
タキサン化学受容性予測試験法
本発明は、癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性に対するタキサンの効果を評価する工程を含む、タキサンに対する癌細胞の化学受容性を決定するための方法を提供する。このような方法は、自動化アナライザーシステムを使用し、CDKIキナーゼ活性、CDK1発現、CDK2キナーゼ活性、CDK2発現、MAD2発現、サイクリンB1、サイクリンE発現、p2l発現、およびCDK6発現などの細胞周期パラメーター(分子)が評価される。本発明は、さらに、タキサンに感受性である癌細胞の細胞周期プロファイルを得る方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
1. 発明の分野
本発明は、一般に癌治療および癌予防の分野に関する。より詳細には、癌患者におけるタキサン化学受容性を予測することに関する。なお、本願は、2003年8月25日に出願された米国仮特許出願第60/497,665号の優先権を主張し、その全ての開示が、具体的に参照として本明細書に組み入れられる。
【背景技術】
【0002】
2. 関連技術の説明
癌化学療法に付随する主な問題のうちの1つは、同じ組織像をもつ個々の患者が、所与の薬剤または所与の治療プロトコルに対して同様に応答しないということである。応答範囲は、乳癌などの化学受容性の腫瘍においてさえも、大部分において異なる可能性がある。薬物用量、薬物の併用、並びに投与計画、患者の年齢および状態、腫瘍局在、その他などの、薬物感受性の多くの決定因子が周知であるが、所与の腫瘍の内因性の感受性は、評価することが依然困難である主な因子である。多数の耐性のメカニズムが同定されており、これらいくつかを腫瘍生検で評価することができるが、これらは、抗癌剤に対する腫瘍感受性の問題を解決するものではない。これは、腫瘍細胞の感受性の関数として薬物療法を個別化しようとするための多くの研究をリードしてきた。
【0003】
患者から得られた腫瘍細胞に対する抗癌剤の感受性を決定するための1つのアプローチには、インビトロ試験であるヒト腫瘍幹細胞アッセイ法(Human Tumor Stem Cell Assay)(HTSCA)を含む。このアッセイ法は、開発された種々のその他のアッセイ法と共に、全て4つの基本工程:1)細胞の単離;2)薬物との細胞のインキュベーション;3)細胞生存の評価;および、4)結果の解釈、を含み、これらは全て、薬物が患者においてどれくらい有効であり得るかについて予測するために使用される。しかし、これらのインビトロでのアッセイ法は、インビボでの抗癌剤に対する化学受容性の不十分な予測法であることが多い。現在では、化学受容性予測試験のための標準的プロトコルは、エキソビボ試験によって行われている。このプロトコルでは、外科的に切除された組織を抗癌剤を含む培地に包埋し、1週間インキュベートする。次いで、組織のサイズまたは生存度を測定して、感受性を決定する。しかし、この方法は、時間がかかり、完全に手動式で、高価である(1試験について600〜1000ドル)。これまでは、化学受容性試験は、臨床使用まで到達しておらず、現在、所与の患者における抗癌剤に対する腫瘍の感受性の好結果の予測をルーチン的に達成することができない。
【0004】
化学受容性を決定することが必要な抗癌剤の1つの群は、パクリタキセルなどのタキサンを含む。パクリタキセル耐性は、紡錘体形成チェックポイントおよびCdk1に関係がある可能性があることが文献に示されている。パクリタキセルが、微小管を安定化して、紡錘体の形成の間に生じる動的な変化を妨害する場合に、紡錘体形成チェックポイントが活性化されて細胞を有糸分裂で停止させる(Horwitz et al., 1982)。有糸分裂チェックポイント/紡錘体形成チェックポイントは、細胞周期としても公知であり、正確な染色体分離をモニターし、ゲノム恒常性を維持するのに重要な役割を果たす。このチェックポイントは、紡錘体に対する染色体の付着および微小管によって発生される姉妹染色分体全体の張力の両方をモニターして、未熟な染色体の分離を防止する。
【0005】
紡錘体形成チェックポイントの分子構成成分は、最初に酵母(Saccharomyces cerevisiae)で同定された。チェックポイント・タンパク質の哺乳類相同体は、Mad1、Mad2、BubR1、Bub3、およびMps1を含む(LiおよびBenezra, 1996; Jin et al., 1998; Taylor et al., 1998; Chan et al., 1999)。チェックポイント機構は、Mad1、Mad2、BubR1、Bub3、およびcdc20で構成されるタンパク質複合体であり、染色体の動原体に位置する。このチェックポイントの標的は、後期促進複合体(APC)およびその活性化補助因子Cdc20である。Mad2およびBubR1は、下流に位置し、この機構の主要タンパク質であると思われ、Cdc20と直接相互作用して、APC活性を協同的に阻害する(Fang et al., 1998; Sudakin et al., 2001; Tang et al., 2001; Fang,2002)。
【0006】
腫瘍細胞では、チェックポイントの欠陥が観察されることが多く、これらはゲノム不安定化を誘導すると考えられている。最近では、Huang et al.(2000)は、MAD2およびCDK1キナーゼが、パクリタキセルで誘導されるアポトーシスに協同的に関与することを示唆した。その他の報告では、カスパーゼ-3活性化を介したアポトーシス誘導のためには、CDK1の活性化が必要とされることが示され(Tan et al., 2002)、p21Waf1(M期でのCDK1阻害剤)は、パクリタキセルで誘導されるアポトーシスの阻害に重要な役割を果たすことが証明された(Fang et al., 2000)。
【0007】
Cdk1は、有糸分裂サイクリンと合わせて、有糸分裂の制御のために必要とされる普遍的なマスター・キナーゼである(Nigg, 2001)。Cdk1活性は、紡錘体形成チェックポイントの活性化に従って最大になる。Cdk阻害剤またはドミナントネガティブCdk1のいずれかを使用する以前の報告では、Cdk1が、パクリタキセルで誘導される細胞死阻害剤またはドミナントネガティブCdkのために重要であることを示したが(MeikrantzおよびSchlegel, 1996; Shen et al., 1998);しかし、Cdk1の活性化が、活性化されたチェックポイント活性化の原因または結果であるかどうかは、不明なままである。従って、紡錘体形成チェックポイントとパクリタキセル感受性との関係は、依然不明である。
【0008】
従って、癌の治療における、タキサンなどの抗癌剤使用に対する新たな、かつより優れたアプローチが必要である。これらの薬剤の有効性は、化学受容性を評価することにより決定される可能性がある。
【発明の開示】
【0009】
発明の概要
癌療法における主要な問題は、化学療法薬に対する癌細胞の耐性である。従って、本発明は、癌細胞または腫瘍組織のタキサン化学受容性を決定するための方法を提供する。本方法は、パクリタキセルおよびドセタキセルなどのタキサンの骨格を有する全ての抗癌剤に対して有効である。
【0010】
従って、特定の態様において、本発明は、癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性に対するタキサンの効果を評価する工程を含む、タキサンに対する癌細胞の化学受容性を決定する方法を提供する。本発明において想定される癌細胞は、患者から得てもよい。本発明の一部の態様において、癌細胞は、生検組織試料、エキソビボで培養された生検組織試料、外科的に切開した組織試料、またはエキソビボで培養された外科的に切開した組織試料などの組織試料であってもよい。
【0011】
本発明の細胞周期分子は、1つもしくは複数のサイクリン依存性キナーゼ(CDK)、サイクリン、CDK阻害剤(CDKI)、p53、または有糸分裂/紡錘体形成チェックポイント分子を含んでいてもよい。一部の態様において、本発明は、1個、2個、3個、4個、5個、6個、7個、8個、9個、10個、11個、12個、13個、14個、15個、16個、17個、18個、19個、20個またはそれ以上の細胞周期分子を評価することを含む。特定の態様において、本発明は、CDK1活性、CDK2活性、CDK4活性、またはCDK6活性などのCDK活性について1つまたは複数の細胞周期分子を評価することを含むが、このようなものに限定されるわけではない。もう一つの特定の態様において、本発明は、CDK1発現、CDK2発現、CDK4発現、またはCDK6発現などの1つまたは複数のCDK分子の発現を評価する工程を含む。さらにもう一つの特定の態様において、本発明は、サイクリンB1発現またはサイクリンE発現などの1つまたは複数のサイクリン分子の発現を評価する工程を含む。さらにもう一つの特定の態様において、本発明は、p16発現、p21/Waf1発現、またはp27/Kip1発現などの1つまたは複数のCDKIの発現を評価する工程を含むが、これらに限定されるわけではない。なおさらなる特定の態様において、本発明は、MAD2発現またはBubR1発現などの1つまたは複数の有糸分裂/紡錘体形成チェックポイント分子の発現を評価する工程を含むが、これらに限定されるわけではない。
【0012】
本発明の特定の態様において、パクリタキセルおよびドセタキセルなどのタキサンの骨格を有する任意の分子もしくは化合物、またはこれらの誘導体もしくは類似体が、本発明において想定される。
【0013】
本発明において想定される癌細胞は、乳癌細胞、前立腺癌細胞、皮膚癌細胞、肺癌細胞、頭頚部癌細胞、膀胱癌細胞、骨癌細胞、骨髄癌細胞、脳癌細胞、大腸癌細胞、食道癌細胞、胃腸癌細胞、歯肉癌細胞、腎臓癌細胞、肝臓癌細胞、上咽頭癌細胞、卵巣癌細胞、胃癌細胞、精巣癌細胞、舌癌細胞、または子宮癌細胞を含んでもよいが、これらに限定されるわけではない。
【0014】
癌細胞は、抗癌治療の適用前に、または抗癌治療の適用後に、患者または被検者から得てもよい。抗癌治療は、化学療法または放射線療法であってもよいが、このようなものに限定されるわけではない。
【0015】
特定の態様において、本発明は、細胞周期プロファイルを得る工程を含む。本発明において想定される細胞周期プロファイルは、タキサン治療を受ける患者の癌細胞において、細胞周期分子(たとえば、タンパク質)の活性もしくは発現を測定するか、決定するか、評価するか、または検出する工程を含む。細胞周期プロファイリングにより、タキサンの感受性を調節する分子を解析してもよい。細胞周期分子の活性または発現は、当業者に公知の方法によって測定し、決定し、評価し、または検出してもよい。一部の態様において、細胞周期プロファイルは、細胞周期もしくは多タンパク質アナライザーなどの自動化アナライザーまたは装置を使用して得てもよい。細胞周期プロファイルを、タキサンに対して感受性である癌細胞の細胞周期プロファイルと比較してもよいことが想定される。細胞周期プロファイルを、タキサンに対して感受性でない癌細胞の細胞周期プロファイルと比較してもよいことも想定される。細胞周期プロファイルは、抗癌治療の適用前に、抗癌治療の適用後の細胞周期プロファイルのものと比較してもよい。
【0016】
一部の態様において、細胞周期プロファイルを得る工程は、以下のパラメーターのうちの1つ、2つ、3つ、4つ、5つ、6つ、7つ、8つ、もしくは9つを測定するか、決定するか、評価するか、または検出することを含む:(1)CDK1キナーゼ活性;(2)CDK1発現(CDK1比活性の算出のために);(3)CDK2キナーゼ活性;(4)CDK2発現(CDK2比活性の算出のために);(5)MAD2発現;(6)サイクリンB1;(7)サイクリンE発現;(8)p21発現;または(9)CDK6発現。一部の態様において、タキサン感受性とタキサン耐性細胞とを区別するために有用な細胞周期プロファイルは、以下のパラメーターのうちの1つ、2つ、3つ、4つ、5つ、6つ、または7つを含む:(1)タキサン治療の24時間後のCDK1比活性;(2)タキサン治療前のCDK2比活性;(3)タキサン治療前のMAD2発現;(4)タキサン治療前のサイクリンB1;(5)タキサン治療前のサイクリンE発現;(6)タキサン治療前のp21発現;および(7)タキサン治療前のCDK6発現。
【0017】
さらにもう一つの態様において、本発明は、癌を有する患者の治療に関する決定を行う工程をさらに含む。なおさらなる態様において、本発明は、癌を有する患者の生存を評価する工程を含む。
【0018】
もう一つの特定の態様において、本発明は、以下の工程を含む患者における癌を治療するか、または予防する方法を提供する:a)患者の癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性に対するタキサンの効果を決定する工程;b)タキサンに対する癌の感受性を評価する工程;および、c)患者にタキサンを投与する工程。タキサンに対する癌の感受性を評価する工程は、細胞周期プロファイルを得る工程を含み、これは自動化アナライザーを使用して得てもよい。
【0019】
本発明において、乳癌、前立腺癌、皮膚癌、肺癌、頭頚部癌、膀胱癌、骨癌、骨髄癌、脳癌、大腸癌、食道癌、胃腸癌、歯肉癌、腎臓癌、肝臓癌、上咽頭癌、卵巣癌、胃癌、精巣癌、舌癌、または子宮癌などの癌が想定されるが、これらに限定されるわけでない。
【0020】
タキサン、これらの誘導体、または類似体は、静脈内もしくは腫瘍内に一回またはそれ以上投与してもよいが、このような投与の方法に、限定されるわけではない。本発明のタキサンは、化学療法または放射線療法などの抗癌治療を適用することをさらに含んでもよい。このような抗癌治療は、タキサンの前に、タキサンの後に、またはタキサンと同時に適用してもよい。抗癌治療は、さらに被検者もしくは患者に一回またはそれ以上適用してもよい。
【0021】
本明細書に記載されている任意の方法または組成物は、本明細書に記載されている他のいかなる方法または組成物に関しても実施することができることが想定される。
【0022】
「ある(one)」の語の使用は、請求項および/または明細書において「含む」という用語と共に使用された場合は、「1つ(a)または1つ(an)」を意味し得るが、「1つまたは複数」、「少なくとも1つ」、および「1つまたは複数」の意味とも一致する。
【0023】
本発明のその他の目的、特徴、および利点は、以下の詳細な説明から明らかになると思われる。しかし、当業者には、本発明の精神および範囲内の種々の変更および修飾が、この詳細な説明から明らかになると考えられるので、詳細な説明および具体例は、本発明の特定の態様を示すと共に、例示目的のみによって与えられることが理解されるはずである。
【0024】
好ましい態様の説明
I. 本発明
本発明は、癌細胞および組織のタキサン化学受容性を決定するための方法を提供する。本発明は、紡錘体形成チェックポイントにおいてタキサン感受性のために必要である分子、したがって、Cdk1またはその他の分子マーカーなどのこのチェックポイントに関与する分子が、タキサン感受性を予測する際に有用であることを決定するということを決定する。従って、本発明は、癌細胞および組織の細胞周期プロファイリングによってタキサン化学受容性を決定するか、または評価する方法を提供する。細胞周期プロファイリングは、キナーゼ活性測定値についてCDK1、CDK2、CDK4、およびCDK6などのいくつかの細胞周期分子(また、本明細書においてパラメーターともいわれる)を;並びにタンパク質発現測定値についてCDK1、CDK2、CDK4、CDK6、サイクリンB1、サイクリンD1、サイクリンE、p21/Waf1、p27/Kip1、p16、p53、およびMAD2を測定することによって行った。細胞周期プロファイリング系のパラメーターは、MAD2発現およびCDK1活性を含むM期調節機構に関与するので、本系は、タキサン感受性の有効な予測法である。
【0025】
II. タキサン
タキサンおよび関連活性成分は、イチイ属(Taxus)種の植物によって産生され、このような植物の異なる部分の成分である。タキソール(パクリタキセル)などのタキサンは、イチイ樹木から得られる環状毒性ジテルペンである。タキサンは、これらが細胞周期のG2/M期において増殖細胞を阻害するという点で、分子に基づいて細胞複製を阻害することが当技術分野において公知である。従って、タキサンは、抗腫瘍効果を有し、一連の癌腫(卵巣癌、乳癌、気管支癌、肺癌)の治療のために次第に使用されている。
【0026】
A. パクリタキセル/タキソール
パクリタキセルは、タキソールとしても知られ、ジテルペンアルカロイドであって、従ってこれは、その構造にタキサン骨格を有する。パクリタキセルは、抗癌活性を有する天然の化合物としてイチイ(タキサス・ブレビフォリア(Taxus brevifolia))の樹皮から抽出される(FuchsおよびJohnson, 1978)。パクリタキセルは、有糸分裂を妨害することによって癌に対して作用する。パクリタキセルは、タキソイド(taxoid)薬であり、原発性および転移性癌の有効な治療として広く使用されている。
【0027】
パクリタキセル(タキソール)は、乳房、卵巣、およびその他の固形腫瘍の治療に広く使用されている。無作為臨床試験では、アントラサイクリン含有補助化学療法に加えてパクリタキセルを受けた原発性乳癌である患者の間で生存優位性を示した(Eifel et al., 2001)。さらに、パクリタキセルは、転移性乳癌(Holmes et al., 1991; Nabholtz et al., 1996; Bishop et al., 1999)および進行卵巣癌(McGuire et al., 1996; Piccart et al., 2000)の両方に対して有効である。パクリタキセルの抗腫瘍活性は、これが微小管構築を促進して微小管を安定化し、このように有糸分裂を防げるために、特有である(Huizing et al., 1995)。パクリタキセルは、チューブリンのBサブユニットに、可逆的かつ特異的に結合して、微小管重合体を形成し、これによりこれらを脱重合に対して安定にし、こうして細胞周期のG2/M期における増殖停止を引き起こすことによってこれを行う(GotaskieおよびAndreassi, 1994)。これにより、微小管分解を生じさせるビンクリスチンおよびビンブラスチンと比較して、タキソールは特有となる(Gatzemeier et al., 1995)。加えて、最近の証拠は、微小管系が種々のサイトカインの放出に必須なこと、およびサイトカイン放出の調整が薬物の抗腫瘍活性において主要な役割を果たしている可能性があることを示す(Smith et al., 1995)。
【0028】
しかし、一部の患者は、パクリタキセル療法に耐性であり、薬物による利益を受ける患者の特徴は、明確ではなかった。パクリタキセル感受性または耐性を予測する分子特性の同定は、この療法を受ける患者を選択する際の助けとなり得る。従って、特定の態様において、本発明は、癌である患者のパクリタキセル感受性に関する。
【0029】
以前の報告では、パクリタキセル耐性が、Bcl-2およびBcl-XLなどの抗アポトーシスBcl-2ファミリー・メンバーのアップレギュレーション(Tang et al., 1994);薬物流出の増大を生じる膜輸送体(たとえば、mdr-1)のアップレギュレーション(Huang et al., 1997);パクリタキセル結合(Giannakakou et al., 1997)の消失を生じるβ-チューブリンの突然変異;および有糸分裂の遅延を生じるサイクリン依存性キナーゼ1(Cdk1)の阻害を介したErbB2(HER2)のアップレギュレーション(Yu et al., 1998)などの種々のメカニズムによるものであることを証明した。
【0030】
パクリタキセルの抗有糸分裂活性により、これは、いくつかの古典的な難治の腫瘍を治療する際に有用な細胞毒である。パクリタキセルは、主に乳癌および卵巣癌を治療するために使用されてきた。また、これは、頭頚部癌、カポジ肉腫および肺癌、小細胞および非小細胞肺癌を治療するために使用してもよい。また、これにより、黒色腫の経過を遅くさせてもよい。タキソール処置に対する応答率は、癌間で変化する。進行した薬物難治卵巣癌は、19〜36%の割合で、以前に治療された転移性乳癌は27〜62%で、および種々の肺癌は21〜37%で応答する。また、タキソールは、いくつかの場合に、完全な腫瘍緩解を生じることが示された(Guchelaar et al., 1994)。
【0031】
パクリタキセルは、これが接触時に皮膚および粘膜を刺激することから、静脈内に与えられる。これは、典型的には、1週あたり3回、3〜24時間の注入によって静脈内に投与される(Guchelaar et al., 1994)。
【0032】
パクリタキセル(これは、たとえばTAXOL(商標)、TAXOTERE(商標)、ドセタキセル、パクリタキセルの10-デスアセチル類似体、およびパクリタキセルの3'N-デスベンゾイル-3'-N-t-ブトキシカルボニル類似体などの製剤、プロドラッグ、類似体、並びに誘導体を含んでもよい)は、当業者に公知の技術を利用して容易に調製してもよい(たとえば、Schiff et al., 1979; LongおよびFairchild, 1994; RingelおよびHorwitz, 1991; Pazdur et al., 1993;PCT出願のWO94/07882;WO94/07881;WO94/07880;WO94/07876;WO93/23555;WO93/10076;WO94/00156;WO93/24476;EP590267;WO94/20089;米国特許第5,294,637号;米国特許第5,283,253号;米国特許第5,279,949号;米国特許第5,274,137号;米国特許第5,202,448号;米国特許第5,200,534号;米国特許第5,229,529号;米国特許第5,254,580号;米国特許第5,412,092号;米国特許第5,395,850号;米国特許第5,380,751;米国特許第5,350,866号;米国特許第4,857,653号;米国特許第5,272,171号;米国特許第5,411,984号;米国特許第5,248,796号;米国特許第5,422,364号;米国特許第5,300,638号;米国特許第5,362,831号;米国特許第5,440,056号;米国特許第4,814,470号;米国特許第5,278,324号;米国特許第5,352,805号;米国特許第5,059,699号;米国特許第4,942,184号を参照されたい);またはたとえばSigma, St. Louis, MO(Taxus brevifoliaからのT7402)を含む種々の市販の供与源から得られる。
【0033】
B. ドセタキセル/キソテール
ドセタキセルは、タキソイドファミリーに属する抗悪性腫瘍薬である。ドセタキセルは、主に乳癌、肺癌、非小細胞肺癌を治療するために使用されてきた。加えて、これは、頭頚部癌、小細胞肺癌、中皮腫、卵巣癌、前立腺癌、および尿路上皮移行細胞癌を治療するために使用してもよい。ドセタキセルは、癌細胞の増殖を妨害し、最終的にこれが破壊される。しかし、パクリタキセル療法に関して上で議論したように、一部の患者は、ドセタキセル療法に耐性である。従って、癌患者におけるこのタキサンの化学受容性を評価するか、または決定することにより、この薬剤は、癌を治療するためにより効率的に使用することができる。
【0034】
ドセタキセルは、欧州イチイ樹木(タキサス・バカタ(Taxus baccata))の針状葉から抽出される前駆体に由来する半合成の薬物である。ドセタキセルの化学名は、5b-20-エポキシ-12a,4,7b,10b,13a-ヘキサヒドロキシタクス-11-エン-9-オン 4-アセテート2-安息香酸三水和物との(2R,3S)-N-カルボキシ-3-フェニルイソセリン,N-tert-ブチルエステル,13-エステルである。これは、有糸分裂および間期の細胞機能に必須である微小管ネットワークを崩壊させることによって作用する。これは、安定な微小管内におけるチューブリンの構築およびこれらの分解の阻害を促進して、細胞分裂の阻害および最終的に細胞死を生じさせる。ドセタキセルおよびパクリタキセルは、両方とも同じ微小管部位に結合するが、ドセタキセルの親和性は、1.9倍高い。ドセタキセルとパクリタキセルとの間の交差耐性は、一貫して生じない。ドセタキセルは、放射線感受性薬である。これは、細胞周期特異的(G2/M期)である。
【0035】
C. その他のタキサン
また、本発明は、抗癌活性を有するタキサン類似体もしくは誘導体を含む、任意のタキサンまたは当技術分野において当業者に周知であるタキサン骨格を有する化合物の化学受容性を試験することを想定する。「タキサン化合物」は、タキソール、タキソールと構造的に類似する化合物、および/またはタキソールの類似体を含んでもよい。また、「タキサン化合物」は、「擬態」を含んでもよい。「擬態」には、タキソールと構造的に類似しないが、インビボでタキソールまたは構造的に類似のタキサン化合物の治療活性を模倣する化合物を含むことが企図される。本発明のタキサン化合物は、癌を有する被検者(患者)における腫瘍成長を阻害するために有用である化合物である。また、タキサン化合物という用語は、化合物の薬学的に許容される塩を含むことが企図される。タキサン化合物は、米国特許5,641,803号、米国特許第5,665,671号、米国特許第5,380,751号、米国特許第5,728,687号、米国特許第5,415,869号、米国特許第5,407,683号、米国特許第5,399,363号、米国特許第5,424,073号、米国特許第5,157,049号、米国特許第5,773,464号、米国特許第5,821,263号、米国特許第5,840,929号、米国特許第4,814,470号、米国特許第5,438,072号、米国特許第5,403,858号、米国特許第4,960,790号、米国特許第5,433,364号、米国特許第4,942,184号、米国特許第5,362,831号、米国特許第5,705,503号、米国特許第5,278,324号、米国特許第5,840,929号、米国特許第5,773,464号、米国特許第5,248,796号、米国特許第5,821,263号、米国特許第4,814,470号、米国特許第5,438,072号、米国特許第4,960,790号、米国特許第4,942,184号、米国特許第5,433,364号、米国特許第5,278,324号、米国特許第6,362,217号、米国特許第6,017,935号、米国特許第5,977,376号、米国特許第5,912,264号、米国特許第5,773,464号、米国特許第5,739,539号、米国特許第5,698,712号、米国特許第6,284,746号;米国特許出願第20030144344号、米国特許出願第20030130341号、米国特許出願第20030134793号、米国特許出願第20030130170号、米国特許出願第20030130178号、米国特許出願第20030124055号、および米国特許出願第20020016356号;並びにPCT出願のWO95/33740、WO96/03394、WO95/33736、WO93/02067、WO94/15929、およびWO94/15599に以前に記載されており、これらの全てが参照として本明細書に組み入れられる。
【0036】
その他のタキサンは、ポリグルタミン酸、ポリアスパラギン酸、またはポリリジンなどの水溶性重合体にパクリタキセルまたはドセタキセルを抱合することによって形成されたパクリタキセルおよびドセタキセルの水溶性組成物を含んでもよい(米国特許出願第20030147807号)。パクリタキセルの誘導体は、様々な程度の薬理活性を有する。このようなパクリタキセル誘導体化合物の合成および評価における研究では、ヒトを含む温血動物の癌の治療のために有用な、安全で、便利で、有効な薬物製剤を開発するための試みがなされてきた。パクリタキセルの発見以来、関連した構造を有する100を超える化合物が種々のイチイ属種から単離され、および/または合成的に作製されてきた。
【0037】
望ましい抗腫瘍特性を有する一つの例示的なパクリタキセル誘導体は、タキサン環のC7位が構造的にパクリタキセルと異なる化合物、7-O-メチルチオメチルパクリタキセル(本明細書において「7-O-MTMパクリタキセル」と称する)である。7-O-MTMパクリタキセルは、現在臨床試験で研究中の公知の抗腫瘍薬である。7-O-MTMパクリタキセルに関する研究では、パクリタキセルがあまり有効ではないことが見いだされた胃腸癌および結腸直腸癌の治療において有望な結果を示した。7-O-MTMパクリタキセルは、合成法によって生成できることが公知である(米国特許第5,646,176号およびWO96/00724;いずれの内容も、それぞれ参照として本明細書に組み入れられる)。
【0038】
本発明において想定されるパクリタキセル誘導体または類似体の代表例は、以下のものを含んでもよいが、これらに限定されるわけではない:7-デオキシ-ドセタキソール、7,8-シクロプロパタキサン、N置換2-アゼチドン、6,7-エポキシパクリタキセル、6,7-修飾パクリタキセル、10-デスアセトキシタキソール、10-デアセチルタキソール(10-デアセチルバッカチンIII由来)、タキソールのホスホノオキシおよびカルボナート誘導体、タキソール2',7-ジ(ナトリウム1,2-ベンゼンジカルボキシラート、10-デスアセトキシ-11,12-ジヒドロタキソール-10,12(18)-ジエン誘導体、10-デスアセトキシタキソール、プロタキソール(2'-および/または7-O-エステル誘導体)、(2'-および/または7-O-カルボナート誘導体)、タキソール側鎖の不斉合成、フッ化タキソール、9-デオキソタキサン、(13-アセチル-9-デオキソバッカチンIII、9-デオキソタキソール、7-デオキシ-9-デオキソタキソール、10-デスアセトキシ-7-デオキシ-9-デオキソタキソール;水素またはアセチル基並びにヒドロキシおよびtert-ブトキシカルボニルアミノを含む誘導体、スルホン化2'-アクリロイルタキソールおよびスルホン化2'-O-アシル酸タキソール誘導体、スクシニルタキソール、2'-γ-アミノブチリルタキソールホルマート、2'-アセチルタキソール、7-アセチルタキソール、7-グリシンカルバメートタキソール、2'-OH-7-PEG(5000)カルバメートタキソール、2'-ベンゾイルおよび2',7-ジベンゾイルタキソール誘導体;その他のプロドラッグ(2'-アセチルタキソール;2',7-ジアセチルタキソール;2'スクシニルタキソール;2'-(β-アラニル)-タキソール);2'γ-アミノブチリルタキソールホルマート;2'-スクシニルタキソールのエチレングリコール誘導体;2'-グルタリルタキソール;2'-(N,N-ジメチルグリシル)タキソール;2'-(2-(N,N-ジメチルアミノ)プロピオニル)タキソール;2'オルトカルボキシベンゾイルタキソール;タキソールの2'脂肪族カルボン酸誘導体;プロドラッグ{2'(N,N-ジエチルアミノプロピオニル)タキソール、2'(N,N-ジメチルグリシル)タキソール、7(N,N-ジメチルグリシル)タキソール、2',7-ジ-(N,N-ジメチルグリシル)タキソール、7(N,N-ジエチルアミノプロピオニル)タキソール、2',7-ジ(N,N-ジエチルアミノプロピオニル)タキソール、2'-(L-グリシル)タキソール、7-(L-グリシル)タキソール、2',7-ジ(L-グリシル)タキソール、2'-(L-アラニル)タキソール、7-(L-アラニル)タキソール、2',7-ジ(L-アラニル)タキソール、2'-(L-ロイシル)タキソール、7-(L-ロイシル)タキソール、2',7-ジ(L-ロイシル)タキソール、2'-(L-イソロイシル)タキソール、7-(L-イソロイシル)タキソール、2',7-ジ(L-イソロイシル)タキソール、2'-(L-バリル)タキソール、7-(L-バリル)タキソール、2',7-ジ(L-バリル)タキソール、2'-(L-フェニルアラニル)タキソール、7-(L-フェニルアラニル)タキソール、2',7-ジ(L-フェニルアラニル)タキソール、2'-(L-プロリル)タキソール、7-(L-プロリル)タキソール、2',7-ジ(L-プロリル)タキソール、2'-(L-リジル)タキソール、7-(L-リジル)タキソール、2',7-ジ(L-リジル)タキソール、2'-(L-グルタミル)タキソール、7-(L-グルタミル)タキソール、2',7-ジ(L-グルタミル)タキソール、2'-(L-アルギニル)タキソール、7-(L-アルギニル)タキソール、2',7-ジ(L-アルギニル)タキソール};修飾フェニルイソセリン側鎖をもつタキソール類似体、タキソテール、(N-デベンゾイル-N-tert-(ブトキシカロニル)-10-デアセチルタキソール、およびタキサン(たとえば、バッカチンIII、セファロマンニン(cephalomannine)、10-デアセチルバッカチンIII、ブレビフォリオール(brevifoliol)、ユナンタクスシン(yunantaxusin)、およびタクスシン(taxusin));並びに以下のものを含むその他のタキサン類似体および誘導体:14-β-ヒドロキシ-10バッカチンIII、デベンゾイル-2-アシルパクリタキセル誘導体、安息香酸パクリタキセル誘導体、ホスホノオキシおよびカルボナートパクリタキセル誘導体、スルホン化2'-アクリロイルタキソール;スルホン化2'-O-アシル酸パクリタキセル誘導体、18位置換パクリタキセル誘導体、塩素化パクリタキセル類似体、C4メトキシエーテルパクリタキセル誘導体、スルフェンアミドタキサン誘導体、ブロム化パクリタキセル類似体、ジラールタキサン誘導体、ニトロフェニルパクリタキセル、10-デアセチル化置換パクリタキセル誘導体、14-β-ヒドロキシ-10デアセチルバッカチンIIIタキサン誘導体、C7タキサン誘導体、C10タキサン誘導体、2-デベンゾイル-2-アシルタキサン誘導体、2-デベンゾイルおよび-2-アシルパクリタキセル誘導体、新たなC2およびC4官能基を有するタキサンおよびバッカチンIII誘導体、n-アシルパクリタキセル類似体、10-デアセチルタキソールA由来の10-デアセチルバッカチンIIIおよび7-保護10-デアセチルバッカチンIII誘導体、10-デアセチルタキソールB、および10-デアセチルタキソール、タキソールの安息香酸の誘導体、2-アロイル-4-アシルパクリタキセル類似体、オルト-エステルパクリタキセル類似体、2-アロイル-4-アシルパクリタキセル類似体、並びに1-デオキシパクリタキセルおよび1-デオキシパクリタキセル類似体。
【0039】
III. 細胞周期プロファイリングのための試料の取得
本発明は、細胞、組織、または器官試料などの試料を得ることを想定する。本発明の試料は、いくつかの手段によって患者から得てもよい。たとえば、本発明の細胞、器官、または組織試料は、生検によって得てもよい。生検は、体から試料を除去することである。本発明に使用してもよい生検は、パンチ生検または針生検を含むが、このようなものに限定されるわけではない。細胞周期分子を含む試料は、当技術分野において公知の任意の方法によって得てもよい。血液または血清試料は、静脈穿刺を使用して収集してもよい。この方法を使用して、血液が単一の静脈に配置された針を介して個体の腕の血管から直接引き出される。次いで、血液をガラスまたはプラスチックチューブに収集してもよい。
【0040】
A. パンチ生検および錐体生検
本発明は、癌試料などの試料を得るための、パンチまたは錐体生検の使用を想定する。パンチ生検は、典型的には皮膚発疹、ほくろ、頚部の小さな組織試料、およびその他の小さな質量の試料を得るために使用される。局所麻酔薬を注射した後、生検パンチ(3mm〜4mmまたは直径0.15インチ)を使用して円柱形部分を皮膚から切断する。開口部は、典型的には縫合で閉じられて、最小限の瘢痕で回復する。
【0041】
一方で、錐体生検は、円柱形または円錐形状である組織を得るために使用される。錐体生検の利点は、これが解析のための大量の組織試料を提供することである。
【0042】
B. 針生検
1. コア針生検
コア針生検(またはコア生検)は、皮膚を通して器官内に小さな中空針を挿入することによって行われる。次いで、針を細胞層に進めて、試料またはコアを除去する。針は、組織試料を除去するのを補助するために、切断先端に設計してもよい。コア生検は、組織試料を除去するのを補助するために、バネを充填した銃を使用して行われることが多い。
【0043】
コア生検は、典型的にはCTイメージング、超音波、または乳房X線撮影などのイメージ・ガイダンス下で行われる。針は、手で、またはサンプリングデバイスの助けを借りて配置する。十分な組織を得るために、複数の挿入が行われることが多く、複数の試料が採取される。組織試料が採取される場合に、サンプリング機器からクリック音が聞こえる場合がある。
【0044】
コア生検は、真空装置で援助された吸引の場合もある(真空補助生検)。この方法は、1つの針挿入だけで複数の試料を除去することができる。コア生検とは異なり、真空補助生検プローブが、ただ一回、小さい皮膚ニックを介して組織内に挿入される。次いで、サンプリング針開口(開口部)の回転を使用して、吸引の助けを借りて、複数の試料が採取される。従って、本発明において、組織試料を得るために、コア針生検または真空補助針生検を使用してもよい。
【0045】
2. 吸引/細針吸引(FNA)生検
吸引生検法は、細針穿刺吸引(FNA)とも称され、注射器に取り付けた細針で行われる。吸引生検またはFNAは、本発明において癌試料を得るために使用してもよい。FNA生検は、経皮的(皮膚を介した)生検である。FNA生検は、典型的には細いゲージ針(22ゲージまたは25ゲージ)で達成される。領域を最初に洗って、次いで通常局所麻酔薬で麻痺させる。針は、関心対象の器官または組織の領域に入れられる。一旦針が配置されたら、注射器で真空を生じさせ、繰り返し針を出し入れする動作を行う。サンプル採取される細胞は、細針を介して注射器に吸い込まれる。通常、3つまたは4つの試料が作製される。
【0046】
膵臓、肺、および肝臓などの容易に到達されない器官は、FNAの優れた候補である。FNA方法は、典型的には超音波またはコンピュータ断層撮影法(CT)イメージングを使用してなされる。
【0047】
C. 内視鏡生検
内視鏡生検は、本発明において癌試料を得るために使用してもよい生検のうちの非常に一般的なタイプである。内視鏡生検は、サンプリング機器と共に体に挿入される内視鏡(体内を見るための光ファイバーケーブル)を介してなされる。内視鏡は、関心対象の器官の裏打ち上の領域を直接可視化すること;試料の内視鏡内部を走る長いケーブルに取り付けられた鉗子で組織の極めて小さな小片を収集し、またはつまみとることができる。内視鏡生検は、消化管(消化管内視鏡検査)、膀胱(膀胱鏡検査)、腹腔(腹腔鏡検査)、関節腔(関節鏡検査)、胸部の中間部分(縦隔鏡)、または気管および気管支系(喉頭鏡検査および気管支鏡検査法)で、天然の体の開口部か、もしくはある小さな手術切開を介して、いずれかで行ってもよい。
【0048】
D. 表面生検
本発明において癌試料を得るために、表面生検を使用してもよい。この技術は、細胞を除去するために、組織もしくは器官の表面のサンプリングすること、または削ることを含む。表面生検は、皮膚のわずかな部分を除去するために行われることが多い。
【0049】
IV. 細胞周期分子(パラメーター)
タキサン化学受容性を評価または決定する際に、本発明は、患者の癌細胞における細胞周期分子もしくは因子の発現レベルおよび活性を評価または決定する。
【0050】
主に、細胞周期制御因子の2つの群が、細胞に存在する。一方は、正の制御因子であるサイクリン依存性キナーゼ(CDK)と称されるキナーゼ群であり、他方は、負の制御因子であるCDK阻害剤(CDKI)群である。CDKは、細胞質に不活性型として存在する。CDKは、活性化されて、たとえばリン酸化されて、細胞の核へ移動する。核では、CDKがサイクリン分子と結合して、サイクリンと複合体を形成し(本明細書において、活性化されたCDKと称する)、細胞周期の種々の段階で細胞周期の進行を正に調節する。一方で、CDKIは、活性化されたCDKまたはCDK単体に結合することによってCDKを不活性化し、これにより負に細胞周期を調節する。
【0051】
CDK1キナーゼは、有糸分裂サイクリンと合わせて、有糸分裂の制御のために必要とされる普遍的マスター・キナーゼであって、パクリタキセルが攻撃するキナーゼである。このキナーゼは、細胞周期のG2期に活性化されて、有糸分裂初期において核膜の破壊、クロマチンの凝縮、および紡錐体装置の形成を促進する。対照的に、中期を出るためには、APC-プロテアソーム経路によって、その構築パートナーのサイクリンBの分解を介してCDK1が不活性化されなければならない。CDK1活性は、紡錘体が動原体に完全に付着されるまで、高いままである。
【0052】
A. サイクリン依存性キナーゼ(CDK)
異なるサイクリンが結合するCDKの7つのタイプ、すなわちCDK1、CDK2、CDK3、CDK4、CDK5、CDK6、およびCDK7が公知である。より詳細には、CDK1は、サイクリンAまたはBと結合し、CDK2は、サイクリンAまたはEと結合し、CDK4およびCDK6は、サイクリンD1、D2、またはD3と結合して活性化される。活性化されたCDKは、細胞周期の特定の期を制御する。従って、細胞周期が制御されて、細胞増殖は、異なるタイプのCDKの活性化によって調節される。活性化されたCDKは、基質としてタンパク質のセリン残基およびスレオニン残基をリン酸化する。インビトロ反応系では、活性化されたCDK1およびCDK2が、基質としてヒストンH1に対して十分に反応し、および活性化されたCDK4およびCDK6が、基質としてRb(網膜芽細胞腫タンパク質)に対して十分に反応する。インビボでの細胞周期制御では、活性化されたCDKは、生理的基質としてRbを必要とすることが考えられるが、その他のどのタンパク質が基質として作用するかは公知でない。
【0053】
上述のとおり、CDKおよびサイクリンは、互いに密接に会合して細胞周期を調節する。サイクリンD1遺伝子の増殖は、食道癌の多くの場合に観察されるが、一方で、サイクリンD1遺伝子の過剰発現は、胃癌および大腸癌の多くの場合に観察される。一方、サイクリンE遺伝子の増殖は、胃癌および大腸癌において観察されるが、食道癌では観察されない。胃および大腸におけるサイクリンEの過剰発現は、腺腫および腺癌の場合に高頻度で生じ、浸潤、段階の進行、転移等の悪性度と有意な相関を示す。CDK1の発現およびキナーゼ活性は、正常な粘膜組織と比較して、胃癌および大腸癌の大部分の場合に著しく促進される。サイクリン遺伝子の発現が増大されることは、種々の癌の進行および悪性度と相関することが公知である(Wataru et al., 2000を参照されたい)。
【0054】
従って、CDKの個体種の発現レベルもしくは活性を評価するか、決定するか、または測定することにより、タキサンに対する患者の癌細胞の細胞周期プロファイリングを提供し、これにより癌を予測するかまたは診断することが考えられる。言い換えると、一般にR位では、CDK2の発現が減少して、細胞周期停止および細胞の分裂が制御されている。しかし、CDK2の発現が、R位で増大する場合には、細胞周期が止まることができないことを意味し、すなわち、癌などの疾患の状態を意味する。
【0055】
通常、CDKの活性は、放射性同位元素を使用して決定される。より詳細には、抗CDK抗体を使用する免疫沈降法によって細胞可溶化物から抽出され、かつその活性が未知であるCDKの存在下において、32P標識されたアデノシン5'-O-(3-三リン酸)(ATP)を基質中でセリン残基またはスレオニン残基と反応させて、32P標識されたATPに由来する一リン酸基を導入する。基質によって取り込まれた32Pの量をオートラジオグラフィーによって、またはシンチレーションカウンターによって検出する。これにより、リン酸化された基質の量を測定して、CDKの活性をリン酸化された基質の量から算出する。
【0056】
B. CDK阻害剤(CDKI)
p16INK4は、p16B、p19、p21WAF1、およびp27KIP1をも含むCDK阻害性タンパク質の新たに記載されたクラスに属する。p16INK4遺伝子は、9p21にマップし、多くの腫瘍型において染色体領域が頻繁に欠失されている。p16INK4遺伝子のホモ接合性欠失および突然変異は、ヒト腫瘍株に頻繁に存在する。これは、p16INK4遺伝子が、癌抑制遺伝子であることを示唆した。しかし、この解釈は、p16INK4遺伝子変化の頻度が、培養された株化細胞におけるよりも、初代無培養腫瘍において非常に低いという知見によって疑いがもたれていた(Caldas et al., 1994;Cheng et al., 1994;Hussussian et al., 1994;Kamb et al., 1994a;Kamb et al., 1994b;Mori et al., 1994;Okamoto et al., 1994;Nobori et al., 1995;Orlow et al., 1994;Arap et al., 1995)。プラスミド発現ベクターでトランスフェクションを行うことによる野生型p16INK4機能の回復により、いくつかのヒト癌株化細胞によるコロニー形成が減少した(Okamoto, 1994;Arap, 1995)。
【0057】
細胞増殖のもう一つの阻害剤は、p16である。真核生物の細胞周期の主な移行は、サイクリン依存性キナーゼ、またはCDKによってトリガーされる。1つのCDK、サイクリン依存性キナーゼ4(CDK4)は、G1を介して進行を調節する。この酵素の活性は、後期のG1にてRbをリン酸化することである可能性がある。CDK4の活性は、サブユニットD型サイクリンを活性化することによって、および阻害性サブユニットによって制御されており、p16INK4は、CDK4に特異的に結合して阻害し、したがって、Rbリン酸化を調節し得るタンパク質として生化学的に特徴づけられた(Serrano et al., 1993;Serrano et al., 1995)。p16INK4タンパク質は、CDK4阻害剤であるので(Serrano, 1993)、この遺伝子の欠失は、CDK4の活性を増大して、Rbタンパク質の過剰リン酸化を生じ得る。また、p16は、CDK6の機能を調節することが公知である。
【0058】
p21WAF1/CIP1は、最初にp53誘導性タンパク質として同定された。p53の減少は、腫瘍細胞における共通の現象であり、放射線療法または化学療法に対する細胞の反応に変化を生じさせる。p53を媒介した細胞周期チェックポイントを欠いた細胞は、ますます遺伝的に不安定で、アポトーシスになりにくくなる。DNA損傷に応答して、p21は、種々のサイクリン/CDK複合体と結合してこれらの複合体の活性を阻害し、p53依存的様式で細胞周期進行の抑制を生じることが証明された。最近では、p21Waf1/CIP1のアップレギュレーションをp53の他のその他のシスまたはトランス・エレメントによって達成することができることも示された。
【0059】
C. 紡錘体形成チェックポイント分子
有糸分裂紡錘体形成チェックポイントでは、未熟な染色体の分裂を防止するために、紡錘体に対する染色体の付着および微小管によって作製される姉妹染色分体全体の緊張の両方をモニターする。パクリタキセルなどの薬物が、微小管を安定化して紡錘体の形成の間に生じる動的変化を妨害する場合に、紡錘体形成チェックポイントが活性化されて、有糸分裂で細胞停止される。
【0060】
紡錘体形成チェックポイントの分子構成成分は、最初に酵母(Saccharomyces cerevisiae)において同定された。チェックポイント・タンパク質の哺乳類相同体は、Mad1、Mad2、BubR1、Bub3、およびMps1を含む(LiおよびBenezra, 1996;Jin et al., 1998;Taylor et al., 1998;Chan et al., 1999)。チェックポイント機構は、Mad1、Mad2、BubR1、Bub3、およびcdc20で構成されたタンパク質複合体であり、染色体の動原体に位置する。このチェックポイントの標的は、後期促進複合体(APC)およびその活性化補助因子Cdc20である。Mad2およびBubR1は、下流に位置し、この機構の主要タンパク質であると思われ、Cdc20と直接相互作用して、APC活性を協同的に阻害する(Fang et al., 1998; Sudakin et al., 2001; Tang et al., 2001; Fang, 2002)。腫瘍細胞では、チェックポイントの欠陥が観察されることが多く、これらはゲノム不安定化を誘導すると考えられている。
【0061】
V. 細胞周期パラメーターの発現または活性を決定する方法
A. 細胞周期プロファイリング系
タキサン化学受容性に関与する細胞周期分子を同定するために、本発明は、細胞周期プロファイリングのために多パラメーター解析を使用する。この系は、ドットブロット技術に基づいた、日本特許出願第200348653号(その全体が本明細書に組み入れられる)に記載されている装置を使用して、同位元素を用いずに正常および癌組織の小さな臨床試料におけるタンパク質発現、並びに活性を迅速に、定量的に、かつ自動的にアッセイし得る。CDKの活性およびその他の種類のタンパク質の発現を測定するためには、2mm3だけの組織試料で十分である。この細胞周期プロファイリング系/装置は、多くの疾患の診断の際の、または分子ターゲティング療法を含む種々の療法に対する予後もしくは患者の感受性を評価する際の、その他のキナーゼのまたはタンパク質の活性の測定など種々の適用可能性を有する。また、この装置/系は、個々の疾患に対する危険因子の診断のために使用してもよい。
【0062】
1. タンパク質発現を測定する方法
本発明に使用されるアッセイ法プロトコルの原理を下記に簡単に説明してある。細胞周期分子(パラメーター)のタンパク質発現は、CPDIBという名で(粗タンパク質直接ブロッティング、米国出願第10/423,892号;その全体が参照として本明細書に組み入れられる)かつドットブロット技術に基づいた新たな技術によって測定してもよい。解析は、3工程だけを含む:疎水性膜に対する粗製細胞可溶化物の直接固定化、一次抗体の反応、および結合した一次抗体の検出。たとえば、ビオチン化二次抗体およびフルオレッセイン標識されたストレプトアビジンの逐次反応による。合計アッセイ時間は、3時間以内である。相対的蛍光単位と標準的な組換えタンパク質の量とは、直線的関連があることが確かめられる。MAD2のためのアッセイ条件は、現在最適化されている。本系は、1つの標的分子に対する1つの特異的なポリクローナル抗体が必要なだけであるので、本系の主な利点は、プロファイリングのための新たなパラメーターを追加することが、新たなサンドイッチELISA系を開発するよりも非常に容易であることである。
【0063】
自動化解析のための活性および発現の両方の測定手順が開発された。外科的に切開した組織の小片(2mm3)の可溶化液は、新しく開発された組織ホモジナイザー(Sysmex, Kobe, Japan)を使用して溶解緩衝液(0.1%のNP-40、20 mM トリス-HCl[pH 7.4]、150 mM NaCl、2% プロテイナーゼ阻害剤カクテル(Sigma, St Louis, MO)で調製してもよい。ホモジナイザーは、フィルターディスク上の不溶性物質を自動的に除去する。タンパク質濃度を解析し(DCキット, Pierce, Rockford, IL)、2.5μgの総タンパク質量を新しく開発した疎水性膜(0.22μm孔を有するPVDF;Millipore, Billerica, MA)を有する5×7cm2ドットブロット装置(Sysmex)の78μlウェル(3mm[w]×5mm[l]×7mm[d])に加える。膜に結合した粗試料中の標的タンパク質は、抗CDK抗体(Santa Cruz Biotechnology, Santa Cruz, CA)、ビオチン化二次抗体(Santa Cruz Biotechnology)、およびフルオレッセイン標識されたストレプトアビジン(Vector, Burlingame, CA)と連続して反応させることによって定量的に検出してもよい。それぞれの反応の間に、ウェルがTBS溶液(25mMのトリス-HCl[pH 7.4]、150 mM NaCl)で自動的に洗浄される。膜の蛍光イメージをイメージアナライザー(Bio-Rad, Hercules, CA, USA)を使用して解析し、ドットの強度を「Quantity One」(Bio-Rad)によって定量化する。相対的蛍光ユニット(RFU)および標準的な組換えタンパク質(Santa Cruz Biotechnology)の量を、標準化された範囲内で直線的に相関させる(たとえば、CDK1、2.5〜25ng/ドット;CDK2、1.0〜10ng/ドット;CDK4、1.0〜10ng/ドット;CDK6、2.5〜25ng/ドット)。
【0064】
2. キナーゼ活性の測定
CDKの酵素アッセイのために、非放射同位元素CDKアッセイ法を使用した(米国特許出願第20020164673号)。本アッセイ法は、以下の工程を含む:対応する抗体(たとえば、抗CDK1、抗CDK2、抗CDK4、または抗CDK6抗体)とプロテインAビーズとによるCDK分子の沈澱。基質のセリンまたはスレオニン残基にモノチオリン酸エステル基を導入するために、タンパク質基質およびアデノシン5'-O-(3-チオ三リン酸エステル)(ATP-γS)のキナーゼ緩衝液の溶液を含む基質混合物を反応させる工程(タンパク質基質として、CDK1およびCDK2のためにはヒストンH1を、並びにCDK4およびCDK6のためには組換えRBタンパク質(アミノ酸769〜921)を使用してもよい)。標識化フルオロフォアまたは標識化酵素を、導入されたモノチオリン酸エステル基の硫黄原子と結合することによって基質を標識する工程。基質を標識する標識化フルオロフォアから蛍光の量を測定する工程か、または基質を標識する標識化酵素を、標識化酵素との反応によって光学的に検出可能な生成物を生じる物質と反応させて、生じる生成物の量を光学的に測定する工程。測定された蛍光の量または測定された生じた生成物の量からサイクリン依存性キナーゼの活性を、予め作製した参照曲線を基準として算出する工程。
【0065】
細胞可溶化物は、発現解析について記載したとおりに調製される。CDK分子を、2μgの対応する抗体(抗CDK1抗体、抗CDK2抗体、抗CDK4抗体、または抗CDK6抗体;Santa Cruz Biotechnology)および20μlのプロテインAビーズ(Amersham Pharmacia, Uppsala, Sweden)で、100μgの可溶化液総タンパク質から、4℃で1時間選択的に沈殿させる。洗浄緩衝液(0.1%のNP-40、50 mM トリス-Cl[pH 7.4])で3回洗浄後、10μgのタンパク質基質、5mMアデノシン5'-O-(3-チオ三リン酸エステル)(Sigma)、20mM、トリス-Cl(pH 7.4)、および0.1%のトリトンX-100を含む50μlの基質混合物をビーズに添加して、連続的振盪下で37℃において10分間インキュベートする。タンパク質基質として、CDK1およびCDK2のためにはヒストンH1(Upstate Biotechnology, Lake Placid, NY)を、CDK4およびCDK6のためには組換えRBタンパク質(アミノ酸769〜921)を使用してもよい。ビーズを除去した後、基質に導入されたモノチオリン酸エステルを、10mMのヨードアセチル-ビオチン(Pierce)と共に、結合緩衝液(100mMのトリス-Cl[pH 8.5] ,1 mM EDTA)中で、暗がりで室温において90分間インキュベーションすることによってさらに標識する。反応をβ-メルカプトエタノールで停止して、0.4μgの基質をドットブロット装置(Sysmex)のウェルに加える。ウェルを4%のウシ血清アルブミン(BSA)で30分間ブロッキングし、次いで37℃において1時間アビジン-FITC(Vector)と共にインキュベートした。膜を洗浄した後、イメージアナライザー(Bio-Rad)を使用してイメージを評価し、ドットの蛍光強度を「Quantity One」(Bio-Rad)を使用して定量化する。癌株化細胞から抽出された0μg、12.5μg、25μg、50μg、100μg、および150μgのタンパク質に対応するCDK活性で調製した検量線によって活性を算出する。1ユニット(U)は、細胞からの1μgの総タンパク質量のキナーゼ活性に匹敵する。
【0066】
VI. 薬学的組成物、送達、および治療の計画
本発明の態様において、癌を有する患者のタキサン化学受容性の評価に基づいて、効率的に癌を治療および予防する方法が想定される。治療のために想定される癌の例は、肺癌、頭頚部癌、乳癌、膵癌、前立腺癌、腎癌、骨癌、精巣癌、子宮頚癌、胃腸癌、リンパ腫、肺における新生物発生前の病変、大腸癌、黒色腫、膀胱癌、および他の任意の腫瘍疾患も含む。
【0067】
薬学的タキサン組成物の有効な量は、一般に、検出可能な程度に、かつ繰り返し、疾患、またはこれらの症状もしくは徴候の範囲を改善するか、減少させるか、最小化するか、または制限するために十分なその量として定義される。疾患の除去、根絶、または治癒を含むより厳密な定義を適用してもよい。好ましくは、患者は、十分な骨髄機能(>2,000/mm3の末梢絶対顆粒球数および100,000/mm3の血小板数として定義される)、十分な肝機能(ビリルビン<1.5mg/dl)および十分な腎機能(クレアチニン<1.5mg/dl)を有する。
【0068】
A. タキサン投与
本発明の方法および組成物を使用して細胞を死滅させ、細胞増殖を阻害し、転移を阻害し、腫瘍または組織サイズを減少し、およびそうでなければ、腫瘍細胞の悪性の表現型を逆転し、または減少させるためには、一般にタキサン化合物と癌細胞を接触させることが考えられる。投与の経路は、必然的に、病変の位置および性質と共に変化し、たとえば、皮内、経皮、非経口的、静脈内、筋肉内、鼻腔内、皮下、経皮的、気管内、腹腔内、腫瘍内、灌流、洗浄液、直接注射、並びに経口投与および製剤を含む。また、癌の治療または診断に関して議論した任意の製剤および投与経路を、腫瘍性疾患および症状に関して使用してもよい。
【0069】
腫瘍内注射、または腫瘍脈管構造への注射は、具体的には、分離した、固体の、アクセスできる腫瘍について想定される。また、局部的、局所的、または全身投与が適切である場合がある。>4cmの腫瘍については、投与される量は、約4〜10ml(好ましくは10ml)であるが、一方、<4cmの腫瘍については、約1〜3ml(好ましくは3ml)の量が使用される。単一用量として送達される複数の注射では、約0.1〜約0.5mlの量を含む。ウイルス粒子は、好都合なことに、腫瘍に対して、約1cmの間隔で間隔を置いて、複数の注射を投与することによって接触してもよい。
【0070】
手術介入の場合、本発明を術前に使用して、手術不能の腫瘍を切除に供してもよい。または、本発明は、手術時に、および/またはその後に、残留するかもしくは転移性の疾患を治療するために使用してもよい。たとえば、切除した腫瘍ベッドを、タキサン化合物を含む製剤で注射または灌流してもよい。灌流は、たとえば、カテーテルを手術部位に移植したままにしておくことによって、切除後に続けてもよい。また、周期的な術後治療も考えられる。
【0071】
必要に応じて、たとえば、腫瘍が切除されており、腫瘍ベッドが残留する顕微鏡学的疾患を除去するための治療をする場合、連続投与を適用してもよい。注射器またはカテーテル処置(catherization)を経た送達が好ましい。このような連続灌流は、治療の開始に続いて少なくとも約1〜2時間、約2〜6時間、約6〜12時間、約12〜24時間、約1〜2日、約1〜2週の期間行ってもよい。一般に、連続灌流を経たタキサン組成物の投与は、一回または複数回の注射によって与えられるものに匹敵し、灌流が生じる期間を通して調節される。本発明のタキサン組成物を投与するために肢灌流を使用してもよいことが、さらに想定される。
【0072】
治療計画は、さらに変更してもよく、たいてい腫瘍型、腫瘍位置、疾患進行、並びに患者の健康および年齢に依存する。明らかであるが、一定タイプの腫瘍は、より攻撃的な治療を必要とするが、一方で同時に、一定の患者は、より負担の重いプロトコルを許容することができない。臨床家は、細胞周期プロファイリング並びにタキサン製剤の公知の有効性および毒性(あるとしても)によるタキサン化学受容性の評価に基づいて、このような決定を行うことが最適である。
【0073】
ある態様において、治療される腫瘍は、少なくとも最初に、切除可能でなくてもよい。治療組成物での治療により、縁が収縮することにより、または一定の特に浸潤部分を除去することによって、腫瘍の切除可能性を増大してもよい。治療に続いて切除が可能である場合がある。切除後の後処置は、腫瘍部位の顕微鏡学的な残留疾患を排除するのに役立つと考えられる。
【0074】
典型的な治療の経過は、原発腫瘍または切除後腫瘍ベッドに対する多回投与を含む。典型的な原発腫瘍治療は、2週間にわたって6用量の適用を含む。2週の処方計画を1回、2回、3回、4回、5回、6回またはそれ以上繰り返してもよい。治療経過の間に、予定の投薬を完了する必要性を再評価してもよい。
【0075】
治療は、種々の「単位用量」を含んでもよい。単位用量は、タキサン組成物の予定された量を含むものとして定義される。投与される量、並びに特定の経路および製剤は、臨床分野の当業者の範囲内である。単位用量は、単回注射として投与される必要はなく、セット期間を通した連続的注入を含んでもよい。
【0076】
B. 注射剤組成物および製剤
本発明の癌細胞に対するタキサン組成物の送達のための好ましい方法は、全身的であるか、または腫瘍内注射を介する。しかし、本明細書に開示された薬学的組成物は、代わりに、米国特許第5,543,158号;米国特許第5,641,515号、および米国特許第5,399,363号(それぞれ、その全体が参照として本明細書に具体的に組み入れられる)に記載されているように、非経口的、静脈内、皮内、筋肉内、経皮的、またはさらに腹腔内に投与してもよい。
【0077】
タキサン組成物の注射は、化合物が、注射のために必要とされる針の特定のゲージを貫通することができる限り、注射器によって、または溶液の注射のために使用される他の任意の方法によって送達してもよい。溶液を保持するためのアンプル・チャンバーと、ノズルから送達部位に溶液を押し出すためのエネルギー装置とを定義するノズルを有する新規の針のない注射システムが記載されている(米国特許第5,846,233号)。また、任意の深度に正確に溶液の予め定められた量の複数回の注射が可能な、遺伝子療法に使用するための注射器系も記載されている(米国特許第5,846,225号)。
【0078】
遊離塩基または薬理学的に許容される塩としての活性化合物の溶液は、ヒドロキシプロピルセルロースなどの界面活性物質と最適に混合した水溶液に調製してもよい。また、分散液は、グリセロール、液体ポリエチレングリコール、およびこれらの混合物の溶液に、並びに油に調製してもよい。通常の貯蔵および使用条件下では、これらの標品は、微生物の増殖を防止するための保存剤を含む。注射用に使用するために適した剤型は、無菌の水溶液または分散液、および無菌の注射用の溶液または分散液の即座の調製のための無菌の粉末を含む(米国特許第5,466,468号、その全体が参照として具体的に本明細書に組み入れられる)。全ての場合において、形態は、無菌でなければならず、かつ簡単に注射可能(syringability)である程度に流動性でなければならない。これは、製造および貯蔵条件下で安定でなければならず、かつ細菌および真菌などの微生物の混入作用を避けて保存されなければならない。担体は、たとえば水、エタノール、ポリオール(たとえば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、これらの適切な混合物、および/または植物油を含む溶媒または分散媒であることができる。たとえば、レシチンなどのコーティングの使用によって、分散の場合には必要とされる粒径を維持することによって、および界面活性物質を使用することによって、適当な流動性が、維持されてもよい。微生物の作用の予防は、種々の抗菌薬および抗真菌薬、たとえばパラベン、クロロブタノール、フェノール、ソルビン酸、チメロサール等によってもたらすことができる。多くの場合、等張性の薬剤、たとえば糖または塩化ナトリウムを含むことが好ましい。注射用組成物の吸収の延長は、吸収を遅延させる薬剤、たとえばモノステアリン酸アルミニウムおよびゼラチンを組成物に使用することによってもたらすことができる。
【0079】
水溶液の非経口投与については、たとえば、必要に応じて溶液を適切に緩衝化し、液状希釈液を最初に十分な食塩水またはグルコースで等張性にするべきである。これらの特定の水溶液は、特に静脈内、筋肉内、皮下、腫瘍内、および腹腔内投与のために適している。この点について、使用することができる無菌水性培地は、本開示の観点から当業者には公知である。たとえば、1用量は、1mlの等張性NaCl溶液に溶解して、1000mlの皮下注入液体に添加するか、または提唱された注入部位のいずれかに注射してもよい(たとえば「Remington's Pharmaceutical Sciences」第15版、1035-1038頁および1570-1580頁を参照されたい)。治療される被検者の症状に応じて、必然的に用量のいくらかの変更が生じる。いずれにしても、投与に対して責任がある人が、個々の被検者に適した用量を決定する。さらに、ヒト投与については、製剤は、FDA生物学的基準局(FDA Office of Biologics standards)によって要求されるとおりの無菌性、発熱原性、一般的安全性、および純度基準を満たすべきである。
【0080】
無菌注射用溶液は、必要に応じて上記で列挙した種々のその他の成分と共に、適切な溶媒中に必要とされる量で活性化合物を組み入れ、続いて濾過滅菌することによって調製される。一般に、分散液は、種々の殺菌された活性成分を、塩基性分散媒と上記で列挙したものから必要とされるその他の成分とを含む無菌媒体に組み入れることによって調製される。無菌注射用溶液の調製のための無菌粉末の場合、好ましい調製法は、これらの事前に滅菌濾過した溶液から、活性成分プラス任意のさらなる所望の成分の粉末を得る、真空乾燥および凍結乾燥技術である。
【0081】
本明細書に開示された組成物は、中性または塩形態に製剤化してもよい。薬学的に許容される塩は、酸付加塩(タンパク質の遊離アミノ基と形成される)を含み、酸付加塩は、たとえば、塩酸もしくはリン酸などの無機酸、または酢酸、シュウ酸、酒石酸、マンデル酸などの有機酸で形成される。また、遊離カルボキシル基で形成された塩は、たとえば、ナトリウム、カリウム、アンモニウム、カルシウム、または水酸化第二鉄などの無機塩基、およびイソプロピルアミン、トリメチルアミン、ヒスチジン、プロカインなどの有機塩基などに由来することができる。製剤に関しては、用量製剤と適合した様式で、治療的に有効であるような量で溶液が投与される。製剤は、注射用溶液、薬物放出カプセルなどの種々の剤形で容易に投与される。
【0082】
本明細書に使用される「担体」は、任意の、並びに全ての溶媒、分散媒、媒体、コーティング、希釈液、抗菌薬および抗真菌薬、等張性の吸収遅延薬、緩衝液、担体溶液、懸濁液、コロイドなどを含む。薬学的に活性な物質のためのこのような媒体および薬剤の使用は、当技術分野において周知である。任意の従来の媒質または薬剤が、活性成分と不適合性である場合以外は、治療組成物におけるその使用が想定される。また、補充活性成分を組成物に組み入れることもできる。
【0083】
「薬学的に許容される」または「薬理学的に許容される」という句は、ヒトに投与される場合に、アレルギー性または同様の有害反応を生じない分子的実体および組成物をいう。活性成分としてタンパク質を含む水性組成物の調製も、当技術分野において周知である。典型的には、このような組成物は、液体溶液または懸濁液のいずれかとして注射剤として調製され;注射前に液体の溶液に、または懸濁液に適した固体形態も調製することができる。
【0084】
VII. 併用治療
本発明の化合物および方法は、癌を含む腫瘍性疾患/症状に関して使用してもよい。癌のタイプは、肺癌、頭頚部癌、乳癌、膵癌、前立腺癌、腎癌、骨癌、精巣癌、子宮頚癌、胃腸癌、リンパ腫、肺における新生物発生前の病変、大腸癌、黒色腫、膀胱癌、および他のいかなる腫瘍疾患も含んでもよい。本発明のタキサン組成物での治療の有効性を増大するために、パクリタキセル、ドセタキセル、またはこれらの類似体などの、これらの疾患および症状の治療に有効なその他の薬剤とこれらの組成物を併用することが望ましい場合がある。たとえば、本発明のタキサン化合物および抗癌剤または手術などのその他の抗癌療法と共に、癌の治療を実行してもよい。
【0085】
種々の組み合わせを使用してもよく;たとえば、タキサン組成物は、「A」、および抗癌療法は、「B」である。

【0086】
患者に対する本発明の治療組成物の投与は、その特定の二次療法を適用するための一般的なプロトコルに従って、もしあれば、タキサン治療の毒性を考慮する。必要に応じて、治療サイクルを繰り返すことが予想される。また、種々の標準療法、並びに外科的介入を、記載した癌細胞と併用して適用してもよいことが想定される。
【0087】
A. 抗癌治療
本発明で使用することが想定される抗癌治療は、たとえば、癌細胞を死滅させるか、癌細胞のアポトーシスを誘導するか、癌細胞の増殖速度を減少させるか、転移の発病率もしくは数を減少させるか、腫瘍サイズを減少させるか、腫瘍成長を阻害するか、腫瘍もしくは癌細胞に対する血液供給を減少させるか、癌細胞もしくは腫瘍に対する免疫応答を促進するか、癌の進行を防げるか、もしくは阻害するか、または癌である被検者の寿命を増大することにより、被検者において癌に負の影響を与えることができると思われる。抗癌治療は、生物学的薬剤(生物療法)、化学療法薬、および放射線療法薬を含む。生物学的療法と化学療法との併用は、生物化学療法として公知である。
【0088】
従って、本発明は、細胞の死滅または増殖を阻害するために有効な量を併用して提供されたタキサン組成物および抗癌剤を想定する。本方法は、細胞をタキサンおよび薬剤または因子と同時に接触させる工程を含んでもよい。これは、細胞をタキサンおよびその他の薬剤の両方を含む単一の組成物もしくは薬理学的製剤と接触させることによって、または細胞を、一方の組成物がタキサンを含み、他方は抗癌剤を含む2つの異なる組成物もしくは製剤と同時に接触させることによって達成してもよい。
【0089】
1. 化学療法
また、本発明において、タキサンを化学療法薬と併用して使用してもよいことが想定される。このような化学療法薬は、たとえば、シスプラチン(CDDP)、カルボプラチン、プロカルバジン、メクロレタミン、シクロホスファミド、カンプトセシン、イホスファミド、メルファラン、クロランブシル、ブスルファン、ニトロソ尿素(nitrosurea)、ダクチノマイシン、ダウノルビシン、ドキソルビシン、ブレオマイシン、プリコマイシン(plicomycin)、マイトマイシン、エトポシド(VP 16)、タモキシフェン、ラロキシフェン、エストロゲン受容体結合剤、タキソール、ゲムシタビン、ナベルビン、ファルネシル-タンパク質トランスフェラーゼ阻害剤、トランスプラチナ(transplatinum)、5-フルオロウラシル、ビンクリスチン、ビンブラスチンおよびメトトレキセート、テマゾロミド(Temazolomide)(DTICの水性型)、またはこれらの任意の類似体もしくは誘導体を含んでもよい。膵癌を治療するために現在使用される化学療法薬の一例は、ゲムシタビンである。その他の研究では、進行した膵癌の治療のために、高用量の5-フルオロウラシル(5-FU)を使用する。
【0090】
また、タキサンは、タンパク質チロシンキナーゼ阻害剤などのその他の化学療法薬と併用して使用してもよい。このような阻害剤は、最適には、イマチニブまたはイマチニブメシレート(STI-571、グリベック(商標);Norvartis, Inc.)、OSI-774(タルセバ(商標);OSI Pharmaceuticals, Inc.)、ZD-1839(イレッサ(登録商標);AstraZeneca, Inc.)、SU-101(Sugen, Inc.)、およびCP-701(Cephalon, Inc.)を含んでもよい。
【0091】
2. 放射線療法
癌を有する患者を治療する際に、タキサンと併用して本発明に使用してもよいもう一つの療法は、放射線療法である。本発明に使用してもよい放射線療法因子は、DNA損傷を生じさせ、かつγ線、X線、および/または腫瘍細胞に対して放射性同位元素を直接送達するものなどの広範囲に使用されてきた因子であることが想定される。また、マイクロ波およびUV照射などのDNAに損害を与える因子のその他の形態も想定される。これらの因子は全て、DNAに対する、DNAの前駆体に対する、DNAの複製および修復に対する、並びに染色体の構築および維持に対する広範な損傷を及ぼす可能性が最も高い。X線の用量範囲は、長期間(3〜4週)にわたって50〜200レントゲンの1日量から2000〜6000レントゲンの一回用量の範囲である。放射性同位元素のための用量範囲は、広く変動し、同位元素の半減期、放射される放射線の強度および型、並びに癌または腫瘍細胞による摂取に依存する。
【0092】
3. 手術
癌である人の約60%は、予防的、診断的または段階的(staging)、治療的、並びに一時的な手術を含む、いくつかのタイプの手術を受けると考えられる。治療的手術は、本発明の治療、化学療法、放射線療法、ホルモン療法、遺伝子療法、免疫療法、および/または代替療法などのその他の療法と共に使用してもよい癌治療法である。
【0093】
治療的手術は、癌組織の全部または一部を物理的に除去し、切除し、および/または破壊する切除を含む。腫瘍切除は、腫瘍の少なくとも一部を物理的に除去することをいう。腫瘍切除に加えて、手術による治療は、レーザー手術、冷凍外科療法、電気外科療法、および顕微鏡的に制御された手術(モース手術(Mohs' surgery))を含む。本発明は、表在癌、前癌、または付随的な正常組織の量の除去と組み合わせて使用してもよいことがさらに想定される。
【0094】
癌細胞、組織、または腫瘍の全ての部分の切除術により、腔が体内に形成されてもよい。治療は、さらなる抗癌療法による領域の灌流、直接注射、または局所投与によって達成してもよい。このような治療は、たとえば1日、2日、3日、4日、5日、6日、もしくは7日毎、または1週、2週、3週、4週、および5週毎、または1ヶ月、2ヶ月、3ヶ月、4ヶ月、5ヶ月、6ヶ月、7ヶ月、8ヶ月、9ヶ月、10ヶ月、11ヶ月、もしくは12ヶ月毎に繰り返してもよい。これらの治療もまた、用量を変化させてもよい。
【0095】
VIII. 実施例
以下の実施例は、本発明の好ましい態様を証明するために含まれる。続く実施例に開示した技術は、本発明の実施において十分に機能することが本発明者らによって発見された技術を示すことが当業者により認識されるはずであり、したがって、その実施のための好ましい様式を構成するとみなすことができる。しかし、当業者であれば、本開示を考慮して、開示された特定の態様に多くの変更を行うことができ、本発明の精神と範囲から逸脱することなく、なおも同様の、または類似の結果を得ることができることを認識するはずである。
【0096】
実施例1
実験手順
株化細胞および細胞培養
本研究に使用される全てのヒト株化細胞組換えプラスミドの開発のためのHEK 293細胞;機能的な紡錘体形成チェックポイントを有することが公知であるMCF-7乳癌およびMCF-10A正常細胞;並びに低Mad2発現のために欠陥チェックポイントを有するT47D乳癌およびOvca432卵嚢癌細胞は、アメリカン・タイプ・カルチャー・コレクション(American Type Culture Collection)(Rockville, MD)から得た。HEK 293細胞、MCF-7細胞、およびT47D細胞は、ダルベッコ改変イーグル培地(DMEM)/F12培地中で培養した。Ovca432細胞は、RPMI 1640中で維持した。DMEM/F12培地およびRPMI 1640は、2mMのL-グルタミン、10%のウシ胎児血清(FBS;100IU/ml)、およびペニシリン・ストレプトマイシン(100mg/ml)を補った。MCF-10A細胞は、5%のウマ血清、0.02μg/mlの上皮細胞成長因子、0.5μg/mlのヒドロコルチゾン、10μg/mlのインスリン、0.1μg/mlのコレラ毒素、100IU/mlのペニシリン、および100mg/mlのストレプトマイシンを補ったDMEM/F12培地中で維持した。
【0097】
低分子干渉RNAトランスフェクション
Mad2配列 5'-
およびBubR1配列 5'-
を標的化するために、21ヌクレオチドのsiRNA二重鎖をDharmacon Research, Inc. (Lafayette, CO)によって合成した。MCF-7細胞のトランスフェクションは、オリゴフェクトアミントランスフェクション試薬(Invitrogen, Carlsbad, CA)を使用して、Dharmacon Researchによって提供されるプロトコルに従って行った。対照研究については、細胞はsiRNAスクランブル二重鎖(Dharmacon Research)でトランスフェクションを行った。siRNAの終濃度は、200nMであった。
【0098】
複製欠陥組換えアデノウイルスの作製
アデノウイルスは、以前に(He et al., 1998)およびStratagene(La Jolla, CA)記載されているプロトコルに従って作製した。簡潔には、最初に、cDNA Mad2の遺伝子をシャトルベクター、pAdTrack-サイトメガロウイルス(CMV)にクローニングした。生じるプラスミドを制限エンドヌクレアーゼPme Iで消化することによって直線化し、その後にアデノウイルス・バックボーン・プラスミドpAdEasy-1(Stratagene, La Jolla, CA)を使用して大腸菌(Escherichia coli.)BJ5183細胞に同時形質転換した。組換え体をカナマイシン耐性について選択し、組換えを制限エンドヌクレアーゼ解析によって確認した。最後に、HEK293細胞を直線化した組換えプラスミドでトランスフェクションを行った。研究については、それぞれの細胞において、細胞変性効果を伴わずに80〜90%の感染効率が得られた。
【0099】
ウェスタンブロット解析
トランスフェクションの24時間、48時間、および72時間後に、細胞を収集してタンパク質免疫ブロット解析に供した。細胞を氷冷リン酸緩衝食塩水中で一度洗浄し、プロテアーゼ阻害剤(1mMのフェニルメタンスルホニルフルオライドおよび10μg/mlアプロチニン)およびホスファターゼ阻害剤(20mMのβ-グリセロリン酸、5 mMのNaF、および100μMのNa3VO4)を含む溶解緩衝液(1%のNP-40、150mMのNaCl、および50mMのトリス-HCl[pH 7.5])で溶解した。氷上で30分後、細胞を4℃において15分間、13,000rpmでの遠心分離に供した。ウェスタンブロット法のために、同量のタンパク質をドデシル硫酸ナトリウム-ポリアクリルアミドゲル電気泳動を使用して分解し、ニトロセルロース膜に転送した。膜をポリクローナル抗Mad2抗体(Covance, Princeton, NJ; 1:500)、 モノクローナル抗BubR1抗体(Chemicon, Temecula, CA; 1:500)、およびモノクローナル抗αチューブリン(Sigma-Aldrich Chemical Co., St. Louis, MO; 1:5000)と共に室温で1時間(または4℃で一晩)インキュベートし、続いて西洋ワサビペルオキシダーゼ結合抗体とインキュベーションした。結果は、化学発光検出系で増強して視覚化した。
【0100】
薬物感受性アッセイ法
細胞をトリプシン処理によって剥離し、96ウェル・マイクロタイタープレートに2.0×103細胞/ウェルで播き、パクリタキセル(1、5、10、50、100、および1000 nM)の種々の濃度で処置した。72時間後に、細胞増殖に対する効果を3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミド(MTT)アッセイ法によって検査し:20μlのMTT溶液(5mg/mlのリン酸塩緩衝食塩水溶液;Sigma Aldrich)をそれぞれのウェルに添加して、細胞を37℃において4時間インキュベートした。代謝的に生存可能な細胞によって形成されたMTT-ホルマザンを100μlの細胞溶解緩衝液に溶解し、570nmの波長でマイクロプレートを使用して蛍光をモニターした。細胞増殖率は、パクリタキセル(対照)で処置されていない細胞の吸収を100%と定義することによって算出した。
【0101】
有糸分裂指数の算出
有糸分裂の凝縮染色質をもつ細胞は、10μg/mlヨウ化プロピジウムと連動して10μMのヘキストで33342色素(Aventis Pharmaceuticals Inc., Bridgewater, NJ)で染色することによって視覚化し;ヨウ化プロピジウムは、死細胞だけに組みこまれた。従って、死細胞は、ヨウ化プロピジウムおよびヘキスト33342色素の両方で染色されるが、有糸分裂細胞は、ヘキスト33342色素のみで凝縮染色質を示した。細胞をトランスフェクションの12時間、24時間、および36時間後に収集して有糸分裂指数を算出した。
【0102】
細胞死解析
細胞死は、トリパンブルー染料排除アッセイ法を使用して評価した。簡潔には、細胞を、トリプシンを使用して収集し、0.4%のトリパンブルー色素(Sigma-Aldrich Chemical Co., St. Louis, MO)で染色した。トリパンブルー陽性および陰性の細胞は、位相差顕微鏡(Fisher Scientific, Pittsburgh, PA)下で、血球計算板(Hausser Scientific, Horsham, PA)を使用して計数した。それぞれのアッセイ結果は、細胞の総数を基準として死細胞の割合に関して表してある。個々の実験は、三回行った。結果は、平均値±標準偏差として報告してある。
【0103】
Cdk1キナーゼ・アッセイ法
Cdk1プロテインキナーゼ・アッセイ法は、SignaTECT cdc2タンパク質アッセイ法系(Promega, Madison, WI)を使用して行った。簡潔には、収集した細胞をプロテアーゼ阻害剤(100μg/mlのアプロチニンおよび0.5mMのフェニルメタンスルホニルフルオライド)およびホスファターゼ阻害剤(50mMのNaF)を含む抽出緩衝液(50mMのトリス[pH 7.4]、150 mMのNaCl、0.1%のトリトンX-100、および1mMのEDTA)で溶解した。これらの可溶化液を、ヒストンH1および[γ-32P] ATPに由来するcdc2特異的ビオチン化ペプチドからなる基質と結合させて、30℃において10分間インキュベートした。これらの放射標識された、リン酸化された基質をストレプトアビジン・マトリックス・ビオチン捕獲膜で回収した(SAM;Promega)。数回の洗浄後、それぞれの捕獲した膜を別々のバイアルに入れて、液体シンチレーションカウンター(Beckman Coulter, Palo Alto, CA)を使用して解析した。
【0104】
細胞周期プロファイリングための細胞可溶化物の調製
細胞周期プロファイリングに供される株化細胞の可溶化液は、以下のとおりに調製した。細胞を10%のFCS(ウシ胎児血清)を含むDMEM(ダルベッコ改変イーグル培地)で培養し、100nMのパクリタキセルで0時間、24時間、48時間、または72時間処置した。処置後、細胞を収集して、PBSで一度洗浄した。次いで、細胞を21Gの針で20回注射することによって溶解緩衝液(0.1%のNP-40、20 mMのトリス-HCl[pH 7.4]、150 mMのNaCl、2% プロテイナーゼ阻害剤反応混液[Sigma, St Louis, MO, USA])で溶解した。不溶性物質を除去するために5分間の15000rpmの遠心分離後、上清のタンパク質濃度を解析し(DCキット, Pierce, Rockford, IL, USA)、使用するまで-80℃で貯蔵した。
【0105】
細胞周期プロファイリング発現解析
細胞周期プロファイリングの発現解析については、2.5μgの総タンパク質を、疎水性膜(0.22μmの孔をもつPVDF;Millipore, Billerica, MA(USA))をもつ5×7cm2のドットブロット装置の78μlのウェル(3mm [w]×5mm [l]×7mm[d])に加えた。膜に結合した粗試料中の標的タンパク質は、抗CDK抗体(Santa Cruz Biotechnology, Santa Cruz, CA, USA)、ビオチン化二次抗体(Santa Cruz Biotechnology)、およびフルオレッセイン標識されたストレプトアビジン(Vector, Burlingame, CA, USA)での連続反応によって定量的に検出した。それぞれの反応間に、ウェルをTBS溶液(25mMのトリス-HCl[pH 7.4]、150 mMのNaCl)で自動洗浄した。膜の蛍光イメージは、イメージアナライザー(Bio-Rad, Hercules, CA, USA)を使用して解析し、ドットの強度は、「Quantity One」ソフトウェア(Bio-Rad)を使用して定量化した。相対的蛍光単位(RFU)および標準的組換えタンパク質(Santa Cruz Biotechnology)の量は、標準化された範囲で直線的に相関させた(CDK1、2.5〜25ng/ドット;CDK2、1.0〜10ng/ドット;CDK4、1.0〜10ng/ドット;CDK6、2.5〜25ng/ドット)。
【0106】
細胞周期プロファイリング酵素活性解析
細胞周期プロファイリングの酵素活性解析は、非放射同位元素法を使用して行った。細胞可溶化物は、発現解析について記載したとおりに調製した。CDK分子は、2μgの対応する抗体(抗CDK1、抗CDK2、抗CDK4、または抗CDK6抗体;Santa Cruz Biotechnology)および20μlのプロテインAビーズ(Amersham Pharmacia, Uppsala, Sweden)で、100μgの可溶化液総タンパク質量から4℃において1時間、選択的に沈殿させた。洗浄緩衝液(0.1%のNP-40、50 mM トリス-Cl[pH 7.4])で3回洗浄後、10μgのタンパク質基質、5mMアデノシン5'-O-(3-チオ三リン酸エステル)(Sigma)、20mM トリス-Cl(pH 7.4)、および0.1%のトリトンX-100を含む50μlの基質混合物をビーズに添加して、37℃において10分間、連続振盪下でインキュベートした。タンパク質基質として、CDK1およびCDK2のためにはヒストンH1(Upstate Biotechnology, Lake Placid, NY, USA)を、並びにCDK4およびCDK6のためには組換えRBタンパク質(アミノ酸769〜921)を使用した。ビーズを除去した後、基質に導入されたモノチオリン酸エステルを、10mMのヨードアセチル-ビオチン(Pierce)と共に、結合緩衝液(100mMのトリス-Cl[pH 8.5]、1 mM EDTA)中で、暗がりで室温において90分間インキュベーションすることによってさらに標識した。反応をβ-メルカプトエタノールでクエンチして、0.4μgの基質をドットブロット装置のウェルに適用した。ウェルを4%のウシ血清アルブミン(BSA)で30分間ブロッキングし、次いで37℃において1時間アビジン-FITC(Vector)と共にインキュベートした。膜を洗浄した後、イメージアナライザー(Bio-Rad)を使用してイメージを評価し、ドットの蛍光強度を「Quantity One」ソフトウェア(Bio-Rad)を使用して定量化した。K562慢性骨髄性白血病株化細胞から抽出された0μg、12.5μg、25μg、50μg、100μg、および150μgのタンパク質に対応するCDK活性で調製した検量線によって活性を算出した。1ユニット(U)は、K562細胞からの1μgの総タンパク質量のキナーゼ活性に匹敵する。
【0107】
実施例2
紡錘体形成チェックポイントの不活性化とCdk1活性の抑制との相関
以前の研究は、紡錘体形成チェックポイントの活性化により、Mad2およびBubR1の両方がCdc20と直接相互作用し、そのAPCを活性化する能力を阻害することを示した(Fang et al., 1998;Sudakin et al., 2001;Tang et al., 2001;Fang, 2002)。株化細胞における一過性ノックダウンを介したMad2またはBubR1の抑制が、パクリタキセルで誘導される紡錘体形成チェックポイントの機能に影響を及ぼすかどうかを決定するために、本発明者らは、siRNA二重鎖を使用して遺伝子サイレンシングを行った。
【0108】
Mad2およびBubR1配列の一部に対する21ヌクレオチドの低分子干渉RNA(siRNA)二重鎖相同体を機能的なチェックポイントを有するMCF-7細胞にトランスフェクションを行うことにより、Mad2およびBubR1タンパク質レベルの劇的な減少を生じ;これらのレベルは、それぞれの株化細胞においてトランスフェクションの24時間、48時間、および72時間後にも低いままであった。また、siRNA/Mad2およびsiRNA/BubR1でのMCF-7細胞の同時トランスフェクションにより、両方の発現が減少し、単一トランスフェクションで得られたものと同様の結果を生じた(図1A)。スクランブルされたsiRNA二重鎖(siRNA/対照)は、Mad2またはBubR1の発現レベルに影響を及ぼさず、siRNAアプローチの特異性が検証された。
【0109】
フローサイトメトリーを使用して、本発明者らは、有糸分裂チェックポイント遺伝子の一過性抑制が、細胞周期分布に影響を及ぼさないという可能性を除外することを試みた。トランスフェクションの72時間後に、Mad2およびBubR1の両方を抑制した細胞における細胞周期分布は、対照細胞における分布と同様であった(図1B)。パクリタキセルによって活性化される紡錘体形成チェックポイントに対するMad2、BubR1、または両方の抑制の効果を試験するために、有糸分裂指数を決定し、Cdk1活性を測定した(これらは両方とも、このチェックポイントの状態を反映する)。パクリタキセル処置の24時間後に、対照細胞の少なくとも80%が有糸分裂で停止され、Cdk1活性の劇的な増大を示し、したがって、紡錘体形成チェックポイントの活性化が検証された(図1Cおよび図1D)。対照的に、有糸分裂指数の蓄積およびCdk1の活性化は、Mad2およびBubR1を抑制した細胞では不完全であった(図1Cおよび図1D)。面白いことに、Mad2およびBubR1の同時抑制により、有糸分裂指数の蓄積およびCdk1の活性化の減少を示し、Mad2またはBubR1のいずれか単独の抑制で得られるものと同様となった(図1Cおよび図1D)。これらの結果は、紡錘体形成チェックポイントの機能を消失させるためには、Mad2またはBubR1単独で十分であったことを示す。この消失は、Cdk1活性の抑制によって反映された。
【0110】
最近の報告は、こうしてこれまでに同定された全ての単一紡錘体形成チェックポイント遺伝子が、有糸分裂停止を維持するために必須であることを明確に証明した(Dobles et al., 2000;Kalitsis et al., 2000;Luo et al., 2002)。Mad2およびBubR1は、紡錘体形成チェックポイントの開始および維持のために、有糸分裂チェックポイント複合体において協調して作用し、本結果と一致すると思われる知見である(Fang, 2002)。そのうえ、以前の研究では、機能的な紡錘体形成チェックポイントを有するMDA-MB-231およびSKBr-3細胞のsiRNA/Mad2、siRNA/BubR1、または両方ともによるトランスフェクションにより;チェックポイントの減少およびパクリタキセルに対する耐性を生じた(データ示さず)。これらの株化細胞は、それぞれ変異されたp53およびHER2/neu過剰発現を有することが公知である。これらの公知の遺伝的相違にもかかわらず、紡錘体形成チェックポイントの消失は、同様のパクリタキセルに対する耐性の知見を与えた。
【0111】
実施例3
紡錘体形成チェックポイントおよびパクリタキセル耐性の減少
パクリタキセル感受性に対するMad2、BubR1、または両方ともの抑制による紡錘体形成チェックポイントの減少の効果を決定するために、MTTアッセイ法を使用してパクリタキセルの細胞生存度を比較した。図2Aに示したように、Mad2、BubR1、または両方ともが抑制されたMCF-7細胞は、対照細胞よりもパクリタキセルに対して耐性であった。次に、これらの耐性がパクリタキセルで誘導される細胞死の減少によるものであったかどうかを決定するために、本発明者らは、トリパンブルー排除法を使用して細胞死の集団を評価した。パクリタキセルでの処置の48時間後に、細胞死のレベルは、対照細胞では高かったが、Mad2、BubR1、または両方ともが抑制された細胞では減少していた(図2B)。これらのデータは、紡錘体形成チェックポイントの減少が、パクリタキセル耐性を増大することを証明する。
【0112】
実施例4
Mad2依存的な、チェックポイント欠陥細胞におけるCdk1活性およびパクリタキセル感受性に対するMad2過剰発現の効果
Mad2突然変異は、チェックポイント欠陥がある癌株化細胞において検出されなかったが(Takahashi et al., 1999)、Mad2タンパク質の発現レベルは、紡錘体形成チェックポイントの応答能と相関しているように見える(Wang et al., 2000;Wang et al., 2002)。ヒト検体におけるMad2タンパク質の発現レベルについての報告は、ほとんど発表されていなかった(Tanaka et al., 2001)。従って、Mad2の過剰発現が、低Mad2発現によって紡錘体形成チェックポイントが機能しなくなった細胞における紡錘体形成チェックポイント活性化を回復するかどうかを決定した。効率的にMad2を発現させるために、Mad2(Ad-EGFP/Mad2)を発現する組換えアデノウイルスを作製した。このアデノウイルスは、2つの独立したCMV駆動転写単位(GFPについて1つ、およびMad2について1つ)を含み、感染効率を直接観察することができる。Ad-EGFP/Mad2は、外来Mad2の高発現を誘導し(図3A)、細胞周期の種々の期の間の細胞の分布に影響を及ぼさなかった(図3B)。
【0113】
最初に、機能しないチェックポイントを有することが示されたMad2-ノックダウンMCF-7細胞を使用した(図1Cおよび図1D)。Mad2の発現は、Ad-EGFP/Mad2の感染により、Mad2-ノックダウン細胞において回復された(図3C)。次いで、チェックポイントの機能をMad2の再発現によって回復することができるかどうかを決定するために、Ad-EGFP/Mad2感染させたMad2-ノックダウン細胞におけるCdk1の活性化をパクリタキセル処置後に評価した。Cdk1活性は、パクリタキセルに対する曝露後でさえもルシフェラーゼAd(-EGFP/Luc)を発現する組換えアデノウイルスに感染させた細胞では増大せず;これらの知見は、図1に示したデータと一致する。対照的に、Cdk1活性は、Ad-EGFP/Mad2感染細胞において回復し、Mad2の過剰発現が紡錘体形成チェックポイントの機能を回復することができることを示した(図3D)。次に、この回復により、パクリタキセルで誘導される細胞死のレベルが増強したかどうかを決定し、パクリタキセルで誘導される細胞死は、実際はAd-EGFP/Luc感染細胞(図3E)において、Ad-EGFP/Mad2感染細胞においてより有意に高かったことが見いだされた。
【0114】
また、Mad2の低発現のために欠陥チェックポイントを示すことが公知であるT47DおよびOvca432細胞を使用した(LiおよびBenezra, 1996)。Ad-EGFP/Mad2によるこれらの株化細胞の感染では、外来Mad2の高発現レベルを誘導し、細胞周期分布に影響を及ぼさなかった(図3Aおよび図3B)。Cdk1活性およびパクリタキセル感受性は、Ad-Luc感染させた細胞(図3Fおよび図3G)において、Ad-EGFP/Mad2感染細胞におけるよりも高かった。従って、これらのデータは、外来Mad2発現が、チェックポイントの機能を回復することができ、Mad2依存的なチェックポイント欠陥細胞における細胞死を増強することを証明する。
【0115】
実施例5
BubR1が抑制された細胞におけるチェックポイント機能およびパクリタキセル感受性に対するMad2過剰発現の効果
次に、Mad2の過剰発現が、Mad2以外の分子のための紡錘体形成チェックポイント欠陥を克服することができるかどうかを決定した。チェックポイントに欠陥のあることが示されたBubR1-ノックダウンMCF-7細胞を使用した(図1Cおよび図1D)。Ad-EGFP/Mad2は、外来Mad2の高発現を誘導し、BubR1-ノックダウン細胞におけるBubR1の発現には影響を及ぼさなかった(図4A)。しかし、Cdk1は、Ad-EGFP/Mad2感染細胞においてアップレギュレートされず、パクリタキセルで誘導される細胞死は、増強されなかった(図4Bおよび図4C)。これらのデータは、Mad2の過剰発現が、Mad2非依存的な欠陥チェックポイントをもつ細胞におけるチェックポイントの機能およびパクリタキセル感受性を克服しなかったことを示す。
【0116】
実施例6
機能的チェックポイントをもつ細胞におけるチェックポイント機能およびパクリタキセル感受性に対するMad2の過剰発現の効果
次に、Mad2の過剰発現が、紡錘体形成チェックポイントの機能(Cdk1活性およびパクリタキセル感受性)を増強するかどうか(チェックポイントが無傷の細胞におけるパクリタキセルで誘導される細胞死が増強されるように移行することができるという知見)を決定した。以前の報告では、一方のMad2対立遺伝子の欠失が、欠陥のある紡錘体形成チェックポイントを生じること(Michel et al., 2001)並びにMad2が、そのMad1との相互作用およびAPC/Cdc20の阻害を経て動原体に動員されなければならないこと(Chen et al., 1998;Waters et al., 1998)を示した。これらの結果は、Mad2が紡錘体形成チェックポイント機構の構成成分として作用するためには、Mad2の一定量および特異的局在化が必要とされること、並びにチェックポイントの機能の増強のために、大量のMad2が必要とされないことを示唆する。
【0117】
MCF-10A細胞およびMCF-7細胞(両方とも機能的なチェックポイントを有することが公知である)では、Ad-EGFP/Mad2が効率的にMad2の高発現レベルを誘導した(図3A)。しかし、Cdk1活性は、いずれの株化細胞のAd-EGFP/Mad2感染細胞においても増大されず、従って、パクリタキセルで誘導される細胞死の増強もなかった(図4Dおよび図4E)。従って、Mad2の過剰発現は、基礎的に機能的な紡錘体形成チェックポイントをもつ細胞におけるチェックポイント機能およびパクリタキセル感受性を増強することができなかった。
【0118】
染色体の喪失または獲得は、ヒトの癌の特徴であるので、紡錘体形成チェックポイントは、臨床的状況では頻繁に失われていると思われる。ヒト肺、結腸直腸、卵巣、およびインビトロでの鼻咽頭癌株化細胞の紡錘体形成チェックポイント欠陥の検出についての多くの報告にもかかわらず(Takahashi et al., 1999; Wang et al., 2000; Cahill et al., 1998; Nakagawa et al., 2002; Saeki et al., 2002)、公知の紡錘体形成チェックポイント遺伝子の突然変異は、ヒト癌では非常にまれにしか生じない(Saeki et al., 2002; Sato et al., 2000; Haruki et al., 2001)。このパラドックスは、チェックポイント複合体に対する種々の転写後または翻訳後修飾によって説明される可能性がある。さらに、紡錘体形成チェックポイント機構は、種々の分子からなるために、遺伝子またはタンパク質発現の突然変異を解析することによってヒト癌試料のチェックポイントの機能を評価することは、非実用的であると思われる。
【0119】
実施例7
パクリタキセルに対する株化細胞の感受性
パクリタキセルに対する4種のヒト乳癌株化細胞の感受性をパクリタキセル細胞障害性試験のグラフ解析に従って判断した(図5)。結果は、MDA-MB231およびMDA-MB435が感受性であり、MCF-7およびT47Dが耐性であったことを示した。パクリタキセル細胞障害性試験は、以下のとおりに行った。細胞を96ウェル培養プレートのウェル内で1nM、5nM、10nM、50nM、または100nMパクリタキセルの存在下において培養した。72時間の培養後、細胞生存をMTTアッセイ法によって測定した。
【0120】
実施例8
インビトロでのバリデーション
インビトロでのバリデーション研究において、全ての株化細胞を100nMパクリタキセルで処置して、処置の0時間、24時間、48時間、および72時間後に収集した。次いで、細胞可溶化物を細胞周期プロファイリングに供した。プロファイリング結果により、7つのパラメーター(細胞周期分子)が、パクリタキセルに対して感受性と耐性の株化細胞とを区別するために有用であることが明らかになった。これらは、(1)処置の24時間後のCDK1比活性;(2)処置前のCDK2比活性;(3)処置前のMAD2発現;(4)処置前のサイクリンB1;(5)処置前のサイクリンE発現;(6)処置前のp21発現;および(7)処置前のCDK6発現である(図6)。結果によれば、パクリタキセル感受性予測試験のためには、9つのパラメーターが決定された。これらは、(1)CDK1キナーゼ活性;(2)CDK1発現(CDK1比活性の算出のため);(3)CDK2キナーゼ活性;(4)CDK2発現(CDK2比活性の算出のため);(5)MAD2発現;(6)サイクリンB1;(7)サイクリンE発現;(8)p21発現;および(9)CDK6発現である。
【0121】
細胞周期プロファイリング技術により、7つのパラメーターが感受性と耐性の株化細胞とを区別するために有用であることが明らかになった。これらは、(1)処置の24時間後のCDK1比活性;(2)処置前のCDK2比活性;(3)処置前のMAD2発現;(4)処置前のサイクリンB1;(5)処置前のサイクリンE発現;(6)処置前のp21発現;および(7)処置前のCDK6発現である(図6)。
【0122】
実施例9
インビボでのバリデーション
上記のインビトロでの結果をさらに確認するために、インビボでのバリデーション研究を行った。感受性および耐性の細胞をヌードマウスに接種した。腫瘍体積が300mm3を超える体積に達した場合に、1日につき20mgのパクリタキセルを5日連続で(接種後34〜38日)腹腔内に投与した。腫瘍サイズを20〜43日に記録した。図7に示したように、感受性の腫瘍は、460mm3〜120mm3に腫瘍体積の有意な減少を示した。対照的に、耐性腫瘍の体積は、43日までに350mm3のままであった。
【0123】
パクリタキセル投与の24時間前後に、腫瘍を有するマウスから腫瘍組織を外科的に切開して、細胞周期プロファイリングに供した。サイクリンB1発現を除いて、プロファイリングは、インビトロ研究とほぼ同一の結果を示した。パクリタキセルに対する感受性と耐性の腫瘍とを区別するためには、6つのパラメーターが有用であった。これらは、(1)処置の24時間後のCDK1比活性;(2)処置前のCDK2比活性;(3)処置前のMAD2発現;(4)処置前のサイクリンE発現;(5)処置前のp21発現;および(6)処置前のCDK6発現である(図8)。
【0124】
インビトロおよびインビボでのバリデーション研究の結果を合わせると、細胞周期プロファイリング技術は、腫瘍組織のタキサン化学受容性を決定するための有効な系であることが判明した。決定のために選択したパラメーターにより、正確な予測がなされることが科学的に証明される。
【0125】
上記の結果は、細胞周期プロファイリング技術が、腫瘍組織のタキサン化学受容性を決定する際に良好なことを強く示す。インビトロおよびインビボでのバリデーション研究の結果を合わせると、細胞周期プロファイリング技術は、腫瘍組織のタキサン化学受容性を決定する際に良好であることが判明した。
【0126】
実施例10
ヌードマウスにおけるパクリタキセルに対する感受性
感受性および耐性の細胞(1×107細胞/マウス)をヌードマウスに接種した。腫瘍体積が300mm3を超える体積に達した場合に、1日につき20mgのパクリタキセルを5日連続で(接種後34〜38日)腹腔内に投与した。腫瘍サイズを20〜43日に記録した。図7に示したように、感受性の腫瘍は、460mm3〜120mm3に腫瘍体積の有意な減少を示した。対照的に、耐性腫瘍の体積は、43日までに350mm3のままであった(図7)。
【0127】
細胞周期プロファイリングのためには、パクリタキセル投与の24時間前後に、腫瘍を有するマウスから腫瘍組織を外科的に切開した。組織の一部(2mm3)の可溶化液を溶解緩衝液(0.1%のNP-40、20mMのトリス-HCl[pH 7.4]、150mM NaCl、2%のプロテイナーゼ阻害剤反応混液(Sigma, St Louis, MO)で組織ホモジナイザーを使用して調製した。ホモジナイザーは、自動的にフィルターディスク上の不溶性物質を除去する。可溶化液のタンパク質濃度を解析し(DCキット, Pierce, Rockford, IL, USA)、使用するまで-80℃で貯蔵した。
【0128】
本明細書に開示され、請求される全ての組成物および方法は、本開示を考慮して過度の実験なしに作製し、実施することができる。本発明の組成物および方法は、好ましい態様に関して記載したが、本発明の概念、精神、および範囲から逸脱することなく、本明細書に記載されている組成物および方法並びに方法の工程または工程の順序にバリエーションを適用してもよいことが、当業者には明らかであると思われる。より詳細には、それは、化学的および生理的に関連する一定の薬剤を、本明細書に記載されており、一方で同じまたは同様の結果が達成されると考えられる薬剤と置換してもよいことが明らかであると思われる。当業者に明らかな全てのこのような同様の置換および修飾は、添付の請求の範囲に定義された本発明の精神、範囲、および概念の範囲内であると考えられる。
【0129】
参照文献
以下の参照文献は、これらが本明細書において記載されるものに対して例示的な手順、またはその他の詳細な補充を提供する範囲で、具体的に参照として本明細書に組み入れられる。
【図面の簡単な説明】
【0130】
以下の図面は、本明細書の一部を形成し、本発明の一定の側面をさらに証明するために含まれる。本発明は、本明細書に示される特定の態様の詳細な説明と合わせて、これらの図面の1つまたは複数を参照することで、よりよく理解され得る。
【図1】Mad2、BubR1、または両方ともの抑制による細胞における紡錘体形成チェックポイントの喪失を示す。図1Aは、低分子干渉RNA(siRNA)を用いたトランスフェクション後のBubR1およびMad2レベルの減少を示す。MCF-7細胞には、siRNAについて200nMの終濃度を生じるsiRNA/Mad2またはsiRNA/BubR1でトランスフェクションを行った。トランスフェクション後の示した時間にて、細胞を集めて、それぞれの抗体およびゲル充填のための対照のためのα-チューブリン抗体によるタンパク質免疫ブロット解析に供した。図1Bは、本明細書に記載した蛍光活性化細胞選別によるDNA含量によって測定されるMad2、BubR1、または両方の抑制による細胞の細胞周期分布を示す。MCF-7細胞をトランスフェクし、トランスフェクションの72時間後に集めた。数は、それぞれの期における細胞の割合を示す。図1Cは、パクリタキセルでの処置後のそれぞれの細胞の分裂指数を示す。トランスフェクションの24時間後に、細胞をパクリタキセル(100nM)で処置して、示した時間に収集した。図1Dは、それぞれの細胞におけるパクリタキセルで処置後のサイクリン依存性キナーゼ-1(Cdk1)の活性を示す。トランスフェクションの24時間後に、細胞をパクリタキセル(100nM)で処置して、示した時間に収集した。可溶化液におけるCdk1の活性は、実験手順に記載されているように決定した。
【図2】紡錘体形成チェックポイントの喪失を介したパクリタキセル耐性の誘導を示す。図2Aは、細胞増殖に対するパクリタキセルの効果を決定するために、siRNA/Mad2、BubR1、または両方でトランスフェクションを行ったMCF-7細胞を、MTTアッセイ法を使用して調べた。トランスフェクションの12時間後に、細胞をトリプシン処置によって分離した。接種の12時間後に、細胞を種々の濃度のパクリタキセルで処置した。バー(標準偏差)。図2Bは、siRNA/Mad2、BubR1、または両方でトランスフェクションを行ったMCF-7細胞を、パクリタキセルによって誘導される細胞死について調べた。トランスフェクションの24時間後に、細胞をパクリタキセル(100nM)で48時間処置した。細胞生存度は、トリパンブルー排除アッセイ法を使用して評価した。バー(標準偏差)。
【図3】Mad2依存的チェックポイント欠陥細胞におけるMad2の過剰発現による紡錘体形成チェックポイントの機能の回復およびパクリタキセル感受性の増強を示す。図3Aは、Ad-EGFP/Mad2での感染によるMCF-7、MCF-10A、T47D、およびOvca432細胞における外来性Mad2発現を示す。25および50の感染効率値での感染の24時間後、20μgのそれぞれの可溶化試料を適用して、抗Mad2抗体でのタンパク質免疫ブロット解析に供した。図3Bは、蛍光活性化細胞選別によるDNA含量によって測定される、Mad2を過剰発現した細胞の細胞周期分布を示す。MCF-7、T47D、およびOvca432細胞を感染の48時間後に収集し、その後DNA含量を検出するために、ヨウ化プロピジウムで染色した。数字は、それぞれの期における細胞の割合を示す。図3Cの上段は、組み合わせた低分子干渉RNA(siRNA)/Mad2トランスフェクションおよびアデノウイルス感染のためのスケジュールを示す。siRNA/Mad2トランスフェクションの24時間後に、アデノウイルスベクター(50の感染効率)を24時間にわたって送達し、細胞をパクリタキセル(100nM)に曝露した。下段は、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたMad2が抑制されたMCF-7細胞におけるMad2の発現を示す。細胞をパクリタキセル処置の0時間、24時間、および48時間後に収集して、それぞれの抗体およびゲル充填のための対照のためのα-チューブリン抗体によるタンパク質免疫ブロット解析に供した。図3Dは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたMad2が抑制されたMCF-7細胞におけるパクリタキセルで処置後のサイクリン依存性キナーゼ-1(Cdk1)の活性を示す。50の感染効率でAd-EGFP/Mad2またはAd-EGFP/Lucのいずれかに感染させた24時間後に、細胞をパクリタキセル(100nM)で処置して、示した時間に収集した。本明細書に記載したとおりに、可溶化液中のCdk1活性を決定した。図3Eは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたMad2の抑制されたMCF-7細胞におけるパクリタキセルで誘導される細胞死を示す。感染の24時間後に、細胞を収集した。パクリタキセルでの処置の48時間後に、細胞生存度をトリパンブルー排除アッセイ法を使用して評価した。バー(標準偏差)。図3Fは、感染させたT47DおよびOvca432細胞におけるパクリタキセルで処置後のサイクリン依存性キナーゼ-1(Cdk1)の活性を示す。50の感染効率でAd-EGFP/Mad2またはAd-EGFP/Lucのいずれかに感染させた24時間後に、細胞をパクリタキセル(100nM)で処置して、示した時間に収集した。本明細書に記載したとおりに、可溶化液中のCdk1活性を決定した。図3Gは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたT47DおよびOvca432細胞におけるパクリタキセルで誘導される細胞死を示す。感染の24時間後に、細胞を収集した。パクリタキセルでの処置の48時間後に、細胞生存度をトリパンブルー排除アッセイ法を使用して評価した。バー(標準偏差)。
【図4】Mad2過剰発現では、Mad2非依存的欠陥があるか、または機能的チェックポイントをもつ細胞におけるチェックポイント機能およびパクリタキセル感受性を増強することができない。図4Aの上段は、組み合わせた低分子干渉RNA(siRNA)/BubR1トランスフェクションおよびアデノウイルス感染のためのスケジュールを示す。siRNA/BubR1のトランスフェクションの24時間後に、アデノウイルスベクター(50の感染効率)を24時間にわたって送達し、細胞をパクリタキセル(100nM)に曝露した。下段は、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたBubR1が抑制されたMCF-7細胞におけるMad2およびBubR1の発現を示す。細胞をパクリタキセル処置後の示した時間に収集して、それぞれの抗体およびゲル充填のための対照のためのα-チューブリン抗体によるタンパク質免疫ブロット解析に供した。図4Bは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたBubR1が抑制されたMCF-7細胞におけるパクリタキセルで処置後のCdk1活性。細胞をパクリタキセル処置後の示した時間に収集した。本明細書に記載したとおりに、可溶化液中のCdk1活性を決定した。図4Cは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたBubR1の抑制されたMCF-7細胞におけるパクリタキセルで誘導される細胞死を示す。細胞をパクリタキセル処置の48時間後に収集した。細胞生存度は、トリパンブルー排除によって評価した。バー(標準偏差)。図4Dは、感染させたMCF-7およびMCF-10A細胞におけるパクリタキセル処置後のサイクリン依存性キナーゼ-1(Cdk1)の活性を示す。50の感染効率でAd-EGFP/Mad2またはAd-EGFP/Lucに感染させた24時間後に、細胞をパクリタキセル(100nM)で処置して、示した時間に収集した。本明細書に記載したとおりに、可溶化液中のCdk1活性を決定した。図4Eは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたMCF-7細胞およびMCF-10A細胞におけるパクリタキセルで誘導される細胞死を示す。感染の24時間後に、細胞を収集した。パクリタキセルでの処置の48時間後に、細胞生存度をトリパンブルー排除アッセイ法を使用して評価した。バー(標準偏差)。
【図5】パクリタキセルに対する株化細胞の感受性をパクリタキセル細胞障害性試験のグラフ解析に従って判断した。結果において、MDA-MB231およびMDA-MB435は、感受性のものと判断され、MCF-7およびT47Dは、耐性と判断された。パクリタキセル細胞障害性試験は、以下のとおりに行った。細胞を96ウェル培養板のウェル中で1nM、5nM、10nM、50nM、または100nMのパクリタキセルの存在下において培養した。72時間の培養後、細胞生存をMTTアッセイ法によって測定した。
【図6】株化細胞の可溶化液を細胞周期プロファイリングに供した。細胞周期プロファイリング技術により、7つのパラメーターが感受性と耐性との株化細胞を区別するために有用であることが明らかになった。これらは、(A1)処置の24時間後のCDK1比活性;(A2)処置前のCDK2比活性;(E5)処置前のMAD2発現;(E2)処置前のサイクリンB1;(E3)処置前のサイクリン E発現;(E4)処置前のp21発現;および(E1)処置前のCDK6発現である。グラフにプロットした値は、それぞれのパラメーターに対して、4つの株化細胞の中で最も大きい生の値で割ることによって算出した。たとえば、サイクリンB1発現(E2、可溶化液のng/μg)の生の値;MDA-MB231について0.135、MDA-MB435について0.094、MCF-7について0.061、およびT47Dについて0.042は、MDA-MB231の0.135で割ることにより、MDA-MB231について1.0、MDA-MB435について0.70、MCF-7について0.45、およびT47Dについて0.31になった。
【図7】感受性および耐性の細胞(1×107細胞/マウス)をヌードマウスに接種した。腫瘍体積が、300mm3を超える体積に達した場合に、1日につき20mgのパクリタキセルを5日連続で腹腔内に投与した(接種後34〜38日)。腫瘍サイズは、20〜43日に記録した。図3に示したように、感受性の腫瘍は、460mm3から120mm3に腫瘍体積の劇的な減少を示した。対照的に、耐性腫瘍の体積は、43日まで350mm3のままであった。
【図8】腫瘍を有するマウスからの腫瘍組織の可溶化液を細胞周期プロファイリングに供した。細胞周期技術は、サイクリンB1発現(E2)を除いて、インビトロ研究とほぼ同一の結果を示した。感受性と耐性との腫瘍を区別するためには、6つのパラメーターが有用であった。これらは、(A1)処置の24時間後のCDK1比活性;(A2)処置前のCDK2比活性;(E5)処置前のMAD2発現;(E3)処置前のサイクリンE発現;(E4)処置前のp21発現;および(E1)処置前のCDK6発現であった。グラフにプロットした値は、それぞれのパラメーターに対する2つの腫瘍間でより大きい生の値で割ることによって算出した。たとえば、CDK1比活性(Al、CDK1のmU/ng)の生の値;MDA-MB231の腫瘍について3.0、およびT47Dの腫瘍について0.2は、MDA-MB231について1.0およびT47Dについて0.067になった。
【図1A】
【図1B】
【図1C】
【図1D】
【技術分野】
【0001】
1. 発明の分野
本発明は、一般に癌治療および癌予防の分野に関する。より詳細には、癌患者におけるタキサン化学受容性を予測することに関する。なお、本願は、2003年8月25日に出願された米国仮特許出願第60/497,665号の優先権を主張し、その全ての開示が、具体的に参照として本明細書に組み入れられる。
【背景技術】
【0002】
2. 関連技術の説明
癌化学療法に付随する主な問題のうちの1つは、同じ組織像をもつ個々の患者が、所与の薬剤または所与の治療プロトコルに対して同様に応答しないということである。応答範囲は、乳癌などの化学受容性の腫瘍においてさえも、大部分において異なる可能性がある。薬物用量、薬物の併用、並びに投与計画、患者の年齢および状態、腫瘍局在、その他などの、薬物感受性の多くの決定因子が周知であるが、所与の腫瘍の内因性の感受性は、評価することが依然困難である主な因子である。多数の耐性のメカニズムが同定されており、これらいくつかを腫瘍生検で評価することができるが、これらは、抗癌剤に対する腫瘍感受性の問題を解決するものではない。これは、腫瘍細胞の感受性の関数として薬物療法を個別化しようとするための多くの研究をリードしてきた。
【0003】
患者から得られた腫瘍細胞に対する抗癌剤の感受性を決定するための1つのアプローチには、インビトロ試験であるヒト腫瘍幹細胞アッセイ法(Human Tumor Stem Cell Assay)(HTSCA)を含む。このアッセイ法は、開発された種々のその他のアッセイ法と共に、全て4つの基本工程:1)細胞の単離;2)薬物との細胞のインキュベーション;3)細胞生存の評価;および、4)結果の解釈、を含み、これらは全て、薬物が患者においてどれくらい有効であり得るかについて予測するために使用される。しかし、これらのインビトロでのアッセイ法は、インビボでの抗癌剤に対する化学受容性の不十分な予測法であることが多い。現在では、化学受容性予測試験のための標準的プロトコルは、エキソビボ試験によって行われている。このプロトコルでは、外科的に切除された組織を抗癌剤を含む培地に包埋し、1週間インキュベートする。次いで、組織のサイズまたは生存度を測定して、感受性を決定する。しかし、この方法は、時間がかかり、完全に手動式で、高価である(1試験について600〜1000ドル)。これまでは、化学受容性試験は、臨床使用まで到達しておらず、現在、所与の患者における抗癌剤に対する腫瘍の感受性の好結果の予測をルーチン的に達成することができない。
【0004】
化学受容性を決定することが必要な抗癌剤の1つの群は、パクリタキセルなどのタキサンを含む。パクリタキセル耐性は、紡錘体形成チェックポイントおよびCdk1に関係がある可能性があることが文献に示されている。パクリタキセルが、微小管を安定化して、紡錘体の形成の間に生じる動的な変化を妨害する場合に、紡錘体形成チェックポイントが活性化されて細胞を有糸分裂で停止させる(Horwitz et al., 1982)。有糸分裂チェックポイント/紡錘体形成チェックポイントは、細胞周期としても公知であり、正確な染色体分離をモニターし、ゲノム恒常性を維持するのに重要な役割を果たす。このチェックポイントは、紡錘体に対する染色体の付着および微小管によって発生される姉妹染色分体全体の張力の両方をモニターして、未熟な染色体の分離を防止する。
【0005】
紡錘体形成チェックポイントの分子構成成分は、最初に酵母(Saccharomyces cerevisiae)で同定された。チェックポイント・タンパク質の哺乳類相同体は、Mad1、Mad2、BubR1、Bub3、およびMps1を含む(LiおよびBenezra, 1996; Jin et al., 1998; Taylor et al., 1998; Chan et al., 1999)。チェックポイント機構は、Mad1、Mad2、BubR1、Bub3、およびcdc20で構成されるタンパク質複合体であり、染色体の動原体に位置する。このチェックポイントの標的は、後期促進複合体(APC)およびその活性化補助因子Cdc20である。Mad2およびBubR1は、下流に位置し、この機構の主要タンパク質であると思われ、Cdc20と直接相互作用して、APC活性を協同的に阻害する(Fang et al., 1998; Sudakin et al., 2001; Tang et al., 2001; Fang,2002)。
【0006】
腫瘍細胞では、チェックポイントの欠陥が観察されることが多く、これらはゲノム不安定化を誘導すると考えられている。最近では、Huang et al.(2000)は、MAD2およびCDK1キナーゼが、パクリタキセルで誘導されるアポトーシスに協同的に関与することを示唆した。その他の報告では、カスパーゼ-3活性化を介したアポトーシス誘導のためには、CDK1の活性化が必要とされることが示され(Tan et al., 2002)、p21Waf1(M期でのCDK1阻害剤)は、パクリタキセルで誘導されるアポトーシスの阻害に重要な役割を果たすことが証明された(Fang et al., 2000)。
【0007】
Cdk1は、有糸分裂サイクリンと合わせて、有糸分裂の制御のために必要とされる普遍的なマスター・キナーゼである(Nigg, 2001)。Cdk1活性は、紡錘体形成チェックポイントの活性化に従って最大になる。Cdk阻害剤またはドミナントネガティブCdk1のいずれかを使用する以前の報告では、Cdk1が、パクリタキセルで誘導される細胞死阻害剤またはドミナントネガティブCdkのために重要であることを示したが(MeikrantzおよびSchlegel, 1996; Shen et al., 1998);しかし、Cdk1の活性化が、活性化されたチェックポイント活性化の原因または結果であるかどうかは、不明なままである。従って、紡錘体形成チェックポイントとパクリタキセル感受性との関係は、依然不明である。
【0008】
従って、癌の治療における、タキサンなどの抗癌剤使用に対する新たな、かつより優れたアプローチが必要である。これらの薬剤の有効性は、化学受容性を評価することにより決定される可能性がある。
【発明の開示】
【0009】
発明の概要
癌療法における主要な問題は、化学療法薬に対する癌細胞の耐性である。従って、本発明は、癌細胞または腫瘍組織のタキサン化学受容性を決定するための方法を提供する。本方法は、パクリタキセルおよびドセタキセルなどのタキサンの骨格を有する全ての抗癌剤に対して有効である。
【0010】
従って、特定の態様において、本発明は、癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性に対するタキサンの効果を評価する工程を含む、タキサンに対する癌細胞の化学受容性を決定する方法を提供する。本発明において想定される癌細胞は、患者から得てもよい。本発明の一部の態様において、癌細胞は、生検組織試料、エキソビボで培養された生検組織試料、外科的に切開した組織試料、またはエキソビボで培養された外科的に切開した組織試料などの組織試料であってもよい。
【0011】
本発明の細胞周期分子は、1つもしくは複数のサイクリン依存性キナーゼ(CDK)、サイクリン、CDK阻害剤(CDKI)、p53、または有糸分裂/紡錘体形成チェックポイント分子を含んでいてもよい。一部の態様において、本発明は、1個、2個、3個、4個、5個、6個、7個、8個、9個、10個、11個、12個、13個、14個、15個、16個、17個、18個、19個、20個またはそれ以上の細胞周期分子を評価することを含む。特定の態様において、本発明は、CDK1活性、CDK2活性、CDK4活性、またはCDK6活性などのCDK活性について1つまたは複数の細胞周期分子を評価することを含むが、このようなものに限定されるわけではない。もう一つの特定の態様において、本発明は、CDK1発現、CDK2発現、CDK4発現、またはCDK6発現などの1つまたは複数のCDK分子の発現を評価する工程を含む。さらにもう一つの特定の態様において、本発明は、サイクリンB1発現またはサイクリンE発現などの1つまたは複数のサイクリン分子の発現を評価する工程を含む。さらにもう一つの特定の態様において、本発明は、p16発現、p21/Waf1発現、またはp27/Kip1発現などの1つまたは複数のCDKIの発現を評価する工程を含むが、これらに限定されるわけではない。なおさらなる特定の態様において、本発明は、MAD2発現またはBubR1発現などの1つまたは複数の有糸分裂/紡錘体形成チェックポイント分子の発現を評価する工程を含むが、これらに限定されるわけではない。
【0012】
本発明の特定の態様において、パクリタキセルおよびドセタキセルなどのタキサンの骨格を有する任意の分子もしくは化合物、またはこれらの誘導体もしくは類似体が、本発明において想定される。
【0013】
本発明において想定される癌細胞は、乳癌細胞、前立腺癌細胞、皮膚癌細胞、肺癌細胞、頭頚部癌細胞、膀胱癌細胞、骨癌細胞、骨髄癌細胞、脳癌細胞、大腸癌細胞、食道癌細胞、胃腸癌細胞、歯肉癌細胞、腎臓癌細胞、肝臓癌細胞、上咽頭癌細胞、卵巣癌細胞、胃癌細胞、精巣癌細胞、舌癌細胞、または子宮癌細胞を含んでもよいが、これらに限定されるわけではない。
【0014】
癌細胞は、抗癌治療の適用前に、または抗癌治療の適用後に、患者または被検者から得てもよい。抗癌治療は、化学療法または放射線療法であってもよいが、このようなものに限定されるわけではない。
【0015】
特定の態様において、本発明は、細胞周期プロファイルを得る工程を含む。本発明において想定される細胞周期プロファイルは、タキサン治療を受ける患者の癌細胞において、細胞周期分子(たとえば、タンパク質)の活性もしくは発現を測定するか、決定するか、評価するか、または検出する工程を含む。細胞周期プロファイリングにより、タキサンの感受性を調節する分子を解析してもよい。細胞周期分子の活性または発現は、当業者に公知の方法によって測定し、決定し、評価し、または検出してもよい。一部の態様において、細胞周期プロファイルは、細胞周期もしくは多タンパク質アナライザーなどの自動化アナライザーまたは装置を使用して得てもよい。細胞周期プロファイルを、タキサンに対して感受性である癌細胞の細胞周期プロファイルと比較してもよいことが想定される。細胞周期プロファイルを、タキサンに対して感受性でない癌細胞の細胞周期プロファイルと比較してもよいことも想定される。細胞周期プロファイルは、抗癌治療の適用前に、抗癌治療の適用後の細胞周期プロファイルのものと比較してもよい。
【0016】
一部の態様において、細胞周期プロファイルを得る工程は、以下のパラメーターのうちの1つ、2つ、3つ、4つ、5つ、6つ、7つ、8つ、もしくは9つを測定するか、決定するか、評価するか、または検出することを含む:(1)CDK1キナーゼ活性;(2)CDK1発現(CDK1比活性の算出のために);(3)CDK2キナーゼ活性;(4)CDK2発現(CDK2比活性の算出のために);(5)MAD2発現;(6)サイクリンB1;(7)サイクリンE発現;(8)p21発現;または(9)CDK6発現。一部の態様において、タキサン感受性とタキサン耐性細胞とを区別するために有用な細胞周期プロファイルは、以下のパラメーターのうちの1つ、2つ、3つ、4つ、5つ、6つ、または7つを含む:(1)タキサン治療の24時間後のCDK1比活性;(2)タキサン治療前のCDK2比活性;(3)タキサン治療前のMAD2発現;(4)タキサン治療前のサイクリンB1;(5)タキサン治療前のサイクリンE発現;(6)タキサン治療前のp21発現;および(7)タキサン治療前のCDK6発現。
【0017】
さらにもう一つの態様において、本発明は、癌を有する患者の治療に関する決定を行う工程をさらに含む。なおさらなる態様において、本発明は、癌を有する患者の生存を評価する工程を含む。
【0018】
もう一つの特定の態様において、本発明は、以下の工程を含む患者における癌を治療するか、または予防する方法を提供する:a)患者の癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性に対するタキサンの効果を決定する工程;b)タキサンに対する癌の感受性を評価する工程;および、c)患者にタキサンを投与する工程。タキサンに対する癌の感受性を評価する工程は、細胞周期プロファイルを得る工程を含み、これは自動化アナライザーを使用して得てもよい。
【0019】
本発明において、乳癌、前立腺癌、皮膚癌、肺癌、頭頚部癌、膀胱癌、骨癌、骨髄癌、脳癌、大腸癌、食道癌、胃腸癌、歯肉癌、腎臓癌、肝臓癌、上咽頭癌、卵巣癌、胃癌、精巣癌、舌癌、または子宮癌などの癌が想定されるが、これらに限定されるわけでない。
【0020】
タキサン、これらの誘導体、または類似体は、静脈内もしくは腫瘍内に一回またはそれ以上投与してもよいが、このような投与の方法に、限定されるわけではない。本発明のタキサンは、化学療法または放射線療法などの抗癌治療を適用することをさらに含んでもよい。このような抗癌治療は、タキサンの前に、タキサンの後に、またはタキサンと同時に適用してもよい。抗癌治療は、さらに被検者もしくは患者に一回またはそれ以上適用してもよい。
【0021】
本明細書に記載されている任意の方法または組成物は、本明細書に記載されている他のいかなる方法または組成物に関しても実施することができることが想定される。
【0022】
「ある(one)」の語の使用は、請求項および/または明細書において「含む」という用語と共に使用された場合は、「1つ(a)または1つ(an)」を意味し得るが、「1つまたは複数」、「少なくとも1つ」、および「1つまたは複数」の意味とも一致する。
【0023】
本発明のその他の目的、特徴、および利点は、以下の詳細な説明から明らかになると思われる。しかし、当業者には、本発明の精神および範囲内の種々の変更および修飾が、この詳細な説明から明らかになると考えられるので、詳細な説明および具体例は、本発明の特定の態様を示すと共に、例示目的のみによって与えられることが理解されるはずである。
【0024】
好ましい態様の説明
I. 本発明
本発明は、癌細胞および組織のタキサン化学受容性を決定するための方法を提供する。本発明は、紡錘体形成チェックポイントにおいてタキサン感受性のために必要である分子、したがって、Cdk1またはその他の分子マーカーなどのこのチェックポイントに関与する分子が、タキサン感受性を予測する際に有用であることを決定するということを決定する。従って、本発明は、癌細胞および組織の細胞周期プロファイリングによってタキサン化学受容性を決定するか、または評価する方法を提供する。細胞周期プロファイリングは、キナーゼ活性測定値についてCDK1、CDK2、CDK4、およびCDK6などのいくつかの細胞周期分子(また、本明細書においてパラメーターともいわれる)を;並びにタンパク質発現測定値についてCDK1、CDK2、CDK4、CDK6、サイクリンB1、サイクリンD1、サイクリンE、p21/Waf1、p27/Kip1、p16、p53、およびMAD2を測定することによって行った。細胞周期プロファイリング系のパラメーターは、MAD2発現およびCDK1活性を含むM期調節機構に関与するので、本系は、タキサン感受性の有効な予測法である。
【0025】
II. タキサン
タキサンおよび関連活性成分は、イチイ属(Taxus)種の植物によって産生され、このような植物の異なる部分の成分である。タキソール(パクリタキセル)などのタキサンは、イチイ樹木から得られる環状毒性ジテルペンである。タキサンは、これらが細胞周期のG2/M期において増殖細胞を阻害するという点で、分子に基づいて細胞複製を阻害することが当技術分野において公知である。従って、タキサンは、抗腫瘍効果を有し、一連の癌腫(卵巣癌、乳癌、気管支癌、肺癌)の治療のために次第に使用されている。
【0026】
A. パクリタキセル/タキソール
パクリタキセルは、タキソールとしても知られ、ジテルペンアルカロイドであって、従ってこれは、その構造にタキサン骨格を有する。パクリタキセルは、抗癌活性を有する天然の化合物としてイチイ(タキサス・ブレビフォリア(Taxus brevifolia))の樹皮から抽出される(FuchsおよびJohnson, 1978)。パクリタキセルは、有糸分裂を妨害することによって癌に対して作用する。パクリタキセルは、タキソイド(taxoid)薬であり、原発性および転移性癌の有効な治療として広く使用されている。
【0027】
パクリタキセル(タキソール)は、乳房、卵巣、およびその他の固形腫瘍の治療に広く使用されている。無作為臨床試験では、アントラサイクリン含有補助化学療法に加えてパクリタキセルを受けた原発性乳癌である患者の間で生存優位性を示した(Eifel et al., 2001)。さらに、パクリタキセルは、転移性乳癌(Holmes et al., 1991; Nabholtz et al., 1996; Bishop et al., 1999)および進行卵巣癌(McGuire et al., 1996; Piccart et al., 2000)の両方に対して有効である。パクリタキセルの抗腫瘍活性は、これが微小管構築を促進して微小管を安定化し、このように有糸分裂を防げるために、特有である(Huizing et al., 1995)。パクリタキセルは、チューブリンのBサブユニットに、可逆的かつ特異的に結合して、微小管重合体を形成し、これによりこれらを脱重合に対して安定にし、こうして細胞周期のG2/M期における増殖停止を引き起こすことによってこれを行う(GotaskieおよびAndreassi, 1994)。これにより、微小管分解を生じさせるビンクリスチンおよびビンブラスチンと比較して、タキソールは特有となる(Gatzemeier et al., 1995)。加えて、最近の証拠は、微小管系が種々のサイトカインの放出に必須なこと、およびサイトカイン放出の調整が薬物の抗腫瘍活性において主要な役割を果たしている可能性があることを示す(Smith et al., 1995)。
【0028】
しかし、一部の患者は、パクリタキセル療法に耐性であり、薬物による利益を受ける患者の特徴は、明確ではなかった。パクリタキセル感受性または耐性を予測する分子特性の同定は、この療法を受ける患者を選択する際の助けとなり得る。従って、特定の態様において、本発明は、癌である患者のパクリタキセル感受性に関する。
【0029】
以前の報告では、パクリタキセル耐性が、Bcl-2およびBcl-XLなどの抗アポトーシスBcl-2ファミリー・メンバーのアップレギュレーション(Tang et al., 1994);薬物流出の増大を生じる膜輸送体(たとえば、mdr-1)のアップレギュレーション(Huang et al., 1997);パクリタキセル結合(Giannakakou et al., 1997)の消失を生じるβ-チューブリンの突然変異;および有糸分裂の遅延を生じるサイクリン依存性キナーゼ1(Cdk1)の阻害を介したErbB2(HER2)のアップレギュレーション(Yu et al., 1998)などの種々のメカニズムによるものであることを証明した。
【0030】
パクリタキセルの抗有糸分裂活性により、これは、いくつかの古典的な難治の腫瘍を治療する際に有用な細胞毒である。パクリタキセルは、主に乳癌および卵巣癌を治療するために使用されてきた。また、これは、頭頚部癌、カポジ肉腫および肺癌、小細胞および非小細胞肺癌を治療するために使用してもよい。また、これにより、黒色腫の経過を遅くさせてもよい。タキソール処置に対する応答率は、癌間で変化する。進行した薬物難治卵巣癌は、19〜36%の割合で、以前に治療された転移性乳癌は27〜62%で、および種々の肺癌は21〜37%で応答する。また、タキソールは、いくつかの場合に、完全な腫瘍緩解を生じることが示された(Guchelaar et al., 1994)。
【0031】
パクリタキセルは、これが接触時に皮膚および粘膜を刺激することから、静脈内に与えられる。これは、典型的には、1週あたり3回、3〜24時間の注入によって静脈内に投与される(Guchelaar et al., 1994)。
【0032】
パクリタキセル(これは、たとえばTAXOL(商標)、TAXOTERE(商標)、ドセタキセル、パクリタキセルの10-デスアセチル類似体、およびパクリタキセルの3'N-デスベンゾイル-3'-N-t-ブトキシカルボニル類似体などの製剤、プロドラッグ、類似体、並びに誘導体を含んでもよい)は、当業者に公知の技術を利用して容易に調製してもよい(たとえば、Schiff et al., 1979; LongおよびFairchild, 1994; RingelおよびHorwitz, 1991; Pazdur et al., 1993;PCT出願のWO94/07882;WO94/07881;WO94/07880;WO94/07876;WO93/23555;WO93/10076;WO94/00156;WO93/24476;EP590267;WO94/20089;米国特許第5,294,637号;米国特許第5,283,253号;米国特許第5,279,949号;米国特許第5,274,137号;米国特許第5,202,448号;米国特許第5,200,534号;米国特許第5,229,529号;米国特許第5,254,580号;米国特許第5,412,092号;米国特許第5,395,850号;米国特許第5,380,751;米国特許第5,350,866号;米国特許第4,857,653号;米国特許第5,272,171号;米国特許第5,411,984号;米国特許第5,248,796号;米国特許第5,422,364号;米国特許第5,300,638号;米国特許第5,362,831号;米国特許第5,440,056号;米国特許第4,814,470号;米国特許第5,278,324号;米国特許第5,352,805号;米国特許第5,059,699号;米国特許第4,942,184号を参照されたい);またはたとえばSigma, St. Louis, MO(Taxus brevifoliaからのT7402)を含む種々の市販の供与源から得られる。
【0033】
B. ドセタキセル/キソテール
ドセタキセルは、タキソイドファミリーに属する抗悪性腫瘍薬である。ドセタキセルは、主に乳癌、肺癌、非小細胞肺癌を治療するために使用されてきた。加えて、これは、頭頚部癌、小細胞肺癌、中皮腫、卵巣癌、前立腺癌、および尿路上皮移行細胞癌を治療するために使用してもよい。ドセタキセルは、癌細胞の増殖を妨害し、最終的にこれが破壊される。しかし、パクリタキセル療法に関して上で議論したように、一部の患者は、ドセタキセル療法に耐性である。従って、癌患者におけるこのタキサンの化学受容性を評価するか、または決定することにより、この薬剤は、癌を治療するためにより効率的に使用することができる。
【0034】
ドセタキセルは、欧州イチイ樹木(タキサス・バカタ(Taxus baccata))の針状葉から抽出される前駆体に由来する半合成の薬物である。ドセタキセルの化学名は、5b-20-エポキシ-12a,4,7b,10b,13a-ヘキサヒドロキシタクス-11-エン-9-オン 4-アセテート2-安息香酸三水和物との(2R,3S)-N-カルボキシ-3-フェニルイソセリン,N-tert-ブチルエステル,13-エステルである。これは、有糸分裂および間期の細胞機能に必須である微小管ネットワークを崩壊させることによって作用する。これは、安定な微小管内におけるチューブリンの構築およびこれらの分解の阻害を促進して、細胞分裂の阻害および最終的に細胞死を生じさせる。ドセタキセルおよびパクリタキセルは、両方とも同じ微小管部位に結合するが、ドセタキセルの親和性は、1.9倍高い。ドセタキセルとパクリタキセルとの間の交差耐性は、一貫して生じない。ドセタキセルは、放射線感受性薬である。これは、細胞周期特異的(G2/M期)である。
【0035】
C. その他のタキサン
また、本発明は、抗癌活性を有するタキサン類似体もしくは誘導体を含む、任意のタキサンまたは当技術分野において当業者に周知であるタキサン骨格を有する化合物の化学受容性を試験することを想定する。「タキサン化合物」は、タキソール、タキソールと構造的に類似する化合物、および/またはタキソールの類似体を含んでもよい。また、「タキサン化合物」は、「擬態」を含んでもよい。「擬態」には、タキソールと構造的に類似しないが、インビボでタキソールまたは構造的に類似のタキサン化合物の治療活性を模倣する化合物を含むことが企図される。本発明のタキサン化合物は、癌を有する被検者(患者)における腫瘍成長を阻害するために有用である化合物である。また、タキサン化合物という用語は、化合物の薬学的に許容される塩を含むことが企図される。タキサン化合物は、米国特許5,641,803号、米国特許第5,665,671号、米国特許第5,380,751号、米国特許第5,728,687号、米国特許第5,415,869号、米国特許第5,407,683号、米国特許第5,399,363号、米国特許第5,424,073号、米国特許第5,157,049号、米国特許第5,773,464号、米国特許第5,821,263号、米国特許第5,840,929号、米国特許第4,814,470号、米国特許第5,438,072号、米国特許第5,403,858号、米国特許第4,960,790号、米国特許第5,433,364号、米国特許第4,942,184号、米国特許第5,362,831号、米国特許第5,705,503号、米国特許第5,278,324号、米国特許第5,840,929号、米国特許第5,773,464号、米国特許第5,248,796号、米国特許第5,821,263号、米国特許第4,814,470号、米国特許第5,438,072号、米国特許第4,960,790号、米国特許第4,942,184号、米国特許第5,433,364号、米国特許第5,278,324号、米国特許第6,362,217号、米国特許第6,017,935号、米国特許第5,977,376号、米国特許第5,912,264号、米国特許第5,773,464号、米国特許第5,739,539号、米国特許第5,698,712号、米国特許第6,284,746号;米国特許出願第20030144344号、米国特許出願第20030130341号、米国特許出願第20030134793号、米国特許出願第20030130170号、米国特許出願第20030130178号、米国特許出願第20030124055号、および米国特許出願第20020016356号;並びにPCT出願のWO95/33740、WO96/03394、WO95/33736、WO93/02067、WO94/15929、およびWO94/15599に以前に記載されており、これらの全てが参照として本明細書に組み入れられる。
【0036】
その他のタキサンは、ポリグルタミン酸、ポリアスパラギン酸、またはポリリジンなどの水溶性重合体にパクリタキセルまたはドセタキセルを抱合することによって形成されたパクリタキセルおよびドセタキセルの水溶性組成物を含んでもよい(米国特許出願第20030147807号)。パクリタキセルの誘導体は、様々な程度の薬理活性を有する。このようなパクリタキセル誘導体化合物の合成および評価における研究では、ヒトを含む温血動物の癌の治療のために有用な、安全で、便利で、有効な薬物製剤を開発するための試みがなされてきた。パクリタキセルの発見以来、関連した構造を有する100を超える化合物が種々のイチイ属種から単離され、および/または合成的に作製されてきた。
【0037】
望ましい抗腫瘍特性を有する一つの例示的なパクリタキセル誘導体は、タキサン環のC7位が構造的にパクリタキセルと異なる化合物、7-O-メチルチオメチルパクリタキセル(本明細書において「7-O-MTMパクリタキセル」と称する)である。7-O-MTMパクリタキセルは、現在臨床試験で研究中の公知の抗腫瘍薬である。7-O-MTMパクリタキセルに関する研究では、パクリタキセルがあまり有効ではないことが見いだされた胃腸癌および結腸直腸癌の治療において有望な結果を示した。7-O-MTMパクリタキセルは、合成法によって生成できることが公知である(米国特許第5,646,176号およびWO96/00724;いずれの内容も、それぞれ参照として本明細書に組み入れられる)。
【0038】
本発明において想定されるパクリタキセル誘導体または類似体の代表例は、以下のものを含んでもよいが、これらに限定されるわけではない:7-デオキシ-ドセタキソール、7,8-シクロプロパタキサン、N置換2-アゼチドン、6,7-エポキシパクリタキセル、6,7-修飾パクリタキセル、10-デスアセトキシタキソール、10-デアセチルタキソール(10-デアセチルバッカチンIII由来)、タキソールのホスホノオキシおよびカルボナート誘導体、タキソール2',7-ジ(ナトリウム1,2-ベンゼンジカルボキシラート、10-デスアセトキシ-11,12-ジヒドロタキソール-10,12(18)-ジエン誘導体、10-デスアセトキシタキソール、プロタキソール(2'-および/または7-O-エステル誘導体)、(2'-および/または7-O-カルボナート誘導体)、タキソール側鎖の不斉合成、フッ化タキソール、9-デオキソタキサン、(13-アセチル-9-デオキソバッカチンIII、9-デオキソタキソール、7-デオキシ-9-デオキソタキソール、10-デスアセトキシ-7-デオキシ-9-デオキソタキソール;水素またはアセチル基並びにヒドロキシおよびtert-ブトキシカルボニルアミノを含む誘導体、スルホン化2'-アクリロイルタキソールおよびスルホン化2'-O-アシル酸タキソール誘導体、スクシニルタキソール、2'-γ-アミノブチリルタキソールホルマート、2'-アセチルタキソール、7-アセチルタキソール、7-グリシンカルバメートタキソール、2'-OH-7-PEG(5000)カルバメートタキソール、2'-ベンゾイルおよび2',7-ジベンゾイルタキソール誘導体;その他のプロドラッグ(2'-アセチルタキソール;2',7-ジアセチルタキソール;2'スクシニルタキソール;2'-(β-アラニル)-タキソール);2'γ-アミノブチリルタキソールホルマート;2'-スクシニルタキソールのエチレングリコール誘導体;2'-グルタリルタキソール;2'-(N,N-ジメチルグリシル)タキソール;2'-(2-(N,N-ジメチルアミノ)プロピオニル)タキソール;2'オルトカルボキシベンゾイルタキソール;タキソールの2'脂肪族カルボン酸誘導体;プロドラッグ{2'(N,N-ジエチルアミノプロピオニル)タキソール、2'(N,N-ジメチルグリシル)タキソール、7(N,N-ジメチルグリシル)タキソール、2',7-ジ-(N,N-ジメチルグリシル)タキソール、7(N,N-ジエチルアミノプロピオニル)タキソール、2',7-ジ(N,N-ジエチルアミノプロピオニル)タキソール、2'-(L-グリシル)タキソール、7-(L-グリシル)タキソール、2',7-ジ(L-グリシル)タキソール、2'-(L-アラニル)タキソール、7-(L-アラニル)タキソール、2',7-ジ(L-アラニル)タキソール、2'-(L-ロイシル)タキソール、7-(L-ロイシル)タキソール、2',7-ジ(L-ロイシル)タキソール、2'-(L-イソロイシル)タキソール、7-(L-イソロイシル)タキソール、2',7-ジ(L-イソロイシル)タキソール、2'-(L-バリル)タキソール、7-(L-バリル)タキソール、2',7-ジ(L-バリル)タキソール、2'-(L-フェニルアラニル)タキソール、7-(L-フェニルアラニル)タキソール、2',7-ジ(L-フェニルアラニル)タキソール、2'-(L-プロリル)タキソール、7-(L-プロリル)タキソール、2',7-ジ(L-プロリル)タキソール、2'-(L-リジル)タキソール、7-(L-リジル)タキソール、2',7-ジ(L-リジル)タキソール、2'-(L-グルタミル)タキソール、7-(L-グルタミル)タキソール、2',7-ジ(L-グルタミル)タキソール、2'-(L-アルギニル)タキソール、7-(L-アルギニル)タキソール、2',7-ジ(L-アルギニル)タキソール};修飾フェニルイソセリン側鎖をもつタキソール類似体、タキソテール、(N-デベンゾイル-N-tert-(ブトキシカロニル)-10-デアセチルタキソール、およびタキサン(たとえば、バッカチンIII、セファロマンニン(cephalomannine)、10-デアセチルバッカチンIII、ブレビフォリオール(brevifoliol)、ユナンタクスシン(yunantaxusin)、およびタクスシン(taxusin));並びに以下のものを含むその他のタキサン類似体および誘導体:14-β-ヒドロキシ-10バッカチンIII、デベンゾイル-2-アシルパクリタキセル誘導体、安息香酸パクリタキセル誘導体、ホスホノオキシおよびカルボナートパクリタキセル誘導体、スルホン化2'-アクリロイルタキソール;スルホン化2'-O-アシル酸パクリタキセル誘導体、18位置換パクリタキセル誘導体、塩素化パクリタキセル類似体、C4メトキシエーテルパクリタキセル誘導体、スルフェンアミドタキサン誘導体、ブロム化パクリタキセル類似体、ジラールタキサン誘導体、ニトロフェニルパクリタキセル、10-デアセチル化置換パクリタキセル誘導体、14-β-ヒドロキシ-10デアセチルバッカチンIIIタキサン誘導体、C7タキサン誘導体、C10タキサン誘導体、2-デベンゾイル-2-アシルタキサン誘導体、2-デベンゾイルおよび-2-アシルパクリタキセル誘導体、新たなC2およびC4官能基を有するタキサンおよびバッカチンIII誘導体、n-アシルパクリタキセル類似体、10-デアセチルタキソールA由来の10-デアセチルバッカチンIIIおよび7-保護10-デアセチルバッカチンIII誘導体、10-デアセチルタキソールB、および10-デアセチルタキソール、タキソールの安息香酸の誘導体、2-アロイル-4-アシルパクリタキセル類似体、オルト-エステルパクリタキセル類似体、2-アロイル-4-アシルパクリタキセル類似体、並びに1-デオキシパクリタキセルおよび1-デオキシパクリタキセル類似体。
【0039】
III. 細胞周期プロファイリングのための試料の取得
本発明は、細胞、組織、または器官試料などの試料を得ることを想定する。本発明の試料は、いくつかの手段によって患者から得てもよい。たとえば、本発明の細胞、器官、または組織試料は、生検によって得てもよい。生検は、体から試料を除去することである。本発明に使用してもよい生検は、パンチ生検または針生検を含むが、このようなものに限定されるわけではない。細胞周期分子を含む試料は、当技術分野において公知の任意の方法によって得てもよい。血液または血清試料は、静脈穿刺を使用して収集してもよい。この方法を使用して、血液が単一の静脈に配置された針を介して個体の腕の血管から直接引き出される。次いで、血液をガラスまたはプラスチックチューブに収集してもよい。
【0040】
A. パンチ生検および錐体生検
本発明は、癌試料などの試料を得るための、パンチまたは錐体生検の使用を想定する。パンチ生検は、典型的には皮膚発疹、ほくろ、頚部の小さな組織試料、およびその他の小さな質量の試料を得るために使用される。局所麻酔薬を注射した後、生検パンチ(3mm〜4mmまたは直径0.15インチ)を使用して円柱形部分を皮膚から切断する。開口部は、典型的には縫合で閉じられて、最小限の瘢痕で回復する。
【0041】
一方で、錐体生検は、円柱形または円錐形状である組織を得るために使用される。錐体生検の利点は、これが解析のための大量の組織試料を提供することである。
【0042】
B. 針生検
1. コア針生検
コア針生検(またはコア生検)は、皮膚を通して器官内に小さな中空針を挿入することによって行われる。次いで、針を細胞層に進めて、試料またはコアを除去する。針は、組織試料を除去するのを補助するために、切断先端に設計してもよい。コア生検は、組織試料を除去するのを補助するために、バネを充填した銃を使用して行われることが多い。
【0043】
コア生検は、典型的にはCTイメージング、超音波、または乳房X線撮影などのイメージ・ガイダンス下で行われる。針は、手で、またはサンプリングデバイスの助けを借りて配置する。十分な組織を得るために、複数の挿入が行われることが多く、複数の試料が採取される。組織試料が採取される場合に、サンプリング機器からクリック音が聞こえる場合がある。
【0044】
コア生検は、真空装置で援助された吸引の場合もある(真空補助生検)。この方法は、1つの針挿入だけで複数の試料を除去することができる。コア生検とは異なり、真空補助生検プローブが、ただ一回、小さい皮膚ニックを介して組織内に挿入される。次いで、サンプリング針開口(開口部)の回転を使用して、吸引の助けを借りて、複数の試料が採取される。従って、本発明において、組織試料を得るために、コア針生検または真空補助針生検を使用してもよい。
【0045】
2. 吸引/細針吸引(FNA)生検
吸引生検法は、細針穿刺吸引(FNA)とも称され、注射器に取り付けた細針で行われる。吸引生検またはFNAは、本発明において癌試料を得るために使用してもよい。FNA生検は、経皮的(皮膚を介した)生検である。FNA生検は、典型的には細いゲージ針(22ゲージまたは25ゲージ)で達成される。領域を最初に洗って、次いで通常局所麻酔薬で麻痺させる。針は、関心対象の器官または組織の領域に入れられる。一旦針が配置されたら、注射器で真空を生じさせ、繰り返し針を出し入れする動作を行う。サンプル採取される細胞は、細針を介して注射器に吸い込まれる。通常、3つまたは4つの試料が作製される。
【0046】
膵臓、肺、および肝臓などの容易に到達されない器官は、FNAの優れた候補である。FNA方法は、典型的には超音波またはコンピュータ断層撮影法(CT)イメージングを使用してなされる。
【0047】
C. 内視鏡生検
内視鏡生検は、本発明において癌試料を得るために使用してもよい生検のうちの非常に一般的なタイプである。内視鏡生検は、サンプリング機器と共に体に挿入される内視鏡(体内を見るための光ファイバーケーブル)を介してなされる。内視鏡は、関心対象の器官の裏打ち上の領域を直接可視化すること;試料の内視鏡内部を走る長いケーブルに取り付けられた鉗子で組織の極めて小さな小片を収集し、またはつまみとることができる。内視鏡生検は、消化管(消化管内視鏡検査)、膀胱(膀胱鏡検査)、腹腔(腹腔鏡検査)、関節腔(関節鏡検査)、胸部の中間部分(縦隔鏡)、または気管および気管支系(喉頭鏡検査および気管支鏡検査法)で、天然の体の開口部か、もしくはある小さな手術切開を介して、いずれかで行ってもよい。
【0048】
D. 表面生検
本発明において癌試料を得るために、表面生検を使用してもよい。この技術は、細胞を除去するために、組織もしくは器官の表面のサンプリングすること、または削ることを含む。表面生検は、皮膚のわずかな部分を除去するために行われることが多い。
【0049】
IV. 細胞周期分子(パラメーター)
タキサン化学受容性を評価または決定する際に、本発明は、患者の癌細胞における細胞周期分子もしくは因子の発現レベルおよび活性を評価または決定する。
【0050】
主に、細胞周期制御因子の2つの群が、細胞に存在する。一方は、正の制御因子であるサイクリン依存性キナーゼ(CDK)と称されるキナーゼ群であり、他方は、負の制御因子であるCDK阻害剤(CDKI)群である。CDKは、細胞質に不活性型として存在する。CDKは、活性化されて、たとえばリン酸化されて、細胞の核へ移動する。核では、CDKがサイクリン分子と結合して、サイクリンと複合体を形成し(本明細書において、活性化されたCDKと称する)、細胞周期の種々の段階で細胞周期の進行を正に調節する。一方で、CDKIは、活性化されたCDKまたはCDK単体に結合することによってCDKを不活性化し、これにより負に細胞周期を調節する。
【0051】
CDK1キナーゼは、有糸分裂サイクリンと合わせて、有糸分裂の制御のために必要とされる普遍的マスター・キナーゼであって、パクリタキセルが攻撃するキナーゼである。このキナーゼは、細胞周期のG2期に活性化されて、有糸分裂初期において核膜の破壊、クロマチンの凝縮、および紡錐体装置の形成を促進する。対照的に、中期を出るためには、APC-プロテアソーム経路によって、その構築パートナーのサイクリンBの分解を介してCDK1が不活性化されなければならない。CDK1活性は、紡錘体が動原体に完全に付着されるまで、高いままである。
【0052】
A. サイクリン依存性キナーゼ(CDK)
異なるサイクリンが結合するCDKの7つのタイプ、すなわちCDK1、CDK2、CDK3、CDK4、CDK5、CDK6、およびCDK7が公知である。より詳細には、CDK1は、サイクリンAまたはBと結合し、CDK2は、サイクリンAまたはEと結合し、CDK4およびCDK6は、サイクリンD1、D2、またはD3と結合して活性化される。活性化されたCDKは、細胞周期の特定の期を制御する。従って、細胞周期が制御されて、細胞増殖は、異なるタイプのCDKの活性化によって調節される。活性化されたCDKは、基質としてタンパク質のセリン残基およびスレオニン残基をリン酸化する。インビトロ反応系では、活性化されたCDK1およびCDK2が、基質としてヒストンH1に対して十分に反応し、および活性化されたCDK4およびCDK6が、基質としてRb(網膜芽細胞腫タンパク質)に対して十分に反応する。インビボでの細胞周期制御では、活性化されたCDKは、生理的基質としてRbを必要とすることが考えられるが、その他のどのタンパク質が基質として作用するかは公知でない。
【0053】
上述のとおり、CDKおよびサイクリンは、互いに密接に会合して細胞周期を調節する。サイクリンD1遺伝子の増殖は、食道癌の多くの場合に観察されるが、一方で、サイクリンD1遺伝子の過剰発現は、胃癌および大腸癌の多くの場合に観察される。一方、サイクリンE遺伝子の増殖は、胃癌および大腸癌において観察されるが、食道癌では観察されない。胃および大腸におけるサイクリンEの過剰発現は、腺腫および腺癌の場合に高頻度で生じ、浸潤、段階の進行、転移等の悪性度と有意な相関を示す。CDK1の発現およびキナーゼ活性は、正常な粘膜組織と比較して、胃癌および大腸癌の大部分の場合に著しく促進される。サイクリン遺伝子の発現が増大されることは、種々の癌の進行および悪性度と相関することが公知である(Wataru et al., 2000を参照されたい)。
【0054】
従って、CDKの個体種の発現レベルもしくは活性を評価するか、決定するか、または測定することにより、タキサンに対する患者の癌細胞の細胞周期プロファイリングを提供し、これにより癌を予測するかまたは診断することが考えられる。言い換えると、一般にR位では、CDK2の発現が減少して、細胞周期停止および細胞の分裂が制御されている。しかし、CDK2の発現が、R位で増大する場合には、細胞周期が止まることができないことを意味し、すなわち、癌などの疾患の状態を意味する。
【0055】
通常、CDKの活性は、放射性同位元素を使用して決定される。より詳細には、抗CDK抗体を使用する免疫沈降法によって細胞可溶化物から抽出され、かつその活性が未知であるCDKの存在下において、32P標識されたアデノシン5'-O-(3-三リン酸)(ATP)を基質中でセリン残基またはスレオニン残基と反応させて、32P標識されたATPに由来する一リン酸基を導入する。基質によって取り込まれた32Pの量をオートラジオグラフィーによって、またはシンチレーションカウンターによって検出する。これにより、リン酸化された基質の量を測定して、CDKの活性をリン酸化された基質の量から算出する。
【0056】
B. CDK阻害剤(CDKI)
p16INK4は、p16B、p19、p21WAF1、およびp27KIP1をも含むCDK阻害性タンパク質の新たに記載されたクラスに属する。p16INK4遺伝子は、9p21にマップし、多くの腫瘍型において染色体領域が頻繁に欠失されている。p16INK4遺伝子のホモ接合性欠失および突然変異は、ヒト腫瘍株に頻繁に存在する。これは、p16INK4遺伝子が、癌抑制遺伝子であることを示唆した。しかし、この解釈は、p16INK4遺伝子変化の頻度が、培養された株化細胞におけるよりも、初代無培養腫瘍において非常に低いという知見によって疑いがもたれていた(Caldas et al., 1994;Cheng et al., 1994;Hussussian et al., 1994;Kamb et al., 1994a;Kamb et al., 1994b;Mori et al., 1994;Okamoto et al., 1994;Nobori et al., 1995;Orlow et al., 1994;Arap et al., 1995)。プラスミド発現ベクターでトランスフェクションを行うことによる野生型p16INK4機能の回復により、いくつかのヒト癌株化細胞によるコロニー形成が減少した(Okamoto, 1994;Arap, 1995)。
【0057】
細胞増殖のもう一つの阻害剤は、p16である。真核生物の細胞周期の主な移行は、サイクリン依存性キナーゼ、またはCDKによってトリガーされる。1つのCDK、サイクリン依存性キナーゼ4(CDK4)は、G1を介して進行を調節する。この酵素の活性は、後期のG1にてRbをリン酸化することである可能性がある。CDK4の活性は、サブユニットD型サイクリンを活性化することによって、および阻害性サブユニットによって制御されており、p16INK4は、CDK4に特異的に結合して阻害し、したがって、Rbリン酸化を調節し得るタンパク質として生化学的に特徴づけられた(Serrano et al., 1993;Serrano et al., 1995)。p16INK4タンパク質は、CDK4阻害剤であるので(Serrano, 1993)、この遺伝子の欠失は、CDK4の活性を増大して、Rbタンパク質の過剰リン酸化を生じ得る。また、p16は、CDK6の機能を調節することが公知である。
【0058】
p21WAF1/CIP1は、最初にp53誘導性タンパク質として同定された。p53の減少は、腫瘍細胞における共通の現象であり、放射線療法または化学療法に対する細胞の反応に変化を生じさせる。p53を媒介した細胞周期チェックポイントを欠いた細胞は、ますます遺伝的に不安定で、アポトーシスになりにくくなる。DNA損傷に応答して、p21は、種々のサイクリン/CDK複合体と結合してこれらの複合体の活性を阻害し、p53依存的様式で細胞周期進行の抑制を生じることが証明された。最近では、p21Waf1/CIP1のアップレギュレーションをp53の他のその他のシスまたはトランス・エレメントによって達成することができることも示された。
【0059】
C. 紡錘体形成チェックポイント分子
有糸分裂紡錘体形成チェックポイントでは、未熟な染色体の分裂を防止するために、紡錘体に対する染色体の付着および微小管によって作製される姉妹染色分体全体の緊張の両方をモニターする。パクリタキセルなどの薬物が、微小管を安定化して紡錘体の形成の間に生じる動的変化を妨害する場合に、紡錘体形成チェックポイントが活性化されて、有糸分裂で細胞停止される。
【0060】
紡錘体形成チェックポイントの分子構成成分は、最初に酵母(Saccharomyces cerevisiae)において同定された。チェックポイント・タンパク質の哺乳類相同体は、Mad1、Mad2、BubR1、Bub3、およびMps1を含む(LiおよびBenezra, 1996;Jin et al., 1998;Taylor et al., 1998;Chan et al., 1999)。チェックポイント機構は、Mad1、Mad2、BubR1、Bub3、およびcdc20で構成されたタンパク質複合体であり、染色体の動原体に位置する。このチェックポイントの標的は、後期促進複合体(APC)およびその活性化補助因子Cdc20である。Mad2およびBubR1は、下流に位置し、この機構の主要タンパク質であると思われ、Cdc20と直接相互作用して、APC活性を協同的に阻害する(Fang et al., 1998; Sudakin et al., 2001; Tang et al., 2001; Fang, 2002)。腫瘍細胞では、チェックポイントの欠陥が観察されることが多く、これらはゲノム不安定化を誘導すると考えられている。
【0061】
V. 細胞周期パラメーターの発現または活性を決定する方法
A. 細胞周期プロファイリング系
タキサン化学受容性に関与する細胞周期分子を同定するために、本発明は、細胞周期プロファイリングのために多パラメーター解析を使用する。この系は、ドットブロット技術に基づいた、日本特許出願第200348653号(その全体が本明細書に組み入れられる)に記載されている装置を使用して、同位元素を用いずに正常および癌組織の小さな臨床試料におけるタンパク質発現、並びに活性を迅速に、定量的に、かつ自動的にアッセイし得る。CDKの活性およびその他の種類のタンパク質の発現を測定するためには、2mm3だけの組織試料で十分である。この細胞周期プロファイリング系/装置は、多くの疾患の診断の際の、または分子ターゲティング療法を含む種々の療法に対する予後もしくは患者の感受性を評価する際の、その他のキナーゼのまたはタンパク質の活性の測定など種々の適用可能性を有する。また、この装置/系は、個々の疾患に対する危険因子の診断のために使用してもよい。
【0062】
1. タンパク質発現を測定する方法
本発明に使用されるアッセイ法プロトコルの原理を下記に簡単に説明してある。細胞周期分子(パラメーター)のタンパク質発現は、CPDIBという名で(粗タンパク質直接ブロッティング、米国出願第10/423,892号;その全体が参照として本明細書に組み入れられる)かつドットブロット技術に基づいた新たな技術によって測定してもよい。解析は、3工程だけを含む:疎水性膜に対する粗製細胞可溶化物の直接固定化、一次抗体の反応、および結合した一次抗体の検出。たとえば、ビオチン化二次抗体およびフルオレッセイン標識されたストレプトアビジンの逐次反応による。合計アッセイ時間は、3時間以内である。相対的蛍光単位と標準的な組換えタンパク質の量とは、直線的関連があることが確かめられる。MAD2のためのアッセイ条件は、現在最適化されている。本系は、1つの標的分子に対する1つの特異的なポリクローナル抗体が必要なだけであるので、本系の主な利点は、プロファイリングのための新たなパラメーターを追加することが、新たなサンドイッチELISA系を開発するよりも非常に容易であることである。
【0063】
自動化解析のための活性および発現の両方の測定手順が開発された。外科的に切開した組織の小片(2mm3)の可溶化液は、新しく開発された組織ホモジナイザー(Sysmex, Kobe, Japan)を使用して溶解緩衝液(0.1%のNP-40、20 mM トリス-HCl[pH 7.4]、150 mM NaCl、2% プロテイナーゼ阻害剤カクテル(Sigma, St Louis, MO)で調製してもよい。ホモジナイザーは、フィルターディスク上の不溶性物質を自動的に除去する。タンパク質濃度を解析し(DCキット, Pierce, Rockford, IL)、2.5μgの総タンパク質量を新しく開発した疎水性膜(0.22μm孔を有するPVDF;Millipore, Billerica, MA)を有する5×7cm2ドットブロット装置(Sysmex)の78μlウェル(3mm[w]×5mm[l]×7mm[d])に加える。膜に結合した粗試料中の標的タンパク質は、抗CDK抗体(Santa Cruz Biotechnology, Santa Cruz, CA)、ビオチン化二次抗体(Santa Cruz Biotechnology)、およびフルオレッセイン標識されたストレプトアビジン(Vector, Burlingame, CA)と連続して反応させることによって定量的に検出してもよい。それぞれの反応の間に、ウェルがTBS溶液(25mMのトリス-HCl[pH 7.4]、150 mM NaCl)で自動的に洗浄される。膜の蛍光イメージをイメージアナライザー(Bio-Rad, Hercules, CA, USA)を使用して解析し、ドットの強度を「Quantity One」(Bio-Rad)によって定量化する。相対的蛍光ユニット(RFU)および標準的な組換えタンパク質(Santa Cruz Biotechnology)の量を、標準化された範囲内で直線的に相関させる(たとえば、CDK1、2.5〜25ng/ドット;CDK2、1.0〜10ng/ドット;CDK4、1.0〜10ng/ドット;CDK6、2.5〜25ng/ドット)。
【0064】
2. キナーゼ活性の測定
CDKの酵素アッセイのために、非放射同位元素CDKアッセイ法を使用した(米国特許出願第20020164673号)。本アッセイ法は、以下の工程を含む:対応する抗体(たとえば、抗CDK1、抗CDK2、抗CDK4、または抗CDK6抗体)とプロテインAビーズとによるCDK分子の沈澱。基質のセリンまたはスレオニン残基にモノチオリン酸エステル基を導入するために、タンパク質基質およびアデノシン5'-O-(3-チオ三リン酸エステル)(ATP-γS)のキナーゼ緩衝液の溶液を含む基質混合物を反応させる工程(タンパク質基質として、CDK1およびCDK2のためにはヒストンH1を、並びにCDK4およびCDK6のためには組換えRBタンパク質(アミノ酸769〜921)を使用してもよい)。標識化フルオロフォアまたは標識化酵素を、導入されたモノチオリン酸エステル基の硫黄原子と結合することによって基質を標識する工程。基質を標識する標識化フルオロフォアから蛍光の量を測定する工程か、または基質を標識する標識化酵素を、標識化酵素との反応によって光学的に検出可能な生成物を生じる物質と反応させて、生じる生成物の量を光学的に測定する工程。測定された蛍光の量または測定された生じた生成物の量からサイクリン依存性キナーゼの活性を、予め作製した参照曲線を基準として算出する工程。
【0065】
細胞可溶化物は、発現解析について記載したとおりに調製される。CDK分子を、2μgの対応する抗体(抗CDK1抗体、抗CDK2抗体、抗CDK4抗体、または抗CDK6抗体;Santa Cruz Biotechnology)および20μlのプロテインAビーズ(Amersham Pharmacia, Uppsala, Sweden)で、100μgの可溶化液総タンパク質から、4℃で1時間選択的に沈殿させる。洗浄緩衝液(0.1%のNP-40、50 mM トリス-Cl[pH 7.4])で3回洗浄後、10μgのタンパク質基質、5mMアデノシン5'-O-(3-チオ三リン酸エステル)(Sigma)、20mM、トリス-Cl(pH 7.4)、および0.1%のトリトンX-100を含む50μlの基質混合物をビーズに添加して、連続的振盪下で37℃において10分間インキュベートする。タンパク質基質として、CDK1およびCDK2のためにはヒストンH1(Upstate Biotechnology, Lake Placid, NY)を、CDK4およびCDK6のためには組換えRBタンパク質(アミノ酸769〜921)を使用してもよい。ビーズを除去した後、基質に導入されたモノチオリン酸エステルを、10mMのヨードアセチル-ビオチン(Pierce)と共に、結合緩衝液(100mMのトリス-Cl[pH 8.5] ,1 mM EDTA)中で、暗がりで室温において90分間インキュベーションすることによってさらに標識する。反応をβ-メルカプトエタノールで停止して、0.4μgの基質をドットブロット装置(Sysmex)のウェルに加える。ウェルを4%のウシ血清アルブミン(BSA)で30分間ブロッキングし、次いで37℃において1時間アビジン-FITC(Vector)と共にインキュベートした。膜を洗浄した後、イメージアナライザー(Bio-Rad)を使用してイメージを評価し、ドットの蛍光強度を「Quantity One」(Bio-Rad)を使用して定量化する。癌株化細胞から抽出された0μg、12.5μg、25μg、50μg、100μg、および150μgのタンパク質に対応するCDK活性で調製した検量線によって活性を算出する。1ユニット(U)は、細胞からの1μgの総タンパク質量のキナーゼ活性に匹敵する。
【0066】
VI. 薬学的組成物、送達、および治療の計画
本発明の態様において、癌を有する患者のタキサン化学受容性の評価に基づいて、効率的に癌を治療および予防する方法が想定される。治療のために想定される癌の例は、肺癌、頭頚部癌、乳癌、膵癌、前立腺癌、腎癌、骨癌、精巣癌、子宮頚癌、胃腸癌、リンパ腫、肺における新生物発生前の病変、大腸癌、黒色腫、膀胱癌、および他の任意の腫瘍疾患も含む。
【0067】
薬学的タキサン組成物の有効な量は、一般に、検出可能な程度に、かつ繰り返し、疾患、またはこれらの症状もしくは徴候の範囲を改善するか、減少させるか、最小化するか、または制限するために十分なその量として定義される。疾患の除去、根絶、または治癒を含むより厳密な定義を適用してもよい。好ましくは、患者は、十分な骨髄機能(>2,000/mm3の末梢絶対顆粒球数および100,000/mm3の血小板数として定義される)、十分な肝機能(ビリルビン<1.5mg/dl)および十分な腎機能(クレアチニン<1.5mg/dl)を有する。
【0068】
A. タキサン投与
本発明の方法および組成物を使用して細胞を死滅させ、細胞増殖を阻害し、転移を阻害し、腫瘍または組織サイズを減少し、およびそうでなければ、腫瘍細胞の悪性の表現型を逆転し、または減少させるためには、一般にタキサン化合物と癌細胞を接触させることが考えられる。投与の経路は、必然的に、病変の位置および性質と共に変化し、たとえば、皮内、経皮、非経口的、静脈内、筋肉内、鼻腔内、皮下、経皮的、気管内、腹腔内、腫瘍内、灌流、洗浄液、直接注射、並びに経口投与および製剤を含む。また、癌の治療または診断に関して議論した任意の製剤および投与経路を、腫瘍性疾患および症状に関して使用してもよい。
【0069】
腫瘍内注射、または腫瘍脈管構造への注射は、具体的には、分離した、固体の、アクセスできる腫瘍について想定される。また、局部的、局所的、または全身投与が適切である場合がある。>4cmの腫瘍については、投与される量は、約4〜10ml(好ましくは10ml)であるが、一方、<4cmの腫瘍については、約1〜3ml(好ましくは3ml)の量が使用される。単一用量として送達される複数の注射では、約0.1〜約0.5mlの量を含む。ウイルス粒子は、好都合なことに、腫瘍に対して、約1cmの間隔で間隔を置いて、複数の注射を投与することによって接触してもよい。
【0070】
手術介入の場合、本発明を術前に使用して、手術不能の腫瘍を切除に供してもよい。または、本発明は、手術時に、および/またはその後に、残留するかもしくは転移性の疾患を治療するために使用してもよい。たとえば、切除した腫瘍ベッドを、タキサン化合物を含む製剤で注射または灌流してもよい。灌流は、たとえば、カテーテルを手術部位に移植したままにしておくことによって、切除後に続けてもよい。また、周期的な術後治療も考えられる。
【0071】
必要に応じて、たとえば、腫瘍が切除されており、腫瘍ベッドが残留する顕微鏡学的疾患を除去するための治療をする場合、連続投与を適用してもよい。注射器またはカテーテル処置(catherization)を経た送達が好ましい。このような連続灌流は、治療の開始に続いて少なくとも約1〜2時間、約2〜6時間、約6〜12時間、約12〜24時間、約1〜2日、約1〜2週の期間行ってもよい。一般に、連続灌流を経たタキサン組成物の投与は、一回または複数回の注射によって与えられるものに匹敵し、灌流が生じる期間を通して調節される。本発明のタキサン組成物を投与するために肢灌流を使用してもよいことが、さらに想定される。
【0072】
治療計画は、さらに変更してもよく、たいてい腫瘍型、腫瘍位置、疾患進行、並びに患者の健康および年齢に依存する。明らかであるが、一定タイプの腫瘍は、より攻撃的な治療を必要とするが、一方で同時に、一定の患者は、より負担の重いプロトコルを許容することができない。臨床家は、細胞周期プロファイリング並びにタキサン製剤の公知の有効性および毒性(あるとしても)によるタキサン化学受容性の評価に基づいて、このような決定を行うことが最適である。
【0073】
ある態様において、治療される腫瘍は、少なくとも最初に、切除可能でなくてもよい。治療組成物での治療により、縁が収縮することにより、または一定の特に浸潤部分を除去することによって、腫瘍の切除可能性を増大してもよい。治療に続いて切除が可能である場合がある。切除後の後処置は、腫瘍部位の顕微鏡学的な残留疾患を排除するのに役立つと考えられる。
【0074】
典型的な治療の経過は、原発腫瘍または切除後腫瘍ベッドに対する多回投与を含む。典型的な原発腫瘍治療は、2週間にわたって6用量の適用を含む。2週の処方計画を1回、2回、3回、4回、5回、6回またはそれ以上繰り返してもよい。治療経過の間に、予定の投薬を完了する必要性を再評価してもよい。
【0075】
治療は、種々の「単位用量」を含んでもよい。単位用量は、タキサン組成物の予定された量を含むものとして定義される。投与される量、並びに特定の経路および製剤は、臨床分野の当業者の範囲内である。単位用量は、単回注射として投与される必要はなく、セット期間を通した連続的注入を含んでもよい。
【0076】
B. 注射剤組成物および製剤
本発明の癌細胞に対するタキサン組成物の送達のための好ましい方法は、全身的であるか、または腫瘍内注射を介する。しかし、本明細書に開示された薬学的組成物は、代わりに、米国特許第5,543,158号;米国特許第5,641,515号、および米国特許第5,399,363号(それぞれ、その全体が参照として本明細書に具体的に組み入れられる)に記載されているように、非経口的、静脈内、皮内、筋肉内、経皮的、またはさらに腹腔内に投与してもよい。
【0077】
タキサン組成物の注射は、化合物が、注射のために必要とされる針の特定のゲージを貫通することができる限り、注射器によって、または溶液の注射のために使用される他の任意の方法によって送達してもよい。溶液を保持するためのアンプル・チャンバーと、ノズルから送達部位に溶液を押し出すためのエネルギー装置とを定義するノズルを有する新規の針のない注射システムが記載されている(米国特許第5,846,233号)。また、任意の深度に正確に溶液の予め定められた量の複数回の注射が可能な、遺伝子療法に使用するための注射器系も記載されている(米国特許第5,846,225号)。
【0078】
遊離塩基または薬理学的に許容される塩としての活性化合物の溶液は、ヒドロキシプロピルセルロースなどの界面活性物質と最適に混合した水溶液に調製してもよい。また、分散液は、グリセロール、液体ポリエチレングリコール、およびこれらの混合物の溶液に、並びに油に調製してもよい。通常の貯蔵および使用条件下では、これらの標品は、微生物の増殖を防止するための保存剤を含む。注射用に使用するために適した剤型は、無菌の水溶液または分散液、および無菌の注射用の溶液または分散液の即座の調製のための無菌の粉末を含む(米国特許第5,466,468号、その全体が参照として具体的に本明細書に組み入れられる)。全ての場合において、形態は、無菌でなければならず、かつ簡単に注射可能(syringability)である程度に流動性でなければならない。これは、製造および貯蔵条件下で安定でなければならず、かつ細菌および真菌などの微生物の混入作用を避けて保存されなければならない。担体は、たとえば水、エタノール、ポリオール(たとえば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、これらの適切な混合物、および/または植物油を含む溶媒または分散媒であることができる。たとえば、レシチンなどのコーティングの使用によって、分散の場合には必要とされる粒径を維持することによって、および界面活性物質を使用することによって、適当な流動性が、維持されてもよい。微生物の作用の予防は、種々の抗菌薬および抗真菌薬、たとえばパラベン、クロロブタノール、フェノール、ソルビン酸、チメロサール等によってもたらすことができる。多くの場合、等張性の薬剤、たとえば糖または塩化ナトリウムを含むことが好ましい。注射用組成物の吸収の延長は、吸収を遅延させる薬剤、たとえばモノステアリン酸アルミニウムおよびゼラチンを組成物に使用することによってもたらすことができる。
【0079】
水溶液の非経口投与については、たとえば、必要に応じて溶液を適切に緩衝化し、液状希釈液を最初に十分な食塩水またはグルコースで等張性にするべきである。これらの特定の水溶液は、特に静脈内、筋肉内、皮下、腫瘍内、および腹腔内投与のために適している。この点について、使用することができる無菌水性培地は、本開示の観点から当業者には公知である。たとえば、1用量は、1mlの等張性NaCl溶液に溶解して、1000mlの皮下注入液体に添加するか、または提唱された注入部位のいずれかに注射してもよい(たとえば「Remington's Pharmaceutical Sciences」第15版、1035-1038頁および1570-1580頁を参照されたい)。治療される被検者の症状に応じて、必然的に用量のいくらかの変更が生じる。いずれにしても、投与に対して責任がある人が、個々の被検者に適した用量を決定する。さらに、ヒト投与については、製剤は、FDA生物学的基準局(FDA Office of Biologics standards)によって要求されるとおりの無菌性、発熱原性、一般的安全性、および純度基準を満たすべきである。
【0080】
無菌注射用溶液は、必要に応じて上記で列挙した種々のその他の成分と共に、適切な溶媒中に必要とされる量で活性化合物を組み入れ、続いて濾過滅菌することによって調製される。一般に、分散液は、種々の殺菌された活性成分を、塩基性分散媒と上記で列挙したものから必要とされるその他の成分とを含む無菌媒体に組み入れることによって調製される。無菌注射用溶液の調製のための無菌粉末の場合、好ましい調製法は、これらの事前に滅菌濾過した溶液から、活性成分プラス任意のさらなる所望の成分の粉末を得る、真空乾燥および凍結乾燥技術である。
【0081】
本明細書に開示された組成物は、中性または塩形態に製剤化してもよい。薬学的に許容される塩は、酸付加塩(タンパク質の遊離アミノ基と形成される)を含み、酸付加塩は、たとえば、塩酸もしくはリン酸などの無機酸、または酢酸、シュウ酸、酒石酸、マンデル酸などの有機酸で形成される。また、遊離カルボキシル基で形成された塩は、たとえば、ナトリウム、カリウム、アンモニウム、カルシウム、または水酸化第二鉄などの無機塩基、およびイソプロピルアミン、トリメチルアミン、ヒスチジン、プロカインなどの有機塩基などに由来することができる。製剤に関しては、用量製剤と適合した様式で、治療的に有効であるような量で溶液が投与される。製剤は、注射用溶液、薬物放出カプセルなどの種々の剤形で容易に投与される。
【0082】
本明細書に使用される「担体」は、任意の、並びに全ての溶媒、分散媒、媒体、コーティング、希釈液、抗菌薬および抗真菌薬、等張性の吸収遅延薬、緩衝液、担体溶液、懸濁液、コロイドなどを含む。薬学的に活性な物質のためのこのような媒体および薬剤の使用は、当技術分野において周知である。任意の従来の媒質または薬剤が、活性成分と不適合性である場合以外は、治療組成物におけるその使用が想定される。また、補充活性成分を組成物に組み入れることもできる。
【0083】
「薬学的に許容される」または「薬理学的に許容される」という句は、ヒトに投与される場合に、アレルギー性または同様の有害反応を生じない分子的実体および組成物をいう。活性成分としてタンパク質を含む水性組成物の調製も、当技術分野において周知である。典型的には、このような組成物は、液体溶液または懸濁液のいずれかとして注射剤として調製され;注射前に液体の溶液に、または懸濁液に適した固体形態も調製することができる。
【0084】
VII. 併用治療
本発明の化合物および方法は、癌を含む腫瘍性疾患/症状に関して使用してもよい。癌のタイプは、肺癌、頭頚部癌、乳癌、膵癌、前立腺癌、腎癌、骨癌、精巣癌、子宮頚癌、胃腸癌、リンパ腫、肺における新生物発生前の病変、大腸癌、黒色腫、膀胱癌、および他のいかなる腫瘍疾患も含んでもよい。本発明のタキサン組成物での治療の有効性を増大するために、パクリタキセル、ドセタキセル、またはこれらの類似体などの、これらの疾患および症状の治療に有効なその他の薬剤とこれらの組成物を併用することが望ましい場合がある。たとえば、本発明のタキサン化合物および抗癌剤または手術などのその他の抗癌療法と共に、癌の治療を実行してもよい。
【0085】
種々の組み合わせを使用してもよく;たとえば、タキサン組成物は、「A」、および抗癌療法は、「B」である。
【0086】
患者に対する本発明の治療組成物の投与は、その特定の二次療法を適用するための一般的なプロトコルに従って、もしあれば、タキサン治療の毒性を考慮する。必要に応じて、治療サイクルを繰り返すことが予想される。また、種々の標準療法、並びに外科的介入を、記載した癌細胞と併用して適用してもよいことが想定される。
【0087】
A. 抗癌治療
本発明で使用することが想定される抗癌治療は、たとえば、癌細胞を死滅させるか、癌細胞のアポトーシスを誘導するか、癌細胞の増殖速度を減少させるか、転移の発病率もしくは数を減少させるか、腫瘍サイズを減少させるか、腫瘍成長を阻害するか、腫瘍もしくは癌細胞に対する血液供給を減少させるか、癌細胞もしくは腫瘍に対する免疫応答を促進するか、癌の進行を防げるか、もしくは阻害するか、または癌である被検者の寿命を増大することにより、被検者において癌に負の影響を与えることができると思われる。抗癌治療は、生物学的薬剤(生物療法)、化学療法薬、および放射線療法薬を含む。生物学的療法と化学療法との併用は、生物化学療法として公知である。
【0088】
従って、本発明は、細胞の死滅または増殖を阻害するために有効な量を併用して提供されたタキサン組成物および抗癌剤を想定する。本方法は、細胞をタキサンおよび薬剤または因子と同時に接触させる工程を含んでもよい。これは、細胞をタキサンおよびその他の薬剤の両方を含む単一の組成物もしくは薬理学的製剤と接触させることによって、または細胞を、一方の組成物がタキサンを含み、他方は抗癌剤を含む2つの異なる組成物もしくは製剤と同時に接触させることによって達成してもよい。
【0089】
1. 化学療法
また、本発明において、タキサンを化学療法薬と併用して使用してもよいことが想定される。このような化学療法薬は、たとえば、シスプラチン(CDDP)、カルボプラチン、プロカルバジン、メクロレタミン、シクロホスファミド、カンプトセシン、イホスファミド、メルファラン、クロランブシル、ブスルファン、ニトロソ尿素(nitrosurea)、ダクチノマイシン、ダウノルビシン、ドキソルビシン、ブレオマイシン、プリコマイシン(plicomycin)、マイトマイシン、エトポシド(VP 16)、タモキシフェン、ラロキシフェン、エストロゲン受容体結合剤、タキソール、ゲムシタビン、ナベルビン、ファルネシル-タンパク質トランスフェラーゼ阻害剤、トランスプラチナ(transplatinum)、5-フルオロウラシル、ビンクリスチン、ビンブラスチンおよびメトトレキセート、テマゾロミド(Temazolomide)(DTICの水性型)、またはこれらの任意の類似体もしくは誘導体を含んでもよい。膵癌を治療するために現在使用される化学療法薬の一例は、ゲムシタビンである。その他の研究では、進行した膵癌の治療のために、高用量の5-フルオロウラシル(5-FU)を使用する。
【0090】
また、タキサンは、タンパク質チロシンキナーゼ阻害剤などのその他の化学療法薬と併用して使用してもよい。このような阻害剤は、最適には、イマチニブまたはイマチニブメシレート(STI-571、グリベック(商標);Norvartis, Inc.)、OSI-774(タルセバ(商標);OSI Pharmaceuticals, Inc.)、ZD-1839(イレッサ(登録商標);AstraZeneca, Inc.)、SU-101(Sugen, Inc.)、およびCP-701(Cephalon, Inc.)を含んでもよい。
【0091】
2. 放射線療法
癌を有する患者を治療する際に、タキサンと併用して本発明に使用してもよいもう一つの療法は、放射線療法である。本発明に使用してもよい放射線療法因子は、DNA損傷を生じさせ、かつγ線、X線、および/または腫瘍細胞に対して放射性同位元素を直接送達するものなどの広範囲に使用されてきた因子であることが想定される。また、マイクロ波およびUV照射などのDNAに損害を与える因子のその他の形態も想定される。これらの因子は全て、DNAに対する、DNAの前駆体に対する、DNAの複製および修復に対する、並びに染色体の構築および維持に対する広範な損傷を及ぼす可能性が最も高い。X線の用量範囲は、長期間(3〜4週)にわたって50〜200レントゲンの1日量から2000〜6000レントゲンの一回用量の範囲である。放射性同位元素のための用量範囲は、広く変動し、同位元素の半減期、放射される放射線の強度および型、並びに癌または腫瘍細胞による摂取に依存する。
【0092】
3. 手術
癌である人の約60%は、予防的、診断的または段階的(staging)、治療的、並びに一時的な手術を含む、いくつかのタイプの手術を受けると考えられる。治療的手術は、本発明の治療、化学療法、放射線療法、ホルモン療法、遺伝子療法、免疫療法、および/または代替療法などのその他の療法と共に使用してもよい癌治療法である。
【0093】
治療的手術は、癌組織の全部または一部を物理的に除去し、切除し、および/または破壊する切除を含む。腫瘍切除は、腫瘍の少なくとも一部を物理的に除去することをいう。腫瘍切除に加えて、手術による治療は、レーザー手術、冷凍外科療法、電気外科療法、および顕微鏡的に制御された手術(モース手術(Mohs' surgery))を含む。本発明は、表在癌、前癌、または付随的な正常組織の量の除去と組み合わせて使用してもよいことがさらに想定される。
【0094】
癌細胞、組織、または腫瘍の全ての部分の切除術により、腔が体内に形成されてもよい。治療は、さらなる抗癌療法による領域の灌流、直接注射、または局所投与によって達成してもよい。このような治療は、たとえば1日、2日、3日、4日、5日、6日、もしくは7日毎、または1週、2週、3週、4週、および5週毎、または1ヶ月、2ヶ月、3ヶ月、4ヶ月、5ヶ月、6ヶ月、7ヶ月、8ヶ月、9ヶ月、10ヶ月、11ヶ月、もしくは12ヶ月毎に繰り返してもよい。これらの治療もまた、用量を変化させてもよい。
【0095】
VIII. 実施例
以下の実施例は、本発明の好ましい態様を証明するために含まれる。続く実施例に開示した技術は、本発明の実施において十分に機能することが本発明者らによって発見された技術を示すことが当業者により認識されるはずであり、したがって、その実施のための好ましい様式を構成するとみなすことができる。しかし、当業者であれば、本開示を考慮して、開示された特定の態様に多くの変更を行うことができ、本発明の精神と範囲から逸脱することなく、なおも同様の、または類似の結果を得ることができることを認識するはずである。
【0096】
実施例1
実験手順
株化細胞および細胞培養
本研究に使用される全てのヒト株化細胞組換えプラスミドの開発のためのHEK 293細胞;機能的な紡錘体形成チェックポイントを有することが公知であるMCF-7乳癌およびMCF-10A正常細胞;並びに低Mad2発現のために欠陥チェックポイントを有するT47D乳癌およびOvca432卵嚢癌細胞は、アメリカン・タイプ・カルチャー・コレクション(American Type Culture Collection)(Rockville, MD)から得た。HEK 293細胞、MCF-7細胞、およびT47D細胞は、ダルベッコ改変イーグル培地(DMEM)/F12培地中で培養した。Ovca432細胞は、RPMI 1640中で維持した。DMEM/F12培地およびRPMI 1640は、2mMのL-グルタミン、10%のウシ胎児血清(FBS;100IU/ml)、およびペニシリン・ストレプトマイシン(100mg/ml)を補った。MCF-10A細胞は、5%のウマ血清、0.02μg/mlの上皮細胞成長因子、0.5μg/mlのヒドロコルチゾン、10μg/mlのインスリン、0.1μg/mlのコレラ毒素、100IU/mlのペニシリン、および100mg/mlのストレプトマイシンを補ったDMEM/F12培地中で維持した。
【0097】
低分子干渉RNAトランスフェクション
Mad2配列 5'-
およびBubR1配列 5'-
を標的化するために、21ヌクレオチドのsiRNA二重鎖をDharmacon Research, Inc. (Lafayette, CO)によって合成した。MCF-7細胞のトランスフェクションは、オリゴフェクトアミントランスフェクション試薬(Invitrogen, Carlsbad, CA)を使用して、Dharmacon Researchによって提供されるプロトコルに従って行った。対照研究については、細胞はsiRNAスクランブル二重鎖(Dharmacon Research)でトランスフェクションを行った。siRNAの終濃度は、200nMであった。
【0098】
複製欠陥組換えアデノウイルスの作製
アデノウイルスは、以前に(He et al., 1998)およびStratagene(La Jolla, CA)記載されているプロトコルに従って作製した。簡潔には、最初に、cDNA Mad2の遺伝子をシャトルベクター、pAdTrack-サイトメガロウイルス(CMV)にクローニングした。生じるプラスミドを制限エンドヌクレアーゼPme Iで消化することによって直線化し、その後にアデノウイルス・バックボーン・プラスミドpAdEasy-1(Stratagene, La Jolla, CA)を使用して大腸菌(Escherichia coli.)BJ5183細胞に同時形質転換した。組換え体をカナマイシン耐性について選択し、組換えを制限エンドヌクレアーゼ解析によって確認した。最後に、HEK293細胞を直線化した組換えプラスミドでトランスフェクションを行った。研究については、それぞれの細胞において、細胞変性効果を伴わずに80〜90%の感染効率が得られた。
【0099】
ウェスタンブロット解析
トランスフェクションの24時間、48時間、および72時間後に、細胞を収集してタンパク質免疫ブロット解析に供した。細胞を氷冷リン酸緩衝食塩水中で一度洗浄し、プロテアーゼ阻害剤(1mMのフェニルメタンスルホニルフルオライドおよび10μg/mlアプロチニン)およびホスファターゼ阻害剤(20mMのβ-グリセロリン酸、5 mMのNaF、および100μMのNa3VO4)を含む溶解緩衝液(1%のNP-40、150mMのNaCl、および50mMのトリス-HCl[pH 7.5])で溶解した。氷上で30分後、細胞を4℃において15分間、13,000rpmでの遠心分離に供した。ウェスタンブロット法のために、同量のタンパク質をドデシル硫酸ナトリウム-ポリアクリルアミドゲル電気泳動を使用して分解し、ニトロセルロース膜に転送した。膜をポリクローナル抗Mad2抗体(Covance, Princeton, NJ; 1:500)、 モノクローナル抗BubR1抗体(Chemicon, Temecula, CA; 1:500)、およびモノクローナル抗αチューブリン(Sigma-Aldrich Chemical Co., St. Louis, MO; 1:5000)と共に室温で1時間(または4℃で一晩)インキュベートし、続いて西洋ワサビペルオキシダーゼ結合抗体とインキュベーションした。結果は、化学発光検出系で増強して視覚化した。
【0100】
薬物感受性アッセイ法
細胞をトリプシン処理によって剥離し、96ウェル・マイクロタイタープレートに2.0×103細胞/ウェルで播き、パクリタキセル(1、5、10、50、100、および1000 nM)の種々の濃度で処置した。72時間後に、細胞増殖に対する効果を3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミド(MTT)アッセイ法によって検査し:20μlのMTT溶液(5mg/mlのリン酸塩緩衝食塩水溶液;Sigma Aldrich)をそれぞれのウェルに添加して、細胞を37℃において4時間インキュベートした。代謝的に生存可能な細胞によって形成されたMTT-ホルマザンを100μlの細胞溶解緩衝液に溶解し、570nmの波長でマイクロプレートを使用して蛍光をモニターした。細胞増殖率は、パクリタキセル(対照)で処置されていない細胞の吸収を100%と定義することによって算出した。
【0101】
有糸分裂指数の算出
有糸分裂の凝縮染色質をもつ細胞は、10μg/mlヨウ化プロピジウムと連動して10μMのヘキストで33342色素(Aventis Pharmaceuticals Inc., Bridgewater, NJ)で染色することによって視覚化し;ヨウ化プロピジウムは、死細胞だけに組みこまれた。従って、死細胞は、ヨウ化プロピジウムおよびヘキスト33342色素の両方で染色されるが、有糸分裂細胞は、ヘキスト33342色素のみで凝縮染色質を示した。細胞をトランスフェクションの12時間、24時間、および36時間後に収集して有糸分裂指数を算出した。
【0102】
細胞死解析
細胞死は、トリパンブルー染料排除アッセイ法を使用して評価した。簡潔には、細胞を、トリプシンを使用して収集し、0.4%のトリパンブルー色素(Sigma-Aldrich Chemical Co., St. Louis, MO)で染色した。トリパンブルー陽性および陰性の細胞は、位相差顕微鏡(Fisher Scientific, Pittsburgh, PA)下で、血球計算板(Hausser Scientific, Horsham, PA)を使用して計数した。それぞれのアッセイ結果は、細胞の総数を基準として死細胞の割合に関して表してある。個々の実験は、三回行った。結果は、平均値±標準偏差として報告してある。
【0103】
Cdk1キナーゼ・アッセイ法
Cdk1プロテインキナーゼ・アッセイ法は、SignaTECT cdc2タンパク質アッセイ法系(Promega, Madison, WI)を使用して行った。簡潔には、収集した細胞をプロテアーゼ阻害剤(100μg/mlのアプロチニンおよび0.5mMのフェニルメタンスルホニルフルオライド)およびホスファターゼ阻害剤(50mMのNaF)を含む抽出緩衝液(50mMのトリス[pH 7.4]、150 mMのNaCl、0.1%のトリトンX-100、および1mMのEDTA)で溶解した。これらの可溶化液を、ヒストンH1および[γ-32P] ATPに由来するcdc2特異的ビオチン化ペプチドからなる基質と結合させて、30℃において10分間インキュベートした。これらの放射標識された、リン酸化された基質をストレプトアビジン・マトリックス・ビオチン捕獲膜で回収した(SAM;Promega)。数回の洗浄後、それぞれの捕獲した膜を別々のバイアルに入れて、液体シンチレーションカウンター(Beckman Coulter, Palo Alto, CA)を使用して解析した。
【0104】
細胞周期プロファイリングための細胞可溶化物の調製
細胞周期プロファイリングに供される株化細胞の可溶化液は、以下のとおりに調製した。細胞を10%のFCS(ウシ胎児血清)を含むDMEM(ダルベッコ改変イーグル培地)で培養し、100nMのパクリタキセルで0時間、24時間、48時間、または72時間処置した。処置後、細胞を収集して、PBSで一度洗浄した。次いで、細胞を21Gの針で20回注射することによって溶解緩衝液(0.1%のNP-40、20 mMのトリス-HCl[pH 7.4]、150 mMのNaCl、2% プロテイナーゼ阻害剤反応混液[Sigma, St Louis, MO, USA])で溶解した。不溶性物質を除去するために5分間の15000rpmの遠心分離後、上清のタンパク質濃度を解析し(DCキット, Pierce, Rockford, IL, USA)、使用するまで-80℃で貯蔵した。
【0105】
細胞周期プロファイリング発現解析
細胞周期プロファイリングの発現解析については、2.5μgの総タンパク質を、疎水性膜(0.22μmの孔をもつPVDF;Millipore, Billerica, MA(USA))をもつ5×7cm2のドットブロット装置の78μlのウェル(3mm [w]×5mm [l]×7mm[d])に加えた。膜に結合した粗試料中の標的タンパク質は、抗CDK抗体(Santa Cruz Biotechnology, Santa Cruz, CA, USA)、ビオチン化二次抗体(Santa Cruz Biotechnology)、およびフルオレッセイン標識されたストレプトアビジン(Vector, Burlingame, CA, USA)での連続反応によって定量的に検出した。それぞれの反応間に、ウェルをTBS溶液(25mMのトリス-HCl[pH 7.4]、150 mMのNaCl)で自動洗浄した。膜の蛍光イメージは、イメージアナライザー(Bio-Rad, Hercules, CA, USA)を使用して解析し、ドットの強度は、「Quantity One」ソフトウェア(Bio-Rad)を使用して定量化した。相対的蛍光単位(RFU)および標準的組換えタンパク質(Santa Cruz Biotechnology)の量は、標準化された範囲で直線的に相関させた(CDK1、2.5〜25ng/ドット;CDK2、1.0〜10ng/ドット;CDK4、1.0〜10ng/ドット;CDK6、2.5〜25ng/ドット)。
【0106】
細胞周期プロファイリング酵素活性解析
細胞周期プロファイリングの酵素活性解析は、非放射同位元素法を使用して行った。細胞可溶化物は、発現解析について記載したとおりに調製した。CDK分子は、2μgの対応する抗体(抗CDK1、抗CDK2、抗CDK4、または抗CDK6抗体;Santa Cruz Biotechnology)および20μlのプロテインAビーズ(Amersham Pharmacia, Uppsala, Sweden)で、100μgの可溶化液総タンパク質量から4℃において1時間、選択的に沈殿させた。洗浄緩衝液(0.1%のNP-40、50 mM トリス-Cl[pH 7.4])で3回洗浄後、10μgのタンパク質基質、5mMアデノシン5'-O-(3-チオ三リン酸エステル)(Sigma)、20mM トリス-Cl(pH 7.4)、および0.1%のトリトンX-100を含む50μlの基質混合物をビーズに添加して、37℃において10分間、連続振盪下でインキュベートした。タンパク質基質として、CDK1およびCDK2のためにはヒストンH1(Upstate Biotechnology, Lake Placid, NY, USA)を、並びにCDK4およびCDK6のためには組換えRBタンパク質(アミノ酸769〜921)を使用した。ビーズを除去した後、基質に導入されたモノチオリン酸エステルを、10mMのヨードアセチル-ビオチン(Pierce)と共に、結合緩衝液(100mMのトリス-Cl[pH 8.5]、1 mM EDTA)中で、暗がりで室温において90分間インキュベーションすることによってさらに標識した。反応をβ-メルカプトエタノールでクエンチして、0.4μgの基質をドットブロット装置のウェルに適用した。ウェルを4%のウシ血清アルブミン(BSA)で30分間ブロッキングし、次いで37℃において1時間アビジン-FITC(Vector)と共にインキュベートした。膜を洗浄した後、イメージアナライザー(Bio-Rad)を使用してイメージを評価し、ドットの蛍光強度を「Quantity One」ソフトウェア(Bio-Rad)を使用して定量化した。K562慢性骨髄性白血病株化細胞から抽出された0μg、12.5μg、25μg、50μg、100μg、および150μgのタンパク質に対応するCDK活性で調製した検量線によって活性を算出した。1ユニット(U)は、K562細胞からの1μgの総タンパク質量のキナーゼ活性に匹敵する。
【0107】
実施例2
紡錘体形成チェックポイントの不活性化とCdk1活性の抑制との相関
以前の研究は、紡錘体形成チェックポイントの活性化により、Mad2およびBubR1の両方がCdc20と直接相互作用し、そのAPCを活性化する能力を阻害することを示した(Fang et al., 1998;Sudakin et al., 2001;Tang et al., 2001;Fang, 2002)。株化細胞における一過性ノックダウンを介したMad2またはBubR1の抑制が、パクリタキセルで誘導される紡錘体形成チェックポイントの機能に影響を及ぼすかどうかを決定するために、本発明者らは、siRNA二重鎖を使用して遺伝子サイレンシングを行った。
【0108】
Mad2およびBubR1配列の一部に対する21ヌクレオチドの低分子干渉RNA(siRNA)二重鎖相同体を機能的なチェックポイントを有するMCF-7細胞にトランスフェクションを行うことにより、Mad2およびBubR1タンパク質レベルの劇的な減少を生じ;これらのレベルは、それぞれの株化細胞においてトランスフェクションの24時間、48時間、および72時間後にも低いままであった。また、siRNA/Mad2およびsiRNA/BubR1でのMCF-7細胞の同時トランスフェクションにより、両方の発現が減少し、単一トランスフェクションで得られたものと同様の結果を生じた(図1A)。スクランブルされたsiRNA二重鎖(siRNA/対照)は、Mad2またはBubR1の発現レベルに影響を及ぼさず、siRNAアプローチの特異性が検証された。
【0109】
フローサイトメトリーを使用して、本発明者らは、有糸分裂チェックポイント遺伝子の一過性抑制が、細胞周期分布に影響を及ぼさないという可能性を除外することを試みた。トランスフェクションの72時間後に、Mad2およびBubR1の両方を抑制した細胞における細胞周期分布は、対照細胞における分布と同様であった(図1B)。パクリタキセルによって活性化される紡錘体形成チェックポイントに対するMad2、BubR1、または両方の抑制の効果を試験するために、有糸分裂指数を決定し、Cdk1活性を測定した(これらは両方とも、このチェックポイントの状態を反映する)。パクリタキセル処置の24時間後に、対照細胞の少なくとも80%が有糸分裂で停止され、Cdk1活性の劇的な増大を示し、したがって、紡錘体形成チェックポイントの活性化が検証された(図1Cおよび図1D)。対照的に、有糸分裂指数の蓄積およびCdk1の活性化は、Mad2およびBubR1を抑制した細胞では不完全であった(図1Cおよび図1D)。面白いことに、Mad2およびBubR1の同時抑制により、有糸分裂指数の蓄積およびCdk1の活性化の減少を示し、Mad2またはBubR1のいずれか単独の抑制で得られるものと同様となった(図1Cおよび図1D)。これらの結果は、紡錘体形成チェックポイントの機能を消失させるためには、Mad2またはBubR1単独で十分であったことを示す。この消失は、Cdk1活性の抑制によって反映された。
【0110】
最近の報告は、こうしてこれまでに同定された全ての単一紡錘体形成チェックポイント遺伝子が、有糸分裂停止を維持するために必須であることを明確に証明した(Dobles et al., 2000;Kalitsis et al., 2000;Luo et al., 2002)。Mad2およびBubR1は、紡錘体形成チェックポイントの開始および維持のために、有糸分裂チェックポイント複合体において協調して作用し、本結果と一致すると思われる知見である(Fang, 2002)。そのうえ、以前の研究では、機能的な紡錘体形成チェックポイントを有するMDA-MB-231およびSKBr-3細胞のsiRNA/Mad2、siRNA/BubR1、または両方ともによるトランスフェクションにより;チェックポイントの減少およびパクリタキセルに対する耐性を生じた(データ示さず)。これらの株化細胞は、それぞれ変異されたp53およびHER2/neu過剰発現を有することが公知である。これらの公知の遺伝的相違にもかかわらず、紡錘体形成チェックポイントの消失は、同様のパクリタキセルに対する耐性の知見を与えた。
【0111】
実施例3
紡錘体形成チェックポイントおよびパクリタキセル耐性の減少
パクリタキセル感受性に対するMad2、BubR1、または両方ともの抑制による紡錘体形成チェックポイントの減少の効果を決定するために、MTTアッセイ法を使用してパクリタキセルの細胞生存度を比較した。図2Aに示したように、Mad2、BubR1、または両方ともが抑制されたMCF-7細胞は、対照細胞よりもパクリタキセルに対して耐性であった。次に、これらの耐性がパクリタキセルで誘導される細胞死の減少によるものであったかどうかを決定するために、本発明者らは、トリパンブルー排除法を使用して細胞死の集団を評価した。パクリタキセルでの処置の48時間後に、細胞死のレベルは、対照細胞では高かったが、Mad2、BubR1、または両方ともが抑制された細胞では減少していた(図2B)。これらのデータは、紡錘体形成チェックポイントの減少が、パクリタキセル耐性を増大することを証明する。
【0112】
実施例4
Mad2依存的な、チェックポイント欠陥細胞におけるCdk1活性およびパクリタキセル感受性に対するMad2過剰発現の効果
Mad2突然変異は、チェックポイント欠陥がある癌株化細胞において検出されなかったが(Takahashi et al., 1999)、Mad2タンパク質の発現レベルは、紡錘体形成チェックポイントの応答能と相関しているように見える(Wang et al., 2000;Wang et al., 2002)。ヒト検体におけるMad2タンパク質の発現レベルについての報告は、ほとんど発表されていなかった(Tanaka et al., 2001)。従って、Mad2の過剰発現が、低Mad2発現によって紡錘体形成チェックポイントが機能しなくなった細胞における紡錘体形成チェックポイント活性化を回復するかどうかを決定した。効率的にMad2を発現させるために、Mad2(Ad-EGFP/Mad2)を発現する組換えアデノウイルスを作製した。このアデノウイルスは、2つの独立したCMV駆動転写単位(GFPについて1つ、およびMad2について1つ)を含み、感染効率を直接観察することができる。Ad-EGFP/Mad2は、外来Mad2の高発現を誘導し(図3A)、細胞周期の種々の期の間の細胞の分布に影響を及ぼさなかった(図3B)。
【0113】
最初に、機能しないチェックポイントを有することが示されたMad2-ノックダウンMCF-7細胞を使用した(図1Cおよび図1D)。Mad2の発現は、Ad-EGFP/Mad2の感染により、Mad2-ノックダウン細胞において回復された(図3C)。次いで、チェックポイントの機能をMad2の再発現によって回復することができるかどうかを決定するために、Ad-EGFP/Mad2感染させたMad2-ノックダウン細胞におけるCdk1の活性化をパクリタキセル処置後に評価した。Cdk1活性は、パクリタキセルに対する曝露後でさえもルシフェラーゼAd(-EGFP/Luc)を発現する組換えアデノウイルスに感染させた細胞では増大せず;これらの知見は、図1に示したデータと一致する。対照的に、Cdk1活性は、Ad-EGFP/Mad2感染細胞において回復し、Mad2の過剰発現が紡錘体形成チェックポイントの機能を回復することができることを示した(図3D)。次に、この回復により、パクリタキセルで誘導される細胞死のレベルが増強したかどうかを決定し、パクリタキセルで誘導される細胞死は、実際はAd-EGFP/Luc感染細胞(図3E)において、Ad-EGFP/Mad2感染細胞においてより有意に高かったことが見いだされた。
【0114】
また、Mad2の低発現のために欠陥チェックポイントを示すことが公知であるT47DおよびOvca432細胞を使用した(LiおよびBenezra, 1996)。Ad-EGFP/Mad2によるこれらの株化細胞の感染では、外来Mad2の高発現レベルを誘導し、細胞周期分布に影響を及ぼさなかった(図3Aおよび図3B)。Cdk1活性およびパクリタキセル感受性は、Ad-Luc感染させた細胞(図3Fおよび図3G)において、Ad-EGFP/Mad2感染細胞におけるよりも高かった。従って、これらのデータは、外来Mad2発現が、チェックポイントの機能を回復することができ、Mad2依存的なチェックポイント欠陥細胞における細胞死を増強することを証明する。
【0115】
実施例5
BubR1が抑制された細胞におけるチェックポイント機能およびパクリタキセル感受性に対するMad2過剰発現の効果
次に、Mad2の過剰発現が、Mad2以外の分子のための紡錘体形成チェックポイント欠陥を克服することができるかどうかを決定した。チェックポイントに欠陥のあることが示されたBubR1-ノックダウンMCF-7細胞を使用した(図1Cおよび図1D)。Ad-EGFP/Mad2は、外来Mad2の高発現を誘導し、BubR1-ノックダウン細胞におけるBubR1の発現には影響を及ぼさなかった(図4A)。しかし、Cdk1は、Ad-EGFP/Mad2感染細胞においてアップレギュレートされず、パクリタキセルで誘導される細胞死は、増強されなかった(図4Bおよび図4C)。これらのデータは、Mad2の過剰発現が、Mad2非依存的な欠陥チェックポイントをもつ細胞におけるチェックポイントの機能およびパクリタキセル感受性を克服しなかったことを示す。
【0116】
実施例6
機能的チェックポイントをもつ細胞におけるチェックポイント機能およびパクリタキセル感受性に対するMad2の過剰発現の効果
次に、Mad2の過剰発現が、紡錘体形成チェックポイントの機能(Cdk1活性およびパクリタキセル感受性)を増強するかどうか(チェックポイントが無傷の細胞におけるパクリタキセルで誘導される細胞死が増強されるように移行することができるという知見)を決定した。以前の報告では、一方のMad2対立遺伝子の欠失が、欠陥のある紡錘体形成チェックポイントを生じること(Michel et al., 2001)並びにMad2が、そのMad1との相互作用およびAPC/Cdc20の阻害を経て動原体に動員されなければならないこと(Chen et al., 1998;Waters et al., 1998)を示した。これらの結果は、Mad2が紡錘体形成チェックポイント機構の構成成分として作用するためには、Mad2の一定量および特異的局在化が必要とされること、並びにチェックポイントの機能の増強のために、大量のMad2が必要とされないことを示唆する。
【0117】
MCF-10A細胞およびMCF-7細胞(両方とも機能的なチェックポイントを有することが公知である)では、Ad-EGFP/Mad2が効率的にMad2の高発現レベルを誘導した(図3A)。しかし、Cdk1活性は、いずれの株化細胞のAd-EGFP/Mad2感染細胞においても増大されず、従って、パクリタキセルで誘導される細胞死の増強もなかった(図4Dおよび図4E)。従って、Mad2の過剰発現は、基礎的に機能的な紡錘体形成チェックポイントをもつ細胞におけるチェックポイント機能およびパクリタキセル感受性を増強することができなかった。
【0118】
染色体の喪失または獲得は、ヒトの癌の特徴であるので、紡錘体形成チェックポイントは、臨床的状況では頻繁に失われていると思われる。ヒト肺、結腸直腸、卵巣、およびインビトロでの鼻咽頭癌株化細胞の紡錘体形成チェックポイント欠陥の検出についての多くの報告にもかかわらず(Takahashi et al., 1999; Wang et al., 2000; Cahill et al., 1998; Nakagawa et al., 2002; Saeki et al., 2002)、公知の紡錘体形成チェックポイント遺伝子の突然変異は、ヒト癌では非常にまれにしか生じない(Saeki et al., 2002; Sato et al., 2000; Haruki et al., 2001)。このパラドックスは、チェックポイント複合体に対する種々の転写後または翻訳後修飾によって説明される可能性がある。さらに、紡錘体形成チェックポイント機構は、種々の分子からなるために、遺伝子またはタンパク質発現の突然変異を解析することによってヒト癌試料のチェックポイントの機能を評価することは、非実用的であると思われる。
【0119】
実施例7
パクリタキセルに対する株化細胞の感受性
パクリタキセルに対する4種のヒト乳癌株化細胞の感受性をパクリタキセル細胞障害性試験のグラフ解析に従って判断した(図5)。結果は、MDA-MB231およびMDA-MB435が感受性であり、MCF-7およびT47Dが耐性であったことを示した。パクリタキセル細胞障害性試験は、以下のとおりに行った。細胞を96ウェル培養プレートのウェル内で1nM、5nM、10nM、50nM、または100nMパクリタキセルの存在下において培養した。72時間の培養後、細胞生存をMTTアッセイ法によって測定した。
【0120】
実施例8
インビトロでのバリデーション
インビトロでのバリデーション研究において、全ての株化細胞を100nMパクリタキセルで処置して、処置の0時間、24時間、48時間、および72時間後に収集した。次いで、細胞可溶化物を細胞周期プロファイリングに供した。プロファイリング結果により、7つのパラメーター(細胞周期分子)が、パクリタキセルに対して感受性と耐性の株化細胞とを区別するために有用であることが明らかになった。これらは、(1)処置の24時間後のCDK1比活性;(2)処置前のCDK2比活性;(3)処置前のMAD2発現;(4)処置前のサイクリンB1;(5)処置前のサイクリンE発現;(6)処置前のp21発現;および(7)処置前のCDK6発現である(図6)。結果によれば、パクリタキセル感受性予測試験のためには、9つのパラメーターが決定された。これらは、(1)CDK1キナーゼ活性;(2)CDK1発現(CDK1比活性の算出のため);(3)CDK2キナーゼ活性;(4)CDK2発現(CDK2比活性の算出のため);(5)MAD2発現;(6)サイクリンB1;(7)サイクリンE発現;(8)p21発現;および(9)CDK6発現である。
【0121】
細胞周期プロファイリング技術により、7つのパラメーターが感受性と耐性の株化細胞とを区別するために有用であることが明らかになった。これらは、(1)処置の24時間後のCDK1比活性;(2)処置前のCDK2比活性;(3)処置前のMAD2発現;(4)処置前のサイクリンB1;(5)処置前のサイクリンE発現;(6)処置前のp21発現;および(7)処置前のCDK6発現である(図6)。
【0122】
実施例9
インビボでのバリデーション
上記のインビトロでの結果をさらに確認するために、インビボでのバリデーション研究を行った。感受性および耐性の細胞をヌードマウスに接種した。腫瘍体積が300mm3を超える体積に達した場合に、1日につき20mgのパクリタキセルを5日連続で(接種後34〜38日)腹腔内に投与した。腫瘍サイズを20〜43日に記録した。図7に示したように、感受性の腫瘍は、460mm3〜120mm3に腫瘍体積の有意な減少を示した。対照的に、耐性腫瘍の体積は、43日までに350mm3のままであった。
【0123】
パクリタキセル投与の24時間前後に、腫瘍を有するマウスから腫瘍組織を外科的に切開して、細胞周期プロファイリングに供した。サイクリンB1発現を除いて、プロファイリングは、インビトロ研究とほぼ同一の結果を示した。パクリタキセルに対する感受性と耐性の腫瘍とを区別するためには、6つのパラメーターが有用であった。これらは、(1)処置の24時間後のCDK1比活性;(2)処置前のCDK2比活性;(3)処置前のMAD2発現;(4)処置前のサイクリンE発現;(5)処置前のp21発現;および(6)処置前のCDK6発現である(図8)。
【0124】
インビトロおよびインビボでのバリデーション研究の結果を合わせると、細胞周期プロファイリング技術は、腫瘍組織のタキサン化学受容性を決定するための有効な系であることが判明した。決定のために選択したパラメーターにより、正確な予測がなされることが科学的に証明される。
【0125】
上記の結果は、細胞周期プロファイリング技術が、腫瘍組織のタキサン化学受容性を決定する際に良好なことを強く示す。インビトロおよびインビボでのバリデーション研究の結果を合わせると、細胞周期プロファイリング技術は、腫瘍組織のタキサン化学受容性を決定する際に良好であることが判明した。
【0126】
実施例10
ヌードマウスにおけるパクリタキセルに対する感受性
感受性および耐性の細胞(1×107細胞/マウス)をヌードマウスに接種した。腫瘍体積が300mm3を超える体積に達した場合に、1日につき20mgのパクリタキセルを5日連続で(接種後34〜38日)腹腔内に投与した。腫瘍サイズを20〜43日に記録した。図7に示したように、感受性の腫瘍は、460mm3〜120mm3に腫瘍体積の有意な減少を示した。対照的に、耐性腫瘍の体積は、43日までに350mm3のままであった(図7)。
【0127】
細胞周期プロファイリングのためには、パクリタキセル投与の24時間前後に、腫瘍を有するマウスから腫瘍組織を外科的に切開した。組織の一部(2mm3)の可溶化液を溶解緩衝液(0.1%のNP-40、20mMのトリス-HCl[pH 7.4]、150mM NaCl、2%のプロテイナーゼ阻害剤反応混液(Sigma, St Louis, MO)で組織ホモジナイザーを使用して調製した。ホモジナイザーは、自動的にフィルターディスク上の不溶性物質を除去する。可溶化液のタンパク質濃度を解析し(DCキット, Pierce, Rockford, IL, USA)、使用するまで-80℃で貯蔵した。
【0128】
本明細書に開示され、請求される全ての組成物および方法は、本開示を考慮して過度の実験なしに作製し、実施することができる。本発明の組成物および方法は、好ましい態様に関して記載したが、本発明の概念、精神、および範囲から逸脱することなく、本明細書に記載されている組成物および方法並びに方法の工程または工程の順序にバリエーションを適用してもよいことが、当業者には明らかであると思われる。より詳細には、それは、化学的および生理的に関連する一定の薬剤を、本明細書に記載されており、一方で同じまたは同様の結果が達成されると考えられる薬剤と置換してもよいことが明らかであると思われる。当業者に明らかな全てのこのような同様の置換および修飾は、添付の請求の範囲に定義された本発明の精神、範囲、および概念の範囲内であると考えられる。
【0129】
参照文献
以下の参照文献は、これらが本明細書において記載されるものに対して例示的な手順、またはその他の詳細な補充を提供する範囲で、具体的に参照として本明細書に組み入れられる。
【図面の簡単な説明】
【0130】
以下の図面は、本明細書の一部を形成し、本発明の一定の側面をさらに証明するために含まれる。本発明は、本明細書に示される特定の態様の詳細な説明と合わせて、これらの図面の1つまたは複数を参照することで、よりよく理解され得る。
【図1】Mad2、BubR1、または両方ともの抑制による細胞における紡錘体形成チェックポイントの喪失を示す。図1Aは、低分子干渉RNA(siRNA)を用いたトランスフェクション後のBubR1およびMad2レベルの減少を示す。MCF-7細胞には、siRNAについて200nMの終濃度を生じるsiRNA/Mad2またはsiRNA/BubR1でトランスフェクションを行った。トランスフェクション後の示した時間にて、細胞を集めて、それぞれの抗体およびゲル充填のための対照のためのα-チューブリン抗体によるタンパク質免疫ブロット解析に供した。図1Bは、本明細書に記載した蛍光活性化細胞選別によるDNA含量によって測定されるMad2、BubR1、または両方の抑制による細胞の細胞周期分布を示す。MCF-7細胞をトランスフェクし、トランスフェクションの72時間後に集めた。数は、それぞれの期における細胞の割合を示す。図1Cは、パクリタキセルでの処置後のそれぞれの細胞の分裂指数を示す。トランスフェクションの24時間後に、細胞をパクリタキセル(100nM)で処置して、示した時間に収集した。図1Dは、それぞれの細胞におけるパクリタキセルで処置後のサイクリン依存性キナーゼ-1(Cdk1)の活性を示す。トランスフェクションの24時間後に、細胞をパクリタキセル(100nM)で処置して、示した時間に収集した。可溶化液におけるCdk1の活性は、実験手順に記載されているように決定した。
【図2】紡錘体形成チェックポイントの喪失を介したパクリタキセル耐性の誘導を示す。図2Aは、細胞増殖に対するパクリタキセルの効果を決定するために、siRNA/Mad2、BubR1、または両方でトランスフェクションを行ったMCF-7細胞を、MTTアッセイ法を使用して調べた。トランスフェクションの12時間後に、細胞をトリプシン処置によって分離した。接種の12時間後に、細胞を種々の濃度のパクリタキセルで処置した。バー(標準偏差)。図2Bは、siRNA/Mad2、BubR1、または両方でトランスフェクションを行ったMCF-7細胞を、パクリタキセルによって誘導される細胞死について調べた。トランスフェクションの24時間後に、細胞をパクリタキセル(100nM)で48時間処置した。細胞生存度は、トリパンブルー排除アッセイ法を使用して評価した。バー(標準偏差)。
【図3】Mad2依存的チェックポイント欠陥細胞におけるMad2の過剰発現による紡錘体形成チェックポイントの機能の回復およびパクリタキセル感受性の増強を示す。図3Aは、Ad-EGFP/Mad2での感染によるMCF-7、MCF-10A、T47D、およびOvca432細胞における外来性Mad2発現を示す。25および50の感染効率値での感染の24時間後、20μgのそれぞれの可溶化試料を適用して、抗Mad2抗体でのタンパク質免疫ブロット解析に供した。図3Bは、蛍光活性化細胞選別によるDNA含量によって測定される、Mad2を過剰発現した細胞の細胞周期分布を示す。MCF-7、T47D、およびOvca432細胞を感染の48時間後に収集し、その後DNA含量を検出するために、ヨウ化プロピジウムで染色した。数字は、それぞれの期における細胞の割合を示す。図3Cの上段は、組み合わせた低分子干渉RNA(siRNA)/Mad2トランスフェクションおよびアデノウイルス感染のためのスケジュールを示す。siRNA/Mad2トランスフェクションの24時間後に、アデノウイルスベクター(50の感染効率)を24時間にわたって送達し、細胞をパクリタキセル(100nM)に曝露した。下段は、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたMad2が抑制されたMCF-7細胞におけるMad2の発現を示す。細胞をパクリタキセル処置の0時間、24時間、および48時間後に収集して、それぞれの抗体およびゲル充填のための対照のためのα-チューブリン抗体によるタンパク質免疫ブロット解析に供した。図3Dは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたMad2が抑制されたMCF-7細胞におけるパクリタキセルで処置後のサイクリン依存性キナーゼ-1(Cdk1)の活性を示す。50の感染効率でAd-EGFP/Mad2またはAd-EGFP/Lucのいずれかに感染させた24時間後に、細胞をパクリタキセル(100nM)で処置して、示した時間に収集した。本明細書に記載したとおりに、可溶化液中のCdk1活性を決定した。図3Eは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたMad2の抑制されたMCF-7細胞におけるパクリタキセルで誘導される細胞死を示す。感染の24時間後に、細胞を収集した。パクリタキセルでの処置の48時間後に、細胞生存度をトリパンブルー排除アッセイ法を使用して評価した。バー(標準偏差)。図3Fは、感染させたT47DおよびOvca432細胞におけるパクリタキセルで処置後のサイクリン依存性キナーゼ-1(Cdk1)の活性を示す。50の感染効率でAd-EGFP/Mad2またはAd-EGFP/Lucのいずれかに感染させた24時間後に、細胞をパクリタキセル(100nM)で処置して、示した時間に収集した。本明細書に記載したとおりに、可溶化液中のCdk1活性を決定した。図3Gは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたT47DおよびOvca432細胞におけるパクリタキセルで誘導される細胞死を示す。感染の24時間後に、細胞を収集した。パクリタキセルでの処置の48時間後に、細胞生存度をトリパンブルー排除アッセイ法を使用して評価した。バー(標準偏差)。
【図4】Mad2過剰発現では、Mad2非依存的欠陥があるか、または機能的チェックポイントをもつ細胞におけるチェックポイント機能およびパクリタキセル感受性を増強することができない。図4Aの上段は、組み合わせた低分子干渉RNA(siRNA)/BubR1トランスフェクションおよびアデノウイルス感染のためのスケジュールを示す。siRNA/BubR1のトランスフェクションの24時間後に、アデノウイルスベクター(50の感染効率)を24時間にわたって送達し、細胞をパクリタキセル(100nM)に曝露した。下段は、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたBubR1が抑制されたMCF-7細胞におけるMad2およびBubR1の発現を示す。細胞をパクリタキセル処置後の示した時間に収集して、それぞれの抗体およびゲル充填のための対照のためのα-チューブリン抗体によるタンパク質免疫ブロット解析に供した。図4Bは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたBubR1が抑制されたMCF-7細胞におけるパクリタキセルで処置後のCdk1活性。細胞をパクリタキセル処置後の示した時間に収集した。本明細書に記載したとおりに、可溶化液中のCdk1活性を決定した。図4Cは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたBubR1の抑制されたMCF-7細胞におけるパクリタキセルで誘導される細胞死を示す。細胞をパクリタキセル処置の48時間後に収集した。細胞生存度は、トリパンブルー排除によって評価した。バー(標準偏差)。図4Dは、感染させたMCF-7およびMCF-10A細胞におけるパクリタキセル処置後のサイクリン依存性キナーゼ-1(Cdk1)の活性を示す。50の感染効率でAd-EGFP/Mad2またはAd-EGFP/Lucに感染させた24時間後に、細胞をパクリタキセル(100nM)で処置して、示した時間に収集した。本明細書に記載したとおりに、可溶化液中のCdk1活性を決定した。図4Eは、Ad-EGFP/Mad2またはAd-EGFP/Lucに感染させたMCF-7細胞およびMCF-10A細胞におけるパクリタキセルで誘導される細胞死を示す。感染の24時間後に、細胞を収集した。パクリタキセルでの処置の48時間後に、細胞生存度をトリパンブルー排除アッセイ法を使用して評価した。バー(標準偏差)。
【図5】パクリタキセルに対する株化細胞の感受性をパクリタキセル細胞障害性試験のグラフ解析に従って判断した。結果において、MDA-MB231およびMDA-MB435は、感受性のものと判断され、MCF-7およびT47Dは、耐性と判断された。パクリタキセル細胞障害性試験は、以下のとおりに行った。細胞を96ウェル培養板のウェル中で1nM、5nM、10nM、50nM、または100nMのパクリタキセルの存在下において培養した。72時間の培養後、細胞生存をMTTアッセイ法によって測定した。
【図6】株化細胞の可溶化液を細胞周期プロファイリングに供した。細胞周期プロファイリング技術により、7つのパラメーターが感受性と耐性との株化細胞を区別するために有用であることが明らかになった。これらは、(A1)処置の24時間後のCDK1比活性;(A2)処置前のCDK2比活性;(E5)処置前のMAD2発現;(E2)処置前のサイクリンB1;(E3)処置前のサイクリン E発現;(E4)処置前のp21発現;および(E1)処置前のCDK6発現である。グラフにプロットした値は、それぞれのパラメーターに対して、4つの株化細胞の中で最も大きい生の値で割ることによって算出した。たとえば、サイクリンB1発現(E2、可溶化液のng/μg)の生の値;MDA-MB231について0.135、MDA-MB435について0.094、MCF-7について0.061、およびT47Dについて0.042は、MDA-MB231の0.135で割ることにより、MDA-MB231について1.0、MDA-MB435について0.70、MCF-7について0.45、およびT47Dについて0.31になった。
【図7】感受性および耐性の細胞(1×107細胞/マウス)をヌードマウスに接種した。腫瘍体積が、300mm3を超える体積に達した場合に、1日につき20mgのパクリタキセルを5日連続で腹腔内に投与した(接種後34〜38日)。腫瘍サイズは、20〜43日に記録した。図3に示したように、感受性の腫瘍は、460mm3から120mm3に腫瘍体積の劇的な減少を示した。対照的に、耐性腫瘍の体積は、43日まで350mm3のままであった。
【図8】腫瘍を有するマウスからの腫瘍組織の可溶化液を細胞周期プロファイリングに供した。細胞周期技術は、サイクリンB1発現(E2)を除いて、インビトロ研究とほぼ同一の結果を示した。感受性と耐性との腫瘍を区別するためには、6つのパラメーターが有用であった。これらは、(A1)処置の24時間後のCDK1比活性;(A2)処置前のCDK2比活性;(E5)処置前のMAD2発現;(E3)処置前のサイクリンE発現;(E4)処置前のp21発現;および(E1)処置前のCDK6発現であった。グラフにプロットした値は、それぞれのパラメーターに対する2つの腫瘍間でより大きい生の値で割ることによって算出した。たとえば、CDK1比活性(Al、CDK1のmU/ng)の生の値;MDA-MB231の腫瘍について3.0、およびT47Dの腫瘍について0.2は、MDA-MB231について1.0およびT47Dについて0.067になった。
【図1A】
【図1B】
【図1C】
【図1D】
【特許請求の範囲】
【請求項1】
癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性に対するタキサンの効果を評価する工程を含むタキサンに対する該癌細胞の化学受容性を決定する方法。
【請求項2】
癌細胞が患者から得られる、請求項1記載の方法。
【請求項3】
癌細胞が組織試料である、請求項2記載の方法。
【請求項4】
組織試料が生検組織試料である、請求項3記載の方法。
【請求項5】
組織試料がエキソビボで培養された生検組織試料である、請求項3記載の方法。
【請求項6】
組織試料が外科的に切開した組織試料である、請求項3記載の方法。
【請求項7】
組織試料がエキソビボで培養された外科的に切開した組織試料である、請求項2記載の方法。
【請求項8】
細胞周期分子が1つもしくは複数のサイクリン依存性キナーゼ(CDK)、サイクリン、CDK阻害剤(CDKI)、p53、または有糸分裂/紡錘体形成チェックポイント分子を含む、請求項1記載の方法。
【請求項9】
2つまたはそれ以上の細胞周期分子を評価する工程を含む、請求項1記載の方法。
【請求項10】
1つまたは複数のCDK分子のキナーゼ活性を評価する工程を含む、請求項8記載の方法。
【請求項11】
CDK分子がCDK1、CDK2、CDK4、またはCDK6である、請求項10記載の方法。
【請求項12】
1つまたは複数のCDK分子の発現を評価する工程を含む、請求項8記載の方法。
【請求項13】
CDK分子がCDK1、CDK2、CDK4、またはCDK6である、請求項12記載の方法。
【請求項14】
1つまたは複数のサイクリン分子の発現を評価する工程を含む、請求項8記載の方法。
【請求項15】
サイクリン分子がサイクリンB1またはサイクリンEである、請求項14記載の方法。
【請求項16】
1つまたは複数のCDKIの発現を評価する工程を含む、請求項8記載の方法。
【請求項17】
CDKIがp16、p21/Waf1、またはp27/Kip1である、請求項16記載の方法。
【請求項18】
1つまたは複数の有糸分裂/紡錘体形成チェックポイント分子の発現を評価することを含む、請求項8記載の方法。
【請求項19】
評価される発現がMAD2発現である、請求項18記載の方法。
【請求項20】
評価される発現がBubR1発現である、請求項18記載の方法。
【請求項21】
タキサンがパクリタキセルである、請求項1記載の方法。
【請求項22】
タキサンがドセタキセルである、請求項1記載の方法。
【請求項23】
癌細胞が乳癌細胞、前立腺癌細胞、皮膚癌細胞、肺癌細胞、頭頚部癌細胞、膀胱癌細胞、骨癌細胞、骨髄癌細胞、脳癌細胞、大腸癌細胞、食道癌細胞、胃腸癌細胞、歯肉癌細胞、腎臓癌細胞、肝臓癌細胞、上咽頭癌細胞、卵巣癌細胞、胃癌細胞、精巣癌細胞、舌癌細胞、または子宮癌細胞である、請求項1記載の方法。
【請求項24】
癌細胞が抗癌治療の適用前に得られる、請求項2記載の方法。
【請求項25】
抗癌治療が化学療法である、請求項24記載の方法。
【請求項26】
抗癌治療が放射線療法である、請求項24記載の方法。
【請求項27】
癌細胞が抗癌治療の適用後に得られる、請求項2記載の方法。
【請求項28】
抗癌治療が化学療法である、請求項27記載の方法。
【請求項29】
抗癌治療が放射線療法である、請求項27記載の方法。
【請求項30】
抗癌治療の適用前の1つもしくは複数の細胞周期分子の発現レベルまたは活性を、抗癌治療の適用後の1つもしくは複数の細胞周期分子の発現レベルまたは活性と比較する工程をさらに含む、請求項1記載の方法。
【請求項31】
細胞周期プロファイルを得る工程を含む、請求項1記載の方法。
【請求項32】
抗癌治療の適用前の細胞周期プロファイルを、抗癌治療の適用後の細胞周期プロファイルと比較する工程をさらに含む、請求項31記載の方法。
【請求項33】
細胞周期プロファイルが自動化アナライザーを使用して得られる、請求項31記載の方法。
【請求項34】
細胞周期プロファイルを、タキサンに感受性である癌細胞の細胞周期プロファイルと比較する工程をさらに含む、請求項31記載の方法。
【請求項35】
細胞周期プロファイルを、タキサンに耐性である癌細胞の細胞周期プロファイルと比較する工程をさらに含む、請求項31記載の方法。
【請求項36】
癌を有する患者の治療に関する決定を行う工程をさらに含む、請求項2記載の方法。
【請求項37】
癌を有する患者の生存を評価する工程をさらに含む、請求項2記載の方法。
【請求項38】
癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性を、タキサンに感受性である癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性と比較する工程をさらに含む、請求項1記載の方法。
【請求項39】
癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性を、タキサンに感受性でない癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性と比較する工程をさらに含む、請求項1記載の方法。
【請求項40】
以下の工程を含む患者における癌を治療するかまたは予防する方法:
a)請求項1記載の患者の癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性に対するタキサンの効果を決定する工程;
b)タキサンに対する癌細胞の感受性を評価する工程;および、
c)患者にタキサンを投与する工程。
【請求項41】
癌細胞の感受性を評価する工程が細胞周期プロファイルを得る工程を含む、請求項48記載の方法。
【請求項42】
細胞周期プロファイルが自動化アナライザーを使用して得られる、請求項41記載の方法。
【請求項43】
癌が乳癌、前立腺癌、皮膚癌、肺癌、頭頚部癌、膀胱癌、骨癌、骨髄癌、脳癌、大腸癌、食道癌、胃腸癌、歯肉癌、腎臓癌、肝臓癌、上咽頭癌、卵巣癌、胃癌、精巣癌、舌癌、または子宮癌である、請求項40記載の方法。
【請求項44】
タキサンがパクリタキセルである、請求項40記載の方法。
【請求項45】
タキサンがドセタキセルである、請求項40記載の方法。
【請求項46】
タキサンが一回またはそれ以上投与される、請求項40記載の方法。
【請求項47】
タキサンが静脈内に、または腫瘍内に投与される、請求項40記載の方法。
【請求項48】
抗癌治療を適用する工程をさらに含む、請求項40記載の方法。
【請求項49】
抗癌治療が化学療法である、請求項48記載の方法。
【請求項50】
抗癌治療が放射線療法である、請求項48記載の方法。
【請求項51】
抗癌治療がタキサンの前に適用される、請求項48記載の方法。
【請求項52】
抗癌治療がタキサンの後に適用される、請求項48記載の方法。
【請求項53】
抗癌治療がタキサンと同時に適用される、請求項48記載の方法。
【請求項54】
細胞周期分子が1つもしくは複数のサイクリン依存性キナーゼ(CDK)、サイクリン、CDK阻害剤(CDKI)、p53、または有糸分裂/紡錘体形成チェックポイント分子を含む、請求項40記載の方法。
【請求項55】
2つまたはそれ以上の細胞周期分子を評価する工程を含む、請求項40記載の方法。
【請求項56】
1つまたは複数のCDK分子のキナーゼ活性を評価する工程を含む、請求項54記載の方法。
【請求項57】
CDK分子がCDK1、CDK2、CDK4、またはCDK6である、請求項56記載の方法。
【請求項58】
1つまたは複数のCDK分子の発現を評価する工程を含む、請求項54記載の方法。
【請求項59】
CDK分子がCDK1、CDK2、CDK4、またはCDK6である、請求項58記載の方法。
【請求項60】
1つまたは複数のサイクリン分子の発現を評価する工程を含む、請求項54記載の方法。
【請求項61】
サイクリン分子がサイクリンB1またはサイクリンEである、請求項60記載の方法。
【請求項62】
1つまたは複数のCDKIの発現を評価する工程を含む、請求項54記載の方法。
【請求項63】
CDKIがp16、p21/Waf1、またはp27/Kip1である、請求項62記載の方法。
【請求項64】
1つまたは複数の有糸分裂/紡錘体形成チェックポイント分子の発現を評価する工程を含む、請求項54記載の方法。
【請求項65】
評価される発現がMAD2発現である、請求項64記載の方法。
【請求項66】
評価される発現がBubR1発現である、請求項64記載の方法。
【請求項1】
癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性に対するタキサンの効果を評価する工程を含むタキサンに対する該癌細胞の化学受容性を決定する方法。
【請求項2】
癌細胞が患者から得られる、請求項1記載の方法。
【請求項3】
癌細胞が組織試料である、請求項2記載の方法。
【請求項4】
組織試料が生検組織試料である、請求項3記載の方法。
【請求項5】
組織試料がエキソビボで培養された生検組織試料である、請求項3記載の方法。
【請求項6】
組織試料が外科的に切開した組織試料である、請求項3記載の方法。
【請求項7】
組織試料がエキソビボで培養された外科的に切開した組織試料である、請求項2記載の方法。
【請求項8】
細胞周期分子が1つもしくは複数のサイクリン依存性キナーゼ(CDK)、サイクリン、CDK阻害剤(CDKI)、p53、または有糸分裂/紡錘体形成チェックポイント分子を含む、請求項1記載の方法。
【請求項9】
2つまたはそれ以上の細胞周期分子を評価する工程を含む、請求項1記載の方法。
【請求項10】
1つまたは複数のCDK分子のキナーゼ活性を評価する工程を含む、請求項8記載の方法。
【請求項11】
CDK分子がCDK1、CDK2、CDK4、またはCDK6である、請求項10記載の方法。
【請求項12】
1つまたは複数のCDK分子の発現を評価する工程を含む、請求項8記載の方法。
【請求項13】
CDK分子がCDK1、CDK2、CDK4、またはCDK6である、請求項12記載の方法。
【請求項14】
1つまたは複数のサイクリン分子の発現を評価する工程を含む、請求項8記載の方法。
【請求項15】
サイクリン分子がサイクリンB1またはサイクリンEである、請求項14記載の方法。
【請求項16】
1つまたは複数のCDKIの発現を評価する工程を含む、請求項8記載の方法。
【請求項17】
CDKIがp16、p21/Waf1、またはp27/Kip1である、請求項16記載の方法。
【請求項18】
1つまたは複数の有糸分裂/紡錘体形成チェックポイント分子の発現を評価することを含む、請求項8記載の方法。
【請求項19】
評価される発現がMAD2発現である、請求項18記載の方法。
【請求項20】
評価される発現がBubR1発現である、請求項18記載の方法。
【請求項21】
タキサンがパクリタキセルである、請求項1記載の方法。
【請求項22】
タキサンがドセタキセルである、請求項1記載の方法。
【請求項23】
癌細胞が乳癌細胞、前立腺癌細胞、皮膚癌細胞、肺癌細胞、頭頚部癌細胞、膀胱癌細胞、骨癌細胞、骨髄癌細胞、脳癌細胞、大腸癌細胞、食道癌細胞、胃腸癌細胞、歯肉癌細胞、腎臓癌細胞、肝臓癌細胞、上咽頭癌細胞、卵巣癌細胞、胃癌細胞、精巣癌細胞、舌癌細胞、または子宮癌細胞である、請求項1記載の方法。
【請求項24】
癌細胞が抗癌治療の適用前に得られる、請求項2記載の方法。
【請求項25】
抗癌治療が化学療法である、請求項24記載の方法。
【請求項26】
抗癌治療が放射線療法である、請求項24記載の方法。
【請求項27】
癌細胞が抗癌治療の適用後に得られる、請求項2記載の方法。
【請求項28】
抗癌治療が化学療法である、請求項27記載の方法。
【請求項29】
抗癌治療が放射線療法である、請求項27記載の方法。
【請求項30】
抗癌治療の適用前の1つもしくは複数の細胞周期分子の発現レベルまたは活性を、抗癌治療の適用後の1つもしくは複数の細胞周期分子の発現レベルまたは活性と比較する工程をさらに含む、請求項1記載の方法。
【請求項31】
細胞周期プロファイルを得る工程を含む、請求項1記載の方法。
【請求項32】
抗癌治療の適用前の細胞周期プロファイルを、抗癌治療の適用後の細胞周期プロファイルと比較する工程をさらに含む、請求項31記載の方法。
【請求項33】
細胞周期プロファイルが自動化アナライザーを使用して得られる、請求項31記載の方法。
【請求項34】
細胞周期プロファイルを、タキサンに感受性である癌細胞の細胞周期プロファイルと比較する工程をさらに含む、請求項31記載の方法。
【請求項35】
細胞周期プロファイルを、タキサンに耐性である癌細胞の細胞周期プロファイルと比較する工程をさらに含む、請求項31記載の方法。
【請求項36】
癌を有する患者の治療に関する決定を行う工程をさらに含む、請求項2記載の方法。
【請求項37】
癌を有する患者の生存を評価する工程をさらに含む、請求項2記載の方法。
【請求項38】
癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性を、タキサンに感受性である癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性と比較する工程をさらに含む、請求項1記載の方法。
【請求項39】
癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性を、タキサンに感受性でない癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性と比較する工程をさらに含む、請求項1記載の方法。
【請求項40】
以下の工程を含む患者における癌を治療するかまたは予防する方法:
a)請求項1記載の患者の癌細胞における1つもしくは複数の細胞周期分子の発現レベルまたは活性に対するタキサンの効果を決定する工程;
b)タキサンに対する癌細胞の感受性を評価する工程;および、
c)患者にタキサンを投与する工程。
【請求項41】
癌細胞の感受性を評価する工程が細胞周期プロファイルを得る工程を含む、請求項48記載の方法。
【請求項42】
細胞周期プロファイルが自動化アナライザーを使用して得られる、請求項41記載の方法。
【請求項43】
癌が乳癌、前立腺癌、皮膚癌、肺癌、頭頚部癌、膀胱癌、骨癌、骨髄癌、脳癌、大腸癌、食道癌、胃腸癌、歯肉癌、腎臓癌、肝臓癌、上咽頭癌、卵巣癌、胃癌、精巣癌、舌癌、または子宮癌である、請求項40記載の方法。
【請求項44】
タキサンがパクリタキセルである、請求項40記載の方法。
【請求項45】
タキサンがドセタキセルである、請求項40記載の方法。
【請求項46】
タキサンが一回またはそれ以上投与される、請求項40記載の方法。
【請求項47】
タキサンが静脈内に、または腫瘍内に投与される、請求項40記載の方法。
【請求項48】
抗癌治療を適用する工程をさらに含む、請求項40記載の方法。
【請求項49】
抗癌治療が化学療法である、請求項48記載の方法。
【請求項50】
抗癌治療が放射線療法である、請求項48記載の方法。
【請求項51】
抗癌治療がタキサンの前に適用される、請求項48記載の方法。
【請求項52】
抗癌治療がタキサンの後に適用される、請求項48記載の方法。
【請求項53】
抗癌治療がタキサンと同時に適用される、請求項48記載の方法。
【請求項54】
細胞周期分子が1つもしくは複数のサイクリン依存性キナーゼ(CDK)、サイクリン、CDK阻害剤(CDKI)、p53、または有糸分裂/紡錘体形成チェックポイント分子を含む、請求項40記載の方法。
【請求項55】
2つまたはそれ以上の細胞周期分子を評価する工程を含む、請求項40記載の方法。
【請求項56】
1つまたは複数のCDK分子のキナーゼ活性を評価する工程を含む、請求項54記載の方法。
【請求項57】
CDK分子がCDK1、CDK2、CDK4、またはCDK6である、請求項56記載の方法。
【請求項58】
1つまたは複数のCDK分子の発現を評価する工程を含む、請求項54記載の方法。
【請求項59】
CDK分子がCDK1、CDK2、CDK4、またはCDK6である、請求項58記載の方法。
【請求項60】
1つまたは複数のサイクリン分子の発現を評価する工程を含む、請求項54記載の方法。
【請求項61】
サイクリン分子がサイクリンB1またはサイクリンEである、請求項60記載の方法。
【請求項62】
1つまたは複数のCDKIの発現を評価する工程を含む、請求項54記載の方法。
【請求項63】
CDKIがp16、p21/Waf1、またはp27/Kip1である、請求項62記載の方法。
【請求項64】
1つまたは複数の有糸分裂/紡錘体形成チェックポイント分子の発現を評価する工程を含む、請求項54記載の方法。
【請求項65】
評価される発現がMAD2発現である、請求項64記載の方法。
【請求項66】
評価される発現がBubR1発現である、請求項64記載の方法。
【図2】

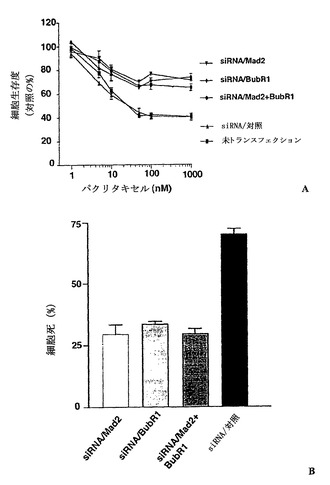
【図3A】
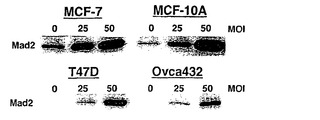
【図3B】
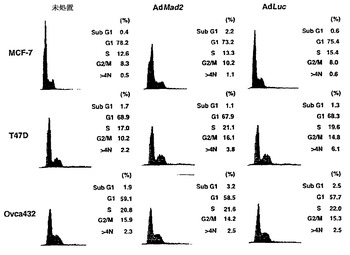
【図3C】
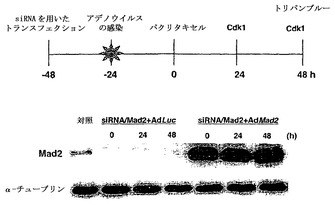
【図3D】
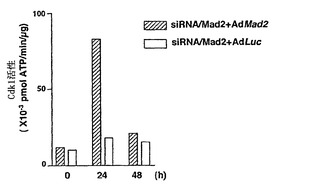
【図3E】
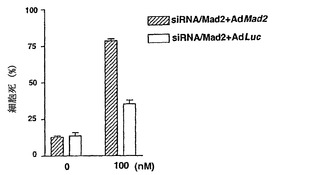
【図3F】
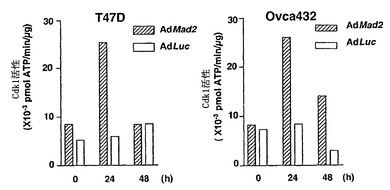
【図3G】
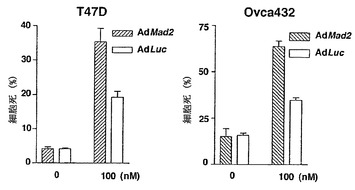
【図4A】
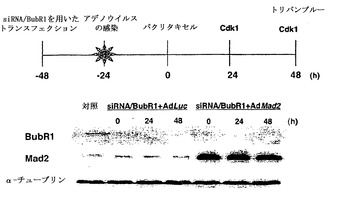
【図4B】
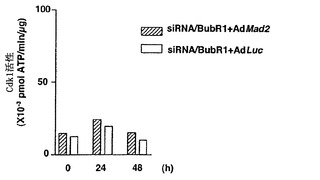
【図4C】
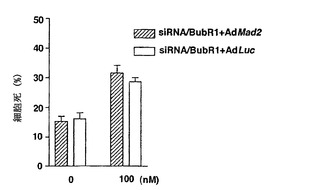
【図4D】
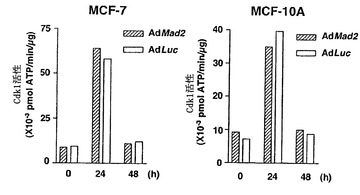
【図4E】
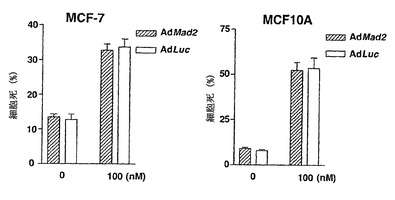
【図5】
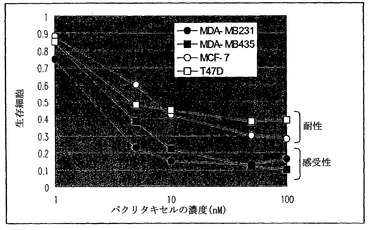
【図6】
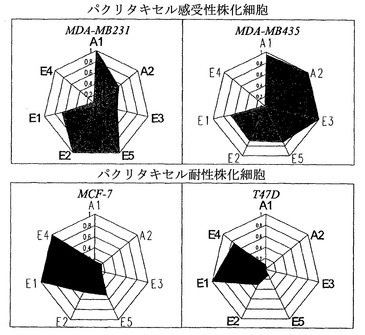
【図7】
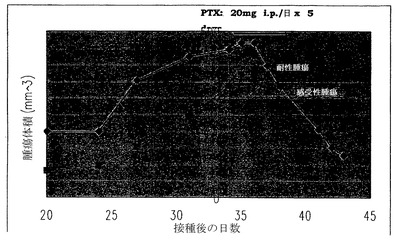
【図8】
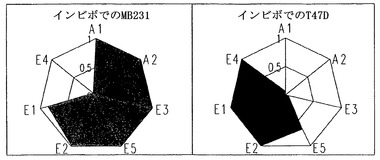
【図3A】
【図3B】
【図3C】
【図3D】
【図3E】
【図3F】
【図3G】
【図4A】
【図4B】
【図4C】
【図4D】
【図4E】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2007−503809(P2007−503809A)
【公表日】平成19年3月1日(2007.3.1)
【国際特許分類】
【出願番号】特願2006−524833(P2006−524833)
【出願日】平成16年8月25日(2004.8.25)
【国際出願番号】PCT/US2004/027661
【国際公開番号】WO2005/020794
【国際公開日】平成17年3月10日(2005.3.10)
【出願人】(503049667)ボード オブ リージェンツ,ザ ユニバーシティ オブ テキサス システム (8)
【出願人】(390014960)シスメックス株式会社 (810)
【Fターム(参考)】
【公表日】平成19年3月1日(2007.3.1)
【国際特許分類】
【出願日】平成16年8月25日(2004.8.25)
【国際出願番号】PCT/US2004/027661
【国際公開番号】WO2005/020794
【国際公開日】平成17年3月10日(2005.3.10)
【出願人】(503049667)ボード オブ リージェンツ,ザ ユニバーシティ オブ テキサス システム (8)
【出願人】(390014960)シスメックス株式会社 (810)
【Fターム(参考)】
[ Back to top ]