリアセチレン膜は、酸素富化膜などへの応用が期待されている。ジフェニルアセチレンの誘導体を配位重合させることによって、高分子量のシス配座リッチな重合体が得られ、これを用いた膜の気体透過性なども報告されている(特許文献1、非特許文献4、非特許文献5)。
【0004】
また、置換ポリアセチレンにイオン解離基を導入して、固体高分子電解質膜を作製する方法が記載されている(特許文献2)。記載されたスルホン化方法は、主に二通りであり、重合したポリアセチレンをクロロスルホン酸、濃硫酸などのスルホン化剤と接触した後に製膜する方法と、スルホン酸基を持つモノマーを重合した後に製膜する方法である。しかしながら、我々の研究においては、ポリマー溶液にスルホン化剤を加え、スルホン化した場合には、得られたポリマーは、N,N−ジメチルスルホキシド、N,N−ジメチルアセトアミド、水、メタノール、アセトン、酢酸エチルなどの一般的な溶媒には不溶となり、製膜は困難であった。さらに、スルホン酸基の導入量を増加させると水溶性となり、製膜は可能であっても、固体電解質特に燃料電池用固体電解質膜としては適用困難であると予想される。さらに、スルホン酸基を持つモノマーを重合又は共重合した場合にも、得られる構造は同様であり、上記のような溶媒に不溶であることが予想される。また、スルホン酸基をアミンなどで保護してスルホンアミドとした後に重合し、加水分解する方法も例示されているが、一般にスルホンアミドは、臭化水素や過塩素酸などの強酸や、ソジウム ナフタレニド、ソジウム アントラセニドといった強塩基でのみ脱離することが知られている。これらの反応を用いる場合には、主鎖の切断などの副反応を生じる可能性があり、好ましい方法とはいえない(非特許文献6)。
【0005】
先に挙げた酸解離性官能基を有するフッ素系高分子電解質は、電解質特性、加工性、機械的強度、化学的安定性に優れるものの、耐熱性が十分でなく、原料及び製造コストが高いという問題があった。また、構造中にフッ素原子を含むため、製造工程あるいは製造物が廃棄される際の環境へのフッ素イオン又はフッ化物の放出が、生体への影響又は環境負荷などの点で危惧されている。そうした社会的側面もうけて、フッ素系に代わる炭化水素系高分子電解質の開発が活発に行われているが、芳香族系高分子電解質、フラーレン含有電解質、共役系高分子電解質は、いずれにおいても、機械的強度が低い、加工性が悪いなどの問題があった。
【0006】
一方、ポリアセチレン誘導体については、電子伝導性、イオン伝導性などの報告があり、また、上記のようにイオン交換基を導入した置換ポリアセチレン電解質も知られているが、導入するイオン交換基がスルホン酸基である場合のスルホン化方法と製膜方法については、上記のように十分な技術であるとはいえなかった。
【0007】
本発明者らは、基材となる置換ポリアセチレンを製膜した後にスルホン化することによってスルホン酸基を導入すれば、上記の不溶化の問題を克服できると考えた。しかしながら、ポリジフェニルアセチレン膜を、濃硫酸等のスルホン化剤に浸漬してスルホン化を行うと、スルホン酸基は、膜の内部には導入されず、膜厚方向に均一にスルホン酸基を導入できないことが明らかとなった。
【特許文献1】特開2002−322293号公報
【特許文献2】特開2004−296141号公報
【非特許文献1】H. Shirakawa, T. Masuda and K. Takeda, The chemistry of triple-bonded functional groups, Chapter 17, pp 945-1016, Ed. By S. Patai, John Wiley & Sons, Chichester, 2004
【非特許文献2】K. Akagi, T. Kadokura and H. Shirakawa, Polymer, 1992, 33, 4058.
【非特許文献3】H. Onouchi, D. Kashiwagi, K. Hayashi, K. Maeda and E. Yashima, Macromolecules, 2004, 37, 5495-5503.
【非特許文献4】K. Nagai, T. Masuda, T. Nakagawa, B. D. Freeman and I. Pinnau, Prog. Polym. Sci., 2001, 26, 721-798.
【非特許文献5】T. Masuda, M. Teraguchi and R. Nomura, Am. Chem. Soc. Sym. Ser., 1999 , 733, 28-37.
【非特許文献6】T. W. Greene, P. G. M. Wuts, PROTECTIVE GROUPS IN ORGANIC SYNTHESIS 3rd ed., pp 605, JOHN WILY & SONS, New York, 1999.
【非特許文献7】T. Masuda, H. Tachimori, Pure Appl. Chem., 1994, A31, 1675-1690.
【非特許文献8】Peter G. M. Wuts, Katherun E. Wilson, Synthesis, 1998, 1593-1595.
【非特許文献9】R. W. Bott, C. Eaborn, Tadashi Hashimoto, J. Organometallic. Chem., 1965, 3, 442-447.
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、上記既知事実に鑑み、電気化学デバイスで使用するのに充分な電解質特性を示し、かつ用途に応じて充分な耐熱性、機械的強度を有し、環境負荷の大きなフッ素などのハロゲン元素を含まず、製膜性など加工性に優れた固体電解質膜、電極接合体及びそれらを用いた電気化学デバイス及び燃料電池を提供するために、均一にスルホン酸基を導入した固体電解質膜、好ましくは、そのイオン交換容量が従来より大きいスルホン酸基を有する置換ポリアセチレン電解質膜の製造方法とそれによって得られる電解質膜を提供することを課題とする。
【課題を解決するための手段】
【0009】
上記課題を解決するために、本発明では、下記式(1)で表される繰り返し単位を有する置換ポリアセチレンを膜状に成形し、該成形物をスルホン化剤に接触させてスルホン化することを特徴とするスルホン酸基を有する置換ポリアセチレン膜の製造方法としたものである。
【化1】
〔ただし、式(1)中、R1,R2は、いずれか一方又はいずれもが下記式(2)で表されるシリル基であり、残りの一方は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、t-ブチルジメチルシリルオキシ基、アセチルオキシ基、又は下記式(3)で表される。〕
【化2】
〔ただし、式(2)中、X1,X2,X3は、独立に炭素数1〜6の直鎖又は分岐のアルキル基を表す。〕
【化3】
〔ただし、式(3)中、R3は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、トリメチルシリル基、t-ブチルジメチルシリルオキシ基又はアセチルオキシ基又は式(2)を表す。〕
【0010】
このとき、モノマー合成の際に用いる出発原料の入手の容易さ、置換ポリアセチレンの重合度又は有機溶媒への溶解性を考慮すると、好ましくは、式(2)において、X1,X2,X3が独立に炭素数1〜4のアルキル基であり、さらに好ましくは、式(2)はトリメチルシリル基である。また、このときに用いる前記スルホン化剤としては、濃硫酸、濃硫酸と溶媒の混合溶液、発煙硫酸、三酸化硫黄−ジオキサン、三酸化硫黄-ピリジン、クロロスルホン酸、亜硫酸から選択されるいずれか1つ又は複数の組み合わせからなるスルホン化剤が特に好ましい。
【0011】
また、本発明では、上記のいずれかに記載の製造方法によって製造され、スルホン酸基の分布が膜厚芳香に均一であるスルホン酸基を有する置換ポリアセチレン膜としたものであり、スルホン酸基が膜内部まで導入され、スルホン酸基の分布が、膜厚方向に均一であるスルホン酸基を有する置換ポリアセチレン膜である。このとき、スルホン酸基の分布が膜厚方向に均一であることの指標として、例えば、SEM−EDSによって測定されるスルホン酸基を構成する硫黄原子由来の特性X線(SKa)強度比を用いることができ、膜中心部の強度が、測定範囲のSKaの最大値の好ましくは、70%以上、より好ましくは80%以上、もっとも好ましくは90%以上である。またさらに、上記のいずれかに記載の製造方法によって製造されるスルホン酸基を有する置換ポリアセチレン膜においては、イオン交換容量が2.0−3.5meq/gであることが特に好ましい。
【0012】
本発明によって得られる上記のスルホン基を有する置換ポリアセチレン膜の好ましい用途としては、このスルホン基を有する置換ポリアセチレン膜に電極を付与した、置換ポリアセチレン膜/電極接合体があり、また、この置換ポリアセチレン膜/電極接合体は、電気化学デバイスに好適に用いることができる。電気化学デバイスとしては、例えば、燃料電池、二次電池、湿度センサー、イオンセンサー、ガスセンサー、デシカント剤等、種々の電気デバイスに好適に用いることができ、特に燃料電池として最も好適に用いることができる。
【0013】
前記本発明は、公知のスルホン化方法である濃硫酸との接触においては、濃硫酸の粘度が比較的大きいために膜内部まで濃硫酸が浸透せず、スルホン化が進行しないものと推測し、その結果に基づき、硫酸(スルホン化剤)が内部に浸透しやすい膜であれば、容易に膜内部までスルホン化できると着想し、そのような構造としてポリマー分子鎖の間隙が大きい膜を用いることを試みた。
例えば、トリメチルシリル基を含有するポリアセチレン膜は、嵩高いトリメチルシリル基の影響でポリマー分子鎖の間隙が大きい構造を有していることが知られている(T. Masuda, H. Tachimori, Pure Appl. Chem., 1994, A31, 1675-1690.)。さらに、トリメチルシリル基を含有する芳香環を、三酸化硫黄-ジオキサン又は三酸化硫黄を用いて、トリメチルシリル基との置換反応によりスルホン化する方法がそれぞれ知られている(Peter G. M. Wuts, Katherun E. Wilson, Synthesis, 1998, 1593-1595. R. W. Bott, C. Eaborn, Tadashi Hashimoto, J. Organometallic. Chem., 1965, 3, 442-447)
【0014】
本発明者らは、これらの知見を応用することにより、本発明の課題を解決するための手段を発案し、本発明を完成するに至った。しかしながら、このような知見を高分子電解質の合成、さらには置換ポリアセチレンのスルホン化に用いた例は存在せず、さらには、膜状基材のスルホン化に関するスルホン酸基導入の均一性については、上記文献には一切例示も示唆もされていない。
すなわち、本発明は、嵩高い炭素数1〜6の直鎖又は分岐の含ケイ素置換基(シリル基)を有するアセチレンモノマーを配位重合すること等により得られる、シリル基を有する置換ポリアセチレンを用い、これを製膜した後に、濃硫酸などの強酸、濃硫酸と溶媒との混合溶液、三酸化硫黄又は三酸化硫黄―ジオキサンなどのスルホン化剤と接触させることにより、内部までスルホン化剤を浸透させ、さらには、シリル基の脱離を伴ってスルホン酸基を膜厚方向に均一に導入する技術である。
【0015】
なお、本発明において、導入されたスルホン酸基が内部まで導入され均一であるとは、SEM-EDS(Scanning Electron Microscope - Energy Dispersive X-ray Spectrometer)によって膜厚方向のスルホン酸基(硫黄原子Sに基づく)分布を調べた場合に、膜中心部の硫黄の特性X線(SKa)の強度が、測定範囲中のSKaの最大値の70%以上、より好ましくは80%以上、もっとも好ましくは90%以上であることを指す。
【0016】
さらに、本発明の方法によって得られたスルホン酸基を有する置換ポリアセチレン膜は、膜厚方向に均一にスルホン酸基を有するため、従来技術で作製された置換ポリアセチレン電解質膜とは異なり、条件によってはイオン交換容量が2.0meq/g以上であっても、十分な膜強度を持ち、且つ水やメタノール水溶液に溶解しないスルホン酸基を有する置換ポリアセチレン膜である。ただし、イオン交換容量が3.5meq/gを越えると、スルホン酸基を有する置換ポリアセチレン膜が水又はメタノール水溶液に溶解する虞がある。これら置換ポリアセチレン膜は、スルホン酸基が均一に導入され、さらには条件によっては大きなイオン交換容量を有するため、プロトン(水素イオン)、リチウムイオンなどのイオン伝導性に優れた固体高分子電解質である。加えて、先述のように大きなイオン交換容量を有する場合には、低湿度状態であっても高いプロトン伝導性を有することが期待できる。勿論、構成する化学構造中に、ハロゲン元素は共有結合で導入されていないため、ハロゲン元素に関連する環境負荷は小さいことが期待される。
【発明の効果】
【0017】
本発明により、膜厚方向にスルホン酸基を均一に導入し、条件によっては従来では合成できなかった、イオン交換容量が2.0−3.5meq/gスルホン酸基を有する置換ポリアセチレン膜を合成することができる。これらスルホン酸基を有する置換アセチレン膜は、プロトン伝導性、イオン伝導性に優れた固体電解質として用いることができる。これらは、電気化学デバイスに用いるのに十分な機械的強度を有し、耐熱性に優れ、かつ化学構造中にハロゲン元素を共有結合で導入していないことから、製造及び廃棄時の環境負荷が小さいことが期待されるため、燃料電池やイオンセンサーなどの電気デバイス又は燃料電池に用いるのに好適である。
【発明を実施するための最良の形態】
【0018】
以下に、本発明をさらに詳細に説明する。本発明に使用される置換ポリアセチレンは、その分子構造中に式(1)の構造を含んでいれば特に制限はなく、一種類のアセチレン誘導体を重合してなる単独重合体でも2種以上のアセチレン誘導体を重合してなる共重合体でもよい。アセチレン誘導体は、所望のシリル基を有するハロゲン化アリーレン化合物とフェニルアセチレンなどのアセチレン化合物から、薗頭-萩原カップリング法などの公知の方法を用いて合成することができる。置換ポリアセチレンは、当該アセチレン誘導体をNb,Ta,Mo,Wなどの遷移金属触媒、又は、これら触媒とテトラブチル錫などの助触媒を用いて、脱水溶媒中で加熱することにより得られるが、公知すべての重合方法を用いることができる。置換ポリアセチレンの分子量は、電解質膜の耐熱性及び機械的強度に大きな影響を与え、分子量が小さすぎると、耐熱性及び機械的強度の低下を招き、分子量が大きすぎると、溶解性の低下、製膜時の溶媒量の増加などを招くので、おおむね1万から1000万の範囲、より望ましくは5万から500万の範囲の分子量の置換ポリアセチレンを用いるとよい。
【0019】
【化4】
〔ただし、式(1)中、R1,R2は、いずれか一方又はいずれもが下記式(2)で表されるシリル基であり、残りの一方は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、t-ブチルジメチルシリルオキシ基、アセチルオキシ基、又は下記式(3)で表される。〕
【化5】
〔ただし、式(2)中、X1,X2,X3は、独立に炭素数1〜6の直鎖又は分岐のアルキル基を表す。〕
【化6】
〔ただし、式(3)中、R3は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、トリメチルシリル基、t-ブチルジメチルシリルオキシ基、アセチルオキシ基又は一般式(2)を表す。〕
【0020】
芳香環上の置換基R1、R2及びR3がアルキル基又はアルコキシ基の場合、具体的には、メチル基、エチル基、n-プロピル基、i-プロピル基、シクロプロピル基、n-ブチル基、t-ブチル基、1-メチルプロピル基、2-メチルプロピル基、シクロブチル基、シクロプロピルメチル基、n-ペンチル基、1-メチルブチル基、2-メチルブチル基、3-メチルブチル基、シクロペンチル基、シクロブチルメチル基、n-ヘキシル基、4-メチルペンチル基、2-エチルブチル基、1-エチル-1-メチルプロピル基、シクロヘキシル基、n-ヘプチル基、1-メチルヘキシル基、シクロヘキシルメチル基、4-メチルシクロヘキシル基、シクロヘプチル基、n-オクチル基、2-エチルヘキシル基、メトキシ基、エトキシ基、n-プロポキシ基、i-プロポキシ基、n-ブトキシ基、t-ブトキシ基、1-メチルプロポキシ基、2-メチルプロポキシ基、シクロプロピルメトキシ基、シクロブトキシ基、n-ペンチルオキシ基、1-メチルブトキシ基、2-メチルブトキシ基、3-メチルブトキシ基、1-エチルプロポキシ基、1,1-ジメチルプロポキシ基、1,2-ジメチルプロポキシ基、2,2-ジメチルプロポキシ基、シクロペンチルオキシ基、1-メチルシクロプロピルメトキシ基、2-メチルシクロプロピルメトキシ基、n-ヘキシルオキシ基、1-メチルペンチルオキシ基、2-メチルペンチルオキシ基、3-メチルペンチルオキシ基、4-メチルペンチルオキシ基、1-エチルブトキシ基、2-エチルブトキシ基、1,1-ジメチルブトキシ基、3,3-ジメチルブトキシ基、1,2-ジメチルブトキシ基、1,3-ジメチルブトキシ基、1,1,2-トリメチルプロポキシ基、1,2,2-トリメチルプロポキシ基、1-メチル-1-エチルプロポキシ基、2-メチル-1-エチルプロポキシ基、シクロヘキシルオキシ基、シクロペンチルメトキシ基、1-メチルシクロペンチルオキシ基、2-メチルシクロペンチルオキシ基、3-メチルシクロペンチルオキシ基、n-ヘプチルオキシ基、1-メチルヘキシルオキシ基、1-エチルペンチルオキシ基、5-エチルペンチルオキシ基、1,1-ジメチルペンチルオキシ基、1,4-ジメチルペンチルオキシ基、1-(1-メチルエチル)ブトキシ基、1,3,3-トリメチルブトキシ基、1-エチル-2,2-ジメチルプロポキシ基、1-エチル-1,2-ジメチルプロポキシ基、1,1-ジエチルプロポキシ基、ジイソプロピルメトキシ基、シクロヘプチルオキシ基、シクロヘキシルメトキシ基、1-シクロペンチルエトキシ基、1-メチルシクロヘキシルオキシ基、2-メチルシクロヘキシルオキシ基、3-メチルシクロヘキシルオキシ基、4-メチルシクロヘキシルオキシ基、n-オクチルオキシ基、1-メチルヘプチルオキシ基、2-エチルヘキシルオキシ基、1,5-ジメチルヘキシルオキシ基、2-プロピルペンチルオキシ基、2-メチル-1-エチルペンチルオキシ基、2,4,4-トリメチルペンチルオキシ基、シクロオクチルオキシ基、1-シクロヘキシルエトキシ基、2-シクロヘキシルエトキシ基、2-エチルシクロヘキシルオキシ基、4-エチルシクロヘキシルオキシ基、2,3-ジメチルシクロヘキシルオキシ基、2,6-ジメチルシクロヘキシルオキシ基、3,5-ジメチルシクロヘキシルオキシ基、3-シクロペンチルプロポキシ基などを例示できる。
【0021】
さらに、式(2)のX1、X2、X3は、具体的には、メチル基、エチル基、n-プロピル基、i-プロピル基、n-ブチル基、1-メチルプロピル基、2-メチルプロピル基、t-ブチル基、n-ペンチル基、1-メチルブチル基、2-メチルブチル基、3-メチルブチル基、1,1-ジメチルプロピル基、1,2-ジメチルプロピル基、2,2-ジメチルプロピル基、3,3-ジメチルプロピル基、1-エチルプロピル基、n-ヘキシル基、1-メチルペンチル基、2-メチルペンチル基、3-メチルペンチル基、4-メチルペンチル基、1,1-ジメチルブチル基、1,2-ジメチルブチル基、1,3-ジメチルブチル基、1,1,2-トリメチルプロピル基、1,2,2-トリメチルプロピル基、1,1,2,2-テトラメチルエチル基、1-エチルブチル基、2-エチルブチル基を挙げることができる。さらに出発原料の入手の容易さ、置換ポリアセチレンの重合度、有機溶媒に対する溶解性を考慮すると、式(2)のX1、X2、X3は、独立に炭素数1〜4の直鎖又は分岐のアルキル基であることが好ましく、具体的にはメチル基、エチル基、n-プロピル基、i-プロピル基、n-ブチル基、1-メチルプロピル基、2-メチルプロピル基、t-ブチル基である。さらに好ましくは式(2)はトリメチルシリル基である。これら置換基を持つ置換ポリアセチレンを公知の方法により製膜し、置換ポリアセチレン膜を得ることができる。
【0022】
公知の製膜方法としては、溶媒キャスト法、スピンコート法、転写法、印刷法等による製膜方法の他、必要に応じて加熱処理や、圧延・延伸などの機械的処理を組み合わせてもよく、膜状に成形できれば特に制限はない。
【0023】
スルホン化方法としては、濃硫酸、濃硫酸と溶媒の混合溶液、発煙硫酸、三酸化硫黄-ジオキサン、三酸化硫黄-ピリジン、クロロスルホン酸又は亜硫酸などのスルホン化剤を用いることが出来る。液相中でスルホン化する場合には、必要に応じて溶媒や界面活性剤を用いることができる。この溶媒及び界面活性剤としては、膜の性状、スルホン化の制御において悪影響を及ぼさなければ特に制限はなく、例えば、溶媒としては、水、炭素数1〜8のアルコール、酢酸エチル、酢酸ブチル、クロロホルム、ジクロロメタン、1,2-ジクロロエタン、ギ酸、酢酸、酪酸、無水酢酸、クロロ酢酸、トリフルオロ酢酸、トリフルオロ酢酸無水物、ニトロベンゼンなどを単独、あるいは2種以上混合して用いることができる。界面活性剤としては、テトラメチルアンモニウム、テトラエチルアンモニウム、テトラプロピルアンモニウム、テトラブチルアンモニウムなどの4級アンモニウムイオンと塩化物イオン、臭化物イオン、ヨウ化物イオン、硫酸水素イオンなどとの塩、又はノナニルベンゼンスルホン酸、ラウリル硫酸、安息香酸などのナトリウム塩やアンモニウム塩といったイオン性界面活性剤、プロピレングリコールやポリオキシエチレングリコールモノラウリルエーテルなどの非イオン性界面活性剤などを、単独又は2種以上混合して用いることができる。気相中で反応させる場合に、例えば亜硫酸ガスに直接膜を曝しても良いが、スルホン化雰囲気の制御又は移動相として、窒素ガス、空気、又は先にあげた溶媒の蒸気などを単独又は2種以上混合して用いることができる。スルホン化試薬の量、溶媒や界面活性剤の量、ガス量、スルホン化処理に要す時間、温度などは、膜の性状への影響、目的の電気化学デバイスで必要な電気化学特性に応じたスルホン化量をもとに決めればよく、生産効率を考慮して、処理時間が数分から数時間になるように決めればよい。
【0024】
スルホン化剤の中では、工業上安価でかつ取り扱いが比較的容易であり、さらには再利用が可能な、濃硫酸又は濃硫酸と溶媒の混合溶液が好ましい。濃硫酸と混合する溶媒としては、濃硫酸と反応しない溶媒であれば特に制限はなく、上記の溶媒を用いることが出来る。濃硫酸と溶媒の濃度は、濃硫酸100〜20重量%、好ましくは100〜50重量%、さらに好ましくは100〜80重量%である。
置換ポリアセチレン膜をこれらスルホン化剤に浸漬させる場合には、予め膜を溶媒に浸漬して膨潤させておいても良い。膨潤させる溶媒は、膜が溶解しなければ特に制限はなく、酢酸エチル、ジエチルエーテルなどが例示できる。さらには、置換ポリアセチレン膜を浸漬させる温度については、用いた溶媒の沸点以下であれば特に制限はないが、好ましくは-30〜200℃、より好ましくは0〜100℃である。
【実施例】
【0025】
以下実施例に基づいて本発明をさらに詳しく説明する。なお、実施例及び比較例において、使用した膜の作製方法をモノマー合成、ポリマー合成、製膜方法、脱シリル化方法の順に記載した。さらに、比較例として、ポリマー溶液にスルホン化剤を加えてポリマーをスルホン化した結果、及び当該シリル基を有しない置換ポリアセチレン膜のスルホン化結果を記載した。なお、1H−NMRスペクトル、FT−IRスペクトル、分子量、膜厚、イオン交換容量、含水率、膨潤度、イオン交換容量及びスルホン酸基分布は以下のようにして求めた。
【0026】
1.1H−NMRスペクトル
核磁気共鳴装置(BurkerBiospin製, 商品名AVNCE DRX 400)を用いて測定した。
2.FT−IRスペクトル
FT-IR測定装置(PerkinElmer製、商品名PARAGON FT-IR)を用いてKBrディスク法により測定した。
3.分子量
分子量は得られたポリマーをテトラヒドロフラン(THF)に溶解し、ゲルパーミエーションクロマトグラフィー(GPC)(東ソー製、商品名HLC-802A)により、数平均分子量及び重量平均分子量を測定した。溶離液にはTHFを用い、標準試料としてポリスチレンを用いた。
4.膜厚
所定の膜を110℃にて16時間真空乾燥した後、膜厚計(Mitutoyo製、商品名クイックマイクロ)を用いて膜の周囲及び中心部5点を測定し、その平均値を算出した。
【0027】
5.イオン交換容量
所定量の膜を110℃にて16時間真空乾燥し、重量測定を行った。その後、膜を0.1mol/l塩化ナトリウム水溶液50mlに浸漬し、16時間緩やかに撹拌した。その後膜を取り出し、1/50N水酸化ナトリウム水溶液にて滴定した。滴定には自動滴定装置(東亜電波工業製、商品名AUT−501)を用い、滴的曲線の変曲点を中和点(終点)として、(式1)に従いイオン交換容量を算出した。
イオン交換容量(meq/g)=〔0.02×ファクター×1/50N水酸化ナトリウム水溶液の消費量(mL)〕/〔膜の重量(g)〕 (式1)
6.含水率
所定量の膜を1.0mol/l硫酸水溶液にて1時間煮沸し、さらに純水にて1時間煮沸したのち、膜の重量を測定した。その後、膜を110℃にて16時間真空乾燥し重量測定を行い、(式2)に従って含水率を算出した。
含水率(%)=〔含水膜の重量(g)−乾燥時の重量(g)〕/〔乾燥時の重量(g)〕×100 (式2)
【0028】
7.膨潤度
所定量の膜を1.0mol/l硫酸水溶液にて1時間煮沸し、さらに純水にて1時間煮沸したのち、膜の大きさ(縦×横×厚さ)を測定した。その後、膜を110℃にて16時間真空乾燥し膜の大きさを測定し、(式3)に従って膨潤度を算出した。
膨潤度(%)=〔膨潤時の体積(mm3)〕/〔乾燥時の体積(mm3)〕× 100 (式3)
8.イオン伝導度
膜を2cm ´ 5cmの大きさに切り、1mol/lの硫酸水溶液で1時間煮沸処理した。続いて蒸留水で1時間煮沸した後、0.5cm離して平行に配置した長さ4cmの金電極上に密着して、恒温恒湿槽内で90 ℃、相対湿度90%に制御しながらインピーダンスアナライザ(東陽テクニカ製、商品名Solartron1260)を用いて周波数0.5 Hz 〜10 MHzの範囲でインピーダンス測定を行った。得られたNyquist Plotよりインピーダンスを求め、(式4)に従いイオン伝導度を算出した。
イオン伝導度(S/cm)=〔0.5 (cm)〕/〔インピーダンス(Ω)´(4 (cm) ´ 膜厚(cm)〕 (式4)
【0029】
9.膜厚方向のスルホン酸基分布測定
膜を小片状に切り出し、サンプルホルダーに固定したのち、PT−Pd蒸着を行い、サンプルの膜厚方向の炭素と硫黄の分布をSEM-EDS(Scanning Electron Microscope - Energy Dispersive X-ray Spectrometer)、(日本電子製、商品名JSM-5800LV)により分析した。図中、CKaは、炭素の特性X線の強度、SKaは、硫黄の特性X線の強度を表す。CKaは、置換ポリアセチレン膜の、SKaは、スルホン酸基の存在位置及び存在量の相対値にそれぞれ相当する。スルホン酸基が均一に導入されていることを示す指標として、SKaの最大値に対する膜中心部のSKaの強度比(a)を用いた。
【0030】
【化7】
(モノマーM-1の合成)
アルゴン雰囲気下、200024ml三口フラスコにビス(トリフェニルホスフィン)パラジウム(II)ジクロライド17 mg (0.024mmol)とヨウ化銅23 mg (0.12mmol)及びトリフェニルホスフィン32mg(0.12mmol)を量り取った。その後、予め水素化カルシウムにて脱水したトリエチルアミン 70ml (0.50mmol)を加えた。さらに、1−ブロモ−4−(トリメチルシリル)ベンゼン 1.6ml (8.0mmol)とフェニルアセチレン 0.90ml(8.0mmol)を加え、90℃にて16時間撹拌した。
【0031】
その後、トリエチルアミンを留去し、ジエチルエーテルを加えろ過した。ろ液を濃縮し、シリカゲルカラムクロマトグラフィー(溶媒:ヘキサン)にて精製した。その後、アルミナカラムクロマトグラフィー(溶媒:ヘキサン)にて精製を行ったところ、透明で粘ちょうな液体1.2 g(収率:61 %)を得た。1H-NMR, IR測定により、M−1であることを確認した。
1H-NMR, d (ppm, CDCl3, 400 MHz): 0.28 (9H, s, CH3´3), 7.33 (2H, m, Ph), 7.35 (1H, m, Ph), 7.50 (4H, s, Ph), 7.53 (2H, m, Ph).
IR, n (KBr disk, cm-1): 3065 (w), 2956 (m, C-H), 2219 (vw, CoC), 1601 (m, arC-C), 1249 (s), 1101 (w), 855 (s, Si-C), 839 (s), 820 (s), 755 (m), 690 (s), 627 (w), 633 (m).
【0032】
(ポリマーP−1の合成)
50mlナスフラスコに、グローブボックス中にて、五塩化タンタル (V)55mg(0.15mmol)及びテトラブチル錫(IV) 0.10ml(0.31mmol)を加えた。さらに、脱水トルエン4.0mlを加え、80℃にて20分間撹拌し、触媒溶液を熟成した。また、アルゴン雰囲気下50 mlナスフラスコにM−11.0g(4.0mmol)を量り取り、脱水トルエン4.0mlを加えた。その後、キャヌラによりモノマー溶液を触媒溶液に加え、80℃にて2時間撹拌した。反応液を希釈し、メタノール中に析出させたところ、黄色繊維 0.70g(収率:73%)を得た。IR測定により、P−1であることを確認した。また、GPC測定により、平均分子量を測定した。
IR, n (KBr disk, cm-1): 3055 (w, arC-H), 3017 (w), 2957 (s, C-H), 1646 (vw, >C=C<), 1597 (w, arC-C), 1494 (w), 1248 (s), 1118 (m), 855 (s, Si-C), 835 (s), 814 (s), 755 (s), 689 (s), 630 (w), 554 (s).
GPC測定結果:数平均分子量= 6.0×105、重量平均分子量=6.1×105
【0033】
(A膜の作成)
300mlナスフラスコにP−1を0.2g量り取り、トルエン50mlを加え100℃にて16時間溶解させた。その後、恒温槽内に水平に設置した10cm×10cmのガラス板に、幅1cmのテフロン(登録商標)枠を取り付け、P−1のトルエン溶液を溜延した。60℃にて3日間静置したところ、膜厚29μmの丈夫な黄色のA−1膜を得た。さらに、P−1の量を変化させることによりその他は同様の手順により、膜厚の異なるA−2膜(膜厚56μm)を合成した。
【0034】
(B膜の作製)(A膜の脱シリル化処理)
【化8】
10mlナスフラスコに、ヘキサン:トリフルオロ酢酸=1:1溶液50mlを加え、A−1膜を含浸させ、室温にて24時間撹拌した。その後膜をヘキサン溶液50mlに含浸させ16時間撹拌した。その後110℃にて16時間膜を乾燥したところ、膜厚28μmの丈夫な黄色のB−1膜を得た。IR測定により脱シリル化されたことを確認した。同様にして、A−2膜を脱シリル化することにより、B−2膜を得た。
IR, n (cm-1, KBr disk): 3085 (w), 3054 (s, arC-H), 3019 (m), 2959 (vw C-H), 2923 (vw, C-H), 1661 (w, >C=C<), 1599 (w, arC-C), 1576 (w), 1494 (s, arC-C), 1442 (s, arC-C), 1250 (w), 1156 (w), 1076 (w), 1030 (w), 902 (m), 830 (w), 769 (s), 691 (s), 553 (s).
【0035】
【化9】
(モノマーM−2の合成)
アルゴン雰囲気下、200ml三口フラスコにビス(トリフェニルホスフィン)パラジウム(II)ジクロライド 62mg (0.089mmlo), ヨウ化銅 85mg (0.44mmol)及びトリフェニルホスフィン0.12g(0.44mmol)を量り取った。その後、予め水素化カルシウムにて脱水したトリエチルアミン30mlを加え、さらにフェニルアセチレン3.3ml(30mmol)及び4−ブロモジフェニルエーテル5.2ml (30mmol)を加え、90℃にて4時間撹拌した。トリエチルアミンを留去した後、ジエチルエーテルを加えて抽出し、ろ過を行った。ろ液を水洗し、さらにエバポレートした後、シリカゲルカラムクロマトグラフィー(溶媒:ヘキサン)にて精製したところ、白色固体3.1g(収率:39 %)を得た。1H-NMR, IR測定により、M-2であることを確認した。
1H-NMR, d (ppm, CDCl3, 400 Mz): 6.97 (2H, d, J=8.8Hz, Ph), 7.20 (2H, d, J=8.8Hz,Ph), 7.15 (1H, t, J=8Hz, Ph), 7.32-7.39 (5H, m, Ph), 7.50-7.54 (4H, m, Ph).
IR, n (cm-1, KBr disk): 3050 (m, arC-H), 2360 (w, C≡C), 1591 (s, arC-C), 1490 (s, arC-C), 1286 (s), 1258 (s, arC-O-arC), 1105 (s), 1071 (s), 838 (s), 751 (s), 691 (s).
【0036】
(ポリマーP-2の合成)
アルゴン雰囲気下、100ml二口フラスコにグローブボックス中にて五塩化タンタル 0.26g(0.69mmol)及びテトラブチル錫(IV) 0.45ml(1.4mmol)を加えた。さらに、脱水トルエン15mlを加え、80℃にて20分間撹拌し、触媒溶液を熟成した。また、アルゴン雰囲気下100mlナスフラスコにM-21.0g(2.9mmol)を量り取り、脱水トルエン15mlを加えた。その後、キャヌラによりモノマー溶液を触媒溶液に加え24時間撹拌した。その後、メタノール中に析出させたところ黄褐色繊維0.62g(収率:62%)を得た。IR測定により、P−2であることを確認した。またGPC測定により、平均分子量を測定した。
IR, ν (cm-1, KBr disk): 3052 (w, arC-H), 1588 (m, arC-C), 1489 (s, arC-C), 1237 (s,ar C-O-arC), 890 (s), 750 (m).
GPC測定結果:数平均分子量=1.4×106、重量平均分子量=1.5×106
【0037】
(C膜の作成)
500mlナスフラスコにP−20.20gを量り取り、トルエン50mlを加え、90℃にて16時間撹拌した。その後、恒温槽内に水平に設置した10cm×10cmのガラス板に、幅1cmのテフロン(登録商標)枠を取り付け、P−2のトルエン溶液を溜延した。その後、50℃にて6時間静置したところ、膜厚29μmの黄色のC−1膜を得た。さらに、P−2の量を変化させることによりその他は同様の手順で、膜厚の異なるC−2膜(膜厚35μm)、C−3膜(膜厚55μm)を合成した。
【0038】
【化10】
(モノマーM−3の合成)
アルゴン雰囲気下、200ml三口フラスコにビス(トリフェニルホスフィン)パラジウム(II)ジクロライド 25mg (0.035mmlo),ヨウ化銅 6.7 mg (0.035mmol),トリフェニルホスフィン 0.047 g (0.18mmol)及び4−エチニルジフェニルエーテル2.3g(12mmol)を量り取った。その後、予め水素化カルシウムにて脱水したトリエチルアミン 5.9mlを加え、さらに1−ブロモ−4−(トリメチルシリル)ベンゼン 2.5ml(12mmol)を加え、90℃にて4時間撹拌した。トリエチルアミンを留去した後、ジエチルエーテルを加えて抽出し、ろ過を行った。ろ液を水洗し、さらにエバポレートした後、シリカゲルカラムクロマトグラフィー(溶媒:ヘキサン)にて精製したところ、白色固体3.0 g(収率:75 %)を得た。1H-NMR, IR測定により、M−3であることを確認した。
1H-NMR, d (ppm, CDCl3, 400 Mz): 0.28 (9H, s, CH3´3), 6.97 (2H, m, Ph), 7.05 (2H, m, Ph), 7.15 (1H, m, Ph), 7.37 (2H, m, Ph), 7.50 (6H, m, Ph).
IR, n (cm-1, KBr disk): 3067 (m, arC-H), 2954 (w, C-H st), 2213 (w, C≡C), 1587 (m, arC-C), 1508 (m), 1487 (s, arC-C), 1244 (s, arC-O-arC), 1163 (m), 1099 (s), 838 (s, Si-C),819 (s), 751 (s), 690 (m), 518 (m).
【0039】
(ポリマーP−3の合成)
アルゴン雰囲気下、100ml二口フラスコにグローブボックス中にて五塩化タンタル 0.45g(1.17mmol)及びテトラブチル錫(IV)0.81ml(2.3mmol)を加えた。さらに、脱水トルエン38mlを加え、80℃にて20分間撹拌し、触媒溶液を熟成した。またアルゴン雰囲気下100mlナスフラスコにM−3を2.0g(5.9mmol)量り取り、脱水トルエン20mlを加えた。その後、キャヌラによりモノマー溶液を触媒溶液に加え24時間撹拌した。その後、メタノール中に析出させたところ黄褐色繊維1.0g(収率:50 %)を得た。IR測定によりP−3であることを確認した。またGPC測定により、平均分子量を測定した。
IR, ν (cm-1, KBr disk): 3064 (w, arC-H), 1590 (s, arC-C), 1488 (s, arC-C), 1241 (s,ar C-O-arC), 836 (s, Si-C), 750 (s), 690 (s).
GPC測定結果:数平均分子量=3.8×106、重量平均分子量=5.8×106
【0040】
(D膜の作成)
500mlナスフラスコにP−3を0.20g量り取り、テトラヒドロフラン60mlを加え、70℃にて16時間撹拌した。その後、恒温槽内に水平に設置した10cm×10cmのガラス板に、幅1cmのテフロン(登録商標)枠を取り付け、P−3のテトラヒドロフラン溶液を溜延した。その後、50℃にて6時間静置したところ、膜厚50μmの黄色のD−1膜を得た。さらに、P−3の量を変えることにより、その他は同様の手順で、膜厚の異なる膜D−2(膜厚:40μm)を合成した。
比較例1 P−1溶液にスルホン化剤を加えたスルホン化
【0041】
P−1の20mgをアルゴン雰囲気下、50mlナスフラスコに量り取り、ジクロロメタン(脱水)7.0mlを加え、室温にて16時間撹拌して、P−1のジクロロメタン溶液を調整した。このポリマー溶液に、クロロスルホン酸 : ジクロロメタン=1:99(容積比)0.5mlを滴下したところ、繊維状の沈殿物が生じた。2時間撹拌した後、反応溶液をエーテル中に加え、沈殿物をろ別し、60℃、16時間真空乾燥した。その後、IR測定を行ったところ、トリメチルシリル基の脱離とスルホン酸基の導入が確認された。一方、沈殿物はN,N-ジメチルスルホキシド、N,N-ジメチルアセトアミド、m-クレゾール、メタノール、アセトン、酢酸エチル、水に不溶であった。
IR, ν (cm-1, KBr disk): 3443 (s), 1637 (m, arC-C), 1216 (m), 1178 (m), 1128 (m, SO3H), 1036 (m, SO3H), 1009 (s), 759 (w), 689 (m), 578 (w)
【0042】
比較例2 P−2溶液にスルホン化剤を加えたP−2のスルホン化
P−2の20mgをアルゴン雰囲気下、50mlナスフラスコに量り取り、ジクロロメタン(脱水)3.5mlを加え、室温にて16時間撹拌して、P−2のジクロロメタン溶液を調整した。このポリマー溶液にクロロスルホン酸:ジクロロメタン=1:99(容積比)0.25mlを滴下したところ繊維状の沈殿物が生じた。2時間撹拌した後、反応溶液をエーテル中に加え、沈殿物をろ別した後、60℃、16時間真空乾燥した。その後IR測定を行ったところ、トリメチルシリル基の脱離とスルホン酸の導入が確認された。一方、沈殿物はN,N−ジメチルスルホキシド、N,N−ジメチルアセトアミド、m−クレゾール、メタノール、アセトン、酢酸エチル、水に不溶であった。
IR, ν (cm-1, KBr disk): 3444 (s), 1637 (m, arC-C), 1490 (m, arC-C), 1241 (m, arC-O-arC), 1169 (m), 1125 (w, SO3H), 1033 (m, SO3H), 1007 (w), 694 (m), 607 (w), 552 (w).
【0043】
比較例3 B−1膜のスルホン化(SB−1膜の合成)
50mlナスフラスコに、濃硫酸(97%)10mlを量り取り、B−1膜の1.8mgを浸漬して室温にて3時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、SB−1膜を得た。IR測定を行ったが、スルホン酸の導入は確認されなかった。
IR, n (cm-1, KBr disk): 3055 (s, arC-H), 1599 (w, arC-C), 1491 (s, arC-C), 1440 (m), 1246 (m), 1162 (w), 903 (m), 832 (w), 754 (m), 687 (s), 548 (s).
得た膜は、膜厚28μm、イオン交換容量は検出限界以下であった。
【0044】
比較例4 B-2膜のスルホン化(SB−2膜の合成)
100mlナスフラスコに、濃硫酸(97%)50mlを量り取り、B−2膜41mgを浸漬して室温にて16時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色のSB−2膜を得た。IR測定により、スルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1632 (m, arC-C), 1490 (s, arC-C), 1440 (s), 1254 (s), 1168 (m), 1128 (w, SO3H), 1032 (w, SO3H), 1003 (w), 906 (w), 829 (w), 755 (s), 690 (s), 567 (m).
得た膜は、膜厚26μm、イオン交換容量1.4meq/g、含水率21%、膨潤度156%、イオン伝導度は5.6 x 10-3S/cmであった(90℃,RH90%)。
【0045】
実施例1 A−1膜のスルホン化(SA−1膜の合成)
100mlナスフラスコに、濃硫酸(97%)50mlを量り取り、A−1膜69mgを浸漬して室温にて3時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色のSA−1膜を得た。IR測定により、脱シリル化反応及びスルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1642 (m, arC-C), 1492 (s, arC-C), 1442 (w), 1218 (s), 1154 (s), 1128 (w, SO3H), 1033 (w, SO3H), 1005 (w), 907 (w), 825 (w), 755 (s), 691 (s), 572 (m).
得た膜は、膜厚29μm、イオン交換容量2.3meq/g、含水率80 %、膨潤度282%、イオン伝導度は3.7x10-1S/cmであった(90℃,RH90%)。
【0046】
実施例2 A−2膜のスルホン化 (SA−2膜の合成)
100mlナスフラスコに、濃硫酸(97%)と酢酸エチルの混合溶液(濃硫酸:酢酸エチル=801:20)50mlを量り取り、A−2膜52mgを浸漬して室温にて16時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色のSA−2膜を得た。IR測定により、脱シリル化反応及びスルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1634 (m, arC-C), 1490 (s, arC-C), 1441 (w), 1215 (s), 1159 (s), 1128 (s, SO3H), 1032 (w, SO3H), 1003 (w), 910 (w), 827 (w), 756 (s), 693 (s), 572 (m).
得た膜は、膜厚56μm、イオン交換容量2.1meq/g、含水率78 %、膨潤度375%、イオン伝導度は2.4x10-1S/cmであった(90℃, RH90%)。
【0047】
比較例5 C−1膜のスルホン化 (SC−1膜の合成) (M48−11)
100mlナスフラスコに、濃硫酸(97%)30mlを量り取り、C−1膜56mgを浸漬して室温にて16分間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色のSC−1膜を得た。IR測定を行ったがスルホン酸の導入は確認されなかった。
IR, n (cm-1, KBr disk): 3053 (m, arC-H), 1588 (m, arC-C), 1487 (s, arC-C), 1238 (s, arC-O-arC), 1164 (s), 869(w), 750 (s), 689 (s).
得た膜は、膜厚28μm、イオン交換容量は検出限界以下であった。
【0048】
比較例6 C−2膜のスルホン化 (SC−2膜の合成)(M45−42)
100mlナスフラスコに、濃硫酸(97%)50mlを量り取り、C−2膜69mgを浸漬して室温にて1時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色のSC−2膜を得た。IR測定により、スルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1588 (m, arC-C), 1489 (s, arC-C), 1240 (s, arC-O-arC), 1166 (s), 1123(s, SO3H), 1030 (s, SO3H), 1003 (s), 831(w), 748 (s), 690 (s).
得た膜は、膜厚34μm、イオン交換容量1.1meq/g、含水率15%、膨潤度106%、イオン伝導度は1.0x10-1S/cmであった(90℃,RH90%)。
【0049】
比較例7 C−3膜のスルホン化 (SC−3膜の合成)(M48−5)
100mlナスフラスコに、濃硫酸(97%)と酢酸エチルの混合溶液(濃硫酸:酢酸エチル=80:20)50mlを量り取り、C−3膜65mgを浸漬して室温にて3時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色の膜SC−3を得た。IR測定により、スルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1588 (m, arC-C), 1489 (s, arC-C), 1239 (s, arC-O-alC), 1164 (s), 1123(s, SO3H), 1029 (s, SO3H), 1004 (s), 832(w), 751 (s), 691 (s).
得た膜は、膜厚56μm、イオン交換容量1.0meq/g、含水率22%、膨潤度127%、イオン伝導度は3.8x10-2S/cmであった(90℃,RH90%)
【0050】
実施例3 D−1膜のスルホン化 (SD−1膜の合成)
100mlナスフラスコに、濃硫酸(97%)50mlを量り取り、D−1膜49mgを浸漬して室温にて16分間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色の膜SD−1を得た。IR測定により、脱シリル化反応及びスルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1588 (m, arC-C), 1489 (s, arC-C), 1238 (s, arC-O-arC), 1164 (s), 1122(s, SO3H), 1028 (s, SO3H), 1002 (s), 830(w), 753 (s), 691 (s).
得た膜は、膜厚50μm、イオン交換容量1.9meq/g、含水率65%、膨潤度151%、イオン伝導度は8.0x 10-2S/cmであった(90℃,RH90%)
【0051】
実施例4 D−2膜のスルホン化(SD−2)膜の合成
100mlナスフラスコに、濃硫酸(97%)と酢酸エチルの混合溶媒(濃硫酸:酢酸エチル=80:20)50mlを量り取り、D−2膜30mgを浸漬して室温にて3時間緩やかに撹拌した。膜を取り出したのち水洗し、さらに純水にて1時間煮沸した。その後、110℃にて16時間真空乾燥し、緑色の膜SD−2を得た。IR測定により、脱シリル化反応及びスルホン化反応が進行していることを確認した。
IR, n (cm-1, KBr disk): 3056 (w, arC-H), 1587 (m, arC-C), 1492 (s, arC-C), 1239 (s, arC-O-arC), 1163 (s), 1122(s, SO3H), 1028 (s, SO3H), 1001 (s), 830(w), 752 (s), 692 (s).
得た膜は、膜厚40μm、イオン交換容量1.5meq/g、含水率63%、膨潤度262%、イオン伝導度は4.7x10-1S/cmであった(90℃,RH90%)
図1〜図7に比較例及び実施例の膜厚方向のSEM−EDS分析結果を示す。さらに、表1に各サンプルのaを示した。比較例に対して実施例の膜中心部の強度は大きく、均一にスルホン酸基が導入されていることが示された。
【0052】
【表1】
【図面の簡単な説明】
【0053】
【図1】SB−2膜のSEM−EDS測定結果に示すグラフ。
【図2】SA−1膜のSEM−EDS測定結果に示すグラフ。
【図3】SA−2膜のSEM−EDS測定結果に示すグラフ。
【図4】SC−2膜のSEM−EDS測定結果に示すグラフ。
【図5】SC−3膜のSEM−EDS測定結果に示すグラフ。
【図6】SD−1膜のSEM−EDS測定結果に示すグラフ。
【図7】SD−2膜のSEM−EDS測定結果に示すグラフ。
【特許請求の範囲】
【請求項1】
下記式(1)で表される繰り返し単位を有する置換ポリアセチレンを膜状に成形し、該成形物をスルホン化剤に接触させてスルホン化することを特徴とするスルホン酸基を有する置換ポリアセチレン膜の製造方法。
【化1】
〔ただし、式(1)中、R1,R2は、いずれか一方又はいずれもが下記式(2)で表されるシリル基であり、残りの一方は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、t-ブチルジメチルシリルオキシ基、アセチルオキシ基、又は下記式(3)で表される。〕
【化2】
〔ただし、式(2)中、X1,X2,X3は、独立に炭素数1〜6の直鎖又は分岐のアルキル基を表す。〕
【化3】
〔ただし、式(3)中、R3は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、トリメチルシリル基、t-ブチルジメチルシリルオキシ基、アセチルオキシ基又は式(2)を表す。〕
【請求項2】
前記式(1)中のR1、R2は、いずれか一方又はいずれもがトリメチルシリル基であることを特徴とする請求項1に記載のスルホン酸基を有する置換ポリアセチレン膜の製造方法。
【請求項3】
前記スルホン化剤は、濃硫酸、濃硫酸と溶媒の混合液、発煙硫酸、三酸化硫黄-ジオキサン、三酸化硫黄-ピリジン、クロロスルホン酸、亜硫酸から選択されるいずれか1つ又は複数の組み合わせからなることを特徴とする請求項1又は2に記載のスルホン酸基を有する置換ポリアセチレン膜の製造方法。
【請求項4】
請求項1〜3のいずれか1項に記載の製造方法によって製造されたスルホン酸基を有する置換ポリアセチレン膜であって、スルホン酸基の分布が膜厚方向に均一であることを特徴とするスルホン酸基を有する置換ポリアセチレン膜。
【請求項5】
請求項4に記載のスルホン基を有する置換ポリアセチレン膜に電極を付与したことを特徴とする置換ポリアセチレン膜/電極接合体。
【請求項6】
請求項5に記載の置換ポリアセチレン膜/電極接合体を用いることを特徴とする電気化学デバイス。
【請求項7】
請求項5に記載の置換ポリアセチレン膜/電極接合体を用いることを特徴とする燃料電池。
【請求項1】
下記式(1)で表される繰り返し単位を有する置換ポリアセチレンを膜状に成形し、該成形物をスルホン化剤に接触させてスルホン化することを特徴とするスルホン酸基を有する置換ポリアセチレン膜の製造方法。
【化1】
〔ただし、式(1)中、R1,R2は、いずれか一方又はいずれもが下記式(2)で表されるシリル基であり、残りの一方は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、t-ブチルジメチルシリルオキシ基、アセチルオキシ基、又は下記式(3)で表される。〕
【化2】
〔ただし、式(2)中、X1,X2,X3は、独立に炭素数1〜6の直鎖又は分岐のアルキル基を表す。〕
【化3】
〔ただし、式(3)中、R3は、水素、水酸基、炭素数1〜8のアルキル基又はアルコキシ基、トリメチルシリル基、t-ブチルジメチルシリルオキシ基、アセチルオキシ基又は式(2)を表す。〕
【請求項2】
前記式(1)中のR1、R2は、いずれか一方又はいずれもがトリメチルシリル基であることを特徴とする請求項1に記載のスルホン酸基を有する置換ポリアセチレン膜の製造方法。
【請求項3】
前記スルホン化剤は、濃硫酸、濃硫酸と溶媒の混合液、発煙硫酸、三酸化硫黄-ジオキサン、三酸化硫黄-ピリジン、クロロスルホン酸、亜硫酸から選択されるいずれか1つ又は複数の組み合わせからなることを特徴とする請求項1又は2に記載のスルホン酸基を有する置換ポリアセチレン膜の製造方法。
【請求項4】
請求項1〜3のいずれか1項に記載の製造方法によって製造されたスルホン酸基を有する置換ポリアセチレン膜であって、スルホン酸基の分布が膜厚方向に均一であることを特徴とするスルホン酸基を有する置換ポリアセチレン膜。
【請求項5】
請求項4に記載のスルホン基を有する置換ポリアセチレン膜に電極を付与したことを特徴とする置換ポリアセチレン膜/電極接合体。
【請求項6】
請求項5に記載の置換ポリアセチレン膜/電極接合体を用いることを特徴とする電気化学デバイス。
【請求項7】
請求項5に記載の置換ポリアセチレン膜/電極接合体を用いることを特徴とする燃料電池。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2007−270028(P2007−270028A)
【公開日】平成19年10月18日(2007.10.18)
【国際特許分類】
【出願番号】特願2006−99390(P2006−99390)
【出願日】平成18年3月31日(2006.3.31)
【出願人】(000000239)株式会社荏原製作所 (1,477)
【Fターム(参考)】
【公開日】平成19年10月18日(2007.10.18)
【国際特許分類】
【出願日】平成18年3月31日(2006.3.31)
【出願人】(000000239)株式会社荏原製作所 (1,477)
【Fターム(参考)】
[ Back to top ]