
セリアック病の腸内の免疫炎症性成分の選択的処置のための化合物
【課題】本発明の主な目的の1つは、セリアック病の局所的免疫炎症性成分に対する選択的作用を与える化合物を提供することである。
【解決手段】一態様において、本発明は、セリアック病の炎症化合物の処置に使用するためのアミノサリチル酸−アミノフェニルプロピオン酸化合物に関する。それらの化合物は、セリアック病において放出されるサイトカインを遮断することによって作用し、特に、食事療法に対して難治性の症状の処置、食事療法の誤り、およびセリアック病の寛解時間の減少に有用である。
【解決手段】一態様において、本発明は、セリアック病の炎症化合物の処置に使用するためのアミノサリチル酸−アミノフェニルプロピオン酸化合物に関する。それらの化合物は、セリアック病において放出されるサイトカインを遮断することによって作用し、特に、食事療法に対して難治性の症状の処置、食事療法の誤り、およびセリアック病の寛解時間の減少に有用である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、セリアック病の腸内の免疫炎症性成分の選択的処置のための化合物に関する。
【0002】
本発明は、小腸の最初の管の粘膜の位置に局在化する炎症性成分を有する疾患、例えばセリアック病の処置のための薬物の分野に関する。
【背景技術】
【0003】
特に、本発明は、セリアック病を罹患している個体における十二指腸および近位空腸の位置で発生する炎症を選択的に減少させるのに適切な一群の分子に関する。
【0004】
セリアックスプルー(celiac sprue)としても公知である、セリアック病は、遺伝的、免疫学的および環境的要素を有する非常によく見られる自己免疫疾患である。
【0005】
セリアック病の根底には、コムギに含まれるグルテンのタンパク質分画である、グリアジン、またはオオムギ、ライムギ、スペルト、カムートおよび他の少数の穀物に含まれる、アルコール(プロラミン)に可溶性の類似のタンパク質分画に対する永続的な過敏症がある。
【0006】
セリアック病において、小腸の粘膜は、抗原(グリアジン)に対する曝露後に損傷を受ける。この疾患において、腸絨毛は平らになる傾向があり、埋め合わせるために陰窩が過剰に増え、腸細胞が管形状よりむしろ立方体になり、腸管腔内のリンパ球の数が増加する。
【0007】
この疾患に付随する症状は、非常に複雑であり、胃腸領域に限定されない。実際に、一般に、下痢、潜在的な出血を伴う腹部の痛み、乳糖不耐症を含む、局所的な位置での典型的な症状は、アフタ性口内炎、骨痛、弱体化を伴う進行性の体重減少を含み得る腸以外での症状と付随して起こる。さらに、未処置または難治性のセリアック病の場合において、胃腸のリンパ腫または同様の癌を発生する危険性が存在する。
【0008】
さらに、しばしば、セリアック病の被験体は、腸の吸収不良によって引き起こされるビタミンA、B12、D、E、Kおよび葉酸の起こり得る欠損とともに、鉄またはフェリチンの欠損を示す場合がある。さらに、排便による脂肪の連続的な損失は、カルシウム欠損を引き起こし、1つは腎臓位置でシュウ酸カルシウム結石の形成、もう1つは骨組織の位置で骨弱化を引き起こす症状である、骨軟化症の発生を伴う起こり得る2つの合併症を発生する場合がある。
【0009】
一定の割合の個体において、この疾患は、完全または部分的に無症状である場合があり、明らかな症状を示さないか、潜在的な形態であり、特定の事象の後に発生しやすい。
【0010】
この疾患を誘発する抗原は公知であるので、グルテンを含む食物の摂取を単に回避することによって、関連症状の完全な寛解を得ることは可能である。
【0011】
疾患の管理は厳重食の順守に基づくため、誰でも知っている、パスタ、パン、オオムギ、スプレトなどのグルテンを含む食物の摂取を回避するだけでは十分ではなく、例えば、増粘剤または構造化剤あるいは加工中に損失する微量物質などの食物の中に少量含まれ得るものの摂取を回避することも必須である。
【0012】
例えば、セリアック病の患者は、バーでエスプレッソコーヒー(これはオオムギで汚染されている可能性がある)、スパイス、粉砂糖、一部の医薬品を摂取することを回避しなければならず、例えば、切手および郵便用封筒に見られる糊にも注意を払わなければならない。
【0013】
いずれの場合においても、セリアック病の患者の食事は、十分に変化に富ませて、コメ、トウモロコシ、ソバ、キビ、アマランサス、肉、魚、野菜、果物、チーズおよびマメなどの食物のみを十分にバランスよく食べる必要がある。
【0014】
いくつかの最近の研究によれば、食物あたり20ppmのグルテンの臨界閾値が存在し、食物含量がそれを超えると、セリアック病の被験体にとって有害になる。コーデックス委員会は、グルテンを含まない食物をラベル付けするために2種類の閾値を想定している。第1のものは100ppmに設定され、出発物質の中にも有害な穀物誘導体、例えばコムギデンプンなどを含む「有害性を中和された(detoxified)」食物に関し、有害な穀物由来の成分を含まない食物に関しては20ppmの閾値に設定される。
【0015】
食事および一部の基本的な行動ルールの厳守は、新しい症状の発症を予防し、一般に、個人の反応による程度の差はあるが示された様式において、発生する症状の寛解を生じる。しかしながら、症状の欠如または血清中の抗体の欠如にもかかわらず、生活全てについて食事を管理されなければならない。
【0016】
しかしながら、食事以外に、セリアック病の被験体に現在利用可能な処置の形態はなく、特に、偶発的でもグルテンを含む食物の摂取に伴う炎症症状を抑え得る製剤が存在しない。
【0017】
今までに試みられた少数の治療的介入の方法は、有意な結果の達成をもたらさなかった。
【0018】
この疾患は、ヒト白血球(HLA)DQ2およびDQ8の抗原をコードする一部の遺伝子と厳密に関連するため、一部の処置形態は、HLA DQ2/DQ8においてグルテンのペプチドの結合を阻害することを目的としている。特に、一部のHLA−DQ2−遮断化合物が試験されているが、有意な結果をもたらしていない。
【0019】
小腸の細胞間結合部の調節に関与するタンパク質であり、その発現が疾患の急性期の間に増加する、ゾヌリン拮抗薬の使用が、セリアック症候群の処置に関する文献に記載されている。しかしながら、このタンパク質に基づいた薬物は現在市場で利用可能ではない。
【0020】
さらに、局所的な粘膜に使用するための抗炎症性薬物は、今まで、腸の末梢管である大腸の位置に局在化する慢性的腸炎の形態の処置以外の適用に見出されていない。この理由のために、クローン病および潰瘍性大腸炎を処置するための市場で利用可能な製剤は、直腸用途のための坐薬または液剤として販売されている。一方、これらの製剤が経口投与を意図される場合、それらは、消化管をインタクトに通過し、結腸のみの位置で活性成分を放出させるように、活性成分の遅延放出のために処方される。
【0021】
従って、これらの薬物は、セリアック病において集中する炎症が局在化する腸の十二指腸部分に対する効果を有さない。
【発明の概要】
【発明が解決しようとする課題】
【0022】
従って、臨床的必要性として、現在、セリアック病において小腸の十二指腸粘膜の位置で発生する症状に基づいた免疫炎症を抑制する薬理学的活性を与えられる物質が求められている。
【0023】
本発明の一般的目的は、薬理学的活性を与える化合物の使用に対する新規の適切な効能を提供することである。
【0024】
従って、本発明の主な目的の1つは、セリアック病の局所的免疫炎症性成分に対する選択的作用を与える化合物を提供することである。
【課題を解決するための手段】
【0025】
これらの目的を考慮して、本発明の一般的態様によれば、セリアック病の炎症性成分の選択的処置に使用するための以下の共通の化学構造
【化1】

(式中、R1およびR2は以下に定義され、Xは
【化2】
から選択され、
Y、ZおよびR3は以下に定義される)
を有する化合物が提供される。
【0026】
一態様において、出願人は、式(A)の化合物および本明細書の以下に記載される式(I)および(II)を有する化合物が、特異的親和性を有すること、例えば、PPARガンマ(PPARγ)受容体についての作動薬であり、その活性化を与えることを見出した。特に、このような受容体の存在は、セリアック病を罹患している患者の十二指腸上皮細胞の位置で検出され、式(A)、IおよびIIの化合物の抗炎症性効果は、炎症性サイトカインの産生の減少により示された。
【0027】
典型的に、式(I)および(II)の化合物は、それぞれ、アミノフェニルプロピオン酸およびアミノサリチル酸様構造を有し、セリアック病で放出されるサイトカインを遮断することにより作用する。
【0028】
一実施形態において、式(I)および(II)の化合物は、食事療法に対する難治性のセリアック病の処置、食事療法の誤り、およびセリアック病の寛解時間の減少において特に有用である。
【0029】
本発明の一態様によれば、セリアック病の免疫炎症性成分を選択的に処置するための医薬を製造するための式(I)の化合物
【化3】
(式中、
R1およびR2は、互いに同じであるか、または異なり、−Hまたは1〜6個の炭素を有する直鎖もしくは分枝アルキル基からなる群より選択されるか、あるいはそれらは一緒に、5または6個の原子を有する芳香環または脂肪族環を形成し、
YおよびZは、互いに同じであるか、または異なり、−H、−OH、−COOH、−OR3、−CH(OR3)COOHからなる群より選択され、
R3は、H、フェニル、ベンジル、−CF3、−CF2CF3、ビニル、アリル、1〜6個の炭素、好ましくは3〜6個の炭素を有する直鎖もしくは分枝アルキル基から選択される)
またはその混合物および薬理学的に許容可能な塩もしくはエステルの使用が提供される。
【0030】
一実施形態において、式(I)の化合物において、
R1およびR2は、互いに同じであるか、または異なり、−Hまたは1〜6個の炭素、好ましくは1〜3個の炭素を有する直鎖もしくは分枝アルキル基からなる群より選択され、
YおよびZは、互いに同じであるか、または異なり、−H、−OH、−COOH、−OR3、−CH(OR3)COOHからなる群より選択され、
R3は、H、1〜6個の炭素、好ましくは3〜6個の炭素を有する直鎖もしくは分枝アルキル基から選択される。
【図面の簡単な説明】
【0031】
【図1】生検試料においてサイトカインをコードするmRNAの存在および量を確認するために、1×TBE中で1%アガロースゲル電気泳動を行った結果のグラフを示す。
【図2】蛍光顕微鏡によって現されるPPARγ受容体の存在を示す。
【図3】蛍光顕微鏡によって現されるPPARγ受容体の存在を示す。
【図4】蛍光顕微鏡によって蛍光が検出されないコントロールを示す。
【図5】蛍光顕微鏡によって蛍光が検出されないコントロールを示す。
【図6】1×アガロースゲルを用いる電気泳動によって得られた異なるバンドから推定される吸光度の値を示す。
【図7】1×アガロースゲルを用いる電気泳動によって得られた異なるバンドから推定される吸光度の値を示す。
【図8】1×アガロースゲルを用いる電気泳動によって得られた異なるバンドから推定される吸光度の値を示す。
【発明を実施するための形態】
【0032】
本発明の範囲内において、前記式(I)の化合物の実施形態は、添付の特許請求の範囲2〜6に与えられる。例として、実施例7および
(±)−2−ヒドロキシ−3−(3’−アミノフェニル)−プロピオン酸、
(±)−2−メトキシ−2−(4’−アミノフェニル)酢酸、
(±)−2−エトキシ−2−(3’−アミノフェニル)酢酸、
(±)−2−エトキシ−2−(4’−アミノフェニル)酢酸、
(±)−2−メトキシ−3−(4’−アミノフェニル)プロピオン酸、
(±)−2−エトキシ−3−(4’−アミノフェニル)プロピオン酸、
(±)−2−エトキシ−3−(3’−アミノフェニル)プロピオン酸
にも例示される化合物H〜Qが、特にプロピオン酸誘導体に関して、本発明による使用に適切である。
【0033】
一態様において、本発明の使用に適切な式(I)の化合物のさらなる例は、国際公開WO2007/010516号に記載され、その内容は参照として完全に本明細書に援用される。
【0034】
本発明の別の態様によれば、セリアック病の免疫炎症性成分を選択的に処置するための医薬を製造するための式(II)の化合物
【化4】
(式中、
R1およびR2は、互いに同じであるか、または異なり、H、−CO−CH3、nが1〜6、好ましくは1〜3の整数である−CnH2n−1、1〜6個、好ましくは1〜3個の炭素を有する直鎖もしくは分枝アルキル基からなる群より選択されるか、あるいはそれらは一緒に、5または6個の原子を有する芳香環または脂肪族環を形成し、
R3は、H、−CO−CH3、−NHOH、−OH、−OR6から選択され、R6は、1〜6個の炭素を有する直鎖もしくは分枝アルキル基であり、
R4は、H、1〜6個の炭素を有する直鎖もしくは分枝アルキル基から選択され、
R5、R7、R8は、H原子であり、
R3およびR4、R4およびR5、またはR7およびR8は、一緒に、ベンゼン環に縮合された環、N、Oからなる群より独立して選択される1〜2個のヘテロ原子を含む5または6個の原子を有する芳香環または脂肪族環のいずれかを形成する)
またはその混合物および薬理学的に許容可能な塩もしくはエステルの使用が提供される。
【0035】
前記式(II)の化合物の実施形態は、添付の特許請求の範囲8〜24に与えられる。
【0036】
例として、実施例5に例示される化合物A〜Gが本発明の使用に適切である。
【0037】
本発明の使用に適切な式(II)の化合物のさらなる例は、国際公開WO2007/010514号に記載され、その内容は参照として完全に本明細書に援用される。
【0038】
出願人は、式IおよびIIの化合物が、セリアック病の被験体の腸粘膜の位置、特に2番目の十二指腸部分の位置で観測される免疫炎症性成分に対する特異的活性を有することを示している。
【0039】
出願人は、式(I)および(II)の化合物によって与えられる局所的な抗炎症活性が、炎症プロセスにおいて重要な役割、および/またはその炎症プロセスが慢性的になるのに重要な役割を果たす物質である、サイトカインの放出に対する阻害活性に関与していることをさらに示している。
【0040】
特に、驚くべきことに、式IおよびIIの化合物が、セリアック病の被験体においてのみ、サイトカインを十分な方法で遮断するか、またはいずれかの場合において、実質的および有意に阻害するが、それらの化合物は、セリアック病以外の特定の胃腸管に影響を与える炎症性疾患を罹患している被験体において有意な活性を有さないことを見出した。
【0041】
特に、式IおよびIIの化合物は、いわゆるプロフロゴジェン(prophlogogen)Th1(ヘルパーT細胞タイプ1)サイトカインを遮断するのに適切である。なぜなら、それらの化合物は、損傷部位における免疫細胞および炎症細胞の動員に有利に働くからである。
【0042】
この特異的活性は、セリアック病において、排除できず、免疫系が耐性を生じることができない抗原(グルテンのグルアジン分画)に対する免疫系の活性化を、Th1タイプの炎症(phlogosis)の持続が反映するという事実に帰する可能性がある。
【0043】
セリアック病の免疫病原性に関与する典型的なプロフロゴジェンサイトカイン(Th1)は、IL−1、IL−2、IL−6(インターロイキン1、2、6)、IFN(インターフェロン)を含む。さらに、Th2(ヘルパーT細胞タイプ2)に関連するサイトカイン、およびTNF−α(腫瘍壊死因子)などのマクロファージ由来のサイトカインもまた、セリアック病に関与する。特に、TNF−αは、プロフロゴジェンサイトカイン(その一部はかなりの毒性を与えられる)の産生を上昇させる能力に起因する慢性的な炎症性疾患の発生に重要な役割を果たす。
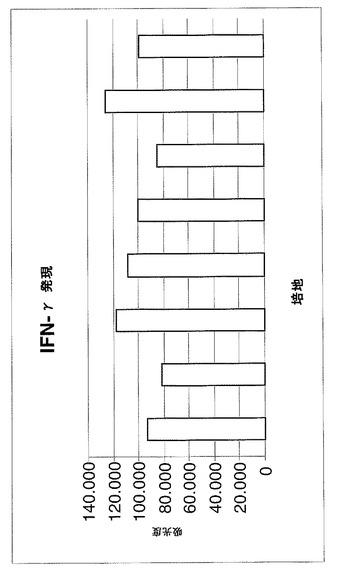
【0044】
特に、セリアック病を罹患している腸粘膜試料由来のサイトカインの産生および放出に対する式(I)および(II)の化合物の効果は、有機培養系を用いて実証されている。特に、培養に対して腸免疫対象物の生検において式(I)(II)の化合物の1つ以上を用いることによるIFN−γ、IL−2、TNF−α値の有意な減少が強調されているため、フロゴシス(phlogosis)の効果的な減少が示されている。
【0045】
本発明の別の態様によれば、難治性セリアック病の被験体における炎症を処置するための医薬を製造するための上記のような式IまたはIIの化合物およびそれらの混合物の使用が提供される。
【0046】
本発明の別の態様によれば、食事療法の誤りによって誘発される、食事療法中のセリアック病の被験体における炎症性反応を処置するための医薬を製造するための上記のような式IまたはIIの化合物およびそれらの混合物の使用が提供される。
【0047】
本発明の別の態様によれば、典型的に、1つ以上の薬理学的に許容可能な賦形剤またはアジュバントと組み合わせて、セリアック病における免疫炎症性成分を処置するため、臨床時間および/または組織学的寛解時間を短縮するための医薬を製造するための式IまたはIIの化合物およびそれらの混合物の使用が提供される。
【0048】
本発明の1つ以上の態様に従って医薬を調製する際に、1つ以上の薬理学的に許容可能な賦形剤に加えて、薬理学的使用のための調製物の範囲内で典型的に使用される滑剤、保湿剤、懸濁化剤、分散剤、防腐剤を使用することも可能である。
【0049】
一実施形態において、式(I)および/または(II)の化合物は、錠剤、カプセル剤、粒状処方物、分散剤および薬理学的分野に典型的な他の処方物などの様々な形態で投与されてもよい。
【0050】
典型的に、活性成分(I)および(II)は、セリアック病における抗炎症性反応を達成するのに有効な量で投与に適切な処方物に組み込まれてもよい。
【0051】
例として、活性成分は、50mg〜2000mgの範囲、好ましくは200〜600mg、より好ましくは250〜500mgの範囲の量で医薬組成物に組み込まれてもよい。
【0052】
式(I)および/または(II)の1つ以上の化合物に基づいた医薬の使用は、患者および医師のコンプライアンスを改善し、寛解時間を減少させることを決定づけることが示されている。
【0053】
別の実施形態において、式(I)および/または(II)の化合物は、抵抗性および/または難治性のセリアック病の処置におけるさらなる治療用途を見出された。
【0054】
以下の実施例は単に本発明の例示のみとして与えられ、添付の特許請求の範囲から明らかなように、保護の範囲を限定することを意図しない。
【実施例】
【0055】
(実施例1)
セリアック病で観測される炎症性成分に対する、本発明の好ましい化合物の1つである、メサラジンの効果を評価した。
【0056】
このために、疾患の活動期中の16人の被験体およびグルテンを含まない食事療法中の臨床的寛解期中の6人の患者を選択した。
【0057】
各研究被験体につき5つの生検断片を、EGDS試験により得た。
【0058】
腸粘膜の各断片を、メサラジン(5ASA)を含むものと、含まないものの両方で、適切な培地中の培養物に入れた。
【0059】
次いで、以下のものを液体培地中で観測した:
異なる培地中で放出されたサイトカインのレベル、
適切に均質化された生検粘膜に存在する同じサイトカインの量。
【0060】
(物質および方法)
(方法)
3群の被験体を評価した。
【0061】
第1(萎縮性)の群は、抗筋肉膜および抗トランスグルタミナーゼ抗体に対して血清試験で陽性であり、セリアック病の新診断をして、グルテンを含む食事をしている29人の被験体を含んだ。
【0062】
第2(寛解)の群は、臨床的寛解において、少なくとも6ヶ月間、グルテンを含まない食事療法の17人のセリアック病の被験体を含んだ。
【0063】
第3(健康的)の群は、セリアック病を罹患していないが、胃腸管の炎症を罹患している被験体を含んだ。
【0064】
(液体培地中のサイトカインアッセイ)
(臓器培養)
この研究患者は、EGDS検査によって十二指腸粘膜の5つの生検を提供した。1つは組織学的アッセイについてのものであり、他の4つ(それぞれ2つの部分に分けた)は、それぞれ、1つの培地のみ(培地C)およびグリアジンペプチドを含む培地ならびにメサラジンの存在下でグリアジンを含む培地(培地+5ASA C)を用いて培養に供した。
【0065】
生検を、少なくとも3回、生理食塩水で2分間洗浄した。その生検を、1mlに相当する体積を有する培地に入れた:
培地(RPM11860+ウシ胎仔血清+ペニシリン/ストレプトマイシン)、
培地+グリアジンまたは31〜43または他の穀物のペプシン−トリプシン消化物、
培地+メサラジン(5−ASA)、
培地+グリアジンのペプシン−トリプシン消化物+5−ASA(培地1mlあたり1.5〜8.0mg)。
【0066】
生検を、4時間〜72時間の間、37℃の温度でO2(95%)およびCO2(5%)雰囲気下でインキュベートする。
【0067】
3つの異なる量の培地を、培地に放出されたTh1およびTh2サイトカインのELISAアッセイについて共有する。
【0068】
(ホモジネートからのサイトカインアッセイ)
37℃の温度で4時間〜72時間のインキュベーション時間の後、生検断片を2分間、生理食塩水で3回洗浄し、生検を機械的または化学的分解により均質化する。遠心分離を実施し、上清を回収し、1.5mlのエッペンドルフに移す。粘膜組織中の固定されたサイトカインをElisaアッセイにより分析する。
【0069】
(結果)
見出されるデータは、培地中のメサラジンの存在が、セリアック病の被験体のみにおいてサイトカインを遮断し、従って、セリアック病の被験体から得た試料において選択的に炎症性成分の減少を決定づけることを示す(表1)。
【0070】
【表1】
【0071】
【表2】
【0072】
【表3】
【0073】
【表4】
【0074】
達成された炎症性成分の減少により、メサラジンの単独の使用または全ての緩慢もしくは困難な臨床的解決の症例における治療支持としての使用が可能になる。
【0075】
(実施例2)
(サイトカインの遺伝子発現)
胃内視鏡検査処置の間に患者から得た生検の十二指腸断片を、5−ASA薬物の非存在下および存在下で、48時間、37℃で培養物中に入れた。培養後、断片を、フェノールおよびグアニジン(Isol−RNA溶解試薬)からなる単相溶液中で均質化した。
【0076】
ホモジナイザーIKA T10−Ultra Turraxを、均質化のために使用した。
【0077】
全ての不溶性物質を遠心分離により溶液から分離し、上清を無菌の試験管に移し、それに、溶解物1mlあたり200μlのクロロホルムを加えた。
【0078】
短時間の機械的攪拌後、試料を室温で数分間静置させ、その後、4℃で15分間、12000rpmで遠心分離した。RNAを含む無機段階を無菌の試験管に移して行った。
【0079】
RNAを、溶解物1mlあたり500μlのイソプロピルアルコールを加えることにより沈殿させた。試料を、室温で10分間インキュベートし、次いで、4℃で15分間、12000rpmで遠心分離した。
【0080】
上清を除去した後に、RNA沈殿物を、溶解物1mlあたり1000μlの75%エタノールで洗浄し、機械的に攪拌し、4℃で5分間、7500rpmで遠心分離した。最終的に、エタノールを除去し、RNA沈殿物を化学フード下で空気乾燥させた。
【0081】
RNAを、適切な量のRNasiを含まない水に再懸濁した。
【0082】
RNAの濃度を、分光光度計で260nmでの吸収を測定することにより決定し、異なるRNA鋳型についてのA260/A280の比を評価した。
【0083】
(RT−PCR)
cDNAを、適切な体積のRNasiを含まない水に希釈した異なる量の抽出したRNAから開始して合成した。関連遺伝子(サイトカイン)のcDNAを、特異的プライマーを使用してPCRにより増幅させた。
【0084】
マスタースクリプト RT−PCR系(5プライマー)をRT−PCRに使用した。
【0085】
生検試料においてサイトカインをコードするmRNAの存在および量を確認するために、1×TBE中で1%アガロースゲル電気泳動を行った。
【0086】
得られた結果を図1のグラフに示す。
【0087】
(実施例3)
(セリアック病患者の十二指腸上皮細胞におけるPPARγ受容体の存在)
ペルオキシソーム増殖因子活性化受容体γは、核内受容体のスーパーファミリーに属し、それは、エストロゲン、糖質コルチコイド、甲状腺ホルモン、ビタミンD3およびレチノイン酸についての受容体ならびに脂質代謝の異なる生成物を結合できる受容体(例えば、PPARおよびLXR受容体)を含む。
【0088】
特に、PPARファミリーは、異なる組織分布および異なるリガンド特異性を有する3つのサブタイプ(PPARα、βまたはδおよびγ)を含む。
【0089】
PPARγおよびαの両方は、分化したヒトマクロファージにおいて発現され、それらは、炎症性反応に関係する遺伝子を調節し、マクロファージの分化を調節する。
【0090】
異なるPPARγ作動薬は、ヒトの単球において炎症性サイトカインの産生を阻害し、活性化マクロファージにおけるTNF−α、IL−6、IL−1b、iNOS、ゼラチンB、スカベンジャー受容体AおよびCOX−2についての遺伝子発現を減少させ、PPARγの抗炎症性の役割を確認する。
【0091】
主な目的の1つは、セリアック病患者の十二指腸上皮細胞においてこのような受容体の存在を検出することであり、式(A)、I、IIの化合物の抗炎症性効果および炎症性サイトカインの産生の減少によって現れる5−ASAの特異性は、以前に確認されている。
【0092】
(免疫蛍光)
十二指腸生検断片を、グルテンを含む食事をしているセリアック病の患者およびセリアック病を罹患していない被験体(コントロール群)の両方からEGDS処置により取得した。生検断片を洗浄し、OCTに配向し、−80℃で保存した。5μmのいくつかの断片を、各々の凍結した生検切片(セリアック病の患者およびその疾患を罹患していない被験体の両方からEGDS工程で得た)から得て、その断片をPPARγ一次抗体に一晩(適切な固定および特異性除去の処置後)、曝露した。
【0093】
PBSで洗浄した後、試料を、約1時間、二次フルオレセイン抗体ALEXA 488とともにインキュベートした。一次抗体がPPARγ受容体に結合する場合において、存在する場合、一次/二次抗体複合体が形成され、顕微鏡によって観測可能な断片上の蛍光として現される。
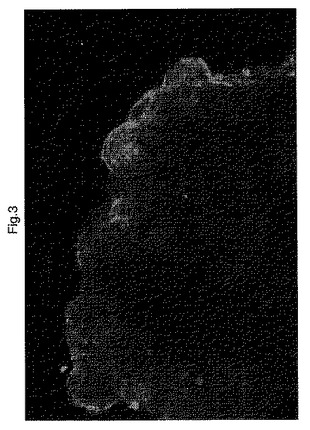
【0094】
蛍光顕微鏡によって現されるPPARγ受容体の存在は、図2および図3(写真診断)から証明される。
【0095】
特に、図2および図3の画像において、受容体の存在は、末梢部分の断片において検出可能な蛍光シグナルによって現される。蛍光により、腸管腔の方を向いている表面上の腸細胞、腸上皮細胞の位置でPPARγの存在が実証される。
【0096】
セリアック病を罹患していない患者の生検は、ネガティブコントロールとして使用され、シグナルが存在せず、その結果として、顕微鏡によって分析された断片で受容体が存在しない。特に、図4および図5(コントロール)は蛍光が検出されないことを示し、これは、コントロールの健康な群の同じ腸内部分の位置において受容体が存在しないことのしるしである。
【0097】
同じように、本明細書の以下に記載される化合物を試験し、同様の結果を得た。
【0098】
(実施例4)
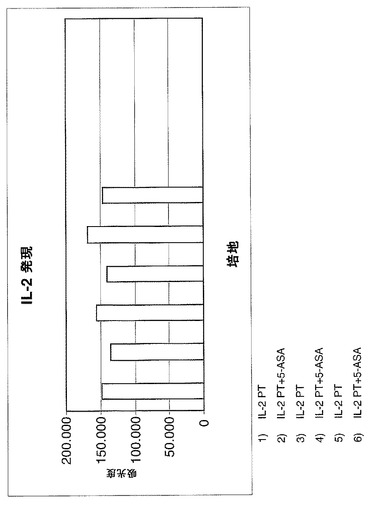
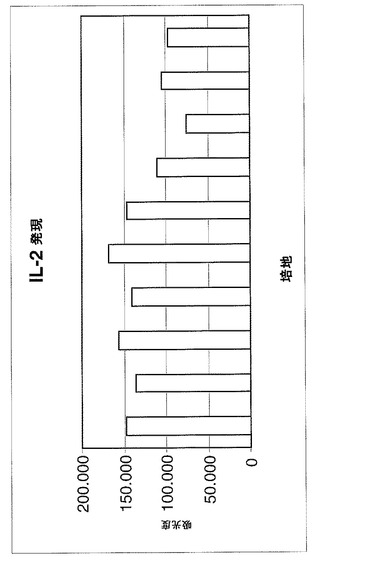
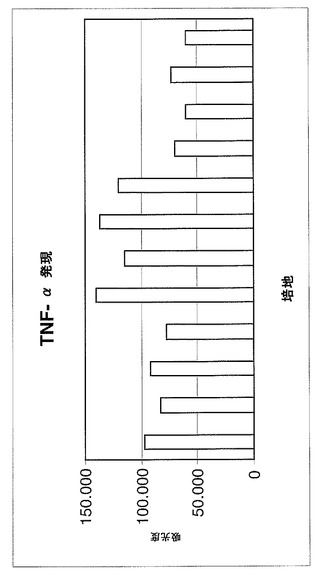
疾患の急性期で放出される炎症性サイトカインIL−2、TFN−αおよびIFN−γの遺伝子発現(mRNA)をモニターした。
【0099】
特に、図6〜8に示される図は、1×アガロースゲルを用いる電気泳動によって得られた異なるバンドから推定される吸光度の値(IMAGE J)を示し、この研究のサイトカイン増幅対象に関連する。
【0100】
サイトカインの遺伝子発現は、セリアック病を罹患している患者の十二指腸の生検の抽出されたRNAのRT−PCRによって評価される。このような生検は、5−ASA薬物が加えられる、グリアジンのトリプシン消化物(PT)を含む培地およびPT培地の両方において、48時間、37℃で増殖を維持されている。
【0101】
(結果)
3つの図により、生検断片が薬物の存在下において培地中に維持され、疾患急性期に放出される炎症性サイトカインIL−2、TFN−αおよびIFN−γの遺伝子発現(mRNA)が十分に減少されることが示される。
【0102】
患者が、薬物でのより持続的かつ一定の処置を受けている場合に、この減少が増加する。
【0103】
(実施例5)
(PPARγ活性化/発現ならびに細胞増殖およびアポトーシスの調節に対する本発明による化合物の効果の研究)
(物質および方法)
化合物A−G
5−ASAを、Sigma−AldrichTM(St Quentin Fallavier,フランス)で購入した。ロシグリタゾンを、Spi BioTM(Massy,フランス)で得た。式(II)の範囲内である以下の化合物A〜Gを試験した。
【化5】
【0104】
(細胞株)
結腸細胞株HT−29 STD(ATCC HTB−38)を、10%のheat−FCSおよび抗生物質を補足したDMEM中で慣例的に増殖させた。細胞を単分子層で増殖させ、5%CO2および95%相対湿度において37℃でインキュベートした。
【0105】
(PPARγでの一過性トランスフェクションおよび細胞の刺激)
HT−29 STD細胞を、製造業者の指示書に従ってEffectene TMトランスフェクション試薬(Qiagen TM)を用いて一過性トランスフェクションした。PPARγ活性化を試験するために、本発明者らは、シトクロムp450 4A(2XCYP)から得た2つのコピーのPPREを含む500ngの最小プロモーター構築物でトランスフェクションを実施した。ウミシイタケルシフェラーゼプラスミド(0.1μg/ウェル)も、トランスフェクション効果をモニターするため、およびホタルルシフェラーゼ活性を正規化するための内部コントロールとしてトランスフェクションした。トランスフェクションした細胞を48時間静置し、37℃でインキュベートした。刺激を、30mMの濃度で化合物A−Gを用いて、3−6−9−12−15−18−24時間の間、細胞のインキュベーション後に実施し、ポジティブコントロールとして使用した2つのPPARγ合成リガンド5−ASA 30mMおよびロシグリタゾン 10〜5Mと比較した。薬物溶液のpHをNaOHで7.4に調整した。全細胞抽出物を、受動的溶解緩衝液(Passive Lysis Buffer)(PromegaTM,Madison,Wis.)を用いて調製した。ルシフェラーゼ活性を、製造業者のプロトコルによるPromegaTMのデュアルルシフェラーゼアッセイシステムを用いて20μlの抽出物中で測定した。トランスフェクションを、少なくとも3つの別々の実験において3連で分析した。ルシフェラーゼ活性を、刺激していない細胞からのルシフェラーゼ活性で割られた異なる分子で処理した細胞で得られた活性の倍数として表した。
【0106】
(ウェスタンブロット解析によるPPARγおよびβ−アクチンの評価)
全タンパク質を、2%のTritonTM、フッ化フェニルメチルスルホニル(PMSF)100mMおよび古典的プロテアーゼ阻害剤カクテルを含むPBSからなる抽出緩衝液中での細胞均質化によって得た。次いで、全タンパク質を、ポリアクリルアミドゲル電気泳動および電気ブロットによって分離した。二フッ化ポリビニリデン(PVDF)膜を、PPARγに対するウサギポリクローナル一次抗体(1/500希釈,TEBU,Le Perray en Yveline,フランス)とともに一晩インキュベートした。β−アクチンを、1/10,000で希釈したウサギモノクローナル一次抗体(Sigma)を用いて検出した。二次ペルオキシダーゼ標識抗体(1/1000,DakoTM,Trappes,フランス)および化学発光を用いる免疫検出を、製造業者のプロトコル(ECLTM、Amersham Pharmacia BiotechTM,Orsay,フランス)に従って実施した。PPARγの吸光度の値を、同じ試料内の内部コントロールβ−アクチンの量に比例して各条件について得た。
【0107】
(Ki−67免疫染色による細胞増殖の分析)
24時間の培養後、HT−29 STD細胞を、30mMで化合物A、B、C、DおよびFを用いて24時間、処理した。5−ASA(30mM)およびロシグリタゾン(10〜5M)を、ポジティブコントロールとして使用した。分子Gは、その不十分な溶解性のために、この実験に含まなかった。薬物溶液のpHをNaOHで7.4に調整した。細胞を、PFA 4%中で固定し、4℃で0.1%のTriton X−100TMを含むPBS中で透過処理し、次いで、抗体の非特異的吸着を最小化するためにヤギ正常血清および遮断バッファー(PBS中の1% BSA)とともにインキュベートした。
【0108】
細胞増殖を、Ki−67に対するマウスモノクローナル一次抗体を用いて核のKi−67染色によって評価した(1:50希釈、一晩;ZYMEDTM,ClinisciencesTM,Montrouge,フランス)。一次抗体を、アクリジン赤蛍光色素(1:100希釈,Molecular ProbesTM,InvitrogenTM,Cergy Pontoise,フランス)と結合したAlexa 594ロバ抗マウスIgGで明らかにした。核を、Hoescht 33342溶液(0.125mg/mL)(Sigma−AldrichTM)で染色し、蛍光顕微鏡(LeicaTM,Bensheim,ドイツ)下で視覚化した。ネガティブコントロールは、特異的抗体の代わりに非特異的マウス血清での染色からなる。少なくとも500細胞/試料のカウントを、1つの実験において体系的に盲目的に実施した。その結果を、染色細胞の数の平均±SEMとして表した。
【0109】
(アポトーシスの検出)
24時間の培養後、HT−29 STD細胞を、24時間の間、30mMの濃度で化合物A、B、C、D、Fを用いて処理した。5−ASA(30mM)およびロシグリタゾン(10〜5M)を、ポジティブコントロールとして使用した。分子EおよびGは、それらの不十分な溶解度のためにこの実験に含まなかった。薬物溶液のpHをNaOHで7.4に調整した。アポトーシスを受けた細胞を、ターミナルトランスフェラーゼdUTPニック末端標識アッセイ(TUNELアッセイ,Roche DiagnosticsTM,Meylan,フランス)を用いてDNA鎖の酵素標識により同定した。少なくとも500細胞/試料のカウントを、1つの実験において体系的に盲目的に実施した。その結果を、染色細胞の数の平均±SEMとして表した。
【0110】
(結果)
分子CおよびFは、PPARγ活性化を誘導することが観測された。化合物Dもまた、PPARγを誘導するが、わずかに少ない程度である。PPAARγの活性化は、ペルオキシソーム増殖因子活性化受容体エレメント(PPRE)と呼ばれる特異的DNA配列エレメントに対する結合を導く反応のカスケードを生じる。
【0111】
本発明者らは、ウミシイタケルシフェラーゼPPREプラスミドでの上皮細胞の一過性トランスフェクションによるPPARγ転写活性を研究した。細胞を、24時間、異なる分子で刺激した。トランスフェクションされたHT−29細胞におけるPPARγ活性の分析により、30mMの濃度で化合物CおよびFが、レポーター遺伝子活性を2倍増加させ、それにより、5−ASAおよびロシグリタゾンと同様の活性を表したことが示された。30mMの濃度で化合物A、BおよびGは、6時間後にPPARγ活性化の研究を制限する上皮細胞に対する急速な細胞毒性効果を与えた。
【0112】
特に、分子C、DおよびFは、PPARγ発現を誘導する。通常、全ての化合物A〜Gは、HT−29細胞株におけるタンパク質レベルでPPARγ発現を誘導する能力を示す。特に、ウェスタンブロットにより定量化したPPARγタンパク質レベルの平均2倍の誘導を、24時間、分子C、DおよびFで処理した細胞において観測した。
【0113】
特に、分子CおよびFは、上皮細胞増殖を阻害する。本発明者は、細胞増殖の調節における分子の役割をHT−29 STD細胞株において評価した。細胞増殖を、増殖細胞中で発現される核タンパク質Ki−67染色により評価し、Ki−67の存在が細胞増殖を維持するのに必須であった。未処理の細胞と比較して、分子CおよびF(30mM)との24時間のHT−29細胞のインキュベーションは、細胞増殖の67〜75%の阻害を生じた。
【0114】
同様の結果を、最適濃度で使用した2つのポジティブコントロールのロシグリタゾン(10〜5M)および5−ASA(30mM)で得た。分子A、BおよびDの潜在的抗分離促進効果の確認は、この濃度での上皮細胞に対するそれらの急速な細胞毒性効果によって制限された。
【0115】
化合物Fもまた、PPARγを介して上皮細胞のアポトーシスを誘発する。ロシグリタゾンおよび5−ASAと同様に、分子Fは、ターミナルトランスフェラーゼdUTPニック末端標識(TUNEL)を用いてDNA鎖の分裂を標識することにより同定された80%の上皮細胞のアポトーシスを示した。以前の実験と同様に、分子A、BおよびDは、30mMの阻害細胞アポトーシス分析において急速な細胞毒性効果を誘発した。
【0116】
(結論)
この実施例は特に、PPARγ発現および活性化を誘発し、上皮細胞増殖およびアポトーシスを調節する化合物CおよびFの能力を示す。さらに、30mMでの化合物A、B、D(およびE〜G)の上皮細胞に対する細胞毒性効果は、多くの種々の酵素と高い親和性を示すことが知られている高い反応性のヒドロキサム酸基の構造の存在と関連し得る。
【0117】
(実施例6)
(分子モデル)
分子モデル研究を、Silicon GraphicsTMワークステーションで作動するSYBYLソフトウェアバージョン6.9.1(Tripos Associates IncTM,ST Louis,MO)を用いて実施した。5−ASAの両性イオン形態の三次元モデルを、標準的なフラグメントライブラリーから構築し、次いで、その形状を、Tripos力場を用いて最適化した。化合物のpKaはまだ知られていないため、SPARCオンライン計算機を、生理学的pH(7.4)で生じる種を決定するために使用した。イオン化化合物の三次元モデルを、標準的なフラグメントライブラリーから構築し、次いで、それらの形状を、GasteigerおよびHuckel原子電荷から計算される静電条件を含むTripos力場を用いて最適化した。Maximin2処理に利用可能なPowell法を、勾配値が0.001kcal/mol.Åより小さくなるまで、エネルギー最小化に使用した。ヒトPPARγリガンド結合ドメインの構造を、RCSBタンパク質データバンク(1171)(4,5)で利用可能なテサグリタザル(AZ242)を用いてその錯体化したX線結晶構造から得た。化合物の受容体活性部位へのフレキシブルなドッキングを、GOLDソフトウェアを用いて実施した。最も安定なドッキングモデルを、GoldScoreおよびX−Score評価関数によって予測された最適に評価された構造に従って選択した。錯体を、勾配値が0.01kcal/mol.Åに到達するまで、Tripos力場および4.0の誘電率でMaximin2処理に利用可能なPowell法を用いてエネルギー最小化をした。アニール機能を使用して、リガンドのホット領域(hot region)(10Å)を規定する。
【0118】
(ドッキング研究)
化合物A〜Gは、分子認識およびPPARγ活性化に必要な主要な決定因子とみなされるHis−323、His−449、Tyr−473およびSer−289との水素結合を介して相互作用するPPARγ−LBDと強く適合した。
【0119】
式(II)に含まれる化合物の抗炎症効果は、上皮細胞によって主に発現されるPPARγにより媒介されることが示された。このドッキング分析により、30mMの濃度で使用される上述の化合物がPPARγを活性化し、腸の上皮細胞によりその発現を誘導し、セリアック病の免疫炎症性成分に対する薬理作用を与えることが明らかにされた。
【0120】
(結論)
アミノサリチル酸構造(5−ASA構造など)との式IIの化合物の抗炎症効果は、PPARγにより媒介され、十二指腸の位置で上皮細胞において発現されることが以前に示されている。ドッキング分析に基づいた化合物A〜Gの合理的開発により、30mMの濃度で使用される前記化合物、特にC、Fが、PPARγを活性化し、腸の上皮細胞によりその発現を誘導することが明らかにされた。その化合物はまた、PPARγ活性化による2つの重要な機構である、上皮細胞増殖を阻害し、アポトーシスを誘導する。特に、分子A、B、D、Gに関して、それらが、PPARγ活性化の分析ならびに細胞増殖およびアポトーシスの調節および評価を妨げる、30mMの濃度での上皮細胞に対する細胞毒性効果に関することが検出されている。
【0121】
(実施例7)
(PPARγ活性化に対する式(I)の化合物H〜Qの効果)
(物質および方法)
(物質)
5−ASAを、Sigma−AldrichTM(St Quentin Fallavier、フランス)で購入した。ロシグリタゾンを、Spi BioTM(Massy、フランス)で得た。式(I)内に含まれる以下の化合物H〜Qを試験した。
【化6】
【化7】
【0122】
(細胞株)
結腸癌細胞株HT−29 STD(ATCC HTB−38)を、10%のheat−FCSおよび抗生物質を補足したDMEM中で慣例的に増殖させた。細胞を単分子層で増殖させ、5%CO2および95%相対湿度において37℃でインキュベートした。
【0123】
(PPARγでの一過性トランスフェクションおよび細胞の刺激)
HT−29 STD細胞を、製造業者の指示書に従ってEffecteneTMトランスフェクション試薬(QiagenTM)を用いて一過性トランスフェクトした。PPARγ活性化を試験するために、本発明者らは、シトクロムp450 4A(2XCYP)から得た2つのコピーのPPREを含む500ngの最小プロモーター構築物でトランスフェクションを実施した。ウミシイタケルシフェラーゼプラスミド(0.1μg/ウェル)も、トランスフェクション効果をモニタリングするため、およびホタルルシフェラーゼ活性を正規化するための内部コントロールとしてトランスフェクションした。トランスフェクションした細胞を、37℃で24時間のインキュベーションのために静置させておいた。刺激を、1mMの濃度で化合物H〜Qを用いて18時間の細胞のインキュベーション後に実施して、ポジティブコントロールとして使用した2つのPPARγ合成リガンド5−ASA(30mM)およびロシグリタゾン(10〜5M)と比較した。薬物溶液のpHをNaOHで7.4に調整した。全細胞抽出物を、受動的溶解緩衝液(Passive Lysis Buffer)(PromegaTM,Madison,Wis.)を用いて調製した。ルシフェラーゼ活性を、製造業者のプロトコルによるPromegaのデュアルルシフェラーゼアッセイシステムを用いて20μlの抽出物中で測定した。トランスフェクションを、少なくとも3つの別々の実験において3連で分析した。ルシフェラーゼ活性を、刺激していない細胞からのルシフェラーゼ活性で割られた異なる分子で処理した細胞で得られた活性の倍数として表した。
【0124】
(結果)
PPARγの活性化は、ペルオキシソーム増殖因子活性化受容体エレメント(PPRE)と呼ばれる特異的DNA配列エレメントに対する結合を導く反応のカスケードを生じる。
【0125】
本発明者らは、ウミシイタケルシフェラーゼおよびPPREプラスミドでの上皮細胞の一過性トランスフェクションによるPPARγ転写活性を調べた。化合物H〜QがPPARγ活性化を刺激する有効性および/または5−ASAより有効かどうかを評価するために、本発明者らは1mMの濃度でこれらの分子を試験した。1mMの濃度での新規の分子の効果を、それぞれ30mMおよび10〜5Mの最適濃度でポジティブコントロールとして使用した5−ASAおよびロシグリタゾンと比較した。細胞を、24時間、異なる分子で刺激した。
【0126】
トランスフェクションされたHT−29細胞中のPPARγ活性の分析により、1mMで前記化合物40が、それぞれ、4.8±0.71倍、2.73±0.31倍、2.64±0.46倍、3.4±0.97倍、受容体遺伝子活性を増加させ、それにより、30mMでの5−ASA(2.8±0.7)および10〜5Mでのロシグリタゾン(3.17±0.29)と同等またはそれらより優れた活性を表したことが示された。
【0127】
トランスフェクションされたHT−29細胞中のPPARγ活性を増加させる本発明の式(I)の化合物に含まれる分子H〜Qの能力は、30mMでの5−ASAおよび10〜5Mでのロシグリタゾンと同等またはそれらより優れた活性を表す。
【0128】
2007年12月24日に出願された、詳細な説明、特許請求の範囲および図面を含む伊国特許出願第MI2007A2429号は、その優先権が本明細書で主張され、完全に参照によって援用される。
【技術分野】
【0001】
本発明は、セリアック病の腸内の免疫炎症性成分の選択的処置のための化合物に関する。
【0002】
本発明は、小腸の最初の管の粘膜の位置に局在化する炎症性成分を有する疾患、例えばセリアック病の処置のための薬物の分野に関する。
【背景技術】
【0003】
特に、本発明は、セリアック病を罹患している個体における十二指腸および近位空腸の位置で発生する炎症を選択的に減少させるのに適切な一群の分子に関する。
【0004】
セリアックスプルー(celiac sprue)としても公知である、セリアック病は、遺伝的、免疫学的および環境的要素を有する非常によく見られる自己免疫疾患である。
【0005】
セリアック病の根底には、コムギに含まれるグルテンのタンパク質分画である、グリアジン、またはオオムギ、ライムギ、スペルト、カムートおよび他の少数の穀物に含まれる、アルコール(プロラミン)に可溶性の類似のタンパク質分画に対する永続的な過敏症がある。
【0006】
セリアック病において、小腸の粘膜は、抗原(グリアジン)に対する曝露後に損傷を受ける。この疾患において、腸絨毛は平らになる傾向があり、埋め合わせるために陰窩が過剰に増え、腸細胞が管形状よりむしろ立方体になり、腸管腔内のリンパ球の数が増加する。
【0007】
この疾患に付随する症状は、非常に複雑であり、胃腸領域に限定されない。実際に、一般に、下痢、潜在的な出血を伴う腹部の痛み、乳糖不耐症を含む、局所的な位置での典型的な症状は、アフタ性口内炎、骨痛、弱体化を伴う進行性の体重減少を含み得る腸以外での症状と付随して起こる。さらに、未処置または難治性のセリアック病の場合において、胃腸のリンパ腫または同様の癌を発生する危険性が存在する。
【0008】
さらに、しばしば、セリアック病の被験体は、腸の吸収不良によって引き起こされるビタミンA、B12、D、E、Kおよび葉酸の起こり得る欠損とともに、鉄またはフェリチンの欠損を示す場合がある。さらに、排便による脂肪の連続的な損失は、カルシウム欠損を引き起こし、1つは腎臓位置でシュウ酸カルシウム結石の形成、もう1つは骨組織の位置で骨弱化を引き起こす症状である、骨軟化症の発生を伴う起こり得る2つの合併症を発生する場合がある。
【0009】
一定の割合の個体において、この疾患は、完全または部分的に無症状である場合があり、明らかな症状を示さないか、潜在的な形態であり、特定の事象の後に発生しやすい。
【0010】
この疾患を誘発する抗原は公知であるので、グルテンを含む食物の摂取を単に回避することによって、関連症状の完全な寛解を得ることは可能である。
【0011】
疾患の管理は厳重食の順守に基づくため、誰でも知っている、パスタ、パン、オオムギ、スプレトなどのグルテンを含む食物の摂取を回避するだけでは十分ではなく、例えば、増粘剤または構造化剤あるいは加工中に損失する微量物質などの食物の中に少量含まれ得るものの摂取を回避することも必須である。
【0012】
例えば、セリアック病の患者は、バーでエスプレッソコーヒー(これはオオムギで汚染されている可能性がある)、スパイス、粉砂糖、一部の医薬品を摂取することを回避しなければならず、例えば、切手および郵便用封筒に見られる糊にも注意を払わなければならない。
【0013】
いずれの場合においても、セリアック病の患者の食事は、十分に変化に富ませて、コメ、トウモロコシ、ソバ、キビ、アマランサス、肉、魚、野菜、果物、チーズおよびマメなどの食物のみを十分にバランスよく食べる必要がある。
【0014】
いくつかの最近の研究によれば、食物あたり20ppmのグルテンの臨界閾値が存在し、食物含量がそれを超えると、セリアック病の被験体にとって有害になる。コーデックス委員会は、グルテンを含まない食物をラベル付けするために2種類の閾値を想定している。第1のものは100ppmに設定され、出発物質の中にも有害な穀物誘導体、例えばコムギデンプンなどを含む「有害性を中和された(detoxified)」食物に関し、有害な穀物由来の成分を含まない食物に関しては20ppmの閾値に設定される。
【0015】
食事および一部の基本的な行動ルールの厳守は、新しい症状の発症を予防し、一般に、個人の反応による程度の差はあるが示された様式において、発生する症状の寛解を生じる。しかしながら、症状の欠如または血清中の抗体の欠如にもかかわらず、生活全てについて食事を管理されなければならない。
【0016】
しかしながら、食事以外に、セリアック病の被験体に現在利用可能な処置の形態はなく、特に、偶発的でもグルテンを含む食物の摂取に伴う炎症症状を抑え得る製剤が存在しない。
【0017】
今までに試みられた少数の治療的介入の方法は、有意な結果の達成をもたらさなかった。
【0018】
この疾患は、ヒト白血球(HLA)DQ2およびDQ8の抗原をコードする一部の遺伝子と厳密に関連するため、一部の処置形態は、HLA DQ2/DQ8においてグルテンのペプチドの結合を阻害することを目的としている。特に、一部のHLA−DQ2−遮断化合物が試験されているが、有意な結果をもたらしていない。
【0019】
小腸の細胞間結合部の調節に関与するタンパク質であり、その発現が疾患の急性期の間に増加する、ゾヌリン拮抗薬の使用が、セリアック症候群の処置に関する文献に記載されている。しかしながら、このタンパク質に基づいた薬物は現在市場で利用可能ではない。
【0020】
さらに、局所的な粘膜に使用するための抗炎症性薬物は、今まで、腸の末梢管である大腸の位置に局在化する慢性的腸炎の形態の処置以外の適用に見出されていない。この理由のために、クローン病および潰瘍性大腸炎を処置するための市場で利用可能な製剤は、直腸用途のための坐薬または液剤として販売されている。一方、これらの製剤が経口投与を意図される場合、それらは、消化管をインタクトに通過し、結腸のみの位置で活性成分を放出させるように、活性成分の遅延放出のために処方される。
【0021】
従って、これらの薬物は、セリアック病において集中する炎症が局在化する腸の十二指腸部分に対する効果を有さない。
【発明の概要】
【発明が解決しようとする課題】
【0022】
従って、臨床的必要性として、現在、セリアック病において小腸の十二指腸粘膜の位置で発生する症状に基づいた免疫炎症を抑制する薬理学的活性を与えられる物質が求められている。
【0023】
本発明の一般的目的は、薬理学的活性を与える化合物の使用に対する新規の適切な効能を提供することである。
【0024】
従って、本発明の主な目的の1つは、セリアック病の局所的免疫炎症性成分に対する選択的作用を与える化合物を提供することである。
【課題を解決するための手段】
【0025】
これらの目的を考慮して、本発明の一般的態様によれば、セリアック病の炎症性成分の選択的処置に使用するための以下の共通の化学構造
【化1】
(式中、R1およびR2は以下に定義され、Xは
【化2】
から選択され、
Y、ZおよびR3は以下に定義される)
を有する化合物が提供される。
【0026】
一態様において、出願人は、式(A)の化合物および本明細書の以下に記載される式(I)および(II)を有する化合物が、特異的親和性を有すること、例えば、PPARガンマ(PPARγ)受容体についての作動薬であり、その活性化を与えることを見出した。特に、このような受容体の存在は、セリアック病を罹患している患者の十二指腸上皮細胞の位置で検出され、式(A)、IおよびIIの化合物の抗炎症性効果は、炎症性サイトカインの産生の減少により示された。
【0027】
典型的に、式(I)および(II)の化合物は、それぞれ、アミノフェニルプロピオン酸およびアミノサリチル酸様構造を有し、セリアック病で放出されるサイトカインを遮断することにより作用する。
【0028】
一実施形態において、式(I)および(II)の化合物は、食事療法に対する難治性のセリアック病の処置、食事療法の誤り、およびセリアック病の寛解時間の減少において特に有用である。
【0029】
本発明の一態様によれば、セリアック病の免疫炎症性成分を選択的に処置するための医薬を製造するための式(I)の化合物
【化3】
(式中、
R1およびR2は、互いに同じであるか、または異なり、−Hまたは1〜6個の炭素を有する直鎖もしくは分枝アルキル基からなる群より選択されるか、あるいはそれらは一緒に、5または6個の原子を有する芳香環または脂肪族環を形成し、
YおよびZは、互いに同じであるか、または異なり、−H、−OH、−COOH、−OR3、−CH(OR3)COOHからなる群より選択され、
R3は、H、フェニル、ベンジル、−CF3、−CF2CF3、ビニル、アリル、1〜6個の炭素、好ましくは3〜6個の炭素を有する直鎖もしくは分枝アルキル基から選択される)
またはその混合物および薬理学的に許容可能な塩もしくはエステルの使用が提供される。
【0030】
一実施形態において、式(I)の化合物において、
R1およびR2は、互いに同じであるか、または異なり、−Hまたは1〜6個の炭素、好ましくは1〜3個の炭素を有する直鎖もしくは分枝アルキル基からなる群より選択され、
YおよびZは、互いに同じであるか、または異なり、−H、−OH、−COOH、−OR3、−CH(OR3)COOHからなる群より選択され、
R3は、H、1〜6個の炭素、好ましくは3〜6個の炭素を有する直鎖もしくは分枝アルキル基から選択される。
【図面の簡単な説明】
【0031】
【図1】生検試料においてサイトカインをコードするmRNAの存在および量を確認するために、1×TBE中で1%アガロースゲル電気泳動を行った結果のグラフを示す。
【図2】蛍光顕微鏡によって現されるPPARγ受容体の存在を示す。
【図3】蛍光顕微鏡によって現されるPPARγ受容体の存在を示す。
【図4】蛍光顕微鏡によって蛍光が検出されないコントロールを示す。
【図5】蛍光顕微鏡によって蛍光が検出されないコントロールを示す。
【図6】1×アガロースゲルを用いる電気泳動によって得られた異なるバンドから推定される吸光度の値を示す。
【図7】1×アガロースゲルを用いる電気泳動によって得られた異なるバンドから推定される吸光度の値を示す。
【図8】1×アガロースゲルを用いる電気泳動によって得られた異なるバンドから推定される吸光度の値を示す。
【発明を実施するための形態】
【0032】
本発明の範囲内において、前記式(I)の化合物の実施形態は、添付の特許請求の範囲2〜6に与えられる。例として、実施例7および
(±)−2−ヒドロキシ−3−(3’−アミノフェニル)−プロピオン酸、
(±)−2−メトキシ−2−(4’−アミノフェニル)酢酸、
(±)−2−エトキシ−2−(3’−アミノフェニル)酢酸、
(±)−2−エトキシ−2−(4’−アミノフェニル)酢酸、
(±)−2−メトキシ−3−(4’−アミノフェニル)プロピオン酸、
(±)−2−エトキシ−3−(4’−アミノフェニル)プロピオン酸、
(±)−2−エトキシ−3−(3’−アミノフェニル)プロピオン酸
にも例示される化合物H〜Qが、特にプロピオン酸誘導体に関して、本発明による使用に適切である。
【0033】
一態様において、本発明の使用に適切な式(I)の化合物のさらなる例は、国際公開WO2007/010516号に記載され、その内容は参照として完全に本明細書に援用される。
【0034】
本発明の別の態様によれば、セリアック病の免疫炎症性成分を選択的に処置するための医薬を製造するための式(II)の化合物
【化4】
(式中、
R1およびR2は、互いに同じであるか、または異なり、H、−CO−CH3、nが1〜6、好ましくは1〜3の整数である−CnH2n−1、1〜6個、好ましくは1〜3個の炭素を有する直鎖もしくは分枝アルキル基からなる群より選択されるか、あるいはそれらは一緒に、5または6個の原子を有する芳香環または脂肪族環を形成し、
R3は、H、−CO−CH3、−NHOH、−OH、−OR6から選択され、R6は、1〜6個の炭素を有する直鎖もしくは分枝アルキル基であり、
R4は、H、1〜6個の炭素を有する直鎖もしくは分枝アルキル基から選択され、
R5、R7、R8は、H原子であり、
R3およびR4、R4およびR5、またはR7およびR8は、一緒に、ベンゼン環に縮合された環、N、Oからなる群より独立して選択される1〜2個のヘテロ原子を含む5または6個の原子を有する芳香環または脂肪族環のいずれかを形成する)
またはその混合物および薬理学的に許容可能な塩もしくはエステルの使用が提供される。
【0035】
前記式(II)の化合物の実施形態は、添付の特許請求の範囲8〜24に与えられる。
【0036】
例として、実施例5に例示される化合物A〜Gが本発明の使用に適切である。
【0037】
本発明の使用に適切な式(II)の化合物のさらなる例は、国際公開WO2007/010514号に記載され、その内容は参照として完全に本明細書に援用される。
【0038】
出願人は、式IおよびIIの化合物が、セリアック病の被験体の腸粘膜の位置、特に2番目の十二指腸部分の位置で観測される免疫炎症性成分に対する特異的活性を有することを示している。
【0039】
出願人は、式(I)および(II)の化合物によって与えられる局所的な抗炎症活性が、炎症プロセスにおいて重要な役割、および/またはその炎症プロセスが慢性的になるのに重要な役割を果たす物質である、サイトカインの放出に対する阻害活性に関与していることをさらに示している。
【0040】
特に、驚くべきことに、式IおよびIIの化合物が、セリアック病の被験体においてのみ、サイトカインを十分な方法で遮断するか、またはいずれかの場合において、実質的および有意に阻害するが、それらの化合物は、セリアック病以外の特定の胃腸管に影響を与える炎症性疾患を罹患している被験体において有意な活性を有さないことを見出した。
【0041】
特に、式IおよびIIの化合物は、いわゆるプロフロゴジェン(prophlogogen)Th1(ヘルパーT細胞タイプ1)サイトカインを遮断するのに適切である。なぜなら、それらの化合物は、損傷部位における免疫細胞および炎症細胞の動員に有利に働くからである。
【0042】
この特異的活性は、セリアック病において、排除できず、免疫系が耐性を生じることができない抗原(グルテンのグルアジン分画)に対する免疫系の活性化を、Th1タイプの炎症(phlogosis)の持続が反映するという事実に帰する可能性がある。
【0043】
セリアック病の免疫病原性に関与する典型的なプロフロゴジェンサイトカイン(Th1)は、IL−1、IL−2、IL−6(インターロイキン1、2、6)、IFN(インターフェロン)を含む。さらに、Th2(ヘルパーT細胞タイプ2)に関連するサイトカイン、およびTNF−α(腫瘍壊死因子)などのマクロファージ由来のサイトカインもまた、セリアック病に関与する。特に、TNF−αは、プロフロゴジェンサイトカイン(その一部はかなりの毒性を与えられる)の産生を上昇させる能力に起因する慢性的な炎症性疾患の発生に重要な役割を果たす。
【0044】
特に、セリアック病を罹患している腸粘膜試料由来のサイトカインの産生および放出に対する式(I)および(II)の化合物の効果は、有機培養系を用いて実証されている。特に、培養に対して腸免疫対象物の生検において式(I)(II)の化合物の1つ以上を用いることによるIFN−γ、IL−2、TNF−α値の有意な減少が強調されているため、フロゴシス(phlogosis)の効果的な減少が示されている。
【0045】
本発明の別の態様によれば、難治性セリアック病の被験体における炎症を処置するための医薬を製造するための上記のような式IまたはIIの化合物およびそれらの混合物の使用が提供される。
【0046】
本発明の別の態様によれば、食事療法の誤りによって誘発される、食事療法中のセリアック病の被験体における炎症性反応を処置するための医薬を製造するための上記のような式IまたはIIの化合物およびそれらの混合物の使用が提供される。
【0047】
本発明の別の態様によれば、典型的に、1つ以上の薬理学的に許容可能な賦形剤またはアジュバントと組み合わせて、セリアック病における免疫炎症性成分を処置するため、臨床時間および/または組織学的寛解時間を短縮するための医薬を製造するための式IまたはIIの化合物およびそれらの混合物の使用が提供される。
【0048】
本発明の1つ以上の態様に従って医薬を調製する際に、1つ以上の薬理学的に許容可能な賦形剤に加えて、薬理学的使用のための調製物の範囲内で典型的に使用される滑剤、保湿剤、懸濁化剤、分散剤、防腐剤を使用することも可能である。
【0049】
一実施形態において、式(I)および/または(II)の化合物は、錠剤、カプセル剤、粒状処方物、分散剤および薬理学的分野に典型的な他の処方物などの様々な形態で投与されてもよい。
【0050】
典型的に、活性成分(I)および(II)は、セリアック病における抗炎症性反応を達成するのに有効な量で投与に適切な処方物に組み込まれてもよい。
【0051】
例として、活性成分は、50mg〜2000mgの範囲、好ましくは200〜600mg、より好ましくは250〜500mgの範囲の量で医薬組成物に組み込まれてもよい。
【0052】
式(I)および/または(II)の1つ以上の化合物に基づいた医薬の使用は、患者および医師のコンプライアンスを改善し、寛解時間を減少させることを決定づけることが示されている。
【0053】
別の実施形態において、式(I)および/または(II)の化合物は、抵抗性および/または難治性のセリアック病の処置におけるさらなる治療用途を見出された。
【0054】
以下の実施例は単に本発明の例示のみとして与えられ、添付の特許請求の範囲から明らかなように、保護の範囲を限定することを意図しない。
【実施例】
【0055】
(実施例1)
セリアック病で観測される炎症性成分に対する、本発明の好ましい化合物の1つである、メサラジンの効果を評価した。
【0056】
このために、疾患の活動期中の16人の被験体およびグルテンを含まない食事療法中の臨床的寛解期中の6人の患者を選択した。
【0057】
各研究被験体につき5つの生検断片を、EGDS試験により得た。
【0058】
腸粘膜の各断片を、メサラジン(5ASA)を含むものと、含まないものの両方で、適切な培地中の培養物に入れた。
【0059】
次いで、以下のものを液体培地中で観測した:
異なる培地中で放出されたサイトカインのレベル、
適切に均質化された生検粘膜に存在する同じサイトカインの量。
【0060】
(物質および方法)
(方法)
3群の被験体を評価した。
【0061】
第1(萎縮性)の群は、抗筋肉膜および抗トランスグルタミナーゼ抗体に対して血清試験で陽性であり、セリアック病の新診断をして、グルテンを含む食事をしている29人の被験体を含んだ。
【0062】
第2(寛解)の群は、臨床的寛解において、少なくとも6ヶ月間、グルテンを含まない食事療法の17人のセリアック病の被験体を含んだ。
【0063】
第3(健康的)の群は、セリアック病を罹患していないが、胃腸管の炎症を罹患している被験体を含んだ。
【0064】
(液体培地中のサイトカインアッセイ)
(臓器培養)
この研究患者は、EGDS検査によって十二指腸粘膜の5つの生検を提供した。1つは組織学的アッセイについてのものであり、他の4つ(それぞれ2つの部分に分けた)は、それぞれ、1つの培地のみ(培地C)およびグリアジンペプチドを含む培地ならびにメサラジンの存在下でグリアジンを含む培地(培地+5ASA C)を用いて培養に供した。
【0065】
生検を、少なくとも3回、生理食塩水で2分間洗浄した。その生検を、1mlに相当する体積を有する培地に入れた:
培地(RPM11860+ウシ胎仔血清+ペニシリン/ストレプトマイシン)、
培地+グリアジンまたは31〜43または他の穀物のペプシン−トリプシン消化物、
培地+メサラジン(5−ASA)、
培地+グリアジンのペプシン−トリプシン消化物+5−ASA(培地1mlあたり1.5〜8.0mg)。
【0066】
生検を、4時間〜72時間の間、37℃の温度でO2(95%)およびCO2(5%)雰囲気下でインキュベートする。
【0067】
3つの異なる量の培地を、培地に放出されたTh1およびTh2サイトカインのELISAアッセイについて共有する。
【0068】
(ホモジネートからのサイトカインアッセイ)
37℃の温度で4時間〜72時間のインキュベーション時間の後、生検断片を2分間、生理食塩水で3回洗浄し、生検を機械的または化学的分解により均質化する。遠心分離を実施し、上清を回収し、1.5mlのエッペンドルフに移す。粘膜組織中の固定されたサイトカインをElisaアッセイにより分析する。
【0069】
(結果)
見出されるデータは、培地中のメサラジンの存在が、セリアック病の被験体のみにおいてサイトカインを遮断し、従って、セリアック病の被験体から得た試料において選択的に炎症性成分の減少を決定づけることを示す(表1)。
【0070】
【表1】
【0071】
【表2】
【0072】
【表3】
【0073】
【表4】
【0074】
達成された炎症性成分の減少により、メサラジンの単独の使用または全ての緩慢もしくは困難な臨床的解決の症例における治療支持としての使用が可能になる。
【0075】
(実施例2)
(サイトカインの遺伝子発現)
胃内視鏡検査処置の間に患者から得た生検の十二指腸断片を、5−ASA薬物の非存在下および存在下で、48時間、37℃で培養物中に入れた。培養後、断片を、フェノールおよびグアニジン(Isol−RNA溶解試薬)からなる単相溶液中で均質化した。
【0076】
ホモジナイザーIKA T10−Ultra Turraxを、均質化のために使用した。
【0077】
全ての不溶性物質を遠心分離により溶液から分離し、上清を無菌の試験管に移し、それに、溶解物1mlあたり200μlのクロロホルムを加えた。
【0078】
短時間の機械的攪拌後、試料を室温で数分間静置させ、その後、4℃で15分間、12000rpmで遠心分離した。RNAを含む無機段階を無菌の試験管に移して行った。
【0079】
RNAを、溶解物1mlあたり500μlのイソプロピルアルコールを加えることにより沈殿させた。試料を、室温で10分間インキュベートし、次いで、4℃で15分間、12000rpmで遠心分離した。
【0080】
上清を除去した後に、RNA沈殿物を、溶解物1mlあたり1000μlの75%エタノールで洗浄し、機械的に攪拌し、4℃で5分間、7500rpmで遠心分離した。最終的に、エタノールを除去し、RNA沈殿物を化学フード下で空気乾燥させた。
【0081】
RNAを、適切な量のRNasiを含まない水に再懸濁した。
【0082】
RNAの濃度を、分光光度計で260nmでの吸収を測定することにより決定し、異なるRNA鋳型についてのA260/A280の比を評価した。
【0083】
(RT−PCR)
cDNAを、適切な体積のRNasiを含まない水に希釈した異なる量の抽出したRNAから開始して合成した。関連遺伝子(サイトカイン)のcDNAを、特異的プライマーを使用してPCRにより増幅させた。
【0084】
マスタースクリプト RT−PCR系(5プライマー)をRT−PCRに使用した。
【0085】
生検試料においてサイトカインをコードするmRNAの存在および量を確認するために、1×TBE中で1%アガロースゲル電気泳動を行った。
【0086】
得られた結果を図1のグラフに示す。
【0087】
(実施例3)
(セリアック病患者の十二指腸上皮細胞におけるPPARγ受容体の存在)
ペルオキシソーム増殖因子活性化受容体γは、核内受容体のスーパーファミリーに属し、それは、エストロゲン、糖質コルチコイド、甲状腺ホルモン、ビタミンD3およびレチノイン酸についての受容体ならびに脂質代謝の異なる生成物を結合できる受容体(例えば、PPARおよびLXR受容体)を含む。
【0088】
特に、PPARファミリーは、異なる組織分布および異なるリガンド特異性を有する3つのサブタイプ(PPARα、βまたはδおよびγ)を含む。
【0089】
PPARγおよびαの両方は、分化したヒトマクロファージにおいて発現され、それらは、炎症性反応に関係する遺伝子を調節し、マクロファージの分化を調節する。
【0090】
異なるPPARγ作動薬は、ヒトの単球において炎症性サイトカインの産生を阻害し、活性化マクロファージにおけるTNF−α、IL−6、IL−1b、iNOS、ゼラチンB、スカベンジャー受容体AおよびCOX−2についての遺伝子発現を減少させ、PPARγの抗炎症性の役割を確認する。
【0091】
主な目的の1つは、セリアック病患者の十二指腸上皮細胞においてこのような受容体の存在を検出することであり、式(A)、I、IIの化合物の抗炎症性効果および炎症性サイトカインの産生の減少によって現れる5−ASAの特異性は、以前に確認されている。
【0092】
(免疫蛍光)
十二指腸生検断片を、グルテンを含む食事をしているセリアック病の患者およびセリアック病を罹患していない被験体(コントロール群)の両方からEGDS処置により取得した。生検断片を洗浄し、OCTに配向し、−80℃で保存した。5μmのいくつかの断片を、各々の凍結した生検切片(セリアック病の患者およびその疾患を罹患していない被験体の両方からEGDS工程で得た)から得て、その断片をPPARγ一次抗体に一晩(適切な固定および特異性除去の処置後)、曝露した。
【0093】
PBSで洗浄した後、試料を、約1時間、二次フルオレセイン抗体ALEXA 488とともにインキュベートした。一次抗体がPPARγ受容体に結合する場合において、存在する場合、一次/二次抗体複合体が形成され、顕微鏡によって観測可能な断片上の蛍光として現される。
【0094】
蛍光顕微鏡によって現されるPPARγ受容体の存在は、図2および図3(写真診断)から証明される。
【0095】
特に、図2および図3の画像において、受容体の存在は、末梢部分の断片において検出可能な蛍光シグナルによって現される。蛍光により、腸管腔の方を向いている表面上の腸細胞、腸上皮細胞の位置でPPARγの存在が実証される。
【0096】
セリアック病を罹患していない患者の生検は、ネガティブコントロールとして使用され、シグナルが存在せず、その結果として、顕微鏡によって分析された断片で受容体が存在しない。特に、図4および図5(コントロール)は蛍光が検出されないことを示し、これは、コントロールの健康な群の同じ腸内部分の位置において受容体が存在しないことのしるしである。
【0097】
同じように、本明細書の以下に記載される化合物を試験し、同様の結果を得た。
【0098】
(実施例4)
疾患の急性期で放出される炎症性サイトカインIL−2、TFN−αおよびIFN−γの遺伝子発現(mRNA)をモニターした。
【0099】
特に、図6〜8に示される図は、1×アガロースゲルを用いる電気泳動によって得られた異なるバンドから推定される吸光度の値(IMAGE J)を示し、この研究のサイトカイン増幅対象に関連する。
【0100】
サイトカインの遺伝子発現は、セリアック病を罹患している患者の十二指腸の生検の抽出されたRNAのRT−PCRによって評価される。このような生検は、5−ASA薬物が加えられる、グリアジンのトリプシン消化物(PT)を含む培地およびPT培地の両方において、48時間、37℃で増殖を維持されている。
【0101】
(結果)
3つの図により、生検断片が薬物の存在下において培地中に維持され、疾患急性期に放出される炎症性サイトカインIL−2、TFN−αおよびIFN−γの遺伝子発現(mRNA)が十分に減少されることが示される。
【0102】
患者が、薬物でのより持続的かつ一定の処置を受けている場合に、この減少が増加する。
【0103】
(実施例5)
(PPARγ活性化/発現ならびに細胞増殖およびアポトーシスの調節に対する本発明による化合物の効果の研究)
(物質および方法)
化合物A−G
5−ASAを、Sigma−AldrichTM(St Quentin Fallavier,フランス)で購入した。ロシグリタゾンを、Spi BioTM(Massy,フランス)で得た。式(II)の範囲内である以下の化合物A〜Gを試験した。
【化5】
【0104】
(細胞株)
結腸細胞株HT−29 STD(ATCC HTB−38)を、10%のheat−FCSおよび抗生物質を補足したDMEM中で慣例的に増殖させた。細胞を単分子層で増殖させ、5%CO2および95%相対湿度において37℃でインキュベートした。
【0105】
(PPARγでの一過性トランスフェクションおよび細胞の刺激)
HT−29 STD細胞を、製造業者の指示書に従ってEffectene TMトランスフェクション試薬(Qiagen TM)を用いて一過性トランスフェクションした。PPARγ活性化を試験するために、本発明者らは、シトクロムp450 4A(2XCYP)から得た2つのコピーのPPREを含む500ngの最小プロモーター構築物でトランスフェクションを実施した。ウミシイタケルシフェラーゼプラスミド(0.1μg/ウェル)も、トランスフェクション効果をモニターするため、およびホタルルシフェラーゼ活性を正規化するための内部コントロールとしてトランスフェクションした。トランスフェクションした細胞を48時間静置し、37℃でインキュベートした。刺激を、30mMの濃度で化合物A−Gを用いて、3−6−9−12−15−18−24時間の間、細胞のインキュベーション後に実施し、ポジティブコントロールとして使用した2つのPPARγ合成リガンド5−ASA 30mMおよびロシグリタゾン 10〜5Mと比較した。薬物溶液のpHをNaOHで7.4に調整した。全細胞抽出物を、受動的溶解緩衝液(Passive Lysis Buffer)(PromegaTM,Madison,Wis.)を用いて調製した。ルシフェラーゼ活性を、製造業者のプロトコルによるPromegaTMのデュアルルシフェラーゼアッセイシステムを用いて20μlの抽出物中で測定した。トランスフェクションを、少なくとも3つの別々の実験において3連で分析した。ルシフェラーゼ活性を、刺激していない細胞からのルシフェラーゼ活性で割られた異なる分子で処理した細胞で得られた活性の倍数として表した。
【0106】
(ウェスタンブロット解析によるPPARγおよびβ−アクチンの評価)
全タンパク質を、2%のTritonTM、フッ化フェニルメチルスルホニル(PMSF)100mMおよび古典的プロテアーゼ阻害剤カクテルを含むPBSからなる抽出緩衝液中での細胞均質化によって得た。次いで、全タンパク質を、ポリアクリルアミドゲル電気泳動および電気ブロットによって分離した。二フッ化ポリビニリデン(PVDF)膜を、PPARγに対するウサギポリクローナル一次抗体(1/500希釈,TEBU,Le Perray en Yveline,フランス)とともに一晩インキュベートした。β−アクチンを、1/10,000で希釈したウサギモノクローナル一次抗体(Sigma)を用いて検出した。二次ペルオキシダーゼ標識抗体(1/1000,DakoTM,Trappes,フランス)および化学発光を用いる免疫検出を、製造業者のプロトコル(ECLTM、Amersham Pharmacia BiotechTM,Orsay,フランス)に従って実施した。PPARγの吸光度の値を、同じ試料内の内部コントロールβ−アクチンの量に比例して各条件について得た。
【0107】
(Ki−67免疫染色による細胞増殖の分析)
24時間の培養後、HT−29 STD細胞を、30mMで化合物A、B、C、DおよびFを用いて24時間、処理した。5−ASA(30mM)およびロシグリタゾン(10〜5M)を、ポジティブコントロールとして使用した。分子Gは、その不十分な溶解性のために、この実験に含まなかった。薬物溶液のpHをNaOHで7.4に調整した。細胞を、PFA 4%中で固定し、4℃で0.1%のTriton X−100TMを含むPBS中で透過処理し、次いで、抗体の非特異的吸着を最小化するためにヤギ正常血清および遮断バッファー(PBS中の1% BSA)とともにインキュベートした。
【0108】
細胞増殖を、Ki−67に対するマウスモノクローナル一次抗体を用いて核のKi−67染色によって評価した(1:50希釈、一晩;ZYMEDTM,ClinisciencesTM,Montrouge,フランス)。一次抗体を、アクリジン赤蛍光色素(1:100希釈,Molecular ProbesTM,InvitrogenTM,Cergy Pontoise,フランス)と結合したAlexa 594ロバ抗マウスIgGで明らかにした。核を、Hoescht 33342溶液(0.125mg/mL)(Sigma−AldrichTM)で染色し、蛍光顕微鏡(LeicaTM,Bensheim,ドイツ)下で視覚化した。ネガティブコントロールは、特異的抗体の代わりに非特異的マウス血清での染色からなる。少なくとも500細胞/試料のカウントを、1つの実験において体系的に盲目的に実施した。その結果を、染色細胞の数の平均±SEMとして表した。
【0109】
(アポトーシスの検出)
24時間の培養後、HT−29 STD細胞を、24時間の間、30mMの濃度で化合物A、B、C、D、Fを用いて処理した。5−ASA(30mM)およびロシグリタゾン(10〜5M)を、ポジティブコントロールとして使用した。分子EおよびGは、それらの不十分な溶解度のためにこの実験に含まなかった。薬物溶液のpHをNaOHで7.4に調整した。アポトーシスを受けた細胞を、ターミナルトランスフェラーゼdUTPニック末端標識アッセイ(TUNELアッセイ,Roche DiagnosticsTM,Meylan,フランス)を用いてDNA鎖の酵素標識により同定した。少なくとも500細胞/試料のカウントを、1つの実験において体系的に盲目的に実施した。その結果を、染色細胞の数の平均±SEMとして表した。
【0110】
(結果)
分子CおよびFは、PPARγ活性化を誘導することが観測された。化合物Dもまた、PPARγを誘導するが、わずかに少ない程度である。PPAARγの活性化は、ペルオキシソーム増殖因子活性化受容体エレメント(PPRE)と呼ばれる特異的DNA配列エレメントに対する結合を導く反応のカスケードを生じる。
【0111】
本発明者らは、ウミシイタケルシフェラーゼPPREプラスミドでの上皮細胞の一過性トランスフェクションによるPPARγ転写活性を研究した。細胞を、24時間、異なる分子で刺激した。トランスフェクションされたHT−29細胞におけるPPARγ活性の分析により、30mMの濃度で化合物CおよびFが、レポーター遺伝子活性を2倍増加させ、それにより、5−ASAおよびロシグリタゾンと同様の活性を表したことが示された。30mMの濃度で化合物A、BおよびGは、6時間後にPPARγ活性化の研究を制限する上皮細胞に対する急速な細胞毒性効果を与えた。
【0112】
特に、分子C、DおよびFは、PPARγ発現を誘導する。通常、全ての化合物A〜Gは、HT−29細胞株におけるタンパク質レベルでPPARγ発現を誘導する能力を示す。特に、ウェスタンブロットにより定量化したPPARγタンパク質レベルの平均2倍の誘導を、24時間、分子C、DおよびFで処理した細胞において観測した。
【0113】
特に、分子CおよびFは、上皮細胞増殖を阻害する。本発明者は、細胞増殖の調節における分子の役割をHT−29 STD細胞株において評価した。細胞増殖を、増殖細胞中で発現される核タンパク質Ki−67染色により評価し、Ki−67の存在が細胞増殖を維持するのに必須であった。未処理の細胞と比較して、分子CおよびF(30mM)との24時間のHT−29細胞のインキュベーションは、細胞増殖の67〜75%の阻害を生じた。
【0114】
同様の結果を、最適濃度で使用した2つのポジティブコントロールのロシグリタゾン(10〜5M)および5−ASA(30mM)で得た。分子A、BおよびDの潜在的抗分離促進効果の確認は、この濃度での上皮細胞に対するそれらの急速な細胞毒性効果によって制限された。
【0115】
化合物Fもまた、PPARγを介して上皮細胞のアポトーシスを誘発する。ロシグリタゾンおよび5−ASAと同様に、分子Fは、ターミナルトランスフェラーゼdUTPニック末端標識(TUNEL)を用いてDNA鎖の分裂を標識することにより同定された80%の上皮細胞のアポトーシスを示した。以前の実験と同様に、分子A、BおよびDは、30mMの阻害細胞アポトーシス分析において急速な細胞毒性効果を誘発した。
【0116】
(結論)
この実施例は特に、PPARγ発現および活性化を誘発し、上皮細胞増殖およびアポトーシスを調節する化合物CおよびFの能力を示す。さらに、30mMでの化合物A、B、D(およびE〜G)の上皮細胞に対する細胞毒性効果は、多くの種々の酵素と高い親和性を示すことが知られている高い反応性のヒドロキサム酸基の構造の存在と関連し得る。
【0117】
(実施例6)
(分子モデル)
分子モデル研究を、Silicon GraphicsTMワークステーションで作動するSYBYLソフトウェアバージョン6.9.1(Tripos Associates IncTM,ST Louis,MO)を用いて実施した。5−ASAの両性イオン形態の三次元モデルを、標準的なフラグメントライブラリーから構築し、次いで、その形状を、Tripos力場を用いて最適化した。化合物のpKaはまだ知られていないため、SPARCオンライン計算機を、生理学的pH(7.4)で生じる種を決定するために使用した。イオン化化合物の三次元モデルを、標準的なフラグメントライブラリーから構築し、次いで、それらの形状を、GasteigerおよびHuckel原子電荷から計算される静電条件を含むTripos力場を用いて最適化した。Maximin2処理に利用可能なPowell法を、勾配値が0.001kcal/mol.Åより小さくなるまで、エネルギー最小化に使用した。ヒトPPARγリガンド結合ドメインの構造を、RCSBタンパク質データバンク(1171)(4,5)で利用可能なテサグリタザル(AZ242)を用いてその錯体化したX線結晶構造から得た。化合物の受容体活性部位へのフレキシブルなドッキングを、GOLDソフトウェアを用いて実施した。最も安定なドッキングモデルを、GoldScoreおよびX−Score評価関数によって予測された最適に評価された構造に従って選択した。錯体を、勾配値が0.01kcal/mol.Åに到達するまで、Tripos力場および4.0の誘電率でMaximin2処理に利用可能なPowell法を用いてエネルギー最小化をした。アニール機能を使用して、リガンドのホット領域(hot region)(10Å)を規定する。
【0118】
(ドッキング研究)
化合物A〜Gは、分子認識およびPPARγ活性化に必要な主要な決定因子とみなされるHis−323、His−449、Tyr−473およびSer−289との水素結合を介して相互作用するPPARγ−LBDと強く適合した。
【0119】
式(II)に含まれる化合物の抗炎症効果は、上皮細胞によって主に発現されるPPARγにより媒介されることが示された。このドッキング分析により、30mMの濃度で使用される上述の化合物がPPARγを活性化し、腸の上皮細胞によりその発現を誘導し、セリアック病の免疫炎症性成分に対する薬理作用を与えることが明らかにされた。
【0120】
(結論)
アミノサリチル酸構造(5−ASA構造など)との式IIの化合物の抗炎症効果は、PPARγにより媒介され、十二指腸の位置で上皮細胞において発現されることが以前に示されている。ドッキング分析に基づいた化合物A〜Gの合理的開発により、30mMの濃度で使用される前記化合物、特にC、Fが、PPARγを活性化し、腸の上皮細胞によりその発現を誘導することが明らかにされた。その化合物はまた、PPARγ活性化による2つの重要な機構である、上皮細胞増殖を阻害し、アポトーシスを誘導する。特に、分子A、B、D、Gに関して、それらが、PPARγ活性化の分析ならびに細胞増殖およびアポトーシスの調節および評価を妨げる、30mMの濃度での上皮細胞に対する細胞毒性効果に関することが検出されている。
【0121】
(実施例7)
(PPARγ活性化に対する式(I)の化合物H〜Qの効果)
(物質および方法)
(物質)
5−ASAを、Sigma−AldrichTM(St Quentin Fallavier、フランス)で購入した。ロシグリタゾンを、Spi BioTM(Massy、フランス)で得た。式(I)内に含まれる以下の化合物H〜Qを試験した。
【化6】
【化7】
【0122】
(細胞株)
結腸癌細胞株HT−29 STD(ATCC HTB−38)を、10%のheat−FCSおよび抗生物質を補足したDMEM中で慣例的に増殖させた。細胞を単分子層で増殖させ、5%CO2および95%相対湿度において37℃でインキュベートした。
【0123】
(PPARγでの一過性トランスフェクションおよび細胞の刺激)
HT−29 STD細胞を、製造業者の指示書に従ってEffecteneTMトランスフェクション試薬(QiagenTM)を用いて一過性トランスフェクトした。PPARγ活性化を試験するために、本発明者らは、シトクロムp450 4A(2XCYP)から得た2つのコピーのPPREを含む500ngの最小プロモーター構築物でトランスフェクションを実施した。ウミシイタケルシフェラーゼプラスミド(0.1μg/ウェル)も、トランスフェクション効果をモニタリングするため、およびホタルルシフェラーゼ活性を正規化するための内部コントロールとしてトランスフェクションした。トランスフェクションした細胞を、37℃で24時間のインキュベーションのために静置させておいた。刺激を、1mMの濃度で化合物H〜Qを用いて18時間の細胞のインキュベーション後に実施して、ポジティブコントロールとして使用した2つのPPARγ合成リガンド5−ASA(30mM)およびロシグリタゾン(10〜5M)と比較した。薬物溶液のpHをNaOHで7.4に調整した。全細胞抽出物を、受動的溶解緩衝液(Passive Lysis Buffer)(PromegaTM,Madison,Wis.)を用いて調製した。ルシフェラーゼ活性を、製造業者のプロトコルによるPromegaのデュアルルシフェラーゼアッセイシステムを用いて20μlの抽出物中で測定した。トランスフェクションを、少なくとも3つの別々の実験において3連で分析した。ルシフェラーゼ活性を、刺激していない細胞からのルシフェラーゼ活性で割られた異なる分子で処理した細胞で得られた活性の倍数として表した。
【0124】
(結果)
PPARγの活性化は、ペルオキシソーム増殖因子活性化受容体エレメント(PPRE)と呼ばれる特異的DNA配列エレメントに対する結合を導く反応のカスケードを生じる。
【0125】
本発明者らは、ウミシイタケルシフェラーゼおよびPPREプラスミドでの上皮細胞の一過性トランスフェクションによるPPARγ転写活性を調べた。化合物H〜QがPPARγ活性化を刺激する有効性および/または5−ASAより有効かどうかを評価するために、本発明者らは1mMの濃度でこれらの分子を試験した。1mMの濃度での新規の分子の効果を、それぞれ30mMおよび10〜5Mの最適濃度でポジティブコントロールとして使用した5−ASAおよびロシグリタゾンと比較した。細胞を、24時間、異なる分子で刺激した。
【0126】
トランスフェクションされたHT−29細胞中のPPARγ活性の分析により、1mMで前記化合物40が、それぞれ、4.8±0.71倍、2.73±0.31倍、2.64±0.46倍、3.4±0.97倍、受容体遺伝子活性を増加させ、それにより、30mMでの5−ASA(2.8±0.7)および10〜5Mでのロシグリタゾン(3.17±0.29)と同等またはそれらより優れた活性を表したことが示された。
【0127】
トランスフェクションされたHT−29細胞中のPPARγ活性を増加させる本発明の式(I)の化合物に含まれる分子H〜Qの能力は、30mMでの5−ASAおよび10〜5Mでのロシグリタゾンと同等またはそれらより優れた活性を表す。
【0128】
2007年12月24日に出願された、詳細な説明、特許請求の範囲および図面を含む伊国特許出願第MI2007A2429号は、その優先権が本明細書で主張され、完全に参照によって援用される。
【特許請求の範囲】
【請求項1】
セリアック病の炎症性成分の処置に使用するための式(I)の化合物
【化8】
(式中、
R1およびR2は、互いに同じであるか、または異なり、−Hまたは1〜6個の炭素を有する直鎖もしくは分枝アルキル基からなる群より選択されるか、あるいはそれらは一緒に、5または6個の原子を有する芳香環または脂肪族環を形成し、
YおよびZは、互いに同じであるか、または異なり、−H、−OH、−COOH、−OR3、−CH(OR3)COOHからなる群より選択され、
ここで、R3は、H、フェニル、ベンジル、−CF3、−CF2CF3、ビニル、アリル、1〜6個の炭素を有する直鎖もしくは分枝アルキル基から選択される)
またはその混合物および薬理学的に許容可能な塩もしくはエステル。
【請求項2】
1〜6個の炭素を有する前記直鎖もしくは分枝アルキル基は、−CH3、−CH2CH3、−CH(CH3)2、−CH2CH2CH3、−CnH2n−1から選択され、nが1〜6の整数である、請求項1に記載の化合物。
【請求項3】
YはHである、請求項1または2に記載の化合物。
【請求項4】
Zは、−CH3(OR3)COOHである、請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
R3は、H、−CH3、−CH2CH3から選択される、請求項1〜4のいずれか1項に記載の化合物。
【請求項6】
前記化合物は、式
【化9】
の(±)3−(3’−アミノフェニル)−2−ヒドロキシ−プロピオン酸、
2−(4−アミノフェニル)−2−メトキシ酢酸、
2−(3−アミノフェニル)−2−エトキシ酢酸、
2−(4−アミノフェニル)−2−エトキシ酢酸、
式
【化10】
の3−(4’−アミノフェニル)−2−メトキシプロピオン酸、
式
【化11】
の3−(4’−アミノフェニル)−2−エトキシプロピオン酸、
式
【化12】
の3−(3’−アミノフェニル)−2−メトキシプロピオン酸、
式
【化13】
の3−(3’−アミノフェニル)−2−エトキシプロピオン酸
からなる群より選択される、請求項1に記載の化合物。
【請求項7】
セリアック病の炎症性成分の処置に使用するための式(II)の化合物
【化14】
(式中、
R1およびR2は、互いに同じであるか、または異なり、−H、−CO−CH3、nが1〜6である−CnH2n−1、1〜6個の炭素を有する直鎖もしくは分枝アルキル基からなる群より選択されるか、あるいはそれらは一緒に、5または6個の原子を有する芳香環または脂肪族環を形成し、
R3は、H、−CO−CH3、−NHOH、−OH、−OR6から選択され、ここでR6は、1〜6個の炭素を有する直鎖もしくは分枝アルキル基であり、
R4は、H、1〜6個の炭素を有する直鎖もしくは分枝アルキル基、フェニル、ベンジル、−CF3、−CF2CF3、ビニルまたはアリルから選択され、
R5、R7、R8は、H原子であるか、あるいは
R3およびR4、R4およびR5、またはR7およびR8は、一緒に、ベンゼン環に縮合された環、N、Oからなる群より独立して選択される1〜2個のヘテロ原子を含む5または6個の原子を有する芳香環または脂肪族環のいずれかを形成する)
またはその混合物およびそれらの薬理学的に許容可能な塩もしくはエステル。
【請求項8】
1〜6個の炭素を有する前記直鎖または分枝アルキル基は、−CH3、−C2H5、イソプロピル、プロピル、−CnH2n−1から選択される、請求項7に記載の化合物。
【請求項9】
R3およびR4は、以下の式(III)
【化15】
による環を形成する、請求項7に記載の化合物。
【請求項10】
R4およびR5は、以下の式(IV)
【化16】
による環を形成する、請求項7に記載の化合物。
【請求項11】
R7およびR8は、以下の式(V)または(VI)
【化17】
による環を形成する、請求項7に記載の化合物。
【請求項12】
前記化合物は、
4−アミノ−N−ヒドロキシ−2−メトキシベンズアミド、
5−アミノ−N−ヒドロキシ−2−メトキシベンズアミド、
5−アミノ−2,3−ジヒドロベンゾフラン−7−カルボン酸、
5−アミノ−2−エトキシ−N−ヒドロキシベンズアミド、
6−アミノ−2,2−ジメチル−4H−ベンゾ[1,3]ジオキシン−4−オン、
1,2,3,4−テトラヒドロ−6−ヒドロキシキノリン−5−カルボン酸、
5−アミノ−2−イソプロポキシ安息香酸、
6−メトキシキノリン−5−カルボン酸、
6−メトキシ−1,2,3,4−テトラヒドロキノリン−5−カルボン酸、
5−ジイソプロピルアミノサリチル酸、
4−ジイソプロピルアミノサリチル酸
からなる群より選択される、請求項7に記載の化合物。
【請求項13】
R1およびR2は、両方とも−CH(CH3)2である、請求項7に記載の化合物。
【請求項14】
前記化合物は、以下の構造
【化18】
を含む、請求項13に記載の化合物。
【請求項15】
R1およびR2は、両方とも−Hである、請求項7に記載の化合物。
【請求項16】
R3は、−NHOHである、請求項7または15に記載の化合物。
【請求項17】
前記化合物は、以下の構造
【化19】
を含む、請求項16に記載の化合物。
【請求項18】
前記化合物は、以下の構造
【化20】
を含む、請求項16に記載の化合物。
【請求項19】
前記化合物は、以下の構造
【化21】
を含む、請求項16に記載の化合物。
【請求項20】
R3は−OHである、請求項15に記載の化合物。
【請求項21】
前記化合物は、以下の構造
【化22】
を含む、請求項20に記載の化合物。
【請求項22】
前記化合物は、以下の構造
【化23】
を含む、請求項20に記載の化合物。
【請求項23】
前記化合物は、以下の構造
【化24】
を含む、請求項9に記載の化合物。
【請求項24】
前記化合物は、メサラジンまたは5−アミノサリチル酸である、請求項7に記載の化合物。
【請求項25】
難治性のセリアック病被験体における免疫炎症の処置に使用するための請求項1〜24のいずれか1項に記載の化合物。
【請求項26】
食事療法の誤りにより誘発される、食事療法中のセリアック病被験体における免疫炎症反応の処置に使用するための請求項1〜24のいずれか1項に記載の化合物。
【請求項27】
臨床的および/または組織学的寛解時間を短縮するためのセリアック病における免疫炎症性成分の処置に使用するための請求項1〜24のいずれか1項に記載の化合物。
【請求項28】
セリアック病におけるサイトカインの放出を選択的に遮断するのに使用するための請求項1〜24のいずれか1項に記載の化合物。
【請求項1】
セリアック病の炎症性成分の処置に使用するための式(I)の化合物
【化8】
(式中、
R1およびR2は、互いに同じであるか、または異なり、−Hまたは1〜6個の炭素を有する直鎖もしくは分枝アルキル基からなる群より選択されるか、あるいはそれらは一緒に、5または6個の原子を有する芳香環または脂肪族環を形成し、
YおよびZは、互いに同じであるか、または異なり、−H、−OH、−COOH、−OR3、−CH(OR3)COOHからなる群より選択され、
ここで、R3は、H、フェニル、ベンジル、−CF3、−CF2CF3、ビニル、アリル、1〜6個の炭素を有する直鎖もしくは分枝アルキル基から選択される)
またはその混合物および薬理学的に許容可能な塩もしくはエステル。
【請求項2】
1〜6個の炭素を有する前記直鎖もしくは分枝アルキル基は、−CH3、−CH2CH3、−CH(CH3)2、−CH2CH2CH3、−CnH2n−1から選択され、nが1〜6の整数である、請求項1に記載の化合物。
【請求項3】
YはHである、請求項1または2に記載の化合物。
【請求項4】
Zは、−CH3(OR3)COOHである、請求項1〜3のいずれか1項に記載の化合物。
【請求項5】
R3は、H、−CH3、−CH2CH3から選択される、請求項1〜4のいずれか1項に記載の化合物。
【請求項6】
前記化合物は、式
【化9】
の(±)3−(3’−アミノフェニル)−2−ヒドロキシ−プロピオン酸、
2−(4−アミノフェニル)−2−メトキシ酢酸、
2−(3−アミノフェニル)−2−エトキシ酢酸、
2−(4−アミノフェニル)−2−エトキシ酢酸、
式
【化10】
の3−(4’−アミノフェニル)−2−メトキシプロピオン酸、
式
【化11】
の3−(4’−アミノフェニル)−2−エトキシプロピオン酸、
式
【化12】
の3−(3’−アミノフェニル)−2−メトキシプロピオン酸、
式
【化13】
の3−(3’−アミノフェニル)−2−エトキシプロピオン酸
からなる群より選択される、請求項1に記載の化合物。
【請求項7】
セリアック病の炎症性成分の処置に使用するための式(II)の化合物
【化14】
(式中、
R1およびR2は、互いに同じであるか、または異なり、−H、−CO−CH3、nが1〜6である−CnH2n−1、1〜6個の炭素を有する直鎖もしくは分枝アルキル基からなる群より選択されるか、あるいはそれらは一緒に、5または6個の原子を有する芳香環または脂肪族環を形成し、
R3は、H、−CO−CH3、−NHOH、−OH、−OR6から選択され、ここでR6は、1〜6個の炭素を有する直鎖もしくは分枝アルキル基であり、
R4は、H、1〜6個の炭素を有する直鎖もしくは分枝アルキル基、フェニル、ベンジル、−CF3、−CF2CF3、ビニルまたはアリルから選択され、
R5、R7、R8は、H原子であるか、あるいは
R3およびR4、R4およびR5、またはR7およびR8は、一緒に、ベンゼン環に縮合された環、N、Oからなる群より独立して選択される1〜2個のヘテロ原子を含む5または6個の原子を有する芳香環または脂肪族環のいずれかを形成する)
またはその混合物およびそれらの薬理学的に許容可能な塩もしくはエステル。
【請求項8】
1〜6個の炭素を有する前記直鎖または分枝アルキル基は、−CH3、−C2H5、イソプロピル、プロピル、−CnH2n−1から選択される、請求項7に記載の化合物。
【請求項9】
R3およびR4は、以下の式(III)
【化15】
による環を形成する、請求項7に記載の化合物。
【請求項10】
R4およびR5は、以下の式(IV)
【化16】
による環を形成する、請求項7に記載の化合物。
【請求項11】
R7およびR8は、以下の式(V)または(VI)
【化17】
による環を形成する、請求項7に記載の化合物。
【請求項12】
前記化合物は、
4−アミノ−N−ヒドロキシ−2−メトキシベンズアミド、
5−アミノ−N−ヒドロキシ−2−メトキシベンズアミド、
5−アミノ−2,3−ジヒドロベンゾフラン−7−カルボン酸、
5−アミノ−2−エトキシ−N−ヒドロキシベンズアミド、
6−アミノ−2,2−ジメチル−4H−ベンゾ[1,3]ジオキシン−4−オン、
1,2,3,4−テトラヒドロ−6−ヒドロキシキノリン−5−カルボン酸、
5−アミノ−2−イソプロポキシ安息香酸、
6−メトキシキノリン−5−カルボン酸、
6−メトキシ−1,2,3,4−テトラヒドロキノリン−5−カルボン酸、
5−ジイソプロピルアミノサリチル酸、
4−ジイソプロピルアミノサリチル酸
からなる群より選択される、請求項7に記載の化合物。
【請求項13】
R1およびR2は、両方とも−CH(CH3)2である、請求項7に記載の化合物。
【請求項14】
前記化合物は、以下の構造
【化18】
を含む、請求項13に記載の化合物。
【請求項15】
R1およびR2は、両方とも−Hである、請求項7に記載の化合物。
【請求項16】
R3は、−NHOHである、請求項7または15に記載の化合物。
【請求項17】
前記化合物は、以下の構造
【化19】
を含む、請求項16に記載の化合物。
【請求項18】
前記化合物は、以下の構造
【化20】
を含む、請求項16に記載の化合物。
【請求項19】
前記化合物は、以下の構造
【化21】
を含む、請求項16に記載の化合物。
【請求項20】
R3は−OHである、請求項15に記載の化合物。
【請求項21】
前記化合物は、以下の構造
【化22】
を含む、請求項20に記載の化合物。
【請求項22】
前記化合物は、以下の構造
【化23】
を含む、請求項20に記載の化合物。
【請求項23】
前記化合物は、以下の構造
【化24】
を含む、請求項9に記載の化合物。
【請求項24】
前記化合物は、メサラジンまたは5−アミノサリチル酸である、請求項7に記載の化合物。
【請求項25】
難治性のセリアック病被験体における免疫炎症の処置に使用するための請求項1〜24のいずれか1項に記載の化合物。
【請求項26】
食事療法の誤りにより誘発される、食事療法中のセリアック病被験体における免疫炎症反応の処置に使用するための請求項1〜24のいずれか1項に記載の化合物。
【請求項27】
臨床的および/または組織学的寛解時間を短縮するためのセリアック病における免疫炎症性成分の処置に使用するための請求項1〜24のいずれか1項に記載の化合物。
【請求項28】
セリアック病におけるサイトカインの放出を選択的に遮断するのに使用するための請求項1〜24のいずれか1項に記載の化合物。
【図1】

【図2】

【図3】
【図4】

【図5】

【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2011−507818(P2011−507818A)
【公表日】平成23年3月10日(2011.3.10)
【国際特許分類】
【出願番号】特願2010−538801(P2010−538801)
【出願日】平成20年12月23日(2008.12.23)
【国際出願番号】PCT/EP2008/068265
【国際公開番号】WO2009/080828
【国際公開日】平成21年7月2日(2009.7.2)
【出願人】(505367017)ジュリアーニ インターナショナル リミテッド (9)
【Fターム(参考)】
【公表日】平成23年3月10日(2011.3.10)
【国際特許分類】
【出願日】平成20年12月23日(2008.12.23)
【国際出願番号】PCT/EP2008/068265
【国際公開番号】WO2009/080828
【国際公開日】平成21年7月2日(2009.7.2)
【出願人】(505367017)ジュリアーニ インターナショナル リミテッド (9)
【Fターム(参考)】
[ Back to top ]