
センダイウイルス温度感受性株由来のウイルスベクター
【課題】細胞障害性がなく、しかも培養細胞にのみでなく、さらに生体内においても長期間持続する遺伝子発現を実現でき、遺伝子治療用ベクター、タンパク質生産用ベクターとして極めて有用な新規ウイルスベクターを提供する。
【解決手段】センダイウイルスの温度感受性株、特にセンダイウイルスCl.151株の全長ゲノムcDNAを用いて、外来遺伝子導入組換えウイルスを得、該組換えウイルスを外来遺伝子の生体内あるいは培養細胞内持続発現用組換えベクターとする。
【解決手段】センダイウイルスの温度感受性株、特にセンダイウイルスCl.151株の全長ゲノムcDNAを用いて、外来遺伝子導入組換えウイルスを得、該組換えウイルスを外来遺伝子の生体内あるいは培養細胞内持続発現用組換えベクターとする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は遺伝子治療等に有用な、生体内において安定して長期間外来遺伝子を発現するウイルスベクターに関する。
【背景技術】
【0002】
遺伝性代謝疾患等の遺伝子治療においては、導入した外来遺伝子の発現が長期間持続することが望まれている。これまでは、レトロウイルスベクターを用いて宿主の染色体に遺伝情報を組み込むことによってこの目標を達成してきたが、組み込んだ遺伝子の影響で細胞がガン化した臨床例が報告され、安全性が問題視されている。このため、染色体とは独立にかつ安定に存在できる遺伝情報発現系の開発が提唱されているが、未だに実現していない。
センダイウイルスはパラミクソウイルス科に属するマイナス一本鎖RNAウイルスで、ヒトに対する病原性がない点、転写や複製は細胞質内で行われ宿主の遺伝情報に影響を与えない点、および遺伝子発現活性が高くて種特異性が低い点等の特徴を持っているため、遺伝子治療用のベクターの素材として注目されている。
【0003】
現在のところ、ワクシニアウイルスベクター、もしくはプラスミドベクターを用いてT7 RNA polymeraseを強制発現した培養細胞に、T7 RNA polymeraseによってセンダイウイルスの全長ゲノムRNAの相補鎖が発現する発現ベクターと、センダイウイルスの転写複製に関与するNP、P、Lの各遺伝子の発現ベクターをトランスフェクションすることによって組換え体センダイウイルスを作製する方法が確立されている。この方法を用いて、外来遺伝子を挿入したセンダイウイルス作製用ベクターから組換え体センダイウイルスが作製されており、またその応用として、センダイウイルスのF、M、HNの各遺伝子を欠損させた組換え体センダイウイルスも作製されている。また、目的のタンパク質をこれらの組換え体センダイウイルスに発現させることによって、タンパク質生産系としての応用も検討されている。
【0004】
これらのセンダイウイルス作製用ベクターから作製された組換え体センダイウイルスを用いて、染色体とは独立に存在できる遺伝情報発現系として、遺伝子治療への応用が多くの研究グループによって試行されている。しかし、これらのセンダイウイルスベクターは細胞障害性のあるZ株をベースにしたものであり、その細胞障害性を抑制させるために該ウイルスの遺伝子を欠損させたものであり、一代で死滅するため、安全性は向上しているものの遺伝子発現の持続する期間は限られている。また、タンパク質発現系として応用する際にも、細胞傷害性のために、一過性の発現系としての応用しかできない。
【0005】
一方、センダイウイルスには種々の性質を有する株が知られており、この中で、温度感受性株として、38°Cで殆どウイルス粒子を産生せず、32°Cでは複製サイクルが働きウイルス粒子を産生する、温度感受性変異株Cl.151株が、現・広島大学の吉田哲也教授らによって1979年に報告されている。また、このほか、センダイウイルスの温度感受性変異株としては、例えばHVJpi株が挙げられるが、この株をマウスに感染させた場合、野生株感染における致死量よりも遥かに多い量を感染させてもマウスは生存し続けることが明らかになっている。また、HVJpi株感染マウスに野生株を感染させると、マウスが生存し続けることから、温度感受性株は野生株に対するワクチンとして機能すると考えられている。しかし、HVJpi株感染マウスの肺において、ウイルス由来タンパク質の発現が約一か月後には確認されなくなることから、生体内において、温度感受性株が持続感染能を有するか否かは明らかではなく、例えば遺伝子治療用のベクターとして、生体内において外来遺伝子の実質的な持続発現能を有するとはいえない状況にあった。
【0006】
【特許文献1】WO97/16359
【特許文献2】特開P2002-272465号公報
【特許文献3】特開P2000-201689号公報
【非特許文献1】T. Yoshida et al. (1979) Virology 92, 139-154.
【非特許文献2】K. Kiyotani et al. (1990) Virology 177, 65-74.
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで、本発明の課題は、従来のセンダイウイルスベクターでは不可能であった、細胞障害性がなく、しかも培養細胞にのみでなく、さらに生体内においても長期間持続する遺伝子発現を実現でき、遺伝子治療用ベクター、タンパク質生産用ベクターとして極めて有用な新規ウイルスベクターを提供することにある。
【課題を解決するための手段】
【0008】
本発明者らは、上記課題を解決するため、新たな遺伝子治療用ベクターの開発に当たって、上記センダイウイルス温度感受性変異体、特にCl.151株が38°Cで殆どウイルス粒子を産生しない点に着目し、生体内において長期間持続する遺伝子発現を実現するセンダイウイルスベクターを構築するために、上記Cl.151株の全長ゲノムcDNAをクローニングした。さらに、この全長ゲノムcDNAに外来遺伝子発現カセットを挿入し、そこから得られる、組換え体センダイウイルスによる外来遺伝子発現の持続性を検討した。
【0009】
その結果、培養細胞における発現は、Z株由来センダイウイルスベクターを用いた場合、感染細胞が死滅するために短期間であるのに対し、Cl.151株由来センダイウイルスベクターを用いた場合、4か月以上発現が持続した。また、ラットの大腸に感染させた場合、Z株由来センダイウイルスベクターでは約2週間で発現が確認されなくなるのに対し、Cl.151株由来センダイウイルスベクターを用いた場合、大腸上皮細胞において2か月以上発現が持続した。このような培養細胞と大腸上皮におけるCl.151株由来センダイウイルスベクターの有意な発現の持続性からみて、生体内の他臓器においても、このセンダイウイルスベクターからの持続的な外来遺伝子の発現が可能であると確信し、さらに、驚くべきことに、通常の発現プラスミドと比較しても、Cl.151由来組換え体センダイウイルスからの、目的タンパク質の発現が、量、持続時間ともに多いとの知見を得て、本発明を完成させるに至ったものである。
すなわち、本発明は以下のとおりである。
【0010】
(1) センダイウイルス温度感受性株の全長ゲノムcDNAおよび外来遺伝子挿入部位を少なくとも有することを特徴とする、組換えセンダイウイルス作製用ベクター。
(2) センダイウイルス温度感受性株がセンダイウイルスCl.151株であることを特徴とする、上記(1)に記載の組換えセンダイウイルス作製用ベクター。
(3) クローニングベクターのクローニングサイトに、センダイウイルスCl.151株の全長ゲノムcDNAを挿入したことを特徴とする、上記(1)に記載の組換えセンダイウイルス作製用ベクター。
(4)上記(1)〜(3)のいずれかに記載のベクターに外来遺伝子DNAが挿入されていることを特徴とする、組換えセンダイウイルス作製用ベクター。
(5) センダイウイルスCl.151株の全長ゲノムRNAに外来遺伝子DNAに対応するRNAが連結されていることを特徴とするRNA。
(6) 外来遺伝子DNAに対応するRNAが連結された、センダイウイルスCl.151株の全長ゲノムRNAを有することを特徴とする、組換えウイルス。
(7)上記(6)に記載の組換えウイルスからなることを特徴とする、外来遺伝子の生体内持続発現用組換えセンダイウイルスベクター
(8)上記(6)に記載の組換えウイルスからなることを特徴とする、遺伝子治療用組換えベクター
(9)上記(6)に記載の組換えウイルスからなることを特徴とする、外来遺伝子の培養細胞内持続発現用ウイルスベクター。
(10)上記(4)に記載の組換えベクターが導入されていることを特徴とする細胞。
(11) 外来遺伝子DNAに対応するRNAが連結されたセンダイウイルスCl.151株の全長ゲノムRNAを含有することを特徴とする細胞。
(12) 上記(10)に記載の細胞を、ウイルス粒子産生温度下で培地に培養し、培養物からウイルス粒子を採取することを特徴とする、上記(6)に記載の組み換えウイルスを製造する方法。
(13)上記(6)に記載の組換えウイルスを培養細胞に感染させ、ウイルス粒子非産生温度下で培地に培養し、外来遺伝子由来の蛋白質を採取することを特徴とする、蛋白質の製造方法。
【発明の効果】
【0011】
本発明は、センダイウイルス温度感受性変異体、特にCl.151株の全長RNAを有し、外来遺伝子を生体内あるいは培養細胞において持続的に発現する、組み換えセンダイウイルスからなる持続発現用組換えベクターに関するものであり、該RNAに連結した外来遺伝子の発現が、培養細胞や生体内において従来のセンダイウイルスベクターと比較して、非常に長期間持続することを明らかにしたものである。本発明のセンダイウイルスベクターを用いることによって、従来の遺伝子治療用ベクターでは不可能であった、生体内で安定して、染色体とは独立に存在できる遺伝情報発現系を実現することが可能になった。
【0012】
また、該センダイウイルスベクターの外来遺伝子発現能の高さは、生体内で極めて効率的に外来遺伝子を発現させることを可能とし、例えば、疾病の治療あるいは予防に必要な蛋白質を生体内において有効に産生させることが可能となる。また、外来遺伝子発現能の高さと持続性を利用すれば、本発明のセンダイウイルスベクターを一度細胞に感染させるだけで、その細胞から長期間目的タンパク質を回収し続けることができるため、簡便で効率的なタンパク生産系としての応用も可能である。
【発明を実施するための最良の形態】
【0013】
センダイウイルスには種々の性質の異なる株が知られ、その中で温度感受性株、特にセンダイウイルスCl.151株は、38℃ではほとんどウイルス粒子を産生せず、32℃では複製サイクルが働き、ウイルス粒子を産生する温度感受性株である。したがって、ヒトをはじめとする哺乳動物の体温ではほとんど細胞障害性を発揮しない。
本発明によれば、センダイウイルスの温度感受性株に外来遺伝子を導入した組換えウイルスからなる、外来遺伝子の持続発現用組換えウイルスベクターが提供できる。
【0014】
このような、組み換えウイルスを得て、生体内あるいは培養細胞内で外来遺伝子を持続発現させるためには、まず、センダイウイルス温度感受性株の全長ゲノムcDNA(配列表の配列番号1)をクローニングし、少なくとも該全長ゲノムcDNAと外来遺伝子挿入部位を構成するDNA配列をクローニングベクタ―に組み込み、組換えセンダイウイルス作製用ベクターとする。次いで、得られた組換えセンダイウイルス作製用ベクターの外来遺伝子挿入部位に、外来遺伝子DNAを挿入し、外来遺伝子が導入された組換えセンダイウイルス作製用ベクターを得る。
ただし、この外来遺伝子導入組換えセンダイウイルス作成用ベクターを得るには、予め、上記温度感受性株の全長ゲノムcDNAと外来遺伝子を連結しておき、これをクローニングベクターに組み込んでもよい。
【0015】
このようにして作成した遺伝子導入組換えセンダイウイルス作成用ベクターは、細胞にトランスフェクションすることにより、細胞内で、外来遺伝子に対応するRNAがゲノム全長RNA(同配列番号2)に連結した組換えウイルスを再構成する。この組換えウイルスは、生体内あるいは培養細胞において、外来遺伝子を長期間発現し続ける。
【0016】
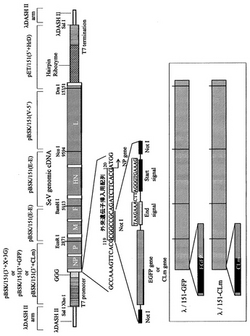
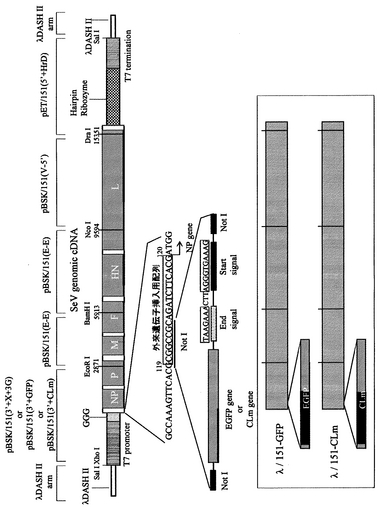
次に、外来遺伝子導入組換えセンダイウイルス作成用ベクターの構築、及びこれを用いた、外来遺伝子導入組換えウイルスを生体内で発現させる場合について、図1を参酌しつつ、温度感受性株として、特にセンダイウイルスCl.151の全長cDNAを使用する場合の例を挙げて具体的に説明する。
【0017】
外来遺伝子導入組換えセンダイウイルス作成用ベクターを得るには、Cl.151株全長cDNAをクローニングし、細胞内で(+)鎖のゲノムRNAが生合成されるように、λDASHII等のクローニングベクターに組み込むとともに、全長cDNAの上流(ゲノムRNAにおける3’末端側)にT7プロモーター配列、3個のグアニジン残基をこの順で配置し、全長cDNAの下流(ゲノムRNAにおける5’末端側)にタバコリングスポットウイルスのヘアピンリボザイム配列、T7 RNA polymerase終止配列をこの順で配置する。
【0018】
なお、T7プロモーター配列はT7 RNA polymeraseによってゲノムRNAにおける3’末端側から(+)鎖ゲノムRNAが生合成されるように、3個のグアニジン残基はT7 RNA polymeraseによるRNA転写効率を上昇させるように(S. Leyrer et al. (1998) J. Virol. Methods 75; 47-58)、タバコリングスポットウイルスのヘアピンリボザイム配列は転写された(+)鎖ゲノムRNAが末端で正確に切断されるように、T7 RNA polymerase終止配列はT7 RNA polymeraseによるRNA転写を正確に終結させるために付加するものである。
【0019】
このようにして得られた、本発明のベクターには、外来遺伝子(図1では、EGFP gene 、CLm geneを例示)を組み込み、外来遺伝子導入組換えセンダイウイルス作成用ベクターとするが、外来遺伝子はベクター上の、センダイウイルスの遺伝子がコードされていない部分に挿入することができる。センダイウイルスの遺伝子発現には、ゲノムRNAの3’末端に近い遺伝子ほど強く発現するという極性効果があるため、NP遺伝子の上流側に挿入した場合最も発現が強く、L遺伝子の下流に挿入した場合最も発現が弱くなる。
挿入する際、外来遺伝子の下流側には、外来遺伝子の翻訳をストップさせる終止配列と、それに続くセンダイウイルス遺伝子の翻訳を開始させる開始配列を設けて、外来遺伝子導入組換えセンダイウイルス作成用ベクターとする。
外来遺伝子としては特に制限はなく、遺伝子治療に用いられているものを使用でき、例えば患者においてその産生が不足あるいは欠損している、酵素、ホルモン、その他の生理活性ペプチドあるいはタンパク質が挙げられる。
【0020】
図1の例においては、まず外来遺伝子挿入部位を作製するために、制限酵素NotI切断部位を含む配列をNP遺伝子の上流側直前に挿入した後に、さらに制限酵素NotI切断部位を両端に有する、外来遺伝子−終止配列−開始配列からなるDNAを、上記本発明のベクターの外来遺伝子挿入部位であるNotI切断部位に挿入する場合が示されている。
本発明の外来遺伝子導入組換えセンダイウイルス作成用ベクターから作製された組換えウイルスを遺伝子治療に用いるには、まず、該ベクターをウイルス産生用細胞に導入するが、この際、T7 RNA polymeraseの給源として、例えば、T7 RNA polymerase発現ワクシニアウイルスを該細胞に感染させるとともに、ウイルスタンパク質の形成を補い、ウイルス粒子を効率的に産生させるために、NP遺伝子、P遺伝子およびL遺伝子を有する発現ベクターも細胞に導入するのが好ましい。
【0021】
使用する細胞としては、センダイウイルスが高効率で増殖する性質を有するものが好ましく、サル腎臓由来のLLC-MK2細胞が最も理想的である。
この例においては、該ベクターが導入されたウイルス産生細胞においては、ベクターDNAがT7 RNA polymeraseによってT7プロモーター以降がRNAに転写されるが、その際、産生するRNA分子はヘアピンリボザイム配列によって、それ以降の配列が切断されて削除され、外来遺伝子DNAに対応するRNAが、終止配列、開始配列及びNot I配列を介して、Cl.151株のゲノム全長RNAと連結したRNA分子(プラス鎖)が形成される。
【0022】
次いで、このようにして形質転換され、外来遺伝子DNAに対応するRNAが連結したCl.151株のゲノム全長RNA分子を含有する細胞を、Cl.151株のウイルス粒子産生温度である32℃で培養する。細胞においては、組換えられたセンダイウイルスのRNA(+)鎖にNP、P、L遺伝子産物が結合した複合体が鋳型となって、ウイルスRNAポリメラーゼにより(−)鎖に転写され、外来遺伝子が導入されたセンダイウイルス粒子が再構成される。このセンダイウイルス粒子をさらに多量に得る場合には、細胞から回収されたセンダイウイルス粒子を例えば鶏卵に接種し、32℃で孵卵することにより増殖させればよい。該センダイウイルス粒子は、Cl.151株由来の持続感染能を有し、ヒトの体温ではウイルス粒子を産生しない持続感染型で存在し、生体内において導入した外来遺伝子を持続的に発現する。
【0023】
なお、本発明は、これら例示中に示したものに限らず、種々の遺伝子工学的材料、手段を使用できる。
例えば、本発明の組換えセンダイウイルス作成用ベクターを作成する際使用するクローニングベクターとしては、λDASH IIの他にもcharon 40等が挙げられ、センダイウイルスのゲノムサイズである約15.4 bpのDNA断片がクローニングできれば利用できる。
また、得られた外来遺伝子導入組換えセンダイウイルス作成用ベクターを培養する細胞としては、サル腎臓由来のLLC-MK2細胞が最も理想的であるが、ハムスター腎臓由来のBHK-21等の細胞も使用できる。
【0024】
一方、本発明の外来遺伝子が導入された組換えセンダイウイルスは、培養細胞において外来遺伝子を持続的に発現する組換えベクターとして、蛋白質の効率的生産に有効に用いられる。これには、上記組換えセンダイウイルス作成用ベクターの外来遺伝子挿入部位に、目的とする蛋白質をコードする遺伝子を挿入して、外来遺伝子導入組換えセンダイウイルス作成用ベクターを得、この組換えベクターを上記と同様に細胞にトランスフェクションして得られた組換えセンダイウイルスを培養細胞に感染させ、感染細胞をウイルス粒子の非産生温度下(センダイウイルスCl.151株由来のウイルスベクターの場合、例えば37℃)で培地に培養する。この感染細胞においては、転写産物として上記外来遺伝子由来のRNAがセンダイウイルスCl.151株のゲノム全長RNAに連結したRNA分子が形成され、該RNAは、形質転換細胞の増殖に伴い各細胞に受け継がれ、持続的に外来遺伝子を発現し、外来遺伝子由来の蛋白質が産生する。次いで、この培養細胞を含む培養物から目的とする蛋白質を採取する。
【0025】
この蛋白質の生産において使用する手段、材料は、ウイルス粒子の作成以降の工程を除いて、上記生体内で外来遺伝子の発現する場合と基本的には同様である。
以下に、本発明の実施例を示す。但し本発明はこれら実施例により限定されるものではない。
【実施例】
【0026】
〔実施例1〕センダイウイルスCl.151株からのゲノムcDNAのクローニング
センダイウイルスCl.151株(広島大学吉田哲也教授から分与)から100 mg/mlプロテアーゼK、0.5% SDS処理によって、ゲノムRNAを分離した。分離したRNAの3’末端にRNAリンカーをライゲーションさせ、このRNAリンカーに相補的なプライマーを用いてゲノムRNAの3’末端をPCRによってクローニングし塩基配列を決定した。また、ゲノムRNAの5’末端付近をPCRによってクローニングし、同様に塩基配列を決定した。決定した塩基配列をもとに作製したゲノムRNAの5’末端に相補的なDNAリンカー
5’-AAATTTAAAAGAATACATATCTCTTAAACTCTTGTCTGGTGCGGCCGCAAAAGGAAAA-3’(同配列番号3)
をゲノムRNAにアニーリングさせ、この「ゲノムRNA-リンカーDNA複合体」を鋳型にして、ゲノムRNAの3’末端に相補的なプライマー
5’-TTTTCCTTTTGCGGCCGCTAATACGACTCACTATAACCAAACAAGAGAAGAAACA-3’(同配列番号4)
を用いて逆転写反応をSuperScript
III First-strand synthesis system for RT-PCR (Invitrogen)によって行った。
【0027】
このようにして得られた一本鎖cDNAを鋳型として、下記の3組のプライマーセット
5’-TTTTCCTTTTGCGGCCGCTAATACGACTCACTATAACCAAACAAGAGAAGAAACA-3’(同配列番号5)、
5’-GGGATCTTGGCTATGGTGAT-3’(同配列番号6) [for 1-2875]
5’-GGGCATAGGAGAGAACACATCATCT-3’(同配列番号7)、 5’-ACTGCATGACACTCGTATAGGGTCT-3’ (同配列番号8) [for 2870-10484]
5’-CCGGAATTCAGTCGTTACTCGCCATTTTCC-3’(同配列番号9)、
5’-TTTTCCTTTTGCGGCCGCACCAGACAAGAG-3’(同配列番号10)[for 10479-15384]
を用いてPfuUltra High-fidelity DNA polymerase (STRATAGENE)によってPCRで増幅し、3本の二本鎖cDNAを得た。各々のcDNAを制限酵素を用いて切断し、SeV: 1-2875, 2870-10484, 10479-15384の3断片に分けてpBluescript II SK(+) (STRATAGENE)にクローニングした。
【0028】
〔実施例2〕クローニングしたゲノムcDNAへの、lDASHIIへのクローニングやゲノムRNA転写のために必要な配列の付加
〔実施例1〕によって得られた3断片のcDNAのうち、SeV: 1-2875を含むもの(pBSK/151(3’-E))の配列の内、センダイウイルスcDNAを含む配列のすぐ上流にT7プロモーター配列、3塩基のグアニジン残基を、この順で挿入した (pBSK/151(3’+X+3G))。SeV:
10479-15384を含むもの(pBSK/151(E-5’))から、SeV: 15351-15384の部分を切り出し、そのすぐ下流に、タバコリングスポットウイルスのヘアピンリボザイム配列、T7 RNA polymerase終止配列をこの順で挿入した形で、pET30a(+) (Novagen)にクローニングし直した(pET/151(5’+HrD))。SeV: 2870-10484を含むもの(pBSK/151(E-E))からSeV: 9015-10479を含む断片を、pBSK/151(E-5’)のSeV: 10479-15384のすぐ上流に挿入した(pBSK/151(V-5’))。
【0029】
〔実施例3〕外来遺伝子挿入センダイウイルスcDNA断片の作製
(1)外来遺伝子挿入部位の組み込み
pBSK/151(3’+X+3G)に対し、外来遺伝子挿入部位作製用プライマーとして、
5’-GCCAAAGTTCACGCGGCCGCAGATCTTCACGATGGCCGGGTTGT-3’(同配列番号11(センス鎖))
5’-ACAACCCGGCCATCGTGAAGATCTGCGGCCGCGTGAACTTTGGC-3’(同配列番号12(アンチセンス鎖))を用いて、Quikchange Site-directed Mutagenesis II (STRATAGENE) によってNot I 認識配列をSeV: 119の後ろに挿入した(pBSK/151(3’+Not))。
【0030】
(2)pBSK/151(Nhe-Not)の作製
外来遺伝子の導入を容易に行うため、〔実施例3〕で得られたpBSK/151(3'’+Not)のNot I サイトの直前5塩基目のT、3塩基目C、2塩基目Aを下記のプライマーを用いてPCRを行うことにより、それぞれC、A、Gに置換して、Nhe I認識配列の導入を行った。
始めに、M13リバースプライマー5’-GGAAACAGCTATGACCATG-3’(同配列番号13(N末端側))と
Nhe I認識配列導入用プライマー1;
5’-CTGCGGCCGCGCTAGCTTTGGCAGCAAAGAA-3’(同配列番号14(C末端側))あるいはNhe I認識配列導入用プライマー2;
5’-AAGCTAGCGCGGCCGCAGATCTTC-3’(同配列番号15(N末端側))
NP C末側プライマー;
5’-CCGGAATTCGTATGATCCTAGATTCCTCCT-3’(同配列番号16(C末端側))
の2種類のプライマーセットを用い、pBSK/151(3’+Not)を鋳型として、それぞれPCRを行った。生じたPCR産物を混合し、M13リバースプライマーとNP C末側プライマーとで再度PCRを行い、Nhe I認識配列が導入されたcl151の3’DNA断片を得た。このPCR産物を制限酵素Sac Iで切断し、同酵素で切断したpBSK/151(3'+Not)に組み込み、pBSK/151(Nhe-Not)を得た。
【0031】
(3)外来遺伝子(EGFP遺伝子、CLm遺伝子、PG-FGF-1遺伝子)の導入
EGFP遺伝子挿入用プライマーとして、
5’-ACTTGCGGCCGCTCGCCACCATGGTGAGCAAGGGCGAGGA-3’(同配列番号17(N末端側))
5’-ACTTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTAGACGGCCGCTTTACTTGTACAGCTCGTCCA-3’(同配列番号18(C末端側))
の2本のプライマーを用いてpEGFP-C1(Clontech)上からEGFP遺伝子を増幅し、得られた二本鎖DNAの末端をNot Iで切断し、pBSK/151(3’+Not)のNot I部位に挿入することによって、pBSK/151(3’+GFP)を得た。
同様の方法でCLm(分泌型ルシフェラーゼ)遺伝子挿入用プライマーとして、
5’-
ACTTGCGGCCGCTCGCCCTTATGAAGACCTTAATTCTTGCC-3’(同配列番号19(N末端側))、
5’-ACTTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTAATTCGCCCTTCTATTTGCATTCATCTGGTA-3’(同配列番号20(C末端側))
の2本のプライマーを用いてpCLm上からCLm遺伝子を増幅し、pBSK/151(3’+Not)のNot I部位に挿入することによって、pBSK/151(3’+CLm)を得た。
【0032】
PG-FGF-1(シンデカン融合ヘパリン結合性増殖因子FGF-1)遺伝子挿入用プライマーとして、
5’-TGGCTAGCTCACCATGGCCCCCGCCCGT-3’(同配列番号21(N末端側))、
5’-TTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTAAGGCTACCTTTAATCAGAAGAGACTGGCAG-3’(同配列番号22(C末端側))
の2本のプライマーを用いてpMKIT-neo-PGFGF-1上からPG-FGF-1遺伝子を増幅し、得られた二本鎖DNAの末端をNhe IとNot Iで切断し、pBSK/151(Nhe-Not)のNhe I、Not I部位に挿入することによって、pBSK/151(3' +PG-FGF-1)を得た。
【0033】
〔実施例4〕λDASH IIへのクローニングによる外来遺伝子導入組換えウイルス作成用ベクターの作製
〔実施例2〕、〔実施例3〕によって得られた各プラスミドのうち、pBSK/151(3’+GFP)、pBSK/151(3’+CLm)もしくはpBSK/151(3’+PG-FGF-1)からT7プロモーター配列−SeV: 1- 2875を、pBSK/151(E-E)からSeV: 2870-5917、SeV: 5913-9598を、pBSK/151(V-5’)からSeV: 9593-15351を、pET/151(5’+HrD)からSeV: 15351-15384−T7 RNA polymerase終止配列を切り出して、lDASH II (STRATAGENE)にこの順番でクローニングし直した。得られたクローンをそれぞれl/151-GFP、l/151-CLmもしくはl/151-PG-FGF-1と名づけ、それらからファージDNAを精製した。
【0034】
〔実施例5〕センダイウイルスの再構成
LLC-MK2細胞を1 x 106 cells / wellで6-wellプレートに蒔き、24時間培養後T7 RNA polymeraseを発現する弱毒性ワクシニアウイルス(MVAGKT7)をM.O.I.=1.0で1時間、37°Cで感染させた。細胞を洗浄した後、lDASH IIにクローニングしたセンダイウイルスcDNA(l/151-GFP、l/151-CLmもしくはl/151-PG-FGF-1)、pGEM/NP、pGEM/P、pGEM/Lをそれぞれ5 mg、2 mg、1 mg、2 mgの量比でOptiMEM 300 mlに懸濁し、10 mlのLipofectamine 2000を含む300 mlのOptiMEMと混合し、20分間室温放置後、細胞に添加して4時間培養した。培養後、20% 血清、80 mg/ml シトシンアラビノシドC(AraC)を含んだ培地を等量加えてさらに32°Cで 48時間培養した。
【0035】
これらの細胞を回収し、ぺレットを500 mlのPBSで懸濁し、凍結融解を4回繰り返した。これらを10日間孵卵させた鶏卵に100 ml接種し、32°Cで5日間孵卵させた後に漿尿液を回収した。ワクシニアフリーにするためにこれら回収した漿尿液をさらに10-4〜10-8希釈して鶏卵に再接種し、同様に回収した。得られた漿尿液を15,000 rpmで30分遠心することによってセンダイウイルス(r151-GFP、r151-CLmもしくはr151-PG-FGF-1)を沈澱させ、組換えセンダイウイルスの感染実験に用いた。
【0036】
〔実施例6〕EGFP遺伝子導入組換えセンダイウイルスからの培養細胞中におけるEGFP遺伝子の持続的発現の解析
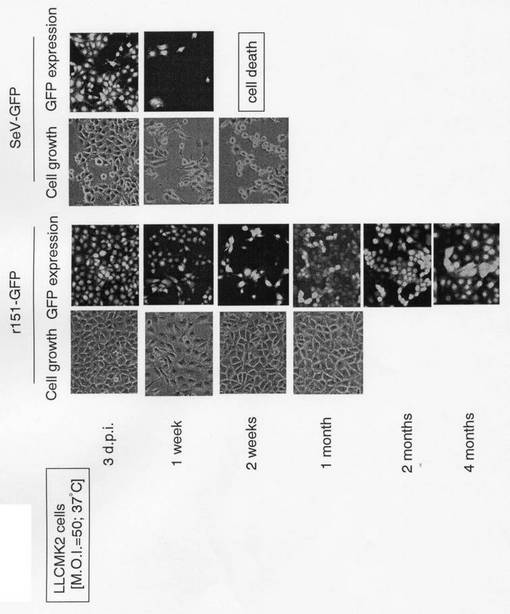
r151-GFPをLLC-MK2細胞にM.O.I.=50で感染させ、感染細胞を蛍光顕微鏡で観察することによって、EGFP遺伝子の発現を経時的に確認した。
図2から明らかなように、持続感染株由来センダイウイルスベクターからのEGFP遺伝子の発現は4ヶ月以上も持続しており、従来のZ株を基本骨格としたセンダイウイルスベクター(SeV-GFP)と比較して、有意に持続していた。
【0037】
〔実施例7〕EGFP遺伝子導入組換えセンダイウイルスの生体中におけるEGFP遺伝子の持続的発現の解析
再構成したr151-GFP、300 mgを3cm長に切った経口ゾンデを用いてラットの大腸に感染させた。
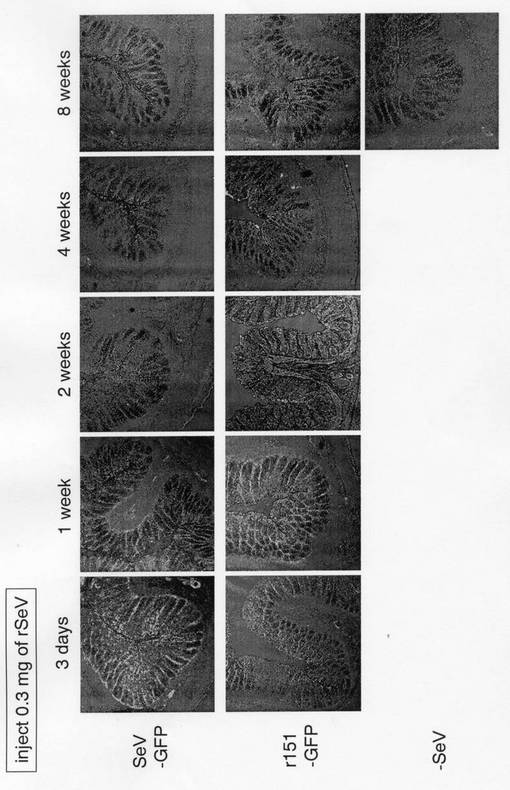
EGFP遺伝子の発現については、凍結包埋させた大腸組織を蛍光顕微鏡で観察することによって、確認を行った。
図3から明らかなように、持続感染型センダイウイルスベクターからのEGFP遺伝子の発現は2ヶ月以上も持続しており、従来のZ株を基本骨格としたセンダイウイルスベクター(SeV-GFP)と比較して、有意に持続していた。
【0038】
〔実施例8〕CLm遺伝子導入組換えセンダイウイルスの培養細胞中におけるCLm遺伝子発現量の検討
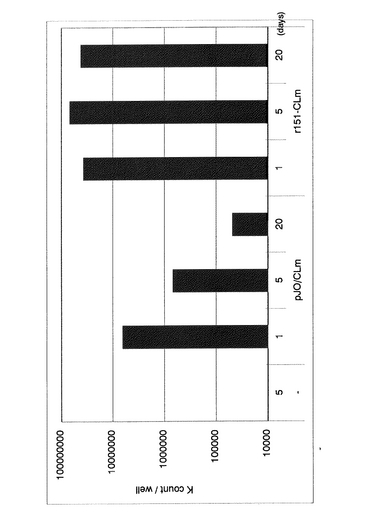
r151-CLmをLLC-MK2細胞にM.O.I.=50で感染させた。また、CMVプロモーターによりCLm遺伝子が発現するCLm発現ベクタープラスミド(pJO/CLm)をトランスフェクションしたLLC-MK2細胞も同様に用意し、これら細胞を37℃で培養した。
各々の細胞の培養上清を希釈し、等量の216 nMルシフェリンを加え、ルミノメーターでルシフェラーゼ活性を測定することによって、CLm遺伝子発現量を定量した。その結果、2日後の発現量は感染細胞のほうが10倍近く高く、さらに、トランスフェクション細胞では数日で発現量が低下するのに対し、感染細胞においては発現が長期間持続した(図4)。
【0039】
〔実施例9〕PG-FGF-1遺伝子導入組換えセンダイウイルスの培養細胞におけるPG-FGF-1遺伝子発現量の検討
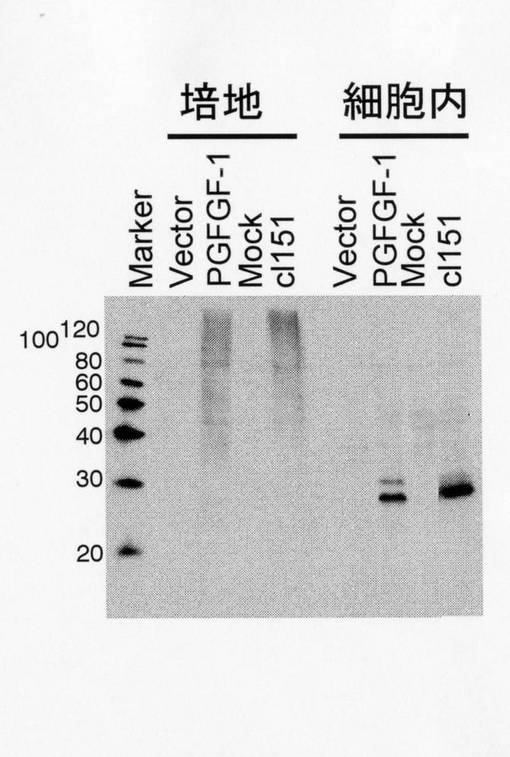
サル腎細胞由来COS細胞に、r151-PG-FGF-1をM.O.I.=50で感染させた場合と、発現プラスミドpMKIT-neo-PGFGF-1をリポフェクトアミン2000(Invitrogen)を用いて導入し、これら細胞を32℃で培養し、PG-FGF-1の発現量を比較した。
感染2日目に培養上清と細胞抽出液を調製しウェスタンブロット法を用いて比較した結果、細胞内において顕著なPG-FGF-1の発現量の増加が認められ、その発現量はトランスフェクションと比較して約3倍であった(図5)。
【図面の簡単な説明】
【0040】
【図1】本発明の外来遺伝子導入組換えセンダイウイルス作成用ベクター構造を示す図である。
【図2】本発明の組換えセンダイウイルス作成用ベクターにEGFP遺伝子を導入し、LLC-MK2細胞に感染させた場合の、EGFP遺伝子発現量と細胞の形態の経時変化を示す蛍光顕微鏡写真である。
【図3】本発明のEGFP遺伝子導入組換えセンダイウイルスをラットの大腸に感染させた場合の、大腸上皮におけるEGFP遺伝子発現量の経時変化を示す蛍光顕微鏡写真である。
【図4】本発明の分泌型ルシフェラーゼ(CLm)遺伝子導入組換えセンダイウイルスをLLC-MK2細胞に感染させた場合(r151-CLm)と、分泌型ルシフェラーゼ導入プラスミド(pJO/CLm)をトランスフェクションさせた場合とにおける、培養上清におけるルシフェラーゼ活性の経時変化を示すグラフである。
【図5】本発明のシンデカン融合ヘパリン結合性増殖因子FGF-1(PG-FGF-1)遺伝子導入組換えセンダイウイルスをCOS細胞に感染させた場合(cl151)と、シンデカン融合ヘパリン結合性増殖因子FGF-1導入プラスミドをトランスフェクションさせた場合(PGFGF-1)とにおける、培地中と細胞内におけるシンデカン融合ヘパリン結合性増殖因子FGF-1の発現量を示すグラフ図である。
【技術分野】
【0001】
本発明は遺伝子治療等に有用な、生体内において安定して長期間外来遺伝子を発現するウイルスベクターに関する。
【背景技術】
【0002】
遺伝性代謝疾患等の遺伝子治療においては、導入した外来遺伝子の発現が長期間持続することが望まれている。これまでは、レトロウイルスベクターを用いて宿主の染色体に遺伝情報を組み込むことによってこの目標を達成してきたが、組み込んだ遺伝子の影響で細胞がガン化した臨床例が報告され、安全性が問題視されている。このため、染色体とは独立にかつ安定に存在できる遺伝情報発現系の開発が提唱されているが、未だに実現していない。
センダイウイルスはパラミクソウイルス科に属するマイナス一本鎖RNAウイルスで、ヒトに対する病原性がない点、転写や複製は細胞質内で行われ宿主の遺伝情報に影響を与えない点、および遺伝子発現活性が高くて種特異性が低い点等の特徴を持っているため、遺伝子治療用のベクターの素材として注目されている。
【0003】
現在のところ、ワクシニアウイルスベクター、もしくはプラスミドベクターを用いてT7 RNA polymeraseを強制発現した培養細胞に、T7 RNA polymeraseによってセンダイウイルスの全長ゲノムRNAの相補鎖が発現する発現ベクターと、センダイウイルスの転写複製に関与するNP、P、Lの各遺伝子の発現ベクターをトランスフェクションすることによって組換え体センダイウイルスを作製する方法が確立されている。この方法を用いて、外来遺伝子を挿入したセンダイウイルス作製用ベクターから組換え体センダイウイルスが作製されており、またその応用として、センダイウイルスのF、M、HNの各遺伝子を欠損させた組換え体センダイウイルスも作製されている。また、目的のタンパク質をこれらの組換え体センダイウイルスに発現させることによって、タンパク質生産系としての応用も検討されている。
【0004】
これらのセンダイウイルス作製用ベクターから作製された組換え体センダイウイルスを用いて、染色体とは独立に存在できる遺伝情報発現系として、遺伝子治療への応用が多くの研究グループによって試行されている。しかし、これらのセンダイウイルスベクターは細胞障害性のあるZ株をベースにしたものであり、その細胞障害性を抑制させるために該ウイルスの遺伝子を欠損させたものであり、一代で死滅するため、安全性は向上しているものの遺伝子発現の持続する期間は限られている。また、タンパク質発現系として応用する際にも、細胞傷害性のために、一過性の発現系としての応用しかできない。
【0005】
一方、センダイウイルスには種々の性質を有する株が知られており、この中で、温度感受性株として、38°Cで殆どウイルス粒子を産生せず、32°Cでは複製サイクルが働きウイルス粒子を産生する、温度感受性変異株Cl.151株が、現・広島大学の吉田哲也教授らによって1979年に報告されている。また、このほか、センダイウイルスの温度感受性変異株としては、例えばHVJpi株が挙げられるが、この株をマウスに感染させた場合、野生株感染における致死量よりも遥かに多い量を感染させてもマウスは生存し続けることが明らかになっている。また、HVJpi株感染マウスに野生株を感染させると、マウスが生存し続けることから、温度感受性株は野生株に対するワクチンとして機能すると考えられている。しかし、HVJpi株感染マウスの肺において、ウイルス由来タンパク質の発現が約一か月後には確認されなくなることから、生体内において、温度感受性株が持続感染能を有するか否かは明らかではなく、例えば遺伝子治療用のベクターとして、生体内において外来遺伝子の実質的な持続発現能を有するとはいえない状況にあった。
【0006】
【特許文献1】WO97/16359
【特許文献2】特開P2002-272465号公報
【特許文献3】特開P2000-201689号公報
【非特許文献1】T. Yoshida et al. (1979) Virology 92, 139-154.
【非特許文献2】K. Kiyotani et al. (1990) Virology 177, 65-74.
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで、本発明の課題は、従来のセンダイウイルスベクターでは不可能であった、細胞障害性がなく、しかも培養細胞にのみでなく、さらに生体内においても長期間持続する遺伝子発現を実現でき、遺伝子治療用ベクター、タンパク質生産用ベクターとして極めて有用な新規ウイルスベクターを提供することにある。
【課題を解決するための手段】
【0008】
本発明者らは、上記課題を解決するため、新たな遺伝子治療用ベクターの開発に当たって、上記センダイウイルス温度感受性変異体、特にCl.151株が38°Cで殆どウイルス粒子を産生しない点に着目し、生体内において長期間持続する遺伝子発現を実現するセンダイウイルスベクターを構築するために、上記Cl.151株の全長ゲノムcDNAをクローニングした。さらに、この全長ゲノムcDNAに外来遺伝子発現カセットを挿入し、そこから得られる、組換え体センダイウイルスによる外来遺伝子発現の持続性を検討した。
【0009】
その結果、培養細胞における発現は、Z株由来センダイウイルスベクターを用いた場合、感染細胞が死滅するために短期間であるのに対し、Cl.151株由来センダイウイルスベクターを用いた場合、4か月以上発現が持続した。また、ラットの大腸に感染させた場合、Z株由来センダイウイルスベクターでは約2週間で発現が確認されなくなるのに対し、Cl.151株由来センダイウイルスベクターを用いた場合、大腸上皮細胞において2か月以上発現が持続した。このような培養細胞と大腸上皮におけるCl.151株由来センダイウイルスベクターの有意な発現の持続性からみて、生体内の他臓器においても、このセンダイウイルスベクターからの持続的な外来遺伝子の発現が可能であると確信し、さらに、驚くべきことに、通常の発現プラスミドと比較しても、Cl.151由来組換え体センダイウイルスからの、目的タンパク質の発現が、量、持続時間ともに多いとの知見を得て、本発明を完成させるに至ったものである。
すなわち、本発明は以下のとおりである。
【0010】
(1) センダイウイルス温度感受性株の全長ゲノムcDNAおよび外来遺伝子挿入部位を少なくとも有することを特徴とする、組換えセンダイウイルス作製用ベクター。
(2) センダイウイルス温度感受性株がセンダイウイルスCl.151株であることを特徴とする、上記(1)に記載の組換えセンダイウイルス作製用ベクター。
(3) クローニングベクターのクローニングサイトに、センダイウイルスCl.151株の全長ゲノムcDNAを挿入したことを特徴とする、上記(1)に記載の組換えセンダイウイルス作製用ベクター。
(4)上記(1)〜(3)のいずれかに記載のベクターに外来遺伝子DNAが挿入されていることを特徴とする、組換えセンダイウイルス作製用ベクター。
(5) センダイウイルスCl.151株の全長ゲノムRNAに外来遺伝子DNAに対応するRNAが連結されていることを特徴とするRNA。
(6) 外来遺伝子DNAに対応するRNAが連結された、センダイウイルスCl.151株の全長ゲノムRNAを有することを特徴とする、組換えウイルス。
(7)上記(6)に記載の組換えウイルスからなることを特徴とする、外来遺伝子の生体内持続発現用組換えセンダイウイルスベクター
(8)上記(6)に記載の組換えウイルスからなることを特徴とする、遺伝子治療用組換えベクター
(9)上記(6)に記載の組換えウイルスからなることを特徴とする、外来遺伝子の培養細胞内持続発現用ウイルスベクター。
(10)上記(4)に記載の組換えベクターが導入されていることを特徴とする細胞。
(11) 外来遺伝子DNAに対応するRNAが連結されたセンダイウイルスCl.151株の全長ゲノムRNAを含有することを特徴とする細胞。
(12) 上記(10)に記載の細胞を、ウイルス粒子産生温度下で培地に培養し、培養物からウイルス粒子を採取することを特徴とする、上記(6)に記載の組み換えウイルスを製造する方法。
(13)上記(6)に記載の組換えウイルスを培養細胞に感染させ、ウイルス粒子非産生温度下で培地に培養し、外来遺伝子由来の蛋白質を採取することを特徴とする、蛋白質の製造方法。
【発明の効果】
【0011】
本発明は、センダイウイルス温度感受性変異体、特にCl.151株の全長RNAを有し、外来遺伝子を生体内あるいは培養細胞において持続的に発現する、組み換えセンダイウイルスからなる持続発現用組換えベクターに関するものであり、該RNAに連結した外来遺伝子の発現が、培養細胞や生体内において従来のセンダイウイルスベクターと比較して、非常に長期間持続することを明らかにしたものである。本発明のセンダイウイルスベクターを用いることによって、従来の遺伝子治療用ベクターでは不可能であった、生体内で安定して、染色体とは独立に存在できる遺伝情報発現系を実現することが可能になった。
【0012】
また、該センダイウイルスベクターの外来遺伝子発現能の高さは、生体内で極めて効率的に外来遺伝子を発現させることを可能とし、例えば、疾病の治療あるいは予防に必要な蛋白質を生体内において有効に産生させることが可能となる。また、外来遺伝子発現能の高さと持続性を利用すれば、本発明のセンダイウイルスベクターを一度細胞に感染させるだけで、その細胞から長期間目的タンパク質を回収し続けることができるため、簡便で効率的なタンパク生産系としての応用も可能である。
【発明を実施するための最良の形態】
【0013】
センダイウイルスには種々の性質の異なる株が知られ、その中で温度感受性株、特にセンダイウイルスCl.151株は、38℃ではほとんどウイルス粒子を産生せず、32℃では複製サイクルが働き、ウイルス粒子を産生する温度感受性株である。したがって、ヒトをはじめとする哺乳動物の体温ではほとんど細胞障害性を発揮しない。
本発明によれば、センダイウイルスの温度感受性株に外来遺伝子を導入した組換えウイルスからなる、外来遺伝子の持続発現用組換えウイルスベクターが提供できる。
【0014】
このような、組み換えウイルスを得て、生体内あるいは培養細胞内で外来遺伝子を持続発現させるためには、まず、センダイウイルス温度感受性株の全長ゲノムcDNA(配列表の配列番号1)をクローニングし、少なくとも該全長ゲノムcDNAと外来遺伝子挿入部位を構成するDNA配列をクローニングベクタ―に組み込み、組換えセンダイウイルス作製用ベクターとする。次いで、得られた組換えセンダイウイルス作製用ベクターの外来遺伝子挿入部位に、外来遺伝子DNAを挿入し、外来遺伝子が導入された組換えセンダイウイルス作製用ベクターを得る。
ただし、この外来遺伝子導入組換えセンダイウイルス作成用ベクターを得るには、予め、上記温度感受性株の全長ゲノムcDNAと外来遺伝子を連結しておき、これをクローニングベクターに組み込んでもよい。
【0015】
このようにして作成した遺伝子導入組換えセンダイウイルス作成用ベクターは、細胞にトランスフェクションすることにより、細胞内で、外来遺伝子に対応するRNAがゲノム全長RNA(同配列番号2)に連結した組換えウイルスを再構成する。この組換えウイルスは、生体内あるいは培養細胞において、外来遺伝子を長期間発現し続ける。
【0016】
次に、外来遺伝子導入組換えセンダイウイルス作成用ベクターの構築、及びこれを用いた、外来遺伝子導入組換えウイルスを生体内で発現させる場合について、図1を参酌しつつ、温度感受性株として、特にセンダイウイルスCl.151の全長cDNAを使用する場合の例を挙げて具体的に説明する。
【0017】
外来遺伝子導入組換えセンダイウイルス作成用ベクターを得るには、Cl.151株全長cDNAをクローニングし、細胞内で(+)鎖のゲノムRNAが生合成されるように、λDASHII等のクローニングベクターに組み込むとともに、全長cDNAの上流(ゲノムRNAにおける3’末端側)にT7プロモーター配列、3個のグアニジン残基をこの順で配置し、全長cDNAの下流(ゲノムRNAにおける5’末端側)にタバコリングスポットウイルスのヘアピンリボザイム配列、T7 RNA polymerase終止配列をこの順で配置する。
【0018】
なお、T7プロモーター配列はT7 RNA polymeraseによってゲノムRNAにおける3’末端側から(+)鎖ゲノムRNAが生合成されるように、3個のグアニジン残基はT7 RNA polymeraseによるRNA転写効率を上昇させるように(S. Leyrer et al. (1998) J. Virol. Methods 75; 47-58)、タバコリングスポットウイルスのヘアピンリボザイム配列は転写された(+)鎖ゲノムRNAが末端で正確に切断されるように、T7 RNA polymerase終止配列はT7 RNA polymeraseによるRNA転写を正確に終結させるために付加するものである。
【0019】
このようにして得られた、本発明のベクターには、外来遺伝子(図1では、EGFP gene 、CLm geneを例示)を組み込み、外来遺伝子導入組換えセンダイウイルス作成用ベクターとするが、外来遺伝子はベクター上の、センダイウイルスの遺伝子がコードされていない部分に挿入することができる。センダイウイルスの遺伝子発現には、ゲノムRNAの3’末端に近い遺伝子ほど強く発現するという極性効果があるため、NP遺伝子の上流側に挿入した場合最も発現が強く、L遺伝子の下流に挿入した場合最も発現が弱くなる。
挿入する際、外来遺伝子の下流側には、外来遺伝子の翻訳をストップさせる終止配列と、それに続くセンダイウイルス遺伝子の翻訳を開始させる開始配列を設けて、外来遺伝子導入組換えセンダイウイルス作成用ベクターとする。
外来遺伝子としては特に制限はなく、遺伝子治療に用いられているものを使用でき、例えば患者においてその産生が不足あるいは欠損している、酵素、ホルモン、その他の生理活性ペプチドあるいはタンパク質が挙げられる。
【0020】
図1の例においては、まず外来遺伝子挿入部位を作製するために、制限酵素NotI切断部位を含む配列をNP遺伝子の上流側直前に挿入した後に、さらに制限酵素NotI切断部位を両端に有する、外来遺伝子−終止配列−開始配列からなるDNAを、上記本発明のベクターの外来遺伝子挿入部位であるNotI切断部位に挿入する場合が示されている。
本発明の外来遺伝子導入組換えセンダイウイルス作成用ベクターから作製された組換えウイルスを遺伝子治療に用いるには、まず、該ベクターをウイルス産生用細胞に導入するが、この際、T7 RNA polymeraseの給源として、例えば、T7 RNA polymerase発現ワクシニアウイルスを該細胞に感染させるとともに、ウイルスタンパク質の形成を補い、ウイルス粒子を効率的に産生させるために、NP遺伝子、P遺伝子およびL遺伝子を有する発現ベクターも細胞に導入するのが好ましい。
【0021】
使用する細胞としては、センダイウイルスが高効率で増殖する性質を有するものが好ましく、サル腎臓由来のLLC-MK2細胞が最も理想的である。
この例においては、該ベクターが導入されたウイルス産生細胞においては、ベクターDNAがT7 RNA polymeraseによってT7プロモーター以降がRNAに転写されるが、その際、産生するRNA分子はヘアピンリボザイム配列によって、それ以降の配列が切断されて削除され、外来遺伝子DNAに対応するRNAが、終止配列、開始配列及びNot I配列を介して、Cl.151株のゲノム全長RNAと連結したRNA分子(プラス鎖)が形成される。
【0022】
次いで、このようにして形質転換され、外来遺伝子DNAに対応するRNAが連結したCl.151株のゲノム全長RNA分子を含有する細胞を、Cl.151株のウイルス粒子産生温度である32℃で培養する。細胞においては、組換えられたセンダイウイルスのRNA(+)鎖にNP、P、L遺伝子産物が結合した複合体が鋳型となって、ウイルスRNAポリメラーゼにより(−)鎖に転写され、外来遺伝子が導入されたセンダイウイルス粒子が再構成される。このセンダイウイルス粒子をさらに多量に得る場合には、細胞から回収されたセンダイウイルス粒子を例えば鶏卵に接種し、32℃で孵卵することにより増殖させればよい。該センダイウイルス粒子は、Cl.151株由来の持続感染能を有し、ヒトの体温ではウイルス粒子を産生しない持続感染型で存在し、生体内において導入した外来遺伝子を持続的に発現する。
【0023】
なお、本発明は、これら例示中に示したものに限らず、種々の遺伝子工学的材料、手段を使用できる。
例えば、本発明の組換えセンダイウイルス作成用ベクターを作成する際使用するクローニングベクターとしては、λDASH IIの他にもcharon 40等が挙げられ、センダイウイルスのゲノムサイズである約15.4 bpのDNA断片がクローニングできれば利用できる。
また、得られた外来遺伝子導入組換えセンダイウイルス作成用ベクターを培養する細胞としては、サル腎臓由来のLLC-MK2細胞が最も理想的であるが、ハムスター腎臓由来のBHK-21等の細胞も使用できる。
【0024】
一方、本発明の外来遺伝子が導入された組換えセンダイウイルスは、培養細胞において外来遺伝子を持続的に発現する組換えベクターとして、蛋白質の効率的生産に有効に用いられる。これには、上記組換えセンダイウイルス作成用ベクターの外来遺伝子挿入部位に、目的とする蛋白質をコードする遺伝子を挿入して、外来遺伝子導入組換えセンダイウイルス作成用ベクターを得、この組換えベクターを上記と同様に細胞にトランスフェクションして得られた組換えセンダイウイルスを培養細胞に感染させ、感染細胞をウイルス粒子の非産生温度下(センダイウイルスCl.151株由来のウイルスベクターの場合、例えば37℃)で培地に培養する。この感染細胞においては、転写産物として上記外来遺伝子由来のRNAがセンダイウイルスCl.151株のゲノム全長RNAに連結したRNA分子が形成され、該RNAは、形質転換細胞の増殖に伴い各細胞に受け継がれ、持続的に外来遺伝子を発現し、外来遺伝子由来の蛋白質が産生する。次いで、この培養細胞を含む培養物から目的とする蛋白質を採取する。
【0025】
この蛋白質の生産において使用する手段、材料は、ウイルス粒子の作成以降の工程を除いて、上記生体内で外来遺伝子の発現する場合と基本的には同様である。
以下に、本発明の実施例を示す。但し本発明はこれら実施例により限定されるものではない。
【実施例】
【0026】
〔実施例1〕センダイウイルスCl.151株からのゲノムcDNAのクローニング
センダイウイルスCl.151株(広島大学吉田哲也教授から分与)から100 mg/mlプロテアーゼK、0.5% SDS処理によって、ゲノムRNAを分離した。分離したRNAの3’末端にRNAリンカーをライゲーションさせ、このRNAリンカーに相補的なプライマーを用いてゲノムRNAの3’末端をPCRによってクローニングし塩基配列を決定した。また、ゲノムRNAの5’末端付近をPCRによってクローニングし、同様に塩基配列を決定した。決定した塩基配列をもとに作製したゲノムRNAの5’末端に相補的なDNAリンカー
5’-AAATTTAAAAGAATACATATCTCTTAAACTCTTGTCTGGTGCGGCCGCAAAAGGAAAA-3’(同配列番号3)
をゲノムRNAにアニーリングさせ、この「ゲノムRNA-リンカーDNA複合体」を鋳型にして、ゲノムRNAの3’末端に相補的なプライマー
5’-TTTTCCTTTTGCGGCCGCTAATACGACTCACTATAACCAAACAAGAGAAGAAACA-3’(同配列番号4)
を用いて逆転写反応をSuperScript
III First-strand synthesis system for RT-PCR (Invitrogen)によって行った。
【0027】
このようにして得られた一本鎖cDNAを鋳型として、下記の3組のプライマーセット
5’-TTTTCCTTTTGCGGCCGCTAATACGACTCACTATAACCAAACAAGAGAAGAAACA-3’(同配列番号5)、
5’-GGGATCTTGGCTATGGTGAT-3’(同配列番号6) [for 1-2875]
5’-GGGCATAGGAGAGAACACATCATCT-3’(同配列番号7)、 5’-ACTGCATGACACTCGTATAGGGTCT-3’ (同配列番号8) [for 2870-10484]
5’-CCGGAATTCAGTCGTTACTCGCCATTTTCC-3’(同配列番号9)、
5’-TTTTCCTTTTGCGGCCGCACCAGACAAGAG-3’(同配列番号10)[for 10479-15384]
を用いてPfuUltra High-fidelity DNA polymerase (STRATAGENE)によってPCRで増幅し、3本の二本鎖cDNAを得た。各々のcDNAを制限酵素を用いて切断し、SeV: 1-2875, 2870-10484, 10479-15384の3断片に分けてpBluescript II SK(+) (STRATAGENE)にクローニングした。
【0028】
〔実施例2〕クローニングしたゲノムcDNAへの、lDASHIIへのクローニングやゲノムRNA転写のために必要な配列の付加
〔実施例1〕によって得られた3断片のcDNAのうち、SeV: 1-2875を含むもの(pBSK/151(3’-E))の配列の内、センダイウイルスcDNAを含む配列のすぐ上流にT7プロモーター配列、3塩基のグアニジン残基を、この順で挿入した (pBSK/151(3’+X+3G))。SeV:
10479-15384を含むもの(pBSK/151(E-5’))から、SeV: 15351-15384の部分を切り出し、そのすぐ下流に、タバコリングスポットウイルスのヘアピンリボザイム配列、T7 RNA polymerase終止配列をこの順で挿入した形で、pET30a(+) (Novagen)にクローニングし直した(pET/151(5’+HrD))。SeV: 2870-10484を含むもの(pBSK/151(E-E))からSeV: 9015-10479を含む断片を、pBSK/151(E-5’)のSeV: 10479-15384のすぐ上流に挿入した(pBSK/151(V-5’))。
【0029】
〔実施例3〕外来遺伝子挿入センダイウイルスcDNA断片の作製
(1)外来遺伝子挿入部位の組み込み
pBSK/151(3’+X+3G)に対し、外来遺伝子挿入部位作製用プライマーとして、
5’-GCCAAAGTTCACGCGGCCGCAGATCTTCACGATGGCCGGGTTGT-3’(同配列番号11(センス鎖))
5’-ACAACCCGGCCATCGTGAAGATCTGCGGCCGCGTGAACTTTGGC-3’(同配列番号12(アンチセンス鎖))を用いて、Quikchange Site-directed Mutagenesis II (STRATAGENE) によってNot I 認識配列をSeV: 119の後ろに挿入した(pBSK/151(3’+Not))。
【0030】
(2)pBSK/151(Nhe-Not)の作製
外来遺伝子の導入を容易に行うため、〔実施例3〕で得られたpBSK/151(3'’+Not)のNot I サイトの直前5塩基目のT、3塩基目C、2塩基目Aを下記のプライマーを用いてPCRを行うことにより、それぞれC、A、Gに置換して、Nhe I認識配列の導入を行った。
始めに、M13リバースプライマー5’-GGAAACAGCTATGACCATG-3’(同配列番号13(N末端側))と
Nhe I認識配列導入用プライマー1;
5’-CTGCGGCCGCGCTAGCTTTGGCAGCAAAGAA-3’(同配列番号14(C末端側))あるいはNhe I認識配列導入用プライマー2;
5’-AAGCTAGCGCGGCCGCAGATCTTC-3’(同配列番号15(N末端側))
NP C末側プライマー;
5’-CCGGAATTCGTATGATCCTAGATTCCTCCT-3’(同配列番号16(C末端側))
の2種類のプライマーセットを用い、pBSK/151(3’+Not)を鋳型として、それぞれPCRを行った。生じたPCR産物を混合し、M13リバースプライマーとNP C末側プライマーとで再度PCRを行い、Nhe I認識配列が導入されたcl151の3’DNA断片を得た。このPCR産物を制限酵素Sac Iで切断し、同酵素で切断したpBSK/151(3'+Not)に組み込み、pBSK/151(Nhe-Not)を得た。
【0031】
(3)外来遺伝子(EGFP遺伝子、CLm遺伝子、PG-FGF-1遺伝子)の導入
EGFP遺伝子挿入用プライマーとして、
5’-ACTTGCGGCCGCTCGCCACCATGGTGAGCAAGGGCGAGGA-3’(同配列番号17(N末端側))
5’-ACTTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTAGACGGCCGCTTTACTTGTACAGCTCGTCCA-3’(同配列番号18(C末端側))
の2本のプライマーを用いてpEGFP-C1(Clontech)上からEGFP遺伝子を増幅し、得られた二本鎖DNAの末端をNot Iで切断し、pBSK/151(3’+Not)のNot I部位に挿入することによって、pBSK/151(3’+GFP)を得た。
同様の方法でCLm(分泌型ルシフェラーゼ)遺伝子挿入用プライマーとして、
5’-
ACTTGCGGCCGCTCGCCCTTATGAAGACCTTAATTCTTGCC-3’(同配列番号19(N末端側))、
5’-ACTTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTAATTCGCCCTTCTATTTGCATTCATCTGGTA-3’(同配列番号20(C末端側))
の2本のプライマーを用いてpCLm上からCLm遺伝子を増幅し、pBSK/151(3’+Not)のNot I部位に挿入することによって、pBSK/151(3’+CLm)を得た。
【0032】
PG-FGF-1(シンデカン融合ヘパリン結合性増殖因子FGF-1)遺伝子挿入用プライマーとして、
5’-TGGCTAGCTCACCATGGCCCCCGCCCGT-3’(同配列番号21(N末端側))、
5’-TTGCGGCCGCGATGAACTTTCACCCTAAGTTTTTCTTAAGGCTACCTTTAATCAGAAGAGACTGGCAG-3’(同配列番号22(C末端側))
の2本のプライマーを用いてpMKIT-neo-PGFGF-1上からPG-FGF-1遺伝子を増幅し、得られた二本鎖DNAの末端をNhe IとNot Iで切断し、pBSK/151(Nhe-Not)のNhe I、Not I部位に挿入することによって、pBSK/151(3' +PG-FGF-1)を得た。
【0033】
〔実施例4〕λDASH IIへのクローニングによる外来遺伝子導入組換えウイルス作成用ベクターの作製
〔実施例2〕、〔実施例3〕によって得られた各プラスミドのうち、pBSK/151(3’+GFP)、pBSK/151(3’+CLm)もしくはpBSK/151(3’+PG-FGF-1)からT7プロモーター配列−SeV: 1- 2875を、pBSK/151(E-E)からSeV: 2870-5917、SeV: 5913-9598を、pBSK/151(V-5’)からSeV: 9593-15351を、pET/151(5’+HrD)からSeV: 15351-15384−T7 RNA polymerase終止配列を切り出して、lDASH II (STRATAGENE)にこの順番でクローニングし直した。得られたクローンをそれぞれl/151-GFP、l/151-CLmもしくはl/151-PG-FGF-1と名づけ、それらからファージDNAを精製した。
【0034】
〔実施例5〕センダイウイルスの再構成
LLC-MK2細胞を1 x 106 cells / wellで6-wellプレートに蒔き、24時間培養後T7 RNA polymeraseを発現する弱毒性ワクシニアウイルス(MVAGKT7)をM.O.I.=1.0で1時間、37°Cで感染させた。細胞を洗浄した後、lDASH IIにクローニングしたセンダイウイルスcDNA(l/151-GFP、l/151-CLmもしくはl/151-PG-FGF-1)、pGEM/NP、pGEM/P、pGEM/Lをそれぞれ5 mg、2 mg、1 mg、2 mgの量比でOptiMEM 300 mlに懸濁し、10 mlのLipofectamine 2000を含む300 mlのOptiMEMと混合し、20分間室温放置後、細胞に添加して4時間培養した。培養後、20% 血清、80 mg/ml シトシンアラビノシドC(AraC)を含んだ培地を等量加えてさらに32°Cで 48時間培養した。
【0035】
これらの細胞を回収し、ぺレットを500 mlのPBSで懸濁し、凍結融解を4回繰り返した。これらを10日間孵卵させた鶏卵に100 ml接種し、32°Cで5日間孵卵させた後に漿尿液を回収した。ワクシニアフリーにするためにこれら回収した漿尿液をさらに10-4〜10-8希釈して鶏卵に再接種し、同様に回収した。得られた漿尿液を15,000 rpmで30分遠心することによってセンダイウイルス(r151-GFP、r151-CLmもしくはr151-PG-FGF-1)を沈澱させ、組換えセンダイウイルスの感染実験に用いた。
【0036】
〔実施例6〕EGFP遺伝子導入組換えセンダイウイルスからの培養細胞中におけるEGFP遺伝子の持続的発現の解析
r151-GFPをLLC-MK2細胞にM.O.I.=50で感染させ、感染細胞を蛍光顕微鏡で観察することによって、EGFP遺伝子の発現を経時的に確認した。
図2から明らかなように、持続感染株由来センダイウイルスベクターからのEGFP遺伝子の発現は4ヶ月以上も持続しており、従来のZ株を基本骨格としたセンダイウイルスベクター(SeV-GFP)と比較して、有意に持続していた。
【0037】
〔実施例7〕EGFP遺伝子導入組換えセンダイウイルスの生体中におけるEGFP遺伝子の持続的発現の解析
再構成したr151-GFP、300 mgを3cm長に切った経口ゾンデを用いてラットの大腸に感染させた。
EGFP遺伝子の発現については、凍結包埋させた大腸組織を蛍光顕微鏡で観察することによって、確認を行った。
図3から明らかなように、持続感染型センダイウイルスベクターからのEGFP遺伝子の発現は2ヶ月以上も持続しており、従来のZ株を基本骨格としたセンダイウイルスベクター(SeV-GFP)と比較して、有意に持続していた。
【0038】
〔実施例8〕CLm遺伝子導入組換えセンダイウイルスの培養細胞中におけるCLm遺伝子発現量の検討
r151-CLmをLLC-MK2細胞にM.O.I.=50で感染させた。また、CMVプロモーターによりCLm遺伝子が発現するCLm発現ベクタープラスミド(pJO/CLm)をトランスフェクションしたLLC-MK2細胞も同様に用意し、これら細胞を37℃で培養した。
各々の細胞の培養上清を希釈し、等量の216 nMルシフェリンを加え、ルミノメーターでルシフェラーゼ活性を測定することによって、CLm遺伝子発現量を定量した。その結果、2日後の発現量は感染細胞のほうが10倍近く高く、さらに、トランスフェクション細胞では数日で発現量が低下するのに対し、感染細胞においては発現が長期間持続した(図4)。
【0039】
〔実施例9〕PG-FGF-1遺伝子導入組換えセンダイウイルスの培養細胞におけるPG-FGF-1遺伝子発現量の検討
サル腎細胞由来COS細胞に、r151-PG-FGF-1をM.O.I.=50で感染させた場合と、発現プラスミドpMKIT-neo-PGFGF-1をリポフェクトアミン2000(Invitrogen)を用いて導入し、これら細胞を32℃で培養し、PG-FGF-1の発現量を比較した。
感染2日目に培養上清と細胞抽出液を調製しウェスタンブロット法を用いて比較した結果、細胞内において顕著なPG-FGF-1の発現量の増加が認められ、その発現量はトランスフェクションと比較して約3倍であった(図5)。
【図面の簡単な説明】
【0040】
【図1】本発明の外来遺伝子導入組換えセンダイウイルス作成用ベクター構造を示す図である。
【図2】本発明の組換えセンダイウイルス作成用ベクターにEGFP遺伝子を導入し、LLC-MK2細胞に感染させた場合の、EGFP遺伝子発現量と細胞の形態の経時変化を示す蛍光顕微鏡写真である。
【図3】本発明のEGFP遺伝子導入組換えセンダイウイルスをラットの大腸に感染させた場合の、大腸上皮におけるEGFP遺伝子発現量の経時変化を示す蛍光顕微鏡写真である。
【図4】本発明の分泌型ルシフェラーゼ(CLm)遺伝子導入組換えセンダイウイルスをLLC-MK2細胞に感染させた場合(r151-CLm)と、分泌型ルシフェラーゼ導入プラスミド(pJO/CLm)をトランスフェクションさせた場合とにおける、培養上清におけるルシフェラーゼ活性の経時変化を示すグラフである。
【図5】本発明のシンデカン融合ヘパリン結合性増殖因子FGF-1(PG-FGF-1)遺伝子導入組換えセンダイウイルスをCOS細胞に感染させた場合(cl151)と、シンデカン融合ヘパリン結合性増殖因子FGF-1導入プラスミドをトランスフェクションさせた場合(PGFGF-1)とにおける、培地中と細胞内におけるシンデカン融合ヘパリン結合性増殖因子FGF-1の発現量を示すグラフ図である。
【特許請求の範囲】
【請求項1】
センダイウイルス温度感受性株の全長ゲノムcDNAおよび外来遺伝子挿入部位を少なくとも有することを特徴とする、組換えセンダイウイルス作製用ベクター。
【請求項2】
センダイウイルス温度感受性株がセンダイウイルスCl.151株であることを特徴とする、請求項1に記載の組換えセンダイウイルス作製用ベクター。
【請求項3】
クローニングベクターのクローニングサイトに、センダイウイルスCl.151株の全長ゲノムcDNAを挿入したことを特徴とする、請求項1に記載の組換えセンダイウイルス作製用ベクター。
【請求項4】
請求項1〜3のいずれかに記載のベクターに外来遺伝子DNAが挿入されていることを特徴とする、組換えセンダイウイルス作製用ベクター。
【請求項5】
センダイウイルスCl.151株の全長ゲノムRNAに外来遺伝子DNAに対応するRNAが連結されていることを特徴とするRNA。
【請求項6】
外来遺伝子DNAに対応するRNAが連結された、センダイウイルスCl.151株の全長ゲノムRNAを有することを特徴とする、組換えウイルス。
【請求項7】
請求項6に記載の組換えウイルスからなることを特徴とする、外来遺伝子の生体内持続発現用組換えセンダイウイルスベクター
【請求項8】
請求項6に記載の組換えウイルスからなることを特徴とする、遺伝子治療用組換えベクター
【請求項9】
請求項6に記載の組換えウイルスからなることを特徴とする、外来遺伝子の培養細胞内持続発現用ウイルスベクター。
【請求項10】
請求項4に記載の組換えベクターが導入されていることを特徴とする細胞。
【請求項11】
外来遺伝子DNAに対応するRNAが連結されたセンダイウイルスCl.151株の全長ゲノムRNAを含有することを特徴とする細胞。
【請求項12】
請求項10に記載の細胞を、ウイルス粒子産生温度下で培地に培養し、培養物からウイルス粒子を採取することを特徴とする、請求項6に記載の組み換えウイルスを製造する方法。
【請求項13】
請求項6に記載の組換えウイルスを培養細胞に感染させ、ウイルス粒子非産生温度下で培地に培養し、外来遺伝子由来の蛋白質を採取することを特徴とする、蛋白質の製造方法。
【請求項1】
センダイウイルス温度感受性株の全長ゲノムcDNAおよび外来遺伝子挿入部位を少なくとも有することを特徴とする、組換えセンダイウイルス作製用ベクター。
【請求項2】
センダイウイルス温度感受性株がセンダイウイルスCl.151株であることを特徴とする、請求項1に記載の組換えセンダイウイルス作製用ベクター。
【請求項3】
クローニングベクターのクローニングサイトに、センダイウイルスCl.151株の全長ゲノムcDNAを挿入したことを特徴とする、請求項1に記載の組換えセンダイウイルス作製用ベクター。
【請求項4】
請求項1〜3のいずれかに記載のベクターに外来遺伝子DNAが挿入されていることを特徴とする、組換えセンダイウイルス作製用ベクター。
【請求項5】
センダイウイルスCl.151株の全長ゲノムRNAに外来遺伝子DNAに対応するRNAが連結されていることを特徴とするRNA。
【請求項6】
外来遺伝子DNAに対応するRNAが連結された、センダイウイルスCl.151株の全長ゲノムRNAを有することを特徴とする、組換えウイルス。
【請求項7】
請求項6に記載の組換えウイルスからなることを特徴とする、外来遺伝子の生体内持続発現用組換えセンダイウイルスベクター
【請求項8】
請求項6に記載の組換えウイルスからなることを特徴とする、遺伝子治療用組換えベクター
【請求項9】
請求項6に記載の組換えウイルスからなることを特徴とする、外来遺伝子の培養細胞内持続発現用ウイルスベクター。
【請求項10】
請求項4に記載の組換えベクターが導入されていることを特徴とする細胞。
【請求項11】
外来遺伝子DNAに対応するRNAが連結されたセンダイウイルスCl.151株の全長ゲノムRNAを含有することを特徴とする細胞。
【請求項12】
請求項10に記載の細胞を、ウイルス粒子産生温度下で培地に培養し、培養物からウイルス粒子を採取することを特徴とする、請求項6に記載の組み換えウイルスを製造する方法。
【請求項13】
請求項6に記載の組換えウイルスを培養細胞に感染させ、ウイルス粒子非産生温度下で培地に培養し、外来遺伝子由来の蛋白質を採取することを特徴とする、蛋白質の製造方法。
【図1】


【図4】
【図2】
【図3】
【図5】
【図4】
【図2】
【図3】
【図5】
【公開番号】特開2006−325531(P2006−325531A)
【公開日】平成18年12月7日(2006.12.7)
【国際特許分類】
【出願番号】特願2005−156446(P2005−156446)
【出願日】平成17年5月27日(2005.5.27)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
【公開日】平成18年12月7日(2006.12.7)
【国際特許分類】
【出願日】平成17年5月27日(2005.5.27)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
[ Back to top ]