
セルラーゼ・ヘミセルラーゼ転写因子
【課題】植物系バイオマスの完全酵素糖化に必要なセルラーゼ酵素群の大量生産を可能にする。
【解決手段】各種セルラーゼ遺伝子及びヘミセルラーゼ遺伝子を組み込んだ高発現ベクターを、A.aculeatus No.F-50などの宿主細胞に組み込み、特定のアミノ酸配列を有するセルラーゼ遺伝子又はヘミセルロース遺伝子の転写因子(タンパク質)を高発現させて、カルボキシメチルセルラーゼI、キシラナーゼIb、セロビオヒドロラーゼI、カルボキシメチラーゼIbの各遺伝子の発現を亢進させる。
【解決手段】各種セルラーゼ遺伝子及びヘミセルラーゼ遺伝子を組み込んだ高発現ベクターを、A.aculeatus No.F-50などの宿主細胞に組み込み、特定のアミノ酸配列を有するセルラーゼ遺伝子又はヘミセルロース遺伝子の転写因子(タンパク質)を高発現させて、カルボキシメチルセルラーゼI、キシラナーゼIb、セロビオヒドロラーゼI、カルボキシメチラーゼIbの各遺伝子の発現を亢進させる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、セルラーゼ・ヘミセルラーゼ遺伝子の発現を正に制御する転写因子に関する。
【背景技術】
【0002】
植物系バイオマスは地球上で最も多く生産される再生可能な資源である。その中でも稲ワラ等のセルロース系バイオマスは食糧資源とも競合しないため、これを原料としたバイオエタノール等のバイオ燃料やポリ乳酸等のバイオ化成品の代替製品を製造する技術革新が進めば、新たなバイオ産業創出につながると期待されている。
【0003】
植物系バイオマスの大部分を占めるセルロースは、グルコースがβ−1,4結合した直鎖状高分子であって、分子内あるいは分子間で広範囲にわたり数多くの水素結合を形成し、非常に強固な結晶構造をとる不溶性物質である。更に、その周りを囲むようにアラビノース、ガラクトース、グルクロニル基、フェルロイル基、アセチル基により部分的に修飾されたキシランを主要成分としたヘミセルロースに覆われている。従って、これらセルロースを中心とするセルロース系バイオマスから単糖を生成することは容易ではなく、石油の代替原料として、バイオ燃料やバイオ化成品を創り出すための障壁の一つとなっている。
【0004】
セルロース系バイオマスを糖化する方法として、塩酸や硫酸などの強酸を用いた酸糖化法がある。しかし、この方法は、単糖の過分解のために低収率である、酸の回収あるいは中和にコストがかる上環境負荷が大きいという難点がある。また、近年開発された注目の糖化法として超臨界水・亜臨界水処理による加水分解による方法がある。この方法は、リグニン除去などバイオマスの前処理の必要がなく、短時間で行える上、水と熱のみの反応でクリーンであるという利点を有する。しかし、多大な熱エネルギーを必要とすることや反応制御が難しいことなど課題が残っている。
【0005】
このような状況下、温和な条件で反応が行うことができる、最も環境負荷が小さいなどの理由により、酵素糖化法が注目されてきている。この方法は、生じた単糖の過分解を生じない利点もあるが、これまでのところ、反応時間が長いことや前処理を必要とすること、また、上述の結晶構造を形成するセルロースやキシランを中心とするヘミセルロースを加水分解するため、様々な基質特異性を有した酵素、例えば結晶性セルロースをグルコースにまで加水分解するには、セロビオヒドロラーゼ、エンドグルカナーゼ、β−グルコシダーゼ等の酵素群が、またヘミセルロースを単糖まで加水分解するにはエンドキシラナーゼ、β−キシロシダーゼ、α−アラビノフラノシダーゼ、β−ガラクトシダーゼ、α−グルクロニダーゼ、フェルロイルエステラーゼ、アセチルキシランエステラーゼ等、種々の基質特異性を有した酵素群が必要な点が短所としてあげられる。
【0006】
セルラーゼは、それぞれ構造が異なるセルロースを加水分解する酵素の総称で、セルラーゼはその基質特異性及び加水分解様式により大きく3種類に分類される。1つ目は結晶性セルロースに作用し、セルロース鎖の末端から二糖(セロビオース)単位で分解するセロビオヒドロラーゼ(cellobiohydrolase:CBH)、2つ目は結晶性セルロースを分解できないが、非結晶性セルロース鎖をランダムに切断するエンドグルカナーゼ(endo-glucanase:EG)、そして3つ目が可溶性のセロビオース並びにセロオリゴ糖に作用してグルコースを生成するβ−グルコシダーゼ(β−glucosidase:BGL)である。
【0007】
糸状菌Trichoderma reeseiが生産するセルラーゼは最強のセルラーゼと言われている。このセルラーゼは結晶性セルロースの分解には優れるものの、単糖を生成する力、すなわちβ‐グルコシダーゼの活性が弱く、糖化液中にはセロオリゴ糖(β−グルカンオリゴ糖)が多く残ってしまうという問題があった。また、セルロース系バイオマスにはセルロース単独で存在することは少なく、ヘミセルロースなどを伴うためセルロースを分解するにはまずこれらを除く必要がある。また、取り除いたヘミセルロースを構成するペントースも、例えば酵母にペントース資化能を付与すれば発酵によってエタノール生産の原料とすることができる。しかし、T. reeseiのセルラーゼはヘミセルラーゼ活性が弱いため、セルロース系バイオマス中に共存するセルロースとヘミセルロースの両方を効率的に分解し構成単糖を効率よく生産するには更にヘミセルラーゼを添加する必要がある。
【0008】
T. reeseiのセルラーゼのこうした弱点を補い、かつセルロース系バイオマスの分解において、T. reeseiのセルラーゼと強い相乗作用を示すセルラーゼ酵素群を生産する微生物として、Aspergillus aculeatus No.F-50株が知られている。この菌の生産するセルラーゼ酵素群は、非常に強い単糖生成能を有することに加え、ヘミセルラーゼ活性も強く、セルロースだけでなくヘミセルロースを含めたセルロース系バイオマスの糖化に非常に有効である。特筆すべき点は、A. aculeatus No.F-50株の生産するセルラーゼ酵素群には、セロビオースを分解してグルコースにするだけでなく、セロペンタオースやセロヘキサオースなど比較的長鎖のセロオリゴ糖に対しても強い分解活性を有するβ−グルコシダーゼ(BGLI)が存在するので、A. aculeatusのセルラーゼ酵素群を添加することによって、セルラーゼの単糖生成力が弱いというT. reeseiの弱点を補うことができる点である(非特許文献1)。
【0009】
ところで、セルラーゼ・ヘミセルラーゼは、セルロースやキシロース等の基質存在下で誘導され、グルコース存在下ではカタボライトリプレッションによりその生成が抑制される。これまでにセルラーゼ・ヘミセルラーゼ遺伝子発現制御機構に関する研究において、A. nigerからキシラナーゼ遺伝子発現制御因子であるXlnRが同定され、当該因子がエンドキシラナーゼだけでなくキシランの化学修飾部位や、セルラーゼ遺伝子の発現も制御していることが解明されている(非特許文献2、3)。また、T. reeseiでは、XlnR遺伝子のホモログ(homolog)であるXyr1遺伝子が全てのセルラーゼ・ヘミセルラーゼ遺伝子発現を制御していることが報告されている(非特許文献4、5)。一方、Aspergillus属糸状菌では、セルラーゼ・ヘミセルラーゼの遺伝子発現はXlnRに依存せず、セルロースやキシロース等の基質に応答して発現が制御される遺伝子であることが知られている(非特許文献6、7)。しかしながらXlnR以外の転写因子で、セルラーゼ・ヘミセルラーゼ遺伝子の発現を正に制御する転写因子について、遺伝子の詳細な配列やその機能については知られていなかった。
【0010】
また、特許文献1には、糸状菌において菌体外プロテアーゼ遺伝子の発現を正に制御する転写因子であるprtTの遺伝子破壊により、α−グルコシダーゼ、β−グルコシダーゼ、セルラーゼやペクチン分解酵素等の生産量を亢進することが開示されている。特許文献2には、Aspergillus属糸状菌において、α−アミラーゼプロモーターによって転写され、α−アミラーゼの転写を制御する転写因子AmyRが開示されている。特許文献3には、T. reeseiにおいて、セロビオヒドロラーゼI(cbhI)、セロビオヒドロラーゼII(cbhII)などのプロモーターによるこれら遺伝子の転写を負に制御する転写因子AceIが開示されている。しかしながら、これらの特許文献においても、セルラーゼ・ヘミセルラーゼ遺伝子の転写を正に制御する転写因子は報告されていない。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特表2003−526374号公報
【特許文献2】特表2000−514292号公報
【特許文献3】特表2004−526440号公報
【非特許文献】
【0012】
【非特許文献1】Murao S. et al., Isolation and Identification of a cellulolytic enzyme producing microorganisms. J. Ferment Techol.,57, 151, 1979
【非特許文献2】van Peij, et al., Isolation and analysis of XlnR, encoding a transcriptional activator co-ordinating xylanolytic expression in Aspergillus niger. Mol. Microbiol. 27:131-142
【非特許文献3】van Peij, et al., The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Appl. Environ. Microbiol. 64:3615-3619
【非特許文献4】Aro, N., et al. A novel transcriptional activator involved in regulation of cellulase and xylanase genes of Trichoderma reesei. J. Biol. Chem., 276:24309-24314
【非特許文献5】Stricker, A. et al., Xyr1 (xylanase regulator 1) regulates both the hydrolytic enzyme system and D-xylose metabolism in Hypocrea jecorina. Eukaryot. Cell. 5:2128-2137
【非特許文献6】Endo, Y., M. et al., Novel promoter sequence required for inductive expression of the Aspergillus nidulans endoglucanase gene eglA. Biosci. Biotechnol. Biochem. 72:312-320
【非特許文献7】Marui, J., N. et al., Transcriptional activator, AoXlnR, mediates cellulose-inductive expression of the xylanolytic and cellulolytic genes in Aspergillus oryzae. FEBS Lett. 528:279-282
【発明の概要】
【発明が解決しようとする課題】
【0013】
上記のように、基質特異性の異なる多様な加水分解酵素群が必要となるセルロース系バイオマスの完全酵素糖化には、これら酵素群中の任意の酵素を同時に大量生産する技術が必要である。しかし、セルロース系バイオマスの完全酵素糖化に、A. aculeatus No.F-50株の生産するセルラーゼ酵素群が有利ではあるものの、当該セルラーゼ酵素群の生産量が低いために実用化には至っていない。また、糖化に必要な多種類の酵素を、それぞれの酵素を一つずつ大量生産する系を構築すること、及び個別、逐一的に各酵素を生産すること自体も多大な労力を要するので、実現性が乏しい。そこで、これらの酵素遺伝子の発現を正に制御している因子を高発現できれば、前記セルラーゼ酵素群の生産量が上昇し、セルロール系バイオマスの酵素糖化を効率よく行わせることができる。
【課題を解決するための手段】
【0014】
本発明の遺伝子は配列番号2で示された塩基配列を有し、セルラーゼ酵素群の酵素、特にカルボキシメチルセルラーゼ1、キシラナーゼIb、セロビオヒドロラーゼI、カルボキシメチルセルラーゼ2の各遺伝子を誘導発現する。この遺伝子を高発現させることにより、これらのセルラーゼやヘミセルラーゼの生産量を高めることができる。
【発明の効果】
【0015】
本発明の遺伝子をA. aculeatusなどの宿主細胞に導入して、当該遺伝子を高発現させることにより、カルボキシメチルセルラーゼ1、キシラナーゼIb、セロビオヒドロラーゼI、カルボキシメチルセルラーゼ2を含むセルラーゼ・ヘミセルラーゼの生産量を高めることができる。
【図面の簡単な説明】
【0016】
【図1】図1はS4-22株におけるT-DNA挿入部位を示す概略図である。
【図2】図2はS4-22株における各種セルラーゼ及びヘミセルラーゼ遺伝子の発現解析による結果を示す図であって、(A)は宿主株(Host)及びS4-22株における半定量的RT−PCRの結果を、(B)はセロビオヒドロラーゼIの発現量を示す定量的PCRの結果を示す。
【図3】図3は破壊株における各種セルラーゼ及びヘミセルラーゼ遺伝子の発現解析による結果を示す図である。
【図4】図4は高発現株により産生されたセルラーゼの活性を示す図である。
【図5】図5は高発現株により産生されたキシラーゼの活性を示す図である。
【図6】図6は制御遺伝子スクリーニング用の宿主株を作出するためのpNCPの模式図である。
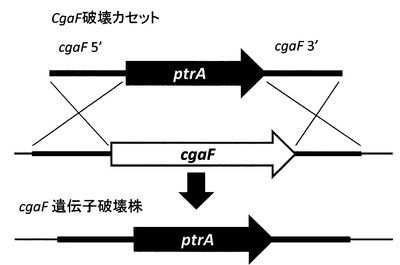
【図7】図7は転写因子(cgaF)が破壊された破壊カセットの模式図である。
【図8】図8は高発現用ベクターの作出に用いられたpTEFの模式図である。
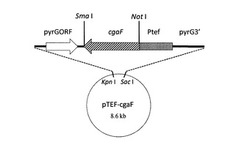
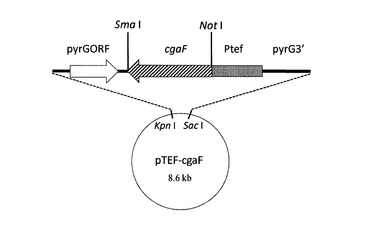
【図9】図9は高発現株を作出するためのpTEF−cgaFの模式図である。
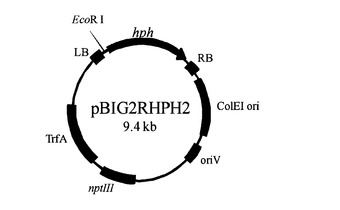
【図10】図10は宿主株を作出ためのバイナリーベクターpBIG2RHPH2の模式図である。
【発明を実施するための形態】
【0017】
本発明の転写因子は、糸状菌においてセルロース存在下でセルラーゼ遺伝子又はヘミセルラーゼ遺伝子の転写を正に制御する制御因子である。セルラーゼ遺伝子は一般的にはセルラーゼ群の酵素をコードする遺伝子を包括した意味で用いられるが、本明細書においては、特段の記載がない限りカルボキシメチルセルラーゼ1遺伝子(cmc1)、セロビオヒドロラーゼI遺伝子(cbhI)、カルボキシメチルセルラーゼ2遺伝子(cmc2)など、本発明の転写因子によりその発現が制御される遺伝子を包括した意味で用いられる。また、ヘミセルラーゼ遺伝子は一般的にはヘミセルラーゼ群の酵素をコードする遺伝子を包括した意味で用いられるが、本明細書においては、特段の記載がない限りキシラナーゼIb(xynIb)遺伝子など、本発明の転写因子によりその発現が制御される遺伝子を包括した意味で用いられる。
【0018】
本発明の転写因子は、カルボキシメチルセルラーゼ1遺伝子(cmc1)、キシラナーゼIb遺伝子(xynIb)、セロビオヒドロラーゼI遺伝子(cbhI)、カルボキシメチルセルラーゼ2遺伝子(cmc2)の少なくとも四つの遺伝子の発現をセルロース性基質の存在下で正に制御する。前記4つの遺伝子の内、xynIb、cmc1の発現はXlnRタンパク質により正に制御され、cbhI、cmc2の発現はXlnRタンパク質により制御されていない。このことから、本発明の転写因子は、XlnRタンパク質の介在の有無に関わらずセルラーゼ・ヘミセルラーゼの発現を正に制御する因子であると言える。本明細書において、セルロース性基質とは、グルコースがβ−1,4結合した高分子化合物であるセルロールやグルコースがβ−1,4結合した二糖類であるセロビオースなど、グルコースがβ−1,4結合したオリゴ糖又はオリゴ糖よりも多くの多糖が結合した高分子化合物を意味する。
【0019】
本発明の転写因子であるタンパク質は、配列番号1で示されたアミノ酸配列を有する。当該タンパク質は、上記セルラーゼ遺伝子又はヘミセルラーゼ遺伝子の転写を正に制御する機能を有している限り、配列番号1のアミノ酸配列から1個ないし数個のアミノ酸が欠失又は置換、挿入されたアミノ酸配列を有していてもよい。
【0020】
本発明の遺伝子は上記転写因子をコードする遺伝子であって、配列番号2に示す塩基配列を有する。また、本発明の遺伝子は、上記転写因子をコードする遺伝子又はその相補鎖とストリンジェントな条件でハイブリダイズする遺伝子でもあり得る。本発明の遺伝子は本発明の上記転写因子を発現できるものであればよく、配列番号2に示す塩基配列と80%以上の同一性、好ましくは90%以上の同一性、さらに望ましくは95%以上の同一性を有する塩基配列を有していればよい。なお、同一性は、ClustalW(Thompson et al., Nucleic Acids Res 24(1997), p.4876-7882)によって行われるアラインメントの結果に従う。
【0021】
本発明の遺伝子は、後の実施例に詳細を述べた方法で取得できる。すなわち、アグロバクテリウム属細菌による形質転換法を利用したT-DNAタギングによりcbhI遺伝子の転写を活性化する遺伝子として取得され得る。また、本発明の遺伝子は前記の方法で取得したcbhI遺伝子の転写を活性化する遺伝子又はその相補鎖をプローブとして、ストリンジェントな条件でハイブリダイズする遺伝子としても取得できる。ここで、ストリンジェントな条件とは、5×SSPE、3%のSDS、200mg/mlの剪断され、変性されたサケ精子DNA及び50%のホルムアミド含む溶液中で42℃でのプレハイブリダイゼーション及びハイブリダイゼーションを行った後、0.2%のSDSを含む2×SSC溶液中で、70℃での30分間の洗浄を行う条件である。
【0022】
本発明の遺伝子は、キシラナーゼ遺伝子の制御因子(XlnR)を介さずにセルビオヒドロラーゼI遺伝子やカルボキシメチルセルラーゼ2遺伝子の発現を制御し、セルビオースを含むセルロース性基質の存在下において、当該遺伝子の発現は、セルビオヒドロラーゼIの産生やカルボキシメチルセルラーゼ2の産生を亢進する。従って、本発明の遺伝子が組み込まれた宿主において、当該遺伝子を高発現させると、セルビオヒロラーゼI及びカルボキシメチルセルラーゼ2の産生量が向上する。
【0023】
また、本発明の遺伝子の発現は、セルロース性基質の存在下において、XlnRを介したキシラナーゼIb及びカルボキシメチルセルラーゼ1の産生を亢進する。従って、本発明の遺伝子が組み込まれた宿主細胞において、当該遺伝子を高発現させると、キシラナーゼIbおよびカルボキシメチルセルラーゼ1を含むヘミセルラーゼ及びセルラーゼの産生量を向上させることができる。なお、高発現とは、遺伝子の転写量ないし発現量を高めることを意味し、例えば、特定のプロモーターや発現ベクターの使用により実現され得る。また、酵素の産生の亢進とは、高発現させる前に比べて酵素の生産量が増加することを意味する。
【0024】
本発明のベクターは転写因子をコードする遺伝子を含み、そのベクターは、転写因子の発現に必要なプロモーターやエンハンサー、ターミネーター、選択マーカ遺伝子などを含み得る。当該遺伝子が組み込まれるプラスミドには、全て公知である、細菌用、糸状菌用、酵母用のプラスミドを用いることができる。公知のプラスミドとして大腸菌用としてpUC18やpKK233およびその誘導体、糸状菌用としてはpAUR316、酵母用としてYIp5、YRp19などが例示される。また、高発現可能なプラスミドが好ましく用いられる。糸状菌において遺伝子を高発現させるプラスミドは、構成的に高発現している当該遺伝子のプロモーターを有する。当該プロモーターは、例えば、トランスレーションエロンゲーション1遺伝子のプロモーターであり得る。従って、このプロモーターの下流に転写因子をコードする遺伝子を組み込むことにより、達成される。また、当該転写因子をコードする遺伝子の上流に、構成的に高発現させるプロモーターを組み込み、そのプロモーターの制御下で転写可能なように当該プロモーターを組み込むためのDNA断片を構築することによっても、達成できる。
【0025】
組換えに利用される宿主はセルラーゼ遺伝子又はキシラーゼ遺伝子を有する宿主であればよく、例えば糸状菌が好ましく用いられる。当該糸状菌は、例えば、アスペルギルス(Aspergillus)属菌であり、トリコデルマ(Tricoderma)属菌であり、フサリウム(Fusarium)属菌であり、ムコル(Mucor)属菌であり、ペニシリウム(Penicillium)属菌であり得る。アスペルギルス属菌は、例えば、A. aculeatusであり、A. awamoriであり、A. japonicusであり、A. nidulansであり、A. nigerであり、A. oryzaeであり得る。トリコデルマ属菌は、例えば、T. reeseiであり、T. harzianumであり、T. koningiiであり、T. virideであり得る。フサリウム属菌は、F. cerealisであり、F. crookwellenseであり、F. culmorumであり、F. oxysporumであり、F. roseumであり、F. reticulatumであり得る。ムコル属菌は、M. meiheiであり、M. javanicsuであり得る。ペニシリン属菌は、P. purpurogenumであり、P. notatumであり得る。本発明の転写因子は、Aspergillus aculeatus由来であるので、宿主としてアスペルギルス属菌が好適であり、望ましくは、A. aculeatus、さらに望ましくはA. aculeatus No.F-50株である。
【0026】
形質転換のための手法は特に制限されることはなく、公知の方法が用いられる。例えば、細胞壁を溶解したプロトプラストに対してポリエチレングリコールの存在下でベクターを導入するプロトプラスト−PEG法、電気パルスを利用したエレクトロポレーション法、アグロバクテリア菌を用いたアグロバクテリウム法であり得る。
【0027】
こうして、形質転換された宿主は基質であるセルロース及び/又はヘミセルロースの存在下でセルラーゼ、ヘミセルラーゼを産生する。特に、カルボキシメチルセルラーゼ1遺伝子(cmc1)、セロビオヒドロラーゼI遺伝子(cbhI)、カルボキシメチルセルラーゼ2遺伝子(cmc2)、キシラナーゼIb遺伝子(xynIb)の転写が亢進される。その結果、宿主の培養液は高いセルラーゼ活性や高いヘミセルラーゼ活性を示す。
【0028】
本発明のセルラーゼ及び/又はヘミセルラーゼ生産方法は、こうして得られた形質転換体を培養することによってセルラーゼやヘミセルラーゼを生産する方法である。培養方法は特に限定されるものではなく、公知の培地や培養条件が適用され得る。使用される培地は、最少培地であり、完全培地であり得る。また、セルラーゼやヘミセルラーゼを誘導するための特別の培地であり得る。特別の培地として、炭素源としてセルロースやキシランを含む培地である。セルロースは、グルコースがβ−1,4結合した直鎖状の高分子であり、例えばアビセル(商品名)として市販されている。また、キシランは、キシロースがβ−1,4結合した直鎖に種々の側鎖が結合したヘテロ糖であり、例えば、キシラン(オート麦・スペルト小麦由来)として市販されている。また、特別の培地は、セルロースやキシランと共に、あるいはセルロールやキシランに替え、両者を含む天然物を含む培地でもあり得る。この天然物は、例えば、木質系バイオマス、草本系バイオマス、コムギのふすまやオートミールであり得る。こうして培養された形質転換体の培養液は、カルボキシメチルセルラーゼ1やセロビオヒドロラーゼI、カルボキシメチルセルラーゼ2のセルラーゼやキシラナーゼIb(xynIb)に限定されず、多種・多量のセルラーゼやヘミセルラーゼを含む。このセルラーゼやヘミセルラーゼを培地から回収して、セルラーゼやヘミセルラーゼ酵素剤を得ることができる。また、当該培養液を濃縮・脱塩してセルラーゼやヘミセルラーゼ酵素剤を製造することもできる。
【0029】
本発明の植物系バイオマスの糖化方法は、上記形質転換体を含む培養液ないし当該培養液から回収したセルラーゼやヘミセルラーゼを含む酵素剤の存在下においてセルロース系バイオマスの糖化を行う方法である。また形質転換体を培養した培養液には、セルラーゼやヘミセルラーゼを多量に含む。特に、A. aculeatus No.F-50株は、カルボキシメチルセルラーゼ1やキシラナーゼIb、セロビオヒドロラーゼI、カルボキシメチルセルラーゼ2の他に多種のセルラーゼやヘミセルラーゼを生産するので、当該培養液ないしそれからセルラーゼやヘミセルラーゼを回収して得た酵素剤を添加することにより効率的にセルロース系バイオマスの糖化を行うことができる。
【0030】
セルロース系バイオマスは、草や木材、農業廃棄物、紙や食料廃棄物などいわゆるゴミのようなバイオマスが例示され、セルロースを含むものであれば特に限定されるものではない。バイオマスの糖化は公知の方法によればよく、バイオマスの粉砕、酸やアルカリを用いた前処理、高温高圧による前処理など各種前処理を経て、上記形質転換体を含む培養液ないし酵素剤の存在下でグルコースに糖化される。
【実施例1】
【0031】
〔遺伝子タギングによる新規転写因子のスクリーニング〕
XlnR非依存的に発現が誘導されるcbhIプロモーターの誘導能を指標に新規セルラーゼ遺伝子発現転写因子(「cellulase-genes-activating factor, CgaF」で示される場合がある。)を同定した。このために、遺伝子タギングの一手法であるAgrobacterium tumefaciens-mediated transformation(AtMT)を採用した。
【0032】
まず、A. aculeatus 二重栄養要求性株(Aspergillus aculeatus(ku80,niaD, pyrG))の硝酸還元酵素遺伝子座に、cbhIプロモーターにオロチジンリン酸脱炭酸酵素(pyrG)をレポーターとして繋いだ断片を1コピー導入した宿主株を作製した。誘導条件下では、pyrGがcbhIプロモーター制御下で発現するため、この宿主株はセルロースを含む培地上で生育可能となる。一方、セルロースだけでなくフルオロオロチン酸(FOA)含有培地では、cbhIプロモーター制御下で生産されたオロチジンリン酸脱炭酸酵素が機能することにより、FOAから毒性物質が菌体内で生産されて、この宿主は生育できなくなる。従って、cbhI遺伝子の発現を制御する因子(cgaFを含む)にアグロバクテリウム形質転換法を用いてT-DNAが挿入され、cbhI遺伝子プロモーターの発現誘導能が消失すると、pyrGは発現せずにこの株はFOA含有培地でも生育が可能となる。こうして、FOA含有培地で生育した株を選択することにより一次スクリーニングを行い、目的の遺伝子変異候補株(cbhI遺伝子の発現を制御する因子にT-DNAが挿入された株)を選択した。
【0033】
次に、一次スクリーニングでの単離株を、セルロース、グルコース、キシランを単一炭素源とした最少培地上で生育させ、セルロース培地上でのみ生育が低下する株を選択し、二次スクリーニングを行った。
【0034】
この二次スクリーニングでの単離株について、cbhI、cmc2遺伝子の発現量を半定量的RT-PCR法を用いて解析し、cbhI、cmc2遺伝子発現量が低下した株を選択し、三次スクリーニングを行った。このようにして6000株のAtMT形質転換体の中から、cbhI発現誘導能が低下した株を一株単離し、S4-22株と命名した。
【0035】
〔新規転写因子(CgaF)の同定〕
S4-22株においてT-DNAの挿入により破壊された遺伝子を同定するために、下記Inverse-PCR法を用いてT-DNAの周辺配列を増幅し、得られたDNA断片のシークエンス解析を行った。その結果、T-DNAはZn(II)2Cys6 DNA結合モチーフを有する遺伝子の5´側に挿入されていることが明らかになった(図1)。この破壊された遺伝子をcellulase-genes-activating factor (cgaF)遺伝子と命名した。cgaF遺伝子の染色体DNAおよびcDNA配列を解読した結果、cgaF遺伝子は四つのイントロンに分断され、727アミノ酸残基のタンパク質をコードしていることが明らかになった。当該タンパク質のアミノ酸配列は配列番号1に、cDNAの塩基配列は配列番号2に示される。
【0036】
〔転写因子CgaFの機能解析〕
cgaF遺伝子の機能を解析するにあたり、宿主株とS4-22株におけるセロビオヒドロラーゼI(cbhI)、カルボキシメチルセルラーゼ2(cmc2)の遺伝子発現量を半定量的RT−PCR法を用いて解析した。各株を24時間培養した菌体を集菌して炭素源を含まない最少培地で洗浄後、1%グルコース(D)、1%アビセル(Av、微結晶セルロース)、1%キシロース(X)を含む最少培地で3時間培養した時点での各遺伝子発現量を解析した。炭素源が変わっても遺伝子の発現量が変わらないグリセルアルデヒド3リン酸脱水素酵素(gpdA)遺伝子の発現量を内在性のコントロールとして用いた。その結果を図2に示す。図2(A)に示されたように、宿主株で観察されたアビセルによるcbhI、cmc2遺伝子発現の誘導は、S4-22株では観察されなかった。また、図2(B)に示されたように、定量的PCR法を用いて同条件下で発現しているcbhIの転写物を定量したところ、cbhI遺伝子発現量は、cgaF遺伝子破壊株において宿主株における発現量の約20%にまで低下していた。これらの結果から、cgaF遺伝子がセルラーゼ遺伝子発現誘導に関与していることが示唆された。
〔cgaF遺伝子破壊株におけるセルラーゼ遺伝子発現誘導への影響〕
【0037】
cgaF遺伝子がセルラーゼ遺伝子発現誘導に関与することを確認するため、cgaF遺伝子が破壊された株を相同組換えにより作製し、セルラーゼ・ヘミセルラーゼ遺伝子発現誘導に与える影響を解析することを目的として、上記方法に準じ、セロビオヒドロラーゼ(cbhI)、カルボキシメチルセルラーゼ2(cmc2)、カルボキシメチルセルラーゼ1(cmc1)、およびキシラナーゼIb(xynIb)遺伝子の発現量を解析した。その結果を図3に示す。この結果から分かるように、cgaF遺伝子破壊株における各遺伝子の発現量は、いずれの遺伝子においても宿主株のそれに比べて低下していたことから、CgaF因子がセルロースによるセルラーゼ・ヘミセルラーゼ遺伝子発現誘導に関与していることが示された。なお図3中act1はアクチン遺伝子である。
【0038】
また、XlnRを介したセルロースによるcmc1、xynIb遺伝子発現誘導も、cbhI、cmc2同様に低下しており、CgaF因子がXlnRを介したセルロースからのシグナル伝達にも関与していることが示された。
【0039】
〔cgaF遺伝子高発現によるセルラーゼ及びキシラナーゼの生産〕
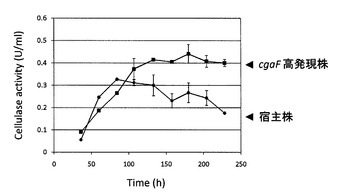
cgaF遺伝子高発現が、セルラーゼ生産量及びキシラナーゼ生産量に与える影響を調べるために、構成的に高発現するトランスレーションエロンゲーションファクター1遺伝子(tef1)のプロモータ(Ptef)を用いてcgaF遺伝子を高発現させた。cgaF高発現株は、cgaF遺伝子破壊株を、cgaF高発現カセットDNAを用いて形質転換することで取得した。cgaF遺伝子を破壊していない宿主株と、cgaF高発現株を4%の小麦ふすまを炭素源とした完全培地で30℃、160rpmで振とう培養した際の各株の培養上清のセルラーゼ活性及びキシラナーゼ活性を測定した。その結果を図4及び図5に示す。
【0040】
アルカリ膨潤セルロースを基質としてセルラーゼ活性を測定した場合、培養84時間目までは両株とも同等のセルラーゼ生産が観察された。しかし、108時間目以降、宿主株では活性が徐々に低下するのに対して、cgaF高発現株では更に酵素が生産され、酵素活性の低下は観察されなかった。さらに、培養228時間目では生産量に2倍の差が生じた(図4)。キシラナーゼ生産量は、培養60時間までは両株ともに生産量に差は観察されなかった。しかし、それ以降、宿主株では生産量が低下したのに対して、cgaF高発現株では生産が亢進し、キシラナーゼ生産量が6倍に向上した(図5)。
【0041】
以上の結果より、cgaF遺伝子を高発現させることにより、セルラーゼおよびキシラナーゼの高生産が可能であることが示された。
【0042】
〔実験手法〕
(1)AtMT宿主株(A. aculeatus(niaD, pyrG, (niaD-PcbhI-pyrG))の作出
cbhIプロモータの遺伝子発現能をA. nidulansオロチジンリン酸脱炭酸酵素遺伝子(pyrG)をレポーターとして評価する為に用いたniaDORF-Pcbh-pyrG-niaDT断片を以下のように構築した。cbhI遺伝子プロモータ(PcbhI)の翻訳開始点の上流661bpとA. nidulans pyrG遺伝子をプライマーAacbhFABとAacbhRE及び、AnpyrGFとAnpyrGRを用いてPCRにより増幅した。得られたDNA断片をそれぞれBamH I-EcoR I及び、EcoR I-Xha Iで消化し、pBSのBamH I-Xha Iサイトに同時にサブクローニングした(Pcbh?pyrG)。このプラスミドを鋳型として、プライマーniaDt-cbhIpFとniaDt-pyrGtRを用いてPCRを行い、PcbhI-pyrG断片を取得した。また、A. aculeatus染色体DNAを鋳型として、プライマーniaDpFとcbhI-niaDtR、及びpyrGt-niaDtFとniaDtRnotを用いて硝酸還元酵素遺伝子ORF領域(niaDORF)とその3´側のniaDT断片を増幅した。これらniaDORF、Pcbh-pyrG、niaDTの三断片を鋳型として、プライマーniaDpFとniaDtRnotを用いてフュージョンPCRを行った。増幅されたDNA断片をpBSのEcoR Vサイトにサブクローニンし、pNCPを取得した(図6)。
【0043】
上記で作製されたpNCPを鋳型として、プライマーniaDpFとniaDtRnotを用いてPCRを行うことで、niaDORF-PcbhI-pyrG-niaDT遺伝子断片を増幅し、形質転換に用いることでPcbhI-pyrGカセットがniaD遺伝子座に1コピー導入された形質転換体を取得した。この株をアグロバクテリウム形質転換の宿主株(A. aculeatus(niaD, pyrG, (niaD-PcbhI-pyrG))に用いた。
【0044】
(2)cgaF遺伝子破壊株(A. aculeatus(ku80, pyrG, cgaF::ptrA))の作出
cgaF遺伝子破壊カセット構築のために、まず、cgaF遺伝子5´および3´側隣接領域をそれぞれ1kbずつPCRにより増幅した。鋳型はA. aculeatus染色体DNAとし、プライマーはそれぞれ、22a-F_Xbaと22aptrA-R2、及び22aDptrA-Fと22aD-xho-Rを用いた。形質転換の選択マーカには、A. oryzaeのピリチアミン耐性遺伝子(ptrA)を用いた。pPTRII(Takara)を鋳型として、22a-ptrA-F2と22a-ptrA-Rプライマーを用いてPCRによりptrAを発現するのに充分な領域を増幅した。得られた三断片のDNAを鋳型として、22aA-F2と22aD-R2プライマーを用いてフュージョンPCRを行うことで、cgaF遺伝子破壊カセットを構築した(図7)。この遺伝子破壊カセットにて宿主株を形質転換して、遺伝子破壊株(A. aculeatus(ku80, pyrG, cgaF::ptrA))を得た。
【0045】
(3)高発現株(A. aculeatus(ku80, pyrG, cgaF::ptrA(Ptef-cgaF-pyrG)))の作出
まず、cgaF遺伝子をトランスレーションエロンゲーションファクター1遺伝子(tef1)プロモータ(Ptef)の制御化で高発現させるためのベクターを構築した。A. aculeatus染色体DNAを鋳型としてFAoe-F1_NotIとFAoe-R1プライマーを用いてPCRによりcgaF遺伝子断片を増幅した。得られたDNA断片をNot Iで消化後、pTEFプラスミド(図8)のNot I-Sma IサイトにサブクローニングすることでpTEF-cgaFを構築した(図9)。このプラスミドを鋳型としてプライマーAapyrGF2とAapyrG3Sを用いてPCRを行い、pyrGORF-cgaF-Ptef-pyrG3´の断片約6kbを増幅した。この増幅断片を用いて、A. aculeatusウリジン要求性株を形質転換し、pyrG遺伝子座に1コピーで上記DNA断片が挿入された高発現株を取得した。
【0046】
(4)Agrobacterium コンピテントセルの作製及び形質転換
大腸菌コンピテントセルの作製法を基に簡略化した塩化カルシウム法(Mandel and Higa 1970)により、Agrobacteriumのコンピテントセルを作製した。
【0047】
形質転換は、1μgのプラスミドDNAを、100μlのAgrobacteriumコンピテントセル(溶液)に添加し、37℃で約5分間融解した。1mlの2xTY培地を加え、28℃で2〜4時間振盪培養し、その後、カナマイシンを含むLBプレート培地にスプレッドして形質転換体を得た。
【0048】
(5)アグロバクテリウム法によるA. aculeatusの形質転換
AtMTは、de Grootらの方法(1998)を改変した方法で行った。共培養は液体誘導培地で行った。京都府立大学辻先生より供与されたバイナリーベクターpBIG2RHPH2(図10)(Tsuji et al., 2003)を保有するA. tumefaciens C58C1を、カナマイシンを含む50mlのLB培地に数コロニー接種し、28℃、200rpmで一晩振とう培養した。この培養液を、カナマイシンを含む100mlのIMに、660nmの吸光度が0.15になるように植菌し、OD440=0.4になるまで、28℃、200rpmで振とう培養した(種培養液)。MMプレート培地で3〜10日生育させたA. aculeatusの分生子を、5%Glycerol溶液(0.9%のNaClを含む)に懸濁し、IMで107個になるように100mlの0.01%のウリジンを含むIM(500ml容羽付きフラスコ)に植菌し、24℃、150rpmで24時間培養した。この培養液に終濃度200μMとなるようにアセトシリンゴンを添加後、上述のアグロバクテリウム種培養液1mlを植菌し、24℃、150rpmで48時間培養後、菌体を集菌、液体選択培地で洗浄し、選択培地に移した。
【0049】
(6)T-DNA周辺 A. aculeatus genomic DNA 配列の取得(Inverse PCR:IPCR)
T-DNA周辺配列を取得するため IPCR(Ochman et al., 1988)を行った。形質転換体の染色体DNAをT-DNAの Left border ニックサイトから857bpにサイトが存在するNco Iと、955bpにサイトが存在するNde Iそれぞれで消化した後、クロロホルム/イソアミルアルコール抽出、エタノール沈澱、70%エタノール洗浄により、DNAを精製し、およそ5μg/μlになるように滅菌水に溶解した。次に、以下の条件でライゲーションし、緩衝液を除くためにエタノール沈澱、70%エタノール洗浄を行い、20μlの滅菌水に溶解した(2.5ng/μl)。
(Ligation reaction mixture)
DNA 50ng
T4 DNA ligase 500U
10x ligation buffer 5μl
滅菌水 up to 50μl
(Ligation condition)
16℃、1時間
【0050】
次に先に作製したDNA溶液を鋳型とし、IPCR反応を行った。プライマーとしてLeft border 側はHAS-4とHAS-2comの組み合わせを、Right border側はHS-3とHS-1com1の組み合わせを使用し、ポリメラーゼとしてPrime STARTM HS DNA polymeraseを使用した。
(PCR reaction mixture)
Template DNA 10μl(25ng)
Forward primer 10pmol(HAS-4 or HS-3)
Reverse primer 10pmol(HAS-2com or HS-1com1)
5x PrimeSTAR buffer 10μl
dNTP (2.5 mM each) 4.0μl(final 0.2mM)
PrimeSTAR(r) HS
DNA polymerase 1.25U
滅菌水 up to 50μl
(PCR condition)
Cycle Thermal settings
1 98℃、2min
30 98℃、10sec、62℃、5sec、72℃、5min
1 72℃、10min
【0051】
PCR産物は場合に応じてアガロースゲルから単一バンドを回収し、illustraTM GFXTM PCR DNA and Gel Band Purification Kitを用いて精製し、シークエンス解析を行った。シークエンス解析はタカラバイオ(株)に委託した。
【0052】
(7)RT−PCR
PCRは、PrimeSTAR HS DNA polymerase(Takara, Japan)と表1に示したプライマーを用いて行った。染色体DNAの調製は、Adachi et al.(2009)の論文に従って行った。RNAの調製は、TRIZOL reagent(Invitrogen)を用いて行った。半定量的RT-PCRは、RNA PCR kit (AMV) ver 3.0(Takara、Japan)とKOD-plus polymerase (Toyobo、Japan)を用いて行った。セロビオヒドロラーゼI遺伝子(cbhI)、カルボキシメチルセルラーゼ1及び2遺伝子(cmc1、cmc2)、キシラナーゼIb遺伝子(xynIb)及びグルセロール3リン酸脱水素酵素遺伝子(gpdA)の発現量解析は、それぞれ、cbhIF-RT/cbhIR-RT、cmc1F-RT/cmc1R-RT、cmc2F-RT/cmc2R-RT、xyn2F-RT/xyn2R-RTとgpdAF/gpdARのプライマーセットを用いて行った(表1)。定量的PCRは、SYBR Premix Ex TaqTM(Perfect Real Time)(Takara、Japan)を用いて行い、ABI 7300 real-time PCR system(Applied Biosystems)を用いて検出した。各実験は三回以上行い、再現した場合にはその代表的なデータを、或はその平均と標準偏差をデータとして示した。
【0053】
(8)セルラーゼ活性、ヘミセルラーゼ活性の測定
宿主株とcgaF高発現株を、0.2%ウリジンを含む最少培地プレートで5日間培養し、それぞれの胞子数2.0×106個を0.2%を添加したセルラーゼ、ヘミセルラーゼ誘導培地200mlに植菌した。植菌後、36時間後から約24時間ごとに培養上清を回収し、上清のエンドグルカナーゼ活性とエンドキシラナーゼ活性をSomogyi-Nelson 法で、β−グルコシダーゼ活性をpNP法で測定した。
【0054】
セルラーゼ活性は、1%アルカリ膨潤セルロース(ASC)溶液を基質溶液とし、培養上清を100mM酢酸緩衝液(pH5.0)で適宜希釈した溶液を酵素溶液として測定した。100μlの酵素溶液に100μlの基質溶液を加え、両者をよく混合し、37℃にて反応させた。反応後、500μlのソモギー液を加えて反応を停止させ、300μlのイオン交換水を加えた。次に、100℃で15分間サンプルを煮沸し、流水中で5分間急冷した。さらに500μlのネルソン試薬を加えて、泡を立てないようにして激しく攪拌し、20分間室温で放置し、よく混合した後、800×gで10分間の遠心分離を行った。その上清の500nmにおける吸光度を測定して、活性を求めた。ブランクとして、100μlの酵素溶液に500μのSomogyi液を加え、酵素反応を行わなかった溶液を用いた。1unitのセルラーゼ活性は1分間に1μmolのグルコースに相当する還元糖を遊離する酵素量と定義し、グルコースを用いた検量線より算出した。
【0055】
エンドキシラナーゼ活性は、1%のBirchwood xylan溶液(100mM酢酸緩衝液、pH5.0)を基質溶液とし、セルラーゼ活性の場合と同様に調整した溶液を酵素溶液として測定した。100μlの酵素溶液に100μlの基質溶液を加え、両者をよく混合し、37℃にて10分間反応させた。反応後、500μlのソモギー液を加えて反応を停止させ、300μlのイオン交換水を加えた。その後、セルラーゼ活性測定と同様の操作を行った。また、1unitのキシラーゼ活性は1分間に1μmolのキシロースに相当する還元糖を遊離する酵素量と定義し、キシロースを用いた検量線より算出した。
【0056】
(9)培地
上記で使用した培地を下記に示した。
(a)完全培地(CM)(per liter)
Salt solution* 50ml
Trace element mixture** 11ml
Glucose 10g
Peptone 2g
Yeast extract 1g
Casein hydrolysate 1g
Vitamin solution*** 1ml(pH6.5)
【0057】
(b)最少培地(MM)(per liter)
Salt solution* 50ml
Trace element mixture** 1ml
Glucose 10g
NaNO3 3g(pH6.5)
【0058】
*Salt solution (per liter)
KCl 26g
MgSO4・4H2O 26g
KH2PO4 76g
**Trace element mixture (per liter)
Na2MoO4・2H2O 0.8g
H3BO3 11.1g
CoCl・6H2O 1.6g
CuSO4・5H2O 1.6g
EDTA 50g
FeSO4・5H2O 5.0g
MnCl2・4H2O 5.0g
ZnSO4・7H2O 22g
***Vitamin solution (per liter)
Biotin 2.5g
Nicotinic acid 2.5g
PABA (p-aminobenzoic acid) 0.8g
Pyridoxine hydrochloride 1.0g
Pantothenate 2.0g
Riboflavin 2.5g
Aneurine hydrochloride 1.5g
Choline chloride 20g
【0059】
(c)セルラーゼ・ヘミセルラーゼ誘導生産培地
Wheat bran 4.0%(w/v)
Liquid Bleached Kraft Pulp 0.2%(w/v)
Sodium Glutamate 0.5%(w/v)
Corn Steep Liquor 3.0%(w/v)
KH2PO4 1.0%(w/v)
MgSO4・7H2O 0.1%(w/v)
NaCl 0.2%(w/v)
Tween-80 0.1%(w/v)
adjusted to pH6.0 with KOH
【0060】
(10)プライマー
上記で使用したプライマーを表1にまとめた。
【0061】
【表1】

【産業上の利用可能性】
【0062】
本発明によると、セルロース酵素群やキシラナーゼの産生量を高めることができる。これにより、植物系バイオマスの酵素糖化の効率化が図られる。
【技術分野】
【0001】
本発明は、セルラーゼ・ヘミセルラーゼ遺伝子の発現を正に制御する転写因子に関する。
【背景技術】
【0002】
植物系バイオマスは地球上で最も多く生産される再生可能な資源である。その中でも稲ワラ等のセルロース系バイオマスは食糧資源とも競合しないため、これを原料としたバイオエタノール等のバイオ燃料やポリ乳酸等のバイオ化成品の代替製品を製造する技術革新が進めば、新たなバイオ産業創出につながると期待されている。
【0003】
植物系バイオマスの大部分を占めるセルロースは、グルコースがβ−1,4結合した直鎖状高分子であって、分子内あるいは分子間で広範囲にわたり数多くの水素結合を形成し、非常に強固な結晶構造をとる不溶性物質である。更に、その周りを囲むようにアラビノース、ガラクトース、グルクロニル基、フェルロイル基、アセチル基により部分的に修飾されたキシランを主要成分としたヘミセルロースに覆われている。従って、これらセルロースを中心とするセルロース系バイオマスから単糖を生成することは容易ではなく、石油の代替原料として、バイオ燃料やバイオ化成品を創り出すための障壁の一つとなっている。
【0004】
セルロース系バイオマスを糖化する方法として、塩酸や硫酸などの強酸を用いた酸糖化法がある。しかし、この方法は、単糖の過分解のために低収率である、酸の回収あるいは中和にコストがかる上環境負荷が大きいという難点がある。また、近年開発された注目の糖化法として超臨界水・亜臨界水処理による加水分解による方法がある。この方法は、リグニン除去などバイオマスの前処理の必要がなく、短時間で行える上、水と熱のみの反応でクリーンであるという利点を有する。しかし、多大な熱エネルギーを必要とすることや反応制御が難しいことなど課題が残っている。
【0005】
このような状況下、温和な条件で反応が行うことができる、最も環境負荷が小さいなどの理由により、酵素糖化法が注目されてきている。この方法は、生じた単糖の過分解を生じない利点もあるが、これまでのところ、反応時間が長いことや前処理を必要とすること、また、上述の結晶構造を形成するセルロースやキシランを中心とするヘミセルロースを加水分解するため、様々な基質特異性を有した酵素、例えば結晶性セルロースをグルコースにまで加水分解するには、セロビオヒドロラーゼ、エンドグルカナーゼ、β−グルコシダーゼ等の酵素群が、またヘミセルロースを単糖まで加水分解するにはエンドキシラナーゼ、β−キシロシダーゼ、α−アラビノフラノシダーゼ、β−ガラクトシダーゼ、α−グルクロニダーゼ、フェルロイルエステラーゼ、アセチルキシランエステラーゼ等、種々の基質特異性を有した酵素群が必要な点が短所としてあげられる。
【0006】
セルラーゼは、それぞれ構造が異なるセルロースを加水分解する酵素の総称で、セルラーゼはその基質特異性及び加水分解様式により大きく3種類に分類される。1つ目は結晶性セルロースに作用し、セルロース鎖の末端から二糖(セロビオース)単位で分解するセロビオヒドロラーゼ(cellobiohydrolase:CBH)、2つ目は結晶性セルロースを分解できないが、非結晶性セルロース鎖をランダムに切断するエンドグルカナーゼ(endo-glucanase:EG)、そして3つ目が可溶性のセロビオース並びにセロオリゴ糖に作用してグルコースを生成するβ−グルコシダーゼ(β−glucosidase:BGL)である。
【0007】
糸状菌Trichoderma reeseiが生産するセルラーゼは最強のセルラーゼと言われている。このセルラーゼは結晶性セルロースの分解には優れるものの、単糖を生成する力、すなわちβ‐グルコシダーゼの活性が弱く、糖化液中にはセロオリゴ糖(β−グルカンオリゴ糖)が多く残ってしまうという問題があった。また、セルロース系バイオマスにはセルロース単独で存在することは少なく、ヘミセルロースなどを伴うためセルロースを分解するにはまずこれらを除く必要がある。また、取り除いたヘミセルロースを構成するペントースも、例えば酵母にペントース資化能を付与すれば発酵によってエタノール生産の原料とすることができる。しかし、T. reeseiのセルラーゼはヘミセルラーゼ活性が弱いため、セルロース系バイオマス中に共存するセルロースとヘミセルロースの両方を効率的に分解し構成単糖を効率よく生産するには更にヘミセルラーゼを添加する必要がある。
【0008】
T. reeseiのセルラーゼのこうした弱点を補い、かつセルロース系バイオマスの分解において、T. reeseiのセルラーゼと強い相乗作用を示すセルラーゼ酵素群を生産する微生物として、Aspergillus aculeatus No.F-50株が知られている。この菌の生産するセルラーゼ酵素群は、非常に強い単糖生成能を有することに加え、ヘミセルラーゼ活性も強く、セルロースだけでなくヘミセルロースを含めたセルロース系バイオマスの糖化に非常に有効である。特筆すべき点は、A. aculeatus No.F-50株の生産するセルラーゼ酵素群には、セロビオースを分解してグルコースにするだけでなく、セロペンタオースやセロヘキサオースなど比較的長鎖のセロオリゴ糖に対しても強い分解活性を有するβ−グルコシダーゼ(BGLI)が存在するので、A. aculeatusのセルラーゼ酵素群を添加することによって、セルラーゼの単糖生成力が弱いというT. reeseiの弱点を補うことができる点である(非特許文献1)。
【0009】
ところで、セルラーゼ・ヘミセルラーゼは、セルロースやキシロース等の基質存在下で誘導され、グルコース存在下ではカタボライトリプレッションによりその生成が抑制される。これまでにセルラーゼ・ヘミセルラーゼ遺伝子発現制御機構に関する研究において、A. nigerからキシラナーゼ遺伝子発現制御因子であるXlnRが同定され、当該因子がエンドキシラナーゼだけでなくキシランの化学修飾部位や、セルラーゼ遺伝子の発現も制御していることが解明されている(非特許文献2、3)。また、T. reeseiでは、XlnR遺伝子のホモログ(homolog)であるXyr1遺伝子が全てのセルラーゼ・ヘミセルラーゼ遺伝子発現を制御していることが報告されている(非特許文献4、5)。一方、Aspergillus属糸状菌では、セルラーゼ・ヘミセルラーゼの遺伝子発現はXlnRに依存せず、セルロースやキシロース等の基質に応答して発現が制御される遺伝子であることが知られている(非特許文献6、7)。しかしながらXlnR以外の転写因子で、セルラーゼ・ヘミセルラーゼ遺伝子の発現を正に制御する転写因子について、遺伝子の詳細な配列やその機能については知られていなかった。
【0010】
また、特許文献1には、糸状菌において菌体外プロテアーゼ遺伝子の発現を正に制御する転写因子であるprtTの遺伝子破壊により、α−グルコシダーゼ、β−グルコシダーゼ、セルラーゼやペクチン分解酵素等の生産量を亢進することが開示されている。特許文献2には、Aspergillus属糸状菌において、α−アミラーゼプロモーターによって転写され、α−アミラーゼの転写を制御する転写因子AmyRが開示されている。特許文献3には、T. reeseiにおいて、セロビオヒドロラーゼI(cbhI)、セロビオヒドロラーゼII(cbhII)などのプロモーターによるこれら遺伝子の転写を負に制御する転写因子AceIが開示されている。しかしながら、これらの特許文献においても、セルラーゼ・ヘミセルラーゼ遺伝子の転写を正に制御する転写因子は報告されていない。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特表2003−526374号公報
【特許文献2】特表2000−514292号公報
【特許文献3】特表2004−526440号公報
【非特許文献】
【0012】
【非特許文献1】Murao S. et al., Isolation and Identification of a cellulolytic enzyme producing microorganisms. J. Ferment Techol.,57, 151, 1979
【非特許文献2】van Peij, et al., Isolation and analysis of XlnR, encoding a transcriptional activator co-ordinating xylanolytic expression in Aspergillus niger. Mol. Microbiol. 27:131-142
【非特許文献3】van Peij, et al., The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Appl. Environ. Microbiol. 64:3615-3619
【非特許文献4】Aro, N., et al. A novel transcriptional activator involved in regulation of cellulase and xylanase genes of Trichoderma reesei. J. Biol. Chem., 276:24309-24314
【非特許文献5】Stricker, A. et al., Xyr1 (xylanase regulator 1) regulates both the hydrolytic enzyme system and D-xylose metabolism in Hypocrea jecorina. Eukaryot. Cell. 5:2128-2137
【非特許文献6】Endo, Y., M. et al., Novel promoter sequence required for inductive expression of the Aspergillus nidulans endoglucanase gene eglA. Biosci. Biotechnol. Biochem. 72:312-320
【非特許文献7】Marui, J., N. et al., Transcriptional activator, AoXlnR, mediates cellulose-inductive expression of the xylanolytic and cellulolytic genes in Aspergillus oryzae. FEBS Lett. 528:279-282
【発明の概要】
【発明が解決しようとする課題】
【0013】
上記のように、基質特異性の異なる多様な加水分解酵素群が必要となるセルロース系バイオマスの完全酵素糖化には、これら酵素群中の任意の酵素を同時に大量生産する技術が必要である。しかし、セルロース系バイオマスの完全酵素糖化に、A. aculeatus No.F-50株の生産するセルラーゼ酵素群が有利ではあるものの、当該セルラーゼ酵素群の生産量が低いために実用化には至っていない。また、糖化に必要な多種類の酵素を、それぞれの酵素を一つずつ大量生産する系を構築すること、及び個別、逐一的に各酵素を生産すること自体も多大な労力を要するので、実現性が乏しい。そこで、これらの酵素遺伝子の発現を正に制御している因子を高発現できれば、前記セルラーゼ酵素群の生産量が上昇し、セルロール系バイオマスの酵素糖化を効率よく行わせることができる。
【課題を解決するための手段】
【0014】
本発明の遺伝子は配列番号2で示された塩基配列を有し、セルラーゼ酵素群の酵素、特にカルボキシメチルセルラーゼ1、キシラナーゼIb、セロビオヒドロラーゼI、カルボキシメチルセルラーゼ2の各遺伝子を誘導発現する。この遺伝子を高発現させることにより、これらのセルラーゼやヘミセルラーゼの生産量を高めることができる。
【発明の効果】
【0015】
本発明の遺伝子をA. aculeatusなどの宿主細胞に導入して、当該遺伝子を高発現させることにより、カルボキシメチルセルラーゼ1、キシラナーゼIb、セロビオヒドロラーゼI、カルボキシメチルセルラーゼ2を含むセルラーゼ・ヘミセルラーゼの生産量を高めることができる。
【図面の簡単な説明】
【0016】
【図1】図1はS4-22株におけるT-DNA挿入部位を示す概略図である。
【図2】図2はS4-22株における各種セルラーゼ及びヘミセルラーゼ遺伝子の発現解析による結果を示す図であって、(A)は宿主株(Host)及びS4-22株における半定量的RT−PCRの結果を、(B)はセロビオヒドロラーゼIの発現量を示す定量的PCRの結果を示す。
【図3】図3は破壊株における各種セルラーゼ及びヘミセルラーゼ遺伝子の発現解析による結果を示す図である。
【図4】図4は高発現株により産生されたセルラーゼの活性を示す図である。
【図5】図5は高発現株により産生されたキシラーゼの活性を示す図である。
【図6】図6は制御遺伝子スクリーニング用の宿主株を作出するためのpNCPの模式図である。
【図7】図7は転写因子(cgaF)が破壊された破壊カセットの模式図である。
【図8】図8は高発現用ベクターの作出に用いられたpTEFの模式図である。
【図9】図9は高発現株を作出するためのpTEF−cgaFの模式図である。
【図10】図10は宿主株を作出ためのバイナリーベクターpBIG2RHPH2の模式図である。
【発明を実施するための形態】
【0017】
本発明の転写因子は、糸状菌においてセルロース存在下でセルラーゼ遺伝子又はヘミセルラーゼ遺伝子の転写を正に制御する制御因子である。セルラーゼ遺伝子は一般的にはセルラーゼ群の酵素をコードする遺伝子を包括した意味で用いられるが、本明細書においては、特段の記載がない限りカルボキシメチルセルラーゼ1遺伝子(cmc1)、セロビオヒドロラーゼI遺伝子(cbhI)、カルボキシメチルセルラーゼ2遺伝子(cmc2)など、本発明の転写因子によりその発現が制御される遺伝子を包括した意味で用いられる。また、ヘミセルラーゼ遺伝子は一般的にはヘミセルラーゼ群の酵素をコードする遺伝子を包括した意味で用いられるが、本明細書においては、特段の記載がない限りキシラナーゼIb(xynIb)遺伝子など、本発明の転写因子によりその発現が制御される遺伝子を包括した意味で用いられる。
【0018】
本発明の転写因子は、カルボキシメチルセルラーゼ1遺伝子(cmc1)、キシラナーゼIb遺伝子(xynIb)、セロビオヒドロラーゼI遺伝子(cbhI)、カルボキシメチルセルラーゼ2遺伝子(cmc2)の少なくとも四つの遺伝子の発現をセルロース性基質の存在下で正に制御する。前記4つの遺伝子の内、xynIb、cmc1の発現はXlnRタンパク質により正に制御され、cbhI、cmc2の発現はXlnRタンパク質により制御されていない。このことから、本発明の転写因子は、XlnRタンパク質の介在の有無に関わらずセルラーゼ・ヘミセルラーゼの発現を正に制御する因子であると言える。本明細書において、セルロース性基質とは、グルコースがβ−1,4結合した高分子化合物であるセルロールやグルコースがβ−1,4結合した二糖類であるセロビオースなど、グルコースがβ−1,4結合したオリゴ糖又はオリゴ糖よりも多くの多糖が結合した高分子化合物を意味する。
【0019】
本発明の転写因子であるタンパク質は、配列番号1で示されたアミノ酸配列を有する。当該タンパク質は、上記セルラーゼ遺伝子又はヘミセルラーゼ遺伝子の転写を正に制御する機能を有している限り、配列番号1のアミノ酸配列から1個ないし数個のアミノ酸が欠失又は置換、挿入されたアミノ酸配列を有していてもよい。
【0020】
本発明の遺伝子は上記転写因子をコードする遺伝子であって、配列番号2に示す塩基配列を有する。また、本発明の遺伝子は、上記転写因子をコードする遺伝子又はその相補鎖とストリンジェントな条件でハイブリダイズする遺伝子でもあり得る。本発明の遺伝子は本発明の上記転写因子を発現できるものであればよく、配列番号2に示す塩基配列と80%以上の同一性、好ましくは90%以上の同一性、さらに望ましくは95%以上の同一性を有する塩基配列を有していればよい。なお、同一性は、ClustalW(Thompson et al., Nucleic Acids Res 24(1997), p.4876-7882)によって行われるアラインメントの結果に従う。
【0021】
本発明の遺伝子は、後の実施例に詳細を述べた方法で取得できる。すなわち、アグロバクテリウム属細菌による形質転換法を利用したT-DNAタギングによりcbhI遺伝子の転写を活性化する遺伝子として取得され得る。また、本発明の遺伝子は前記の方法で取得したcbhI遺伝子の転写を活性化する遺伝子又はその相補鎖をプローブとして、ストリンジェントな条件でハイブリダイズする遺伝子としても取得できる。ここで、ストリンジェントな条件とは、5×SSPE、3%のSDS、200mg/mlの剪断され、変性されたサケ精子DNA及び50%のホルムアミド含む溶液中で42℃でのプレハイブリダイゼーション及びハイブリダイゼーションを行った後、0.2%のSDSを含む2×SSC溶液中で、70℃での30分間の洗浄を行う条件である。
【0022】
本発明の遺伝子は、キシラナーゼ遺伝子の制御因子(XlnR)を介さずにセルビオヒドロラーゼI遺伝子やカルボキシメチルセルラーゼ2遺伝子の発現を制御し、セルビオースを含むセルロース性基質の存在下において、当該遺伝子の発現は、セルビオヒドロラーゼIの産生やカルボキシメチルセルラーゼ2の産生を亢進する。従って、本発明の遺伝子が組み込まれた宿主において、当該遺伝子を高発現させると、セルビオヒロラーゼI及びカルボキシメチルセルラーゼ2の産生量が向上する。
【0023】
また、本発明の遺伝子の発現は、セルロース性基質の存在下において、XlnRを介したキシラナーゼIb及びカルボキシメチルセルラーゼ1の産生を亢進する。従って、本発明の遺伝子が組み込まれた宿主細胞において、当該遺伝子を高発現させると、キシラナーゼIbおよびカルボキシメチルセルラーゼ1を含むヘミセルラーゼ及びセルラーゼの産生量を向上させることができる。なお、高発現とは、遺伝子の転写量ないし発現量を高めることを意味し、例えば、特定のプロモーターや発現ベクターの使用により実現され得る。また、酵素の産生の亢進とは、高発現させる前に比べて酵素の生産量が増加することを意味する。
【0024】
本発明のベクターは転写因子をコードする遺伝子を含み、そのベクターは、転写因子の発現に必要なプロモーターやエンハンサー、ターミネーター、選択マーカ遺伝子などを含み得る。当該遺伝子が組み込まれるプラスミドには、全て公知である、細菌用、糸状菌用、酵母用のプラスミドを用いることができる。公知のプラスミドとして大腸菌用としてpUC18やpKK233およびその誘導体、糸状菌用としてはpAUR316、酵母用としてYIp5、YRp19などが例示される。また、高発現可能なプラスミドが好ましく用いられる。糸状菌において遺伝子を高発現させるプラスミドは、構成的に高発現している当該遺伝子のプロモーターを有する。当該プロモーターは、例えば、トランスレーションエロンゲーション1遺伝子のプロモーターであり得る。従って、このプロモーターの下流に転写因子をコードする遺伝子を組み込むことにより、達成される。また、当該転写因子をコードする遺伝子の上流に、構成的に高発現させるプロモーターを組み込み、そのプロモーターの制御下で転写可能なように当該プロモーターを組み込むためのDNA断片を構築することによっても、達成できる。
【0025】
組換えに利用される宿主はセルラーゼ遺伝子又はキシラーゼ遺伝子を有する宿主であればよく、例えば糸状菌が好ましく用いられる。当該糸状菌は、例えば、アスペルギルス(Aspergillus)属菌であり、トリコデルマ(Tricoderma)属菌であり、フサリウム(Fusarium)属菌であり、ムコル(Mucor)属菌であり、ペニシリウム(Penicillium)属菌であり得る。アスペルギルス属菌は、例えば、A. aculeatusであり、A. awamoriであり、A. japonicusであり、A. nidulansであり、A. nigerであり、A. oryzaeであり得る。トリコデルマ属菌は、例えば、T. reeseiであり、T. harzianumであり、T. koningiiであり、T. virideであり得る。フサリウム属菌は、F. cerealisであり、F. crookwellenseであり、F. culmorumであり、F. oxysporumであり、F. roseumであり、F. reticulatumであり得る。ムコル属菌は、M. meiheiであり、M. javanicsuであり得る。ペニシリン属菌は、P. purpurogenumであり、P. notatumであり得る。本発明の転写因子は、Aspergillus aculeatus由来であるので、宿主としてアスペルギルス属菌が好適であり、望ましくは、A. aculeatus、さらに望ましくはA. aculeatus No.F-50株である。
【0026】
形質転換のための手法は特に制限されることはなく、公知の方法が用いられる。例えば、細胞壁を溶解したプロトプラストに対してポリエチレングリコールの存在下でベクターを導入するプロトプラスト−PEG法、電気パルスを利用したエレクトロポレーション法、アグロバクテリア菌を用いたアグロバクテリウム法であり得る。
【0027】
こうして、形質転換された宿主は基質であるセルロース及び/又はヘミセルロースの存在下でセルラーゼ、ヘミセルラーゼを産生する。特に、カルボキシメチルセルラーゼ1遺伝子(cmc1)、セロビオヒドロラーゼI遺伝子(cbhI)、カルボキシメチルセルラーゼ2遺伝子(cmc2)、キシラナーゼIb遺伝子(xynIb)の転写が亢進される。その結果、宿主の培養液は高いセルラーゼ活性や高いヘミセルラーゼ活性を示す。
【0028】
本発明のセルラーゼ及び/又はヘミセルラーゼ生産方法は、こうして得られた形質転換体を培養することによってセルラーゼやヘミセルラーゼを生産する方法である。培養方法は特に限定されるものではなく、公知の培地や培養条件が適用され得る。使用される培地は、最少培地であり、完全培地であり得る。また、セルラーゼやヘミセルラーゼを誘導するための特別の培地であり得る。特別の培地として、炭素源としてセルロースやキシランを含む培地である。セルロースは、グルコースがβ−1,4結合した直鎖状の高分子であり、例えばアビセル(商品名)として市販されている。また、キシランは、キシロースがβ−1,4結合した直鎖に種々の側鎖が結合したヘテロ糖であり、例えば、キシラン(オート麦・スペルト小麦由来)として市販されている。また、特別の培地は、セルロースやキシランと共に、あるいはセルロールやキシランに替え、両者を含む天然物を含む培地でもあり得る。この天然物は、例えば、木質系バイオマス、草本系バイオマス、コムギのふすまやオートミールであり得る。こうして培養された形質転換体の培養液は、カルボキシメチルセルラーゼ1やセロビオヒドロラーゼI、カルボキシメチルセルラーゼ2のセルラーゼやキシラナーゼIb(xynIb)に限定されず、多種・多量のセルラーゼやヘミセルラーゼを含む。このセルラーゼやヘミセルラーゼを培地から回収して、セルラーゼやヘミセルラーゼ酵素剤を得ることができる。また、当該培養液を濃縮・脱塩してセルラーゼやヘミセルラーゼ酵素剤を製造することもできる。
【0029】
本発明の植物系バイオマスの糖化方法は、上記形質転換体を含む培養液ないし当該培養液から回収したセルラーゼやヘミセルラーゼを含む酵素剤の存在下においてセルロース系バイオマスの糖化を行う方法である。また形質転換体を培養した培養液には、セルラーゼやヘミセルラーゼを多量に含む。特に、A. aculeatus No.F-50株は、カルボキシメチルセルラーゼ1やキシラナーゼIb、セロビオヒドロラーゼI、カルボキシメチルセルラーゼ2の他に多種のセルラーゼやヘミセルラーゼを生産するので、当該培養液ないしそれからセルラーゼやヘミセルラーゼを回収して得た酵素剤を添加することにより効率的にセルロース系バイオマスの糖化を行うことができる。
【0030】
セルロース系バイオマスは、草や木材、農業廃棄物、紙や食料廃棄物などいわゆるゴミのようなバイオマスが例示され、セルロースを含むものであれば特に限定されるものではない。バイオマスの糖化は公知の方法によればよく、バイオマスの粉砕、酸やアルカリを用いた前処理、高温高圧による前処理など各種前処理を経て、上記形質転換体を含む培養液ないし酵素剤の存在下でグルコースに糖化される。
【実施例1】
【0031】
〔遺伝子タギングによる新規転写因子のスクリーニング〕
XlnR非依存的に発現が誘導されるcbhIプロモーターの誘導能を指標に新規セルラーゼ遺伝子発現転写因子(「cellulase-genes-activating factor, CgaF」で示される場合がある。)を同定した。このために、遺伝子タギングの一手法であるAgrobacterium tumefaciens-mediated transformation(AtMT)を採用した。
【0032】
まず、A. aculeatus 二重栄養要求性株(Aspergillus aculeatus(ku80,niaD, pyrG))の硝酸還元酵素遺伝子座に、cbhIプロモーターにオロチジンリン酸脱炭酸酵素(pyrG)をレポーターとして繋いだ断片を1コピー導入した宿主株を作製した。誘導条件下では、pyrGがcbhIプロモーター制御下で発現するため、この宿主株はセルロースを含む培地上で生育可能となる。一方、セルロースだけでなくフルオロオロチン酸(FOA)含有培地では、cbhIプロモーター制御下で生産されたオロチジンリン酸脱炭酸酵素が機能することにより、FOAから毒性物質が菌体内で生産されて、この宿主は生育できなくなる。従って、cbhI遺伝子の発現を制御する因子(cgaFを含む)にアグロバクテリウム形質転換法を用いてT-DNAが挿入され、cbhI遺伝子プロモーターの発現誘導能が消失すると、pyrGは発現せずにこの株はFOA含有培地でも生育が可能となる。こうして、FOA含有培地で生育した株を選択することにより一次スクリーニングを行い、目的の遺伝子変異候補株(cbhI遺伝子の発現を制御する因子にT-DNAが挿入された株)を選択した。
【0033】
次に、一次スクリーニングでの単離株を、セルロース、グルコース、キシランを単一炭素源とした最少培地上で生育させ、セルロース培地上でのみ生育が低下する株を選択し、二次スクリーニングを行った。
【0034】
この二次スクリーニングでの単離株について、cbhI、cmc2遺伝子の発現量を半定量的RT-PCR法を用いて解析し、cbhI、cmc2遺伝子発現量が低下した株を選択し、三次スクリーニングを行った。このようにして6000株のAtMT形質転換体の中から、cbhI発現誘導能が低下した株を一株単離し、S4-22株と命名した。
【0035】
〔新規転写因子(CgaF)の同定〕
S4-22株においてT-DNAの挿入により破壊された遺伝子を同定するために、下記Inverse-PCR法を用いてT-DNAの周辺配列を増幅し、得られたDNA断片のシークエンス解析を行った。その結果、T-DNAはZn(II)2Cys6 DNA結合モチーフを有する遺伝子の5´側に挿入されていることが明らかになった(図1)。この破壊された遺伝子をcellulase-genes-activating factor (cgaF)遺伝子と命名した。cgaF遺伝子の染色体DNAおよびcDNA配列を解読した結果、cgaF遺伝子は四つのイントロンに分断され、727アミノ酸残基のタンパク質をコードしていることが明らかになった。当該タンパク質のアミノ酸配列は配列番号1に、cDNAの塩基配列は配列番号2に示される。
【0036】
〔転写因子CgaFの機能解析〕
cgaF遺伝子の機能を解析するにあたり、宿主株とS4-22株におけるセロビオヒドロラーゼI(cbhI)、カルボキシメチルセルラーゼ2(cmc2)の遺伝子発現量を半定量的RT−PCR法を用いて解析した。各株を24時間培養した菌体を集菌して炭素源を含まない最少培地で洗浄後、1%グルコース(D)、1%アビセル(Av、微結晶セルロース)、1%キシロース(X)を含む最少培地で3時間培養した時点での各遺伝子発現量を解析した。炭素源が変わっても遺伝子の発現量が変わらないグリセルアルデヒド3リン酸脱水素酵素(gpdA)遺伝子の発現量を内在性のコントロールとして用いた。その結果を図2に示す。図2(A)に示されたように、宿主株で観察されたアビセルによるcbhI、cmc2遺伝子発現の誘導は、S4-22株では観察されなかった。また、図2(B)に示されたように、定量的PCR法を用いて同条件下で発現しているcbhIの転写物を定量したところ、cbhI遺伝子発現量は、cgaF遺伝子破壊株において宿主株における発現量の約20%にまで低下していた。これらの結果から、cgaF遺伝子がセルラーゼ遺伝子発現誘導に関与していることが示唆された。
〔cgaF遺伝子破壊株におけるセルラーゼ遺伝子発現誘導への影響〕
【0037】
cgaF遺伝子がセルラーゼ遺伝子発現誘導に関与することを確認するため、cgaF遺伝子が破壊された株を相同組換えにより作製し、セルラーゼ・ヘミセルラーゼ遺伝子発現誘導に与える影響を解析することを目的として、上記方法に準じ、セロビオヒドロラーゼ(cbhI)、カルボキシメチルセルラーゼ2(cmc2)、カルボキシメチルセルラーゼ1(cmc1)、およびキシラナーゼIb(xynIb)遺伝子の発現量を解析した。その結果を図3に示す。この結果から分かるように、cgaF遺伝子破壊株における各遺伝子の発現量は、いずれの遺伝子においても宿主株のそれに比べて低下していたことから、CgaF因子がセルロースによるセルラーゼ・ヘミセルラーゼ遺伝子発現誘導に関与していることが示された。なお図3中act1はアクチン遺伝子である。
【0038】
また、XlnRを介したセルロースによるcmc1、xynIb遺伝子発現誘導も、cbhI、cmc2同様に低下しており、CgaF因子がXlnRを介したセルロースからのシグナル伝達にも関与していることが示された。
【0039】
〔cgaF遺伝子高発現によるセルラーゼ及びキシラナーゼの生産〕
cgaF遺伝子高発現が、セルラーゼ生産量及びキシラナーゼ生産量に与える影響を調べるために、構成的に高発現するトランスレーションエロンゲーションファクター1遺伝子(tef1)のプロモータ(Ptef)を用いてcgaF遺伝子を高発現させた。cgaF高発現株は、cgaF遺伝子破壊株を、cgaF高発現カセットDNAを用いて形質転換することで取得した。cgaF遺伝子を破壊していない宿主株と、cgaF高発現株を4%の小麦ふすまを炭素源とした完全培地で30℃、160rpmで振とう培養した際の各株の培養上清のセルラーゼ活性及びキシラナーゼ活性を測定した。その結果を図4及び図5に示す。
【0040】
アルカリ膨潤セルロースを基質としてセルラーゼ活性を測定した場合、培養84時間目までは両株とも同等のセルラーゼ生産が観察された。しかし、108時間目以降、宿主株では活性が徐々に低下するのに対して、cgaF高発現株では更に酵素が生産され、酵素活性の低下は観察されなかった。さらに、培養228時間目では生産量に2倍の差が生じた(図4)。キシラナーゼ生産量は、培養60時間までは両株ともに生産量に差は観察されなかった。しかし、それ以降、宿主株では生産量が低下したのに対して、cgaF高発現株では生産が亢進し、キシラナーゼ生産量が6倍に向上した(図5)。
【0041】
以上の結果より、cgaF遺伝子を高発現させることにより、セルラーゼおよびキシラナーゼの高生産が可能であることが示された。
【0042】
〔実験手法〕
(1)AtMT宿主株(A. aculeatus(niaD, pyrG, (niaD-PcbhI-pyrG))の作出
cbhIプロモータの遺伝子発現能をA. nidulansオロチジンリン酸脱炭酸酵素遺伝子(pyrG)をレポーターとして評価する為に用いたniaDORF-Pcbh-pyrG-niaDT断片を以下のように構築した。cbhI遺伝子プロモータ(PcbhI)の翻訳開始点の上流661bpとA. nidulans pyrG遺伝子をプライマーAacbhFABとAacbhRE及び、AnpyrGFとAnpyrGRを用いてPCRにより増幅した。得られたDNA断片をそれぞれBamH I-EcoR I及び、EcoR I-Xha Iで消化し、pBSのBamH I-Xha Iサイトに同時にサブクローニングした(Pcbh?pyrG)。このプラスミドを鋳型として、プライマーniaDt-cbhIpFとniaDt-pyrGtRを用いてPCRを行い、PcbhI-pyrG断片を取得した。また、A. aculeatus染色体DNAを鋳型として、プライマーniaDpFとcbhI-niaDtR、及びpyrGt-niaDtFとniaDtRnotを用いて硝酸還元酵素遺伝子ORF領域(niaDORF)とその3´側のniaDT断片を増幅した。これらniaDORF、Pcbh-pyrG、niaDTの三断片を鋳型として、プライマーniaDpFとniaDtRnotを用いてフュージョンPCRを行った。増幅されたDNA断片をpBSのEcoR Vサイトにサブクローニンし、pNCPを取得した(図6)。
【0043】
上記で作製されたpNCPを鋳型として、プライマーniaDpFとniaDtRnotを用いてPCRを行うことで、niaDORF-PcbhI-pyrG-niaDT遺伝子断片を増幅し、形質転換に用いることでPcbhI-pyrGカセットがniaD遺伝子座に1コピー導入された形質転換体を取得した。この株をアグロバクテリウム形質転換の宿主株(A. aculeatus(niaD, pyrG, (niaD-PcbhI-pyrG))に用いた。
【0044】
(2)cgaF遺伝子破壊株(A. aculeatus(ku80, pyrG, cgaF::ptrA))の作出
cgaF遺伝子破壊カセット構築のために、まず、cgaF遺伝子5´および3´側隣接領域をそれぞれ1kbずつPCRにより増幅した。鋳型はA. aculeatus染色体DNAとし、プライマーはそれぞれ、22a-F_Xbaと22aptrA-R2、及び22aDptrA-Fと22aD-xho-Rを用いた。形質転換の選択マーカには、A. oryzaeのピリチアミン耐性遺伝子(ptrA)を用いた。pPTRII(Takara)を鋳型として、22a-ptrA-F2と22a-ptrA-Rプライマーを用いてPCRによりptrAを発現するのに充分な領域を増幅した。得られた三断片のDNAを鋳型として、22aA-F2と22aD-R2プライマーを用いてフュージョンPCRを行うことで、cgaF遺伝子破壊カセットを構築した(図7)。この遺伝子破壊カセットにて宿主株を形質転換して、遺伝子破壊株(A. aculeatus(ku80, pyrG, cgaF::ptrA))を得た。
【0045】
(3)高発現株(A. aculeatus(ku80, pyrG, cgaF::ptrA(Ptef-cgaF-pyrG)))の作出
まず、cgaF遺伝子をトランスレーションエロンゲーションファクター1遺伝子(tef1)プロモータ(Ptef)の制御化で高発現させるためのベクターを構築した。A. aculeatus染色体DNAを鋳型としてFAoe-F1_NotIとFAoe-R1プライマーを用いてPCRによりcgaF遺伝子断片を増幅した。得られたDNA断片をNot Iで消化後、pTEFプラスミド(図8)のNot I-Sma IサイトにサブクローニングすることでpTEF-cgaFを構築した(図9)。このプラスミドを鋳型としてプライマーAapyrGF2とAapyrG3Sを用いてPCRを行い、pyrGORF-cgaF-Ptef-pyrG3´の断片約6kbを増幅した。この増幅断片を用いて、A. aculeatusウリジン要求性株を形質転換し、pyrG遺伝子座に1コピーで上記DNA断片が挿入された高発現株を取得した。
【0046】
(4)Agrobacterium コンピテントセルの作製及び形質転換
大腸菌コンピテントセルの作製法を基に簡略化した塩化カルシウム法(Mandel and Higa 1970)により、Agrobacteriumのコンピテントセルを作製した。
【0047】
形質転換は、1μgのプラスミドDNAを、100μlのAgrobacteriumコンピテントセル(溶液)に添加し、37℃で約5分間融解した。1mlの2xTY培地を加え、28℃で2〜4時間振盪培養し、その後、カナマイシンを含むLBプレート培地にスプレッドして形質転換体を得た。
【0048】
(5)アグロバクテリウム法によるA. aculeatusの形質転換
AtMTは、de Grootらの方法(1998)を改変した方法で行った。共培養は液体誘導培地で行った。京都府立大学辻先生より供与されたバイナリーベクターpBIG2RHPH2(図10)(Tsuji et al., 2003)を保有するA. tumefaciens C58C1を、カナマイシンを含む50mlのLB培地に数コロニー接種し、28℃、200rpmで一晩振とう培養した。この培養液を、カナマイシンを含む100mlのIMに、660nmの吸光度が0.15になるように植菌し、OD440=0.4になるまで、28℃、200rpmで振とう培養した(種培養液)。MMプレート培地で3〜10日生育させたA. aculeatusの分生子を、5%Glycerol溶液(0.9%のNaClを含む)に懸濁し、IMで107個になるように100mlの0.01%のウリジンを含むIM(500ml容羽付きフラスコ)に植菌し、24℃、150rpmで24時間培養した。この培養液に終濃度200μMとなるようにアセトシリンゴンを添加後、上述のアグロバクテリウム種培養液1mlを植菌し、24℃、150rpmで48時間培養後、菌体を集菌、液体選択培地で洗浄し、選択培地に移した。
【0049】
(6)T-DNA周辺 A. aculeatus genomic DNA 配列の取得(Inverse PCR:IPCR)
T-DNA周辺配列を取得するため IPCR(Ochman et al., 1988)を行った。形質転換体の染色体DNAをT-DNAの Left border ニックサイトから857bpにサイトが存在するNco Iと、955bpにサイトが存在するNde Iそれぞれで消化した後、クロロホルム/イソアミルアルコール抽出、エタノール沈澱、70%エタノール洗浄により、DNAを精製し、およそ5μg/μlになるように滅菌水に溶解した。次に、以下の条件でライゲーションし、緩衝液を除くためにエタノール沈澱、70%エタノール洗浄を行い、20μlの滅菌水に溶解した(2.5ng/μl)。
(Ligation reaction mixture)
DNA 50ng
T4 DNA ligase 500U
10x ligation buffer 5μl
滅菌水 up to 50μl
(Ligation condition)
16℃、1時間
【0050】
次に先に作製したDNA溶液を鋳型とし、IPCR反応を行った。プライマーとしてLeft border 側はHAS-4とHAS-2comの組み合わせを、Right border側はHS-3とHS-1com1の組み合わせを使用し、ポリメラーゼとしてPrime STARTM HS DNA polymeraseを使用した。
(PCR reaction mixture)
Template DNA 10μl(25ng)
Forward primer 10pmol(HAS-4 or HS-3)
Reverse primer 10pmol(HAS-2com or HS-1com1)
5x PrimeSTAR buffer 10μl
dNTP (2.5 mM each) 4.0μl(final 0.2mM)
PrimeSTAR(r) HS
DNA polymerase 1.25U
滅菌水 up to 50μl
(PCR condition)
Cycle Thermal settings
1 98℃、2min
30 98℃、10sec、62℃、5sec、72℃、5min
1 72℃、10min
【0051】
PCR産物は場合に応じてアガロースゲルから単一バンドを回収し、illustraTM GFXTM PCR DNA and Gel Band Purification Kitを用いて精製し、シークエンス解析を行った。シークエンス解析はタカラバイオ(株)に委託した。
【0052】
(7)RT−PCR
PCRは、PrimeSTAR HS DNA polymerase(Takara, Japan)と表1に示したプライマーを用いて行った。染色体DNAの調製は、Adachi et al.(2009)の論文に従って行った。RNAの調製は、TRIZOL reagent(Invitrogen)を用いて行った。半定量的RT-PCRは、RNA PCR kit (AMV) ver 3.0(Takara、Japan)とKOD-plus polymerase (Toyobo、Japan)を用いて行った。セロビオヒドロラーゼI遺伝子(cbhI)、カルボキシメチルセルラーゼ1及び2遺伝子(cmc1、cmc2)、キシラナーゼIb遺伝子(xynIb)及びグルセロール3リン酸脱水素酵素遺伝子(gpdA)の発現量解析は、それぞれ、cbhIF-RT/cbhIR-RT、cmc1F-RT/cmc1R-RT、cmc2F-RT/cmc2R-RT、xyn2F-RT/xyn2R-RTとgpdAF/gpdARのプライマーセットを用いて行った(表1)。定量的PCRは、SYBR Premix Ex TaqTM(Perfect Real Time)(Takara、Japan)を用いて行い、ABI 7300 real-time PCR system(Applied Biosystems)を用いて検出した。各実験は三回以上行い、再現した場合にはその代表的なデータを、或はその平均と標準偏差をデータとして示した。
【0053】
(8)セルラーゼ活性、ヘミセルラーゼ活性の測定
宿主株とcgaF高発現株を、0.2%ウリジンを含む最少培地プレートで5日間培養し、それぞれの胞子数2.0×106個を0.2%を添加したセルラーゼ、ヘミセルラーゼ誘導培地200mlに植菌した。植菌後、36時間後から約24時間ごとに培養上清を回収し、上清のエンドグルカナーゼ活性とエンドキシラナーゼ活性をSomogyi-Nelson 法で、β−グルコシダーゼ活性をpNP法で測定した。
【0054】
セルラーゼ活性は、1%アルカリ膨潤セルロース(ASC)溶液を基質溶液とし、培養上清を100mM酢酸緩衝液(pH5.0)で適宜希釈した溶液を酵素溶液として測定した。100μlの酵素溶液に100μlの基質溶液を加え、両者をよく混合し、37℃にて反応させた。反応後、500μlのソモギー液を加えて反応を停止させ、300μlのイオン交換水を加えた。次に、100℃で15分間サンプルを煮沸し、流水中で5分間急冷した。さらに500μlのネルソン試薬を加えて、泡を立てないようにして激しく攪拌し、20分間室温で放置し、よく混合した後、800×gで10分間の遠心分離を行った。その上清の500nmにおける吸光度を測定して、活性を求めた。ブランクとして、100μlの酵素溶液に500μのSomogyi液を加え、酵素反応を行わなかった溶液を用いた。1unitのセルラーゼ活性は1分間に1μmolのグルコースに相当する還元糖を遊離する酵素量と定義し、グルコースを用いた検量線より算出した。
【0055】
エンドキシラナーゼ活性は、1%のBirchwood xylan溶液(100mM酢酸緩衝液、pH5.0)を基質溶液とし、セルラーゼ活性の場合と同様に調整した溶液を酵素溶液として測定した。100μlの酵素溶液に100μlの基質溶液を加え、両者をよく混合し、37℃にて10分間反応させた。反応後、500μlのソモギー液を加えて反応を停止させ、300μlのイオン交換水を加えた。その後、セルラーゼ活性測定と同様の操作を行った。また、1unitのキシラーゼ活性は1分間に1μmolのキシロースに相当する還元糖を遊離する酵素量と定義し、キシロースを用いた検量線より算出した。
【0056】
(9)培地
上記で使用した培地を下記に示した。
(a)完全培地(CM)(per liter)
Salt solution* 50ml
Trace element mixture** 11ml
Glucose 10g
Peptone 2g
Yeast extract 1g
Casein hydrolysate 1g
Vitamin solution*** 1ml(pH6.5)
【0057】
(b)最少培地(MM)(per liter)
Salt solution* 50ml
Trace element mixture** 1ml
Glucose 10g
NaNO3 3g(pH6.5)
【0058】
*Salt solution (per liter)
KCl 26g
MgSO4・4H2O 26g
KH2PO4 76g
**Trace element mixture (per liter)
Na2MoO4・2H2O 0.8g
H3BO3 11.1g
CoCl・6H2O 1.6g
CuSO4・5H2O 1.6g
EDTA 50g
FeSO4・5H2O 5.0g
MnCl2・4H2O 5.0g
ZnSO4・7H2O 22g
***Vitamin solution (per liter)
Biotin 2.5g
Nicotinic acid 2.5g
PABA (p-aminobenzoic acid) 0.8g
Pyridoxine hydrochloride 1.0g
Pantothenate 2.0g
Riboflavin 2.5g
Aneurine hydrochloride 1.5g
Choline chloride 20g
【0059】
(c)セルラーゼ・ヘミセルラーゼ誘導生産培地
Wheat bran 4.0%(w/v)
Liquid Bleached Kraft Pulp 0.2%(w/v)
Sodium Glutamate 0.5%(w/v)
Corn Steep Liquor 3.0%(w/v)
KH2PO4 1.0%(w/v)
MgSO4・7H2O 0.1%(w/v)
NaCl 0.2%(w/v)
Tween-80 0.1%(w/v)
adjusted to pH6.0 with KOH
【0060】
(10)プライマー
上記で使用したプライマーを表1にまとめた。
【0061】
【表1】
【産業上の利用可能性】
【0062】
本発明によると、セルロース酵素群やキシラナーゼの産生量を高めることができる。これにより、植物系バイオマスの酵素糖化の効率化が図られる。
【特許請求の範囲】
【請求項1】
糸状菌においてセルラーゼ遺伝子及びヘミセルラーゼ遺伝子の転写を正に制御する活性を有し、かつ以下のいずれかのアミノ酸配列からなる転写因子。
(1)配列番号1に示されたアミノ配列。
(2)配列番号1に示されたアミノ配列から1から数個のアミノ酸が欠失、置換または付加した配列を有するアミノ酸配列。
【請求項2】
Aspergillus aculeatus由来である請求項1に記載の転写因子。
【請求項3】
請求項1又は請求項2に記載の転写因子をコードする遺伝子、又は、当該遺伝子若しくはその相補鎖とストリンジェントな条件でハイブリダイズする遺伝子。
【請求項4】
請求項3に記載の遺伝子が人工的に組み込まれたベクター。
【請求項5】
前記ベクターが、遺伝子が高発現可能な高発現ベクターである請求項4に記載のベクター。
【請求項6】
前記請求項3に記載の遺伝子が、構成的に高発現されるプロモーターの制御下で転写されるように組み込まれた請求項4又は5に記載のベクター。
【請求項7】
トランスレーションエロンゲーションファクター1遺伝子プロモーターの制御下で請求項3に記載の遺伝子が転写される請求項4〜6の何れか一項に記載のベクター。
【請求項8】
請求項4〜7の何れか1項に記載のベクターを含む形質転換体。
【請求項9】
前記形質転換体が糸状菌である請求項8に記載の形質転換体。
【請求項10】
前記糸状菌がAspergillus aculeatusである請求項9に記載の形質転換体。
【請求項11】
セルラーゼ及び/又はキシラナーゼを生産する方法であって、請求項8〜10に記載の形質転換体を培養する工程を有する方法。
【請求項12】
前記セルラーゼが、カルボキシメチルセルラーゼ1、セロビオヒドロラーゼI、カルボキシメチルセルラーゼ2の少なくとも1種であり、前記キシラーゼがキシラナーゼIbである請求項11に記載の方法。
【請求項13】
請求項8〜10に記載の形質転換体を含む培養液の存在下において、植物系バイオマスを糖化する方法。
【請求項14】
糸状菌において、請求項1記載の転写因子を高度に発現させて、セルラーゼ及び/又はキシラナーゼの産生量を高める方法。
【請求項15】
前記糸状菌は、Aspergillus aculeatusである請求項14に記載の方法。
【請求項1】
糸状菌においてセルラーゼ遺伝子及びヘミセルラーゼ遺伝子の転写を正に制御する活性を有し、かつ以下のいずれかのアミノ酸配列からなる転写因子。
(1)配列番号1に示されたアミノ配列。
(2)配列番号1に示されたアミノ配列から1から数個のアミノ酸が欠失、置換または付加した配列を有するアミノ酸配列。
【請求項2】
Aspergillus aculeatus由来である請求項1に記載の転写因子。
【請求項3】
請求項1又は請求項2に記載の転写因子をコードする遺伝子、又は、当該遺伝子若しくはその相補鎖とストリンジェントな条件でハイブリダイズする遺伝子。
【請求項4】
請求項3に記載の遺伝子が人工的に組み込まれたベクター。
【請求項5】
前記ベクターが、遺伝子が高発現可能な高発現ベクターである請求項4に記載のベクター。
【請求項6】
前記請求項3に記載の遺伝子が、構成的に高発現されるプロモーターの制御下で転写されるように組み込まれた請求項4又は5に記載のベクター。
【請求項7】
トランスレーションエロンゲーションファクター1遺伝子プロモーターの制御下で請求項3に記載の遺伝子が転写される請求項4〜6の何れか一項に記載のベクター。
【請求項8】
請求項4〜7の何れか1項に記載のベクターを含む形質転換体。
【請求項9】
前記形質転換体が糸状菌である請求項8に記載の形質転換体。
【請求項10】
前記糸状菌がAspergillus aculeatusである請求項9に記載の形質転換体。
【請求項11】
セルラーゼ及び/又はキシラナーゼを生産する方法であって、請求項8〜10に記載の形質転換体を培養する工程を有する方法。
【請求項12】
前記セルラーゼが、カルボキシメチルセルラーゼ1、セロビオヒドロラーゼI、カルボキシメチルセルラーゼ2の少なくとも1種であり、前記キシラーゼがキシラナーゼIbである請求項11に記載の方法。
【請求項13】
請求項8〜10に記載の形質転換体を含む培養液の存在下において、植物系バイオマスを糖化する方法。
【請求項14】
糸状菌において、請求項1記載の転写因子を高度に発現させて、セルラーゼ及び/又はキシラナーゼの産生量を高める方法。
【請求項15】
前記糸状菌は、Aspergillus aculeatusである請求項14に記載の方法。
【図1】


【図4】
【図5】

【図6】

【図7】
【図8】

【図9】
【図10】
【図2】

【図3】

【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【公開番号】特開2012−200184(P2012−200184A)
【公開日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願番号】特願2011−66797(P2011−66797)
【出願日】平成23年3月24日(2011.3.24)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り (1)刊行物名 第62回日本生物工学会大会講演要旨集 (2)発行日 平成22年9月25日 (3)発行所 社団法人 日本生物工学会 (4)該当ページ 第61ページ
【出願人】(505127721)公立大学法人大阪府立大学 (688)
【Fターム(参考)】
【公開日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願日】平成23年3月24日(2011.3.24)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り (1)刊行物名 第62回日本生物工学会大会講演要旨集 (2)発行日 平成22年9月25日 (3)発行所 社団法人 日本生物工学会 (4)該当ページ 第61ページ
【出願人】(505127721)公立大学法人大阪府立大学 (688)
【Fターム(参考)】
[ Back to top ]