
タバコ属(Nicotiana)からのチトクロムp450遺伝子のクローニング
【課題】タバコ属(Nicotiana)におけるp450酵素およびp450酵素をコードする核酸配列、並びにこれらの酵素および核酸配列を用いて、植物表現型を改変する方法を提供する。
【解決手段】トランスジェニック植物を産生する方法であって、前記核酸分子を、前記植物において機能するプロモーターと、機能可能であるように連結して、植物形質転換ベクターを生成し、前記植物を、形質転換し、形質転換植物細胞から形質転換植物を再生する方法。前記核酸分子がRNA干渉方向である方法。これにより、前期植物におけるノルニコチンレベルを増加させるかまたは減少させる方法。
【解決手段】トランスジェニック植物を産生する方法であって、前記核酸分子を、前記植物において機能するプロモーターと、機能可能であるように連結して、植物形質転換ベクターを生成し、前記植物を、形質転換し、形質転換植物細胞から形質転換植物を再生する方法。前記核酸分子がRNA干渉方向である方法。これにより、前期植物におけるノルニコチンレベルを増加させるかまたは減少させる方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、タバコ属(Nicotiana)植物においてチトクロムP450酵素(以後、p450およびp450酵素と称する)をコードする核酸配列、およびこうした核酸配列を用いて植物表現型を改変する方法に関する。
【背景技術】
【0002】
背景
チトクロムp450は、内因性基質および生体異物基質の酸化代謝、過酸化代謝および還元代謝を含む、多様な範囲の化学的に異なる基質の酵素反応を触媒する。植物において、p450は、フェニルプロパノイド、アルカロイド、テルペノイド、脂質、青酸グリコシド、およびグルコシノレートなどの植物産物の合成を含む、生化学的経路に関与する(Chappel, Annu. Rev. Plant Physiol. Plant Mol. Biol. 198, 49:311−343(非特許文献1))。チトクロムp450は、p450ヘム−チオレート・タンパク質としても知られ、通常、p450含有モノオキシゲナーゼ系と呼ばれる多構成要素電子移動連鎖の最後のオキシダーゼとして作用する。触媒される特定の反応には、脱メチル化、水酸化、エポキシ化、N−酸化、スルホ酸化(sulfooxidation)、N−、S−、およびO−脱アルキル化、脱硫酸化、脱アミノ化、並びにアゾ、ニトロ、およびN−オキシド基の還元が含まれる。
【0003】
タバコ属植物p450酵素の多様な役割が、フェニルプロパノイド、アルカロイド、テルペノイド、脂質、青酸グリコシド、グルコシノレートおよび多くの他の化学実体などの多様な植物代謝産物を生じるのに関連付けられてきている。近年、いくつかのp450酵素が、植物において植物代謝産物の組成に影響を及ぼしうることが明らかになってきている。例えば、選択される脂肪酸のプロフィールを、育種を通じて改変することによって、特定の植物のフレーバーおよび香りを改善することが以前から望まれてきた;が、これらの葉の構成要素のレベルを調節するのに関与する機構に関してはほとんどわかっていない。脂肪酸の修飾に関与するp450酵素を下方制御すると、より好ましい葉の表現型特性を提供する、望ましい脂肪酸の集積が促進されることも可能である。植物構成要素におけるp450酵素の機能およびその広い役割は、なお発見される途上にある。例えば、特殊な種類のp450酵素が、脂肪酸を分解して、果物および野菜の「新鮮な緑」のにおいに主に貢献する、揮発性C6およびC9アルデヒドおよびアルコールにするのを触媒することが見出された。タバコ属の葉において、脂質構成要素および関連する分解代謝産物を修飾することによって、標的とされる他の新規p450のレベルを改変して、葉の構成要素の品質を増進することも可能である。葉におけるこれらの構成要素のいくつかが、葉の品質特性の成熟を刺激する老化によって影響を受ける。さらに他の報告によって、p450酵素が、植物−病原体相互作用および疾患抵抗性に関与する脂肪酸を改変する際に機能的役割を果たすことが示されてきている。
【0004】
他の例において、p450酵素が、アルカロイド生合成に関与することも示唆されてきている。ノルニコチンは、タバコ(Nicotiana tabaceum)に見られる微量アルカロイドである。ノルニコチンは、p450が仲介してニコチンが脱メチル化され、その後、N位でアシル化およびニトロソ化が起こり、それによって一連のN−アシルノニコチン(N−acylnonicotines)およびN−ニトロソノルニコチンが生じることによって産生されると推論されてきている。推定上のp450脱メチル化酵素(demethylase)に触媒されるN−脱メチル化は、タバコ属におけるノルニコチン生合成の主な供給源であると考えられる。該酵素はミクロソーム性であると考えられ
るが、これまで、ニコチン脱メチル化酵素の精製は成功しておらず、関与する遺伝子も単離されていない。
【0005】
さらに、p450酵素の活性が、遺伝的に調節され、そしてまた、環境要因によって強く影響されると仮定されているが、証明されていない。例えば、タバコ属におけるニコチンの脱メチル化は、植物が成熟段階に達したときに実質的に増加すると考えられている。さらに、RNAが存在する場合、その翻訳を阻害することも可能な転移可能要素を、脱メチル化酵素遺伝子が含有すると仮定されるが、証明はされていない。
【0006】
p450酵素型が非常に多様であり、構造および機能が異なることから、タバコ属p450酵素の研究は、本発明以前には非常に困難であった。さらに、少なくとも部分的に、膜に局在するこれらのp450酵素が、典型的には存在量が少なく、そしてしばしば、精製するには不安定であるため、これらのタンパク質のクローニングが妨害されてきた。したがって、植物におけるp450酵素、およびこれらのp450酵素に関連する核酸配列を同定する必要性が存在する。特に、タバコ属においては、いくつかのチトクロムp450タンパク質しか報告されてきていない。本明細書に記載する発明は、配列同一性に基づいて、p450種のいくつかのグループに対応する、かなりの数のチトクロムp450断片の発見を含む。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Chappel, Annu. Rev. Plant Physiol. Plant Mol. Biol. 198, 49:311−343
【発明の概要】
【課題を解決するための手段】
【0008】
概要
本発明は植物p450酵素に関する。本発明はさらに、タバコ属由来の植物p450酵素に関する。本発明はまた、その発現がエチレンおよび/または植物老化によって誘導される、植物におけるp450酵素にも関する。本発明はさらに、酵素活性、例えばオキシゲナーゼ、脱メチル化酵素等、またはその他に分類される酵素活性を有する、植物中の核酸配列、およびこれらの酵素の発現または過剰発現を減少させるかまたはサイレンシングするための、これらの配列の使用にも関する。本発明はまた、より低いノルニコチンレベルを示す植物より、より高いノルニコチンレベルを含有する植物で見られるp450酵素にも関する。
【0009】
1つの側面において、本発明は、配列番号1、3、5、7、9、11、13、15、17、19、21、23、25、27、29、31、33、35、37、39、41、43、45、47、49、51、53、55、57、59、61、63、65、67、69、71、73、75、77、79、81、83、85、87、89、91、95、97、99、101、103、105、107、109、111、113、115、117、119、121、123、125、127、129、131、133、135、137、139、143、145、147、149、151、153、155、157、159、161、163、165、167、169、171、173、175、177、179、181、183、185、187、189、191、193、195、197、199、201、203、205、207、209、211、213、215、217、219、221、223、225、227、229、231、233、235、237、239、241、243、245、247、249、251、253、255、257、259、261、263、265、267、269、271、273、275、277、279、281、283、285、287、289、291、293、295および297に示す核酸配列に関する。
【0010】
第二の関連する側面において、核酸配列において75%を超える同一性を含有する断片を、チトクロムp450モチーフGXRXCX(G/A)に続く最初の核酸から停止コドンまでに対応する領域における同一性に応じたグループに入れた。代表的な核酸グループ
およびそれぞれの種を表Iに示す。
【0011】
第三の側面において、本発明は、配列番号2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、32、34、36、38、40、42、44、46、48、50、52、54、56、58、60、62、64、66、68、70、72、74、76、78、80、82、84、86、88、90、92、96、98、100、102、104、106、108、110、112、114、116、118、120、122、124、126、128、130、132、134、136、138、140、144、146、148、150、152、154、156、158、160、162、164、166、168、170、172、174、176、178、180、182、184、186、188、190、192、194、196、198、200、202、204、206、208、210、212、214、216、218、220、222、224、226、228、230、232、234、236、238、240、242、244、246、248、250、252、254、256、258、260、262、264、266、268、270、272、274、276、278、280、282、284、286、288、290、292、294、296および298に示すアミノ酸配列に関する。
【0012】
第四の関連する側面において、アミノ酸配列において71%を超える同一性を含有する断片を、チトクロムp450モチーフGXRXCX(G/A)に続く最初のアミノ酸から停止コドンまでに対応する領域における、互いに対する同一性に応じたグループに入れた。代表的なアミノ酸グループおよびそれぞれの種を表IIに示す。
【0013】
第五の側面において、本発明は、配列番号150、152、154、156、158、160、162、164、166、168、170、172、174、176、178、180、182、184、186、188、190、192、194、196、198、200、202、204、206、208、210、212、214、216、218、220、222、224、226、228、230、232、234、236、238、240、242、244、246、248、250、252、254、256、258、260、262、264、266、268、270、272、274、276、278、280、282、284、286、288、290、292、294、296および298に示す全長遺伝子のアミノ酸配列に関する。
【0014】
第六の関連する側面において、アミノ酸配列において85%以上の同一性を含有する全長遺伝子を、互いに対する同一性に応じたグループに入れた。代表的なアミノ酸グループおよびそれぞれの種を表IIIに示す。
【0015】
第七の側面において、本発明は、配列番号299〜357に示す断片のアミノ酸配列に関する。
第八の関連する側面において、アミノ酸配列において90%以上の同一性を含有する断片を、最初のチトクロムp450ドメイン、UXXRXXZから、第三のチトクロム・ドメイン、GXRXO、ここでUはEまたはKであり、Xはアミノ酸いずれかであり、そしてZはR、T、SまたはMである、までに対応する領域における、互いに対する同一性に応じたグループに入れた。代表的なアミノ酸グループおよびそれぞれの種を表IVに示す。
【0016】
第九の関連する側面において、RNAウイルス系を用いて、タバコ属植物におけるp450酵素の減少または除去または過剰発現を一過性に達成することも可能である。
一般の当業者に通常利用可能な技術を用いて、限定されるわけではないが、内因性p450 RNA転写物、発現されたp450ペプチド、および植物代謝産物濃度の解析を含
む、表現型変化に関して、生じた形質転換植物または感染植物を評価する。
【0017】
第十の重要な側面において、本発明はまた、改変されたp450酵素活性レベルを有するトランスジェニック・タバコ属系統の生成にも関する。本発明にしたがって、これらのトランスジェニック系統には、特定の酵素の発現を減少させるかまたはサイレンシングするかまたは増加させ、したがってタバコ属内で表現型効果を生じるのに有効である核酸配列が含まれる。こうした核酸配列には、配列番号1、3、5、7、9、11、13、15、17、19、21、23、25、27、29、31、33、35、37、39、41、43、45、47、49、51、53、55、57、59、61、63、65、67、69、71、73、75、77、79、81、83、85、87、89、91、95、97、99、101、103、105、107、109、111、113、115、117、119、121、123、125、127、129、131、133、135、137、139、143、145、147、149、151、153、155、157、159、161、163、165、167、169、171、173、175、177、179、181、183、185、187、189、191、193、195、197、199、201、203、205、207、209、211、213、215、217、219、221、223、225、227、229、231、233、235、237、239、241、243、245、247、249、251、253、255、257、259、261、263、265、267、269、271、273、275、277、279、281、283、285、287、289、291、293、295および297が含まれる。
【0018】
本発明の非常に重要な第十一の側面において、全長遺伝子またはその断片のいずれかを用いて下方制御能力がある、あるいは全長遺伝子を用いて過剰発現能力がある、本発明の核酸を含む植物品種は、対照植物に比較して、改変された代謝産物プロフィールを有するであろう。
【0019】
本発明の第十二の側面において、本発明の核酸を含む植物品種は、植物または植物外部に由来する代謝産物の生合成または分解を修飾する際に、全長遺伝子またはその断片のいずれかを用いて、特定の外因性化学薬品または植物疫病に耐容性を示すのに使用を有するであろう。こうした核酸配列には、配列番号1、3、5、7、9、11、13、15、17、19、21、23、25、27、29、31、33、35、37、39、41、43、45、47、49、51、53、55、57、59、61、63、65、67、69、71、73、75、77、79、81、83、85、87、89、91、95、97、99、101、103、105、107、109、111、113、115、117、119、121、123、125、127、129、131、133、135、137、139、143、145、147、149、151、153、155、157、159、161、163、165、167、169、171、173、175、177、179、181、183、185、187、189、191、193、195、197、199、201、203、205、207、209、211、213、215、217、219、221、223、225、227、229、231、233、235、237、239、241、243、245、247、249、251、253、255、257、259、261、263、265、267、269、271、273、275、277、279、281、283、285、287、289、291、293、295および297が含まれる。
【0020】
第十三の側面において、本発明は、解説される核酸配列に実質的な核酸同一性を有する遺伝子を含有する植物、より好ましくはタバコ属のスクリーニングに関する。本発明の使用は、こうした植物が伝統的な品種またはトランスジェニック品種の育種プログラム、突然変異誘発プログラム、あるいは天然存在の多様な植物集団の一部である場合、厳密なま
たは実質的な同一性を持つ核酸配列を含有する植物を同定し、そして選択するのに好適であろう。実質的な核酸同一性に関する植物のスクリーニングは、限定されるわけではないが、核酸ハイブリダイゼーションおよびPCR解析を含む核酸検出プロトコルと組み合わせて、核酸プローブを用い、植物核酸物質を評価することによって、達成可能である。核酸プローブは、配列番号1、3、5、7、9、11、13、15、17、19、21、23、25、27、29、31、33、35、37、39、41、43、45、47、49、51、53、55、57、59、61、63、65、67、69、71、73、75、77、79、81、83、85、87、89、91、95、97、99、101、103、105、107、109、111、113、115、117、119、121、123、125、127、129、131、133、135、137、139、143、145、147、149、151、153、155、157、159、161、163、165、167、169、171、173、175、177、179、181、183、185、187、189、191、193、195、197、199、201、203、205、207、209、211、213、215、217、219、221、223、225、227、229、231、233、235、237、239、241、243、245、247、249、251、253、255、257、259、261、263、265、267、269、271、273、275、277、279、281、283、285、287、289、291、293、295および297に対応する、解説される核酸配列またはその断片からなることも可能である。
【0021】
第十四の側面において、本発明は、解説される核酸配列に対応する実質的なアミノ酸同一性を共有する植物遺伝子、より好ましくはタバコ属遺伝子の同定に関する。cDNAおよびゲノムクローン両方を含む植物遺伝子、より好ましくはタバコ属由来のcDNAおよびゲノムクローンを含む植物遺伝子の同定は、限定されるわけではないが、核酸ハイブリダイゼーションおよびPCR解析を含む核酸検出プロトコルと組み合わせて核酸プローブを用い、植物cDNAライブラリーをスクリーニングすることによって、達成可能である。核酸プローブは、配列番号1、3、5、7、9、11、13、15、17、19、21、23、25、27、29、31、33、35、37、39、41、43、45、47、49、51、53、55、57、59、61、63、65、67、69、71、73、75、77、79、81、83、85、87、89、91、95、97、99、101、103、105、107、109、111、113、115、117、119、121、123、125、127、129、131、133、135、137、139、143、145および147に対応する核酸配列またはその断片で構成されることも可能である。
【0022】
別の第十五の側面において、解説されるアミノ酸配列の一部またはすべてに向けられる抗体を用いて、ペプチドを発現するcDNA発現ライブラリーをスクリーニングすることも可能である。こうしたアミノ酸配列には、配列番号2、4、8、9、10、12、14、16、18、20、22、24、26、28、30、32、34、36、38、40、42、44、46、48、50、52、54、56、58、60、62、64、66、68、70、72、74、76、78、80、82、84、86、88、90、92、96、98、100、102、104、106、108、110、112、114、116、118、120、122、124、126、128、130、132、134、136、138、140、144、146、148が含まれる。
【0023】
第十六の重要な側面において、本発明はまた、p450酵素活性レベルの過剰発現を有するトランスジェニック・タバコ属系統の生成にも関する。本発明にしたがって、これらのトランスジェニック系統には、特定の酵素の発現を増加させるのに有効であり、したがってタバコ属内で表現型効果を生じるのに有効である全長遺伝子のアミノ酸配列をコードするすべての核酸配列が含まれる。こうしたアミノ酸配列には、配列番号150、152、154、156、158、160、162、164、166、168、170、172
、174、176、178、180、182、184、186、188、190、192、194、196、198、200、202、204、206、208、210、212、214、216、218、220、222、224、226、228、230、232、234、236、238、240、242、244、246、248、250、252、254、256、258、260、262、264、266、268、270、272、274、276、278、280、282、284、286、288、290、292、294、296および298が含まれる。
【0024】
減少した量のノルニコチンを有するタバコ葉(ラミナおよび/または茎)を含むタバコ製品もまた提供する。タバコ製品には、本明細書記載の配列を含む植物、あるいはタバコ特異的ニトロソアミンをコードする遺伝子が除去または抑制されている植物由来のタバコ(ラミナおよび/または茎を含むタバコ葉)が含まれる。タバコ特異的ニトロソアミンをコードする遺伝子の除去または抑制は、タバコ特異的ニトロソアミンをコードする遺伝子が除去または抑制されていないタバコ植物から作成されたタバコ製品に比較して、タバコ製品中のタバコ特異的ニトロソアミンを、約5〜約10%、別の側面においては約10〜20%、別の側面においては約20〜30%、そして別の側面においては30%を超えて、減少させるのに有効である。本明細書において、タバコ製品は、巻きタバコ、葉巻、パイプタバコ、嗅ぎタバコ、噛みタバコ、タバコ製品とブレンドした製品、およびこれらの混合物を含むことも可能である。
【0025】
詳細な説明
定義
別に定義しない限り、本明細書で用いるすべての技術的用語および科学的用語は、本発明が属する当該技術分野の一般の当業者に一般的に理解されるのと同じ意味を有する。Singletonら(1994)Dictionary of Microbiology and Molecular Biology, 第2版, John Wiley
and Sons(ニューヨーク)は、本発明で用いる用語の多くの一般的辞書とともに、技術の1つを提供する。本明細書に引用する特許および刊行物はすべて本明細書に援用される。本発明の目的のため、次の用語を以下に定義する。
【0026】
「酵素活性」は、脱メチル化、水酸化、エポキシ化、N−酸化、スルホ酸化、N−、S−、およびO−脱アルキル化、脱硫酸化、脱アミノ化、並びにアゾ、ニトロ、およびN−オキシド基の還元を含むことを意味される。用語「核酸」は、一本鎖または二本鎖型いずれか、あるいはセンスまたはアンチセンスのデオキシリボヌクレオチドまたはリボヌクレオチドポリマーを指し、そして別に限定されない限り、天然存在ヌクレオチドに似た方式で、核酸にハイブリダイズする天然ヌクレオチドの既知の類似体(analogue)を含む。別に示さない限り、特定の核酸配列には、その相補配列が含まれる。
【0027】
用語「機能可能であるように連結される」、「機能可能な組み合わせ」および「機能可能な順序」は、核酸発現調節配列(プロモーター、シグナル配列、または転写因子結合部位の列など)および第二の核酸配列の間の機能する連結を指し、ここで発現調節配列は、第二の配列に対応する核酸の転写および/または翻訳に影響を及ぼす。
【0028】
用語「組換え」は、細胞に関して用いた場合、細胞が異種核酸を複製するか、前記核酸を発現するか、あるいは異種核酸にコードされるペプチド、異種ペプチド、またはタンパク質を発現することを示す。組換え細胞は、細胞の天然(非組換え)型には見られないセンス型またはアンチセンス型いずれかの遺伝子または遺伝子断片を発現することも可能である。組換え細胞はまた、細胞の天然型に見られるが、修飾され、そして人工的手段によって細胞に再度導入されている遺伝子を発現することもまた可能である。
【0029】
「構造遺伝子」は、タンパク質、ポリペプチドまたはその一部をコードするDNAセグメントを含み、そして転写開始を駆動する5’配列を除く遺伝子の部分である。あるいは構造遺伝子は翻訳不能産物をコードすることも可能である。構造遺伝子は、細胞に通常見られるもの、あるいは導入される細胞または細胞位置で通常は見られないものであることも可能であり、この場合、該遺伝子は「異種遺伝子」と呼ばれる。異種遺伝子は、細菌ゲノムまたはエピソーム、真核、核またはプラスミドDNA、cDNA、ウイルスDNAあるいは化学的に合成されたDNAを含む、当該技術分野に知られるいかなる供給源に、すべてまたは一部、由来していることも可能である。構造遺伝子は、生物学的活性またはその特性、発現産物の生物学的活性または化学構造、発現率または発現調節の方式を達成可能な1以上の修飾を含有することも可能である。こうした修飾には、限定されるわけではないが、1以上のヌクレオチドの突然変異、挿入、欠失および置換が含まれる。構造遺伝子は、中断されないコード配列を構成することも可能であるし、または適切なスプライス接合部によって結合される1以上のイントロンを含むことも可能である。構造遺伝子は翻訳可能または翻訳不能であることも可能であり、アンチセンス方向にあるものを含む。構造遺伝子は複数の供給源由来および複数の遺伝子配列(天然存在または合成、ここで合成は化学的に合成されたDNAを指す)由来のセグメントの合成物(composite)であることも可能である。
【0030】
「由来する」は、供給源(化学的および/または生物学的)から採取されるか、得られるか、受け取られるか、帰着するか、複製されるか、または伝わる(descend)ことを意味するよう用いられる。派生物(derivative)は、元来の供給源の化学的操作または生物学的操作(限定されるわけではないが、置換、付加、挿入、欠失、抽出、単離、突然変異および複製を含む)によって産生されることも可能である。
【0031】
「化学的に合成された」は、DNAの配列に関する場合、構成要素ヌクレオチドの一部がin vitroで組み立てられたことを意味する。よく確立された方法(Caruthers, Methodology of DNA and RNA Sequencing, (1983), Weissman(監修), Praeger Publishers, ニューヨーク, 第1章)を用いて、DNAの手動の化学的合成を達成することも可能であり;いくつかの商業的に入手可能な機械の1つを用いて、自動化化学合成を行うことも可能である。
【0032】
SmithおよびWaterman, Adv. Appl. Math. 2:482(1981)の局所相同性アルゴリズムによって、NeedlemanおよびWunsch, J. Mol. Biol. 48:443(1970)の相同性並列アルゴリズムによって、PearsonおよびLipman Proc. Natl. Acad. Sci.(U.S.A.)85:2444(1988)の類似性法に関する検索によって、これらのアルゴリズムのコンピュータ化実行(ウィスコンシン遺伝学ソフトウェアパッケージ、遺伝学コンピュータグループ、575 Science Dr., Madison, Wis.のGAP、BESTFIT、FASTA、およびTFASTA)によって、または視診によって、比較のための配列の最適並列を行うことも可能である。
【0033】
NCBI基本的局所並列検索ツール(BLAST)(Altschulら、1990)は、米国生物学情報センター(NCBI、メリーランド州ベセスダ)を含むいくつかの供給源から入手可能であり、そして配列解析プログラムblastp、blastn、blastx、tblastnおよびtblastxと関連付けて使用するため、インターネット上で入手可能である。該ツールは、htp://www.ncbi.nlm.nih.gov/BLAST/でアクセス可能である。このプログラムを用いて配列同一性を決定する方法の説明が、http://www.ncbi.nlm.nih.gov/BLAST/blast help.htmlで入手可能である。
【0034】
用語「実質的なアミノ酸同一性」または「実質的なアミノ酸配列同一性」は、アミノ酸配列に適用した場合、そして本明細書で用いる場合、ペプチドが、翻訳されたペプチドのチトクロムp450モチーフGXRXCX(G/A)に続く最初のアミノ酸から停止コドンまでに対応する領域に渡って、参照群に比較した際、少なくとも70パーセントの配列同一性、好ましくは80パーセントのアミノ酸配列同一性、より好ましくは90パーセントのアミノ酸配列同一性、そして最も好ましくは少なくとも99〜100パーセントの配列同一性を有する配列を含む、ポリペプチドの特性を示す。
【0035】
用語「実質的な核酸同一性」または「実質的な核酸配列同一性」は、核酸配列に適用した場合、そして本明細書で用いる場合、ポリヌクレオチドが、翻訳されたペプチドのチトクロムp450モチーフGXRXCX(G/A)に続く最初の核酸から停止コドンまでに対応する領域に渡って、参照群に比較した際、少なくとも75パーセントの配列同一性、好ましくは81パーセントの配列同一性、より好ましくは少なくとも91パーセントの配列同一性、そして最も好ましくは少なくとも99〜100パーセントの配列同一性を有する配列を含む、ポリヌクレオチド配列の特性を示す。
【0036】
ヌクレオチド配列が実質的に同一であることの別の指標は、2つの分子がストリンジェントな条件下で互いにハイブリダイズする場合である。ストリンジェントな条件は配列依存性であり、そして異なる環境において異なるであろう。一般的に、ストリンジェントな条件は、特定の配列に関する、定義されるイオン強度およびpHでの熱融点(Tm)より約5℃〜約20℃、通常、約10℃〜約15℃低いように選択される。Tmは、(定義されるイオン強度およびpHで)標的配列の50%が、マッチしたプローブにハイブリダイズする温度である。典型的には、ストリンジェントな条件は、pH7で塩濃度が約0.02モル濃度であり、そして温度が少なくとも約60℃であるものであろう。例えば標準的サザンハイブリダイゼーション法において、ストリンジェントな条件は、42℃の6xSSC中の最初の洗浄、続いて少なくとも約55℃、典型的には約60℃、そしてしばしば約65℃の温度での0.2xSSC中の1以上のさらなる洗浄を含むであろう。
【0037】
ヌクレオチド配列はまた、コードするポリペプチドおよび/またはタンパク質が実質的に同一である場合、本発明の目的のためには、実質的に同一である。したがって、1つの核酸配列が第二の核酸配列と本質的に同じポリペプチドをコードする場合、遺伝暗号に許容される縮重のため、ストリンジェントな条件下でハイブリダイズしない場合であっても、2つの核酸配列は実質的に同一である(コドン縮重および遺伝暗号の説明に関しては、Darnellら(1990)Molecular Cell Biology, 第2版 Scientific American Books W. H. Freeman and Company ニューヨークを参照されたい)。タンパク質試料のポリアクリルアミドゲル電気泳動、その後、染色での視覚化などの、当該技術分野に周知のいくつかの手段によって、タンパク質純度または均一性を示すことも可能である。特定の目的のため、高分解能が必要とされる可能性もあり、そして精製のためにHPLCまたは類似の手段を利用することも可能である。
【0038】
本明細書において、細胞にDNAセグメント(単数または複数)をトランスファーする核酸分子に関して、用語「ベクター」を用いる。ベクターはDNAを複製するよう作用することも可能であり、そして宿主細胞において、独立に複製可能である。用語「ビヒクル」は、ときに、「ベクター」と交換可能に用いられる。用語「発現ベクター」は、本明細書において、望ましいコード配列、および特定の宿主生物において、機能可能であるように連結されたコード配列の発現に必要な適切な核酸配列を含有する、組換えDNA分子を指す。原核生物における発現に必要な核酸配列には、通常、プロモーター、オペレーター(場合による)、およびリボソーム結合部位に、しばしば他の配列が伴って、含まれる。
真核細胞は、プロモーター、エンハンサー、および終結シグナルおよびポリアデニル化シグナルを利用することが知られる。
【0039】
根がある、完全に遺伝子操作された植物を再生するため、in vivo接種などの技術いずれかによって、または完全な植物に再生可能な形質転換植物細胞を産生する、既知のin vitro組織培養技術のいずれかによって、核酸を植物細胞に挿入することも可能である。したがって、例えば、植物細胞への挿入は、病原性または非病原性A.ツメファシエンス(A. tumefaciens)によるin vitro接種によることも可能である。他のこうした組織培養技術もまた、使用可能である。
【0040】
「植物組織」には、植物の分化組織および未分化組織が含まれ、限定されるわけではないが、根、芽、葉、花粉、種子、腫瘍組織および培養中の多様な型の細胞、例えば単細胞、プロトプラスト、胚およびカルス組織が含まれる。植物組織は、植物中(in planta)または器官中にあることも、組織または細胞培養物であることも可能である。
【0041】
「植物細胞」には、本明細書において、植物中の植物細胞、並びに培養中の植物細胞およびプロトプラストが含まれる。
「cDNA」または「相補DNA」は、一般的に、RNA分子に相補的なヌクレオチド配列を持つ一本鎖DNA分子を指す。cDNAは、RNAテンプレートに対する逆転写酵素の作用によって形成される。
【0042】
核酸配列を得る戦略
本発明にしたがって、変換(converter)および非変換タバコ属系統のタバコ属組織からRNAを抽出した。次いで、抽出したRNAを用いてcDNAを生成した。次いで、2つの戦略を用いて、本発明の核酸配列を生成した。
【0043】
第一の戦略では、植物組織からポリA濃縮RNAを抽出し、そして逆転写PCRによってcDNAを作成した。次いで、一本鎖cDNAを用い、縮重プライマーに加えて、オリゴd(T)逆方向プライマーを用いて、p450特異的PCR集団を生成した。プライマー設計は、p450の高度に保存されたモチーフに基づいた。特異的縮重プライマーの例を図1に示す。適切なサイズの挿入物を含有するプラスミド由来の配列断片をさらに解析した。これらのサイズの挿入物は、どのプライマーを用いたかに応じて、典型的には、約300〜約800ヌクレオチドの範囲であった。
【0044】
第二の戦略では、まず、cDNAライブラリーを構築した。プラスミド中のcDNAを用い、縮重プライマーに加えて、逆方向プライマーとしてプラスミド上のT7プライマーを用いて、p450特異的PCR集団を生成した。第一の戦略におけるように、適切なサイズの挿入物を含有するプラスミド由来の配列断片をさらに解析した。
【0045】
高レベルのノルニコチンを生じることが知られるタバコ属植物系統(変換系統)、および検出不能なレベルのノルニコチンを有する植物系統を出発材料として使用することも可能である。
【0046】
次いで、植物から葉を取り除き、そしてエチレンで処理して、本明細書に定義するp450酵素活性を活性化することも可能である。当該技術分野に知られる技術を用いて、総RNAを抽出する。次いで、図153に記載するようなオリゴd(T)プライマーを用いたPCR(RT−PCR)を用いて、cDNA断片を生成することも可能である。次いで、本明細書の実施例に、より完全に記載する、cDNAライブラリーを構築することも可能である。
【0047】
p450型酵素に保存される領域を縮重プライマーのテンプレートとして用いることも可能である(図75)。縮重プライマーを用いて、p450特異的バンドをPCRによって増幅することも可能である。p450様酵素を示すバンドをDNA配列決定によって同定することも可能である。BLAST検索、並列、または適切な候補を同定する他のツールを用いて、PCR断片を性質決定することも可能である。
【0048】
同定された断片からの配列情報を用いて、PCRプライマーを発展させることも可能である。cDNAライブラリー中のプラスミドプライマーと組み合わせてこれらのプライマーを用い、全長p450遺伝子をクローニングした。大規模サザン逆解析を行って、得られたすべての断片クローン、およびいくつかの場合、全長クローンに関して、示差発現を調べた。本発明のこの側面において、クローニングされた挿入物すべてをスクリーニングするため、クローニングされたDNA断片とハイブリダイズするプローブとして、異なる組織由来の標識総cDNAを用いて、これらの大規模逆サザンアッセイを行うことも可能である。
【0049】
非放射性および放射性(P32)ノーザンブロッティングアッセイもまた用いて、クローンp450断片および全長クローンを性質決定した。
全長クローンのアミノ酸配列を得て、そして抗原性であり、そして他のクローンに比較してユニークであるペプチド領域を選択することによって、いくつかの全長クローンに対してペプチド特異的抗体を作成した。キャリアータンパク質にコンジュゲート化した合成ペプチドに対して、ウサギ抗体を作成した。これらの抗体を用いて、植物組織に対して、ウェスタンブロッティング解析または他の免疫学的方法を行った。
【0050】
ウイルス誘導性遺伝子サイレンシング技術を用いることによって、上述のように同定された核酸配列を調べることも可能である(VIGS, Baulcombe, Current Opinions in Plant Biology, 1999, 2:109−113)。
【0051】
全長クローンのアミノ酸配列を得て、そして潜在的に抗原性であり、そして他のクローンに比較してユニークであるペプチド領域を選択することによって、いくつかの全長クローンに対してペプチド特異的抗体を作成した。キャリアータンパク質にコンジュゲート化した合成ペプチドに対して、ウサギ抗体を作成した。これらの抗体を用いて、ウェスタンブロッティング解析を行った。
【0052】
本発明の別の側面において、本発明のタバコ属植物におけるチトクロムp450酵素活性をさらに性質決定するため、RNA干渉技術(RNAi)を用いる。この技術を説明する以下の参考文献、Smithら, Nature, 2000, 407:319−320;Fireら, Nature, 1998, 391:306−311;Waterhouseら, PNAS, 1998, 95:13959−13964;Stalbergら, Plant Molecular Biology, 1993, 23:671−683;Baulcombe, Current Opinions in Plant Biology, 1999, 2:109−113;およびBrignetiら, EMBO Journal, 1998, 17(22):6739−6746が本明細書に援用される。RNAi技術、アンチセンス技術、または記載される多様な他の方法を用いて、植物を形質転換することも可能である。
【0053】
植物細胞に外来(foreign)遺伝物質を導入するための、そして導入された遺伝子を安定して維持し、そして発現する植物を得るための、いくつかの技術が存在する。こうした技術には、微小粒子上にコーティングした遺伝物質を細胞内に直接加速することが含まれる(Cornellに対する米国特許4,945,050およびDowElanc
oに対する5,141,131)。アグロバクテリウム(Agrobacterium)技術を用いて、植物を形質転換することも可能であり、トレド大学に対する米国特許5,177,010、Texax A&Mに対する5,104,310、Schilperootに対する欧州特許出願0131624B1、欧州特許出願120516、159418B1、欧州特許出願120516、159418B1および176,112、Schilperootに対する米国特許5,149,645、5,469,976、5,464,763および4,940,838および4,693,976、すべてMaxPlanckに対する欧州特許出願116718、290799、320500、Japan Nicotianaに対する欧州特許出願604662および627752、すべてCiba
Geigyに対する欧州特許出願0267159および0292435および米国特許5,231,019、どちらもCalgeneに対する米国特許5,463,174および4,762,785、並びにどちらもAgracetusに対する米国特許5,004,863および5,159,135を参照されたい。他の形質転換技術には、ウィスカー技術が含まれ、どちらもZenecaに対する米国特許5,302,523および5,464,765を参照されたい。植物を形質転換するにはエレクトロポレーション技術もまた用いられてきており、Boyce Thompson Instituteに対するWO 87/06614、どちらもDekalbに対する5,472,869および5,384,253、どちらもPGSに対するWO9209696およびWO9321335を参照されたい。これらの形質転換特許および刊行物はすべて、本明細書に援用される。植物を形質転換する多くの技術に加えて、外来遺伝子と接触させる組織の種類もまた、多様であることも可能である。こうした組織には、限定されるわけではないが、胚形成組織、カルス組織I型およびII型、胚軸、成長点等が含まれるであろう。当業者の技術内の適切な技術を用いて、脱分化中の、ほぼすべての植物組織を形質転換することも可能である。
【0054】
植物に導入される外来遺伝物質には、選択可能マーカーが含まれることも可能である。特定のマーカーを優先するのは、当業者の自由裁量であるが、以下の選択可能マーカーのいずれかとともに、選択可能マーカーとして機能可能な、本明細書に列挙されていない他の遺伝子のいずれも使用可能である。こうした選択可能マーカーには、限定されるわけではないが、抗生物質カナマイシン、ネオマイシンおよびG418に対する抵抗性をコードするトランスポゾンTn5のアミノグリコシド・ホスホトランスフェラーゼ遺伝子(AphII)とともに、グリフォセート;ハイグロマイシン;メトトレキセート;フォスフィノスリシン(bar);イミダゾリノン、スルホニル尿素およびトリアゾロピリミジン除草剤、例えばクロロスルフロン;ブロモキシニル、ダラポン等に対する抵抗性または耐性をコードする遺伝子が含まれる。
【0055】
選択可能マーカーに加えて、レポーター遺伝子を用いることが望ましい可能性もある。いくつかの例において、選択可能マーカーを伴わずに、レポーター遺伝子を用いることも可能である。レポーター遺伝子は、レシピエント生物または組織に典型的には存在しないかまたはこれらで発現されていない遺伝子である。レポーター遺伝子は、典型的には何らかの表現型変化または酵素特性を提供するタンパク質をコードする。こうした遺伝子の例が、本明細書に援用されるK. Weisingら Ann. Rev. Genetics, 22, 421(1988)に提供される。好ましいレポーター遺伝子には、限定なしに、グルクロニダーゼ(GUS)遺伝子およびGFP遺伝子が含まれる。
【0056】
植物組織にひとたび導入されたならば、当該技術分野に知られるいかなる手段によって構造遺伝子の発現をアッセイすることも可能であり、そして転写されるmRNA、合成されるタンパク質、または生じる遺伝子サイレンシングの量として、発現を測定することも可能である(本明細書に援用される米国特許第5,583,021号を参照されたい)。植物組織のin vitro培養のための技術が知られ、そしていくつかの場合、全植物
への再生の技術が知られる(EP出願第88810309.0)。導入された発現複合体を商業的に有用な品種にトランスファーするための方法が、当業者に知られる。
【0057】
望ましいレベルのp450酵素を発現する植物細胞がひとたび得られたら、当該技術分野に周知の方法および技術を用いて、そこから植物組織および全植物を再生することも可能である。次いで、再生された植物を、慣用的手段によって、繁殖させ、そして慣用的な植物育種技術によって、導入された遺伝子を他の系統および品種にトランスファーすることも可能である。
【0058】
以下の実施例は、本発明を実行する方法を例示し、そして付随する請求項に定義される本発明の範囲を例示するが、これを限定しないことが理解されなければならない。
【図面の簡単な説明】
【0059】
【図1】図1は、核酸配列番号1およびアミノ酸配列番号2を示す。
【図2】図2は、核酸配列番号3およびアミノ酸配列番号4を示す。
【図3】図3は、核酸配列番号5およびアミノ酸配列番号6を示す。
【図4】図4は、核酸配列番号7およびアミノ酸配列番号8を示す。
【図5】図5は、核酸配列番号9およびアミノ酸配列番号10を示す。
【図6】図6は、核酸配列番号11およびアミノ酸配列番号12を示す。
【図7】図7は、核酸配列番号13およびアミノ酸配列番号14を示す。
【図8】図8は、核酸配列番号15およびアミノ酸配列番号16を示す。
【図9】図9は、核酸配列番号17およびアミノ酸配列番号18を示す。
【図10】図10は、核酸配列番号19およびアミノ酸配列番号20を示す。
【図11】図11は、核酸配列番号21およびアミノ酸配列番号22を示す。
【図12】図12は、核酸配列番号23およびアミノ酸配列番号24を示す。
【図13】図13は、核酸配列番号25およびアミノ酸配列番号26を示す。
【図14】図14は、核酸配列番号27およびアミノ酸配列番号28を示す。
【図15】図15は、核酸配列番号29およびアミノ酸配列番号30を示す。
【図16】図16は、核酸配列番号31およびアミノ酸配列番号32を示す。
【図17】図17は、核酸配列番号33およびアミノ酸配列番号34を示す。
【図18】図18は、核酸配列番号35およびアミノ酸配列番号36を示す。
【図19】図19は、核酸配列番号37およびアミノ酸配列番号38を示す。
【図20】図20は、核酸配列番号39およびアミノ酸配列番号40を示す。
【図21】図21は、核酸配列番号41およびアミノ酸配列番号42を示す。
【図22】図22は、核酸配列番号43およびアミノ酸配列番号44を示す。
【図23】図23は、核酸配列番号45およびアミノ酸配列番号46を示す。
【図24】図24は、核酸配列番号47およびアミノ酸配列番号48を示す。
【図25】図25は、核酸配列番号49およびアミノ酸配列番号50を示す。
【図26】図26は、核酸配列番号51およびアミノ酸配列番号52を示す。
【図27】図27は、核酸配列番号53およびアミノ酸配列番号54を示す。
【図28】図28は、核酸配列番号55およびアミノ酸配列番号56を示す。
【図29】図29は、核酸配列番号57およびアミノ酸配列番号58を示す。
【図30】図30は、核酸配列番号59およびアミノ酸配列番号60を示す。
【図31】図31は、核酸配列番号61およびアミノ酸配列番号62を示す。
【図32】図32は、核酸配列番号63およびアミノ酸配列番号64を示す。
【図33】図33は、核酸配列番号65およびアミノ酸配列番号66を示す。
【図34】図34は、核酸配列番号67およびアミノ酸配列番号68を示す。
【図35】図35は、核酸配列番号69およびアミノ酸配列番号70を示す。
【図36】図36は、核酸配列番号71およびアミノ酸配列番号72を示す。
【図37】図37は、核酸配列番号73およびアミノ酸配列番号74を示す。
【図38】図38は、核酸配列番号75およびアミノ酸配列番号76を示す。
【図39】図39は、核酸配列番号77およびアミノ酸配列番号78を示す。
【図40】図40は、核酸配列番号79およびアミノ酸配列番号80を示す。
【図41】図41は、核酸配列番号81およびアミノ酸配列番号82を示す。
【図42】図42は、核酸配列番号83およびアミノ酸配列番号84を示す。
【図43】図43は、核酸配列番号85およびアミノ酸配列番号86を示す。
【図44】図44は、核酸配列番号87およびアミノ酸配列番号88を示す。
【図45】図45は、核酸配列番号89およびアミノ酸配列番号90を示す。
【図46】図46は、核酸配列番号91およびアミノ酸配列番号92を示す。
【図47】図47は、核酸配列番号92およびアミノ酸配列番号93を示す。
【図48】図48は、核酸配列番号95およびアミノ酸配列番号96を示す。
【図49】図49は、核酸配列番号97およびアミノ酸配列番号98を示す。
【図50】図50は、核酸配列番号99およびアミノ酸配列番号100を示す。
【図51】図51は、核酸配列番号101およびアミノ酸配列番号102を示す。
【図52】図52は、核酸配列番号103およびアミノ酸配列番号104を示す。
【図53】図53は、核酸配列番号105およびアミノ酸配列番号106を示す。
【図54】図54は、核酸配列番号107およびアミノ酸配列番号108を示す。
【図55】図55は、核酸配列番号109およびアミノ酸配列番号110を示す。
【図56】図56は、核酸配列番号111およびアミノ酸配列番号112を示す。
【図57】図57は、核酸配列番号113およびアミノ酸配列番号114を示す。
【図58】図58は、核酸配列番号115およびアミノ酸配列番号116を示す。
【図59】図59は、核酸配列番号117およびアミノ酸配列番号118を示す。
【図60】図60は、核酸配列番号119およびアミノ酸配列番号120を示す。
【図61】図61は、核酸配列番号121およびアミノ酸配列番号122を示す。
【図62】図62は、核酸配列番号123およびアミノ酸配列番号124を示す。
【図63】図63は、核酸配列番号125およびアミノ酸配列番号126を示す。
【図64】図64は、核酸配列番号127およびアミノ酸配列番号128を示す。
【図65】図65は、核酸配列番号129およびアミノ酸配列番号130を示す。
【図66】図66は、核酸配列番号131およびアミノ酸配列番号132を示す。
【図67】図67は、核酸配列番号133およびアミノ酸配列番号134を示す。
【図68】図68は、核酸配列番号135およびアミノ酸配列番号136を示す。
【図69】図69は、核酸配列番号137およびアミノ酸配列番号138を示す。
【図70】図70は、核酸配列番号139およびアミノ酸配列番号140を示す。
【図71】図71は、核酸配列番号141およびアミノ酸配列番号142を示す。
【図72】図72は、核酸配列番号143およびアミノ酸配列番号144を示す。
【図73】図73は、核酸配列番号145およびアミノ酸配列番号146を示す。
【図74】図74は、核酸配列番号147およびアミノ酸配列番号148を示す。
【図75】図75は、核酸配列番号149およびアミノ酸配列番号150を示す。
【図76】図76は、核酸配列番号151およびアミノ酸配列番号152を示す。
【図77】図77は、核酸配列番号153およびアミノ酸配列番号154を示す。
【図78】図78は、核酸配列番号155およびアミノ酸配列番号156を示す。
【図79】図79は、核酸配列番号157およびアミノ酸配列番号158を示す。
【図80】図80は、核酸配列番号159およびアミノ酸配列番号160を示す。
【図81】図81は、核酸配列番号161およびアミノ酸配列番号162を示す。
【図82】図82は、核酸配列番号163およびアミノ酸配列番号164を示す。
【図83】図83は、核酸配列番号165およびアミノ酸配列番号166を示す。
【図84】図84は、核酸配列番号167およびアミノ酸配列番号168を示す。
【図85】図85は、核酸配列番号169およびアミノ酸配列番号170を示す。
【図86】図86は、核酸配列番号171およびアミノ酸配列番号172を示す。
【図87】図87は、核酸配列番号173およびアミノ酸配列番号174を示す。
【図88】図88は、核酸配列番号175およびアミノ酸配列番号176を示す。
【図89】図89は、核酸配列番号177およびアミノ酸配列番号178を示す。
【図90】図90は、核酸配列番号179およびアミノ酸配列番号180を示す。
【図91】図91は、核酸配列番号181およびアミノ酸配列番号182を示す。
【図92】図92は、核酸配列番号183およびアミノ酸配列番号184を示す。
【図93】図93は、核酸配列番号185およびアミノ酸配列番号186を示す。
【図94】図94は、核酸配列番号187およびアミノ酸配列番号188を示す。
【図95】図95は、核酸配列番号189およびアミノ酸配列番号190を示す。
【図96】図96は、核酸配列番号191およびアミノ酸配列番号192を示す。
【図97】図97は、核酸配列番号193およびアミノ酸配列番号194を示す。
【図98】図98は、核酸配列番号195およびアミノ酸配列番号196を示す。
【図99】図99は、核酸配列番号197およびアミノ酸配列番号198を示す。
【図100】図100は、核酸配列番号199およびアミノ酸配列番号200を示す。
【図101】図101は、核酸配列番号201およびアミノ酸配列番号202を示す。
【図102】図102は、核酸配列番号203およびアミノ酸配列番号204を示す。
【図103】図103は、核酸配列番号205およびアミノ酸配列番号206を示す。
【図104】図104は、核酸配列番号207およびアミノ酸配列番号208を示す。
【図105】図105は、核酸配列番号209およびアミノ酸配列番号210を示す。
【図106】図106は、核酸配列番号211およびアミノ酸配列番号212を示す。
【図107】図107は、核酸配列番号213およびアミノ酸配列番号214を示す。
【図108】図108は、核酸配列番号215およびアミノ酸配列番号216を示す。
【図109】図109は、核酸配列番号217およびアミノ酸配列番号218を示す。
【図110】図110は、核酸配列番号219およびアミノ酸配列番号220を示す。
【図111】図111は、核酸配列番号221およびアミノ酸配列番号222を示す。
【図112】図112は、核酸配列番号223およびアミノ酸配列番号224を示す。
【図113】図113は、核酸配列番号225およびアミノ酸配列番号226を示す。
【図114】図114は、核酸配列番号227およびアミノ酸配列番号228を示す。
【図115】図115は、核酸配列番号229およびアミノ酸配列番号230を示す。
【図116】図116は、核酸配列番号231およびアミノ酸配列番号232を示す。
【図117】図117は、核酸配列番号233およびアミノ酸配列番号234を示す。
【図118】図118は、核酸配列番号235およびアミノ酸配列番号236を示す。
【図119】図119は、核酸配列番号237およびアミノ酸配列番号238を示す。
【図120】図120は、核酸配列番号239およびアミノ酸配列番号240を示す。
【図121】図121は、核酸配列番号241およびアミノ酸配列番号242を示す。
【図122】図122は、核酸配列番号243およびアミノ酸配列番号244を示す。
【図123】図123は、核酸配列番号245およびアミノ酸配列番号246を示す。
【図124】図124は、核酸配列番号247およびアミノ酸配列番号248を示す。
【図125】図125は、核酸配列番号249およびアミノ酸配列番号250を示す。
【図126】図126は、核酸配列番号251およびアミノ酸配列番号252を示す。
【図127】図127は、核酸配列番号253およびアミノ酸配列番号254を示す。
【図128】図128は、核酸配列番号255およびアミノ酸配列番号256を示す。
【図129】図129は、核酸配列番号257およびアミノ酸配列番号258を示す。
【図130】図130は、核酸配列番号259およびアミノ酸配列番号260を示す。
【図131】図131は、核酸配列番号261およびアミノ酸配列番号262を示す。
【図132】図132は、核酸配列番号263およびアミノ酸配列番号264を示す。
【図133】図133は、核酸配列番号265およびアミノ酸配列番号266を示す。
【図134】図134は、核酸配列番号267およびアミノ酸配列番号268を示す。
【図135】図135は、核酸配列番号269およびアミノ酸配列番号270を示す。
【図136】図136は、核酸配列番号271およびアミノ酸配列番号272を示す。
【図137】図137は、核酸配列番号273およびアミノ酸配列番号274を示す。
【図138】図138は、核酸配列番号275およびアミノ酸配列番号276を示す。
【図139】図139は、核酸配列番号277およびアミノ酸配列番号278を示す。
【図140】図140は、核酸配列番号279およびアミノ酸配列番号280を示す。
【図141】図141は、核酸配列番号281およびアミノ酸配列番号282を示す。
【図142】図142は、核酸配列番号283およびアミノ酸配列番号284を示す。
【図143】図143は、核酸配列番号285およびアミノ酸配列番号286を示す。
【図144】図144は、核酸配列番号287およびアミノ酸配列番号288を示す。
【図145】図145は、核酸配列番号289およびアミノ酸配列番号290を示す。
【図146】図146は、核酸配列番号291およびアミノ酸配列番号292を示す。
【図147】図147は、核酸配列番号293およびアミノ酸配列番号294を示す。
【図148】図148は、核酸配列番号295およびアミノ酸配列番号296を示す。
【図149】図149は、核酸配列番号297およびアミノ酸配列番号298を示す。
【図151−1】図151は、配列グループの比較を示す。
【図151−2】図151は、配列グループの比較を示す。
【図151−3】図151は、配列グループの比較を示す。
【図151−4】図151は、配列グループの比較を示す。
【図151−5】図151は、配列グループの比較を示す。
【図151−6】図151は、配列グループの比較を示す。
【図151−7】図151は、配列グループの比較を示す。
【図151−8】図151は、配列グループの比較を示す。
【図151−9】図151は、配列グループの比較を示す。
【図151−10】図151は、配列グループの比較を示す。
【図151−11】図151は、配列グループの比較を示す。
【図152−1】図152は、全長クローンの並列を例示する。
【図152−2】図152は、全長クローンの並列を例示する。
【図152−3】図152は、全長クローンの並列を例示する。
【図152−4】図152は、全長クローンの並列を例示する。
【図152−5】図152は、全長クローンの並列を例示する。
【図153】図153は、PCRによるチトクロムp450 cDNAのクローニングに用いた方法を示す。
【実施例】
【0060】
(実施例1)
植物組織の発育およびエチレン処理
植物栽培
植物の種子を植木鉢に蒔き、そして温室で4週間栽培した。4週齢の苗を個々の植木鉢に移植し、そして温室で2ヶ月栽培した。栽培中、植物に1日2回、150ppm NPK肥料を含有する水を与えた。広がった緑の葉を植物から分離して、以下に記載するエチレン処理を行った。
【0061】
細胞株78379
ケンタッキー大学から譲渡されたバレー種タバコ系統であるタバコ系統78379を、植物材料の供給源として用いた。当該技術分野における標準のように、100の植物を栽培し、そして移植して、そして別個の番号(1〜100)でタグ付けした。推奨されるように、受精およびフィールド管理を行った。
【0062】
100の植物の4分の3が、ニコチンの20〜100%をノルニコチンに変換した。100の植物の4分の1が、ニコチンの5%未満をノルニコチンに変換した。87番の植物が最低の変換を有し(2%)、一方、21番の植物が100%の変換を有した。3%未満を変換する植物を非変換系統と分類した。87番の植物および21番の植物の自家受粉種子とともに交雑(21x87および87x21)種子を作成して、遺伝子相違および表現型相違を研究した。21番の自家受粉由来の植物は変換系統であり、そして87番の自家受粉由来の99%が非変換系統であった。87番由来の植物の残り1%は、低変換(5〜15%)を示した。相互交雑由来の植物は、すべて変換系統であった。
【0063】
細胞株4407
バレー種系統であるタバコ属系統4407を植物材料の供給源として用いた。均質でそして代表的な植物(100)を選択し、そしてタグ付けした。100の植物のうち97が非転換系統であり、そして3つが変換系統であった。56番の植物が最小量の変換を有し(1.2%)、そして58番の植物が最高レベルの変換を有した(96%)。これらの2つの植物を用いて、自家受粉種子および交雑種子を作成した。
【0064】
58番の自家受粉由来の植物は、変換系統対非変換系統比3:1に分かれた。58−33および58−25の植物が、それぞれ、ホモ接合体変換植物系統および非変換植物系統と同定された。次世代の子孫を解析することによって、58−33が安定変換系統であることが確認された。
【0065】
細胞株PBLB01
PBLB01は、ProfiGen, Inc.に開発されたバレー種系統であり、そ
してこれを植物材料の供給源として用いた。PBLB01の原種(foundation)種子から、変換系統植物を選択した。
【0066】
エチレン処理法
温室で2〜3ヶ月栽培した植物から緑の葉を分離し、そして0.3%のエチレン溶液(調製商標、エテフォン(Rhone−Poulenc))をスプレーした。スプレーした葉を各々、加湿器を備えた養生ラックに吊るし、そしてプラスチックで覆った。処理中、試料の葉に定期的にエチレン溶液をスプレーした。エチレン処理のおよそ24〜48時間後、RNA抽出のため、葉を収集した。代謝構成要素解析のため、別の下位試料を採取して、葉の代謝産物、および多様なアルカロイドなどのより具体的な目的の構成要素の濃度を測定した。
【0067】
例えば、以下のようにアルカロイド分析を行うことも可能である。試料(0.1g)を、0.5mlの2N NaOH、並びに内部標準としてのキノリン、およびメチルt−ブチルエーテルを含有する5mlの抽出溶液とともに、150rpmで振盪した。FID検出装置を備えたHP6890GC上で試料を分析した。検出装置および注入装置には、250℃の温度を用いた。1分あたり10℃、110〜185℃の温度勾配で、5%フェノールおよび95%メチルシリコンで架橋した融解石英からなるHPカラム(30m−0.32nm−1m)を用いた。キャリアーガスとしてヘリウムを用いて、100℃、流速1.7cm3/分、40:1の分割比、2・1注入体積で、カラムを操作した。
【0068】
(実施例2)
RNA単離
RNA抽出のため、2ヶ月齢の温室栽培植物の中央部の葉を、記載するようにエチレン処理した。0時間および24〜48時間の試料をRNA抽出に用いた。いくつかの場合、花頭除去10日後の植物から、老化過程にある葉の試料を採取した。これらの試料もまた、抽出に用いた。製造者のプロトコルにしたがって、Rneasy Plant Mini Kit(登録商標)(Qiagen, Inc.、カリフォルニア州バレンシア)を用い、総RNAを単離した。
【0069】
液体窒素下で、DEPC処理した乳鉢および乳棒を用いて、組織試料を細かい粉末にすりつぶした。すりつぶした組織およそ100ミリグラムを、滅菌1.5mlエッペンドルフ試験管に移した。すべての試料を収集し終わるまで、この試料試験管を液体窒素中に入れた。次いで、キットに提供される緩衝液RLT 450μl(メルカプトエタノールを添加したもの)を個々の各試験管に添加した。試料を激しくボルテックスし、そして56℃で3分間インキュベーションした。次いで、2ml収集試験管に入れたQIAshredderTMスピンカラムに溶解物を適用し、そして最大速度で2分間遠心分離した。フロースルーを収集し、そして透明な溶解物に0.5体積のエタノールを添加した。試料をよく混合し、そして2ml収集試験管に入れたRneasy(登録商標)ミニスピンカラムに移した。試料を10,000rpmで1分間遠心分離した。次に、700μlの緩衝液RW1をRneasy(登録商標)カラム上にピペッティングし、そして10,000rpmで1分間遠心分離した。緩衝液RPEを新しい収集試験管中のRneasy(登録商標)カラム上にピペッティングし、そして10,000rpmで1分間遠心分離した。緩衝液RPEを再びRneasy(登録商標)スピンカラム上に添加し、そして最大速度で2分間遠心分離して、膜を乾燥させた。エタノールのキャリー・オーバーをすべて取り除くため、別個の収集試験管中に膜を入れ、そして最大速度でさらに1分間遠心分離した。Rneasy(登録商標)カラムを新しい1.5ml収集試験管に移し、そして40μlのRnase不含水を、Rneasy(登録商標)膜上に直接ピペッティングした。この最終溶出液試験管を10,000rpmで1分間遠心分離した。変性ホルムアルデヒドゲルおよび分光光度計によって、総RNAの品質および量を解析した。
【0070】
製造者のプロトコルにしたがって、OligotexTMポリA+ RNA精製キット(Qiagen Inc.)を用いてポリ(A)RNAを単離した。最大体積250μl中、約200μgの総RNAを用いた。250μlの体積の緩衝液OBBおよび15μlのOligotexTM懸濁物を250μlの総RNAに添加した。ピペッティングによって内容物を完全に混合し、そして加熱ブロック上、70℃で3分間インキュベーションした。次いで、試料をおよそ20分間室温に置いた。最大速度で2分間遠心分離することによって、Oligotex:mRNA複合体をペレットにした。50μlを残してすべての上清を微量遠心管から取り除いた。OBB緩衝液によって試料をさらに処理した。ボルテックスによって、Oligotex:mRNAペレットを400μlの緩衝液OW2に再懸濁した。新しい試験管に入れた小さいスピンカラム上にこの混合物を移して、そして最大速度で1分間遠心分離した。スピンカラムを新しい試験管に移して、そしてさらに400μlの緩衝液OW2をカラムに添加した。次いで、試験管を最大速度で1分間遠心分離した。スピンカラムを最後の1.5ml微量遠心管に移した。60μlの熱い(70℃)緩衝液OEBで試料を溶出した。変性ホルムアルデヒドゲルおよび分光光度解析によって、ポリA産物を解析した。
【0071】
(実施例3)
逆転写−PCR
製造者のプロトコル(Invitrogen、カリフォルニア州カールスバッド)にしたがってSuperScript逆転写酵素を用いて、第一鎖cDNAを産生した。ポリA+濃縮RNA/オリゴdTプライマー混合物は、5μg未満の総RNA、1μlの10mM dNTP混合物、1μlのオリゴd(T)12−18(0.5μg/μl)、および10μlまでのDEPC処理水からなった。各試料を65℃で5分間インキュベーションし、次いで氷上に少なくとも1分間置いた。以下の構成要素の各々を順番に添加することによって、反応混合物を調製した:2μlの10xRT緩衝液、4μlの25mM MgCl2、2μlの0.1M DTT、および1μlのRNase OUT組換えRNase阻害剤。9μlの反応混合物を各RNA/プライマー混合物にピペッティングして添加し、そして穏やかに混合した。これを42℃で2分間インキュベーションし、そして1μlのSuperScript IITM RTを各試験管に添加した。試験管を42℃で50分間インキュベーションした。70℃で15分間処理して反応を終結させ、そして氷上で冷却した。遠心分離によって試料を収集し、そして1μlのRNase Hを各試験管に添加し、そして37℃で20分間インキュベーションした。200pmolの順方向プライマー(図75、配列番号149〜156に示すような縮重プライマー)および100pmolの逆方向プライマー(18ntオリゴd(T)の後に1つのランダム塩基が続く混合物)を用いて、第二のPCRを行った。
【0072】
反応条件は、94℃で2分間であり、そして次いで94℃1分間、45℃〜60℃2分間、72℃3分間の40サイクルのPCRを行い、72℃でさらに10分間伸長を行った。
【0073】
1%アガロースゲルを用いた電気泳動によって、10μlの増幅試料を解析した。アガロースゲルから正しいサイズの断片を精製した。
【0074】
(実施例4)
PCR断片集団の生成
製造者の指示にしたがって、実施例3由来のPCR断片をpGEM−T(登録商標)Easyベクター(Promega、ウィスコンシン州マディソン)に連結した。連結した産物をJM109コンピテント細胞に形質転換し、そしてブルー/ホワイトセレクションのため、LB培地プレート上に蒔いた。コロニーを選択し、そして1.2mlのLB培地
を含む96ウェルプレート中で37℃で一晩増殖させた。すべての選択されるコロニーに関して、凍結ストックを生成した。Wizard SV Miniprep(登録商標)キット(Promega)とともに、BeckmanのBiomeck 2000ミニプレップ・ロボットを用いて、プレートからプラスミドDNAを精製した。100μlの水でプラスミドDNAを溶出し、そして96ウェルプレート中に保存した。EcoR1でプラスミドを消化し、そして1%アガロースゲルを用いて解析して、DNA量および挿入物のサイズを確認した。CEQ 2000配列決定装置(Beckman、カリフォルニア州フラートン)を用いて、400〜600bpの挿入物を含有するプラスミドの配列を決定した。BLAST検索によって、GenBankデータベースと該配列を並列した。p450関連断片を同定し、そしてさらに解析した。あるいは、サブトラクション・ライブラリーからp450断片を単離した。これらの断片もまた、上述のように解析した。
【0075】
(実施例5)
cDNAライブラリーの構築
以下のように、エチレン処理した葉から、総RNAを調製することによって、cDNAライブラリーを構築した。まず、修飾した酸フェノールおよびクロロホルム抽出プロトコルを用いて、タバコ系統58−33のエチレン処理した葉から、総RNAを抽出した。1グラムの組織を用いて、これをすりつぶし、そして続いて5mlの抽出緩衝液(100mM Tris−HCl、pH8.5;200mM NaCl;10mM EDTA;0.5%SDS)中でボルテックスし、これに5mlのフェノール(pH5.5)および5mlのクロロホルムを添加するように、プロトコルを修飾した。抽出した試料を遠心分離し、そして上清を取り置いた。上清が透明に見えるようになるまで、この抽出工程をさらに2〜3回反復した。およそ5mlのクロロホルムを添加して、微量のフェノールを取り除いた。3倍体積のEtOHおよび1/10体積の3M NaOAc(pH5.2)を添加し、そして−20℃に1時間保存することによって、合わせた上清分画からRNAを沈殿させた。Corexガラス容器に移した後、RNA分画を、4℃、9,000RPMで45分間遠心分離した。ペレットを70%エタノールで洗浄し、そして4℃、9,000RPMで5分間回転させた。ペレットを乾燥させた後、ペレットにしたRNAを0.5ml
RNase不含水に溶解した。ペレットにしたRNAを0.5ml RNase不含水に溶解した。変性ホルムアルデヒドゲルおよび分光光度計によって、それぞれ、総RNAの品質および量を解析した。
【0076】
以下のプロトコルによって、オリゴ(dT)セルロース・プロトコル(Invitrogen)および微量遠心分離装置スピンカラム(Invitrogen)を用いて、ポリA+ RNAに関して、生じた総RNAを単離した。およそ20mgの総RNAを、2回の精製に供して、高品質のポリA+ RNAを得た。mRNAが高品質であることを確実にするため、変性ホルムアルデヒドゲル、そして続いて既知の全長遺伝子のRT−PCRを行うことによって、ポリA+ RNA産物を解析した。
【0077】
次に、ポリA+ RNAをテンプレートとして用いて、cDNA合成キット、ZAP−cDNA(登録商標)合成キット、およびZAP−cDNA(登録商標)Gigapack(登録商標)IIIゴールド・クローニングキット(Stratagene、カリフォルニア州ラホヤ)を使用して、cDNAライブラリーを産生した。方法は、明記するような以下の製造者のプロトコルを含んだ。およそ8μgのポリA+ RNAを用いて、cDNAライブラリーを構築した。一次ライブラリーの解析によって、約2.5x106〜1x107pfuであることが明らかになった。IPTGおよびX−galを用いた補完アッセイによって、ライブラリーの品質バックグラウンド試験を完了し、ここで組換えプラークは、バックグラウンド反応より100倍を超えて発現していた。
【0078】
ランダムPCRによる、ライブラリーのより定量的な解析によって、挿入cDNAの平
均サイズがおよそ1.2kbであることが示された。該方法は、以下のような2工程PCR法を用いた。第一の工程のため、p450断片から得た予備的配列情報に基づいて、逆プライマーを設計した。設計した逆方向プライマーおよびT3(順方向)プライマーを用いて、cDNAライブラリーから、対応する遺伝子を増幅した。PCR反応をアガロース電気泳動に供して、そして高分子量の対応するバンドを切り出し、精製し、クローニングし、そして配列決定した。第二の工程において、p450の5’UTRまたは開始コード領域から順方向プライマーとして設計した新規プライマーを、逆方向プライマー(p450の3’UTRから設計)とともに、続くPCRで用いて、全長p450クローンを得た。
【0079】
逆方向プライマーを除いて、実施例3に記載するように構築したcDNAライブラリーから、PCR増幅によって、p450断片を生成した。プラスミド上でcDNA挿入物(図75を参照されたい)の下流に位置するT7プライマーを逆方向プライマーとして用いた。実施例4に記載するように、PCR断片を単離し、クローニングし、そして配列決定した。
【0080】
構築したcDNAライブラリーから、PCR法によって、全長p450遺伝子を単離した。遺伝子特異的逆方向プライマー(p450断片の下流配列から設計)および順方向プライマー(ライブラリープラスミド上のT3)を用いて、全長遺伝子をクローニングした。PCR断片を単離し、クローニングし、そして配列決定した。必要であれば、第二の工程のPCRを適用した。第二の工程において、クローニングしたp450の5’UTRから設計した新規順方向プライマーとともに、p450クローンの3’UTRから設計した逆方向プライマーを、続くPCR反応で用いて、全長p450クローンを得た。続いてクローンの配列を決定した。
【0081】
(実施例6)
クローニングした断片の性質決定−逆サザンブロッティング解析
上記実施例で同定したp450クローンすべてに対して、非放射性大規模逆サザンブロッティングアッセイを行って、示差発現を検出した。異なるp450クラスターの間で発現レベルが非常に異なることが観察された。高発現のものに対して、さらなるリアルタイム検出を行った。
【0082】
非放射性サザンブロッティング法を以下のように行った。
1)実施例2に記載するように、Qiagen Rnaeasyキットを用いて、エチレン処理および非処理変換系統(58−33)および非変換系統(58−25)の葉から総RNAを抽出した。
【0083】
2)上記工程で生成したポリA+濃縮RNA由来の一本鎖cDNAをビオチン−テール標識することによって、プローブを産生した。ビオチン化オリゴdTをプライマーとして(Promega)用いることを除いて、実施例3に記載するように、変換系統および非変換系統総RNAのRT−PCR(Invitrogen)によって、この標識一本鎖cDNAを生成した。これらをプローブとして用いて、クローニングしたDNAとハイブリダイズさせた。
【0084】
3)プラスミドDNAを制限酵素EcoR1で消化して、そしてアガロースゲル上で泳動した。ゲルを乾燥させて、そして同時に2つのナイロン膜にトランスファーした(Biodyne B(登録商標))。一方の膜を変換系統プローブとハイブリダイズさせ、そして他方を非変換系統プローブとハイブリダイズさせた。ハイブリダイゼーション前に、膜をUV架橋した(自動架橋セッティング、254nm、Stratagene、Stratalinker)。
【0085】
あるいは、p−GEMプラスミドの両方のアームに位置する配列、T3およびSP6をプライマーとして用いて、各プラスミドから挿入物をPCR増幅した。96ウェルReady−to−runアガロースゲル上で泳動することによって、PCR産物を解析した。確認した挿入物を2つのナイロン膜上にドットした。一方の膜を変換系統プローブとハイブリダイズさせ、そして他方を非変換プローブとハイブリダイズさせた。
【0086】
4)洗浄ストリンジェンシーを修飾し、製造者の指示にしたがって、膜をハイブリダイズさせ、そして洗浄した(Enzo MaxSenceTMキット、Enzo Diagnostics, Inc、ニューヨーク州ファーミンデール)。膜を、ハイブリダイゼーション緩衝液(界面活性剤およびハイブリダイゼーション増進剤を含有する、2xSSC緩衝ホルムアミド)と42℃で30分間プレハイブリダイズさせ、そして10μl変性プローブと42℃で一晩ハイブリダイズさせた。次いで、膜を1xハイブリダイゼーション洗浄緩衝液で、室温で10分間1回洗浄し、そして68℃で15分間4回洗浄した。膜の検出準備が出来た。
【0087】
5)製造者の検出法に記載されるように(Enzo Diagnostics, Inc.)、アルカリホスファターゼ標識、その後、NBT/BCIP比色検出によって、洗浄した膜を検出した。1xブロッキング溶液を用いて、膜を室温で1時間ブロッキングし、1x検出試薬で10分間3回洗浄し、1x現像前反応緩衝液で5分間2回洗浄し、そして次いで、ドットが現れるまで、現像溶液中でブロットを30〜45分間現像した。すべての試薬は、製造者(Enzo Diagnostics, Inc)に提供された。さらに、製造者の指示にしたがって、KPLサザンハイブリダイゼーションおよび検出キットTM(KPL、メリーランド州ガイザーズバーグ)を用いてもまた、大規模逆サザンアッセイを行った。
【0088】
(実施例7)
クローンの性質決定−ノーザンブロット解析
サザンブロット解析の代わりに、ノーザンブロッティングアッセイの例に記載されるように、いくつかの膜をハイブリダイズさせ、そして検出した。以下のように、ノーザンハイブリダイゼーションを用いて、タバコ属において、示差発現されるmRNAを検出した。
【0089】
ランダムプライミング法を用いて、クローニングしたp450からプローブを調製した(MegaprimeTM DNA標識系、Amersham Biosciences)。
【0090】
以下の構成要素を混合した:25ng変性DNAテンプレート;各4μlの非標識dTTP、dGTPおよびdCTP;5μlの反応緩衝液;P32−標識dATPおよび2μlのクレノウI;並びに反応を50μlにするH2O。混合物を37℃で1〜4時間インキュベーションし、次いで2μlの0.5M EDTAで反応を停止した。使用前にプローブを95℃で5分間インキュベーションすることによって、変性させた。
【0091】
いくつかの対のタバコ系統の新鮮なエチレン処理葉および非処理葉から、RNA試料を調製した。いくつかの場合、ポリA+濃縮RNAを用いた。およそ15μgの総RNAまたは1.8μgのmRNA(実施例5に記載するようなRNAおよびmRNA抽出法)をDEPC H2O(5〜10μl)で等体積にした。同体積の装填緩衝液(1xMOPS;18.5%ホルムアルデヒド;50%ホルムアミド;4% Ficoll400;ブロモフェノールブルー)および0.5μl EtBr(0.5μg/μl)を添加した。続いて、電気泳動によってRNAを分離するため、調製物中の試料を変性させた。
【0092】
1xMOP緩衝液(0.4Mモルホリノプロパンスルホン酸;0.1M Na−酢酸−3xH2O;10mM EDTA;NaOHでpH7.2に調整)を用いて、ホルムアルデヒドゲル(1%アガロース、1xMOPS、0.6Mホルムアルデヒド)上で、試料を電気泳動に供した。10xSSC緩衝液(1.5M NaCl;0.15Mクエン酸ナトリウム)中で24時間キャピラリー法を行うことによって、RNAをHybond−N+膜(ナイロン、Amersham Pharmacia Biotech)にトランスファーした。RNA試料を含む膜を、ハイブリダイゼーション前にUV架橋した(自動架橋セッティング、254nm、Stratagene、Stratalinker)。
【0093】
膜を、5〜10mlのプレハイブリダイゼーション緩衝液(5xSSC;50%ホルムアミド;5xデンハルト溶液;1% SDS;100μg/ml熱変性剪断非相同DNA)と42℃で1〜4時間プレハイブリダイズさせた。古いプレハイブリダイゼーション緩衝液を廃棄し、そして新しいプレハイブリダイゼーション緩衝液およびプローブを添加した。ハイブリダイゼーションを42℃で一晩行った。2xSSCで膜を室温で15分間洗浄し、次いで2xSSCで洗浄した。
【0094】
本発明の主要な焦点は、エチレン処理の結果誘導可能であるか、またはタバコ葉の品質および構成要素に重要な役割を果たすことも可能な、新規遺伝子の発見であった。以下の表に例示するように、ノーザンブロットおよび逆サザンブロットは、非誘導植物に比較して、どの遺伝子がエチレン処理によって誘導されたのかを決定するのに有用であった。興味深いことに、変換系統および非変換系統で、すべての断片が同様に影響を受けるのではなかった。注目されるチトクロムp450断片を部分的に配列決定して、構造的関連性を決定した。この情報を用いて、注目される全長遺伝子クローンを続いて単離し、そして性質決定した。
【0095】
【表1】

【0096】
エチレン処理で誘導した、変換および非変換バレー種系統から得たタバコ組織に対して、全長クローンを用いて、ノーザン解析を行った。目的は、エチレン誘導非変換バレー種系統に比較して、エチレン誘導変換系統に比較して、エチレン誘導変換系統で発現上昇を示す全長クローンを同定することであった。そうすることによって、変換系統および非変換系統間の葉構成要素の生化学的相違を比較することにより、全長クローンの機能的関係を決定することも可能である。以下の表に示すように、+で示す非変換系統エチレン処理
組織のものより、変換系統エチレン処理組織において、++および+++で示すように、6つのクローンが有意により高い発現を示した。これらのクローンはすべて、エチレン処理しない変換系統および非変換系統で、ほとんどまたはまったく発現を示さなかった。
【0097】
【表2】
【0098】
(実施例8)
クローニングした遺伝子にコードされるp450の免疫検出
3つのp450クローンから、1)他のクローンに対してより低い相同性を有するかまたはまったく相同性を持たず、そして2)優れた親水性および抗原性を有する、長さ20〜22アミノ酸に対応するペプチド領域を選択した。それぞれのp450クローンから選択したペプチド領域のアミノ酸配列を以下に列挙する。合成したペプチドをKHLとコンジュゲート化し、そして次いでウサギに注射した。第4回の注射の2週間後および4週間後に抗血清を収集した(Alpha Diagnostic Intl. Inc.、テキサス州サンアントニオ)。
【0099】
D234−AD1 DIDGSKSKLVKAHRKIDEILG
D90a−BB3 RDAFREKETFDENDVEELNY
D89−AB1 FKNNGDEDRHFSQKLGDLADKY
ウェスタンブロット解析によって、タバコ植物組織由来のタンパク質を標的とする交差反応性に関して、抗血清を調べた。エチレン処理した(0〜40時間)、変換系統および非変換系統の中央部の葉から、未精製タンパク質抽出物を得た。製造者のプロトコルにしたがって、RC DCタンパク質アッセイキット(BIO−RAD)を用いて、抽出物のタンパク質濃度を測定した。
【0100】
各レーンに2マイクログラムのタンパク質を装填し、そしてLaemmli SDS−PAGE系を用いて、10%〜20%勾配ゲル上でタンパク質を分離した。Trans−Blot(登録商標)セミドライセル(BIO−RAD)を用いて、PROTRAN(登録商標)ニトロセルロース・トランスファー膜(Schleicher & Schuell)にゲルからタンパク質をトランスファーした。ECL AdvanceTMウェスタンブロッティング検出キット(Amersham Biosciences)を用いて、標的p450タンパク質を検出し、そして視覚化した。ウサギにおいて、合成KLHコンジュゲートに対する一次抗体を作成した。ペルオキシダーゼとカップリングした、ウサギIgGに対する二次抗体をSigmaから購入した。一次抗体および二次抗体のどちらも、1:1000希釈で用いた。抗体は、ウェスタンブロット上で、単一のバンドに強い反応性を示し、抗血清が、目的の標的ペプチドに単一特異的であることを示した。抗血清はまた、KLHにコンジュゲート化した合成ペプチドとも交差反応性であった。
【0101】
(実施例9)
単離核酸断片の核酸同一性および構造関連性
ノーザンブロット解析と組み合わせて、クローニングした100を超えるp450断片を配列決定して、構造的関連性を決定した。用いたアプローチは、p450遺伝子のカルボキシル末端近くに位置する2つの一般的なp450モチーフのいずれかに基づく順方向プライマーを利用した。順方向プライマーは、図1に示すような、チトクロムp450モチーフFXPERFまたはGRRXCP(A/G)に相当した。逆方向プライマーは、プラスミド、pGEMTMプラスミドの両方のアーム上に位置するSP6またはT7、あるいはポリAテールのいずれか由来の、標準的プライマーを用いた。用いたプロトコルを以下に記載する。
【0102】
分光光度測定を用い、製造者のプロトコル(Beckman Coulter)にしたがって、出発二本鎖DNAの濃度を概算した。テンプレートを水で適切な濃度に希釈し、95℃で2分間加熱し、そして続いて氷上に置くことによって変性させた。0.5〜10μlの変性DNAテンプレート、2μlの1.6pmol順方向プライマー、8μlのDTCSクイックスタートマスターミックス、および総体積を20μlにする水を用いて、配列決定反応物を氷上で調製した。熱周期プログラムは、30サイクルの以下の周期からなった:96℃20秒間、50℃20秒間、および60℃4分間、その後、4℃で維持。
【0103】
5μlの反応停止緩衝液(等体積の3M NaOAcおよび100mM EDTA、並びに1μlの20mg/mlグリコーゲン)を添加することによって、配列決定反応を停止した。60μlの冷95%エタノールで試料を沈殿させ、そして6000gで6分間遠心分離した。エタノールを廃棄した。ペレットを200μlの冷70%エタノールで2回洗浄した。ペレットを乾燥させた後、40μlのSLS溶液を添加し、そしてペレットを再懸濁した。ミネラルオイルの層を重層した。次いで、さらに解析するため、試料をCEQ 8000自動化配列決定装置に入れた。
【0104】
核酸配列を検証するため、p450遺伝子のFXPERFまたはGRRXCP(A/G)領域に対する順方向プライマー、あるいはプラスミドまたはポリAテールいずれかに対する逆方向プライマーを用いて、核酸配列を、両方向に再配列決定した。すべての配列決定を、両方向に、少なくとも2回行った。
【0105】
チトクロムp450断片の核酸配列を、GRRXCP(A/G)モチーフをコードする領域の後の最初の核酸から停止コドンまでに対応するコード領域で、互いに比較した。この領域を、p450タンパク質間の遺伝子多様性の指標として選択した。他の植物種と同様、多数の遺伝的に別個のp450遺伝子が70遺伝子を超えて観察された。核酸配列の比較に際して、配列同一性に基づいて、遺伝子を別個の配列グループに入れることが可能であることが見出された。p450メンバーの最適なユニークなグループ分けが、75%以上の核酸同一性を持つ配列であると決定されることが見出された(表Iに示す)。同一性パーセントを減少させると、有意により大きいグループとなった。81%以上の核酸同一性を持つ配列に関して、好ましいグループ分けが観察され、より好ましいグループ分けは91%以上の核酸同一性のものであり、そして最も好ましいグループ分けは99%以上の核酸同一性の配列であった。グループの大部分は少なくとも2つのメンバーを含有し、そしてしばしば3以上のメンバーを含有した。他のものは反復して発見されず、ここで取ったアプローチが、用いた組織で低発現されるmRNAおよび高発現されるmRNAの両方を単離可能であったことが示唆される。
【0106】
75%以上の核酸同一性に基づいて、2つのチトクロムp450グループが、グループ内にあるものとは遺伝的に異なる、先のタバコ・チトクロム遺伝子に核酸配列同一性を含有することが見出された。グループ23は、表Iに用いたパラメーター内で、それぞれCzernicらおよびRalstonらによる、GI:1171579(CAA64635)およびGI:14423327(またはAAK62346)の先のGenBank配列に、核酸同一性を示した。GI:1171579は、グループ23のメンバーに、96.9%〜99.5%の範囲の核酸同一性を示し、一方、GI:14423327は、このグループに、95.4%〜96.9%の範囲の同一性を有した。グループ31のメンバーは、RalstonらによるGI:14423319(AAK62342)のGenBankに報告された配列に、76.7%〜97.8%の範囲の核酸同一性を示した。表1の他のp450同一性グループはいずれも、Ralstonら、Czernicら、Wangら、またはLaRosaおよびSmigockiに報告されるタバコ属p450遺伝子に、表1で用いたようなパラメーター同一性を含有しなかった。
【0107】
図76に示すように、グループに関して、適切な核酸縮重プローブとともにコンセンサス配列を得て、タバコ属植物からの各グループのさらなるメンバーを優先的に同定し、そして単離することも可能である。
【0108】
【表3−1】
【0109】
【表3−2】
【0110】
(実施例10)
単離核酸断片の関連アミノ酸配列同一性
実施例8由来のチトクロムp450断片に関して得た核酸配列のアミノ酸配列を推定した。推定した領域は、GXRXCP(A/G)配列モチーフの直後のアミノ酸からカルボキシル末端の終わり、または停止コドンまでに相当した。断片の配列同一性の比較に際して、70%以上のアミノ酸同一性を持つ配列に関して、ユニークなグループ分けが観察された。80%以上のアミノ酸同一性を持つ配列に関して、好ましいグループ分けが観察され、より好ましいグループ分けは90%以上のアミノ酸同一性のものであり、そして最も好ましいグループ分けは99%以上のアミノ酸同一性の配列であった。グループおよびグループメンバーの対応するアミノ酸配列を図2に示す。ユニークな核酸配列のいくつかは、他の断片と完全なアミノ酸同一性を有することが見出され、そしてしたがって、同一アミノ酸を持つ1つのメンバーのみが報告された。
【0111】
表IIのグループ19のアミノ酸同一性は、核酸配列に基づいて、3つの別個のグループに対応した。各グループメンバーのアミノ酸配列およびその同一性を図77に示す。アミノ酸相違に適切に印を付ける。
【0112】
遺伝子クローニングおよび植物を用いた機能研究のため、各アミノ酸同一性グループの少なくとも1つのメンバーを選択した。さらに、ノーザンおよびサザン解析によって評価されるように、エチレン処理または他の生物学的相違によって異なって影響を受けるグループメンバーを、遺伝子クローニングおよび機能研究のため、選択した。遺伝子クローニング、発現研究および全植物評価を補助するため、配列同一性および示差配列に関して、ペプチド特異的抗体が調製されるであろう。
【0113】
【表4−1】
【0114】
【表4−2】
【0115】
(実施例11)
全長クローンの関連アミノ酸配列同一性
実施例5でクローニングした全長タバコ属遺伝子の核酸配列を、全アミノ酸配列に関して推定した。チトクロムp450遺伝子は、3つの保存されるp450ドメインモチーフの存在によって同定され、該モチーフは、カルボキシル末端のUXXRXXZ、PXRFXFまたはGXRXC、ここでUはEまたはKであり、Xはアミノ酸いずれかであり、そしてZはP、T、SまたはMである、に対応した。クローンのうち2つ、D130−AA1およびD101−BA2は、ほぼ完全であるようだったが、適切な停止コドンを欠き、しかしどちらも3つのp450チトクロムドメインすべてを含有した。BLASTプログラムを用いて、全長配列を互いに、そして既知のタバコ遺伝子に比較して、p450遺伝子すべてをアミノ酸同一性に関して性質決定した。該プログラムは、NCBI特別BLASTツール(2配列並列(b12seq)、http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html)を用いた。核酸配列に関してはフィルターをかけずにBLASTNで、そしてアミノ酸配列に関してはBLASTPで、2つの配列を並列させた。アミノ酸同一性パーセントに基づいて、各配列を同一性グループにグループ分けし、グループ分けは、別のメンバーと少なくとも85%の同一性を共有するメンバーを含有した。90%以上のアミノ酸同一性を持つ配列に関して、好ましいグループ分けが観察され、より好ましいグループ分けは95%以上のアミノ酸同一性のものであり、そして最も好ましいグループ分けは99%以上のアミノ酸同一性の配列を有した。これらの基準を用いて、25のユニークなグループが同定され、そしてこれらを表IIIに示す。
【0116】
アミノ酸同一性に関する表IIIに用いたパラメーター内で、3つのグループが、既知のタバコ遺伝子に対して85%以上の同一性を含有することが見出された。グループ5のメンバーは、RalstonらによるGI:14423327(またはAAK62346)の先のGenBank配列に対して、全長配列に関して最大96%のアミノ酸同一性を有した。グループ23は、RalstonらによるGI:14423328(またはAAK62347)に対して、最大93%のアミノ酸同一性を有し、そしてグループ24は、RalstonらによるGI:14423318(またはAAK62343)に対して、92%の同一性を有した。
【0117】
【表5−1】
【0118】
【表5−2】
【0119】
カルボキシル末端近くのUXXRXXZ p450ドメインおよびGXRXC p450ドメインの間の非常に保存されるアミノ酸相同性に基づいて、全長遺伝子をさらにグループ分けした。図3に示すように、互いに比較して、保存されるドメイン間の配列相同性に関して、個々のクローンを並列させ、そして別個の同一性グループに入れた。いくつかの場合で、クローンの核酸配列はユニークであったが、該領域のアミノ酸配列は同一であった。90%以上のアミノ酸同一性を持つ配列に関して、好ましいグループ分けが観察され、より好ましいグループは95%以上のアミノ酸同一性を有し、そして最も好ましいグループ分けは99%以上のアミノ酸同一性の配列を有した。最後のグループ分けは、クローンの全アミノ酸配列に関する同一性パーセントに基づくものと同様であったが、(表IIIの)グループ17だけは例外であり、このグループは2つの別個のグループに分けられた。
【0120】
表IVのアミノ酸同一性に関して用いたパラメーター内で、3つのグループが、既知のタバコ属遺伝子に対して90%以上の同一性を含有することが見出された。グループ5のメンバーは、RalstonらによるGI:14423326(AAK62346)の先のGenBank配列に対して、全長配列に関して最大93.4%のアミノ酸同一性を有した。グループ23は、RalstonらによるGI:14423328(またはAAK62347)に対して、最大91.8%のアミノ酸同一性を有し、そしてグループ24は、RalstonらによるGI:14423318(またはAAK62342)に対して、98.8%の同一性を有した。
【0121】
【表6−1】
【0122】
【表6−2】
【0123】
(実施例12)
1以上のタバコ・チトクロムp450特異的ドメインを欠く、タバコ属チトクロムp450クローン
4つのクローンは、表IIIに報告する他のタバコ・チトクロム遺伝子に、90%〜99%の核酸相同性の範囲の、高い核酸相同性を有した。4つのクローンには、D136−AD5、D138−AD12、D243−AB3およびD250−AC11が含まれた。しかし、ヌクレオチド・フレームシフトのため、これらの遺伝子は、3つのC末端チトクロムp450ドメインの1以上を含有せず、そして表IIIまたは表IVに示す同一性グループからは除外された。
【0124】
1つのクローン、D95−AG1のアミノ酸同一性は、表IIIまたは表IVでp450タバコ遺伝子をグループ分けするのに用いた、第三のドメイン、GXRXCを含有しなかった。このクローンの核酸相同性は、他のタバコ・チトクロム遺伝子に低い相同性を有した。このクローンは、タバコ属のチトクロムp450遺伝子の新規のそして異なるグループに相当する。
【0125】
(実施例13)
タバコ特性の制御改変におけるタバコ属チトクロムp450断片およびクローンの使用
タバコp450核酸断片または全遺伝子の使用は、改変されたタバコ表現型またはタバコ構成要素、そしてより重要なことに、改変された代謝産物を有する植物を同定し、そして選択する際に有用である。下方制御のため、例えばアンチセンス方向で、または過剰発現のため、例えばセンス方向で、本明細書に報告するものから選択される核酸断片または全長遺伝子を取り込む、多様な形質転換系によって、トランスジェニック・タバコ植物を生成する。全長遺伝子を過剰発現するため、特定の酵素の発現を増加させるのに有効であり、そしてしたがってタバコ属内で表現型効果を生じる、本発明に記載する全長遺伝子のすべてまたは機能する部分またはアミノ酸配列をコードする核酸配列いずれかが望ましい。一連の戻し交雑を通じて、ホモ接合体系統であるタバコ属系統を得て、そして一般の当業者に一般的に利用可能な技術を用いて、限定されるわけではないが、内因性p450 RNA、転写物、p450発現ペプチドおよび植物代謝産物の濃度の解析を含む、表現型変化に関して評価する。タバコ植物で示される変化は、目的の選択される遺伝子の機能的役割に関する情報を提供するか、または好ましいタバコ属植物種として有用性を有する。
【0126】
(実施例14)
エチレン処理した変換系統で誘導される遺伝子の同定
遺伝子発現の定量的で高度に並行の測定のために、高密度オリゴヌクレオチドアレイ技
術であるAffymetrix GeneChip(登録商標)(Affymetrix
Inc.、カリフォルニア州サンタクララ)アレイを用いた。この技術を用いる際、固体表面上でオリゴヌクレオチドを直接合成することによって、核酸アレイを組み立てた。この固相化学反応は、GeneChip(登録商標)と称されるチップ上に、非常に高密度に充填された数十万のオリゴヌクレオチドプローブを含有するアレイを産生可能である。単一のハイブリダイゼーションから、数千の遺伝子を同時にスクリーニングすることも可能である。各遺伝子は、典型的には、サイズに応じて、11〜25対のプローブの組によって表される。感度、特異性、および再現性を最大にするようにプローブを設計して、特異的シグナルおよびバックグラウンドシグナル間、並びに緊密に関連する標的配列間の一貫した区別を可能にする。
【0127】
Affymetrix GeneChipハイブリダイゼーション実験は、以下の工程を伴う:アレイの設計および産生、生物学的標本から単離したRNAからの蛍光標識標的の調製、GeneChipへの標識した標的のハイブリダイゼーション、アレイのスクリーニング、並びにスキャンした画像の解析および遺伝子発現プロフィールの生成。
【0128】
A. Affymetrix GeneChipの設計およびオーダーメード(custom making)
GeneChip CustomExpress Advantageアレイは、Affymetrix Inc.(カリフォルニア州サンタクララ)によってオーダーメードされた。チップサイズは18ミクロンであり、そしてアレイ形式は100〜2187であって、528のプローブセット(11,628プローブ)に適応可能である。GenBank由来核酸配列を除いて、我々が先に同定したタバコクローンからすべての配列を選択し、そしてプローブはすべてあつらえて設計した。総数400のタバコ遺伝子または断片がGeneChip上に含まれるように選択した。選択したオリゴヌクレオチドの配列は、遺伝子の3’端のユニークな領域に基づいた。選択した核酸配列は、(特許出願)に記載される、タバコからクローニングされた、56の全長p450遺伝子および71のp450断片からなった。他のタバコ配列には、Clontech SSHキット(BD Biosciences、カリフォルニア州パロアルト)を用いた抑制サブトラクション・ライブラリーから生成した、270のタバコESTが含まれた。これらの遺伝子の中で、GenBankに列挙されるチトクロムP450遺伝子から、いくつかのオリゴヌクレオチド配列を選択した。各全長遺伝子に関して、最大25のプローブを用い、そして各断片に関して11のプローブを用いた。いくつかのクローンに関しては、ユニークな高品質のプローブがないため、減少した数のプローブを用いた。適切な対照配列もまた、GeneChip(登録商標)上に含んだ。
【0129】
プローブアレイは、25量体オリゴヌクレオチドであり、半導体に基づくフォトリソグラフィーおよび固相化学合成技術の組み合わせによって、ガラス・ウェハー上に直接合成された。各アレイは、最大100,000の異なるオリゴヌクレオチドプローブを含有した。オリゴヌクレオチドプローブは、アレイ上の既知の位置で合成されるため、Affymetrix Microarray Suite(登録商標)ソフトウェアによって、ハイブリダイゼーションパターンおよびシグナル強度を、遺伝子同一性および相対発現レベルに関して解釈することも可能である。各プローブ対は、完全マッチオリゴヌクレオチドおよびミスマッチオリゴヌクレオチドからなる。完全マッチプローブは、特定の遺伝子に正確に相補的な配列を有し、そしてしたがって遺伝子の発現を測定する。ミスマッチプローブは、中央の塩基位での単一塩基置換によって、完全マッチプローブとは異なり、この塩基置換が標的遺伝子転写物の結合を妨げる。ミスマッチは、非特異的ハイブリダイゼーションシグナルまたはバックグラウンドシグナルを生じ、これを、完全マッチオリゴヌクレオチドに関して測定されるシグナルに比較した。
【0130】
B.試料調製
Genome Explorations, Inc.(テネシー州メンフィス)によって、ハイブリダイゼーション実験を行った。ハイブリダイゼーションで用いるRNA試料は、エチレン処理によって誘導される、6対の非変換/変換同系系統からなった。試料には、1対の4407−25/4407−33非処理バレー種タバコ試料、3対のエチレン処理4407−25/4407−33試料、1対のエチレン処理した黒(dark)タバコNL Madole/181および1対のエチレン処理バレー品種PBLB01/178が含まれた。エチレン処理は、実施例1に記載するとおりであった。
【0131】
修飾した酸フェノールおよびクロロホルム抽出プロトコルを用いて、上述のエチレン処理葉および非処理葉から、総RNAを抽出した。1グラムの組織を用いて、これをすりつぶし、そして続いて5mlの抽出緩衝液(100mM Tris−HCl、pH8.5;200mM NaCl;10mM EDTA;0.5%SDS)中でボルテックスし、これに5mlのフェノール(pH5.5)および5mlのクロロホルムを添加するように、プロトコルを修飾した。抽出した試料を遠心分離し、そして上清を取り置いた。上清が透明に見えるようになるまで、この抽出工程をさらに2〜3回反復した。およそ5mlのクロロホルムを添加して、微量のフェノールを取り除いた。3倍体積のEtOHおよび1/10体積の3M NaOAc(pH5.2)を添加し、そして−20℃に1時間保存することによって、合わせた上清分画からRNAを沈殿させた。Corexガラス容器に移した後、RNA分画を、4℃、9,000RPMで45分間遠心分離した。ペレットを70%エタノールで洗浄し、そして4℃、9,000RPMで5分間回転させた。ペレットを乾燥させた後、ペレットにしたRNAを0.5ml RNase不含水に溶解した。ペレットにしたRNAを0.5ml RNase不含水に溶解した。変性ホルムアルデヒドゲルおよび分光光度計によって、それぞれ、総RNAの品質および量を解析した。3〜5μg/μlの総RNA試料をGenome Explorations, Inc.に送ってハイブリダイゼーションを行った。
【0132】
C.ハイブリダイゼーション、検出およびデータ・アウトプット
標識cRNA物質の調製を以下のように行った。製造者の指示にしたがって、SuperScript二本鎖cDNA合成キット(Gibco Life Technologies)およびオリゴ−dT24−T7(5’− GGC CAG TGA ATT GTA ATA CGA CTC ACT ATA GGG AGG CGG−3’)プライマーを用いて、5〜15μgの総RNAから、第一鎖および第二鎖cDNAを合成した。
【0133】
T7プロモーターがカップリングした二本鎖cDNAをテンプレートとして、そしてT7 RNA転写物標識キット(ENZO Diagnostics Inc.)を用いた、in vitro転写によって、cRNAを合成し、そして同時にビオチン化UTPおよびCTPで標識化した。簡潔には、先の工程から合成した二本鎖cDNAを70%エタノールで2回洗浄し、そして22μlのRnase不含H2Oに再懸濁した。cDNAを4μlの10x各反応緩衝液、ビオチン標識リボヌクレオチド、DTT、Rnase阻害剤混合物、および2μlの20xT7 RNAポリメラーゼと37℃で5時間インキュベーションした。CHROMA SPIN−100カラム(Clontech)を通過させ、そして−20℃で1時間〜一晩沈殿させることによって、取り込まれていないリボヌクレオチドから、標識cRNAを分離した。
【0134】
オリゴヌクレオチドアレイ・ハイブリダイゼーションおよび解析を以下のように行った。cRNAペレットを10μlのRnase不含H2Oに再懸濁し、そして200mM Tris−酢酸、pH8.1、500mM KOAc、150mM MgOAc中で95℃で35分間、熱およびイオンが仲介する加水分解によって、10.0μgを断片化した
。断片化したcRNAを、〜12,500のアノテートした全長遺伝子とともに、EST配列に相当するように設計されたさらなるプローブセットを含有する、HG U95Av2オリゴヌクレオチドアレイ(Affymetrix)に45℃で16時間ハイブリダイズさせた。6xSSPE(0.9M NaCl、60mM NaH2PO4、6mM EDTA+0.01% Tween20)を用いてアレイを25℃で洗浄し、次いで100mM MES、0.1M[Na+]、0.01% Tween20を用いて、50℃でストリンジェントな洗浄を行った。アレイをフィコエリトリン・コンジュゲート化ストレプトアビジン(Molecular Probes)で染色し、そしてレーザー共焦点スキャナー(Hewlett−Packard)を用いて、蛍光強度を測定した。Microarrayソフトウェア(Affymetrix)を用いて、スキャンした画像を解析した。用いたすべてのアレイに関して、アレイ上のすべての遺伝子の蛍光強度の平均を、一定の標的強度(250)にスケーリングすることによって、試料装填および染色における変動を標準化した。ユーザー指針にしたがって、Microarray Suite 5.0(Affymetrix)を用いて、データ解析を行った。[Σ(PM−MM)/(プローブ対の数)]、式中、PMおよびMMは完全マッチおよび不完全マッチを示す、に表される、平均強度相違として各遺伝子のシグナル強度を計算した。
【0135】
D.データ解析および結果
Genome Explorationsの検出装置を用いて生成した発現レポートに立証されるように、12組のハイブリダイゼーションに成功した。レポートの主なパラメーターには、ノイズ、スケール係数、バックグラウンド、総プローブセット、存在および非存在プローブセットの数およびパーセンテージ、ハウスキーピング対照のシグナル強度が含まれた。続いて、他のMicrosoftソフトウェアと組み合わせてソフトフェアGCOSを用いて、データを解析し、そして提示した。処理対間のシグナル比較を解析した。複製を含む、異なる処理各々の遺伝子および断片に対応するそれぞれのプローブすべてに関する全体のデータをコンパイルし、そしてコンパイルした発現データ、例えば変化のコールおよびシグナルlog2比の変化を解析した。
【0136】
GeneChip技術の典型的な適用は、異なる組織で差次的に発現される遺伝子を発見することである。本出願において、4407−25/4407−33バレー品種、PBLB01/178バレー品種、およびNL Madole/181黒タバコ品種を含む、変換および非変換タバコ系統対に関して、エチレン処理によって引き起こされる遺伝子発現変動を決定した。これらの解析によって、生物学的変動のため、発現が有意に改変されている遺伝子のみが検出された。これらの解析は、誘導された遺伝子を同定する主な基準として、倍変化(シグナル比)を使用した。他のパラメーター、例えばシグナル強度、存在/非存在コールもまた、考慮した。
【0137】
およそ400遺伝子に関して、変換および非変換対の試料の発現相違に関して、データを解析した後、シグナル強度に基づく結果によって、2つの遺伝子、D121−AA8、およびD120−AH4、並びに1つの断片、D121−AA8の部分的断片であるD35−BG11のみが、エチレン処理変換系統対非変換系統において、再現可能な誘導を有した。これらの遺伝子の示差発現を例示すると、データは以下のように示された。表Vに示すように、変換系統、例えばバレー種タバコ品種、4407−33の遺伝子のシグナルを、関連する非変換同系系統、4407−25のシグナルに対する比として決定した。エチレン処理なしでは、すべての遺伝子の変換系統対非変換系統のシグナルの比は、1.00に近づいた。エチレン処理すると、同系バレー系統を用いた3回の独立の解析によって決定されるように、非変換系統に比較して、変換系統において、2つの遺伝子、D121−AA8およびD120−AH4が誘導された。これらの遺伝子は、互いに非常に高い相同性を有し、およそ99.8%以上の核酸配列相同性を有した。表Vに示すように、変換品種における相対的ハイブリダイゼーションシグナルは、変換系統において、非変換対応
物におけるシグナルのおよそ2〜12倍高い範囲であった。これと比較して、内部対照である2つのアクチン様対照クローンは、標準化した比に基づくと、変換系統で誘導されていないことが見出された。さらに、コード領域中の配列が完全にD121−AA8およびD120−AH4遺伝子両方に含有される断片(D35−BG11)は、対の同系変換系統および非変換系統の同じ試料で、高度に誘導された。バレー種タバコ品種、PBLB01および178の別の同系対が、エチレン誘導下、変換系統試料で誘導される、同じ遺伝子、D121−AA8およびD120−AH4を有することが示された。さらに、D121−AA8およびD120−AH4遺伝子は、同系黒タバコ対、NL Madoleおよび181の変換系統で優先的に誘導され、変換系統におけるこれらの遺伝子のエチレン誘導が、バレー種タバコ品種に限定されないことが立証された。すべての場合で、D35−BG11断片が、非変換対系統に比較して、変換系統で、最も高く誘導された。
【0138】
【表7】
【0139】
(実施例15)
タバコ変換系統におけるミクロソーム・ニコチン脱メチル化酵素のエチレン誘導
変換および非変換タバコ系統のエチレン処理および非処理対のミクロソーム濃縮分画において、脱メチル化酵素活性の生化学的解析を、以下のように行った。
【0140】
A.ミクロソームの調製
ミクロソームを4℃で単離した。50mM N−(2−ヒドロオキシエチル)ピペラジン−N’−(2−エタンスルホン酸)(HEPES)、pH7.5、3mM DL−ジチオスレイトール(DTT)およびプロテアーゼ阻害剤カクテル(Roche)を1錠剤/50mlからなる緩衝液中で、タバコ葉を抽出した。四層のチーズクロスを通じて未精製抽出物をろ過して、破壊されていない組織を取り除き、そしてろ液を20,000xgで20分間遠心分離して、細胞破片を取り除いた。上清を100,000xgで60分間、超遠心に供して、そして生じたペレットはミクロソーム分画を含有した。抽出緩衝液にミクロソーム分画を懸濁し、そして超遠心工程に適用し、ここで抽出緩衝液中の0.5Mスクロースの不連続スクロース勾配を用いた。凍結保護剤として10%(w/v)グリセロールを補った抽出緩衝液に、精製ミクロソームを再懸濁した。ミクロソーム調製物を使用
するまで液体窒素フリーザー中に保存した。
【0141】
B.タンパク質濃度測定
アセトン中の10%トリクロロ酢酸(TCA)(w/v)を用いて、ミクロソームタンパク質を沈殿させ、そして製造者のプロトコルにしたがってRC DCタンパク質アッセイキット(BIO−RAD)を用いて、ミクロソームのタンパク質濃度を測定した。
【0142】
3)ニコチン脱メチル化酵素活性アッセイ
Moravek BiochemicalsからDL−ニコチン(ピロリジン−2−14C)を得て、そしてこれは54mCi/mmolの比活性を有した。どちらもp450阻害剤であるクロルプロマジン(CPZ)および酸化チトクロムc(cyt.C)を、Sigmaから購入した。ニコチンアミドアデニンジヌクレオチドホスフェートの還元型(NADPH)は、NADPH:チトクロムP450還元酵素を介したチトクロムP450の典型的な電子供与体である。対照インキュベーションに関してはNADPHを省いた。日常的な酵素アッセイは、ミクロソームタンパク質(およそ2mg/ml)、6mM NADPH、55μM 14C標識ニコチンからなった。CPZおよびCyt.Cの濃度は、これらを用いる場合には、それぞれ1mMおよび100μMであった。25℃で1時間反応を行い、そして各25μl反応混合物に300μlメタノールを添加して反応を停止した。回転後、メタノール抽出物の20μlをVarianのInertsil ODS−3 3μ(150x4.6mm)カラムを用いて、逆相高性能液体クロマトグラフィー(HPLC)系(Agilent)で分離した。アイソクラチック可動相は、60:40(v/v)の比のメタノールおよび50mMリン酸カリウム緩衝液、pH6.25の混合物であり、そして流速は1ml/分であった。真性の非標識ノルニコチンとの比較によって決定されるようなノルニコチンピークを収集し、そして定量化のため、2900 tri−carb液体シンチレーションカウンター(LSC)(Perkin Elmer)に供した。1時間インキュベーションに渡る、14C標識ノルニコチンの産生に基づいて、ニコチン脱メチル化酵素の活性を計算する。
【0143】
エチレン処理したかまたはしなかった、バレー種変換(系統4407−33)および非変換(系統4407−25)タバコ系統の対から試料を得た。すべての未処理試料は、検出可能なミクロソーム・ニコチン脱メチル化酵素活性をまったく持たなかった。対照的に、エチレン処理した変換系統から得たミクロソーム試料は、有意なレベルのニコチン脱メチル化酵素活性を含有することが見出された。ニコチン脱メチル化酵素活性は、P450特異的阻害剤によって阻害されることが示され、脱メチル化酵素活性が、P450ミクロソーム由来酵素に一致することが立証された。バレー種変換タバコ系統に関して得られる、典型的な酵素アッセイ結果の組を表VIに示す。対照的に、エチレン処理した非変換系統タバコから得た試料は、ニコチン脱メチル化酵素活性をまったく含有しなかった。これらの結果によって、ニコチン脱メチル化酵素活性は、変換系統においてエチレン処理で誘導されたが、対応する同系非変換系統では誘導されないことが立証された。同系黒タバコ品種対に関しても同様の結果が得られ、この場合、ミクロソーム・ニコチン脱メチル化酵素活性は、変換系統では誘導され、そして非変換対系統では検出不能であった。これらの実験を総合すると、ミクロソーム・ニコチン脱メチル化酵素活性は、変換系統でエチレン処理に際して誘導される一方、対の同系非変換系統では誘導されないことが立証された。P450由来遺伝子であり、そして対の非変換系統に比較して、変換系統で優先的に誘導される遺伝子は、ニコチン脱メチル化酵素をコードする遺伝子の候補である。
【0144】
【表8】
【0145】
(実施例16)
ニコチン脱メチル化酵素としてのD121−AA8の機能的同定
酵母細胞において、異種発現されるP450の酵素活性をアッセイすることによって、ニコチン脱メチル化酵素のコード遺伝子としての候補クローン(D121−AA8)の機能を確認した。
【0146】
1.酵母発現ベクターの構築
P450コードcDNA(121AA8)の推定上のタンパク質コード配列を、酵母発現ベクターpYeDP60にクローニングした。適切なBamHIおよびMfeI部位(下線)を含有するPCRプライマーを介して、これらの配列を、翻訳開始コドン(ATG)の上流または停止コドン(TAA)の下流に導入した。増幅されたPCR産物上のMfeIは、ベクター上のEcoRI部位と適合する。121AA8 cDNAを増幅するのに用いたプライマーは、
【0147】
【化1】
【0148】
であった。6つのヒスチジンを含めて、タンパク質のC末端に9つの余分なアミノ酸をコードする配列セグメントを逆方向プライマーに取り込んだ。これによって、誘導に際して、6xHisをタグ付けしたP450の発現が促進される。酵素消化した後、PCR産物をGAL10−CYC1プロモーターに関してセンス方向でpYeDP60ベクターに連結した。酵素制限およびDNA配列決定によって、構築物を確認した。
【0149】
2.酵母形質転換
シロイヌナズナ属(Arabidopsis)NADPH−チトクロムP450還元酵素ATR1を発現するように修飾したWAT11酵母株を、構築物pYeDP60−P450 cDNAプラスミドで形質転換した。0.2cm電極ギャップのキュベット中で、50マイクロリットルのWAT11酵母細胞懸濁物を〜1μgのプラスミドDNAと混合した。Eppendorfエレクトロポレーション装置(モデル2510)によって、2
.0kVでパルスを1回適用した。SGIプレート(5g/lバクトカザミノ酸、6.7g/lのアミノ酸不含酵母窒素基剤、20g/lグルコース、40mg/l DL−トリプトファン、20g/l寒天)上に細胞を広げた。ランダムに選択したコロニーに対して、PCR解析を直接行うことによって、形質転換体を確認した。
【0150】
3.形質転換酵母細胞におけるP450発現
単一酵母コロニーを用いて、30ml SGI培地(5g/lバクトカザミノ酸、6.7g/lのアミノ酸不含酵母窒素基剤、20g/lグルコース、40mg/l DL−トリプトファン)に接種し、そして30℃で約24時間増殖させた。この培養物のアリコットを、1000mlのYPGE培地(10g/l酵母エキス、20g/lバクトペプトン、5g/lグルコース、30ml/lエタノール)に1:50で希釈し、そしてDiastix検尿試薬紙(Bayer、インディアナ州エルクハート)の色変化によって示されるように、グルコースが完全に消費されるまで増殖させた。最終濃度2%までDL−ガラクトースを添加することによって、クローニングしたP450の誘導を開始した。in vivo活性アッセイまたはミクロソーム調製に用いる前に、培養物をさらに20時間増殖させた。
【0151】
pYeDP60−CYP71D20(タバコ(Nicotiana tabacum)の5−エピ−アリストロチェンおよび1−デオキシカプシジオールの水酸化を触媒するP450)を発現するWAT11酵母細胞を、P450発現および酵素活性アッセイの対照として用いた。
【0152】
4. in vivo酵素活性
酵母培養物にDL−ニコチン(ピロリジン−2−14C)を供給することによって、形質転換酵母細胞におけるニコチン脱メチル化酵素活性をアッセイした。75μlのガラクトース誘導培養物に、14C標識ニコチン(54mCi/mmol)を最終濃度55μMまで添加した。14mlポリプロピレン試験管中、アッセイ培養物を振盪しながら6時間インキュベーションし、そして900μlメタノールで抽出した。回転後、20μlのメタノール抽出物をrp−HPLCで分離して、そしてLSCによってノルニコチン分画を定量化した。
【0153】
WAT11(pYeDP60−CYP71D20)の対照培養物はニコチンをノルニコチンに変換せず、WAT11酵母株が、ニコチンをノルニコチンに生物変換する工程を触媒可能な内因性酵素活性を含有しないことが示された。対照的に、121AA8遺伝子を発現する酵母は、検出可能な量のノルニコチンを産生し、このP450酵素がニコチン脱メチル化酵素活性を持つことが示された。
【0154】
5.酵母ミクロソーム調製
ガラクトースで20時間誘導した後、遠心分離によって酵母細胞を収集し、そしてTES−M緩衝液(50mM Tris−HCl、pH7.5、1mM EDTA、0.6Mソルビトール、10mM 2−メルカプトエタノール)で2回洗浄した。ペレットを抽出懸濁液(50mM Tris−HCl、pH7.5、1mM EDTA、0.6Mソルビトール、2mM 2−メルカプトエタノール、1%ウシ血清アルブミン、プロテアーゼ阻害剤カクテル(Roche)1錠剤/50ml)に再懸濁した。次いで、細胞をガラスビーズ(直径0.5mm、Sigma)で破壊した。細胞抽出物を20,000xgで20分間遠心分離して、細胞破片を取り除いた。上清を100,000xg、60分間の超遠心に供して、そして生じたペレットはミクロソーム分画を含有した。ミクロソーム分画をTEG−M緩衝液(50mM Tris−HCl、pH7.5、1mM EDTA、20%グリセロールおよび1.5mM 2−メルカプトエタノール)に、タンパク質濃度1mg/mlで懸濁した。ミクロソーム調製物を使用するまで液体窒素フリーザー中に保存し
た。
【0155】
6.酵母ミクロソーム調製物における酵素活性アッセイ
酵母ミクロソーム調製物を用いて、タンパク質濃度が1mg/mlで一定であったことを除いて、タバコ葉からのミクロソーム調製物と同じ方式で(実施例15)、ニコチン脱メチル化酵素活性アッセイを行った。
【0156】
CYP71D20を発現する対照酵母細胞由来のミクロソーム調製物は、検出可能なミクロソーム・ニコチン脱メチル化酵素活性をまったく持たなかった。対照的に、121AA8遺伝子を発現する酵母細胞から得たミクロソーム試料は、有意なレベルのニコチン脱メチル化酵素活性を示した。ニコチン脱メチル化酵素活性は、NADPHに必要であり、そしてP450特異的阻害剤によって阻害されることが示されており、これは研究中のP450に一致する。酵母細胞に関して得られる、典型的な酵素アッセイ結果の組を表VIIに示す。
【0157】
【表9】
【0158】
これらの実験を総合すると、クローニングされた全長遺伝子D121−AA8が、酵母で発現させた際、ニコチンのノルニコチンへの変換を触媒するチトクロムP450タンパク質をコードすることが立証された。
【0159】
本発明の実施にあたり、本発明の前述の詳細な説明を考慮すると、多くの修飾および変動が当業者には思い浮かぶと期待される。その結果、こうした修飾および変動は、付随する請求項の範囲内に含まれると意図される。
【技術分野】
【0001】
本発明は、タバコ属(Nicotiana)植物においてチトクロムP450酵素(以後、p450およびp450酵素と称する)をコードする核酸配列、およびこうした核酸配列を用いて植物表現型を改変する方法に関する。
【背景技術】
【0002】
背景
チトクロムp450は、内因性基質および生体異物基質の酸化代謝、過酸化代謝および還元代謝を含む、多様な範囲の化学的に異なる基質の酵素反応を触媒する。植物において、p450は、フェニルプロパノイド、アルカロイド、テルペノイド、脂質、青酸グリコシド、およびグルコシノレートなどの植物産物の合成を含む、生化学的経路に関与する(Chappel, Annu. Rev. Plant Physiol. Plant Mol. Biol. 198, 49:311−343(非特許文献1))。チトクロムp450は、p450ヘム−チオレート・タンパク質としても知られ、通常、p450含有モノオキシゲナーゼ系と呼ばれる多構成要素電子移動連鎖の最後のオキシダーゼとして作用する。触媒される特定の反応には、脱メチル化、水酸化、エポキシ化、N−酸化、スルホ酸化(sulfooxidation)、N−、S−、およびO−脱アルキル化、脱硫酸化、脱アミノ化、並びにアゾ、ニトロ、およびN−オキシド基の還元が含まれる。
【0003】
タバコ属植物p450酵素の多様な役割が、フェニルプロパノイド、アルカロイド、テルペノイド、脂質、青酸グリコシド、グルコシノレートおよび多くの他の化学実体などの多様な植物代謝産物を生じるのに関連付けられてきている。近年、いくつかのp450酵素が、植物において植物代謝産物の組成に影響を及ぼしうることが明らかになってきている。例えば、選択される脂肪酸のプロフィールを、育種を通じて改変することによって、特定の植物のフレーバーおよび香りを改善することが以前から望まれてきた;が、これらの葉の構成要素のレベルを調節するのに関与する機構に関してはほとんどわかっていない。脂肪酸の修飾に関与するp450酵素を下方制御すると、より好ましい葉の表現型特性を提供する、望ましい脂肪酸の集積が促進されることも可能である。植物構成要素におけるp450酵素の機能およびその広い役割は、なお発見される途上にある。例えば、特殊な種類のp450酵素が、脂肪酸を分解して、果物および野菜の「新鮮な緑」のにおいに主に貢献する、揮発性C6およびC9アルデヒドおよびアルコールにするのを触媒することが見出された。タバコ属の葉において、脂質構成要素および関連する分解代謝産物を修飾することによって、標的とされる他の新規p450のレベルを改変して、葉の構成要素の品質を増進することも可能である。葉におけるこれらの構成要素のいくつかが、葉の品質特性の成熟を刺激する老化によって影響を受ける。さらに他の報告によって、p450酵素が、植物−病原体相互作用および疾患抵抗性に関与する脂肪酸を改変する際に機能的役割を果たすことが示されてきている。
【0004】
他の例において、p450酵素が、アルカロイド生合成に関与することも示唆されてきている。ノルニコチンは、タバコ(Nicotiana tabaceum)に見られる微量アルカロイドである。ノルニコチンは、p450が仲介してニコチンが脱メチル化され、その後、N位でアシル化およびニトロソ化が起こり、それによって一連のN−アシルノニコチン(N−acylnonicotines)およびN−ニトロソノルニコチンが生じることによって産生されると推論されてきている。推定上のp450脱メチル化酵素(demethylase)に触媒されるN−脱メチル化は、タバコ属におけるノルニコチン生合成の主な供給源であると考えられる。該酵素はミクロソーム性であると考えられ
るが、これまで、ニコチン脱メチル化酵素の精製は成功しておらず、関与する遺伝子も単離されていない。
【0005】
さらに、p450酵素の活性が、遺伝的に調節され、そしてまた、環境要因によって強く影響されると仮定されているが、証明されていない。例えば、タバコ属におけるニコチンの脱メチル化は、植物が成熟段階に達したときに実質的に増加すると考えられている。さらに、RNAが存在する場合、その翻訳を阻害することも可能な転移可能要素を、脱メチル化酵素遺伝子が含有すると仮定されるが、証明はされていない。
【0006】
p450酵素型が非常に多様であり、構造および機能が異なることから、タバコ属p450酵素の研究は、本発明以前には非常に困難であった。さらに、少なくとも部分的に、膜に局在するこれらのp450酵素が、典型的には存在量が少なく、そしてしばしば、精製するには不安定であるため、これらのタンパク質のクローニングが妨害されてきた。したがって、植物におけるp450酵素、およびこれらのp450酵素に関連する核酸配列を同定する必要性が存在する。特に、タバコ属においては、いくつかのチトクロムp450タンパク質しか報告されてきていない。本明細書に記載する発明は、配列同一性に基づいて、p450種のいくつかのグループに対応する、かなりの数のチトクロムp450断片の発見を含む。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Chappel, Annu. Rev. Plant Physiol. Plant Mol. Biol. 198, 49:311−343
【発明の概要】
【課題を解決するための手段】
【0008】
概要
本発明は植物p450酵素に関する。本発明はさらに、タバコ属由来の植物p450酵素に関する。本発明はまた、その発現がエチレンおよび/または植物老化によって誘導される、植物におけるp450酵素にも関する。本発明はさらに、酵素活性、例えばオキシゲナーゼ、脱メチル化酵素等、またはその他に分類される酵素活性を有する、植物中の核酸配列、およびこれらの酵素の発現または過剰発現を減少させるかまたはサイレンシングするための、これらの配列の使用にも関する。本発明はまた、より低いノルニコチンレベルを示す植物より、より高いノルニコチンレベルを含有する植物で見られるp450酵素にも関する。
【0009】
1つの側面において、本発明は、配列番号1、3、5、7、9、11、13、15、17、19、21、23、25、27、29、31、33、35、37、39、41、43、45、47、49、51、53、55、57、59、61、63、65、67、69、71、73、75、77、79、81、83、85、87、89、91、95、97、99、101、103、105、107、109、111、113、115、117、119、121、123、125、127、129、131、133、135、137、139、143、145、147、149、151、153、155、157、159、161、163、165、167、169、171、173、175、177、179、181、183、185、187、189、191、193、195、197、199、201、203、205、207、209、211、213、215、217、219、221、223、225、227、229、231、233、235、237、239、241、243、245、247、249、251、253、255、257、259、261、263、265、267、269、271、273、275、277、279、281、283、285、287、289、291、293、295および297に示す核酸配列に関する。
【0010】
第二の関連する側面において、核酸配列において75%を超える同一性を含有する断片を、チトクロムp450モチーフGXRXCX(G/A)に続く最初の核酸から停止コドンまでに対応する領域における同一性に応じたグループに入れた。代表的な核酸グループ
およびそれぞれの種を表Iに示す。
【0011】
第三の側面において、本発明は、配列番号2、4、6、8、10、12、14、16、18、20、22、24、26、28、30、32、34、36、38、40、42、44、46、48、50、52、54、56、58、60、62、64、66、68、70、72、74、76、78、80、82、84、86、88、90、92、96、98、100、102、104、106、108、110、112、114、116、118、120、122、124、126、128、130、132、134、136、138、140、144、146、148、150、152、154、156、158、160、162、164、166、168、170、172、174、176、178、180、182、184、186、188、190、192、194、196、198、200、202、204、206、208、210、212、214、216、218、220、222、224、226、228、230、232、234、236、238、240、242、244、246、248、250、252、254、256、258、260、262、264、266、268、270、272、274、276、278、280、282、284、286、288、290、292、294、296および298に示すアミノ酸配列に関する。
【0012】
第四の関連する側面において、アミノ酸配列において71%を超える同一性を含有する断片を、チトクロムp450モチーフGXRXCX(G/A)に続く最初のアミノ酸から停止コドンまでに対応する領域における、互いに対する同一性に応じたグループに入れた。代表的なアミノ酸グループおよびそれぞれの種を表IIに示す。
【0013】
第五の側面において、本発明は、配列番号150、152、154、156、158、160、162、164、166、168、170、172、174、176、178、180、182、184、186、188、190、192、194、196、198、200、202、204、206、208、210、212、214、216、218、220、222、224、226、228、230、232、234、236、238、240、242、244、246、248、250、252、254、256、258、260、262、264、266、268、270、272、274、276、278、280、282、284、286、288、290、292、294、296および298に示す全長遺伝子のアミノ酸配列に関する。
【0014】
第六の関連する側面において、アミノ酸配列において85%以上の同一性を含有する全長遺伝子を、互いに対する同一性に応じたグループに入れた。代表的なアミノ酸グループおよびそれぞれの種を表IIIに示す。
【0015】
第七の側面において、本発明は、配列番号299〜357に示す断片のアミノ酸配列に関する。
第八の関連する側面において、アミノ酸配列において90%以上の同一性を含有する断片を、最初のチトクロムp450ドメイン、UXXRXXZから、第三のチトクロム・ドメイン、GXRXO、ここでUはEまたはKであり、Xはアミノ酸いずれかであり、そしてZはR、T、SまたはMである、までに対応する領域における、互いに対する同一性に応じたグループに入れた。代表的なアミノ酸グループおよびそれぞれの種を表IVに示す。
【0016】
第九の関連する側面において、RNAウイルス系を用いて、タバコ属植物におけるp450酵素の減少または除去または過剰発現を一過性に達成することも可能である。
一般の当業者に通常利用可能な技術を用いて、限定されるわけではないが、内因性p450 RNA転写物、発現されたp450ペプチド、および植物代謝産物濃度の解析を含
む、表現型変化に関して、生じた形質転換植物または感染植物を評価する。
【0017】
第十の重要な側面において、本発明はまた、改変されたp450酵素活性レベルを有するトランスジェニック・タバコ属系統の生成にも関する。本発明にしたがって、これらのトランスジェニック系統には、特定の酵素の発現を減少させるかまたはサイレンシングするかまたは増加させ、したがってタバコ属内で表現型効果を生じるのに有効である核酸配列が含まれる。こうした核酸配列には、配列番号1、3、5、7、9、11、13、15、17、19、21、23、25、27、29、31、33、35、37、39、41、43、45、47、49、51、53、55、57、59、61、63、65、67、69、71、73、75、77、79、81、83、85、87、89、91、95、97、99、101、103、105、107、109、111、113、115、117、119、121、123、125、127、129、131、133、135、137、139、143、145、147、149、151、153、155、157、159、161、163、165、167、169、171、173、175、177、179、181、183、185、187、189、191、193、195、197、199、201、203、205、207、209、211、213、215、217、219、221、223、225、227、229、231、233、235、237、239、241、243、245、247、249、251、253、255、257、259、261、263、265、267、269、271、273、275、277、279、281、283、285、287、289、291、293、295および297が含まれる。
【0018】
本発明の非常に重要な第十一の側面において、全長遺伝子またはその断片のいずれかを用いて下方制御能力がある、あるいは全長遺伝子を用いて過剰発現能力がある、本発明の核酸を含む植物品種は、対照植物に比較して、改変された代謝産物プロフィールを有するであろう。
【0019】
本発明の第十二の側面において、本発明の核酸を含む植物品種は、植物または植物外部に由来する代謝産物の生合成または分解を修飾する際に、全長遺伝子またはその断片のいずれかを用いて、特定の外因性化学薬品または植物疫病に耐容性を示すのに使用を有するであろう。こうした核酸配列には、配列番号1、3、5、7、9、11、13、15、17、19、21、23、25、27、29、31、33、35、37、39、41、43、45、47、49、51、53、55、57、59、61、63、65、67、69、71、73、75、77、79、81、83、85、87、89、91、95、97、99、101、103、105、107、109、111、113、115、117、119、121、123、125、127、129、131、133、135、137、139、143、145、147、149、151、153、155、157、159、161、163、165、167、169、171、173、175、177、179、181、183、185、187、189、191、193、195、197、199、201、203、205、207、209、211、213、215、217、219、221、223、225、227、229、231、233、235、237、239、241、243、245、247、249、251、253、255、257、259、261、263、265、267、269、271、273、275、277、279、281、283、285、287、289、291、293、295および297が含まれる。
【0020】
第十三の側面において、本発明は、解説される核酸配列に実質的な核酸同一性を有する遺伝子を含有する植物、より好ましくはタバコ属のスクリーニングに関する。本発明の使用は、こうした植物が伝統的な品種またはトランスジェニック品種の育種プログラム、突然変異誘発プログラム、あるいは天然存在の多様な植物集団の一部である場合、厳密なま
たは実質的な同一性を持つ核酸配列を含有する植物を同定し、そして選択するのに好適であろう。実質的な核酸同一性に関する植物のスクリーニングは、限定されるわけではないが、核酸ハイブリダイゼーションおよびPCR解析を含む核酸検出プロトコルと組み合わせて、核酸プローブを用い、植物核酸物質を評価することによって、達成可能である。核酸プローブは、配列番号1、3、5、7、9、11、13、15、17、19、21、23、25、27、29、31、33、35、37、39、41、43、45、47、49、51、53、55、57、59、61、63、65、67、69、71、73、75、77、79、81、83、85、87、89、91、95、97、99、101、103、105、107、109、111、113、115、117、119、121、123、125、127、129、131、133、135、137、139、143、145、147、149、151、153、155、157、159、161、163、165、167、169、171、173、175、177、179、181、183、185、187、189、191、193、195、197、199、201、203、205、207、209、211、213、215、217、219、221、223、225、227、229、231、233、235、237、239、241、243、245、247、249、251、253、255、257、259、261、263、265、267、269、271、273、275、277、279、281、283、285、287、289、291、293、295および297に対応する、解説される核酸配列またはその断片からなることも可能である。
【0021】
第十四の側面において、本発明は、解説される核酸配列に対応する実質的なアミノ酸同一性を共有する植物遺伝子、より好ましくはタバコ属遺伝子の同定に関する。cDNAおよびゲノムクローン両方を含む植物遺伝子、より好ましくはタバコ属由来のcDNAおよびゲノムクローンを含む植物遺伝子の同定は、限定されるわけではないが、核酸ハイブリダイゼーションおよびPCR解析を含む核酸検出プロトコルと組み合わせて核酸プローブを用い、植物cDNAライブラリーをスクリーニングすることによって、達成可能である。核酸プローブは、配列番号1、3、5、7、9、11、13、15、17、19、21、23、25、27、29、31、33、35、37、39、41、43、45、47、49、51、53、55、57、59、61、63、65、67、69、71、73、75、77、79、81、83、85、87、89、91、95、97、99、101、103、105、107、109、111、113、115、117、119、121、123、125、127、129、131、133、135、137、139、143、145および147に対応する核酸配列またはその断片で構成されることも可能である。
【0022】
別の第十五の側面において、解説されるアミノ酸配列の一部またはすべてに向けられる抗体を用いて、ペプチドを発現するcDNA発現ライブラリーをスクリーニングすることも可能である。こうしたアミノ酸配列には、配列番号2、4、8、9、10、12、14、16、18、20、22、24、26、28、30、32、34、36、38、40、42、44、46、48、50、52、54、56、58、60、62、64、66、68、70、72、74、76、78、80、82、84、86、88、90、92、96、98、100、102、104、106、108、110、112、114、116、118、120、122、124、126、128、130、132、134、136、138、140、144、146、148が含まれる。
【0023】
第十六の重要な側面において、本発明はまた、p450酵素活性レベルの過剰発現を有するトランスジェニック・タバコ属系統の生成にも関する。本発明にしたがって、これらのトランスジェニック系統には、特定の酵素の発現を増加させるのに有効であり、したがってタバコ属内で表現型効果を生じるのに有効である全長遺伝子のアミノ酸配列をコードするすべての核酸配列が含まれる。こうしたアミノ酸配列には、配列番号150、152、154、156、158、160、162、164、166、168、170、172
、174、176、178、180、182、184、186、188、190、192、194、196、198、200、202、204、206、208、210、212、214、216、218、220、222、224、226、228、230、232、234、236、238、240、242、244、246、248、250、252、254、256、258、260、262、264、266、268、270、272、274、276、278、280、282、284、286、288、290、292、294、296および298が含まれる。
【0024】
減少した量のノルニコチンを有するタバコ葉(ラミナおよび/または茎)を含むタバコ製品もまた提供する。タバコ製品には、本明細書記載の配列を含む植物、あるいはタバコ特異的ニトロソアミンをコードする遺伝子が除去または抑制されている植物由来のタバコ(ラミナおよび/または茎を含むタバコ葉)が含まれる。タバコ特異的ニトロソアミンをコードする遺伝子の除去または抑制は、タバコ特異的ニトロソアミンをコードする遺伝子が除去または抑制されていないタバコ植物から作成されたタバコ製品に比較して、タバコ製品中のタバコ特異的ニトロソアミンを、約5〜約10%、別の側面においては約10〜20%、別の側面においては約20〜30%、そして別の側面においては30%を超えて、減少させるのに有効である。本明細書において、タバコ製品は、巻きタバコ、葉巻、パイプタバコ、嗅ぎタバコ、噛みタバコ、タバコ製品とブレンドした製品、およびこれらの混合物を含むことも可能である。
【0025】
詳細な説明
定義
別に定義しない限り、本明細書で用いるすべての技術的用語および科学的用語は、本発明が属する当該技術分野の一般の当業者に一般的に理解されるのと同じ意味を有する。Singletonら(1994)Dictionary of Microbiology and Molecular Biology, 第2版, John Wiley
and Sons(ニューヨーク)は、本発明で用いる用語の多くの一般的辞書とともに、技術の1つを提供する。本明細書に引用する特許および刊行物はすべて本明細書に援用される。本発明の目的のため、次の用語を以下に定義する。
【0026】
「酵素活性」は、脱メチル化、水酸化、エポキシ化、N−酸化、スルホ酸化、N−、S−、およびO−脱アルキル化、脱硫酸化、脱アミノ化、並びにアゾ、ニトロ、およびN−オキシド基の還元を含むことを意味される。用語「核酸」は、一本鎖または二本鎖型いずれか、あるいはセンスまたはアンチセンスのデオキシリボヌクレオチドまたはリボヌクレオチドポリマーを指し、そして別に限定されない限り、天然存在ヌクレオチドに似た方式で、核酸にハイブリダイズする天然ヌクレオチドの既知の類似体(analogue)を含む。別に示さない限り、特定の核酸配列には、その相補配列が含まれる。
【0027】
用語「機能可能であるように連結される」、「機能可能な組み合わせ」および「機能可能な順序」は、核酸発現調節配列(プロモーター、シグナル配列、または転写因子結合部位の列など)および第二の核酸配列の間の機能する連結を指し、ここで発現調節配列は、第二の配列に対応する核酸の転写および/または翻訳に影響を及ぼす。
【0028】
用語「組換え」は、細胞に関して用いた場合、細胞が異種核酸を複製するか、前記核酸を発現するか、あるいは異種核酸にコードされるペプチド、異種ペプチド、またはタンパク質を発現することを示す。組換え細胞は、細胞の天然(非組換え)型には見られないセンス型またはアンチセンス型いずれかの遺伝子または遺伝子断片を発現することも可能である。組換え細胞はまた、細胞の天然型に見られるが、修飾され、そして人工的手段によって細胞に再度導入されている遺伝子を発現することもまた可能である。
【0029】
「構造遺伝子」は、タンパク質、ポリペプチドまたはその一部をコードするDNAセグメントを含み、そして転写開始を駆動する5’配列を除く遺伝子の部分である。あるいは構造遺伝子は翻訳不能産物をコードすることも可能である。構造遺伝子は、細胞に通常見られるもの、あるいは導入される細胞または細胞位置で通常は見られないものであることも可能であり、この場合、該遺伝子は「異種遺伝子」と呼ばれる。異種遺伝子は、細菌ゲノムまたはエピソーム、真核、核またはプラスミドDNA、cDNA、ウイルスDNAあるいは化学的に合成されたDNAを含む、当該技術分野に知られるいかなる供給源に、すべてまたは一部、由来していることも可能である。構造遺伝子は、生物学的活性またはその特性、発現産物の生物学的活性または化学構造、発現率または発現調節の方式を達成可能な1以上の修飾を含有することも可能である。こうした修飾には、限定されるわけではないが、1以上のヌクレオチドの突然変異、挿入、欠失および置換が含まれる。構造遺伝子は、中断されないコード配列を構成することも可能であるし、または適切なスプライス接合部によって結合される1以上のイントロンを含むことも可能である。構造遺伝子は翻訳可能または翻訳不能であることも可能であり、アンチセンス方向にあるものを含む。構造遺伝子は複数の供給源由来および複数の遺伝子配列(天然存在または合成、ここで合成は化学的に合成されたDNAを指す)由来のセグメントの合成物(composite)であることも可能である。
【0030】
「由来する」は、供給源(化学的および/または生物学的)から採取されるか、得られるか、受け取られるか、帰着するか、複製されるか、または伝わる(descend)ことを意味するよう用いられる。派生物(derivative)は、元来の供給源の化学的操作または生物学的操作(限定されるわけではないが、置換、付加、挿入、欠失、抽出、単離、突然変異および複製を含む)によって産生されることも可能である。
【0031】
「化学的に合成された」は、DNAの配列に関する場合、構成要素ヌクレオチドの一部がin vitroで組み立てられたことを意味する。よく確立された方法(Caruthers, Methodology of DNA and RNA Sequencing, (1983), Weissman(監修), Praeger Publishers, ニューヨーク, 第1章)を用いて、DNAの手動の化学的合成を達成することも可能であり;いくつかの商業的に入手可能な機械の1つを用いて、自動化化学合成を行うことも可能である。
【0032】
SmithおよびWaterman, Adv. Appl. Math. 2:482(1981)の局所相同性アルゴリズムによって、NeedlemanおよびWunsch, J. Mol. Biol. 48:443(1970)の相同性並列アルゴリズムによって、PearsonおよびLipman Proc. Natl. Acad. Sci.(U.S.A.)85:2444(1988)の類似性法に関する検索によって、これらのアルゴリズムのコンピュータ化実行(ウィスコンシン遺伝学ソフトウェアパッケージ、遺伝学コンピュータグループ、575 Science Dr., Madison, Wis.のGAP、BESTFIT、FASTA、およびTFASTA)によって、または視診によって、比較のための配列の最適並列を行うことも可能である。
【0033】
NCBI基本的局所並列検索ツール(BLAST)(Altschulら、1990)は、米国生物学情報センター(NCBI、メリーランド州ベセスダ)を含むいくつかの供給源から入手可能であり、そして配列解析プログラムblastp、blastn、blastx、tblastnおよびtblastxと関連付けて使用するため、インターネット上で入手可能である。該ツールは、htp://www.ncbi.nlm.nih.gov/BLAST/でアクセス可能である。このプログラムを用いて配列同一性を決定する方法の説明が、http://www.ncbi.nlm.nih.gov/BLAST/blast help.htmlで入手可能である。
【0034】
用語「実質的なアミノ酸同一性」または「実質的なアミノ酸配列同一性」は、アミノ酸配列に適用した場合、そして本明細書で用いる場合、ペプチドが、翻訳されたペプチドのチトクロムp450モチーフGXRXCX(G/A)に続く最初のアミノ酸から停止コドンまでに対応する領域に渡って、参照群に比較した際、少なくとも70パーセントの配列同一性、好ましくは80パーセントのアミノ酸配列同一性、より好ましくは90パーセントのアミノ酸配列同一性、そして最も好ましくは少なくとも99〜100パーセントの配列同一性を有する配列を含む、ポリペプチドの特性を示す。
【0035】
用語「実質的な核酸同一性」または「実質的な核酸配列同一性」は、核酸配列に適用した場合、そして本明細書で用いる場合、ポリヌクレオチドが、翻訳されたペプチドのチトクロムp450モチーフGXRXCX(G/A)に続く最初の核酸から停止コドンまでに対応する領域に渡って、参照群に比較した際、少なくとも75パーセントの配列同一性、好ましくは81パーセントの配列同一性、より好ましくは少なくとも91パーセントの配列同一性、そして最も好ましくは少なくとも99〜100パーセントの配列同一性を有する配列を含む、ポリヌクレオチド配列の特性を示す。
【0036】
ヌクレオチド配列が実質的に同一であることの別の指標は、2つの分子がストリンジェントな条件下で互いにハイブリダイズする場合である。ストリンジェントな条件は配列依存性であり、そして異なる環境において異なるであろう。一般的に、ストリンジェントな条件は、特定の配列に関する、定義されるイオン強度およびpHでの熱融点(Tm)より約5℃〜約20℃、通常、約10℃〜約15℃低いように選択される。Tmは、(定義されるイオン強度およびpHで)標的配列の50%が、マッチしたプローブにハイブリダイズする温度である。典型的には、ストリンジェントな条件は、pH7で塩濃度が約0.02モル濃度であり、そして温度が少なくとも約60℃であるものであろう。例えば標準的サザンハイブリダイゼーション法において、ストリンジェントな条件は、42℃の6xSSC中の最初の洗浄、続いて少なくとも約55℃、典型的には約60℃、そしてしばしば約65℃の温度での0.2xSSC中の1以上のさらなる洗浄を含むであろう。
【0037】
ヌクレオチド配列はまた、コードするポリペプチドおよび/またはタンパク質が実質的に同一である場合、本発明の目的のためには、実質的に同一である。したがって、1つの核酸配列が第二の核酸配列と本質的に同じポリペプチドをコードする場合、遺伝暗号に許容される縮重のため、ストリンジェントな条件下でハイブリダイズしない場合であっても、2つの核酸配列は実質的に同一である(コドン縮重および遺伝暗号の説明に関しては、Darnellら(1990)Molecular Cell Biology, 第2版 Scientific American Books W. H. Freeman and Company ニューヨークを参照されたい)。タンパク質試料のポリアクリルアミドゲル電気泳動、その後、染色での視覚化などの、当該技術分野に周知のいくつかの手段によって、タンパク質純度または均一性を示すことも可能である。特定の目的のため、高分解能が必要とされる可能性もあり、そして精製のためにHPLCまたは類似の手段を利用することも可能である。
【0038】
本明細書において、細胞にDNAセグメント(単数または複数)をトランスファーする核酸分子に関して、用語「ベクター」を用いる。ベクターはDNAを複製するよう作用することも可能であり、そして宿主細胞において、独立に複製可能である。用語「ビヒクル」は、ときに、「ベクター」と交換可能に用いられる。用語「発現ベクター」は、本明細書において、望ましいコード配列、および特定の宿主生物において、機能可能であるように連結されたコード配列の発現に必要な適切な核酸配列を含有する、組換えDNA分子を指す。原核生物における発現に必要な核酸配列には、通常、プロモーター、オペレーター(場合による)、およびリボソーム結合部位に、しばしば他の配列が伴って、含まれる。
真核細胞は、プロモーター、エンハンサー、および終結シグナルおよびポリアデニル化シグナルを利用することが知られる。
【0039】
根がある、完全に遺伝子操作された植物を再生するため、in vivo接種などの技術いずれかによって、または完全な植物に再生可能な形質転換植物細胞を産生する、既知のin vitro組織培養技術のいずれかによって、核酸を植物細胞に挿入することも可能である。したがって、例えば、植物細胞への挿入は、病原性または非病原性A.ツメファシエンス(A. tumefaciens)によるin vitro接種によることも可能である。他のこうした組織培養技術もまた、使用可能である。
【0040】
「植物組織」には、植物の分化組織および未分化組織が含まれ、限定されるわけではないが、根、芽、葉、花粉、種子、腫瘍組織および培養中の多様な型の細胞、例えば単細胞、プロトプラスト、胚およびカルス組織が含まれる。植物組織は、植物中(in planta)または器官中にあることも、組織または細胞培養物であることも可能である。
【0041】
「植物細胞」には、本明細書において、植物中の植物細胞、並びに培養中の植物細胞およびプロトプラストが含まれる。
「cDNA」または「相補DNA」は、一般的に、RNA分子に相補的なヌクレオチド配列を持つ一本鎖DNA分子を指す。cDNAは、RNAテンプレートに対する逆転写酵素の作用によって形成される。
【0042】
核酸配列を得る戦略
本発明にしたがって、変換(converter)および非変換タバコ属系統のタバコ属組織からRNAを抽出した。次いで、抽出したRNAを用いてcDNAを生成した。次いで、2つの戦略を用いて、本発明の核酸配列を生成した。
【0043】
第一の戦略では、植物組織からポリA濃縮RNAを抽出し、そして逆転写PCRによってcDNAを作成した。次いで、一本鎖cDNAを用い、縮重プライマーに加えて、オリゴd(T)逆方向プライマーを用いて、p450特異的PCR集団を生成した。プライマー設計は、p450の高度に保存されたモチーフに基づいた。特異的縮重プライマーの例を図1に示す。適切なサイズの挿入物を含有するプラスミド由来の配列断片をさらに解析した。これらのサイズの挿入物は、どのプライマーを用いたかに応じて、典型的には、約300〜約800ヌクレオチドの範囲であった。
【0044】
第二の戦略では、まず、cDNAライブラリーを構築した。プラスミド中のcDNAを用い、縮重プライマーに加えて、逆方向プライマーとしてプラスミド上のT7プライマーを用いて、p450特異的PCR集団を生成した。第一の戦略におけるように、適切なサイズの挿入物を含有するプラスミド由来の配列断片をさらに解析した。
【0045】
高レベルのノルニコチンを生じることが知られるタバコ属植物系統(変換系統)、および検出不能なレベルのノルニコチンを有する植物系統を出発材料として使用することも可能である。
【0046】
次いで、植物から葉を取り除き、そしてエチレンで処理して、本明細書に定義するp450酵素活性を活性化することも可能である。当該技術分野に知られる技術を用いて、総RNAを抽出する。次いで、図153に記載するようなオリゴd(T)プライマーを用いたPCR(RT−PCR)を用いて、cDNA断片を生成することも可能である。次いで、本明細書の実施例に、より完全に記載する、cDNAライブラリーを構築することも可能である。
【0047】
p450型酵素に保存される領域を縮重プライマーのテンプレートとして用いることも可能である(図75)。縮重プライマーを用いて、p450特異的バンドをPCRによって増幅することも可能である。p450様酵素を示すバンドをDNA配列決定によって同定することも可能である。BLAST検索、並列、または適切な候補を同定する他のツールを用いて、PCR断片を性質決定することも可能である。
【0048】
同定された断片からの配列情報を用いて、PCRプライマーを発展させることも可能である。cDNAライブラリー中のプラスミドプライマーと組み合わせてこれらのプライマーを用い、全長p450遺伝子をクローニングした。大規模サザン逆解析を行って、得られたすべての断片クローン、およびいくつかの場合、全長クローンに関して、示差発現を調べた。本発明のこの側面において、クローニングされた挿入物すべてをスクリーニングするため、クローニングされたDNA断片とハイブリダイズするプローブとして、異なる組織由来の標識総cDNAを用いて、これらの大規模逆サザンアッセイを行うことも可能である。
【0049】
非放射性および放射性(P32)ノーザンブロッティングアッセイもまた用いて、クローンp450断片および全長クローンを性質決定した。
全長クローンのアミノ酸配列を得て、そして抗原性であり、そして他のクローンに比較してユニークであるペプチド領域を選択することによって、いくつかの全長クローンに対してペプチド特異的抗体を作成した。キャリアータンパク質にコンジュゲート化した合成ペプチドに対して、ウサギ抗体を作成した。これらの抗体を用いて、植物組織に対して、ウェスタンブロッティング解析または他の免疫学的方法を行った。
【0050】
ウイルス誘導性遺伝子サイレンシング技術を用いることによって、上述のように同定された核酸配列を調べることも可能である(VIGS, Baulcombe, Current Opinions in Plant Biology, 1999, 2:109−113)。
【0051】
全長クローンのアミノ酸配列を得て、そして潜在的に抗原性であり、そして他のクローンに比較してユニークであるペプチド領域を選択することによって、いくつかの全長クローンに対してペプチド特異的抗体を作成した。キャリアータンパク質にコンジュゲート化した合成ペプチドに対して、ウサギ抗体を作成した。これらの抗体を用いて、ウェスタンブロッティング解析を行った。
【0052】
本発明の別の側面において、本発明のタバコ属植物におけるチトクロムp450酵素活性をさらに性質決定するため、RNA干渉技術(RNAi)を用いる。この技術を説明する以下の参考文献、Smithら, Nature, 2000, 407:319−320;Fireら, Nature, 1998, 391:306−311;Waterhouseら, PNAS, 1998, 95:13959−13964;Stalbergら, Plant Molecular Biology, 1993, 23:671−683;Baulcombe, Current Opinions in Plant Biology, 1999, 2:109−113;およびBrignetiら, EMBO Journal, 1998, 17(22):6739−6746が本明細書に援用される。RNAi技術、アンチセンス技術、または記載される多様な他の方法を用いて、植物を形質転換することも可能である。
【0053】
植物細胞に外来(foreign)遺伝物質を導入するための、そして導入された遺伝子を安定して維持し、そして発現する植物を得るための、いくつかの技術が存在する。こうした技術には、微小粒子上にコーティングした遺伝物質を細胞内に直接加速することが含まれる(Cornellに対する米国特許4,945,050およびDowElanc
oに対する5,141,131)。アグロバクテリウム(Agrobacterium)技術を用いて、植物を形質転換することも可能であり、トレド大学に対する米国特許5,177,010、Texax A&Mに対する5,104,310、Schilperootに対する欧州特許出願0131624B1、欧州特許出願120516、159418B1、欧州特許出願120516、159418B1および176,112、Schilperootに対する米国特許5,149,645、5,469,976、5,464,763および4,940,838および4,693,976、すべてMaxPlanckに対する欧州特許出願116718、290799、320500、Japan Nicotianaに対する欧州特許出願604662および627752、すべてCiba
Geigyに対する欧州特許出願0267159および0292435および米国特許5,231,019、どちらもCalgeneに対する米国特許5,463,174および4,762,785、並びにどちらもAgracetusに対する米国特許5,004,863および5,159,135を参照されたい。他の形質転換技術には、ウィスカー技術が含まれ、どちらもZenecaに対する米国特許5,302,523および5,464,765を参照されたい。植物を形質転換するにはエレクトロポレーション技術もまた用いられてきており、Boyce Thompson Instituteに対するWO 87/06614、どちらもDekalbに対する5,472,869および5,384,253、どちらもPGSに対するWO9209696およびWO9321335を参照されたい。これらの形質転換特許および刊行物はすべて、本明細書に援用される。植物を形質転換する多くの技術に加えて、外来遺伝子と接触させる組織の種類もまた、多様であることも可能である。こうした組織には、限定されるわけではないが、胚形成組織、カルス組織I型およびII型、胚軸、成長点等が含まれるであろう。当業者の技術内の適切な技術を用いて、脱分化中の、ほぼすべての植物組織を形質転換することも可能である。
【0054】
植物に導入される外来遺伝物質には、選択可能マーカーが含まれることも可能である。特定のマーカーを優先するのは、当業者の自由裁量であるが、以下の選択可能マーカーのいずれかとともに、選択可能マーカーとして機能可能な、本明細書に列挙されていない他の遺伝子のいずれも使用可能である。こうした選択可能マーカーには、限定されるわけではないが、抗生物質カナマイシン、ネオマイシンおよびG418に対する抵抗性をコードするトランスポゾンTn5のアミノグリコシド・ホスホトランスフェラーゼ遺伝子(AphII)とともに、グリフォセート;ハイグロマイシン;メトトレキセート;フォスフィノスリシン(bar);イミダゾリノン、スルホニル尿素およびトリアゾロピリミジン除草剤、例えばクロロスルフロン;ブロモキシニル、ダラポン等に対する抵抗性または耐性をコードする遺伝子が含まれる。
【0055】
選択可能マーカーに加えて、レポーター遺伝子を用いることが望ましい可能性もある。いくつかの例において、選択可能マーカーを伴わずに、レポーター遺伝子を用いることも可能である。レポーター遺伝子は、レシピエント生物または組織に典型的には存在しないかまたはこれらで発現されていない遺伝子である。レポーター遺伝子は、典型的には何らかの表現型変化または酵素特性を提供するタンパク質をコードする。こうした遺伝子の例が、本明細書に援用されるK. Weisingら Ann. Rev. Genetics, 22, 421(1988)に提供される。好ましいレポーター遺伝子には、限定なしに、グルクロニダーゼ(GUS)遺伝子およびGFP遺伝子が含まれる。
【0056】
植物組織にひとたび導入されたならば、当該技術分野に知られるいかなる手段によって構造遺伝子の発現をアッセイすることも可能であり、そして転写されるmRNA、合成されるタンパク質、または生じる遺伝子サイレンシングの量として、発現を測定することも可能である(本明細書に援用される米国特許第5,583,021号を参照されたい)。植物組織のin vitro培養のための技術が知られ、そしていくつかの場合、全植物
への再生の技術が知られる(EP出願第88810309.0)。導入された発現複合体を商業的に有用な品種にトランスファーするための方法が、当業者に知られる。
【0057】
望ましいレベルのp450酵素を発現する植物細胞がひとたび得られたら、当該技術分野に周知の方法および技術を用いて、そこから植物組織および全植物を再生することも可能である。次いで、再生された植物を、慣用的手段によって、繁殖させ、そして慣用的な植物育種技術によって、導入された遺伝子を他の系統および品種にトランスファーすることも可能である。
【0058】
以下の実施例は、本発明を実行する方法を例示し、そして付随する請求項に定義される本発明の範囲を例示するが、これを限定しないことが理解されなければならない。
【図面の簡単な説明】
【0059】
【図1】図1は、核酸配列番号1およびアミノ酸配列番号2を示す。
【図2】図2は、核酸配列番号3およびアミノ酸配列番号4を示す。
【図3】図3は、核酸配列番号5およびアミノ酸配列番号6を示す。
【図4】図4は、核酸配列番号7およびアミノ酸配列番号8を示す。
【図5】図5は、核酸配列番号9およびアミノ酸配列番号10を示す。
【図6】図6は、核酸配列番号11およびアミノ酸配列番号12を示す。
【図7】図7は、核酸配列番号13およびアミノ酸配列番号14を示す。
【図8】図8は、核酸配列番号15およびアミノ酸配列番号16を示す。
【図9】図9は、核酸配列番号17およびアミノ酸配列番号18を示す。
【図10】図10は、核酸配列番号19およびアミノ酸配列番号20を示す。
【図11】図11は、核酸配列番号21およびアミノ酸配列番号22を示す。
【図12】図12は、核酸配列番号23およびアミノ酸配列番号24を示す。
【図13】図13は、核酸配列番号25およびアミノ酸配列番号26を示す。
【図14】図14は、核酸配列番号27およびアミノ酸配列番号28を示す。
【図15】図15は、核酸配列番号29およびアミノ酸配列番号30を示す。
【図16】図16は、核酸配列番号31およびアミノ酸配列番号32を示す。
【図17】図17は、核酸配列番号33およびアミノ酸配列番号34を示す。
【図18】図18は、核酸配列番号35およびアミノ酸配列番号36を示す。
【図19】図19は、核酸配列番号37およびアミノ酸配列番号38を示す。
【図20】図20は、核酸配列番号39およびアミノ酸配列番号40を示す。
【図21】図21は、核酸配列番号41およびアミノ酸配列番号42を示す。
【図22】図22は、核酸配列番号43およびアミノ酸配列番号44を示す。
【図23】図23は、核酸配列番号45およびアミノ酸配列番号46を示す。
【図24】図24は、核酸配列番号47およびアミノ酸配列番号48を示す。
【図25】図25は、核酸配列番号49およびアミノ酸配列番号50を示す。
【図26】図26は、核酸配列番号51およびアミノ酸配列番号52を示す。
【図27】図27は、核酸配列番号53およびアミノ酸配列番号54を示す。
【図28】図28は、核酸配列番号55およびアミノ酸配列番号56を示す。
【図29】図29は、核酸配列番号57およびアミノ酸配列番号58を示す。
【図30】図30は、核酸配列番号59およびアミノ酸配列番号60を示す。
【図31】図31は、核酸配列番号61およびアミノ酸配列番号62を示す。
【図32】図32は、核酸配列番号63およびアミノ酸配列番号64を示す。
【図33】図33は、核酸配列番号65およびアミノ酸配列番号66を示す。
【図34】図34は、核酸配列番号67およびアミノ酸配列番号68を示す。
【図35】図35は、核酸配列番号69およびアミノ酸配列番号70を示す。
【図36】図36は、核酸配列番号71およびアミノ酸配列番号72を示す。
【図37】図37は、核酸配列番号73およびアミノ酸配列番号74を示す。
【図38】図38は、核酸配列番号75およびアミノ酸配列番号76を示す。
【図39】図39は、核酸配列番号77およびアミノ酸配列番号78を示す。
【図40】図40は、核酸配列番号79およびアミノ酸配列番号80を示す。
【図41】図41は、核酸配列番号81およびアミノ酸配列番号82を示す。
【図42】図42は、核酸配列番号83およびアミノ酸配列番号84を示す。
【図43】図43は、核酸配列番号85およびアミノ酸配列番号86を示す。
【図44】図44は、核酸配列番号87およびアミノ酸配列番号88を示す。
【図45】図45は、核酸配列番号89およびアミノ酸配列番号90を示す。
【図46】図46は、核酸配列番号91およびアミノ酸配列番号92を示す。
【図47】図47は、核酸配列番号92およびアミノ酸配列番号93を示す。
【図48】図48は、核酸配列番号95およびアミノ酸配列番号96を示す。
【図49】図49は、核酸配列番号97およびアミノ酸配列番号98を示す。
【図50】図50は、核酸配列番号99およびアミノ酸配列番号100を示す。
【図51】図51は、核酸配列番号101およびアミノ酸配列番号102を示す。
【図52】図52は、核酸配列番号103およびアミノ酸配列番号104を示す。
【図53】図53は、核酸配列番号105およびアミノ酸配列番号106を示す。
【図54】図54は、核酸配列番号107およびアミノ酸配列番号108を示す。
【図55】図55は、核酸配列番号109およびアミノ酸配列番号110を示す。
【図56】図56は、核酸配列番号111およびアミノ酸配列番号112を示す。
【図57】図57は、核酸配列番号113およびアミノ酸配列番号114を示す。
【図58】図58は、核酸配列番号115およびアミノ酸配列番号116を示す。
【図59】図59は、核酸配列番号117およびアミノ酸配列番号118を示す。
【図60】図60は、核酸配列番号119およびアミノ酸配列番号120を示す。
【図61】図61は、核酸配列番号121およびアミノ酸配列番号122を示す。
【図62】図62は、核酸配列番号123およびアミノ酸配列番号124を示す。
【図63】図63は、核酸配列番号125およびアミノ酸配列番号126を示す。
【図64】図64は、核酸配列番号127およびアミノ酸配列番号128を示す。
【図65】図65は、核酸配列番号129およびアミノ酸配列番号130を示す。
【図66】図66は、核酸配列番号131およびアミノ酸配列番号132を示す。
【図67】図67は、核酸配列番号133およびアミノ酸配列番号134を示す。
【図68】図68は、核酸配列番号135およびアミノ酸配列番号136を示す。
【図69】図69は、核酸配列番号137およびアミノ酸配列番号138を示す。
【図70】図70は、核酸配列番号139およびアミノ酸配列番号140を示す。
【図71】図71は、核酸配列番号141およびアミノ酸配列番号142を示す。
【図72】図72は、核酸配列番号143およびアミノ酸配列番号144を示す。
【図73】図73は、核酸配列番号145およびアミノ酸配列番号146を示す。
【図74】図74は、核酸配列番号147およびアミノ酸配列番号148を示す。
【図75】図75は、核酸配列番号149およびアミノ酸配列番号150を示す。
【図76】図76は、核酸配列番号151およびアミノ酸配列番号152を示す。
【図77】図77は、核酸配列番号153およびアミノ酸配列番号154を示す。
【図78】図78は、核酸配列番号155およびアミノ酸配列番号156を示す。
【図79】図79は、核酸配列番号157およびアミノ酸配列番号158を示す。
【図80】図80は、核酸配列番号159およびアミノ酸配列番号160を示す。
【図81】図81は、核酸配列番号161およびアミノ酸配列番号162を示す。
【図82】図82は、核酸配列番号163およびアミノ酸配列番号164を示す。
【図83】図83は、核酸配列番号165およびアミノ酸配列番号166を示す。
【図84】図84は、核酸配列番号167およびアミノ酸配列番号168を示す。
【図85】図85は、核酸配列番号169およびアミノ酸配列番号170を示す。
【図86】図86は、核酸配列番号171およびアミノ酸配列番号172を示す。
【図87】図87は、核酸配列番号173およびアミノ酸配列番号174を示す。
【図88】図88は、核酸配列番号175およびアミノ酸配列番号176を示す。
【図89】図89は、核酸配列番号177およびアミノ酸配列番号178を示す。
【図90】図90は、核酸配列番号179およびアミノ酸配列番号180を示す。
【図91】図91は、核酸配列番号181およびアミノ酸配列番号182を示す。
【図92】図92は、核酸配列番号183およびアミノ酸配列番号184を示す。
【図93】図93は、核酸配列番号185およびアミノ酸配列番号186を示す。
【図94】図94は、核酸配列番号187およびアミノ酸配列番号188を示す。
【図95】図95は、核酸配列番号189およびアミノ酸配列番号190を示す。
【図96】図96は、核酸配列番号191およびアミノ酸配列番号192を示す。
【図97】図97は、核酸配列番号193およびアミノ酸配列番号194を示す。
【図98】図98は、核酸配列番号195およびアミノ酸配列番号196を示す。
【図99】図99は、核酸配列番号197およびアミノ酸配列番号198を示す。
【図100】図100は、核酸配列番号199およびアミノ酸配列番号200を示す。
【図101】図101は、核酸配列番号201およびアミノ酸配列番号202を示す。
【図102】図102は、核酸配列番号203およびアミノ酸配列番号204を示す。
【図103】図103は、核酸配列番号205およびアミノ酸配列番号206を示す。
【図104】図104は、核酸配列番号207およびアミノ酸配列番号208を示す。
【図105】図105は、核酸配列番号209およびアミノ酸配列番号210を示す。
【図106】図106は、核酸配列番号211およびアミノ酸配列番号212を示す。
【図107】図107は、核酸配列番号213およびアミノ酸配列番号214を示す。
【図108】図108は、核酸配列番号215およびアミノ酸配列番号216を示す。
【図109】図109は、核酸配列番号217およびアミノ酸配列番号218を示す。
【図110】図110は、核酸配列番号219およびアミノ酸配列番号220を示す。
【図111】図111は、核酸配列番号221およびアミノ酸配列番号222を示す。
【図112】図112は、核酸配列番号223およびアミノ酸配列番号224を示す。
【図113】図113は、核酸配列番号225およびアミノ酸配列番号226を示す。
【図114】図114は、核酸配列番号227およびアミノ酸配列番号228を示す。
【図115】図115は、核酸配列番号229およびアミノ酸配列番号230を示す。
【図116】図116は、核酸配列番号231およびアミノ酸配列番号232を示す。
【図117】図117は、核酸配列番号233およびアミノ酸配列番号234を示す。
【図118】図118は、核酸配列番号235およびアミノ酸配列番号236を示す。
【図119】図119は、核酸配列番号237およびアミノ酸配列番号238を示す。
【図120】図120は、核酸配列番号239およびアミノ酸配列番号240を示す。
【図121】図121は、核酸配列番号241およびアミノ酸配列番号242を示す。
【図122】図122は、核酸配列番号243およびアミノ酸配列番号244を示す。
【図123】図123は、核酸配列番号245およびアミノ酸配列番号246を示す。
【図124】図124は、核酸配列番号247およびアミノ酸配列番号248を示す。
【図125】図125は、核酸配列番号249およびアミノ酸配列番号250を示す。
【図126】図126は、核酸配列番号251およびアミノ酸配列番号252を示す。
【図127】図127は、核酸配列番号253およびアミノ酸配列番号254を示す。
【図128】図128は、核酸配列番号255およびアミノ酸配列番号256を示す。
【図129】図129は、核酸配列番号257およびアミノ酸配列番号258を示す。
【図130】図130は、核酸配列番号259およびアミノ酸配列番号260を示す。
【図131】図131は、核酸配列番号261およびアミノ酸配列番号262を示す。
【図132】図132は、核酸配列番号263およびアミノ酸配列番号264を示す。
【図133】図133は、核酸配列番号265およびアミノ酸配列番号266を示す。
【図134】図134は、核酸配列番号267およびアミノ酸配列番号268を示す。
【図135】図135は、核酸配列番号269およびアミノ酸配列番号270を示す。
【図136】図136は、核酸配列番号271およびアミノ酸配列番号272を示す。
【図137】図137は、核酸配列番号273およびアミノ酸配列番号274を示す。
【図138】図138は、核酸配列番号275およびアミノ酸配列番号276を示す。
【図139】図139は、核酸配列番号277およびアミノ酸配列番号278を示す。
【図140】図140は、核酸配列番号279およびアミノ酸配列番号280を示す。
【図141】図141は、核酸配列番号281およびアミノ酸配列番号282を示す。
【図142】図142は、核酸配列番号283およびアミノ酸配列番号284を示す。
【図143】図143は、核酸配列番号285およびアミノ酸配列番号286を示す。
【図144】図144は、核酸配列番号287およびアミノ酸配列番号288を示す。
【図145】図145は、核酸配列番号289およびアミノ酸配列番号290を示す。
【図146】図146は、核酸配列番号291およびアミノ酸配列番号292を示す。
【図147】図147は、核酸配列番号293およびアミノ酸配列番号294を示す。
【図148】図148は、核酸配列番号295およびアミノ酸配列番号296を示す。
【図149】図149は、核酸配列番号297およびアミノ酸配列番号298を示す。
【図151−1】図151は、配列グループの比較を示す。
【図151−2】図151は、配列グループの比較を示す。
【図151−3】図151は、配列グループの比較を示す。
【図151−4】図151は、配列グループの比較を示す。
【図151−5】図151は、配列グループの比較を示す。
【図151−6】図151は、配列グループの比較を示す。
【図151−7】図151は、配列グループの比較を示す。
【図151−8】図151は、配列グループの比較を示す。
【図151−9】図151は、配列グループの比較を示す。
【図151−10】図151は、配列グループの比較を示す。
【図151−11】図151は、配列グループの比較を示す。
【図152−1】図152は、全長クローンの並列を例示する。
【図152−2】図152は、全長クローンの並列を例示する。
【図152−3】図152は、全長クローンの並列を例示する。
【図152−4】図152は、全長クローンの並列を例示する。
【図152−5】図152は、全長クローンの並列を例示する。
【図153】図153は、PCRによるチトクロムp450 cDNAのクローニングに用いた方法を示す。
【実施例】
【0060】
(実施例1)
植物組織の発育およびエチレン処理
植物栽培
植物の種子を植木鉢に蒔き、そして温室で4週間栽培した。4週齢の苗を個々の植木鉢に移植し、そして温室で2ヶ月栽培した。栽培中、植物に1日2回、150ppm NPK肥料を含有する水を与えた。広がった緑の葉を植物から分離して、以下に記載するエチレン処理を行った。
【0061】
細胞株78379
ケンタッキー大学から譲渡されたバレー種タバコ系統であるタバコ系統78379を、植物材料の供給源として用いた。当該技術分野における標準のように、100の植物を栽培し、そして移植して、そして別個の番号(1〜100)でタグ付けした。推奨されるように、受精およびフィールド管理を行った。
【0062】
100の植物の4分の3が、ニコチンの20〜100%をノルニコチンに変換した。100の植物の4分の1が、ニコチンの5%未満をノルニコチンに変換した。87番の植物が最低の変換を有し(2%)、一方、21番の植物が100%の変換を有した。3%未満を変換する植物を非変換系統と分類した。87番の植物および21番の植物の自家受粉種子とともに交雑(21x87および87x21)種子を作成して、遺伝子相違および表現型相違を研究した。21番の自家受粉由来の植物は変換系統であり、そして87番の自家受粉由来の99%が非変換系統であった。87番由来の植物の残り1%は、低変換(5〜15%)を示した。相互交雑由来の植物は、すべて変換系統であった。
【0063】
細胞株4407
バレー種系統であるタバコ属系統4407を植物材料の供給源として用いた。均質でそして代表的な植物(100)を選択し、そしてタグ付けした。100の植物のうち97が非転換系統であり、そして3つが変換系統であった。56番の植物が最小量の変換を有し(1.2%)、そして58番の植物が最高レベルの変換を有した(96%)。これらの2つの植物を用いて、自家受粉種子および交雑種子を作成した。
【0064】
58番の自家受粉由来の植物は、変換系統対非変換系統比3:1に分かれた。58−33および58−25の植物が、それぞれ、ホモ接合体変換植物系統および非変換植物系統と同定された。次世代の子孫を解析することによって、58−33が安定変換系統であることが確認された。
【0065】
細胞株PBLB01
PBLB01は、ProfiGen, Inc.に開発されたバレー種系統であり、そ
してこれを植物材料の供給源として用いた。PBLB01の原種(foundation)種子から、変換系統植物を選択した。
【0066】
エチレン処理法
温室で2〜3ヶ月栽培した植物から緑の葉を分離し、そして0.3%のエチレン溶液(調製商標、エテフォン(Rhone−Poulenc))をスプレーした。スプレーした葉を各々、加湿器を備えた養生ラックに吊るし、そしてプラスチックで覆った。処理中、試料の葉に定期的にエチレン溶液をスプレーした。エチレン処理のおよそ24〜48時間後、RNA抽出のため、葉を収集した。代謝構成要素解析のため、別の下位試料を採取して、葉の代謝産物、および多様なアルカロイドなどのより具体的な目的の構成要素の濃度を測定した。
【0067】
例えば、以下のようにアルカロイド分析を行うことも可能である。試料(0.1g)を、0.5mlの2N NaOH、並びに内部標準としてのキノリン、およびメチルt−ブチルエーテルを含有する5mlの抽出溶液とともに、150rpmで振盪した。FID検出装置を備えたHP6890GC上で試料を分析した。検出装置および注入装置には、250℃の温度を用いた。1分あたり10℃、110〜185℃の温度勾配で、5%フェノールおよび95%メチルシリコンで架橋した融解石英からなるHPカラム(30m−0.32nm−1m)を用いた。キャリアーガスとしてヘリウムを用いて、100℃、流速1.7cm3/分、40:1の分割比、2・1注入体積で、カラムを操作した。
【0068】
(実施例2)
RNA単離
RNA抽出のため、2ヶ月齢の温室栽培植物の中央部の葉を、記載するようにエチレン処理した。0時間および24〜48時間の試料をRNA抽出に用いた。いくつかの場合、花頭除去10日後の植物から、老化過程にある葉の試料を採取した。これらの試料もまた、抽出に用いた。製造者のプロトコルにしたがって、Rneasy Plant Mini Kit(登録商標)(Qiagen, Inc.、カリフォルニア州バレンシア)を用い、総RNAを単離した。
【0069】
液体窒素下で、DEPC処理した乳鉢および乳棒を用いて、組織試料を細かい粉末にすりつぶした。すりつぶした組織およそ100ミリグラムを、滅菌1.5mlエッペンドルフ試験管に移した。すべての試料を収集し終わるまで、この試料試験管を液体窒素中に入れた。次いで、キットに提供される緩衝液RLT 450μl(メルカプトエタノールを添加したもの)を個々の各試験管に添加した。試料を激しくボルテックスし、そして56℃で3分間インキュベーションした。次いで、2ml収集試験管に入れたQIAshredderTMスピンカラムに溶解物を適用し、そして最大速度で2分間遠心分離した。フロースルーを収集し、そして透明な溶解物に0.5体積のエタノールを添加した。試料をよく混合し、そして2ml収集試験管に入れたRneasy(登録商標)ミニスピンカラムに移した。試料を10,000rpmで1分間遠心分離した。次に、700μlの緩衝液RW1をRneasy(登録商標)カラム上にピペッティングし、そして10,000rpmで1分間遠心分離した。緩衝液RPEを新しい収集試験管中のRneasy(登録商標)カラム上にピペッティングし、そして10,000rpmで1分間遠心分離した。緩衝液RPEを再びRneasy(登録商標)スピンカラム上に添加し、そして最大速度で2分間遠心分離して、膜を乾燥させた。エタノールのキャリー・オーバーをすべて取り除くため、別個の収集試験管中に膜を入れ、そして最大速度でさらに1分間遠心分離した。Rneasy(登録商標)カラムを新しい1.5ml収集試験管に移し、そして40μlのRnase不含水を、Rneasy(登録商標)膜上に直接ピペッティングした。この最終溶出液試験管を10,000rpmで1分間遠心分離した。変性ホルムアルデヒドゲルおよび分光光度計によって、総RNAの品質および量を解析した。
【0070】
製造者のプロトコルにしたがって、OligotexTMポリA+ RNA精製キット(Qiagen Inc.)を用いてポリ(A)RNAを単離した。最大体積250μl中、約200μgの総RNAを用いた。250μlの体積の緩衝液OBBおよび15μlのOligotexTM懸濁物を250μlの総RNAに添加した。ピペッティングによって内容物を完全に混合し、そして加熱ブロック上、70℃で3分間インキュベーションした。次いで、試料をおよそ20分間室温に置いた。最大速度で2分間遠心分離することによって、Oligotex:mRNA複合体をペレットにした。50μlを残してすべての上清を微量遠心管から取り除いた。OBB緩衝液によって試料をさらに処理した。ボルテックスによって、Oligotex:mRNAペレットを400μlの緩衝液OW2に再懸濁した。新しい試験管に入れた小さいスピンカラム上にこの混合物を移して、そして最大速度で1分間遠心分離した。スピンカラムを新しい試験管に移して、そしてさらに400μlの緩衝液OW2をカラムに添加した。次いで、試験管を最大速度で1分間遠心分離した。スピンカラムを最後の1.5ml微量遠心管に移した。60μlの熱い(70℃)緩衝液OEBで試料を溶出した。変性ホルムアルデヒドゲルおよび分光光度解析によって、ポリA産物を解析した。
【0071】
(実施例3)
逆転写−PCR
製造者のプロトコル(Invitrogen、カリフォルニア州カールスバッド)にしたがってSuperScript逆転写酵素を用いて、第一鎖cDNAを産生した。ポリA+濃縮RNA/オリゴdTプライマー混合物は、5μg未満の総RNA、1μlの10mM dNTP混合物、1μlのオリゴd(T)12−18(0.5μg/μl)、および10μlまでのDEPC処理水からなった。各試料を65℃で5分間インキュベーションし、次いで氷上に少なくとも1分間置いた。以下の構成要素の各々を順番に添加することによって、反応混合物を調製した:2μlの10xRT緩衝液、4μlの25mM MgCl2、2μlの0.1M DTT、および1μlのRNase OUT組換えRNase阻害剤。9μlの反応混合物を各RNA/プライマー混合物にピペッティングして添加し、そして穏やかに混合した。これを42℃で2分間インキュベーションし、そして1μlのSuperScript IITM RTを各試験管に添加した。試験管を42℃で50分間インキュベーションした。70℃で15分間処理して反応を終結させ、そして氷上で冷却した。遠心分離によって試料を収集し、そして1μlのRNase Hを各試験管に添加し、そして37℃で20分間インキュベーションした。200pmolの順方向プライマー(図75、配列番号149〜156に示すような縮重プライマー)および100pmolの逆方向プライマー(18ntオリゴd(T)の後に1つのランダム塩基が続く混合物)を用いて、第二のPCRを行った。
【0072】
反応条件は、94℃で2分間であり、そして次いで94℃1分間、45℃〜60℃2分間、72℃3分間の40サイクルのPCRを行い、72℃でさらに10分間伸長を行った。
【0073】
1%アガロースゲルを用いた電気泳動によって、10μlの増幅試料を解析した。アガロースゲルから正しいサイズの断片を精製した。
【0074】
(実施例4)
PCR断片集団の生成
製造者の指示にしたがって、実施例3由来のPCR断片をpGEM−T(登録商標)Easyベクター(Promega、ウィスコンシン州マディソン)に連結した。連結した産物をJM109コンピテント細胞に形質転換し、そしてブルー/ホワイトセレクションのため、LB培地プレート上に蒔いた。コロニーを選択し、そして1.2mlのLB培地
を含む96ウェルプレート中で37℃で一晩増殖させた。すべての選択されるコロニーに関して、凍結ストックを生成した。Wizard SV Miniprep(登録商標)キット(Promega)とともに、BeckmanのBiomeck 2000ミニプレップ・ロボットを用いて、プレートからプラスミドDNAを精製した。100μlの水でプラスミドDNAを溶出し、そして96ウェルプレート中に保存した。EcoR1でプラスミドを消化し、そして1%アガロースゲルを用いて解析して、DNA量および挿入物のサイズを確認した。CEQ 2000配列決定装置(Beckman、カリフォルニア州フラートン)を用いて、400〜600bpの挿入物を含有するプラスミドの配列を決定した。BLAST検索によって、GenBankデータベースと該配列を並列した。p450関連断片を同定し、そしてさらに解析した。あるいは、サブトラクション・ライブラリーからp450断片を単離した。これらの断片もまた、上述のように解析した。
【0075】
(実施例5)
cDNAライブラリーの構築
以下のように、エチレン処理した葉から、総RNAを調製することによって、cDNAライブラリーを構築した。まず、修飾した酸フェノールおよびクロロホルム抽出プロトコルを用いて、タバコ系統58−33のエチレン処理した葉から、総RNAを抽出した。1グラムの組織を用いて、これをすりつぶし、そして続いて5mlの抽出緩衝液(100mM Tris−HCl、pH8.5;200mM NaCl;10mM EDTA;0.5%SDS)中でボルテックスし、これに5mlのフェノール(pH5.5)および5mlのクロロホルムを添加するように、プロトコルを修飾した。抽出した試料を遠心分離し、そして上清を取り置いた。上清が透明に見えるようになるまで、この抽出工程をさらに2〜3回反復した。およそ5mlのクロロホルムを添加して、微量のフェノールを取り除いた。3倍体積のEtOHおよび1/10体積の3M NaOAc(pH5.2)を添加し、そして−20℃に1時間保存することによって、合わせた上清分画からRNAを沈殿させた。Corexガラス容器に移した後、RNA分画を、4℃、9,000RPMで45分間遠心分離した。ペレットを70%エタノールで洗浄し、そして4℃、9,000RPMで5分間回転させた。ペレットを乾燥させた後、ペレットにしたRNAを0.5ml
RNase不含水に溶解した。ペレットにしたRNAを0.5ml RNase不含水に溶解した。変性ホルムアルデヒドゲルおよび分光光度計によって、それぞれ、総RNAの品質および量を解析した。
【0076】
以下のプロトコルによって、オリゴ(dT)セルロース・プロトコル(Invitrogen)および微量遠心分離装置スピンカラム(Invitrogen)を用いて、ポリA+ RNAに関して、生じた総RNAを単離した。およそ20mgの総RNAを、2回の精製に供して、高品質のポリA+ RNAを得た。mRNAが高品質であることを確実にするため、変性ホルムアルデヒドゲル、そして続いて既知の全長遺伝子のRT−PCRを行うことによって、ポリA+ RNA産物を解析した。
【0077】
次に、ポリA+ RNAをテンプレートとして用いて、cDNA合成キット、ZAP−cDNA(登録商標)合成キット、およびZAP−cDNA(登録商標)Gigapack(登録商標)IIIゴールド・クローニングキット(Stratagene、カリフォルニア州ラホヤ)を使用して、cDNAライブラリーを産生した。方法は、明記するような以下の製造者のプロトコルを含んだ。およそ8μgのポリA+ RNAを用いて、cDNAライブラリーを構築した。一次ライブラリーの解析によって、約2.5x106〜1x107pfuであることが明らかになった。IPTGおよびX−galを用いた補完アッセイによって、ライブラリーの品質バックグラウンド試験を完了し、ここで組換えプラークは、バックグラウンド反応より100倍を超えて発現していた。
【0078】
ランダムPCRによる、ライブラリーのより定量的な解析によって、挿入cDNAの平
均サイズがおよそ1.2kbであることが示された。該方法は、以下のような2工程PCR法を用いた。第一の工程のため、p450断片から得た予備的配列情報に基づいて、逆プライマーを設計した。設計した逆方向プライマーおよびT3(順方向)プライマーを用いて、cDNAライブラリーから、対応する遺伝子を増幅した。PCR反応をアガロース電気泳動に供して、そして高分子量の対応するバンドを切り出し、精製し、クローニングし、そして配列決定した。第二の工程において、p450の5’UTRまたは開始コード領域から順方向プライマーとして設計した新規プライマーを、逆方向プライマー(p450の3’UTRから設計)とともに、続くPCRで用いて、全長p450クローンを得た。
【0079】
逆方向プライマーを除いて、実施例3に記載するように構築したcDNAライブラリーから、PCR増幅によって、p450断片を生成した。プラスミド上でcDNA挿入物(図75を参照されたい)の下流に位置するT7プライマーを逆方向プライマーとして用いた。実施例4に記載するように、PCR断片を単離し、クローニングし、そして配列決定した。
【0080】
構築したcDNAライブラリーから、PCR法によって、全長p450遺伝子を単離した。遺伝子特異的逆方向プライマー(p450断片の下流配列から設計)および順方向プライマー(ライブラリープラスミド上のT3)を用いて、全長遺伝子をクローニングした。PCR断片を単離し、クローニングし、そして配列決定した。必要であれば、第二の工程のPCRを適用した。第二の工程において、クローニングしたp450の5’UTRから設計した新規順方向プライマーとともに、p450クローンの3’UTRから設計した逆方向プライマーを、続くPCR反応で用いて、全長p450クローンを得た。続いてクローンの配列を決定した。
【0081】
(実施例6)
クローニングした断片の性質決定−逆サザンブロッティング解析
上記実施例で同定したp450クローンすべてに対して、非放射性大規模逆サザンブロッティングアッセイを行って、示差発現を検出した。異なるp450クラスターの間で発現レベルが非常に異なることが観察された。高発現のものに対して、さらなるリアルタイム検出を行った。
【0082】
非放射性サザンブロッティング法を以下のように行った。
1)実施例2に記載するように、Qiagen Rnaeasyキットを用いて、エチレン処理および非処理変換系統(58−33)および非変換系統(58−25)の葉から総RNAを抽出した。
【0083】
2)上記工程で生成したポリA+濃縮RNA由来の一本鎖cDNAをビオチン−テール標識することによって、プローブを産生した。ビオチン化オリゴdTをプライマーとして(Promega)用いることを除いて、実施例3に記載するように、変換系統および非変換系統総RNAのRT−PCR(Invitrogen)によって、この標識一本鎖cDNAを生成した。これらをプローブとして用いて、クローニングしたDNAとハイブリダイズさせた。
【0084】
3)プラスミドDNAを制限酵素EcoR1で消化して、そしてアガロースゲル上で泳動した。ゲルを乾燥させて、そして同時に2つのナイロン膜にトランスファーした(Biodyne B(登録商標))。一方の膜を変換系統プローブとハイブリダイズさせ、そして他方を非変換系統プローブとハイブリダイズさせた。ハイブリダイゼーション前に、膜をUV架橋した(自動架橋セッティング、254nm、Stratagene、Stratalinker)。
【0085】
あるいは、p−GEMプラスミドの両方のアームに位置する配列、T3およびSP6をプライマーとして用いて、各プラスミドから挿入物をPCR増幅した。96ウェルReady−to−runアガロースゲル上で泳動することによって、PCR産物を解析した。確認した挿入物を2つのナイロン膜上にドットした。一方の膜を変換系統プローブとハイブリダイズさせ、そして他方を非変換プローブとハイブリダイズさせた。
【0086】
4)洗浄ストリンジェンシーを修飾し、製造者の指示にしたがって、膜をハイブリダイズさせ、そして洗浄した(Enzo MaxSenceTMキット、Enzo Diagnostics, Inc、ニューヨーク州ファーミンデール)。膜を、ハイブリダイゼーション緩衝液(界面活性剤およびハイブリダイゼーション増進剤を含有する、2xSSC緩衝ホルムアミド)と42℃で30分間プレハイブリダイズさせ、そして10μl変性プローブと42℃で一晩ハイブリダイズさせた。次いで、膜を1xハイブリダイゼーション洗浄緩衝液で、室温で10分間1回洗浄し、そして68℃で15分間4回洗浄した。膜の検出準備が出来た。
【0087】
5)製造者の検出法に記載されるように(Enzo Diagnostics, Inc.)、アルカリホスファターゼ標識、その後、NBT/BCIP比色検出によって、洗浄した膜を検出した。1xブロッキング溶液を用いて、膜を室温で1時間ブロッキングし、1x検出試薬で10分間3回洗浄し、1x現像前反応緩衝液で5分間2回洗浄し、そして次いで、ドットが現れるまで、現像溶液中でブロットを30〜45分間現像した。すべての試薬は、製造者(Enzo Diagnostics, Inc)に提供された。さらに、製造者の指示にしたがって、KPLサザンハイブリダイゼーションおよび検出キットTM(KPL、メリーランド州ガイザーズバーグ)を用いてもまた、大規模逆サザンアッセイを行った。
【0088】
(実施例7)
クローンの性質決定−ノーザンブロット解析
サザンブロット解析の代わりに、ノーザンブロッティングアッセイの例に記載されるように、いくつかの膜をハイブリダイズさせ、そして検出した。以下のように、ノーザンハイブリダイゼーションを用いて、タバコ属において、示差発現されるmRNAを検出した。
【0089】
ランダムプライミング法を用いて、クローニングしたp450からプローブを調製した(MegaprimeTM DNA標識系、Amersham Biosciences)。
【0090】
以下の構成要素を混合した:25ng変性DNAテンプレート;各4μlの非標識dTTP、dGTPおよびdCTP;5μlの反応緩衝液;P32−標識dATPおよび2μlのクレノウI;並びに反応を50μlにするH2O。混合物を37℃で1〜4時間インキュベーションし、次いで2μlの0.5M EDTAで反応を停止した。使用前にプローブを95℃で5分間インキュベーションすることによって、変性させた。
【0091】
いくつかの対のタバコ系統の新鮮なエチレン処理葉および非処理葉から、RNA試料を調製した。いくつかの場合、ポリA+濃縮RNAを用いた。およそ15μgの総RNAまたは1.8μgのmRNA(実施例5に記載するようなRNAおよびmRNA抽出法)をDEPC H2O(5〜10μl)で等体積にした。同体積の装填緩衝液(1xMOPS;18.5%ホルムアルデヒド;50%ホルムアミド;4% Ficoll400;ブロモフェノールブルー)および0.5μl EtBr(0.5μg/μl)を添加した。続いて、電気泳動によってRNAを分離するため、調製物中の試料を変性させた。
【0092】
1xMOP緩衝液(0.4Mモルホリノプロパンスルホン酸;0.1M Na−酢酸−3xH2O;10mM EDTA;NaOHでpH7.2に調整)を用いて、ホルムアルデヒドゲル(1%アガロース、1xMOPS、0.6Mホルムアルデヒド)上で、試料を電気泳動に供した。10xSSC緩衝液(1.5M NaCl;0.15Mクエン酸ナトリウム)中で24時間キャピラリー法を行うことによって、RNAをHybond−N+膜(ナイロン、Amersham Pharmacia Biotech)にトランスファーした。RNA試料を含む膜を、ハイブリダイゼーション前にUV架橋した(自動架橋セッティング、254nm、Stratagene、Stratalinker)。
【0093】
膜を、5〜10mlのプレハイブリダイゼーション緩衝液(5xSSC;50%ホルムアミド;5xデンハルト溶液;1% SDS;100μg/ml熱変性剪断非相同DNA)と42℃で1〜4時間プレハイブリダイズさせた。古いプレハイブリダイゼーション緩衝液を廃棄し、そして新しいプレハイブリダイゼーション緩衝液およびプローブを添加した。ハイブリダイゼーションを42℃で一晩行った。2xSSCで膜を室温で15分間洗浄し、次いで2xSSCで洗浄した。
【0094】
本発明の主要な焦点は、エチレン処理の結果誘導可能であるか、またはタバコ葉の品質および構成要素に重要な役割を果たすことも可能な、新規遺伝子の発見であった。以下の表に例示するように、ノーザンブロットおよび逆サザンブロットは、非誘導植物に比較して、どの遺伝子がエチレン処理によって誘導されたのかを決定するのに有用であった。興味深いことに、変換系統および非変換系統で、すべての断片が同様に影響を受けるのではなかった。注目されるチトクロムp450断片を部分的に配列決定して、構造的関連性を決定した。この情報を用いて、注目される全長遺伝子クローンを続いて単離し、そして性質決定した。
【0095】
【表1】
【0096】
エチレン処理で誘導した、変換および非変換バレー種系統から得たタバコ組織に対して、全長クローンを用いて、ノーザン解析を行った。目的は、エチレン誘導非変換バレー種系統に比較して、エチレン誘導変換系統に比較して、エチレン誘導変換系統で発現上昇を示す全長クローンを同定することであった。そうすることによって、変換系統および非変換系統間の葉構成要素の生化学的相違を比較することにより、全長クローンの機能的関係を決定することも可能である。以下の表に示すように、+で示す非変換系統エチレン処理
組織のものより、変換系統エチレン処理組織において、++および+++で示すように、6つのクローンが有意により高い発現を示した。これらのクローンはすべて、エチレン処理しない変換系統および非変換系統で、ほとんどまたはまったく発現を示さなかった。
【0097】
【表2】
【0098】
(実施例8)
クローニングした遺伝子にコードされるp450の免疫検出
3つのp450クローンから、1)他のクローンに対してより低い相同性を有するかまたはまったく相同性を持たず、そして2)優れた親水性および抗原性を有する、長さ20〜22アミノ酸に対応するペプチド領域を選択した。それぞれのp450クローンから選択したペプチド領域のアミノ酸配列を以下に列挙する。合成したペプチドをKHLとコンジュゲート化し、そして次いでウサギに注射した。第4回の注射の2週間後および4週間後に抗血清を収集した(Alpha Diagnostic Intl. Inc.、テキサス州サンアントニオ)。
【0099】
D234−AD1 DIDGSKSKLVKAHRKIDEILG
D90a−BB3 RDAFREKETFDENDVEELNY
D89−AB1 FKNNGDEDRHFSQKLGDLADKY
ウェスタンブロット解析によって、タバコ植物組織由来のタンパク質を標的とする交差反応性に関して、抗血清を調べた。エチレン処理した(0〜40時間)、変換系統および非変換系統の中央部の葉から、未精製タンパク質抽出物を得た。製造者のプロトコルにしたがって、RC DCタンパク質アッセイキット(BIO−RAD)を用いて、抽出物のタンパク質濃度を測定した。
【0100】
各レーンに2マイクログラムのタンパク質を装填し、そしてLaemmli SDS−PAGE系を用いて、10%〜20%勾配ゲル上でタンパク質を分離した。Trans−Blot(登録商標)セミドライセル(BIO−RAD)を用いて、PROTRAN(登録商標)ニトロセルロース・トランスファー膜(Schleicher & Schuell)にゲルからタンパク質をトランスファーした。ECL AdvanceTMウェスタンブロッティング検出キット(Amersham Biosciences)を用いて、標的p450タンパク質を検出し、そして視覚化した。ウサギにおいて、合成KLHコンジュゲートに対する一次抗体を作成した。ペルオキシダーゼとカップリングした、ウサギIgGに対する二次抗体をSigmaから購入した。一次抗体および二次抗体のどちらも、1:1000希釈で用いた。抗体は、ウェスタンブロット上で、単一のバンドに強い反応性を示し、抗血清が、目的の標的ペプチドに単一特異的であることを示した。抗血清はまた、KLHにコンジュゲート化した合成ペプチドとも交差反応性であった。
【0101】
(実施例9)
単離核酸断片の核酸同一性および構造関連性
ノーザンブロット解析と組み合わせて、クローニングした100を超えるp450断片を配列決定して、構造的関連性を決定した。用いたアプローチは、p450遺伝子のカルボキシル末端近くに位置する2つの一般的なp450モチーフのいずれかに基づく順方向プライマーを利用した。順方向プライマーは、図1に示すような、チトクロムp450モチーフFXPERFまたはGRRXCP(A/G)に相当した。逆方向プライマーは、プラスミド、pGEMTMプラスミドの両方のアーム上に位置するSP6またはT7、あるいはポリAテールのいずれか由来の、標準的プライマーを用いた。用いたプロトコルを以下に記載する。
【0102】
分光光度測定を用い、製造者のプロトコル(Beckman Coulter)にしたがって、出発二本鎖DNAの濃度を概算した。テンプレートを水で適切な濃度に希釈し、95℃で2分間加熱し、そして続いて氷上に置くことによって変性させた。0.5〜10μlの変性DNAテンプレート、2μlの1.6pmol順方向プライマー、8μlのDTCSクイックスタートマスターミックス、および総体積を20μlにする水を用いて、配列決定反応物を氷上で調製した。熱周期プログラムは、30サイクルの以下の周期からなった:96℃20秒間、50℃20秒間、および60℃4分間、その後、4℃で維持。
【0103】
5μlの反応停止緩衝液(等体積の3M NaOAcおよび100mM EDTA、並びに1μlの20mg/mlグリコーゲン)を添加することによって、配列決定反応を停止した。60μlの冷95%エタノールで試料を沈殿させ、そして6000gで6分間遠心分離した。エタノールを廃棄した。ペレットを200μlの冷70%エタノールで2回洗浄した。ペレットを乾燥させた後、40μlのSLS溶液を添加し、そしてペレットを再懸濁した。ミネラルオイルの層を重層した。次いで、さらに解析するため、試料をCEQ 8000自動化配列決定装置に入れた。
【0104】
核酸配列を検証するため、p450遺伝子のFXPERFまたはGRRXCP(A/G)領域に対する順方向プライマー、あるいはプラスミドまたはポリAテールいずれかに対する逆方向プライマーを用いて、核酸配列を、両方向に再配列決定した。すべての配列決定を、両方向に、少なくとも2回行った。
【0105】
チトクロムp450断片の核酸配列を、GRRXCP(A/G)モチーフをコードする領域の後の最初の核酸から停止コドンまでに対応するコード領域で、互いに比較した。この領域を、p450タンパク質間の遺伝子多様性の指標として選択した。他の植物種と同様、多数の遺伝的に別個のp450遺伝子が70遺伝子を超えて観察された。核酸配列の比較に際して、配列同一性に基づいて、遺伝子を別個の配列グループに入れることが可能であることが見出された。p450メンバーの最適なユニークなグループ分けが、75%以上の核酸同一性を持つ配列であると決定されることが見出された(表Iに示す)。同一性パーセントを減少させると、有意により大きいグループとなった。81%以上の核酸同一性を持つ配列に関して、好ましいグループ分けが観察され、より好ましいグループ分けは91%以上の核酸同一性のものであり、そして最も好ましいグループ分けは99%以上の核酸同一性の配列であった。グループの大部分は少なくとも2つのメンバーを含有し、そしてしばしば3以上のメンバーを含有した。他のものは反復して発見されず、ここで取ったアプローチが、用いた組織で低発現されるmRNAおよび高発現されるmRNAの両方を単離可能であったことが示唆される。
【0106】
75%以上の核酸同一性に基づいて、2つのチトクロムp450グループが、グループ内にあるものとは遺伝的に異なる、先のタバコ・チトクロム遺伝子に核酸配列同一性を含有することが見出された。グループ23は、表Iに用いたパラメーター内で、それぞれCzernicらおよびRalstonらによる、GI:1171579(CAA64635)およびGI:14423327(またはAAK62346)の先のGenBank配列に、核酸同一性を示した。GI:1171579は、グループ23のメンバーに、96.9%〜99.5%の範囲の核酸同一性を示し、一方、GI:14423327は、このグループに、95.4%〜96.9%の範囲の同一性を有した。グループ31のメンバーは、RalstonらによるGI:14423319(AAK62342)のGenBankに報告された配列に、76.7%〜97.8%の範囲の核酸同一性を示した。表1の他のp450同一性グループはいずれも、Ralstonら、Czernicら、Wangら、またはLaRosaおよびSmigockiに報告されるタバコ属p450遺伝子に、表1で用いたようなパラメーター同一性を含有しなかった。
【0107】
図76に示すように、グループに関して、適切な核酸縮重プローブとともにコンセンサス配列を得て、タバコ属植物からの各グループのさらなるメンバーを優先的に同定し、そして単離することも可能である。
【0108】
【表3−1】
【0109】
【表3−2】
【0110】
(実施例10)
単離核酸断片の関連アミノ酸配列同一性
実施例8由来のチトクロムp450断片に関して得た核酸配列のアミノ酸配列を推定した。推定した領域は、GXRXCP(A/G)配列モチーフの直後のアミノ酸からカルボキシル末端の終わり、または停止コドンまでに相当した。断片の配列同一性の比較に際して、70%以上のアミノ酸同一性を持つ配列に関して、ユニークなグループ分けが観察された。80%以上のアミノ酸同一性を持つ配列に関して、好ましいグループ分けが観察され、より好ましいグループ分けは90%以上のアミノ酸同一性のものであり、そして最も好ましいグループ分けは99%以上のアミノ酸同一性の配列であった。グループおよびグループメンバーの対応するアミノ酸配列を図2に示す。ユニークな核酸配列のいくつかは、他の断片と完全なアミノ酸同一性を有することが見出され、そしてしたがって、同一アミノ酸を持つ1つのメンバーのみが報告された。
【0111】
表IIのグループ19のアミノ酸同一性は、核酸配列に基づいて、3つの別個のグループに対応した。各グループメンバーのアミノ酸配列およびその同一性を図77に示す。アミノ酸相違に適切に印を付ける。
【0112】
遺伝子クローニングおよび植物を用いた機能研究のため、各アミノ酸同一性グループの少なくとも1つのメンバーを選択した。さらに、ノーザンおよびサザン解析によって評価されるように、エチレン処理または他の生物学的相違によって異なって影響を受けるグループメンバーを、遺伝子クローニングおよび機能研究のため、選択した。遺伝子クローニング、発現研究および全植物評価を補助するため、配列同一性および示差配列に関して、ペプチド特異的抗体が調製されるであろう。
【0113】
【表4−1】
【0114】
【表4−2】
【0115】
(実施例11)
全長クローンの関連アミノ酸配列同一性
実施例5でクローニングした全長タバコ属遺伝子の核酸配列を、全アミノ酸配列に関して推定した。チトクロムp450遺伝子は、3つの保存されるp450ドメインモチーフの存在によって同定され、該モチーフは、カルボキシル末端のUXXRXXZ、PXRFXFまたはGXRXC、ここでUはEまたはKであり、Xはアミノ酸いずれかであり、そしてZはP、T、SまたはMである、に対応した。クローンのうち2つ、D130−AA1およびD101−BA2は、ほぼ完全であるようだったが、適切な停止コドンを欠き、しかしどちらも3つのp450チトクロムドメインすべてを含有した。BLASTプログラムを用いて、全長配列を互いに、そして既知のタバコ遺伝子に比較して、p450遺伝子すべてをアミノ酸同一性に関して性質決定した。該プログラムは、NCBI特別BLASTツール(2配列並列(b12seq)、http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html)を用いた。核酸配列に関してはフィルターをかけずにBLASTNで、そしてアミノ酸配列に関してはBLASTPで、2つの配列を並列させた。アミノ酸同一性パーセントに基づいて、各配列を同一性グループにグループ分けし、グループ分けは、別のメンバーと少なくとも85%の同一性を共有するメンバーを含有した。90%以上のアミノ酸同一性を持つ配列に関して、好ましいグループ分けが観察され、より好ましいグループ分けは95%以上のアミノ酸同一性のものであり、そして最も好ましいグループ分けは99%以上のアミノ酸同一性の配列を有した。これらの基準を用いて、25のユニークなグループが同定され、そしてこれらを表IIIに示す。
【0116】
アミノ酸同一性に関する表IIIに用いたパラメーター内で、3つのグループが、既知のタバコ遺伝子に対して85%以上の同一性を含有することが見出された。グループ5のメンバーは、RalstonらによるGI:14423327(またはAAK62346)の先のGenBank配列に対して、全長配列に関して最大96%のアミノ酸同一性を有した。グループ23は、RalstonらによるGI:14423328(またはAAK62347)に対して、最大93%のアミノ酸同一性を有し、そしてグループ24は、RalstonらによるGI:14423318(またはAAK62343)に対して、92%の同一性を有した。
【0117】
【表5−1】
【0118】
【表5−2】
【0119】
カルボキシル末端近くのUXXRXXZ p450ドメインおよびGXRXC p450ドメインの間の非常に保存されるアミノ酸相同性に基づいて、全長遺伝子をさらにグループ分けした。図3に示すように、互いに比較して、保存されるドメイン間の配列相同性に関して、個々のクローンを並列させ、そして別個の同一性グループに入れた。いくつかの場合で、クローンの核酸配列はユニークであったが、該領域のアミノ酸配列は同一であった。90%以上のアミノ酸同一性を持つ配列に関して、好ましいグループ分けが観察され、より好ましいグループは95%以上のアミノ酸同一性を有し、そして最も好ましいグループ分けは99%以上のアミノ酸同一性の配列を有した。最後のグループ分けは、クローンの全アミノ酸配列に関する同一性パーセントに基づくものと同様であったが、(表IIIの)グループ17だけは例外であり、このグループは2つの別個のグループに分けられた。
【0120】
表IVのアミノ酸同一性に関して用いたパラメーター内で、3つのグループが、既知のタバコ属遺伝子に対して90%以上の同一性を含有することが見出された。グループ5のメンバーは、RalstonらによるGI:14423326(AAK62346)の先のGenBank配列に対して、全長配列に関して最大93.4%のアミノ酸同一性を有した。グループ23は、RalstonらによるGI:14423328(またはAAK62347)に対して、最大91.8%のアミノ酸同一性を有し、そしてグループ24は、RalstonらによるGI:14423318(またはAAK62342)に対して、98.8%の同一性を有した。
【0121】
【表6−1】
【0122】
【表6−2】
【0123】
(実施例12)
1以上のタバコ・チトクロムp450特異的ドメインを欠く、タバコ属チトクロムp450クローン
4つのクローンは、表IIIに報告する他のタバコ・チトクロム遺伝子に、90%〜99%の核酸相同性の範囲の、高い核酸相同性を有した。4つのクローンには、D136−AD5、D138−AD12、D243−AB3およびD250−AC11が含まれた。しかし、ヌクレオチド・フレームシフトのため、これらの遺伝子は、3つのC末端チトクロムp450ドメインの1以上を含有せず、そして表IIIまたは表IVに示す同一性グループからは除外された。
【0124】
1つのクローン、D95−AG1のアミノ酸同一性は、表IIIまたは表IVでp450タバコ遺伝子をグループ分けするのに用いた、第三のドメイン、GXRXCを含有しなかった。このクローンの核酸相同性は、他のタバコ・チトクロム遺伝子に低い相同性を有した。このクローンは、タバコ属のチトクロムp450遺伝子の新規のそして異なるグループに相当する。
【0125】
(実施例13)
タバコ特性の制御改変におけるタバコ属チトクロムp450断片およびクローンの使用
タバコp450核酸断片または全遺伝子の使用は、改変されたタバコ表現型またはタバコ構成要素、そしてより重要なことに、改変された代謝産物を有する植物を同定し、そして選択する際に有用である。下方制御のため、例えばアンチセンス方向で、または過剰発現のため、例えばセンス方向で、本明細書に報告するものから選択される核酸断片または全長遺伝子を取り込む、多様な形質転換系によって、トランスジェニック・タバコ植物を生成する。全長遺伝子を過剰発現するため、特定の酵素の発現を増加させるのに有効であり、そしてしたがってタバコ属内で表現型効果を生じる、本発明に記載する全長遺伝子のすべてまたは機能する部分またはアミノ酸配列をコードする核酸配列いずれかが望ましい。一連の戻し交雑を通じて、ホモ接合体系統であるタバコ属系統を得て、そして一般の当業者に一般的に利用可能な技術を用いて、限定されるわけではないが、内因性p450 RNA、転写物、p450発現ペプチドおよび植物代謝産物の濃度の解析を含む、表現型変化に関して評価する。タバコ植物で示される変化は、目的の選択される遺伝子の機能的役割に関する情報を提供するか、または好ましいタバコ属植物種として有用性を有する。
【0126】
(実施例14)
エチレン処理した変換系統で誘導される遺伝子の同定
遺伝子発現の定量的で高度に並行の測定のために、高密度オリゴヌクレオチドアレイ技
術であるAffymetrix GeneChip(登録商標)(Affymetrix
Inc.、カリフォルニア州サンタクララ)アレイを用いた。この技術を用いる際、固体表面上でオリゴヌクレオチドを直接合成することによって、核酸アレイを組み立てた。この固相化学反応は、GeneChip(登録商標)と称されるチップ上に、非常に高密度に充填された数十万のオリゴヌクレオチドプローブを含有するアレイを産生可能である。単一のハイブリダイゼーションから、数千の遺伝子を同時にスクリーニングすることも可能である。各遺伝子は、典型的には、サイズに応じて、11〜25対のプローブの組によって表される。感度、特異性、および再現性を最大にするようにプローブを設計して、特異的シグナルおよびバックグラウンドシグナル間、並びに緊密に関連する標的配列間の一貫した区別を可能にする。
【0127】
Affymetrix GeneChipハイブリダイゼーション実験は、以下の工程を伴う:アレイの設計および産生、生物学的標本から単離したRNAからの蛍光標識標的の調製、GeneChipへの標識した標的のハイブリダイゼーション、アレイのスクリーニング、並びにスキャンした画像の解析および遺伝子発現プロフィールの生成。
【0128】
A. Affymetrix GeneChipの設計およびオーダーメード(custom making)
GeneChip CustomExpress Advantageアレイは、Affymetrix Inc.(カリフォルニア州サンタクララ)によってオーダーメードされた。チップサイズは18ミクロンであり、そしてアレイ形式は100〜2187であって、528のプローブセット(11,628プローブ)に適応可能である。GenBank由来核酸配列を除いて、我々が先に同定したタバコクローンからすべての配列を選択し、そしてプローブはすべてあつらえて設計した。総数400のタバコ遺伝子または断片がGeneChip上に含まれるように選択した。選択したオリゴヌクレオチドの配列は、遺伝子の3’端のユニークな領域に基づいた。選択した核酸配列は、(特許出願)に記載される、タバコからクローニングされた、56の全長p450遺伝子および71のp450断片からなった。他のタバコ配列には、Clontech SSHキット(BD Biosciences、カリフォルニア州パロアルト)を用いた抑制サブトラクション・ライブラリーから生成した、270のタバコESTが含まれた。これらの遺伝子の中で、GenBankに列挙されるチトクロムP450遺伝子から、いくつかのオリゴヌクレオチド配列を選択した。各全長遺伝子に関して、最大25のプローブを用い、そして各断片に関して11のプローブを用いた。いくつかのクローンに関しては、ユニークな高品質のプローブがないため、減少した数のプローブを用いた。適切な対照配列もまた、GeneChip(登録商標)上に含んだ。
【0129】
プローブアレイは、25量体オリゴヌクレオチドであり、半導体に基づくフォトリソグラフィーおよび固相化学合成技術の組み合わせによって、ガラス・ウェハー上に直接合成された。各アレイは、最大100,000の異なるオリゴヌクレオチドプローブを含有した。オリゴヌクレオチドプローブは、アレイ上の既知の位置で合成されるため、Affymetrix Microarray Suite(登録商標)ソフトウェアによって、ハイブリダイゼーションパターンおよびシグナル強度を、遺伝子同一性および相対発現レベルに関して解釈することも可能である。各プローブ対は、完全マッチオリゴヌクレオチドおよびミスマッチオリゴヌクレオチドからなる。完全マッチプローブは、特定の遺伝子に正確に相補的な配列を有し、そしてしたがって遺伝子の発現を測定する。ミスマッチプローブは、中央の塩基位での単一塩基置換によって、完全マッチプローブとは異なり、この塩基置換が標的遺伝子転写物の結合を妨げる。ミスマッチは、非特異的ハイブリダイゼーションシグナルまたはバックグラウンドシグナルを生じ、これを、完全マッチオリゴヌクレオチドに関して測定されるシグナルに比較した。
【0130】
B.試料調製
Genome Explorations, Inc.(テネシー州メンフィス)によって、ハイブリダイゼーション実験を行った。ハイブリダイゼーションで用いるRNA試料は、エチレン処理によって誘導される、6対の非変換/変換同系系統からなった。試料には、1対の4407−25/4407−33非処理バレー種タバコ試料、3対のエチレン処理4407−25/4407−33試料、1対のエチレン処理した黒(dark)タバコNL Madole/181および1対のエチレン処理バレー品種PBLB01/178が含まれた。エチレン処理は、実施例1に記載するとおりであった。
【0131】
修飾した酸フェノールおよびクロロホルム抽出プロトコルを用いて、上述のエチレン処理葉および非処理葉から、総RNAを抽出した。1グラムの組織を用いて、これをすりつぶし、そして続いて5mlの抽出緩衝液(100mM Tris−HCl、pH8.5;200mM NaCl;10mM EDTA;0.5%SDS)中でボルテックスし、これに5mlのフェノール(pH5.5)および5mlのクロロホルムを添加するように、プロトコルを修飾した。抽出した試料を遠心分離し、そして上清を取り置いた。上清が透明に見えるようになるまで、この抽出工程をさらに2〜3回反復した。およそ5mlのクロロホルムを添加して、微量のフェノールを取り除いた。3倍体積のEtOHおよび1/10体積の3M NaOAc(pH5.2)を添加し、そして−20℃に1時間保存することによって、合わせた上清分画からRNAを沈殿させた。Corexガラス容器に移した後、RNA分画を、4℃、9,000RPMで45分間遠心分離した。ペレットを70%エタノールで洗浄し、そして4℃、9,000RPMで5分間回転させた。ペレットを乾燥させた後、ペレットにしたRNAを0.5ml RNase不含水に溶解した。ペレットにしたRNAを0.5ml RNase不含水に溶解した。変性ホルムアルデヒドゲルおよび分光光度計によって、それぞれ、総RNAの品質および量を解析した。3〜5μg/μlの総RNA試料をGenome Explorations, Inc.に送ってハイブリダイゼーションを行った。
【0132】
C.ハイブリダイゼーション、検出およびデータ・アウトプット
標識cRNA物質の調製を以下のように行った。製造者の指示にしたがって、SuperScript二本鎖cDNA合成キット(Gibco Life Technologies)およびオリゴ−dT24−T7(5’− GGC CAG TGA ATT GTA ATA CGA CTC ACT ATA GGG AGG CGG−3’)プライマーを用いて、5〜15μgの総RNAから、第一鎖および第二鎖cDNAを合成した。
【0133】
T7プロモーターがカップリングした二本鎖cDNAをテンプレートとして、そしてT7 RNA転写物標識キット(ENZO Diagnostics Inc.)を用いた、in vitro転写によって、cRNAを合成し、そして同時にビオチン化UTPおよびCTPで標識化した。簡潔には、先の工程から合成した二本鎖cDNAを70%エタノールで2回洗浄し、そして22μlのRnase不含H2Oに再懸濁した。cDNAを4μlの10x各反応緩衝液、ビオチン標識リボヌクレオチド、DTT、Rnase阻害剤混合物、および2μlの20xT7 RNAポリメラーゼと37℃で5時間インキュベーションした。CHROMA SPIN−100カラム(Clontech)を通過させ、そして−20℃で1時間〜一晩沈殿させることによって、取り込まれていないリボヌクレオチドから、標識cRNAを分離した。
【0134】
オリゴヌクレオチドアレイ・ハイブリダイゼーションおよび解析を以下のように行った。cRNAペレットを10μlのRnase不含H2Oに再懸濁し、そして200mM Tris−酢酸、pH8.1、500mM KOAc、150mM MgOAc中で95℃で35分間、熱およびイオンが仲介する加水分解によって、10.0μgを断片化した
。断片化したcRNAを、〜12,500のアノテートした全長遺伝子とともに、EST配列に相当するように設計されたさらなるプローブセットを含有する、HG U95Av2オリゴヌクレオチドアレイ(Affymetrix)に45℃で16時間ハイブリダイズさせた。6xSSPE(0.9M NaCl、60mM NaH2PO4、6mM EDTA+0.01% Tween20)を用いてアレイを25℃で洗浄し、次いで100mM MES、0.1M[Na+]、0.01% Tween20を用いて、50℃でストリンジェントな洗浄を行った。アレイをフィコエリトリン・コンジュゲート化ストレプトアビジン(Molecular Probes)で染色し、そしてレーザー共焦点スキャナー(Hewlett−Packard)を用いて、蛍光強度を測定した。Microarrayソフトウェア(Affymetrix)を用いて、スキャンした画像を解析した。用いたすべてのアレイに関して、アレイ上のすべての遺伝子の蛍光強度の平均を、一定の標的強度(250)にスケーリングすることによって、試料装填および染色における変動を標準化した。ユーザー指針にしたがって、Microarray Suite 5.0(Affymetrix)を用いて、データ解析を行った。[Σ(PM−MM)/(プローブ対の数)]、式中、PMおよびMMは完全マッチおよび不完全マッチを示す、に表される、平均強度相違として各遺伝子のシグナル強度を計算した。
【0135】
D.データ解析および結果
Genome Explorationsの検出装置を用いて生成した発現レポートに立証されるように、12組のハイブリダイゼーションに成功した。レポートの主なパラメーターには、ノイズ、スケール係数、バックグラウンド、総プローブセット、存在および非存在プローブセットの数およびパーセンテージ、ハウスキーピング対照のシグナル強度が含まれた。続いて、他のMicrosoftソフトウェアと組み合わせてソフトフェアGCOSを用いて、データを解析し、そして提示した。処理対間のシグナル比較を解析した。複製を含む、異なる処理各々の遺伝子および断片に対応するそれぞれのプローブすべてに関する全体のデータをコンパイルし、そしてコンパイルした発現データ、例えば変化のコールおよびシグナルlog2比の変化を解析した。
【0136】
GeneChip技術の典型的な適用は、異なる組織で差次的に発現される遺伝子を発見することである。本出願において、4407−25/4407−33バレー品種、PBLB01/178バレー品種、およびNL Madole/181黒タバコ品種を含む、変換および非変換タバコ系統対に関して、エチレン処理によって引き起こされる遺伝子発現変動を決定した。これらの解析によって、生物学的変動のため、発現が有意に改変されている遺伝子のみが検出された。これらの解析は、誘導された遺伝子を同定する主な基準として、倍変化(シグナル比)を使用した。他のパラメーター、例えばシグナル強度、存在/非存在コールもまた、考慮した。
【0137】
およそ400遺伝子に関して、変換および非変換対の試料の発現相違に関して、データを解析した後、シグナル強度に基づく結果によって、2つの遺伝子、D121−AA8、およびD120−AH4、並びに1つの断片、D121−AA8の部分的断片であるD35−BG11のみが、エチレン処理変換系統対非変換系統において、再現可能な誘導を有した。これらの遺伝子の示差発現を例示すると、データは以下のように示された。表Vに示すように、変換系統、例えばバレー種タバコ品種、4407−33の遺伝子のシグナルを、関連する非変換同系系統、4407−25のシグナルに対する比として決定した。エチレン処理なしでは、すべての遺伝子の変換系統対非変換系統のシグナルの比は、1.00に近づいた。エチレン処理すると、同系バレー系統を用いた3回の独立の解析によって決定されるように、非変換系統に比較して、変換系統において、2つの遺伝子、D121−AA8およびD120−AH4が誘導された。これらの遺伝子は、互いに非常に高い相同性を有し、およそ99.8%以上の核酸配列相同性を有した。表Vに示すように、変換品種における相対的ハイブリダイゼーションシグナルは、変換系統において、非変換対応
物におけるシグナルのおよそ2〜12倍高い範囲であった。これと比較して、内部対照である2つのアクチン様対照クローンは、標準化した比に基づくと、変換系統で誘導されていないことが見出された。さらに、コード領域中の配列が完全にD121−AA8およびD120−AH4遺伝子両方に含有される断片(D35−BG11)は、対の同系変換系統および非変換系統の同じ試料で、高度に誘導された。バレー種タバコ品種、PBLB01および178の別の同系対が、エチレン誘導下、変換系統試料で誘導される、同じ遺伝子、D121−AA8およびD120−AH4を有することが示された。さらに、D121−AA8およびD120−AH4遺伝子は、同系黒タバコ対、NL Madoleおよび181の変換系統で優先的に誘導され、変換系統におけるこれらの遺伝子のエチレン誘導が、バレー種タバコ品種に限定されないことが立証された。すべての場合で、D35−BG11断片が、非変換対系統に比較して、変換系統で、最も高く誘導された。
【0138】
【表7】
【0139】
(実施例15)
タバコ変換系統におけるミクロソーム・ニコチン脱メチル化酵素のエチレン誘導
変換および非変換タバコ系統のエチレン処理および非処理対のミクロソーム濃縮分画において、脱メチル化酵素活性の生化学的解析を、以下のように行った。
【0140】
A.ミクロソームの調製
ミクロソームを4℃で単離した。50mM N−(2−ヒドロオキシエチル)ピペラジン−N’−(2−エタンスルホン酸)(HEPES)、pH7.5、3mM DL−ジチオスレイトール(DTT)およびプロテアーゼ阻害剤カクテル(Roche)を1錠剤/50mlからなる緩衝液中で、タバコ葉を抽出した。四層のチーズクロスを通じて未精製抽出物をろ過して、破壊されていない組織を取り除き、そしてろ液を20,000xgで20分間遠心分離して、細胞破片を取り除いた。上清を100,000xgで60分間、超遠心に供して、そして生じたペレットはミクロソーム分画を含有した。抽出緩衝液にミクロソーム分画を懸濁し、そして超遠心工程に適用し、ここで抽出緩衝液中の0.5Mスクロースの不連続スクロース勾配を用いた。凍結保護剤として10%(w/v)グリセロールを補った抽出緩衝液に、精製ミクロソームを再懸濁した。ミクロソーム調製物を使用
するまで液体窒素フリーザー中に保存した。
【0141】
B.タンパク質濃度測定
アセトン中の10%トリクロロ酢酸(TCA)(w/v)を用いて、ミクロソームタンパク質を沈殿させ、そして製造者のプロトコルにしたがってRC DCタンパク質アッセイキット(BIO−RAD)を用いて、ミクロソームのタンパク質濃度を測定した。
【0142】
3)ニコチン脱メチル化酵素活性アッセイ
Moravek BiochemicalsからDL−ニコチン(ピロリジン−2−14C)を得て、そしてこれは54mCi/mmolの比活性を有した。どちらもp450阻害剤であるクロルプロマジン(CPZ)および酸化チトクロムc(cyt.C)を、Sigmaから購入した。ニコチンアミドアデニンジヌクレオチドホスフェートの還元型(NADPH)は、NADPH:チトクロムP450還元酵素を介したチトクロムP450の典型的な電子供与体である。対照インキュベーションに関してはNADPHを省いた。日常的な酵素アッセイは、ミクロソームタンパク質(およそ2mg/ml)、6mM NADPH、55μM 14C標識ニコチンからなった。CPZおよびCyt.Cの濃度は、これらを用いる場合には、それぞれ1mMおよび100μMであった。25℃で1時間反応を行い、そして各25μl反応混合物に300μlメタノールを添加して反応を停止した。回転後、メタノール抽出物の20μlをVarianのInertsil ODS−3 3μ(150x4.6mm)カラムを用いて、逆相高性能液体クロマトグラフィー(HPLC)系(Agilent)で分離した。アイソクラチック可動相は、60:40(v/v)の比のメタノールおよび50mMリン酸カリウム緩衝液、pH6.25の混合物であり、そして流速は1ml/分であった。真性の非標識ノルニコチンとの比較によって決定されるようなノルニコチンピークを収集し、そして定量化のため、2900 tri−carb液体シンチレーションカウンター(LSC)(Perkin Elmer)に供した。1時間インキュベーションに渡る、14C標識ノルニコチンの産生に基づいて、ニコチン脱メチル化酵素の活性を計算する。
【0143】
エチレン処理したかまたはしなかった、バレー種変換(系統4407−33)および非変換(系統4407−25)タバコ系統の対から試料を得た。すべての未処理試料は、検出可能なミクロソーム・ニコチン脱メチル化酵素活性をまったく持たなかった。対照的に、エチレン処理した変換系統から得たミクロソーム試料は、有意なレベルのニコチン脱メチル化酵素活性を含有することが見出された。ニコチン脱メチル化酵素活性は、P450特異的阻害剤によって阻害されることが示され、脱メチル化酵素活性が、P450ミクロソーム由来酵素に一致することが立証された。バレー種変換タバコ系統に関して得られる、典型的な酵素アッセイ結果の組を表VIに示す。対照的に、エチレン処理した非変換系統タバコから得た試料は、ニコチン脱メチル化酵素活性をまったく含有しなかった。これらの結果によって、ニコチン脱メチル化酵素活性は、変換系統においてエチレン処理で誘導されたが、対応する同系非変換系統では誘導されないことが立証された。同系黒タバコ品種対に関しても同様の結果が得られ、この場合、ミクロソーム・ニコチン脱メチル化酵素活性は、変換系統では誘導され、そして非変換対系統では検出不能であった。これらの実験を総合すると、ミクロソーム・ニコチン脱メチル化酵素活性は、変換系統でエチレン処理に際して誘導される一方、対の同系非変換系統では誘導されないことが立証された。P450由来遺伝子であり、そして対の非変換系統に比較して、変換系統で優先的に誘導される遺伝子は、ニコチン脱メチル化酵素をコードする遺伝子の候補である。
【0144】
【表8】
【0145】
(実施例16)
ニコチン脱メチル化酵素としてのD121−AA8の機能的同定
酵母細胞において、異種発現されるP450の酵素活性をアッセイすることによって、ニコチン脱メチル化酵素のコード遺伝子としての候補クローン(D121−AA8)の機能を確認した。
【0146】
1.酵母発現ベクターの構築
P450コードcDNA(121AA8)の推定上のタンパク質コード配列を、酵母発現ベクターpYeDP60にクローニングした。適切なBamHIおよびMfeI部位(下線)を含有するPCRプライマーを介して、これらの配列を、翻訳開始コドン(ATG)の上流または停止コドン(TAA)の下流に導入した。増幅されたPCR産物上のMfeIは、ベクター上のEcoRI部位と適合する。121AA8 cDNAを増幅するのに用いたプライマーは、
【0147】
【化1】
【0148】
であった。6つのヒスチジンを含めて、タンパク質のC末端に9つの余分なアミノ酸をコードする配列セグメントを逆方向プライマーに取り込んだ。これによって、誘導に際して、6xHisをタグ付けしたP450の発現が促進される。酵素消化した後、PCR産物をGAL10−CYC1プロモーターに関してセンス方向でpYeDP60ベクターに連結した。酵素制限およびDNA配列決定によって、構築物を確認した。
【0149】
2.酵母形質転換
シロイヌナズナ属(Arabidopsis)NADPH−チトクロムP450還元酵素ATR1を発現するように修飾したWAT11酵母株を、構築物pYeDP60−P450 cDNAプラスミドで形質転換した。0.2cm電極ギャップのキュベット中で、50マイクロリットルのWAT11酵母細胞懸濁物を〜1μgのプラスミドDNAと混合した。Eppendorfエレクトロポレーション装置(モデル2510)によって、2
.0kVでパルスを1回適用した。SGIプレート(5g/lバクトカザミノ酸、6.7g/lのアミノ酸不含酵母窒素基剤、20g/lグルコース、40mg/l DL−トリプトファン、20g/l寒天)上に細胞を広げた。ランダムに選択したコロニーに対して、PCR解析を直接行うことによって、形質転換体を確認した。
【0150】
3.形質転換酵母細胞におけるP450発現
単一酵母コロニーを用いて、30ml SGI培地(5g/lバクトカザミノ酸、6.7g/lのアミノ酸不含酵母窒素基剤、20g/lグルコース、40mg/l DL−トリプトファン)に接種し、そして30℃で約24時間増殖させた。この培養物のアリコットを、1000mlのYPGE培地(10g/l酵母エキス、20g/lバクトペプトン、5g/lグルコース、30ml/lエタノール)に1:50で希釈し、そしてDiastix検尿試薬紙(Bayer、インディアナ州エルクハート)の色変化によって示されるように、グルコースが完全に消費されるまで増殖させた。最終濃度2%までDL−ガラクトースを添加することによって、クローニングしたP450の誘導を開始した。in vivo活性アッセイまたはミクロソーム調製に用いる前に、培養物をさらに20時間増殖させた。
【0151】
pYeDP60−CYP71D20(タバコ(Nicotiana tabacum)の5−エピ−アリストロチェンおよび1−デオキシカプシジオールの水酸化を触媒するP450)を発現するWAT11酵母細胞を、P450発現および酵素活性アッセイの対照として用いた。
【0152】
4. in vivo酵素活性
酵母培養物にDL−ニコチン(ピロリジン−2−14C)を供給することによって、形質転換酵母細胞におけるニコチン脱メチル化酵素活性をアッセイした。75μlのガラクトース誘導培養物に、14C標識ニコチン(54mCi/mmol)を最終濃度55μMまで添加した。14mlポリプロピレン試験管中、アッセイ培養物を振盪しながら6時間インキュベーションし、そして900μlメタノールで抽出した。回転後、20μlのメタノール抽出物をrp−HPLCで分離して、そしてLSCによってノルニコチン分画を定量化した。
【0153】
WAT11(pYeDP60−CYP71D20)の対照培養物はニコチンをノルニコチンに変換せず、WAT11酵母株が、ニコチンをノルニコチンに生物変換する工程を触媒可能な内因性酵素活性を含有しないことが示された。対照的に、121AA8遺伝子を発現する酵母は、検出可能な量のノルニコチンを産生し、このP450酵素がニコチン脱メチル化酵素活性を持つことが示された。
【0154】
5.酵母ミクロソーム調製
ガラクトースで20時間誘導した後、遠心分離によって酵母細胞を収集し、そしてTES−M緩衝液(50mM Tris−HCl、pH7.5、1mM EDTA、0.6Mソルビトール、10mM 2−メルカプトエタノール)で2回洗浄した。ペレットを抽出懸濁液(50mM Tris−HCl、pH7.5、1mM EDTA、0.6Mソルビトール、2mM 2−メルカプトエタノール、1%ウシ血清アルブミン、プロテアーゼ阻害剤カクテル(Roche)1錠剤/50ml)に再懸濁した。次いで、細胞をガラスビーズ(直径0.5mm、Sigma)で破壊した。細胞抽出物を20,000xgで20分間遠心分離して、細胞破片を取り除いた。上清を100,000xg、60分間の超遠心に供して、そして生じたペレットはミクロソーム分画を含有した。ミクロソーム分画をTEG−M緩衝液(50mM Tris−HCl、pH7.5、1mM EDTA、20%グリセロールおよび1.5mM 2−メルカプトエタノール)に、タンパク質濃度1mg/mlで懸濁した。ミクロソーム調製物を使用するまで液体窒素フリーザー中に保存し
た。
【0155】
6.酵母ミクロソーム調製物における酵素活性アッセイ
酵母ミクロソーム調製物を用いて、タンパク質濃度が1mg/mlで一定であったことを除いて、タバコ葉からのミクロソーム調製物と同じ方式で(実施例15)、ニコチン脱メチル化酵素活性アッセイを行った。
【0156】
CYP71D20を発現する対照酵母細胞由来のミクロソーム調製物は、検出可能なミクロソーム・ニコチン脱メチル化酵素活性をまったく持たなかった。対照的に、121AA8遺伝子を発現する酵母細胞から得たミクロソーム試料は、有意なレベルのニコチン脱メチル化酵素活性を示した。ニコチン脱メチル化酵素活性は、NADPHに必要であり、そしてP450特異的阻害剤によって阻害されることが示されており、これは研究中のP450に一致する。酵母細胞に関して得られる、典型的な酵素アッセイ結果の組を表VIIに示す。
【0157】
【表9】
【0158】
これらの実験を総合すると、クローニングされた全長遺伝子D121−AA8が、酵母で発現させた際、ニコチンのノルニコチンへの変換を触媒するチトクロムP450タンパク質をコードすることが立証された。
【0159】
本発明の実施にあたり、本発明の前述の詳細な説明を考慮すると、多くの修飾および変動が当業者には思い浮かぶと期待される。その結果、こうした修飾および変動は、付随する請求項の範囲内に含まれると意図される。
【特許請求の範囲】
【請求項1】
配列番号181である、タバコ属(Nicotiana)由来の単離核酸分子。
【請求項2】
配列番号181に少なくとも81%の配列同一性を有する、タバコ属由来の単離核酸分子。
【請求項3】
配列番号181に少なくとも91%の配列同一性を有する、タバコ属由来の単離核酸分子。
【請求項4】
配列番号182を含む、タバコ属由来の単離タンパク質。
【請求項5】
配列番号182に少なくとも80パーセントの配列同一性を有する、タバコ属由来の単離タンパク質。
【請求項6】
配列番号182に少なくとも90パーセントの配列同一性を有する、タバコ属由来の単離タンパク質。
【請求項7】
請求項1、2または3の核酸分子を含む、トランスジェニック植物。
【請求項8】
タバコ植物である、請求項7のトランスジェニック植物。
【請求項9】
トランスジェニック植物を産生する方法であって:
(i)請求項1、2または3の前記核酸分子を、前記植物において機能するプロモーターと、機能可能であるように連結して、植物形質転換ベクターを生成し;
(ii)前記植物を、工程(i)の前記植物形質転換ベクターで形質転換し;
(iii)前記形質転換ベクターで形質転換された植物細胞を選択し;そして
(iv)前記形質転換植物細胞から形質転換植物を再生する;
工程を含む、前記方法。
【請求項10】
植物が、減少したレベルのノルニコチンを有する、請求項9の方法。
【請求項11】
前記核酸分子がアンチセンス方向である、請求項9の方法。
【請求項12】
前記核酸分子がセンス方向である、請求項9の方法。
【請求項13】
前記核酸分子がRNA干渉方向である、請求項9の方法。
【請求項14】
前記核酸分子が二本鎖RNA分子として発現される、請求項9の方法。
【請求項15】
前記トランスジェニック植物がタバコ植物である、請求項9の方法。
【請求項16】
核酸分子を含有する植物を選択する方法であって、請求項1、2または3の核酸配列の存在に関して、前記植物を解析する、前記方法。
【請求項17】
前記植物をDNAハイブリダイゼーションによって解析する、請求項16の植物を選択する方法。
【請求項18】
前記DNAハイブリダイゼーションがサザンブロット解析である、請求項17の植物を選択する方法。
【請求項19】
前記DNAハイブリダイゼーションがノーザンブロット解析である、請求項17の植物を選択する方法。
【請求項20】
前記植物をPCR検出によって解析する、請求項16の植物を選択する方法。
【請求項21】
前記植物がタバコ植物である、請求項16の方法。
【請求項22】
植物におけるノルニコチンレベルを増加させるかまたは減少させる方法であって:
(i)請求項1、2または3の前記核酸分子を、前記植物において機能するプロモーターと、機能可能であるように連結して、植物形質転換ベクターを生成し;
(ii)前記植物を、工程(i)の前記植物形質転換ベクターで形質転換し;
(iii)前記形質転換ベクターで形質転換された植物細胞を選択し;そして
(iv)前記形質転換植物細胞から形質転換植物を再生する
工程を含む、前記方法。
【請求項23】
前記核酸分子がアンチセンス方向である、請求項22の方法。
【請求項24】
前記核酸分子がセンス方向である、請求項22の方法。
【請求項25】
前記核酸分子がRNA干渉方向である、請求項22の方法。
【請求項26】
前記核酸分子が二本鎖RNA分子として発現される、請求項22の方法。
【請求項27】
前記トランスジェニック植物がタバコ植物である、請求項22の方法。
【請求項28】
減少した量のノルニコチンレベルを有するタバコ製品であって、請求項7の植物由来のタバコを含む、前記タバコ製品。
【請求項29】
タバコ製品が、巻きタバコ、葉巻、パイプタバコ、嗅ぎタバコ、噛みタバコ、タバコ製品とブレンドした製品、およびこれらの混合物からなる群より選択される、請求項27のタバコ製品。
【請求項30】
ノルニコチンレベルが、約5〜約10%減少している、請求項28のタバコ製品。
【請求項31】
ノルニコチンレベルが、約10〜約20%減少している、請求項28のタバコ製品。
【請求項32】
ノルニコチンレベルが、約20〜約30%減少している、請求項28のタバコ製品。
【請求項33】
ノルニコチンレベルが、約30%を超えて減少している、請求項28のタバコ製品。
【請求項34】
請求項7の植物由来のタバコ葉を含む、減少した量のノルニコチンレベルを有するタバコ葉。
【請求項35】
タバコ製品が形成される請求項34のタバコ葉であって、該タバコ製品が、巻きタバコ、葉巻、パイプタバコ、嗅ぎタバコ、噛みタバコ、タバコ製品とブレンドした製品、およびこれらの混合物からなる群より選択される、前記タバコ葉。
【請求項36】
請求項1、2または3の単離核酸を用いて、植物から遺伝子を単離する方法。
【請求項1】
配列番号181である、タバコ属(Nicotiana)由来の単離核酸分子。
【請求項2】
配列番号181に少なくとも81%の配列同一性を有する、タバコ属由来の単離核酸分子。
【請求項3】
配列番号181に少なくとも91%の配列同一性を有する、タバコ属由来の単離核酸分子。
【請求項4】
配列番号182を含む、タバコ属由来の単離タンパク質。
【請求項5】
配列番号182に少なくとも80パーセントの配列同一性を有する、タバコ属由来の単離タンパク質。
【請求項6】
配列番号182に少なくとも90パーセントの配列同一性を有する、タバコ属由来の単離タンパク質。
【請求項7】
請求項1、2または3の核酸分子を含む、トランスジェニック植物。
【請求項8】
タバコ植物である、請求項7のトランスジェニック植物。
【請求項9】
トランスジェニック植物を産生する方法であって:
(i)請求項1、2または3の前記核酸分子を、前記植物において機能するプロモーターと、機能可能であるように連結して、植物形質転換ベクターを生成し;
(ii)前記植物を、工程(i)の前記植物形質転換ベクターで形質転換し;
(iii)前記形質転換ベクターで形質転換された植物細胞を選択し;そして
(iv)前記形質転換植物細胞から形質転換植物を再生する;
工程を含む、前記方法。
【請求項10】
植物が、減少したレベルのノルニコチンを有する、請求項9の方法。
【請求項11】
前記核酸分子がアンチセンス方向である、請求項9の方法。
【請求項12】
前記核酸分子がセンス方向である、請求項9の方法。
【請求項13】
前記核酸分子がRNA干渉方向である、請求項9の方法。
【請求項14】
前記核酸分子が二本鎖RNA分子として発現される、請求項9の方法。
【請求項15】
前記トランスジェニック植物がタバコ植物である、請求項9の方法。
【請求項16】
核酸分子を含有する植物を選択する方法であって、請求項1、2または3の核酸配列の存在に関して、前記植物を解析する、前記方法。
【請求項17】
前記植物をDNAハイブリダイゼーションによって解析する、請求項16の植物を選択する方法。
【請求項18】
前記DNAハイブリダイゼーションがサザンブロット解析である、請求項17の植物を選択する方法。
【請求項19】
前記DNAハイブリダイゼーションがノーザンブロット解析である、請求項17の植物を選択する方法。
【請求項20】
前記植物をPCR検出によって解析する、請求項16の植物を選択する方法。
【請求項21】
前記植物がタバコ植物である、請求項16の方法。
【請求項22】
植物におけるノルニコチンレベルを増加させるかまたは減少させる方法であって:
(i)請求項1、2または3の前記核酸分子を、前記植物において機能するプロモーターと、機能可能であるように連結して、植物形質転換ベクターを生成し;
(ii)前記植物を、工程(i)の前記植物形質転換ベクターで形質転換し;
(iii)前記形質転換ベクターで形質転換された植物細胞を選択し;そして
(iv)前記形質転換植物細胞から形質転換植物を再生する
工程を含む、前記方法。
【請求項23】
前記核酸分子がアンチセンス方向である、請求項22の方法。
【請求項24】
前記核酸分子がセンス方向である、請求項22の方法。
【請求項25】
前記核酸分子がRNA干渉方向である、請求項22の方法。
【請求項26】
前記核酸分子が二本鎖RNA分子として発現される、請求項22の方法。
【請求項27】
前記トランスジェニック植物がタバコ植物である、請求項22の方法。
【請求項28】
減少した量のノルニコチンレベルを有するタバコ製品であって、請求項7の植物由来のタバコを含む、前記タバコ製品。
【請求項29】
タバコ製品が、巻きタバコ、葉巻、パイプタバコ、嗅ぎタバコ、噛みタバコ、タバコ製品とブレンドした製品、およびこれらの混合物からなる群より選択される、請求項27のタバコ製品。
【請求項30】
ノルニコチンレベルが、約5〜約10%減少している、請求項28のタバコ製品。
【請求項31】
ノルニコチンレベルが、約10〜約20%減少している、請求項28のタバコ製品。
【請求項32】
ノルニコチンレベルが、約20〜約30%減少している、請求項28のタバコ製品。
【請求項33】
ノルニコチンレベルが、約30%を超えて減少している、請求項28のタバコ製品。
【請求項34】
請求項7の植物由来のタバコ葉を含む、減少した量のノルニコチンレベルを有するタバコ葉。
【請求項35】
タバコ製品が形成される請求項34のタバコ葉であって、該タバコ製品が、巻きタバコ、葉巻、パイプタバコ、嗅ぎタバコ、噛みタバコ、タバコ製品とブレンドした製品、およびこれらの混合物からなる群より選択される、前記タバコ葉。
【請求項36】
請求項1、2または3の単離核酸を用いて、植物から遺伝子を単離する方法。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図17】

【図18】

【図19】

【図20】

【図21】

【図22】

【図23】

【図24】

【図25】

【図26】

【図27】

【図28】

【図29】

【図30】

【図31】

【図32】

【図33】

【図34】

【図35】

【図36】

【図37】

【図38】

【図39】

【図40】

【図41】

【図42】

【図43】

【図44】

【図45】

【図46】

【図47】

【図48】

【図49】

【図50】

【図51】

【図52】

【図53】

【図54】

【図55】

【図56】

【図57】

【図58】

【図59】

【図60】

【図61】

【図62】

【図63】

【図64】

【図65】

【図66】

【図67】

【図68】

【図69】

【図70】

【図71】

【図72】

【図73】

【図74】

【図75】
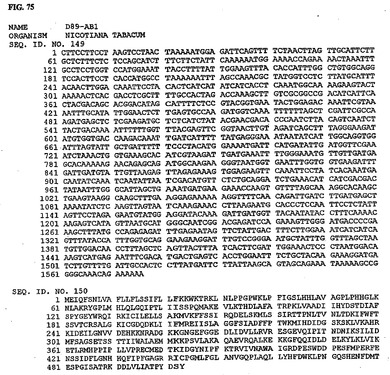
【図76】

【図77】

【図78】

【図79】

【図80】

【図81】

【図82】

【図83】

【図84】

【図85】

【図86】

【図87】

【図88】

【図89】

【図90】
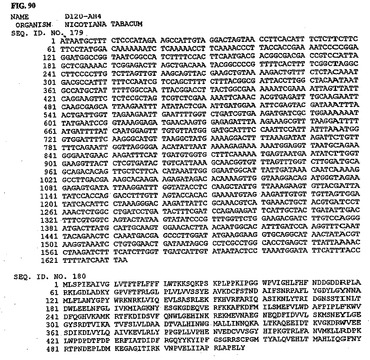
【図91】

【図92】

【図93】

【図94】
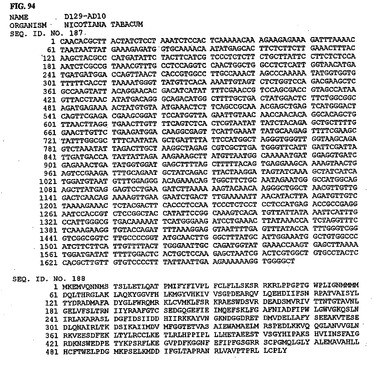
【図95】
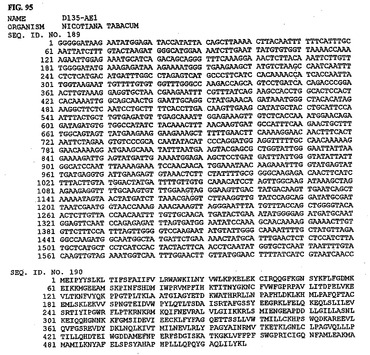
【図96】

【図97】
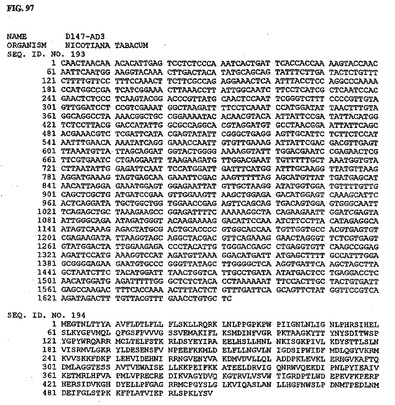
【図98】

【図99】

【図100】

【図101】

【図102】
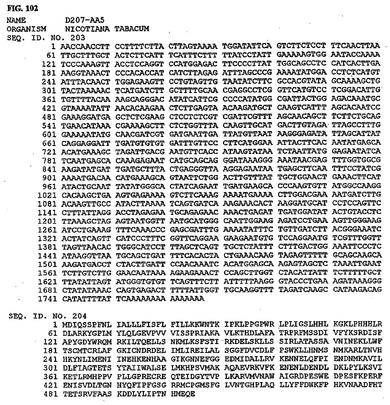
【図103】

【図104】
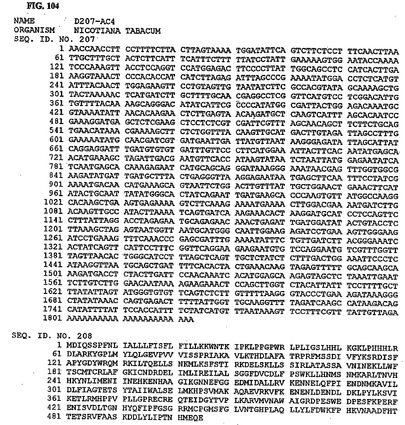
【図105】
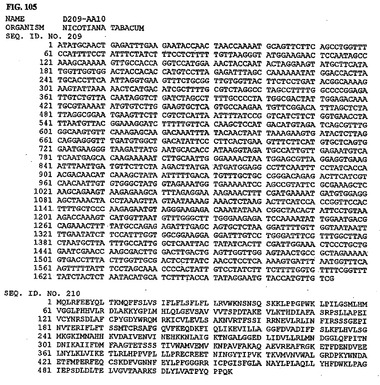
【図106】

【図107】

【図108】
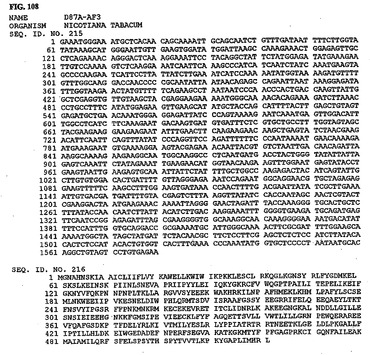
【図109】

【図110】

【図111】

【図112】
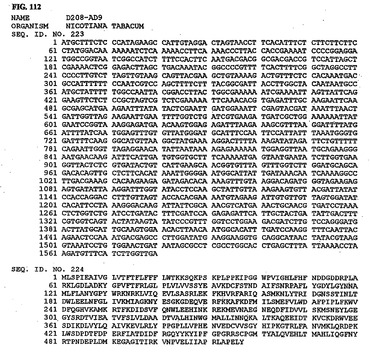
【図113】

【図114】

【図115】

【図116】
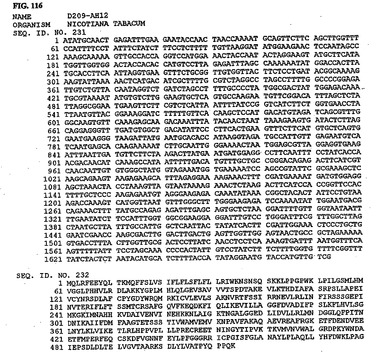
【図117】

【図118】
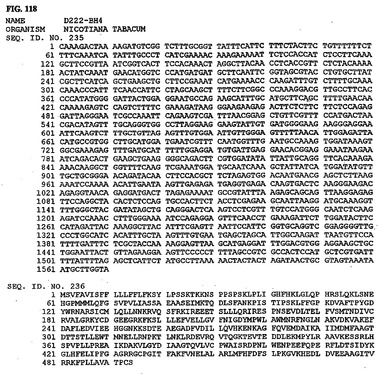
【図119】

【図120】

【図121】

【図122】

【図123】
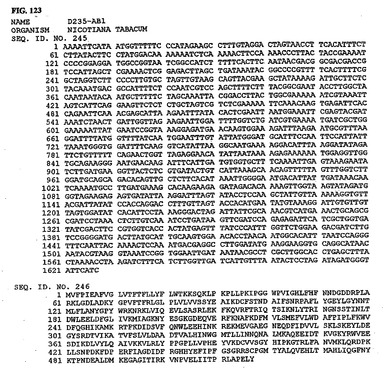
【図124】

【図125】

【図126】

【図127】

【図128】

【図129】

【図130】

【図131】

【図132】

【図133】

【図134】

【図135】

【図136】

【図137】
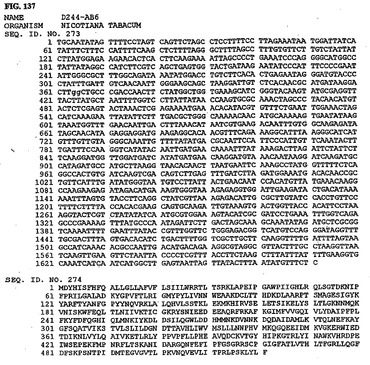
【図138】

【図139】
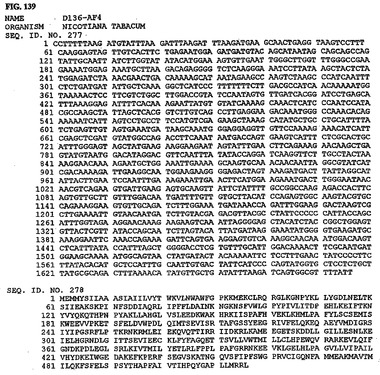
【図140】
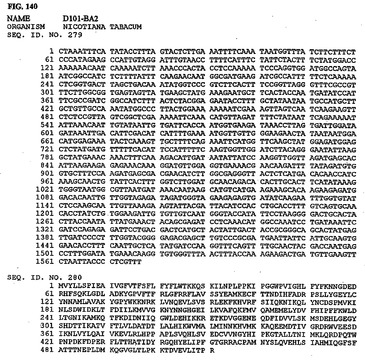
【図141】

【図142】
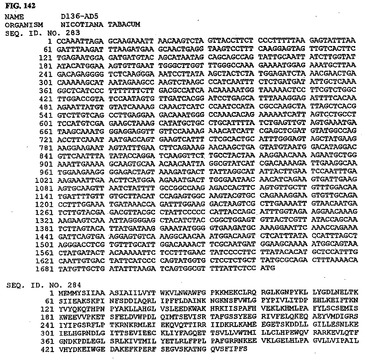
【図143】

【図144】

【図145】

【図146】

【図147】

【図148】

【図149】
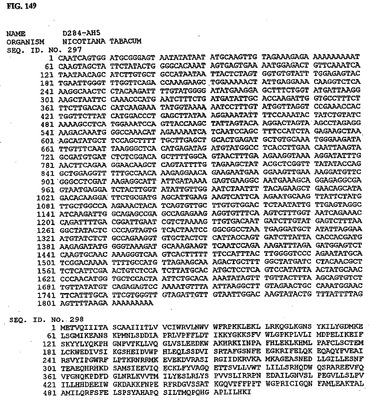
【図151−1】

【図151−2】
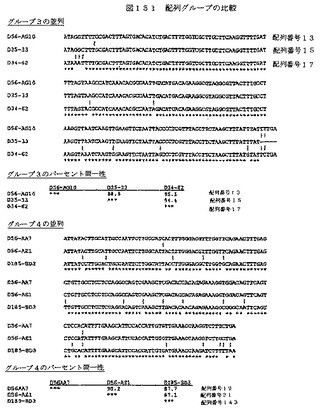
【図151−3】

【図151−4】

【図151−5】

【図151−6】
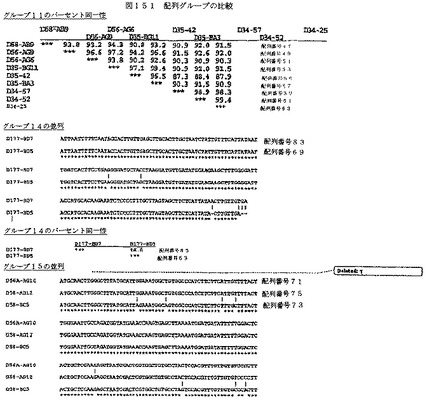
【図151−7】

【図151−8】

【図151−9】

【図151−10】
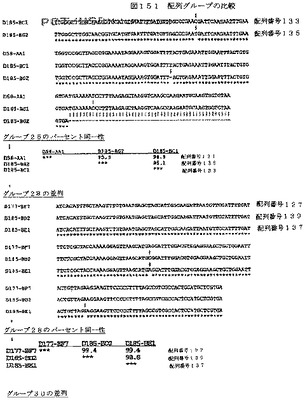
【図151−11】

【図152−1】

【図152−2】

【図152−3】

【図152−4】

【図152−5】

【図153】
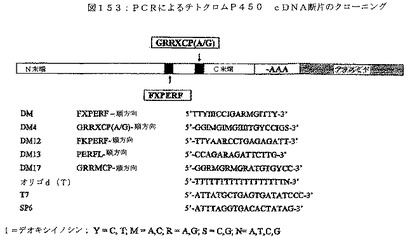
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【図49】
【図50】
【図51】
【図52】
【図53】
【図54】
【図55】
【図56】
【図57】
【図58】
【図59】
【図60】
【図61】
【図62】
【図63】
【図64】
【図65】
【図66】
【図67】
【図68】
【図69】
【図70】
【図71】
【図72】
【図73】
【図74】
【図75】
【図76】
【図77】
【図78】
【図79】
【図80】
【図81】
【図82】
【図83】
【図84】
【図85】
【図86】
【図87】
【図88】
【図89】
【図90】
【図91】
【図92】
【図93】
【図94】
【図95】
【図96】
【図97】
【図98】
【図99】
【図100】
【図101】
【図102】
【図103】
【図104】
【図105】
【図106】
【図107】
【図108】
【図109】
【図110】
【図111】
【図112】
【図113】
【図114】
【図115】
【図116】
【図117】
【図118】
【図119】
【図120】
【図121】
【図122】
【図123】
【図124】
【図125】
【図126】
【図127】
【図128】
【図129】
【図130】
【図131】
【図132】
【図133】
【図134】
【図135】
【図136】
【図137】
【図138】
【図139】
【図140】
【図141】
【図142】
【図143】
【図144】
【図145】
【図146】
【図147】
【図148】
【図149】
【図151−1】
【図151−2】
【図151−3】
【図151−4】
【図151−5】
【図151−6】
【図151−7】
【図151−8】
【図151−9】
【図151−10】
【図151−11】
【図152−1】
【図152−2】
【図152−3】
【図152−4】
【図152−5】
【図153】
【公開番号】特開2010−99074(P2010−99074A)
【公開日】平成22年5月6日(2010.5.6)
【国際特許分類】
【出願番号】特願2009−276020(P2009−276020)
【出願日】平成21年12月4日(2009.12.4)
【分割の表示】特願2006−535378(P2006−535378)の分割
【原出願日】平成16年10月15日(2004.10.15)
【出願人】(398069229)ユーエス スモークレス タバコ カンパニー (20)
【Fターム(参考)】
【公開日】平成22年5月6日(2010.5.6)
【国際特許分類】
【出願日】平成21年12月4日(2009.12.4)
【分割の表示】特願2006−535378(P2006−535378)の分割
【原出願日】平成16年10月15日(2004.10.15)
【出願人】(398069229)ユーエス スモークレス タバコ カンパニー (20)
【Fターム(参考)】
[ Back to top ]