
ゾレドロン酸の結晶性三水和物
ゾレドロン酸三水和物、その調製方法、およびゾレドロン酸一水和物への変換。本発明の一態様は、その単結晶X線回折図形(XRD)、X線粉末回折(XRPD)パターン、赤外(IR)吸収スペクトル、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線によって特徴付けられる結晶性ゾレドロン酸三水和物を提供する。もう1つの態様において、本発明は結晶性ゾレドロン酸三水和物のロバストで再現可能な調製方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の序論)
本発明は、結晶性ゾレドロン酸三水和物およびその調製方法に関する。
【背景技術】
【0002】
ゾレドロン酸の化学名は、(1−ヒドロキシ−2−イミダゾール−1−イルホスホノエチル)リン酸であり、この化合物は、式Iにより構造を示すことができる。
【0003】
【化2】

ゾレドロン酸は、イミダゾール環を含む側鎖を特徴とする、第3世代のビスホスホネート誘導体である。ゾレドロン酸は、破骨細胞の骨吸収を阻害し、腫瘍性高カルシウム血症の治療に使用される。ゾレドロン酸は、ゾメタ(ZOMETA)(商標)という商品名で販売されている、バイアル中に静脈内注入用の滅菌粉末または溶液として市販されている。各バイアルは4mgのゾレドロン酸(無水)を含み、それは4.264mgのゾレドロン酸一水和物に相当する。
【0004】
ゾレドロン酸の化学合成は、現在までその一水和物の物質の調製に向けられている。特許文献1は、ゾレドロン酸を開示し、実施例10においてスキーム1に示すようなゾレドロン酸の調製方法を開示している。
【0005】
【化3】
手短に言うと、その方法は、三塩化リンと塩酸の存在下で、2−(1−イミダゾリル)酢酸塩酸塩をリン酸と反応させてゾレドロン酸を得ることを含み、ゾレドロン酸はアセトンで希釈して沈殿させる。このようにして得られた粗製ゾレドロン酸を水中で再結晶化する。水からの粗物質を再結晶化する最終工程は、ゾレドロン酸一水和物を提供する。
【0006】
特許文献2もまた、最終工程において水から同様の再結晶化によりゾレドロン酸一水和物の化合物を提供することを含む。
【0007】
特許文献3は、ゾレドロン酸の様々な結晶形態、およびそのナトリウム塩、ならびにそれらの調製方法を開示している。ゾレドロン酸の一水和物である結晶形態I、II、XIIおよびXVIII、ならびにゾレドロン酸の無水和物の形である結晶形態XV、XX、およびXXVIの調製が記載されている。ゾレドロン酸の一ナトリウム塩および二ナトリウム塩の様々な水和物および無水和物の形もまた記載しており、非晶質ゾレドロン酸一ナトリウム塩、二ナトリウム塩および三ナトリウム塩もまた記載している。
【0008】
ゾレドロン酸の多形性の特性決定についてかなりの数の研究が行われているが、生成できる他の形態を同定する必要性が残っている。
【0009】
上記特許のうち2つの特許は一水和物の調製を記載しているが、その方法の完全なる詳細についてはいずれも示していない。特許文献1では、実施例1において単純に、その生成物は水中で再結晶化させると述べているが、再結晶化の条件は示していない。特許文献2は、その粗製ゾレドロン酸を90から95℃で2から3時間水に溶解させ、次いで高温下で炭素処理を行い、そして再結晶化のためにその反応塊を25から35℃に冷却することによる水中の粗製ゾレドロン酸の再結晶を伴う一水和物の調製方法を例示している。
【0010】
上記いずれの特許も水からの再結晶化の間の一水和物形成のために重要なパラメータを述べていない。上記方法に従って一水和物の製造のためにバッチをスケールアップする間に、他の結晶形態による汚染がしばしば観察されている。
【0011】
世界中の監督官庁が、同じ活性化合物のすべての可能な結晶形態を合成し、できるだけ完全に特性決定することを要求している。さらに、市販の製品は、微量の他の形態のいずれも含むべきではなく、存在する場合、その各形態の割合は、貯蔵中に原薬の溶解特性および生物学的利用態の特性の変化を避けるのに十分に特性決定されることを要求している。
【特許文献1】米国特許第4939130号明細書
【特許文献2】国際公開第2005/063717号パンフレット
【特許文献3】国際公開第2005/005447号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0012】
このように、引き続き、ゾレドロン酸などの商業的興味のある薬理学的に活性な化合物の新しい多形形態を調製する必要があり、それは製剤学者に、それらの異なる物理化学的特徴に基づいて、選択するための活性成分のより広い範囲の結晶形態を提供する。
【0013】
多形形態の調製方法は、その方法が工場で簡単にスケールアップできるように、ロバストであり、再現可能であることも重要である。このようにして、ゾレドロン酸の製造における改良が必要とされている。
【課題を解決するための手段】
【0014】
(発明の要旨)
本発明は、結晶性ゾレドロン酸三水和物、およびロバストで再現可能なその調製方法に関する。
【0015】
本発明の一態様は、その単結晶X線回折図形(XRD)、X線粉末回折(XRPD)パターン、赤外(IR)吸収スペクトル、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線によって特徴付けられる結晶性ゾレドロン酸三水和物を提供する。
【0016】
もう1つの態様において、本発明は結晶性ゾレドロン酸三水和物のロバストで再現可能な調製方法を提供する。
【0017】
一実施形態において、結晶性ゾレドロン酸三水和物の調製方法は、
a)ゾレドロン酸溶液を供給すること、
b)その溶液から固体を結晶化すること、および
c)分離したゾレドロン酸三水和物結晶を回収すること
を含む。
【0018】
本発明の別の態様は、ゾレドロン酸一水和物とゾレドロン酸三水和物の混合物をゾレドロン酸一水和物に変換する方法を提供する。
【0019】
本発明のさらに別の態様は、ゾレドロン酸三水和物からゾレドロン酸一水和物を調製する方法を提供する。
【0020】
本発明のさらなる態様は、ゾレドロン酸一水和物と実質的に同等の溶解性を有する結晶性ゾレドロン酸三水和物を提供する。
【0021】
本発明のさらなる態様は、約300μm未満の粒径を有する結晶性ゾレドロン酸三水和物を提供する。
【0022】
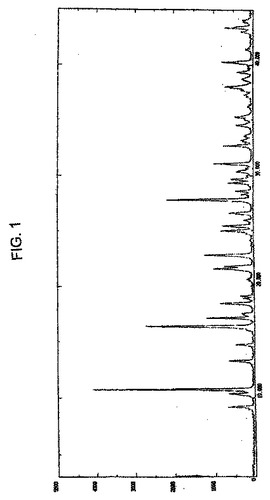
ゾレドロン酸三水和物は、そのXRPDパターンが、実質的に図1に従うことを特徴とすることができる。
【0023】
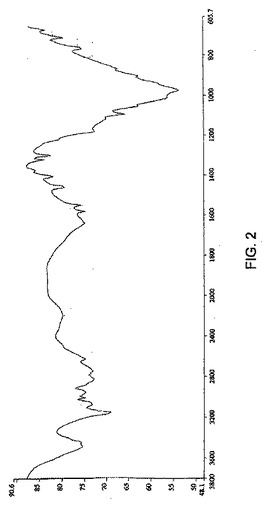
また、ゾレドロン酸三水和物は、そのIRスペクトルが、実質的に図2に従うことを特徴とすることができる。
【0024】
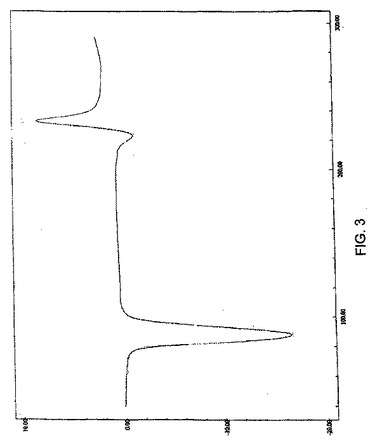
また、ゾレドロン酸三水和物は、そのDSC曲線が、実質的に図3に従うことを特徴とすることができる。
【0025】
一実施形態において、ゾレドロン酸三水和物の調製方法は、約60から80℃の温度の水を含む溶媒中のゾレドロン酸溶液を供給すること、およびその溶液を冷却してゾレドロン酸三水和物を結晶化することを含む。
【0026】
もう1つの実施形態において、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換する方法は、ゾレドロン酸三水和物を約40から90℃の温度で乾燥することを含む。
【0027】
さらなる実施形態において、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換する方法は、ケトン中でゾレドロン酸三水和物のスラリーを形成することを含む。
【0028】
なお一層さらなる実施形態において、ゾレドロン酸一水和物の調製方法は、ゾレドロン酸の水溶液を供給すること、およびゾレドロン酸に貧溶媒を添加することを含む。
【0029】
その上さらなる態様において、本発明はゾレドロン酸三水和物を1種またはそれ以上の薬学的に許容できる担体、賦形剤、または希釈剤と一緒に含む医薬組成物を提供する。
【発明を実施するための最良の形態】
【0030】
(発明の詳細な説明)
本発明の一態様は、結晶性ゾレドロン酸三水和物を包含する。
【0031】
結晶性ゾレドロン酸三水和物は、その粉末X線回折(「XRPD」)パターン、単結晶X線回折(「XRD」)パラメータ、赤外吸収(「IR」)スペクトル、示差走査熱量測定(「DSC」)曲線、および熱重量分析(「TGA」)曲線のいずれかによって特性決定する。
【0032】
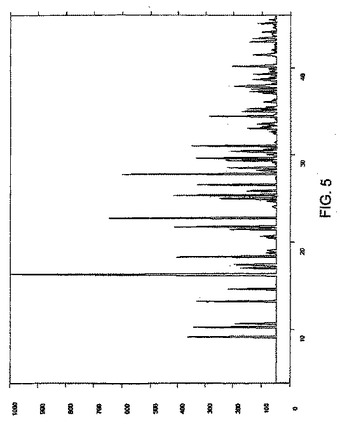
単結晶X線回折データは、グラファイト単色光Mo−Kα放射線を用いてRigaku Mercury CCD領域検出器で収集した。その構造は、直接法および(SIR92)によって解析し、最小二乗法により精密化した。2110について観察された反射では、R値が0.038、Rw=0.039である。単結晶データからシミュレートした粉末回折パターンを図5に示す。
【0033】
ゾレドロン酸三水和物は、そのXRPDパターンにより特性決定し、このパターンは既知の形態との違いを示している。本明細書で報告されるXRPDデータは、波長が1.541ÅのCu Kα−1放射線を用いて得、Bruker Axe、D8高度粉末X線回折装置を用いて測定した。
【0034】
結晶性ゾレドロン酸三水和物は、図1のパターンに実質的に従うXRPDパターンを特徴とする。結晶性ゾレドロン酸三水和物はまた、約10.8、16.4、17.1、18.4、21.6、24.9、25.4、27.8、31.0、および32.6の±0.2°2θに顕著なピークを有するXRPDパターンを特徴とする。また、約38.0、40.2、21.8、9.2、10.3、および43.4の±0.2°2θのさらなるXRPDピークを特徴とする。
【0035】
ゾレドロン酸三水和物はまた、その格子パラメータが、単結晶X線回折によって決定された結晶構造を特徴とする。
【0036】
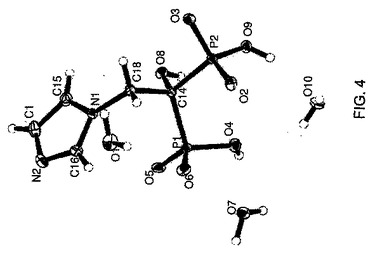
ゾレドロン酸三水和物の結晶構造を図4に示す。その三水和物は、表1に示す単位セルパラメータで三斜晶空間群P1において結晶化する。
【0037】
【化4】
三次元の充填は、表2に示す強い分子外および分子内水素結合により安定化する。
【0038】
【化5】
結晶性ゾレドロン酸三水和物の単結晶の情報から、シミュレートした粉末回折図形(理論的な回折図形)は、実験的に得られたものに匹敵する回折図形が得られた。理論的な回折図形と実験的な回折図形との間にみられる類似性が非常に高いことは、粉末に含まれるその構造は、その単結晶において測定されるものに対応しており、その構造が唯一である、すなわち、結晶性ゾレドロン酸三水和物に混合している他の多形形態が無いことを意味している。
【0039】
結晶性ゾレドロン酸三水和物の赤外スペクトルは、Perkin Elmer System 200 FT−IR分光光度計を用いて、400cm−1と4000cm−1との間で、4cm−1の分解能、試験化合物が0.5質量%の濃度である臭化カリウムのペレット中で記録されている。
【0040】
結晶性ゾレドロン酸三水和物はさらに、約671、712、766、975、1301、1323、1406、1460、1550、2826、3154、および3484の±5cm−1にピークを含む赤外吸収スペクトルを特徴とする。結晶性ゾレドロン酸三水和物三水和物はまた、図2のスペクトルに実質的に従う赤外吸収スペクトルを特徴とする。
【0041】
結晶性ゾレドロン酸三水和物は、さらにまた、図3の曲線に、実質的に従うその示差走査熱量測定曲線を特徴とする。結晶性ゾレドロン酸三水和物は、また、約234℃で発熱、約224℃および約88℃で吸熱を示すDSC曲線を特徴とする。
【0042】
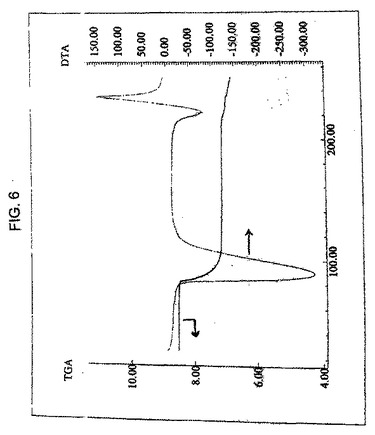
結晶性ゾレドロン酸三水和物は、さらにまた、水3分子の損失を示す図6のDTA曲線に実質的に従う熱重量分析曲線を特徴とする。図6において、左の縦軸はサンプル(ミリグラム)であり、右の縦軸は熱電対(ミリボルト)であり、横軸は温度(℃)である。
【0043】
ゾレドロン酸の水分含量は、15から18重量%の範囲であることが可能である。
【0044】
別の態様において、本発明は、結晶性ゾレドロン酸三水和物のロバストで再現可能である調製方法を提供する。
【0045】
一実施形態において、その三水和物の調製方法は、
a)ゾレドロン酸溶液を供給すること、
b)その溶液から固体を結晶化すること、および
c)分離したゾレドロン酸三水和物結晶を回収すること
を含む。
【0046】
工程a)は、ゾレドロン酸溶液を供給することを含む。
【0047】
ゾレドロン酸溶液はゾレドロン酸を適当な溶媒に溶解することによって得ることができ、またはそのような溶液はゾレドロン酸が形成される反応から直接得ることもできる。
【0048】
適当な溶媒にゾレドロン酸を溶解することによってその溶液を調製する場合には、ゾレドロン酸の任意の塩、溶媒和物、および水和物を含む、結晶形態または非晶質形などの、ゾレドロン酸の任意の形をその溶液の調製に利用することができる。
【0049】
ゾレドロン酸三水和物の調製に有用である適切な溶媒には、水のみ、または例えば、メタノール、エタノール、プロパノール、第3級ブタノール、n−ブタノールなどのアルコール、アセトン、プロパノンなどのケトン、アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、ジオキサンなど、およびそれらの混合物などの有機溶媒との組合せを含む。
【0050】
関連する実施形態において、本発明は、ゾレドロン酸の溶媒または溶媒混合物中溶液をほぼ周囲温度から約80℃、または約60から80℃、あるいは約70から75℃の温度に加熱し、透明な溶液を得ることを含む。ゾレドロン酸三水和物溶液の調製には、透明な溶液が得られる限り約80℃未満の任意の温度を使用することができる。これらの範囲において温度が高いほど、より高濃度の溶質を提供し、一般に高い処理効率をもたらす。
【0051】
ゾレドロン酸の溶解のための最高温度は、得られるゾレドロン酸の多形形態を決定するので重要である。その溶液を約90℃より高い温度に加熱すると、結晶性一水和物がもたらされる。溶液をより低い温度、例えば約40から80℃、または約70から75℃の範囲に加熱すると、結晶性ゾレドロン酸三水和物がもたらされる。
【0052】
その溶液は、その温度で約1分から任意の所望の時間維持される。混合物を約75℃に加熱する場合、冷却を始める前にその高温での必要とされる最小の維持時間はごくわずかである。
【0053】
溶液は、任意でろ紙、ガラス繊維、または他の膜物質を、またはセライトなどの清澄剤を通過させることによってろ過することもできる。使用する装置、ならびにその溶液の濃度および温度に応じて、早すぎる結晶化を避けるために、ろ過装置を余熱する必要があることがある。
【0054】
その溶質の濃度は、溶媒中に約0.1g/mlから約20g/mlであることが可能であり、または1g/mlから5g/mlの範囲であることが可能である。
【0055】
工程b)はろ液から固体を結晶化することを含む。
【0056】
結晶化は、通常、溶解温度よりも低い温度で行われる。結晶化の温度は約40℃未満、または30℃未満であってよい。
【0057】
結晶化は、所望の結晶収率が得られるまで、例えば約1時間から約72時間攪拌して行うことができる。結晶化工程はさらに当業者に公知の簡易手段を含むことができる。例えば、結晶化工程はさらに、その溶液を冷却すること、その溶液を加熱すること、または沈殿を引き起こす薬剤を加えることを含むことができる。
【0058】
溶液の温度は外部の冷却を用いて結晶化が急速に起こるように下げることができ、またはその分離温度に自然に冷やすこともできる。一般に、1から5kgまたはそれより大規模のバッチでは、反応塊を自然に冷やすとすると不都合な時間かかってしまうことがあるため、外部の冷却を反応塊にしばしば用いてその必要とするレベルにまでその温度を下げる。
【0059】
さらに冷却時間の延長は、作業経費の増加の他には欠点は無く、所与のバッチサイズに適切な時間は、わずかな努力によって当業者が決定することができる。溶液の冷却は、攪拌を伴う大気条件下の単なる放射冷却によって、または例えばジャケット容器内での冷却媒体の循環などの制御された冷却装置の使用などによって達成される。そのような急速冷却および徐冷のための技術は当業者に公知であって、限定することなくすべて本明細書に含まれる。
【0060】
90から95℃のより高温で溶媒にゾレドロン酸を溶解し、続いて低温で固体を分離することを含む、結晶性ゾレドロン酸一水和物の調製方法と比べて、ゾレドロン酸三水和物用の方法はロバストであり、再現可能である。一水和物の調製は、分離の間のゾレドロン酸溶液の冷却速度や溶解の間の溶液の維持温度などの変数に依存する。
【0061】
溶解中の90から95℃を超える温度でのゾレドロン酸溶液の不適切な維持により、一水和物と三水和物の混合物をもたらすことがある。また、反応塊が溶解温度から結晶化温度に急速に冷却されると、その結果はゾレドロン酸の一水和物と三水和物の混合物となる。
【0062】
大規模の一水和物の調製の間には、多くのプロセス方法を行う必要がある。
【0063】
工程c)は、分離されたゾレドロン酸三水和物の結晶の回収を含む。
【0064】
回収は、それだけに限定されないが、ろ過、遠心分離、デカントを含む任意の手段によって行うことができる。その結晶形態は、その結晶形態と、それだけに限定されない、懸濁液、溶液、スラリーおよび乳液を含む溶媒(1またはそれ以上)とを含む任意の組成物から回収することができる。
【0065】
得られた化合物は、さらに周囲圧力または減圧下で乾燥させることができる。例えば、乾燥は、減圧下または大気圧下で、約40℃から60℃、または70℃から80℃、あるいはそれ以上の温度で行うことができる。乾燥は所望の残留溶媒量が得られるまで、例えば約2時間から24時間、または約3から6時間の間行うことができる。
【0066】
本発明のさらに別の態様は、ゾレドロン酸の一水和物と三水和物の混合物をゾレドロン酸一水和物へ変換するための方法を提供する。
【0067】
公知のように、ゾレドロン酸一水和物の調製方法は、ロバストではなく、大規模の製造の間では、もし決定的なパラメータが用いられないならば、ゾレドロン酸の一水和物と三水和物の混合物になる可能性がある。
【0068】
本発明は、ゾレドロン酸三水和物と一水和物の混合物をゾレドロン酸一水和物へ変換する方法を提供する。
【0069】
一実施形態において、ゾレドロン酸一水和物および三水和物の混合物をゾレドロン酸一水和物へ変換する方法は、三水和物を含む物質を50℃より高い温度で真空下でさらに乾燥する工程、または有機溶媒中に三水和物の物質を含むスラリーを形成する工程のいずれか1つの工程を含む。
【0070】
乾燥温度は40から90℃、または60から70℃、あるいは55から60℃の範囲であることが可能であり、その化合物は周囲圧または減圧下で乾燥することができる。例えば、乾燥は減圧下でまたは周囲圧下で、空気乾燥機、真空乾燥機または箱型乾燥機などのいずれかを用いて行うことができる。場合により、不活性雰囲気下で乾燥を行うことができる。
【0071】
三水和物をスラリーするために用いることができる適当な溶媒は、アセトン、エチルメチルケトン、プロパノンなどのケトンである。
【0072】
スラリー形成は、攪拌を伴ってもよく、約1時間から約10時間またはそれ以上の期間行うことができる。
【0073】
スラリーを形成する目的で約5から100倍の範囲の任意の量の溶媒を用いることができる。
【0074】
本発明のさらなる態様は、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換することを含む。
【0075】
別の実施形態において、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換する方法は、溶媒−貧溶媒技術によって再結晶化することを含む。その方法はゾレドロン酸と適当な溶媒を供給すること、その混合物を加熱して透明な溶液を得ること、続いて貧溶媒を添加して必要とする生成物の沈殿を得ることを含む。
【0076】
溶解のために用いることができる適当な溶媒には、例えば、水、メタノール、エタノール、プロパノール、n−ブタノールなどのアルコール、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフランなど、およびそれらの混合物が含まれる。
【0077】
用いることができる貧溶媒には、例えば、n−ヘキサン、n−ヘプタン、およびトルエンなどの炭化水素、アセトン、プロパノン、エチルメチルケトン、およびブタノンなどのケトン、ジエチルエーテル、イソプロピルエーテルなどのエーテル、酢酸エチル、第3級酢酸ブチルなどのエステル、ジクロロメタン、1,2−ジクロロエタン、クロロホルム、四塩化炭素などのハロゲン化炭水化物、およびそれらの混合物が含まれる。
【0078】
溶解の手順は、約95から120℃の範囲の高温で行うことができる。加熱は、それだけに限定されない機械的な手段および磁力の手段を含む任意の手段で連続してまたは時々攪拌またはかき混ぜることにより為し遂げることができる。溶媒の量は、ゾレドロン酸を溶解して濃縮溶液を形成するのに十分な量であるべきである。
【0079】
貧溶媒のゾレドロン酸溶液への添加は、約0から120℃、もしくは60から90℃の温度、または周囲温度、あるいは約0から15℃の範囲のより低い温度で行うことができる。
【0080】
分離した固体の回収はそれだけに限定されない、ろ過、遠心分離およびデカントを含む任意の手段によって行うことができる。その結晶形態は、その結晶形態と、それだけに限定されない、懸濁液、溶液、スラリーおよび乳液を含む溶媒(1またはそれ以上)とを含む任意の組成物から回収される。
【0081】
得られた化合物は、周囲圧または減圧下でさらに乾燥させることができる。例えば、乾燥は減圧下または大気圧下で約40℃から60℃、または70℃から80℃、あるいはより高い温度で行うことができる。乾燥は使用される乾燥条件および許容可能である残留溶媒分の量に応じて、約2時間まで、または約5時間まで、またはより長時間の持続時間で行うことができる。
【0082】
したがって、本発明は、医薬品を製造するために使用できる、純粋なゾレドロン酸三水和物を調製するための再現可能な方法を提供する。しかしながら、所望により、ゾレドロン酸三水和物は、容易に純粋なゾレドロン酸一水和物に変換し、医薬品を製造するために使用することもできる。本発明の利点は、所望の純粋な形態のゾレドロン酸を予想通りに調製する能力を提供することである。
【0083】
本発明のさらに別の態様は、ゾレドロン酸一水和物と実質的に同等の溶解性を有する結晶性ゾレドロン酸三水和物を提供する。このゾレドロン酸の溶解度は、ゾレドロン酸一水和物と同程度である。このことは医薬組成物におけるゾレドロン酸三水和物の使用を容易にする。
【0084】
本発明のなお一層さらなる態様は、粒径が300μm未満の結晶性ゾレドロン酸三水和物を提供する。
【0085】
D10、D50、およびD90値は、粒度分布を示すのに有用な方法である。D90は、粒子の少なくとも90容量%がそのD90値よりも小さい粒径である値を意味する。同様にD50およびD10は、粒子の50容量%および10容量%がその値よりも小さい粒径である値を意味する。D50値は粉末の平均粒径とみなすことができる。D10、D50およびD90を決定する方法には、マルバーン装置を使用したレーザー回折を含む。
【0086】
本発明の結晶性ゾレドロン酸三水和物は、10μm未満または20μm未満のD10、100μm未満または150μm未満のD50、および200μm未満または300μm未満のD90を有する。いずれのD値においても特定の下限値は無い。
【0087】
その上さらなる態様において、本発明は、ゾレドロン酸三水和物を1種またはそれ以上の薬学的に許容できる担体、賦形剤または希釈剤と一緒に含む、医薬組成物を提供する。
【0088】
本発明のゾレドロン酸三水和物を1種またはそれ以上の薬学的に許容可能である担体と一緒に含む医薬組成物は、さらに以下のように製剤することができる:例えば、それだけに限定されないが、粉末、顆粒、ペレット、錠剤およびカプセルなどの固体経口剤型、例えば、それだけに限定されないが、シロップ、懸濁液、分散液および乳液などの液体経口剤型、および例えば、それだけに限定されないが、溶液、分散液および凍結乾燥組成物などの注射製剤。製剤は、即時放出、遅延放出、または放出調整された形態とすることができる。さらに、即時放出性組成物は、従来の製剤、分散可能な製剤、チュアブル製剤、口内溶解製剤または瞬時溶解製剤であることが可能であり、放出調整された組成物は、親水性または疎水性、あるいは親水性および疎水性の組合せの放出速度を制御する物質を含み、マトリックスまたはレザーバ、あるいはマトリックスおよびレザーバの組合せを形成してもよい。組成物は直接混合、乾式造粒または湿式造粒によって、あるいは射出および球形化によって調製することができる。組成物は、非被覆、フィルム被覆、糖被覆、粉末被覆、腸溶性被覆、または放出調整された被覆として存在可能である。本発明の組成物は、さらに1種またはそれ以上の薬学的に許容できる賦形剤を含むことができる。
【0089】
本発明において使用される薬学的に許容できる賦形剤には、それだけに限定されないが、希釈剤(例えば、でんぷん、アルファ化でんぷん、ラクトース、粉末セルロース、結晶セルロース、リン酸二カルシウム、リン酸三カルシウム、マンニトール、ソルビトール、糖など)、結合剤(例えば、アカシア、グアーガム、トラガカント、ゼラチン、ポリビニルピロリドン、ヒドロキシプロピルセルロース、ヒロドキシプロピルメチルセルロース、アルファ化でんぷんなど)、崩壊剤(例えば、でんぷん、グリコール酸ナトリウムでんぷん、アルファ化でんぷん、クロスポビドン、クロスカルメロースナトリウム、コロイド状二酸化ケイ素など)、潤滑剤(例えば、ステアリン酸、ステアリン酸マグネシウム、ステアリン酸亜鉛など)、流動促進剤(例えば、コロイド状二酸化ケイ素など)、溶解性またはぬれ性増強剤(例えば、陰イオン性、陽イオン性または中性の界面活性剤)、錯体形成剤(例えば、種々の等級のシクロデキストリン、樹脂)、放出速度調整剤(例えば、ヒドロキシプロピルセルロース、ヒドロキシメチルセルロース、ヒドロキシプロピルメチルセルロース、エチルセルロース、メチルセルロース、種々の等級のメタクリル酸メチル、ワックス等)が含まれる。他の薬学的に許容できる賦形剤には、それだけに限定されないが、フィルム形成剤、可塑剤、着色剤、香味料、甘味料、粘性増強剤、保存剤、抗酸化剤などが含まれる。
【0090】
本発明の組成物において、ゾレドロン酸三水和物は、0.5mgから50mg、または1mgから25mgの範囲において有用な活性成分である。
【0091】
本発明のある特定の態様および実施形態は、さらに詳細に以下の実施例によって記載される。その実施例は、いかなる方法においても、付随する請求項の範囲を限定する意図は無い。
【実施例】
【0092】
(実施例1)
ゾレドロン酸三水和物の調製
5gの無水ゾレドロン酸を、マグネチックスターラー、コンデンサー、およびオイルバスを備えた丸底フラスコに入れ、次いで150mlの水をそれに加えた。反応塊をゆっくりと73℃に加熱し、透明な溶液を得た。溶液を熱い間にろ過し、粒子が無い溶液にした。透明なろ液を新しい丸底フラスコに入れ、30℃に冷却した。その反応塊を、30℃で10分間攪拌した。分離した固体を真空下でろ過した。その化合物を600mmHgの減圧下で10分間吸引乾燥し、3.6gの表題化合物を得た。
【0093】
この生成物のサンプルを分析し、図1から6すべてを作成した。
【0094】
湿度:15.5%(w/w)、カールフィッシャー法による
融点:238±3℃。
【0095】
(実施例2)
乾燥による三水和物と一水和物の混合物の一水和物への変換
1gのゾレドロン酸三水和物を清潔なペトリ皿に入れた。その化合物を次いで真空オーブン内において、60℃で600mgHgの減圧下で16時間乾燥し、ゾレドロン酸一水和物を得た。
【0096】
(実施例3)
スラリー化による一水和物と三水和物の混合物の一水和物への変換
5mlのアセトンを0.5gのゾレドロン酸三水和物と一緒に丸底フラスコに入れた。次いで混合物を28℃で30分間攪拌した。混合物を600mmHgの減圧下でろ過し、固体を最後に真空下、28℃で乾燥してゾレドロン酸一水和物を得た。
【0097】
(実施例4)
溶媒−貧溶媒技術を用いた三水和物の一水和物への変換
30mlの水を1gのゾレドロン酸三水和物と一緒に丸底フラスコに入れた。混合物を約10から20分間、28℃で攪拌し、次いで99℃に加熱し、99℃でさらに15分間維持した。次いでその塊を放射により67℃に冷却した。この温度で10mlのメタノールを添加して生成物を沈殿させ、さらにその塊を28℃に冷却するまで攪拌した。分離した固体を真空下でろ過し、10mlの水で洗浄した。次いで固体を600mgHgの減圧下で30分間、28℃で吸引乾燥し、最後に59℃で600mmHgの減圧下で12時間乾燥し、結晶性ゾレドロン酸一水和物を得た。
【0098】
(実施例5)
溶媒−貧溶媒技術を用いた三水和物の一水和物への変換
30mlの水を1gのゾレドロン酸三水和物と一緒に丸底フラスコに入れた。混合物を約10分間28℃で攪拌し、続いて99℃に加熱し、99℃でさらに30分間維持した。次いで、混合物を放射により57℃に冷却した。この温度で10mlのアセトンを加えて、生成物を沈殿させた。続いて、混合物を28℃になるまで攪拌した。塊を28℃で3時間維持した。続いて、分離した固体を600mmHgの減圧下でろ過した。固体を45分間吸引乾燥し、最後に600mmHgの減圧下、60℃で約3時間乾燥して、結晶性ゾレドロン酸一水和物を得た。
【図面の簡単な説明】
【0099】
【図1】図1は、実施例1で調製した結晶性ゾレドロン酸三水和物のXRPDパターンである。
【図2】図2は、実施例1で調製した結晶性ゾレドロン酸三水和物のIRスペクトルである。
【図3】図3は、実施例1で調製した結晶性ゾレドロン酸三水和物のDSC曲線である。
【図4】図4は、実施例1で調製した結晶性ゾレドロン酸三水和物の単結晶構造である。
【図5】図5は、実施例1で調製した結晶性ゾレドロン酸三水和物の単結晶データからシミュレートしたXRDパターンである。
【図6】図6は、結晶性ゾレドロン酸三水和物のTGA曲線であり、実施例1で調製した化合物のDSC曲線と重ね合わせている。
【技術分野】
【0001】
(発明の序論)
本発明は、結晶性ゾレドロン酸三水和物およびその調製方法に関する。
【背景技術】
【0002】
ゾレドロン酸の化学名は、(1−ヒドロキシ−2−イミダゾール−1−イルホスホノエチル)リン酸であり、この化合物は、式Iにより構造を示すことができる。
【0003】
【化2】
ゾレドロン酸は、イミダゾール環を含む側鎖を特徴とする、第3世代のビスホスホネート誘導体である。ゾレドロン酸は、破骨細胞の骨吸収を阻害し、腫瘍性高カルシウム血症の治療に使用される。ゾレドロン酸は、ゾメタ(ZOMETA)(商標)という商品名で販売されている、バイアル中に静脈内注入用の滅菌粉末または溶液として市販されている。各バイアルは4mgのゾレドロン酸(無水)を含み、それは4.264mgのゾレドロン酸一水和物に相当する。
【0004】
ゾレドロン酸の化学合成は、現在までその一水和物の物質の調製に向けられている。特許文献1は、ゾレドロン酸を開示し、実施例10においてスキーム1に示すようなゾレドロン酸の調製方法を開示している。
【0005】
【化3】
手短に言うと、その方法は、三塩化リンと塩酸の存在下で、2−(1−イミダゾリル)酢酸塩酸塩をリン酸と反応させてゾレドロン酸を得ることを含み、ゾレドロン酸はアセトンで希釈して沈殿させる。このようにして得られた粗製ゾレドロン酸を水中で再結晶化する。水からの粗物質を再結晶化する最終工程は、ゾレドロン酸一水和物を提供する。
【0006】
特許文献2もまた、最終工程において水から同様の再結晶化によりゾレドロン酸一水和物の化合物を提供することを含む。
【0007】
特許文献3は、ゾレドロン酸の様々な結晶形態、およびそのナトリウム塩、ならびにそれらの調製方法を開示している。ゾレドロン酸の一水和物である結晶形態I、II、XIIおよびXVIII、ならびにゾレドロン酸の無水和物の形である結晶形態XV、XX、およびXXVIの調製が記載されている。ゾレドロン酸の一ナトリウム塩および二ナトリウム塩の様々な水和物および無水和物の形もまた記載しており、非晶質ゾレドロン酸一ナトリウム塩、二ナトリウム塩および三ナトリウム塩もまた記載している。
【0008】
ゾレドロン酸の多形性の特性決定についてかなりの数の研究が行われているが、生成できる他の形態を同定する必要性が残っている。
【0009】
上記特許のうち2つの特許は一水和物の調製を記載しているが、その方法の完全なる詳細についてはいずれも示していない。特許文献1では、実施例1において単純に、その生成物は水中で再結晶化させると述べているが、再結晶化の条件は示していない。特許文献2は、その粗製ゾレドロン酸を90から95℃で2から3時間水に溶解させ、次いで高温下で炭素処理を行い、そして再結晶化のためにその反応塊を25から35℃に冷却することによる水中の粗製ゾレドロン酸の再結晶を伴う一水和物の調製方法を例示している。
【0010】
上記いずれの特許も水からの再結晶化の間の一水和物形成のために重要なパラメータを述べていない。上記方法に従って一水和物の製造のためにバッチをスケールアップする間に、他の結晶形態による汚染がしばしば観察されている。
【0011】
世界中の監督官庁が、同じ活性化合物のすべての可能な結晶形態を合成し、できるだけ完全に特性決定することを要求している。さらに、市販の製品は、微量の他の形態のいずれも含むべきではなく、存在する場合、その各形態の割合は、貯蔵中に原薬の溶解特性および生物学的利用態の特性の変化を避けるのに十分に特性決定されることを要求している。
【特許文献1】米国特許第4939130号明細書
【特許文献2】国際公開第2005/063717号パンフレット
【特許文献3】国際公開第2005/005447号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0012】
このように、引き続き、ゾレドロン酸などの商業的興味のある薬理学的に活性な化合物の新しい多形形態を調製する必要があり、それは製剤学者に、それらの異なる物理化学的特徴に基づいて、選択するための活性成分のより広い範囲の結晶形態を提供する。
【0013】
多形形態の調製方法は、その方法が工場で簡単にスケールアップできるように、ロバストであり、再現可能であることも重要である。このようにして、ゾレドロン酸の製造における改良が必要とされている。
【課題を解決するための手段】
【0014】
(発明の要旨)
本発明は、結晶性ゾレドロン酸三水和物、およびロバストで再現可能なその調製方法に関する。
【0015】
本発明の一態様は、その単結晶X線回折図形(XRD)、X線粉末回折(XRPD)パターン、赤外(IR)吸収スペクトル、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線によって特徴付けられる結晶性ゾレドロン酸三水和物を提供する。
【0016】
もう1つの態様において、本発明は結晶性ゾレドロン酸三水和物のロバストで再現可能な調製方法を提供する。
【0017】
一実施形態において、結晶性ゾレドロン酸三水和物の調製方法は、
a)ゾレドロン酸溶液を供給すること、
b)その溶液から固体を結晶化すること、および
c)分離したゾレドロン酸三水和物結晶を回収すること
を含む。
【0018】
本発明の別の態様は、ゾレドロン酸一水和物とゾレドロン酸三水和物の混合物をゾレドロン酸一水和物に変換する方法を提供する。
【0019】
本発明のさらに別の態様は、ゾレドロン酸三水和物からゾレドロン酸一水和物を調製する方法を提供する。
【0020】
本発明のさらなる態様は、ゾレドロン酸一水和物と実質的に同等の溶解性を有する結晶性ゾレドロン酸三水和物を提供する。
【0021】
本発明のさらなる態様は、約300μm未満の粒径を有する結晶性ゾレドロン酸三水和物を提供する。
【0022】
ゾレドロン酸三水和物は、そのXRPDパターンが、実質的に図1に従うことを特徴とすることができる。
【0023】
また、ゾレドロン酸三水和物は、そのIRスペクトルが、実質的に図2に従うことを特徴とすることができる。
【0024】
また、ゾレドロン酸三水和物は、そのDSC曲線が、実質的に図3に従うことを特徴とすることができる。
【0025】
一実施形態において、ゾレドロン酸三水和物の調製方法は、約60から80℃の温度の水を含む溶媒中のゾレドロン酸溶液を供給すること、およびその溶液を冷却してゾレドロン酸三水和物を結晶化することを含む。
【0026】
もう1つの実施形態において、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換する方法は、ゾレドロン酸三水和物を約40から90℃の温度で乾燥することを含む。
【0027】
さらなる実施形態において、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換する方法は、ケトン中でゾレドロン酸三水和物のスラリーを形成することを含む。
【0028】
なお一層さらなる実施形態において、ゾレドロン酸一水和物の調製方法は、ゾレドロン酸の水溶液を供給すること、およびゾレドロン酸に貧溶媒を添加することを含む。
【0029】
その上さらなる態様において、本発明はゾレドロン酸三水和物を1種またはそれ以上の薬学的に許容できる担体、賦形剤、または希釈剤と一緒に含む医薬組成物を提供する。
【発明を実施するための最良の形態】
【0030】
(発明の詳細な説明)
本発明の一態様は、結晶性ゾレドロン酸三水和物を包含する。
【0031】
結晶性ゾレドロン酸三水和物は、その粉末X線回折(「XRPD」)パターン、単結晶X線回折(「XRD」)パラメータ、赤外吸収(「IR」)スペクトル、示差走査熱量測定(「DSC」)曲線、および熱重量分析(「TGA」)曲線のいずれかによって特性決定する。
【0032】
単結晶X線回折データは、グラファイト単色光Mo−Kα放射線を用いてRigaku Mercury CCD領域検出器で収集した。その構造は、直接法および(SIR92)によって解析し、最小二乗法により精密化した。2110について観察された反射では、R値が0.038、Rw=0.039である。単結晶データからシミュレートした粉末回折パターンを図5に示す。
【0033】
ゾレドロン酸三水和物は、そのXRPDパターンにより特性決定し、このパターンは既知の形態との違いを示している。本明細書で報告されるXRPDデータは、波長が1.541ÅのCu Kα−1放射線を用いて得、Bruker Axe、D8高度粉末X線回折装置を用いて測定した。
【0034】
結晶性ゾレドロン酸三水和物は、図1のパターンに実質的に従うXRPDパターンを特徴とする。結晶性ゾレドロン酸三水和物はまた、約10.8、16.4、17.1、18.4、21.6、24.9、25.4、27.8、31.0、および32.6の±0.2°2θに顕著なピークを有するXRPDパターンを特徴とする。また、約38.0、40.2、21.8、9.2、10.3、および43.4の±0.2°2θのさらなるXRPDピークを特徴とする。
【0035】
ゾレドロン酸三水和物はまた、その格子パラメータが、単結晶X線回折によって決定された結晶構造を特徴とする。
【0036】
ゾレドロン酸三水和物の結晶構造を図4に示す。その三水和物は、表1に示す単位セルパラメータで三斜晶空間群P1において結晶化する。
【0037】
【化4】
三次元の充填は、表2に示す強い分子外および分子内水素結合により安定化する。
【0038】
【化5】
結晶性ゾレドロン酸三水和物の単結晶の情報から、シミュレートした粉末回折図形(理論的な回折図形)は、実験的に得られたものに匹敵する回折図形が得られた。理論的な回折図形と実験的な回折図形との間にみられる類似性が非常に高いことは、粉末に含まれるその構造は、その単結晶において測定されるものに対応しており、その構造が唯一である、すなわち、結晶性ゾレドロン酸三水和物に混合している他の多形形態が無いことを意味している。
【0039】
結晶性ゾレドロン酸三水和物の赤外スペクトルは、Perkin Elmer System 200 FT−IR分光光度計を用いて、400cm−1と4000cm−1との間で、4cm−1の分解能、試験化合物が0.5質量%の濃度である臭化カリウムのペレット中で記録されている。
【0040】
結晶性ゾレドロン酸三水和物はさらに、約671、712、766、975、1301、1323、1406、1460、1550、2826、3154、および3484の±5cm−1にピークを含む赤外吸収スペクトルを特徴とする。結晶性ゾレドロン酸三水和物三水和物はまた、図2のスペクトルに実質的に従う赤外吸収スペクトルを特徴とする。
【0041】
結晶性ゾレドロン酸三水和物は、さらにまた、図3の曲線に、実質的に従うその示差走査熱量測定曲線を特徴とする。結晶性ゾレドロン酸三水和物は、また、約234℃で発熱、約224℃および約88℃で吸熱を示すDSC曲線を特徴とする。
【0042】
結晶性ゾレドロン酸三水和物は、さらにまた、水3分子の損失を示す図6のDTA曲線に実質的に従う熱重量分析曲線を特徴とする。図6において、左の縦軸はサンプル(ミリグラム)であり、右の縦軸は熱電対(ミリボルト)であり、横軸は温度(℃)である。
【0043】
ゾレドロン酸の水分含量は、15から18重量%の範囲であることが可能である。
【0044】
別の態様において、本発明は、結晶性ゾレドロン酸三水和物のロバストで再現可能である調製方法を提供する。
【0045】
一実施形態において、その三水和物の調製方法は、
a)ゾレドロン酸溶液を供給すること、
b)その溶液から固体を結晶化すること、および
c)分離したゾレドロン酸三水和物結晶を回収すること
を含む。
【0046】
工程a)は、ゾレドロン酸溶液を供給することを含む。
【0047】
ゾレドロン酸溶液はゾレドロン酸を適当な溶媒に溶解することによって得ることができ、またはそのような溶液はゾレドロン酸が形成される反応から直接得ることもできる。
【0048】
適当な溶媒にゾレドロン酸を溶解することによってその溶液を調製する場合には、ゾレドロン酸の任意の塩、溶媒和物、および水和物を含む、結晶形態または非晶質形などの、ゾレドロン酸の任意の形をその溶液の調製に利用することができる。
【0049】
ゾレドロン酸三水和物の調製に有用である適切な溶媒には、水のみ、または例えば、メタノール、エタノール、プロパノール、第3級ブタノール、n−ブタノールなどのアルコール、アセトン、プロパノンなどのケトン、アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド、ジオキサンなど、およびそれらの混合物などの有機溶媒との組合せを含む。
【0050】
関連する実施形態において、本発明は、ゾレドロン酸の溶媒または溶媒混合物中溶液をほぼ周囲温度から約80℃、または約60から80℃、あるいは約70から75℃の温度に加熱し、透明な溶液を得ることを含む。ゾレドロン酸三水和物溶液の調製には、透明な溶液が得られる限り約80℃未満の任意の温度を使用することができる。これらの範囲において温度が高いほど、より高濃度の溶質を提供し、一般に高い処理効率をもたらす。
【0051】
ゾレドロン酸の溶解のための最高温度は、得られるゾレドロン酸の多形形態を決定するので重要である。その溶液を約90℃より高い温度に加熱すると、結晶性一水和物がもたらされる。溶液をより低い温度、例えば約40から80℃、または約70から75℃の範囲に加熱すると、結晶性ゾレドロン酸三水和物がもたらされる。
【0052】
その溶液は、その温度で約1分から任意の所望の時間維持される。混合物を約75℃に加熱する場合、冷却を始める前にその高温での必要とされる最小の維持時間はごくわずかである。
【0053】
溶液は、任意でろ紙、ガラス繊維、または他の膜物質を、またはセライトなどの清澄剤を通過させることによってろ過することもできる。使用する装置、ならびにその溶液の濃度および温度に応じて、早すぎる結晶化を避けるために、ろ過装置を余熱する必要があることがある。
【0054】
その溶質の濃度は、溶媒中に約0.1g/mlから約20g/mlであることが可能であり、または1g/mlから5g/mlの範囲であることが可能である。
【0055】
工程b)はろ液から固体を結晶化することを含む。
【0056】
結晶化は、通常、溶解温度よりも低い温度で行われる。結晶化の温度は約40℃未満、または30℃未満であってよい。
【0057】
結晶化は、所望の結晶収率が得られるまで、例えば約1時間から約72時間攪拌して行うことができる。結晶化工程はさらに当業者に公知の簡易手段を含むことができる。例えば、結晶化工程はさらに、その溶液を冷却すること、その溶液を加熱すること、または沈殿を引き起こす薬剤を加えることを含むことができる。
【0058】
溶液の温度は外部の冷却を用いて結晶化が急速に起こるように下げることができ、またはその分離温度に自然に冷やすこともできる。一般に、1から5kgまたはそれより大規模のバッチでは、反応塊を自然に冷やすとすると不都合な時間かかってしまうことがあるため、外部の冷却を反応塊にしばしば用いてその必要とするレベルにまでその温度を下げる。
【0059】
さらに冷却時間の延長は、作業経費の増加の他には欠点は無く、所与のバッチサイズに適切な時間は、わずかな努力によって当業者が決定することができる。溶液の冷却は、攪拌を伴う大気条件下の単なる放射冷却によって、または例えばジャケット容器内での冷却媒体の循環などの制御された冷却装置の使用などによって達成される。そのような急速冷却および徐冷のための技術は当業者に公知であって、限定することなくすべて本明細書に含まれる。
【0060】
90から95℃のより高温で溶媒にゾレドロン酸を溶解し、続いて低温で固体を分離することを含む、結晶性ゾレドロン酸一水和物の調製方法と比べて、ゾレドロン酸三水和物用の方法はロバストであり、再現可能である。一水和物の調製は、分離の間のゾレドロン酸溶液の冷却速度や溶解の間の溶液の維持温度などの変数に依存する。
【0061】
溶解中の90から95℃を超える温度でのゾレドロン酸溶液の不適切な維持により、一水和物と三水和物の混合物をもたらすことがある。また、反応塊が溶解温度から結晶化温度に急速に冷却されると、その結果はゾレドロン酸の一水和物と三水和物の混合物となる。
【0062】
大規模の一水和物の調製の間には、多くのプロセス方法を行う必要がある。
【0063】
工程c)は、分離されたゾレドロン酸三水和物の結晶の回収を含む。
【0064】
回収は、それだけに限定されないが、ろ過、遠心分離、デカントを含む任意の手段によって行うことができる。その結晶形態は、その結晶形態と、それだけに限定されない、懸濁液、溶液、スラリーおよび乳液を含む溶媒(1またはそれ以上)とを含む任意の組成物から回収することができる。
【0065】
得られた化合物は、さらに周囲圧力または減圧下で乾燥させることができる。例えば、乾燥は、減圧下または大気圧下で、約40℃から60℃、または70℃から80℃、あるいはそれ以上の温度で行うことができる。乾燥は所望の残留溶媒量が得られるまで、例えば約2時間から24時間、または約3から6時間の間行うことができる。
【0066】
本発明のさらに別の態様は、ゾレドロン酸の一水和物と三水和物の混合物をゾレドロン酸一水和物へ変換するための方法を提供する。
【0067】
公知のように、ゾレドロン酸一水和物の調製方法は、ロバストではなく、大規模の製造の間では、もし決定的なパラメータが用いられないならば、ゾレドロン酸の一水和物と三水和物の混合物になる可能性がある。
【0068】
本発明は、ゾレドロン酸三水和物と一水和物の混合物をゾレドロン酸一水和物へ変換する方法を提供する。
【0069】
一実施形態において、ゾレドロン酸一水和物および三水和物の混合物をゾレドロン酸一水和物へ変換する方法は、三水和物を含む物質を50℃より高い温度で真空下でさらに乾燥する工程、または有機溶媒中に三水和物の物質を含むスラリーを形成する工程のいずれか1つの工程を含む。
【0070】
乾燥温度は40から90℃、または60から70℃、あるいは55から60℃の範囲であることが可能であり、その化合物は周囲圧または減圧下で乾燥することができる。例えば、乾燥は減圧下でまたは周囲圧下で、空気乾燥機、真空乾燥機または箱型乾燥機などのいずれかを用いて行うことができる。場合により、不活性雰囲気下で乾燥を行うことができる。
【0071】
三水和物をスラリーするために用いることができる適当な溶媒は、アセトン、エチルメチルケトン、プロパノンなどのケトンである。
【0072】
スラリー形成は、攪拌を伴ってもよく、約1時間から約10時間またはそれ以上の期間行うことができる。
【0073】
スラリーを形成する目的で約5から100倍の範囲の任意の量の溶媒を用いることができる。
【0074】
本発明のさらなる態様は、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換することを含む。
【0075】
別の実施形態において、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換する方法は、溶媒−貧溶媒技術によって再結晶化することを含む。その方法はゾレドロン酸と適当な溶媒を供給すること、その混合物を加熱して透明な溶液を得ること、続いて貧溶媒を添加して必要とする生成物の沈殿を得ることを含む。
【0076】
溶解のために用いることができる適当な溶媒には、例えば、水、メタノール、エタノール、プロパノール、n−ブタノールなどのアルコール、ジメチルホルムアミド、ジメチルスルホキシド、テトラヒドロフランなど、およびそれらの混合物が含まれる。
【0077】
用いることができる貧溶媒には、例えば、n−ヘキサン、n−ヘプタン、およびトルエンなどの炭化水素、アセトン、プロパノン、エチルメチルケトン、およびブタノンなどのケトン、ジエチルエーテル、イソプロピルエーテルなどのエーテル、酢酸エチル、第3級酢酸ブチルなどのエステル、ジクロロメタン、1,2−ジクロロエタン、クロロホルム、四塩化炭素などのハロゲン化炭水化物、およびそれらの混合物が含まれる。
【0078】
溶解の手順は、約95から120℃の範囲の高温で行うことができる。加熱は、それだけに限定されない機械的な手段および磁力の手段を含む任意の手段で連続してまたは時々攪拌またはかき混ぜることにより為し遂げることができる。溶媒の量は、ゾレドロン酸を溶解して濃縮溶液を形成するのに十分な量であるべきである。
【0079】
貧溶媒のゾレドロン酸溶液への添加は、約0から120℃、もしくは60から90℃の温度、または周囲温度、あるいは約0から15℃の範囲のより低い温度で行うことができる。
【0080】
分離した固体の回収はそれだけに限定されない、ろ過、遠心分離およびデカントを含む任意の手段によって行うことができる。その結晶形態は、その結晶形態と、それだけに限定されない、懸濁液、溶液、スラリーおよび乳液を含む溶媒(1またはそれ以上)とを含む任意の組成物から回収される。
【0081】
得られた化合物は、周囲圧または減圧下でさらに乾燥させることができる。例えば、乾燥は減圧下または大気圧下で約40℃から60℃、または70℃から80℃、あるいはより高い温度で行うことができる。乾燥は使用される乾燥条件および許容可能である残留溶媒分の量に応じて、約2時間まで、または約5時間まで、またはより長時間の持続時間で行うことができる。
【0082】
したがって、本発明は、医薬品を製造するために使用できる、純粋なゾレドロン酸三水和物を調製するための再現可能な方法を提供する。しかしながら、所望により、ゾレドロン酸三水和物は、容易に純粋なゾレドロン酸一水和物に変換し、医薬品を製造するために使用することもできる。本発明の利点は、所望の純粋な形態のゾレドロン酸を予想通りに調製する能力を提供することである。
【0083】
本発明のさらに別の態様は、ゾレドロン酸一水和物と実質的に同等の溶解性を有する結晶性ゾレドロン酸三水和物を提供する。このゾレドロン酸の溶解度は、ゾレドロン酸一水和物と同程度である。このことは医薬組成物におけるゾレドロン酸三水和物の使用を容易にする。
【0084】
本発明のなお一層さらなる態様は、粒径が300μm未満の結晶性ゾレドロン酸三水和物を提供する。
【0085】
D10、D50、およびD90値は、粒度分布を示すのに有用な方法である。D90は、粒子の少なくとも90容量%がそのD90値よりも小さい粒径である値を意味する。同様にD50およびD10は、粒子の50容量%および10容量%がその値よりも小さい粒径である値を意味する。D50値は粉末の平均粒径とみなすことができる。D10、D50およびD90を決定する方法には、マルバーン装置を使用したレーザー回折を含む。
【0086】
本発明の結晶性ゾレドロン酸三水和物は、10μm未満または20μm未満のD10、100μm未満または150μm未満のD50、および200μm未満または300μm未満のD90を有する。いずれのD値においても特定の下限値は無い。
【0087】
その上さらなる態様において、本発明は、ゾレドロン酸三水和物を1種またはそれ以上の薬学的に許容できる担体、賦形剤または希釈剤と一緒に含む、医薬組成物を提供する。
【0088】
本発明のゾレドロン酸三水和物を1種またはそれ以上の薬学的に許容可能である担体と一緒に含む医薬組成物は、さらに以下のように製剤することができる:例えば、それだけに限定されないが、粉末、顆粒、ペレット、錠剤およびカプセルなどの固体経口剤型、例えば、それだけに限定されないが、シロップ、懸濁液、分散液および乳液などの液体経口剤型、および例えば、それだけに限定されないが、溶液、分散液および凍結乾燥組成物などの注射製剤。製剤は、即時放出、遅延放出、または放出調整された形態とすることができる。さらに、即時放出性組成物は、従来の製剤、分散可能な製剤、チュアブル製剤、口内溶解製剤または瞬時溶解製剤であることが可能であり、放出調整された組成物は、親水性または疎水性、あるいは親水性および疎水性の組合せの放出速度を制御する物質を含み、マトリックスまたはレザーバ、あるいはマトリックスおよびレザーバの組合せを形成してもよい。組成物は直接混合、乾式造粒または湿式造粒によって、あるいは射出および球形化によって調製することができる。組成物は、非被覆、フィルム被覆、糖被覆、粉末被覆、腸溶性被覆、または放出調整された被覆として存在可能である。本発明の組成物は、さらに1種またはそれ以上の薬学的に許容できる賦形剤を含むことができる。
【0089】
本発明において使用される薬学的に許容できる賦形剤には、それだけに限定されないが、希釈剤(例えば、でんぷん、アルファ化でんぷん、ラクトース、粉末セルロース、結晶セルロース、リン酸二カルシウム、リン酸三カルシウム、マンニトール、ソルビトール、糖など)、結合剤(例えば、アカシア、グアーガム、トラガカント、ゼラチン、ポリビニルピロリドン、ヒドロキシプロピルセルロース、ヒロドキシプロピルメチルセルロース、アルファ化でんぷんなど)、崩壊剤(例えば、でんぷん、グリコール酸ナトリウムでんぷん、アルファ化でんぷん、クロスポビドン、クロスカルメロースナトリウム、コロイド状二酸化ケイ素など)、潤滑剤(例えば、ステアリン酸、ステアリン酸マグネシウム、ステアリン酸亜鉛など)、流動促進剤(例えば、コロイド状二酸化ケイ素など)、溶解性またはぬれ性増強剤(例えば、陰イオン性、陽イオン性または中性の界面活性剤)、錯体形成剤(例えば、種々の等級のシクロデキストリン、樹脂)、放出速度調整剤(例えば、ヒドロキシプロピルセルロース、ヒドロキシメチルセルロース、ヒドロキシプロピルメチルセルロース、エチルセルロース、メチルセルロース、種々の等級のメタクリル酸メチル、ワックス等)が含まれる。他の薬学的に許容できる賦形剤には、それだけに限定されないが、フィルム形成剤、可塑剤、着色剤、香味料、甘味料、粘性増強剤、保存剤、抗酸化剤などが含まれる。
【0090】
本発明の組成物において、ゾレドロン酸三水和物は、0.5mgから50mg、または1mgから25mgの範囲において有用な活性成分である。
【0091】
本発明のある特定の態様および実施形態は、さらに詳細に以下の実施例によって記載される。その実施例は、いかなる方法においても、付随する請求項の範囲を限定する意図は無い。
【実施例】
【0092】
(実施例1)
ゾレドロン酸三水和物の調製
5gの無水ゾレドロン酸を、マグネチックスターラー、コンデンサー、およびオイルバスを備えた丸底フラスコに入れ、次いで150mlの水をそれに加えた。反応塊をゆっくりと73℃に加熱し、透明な溶液を得た。溶液を熱い間にろ過し、粒子が無い溶液にした。透明なろ液を新しい丸底フラスコに入れ、30℃に冷却した。その反応塊を、30℃で10分間攪拌した。分離した固体を真空下でろ過した。その化合物を600mmHgの減圧下で10分間吸引乾燥し、3.6gの表題化合物を得た。
【0093】
この生成物のサンプルを分析し、図1から6すべてを作成した。
【0094】
湿度:15.5%(w/w)、カールフィッシャー法による
融点:238±3℃。
【0095】
(実施例2)
乾燥による三水和物と一水和物の混合物の一水和物への変換
1gのゾレドロン酸三水和物を清潔なペトリ皿に入れた。その化合物を次いで真空オーブン内において、60℃で600mgHgの減圧下で16時間乾燥し、ゾレドロン酸一水和物を得た。
【0096】
(実施例3)
スラリー化による一水和物と三水和物の混合物の一水和物への変換
5mlのアセトンを0.5gのゾレドロン酸三水和物と一緒に丸底フラスコに入れた。次いで混合物を28℃で30分間攪拌した。混合物を600mmHgの減圧下でろ過し、固体を最後に真空下、28℃で乾燥してゾレドロン酸一水和物を得た。
【0097】
(実施例4)
溶媒−貧溶媒技術を用いた三水和物の一水和物への変換
30mlの水を1gのゾレドロン酸三水和物と一緒に丸底フラスコに入れた。混合物を約10から20分間、28℃で攪拌し、次いで99℃に加熱し、99℃でさらに15分間維持した。次いでその塊を放射により67℃に冷却した。この温度で10mlのメタノールを添加して生成物を沈殿させ、さらにその塊を28℃に冷却するまで攪拌した。分離した固体を真空下でろ過し、10mlの水で洗浄した。次いで固体を600mgHgの減圧下で30分間、28℃で吸引乾燥し、最後に59℃で600mmHgの減圧下で12時間乾燥し、結晶性ゾレドロン酸一水和物を得た。
【0098】
(実施例5)
溶媒−貧溶媒技術を用いた三水和物の一水和物への変換
30mlの水を1gのゾレドロン酸三水和物と一緒に丸底フラスコに入れた。混合物を約10分間28℃で攪拌し、続いて99℃に加熱し、99℃でさらに30分間維持した。次いで、混合物を放射により57℃に冷却した。この温度で10mlのアセトンを加えて、生成物を沈殿させた。続いて、混合物を28℃になるまで攪拌した。塊を28℃で3時間維持した。続いて、分離した固体を600mmHgの減圧下でろ過した。固体を45分間吸引乾燥し、最後に600mmHgの減圧下、60℃で約3時間乾燥して、結晶性ゾレドロン酸一水和物を得た。
【図面の簡単な説明】
【0099】
【図1】図1は、実施例1で調製した結晶性ゾレドロン酸三水和物のXRPDパターンである。
【図2】図2は、実施例1で調製した結晶性ゾレドロン酸三水和物のIRスペクトルである。
【図3】図3は、実施例1で調製した結晶性ゾレドロン酸三水和物のDSC曲線である。
【図4】図4は、実施例1で調製した結晶性ゾレドロン酸三水和物の単結晶構造である。
【図5】図5は、実施例1で調製した結晶性ゾレドロン酸三水和物の単結晶データからシミュレートしたXRDパターンである。
【図6】図6は、結晶性ゾレドロン酸三水和物のTGA曲線であり、実施例1で調製した化合物のDSC曲線と重ね合わせている。
【特許請求の範囲】
【請求項1】
ゾレドロン酸三水和物。
【請求項2】
図1に実質的に従うCu Kα放射線を用いた粉末X線回折パターンを有する、請求項1に記載のゾレドロン酸三水和物。
【請求項3】
約10.8、16.4、17.1、18.4、21.6、24.9、25.4、27.8、31.0、および32.6の±0.2°2θのピークを含む、Cu Kα放射線を用いた粉末X線回折パターンを有する、請求項1に記載のゾレドロン酸三水和物。
【請求項4】
さらに約38.0、40.2、21.8、9.2、10.3、および43.4の±0.2°2θのピークを含む、Cu Kα放射線を用いた粉末X線回折パターンを有する、請求項2に記載のゾレドロン酸三水和物。
【請求項5】
図2に実質的に従う赤外吸収スペクトルを有する、請求項1に記載のゾレドロン酸三水和物。
【請求項6】
約671、712、766、975、1301、1323、1406、1460、1550、2826、3154、および3484の±5cm−1のピークを含む赤外吸収スペクトルを有する、請求項1に記載のゾレドロン酸三水和物。
【請求項7】
図3に実質的に従う示差走査熱量測定曲線を有する、請求項1に記載のゾレドロン酸三水和物。
【請求項8】
約234℃で発熱、約224℃および約88℃で吸熱を示す示差走査熱量測定曲線を有する、請求項1に記載のゾレドロン酸三水和物。
【請求項9】
約80℃未満の温度の水を含む溶媒中のゾレドロン酸の溶液を供給すること、およびその溶液を冷却してゾレドロン酸三水和物を結晶化することを含む、ゾレドロン酸三水和物の調製方法。
【請求項10】
ゾレドロン酸の溶液が約70から75℃の温度である、請求項9に記載の方法。
【請求項11】
溶媒が水および有機溶媒を含む、請求項9に記載の方法。
【請求項12】
ゾレドロン酸三水和物を約40から90℃の温度で乾燥することを含む、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換する方法。
【請求項13】
ケトン中でゾレドロン酸三水和物のスラリーを形成することを含む、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換する方法。
【請求項14】
ゾレドロン酸の水溶液を供給すること、およびゾレドロン酸の貧溶媒を添加することを含む、ゾレドロン酸一水和物の調製方法。
【請求項15】
貧溶媒が、炭化水素、ケトン、エーテル、エステルおよびハロゲン化炭化水素の1種またはそれ以上を含む、請求項14に記載の方法。
【請求項16】
貧溶媒がケトンを含む、請求項14に記載の方法。
【請求項17】
単結晶パラメータが、およそ、X線回折により定義された、
【化1】
を有する、ゾレドロン酸三水和物。
【請求項1】
ゾレドロン酸三水和物。
【請求項2】
図1に実質的に従うCu Kα放射線を用いた粉末X線回折パターンを有する、請求項1に記載のゾレドロン酸三水和物。
【請求項3】
約10.8、16.4、17.1、18.4、21.6、24.9、25.4、27.8、31.0、および32.6の±0.2°2θのピークを含む、Cu Kα放射線を用いた粉末X線回折パターンを有する、請求項1に記載のゾレドロン酸三水和物。
【請求項4】
さらに約38.0、40.2、21.8、9.2、10.3、および43.4の±0.2°2θのピークを含む、Cu Kα放射線を用いた粉末X線回折パターンを有する、請求項2に記載のゾレドロン酸三水和物。
【請求項5】
図2に実質的に従う赤外吸収スペクトルを有する、請求項1に記載のゾレドロン酸三水和物。
【請求項6】
約671、712、766、975、1301、1323、1406、1460、1550、2826、3154、および3484の±5cm−1のピークを含む赤外吸収スペクトルを有する、請求項1に記載のゾレドロン酸三水和物。
【請求項7】
図3に実質的に従う示差走査熱量測定曲線を有する、請求項1に記載のゾレドロン酸三水和物。
【請求項8】
約234℃で発熱、約224℃および約88℃で吸熱を示す示差走査熱量測定曲線を有する、請求項1に記載のゾレドロン酸三水和物。
【請求項9】
約80℃未満の温度の水を含む溶媒中のゾレドロン酸の溶液を供給すること、およびその溶液を冷却してゾレドロン酸三水和物を結晶化することを含む、ゾレドロン酸三水和物の調製方法。
【請求項10】
ゾレドロン酸の溶液が約70から75℃の温度である、請求項9に記載の方法。
【請求項11】
溶媒が水および有機溶媒を含む、請求項9に記載の方法。
【請求項12】
ゾレドロン酸三水和物を約40から90℃の温度で乾燥することを含む、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換する方法。
【請求項13】
ケトン中でゾレドロン酸三水和物のスラリーを形成することを含む、ゾレドロン酸三水和物をゾレドロン酸一水和物に変換する方法。
【請求項14】
ゾレドロン酸の水溶液を供給すること、およびゾレドロン酸の貧溶媒を添加することを含む、ゾレドロン酸一水和物の調製方法。
【請求項15】
貧溶媒が、炭化水素、ケトン、エーテル、エステルおよびハロゲン化炭化水素の1種またはそれ以上を含む、請求項14に記載の方法。
【請求項16】
貧溶媒がケトンを含む、請求項14に記載の方法。
【請求項17】
単結晶パラメータが、およそ、X線回折により定義された、
【化1】
を有する、ゾレドロン酸三水和物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2009−507831(P2009−507831A)
【公表日】平成21年2月26日(2009.2.26)
【国際特許分類】
【出願番号】特願2008−529983(P2008−529983)
【出願日】平成18年7月6日(2006.7.6)
【国際出願番号】PCT/US2006/026153
【国際公開番号】WO2007/032808
【国際公開日】平成19年3月22日(2007.3.22)
【出願人】(506159459)ドクター レディズ ラボラトリーズ リミテッド (9)
【出願人】(506017137)ドクター レディズ ラボラトリーズ, インコーポレイテッド (24)
【Fターム(参考)】
【公表日】平成21年2月26日(2009.2.26)
【国際特許分類】
【出願日】平成18年7月6日(2006.7.6)
【国際出願番号】PCT/US2006/026153
【国際公開番号】WO2007/032808
【国際公開日】平成19年3月22日(2007.3.22)
【出願人】(506159459)ドクター レディズ ラボラトリーズ リミテッド (9)
【出願人】(506017137)ドクター レディズ ラボラトリーズ, インコーポレイテッド (24)
【Fターム(参考)】
[ Back to top ]