
センダイウイルスベクターを用いたAIDSウイルスワクチン
【課題】免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチンの提供。
【解決手段】免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスをマカクサルに鼻腔内投与することにより、免疫不全ウイルスに対する防御免疫を効率的に誘発できる。鼻腔内へのワクチン接種により、センダイウイルスベクターを介した抗原遺伝子の発現が鼻腔粘膜および局所リンパ節で検出され、有意なレベルの抗原特異的細胞性免疫応答が誘導された。ワクチネーションによる病理学的徴候は見られなかった。ワクチネーション後にサル免疫不全ウイルスを暴露させて効果を検討したところ、対照動物に比べ血漿中のウイルス量の有意な低下が認められた。ワクチンは、AIDSワクチンとして有望である。
【解決手段】免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスをマカクサルに鼻腔内投与することにより、免疫不全ウイルスに対する防御免疫を効率的に誘発できる。鼻腔内へのワクチン接種により、センダイウイルスベクターを介した抗原遺伝子の発現が鼻腔粘膜および局所リンパ節で検出され、有意なレベルの抗原特異的細胞性免疫応答が誘導された。ワクチネーションによる病理学的徴候は見られなかった。ワクチネーション後にサル免疫不全ウイルスを暴露させて効果を検討したところ、対照動物に比べ血漿中のウイルス量の有意な低下が認められた。ワクチンは、AIDSワクチンとして有望である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、センダイウイルスベクターを用いたAIDSウイルスワクチンに関する。また本発明は、センダイウイルスベクターを用いたワクチネーションの方法に関する。
【背景技術】
【0002】
ヒト免疫不全ウイルス(HIV)の世界的な伝播を防ぐため、HIVの感染に対する効果的かつ安全なワクチンの開発が早急に求められている。これまで複数の後天性免疫不全症候群(AIDS)ワクチンの手法が霊長類のレンチウイルス感染モデルにおいて評価されてきたが、AIDSに対して一貫して十分な抵抗性を示すワクチンは未だ知られていない(Almond, N. M. and Heeney, J. L., 1998, AIDS 12 (Suppl. A): S133-140(非特許文献1); Burton, D. R. and Moore, J. P., 1998, Nat. Med. 4: 495-498(非特許文献2); Hulskotte, E. Q. et al., 1998, Vaccine 16: 904-915(非特許文献3); Letvin, N. L., 1998, Science 280: 1875-1880(非特許文献4))。
【0003】
生きたウイルスベクターを基にしたワクチンは、抗原の十分な発現が持続的に誘導されるため、有望な予防戦略の1つと見なされている(Cairns, J. S. and Sarver, N., 1998, AIDS Res. Hum. Retroviruses 14: 1501-1508(非特許文献5); Hirsch, V. M. et al., 1996, J. Virol. 70: 3741-3752(非特許文献6); Buge, S. L. et al., 1997, J. Virol. 71: 8531-8541(非特許文献7))。最もよく使われてきたのはポックスウイルス(poxvirus)であり、また、アデノウイルスなど他の選択肢も多数存在している。しかしながら、これらのベクターを用いた試みは、HIVに対する十分な防御免疫を誘発することに成功していない。十分な防御免疫の誘発には、抗原発現のレベルや期間、ベクターウイルスの複製のカイネティクス、およびベクターウイルスのトロピズムや病原性と言った多数の要因が影響しているものと考えられる。現在使用することができるウイルスベクターはいずれも、有利な側面と不利な側面の両面を持っている。最適なベクターを見つけ出すためには、それぞれのウイルスベクターの正確な評価と比較が必要である。
【0004】
ウイルスベクターを基にしたワクチン戦略における決定的なデメリットの1つは、標的とすべき抗原よりもむしろ、ベクターウイルスに由来する抗原に対して強力な免疫応答が誘導されてしまうことである。この問題は、初回免疫と追加免疫のそれぞれにおいて、2つまたはそれ以上の異なる種類のウイルスベクターを用いることにより解決することができる。DNAワクチンを基にした初回免疫後にウイルスベクターを基にした追加免疫を行うことも、好ましい戦略の1つである(Hanke, T. et al. 1999, J. Virol. 73: 7524-7532(非特許文献8); Robinson, H. L. et al., 1999, Nat. Med. 5: 526-534(非特許文献9))。このことから、新しいクラスのウイルスベクターの開発が、現在でも求められている。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Almond, N. M. and Heeney, J. L., 1998, AIDS 12 (Suppl. A): S133-140
【非特許文献2】Burton, D. R. and Moore, J. P., 1998, Nat. Med. 4: 495-498
【非特許文献3】Hulskotte, E. Q. et al., 1998, Vaccine 16: 904-915
【非特許文献4】Letvin, N. L., 1998, Science 280: 1875-1880
【非特許文献5】Cairns, J. S. and Sarver, N., 1998, AIDS Res. Hum. Retroviruses 14: 1501-1508
【非特許文献6】Hirsch, V. M. et al., 1996, J. Virol. 70: 3741-3752
【非特許文献7】Buge, S. L. et al., 1997, J. Virol. 71: 8531-8541
【非特許文献8】Hanke, T. et al. 1999, J. Virol. 73: 7524-7532
【非特許文献9】Robinson, H. L. et al., 1999, Nat. Med. 5: 526-534
【発明の概要】
【0006】
本発明者らは以前、組み換えセンダイウイルス(SeV)を用いて、効率的に抗原を発現させる系を確立させた(Kato, A. et al. 1996. Genes Cells 1:569-579)。マウスパラインフルエンザウイルス 1型であるSeVは非分節型ネガティブ鎖RNAゲノムを持つエンベロープウイルスの1つで、パラミクソウイルス科(Paramyxoviridae)に属す(Nagai, Y. 1999. Rev. Med. Virol. 9:83-99)。このウイルスはマウスに対しては致命的な呼吸器疾患を引き起こすが、非ヒト霊長類およびヒトには病原性がないとされている(Nagai, Y. 1999. Rev. Med. Virol. 9:83-99; Hurwitz, J.L. et al., Vaccine 15: 533-540, 1997)。
【0007】
SeVの複製は細胞質で行われ、核のフェーズは存在しないため、Gag、Pol、それにEnvなどのレンチウイルスの構造蛋白であっても、組み換えSeVを用いることにより、Rev非依存的に効率良く発現されることが期待される。より重要なことは、SeVベクターは非分裂細胞にも感染でき、外来遺伝子を強力に発現することである。例えば、組み換えSeVベクター(V(-)SeV)によるヒト免疫不全ウイルス type 1(HIV-1)の Env gp120 の発現量は培養上清において 6μg/ml(106 細胞あたり 6μg 相当)にも達するが、これは哺乳動物細胞で現在使用可能なベクター系の中では最も高い(Yu, D. et al. 1997. Genes Cells 2:457-466)。そこで本発明者らは、センダイウイルス(SeV)の系を用いて、この系がサル免疫不全ウイルス(SIV)のマカクサルへの感染に対して、防御的免疫を誘発できるかを検討し、組み換えSeVベクターをAIDSワクチンとして用いることの有効性を評価した。このために、まず、SeVのアクセサリー遺伝子の1つであるV遺伝子をノックアウトした変異SeVである V(-)SeV を構築した(Kato, A. et al. 1997. J. Virol. 71:7266-7272; Kato, A. et al. 1997. EMBO J. 16:578-587)。マウスにおいて、この V(-)SeV は劇的に弱毒化されるが、その遺伝子発現はかえって増強される。安全および効率の両面から、本発明者らはこの V(-) 型をベクターのバックボーンとして用いた。V(-)SeVを用いて、サル免疫不全ウイルスmac239株(SIVmac239)のGagを発現する組み換え V(-)SeV であるSeV/SIVgagを構築し、マカクサルにおいて抗SIV免疫を誘導する効力について試験し、マカクサルのAIDSモデル(Johnson, R.P. 1996. Curr. Opin. Immunol. 8:554-560; Almond, N.M., and Heeney, J.L. 1998. AIDS 12:133-140)でのワクチネーションによる防御効果を評価した。
【0008】
回収したSeV/SIVgagを用いて、このベクターを介したGag発現により誘導されるSIV Gag特異的な細胞性免疫応答のin vitro試験を行った。その結果、細胞傷害性Tリンパ球(CTL)アッセイにより、SeV/SIVgagを感染させた細胞がGag特異的CTLの標的として働くことが判明した。また、末梢血リンパ球の細胞培養では、SeV/SIVgag を感染させた細胞はGag特異的CTLの細胞集団の増殖を誘導した。動物実験では、4個体のカニクイザル(cynomolgus macaques)を用い、2個体はSeV/SIVgagの鼻腔内接種によりワクチネーションを行い、1個体はコントロールSeVを接種、残る1個体は非ワクチネーションコントロールとした。最初のワクチン接種から 22週後にこれらのマカクサルに100 TCID50 のSIVmac239 を静脈注射により暴露した。SIVに暴露させると、急性期の間は、全てのマカクサルにおいて血漿中のSIV量は同様に高値を示したが、SeV/SIVgagでワクチネーションを行ったマカクサルでは、その後のウイルス量はコントロールのマカクサルの 105 copies/ml 前後に比べはるかに低くなり、最終的に検出可能なレベル(100 copies/ml)を前後した。このように、SIVの構造蛋白質を発現するSeVでワクチネーションを行うことによって、SIV暴露後の慢性期(set-point)におけるウイルス量を顕著に減少させることが可能であることが示された。この結果は、センダイウイルスベクターは新しいAIDSワクチンとして有用であり、同時に細胞免疫応答の評価にも使用できることを示している。
【0009】
本発明の目的の1つは、組み換えセンダイウイルスベクターの系を用いることにより効率的なSIV Gagの発現が誘導されることを示し、この系をAIDSワクチンの開発に適用することの有効性を実証することにある。AIDSワクチンの研究に広く使用されているポックスウイルスと比べ、SeVベクターは哺乳動物細胞における細胞傷害性が低く、抗原の発現レベルは明らかに高い(Yu, D. et al., 1997, Genes Cells 2: 457-466)。また、実施例で用いられたSeVベクターの特徴の1つは、挿入遺伝子がSeVゲノムの3'端近くに収容されているため、その発現がベクターに由来する他のいずれのSeV特異的遺伝子よりも迅速で高率であることである(Nagai, Y., 1999, Rev. Med. Virol. 9: 83-99)。このことは、標的抗原に特異的な免疫応答の効率的な誘導に有利なはずである。
【0010】
CTLアッセイにおいて、SeV/SIVgagを感染させたB-LCLはSIV Gag特異的CTLの標的として良好に働いた。それに加え、SeV/SIVgagを感染させたPBMC(peripheral blood mononuclear cells; 末梢血単核細胞)と正常PBMCとの共培養はIFN-γの誘導およびSIV Gag特異的CTLの増殖をもたらした。これらの結果は、SeVの系が抗原特異的な細胞性免疫応答のアッセイに有用であることを示している。SeVの不活性なエンベロープ蛋白質の前駆体から活性型へのプロセッシングにはトリプシン様プロテアーゼが必要であり(Nagai, Y., 1993, Trends Microbiol. 1: 81-87)、外から加えられるこのプロテアーゼが存在しなければSeVは共培養細胞へ感染できないため、組み換えSeVベクターを用いることによって、固定やUV照射をすることなくCTL増殖のための共培養を容易に構築することが可能である。
【0011】
最近の報告が示すように、ウイルス特異的な細胞性免疫応答の誘導は、HIV-1感染に対する防御にとって有効となり得る(Geretti, A. M. et al., 1998, J. Gen. Virol. 79: 415-421; Matano, T. et al., 1998, J. Virol. 72: 164-169; Ogg, G. S. et al., 1998, Science 279: 2103-2106; Rowland-Jones, S. L. et al., 1998, J. Clin. Invest. 102: 1758-1765; Jin, X. et al., 1999, J. Exp. Med. 189: 991-998; Schmitz, J. E. et al., 1999, Science 283: 857-860)。SeV/SIVgagを感染させたPBMCはSIV Gag特異的CTLの増殖を誘導した。これは、この系が抗原特異的な細胞性免疫応答をin vivoで誘導することを示唆している。
【0012】
SIVmac239の暴露実験では、SeV/SIVgagでワクチネーションを行ったカニクイザルの血漿SIV量は、感染の急性期の間はコントロールに匹敵していた。しかしながら、その後SeV/SIVgagでワクチネーションを行ったカニクイザルは有意に低いSIV量を示した。特に目立ったのは、残存するウイルス量が検出可能なレベルまたはそれ以下であったことである。1つの抗原成分(Gag)を用いるだけで顕著な感染防御作用が得られたことも注目に値する。慢性期(set-point)の血漿ウイルス量の減少は、高頻度のSIV Gag特異的なT細胞前駆細胞の活性化によるものと考えられる。T細胞前駆細胞の頻度は、ウイルス感染の自然経過において最初のバースト量(initial burst size)に大きく依存している。外来抗原の産生に関してSeVが極めて高い性能を持っていることが、推定される高いT細胞前駆細胞の頻度に貢献し得るだろう。しかしながら、同じ組み換えウイルスを再度投与しても、追加的なGag特異的応答を引き起こすには不十分である可能性がある。従って、SeV/SIVgagで初回免疫を行い、追加免疫に異なるウイルスベクターまたはDNAワクチン等を用いるのが有効である。本明細書に示される実施例ではGag抗原のみを免疫に用いたが、組み換えSeVベクターを用いて複数の抗原を発現させれば、防御の効力をより向上させることができるかも知れない。
【0013】
SeVは自身の複製にエンベロープをプロセシングするプロテアーゼを必要とするため、複製のトロピズムは気道上皮などの特定に組織に限られる(Nagai, Y., 1993, Trends Microbiol. 1: 81-87)。気道を超えて他の組織に広がることが予想されないため、SeVベクターは例え複製型であっても安全面で利点を有することが示唆される。さらに、組み換えSeVベクターの鼻腔内投与は全身の粘膜免疫の誘導も期待できる。これはHIV-1感染に対する予防においてSeVベクターが有するもう1つの利点となり得る。
【0014】
また、本発明は、霊長類におけるSeV初期複製の解析を初めて開示する。ウイルスベクターをベースにした遺伝子導入の効率は宿主に依存するため、臨床研究に先だって霊長類でその効率、有効性、および安全性を評価することは必須である。霊長類においては、アフリカミドリザルで鼻腔内へのSeVの接種後の鼻腔拭い液試料の試験結果が報告されている(Hurwitz, J.L. et al. 1997. Vaccine 15:533-540)ものの、SeVの複製はこれまで詳細には解析されていなかった。また本発明者らは、SeV/SIVgag接種後1年以上経過したマカクサルの剖検で得られた鼻腔粘膜および肺を含む組織において、SeV/SIVgagの発現は検出されないことを見出した(データ省略)。そこで、本発明者らは、組み換えセンダイウイルスベクターでワクチネーションを行ったマカクサルにおける初期段階の抗原発現および細胞性免疫応答の解析を実施した。SIV Gagを発現する組み換えSeV(SeV/SIVgag )をマカクサルの鼻腔内に接種後、様々な組織におけるgag発現および細胞性免疫応答を調べた。SeVの複製は制御されており、Gagの発現は主に鼻腔粘膜およびその近傍リンパ節(lymph nodes; LN)に限定されていた。すなわち、gagの強い発現が鼻腔粘膜で観察され、それよりは低いが有意レベルのgagの発現が局所の咽頭後リンパ節(retropharyngeal LN)および顎下リンパ節(submandibular LN)で検出された。発現は免疫後1週間以内にピークに達し、少なくとも13日まで持続した。SeV/SIVgag は4日目(day 4)の鼻腔拭い液から確実に単離されたが、7日目(day 7)では頻度が低下し、13日目(day 13)では全く単離されなかった。これに対し、胸腺、脾臓、および鼠径LNなどの離れたリンパ組織では、検出可能な抗原の発現は見られなかった。本来の宿主であるマウスでは、SeVの全般的な感染は見られず、気道上皮に局在した急性の粘膜感染を示すが、サルでのSeV発現発現の組織の制限および発現の時間変化は、このマウスにおけるSeVの複製パターンと一致していた。しかしながら、抗原は気管や肺にはほとんど存在していないことから、マカクサルではウイルスの伝播はマウスの場合より制限されていることが示された。7日目(day 7)における細胞性免疫応答の解析では、SeV特異的 CD8+ T細胞の迅速な出現が示された。免疫後に明白な臨床症状を示すサルはなかった。マカクサル鼻腔内へのSeV/SIVgag の接種は、Gag特異的CD8+ T細胞の効率的な誘導をもたらした。Gag特異的CD8+ T細胞が、咽頭後LN中だけでなく末梢血単核細胞でも有意な頻度で検出されたことから、SeVが持つ抗原特異的な細胞性免疫応答の誘導能力は、霊長類においても効率的に働くことが示された。このように、免疫後に全身および局所の両方で、Gag特異的 CD8+ T細胞が著しく高頻度で検出されることが判明した。これらの結果から、組み換えSeVベクターをAIDSワクチンとして使用することの可能性がさらに強められた。
【0015】
上記のように、SeV/SIVgagでワクチネーションを行ったマカクサルにおいて、SeV/SIVgagは効率的に発現し、鼻腔粘膜およびその局所LNで強いことが示された。検出されるgag RNAはゲノムRNAとmRNAからなる可能性がある。そこで、以前確立した、SeV N mRNAだけを検出し、SeVゲノムの N RNAを検出しない系(Kato, A. et al. 2001. J. Virol. In press)を利用して、mRNAの発現を確認した。gag RNAレベルはSeV N mRNAレベルよりも高く、これは前者がmRNAに加えゲノムRNAも含んでいるからであろう。もう一つの説明としては、ゲノムにおいてgagの位置はNの位置よりも上流であるため、gag mRNAレベルはN mRNAレベルよりも高いことが期待される(Nagai, Y. 1999. Rev. Med. Virol. 9:83-99)。
【0016】
鼻腔内接種は、粘膜免疫応答を誘導するという利点も持っている。咽頭後LN(retropharyngeal LN)および顎下LN(submandibular LN)は鼻腔内からのリンパ球が最初に流入するLNである(Suen, J.Y., and Stern, S.J. 1996. Cancer of the Neck. In Cancer of the Head and Neck, 3rd ed. E. N. Myers and J. Y. Suen, editors. W. B. Saunders Company, Philadelphia. 462-484)。これらのLNは粘膜免疫応答に関与している可能性が高い。最近、マウスにおいて NALT(nasal-associated lymphoid tissue; 鼻関連リンパ系組織)が粘膜免疫応答に機能していることが示された(Yanagita, M. et al., 1999, J. Immunol. 162:3559-3565)。マウス NALT に対応する Waldeyer's ring から調製した細胞の解析により、この組織におけるSeV/SIVgagの発現および免疫応答を確認することができる。咽頭後LN(retropharyngeal LN)および顎下LN(submandibular LN)におけるSeV/SIVgagの発現の検出では、両方の組織で有意なレベルのgag RNAが検出された。これらのLNにはSeV蛋白のプロセッシングに必須のプロテアーゼが存在しないことから(Nagai, Y. 1993. Trends Microbiol. 1:81-87)、LN内でSeVの複製は起こらないと予想され、これらのLNのgag mRNAは、鼻腔内から流入したSeV/SIVgag感染リンパ球に由来するものであろう。SeV/SIVgagの鼻腔内接種により、鼻腔粘膜のみならず局所LNでも効率的な抗原の発現が見られたことから、SeVの粘膜免疫誘導能が示唆される(Gallichan, W.S., and Rosenthal, K.L. 1996. J. Exp. Med. 184:1879-90)。
【0017】
細胞性免疫応答は、HIV-1やSIVなどのヒトおよび非ヒト霊長類レンチウイルスの制御に重要な役割を果たしていることが示されている(Ogg, G.S. et al. 1998. Science 279:2103-2106; Rowland-Jones, S.L. et al. 1998. J. Clin. Invest. 102:1758-1765; Brander, C., and Walker, B.D. 1999. Curr. Opin. Immunol. 11:451-459; Seder, R.A., and Hill, A.V.S. 2000. Nature 406:793-798)。マカクサルのAIDSモデルにおいて、抗CD8抗体を用いた in vivo によるCD8+ T細胞欠損実験により、CD8+ T細胞が初期感染および慢性感染の両方の制御に重要であることが示されている(Matano, T. et al. 1998. J. Virol. 72:164-169; Schmitz, J.E. et al. 1999. Science 283:857-860; Jin, X. et al. 1999. J. Exp. Med. 189:991-998)。従って、ウイルス特異的なCD8+ T細胞応答の誘導はHIV-1感染に対する防御に非常に有効であり得る。フローサイトメトリーによる細胞内サイトカイン誘導の解析により検出された抗原特異的T細胞の頻度は、常に抗原特異的な細胞傷害活性と相関するわけではないものの、抗原特異的な細胞性免疫応答の指標と考えられている(Lavini, A. et al. 1997. J. Exp. Med. 186:859-865; Butz, E.A., and Bevan, M.J. 1998. Immunity 8:167-175; Murali-Krishna, K. et al. 1998. Immunity 8:177-187; Donahoe, S.M. et al. 2000. Virology 272:347-356; Appay, V. et al. 2000. J. Exp. Med. 192:63-75)。この方法を用いた解析により、SeV/SIVgagに感染した細胞が in vitro でGag特異的 CD8+ T細胞を刺激できることが示された。また、SeV/SIVgagでワクチネーションを行った3頭全てのマカクサルのPBMCで、高レベルの Gag特異的 CD8+ T細胞が検出された。咽頭後LNでも、Gag特異的 CD8+ T細胞の効率的な誘導が観察された。これらの結果は、SeV/SIVgagワクチネーションにより、Gag特異的な細胞性免疫応答が全身および局所の両方で誘導されることを示している。
【0018】
鼻腔内へのSeV/SIVgagのワクチネーション後の観察では、明白な臨床症状を示すサルはいなかった。SeV/SIVgagの発現はほとんど鼻腔内およびその局所LNに局在しており、発現レベルはワクチネーションから1週間以内にピークに達した。このような発現パターンは天然の宿主であるマウスにおけるSeVの性質に一致していた。しかしながら、気管および肺では抗原の発現がみられないことから、マカクサルにおいてはウイルスの伝播はより厳格に制限されていることが示される。抗原特異的IFN-γ誘導の解析において、SeV特異的CD8+ T細胞は1週目(week 1)で迅速に出現したことから、細胞性免疫応答がSeV複製を制御している可能性が示唆される。すなわち、マカクサルではSeV/SIVgagの複製はよく局在化されており、またよく制御されている。これらの結果は、本発明の系が霊長類においても安全であることを支持している。また、複製能を欠損したSeVを用いてより安全な系を構築することもできる。
【0019】
すなわち本発明は、組み換えSeVを介した免疫によるマカクサルの初期抗原発現および細胞性免疫応答を初めて開示する。本発明者らは、鼻腔内免疫したマカクサルの全てにおいて、効率的な抗原の発現のみならず、抗原特異的な細胞性免疫応答が効率的に誘導されることを示した。抗原の発現パターンは局在化され、またよく制御されており、霊長類におけるベクターの安全性が支持される。これらのことから、SeVの系はAIDSワクチンのためのツールとして有望であることがさらに裏付けられた。
【0020】
本発明は、免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチンを提供することを課題とする。本発明のワクチンは、AIDSワクチンとして、AIDSの予防および治療に極めて有用である。また本発明は、本発明のワクチンを投与することによるワクチネーションの方法を提供することを課題とする。より具体的には、本発明は、
(1)免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチン、
(2)ウイルス蛋白質がGag蛋白質またはその部分を含む、(1)に記載のワクチン、
(3)センダイウイルスがV遺伝子欠損型である、(1)または(2)に記載のワクチン、
(4)免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチンを接種する工程を含むワクチンネーションの方法、
(5)接種が鼻腔内投与である、(4)に記載の方法、
(6)複数回のワクチン接種において、免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチンの接種を少なくとも1回行う、(4)または(5)に記載の方法、
(7)(a)免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを抗原提示細胞に導入する工程、並びに(b)該導入細胞とTヘルパー細胞および細胞傷害性T細胞とを接触させる工程、を含む、免疫不全ウイルスのウイルス蛋白質特異的細胞性免疫応答を誘導する方法、に関する。
【0021】
本発明において「ワクチン」とは、伝染病の予防または治療のために使用される組成物を言う。ワクチンは抗原を含んでいるか、または抗原を発現可能であり、これにより抗原に対する免疫応答を誘導することができる。病原微生物の感染、伝播、および流行の予防または治療のために、本発明のセンダイウイルスベクターを含むワクチンは、所望の形態で用いることができる。
【0022】
「ワクチネーション(vaccination)」とはワクチンの接種により生物の体内または培養系に能動的に免疫(体液性免疫・細胞性免疫、または両者)を作らせることを意味する。これにより病原体の感染、増殖、伝播、および/または流行等を阻止することができる。また、病原体感染後の症状の発症および/または進展を抑制することができる。
【0023】
「抗原」とは、1つまたはそれ以上のエピトープを含む分子であり、宿主の免疫系を刺激して抗原特異的な免疫応答を誘導し得るものを言う。免疫応答は、体液性免疫応答および/または細胞性免疫応答であってよい。3個〜数個程度のアミノ酸でも1つのエピトープとなり得るが、通常、蛋白質中の1つのエピトープは、約7〜約15アミノ酸、例えば8、9、10、12、または14アミノ酸を含んでいる。抗原は免疫原とも言う。なお、抗原蛋白質をコードするポリヌクレオチドまたはベクターを用いて抗原を発現させる場合、本発明においては該ポリヌクレオチドおよびベクターも抗原と称し、これらはワクチン成分としても用いられ得る。
【0024】
「免疫応答」および「免疫学的応答」とは、抗原またはワクチンに対する体液性および/または細胞性免疫応答を言う。体液性免疫応答とは、抗体分子により媒介される免疫応答を言う。細胞性免疫応答とは、Tリンパ球および/または他の白血球により媒介される免疫応答を言う。細胞性免疫応答には、例えばCTL産生、あるいはヘルパーT細胞の産生または活性化等が含まれる。細胞性免疫応答は、例えばCD8+ T細胞などの活性化T細胞またはその他の白血球から産生されるサイトカインまたはケモカインを調べることにより検出することができる。また、公知のリンパ球増殖アッセイ、CTLアッセイ、または抗原特異的T細胞のアッセイ等により決定することもできる。
【0025】
「組み換え」体とは、組み換えポリヌクレオチドを介して生成した化合物または組成物を言う。組み換えポリヌクレオチドとは、自然の状態と同じようには結合していないポリヌクレオチドを言う。組み換え蛋白質は、組み換えポリヌクレオチドを発現させて得ることができる。また「組み換え」ウイルスベクターとは、遺伝子操作により組み換えポリヌクレオチドを介して構築されたウイルスベクターまたはそれを増幅して得られるウイルスベクターを言う。
【0026】
本発明において「パラミクソウイルス」とはパラミクソウイルス科(Paramyxoviridae)に属するウイルスを指す。パラミクソウイルスとしては、例えばセンダイウイルス(Sendai virus)、ニューカッスル病ウイルス(Newcastle disease virus)、おたふくかぜウイルス(Mumps virus)、麻疹ウイルス(Measles virus)、RSウイルス(Respiratory syncytial virus)、牛疫ウイルス(rinderpest virus)、ジステンパーウイルス(distemper virus)、サルパラインフルエンザウイルス(SV5)、ヒトパラインフルエンザウイルス1,2,3型等が挙げられる。センダイウイルスは、野生株、変異株、ラボ継代株、および人為的に構築された株などが含まれる。DI粒子(J. Virol. 68, 8413-8417(1994))等の不完全ウイルスや、合成したオリゴヌクレオチド等も、本発明のワクチンを製造するための材料として使用することができる。
【0027】
パラミクソウイルスのウイルスタンパク質をコードする遺伝子としては、NP、P、M、F、HN、およびL遺伝子が含まれる。「NP、P、M、F、HN、およびL遺伝子」とは、それぞれヌクレオキャプシド、ホスホ、マトリックス、フュージョン、ヘマグルチニン-ノイラミニダーゼ、およびラージ蛋白質をコードする遺伝子のことを指す。パラミクソウイルス亜科に属する各ウイルスにおける各遺伝子は、一般に次のように表記される。一般に、NP遺伝子は「N遺伝子」と表記されることもある。
【0028】
パラミクソウイルス属 NP P/C/V M F HN - L
ルブラウイルス属 NP P/V M F HN (SH) L
モービリウイルス属 NP P/C/V M F H - L
【0029】
例えばパラミクソウイルス科(Paramyxoviridae)のレスピロウイルス属(Respirovirus)に分類されるセンダイウイルスの各遺伝子の塩基配列のデータベースのアクセッション番号は、NP遺伝子については M29343、M30202, M30203, M30204, M51331, M55565, M69046, X17218、P遺伝子については M30202, M30203, M30204, M55565, M69046, X00583, X17007, X17008、M遺伝子については D11446, K02742, M30202, M30203, M30204, M69046, U31956, X00584, X53056、F遺伝子については D00152, D11446, D17334, D17335, M30202, M30203, M30204, M69046, X00152, X02131、HN遺伝子については D26475, M12397, M30202, M30203, M30204, M69046, X00586, X02808, X56131、L遺伝子については D00053, M30202, M30203, M30204, M69040, X00587, X58886を参照のこと。
【0030】
本発明において「遺伝子」とは遺伝物質を指し、RNAおよびDNA等の核酸が含まれる。遺伝子は天然由来または人為的に設計された配列を持つものであり得る。本発明で用いられるパラミクソウイルスベクターは、免疫不全ウイルスのウイルス蛋白質またはその部分をコードする外来遺伝子を有する。該外来遺伝子は、天然の免疫不全ウイルスが有する遺伝子またはその断片であってもよく、また、例えば、天然のウイルス蛋白質の欠失体、変異体、不活化蛋白質、他の蛋白質との融合蛋白質などをコードする核酸であってもよい。また、本発明において「DNA」とは、一本鎖DNAおよび二本鎖DNAを含む。
【0031】
本発明において免疫不全ウイルスとは、ヒトまたは動物に免疫不全症候群を引き起こすウイルスを言う。免疫不全症候群とは、免疫系を構成する細胞要素の一部あるいは幾つかの欠損あるいは機能不全によって、正常な免疫機構に障害を生じた病態を言う。免疫不全ウイルスによる病原性は、主にCD4陽性 T細胞を中心とする免疫担当細胞の破壊を伴う。免疫不全ウイルスとしては、特にレンチウイルス亜科に属するヒト免疫不全ウイルス(human immunodeficiency virus; HIV)およびサル免疫不全ウイルス(simian immunodeficiency virus; SIV)等が挙げられる。HIVはヒトの後天性免疫不全症候群(AIDS)を引き起こすウイルスである。HIVには、I型(HIV-1)および II型(HIV-2)が存在する。本発明において免疫不全ウイルスには、HIV-1およびHIV-2の全ての株およびサブタイプが含まれる。また本発明において免疫不全ウイルスとしては、SIVの全ての株およびサブタイプを含む。SIVの単離株には、SIVmac、SIVagm、および SIVsmなどを含む多数の株が知られている。その他、免疫不全ウイルスとしては、ネコ免疫不全ウイルス(feline immunodeficiency virus; FIV)等を例示できる。
【0032】
ウイルス蛋白質とは、ウイルスに含まれる蛋白質を言う。ウイルス蛋白質としては、構造蛋白質、調節蛋白質、およびアクセサリー蛋白質が含まれる。例えば、レンチウイルスの主要な構造蛋白質としては、例えば Gag、Pol、および Env が挙げられ、調節蛋白質としては、例えば Tat および Rev、アクセサリー蛋白質としては、例えば Vpu、Vpr、Vif、および Nef が挙げられる。本発明においては、これらの蛋白質のいずれか、その部分、あるいはそれらの組み合わせを発現するSeVベクターが好適に用いられる。
【0033】
本発明において naked(ネイキッド)DNAとは、蛋白質に包装されていない裸のDNAを言う。通常、naked DNAは精製されている。「精製された」とは、天然における状態よりも純度が高いことを言い、好ましくはそれが存在する試料中の成分として主要な割合を占めることを言う。DNAの精製は、フェノールおよび/またはクロロホルム抽出、エタノール沈澱、PEG沈澱(PEG/NaCl沈澱)などの他、電気泳動、塩化セシウム超遠心、逆相カラム、ゲル濾過、HPLC、シリカ吸着などの公知の方法を適宜組み合わせて行うことができる。naked DNA は直鎖状または環状であり得る。例えばプラスミド、ポリメラーゼ連鎖反応(PCR)産物、ウイルスや細胞等から抽出した精製または粗精製DNA等であってよい。naked DNAは緩衝剤、塩、脂質、蛋白質等と組み合わされた人工的な複合物にできる。例えば、カチオン性脂質等のトランスフェクション試薬と組み合わせた組成物としてもよい。DNAワクチンとは、naked DNAを成分として含むワクチンを言う。
【0034】
本発明において「センダイウイルスベクター」とは、センダイウイルスに由来し遺伝子を宿主細胞に導入するベクター(担体)を指す。センダイウイルスベクターはリボ核タンパク質(RNP)であってもよく、また、感染力を持つウイルス粒子であってもよい。ここで「感染力」とは、組み換えセンダイウイルスベクターが細胞への接着能および膜融合能を保持していることにより、接着した細胞の内部にベクター内部の遺伝子を導入することのできる能力を言う。本発明におけるセンダイウイルスベクターは、抗原となる免疫不全ウイルス蛋白質をコードする外来遺伝子を発現することができるように保持する。センダイウイルスベクターは野生型と同等の複製能力を有していてもよく、また遺伝子変異により弱毒化されていてもよい。また、本発明におけるセンダイウイルスベクターは、複製能を有さない欠損型ベクターであってもよい。「複製能を有する」とは、ウイルスベクターが宿主細胞に感染した場合、該細胞においてウイルスが複製され、感染性ウイルス粒子が産生されることを指す。
【0035】
本発明は、免疫不全ウイルスのウイルス蛋白質またはその一部を発現可能に保持するセンダイウイルスベクターを含むワクチンを提供する。センダイウイルスにコードされる免疫不全ウイルスのウイルス蛋白質としては、免疫原性を有する限り特に制限はない。免疫不全ウイルスのウイルス蛋白質には、該ウイルスの構造蛋白質、調節蛋白質、およびアクセサリー蛋白質が含まれる。例えば、HIV-1を含むレンチウイルスの主要な構造蛋白質としては、例えば Gag、Pol、および Env が挙げられ、調節蛋白質としては、例えば Tat および Rev、アクセサリー蛋白質としては、例えば Vpu、Vpr、Vif、および Nef が挙げられる。ワクチンの製造には、これらの蛋白質またはその部分ペプチド等を用いる。上記の蛋白質またはその部分を発現するセンダイウイルスベクターを構築することにより、ワクチンの製造を行うことができる。これらのタンパク質は単独で用いても、また複数を組み合わせて用いてもよい。本発明においては、特に免疫不全ウイルスの構造蛋白質を発現するSeVを用いることが好ましい。具体的には、Gag蛋白質全長、Gag-Pol融合蛋白質、またはそれらの断片などを発現するSeVを用いることができる。
【0036】
本発明者等により、鼻腔内接種でマカクサルに導入された組み換えSeVベクターからの遺伝子の発現は、接種後1週間以内にピークに達し、少なくとも13日まで持続することが示された。また、反復投与により発現を持続することも可能である。このことは、組み換えSeVベクターを利用してワクチネーションを行った場合に、早急かつ持続的な治療効果を得ることができるという利点をもたらす。
【0037】
また、安全性の面においても、SeVベクターはヒトへの臨床適用に好適に用いられうる可能性が示唆される。第一に、外来遺伝子の発現は、多くのベクター場合、導入したDNAの核局在化が必要であることが、遺伝子導入の成功率を低下させる主要な障害になっている。しかし、例えばSeVなどの場合、外来遺伝子の発現は、細胞質内において細胞性チューブリンおよび自身が持つRNAポリメラーゼ(L蛋白質)の両方によって駆動される。これは、SeVが宿主のゲノムと相互作用しないことを示しており、癌化などの安全面における問題が生じないと考えられる。第二に、SeVは齧歯類にとっては病原性で肺炎を生じることが知られているが、人には病原性ではない。これはまた、野生型SeVの経鼻的投与によって非ヒト霊長類において重篤な有害作用を示さないというこれまでの報告によっても支持されている(Hurwitz, J.L. et al., Vaccine 15: 533-540, 1997)。SeVのこれらの特徴は、SeVベクターが、ヒトへの応用に際して高い安全性を有することを示唆し、ワクチネーションのための抗原蛋白の発現を目的としたベクターの有望な選択肢の一つとなることを支持するものである。実際、本発明においてSeVを接種された霊長類マカクサルでは、抗原の有意な発現が検出された。またSeVベクター接種自体による明確な病理学的症状は見られず、末梢のCD4またはCD8細胞数の有意な減少も観察されなかった。
【0038】
本発明のワクチンは、特にAIDSウイルスを対象とするワクチネーションに好適に利用され得る。すなわち、本発明のワクチンを接種することにより、免疫不全ウイルスに対する免疫を誘導し、該ウイルスの感染および/または増殖を防止することが可能になる。本発明のワクチンは、免疫不全ウイルスの感染前に行う予防、および感染後に行う治療において好適に用いられる。
【0039】
本発明においてワクチンに用いられるSeVベクターとしては特に制限はない。好適なSeVベクターとして、例えば、複製能を有し、自立的に増殖するようなベクターが挙げられる。例えば、一般に天然型SeVのゲノムは、3'の短いリーダー領域に続き、N(ヌクレオキャプシド)、P(ホスホ)、M(マトリックス)、F(フュージョン)、HN(ヘマグルチニン-ノイラミニダーゼ)、およびL(ラージ)蛋白質をコードする6つの遺伝子が並んでおり、短い5'トレイラー領域を他端に有する。これと同様の構造を有するゲノムを設計することにより、自律複製可能なベクターを製造することができる。ゲノム内に外来遺伝子を挿入することにより、外来遺伝子を発現するベクターを製造することができる。なお、SeVベクターにおいては、SeV遺伝子の配置は野生型ウイルスから改変されていてもよい。
【0040】
また、本発明においてワクチンに用いられるSeVベクターとしては、野生型SeVが持つ遺伝子のいずれかを欠損したものであってもよい。例えば、SeVベクターを再構成し、遺伝子を発現させるためには、NP、P/CおよびL遺伝子から作られる蛋白質群が必要だと考えられているので、該蛋白質群をコードする遺伝子がSeVベクターに含まれている必要がある。しかし、M、F、および HN 蛋白質はトランスに供給してSeVベクターを再構成することができ、ベクターからの遺伝子発現が可能である。これらの蛋白質群をコードする遺伝子を有する発現ベクターを、SeVベクターゲノムをコードする発現ベクターと共に宿主細胞にトランスフェクションすることにより、ベクターの再構成を行うことができる。また、該蛋白質群をコードする遺伝子を有する宿主細胞にベクターゲノムをコードする発現ベクターを導入し、該宿主細胞から該蛋白質群を供給して再構成を行ってもよい。これらの蛋白質群のアミノ酸配列は、ウイルス由来の配列そのままでなくとも、核酸の導入における活性が天然型のそれと同等かそれ以上ならば、変異を導入したり、あるいは他のウイルスの相同遺伝子で代用してもよい。
【0041】
また、SeVベクターをRNPとして調製する場合は、SeVベクターが細胞に伝播してゆくために必要と考えられている M、FおよびHN遺伝子から作られる蛋白質は必要ない。RNPに含まれるゲノムに、M、FおよびHN遺伝子が含まれていれば、宿主に導入された時に、これらの遺伝子産物が生産され、感染性のあるウイルス粒子が形成される。感染性ウイルスを産生するRNPベクターとしては、例えば N、P、M、F、HN、およびL遺伝子をコードするウイルスゲノムRNAと、N蛋白質、P蛋白質、およびL蛋白質とを含むRNPが挙げられる。このようなRNPを細胞内に導入すると、N蛋白質、P蛋白質、およびL蛋白質の働きによりウイルスゲノムが発現、複製され、感染性ウイルスベクターが増幅する。
【0042】
RNPを細胞に導入するには、例えばリポフェクトアミンやポリカチオニックリポソームなどと共に複合体を形成させて導入することが可能である。具体的には、種々のトランスフェクション試薬が利用できる。例えば、DOTMA(Boehringer)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Boehringer #1811169)などが挙げられる。エンドソーム中での分解を防ぐため、クロロキンを加えることもできる(Calos, M.P., 1983, Proc. Natl. Acad. Sci. USA 80: 3015)。複製型ウイルスの場合、産生されたウイルスは、培養細胞、鶏卵、個体(例えばマウスなどの哺乳動物)などに再感染させて増幅または継代することができる。
【0043】
逆に、M、Fおよび/またはHN遺伝子が含まれていないSeVベクターも、本発明のワクチンに用いるSeVベクターとして好適である。このようなウイルスベクターの再構成は、例えば、欠損している遺伝子産物を外来的に供給することにより行うことができる。このようにして製造されたウイルスベクターは、野生型ウイルスと同様に宿主細胞に接着して細胞融合を起こすが、細胞に導入されたベクターゲノムはこれらのいずれかの遺伝子を欠損しているため、最初と同じような感染力を持つ娘ウイルス粒子は形成されない。このため、一回限りの遺伝子導入力を持つ安全なウイルスベクターとして有用である。ゲノムから欠損させる遺伝子としては、例えばF遺伝子および/またはHN遺伝子が挙げられる。例えば、F遺伝子が欠損した組み換えパラミクソウイルスベクターゲノムを発現するプラスミドを、F蛋白質の発現ベクターならびにNP、P/CおよびL蛋白質の発現ベクターと共に宿主細胞にトランスフェクションすることにより、ウイルスベクターの再構成を行うことができる(国際公開番号 WO00/70055 および WO00/70070)。また、例えば、F遺伝子が染色体に組み込まれた宿主細胞を用いて製造することもできる。これらの蛋白質群を外から供給する場合、そのアミノ酸配列はウイルス由来の配列そのままでなくとも、核酸の導入における活性が天然型のそれと同等かそれ以上ならば、変異を導入したり、あるいは他のウイルスの相同遺伝子で代用してもよい。
【0044】
また、ベクターゲノムが由来するウイルスのエンベロープ蛋白質とは異なる蛋白質をベクターのエンベロープに含むSeVベクターを作製することもできる。このようなタンパク質に特に制限はない。例えば、他のウイルスのエンベロープタンパク質、例えば水疱性口内炎ウイルス(VSV)のGタンパク質(VSV-G)を挙げることができる。本発明のワクチンを構成するSeVベクターとしては、VSV-Gタンパク質などのように、ゲノムが由来するウイルス以外のウイルスに由来するエンベロープタンパク質を含むシュードタイプウイルスベクターが含まれる。
【0045】
また、本発明のワクチンに用いられるSeVベクターは、例えば、エンベロープ表面に特定の細胞に接着しうるような接着因子、リガンド、受容体等の蛋白質、あるいはこれらの蛋白質を細胞外領域に有し、ウイルスエンベロープ由来のポリペプチドを細胞内領域に有するキメラタンパク質などを含むものであってもよい。これにより、特定の組織を標的とするベクターを作り出すこともできる。これらはウイルスゲノムにコードされていてもよいし、ウイルスベクターの再構成時に、ウイルスゲノム以外の遺伝子(例えば別の発現ベクターまたは宿主染色体などの遺伝子)の発現により供給されてもよい。
【0046】
また、本発明のワクチンに用いられるSeVベクターは、例えばSeV蛋白質に対する免疫原性を低下させるために、またはRNAの転写効率や複製効率を高めるために、ベクターに含まれるウイルス遺伝子が改変されたものであってもよい。具体的には、例えば複製因子であるNP遺伝子、P/C遺伝子およびL遺伝子の少なくとも一つを改変し、転写または複製の機能を高めることが考えられる。また、構造体蛋白質の1つであるHN蛋白質は、赤血球凝集素であるヘマグルチニン(hemagglutinin)活性とノイラミニダーゼ(neuraminidase)活性との両者の活性を有するが、例えば前者の活性を弱めることができれば、血液中でのウイルスの安定性を向上させることが可能であろうし、例えば後者の活性を改変することにより、感染能を調節することも可能である。また、膜融合に関わるF蛋白質を改変することにより、膜融合リポソームの融合能を調節することもできる。また、例えば、細胞表面の抗原分子となりうるF蛋白質やHN蛋白質の抗原提示エピトープ等を解析し、これを利用してこれらの蛋白質に関する抗原提示能を弱めたSeVを作製することもできる。
【0047】
また、本発明のワクチンに用いられるSeVは、アクセサリー遺伝子が欠損したものであってよい。例えばSeVのアクセサリー遺伝子の1つであるV遺伝子をノックアウトすることにより、培養細胞における遺伝子発現や複製は障害されることなく、マウスに対するSeVの病原性が顕著に減少する(Kato, A. et al. 1997. J. Virol. 71:7266-7272; Kato, A. et al. 1997. EMBO J. 16:578-587; Curran, J. et al., WO01/04272, EP1067179)。このような弱毒化ベクターは、本発明のワクチンを構成するベクターとして特に好適である。
【0048】
本発明のワクチンに用いられるウイルスベクターは、ゲノムRNA中に免疫不全ウイルスのウイルス蛋白質またはその部分をコードする。このような外来遺伝子を発現する組み換えSeVベクターは、上記のSeVベクターゲノムに外来遺伝子を挿入することによって得られる。外来遺伝子としては、免疫不全ウイルスのウイルス蛋白質またはその部分をコードする遺伝子断片を用いることができる。該遺伝子断片は、免疫不全ウイルス蛋白質をコードする天然由来の遺伝子断片であってもよく、また天然型蛋白質と少なくとも部分的に同等の抗原性を有する蛋白質をコードする限り、欠失、置換または挿入等により天然型蛋白質を改変した蛋白質をコードする遺伝子であってもよい。
【0049】
HIVを含む免疫不全ウイルスのウイルス蛋白質としては、構造蛋白質、調節蛋白質、およびアクセサリー蛋白質が含まれる。例えば、レンチウイルスの主要な構造蛋白質としては、例えば Gag、Pol、および Env が挙げられ、調節蛋白質としては、例えば Tat および Rev、アクセサリー蛋白質としては、例えば Vpu、Vpr、Vif、および Nef が挙げられる。本発明においては、これらの蛋白質、その部分、あるいはそれらの組み合わせを発現するSeVベクターが好適に用いられる。
【0050】
例えばHIV-1においては、Gagは p55 と呼ばれる約55kDの前駆蛋白質として発現され、pol遺伝子がコードするプロテアーゼにより切断され MA(matrix; マトリックス [p17])、CA(capsid, キャプシド [p24])、NC(nucleocapsid, ヌクレオキャプシド [p9])および p6 が生じる(Gottlinger, H. G. et al., Proc. Natl. Acad. Sci. USA 1989; 86: 5781-5785)。pol遺伝子は、ウイルスのプロテアーゼ(Pro)、インテグラーゼ(IN)、RNaseH、および逆転写酵素(RT)をコードし、まず Gag-Pol融合蛋白質として発現される(Jacks, T. et al., Nature 1988; 331: 280-283)。Gag-Pol前駆体(p160)は、リボソームによる翻訳時に約5%の頻度で生じるフレームシフトにより生成する。Polポリペプチドは、ウイルスがコードするプロテアーゼによりGagから切断され、さらにプロテアーゼ(p10)、RT(p50)、RNaseH(p15)、およびインテグラーゼ(p31)に分断される。また、例えばRT蛋白とRNaseHが繋がったポリペプチド(p65)などの、完全に分断されない断片も存在する。160kDのEnv(gp160)は、細胞のプロテアーゼによりgp41およびgp120に切断される。gp120はウイルス感染において、標的細胞表面に存在するレセプター CD4 およびコレセプターである CCR5などと相互作用する(Berger, E. A. et al., Annu. Rev. Immunol. 1999; 17: 657-700)。また、gp120には V1〜V5 と呼ばれるハイパーバリアブル領域があり、これらは単離株により比較的大きく異なっており、中でもV3 loopと呼ばれる領域はウイルスのトロピズムに影響する(Hwang, S. S. et al., Science 1991; 253: 71-74)。V3 loopは、HIV感染を防ぐための中和抗体主要なターゲットでもある(Goudsmit, J, et al., Proc. Natl. Acad. Sci. USA 1988; 85: 4478-4482)。
【0051】
Tatはウイルス複製に必須の転写のトランスアクチベーターであり、長さに違う複数のペプチドが発現される(Ruben, S. et al., J. Virol. 1989; 63: 1-8)。Revは約13kDの配列特異的なRNA結合蛋白質であり(Zapp, M. L. and Green, M. R., Nature 1989; 342: 714-716)、Rev応答配列(RRE)に結合しウイルス遺伝子発現のフェーズを制御する。これらの遺伝子に加え、HIV-1にはアクセサリー蛋白質をコードする nef、vif、vpr、および vpu の4つの遺伝子が存在する。HIV-2には、vpu の代わりに vpx を持つ。
【0052】
SeVベクターは、これらのウイルス蛋白質(プロセシングを受けた蛋白質および受けていない蛋白質を含む)のいずれかの全長またはその部分、あるいはそれらの組み合わせを発現するように構築される。部分としては、抗原となる活性を有する限り長さや位置等に制限はない。例えば、1つまたはそれ以上のエピトープを含む部分ペプチドであってよい。このような部分ペプチドは、通常、ウイルス蛋白質のアミノ酸配列中、連続した少なくとも3個〜数個程度のアミノ酸を含むペプチドである。好ましくは、ウイルス蛋白質のアミノ酸配列中、連続した約7〜約15アミノ酸、例えば8、9、10、12、または14アミノ酸を含んでいる。
【0053】
ワクチンは、免疫不全ウイルスのウイルス蛋白質のうち少なくとも1種類を用いる。本発明によれば、Gag抗原のみを発現させた場合でも、効率的な免疫応答を誘導することが可能であった。また、複数のタイプの蛋白質を抗原として用いれば、より高い免疫を獲得させることが可能となる。
【0054】
また、ワクチンは、免疫不全ウイルスのうち、1種類のウイルス由来のウイルス蛋白質を用いてもよいが、複数のタイプのウイルス由来のウイルス蛋白質を抗原として用いれば、より広い株またはサブタイプの免疫不全ウイルスに対する免疫を獲得させることができる。複数のタイプの免疫不全ウイルスを抗原として用いる場合、その組み合わせに特に制限はなく、例えば、HIV-1、HIV-2、またはSIVの各種単離株由来の遺伝子を用いてワクチンを製造することができる。複数の免疫不全ウイルスの遺伝子は、それぞれ別々のSeVベクターゲノムに組み込んでSeVを構築後、それらを組み合わせたり、混合したりしてワクチンを製造することもできれば、同じSeVベクターゲノムに複数の遺伝子を組み込んで発現させることもできる。
【0055】
例えば、HIV-1としては、全てのメジャー(M)サブタイプ(AからJを含む)、Nおよび outlier(O)が含まれる(Hu, D. J. et al., JAMA 1996; 275: 210-216; Zhu, T. et al., Nature 1998, 5; 391(6667): 594-7; Simon, F. et al., Nat. Med. 1998, 4(9): 1032-7)。SIV単離株としては、SIVagm、SIVcpz、SIVmac、SIVmnd、SIVsnm、SIVsyk等が例示できる。
【0056】
本発明のワクチンに用いられるウイルス蛋白質が由来する免疫不全ウイルスとしては、特に霊長類を宿主とする免疫不全ウイルスが好ましい。このようなウイルスとしては、例えばHIV-1、HIV-2、およびSIVが挙げられる。
【0057】
異なる株またはサブタイプのウイルスであっても、その蛋白質のアミノ酸配列が高い相同性を有していれば、ある株に属する1種の免疫不全ウイルスのウイルス蛋白質を抗原としたワクチネーションにより、異なる株またはサブタイプに対する免疫もある程度獲得できる可能性が高い。
【0058】
免疫不全ウイルスのウイルス蛋白質を発現するSeVを構築するには、例えば、SeVゲノムをコードするDNA(SeVベクターDNA)に目的の免疫不全ウイルスのウイルス蛋白質をコードする遺伝子を挿入する。ウイルスベクターDNAに外来遺伝子を導入する場合は、例えば、SeVベクターDNAにおいては、転写終結(E)配列と転写開始(S)配列との間などに、6の倍数の塩基数を有する配列を挿入することが望ましい(Journal of Virology, Vol.67, No.8, 1993, p.4822-4830)。外来遺伝子は、SeVの各遺伝子(NP、P、M、F、HN、およびL遺伝子)の前および/または後ろに挿入することができる。前後の遺伝子の発現を妨げないようにするため、外来遺伝子の前または後ろに適宜 E-I-S配列(転写終結配列−介在配列−転写開始配列)またはその部分を挿入し、各遺伝子の間にE-I-S配列を配置する。あるいは、IRESを挿入して外来遺伝子を発現し得る。
【0059】
挿入した外来性遺伝子の発現量は、外来遺伝子の上流に付加する転写開始配列の種類により調節することができる。また、遺伝子挿入の位置、および遺伝子の前後の塩基配列により調節しうる。例えば、SeVにおいては、挿入位置がウイルスゲノムのネガティブ鎖RNAの3'端に近いほど(野生型ウイルスのゲノム上の遺伝子配置においては、NP遺伝子に近いほど)、挿入された遺伝子の発現量が高い。外来遺伝子の高い発現を得るためには、外来遺伝子をNP遺伝子の上流(マイナス鎖においては3'側)またはNP遺伝子とP遺伝子の間など、ネガティブ鎖ゲノムにおいて上流領域に挿入することが好ましい。逆に、挿入位置がネガティブ鎖RNAの5'端に近いほど(野生型ウイルスのゲノム上の遺伝子配置においては、L遺伝子に近いほど)、挿入された遺伝子の発現量が低くなる。外来遺伝子の発現を低く抑えるためには、例えばネガティブ鎖の最も5'側、すなわち野生型ウイルスゲノムにおいてはL遺伝子の下流(ネガティブ鎖においてはL遺伝子の5'隣接部位)、またはL遺伝子の上流(ネガティブ鎖においてはL遺伝子の3'隣接部位)に外来遺伝子を挿入する。このように、外来遺伝子の挿入位置は、該遺伝子の所望の発現量を得るために、また前後のウイルスタンパク質をコードする遺伝子との組み合わせが最適となる様に適宜調節することができる。例えば、高力価ウイルスベクターの投与による導入遺伝子の高発現が毒性を示す場合は、投与するウイルス力価を制限することができる他、例えばベクターにおける外来遺伝子の挿入位置をネガティブ鎖のなるべく5'側に設定したり、転写開始配列を効率の低いものにするなどして、個々のSeVベクターからの発現レベルを低く抑えることで適切な効果が得られるすることも可能である。
【0060】
一般に、細胞毒性を示さない限りにおいて、抗原蛋白質の高い発現が得られれば、免疫獲得に有利と考えられるため、抗原蛋白質をコードする遺伝子は、効率の高い転写開始配列に連結し、ネガティブ鎖ゲノムの3'端近くに挿入することが好ましい。好適なベクターの例としては、免疫不全ウイルスのウイルス蛋白質が、パラミクソウイルスベクターのネガティブ鎖ゲノムにおいて、該パラミクソウイルスのウイルス蛋白質のいずれよりも3'側にコードされているベクターが挙げられる。例えばN遺伝子の上流(ネガティブ鎖の3'側)に抗原遺伝子が挿入されたベクターが好ましい。あるいは、N遺伝子のすぐ下流に挿入してもよい。
【0061】
外来遺伝子を容易に挿入できるようにするために、挿入部位にクローニングサイトを設計することができる。クローニングサイトは、例えば制限酵素の認識配列とすることができる。ゲノムをコードするベクターDNA中の当該制限酵素部位に外来遺伝子断片を挿入することができる。クローニングサイトは、複数の制限酵素認識配列を有する、いわゆるマルチクローニングサイトとしてもよい。また、本発明のワクチンに用いられるベクターは、このように免疫不全ウイルスのウイルス蛋白質をコードする遺伝子を挿入した以外に位置に他の外来遺伝子を保持していてもよい。このような外来遺伝子としては制限はなく、免疫誘導に関与するサイトカインまたはケモカイン遺伝子等であってもよく、また、その他の遺伝子であってもよい。
【0062】
外来遺伝子を有する組み換えセンダイウイルスベクターは、例えば、Hasan, M.K. et al. J. Gen. Virol. 78: 2813-2820, 1997 及び Yu, D. et al., 1997, Genes Cells 2: 457-466の記載に準じて、次のようにして構築することができる。
【0063】
まず、所望の外来遺伝子のcDNA塩基配列を含むDNA試料を用意する。DNA試料は、25ng/μl以上の濃度で電気泳動的に単一のプラスミドと確認できることが好ましい。以下、外来遺伝子を、NotI部位を利用してウイルスゲノムをコードするDNAに挿入する場合を例にとって説明する。挿入したいcDNA塩基配列の中にNotI認識部位が含まれる場合は、部位特異的変異挿入法などを用いて、コードするアミノ酸配列を変化させないように塩基配列を改変し、NotI部位を予め除去しておくことが好ましい。この試料から所望の遺伝子断片をPCRにより増幅回収する。増幅された断片の両端をNotI部位とし、さらに一端にセンダイウイルスの転写終結配列(E)、介在配列(I)及び転写開始配列(S)(EIS配列)のコピーを付加するために、NotI制限酵素切断部位配列及び転写終結配列(E)、介在配列(I)及び転写開始配列(S)と目的遺伝子の一部の配列を含むプライマー対として、フォワード側(センス鎖)合成DNA配列及びリバース側(アンチセンス鎖)合成DNA配列(プライマーセット)を作成する。
【0064】
例えば、フォワード側合成DNA配列は、NotIによる切断を保証するために 5'側に任意の2以上のヌクレオチド(好ましくはGCGおよびGCCなどのNotI認識部位由来の配列が含まれない4塩基、更に好ましくはACTT)を選択し、その3'側にNotI認識部位gcggccgcを付加し、さらにその3'側にスペーサー配列として任意の9塩基または9に6の倍数を加えた数の塩基を付加し、さらにその3'側に所望のcDNAの開始コドンATGからこれを含めてORFの約25塩基相当の配列を付加した形態とする。最後の塩基はGまたはCとなるように該所望のcDNAから約25塩基を選択してフォワード側合成オリゴDNAの3'の末端とすることが好ましい。
【0065】
リバース側合成DNA配列は5'側から任意の2以上のヌクレオチド(好ましくはGCGおよびGCCなどのNotI認識部位由来の配列が含まれない4塩基、更に好ましくはACTT)を選択し、その3'側にNotI認識部位gcggccgcを付加し、さらにその3'側に長さを調節するための挿入断片のオリゴDNAを付加する。このオリゴDNAの長さは、NotI認識部位gcggccgcを含め、cDNAの相補鎖塩基配列と後述するセンダイウイルスに由来するセンダイウイルスゲノムのEIS塩基配列の合計が6の倍数になるように塩基数を設計する(いわゆる「6のルール(rule of six)」; Kolakofski, D. et al., J. Virol. 72: 891-899, 1998; Calain, P. and Roux, L., J. Virol. 67: 4822-4830, 1993)。さらに挿入断片の3'側にセンダイウイルスのS配列の相補鎖配列、好ましくは5'-CTTTCACCCT-3'(配列番号:1)、I配列、好ましくは5'-AAG-3'、E配列の相補鎖配列、好ましくは5'-TTTTTCTTACTACGG-3'(配列番号:2)、さらにその3'側に所望のcDNA配列の終始コドンから逆に数えて約25塩基相当の相補鎖の最後の塩基がGまたはCになるように長さを選択して配列を付加し、リバース側合成オリゴDNAの3'の末端とする。
【0066】
PCRは、例えば、ExTaqポリメラーゼ(宝酒造)を用いる通常の方法を用いることができる。好ましくはVentポリメラーゼ(NEB)を用いて行い、増幅したDNA断片はNotIで消化した後、プラスミドベクターpBluescriptのNotI部位に挿入する。得られたPCR産物の塩基配列をシークエンサーで確認し、正しい配列のプラスミドを選択する。このプラスミドから挿入断片をNotIで切り出し、パラミクソウイルスゲノムcDNAを含むプラスミドのNotI部位にクローニングする。またプラスミドベクターpBluescriptを介さずにNotI部位に直接挿入し、組み換えセンダイウイルスcDNAを得ることも可能である。
【0067】
例えば、組み換えセンダイウイルスゲノムcDNAであれば、文献記載の方法に準じて構築することができる(Yu, D. et al., Genes Cells 2: 457-466, 1997; Hasan, M.K. et al. J. Gen. Virol. 78: 2813-2820, 1997)。例えば、まずNotI制限部位を有する18bpのスペーサー配列(5'-(G)-CGGCCGCAGATCTTCACG-3')(配列番号:3)を、クローニングされたセンダイウイルスゲノムcDNA(pSeV(+))のリーダー配列とN-タンパク質をコードする配列の5'末端との間の隣接遺伝子座に挿入し、デルタ肝炎ウイルスのアンチゲノム鎖(antigenomic strand)由来の自己開裂リボザイム部位を含むプラスミドpSeV18+b(+)を得る(Hasan, M. K. et al., 1997, J. General Virology 78: 2813-2820)。pSeV18+b(+)のNotI部位に外来遺伝子断片を挿入し、所望の外来遺伝子が組込まれた組み換えセンダイウイルスcDNAを得ることができる。
【0068】
このようにして作製した組み換えパラミクソウイルスベクターDNAを試験管内または細胞内で転写させ、ウイルスのL、P、およびNPタンパク質の共存下でRNPを再構成させると、このRNPを含むウイルスベクターを生成させることができる。本発明は、免疫不全ウイルスのウイルス蛋白質をコードするパラミクソウイルスベクターのゲノムDNAを転写させる工程を含む、該ベクターを含むワクチンの製造方法を提供する。また本発明は、該DNAからなる、本発明のワクチンの成分として用いるパラミクソウイルスベクター製造用DNAを提供する。また本発明は、本発明のワクチンの成分となるパラミクソウイルスベクターを製造するための、該ベクターのゲノムをコードするDNAの使用に関する。ウイルスベクターDNAからのウイルスの再構成は公知の方法に従って行うことができる(国際公開97/16539号; 国際公開97/16538号; Durbin, A.P. et al., 1997, Virology 235: 323-332; Whelan, S.P. et al., 1995, Proc. Natl. Acad. Sci. USA 92: 8388-8392; Schnell. M.J. et al., 1994, EMBO J. 13: 4195-4203; Radecke, F. et al., 1995, EMBO J. 14: 5773-5784; Lawson, N.D. et al., Proc. Natl. Acad. Sci. USA 92: 4477-4481; Garcin, D. et al., 1995, EMBO J. 14: 6087-6094; Kato, A. et al., 1996, Genes Cells 1: 569-579; Baron, M.D. and Barrett, T., 1997, J. Virol. 71: 1265-1271; Bridgen, A. and Elliott, R.M., 1996, Proc. Natl. Acad. Sci. USA 93: 15400-15404)。これらの方法により、パラインフルエンザ、水疱性口内炎ウイルス、狂犬病ウイルス、麻疹ウイルス、リンダーペストウイルス、センダイウイルスなどを含むパラミクソウイルスベクターをDNAからウイルスを再構成させることができる。ウイルスベクターDNAにおいて、F遺伝子、HN遺伝子、および/またはM遺伝子を欠失させた場合には、そのままでは感染性のウイルス粒子を形成しないが、宿主細胞に、これら欠失させた遺伝子および/または他のウイルスのエンベロープ蛋白質をコードする遺伝子などを別途、導入し発現させることにより、感染性のウイルス粒子を形成させることが可能である。
【0069】
例えば、ベクターDNAを細胞内に導入する方法には、次のような方法、マル1 目的の細胞が取り込めるようなDNA沈殿物を作る方法、マル2 目的の細胞による取りこみに適し、かつ細胞毒性の少ない陽電荷特性を持つDNAを含む複合体を作る方法、マル3 目的の細胞膜に、DNA分子が通り抜けられるだけに十分な穴を電気パルスによって瞬間的に開ける方法などがある。
【0070】
マル2 としては、種々のトランスフェクション試薬が利用できる。例えば、DOTMA(Boehringer)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Boehringer #1811169)などが挙げられる。マル1 としては例えばリン酸カルシウムを用いたトランスフェクション法が挙げられ、この方法によって細胞内に入ったDNAは貧食小胞に取り込まれるが、核内にも十分な量のDNAが入ることが知られている(Graham, F.L. and Van Der Eb, J., 1973, Virology 52: 456; Wigler, M. and Silverstein, S., 1977, Cell 11: 223)。ChenおよびOkayamaはトランスファー技術の最適化を検討し、1) 細胞を共沈殿物のインキュベーション条件を 2〜4% CO2 、35℃、15〜24時間、2) DNAは直鎖状より環状のものが活性が高く、3) 沈殿混液中のDNA濃度が 20〜30μg/mlのとき最適な沈殿が得られると報告している(Chen, C. and Okayama, H., 1987, Mol. Cell. Biol. 7: 2745)。マル2 の方法は、一過的なトランスフェクションに適している。古くはDEAE-デキストラン(Sigma #D-9885 M.W. 5×105 )混液を所望のDNA濃度比で調製し、トランスフェクションを行う方法が知られている。複合体の多くはエンドソームの中で分解されてしまうため、効果を高めるためにクロロキンを加えることもできる(Calos, M.P., 1983, Proc. Natl. Acad. Sci. USA 80: 3015)。マル3 の方法は電気穿孔法と呼ばれる方法で、細胞選択性がないという点でマル1 やマル2 の方法に比べて汎用性が高い。効率はパルス電流の持続時間、パルスの形、電界(電極間のギャップ、電圧)の強さ、バッファーの導電率、DNA濃度、細胞密度の最適条件下で良いとされている。
【0071】
以上、3つのカテゴリーの中でマル2 の方法は操作が簡便で多量の細胞を用いて多数の検体を検討することができるので、ベクター再構成のためのDNAの細胞への導入にはトランスフェクション試薬が適している。好適には Superfect Transfection Ragent(QIAGEN, Cat No. 301305)、または DOSPER Liposomal Transfection Reagent(Boehringer Mannheim, Cat No. 1811169)が用いられる。
【0072】
cDNAからの再構成は具体的には次のようにして行うことができる。
【0073】
24穴から6穴程度のプラスチックプレートまたは100mmペトリ皿上で、10%ウシ胎児血清(FCS)および抗生物質(100 units/ml ペニシリンGおよび100μg/ml ストレプトマイシン)を含む最少必須培地(MEM)を用いてサル腎臓由来細胞株LLC-MK2を70〜80%コンフルエントになるまで培養し、例えば 1μg/ml psoralen(ソラレン)存在下 UV照射処理を20分処理で不活化した、T7ポリメラーゼを発現する組み換えワクシニアウイルスvTF7-3(Fuerst, T.R. et al., Proc. Natl. Acad. Sci. USA 83: 8122-8126,1986、Kato, A. et al., Genes Cells 1: 569-579, 1996)を2 PFU/細胞で感染させる。ソラレンの添加量およびUV照射時間が適宜調整することができる。感染1時間後、2〜60μg、より好ましくは3〜5μgの上記の組み換えセンダイウイルスcDNAを、全長センダイウイルスゲノムの生成に必須なトランスに作用するウイルスタンパク質を発現するプラスミド(例えば24-0.5μgのpGEM-N、12-0.25μgのpGEM-P、および24-0.5μgのpGEM-L、より好ましくは例えば1μgのpGEM-N、0.5μgのpGEM-P、および1μgのpGEM-L)(Kato, A. et al., Genes Cells 1: 569-579,1996)と共にSuperfect(QIAGEN社)を用いたリポフェクション法等によりトランスフェクションする。トランスフェクションを行った細胞は、所望により100μg/mlのリファンピシン(Sigma)及びシトシンアラビノシド(AraC)、より好ましくは40μg/mlのシトシンアラビノシド(AraC)(Sigma)のみを含む血清不含のMEMで培養し、ワクシニアウイルスによる細胞毒性を最少にとどめ、ウイルスの回収率を最大にするように薬剤の最適濃度を設定する(Kato, A. et al., 1996, Genes Cells 1: 569-579)。トランスフェクションから48〜72時間程度培養後、細胞を回収し、凍結融解を3回繰り返して細胞を破砕した後、LLC-MK2細胞にトランスフェクションして培養する。培養3〜7日後に培養液を回収する。エンベロープ蛋白質をコードする遺伝子を欠損した複製能を持たないウイルスベクターを再構成させるには、エンベロープタンパク質を発現するLLC-MK2細胞をトランスフェクションに使用するか、またはエンベロープ発現プラスミドを共にトランスフェクションすればよい。また、トランスフェクションを行った細胞にエンベロープタンパク質を発現するLLC-MK2細胞に重層して培養することによって欠損型ウイルスベクターを増幅することもできる(国際公開番号 WO00/70055 および WO00/70070参照)。培養上清に含まれるウイルス力価は赤血球凝集活性(HA)を測定することにより決定することができる。HAは「endo-point 希釈法」(Kato, A. et al., 1996, Genes Cells 1: 569-579; Yonemitsu, Y. & Kaneda, Y., Hemaggulutinating virus of Japan-liposome-mediated gene delivery to vascular cells. Ed. by Baker AH. Molecular Biology of Vascular Diseases. Method in Molecular Medicine: Humana Press: pp. 295-306, 1999)により決定することができる。混入し得るワクシニアウイルスvTF7-3の除去は、得られた尿液試料を適宜希釈(例えば106 倍)して vTF7-3 を除去した後、ベクターウイルスを鶏卵で再増幅させることで可能である。再増幅は、例えば3回上繰り返すことができる。得られたウイルスストックは-80℃で保存することができる。
【0074】
ウイルスベクターが再構成する限り、再構成に用いる宿主細胞は特に制限されない。例えば、LLCMK2細胞、サル腎由来のCV-1細胞、ハムスター腎由来のBHK細胞などの培養細胞、ヒト由来細胞等を使うことができる。また、大量にセンダイウイルスベクターを得るために、上記の宿主から得られたウイルスベクターを発育鶏卵に感染させ、該ベクターを増幅することができる。鶏卵を使ったウイルスベクターの製造方法は既に開発されている(中西ら編,(1993),「神経科学研究の先端技術プロトコールIII, 分子神経細胞生理学」, 厚生社, 大阪, pp.153-172)。具体的には、例えば、受精卵を培養器に入れ9〜12日間 37〜38℃で培養し、胚を成長させる。ウイルスベクターを尿膜腔へ接種し、数日間卵を培養してウイルスベクターを増殖させる。培養期間等の条件は、使用する組み換えセンダイウイルスにより変わり得る。その後、ウイルスを含んだ尿液を回収する。尿液からのセンダイウイルスベクターの分離・精製は常法に従って行うことができる(田代眞人,「ウイルス実験プロトコール」, 永井、石浜監修, メジカルビュー社, pp.68-73,(1995))。
【0075】
例えば、F蛋白質を欠失したセンダイウイルスベクターの構築と調製は、以下のように行うことができる(国際公開番号 WO00/70055 および WO00/70070参照)。
<1> F欠失型センダイウイルスゲノムcDNAおよびF発現プラスミドの構築
【0076】
センダイウイルス(SeV)全長ゲノムcDNA、pSeV18+ b(+)(Hasan, M. K. et al., 1997, J. General Virology 78: 2813-2820)(「pSeV18+ b(+)」は「pSeV18+」ともいう)のcDNAをSphI/KpnIで消化してフラグメント(14673bp)を回収し、pUC18にクローニングしてプラスミドpUC18/KSとする。F欠損部位の構築はこのpUC18/KS上で行う。F遺伝子の欠損は、PCR-ライゲーション方法の組み合わせで行い、結果としてF遺伝子のORF(ATG-TGA=1698bp)を除いてatgcatgccggcagatga(配列番号:4)で連結し、F欠失型SeVゲノムcDNA(pSeV18+/ΔF)を構築する。PCRは、Fの上流には(forward: 5'-gttgagtactgcaagagc/配列番号:5, reverse: 5'-tttgccggcatgcatgtttcccaaggggagagttttgcaacc/配列番号:6)、F遺伝子の下流には(forward: 5'-atgcatgccggcagatga/配列番号:7, reverse: 5'-tgggtgaatgagagaatcagc/配列番号:8)のPCR産物をEcoT22Iで連結する。このように得られたプラスミドをSacIとSalIで消化して、F欠損部位を含む領域の断片(4931bp)を回収してpUC18にクローニングし、pUC18/dFSSとする。このpUC18/dFSSをDraIIIで消化して、断片を回収してpSeV18+のF遺伝子を含む領域のDraIII断片と置き換え、ライゲーションしてプラスミドpSeV18+/ΔF を得る。
【0077】
外来遺伝子は、pUC18/dFSSのF欠失部位にある制限酵素 NsiI および NgoMIV 部位に挿入する。このためには、例えば外来遺伝子断片を、NsiI-tailedプライマーおよびNgoMIV-tailedプライマーで増幅すればよい。
<2> SeV-F蛋白を誘導発現するヘルパー細胞の作製
【0078】
センダイウイルスのF遺伝子(SeV-F)を発現するCre/loxP誘導型発現プラスミドの構築のためには、SeV-F遺伝子をPCRで増幅し、Cre DNAリコンビナーゼにより遺伝子産物を誘導発現されるように設計されたプラスミドpCALNdlw(Araiら, J. Virology 72,1998, p1115-1121)のユニークサイト SwaI部位に挿入し、プラスミドpCALNdLw/Fを構築する。
【0079】
F欠損ゲノムから感染ウイルス粒子を回収するため、SeV-F蛋白を発現するヘルパー細胞株を樹立する。細胞は、例えばSeVの増殖によく用いられているサル腎臓由来細胞株LLC-MK2細胞を用いることができる。LLC-MK2細胞は、10%の熱処理した不動化ウシ胎児血清(FBS)、ペニシリンGナトリウム 50単位/ml、およびストレプトマイシン 50μg/mlを添加したMEMで37℃、5% CO2で培養する。SeV-F遺伝子産物は細胞傷害性を有するため、Cre DNAリコンビナーゼによりF遺伝子産物を誘導発現されるように設計された上記プラスミドpCALNdLw/Fを、リン酸カルシウム法(mammalian transfection kit (Stratagene))により、周知のプロトコールに従ってLLC-MK2細胞に遺伝子導入を行う。
【0080】
10cmプレートを用い、40%コンフルエントまで生育したLLC-MK2細胞に10μgのプラスミドpCALNdLw/Fを導入後、10mlの10% FBSを含むMEM培地にて、37℃の5%CO2 インキュベーター中で24時間培養する。24時間後に細胞をはがし、10ml培地に懸濁後、10cmシャーレ5枚を用い、5ml 1枚、2ml 2枚、0.2ml 2枚に蒔き、G418 (GIBCO-BRL)を1200μg/mlを含む10mlの10%FBSを含むMEM培地にて培養を行い、2日毎に培地交換しながら、14日間培養し、遺伝子の安定導入株の選択を行う。該培地により生育してきたG418に耐性を示す細胞はクローニングリングを用いて回収する。回収した各クローンは10cmプレートでコンフルエントになるまで拡大培養を続ける。
【0081】
F蛋白質の発現誘導は、細胞を6cmシャーレにてコンフルエントまで生育させた後、アデノウイルスAxCANCreを斉藤らの方法(Saito et al., Nucl. Acids Res. 23: 3816-3821 (1995); Arai, T.et al., J. Virol. 72,1115-1121 (1998))により moi=3 で感染させて行う。
<3> F欠失SeVウイルスの再構築及び増幅
【0082】
上記 pSeV18+/ΔF の外来遺伝子が挿入されたプラスミドを以下のようにしてLLC-MK2細胞にトランスフェクションする。LLC-MK2 細胞を5×106cells/dish で 100mm ペトリ皿に蒔き、24時間培養後、ソラレンと長波長紫外線(365nm)で 20 分間処理したT7 RNAポリメラーゼを発現するリコンビナントワクシニアウイルス(Fuerst, T.R. et al., Proc. Natl. Acad. Sci. USA 83, 8122-8126 (1986))に室温で1時間感染させる(moi=2〜3、好適にはmoi=2が用いられる)。ワクシニアウイルスへの紫外線照射には、例えば15ワットバルブを5本が装備された UV Stratalinker 2400(カタログ番号 400676 (100V), ストラタジーン社, La Jolla, CA, USA)を用いる。細胞を3回洗浄してからプラスミド pSeV18+/ΔF-GFP, pGEM/NP, pGEM/P, 及びpGEM/L(Kato, A. et al., Genes cells 1, 569-579 (1996))をそれぞれ12μg, 4μg, 2μg, 及び4μg /dish の量比でOptiMEM(GIBCO)に懸濁し、SuperFect transfection reagent(1μg DNA/5μl の SuperFect, QIAGEN)を入れて混合し、室温で10 分間放置後、最終的に3%FBSを含むOptiMEM 3mlに入れ、細胞に添加して培養する。3時間培養後、細胞を、血清を含まないMEM で2回洗浄し、シトシンβ-D-アラビノフラノシド40μg/ml (AraC, Sigma), トリプシン7.5μg/ml (GIBCO) を含むMEMで70時間培養する。これらの細胞を回収し、ペレットをOptiMEM に懸濁する(107 cells/ ml)。凍結融解を3回繰り返してlipofection reagent DOSPER(Boehringer mannheim)と混合し(106 cells/25μl DOSPER)室温で15分放置した後、上記でクローニングしたF発現ヘルパー細胞の一つ、例えばLLC-MK2/F7細胞にトランスフェクション(106cells /well 12-well-plate)し、血清を含まないMEM(40μg/ml AraC, 7.5μg/ml トリプシンを含む)で培養し、上清を回収する。混入し得るワクシニアウイルスは、得られた上清を希釈してLLC-MK2/F7細胞に感染させ、上清を回収する操作を数回繰り返すことで除去することができる。
【0083】
欠損型ウイルスベクターを調製する場合、例えば、ウイルスゲノム上で欠損しているエンベロープ遺伝子が異なる2種のベクターを同じ細胞に導入すれば、それぞれで欠損するエンベロープタンパク質が、もう一方のベクターからの発現により供給されるため、互いに相補しあって感染力のあるウイルス粒子が形成され、複製サイクルがまわりウイルスベクターが増幅される。すなわち、2種またはそれ以上のベクターを、エンベロープタンパク質を相補する組み合わせで接種すれば、それぞれのエンベロープ遺伝子欠損型ウイルスベクターの混合物を大量かつ低コストで生産することができる。これらのウイルスは、エンベロープ遺伝子が欠損しているため、エンベロープ遺伝子を欠損していないウイルスに比べゲノムサイズが小さくなり長い外来遺伝子を保持することができる。また、元々感染性のないこれらのウイルスは細胞外で希釈され共感染の維持が困難であることから、不稔化するため、環境放出管理上の利点がある。
【0084】
回収したパラミクソウイルスは実質的に純粋になるよう精製することができる。精製方法はフィルトレーション、遠心分離、およびカラム精製等を含む公知の精製・分離方法またはその組み合わせにより行うことができる。「実質的に純粋」とは、単離した物質(化合物、ポリペプチド、またはウイルス等)が、それが存在する試料中の成分として主要な割合を占めることを言う。典型的には、試料中に存在する実質的に純粋な成分は、試料中の他の成分を合わせた全体の50%以上、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上を占める。割合は当業者に公知の手順により、例えば重量比率[w/w]等で算出される。当然溶媒、塩、添加化合物などを除いて算出されるべきである。パラミクソウイルスの具体的な精製方法としては、例えばセルロース硫酸エステルまたは架橋ポリサッカライド硫酸エステルを用いる方法(特公昭62-30752号公報、特公昭62-33879号公報、および特公昭62-30753号公報)、およびフコース硫酸含有多糖および/またはその分解物に吸着させる方法(WO97/32010)等を例示することができる。
【0085】
回収されたSeVベクターは、生組み換えワクチン(live recombinant vaccine)として使うことができる。本発明において生ワクチンとは、ウイルスベクターを投与された個体の細胞中でベクターゲノムが増幅し、抗原蛋白質を発現させ免疫を獲得させる組成物を言う。実施例に示すように、SeVベクターを用いたワクチネーションはマカクサルにおいて免疫を効率よく誘導し、有意な臨床症状を示さないことから、生ワクチンとして好適に用いられる。このような生ワクチンを接種する対象に制限はなく、ヒト、サル、ネコ、イヌ、ブタ、ウマ、ウシ等免疫不全ウイルスが感染しうるあらゆる動物が含まれる。また、前述の伝播力が欠損したSeVベクターを用いれば、生ワクチンであってもベクターが伝播しないワクチンを製造することができる。
【0086】
また、発現蛋白質がSeV粒子に取りこまれる場合には、SeVベクターを不活化全粒子ワクチンとして利用することもできる。あるいは、SeVベクターを導入した細胞から、または発現蛋白質がSeV粒子に取りこまれる場合にはSeVベクターから、発現させた免疫不全ウイルス蛋白質を分離・精製してワクチンとすることもできる。SeVベクターには、限られた種類の蛋白質しか含まれないため、例えば発現ベクター等を用いて細胞内で発現させた免疫不全ウイルス蛋白質を細胞全抽出物から分離するのに比べ、格段に精製が容易である。蛋白質の精製には周知の分離技術を使用することができる。例えば、免疫不全ウイルス蛋白質に対する抗体を用いて、イムノアフィニティーカラム等により精製することも可能である。精製蛋白質をワクチンとして使うことで、生ワクチンや不活化ワクチンに比べ接種後の発熱や局所反応が起きる頻度が抑えられることが期待できる。
【0087】
SeVベクターを含むワクチンは、必要に応じて薬理学的に許容される所望の担体または媒体と組み合わせることができる。「薬学的に許容される担体」とは、ベクターと共に投与することが可能であり、ベクターによる遺伝子導入を有意に阻害しない材料である。例えばSeVベクターを生理食塩水やリン酸緩衝生理食塩水(PBS)などで適宜希釈して組成物とすることができる。SeVベクターを鶏卵で増殖させた場合等においては尿液を含んでよい。またSeVベクターを含むワクチン組成物は、脱イオン水、5%デキストロース水溶液等の媒体を含んでいてもよい。さらに、その他にも、植物油、懸濁剤、界面活性剤、安定剤、殺生物剤等が含有されていてもよい。また保存剤やその他の添加剤を添加することができる。また、免疫原性を高めるために、サイトカイン、コレラ毒素、サルモネラ毒素等の免疫促進剤を添加することもできる。またワクチンには、ミョウバン、不完全Freund'sアジュバント、MF59 (オイルエマルジョン)、MTP-PE (マイコバクテリア細胞壁由来の muramyl tripeptide)、および QS-21 (soapbark tree Quilaja saponaria 由来)などのアジュバントを組み合わせることもできる。
【0088】
本発明のワクチンを用いたワクチネーションは、免疫不全ウイルスの感染の防止および/または感染後のウイルスの排除またはウイルス増殖の抑制のために用いられ得る。また、免疫不全症候群発症の予防または発症後の治療のために用いられ得る。また、免疫不全ウイルスの感染モデル等における予防および/または治療方法の開発または評価においても有用である。
【0089】
本発明のワクチンは、有効量のベクターが対象組織の細胞に導入されるのに十分な量を投与される。「有効量」とは、所望の免疫応答を少なくとも部分的にもたらすように対象組織の細胞に遺伝子が導入される量を言う。所望の遺伝子を含むSeVベクターの有効量が投与されることにより、ベクターが導入された細胞から導入遺伝子産物が産生される。好ましくは、所望の導入遺伝子を有するSeVベクターの有効量が投与されることにより、投与された組織中または血中で有意なレベルの導入遺伝子の発現が検出される。「有意なレベル」とは、SeVベクターにより導入された遺伝子の発現(転写産物または翻訳産物の量)が検出可能であることを指す。但し、導入遺伝子の発現量は、その有効発現量レベルおよび中毒レベルを考慮して決められるべきである。
【0090】
細胞に導入された遺伝子の発現量は、当業者に公知の方法によりアッセイすることが可能である。遺伝子の転写産物は、例えば、ノーザンハイブリダイゼーション、RT-PCR、RNAプロテクションアッセイ等により検出・定量することができる。ノーザンハイブリダイゼーションやRT-PCR等による検出は in situ でも行い得る。また、翻訳産物を検出するには、抗体を用いたウェスタンブロット、免疫沈降、RIA、ELISA、プルダウンアッセイ等により行うことができる。また、導入遺伝子発現の検出を容易にするため、発現させる蛋白質にタグを付加したり、レポーター遺伝子を発現するように組み込んでおくことも可能である。レポーター遺伝子は、βガラクトシダーゼ、CAT、アルカリホスファターゼ、またはGFPをコードする遺伝子等が挙げられるがこれらに制限されない。
【0091】
免疫応答の検出は、抗体や免疫細胞の検出により行うことができる。例えば免疫不全ウイルスに対する体液性免疫応答のアッセイは、公知の種々のアッセイ方法に従って行うことが可能であり、例えばウイルス蛋白質への結合(ELISAまたはウェスタンブロッティング等によるアッセイ)、シンシチウム形成の阻害の検出、補体結合反応(complement fixation)、ADCC(antibody-dependent cell-mediated cytotoxicity; 抗体依存性の細胞を介した細胞傷害)能、感染または細胞融合に対する中和能、およびCD4とgp120との相互作用の阻害などを試験することによりアッセイすることができる。
【0092】
細胞性免疫応答の検出は、例えば抗原特異的CTL活性、CTL産生、あるいはヘルパーT細胞の産生または活性などを試験することによりアッセイすることができる。また、例えばCD8+ T細胞などの活性化T細胞またはその他の白血球から産生されるサイトカインまたはケモカインを調べることにより検出することができる。また、公知のリンパ球増殖アッセイ、CTLアッセイ、または抗原特異的T細胞のアッセイ等により決定することもできる。
【0093】
ベクターの投与量は、疾患、患者の体重、年齢、性別、症状、投与目的、ワクチンの形態や投与方法等により異なるが、当業者であれば適宜決定することが可能である。好ましくは、ワクチンに含有されるベクター量は約105 pfu/mlから約1011 pfu/mlの範囲内であるとよい。より好ましくは、ワクチンに含有されるベクターの量は約107 pfu/mlから約109 pfu/mlの範囲内であるとよい。最も好ましくは、約1×108 pfu/mlから約5×108 pfu/mlの範囲内の量を薬学上容認可能な担体中で投与することが好ましい。
【0094】
ワクチンの接種は、経皮内、経皮下、鼻腔内、経気管支、筋内、静脈内、または経口的に行われうる。例えば上気道の周辺、すなわち鼻腔内粘膜や上気道へのワクチン接種により粘膜免疫を誘導することができる。そのためには、SeVワクチンを鼻内噴霧等により気道へ接種することが有効と考えられる。鼻腔内投与は、例えばカテーテルを介した投与などにより行うこともできる。また、SeVを細胞に導入し、この細胞をワクチンとして接種することも可能である。例えばワクチン接種の対象個体由来の細胞にSeVを感染させ、ex vivo 投与によりワクチネーションを行うこともできる。
【0095】
また、ワクチンは単回投与(single dose administration)のみならず、例えばワクチンの2回接種または複数回接種により十分な免疫を誘導することも有効であると考えられる。ヒトの場合、複数回接種の間隔は通常2〜4週間である。
【0096】
複数回接種においては、SeVを含む本発明のワクチンを複数回投与することもできるが、SeVワクチンと他のワクチンとを組み合わせて用いることも好適である。上述のように、ウイルスベクターを基にしたワクチン戦略におけるデメリットの1つは、標的とすべき抗原よりもむしろ、ベクターウイルスに由来する抗原に対して強力な免疫応答が誘導されてしまうことである。この問題は、初回免疫と追加免疫のそれぞれにおいて、2つまたはそれ以上の異なる種類のウイルスベクターを用いることにより解決することができる。従って、先に述べたように、DNAワクチンを基にした初回免疫後にウイルスベクターを基にした追加免疫を行うことも、好ましい戦略の1つである。また、同じ組み換えウイルスを再度接種しても、追加的な抗原特異的応答を引き起こすには不十分である可能性がある。従って、まずSeVベクターによるワクチンで初回免疫を行い、追加免疫に異なるウイルスベクターまたはDNAワクチンを用いるのも有効である。また、組み換えSeVベクターを用いて複数の抗原を発現させれば、防御の効力をより向上させ得る。
【0097】
このように、異なる種類のワクチンを用いたプライム-ブーストプロトコルにおいてSeVベクターによるワクチンと組み合わされるワクチンに特に制限はなく、所望のワクチンを用いることができる。例えば、組み換えサブユニットワクチン、SeV以外のウイルスや微生物をベースにした生組み換えワクチン、全粒子の不活化免疫不全ウイルス、免疫不全ウイルスのシュードビリオンやウイルス様パーティクル、ペプチドワクチン、弱毒化免疫不全ウイルスの生ワクチン、DNAワクチンなどが挙げられるが、これらに制限されない。サブユニットワクチンとは、対象となる免疫不全ウイルス由来の抗原の全てを有さず、1つまたは複数の選択された蛋白抗原を含むワクチンを言う。このようなワクチンは、ウイルスの他の成分や感染細胞由来の成分から少なくとも部分的に切り離されている。サブユニットワクチンは免疫不全ウイルス蛋白質を少なくとも部分的に精製することにより調製することが可能である。また、組み換えによる生産または合成により作り出すことも可能である。生組み換えワクチンのベースとなるウイルスや微生物としては、例えばポックスウイルス、アデノウイルス、サルモネラ、ポリオウイルス、マイコバクテリア、インフルエンザウイルス、およびセムリキフォレストウイルスなどが挙げられるが、これらに限定されない。また、免疫不全ウイルスとして、env遺伝子の置換により複製能を限局させた限局的複製型ウイルス(Matano, T. et al. 2000. Vaccine 18:3310-3318)をワクチンとして用い、本発明のワクチンと組み合わせて追加免疫を行うことも可能である。SeVワクチンと、これら他のワクチンの接種の順番は特に制限はなく、SeVワクチンを接種した後に他のワクチンを接種することもできるし、逆に他のワクチンを接種した後にSeVワクチンを接種することもできる。
【0098】
一例を示せば、DNAワクチンによりプライミングした後、SeVワクチンによりブーストをかけることができる。このようなワクチネーションは、(a)DNAワクチンを投与する工程、およびその後(b)免疫不全ウイルスのウイルス蛋白質をコードするパラミクソウイルスベクターを投与する工程、を含む方法である。DNAワクチンとしては、例えば免疫不全ウイルスゲノムをコードするDNA等を用いることができる。DNAワクチンの接種は、例えば筋肉内投与および/またはジーンガンによる投与により行い得る。例えば、DNAワクチンの接種を数回行った後、本発明のSeVベースのワクチンを接種することができる。接種の間隔は通常、数日から数週間で行う。
【0099】
ワクチン接種が可能な動物としては、免疫系を有し、かつ免疫不全ウイルスに感染し得るあらゆる宿主が挙げられ、ヒト、サル、マウス、ラット、ウサギ、ヒツジ、ブタ、ウシ、ウマ、トリなどを含む、全ての哺乳動物等が含まれる。本発明のワクチンが接種される動物は、好ましくは霊長類があげられる。例えば、ヒト以外に本発明のワクチンの接種の対象となる霊長類(非ヒト霊長類)としてはレムール、ロリス、およびメガネザル等の原猿、広鼻猿類および狭鼻猿類を含む真猿、ならびにテナガザル、オランウータン、ゴリラ、チンパンジー、およびボノボなどを含む類人猿が挙げられる。狭鼻猿類の中では、特にオナガザル亜科、具体的にはニホンザル、カニクイザル、アカゲザル、ボンネットザル、ブタオザル、ベニガオザル、およびアッサムモンキー等を含むマカカ属などが挙げられる。非ヒト霊長類へのワクチン接種は、ヒトへの臨床適用に向けたAIDSワクチンの開発および評価に極めて有用である。
【0100】
免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターを含むワクチンは、宿主の免疫応答を局所および全身で誘導することができる。特に、該ベクターが導入された細胞は、抗原特異的な免疫応答のスティミュレーター(刺激細胞)として機能し、細胞性免疫応答を誘導する。本発明は、(a)免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを抗原提示細胞に導入する工程、(b)該導入細胞とTヘルパー細胞および細胞傷害性T細胞とを接触させる工程、を含む、免疫不全ウイルスのウイルス蛋白質特異的細胞性免疫応答を誘導する方法を提供する。ここで細胞を「接触させる」とは細胞の接触を許すことも含む。すなわち、例えばベクターを導入した細胞を血中に注入したり(体内のTヘルパー細胞および細胞傷害性T細胞と接触し得る)、あるいは同一培地中にベクター導入細胞とTヘルパー細胞および細胞傷害性T細胞とを共培養するなど、細胞が接触し得るようにすることであってよい。また、抗原特異的な「細胞性免疫応答の誘導」とは、該細胞性免疫応答の過程の少なくとも一部の誘導を意味する。例えば、抗原特異的CTLの刺激、該CTLの頻度上昇または活性(例えば細胞傷害性)の上昇などであってよい。
【0101】
抗原提示細胞とは、主要組織適合性複合体(MHC)class I または MHC class II 分子が提示されている細胞で、抗原蛋白のペプチドを各分子に結合できる能力を有する細胞を言う。抗原提示細胞としては、例えば樹状細胞(DC)が挙げられる。MHC class I 分子とは、抗原ペプチドに結合して細胞傷害性T細胞(CD8+)に提示する分子である。MHC class II分子とは、抗原ペプチドに結合してTヘルパー細胞(CD4+)に提示する分子である。Tヘルパー細胞とは、T細胞の系譜の一群であり、MHC class II分子により提示された抗原を認識し、免疫応答のカスケードを組織化する細胞である。細胞傷害性T細胞とは、T細胞の系譜の一群であり、MHC class I分子により提示された抗原を認識し、ウイルス感染細胞、癌、および移植片などの細胞を殺す細胞である(Xu M. et al., Trends Biotechnol. 18(4):167-72, 2000参照)。
【0102】
例えば末梢血単核細胞(PBMC)などに免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターを導入し、in vitro でPBMCと共培養することにより、IFM-γ産生の誘導および免疫不全ウイルスのウイルス蛋白質特異的CTLの増殖などの細胞性免疫応答を誘導することができる。また in vivo で投与すれば、宿主における抗原特異的な細胞性免疫応答を誘導することができる。
【0103】
細胞性免疫応答は、例えばIFN-γ量のアッセイやCD8+ IFN-γ+ T細胞の頻度測定などにより確認することができる。また、免疫不全ウイルスのウイルス蛋白質を発現させた細胞を標的として、標的細胞のリシスを測定することによってCTL活性をアッセイすることもできる。このような標的細胞は、上記SeVベクターを導入することにより作製することができる。例えば自家Herpesvirus papio不死化Bリンパ芽球細胞株(BLC)などに免疫不全ウイルスのウイルス蛋白質を発現するSeVを導入し、CTLを含むことが予想される試料と共にインキュベートして、該BLCのリシスを51Cr放出などを指標に測定することができる。また、不死化した細胞株 H9(ヒトT細胞由来)なども例示することができる。
【0104】
本発明は、免疫不全ウイルスのウイルス蛋白質特異的細胞性免疫応答を誘導または検出するための、免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターまたは該ベクターの導入細胞の使用に関する。また本発明は、免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターを導入した細胞を含む、免疫不全ウイルスのウイルス蛋白質特異的な細胞性免疫応答の刺激細胞に関する。さらに本発明は、免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターを導入した細胞を含む、免疫不全ウイルスのウイルス蛋白質特異的な細胞性免疫応答の標的細胞に関する。また本発明は、該刺激細胞または該標的細胞において免疫不全ウイルスのウイルス蛋白質を発現させるための、免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターの使用に関する。
【0105】
SeVベクターがコードする免疫不全ウイルスのウイルス蛋白質に制限はなく、上記と同等にウイルスの構造蛋白質、調節蛋白質、アクセサリー蛋白質等であってよい。構造蛋白質としては、Gag、Pol、Env等が挙げられる。例えば、免疫不全ウイルスのGag蛋白質をコードするSeVを用いて、Gag特異的細胞性免疫応答の誘導を行うことができる。
【図面の簡単な説明】
【0106】
【図1】図1は、SeV/SIVgagを用いたSIV Gag発現を示す図および写真である。(A) SeV/SIVgag アンチゲノムRNAを生成する構築プラスミドであるpSeV(+)18bV(-)/SIVgagの構造。(B) モノクローナルマウス抗p27抗体を用いたウェスタンブロット解析。偽感染(レーン1)、SeV/control感染(レーン2)、またはSeV/SIVgag感染(レーン3)の24後にCV1細胞を溶解した。(C) 抗p27抗体およびFITC結合抗マウスIgG抗体を用いた免疫染色。偽トランスフェクト(a)、またはSHIVMD14YE DNA(Shibata, R. et al. 1997. J. Infect. Dis. 176:362-373)をトランスフェクト(b)、あるいはSeV/control(c)またはSeV/SIVgag(d)を感染(MOI=0.05)させたCV1細胞。(D) トリプシン(7.5μg/ml)存在下の多サイクル増殖条件(Sakai, Y. et al. 1999. FEBS Lett. 456: 221-226)でのCV1細胞におけるSeV/controlおよびSeV/SIVgagの複製カイネティクス。初期感染において、細胞にSeV/controlまたはSeV/SIVgagを、m.o.iを0.05で感染させた。
【図2】図2は、SeV/SIVgagを介したGag発現により誘導されるSIV Gag特異的細胞性免疫応答を示す図である。(A) VV/SIVgag(左パネル, 黒四角)またはSeV/SIVgag(右パネル, 黒丸)を感染させたB細胞をSIV Gag特異的リシスの標的として用いたCTLアッセイ。VV/WR(左パネル, 白四角)またはSeV/control(右パネル, 白丸)を感染させたB細胞を非特異的リシスの測定のため用いた。SIV感染の3週目に新たに調製した Rh018 PBMC をエフェクターとして用いた。(B) SeV/SIVgag感染細胞と共培養したPBMCにおけるIFN-γ産生。12週目に調製した Rh018 PBMC を、SeV/control感染細胞による非特異的刺激共培養(左レーン)またはSeV/SIVgag感染細胞によるGag特異的刺激共培養(右レーン)に用いた。それらの培養上清中のIFN-γ濃度をELISAでアッセイした。(C) SeV/SIVgag感染細胞を用いたPBMC共培養におけるSIV Gag特異的CTLの増殖。上記の非特異的(左レーン)またはGag特異的(右レーン)刺激共培養中のPBMCをエフェクターとして用いた。VV/SIVgag感染B細胞をGag特異的リシス(黒四角)の標的として、またVV/WR感染B細胞を非特異的リシス(白四角)の標的として用いた。
【図3】図3は、ワクチネーション後の免疫応答を示す図である。矢印は初回、2回目、および3回目のワクチネーションの時点を示す。(A) 末梢血中のCD4陽性(黒丸)またはCD8陽性(白四角)リンパ球数(cells/μl)。(B) 抗SeV(黒丸)または抗p27(白四角)抗体ELISAにおける405nmの光学密度(OD)。
【図4】図4は、SIV暴露後の血漿SIV RNAコピー数(copies/ml)を示す図である。カニクイザル鼻腔内に 108 CIUのSeVベクターによる3回のワクチネーションを行った。最初のワクチネーション後22週目に、それらのカニクイザルに100 TCID50 の SIVmac239 を静脈注射により暴露させた。
【図5】図5は、グループ I カニクイザルのSeV/SIVgagRNA発現を示す図である。(A) 鼻腔粘膜から調製した細胞のSIV gag RNAレベル。(B) 鼻腔粘膜から調製した細胞のSeV N mRNAレベル。(C) 咽頭後LNから調製した細胞のSIV gag RNAレベル。(D) 顎下LNから調製した細胞のSIV gag RNAレベル。4日目(day 4)における C3880 または C4325 のレベルは未決定。
【図6】図6は、SeV/SIVgagを感染させた細胞で刺激後の細胞内IFN-γ誘導のフローサイトメトリーによる検出を示す図である。SIVを感染させたアカゲザルの PBMC を、それぞれ SeV/control (A)、SeV/SIVgag (B)、および Vv-Gag (C) を感染させた自家BLCと共に共培養した。CD3+CD8+ リンパ球をゲーティングしたドットプロットを示す。各ドットプロットにおいて、ゲーティングされた細胞数に対する、四角で囲んだ細胞数の割合(%IFN-γ+)を示した。
【図7】図7は、ワクチネーションを行ったアカゲザルにおける Gag特異的 CD8+IFN-γ+ T細胞を頻度を示す図である。(A) グループ II アカゲザルの3週目(week 3)の PBMC を、Vv-control(白いボックス)または Vv-Gag(黒いボックス)を感染させたBLCと共培養し、フローサイトメトリーによる解析を行った。ND, 未決定。(B) 2週目(week 2)のグループ I-C アカゲザルの咽頭後LN由来の細胞を、SIV Gag p27 の非存在下(白いボックス)または存在下(黒いボックス)でインキュベートし、フローサイトメトリーによる解析を行った。
【図8】図8は、ワクチネーションを行ったアカゲザルにおける SeV特異的 CD8+IFN-γ+ T細胞の頻度を示す図である。グループ II アカゲザルの1週目(week 1)(A) または 3週目(week 3)(B) の PBMC を、Vv-control(白いボックス)または SeV/control(黒いボックス)を感染させたBLCと共培養し、フローサイトメトリーによる解析を行った。ND, 未決定。
【図9】図9は、ワクチネーションを行ったアカゲザルにおける血漿中の抗SeV抗体レベルのELISAによる決定を示す図である。1:1000 に希釈した血漿で得られた吸収のカイネティクスを示す。OD, 光学密度。
【図10】図10は、DNAワクチン + SeV/SIVgagワクチンによるSIV Gag特異的CTLの誘導を示す図である。SIV Gag特異的 51Cr-放出(%)を示す。SeV/SIVgag投与(week 12)による追加免疫(booster)の効果を示している。12週目と14週目とを比較のこと。
【図11】図11は、DNAワクチン + SeV/SIVgagワクチンによる病原性SHIV暴露(challenge)に対する感染防御を示す図である。末梢血CD4 T細胞のcountの変化を示す。control群4頭全てにおいて、challenge後2週目でCD4-depletionが認められたが、DNAワクチン + SeV/SIVgagワクチン投与群では、4頭全てにおいてCD4-depletionから保護された。
【図12】図12は、DNAワクチン + SeV/SIVgagワクチンによる病原性SHIV暴露に対する感染防御を示す図である。血漿中のSHIV RNAコピー数の変化を示す。コントロール群と比較して、DNAワクチン + SeV/SIVgagワクチン投与群のSHIV RNA コピー数は、感染急性期より有意に低値を示した。
【発明を実施するための形態】
【0107】
以下実施例により本発明を具体的に説明するが、本発明はこれらの実施例に限定されるものではない。なお、本明細書全体を通じて引用された文献の内容はすべて、本明細書に組み込まれる。
【実施例】
【0108】
[実施例1] SeV/SIVgagの回収
安全面でより優れているVノックアウト型のSeV(V[-]SeV)(Kato, A. et al. 1997. J. Virol. 71:7266-7272; Kato, A. et al. 1997. EMBO J. 16:578-587)を使用してSIV Gagを発現する組み換えSeVであるSeV/SIVgagを構築した。SeVのアクセサリー遺伝子の1つであるV遺伝子を組み換えによりノックアウトすると、培養細胞におけるウイルス遺伝子の発現や複製は障害されることなく、マウスに対するSeVの病原性が顕著に弱毒化される。弱毒化V欠損SeVゲノムcDNAの全長を含むプラスミド pSeV(+)18bV(-) は以前記載されている(Kato, A. et al. 1996. Genes Cells 1:569-579; Hasan, M.K. et al. 1997. J. Gen. Virol. 78:2813-2820)。SIVmac239 Gag(ヌクレオチド 1306−2845 [GenBank accession number: M33262](Kestler, H. et al. 1990. Science 248:1109-1112))をコードする遺伝子断片はPCR増幅により調製し、pSeV(+)18bV(-) に導入して pSeV(+)18bV(-)/SIVgag を得た。PCRで用いたプライマーは 5'-AAG CGG CCG CGA GAT GGG CGT GAG AAA CTC CG-3'(配列番号:9)および 5'-TTG CGG CCG CGA TGA ACT TTC ACC CTA AGT TTT TCT TAC TGT GAC TAC TGG TCT CCT CCA AAG-3'(配列番号:10)である。gag遺伝子断片は、pSeV(+)18bV(-)のNのコード領域の直ぐ上流にあるNotI部位に挿入した(図1A)。このプラスミド pSeV(+)18bV(-)/SIVgag は T7 RNA ポリメラーゼの存在下でSeV/SIVgagアンチゲノムRNAの全長を生じる。組み換えSeV, SeV/SIVgag, は、pSeV(+)18bV(-)/SIVgagから以前記載したようにして回収した(Kato, A. et al. 1996. Genes Cells 1:569-579)。具体的には、T7 RNAポリメラーゼを発現する組み換えワクシニアウイルス(VV)ベクターであるvTF7-3(Fuerst, T. R. et al., 1986, Proc. Natl. Acad. Sci. USA 83: 8122-8126)をLLCMK2細胞に感染させ、pSeV(+)18bV(-)/SIVgag、pGEM-N、pGEM-P、および pGEM-L(Garcin, D. et al., 1995, EMBO J. 14: 6087-6094)をコトランスフェクトした。トランスフェクションから40時間後に、細胞を回収し鶏卵の尿膜腔に注入した。2回継代後、尿液を回収しSeV/SIVgagとした。同様にpSeV(+)18bV(-)からコントロールSeVであるSeV/controlを得た。SeVタイター(CIU [cell infectious unit] /ml)は、以前記載されたように(Kiyotani, K. et al. 1990. Virology 177:65-74)、CV1細胞使って抗SeV抗体を用いた免疫染色により測定した。
【0109】
[実施例2] SeV/SIVgagを用いた in vitro でのSIV Gag発現
プラスミド pSeV(+)18bV(-)/SIVgag(図1A)から、標準的なトランスフェクションプロトコルに従って感染性SeV/SIVgagをレスキューさせた(実施例1参照)。このSeV/SIVgagを細胞に感染させ、発現した蛋白質の解析を以下のように行った。細胞ライセートを回収するため、6-wellプレートの各ウェルに 4×105 細胞の密度でCV-1細胞を撒き、一晩培養後、SeV/controlまたはSeV/SIVgagを m.o.i.を 5 で感染させた。1日後、細胞を600μlのリシスバッファー(50 mM Tris-HCl, pH8.0, 150 mM NaCl, 0.02% sodium azide, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 0.1 mg/ml phenylmethylsulfonyl fluoride, 1% Triton X-100)中でリシスさせた。各レーンに10μlの細胞ライセートをロードした。ウェスタンブロット解析はモノクローナルマウス抗p27抗体を用いて以前の記載に従って行った(Matano, T. et al. 1993. J. Virol. 67:2026-2033)。免疫染色は抗p27抗体およびフルオレセイン結合ヤギ抗マウスイムノグロブリンG(IgG)抗体を用いて記載されたようにして行った(Matano, T. et al. 1993. J. Virol. 67:2026-2033)。SeV/SIVgagを感染させたCV1細胞において、プロセッシングを受けていないSIV Gagであるp55の発現が、抗SIV Gag p27モノクローナル抗体を用いたウェスタンブロッティングおよび免疫染色の両者において確認された(図1Bおよび1C)。組み換えウイルスは、SeV/controlに比べ僅かに遅い増殖カイネティクスを示した(図1D)。この増殖の遅延はゲノム長の増加による可能性がある(SeV/control, 15.4kb; SeV/SIVgag, 17.0kb)(Yu, D. et al. 1997. Genes Cells 2:457-466; Hasan, M.K. et al. 1997. J. Gen. Virol. 78:2813-2820; Sakai, Y. et al. 1999. FEBS Lett. 456: 221-226)。
【0110】
[実施例3] SeV/SIVgagによる in vitro でのSIV Gag特異的細胞応答の誘導
SeV/SIVgagを介したGag発現によりSIV Gag特異的応答が誘導されるかについて、1頭のアカゲザル(Macaca mulatta)から調製したPBMCを用いて in vitro で細胞性免疫応答の試験を行った。このアカゲザルは別の実験において前もってSIV抗原を発現するnaked DNA(Gagを含むSIV抗原を発現するプラスミドDNA)でワクチネーションを行い、SIVmac239を暴露した。SIVmac239暴露用ストックは以前に記載に従ってアカゲザルPBMCを用いて調製した(Kestler, H. et al., 1990, Science 248: 1109-1112; Shibata, R. et al., 1997, J. Infect. Dis. 176: 362-373)。ストックの感染力価はMT-4細胞でアッセイした。なお、このアカゲザルは実験に用いる前にSeV、SIV、およびサルtype Dレトロウイルスの陰性試験を行い、実験動物に関する研究所のガイダンスに従って維持した。採血、ワクチネーション、およびウイルス接種はケタミン麻酔下で行った。
【0111】
SIV暴露後3週目(week 3)にこのアカゲザルからPBMCを新たに調製し、このCTLアッセイのエフェクターとして用いた。末梢血単核細胞(PBMC)は全血試料から以前の記載に従って調製した(Shibata, R. et al., 1997, J. Infect. Dis. 176: 362-373)。PBMCは10%ウシ胎児血清(Hyclone)を含むRPMI1640(Life Technologies)で培養した。PBMCにSeV/controlまたはSeV/SIVgagをMOI 10で感染させ、2時間インキュベート後、共培養に用いた。SIV Gag特異的刺激を行う共培養では、U-bottomの96ウェルプレートにおいて、1ウェル当たり 5×105 PBMC と 1×105 SeV/SIVgag感染PBMCとを混ぜ合わせた。非特異的刺激を行う共培養では、5×105 PBMC と 1×105 SeV/control感染PBMCとを混ぜ合わせた。共培養を3日行った後、培養上清を回収し、細胞は10 unit/mlの組み換えヒトインターロイキン-2(Boehringer Mannheim)を含む培地中で培養した。上清中のインターフェロン-γ(IFN-γ)濃度は酵素結合イムノソルベントアッセイ(ELISA)(Biosource)で試験した。このアッセイの検出下限は 15pg/mlである。SIV Gag特異的刺激を行った共培養におけるPBMCを細胞傷害性Tリンパ球(CTL)アッセイにおけるエフェクター細胞として用いた。あるいは、別途調製したばかりのPBMCをエフェクターとして用いた。CTLアッセイ(51Cr-放出アッセイ)は以前の記載に従って行った(Voss, Q. et al., 1992, J. Virol. Methods 39: 185-195; Voss, Q. et al., 1995, Virology 208: 770-775)。SIV Gagを発現する組み換えワクシニアウイルス(VV)である VV/SIVgag を感染させた自家Herpesvirus papio不死化Bリンパ芽球細胞株(B-LCL)をSIV Gag特異的リシスの標的として用いた。あるいは、SeV/SIVgagを感染させたB-LCLを用いた。コントロールVV(VV/WR)または SeV/control を感染させた B-LCL を非特異的リシスの測定のために用いた。51Cr-放出の割合を次のようにして計算した: %51Cr放出 = (試験放出 - 自発的放出) / (最大放出 - 自発的放出)×100。その後、Gag特異的51Cr放出の割合を、Gag特異的リシスにおける %51Cr放出から非特異的リシスにおける放出を差し引くことによって算出した。Gag特異的 51Cr放出が 10%を超えた場合、Gag特異的CTL活性を陽性とした。
【0112】
SeV/SIVgagまたはVV/SIVgagを感染させた自家B-LCLを標的として用いた。図2Aに示したように、このアッセイでGag特異的リシスがSeV/SIVgagを感染させたB-LCLおよびVV/SIVgagを感染させたB-LCLの両細胞で観察されたことから、SeV/SIVgagを感染させたB-LCLはSIV Gag特異的CTLの標的として働くことが示された。
【0113】
次に、SeV/SIVgagを感染させた細胞がSIV Gag特異的CTLの増殖を誘導するかを調べた。12週(week 12)のアカゲザルRh018から調製したPBMCをSeV/SIVgagに感染させたPBMCと共に共培養すると、Th1サイトカインの一種である IFN-γの産生が観察された(図2B)。これとは対照的に、SeV/controlを感染させたPBMCとのPBMC共培養では検出可能なIFN-γは誘導されなかった。前者(SeV/SIVgagに感染させたPBMCとの共培養)においてはSIV Gag特異的CTL活性が見られたが、後者(SeV/controlを感染させたPBMCとの共培養)の対照共培養では見られなかった(図2C)ことから、SeV/SIVgag感染細胞によるGag特異的CTLの増殖が示された。
【0114】
[実施例4] カニクイザルにおけるSeV/SIVgagのワクチネーション
表1に概要を示したプロトコルに従って、4頭のカニクイザル(Macaca fascicularis)を用いてワクチネーション実験を行った。4頭のカニクイザルは実験に用いる前にSeV、SIV、およびサルtype Dレトロウイルスの陰性試験を行い、実験動物に関する研究所のガイダンスに従って維持した。採血、鼻腔拭い液(nasal swab)の採取、ワクチネーション、およびウイルス接種を行う場合はケタミン麻酔下で行った。カニクイザル 1329605029(Cy029)はワクチネーションを行わず、未処理のコントロールとした。カニクイザル 1429407002(Cy002)は、108 CIUの SeV/controlを初回免疫(0週目)、その後4週目および14週目の計3回、鼻腔内に接種してコントロールのワクチネーションを行った。同じスケジュールで2頭のカニクイザル 1129307001(Cy001)および 1128206162(Cy162)に 108 CIUの SeV/SIVgagを鼻腔内に3回のワクチネーションを行った。これらのカニクイザルはいずれも、ワクチネーション後に体重減少を含む病理学的兆候は見られなかった。また、末梢のCD4またはCD8細胞数の有意な減少はいずれも観察されなかった(図3A)。
【0115】
表1 ワクチネーションプロトコルおよび in vivo SeV/SIVgag発現

【0116】
気道におけるSeV/SIVgagの複製および発現を鼻腔拭い液(nasal swab)により調べた(表1)。鼻腔拭い液の採取は以前の記載に従って行った(Hurwitz, J. L. et al., 1997, Vaccine 15: 533-540)。拭い液試料は培地で希釈し、SeVを回収するため鶏卵の尿膜腔に注入した。48時間インキュベート後、尿液を回収し、ヘマグルチネーション(HA)アッセイに供しSeVを検出した。SIV gagの発現を調べるためには、High Pure Viral RNA kit(Boehringer Mannheim)を用いて拭い液試料からRNAを抽出した。SIV gag特異的プライマーを用いてnested RT-PCRを行った。最初のRT-PCRに用いたプライマーは5'-AGA AAC TCC GTC TTG TCA GG-3'(配列番号:11)および5'-TGA TAA TCT GCA TAG CCG C-3'(配列番号:12)、2回目のPCRに用いたプライマーは5'-GAT TAG CAG AAA GCC TGT TGG-3'(配列番号:13)および5'-TGC AAC CTT CTG ACA GTG C-3'(配列番号:14)である。
【0117】
カニクイザル Cy002、Cy001、および Cy162 では、拭い液を接種した鶏卵の尿液中に蓄積したSeV HA活性から分かるように、1週目においてSeV(SeV/controlまたはSeV/SIVgag)の複製が検出された(表1)。2週目では検出可能なSeVは回収されなかった。2回目または3回目のワクチネーションではいずれも、拭い液中に検出可能なSeVは見られなかった。SIV gag特異的プライマーを用いたnested PCRにより、SIV gagの発現がSeV/SIVgagによるワクチネーションを行ったカニクイザル(Cy001およびCy162)の両者の鼻拭い液で検出されたが、SeV/controlによるワクチネーションを行ったカニクイザル(Cy002)では検出されなかった(表1)。
【0118】
これら3頭のカニクイザルのワクチネーション後の免疫応答を調べた。ELISAによる体液性免疫応答試験の結果、3頭の全てのカニクイザルで血漿中に高いレベルで抗SeV抗体が誘導されていることが示された(図3B)。2回目および3回目のワクチネーションはそれらのレベルの急速な上昇を誘導したことから、追加免疫の効果が示唆された。対照的に、血漿中の抗SIV Gag p27抗体の有意な誘導は観察されなかった。なお、抗SeV抗体ELISAは不活化したSeVを用いて行った。抗SIV Gag p27抗体ELISAは組み換えp27抗原(ImmunoDiagnostics)を用いて行った。抗SeV抗体ELISAでは血漿試料を1000倍に、抗p27抗体ELISAでは100倍に希釈した。
【0119】
未処理のカニクイザル(Cy029)、SeV/contorolでワクチネーションを行ったカニクイザル(Cy002)、およびSeV/SIVgagでワクチネーションを行ったカニクイザル(Cy001およびCy162)において、22週目に多量(100 TCID50)のSIVmac239を静脈注射により暴露させた。具体的には、カニクイザル鼻腔内に 108 CIUのSeV/SIVgagによる3回のワクチネーションを行い(0、4、および 14 週目)、最初のワクチネーション後22週目に、それらのカニクイザルに100 TCID50 の SIVmac239 を静脈注射により暴露させ、血漿中のSIV RNAを測定した。SeV/controlを接種したカニクイザルおよびワクチネーションを行わないカニクイザルを対照とした。
【0120】
これらのカニクイザルの血漿SIV RNA量の測定を以下のようにして行った。High Pure Viral RNA kitを用いて血漿RNAを抽出した。SIV env特異的プライマーを用いてnested PCRを行った。最初のRT-PCRに用いたプライマーは 5'-ATG GGA TGT CTT GGG AAT C-3'(配列番号:15)および 5'-CCA AAT CTG CAG AGT ACC AAG-3'(配列番号:16)、2回目のPCRに用いたプライマーは 5'-CAG CTT GGA GGA ATG CG-3'(配列番号:17)および 5'-CTT GTT CCA AGC CTG TGC-3'(配列番号:18)である。5倍希釈したRNA試料を4連で増幅し、以前の記載にように定量した(Shibata, R. et al., 1997, J. Infect. Dis. 176: 362-373; Reed, L. J. and Muench, H., 1938, Am. J. Hyg. 27: 493-497)。このアッセイの検出下限は 1.0×102 copies/mlである。
【0121】
これら4頭のカニクイザルは全て、暴露後2週目に同様のレベルで血漿SIV量のピークを示した(図4)。その後、コントロールのカニクイザルでは2頭とも血漿SIV RNAレベルは減少したが、約105 copies/mlが残存した。これに対しSeV/SIVgagでワクチネーションを行ったカニクイザルでは、2頭とも初期のウイルス血症の後、ウイルス量は有意に低下した(102から103 copies/ml)。より重要なことは、コントロールのカニクイザルの血漿ウイルス量は 105 copies/ml 周辺の高い値に留まったのに対し、免疫したカニクイザルの血漿ウイルス量は最終的に検出可能なレベル(100 copies/ml)を下回ったことである(図4)。
【0122】
[実施例5] カニクイザルの各組織におけるSeV/SIVgagの発現
カニクイザルにおける組み換えSeVの初期複製を調べるため、6頭のカニクイザルの鼻腔内にSeV/SIVgagを接種した(表2、グループ I)。カニクイザルは実験動物に関する研究所のガイドラインに従って維持した。実験に用いる前にSeVおよびSIVの陰性試験を行った。採血、鼻腔拭い液(nasal swab)の採取、およびワクチネーションを行う場合はケタミン麻酔下で行った。これらのサルはいずれも、接種後に明白な臨床症状は示さなかった。接種後4日目(day 4)にグループ I-A 中の2頭(C3880およびC4325)を、7日目(day 7)にグループ I-B 中の2頭(C3993およびC4240)を、13日目(day 13)にグループ I-C 中の2頭(C3882およびC4324)をそれぞれ安楽死させた。剖検により採取した各組織から細胞を調製し、細胞からRNAを抽出した。
【0123】
具体的には、LN、胸腺、および脾臓から、組織をミンスして細胞を調製した。鼻腔粘膜、口蓋扁桃、気管、および肺からは、コラゲナーゼおよびディスパーゼ処理後に細胞を調製した。これらの細胞はRNA抽出前にPBSで3回洗浄した。末梢血単核細胞(PBMC)は全血試料から Ficoll-Paque Plus(Amersham Pharmacia Biotech)を用いて文献(Shibata, R. et al. 1997. J. Infect. Dis. 176:362-373)に従って調製した。RNAは細胞からRNA-extraction kit(Qiagen)を用いて抽出した。nested RT-PCR(逆転写およびnested PCR)はSIV gag 特異的プライマーを用いて行い、RNA試料を限界希釈し、文献(Shibata, R. et al. 1997. J. Infect. Dis. 176:362-373)に従って希釈限界を決定することによりgag RNAレベルを定量した。SeV N mRNAレベルは TaqMan PCR system(ABI PRISM 7700, Applied Biosystems Japan)を用いた定量PCRにより定量した。
【0124】
表2 動物モデル
【0125】
5−1.カニクイザルの鼻腔粘膜におけるSeV/SIVgagの発現
定量RT-PCRにより、6頭のカニクイザル全ての鼻腔粘膜から調製した細胞において、有意なレベルのSIV gag RNAが検出された(図5A)。グループ I-A の4日目(day 4)においては、106 細胞当たり約 1.7×104 または 2.6×105 コピーの gag RNAが検出された。7日目(day 7)および 13日目(day 13)の発現レベルは4日目(day 4)に比べて低下した。さらに、定量RT-PCRにより鼻腔粘膜におけるSeV N mRNAの発現レベルを検出した(図5B)。ほとんどのカニクイザルにおいて、SeV N mRNAレベル(図5Bに示す)は gag RNAレベル(図5Aに示す)の1/4または1/3に相当した。これらの結果から、SeV/SIVgagを接種したカニクイザルの鼻腔粘膜において、SeV/SIVgagが有意に発現することが確認された。
【0126】
次に、鼻腔拭い液からSeVが回収されるかを調べた。鼻腔拭い液の採取は上記と同様にして行った。拭い液試料はRPMI-1640 で希釈し、SeVを回収するため鶏卵の尿膜腔に注入した。鼻腔粘膜から調製した細胞からのSeVの回収においては、細胞の凍結融解を2回行った後、RPMI-1640中に懸濁させた 1×105 細胞を鶏卵の尿膜腔に注入した。48時間インキュベート後、尿液を回収し、文献(Kato, A. et al. 1996. Genes Cells 1:569-579)に従ってヘマグルチネーション(HA)アッセイに供しSeVを検出した。
【0127】
表3に示すように、4日目(day 4)において6頭全ての鼻腔拭い液からウイルスが回収された。7日目(day 7)では、4頭のカニクイザルのうち2頭からウイルスが回収されたが、この2頭はいずれも、鼻腔粘膜においてより高いgag RNAレベルを示していた。13日目(day 13)では、両方のサルでウイルスは回収されなかった。さらに、鼻腔粘膜から調製した細胞 1×105 からウイルスが回収されるかを調べた(表3)。4日目(day 4)の試料からはウイルスが回収されたが、7日目(day 7)または 13日目(day 13)の試料からはウイルスは回収されなかった。これらの結果は、SeV/SIVgagの複製レベルは1週間以内にピークに達することを示している。
【0128】
表3 SeVの回収
【0129】
5−2.カニクイザルの鼻腔の近傍リンパ節でのSeV/SIVgagの発現
有意なレベルのgag RNAが、咽頭後LN(retropharyngeal LN)および顎下LN(submandibular LN)でも見出された(図5Cおよび図5D)。これらはいずれも鼻腔からのリンパ球が最初に流入するリンパ節である。7日目(day 7)および 13日目(day 13)の各動物におけるLNでのgag RNAレベルは、鼻腔粘膜の場合の約1/20であった。しかし4日目(day 4)では、各動物における咽頭後LNのgag RNAレベルは鼻腔粘膜の場合の1/50以下であった。従って、4日目(day 4)と7日目(day 7)の間でこのLN中のレベルの有意な違いは観察されなかった。
【0130】
5−3.カニクイザルの他の組織でのSeV/SIVgagの発現
gagの発現を表4に示すように他の組織においても調べた。口蓋扁桃では十分な細胞が得られなかったが、その発現レベルは咽頭後LNの発現レベルよりも低いことが示された。気管での発現は2点、すなわち 4日目(day 4)の C3880 および 7日目(day 7)の C4240 で検出可能であった。前者の発現は後者の発現よりずっと高かった。肺での発現は C3880[4日目(day 4)]でのみ検出された。胸腺または鼠径LNでは発現は観察されなかった。脾臓では発現はほとんど検出されず、わずかな発現が1個体のみで検出された[7日目(day 7)]。PBMCにおいては、7日目(day 7)において4頭のカニクイザル中2頭でgagの発現が示されたが、4日目(day 4)または13日目(day 13)では発現は観察されなかった。表4で調べた全ての組織において、gagの発現は鼻腔粘膜、咽頭後LN、および顎下LNに比べずっと低かった。
【0131】
表4 gag RNA発現
【0132】
[実施例6] SeV/SIVgagを感染させた細胞によるGag特異的CD8+ T細胞の刺激
SeV/SIVgagを感染させた細胞が、実際にGag特異的CD8+ T細胞をin vitroで刺激できるかを調べるため、抗原特異的な細胞内IFN-γ誘導をフローサイトメトリーにより解析した。SIVに慢性的に感染したアカゲザル由来のPBMCを用いた。アカゲザル(Macaca mulatta)は、実験動物に関する研究所のガイドラインに従って維持した。実験に用いる前にSeVおよびSIVの陰性試験を行った。採血、鼻腔拭い液(nasal swab)の採取、およびワクチネーションを行う場合はケタミン麻酔下で行った。文献(Matano, T. et al. 2000. Vaccine 18:3310-3318)に記載のように、この動物は予めプロウイルスDNAワクチネーションを受け、その後SIVmac239(Kestler, H. et al. 1990. Science 248:1109-1112)の暴露による感染を受けている。51Cr-放出アッセイにより、SIV感染の慢性期の間、SIV-Gag特異的細胞傷害性Tリンパ球(CTL)活性が確認された(データ省略)。
【0133】
抗原特異的インターフェロン-γ(IFN-γ)誘導のフローサイトメトリーによる検出は、以下のようにして行った。まず、自家Herpesvirus papio不死化Bリンパ芽球細胞株(BLC)(Voss, G. et al. 1992. J. Virol. Methods 39:185-195)にコントロールのワクシニアウイルスベクター(Vv-control)(Mackett, M. et al. Proc. Natl. Acad. Sci. USA 79:7415-7419)、SIV Gagを発現する組み換えワクシニアウイルスベクター(Vv-Gag)、またはコントロールSeVベクター(SeV/control)を m.o.i. を 5 で感染させ、1日後、文献に記載のように(Gea-Banacloche, J.C. et al. 2000. J. Immunol. 165:1082-1092)、それぞれ対照の非特異的、Gag特異的、またはSeV特異的スティミュレーター(stimulator; 刺激細胞)として使用した。
【0134】
培養管(Falcon #3033)に 1×106 の PBMC を 1×103 の上記のスティミュレーター細胞と共に 10%FBSを含む 1ml の RPMI-1640 中で共培養した。1時間インキュベートした後、細胞に 0.75μl/mlの GolgiStop(monesin)(Pharmingen, San Diego, CA)を加え、さらに5時間インキュベートした。
【0135】
p27による刺激(実施例7)の場合、1×106 の PBMC は 2μg の抗CD28モノクローナル抗体(Becton Dickinson, San Jose, CA)および 10μg の組み換えSIV Gag p27蛋白質を含む、10%FBSを含む 1ml の RPMI-1640 中で、培養管中でインキュベートした。3時間インキュベートした後、細胞に 0.75μl/mlの GolgiStop を加え、さらに15時間インキュベートした。
【0136】
その後、Cytofix-Cytoperm kit(Pharmingen)を用い、説明書に従って細胞内IFN-γ染色を行った。具体的には、刺激した細胞を回収し、表面分子に対する抗体の混合液と共に室温で20分染色した。抗体には、抗ヒトCD8 PerCP(Becton Dickinson)および抗ヒトCD3 APC(Pharmingen)を用いた。その後 0.5%ウシ血清アルブミンを含むPBS(PBS-BSA)で細胞を洗浄し、Cytofix-Cytopermにより固定、透過化処理を行い、抗ヒトIFN-γ PE(Pharmingen)で4℃で30分染色した。染色した試料をFACScaliburで回収し、CellQuestソフトウェア(Becton Dickinson)を用いて解析した。各試料において、BLCによる刺激の場合の全事象(total events)は 100,000から200,000であり、組み換え蛋白質による刺激の場合の全事象は 50,000から100,000であった。ゲーティングは単核細胞において行い、その後 CD3+CD8+のサブポピュレーションについて行った。単核細胞数に対する CD3+CD8+IFN-γ+ 細胞数の比を評価し、 1×106 リンパ球当たりの CD8+IFN-γ+ T細胞数として示した。
【0137】
SIV Gagを発現する組み換えワクシニアウイルス(Vv-Gag)を感染させた自家BLCとPBMCとの共培養では、CD8+ T細胞におけるSIV Gag特異的細胞内IFN-γ誘導が示された(図6C)。SeV/SIVgagを感染させたBLCとPBMCとの共培養でも、CD8+IFN-γ+ T細胞が同様の頻度で観察された(図6B)。SeV/controlを感染させたBLCとPBMCとの共培養では、CD8+IFN-γ+ T細胞の有意な誘導は見出されなかった(図6A)。これらの結果は、SeV/SIVgagを感染させたBLCはGag特異的 CD8+ T細胞を効率的に刺激することを示している。
【0138】
[実施例7] アカゲザルのGag特異的 CD8+ T細胞の頻度
SeVワクチネーション後の初期免疫応答を解析するため、5頭のアカゲザルを用いて別のの実験を行った(表2、グループ II)。グループII-A のうち2頭(R004およびR014)はSeV/controlを鼻腔内に接種し、グループII-Bの3頭(R013、R015、および R017)はSeV/SIVgagを接種した。R010はSeV/controlワクチネーションの12週から6週間前に、他の実験のためにコントロールDNAワクチネーション(筋肉内接種により800μgのpCMVN DNA[Matano, T. et al. 2000. Vaccine 18:3310-3318]およびgene gunにより10μgのpCMVN DNAを4回)を受けている。これらのサルはいずれも、接種後に明白な臨床症状は示さなかった。
【0139】
3週目(week 3)に得たPBMCを、Vv-Gagに感染させた自家BLCと共培養し、CD8+ T細胞におけるIFN-γ誘導を調べた。SeV/SIVgagを接種したグループII-Bのアカゲザルは全て、Gag特異的刺激後に CD8+IFN-γ+ T細胞が有意な頻度で検出されたが、非特異的な刺激では検出されなかった(図7A)。これに対し、SeV/controlを接種したグループII-Aのアカゲザルでは、Gag特異的刺激後も、CD8+ T細胞においてIFN-γの有意な誘導は観察されなかった。これらの結果は、全身性免疫応答、すなわちGag特異的 CD8+ T細胞の誘導が、SeV/SIVgagでワクチネーションを行ったアカゲザルで起こることを示している。
【0140】
次に、鼻腔周囲の局所的な免疫応答を、グループ I-Cの咽頭後LNから調製した細胞を用いて調べた。グループ Iのアカゲザルからの自家BLCの調製がうまく行われなかったため、組み換えSIV Gag p27蛋白質を用いてGag特異的刺激を行った。グループ I-Cの両方のアカゲザルにおいて、p27と反応する CD8+IFN-γ+ T細胞が有意な頻度で検出されたことから、このLNにおいてp27特異的 CD8+ T細胞が誘導されていることが示された。
【0141】
[実施例8] アカゲザルにおけるSeV特異的CD8+ T細胞の頻度
SeV特異的な細胞性免疫応答を、SeV/controlまたはSeV/SIVgagを接種したアカゲザルで調べた。グループIIの全ての動物のPBMCにおいて、3週目(week 3)にSeV特異的 CD8+ T細胞が高い頻度で検出された(図8B)。また、これらの全てにおいて、1週目(week 1)で既にSeV特異的 CD8+ T細胞が有意な頻度で出現していることが示された(図8A)。従って、大規模なSeV特異的細胞性免疫応答が、ワクチネーション後に迅速に誘導されることが判明した。
【0142】
抗体ELISAにより、血漿中の蛋白質レベルを評価した。抗SeV抗体ELISAは不活化したSeVを用いて行った(Watanabe, T. et al. 2000. Arch. Dermatol. 136:1518-1522)。抗SIV Gag p27抗体ELISAは組み換えp27蛋白質(ImmunoDiagnostics, Woburn, MA)を用いて行った。抗SeV抗体ELISAでは血漿試料を1000倍に、抗p27抗体ELISAでは100倍に希釈した。
【0143】
グループIIの全てのアカゲザルにおいて、血漿中の抗SeV抗体はワクチネーション後 1週目(week 1)では検出されないが、2週目(week 2)では有意なレベルの抗体が出現していた(図9)。これに対して血漿中の抗Gag抗体は、SeV/SIVgagを接種したグループII-Bのアカゲザルにおいても検出されなかった(データ省略)。
【0144】
[実施例9] DNAワクチンとの組み合わせによるプライム−ブースト効果
prime-boostによるワクチネーションにおいて、DNAワクチンとの組み合わせの効果を試験した。アカゲザルを用いた実験のプロトコルを表5に示した。DNAワクチネーションは文献(Matano, T. et al., Vaccine 18: 3310-3318, 2000 参照)に従って、筋肉内(intramuscular; i.m.)接種およびジーンガン(Bio-Rad)により行った。i.m.接種では、PBSに溶解した0.25μg/μlのDNA溶液を各回200μlずつ、計800μl四頭筋に注入した。また、ジーンガンによる接種は大腿の前部皮膚中への注入により行った(Matano, T. et al., Vaccine 18: 3310-3318, 2000 参照)。control群4頭、DNAワクチン + SeV/SIVgagワクチン投与群4頭とした。DNAワクチンの詳細は、文献(Donnelly, J. J. et al., Annu. Rev. Immunol. 15: 617-648, 1997; Lu, S. et al., J. Virol. 70: 3987-3991, 1996; Matano, T. et al., Vaccine 18: 3310-3318, 2000)を参照。SeV/SIVgag投与は、上記と同様で、108 CIU 鼻腔投与により行った。ワクチネーション開始の26週目に 10 TCID50 のサル-ヒト免疫不全ウイルス(SHIV 89.6-PD)(Lu, Y. et al., J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 19(1):6-18, 1998)を静脈注射により暴露した。DNAワクチン + SeV/SIVgagワクチンにより、SIV Gag特異的CTLの誘導が観察され、末梢血CD4 countの変化から、投与群4頭の全てで病原性SHIV暴露(challenge)に対する感染防御が認められた(図10および11)。また、DNAワクチン + SeV/SIVgagワクチン接種群の血漿中のSHIV RNAコピー数は、コントロール群と比較して、感染急性期より有意に低値を示した(図12)。
【0145】
表5 マカクサルプロトコル:DNA + SeV
【産業上の利用可能性】
【0146】
本発明により、免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチンが提供された。センダイウイルスベクターは毒性が低く、また鶏卵を使って大量に製造できるため、安全で簡便な生組み換えワクチンの製造が可能である。本発明のワクチンにより、AIDSウイルスの感染および/またはAIDSの発症・進行を抑制する有望なワクチン戦略が提供される。
【技術分野】
【0001】
本発明は、センダイウイルスベクターを用いたAIDSウイルスワクチンに関する。また本発明は、センダイウイルスベクターを用いたワクチネーションの方法に関する。
【背景技術】
【0002】
ヒト免疫不全ウイルス(HIV)の世界的な伝播を防ぐため、HIVの感染に対する効果的かつ安全なワクチンの開発が早急に求められている。これまで複数の後天性免疫不全症候群(AIDS)ワクチンの手法が霊長類のレンチウイルス感染モデルにおいて評価されてきたが、AIDSに対して一貫して十分な抵抗性を示すワクチンは未だ知られていない(Almond, N. M. and Heeney, J. L., 1998, AIDS 12 (Suppl. A): S133-140(非特許文献1); Burton, D. R. and Moore, J. P., 1998, Nat. Med. 4: 495-498(非特許文献2); Hulskotte, E. Q. et al., 1998, Vaccine 16: 904-915(非特許文献3); Letvin, N. L., 1998, Science 280: 1875-1880(非特許文献4))。
【0003】
生きたウイルスベクターを基にしたワクチンは、抗原の十分な発現が持続的に誘導されるため、有望な予防戦略の1つと見なされている(Cairns, J. S. and Sarver, N., 1998, AIDS Res. Hum. Retroviruses 14: 1501-1508(非特許文献5); Hirsch, V. M. et al., 1996, J. Virol. 70: 3741-3752(非特許文献6); Buge, S. L. et al., 1997, J. Virol. 71: 8531-8541(非特許文献7))。最もよく使われてきたのはポックスウイルス(poxvirus)であり、また、アデノウイルスなど他の選択肢も多数存在している。しかしながら、これらのベクターを用いた試みは、HIVに対する十分な防御免疫を誘発することに成功していない。十分な防御免疫の誘発には、抗原発現のレベルや期間、ベクターウイルスの複製のカイネティクス、およびベクターウイルスのトロピズムや病原性と言った多数の要因が影響しているものと考えられる。現在使用することができるウイルスベクターはいずれも、有利な側面と不利な側面の両面を持っている。最適なベクターを見つけ出すためには、それぞれのウイルスベクターの正確な評価と比較が必要である。
【0004】
ウイルスベクターを基にしたワクチン戦略における決定的なデメリットの1つは、標的とすべき抗原よりもむしろ、ベクターウイルスに由来する抗原に対して強力な免疫応答が誘導されてしまうことである。この問題は、初回免疫と追加免疫のそれぞれにおいて、2つまたはそれ以上の異なる種類のウイルスベクターを用いることにより解決することができる。DNAワクチンを基にした初回免疫後にウイルスベクターを基にした追加免疫を行うことも、好ましい戦略の1つである(Hanke, T. et al. 1999, J. Virol. 73: 7524-7532(非特許文献8); Robinson, H. L. et al., 1999, Nat. Med. 5: 526-534(非特許文献9))。このことから、新しいクラスのウイルスベクターの開発が、現在でも求められている。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Almond, N. M. and Heeney, J. L., 1998, AIDS 12 (Suppl. A): S133-140
【非特許文献2】Burton, D. R. and Moore, J. P., 1998, Nat. Med. 4: 495-498
【非特許文献3】Hulskotte, E. Q. et al., 1998, Vaccine 16: 904-915
【非特許文献4】Letvin, N. L., 1998, Science 280: 1875-1880
【非特許文献5】Cairns, J. S. and Sarver, N., 1998, AIDS Res. Hum. Retroviruses 14: 1501-1508
【非特許文献6】Hirsch, V. M. et al., 1996, J. Virol. 70: 3741-3752
【非特許文献7】Buge, S. L. et al., 1997, J. Virol. 71: 8531-8541
【非特許文献8】Hanke, T. et al. 1999, J. Virol. 73: 7524-7532
【非特許文献9】Robinson, H. L. et al., 1999, Nat. Med. 5: 526-534
【発明の概要】
【0006】
本発明者らは以前、組み換えセンダイウイルス(SeV)を用いて、効率的に抗原を発現させる系を確立させた(Kato, A. et al. 1996. Genes Cells 1:569-579)。マウスパラインフルエンザウイルス 1型であるSeVは非分節型ネガティブ鎖RNAゲノムを持つエンベロープウイルスの1つで、パラミクソウイルス科(Paramyxoviridae)に属す(Nagai, Y. 1999. Rev. Med. Virol. 9:83-99)。このウイルスはマウスに対しては致命的な呼吸器疾患を引き起こすが、非ヒト霊長類およびヒトには病原性がないとされている(Nagai, Y. 1999. Rev. Med. Virol. 9:83-99; Hurwitz, J.L. et al., Vaccine 15: 533-540, 1997)。
【0007】
SeVの複製は細胞質で行われ、核のフェーズは存在しないため、Gag、Pol、それにEnvなどのレンチウイルスの構造蛋白であっても、組み換えSeVを用いることにより、Rev非依存的に効率良く発現されることが期待される。より重要なことは、SeVベクターは非分裂細胞にも感染でき、外来遺伝子を強力に発現することである。例えば、組み換えSeVベクター(V(-)SeV)によるヒト免疫不全ウイルス type 1(HIV-1)の Env gp120 の発現量は培養上清において 6μg/ml(106 細胞あたり 6μg 相当)にも達するが、これは哺乳動物細胞で現在使用可能なベクター系の中では最も高い(Yu, D. et al. 1997. Genes Cells 2:457-466)。そこで本発明者らは、センダイウイルス(SeV)の系を用いて、この系がサル免疫不全ウイルス(SIV)のマカクサルへの感染に対して、防御的免疫を誘発できるかを検討し、組み換えSeVベクターをAIDSワクチンとして用いることの有効性を評価した。このために、まず、SeVのアクセサリー遺伝子の1つであるV遺伝子をノックアウトした変異SeVである V(-)SeV を構築した(Kato, A. et al. 1997. J. Virol. 71:7266-7272; Kato, A. et al. 1997. EMBO J. 16:578-587)。マウスにおいて、この V(-)SeV は劇的に弱毒化されるが、その遺伝子発現はかえって増強される。安全および効率の両面から、本発明者らはこの V(-) 型をベクターのバックボーンとして用いた。V(-)SeVを用いて、サル免疫不全ウイルスmac239株(SIVmac239)のGagを発現する組み換え V(-)SeV であるSeV/SIVgagを構築し、マカクサルにおいて抗SIV免疫を誘導する効力について試験し、マカクサルのAIDSモデル(Johnson, R.P. 1996. Curr. Opin. Immunol. 8:554-560; Almond, N.M., and Heeney, J.L. 1998. AIDS 12:133-140)でのワクチネーションによる防御効果を評価した。
【0008】
回収したSeV/SIVgagを用いて、このベクターを介したGag発現により誘導されるSIV Gag特異的な細胞性免疫応答のin vitro試験を行った。その結果、細胞傷害性Tリンパ球(CTL)アッセイにより、SeV/SIVgagを感染させた細胞がGag特異的CTLの標的として働くことが判明した。また、末梢血リンパ球の細胞培養では、SeV/SIVgag を感染させた細胞はGag特異的CTLの細胞集団の増殖を誘導した。動物実験では、4個体のカニクイザル(cynomolgus macaques)を用い、2個体はSeV/SIVgagの鼻腔内接種によりワクチネーションを行い、1個体はコントロールSeVを接種、残る1個体は非ワクチネーションコントロールとした。最初のワクチン接種から 22週後にこれらのマカクサルに100 TCID50 のSIVmac239 を静脈注射により暴露した。SIVに暴露させると、急性期の間は、全てのマカクサルにおいて血漿中のSIV量は同様に高値を示したが、SeV/SIVgagでワクチネーションを行ったマカクサルでは、その後のウイルス量はコントロールのマカクサルの 105 copies/ml 前後に比べはるかに低くなり、最終的に検出可能なレベル(100 copies/ml)を前後した。このように、SIVの構造蛋白質を発現するSeVでワクチネーションを行うことによって、SIV暴露後の慢性期(set-point)におけるウイルス量を顕著に減少させることが可能であることが示された。この結果は、センダイウイルスベクターは新しいAIDSワクチンとして有用であり、同時に細胞免疫応答の評価にも使用できることを示している。
【0009】
本発明の目的の1つは、組み換えセンダイウイルスベクターの系を用いることにより効率的なSIV Gagの発現が誘導されることを示し、この系をAIDSワクチンの開発に適用することの有効性を実証することにある。AIDSワクチンの研究に広く使用されているポックスウイルスと比べ、SeVベクターは哺乳動物細胞における細胞傷害性が低く、抗原の発現レベルは明らかに高い(Yu, D. et al., 1997, Genes Cells 2: 457-466)。また、実施例で用いられたSeVベクターの特徴の1つは、挿入遺伝子がSeVゲノムの3'端近くに収容されているため、その発現がベクターに由来する他のいずれのSeV特異的遺伝子よりも迅速で高率であることである(Nagai, Y., 1999, Rev. Med. Virol. 9: 83-99)。このことは、標的抗原に特異的な免疫応答の効率的な誘導に有利なはずである。
【0010】
CTLアッセイにおいて、SeV/SIVgagを感染させたB-LCLはSIV Gag特異的CTLの標的として良好に働いた。それに加え、SeV/SIVgagを感染させたPBMC(peripheral blood mononuclear cells; 末梢血単核細胞)と正常PBMCとの共培養はIFN-γの誘導およびSIV Gag特異的CTLの増殖をもたらした。これらの結果は、SeVの系が抗原特異的な細胞性免疫応答のアッセイに有用であることを示している。SeVの不活性なエンベロープ蛋白質の前駆体から活性型へのプロセッシングにはトリプシン様プロテアーゼが必要であり(Nagai, Y., 1993, Trends Microbiol. 1: 81-87)、外から加えられるこのプロテアーゼが存在しなければSeVは共培養細胞へ感染できないため、組み換えSeVベクターを用いることによって、固定やUV照射をすることなくCTL増殖のための共培養を容易に構築することが可能である。
【0011】
最近の報告が示すように、ウイルス特異的な細胞性免疫応答の誘導は、HIV-1感染に対する防御にとって有効となり得る(Geretti, A. M. et al., 1998, J. Gen. Virol. 79: 415-421; Matano, T. et al., 1998, J. Virol. 72: 164-169; Ogg, G. S. et al., 1998, Science 279: 2103-2106; Rowland-Jones, S. L. et al., 1998, J. Clin. Invest. 102: 1758-1765; Jin, X. et al., 1999, J. Exp. Med. 189: 991-998; Schmitz, J. E. et al., 1999, Science 283: 857-860)。SeV/SIVgagを感染させたPBMCはSIV Gag特異的CTLの増殖を誘導した。これは、この系が抗原特異的な細胞性免疫応答をin vivoで誘導することを示唆している。
【0012】
SIVmac239の暴露実験では、SeV/SIVgagでワクチネーションを行ったカニクイザルの血漿SIV量は、感染の急性期の間はコントロールに匹敵していた。しかしながら、その後SeV/SIVgagでワクチネーションを行ったカニクイザルは有意に低いSIV量を示した。特に目立ったのは、残存するウイルス量が検出可能なレベルまたはそれ以下であったことである。1つの抗原成分(Gag)を用いるだけで顕著な感染防御作用が得られたことも注目に値する。慢性期(set-point)の血漿ウイルス量の減少は、高頻度のSIV Gag特異的なT細胞前駆細胞の活性化によるものと考えられる。T細胞前駆細胞の頻度は、ウイルス感染の自然経過において最初のバースト量(initial burst size)に大きく依存している。外来抗原の産生に関してSeVが極めて高い性能を持っていることが、推定される高いT細胞前駆細胞の頻度に貢献し得るだろう。しかしながら、同じ組み換えウイルスを再度投与しても、追加的なGag特異的応答を引き起こすには不十分である可能性がある。従って、SeV/SIVgagで初回免疫を行い、追加免疫に異なるウイルスベクターまたはDNAワクチン等を用いるのが有効である。本明細書に示される実施例ではGag抗原のみを免疫に用いたが、組み換えSeVベクターを用いて複数の抗原を発現させれば、防御の効力をより向上させることができるかも知れない。
【0013】
SeVは自身の複製にエンベロープをプロセシングするプロテアーゼを必要とするため、複製のトロピズムは気道上皮などの特定に組織に限られる(Nagai, Y., 1993, Trends Microbiol. 1: 81-87)。気道を超えて他の組織に広がることが予想されないため、SeVベクターは例え複製型であっても安全面で利点を有することが示唆される。さらに、組み換えSeVベクターの鼻腔内投与は全身の粘膜免疫の誘導も期待できる。これはHIV-1感染に対する予防においてSeVベクターが有するもう1つの利点となり得る。
【0014】
また、本発明は、霊長類におけるSeV初期複製の解析を初めて開示する。ウイルスベクターをベースにした遺伝子導入の効率は宿主に依存するため、臨床研究に先だって霊長類でその効率、有効性、および安全性を評価することは必須である。霊長類においては、アフリカミドリザルで鼻腔内へのSeVの接種後の鼻腔拭い液試料の試験結果が報告されている(Hurwitz, J.L. et al. 1997. Vaccine 15:533-540)ものの、SeVの複製はこれまで詳細には解析されていなかった。また本発明者らは、SeV/SIVgag接種後1年以上経過したマカクサルの剖検で得られた鼻腔粘膜および肺を含む組織において、SeV/SIVgagの発現は検出されないことを見出した(データ省略)。そこで、本発明者らは、組み換えセンダイウイルスベクターでワクチネーションを行ったマカクサルにおける初期段階の抗原発現および細胞性免疫応答の解析を実施した。SIV Gagを発現する組み換えSeV(SeV/SIVgag )をマカクサルの鼻腔内に接種後、様々な組織におけるgag発現および細胞性免疫応答を調べた。SeVの複製は制御されており、Gagの発現は主に鼻腔粘膜およびその近傍リンパ節(lymph nodes; LN)に限定されていた。すなわち、gagの強い発現が鼻腔粘膜で観察され、それよりは低いが有意レベルのgagの発現が局所の咽頭後リンパ節(retropharyngeal LN)および顎下リンパ節(submandibular LN)で検出された。発現は免疫後1週間以内にピークに達し、少なくとも13日まで持続した。SeV/SIVgag は4日目(day 4)の鼻腔拭い液から確実に単離されたが、7日目(day 7)では頻度が低下し、13日目(day 13)では全く単離されなかった。これに対し、胸腺、脾臓、および鼠径LNなどの離れたリンパ組織では、検出可能な抗原の発現は見られなかった。本来の宿主であるマウスでは、SeVの全般的な感染は見られず、気道上皮に局在した急性の粘膜感染を示すが、サルでのSeV発現発現の組織の制限および発現の時間変化は、このマウスにおけるSeVの複製パターンと一致していた。しかしながら、抗原は気管や肺にはほとんど存在していないことから、マカクサルではウイルスの伝播はマウスの場合より制限されていることが示された。7日目(day 7)における細胞性免疫応答の解析では、SeV特異的 CD8+ T細胞の迅速な出現が示された。免疫後に明白な臨床症状を示すサルはなかった。マカクサル鼻腔内へのSeV/SIVgag の接種は、Gag特異的CD8+ T細胞の効率的な誘導をもたらした。Gag特異的CD8+ T細胞が、咽頭後LN中だけでなく末梢血単核細胞でも有意な頻度で検出されたことから、SeVが持つ抗原特異的な細胞性免疫応答の誘導能力は、霊長類においても効率的に働くことが示された。このように、免疫後に全身および局所の両方で、Gag特異的 CD8+ T細胞が著しく高頻度で検出されることが判明した。これらの結果から、組み換えSeVベクターをAIDSワクチンとして使用することの可能性がさらに強められた。
【0015】
上記のように、SeV/SIVgagでワクチネーションを行ったマカクサルにおいて、SeV/SIVgagは効率的に発現し、鼻腔粘膜およびその局所LNで強いことが示された。検出されるgag RNAはゲノムRNAとmRNAからなる可能性がある。そこで、以前確立した、SeV N mRNAだけを検出し、SeVゲノムの N RNAを検出しない系(Kato, A. et al. 2001. J. Virol. In press)を利用して、mRNAの発現を確認した。gag RNAレベルはSeV N mRNAレベルよりも高く、これは前者がmRNAに加えゲノムRNAも含んでいるからであろう。もう一つの説明としては、ゲノムにおいてgagの位置はNの位置よりも上流であるため、gag mRNAレベルはN mRNAレベルよりも高いことが期待される(Nagai, Y. 1999. Rev. Med. Virol. 9:83-99)。
【0016】
鼻腔内接種は、粘膜免疫応答を誘導するという利点も持っている。咽頭後LN(retropharyngeal LN)および顎下LN(submandibular LN)は鼻腔内からのリンパ球が最初に流入するLNである(Suen, J.Y., and Stern, S.J. 1996. Cancer of the Neck. In Cancer of the Head and Neck, 3rd ed. E. N. Myers and J. Y. Suen, editors. W. B. Saunders Company, Philadelphia. 462-484)。これらのLNは粘膜免疫応答に関与している可能性が高い。最近、マウスにおいて NALT(nasal-associated lymphoid tissue; 鼻関連リンパ系組織)が粘膜免疫応答に機能していることが示された(Yanagita, M. et al., 1999, J. Immunol. 162:3559-3565)。マウス NALT に対応する Waldeyer's ring から調製した細胞の解析により、この組織におけるSeV/SIVgagの発現および免疫応答を確認することができる。咽頭後LN(retropharyngeal LN)および顎下LN(submandibular LN)におけるSeV/SIVgagの発現の検出では、両方の組織で有意なレベルのgag RNAが検出された。これらのLNにはSeV蛋白のプロセッシングに必須のプロテアーゼが存在しないことから(Nagai, Y. 1993. Trends Microbiol. 1:81-87)、LN内でSeVの複製は起こらないと予想され、これらのLNのgag mRNAは、鼻腔内から流入したSeV/SIVgag感染リンパ球に由来するものであろう。SeV/SIVgagの鼻腔内接種により、鼻腔粘膜のみならず局所LNでも効率的な抗原の発現が見られたことから、SeVの粘膜免疫誘導能が示唆される(Gallichan, W.S., and Rosenthal, K.L. 1996. J. Exp. Med. 184:1879-90)。
【0017】
細胞性免疫応答は、HIV-1やSIVなどのヒトおよび非ヒト霊長類レンチウイルスの制御に重要な役割を果たしていることが示されている(Ogg, G.S. et al. 1998. Science 279:2103-2106; Rowland-Jones, S.L. et al. 1998. J. Clin. Invest. 102:1758-1765; Brander, C., and Walker, B.D. 1999. Curr. Opin. Immunol. 11:451-459; Seder, R.A., and Hill, A.V.S. 2000. Nature 406:793-798)。マカクサルのAIDSモデルにおいて、抗CD8抗体を用いた in vivo によるCD8+ T細胞欠損実験により、CD8+ T細胞が初期感染および慢性感染の両方の制御に重要であることが示されている(Matano, T. et al. 1998. J. Virol. 72:164-169; Schmitz, J.E. et al. 1999. Science 283:857-860; Jin, X. et al. 1999. J. Exp. Med. 189:991-998)。従って、ウイルス特異的なCD8+ T細胞応答の誘導はHIV-1感染に対する防御に非常に有効であり得る。フローサイトメトリーによる細胞内サイトカイン誘導の解析により検出された抗原特異的T細胞の頻度は、常に抗原特異的な細胞傷害活性と相関するわけではないものの、抗原特異的な細胞性免疫応答の指標と考えられている(Lavini, A. et al. 1997. J. Exp. Med. 186:859-865; Butz, E.A., and Bevan, M.J. 1998. Immunity 8:167-175; Murali-Krishna, K. et al. 1998. Immunity 8:177-187; Donahoe, S.M. et al. 2000. Virology 272:347-356; Appay, V. et al. 2000. J. Exp. Med. 192:63-75)。この方法を用いた解析により、SeV/SIVgagに感染した細胞が in vitro でGag特異的 CD8+ T細胞を刺激できることが示された。また、SeV/SIVgagでワクチネーションを行った3頭全てのマカクサルのPBMCで、高レベルの Gag特異的 CD8+ T細胞が検出された。咽頭後LNでも、Gag特異的 CD8+ T細胞の効率的な誘導が観察された。これらの結果は、SeV/SIVgagワクチネーションにより、Gag特異的な細胞性免疫応答が全身および局所の両方で誘導されることを示している。
【0018】
鼻腔内へのSeV/SIVgagのワクチネーション後の観察では、明白な臨床症状を示すサルはいなかった。SeV/SIVgagの発現はほとんど鼻腔内およびその局所LNに局在しており、発現レベルはワクチネーションから1週間以内にピークに達した。このような発現パターンは天然の宿主であるマウスにおけるSeVの性質に一致していた。しかしながら、気管および肺では抗原の発現がみられないことから、マカクサルにおいてはウイルスの伝播はより厳格に制限されていることが示される。抗原特異的IFN-γ誘導の解析において、SeV特異的CD8+ T細胞は1週目(week 1)で迅速に出現したことから、細胞性免疫応答がSeV複製を制御している可能性が示唆される。すなわち、マカクサルではSeV/SIVgagの複製はよく局在化されており、またよく制御されている。これらの結果は、本発明の系が霊長類においても安全であることを支持している。また、複製能を欠損したSeVを用いてより安全な系を構築することもできる。
【0019】
すなわち本発明は、組み換えSeVを介した免疫によるマカクサルの初期抗原発現および細胞性免疫応答を初めて開示する。本発明者らは、鼻腔内免疫したマカクサルの全てにおいて、効率的な抗原の発現のみならず、抗原特異的な細胞性免疫応答が効率的に誘導されることを示した。抗原の発現パターンは局在化され、またよく制御されており、霊長類におけるベクターの安全性が支持される。これらのことから、SeVの系はAIDSワクチンのためのツールとして有望であることがさらに裏付けられた。
【0020】
本発明は、免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチンを提供することを課題とする。本発明のワクチンは、AIDSワクチンとして、AIDSの予防および治療に極めて有用である。また本発明は、本発明のワクチンを投与することによるワクチネーションの方法を提供することを課題とする。より具体的には、本発明は、
(1)免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチン、
(2)ウイルス蛋白質がGag蛋白質またはその部分を含む、(1)に記載のワクチン、
(3)センダイウイルスがV遺伝子欠損型である、(1)または(2)に記載のワクチン、
(4)免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチンを接種する工程を含むワクチンネーションの方法、
(5)接種が鼻腔内投与である、(4)に記載の方法、
(6)複数回のワクチン接種において、免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチンの接種を少なくとも1回行う、(4)または(5)に記載の方法、
(7)(a)免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを抗原提示細胞に導入する工程、並びに(b)該導入細胞とTヘルパー細胞および細胞傷害性T細胞とを接触させる工程、を含む、免疫不全ウイルスのウイルス蛋白質特異的細胞性免疫応答を誘導する方法、に関する。
【0021】
本発明において「ワクチン」とは、伝染病の予防または治療のために使用される組成物を言う。ワクチンは抗原を含んでいるか、または抗原を発現可能であり、これにより抗原に対する免疫応答を誘導することができる。病原微生物の感染、伝播、および流行の予防または治療のために、本発明のセンダイウイルスベクターを含むワクチンは、所望の形態で用いることができる。
【0022】
「ワクチネーション(vaccination)」とはワクチンの接種により生物の体内または培養系に能動的に免疫(体液性免疫・細胞性免疫、または両者)を作らせることを意味する。これにより病原体の感染、増殖、伝播、および/または流行等を阻止することができる。また、病原体感染後の症状の発症および/または進展を抑制することができる。
【0023】
「抗原」とは、1つまたはそれ以上のエピトープを含む分子であり、宿主の免疫系を刺激して抗原特異的な免疫応答を誘導し得るものを言う。免疫応答は、体液性免疫応答および/または細胞性免疫応答であってよい。3個〜数個程度のアミノ酸でも1つのエピトープとなり得るが、通常、蛋白質中の1つのエピトープは、約7〜約15アミノ酸、例えば8、9、10、12、または14アミノ酸を含んでいる。抗原は免疫原とも言う。なお、抗原蛋白質をコードするポリヌクレオチドまたはベクターを用いて抗原を発現させる場合、本発明においては該ポリヌクレオチドおよびベクターも抗原と称し、これらはワクチン成分としても用いられ得る。
【0024】
「免疫応答」および「免疫学的応答」とは、抗原またはワクチンに対する体液性および/または細胞性免疫応答を言う。体液性免疫応答とは、抗体分子により媒介される免疫応答を言う。細胞性免疫応答とは、Tリンパ球および/または他の白血球により媒介される免疫応答を言う。細胞性免疫応答には、例えばCTL産生、あるいはヘルパーT細胞の産生または活性化等が含まれる。細胞性免疫応答は、例えばCD8+ T細胞などの活性化T細胞またはその他の白血球から産生されるサイトカインまたはケモカインを調べることにより検出することができる。また、公知のリンパ球増殖アッセイ、CTLアッセイ、または抗原特異的T細胞のアッセイ等により決定することもできる。
【0025】
「組み換え」体とは、組み換えポリヌクレオチドを介して生成した化合物または組成物を言う。組み換えポリヌクレオチドとは、自然の状態と同じようには結合していないポリヌクレオチドを言う。組み換え蛋白質は、組み換えポリヌクレオチドを発現させて得ることができる。また「組み換え」ウイルスベクターとは、遺伝子操作により組み換えポリヌクレオチドを介して構築されたウイルスベクターまたはそれを増幅して得られるウイルスベクターを言う。
【0026】
本発明において「パラミクソウイルス」とはパラミクソウイルス科(Paramyxoviridae)に属するウイルスを指す。パラミクソウイルスとしては、例えばセンダイウイルス(Sendai virus)、ニューカッスル病ウイルス(Newcastle disease virus)、おたふくかぜウイルス(Mumps virus)、麻疹ウイルス(Measles virus)、RSウイルス(Respiratory syncytial virus)、牛疫ウイルス(rinderpest virus)、ジステンパーウイルス(distemper virus)、サルパラインフルエンザウイルス(SV5)、ヒトパラインフルエンザウイルス1,2,3型等が挙げられる。センダイウイルスは、野生株、変異株、ラボ継代株、および人為的に構築された株などが含まれる。DI粒子(J. Virol. 68, 8413-8417(1994))等の不完全ウイルスや、合成したオリゴヌクレオチド等も、本発明のワクチンを製造するための材料として使用することができる。
【0027】
パラミクソウイルスのウイルスタンパク質をコードする遺伝子としては、NP、P、M、F、HN、およびL遺伝子が含まれる。「NP、P、M、F、HN、およびL遺伝子」とは、それぞれヌクレオキャプシド、ホスホ、マトリックス、フュージョン、ヘマグルチニン-ノイラミニダーゼ、およびラージ蛋白質をコードする遺伝子のことを指す。パラミクソウイルス亜科に属する各ウイルスにおける各遺伝子は、一般に次のように表記される。一般に、NP遺伝子は「N遺伝子」と表記されることもある。
【0028】
パラミクソウイルス属 NP P/C/V M F HN - L
ルブラウイルス属 NP P/V M F HN (SH) L
モービリウイルス属 NP P/C/V M F H - L
【0029】
例えばパラミクソウイルス科(Paramyxoviridae)のレスピロウイルス属(Respirovirus)に分類されるセンダイウイルスの各遺伝子の塩基配列のデータベースのアクセッション番号は、NP遺伝子については M29343、M30202, M30203, M30204, M51331, M55565, M69046, X17218、P遺伝子については M30202, M30203, M30204, M55565, M69046, X00583, X17007, X17008、M遺伝子については D11446, K02742, M30202, M30203, M30204, M69046, U31956, X00584, X53056、F遺伝子については D00152, D11446, D17334, D17335, M30202, M30203, M30204, M69046, X00152, X02131、HN遺伝子については D26475, M12397, M30202, M30203, M30204, M69046, X00586, X02808, X56131、L遺伝子については D00053, M30202, M30203, M30204, M69040, X00587, X58886を参照のこと。
【0030】
本発明において「遺伝子」とは遺伝物質を指し、RNAおよびDNA等の核酸が含まれる。遺伝子は天然由来または人為的に設計された配列を持つものであり得る。本発明で用いられるパラミクソウイルスベクターは、免疫不全ウイルスのウイルス蛋白質またはその部分をコードする外来遺伝子を有する。該外来遺伝子は、天然の免疫不全ウイルスが有する遺伝子またはその断片であってもよく、また、例えば、天然のウイルス蛋白質の欠失体、変異体、不活化蛋白質、他の蛋白質との融合蛋白質などをコードする核酸であってもよい。また、本発明において「DNA」とは、一本鎖DNAおよび二本鎖DNAを含む。
【0031】
本発明において免疫不全ウイルスとは、ヒトまたは動物に免疫不全症候群を引き起こすウイルスを言う。免疫不全症候群とは、免疫系を構成する細胞要素の一部あるいは幾つかの欠損あるいは機能不全によって、正常な免疫機構に障害を生じた病態を言う。免疫不全ウイルスによる病原性は、主にCD4陽性 T細胞を中心とする免疫担当細胞の破壊を伴う。免疫不全ウイルスとしては、特にレンチウイルス亜科に属するヒト免疫不全ウイルス(human immunodeficiency virus; HIV)およびサル免疫不全ウイルス(simian immunodeficiency virus; SIV)等が挙げられる。HIVはヒトの後天性免疫不全症候群(AIDS)を引き起こすウイルスである。HIVには、I型(HIV-1)および II型(HIV-2)が存在する。本発明において免疫不全ウイルスには、HIV-1およびHIV-2の全ての株およびサブタイプが含まれる。また本発明において免疫不全ウイルスとしては、SIVの全ての株およびサブタイプを含む。SIVの単離株には、SIVmac、SIVagm、および SIVsmなどを含む多数の株が知られている。その他、免疫不全ウイルスとしては、ネコ免疫不全ウイルス(feline immunodeficiency virus; FIV)等を例示できる。
【0032】
ウイルス蛋白質とは、ウイルスに含まれる蛋白質を言う。ウイルス蛋白質としては、構造蛋白質、調節蛋白質、およびアクセサリー蛋白質が含まれる。例えば、レンチウイルスの主要な構造蛋白質としては、例えば Gag、Pol、および Env が挙げられ、調節蛋白質としては、例えば Tat および Rev、アクセサリー蛋白質としては、例えば Vpu、Vpr、Vif、および Nef が挙げられる。本発明においては、これらの蛋白質のいずれか、その部分、あるいはそれらの組み合わせを発現するSeVベクターが好適に用いられる。
【0033】
本発明において naked(ネイキッド)DNAとは、蛋白質に包装されていない裸のDNAを言う。通常、naked DNAは精製されている。「精製された」とは、天然における状態よりも純度が高いことを言い、好ましくはそれが存在する試料中の成分として主要な割合を占めることを言う。DNAの精製は、フェノールおよび/またはクロロホルム抽出、エタノール沈澱、PEG沈澱(PEG/NaCl沈澱)などの他、電気泳動、塩化セシウム超遠心、逆相カラム、ゲル濾過、HPLC、シリカ吸着などの公知の方法を適宜組み合わせて行うことができる。naked DNA は直鎖状または環状であり得る。例えばプラスミド、ポリメラーゼ連鎖反応(PCR)産物、ウイルスや細胞等から抽出した精製または粗精製DNA等であってよい。naked DNAは緩衝剤、塩、脂質、蛋白質等と組み合わされた人工的な複合物にできる。例えば、カチオン性脂質等のトランスフェクション試薬と組み合わせた組成物としてもよい。DNAワクチンとは、naked DNAを成分として含むワクチンを言う。
【0034】
本発明において「センダイウイルスベクター」とは、センダイウイルスに由来し遺伝子を宿主細胞に導入するベクター(担体)を指す。センダイウイルスベクターはリボ核タンパク質(RNP)であってもよく、また、感染力を持つウイルス粒子であってもよい。ここで「感染力」とは、組み換えセンダイウイルスベクターが細胞への接着能および膜融合能を保持していることにより、接着した細胞の内部にベクター内部の遺伝子を導入することのできる能力を言う。本発明におけるセンダイウイルスベクターは、抗原となる免疫不全ウイルス蛋白質をコードする外来遺伝子を発現することができるように保持する。センダイウイルスベクターは野生型と同等の複製能力を有していてもよく、また遺伝子変異により弱毒化されていてもよい。また、本発明におけるセンダイウイルスベクターは、複製能を有さない欠損型ベクターであってもよい。「複製能を有する」とは、ウイルスベクターが宿主細胞に感染した場合、該細胞においてウイルスが複製され、感染性ウイルス粒子が産生されることを指す。
【0035】
本発明は、免疫不全ウイルスのウイルス蛋白質またはその一部を発現可能に保持するセンダイウイルスベクターを含むワクチンを提供する。センダイウイルスにコードされる免疫不全ウイルスのウイルス蛋白質としては、免疫原性を有する限り特に制限はない。免疫不全ウイルスのウイルス蛋白質には、該ウイルスの構造蛋白質、調節蛋白質、およびアクセサリー蛋白質が含まれる。例えば、HIV-1を含むレンチウイルスの主要な構造蛋白質としては、例えば Gag、Pol、および Env が挙げられ、調節蛋白質としては、例えば Tat および Rev、アクセサリー蛋白質としては、例えば Vpu、Vpr、Vif、および Nef が挙げられる。ワクチンの製造には、これらの蛋白質またはその部分ペプチド等を用いる。上記の蛋白質またはその部分を発現するセンダイウイルスベクターを構築することにより、ワクチンの製造を行うことができる。これらのタンパク質は単独で用いても、また複数を組み合わせて用いてもよい。本発明においては、特に免疫不全ウイルスの構造蛋白質を発現するSeVを用いることが好ましい。具体的には、Gag蛋白質全長、Gag-Pol融合蛋白質、またはそれらの断片などを発現するSeVを用いることができる。
【0036】
本発明者等により、鼻腔内接種でマカクサルに導入された組み換えSeVベクターからの遺伝子の発現は、接種後1週間以内にピークに達し、少なくとも13日まで持続することが示された。また、反復投与により発現を持続することも可能である。このことは、組み換えSeVベクターを利用してワクチネーションを行った場合に、早急かつ持続的な治療効果を得ることができるという利点をもたらす。
【0037】
また、安全性の面においても、SeVベクターはヒトへの臨床適用に好適に用いられうる可能性が示唆される。第一に、外来遺伝子の発現は、多くのベクター場合、導入したDNAの核局在化が必要であることが、遺伝子導入の成功率を低下させる主要な障害になっている。しかし、例えばSeVなどの場合、外来遺伝子の発現は、細胞質内において細胞性チューブリンおよび自身が持つRNAポリメラーゼ(L蛋白質)の両方によって駆動される。これは、SeVが宿主のゲノムと相互作用しないことを示しており、癌化などの安全面における問題が生じないと考えられる。第二に、SeVは齧歯類にとっては病原性で肺炎を生じることが知られているが、人には病原性ではない。これはまた、野生型SeVの経鼻的投与によって非ヒト霊長類において重篤な有害作用を示さないというこれまでの報告によっても支持されている(Hurwitz, J.L. et al., Vaccine 15: 533-540, 1997)。SeVのこれらの特徴は、SeVベクターが、ヒトへの応用に際して高い安全性を有することを示唆し、ワクチネーションのための抗原蛋白の発現を目的としたベクターの有望な選択肢の一つとなることを支持するものである。実際、本発明においてSeVを接種された霊長類マカクサルでは、抗原の有意な発現が検出された。またSeVベクター接種自体による明確な病理学的症状は見られず、末梢のCD4またはCD8細胞数の有意な減少も観察されなかった。
【0038】
本発明のワクチンは、特にAIDSウイルスを対象とするワクチネーションに好適に利用され得る。すなわち、本発明のワクチンを接種することにより、免疫不全ウイルスに対する免疫を誘導し、該ウイルスの感染および/または増殖を防止することが可能になる。本発明のワクチンは、免疫不全ウイルスの感染前に行う予防、および感染後に行う治療において好適に用いられる。
【0039】
本発明においてワクチンに用いられるSeVベクターとしては特に制限はない。好適なSeVベクターとして、例えば、複製能を有し、自立的に増殖するようなベクターが挙げられる。例えば、一般に天然型SeVのゲノムは、3'の短いリーダー領域に続き、N(ヌクレオキャプシド)、P(ホスホ)、M(マトリックス)、F(フュージョン)、HN(ヘマグルチニン-ノイラミニダーゼ)、およびL(ラージ)蛋白質をコードする6つの遺伝子が並んでおり、短い5'トレイラー領域を他端に有する。これと同様の構造を有するゲノムを設計することにより、自律複製可能なベクターを製造することができる。ゲノム内に外来遺伝子を挿入することにより、外来遺伝子を発現するベクターを製造することができる。なお、SeVベクターにおいては、SeV遺伝子の配置は野生型ウイルスから改変されていてもよい。
【0040】
また、本発明においてワクチンに用いられるSeVベクターとしては、野生型SeVが持つ遺伝子のいずれかを欠損したものであってもよい。例えば、SeVベクターを再構成し、遺伝子を発現させるためには、NP、P/CおよびL遺伝子から作られる蛋白質群が必要だと考えられているので、該蛋白質群をコードする遺伝子がSeVベクターに含まれている必要がある。しかし、M、F、および HN 蛋白質はトランスに供給してSeVベクターを再構成することができ、ベクターからの遺伝子発現が可能である。これらの蛋白質群をコードする遺伝子を有する発現ベクターを、SeVベクターゲノムをコードする発現ベクターと共に宿主細胞にトランスフェクションすることにより、ベクターの再構成を行うことができる。また、該蛋白質群をコードする遺伝子を有する宿主細胞にベクターゲノムをコードする発現ベクターを導入し、該宿主細胞から該蛋白質群を供給して再構成を行ってもよい。これらの蛋白質群のアミノ酸配列は、ウイルス由来の配列そのままでなくとも、核酸の導入における活性が天然型のそれと同等かそれ以上ならば、変異を導入したり、あるいは他のウイルスの相同遺伝子で代用してもよい。
【0041】
また、SeVベクターをRNPとして調製する場合は、SeVベクターが細胞に伝播してゆくために必要と考えられている M、FおよびHN遺伝子から作られる蛋白質は必要ない。RNPに含まれるゲノムに、M、FおよびHN遺伝子が含まれていれば、宿主に導入された時に、これらの遺伝子産物が生産され、感染性のあるウイルス粒子が形成される。感染性ウイルスを産生するRNPベクターとしては、例えば N、P、M、F、HN、およびL遺伝子をコードするウイルスゲノムRNAと、N蛋白質、P蛋白質、およびL蛋白質とを含むRNPが挙げられる。このようなRNPを細胞内に導入すると、N蛋白質、P蛋白質、およびL蛋白質の働きによりウイルスゲノムが発現、複製され、感染性ウイルスベクターが増幅する。
【0042】
RNPを細胞に導入するには、例えばリポフェクトアミンやポリカチオニックリポソームなどと共に複合体を形成させて導入することが可能である。具体的には、種々のトランスフェクション試薬が利用できる。例えば、DOTMA(Boehringer)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Boehringer #1811169)などが挙げられる。エンドソーム中での分解を防ぐため、クロロキンを加えることもできる(Calos, M.P., 1983, Proc. Natl. Acad. Sci. USA 80: 3015)。複製型ウイルスの場合、産生されたウイルスは、培養細胞、鶏卵、個体(例えばマウスなどの哺乳動物)などに再感染させて増幅または継代することができる。
【0043】
逆に、M、Fおよび/またはHN遺伝子が含まれていないSeVベクターも、本発明のワクチンに用いるSeVベクターとして好適である。このようなウイルスベクターの再構成は、例えば、欠損している遺伝子産物を外来的に供給することにより行うことができる。このようにして製造されたウイルスベクターは、野生型ウイルスと同様に宿主細胞に接着して細胞融合を起こすが、細胞に導入されたベクターゲノムはこれらのいずれかの遺伝子を欠損しているため、最初と同じような感染力を持つ娘ウイルス粒子は形成されない。このため、一回限りの遺伝子導入力を持つ安全なウイルスベクターとして有用である。ゲノムから欠損させる遺伝子としては、例えばF遺伝子および/またはHN遺伝子が挙げられる。例えば、F遺伝子が欠損した組み換えパラミクソウイルスベクターゲノムを発現するプラスミドを、F蛋白質の発現ベクターならびにNP、P/CおよびL蛋白質の発現ベクターと共に宿主細胞にトランスフェクションすることにより、ウイルスベクターの再構成を行うことができる(国際公開番号 WO00/70055 および WO00/70070)。また、例えば、F遺伝子が染色体に組み込まれた宿主細胞を用いて製造することもできる。これらの蛋白質群を外から供給する場合、そのアミノ酸配列はウイルス由来の配列そのままでなくとも、核酸の導入における活性が天然型のそれと同等かそれ以上ならば、変異を導入したり、あるいは他のウイルスの相同遺伝子で代用してもよい。
【0044】
また、ベクターゲノムが由来するウイルスのエンベロープ蛋白質とは異なる蛋白質をベクターのエンベロープに含むSeVベクターを作製することもできる。このようなタンパク質に特に制限はない。例えば、他のウイルスのエンベロープタンパク質、例えば水疱性口内炎ウイルス(VSV)のGタンパク質(VSV-G)を挙げることができる。本発明のワクチンを構成するSeVベクターとしては、VSV-Gタンパク質などのように、ゲノムが由来するウイルス以外のウイルスに由来するエンベロープタンパク質を含むシュードタイプウイルスベクターが含まれる。
【0045】
また、本発明のワクチンに用いられるSeVベクターは、例えば、エンベロープ表面に特定の細胞に接着しうるような接着因子、リガンド、受容体等の蛋白質、あるいはこれらの蛋白質を細胞外領域に有し、ウイルスエンベロープ由来のポリペプチドを細胞内領域に有するキメラタンパク質などを含むものであってもよい。これにより、特定の組織を標的とするベクターを作り出すこともできる。これらはウイルスゲノムにコードされていてもよいし、ウイルスベクターの再構成時に、ウイルスゲノム以外の遺伝子(例えば別の発現ベクターまたは宿主染色体などの遺伝子)の発現により供給されてもよい。
【0046】
また、本発明のワクチンに用いられるSeVベクターは、例えばSeV蛋白質に対する免疫原性を低下させるために、またはRNAの転写効率や複製効率を高めるために、ベクターに含まれるウイルス遺伝子が改変されたものであってもよい。具体的には、例えば複製因子であるNP遺伝子、P/C遺伝子およびL遺伝子の少なくとも一つを改変し、転写または複製の機能を高めることが考えられる。また、構造体蛋白質の1つであるHN蛋白質は、赤血球凝集素であるヘマグルチニン(hemagglutinin)活性とノイラミニダーゼ(neuraminidase)活性との両者の活性を有するが、例えば前者の活性を弱めることができれば、血液中でのウイルスの安定性を向上させることが可能であろうし、例えば後者の活性を改変することにより、感染能を調節することも可能である。また、膜融合に関わるF蛋白質を改変することにより、膜融合リポソームの融合能を調節することもできる。また、例えば、細胞表面の抗原分子となりうるF蛋白質やHN蛋白質の抗原提示エピトープ等を解析し、これを利用してこれらの蛋白質に関する抗原提示能を弱めたSeVを作製することもできる。
【0047】
また、本発明のワクチンに用いられるSeVは、アクセサリー遺伝子が欠損したものであってよい。例えばSeVのアクセサリー遺伝子の1つであるV遺伝子をノックアウトすることにより、培養細胞における遺伝子発現や複製は障害されることなく、マウスに対するSeVの病原性が顕著に減少する(Kato, A. et al. 1997. J. Virol. 71:7266-7272; Kato, A. et al. 1997. EMBO J. 16:578-587; Curran, J. et al., WO01/04272, EP1067179)。このような弱毒化ベクターは、本発明のワクチンを構成するベクターとして特に好適である。
【0048】
本発明のワクチンに用いられるウイルスベクターは、ゲノムRNA中に免疫不全ウイルスのウイルス蛋白質またはその部分をコードする。このような外来遺伝子を発現する組み換えSeVベクターは、上記のSeVベクターゲノムに外来遺伝子を挿入することによって得られる。外来遺伝子としては、免疫不全ウイルスのウイルス蛋白質またはその部分をコードする遺伝子断片を用いることができる。該遺伝子断片は、免疫不全ウイルス蛋白質をコードする天然由来の遺伝子断片であってもよく、また天然型蛋白質と少なくとも部分的に同等の抗原性を有する蛋白質をコードする限り、欠失、置換または挿入等により天然型蛋白質を改変した蛋白質をコードする遺伝子であってもよい。
【0049】
HIVを含む免疫不全ウイルスのウイルス蛋白質としては、構造蛋白質、調節蛋白質、およびアクセサリー蛋白質が含まれる。例えば、レンチウイルスの主要な構造蛋白質としては、例えば Gag、Pol、および Env が挙げられ、調節蛋白質としては、例えば Tat および Rev、アクセサリー蛋白質としては、例えば Vpu、Vpr、Vif、および Nef が挙げられる。本発明においては、これらの蛋白質、その部分、あるいはそれらの組み合わせを発現するSeVベクターが好適に用いられる。
【0050】
例えばHIV-1においては、Gagは p55 と呼ばれる約55kDの前駆蛋白質として発現され、pol遺伝子がコードするプロテアーゼにより切断され MA(matrix; マトリックス [p17])、CA(capsid, キャプシド [p24])、NC(nucleocapsid, ヌクレオキャプシド [p9])および p6 が生じる(Gottlinger, H. G. et al., Proc. Natl. Acad. Sci. USA 1989; 86: 5781-5785)。pol遺伝子は、ウイルスのプロテアーゼ(Pro)、インテグラーゼ(IN)、RNaseH、および逆転写酵素(RT)をコードし、まず Gag-Pol融合蛋白質として発現される(Jacks, T. et al., Nature 1988; 331: 280-283)。Gag-Pol前駆体(p160)は、リボソームによる翻訳時に約5%の頻度で生じるフレームシフトにより生成する。Polポリペプチドは、ウイルスがコードするプロテアーゼによりGagから切断され、さらにプロテアーゼ(p10)、RT(p50)、RNaseH(p15)、およびインテグラーゼ(p31)に分断される。また、例えばRT蛋白とRNaseHが繋がったポリペプチド(p65)などの、完全に分断されない断片も存在する。160kDのEnv(gp160)は、細胞のプロテアーゼによりgp41およびgp120に切断される。gp120はウイルス感染において、標的細胞表面に存在するレセプター CD4 およびコレセプターである CCR5などと相互作用する(Berger, E. A. et al., Annu. Rev. Immunol. 1999; 17: 657-700)。また、gp120には V1〜V5 と呼ばれるハイパーバリアブル領域があり、これらは単離株により比較的大きく異なっており、中でもV3 loopと呼ばれる領域はウイルスのトロピズムに影響する(Hwang, S. S. et al., Science 1991; 253: 71-74)。V3 loopは、HIV感染を防ぐための中和抗体主要なターゲットでもある(Goudsmit, J, et al., Proc. Natl. Acad. Sci. USA 1988; 85: 4478-4482)。
【0051】
Tatはウイルス複製に必須の転写のトランスアクチベーターであり、長さに違う複数のペプチドが発現される(Ruben, S. et al., J. Virol. 1989; 63: 1-8)。Revは約13kDの配列特異的なRNA結合蛋白質であり(Zapp, M. L. and Green, M. R., Nature 1989; 342: 714-716)、Rev応答配列(RRE)に結合しウイルス遺伝子発現のフェーズを制御する。これらの遺伝子に加え、HIV-1にはアクセサリー蛋白質をコードする nef、vif、vpr、および vpu の4つの遺伝子が存在する。HIV-2には、vpu の代わりに vpx を持つ。
【0052】
SeVベクターは、これらのウイルス蛋白質(プロセシングを受けた蛋白質および受けていない蛋白質を含む)のいずれかの全長またはその部分、あるいはそれらの組み合わせを発現するように構築される。部分としては、抗原となる活性を有する限り長さや位置等に制限はない。例えば、1つまたはそれ以上のエピトープを含む部分ペプチドであってよい。このような部分ペプチドは、通常、ウイルス蛋白質のアミノ酸配列中、連続した少なくとも3個〜数個程度のアミノ酸を含むペプチドである。好ましくは、ウイルス蛋白質のアミノ酸配列中、連続した約7〜約15アミノ酸、例えば8、9、10、12、または14アミノ酸を含んでいる。
【0053】
ワクチンは、免疫不全ウイルスのウイルス蛋白質のうち少なくとも1種類を用いる。本発明によれば、Gag抗原のみを発現させた場合でも、効率的な免疫応答を誘導することが可能であった。また、複数のタイプの蛋白質を抗原として用いれば、より高い免疫を獲得させることが可能となる。
【0054】
また、ワクチンは、免疫不全ウイルスのうち、1種類のウイルス由来のウイルス蛋白質を用いてもよいが、複数のタイプのウイルス由来のウイルス蛋白質を抗原として用いれば、より広い株またはサブタイプの免疫不全ウイルスに対する免疫を獲得させることができる。複数のタイプの免疫不全ウイルスを抗原として用いる場合、その組み合わせに特に制限はなく、例えば、HIV-1、HIV-2、またはSIVの各種単離株由来の遺伝子を用いてワクチンを製造することができる。複数の免疫不全ウイルスの遺伝子は、それぞれ別々のSeVベクターゲノムに組み込んでSeVを構築後、それらを組み合わせたり、混合したりしてワクチンを製造することもできれば、同じSeVベクターゲノムに複数の遺伝子を組み込んで発現させることもできる。
【0055】
例えば、HIV-1としては、全てのメジャー(M)サブタイプ(AからJを含む)、Nおよび outlier(O)が含まれる(Hu, D. J. et al., JAMA 1996; 275: 210-216; Zhu, T. et al., Nature 1998, 5; 391(6667): 594-7; Simon, F. et al., Nat. Med. 1998, 4(9): 1032-7)。SIV単離株としては、SIVagm、SIVcpz、SIVmac、SIVmnd、SIVsnm、SIVsyk等が例示できる。
【0056】
本発明のワクチンに用いられるウイルス蛋白質が由来する免疫不全ウイルスとしては、特に霊長類を宿主とする免疫不全ウイルスが好ましい。このようなウイルスとしては、例えばHIV-1、HIV-2、およびSIVが挙げられる。
【0057】
異なる株またはサブタイプのウイルスであっても、その蛋白質のアミノ酸配列が高い相同性を有していれば、ある株に属する1種の免疫不全ウイルスのウイルス蛋白質を抗原としたワクチネーションにより、異なる株またはサブタイプに対する免疫もある程度獲得できる可能性が高い。
【0058】
免疫不全ウイルスのウイルス蛋白質を発現するSeVを構築するには、例えば、SeVゲノムをコードするDNA(SeVベクターDNA)に目的の免疫不全ウイルスのウイルス蛋白質をコードする遺伝子を挿入する。ウイルスベクターDNAに外来遺伝子を導入する場合は、例えば、SeVベクターDNAにおいては、転写終結(E)配列と転写開始(S)配列との間などに、6の倍数の塩基数を有する配列を挿入することが望ましい(Journal of Virology, Vol.67, No.8, 1993, p.4822-4830)。外来遺伝子は、SeVの各遺伝子(NP、P、M、F、HN、およびL遺伝子)の前および/または後ろに挿入することができる。前後の遺伝子の発現を妨げないようにするため、外来遺伝子の前または後ろに適宜 E-I-S配列(転写終結配列−介在配列−転写開始配列)またはその部分を挿入し、各遺伝子の間にE-I-S配列を配置する。あるいは、IRESを挿入して外来遺伝子を発現し得る。
【0059】
挿入した外来性遺伝子の発現量は、外来遺伝子の上流に付加する転写開始配列の種類により調節することができる。また、遺伝子挿入の位置、および遺伝子の前後の塩基配列により調節しうる。例えば、SeVにおいては、挿入位置がウイルスゲノムのネガティブ鎖RNAの3'端に近いほど(野生型ウイルスのゲノム上の遺伝子配置においては、NP遺伝子に近いほど)、挿入された遺伝子の発現量が高い。外来遺伝子の高い発現を得るためには、外来遺伝子をNP遺伝子の上流(マイナス鎖においては3'側)またはNP遺伝子とP遺伝子の間など、ネガティブ鎖ゲノムにおいて上流領域に挿入することが好ましい。逆に、挿入位置がネガティブ鎖RNAの5'端に近いほど(野生型ウイルスのゲノム上の遺伝子配置においては、L遺伝子に近いほど)、挿入された遺伝子の発現量が低くなる。外来遺伝子の発現を低く抑えるためには、例えばネガティブ鎖の最も5'側、すなわち野生型ウイルスゲノムにおいてはL遺伝子の下流(ネガティブ鎖においてはL遺伝子の5'隣接部位)、またはL遺伝子の上流(ネガティブ鎖においてはL遺伝子の3'隣接部位)に外来遺伝子を挿入する。このように、外来遺伝子の挿入位置は、該遺伝子の所望の発現量を得るために、また前後のウイルスタンパク質をコードする遺伝子との組み合わせが最適となる様に適宜調節することができる。例えば、高力価ウイルスベクターの投与による導入遺伝子の高発現が毒性を示す場合は、投与するウイルス力価を制限することができる他、例えばベクターにおける外来遺伝子の挿入位置をネガティブ鎖のなるべく5'側に設定したり、転写開始配列を効率の低いものにするなどして、個々のSeVベクターからの発現レベルを低く抑えることで適切な効果が得られるすることも可能である。
【0060】
一般に、細胞毒性を示さない限りにおいて、抗原蛋白質の高い発現が得られれば、免疫獲得に有利と考えられるため、抗原蛋白質をコードする遺伝子は、効率の高い転写開始配列に連結し、ネガティブ鎖ゲノムの3'端近くに挿入することが好ましい。好適なベクターの例としては、免疫不全ウイルスのウイルス蛋白質が、パラミクソウイルスベクターのネガティブ鎖ゲノムにおいて、該パラミクソウイルスのウイルス蛋白質のいずれよりも3'側にコードされているベクターが挙げられる。例えばN遺伝子の上流(ネガティブ鎖の3'側)に抗原遺伝子が挿入されたベクターが好ましい。あるいは、N遺伝子のすぐ下流に挿入してもよい。
【0061】
外来遺伝子を容易に挿入できるようにするために、挿入部位にクローニングサイトを設計することができる。クローニングサイトは、例えば制限酵素の認識配列とすることができる。ゲノムをコードするベクターDNA中の当該制限酵素部位に外来遺伝子断片を挿入することができる。クローニングサイトは、複数の制限酵素認識配列を有する、いわゆるマルチクローニングサイトとしてもよい。また、本発明のワクチンに用いられるベクターは、このように免疫不全ウイルスのウイルス蛋白質をコードする遺伝子を挿入した以外に位置に他の外来遺伝子を保持していてもよい。このような外来遺伝子としては制限はなく、免疫誘導に関与するサイトカインまたはケモカイン遺伝子等であってもよく、また、その他の遺伝子であってもよい。
【0062】
外来遺伝子を有する組み換えセンダイウイルスベクターは、例えば、Hasan, M.K. et al. J. Gen. Virol. 78: 2813-2820, 1997 及び Yu, D. et al., 1997, Genes Cells 2: 457-466の記載に準じて、次のようにして構築することができる。
【0063】
まず、所望の外来遺伝子のcDNA塩基配列を含むDNA試料を用意する。DNA試料は、25ng/μl以上の濃度で電気泳動的に単一のプラスミドと確認できることが好ましい。以下、外来遺伝子を、NotI部位を利用してウイルスゲノムをコードするDNAに挿入する場合を例にとって説明する。挿入したいcDNA塩基配列の中にNotI認識部位が含まれる場合は、部位特異的変異挿入法などを用いて、コードするアミノ酸配列を変化させないように塩基配列を改変し、NotI部位を予め除去しておくことが好ましい。この試料から所望の遺伝子断片をPCRにより増幅回収する。増幅された断片の両端をNotI部位とし、さらに一端にセンダイウイルスの転写終結配列(E)、介在配列(I)及び転写開始配列(S)(EIS配列)のコピーを付加するために、NotI制限酵素切断部位配列及び転写終結配列(E)、介在配列(I)及び転写開始配列(S)と目的遺伝子の一部の配列を含むプライマー対として、フォワード側(センス鎖)合成DNA配列及びリバース側(アンチセンス鎖)合成DNA配列(プライマーセット)を作成する。
【0064】
例えば、フォワード側合成DNA配列は、NotIによる切断を保証するために 5'側に任意の2以上のヌクレオチド(好ましくはGCGおよびGCCなどのNotI認識部位由来の配列が含まれない4塩基、更に好ましくはACTT)を選択し、その3'側にNotI認識部位gcggccgcを付加し、さらにその3'側にスペーサー配列として任意の9塩基または9に6の倍数を加えた数の塩基を付加し、さらにその3'側に所望のcDNAの開始コドンATGからこれを含めてORFの約25塩基相当の配列を付加した形態とする。最後の塩基はGまたはCとなるように該所望のcDNAから約25塩基を選択してフォワード側合成オリゴDNAの3'の末端とすることが好ましい。
【0065】
リバース側合成DNA配列は5'側から任意の2以上のヌクレオチド(好ましくはGCGおよびGCCなどのNotI認識部位由来の配列が含まれない4塩基、更に好ましくはACTT)を選択し、その3'側にNotI認識部位gcggccgcを付加し、さらにその3'側に長さを調節するための挿入断片のオリゴDNAを付加する。このオリゴDNAの長さは、NotI認識部位gcggccgcを含め、cDNAの相補鎖塩基配列と後述するセンダイウイルスに由来するセンダイウイルスゲノムのEIS塩基配列の合計が6の倍数になるように塩基数を設計する(いわゆる「6のルール(rule of six)」; Kolakofski, D. et al., J. Virol. 72: 891-899, 1998; Calain, P. and Roux, L., J. Virol. 67: 4822-4830, 1993)。さらに挿入断片の3'側にセンダイウイルスのS配列の相補鎖配列、好ましくは5'-CTTTCACCCT-3'(配列番号:1)、I配列、好ましくは5'-AAG-3'、E配列の相補鎖配列、好ましくは5'-TTTTTCTTACTACGG-3'(配列番号:2)、さらにその3'側に所望のcDNA配列の終始コドンから逆に数えて約25塩基相当の相補鎖の最後の塩基がGまたはCになるように長さを選択して配列を付加し、リバース側合成オリゴDNAの3'の末端とする。
【0066】
PCRは、例えば、ExTaqポリメラーゼ(宝酒造)を用いる通常の方法を用いることができる。好ましくはVentポリメラーゼ(NEB)を用いて行い、増幅したDNA断片はNotIで消化した後、プラスミドベクターpBluescriptのNotI部位に挿入する。得られたPCR産物の塩基配列をシークエンサーで確認し、正しい配列のプラスミドを選択する。このプラスミドから挿入断片をNotIで切り出し、パラミクソウイルスゲノムcDNAを含むプラスミドのNotI部位にクローニングする。またプラスミドベクターpBluescriptを介さずにNotI部位に直接挿入し、組み換えセンダイウイルスcDNAを得ることも可能である。
【0067】
例えば、組み換えセンダイウイルスゲノムcDNAであれば、文献記載の方法に準じて構築することができる(Yu, D. et al., Genes Cells 2: 457-466, 1997; Hasan, M.K. et al. J. Gen. Virol. 78: 2813-2820, 1997)。例えば、まずNotI制限部位を有する18bpのスペーサー配列(5'-(G)-CGGCCGCAGATCTTCACG-3')(配列番号:3)を、クローニングされたセンダイウイルスゲノムcDNA(pSeV(+))のリーダー配列とN-タンパク質をコードする配列の5'末端との間の隣接遺伝子座に挿入し、デルタ肝炎ウイルスのアンチゲノム鎖(antigenomic strand)由来の自己開裂リボザイム部位を含むプラスミドpSeV18+b(+)を得る(Hasan, M. K. et al., 1997, J. General Virology 78: 2813-2820)。pSeV18+b(+)のNotI部位に外来遺伝子断片を挿入し、所望の外来遺伝子が組込まれた組み換えセンダイウイルスcDNAを得ることができる。
【0068】
このようにして作製した組み換えパラミクソウイルスベクターDNAを試験管内または細胞内で転写させ、ウイルスのL、P、およびNPタンパク質の共存下でRNPを再構成させると、このRNPを含むウイルスベクターを生成させることができる。本発明は、免疫不全ウイルスのウイルス蛋白質をコードするパラミクソウイルスベクターのゲノムDNAを転写させる工程を含む、該ベクターを含むワクチンの製造方法を提供する。また本発明は、該DNAからなる、本発明のワクチンの成分として用いるパラミクソウイルスベクター製造用DNAを提供する。また本発明は、本発明のワクチンの成分となるパラミクソウイルスベクターを製造するための、該ベクターのゲノムをコードするDNAの使用に関する。ウイルスベクターDNAからのウイルスの再構成は公知の方法に従って行うことができる(国際公開97/16539号; 国際公開97/16538号; Durbin, A.P. et al., 1997, Virology 235: 323-332; Whelan, S.P. et al., 1995, Proc. Natl. Acad. Sci. USA 92: 8388-8392; Schnell. M.J. et al., 1994, EMBO J. 13: 4195-4203; Radecke, F. et al., 1995, EMBO J. 14: 5773-5784; Lawson, N.D. et al., Proc. Natl. Acad. Sci. USA 92: 4477-4481; Garcin, D. et al., 1995, EMBO J. 14: 6087-6094; Kato, A. et al., 1996, Genes Cells 1: 569-579; Baron, M.D. and Barrett, T., 1997, J. Virol. 71: 1265-1271; Bridgen, A. and Elliott, R.M., 1996, Proc. Natl. Acad. Sci. USA 93: 15400-15404)。これらの方法により、パラインフルエンザ、水疱性口内炎ウイルス、狂犬病ウイルス、麻疹ウイルス、リンダーペストウイルス、センダイウイルスなどを含むパラミクソウイルスベクターをDNAからウイルスを再構成させることができる。ウイルスベクターDNAにおいて、F遺伝子、HN遺伝子、および/またはM遺伝子を欠失させた場合には、そのままでは感染性のウイルス粒子を形成しないが、宿主細胞に、これら欠失させた遺伝子および/または他のウイルスのエンベロープ蛋白質をコードする遺伝子などを別途、導入し発現させることにより、感染性のウイルス粒子を形成させることが可能である。
【0069】
例えば、ベクターDNAを細胞内に導入する方法には、次のような方法、マル1 目的の細胞が取り込めるようなDNA沈殿物を作る方法、マル2 目的の細胞による取りこみに適し、かつ細胞毒性の少ない陽電荷特性を持つDNAを含む複合体を作る方法、マル3 目的の細胞膜に、DNA分子が通り抜けられるだけに十分な穴を電気パルスによって瞬間的に開ける方法などがある。
【0070】
マル2 としては、種々のトランスフェクション試薬が利用できる。例えば、DOTMA(Boehringer)、Superfect(QIAGEN #301305)、DOTAP、DOPE、DOSPER(Boehringer #1811169)などが挙げられる。マル1 としては例えばリン酸カルシウムを用いたトランスフェクション法が挙げられ、この方法によって細胞内に入ったDNAは貧食小胞に取り込まれるが、核内にも十分な量のDNAが入ることが知られている(Graham, F.L. and Van Der Eb, J., 1973, Virology 52: 456; Wigler, M. and Silverstein, S., 1977, Cell 11: 223)。ChenおよびOkayamaはトランスファー技術の最適化を検討し、1) 細胞を共沈殿物のインキュベーション条件を 2〜4% CO2 、35℃、15〜24時間、2) DNAは直鎖状より環状のものが活性が高く、3) 沈殿混液中のDNA濃度が 20〜30μg/mlのとき最適な沈殿が得られると報告している(Chen, C. and Okayama, H., 1987, Mol. Cell. Biol. 7: 2745)。マル2 の方法は、一過的なトランスフェクションに適している。古くはDEAE-デキストラン(Sigma #D-9885 M.W. 5×105 )混液を所望のDNA濃度比で調製し、トランスフェクションを行う方法が知られている。複合体の多くはエンドソームの中で分解されてしまうため、効果を高めるためにクロロキンを加えることもできる(Calos, M.P., 1983, Proc. Natl. Acad. Sci. USA 80: 3015)。マル3 の方法は電気穿孔法と呼ばれる方法で、細胞選択性がないという点でマル1 やマル2 の方法に比べて汎用性が高い。効率はパルス電流の持続時間、パルスの形、電界(電極間のギャップ、電圧)の強さ、バッファーの導電率、DNA濃度、細胞密度の最適条件下で良いとされている。
【0071】
以上、3つのカテゴリーの中でマル2 の方法は操作が簡便で多量の細胞を用いて多数の検体を検討することができるので、ベクター再構成のためのDNAの細胞への導入にはトランスフェクション試薬が適している。好適には Superfect Transfection Ragent(QIAGEN, Cat No. 301305)、または DOSPER Liposomal Transfection Reagent(Boehringer Mannheim, Cat No. 1811169)が用いられる。
【0072】
cDNAからの再構成は具体的には次のようにして行うことができる。
【0073】
24穴から6穴程度のプラスチックプレートまたは100mmペトリ皿上で、10%ウシ胎児血清(FCS)および抗生物質(100 units/ml ペニシリンGおよび100μg/ml ストレプトマイシン)を含む最少必須培地(MEM)を用いてサル腎臓由来細胞株LLC-MK2を70〜80%コンフルエントになるまで培養し、例えば 1μg/ml psoralen(ソラレン)存在下 UV照射処理を20分処理で不活化した、T7ポリメラーゼを発現する組み換えワクシニアウイルスvTF7-3(Fuerst, T.R. et al., Proc. Natl. Acad. Sci. USA 83: 8122-8126,1986、Kato, A. et al., Genes Cells 1: 569-579, 1996)を2 PFU/細胞で感染させる。ソラレンの添加量およびUV照射時間が適宜調整することができる。感染1時間後、2〜60μg、より好ましくは3〜5μgの上記の組み換えセンダイウイルスcDNAを、全長センダイウイルスゲノムの生成に必須なトランスに作用するウイルスタンパク質を発現するプラスミド(例えば24-0.5μgのpGEM-N、12-0.25μgのpGEM-P、および24-0.5μgのpGEM-L、より好ましくは例えば1μgのpGEM-N、0.5μgのpGEM-P、および1μgのpGEM-L)(Kato, A. et al., Genes Cells 1: 569-579,1996)と共にSuperfect(QIAGEN社)を用いたリポフェクション法等によりトランスフェクションする。トランスフェクションを行った細胞は、所望により100μg/mlのリファンピシン(Sigma)及びシトシンアラビノシド(AraC)、より好ましくは40μg/mlのシトシンアラビノシド(AraC)(Sigma)のみを含む血清不含のMEMで培養し、ワクシニアウイルスによる細胞毒性を最少にとどめ、ウイルスの回収率を最大にするように薬剤の最適濃度を設定する(Kato, A. et al., 1996, Genes Cells 1: 569-579)。トランスフェクションから48〜72時間程度培養後、細胞を回収し、凍結融解を3回繰り返して細胞を破砕した後、LLC-MK2細胞にトランスフェクションして培養する。培養3〜7日後に培養液を回収する。エンベロープ蛋白質をコードする遺伝子を欠損した複製能を持たないウイルスベクターを再構成させるには、エンベロープタンパク質を発現するLLC-MK2細胞をトランスフェクションに使用するか、またはエンベロープ発現プラスミドを共にトランスフェクションすればよい。また、トランスフェクションを行った細胞にエンベロープタンパク質を発現するLLC-MK2細胞に重層して培養することによって欠損型ウイルスベクターを増幅することもできる(国際公開番号 WO00/70055 および WO00/70070参照)。培養上清に含まれるウイルス力価は赤血球凝集活性(HA)を測定することにより決定することができる。HAは「endo-point 希釈法」(Kato, A. et al., 1996, Genes Cells 1: 569-579; Yonemitsu, Y. & Kaneda, Y., Hemaggulutinating virus of Japan-liposome-mediated gene delivery to vascular cells. Ed. by Baker AH. Molecular Biology of Vascular Diseases. Method in Molecular Medicine: Humana Press: pp. 295-306, 1999)により決定することができる。混入し得るワクシニアウイルスvTF7-3の除去は、得られた尿液試料を適宜希釈(例えば106 倍)して vTF7-3 を除去した後、ベクターウイルスを鶏卵で再増幅させることで可能である。再増幅は、例えば3回上繰り返すことができる。得られたウイルスストックは-80℃で保存することができる。
【0074】
ウイルスベクターが再構成する限り、再構成に用いる宿主細胞は特に制限されない。例えば、LLCMK2細胞、サル腎由来のCV-1細胞、ハムスター腎由来のBHK細胞などの培養細胞、ヒト由来細胞等を使うことができる。また、大量にセンダイウイルスベクターを得るために、上記の宿主から得られたウイルスベクターを発育鶏卵に感染させ、該ベクターを増幅することができる。鶏卵を使ったウイルスベクターの製造方法は既に開発されている(中西ら編,(1993),「神経科学研究の先端技術プロトコールIII, 分子神経細胞生理学」, 厚生社, 大阪, pp.153-172)。具体的には、例えば、受精卵を培養器に入れ9〜12日間 37〜38℃で培養し、胚を成長させる。ウイルスベクターを尿膜腔へ接種し、数日間卵を培養してウイルスベクターを増殖させる。培養期間等の条件は、使用する組み換えセンダイウイルスにより変わり得る。その後、ウイルスを含んだ尿液を回収する。尿液からのセンダイウイルスベクターの分離・精製は常法に従って行うことができる(田代眞人,「ウイルス実験プロトコール」, 永井、石浜監修, メジカルビュー社, pp.68-73,(1995))。
【0075】
例えば、F蛋白質を欠失したセンダイウイルスベクターの構築と調製は、以下のように行うことができる(国際公開番号 WO00/70055 および WO00/70070参照)。
<1> F欠失型センダイウイルスゲノムcDNAおよびF発現プラスミドの構築
【0076】
センダイウイルス(SeV)全長ゲノムcDNA、pSeV18+ b(+)(Hasan, M. K. et al., 1997, J. General Virology 78: 2813-2820)(「pSeV18+ b(+)」は「pSeV18+」ともいう)のcDNAをSphI/KpnIで消化してフラグメント(14673bp)を回収し、pUC18にクローニングしてプラスミドpUC18/KSとする。F欠損部位の構築はこのpUC18/KS上で行う。F遺伝子の欠損は、PCR-ライゲーション方法の組み合わせで行い、結果としてF遺伝子のORF(ATG-TGA=1698bp)を除いてatgcatgccggcagatga(配列番号:4)で連結し、F欠失型SeVゲノムcDNA(pSeV18+/ΔF)を構築する。PCRは、Fの上流には(forward: 5'-gttgagtactgcaagagc/配列番号:5, reverse: 5'-tttgccggcatgcatgtttcccaaggggagagttttgcaacc/配列番号:6)、F遺伝子の下流には(forward: 5'-atgcatgccggcagatga/配列番号:7, reverse: 5'-tgggtgaatgagagaatcagc/配列番号:8)のPCR産物をEcoT22Iで連結する。このように得られたプラスミドをSacIとSalIで消化して、F欠損部位を含む領域の断片(4931bp)を回収してpUC18にクローニングし、pUC18/dFSSとする。このpUC18/dFSSをDraIIIで消化して、断片を回収してpSeV18+のF遺伝子を含む領域のDraIII断片と置き換え、ライゲーションしてプラスミドpSeV18+/ΔF を得る。
【0077】
外来遺伝子は、pUC18/dFSSのF欠失部位にある制限酵素 NsiI および NgoMIV 部位に挿入する。このためには、例えば外来遺伝子断片を、NsiI-tailedプライマーおよびNgoMIV-tailedプライマーで増幅すればよい。
<2> SeV-F蛋白を誘導発現するヘルパー細胞の作製
【0078】
センダイウイルスのF遺伝子(SeV-F)を発現するCre/loxP誘導型発現プラスミドの構築のためには、SeV-F遺伝子をPCRで増幅し、Cre DNAリコンビナーゼにより遺伝子産物を誘導発現されるように設計されたプラスミドpCALNdlw(Araiら, J. Virology 72,1998, p1115-1121)のユニークサイト SwaI部位に挿入し、プラスミドpCALNdLw/Fを構築する。
【0079】
F欠損ゲノムから感染ウイルス粒子を回収するため、SeV-F蛋白を発現するヘルパー細胞株を樹立する。細胞は、例えばSeVの増殖によく用いられているサル腎臓由来細胞株LLC-MK2細胞を用いることができる。LLC-MK2細胞は、10%の熱処理した不動化ウシ胎児血清(FBS)、ペニシリンGナトリウム 50単位/ml、およびストレプトマイシン 50μg/mlを添加したMEMで37℃、5% CO2で培養する。SeV-F遺伝子産物は細胞傷害性を有するため、Cre DNAリコンビナーゼによりF遺伝子産物を誘導発現されるように設計された上記プラスミドpCALNdLw/Fを、リン酸カルシウム法(mammalian transfection kit (Stratagene))により、周知のプロトコールに従ってLLC-MK2細胞に遺伝子導入を行う。
【0080】
10cmプレートを用い、40%コンフルエントまで生育したLLC-MK2細胞に10μgのプラスミドpCALNdLw/Fを導入後、10mlの10% FBSを含むMEM培地にて、37℃の5%CO2 インキュベーター中で24時間培養する。24時間後に細胞をはがし、10ml培地に懸濁後、10cmシャーレ5枚を用い、5ml 1枚、2ml 2枚、0.2ml 2枚に蒔き、G418 (GIBCO-BRL)を1200μg/mlを含む10mlの10%FBSを含むMEM培地にて培養を行い、2日毎に培地交換しながら、14日間培養し、遺伝子の安定導入株の選択を行う。該培地により生育してきたG418に耐性を示す細胞はクローニングリングを用いて回収する。回収した各クローンは10cmプレートでコンフルエントになるまで拡大培養を続ける。
【0081】
F蛋白質の発現誘導は、細胞を6cmシャーレにてコンフルエントまで生育させた後、アデノウイルスAxCANCreを斉藤らの方法(Saito et al., Nucl. Acids Res. 23: 3816-3821 (1995); Arai, T.et al., J. Virol. 72,1115-1121 (1998))により moi=3 で感染させて行う。
<3> F欠失SeVウイルスの再構築及び増幅
【0082】
上記 pSeV18+/ΔF の外来遺伝子が挿入されたプラスミドを以下のようにしてLLC-MK2細胞にトランスフェクションする。LLC-MK2 細胞を5×106cells/dish で 100mm ペトリ皿に蒔き、24時間培養後、ソラレンと長波長紫外線(365nm)で 20 分間処理したT7 RNAポリメラーゼを発現するリコンビナントワクシニアウイルス(Fuerst, T.R. et al., Proc. Natl. Acad. Sci. USA 83, 8122-8126 (1986))に室温で1時間感染させる(moi=2〜3、好適にはmoi=2が用いられる)。ワクシニアウイルスへの紫外線照射には、例えば15ワットバルブを5本が装備された UV Stratalinker 2400(カタログ番号 400676 (100V), ストラタジーン社, La Jolla, CA, USA)を用いる。細胞を3回洗浄してからプラスミド pSeV18+/ΔF-GFP, pGEM/NP, pGEM/P, 及びpGEM/L(Kato, A. et al., Genes cells 1, 569-579 (1996))をそれぞれ12μg, 4μg, 2μg, 及び4μg /dish の量比でOptiMEM(GIBCO)に懸濁し、SuperFect transfection reagent(1μg DNA/5μl の SuperFect, QIAGEN)を入れて混合し、室温で10 分間放置後、最終的に3%FBSを含むOptiMEM 3mlに入れ、細胞に添加して培養する。3時間培養後、細胞を、血清を含まないMEM で2回洗浄し、シトシンβ-D-アラビノフラノシド40μg/ml (AraC, Sigma), トリプシン7.5μg/ml (GIBCO) を含むMEMで70時間培養する。これらの細胞を回収し、ペレットをOptiMEM に懸濁する(107 cells/ ml)。凍結融解を3回繰り返してlipofection reagent DOSPER(Boehringer mannheim)と混合し(106 cells/25μl DOSPER)室温で15分放置した後、上記でクローニングしたF発現ヘルパー細胞の一つ、例えばLLC-MK2/F7細胞にトランスフェクション(106cells /well 12-well-plate)し、血清を含まないMEM(40μg/ml AraC, 7.5μg/ml トリプシンを含む)で培養し、上清を回収する。混入し得るワクシニアウイルスは、得られた上清を希釈してLLC-MK2/F7細胞に感染させ、上清を回収する操作を数回繰り返すことで除去することができる。
【0083】
欠損型ウイルスベクターを調製する場合、例えば、ウイルスゲノム上で欠損しているエンベロープ遺伝子が異なる2種のベクターを同じ細胞に導入すれば、それぞれで欠損するエンベロープタンパク質が、もう一方のベクターからの発現により供給されるため、互いに相補しあって感染力のあるウイルス粒子が形成され、複製サイクルがまわりウイルスベクターが増幅される。すなわち、2種またはそれ以上のベクターを、エンベロープタンパク質を相補する組み合わせで接種すれば、それぞれのエンベロープ遺伝子欠損型ウイルスベクターの混合物を大量かつ低コストで生産することができる。これらのウイルスは、エンベロープ遺伝子が欠損しているため、エンベロープ遺伝子を欠損していないウイルスに比べゲノムサイズが小さくなり長い外来遺伝子を保持することができる。また、元々感染性のないこれらのウイルスは細胞外で希釈され共感染の維持が困難であることから、不稔化するため、環境放出管理上の利点がある。
【0084】
回収したパラミクソウイルスは実質的に純粋になるよう精製することができる。精製方法はフィルトレーション、遠心分離、およびカラム精製等を含む公知の精製・分離方法またはその組み合わせにより行うことができる。「実質的に純粋」とは、単離した物質(化合物、ポリペプチド、またはウイルス等)が、それが存在する試料中の成分として主要な割合を占めることを言う。典型的には、試料中に存在する実質的に純粋な成分は、試料中の他の成分を合わせた全体の50%以上、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上を占める。割合は当業者に公知の手順により、例えば重量比率[w/w]等で算出される。当然溶媒、塩、添加化合物などを除いて算出されるべきである。パラミクソウイルスの具体的な精製方法としては、例えばセルロース硫酸エステルまたは架橋ポリサッカライド硫酸エステルを用いる方法(特公昭62-30752号公報、特公昭62-33879号公報、および特公昭62-30753号公報)、およびフコース硫酸含有多糖および/またはその分解物に吸着させる方法(WO97/32010)等を例示することができる。
【0085】
回収されたSeVベクターは、生組み換えワクチン(live recombinant vaccine)として使うことができる。本発明において生ワクチンとは、ウイルスベクターを投与された個体の細胞中でベクターゲノムが増幅し、抗原蛋白質を発現させ免疫を獲得させる組成物を言う。実施例に示すように、SeVベクターを用いたワクチネーションはマカクサルにおいて免疫を効率よく誘導し、有意な臨床症状を示さないことから、生ワクチンとして好適に用いられる。このような生ワクチンを接種する対象に制限はなく、ヒト、サル、ネコ、イヌ、ブタ、ウマ、ウシ等免疫不全ウイルスが感染しうるあらゆる動物が含まれる。また、前述の伝播力が欠損したSeVベクターを用いれば、生ワクチンであってもベクターが伝播しないワクチンを製造することができる。
【0086】
また、発現蛋白質がSeV粒子に取りこまれる場合には、SeVベクターを不活化全粒子ワクチンとして利用することもできる。あるいは、SeVベクターを導入した細胞から、または発現蛋白質がSeV粒子に取りこまれる場合にはSeVベクターから、発現させた免疫不全ウイルス蛋白質を分離・精製してワクチンとすることもできる。SeVベクターには、限られた種類の蛋白質しか含まれないため、例えば発現ベクター等を用いて細胞内で発現させた免疫不全ウイルス蛋白質を細胞全抽出物から分離するのに比べ、格段に精製が容易である。蛋白質の精製には周知の分離技術を使用することができる。例えば、免疫不全ウイルス蛋白質に対する抗体を用いて、イムノアフィニティーカラム等により精製することも可能である。精製蛋白質をワクチンとして使うことで、生ワクチンや不活化ワクチンに比べ接種後の発熱や局所反応が起きる頻度が抑えられることが期待できる。
【0087】
SeVベクターを含むワクチンは、必要に応じて薬理学的に許容される所望の担体または媒体と組み合わせることができる。「薬学的に許容される担体」とは、ベクターと共に投与することが可能であり、ベクターによる遺伝子導入を有意に阻害しない材料である。例えばSeVベクターを生理食塩水やリン酸緩衝生理食塩水(PBS)などで適宜希釈して組成物とすることができる。SeVベクターを鶏卵で増殖させた場合等においては尿液を含んでよい。またSeVベクターを含むワクチン組成物は、脱イオン水、5%デキストロース水溶液等の媒体を含んでいてもよい。さらに、その他にも、植物油、懸濁剤、界面活性剤、安定剤、殺生物剤等が含有されていてもよい。また保存剤やその他の添加剤を添加することができる。また、免疫原性を高めるために、サイトカイン、コレラ毒素、サルモネラ毒素等の免疫促進剤を添加することもできる。またワクチンには、ミョウバン、不完全Freund'sアジュバント、MF59 (オイルエマルジョン)、MTP-PE (マイコバクテリア細胞壁由来の muramyl tripeptide)、および QS-21 (soapbark tree Quilaja saponaria 由来)などのアジュバントを組み合わせることもできる。
【0088】
本発明のワクチンを用いたワクチネーションは、免疫不全ウイルスの感染の防止および/または感染後のウイルスの排除またはウイルス増殖の抑制のために用いられ得る。また、免疫不全症候群発症の予防または発症後の治療のために用いられ得る。また、免疫不全ウイルスの感染モデル等における予防および/または治療方法の開発または評価においても有用である。
【0089】
本発明のワクチンは、有効量のベクターが対象組織の細胞に導入されるのに十分な量を投与される。「有効量」とは、所望の免疫応答を少なくとも部分的にもたらすように対象組織の細胞に遺伝子が導入される量を言う。所望の遺伝子を含むSeVベクターの有効量が投与されることにより、ベクターが導入された細胞から導入遺伝子産物が産生される。好ましくは、所望の導入遺伝子を有するSeVベクターの有効量が投与されることにより、投与された組織中または血中で有意なレベルの導入遺伝子の発現が検出される。「有意なレベル」とは、SeVベクターにより導入された遺伝子の発現(転写産物または翻訳産物の量)が検出可能であることを指す。但し、導入遺伝子の発現量は、その有効発現量レベルおよび中毒レベルを考慮して決められるべきである。
【0090】
細胞に導入された遺伝子の発現量は、当業者に公知の方法によりアッセイすることが可能である。遺伝子の転写産物は、例えば、ノーザンハイブリダイゼーション、RT-PCR、RNAプロテクションアッセイ等により検出・定量することができる。ノーザンハイブリダイゼーションやRT-PCR等による検出は in situ でも行い得る。また、翻訳産物を検出するには、抗体を用いたウェスタンブロット、免疫沈降、RIA、ELISA、プルダウンアッセイ等により行うことができる。また、導入遺伝子発現の検出を容易にするため、発現させる蛋白質にタグを付加したり、レポーター遺伝子を発現するように組み込んでおくことも可能である。レポーター遺伝子は、βガラクトシダーゼ、CAT、アルカリホスファターゼ、またはGFPをコードする遺伝子等が挙げられるがこれらに制限されない。
【0091】
免疫応答の検出は、抗体や免疫細胞の検出により行うことができる。例えば免疫不全ウイルスに対する体液性免疫応答のアッセイは、公知の種々のアッセイ方法に従って行うことが可能であり、例えばウイルス蛋白質への結合(ELISAまたはウェスタンブロッティング等によるアッセイ)、シンシチウム形成の阻害の検出、補体結合反応(complement fixation)、ADCC(antibody-dependent cell-mediated cytotoxicity; 抗体依存性の細胞を介した細胞傷害)能、感染または細胞融合に対する中和能、およびCD4とgp120との相互作用の阻害などを試験することによりアッセイすることができる。
【0092】
細胞性免疫応答の検出は、例えば抗原特異的CTL活性、CTL産生、あるいはヘルパーT細胞の産生または活性などを試験することによりアッセイすることができる。また、例えばCD8+ T細胞などの活性化T細胞またはその他の白血球から産生されるサイトカインまたはケモカインを調べることにより検出することができる。また、公知のリンパ球増殖アッセイ、CTLアッセイ、または抗原特異的T細胞のアッセイ等により決定することもできる。
【0093】
ベクターの投与量は、疾患、患者の体重、年齢、性別、症状、投与目的、ワクチンの形態や投与方法等により異なるが、当業者であれば適宜決定することが可能である。好ましくは、ワクチンに含有されるベクター量は約105 pfu/mlから約1011 pfu/mlの範囲内であるとよい。より好ましくは、ワクチンに含有されるベクターの量は約107 pfu/mlから約109 pfu/mlの範囲内であるとよい。最も好ましくは、約1×108 pfu/mlから約5×108 pfu/mlの範囲内の量を薬学上容認可能な担体中で投与することが好ましい。
【0094】
ワクチンの接種は、経皮内、経皮下、鼻腔内、経気管支、筋内、静脈内、または経口的に行われうる。例えば上気道の周辺、すなわち鼻腔内粘膜や上気道へのワクチン接種により粘膜免疫を誘導することができる。そのためには、SeVワクチンを鼻内噴霧等により気道へ接種することが有効と考えられる。鼻腔内投与は、例えばカテーテルを介した投与などにより行うこともできる。また、SeVを細胞に導入し、この細胞をワクチンとして接種することも可能である。例えばワクチン接種の対象個体由来の細胞にSeVを感染させ、ex vivo 投与によりワクチネーションを行うこともできる。
【0095】
また、ワクチンは単回投与(single dose administration)のみならず、例えばワクチンの2回接種または複数回接種により十分な免疫を誘導することも有効であると考えられる。ヒトの場合、複数回接種の間隔は通常2〜4週間である。
【0096】
複数回接種においては、SeVを含む本発明のワクチンを複数回投与することもできるが、SeVワクチンと他のワクチンとを組み合わせて用いることも好適である。上述のように、ウイルスベクターを基にしたワクチン戦略におけるデメリットの1つは、標的とすべき抗原よりもむしろ、ベクターウイルスに由来する抗原に対して強力な免疫応答が誘導されてしまうことである。この問題は、初回免疫と追加免疫のそれぞれにおいて、2つまたはそれ以上の異なる種類のウイルスベクターを用いることにより解決することができる。従って、先に述べたように、DNAワクチンを基にした初回免疫後にウイルスベクターを基にした追加免疫を行うことも、好ましい戦略の1つである。また、同じ組み換えウイルスを再度接種しても、追加的な抗原特異的応答を引き起こすには不十分である可能性がある。従って、まずSeVベクターによるワクチンで初回免疫を行い、追加免疫に異なるウイルスベクターまたはDNAワクチンを用いるのも有効である。また、組み換えSeVベクターを用いて複数の抗原を発現させれば、防御の効力をより向上させ得る。
【0097】
このように、異なる種類のワクチンを用いたプライム-ブーストプロトコルにおいてSeVベクターによるワクチンと組み合わされるワクチンに特に制限はなく、所望のワクチンを用いることができる。例えば、組み換えサブユニットワクチン、SeV以外のウイルスや微生物をベースにした生組み換えワクチン、全粒子の不活化免疫不全ウイルス、免疫不全ウイルスのシュードビリオンやウイルス様パーティクル、ペプチドワクチン、弱毒化免疫不全ウイルスの生ワクチン、DNAワクチンなどが挙げられるが、これらに制限されない。サブユニットワクチンとは、対象となる免疫不全ウイルス由来の抗原の全てを有さず、1つまたは複数の選択された蛋白抗原を含むワクチンを言う。このようなワクチンは、ウイルスの他の成分や感染細胞由来の成分から少なくとも部分的に切り離されている。サブユニットワクチンは免疫不全ウイルス蛋白質を少なくとも部分的に精製することにより調製することが可能である。また、組み換えによる生産または合成により作り出すことも可能である。生組み換えワクチンのベースとなるウイルスや微生物としては、例えばポックスウイルス、アデノウイルス、サルモネラ、ポリオウイルス、マイコバクテリア、インフルエンザウイルス、およびセムリキフォレストウイルスなどが挙げられるが、これらに限定されない。また、免疫不全ウイルスとして、env遺伝子の置換により複製能を限局させた限局的複製型ウイルス(Matano, T. et al. 2000. Vaccine 18:3310-3318)をワクチンとして用い、本発明のワクチンと組み合わせて追加免疫を行うことも可能である。SeVワクチンと、これら他のワクチンの接種の順番は特に制限はなく、SeVワクチンを接種した後に他のワクチンを接種することもできるし、逆に他のワクチンを接種した後にSeVワクチンを接種することもできる。
【0098】
一例を示せば、DNAワクチンによりプライミングした後、SeVワクチンによりブーストをかけることができる。このようなワクチネーションは、(a)DNAワクチンを投与する工程、およびその後(b)免疫不全ウイルスのウイルス蛋白質をコードするパラミクソウイルスベクターを投与する工程、を含む方法である。DNAワクチンとしては、例えば免疫不全ウイルスゲノムをコードするDNA等を用いることができる。DNAワクチンの接種は、例えば筋肉内投与および/またはジーンガンによる投与により行い得る。例えば、DNAワクチンの接種を数回行った後、本発明のSeVベースのワクチンを接種することができる。接種の間隔は通常、数日から数週間で行う。
【0099】
ワクチン接種が可能な動物としては、免疫系を有し、かつ免疫不全ウイルスに感染し得るあらゆる宿主が挙げられ、ヒト、サル、マウス、ラット、ウサギ、ヒツジ、ブタ、ウシ、ウマ、トリなどを含む、全ての哺乳動物等が含まれる。本発明のワクチンが接種される動物は、好ましくは霊長類があげられる。例えば、ヒト以外に本発明のワクチンの接種の対象となる霊長類(非ヒト霊長類)としてはレムール、ロリス、およびメガネザル等の原猿、広鼻猿類および狭鼻猿類を含む真猿、ならびにテナガザル、オランウータン、ゴリラ、チンパンジー、およびボノボなどを含む類人猿が挙げられる。狭鼻猿類の中では、特にオナガザル亜科、具体的にはニホンザル、カニクイザル、アカゲザル、ボンネットザル、ブタオザル、ベニガオザル、およびアッサムモンキー等を含むマカカ属などが挙げられる。非ヒト霊長類へのワクチン接種は、ヒトへの臨床適用に向けたAIDSワクチンの開発および評価に極めて有用である。
【0100】
免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターを含むワクチンは、宿主の免疫応答を局所および全身で誘導することができる。特に、該ベクターが導入された細胞は、抗原特異的な免疫応答のスティミュレーター(刺激細胞)として機能し、細胞性免疫応答を誘導する。本発明は、(a)免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを抗原提示細胞に導入する工程、(b)該導入細胞とTヘルパー細胞および細胞傷害性T細胞とを接触させる工程、を含む、免疫不全ウイルスのウイルス蛋白質特異的細胞性免疫応答を誘導する方法を提供する。ここで細胞を「接触させる」とは細胞の接触を許すことも含む。すなわち、例えばベクターを導入した細胞を血中に注入したり(体内のTヘルパー細胞および細胞傷害性T細胞と接触し得る)、あるいは同一培地中にベクター導入細胞とTヘルパー細胞および細胞傷害性T細胞とを共培養するなど、細胞が接触し得るようにすることであってよい。また、抗原特異的な「細胞性免疫応答の誘導」とは、該細胞性免疫応答の過程の少なくとも一部の誘導を意味する。例えば、抗原特異的CTLの刺激、該CTLの頻度上昇または活性(例えば細胞傷害性)の上昇などであってよい。
【0101】
抗原提示細胞とは、主要組織適合性複合体(MHC)class I または MHC class II 分子が提示されている細胞で、抗原蛋白のペプチドを各分子に結合できる能力を有する細胞を言う。抗原提示細胞としては、例えば樹状細胞(DC)が挙げられる。MHC class I 分子とは、抗原ペプチドに結合して細胞傷害性T細胞(CD8+)に提示する分子である。MHC class II分子とは、抗原ペプチドに結合してTヘルパー細胞(CD4+)に提示する分子である。Tヘルパー細胞とは、T細胞の系譜の一群であり、MHC class II分子により提示された抗原を認識し、免疫応答のカスケードを組織化する細胞である。細胞傷害性T細胞とは、T細胞の系譜の一群であり、MHC class I分子により提示された抗原を認識し、ウイルス感染細胞、癌、および移植片などの細胞を殺す細胞である(Xu M. et al., Trends Biotechnol. 18(4):167-72, 2000参照)。
【0102】
例えば末梢血単核細胞(PBMC)などに免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターを導入し、in vitro でPBMCと共培養することにより、IFM-γ産生の誘導および免疫不全ウイルスのウイルス蛋白質特異的CTLの増殖などの細胞性免疫応答を誘導することができる。また in vivo で投与すれば、宿主における抗原特異的な細胞性免疫応答を誘導することができる。
【0103】
細胞性免疫応答は、例えばIFN-γ量のアッセイやCD8+ IFN-γ+ T細胞の頻度測定などにより確認することができる。また、免疫不全ウイルスのウイルス蛋白質を発現させた細胞を標的として、標的細胞のリシスを測定することによってCTL活性をアッセイすることもできる。このような標的細胞は、上記SeVベクターを導入することにより作製することができる。例えば自家Herpesvirus papio不死化Bリンパ芽球細胞株(BLC)などに免疫不全ウイルスのウイルス蛋白質を発現するSeVを導入し、CTLを含むことが予想される試料と共にインキュベートして、該BLCのリシスを51Cr放出などを指標に測定することができる。また、不死化した細胞株 H9(ヒトT細胞由来)なども例示することができる。
【0104】
本発明は、免疫不全ウイルスのウイルス蛋白質特異的細胞性免疫応答を誘導または検出するための、免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターまたは該ベクターの導入細胞の使用に関する。また本発明は、免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターを導入した細胞を含む、免疫不全ウイルスのウイルス蛋白質特異的な細胞性免疫応答の刺激細胞に関する。さらに本発明は、免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターを導入した細胞を含む、免疫不全ウイルスのウイルス蛋白質特異的な細胞性免疫応答の標的細胞に関する。また本発明は、該刺激細胞または該標的細胞において免疫不全ウイルスのウイルス蛋白質を発現させるための、免疫不全ウイルスのウイルス蛋白質をコードするSeVベクターの使用に関する。
【0105】
SeVベクターがコードする免疫不全ウイルスのウイルス蛋白質に制限はなく、上記と同等にウイルスの構造蛋白質、調節蛋白質、アクセサリー蛋白質等であってよい。構造蛋白質としては、Gag、Pol、Env等が挙げられる。例えば、免疫不全ウイルスのGag蛋白質をコードするSeVを用いて、Gag特異的細胞性免疫応答の誘導を行うことができる。
【図面の簡単な説明】
【0106】
【図1】図1は、SeV/SIVgagを用いたSIV Gag発現を示す図および写真である。(A) SeV/SIVgag アンチゲノムRNAを生成する構築プラスミドであるpSeV(+)18bV(-)/SIVgagの構造。(B) モノクローナルマウス抗p27抗体を用いたウェスタンブロット解析。偽感染(レーン1)、SeV/control感染(レーン2)、またはSeV/SIVgag感染(レーン3)の24後にCV1細胞を溶解した。(C) 抗p27抗体およびFITC結合抗マウスIgG抗体を用いた免疫染色。偽トランスフェクト(a)、またはSHIVMD14YE DNA(Shibata, R. et al. 1997. J. Infect. Dis. 176:362-373)をトランスフェクト(b)、あるいはSeV/control(c)またはSeV/SIVgag(d)を感染(MOI=0.05)させたCV1細胞。(D) トリプシン(7.5μg/ml)存在下の多サイクル増殖条件(Sakai, Y. et al. 1999. FEBS Lett. 456: 221-226)でのCV1細胞におけるSeV/controlおよびSeV/SIVgagの複製カイネティクス。初期感染において、細胞にSeV/controlまたはSeV/SIVgagを、m.o.iを0.05で感染させた。
【図2】図2は、SeV/SIVgagを介したGag発現により誘導されるSIV Gag特異的細胞性免疫応答を示す図である。(A) VV/SIVgag(左パネル, 黒四角)またはSeV/SIVgag(右パネル, 黒丸)を感染させたB細胞をSIV Gag特異的リシスの標的として用いたCTLアッセイ。VV/WR(左パネル, 白四角)またはSeV/control(右パネル, 白丸)を感染させたB細胞を非特異的リシスの測定のため用いた。SIV感染の3週目に新たに調製した Rh018 PBMC をエフェクターとして用いた。(B) SeV/SIVgag感染細胞と共培養したPBMCにおけるIFN-γ産生。12週目に調製した Rh018 PBMC を、SeV/control感染細胞による非特異的刺激共培養(左レーン)またはSeV/SIVgag感染細胞によるGag特異的刺激共培養(右レーン)に用いた。それらの培養上清中のIFN-γ濃度をELISAでアッセイした。(C) SeV/SIVgag感染細胞を用いたPBMC共培養におけるSIV Gag特異的CTLの増殖。上記の非特異的(左レーン)またはGag特異的(右レーン)刺激共培養中のPBMCをエフェクターとして用いた。VV/SIVgag感染B細胞をGag特異的リシス(黒四角)の標的として、またVV/WR感染B細胞を非特異的リシス(白四角)の標的として用いた。
【図3】図3は、ワクチネーション後の免疫応答を示す図である。矢印は初回、2回目、および3回目のワクチネーションの時点を示す。(A) 末梢血中のCD4陽性(黒丸)またはCD8陽性(白四角)リンパ球数(cells/μl)。(B) 抗SeV(黒丸)または抗p27(白四角)抗体ELISAにおける405nmの光学密度(OD)。
【図4】図4は、SIV暴露後の血漿SIV RNAコピー数(copies/ml)を示す図である。カニクイザル鼻腔内に 108 CIUのSeVベクターによる3回のワクチネーションを行った。最初のワクチネーション後22週目に、それらのカニクイザルに100 TCID50 の SIVmac239 を静脈注射により暴露させた。
【図5】図5は、グループ I カニクイザルのSeV/SIVgagRNA発現を示す図である。(A) 鼻腔粘膜から調製した細胞のSIV gag RNAレベル。(B) 鼻腔粘膜から調製した細胞のSeV N mRNAレベル。(C) 咽頭後LNから調製した細胞のSIV gag RNAレベル。(D) 顎下LNから調製した細胞のSIV gag RNAレベル。4日目(day 4)における C3880 または C4325 のレベルは未決定。
【図6】図6は、SeV/SIVgagを感染させた細胞で刺激後の細胞内IFN-γ誘導のフローサイトメトリーによる検出を示す図である。SIVを感染させたアカゲザルの PBMC を、それぞれ SeV/control (A)、SeV/SIVgag (B)、および Vv-Gag (C) を感染させた自家BLCと共に共培養した。CD3+CD8+ リンパ球をゲーティングしたドットプロットを示す。各ドットプロットにおいて、ゲーティングされた細胞数に対する、四角で囲んだ細胞数の割合(%IFN-γ+)を示した。
【図7】図7は、ワクチネーションを行ったアカゲザルにおける Gag特異的 CD8+IFN-γ+ T細胞を頻度を示す図である。(A) グループ II アカゲザルの3週目(week 3)の PBMC を、Vv-control(白いボックス)または Vv-Gag(黒いボックス)を感染させたBLCと共培養し、フローサイトメトリーによる解析を行った。ND, 未決定。(B) 2週目(week 2)のグループ I-C アカゲザルの咽頭後LN由来の細胞を、SIV Gag p27 の非存在下(白いボックス)または存在下(黒いボックス)でインキュベートし、フローサイトメトリーによる解析を行った。
【図8】図8は、ワクチネーションを行ったアカゲザルにおける SeV特異的 CD8+IFN-γ+ T細胞の頻度を示す図である。グループ II アカゲザルの1週目(week 1)(A) または 3週目(week 3)(B) の PBMC を、Vv-control(白いボックス)または SeV/control(黒いボックス)を感染させたBLCと共培養し、フローサイトメトリーによる解析を行った。ND, 未決定。
【図9】図9は、ワクチネーションを行ったアカゲザルにおける血漿中の抗SeV抗体レベルのELISAによる決定を示す図である。1:1000 に希釈した血漿で得られた吸収のカイネティクスを示す。OD, 光学密度。
【図10】図10は、DNAワクチン + SeV/SIVgagワクチンによるSIV Gag特異的CTLの誘導を示す図である。SIV Gag特異的 51Cr-放出(%)を示す。SeV/SIVgag投与(week 12)による追加免疫(booster)の効果を示している。12週目と14週目とを比較のこと。
【図11】図11は、DNAワクチン + SeV/SIVgagワクチンによる病原性SHIV暴露(challenge)に対する感染防御を示す図である。末梢血CD4 T細胞のcountの変化を示す。control群4頭全てにおいて、challenge後2週目でCD4-depletionが認められたが、DNAワクチン + SeV/SIVgagワクチン投与群では、4頭全てにおいてCD4-depletionから保護された。
【図12】図12は、DNAワクチン + SeV/SIVgagワクチンによる病原性SHIV暴露に対する感染防御を示す図である。血漿中のSHIV RNAコピー数の変化を示す。コントロール群と比較して、DNAワクチン + SeV/SIVgagワクチン投与群のSHIV RNA コピー数は、感染急性期より有意に低値を示した。
【発明を実施するための形態】
【0107】
以下実施例により本発明を具体的に説明するが、本発明はこれらの実施例に限定されるものではない。なお、本明細書全体を通じて引用された文献の内容はすべて、本明細書に組み込まれる。
【実施例】
【0108】
[実施例1] SeV/SIVgagの回収
安全面でより優れているVノックアウト型のSeV(V[-]SeV)(Kato, A. et al. 1997. J. Virol. 71:7266-7272; Kato, A. et al. 1997. EMBO J. 16:578-587)を使用してSIV Gagを発現する組み換えSeVであるSeV/SIVgagを構築した。SeVのアクセサリー遺伝子の1つであるV遺伝子を組み換えによりノックアウトすると、培養細胞におけるウイルス遺伝子の発現や複製は障害されることなく、マウスに対するSeVの病原性が顕著に弱毒化される。弱毒化V欠損SeVゲノムcDNAの全長を含むプラスミド pSeV(+)18bV(-) は以前記載されている(Kato, A. et al. 1996. Genes Cells 1:569-579; Hasan, M.K. et al. 1997. J. Gen. Virol. 78:2813-2820)。SIVmac239 Gag(ヌクレオチド 1306−2845 [GenBank accession number: M33262](Kestler, H. et al. 1990. Science 248:1109-1112))をコードする遺伝子断片はPCR増幅により調製し、pSeV(+)18bV(-) に導入して pSeV(+)18bV(-)/SIVgag を得た。PCRで用いたプライマーは 5'-AAG CGG CCG CGA GAT GGG CGT GAG AAA CTC CG-3'(配列番号:9)および 5'-TTG CGG CCG CGA TGA ACT TTC ACC CTA AGT TTT TCT TAC TGT GAC TAC TGG TCT CCT CCA AAG-3'(配列番号:10)である。gag遺伝子断片は、pSeV(+)18bV(-)のNのコード領域の直ぐ上流にあるNotI部位に挿入した(図1A)。このプラスミド pSeV(+)18bV(-)/SIVgag は T7 RNA ポリメラーゼの存在下でSeV/SIVgagアンチゲノムRNAの全長を生じる。組み換えSeV, SeV/SIVgag, は、pSeV(+)18bV(-)/SIVgagから以前記載したようにして回収した(Kato, A. et al. 1996. Genes Cells 1:569-579)。具体的には、T7 RNAポリメラーゼを発現する組み換えワクシニアウイルス(VV)ベクターであるvTF7-3(Fuerst, T. R. et al., 1986, Proc. Natl. Acad. Sci. USA 83: 8122-8126)をLLCMK2細胞に感染させ、pSeV(+)18bV(-)/SIVgag、pGEM-N、pGEM-P、および pGEM-L(Garcin, D. et al., 1995, EMBO J. 14: 6087-6094)をコトランスフェクトした。トランスフェクションから40時間後に、細胞を回収し鶏卵の尿膜腔に注入した。2回継代後、尿液を回収しSeV/SIVgagとした。同様にpSeV(+)18bV(-)からコントロールSeVであるSeV/controlを得た。SeVタイター(CIU [cell infectious unit] /ml)は、以前記載されたように(Kiyotani, K. et al. 1990. Virology 177:65-74)、CV1細胞使って抗SeV抗体を用いた免疫染色により測定した。
【0109】
[実施例2] SeV/SIVgagを用いた in vitro でのSIV Gag発現
プラスミド pSeV(+)18bV(-)/SIVgag(図1A)から、標準的なトランスフェクションプロトコルに従って感染性SeV/SIVgagをレスキューさせた(実施例1参照)。このSeV/SIVgagを細胞に感染させ、発現した蛋白質の解析を以下のように行った。細胞ライセートを回収するため、6-wellプレートの各ウェルに 4×105 細胞の密度でCV-1細胞を撒き、一晩培養後、SeV/controlまたはSeV/SIVgagを m.o.i.を 5 で感染させた。1日後、細胞を600μlのリシスバッファー(50 mM Tris-HCl, pH8.0, 150 mM NaCl, 0.02% sodium azide, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 0.1 mg/ml phenylmethylsulfonyl fluoride, 1% Triton X-100)中でリシスさせた。各レーンに10μlの細胞ライセートをロードした。ウェスタンブロット解析はモノクローナルマウス抗p27抗体を用いて以前の記載に従って行った(Matano, T. et al. 1993. J. Virol. 67:2026-2033)。免疫染色は抗p27抗体およびフルオレセイン結合ヤギ抗マウスイムノグロブリンG(IgG)抗体を用いて記載されたようにして行った(Matano, T. et al. 1993. J. Virol. 67:2026-2033)。SeV/SIVgagを感染させたCV1細胞において、プロセッシングを受けていないSIV Gagであるp55の発現が、抗SIV Gag p27モノクローナル抗体を用いたウェスタンブロッティングおよび免疫染色の両者において確認された(図1Bおよび1C)。組み換えウイルスは、SeV/controlに比べ僅かに遅い増殖カイネティクスを示した(図1D)。この増殖の遅延はゲノム長の増加による可能性がある(SeV/control, 15.4kb; SeV/SIVgag, 17.0kb)(Yu, D. et al. 1997. Genes Cells 2:457-466; Hasan, M.K. et al. 1997. J. Gen. Virol. 78:2813-2820; Sakai, Y. et al. 1999. FEBS Lett. 456: 221-226)。
【0110】
[実施例3] SeV/SIVgagによる in vitro でのSIV Gag特異的細胞応答の誘導
SeV/SIVgagを介したGag発現によりSIV Gag特異的応答が誘導されるかについて、1頭のアカゲザル(Macaca mulatta)から調製したPBMCを用いて in vitro で細胞性免疫応答の試験を行った。このアカゲザルは別の実験において前もってSIV抗原を発現するnaked DNA(Gagを含むSIV抗原を発現するプラスミドDNA)でワクチネーションを行い、SIVmac239を暴露した。SIVmac239暴露用ストックは以前に記載に従ってアカゲザルPBMCを用いて調製した(Kestler, H. et al., 1990, Science 248: 1109-1112; Shibata, R. et al., 1997, J. Infect. Dis. 176: 362-373)。ストックの感染力価はMT-4細胞でアッセイした。なお、このアカゲザルは実験に用いる前にSeV、SIV、およびサルtype Dレトロウイルスの陰性試験を行い、実験動物に関する研究所のガイダンスに従って維持した。採血、ワクチネーション、およびウイルス接種はケタミン麻酔下で行った。
【0111】
SIV暴露後3週目(week 3)にこのアカゲザルからPBMCを新たに調製し、このCTLアッセイのエフェクターとして用いた。末梢血単核細胞(PBMC)は全血試料から以前の記載に従って調製した(Shibata, R. et al., 1997, J. Infect. Dis. 176: 362-373)。PBMCは10%ウシ胎児血清(Hyclone)を含むRPMI1640(Life Technologies)で培養した。PBMCにSeV/controlまたはSeV/SIVgagをMOI 10で感染させ、2時間インキュベート後、共培養に用いた。SIV Gag特異的刺激を行う共培養では、U-bottomの96ウェルプレートにおいて、1ウェル当たり 5×105 PBMC と 1×105 SeV/SIVgag感染PBMCとを混ぜ合わせた。非特異的刺激を行う共培養では、5×105 PBMC と 1×105 SeV/control感染PBMCとを混ぜ合わせた。共培養を3日行った後、培養上清を回収し、細胞は10 unit/mlの組み換えヒトインターロイキン-2(Boehringer Mannheim)を含む培地中で培養した。上清中のインターフェロン-γ(IFN-γ)濃度は酵素結合イムノソルベントアッセイ(ELISA)(Biosource)で試験した。このアッセイの検出下限は 15pg/mlである。SIV Gag特異的刺激を行った共培養におけるPBMCを細胞傷害性Tリンパ球(CTL)アッセイにおけるエフェクター細胞として用いた。あるいは、別途調製したばかりのPBMCをエフェクターとして用いた。CTLアッセイ(51Cr-放出アッセイ)は以前の記載に従って行った(Voss, Q. et al., 1992, J. Virol. Methods 39: 185-195; Voss, Q. et al., 1995, Virology 208: 770-775)。SIV Gagを発現する組み換えワクシニアウイルス(VV)である VV/SIVgag を感染させた自家Herpesvirus papio不死化Bリンパ芽球細胞株(B-LCL)をSIV Gag特異的リシスの標的として用いた。あるいは、SeV/SIVgagを感染させたB-LCLを用いた。コントロールVV(VV/WR)または SeV/control を感染させた B-LCL を非特異的リシスの測定のために用いた。51Cr-放出の割合を次のようにして計算した: %51Cr放出 = (試験放出 - 自発的放出) / (最大放出 - 自発的放出)×100。その後、Gag特異的51Cr放出の割合を、Gag特異的リシスにおける %51Cr放出から非特異的リシスにおける放出を差し引くことによって算出した。Gag特異的 51Cr放出が 10%を超えた場合、Gag特異的CTL活性を陽性とした。
【0112】
SeV/SIVgagまたはVV/SIVgagを感染させた自家B-LCLを標的として用いた。図2Aに示したように、このアッセイでGag特異的リシスがSeV/SIVgagを感染させたB-LCLおよびVV/SIVgagを感染させたB-LCLの両細胞で観察されたことから、SeV/SIVgagを感染させたB-LCLはSIV Gag特異的CTLの標的として働くことが示された。
【0113】
次に、SeV/SIVgagを感染させた細胞がSIV Gag特異的CTLの増殖を誘導するかを調べた。12週(week 12)のアカゲザルRh018から調製したPBMCをSeV/SIVgagに感染させたPBMCと共に共培養すると、Th1サイトカインの一種である IFN-γの産生が観察された(図2B)。これとは対照的に、SeV/controlを感染させたPBMCとのPBMC共培養では検出可能なIFN-γは誘導されなかった。前者(SeV/SIVgagに感染させたPBMCとの共培養)においてはSIV Gag特異的CTL活性が見られたが、後者(SeV/controlを感染させたPBMCとの共培養)の対照共培養では見られなかった(図2C)ことから、SeV/SIVgag感染細胞によるGag特異的CTLの増殖が示された。
【0114】
[実施例4] カニクイザルにおけるSeV/SIVgagのワクチネーション
表1に概要を示したプロトコルに従って、4頭のカニクイザル(Macaca fascicularis)を用いてワクチネーション実験を行った。4頭のカニクイザルは実験に用いる前にSeV、SIV、およびサルtype Dレトロウイルスの陰性試験を行い、実験動物に関する研究所のガイダンスに従って維持した。採血、鼻腔拭い液(nasal swab)の採取、ワクチネーション、およびウイルス接種を行う場合はケタミン麻酔下で行った。カニクイザル 1329605029(Cy029)はワクチネーションを行わず、未処理のコントロールとした。カニクイザル 1429407002(Cy002)は、108 CIUの SeV/controlを初回免疫(0週目)、その後4週目および14週目の計3回、鼻腔内に接種してコントロールのワクチネーションを行った。同じスケジュールで2頭のカニクイザル 1129307001(Cy001)および 1128206162(Cy162)に 108 CIUの SeV/SIVgagを鼻腔内に3回のワクチネーションを行った。これらのカニクイザルはいずれも、ワクチネーション後に体重減少を含む病理学的兆候は見られなかった。また、末梢のCD4またはCD8細胞数の有意な減少はいずれも観察されなかった(図3A)。
【0115】
表1 ワクチネーションプロトコルおよび in vivo SeV/SIVgag発現
【0116】
気道におけるSeV/SIVgagの複製および発現を鼻腔拭い液(nasal swab)により調べた(表1)。鼻腔拭い液の採取は以前の記載に従って行った(Hurwitz, J. L. et al., 1997, Vaccine 15: 533-540)。拭い液試料は培地で希釈し、SeVを回収するため鶏卵の尿膜腔に注入した。48時間インキュベート後、尿液を回収し、ヘマグルチネーション(HA)アッセイに供しSeVを検出した。SIV gagの発現を調べるためには、High Pure Viral RNA kit(Boehringer Mannheim)を用いて拭い液試料からRNAを抽出した。SIV gag特異的プライマーを用いてnested RT-PCRを行った。最初のRT-PCRに用いたプライマーは5'-AGA AAC TCC GTC TTG TCA GG-3'(配列番号:11)および5'-TGA TAA TCT GCA TAG CCG C-3'(配列番号:12)、2回目のPCRに用いたプライマーは5'-GAT TAG CAG AAA GCC TGT TGG-3'(配列番号:13)および5'-TGC AAC CTT CTG ACA GTG C-3'(配列番号:14)である。
【0117】
カニクイザル Cy002、Cy001、および Cy162 では、拭い液を接種した鶏卵の尿液中に蓄積したSeV HA活性から分かるように、1週目においてSeV(SeV/controlまたはSeV/SIVgag)の複製が検出された(表1)。2週目では検出可能なSeVは回収されなかった。2回目または3回目のワクチネーションではいずれも、拭い液中に検出可能なSeVは見られなかった。SIV gag特異的プライマーを用いたnested PCRにより、SIV gagの発現がSeV/SIVgagによるワクチネーションを行ったカニクイザル(Cy001およびCy162)の両者の鼻拭い液で検出されたが、SeV/controlによるワクチネーションを行ったカニクイザル(Cy002)では検出されなかった(表1)。
【0118】
これら3頭のカニクイザルのワクチネーション後の免疫応答を調べた。ELISAによる体液性免疫応答試験の結果、3頭の全てのカニクイザルで血漿中に高いレベルで抗SeV抗体が誘導されていることが示された(図3B)。2回目および3回目のワクチネーションはそれらのレベルの急速な上昇を誘導したことから、追加免疫の効果が示唆された。対照的に、血漿中の抗SIV Gag p27抗体の有意な誘導は観察されなかった。なお、抗SeV抗体ELISAは不活化したSeVを用いて行った。抗SIV Gag p27抗体ELISAは組み換えp27抗原(ImmunoDiagnostics)を用いて行った。抗SeV抗体ELISAでは血漿試料を1000倍に、抗p27抗体ELISAでは100倍に希釈した。
【0119】
未処理のカニクイザル(Cy029)、SeV/contorolでワクチネーションを行ったカニクイザル(Cy002)、およびSeV/SIVgagでワクチネーションを行ったカニクイザル(Cy001およびCy162)において、22週目に多量(100 TCID50)のSIVmac239を静脈注射により暴露させた。具体的には、カニクイザル鼻腔内に 108 CIUのSeV/SIVgagによる3回のワクチネーションを行い(0、4、および 14 週目)、最初のワクチネーション後22週目に、それらのカニクイザルに100 TCID50 の SIVmac239 を静脈注射により暴露させ、血漿中のSIV RNAを測定した。SeV/controlを接種したカニクイザルおよびワクチネーションを行わないカニクイザルを対照とした。
【0120】
これらのカニクイザルの血漿SIV RNA量の測定を以下のようにして行った。High Pure Viral RNA kitを用いて血漿RNAを抽出した。SIV env特異的プライマーを用いてnested PCRを行った。最初のRT-PCRに用いたプライマーは 5'-ATG GGA TGT CTT GGG AAT C-3'(配列番号:15)および 5'-CCA AAT CTG CAG AGT ACC AAG-3'(配列番号:16)、2回目のPCRに用いたプライマーは 5'-CAG CTT GGA GGA ATG CG-3'(配列番号:17)および 5'-CTT GTT CCA AGC CTG TGC-3'(配列番号:18)である。5倍希釈したRNA試料を4連で増幅し、以前の記載にように定量した(Shibata, R. et al., 1997, J. Infect. Dis. 176: 362-373; Reed, L. J. and Muench, H., 1938, Am. J. Hyg. 27: 493-497)。このアッセイの検出下限は 1.0×102 copies/mlである。
【0121】
これら4頭のカニクイザルは全て、暴露後2週目に同様のレベルで血漿SIV量のピークを示した(図4)。その後、コントロールのカニクイザルでは2頭とも血漿SIV RNAレベルは減少したが、約105 copies/mlが残存した。これに対しSeV/SIVgagでワクチネーションを行ったカニクイザルでは、2頭とも初期のウイルス血症の後、ウイルス量は有意に低下した(102から103 copies/ml)。より重要なことは、コントロールのカニクイザルの血漿ウイルス量は 105 copies/ml 周辺の高い値に留まったのに対し、免疫したカニクイザルの血漿ウイルス量は最終的に検出可能なレベル(100 copies/ml)を下回ったことである(図4)。
【0122】
[実施例5] カニクイザルの各組織におけるSeV/SIVgagの発現
カニクイザルにおける組み換えSeVの初期複製を調べるため、6頭のカニクイザルの鼻腔内にSeV/SIVgagを接種した(表2、グループ I)。カニクイザルは実験動物に関する研究所のガイドラインに従って維持した。実験に用いる前にSeVおよびSIVの陰性試験を行った。採血、鼻腔拭い液(nasal swab)の採取、およびワクチネーションを行う場合はケタミン麻酔下で行った。これらのサルはいずれも、接種後に明白な臨床症状は示さなかった。接種後4日目(day 4)にグループ I-A 中の2頭(C3880およびC4325)を、7日目(day 7)にグループ I-B 中の2頭(C3993およびC4240)を、13日目(day 13)にグループ I-C 中の2頭(C3882およびC4324)をそれぞれ安楽死させた。剖検により採取した各組織から細胞を調製し、細胞からRNAを抽出した。
【0123】
具体的には、LN、胸腺、および脾臓から、組織をミンスして細胞を調製した。鼻腔粘膜、口蓋扁桃、気管、および肺からは、コラゲナーゼおよびディスパーゼ処理後に細胞を調製した。これらの細胞はRNA抽出前にPBSで3回洗浄した。末梢血単核細胞(PBMC)は全血試料から Ficoll-Paque Plus(Amersham Pharmacia Biotech)を用いて文献(Shibata, R. et al. 1997. J. Infect. Dis. 176:362-373)に従って調製した。RNAは細胞からRNA-extraction kit(Qiagen)を用いて抽出した。nested RT-PCR(逆転写およびnested PCR)はSIV gag 特異的プライマーを用いて行い、RNA試料を限界希釈し、文献(Shibata, R. et al. 1997. J. Infect. Dis. 176:362-373)に従って希釈限界を決定することによりgag RNAレベルを定量した。SeV N mRNAレベルは TaqMan PCR system(ABI PRISM 7700, Applied Biosystems Japan)を用いた定量PCRにより定量した。
【0124】
表2 動物モデル
【0125】
5−1.カニクイザルの鼻腔粘膜におけるSeV/SIVgagの発現
定量RT-PCRにより、6頭のカニクイザル全ての鼻腔粘膜から調製した細胞において、有意なレベルのSIV gag RNAが検出された(図5A)。グループ I-A の4日目(day 4)においては、106 細胞当たり約 1.7×104 または 2.6×105 コピーの gag RNAが検出された。7日目(day 7)および 13日目(day 13)の発現レベルは4日目(day 4)に比べて低下した。さらに、定量RT-PCRにより鼻腔粘膜におけるSeV N mRNAの発現レベルを検出した(図5B)。ほとんどのカニクイザルにおいて、SeV N mRNAレベル(図5Bに示す)は gag RNAレベル(図5Aに示す)の1/4または1/3に相当した。これらの結果から、SeV/SIVgagを接種したカニクイザルの鼻腔粘膜において、SeV/SIVgagが有意に発現することが確認された。
【0126】
次に、鼻腔拭い液からSeVが回収されるかを調べた。鼻腔拭い液の採取は上記と同様にして行った。拭い液試料はRPMI-1640 で希釈し、SeVを回収するため鶏卵の尿膜腔に注入した。鼻腔粘膜から調製した細胞からのSeVの回収においては、細胞の凍結融解を2回行った後、RPMI-1640中に懸濁させた 1×105 細胞を鶏卵の尿膜腔に注入した。48時間インキュベート後、尿液を回収し、文献(Kato, A. et al. 1996. Genes Cells 1:569-579)に従ってヘマグルチネーション(HA)アッセイに供しSeVを検出した。
【0127】
表3に示すように、4日目(day 4)において6頭全ての鼻腔拭い液からウイルスが回収された。7日目(day 7)では、4頭のカニクイザルのうち2頭からウイルスが回収されたが、この2頭はいずれも、鼻腔粘膜においてより高いgag RNAレベルを示していた。13日目(day 13)では、両方のサルでウイルスは回収されなかった。さらに、鼻腔粘膜から調製した細胞 1×105 からウイルスが回収されるかを調べた(表3)。4日目(day 4)の試料からはウイルスが回収されたが、7日目(day 7)または 13日目(day 13)の試料からはウイルスは回収されなかった。これらの結果は、SeV/SIVgagの複製レベルは1週間以内にピークに達することを示している。
【0128】
表3 SeVの回収
【0129】
5−2.カニクイザルの鼻腔の近傍リンパ節でのSeV/SIVgagの発現
有意なレベルのgag RNAが、咽頭後LN(retropharyngeal LN)および顎下LN(submandibular LN)でも見出された(図5Cおよび図5D)。これらはいずれも鼻腔からのリンパ球が最初に流入するリンパ節である。7日目(day 7)および 13日目(day 13)の各動物におけるLNでのgag RNAレベルは、鼻腔粘膜の場合の約1/20であった。しかし4日目(day 4)では、各動物における咽頭後LNのgag RNAレベルは鼻腔粘膜の場合の1/50以下であった。従って、4日目(day 4)と7日目(day 7)の間でこのLN中のレベルの有意な違いは観察されなかった。
【0130】
5−3.カニクイザルの他の組織でのSeV/SIVgagの発現
gagの発現を表4に示すように他の組織においても調べた。口蓋扁桃では十分な細胞が得られなかったが、その発現レベルは咽頭後LNの発現レベルよりも低いことが示された。気管での発現は2点、すなわち 4日目(day 4)の C3880 および 7日目(day 7)の C4240 で検出可能であった。前者の発現は後者の発現よりずっと高かった。肺での発現は C3880[4日目(day 4)]でのみ検出された。胸腺または鼠径LNでは発現は観察されなかった。脾臓では発現はほとんど検出されず、わずかな発現が1個体のみで検出された[7日目(day 7)]。PBMCにおいては、7日目(day 7)において4頭のカニクイザル中2頭でgagの発現が示されたが、4日目(day 4)または13日目(day 13)では発現は観察されなかった。表4で調べた全ての組織において、gagの発現は鼻腔粘膜、咽頭後LN、および顎下LNに比べずっと低かった。
【0131】
表4 gag RNA発現
【0132】
[実施例6] SeV/SIVgagを感染させた細胞によるGag特異的CD8+ T細胞の刺激
SeV/SIVgagを感染させた細胞が、実際にGag特異的CD8+ T細胞をin vitroで刺激できるかを調べるため、抗原特異的な細胞内IFN-γ誘導をフローサイトメトリーにより解析した。SIVに慢性的に感染したアカゲザル由来のPBMCを用いた。アカゲザル(Macaca mulatta)は、実験動物に関する研究所のガイドラインに従って維持した。実験に用いる前にSeVおよびSIVの陰性試験を行った。採血、鼻腔拭い液(nasal swab)の採取、およびワクチネーションを行う場合はケタミン麻酔下で行った。文献(Matano, T. et al. 2000. Vaccine 18:3310-3318)に記載のように、この動物は予めプロウイルスDNAワクチネーションを受け、その後SIVmac239(Kestler, H. et al. 1990. Science 248:1109-1112)の暴露による感染を受けている。51Cr-放出アッセイにより、SIV感染の慢性期の間、SIV-Gag特異的細胞傷害性Tリンパ球(CTL)活性が確認された(データ省略)。
【0133】
抗原特異的インターフェロン-γ(IFN-γ)誘導のフローサイトメトリーによる検出は、以下のようにして行った。まず、自家Herpesvirus papio不死化Bリンパ芽球細胞株(BLC)(Voss, G. et al. 1992. J. Virol. Methods 39:185-195)にコントロールのワクシニアウイルスベクター(Vv-control)(Mackett, M. et al. Proc. Natl. Acad. Sci. USA 79:7415-7419)、SIV Gagを発現する組み換えワクシニアウイルスベクター(Vv-Gag)、またはコントロールSeVベクター(SeV/control)を m.o.i. を 5 で感染させ、1日後、文献に記載のように(Gea-Banacloche, J.C. et al. 2000. J. Immunol. 165:1082-1092)、それぞれ対照の非特異的、Gag特異的、またはSeV特異的スティミュレーター(stimulator; 刺激細胞)として使用した。
【0134】
培養管(Falcon #3033)に 1×106 の PBMC を 1×103 の上記のスティミュレーター細胞と共に 10%FBSを含む 1ml の RPMI-1640 中で共培養した。1時間インキュベートした後、細胞に 0.75μl/mlの GolgiStop(monesin)(Pharmingen, San Diego, CA)を加え、さらに5時間インキュベートした。
【0135】
p27による刺激(実施例7)の場合、1×106 の PBMC は 2μg の抗CD28モノクローナル抗体(Becton Dickinson, San Jose, CA)および 10μg の組み換えSIV Gag p27蛋白質を含む、10%FBSを含む 1ml の RPMI-1640 中で、培養管中でインキュベートした。3時間インキュベートした後、細胞に 0.75μl/mlの GolgiStop を加え、さらに15時間インキュベートした。
【0136】
その後、Cytofix-Cytoperm kit(Pharmingen)を用い、説明書に従って細胞内IFN-γ染色を行った。具体的には、刺激した細胞を回収し、表面分子に対する抗体の混合液と共に室温で20分染色した。抗体には、抗ヒトCD8 PerCP(Becton Dickinson)および抗ヒトCD3 APC(Pharmingen)を用いた。その後 0.5%ウシ血清アルブミンを含むPBS(PBS-BSA)で細胞を洗浄し、Cytofix-Cytopermにより固定、透過化処理を行い、抗ヒトIFN-γ PE(Pharmingen)で4℃で30分染色した。染色した試料をFACScaliburで回収し、CellQuestソフトウェア(Becton Dickinson)を用いて解析した。各試料において、BLCによる刺激の場合の全事象(total events)は 100,000から200,000であり、組み換え蛋白質による刺激の場合の全事象は 50,000から100,000であった。ゲーティングは単核細胞において行い、その後 CD3+CD8+のサブポピュレーションについて行った。単核細胞数に対する CD3+CD8+IFN-γ+ 細胞数の比を評価し、 1×106 リンパ球当たりの CD8+IFN-γ+ T細胞数として示した。
【0137】
SIV Gagを発現する組み換えワクシニアウイルス(Vv-Gag)を感染させた自家BLCとPBMCとの共培養では、CD8+ T細胞におけるSIV Gag特異的細胞内IFN-γ誘導が示された(図6C)。SeV/SIVgagを感染させたBLCとPBMCとの共培養でも、CD8+IFN-γ+ T細胞が同様の頻度で観察された(図6B)。SeV/controlを感染させたBLCとPBMCとの共培養では、CD8+IFN-γ+ T細胞の有意な誘導は見出されなかった(図6A)。これらの結果は、SeV/SIVgagを感染させたBLCはGag特異的 CD8+ T細胞を効率的に刺激することを示している。
【0138】
[実施例7] アカゲザルのGag特異的 CD8+ T細胞の頻度
SeVワクチネーション後の初期免疫応答を解析するため、5頭のアカゲザルを用いて別のの実験を行った(表2、グループ II)。グループII-A のうち2頭(R004およびR014)はSeV/controlを鼻腔内に接種し、グループII-Bの3頭(R013、R015、および R017)はSeV/SIVgagを接種した。R010はSeV/controlワクチネーションの12週から6週間前に、他の実験のためにコントロールDNAワクチネーション(筋肉内接種により800μgのpCMVN DNA[Matano, T. et al. 2000. Vaccine 18:3310-3318]およびgene gunにより10μgのpCMVN DNAを4回)を受けている。これらのサルはいずれも、接種後に明白な臨床症状は示さなかった。
【0139】
3週目(week 3)に得たPBMCを、Vv-Gagに感染させた自家BLCと共培養し、CD8+ T細胞におけるIFN-γ誘導を調べた。SeV/SIVgagを接種したグループII-Bのアカゲザルは全て、Gag特異的刺激後に CD8+IFN-γ+ T細胞が有意な頻度で検出されたが、非特異的な刺激では検出されなかった(図7A)。これに対し、SeV/controlを接種したグループII-Aのアカゲザルでは、Gag特異的刺激後も、CD8+ T細胞においてIFN-γの有意な誘導は観察されなかった。これらの結果は、全身性免疫応答、すなわちGag特異的 CD8+ T細胞の誘導が、SeV/SIVgagでワクチネーションを行ったアカゲザルで起こることを示している。
【0140】
次に、鼻腔周囲の局所的な免疫応答を、グループ I-Cの咽頭後LNから調製した細胞を用いて調べた。グループ Iのアカゲザルからの自家BLCの調製がうまく行われなかったため、組み換えSIV Gag p27蛋白質を用いてGag特異的刺激を行った。グループ I-Cの両方のアカゲザルにおいて、p27と反応する CD8+IFN-γ+ T細胞が有意な頻度で検出されたことから、このLNにおいてp27特異的 CD8+ T細胞が誘導されていることが示された。
【0141】
[実施例8] アカゲザルにおけるSeV特異的CD8+ T細胞の頻度
SeV特異的な細胞性免疫応答を、SeV/controlまたはSeV/SIVgagを接種したアカゲザルで調べた。グループIIの全ての動物のPBMCにおいて、3週目(week 3)にSeV特異的 CD8+ T細胞が高い頻度で検出された(図8B)。また、これらの全てにおいて、1週目(week 1)で既にSeV特異的 CD8+ T細胞が有意な頻度で出現していることが示された(図8A)。従って、大規模なSeV特異的細胞性免疫応答が、ワクチネーション後に迅速に誘導されることが判明した。
【0142】
抗体ELISAにより、血漿中の蛋白質レベルを評価した。抗SeV抗体ELISAは不活化したSeVを用いて行った(Watanabe, T. et al. 2000. Arch. Dermatol. 136:1518-1522)。抗SIV Gag p27抗体ELISAは組み換えp27蛋白質(ImmunoDiagnostics, Woburn, MA)を用いて行った。抗SeV抗体ELISAでは血漿試料を1000倍に、抗p27抗体ELISAでは100倍に希釈した。
【0143】
グループIIの全てのアカゲザルにおいて、血漿中の抗SeV抗体はワクチネーション後 1週目(week 1)では検出されないが、2週目(week 2)では有意なレベルの抗体が出現していた(図9)。これに対して血漿中の抗Gag抗体は、SeV/SIVgagを接種したグループII-Bのアカゲザルにおいても検出されなかった(データ省略)。
【0144】
[実施例9] DNAワクチンとの組み合わせによるプライム−ブースト効果
prime-boostによるワクチネーションにおいて、DNAワクチンとの組み合わせの効果を試験した。アカゲザルを用いた実験のプロトコルを表5に示した。DNAワクチネーションは文献(Matano, T. et al., Vaccine 18: 3310-3318, 2000 参照)に従って、筋肉内(intramuscular; i.m.)接種およびジーンガン(Bio-Rad)により行った。i.m.接種では、PBSに溶解した0.25μg/μlのDNA溶液を各回200μlずつ、計800μl四頭筋に注入した。また、ジーンガンによる接種は大腿の前部皮膚中への注入により行った(Matano, T. et al., Vaccine 18: 3310-3318, 2000 参照)。control群4頭、DNAワクチン + SeV/SIVgagワクチン投与群4頭とした。DNAワクチンの詳細は、文献(Donnelly, J. J. et al., Annu. Rev. Immunol. 15: 617-648, 1997; Lu, S. et al., J. Virol. 70: 3987-3991, 1996; Matano, T. et al., Vaccine 18: 3310-3318, 2000)を参照。SeV/SIVgag投与は、上記と同様で、108 CIU 鼻腔投与により行った。ワクチネーション開始の26週目に 10 TCID50 のサル-ヒト免疫不全ウイルス(SHIV 89.6-PD)(Lu, Y. et al., J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 19(1):6-18, 1998)を静脈注射により暴露した。DNAワクチン + SeV/SIVgagワクチンにより、SIV Gag特異的CTLの誘導が観察され、末梢血CD4 countの変化から、投与群4頭の全てで病原性SHIV暴露(challenge)に対する感染防御が認められた(図10および11)。また、DNAワクチン + SeV/SIVgagワクチン接種群の血漿中のSHIV RNAコピー数は、コントロール群と比較して、感染急性期より有意に低値を示した(図12)。
【0145】
表5 マカクサルプロトコル:DNA + SeV
【産業上の利用可能性】
【0146】
本発明により、免疫不全ウイルスのウイルス蛋白質をコードするセンダイウイルスベクターを含むワクチンが提供された。センダイウイルスベクターは毒性が低く、また鶏卵を使って大量に製造できるため、安全で簡便な生組み換えワクチンの製造が可能である。本発明のワクチンにより、AIDSウイルスの感染および/またはAIDSの発症・進行を抑制する有望なワクチン戦略が提供される。
【特許請求の範囲】
【請求項1】
免疫不全ウイルスのGag蛋白質またはその部分をコードするセンダイウイルスベクターを含む、免疫不全ウイルスの感染、後天性免疫不全症候群の発症、および/または進行を抑制するワクチン。
【請求項2】
センダイウイルスがV遺伝子欠損型である、請求項1に記載のワクチン。
【請求項3】
ワクチンが鼻腔内投与用である、請求項1または2に記載のワクチン。
【請求項1】
免疫不全ウイルスのGag蛋白質またはその部分をコードするセンダイウイルスベクターを含む、免疫不全ウイルスの感染、後天性免疫不全症候群の発症、および/または進行を抑制するワクチン。
【請求項2】
センダイウイルスがV遺伝子欠損型である、請求項1に記載のワクチン。
【請求項3】
ワクチンが鼻腔内投与用である、請求項1または2に記載のワクチン。
【図2】

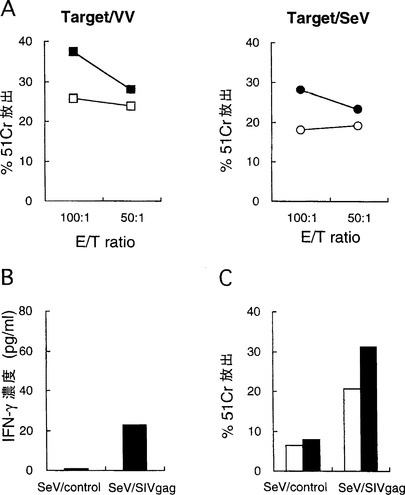
【図3】
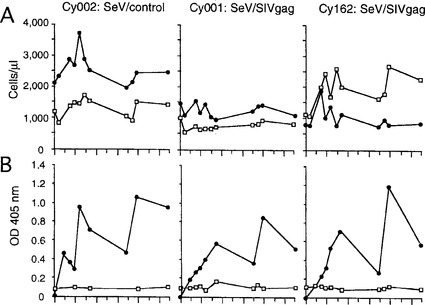
【図4】
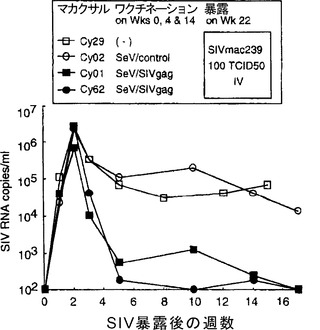
【図5】
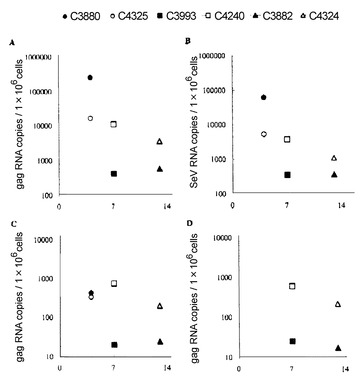
【図6】
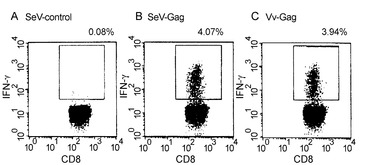
【図7】
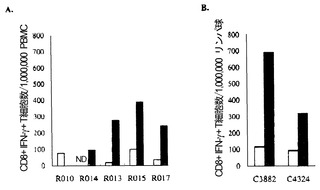
【図8】
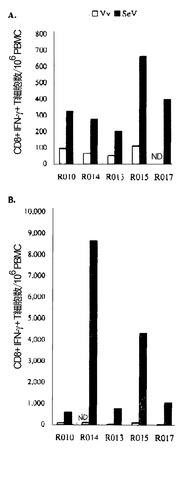
【図9】
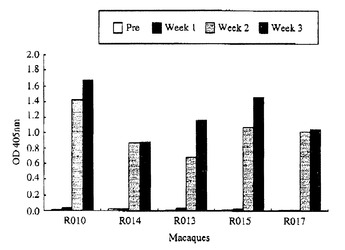
【図10】
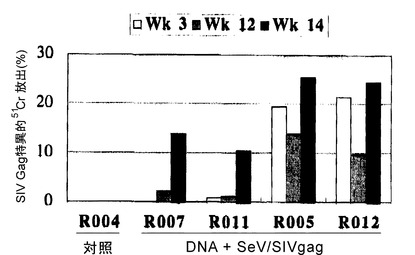
【図11】
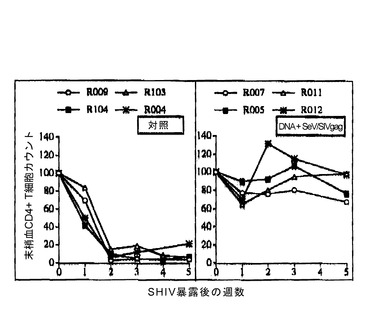
【図12】
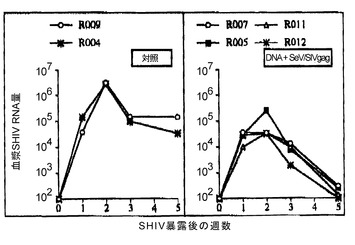
【図1】
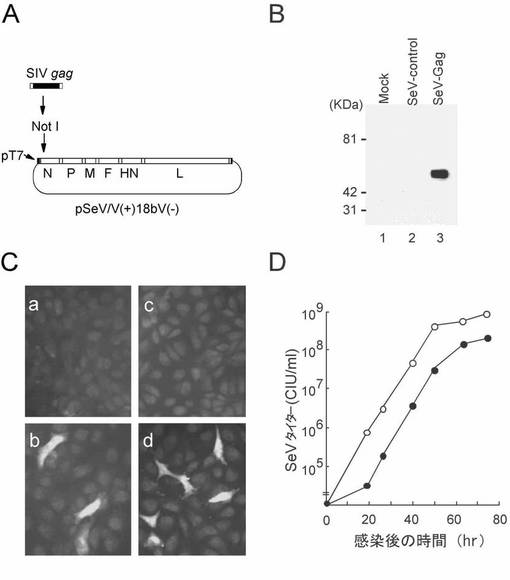
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図1】
【公開番号】特開2012−144541(P2012−144541A)
【公開日】平成24年8月2日(2012.8.2)
【国際特許分類】
【出願番号】特願2012−46108(P2012−46108)
【出願日】平成24年3月2日(2012.3.2)
【分割の表示】特願2001−570301(P2001−570301)の分割
【原出願日】平成13年3月30日(2001.3.30)
【出願人】(595155107)株式会社ディナベック研究所 (22)
【出願人】(591222245)国立感染症研究所長 (48)
【Fターム(参考)】
【公開日】平成24年8月2日(2012.8.2)
【国際特許分類】
【出願日】平成24年3月2日(2012.3.2)
【分割の表示】特願2001−570301(P2001−570301)の分割
【原出願日】平成13年3月30日(2001.3.30)
【出願人】(595155107)株式会社ディナベック研究所 (22)
【出願人】(591222245)国立感染症研究所長 (48)
【Fターム(参考)】
[ Back to top ]