
セリンプロテアーゼ酵素の大環状阻害剤
本発明は、セリンプロテアーゼ酵素に結合し、そして/またはその阻害剤である、新規な大環状化合物およびその塩に関する。本発明は、これらの化合物の中間体、これらの化合物を含有する医薬組成物、および該化合物を使用する方法にも関する。これらの化合物は、増殖過剰性障害、詳細には、腫瘍転移、炎症性障害、皮膚および組織障害、心臓血管障害、呼吸器障害ならびにウイルス感染を特徴とするそれらの障害をはじめとする一定範囲の適応症の処置および予防のための治療薬として有用である。
【発明の詳細な説明】
【技術分野】
【0001】
[関連出願の相互参照]
本願は、ここに引用することで本明細書の一部をなすもとする2009年10月23日出願の米国特許仮出願第61/254,434号の優先権を主張するものである。
【0002】
[発明の分野]
本発明は、セリンプロテアーゼ酵素に結合し、そして/またはその調整物質、詳細には阻害剤である、新規な大環状化合物および薬学的に許容しうるその塩に関する。本発明は、これらの化合物の中間体、これらの化合物を含有する医薬組成物および該化合物を使用する方法にも関する。該化合物は、増殖過剰性障害、詳細には、腫瘍転移、炎症性障害、皮膚および組織障害、心臓血管障害、呼吸器障害ならびにウイルス感染を特徴とするそれらの障害をはじめとする一定範囲の適応症の処置および予防のための治療薬として有用である。
【背景技術】
【0003】
発明の背景
セリンプロテアーゼ酵素は、ホ乳類、ウイルス、細菌および他の生物体における複数の重要な生理学的プロセスに関与し、組織のホメオスタシスおよび修復、発達、免疫性ならびに生殖能力などのような多様な機能を調節する。これらのプロテアーゼは、生化学的レベルでは、ホルモン、成長因子、サイトカインおよび他の内在性の生理学的送達物質の活性化、イオンチャネルの調節、受容体の活性化ならびに細胞透過性の制御を担う。
【0004】
この種の作用により、セリンプロテアーゼは、医薬品開発のターゲットとなった(Drews, J.; Ryser, S. Nat. Biotech. 1997, 15, 1318-1319; Imming, P.; Sinning, C.; Meyer, A. Nat. Rev. Drug Disc. 2006, 5, 821-834.)。実際に、新薬開発につながる生物学的ターゲット全体の3〜4%が、この分類の一種と推定された(Hopkins, A.L.; Groom, C.R. Nat. Rev. Drug Disc. 2002, 1, 727-730.)。具体的には、これらの酵素の阻害剤が、効果的心臓血管調整物質、呼吸器疾患処置剤、抗炎症剤、抗ウイルス薬およびCNS薬として広範囲の薬学的関連活性を有することが立証された。加えて、様々な組織の維持プロセスにおいてセリンプロテアーゼが密接に関与することで、それらが癌(Bialas, A.; Kafarski, P. Anti-cancer Agents Med. Chem. 2009, 9, 728-762) 、ならびに皮膚疾患および障害(Meyer-Hoffert, U. Arch. Immunol. Ther. Exp. 2009, 57, 345-354) の新しいターゲットとなった。
【0005】
とりわけ、癌細胞のより潜行的特徴は、体内の他の部位へ拡大または転移する能力である。多くの例では、高い転移性を有する腫瘍は転帰が不良であるため、腫瘍の転移能力は予後に相関する。タンパク質分解活性の上昇は、癌の進行および転移に関連づけられてきた。数あるタンパク質分解酵素の中でもセリンプロテアーゼは、細胞構造の分解および組織リモデリングに寄与し、それにより癌浸潤および拡大の一助となる。更にプロテアーゼは、癌細胞の増殖または血管新生を刺激するのに必要となる増殖因子の宿主の活性化に関与する。このプロセスに関与するセリンプロテアーゼの幾つかが、ウロキナーゼ、プラスミン、エラスターゼ、トロンビンおよびカテプシンGである。癌に関与するプロテアーゼごとに異なる基質特異性が見出され、これらのプロテアーゼの選択的ターゲッティングが可能であることが示唆された(Beliveau, F.; Desilets, A.; Leduc, R. FEBS J. 2009, 276, 2213-2226.)。加えて、II型膜貫通性セリンプロテアーゼ(TTSP)と呼ばれるセリンプロテアーゼの新しい種類が、組織ホメオスタシスおよび癌、詳細には腫瘍転移に重要であることが見出された(Wu, Q. Curr. Top. Develop. Biol. 2003, 54, 167-206; Qui, D.; Owen, K.; Gray, K.; Bass, R.; Ellis, V. Biochem. Soc. Trans. 2007, 35, 583-587.)。TTSPファミリーの一種は、消化、心臓機能、血圧調節および聴力と同様に多様な生理学的プロセスにも役割を有する(Bugge, T.H.; Antalis, T.M.; Wu, Q. J. Biol. Chem. 2009, 284, 23177-23181.)。これらの役割では、TTSPは、典型的にはホメオスタシスを保持する働きがあり、多くの場合、ホルモンもしくは成長因子の活性化、またはタンパク質分解カスケードの開始に関与する。加えて、より近年の知見で、インフルエンザおよび他の呼吸器系ウイルス、例えばヒトメタニューモウイルスが、TTSPを活用してその伝播を促進し、これらのプロテアーゼがウイルス感染に介入する潜在的ターゲットとなることが示唆された(Choi, S.-Y.; Bertram, S.; Glowacka, I.; Park, Y.W.; Pohlmann, S. Trends Mol. Med. 2009, 15, 303-312.)。
【0006】
TTSPは、細胞質、膜貫通領域、C末端のリガンド結合ドメインおよびセリンプロテアーゼドメイン中に保持される短いN末端テールを特徴とする。そのような特性により、それらは他の細胞表面タンパク質および隣接細胞の成分と相互作用するのに理想的である。
【0007】
この酵素類の一種であるマトリプターゼ(マトリプターゼ−1、MT−SP1、TADG−15、エピチン、ST14)は、上皮の起源細胞により発現され、非常に様々なヒト癌において過剰発現されるトリプシン様セリンプロテアーゼである(US 5,482,848; US 5,792,616; US 5,972,616; US 6,649,741; US 7,030,231; US 7,227,009; US 7,276,364; US 7,291,462; WO 99/42120; WO 00/53232; WO 01/23524; WO 01/29056; WO 01/57194; WO 01/36604; US 2003/0119168; US 2006/0099625; US 2008/0051559; Takeuchi, T.; Shuman, M.A.; Craik, C.S. Proc. Natl. Acad. Sci. 1999, 96, 11054-11061; Lin, C.Y.; Anders, J.; Johnson, M.; Sang, Q.A.; Dickson, R.B.; J. Biol. Chem. 2001, 274, 18231-18236; Oberst, M.; Johnson, M.; Dickson, R.B.; Lin, C.-Y. Recent Res. Develop. Biochem. 2002, 3, 169-190; Lin, C.-Y.; Oberst, M.; Johnson, M.; Dickson, R.B. Handbook of Proteolytic Enzymes, 2nded., Barrett, A.J.; Rawlings, N.D.; Woessner, J.F., Elsevier: London, 2004, pp 1559-1561; List, K.; Bugge, T.H.; Szabo, R. Mol. Med. 2006, 12, 1-7; Lee, M.-S.; Johnson, M.D.; Lin, C.-Y. J. Cancer Mol. 2006, 2, 183-190; Uhland, K. Cell. Mol. Life Sci. 2006, 63, 2968-2978; List, K. Future Oncol. 2009, 5, 97-104.)。TTSPとしてのマトリプターゼは、細胞内から分泌されるか、または細胞内に保持されるほとんどのプロテアーゼとは異なり、細胞表面に容易に接近することができ、このためワクチン、モノクローナル抗体および小分子化合物をはじめとする様々な治療薬の良好なターゲットである。その酵素を阻害すると、腫瘍形成における2種の不可欠な媒介物質、つまり肝細胞増殖因子(HGF)およびウロキナーゼ型プラスミノーゲン活性化物質(uPA)の同時阻害が得られる。HGFおよびuPAは、癌浸潤および転移における、細胞運動性、細胞外マトリックス分解および腫瘍血管分布での役割について関連づけられてきた。
【0008】
マトリプターゼ活性は、広範囲のトリプシン様セリンプロテアーゼと拮抗する活性を示す上皮クニッツ型膜貫通阻害剤である、内在性物質の肝細胞増殖因子活性化因子阻害因子(HAI−1)により調節される(Oberst, M.D.; Chen, L.-Y.L.; Kiyomiya, K.-I.; Williams, C.A.; Lee, M.-S.; Johnson, M.D.; Dickson, R.B.; Lin, C.-Y. Am. J. Physiol. 2005, 289, C462-C470; Kojima, K.; Tsuzuki, S.; Fushiki, T.; Inouye, K. J. Biol. Chem. 2008, 283, 2478-2487.)。
【0009】
マトリプターゼは、細胞外マトリックスの分解において役割を果たし、腫瘍転移を促進することが見出された(WO 00/53232; WO 01/97794; WO 02/08392; Hooper, J. Biol. Chem. 2001, 276, 857-860.)。この活性は、ストロムテリシンおよびIV型コラーゲナーゼをはじめとする特定のマトリックスメタロプロテアーゼ酵素(MMP)で認められるものと類似している。マトリプターゼ−1発現の低下は、マウスモデルの前立腺癌における攻撃性および進行の低下に関連した。(Sanders, A.J.; Parr, C.; Davies, G.; et al. J. Exp. Ther. Oncol. 2006, 6, 39-48.)
【0010】
加えて、マトリプターゼは、全身の上皮ホメオスタシスおよび末端上皮分化を担う細胞周囲タンパク質分解経路において役割を果たす(List, K.; Kosa, P.; Szabo, R.; et al. Am. J. Pathol. 2009, 175, 1453-1463.)。マトリプターゼは、PAR−2の活性化を介した、内皮細胞中の炎症性サイトカインの放出も誘導する。それゆえ阻害剤は、抗炎症薬としての用途を有するであろう。更に該プロテアーゼは、単球中で発現され、それとPAR−2との相互作用はアテローム性硬化症に寄与する。つまりマトリプターゼの阻害剤は、アテローム性硬化症の処置および予防にも用途を有する(Seitz, I.; Hess, S.; Schulz, H.; Eckl, R.; Busch, G.; et al. Arterioscler. Throm. Vasc. Biol. 2007, 27, 769-775.)。
【0011】
マトリプターゼの遺伝子発現は、骨関節炎において有意に上昇することが見出され、その酵素は、軟骨マトリックス分解をもたらす多重機構の開始に関与する(Milner, J.A.; Patel, A.; Davidson, R.K.; et al. Arthr. Rheum. 2010, 62, 1955-1966.)。それゆえその酵素の阻害は、この適応症の治療の1つのアプローチとなろう。
【0012】
マトリプターゼ−2(TMPRSS6)は、肝臓により発現されるTTSPである(WO 2008/009895; Ramsay, A.J.; Reid, J.C.; Velasco, G.; Quigley, J.P.; Hooper, J.D. Front. Biosci. 2008, 13, 569-579.)。マトリプターゼ−2は、正常な状態では、腸における鉄吸収とマクロファージからの鉄放出を阻害するホルモンのヘピシジンをダウンレギュレートする作用がある。この酵素が遺伝子内で突然変異を起こすと、高レベルのへプシジンにより、ヒトにおける鉄不応性鉄欠乏性貧血(IRIDA)に関連した、異常なタンパク質分解活性をもたらす(Folgueras, A.R.; Martin de Lara, F.; Pendas, A.M.; Garabaya, C.; et al. Blood 2008, 112, 2539-3545; Anderson, G.J.; Frazer, D.M.; McLaren, G.D. Curr. Opin. Gastroenterol. 2009, 25, 129-135; Ramsay, A.J.; Hooper, J.D.; Folgueras, A.R.; Velasco, G.; Lopez-Otin, C. Haematologica 2009, 94, 840-849; Finberg, K.E. Semin. Hematol. 2009, 46, 378-386; Cui, Y.; Wu, Q.; Zhou, Y. Kidney Intl. 2009, 76, 1137-1141; Lee, P. Acta Haematologica 2009, 122, 87-96; deFalco, L.; Totaro, F.; Nai, A.; et al. Human Mut. 2010, 31, e1390-e1405.)。この酵素は、マトリプターゼ−1に対して35%の配列相同性を有する。
【0013】
マトリプターゼ−2は、マトリプターゼ−1の作用とは異なり、乳房腫瘍増殖および浸潤を血漿レベルで阻害し、好適な予後に相関する(Parr, C.; Sanders, A.J.; Davies, G.; et al. Clin. Cancer Res. 2007, 13, 3568-3576.)。治療的アプローチとしての、癌の発生および進行ならびに調整能力におけるこの酵素の役割は、依然として活発な研究領域である(Sanders, A.J.; Webb, S.L.; Parr, C.; Mason, M.D.; Jiang, W.G. Anti-cancer Agents Med. Chem. 2010, 10, 64-69.)。マトリプターゼ−2およびそれに由来する薬剤は、前立腺癌の処置薬としても報告された(WO 2009/009895)。
【0014】
マトリプターゼ−3は、多くの種で保存され、広範囲のセルピン活性を示すが、他のTTSPに類を見ない発現パターンおよび調節ネットワークを有する(Szabo, R.; Netzel-Arnett, S.; Hobson, J.P.; Antalis, T.M. Bugge, T.H. Biochem. J. 2005, 390, 231-242.)。
【0015】
マトリプターゼ酵素に加えて、他のTTSPとしては、非限定的に、ヘプシン(TMPRSS1)、TMPRSS2、TMPRSS3/TADG−12、TMPRSS4、モザイクセリンプロテアーゼ大型(MSPL)、TMPRSS11A、ヒト気道トリプシン(HAT)様プロテアーゼ、HAT−like−2、HAT−like−3、HAT−like−4、HAT−like−5、ポリセラーゼ−1、スピネシン、エンテロペプチダーゼ、コリンおよび扁平上皮癌における差異的発現遺伝子1(differentially expressed in squamous cell carcinoma 1)(DESC1)が挙げられる。TTSP遺伝子の突然変異は、ヒトにおける複数の遺伝子障害の潜在的原因として確証されており、TTSP遺伝子発現の変化はヒト癌腫に関連する。
【0016】
プロテアーゼは、様々な有害な皮膚状態の誘発にも関与する。それは、表皮の分化(Zeeuwen, P.L.J.M.; Eur. J. Cell Biol. 2004, 83, 761-773)と、上皮の発育(Bugge, T.H.; List, K.; Szabo, R. Front. Biosci. 2007, 12, 5060-5070)の両方において役割を果たす。セリンプロテアーゼを含むシグナル伝達カスケードは、表皮のホメオスタシスにおいて重大な役割を果たす(Ovaere, P.; Lippens, S.; Vandenabeele, P.; Declercq, W. Trends Biochem. Sci. 2009, 34, 453-463.)。これらには、マトリプターゼ−1に加えて、フリン、プロスタシン、カリクレイン関連ペプチダーゼ4(KLK4、プロスターゼ)、角質層トリプシン様酵素(SCTE、カリクレイン関連ペプチダーゼ5、KLK5)、カリクレイン関連ペプチダーゼ6(KLK6、プロテアーゼM)、角質層キモトリプシン様酵素(SCCE、カリクレイン関連ペプチダーゼ7、KLK7)、カリクレイン関連ペプチダーゼ8(KLK8、ニューロプシン)、カリクレイン関連ペプチダーゼ10(KLK10)、カリクレイン関連ペプチダーゼ11(KLK11)、カリクレイン関連ペプチダーゼ13(KLK13)、カリクレイン関連ペプチダーゼ14(KLK14)が挙げられる。例えば、疾患発症中にマトリプターゼにより活性化されるプロカリクレイン経路の関与が、ネザートン症候群のマウスモデルで確認された(Sales, K.U.; Masedunskas, A.; Bey, A.L.; et al. Nat. Genetics 2010, 42, 676-683.)。これらのプロテアーゼ酵素が皮膚層を含む保護組織を破壊し始めると、それらは炎症反応を誘発する。加えて、皮膚のタンパク質分解バランスの変化が炎症をもたらして、発赤、鱗屑および痒みが生じる可能性がある。事実、フィラグリン、プロテアーゼ活性化受容体(PAR)およびコルネオデスモシンをはじめとするプロテアーゼ、その阻害物質およびターゲットタンパク質は、皮膚組織のための調節ネットワークを含み、皮膚の統合性およびバリア機能に寄与する(Meyer-Hoffert, U. Arch. Immunol. Ther. Exp. 2009, 57, 345-354.)。阻害剤は、これらの炎症事象を低減して、様々な皮膚および組織障害を処置するのに有用となろう。
【0017】
マトリプターゼは、皮膚に加えて、腸における上皮バリア形成および透過性の調節において重要な役割を果たす(Buzza, M.S.; Netzel-Arnett, S.; Shea-Donohue, T.; et al. Proc. Nat. Acad. Sci. 2010, 107, 4200-4205.)。
【0018】
プロテアーゼは、上皮ナトリウムチャネル(ENaC)の調節も担う(Planes, C.; Caughey, G.H.; Curr. Top. Development. Biol. 2007, 78, 23-46; Frateschi, S.; Charles, R.-P.; Hummler, E. Open Derm. 2010, 4, 27-35.)。ENaCの調整に関与するチャネル活性化プロテアーゼ(CAP)としては、プロスタシン(CAP1、PRSS8)、PRSS22、TMPRSS11B、TMPRSS11E、TMPRSS2、TMPRSS3、TMPRSS4、(MT−SP2)、MT−SP1、CAP2、CAP3、トリプシン、カテプシンAおよび好中球エラスターゼが挙げられる。CAPの阻害剤が、図示されたピロリジン塩基骨格の周囲を基盤とする化学構造と共に開示された(WO 2007/137080; WO 2007/140117; WO 2008/085608; WO 2008/097673; WO 2008/097676)。
【化1】

【0019】
今日まで、マトリプターゼの阻害剤は、わずかな数しか記載されていない。これらは、小分子、例えば第二級アミノ酸アミドのメタ置換スルホニルアミドを含む(WO 2008/107176; Steinmetzer, T.; Doennecke, D.; Korsonewski, M.; Neuwirth, C.; Steinmetzer, P.; Schulze, A.; Saupe, S.M.; Schweinitz, A. Bioorg. Med. Chem. Lett. 2009, 19, 67-73; Schweinitz, A.; Doennecke, D.; Ludwig, A.; Steinmetzer, P.; Schulze, A.; Kotthaus, J.;Wein, S.; Clement, B.; Steinmetzer, T. Bioorg. Med. Chem. Lett. 2009, 19, 1960-1965.)。
【化2】
【0020】
マトリプターゼ阻害剤の別の構造類は、N−スルホニル化アミノ酸誘導体に基づいている(WO 2004/101507; US 2007/0055065; Steinmetzer, T.; Schweinitz, A.; Stuerzbecher, A.; et al. J. Med. Chem. 2006, 49, 4116-4126)。
【化3】
【0021】
直鎖状ペプチド(US 6,797,504; US 7,157,596; WO 02/020475)およびペプチド模倣体(US 7,019,019; WO 2004/058688)阻害剤が、開示された。
【化4】
【0022】
これらのペプチド模倣体マトリプターゼ阻害剤の一種であるCVS−3983が、腫瘍転移のインビボモデルにおいて活性を示した(Galkin, A.V.; Mullen, L.; Fox, W.D.; Brown, J.; et al. Prostate 2004, 61, 228-235.)。
【化5】
【0023】
他の2種のペプチド模倣体阻害剤、CJ−1737およびCJ−672の代謝および分布に関する研究で、この種の分子についての動物とヒトとの重大な代謝的差異が明らかとなった(Kotthaus, J.; Steinmetzer, T.; Kotthaus, J.; Schade, D.; van de Locht, A.; Clement, B. Xenobiotica 2010, 40, 93-101.)。
【化6】
【0024】
より近年になり、4−アミジノベンジルアミドを含有するN−保護ジペプチドが、マトリプターゼ−1およびマトリプターゼ−2阻害剤として報告された(Sisay, M.T.; Steinmetzer, T.; Stirnberg, M.; Maurer, E.; Hammami, M.; Bajorath, J.; Guetschow, M. J. Med. Chem. 2010, 53, 5523-5535.)。化合物1は、マトリプターゼ−1の阻害に関し、マトリプターゼ−2の50倍の選択性を示した。これらの、マトリプターゼ−2の最初の小分子阻害剤は、鉄障害、例えば、ヘプシジンレベルが過度に低いヘモクロマトーシスおよび鉄負荷性貧血(iron loading anemias)を処置するための潜在的治療薬として示唆される。
【化7】
【0025】
エグリンC変異体であるより長い直鎖ペプチドも、マトリプターゼ阻害剤として公知である(Desilets, A.; Longpre, J.-M.; Beaulieu, M.-E.; Leduc, R. FEBS Lett. 2006, 580, 2227-2232.)。
【0026】
アミノ酸残基が14個の二環式ペプチドであるヒマワリトリプシン阻害剤(SFTI−1)が、マトリプターゼおよびカテプシンGの阻害剤として同定された。この阻害剤は、エラスターゼ、トロンビンおよびXa因子をはじめとする他のプロテアーゼ酵素に対比して選択性を有する。(Luckett, J. Mol. Biol. 1999, 290, 525.)。不幸にもSFTI−1は、インビボでは相対的に急速に分解され、重要な生理学的セリンプロテアーゼ、トリプシンおよびキモトリプシンを上回る選択性を示さず、そのため医薬剤としての使用に適さない。
【化8】
【0027】
同じく本質的に二環式であるSFTI−1類似体および模倣体も、報告された(US 7,439,226; WO 2006/043933; Long, Y.-Q.; Lee, S.-L.; Lin, C.-Y.; Enyedy, I.J.; Wang, S.; Li, P.; Dickson, R.B.; Roller, P.P. Bioorg. Med. Chem. Lett. 2001, 11, 2515-2519; Jiang, S.; Li, P.; Lee, S.-L.L.; Lin, C.-Y.; Long, Y.-Q.; Johnson, M.D.; Dickson, R.B. Roller, P.B. Org. Lett. 2007, 9, 9-12; Li, P.; Jiang, S.; Lee, S.-L.L.; Lin, C.-Y.; Johnson, M.D.; Dickson, R.B.; Michejda, C.J.; Roller, P.J. J. Med. Chem. 2007, 50, 5976-5983.)。
【0028】
アミノ酸を11または14個含有する環状ペプチド、およびマトリプターゼをはじめとするセリンプロテアーゼの阻害により作用する皮膚刺激の予防または処置のための使用方法が、US 7,217,690に開示された。
【0029】
天然および合成プロテアーゼ阻害剤(Yamasaki, Y.; Satomi, S.; Murai, N.; Tsuzuki, A.; Fushiki, T. J. Nutr. Sci. Vitamin. 2003, 49, 27-32)、ならびに合成クンツ型阻害剤(WO 2007/079096)が、マトリプターゼをはじめとする複数のプロテアーゼ酵素に拮抗する活性を示した。
【0030】
事実、特定のプロテアーゼ類のうち、該酵素は、共通の化学的および構造的特性を利用してそれらの基質と相互作用し、つまり阻害剤は、多くの場合、その分類の他の酵素も阻害することができる。もちろん酵素間の選択性が、特異的副作用を抑制するなどで重要となる場合、これは克服すべき難題も生じる。
【0031】
構造を基にした設計から得られる、図示されたアミジンまたは代替物など、2つ以上の重要な塩基性相互作用部分を分離する直鎖構造を有する一連のマトリプターゼを阻害剤が、報告された(US 6,677,377; WO 01/097784; Enyedy, I.J.; Lee, S.-L.; Kuo, A.H.; Dickson, R.B.; Lin, C.-Y.; Wang, S. J. Med. Chem. 2001, 44, 1349-1355)。これらの化合物において、Zは、1個以上の酸素または硫黄原子で場合により置換された炭素原子の直鎖、または芳香族もしくは複素環芳香族のスペーサー成分のいずれかを表す。
【化9】
【0032】
マトリプターゼを対象とするヒトモノクローナル抗体が、癌の診断、予防または処置に関して開示された(US 7,572,444; WO 2006/068975; Farady, C.J.; Sun, J.; Derragh, M.R.; Miller, S.M.; Craik, C.S. J. Mol. Biol. 2007, 369, 1041-1051; Farady, C.J.; Egea, P.F.; Schneider, E.L.; Darragh, M.R.; Craik, C.S. J. Mol. Biol. 2008, 380, 351-360.)。マトリプターゼマウス抗体を有することから(US 7,355,015)、様々なタイプの癌の処置、スクリーニング、診断、予後および治療に使用される、マトリプターゼタンパク質由来の他の抗体も、記載された(WO 2009/020645; US 2003/270245; US 2009/0155248)。マトリプターゼの検出用抗体キットは、US 7,022,821の主題である。
【0033】
診断または治療的目的で抗体を生成するのに用いうるマトリプターゼおよび他の癌関連プロテアーゼの部分的配列を含む抗原性ペプチドが、WO 2008/066749に提供されている。
【0034】
マトリプターゼ発現を刺激する薬剤が、美容目的で有用であるとして開示された(WO 2008/034821)。
【0035】
今日まで、マトリプターゼ阻害剤で臨床開発に至ったものはなく、そのため薬理学的物質としての追跡で既に研究されたものとは異なる構造を有する新規なマトリプターゼ阻害剤が、依然として求められている。
【発明の概要】
【発明が解決しようとする課題】
【0036】
発明の概要
本発明は、コンホメーションが定義された新規な大環状化合物を提供する。これらの化合物は、セリンプロテアーゼ酵素の調整物質、詳細には阻害剤として機能することができる。
【課題を解決するための手段】
【0037】
本発明の態様によれば、本発明は、式(I):
【化10】
(式中、
R1は、−H、−CH3、−CH2CH3、−(CH2)2CH3および−CH(CH3)2からなる群より選択され、
R2は、−H、−CH3および−CH2CH3からなる群より選択され、
R3は、任意で存在し、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より選択され、
mは、1、2、3、4または5であり、
X1は、アミジノ、ウレイドおよびグアニジノからなる群より選択され、
Wは、CR4aR4bからなる群より選択され、ここでR4aおよびR4bは、水素、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、
Z1は、CR5aR5bからなる群より選択され、ここでR5aおよびR5bは、水素、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、
Tは、
【化11】
からなる群より選択され、
ここでM1は、Oおよび(CH2)qからなる群より選択され、ここでqは、1、2、3、4または5であり、M2は、O、S、NR6およびCR7aR7bからなる群より選択され、ここでR6は、水素、アルキル、ホルミル、アシル、カルボキシアルキル、カルボキシアリール、アミド、スルホニルおよびスルホンアミドからなる群より選択され、R7aおよびR7bは、水素、ヒドロキシル、アルコキシ、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、p1およびp2は、独立して0、1、2または3であり、そしてp3、p4およびp5は、独立して0、1または2であり、
(W)は、Wの付加された炭素原子への結合部位を示し、
(Z)は、Z1の付加された炭素原子への結合部位を示す)
による化合物および薬学的に許容し得るその塩に関する。
【0038】
本発明の更なる態様は、式(II):
【化12】
(式中、
R11は、−H、−CH3、−CH2CH3、−(CH2)2CH3および−CH(CH3)2からなる群より選択され、
R12は、−H、−CH3および−CH2CH3からなる群より選択され、
R13は、−(CH2)r1NR18aR18b、−(CH2)r2CONR19aR19b、
【化13】
からなる群より選択され、
ここでr1は、1、2、3、4または5であり、r2は、1、2または3であり、R18a、R19aおよびR19bは、水素およびC1〜C4アルキルからなる群より独立して選択され、R18bは、水素、C1〜C4アルキル、ホルミル、アシル、アミド、アミジノおよびスルホンアミドからなる群より選択され、A1、A4、A7、A9、A12、A14、A17、A19、A23、A35、A37およびA39は、それぞれ任意で存在し、ハロゲン、トリフルオロメチル、アミジノ、ウレイド、グアニジノ、ヒドロキシル、アルコキシおよびC1〜C4アルキルからなる群より独立して選択され、A2、A3、A5、A6、A8、A10、A11、A13、A15、A16、A18、A20、A21、A24、A25、A36、A38およびA40は、それぞれ任意で存在し、ハロゲン、トリフルオロメチル、ヒドロキシル、アルコキシおよびC1〜C4アルキルからなる群より独立して選択され、A22、A26、A27、A29、A31およびA33は、それぞれ任意で存在し、トリフルオロメチル、アミジノ、ウレイド、グアニジノおよびC1〜C4アルキルからなる群より独立して選択され、A28、A30、A32およびA34は、それぞれ任意で存在し、トリフルオロメチルおよびC1〜C4アルキルからなる群より独立して選択され、そしてB1、B2、B3、B4、B5およびB7は、独立してNR20、SまたはOであり、ここでR20は、水素、アルキル、ホルミル、アシル、カルボキシアルキル、カルボキシアリール、アミド、スルホニルおよびスルホンアミドからなる群より選択され、B6およびB8は、独立してNまたはCHであり、
R14は、アミノ、ヒドロキシル、アルコキシ、カルボキシ、ウレイド、アミジノ、またはグアニジンで場合により置換されたC1〜C4アルキルと、アルキル、ヒドロキシルまたはアルコキシで場合により置換されたC3〜C7シクロアルキルとからなる群より選択され、
R15およびR16は、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より独立して選択され、
R17は、水素およびC1〜C4アルキルからなる群より選択され、
nは、1、2、3、4または5であり、
Z2は、CHR21aCHR22a、CR21b=CR22bおよびC≡Cからなる群より選択され、ここでR21aおよびR22aは、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より独立して選択されるか、またはR21aとR22aとが、結合する炭素と一緒になって、3員環を形成しており、そしてR21bおよびR22bは、水素およびC1〜C4アルキルからなる群より独立して選択され、
X2は、水素、ハロゲン、アミジノ、ウレイドおよびグアニジノからなる群より選択され、
X3は、水素、ヒドロキシル、アルコキシ、アミノ、ハロゲン、トリフルオロメチルおよびC1〜C4アルキルからなる群より選択され、
L2は、OおよびCR23aR23bからなる群より選択され、ここでR23aは、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より選択され、そしてR23bは、水素およびC1〜C4アルキルからなる群より選択され、
L3は、CX4およびNからなる群より選択され、ここでX4は、水素、ハロゲン、ヒドロキシル、アルコキシ、アミノ、ハロゲン、トリフルオロメチル、アミジノ、ウレイドおよびグアニジノからなる群より選択され、そして
L4は、CX5およびNからなる群より選択され、ここでX5は、水素、ハロゲン、トリフルオロメチル、ヒドロキシル、アルコキシ、アミノ、アミジノ、ウレイドおよびグアニジノからなる群より選択される)
による化合物または薬学的に許容し得るその塩に関する。
【0039】
本発明の新規な大環状化合物は、セリンプロテアーゼ酵素の調整物質、詳細には阻害剤として有用である。この阻害剤により、複数の異なる癌、詳細には腫瘍転移を特徴とする癌に取り組むことができる。加えてセリンプロテアーゼの阻害剤、例えば本発明の化合物を、皮膚障害、例えばアトピー性皮膚炎、酒さ、乾癬、魚鱗癬、濾泡性皮膚委縮、過角化症、減毛症、ネザートン症候群などの処置または予防に用いることができる。
【0040】
本発明の特定の実施形態において、セリンプロテアーゼ酵素は、マトリプターゼ−1(MTSP−1、ST14、TADG−15、エピチン)、マトリプターゼ−2(TMPRSS6)、マトリプターゼ−3、MTSP−4、MTSP−6、MTSP−7、MTSP−9、MTSP−10、PRSS22、TMPRSS11A、TMPRSS11C、TMPRSS2、TMPRSS3、TMPRSS4、TMPRSS5(スピネシン)、モザイクセリンプロテアーゼ大型(MSPL)、エンテロペプチダーゼ、ポリセラーゼ−1、コリン、ヒト気道トリプシン様プロテアーゼ(HAT)、HAT−like2、HAT−like3、HAT−like4、HAT−like5、プロスタシン(CAP1、PRSS8)、CAP2、CAP3、トリプシン、カテプシンA、好中球エラスターゼ、ヘプシン、角質層トリプシン様酵素(SCTE、カリクレイン関連ペプチダーゼ5、KLK5)、角質層キモトリプシン様酵素(SCCE、カリクレイン関連ペプチダーゼ7、KLK7)、カリクレイン関連ペプチダーゼ4(KLK4、プロスターゼ)、カリクレイン関連ペプチダーゼ8(KLK8、ニューロプシン)、カリクレイン関連ペプチダーゼ11(KLK11)、カリクレイン関連ペプチダーゼ13(KLK13)、カリクレイン関連ペプチダーゼ14(KLK14)、カリクレイン関連ペプチダーゼ6(KLK6、プロテアーゼM)、カリクレイン関連ペプチダーゼ10(KLK10)、グランザイムB、カルシウムシグナル伝達物質1、カルシウムシグナル伝達物質2、クローディン3、クローディン4、フリン、ラジニン、ラルニニン、プラスミン、ストラチフィン、SI00A2、CD24、リポカリン2、オステオポンチン、組織型プラスミノーゲン活性化因子、ウロキナーゼ型プラスミノーゲン活性化因子または扁平上皮癌における差異的発現遺伝子1(DESC1)である。
【0041】
本発明の化合物は、異常な新血管形成または血管新生を特徴とする病的状態にも有用である。そのような状態の例としては、非限定的に、目の新生血管疾患、血管腫、ならびに関節リウマチおよびクローン病をはじめとする慢性炎症を特徴とする障害が挙げられる。
【0042】
本発明の別の態様において、本発明の化合物は、特定の実施形態において、鉄不応性鉄欠乏性貧血(IRIDA)、全身鉄過負荷(ヘモクロマトーシス)または鉄負荷性貧血をはじめとする、鉄ホメオスタシスの調節解除を特徴とする病的状態を処置するのに用いることができる。
【0043】
本発明の更なる態様は、式(I)で示される化合物または式(II)で示される化合物と、薬学的に許容しうる担体、賦形剤または希釈剤とを含む医薬組成物を更に提供する。
【0044】
本発明の別の態様は、必要とする対象に有効量の式(I)または式(II)で示される化合物を投与することを含む、増殖過剰性障害、炎症性障害、組織障害、心臓血管障害、呼吸器障害またはウイルス感染を処置する方法を提供する。
【0045】
本発明の更なる態様は、使用するための任意の使用説明書と共に包装された有効量の本発明の化合物を1種以上含む医薬投与単位を含有する容器1個以上を含むキットを提供する。
【0046】
本発明の更なる態様は、式(I)および式(II)で示される化合物を製造する方法に関する。
【0047】
本発明の態様は、更に、本明細書に記載された障害、詳細には、病的状態、増殖過剰性障害、組織障害、炎症性障害、呼吸器障害およびウイルス感染を予防および/または処置する方法に関する。
【0048】
特定の実施形態において、増殖過剰性障害は、CMLをはじめとする白血病、リンパ腫、乳癌、胃腸癌、食道癌、胃癌(stomach cancer)、胃癌(gastric cancer)、結腸癌、腸癌、大腸癌、前立腺癌、膀胱癌、精巣癌、卵巣癌、子宮癌、子宮頸癌、子宮内膜癌、上皮癌、頭頸部癌、脳癌、肺癌、肝臓癌、腎臓癌、気管支癌、膵臓癌、甲状腺癌、骨癌および皮膚癌である。
【0049】
別の特定の実施形態において、増殖過剰性障害は、腫瘍転移を特徴とし、その腫瘍は、乳房、脳、卵巣、結腸、直腸、胃、肝臓、腎臓、腸、口、喉、食道、前立腺、精巣、膀胱、子宮、子宮頸部、肺、膵臓、骨、甲状腺または皮膚に見出される。
【0050】
別の特定の実施形態において、増殖過剰性障害は、前立腺癌、卵巣癌腫、子宮頸部新生物、小細胞肺癌、非小細胞肺癌、腎細胞癌、膵管腺癌、子宮平滑筋肉腫、移行上皮細胞腺癌、非メラノーマ皮膚癌、扁平上皮癌、悪性中皮腫または神経膠芽腫である。
【0051】
追加の実施形態において、本発明の化合物は、特定の実施形態において、アトピー性皮膚炎、酒さ、乾癬、魚鱗癬、濾泡性皮膚委縮、過角化症、減毛症、ネザートン症候群などをはじめとする組織または皮膚の障害の処置または予防に用いることができる。
【0052】
更に別の特定の実施形態において、炎症性障害は、関節リウマチ、骨関節炎、クローン病、潰瘍性大腸炎またはアテローム性硬化症である。
【0053】
更なる特定の実施形態において、病的状態は、上皮細胞増殖または異常な新血管形成を特徴とする。
【0054】
追加の特定の実施形態において、呼吸器障害は、嚢胞性線維症、気管支炎、慢性閉塞性肺疾患(COPD)、喘息、アレルギー性鼻炎、線毛機能不全、肺癌腫、肺炎または呼吸器感染である。
【0055】
更に別の特定の実施形態において、ウイルス感染は、インフルエンザウイルスまたはメタニューモウイルスにより誘発される。
【0056】
本発明は、本明細書に記載された障害の予防および/または処置用の医薬品の調製に使用される式(I)または(II)で示される化合物にも関する。
【0057】
本発明の前述および他の態様は、以下に示される記述により詳細に説明される。
【図面の簡単な説明】
【0058】
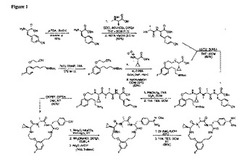
【図1】代表的な本発明の化合物を合成するための反応スキームを示す。
【図2】複数の代表的な本発明の化合物を同時合成するための反応スキームを示す。
【図3】複数の代表的な本発明の化合物を同時合成するための別の反応スキームを示す。
【図4】テザーT32を合成するための反応スキームを示す。
【図5】テザーT201を合成するための反応スキームを示す。
【発明を実施するための形態】
【0059】
ここに、本発明の前述および別の態様を、本明細書に記載された別の実施形態に関連させて、より詳細に記載する。本発明を異なる形態で具体化することができ、本明細書に示された実施形態に限定されると解釈してはならないことを、理解しなければならない。むしろこれらの実施形態を提供することで、この開示が徹底され完全となり、それが本発明の範囲を当業者に完全に伝えることになる。
【0060】
本明細書内の本発明の記載に用いられる用語法は、特定の実施形態のみの記載を目的とし、本発明を限定する意図はない。本発明の記載および添付の特許請求の範囲に用いられる単数形「a」、「an」および「the」は、他に明確に示されない限り、複数形も包含するものとする。加えて、本明細書で用いられる用語「および/または」は、関連して列挙された事柄1つ以上とのあらゆる組合せを包含しており、「/」と略されていてもよい。
【0061】
他に定義されない限り、本明細書で用いられる全ての技術的および科学的用語は、本発明の当業者により一般に理解されるものと同じ意味を有する。
【0062】
本明細書で引用された全ての発行物、米国特許出願、米国特許および他の参考資料は、その全体が参照により本明細書に組み入れられる。
【0063】
用語「アルキル」は、炭素原子を1〜20個、幾つかの例において炭素原子を1〜8個有する直鎖または分枝鎖飽和または部分不飽和炭化水素基を指す。用語「低級アルキル」は、炭素原子を1〜6個含有するアルキル基を指す。アルキル基の例としては、非限定的に、メチル、エチル、イソプロピル、tert−ブチル、3−ヘキセニル、および2−ブチニルが挙げられる。「不飽和」とは、二重結合もしくは三重結合またはその2種の組み合わせが1、2または3個存在することを意味する。そのようなアルキル基は、以下に記載される通り、場合により置換されていてもよい。
【0064】
本明細書に定義されたアルキルまたは他の炭化水素基に関連して下付き文字が用いられる場合、下付き文字は、その基が含みうる炭素原子数を指す。例えば、C2〜C4アルキルは、炭素原子が2、3または4個のアルキル基を示す。
【0065】
用語「シクロアルキル」は、環内に炭素原子を3〜15個、幾つかの例において3〜7個有する飽和または部分不飽和環状炭化水素基、および前記環状炭化水素基を含むアルキル基を指す。シクロアルキル基の例としては、非限定的に、シクロプロピル、シクロプロピルメチル、シクロペンチル、2−(シクロヘキシル)エチル、シクロヘプチル、およびシクロヘキセニルが挙げられる。本明細書で定義されたシクロアルキルとしては、複数の炭素環を有し、そのそれぞれが飽和または部分不飽和であってもよい基、例えば、デカリニル、[2.2.1]−ビシクロヘプタニルまたはアダマンタニルも挙げられる。そのようなシクロアルキル基は全て、以下に記載されたとおり場合により置換されていてもよい。
【0066】
用語「芳香族」は、電子を4n+2個(ここでnは、1以上の整数である)含む共役π電子系を有する不飽和環状炭化水素基を指す。芳香族分子は、典型的には安定しており、交互の二重結合および単結合からなる共鳴構造を有する原子の平面環、例えばベンゼンまたはナフタレンとして表わされる。
【0067】
用語「アリール」は、環原子を6〜15個、幾つかの例において6〜10個有する単環または縮合環の炭素環系の芳香族基、および前記芳香族基を含むアルキル基を指す。アリール基の例としては、非限定的に、フェニル、1−ナフチル、2−ナフチルおよびベンジルが挙げられる。本明細書で定義されたアリールは、ナフチルおよびアントラセニルと同様に縮合であってもよく、またはビフェニルおよびテルフェニルと同様に非縮合であってもよい、複数のアリール基を有する基も挙げられる。アリールは、環の1つが芳香族であり、その他が飽和、部分不飽和、または芳香族であってもよい、二環式または三環式炭素環、例えば、インダニルまたはテトラヒドロナフチル(テトラリニル)も指す。そのようなアリール基は全て、以下に記載される通り場合により置換されていてもよい。
【0068】
用語「複素環」または「複素環式」は、原子を3〜15個、幾つかの例において3〜7個有し、少なくとも1個の環の中にヘテロ原子を少なくとも1個有し、前記ヘテロ原子がO、SまたはNから選択される、飽和または部分不飽和単環、二環または三環基を指す。複素環基の各環は、各環内のヘテロ原子数の合計が4個以下であること、そして各環が炭素原子を少なくとも1個含むことを前提に、O原子を1または2個、S原子を1または2個、N原子を1〜4個含むことができる。二環式または三環式複素環基を完成する縮合環は、炭素環のみを含んでいてもよく、飽和または部分不飽和であってもよい。NおよびS原子は、場合により酸化されていてもよく、N原子は、場合により第四級化されていてもよい。複素環は、前記単環、二環または三環式複素環基を含むアルキルも指す。複素環の例としては、非限定的に、2−または3−ピペリジニル、2−または3−ピペラジニル、2−または3−モルホリニルが挙げられる。そのような複素環基は全て、以下に記載される通り場合により置換されていてもよい。
【0069】
用語「ヘテロアリール」は、環原子を5〜15個、幾つかの例において5〜10個有し、少なくとも1個の環の中にヘテロ原子を少なくとも1個有し、前記ヘテロ原子がO、SまたはNから選択される、単環または縮合環系の芳香族基を指す。ヘテロアリール基の各環は、各環内のヘテロ原子数の合計が4個以下であること、そして各環が炭素原子を少なくとも1個含むことを前提に、O原子を1または2個、S原子を1または2個、N原子を1〜4個含むことができる。二環または三環基を完成させる縮合環は、炭素原子のみを含んでいてもよく、飽和、部分不飽和または芳香族であってもよい。窒素原子の電子の孤立電子対が芳香族π電子系の完成に関与していない構造では、N原子は場合により第四級化または窒素酸化物に酸化されていてもよい。ヘテロアリールは、前記環状基を含むアルキル基も指す。単環式ヘテロアリール基の例としては、非限定的に、ピロリル、ピラゾリル、ピラゾリニル、イミダゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、チアジアゾリル、イソチアゾリル、フラニル、チエニル、オキサジアゾリル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、およびトリアジニルが挙げられる。二環式ヘテロアリールの例としては、非限定的に、インドリル、ベンゾチアゾリル、ベンゾオキサゾリル、ベンゾチエニル、キノリニル、テトラヒドロイソキノリニル、イソキノリニル、ベンゾイミダゾリル、ベンゾピラニル、インドリジニル、ベンゾフラニル、イソベンゾフラニル、クロモニル、クマリニル、ベンゾピラニル、シンノリニル、キノキサリニル、インダゾリル、プリニル、ピロロピリジニル、フロピリジニル、チエノピリジニル、ジヒドロイソインドリル、およびテトラヒドロキノリニルが挙げられる。三環式ヘテロアリール基の例としては、非限定的に、カルバゾリル、ベンゾインドリル、フェナントロリニル、アクリジニル、フェナントリジニル、およびキサンテニルが挙げられる。そのようなヘテロアリール基は全て、以下に記載される通り場合により置換されていてもよい。
【0070】
用語「ヒドロキシ」は、基−OHを指す。
【0071】
用語「アルコキシ」は、基−ORa(ここでRaは、アルキル、シクロアルキルまたは複素環である)を指す。例としては、非限定的に、メトキシ、エトキシ、tert−ブトキシ、シクロヘキシルオキシおよびテトラヒドロピラニルオキシが挙げられる。
【0072】
用語「アリールオキシ」は、基−ORb(ここでRbは、アリールまたはヘテロアリールである)を指す。例としては、非限定的に、フェノキシ、ベンジルオキシおよび2−ナフチルオキシが挙げられる。
【0073】
用語「アシル」は、基−C(=O)−Rc(ここでRcは、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールである)を指す。例としては、非限定的に、アセチル、ベンゾイル、およびフロイルが挙げられる。
【0074】
用語「アミノアシル」は、アミノ酸から誘導されたアシル基を示す。
【0075】
用語「アミノ」は、−NRdRe基(ここでRdおよびReは、水素、アルキル、シクロアルキル、複素環、アリールおよびヘテロアリールからなる群より独立して選択される)を指す。あるいはRdとReとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。
【0076】
用語「アミド」は、基−C(=O)NRfRg(ここでRfおよびRgは、水素、アルキル、シクロアルキル、複素環、アリールおよびヘテロアリールからなる群より独立して選択される)を指す。あるいはRfとRgとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。
【0077】
用語「アミジノ」は、基−C(=NRh)NRiRj(ここでRhは、水素、アルキル、シクロアルキル、複素環、アリールおよびヘテロアリールからなる群より選択され、RiおよびRjは、水素、アルキル、シクロアルキル、複素環、アリールおよびヘテロアリールからなる群より独立して選択される)を指す。あるいはRiとRjとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。
【0078】
用語「カルボキシ」は、基−CO2Hを指す。
【0079】
用語「カルボキシアルキル」は、基−CO2Rk(ここでRkは、アルキル、シクロアルキルまたは複素環である)を指す。
【0080】
用語「カルボキシアリール」は、基−CO2Rm(ここでRmは、アリールまたはヘテロアリールである)を指す。
【0081】
用語「シアノ」は、基−CNを指す。
【0082】
用語「ホルミル」は、基−C(=O)Hを指し、−CHOとも示される。
【0083】
用語「ハロ」、「ハロゲン」または「ハロゲン化物」は、それぞれフルオロ、フッ素またはフッ化物、クロロ、塩素または塩化物、ブロモ、臭素または臭化物、およびヨード、ヨウ素またはヨウ化物を指す。
【0084】
用語「オキソ」は、同じ炭素上の2個の水素原子の代わりに置換されていてカルボニル基を形成している二価基=Oを指す。
【0085】
用語「メルカプト」は、基−SRn(ここでRnは、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールである)を指す。
【0086】
用語「ニトロ」は、基−NO2を指す。
【0087】
用語「トリフルオロメチル」は、基−CF3を指す。
【0088】
用語「スルフィニル」は、基−S(=O)Rp(ここでRpは、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールである)を指す。
【0089】
用語「スルホニル」は、基−S(=O)2−Rq1(ここでRq1は、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールである)を指す。
【0090】
用語「アミノスルホニル」は、基−NRq2−S(=O)2−Rq3(ここでRq2は、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールであり、Rq3は、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールである)を指す。
【0091】
用語「スルホンアミド」は、基−S(=O)2−NRrRs(ここでRrおよびRsは、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールからなる群より独立して選択される)を指す。あるいはRrとRsとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。
【0092】
用語「カルバモイル」は、式−N(Rt)−C(=O)−ORu(式中、Rtは、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールから選択され、Ruは、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールから選択される)で示される基を指す。
【0093】
用語「グアニジノ」は、式−N(Rv)−C(=NRw)−NRxRy(式中、Rv、Rw、RxおよびRyは、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールから独立して選択される)で示される基を指す。あるいはRxとRyとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、複素環または3〜8員を形成している。
【0094】
用語「ウレイド」は、式−N(Rz)−C(=O)−NRaaRbb(式中、Rz、RaaおよびRbbは、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールから独立して選択される)で示される基を指す。あるいはRaaとRbbとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。
【0095】
用語「場合により置換された」は、任意の置換基が明確に示されていれば、該用語は、特定の基が非置換であるか、または特定の置換基で置換されていることを示すが、その場合を除けば、特定の基が非置換であるか、または適切な置換基1個以上で置換されていることを明確に示すものとする。先に定義された通り、本明細書に他に断りがなければ(例えば、特定の基が非置換であることを示すことにより)、様々な基が非置換であるか、または置換されていてもよい(即ち、それらは場合により置換されている)。
【0096】
用語アルキル、シクロアルキル、複素環、アリールおよびヘテロアリールと共に用いられる場合の、用語「置換された」は、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、ハロ、オキソ、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノ、ウレイド、および式−NRccC(=O)Rdd、−NReeC(=NRff)Rgg、−OC(=O)NRhhRii、−OC(=O)Rjj、−OC(=O)ORkk、−NRmmSO2Rnnまたは−NRppSO2NRqqRrr(ここで、Rcc、Rdd、Ree、Rff、Rgg、Rhh、Rii、Rjj、Rmm、Rpp、RqqおよびRrrは、水素、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリールまたは非置換ヘテロアリールから独立して選択され、RkkおよびRnnは、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリールまたは非置換ヘテロアリールから独立して選択される)で示される基から独立して選択される置換基により置換された基の水素原子を1個以上有するアルキル、シクロアルキル、複素環、アリールまたはヘテロアリール基を指す。あるいはRggとRhh、RjjとRkk、またはRppとRqqとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。加えて、アリールおよびヘテロアリール基に関する用語「置換された」は、シアノ、ニトロまたはトリフルオロメチルにより置換された基の水素原子を1個有することを、選択肢として包含する。
【0097】
任意の原子の通常の原子価を超えないこと、および置換により安定した化合物が得られることを前提として、置換が行われる。一般に、基の置換された形態が存在する場合、そのような置換された基は、好ましくは更に置換されないか、または置換される場合には、置換基が置換された基をわずかな数しか含まず、幾つかの例において1、2、3または4個のそのような置換基を含む。
【0098】
任意の変数が本明細書の任意の成分または任意の式中に1回を超えて出現する場合、各出現ごとのその定義は、それぞれ他の出現の際の定義とは独立している。同じく置換基および/または変数の組み合わせは、安定した化合物が得られる場合のみ、許容される。
【0099】
「安定した化合物」または「安定した構造」は、有用な純度への単離、および効能のある治療薬への配合を持続させるのに十分な堅牢性がある化合物を指す。
【0100】
用語「アミノ酸」は、当業者に公知の、一般的な天然(遺伝子コードされた)または合成アミノ酸およびその一般的誘導体を指す。アミノ酸に適用される「標準の」または「タンパク質原性」は、天然形態の遺伝子コードされた20種のアミノ酸を指す。同様にアミノ酸に適用される「非天然」または「異常」は、非天然、希少または合成アミノ酸の広範での選択、例えばHunt, S.の Chemistry and Biochemistry of the Amino Acids, Barrett, G.C., Ed., Chapman and Hall: New York, 1985に記載されたものを指す。
【0101】
アミノ酸またはアミノ酸誘導体に関連する用語「残基」は、式:
【化14】
(式中、RAAは、アミノ酸側鎖であり、そしてこの例ではn=0、1または2である)
で示される基を指す。
【0102】
ジペプチド、トリペプチドまたはより高次のペプチド誘導体に関連する用語「フラグメント」は、それぞれ2、3またはそれを超えるアミノ酸残基を含む基を指す。
【0103】
用語「アミノ酸側鎖」は、標準または非天然アミノ酸からの任意の側鎖を指し、RAAで示される。例えば、アラニンの側鎖はメチルであり、バリンの側鎖はイソプロピルであり、トリプトファンの側鎖は3−インドリルメチルである。
【0104】
用語「アゴニスト」は、タンパク質、受容体、酵素などの内在性リガンドの効果の少なくとも幾つかを複製する化合物を指す。
【0105】
用語「アンタゴニスト」は、タンパク質、受容体、酵素などの内在性リガンドの効果の少なくとも幾つかを阻害する化合物を指す。
【0106】
用語「阻害剤」は、タンパク質または酵素の活性を低減する化合物を指す。
【0107】
用語「癌性状態」は、対象が進行性の癌、例えば、白血病、リンパ腫、メラノーマ、乳癌、胃腸癌、食道癌、胃癌、結腸癌、腸癌、大腸癌、直腸癌、前立腺癌、膀胱癌、精巣癌、卵巣癌、子宮癌、子宮頸癌、脳癌、肺癌、気管支癌、喉頭癌、咽頭癌、膵臓癌、甲状腺癌、骨癌および皮膚癌を有する状態である。
【0108】
用語「チャネル活性化プロテアーゼ」またはCAPは、典型的には細胞の細胞外膜上に分泌されるが、内部に分泌することもでき、アミロライド感受性上皮ナトリウムチャネル(ENaC)の活性を刺激することができる膜結合性プロテアーゼを指す。そのようなCAPの非限定的例は、プロスタシン(PRSS**)、マトリプターゼ、CAP2、CAP3、トリプシン、PRSS22、TMPRSS2、TMPRSS3、TMPRSS4(マトリプターゼ−2)、TMPRSS11、カテプシンA、好中球エラスターゼおよびそのアイソフォームである。
【0109】
用語「腫瘍」は、非制御的細胞複製から得られる組織の異常な増殖を指す。そのような異常な増殖は、多くの場合、癌に関連する。腫瘍は、新生物とも呼ばれる。
【0110】
用語「転移」は、対象の体内の発生部位から他の一ヶ所以上の場所への癌または腫瘍の拡大を指す。
【0111】
用語「調整または調整すること」は、化合物を用いる生物学的または化学的プロセスまたはメカニズムへの影響を付与することを指す。例えば調整することで、生物学的または化学的プロセスまたはメカニズムの上昇、促進、アップレギュレート、活性化、阻害、低減、遮断、予防、遅延、脱感作、脱活性化、ダウンレギュレートなどを行ってもよい。したがって、調整する化合物は、「アゴニスト」または「アンタゴニスト」であってもよい。調整することにより影響を受ける例示的な生物学的プロセスまたはメカニズムとしては、非限定的に、受容体活性化、結合および/またはホルモン放出または分泌、鉄チャネル調節、細胞透過性、リン酸化または脱リン酸化、組織ホメオスタシス、セカンドメッセンジャーシグナル伝達ならびに遺伝子調節が挙げられる。調整することにより影響を受ける例示的な化学的プロセスまたはメカニズムとしては、非限定的に、触媒および加水分解が挙げられる。本明細書で用いられる、調整する化合物は、「調整物質」と呼ばれる。
【0112】
受容体に適用される用語「変異体」は、二量体、三量体、四量体、五量体および複数の成分を含有する他の生物学的複合体の包含を意味する。これらの成分は、同一または異なっていてもよい。
【0113】
用語「ペプチド」は、互いに共有結合する2個以上のアミノ酸で構成された化学的化合物を指す。
【0114】
用語「ペプチド模倣体」は、ペプチドを模倣するように設計されているが、活性または他の性質、例えば、溶解度、代謝安定性、経口生物学的利用度、親油性、透過性などを調整するための、ペプチドの官能基を1個以上付加または置換することによる構造的差異を含む化学的化合物を指す。これは、ペプチド結合の置換、側鎖の修飾、トランケーション、官能基の付加などを含んでいてもよい。化学構造がペプチドに由来していないが、その活性を模倣している場合には、それは多くの場合、「非ペプチド性ペプチド模倣体」と呼ばれる。
【0115】
用語「ペプチド結合」は、個々のアミノ酸が、典型的にはペプチド内で互いに共有結合しているアミド[−C(=O)−NH−]官能基を指す。
【0116】
用語「保護基」は、化学変化が分子内の他の部分で起こる時に、分子上の潜在的に反応性の官能基、例えばアミン、ヒドロキシルまたはカルボキシルが化学反応を受けるのを防ぐために用いられうる任意の化学的化合物を指す。複数のそのような保護基が、当業者に公知であり、例は、“Protective Groups in Organic Synthesis,” Theodora W. Greene and Peter G. Wuts,編 John Wiley & Sons, New York, 3rdedition, 1999 [ISBN 0471160199]に見出すことができる。アミノ保護基の例としては、非限定的に、フタルイミド、トリクロロアセチル、ベンジルオキシカルボニル、tert−ブトキシカルボニル、およびアダマンチルオキシカルボニルが挙げられる。幾つかの実施形態において、アミノ保護基は、カルバマートアミノ保護基であり、それは、アミノ基に結合するとカルバマートを形成するアミノ保護基と定義される。他の実施形態において、アミノカルバマート保護基は、アリルオキシカルボニル(AllocまたはAloc)、ベンジルオキシカルボニル(Cbz)、9−フルオレニルメトキシカルボニル(Fmoc)、tert−ブトキシカルボニル(Boc)およびα,α−ジメチル−3,5−ジメトキシベンジルオキシカルボニル(Ddz)である。より新規な窒素保護基の近年の議論について:Theodoridis, G. Tetrahedron 2000, 56, 2339-2358。ヒドロキシル保護基の例としては、非限定的に、アセチル、tert−ブチルジメチルシリル(TBDMS)、トリチル(Trt)、tert−ブチル、およびテトラヒドロピラニル(THP)が挙げられる。カルボキシル保護基の例としては、非限定的に、メチルエステル、tert−ブチルエステル、ベンジルエステル、トリメチルシリルエチルエステル、および2,2,2−トリクロロエチルエステルが挙げられる。
【0117】
用語「固相化学」は、反応成分の1つが高分子材料(以下に定義される固形担体)に共有結合している化学反応の行為を指す。固相上で化学作用を実施するための反応方法は、ペプチドおよびオリゴヌクレオチド化学の伝統的分野以外でより広範に知られ確立されている。
【0118】
用語「固形担体」、「固相」または「樹脂」は、固相化学を実行するのに用いられる機械的および化学的に安定した高分子マトリックスを指す。これは「樹脂」、「P−」または以下の記号で示される:
【化15】
【0119】
適切な高分子材料の例としては、非限定的に、ポリスチレン、ポリエチレン、ポリエチレングリコール、ポリスチレンにグラフト化または共有結合されたポリエチレングリコール(PEG−ポリスチレン、TentaGel(商標)とも呼ばれる。Rapp, W.; Zhang, L.; Bayer, E.、Innovations and Perspectives in Solid Phase Synthesis. Peptides, Polypeptides and Oligonucleotides; Epton, R., Ed. p 205; 英国バーミンガムのSPCC Ltd.)、ポリアクリラート(CLEAR(商標))、ポリアクリルアミド、ポリウレタン、PEGA[ポリエチレングリコールポリ(N,N−ジメチルアクリルアミド)コポリマー、Meldal, M. Tetrahedron Lett. 1992, 33, 3077-3080]、セルロースなどが挙げられる。これらの材料は、架橋結合を形成して構造を機械的に安定化させる追加の化学剤、例えば、ジビニルベンゼン(DVB、通常は0.1〜5%、好ましくは0.5〜2%)と架橋したポリスチレンを場合により含有していてもよい。この固形担体は、更なる誘導体化または反応のために、非限定的例として、アミノメチルポリスチレン、ヒドロキシメチルポリスチレン、ベンズヒドリルアミンポリスチレン(BHA)、メチルベンズヒドリルアミン(MBHA)ポリスチレン、および遊離化学的官能基、最も典型的には−NH2または−OHを含む他の高分子バックボーンを含むことができる。その用語は、これらの官能基を高率で(増量して)有する「ウルトラレジン(Ultraresins)」、例えばポリエチレンイミンと架橋分子とから調製されたものを含むことも意味する(Barth, M.; Rademann, J. J. Comb. Chem. 2004, 6, 340-349)。合成の最後には、樹脂は典型的には廃棄されるが、Frechet, J.M.J.; Haque, K.E. Tetrahedron Lett. 1975, 16, 3055に記載される通り、再使用しうることが示されている。
【0120】
一般に樹脂として用いられる材料は、不溶性高分子であるが、特定の高分子は、溶媒に応じて溶解度差(differential solubility)を有し、固相化学で用いることができる。例えばポリエチレングリコールは、化学反応を実施しうる多くの有機溶媒に可溶性であり、他のもの、例えばジエチルエーテルには不溶性であるため、この手法で用いることができる。つまり、均質に溶解して反応を実施し、その後、ジエチルエーテルの添加により高分子上での生成物を沈殿させて、固体として処理することができる。これは、「液相」化学と呼ばれてきた。
【0121】
固相化学に関連して用いられる用語「リンカー」は、典型的には固形担体から基質を遊離(切断)させるために、固形担体に共有結合して担体と基質の間を付加させる化学基を指す。しかしそれは、固形担体への結合に安定性を付与するため、または単にスペーサー要素として、用いることもできる。多くの固形担体は、既に付加されたリンカーと共に市販されている。
【0122】
アミノ酸に用いられる略語およびペプチドの名称は、J. Biol. Chem. 1972, 247, 977-983中のIUPAC-IUB Commission of Biochemical Nomenclatureの規則に従う。この文書は、改訂された:Biochem. J., 1984, 219, 345-373; Eur. J. Biochem., 1984, 138, 9-37; 1985, 152, 1; Internat. J. Pept. Prot. Res., 1984, 24, p 84以降; J. Biol. Chem., 1985, 260, 14-42; Pure Appl. Chem., 1984, 56, 595-624; Amino Acids and Peptides, 1985, 16, 387-410; およびBiochemical Nomenclature and Related Documents, 2nd edition, Portland Press, 1992, pp 39-67。規則の追加分は、JCBN/NC-IUB Newsletter 1985, 1986, 1989に発表されており;Biochemical Nomenclature and Related Documents, 2nd edition, Portland Press, 1992, pp 68-69を参照されたい。
【0123】
用語「有効量」または「効果的」は、臨床検査および評価、患者観察などを介して記録された疾患もしくは障害の症状を緩和させる用量、ならびに/または生物活性もしくは化学活性の検出可能な変化を起こす用量を示すものとする。関連するメカニズムまたはプロセスについて、検出可能な変化は、当業者により検出され、そして/または更に定量されてもよい。当該技術分野で一般に理解される通り、投与量は、投与経路、症状および患者の体重に応じて変動するが、投与される化合物にも依存する。
【0124】
2種以上の化合物の「併用」での投与は、一方の存在が他方の生物学的効果を変化させるのに十分、時間的に接近させて2種の化合物を投与することを意味する。2種の化合物を、同時に(同時的に(concurrently))または連続して投与することができる。同時投与は、投与前に化合物を混合することにより、または異なる解剖学的部位で、もしくは異なる投与経路を用いて、該化合物を同じ時点で投与することにより、実施することができる。本明細書で用いられる語句「同時的投与」、「併用投与」、「同時投与」または「同時に投与される」は、該化合物を互いに同じ時点で、または互いに直後に投与することを意味する。後者の例では、観察された結果が化合物を同じ時点で投与する場合に実現される結果と識別不能な程、十分に接近した時間に2種の化合物が投与される。
【0125】
用語「薬学的に活性の代謝産物」は、特定の化合物の体内での代謝を介して生成される薬理学的活性生成物を意味するものとする。
【0126】
用語「溶媒和物」は、生物学的有効性を保持する特定の化合物の薬学的に許容しうる溶媒和物形態を意味する。溶媒和物の例は、非限定的に、水、イソプロパノール、エタノール、メタノール、DMSO、酢酸エチル、酢酸またはエタノールアミンと併用される本発明の化合物が挙げられる。
【0127】
1.化合物
本発明の新規な大環状化合物は、環化を受けて該大環状化合物を形成する、テザー成分を含むビルディングブロック構造を含む大環状化合物を包含する。ビルディングブロック構造は、アミノ酸(標準および非天然)、ヒドロキシ酸、ヒドラジノ酸、アザアミノ酸、特定の部分、例えば、ペプチド代理物およびイソステレスの導入に役割を果たすもの、および本明細書に記載されたテザー成分を含むことができる。
【0128】
本発明は、単離された化合物を包含する。単離された化合物は、幾つかの実施形態において、混合物の化合物を少なくとも10%、少なくとも25%、少なくとも50%または少なくとも70%含む化合物を指す。幾つかの実施形態において、該化合物、薬学的に許容しうるその塩または該化合物を含有する医薬組成物は、ヒトグレリン受容体で生物学的アッセイで検査した場合に、統計学的に有意な結合および/またはアンタゴニスト活性を示す。
【0129】
固体の化合物、塩、または溶媒和物の例では、本発明の化合物、塩、および溶媒和物が異なる血漿または多形形態で存在することができ、その全てが本発明および特定の式の範囲内であることは、当業者に理解されよう。
【0130】
本明細書に開示された化合物は、不斉中心を有していてもよい。本発明の化合物は、単一の立体異性体、ラセミ体、ならびに/または鏡像異性体および/もしくはジアステレオマーの混合物として存在してもよい。そのような単一の立体異性体、ラセミ体、およびその混合物は全て、本発明の範囲内のものとする。しかし特定の実施形態において、本発明の化合物は、光学的に純粋な形態で用いられる。本明細書で用いられる用語「S」および「R」形態は、IUPAC 1974 Recommendations for Section E, Fundamentals of Stereochemistry (Pure Appl. Chem. 1976, 45, 13-30)に定義された通りである。
【0131】
特定の配向があることが他に示されない限り、本発明は、全ての立体異性体形態で説明される。該化合物は、単一の立体異性体または立体異性体混合物として調製してもよい。非ラセミ形態を、合成または分割のいずれかにより得てもよい。標準的技術、例えば塩形成を介したジアステレオマー対の形成により、該化合物を成分の鏡像異性体に分割してもよい。同じく、キラル部分への共有結合により、該化合物を分割してもよい。その後、クロマトグラフィー分離および/または結晶学的分離により、ジアステレオマーを分割してもよい。キラル補助部分の場合には、その後、それを除去することができる。代替法として、キラルクロマトグラフィーの利用により、該化合物を分割することができる。分割の酵素的方法も、特定の例で用いることができる。
【0132】
当業者に一般に理解される通り、「光学的に純粋な」化合物は、単一鏡像異性体のみを含むものである。本明細書で用いられる用語「光学活性」は、混合物が平面偏光を回転させるように、1つの鏡像異性体を他のものよりも少なくとも十分に過剰に含む化合物を意味するものとする。光学活性化合物は、偏光の平面を回転する能力を有する。他よりも過剰な1つの鏡像異性体は、典型的には鏡像異性体過剰(e.e.)として表わされる。光学活性化合物を記載する際に、接頭辞DとL、またはRとSは、キラル中心の周りの分子の絶対配置を示すのに用いられる。接頭辞「d」と「l」、または「+」と(−)は、化合物の回旋(即ち、偏光面が光学活性化合物により回転する方向)を示すのに用いられる。「l」または(−)の接頭辞は、化合物が左旋性(即ち、偏光面を左または反時計回りに回転させる)であることを示し、「d」または(+)の接頭辞は、化合物が右旋性(即ち、偏光面を右または時計回りに回転させる)であることを意味する。回旋記号(−)および(+)は、分子の絶対配置RおよびSには関係しない。
【0133】
所望の薬理学的性質を有する本発明の化合物は、光学活性であり、少なくとも90%(80%e.e.)、少なくとも95%(90%e.e.)、少なくとも97.5%(95%e.e.)または少なくとも99%(98%e.e.)の単一異性体で構成させることができる。
【0134】
同様に、二重結合などの多くの幾何異性体も、本明細書に開示された化合物中に存在することができ、他に断りがなければ、そのような安定した異性体は全て、本発明に包含される。同じく本発明に包含されるのは、該化合物の互変異性体および回転異性体である。
【0135】
次の右側の記号の使用は、示された環の水素原子1個以上が定義された置換基Rで置換されていることを指す。
【化16】
【0136】
次の記号の使用は、単結合または任意の二重結合:----を示す。
【0137】
本発明の実施形態は、本明細書に記載された合成方法により形成されて、式Iおよび/またはIIで示される化合物を提供する中間体化合物を更に提供する。中間体化合物は、本明細書に記載された一定範囲の適応症の治療薬として、そして/または更なる方法および反応の試薬としての用途を有していてもよい。
【0138】
2.合成方法
本発明の化合物を、伝統的な溶液合成技術または固相化学法を用いて合成することができる。いずれかにおいて、構築は4つの相を含み:最初は、コンホメーションの制御および定義のための、生物学的ターゲット受容体の認識要素と1つのテザー部分とを含むビルディングブロックの合成である。これらのビルディングブロックは、標準の化学的変換を用いる第二相において、典型的には連続様式で、一緒に組み立てられる。アセンブリの前駆体を、その後、第三段階で環化して、大環状構造を提供する。最後に、保護基の除去および任意の精製を含む、環化後工程の第四段階は、所望の最終化合物を提供する。この一般的タイプの大環状構造の合成方法は、WO 2004/111077およびWO 2005/012331に記載された精製手順を含み、国際特許出願WO 01/25257、WO 2004/111077、WO 2005/012331、WO 2005/012332、WO 2008/033328およびWO2008/130464に記載されている。米国特許第7,476,653および同第7,491,695も参照されたい。
【0139】
本発明の幾つかの実施形態において、先に定義された可溶性または不溶性高分子マトリックス上の固相化学を利用して、大環状化合物を合成してもよい。固相化学では、樹脂への「負荷」とも呼ばれる、第一のビルディングブロックの付加を含む予備的段階を、実施しなければならない。本発明で用いられる樹脂は、優先的にリンカー部分Lを付加している。これらのリンカーは、該技術分野で公知の標準的反応法、例えばエステルまたはアミド結合の形成のために開発された多数の反応条件のいずれかを介して、塩基樹脂上の適切な遊離化学的官能基、通常はアルコールまたはアミンに付加させるが、他の物質も可能である。本発明のための幾つかのリンカー部分が、一般に「環化遊離(cyclization−release)」と呼ばれるプロセスで、樹脂から同時に切断されて大環状化合物を形成するように設計されている(van Maarseveen, J.H. Solid phase synthesis of heterocycles by cyclization/cleavage methodologies. Comb. Chem. High Throughput Screen. 1998, 1, 185-214; Ian W. James, Linkers for solid phase organic synthesis. Tetrahedron 1999, 55, 4855-4946; Eggenweiler, H.-M. Linkers for solid-phase synthesis of small molecules: coupling and cleavage techniques. Drug Discovery Today 1998, 3, 552-560; Backes, B.J.; Ellman, J.A. Solid support linker strategies. Curr. Opin. Chem. Biol. 1997, 1, 86-93.本発明の化合物ではこれに関連して特に有用なものが、3−チオプロピオン酸リンカーである (Hojo, H.; Aimoto, S. Bull. Chem. Soc. Jpn. 1991, 64, 111-117; Zhang, L.; Tam, J. J. Am. Chem. Soc. 1999, 121, 3311-3320.)。
【0140】
そのようなプロセスは、環状生成物のみが固形担体から遊離し、溶液相で起こるような直鎖状前駆体の混入が生じないため、より高い純度の材料を提供する。公知または標準の反応化学を利用してビルディングブロックおよびテザーの全てを直鎖状前駆体に連続して組み立てた後、テザー・ビルディングブロックの一部である適切な求核官能基により、このリンカーに付加されたカルボニル上での塩基介在性の分子内攻撃が、アミドまたはエステル結合を形成して図示された環状構造を完成させる(スキーム1)。溶液相に適合する類似の方法論は、大規模適用に好ましいと思われるため、それを適用することも可能である。
【0141】
スキーム1.環化−遊離ストラテジー
【化17】
【0142】
この記載は、本発明の方法の1つの経路を正確に表わしているが、テザー成分を実際に環化ステップで組み立てる改変経路では、閉環メタセシス(RCM)の1種である本発明の別法チオエステルストラテジーが進行する。しかし、RCM方法論でも、ビルディングブロックの組立が連続して進行した後、環化される(そして固相であれば樹脂から遊離する)。RCM反応の特定の副生成物を除去するのに、追加の環化後処理ステップが必要となるが、次の残りの工程は、チオエステルまたは類似の塩基介在性環化ストラテジーと同様の手法で実施される。
【0143】
その上、本明細書に提供された方法を含むステップを、独立して実施してもよく、または少なくとも2つのステップを組み合わせてもよいことは、理解されよう。加えて、本明細書に提供された方法を含むステップは、独立して、または組み合わせて実施される場合、同じ温度または異なる温度で実施してもよく、それは本発明の教示から逸脱しない。
【0144】
本発明の新規な大環状化合物は、ビルディングブロック構造の環化を含む新規なプロセスにより形成されて、本明細書に記載されたテザー成分を含む大環状化合物を形成するものを包含する。したがって本発明は、(a)ビルディングブロック構造を組み立てること、(b)ビルディングブロック構造を化学変換すること、(c)テザー成分を含むビルディングブロック構造を環化させること、(d)保護基をビルディングブロックから除去すること、および(e)ステップ(d)から得られた生成物を任意で精製すること、を含む、本発明の化合物を製造する方法を提供する。幾つかの実施形態において、ビルディングブロック構造の組み立ては、連続的であってもよい。更なる実施形態において、合成方法は、伝統的な溶液合成技術または固相化学技術を用いて実施される。
【0145】
A.概論
他に断りがなければ、試薬および溶媒は、試薬品質以上のものであり、様々な販売業者から得て使用した。用いられたDMF、DCM(CH2Cl2)、DME、CH3CNおよびTHFは、(i)脱保護、(ii)樹脂キャッピング反応、および(iii)洗浄、以外は、DriSolv(登録商標)(ドイツ、ダルムスタットのMerck KGaAの一部であるEMD Chemicals, Inc.)のものであるか、または合成等級である。アミノ酸(AA)カップリング反応に用いられるNMPは、分析等級である。DMFは、使用前に真空下に最短で30分間配置することにより、適宜、脱気した。Strem Chemicals, Inc. (米国マサチューセッツ州、ニューベリーポート)から、均一触媒を得た。N−メチルおよび非天然アミノ酸のものを含む、Cbz−、Boc−およびFmoc−保護アミノ酸および側鎖保護誘導体は、販売業者から得るか、または当業者に公知の標準的方法論により合成した。Ddzアミノ酸は、標準法により合成するか、またはOrpegen (ドイツ、ハイデルベルグ)もしくはAdvanced ChemTech (米国ケンタッキー州ルイビル)から市販されていた。Bts−アミノ酸は、確立された手順により合成した。ヒドロキシ酸は、販売業者から得るか、または文献に記載された通り対応するアミノ酸から合成した(Tetrahedron 1989, 45, 1639-1646; Tetrahedron 1990, 46, 6623-6632; J. Org. Chem. 1992, 57, 6239-6256.; J. Am. Chem. Soc. 1999, 121, 6197-6205)。蛍光指示薬を含むシリカゲル60F254のプレコートプレート(0.25mm厚)で、分析TLCを実施し、示された方法および試薬を用いて、例えば紫外線(UV)および/またはモリブデン酸セリウム(CMA)溶液(硫酸100mL、硫酸セリウムアンモニウム10gおよびモリブデン酸アンモニウム25gを混合することにより調製)を用いて、視覚化した。
【0146】
用語「減圧濃縮/蒸発/除去」は、除去される溶媒に応じて適宜、水吸引圧下(典型的には10〜30トル)、または機械的油圧真空ポンプにより提供されるより強い真空下(「高真空」、典型的には1トル以下)で、ロータリーエバポレータを利用して溶媒および揮発成分を除去することを示す。「真空」または「高真空」下での化合物の乾燥は、低圧(1トル以下)の油圧真空ポンプを用いた乾燥を指す。シリカゲル60(230〜400メッシュ、EMD Chemicals, Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923-2925)を用いて「フラッシュクロマトグラフィー」を実施したが、それは当業者に周知の手順である。「ドライパック」は、溶媒での前処理をしていないシリカゲルのクロマトグラフィーを示しており、一般に所望の生成物と任意の不純物の間で、Rfに大きな差が存在する場合の精製に大規模に適用される。固相化学のプロセスでは、「標準的手法で乾燥」は、樹脂を、最初は風乾させ(1時間)、続いて完全な乾燥に至るまで真空乾燥(通常は油圧ポンプ)させる(約30分〜一晩)ことである。空気および水感受性反応で用いられるガラス製品は、オーブンで少なくとも一晩乾燥させて、使用前にデシケータ内で冷却させた。
【0147】
B.アミノ酸
N−メチルおよび非天然アミノ酸のものを含む、アミノ酸、Boc−およびFmoc−保護アミノ酸、ならびに側鎖保護誘導体は、販売業者から得るか[例えば、Advanced ChemTech(米国ケンタッキー州ルイビル)、Astatech (米国ペンシルバニア州ブリストル)、Bachem(スイス、ブーベンドルフ)、ChemImpex (米国イリノイ州ウッドデール)、Novabiochem(Merck KGaAの子会社、ドイツのダルムシュタット)、PepTech(米国マサチューセッツ州バーリントン)、 Synthetech (米国オレゴン州アルバニー)]、または当業者に公知の標準的方法論により合成した。Ddz−アミノ酸は、Orpegen (ドイツ、ハイデルベルグ)もしくはAdvanced ChemTech (米国ケンタッキー州ルイビル)から購入するか、またはDdz−OPhもしくはDdz−N3を用いて標準法を利用して合成した(Birr, C.; Lochinger, W.; Stahnke, G.; Lang, P. Justus Liebigs Ann. Chem. 1972, 763, 162-172.)。Bts−アミノ酸は、公知の方法により合成した(Vedejs, E.; Lin, S.; Klapara, A.; Wang, J. J. Am. Chem. Soc. 1996, 118, 9796-9797. 同じくWO 01/25257, WO 2004/111077)。加えて、N−アルキルアミノ酸誘導体を、文献の方法により入手した(Hansen, D. W., Jr.; Pilipauskas, D. J. Org. Chem. 1985, 50, 945-950.)。
【0148】
C.テザー
テザーは、過去に国際特許出願WO 01/25257、WO 2004/111077、WO 2005/012331、WO 2008/033328およびWO2008/130464に記載された方法で得た。米国特許第7,476,653および同第7,491,695も参照されたい。更なるテザーは、米国特許仮出願第61/256,727に記載されている。更なるテザーの調製は、実施例に示されている。
【0149】
以下は、本発明の化合物の合成で用いられる特定のテザー中間体であり、PGは、窒素保護基、例えば非限定的に、Boc、Fmoc、Ddz、CbzまたはAllocを示す:
【化18】
【0150】
D.固相および溶液相技術
本発明の大環状化合物を合成するための特定の固相技術は、WO 01/25257、WO 2004/111077、WO 2005/012331、WO 2005/012332、WO 2008/033328、WO 2008/130464および米国特許仮出願61/256,727に記載された。大規模製造を受け入れられる方法を含む溶液相合成経路は、米国特許出願第2006/025566およびUS 2007/0021331に記載された。
【0151】
以下の表は、標準法を用いた代表的な本発明の化合物の合成に使用されるビルディングブロックに関する情報を提供している。これらは、固相合成に直接適用可能である。溶液相合成では、典型的には例示のものからの改変された保護ストラテジーにより、収束的アプローチの使用が可能になる。代表的な本発明の大環状化合物を溶液相で構築するための追加的な合成の詳細を、実施例に示している。
【0152】
表の合成では、実施例9Bに概説された方法論を用いた。テザー上にアミジン部分を有する化合物では、実施例8Hに記載された通り例示された代替的なストラテジーを用いることもできる。
【表1A】
【表1B】
【0153】
3.分析法
他に断りがなければ、1Hおよび13C NMRスペクトルを、Varian Mercury 300MHz分光計(カリフォルニア州パロアルトのVarian, Inc.)で記録し、溶媒の残留プロトンシグナルに関して内部参照する。以下の通り、標準的な略語を用いて、1H NMRデータを表す:ppmでの化学シフト(δ)(多重度、積分値、結合定数)。シグナルの多重度を示すために、以下の略語を用いた:s=一重線、d=二重線、t=三重線、q=四重線、quint=五重線、bまたはbr=ブロード、およびm=多重線。溶液中の分子のコンホメーションについての情報は、当業者に公知の適切な二次元NMR技術を用いて決定することができる(Martin, G.E.; Zektzer, A.S. Two-Dimensional NMR Methods for Establishing Molecular Connectivity: A Chemist's Guide to Experiment Selection, Performance, and Interpretation, John Wiley & Sons: New York, 1988, ISBN 0471187070.)。
【0154】
XTerra(登録商標)MS C18カラム(または同等物)4.6×50mm(3.5μm)および指定の勾配法を用いて、1mL/分で運転したWater Alliance(登録商標)システム2695で、HPLC分析を実施した。Waters 996 PDAにより、純度評価用のUVデータを提供した(マサチューセッツ州ミルフォードのWaters Corporation)。特定の分析では、LCPackings(カリフォルニア州サニーベールのDionex Corporation)スプリッタ(50:40:10)により、流れを3つの部分に分離させた。第一の部分(50%)は、同一性を確認するために質量分析計(APCIプローブを備えたMicromass(登録商標)Platform II MS)へ給送した。第二の部分(40%)は、純度評価のために、蒸発光散乱検出器(ELSD, Polymer Laboratories, 現在はカリフォルニア州パロアルトのVarian Inc.,の一部、PL−ELS−1000(商標))へ送られ、最後の部分(10%)は、定量および純度評価のために、化学発光窒素検出器(CLND, Antek(登録商標)Model 8060、テキサス州ヒューストンのAntek Instruments(ジョージア州ドゥルースのRoper Industries, Inc.,の一部))へ送られた。必要とされる分析の性質に応じて、各検出器を別個に用いることもできる。最も新しいバージョンのWaters Millennium(登録商標)ソフトウエアパッケージを用いて、データを獲得および処理した。
【0155】
本発明の化合物の分析に用いられた代表的な標準HPLC条件を、以下に表わす:
典型的なクロマトグラフィー条件
カラム: XTerra PR18、3.5μm、4.6×100mm(または同等物)
検出(PDA): 220〜320nm
カラム温度: 35±10℃
注入容量: 10μL
流速: 1mL/分
運転時間: 20.0分
データ獲得時間:17.0分
移動相A: メタノール(またはアセトニトリル)
移動相B: 水
移動相C: 水中の10%TFA
勾配A4
【表2】
勾配B4
【表3】
【0156】
以下の表は、代表的な本発明の化合物のHPLC保持時間を要約している。
【表4】
【0157】
鏡像異性体およびジアステレオマーの純度を、Waters Breezeシステム(または同等物)を用いた適切なキラルHPLCカラムを用いて評価した。他のパッキング材料を用いることができるが、これらの分析に特に有用なカラムは、Chiralpak AS−RHおよびChiralcel OD−RH (米国ペンシルバニア州ウエストチェスターのChiral Technologies)である。
【0158】
XTerra(登録商標)MS C18カラム(または同等物)19×100mm(5μm)でのWaters FractionLynx(登録商標)システムを用いて、最終的な脱保護大環状化合物で分取HPLC精製を実施した。2mL/分で運転するWaters 2767インジェクタ/コレクタおよびWaters 515ポンプを有するアットカラム希釈構成を利用して、注入を実施した。質量分析計、HPLC、および質量分析指向(mass directed)フラクションコレクションを、FractionLynx (登録商標)を含むMassLynx(登録商標)ソフトウエアバージョン3.5で制御する。MS分析により示された、生成物を含むフラクション(13×125mm試験管)を、最も典型的には遠心エバポレータシステム(Genevac(登録商標)HT−4 (ニューヨーク州バレーコテージのGenevac Inc), ThermoSavant Discovery(登録商標)、SpeedVac(登録商標)もしくは同等物 (マサチューセッツ州ウォルサムのThermo Electron Corporation)で、減圧蒸発するか、または別法として凍結乾燥させた。その後、化合物を、同一性確認、純度および定量評価のために、LC−MC−UV−ELSD−CLND分析により完全に分析した。
【0159】
試料16本まで同時に運転することが可能なディスポーザブルシリカまたはC18カートリッジを有するIsco CombiFlash(登録商標)16xシステム (ネブラスカ州リンカーンのTeledyne Isco, Inc.)で、自動中圧クロマトグラフィー精製を実施した。Waters Micromass(登録商標)Platform IIまたはZQ(商標)システムで、MSスペクトルを記録した。VG Micromass ZAB−ZF分光計で、HRMSスペクトルを記録した。ActivityBase(登録商標)データベースソフトウエア (英国サリー州ギルフォードのID Business Solutions Ltd.)を用いて、化学的および生物学的情報を保存および分析した。
【0160】
下表は、代表的な本発明の化合物の分析データを表す。
【表5A】
【表5B】
【0161】
3.生物学的方法
本発明の化合物を、セリンプロテアーゼ酵素と相互作用する能力に関して評価することができる。そのような方法は十分に確立されており、当業者に公知である。加えて、マトリプターゼの活性を、時間領域近赤外蛍光(NIRF)イメージングを用いて特異的に検査して、阻害活性のインビトロおよびインビボ評価を実施することができる(Napp, J.; Dullin, C.; Mueller, F.; et al. Int. J. Cancer 2010, 127, 1958-1974.)。腫瘍中のマトリプターゼ−1の活性をイメージングする類似の方法は、蛍光顕微鏡法および標識抗体の使用を含む(Darragh, M.R.; Schneider, E.L.; Lou, J.; et al. Canc. Res. 2010, 70, 1505-1512.)。マトリプターゼ−1をコードするSt14遺伝子を欠損した遺伝子変性マウスは、該酵素の調整の効果を検討するための動物モデルを提供する。List, K.; Kosa, P.; Szabo, R.; Bey, A.L.; Wang, C.B.; Molinolo, A.; Bugge, T.H. Am. J. Pathol. 2009, 175, 1453-1463.)。
【0162】
A.阻害アッセイ
セリンプロテアーゼ酵素の阻害レベルを研究するのに、複数の文献での方法を利用することができる。一例として(Sisay, M.T.; Steinmetzer, T.; Stirnberg, M.; et al. J. Med. Chem. 2010, 53, 5523-5535)、HEK−MT2細胞のコンディション培地中のマトリプターゼ−1またはマトリプターゼ−2の活性、マトリプターゼ−2の精製された触媒ドメインの活性、および組換えマトリプターゼの活性(触媒ドメイン;ドイツ、レラハのEnzo Life Sciences)を、Cary 100UV−VIS分光光度計(ドイツダルムスタットのVarian)を用いて、37℃のTris生理食塩緩衝液(50mM Tris、150mM NaCl、pH8.0)中で、色素生成基質Boc−Gln−Ala−Arg−パラニトロアニリド(スイス、ブッベンドルフのBachem)からのパラニトロアニリンの遊離を405nmでモニタリングすることによりアッセイする。三重実験での8種の異なる基質濃度で、Km値を決定する。3種(マトリプターゼ−2の場合)または少なくとも5種(他の実験)の異なる阻害剤濃度での二重また三重測定で、阻害アッセイを実施する。式v = v0/(1+[I]/IC50)による非線形回帰により、IC50値を得た。その後、1〜4およびレイペプチン(ドイツ、ダルムスタットのCalbiochem) の10mM阻害剤原液ならびにBoc−Gln−Ala−Arg−パラニトロアニリドの100mM原液をDMSO中で調製し、アプロチニン (ドイツ、カールスルーエのCarl Roth) の1mM原液をH2O中で調製する。基質の最終濃度は400μMであり、DMSOの最終濃度は1.5%であった。予め加温したアッセイ緩衝液979μLを含むキュベットに、検査試料溶液11μLおよび基質溶液4μLを添加して、完全に混合する。酵素溶液(HEK−MT2細胞のコンディション培地の総タンパク質 5μg/6μL;マトリプターゼ−2の精製された触媒ドメイン 28ng/6μL;マトリプターゼ 3ng/6μL)6μLを添加することにより反応を開始させて、20分間追跡した。
【0163】
代表的な本発明の化合物による、代表的なセリンプロテアーゼのマトリプターゼ−1の阻害を決定する別の方法の使用が、以下の実施例に示されている。
【0164】
B.代表的な本発明の化合物の薬物動態分析
本発明の化合物の薬物動態的挙動を、当業者に周知の方法により確認することができる(Wilkinson, G. R. “Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination” in Goodman & Gilman's The Pharmacological Basis of Therapeutics, Tenth Edition, Hardman, J.G.; Limbird, L.E., Eds., McGraw Hill, Columbus, OH, 2001, Chapter 1.)。以下の方法を用いて、本発明の化合物の静脈内、皮下および経口投与の薬物動態パラメータ(排出半減期、総血漿クリアランスなど)を検査した。国際特許出願WO 2008/033328およびWO 2008/130464 ならびに米国特許第7,476,653および同第7,491,695も参照されたい。
【0165】
C.癌および転移モデル
全てのタイプの癌を処置する本発明の化合物のインビボ有効性を決定するのに、莫大な数の異なる動物モデルを入手することができる。これらには、非限定的に、マウスモデル(Cespedes, M.V.; Casanova, I.; Parreno, M.; Mangues, R. Clin. Transl. Oncol. 2006, 8, 318-329)、ヒト異種移植片モデル(Kerbel, R.S. Cancer Biol. Ther. 2003, 2, S134-S139)、遺伝子組換えマウスモデル(Walrath, J.C.; Hawes, J.J.; Van Dyke, T.; Reilly, K.M. Adv. Cancer Res. 2010, 106, 113-164)およびげっ歯類の転移モデル(Eccles, S.A.; Box, G.; Court, W.; Sandle, J.; Dean, C.J. Cell. Biophys. 1994, 279-291; Hoffman, R.M. Invest. New Drugs 1999, 17, 343-359.Man, S.; Munoz, R.; Kerbel, R.S. Cancer Metastasis Rev. 2007, 26, 737-747)が挙げられる。本発明の化合物に適用可能な幾つかの特定の方法を、実施例に表わす。
【0166】
D.皮膚疾患モデル
皮膚および組織障害を処置する本発明の化合物の効果を研究するための動物モデル、詳細にはげっ歯類を、入手することができる(Magin, T.M. Exp. Dermatol. 2004, 13, 659-660.)。炎症性皮膚疾患の遺伝子修飾マウスモデルは開発されており、該化合物の有効性を検証しうる他の系を提供する(Haase, I.; Pasparakis, M.; Krieg, T. J. Dermatol. 2004, 31, 704-719.)。
【0167】
E.炎症性疾患モデル
炎症性障害の処置における本発明の化合物の用途を決定するために、それらを適切な動物疾患モデルで試験することができる(Brodmerkel, C.M.; Vaddi, K. Curr. Opin. Biotechnol. 2003, 14, 652-658.)。関節リウマチ用(Hegen, M.; Keith, J.C. Jr.; Collins, M.; Nickerson-Nutter, C.L. Ann. Rheum. Dis. 2008, 67, 1505-1515)、骨関節炎用(Bendele, A.M. J. Musculoskelet. Neuronal. Interact. 2001, 1, 363-376; van den Berg, W.B. Curr. Rheumatol. Rep. 2008, 10, 26-29)、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎用(Wirtz, S.; Neurath, M.F. Int. J. Colorectal. Dis. 2000, 15, 144-160; Wirtz, S.; Neurath, M.F. Adv. Drug Deliv. Rev. 2007, 59, 1073-1083)ならびにアテローム性硬化症用(Russell, J.C.; Proctor, S.D. Cardiovasc. Pathol. 2006, 15, 318-330; Zadelaar, S.; Kleemann, R.; Verschuren, L.; et al. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1706-1721)のものなど、そのようなモデルの宿主は公知である。
【0168】
F.呼吸器疾患モデル
COPD(Fox, J.C.; Fitzgerald, M.F. Curr. Opin. Pharmacol. 2009, 9, 231-242.)、喘息(Nials, A.T.; Uddin, S. Dis. Model Mech. 2008, 1, 213-220)、嚢胞性線維症(Carvalho-Oliveira, I.; Scholte, B.J.; Penque, D. Expert Rev. Mol. Diagn. 2007, 7, 407-417)、気管支炎(Nikula, K.J.; Green, F.H. Inhal. Toxicol. 2000, 12, 123-153)、慢性呼吸器感染(Kukavica-Ibrulj, I.; Levesque, R.C. Lab. Anim. 2008, 42, 389-412)、および呼吸器アレルギー (Pauluhn, J.; Mohr, U. Exp. Toxicol. Pathol. 2005, 56, 203-234)の処置における本発明の化合物の有効性を評価するのに用いうる複数の動物モデル系が、公知である。
【0169】
ヒツジモデルが、喘息、COPD、アレルギー性鼻炎および嚢胞性線維症をはじめとする複数の呼吸器障害に効果的であることが、立証された。(Abraham, W.M. Pulm. Pharmacol. Ther. 2008, 21, 743-754.)。
【0170】
G.鉄ホメオスタシスモデル
動物モデルが、鉄輸送障害(Andrews, N.C. Adv. Exp. Med. Biol. 2002, 509, 1-17)および鉄代謝を含む疾患の研究(Latunde-Dada, G.O.; McKie, A.T.; Simpson, R.J. Biochim. Biophys. Acta 2006, 1762, 414-423)に向けて開発された。
【0171】
4.医薬組成物
本発明の大環状化合物または本発明によるその薬理学的に許容しうる塩を、様々な投与形態の医薬組成物に配合させてもよい。本発明の医薬組成物を調製するために、有効成分としての、光学異性体、鏡像異性体、ジアステレオマー、ラセミ体もしくは立体化学混合物を含む1種以上の化合物、または薬学的に許容しうるその塩を、医薬配合剤の当業者に公知の技術により、適切な担体および添加剤と細密に混合する。
【0172】
薬学的に許容しうる塩は、医薬品としての使用または配合を可能にするための、特定の化合物の遊離酸および塩基の生物学的効果を保持し、生物学的にも他についても不適当でない、本発明の化合物の塩形態を指す。そのような塩の例は、Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wermuth, C.G. and Stahl, P.H. (編), Wiley-Verlag Helvetica Acta, Zurich, 2002 [ISBN 3-906390-26-8]に記載されている。そのような塩の例としては、遊離酸および塩基のアルカリ金属塩および付加塩が挙げられる。非限定的な薬学的に許容しうる塩の例としては、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、リン酸一水素塩、リン酸二水素塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ギ酸塩、イソ酪酸塩、カプロン酸塩、ヘプタン酸塩、プロピオール酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−ジオン酸塩、ヘキシン−1,6−ジオン酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、γ−ヒドロキシ酪酸塩、グリコール酸塩、酒石酸塩、メタンスルホン酸塩、エタンスルホン酸塩、プロパンスルホン酸塩、トルエンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スルホン酸塩、およびマンデル酸塩が挙げられる。
【0173】
本発明の化合物が塩基である場合、所望の塩を、例えば非限定的に塩酸、臭化水素酸、ヨウ化水素酸、炭酸、硫酸、硝酸、リン酸などの無機酸、または非限定的にギ酸、酢酸、プロピオン酸、マレイン酸、コハク酸、マンデル酸、フマル酸、マロン酸、ピルビン酸、シュウ酸、ステアリン酸、アスコルビン酸、グリコール酸、サリチル酸、ピラノシジル酸、例えばグルクロン酸もしくはガラクツロン酸、α−ヒドロキシ酸、例えばクエン酸もしくは酒石酸、アミノ酸、例えばアスパラギン酸もしくはグルタミン酸、芳香族酸、例えば安息香酸もしくは桂皮酸、スルホン酸、例えばp−トルエンスルホン酸、メタンスルホン酸、エタンスルホン酸、2−ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、シクロヘキシルアミノスルホンなどをはじめとする有機酸、での遊離塩基の処理をはじめとする、当業者に公知の任意で適切な方法により調製してもよい。
【0174】
本発明の化合物が酸である場合、所望の塩は、無機または有機塩基、例えばアミン(第一級、第二級、または第三級);アルカリ金属またはアルカリ土類金属水酸化物などでの遊離酸の処理をはじめとする、当該技術分野で公知の任意の適切な方法により調製してもよい。適切な塩の例示としては、アミノ酸、例えばグリシン、リシンおよびアルギニン;アンモニア;第一級、第二級および第三級アミン、例えばエチレンジアミン、N,N’−ジベンジルエチレンジアミン、ジエタノールアミン、コリン、および環状アミン、例えばピペリジン、モルホリン、およびピペラジンから得られる有機塩;ならびにナトリウム、カルシウム、カリウム、マグネシウム、マンガン、鉄、銅、亜鉛、アルミニウム、およびリチウムから得られる無機塩が挙げられる。
【0175】
そのような医薬組成物に用いられる担体および添加剤は、予測される投与様式に応じて、様々な形態をとることができる。つまり経口投与用の組成物は、例えば、デンプン、糖、結合剤、希釈剤、造粒剤、滑剤、崩壊剤などの適切な担体および添加剤を含む、固体調製物、例えば錠剤、糖衣錠、ハードカプセル、ソフトカプセル、顆粒、粉末などであってもよい。錠剤およびカプセルは、使用が容易であり患者の服薬遵守が高いことから、多くの医療条件で最も有利な経口投与形態を示す。
【0176】
同様に、液体調製物用の組成物としては、水、アルコール、油、グリコール、保存剤、着香剤、着色剤、懸濁剤などの適切な担体および添加剤を含む、溶液、エマルジョン、分散体、懸濁物、シロップ、エリキシルなどが挙げられる。非経口投与用の典型的な調製物は、有効成分を、担体、例えば滅菌水または非経口的に許容しうる油、例えばポリエチレングリコール、ポリビニルピロリドン、レシチン、落花生油またはゴマ油と共に含み、溶解度または防腐性を補助する他の添加剤を含んでいてもよい。溶液の場合、それを粉末に凍結乾燥し、その後、使用直前に再構成させることができる。分散体および懸濁物では、適切な担体および添加剤としては、水性ゴム、セルロース、ケイ酸塩または油が挙げられる。
【0177】
本発明の実施形態による医薬組成物としては、経口、経直腸、局所、吸入(例えば、エアロゾルを介して)、口腔(例えば、舌下)、経膣、局所(即ち、気道表面を含む、皮膚および粘膜表面の両方)、経皮投与および非経口投与(例えば、皮下、筋肉内、皮膚内、関節内、肋膜内、腹腔内、髄腔内、脳内、頭蓋内、動脈内または静脈内)に適したものが挙げられるが、任意の所定の例に最も適した経路は、処置される状態の性質および重症度、ならびに用いられる特定の活性剤の性質に依存する。
【0178】
注射用の組成物は、有効成分を、プロピレングリコール−アルコール−水、等張水、注射用滅菌水(USP)、Emulphor(商標)−アルコール−水、Cremophor−EL(商標)または当業者に公知の他の適切な担体をはじめとする適切な担体と共に含む。これらの担体は、単独で、または他の従来の可溶化剤、例えば、エタノール、プロピレングリコール、もしくは当業者に公知の他の薬剤と併用で使用してもよい。
【0179】
本発明の大環状化合物が、溶液または注射の形態で適用される場合、該化合物を、任意の従来の希釈剤に溶解または懸濁させることにより用いてもよい。希釈剤は、例えば、生理食塩水、リンガー液、水性グルコース溶液、水性デキストロース溶液、アルコール、脂肪酸エステル、グリセロール、グリコール、植物または動物供給源から得られる油、パラフィンなどを含んでいてもよい。これらの調製物は、当業者に公知の任意の従来法により調製してもよい。
【0180】
経鼻投与用の組成物は、エアロゾル、滴下剤、粉末およびゲルとして配合させてもよい。エアロゾル配合剤は、典型的には生理学的に許容しうる水性または非水性溶媒中の有効成分の溶液または微細懸濁物を含む。そのような配合剤は、典型的には密閉容器中に滅菌形態の単回投与量または反復投与量で存在する。密閉容器は、噴霧装置で使用されるカートリッジまたはレフィルであってもよい。あるいは密閉容器は、単回使用の経鼻吸入器などの単回分注装置、または治療上効果的な量を送達するように設定されていて、内容物が完全に使用されると廃棄される調量弁付きのポンプ式アトマイザもしくはエアロゾルディスペンサであってもよい。投与形態がエアロゾルディスペンサを含む場合、それは噴射剤、例えば圧縮気体、例として空気、またはフルオロクロロ炭化水素もしくはフルオロ炭化水素をはじめとする有機噴射剤を含むであろう。
【0181】
口腔または舌下投与に適した組成物としては、錠剤、トローチおよび香剤が挙げられ、その有効成分は、担体、例えば糖と、アカシア、トラガカントまたはゼラチンと、グリセリンと配合される。
【0182】
経直腸投与用の組成物としては、従来の坐剤ベース、例えばココアバターを含む坐剤が挙げられる。
【0183】
経皮投与に適した組成物は、軟膏、ゲルおよび貼付薬が挙げられる。
【0184】
当業者に公知の他の組成物を、例えば膏薬など、経皮または皮下投与に適用することができる。
【0185】
更に、組成物の配合に必要な成分と混和される単一または複数の有効成分を含むそのような医薬組成物を調製する際に、他の従来の薬理学的に許容しうる添加剤を、例えば賦形剤、安定化剤、防腐剤、湿潤剤、乳化剤、滑剤、甘味剤、着色剤、着香剤、等張剤、緩衝剤、抗酸化剤などを組み込んでもよい。添加剤として、例えば、デンプン、スクロース、フルクトース、デキストロース、ラクトース、グルコース、マンニトール、ソルビトール、沈降炭酸カルシウム、結晶セルロース、カルボキシメチルセルロース、デキストリン、ゼラチン、アカシア、EDTA、ステアリン酸マグネシウム、タルク、ヒドロキシプロピルメチルセルロース、メタ重亜硫酸ナトリウムなどを挙げることができる。
【0186】
幾つかの実施形態において、該組成物は、単位投与形態、例えば錠剤またはカプセル中で提供される。
【0187】
更なる実施形態において、本発明は、有効量の本発明の化合物1種以上を含む医薬投与単位を含む容器を1個以上含むキットを提供する。
【0188】
本発明は、本明細書に記載された化合物を含むプロドラッグを更に提供する。用語「プロドラッグ」は、生理学的条件下で、または加溶媒分解により、または代謝的に、薬学的に活性のある特異的化合物に変換される化合物を意味するものとする。「プロドラッグ」は、化学的に誘導体化されることで、(i)親薬物化合物の生物活性の幾つか、もしくは全てを保持するか、または全く保持しなくなり、そして(ii)対象において代謝されて親薬物化合物を生成する、本発明の化合物であってもよい。本発明のプロドラッグは、化合物が化学的に誘導体化されることで、(i)親薬物化合物の生物活性の幾つか、もしくは全てを保持するか、または全く保持しなくなり、そして(ii)対象において代謝されて該化合物の生物活性誘導体を生成する、「部分プロドラッグ」であってもよい。化合物を誘導体化してプロドラッグを提供するための公知の技術を、用いることができる。そのような方法を利用して、化合物の加水分解可能なカップリングを形成してもよい。
【0189】
本発明は、本発明の化合物が代謝および/もしくは内分泌障害、胃腸障害、心臓血管障害、肥満および肥満関連障害、中枢神経系障害、骨障害、遺伝子障害、増殖過剰性障害ならびに炎症性障害を予防ならびに/または処置するのに用いられる治療剤と併用して投与してもよいことを更に提供する。例示的な薬剤としては、鎮痛剤(オピオイド系鎮痛剤を含む)、麻酔剤、抗真菌剤、抗生物質、抗炎症剤(非ステロイド系抗炎症剤を含む)、駆虫薬、鎮吐薬、抗ヒスタミン剤、血圧降下剤、抗精神病薬、抗関節炎薬、鎮咳薬、抗ウイルス薬、心臓作用薬、下剤、化学療法剤(例えば、DNA相互作用薬、代謝拮抗薬、チューブリン相互作用薬、ホルモン剤、およびアスパラギナーゼまたはヒドロキシ尿素などの薬剤)、コルチコイド(ステロイド)、抗うつ剤、抑制薬、利尿薬、催眠薬、ミネラル物質、栄養補助剤、副交感神経興奮薬、ホルモン(例えば、コルチコトロピン放出ホルモン、アドレノコルチコトロピン、成長ホルモン放出ホルモン、成長ホルモン、チロトロピン放出ホルモンおよび甲状腺刺激ホルモン)、鎮静剤、スルホンアミド、興奮剤、交感神経興奮薬、睡眠薬、血管収縮剤、血管弛緩剤、ビタミン剤およびキサンチン誘導体が挙げられる。
【0190】
本発明により処置されるのに適した対象としては、非限定的に、鳥類およびホ乳類対象が挙げられ、好ましくはホ乳類である。本発明のホ乳類としては、非限定的に、イヌ科、ネコ科、ウシ属、ヤギ、ウマ、ヒツジ、ブタ、げっ歯類(例えば、ラットおよびマウス)、ウサギ、霊長類、ヒトなど、および子宮内のホ乳類が挙げられる。本発明により処置される必要がある任意のホ乳類対象が、適している。ヒト対象が、好ましい。両方の性および発達の任意段階のヒト対象(即ち、新生児、幼児、児童、青年、成人)を、本発明により処置することができる。
【0191】
本発明による例示的鳥類としては、ニワトリ、アヒル、七面鳥、ガン、ウズラ、ギジ、平胸類(例えば、オーストリッチ)および家庭で飼育される鳥(例えば、オウムおよびカナリア)、ならびに卵内の鳥が挙げられる。
【0192】
本発明は、第一に、ヒト対象の処置に関係するが、本発明は獣医学的目的ならびに薬物スクリーニングおよび薬物開発目的で、動物対象、詳細にはホ乳類対象、例えば、マウス、ラット、イヌ、猫、家畜およびウマで実施することもできる。
【0193】
グレリン受容体の調整物質、例えばアゴニストが効果的となるホ乳類(即ち、ヒトまたは動物)の状態を処置するための治療的使用において、本発明の化合物またはその適切な医薬組成物を、有効量で投与してもよい。該化合物の活性および治療効果の程度が変動するため、投与される実際の投与量は、一般に認識された因子、例えば年齢、対象の状態、送達経路および対象の体重に基づいて決定されるであろう。投与量は、1日あたり1〜4回の経口投与で約0.1〜約100mg/kgであってもよい。加えて、1日あたり1〜4回の投与で、化合物を注射により投与あたりにおよそ0.01〜20mg/kg投与してもよい。処置を数週間、数ヶ月またはそれを超えて継続することができる。特定の状況での最適投与量の決定は、当業者の能力内である。
【0194】
5.使用方法
本発明の化合物は、本明細書に記載されたものおよび更に非限定的に増殖過剰性障害、炎症性障害、組織障害、心臓血管障害、呼吸器障害、ウイルス感染およびそれらの組み合わせをはじめとする一定範囲の医療状態の予防および処置に用いることができ、その障害は、複数の病気に罹患した結果であってもよい。特定の実施形態において、疾患または障害は、癌である。
【0195】
本発明の更なる態様によれば、増殖過剰性障害、例えば腫瘍、癌および新生物障害、ならびに前悪性および非新生物または非悪性の増殖過剰性障害の処置の方法が提供される。詳細には、本発明により処置されうる腫瘍、癌、および新生物組織としては、非限定的に、悪性障害、例えば乳癌、骨肉腫、血管肉腫、線維肉腫および他の肉腫、白血病、リンパ腫、洞腫瘍、卵巣癌、子宮癌、膀胱癌、前立腺癌および他の泌尿生殖器癌、結腸癌、食道癌、胃癌および他の胃腸癌、肺癌、骨髄腫、膵臓癌、肝臓癌、腎臓癌、内分泌癌、皮膚癌、ならびにグリオーマおよび神経芽細胞腫をはじめとする悪性または良性の脳または中枢(CNS)および末梢神経系の腫瘍が挙げられる。
【0196】
本明細書で用いられる「処置」は、それに関連する障害または症状の治癒または完全な消滅を付与することを必ずしも意味していない。
【0197】
本発明の化合物は、更に非限定的に、増殖過剰性障害、炎症性障害、呼吸器障害およびウイルス感染をはじめとする一定範囲の医療状態の処置のための医薬品を調製するために使用することができる。
【0198】
本発明の更なる実施形態を、ここに以下の実施例を参照して記載する。これらの実施形態が本発明の実施形態の例示を目的とするもので、本発明の範囲を限定するものではないことを理解しなければならない。
【実施例】
【0199】
実施例1
代表的なセリンプロテアーゼの阻害のためのアッセイ
以下に、代表的なセリンプロテアーゼとしてマトリプターゼのアッセイを記載するが、それは報告された方法に基づいている(Desilets, A.; Longpre, J.-M.; Beaulieu, M.-E.; Leduc, R. FEBS Lett. 2006, 580, 2227-2232.)。類似のアッセイを、他のセリンプロテアーゼ用に適用および入手することができる。
【0200】
FLX−800 TBEマイクロプレートリーダー(米国バーモント州ウィヌースキのBio−Tek Instruments)においてAMC−結合ペプチドからの蛍光の放出を測定することにより(励起:360nm、発光:441nm)、酵素活性をモニタリングした。精製ヒトマトリプターゼを、バースト滴定試薬(burst titrant)4−メチルウンベリフェリル−p−グアニジノ安息香酸塩(MUGB)で活性部位滴定した。500lg/mL BSA、pH9を含有する100mM Tris−HCl中で、マトリプターゼを用いた酵素的アッセイを実施した。文献に記載された通り、ヒト可溶性フリンを発現させ、精製、滴定およびアッセイした(Denault, J.B.; Lazure, C.; Day, R; Leduc, R. Protein Expr. Purif. 2000, 19, 113-124.)。精製されたHATタンパク質を、MUGBで活性部位滴定した。pH8.6の50mM Tris−HCl中で、HATでのアッセイを実施した。
【0201】
酵素を、フリンでは4〜12.5nMの範囲の濃度、マトリプターゼでは2〜7nMの範囲の濃度、そしてHATでは20pMの濃度に希釈して、10μMと共に37℃でインキュベートするか(初期スクリーニングの場合)、または被験化合物を適切に希釈して(反応速度分析の場合)、例えば0、500、1000、2000nMとして、もしくは0、250、500、1000、2500、5000nMで室温で15分間インキュベートした。蛍光発生基質(フリンでは4μM Boc−Arg−Val−Arg−Arg−AMC、マトリプターゼではBoc−Gln−Ala−Arg−AMC、そしてHATで4μg Boc−Val−Pro−Arg−AMC)の加水分解を追跡することにより、残留する酵素活性を測定した(米国ペンシルバニア州キングオブプリシアのBachem Bioscience)。濃度を上昇させる被験化合物の存在下で、飽和曲線を作成した。3回以上の独立した実験のデータを、典型的には平均化して、残留速度を、被験化合物濃度の関数としてプロットした。Enzfitterソフトウェア(米国ミズーリ州ファーガソンのBiosoft)を用いた式(1)への非線形回帰分析により、データを適合させた(Bieth, J.G. Methods Enzymol. 1995, 248, 59-84.)。
式(1):
【数1】
(式中、v0およびviは、それぞれ阻害剤の非存在下および存在下での基質加水分解の定常状態速度であり、[E]0は酵素の初期濃度、[I]0は阻害剤の初期濃度、そしてKi(app)は基質依存性の平衡解離定数である)。基質依存性定数Kiは、式(2)を用いて計算した(Bieth, J.G. Methods Enzymol. 1995, 248, 59-84.)。
式(2):
【数2】
(式中、[S]0は基質の初期濃度であり、Kmは、酵素−基質相互作用でのミカエリス・メンテン定数である)。被験化合物の安定性を調査するために、10μM被験化合物を特定濃度のマトリプターゼまたはHATと共に室温で特定時間インキュベートした。その後、タンパク質をSDS−PAGEにより分解して、Gel Code青色染色試薬を用いて明らかにした(米国イリノイ州ロックフォードのPierce Biotechnology)。
【0202】
表に、代表的な本発明の化合物のマトリプターゼ阻害の結果を表す。
【表6】
【0203】
Kiは、非線形回帰分析を用いた速度から計算することができる。用いられたモデルは、競合酵素阻害式、Km0bs=Km*(1+[I]/Ki)およびY=Vmax*X/(KmObs+X)である。Xは、基質濃度である。Yは、速度である(Copeland, R.A. Enzymes, 2nd edition, Wiley, 2000の式8.11)。Kiは、GraphPad Prism5ソフトウェア(米国カリフォルニア州サンディエゴのGraphPad Software)を用いて計算した。例えば化合物451は、Ki=1.46μMを有し、化合物459は、Ki=245nMを有する。
【0204】
阻害の選択性を決定するために、TTSPおよび他のセリンプロテアーゼを、蛍光発生ペプチドBoc−Gln−Ala−Arg−AMCの存在下で被験化合物とインキュベートした。活性を37℃で20分間測定した。
【0205】
実施例2
プロテアーゼ阻害アッセイ
(Li, P.; Jiang, S.; Lee, S.-L.; et al. J. Med. Chem. 2007, 50, 5976-5983.)。ウシトロンビン、ボーマン・バーク型阻害剤(BBI)、および蛍光基質は、市販されていた(ミズーリ州セントルイスのSigma Chemical Co.)。プロテアーゼに対する本発明の化合物の阻害活性を、2種の異なる系で室温で測定した。第一のアッセイ系では、100mg/mL ウシ血清アルブミンを含有する100mMTris−HCl(pH8.5)の反応緩衝液を用いた。反応緩衝液170μLを含有するキュベットに、酵素溶液10μLおよび阻害剤溶液10μLを添加した。プレインキュベーションの後、蛍光ペプチド基質の溶液(10μL)を添加して、キュベットの内容物を完全に混合した。蛍光分光光度計(日立F4500)を励起波長360nmおよび発光波長480nmで用いて、基質の加水分解により放出された蛍光の変化を追跡することにより、残留酵素活性を決定した。例えば蛍光ペプチドBoc−Gln−Ala−Arg−AMCを、マトリプターゼ用の基質として用いた。ペプチドBoc−Leu−Arg−Arg−AMCを、トロンビン用の基質として用いた。6〜7種の異なる濃度の被験化合物の存在下で、加水分解速度を記録した。異なる基質濃度での2組のデータからのディクソンプロットにより、Ki値を決定した。
【0206】
70kDa活性化マトリプターゼを、記載された通り単離した(Lin, C.-Y.; Anders, J.; Johnson, M.D.; Dickson, R.B. J. Biol. Chem. 1997, 272, 27558-27564; Lin, C.-Y.; Anders, J.; Johnson, M.; Sang, Q. A.; Dickson, R. B. J. Biol. Chem. 1999, 274, 18231-18236.)。第二のアッセイ系は、本質的に同一の結果をもたらし、100mg/mL BSAを含有する100mM Tris(pH8.3)の緩衝液中のマトリプターゼ用の基質としてBoc−Gln−Ala−Arg−AFCペプチドを使用した。Tecan Ultra蛍光分光計(ノースカロライナ州ダーラムのTecan)を用いて黒色96穴プレート中で、合計容量200μL中の精製マトリプターゼでアッセイを実施した。
【0207】
実施例3
代表的なセリンプロテアーゼの阻害に関する細胞培養アッセイ
マトリプターゼcDNAでトランスフェクトされたHEK293細胞中でのマトリプターゼ活性を阻害する能力について、被験化合物を検査した。被験化合物をモック細胞およびマトリプターゼをトランスフェクトした細胞と共に18時間インキュベートした。溶媒中のタンパク質分解活性を、蛍光発生ペプチドBoc−Gln−Ala−Arg−AMCを用いて測定した。
【0208】
実施例4
腫瘍転移についてのインビトロアッセイ
(Galkin, A.V.; Mullen, L.; Fox, W.D.; Brown, J.; et al. Prostate 2004, 61, 228-235)。CWR22RV1細胞を、ATCC(メリーランド州ロックビル)から得て、7%ウシ胎仔血清(カリフォルニア州タルザナのOmega Scientific)、1%ペニシリン−ストレプトマイシンおよび1%L−グルタミン(ニューヨーク州グランドアイランドのGibco)を補足したRPMI−1640培地中で培養する。CWR22RV1細胞増殖速度に対する本発明の化合物の影響を試験するために、播種された細胞を4群に分割して、最初の播種後1、3、および5日目に、1、10、または25mM濃度の被験化合物またはビヒクル溶液で処置する。実験では、1日および1群あたり三枚ずつプレートを使用する。3、5、および7日目に、血球測定装置で細胞を計数する。細胞浸潤性アッセイ(カリフォルニア州テメキュラのChemicon)を利用して、タンパク質の組換え基底膜マトリックス(ECMatrix、Chemicon)を介したCWR22RV1細胞浸潤に対する本発明の化合物の影響を評価する。ECMatixの再水和の後、25mM被験化合物を含む、または含まない無血清培地0.4mL中のCWR22RV1細胞(2×105)を上のチャンバーに添加して、10%ウシ胎仔血清を含有し25mM被験化合物を含む、または含まない培地0.75mLを予め充填した下のチャンバーに入れて、378℃で48時間インキュベートする。インキュベーションの終了時に、培地および任意の非浸潤細胞を除去して、供給したクリスタルバイオレット溶液で膜を染色する。その後、膜をスライドガラスに載せて、細胞を光学顕微鏡で検査する。一群あたり膜6枚(±被験化合物処置)を倍率100倍で分析する。膜あたり5ヶ所のフィールドを無作為に選択して、利用可能な孔の合計数のうちの平均浸潤細胞数を計数する。観察されたフィールドあたりの浸潤細胞の割合を計算する。実験は、二重測定で実施する。
【0209】
実施例5
(Galkin, A.V.; Mullen, L.; Fox, W.D.; Brown, J.; et al. Prostate 2004, 61, 228-235.)。4〜6週齢雌無胸腺BALB/cヌードマウス(Charles Rivers Laboratories)を、病原体フリーの条件下で飼育する。確立されたアンドロゲン非依存性(AI)の3種のCWR22RおよびCWRSA6異種移植片細胞系から得た組換え基底膜 (Matrigel;マサチューセッツ州ベッドフォードのCollaborative Research)と共に、細かく刻んだ腫瘍組織をマウスに皮下接種する。4〜10日後に、およそ5×5mm3の腫瘍を生成したマウスに、生理食塩水中の被験化合物(50または25mg/kgを1日2回、1週間に7回、腹腔内)またはビヒクルのみのいずれかを、同一の投与スケジュールで投与する。腫瘍を週2回、ノギスで計測し、腫瘍容積を式(π/6)×(大径)×(小径)2により計算する(Press, M.F.; Bernstein, L.; Thomas, P.A.; Meisner, L.F.; Zhou, J.Y.; Ma, Y.; Hung, G.; Robinson, R.A.; Harris, C.; El-Naggar, A.; Slamon, D.J.; Phillips, R.N.; Ross, J.S.; Wolman, S.R.; Flom, K.J. J. Clin. Oncol. 1997, 15, 2894-2904.)。腫瘍接種の18〜25日後に動物を殺処分して、腫瘍組織を分析のために急速冷凍する。
【0210】
実施例6
腫瘍転移のインビボアッセイ
6週齢ケラチン−5−マトリプターゼ・トランスジェニックマウスおよび同腹子対照マウス(List, K.; Szabo, R.; Molinolo, A.; Sriuranpong, V.; Redeye, V.; Murdock, T.; Burke, B.; Nielsen, B.S.; Gutkind, J.S.; Bugge, T.H. Genes Dev. 2005, 19, 1934-1950.)を、1種以上の濃度の被験化合物で処置する。その後、腰部中央の表皮ケラチノサイトの増殖速度に対する被験化合物の影響を、ビヒクル対照での処理結果と比較することにより決定する。
【0211】
実施例7
鶏卵尿漿膜モデル
文献の方法を用いて、本発明の化合物の血管形成を阻害する能力を測定することができる(Ghiso, J.A.A.; et al. J. Cell. Biol. 1999, 147, 89-104.)。
【0212】
実施例8
テザーの合成
A.テザーT5の合成に関する標準的手順
【化19】
【0213】
ステップ5−1. 蒸留水(5L)中の3−メチル安息香酸エチル(5−0、300g、1.83mol、1当量)の溶液に、臭素(292.5g、1.83mol)を一度に添加した。この混合物に、200Wのランプ2台を照射した。ランプをフラスコ中央の外側に置いて、フラスコの周りに箱を置いた。照射の間、溶液を激しく撹拌した。温度を45℃に上昇させると、溶液が反応の間に橙色から黄色へ、そしてほとんど無色へ変色した。4時間後に(本質的に無色の溶液)、ランプを消灯して、混合物を室温まで放冷した。混合物をDCM 2Lで希釈し、その後、水相をDCM 500mLで抽出した。ひとまとめにした有機相をブラインで、その後、10%チオ硫酸ナトリウム溶液で、そして最後に再度ブライン(pH=5)で洗浄した。橙色の相をMgSO4で乾燥させ、ろ過してろ液を減圧濃縮して、次のステップで用いるのに十分な品質の5−1を液体として収率96%で得た。
TLC(15% EtOAc/ヘキサン): Rf:0.58、検出:UV
【0214】
ステップ5−2. 室温で撹拌したエタノール(95%、1L)中の5−1(149g、0.611mol)の混合物に、蒸留水(300mL)中のシアン化カリウム(68g、1.7当量)の溶液を、滴加漏斗を用いて滴加した(警告:毒物あり! シアン化カリウムは公知の毒物であり、十分に喚起されたドラフトチャンバー内で適切に保護しながら取り扱わなければならない。HCN検出器の存在下で反応を実施する。ガラス製品は全て反応後に水およびアセトンで洗浄し、洗浄溶液は、「シアン化物」であり「危険」であることが明確に判別される容器に入れて、正しく廃棄しなければならない)。溶液は、添加の間に黄色になった。添加が完了した後、反応混合物を60℃まで2時間加熱し、その後、室温で一晩撹拌した(TLC:10%EcOAc/90%ヘキサン;検出:UV、CMAにより反応をモニタリング)。溶液を水(900mL)で希釈し、その後、Et2Oで抽出した(900mLで3回)。ひとまとめにした有機相をブラインで2回洗浄し(2回)、MgSO2で乾燥させ、ろ過してろ液を減圧蒸発させて、橙色油状物を与えた。油状残渣をシリカゲルのドライパックにより、EtOAc/ヘキサンを用いて精製して(勾配、5/95から15/85へ)、5−2を黄色固体として得た(66g、2段階で59%)。
TLC(30/70 EtOAc/ヘキサン):Rf:0.45、検出: UV;
1H NMR: δ 1.6 ppm (2H, 三重線), 3.8 ppm (3H, s), 4.4 ppm (2H, 四重線),7.4−7.6 ppm (2H, m), 8.0−8.1 ppm (2H, m)
【0215】
ステップ5−3.室温のTHF/水(4.6L/2.3L)中の5−2(220g、1.17mol)の溶液に、塩化コバルト(54.7g、0.23mol)を添加して、水素化ホウ素ナトリウム(132g、3.5mol)を少しずつ添加した。水素発生が、観察される。添加後に、反応物を室温で一晩撹拌した。混合物をCelite(登録商標)でろ過して、THF 1Lで洗浄した。THFを減圧蒸発により除去し、その後、水酸化ナトリウムの溶液(0.5N、2L)を添加して、混合物をEt2Oで抽出した(3回)。ひとまとめにした有機相をブラインで洗浄し(2回)、Na2SO4で乾燥させ、ろ過してろ液を減圧濃縮して、次のステップで直接使用されるのに適切な品質の粗液を5−2から52%得た。
TLC(50/50 EtOAc/ヘキサン):Rf:ベースライン、検出:UV、ニンヒドリン
【0216】
ステップ5−4. 脱気したDMF(200mL)中の5−3(118g、0.61mol)、Ddz−OPh(213g、0.67mol)およびトリエチルアミン(85mL、0.61mol)の溶液を、窒素雰囲気下、50℃で2日間撹拌した。その後、混合物を水2.5Lで希釈した。水相をEt2Oで抽出した(3回)。ひとまとめにした有機相を水、水酸化ナトリウム(0.5N、2回)およびブライン(2回)で引き続き洗浄し、MgSO4で乾燥させ、ろ過してろ液を減圧濃縮して、褐色油状物を得た。粗材料をドライパック(勾配、15%EtOAc/ヘキサン、0.5%Et3Nから25%EtOAc/ヘキサン、0.5%Et3Nへ;検出:UV+CMA)で精製して、5−4を156g(62%)得た。
TLC(25/75 EtOAc/ヘキサン):Rf:0.23、検出:UV + CMA
【0217】
ステップ5−5の還元と適合性のある他の保護基をこの段階で用い、標準的反応条件を用いて付加させることもできる。
【0218】
ステップ5−5. −78℃のDCM(2.1L)中の5−4(291.5g、0.7mol)の溶液に、水素化ジイソブチルアルミニウム(DIBAL−H、DCM中に1.0M、2.1L、2.1mol)を滴加漏斗で添加した。添加が完了したら、溶液を−78℃で2時間撹拌するか、またはTLCのモニタリング(50%EtOAc/ヘキサン;検出:UV、ニンヒドリン)により完了が示されるまで撹拌した。その後、反応混合物を酒石酸の溶液(1.0M、4L)に緩やかに滴加することにより、クエンチした。得られた混合物をDCMで抽出した(3回)。ひとまとめにした有機相をEDTA四ナトリウム塩の0.6M溶液(2Lで1回)およびブライン(2Lで1回)で引き続き洗浄し、MgSO4で乾燥させ、ろ過してろ液を減圧濃縮して、生成物Ddz−T5を黄色油状物(251.4g、96%)として得た。
TLC(50/50, EtOAc/ヘキサン):Rf:0.25、検出: UV、ニンヒドリン;
1H NMR: δ (1.7 ppm (s, 6H, 2 x CH3), 2.8 ppm (t, 2H,2 x CH2), 3.4 ppm (四重線, 2H, 2 x CH2), 3.8 ppm (s, 6H, 2 x OCH3), 4.7 ppm (s, 2H, CH2), 6.3−6.5 ppm (m, 3H, 3 x CH), 7.0−7.4 ppm (m, 4H, 4 x CH)。
【0219】
B.テザーT−28の合成の標準的手順
米国特許第7,521,420も参照されたい。
【化20】
【0220】
ステップ28−1. (Tius, M.A. J. Am. Chem. Soc. 1992, 114, 5959.)。酢酸(115mL)中のサリチルアルデヒド(28−0、23.4g、0.19mol、1.0当量)の溶液に、酢酸アンモニウム(17g、0.22mol、1.15当量)およびニトロメタン(39.5mL、0.73mol、3.8当量)を添加した。混合物を110℃で4.5時間加熱し、その後室温で冷却した。溶媒を真空除去してDCMで希釈し、ブラインで洗浄して(3回)、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させた。残渣をフラッシュクロマトグラフィーで精製して(勾配、EtOAc/ヘキサンを10%、その後20%、その後25%)、28−1を14.5g(45.8%)得た。
TLC (25/75 EtOAc/ヘキサン):Rf= 0.21,検出 UV, CMA;
1H NMR (CDCl3): δ8.16−8.11 (d、 1H), 7.98−7.93 (d、 1H), 7.44 (d、 1H), 7.43−7.32 (m, 1H), 7.32−6.98 (t, 1H), 6.87 (d, 1H)。
【0221】
ステップ28−2. 0℃のTHF/MeOH(7/1、500mL)中の28−1(14.5g、0.088mol、1.0当量)の溶液に、水素化ホウ素ナトリウム(10.0g、26.0mol、3.0当量)を少しずつ添加した。反応物を室温で温めて、完了するまでTLCによりモニタリングした。水を緩やかに添加することにより、反応物をクエンチした。pHを1M HClでpH7〜8に調整した。THFを真空除去し、その後、残りの混合物をエーテルで抽出した(3回)。有機相をブラインで洗浄し(1回)、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させて、次のステップで使用されるのに十分な純度の28−2を9.6g(66%)得た。
TLC (25/75 EtOAc/ヘキサン):Rf= 0.23, 検出: UV, CMA
【0222】
ステップ28−3. 95%EtOH(200mL)中の28−2(9.6g、0.058mol、1.0当量)の溶液に、10%Pd/Cを添加して、水素ガスを一晩バブリングした。混合物をCelite(登録商標)でろ過して、溶媒を減圧蒸発させた。生成物のEtOAcを同時蒸発させた。残渣(7.9g)の28−3は、任意の更なる精製を行わずに次にステップで使用した。
TLC (25/75 EtOAc/ヘキサン):Rf= 0.0,検出: UV, CMA。
【0223】
ステップ28−4. 0℃のDCM中の28−3(7.9g、0.058mol、1.0当量)およびEt3N(16.2mL、0.12mol、2.0当量)の溶液に、DCM中のBoc2O(12.7g、0.058mol、1.0当量)の溶液を滴加した。反応混合物を、一晩撹拌した。反応混合物をクエン酸緩衝液(2回)およびブライン(2回)で洗浄して、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させた。粗残渣をフラッシュクロマトグラフィーにより精製して(勾配、EtOAc/ヘキサンを20%、その後25%)、28−4(7.4g、54%、2段階)を得た。
TLC (25/75 EtOAc/ヘキサン):Rf= 0.36, 検出: UV, CMA
【0224】
ステップ28−5. THF(200mL)中の2−ブロモエタノール(2.29g、42.3mmol、1.0当量)の溶液に、イミダゾール(7.2g、105.8mmol、2.5当量)を、その後TBDMSCI(6.7g、44.4mmol、1.05当量)を添加した。反応混合物を4時間撹拌すると、白色沈殿物が2〜5分後に形成し始めた。エーテル(200mL)を添加して、有機相を塩化アンモニウムの飽和溶液(2回)、重炭酸ナトリウムの飽和溶液(1回)およびブライン(1回)で引き続き洗浄して、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させた。こうして得られた生成物(28−A、8.7g、86%)を、次の反応に直接使用した。
TLC (25/75 EtOAc/ヘキサン):Rf= 0.80,検出: UV, CMA
【0225】
DMF(40mL)中の28−4(4.2g、17.8mmol、1.0当量)、28−A(6.4g、26.7mmol、1.5当量)およびヨウ化カリウム(591mg、3.6mmol、0.2当量)の溶液に、炭酸カリウム(2.7g、19.6mmol、1.1当量)を添加して、混合物を75℃で一晩加熱した。その期間の後、TLCで反応が完了していないことが示され、そのため28−Aおよび炭酸カリウムを更に1当量添加して、混合物を追加で一晩撹拌した。DMFを真空除去した(油圧ポンプ)。油状残渣を水で希釈して、生成物をエーテルで抽出した(3回)。有機相をブラインで洗浄し(2回)、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させた。生成物をフラッシュクロマトグラフィーにより精製して(15%EtOAc/ヘキサン)、28−5を5.2g(74%)得た。
TLC (35/65 EtOAc/ヘキサン):Rf= 0.79, 検出: UV, ニンヒドリン
1H NMR (CDCl3): δ 7.05 (m, 2H), 6.78 (m, 2H), 4.6 (bs, 1H), 3.95 (m, 2H), 3.88 (m, 2H), 3.28 (bq, 2H), 2.72 (t, 2H), 1.3 (s, 9H), 0.8 (s, 9H), 0.0 (s, 6H)。
【0226】
ステップ28−6. THF(20mL)中の28−5(2.5g、13.3mmol、1.0当量)の溶液に、THF中の1.0M TBAF(15.9mL、15.9mmol、1.2当量)を添加して、反応物を室温で30分間撹拌した。反応混合物をエーテル(150mL)で希釈し、その後、塩化アンモニウムの飽和溶液(2回)およびブライン(1回)で洗浄して、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させた。生成物をフラッシュクロマトグラフィーにより精製して(勾配、EtOAc/ヘキサンを25%から40%へ)、Boc−T28を3.5g(94.6%)を得た。
TLC (5/65 EtOAc/ヘキサン):Rf= 0.21,検出: UV,ニンヒドリン;
1H NMR(CDCl3):δ 7.3 (td, 1H), 7.1 (dd, 1H), 6.86 (m, 2H), 4.9 (bs, 1H), 4.1 (m, 2H), 4.0 (m, 2H), 3.3 (bs, 2H), 2.8 (t, 2H), 1.4 (m, 9H);
13C NMR (CDCl3):δ 157.2, 156.6, 130.8, 128.0, 127.6, 120.9, 111.4, 79.7, 69.8, 61.4, 40.9, 32.6, 28.6;
LC−MS (勾配 A4): tR: 10.2 分; (M+H)+ 281, (M+H+Na)+ 304
【0227】
C.テザーT29の合成の標準的手順
【化21】
【0228】
ステップ29−1: 0℃のTHF(DriSolv等級)中の水素化リチウムアルミニウム(LAH、3mol当量)の溶液に、3−シアノベンゾアルデヒド(29−0、1当量)を少しずつ添加した。混合物を0℃で1時間(または出発原料が消失するまで)撹拌し、その後、窒素雰囲気下のオイルバス中で一晩、加熱還流した(70℃)。反応をクエンチするために、溶液を窒素下で0℃に冷却し、次のものを引き続き添加した:水、NaOH(15%)、その後、水(それぞれLAH 5gに対して5mL:5mL:15mLの比で用いなければならない)(警告:水素ガス発生)。溶液をろ過し、塩をTHFで洗浄し、ひとまとめにしたろ液を減圧濃縮して、粗アミノアルコールを、典型的には次のステップで用いられるのに十分な純度で得た(Rf:ベースライン, 30/70, EtOAc/ヘキサン,検出: UV,ニンヒドリン)。
【0229】
ステップ29−2: 窒素雰囲気下、0℃で脱気されたDMF中のステップ29−1の生成物(1当量)およびDdz−N3(1.05当量)の溶液に、テトラメチルグアニジン(TMG、1.05当量)を添加した。10分後に、DIPEA(1.05当量)を添加し、その後、混合物を50℃のオイルバスで一晩撹拌した。混合物を減圧濃縮して(油圧ポンプ)、DMFを除去し、その後、残渣をDCMに溶解して、クエン酸緩衝液(2回)、飽和重炭酸ナトリウム(1回)、およびブライン(2回)で引き続き洗浄し、その後、MgSO4で乾燥させ、ろ過してろ液を減圧濃縮した。こうして得られた粗材料をフラッシュクロマトグラフィーにより精製して(勾配、50%EtOAc/ヘキサン、0.5%Et3Nから60%EtOAc/ヘキサン、0.5%Et3Nへ;DCMを混合物に添加して、開始時に残渣を溶解した)、所望の化合物、DdZ−T29(TLC:50%ヘキサン/EtOAc;検出:UV,ニンヒドリン)を得た。
【0230】
他の典型的な窒素保護基、例えば、Fmoc、Boc、Cbzを、標準の反応条件を用いてステップ29−2で配置することもできる。別法として、水素化ホウ素ナトリウムを塩化コバルトと共に用い、その後、第一級アミンをBoc(図示しない)または他の適切なN−保護基で選択的に保護することにより、ステップ29−1における還元を実施することができる。
【化22】
【0231】
D. テザーT−30の合成の標準的手順
【化23】
【0232】
ステップ30−1. THF/H2O(1:1)125mL中の2−ブロモフェネチルアミン(30−0、5.0g、25.0mmol、1.0当量)の溶液に、重炭酸ナトリウム(2.3g、27.5mmol、1.1当量)を添加した。その後、混合物を0℃に冷却し、Boc無水物(5.5g、25.0mmol、1.0当量)を一度に添加した。混合物を0℃で1時間撹拌し、その後、室温まで昇温させて、一晩撹拌した。溶媒を減圧蒸発させて、残渣をEtOAc/H2O(1:1)に溶解した。分離された有機相をH2O(2回)および飽和塩化ナトリウム(2回)で洗浄し、硫酸マグネシウムで乾燥させ、ろ過してろ液を減圧蒸発させた。得られた黄色油状物をDCMで希釈し、減圧蒸発させて(手順を3回繰り返した)、30−1 7.5g(100%)を白色固体として得た。
TLC (ヘキサン/EtOAc, 7:3): Rf= 0.75,検出:UV,ニンヒドリン
【0233】
ステップ30−2. アルゴン雰囲気下で火力乾燥させたフラスコに、30−1(6.3g、21.0mmol、1.0当量)、再結晶化ヨウ化銅(I)(80.0mg、0.42mmol、0.02当量、Organometallics in Synthesis, 2ndedition, Manfred Schlosser, Ed., 2002, p 669の手順を参照)およびジクロロビス(ベンゾニトリル)パラジウム(II)(242mg、0.63mmol、0.03当量)を添加した。フラスコをアルゴンでパージし(5〜10分間)、無水1,4−ジオキサン 20mLを添加した後、トリ−tert−ブチルホスフィン(ヘキサン中の10%(w/w)溶液、385μL、1.26mmol、0.06当量)およびジイソプロピルアミン(3.6mL、25.2mmol、1.2当量)を添加した。その後、混合物をアルゴンで再度パージして(5〜10分間)、3−ブチノール(30−A、2.4mL、31.5mmol、1.5当量)を混合物に滴加して、TLCモニタリングを行いながら、アルゴン下、室温で24時間撹拌した。混合物をEtOAcで希釈し、シリカゲルパッドでろ過して、TLCにより更なる材料溶出が示されなくなるまでEtOAcで洗浄した。ろ液を減圧蒸発させて、残渣をフラッシュクロマトグラフィーにより精製し(ヘキサン:EtOAc,7:3)、30−2を薄黄色油状物として5.5g(90%)得た。
TLC (ヘキサン/EtOAc, 7:3): Rf= 0.20,検出: UV, CMA
【0234】
ステップ30−3. 窒素下、95%EtOH中のBoc−アミノアルコール30−2(6.1g、21.1mmol、1.0当量)の溶液に、酸化白金(IV)(445mg、2.11mmol、0.1当量)を添加した。混合物を80psi H2で16時間撹拌した(引き続き反応を1気圧H2の室温で24〜36時間実施した)。少量のアリコットを取り出して、1H NMRにより反応をモニタリングした。反応が完了したら、窒素を混合物に10分間バブリングして過剰な水素を除去した。溶媒を減圧蒸発させて、EtOAcで希釈し、シリカゲルパッドでろ過して、TLCにより更なる材料溶出が示されなくなるまでEtOAcで洗浄した。ろ液を減圧蒸発させて、残渣をフラッシュクロマトグラフィーにより精製し(ヘキサン:EtOAc,7:3)、Boc−T30を薄黄色油状物として4.5g(75%)得た。
1H NMR (CDCl3): d 7.18-7.11, (m, 4H), 4.65, (bs, 1H), 3.72-3.65, (t, 2H), 3.32 (bs, 2H), 2.85-2.80, (t, 2H), 2.70-2.65, (t, 2H), 1.71-1.66 (m, 4H), 1.44 (s, 9H)
【0235】
ステップ30−2および30−3の反応系列に適合性のある他のN−保護基を、用いることもできる。
【0236】
E.テザーT−32の合成の標準的手順
T−32の反応スキームを、図4に表わす。
【0237】
ステップ32−1. −30℃のCH3CN(500mL)中の4−ヒドロキシベンゾニトリル(32−0、15.0g、109mmol、1.0当量)の溶液に、トリフリン酸(11.6mL、131mmol、1.2当量)を添加した。NBS(20.3g、117mmol、1.05当量)を少しずつ添加することで、温度が−10℃を超えないようにした。懸濁物が得られ、その溶液が数分後に均質になった。反応混合物を室温に保持し、一晩撹拌した。溶液を水性飽和NaHCO3で処理して、水相をEtOAcで抽出した(1回)。水相を6M HClで酸性化して、EtOAcで抽出した。その後、有機相を水性飽和NH4Clで抽出した(2回)。有機相をMgSO4で乾燥させ、ろ過してろ液を減圧濃縮した。最終化合物が過剰の(1H NMRで10%を超える)スクシンイミド副生成物を含有することが見出されれば、固形残渣を水中で一晩撹拌し、沈殿物をろ過して、一晩真空乾燥させた(油圧ポンプ)。1H NMRにより、所望の化合物32−1の同一性を確証した。生成物は、更に精製せずに次のステップで使用するのに適していた(収率:94%)。
TLC (80%EtOAc, 20%ヘキサン): Rf= 0.47,検出:UVおよびKMnO4
【0238】
ステップ32−2. DMF(300mL)中の32−1(11.3g、57.1mmol、1.0当量)の溶液に、炭酸カリウム(8.7g、62.8mmol、1.1当量)、ヨウ化カリウム(1.9g、11.4mmol、0.2当量)およびTBDMS−ブロモエタノール(32−A、20.5g、85.7mmol、1.5当量)を添加した。得られた混合物70℃で一晩撹拌した。混合物を室温まで冷却し、ブラインを添加して、相を分離した。水相をエーテルで抽出して、ひとまとめにした有機相をブラインで抽出した(2回)。有機相をMgSO4で乾燥させて、減圧濃縮した。残渣をフラッシュクロマトグラフィーにより精製して(20%EtOAc、60%ヘキサン)、32−2を黄色固体として得た(収率100%)。
TLC (40%EtOAc, 60%ヘキサン): Rf= 0.63,検出: UVおよびKMnO4
【0239】
ステップ32−3. 0℃のTHF(10mL)中の32−2(548mg、1.5mmol、1当量)の溶液に、リチウムヘキサメチルジシラジド(LHMDS、THF中の1M、3.0mL、3.0mmol、2.0当量)を添加し、その後、混合物を室温で2時間撹拌した。クエン酸(1M、5mL)を添加して、混合物を1時間撹拌した。エーテルを添加して、相を分離し、その後、有機相を1Mクエン酸で抽出した(2回)。ひとまとめにした水相を3M NaOHでpH=14に調整し、その後、CH2Cl2で抽出した(4回)。有機相をK2CO3で乾燥させて、減圧濃縮した。こうして得られた32−2を、次のステップで直接使用した。
TLC (20% EtOAc, 80%ヘキサン): Rf=ベースライン;検出:UVおよびKMnO4
【0240】
ステップ32−4. THF(6mL)中の32−3(1.5mmol、1.0当量)の溶液に、(Boc)2O(371mg、1.7mmol、1.1当量)およびDMAP(18mg、0.15mmol、0.1当量)を添加して、混合物を3時間撹拌した。ブラインを添加して、水相をエーテルで抽出した(3回)。ひとまとめにした有機相をMgSO4で乾燥させて、減圧濃縮した。残渣をフラッシュクロマトグラフィーにより精製して(30%EtOAc、70%ヘキサン)、白色固体の32−4を得た(収率:67%、2段階)。ジオキサン中の水性水酸化ナトリウム(1N)を、このステップで基剤として用いて、同等の収率を得ることもできる。
TLC (30% EtOAc, 70%ヘキサン): Rf= 0.37;検出: UVおよびKMnO4
【0241】
ステップ32−5. ジイソプロピルアミン(100mL)中の32−4(8.2g、17.3mmol、1.0当量)の溶液に、Ddz−プロパルギルアミン(32−B、9.6g、34.6mmol、2.0当量)を添加して、混合物をArで20〜30分間脱気した。PPh3(546mg、2.08mmol、0.12当量)、PdCl2(PPh3)2(730mg、1.04mmol、0.06当量)およびCuI(131mg、0.69mmol、0.04当量)を添加して、得られた混合物を70℃で一晩撹拌した。溶液をシリカゲルパッドでろ過して、EtOAcですすぎ、その後、溶媒を減圧蒸発させた。得られた残渣をフラッシュクロマトグラフィーで精製して(40%EtOAc、60%ヘキサン)、32−5を橙色固体として得た(収率100%)。
TLC (40%EtOAc, 60%ヘキサン):Rf= 0.27;検出:UVおよびCMA
【0242】
ステップ32−6. 95%エタノール(100mL)中の32−5(15.0g、22.2mmol、1.0当量)の溶液に、PtO2(500mg、2.2mmol、0.1当量)を添加して、水素ガスを溶液に1時間バブリングした。得られた混合物を、室温で一晩撹拌した。その時点で反応が終了していなければ(1H NMR)、PtO2を更に0.1当量添加して、水素ガスを溶液にバブリングし、混合物を再度一晩撹拌した。Arを反応物にバブリングして過剰の水素を排出させ、溶液をシリカゲルパッドでろ過して、パッドをEtOAcですすいだ。ひとまとめにした溶媒を、減圧蒸発させた。得られた32−6を次のステップで用いた(収率100%)。
【0243】
ステップ32−7. THF(100mL)中の32−6(14.5g、21.5mmol、1.0当量)の溶液に、THF中の1M TBAF(32.3mL、32.3mmol、1.5当量)を添加して、混合物を1時間撹拌した。ブラインを添加して、水相をEtOAcで抽出した。ひとまとめにした有機相をMgSO4で乾燥させて、ろ過してろ液を減圧濃縮した。残渣をフラッシュクロマトグラフィーにより精製して(100%EtOAc)、Ddz−T32(Boc)を得た(収率:88%)。
TLC (100% EtOAc):Rf= 0.24;検出:UVおよびCMA。
1H NMR (CDCl3): δ 7.74 (1H, dd), 7.35 (1H, d), 6.72 (1H, d), 6.53-6.49 (2H, m), 3.61-3.29 (1H, m), 5.06 (1H, t), 4.25-4.01 (2H, m), 3.91-3.89 (2H, m), 3.73 (3H, s), 2.99 (2H, dd), 2.63 (2H, t), 1.71 (8H, ブロード), 1.53 (9H, s);
13C NMR (CDCl3, ppm): δ 163.8, 162.2, 161.0, 159.7, 155.9, 149.4, 130.0, 129.1, 128.0, 126.8, 110.8, 98.1, 80.9, 79.3, 69.7, 61.3, 55.5, 39.1, 29.3, 28.5, 26.7。
【0244】
F.テザーT52およびテザーT53の合成の標準的手順
【化24】
【0245】
ステップT52−1. DMF(DriSolv(登録商標))中の3−ヨードフェノール(52−0、1.0当量)の溶液に、水素化ナトリウム(鉱物油中の60%、0.1当量)を少しずつ添加する(警告!水素ガスが発生すると思われる)。反応物を窒素下、100℃で1時間加熱し、その後、炭酸エチレンを添加して、反応混合物を100℃で一晩加熱する。反応をTLCでモニタリングする(条件:25/75 EtOAc/ヘキサン)。反応混合物を放冷し、その後、溶媒を減圧蒸発させる。残渣の油状物をEt2O(1.5L)で希釈し、その後、1N水酸化ナトリウム(3回)およびブライン(2回)で引き続き洗浄して、MgSO4で乾燥させて、ろ過してろ液を減圧蒸発させる。粗生成物を真空蒸留するか、またはフラッシュクロマトグラフィーにより精製して、52−1を得る。
【0246】
ステップT52−2. CH3CN中の52−1(1.0当量)およびBoc−アリルアミン(1.3当量)の溶液に、アルゴンを20〜30分間バブリングする。新たに蒸留されたEt3N(CaH2と共に4時間還流し、その後蒸留したもの、3.6当量)を添加して、アルゴンを10〜15分間バブリングする。その後、トリス(o−トリル)ホスフィン(0.03当量)およびPd(OAc)2(0.03当量)を添加する。反応物をTLCでモニタリングしながら、還流雰囲気で2時間撹拌する。反応が完了しなければ、より長時間利用することができる。揮発物を減圧除去して、残渣をフラッシュカラムクロマトグラフィーで精製して、Boc−T52を得る。
【0247】
ステップT52−3. Boc−T52(1.0当量)を10%Pd/C(15重量%)および95%EtOHを添加する。混合物を水素ガスの圧力下、水素添加装置(例えば、Parr)内に24時間配置する。LC−MSまたは1H NMRにより、モニタリングを実施することができる。混合物をCelite(登録商標)パッドでろ過し、その後減圧濃縮して、Boc−T53を得て、それをフラッシュクロマトグラフィーにより精製することができる。
【0248】
G.テザーT201の標準的手順
T201の反応スキームを、図5に表わす。
【0249】
ステップ201−1. −30℃のトルエン(320mL)中のt−ブチルアミン(40mL、378mmol、3.0当量)の溶液に、Br2(7.1mL、139mmol、1.1当量)を緩やかに添加した(10分間)。混合物を−78℃に冷却して、2−ヒドロキシベンゾニトリル(201−0、15.0g、126mmol、1.0当量)をCH2Cl2(80mL)中で添加した。2−ヒドロキシベンゾニトリルは、DCMにあまり溶解されず、懸濁物としての反応物にピペットで添加した。不均質混合物を緩やかに室温で冷却して、一晩撹拌した。ブラインを添加し、相を分離して、水相を酢酸エチルで抽出した。有機相をひとまとめにして、10%NaOHで抽出した(2回)。水相を6N HClで酸性化して、CH2Cl2で抽出した。有機相をMgSO4で乾燥させて、減圧濃縮し、201−1を得た(収率:90%)。
TLC (60% EtOAc, 40%ヘキサン): Rf= 0.32;検出:UVおよびKMnO4。
【0250】
ステップ201−2. TBDMS−ブロモエタノール(32−A)でのアルキル化による201−1から201−2への変換を、本質的にはステップ32−2の32−2の合成で記載された通り実施した。
【0251】
ステップ201−3. LHMDS 3当量が変換に用いられ、反応時間が2〜3日であったこと以外は、本質的にはステップ32−3の201−3の合成に記載された通り、201−2からのアミジン201−3の形成を実施した。
【0252】
ステップ201−4. Bocによる201−3のアミジン基の保護を、本質的にはステップ32−4の32−4の合成に記載された通り実行した。
【0253】
ステップ201−5. 201−5を得るための、201−4とDdz−プロパルギルアミン(32−B)との薗頭カップリング反応を、本質的にはステップ32−5の32−5の合成に記載された通り実施した。しかしカップリング反応は完了せず、出発原料を2回目に同条件下で処理して、生成物を得た。
【0254】
ステップ201−6. 201−5の水素化および脱保護を、本質的にはステップ32−6のDdz−T32(Boc)の合成に記載された通り実施して、Ddz−T201(Boc)を得た。
1H NMR (CDCl3): δ 7.87 (1H, d), 7.28−7.25 (1H, m), 7.10 (1H, t), 6.51−6.46 (2H, m), 6.31 (1H, t), 5.30−5.20 (1H, m), 3.90−3.85 (2H, m), 3.85−3.80 (2H, m), 3.74 (6H, s), 3.15−3.05 (2H, m), 2.67 (2H, t), 1.85−1.71 (2H, m), 1.71 (6H, s), 1.53 (9H, s);
13C NMR (CDCl3):δ 160.8, 155.6, 155.5, 135.6, 133.9, 129.9, 127.9, 125.0, 103.3, 98.2, 80.8, 79.8, 61.9, 60.6, 55.5, 40.2, 31.3, 29.5, 28.5, 27.1, 14.4。
【0255】
H.テザーT202およびT203の標準的手順
アミジン部分をテザーに組み入れた後で、テザーT32およびT201に関して既に記載された通り分子の残り部分に付加するか、またはマスキングされたアミジン基としてニトリルを用い、その後、ニトリルをアミジンに変換することにより、これらのテザーを調製することができる。前者のアプローチでは、2−ブロモ−5−シアノフェノールから出発すればT202に到達することができ、2−ブロモ−3−シアノフェノールから出発すればT203に到達することができる。
【化25】
【0256】
後者では、化合物451に関して記載された変換を、以下に例示する適切な大環状ニトリル上で用いることができる。
【化26】
【0257】
実施例9
大環状化合物の合成
A.代表的な本発明の大環状化合物を合成するための標準的手順
化合物451の反応スキームを、図1に表わす。
【0258】
ステップ451−1. H−Phe(4CN)−OBnの合成 H−Phe(4CN)−OH(2.85g、15mmol、1.0当量)のトルエン(75mL)溶液に、p−TSA(3.42g、18mmol、1.2当量)、BnOH(7.8mL、75mmol、5.0当量)を添加した。H2Oをディーンスタークトラップで除去しながら、混合物を4時間加熱還流した。混合物を室温まで放冷し、Et2Oで希釈して0℃(アイスバス)で45分間撹拌した。得られた白色沈殿物をろ過して、低温Et2Oですすいだ。白色固体を1M Na2CO3溶液に溶解し、その後、室温で30分間撹拌した。得られた水相をEtOAcで洗浄した(4回)。ひとまとめにした有機相をブラインで洗浄し、Na2SO4で乾燥させて、ろ過して減圧蒸発させ、薄橙色油状物を得た(3.10g、収率70%)。
1H NMR: δ 1.60 (br s, 2H), 3.02 (dq, 2H), 3.77 (t, 1H), 5.13 (q, 2H), 7.21−7.52 (m, 9H)。
【0259】
ステップ451−2 ジペプチドの形成. THF−DCM混合物(1:1、25mL)中のH−Phe(4CN)−OBn(2.9g、10.27mmol、1.0当量)の溶液に、Boc−NMeAla(2.15g、10.6mmol、1.03当量)、6−Cl−HOBt(1.74g、10.3mmol、1.1当量)を0℃(アイスバス)で添加した。DIPEA(8,94mL、51.35mmol、5.0当量)およびその後、EDCI(2.17g、11.3mmol、1.1当量)を添加して、混合物を室温で一晩撹拌した。揮発物を減圧蒸発させて、得られた粗油状物をEtOAcに溶解した。その溶液を1Mクエン酸緩衝液(pH=3.5、2回)、H2O、飽和NaHCO3およびブラインで引き続き洗浄し、その後、Na2SO4で乾燥させ、ろ過して減圧蒸発させた。ひとまとめにした有機相をH2O、飽和NH4Cl、ブラインで洗浄して、Na2SO4で乾燥させて、ろ過して真空蒸発させた。粗生成物をフラッシュクロマトグラフィーにより精製して(勾配、EtOAc/ヘキサンで40%その後、50%)、保護されたジペプチド4.50g(93%)を得た。
TLC(50% EtOAc/ヘキサン):Rf: = 0.15, 検出: UV,ニンヒドリン。
【0260】
保護されたジペプチド(4.46g、9.6mmol、1.0当量)を、MeOH中の3.3N HCl溶液(30mL、96mmol、10当量)に溶解した。混合物を室温で1時間撹拌した。その後、揮発物を減圧蒸発させ、得られた粗油状物を真空乾燥させて(油圧ポンプ)、所望の化合物を非晶質固体として得た(3.50g、100%)。
【0261】
ステップ451−3 Boc−T69−OTsの合成. Boc−T69(4.94g、14.7mmol、1.05当量)のDCM(36mL)溶液に、DMAP(342mg、2.8mmol、0.2当量)およびEt3N(9.8mL、70mmol、5.0当量)を添加して、混合物を0℃(アイスバス)で15分間撹拌した。その後、TsCl(2.67g、14mmol、1.0当量)のDCM溶液(24mL)を0℃で少しずつ添加した。混合物を0℃で45分間、その後、室温で一晩撹拌した。NH4Clの飽和溶液を添加して、二相に分離させて、水相をDCMで洗浄した(3回)。ひとまとめにした有機相を、1M HCl(2回)およびブラインで洗浄して、Na2SO4で乾燥させて、ろ過して減圧蒸発させた。粗生成物を、更に精製せずに次のステップで用いた(6.90g、100%)。
1H NMR (CDCl3): δ1.34 (s, 9H), 1.60 (m, 2H), 2.36 (s, 3H), 2.44 (m, 2H), 2.99 (m, 3H), 4.04 (m, 2H), 4.30 (m, 2H), 4.59 (br s, 1 H), 6.35 (m, 1H), 6.50 (m, 1H), 6.94 (m, 1H), 7.26 (d、 J=8.4 Hz, 2H), 7.72 (d、 J=8.4 Hz, 2H)。
【0262】
ステップ451−4 Boc−T69−Cpg−OMeの合成. EtCN/DMF混合物(3:1、20mL)中のBoc−T69−OTs(6.9g、14.7mmol、1当量)の溶液に、H−Cpg−OMe・HCl(3.65g,22.1mmol,1.5当量)、KI(オーブンで一晩乾燥させたもの、6.09g、36.7mmol、2.5当量)およびDIPEA(7.7mL、44.1mmol、3.0当量)を室温で添加した。LC−MSによりモニタリングしながら、反応混合物を108℃で30時間撹拌した。反応物を室温まで放冷し、その後、H2Oでクエンチした。混合物をEtOAcで希釈して、水相をEtOAcで洗浄した(3回)。ひとまとめにした有機相を、1Mクエン酸緩衝液(pH=3.5)、H2O、飽和NaHCO3およびブラインで引き続き洗浄して、Na2SO4で乾燥させて、ろ過して減圧蒸発させた。粗生成物を、更に精製せずに次にステップで用いた(5.98g、96%)。
LC−MS:tR= 6.24分(A4b),[M+H]+ 425
【0263】
ステップ451−5 Boc−T69−Cpg−OHの合成.DCM/MeOH混合物(9:1、90mL)中のBoc−T69−Cpg−OMe(5.98g、14.0mmol、1.0当量)の溶液に、MeOH中の2M NaOH溶液(14.1mL、28.2mmol、2.0当量)を添加した。混合物を室温で48〜72時間撹拌した。揮発物を減圧蒸発させて、残渣を水で希釈した。水相をEt2Oで洗浄し、その後、pH=1〜2に酸性化した。酸性相をEtOAcで洗浄した(3回)。ひとまとめにした有機相を飽和NH4Clおよびブラインで洗浄し、Na2SO4で乾燥させて、ろ過して減圧蒸発させた。粗固体をヘキサン/DCM混合物(9:1)と共に磨砕して、白色固体を得た(3.76g、65%)。
LC−MS: tR = 6.12分(A4b), [M+H]+ 411
【0264】
ステップ451−6 フラグメントのカップリング. DCM/THF混合物(1:1、90mL)中のBoc−T69−Cpg−OH(3.60g、9.2mmol、1.0当量)の溶液に、H−NMeAla−Phe(4CN)−OBn・HCl(3.36g、9,20mmol、1.05当量)を添加して、混合物を0℃(アイスバス)で15分間撹拌した。DIPEA(9.23mL、53mmol、6.0当量)と、その後、HATU(3.50g、9.20mmol、1.05当量)を添加して、LC−MSでモニタリングしながら、混合物を室温で48〜72時間。混合物をEtOAcで希釈して、1Mクエン酸緩衝液(pH=3.5)、H2O、飽和NaHCO3およびブラインで引き続き洗浄した。有機相をNa2SO4で乾燥させてろ過し、減圧下で蒸発させた。粗生成物をフラッシュクロマトグラフィーにより精製して(勾配、50%EtOAc/ヘキサン、その後100%EtOAc)、カップリングされた生成物を得た(4.35g、62%)。
TLC (50% EtOAc/ヘキサン): Rf: = 0.10,検出: UV,ニンヒドリン
LC−MS:tR= 7.95分(A4b), [M+H]+ 758
【0265】
ステップ451−7 脱保護. トリペプチド−テザー(4.0g、5.28mmol、1.0当量)のDCM(53mL)溶液に、Pd(OAc)2(60mg、0.264mmol、0.05当量)、Et3N(95μL、0.68mmol、0.13当量)を添加した。混合物をAr/真空のサイクルで30分間脱気し、アルゴン下、室温で一晩撹拌した。揮発物を減圧下で蒸発させて、暗色の粗油状物をFlorisil(登録商標)の短いパッドにより、最初EtOAcで、その後MeOHで溶出してろ過し、ひとまとめにしたろ液を減圧濃縮した。粗生成物を薄黄色油状物として得た(3.11g、90%)。
LC−MS:tR= 6.64分(A4b), [M+H]+ 668
【0266】
DCM/TFA/TES混合物(64;33;3、30mL)中の粗油状物(3.1g、4.57mmol、1.0当量)の溶液を、室温で45分間撹拌した。揮発物を減圧下で蒸発させた。残渣をDCM/トルエン混合物(1:1、15mL)に溶解して、減圧濃縮した。得られた油状物を更に精製せずに次のステップで用いた。
【0267】
ステップ451−8 大環状化. 先の粗油状物(3.1g、4.57mmol、1.0当量)を含有するTHF(457mL、c=0.01M)溶液に、DIPEA(5.60mL、32.0mmol、7.0当量)および最後にDEPBT(1.5g、5.03mmol、1.1当量)を添加した。混合物を室温で一晩撹拌した。揮発物を減圧蒸発させて、得られた粗油状物をEtOAc/NaHCO3(飽和)(1:1)の混合物に溶解した。水相をEtOAcで洗浄した(3回)。ひとまとめにした有機相をブラインで洗浄して、Na2SO4で乾燥させ、ろ過して減圧蒸発させた。粗生成物をフラッシュクロマトグラフィーにより精製して(勾配、0.5%/3%/96.5% AcOH/MeOH/EtOAc,100%EtOAc、その後、0.5%/3%/96.5% NH4OH/MeOH/EtOAc))、環状生成物を得た(2.0g、80%)。TLC(3%EtOAc/MeOH):Rf:=0.75,検出:UV,ニンヒドリン
LC−MS:tR= 5.59分(A4b), [M+H]+ 550
【0268】
ステップ451−9 Boc保護. THF/H2O混合物(1:1、40mL)中の大環状化合物(2.0g、3.64mmol、1.0当量)の溶液に、Na2CO3(1.93g、18.2mmol、5.0当量)およびBoc2O(5.01mL、21.84mmol、6.0当量)を添加して、混合物を室温で48〜72時間撹拌した。混合物をNH4Cl(飽和)でクエンチし、その後、水相をEtOAcで洗浄した(3回)。ひとまとめにした有機相をブラインで洗浄し、Na2SO4で乾燥させてろ過し、減圧蒸発させた。Boc−保護大環状化合物を得られた状態で次のステップで用いた。
LC−MS:tR= 9.14分(A4b), [M+H]+ 650
【0269】
ステップ451−10;N−ヒドロキシアミジンの形成. 絶対EtOH(35mL)中の大環状化合物(2.2g、3.35mmol、1.0当量)の溶液に、NH2OH・HCl(0.750g、10.74mmol、3.2当量)およびDIPEA(2.04mL、11.72mmol、3.5当量)を添加して、得られた混合物を一晩、加熱還流した。混合物を室温まで放冷し、その後、揮発物を減圧蒸発させた。得られた黄色透明油状物を次のステップで直接用いた。
LC−MS:tR= 7.20分(A4b), [M+H]+ 683
【0270】
ステップ451−11 N−アセトキシアミジンの形成. 10分間撹拌したAcOH(35mL)中の大環状化合物(2.2g、3.35mmol、1.0当量)の溶液に、Ac2O(2mL、16.75mmol、5.0当量)を添加した。得られた混合物を室温で2.5時間撹拌した。揮発物を減圧蒸発させた。得られた粗油状物をフラッシュクロマトグラフィーにより精製して(10%MeOH/EtOAc)、所望の生成物を得た(1.80g、3ステップ全体で74%)。
LC−MS:tR= 13.12分(A4b), [M+H]+ 725;[M+2H−Boc]+625
【0271】
ステップ451−12 アミジンの形成. AcOH(35mL)中の先のステップからの大環状化合物(1.40g、1.93mmol、1.0当量)の溶液に、亜鉛末(1.26g、19.3mmol、10.0当量)を添加した。得られた混合物を55℃で一晩撹拌した。混合物を室温まで放冷し、その後、混合物を綿の短いパッドでろ過した。綿をAcOHで、そして最後にEtOAcで溶出した。揮発物を減圧蒸発させた。得られた黄色透明油状物は、次のステップで直接用いることができた。
【0272】
ステップ451−13 Bocの切断. 大環状化合物(1.40g、1.93mmol、1.0当量)をDCM−TFA−TES混合物(64%−33%−3%、20mL)に溶解して、室温で1.5時間撹拌した。混合物を真空濃縮した。粗油状物をTHFに溶解し、その後、溶媒を減圧蒸発させた。この手順を、溶媒としてトルエンを用い、その後、EtOAcを用いて繰り返した。得られた粗油状物をフラッシュクロマトグラフィーにより精製した(20%MeOH/DCMおよび0.5%TFA、その後、30%MeOH/DCMおよび0.5%TFA)。
TLC (30% MeOH/DCMおよび0.5% TFA):Rf: 0.61,検出:UV,ニンヒドリン
【0273】
大環状化合物・TFA塩をEtOAcに溶解し、その後、水性1M Na2CO3溶液を添加した。水相をEtOAcで抽出した(3回)。ひとまとめにした有機相をブラインで洗浄し、Na2SO4で乾燥させて、ろ過して減圧蒸発させた。所望の大環状化合物を、白色固体として得た(0.90g、82%)。1種のジアステレオマーのみを、1H NMRにより観察した。不純物がLC−MSに見出されれば、THFまたはCH3CNでの磨砕を利用して、純度を改善することができた。
LC−MS: tR = 4.49分(A4b), [M+H]+ 567
【0274】
脱保護は、ジオキサン中の4M HClで処理することにより実現することもできた。その場合の粗大環状化合物を、フラッシュクロマトグラフィーにより精製した(30%MeOH/DCMおよび0.5%TFA)。120mgの規模で、2段階全体で66%の収率が得られた。
【0275】
ステップ451−14 HCl塩の形成:化合物をアセトニトリルに溶解し、その後、0.1N HCl(4当量)を添加して、溶液を一晩凍結乾燥した。得られた固体をTHFと共に摩砕した。
LC−MS:tR= 6.14分(B4), [M+H]+ 567
【0276】
あるいはアミジノ基は、図示された通り大環状化合物の第二級アミン上のBoc−保護を利用せずに、合成することができた。
【化27】
【0277】
追加の代替的なアプローチは、以下の条件を利用して対応するシアノ前駆体から直接アミジノ含有大環状化合物を合成することである(Garigipati, R. S. Tetrahedron Lett. 1990, 31, 1969.)。
【化28】
【0278】
B.複数の代表的な本発明の化合物を同時合成するための標準的手順
標準的反応スキームを、図2および3に表わす。
【0279】
以下の手順は、複数の個々の化合物で同時に実施する多重反応の追跡を容易にする、無線周波数タグを含む特定の技術を利用する。しかしこれは必要ではなく、固相合成を個々の反応容器内で同時に実施することもできる。
【0280】
ステップB−1 AA3負荷. 塩化2−クロロトリチル樹脂をMiniKan(または他の適切な分離可能な反応容器)に加えて、DCMで15分間洗浄した。DCMを除去して、DIPEA(4当量)およびFmoc−NH−AA3(2当量)の溶液を添加した(別個のAA3それぞれに、MiniKanを含む別個の容器を用いた)。反応混合物を、オービタルシェーカで室温で一晩かき混ぜた。MiniKanを以下のサイクルで2回洗浄した:DCM、iPrOH、DCM、その後、N2気流の下で乾燥させた。
【0281】
MiniKan(QC用)1個、またはMiniKan1個から取り出した樹脂の一部を、HFIP:DCM(1:4、5mL)混合物中で反応させてオービタルシェーカで室温で少なくとも30分間かき混ぜた。樹脂をDCMで洗浄し、揮発物を減圧蒸発させた。そのようにして得られた粗油状物を、その後、負荷効率の推定のために、定量的QC分析に供した。
【0282】
ステップB−2 Fmoc−脱保護. MiniKanをNMP中の20%ピペリジン溶液(3.5mL/MiniKan)で処理し、その後、オービタルシェーカで30分間かき混ぜた。その後、この処理を繰り返した。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、DCM、IPA、DCM(3回)、その後、N2気流の下で乾燥させた。
【0283】
ステップB−3 AA2カップリング. Fmoc−NR−AA2−OH(2.5当量)を、NMPに溶解し、その後、DIPEA(5当量)、続いてHATU(2.5当量)を添加した。混合物を室温で10分間撹拌し、その後、適切な組み合わせのMiniKanに移し(AA2により別個の容器に分離し)、オービタルシェーカで室温で一晩かき混ぜた。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、DCM、IPA、DCM(3回)、その後、N2気流の下で乾燥させた。
【0284】
ステップB−4 Fmoc−脱保護. MiniKanをNMP中の20%ピペリジン溶液(3.5mL/MiniKan)で処理し、その後、オービタルシェーカで30分間かき混ぜた。その後、この処理を繰り返した。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、DCM、IPA、DCM(3回)、その後、N2気流の下で乾燥させた。
【0285】
ステップB−5 AA1カップリング. Fmoc−NH−AA1−OH(2.5当量)を、NMPに溶解し、その後、DIPEA(5当量)、続いてHATU(2.5当量)を添加した。混合物を室温で10分間撹拌し、その後、適切な組み合わせのMiniKanに移して(AA1により別個の容器に分離した)、オービタルシェーカで室温で一晩かき混ぜた。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、DCM、IPA、DCM(3回)、その後、N2気流の下で乾燥させた。
【0286】
ステップB−6A テザーの酸化. テザーのDMSO溶液に、IBX(1.5当量)を添加した。不均質混合物を室温で5分間撹拌し、その後、H2Oを添加して、室温で一晩撹拌し続けた。混合物を水でクエンチし(白色沈殿物が形成された)、その溶液を室温で20分間撹拌した。固体をろ過により除去し、EtOAcで洗浄して、得られた溶液を水性NaHCO3およびブラインで洗浄し、MgSO4で乾燥させ、その後、減圧濃縮した。粗アルデヒドを真空乾燥させて、構造を1H NMRにより確認し、その後、その状態で次のステップで用いた。
【0287】
ステップB−6B 還元アミノ化. MiniKanをNMP中の20%ピペリジン溶液(3.5mL/MiniKan)で処理し、その後、オービタルシェーカで30分間かき混ぜた。その後、この処理を繰り返した。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、DCM、IPA、DCM(3回)、その後、N2気流の下で乾燥させた。ステップ6Aからの粗テザーアルデヒドをTMOF−MeOHの混合物(1:3)に溶解した。得られた溶液を、適切なMiniKanを含む容器に移し(テザーにより分離)、オービタルシェーカで室温で10分間かき混ぜた。BAP試薬(2当量)を添加して、室温で一晩かき混ぜ続けた。[気体が発生することに注意し、容器をしっかりと密閉して(または開孔して)溶媒の損失を回避しなければならない]。MiniKanを以下の順序で洗浄した:DCM(2回)、THF−DCM/MeOH(3:1)、THF/MeOH(3:1)、DCM(3回)、その後、N2気流の下で乾燥させた。
【0288】
ステップB−7 N−ヒドロキシアミジンの形成 最初に、NMP中のNH2OHの1M溶液を、以下の通り調製した:NH2OH・HCl 3.51gをDIPEA(9.2mL)に溶解し、その後、NMPを用いて容量を50mLに調整した。残留する塩が完全に解離するまで、不均質混合物を室温で撹拌した。MiniKanをNMP(4mL/MiniKan)で処理して、溶液をN2/真空サイクルで脱気し(30分間)、その後、NH2OHの1M NMP溶液を添加して(2mL/MiniKan)、混合物を50℃(オイルバス)で24時間撹拌した。溶液を室温まで放冷した。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、NMP、IPA、THF−DCM/MeOH(3:1)、DCM(3回)、その後、N2気流の下で乾燥させた。
【0289】
ステップB−8 樹脂からの開裂 樹脂を個々のMiniKanから除去して、別個の20mL反応器に導入した。HFIP/DCM(1;4)の溶液を添加して、得られた赤色溶液をオービタルシェーカで1時間かき混ぜた。樹脂をろ過により除去して、DCMで洗浄し、揮発物を真空蒸発させた(複数の試料のためにSpeedVac遠心エバポレータを用いた)。
【0290】
ステップB−9 N−アセトキシアミジンの形成 ステップB−9〜B−11に表わされた反応速度が、250μmolトリペプチド(理論的収量)に基づいていて、他の量の場合には比例的に調整しうることに留意されたい。ステップ8からの個々の油状物をAcOH(2.5mL)に溶解し、溶液を室温で10分間撹拌し、その後、Ac2Oを添加して(0.15mL、1.25mmol、5当量)、45分間撹拌し続けた。揮発物を真空蒸発させた(複数の試料のためにSpeedVac遠心エバポレータを用いた)。
【0291】
ステップB−10 テザーの脱保護および大環状化. ステップB−9からの個々の残渣を、TES−TFA−DCM混合物に溶解し(3:33:64、5mL)、溶液を室温で45分間撹拌した。揮発物を真空蒸発させ(複数の試料のためにSpeedVac遠心エバポレータを用いた)、その後、残渣をトルエンに溶解して、再度、真空濃縮した(SpeedVacによる)。
【0292】
Ddz保護されたテザーでは、TFA−TES−DCM(2:3:95)の混合物を脱保護ステップに用いた。Boc側鎖の脱保護が起こる可能性があるため、Ddzの脱保護が1時間を超えないことが重要である。
【0293】
個々の油状物をTHF(25mL)に溶解し、その後、DIPEA(300μL、1.75mmol、7当量)、続いてDEPBT(0.150g、0.50mmol、2当量)を添加した。黄色溶液をオービタルシェーカで室温で一晩かき混ぜた。Si−Trisアミン樹脂を導入し(3.5g/反応)、得られた混合物をオービタルシェーカで室温で2時間かき混ぜた。樹脂をろ過により除去してTHFで洗浄し、揮発物を真空蒸発させた(複数の試料のためにSpeedVac遠心エバポレータを用いた)。
【0294】
ステップB−11 アミジンの形成. ステップ10からの油状物をAcOH(3mL)に溶解し、その後、亜鉛末(0.163g、2.5mmol、10当量)を添加して、溶液をオービタルシェーカで室温で一晩かき混ぜた。綿の短いパッドを用いて過剰の亜鉛末を除去し、その後、AcOHで溶出した。揮発物を真空蒸発させ(複数の試料のためにSpeedVac遠心エバポレータを用い)、その後、残渣をFraction Lynxでの精製に供して、所望の生成物を得た。
【0295】
所望の大環状化合物がアミジノ基を有さない場合には、ステップB−9およびB−11を省略した。他の特定の順序では、AA3位のBoc側鎖脱保護を、TFA−TES−DCM系を用いた標準条件下で実施した。加えてAA1位のTrt側鎖脱保護は、TFA−TES(95:5)を用いた標準条件下で実施した。
【0296】
前述は本発明の例示であり、その限定と解釈すべきではない。本発明は以下の特許請求の範囲と本明細書に包含される特許請求の範囲の均等物により定義される。
【技術分野】
【0001】
[関連出願の相互参照]
本願は、ここに引用することで本明細書の一部をなすもとする2009年10月23日出願の米国特許仮出願第61/254,434号の優先権を主張するものである。
【0002】
[発明の分野]
本発明は、セリンプロテアーゼ酵素に結合し、そして/またはその調整物質、詳細には阻害剤である、新規な大環状化合物および薬学的に許容しうるその塩に関する。本発明は、これらの化合物の中間体、これらの化合物を含有する医薬組成物および該化合物を使用する方法にも関する。該化合物は、増殖過剰性障害、詳細には、腫瘍転移、炎症性障害、皮膚および組織障害、心臓血管障害、呼吸器障害ならびにウイルス感染を特徴とするそれらの障害をはじめとする一定範囲の適応症の処置および予防のための治療薬として有用である。
【背景技術】
【0003】
発明の背景
セリンプロテアーゼ酵素は、ホ乳類、ウイルス、細菌および他の生物体における複数の重要な生理学的プロセスに関与し、組織のホメオスタシスおよび修復、発達、免疫性ならびに生殖能力などのような多様な機能を調節する。これらのプロテアーゼは、生化学的レベルでは、ホルモン、成長因子、サイトカインおよび他の内在性の生理学的送達物質の活性化、イオンチャネルの調節、受容体の活性化ならびに細胞透過性の制御を担う。
【0004】
この種の作用により、セリンプロテアーゼは、医薬品開発のターゲットとなった(Drews, J.; Ryser, S. Nat. Biotech. 1997, 15, 1318-1319; Imming, P.; Sinning, C.; Meyer, A. Nat. Rev. Drug Disc. 2006, 5, 821-834.)。実際に、新薬開発につながる生物学的ターゲット全体の3〜4%が、この分類の一種と推定された(Hopkins, A.L.; Groom, C.R. Nat. Rev. Drug Disc. 2002, 1, 727-730.)。具体的には、これらの酵素の阻害剤が、効果的心臓血管調整物質、呼吸器疾患処置剤、抗炎症剤、抗ウイルス薬およびCNS薬として広範囲の薬学的関連活性を有することが立証された。加えて、様々な組織の維持プロセスにおいてセリンプロテアーゼが密接に関与することで、それらが癌(Bialas, A.; Kafarski, P. Anti-cancer Agents Med. Chem. 2009, 9, 728-762) 、ならびに皮膚疾患および障害(Meyer-Hoffert, U. Arch. Immunol. Ther. Exp. 2009, 57, 345-354) の新しいターゲットとなった。
【0005】
とりわけ、癌細胞のより潜行的特徴は、体内の他の部位へ拡大または転移する能力である。多くの例では、高い転移性を有する腫瘍は転帰が不良であるため、腫瘍の転移能力は予後に相関する。タンパク質分解活性の上昇は、癌の進行および転移に関連づけられてきた。数あるタンパク質分解酵素の中でもセリンプロテアーゼは、細胞構造の分解および組織リモデリングに寄与し、それにより癌浸潤および拡大の一助となる。更にプロテアーゼは、癌細胞の増殖または血管新生を刺激するのに必要となる増殖因子の宿主の活性化に関与する。このプロセスに関与するセリンプロテアーゼの幾つかが、ウロキナーゼ、プラスミン、エラスターゼ、トロンビンおよびカテプシンGである。癌に関与するプロテアーゼごとに異なる基質特異性が見出され、これらのプロテアーゼの選択的ターゲッティングが可能であることが示唆された(Beliveau, F.; Desilets, A.; Leduc, R. FEBS J. 2009, 276, 2213-2226.)。加えて、II型膜貫通性セリンプロテアーゼ(TTSP)と呼ばれるセリンプロテアーゼの新しい種類が、組織ホメオスタシスおよび癌、詳細には腫瘍転移に重要であることが見出された(Wu, Q. Curr. Top. Develop. Biol. 2003, 54, 167-206; Qui, D.; Owen, K.; Gray, K.; Bass, R.; Ellis, V. Biochem. Soc. Trans. 2007, 35, 583-587.)。TTSPファミリーの一種は、消化、心臓機能、血圧調節および聴力と同様に多様な生理学的プロセスにも役割を有する(Bugge, T.H.; Antalis, T.M.; Wu, Q. J. Biol. Chem. 2009, 284, 23177-23181.)。これらの役割では、TTSPは、典型的にはホメオスタシスを保持する働きがあり、多くの場合、ホルモンもしくは成長因子の活性化、またはタンパク質分解カスケードの開始に関与する。加えて、より近年の知見で、インフルエンザおよび他の呼吸器系ウイルス、例えばヒトメタニューモウイルスが、TTSPを活用してその伝播を促進し、これらのプロテアーゼがウイルス感染に介入する潜在的ターゲットとなることが示唆された(Choi, S.-Y.; Bertram, S.; Glowacka, I.; Park, Y.W.; Pohlmann, S. Trends Mol. Med. 2009, 15, 303-312.)。
【0006】
TTSPは、細胞質、膜貫通領域、C末端のリガンド結合ドメインおよびセリンプロテアーゼドメイン中に保持される短いN末端テールを特徴とする。そのような特性により、それらは他の細胞表面タンパク質および隣接細胞の成分と相互作用するのに理想的である。
【0007】
この酵素類の一種であるマトリプターゼ(マトリプターゼ−1、MT−SP1、TADG−15、エピチン、ST14)は、上皮の起源細胞により発現され、非常に様々なヒト癌において過剰発現されるトリプシン様セリンプロテアーゼである(US 5,482,848; US 5,792,616; US 5,972,616; US 6,649,741; US 7,030,231; US 7,227,009; US 7,276,364; US 7,291,462; WO 99/42120; WO 00/53232; WO 01/23524; WO 01/29056; WO 01/57194; WO 01/36604; US 2003/0119168; US 2006/0099625; US 2008/0051559; Takeuchi, T.; Shuman, M.A.; Craik, C.S. Proc. Natl. Acad. Sci. 1999, 96, 11054-11061; Lin, C.Y.; Anders, J.; Johnson, M.; Sang, Q.A.; Dickson, R.B.; J. Biol. Chem. 2001, 274, 18231-18236; Oberst, M.; Johnson, M.; Dickson, R.B.; Lin, C.-Y. Recent Res. Develop. Biochem. 2002, 3, 169-190; Lin, C.-Y.; Oberst, M.; Johnson, M.; Dickson, R.B. Handbook of Proteolytic Enzymes, 2nded., Barrett, A.J.; Rawlings, N.D.; Woessner, J.F., Elsevier: London, 2004, pp 1559-1561; List, K.; Bugge, T.H.; Szabo, R. Mol. Med. 2006, 12, 1-7; Lee, M.-S.; Johnson, M.D.; Lin, C.-Y. J. Cancer Mol. 2006, 2, 183-190; Uhland, K. Cell. Mol. Life Sci. 2006, 63, 2968-2978; List, K. Future Oncol. 2009, 5, 97-104.)。TTSPとしてのマトリプターゼは、細胞内から分泌されるか、または細胞内に保持されるほとんどのプロテアーゼとは異なり、細胞表面に容易に接近することができ、このためワクチン、モノクローナル抗体および小分子化合物をはじめとする様々な治療薬の良好なターゲットである。その酵素を阻害すると、腫瘍形成における2種の不可欠な媒介物質、つまり肝細胞増殖因子(HGF)およびウロキナーゼ型プラスミノーゲン活性化物質(uPA)の同時阻害が得られる。HGFおよびuPAは、癌浸潤および転移における、細胞運動性、細胞外マトリックス分解および腫瘍血管分布での役割について関連づけられてきた。
【0008】
マトリプターゼ活性は、広範囲のトリプシン様セリンプロテアーゼと拮抗する活性を示す上皮クニッツ型膜貫通阻害剤である、内在性物質の肝細胞増殖因子活性化因子阻害因子(HAI−1)により調節される(Oberst, M.D.; Chen, L.-Y.L.; Kiyomiya, K.-I.; Williams, C.A.; Lee, M.-S.; Johnson, M.D.; Dickson, R.B.; Lin, C.-Y. Am. J. Physiol. 2005, 289, C462-C470; Kojima, K.; Tsuzuki, S.; Fushiki, T.; Inouye, K. J. Biol. Chem. 2008, 283, 2478-2487.)。
【0009】
マトリプターゼは、細胞外マトリックスの分解において役割を果たし、腫瘍転移を促進することが見出された(WO 00/53232; WO 01/97794; WO 02/08392; Hooper, J. Biol. Chem. 2001, 276, 857-860.)。この活性は、ストロムテリシンおよびIV型コラーゲナーゼをはじめとする特定のマトリックスメタロプロテアーゼ酵素(MMP)で認められるものと類似している。マトリプターゼ−1発現の低下は、マウスモデルの前立腺癌における攻撃性および進行の低下に関連した。(Sanders, A.J.; Parr, C.; Davies, G.; et al. J. Exp. Ther. Oncol. 2006, 6, 39-48.)
【0010】
加えて、マトリプターゼは、全身の上皮ホメオスタシスおよび末端上皮分化を担う細胞周囲タンパク質分解経路において役割を果たす(List, K.; Kosa, P.; Szabo, R.; et al. Am. J. Pathol. 2009, 175, 1453-1463.)。マトリプターゼは、PAR−2の活性化を介した、内皮細胞中の炎症性サイトカインの放出も誘導する。それゆえ阻害剤は、抗炎症薬としての用途を有するであろう。更に該プロテアーゼは、単球中で発現され、それとPAR−2との相互作用はアテローム性硬化症に寄与する。つまりマトリプターゼの阻害剤は、アテローム性硬化症の処置および予防にも用途を有する(Seitz, I.; Hess, S.; Schulz, H.; Eckl, R.; Busch, G.; et al. Arterioscler. Throm. Vasc. Biol. 2007, 27, 769-775.)。
【0011】
マトリプターゼの遺伝子発現は、骨関節炎において有意に上昇することが見出され、その酵素は、軟骨マトリックス分解をもたらす多重機構の開始に関与する(Milner, J.A.; Patel, A.; Davidson, R.K.; et al. Arthr. Rheum. 2010, 62, 1955-1966.)。それゆえその酵素の阻害は、この適応症の治療の1つのアプローチとなろう。
【0012】
マトリプターゼ−2(TMPRSS6)は、肝臓により発現されるTTSPである(WO 2008/009895; Ramsay, A.J.; Reid, J.C.; Velasco, G.; Quigley, J.P.; Hooper, J.D. Front. Biosci. 2008, 13, 569-579.)。マトリプターゼ−2は、正常な状態では、腸における鉄吸収とマクロファージからの鉄放出を阻害するホルモンのヘピシジンをダウンレギュレートする作用がある。この酵素が遺伝子内で突然変異を起こすと、高レベルのへプシジンにより、ヒトにおける鉄不応性鉄欠乏性貧血(IRIDA)に関連した、異常なタンパク質分解活性をもたらす(Folgueras, A.R.; Martin de Lara, F.; Pendas, A.M.; Garabaya, C.; et al. Blood 2008, 112, 2539-3545; Anderson, G.J.; Frazer, D.M.; McLaren, G.D. Curr. Opin. Gastroenterol. 2009, 25, 129-135; Ramsay, A.J.; Hooper, J.D.; Folgueras, A.R.; Velasco, G.; Lopez-Otin, C. Haematologica 2009, 94, 840-849; Finberg, K.E. Semin. Hematol. 2009, 46, 378-386; Cui, Y.; Wu, Q.; Zhou, Y. Kidney Intl. 2009, 76, 1137-1141; Lee, P. Acta Haematologica 2009, 122, 87-96; deFalco, L.; Totaro, F.; Nai, A.; et al. Human Mut. 2010, 31, e1390-e1405.)。この酵素は、マトリプターゼ−1に対して35%の配列相同性を有する。
【0013】
マトリプターゼ−2は、マトリプターゼ−1の作用とは異なり、乳房腫瘍増殖および浸潤を血漿レベルで阻害し、好適な予後に相関する(Parr, C.; Sanders, A.J.; Davies, G.; et al. Clin. Cancer Res. 2007, 13, 3568-3576.)。治療的アプローチとしての、癌の発生および進行ならびに調整能力におけるこの酵素の役割は、依然として活発な研究領域である(Sanders, A.J.; Webb, S.L.; Parr, C.; Mason, M.D.; Jiang, W.G. Anti-cancer Agents Med. Chem. 2010, 10, 64-69.)。マトリプターゼ−2およびそれに由来する薬剤は、前立腺癌の処置薬としても報告された(WO 2009/009895)。
【0014】
マトリプターゼ−3は、多くの種で保存され、広範囲のセルピン活性を示すが、他のTTSPに類を見ない発現パターンおよび調節ネットワークを有する(Szabo, R.; Netzel-Arnett, S.; Hobson, J.P.; Antalis, T.M. Bugge, T.H. Biochem. J. 2005, 390, 231-242.)。
【0015】
マトリプターゼ酵素に加えて、他のTTSPとしては、非限定的に、ヘプシン(TMPRSS1)、TMPRSS2、TMPRSS3/TADG−12、TMPRSS4、モザイクセリンプロテアーゼ大型(MSPL)、TMPRSS11A、ヒト気道トリプシン(HAT)様プロテアーゼ、HAT−like−2、HAT−like−3、HAT−like−4、HAT−like−5、ポリセラーゼ−1、スピネシン、エンテロペプチダーゼ、コリンおよび扁平上皮癌における差異的発現遺伝子1(differentially expressed in squamous cell carcinoma 1)(DESC1)が挙げられる。TTSP遺伝子の突然変異は、ヒトにおける複数の遺伝子障害の潜在的原因として確証されており、TTSP遺伝子発現の変化はヒト癌腫に関連する。
【0016】
プロテアーゼは、様々な有害な皮膚状態の誘発にも関与する。それは、表皮の分化(Zeeuwen, P.L.J.M.; Eur. J. Cell Biol. 2004, 83, 761-773)と、上皮の発育(Bugge, T.H.; List, K.; Szabo, R. Front. Biosci. 2007, 12, 5060-5070)の両方において役割を果たす。セリンプロテアーゼを含むシグナル伝達カスケードは、表皮のホメオスタシスにおいて重大な役割を果たす(Ovaere, P.; Lippens, S.; Vandenabeele, P.; Declercq, W. Trends Biochem. Sci. 2009, 34, 453-463.)。これらには、マトリプターゼ−1に加えて、フリン、プロスタシン、カリクレイン関連ペプチダーゼ4(KLK4、プロスターゼ)、角質層トリプシン様酵素(SCTE、カリクレイン関連ペプチダーゼ5、KLK5)、カリクレイン関連ペプチダーゼ6(KLK6、プロテアーゼM)、角質層キモトリプシン様酵素(SCCE、カリクレイン関連ペプチダーゼ7、KLK7)、カリクレイン関連ペプチダーゼ8(KLK8、ニューロプシン)、カリクレイン関連ペプチダーゼ10(KLK10)、カリクレイン関連ペプチダーゼ11(KLK11)、カリクレイン関連ペプチダーゼ13(KLK13)、カリクレイン関連ペプチダーゼ14(KLK14)が挙げられる。例えば、疾患発症中にマトリプターゼにより活性化されるプロカリクレイン経路の関与が、ネザートン症候群のマウスモデルで確認された(Sales, K.U.; Masedunskas, A.; Bey, A.L.; et al. Nat. Genetics 2010, 42, 676-683.)。これらのプロテアーゼ酵素が皮膚層を含む保護組織を破壊し始めると、それらは炎症反応を誘発する。加えて、皮膚のタンパク質分解バランスの変化が炎症をもたらして、発赤、鱗屑および痒みが生じる可能性がある。事実、フィラグリン、プロテアーゼ活性化受容体(PAR)およびコルネオデスモシンをはじめとするプロテアーゼ、その阻害物質およびターゲットタンパク質は、皮膚組織のための調節ネットワークを含み、皮膚の統合性およびバリア機能に寄与する(Meyer-Hoffert, U. Arch. Immunol. Ther. Exp. 2009, 57, 345-354.)。阻害剤は、これらの炎症事象を低減して、様々な皮膚および組織障害を処置するのに有用となろう。
【0017】
マトリプターゼは、皮膚に加えて、腸における上皮バリア形成および透過性の調節において重要な役割を果たす(Buzza, M.S.; Netzel-Arnett, S.; Shea-Donohue, T.; et al. Proc. Nat. Acad. Sci. 2010, 107, 4200-4205.)。
【0018】
プロテアーゼは、上皮ナトリウムチャネル(ENaC)の調節も担う(Planes, C.; Caughey, G.H.; Curr. Top. Development. Biol. 2007, 78, 23-46; Frateschi, S.; Charles, R.-P.; Hummler, E. Open Derm. 2010, 4, 27-35.)。ENaCの調整に関与するチャネル活性化プロテアーゼ(CAP)としては、プロスタシン(CAP1、PRSS8)、PRSS22、TMPRSS11B、TMPRSS11E、TMPRSS2、TMPRSS3、TMPRSS4、(MT−SP2)、MT−SP1、CAP2、CAP3、トリプシン、カテプシンAおよび好中球エラスターゼが挙げられる。CAPの阻害剤が、図示されたピロリジン塩基骨格の周囲を基盤とする化学構造と共に開示された(WO 2007/137080; WO 2007/140117; WO 2008/085608; WO 2008/097673; WO 2008/097676)。
【化1】
【0019】
今日まで、マトリプターゼの阻害剤は、わずかな数しか記載されていない。これらは、小分子、例えば第二級アミノ酸アミドのメタ置換スルホニルアミドを含む(WO 2008/107176; Steinmetzer, T.; Doennecke, D.; Korsonewski, M.; Neuwirth, C.; Steinmetzer, P.; Schulze, A.; Saupe, S.M.; Schweinitz, A. Bioorg. Med. Chem. Lett. 2009, 19, 67-73; Schweinitz, A.; Doennecke, D.; Ludwig, A.; Steinmetzer, P.; Schulze, A.; Kotthaus, J.;Wein, S.; Clement, B.; Steinmetzer, T. Bioorg. Med. Chem. Lett. 2009, 19, 1960-1965.)。
【化2】
【0020】
マトリプターゼ阻害剤の別の構造類は、N−スルホニル化アミノ酸誘導体に基づいている(WO 2004/101507; US 2007/0055065; Steinmetzer, T.; Schweinitz, A.; Stuerzbecher, A.; et al. J. Med. Chem. 2006, 49, 4116-4126)。
【化3】
【0021】
直鎖状ペプチド(US 6,797,504; US 7,157,596; WO 02/020475)およびペプチド模倣体(US 7,019,019; WO 2004/058688)阻害剤が、開示された。
【化4】
【0022】
これらのペプチド模倣体マトリプターゼ阻害剤の一種であるCVS−3983が、腫瘍転移のインビボモデルにおいて活性を示した(Galkin, A.V.; Mullen, L.; Fox, W.D.; Brown, J.; et al. Prostate 2004, 61, 228-235.)。
【化5】
【0023】
他の2種のペプチド模倣体阻害剤、CJ−1737およびCJ−672の代謝および分布に関する研究で、この種の分子についての動物とヒトとの重大な代謝的差異が明らかとなった(Kotthaus, J.; Steinmetzer, T.; Kotthaus, J.; Schade, D.; van de Locht, A.; Clement, B. Xenobiotica 2010, 40, 93-101.)。
【化6】
【0024】
より近年になり、4−アミジノベンジルアミドを含有するN−保護ジペプチドが、マトリプターゼ−1およびマトリプターゼ−2阻害剤として報告された(Sisay, M.T.; Steinmetzer, T.; Stirnberg, M.; Maurer, E.; Hammami, M.; Bajorath, J.; Guetschow, M. J. Med. Chem. 2010, 53, 5523-5535.)。化合物1は、マトリプターゼ−1の阻害に関し、マトリプターゼ−2の50倍の選択性を示した。これらの、マトリプターゼ−2の最初の小分子阻害剤は、鉄障害、例えば、ヘプシジンレベルが過度に低いヘモクロマトーシスおよび鉄負荷性貧血(iron loading anemias)を処置するための潜在的治療薬として示唆される。
【化7】
【0025】
エグリンC変異体であるより長い直鎖ペプチドも、マトリプターゼ阻害剤として公知である(Desilets, A.; Longpre, J.-M.; Beaulieu, M.-E.; Leduc, R. FEBS Lett. 2006, 580, 2227-2232.)。
【0026】
アミノ酸残基が14個の二環式ペプチドであるヒマワリトリプシン阻害剤(SFTI−1)が、マトリプターゼおよびカテプシンGの阻害剤として同定された。この阻害剤は、エラスターゼ、トロンビンおよびXa因子をはじめとする他のプロテアーゼ酵素に対比して選択性を有する。(Luckett, J. Mol. Biol. 1999, 290, 525.)。不幸にもSFTI−1は、インビボでは相対的に急速に分解され、重要な生理学的セリンプロテアーゼ、トリプシンおよびキモトリプシンを上回る選択性を示さず、そのため医薬剤としての使用に適さない。
【化8】
【0027】
同じく本質的に二環式であるSFTI−1類似体および模倣体も、報告された(US 7,439,226; WO 2006/043933; Long, Y.-Q.; Lee, S.-L.; Lin, C.-Y.; Enyedy, I.J.; Wang, S.; Li, P.; Dickson, R.B.; Roller, P.P. Bioorg. Med. Chem. Lett. 2001, 11, 2515-2519; Jiang, S.; Li, P.; Lee, S.-L.L.; Lin, C.-Y.; Long, Y.-Q.; Johnson, M.D.; Dickson, R.B. Roller, P.B. Org. Lett. 2007, 9, 9-12; Li, P.; Jiang, S.; Lee, S.-L.L.; Lin, C.-Y.; Johnson, M.D.; Dickson, R.B.; Michejda, C.J.; Roller, P.J. J. Med. Chem. 2007, 50, 5976-5983.)。
【0028】
アミノ酸を11または14個含有する環状ペプチド、およびマトリプターゼをはじめとするセリンプロテアーゼの阻害により作用する皮膚刺激の予防または処置のための使用方法が、US 7,217,690に開示された。
【0029】
天然および合成プロテアーゼ阻害剤(Yamasaki, Y.; Satomi, S.; Murai, N.; Tsuzuki, A.; Fushiki, T. J. Nutr. Sci. Vitamin. 2003, 49, 27-32)、ならびに合成クンツ型阻害剤(WO 2007/079096)が、マトリプターゼをはじめとする複数のプロテアーゼ酵素に拮抗する活性を示した。
【0030】
事実、特定のプロテアーゼ類のうち、該酵素は、共通の化学的および構造的特性を利用してそれらの基質と相互作用し、つまり阻害剤は、多くの場合、その分類の他の酵素も阻害することができる。もちろん酵素間の選択性が、特異的副作用を抑制するなどで重要となる場合、これは克服すべき難題も生じる。
【0031】
構造を基にした設計から得られる、図示されたアミジンまたは代替物など、2つ以上の重要な塩基性相互作用部分を分離する直鎖構造を有する一連のマトリプターゼを阻害剤が、報告された(US 6,677,377; WO 01/097784; Enyedy, I.J.; Lee, S.-L.; Kuo, A.H.; Dickson, R.B.; Lin, C.-Y.; Wang, S. J. Med. Chem. 2001, 44, 1349-1355)。これらの化合物において、Zは、1個以上の酸素または硫黄原子で場合により置換された炭素原子の直鎖、または芳香族もしくは複素環芳香族のスペーサー成分のいずれかを表す。
【化9】
【0032】
マトリプターゼを対象とするヒトモノクローナル抗体が、癌の診断、予防または処置に関して開示された(US 7,572,444; WO 2006/068975; Farady, C.J.; Sun, J.; Derragh, M.R.; Miller, S.M.; Craik, C.S. J. Mol. Biol. 2007, 369, 1041-1051; Farady, C.J.; Egea, P.F.; Schneider, E.L.; Darragh, M.R.; Craik, C.S. J. Mol. Biol. 2008, 380, 351-360.)。マトリプターゼマウス抗体を有することから(US 7,355,015)、様々なタイプの癌の処置、スクリーニング、診断、予後および治療に使用される、マトリプターゼタンパク質由来の他の抗体も、記載された(WO 2009/020645; US 2003/270245; US 2009/0155248)。マトリプターゼの検出用抗体キットは、US 7,022,821の主題である。
【0033】
診断または治療的目的で抗体を生成するのに用いうるマトリプターゼおよび他の癌関連プロテアーゼの部分的配列を含む抗原性ペプチドが、WO 2008/066749に提供されている。
【0034】
マトリプターゼ発現を刺激する薬剤が、美容目的で有用であるとして開示された(WO 2008/034821)。
【0035】
今日まで、マトリプターゼ阻害剤で臨床開発に至ったものはなく、そのため薬理学的物質としての追跡で既に研究されたものとは異なる構造を有する新規なマトリプターゼ阻害剤が、依然として求められている。
【発明の概要】
【発明が解決しようとする課題】
【0036】
発明の概要
本発明は、コンホメーションが定義された新規な大環状化合物を提供する。これらの化合物は、セリンプロテアーゼ酵素の調整物質、詳細には阻害剤として機能することができる。
【課題を解決するための手段】
【0037】
本発明の態様によれば、本発明は、式(I):
【化10】
(式中、
R1は、−H、−CH3、−CH2CH3、−(CH2)2CH3および−CH(CH3)2からなる群より選択され、
R2は、−H、−CH3および−CH2CH3からなる群より選択され、
R3は、任意で存在し、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より選択され、
mは、1、2、3、4または5であり、
X1は、アミジノ、ウレイドおよびグアニジノからなる群より選択され、
Wは、CR4aR4bからなる群より選択され、ここでR4aおよびR4bは、水素、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、
Z1は、CR5aR5bからなる群より選択され、ここでR5aおよびR5bは、水素、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、
Tは、
【化11】
からなる群より選択され、
ここでM1は、Oおよび(CH2)qからなる群より選択され、ここでqは、1、2、3、4または5であり、M2は、O、S、NR6およびCR7aR7bからなる群より選択され、ここでR6は、水素、アルキル、ホルミル、アシル、カルボキシアルキル、カルボキシアリール、アミド、スルホニルおよびスルホンアミドからなる群より選択され、R7aおよびR7bは、水素、ヒドロキシル、アルコキシ、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、p1およびp2は、独立して0、1、2または3であり、そしてp3、p4およびp5は、独立して0、1または2であり、
(W)は、Wの付加された炭素原子への結合部位を示し、
(Z)は、Z1の付加された炭素原子への結合部位を示す)
による化合物および薬学的に許容し得るその塩に関する。
【0038】
本発明の更なる態様は、式(II):
【化12】
(式中、
R11は、−H、−CH3、−CH2CH3、−(CH2)2CH3および−CH(CH3)2からなる群より選択され、
R12は、−H、−CH3および−CH2CH3からなる群より選択され、
R13は、−(CH2)r1NR18aR18b、−(CH2)r2CONR19aR19b、
【化13】
からなる群より選択され、
ここでr1は、1、2、3、4または5であり、r2は、1、2または3であり、R18a、R19aおよびR19bは、水素およびC1〜C4アルキルからなる群より独立して選択され、R18bは、水素、C1〜C4アルキル、ホルミル、アシル、アミド、アミジノおよびスルホンアミドからなる群より選択され、A1、A4、A7、A9、A12、A14、A17、A19、A23、A35、A37およびA39は、それぞれ任意で存在し、ハロゲン、トリフルオロメチル、アミジノ、ウレイド、グアニジノ、ヒドロキシル、アルコキシおよびC1〜C4アルキルからなる群より独立して選択され、A2、A3、A5、A6、A8、A10、A11、A13、A15、A16、A18、A20、A21、A24、A25、A36、A38およびA40は、それぞれ任意で存在し、ハロゲン、トリフルオロメチル、ヒドロキシル、アルコキシおよびC1〜C4アルキルからなる群より独立して選択され、A22、A26、A27、A29、A31およびA33は、それぞれ任意で存在し、トリフルオロメチル、アミジノ、ウレイド、グアニジノおよびC1〜C4アルキルからなる群より独立して選択され、A28、A30、A32およびA34は、それぞれ任意で存在し、トリフルオロメチルおよびC1〜C4アルキルからなる群より独立して選択され、そしてB1、B2、B3、B4、B5およびB7は、独立してNR20、SまたはOであり、ここでR20は、水素、アルキル、ホルミル、アシル、カルボキシアルキル、カルボキシアリール、アミド、スルホニルおよびスルホンアミドからなる群より選択され、B6およびB8は、独立してNまたはCHであり、
R14は、アミノ、ヒドロキシル、アルコキシ、カルボキシ、ウレイド、アミジノ、またはグアニジンで場合により置換されたC1〜C4アルキルと、アルキル、ヒドロキシルまたはアルコキシで場合により置換されたC3〜C7シクロアルキルとからなる群より選択され、
R15およびR16は、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より独立して選択され、
R17は、水素およびC1〜C4アルキルからなる群より選択され、
nは、1、2、3、4または5であり、
Z2は、CHR21aCHR22a、CR21b=CR22bおよびC≡Cからなる群より選択され、ここでR21aおよびR22aは、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より独立して選択されるか、またはR21aとR22aとが、結合する炭素と一緒になって、3員環を形成しており、そしてR21bおよびR22bは、水素およびC1〜C4アルキルからなる群より独立して選択され、
X2は、水素、ハロゲン、アミジノ、ウレイドおよびグアニジノからなる群より選択され、
X3は、水素、ヒドロキシル、アルコキシ、アミノ、ハロゲン、トリフルオロメチルおよびC1〜C4アルキルからなる群より選択され、
L2は、OおよびCR23aR23bからなる群より選択され、ここでR23aは、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より選択され、そしてR23bは、水素およびC1〜C4アルキルからなる群より選択され、
L3は、CX4およびNからなる群より選択され、ここでX4は、水素、ハロゲン、ヒドロキシル、アルコキシ、アミノ、ハロゲン、トリフルオロメチル、アミジノ、ウレイドおよびグアニジノからなる群より選択され、そして
L4は、CX5およびNからなる群より選択され、ここでX5は、水素、ハロゲン、トリフルオロメチル、ヒドロキシル、アルコキシ、アミノ、アミジノ、ウレイドおよびグアニジノからなる群より選択される)
による化合物または薬学的に許容し得るその塩に関する。
【0039】
本発明の新規な大環状化合物は、セリンプロテアーゼ酵素の調整物質、詳細には阻害剤として有用である。この阻害剤により、複数の異なる癌、詳細には腫瘍転移を特徴とする癌に取り組むことができる。加えてセリンプロテアーゼの阻害剤、例えば本発明の化合物を、皮膚障害、例えばアトピー性皮膚炎、酒さ、乾癬、魚鱗癬、濾泡性皮膚委縮、過角化症、減毛症、ネザートン症候群などの処置または予防に用いることができる。
【0040】
本発明の特定の実施形態において、セリンプロテアーゼ酵素は、マトリプターゼ−1(MTSP−1、ST14、TADG−15、エピチン)、マトリプターゼ−2(TMPRSS6)、マトリプターゼ−3、MTSP−4、MTSP−6、MTSP−7、MTSP−9、MTSP−10、PRSS22、TMPRSS11A、TMPRSS11C、TMPRSS2、TMPRSS3、TMPRSS4、TMPRSS5(スピネシン)、モザイクセリンプロテアーゼ大型(MSPL)、エンテロペプチダーゼ、ポリセラーゼ−1、コリン、ヒト気道トリプシン様プロテアーゼ(HAT)、HAT−like2、HAT−like3、HAT−like4、HAT−like5、プロスタシン(CAP1、PRSS8)、CAP2、CAP3、トリプシン、カテプシンA、好中球エラスターゼ、ヘプシン、角質層トリプシン様酵素(SCTE、カリクレイン関連ペプチダーゼ5、KLK5)、角質層キモトリプシン様酵素(SCCE、カリクレイン関連ペプチダーゼ7、KLK7)、カリクレイン関連ペプチダーゼ4(KLK4、プロスターゼ)、カリクレイン関連ペプチダーゼ8(KLK8、ニューロプシン)、カリクレイン関連ペプチダーゼ11(KLK11)、カリクレイン関連ペプチダーゼ13(KLK13)、カリクレイン関連ペプチダーゼ14(KLK14)、カリクレイン関連ペプチダーゼ6(KLK6、プロテアーゼM)、カリクレイン関連ペプチダーゼ10(KLK10)、グランザイムB、カルシウムシグナル伝達物質1、カルシウムシグナル伝達物質2、クローディン3、クローディン4、フリン、ラジニン、ラルニニン、プラスミン、ストラチフィン、SI00A2、CD24、リポカリン2、オステオポンチン、組織型プラスミノーゲン活性化因子、ウロキナーゼ型プラスミノーゲン活性化因子または扁平上皮癌における差異的発現遺伝子1(DESC1)である。
【0041】
本発明の化合物は、異常な新血管形成または血管新生を特徴とする病的状態にも有用である。そのような状態の例としては、非限定的に、目の新生血管疾患、血管腫、ならびに関節リウマチおよびクローン病をはじめとする慢性炎症を特徴とする障害が挙げられる。
【0042】
本発明の別の態様において、本発明の化合物は、特定の実施形態において、鉄不応性鉄欠乏性貧血(IRIDA)、全身鉄過負荷(ヘモクロマトーシス)または鉄負荷性貧血をはじめとする、鉄ホメオスタシスの調節解除を特徴とする病的状態を処置するのに用いることができる。
【0043】
本発明の更なる態様は、式(I)で示される化合物または式(II)で示される化合物と、薬学的に許容しうる担体、賦形剤または希釈剤とを含む医薬組成物を更に提供する。
【0044】
本発明の別の態様は、必要とする対象に有効量の式(I)または式(II)で示される化合物を投与することを含む、増殖過剰性障害、炎症性障害、組織障害、心臓血管障害、呼吸器障害またはウイルス感染を処置する方法を提供する。
【0045】
本発明の更なる態様は、使用するための任意の使用説明書と共に包装された有効量の本発明の化合物を1種以上含む医薬投与単位を含有する容器1個以上を含むキットを提供する。
【0046】
本発明の更なる態様は、式(I)および式(II)で示される化合物を製造する方法に関する。
【0047】
本発明の態様は、更に、本明細書に記載された障害、詳細には、病的状態、増殖過剰性障害、組織障害、炎症性障害、呼吸器障害およびウイルス感染を予防および/または処置する方法に関する。
【0048】
特定の実施形態において、増殖過剰性障害は、CMLをはじめとする白血病、リンパ腫、乳癌、胃腸癌、食道癌、胃癌(stomach cancer)、胃癌(gastric cancer)、結腸癌、腸癌、大腸癌、前立腺癌、膀胱癌、精巣癌、卵巣癌、子宮癌、子宮頸癌、子宮内膜癌、上皮癌、頭頸部癌、脳癌、肺癌、肝臓癌、腎臓癌、気管支癌、膵臓癌、甲状腺癌、骨癌および皮膚癌である。
【0049】
別の特定の実施形態において、増殖過剰性障害は、腫瘍転移を特徴とし、その腫瘍は、乳房、脳、卵巣、結腸、直腸、胃、肝臓、腎臓、腸、口、喉、食道、前立腺、精巣、膀胱、子宮、子宮頸部、肺、膵臓、骨、甲状腺または皮膚に見出される。
【0050】
別の特定の実施形態において、増殖過剰性障害は、前立腺癌、卵巣癌腫、子宮頸部新生物、小細胞肺癌、非小細胞肺癌、腎細胞癌、膵管腺癌、子宮平滑筋肉腫、移行上皮細胞腺癌、非メラノーマ皮膚癌、扁平上皮癌、悪性中皮腫または神経膠芽腫である。
【0051】
追加の実施形態において、本発明の化合物は、特定の実施形態において、アトピー性皮膚炎、酒さ、乾癬、魚鱗癬、濾泡性皮膚委縮、過角化症、減毛症、ネザートン症候群などをはじめとする組織または皮膚の障害の処置または予防に用いることができる。
【0052】
更に別の特定の実施形態において、炎症性障害は、関節リウマチ、骨関節炎、クローン病、潰瘍性大腸炎またはアテローム性硬化症である。
【0053】
更なる特定の実施形態において、病的状態は、上皮細胞増殖または異常な新血管形成を特徴とする。
【0054】
追加の特定の実施形態において、呼吸器障害は、嚢胞性線維症、気管支炎、慢性閉塞性肺疾患(COPD)、喘息、アレルギー性鼻炎、線毛機能不全、肺癌腫、肺炎または呼吸器感染である。
【0055】
更に別の特定の実施形態において、ウイルス感染は、インフルエンザウイルスまたはメタニューモウイルスにより誘発される。
【0056】
本発明は、本明細書に記載された障害の予防および/または処置用の医薬品の調製に使用される式(I)または(II)で示される化合物にも関する。
【0057】
本発明の前述および他の態様は、以下に示される記述により詳細に説明される。
【図面の簡単な説明】
【0058】
【図1】代表的な本発明の化合物を合成するための反応スキームを示す。
【図2】複数の代表的な本発明の化合物を同時合成するための反応スキームを示す。
【図3】複数の代表的な本発明の化合物を同時合成するための別の反応スキームを示す。
【図4】テザーT32を合成するための反応スキームを示す。
【図5】テザーT201を合成するための反応スキームを示す。
【発明を実施するための形態】
【0059】
ここに、本発明の前述および別の態様を、本明細書に記載された別の実施形態に関連させて、より詳細に記載する。本発明を異なる形態で具体化することができ、本明細書に示された実施形態に限定されると解釈してはならないことを、理解しなければならない。むしろこれらの実施形態を提供することで、この開示が徹底され完全となり、それが本発明の範囲を当業者に完全に伝えることになる。
【0060】
本明細書内の本発明の記載に用いられる用語法は、特定の実施形態のみの記載を目的とし、本発明を限定する意図はない。本発明の記載および添付の特許請求の範囲に用いられる単数形「a」、「an」および「the」は、他に明確に示されない限り、複数形も包含するものとする。加えて、本明細書で用いられる用語「および/または」は、関連して列挙された事柄1つ以上とのあらゆる組合せを包含しており、「/」と略されていてもよい。
【0061】
他に定義されない限り、本明細書で用いられる全ての技術的および科学的用語は、本発明の当業者により一般に理解されるものと同じ意味を有する。
【0062】
本明細書で引用された全ての発行物、米国特許出願、米国特許および他の参考資料は、その全体が参照により本明細書に組み入れられる。
【0063】
用語「アルキル」は、炭素原子を1〜20個、幾つかの例において炭素原子を1〜8個有する直鎖または分枝鎖飽和または部分不飽和炭化水素基を指す。用語「低級アルキル」は、炭素原子を1〜6個含有するアルキル基を指す。アルキル基の例としては、非限定的に、メチル、エチル、イソプロピル、tert−ブチル、3−ヘキセニル、および2−ブチニルが挙げられる。「不飽和」とは、二重結合もしくは三重結合またはその2種の組み合わせが1、2または3個存在することを意味する。そのようなアルキル基は、以下に記載される通り、場合により置換されていてもよい。
【0064】
本明細書に定義されたアルキルまたは他の炭化水素基に関連して下付き文字が用いられる場合、下付き文字は、その基が含みうる炭素原子数を指す。例えば、C2〜C4アルキルは、炭素原子が2、3または4個のアルキル基を示す。
【0065】
用語「シクロアルキル」は、環内に炭素原子を3〜15個、幾つかの例において3〜7個有する飽和または部分不飽和環状炭化水素基、および前記環状炭化水素基を含むアルキル基を指す。シクロアルキル基の例としては、非限定的に、シクロプロピル、シクロプロピルメチル、シクロペンチル、2−(シクロヘキシル)エチル、シクロヘプチル、およびシクロヘキセニルが挙げられる。本明細書で定義されたシクロアルキルとしては、複数の炭素環を有し、そのそれぞれが飽和または部分不飽和であってもよい基、例えば、デカリニル、[2.2.1]−ビシクロヘプタニルまたはアダマンタニルも挙げられる。そのようなシクロアルキル基は全て、以下に記載されたとおり場合により置換されていてもよい。
【0066】
用語「芳香族」は、電子を4n+2個(ここでnは、1以上の整数である)含む共役π電子系を有する不飽和環状炭化水素基を指す。芳香族分子は、典型的には安定しており、交互の二重結合および単結合からなる共鳴構造を有する原子の平面環、例えばベンゼンまたはナフタレンとして表わされる。
【0067】
用語「アリール」は、環原子を6〜15個、幾つかの例において6〜10個有する単環または縮合環の炭素環系の芳香族基、および前記芳香族基を含むアルキル基を指す。アリール基の例としては、非限定的に、フェニル、1−ナフチル、2−ナフチルおよびベンジルが挙げられる。本明細書で定義されたアリールは、ナフチルおよびアントラセニルと同様に縮合であってもよく、またはビフェニルおよびテルフェニルと同様に非縮合であってもよい、複数のアリール基を有する基も挙げられる。アリールは、環の1つが芳香族であり、その他が飽和、部分不飽和、または芳香族であってもよい、二環式または三環式炭素環、例えば、インダニルまたはテトラヒドロナフチル(テトラリニル)も指す。そのようなアリール基は全て、以下に記載される通り場合により置換されていてもよい。
【0068】
用語「複素環」または「複素環式」は、原子を3〜15個、幾つかの例において3〜7個有し、少なくとも1個の環の中にヘテロ原子を少なくとも1個有し、前記ヘテロ原子がO、SまたはNから選択される、飽和または部分不飽和単環、二環または三環基を指す。複素環基の各環は、各環内のヘテロ原子数の合計が4個以下であること、そして各環が炭素原子を少なくとも1個含むことを前提に、O原子を1または2個、S原子を1または2個、N原子を1〜4個含むことができる。二環式または三環式複素環基を完成する縮合環は、炭素環のみを含んでいてもよく、飽和または部分不飽和であってもよい。NおよびS原子は、場合により酸化されていてもよく、N原子は、場合により第四級化されていてもよい。複素環は、前記単環、二環または三環式複素環基を含むアルキルも指す。複素環の例としては、非限定的に、2−または3−ピペリジニル、2−または3−ピペラジニル、2−または3−モルホリニルが挙げられる。そのような複素環基は全て、以下に記載される通り場合により置換されていてもよい。
【0069】
用語「ヘテロアリール」は、環原子を5〜15個、幾つかの例において5〜10個有し、少なくとも1個の環の中にヘテロ原子を少なくとも1個有し、前記ヘテロ原子がO、SまたはNから選択される、単環または縮合環系の芳香族基を指す。ヘテロアリール基の各環は、各環内のヘテロ原子数の合計が4個以下であること、そして各環が炭素原子を少なくとも1個含むことを前提に、O原子を1または2個、S原子を1または2個、N原子を1〜4個含むことができる。二環または三環基を完成させる縮合環は、炭素原子のみを含んでいてもよく、飽和、部分不飽和または芳香族であってもよい。窒素原子の電子の孤立電子対が芳香族π電子系の完成に関与していない構造では、N原子は場合により第四級化または窒素酸化物に酸化されていてもよい。ヘテロアリールは、前記環状基を含むアルキル基も指す。単環式ヘテロアリール基の例としては、非限定的に、ピロリル、ピラゾリル、ピラゾリニル、イミダゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、チアジアゾリル、イソチアゾリル、フラニル、チエニル、オキサジアゾリル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、およびトリアジニルが挙げられる。二環式ヘテロアリールの例としては、非限定的に、インドリル、ベンゾチアゾリル、ベンゾオキサゾリル、ベンゾチエニル、キノリニル、テトラヒドロイソキノリニル、イソキノリニル、ベンゾイミダゾリル、ベンゾピラニル、インドリジニル、ベンゾフラニル、イソベンゾフラニル、クロモニル、クマリニル、ベンゾピラニル、シンノリニル、キノキサリニル、インダゾリル、プリニル、ピロロピリジニル、フロピリジニル、チエノピリジニル、ジヒドロイソインドリル、およびテトラヒドロキノリニルが挙げられる。三環式ヘテロアリール基の例としては、非限定的に、カルバゾリル、ベンゾインドリル、フェナントロリニル、アクリジニル、フェナントリジニル、およびキサンテニルが挙げられる。そのようなヘテロアリール基は全て、以下に記載される通り場合により置換されていてもよい。
【0070】
用語「ヒドロキシ」は、基−OHを指す。
【0071】
用語「アルコキシ」は、基−ORa(ここでRaは、アルキル、シクロアルキルまたは複素環である)を指す。例としては、非限定的に、メトキシ、エトキシ、tert−ブトキシ、シクロヘキシルオキシおよびテトラヒドロピラニルオキシが挙げられる。
【0072】
用語「アリールオキシ」は、基−ORb(ここでRbは、アリールまたはヘテロアリールである)を指す。例としては、非限定的に、フェノキシ、ベンジルオキシおよび2−ナフチルオキシが挙げられる。
【0073】
用語「アシル」は、基−C(=O)−Rc(ここでRcは、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールである)を指す。例としては、非限定的に、アセチル、ベンゾイル、およびフロイルが挙げられる。
【0074】
用語「アミノアシル」は、アミノ酸から誘導されたアシル基を示す。
【0075】
用語「アミノ」は、−NRdRe基(ここでRdおよびReは、水素、アルキル、シクロアルキル、複素環、アリールおよびヘテロアリールからなる群より独立して選択される)を指す。あるいはRdとReとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。
【0076】
用語「アミド」は、基−C(=O)NRfRg(ここでRfおよびRgは、水素、アルキル、シクロアルキル、複素環、アリールおよびヘテロアリールからなる群より独立して選択される)を指す。あるいはRfとRgとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。
【0077】
用語「アミジノ」は、基−C(=NRh)NRiRj(ここでRhは、水素、アルキル、シクロアルキル、複素環、アリールおよびヘテロアリールからなる群より選択され、RiおよびRjは、水素、アルキル、シクロアルキル、複素環、アリールおよびヘテロアリールからなる群より独立して選択される)を指す。あるいはRiとRjとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。
【0078】
用語「カルボキシ」は、基−CO2Hを指す。
【0079】
用語「カルボキシアルキル」は、基−CO2Rk(ここでRkは、アルキル、シクロアルキルまたは複素環である)を指す。
【0080】
用語「カルボキシアリール」は、基−CO2Rm(ここでRmは、アリールまたはヘテロアリールである)を指す。
【0081】
用語「シアノ」は、基−CNを指す。
【0082】
用語「ホルミル」は、基−C(=O)Hを指し、−CHOとも示される。
【0083】
用語「ハロ」、「ハロゲン」または「ハロゲン化物」は、それぞれフルオロ、フッ素またはフッ化物、クロロ、塩素または塩化物、ブロモ、臭素または臭化物、およびヨード、ヨウ素またはヨウ化物を指す。
【0084】
用語「オキソ」は、同じ炭素上の2個の水素原子の代わりに置換されていてカルボニル基を形成している二価基=Oを指す。
【0085】
用語「メルカプト」は、基−SRn(ここでRnは、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールである)を指す。
【0086】
用語「ニトロ」は、基−NO2を指す。
【0087】
用語「トリフルオロメチル」は、基−CF3を指す。
【0088】
用語「スルフィニル」は、基−S(=O)Rp(ここでRpは、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールである)を指す。
【0089】
用語「スルホニル」は、基−S(=O)2−Rq1(ここでRq1は、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールである)を指す。
【0090】
用語「アミノスルホニル」は、基−NRq2−S(=O)2−Rq3(ここでRq2は、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールであり、Rq3は、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールである)を指す。
【0091】
用語「スルホンアミド」は、基−S(=O)2−NRrRs(ここでRrおよびRsは、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールからなる群より独立して選択される)を指す。あるいはRrとRsとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。
【0092】
用語「カルバモイル」は、式−N(Rt)−C(=O)−ORu(式中、Rtは、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールから選択され、Ruは、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールから選択される)で示される基を指す。
【0093】
用語「グアニジノ」は、式−N(Rv)−C(=NRw)−NRxRy(式中、Rv、Rw、RxおよびRyは、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールから独立して選択される)で示される基を指す。あるいはRxとRyとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、複素環または3〜8員を形成している。
【0094】
用語「ウレイド」は、式−N(Rz)−C(=O)−NRaaRbb(式中、Rz、RaaおよびRbbは、水素、アルキル、シクロアルキル、複素環、アリールまたはヘテロアリールから独立して選択される)で示される基を指す。あるいはRaaとRbbとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。
【0095】
用語「場合により置換された」は、任意の置換基が明確に示されていれば、該用語は、特定の基が非置換であるか、または特定の置換基で置換されていることを示すが、その場合を除けば、特定の基が非置換であるか、または適切な置換基1個以上で置換されていることを明確に示すものとする。先に定義された通り、本明細書に他に断りがなければ(例えば、特定の基が非置換であることを示すことにより)、様々な基が非置換であるか、または置換されていてもよい(即ち、それらは場合により置換されている)。
【0096】
用語アルキル、シクロアルキル、複素環、アリールおよびヘテロアリールと共に用いられる場合の、用語「置換された」は、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、ハロ、オキソ、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノ、ウレイド、および式−NRccC(=O)Rdd、−NReeC(=NRff)Rgg、−OC(=O)NRhhRii、−OC(=O)Rjj、−OC(=O)ORkk、−NRmmSO2Rnnまたは−NRppSO2NRqqRrr(ここで、Rcc、Rdd、Ree、Rff、Rgg、Rhh、Rii、Rjj、Rmm、Rpp、RqqおよびRrrは、水素、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリールまたは非置換ヘテロアリールから独立して選択され、RkkおよびRnnは、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリールまたは非置換ヘテロアリールから独立して選択される)で示される基から独立して選択される置換基により置換された基の水素原子を1個以上有するアルキル、シクロアルキル、複素環、アリールまたはヘテロアリール基を指す。あるいはRggとRhh、RjjとRkk、またはRppとRqqとが一緒になって、非置換アルキル、非置換シクロアルキル、非置換複素環、非置換アリール、非置換ヘテロアリール、ヒドロキシ、アルコキシ、アリールオキシ、アシル、アミノ、アミド、カルボキシ、カルボキシアルキル、カルボキシアリール、メルカプト、スルフィニル、スルホニル、スルホンアミド、アミジノ、カルバモイル、グアニジノまたはウレイドで場合により置換されていて、O、SまたはNから選択される追加のヘテロ原子1〜3個を場合により含む、3〜8員の複素環を形成している。加えて、アリールおよびヘテロアリール基に関する用語「置換された」は、シアノ、ニトロまたはトリフルオロメチルにより置換された基の水素原子を1個有することを、選択肢として包含する。
【0097】
任意の原子の通常の原子価を超えないこと、および置換により安定した化合物が得られることを前提として、置換が行われる。一般に、基の置換された形態が存在する場合、そのような置換された基は、好ましくは更に置換されないか、または置換される場合には、置換基が置換された基をわずかな数しか含まず、幾つかの例において1、2、3または4個のそのような置換基を含む。
【0098】
任意の変数が本明細書の任意の成分または任意の式中に1回を超えて出現する場合、各出現ごとのその定義は、それぞれ他の出現の際の定義とは独立している。同じく置換基および/または変数の組み合わせは、安定した化合物が得られる場合のみ、許容される。
【0099】
「安定した化合物」または「安定した構造」は、有用な純度への単離、および効能のある治療薬への配合を持続させるのに十分な堅牢性がある化合物を指す。
【0100】
用語「アミノ酸」は、当業者に公知の、一般的な天然(遺伝子コードされた)または合成アミノ酸およびその一般的誘導体を指す。アミノ酸に適用される「標準の」または「タンパク質原性」は、天然形態の遺伝子コードされた20種のアミノ酸を指す。同様にアミノ酸に適用される「非天然」または「異常」は、非天然、希少または合成アミノ酸の広範での選択、例えばHunt, S.の Chemistry and Biochemistry of the Amino Acids, Barrett, G.C., Ed., Chapman and Hall: New York, 1985に記載されたものを指す。
【0101】
アミノ酸またはアミノ酸誘導体に関連する用語「残基」は、式:
【化14】
(式中、RAAは、アミノ酸側鎖であり、そしてこの例ではn=0、1または2である)
で示される基を指す。
【0102】
ジペプチド、トリペプチドまたはより高次のペプチド誘導体に関連する用語「フラグメント」は、それぞれ2、3またはそれを超えるアミノ酸残基を含む基を指す。
【0103】
用語「アミノ酸側鎖」は、標準または非天然アミノ酸からの任意の側鎖を指し、RAAで示される。例えば、アラニンの側鎖はメチルであり、バリンの側鎖はイソプロピルであり、トリプトファンの側鎖は3−インドリルメチルである。
【0104】
用語「アゴニスト」は、タンパク質、受容体、酵素などの内在性リガンドの効果の少なくとも幾つかを複製する化合物を指す。
【0105】
用語「アンタゴニスト」は、タンパク質、受容体、酵素などの内在性リガンドの効果の少なくとも幾つかを阻害する化合物を指す。
【0106】
用語「阻害剤」は、タンパク質または酵素の活性を低減する化合物を指す。
【0107】
用語「癌性状態」は、対象が進行性の癌、例えば、白血病、リンパ腫、メラノーマ、乳癌、胃腸癌、食道癌、胃癌、結腸癌、腸癌、大腸癌、直腸癌、前立腺癌、膀胱癌、精巣癌、卵巣癌、子宮癌、子宮頸癌、脳癌、肺癌、気管支癌、喉頭癌、咽頭癌、膵臓癌、甲状腺癌、骨癌および皮膚癌を有する状態である。
【0108】
用語「チャネル活性化プロテアーゼ」またはCAPは、典型的には細胞の細胞外膜上に分泌されるが、内部に分泌することもでき、アミロライド感受性上皮ナトリウムチャネル(ENaC)の活性を刺激することができる膜結合性プロテアーゼを指す。そのようなCAPの非限定的例は、プロスタシン(PRSS**)、マトリプターゼ、CAP2、CAP3、トリプシン、PRSS22、TMPRSS2、TMPRSS3、TMPRSS4(マトリプターゼ−2)、TMPRSS11、カテプシンA、好中球エラスターゼおよびそのアイソフォームである。
【0109】
用語「腫瘍」は、非制御的細胞複製から得られる組織の異常な増殖を指す。そのような異常な増殖は、多くの場合、癌に関連する。腫瘍は、新生物とも呼ばれる。
【0110】
用語「転移」は、対象の体内の発生部位から他の一ヶ所以上の場所への癌または腫瘍の拡大を指す。
【0111】
用語「調整または調整すること」は、化合物を用いる生物学的または化学的プロセスまたはメカニズムへの影響を付与することを指す。例えば調整することで、生物学的または化学的プロセスまたはメカニズムの上昇、促進、アップレギュレート、活性化、阻害、低減、遮断、予防、遅延、脱感作、脱活性化、ダウンレギュレートなどを行ってもよい。したがって、調整する化合物は、「アゴニスト」または「アンタゴニスト」であってもよい。調整することにより影響を受ける例示的な生物学的プロセスまたはメカニズムとしては、非限定的に、受容体活性化、結合および/またはホルモン放出または分泌、鉄チャネル調節、細胞透過性、リン酸化または脱リン酸化、組織ホメオスタシス、セカンドメッセンジャーシグナル伝達ならびに遺伝子調節が挙げられる。調整することにより影響を受ける例示的な化学的プロセスまたはメカニズムとしては、非限定的に、触媒および加水分解が挙げられる。本明細書で用いられる、調整する化合物は、「調整物質」と呼ばれる。
【0112】
受容体に適用される用語「変異体」は、二量体、三量体、四量体、五量体および複数の成分を含有する他の生物学的複合体の包含を意味する。これらの成分は、同一または異なっていてもよい。
【0113】
用語「ペプチド」は、互いに共有結合する2個以上のアミノ酸で構成された化学的化合物を指す。
【0114】
用語「ペプチド模倣体」は、ペプチドを模倣するように設計されているが、活性または他の性質、例えば、溶解度、代謝安定性、経口生物学的利用度、親油性、透過性などを調整するための、ペプチドの官能基を1個以上付加または置換することによる構造的差異を含む化学的化合物を指す。これは、ペプチド結合の置換、側鎖の修飾、トランケーション、官能基の付加などを含んでいてもよい。化学構造がペプチドに由来していないが、その活性を模倣している場合には、それは多くの場合、「非ペプチド性ペプチド模倣体」と呼ばれる。
【0115】
用語「ペプチド結合」は、個々のアミノ酸が、典型的にはペプチド内で互いに共有結合しているアミド[−C(=O)−NH−]官能基を指す。
【0116】
用語「保護基」は、化学変化が分子内の他の部分で起こる時に、分子上の潜在的に反応性の官能基、例えばアミン、ヒドロキシルまたはカルボキシルが化学反応を受けるのを防ぐために用いられうる任意の化学的化合物を指す。複数のそのような保護基が、当業者に公知であり、例は、“Protective Groups in Organic Synthesis,” Theodora W. Greene and Peter G. Wuts,編 John Wiley & Sons, New York, 3rdedition, 1999 [ISBN 0471160199]に見出すことができる。アミノ保護基の例としては、非限定的に、フタルイミド、トリクロロアセチル、ベンジルオキシカルボニル、tert−ブトキシカルボニル、およびアダマンチルオキシカルボニルが挙げられる。幾つかの実施形態において、アミノ保護基は、カルバマートアミノ保護基であり、それは、アミノ基に結合するとカルバマートを形成するアミノ保護基と定義される。他の実施形態において、アミノカルバマート保護基は、アリルオキシカルボニル(AllocまたはAloc)、ベンジルオキシカルボニル(Cbz)、9−フルオレニルメトキシカルボニル(Fmoc)、tert−ブトキシカルボニル(Boc)およびα,α−ジメチル−3,5−ジメトキシベンジルオキシカルボニル(Ddz)である。より新規な窒素保護基の近年の議論について:Theodoridis, G. Tetrahedron 2000, 56, 2339-2358。ヒドロキシル保護基の例としては、非限定的に、アセチル、tert−ブチルジメチルシリル(TBDMS)、トリチル(Trt)、tert−ブチル、およびテトラヒドロピラニル(THP)が挙げられる。カルボキシル保護基の例としては、非限定的に、メチルエステル、tert−ブチルエステル、ベンジルエステル、トリメチルシリルエチルエステル、および2,2,2−トリクロロエチルエステルが挙げられる。
【0117】
用語「固相化学」は、反応成分の1つが高分子材料(以下に定義される固形担体)に共有結合している化学反応の行為を指す。固相上で化学作用を実施するための反応方法は、ペプチドおよびオリゴヌクレオチド化学の伝統的分野以外でより広範に知られ確立されている。
【0118】
用語「固形担体」、「固相」または「樹脂」は、固相化学を実行するのに用いられる機械的および化学的に安定した高分子マトリックスを指す。これは「樹脂」、「P−」または以下の記号で示される:
【化15】
【0119】
適切な高分子材料の例としては、非限定的に、ポリスチレン、ポリエチレン、ポリエチレングリコール、ポリスチレンにグラフト化または共有結合されたポリエチレングリコール(PEG−ポリスチレン、TentaGel(商標)とも呼ばれる。Rapp, W.; Zhang, L.; Bayer, E.、Innovations and Perspectives in Solid Phase Synthesis. Peptides, Polypeptides and Oligonucleotides; Epton, R., Ed. p 205; 英国バーミンガムのSPCC Ltd.)、ポリアクリラート(CLEAR(商標))、ポリアクリルアミド、ポリウレタン、PEGA[ポリエチレングリコールポリ(N,N−ジメチルアクリルアミド)コポリマー、Meldal, M. Tetrahedron Lett. 1992, 33, 3077-3080]、セルロースなどが挙げられる。これらの材料は、架橋結合を形成して構造を機械的に安定化させる追加の化学剤、例えば、ジビニルベンゼン(DVB、通常は0.1〜5%、好ましくは0.5〜2%)と架橋したポリスチレンを場合により含有していてもよい。この固形担体は、更なる誘導体化または反応のために、非限定的例として、アミノメチルポリスチレン、ヒドロキシメチルポリスチレン、ベンズヒドリルアミンポリスチレン(BHA)、メチルベンズヒドリルアミン(MBHA)ポリスチレン、および遊離化学的官能基、最も典型的には−NH2または−OHを含む他の高分子バックボーンを含むことができる。その用語は、これらの官能基を高率で(増量して)有する「ウルトラレジン(Ultraresins)」、例えばポリエチレンイミンと架橋分子とから調製されたものを含むことも意味する(Barth, M.; Rademann, J. J. Comb. Chem. 2004, 6, 340-349)。合成の最後には、樹脂は典型的には廃棄されるが、Frechet, J.M.J.; Haque, K.E. Tetrahedron Lett. 1975, 16, 3055に記載される通り、再使用しうることが示されている。
【0120】
一般に樹脂として用いられる材料は、不溶性高分子であるが、特定の高分子は、溶媒に応じて溶解度差(differential solubility)を有し、固相化学で用いることができる。例えばポリエチレングリコールは、化学反応を実施しうる多くの有機溶媒に可溶性であり、他のもの、例えばジエチルエーテルには不溶性であるため、この手法で用いることができる。つまり、均質に溶解して反応を実施し、その後、ジエチルエーテルの添加により高分子上での生成物を沈殿させて、固体として処理することができる。これは、「液相」化学と呼ばれてきた。
【0121】
固相化学に関連して用いられる用語「リンカー」は、典型的には固形担体から基質を遊離(切断)させるために、固形担体に共有結合して担体と基質の間を付加させる化学基を指す。しかしそれは、固形担体への結合に安定性を付与するため、または単にスペーサー要素として、用いることもできる。多くの固形担体は、既に付加されたリンカーと共に市販されている。
【0122】
アミノ酸に用いられる略語およびペプチドの名称は、J. Biol. Chem. 1972, 247, 977-983中のIUPAC-IUB Commission of Biochemical Nomenclatureの規則に従う。この文書は、改訂された:Biochem. J., 1984, 219, 345-373; Eur. J. Biochem., 1984, 138, 9-37; 1985, 152, 1; Internat. J. Pept. Prot. Res., 1984, 24, p 84以降; J. Biol. Chem., 1985, 260, 14-42; Pure Appl. Chem., 1984, 56, 595-624; Amino Acids and Peptides, 1985, 16, 387-410; およびBiochemical Nomenclature and Related Documents, 2nd edition, Portland Press, 1992, pp 39-67。規則の追加分は、JCBN/NC-IUB Newsletter 1985, 1986, 1989に発表されており;Biochemical Nomenclature and Related Documents, 2nd edition, Portland Press, 1992, pp 68-69を参照されたい。
【0123】
用語「有効量」または「効果的」は、臨床検査および評価、患者観察などを介して記録された疾患もしくは障害の症状を緩和させる用量、ならびに/または生物活性もしくは化学活性の検出可能な変化を起こす用量を示すものとする。関連するメカニズムまたはプロセスについて、検出可能な変化は、当業者により検出され、そして/または更に定量されてもよい。当該技術分野で一般に理解される通り、投与量は、投与経路、症状および患者の体重に応じて変動するが、投与される化合物にも依存する。
【0124】
2種以上の化合物の「併用」での投与は、一方の存在が他方の生物学的効果を変化させるのに十分、時間的に接近させて2種の化合物を投与することを意味する。2種の化合物を、同時に(同時的に(concurrently))または連続して投与することができる。同時投与は、投与前に化合物を混合することにより、または異なる解剖学的部位で、もしくは異なる投与経路を用いて、該化合物を同じ時点で投与することにより、実施することができる。本明細書で用いられる語句「同時的投与」、「併用投与」、「同時投与」または「同時に投与される」は、該化合物を互いに同じ時点で、または互いに直後に投与することを意味する。後者の例では、観察された結果が化合物を同じ時点で投与する場合に実現される結果と識別不能な程、十分に接近した時間に2種の化合物が投与される。
【0125】
用語「薬学的に活性の代謝産物」は、特定の化合物の体内での代謝を介して生成される薬理学的活性生成物を意味するものとする。
【0126】
用語「溶媒和物」は、生物学的有効性を保持する特定の化合物の薬学的に許容しうる溶媒和物形態を意味する。溶媒和物の例は、非限定的に、水、イソプロパノール、エタノール、メタノール、DMSO、酢酸エチル、酢酸またはエタノールアミンと併用される本発明の化合物が挙げられる。
【0127】
1.化合物
本発明の新規な大環状化合物は、環化を受けて該大環状化合物を形成する、テザー成分を含むビルディングブロック構造を含む大環状化合物を包含する。ビルディングブロック構造は、アミノ酸(標準および非天然)、ヒドロキシ酸、ヒドラジノ酸、アザアミノ酸、特定の部分、例えば、ペプチド代理物およびイソステレスの導入に役割を果たすもの、および本明細書に記載されたテザー成分を含むことができる。
【0128】
本発明は、単離された化合物を包含する。単離された化合物は、幾つかの実施形態において、混合物の化合物を少なくとも10%、少なくとも25%、少なくとも50%または少なくとも70%含む化合物を指す。幾つかの実施形態において、該化合物、薬学的に許容しうるその塩または該化合物を含有する医薬組成物は、ヒトグレリン受容体で生物学的アッセイで検査した場合に、統計学的に有意な結合および/またはアンタゴニスト活性を示す。
【0129】
固体の化合物、塩、または溶媒和物の例では、本発明の化合物、塩、および溶媒和物が異なる血漿または多形形態で存在することができ、その全てが本発明および特定の式の範囲内であることは、当業者に理解されよう。
【0130】
本明細書に開示された化合物は、不斉中心を有していてもよい。本発明の化合物は、単一の立体異性体、ラセミ体、ならびに/または鏡像異性体および/もしくはジアステレオマーの混合物として存在してもよい。そのような単一の立体異性体、ラセミ体、およびその混合物は全て、本発明の範囲内のものとする。しかし特定の実施形態において、本発明の化合物は、光学的に純粋な形態で用いられる。本明細書で用いられる用語「S」および「R」形態は、IUPAC 1974 Recommendations for Section E, Fundamentals of Stereochemistry (Pure Appl. Chem. 1976, 45, 13-30)に定義された通りである。
【0131】
特定の配向があることが他に示されない限り、本発明は、全ての立体異性体形態で説明される。該化合物は、単一の立体異性体または立体異性体混合物として調製してもよい。非ラセミ形態を、合成または分割のいずれかにより得てもよい。標準的技術、例えば塩形成を介したジアステレオマー対の形成により、該化合物を成分の鏡像異性体に分割してもよい。同じく、キラル部分への共有結合により、該化合物を分割してもよい。その後、クロマトグラフィー分離および/または結晶学的分離により、ジアステレオマーを分割してもよい。キラル補助部分の場合には、その後、それを除去することができる。代替法として、キラルクロマトグラフィーの利用により、該化合物を分割することができる。分割の酵素的方法も、特定の例で用いることができる。
【0132】
当業者に一般に理解される通り、「光学的に純粋な」化合物は、単一鏡像異性体のみを含むものである。本明細書で用いられる用語「光学活性」は、混合物が平面偏光を回転させるように、1つの鏡像異性体を他のものよりも少なくとも十分に過剰に含む化合物を意味するものとする。光学活性化合物は、偏光の平面を回転する能力を有する。他よりも過剰な1つの鏡像異性体は、典型的には鏡像異性体過剰(e.e.)として表わされる。光学活性化合物を記載する際に、接頭辞DとL、またはRとSは、キラル中心の周りの分子の絶対配置を示すのに用いられる。接頭辞「d」と「l」、または「+」と(−)は、化合物の回旋(即ち、偏光面が光学活性化合物により回転する方向)を示すのに用いられる。「l」または(−)の接頭辞は、化合物が左旋性(即ち、偏光面を左または反時計回りに回転させる)であることを示し、「d」または(+)の接頭辞は、化合物が右旋性(即ち、偏光面を右または時計回りに回転させる)であることを意味する。回旋記号(−)および(+)は、分子の絶対配置RおよびSには関係しない。
【0133】
所望の薬理学的性質を有する本発明の化合物は、光学活性であり、少なくとも90%(80%e.e.)、少なくとも95%(90%e.e.)、少なくとも97.5%(95%e.e.)または少なくとも99%(98%e.e.)の単一異性体で構成させることができる。
【0134】
同様に、二重結合などの多くの幾何異性体も、本明細書に開示された化合物中に存在することができ、他に断りがなければ、そのような安定した異性体は全て、本発明に包含される。同じく本発明に包含されるのは、該化合物の互変異性体および回転異性体である。
【0135】
次の右側の記号の使用は、示された環の水素原子1個以上が定義された置換基Rで置換されていることを指す。
【化16】
【0136】
次の記号の使用は、単結合または任意の二重結合:----を示す。
【0137】
本発明の実施形態は、本明細書に記載された合成方法により形成されて、式Iおよび/またはIIで示される化合物を提供する中間体化合物を更に提供する。中間体化合物は、本明細書に記載された一定範囲の適応症の治療薬として、そして/または更なる方法および反応の試薬としての用途を有していてもよい。
【0138】
2.合成方法
本発明の化合物を、伝統的な溶液合成技術または固相化学法を用いて合成することができる。いずれかにおいて、構築は4つの相を含み:最初は、コンホメーションの制御および定義のための、生物学的ターゲット受容体の認識要素と1つのテザー部分とを含むビルディングブロックの合成である。これらのビルディングブロックは、標準の化学的変換を用いる第二相において、典型的には連続様式で、一緒に組み立てられる。アセンブリの前駆体を、その後、第三段階で環化して、大環状構造を提供する。最後に、保護基の除去および任意の精製を含む、環化後工程の第四段階は、所望の最終化合物を提供する。この一般的タイプの大環状構造の合成方法は、WO 2004/111077およびWO 2005/012331に記載された精製手順を含み、国際特許出願WO 01/25257、WO 2004/111077、WO 2005/012331、WO 2005/012332、WO 2008/033328およびWO2008/130464に記載されている。米国特許第7,476,653および同第7,491,695も参照されたい。
【0139】
本発明の幾つかの実施形態において、先に定義された可溶性または不溶性高分子マトリックス上の固相化学を利用して、大環状化合物を合成してもよい。固相化学では、樹脂への「負荷」とも呼ばれる、第一のビルディングブロックの付加を含む予備的段階を、実施しなければならない。本発明で用いられる樹脂は、優先的にリンカー部分Lを付加している。これらのリンカーは、該技術分野で公知の標準的反応法、例えばエステルまたはアミド結合の形成のために開発された多数の反応条件のいずれかを介して、塩基樹脂上の適切な遊離化学的官能基、通常はアルコールまたはアミンに付加させるが、他の物質も可能である。本発明のための幾つかのリンカー部分が、一般に「環化遊離(cyclization−release)」と呼ばれるプロセスで、樹脂から同時に切断されて大環状化合物を形成するように設計されている(van Maarseveen, J.H. Solid phase synthesis of heterocycles by cyclization/cleavage methodologies. Comb. Chem. High Throughput Screen. 1998, 1, 185-214; Ian W. James, Linkers for solid phase organic synthesis. Tetrahedron 1999, 55, 4855-4946; Eggenweiler, H.-M. Linkers for solid-phase synthesis of small molecules: coupling and cleavage techniques. Drug Discovery Today 1998, 3, 552-560; Backes, B.J.; Ellman, J.A. Solid support linker strategies. Curr. Opin. Chem. Biol. 1997, 1, 86-93.本発明の化合物ではこれに関連して特に有用なものが、3−チオプロピオン酸リンカーである (Hojo, H.; Aimoto, S. Bull. Chem. Soc. Jpn. 1991, 64, 111-117; Zhang, L.; Tam, J. J. Am. Chem. Soc. 1999, 121, 3311-3320.)。
【0140】
そのようなプロセスは、環状生成物のみが固形担体から遊離し、溶液相で起こるような直鎖状前駆体の混入が生じないため、より高い純度の材料を提供する。公知または標準の反応化学を利用してビルディングブロックおよびテザーの全てを直鎖状前駆体に連続して組み立てた後、テザー・ビルディングブロックの一部である適切な求核官能基により、このリンカーに付加されたカルボニル上での塩基介在性の分子内攻撃が、アミドまたはエステル結合を形成して図示された環状構造を完成させる(スキーム1)。溶液相に適合する類似の方法論は、大規模適用に好ましいと思われるため、それを適用することも可能である。
【0141】
スキーム1.環化−遊離ストラテジー
【化17】
【0142】
この記載は、本発明の方法の1つの経路を正確に表わしているが、テザー成分を実際に環化ステップで組み立てる改変経路では、閉環メタセシス(RCM)の1種である本発明の別法チオエステルストラテジーが進行する。しかし、RCM方法論でも、ビルディングブロックの組立が連続して進行した後、環化される(そして固相であれば樹脂から遊離する)。RCM反応の特定の副生成物を除去するのに、追加の環化後処理ステップが必要となるが、次の残りの工程は、チオエステルまたは類似の塩基介在性環化ストラテジーと同様の手法で実施される。
【0143】
その上、本明細書に提供された方法を含むステップを、独立して実施してもよく、または少なくとも2つのステップを組み合わせてもよいことは、理解されよう。加えて、本明細書に提供された方法を含むステップは、独立して、または組み合わせて実施される場合、同じ温度または異なる温度で実施してもよく、それは本発明の教示から逸脱しない。
【0144】
本発明の新規な大環状化合物は、ビルディングブロック構造の環化を含む新規なプロセスにより形成されて、本明細書に記載されたテザー成分を含む大環状化合物を形成するものを包含する。したがって本発明は、(a)ビルディングブロック構造を組み立てること、(b)ビルディングブロック構造を化学変換すること、(c)テザー成分を含むビルディングブロック構造を環化させること、(d)保護基をビルディングブロックから除去すること、および(e)ステップ(d)から得られた生成物を任意で精製すること、を含む、本発明の化合物を製造する方法を提供する。幾つかの実施形態において、ビルディングブロック構造の組み立ては、連続的であってもよい。更なる実施形態において、合成方法は、伝統的な溶液合成技術または固相化学技術を用いて実施される。
【0145】
A.概論
他に断りがなければ、試薬および溶媒は、試薬品質以上のものであり、様々な販売業者から得て使用した。用いられたDMF、DCM(CH2Cl2)、DME、CH3CNおよびTHFは、(i)脱保護、(ii)樹脂キャッピング反応、および(iii)洗浄、以外は、DriSolv(登録商標)(ドイツ、ダルムスタットのMerck KGaAの一部であるEMD Chemicals, Inc.)のものであるか、または合成等級である。アミノ酸(AA)カップリング反応に用いられるNMPは、分析等級である。DMFは、使用前に真空下に最短で30分間配置することにより、適宜、脱気した。Strem Chemicals, Inc. (米国マサチューセッツ州、ニューベリーポート)から、均一触媒を得た。N−メチルおよび非天然アミノ酸のものを含む、Cbz−、Boc−およびFmoc−保護アミノ酸および側鎖保護誘導体は、販売業者から得るか、または当業者に公知の標準的方法論により合成した。Ddzアミノ酸は、標準法により合成するか、またはOrpegen (ドイツ、ハイデルベルグ)もしくはAdvanced ChemTech (米国ケンタッキー州ルイビル)から市販されていた。Bts−アミノ酸は、確立された手順により合成した。ヒドロキシ酸は、販売業者から得るか、または文献に記載された通り対応するアミノ酸から合成した(Tetrahedron 1989, 45, 1639-1646; Tetrahedron 1990, 46, 6623-6632; J. Org. Chem. 1992, 57, 6239-6256.; J. Am. Chem. Soc. 1999, 121, 6197-6205)。蛍光指示薬を含むシリカゲル60F254のプレコートプレート(0.25mm厚)で、分析TLCを実施し、示された方法および試薬を用いて、例えば紫外線(UV)および/またはモリブデン酸セリウム(CMA)溶液(硫酸100mL、硫酸セリウムアンモニウム10gおよびモリブデン酸アンモニウム25gを混合することにより調製)を用いて、視覚化した。
【0146】
用語「減圧濃縮/蒸発/除去」は、除去される溶媒に応じて適宜、水吸引圧下(典型的には10〜30トル)、または機械的油圧真空ポンプにより提供されるより強い真空下(「高真空」、典型的には1トル以下)で、ロータリーエバポレータを利用して溶媒および揮発成分を除去することを示す。「真空」または「高真空」下での化合物の乾燥は、低圧(1トル以下)の油圧真空ポンプを用いた乾燥を指す。シリカゲル60(230〜400メッシュ、EMD Chemicals, Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923-2925)を用いて「フラッシュクロマトグラフィー」を実施したが、それは当業者に周知の手順である。「ドライパック」は、溶媒での前処理をしていないシリカゲルのクロマトグラフィーを示しており、一般に所望の生成物と任意の不純物の間で、Rfに大きな差が存在する場合の精製に大規模に適用される。固相化学のプロセスでは、「標準的手法で乾燥」は、樹脂を、最初は風乾させ(1時間)、続いて完全な乾燥に至るまで真空乾燥(通常は油圧ポンプ)させる(約30分〜一晩)ことである。空気および水感受性反応で用いられるガラス製品は、オーブンで少なくとも一晩乾燥させて、使用前にデシケータ内で冷却させた。
【0147】
B.アミノ酸
N−メチルおよび非天然アミノ酸のものを含む、アミノ酸、Boc−およびFmoc−保護アミノ酸、ならびに側鎖保護誘導体は、販売業者から得るか[例えば、Advanced ChemTech(米国ケンタッキー州ルイビル)、Astatech (米国ペンシルバニア州ブリストル)、Bachem(スイス、ブーベンドルフ)、ChemImpex (米国イリノイ州ウッドデール)、Novabiochem(Merck KGaAの子会社、ドイツのダルムシュタット)、PepTech(米国マサチューセッツ州バーリントン)、 Synthetech (米国オレゴン州アルバニー)]、または当業者に公知の標準的方法論により合成した。Ddz−アミノ酸は、Orpegen (ドイツ、ハイデルベルグ)もしくはAdvanced ChemTech (米国ケンタッキー州ルイビル)から購入するか、またはDdz−OPhもしくはDdz−N3を用いて標準法を利用して合成した(Birr, C.; Lochinger, W.; Stahnke, G.; Lang, P. Justus Liebigs Ann. Chem. 1972, 763, 162-172.)。Bts−アミノ酸は、公知の方法により合成した(Vedejs, E.; Lin, S.; Klapara, A.; Wang, J. J. Am. Chem. Soc. 1996, 118, 9796-9797. 同じくWO 01/25257, WO 2004/111077)。加えて、N−アルキルアミノ酸誘導体を、文献の方法により入手した(Hansen, D. W., Jr.; Pilipauskas, D. J. Org. Chem. 1985, 50, 945-950.)。
【0148】
C.テザー
テザーは、過去に国際特許出願WO 01/25257、WO 2004/111077、WO 2005/012331、WO 2008/033328およびWO2008/130464に記載された方法で得た。米国特許第7,476,653および同第7,491,695も参照されたい。更なるテザーは、米国特許仮出願第61/256,727に記載されている。更なるテザーの調製は、実施例に示されている。
【0149】
以下は、本発明の化合物の合成で用いられる特定のテザー中間体であり、PGは、窒素保護基、例えば非限定的に、Boc、Fmoc、Ddz、CbzまたはAllocを示す:
【化18】
【0150】
D.固相および溶液相技術
本発明の大環状化合物を合成するための特定の固相技術は、WO 01/25257、WO 2004/111077、WO 2005/012331、WO 2005/012332、WO 2008/033328、WO 2008/130464および米国特許仮出願61/256,727に記載された。大規模製造を受け入れられる方法を含む溶液相合成経路は、米国特許出願第2006/025566およびUS 2007/0021331に記載された。
【0151】
以下の表は、標準法を用いた代表的な本発明の化合物の合成に使用されるビルディングブロックに関する情報を提供している。これらは、固相合成に直接適用可能である。溶液相合成では、典型的には例示のものからの改変された保護ストラテジーにより、収束的アプローチの使用が可能になる。代表的な本発明の大環状化合物を溶液相で構築するための追加的な合成の詳細を、実施例に示している。
【0152】
表の合成では、実施例9Bに概説された方法論を用いた。テザー上にアミジン部分を有する化合物では、実施例8Hに記載された通り例示された代替的なストラテジーを用いることもできる。
【表1A】
【表1B】
【0153】
3.分析法
他に断りがなければ、1Hおよび13C NMRスペクトルを、Varian Mercury 300MHz分光計(カリフォルニア州パロアルトのVarian, Inc.)で記録し、溶媒の残留プロトンシグナルに関して内部参照する。以下の通り、標準的な略語を用いて、1H NMRデータを表す:ppmでの化学シフト(δ)(多重度、積分値、結合定数)。シグナルの多重度を示すために、以下の略語を用いた:s=一重線、d=二重線、t=三重線、q=四重線、quint=五重線、bまたはbr=ブロード、およびm=多重線。溶液中の分子のコンホメーションについての情報は、当業者に公知の適切な二次元NMR技術を用いて決定することができる(Martin, G.E.; Zektzer, A.S. Two-Dimensional NMR Methods for Establishing Molecular Connectivity: A Chemist's Guide to Experiment Selection, Performance, and Interpretation, John Wiley & Sons: New York, 1988, ISBN 0471187070.)。
【0154】
XTerra(登録商標)MS C18カラム(または同等物)4.6×50mm(3.5μm)および指定の勾配法を用いて、1mL/分で運転したWater Alliance(登録商標)システム2695で、HPLC分析を実施した。Waters 996 PDAにより、純度評価用のUVデータを提供した(マサチューセッツ州ミルフォードのWaters Corporation)。特定の分析では、LCPackings(カリフォルニア州サニーベールのDionex Corporation)スプリッタ(50:40:10)により、流れを3つの部分に分離させた。第一の部分(50%)は、同一性を確認するために質量分析計(APCIプローブを備えたMicromass(登録商標)Platform II MS)へ給送した。第二の部分(40%)は、純度評価のために、蒸発光散乱検出器(ELSD, Polymer Laboratories, 現在はカリフォルニア州パロアルトのVarian Inc.,の一部、PL−ELS−1000(商標))へ送られ、最後の部分(10%)は、定量および純度評価のために、化学発光窒素検出器(CLND, Antek(登録商標)Model 8060、テキサス州ヒューストンのAntek Instruments(ジョージア州ドゥルースのRoper Industries, Inc.,の一部))へ送られた。必要とされる分析の性質に応じて、各検出器を別個に用いることもできる。最も新しいバージョンのWaters Millennium(登録商標)ソフトウエアパッケージを用いて、データを獲得および処理した。
【0155】
本発明の化合物の分析に用いられた代表的な標準HPLC条件を、以下に表わす:
典型的なクロマトグラフィー条件
カラム: XTerra PR18、3.5μm、4.6×100mm(または同等物)
検出(PDA): 220〜320nm
カラム温度: 35±10℃
注入容量: 10μL
流速: 1mL/分
運転時間: 20.0分
データ獲得時間:17.0分
移動相A: メタノール(またはアセトニトリル)
移動相B: 水
移動相C: 水中の10%TFA
勾配A4
【表2】
勾配B4
【表3】
【0156】
以下の表は、代表的な本発明の化合物のHPLC保持時間を要約している。
【表4】
【0157】
鏡像異性体およびジアステレオマーの純度を、Waters Breezeシステム(または同等物)を用いた適切なキラルHPLCカラムを用いて評価した。他のパッキング材料を用いることができるが、これらの分析に特に有用なカラムは、Chiralpak AS−RHおよびChiralcel OD−RH (米国ペンシルバニア州ウエストチェスターのChiral Technologies)である。
【0158】
XTerra(登録商標)MS C18カラム(または同等物)19×100mm(5μm)でのWaters FractionLynx(登録商標)システムを用いて、最終的な脱保護大環状化合物で分取HPLC精製を実施した。2mL/分で運転するWaters 2767インジェクタ/コレクタおよびWaters 515ポンプを有するアットカラム希釈構成を利用して、注入を実施した。質量分析計、HPLC、および質量分析指向(mass directed)フラクションコレクションを、FractionLynx (登録商標)を含むMassLynx(登録商標)ソフトウエアバージョン3.5で制御する。MS分析により示された、生成物を含むフラクション(13×125mm試験管)を、最も典型的には遠心エバポレータシステム(Genevac(登録商標)HT−4 (ニューヨーク州バレーコテージのGenevac Inc), ThermoSavant Discovery(登録商標)、SpeedVac(登録商標)もしくは同等物 (マサチューセッツ州ウォルサムのThermo Electron Corporation)で、減圧蒸発するか、または別法として凍結乾燥させた。その後、化合物を、同一性確認、純度および定量評価のために、LC−MC−UV−ELSD−CLND分析により完全に分析した。
【0159】
試料16本まで同時に運転することが可能なディスポーザブルシリカまたはC18カートリッジを有するIsco CombiFlash(登録商標)16xシステム (ネブラスカ州リンカーンのTeledyne Isco, Inc.)で、自動中圧クロマトグラフィー精製を実施した。Waters Micromass(登録商標)Platform IIまたはZQ(商標)システムで、MSスペクトルを記録した。VG Micromass ZAB−ZF分光計で、HRMSスペクトルを記録した。ActivityBase(登録商標)データベースソフトウエア (英国サリー州ギルフォードのID Business Solutions Ltd.)を用いて、化学的および生物学的情報を保存および分析した。
【0160】
下表は、代表的な本発明の化合物の分析データを表す。
【表5A】
【表5B】
【0161】
3.生物学的方法
本発明の化合物を、セリンプロテアーゼ酵素と相互作用する能力に関して評価することができる。そのような方法は十分に確立されており、当業者に公知である。加えて、マトリプターゼの活性を、時間領域近赤外蛍光(NIRF)イメージングを用いて特異的に検査して、阻害活性のインビトロおよびインビボ評価を実施することができる(Napp, J.; Dullin, C.; Mueller, F.; et al. Int. J. Cancer 2010, 127, 1958-1974.)。腫瘍中のマトリプターゼ−1の活性をイメージングする類似の方法は、蛍光顕微鏡法および標識抗体の使用を含む(Darragh, M.R.; Schneider, E.L.; Lou, J.; et al. Canc. Res. 2010, 70, 1505-1512.)。マトリプターゼ−1をコードするSt14遺伝子を欠損した遺伝子変性マウスは、該酵素の調整の効果を検討するための動物モデルを提供する。List, K.; Kosa, P.; Szabo, R.; Bey, A.L.; Wang, C.B.; Molinolo, A.; Bugge, T.H. Am. J. Pathol. 2009, 175, 1453-1463.)。
【0162】
A.阻害アッセイ
セリンプロテアーゼ酵素の阻害レベルを研究するのに、複数の文献での方法を利用することができる。一例として(Sisay, M.T.; Steinmetzer, T.; Stirnberg, M.; et al. J. Med. Chem. 2010, 53, 5523-5535)、HEK−MT2細胞のコンディション培地中のマトリプターゼ−1またはマトリプターゼ−2の活性、マトリプターゼ−2の精製された触媒ドメインの活性、および組換えマトリプターゼの活性(触媒ドメイン;ドイツ、レラハのEnzo Life Sciences)を、Cary 100UV−VIS分光光度計(ドイツダルムスタットのVarian)を用いて、37℃のTris生理食塩緩衝液(50mM Tris、150mM NaCl、pH8.0)中で、色素生成基質Boc−Gln−Ala−Arg−パラニトロアニリド(スイス、ブッベンドルフのBachem)からのパラニトロアニリンの遊離を405nmでモニタリングすることによりアッセイする。三重実験での8種の異なる基質濃度で、Km値を決定する。3種(マトリプターゼ−2の場合)または少なくとも5種(他の実験)の異なる阻害剤濃度での二重また三重測定で、阻害アッセイを実施する。式v = v0/(1+[I]/IC50)による非線形回帰により、IC50値を得た。その後、1〜4およびレイペプチン(ドイツ、ダルムスタットのCalbiochem) の10mM阻害剤原液ならびにBoc−Gln−Ala−Arg−パラニトロアニリドの100mM原液をDMSO中で調製し、アプロチニン (ドイツ、カールスルーエのCarl Roth) の1mM原液をH2O中で調製する。基質の最終濃度は400μMであり、DMSOの最終濃度は1.5%であった。予め加温したアッセイ緩衝液979μLを含むキュベットに、検査試料溶液11μLおよび基質溶液4μLを添加して、完全に混合する。酵素溶液(HEK−MT2細胞のコンディション培地の総タンパク質 5μg/6μL;マトリプターゼ−2の精製された触媒ドメイン 28ng/6μL;マトリプターゼ 3ng/6μL)6μLを添加することにより反応を開始させて、20分間追跡した。
【0163】
代表的な本発明の化合物による、代表的なセリンプロテアーゼのマトリプターゼ−1の阻害を決定する別の方法の使用が、以下の実施例に示されている。
【0164】
B.代表的な本発明の化合物の薬物動態分析
本発明の化合物の薬物動態的挙動を、当業者に周知の方法により確認することができる(Wilkinson, G. R. “Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination” in Goodman & Gilman's The Pharmacological Basis of Therapeutics, Tenth Edition, Hardman, J.G.; Limbird, L.E., Eds., McGraw Hill, Columbus, OH, 2001, Chapter 1.)。以下の方法を用いて、本発明の化合物の静脈内、皮下および経口投与の薬物動態パラメータ(排出半減期、総血漿クリアランスなど)を検査した。国際特許出願WO 2008/033328およびWO 2008/130464 ならびに米国特許第7,476,653および同第7,491,695も参照されたい。
【0165】
C.癌および転移モデル
全てのタイプの癌を処置する本発明の化合物のインビボ有効性を決定するのに、莫大な数の異なる動物モデルを入手することができる。これらには、非限定的に、マウスモデル(Cespedes, M.V.; Casanova, I.; Parreno, M.; Mangues, R. Clin. Transl. Oncol. 2006, 8, 318-329)、ヒト異種移植片モデル(Kerbel, R.S. Cancer Biol. Ther. 2003, 2, S134-S139)、遺伝子組換えマウスモデル(Walrath, J.C.; Hawes, J.J.; Van Dyke, T.; Reilly, K.M. Adv. Cancer Res. 2010, 106, 113-164)およびげっ歯類の転移モデル(Eccles, S.A.; Box, G.; Court, W.; Sandle, J.; Dean, C.J. Cell. Biophys. 1994, 279-291; Hoffman, R.M. Invest. New Drugs 1999, 17, 343-359.Man, S.; Munoz, R.; Kerbel, R.S. Cancer Metastasis Rev. 2007, 26, 737-747)が挙げられる。本発明の化合物に適用可能な幾つかの特定の方法を、実施例に表わす。
【0166】
D.皮膚疾患モデル
皮膚および組織障害を処置する本発明の化合物の効果を研究するための動物モデル、詳細にはげっ歯類を、入手することができる(Magin, T.M. Exp. Dermatol. 2004, 13, 659-660.)。炎症性皮膚疾患の遺伝子修飾マウスモデルは開発されており、該化合物の有効性を検証しうる他の系を提供する(Haase, I.; Pasparakis, M.; Krieg, T. J. Dermatol. 2004, 31, 704-719.)。
【0167】
E.炎症性疾患モデル
炎症性障害の処置における本発明の化合物の用途を決定するために、それらを適切な動物疾患モデルで試験することができる(Brodmerkel, C.M.; Vaddi, K. Curr. Opin. Biotechnol. 2003, 14, 652-658.)。関節リウマチ用(Hegen, M.; Keith, J.C. Jr.; Collins, M.; Nickerson-Nutter, C.L. Ann. Rheum. Dis. 2008, 67, 1505-1515)、骨関節炎用(Bendele, A.M. J. Musculoskelet. Neuronal. Interact. 2001, 1, 363-376; van den Berg, W.B. Curr. Rheumatol. Rep. 2008, 10, 26-29)、炎症性腸疾患、例えばクローン病および潰瘍性大腸炎用(Wirtz, S.; Neurath, M.F. Int. J. Colorectal. Dis. 2000, 15, 144-160; Wirtz, S.; Neurath, M.F. Adv. Drug Deliv. Rev. 2007, 59, 1073-1083)ならびにアテローム性硬化症用(Russell, J.C.; Proctor, S.D. Cardiovasc. Pathol. 2006, 15, 318-330; Zadelaar, S.; Kleemann, R.; Verschuren, L.; et al. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1706-1721)のものなど、そのようなモデルの宿主は公知である。
【0168】
F.呼吸器疾患モデル
COPD(Fox, J.C.; Fitzgerald, M.F. Curr. Opin. Pharmacol. 2009, 9, 231-242.)、喘息(Nials, A.T.; Uddin, S. Dis. Model Mech. 2008, 1, 213-220)、嚢胞性線維症(Carvalho-Oliveira, I.; Scholte, B.J.; Penque, D. Expert Rev. Mol. Diagn. 2007, 7, 407-417)、気管支炎(Nikula, K.J.; Green, F.H. Inhal. Toxicol. 2000, 12, 123-153)、慢性呼吸器感染(Kukavica-Ibrulj, I.; Levesque, R.C. Lab. Anim. 2008, 42, 389-412)、および呼吸器アレルギー (Pauluhn, J.; Mohr, U. Exp. Toxicol. Pathol. 2005, 56, 203-234)の処置における本発明の化合物の有効性を評価するのに用いうる複数の動物モデル系が、公知である。
【0169】
ヒツジモデルが、喘息、COPD、アレルギー性鼻炎および嚢胞性線維症をはじめとする複数の呼吸器障害に効果的であることが、立証された。(Abraham, W.M. Pulm. Pharmacol. Ther. 2008, 21, 743-754.)。
【0170】
G.鉄ホメオスタシスモデル
動物モデルが、鉄輸送障害(Andrews, N.C. Adv. Exp. Med. Biol. 2002, 509, 1-17)および鉄代謝を含む疾患の研究(Latunde-Dada, G.O.; McKie, A.T.; Simpson, R.J. Biochim. Biophys. Acta 2006, 1762, 414-423)に向けて開発された。
【0171】
4.医薬組成物
本発明の大環状化合物または本発明によるその薬理学的に許容しうる塩を、様々な投与形態の医薬組成物に配合させてもよい。本発明の医薬組成物を調製するために、有効成分としての、光学異性体、鏡像異性体、ジアステレオマー、ラセミ体もしくは立体化学混合物を含む1種以上の化合物、または薬学的に許容しうるその塩を、医薬配合剤の当業者に公知の技術により、適切な担体および添加剤と細密に混合する。
【0172】
薬学的に許容しうる塩は、医薬品としての使用または配合を可能にするための、特定の化合物の遊離酸および塩基の生物学的効果を保持し、生物学的にも他についても不適当でない、本発明の化合物の塩形態を指す。そのような塩の例は、Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wermuth, C.G. and Stahl, P.H. (編), Wiley-Verlag Helvetica Acta, Zurich, 2002 [ISBN 3-906390-26-8]に記載されている。そのような塩の例としては、遊離酸および塩基のアルカリ金属塩および付加塩が挙げられる。非限定的な薬学的に許容しうる塩の例としては、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、リン酸一水素塩、リン酸二水素塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ギ酸塩、イソ酪酸塩、カプロン酸塩、ヘプタン酸塩、プロピオール酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−ジオン酸塩、ヘキシン−1,6−ジオン酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、γ−ヒドロキシ酪酸塩、グリコール酸塩、酒石酸塩、メタンスルホン酸塩、エタンスルホン酸塩、プロパンスルホン酸塩、トルエンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スルホン酸塩、およびマンデル酸塩が挙げられる。
【0173】
本発明の化合物が塩基である場合、所望の塩を、例えば非限定的に塩酸、臭化水素酸、ヨウ化水素酸、炭酸、硫酸、硝酸、リン酸などの無機酸、または非限定的にギ酸、酢酸、プロピオン酸、マレイン酸、コハク酸、マンデル酸、フマル酸、マロン酸、ピルビン酸、シュウ酸、ステアリン酸、アスコルビン酸、グリコール酸、サリチル酸、ピラノシジル酸、例えばグルクロン酸もしくはガラクツロン酸、α−ヒドロキシ酸、例えばクエン酸もしくは酒石酸、アミノ酸、例えばアスパラギン酸もしくはグルタミン酸、芳香族酸、例えば安息香酸もしくは桂皮酸、スルホン酸、例えばp−トルエンスルホン酸、メタンスルホン酸、エタンスルホン酸、2−ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、シクロヘキシルアミノスルホンなどをはじめとする有機酸、での遊離塩基の処理をはじめとする、当業者に公知の任意で適切な方法により調製してもよい。
【0174】
本発明の化合物が酸である場合、所望の塩は、無機または有機塩基、例えばアミン(第一級、第二級、または第三級);アルカリ金属またはアルカリ土類金属水酸化物などでの遊離酸の処理をはじめとする、当該技術分野で公知の任意の適切な方法により調製してもよい。適切な塩の例示としては、アミノ酸、例えばグリシン、リシンおよびアルギニン;アンモニア;第一級、第二級および第三級アミン、例えばエチレンジアミン、N,N’−ジベンジルエチレンジアミン、ジエタノールアミン、コリン、および環状アミン、例えばピペリジン、モルホリン、およびピペラジンから得られる有機塩;ならびにナトリウム、カルシウム、カリウム、マグネシウム、マンガン、鉄、銅、亜鉛、アルミニウム、およびリチウムから得られる無機塩が挙げられる。
【0175】
そのような医薬組成物に用いられる担体および添加剤は、予測される投与様式に応じて、様々な形態をとることができる。つまり経口投与用の組成物は、例えば、デンプン、糖、結合剤、希釈剤、造粒剤、滑剤、崩壊剤などの適切な担体および添加剤を含む、固体調製物、例えば錠剤、糖衣錠、ハードカプセル、ソフトカプセル、顆粒、粉末などであってもよい。錠剤およびカプセルは、使用が容易であり患者の服薬遵守が高いことから、多くの医療条件で最も有利な経口投与形態を示す。
【0176】
同様に、液体調製物用の組成物としては、水、アルコール、油、グリコール、保存剤、着香剤、着色剤、懸濁剤などの適切な担体および添加剤を含む、溶液、エマルジョン、分散体、懸濁物、シロップ、エリキシルなどが挙げられる。非経口投与用の典型的な調製物は、有効成分を、担体、例えば滅菌水または非経口的に許容しうる油、例えばポリエチレングリコール、ポリビニルピロリドン、レシチン、落花生油またはゴマ油と共に含み、溶解度または防腐性を補助する他の添加剤を含んでいてもよい。溶液の場合、それを粉末に凍結乾燥し、その後、使用直前に再構成させることができる。分散体および懸濁物では、適切な担体および添加剤としては、水性ゴム、セルロース、ケイ酸塩または油が挙げられる。
【0177】
本発明の実施形態による医薬組成物としては、経口、経直腸、局所、吸入(例えば、エアロゾルを介して)、口腔(例えば、舌下)、経膣、局所(即ち、気道表面を含む、皮膚および粘膜表面の両方)、経皮投与および非経口投与(例えば、皮下、筋肉内、皮膚内、関節内、肋膜内、腹腔内、髄腔内、脳内、頭蓋内、動脈内または静脈内)に適したものが挙げられるが、任意の所定の例に最も適した経路は、処置される状態の性質および重症度、ならびに用いられる特定の活性剤の性質に依存する。
【0178】
注射用の組成物は、有効成分を、プロピレングリコール−アルコール−水、等張水、注射用滅菌水(USP)、Emulphor(商標)−アルコール−水、Cremophor−EL(商標)または当業者に公知の他の適切な担体をはじめとする適切な担体と共に含む。これらの担体は、単独で、または他の従来の可溶化剤、例えば、エタノール、プロピレングリコール、もしくは当業者に公知の他の薬剤と併用で使用してもよい。
【0179】
本発明の大環状化合物が、溶液または注射の形態で適用される場合、該化合物を、任意の従来の希釈剤に溶解または懸濁させることにより用いてもよい。希釈剤は、例えば、生理食塩水、リンガー液、水性グルコース溶液、水性デキストロース溶液、アルコール、脂肪酸エステル、グリセロール、グリコール、植物または動物供給源から得られる油、パラフィンなどを含んでいてもよい。これらの調製物は、当業者に公知の任意の従来法により調製してもよい。
【0180】
経鼻投与用の組成物は、エアロゾル、滴下剤、粉末およびゲルとして配合させてもよい。エアロゾル配合剤は、典型的には生理学的に許容しうる水性または非水性溶媒中の有効成分の溶液または微細懸濁物を含む。そのような配合剤は、典型的には密閉容器中に滅菌形態の単回投与量または反復投与量で存在する。密閉容器は、噴霧装置で使用されるカートリッジまたはレフィルであってもよい。あるいは密閉容器は、単回使用の経鼻吸入器などの単回分注装置、または治療上効果的な量を送達するように設定されていて、内容物が完全に使用されると廃棄される調量弁付きのポンプ式アトマイザもしくはエアロゾルディスペンサであってもよい。投与形態がエアロゾルディスペンサを含む場合、それは噴射剤、例えば圧縮気体、例として空気、またはフルオロクロロ炭化水素もしくはフルオロ炭化水素をはじめとする有機噴射剤を含むであろう。
【0181】
口腔または舌下投与に適した組成物としては、錠剤、トローチおよび香剤が挙げられ、その有効成分は、担体、例えば糖と、アカシア、トラガカントまたはゼラチンと、グリセリンと配合される。
【0182】
経直腸投与用の組成物としては、従来の坐剤ベース、例えばココアバターを含む坐剤が挙げられる。
【0183】
経皮投与に適した組成物は、軟膏、ゲルおよび貼付薬が挙げられる。
【0184】
当業者に公知の他の組成物を、例えば膏薬など、経皮または皮下投与に適用することができる。
【0185】
更に、組成物の配合に必要な成分と混和される単一または複数の有効成分を含むそのような医薬組成物を調製する際に、他の従来の薬理学的に許容しうる添加剤を、例えば賦形剤、安定化剤、防腐剤、湿潤剤、乳化剤、滑剤、甘味剤、着色剤、着香剤、等張剤、緩衝剤、抗酸化剤などを組み込んでもよい。添加剤として、例えば、デンプン、スクロース、フルクトース、デキストロース、ラクトース、グルコース、マンニトール、ソルビトール、沈降炭酸カルシウム、結晶セルロース、カルボキシメチルセルロース、デキストリン、ゼラチン、アカシア、EDTA、ステアリン酸マグネシウム、タルク、ヒドロキシプロピルメチルセルロース、メタ重亜硫酸ナトリウムなどを挙げることができる。
【0186】
幾つかの実施形態において、該組成物は、単位投与形態、例えば錠剤またはカプセル中で提供される。
【0187】
更なる実施形態において、本発明は、有効量の本発明の化合物1種以上を含む医薬投与単位を含む容器を1個以上含むキットを提供する。
【0188】
本発明は、本明細書に記載された化合物を含むプロドラッグを更に提供する。用語「プロドラッグ」は、生理学的条件下で、または加溶媒分解により、または代謝的に、薬学的に活性のある特異的化合物に変換される化合物を意味するものとする。「プロドラッグ」は、化学的に誘導体化されることで、(i)親薬物化合物の生物活性の幾つか、もしくは全てを保持するか、または全く保持しなくなり、そして(ii)対象において代謝されて親薬物化合物を生成する、本発明の化合物であってもよい。本発明のプロドラッグは、化合物が化学的に誘導体化されることで、(i)親薬物化合物の生物活性の幾つか、もしくは全てを保持するか、または全く保持しなくなり、そして(ii)対象において代謝されて該化合物の生物活性誘導体を生成する、「部分プロドラッグ」であってもよい。化合物を誘導体化してプロドラッグを提供するための公知の技術を、用いることができる。そのような方法を利用して、化合物の加水分解可能なカップリングを形成してもよい。
【0189】
本発明は、本発明の化合物が代謝および/もしくは内分泌障害、胃腸障害、心臓血管障害、肥満および肥満関連障害、中枢神経系障害、骨障害、遺伝子障害、増殖過剰性障害ならびに炎症性障害を予防ならびに/または処置するのに用いられる治療剤と併用して投与してもよいことを更に提供する。例示的な薬剤としては、鎮痛剤(オピオイド系鎮痛剤を含む)、麻酔剤、抗真菌剤、抗生物質、抗炎症剤(非ステロイド系抗炎症剤を含む)、駆虫薬、鎮吐薬、抗ヒスタミン剤、血圧降下剤、抗精神病薬、抗関節炎薬、鎮咳薬、抗ウイルス薬、心臓作用薬、下剤、化学療法剤(例えば、DNA相互作用薬、代謝拮抗薬、チューブリン相互作用薬、ホルモン剤、およびアスパラギナーゼまたはヒドロキシ尿素などの薬剤)、コルチコイド(ステロイド)、抗うつ剤、抑制薬、利尿薬、催眠薬、ミネラル物質、栄養補助剤、副交感神経興奮薬、ホルモン(例えば、コルチコトロピン放出ホルモン、アドレノコルチコトロピン、成長ホルモン放出ホルモン、成長ホルモン、チロトロピン放出ホルモンおよび甲状腺刺激ホルモン)、鎮静剤、スルホンアミド、興奮剤、交感神経興奮薬、睡眠薬、血管収縮剤、血管弛緩剤、ビタミン剤およびキサンチン誘導体が挙げられる。
【0190】
本発明により処置されるのに適した対象としては、非限定的に、鳥類およびホ乳類対象が挙げられ、好ましくはホ乳類である。本発明のホ乳類としては、非限定的に、イヌ科、ネコ科、ウシ属、ヤギ、ウマ、ヒツジ、ブタ、げっ歯類(例えば、ラットおよびマウス)、ウサギ、霊長類、ヒトなど、および子宮内のホ乳類が挙げられる。本発明により処置される必要がある任意のホ乳類対象が、適している。ヒト対象が、好ましい。両方の性および発達の任意段階のヒト対象(即ち、新生児、幼児、児童、青年、成人)を、本発明により処置することができる。
【0191】
本発明による例示的鳥類としては、ニワトリ、アヒル、七面鳥、ガン、ウズラ、ギジ、平胸類(例えば、オーストリッチ)および家庭で飼育される鳥(例えば、オウムおよびカナリア)、ならびに卵内の鳥が挙げられる。
【0192】
本発明は、第一に、ヒト対象の処置に関係するが、本発明は獣医学的目的ならびに薬物スクリーニングおよび薬物開発目的で、動物対象、詳細にはホ乳類対象、例えば、マウス、ラット、イヌ、猫、家畜およびウマで実施することもできる。
【0193】
グレリン受容体の調整物質、例えばアゴニストが効果的となるホ乳類(即ち、ヒトまたは動物)の状態を処置するための治療的使用において、本発明の化合物またはその適切な医薬組成物を、有効量で投与してもよい。該化合物の活性および治療効果の程度が変動するため、投与される実際の投与量は、一般に認識された因子、例えば年齢、対象の状態、送達経路および対象の体重に基づいて決定されるであろう。投与量は、1日あたり1〜4回の経口投与で約0.1〜約100mg/kgであってもよい。加えて、1日あたり1〜4回の投与で、化合物を注射により投与あたりにおよそ0.01〜20mg/kg投与してもよい。処置を数週間、数ヶ月またはそれを超えて継続することができる。特定の状況での最適投与量の決定は、当業者の能力内である。
【0194】
5.使用方法
本発明の化合物は、本明細書に記載されたものおよび更に非限定的に増殖過剰性障害、炎症性障害、組織障害、心臓血管障害、呼吸器障害、ウイルス感染およびそれらの組み合わせをはじめとする一定範囲の医療状態の予防および処置に用いることができ、その障害は、複数の病気に罹患した結果であってもよい。特定の実施形態において、疾患または障害は、癌である。
【0195】
本発明の更なる態様によれば、増殖過剰性障害、例えば腫瘍、癌および新生物障害、ならびに前悪性および非新生物または非悪性の増殖過剰性障害の処置の方法が提供される。詳細には、本発明により処置されうる腫瘍、癌、および新生物組織としては、非限定的に、悪性障害、例えば乳癌、骨肉腫、血管肉腫、線維肉腫および他の肉腫、白血病、リンパ腫、洞腫瘍、卵巣癌、子宮癌、膀胱癌、前立腺癌および他の泌尿生殖器癌、結腸癌、食道癌、胃癌および他の胃腸癌、肺癌、骨髄腫、膵臓癌、肝臓癌、腎臓癌、内分泌癌、皮膚癌、ならびにグリオーマおよび神経芽細胞腫をはじめとする悪性または良性の脳または中枢(CNS)および末梢神経系の腫瘍が挙げられる。
【0196】
本明細書で用いられる「処置」は、それに関連する障害または症状の治癒または完全な消滅を付与することを必ずしも意味していない。
【0197】
本発明の化合物は、更に非限定的に、増殖過剰性障害、炎症性障害、呼吸器障害およびウイルス感染をはじめとする一定範囲の医療状態の処置のための医薬品を調製するために使用することができる。
【0198】
本発明の更なる実施形態を、ここに以下の実施例を参照して記載する。これらの実施形態が本発明の実施形態の例示を目的とするもので、本発明の範囲を限定するものではないことを理解しなければならない。
【実施例】
【0199】
実施例1
代表的なセリンプロテアーゼの阻害のためのアッセイ
以下に、代表的なセリンプロテアーゼとしてマトリプターゼのアッセイを記載するが、それは報告された方法に基づいている(Desilets, A.; Longpre, J.-M.; Beaulieu, M.-E.; Leduc, R. FEBS Lett. 2006, 580, 2227-2232.)。類似のアッセイを、他のセリンプロテアーゼ用に適用および入手することができる。
【0200】
FLX−800 TBEマイクロプレートリーダー(米国バーモント州ウィヌースキのBio−Tek Instruments)においてAMC−結合ペプチドからの蛍光の放出を測定することにより(励起:360nm、発光:441nm)、酵素活性をモニタリングした。精製ヒトマトリプターゼを、バースト滴定試薬(burst titrant)4−メチルウンベリフェリル−p−グアニジノ安息香酸塩(MUGB)で活性部位滴定した。500lg/mL BSA、pH9を含有する100mM Tris−HCl中で、マトリプターゼを用いた酵素的アッセイを実施した。文献に記載された通り、ヒト可溶性フリンを発現させ、精製、滴定およびアッセイした(Denault, J.B.; Lazure, C.; Day, R; Leduc, R. Protein Expr. Purif. 2000, 19, 113-124.)。精製されたHATタンパク質を、MUGBで活性部位滴定した。pH8.6の50mM Tris−HCl中で、HATでのアッセイを実施した。
【0201】
酵素を、フリンでは4〜12.5nMの範囲の濃度、マトリプターゼでは2〜7nMの範囲の濃度、そしてHATでは20pMの濃度に希釈して、10μMと共に37℃でインキュベートするか(初期スクリーニングの場合)、または被験化合物を適切に希釈して(反応速度分析の場合)、例えば0、500、1000、2000nMとして、もしくは0、250、500、1000、2500、5000nMで室温で15分間インキュベートした。蛍光発生基質(フリンでは4μM Boc−Arg−Val−Arg−Arg−AMC、マトリプターゼではBoc−Gln−Ala−Arg−AMC、そしてHATで4μg Boc−Val−Pro−Arg−AMC)の加水分解を追跡することにより、残留する酵素活性を測定した(米国ペンシルバニア州キングオブプリシアのBachem Bioscience)。濃度を上昇させる被験化合物の存在下で、飽和曲線を作成した。3回以上の独立した実験のデータを、典型的には平均化して、残留速度を、被験化合物濃度の関数としてプロットした。Enzfitterソフトウェア(米国ミズーリ州ファーガソンのBiosoft)を用いた式(1)への非線形回帰分析により、データを適合させた(Bieth, J.G. Methods Enzymol. 1995, 248, 59-84.)。
式(1):
【数1】
(式中、v0およびviは、それぞれ阻害剤の非存在下および存在下での基質加水分解の定常状態速度であり、[E]0は酵素の初期濃度、[I]0は阻害剤の初期濃度、そしてKi(app)は基質依存性の平衡解離定数である)。基質依存性定数Kiは、式(2)を用いて計算した(Bieth, J.G. Methods Enzymol. 1995, 248, 59-84.)。
式(2):
【数2】
(式中、[S]0は基質の初期濃度であり、Kmは、酵素−基質相互作用でのミカエリス・メンテン定数である)。被験化合物の安定性を調査するために、10μM被験化合物を特定濃度のマトリプターゼまたはHATと共に室温で特定時間インキュベートした。その後、タンパク質をSDS−PAGEにより分解して、Gel Code青色染色試薬を用いて明らかにした(米国イリノイ州ロックフォードのPierce Biotechnology)。
【0202】
表に、代表的な本発明の化合物のマトリプターゼ阻害の結果を表す。
【表6】
【0203】
Kiは、非線形回帰分析を用いた速度から計算することができる。用いられたモデルは、競合酵素阻害式、Km0bs=Km*(1+[I]/Ki)およびY=Vmax*X/(KmObs+X)である。Xは、基質濃度である。Yは、速度である(Copeland, R.A. Enzymes, 2nd edition, Wiley, 2000の式8.11)。Kiは、GraphPad Prism5ソフトウェア(米国カリフォルニア州サンディエゴのGraphPad Software)を用いて計算した。例えば化合物451は、Ki=1.46μMを有し、化合物459は、Ki=245nMを有する。
【0204】
阻害の選択性を決定するために、TTSPおよび他のセリンプロテアーゼを、蛍光発生ペプチドBoc−Gln−Ala−Arg−AMCの存在下で被験化合物とインキュベートした。活性を37℃で20分間測定した。
【0205】
実施例2
プロテアーゼ阻害アッセイ
(Li, P.; Jiang, S.; Lee, S.-L.; et al. J. Med. Chem. 2007, 50, 5976-5983.)。ウシトロンビン、ボーマン・バーク型阻害剤(BBI)、および蛍光基質は、市販されていた(ミズーリ州セントルイスのSigma Chemical Co.)。プロテアーゼに対する本発明の化合物の阻害活性を、2種の異なる系で室温で測定した。第一のアッセイ系では、100mg/mL ウシ血清アルブミンを含有する100mMTris−HCl(pH8.5)の反応緩衝液を用いた。反応緩衝液170μLを含有するキュベットに、酵素溶液10μLおよび阻害剤溶液10μLを添加した。プレインキュベーションの後、蛍光ペプチド基質の溶液(10μL)を添加して、キュベットの内容物を完全に混合した。蛍光分光光度計(日立F4500)を励起波長360nmおよび発光波長480nmで用いて、基質の加水分解により放出された蛍光の変化を追跡することにより、残留酵素活性を決定した。例えば蛍光ペプチドBoc−Gln−Ala−Arg−AMCを、マトリプターゼ用の基質として用いた。ペプチドBoc−Leu−Arg−Arg−AMCを、トロンビン用の基質として用いた。6〜7種の異なる濃度の被験化合物の存在下で、加水分解速度を記録した。異なる基質濃度での2組のデータからのディクソンプロットにより、Ki値を決定した。
【0206】
70kDa活性化マトリプターゼを、記載された通り単離した(Lin, C.-Y.; Anders, J.; Johnson, M.D.; Dickson, R.B. J. Biol. Chem. 1997, 272, 27558-27564; Lin, C.-Y.; Anders, J.; Johnson, M.; Sang, Q. A.; Dickson, R. B. J. Biol. Chem. 1999, 274, 18231-18236.)。第二のアッセイ系は、本質的に同一の結果をもたらし、100mg/mL BSAを含有する100mM Tris(pH8.3)の緩衝液中のマトリプターゼ用の基質としてBoc−Gln−Ala−Arg−AFCペプチドを使用した。Tecan Ultra蛍光分光計(ノースカロライナ州ダーラムのTecan)を用いて黒色96穴プレート中で、合計容量200μL中の精製マトリプターゼでアッセイを実施した。
【0207】
実施例3
代表的なセリンプロテアーゼの阻害に関する細胞培養アッセイ
マトリプターゼcDNAでトランスフェクトされたHEK293細胞中でのマトリプターゼ活性を阻害する能力について、被験化合物を検査した。被験化合物をモック細胞およびマトリプターゼをトランスフェクトした細胞と共に18時間インキュベートした。溶媒中のタンパク質分解活性を、蛍光発生ペプチドBoc−Gln−Ala−Arg−AMCを用いて測定した。
【0208】
実施例4
腫瘍転移についてのインビトロアッセイ
(Galkin, A.V.; Mullen, L.; Fox, W.D.; Brown, J.; et al. Prostate 2004, 61, 228-235)。CWR22RV1細胞を、ATCC(メリーランド州ロックビル)から得て、7%ウシ胎仔血清(カリフォルニア州タルザナのOmega Scientific)、1%ペニシリン−ストレプトマイシンおよび1%L−グルタミン(ニューヨーク州グランドアイランドのGibco)を補足したRPMI−1640培地中で培養する。CWR22RV1細胞増殖速度に対する本発明の化合物の影響を試験するために、播種された細胞を4群に分割して、最初の播種後1、3、および5日目に、1、10、または25mM濃度の被験化合物またはビヒクル溶液で処置する。実験では、1日および1群あたり三枚ずつプレートを使用する。3、5、および7日目に、血球測定装置で細胞を計数する。細胞浸潤性アッセイ(カリフォルニア州テメキュラのChemicon)を利用して、タンパク質の組換え基底膜マトリックス(ECMatrix、Chemicon)を介したCWR22RV1細胞浸潤に対する本発明の化合物の影響を評価する。ECMatixの再水和の後、25mM被験化合物を含む、または含まない無血清培地0.4mL中のCWR22RV1細胞(2×105)を上のチャンバーに添加して、10%ウシ胎仔血清を含有し25mM被験化合物を含む、または含まない培地0.75mLを予め充填した下のチャンバーに入れて、378℃で48時間インキュベートする。インキュベーションの終了時に、培地および任意の非浸潤細胞を除去して、供給したクリスタルバイオレット溶液で膜を染色する。その後、膜をスライドガラスに載せて、細胞を光学顕微鏡で検査する。一群あたり膜6枚(±被験化合物処置)を倍率100倍で分析する。膜あたり5ヶ所のフィールドを無作為に選択して、利用可能な孔の合計数のうちの平均浸潤細胞数を計数する。観察されたフィールドあたりの浸潤細胞の割合を計算する。実験は、二重測定で実施する。
【0209】
実施例5
(Galkin, A.V.; Mullen, L.; Fox, W.D.; Brown, J.; et al. Prostate 2004, 61, 228-235.)。4〜6週齢雌無胸腺BALB/cヌードマウス(Charles Rivers Laboratories)を、病原体フリーの条件下で飼育する。確立されたアンドロゲン非依存性(AI)の3種のCWR22RおよびCWRSA6異種移植片細胞系から得た組換え基底膜 (Matrigel;マサチューセッツ州ベッドフォードのCollaborative Research)と共に、細かく刻んだ腫瘍組織をマウスに皮下接種する。4〜10日後に、およそ5×5mm3の腫瘍を生成したマウスに、生理食塩水中の被験化合物(50または25mg/kgを1日2回、1週間に7回、腹腔内)またはビヒクルのみのいずれかを、同一の投与スケジュールで投与する。腫瘍を週2回、ノギスで計測し、腫瘍容積を式(π/6)×(大径)×(小径)2により計算する(Press, M.F.; Bernstein, L.; Thomas, P.A.; Meisner, L.F.; Zhou, J.Y.; Ma, Y.; Hung, G.; Robinson, R.A.; Harris, C.; El-Naggar, A.; Slamon, D.J.; Phillips, R.N.; Ross, J.S.; Wolman, S.R.; Flom, K.J. J. Clin. Oncol. 1997, 15, 2894-2904.)。腫瘍接種の18〜25日後に動物を殺処分して、腫瘍組織を分析のために急速冷凍する。
【0210】
実施例6
腫瘍転移のインビボアッセイ
6週齢ケラチン−5−マトリプターゼ・トランスジェニックマウスおよび同腹子対照マウス(List, K.; Szabo, R.; Molinolo, A.; Sriuranpong, V.; Redeye, V.; Murdock, T.; Burke, B.; Nielsen, B.S.; Gutkind, J.S.; Bugge, T.H. Genes Dev. 2005, 19, 1934-1950.)を、1種以上の濃度の被験化合物で処置する。その後、腰部中央の表皮ケラチノサイトの増殖速度に対する被験化合物の影響を、ビヒクル対照での処理結果と比較することにより決定する。
【0211】
実施例7
鶏卵尿漿膜モデル
文献の方法を用いて、本発明の化合物の血管形成を阻害する能力を測定することができる(Ghiso, J.A.A.; et al. J. Cell. Biol. 1999, 147, 89-104.)。
【0212】
実施例8
テザーの合成
A.テザーT5の合成に関する標準的手順
【化19】
【0213】
ステップ5−1. 蒸留水(5L)中の3−メチル安息香酸エチル(5−0、300g、1.83mol、1当量)の溶液に、臭素(292.5g、1.83mol)を一度に添加した。この混合物に、200Wのランプ2台を照射した。ランプをフラスコ中央の外側に置いて、フラスコの周りに箱を置いた。照射の間、溶液を激しく撹拌した。温度を45℃に上昇させると、溶液が反応の間に橙色から黄色へ、そしてほとんど無色へ変色した。4時間後に(本質的に無色の溶液)、ランプを消灯して、混合物を室温まで放冷した。混合物をDCM 2Lで希釈し、その後、水相をDCM 500mLで抽出した。ひとまとめにした有機相をブラインで、その後、10%チオ硫酸ナトリウム溶液で、そして最後に再度ブライン(pH=5)で洗浄した。橙色の相をMgSO4で乾燥させ、ろ過してろ液を減圧濃縮して、次のステップで用いるのに十分な品質の5−1を液体として収率96%で得た。
TLC(15% EtOAc/ヘキサン): Rf:0.58、検出:UV
【0214】
ステップ5−2. 室温で撹拌したエタノール(95%、1L)中の5−1(149g、0.611mol)の混合物に、蒸留水(300mL)中のシアン化カリウム(68g、1.7当量)の溶液を、滴加漏斗を用いて滴加した(警告:毒物あり! シアン化カリウムは公知の毒物であり、十分に喚起されたドラフトチャンバー内で適切に保護しながら取り扱わなければならない。HCN検出器の存在下で反応を実施する。ガラス製品は全て反応後に水およびアセトンで洗浄し、洗浄溶液は、「シアン化物」であり「危険」であることが明確に判別される容器に入れて、正しく廃棄しなければならない)。溶液は、添加の間に黄色になった。添加が完了した後、反応混合物を60℃まで2時間加熱し、その後、室温で一晩撹拌した(TLC:10%EcOAc/90%ヘキサン;検出:UV、CMAにより反応をモニタリング)。溶液を水(900mL)で希釈し、その後、Et2Oで抽出した(900mLで3回)。ひとまとめにした有機相をブラインで2回洗浄し(2回)、MgSO2で乾燥させ、ろ過してろ液を減圧蒸発させて、橙色油状物を与えた。油状残渣をシリカゲルのドライパックにより、EtOAc/ヘキサンを用いて精製して(勾配、5/95から15/85へ)、5−2を黄色固体として得た(66g、2段階で59%)。
TLC(30/70 EtOAc/ヘキサン):Rf:0.45、検出: UV;
1H NMR: δ 1.6 ppm (2H, 三重線), 3.8 ppm (3H, s), 4.4 ppm (2H, 四重線),7.4−7.6 ppm (2H, m), 8.0−8.1 ppm (2H, m)
【0215】
ステップ5−3.室温のTHF/水(4.6L/2.3L)中の5−2(220g、1.17mol)の溶液に、塩化コバルト(54.7g、0.23mol)を添加して、水素化ホウ素ナトリウム(132g、3.5mol)を少しずつ添加した。水素発生が、観察される。添加後に、反応物を室温で一晩撹拌した。混合物をCelite(登録商標)でろ過して、THF 1Lで洗浄した。THFを減圧蒸発により除去し、その後、水酸化ナトリウムの溶液(0.5N、2L)を添加して、混合物をEt2Oで抽出した(3回)。ひとまとめにした有機相をブラインで洗浄し(2回)、Na2SO4で乾燥させ、ろ過してろ液を減圧濃縮して、次のステップで直接使用されるのに適切な品質の粗液を5−2から52%得た。
TLC(50/50 EtOAc/ヘキサン):Rf:ベースライン、検出:UV、ニンヒドリン
【0216】
ステップ5−4. 脱気したDMF(200mL)中の5−3(118g、0.61mol)、Ddz−OPh(213g、0.67mol)およびトリエチルアミン(85mL、0.61mol)の溶液を、窒素雰囲気下、50℃で2日間撹拌した。その後、混合物を水2.5Lで希釈した。水相をEt2Oで抽出した(3回)。ひとまとめにした有機相を水、水酸化ナトリウム(0.5N、2回)およびブライン(2回)で引き続き洗浄し、MgSO4で乾燥させ、ろ過してろ液を減圧濃縮して、褐色油状物を得た。粗材料をドライパック(勾配、15%EtOAc/ヘキサン、0.5%Et3Nから25%EtOAc/ヘキサン、0.5%Et3Nへ;検出:UV+CMA)で精製して、5−4を156g(62%)得た。
TLC(25/75 EtOAc/ヘキサン):Rf:0.23、検出:UV + CMA
【0217】
ステップ5−5の還元と適合性のある他の保護基をこの段階で用い、標準的反応条件を用いて付加させることもできる。
【0218】
ステップ5−5. −78℃のDCM(2.1L)中の5−4(291.5g、0.7mol)の溶液に、水素化ジイソブチルアルミニウム(DIBAL−H、DCM中に1.0M、2.1L、2.1mol)を滴加漏斗で添加した。添加が完了したら、溶液を−78℃で2時間撹拌するか、またはTLCのモニタリング(50%EtOAc/ヘキサン;検出:UV、ニンヒドリン)により完了が示されるまで撹拌した。その後、反応混合物を酒石酸の溶液(1.0M、4L)に緩やかに滴加することにより、クエンチした。得られた混合物をDCMで抽出した(3回)。ひとまとめにした有機相をEDTA四ナトリウム塩の0.6M溶液(2Lで1回)およびブライン(2Lで1回)で引き続き洗浄し、MgSO4で乾燥させ、ろ過してろ液を減圧濃縮して、生成物Ddz−T5を黄色油状物(251.4g、96%)として得た。
TLC(50/50, EtOAc/ヘキサン):Rf:0.25、検出: UV、ニンヒドリン;
1H NMR: δ (1.7 ppm (s, 6H, 2 x CH3), 2.8 ppm (t, 2H,2 x CH2), 3.4 ppm (四重線, 2H, 2 x CH2), 3.8 ppm (s, 6H, 2 x OCH3), 4.7 ppm (s, 2H, CH2), 6.3−6.5 ppm (m, 3H, 3 x CH), 7.0−7.4 ppm (m, 4H, 4 x CH)。
【0219】
B.テザーT−28の合成の標準的手順
米国特許第7,521,420も参照されたい。
【化20】
【0220】
ステップ28−1. (Tius, M.A. J. Am. Chem. Soc. 1992, 114, 5959.)。酢酸(115mL)中のサリチルアルデヒド(28−0、23.4g、0.19mol、1.0当量)の溶液に、酢酸アンモニウム(17g、0.22mol、1.15当量)およびニトロメタン(39.5mL、0.73mol、3.8当量)を添加した。混合物を110℃で4.5時間加熱し、その後室温で冷却した。溶媒を真空除去してDCMで希釈し、ブラインで洗浄して(3回)、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させた。残渣をフラッシュクロマトグラフィーで精製して(勾配、EtOAc/ヘキサンを10%、その後20%、その後25%)、28−1を14.5g(45.8%)得た。
TLC (25/75 EtOAc/ヘキサン):Rf= 0.21,検出 UV, CMA;
1H NMR (CDCl3): δ8.16−8.11 (d、 1H), 7.98−7.93 (d、 1H), 7.44 (d、 1H), 7.43−7.32 (m, 1H), 7.32−6.98 (t, 1H), 6.87 (d, 1H)。
【0221】
ステップ28−2. 0℃のTHF/MeOH(7/1、500mL)中の28−1(14.5g、0.088mol、1.0当量)の溶液に、水素化ホウ素ナトリウム(10.0g、26.0mol、3.0当量)を少しずつ添加した。反応物を室温で温めて、完了するまでTLCによりモニタリングした。水を緩やかに添加することにより、反応物をクエンチした。pHを1M HClでpH7〜8に調整した。THFを真空除去し、その後、残りの混合物をエーテルで抽出した(3回)。有機相をブラインで洗浄し(1回)、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させて、次のステップで使用されるのに十分な純度の28−2を9.6g(66%)得た。
TLC (25/75 EtOAc/ヘキサン):Rf= 0.23, 検出: UV, CMA
【0222】
ステップ28−3. 95%EtOH(200mL)中の28−2(9.6g、0.058mol、1.0当量)の溶液に、10%Pd/Cを添加して、水素ガスを一晩バブリングした。混合物をCelite(登録商標)でろ過して、溶媒を減圧蒸発させた。生成物のEtOAcを同時蒸発させた。残渣(7.9g)の28−3は、任意の更なる精製を行わずに次にステップで使用した。
TLC (25/75 EtOAc/ヘキサン):Rf= 0.0,検出: UV, CMA。
【0223】
ステップ28−4. 0℃のDCM中の28−3(7.9g、0.058mol、1.0当量)およびEt3N(16.2mL、0.12mol、2.0当量)の溶液に、DCM中のBoc2O(12.7g、0.058mol、1.0当量)の溶液を滴加した。反応混合物を、一晩撹拌した。反応混合物をクエン酸緩衝液(2回)およびブライン(2回)で洗浄して、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させた。粗残渣をフラッシュクロマトグラフィーにより精製して(勾配、EtOAc/ヘキサンを20%、その後25%)、28−4(7.4g、54%、2段階)を得た。
TLC (25/75 EtOAc/ヘキサン):Rf= 0.36, 検出: UV, CMA
【0224】
ステップ28−5. THF(200mL)中の2−ブロモエタノール(2.29g、42.3mmol、1.0当量)の溶液に、イミダゾール(7.2g、105.8mmol、2.5当量)を、その後TBDMSCI(6.7g、44.4mmol、1.05当量)を添加した。反応混合物を4時間撹拌すると、白色沈殿物が2〜5分後に形成し始めた。エーテル(200mL)を添加して、有機相を塩化アンモニウムの飽和溶液(2回)、重炭酸ナトリウムの飽和溶液(1回)およびブライン(1回)で引き続き洗浄して、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させた。こうして得られた生成物(28−A、8.7g、86%)を、次の反応に直接使用した。
TLC (25/75 EtOAc/ヘキサン):Rf= 0.80,検出: UV, CMA
【0225】
DMF(40mL)中の28−4(4.2g、17.8mmol、1.0当量)、28−A(6.4g、26.7mmol、1.5当量)およびヨウ化カリウム(591mg、3.6mmol、0.2当量)の溶液に、炭酸カリウム(2.7g、19.6mmol、1.1当量)を添加して、混合物を75℃で一晩加熱した。その期間の後、TLCで反応が完了していないことが示され、そのため28−Aおよび炭酸カリウムを更に1当量添加して、混合物を追加で一晩撹拌した。DMFを真空除去した(油圧ポンプ)。油状残渣を水で希釈して、生成物をエーテルで抽出した(3回)。有機相をブラインで洗浄し(2回)、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させた。生成物をフラッシュクロマトグラフィーにより精製して(15%EtOAc/ヘキサン)、28−5を5.2g(74%)得た。
TLC (35/65 EtOAc/ヘキサン):Rf= 0.79, 検出: UV, ニンヒドリン
1H NMR (CDCl3): δ 7.05 (m, 2H), 6.78 (m, 2H), 4.6 (bs, 1H), 3.95 (m, 2H), 3.88 (m, 2H), 3.28 (bq, 2H), 2.72 (t, 2H), 1.3 (s, 9H), 0.8 (s, 9H), 0.0 (s, 6H)。
【0226】
ステップ28−6. THF(20mL)中の28−5(2.5g、13.3mmol、1.0当量)の溶液に、THF中の1.0M TBAF(15.9mL、15.9mmol、1.2当量)を添加して、反応物を室温で30分間撹拌した。反応混合物をエーテル(150mL)で希釈し、その後、塩化アンモニウムの飽和溶液(2回)およびブライン(1回)で洗浄して、MgSO4で乾燥させ、ろ過して溶媒を減圧蒸発させた。生成物をフラッシュクロマトグラフィーにより精製して(勾配、EtOAc/ヘキサンを25%から40%へ)、Boc−T28を3.5g(94.6%)を得た。
TLC (5/65 EtOAc/ヘキサン):Rf= 0.21,検出: UV,ニンヒドリン;
1H NMR(CDCl3):δ 7.3 (td, 1H), 7.1 (dd, 1H), 6.86 (m, 2H), 4.9 (bs, 1H), 4.1 (m, 2H), 4.0 (m, 2H), 3.3 (bs, 2H), 2.8 (t, 2H), 1.4 (m, 9H);
13C NMR (CDCl3):δ 157.2, 156.6, 130.8, 128.0, 127.6, 120.9, 111.4, 79.7, 69.8, 61.4, 40.9, 32.6, 28.6;
LC−MS (勾配 A4): tR: 10.2 分; (M+H)+ 281, (M+H+Na)+ 304
【0227】
C.テザーT29の合成の標準的手順
【化21】
【0228】
ステップ29−1: 0℃のTHF(DriSolv等級)中の水素化リチウムアルミニウム(LAH、3mol当量)の溶液に、3−シアノベンゾアルデヒド(29−0、1当量)を少しずつ添加した。混合物を0℃で1時間(または出発原料が消失するまで)撹拌し、その後、窒素雰囲気下のオイルバス中で一晩、加熱還流した(70℃)。反応をクエンチするために、溶液を窒素下で0℃に冷却し、次のものを引き続き添加した:水、NaOH(15%)、その後、水(それぞれLAH 5gに対して5mL:5mL:15mLの比で用いなければならない)(警告:水素ガス発生)。溶液をろ過し、塩をTHFで洗浄し、ひとまとめにしたろ液を減圧濃縮して、粗アミノアルコールを、典型的には次のステップで用いられるのに十分な純度で得た(Rf:ベースライン, 30/70, EtOAc/ヘキサン,検出: UV,ニンヒドリン)。
【0229】
ステップ29−2: 窒素雰囲気下、0℃で脱気されたDMF中のステップ29−1の生成物(1当量)およびDdz−N3(1.05当量)の溶液に、テトラメチルグアニジン(TMG、1.05当量)を添加した。10分後に、DIPEA(1.05当量)を添加し、その後、混合物を50℃のオイルバスで一晩撹拌した。混合物を減圧濃縮して(油圧ポンプ)、DMFを除去し、その後、残渣をDCMに溶解して、クエン酸緩衝液(2回)、飽和重炭酸ナトリウム(1回)、およびブライン(2回)で引き続き洗浄し、その後、MgSO4で乾燥させ、ろ過してろ液を減圧濃縮した。こうして得られた粗材料をフラッシュクロマトグラフィーにより精製して(勾配、50%EtOAc/ヘキサン、0.5%Et3Nから60%EtOAc/ヘキサン、0.5%Et3Nへ;DCMを混合物に添加して、開始時に残渣を溶解した)、所望の化合物、DdZ−T29(TLC:50%ヘキサン/EtOAc;検出:UV,ニンヒドリン)を得た。
【0230】
他の典型的な窒素保護基、例えば、Fmoc、Boc、Cbzを、標準の反応条件を用いてステップ29−2で配置することもできる。別法として、水素化ホウ素ナトリウムを塩化コバルトと共に用い、その後、第一級アミンをBoc(図示しない)または他の適切なN−保護基で選択的に保護することにより、ステップ29−1における還元を実施することができる。
【化22】
【0231】
D. テザーT−30の合成の標準的手順
【化23】
【0232】
ステップ30−1. THF/H2O(1:1)125mL中の2−ブロモフェネチルアミン(30−0、5.0g、25.0mmol、1.0当量)の溶液に、重炭酸ナトリウム(2.3g、27.5mmol、1.1当量)を添加した。その後、混合物を0℃に冷却し、Boc無水物(5.5g、25.0mmol、1.0当量)を一度に添加した。混合物を0℃で1時間撹拌し、その後、室温まで昇温させて、一晩撹拌した。溶媒を減圧蒸発させて、残渣をEtOAc/H2O(1:1)に溶解した。分離された有機相をH2O(2回)および飽和塩化ナトリウム(2回)で洗浄し、硫酸マグネシウムで乾燥させ、ろ過してろ液を減圧蒸発させた。得られた黄色油状物をDCMで希釈し、減圧蒸発させて(手順を3回繰り返した)、30−1 7.5g(100%)を白色固体として得た。
TLC (ヘキサン/EtOAc, 7:3): Rf= 0.75,検出:UV,ニンヒドリン
【0233】
ステップ30−2. アルゴン雰囲気下で火力乾燥させたフラスコに、30−1(6.3g、21.0mmol、1.0当量)、再結晶化ヨウ化銅(I)(80.0mg、0.42mmol、0.02当量、Organometallics in Synthesis, 2ndedition, Manfred Schlosser, Ed., 2002, p 669の手順を参照)およびジクロロビス(ベンゾニトリル)パラジウム(II)(242mg、0.63mmol、0.03当量)を添加した。フラスコをアルゴンでパージし(5〜10分間)、無水1,4−ジオキサン 20mLを添加した後、トリ−tert−ブチルホスフィン(ヘキサン中の10%(w/w)溶液、385μL、1.26mmol、0.06当量)およびジイソプロピルアミン(3.6mL、25.2mmol、1.2当量)を添加した。その後、混合物をアルゴンで再度パージして(5〜10分間)、3−ブチノール(30−A、2.4mL、31.5mmol、1.5当量)を混合物に滴加して、TLCモニタリングを行いながら、アルゴン下、室温で24時間撹拌した。混合物をEtOAcで希釈し、シリカゲルパッドでろ過して、TLCにより更なる材料溶出が示されなくなるまでEtOAcで洗浄した。ろ液を減圧蒸発させて、残渣をフラッシュクロマトグラフィーにより精製し(ヘキサン:EtOAc,7:3)、30−2を薄黄色油状物として5.5g(90%)得た。
TLC (ヘキサン/EtOAc, 7:3): Rf= 0.20,検出: UV, CMA
【0234】
ステップ30−3. 窒素下、95%EtOH中のBoc−アミノアルコール30−2(6.1g、21.1mmol、1.0当量)の溶液に、酸化白金(IV)(445mg、2.11mmol、0.1当量)を添加した。混合物を80psi H2で16時間撹拌した(引き続き反応を1気圧H2の室温で24〜36時間実施した)。少量のアリコットを取り出して、1H NMRにより反応をモニタリングした。反応が完了したら、窒素を混合物に10分間バブリングして過剰な水素を除去した。溶媒を減圧蒸発させて、EtOAcで希釈し、シリカゲルパッドでろ過して、TLCにより更なる材料溶出が示されなくなるまでEtOAcで洗浄した。ろ液を減圧蒸発させて、残渣をフラッシュクロマトグラフィーにより精製し(ヘキサン:EtOAc,7:3)、Boc−T30を薄黄色油状物として4.5g(75%)得た。
1H NMR (CDCl3): d 7.18-7.11, (m, 4H), 4.65, (bs, 1H), 3.72-3.65, (t, 2H), 3.32 (bs, 2H), 2.85-2.80, (t, 2H), 2.70-2.65, (t, 2H), 1.71-1.66 (m, 4H), 1.44 (s, 9H)
【0235】
ステップ30−2および30−3の反応系列に適合性のある他のN−保護基を、用いることもできる。
【0236】
E.テザーT−32の合成の標準的手順
T−32の反応スキームを、図4に表わす。
【0237】
ステップ32−1. −30℃のCH3CN(500mL)中の4−ヒドロキシベンゾニトリル(32−0、15.0g、109mmol、1.0当量)の溶液に、トリフリン酸(11.6mL、131mmol、1.2当量)を添加した。NBS(20.3g、117mmol、1.05当量)を少しずつ添加することで、温度が−10℃を超えないようにした。懸濁物が得られ、その溶液が数分後に均質になった。反応混合物を室温に保持し、一晩撹拌した。溶液を水性飽和NaHCO3で処理して、水相をEtOAcで抽出した(1回)。水相を6M HClで酸性化して、EtOAcで抽出した。その後、有機相を水性飽和NH4Clで抽出した(2回)。有機相をMgSO4で乾燥させ、ろ過してろ液を減圧濃縮した。最終化合物が過剰の(1H NMRで10%を超える)スクシンイミド副生成物を含有することが見出されれば、固形残渣を水中で一晩撹拌し、沈殿物をろ過して、一晩真空乾燥させた(油圧ポンプ)。1H NMRにより、所望の化合物32−1の同一性を確証した。生成物は、更に精製せずに次のステップで使用するのに適していた(収率:94%)。
TLC (80%EtOAc, 20%ヘキサン): Rf= 0.47,検出:UVおよびKMnO4
【0238】
ステップ32−2. DMF(300mL)中の32−1(11.3g、57.1mmol、1.0当量)の溶液に、炭酸カリウム(8.7g、62.8mmol、1.1当量)、ヨウ化カリウム(1.9g、11.4mmol、0.2当量)およびTBDMS−ブロモエタノール(32−A、20.5g、85.7mmol、1.5当量)を添加した。得られた混合物70℃で一晩撹拌した。混合物を室温まで冷却し、ブラインを添加して、相を分離した。水相をエーテルで抽出して、ひとまとめにした有機相をブラインで抽出した(2回)。有機相をMgSO4で乾燥させて、減圧濃縮した。残渣をフラッシュクロマトグラフィーにより精製して(20%EtOAc、60%ヘキサン)、32−2を黄色固体として得た(収率100%)。
TLC (40%EtOAc, 60%ヘキサン): Rf= 0.63,検出: UVおよびKMnO4
【0239】
ステップ32−3. 0℃のTHF(10mL)中の32−2(548mg、1.5mmol、1当量)の溶液に、リチウムヘキサメチルジシラジド(LHMDS、THF中の1M、3.0mL、3.0mmol、2.0当量)を添加し、その後、混合物を室温で2時間撹拌した。クエン酸(1M、5mL)を添加して、混合物を1時間撹拌した。エーテルを添加して、相を分離し、その後、有機相を1Mクエン酸で抽出した(2回)。ひとまとめにした水相を3M NaOHでpH=14に調整し、その後、CH2Cl2で抽出した(4回)。有機相をK2CO3で乾燥させて、減圧濃縮した。こうして得られた32−2を、次のステップで直接使用した。
TLC (20% EtOAc, 80%ヘキサン): Rf=ベースライン;検出:UVおよびKMnO4
【0240】
ステップ32−4. THF(6mL)中の32−3(1.5mmol、1.0当量)の溶液に、(Boc)2O(371mg、1.7mmol、1.1当量)およびDMAP(18mg、0.15mmol、0.1当量)を添加して、混合物を3時間撹拌した。ブラインを添加して、水相をエーテルで抽出した(3回)。ひとまとめにした有機相をMgSO4で乾燥させて、減圧濃縮した。残渣をフラッシュクロマトグラフィーにより精製して(30%EtOAc、70%ヘキサン)、白色固体の32−4を得た(収率:67%、2段階)。ジオキサン中の水性水酸化ナトリウム(1N)を、このステップで基剤として用いて、同等の収率を得ることもできる。
TLC (30% EtOAc, 70%ヘキサン): Rf= 0.37;検出: UVおよびKMnO4
【0241】
ステップ32−5. ジイソプロピルアミン(100mL)中の32−4(8.2g、17.3mmol、1.0当量)の溶液に、Ddz−プロパルギルアミン(32−B、9.6g、34.6mmol、2.0当量)を添加して、混合物をArで20〜30分間脱気した。PPh3(546mg、2.08mmol、0.12当量)、PdCl2(PPh3)2(730mg、1.04mmol、0.06当量)およびCuI(131mg、0.69mmol、0.04当量)を添加して、得られた混合物を70℃で一晩撹拌した。溶液をシリカゲルパッドでろ過して、EtOAcですすぎ、その後、溶媒を減圧蒸発させた。得られた残渣をフラッシュクロマトグラフィーで精製して(40%EtOAc、60%ヘキサン)、32−5を橙色固体として得た(収率100%)。
TLC (40%EtOAc, 60%ヘキサン):Rf= 0.27;検出:UVおよびCMA
【0242】
ステップ32−6. 95%エタノール(100mL)中の32−5(15.0g、22.2mmol、1.0当量)の溶液に、PtO2(500mg、2.2mmol、0.1当量)を添加して、水素ガスを溶液に1時間バブリングした。得られた混合物を、室温で一晩撹拌した。その時点で反応が終了していなければ(1H NMR)、PtO2を更に0.1当量添加して、水素ガスを溶液にバブリングし、混合物を再度一晩撹拌した。Arを反応物にバブリングして過剰の水素を排出させ、溶液をシリカゲルパッドでろ過して、パッドをEtOAcですすいだ。ひとまとめにした溶媒を、減圧蒸発させた。得られた32−6を次のステップで用いた(収率100%)。
【0243】
ステップ32−7. THF(100mL)中の32−6(14.5g、21.5mmol、1.0当量)の溶液に、THF中の1M TBAF(32.3mL、32.3mmol、1.5当量)を添加して、混合物を1時間撹拌した。ブラインを添加して、水相をEtOAcで抽出した。ひとまとめにした有機相をMgSO4で乾燥させて、ろ過してろ液を減圧濃縮した。残渣をフラッシュクロマトグラフィーにより精製して(100%EtOAc)、Ddz−T32(Boc)を得た(収率:88%)。
TLC (100% EtOAc):Rf= 0.24;検出:UVおよびCMA。
1H NMR (CDCl3): δ 7.74 (1H, dd), 7.35 (1H, d), 6.72 (1H, d), 6.53-6.49 (2H, m), 3.61-3.29 (1H, m), 5.06 (1H, t), 4.25-4.01 (2H, m), 3.91-3.89 (2H, m), 3.73 (3H, s), 2.99 (2H, dd), 2.63 (2H, t), 1.71 (8H, ブロード), 1.53 (9H, s);
13C NMR (CDCl3, ppm): δ 163.8, 162.2, 161.0, 159.7, 155.9, 149.4, 130.0, 129.1, 128.0, 126.8, 110.8, 98.1, 80.9, 79.3, 69.7, 61.3, 55.5, 39.1, 29.3, 28.5, 26.7。
【0244】
F.テザーT52およびテザーT53の合成の標準的手順
【化24】
【0245】
ステップT52−1. DMF(DriSolv(登録商標))中の3−ヨードフェノール(52−0、1.0当量)の溶液に、水素化ナトリウム(鉱物油中の60%、0.1当量)を少しずつ添加する(警告!水素ガスが発生すると思われる)。反応物を窒素下、100℃で1時間加熱し、その後、炭酸エチレンを添加して、反応混合物を100℃で一晩加熱する。反応をTLCでモニタリングする(条件:25/75 EtOAc/ヘキサン)。反応混合物を放冷し、その後、溶媒を減圧蒸発させる。残渣の油状物をEt2O(1.5L)で希釈し、その後、1N水酸化ナトリウム(3回)およびブライン(2回)で引き続き洗浄して、MgSO4で乾燥させて、ろ過してろ液を減圧蒸発させる。粗生成物を真空蒸留するか、またはフラッシュクロマトグラフィーにより精製して、52−1を得る。
【0246】
ステップT52−2. CH3CN中の52−1(1.0当量)およびBoc−アリルアミン(1.3当量)の溶液に、アルゴンを20〜30分間バブリングする。新たに蒸留されたEt3N(CaH2と共に4時間還流し、その後蒸留したもの、3.6当量)を添加して、アルゴンを10〜15分間バブリングする。その後、トリス(o−トリル)ホスフィン(0.03当量)およびPd(OAc)2(0.03当量)を添加する。反応物をTLCでモニタリングしながら、還流雰囲気で2時間撹拌する。反応が完了しなければ、より長時間利用することができる。揮発物を減圧除去して、残渣をフラッシュカラムクロマトグラフィーで精製して、Boc−T52を得る。
【0247】
ステップT52−3. Boc−T52(1.0当量)を10%Pd/C(15重量%)および95%EtOHを添加する。混合物を水素ガスの圧力下、水素添加装置(例えば、Parr)内に24時間配置する。LC−MSまたは1H NMRにより、モニタリングを実施することができる。混合物をCelite(登録商標)パッドでろ過し、その後減圧濃縮して、Boc−T53を得て、それをフラッシュクロマトグラフィーにより精製することができる。
【0248】
G.テザーT201の標準的手順
T201の反応スキームを、図5に表わす。
【0249】
ステップ201−1. −30℃のトルエン(320mL)中のt−ブチルアミン(40mL、378mmol、3.0当量)の溶液に、Br2(7.1mL、139mmol、1.1当量)を緩やかに添加した(10分間)。混合物を−78℃に冷却して、2−ヒドロキシベンゾニトリル(201−0、15.0g、126mmol、1.0当量)をCH2Cl2(80mL)中で添加した。2−ヒドロキシベンゾニトリルは、DCMにあまり溶解されず、懸濁物としての反応物にピペットで添加した。不均質混合物を緩やかに室温で冷却して、一晩撹拌した。ブラインを添加し、相を分離して、水相を酢酸エチルで抽出した。有機相をひとまとめにして、10%NaOHで抽出した(2回)。水相を6N HClで酸性化して、CH2Cl2で抽出した。有機相をMgSO4で乾燥させて、減圧濃縮し、201−1を得た(収率:90%)。
TLC (60% EtOAc, 40%ヘキサン): Rf= 0.32;検出:UVおよびKMnO4。
【0250】
ステップ201−2. TBDMS−ブロモエタノール(32−A)でのアルキル化による201−1から201−2への変換を、本質的にはステップ32−2の32−2の合成で記載された通り実施した。
【0251】
ステップ201−3. LHMDS 3当量が変換に用いられ、反応時間が2〜3日であったこと以外は、本質的にはステップ32−3の201−3の合成に記載された通り、201−2からのアミジン201−3の形成を実施した。
【0252】
ステップ201−4. Bocによる201−3のアミジン基の保護を、本質的にはステップ32−4の32−4の合成に記載された通り実行した。
【0253】
ステップ201−5. 201−5を得るための、201−4とDdz−プロパルギルアミン(32−B)との薗頭カップリング反応を、本質的にはステップ32−5の32−5の合成に記載された通り実施した。しかしカップリング反応は完了せず、出発原料を2回目に同条件下で処理して、生成物を得た。
【0254】
ステップ201−6. 201−5の水素化および脱保護を、本質的にはステップ32−6のDdz−T32(Boc)の合成に記載された通り実施して、Ddz−T201(Boc)を得た。
1H NMR (CDCl3): δ 7.87 (1H, d), 7.28−7.25 (1H, m), 7.10 (1H, t), 6.51−6.46 (2H, m), 6.31 (1H, t), 5.30−5.20 (1H, m), 3.90−3.85 (2H, m), 3.85−3.80 (2H, m), 3.74 (6H, s), 3.15−3.05 (2H, m), 2.67 (2H, t), 1.85−1.71 (2H, m), 1.71 (6H, s), 1.53 (9H, s);
13C NMR (CDCl3):δ 160.8, 155.6, 155.5, 135.6, 133.9, 129.9, 127.9, 125.0, 103.3, 98.2, 80.8, 79.8, 61.9, 60.6, 55.5, 40.2, 31.3, 29.5, 28.5, 27.1, 14.4。
【0255】
H.テザーT202およびT203の標準的手順
アミジン部分をテザーに組み入れた後で、テザーT32およびT201に関して既に記載された通り分子の残り部分に付加するか、またはマスキングされたアミジン基としてニトリルを用い、その後、ニトリルをアミジンに変換することにより、これらのテザーを調製することができる。前者のアプローチでは、2−ブロモ−5−シアノフェノールから出発すればT202に到達することができ、2−ブロモ−3−シアノフェノールから出発すればT203に到達することができる。
【化25】
【0256】
後者では、化合物451に関して記載された変換を、以下に例示する適切な大環状ニトリル上で用いることができる。
【化26】
【0257】
実施例9
大環状化合物の合成
A.代表的な本発明の大環状化合物を合成するための標準的手順
化合物451の反応スキームを、図1に表わす。
【0258】
ステップ451−1. H−Phe(4CN)−OBnの合成 H−Phe(4CN)−OH(2.85g、15mmol、1.0当量)のトルエン(75mL)溶液に、p−TSA(3.42g、18mmol、1.2当量)、BnOH(7.8mL、75mmol、5.0当量)を添加した。H2Oをディーンスタークトラップで除去しながら、混合物を4時間加熱還流した。混合物を室温まで放冷し、Et2Oで希釈して0℃(アイスバス)で45分間撹拌した。得られた白色沈殿物をろ過して、低温Et2Oですすいだ。白色固体を1M Na2CO3溶液に溶解し、その後、室温で30分間撹拌した。得られた水相をEtOAcで洗浄した(4回)。ひとまとめにした有機相をブラインで洗浄し、Na2SO4で乾燥させて、ろ過して減圧蒸発させ、薄橙色油状物を得た(3.10g、収率70%)。
1H NMR: δ 1.60 (br s, 2H), 3.02 (dq, 2H), 3.77 (t, 1H), 5.13 (q, 2H), 7.21−7.52 (m, 9H)。
【0259】
ステップ451−2 ジペプチドの形成. THF−DCM混合物(1:1、25mL)中のH−Phe(4CN)−OBn(2.9g、10.27mmol、1.0当量)の溶液に、Boc−NMeAla(2.15g、10.6mmol、1.03当量)、6−Cl−HOBt(1.74g、10.3mmol、1.1当量)を0℃(アイスバス)で添加した。DIPEA(8,94mL、51.35mmol、5.0当量)およびその後、EDCI(2.17g、11.3mmol、1.1当量)を添加して、混合物を室温で一晩撹拌した。揮発物を減圧蒸発させて、得られた粗油状物をEtOAcに溶解した。その溶液を1Mクエン酸緩衝液(pH=3.5、2回)、H2O、飽和NaHCO3およびブラインで引き続き洗浄し、その後、Na2SO4で乾燥させ、ろ過して減圧蒸発させた。ひとまとめにした有機相をH2O、飽和NH4Cl、ブラインで洗浄して、Na2SO4で乾燥させて、ろ過して真空蒸発させた。粗生成物をフラッシュクロマトグラフィーにより精製して(勾配、EtOAc/ヘキサンで40%その後、50%)、保護されたジペプチド4.50g(93%)を得た。
TLC(50% EtOAc/ヘキサン):Rf: = 0.15, 検出: UV,ニンヒドリン。
【0260】
保護されたジペプチド(4.46g、9.6mmol、1.0当量)を、MeOH中の3.3N HCl溶液(30mL、96mmol、10当量)に溶解した。混合物を室温で1時間撹拌した。その後、揮発物を減圧蒸発させ、得られた粗油状物を真空乾燥させて(油圧ポンプ)、所望の化合物を非晶質固体として得た(3.50g、100%)。
【0261】
ステップ451−3 Boc−T69−OTsの合成. Boc−T69(4.94g、14.7mmol、1.05当量)のDCM(36mL)溶液に、DMAP(342mg、2.8mmol、0.2当量)およびEt3N(9.8mL、70mmol、5.0当量)を添加して、混合物を0℃(アイスバス)で15分間撹拌した。その後、TsCl(2.67g、14mmol、1.0当量)のDCM溶液(24mL)を0℃で少しずつ添加した。混合物を0℃で45分間、その後、室温で一晩撹拌した。NH4Clの飽和溶液を添加して、二相に分離させて、水相をDCMで洗浄した(3回)。ひとまとめにした有機相を、1M HCl(2回)およびブラインで洗浄して、Na2SO4で乾燥させて、ろ過して減圧蒸発させた。粗生成物を、更に精製せずに次のステップで用いた(6.90g、100%)。
1H NMR (CDCl3): δ1.34 (s, 9H), 1.60 (m, 2H), 2.36 (s, 3H), 2.44 (m, 2H), 2.99 (m, 3H), 4.04 (m, 2H), 4.30 (m, 2H), 4.59 (br s, 1 H), 6.35 (m, 1H), 6.50 (m, 1H), 6.94 (m, 1H), 7.26 (d、 J=8.4 Hz, 2H), 7.72 (d、 J=8.4 Hz, 2H)。
【0262】
ステップ451−4 Boc−T69−Cpg−OMeの合成. EtCN/DMF混合物(3:1、20mL)中のBoc−T69−OTs(6.9g、14.7mmol、1当量)の溶液に、H−Cpg−OMe・HCl(3.65g,22.1mmol,1.5当量)、KI(オーブンで一晩乾燥させたもの、6.09g、36.7mmol、2.5当量)およびDIPEA(7.7mL、44.1mmol、3.0当量)を室温で添加した。LC−MSによりモニタリングしながら、反応混合物を108℃で30時間撹拌した。反応物を室温まで放冷し、その後、H2Oでクエンチした。混合物をEtOAcで希釈して、水相をEtOAcで洗浄した(3回)。ひとまとめにした有機相を、1Mクエン酸緩衝液(pH=3.5)、H2O、飽和NaHCO3およびブラインで引き続き洗浄して、Na2SO4で乾燥させて、ろ過して減圧蒸発させた。粗生成物を、更に精製せずに次にステップで用いた(5.98g、96%)。
LC−MS:tR= 6.24分(A4b),[M+H]+ 425
【0263】
ステップ451−5 Boc−T69−Cpg−OHの合成.DCM/MeOH混合物(9:1、90mL)中のBoc−T69−Cpg−OMe(5.98g、14.0mmol、1.0当量)の溶液に、MeOH中の2M NaOH溶液(14.1mL、28.2mmol、2.0当量)を添加した。混合物を室温で48〜72時間撹拌した。揮発物を減圧蒸発させて、残渣を水で希釈した。水相をEt2Oで洗浄し、その後、pH=1〜2に酸性化した。酸性相をEtOAcで洗浄した(3回)。ひとまとめにした有機相を飽和NH4Clおよびブラインで洗浄し、Na2SO4で乾燥させて、ろ過して減圧蒸発させた。粗固体をヘキサン/DCM混合物(9:1)と共に磨砕して、白色固体を得た(3.76g、65%)。
LC−MS: tR = 6.12分(A4b), [M+H]+ 411
【0264】
ステップ451−6 フラグメントのカップリング. DCM/THF混合物(1:1、90mL)中のBoc−T69−Cpg−OH(3.60g、9.2mmol、1.0当量)の溶液に、H−NMeAla−Phe(4CN)−OBn・HCl(3.36g、9,20mmol、1.05当量)を添加して、混合物を0℃(アイスバス)で15分間撹拌した。DIPEA(9.23mL、53mmol、6.0当量)と、その後、HATU(3.50g、9.20mmol、1.05当量)を添加して、LC−MSでモニタリングしながら、混合物を室温で48〜72時間。混合物をEtOAcで希釈して、1Mクエン酸緩衝液(pH=3.5)、H2O、飽和NaHCO3およびブラインで引き続き洗浄した。有機相をNa2SO4で乾燥させてろ過し、減圧下で蒸発させた。粗生成物をフラッシュクロマトグラフィーにより精製して(勾配、50%EtOAc/ヘキサン、その後100%EtOAc)、カップリングされた生成物を得た(4.35g、62%)。
TLC (50% EtOAc/ヘキサン): Rf: = 0.10,検出: UV,ニンヒドリン
LC−MS:tR= 7.95分(A4b), [M+H]+ 758
【0265】
ステップ451−7 脱保護. トリペプチド−テザー(4.0g、5.28mmol、1.0当量)のDCM(53mL)溶液に、Pd(OAc)2(60mg、0.264mmol、0.05当量)、Et3N(95μL、0.68mmol、0.13当量)を添加した。混合物をAr/真空のサイクルで30分間脱気し、アルゴン下、室温で一晩撹拌した。揮発物を減圧下で蒸発させて、暗色の粗油状物をFlorisil(登録商標)の短いパッドにより、最初EtOAcで、その後MeOHで溶出してろ過し、ひとまとめにしたろ液を減圧濃縮した。粗生成物を薄黄色油状物として得た(3.11g、90%)。
LC−MS:tR= 6.64分(A4b), [M+H]+ 668
【0266】
DCM/TFA/TES混合物(64;33;3、30mL)中の粗油状物(3.1g、4.57mmol、1.0当量)の溶液を、室温で45分間撹拌した。揮発物を減圧下で蒸発させた。残渣をDCM/トルエン混合物(1:1、15mL)に溶解して、減圧濃縮した。得られた油状物を更に精製せずに次のステップで用いた。
【0267】
ステップ451−8 大環状化. 先の粗油状物(3.1g、4.57mmol、1.0当量)を含有するTHF(457mL、c=0.01M)溶液に、DIPEA(5.60mL、32.0mmol、7.0当量)および最後にDEPBT(1.5g、5.03mmol、1.1当量)を添加した。混合物を室温で一晩撹拌した。揮発物を減圧蒸発させて、得られた粗油状物をEtOAc/NaHCO3(飽和)(1:1)の混合物に溶解した。水相をEtOAcで洗浄した(3回)。ひとまとめにした有機相をブラインで洗浄して、Na2SO4で乾燥させ、ろ過して減圧蒸発させた。粗生成物をフラッシュクロマトグラフィーにより精製して(勾配、0.5%/3%/96.5% AcOH/MeOH/EtOAc,100%EtOAc、その後、0.5%/3%/96.5% NH4OH/MeOH/EtOAc))、環状生成物を得た(2.0g、80%)。TLC(3%EtOAc/MeOH):Rf:=0.75,検出:UV,ニンヒドリン
LC−MS:tR= 5.59分(A4b), [M+H]+ 550
【0268】
ステップ451−9 Boc保護. THF/H2O混合物(1:1、40mL)中の大環状化合物(2.0g、3.64mmol、1.0当量)の溶液に、Na2CO3(1.93g、18.2mmol、5.0当量)およびBoc2O(5.01mL、21.84mmol、6.0当量)を添加して、混合物を室温で48〜72時間撹拌した。混合物をNH4Cl(飽和)でクエンチし、その後、水相をEtOAcで洗浄した(3回)。ひとまとめにした有機相をブラインで洗浄し、Na2SO4で乾燥させてろ過し、減圧蒸発させた。Boc−保護大環状化合物を得られた状態で次のステップで用いた。
LC−MS:tR= 9.14分(A4b), [M+H]+ 650
【0269】
ステップ451−10;N−ヒドロキシアミジンの形成. 絶対EtOH(35mL)中の大環状化合物(2.2g、3.35mmol、1.0当量)の溶液に、NH2OH・HCl(0.750g、10.74mmol、3.2当量)およびDIPEA(2.04mL、11.72mmol、3.5当量)を添加して、得られた混合物を一晩、加熱還流した。混合物を室温まで放冷し、その後、揮発物を減圧蒸発させた。得られた黄色透明油状物を次のステップで直接用いた。
LC−MS:tR= 7.20分(A4b), [M+H]+ 683
【0270】
ステップ451−11 N−アセトキシアミジンの形成. 10分間撹拌したAcOH(35mL)中の大環状化合物(2.2g、3.35mmol、1.0当量)の溶液に、Ac2O(2mL、16.75mmol、5.0当量)を添加した。得られた混合物を室温で2.5時間撹拌した。揮発物を減圧蒸発させた。得られた粗油状物をフラッシュクロマトグラフィーにより精製して(10%MeOH/EtOAc)、所望の生成物を得た(1.80g、3ステップ全体で74%)。
LC−MS:tR= 13.12分(A4b), [M+H]+ 725;[M+2H−Boc]+625
【0271】
ステップ451−12 アミジンの形成. AcOH(35mL)中の先のステップからの大環状化合物(1.40g、1.93mmol、1.0当量)の溶液に、亜鉛末(1.26g、19.3mmol、10.0当量)を添加した。得られた混合物を55℃で一晩撹拌した。混合物を室温まで放冷し、その後、混合物を綿の短いパッドでろ過した。綿をAcOHで、そして最後にEtOAcで溶出した。揮発物を減圧蒸発させた。得られた黄色透明油状物は、次のステップで直接用いることができた。
【0272】
ステップ451−13 Bocの切断. 大環状化合物(1.40g、1.93mmol、1.0当量)をDCM−TFA−TES混合物(64%−33%−3%、20mL)に溶解して、室温で1.5時間撹拌した。混合物を真空濃縮した。粗油状物をTHFに溶解し、その後、溶媒を減圧蒸発させた。この手順を、溶媒としてトルエンを用い、その後、EtOAcを用いて繰り返した。得られた粗油状物をフラッシュクロマトグラフィーにより精製した(20%MeOH/DCMおよび0.5%TFA、その後、30%MeOH/DCMおよび0.5%TFA)。
TLC (30% MeOH/DCMおよび0.5% TFA):Rf: 0.61,検出:UV,ニンヒドリン
【0273】
大環状化合物・TFA塩をEtOAcに溶解し、その後、水性1M Na2CO3溶液を添加した。水相をEtOAcで抽出した(3回)。ひとまとめにした有機相をブラインで洗浄し、Na2SO4で乾燥させて、ろ過して減圧蒸発させた。所望の大環状化合物を、白色固体として得た(0.90g、82%)。1種のジアステレオマーのみを、1H NMRにより観察した。不純物がLC−MSに見出されれば、THFまたはCH3CNでの磨砕を利用して、純度を改善することができた。
LC−MS: tR = 4.49分(A4b), [M+H]+ 567
【0274】
脱保護は、ジオキサン中の4M HClで処理することにより実現することもできた。その場合の粗大環状化合物を、フラッシュクロマトグラフィーにより精製した(30%MeOH/DCMおよび0.5%TFA)。120mgの規模で、2段階全体で66%の収率が得られた。
【0275】
ステップ451−14 HCl塩の形成:化合物をアセトニトリルに溶解し、その後、0.1N HCl(4当量)を添加して、溶液を一晩凍結乾燥した。得られた固体をTHFと共に摩砕した。
LC−MS:tR= 6.14分(B4), [M+H]+ 567
【0276】
あるいはアミジノ基は、図示された通り大環状化合物の第二級アミン上のBoc−保護を利用せずに、合成することができた。
【化27】
【0277】
追加の代替的なアプローチは、以下の条件を利用して対応するシアノ前駆体から直接アミジノ含有大環状化合物を合成することである(Garigipati, R. S. Tetrahedron Lett. 1990, 31, 1969.)。
【化28】
【0278】
B.複数の代表的な本発明の化合物を同時合成するための標準的手順
標準的反応スキームを、図2および3に表わす。
【0279】
以下の手順は、複数の個々の化合物で同時に実施する多重反応の追跡を容易にする、無線周波数タグを含む特定の技術を利用する。しかしこれは必要ではなく、固相合成を個々の反応容器内で同時に実施することもできる。
【0280】
ステップB−1 AA3負荷. 塩化2−クロロトリチル樹脂をMiniKan(または他の適切な分離可能な反応容器)に加えて、DCMで15分間洗浄した。DCMを除去して、DIPEA(4当量)およびFmoc−NH−AA3(2当量)の溶液を添加した(別個のAA3それぞれに、MiniKanを含む別個の容器を用いた)。反応混合物を、オービタルシェーカで室温で一晩かき混ぜた。MiniKanを以下のサイクルで2回洗浄した:DCM、iPrOH、DCM、その後、N2気流の下で乾燥させた。
【0281】
MiniKan(QC用)1個、またはMiniKan1個から取り出した樹脂の一部を、HFIP:DCM(1:4、5mL)混合物中で反応させてオービタルシェーカで室温で少なくとも30分間かき混ぜた。樹脂をDCMで洗浄し、揮発物を減圧蒸発させた。そのようにして得られた粗油状物を、その後、負荷効率の推定のために、定量的QC分析に供した。
【0282】
ステップB−2 Fmoc−脱保護. MiniKanをNMP中の20%ピペリジン溶液(3.5mL/MiniKan)で処理し、その後、オービタルシェーカで30分間かき混ぜた。その後、この処理を繰り返した。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、DCM、IPA、DCM(3回)、その後、N2気流の下で乾燥させた。
【0283】
ステップB−3 AA2カップリング. Fmoc−NR−AA2−OH(2.5当量)を、NMPに溶解し、その後、DIPEA(5当量)、続いてHATU(2.5当量)を添加した。混合物を室温で10分間撹拌し、その後、適切な組み合わせのMiniKanに移し(AA2により別個の容器に分離し)、オービタルシェーカで室温で一晩かき混ぜた。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、DCM、IPA、DCM(3回)、その後、N2気流の下で乾燥させた。
【0284】
ステップB−4 Fmoc−脱保護. MiniKanをNMP中の20%ピペリジン溶液(3.5mL/MiniKan)で処理し、その後、オービタルシェーカで30分間かき混ぜた。その後、この処理を繰り返した。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、DCM、IPA、DCM(3回)、その後、N2気流の下で乾燥させた。
【0285】
ステップB−5 AA1カップリング. Fmoc−NH−AA1−OH(2.5当量)を、NMPに溶解し、その後、DIPEA(5当量)、続いてHATU(2.5当量)を添加した。混合物を室温で10分間撹拌し、その後、適切な組み合わせのMiniKanに移して(AA1により別個の容器に分離した)、オービタルシェーカで室温で一晩かき混ぜた。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、DCM、IPA、DCM(3回)、その後、N2気流の下で乾燥させた。
【0286】
ステップB−6A テザーの酸化. テザーのDMSO溶液に、IBX(1.5当量)を添加した。不均質混合物を室温で5分間撹拌し、その後、H2Oを添加して、室温で一晩撹拌し続けた。混合物を水でクエンチし(白色沈殿物が形成された)、その溶液を室温で20分間撹拌した。固体をろ過により除去し、EtOAcで洗浄して、得られた溶液を水性NaHCO3およびブラインで洗浄し、MgSO4で乾燥させ、その後、減圧濃縮した。粗アルデヒドを真空乾燥させて、構造を1H NMRにより確認し、その後、その状態で次のステップで用いた。
【0287】
ステップB−6B 還元アミノ化. MiniKanをNMP中の20%ピペリジン溶液(3.5mL/MiniKan)で処理し、その後、オービタルシェーカで30分間かき混ぜた。その後、この処理を繰り返した。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、DCM、IPA、DCM(3回)、その後、N2気流の下で乾燥させた。ステップ6Aからの粗テザーアルデヒドをTMOF−MeOHの混合物(1:3)に溶解した。得られた溶液を、適切なMiniKanを含む容器に移し(テザーにより分離)、オービタルシェーカで室温で10分間かき混ぜた。BAP試薬(2当量)を添加して、室温で一晩かき混ぜ続けた。[気体が発生することに注意し、容器をしっかりと密閉して(または開孔して)溶媒の損失を回避しなければならない]。MiniKanを以下の順序で洗浄した:DCM(2回)、THF−DCM/MeOH(3:1)、THF/MeOH(3:1)、DCM(3回)、その後、N2気流の下で乾燥させた。
【0288】
ステップB−7 N−ヒドロキシアミジンの形成 最初に、NMP中のNH2OHの1M溶液を、以下の通り調製した:NH2OH・HCl 3.51gをDIPEA(9.2mL)に溶解し、その後、NMPを用いて容量を50mLに調整した。残留する塩が完全に解離するまで、不均質混合物を室温で撹拌した。MiniKanをNMP(4mL/MiniKan)で処理して、溶液をN2/真空サイクルで脱気し(30分間)、その後、NH2OHの1M NMP溶液を添加して(2mL/MiniKan)、混合物を50℃(オイルバス)で24時間撹拌した。溶液を室温まで放冷した。MiniKanを以下の順序で洗浄した:NMP(2回)、IPA、NMP、IPA、THF−DCM/MeOH(3:1)、DCM(3回)、その後、N2気流の下で乾燥させた。
【0289】
ステップB−8 樹脂からの開裂 樹脂を個々のMiniKanから除去して、別個の20mL反応器に導入した。HFIP/DCM(1;4)の溶液を添加して、得られた赤色溶液をオービタルシェーカで1時間かき混ぜた。樹脂をろ過により除去して、DCMで洗浄し、揮発物を真空蒸発させた(複数の試料のためにSpeedVac遠心エバポレータを用いた)。
【0290】
ステップB−9 N−アセトキシアミジンの形成 ステップB−9〜B−11に表わされた反応速度が、250μmolトリペプチド(理論的収量)に基づいていて、他の量の場合には比例的に調整しうることに留意されたい。ステップ8からの個々の油状物をAcOH(2.5mL)に溶解し、溶液を室温で10分間撹拌し、その後、Ac2Oを添加して(0.15mL、1.25mmol、5当量)、45分間撹拌し続けた。揮発物を真空蒸発させた(複数の試料のためにSpeedVac遠心エバポレータを用いた)。
【0291】
ステップB−10 テザーの脱保護および大環状化. ステップB−9からの個々の残渣を、TES−TFA−DCM混合物に溶解し(3:33:64、5mL)、溶液を室温で45分間撹拌した。揮発物を真空蒸発させ(複数の試料のためにSpeedVac遠心エバポレータを用いた)、その後、残渣をトルエンに溶解して、再度、真空濃縮した(SpeedVacによる)。
【0292】
Ddz保護されたテザーでは、TFA−TES−DCM(2:3:95)の混合物を脱保護ステップに用いた。Boc側鎖の脱保護が起こる可能性があるため、Ddzの脱保護が1時間を超えないことが重要である。
【0293】
個々の油状物をTHF(25mL)に溶解し、その後、DIPEA(300μL、1.75mmol、7当量)、続いてDEPBT(0.150g、0.50mmol、2当量)を添加した。黄色溶液をオービタルシェーカで室温で一晩かき混ぜた。Si−Trisアミン樹脂を導入し(3.5g/反応)、得られた混合物をオービタルシェーカで室温で2時間かき混ぜた。樹脂をろ過により除去してTHFで洗浄し、揮発物を真空蒸発させた(複数の試料のためにSpeedVac遠心エバポレータを用いた)。
【0294】
ステップB−11 アミジンの形成. ステップ10からの油状物をAcOH(3mL)に溶解し、その後、亜鉛末(0.163g、2.5mmol、10当量)を添加して、溶液をオービタルシェーカで室温で一晩かき混ぜた。綿の短いパッドを用いて過剰の亜鉛末を除去し、その後、AcOHで溶出した。揮発物を真空蒸発させ(複数の試料のためにSpeedVac遠心エバポレータを用い)、その後、残渣をFraction Lynxでの精製に供して、所望の生成物を得た。
【0295】
所望の大環状化合物がアミジノ基を有さない場合には、ステップB−9およびB−11を省略した。他の特定の順序では、AA3位のBoc側鎖脱保護を、TFA−TES−DCM系を用いた標準条件下で実施した。加えてAA1位のTrt側鎖脱保護は、TFA−TES(95:5)を用いた標準条件下で実施した。
【0296】
前述は本発明の例示であり、その限定と解釈すべきではない。本発明は以下の特許請求の範囲と本明細書に包含される特許請求の範囲の均等物により定義される。
【特許請求の範囲】
【請求項1】
式(I):
【化1】
(式中、
R1は、−H、−CH3、−CH2CH3、−(CH2)2CH3および−CH(CH3)2からなる群より選択され、
R2は、−H、−CH3および−CH2CH3からなる群より選択され、
R3は、任意で存在し、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より選択され、
mは、1、2、3、4または5であり、
X1は、アミジノ、ウレイドおよびグアニジノからなる群より選択され、
Wは、CR4aR4bからなる群より選択され、ここでR4aおよびR4bは、水素、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、
Z1は、CR5aR5bからなる群より選択され、ここでR5aおよびR5bは、水素、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、
Tは、
【化2】
からなる群より選択され、
ここでM1は、Oおよび(CH2)qからなる群より選択され、ここでqは、1、2、3、4または5であり、M2は、O、S、NR6およびCR7aR7bからなる群より選択され、ここでR6は、水素、アルキル、ホルミル、アシル、カルボキシアルキル、カルボキシアリール、アミド、スルホニルおよびスルホンアミドからなる群より選択され、R7aおよびR7bは、水素、ヒドロキシル、アルコキシ、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、p1およびp2は、独立して0、1、2または3であり、そしてp3、p4およびp5は、独立して0、1または2であり、
(W)は、Wの付加された炭素原子への結合部位を示し、
(Z)は、Z1の付加された炭素原子への結合部位を示す)
で示される化合物または薬学的に許容し得るその塩。
【請求項2】
構造
【化3】
を有する請求項1記載の化合物。
【請求項3】
(a)請求項1記載の式(I)で示される化合物と、
(b)薬学的に許容しうる担体、賦形剤または希釈剤と
を含む医薬組成物。
【請求項4】
(a)請求項2記載の化合物と、
(b)薬学的に許容しうる担体、賦形剤または希釈剤と
を含む医薬組成物。
【請求項5】
式(II):
【化4】
(式中、
R11は、−H、−CH3、−CH2CH3、−(CH2)2CH3および−CH(CH3)2からなる群より選択され、
R12は、−H、−CH3および−CH2CH3からなる群より選択され、
R13は、−(CH2)r1NR18aR18b、−(CH2)r2CONR19aR19b、
【化5】
からなる群より選択され、
ここでr1は、1、2、3、4または5であり、r2は、1、2または3であり、R18a、R19aおよびR19bは、水素およびC1〜C4アルキルからなる群より独立して選択され、R18bは、水素、C1〜C4アルキル、アシル、アミド、アミジノ、スルホンアミドからなる群より選択され、A1、A4、A7、A9、A12、A14、A17、A19、A23、A35、A37およびA39は、それぞれ任意で存在し、ハロゲン、トリフルオロメチル、アミジノ、ウレイド、グアニジノ、ヒドロキシル、アルコキシおよびC1〜C4アルキルからなる群より独立して選択され、A2、A3、A5、A6、A8、A10、A11、A13、A15、A16、A18、A20、A21、A24、A25、A36、A38およびA40は、それぞれ任意で存在し、ハロゲン、トリフルオロメチル、ヒドロキシル、アルコキシおよびC1〜C4アルキルからなる群より独立して選択され、A22、A26、A27、A29、A31およびA33は、それぞれ任意で存在し、トリフルオロメチル、アミジノ、ウレイド、グアニジノおよびC1〜C4アルキルからなる群より独立して選択され、A28、A30、A32およびA34は、それぞれ任意で存在し、トリフルオロメチルおよびC1〜C4アルキルからなる群より独立して選択され、そしてB1、B2、B3、B4、B5およびB7は、独立してNR20、SまたはOであり、ここでR20は、水素、アルキル、ホルミル、アシル、カルボキシアルキル、カルボキシアリール、アミド、スルホニルおよびスルホンアミドからなる群より選択され、B6およびB8は、独立してNまたはCHであり、
R14は、アミノ、ヒドロキシル、アルコキシ、カルボキシ、ウレイド、アミジノ、またはグアニジンで場合により置換されたC1〜C4アルキルと、アルキル、ヒドロキシルまたはアルコキシで場合により置換されたC3〜C7シクロアルキルとからなる群より選択され、
R15およびR16は、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より独立して選択され、
R17は、水素およびC1〜C4アルキルからなる群より選択され、
nは、1、2、3、4または5であり、
Z2は、CHR21aCHR22a、CR21b=CR22bおよびC≡Cからなる群より選択され、ここでR21aおよびR22aは、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より独立して選択されるか、またはR21aとR22aとが、結合する炭素と一緒になって、3員環を形成しており、そしてR21bおよびR22bは、水素およびC1〜C4アルキルからなる群より独立して選択され、
X2は、水素、ハロゲン、アミジノ、ウレイドおよびグアニジノからなる群より選択され、
X3は、水素、ヒドロキシル、アルコキシ、アミノ、ハロゲン、トリフルオロメチルおよびC1〜C4アルキルからなる群より選択され、
L2は、OおよびCR23aR23bからなる群より選択され、ここでR23bは、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より選択され、そしてR23bは、水素およびC1〜C4アルキルからなる群より選択され、
L3は、CX4およびNからなる群より選択され、ここでX4は、水素、ハロゲン、ヒドロキシル、アルコキシ、アミノ、ハロゲン、トリフルオロメチル、アミジノ、ウレイドおよびグアニジノからなる群より選択され、そして
L4は、CX5およびNからなる群より選択され、ここでX5は、水素、ハロゲン、トリフルオロメチル、ヒドロキシル、アルコキシ、アミノ、アミジノ、ウレイドおよびグアニジノからなる群より選択される)
で示される化合物または薬学的に許容し得るその塩。
【請求項6】
構造
【化6A】
【化6B】
【化6C】
を有する請求項5記載の化合物。
【請求項7】
(a)請求項5記載の式(II)で示される化合物と、
(b)薬学的に許容しうる担体、賦形剤または希釈剤と
を含む医薬組成物。
【請求項8】
(a)請求項6記載の化合物と、
(b)薬学的に許容しうる担体、賦形剤または希釈剤と
を含む医薬組成物。
【請求項1】
式(I):
【化1】
(式中、
R1は、−H、−CH3、−CH2CH3、−(CH2)2CH3および−CH(CH3)2からなる群より選択され、
R2は、−H、−CH3および−CH2CH3からなる群より選択され、
R3は、任意で存在し、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より選択され、
mは、1、2、3、4または5であり、
X1は、アミジノ、ウレイドおよびグアニジノからなる群より選択され、
Wは、CR4aR4bからなる群より選択され、ここでR4aおよびR4bは、水素、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、
Z1は、CR5aR5bからなる群より選択され、ここでR5aおよびR5bは、水素、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、
Tは、
【化2】
からなる群より選択され、
ここでM1は、Oおよび(CH2)qからなる群より選択され、ここでqは、1、2、3、4または5であり、M2は、O、S、NR6およびCR7aR7bからなる群より選択され、ここでR6は、水素、アルキル、ホルミル、アシル、カルボキシアルキル、カルボキシアリール、アミド、スルホニルおよびスルホンアミドからなる群より選択され、R7aおよびR7bは、水素、ヒドロキシル、アルコキシ、C1〜C4アルキルおよびトリフルオロメチルからなる群より独立して選択され、p1およびp2は、独立して0、1、2または3であり、そしてp3、p4およびp5は、独立して0、1または2であり、
(W)は、Wの付加された炭素原子への結合部位を示し、
(Z)は、Z1の付加された炭素原子への結合部位を示す)
で示される化合物または薬学的に許容し得るその塩。
【請求項2】
構造
【化3】
を有する請求項1記載の化合物。
【請求項3】
(a)請求項1記載の式(I)で示される化合物と、
(b)薬学的に許容しうる担体、賦形剤または希釈剤と
を含む医薬組成物。
【請求項4】
(a)請求項2記載の化合物と、
(b)薬学的に許容しうる担体、賦形剤または希釈剤と
を含む医薬組成物。
【請求項5】
式(II):
【化4】
(式中、
R11は、−H、−CH3、−CH2CH3、−(CH2)2CH3および−CH(CH3)2からなる群より選択され、
R12は、−H、−CH3および−CH2CH3からなる群より選択され、
R13は、−(CH2)r1NR18aR18b、−(CH2)r2CONR19aR19b、
【化5】
からなる群より選択され、
ここでr1は、1、2、3、4または5であり、r2は、1、2または3であり、R18a、R19aおよびR19bは、水素およびC1〜C4アルキルからなる群より独立して選択され、R18bは、水素、C1〜C4アルキル、アシル、アミド、アミジノ、スルホンアミドからなる群より選択され、A1、A4、A7、A9、A12、A14、A17、A19、A23、A35、A37およびA39は、それぞれ任意で存在し、ハロゲン、トリフルオロメチル、アミジノ、ウレイド、グアニジノ、ヒドロキシル、アルコキシおよびC1〜C4アルキルからなる群より独立して選択され、A2、A3、A5、A6、A8、A10、A11、A13、A15、A16、A18、A20、A21、A24、A25、A36、A38およびA40は、それぞれ任意で存在し、ハロゲン、トリフルオロメチル、ヒドロキシル、アルコキシおよびC1〜C4アルキルからなる群より独立して選択され、A22、A26、A27、A29、A31およびA33は、それぞれ任意で存在し、トリフルオロメチル、アミジノ、ウレイド、グアニジノおよびC1〜C4アルキルからなる群より独立して選択され、A28、A30、A32およびA34は、それぞれ任意で存在し、トリフルオロメチルおよびC1〜C4アルキルからなる群より独立して選択され、そしてB1、B2、B3、B4、B5およびB7は、独立してNR20、SまたはOであり、ここでR20は、水素、アルキル、ホルミル、アシル、カルボキシアルキル、カルボキシアリール、アミド、スルホニルおよびスルホンアミドからなる群より選択され、B6およびB8は、独立してNまたはCHであり、
R14は、アミノ、ヒドロキシル、アルコキシ、カルボキシ、ウレイド、アミジノ、またはグアニジンで場合により置換されたC1〜C4アルキルと、アルキル、ヒドロキシルまたはアルコキシで場合により置換されたC3〜C7シクロアルキルとからなる群より選択され、
R15およびR16は、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より独立して選択され、
R17は、水素およびC1〜C4アルキルからなる群より選択され、
nは、1、2、3、4または5であり、
Z2は、CHR21aCHR22a、CR21b=CR22bおよびC≡Cからなる群より選択され、ここでR21aおよびR22aは、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より独立して選択されるか、またはR21aとR22aとが、結合する炭素と一緒になって、3員環を形成しており、そしてR21bおよびR22bは、水素およびC1〜C4アルキルからなる群より独立して選択され、
X2は、水素、ハロゲン、アミジノ、ウレイドおよびグアニジノからなる群より選択され、
X3は、水素、ヒドロキシル、アルコキシ、アミノ、ハロゲン、トリフルオロメチルおよびC1〜C4アルキルからなる群より選択され、
L2は、OおよびCR23aR23bからなる群より選択され、ここでR23bは、水素、C1〜C4アルキル、ヒドロキシルおよびアルコキシからなる群より選択され、そしてR23bは、水素およびC1〜C4アルキルからなる群より選択され、
L3は、CX4およびNからなる群より選択され、ここでX4は、水素、ハロゲン、ヒドロキシル、アルコキシ、アミノ、ハロゲン、トリフルオロメチル、アミジノ、ウレイドおよびグアニジノからなる群より選択され、そして
L4は、CX5およびNからなる群より選択され、ここでX5は、水素、ハロゲン、トリフルオロメチル、ヒドロキシル、アルコキシ、アミノ、アミジノ、ウレイドおよびグアニジノからなる群より選択される)
で示される化合物または薬学的に許容し得るその塩。
【請求項6】
構造
【化6A】
【化6B】
【化6C】
を有する請求項5記載の化合物。
【請求項7】
(a)請求項5記載の式(II)で示される化合物と、
(b)薬学的に許容しうる担体、賦形剤または希釈剤と
を含む医薬組成物。
【請求項8】
(a)請求項6記載の化合物と、
(b)薬学的に許容しうる担体、賦形剤または希釈剤と
を含む医薬組成物。
【図1】

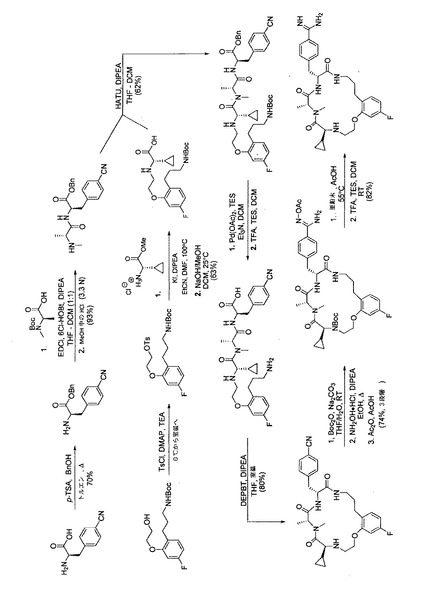
【図2】
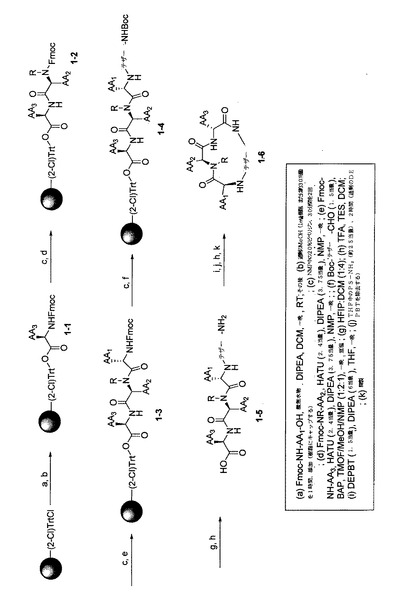
【図3】
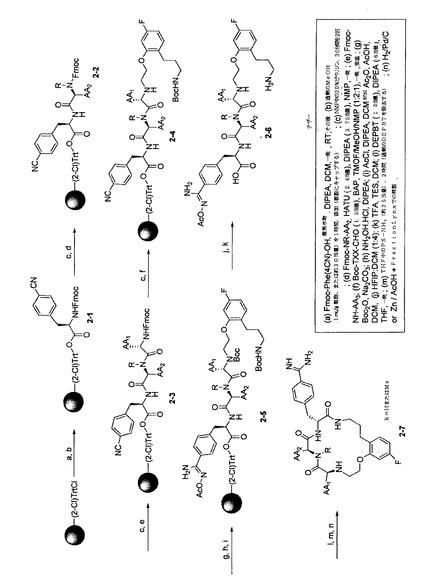
【図4】
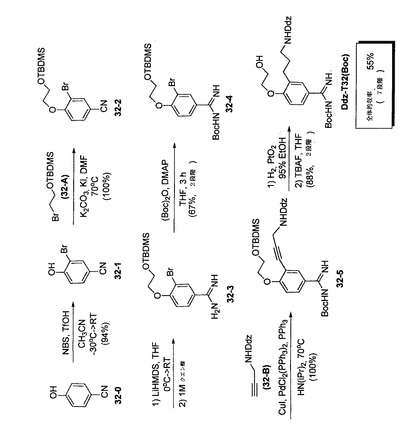
【図5】
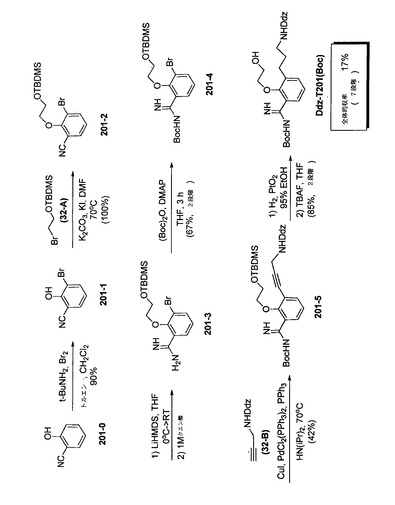
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2013−508409(P2013−508409A)
【公表日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願番号】特願2012−535415(P2012−535415)
【出願日】平成22年10月22日(2010.10.22)
【国際出願番号】PCT/US2010/053754
【国際公開番号】WO2011/050270
【国際公開日】平成23年4月28日(2011.4.28)
【出願人】(506419489)トランザイム・ファーマ,インコーポレイテッド (11)
【Fターム(参考)】
【公表日】平成25年3月7日(2013.3.7)
【国際特許分類】
【出願日】平成22年10月22日(2010.10.22)
【国際出願番号】PCT/US2010/053754
【国際公開番号】WO2011/050270
【国際公開日】平成23年4月28日(2011.4.28)
【出願人】(506419489)トランザイム・ファーマ,インコーポレイテッド (11)
【Fターム(参考)】
[ Back to top ]