
タバコ由来糖鎖付加酵素遺伝子及びその抑制
【課題】汎用的な形質転換体宿主であるタバコ由来のセリンガラクトース転移酵素(SGT)遺伝子をクローニングし、当該遺伝子による形質転換体を用いたO−結合型ガラクトース含有ポリペプチドの製造方法を提供すること、及び当該遺伝子発現を抑制する方法を提供し、形質転換タバコ植物におけるO−結合型糖鎖の付加を抑制したヒト有用タンパク質の製造方法を提供する。
【解決手段】特定のアミノ配列を含むタバコ由来のセリンO−結合型ガラクトース転移酵素をコードする遺伝子をクローニングし、当該遺伝子による形質転換体を用いたO−結合型ガラクトース付加法、当該遺伝子発現を抑制することによる形質転換タバコ植物におけるO−結合型糖鎖の付加抑制方法。
【解決手段】特定のアミノ配列を含むタバコ由来のセリンO−結合型ガラクトース転移酵素をコードする遺伝子をクローニングし、当該遺伝子による形質転換体を用いたO−結合型ガラクトース付加法、当該遺伝子発現を抑制することによる形質転換タバコ植物におけるO−結合型糖鎖の付加抑制方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、タバコ由来のO−結合型糖鎖付加関連酵素をコードする遺伝子の同定、当該遺伝子を用いたO−結合型糖鎖付加ポリペプチドの製造、当該遺伝子発現の抑制技術、及び形質転換植物におけるO−結合型糖鎖の付加を抑制したヒト有用タンパク質の製造方法に関する。
【背景技術】
【0002】
抗体やサイトカイン等のバイオ医薬の生産は、現在、CHO細胞など動物培養細胞を用いた生産系が主流であるが、生産コストが高く、大量生産も難しく、ウィルスの混入の危険もある。大腸菌や酵母など微生物を利用した生産系も開発されているが、糖鎖構造が大きく異なっていることが、課題となっている。
植物は、生産コストが微生物並みに安価であって、大量生産も可能であり、ウィルス混入の危険性もなく、糖鎖構造がN−結合型糖鎖の場合は特にヒトにかなり似ているため、バイオ医薬生産系として有望であることが、最近製薬企業に注目されている。
既に、植物培養細胞を用いて、GM-CSF(非特許文献1)、IL-2およびIL-4(非特許文献2)、免疫グロブリン(非特許文献3)、エリスロポエチン(非特許文献4)、α1−アンチトリプシン(非特許文献5)などヒト有用タンパク質の製造が行われている。
【0003】
タンパク質の翻訳後修飾として、糖鎖修飾は主要なものであり、大きく分けて、N−結合型糖鎖と、O−結合型糖鎖の2種類があるが、これらいずれについても、ヒトを含む哺乳動物に無く、植物にのみ存在する植物特有の糖鎖修飾がある。N−結合型糖鎖修飾に関しては、根元のNアセチルグルコサミンに付加するα1,3−フコース、βマンノースに付加するβ1,2−キシロースが、植物特有の糖鎖構造であり、ヒトにおいて抗原となることも知られているが、従来から研究が進んでおり、除去方法も開発されている。具体的には、N−結合型糖鎖修飾におけるフコース転移酵素遺伝子およびキシロース転移酵素遺伝子が同定されており、これら酵素の発現を抑制することにより、α1,3−フコースおよびβ1,2−キシロースの付加の無いN−結合型糖鎖を有する糖タンパク質を植物細胞で生産することが可能となっている(非特許文献6〜9、20)。
【0004】
一方、植物のO−結合型糖鎖修飾に関連する研究開発は遅れており、O−結合型糖鎖付加に関連した酵素遺伝子の解析は重大な課題として残されていた。
O−結合型糖鎖は、ヒトを含む哺乳動物では、タンパク質のセリンあるいはスレオニン残基にNアセチルガラクトサミン(GalNAc)が付加するのが一般的であり、その他、セリンあるいはスレオニン残基にマンノース、フコース、グルコース、Nアセチルグルコサミン、キシロースの付加が知られている。植物細胞ではこのようなO−結合型糖鎖構造は知られておらず、全く異なった2種類のO−結合型糖鎖構造が知られている。ひとつは、タンパク質のアミノ酸配列中の複数のプロリン残基が水酸化されてヒドロキシプロリンになり、そこにガラクトースあるいはアラビノースが付加する構造である。典型的な配列として、エクステンシンの配列「VYKSOOOOV(Oはヒドロキシプロリン)」がよく知られており、この複数のヒドロキシプロリン基が有するヒドロキシル基が糖修飾された構造であるが、この構造もアレルゲンとなることが報告されている(非特許文献10、11)。これらの糖鎖構造を作るガラクトース転移酵素(HGT)およびアラビノース転移酵素は未だに不明であるが、プロリン水酸化酵素はすでに同定されており、その発現の抑制が試みられており、形質転換植物内での外来タンパク質のO−結合型糖鎖付加も阻害されることが確認されている(非特許文献12〜14)。HGT酵素活性は、FITCで蛍光標識した植物のアラビノガラクタンタンパク質14(AGP14)ペプチド配列「FITC-VDAOAOSOTS」を受容体基質として、酵素活性が測定できている(非特許文献15)。
【0005】
しかしながら、もう一つの植物特有のO−結合型糖鎖構造として、タンパク質のアミノ酸配列中のセリン残基に対して直接ガラクトースが付加される場合がある。典型的な配列として、植物細胞壁に存在する糖タンパク質であるエクステンシンの繰り返し配列「VYKSOOOOV」のセリン残基へのガラクトース付加がよく知られている。また、サツマイモの貯蔵タンパク質であるスポラミン「PTTHEPASSETPVL」をタバコ細胞で発現させたときにも、2番目のセリン残基へのガラクトース付加が確認されている。このセリン残基がガラクトース修飾された構造も抗原性があるため、当該ガラクトース修飾に関連する酵素発現を抑制する必要があるが、この構造を作る酵素および酵素遺伝子についての解析は、全くなされていない状況である(非特許文献16〜18)。
このように、植物をバイオ医薬の生産系として確立するためには、O−結合型糖鎖付加に関連する糖転移酵素のうちでも特に、セリン残基のヒドロキシル基にガラクトースを付加する作用を有するガラクトース転移酵素遺伝子を同定し、当該ガラクトース転移酵素発現を抑制する技術の開発が強く望まれていた。そして、そのためにはセリンO−結合型ガラクトース転移酵素(SGT)単独の活性を正確に測定する酵素活性測定法の開発が急務となっていた。とりわけ実用的には、形質転換体宿主として一般に用いられているタバコなどのナス科植物に直接適用できるタバコ由来のSGT遺伝子をターゲットとすることが好ましいが、タバコの場合は全ゲノム配列が公開されていないため、タバコのゲノムライブラリーから直接クローニングすることは難しいという問題がある。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】James EA, Wang C, Wang Z, Reeves R, Shin JH, Magnuson NS, Lee JM, Production and characterization of biologically active human GM-CSF secreted by genetically modified plant cells. Protein Expr Purif. 19, 131-138 (2000)
【非特許文献2】Magnuson NS, Linzmaier PM, Reeves R, An G, HayGlass K, Lee JM, Secretion of biologically active human interleukin-2 and interleukin-4 from genetically modified tobacco cells in suspension culture. Protein Expr Purif. 13, 45-52 (1998)
【非特許文献3】Magnuson NS, Linzmaier PM, Gao JW, Reeves R, An G, Lee JM, Enhanced recovery of a secreted mammalian protein from suspension culture of genetically modified tobacco cells. Protein Expr Purif. 7, 220-228 (1996)
【非特許文献4】Matsumoto S, Ishii A, Ikura K, Ueda M, Sasaki R, Expression of human erythropoietin in cultured tobacco cells, Biosci. Biotechnol. Biochem. 57, 1249-1252 (1993)
【非特許文献5】Terashima M, Murai Y, Kawamura M, Nakanishi S, Stoltz T, Chen L, Drohan W, Rodriguez RL, Katoh S, Production of functional human alpha 1-antitrypsin by plant cell culture. Appl Microbiol Biotechnol. 52, 516-523 (1999)
【非特許文献6】Fotisch K, Altmann F, Haustein D, Vieths S, Involvement of carbohydrate epitopes in the IgE response of celery-allergic patients. Int Arch Allergy Immunol. 120, 30-42 (1999)
【非特許文献7】Wilson IB, Harthill JE, Mullin NP, Ashford DA, Altmann F, Core alphal, 3-fucose is a key part of the epitope recognized by antibodies reacting against plant N-linked oligosaccharides and is present in a wide variety of plant extracts. Glycobiology. 8, 651-661 (1998)
【非特許文献8】van Ree R, Cabanes-Macheteau M, Akkerdaas J, Milazzo JP, Loutelier-Bourhis C, Rayon C, Villalba M, Koppelman S, Aalberse R, Rodriguez R, Faye L, Lerouge P, Beta(1,2)-xylose and alpha(1,3)-fucose residues have a strong contribution in IgE binding to plant glycoallergens. J Biol Chem. 2000 Apr. 14;275 (15):11451-11458
【非特許文献9】Strasser R, Stadlmann J, Schahs M, Stiegler G, Quendler H, Mach L, Glossl J, Weterings K, Pabst M, and Steinkellner H, Generation of glyco-engineered Nicotiana bethamiana for the production of monoclonal antibodies with a homogeneous human-like N-glycan structure. Plant Biotech. J. 6, 392-402 (2008)
【非特許文献10】Leonard R, Petersen B.O, Himly M, Kaar W, Wopfner N, Kolarich D, van Ree R, Ebner C, Duus J, Ferreira F, and Altmann R, J. Biol. Chem, 280, 7932-7940 (2005)
【非特許文献11】Leonard R, et al, Carbohydr. Res, 340, 657 (2005)
【非特許文献12】松岡健、バイオサイエンスとインダストリー、63、303 (2005)
【非特許文献13】Tiainen P, et al, J. Biol. Chem., 280, 1142 (2005)
【非特許文献14】Yuasa K, et al, Plant J., 41, 81 (2005)
【非特許文献15】Oka T, et al., Characterization of ER localized UDP-D-galactose: hydroxyproline O-galactosyltransferase using synthetic peptide substrates in Arabidopsis thaliana. Plant Physiol.(2009)
【非特許文献16】Lamport DT, Katona L, Roerig S, Galactosylserine in extensin.. Biochem J. 133, 125-32 (1973)
【非特許文献17】Allen AK, Desai NN, Neuberger A, Creeth J, Properties of potate lectin and the nature of its glycoprotein linkages , Biochem J. 17, 665-74 (1978)
【非特許文献18】Matsuoka K, Watanabe N, Nakamura K. O-glycosylation of a precursor to a sweet potato vacuolar protein, sporamin, expressed in tobacco cells Plant J. 8, 877-89 (1995).
【非特許文献19】Busch C, Hofmann F, Selzer J, Munro S, Jeckel D, Aktories K. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J Biol Chem. 1998 Jul 31;273(31):19566-72.
【非特許文献20】Karg SR and Kallio PT (2009) The production of biopharmaceuticals in plant system. Biotech. Advances, 27, 879-894.
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、セリン残基のヒドロキシル基にガラクトースを付加する作用のガラクトース転移酵素(SGT)遺伝子のうちで、実用的な観点からみて、汎用的な形質転換体宿主であるタバコ由来のSGT遺伝子をターゲットとすることにした。すなわち、タバコ由来SGT遺伝子をクローニングし、当該遺伝子による形質転換体を用いたO−結合型ガラクトース含有ポリペプチドの製造方法を提供すること、及び当該遺伝子発現を抑制する方法を提供し、形質転換植物におけるO−結合型糖鎖の付加を抑制したヒト有用タンパク質の製造方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
タバコ由来SGT遺伝子をクローニングするにあたり、タバコの場合は未だ全ゲノム配列が公開されていないため、タバコのゲノムライブラリーから直接クローニングすることは難しい。そこで、まずは全ゲノム配列情報が公開されていて各ORF情報が入手可能なシロイヌナズナなどの他の植物及び藻類のSGT遺伝子を同定し、次いで当該遺伝子の配列情報を利用してタバコ由来のSGT遺伝子をクローニングすることとした。
【0009】
まず、シロイヌナズナの細胞抽出液からセリンO−結合型ガラクトース転移酵素(Peptidyl serine α-galactosyltransferase;SGT)の酵素活性の測定を、受容体基質としてスポラミン配列「FITC-PTTHEPASSETPVL」およびエクステンシン配列「VYKSPPPPV」を用いて試みたが、ガラクトース転移酵素活性は確認できなかった。プロリンを水酸化したスポラミン配列「FITC-PTTHEOASSETPVL」を受容体基質としても酵素活性は検出できなかったが、プロリンを水酸化したエクステンシン配列「VYKSOOOOV」を受容体基質としたところ、HPLCで酵素反応物のピークを検出でき、ガラクトース転移酵素活性を確認できた。したがって、「FITC-VYKSOOOOV」を受容体基質とすることによるSGT酵素活性測定が可能となった。そこで、SGT酵素を精製し、アミノ酸配列を解析することで、SGT酵素をコードする遺伝子の同定を試みたが、シロイヌナズナのSGT糖転移酵素活性がHGT酵素をはじめとした他のタンパク質と共に不溶性の膜画分に含まれ、可溶化できない状態であったため、SGT酵素を単離することができなかった。
【0010】
そこで、本発明者らは、全ての高等植物の祖先といわれる単細胞の緑藻の1種であるクラミドモナスが、高等植物と同様のO−結合型糖鎖構造を有する植物タンパク質を生産していることに着目し、「FITC-VYKSOOOOV」を用いて同様のガラクトース転移酵素活性を確認した。ヒドロキシプロリンへのガラクトース付加は、ペプチド中にとびとびにヒドロキシプロリンが存在することが必要であり、連続するヒドロキシプロリンにはアラビノースが付加し、ガラクトースは付加しないと考えられていた。しかし、予想に反して、セリン残基は一つしかないにもかかわらず、HPLC解析により複数のピークが確認されたことから、受容体基質ペプチドの2カ所にガラクトース付加が検出され、ヒドロキシプロリンへのガラクトース転移酵素(HGT)の活性も検出されていると考えられた。βガラクトシダーゼ処理およびβエリミネーション処理による解析を行った結果、HPLCでSGT酵素産物のピークとHGT酵素産物のピークを同定することができたことから、セリンO−結合型のガラクトース転移酵素活性のみを正確に測定することが可能となった。一方で、ガラクトース転移酵素の可溶化も可能であることを見出し、SGT酵素とHGT酵素を可溶化条件やカラムによる分画で分離できることが確認できたので、クラミドモナスからSGTを単離することとした。
クラミドモナスから、SGTを精製し、そのアミノ酸配列解析の結果から、SGTをコードする遺伝子の同定に成功し、当該遺伝子を異種である酵母細胞で発現させることでSGTをコードする遺伝子であることを証明したとともに、詳細な酵素活性を確認した。さらに、当該遺伝子との相同性検索により、各種植物のSGT遺伝子を同定し、この遺伝子が、緑藻から高等植物まで存在しており、動物や真菌には類似遺伝子が全く存在しないこと、及び従来の糖転移酵素遺伝子とも全く類似性がないため、糖転移酵素遺伝子であると予測されていない新規な遺伝子ファミリーに属していることを見出した。シロイヌナズナSGT遺伝子についても、その遺伝子を単離して酵母で発現させたところ、クラミドモナスSGTと同様のSGT活性を確認することができた。本出願人はこれらの知見をもとに特許出願をしている(PCT/JP2010/071598)。
【0011】
次いで、シロイヌナズナのSGT1配列をクエリーにして、タバコのESTデータベースを検索し、シグナル配列、膜貫通領域、塩基性アミノ酸に富む領域を欠く2074bpからなる部分配列を見出した。当該配列を利用して設計したプライマーを用いて、タバコBY-2細胞から抽出した全RNAを鋳型とし、RT-PCR、5’RACE、3’RACE法によりタバコSGT1(NtSGT1)遺伝子に対応する全長3,336bpからなるcDNAをクローニングした(配列番号20)。タバコSGT1アミノ酸配列は配列番号21で表され、シロイヌナズナSGT1とは80%以上のアミノ酸配列同一性を示し、SGT活性ドメイン1(配列番号22)及び活性ドメイン2(配列番号23)を有する。このタバコSGT1のシグナル配列および膜貫通領域を除いた主要部分を酵母で発現させて、SGT酵素活性を確認した。
さらに、当該SGT酵素活性が一般的な発現抑制技術で抑制可能かどうかを確認するために、タバコ培養細胞BY-2に対してヘアピン型二本鎖RNA(dsRNA)発現プラスミドを導入して誘導したRNAi実験により、SGT1酵素活性が大幅に抑制されることを観測した。このことは、当該SGT遺伝子がタバコ植物体内でのセリンO−結合型ガラクトース付加遺伝子であることが確認できたことであり、かつ当該SGT遺伝子をターゲットとしてその遺伝子発現を抑制すれば、タバコ植物体内でのタンパク質へのセリンO−結合型のガラクトース付加抑制が可能であることを示すものでもある。
RNAiをはじめ植物内での糖転移酵素の発現抑制技術自体は、従来から周知であるから、タバコなど植物体内に本来存在する本発明のSGT遺伝子をターゲットとして、RNAiなど周知抑制手法を適用することでSGTの発現抑制が可能である。当該タバコなどの植物を形質転換植物宿主とすることで、外来遺伝子に対するセリンO−結合型ガラクトース付加を抑制することができるから、バイオ医薬として期待される抗体、生理活性糖タンパク質などのヒト有用糖タンパク質にとってきわめて有力なツールを提供できたことになる。特に、形質転換植物において、当該SGT遺伝子の発現抑制を、プロリン水酸化酵素遺伝子および他のO−結合型糖転移酵素遺伝子の抑制と同時に行うことで、外来タンパク質に対する植物特有のO−結合型糖修飾を完全に抑制することも可能である。
以上の知見を得たことで、本発明を完成するに至った。
【0012】
すなわち、本発明は以下を包含する。
〔1〕 下記の(1)〜(4)のいずれかのアミノ酸配列を含む、セリンO−結合型ガラクトース転移酵素活性(SGT酵素活性)を有するポリペプチド;
(1)配列番号21に示されるアミノ酸配列、
(2)配列番号21に示されるアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列。
(3)配列番号21と80%以上の同一性を有しているアミノ酸配列からなり、かつそのSGT酵素活性ドメインとして、DXDモチーフを有し、配列番号22又は23と90%以上の同一性を有するアミノ酸配列からなる部分配列を有しているアミノ酸配列、
(4)植物のゲノム中の塩基配列によりコードされるアミノ酸配列であって、配列番号21に示されるアミノ酸配列と80%以上の同一性を有するアミノ酸配列からなり、かつDXDモチーフを含むアミノ酸配列。
〔2〕 下記(5)〜(7)に記載のアミノ酸配列を含む、セリンO−結合型ガラクトース転移酵素活性(SGT酵素活性)を有するSGT酵素活性ドメインポリペプチド;
(5)配列番号22又は23に示されるアミノ酸配列、
(6)配列番号22又は23に示されるアミノ酸配列のいずれかにおいて1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列、
(7)配列番号22又は23に示されるアミノ酸配列と少なくとも90%以上の同一性を有するアミノ酸配列であって、かつDXDモチーフを含むアミノ酸配列。
〔3〕 前記〔1〕又は〔2〕に記載のポリペプチドをコードする塩基配列からなる核酸。
〔4〕 セリンO−結合型ガラクトース転移酵素活性を有するポリペプチドをコードする核酸であって、配列番号20に示される塩基配列、又は当該塩基配列の相補配列とストリンジェントな条件下でハイブリダイズする塩基配列からなる核酸。
〔5〕 前記〔3〕又は〔4〕に記載の核酸を組み込んだ発現ベクター。
〔6〕 前記〔3〕又は〔4〕に記載の核酸が発現可能に組みこまれた形質転換体。
〔7〕 前記〔6〕に記載の形質転換体を用いたO−結合型糖鎖を有する組換え糖タンパク質の製造方法であって、当該形質転換体が外来性糖タンパク質をコードする核酸により形質転換されていることを特徴とする、セリンO−結合型ガラクトースを含む糖鎖を有する組換え糖タンパク質の製造方法。
〔8〕 前記〔2〕に記載された、SGT酵素活性を有する活性ドメインポリペプチドに結合する阻害物質により当該ポリペプチドのSGT酵素活性を阻害することを特徴とする、形質転換植物細胞または形質転換植物における外来タンパク質のセリン残基へのO−結合型ガラクトース付加抑制方法。
〔9〕 形質転換植物細胞または形質転換植物により外来タンパク質を産生する際に、外来タンパク質に含まれるセリン残基へのO−結合型ガラクトース付加を抑制する方法であって、形質転換宿主となる植物細胞が本来有しているSGT遺伝子である、前記[3]又は[4]に記載されたSGTをコードする核酸の発現を抑制することを特徴とする、形質転換植物細胞または形質転換植物における外来タンパク質のセリン残基へのO−結合型ガラクトース付加抑制方法。
〔10〕 前記〔8〕又は〔9〕に記載の方法により外来タンパク質のセリン残基へのO−結合型ガラクトース付加が抑制された、形質転換植物細胞または形質転換植物。
〔11〕 形質転換植物細胞または形質転換植物がタバコである前記〔10〕に記載の形質転換植物細胞または形質転換植物。
【発明の効果】
【0013】
本発明によって、汎用的な形質転換宿主であるタバコ由来のSGT遺伝子が提供できたので、外来遺伝子を宿主細胞に導入する際に、本発明のタバコ由来のSGT遺伝子を同時に導入するか、又は前もって導入しておくことで、O−結合型ガラクトースが高付加された外来タンパク質を生産することができる。
反対に、本発明においては、ヒトなど哺乳動物由来タンパク質をタバコなどの形質転換植物により大量生産させる際に問題となる、O−結合型ガラクトースの付加を抑制するためのターゲット遺伝子が特定できたことであるから、RNAi法などの周知の植物体内の酵素遺伝子発現の抑制化技術を適用することで、SGT遺伝子の発現を抑え、外来タンパク質のセリンにO−結合型ガラクトースが付加するのを抑制することができる。公知のプロリン水酸化酵素発現、植物特有のN−結合型の各種糖鎖転移酵素の発現も同時に抑制することで、形質転換植物体内で発現する植物特有の糖鎖結合が阻止できるから、ヒトの生理活性タンパク質なども形質転換植物で安価に大量生産可能となる。抗原性のないバイオ医薬の安価な生産の可能性を開く技術である。
【図面の簡単な説明】
【0014】
【図1】[SGT粗酵素の活性測定] クラミドモナスの細胞破砕液をSGT粗酵素液として用いて、「(HYP)AtEXT4:VYKSOOOOV」を基質ペプチドとして、UDP-ガラクトースを供与体基質として、12時間反応した場合、3種類のプロダクトピーク#1、#2及び#3が確認できた。
【図2】[ガラクトース付加部位の特定(MS解析、β−ガラクトシダーゼ処理)] クラミドモナスSGT酵素反応産物を、MS解析した結果、162Daのピークのシフトが観察され、プロダクト#1及び#2は、基質ペプチドに1残基のヘキソース(ガラクトース)の付加が、プロダクト#3は2残基のヘキソース(ガラクトース)の付加が確認できた。SGT酵素反応産物を、β−ガラクトシダーゼ処理したところ、プロダクト#3は#1に、#2は基質ペプチドの溶出位置にピークがシフトした。これらのことから、プロダクト#1は基質ペプチドのセリン残基(S)にガラクトースが1残基付加したもの、プロダクト#2は、いずれかのヒドロキシプロリン残基(O)にガラクトースが1残基付加したもの、プロダクト#3は、セリン残基とヒドロキシプロリン残基にそれぞれ1残基ずつガラクトースが付加したものであることが明らかになった。
【図3AB】[プロダクト#1のガラクトース付加部位の特定(β−エリミネーション法による)] β−エリミネーション法は、O−結合型糖鎖をペプチドから切り離す反応であることが一般的に知られているが、ヒドロキシプロリンに結合したO−結合型糖鎖は切り離さないことが知られているので、β−エリミネーション処理することにより、ガラクトースがセリン残基あるいはヒドロキシプロリン残基のどちらに結合しているかを区別することができる。(A)では、プロダクト#1は基質ペプチドの分子量にガラクトースの分子量162Daの分子量シフトが観察され、(B)においてβ−エリミネーション処理した結果、ガラクトースが切れて、元の基質ペプチドの分子量のピークが現れたこと、さらに、DTT処理によりβ−エリミネーションにより切れたアミノ酸へのDTT付加が観察された。このことから、プロダクト#1は、セリンにガラクトースが付加していることが明らかになった。
【図3CD】[プロダクト#2のガラクトース付加部位の特定](D)は、(C)のプロダクト#2をβ−エリミネーション処理したものであるが、ピークのシフトは観察されず、ガラクトースは切られなかったことが分かることから、ヒドロキシプロリン残基にガラクトースが付加していたことが明らかとなった。
【図4】[クラミドモナスSGT(クラミドモナス細胞破砕液)を用いたAtEXTペプチドのSer残基に対するSGT酵素活性への最適反応条件の検討](A)ドナー基質の選択性−ドナー基質としては種々の糖ヌクレオチドのうちでUDP-ガラクトース(Gal)が選択的に利用される。ここで、Arafはアラビノフラクトース、GlcNacはN-アセチルグルコサミン、GlcAはグルクロン酸、Glcはグルコース、Xylはキシロースを示す。(B)2価金属イオン要求性−Mn2+が際だって高かった。(C)至適pH−図中、◆は、0.1Mの酢酸ナトリウム、■は、0.1MのMES-NaOH、▲は、0.1MのMOPS-NaOH、●は、0.1MのTris-HClをそれぞれ含有した緩衝液。(D)至適温度 20〜60℃条件でインキュベートした。至適温度は30℃。
【図5A】[クラミドモナスSGTの精製 (step2)]1st DEAE Sepharoseカラムによる分画後、SDS-PAGEでタンパク質バンドを検出。
【図5B】[クラミドモナスSGTの精製 (step3)]2nd DEAE Sepharoseカラムによる分画後、SDS-PAGEでタンパク質バンドを検出。
【図5C】[クラミドモナスSGTの精製 (step5)]2nd Sephadex G-200カラムによる分画後、SDS-PAGEでタンパク質バンドを検出。
【図6A】[精製クラミドモナスSGTタンパク質バンドの切り出し]四角で囲んだ精製SGTの4サンプルのバンドをゲルから切り出した。この4サンプルに対し、MS/MS分析を行い、ペプチド配列を決定し、クラミドモナスのゲノムデータベースに100%一致するORFを見出した。
【図6B】[精製SGTタンパク質バンドの切り出し]同定されたSGTのアミノ酸配列についてハイドロパシー解析を行ったところ、C末端に疎水領域のあるタンパク質と予測された。
【図7A】[タバコSGTと各種植物由来のSGTとのアミノ酸配列アラインメント] タバコ(Nt)由来SGTアミノ酸配列と、クラミドモナス(Cr)、ニセツリガネゴケ(Pp)、シロイヌナズナ(At)とのアラインメントを示す。太字はC末端側にある膜貫通領域を示す。直線及び二重線による下線は、それぞれCD1及びCD2領域を、波線は機能不明な領域を示す。濃い直線部はシグナル配列であり、四角で囲った領域は、DXDモチーフに相当する。
【図7B】[各種植物由来のSGTのアミノ酸配列アラインメント-1(参考)] クラミドモナス、プラシノ藻、ニセツリカネゴケ、イネ、シロイヌナズナ、ブドウ、ポプラ、トウゴマのSGT遺伝子から予測されるアミノ酸配列のアライメント。クラミドモナスのSGTと他の植物のSGTは、30%のアミノ酸同一性を示した。
【図7C】各種植物由来のSGTのアミノ酸配列アラインメント-2(参考)
【図7D】[タバコを含む各種植物由来のSGTの活性ドメイン(SGT1-CD)部分のアライメント] 図中、Ntはタバコ(Nicotiana tabacum)、Crはクラミドモナス(Chlamydomonas reinhardti)、Otはブラシノ藻(Ostreococcus tauri)、Ppはニセツリガネゴケ(Physcomitrella patens)、Smはイヌタカヒバ(Selaginella moellendorffii)、Osはイネ(Oryza sativa)、Atはシロイヌナズナ(Arabidopsis thaliana)、Ptはポプラ(Populus trichocarpa)。以下同様。
【図7E】SGTの活性ドメイン(SGT1-CD)の系統樹
【図8】[SGT1の推定ドメイン構造] 各種植物のSGT1の推定ドメイン構造を図示した。N末端に分泌系のシグナルペプチド、C末端側に膜貫通領域があり、I型膜タンパク質であった。クラミドモナスおよびブラシノ藻は活性ドメインが1つだけであったが、陸上植物では2つずつあり、N末端側をSGT1-CD1、C末端側をSGT1-CD2とした。図中、SSはシグナルペプチド、CDはSGT酵素活性ドメイン、Sはスペイサー、TはTOX-1ドメイン、Pはプロリンリッチドメイン、TMは膜貫通領域、Bは塩基性アミノ酸リッチ領域。
【図9】[クラミドモナスSGT1の酵母における発現] クラミドモナスSGT1(CrSGT1、又はChSGT1ともいう。)を酵母細胞壁タンパク質PIR4に融合して発現させ、SGT酵素活性を測定した。Chlamy-S10-12hrsはクラミドモナス細胞破砕液の酵素活性。PIR4-HA-ΔSSΔTMは、シグナルペプチドと膜貫通領域を削除したChSGT1をPIR4に融合して発現。プロダクト#1だけが検出され、ChSGT1遺伝子がSGT活性だけを持つことを確認。
【図10】[シロイヌナズナのSGT1の酵母における発現] シロイヌナズナSGT1(AtSGT1)を酵母細胞壁タンパク質PIR4に融合して発現させ、SGT酵素活性を測定した。PIR4-HA-ΔSSΔTMは、シグナルペプチドと膜貫通領域を削除した活性ドメインを2つとも含むAtSGT1をPIR4に融合して発現。プロダクト#1の位置に小さなピークが検出されたので、AtSGT1遺伝子はSGT活性を持つことを確認した。PIR4-HA-CTΔTMは、2つの活性ドメインのうちAtSGT1-CD1を削除し、AtSGT1-CD2の1つのみであり、プロダクト#1の位置にピークが検出されたことから、SGT活性ドメインは1つだけでもSGT酵素活性を持つことが分かった。
【図11】[シロイヌナズナSGT1変異株における酵素活性の減少(HPLC解析)] 図中WT1〜3は正常株、Δsgtl1〜3はSGT1遺伝子のT-DNA変異株、CrSGT1は、図9、10のChlamy-S10-12hrsと同じクラミドモナス細胞破砕液の酵素活性を示す。
【図12】[シロイヌナズナSGT1変異株におけるO結合型ガラクトース付加の減少]A:糖鎖から遊離されたαガラクトース量、B:糖鎖から遊離されたβガラクトース量。図中、●は正常株、▲はSGT1遺伝子のT-DNA変異株。
【図13】[タバコSGT1の酵母における発現] 図中、NtSGTΔSSΔTMは、シグナルペプチドと膜貫通領域を削除した活性ドメインを2つとも含むNtSGT1をPIR4に融合して発現したものであり、NtSGT-NTΔSSはCD1だけを含む融合遺伝子であるが、いずれもプロダクト#1の位置にSGT酵素産物は見られなかった。一方、NtSGT-CTΔTMは、CD2だけを含む融合遺伝子であるが、プロダクト#1が検出されており、CD2は単独でSGT酵素活性があることが確認できた。シロイヌナズナでもほぼ同様の結果を得ていることから、CD2は単独でSGT酵素活性があるのに対して、CD1の場合は、まだ理由は不明であるが、何らかの制御機構によりSGT活性が抑制的に制御されている可能性が示唆される。
【図14】[RNAiによるSGT転写抑制効果] A:RT-PCRの電気泳動結果−図中、RNAi1〜3はdsRNA発現ベクターを導入したタバコ培養細胞BY-2の3種類のクローンであり、wt1〜3はコントロールとして用いた非形質転換細胞。B:Aで検出したPCR産物のシグナル強度のグラフ化。C:BY-2細胞のマイクロソーム画分におけるSGT活性の減少。
【発明を実施するための形態】
【0015】
以下、本発明についてさらに詳細に説明する。
1.セリンO−結合型ガラクトース転移酵素(Peptidyl serine α-galactosyltransferase;SGT)について
(1−1)植物のO−結合型糖鎖
植物のO−結合型糖鎖構造は、大きく分けて2種類あることが知られている。ひとつは、タンパク質のアミノ酸配列中の複数のプロリン残基が水酸化されてヒドロキシプロリンになり、そこにガラクトースまたはアラビノースが付加する構造である。典型的な配列として、エクステンシンの配列「VYKSOOOOV(Oはヒドロキシプロリン)」がよく知られており、この複数のヒドロキシプロリン基のヒドロキシル基が糖修飾される。プロリン水酸化酵素はすでに同定されているが、ヒドロキシプロリンにガラクトースまたはアラビノースを付加するガラクトース転移酵素(HGT)及びアラビノース転移酵素は未だに単離されていない。
そして、もう一つの植物特有のO−結合型糖鎖構造が、タンパク質のアミノ酸配列中のセリン残基に対して直接ガラクトースが付加された糖鎖構造であり、この糖鎖修飾を行っている酵素が、本発明のセリンO−結合型ガラクトース転移酵素(SGT)であり、本発明においては、そのうちの特にタバコ由来のSGT(NtSGT)を対象とする。
【0016】
(1−2)本発明のタバコ由来SGT酵素及びそれをコードする遺伝子の配列
タバコは、典型的な遺伝子組換え宿主植物ではあるが、そのゲノム配列は公表されていないため、タバコのゲノムライブラリーなどから直接クローニングすることは難しいため、下記(1−3)に記載のシロイヌナズナなどの他の植物又は藻類のSGT遺伝子を同定し、次いでシロイヌナズナのSGT遺伝子(配列番号4)に基づいてプライマー、プローブを作製し、タバコ植物ゲノムに対して適用し、2箇所の活性ドメイン領域を有するタバコSGT酵素(配列番号21)の遺伝子クローニングに成功し、対応するタバコSGT酵素のアミノ酸配列(配列番号20)を決定したが、これらはいずれも新規酵素遺伝子及び新規ポリペプチドであるといえる。タバコSGT酵素遺伝子の登録番号としては、AB617524が付与された。
タバコSGT酵素活性ドメイン領域は、CD1が配列番号22、CD2が配列番号23で示され、特にCD2は単独でもSGT酵素活性を有することが実証されており、いずれも有用性のある新規ポリペプチドである。
進化系統樹的に最も近いSGT遺伝子はシロイヌナズナ及びポプラ由来SGT遺伝子であり、活性ドメインCD1ではそれぞれアミノ酸配列レベルで81%及び86%、CD2では80%及び83%と高い(下記表3)ものの、SGT遺伝子全体のホモロジーは72%及び76%とさほど高くない。
タバコ植物においても多品種が存在しているので、これら個体差に基づく相同遺伝子配列も含めると、タバコ由来SGT酵素活性を有するポリペプチド及び当該ポリペプチドをコードする遺伝子は、以下のように表すことができる。
すなわち、タバコ由来SGT酵素活性を有するポリペプチドは、以下のように表すことができる。
(1)配列番号21に示されるアミノ酸配列を含むポリペプチド。
(2)配列番号21に示されるアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、SGT酵素活性を有するポリペプチド。なお、ここで「1若しくは数個」というときの数個は、自然に変異が導入される程度の数値範囲を示すので、2〜15個、または2〜8個、2〜5個程度の数値を表す。
(3)配列番号21と80%以上好ましくは90%以上の同一性を有しているアミノ酸配列からなるポリペプチドであって、かつそのSGT酵素活性ドメインとして、DXDモチーフを有し、配列番号22又は23と90%以上好ましくは95%以上の同一性を有するアミノ酸配列からなる部分配列を有しているアミノ酸配列からなり、SGT酵素活性を有するポリペプチド。
(4)植物のゲノム中の塩基配列によりコードされるアミノ酸配列であって、配列番号21に示されるアミノ酸配列と80%以上好ましくは90%以上の同一性を有するアミノ酸配列からなり、かつDXDモチーフを含むアミノ酸配列からなり、SGT酵素活性を有するポリペプチド。
また、その「SGT酵素活性ドメイン」のCD1領域及びCD2領域についても、特にCD2領域は単独でSGT酵素活性を有することが実証されているから、以下のように表現できる。
(5)配列番号22又は23に示されるアミノ酸配列を含むポリペプチド。
(6)配列番号22又は23に示されるアミノ酸配列のいずれかにおいて1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、SGT酵素活性を有するポリペプチド。なお、ここで「1若しくは数個」というときの数個は、上記(2)の定義と同様である。
(7)配列番号22又は23に示されるアミノ酸配列と少なくとも90%以上好ましくは95%以上の同一性を有するアミノ酸配列であって、かつDXDモチーフを含むアミノ酸配列からなり、SGT酵素活性を有するポリペプチド。
【0017】
一方、本発明のタバコ由来SGT酵素遺伝子などのSGT酵素活性を有するポリペプチドをコードする核酸は、以下のように表現できる。
(8)上記(1)〜(7)に記載のポリペプチドをコードする塩基配列を含むSGT酵素活性を有するポリペプチドをコードする核酸。
(9)配列番号20に示される塩基配列、又は当該塩基配列の相補配列とストリンジェントな条件下でハイブリダイズする塩基配列からなる核酸であって、セリンO−結合型ガラクトース転移酵素活性を有するポリペプチドをコードする核酸。なお、ここで、ストリンジェントな条件とは、「Molecular Cloning 3rd edition Sambrook J ら、Cold Spring Harbor Laboratory Press, 2001」などの教科書にストリンジェンシーの高いハイブリダイズ条件として示されている条件を指す。
【0018】
(1−3)本発明のタバコ由来SGT酵素遺伝子及び当該SGT遺伝子と関連するSGT遺伝子一般について
本発明のタバコ由来SGT酵素遺伝子と関連したSGT遺伝子としては、クラミドモナス(Chlamdomonas reinhardtii)では、配列番号1の塩基配列(A8J5Z4_CHLRE配列)からなる遺伝子およびシロイヌナズナ由来の配列番号4の塩基配列(Q8VYF9、At3g01720配列)からなる遺伝子などがあるが、SGT遺伝子は単細胞藻類のクラミドモナスから、被子植物に至るまで植物全般で広く保存された遺伝子ファミリーである。動物や真菌など植物以外の生物種にはホモロジーのある遺伝子が存在しない、植物特有の遺伝子ファミリーであり、他のどの糖転移酵素とも類似性を持たず、全く新規な糖転移酵素遺伝子ファミリーである。
具体的には、クラミドモナス由来の配列番号1の塩基配列に対するBlast検索結果によれば、シロイヌナズナ(Arabidopsis thaliana)、イネ(Oryza sativa)、トウゴマ(Ricinus communis)、ブドウ(Vitis vinifera)、ポプラ(Populus trichocarpa)、ニセツリガネゴケ(Physcomitrella patens)、ブラシノ藻(Ostreococcus tauri)などに、約30%identityのホモログORFが1ゲノムあたりそれぞれ1ORFずつ存在していた(表4)。広葉植物同士では、70%程度のアミノ酸の同一性があり、被子植物間では60%程度、被子植物と単細胞緑藻の間では30%のアミノ酸配列の同一性が認められた。しかし、動物や真菌にはホモロジーのある遺伝子は見つからなかった(表4)。上記各種植物、藻類のSGT1遺伝子に対して付与された登録番号は、以下の通りである。クラミドモナス:AB617522、シロイヌナズナ:AB617523、イネ:BR000899、ポプラ:BR000900、ブドウ:BR000901、トウゴマ:BR000902、ニセツリガネゴケ:BR000904およびBR000905、ブラシノ藻:BR000906。またその他のSGT1遺伝子に対しても下記の登録番号が付与された。ソルグム(Sorghum bicolor ):BR000903、イヌタカヒバ(Selaginella moellendorffii):BR000909、ミヤマハタザオ(Arabidopsis lyrata):BR000908、ボルボックス(Volvox carteri):BR000907、クロレラ(Chlorella variabilis):BR000910。SGT1のドメイン構造は、糖転移酵素としては珍しく、I型膜タンパク質のトポロジーであり、膜貫通領域がC末側に存在する(図8)。また、クラミドモナスのSGT1のN末端側のホモロジーの高い領域(配列番号17)のみでも活性があり、当該領域中に活性ドメインがあると推定される(Chla組換えSGT1)。図7での各植物間のアラインメントの検討の結果、図8に示すように共通した保存配列としての活性ドメイン領域が明らかとなった。
また、すべての植物のSGT1-CD1及びSGT1-CD2ドメイン内では、糖ヌクレオチドを基質とする糖転移酵素に広く保存されているDXDモチーフ(Mn2+イオンを介して糖ヌクレオチドとの結合部位)(非特許文献19)が保存されていることからも、当該領域が各植物共通の活性ドメインであることを裏付けている(図7D)。
該SGT1活性ドメイン(CD)は、単細胞藻類のクラミドモナス及びブラシノ藻ではN末側の1箇所である(SGT1-CD1)が、ニセツリガネゴケからシロイヌナズナなどの高等植物では、ホモロジーの高い活性ドメイン領域がN末端側とC末端側にタンデムに重複して存在していた(SGT1-CD1及びSGT1-CD2)。各高等植物におけるSGT1-CD1及びSGT1-CD2領域との領域同士のホモロジーは、アミノ酸配列で58%程度の同一性であった。シロイヌナズナにおいてSGT1-CD1及びSGT1-CD2領域それぞれのSGT活性を測定したところ、シロイヌナズナ活性領域においても、SGT1-CD2領域(PIR4-HA-CTΔTM、配列番号18)だけでも単独でSGT活性があることが確認された(図9、At組換えSGT1)。
クラミドモナスのSGT1には、高等植物のプロリン水酸化酵素のER-ゴルジ局在配列Tox1が存在し、ER-ゴルジの局在が示唆された。このことから、セリン残基へのガラクトース付加は、糖タンパク質合成の早い段階で付加が起きると考えられる。高等植物のSGT1には、Tox1配列は見つかっていない。
SGT1をコードする塩基配列としては、クラミドモナスおよびブラシノ藻では、複数のホモロジーのあるORFが検索されるが、ニセツリガネゴケからシロイヌナズナなどの各植物のSGT遺伝子についてデータベースの配列情報を検索するといくつかの異なったデータが存在するが、DNA配列及び遺伝子座を詳細に検討してみると、各データ間の配列上の相違は、オールタナティブスプライシング、遺伝子の欠失挿入変異、シークエンス解析のミス、品種による遺伝子の多型などが原因であり、結局、各植物ゲノムあたり1種類の遺伝子しかないことが確認された。
【0019】
(1−4)本発明のタバコ由来SGT酵素遺伝子クローニング
本発明者は、本実施例で詳細に述べるように、先の出願(PCT/JP2010/071598)で取得したシロイヌナズナのSGT遺伝子(配列番号4)に基づいてプライマー、プローブを作製し、アミノ酸配列、及び塩基配列が公開されていないタバコ植物ゲノムに対して適用したところ、2箇所の活性ドメイン領域を有するタバコSGT酵素(配列番号21)の遺伝子クローニングに成功した。
タバコは、典型的な遺伝子組換え宿主植物ではあるが、そのゲノム配列は公表されていないため、上記(1−2)で述べたように、今回クローニングしたタバコSGT酵素のアミノ酸配列(配列番号20)及びその遺伝子配列(配列番号21)、並びにそのSGT酵素活性ドメインCD1及びCD2のアミノ酸配列(配列番号22、23)はいずれも新規ポリペプチド及び新規遺伝子であるといえる。
そして、下記(1−6)、(1−7)に記載したように、これらポリペプチド及びポリヌクレオチドは、タンパク質のセリン残基にガラクトースを付加する酵素製剤及び形質転換宿主細胞内のタンパク質、特に外来タンパク質に対して、そのセリン残基にガラクトースを付加するポリヌクレオチド製剤の有効成分として用いることができる。
【0020】
(1−5)本発明のSGT酵素の作用及び基質特異性
本発明のタバコ由来SGTを含めSGT酵素の作用は、下記の反応式で表される。
【0021】
【化1】

【0022】
基質特異性としては、セリンを含有するポリペプチドであり、好ましくは上記式中のR2としてヒドロキシプロリンが複数存在することが好ましい。
至適pHはpH6.0、至適温度は30℃で、反応温度は50℃以下である(図4)。等電点は中性付近であり、2価金属イオン要求性としては、マンガンイオン(Mn2+)要求性が観察された。
【0023】
(1−6)本発明におけるSGT酵素活性ポリペプチド及びSGT酵素活性酵素製剤
以上のことから、本発明において「SGT酵素活性」というとき、Mn2+イオン存在下でポリペプチド中のセリン残基のヒドロキシル基に対してUDP-ガラクトースからガラクトースを転移する酵素活性を指す。
そして、本発明のSGT酵素は、対象のタンパク質に対して、Mn2+イオン存在下でアミノ酸配列中のセリン残基におけるヒドロキシル基にガラクトースを付加する作用を有するので、当該酵素を有効成分とする酵素製剤として用いることができる。
本発明のSGT酵素遺伝子と関連した各植物のSGT酵素遺伝子は、緑藻から高等陸上植物まで高い相同性の活性ドメインがあるので、データベース上でも、またはcDNAライブラリー、もしくはゲノムDNAライブラリー中でもきわめて検索しやすく、しかも、各ゲノム中でそれぞれ1種類しかないので、SGT遺伝子を同定しやすい。
具体的には、各植物のSGT酵素の活性ドメインは、クラミドモナスSGT酵素の活性ドメイン(配列番号3)又はシロイヌナズナSGT酵素の活性ドメインAtSGT1-CD1(配列番号6)、AtSGT1-CD2(配列番号7)、イネSGT酵素の活性ドメインOryzSGT1-CD1(配列番号10)、OryzSGT1-CD2(配列番号11)、ニセツリガネゴケSGT酵素の活性ドメインPhysSGT1-CD1(配列番号13)、PhysSGT1-CD2(配列番号14)、ブラシノ藻SGT酵素の活性ドメインOstreoSGT1-CD1(配列番号16)として同定された(本出願人の先願であるPCT/JP2010/071598)。上述のように、各SGT酵素内のCD1領域とCD2領域とのホモロジーがアミノ酸配列で57%〜60%であるが、各植物のCD1領域同士のホモロジーは37%〜63%程度であることから、さらには各陸上植物のCD1領域同士のホモロジーは60%〜100%程度であることから、本発明の「CD領域」といえるためには、少なくともアミノ酸配列で50%(好ましくは60%〜100%)のホモロジー(なお、本発明でのホモロジー又は相同性の数値は、塩基配列はBlast検索により、アミノ酸配列の場合はFasta検索結果における「同一性」の数値を示している。)を有し、かつDXDモチーフを有するものを指す。単細胞藻類の場合は当該活性化ドメインを1箇所、多細胞植物であれば2箇所有している。したがって、ここに記載した植物以外の植物由来であっても、上記配列番号3,6,7,10,11,13,14又は16に示されるいずれかのアミノ酸配列と50%以上、好ましくは60%以上、より好ましくは70%以上の相同性を有し、かつDXDモチーフを有するアミノ酸配列であって、ゲノム中に1又は2箇所しか対応する塩基配列が存在しない場合は、SGT酵素の活性ドメインであると考えられる。
一方、上記各植物のSGT酵素の全長は、クラミドモナスSGT酵素のアミノ酸配列(配列番号2)、シロイヌナズナSGT酵素のアミノ酸配列(配列番号5)、イネのSGT酵素のアミノ酸配列(配列番号9)、ニセツリガネゴケSGT酵素(配列番号12)、ブラシノ藻SGT酵素(配列番号15)であると同定され、一般に各植物の全長SGT酵素のアミノ酸配列の相同性は、まれには70%以上のこともあるが、通常30%〜50%の範囲内である。つまり、上記植物以外の植物由来の場合にも、ゲノム中に対応する相同配列が1箇所しかなく、上記配列番号2,5,9,12もしくは15のアミノ酸配列と30%以上、好ましくは50%以上、より好ましくは70%以上の範囲内にあって、かつ上記SGT酵素の活性ドメインに相当する配列が1又は2箇所存在する場合ば、ほぼ確実にSGT酵素であると考えられる。
【0024】
(1−7)本発明のタバコSGT遺伝子を用いるポリヌクレオチド製剤
上記(1−2)で定義された本発明のタバコ由来SGT遺伝子、又は当該遺伝子を含む発現ベクターを用いて宿主細胞を形質転換することにより、哺乳動物細胞など本来SGT酵素活性を持たない細胞に対してSGT酵素活性を付与する、又は植物細胞に対してSGT活性を増強することができる。すなわち、上記SGT遺伝子又は当該遺伝子を含む発現ベクターを有効成分とするポリヌクレオチド製剤は、形質転換宿主細胞内のタンパク質、特に外来タンパク質に対して、そのセリン残基にガラクトースを付加する作用を有するものである。
【0025】
2.本発明におけるSGT酵素又はSGT酵素遺伝子を用いた、セリンO−結合型ガラクトース含有ポリペプチドの製造方法
(2−1)本発明のタバコSGT酵素又はSGT酵素活性ポリペプチドを用いたセリンO−結合型ガラクトース含有ポリペプチドの製造方法
本発明のSGT酵素又はSGT酵素活性ポリペプチドは、各種タバコ植物から単離精製して用いてもよいが、細胞を破砕し、不溶性の膜画分を再懸濁した細胞破砕液のままで用いることもできる。
2〜5mM Mn2+イオンを含有させた溶液中に糖鎖付加をしたいポリペプチドを0.1mM程度含有させ、ガラクトース供与体としてUDP-ガラクトースを1〜5mM含有させ、pH5.5〜7.0程度に調整した溶液に対して、精製酵素又は精製ポリペプチドが0.01〜0.05mg/l程度になるように添加し、20〜35℃で1〜12時間程度作用させればよい。
【0026】
(2−2)タバコSGT酵素又はSGT酵素活性ポリペプチドをコードする遺伝子を用いたセリンO−結合型ガラクトース含有ポリペプチドの製造方法
本発明のタバコSGT酵素又はSGT酵素活性ポリペプチドをコードする遺伝子を導入し、発現させるための形質転換体としては、典型的にはサッカロミセス・セレビッシエ酵母が挙げられるが、糖鎖付加機構を持たない大腸菌などの細菌類以外であれば、各種植物細胞、CHO細胞などの哺乳動物細胞、昆虫細胞などを宿主として選択することができる。その際の導入ベクターは、周知の発現ベクターを適宜選択できる。
本発明のタバコSGT酵素又はSGT酵素活性ポリペプチドによりセリンO−結合型ガラクトースを付加させる対象となるポリペプチドとしては、セリンを分子内に含めばどのようなポリペプチドであっても良い。例えば、エクステンシン、スポラミン、ムチンコアタンパク質などが考えられる。
【0027】
3.本発明におけるSGT酵素活性の抑制方法
(3−1)SGT酵素活性阻害剤を用いたSGT酵素活性の抑制
このようなSGT酵素活性を阻害するためには、活性ドメインをエピトープとする抗体、すなわち抗NtSGT1-CD1抗体、抗NtSGT1CD2抗体又はそれらのフラグメントを用いることができる。また、タバコSGT酵素の活性ドメインに結合するUDP-ガラクトースのアンタゴニスト物質であればよく、例えば各種のガラクトース誘導体などが阻害剤として働く。
【0028】
(3−2)SGT酵素遺伝子発現抑制剤を用いたSGT酵素活性の抑制
本発明のSGT酵素遺伝子発現抑制のためには、活性ドメインの遺伝子配列をターゲットとして、相同組換え法、各種RNAi物質(siRNA, miRNA,dsRNA)による抑制方法、アンチセンス法、トランスポゾン挿入法、T-DNA挿入法による遺伝子の破壊株を用いる方法等を用いてもよく、特にRNAi法が簡便で好ましい。
本発明の実施例では、タバコ植物細胞内のSGT遺伝子の転写をヘアピン型二本鎖RNA(dsRNA)発現プラスミドを用いたRNAiにより抑制できることを実証したが、この手法に限られるものではない。
例えば、植物型N型糖鎖転移酵素に対して用いられた各種方法(非特許文献20)が適用できる。具体的には、非特許文献20のレビュー文献中には、シロイヌナズナのN型糖鎖にα1,3−フコースあるいはβ1,2−キシロースを付加する糖転移酵素遺伝子が、アグロバクテリウムのT-DNAをランダムに挿入した変異株ライブラリーから当該遺伝子にT-DNAが挿入された株を選択し、さらに掛け合わせによりホモにT-DNAが挿入された株を作製する変異株作製法を用いることで、α1,3−フコースあるいはβ1,2−キシロースを付加が達成できることが示されている。また、ウキクサにおいて、RNAi法を用いた例として、α1,3−フコースおよびβ1,2−キシロースを付加する糖転移酵素活性が抑制され、当該形質転換植物体で発現されたヒトCD30には、α1,3−フコースおよびβ1,2−キシロースが付加されないことも示されている。さらに、RNAi法は、タバコ、アルファルファにおいてもα1,3−フコースおよびβ1,2−キシロースの付加が抑制されたことが示されている。
本発明において、汎用的な形質転換宿主となるタバコ由来のSGT酵素という標的遺伝子が提供され、しかもそのターゲットとなる領域としての活性ドメイン配列が特定されたことにより、これら周知の糖転移酵素遺伝子抑制方法が、そのまま本発明においても適用できることとなった。
【0029】
(3−3)SGT酵素遺伝子発現が抑制された形質転換植物及び形質転換植物におけるO−結合型糖鎖の付加を抑制した外来タンパク質の製造技術
本発明で用いる形質転換植物又は形質転換植物細胞としては、タバコ植物が好ましいが、ナス科植物などの双子葉植物の他、イネなどの単子葉植物に適用してもよい。
本発明で、タバコなどの植物細胞又は植物体から製造しようとするO−結合型糖鎖の付加を抑制した外来タンパク質の典型例は、ヒト由来のホルモン、サイトカイン、抗体などのバイオ医薬としての用途が期待されている生理活性タンパク質であるが、酵素等幅広く応用可能である。
【0030】
4.本発明におけるSGT酵素活性測定方法
本発明のSGT酵素活性を測定する際に用いた方法は、本出願人の先のPCT出願(PCT/JP2010/071598)に記載の方法と同様の手法であり、夾雑物が含まれる粗抽出液の状態でもHGT酵素活性など他の酵素活性に左右されず、SGT酵素活性のみを測定できる手法である。そのために、受容体基質ポリペプチドとして、「KSOOOO」を含む、例えば「VYKSOOOO」(配列番号19)からなるポリペプチドを標識化して用いる。標識としては典型的には蛍光色素(FITC)を用いるので、FITC-KSOOOOなどと表記される。
また、例えば、「FITC-KSOOOO」の場合に、蛍光が弱ければ、あらかじめ「FITC-KSOOOOV」を用いて、HGT酵素活性+SGT酵素活性を検出しておき、次いで「FITC-KSOOOO」を用いることで、確実なSGT酵素活性の測定が可能となる。
また、SGT酵素活性用キットとするときは、糖基質としてUDP-ガラクトース及びMn2+イオン供給化合物、例えばMnCl2などを用いる。
本発明のSGT酵素活性測定方法において、測定対象となる植物由来の酵素源は、細胞の破砕液、細胞から調製した膜画分の再懸濁液、膜画分を界面活性剤などで可溶化した粗酵素液、SGT酵素の精製品、SGT1遺伝子を宿主細胞あるいは無細胞発現系で発現させたリコンビナントSGT酵素など、いずれでもよい。
一般的には、2〜5mM Mn2+イオンを含有させた溶液中に、「FITC-VYKSOOOOV」などの受容体基質ポリペプチドを0.1mM程度含有させ、ガラクトース供与体としてUDP-ガラクトースを1〜5mM含有させ、pH5.5〜7.0程度に調整した溶液に対して、SGT酵素を添加し、合計液量25μlで、20〜35℃で1〜12時間程度作用させればよい。
また、RNAi法などにより、植物細胞内でのSGT酵素活性が抑制されたかどうかを評価する際にも上記方法が適用できる。
SGT遺伝子転写抑制効果を直接判定してもよく、その際の典型的な手法としてはRT-PCRによりSGT遺伝子の転写産物量を測定する手法が、定量的な評価もできるため好ましい。
【実施例】
【0031】
以下、実施例を用いて本発明を詳細に説明するが、本発明の技術的範囲は以下の実施例に限定されるものではない。なお、特に断りがない場合は、公知の方法、例えばMolecular Cloning 3rd edition Sambrook J ら、Cold Spring Harbor Laboratory Press, 2001、細胞工学別冊「バイオ実験イラストレイテッド」(秀潤社、2001年)などの記載、又は市販の試薬やキット等の指示書に従って実施可能である。また、本明細書で引用した文献中の記載は本明細書中の記載として組み入れるものとする。
【0032】
〔実施例1〕高等植物細胞抽出液の調製
シロイヌナズナ(Arabidopsis thaliana)の培養細胞はT87を用いた。植物細胞の培養は、Murashige and Skoog培地(2,4-dichlorophenoxyacetic acid and 3% sucroseを添加)を用い、200mlずつを1リットルのフラスコで、2週間、25℃で120 rpm震盪培養した。細胞をフィルターで集め、乳鉢にうつして液体窒素を加えながら、乳棒で破砕して、粉状に破砕した。破砕した細胞は、Buffer A (100 mM MOPS-NaOH (pH7.0), 1 mM MnCl2 with the Complete, EDTA-free protease inhibitor (1 tablet of Complete/50 ml, Roche, Mannheim, Germany)に懸濁し、さらに4℃で10分間 3,000 ×g.で遠心し、破砕残渣を取り除いて、上清を細胞破砕液とした。細胞破砕液をさらに4℃で10分間160,000×g.で遠心し、その沈殿をBuffer A に再懸濁した。さらに10 mM ammonium-acetate buffer (pH 7.0)に対して4℃で一晩、透析した。タンパク質の量はBCA法で測定した。これをシロイヌナズナ植物培養細胞の細胞抽出液ミクロソーム画分とした。
同様の手法で、タバコからの細胞抽出液ミクロソーム画分を得た。
【0033】
〔実施例2〕クラミドモナスの細胞壁レス変異株からの細胞抽出液の調製
全ての高等植物の祖先といわれる単細胞の緑藻の1種であるクラミドモナス(Chlamydomonas reinhardtii)は、様々な生化学的解析法、遺伝学およびゲノム解析が発展しており、多くの変異株の利用も可能であり、研究開発が容易な植物の一つである。クラミドモナスの細胞壁レス変異株(Cell wall-less mutant; CC-503, CC-2656, CC-3491)は、細胞壁がほとんどなく、簡単な操作で細胞を破砕しやすいため、CC-503は界面活性剤で破砕が可能であった。比較のためクラミドモナス正常株(CC-125, CC1690)も使用したが、正常株では界面活性剤で破砕はできず、超音波による破砕が必要であった。また、クラミドモナスは高等植物と同様のO−結合型糖鎖構造を有する植物タンパク質であるエクステンシンを生産しており、しかも、エクステンシン産生量が多いことが知られている。エクステンシンは、細胞壁を構成するタンパク質であるが、クラミドモナスの細胞壁レス変異株では、細胞壁不全の細胞壁を補強しようとしてエクステンシン産生量が多くなり、そのため、SGTを含めO−結合型糖鎖付加酵素の遺伝子を過剰発現している可能性が高いと考えられるので、まずSGT高含有粗酵素液を得るために、クラミドモナスCC-503株の細胞抽出液を得ることとした。
クラミドモナス細胞壁レス変異株は、クラミドモナス培地(0.2% Trypton、0.1% Bacto peptone、0.2% Yeast extract、0.1% Sodium acetate、0.001% CaCl2)で前培養を1リットルずつ1週間、本培養を10リットルずつ25℃で1週間培養した。培養したクラミドモナス細胞を、10分間 10,000×g.の遠心で集めた。集菌したクラミドモナスの細胞は、-80℃で保存可能である。クラミドモナス細胞を、400mlのバッファー(10mM Tris-HCl pH7.6, 0.1% Triton X-100)中で溶菌した。溶菌液を4℃で10分間 10,000×g.で遠心し、沈殿の破砕残渣を取り除いて、上清(S10)をクラミドモナスの細胞破砕液とした。
クラミドモナスの細胞破砕液(S10)画分を、超遠心(160,000×g.)60分間4℃で行い、ゴルジ体を多く含む沈殿画分P100を調製した。P100を400mlバッファー(10mM Tris-HCl pH7.6 , 0.2% Triton X-100)に懸濁し、4℃で8時間振とうして、SGTを可溶化した。これを再び超遠心(160,000×g.)60分間4℃で行い、上清を粗酵素液SL-S100 画分とした。
【0034】
〔実施例3〕クラミドモナスおよびシロイヌナズナの粗酵素液を用いたO−結合型ガラクトース活性の測定
実施例1で得られたクラミドモナスの粗酵素液SL-S100 画分およびシロイヌナズナの細胞抽出液ミクロソーム画分をSGTの粗酵素液として以下の実験に用いた。
糖転移酵素は、一般に基質特異性が厳格であることが知られており、適切な供与体基質と受容体基質を選択しないと、酵素活性を検出することができない。従来知られているO−結合型糖鎖合成に関与する糖転移酵素では、供与体基質としては、UDP、GDP、CMPなどのヌクレオチドが糖に結合した糖ヌクレオチド、及び脂質の一種であるドリコール−二リン酸に糖が結合したドリコール−二リン酸糖が知られている。小胞体に局在し、複数回膜貫通領域を持ち、糖鎖合成の早い段階で機能する糖転移酵素にドリコール−リン酸糖を供与体基質とするものが多いことが知られている。一方、小胞体からゴルジ体に局在し、N末端側に1回の膜貫通領域を持つ糖転移酵素は、糖ヌクレオチドを供与体基質とするものが多いことが知られている。
シロイヌナズナの細胞壁タンパク質AGP14のヒドロキシプロリンにO−ガラクトースを転移するHGTは、UDP-ガラクトースを供与体基質としていることが解明されていたので、SGTも同様の基質特異性をもつ可能性が高いと考えられたため、SGT活性測定の供与体基質として、UDP-ガラクトースを用いた。
タバコからの細胞抽出液ミクロソーム画分に対しても、シロイヌナズナの場合と同様にSGT活性を測定し、O−ガラクトース付加活性の存在を確認した(図示せず)。
受容体基質として、他のO−結合型糖鎖を合成する糖転移酵素の活性測定法と同様に、合成ペプチドを用いた。ペプチドは、HPLC等による活性測定で検出を容易にするために、FITCで蛍光標識をした。合成ペプチドのアミノ酸配列は、当初、セリン残基を含み、かつ植物においてセリン残基にO−ガラクトース付加の報告があるものとして、O−ガラクトース付加をタバコ細胞で確認していたスポラミン配列「FITC-PTTHEPASSETPVL」および植物細胞壁タンパク質の解析でセリン残基にガラクトース付加が知られていたエクステンシン配列「VYKSPPPPV」、哺乳動物由来のppGalNAcTの酵素活性測定で実績のある動物細胞のムチンコアタンパク質配列、等を受容体基質として用いてSGT酵素活性の検出を試みたが、ガラクトース転移酵素活性は確認できなかった。プロリンを水酸化したスポラミン配列「FITC-PTTHEOASSETPVL」を受容体基質としても酵素活性は検出できなかったが、プロリンを水酸化したエクステンシン配列「VYKSOOOOV」を受容体基質としたところ、HPLCで酵素反応物のピークを検出でき、ガラクトース転移酵素活性を確認できた。したがって、「FITC-VYKSOOOOV」を受容体基質とすることによるガラクトース転移酵素活性測定が可能となった。ただし、この段階では、ガラクトースがセリン残基あるいはヒドロキシプロリン残基に付加したのかは不明であった。連続するヒドロキシプロリン残基には、O−アラビノースの付加の報告があるだけで、O−ガラクトースの付加はこれまで報告がなかった。しかし、FITC-VYKSOOOOVには、予想に反して2残基のガラクトース付加が認められ、詳細に解析したところ、セリン残基へのガラクトース付加だけでなく、ヒドロキシプロリン残基へのガラクトース付加の存在が明らかとなった。HPLC解析でSGT活性とHGT活性のピークを区別して検出することができたため、SGT酵素活性測定に初めて成功した。
【0035】
SGT酵素活性測定の詳細な条件として、酵素反応を行う溶液の構成は、100mM MOPS緩衝液(pH7.0)、4mM MnCl2、10mM UDP-ガラクトース、0.1mM FITC標識ペプチド、SGT粗酵素液5μlを加え、合計25μlとした。あるいは100mM MOPS緩衝液(pH7.0)、5mM MnCl2、5mM UDP−ガラクトース、25μM FITC標識ペプチド、2mM EDTA、5×Protease Inhibitor Cocktail、0.1% TritonX-100、酵素液を加えて6.25μl、あるいは0.2M MOPS pH7.0、0.1mM FITC-Peptide(VYKSOOOOV-Ac1)、0.2% TritonX-100、10mM UDP-Galactose、4mM MnCl2、2mM EDTA、5×Inhibitor cocktailに酵素液を加えて12.5μl とした。これらを30℃で1〜12時間保温して酵素反応を行った。酵素反応は、95℃3分の熱処理を行って停止した。
酵素反応産物はHPLC(島津製作所RF-10A XL)で蛍光検出して解析した。逆相カラム5C18-AR-II(250×4.6 mm; Nacalai Tesque, Kyoto, Japan)を用い、カラムを0.1%トリフルオロ酢酸を含む22.0%アセトニトリルで平衡後、22.0%から22.5%の濃度勾配を流速1ml/minで30分かけて溶出し、FITCを488nmで励起し、530nmで検出した。受容体基質のペプチドは、この条件で、約19.0分に溶出し、セリン残基にガラクトースが1残基付加した酵素反応産物であるプロダクト#1は約15.5分に溶出された。ヒドロキシプロリン残基に1残基のガラクトースが付加したプロダクト#2は約16.5分に、セリンとヒドロキシプロリンに1残基ずつ合計2残基のガラクトース付加したプロダクト#3は約14.5分に溶出した。(図1)
【0036】
〔実施例4〕SGT酵素反応産物セリンOガラクトースの構造解析
SGT酵素反応産物をそれぞれ分取してMS解析を行った結果、#1および#2は、ヘキソースが1残基深し、#3はヘキソースが2残基付加していることが確認された。高等植物のセリン残基へのガラクトース付加は、α結合であり、ヒドロキシプロリン残基へのガラクトース付加はβ結合であることが分かっているので、アスペルジルス由来βガラクトシダーゼ処理をしたところ、#1は変化せず、#2と#3は消失した(図2)。ピークを分取して、それぞれβガラクトシダーゼを処理すると、プロダクト#2はペプチド基質に、プロダクト#3はプロダクト#1の位置にピークがシフトした。このことから、ヒドロキシプロリン残基に付加していたガラクトースが消化されたことが示された。
βエリミネーションは、O−結合型糖鎖を切り離すのに一般的に使われる手法であるが、環状アミノ酸であるヒドロキシプロリンにO−ガラクトースが付加したものは、糖を離脱しないことが知られている(Wells et al 2002)。#2をβエリミネーションしても、切れなかった(図3CD)。#3をβエリミネーションすると、切れたものと、DTT付加したものが検出された。#1をβエリミネーションすると、切れて、DTT付加するものが検出された(図3A、B)。エクステンシンのセリンをアラニンに換えたペプチドでは、ピークが一つだけ検出され、βガラクトシダーゼで切れた。
これらの結果を総合すると、プロダクト#1は、SGTによりセリン残基にO−ガラクトースが付加したものであり、プロダクト#2は、ヒドロキシプロリン−O−ガラクトース転移酵素(HGT)によりヒドロキシプロリン残基にO−ガラクトースが付加したものであり、プロダクト#3は、SGTとHGTによりO−ガラクトースが2残基付加したものであることが分かった。
このことから、この細胞破砕液には、SGTに加え、HGTも含まれていることがわかり、SGT及びHGT酵素活性を区別して測定し、セリン残基にO−ガラクトース付加およびヒドロキシプロリンにO−ガラクトース付加したペプチドをそれぞれ分離して検出する方法が確立できた。
【0037】
〔実施例5〕SGTの基質特異性
(5−1)クラミドモナスSGT粗酵素液を用いた基質特異性
供与体基質は、UDP-ガラクトースを用いた。受容体基質は、FITCで蛍光標識をした合成ペプチドのアミノ酸配列を以下に記述するように様々なものを合成して用いた。ペプチドのアミノ酸配列は、クラミドモナスおよびシロイヌナズナでSGT活性を確認できている、エクステンシンの配列、すなわちFITC-VYKSOOOOV(アミノ酸の一文字表記。Oはヒドロキシプロリン)の他に、さらに、ペプチドのアミノ酸配列特異性を検討するために、ヒドロキシル化されていないFITC-VYKSPPPPV、セリン残基をアラニン残基に変えたFITC-VYKAOOOOV、C末端のバリン残基を削除したFITC-KSOOOO、ヒドロキシル化していない3回繰り返し配列を含むFITC-KSPPPPVYKSPPPPVKHYSPPPPVYK、ヒドロキシル化した3回繰り返し配列を含むFITC-SOOOOSOOOOSOOOOSOOOOSP、ヒドロキシル化していないスポラミン配列FITC-PTTHEPASSETPVLおよびヒドロキシル化したスポラミン配列FITC-PTTHEOASSETPVL、AGP14配列FITC-VDAOAOSOTS、FITC-VDAOAOAOAA、セリン繰り返し配列FITC-PSSSSSOSSSSSPなどを用いた。
これらの合成ペプチドを受容体基質としてSGT酵素活性を測定した結果、特にFITC-VYKSOOOOVにおいて強いガラクトース転移酵素活性が測定された。一方で、FITC-VYKSPPPPV、受容体基質とした場合には、ごく弱いSGT酵素活性が検出された。そしてFITC-VYKAOOOOVを受容体基質とした場合には、SGT酵素活性は全く検出されなかった。特筆すべきことは、FITC-KSOOOOを受容体基質とした場合には、強いSGT酵素活性が検出されたが、酵素反応産物のピークは一つだけであり、それはセリン残基にガラクトースが付加したものであったことから、SGT活性だけが検出され、HGT活性は検出されなかったことが分かった。これらの結果から、SGT酵素により、FITC-VYKSOOOOVのSにガラクトースが転移されることが証明され、近隣のアミノ酸がPではなくOであることがSGT活性に重要であることが示唆された。また、HGT酵素によるOへのガラクトースの転移には、FITC-VYKSOOOOVの最後のVが重要であることが示唆された。このことは、FITC-KSOOOOを受容体基質と用いることで、HGT活性の影響を受けずに、SGT活性だけを測定することができることである。すなわち、標識化されたKSOOOOを含む、例えばFITC-VYKSOOOO(配列番号19)を受容体基質として用いるSGT酵素活性測定系を確立できた。
【0038】
(5−2)シロイヌナズナT87細胞抽出液を用いた場合
FITC-VYKSOOOOVを受容体基質とした時、シロイヌナズナT87のSGT粗酵素液において、ガラクトース転移酵素活性を検出したが、FITC-KSOOOOおよびFITC-VYKSPPPPV、あるいはAGP14配列、MUC1AC配列を受容体基質とした場合では、SGT酵素活性は認められなかった。シロイヌナズナの粗酵素液では、SGTの活性が濃縮できておらず、弱い活性が検出できなかったためであると考えられるが、基本的に、クラミドモナスのSGTと同様の基質特異性を示していた。
【0039】
〔実施例6〕SGT遺伝子の発現と酵素活性の確認
SGTの2価金属イオン要求性については、金属イオンをキレートするEDTAを2mMを反応液中に添加するとSGT酵素活性は全く消失したことから、金属イオンを必要とすることが分かった。どの金属イオンを要求するかについて2mMのEDTAに加え、4mMの各種金属イオンを添加して検討したところ、他の糖転移酵素と同様に、Mnを要求し、Fe、Co、Znでは弱い要求性が見られたが、Mg、Caでは全く活性化が認められなかった。至適pHはpH6.0、至適温度は30℃で、50℃では活性は失われた(図4)。
SGTは、陽イオン交換カラムではpH4.5、陰イオン交換カラムではpH8.5で吸着した。HGTは、陽イオン交換カラムではpH5.5で吸着したが、陰イオン交換カラムではpH9でも吸着しなかった。HGTは、高い等電点をもつことが示唆された。SGTは、中性付近の等電点をもつことが示唆された。
【0040】
〔実施例7〕SGTタンパク質の精製法
クラミドモナスの粗酵素液SL-S100を、Ni Chlating Sepharoseカラム(φ5×5cm、結合バッファー;25mM Tris-HCl, 10% Glycerol, 0.2% Triton X-100, 0.5M NaCl, pH7.6、溶出バッファー;25mM Tris-HCl, 10% Glycerol, 0.2% Triton X-100, 0.5M NaCl, 0.05M Glycine, ph7.6)、DEAE Sepharoseカラムに2回(1回目:φ5×11cm、結合バッファー;20mM Tris-HCl, 10% Glycerol, 0.1% Triton X-100, ph8.5、溶出バッファー;20mM Tris-HCl, 10% Glycerol, 0.1% Triton X-100, 0〜0.2M NaCl, 0.05M Glycine, pH7.6、2回目:結合バッファー;25mM Methyl-piperadine, 10% Glycerol, 0.1% Triton X-100, ph8.5、溶出バッファー;25mM Methyl-piperadine, 10% Glycerol, 0.1% Triton X-100, 0〜0.2M NaCl, pH8.5)、Sephadex G-200カラム(φ2.5×5cm、溶出バッファー;25mM MOPS, 10% Glycerol, 0.5% CHAPS, 0.5M NaCl, pH7.0)に2回かけ、Native-PAGE(6%ポリアクリルアミドゲル、泳動バッファー;25mM Tris, 192mM Glycine)にかけた。CBB染色を行い、タンパク質のバンドを検出し、ゲルからバンドを切り出して、酵素活性を測定したところ、ゲル断片No.4とNo.5に高い酵素活性が確認された。そこでNo.4とNo.5からタンパク質を抽出して、さらにIEF-PAGE(pH3〜10キャリアアンフォライト使用ポリアクリルアミドゲル、泳動バッファー;陽極7mM Phosphate, 陰極20mM Argine, 20mM Lysine)にかけた(表1)。CBB染色を行い、再びゲルからバンドを切り出して、酵素活性を測定したところ、ゲル断片No.8、9およびNo.9、10に強い酵素活性が認められた(図5)。それぞれのゲル断片をSDS-PAGEにかけたところ、IEF-PAGEの溶出パターンと挙動をともにするバンドとして、四角で示したバンドが見出された(図6A)。
【0041】
以下に、表1として、クラミドモナス細胞破砕液の精製ステップごとのSGT酵素の比活性を示す。
【0042】
【表1】
【0043】
〔実施例8〕SGT遺伝子ファミリーの同定
SDS-PAGEゲルから切り出したバンドを、株式会社セラバリューズに依頼分析し、LC-MS/MS解析後、MASCOT検索をしたところ、7つのペプチド断片のアミノ酸配列が明らかとなり、それらはすべて、クラミドモナスのデータベースの遺伝子配列CHLREDRAFT_150807 (A8J5Z4_CHLRE) に100%マッチしていることが見出された。A8J5Z4_CHLRE は717アミノ酸をコードする遺伝子であった。(図6、7)
【0044】
【表2】
【0045】
〔実施例9〕SGT遺伝子ファミリーの配列の類似性
(9−1)種間類似性
クラミドモナスのA8J5Z4_CHLRE配列をクエリーにして、Blast検索をしたところ、シロイヌナズナ、イネ、トウゴマ、ブドウ、ポプラ、ニセツリガネゴケ、ブラシノ藻に、約30%identityのホモログORFがそれぞれ1ORFずつ見つかり、植物に広く保存された遺伝子ファミリーであることが分かった(図7)。広葉植物同士では、70%程度のアミノ酸の同一性があり、被子植物間では60%程度、被子植物と単細胞緑藻の間では30%のアミノ酸配列の同一性が認められた。しかし、動物や真菌にはホモロジーのある遺伝子は見つからず、植物界に特異的に保存されている遺伝子ファミリーであることが分かった(表2)。しかも、SGTの配列は、他のどの糖転移酵素とも類似性を持たず、全く新規な糖転移酵素遺伝子ファミリーであった。
SGTは、糖転移酵素としては珍しく、I型膜タンパク質のトポロジーであった(図8)。クラミドモナスのSGT1のN末端側のホモロジーの高い領域だけでも活性があった。SGTの活性ドメインであると考えられるニセツリガネゴケからシロイヌナズナなどの高等植物のSGT1には、ホモロジーの高い領域がタンデムに重複して存在していた。N末端側とC末端側のホモロジーの高い領域同士では、53%程度のアミノ酸配列の同一性であった。シロイヌナズナのSGTのC末端側のホモロジーの高い領域だけでもSGT活性はあった。すべてのSGT1で糖ヌクレオチドを基質とする糖転移酵素に広く保存されているDXDモチーフが保存されていた。クラミドモナスおよびブラシノ藻では、複数のホモロジーのあるORFが検索されるが、ニセツリガネゴケからシロイヌナズナなどの高等植物のSGT1には、ゲノム中に一つずつしかホモログはなかった。クラミドモナスのSGT1には、高等植物のプロリン水酸化酵素のER-ゴルジ局在配列Tox1が存在し、ER-ゴルジの局在が示唆された。
【0046】
各植物のSGT1の活性ドメイン(CD)のIdentity(アミノ酸配列間ホモロジー)を下記表3に示す。
【0047】
【表3】
【0048】
なお、表中のCrはクラミドモナス、Otはブラシノ藻、Ppはニセツリガネゴケ、Smはイヌタカヒバ、Osはイネ、Atはシロイヌナズナ、Ntはタバコ、Ptはポプラ由来SGTであることを表す。
【0049】
(9−2)植物ゲノム中のSGT遺伝子の数
各生物のSGT遺伝子の配列情報は、データベース上はいくつかの異なったデータが登録されてあったが、DNA配列等を比較し、遺伝子座を調べると、オールタナティブスプライシング、遺伝子の欠失挿入変異、シークエンス解析のミス、品種による遺伝子の多型などが原因であり、各ゲノムあたり1種類のSGT遺伝子が1コピーしかなく、SGT酵素活性をコードする遺伝子はSGT1以外にはないことがわかった。
【0050】
〔実施例10〕緑藻と高等植物のSGT遺伝子とSGT酵素活性
クラミドモナスは、すべての緑色植物の祖先であるとされている単細胞の緑藻の一種であり、一方、シロイヌナズナは、双子葉植物であり、陸上の高等植物の一種である。この2種の植物において、SGT遺伝子はアミノ酸配列にして30%の同一性を示すほど高い類似性を持っており、双方とも、同じ基質特異性で、同じ反応産物を生ずる酵素活性を持っていることが確認されたため、相同性遺伝子であることが証明された。Blast等による相同性検索によって、シロイヌナズナと同じ双子葉植物であるトウゴマ、ブドウ、ポプラでも70%のアミノ酸配列同一性を示すSGT遺伝子があり、単子葉植物であるイネでも60%のアミノ酸配列同一性を示すSGT遺伝子が見つかった。さらに、コケ類であるニセツリガネゴケでも50%を超えるアミノ酸配列同一性を示すSGT遺伝子が見つかった。これらのことから、この一群のSGT遺伝子ファミリーはすべて同様のSGT酵素活性を保持しているものと考えられる。逆に言えば、クラミドモナスおよびシロイヌナズナのSGT遺伝子配列に30%以上の類似性を示す遺伝子が、各種植物種のDNA配列から発見されば場合は、すべてSGT遺伝子であると考えられ、SGT酵素活性も保持しているものと考えられる。
【0051】
【表4】
【0052】
〔実施例11〕酵母での発現
クラミドモナスのSGT1遺伝子は、活性ドメインを一つだけ持ち、N末端に分泌経路に入るためのシグナルシークエンスがあり、C末端側に膜に局在するための膜貫通領域がある。ChSGT1のシグナルシークエンスおよび膜貫通領域以外の部分を、HAタグを付けた酵母PIR細胞壁タンパク質に結合し、酵母細胞壁に発現させて(Abe et al, 2004, Shimma et al, 2006)、SGT酵素活性を測定したところ、HPLCで同じ溶出ピークが得られた。このことから、ChSGT1遺伝子がSGT酵素をコードしていることが証明された(図9)。
アラビドプシスのSGT1遺伝子は、他の陸上植物のSGTと同様に、活性ドメインが2つあり、N末端に分泌経路に入るためのシグナルシークエンスがあり、C末端側に膜に局在するための膜貫通領域がある(図8)。同様に、AtSGT1のシグナルシークエンスおよび膜貫通領域以外の部分を、HAタグを付けた酵母PIR細胞壁タンパク質に結合し、酵母細胞壁に発現させてSGT酵素活性を測定したところ、HPLCで同じ溶出ピークが得られた。このことから、AtSGT1遺伝子もSGT酵素をコードしていることが証明された(図10)。
【0053】
〔実施例12〕SGTの酵素活性の必要領域
C末端側の活性ドメインだけをもつ融合タンパク質を同様に酵母で発現させてSGT酵素活性を測定したところ、HPLCで同じ溶出ピークが得られた(図10)。このことから、クラミドモナスでも高等植物でも、活性ドメインが一つでもあれば、SGT酵素活性があることが分かった。
【0054】
〔実施例13〕SGTの抑制法
現在、ゲノムDNA配列情報が解読され、公開されている生物種については、SGT1遺伝子配列情報をホモロジー検索により容易に取得することができるが、今後明らかにされる生物種においても、同様の方法によりその生物種のSGT遺伝子の取得が可能である。それらのSGT遺伝子情報に基づいて、SGTを抑制することができる。1)SGT遺伝子DNA配列情報を元にして、相同性組み換え法による遺伝子破壊法(ノックアウト)、相補的核酸配列を発現させるアンチRNA法、RNAi法、トランスポゾン挿入法、T-DNA変異株作製法など、既知の方法でSGT遺伝子の破壊あるいは機能抑制を行うことができる。一例として、シロイヌナズナのT-DNA変異株が多数作製されており、そのうちAtSGT1に関するものは、SALK_054682、SALK_059879C、WiscDsLox451B08であり、これらは、AtSGT遺伝子にT-DNAの挿入、あるいはトランスポゾンの挿入によりORFが破壊されており、SGT活性が損なわれている。これらAtSGT1遺伝子(At3g01720)の変異を持つシロイヌナズナの種子をThe Arabidopsis Information Resource (TAIR) (http://www.arabidopsis.org/)から購入し、発芽させ、植物体を成育させ、あるいは、植物体よりカルスを誘導して、細胞培養を行った。FLAGタグを融合したエクステンシン繰り返し配列(VYKSOOOOV)を含むペプチドをコードする遺伝子を、植物体あるいはカルスに形質転換し、FLAGタグを利用して、エクステンシン配列を抗体で精製して、セリン残基へのO−ガラクトース付加が抑制されていることを検証している。
前述したように、コケ以上の陸上植物では、SGT遺伝子はゲノムあたり1遺伝子しかないことが分かっており、上記の方法で同定したSGT遺伝子を破壊あるいは抑制することにより、ペプチドのセリン残基へのO−ガラクトースの付加を抑制することができる。
【0055】
〔実施例14〕SGT1変異株における酵素活性の減少
シロイヌナズナにおいては、T-DNA挿入変異株のシリーズが作成されており、各ORFの変異株を購入することができる。SGT1(At3g01720)遺伝子に対応する位置のORFにT-DNAが挿入された変異株(Δsgtl1〜3)をオハイオ大学のアラビドプシスセンターから入手した。正常株(コロンビア)及び上記SGT1変異植物株の種子を発芽させ、茎を寒天培地上で培養してカルスを誘導し、細胞培養した。
約1gの正常株及び変異株のカルス細胞を集めて、乳鉢で破砕し、膜画分を調製した。実施例11と同様のHPLC解析によりSGT酵素活性を測定した結果、正常株ではSGT酵素活性が確認できたが、SGT1変異株では検出されなかった。(図11)
このことから、SGT1遺伝子のT-DNA挿入変異株では、SGT酵素活性が消失していることが示された。
【0056】
〔実施例15〕SGT1変異株における糖鎖付加の減少
正常株及び変異株のカルス細胞を破砕し、細胞壁画分を調製した。具体的には、カルス細胞1gを乳鉢で破砕し、1,000×gで遠心した沈殿を、クエン酸緩衝液(20mM,pH7.0)に懸濁してオートクレーブに2時間かけ、1,000×gで遠心した沈殿を細胞壁画分とした。SGT酵素によりペプチドにα結合で付加されたガラクトースはαガラクトシダーゼで遊離することが分かっており、一方で、ガラクタンおよびアラビノガラクタンに含まれるガラクトースはβ結合で、ガラクトシダーゼにより分離されるから、細胞壁画分0.1ODを、100μlの20mM酢酸アンモニウム(pH4.5)、αガラクトシダーゼ(Guar,5U,40℃)あるいはβガラクトシダーゼ(Oryzae,1U,30℃)処理を行って、ガラクトースを遊離させた。反応液をエタノール沈殿し、上清を乾燥させた。遊離した単糖をABEE標識後、HPLCで解析した。
その結果、正常株の細胞壁から遊離されるαガラクトースの量を100としたときに、変異株から遊離されるαガラクトースの量が約60に有意に減少していた。一方、βガラクトシダーゼにより遊離されたガラクトースの量は、正常株と変異株で差はなかった。
このことから、SGT1遺伝子のT-DNA挿入変異株では、糖タンパク質あるいはペプチドのセリン残基へのO−結合型ガラクトース付加が減少していることが示された。(図12)
【0057】
〔実施例16〕タバコからのSGT1遺伝子クローニング
(16−1)タバコのESTデータベースの検索
ゲノム配列が非公開の実用植物であるタバコから、PCRによりSGT1遺伝子に対応するcDNAをクローニングした。シロイヌナズナのSGT1配列(AtSGT1)をクエリーにして、公共データベース(http://blast.ncbi.nlm.nih.gov/Blast.cgi)にあるタバコのESTを検索したところ、FG201805とFG200221、FG146884という3つのEST配列を見いだした。遺伝情報処理ソフトウェア GENETYX-MAC Ver12((株)ゼネティックス)を用いて、これらの3つのEST配列を繋げて、シグナル配列、膜貫通領域、塩基性アミノ酸に富む領域を欠く2074bpからなるタバコSGT1遺伝子(NtSGT1)の部分配列を見出した。この配列をもとに遺伝子特異的プライマーNtSGTFw-1 (5’-TTGCACTGATGAAGAGAGGAA-3’;配列番号24)及びNtSGTRv-2 (5’- ATCATTGGCTCTAGTTTCAG-3’;配列番号25)をそれぞれフォーワードプライマー、リバースプライマーとして設計して合成した。
【0058】
(16−2)タバコSGT1遺伝子(NtSGT1)の全長クローニング
培養4日目のタバコ培養細胞BY-2からRNeasy Plant Mini Kit ((株)キアゲン)を用いてTotal RNAを抽出した。さらにGenElute mRNA Miniprep Kit (SIGMA)を用いてTotal RNAからmRNAを精製した。
得られたmRNA 500ngを鋳型として、TaKaRa RNA PCR Kit (AMV) ver. 3.0(タカラバイオ(株))を用いてRT-PCRを行った。逆転写はTaKaRa RNA PCR Kit (AMV) ver. 3.0に付属のOligo dT-Adaptor Primer 1.25 pmolを用いて42℃、1時間、次いで95℃、5分反応した。PCRはNtSGTFw-1とNtSGTRv-2を各10 pmol 用いて、94℃、30秒、53℃、30秒、72℃、2分10秒を1サイクルとする、計30サイクルを行った。このPCR産物についてアガロース電気泳動を行ったところ、予測される増幅DNAに相当する約2-kbのPCR産物が確認できたため、この部分を切り出してDNAを精製し、プラスミドpUC19に導入し、大腸菌DH5αを形質転換した。得られたクローンを鋳型とし、DNA配列(NtSGT1部分配列)を決定した。
【0059】
次に3’末端配列をクローニングするため、TaKaRa RNA PCR Kit (AMV) ver. 3.0 (タカラバイオ(株))を用いた3’ RACEを行った。上記で決定したNtSGT1部分配列をもとに遺伝子特異的プライマーNtSGTFw-19 (5’-TCCTCCTGATCCATCGTCACTTG-3’;配列番号26)を3’RACE用アンチセンスプライマーとして設計して合成した。逆転写はTaKaRa RNA PCR Kit (AMV) ver. 3.0に付属のOligo dT-Adaptor Primer 1.25 pmolを用いて42℃、1時間、次いで95℃、5分反応した。PCRはNtSGTFw-19とNtSGTRv-2を各10 pmol 用いて、94℃、30秒、50℃、30秒、72℃、2分を1サイクルとする、計30サイクルを行った。このPCR産物についてアガロース電気泳動を行ったところ、約1.2-kbのPCR産物が確認できたため、この部分を切り出してDNAを精製し、プラスミドpUC19に導入し、大腸菌DH5αを形質転換した。得られたクローンを鋳型とし、DNA塩基配列を決定した。
【0060】
次いで、5’末端配列をクローニングするため、5'-Full RACE Core Set (タカラバイオ(株))を用いた5’ RACEを行った。
上記で決定したNtSGT1部分配列をもとに5’末端リン酸化逆転写反応用プライマーNtSGT1-5phos (5’- [5’リン酸化]AACACCCTCTCGTGG-3’:配列番号39)、1st PCR用センスプライマーNtSGT1-S1 (5’-ATAAACAAACCAGCTGGAGT-3’:配列番号40)、1st PCR用アンチセンスプライマーNtSGT1-A1 (5’-CCAGTTTTGGGATGTCTGCT-3’:配列番号41)、2nd PCR用センスプライマーNtSGT1-S2 (5’-ACAGCAAAGAGGCACAAAAT-3’:配列番号42)、2nd PCR用アンチセンスプライマーNtSGT1-A2 (5’-AAAGTGGGAGCCAACTCCAT-3’:配列番号43)を設計し、合成した。上記で単離したタバコ培養細胞BY-2のmRNA 1μgを鋳型とし、NtSGT1-5phos 500 pmolを用いて30℃、10分、42℃、1時間、次いで80℃、2分反応した。得られたRNA/DNAハイブリッド中のRNAを5'-Full RACE Core Set 付属のRNaseHで分解し、得られたシングルストランドcDNAをエタノール沈殿後、付属のT4 RNA ligase でライゲーションした。このライゲーション液1μlを鋳型としてNtSGT1-S1 とNtSGT1-A1を各15 pmol用いてPrimeSTAR GXL DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、50℃、15秒、68℃、2分30秒を1サイクルとする、計30サイクルを行った。次にこのPCR反応液1μlを鋳型としてNtSGT1-S2 とNtSGT1-A2を各15 pmol用いてPrimeSTAR GXL DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、50℃、15秒、68℃、2分30秒を1サイクルとする、計30サイクルを行った。このPCR産物についてアガロース電気泳動を行ったところ、約700-bpのPCR産物が確認できたため、この部分を切り出してDNAを精製し、プラスミドpUC19に導入し、大腸菌DH5αを形質転換した。得られたクローンを鋳型とし、DNA配列を決定した。
【0061】
3’RACEと5’RACEにより得られた塩基配列をもとに、NtSGT1の全長を増幅するようにNtSGTFw-5term (5’-GGACACACAAACCTTTGACT-3’:配列番号44)とNtSGTRv-3term (TGGAGCTCTTTTTTTTTTTTTTTTTTTTTTTTVN-3’:配列番号45)を設計し、合成した。mRNA 500ngを鋳型として、TaKaRa RNA PCR Kit (AMV) ver. 3.0(タカラバイオ(株))を用いてRT-PCRを行った。逆転写はTaKaRa RNA PCR Kit (AMV) ver. 3.0に付属のOligo dT-Adaptor Primer 1.25 pmolを用いて42℃、1時間、次いで95℃、5分反応した。PCRはNtSGTFw-5termとNtSGTRv-3termを各10 pmol 用いてPrimeSTAR GXL DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、52℃、15秒、68℃、3分30秒を1サイクルとする、計30サイクルを行った。このPCR産物についてアガロース電気泳動を行ったところ、予測される増幅DNAに相当する約3.3-kbのPCR産物が確認できたため、この部分を切り出してDNAを精製し、プラスミドpUC19に導入し、大腸菌DH5αを形質転換した。得られたクローンを鋳型とし、DNA配列を決定した。
得られた完全長クローン(配列番号20)は3,336bpであり、899aa(配列番号21)をコードし、シロイヌナズナSGT1と80%以上のアミノ酸配列同一性を示していた。
タバコのSGT活性ドメイン1及び2のアミノ酸配列は、それぞれ配列番号22及び23で表すことができる。ドメイン1は、34aa〜337aa、ドメイン2は、398aa〜697aaである。
【0062】
〔実施例17〕タバコSGT1遺伝子を用いた形質転換酵母によるSGT酵素活性の発現
上記〔実施例11〕と同様に、タバコSGT1のシグナル配列および膜貫通領域を除いた主要部分(配列番号21に示されたNtSGT1の400〜720番目の領域)を、HAタグを付けた酵母PIR細胞壁タンパク質に結合して酵母細胞壁に発現させ、SGT酵素活性を測定したところ、HPLCでプロダクト#1と同じ溶出ピークが得られた。すなわち、タバコSGT1についても、FITC-VYKSOOOO基質のセリンに対してガラクトースを転位させることができることを確認した(図11)。
【0063】
〔実施例18〕RNAiを用いたタバコSGT1遺伝子発現の抑制
〔18−1〕dsRNAの設計及び発現プラスミドの構築
ヘアピン型二本鎖RNA(dsRNA)発現プラスミドを用いたRNAiにより、NtSGT1の制御を行うために、まずNtSGT1をターゲットとして549-bpのdsRNAの発現プラスミドを次の手順で構築した。
(1)ターゲット領域(センス配列およびアンチセンス配列)とリンカー配列のPCR増幅
ターゲット領域としてNtSGT1の549-bp断片(296番目のTから844番目のTまで)を選択した。PCRによりターゲット領域のセンス配列とアンチセンス配列を増幅した。
センス配列の増幅はプライマーSGTsenseFw (5’-AGATTGTCGACTTGCACTGATGAAGAGAGGAAG-3’ :配列番号46、下線部はSalIサイト)およびSGTsenseRv (5’-GAATAGCATGC AGATCTATCATCAAGTTATCATTAATC-3’:配列番号47、1つ目の下線部はSphIサイト、2つ目の下線部はBglIIサイト)を用いた。これらのプライマーを各々10 pmol用いてPrimeSTAR HS DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、53℃、5秒、72℃、40秒を1サイクルとする計5サイクルを行い、続いて98℃、10秒、59℃、5秒、72℃、40秒を1サイクルとする計25サイクルを行った。
アンチセンス配列の増幅はプライマーSGTantiFw (5’-AGATTGGTACCTTGCACT
GATGAAGAGAGGAAG-3’:配列番号48、下線部はKpnIサイト)およびSGTantiRv (5’-GAATAGAATTC AAGCTTATCATCAAGTTATCATTAATC:配列番号49、1つ目の下線部はEcoRIサイト、2つ目の下線部はHindIIIサイト)を用いた。これらのプライマーを各々10 pmol用いてPrimeSTAR HS DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、51℃、5秒、72℃、40秒を1サイクルとする計5サイクルを行い、続いて98℃、10秒、57℃、5秒、72℃、40秒を1サイクルとする計25サイクルを行った。
また、pIG221(Ohta et al., Plant Cell Physiol. 31: 805-831, 1990)からcastor bean catalase 遺伝子(CAT-1)の第一イントロン遺伝子201-bpをRNAiコンストラクトのリンカー配列として増幅した。プライマーIntron1Fw (5’- GGCGGGGATCCCTACAGG
GTAAATTTCT-3’:配列番号50、下線部はBamHIサイト)およびIntron1Rv (5’- GGCGGGGTACCGGTTCTGTAAC-3’:配列番号51、下線部はKpnIサイト) を用いた。これらのプライマーを各々10 pmol用いてPrimeSTAR HS DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、51℃、5秒、72℃、40秒を1サイクルとする計5サイクルを行い、続いて98℃、10秒、56℃、5秒、72℃、30秒を1サイクルとする計25サイクルを行った。
【0064】
(2)dsRNA発現ベクターの構築
リンカー配列を制限酵素(BamHIおよびKpnI)で切断した後、プラスミドpUC19上のBamHIおよびKpnIサイトに導入し、プラスミドpUC19-Intを得た。次にターゲット領域のアンチセンス配列を制限酵素(SphIおよびSalI)で切断した後、プラスミドpUC19-Int上のSphIおよびSalIサイトに導入し、プラスミドpUC19-SGTanti-Intを得た。次にターゲット領域のセンス配列を制限酵素(KpnIおよびEcoRI)で切断した後、プラスミドpUC19-SGTanti-Int上のKpnIおよびEcoRIに導入し、プラスミドpUC19-SGT RNAi296を得た。
植物導入用の遺伝子発現ベクターには様々な種類があり、それらを適宜用いることができるが、本実施例では pMAT037 (Matsuoka and Nakamura, Proc. Natl. Acad. Sci. USA. 88:834-838, 1991)を用いた。pUC19-SGT RNAi296を制限酵素(BglIIおよびHindIII)で切断し、アンチセンス配列とリンカー配列およびセンス配列を含む約1.2-kbの断片を切り出して、pMAT037のCaMV35Sプロモーター後のマルチクローニングサイト中のBglIIおよびHindIIIサイトに導入し、プラスミドpMAT- SGT RNAi296を得た。
【0065】
〔18−2〕タバコ培養細胞BY-2へのアグロバクテリウム属細菌による遺伝子導入
タバコ培養細胞BY-2への遺伝子導入に先立ち、プラスミドpMAT- SGT RNAi296をエレクトロポーレーション法によりアグロバクテリウム細菌EHA101に組み込んだ。プラスミドを組み込んだアグロバクテリウム細菌を抗生物質であるカナマイシン(12.5 mg/l)およびテトラサイクリン(5 mg/l)を含む培地で増殖させ、600 nmにおける光学濃度(O.D.)値約1のアグロバクテリウム懸濁液を調製した。この懸濁液と培養3日目のBY-2細胞をシャーレに加え、シャーレを軽く旋回することで混合すると同時に培養液をシャーレ全面に広げる。野菜把捉テープでシャーレをシールし26℃、暗所で72時間静置培養した。ピペットを用いてシャーレ上のBY-2を15 mlの滅菌済みチューブに移し、5 mlのBY-2細胞用液体培地を加え、10回程度ピペッティングを行った。これを800 rpm、1分遠心しBY-2細胞を沈殿させる。アグロバクテリウムで濁った培地を除去し、新しい培地を5 ml加えBY-2細胞と上記の方法で混合し遠心によりBY-2細胞を回収した。この操作をさらに4回繰り返した後、BY-2の沈殿を得た。BY-2細胞の体積に対して4倍量または9倍量の新しい培地を加え、よく混合し、各々1 mlを形質転換用の固形培地に広げた。シャーレを傾けて液体培地を一方の隅に集めピペットで吸い取った後、野菜把捉テープでシャーレをシールし28℃、暗所で2週間静置培養し、培地上に出現した形質転換体を得た。
【0066】
〔18−3〕RNAi によるSGT転写抑制効果(RT-PCRによる転写産物量の測定)
RNAiコンストラクトを導入したタバコ培養細胞BY-2と、コントロールとして形質転換を行っていないタバコ培養細胞BY-2を8日間培養した後、培養液を2号濾紙(2枚重ねる)で濾過してBY-2細胞を回収した。このBY-2細胞100 mgを液体窒素で凍結させながら乳鉢で粉砕した。この粉砕したBY-2細胞からRNeasy Plant Mini Kit ((株)キアゲン)を用いてTotal RNAを抽出した。また抽出操作中に ((株)キアゲン)を用いてDNase処理を行った。得られた Total RNA 500ngを鋳型として、TaKaRa RNA PCR Kit (AMV) ver. 3.0(タカラバイオ(株))を用いてRT-PCRを行った。逆転写はTaKaRa RNA PCR Kit (AMV) ver. 3.0に付属のOligo dT-Adaptor Primer 1.25 pmolを用いて42℃、1時間、次いで95℃、5分反応した。PCRに用いた配列と反応条件は以下のとおりである。NtSGT1 mRNAの増幅にはプライマーNtSGTFw-3(5’-GGACACACAAACCTTTGACT-3’:配列番号52)、NtSGTRv-4(5’-CCAGTTTTGGGATGTCTGCT-3’:配列番号53)を各10 pmol用いて、94℃、30秒、53℃、30秒、72℃、30秒を1サイクルとする、計30サイクルを行った。またRT-PCRの内部標準として用いたactin mRNAの増幅にはBY-2 actin Fw(5’-CAATTCTTCGGTTGGATCTTG-3’:配列番号54)、BY-2 actin Rv(5’-CTTCATGCTGCTGGGAG-3’配列番号55)を各10 pmol用いて、94℃、30秒、53℃、30秒、72℃、30秒を1サイクルとする、計20サイクルを行った。RT-PCRは4回繰り返し実験を行い、得られたRT-PCR産物はDNAの蛍光検出剤であるSYBR Safe DNA gel stain(Invitrogen)を添加したアガロースゲルを用いて電気泳動に供し、スキャン型イメージャーであるTyphoon 9400(GEヘルスケア)で検出した。予測される増幅DNAの長さはNtSGT1においては約370-bp、actinにおいては約460-bpであるが、これらに相当するPCR産物が確認できたため、ソフトウェアImage Quant TL(GEヘルスケア・ジャパン)を用いてバンド強度を測定した。図12にRNAi によるSGT転写抑制効果を示した。図12AはRT-PCRの電気泳動結果。図12Bは、図12Aで検出したPCR産物のシグナル強度をグラフ化したものである。RNAiコンストラクト導入BY-2(RNAi1、 RNAi2、RNAi3)では、転写産物量が非形質転換体の50%未満に減少しており、RNAiによるSGT転写抑制効果が認められた。student testを行い、RNAiコンストラクト導入BY-2は全ての非形質転換体に対して p<0.01で有意であった。
【0067】
〔18−4〕RNAi によるSGT転写抑制効果(SGT活性の測定)
(1)マイクロソームの調製
前記実施例1でも用いたタバコ細胞抽出液マイクロソームと同様の調製を行った。具体的には、7日間培養したタバコ培養細胞BY-2の培養液を2号濾紙(2枚重ねる)で濾過してBY-2細胞を回収した。BY-2細胞1 gあたり破砕バッファー1 mlを添加して、ホモジナイザーを用いて、3,700 rpmから4,000 rpmの回転数で4回上下させ、破砕した。破砕バッファーの組成は、40 mM HEPES-KOH (pH7.4)、400 mMソルビトール、2 mM ジチオスレイトールである。この破砕液を1,000×g、10分、4℃で遠心し、上清を回収した。この上清を10,000×g、10分、4℃で遠心し、上清を回収した。この上清を100,000×g、10分、4℃で遠心し、沈殿を回収し、マイクロソーム画分とした。
【0068】
(2)SGT活性測定
上記で得たマイクロソーム画分を用いてSGT活性を測定した。活性測定に用いた組成は、50 μgマイクロソーム画分、0.1 M MES (pH6.0)、12.5 μM VYKSO4-Ac1 peptide、5 mM UDP-Galactose、5 mM MnCl2、2 mM EDTA、5×Protease inhibitor cocktail、0.10 % TritonX-100であり、総量6.25μlとした。反応温度は30℃であった。
測定結果を図12Cに示した。RNAiコンストラクト導入BY-2(RNAi1、 RNAi2、RNAi3)では、SGT活性が非形質転換体の50%未満に減少しており、RNAiによるSGT転写抑制効果が認められた。SGT転写産物量とSGT活性の両方においてRNAiによるSGT転写抑制効果が認められた。
【配列表フリーテキスト】
【0069】
1.クラミドモナス SGT遺伝子配列 Chla(g)
2.クラミドモナス SGTアミノ酸配列 Chla(a)
3.クラミドモナス SGT活性ドメインのアミノ酸配列 Chla-1
4.シロイヌナズナ SGT遺伝子配列 Arab(g)
5.シロイヌナズナ SGTアミノ酸配列 Arab(a)
6.シロイヌナズナ SGT活性ドメイン1のアミノ酸配列 Arab-1
7.シロイヌナズナ SGT活性ドメイン2のアミノ酸配列 Arab-2
8.イネ SGT遺伝子配列 Oryz(g)
9.イネ SGTのアミノ酸配列 Oryz(a)
10.イネ SGT活性ドメイン1のアミノ酸配列 Oryz-1
11.イネ SGT活性ドメイン2のアミノ酸配列 Oryz-2
12.ニセツリガネゴケ SGTのアミノ酸配列 Phys(a)
13.ニセツリガネゴケ SGT活性ドメイン1のアミノ酸配列 Phys-1
14.ニセツリガネゴケ SGT活性ドメイン2のアミノ酸配列 Phys-2
15.ブラシノ藻 SGTアミノ酸配列 Ostreo (a)
16.ブラシノ藻 SGT活性ドメインのアミノ酸配列 Ostreo -1
17.クラミドモナス SGT活性ドメイン1を含む活性化領域 Chla(act)1
18.シロイヌナズナ SGT活性ドメイン2を含む活性化領域 Arab(act)2
19.SGT活性測定用受容体基質ペプチド R-peptide
20.タバコ(nicotiana) SGT遺伝子配列
21.タバコ(nicotiana) SGTアミノ酸配列
22.タバコSGT CD1:
23.タバコSGT CD2:
24.タバコSGT 3’-RACE Forward primer(NtSGTFw-1)
25.タバコSGT 3’-RACE Reverse primer(NtSGTRv-2)
26.タバコSGT 3’-RACE antisense primer(NtSGTFw-19)
27.スポラミン配列(Sporamin sequence)
28.エクステンシン配列(extension sequence)
29.改変エクステンシン配列-1(Extension sequence(mod-1))
30.プロリンが水酸化されたエクステンシン配列(Hydroxyl-proline-extension sequence)
31.プロリンが水酸化された改変エクステンシン配列-1(Hydroxyl-prolin-extension sequence(mod-1))
32.プロリンが水酸化された改変エクステンシン配列-2(Hydroxyl-proline-extension sequence(mod-2))
33.プロリンが水酸化された改変エクステンシン配列-3(Hydroxyl-proline-extension sequence(mod-3))
34.プロリンが水酸化されたスポラミン配列(Hydroxyl-proline-sporamin sequence)
35.プロリンが水酸化されたAGP14配列(Hydroxyl-proline arabinogalactan protein14 (AGP14) sequence)
36.プロリンが水酸化された改変AGP14配列(Hydroxyl-proline AGP14 sequence(mod))
37.Ser繰り返し配列(Ser repetitive sequence)
38.タバコSGT mRNA(Nicotiana tabacum SGTmRNA)
39.5’末端リン酸化逆転写反応用プライマー(phosphorylation RT-PCR primer)(NtSGT1-5phos)
40.1st PCR用センスプライマーNtSGT1-S1(forward primer-1)
41.1st PCR用アンチセンスプライマーNtSGT1-A1(reverse primer-1)
42.2nd PCR用センスプライマーNtSGT1-S2(forward primer-2)
43.2nd PCR用アンチセンスプライマーNtSGT1-A2(reverse primer-2)
44.mRNA全長用NtSGTFw-5term(NtSGT mRNA forward primer)
5’-GGACACACAAACCTTTGACT-3’
45.mRNA全長用NtSGTRv-3term(NtSGT mRNA reverse primer)
TGGAGCTCTTTTTTTTTTTTTTTTTTTTTTTTVN-3’
46.NtSGT1ターゲット領域センス配列増幅用プライマーSGTsenseFw
5’-AGATTGTCGACTTGCACTGATGAAGAGAGGAAG-3’
47.NtSGT1ターゲット領域センス配列増幅用プライマーSGTsenseRv
5’-GAATAGCATGCAGATCTATCATCAAGTTATCATTAATC-3’
48.NtSGT1ターゲット領域アンチセンス配列増幅用プライマーSGTantiFw
5’-AGATTGGTACCTTGCACT
49.NtSGT1ターゲット領域アンチセンス配列増幅用プライマーSGTantiRv
5’-GAATAGAATTCAAGCTTATCATCAAGTTATCATTAATC
50.RNAiコンストラクトリンカー配列増幅用プライマーIntron1Fw
5’- GGCGGGGATCCCTACAGGGTAAATTTCT-3’
51.RNAiコンストラクトリンカー配列増幅用プライマーIntron1Rv
5’- GGCGGGGTACCGGTTCTGTAAC-3’
52.NtSGT1 mRNA増幅用プライマーNtSGTFw-3=配列番号44
5’-GGACACACAAACCTTTGACT-3’
53.NtSGT1 mRNA増幅用プライマーNtSGTRv-4
5’-CCAGTTTTGGGATGTCTGCT-3’
54.RT-PCRの内部標準actin mRNA増幅用プライマーBY-2 actin Fw
5’-CAATTCTTCGGTTGGATCTTG-3’
55.RT-PCRの内部標準actin mRNA増幅用プライマーBY-2 actin Rv
5’-CTTCATGCTGCTGGGAG-3’
【技術分野】
【0001】
本発明は、タバコ由来のO−結合型糖鎖付加関連酵素をコードする遺伝子の同定、当該遺伝子を用いたO−結合型糖鎖付加ポリペプチドの製造、当該遺伝子発現の抑制技術、及び形質転換植物におけるO−結合型糖鎖の付加を抑制したヒト有用タンパク質の製造方法に関する。
【背景技術】
【0002】
抗体やサイトカイン等のバイオ医薬の生産は、現在、CHO細胞など動物培養細胞を用いた生産系が主流であるが、生産コストが高く、大量生産も難しく、ウィルスの混入の危険もある。大腸菌や酵母など微生物を利用した生産系も開発されているが、糖鎖構造が大きく異なっていることが、課題となっている。
植物は、生産コストが微生物並みに安価であって、大量生産も可能であり、ウィルス混入の危険性もなく、糖鎖構造がN−結合型糖鎖の場合は特にヒトにかなり似ているため、バイオ医薬生産系として有望であることが、最近製薬企業に注目されている。
既に、植物培養細胞を用いて、GM-CSF(非特許文献1)、IL-2およびIL-4(非特許文献2)、免疫グロブリン(非特許文献3)、エリスロポエチン(非特許文献4)、α1−アンチトリプシン(非特許文献5)などヒト有用タンパク質の製造が行われている。
【0003】
タンパク質の翻訳後修飾として、糖鎖修飾は主要なものであり、大きく分けて、N−結合型糖鎖と、O−結合型糖鎖の2種類があるが、これらいずれについても、ヒトを含む哺乳動物に無く、植物にのみ存在する植物特有の糖鎖修飾がある。N−結合型糖鎖修飾に関しては、根元のNアセチルグルコサミンに付加するα1,3−フコース、βマンノースに付加するβ1,2−キシロースが、植物特有の糖鎖構造であり、ヒトにおいて抗原となることも知られているが、従来から研究が進んでおり、除去方法も開発されている。具体的には、N−結合型糖鎖修飾におけるフコース転移酵素遺伝子およびキシロース転移酵素遺伝子が同定されており、これら酵素の発現を抑制することにより、α1,3−フコースおよびβ1,2−キシロースの付加の無いN−結合型糖鎖を有する糖タンパク質を植物細胞で生産することが可能となっている(非特許文献6〜9、20)。
【0004】
一方、植物のO−結合型糖鎖修飾に関連する研究開発は遅れており、O−結合型糖鎖付加に関連した酵素遺伝子の解析は重大な課題として残されていた。
O−結合型糖鎖は、ヒトを含む哺乳動物では、タンパク質のセリンあるいはスレオニン残基にNアセチルガラクトサミン(GalNAc)が付加するのが一般的であり、その他、セリンあるいはスレオニン残基にマンノース、フコース、グルコース、Nアセチルグルコサミン、キシロースの付加が知られている。植物細胞ではこのようなO−結合型糖鎖構造は知られておらず、全く異なった2種類のO−結合型糖鎖構造が知られている。ひとつは、タンパク質のアミノ酸配列中の複数のプロリン残基が水酸化されてヒドロキシプロリンになり、そこにガラクトースあるいはアラビノースが付加する構造である。典型的な配列として、エクステンシンの配列「VYKSOOOOV(Oはヒドロキシプロリン)」がよく知られており、この複数のヒドロキシプロリン基が有するヒドロキシル基が糖修飾された構造であるが、この構造もアレルゲンとなることが報告されている(非特許文献10、11)。これらの糖鎖構造を作るガラクトース転移酵素(HGT)およびアラビノース転移酵素は未だに不明であるが、プロリン水酸化酵素はすでに同定されており、その発現の抑制が試みられており、形質転換植物内での外来タンパク質のO−結合型糖鎖付加も阻害されることが確認されている(非特許文献12〜14)。HGT酵素活性は、FITCで蛍光標識した植物のアラビノガラクタンタンパク質14(AGP14)ペプチド配列「FITC-VDAOAOSOTS」を受容体基質として、酵素活性が測定できている(非特許文献15)。
【0005】
しかしながら、もう一つの植物特有のO−結合型糖鎖構造として、タンパク質のアミノ酸配列中のセリン残基に対して直接ガラクトースが付加される場合がある。典型的な配列として、植物細胞壁に存在する糖タンパク質であるエクステンシンの繰り返し配列「VYKSOOOOV」のセリン残基へのガラクトース付加がよく知られている。また、サツマイモの貯蔵タンパク質であるスポラミン「PTTHEPASSETPVL」をタバコ細胞で発現させたときにも、2番目のセリン残基へのガラクトース付加が確認されている。このセリン残基がガラクトース修飾された構造も抗原性があるため、当該ガラクトース修飾に関連する酵素発現を抑制する必要があるが、この構造を作る酵素および酵素遺伝子についての解析は、全くなされていない状況である(非特許文献16〜18)。
このように、植物をバイオ医薬の生産系として確立するためには、O−結合型糖鎖付加に関連する糖転移酵素のうちでも特に、セリン残基のヒドロキシル基にガラクトースを付加する作用を有するガラクトース転移酵素遺伝子を同定し、当該ガラクトース転移酵素発現を抑制する技術の開発が強く望まれていた。そして、そのためにはセリンO−結合型ガラクトース転移酵素(SGT)単独の活性を正確に測定する酵素活性測定法の開発が急務となっていた。とりわけ実用的には、形質転換体宿主として一般に用いられているタバコなどのナス科植物に直接適用できるタバコ由来のSGT遺伝子をターゲットとすることが好ましいが、タバコの場合は全ゲノム配列が公開されていないため、タバコのゲノムライブラリーから直接クローニングすることは難しいという問題がある。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】James EA, Wang C, Wang Z, Reeves R, Shin JH, Magnuson NS, Lee JM, Production and characterization of biologically active human GM-CSF secreted by genetically modified plant cells. Protein Expr Purif. 19, 131-138 (2000)
【非特許文献2】Magnuson NS, Linzmaier PM, Reeves R, An G, HayGlass K, Lee JM, Secretion of biologically active human interleukin-2 and interleukin-4 from genetically modified tobacco cells in suspension culture. Protein Expr Purif. 13, 45-52 (1998)
【非特許文献3】Magnuson NS, Linzmaier PM, Gao JW, Reeves R, An G, Lee JM, Enhanced recovery of a secreted mammalian protein from suspension culture of genetically modified tobacco cells. Protein Expr Purif. 7, 220-228 (1996)
【非特許文献4】Matsumoto S, Ishii A, Ikura K, Ueda M, Sasaki R, Expression of human erythropoietin in cultured tobacco cells, Biosci. Biotechnol. Biochem. 57, 1249-1252 (1993)
【非特許文献5】Terashima M, Murai Y, Kawamura M, Nakanishi S, Stoltz T, Chen L, Drohan W, Rodriguez RL, Katoh S, Production of functional human alpha 1-antitrypsin by plant cell culture. Appl Microbiol Biotechnol. 52, 516-523 (1999)
【非特許文献6】Fotisch K, Altmann F, Haustein D, Vieths S, Involvement of carbohydrate epitopes in the IgE response of celery-allergic patients. Int Arch Allergy Immunol. 120, 30-42 (1999)
【非特許文献7】Wilson IB, Harthill JE, Mullin NP, Ashford DA, Altmann F, Core alphal, 3-fucose is a key part of the epitope recognized by antibodies reacting against plant N-linked oligosaccharides and is present in a wide variety of plant extracts. Glycobiology. 8, 651-661 (1998)
【非特許文献8】van Ree R, Cabanes-Macheteau M, Akkerdaas J, Milazzo JP, Loutelier-Bourhis C, Rayon C, Villalba M, Koppelman S, Aalberse R, Rodriguez R, Faye L, Lerouge P, Beta(1,2)-xylose and alpha(1,3)-fucose residues have a strong contribution in IgE binding to plant glycoallergens. J Biol Chem. 2000 Apr. 14;275 (15):11451-11458
【非特許文献9】Strasser R, Stadlmann J, Schahs M, Stiegler G, Quendler H, Mach L, Glossl J, Weterings K, Pabst M, and Steinkellner H, Generation of glyco-engineered Nicotiana bethamiana for the production of monoclonal antibodies with a homogeneous human-like N-glycan structure. Plant Biotech. J. 6, 392-402 (2008)
【非特許文献10】Leonard R, Petersen B.O, Himly M, Kaar W, Wopfner N, Kolarich D, van Ree R, Ebner C, Duus J, Ferreira F, and Altmann R, J. Biol. Chem, 280, 7932-7940 (2005)
【非特許文献11】Leonard R, et al, Carbohydr. Res, 340, 657 (2005)
【非特許文献12】松岡健、バイオサイエンスとインダストリー、63、303 (2005)
【非特許文献13】Tiainen P, et al, J. Biol. Chem., 280, 1142 (2005)
【非特許文献14】Yuasa K, et al, Plant J., 41, 81 (2005)
【非特許文献15】Oka T, et al., Characterization of ER localized UDP-D-galactose: hydroxyproline O-galactosyltransferase using synthetic peptide substrates in Arabidopsis thaliana. Plant Physiol.(2009)
【非特許文献16】Lamport DT, Katona L, Roerig S, Galactosylserine in extensin.. Biochem J. 133, 125-32 (1973)
【非特許文献17】Allen AK, Desai NN, Neuberger A, Creeth J, Properties of potate lectin and the nature of its glycoprotein linkages , Biochem J. 17, 665-74 (1978)
【非特許文献18】Matsuoka K, Watanabe N, Nakamura K. O-glycosylation of a precursor to a sweet potato vacuolar protein, sporamin, expressed in tobacco cells Plant J. 8, 877-89 (1995).
【非特許文献19】Busch C, Hofmann F, Selzer J, Munro S, Jeckel D, Aktories K. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J Biol Chem. 1998 Jul 31;273(31):19566-72.
【非特許文献20】Karg SR and Kallio PT (2009) The production of biopharmaceuticals in plant system. Biotech. Advances, 27, 879-894.
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、セリン残基のヒドロキシル基にガラクトースを付加する作用のガラクトース転移酵素(SGT)遺伝子のうちで、実用的な観点からみて、汎用的な形質転換体宿主であるタバコ由来のSGT遺伝子をターゲットとすることにした。すなわち、タバコ由来SGT遺伝子をクローニングし、当該遺伝子による形質転換体を用いたO−結合型ガラクトース含有ポリペプチドの製造方法を提供すること、及び当該遺伝子発現を抑制する方法を提供し、形質転換植物におけるO−結合型糖鎖の付加を抑制したヒト有用タンパク質の製造方法を提供することを目的とする。
【課題を解決するための手段】
【0008】
タバコ由来SGT遺伝子をクローニングするにあたり、タバコの場合は未だ全ゲノム配列が公開されていないため、タバコのゲノムライブラリーから直接クローニングすることは難しい。そこで、まずは全ゲノム配列情報が公開されていて各ORF情報が入手可能なシロイヌナズナなどの他の植物及び藻類のSGT遺伝子を同定し、次いで当該遺伝子の配列情報を利用してタバコ由来のSGT遺伝子をクローニングすることとした。
【0009】
まず、シロイヌナズナの細胞抽出液からセリンO−結合型ガラクトース転移酵素(Peptidyl serine α-galactosyltransferase;SGT)の酵素活性の測定を、受容体基質としてスポラミン配列「FITC-PTTHEPASSETPVL」およびエクステンシン配列「VYKSPPPPV」を用いて試みたが、ガラクトース転移酵素活性は確認できなかった。プロリンを水酸化したスポラミン配列「FITC-PTTHEOASSETPVL」を受容体基質としても酵素活性は検出できなかったが、プロリンを水酸化したエクステンシン配列「VYKSOOOOV」を受容体基質としたところ、HPLCで酵素反応物のピークを検出でき、ガラクトース転移酵素活性を確認できた。したがって、「FITC-VYKSOOOOV」を受容体基質とすることによるSGT酵素活性測定が可能となった。そこで、SGT酵素を精製し、アミノ酸配列を解析することで、SGT酵素をコードする遺伝子の同定を試みたが、シロイヌナズナのSGT糖転移酵素活性がHGT酵素をはじめとした他のタンパク質と共に不溶性の膜画分に含まれ、可溶化できない状態であったため、SGT酵素を単離することができなかった。
【0010】
そこで、本発明者らは、全ての高等植物の祖先といわれる単細胞の緑藻の1種であるクラミドモナスが、高等植物と同様のO−結合型糖鎖構造を有する植物タンパク質を生産していることに着目し、「FITC-VYKSOOOOV」を用いて同様のガラクトース転移酵素活性を確認した。ヒドロキシプロリンへのガラクトース付加は、ペプチド中にとびとびにヒドロキシプロリンが存在することが必要であり、連続するヒドロキシプロリンにはアラビノースが付加し、ガラクトースは付加しないと考えられていた。しかし、予想に反して、セリン残基は一つしかないにもかかわらず、HPLC解析により複数のピークが確認されたことから、受容体基質ペプチドの2カ所にガラクトース付加が検出され、ヒドロキシプロリンへのガラクトース転移酵素(HGT)の活性も検出されていると考えられた。βガラクトシダーゼ処理およびβエリミネーション処理による解析を行った結果、HPLCでSGT酵素産物のピークとHGT酵素産物のピークを同定することができたことから、セリンO−結合型のガラクトース転移酵素活性のみを正確に測定することが可能となった。一方で、ガラクトース転移酵素の可溶化も可能であることを見出し、SGT酵素とHGT酵素を可溶化条件やカラムによる分画で分離できることが確認できたので、クラミドモナスからSGTを単離することとした。
クラミドモナスから、SGTを精製し、そのアミノ酸配列解析の結果から、SGTをコードする遺伝子の同定に成功し、当該遺伝子を異種である酵母細胞で発現させることでSGTをコードする遺伝子であることを証明したとともに、詳細な酵素活性を確認した。さらに、当該遺伝子との相同性検索により、各種植物のSGT遺伝子を同定し、この遺伝子が、緑藻から高等植物まで存在しており、動物や真菌には類似遺伝子が全く存在しないこと、及び従来の糖転移酵素遺伝子とも全く類似性がないため、糖転移酵素遺伝子であると予測されていない新規な遺伝子ファミリーに属していることを見出した。シロイヌナズナSGT遺伝子についても、その遺伝子を単離して酵母で発現させたところ、クラミドモナスSGTと同様のSGT活性を確認することができた。本出願人はこれらの知見をもとに特許出願をしている(PCT/JP2010/071598)。
【0011】
次いで、シロイヌナズナのSGT1配列をクエリーにして、タバコのESTデータベースを検索し、シグナル配列、膜貫通領域、塩基性アミノ酸に富む領域を欠く2074bpからなる部分配列を見出した。当該配列を利用して設計したプライマーを用いて、タバコBY-2細胞から抽出した全RNAを鋳型とし、RT-PCR、5’RACE、3’RACE法によりタバコSGT1(NtSGT1)遺伝子に対応する全長3,336bpからなるcDNAをクローニングした(配列番号20)。タバコSGT1アミノ酸配列は配列番号21で表され、シロイヌナズナSGT1とは80%以上のアミノ酸配列同一性を示し、SGT活性ドメイン1(配列番号22)及び活性ドメイン2(配列番号23)を有する。このタバコSGT1のシグナル配列および膜貫通領域を除いた主要部分を酵母で発現させて、SGT酵素活性を確認した。
さらに、当該SGT酵素活性が一般的な発現抑制技術で抑制可能かどうかを確認するために、タバコ培養細胞BY-2に対してヘアピン型二本鎖RNA(dsRNA)発現プラスミドを導入して誘導したRNAi実験により、SGT1酵素活性が大幅に抑制されることを観測した。このことは、当該SGT遺伝子がタバコ植物体内でのセリンO−結合型ガラクトース付加遺伝子であることが確認できたことであり、かつ当該SGT遺伝子をターゲットとしてその遺伝子発現を抑制すれば、タバコ植物体内でのタンパク質へのセリンO−結合型のガラクトース付加抑制が可能であることを示すものでもある。
RNAiをはじめ植物内での糖転移酵素の発現抑制技術自体は、従来から周知であるから、タバコなど植物体内に本来存在する本発明のSGT遺伝子をターゲットとして、RNAiなど周知抑制手法を適用することでSGTの発現抑制が可能である。当該タバコなどの植物を形質転換植物宿主とすることで、外来遺伝子に対するセリンO−結合型ガラクトース付加を抑制することができるから、バイオ医薬として期待される抗体、生理活性糖タンパク質などのヒト有用糖タンパク質にとってきわめて有力なツールを提供できたことになる。特に、形質転換植物において、当該SGT遺伝子の発現抑制を、プロリン水酸化酵素遺伝子および他のO−結合型糖転移酵素遺伝子の抑制と同時に行うことで、外来タンパク質に対する植物特有のO−結合型糖修飾を完全に抑制することも可能である。
以上の知見を得たことで、本発明を完成するに至った。
【0012】
すなわち、本発明は以下を包含する。
〔1〕 下記の(1)〜(4)のいずれかのアミノ酸配列を含む、セリンO−結合型ガラクトース転移酵素活性(SGT酵素活性)を有するポリペプチド;
(1)配列番号21に示されるアミノ酸配列、
(2)配列番号21に示されるアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列。
(3)配列番号21と80%以上の同一性を有しているアミノ酸配列からなり、かつそのSGT酵素活性ドメインとして、DXDモチーフを有し、配列番号22又は23と90%以上の同一性を有するアミノ酸配列からなる部分配列を有しているアミノ酸配列、
(4)植物のゲノム中の塩基配列によりコードされるアミノ酸配列であって、配列番号21に示されるアミノ酸配列と80%以上の同一性を有するアミノ酸配列からなり、かつDXDモチーフを含むアミノ酸配列。
〔2〕 下記(5)〜(7)に記載のアミノ酸配列を含む、セリンO−結合型ガラクトース転移酵素活性(SGT酵素活性)を有するSGT酵素活性ドメインポリペプチド;
(5)配列番号22又は23に示されるアミノ酸配列、
(6)配列番号22又は23に示されるアミノ酸配列のいずれかにおいて1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列、
(7)配列番号22又は23に示されるアミノ酸配列と少なくとも90%以上の同一性を有するアミノ酸配列であって、かつDXDモチーフを含むアミノ酸配列。
〔3〕 前記〔1〕又は〔2〕に記載のポリペプチドをコードする塩基配列からなる核酸。
〔4〕 セリンO−結合型ガラクトース転移酵素活性を有するポリペプチドをコードする核酸であって、配列番号20に示される塩基配列、又は当該塩基配列の相補配列とストリンジェントな条件下でハイブリダイズする塩基配列からなる核酸。
〔5〕 前記〔3〕又は〔4〕に記載の核酸を組み込んだ発現ベクター。
〔6〕 前記〔3〕又は〔4〕に記載の核酸が発現可能に組みこまれた形質転換体。
〔7〕 前記〔6〕に記載の形質転換体を用いたO−結合型糖鎖を有する組換え糖タンパク質の製造方法であって、当該形質転換体が外来性糖タンパク質をコードする核酸により形質転換されていることを特徴とする、セリンO−結合型ガラクトースを含む糖鎖を有する組換え糖タンパク質の製造方法。
〔8〕 前記〔2〕に記載された、SGT酵素活性を有する活性ドメインポリペプチドに結合する阻害物質により当該ポリペプチドのSGT酵素活性を阻害することを特徴とする、形質転換植物細胞または形質転換植物における外来タンパク質のセリン残基へのO−結合型ガラクトース付加抑制方法。
〔9〕 形質転換植物細胞または形質転換植物により外来タンパク質を産生する際に、外来タンパク質に含まれるセリン残基へのO−結合型ガラクトース付加を抑制する方法であって、形質転換宿主となる植物細胞が本来有しているSGT遺伝子である、前記[3]又は[4]に記載されたSGTをコードする核酸の発現を抑制することを特徴とする、形質転換植物細胞または形質転換植物における外来タンパク質のセリン残基へのO−結合型ガラクトース付加抑制方法。
〔10〕 前記〔8〕又は〔9〕に記載の方法により外来タンパク質のセリン残基へのO−結合型ガラクトース付加が抑制された、形質転換植物細胞または形質転換植物。
〔11〕 形質転換植物細胞または形質転換植物がタバコである前記〔10〕に記載の形質転換植物細胞または形質転換植物。
【発明の効果】
【0013】
本発明によって、汎用的な形質転換宿主であるタバコ由来のSGT遺伝子が提供できたので、外来遺伝子を宿主細胞に導入する際に、本発明のタバコ由来のSGT遺伝子を同時に導入するか、又は前もって導入しておくことで、O−結合型ガラクトースが高付加された外来タンパク質を生産することができる。
反対に、本発明においては、ヒトなど哺乳動物由来タンパク質をタバコなどの形質転換植物により大量生産させる際に問題となる、O−結合型ガラクトースの付加を抑制するためのターゲット遺伝子が特定できたことであるから、RNAi法などの周知の植物体内の酵素遺伝子発現の抑制化技術を適用することで、SGT遺伝子の発現を抑え、外来タンパク質のセリンにO−結合型ガラクトースが付加するのを抑制することができる。公知のプロリン水酸化酵素発現、植物特有のN−結合型の各種糖鎖転移酵素の発現も同時に抑制することで、形質転換植物体内で発現する植物特有の糖鎖結合が阻止できるから、ヒトの生理活性タンパク質なども形質転換植物で安価に大量生産可能となる。抗原性のないバイオ医薬の安価な生産の可能性を開く技術である。
【図面の簡単な説明】
【0014】
【図1】[SGT粗酵素の活性測定] クラミドモナスの細胞破砕液をSGT粗酵素液として用いて、「(HYP)AtEXT4:VYKSOOOOV」を基質ペプチドとして、UDP-ガラクトースを供与体基質として、12時間反応した場合、3種類のプロダクトピーク#1、#2及び#3が確認できた。
【図2】[ガラクトース付加部位の特定(MS解析、β−ガラクトシダーゼ処理)] クラミドモナスSGT酵素反応産物を、MS解析した結果、162Daのピークのシフトが観察され、プロダクト#1及び#2は、基質ペプチドに1残基のヘキソース(ガラクトース)の付加が、プロダクト#3は2残基のヘキソース(ガラクトース)の付加が確認できた。SGT酵素反応産物を、β−ガラクトシダーゼ処理したところ、プロダクト#3は#1に、#2は基質ペプチドの溶出位置にピークがシフトした。これらのことから、プロダクト#1は基質ペプチドのセリン残基(S)にガラクトースが1残基付加したもの、プロダクト#2は、いずれかのヒドロキシプロリン残基(O)にガラクトースが1残基付加したもの、プロダクト#3は、セリン残基とヒドロキシプロリン残基にそれぞれ1残基ずつガラクトースが付加したものであることが明らかになった。
【図3AB】[プロダクト#1のガラクトース付加部位の特定(β−エリミネーション法による)] β−エリミネーション法は、O−結合型糖鎖をペプチドから切り離す反応であることが一般的に知られているが、ヒドロキシプロリンに結合したO−結合型糖鎖は切り離さないことが知られているので、β−エリミネーション処理することにより、ガラクトースがセリン残基あるいはヒドロキシプロリン残基のどちらに結合しているかを区別することができる。(A)では、プロダクト#1は基質ペプチドの分子量にガラクトースの分子量162Daの分子量シフトが観察され、(B)においてβ−エリミネーション処理した結果、ガラクトースが切れて、元の基質ペプチドの分子量のピークが現れたこと、さらに、DTT処理によりβ−エリミネーションにより切れたアミノ酸へのDTT付加が観察された。このことから、プロダクト#1は、セリンにガラクトースが付加していることが明らかになった。
【図3CD】[プロダクト#2のガラクトース付加部位の特定](D)は、(C)のプロダクト#2をβ−エリミネーション処理したものであるが、ピークのシフトは観察されず、ガラクトースは切られなかったことが分かることから、ヒドロキシプロリン残基にガラクトースが付加していたことが明らかとなった。
【図4】[クラミドモナスSGT(クラミドモナス細胞破砕液)を用いたAtEXTペプチドのSer残基に対するSGT酵素活性への最適反応条件の検討](A)ドナー基質の選択性−ドナー基質としては種々の糖ヌクレオチドのうちでUDP-ガラクトース(Gal)が選択的に利用される。ここで、Arafはアラビノフラクトース、GlcNacはN-アセチルグルコサミン、GlcAはグルクロン酸、Glcはグルコース、Xylはキシロースを示す。(B)2価金属イオン要求性−Mn2+が際だって高かった。(C)至適pH−図中、◆は、0.1Mの酢酸ナトリウム、■は、0.1MのMES-NaOH、▲は、0.1MのMOPS-NaOH、●は、0.1MのTris-HClをそれぞれ含有した緩衝液。(D)至適温度 20〜60℃条件でインキュベートした。至適温度は30℃。
【図5A】[クラミドモナスSGTの精製 (step2)]1st DEAE Sepharoseカラムによる分画後、SDS-PAGEでタンパク質バンドを検出。
【図5B】[クラミドモナスSGTの精製 (step3)]2nd DEAE Sepharoseカラムによる分画後、SDS-PAGEでタンパク質バンドを検出。
【図5C】[クラミドモナスSGTの精製 (step5)]2nd Sephadex G-200カラムによる分画後、SDS-PAGEでタンパク質バンドを検出。
【図6A】[精製クラミドモナスSGTタンパク質バンドの切り出し]四角で囲んだ精製SGTの4サンプルのバンドをゲルから切り出した。この4サンプルに対し、MS/MS分析を行い、ペプチド配列を決定し、クラミドモナスのゲノムデータベースに100%一致するORFを見出した。
【図6B】[精製SGTタンパク質バンドの切り出し]同定されたSGTのアミノ酸配列についてハイドロパシー解析を行ったところ、C末端に疎水領域のあるタンパク質と予測された。
【図7A】[タバコSGTと各種植物由来のSGTとのアミノ酸配列アラインメント] タバコ(Nt)由来SGTアミノ酸配列と、クラミドモナス(Cr)、ニセツリガネゴケ(Pp)、シロイヌナズナ(At)とのアラインメントを示す。太字はC末端側にある膜貫通領域を示す。直線及び二重線による下線は、それぞれCD1及びCD2領域を、波線は機能不明な領域を示す。濃い直線部はシグナル配列であり、四角で囲った領域は、DXDモチーフに相当する。
【図7B】[各種植物由来のSGTのアミノ酸配列アラインメント-1(参考)] クラミドモナス、プラシノ藻、ニセツリカネゴケ、イネ、シロイヌナズナ、ブドウ、ポプラ、トウゴマのSGT遺伝子から予測されるアミノ酸配列のアライメント。クラミドモナスのSGTと他の植物のSGTは、30%のアミノ酸同一性を示した。
【図7C】各種植物由来のSGTのアミノ酸配列アラインメント-2(参考)
【図7D】[タバコを含む各種植物由来のSGTの活性ドメイン(SGT1-CD)部分のアライメント] 図中、Ntはタバコ(Nicotiana tabacum)、Crはクラミドモナス(Chlamydomonas reinhardti)、Otはブラシノ藻(Ostreococcus tauri)、Ppはニセツリガネゴケ(Physcomitrella patens)、Smはイヌタカヒバ(Selaginella moellendorffii)、Osはイネ(Oryza sativa)、Atはシロイヌナズナ(Arabidopsis thaliana)、Ptはポプラ(Populus trichocarpa)。以下同様。
【図7E】SGTの活性ドメイン(SGT1-CD)の系統樹
【図8】[SGT1の推定ドメイン構造] 各種植物のSGT1の推定ドメイン構造を図示した。N末端に分泌系のシグナルペプチド、C末端側に膜貫通領域があり、I型膜タンパク質であった。クラミドモナスおよびブラシノ藻は活性ドメインが1つだけであったが、陸上植物では2つずつあり、N末端側をSGT1-CD1、C末端側をSGT1-CD2とした。図中、SSはシグナルペプチド、CDはSGT酵素活性ドメイン、Sはスペイサー、TはTOX-1ドメイン、Pはプロリンリッチドメイン、TMは膜貫通領域、Bは塩基性アミノ酸リッチ領域。
【図9】[クラミドモナスSGT1の酵母における発現] クラミドモナスSGT1(CrSGT1、又はChSGT1ともいう。)を酵母細胞壁タンパク質PIR4に融合して発現させ、SGT酵素活性を測定した。Chlamy-S10-12hrsはクラミドモナス細胞破砕液の酵素活性。PIR4-HA-ΔSSΔTMは、シグナルペプチドと膜貫通領域を削除したChSGT1をPIR4に融合して発現。プロダクト#1だけが検出され、ChSGT1遺伝子がSGT活性だけを持つことを確認。
【図10】[シロイヌナズナのSGT1の酵母における発現] シロイヌナズナSGT1(AtSGT1)を酵母細胞壁タンパク質PIR4に融合して発現させ、SGT酵素活性を測定した。PIR4-HA-ΔSSΔTMは、シグナルペプチドと膜貫通領域を削除した活性ドメインを2つとも含むAtSGT1をPIR4に融合して発現。プロダクト#1の位置に小さなピークが検出されたので、AtSGT1遺伝子はSGT活性を持つことを確認した。PIR4-HA-CTΔTMは、2つの活性ドメインのうちAtSGT1-CD1を削除し、AtSGT1-CD2の1つのみであり、プロダクト#1の位置にピークが検出されたことから、SGT活性ドメインは1つだけでもSGT酵素活性を持つことが分かった。
【図11】[シロイヌナズナSGT1変異株における酵素活性の減少(HPLC解析)] 図中WT1〜3は正常株、Δsgtl1〜3はSGT1遺伝子のT-DNA変異株、CrSGT1は、図9、10のChlamy-S10-12hrsと同じクラミドモナス細胞破砕液の酵素活性を示す。
【図12】[シロイヌナズナSGT1変異株におけるO結合型ガラクトース付加の減少]A:糖鎖から遊離されたαガラクトース量、B:糖鎖から遊離されたβガラクトース量。図中、●は正常株、▲はSGT1遺伝子のT-DNA変異株。
【図13】[タバコSGT1の酵母における発現] 図中、NtSGTΔSSΔTMは、シグナルペプチドと膜貫通領域を削除した活性ドメインを2つとも含むNtSGT1をPIR4に融合して発現したものであり、NtSGT-NTΔSSはCD1だけを含む融合遺伝子であるが、いずれもプロダクト#1の位置にSGT酵素産物は見られなかった。一方、NtSGT-CTΔTMは、CD2だけを含む融合遺伝子であるが、プロダクト#1が検出されており、CD2は単独でSGT酵素活性があることが確認できた。シロイヌナズナでもほぼ同様の結果を得ていることから、CD2は単独でSGT酵素活性があるのに対して、CD1の場合は、まだ理由は不明であるが、何らかの制御機構によりSGT活性が抑制的に制御されている可能性が示唆される。
【図14】[RNAiによるSGT転写抑制効果] A:RT-PCRの電気泳動結果−図中、RNAi1〜3はdsRNA発現ベクターを導入したタバコ培養細胞BY-2の3種類のクローンであり、wt1〜3はコントロールとして用いた非形質転換細胞。B:Aで検出したPCR産物のシグナル強度のグラフ化。C:BY-2細胞のマイクロソーム画分におけるSGT活性の減少。
【発明を実施するための形態】
【0015】
以下、本発明についてさらに詳細に説明する。
1.セリンO−結合型ガラクトース転移酵素(Peptidyl serine α-galactosyltransferase;SGT)について
(1−1)植物のO−結合型糖鎖
植物のO−結合型糖鎖構造は、大きく分けて2種類あることが知られている。ひとつは、タンパク質のアミノ酸配列中の複数のプロリン残基が水酸化されてヒドロキシプロリンになり、そこにガラクトースまたはアラビノースが付加する構造である。典型的な配列として、エクステンシンの配列「VYKSOOOOV(Oはヒドロキシプロリン)」がよく知られており、この複数のヒドロキシプロリン基のヒドロキシル基が糖修飾される。プロリン水酸化酵素はすでに同定されているが、ヒドロキシプロリンにガラクトースまたはアラビノースを付加するガラクトース転移酵素(HGT)及びアラビノース転移酵素は未だに単離されていない。
そして、もう一つの植物特有のO−結合型糖鎖構造が、タンパク質のアミノ酸配列中のセリン残基に対して直接ガラクトースが付加された糖鎖構造であり、この糖鎖修飾を行っている酵素が、本発明のセリンO−結合型ガラクトース転移酵素(SGT)であり、本発明においては、そのうちの特にタバコ由来のSGT(NtSGT)を対象とする。
【0016】
(1−2)本発明のタバコ由来SGT酵素及びそれをコードする遺伝子の配列
タバコは、典型的な遺伝子組換え宿主植物ではあるが、そのゲノム配列は公表されていないため、タバコのゲノムライブラリーなどから直接クローニングすることは難しいため、下記(1−3)に記載のシロイヌナズナなどの他の植物又は藻類のSGT遺伝子を同定し、次いでシロイヌナズナのSGT遺伝子(配列番号4)に基づいてプライマー、プローブを作製し、タバコ植物ゲノムに対して適用し、2箇所の活性ドメイン領域を有するタバコSGT酵素(配列番号21)の遺伝子クローニングに成功し、対応するタバコSGT酵素のアミノ酸配列(配列番号20)を決定したが、これらはいずれも新規酵素遺伝子及び新規ポリペプチドであるといえる。タバコSGT酵素遺伝子の登録番号としては、AB617524が付与された。
タバコSGT酵素活性ドメイン領域は、CD1が配列番号22、CD2が配列番号23で示され、特にCD2は単独でもSGT酵素活性を有することが実証されており、いずれも有用性のある新規ポリペプチドである。
進化系統樹的に最も近いSGT遺伝子はシロイヌナズナ及びポプラ由来SGT遺伝子であり、活性ドメインCD1ではそれぞれアミノ酸配列レベルで81%及び86%、CD2では80%及び83%と高い(下記表3)ものの、SGT遺伝子全体のホモロジーは72%及び76%とさほど高くない。
タバコ植物においても多品種が存在しているので、これら個体差に基づく相同遺伝子配列も含めると、タバコ由来SGT酵素活性を有するポリペプチド及び当該ポリペプチドをコードする遺伝子は、以下のように表すことができる。
すなわち、タバコ由来SGT酵素活性を有するポリペプチドは、以下のように表すことができる。
(1)配列番号21に示されるアミノ酸配列を含むポリペプチド。
(2)配列番号21に示されるアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、SGT酵素活性を有するポリペプチド。なお、ここで「1若しくは数個」というときの数個は、自然に変異が導入される程度の数値範囲を示すので、2〜15個、または2〜8個、2〜5個程度の数値を表す。
(3)配列番号21と80%以上好ましくは90%以上の同一性を有しているアミノ酸配列からなるポリペプチドであって、かつそのSGT酵素活性ドメインとして、DXDモチーフを有し、配列番号22又は23と90%以上好ましくは95%以上の同一性を有するアミノ酸配列からなる部分配列を有しているアミノ酸配列からなり、SGT酵素活性を有するポリペプチド。
(4)植物のゲノム中の塩基配列によりコードされるアミノ酸配列であって、配列番号21に示されるアミノ酸配列と80%以上好ましくは90%以上の同一性を有するアミノ酸配列からなり、かつDXDモチーフを含むアミノ酸配列からなり、SGT酵素活性を有するポリペプチド。
また、その「SGT酵素活性ドメイン」のCD1領域及びCD2領域についても、特にCD2領域は単独でSGT酵素活性を有することが実証されているから、以下のように表現できる。
(5)配列番号22又は23に示されるアミノ酸配列を含むポリペプチド。
(6)配列番号22又は23に示されるアミノ酸配列のいずれかにおいて1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、SGT酵素活性を有するポリペプチド。なお、ここで「1若しくは数個」というときの数個は、上記(2)の定義と同様である。
(7)配列番号22又は23に示されるアミノ酸配列と少なくとも90%以上好ましくは95%以上の同一性を有するアミノ酸配列であって、かつDXDモチーフを含むアミノ酸配列からなり、SGT酵素活性を有するポリペプチド。
【0017】
一方、本発明のタバコ由来SGT酵素遺伝子などのSGT酵素活性を有するポリペプチドをコードする核酸は、以下のように表現できる。
(8)上記(1)〜(7)に記載のポリペプチドをコードする塩基配列を含むSGT酵素活性を有するポリペプチドをコードする核酸。
(9)配列番号20に示される塩基配列、又は当該塩基配列の相補配列とストリンジェントな条件下でハイブリダイズする塩基配列からなる核酸であって、セリンO−結合型ガラクトース転移酵素活性を有するポリペプチドをコードする核酸。なお、ここで、ストリンジェントな条件とは、「Molecular Cloning 3rd edition Sambrook J ら、Cold Spring Harbor Laboratory Press, 2001」などの教科書にストリンジェンシーの高いハイブリダイズ条件として示されている条件を指す。
【0018】
(1−3)本発明のタバコ由来SGT酵素遺伝子及び当該SGT遺伝子と関連するSGT遺伝子一般について
本発明のタバコ由来SGT酵素遺伝子と関連したSGT遺伝子としては、クラミドモナス(Chlamdomonas reinhardtii)では、配列番号1の塩基配列(A8J5Z4_CHLRE配列)からなる遺伝子およびシロイヌナズナ由来の配列番号4の塩基配列(Q8VYF9、At3g01720配列)からなる遺伝子などがあるが、SGT遺伝子は単細胞藻類のクラミドモナスから、被子植物に至るまで植物全般で広く保存された遺伝子ファミリーである。動物や真菌など植物以外の生物種にはホモロジーのある遺伝子が存在しない、植物特有の遺伝子ファミリーであり、他のどの糖転移酵素とも類似性を持たず、全く新規な糖転移酵素遺伝子ファミリーである。
具体的には、クラミドモナス由来の配列番号1の塩基配列に対するBlast検索結果によれば、シロイヌナズナ(Arabidopsis thaliana)、イネ(Oryza sativa)、トウゴマ(Ricinus communis)、ブドウ(Vitis vinifera)、ポプラ(Populus trichocarpa)、ニセツリガネゴケ(Physcomitrella patens)、ブラシノ藻(Ostreococcus tauri)などに、約30%identityのホモログORFが1ゲノムあたりそれぞれ1ORFずつ存在していた(表4)。広葉植物同士では、70%程度のアミノ酸の同一性があり、被子植物間では60%程度、被子植物と単細胞緑藻の間では30%のアミノ酸配列の同一性が認められた。しかし、動物や真菌にはホモロジーのある遺伝子は見つからなかった(表4)。上記各種植物、藻類のSGT1遺伝子に対して付与された登録番号は、以下の通りである。クラミドモナス:AB617522、シロイヌナズナ:AB617523、イネ:BR000899、ポプラ:BR000900、ブドウ:BR000901、トウゴマ:BR000902、ニセツリガネゴケ:BR000904およびBR000905、ブラシノ藻:BR000906。またその他のSGT1遺伝子に対しても下記の登録番号が付与された。ソルグム(Sorghum bicolor ):BR000903、イヌタカヒバ(Selaginella moellendorffii):BR000909、ミヤマハタザオ(Arabidopsis lyrata):BR000908、ボルボックス(Volvox carteri):BR000907、クロレラ(Chlorella variabilis):BR000910。SGT1のドメイン構造は、糖転移酵素としては珍しく、I型膜タンパク質のトポロジーであり、膜貫通領域がC末側に存在する(図8)。また、クラミドモナスのSGT1のN末端側のホモロジーの高い領域(配列番号17)のみでも活性があり、当該領域中に活性ドメインがあると推定される(Chla組換えSGT1)。図7での各植物間のアラインメントの検討の結果、図8に示すように共通した保存配列としての活性ドメイン領域が明らかとなった。
また、すべての植物のSGT1-CD1及びSGT1-CD2ドメイン内では、糖ヌクレオチドを基質とする糖転移酵素に広く保存されているDXDモチーフ(Mn2+イオンを介して糖ヌクレオチドとの結合部位)(非特許文献19)が保存されていることからも、当該領域が各植物共通の活性ドメインであることを裏付けている(図7D)。
該SGT1活性ドメイン(CD)は、単細胞藻類のクラミドモナス及びブラシノ藻ではN末側の1箇所である(SGT1-CD1)が、ニセツリガネゴケからシロイヌナズナなどの高等植物では、ホモロジーの高い活性ドメイン領域がN末端側とC末端側にタンデムに重複して存在していた(SGT1-CD1及びSGT1-CD2)。各高等植物におけるSGT1-CD1及びSGT1-CD2領域との領域同士のホモロジーは、アミノ酸配列で58%程度の同一性であった。シロイヌナズナにおいてSGT1-CD1及びSGT1-CD2領域それぞれのSGT活性を測定したところ、シロイヌナズナ活性領域においても、SGT1-CD2領域(PIR4-HA-CTΔTM、配列番号18)だけでも単独でSGT活性があることが確認された(図9、At組換えSGT1)。
クラミドモナスのSGT1には、高等植物のプロリン水酸化酵素のER-ゴルジ局在配列Tox1が存在し、ER-ゴルジの局在が示唆された。このことから、セリン残基へのガラクトース付加は、糖タンパク質合成の早い段階で付加が起きると考えられる。高等植物のSGT1には、Tox1配列は見つかっていない。
SGT1をコードする塩基配列としては、クラミドモナスおよびブラシノ藻では、複数のホモロジーのあるORFが検索されるが、ニセツリガネゴケからシロイヌナズナなどの各植物のSGT遺伝子についてデータベースの配列情報を検索するといくつかの異なったデータが存在するが、DNA配列及び遺伝子座を詳細に検討してみると、各データ間の配列上の相違は、オールタナティブスプライシング、遺伝子の欠失挿入変異、シークエンス解析のミス、品種による遺伝子の多型などが原因であり、結局、各植物ゲノムあたり1種類の遺伝子しかないことが確認された。
【0019】
(1−4)本発明のタバコ由来SGT酵素遺伝子クローニング
本発明者は、本実施例で詳細に述べるように、先の出願(PCT/JP2010/071598)で取得したシロイヌナズナのSGT遺伝子(配列番号4)に基づいてプライマー、プローブを作製し、アミノ酸配列、及び塩基配列が公開されていないタバコ植物ゲノムに対して適用したところ、2箇所の活性ドメイン領域を有するタバコSGT酵素(配列番号21)の遺伝子クローニングに成功した。
タバコは、典型的な遺伝子組換え宿主植物ではあるが、そのゲノム配列は公表されていないため、上記(1−2)で述べたように、今回クローニングしたタバコSGT酵素のアミノ酸配列(配列番号20)及びその遺伝子配列(配列番号21)、並びにそのSGT酵素活性ドメインCD1及びCD2のアミノ酸配列(配列番号22、23)はいずれも新規ポリペプチド及び新規遺伝子であるといえる。
そして、下記(1−6)、(1−7)に記載したように、これらポリペプチド及びポリヌクレオチドは、タンパク質のセリン残基にガラクトースを付加する酵素製剤及び形質転換宿主細胞内のタンパク質、特に外来タンパク質に対して、そのセリン残基にガラクトースを付加するポリヌクレオチド製剤の有効成分として用いることができる。
【0020】
(1−5)本発明のSGT酵素の作用及び基質特異性
本発明のタバコ由来SGTを含めSGT酵素の作用は、下記の反応式で表される。
【0021】
【化1】
【0022】
基質特異性としては、セリンを含有するポリペプチドであり、好ましくは上記式中のR2としてヒドロキシプロリンが複数存在することが好ましい。
至適pHはpH6.0、至適温度は30℃で、反応温度は50℃以下である(図4)。等電点は中性付近であり、2価金属イオン要求性としては、マンガンイオン(Mn2+)要求性が観察された。
【0023】
(1−6)本発明におけるSGT酵素活性ポリペプチド及びSGT酵素活性酵素製剤
以上のことから、本発明において「SGT酵素活性」というとき、Mn2+イオン存在下でポリペプチド中のセリン残基のヒドロキシル基に対してUDP-ガラクトースからガラクトースを転移する酵素活性を指す。
そして、本発明のSGT酵素は、対象のタンパク質に対して、Mn2+イオン存在下でアミノ酸配列中のセリン残基におけるヒドロキシル基にガラクトースを付加する作用を有するので、当該酵素を有効成分とする酵素製剤として用いることができる。
本発明のSGT酵素遺伝子と関連した各植物のSGT酵素遺伝子は、緑藻から高等陸上植物まで高い相同性の活性ドメインがあるので、データベース上でも、またはcDNAライブラリー、もしくはゲノムDNAライブラリー中でもきわめて検索しやすく、しかも、各ゲノム中でそれぞれ1種類しかないので、SGT遺伝子を同定しやすい。
具体的には、各植物のSGT酵素の活性ドメインは、クラミドモナスSGT酵素の活性ドメイン(配列番号3)又はシロイヌナズナSGT酵素の活性ドメインAtSGT1-CD1(配列番号6)、AtSGT1-CD2(配列番号7)、イネSGT酵素の活性ドメインOryzSGT1-CD1(配列番号10)、OryzSGT1-CD2(配列番号11)、ニセツリガネゴケSGT酵素の活性ドメインPhysSGT1-CD1(配列番号13)、PhysSGT1-CD2(配列番号14)、ブラシノ藻SGT酵素の活性ドメインOstreoSGT1-CD1(配列番号16)として同定された(本出願人の先願であるPCT/JP2010/071598)。上述のように、各SGT酵素内のCD1領域とCD2領域とのホモロジーがアミノ酸配列で57%〜60%であるが、各植物のCD1領域同士のホモロジーは37%〜63%程度であることから、さらには各陸上植物のCD1領域同士のホモロジーは60%〜100%程度であることから、本発明の「CD領域」といえるためには、少なくともアミノ酸配列で50%(好ましくは60%〜100%)のホモロジー(なお、本発明でのホモロジー又は相同性の数値は、塩基配列はBlast検索により、アミノ酸配列の場合はFasta検索結果における「同一性」の数値を示している。)を有し、かつDXDモチーフを有するものを指す。単細胞藻類の場合は当該活性化ドメインを1箇所、多細胞植物であれば2箇所有している。したがって、ここに記載した植物以外の植物由来であっても、上記配列番号3,6,7,10,11,13,14又は16に示されるいずれかのアミノ酸配列と50%以上、好ましくは60%以上、より好ましくは70%以上の相同性を有し、かつDXDモチーフを有するアミノ酸配列であって、ゲノム中に1又は2箇所しか対応する塩基配列が存在しない場合は、SGT酵素の活性ドメインであると考えられる。
一方、上記各植物のSGT酵素の全長は、クラミドモナスSGT酵素のアミノ酸配列(配列番号2)、シロイヌナズナSGT酵素のアミノ酸配列(配列番号5)、イネのSGT酵素のアミノ酸配列(配列番号9)、ニセツリガネゴケSGT酵素(配列番号12)、ブラシノ藻SGT酵素(配列番号15)であると同定され、一般に各植物の全長SGT酵素のアミノ酸配列の相同性は、まれには70%以上のこともあるが、通常30%〜50%の範囲内である。つまり、上記植物以外の植物由来の場合にも、ゲノム中に対応する相同配列が1箇所しかなく、上記配列番号2,5,9,12もしくは15のアミノ酸配列と30%以上、好ましくは50%以上、より好ましくは70%以上の範囲内にあって、かつ上記SGT酵素の活性ドメインに相当する配列が1又は2箇所存在する場合ば、ほぼ確実にSGT酵素であると考えられる。
【0024】
(1−7)本発明のタバコSGT遺伝子を用いるポリヌクレオチド製剤
上記(1−2)で定義された本発明のタバコ由来SGT遺伝子、又は当該遺伝子を含む発現ベクターを用いて宿主細胞を形質転換することにより、哺乳動物細胞など本来SGT酵素活性を持たない細胞に対してSGT酵素活性を付与する、又は植物細胞に対してSGT活性を増強することができる。すなわち、上記SGT遺伝子又は当該遺伝子を含む発現ベクターを有効成分とするポリヌクレオチド製剤は、形質転換宿主細胞内のタンパク質、特に外来タンパク質に対して、そのセリン残基にガラクトースを付加する作用を有するものである。
【0025】
2.本発明におけるSGT酵素又はSGT酵素遺伝子を用いた、セリンO−結合型ガラクトース含有ポリペプチドの製造方法
(2−1)本発明のタバコSGT酵素又はSGT酵素活性ポリペプチドを用いたセリンO−結合型ガラクトース含有ポリペプチドの製造方法
本発明のSGT酵素又はSGT酵素活性ポリペプチドは、各種タバコ植物から単離精製して用いてもよいが、細胞を破砕し、不溶性の膜画分を再懸濁した細胞破砕液のままで用いることもできる。
2〜5mM Mn2+イオンを含有させた溶液中に糖鎖付加をしたいポリペプチドを0.1mM程度含有させ、ガラクトース供与体としてUDP-ガラクトースを1〜5mM含有させ、pH5.5〜7.0程度に調整した溶液に対して、精製酵素又は精製ポリペプチドが0.01〜0.05mg/l程度になるように添加し、20〜35℃で1〜12時間程度作用させればよい。
【0026】
(2−2)タバコSGT酵素又はSGT酵素活性ポリペプチドをコードする遺伝子を用いたセリンO−結合型ガラクトース含有ポリペプチドの製造方法
本発明のタバコSGT酵素又はSGT酵素活性ポリペプチドをコードする遺伝子を導入し、発現させるための形質転換体としては、典型的にはサッカロミセス・セレビッシエ酵母が挙げられるが、糖鎖付加機構を持たない大腸菌などの細菌類以外であれば、各種植物細胞、CHO細胞などの哺乳動物細胞、昆虫細胞などを宿主として選択することができる。その際の導入ベクターは、周知の発現ベクターを適宜選択できる。
本発明のタバコSGT酵素又はSGT酵素活性ポリペプチドによりセリンO−結合型ガラクトースを付加させる対象となるポリペプチドとしては、セリンを分子内に含めばどのようなポリペプチドであっても良い。例えば、エクステンシン、スポラミン、ムチンコアタンパク質などが考えられる。
【0027】
3.本発明におけるSGT酵素活性の抑制方法
(3−1)SGT酵素活性阻害剤を用いたSGT酵素活性の抑制
このようなSGT酵素活性を阻害するためには、活性ドメインをエピトープとする抗体、すなわち抗NtSGT1-CD1抗体、抗NtSGT1CD2抗体又はそれらのフラグメントを用いることができる。また、タバコSGT酵素の活性ドメインに結合するUDP-ガラクトースのアンタゴニスト物質であればよく、例えば各種のガラクトース誘導体などが阻害剤として働く。
【0028】
(3−2)SGT酵素遺伝子発現抑制剤を用いたSGT酵素活性の抑制
本発明のSGT酵素遺伝子発現抑制のためには、活性ドメインの遺伝子配列をターゲットとして、相同組換え法、各種RNAi物質(siRNA, miRNA,dsRNA)による抑制方法、アンチセンス法、トランスポゾン挿入法、T-DNA挿入法による遺伝子の破壊株を用いる方法等を用いてもよく、特にRNAi法が簡便で好ましい。
本発明の実施例では、タバコ植物細胞内のSGT遺伝子の転写をヘアピン型二本鎖RNA(dsRNA)発現プラスミドを用いたRNAiにより抑制できることを実証したが、この手法に限られるものではない。
例えば、植物型N型糖鎖転移酵素に対して用いられた各種方法(非特許文献20)が適用できる。具体的には、非特許文献20のレビュー文献中には、シロイヌナズナのN型糖鎖にα1,3−フコースあるいはβ1,2−キシロースを付加する糖転移酵素遺伝子が、アグロバクテリウムのT-DNAをランダムに挿入した変異株ライブラリーから当該遺伝子にT-DNAが挿入された株を選択し、さらに掛け合わせによりホモにT-DNAが挿入された株を作製する変異株作製法を用いることで、α1,3−フコースあるいはβ1,2−キシロースを付加が達成できることが示されている。また、ウキクサにおいて、RNAi法を用いた例として、α1,3−フコースおよびβ1,2−キシロースを付加する糖転移酵素活性が抑制され、当該形質転換植物体で発現されたヒトCD30には、α1,3−フコースおよびβ1,2−キシロースが付加されないことも示されている。さらに、RNAi法は、タバコ、アルファルファにおいてもα1,3−フコースおよびβ1,2−キシロースの付加が抑制されたことが示されている。
本発明において、汎用的な形質転換宿主となるタバコ由来のSGT酵素という標的遺伝子が提供され、しかもそのターゲットとなる領域としての活性ドメイン配列が特定されたことにより、これら周知の糖転移酵素遺伝子抑制方法が、そのまま本発明においても適用できることとなった。
【0029】
(3−3)SGT酵素遺伝子発現が抑制された形質転換植物及び形質転換植物におけるO−結合型糖鎖の付加を抑制した外来タンパク質の製造技術
本発明で用いる形質転換植物又は形質転換植物細胞としては、タバコ植物が好ましいが、ナス科植物などの双子葉植物の他、イネなどの単子葉植物に適用してもよい。
本発明で、タバコなどの植物細胞又は植物体から製造しようとするO−結合型糖鎖の付加を抑制した外来タンパク質の典型例は、ヒト由来のホルモン、サイトカイン、抗体などのバイオ医薬としての用途が期待されている生理活性タンパク質であるが、酵素等幅広く応用可能である。
【0030】
4.本発明におけるSGT酵素活性測定方法
本発明のSGT酵素活性を測定する際に用いた方法は、本出願人の先のPCT出願(PCT/JP2010/071598)に記載の方法と同様の手法であり、夾雑物が含まれる粗抽出液の状態でもHGT酵素活性など他の酵素活性に左右されず、SGT酵素活性のみを測定できる手法である。そのために、受容体基質ポリペプチドとして、「KSOOOO」を含む、例えば「VYKSOOOO」(配列番号19)からなるポリペプチドを標識化して用いる。標識としては典型的には蛍光色素(FITC)を用いるので、FITC-KSOOOOなどと表記される。
また、例えば、「FITC-KSOOOO」の場合に、蛍光が弱ければ、あらかじめ「FITC-KSOOOOV」を用いて、HGT酵素活性+SGT酵素活性を検出しておき、次いで「FITC-KSOOOO」を用いることで、確実なSGT酵素活性の測定が可能となる。
また、SGT酵素活性用キットとするときは、糖基質としてUDP-ガラクトース及びMn2+イオン供給化合物、例えばMnCl2などを用いる。
本発明のSGT酵素活性測定方法において、測定対象となる植物由来の酵素源は、細胞の破砕液、細胞から調製した膜画分の再懸濁液、膜画分を界面活性剤などで可溶化した粗酵素液、SGT酵素の精製品、SGT1遺伝子を宿主細胞あるいは無細胞発現系で発現させたリコンビナントSGT酵素など、いずれでもよい。
一般的には、2〜5mM Mn2+イオンを含有させた溶液中に、「FITC-VYKSOOOOV」などの受容体基質ポリペプチドを0.1mM程度含有させ、ガラクトース供与体としてUDP-ガラクトースを1〜5mM含有させ、pH5.5〜7.0程度に調整した溶液に対して、SGT酵素を添加し、合計液量25μlで、20〜35℃で1〜12時間程度作用させればよい。
また、RNAi法などにより、植物細胞内でのSGT酵素活性が抑制されたかどうかを評価する際にも上記方法が適用できる。
SGT遺伝子転写抑制効果を直接判定してもよく、その際の典型的な手法としてはRT-PCRによりSGT遺伝子の転写産物量を測定する手法が、定量的な評価もできるため好ましい。
【実施例】
【0031】
以下、実施例を用いて本発明を詳細に説明するが、本発明の技術的範囲は以下の実施例に限定されるものではない。なお、特に断りがない場合は、公知の方法、例えばMolecular Cloning 3rd edition Sambrook J ら、Cold Spring Harbor Laboratory Press, 2001、細胞工学別冊「バイオ実験イラストレイテッド」(秀潤社、2001年)などの記載、又は市販の試薬やキット等の指示書に従って実施可能である。また、本明細書で引用した文献中の記載は本明細書中の記載として組み入れるものとする。
【0032】
〔実施例1〕高等植物細胞抽出液の調製
シロイヌナズナ(Arabidopsis thaliana)の培養細胞はT87を用いた。植物細胞の培養は、Murashige and Skoog培地(2,4-dichlorophenoxyacetic acid and 3% sucroseを添加)を用い、200mlずつを1リットルのフラスコで、2週間、25℃で120 rpm震盪培養した。細胞をフィルターで集め、乳鉢にうつして液体窒素を加えながら、乳棒で破砕して、粉状に破砕した。破砕した細胞は、Buffer A (100 mM MOPS-NaOH (pH7.0), 1 mM MnCl2 with the Complete, EDTA-free protease inhibitor (1 tablet of Complete/50 ml, Roche, Mannheim, Germany)に懸濁し、さらに4℃で10分間 3,000 ×g.で遠心し、破砕残渣を取り除いて、上清を細胞破砕液とした。細胞破砕液をさらに4℃で10分間160,000×g.で遠心し、その沈殿をBuffer A に再懸濁した。さらに10 mM ammonium-acetate buffer (pH 7.0)に対して4℃で一晩、透析した。タンパク質の量はBCA法で測定した。これをシロイヌナズナ植物培養細胞の細胞抽出液ミクロソーム画分とした。
同様の手法で、タバコからの細胞抽出液ミクロソーム画分を得た。
【0033】
〔実施例2〕クラミドモナスの細胞壁レス変異株からの細胞抽出液の調製
全ての高等植物の祖先といわれる単細胞の緑藻の1種であるクラミドモナス(Chlamydomonas reinhardtii)は、様々な生化学的解析法、遺伝学およびゲノム解析が発展しており、多くの変異株の利用も可能であり、研究開発が容易な植物の一つである。クラミドモナスの細胞壁レス変異株(Cell wall-less mutant; CC-503, CC-2656, CC-3491)は、細胞壁がほとんどなく、簡単な操作で細胞を破砕しやすいため、CC-503は界面活性剤で破砕が可能であった。比較のためクラミドモナス正常株(CC-125, CC1690)も使用したが、正常株では界面活性剤で破砕はできず、超音波による破砕が必要であった。また、クラミドモナスは高等植物と同様のO−結合型糖鎖構造を有する植物タンパク質であるエクステンシンを生産しており、しかも、エクステンシン産生量が多いことが知られている。エクステンシンは、細胞壁を構成するタンパク質であるが、クラミドモナスの細胞壁レス変異株では、細胞壁不全の細胞壁を補強しようとしてエクステンシン産生量が多くなり、そのため、SGTを含めO−結合型糖鎖付加酵素の遺伝子を過剰発現している可能性が高いと考えられるので、まずSGT高含有粗酵素液を得るために、クラミドモナスCC-503株の細胞抽出液を得ることとした。
クラミドモナス細胞壁レス変異株は、クラミドモナス培地(0.2% Trypton、0.1% Bacto peptone、0.2% Yeast extract、0.1% Sodium acetate、0.001% CaCl2)で前培養を1リットルずつ1週間、本培養を10リットルずつ25℃で1週間培養した。培養したクラミドモナス細胞を、10分間 10,000×g.の遠心で集めた。集菌したクラミドモナスの細胞は、-80℃で保存可能である。クラミドモナス細胞を、400mlのバッファー(10mM Tris-HCl pH7.6, 0.1% Triton X-100)中で溶菌した。溶菌液を4℃で10分間 10,000×g.で遠心し、沈殿の破砕残渣を取り除いて、上清(S10)をクラミドモナスの細胞破砕液とした。
クラミドモナスの細胞破砕液(S10)画分を、超遠心(160,000×g.)60分間4℃で行い、ゴルジ体を多く含む沈殿画分P100を調製した。P100を400mlバッファー(10mM Tris-HCl pH7.6 , 0.2% Triton X-100)に懸濁し、4℃で8時間振とうして、SGTを可溶化した。これを再び超遠心(160,000×g.)60分間4℃で行い、上清を粗酵素液SL-S100 画分とした。
【0034】
〔実施例3〕クラミドモナスおよびシロイヌナズナの粗酵素液を用いたO−結合型ガラクトース活性の測定
実施例1で得られたクラミドモナスの粗酵素液SL-S100 画分およびシロイヌナズナの細胞抽出液ミクロソーム画分をSGTの粗酵素液として以下の実験に用いた。
糖転移酵素は、一般に基質特異性が厳格であることが知られており、適切な供与体基質と受容体基質を選択しないと、酵素活性を検出することができない。従来知られているO−結合型糖鎖合成に関与する糖転移酵素では、供与体基質としては、UDP、GDP、CMPなどのヌクレオチドが糖に結合した糖ヌクレオチド、及び脂質の一種であるドリコール−二リン酸に糖が結合したドリコール−二リン酸糖が知られている。小胞体に局在し、複数回膜貫通領域を持ち、糖鎖合成の早い段階で機能する糖転移酵素にドリコール−リン酸糖を供与体基質とするものが多いことが知られている。一方、小胞体からゴルジ体に局在し、N末端側に1回の膜貫通領域を持つ糖転移酵素は、糖ヌクレオチドを供与体基質とするものが多いことが知られている。
シロイヌナズナの細胞壁タンパク質AGP14のヒドロキシプロリンにO−ガラクトースを転移するHGTは、UDP-ガラクトースを供与体基質としていることが解明されていたので、SGTも同様の基質特異性をもつ可能性が高いと考えられたため、SGT活性測定の供与体基質として、UDP-ガラクトースを用いた。
タバコからの細胞抽出液ミクロソーム画分に対しても、シロイヌナズナの場合と同様にSGT活性を測定し、O−ガラクトース付加活性の存在を確認した(図示せず)。
受容体基質として、他のO−結合型糖鎖を合成する糖転移酵素の活性測定法と同様に、合成ペプチドを用いた。ペプチドは、HPLC等による活性測定で検出を容易にするために、FITCで蛍光標識をした。合成ペプチドのアミノ酸配列は、当初、セリン残基を含み、かつ植物においてセリン残基にO−ガラクトース付加の報告があるものとして、O−ガラクトース付加をタバコ細胞で確認していたスポラミン配列「FITC-PTTHEPASSETPVL」および植物細胞壁タンパク質の解析でセリン残基にガラクトース付加が知られていたエクステンシン配列「VYKSPPPPV」、哺乳動物由来のppGalNAcTの酵素活性測定で実績のある動物細胞のムチンコアタンパク質配列、等を受容体基質として用いてSGT酵素活性の検出を試みたが、ガラクトース転移酵素活性は確認できなかった。プロリンを水酸化したスポラミン配列「FITC-PTTHEOASSETPVL」を受容体基質としても酵素活性は検出できなかったが、プロリンを水酸化したエクステンシン配列「VYKSOOOOV」を受容体基質としたところ、HPLCで酵素反応物のピークを検出でき、ガラクトース転移酵素活性を確認できた。したがって、「FITC-VYKSOOOOV」を受容体基質とすることによるガラクトース転移酵素活性測定が可能となった。ただし、この段階では、ガラクトースがセリン残基あるいはヒドロキシプロリン残基に付加したのかは不明であった。連続するヒドロキシプロリン残基には、O−アラビノースの付加の報告があるだけで、O−ガラクトースの付加はこれまで報告がなかった。しかし、FITC-VYKSOOOOVには、予想に反して2残基のガラクトース付加が認められ、詳細に解析したところ、セリン残基へのガラクトース付加だけでなく、ヒドロキシプロリン残基へのガラクトース付加の存在が明らかとなった。HPLC解析でSGT活性とHGT活性のピークを区別して検出することができたため、SGT酵素活性測定に初めて成功した。
【0035】
SGT酵素活性測定の詳細な条件として、酵素反応を行う溶液の構成は、100mM MOPS緩衝液(pH7.0)、4mM MnCl2、10mM UDP-ガラクトース、0.1mM FITC標識ペプチド、SGT粗酵素液5μlを加え、合計25μlとした。あるいは100mM MOPS緩衝液(pH7.0)、5mM MnCl2、5mM UDP−ガラクトース、25μM FITC標識ペプチド、2mM EDTA、5×Protease Inhibitor Cocktail、0.1% TritonX-100、酵素液を加えて6.25μl、あるいは0.2M MOPS pH7.0、0.1mM FITC-Peptide(VYKSOOOOV-Ac1)、0.2% TritonX-100、10mM UDP-Galactose、4mM MnCl2、2mM EDTA、5×Inhibitor cocktailに酵素液を加えて12.5μl とした。これらを30℃で1〜12時間保温して酵素反応を行った。酵素反応は、95℃3分の熱処理を行って停止した。
酵素反応産物はHPLC(島津製作所RF-10A XL)で蛍光検出して解析した。逆相カラム5C18-AR-II(250×4.6 mm; Nacalai Tesque, Kyoto, Japan)を用い、カラムを0.1%トリフルオロ酢酸を含む22.0%アセトニトリルで平衡後、22.0%から22.5%の濃度勾配を流速1ml/minで30分かけて溶出し、FITCを488nmで励起し、530nmで検出した。受容体基質のペプチドは、この条件で、約19.0分に溶出し、セリン残基にガラクトースが1残基付加した酵素反応産物であるプロダクト#1は約15.5分に溶出された。ヒドロキシプロリン残基に1残基のガラクトースが付加したプロダクト#2は約16.5分に、セリンとヒドロキシプロリンに1残基ずつ合計2残基のガラクトース付加したプロダクト#3は約14.5分に溶出した。(図1)
【0036】
〔実施例4〕SGT酵素反応産物セリンOガラクトースの構造解析
SGT酵素反応産物をそれぞれ分取してMS解析を行った結果、#1および#2は、ヘキソースが1残基深し、#3はヘキソースが2残基付加していることが確認された。高等植物のセリン残基へのガラクトース付加は、α結合であり、ヒドロキシプロリン残基へのガラクトース付加はβ結合であることが分かっているので、アスペルジルス由来βガラクトシダーゼ処理をしたところ、#1は変化せず、#2と#3は消失した(図2)。ピークを分取して、それぞれβガラクトシダーゼを処理すると、プロダクト#2はペプチド基質に、プロダクト#3はプロダクト#1の位置にピークがシフトした。このことから、ヒドロキシプロリン残基に付加していたガラクトースが消化されたことが示された。
βエリミネーションは、O−結合型糖鎖を切り離すのに一般的に使われる手法であるが、環状アミノ酸であるヒドロキシプロリンにO−ガラクトースが付加したものは、糖を離脱しないことが知られている(Wells et al 2002)。#2をβエリミネーションしても、切れなかった(図3CD)。#3をβエリミネーションすると、切れたものと、DTT付加したものが検出された。#1をβエリミネーションすると、切れて、DTT付加するものが検出された(図3A、B)。エクステンシンのセリンをアラニンに換えたペプチドでは、ピークが一つだけ検出され、βガラクトシダーゼで切れた。
これらの結果を総合すると、プロダクト#1は、SGTによりセリン残基にO−ガラクトースが付加したものであり、プロダクト#2は、ヒドロキシプロリン−O−ガラクトース転移酵素(HGT)によりヒドロキシプロリン残基にO−ガラクトースが付加したものであり、プロダクト#3は、SGTとHGTによりO−ガラクトースが2残基付加したものであることが分かった。
このことから、この細胞破砕液には、SGTに加え、HGTも含まれていることがわかり、SGT及びHGT酵素活性を区別して測定し、セリン残基にO−ガラクトース付加およびヒドロキシプロリンにO−ガラクトース付加したペプチドをそれぞれ分離して検出する方法が確立できた。
【0037】
〔実施例5〕SGTの基質特異性
(5−1)クラミドモナスSGT粗酵素液を用いた基質特異性
供与体基質は、UDP-ガラクトースを用いた。受容体基質は、FITCで蛍光標識をした合成ペプチドのアミノ酸配列を以下に記述するように様々なものを合成して用いた。ペプチドのアミノ酸配列は、クラミドモナスおよびシロイヌナズナでSGT活性を確認できている、エクステンシンの配列、すなわちFITC-VYKSOOOOV(アミノ酸の一文字表記。Oはヒドロキシプロリン)の他に、さらに、ペプチドのアミノ酸配列特異性を検討するために、ヒドロキシル化されていないFITC-VYKSPPPPV、セリン残基をアラニン残基に変えたFITC-VYKAOOOOV、C末端のバリン残基を削除したFITC-KSOOOO、ヒドロキシル化していない3回繰り返し配列を含むFITC-KSPPPPVYKSPPPPVKHYSPPPPVYK、ヒドロキシル化した3回繰り返し配列を含むFITC-SOOOOSOOOOSOOOOSOOOOSP、ヒドロキシル化していないスポラミン配列FITC-PTTHEPASSETPVLおよびヒドロキシル化したスポラミン配列FITC-PTTHEOASSETPVL、AGP14配列FITC-VDAOAOSOTS、FITC-VDAOAOAOAA、セリン繰り返し配列FITC-PSSSSSOSSSSSPなどを用いた。
これらの合成ペプチドを受容体基質としてSGT酵素活性を測定した結果、特にFITC-VYKSOOOOVにおいて強いガラクトース転移酵素活性が測定された。一方で、FITC-VYKSPPPPV、受容体基質とした場合には、ごく弱いSGT酵素活性が検出された。そしてFITC-VYKAOOOOVを受容体基質とした場合には、SGT酵素活性は全く検出されなかった。特筆すべきことは、FITC-KSOOOOを受容体基質とした場合には、強いSGT酵素活性が検出されたが、酵素反応産物のピークは一つだけであり、それはセリン残基にガラクトースが付加したものであったことから、SGT活性だけが検出され、HGT活性は検出されなかったことが分かった。これらの結果から、SGT酵素により、FITC-VYKSOOOOVのSにガラクトースが転移されることが証明され、近隣のアミノ酸がPではなくOであることがSGT活性に重要であることが示唆された。また、HGT酵素によるOへのガラクトースの転移には、FITC-VYKSOOOOVの最後のVが重要であることが示唆された。このことは、FITC-KSOOOOを受容体基質と用いることで、HGT活性の影響を受けずに、SGT活性だけを測定することができることである。すなわち、標識化されたKSOOOOを含む、例えばFITC-VYKSOOOO(配列番号19)を受容体基質として用いるSGT酵素活性測定系を確立できた。
【0038】
(5−2)シロイヌナズナT87細胞抽出液を用いた場合
FITC-VYKSOOOOVを受容体基質とした時、シロイヌナズナT87のSGT粗酵素液において、ガラクトース転移酵素活性を検出したが、FITC-KSOOOOおよびFITC-VYKSPPPPV、あるいはAGP14配列、MUC1AC配列を受容体基質とした場合では、SGT酵素活性は認められなかった。シロイヌナズナの粗酵素液では、SGTの活性が濃縮できておらず、弱い活性が検出できなかったためであると考えられるが、基本的に、クラミドモナスのSGTと同様の基質特異性を示していた。
【0039】
〔実施例6〕SGT遺伝子の発現と酵素活性の確認
SGTの2価金属イオン要求性については、金属イオンをキレートするEDTAを2mMを反応液中に添加するとSGT酵素活性は全く消失したことから、金属イオンを必要とすることが分かった。どの金属イオンを要求するかについて2mMのEDTAに加え、4mMの各種金属イオンを添加して検討したところ、他の糖転移酵素と同様に、Mnを要求し、Fe、Co、Znでは弱い要求性が見られたが、Mg、Caでは全く活性化が認められなかった。至適pHはpH6.0、至適温度は30℃で、50℃では活性は失われた(図4)。
SGTは、陽イオン交換カラムではpH4.5、陰イオン交換カラムではpH8.5で吸着した。HGTは、陽イオン交換カラムではpH5.5で吸着したが、陰イオン交換カラムではpH9でも吸着しなかった。HGTは、高い等電点をもつことが示唆された。SGTは、中性付近の等電点をもつことが示唆された。
【0040】
〔実施例7〕SGTタンパク質の精製法
クラミドモナスの粗酵素液SL-S100を、Ni Chlating Sepharoseカラム(φ5×5cm、結合バッファー;25mM Tris-HCl, 10% Glycerol, 0.2% Triton X-100, 0.5M NaCl, pH7.6、溶出バッファー;25mM Tris-HCl, 10% Glycerol, 0.2% Triton X-100, 0.5M NaCl, 0.05M Glycine, ph7.6)、DEAE Sepharoseカラムに2回(1回目:φ5×11cm、結合バッファー;20mM Tris-HCl, 10% Glycerol, 0.1% Triton X-100, ph8.5、溶出バッファー;20mM Tris-HCl, 10% Glycerol, 0.1% Triton X-100, 0〜0.2M NaCl, 0.05M Glycine, pH7.6、2回目:結合バッファー;25mM Methyl-piperadine, 10% Glycerol, 0.1% Triton X-100, ph8.5、溶出バッファー;25mM Methyl-piperadine, 10% Glycerol, 0.1% Triton X-100, 0〜0.2M NaCl, pH8.5)、Sephadex G-200カラム(φ2.5×5cm、溶出バッファー;25mM MOPS, 10% Glycerol, 0.5% CHAPS, 0.5M NaCl, pH7.0)に2回かけ、Native-PAGE(6%ポリアクリルアミドゲル、泳動バッファー;25mM Tris, 192mM Glycine)にかけた。CBB染色を行い、タンパク質のバンドを検出し、ゲルからバンドを切り出して、酵素活性を測定したところ、ゲル断片No.4とNo.5に高い酵素活性が確認された。そこでNo.4とNo.5からタンパク質を抽出して、さらにIEF-PAGE(pH3〜10キャリアアンフォライト使用ポリアクリルアミドゲル、泳動バッファー;陽極7mM Phosphate, 陰極20mM Argine, 20mM Lysine)にかけた(表1)。CBB染色を行い、再びゲルからバンドを切り出して、酵素活性を測定したところ、ゲル断片No.8、9およびNo.9、10に強い酵素活性が認められた(図5)。それぞれのゲル断片をSDS-PAGEにかけたところ、IEF-PAGEの溶出パターンと挙動をともにするバンドとして、四角で示したバンドが見出された(図6A)。
【0041】
以下に、表1として、クラミドモナス細胞破砕液の精製ステップごとのSGT酵素の比活性を示す。
【0042】
【表1】
【0043】
〔実施例8〕SGT遺伝子ファミリーの同定
SDS-PAGEゲルから切り出したバンドを、株式会社セラバリューズに依頼分析し、LC-MS/MS解析後、MASCOT検索をしたところ、7つのペプチド断片のアミノ酸配列が明らかとなり、それらはすべて、クラミドモナスのデータベースの遺伝子配列CHLREDRAFT_150807 (A8J5Z4_CHLRE) に100%マッチしていることが見出された。A8J5Z4_CHLRE は717アミノ酸をコードする遺伝子であった。(図6、7)
【0044】
【表2】
【0045】
〔実施例9〕SGT遺伝子ファミリーの配列の類似性
(9−1)種間類似性
クラミドモナスのA8J5Z4_CHLRE配列をクエリーにして、Blast検索をしたところ、シロイヌナズナ、イネ、トウゴマ、ブドウ、ポプラ、ニセツリガネゴケ、ブラシノ藻に、約30%identityのホモログORFがそれぞれ1ORFずつ見つかり、植物に広く保存された遺伝子ファミリーであることが分かった(図7)。広葉植物同士では、70%程度のアミノ酸の同一性があり、被子植物間では60%程度、被子植物と単細胞緑藻の間では30%のアミノ酸配列の同一性が認められた。しかし、動物や真菌にはホモロジーのある遺伝子は見つからず、植物界に特異的に保存されている遺伝子ファミリーであることが分かった(表2)。しかも、SGTの配列は、他のどの糖転移酵素とも類似性を持たず、全く新規な糖転移酵素遺伝子ファミリーであった。
SGTは、糖転移酵素としては珍しく、I型膜タンパク質のトポロジーであった(図8)。クラミドモナスのSGT1のN末端側のホモロジーの高い領域だけでも活性があった。SGTの活性ドメインであると考えられるニセツリガネゴケからシロイヌナズナなどの高等植物のSGT1には、ホモロジーの高い領域がタンデムに重複して存在していた。N末端側とC末端側のホモロジーの高い領域同士では、53%程度のアミノ酸配列の同一性であった。シロイヌナズナのSGTのC末端側のホモロジーの高い領域だけでもSGT活性はあった。すべてのSGT1で糖ヌクレオチドを基質とする糖転移酵素に広く保存されているDXDモチーフが保存されていた。クラミドモナスおよびブラシノ藻では、複数のホモロジーのあるORFが検索されるが、ニセツリガネゴケからシロイヌナズナなどの高等植物のSGT1には、ゲノム中に一つずつしかホモログはなかった。クラミドモナスのSGT1には、高等植物のプロリン水酸化酵素のER-ゴルジ局在配列Tox1が存在し、ER-ゴルジの局在が示唆された。
【0046】
各植物のSGT1の活性ドメイン(CD)のIdentity(アミノ酸配列間ホモロジー)を下記表3に示す。
【0047】
【表3】
【0048】
なお、表中のCrはクラミドモナス、Otはブラシノ藻、Ppはニセツリガネゴケ、Smはイヌタカヒバ、Osはイネ、Atはシロイヌナズナ、Ntはタバコ、Ptはポプラ由来SGTであることを表す。
【0049】
(9−2)植物ゲノム中のSGT遺伝子の数
各生物のSGT遺伝子の配列情報は、データベース上はいくつかの異なったデータが登録されてあったが、DNA配列等を比較し、遺伝子座を調べると、オールタナティブスプライシング、遺伝子の欠失挿入変異、シークエンス解析のミス、品種による遺伝子の多型などが原因であり、各ゲノムあたり1種類のSGT遺伝子が1コピーしかなく、SGT酵素活性をコードする遺伝子はSGT1以外にはないことがわかった。
【0050】
〔実施例10〕緑藻と高等植物のSGT遺伝子とSGT酵素活性
クラミドモナスは、すべての緑色植物の祖先であるとされている単細胞の緑藻の一種であり、一方、シロイヌナズナは、双子葉植物であり、陸上の高等植物の一種である。この2種の植物において、SGT遺伝子はアミノ酸配列にして30%の同一性を示すほど高い類似性を持っており、双方とも、同じ基質特異性で、同じ反応産物を生ずる酵素活性を持っていることが確認されたため、相同性遺伝子であることが証明された。Blast等による相同性検索によって、シロイヌナズナと同じ双子葉植物であるトウゴマ、ブドウ、ポプラでも70%のアミノ酸配列同一性を示すSGT遺伝子があり、単子葉植物であるイネでも60%のアミノ酸配列同一性を示すSGT遺伝子が見つかった。さらに、コケ類であるニセツリガネゴケでも50%を超えるアミノ酸配列同一性を示すSGT遺伝子が見つかった。これらのことから、この一群のSGT遺伝子ファミリーはすべて同様のSGT酵素活性を保持しているものと考えられる。逆に言えば、クラミドモナスおよびシロイヌナズナのSGT遺伝子配列に30%以上の類似性を示す遺伝子が、各種植物種のDNA配列から発見されば場合は、すべてSGT遺伝子であると考えられ、SGT酵素活性も保持しているものと考えられる。
【0051】
【表4】
【0052】
〔実施例11〕酵母での発現
クラミドモナスのSGT1遺伝子は、活性ドメインを一つだけ持ち、N末端に分泌経路に入るためのシグナルシークエンスがあり、C末端側に膜に局在するための膜貫通領域がある。ChSGT1のシグナルシークエンスおよび膜貫通領域以外の部分を、HAタグを付けた酵母PIR細胞壁タンパク質に結合し、酵母細胞壁に発現させて(Abe et al, 2004, Shimma et al, 2006)、SGT酵素活性を測定したところ、HPLCで同じ溶出ピークが得られた。このことから、ChSGT1遺伝子がSGT酵素をコードしていることが証明された(図9)。
アラビドプシスのSGT1遺伝子は、他の陸上植物のSGTと同様に、活性ドメインが2つあり、N末端に分泌経路に入るためのシグナルシークエンスがあり、C末端側に膜に局在するための膜貫通領域がある(図8)。同様に、AtSGT1のシグナルシークエンスおよび膜貫通領域以外の部分を、HAタグを付けた酵母PIR細胞壁タンパク質に結合し、酵母細胞壁に発現させてSGT酵素活性を測定したところ、HPLCで同じ溶出ピークが得られた。このことから、AtSGT1遺伝子もSGT酵素をコードしていることが証明された(図10)。
【0053】
〔実施例12〕SGTの酵素活性の必要領域
C末端側の活性ドメインだけをもつ融合タンパク質を同様に酵母で発現させてSGT酵素活性を測定したところ、HPLCで同じ溶出ピークが得られた(図10)。このことから、クラミドモナスでも高等植物でも、活性ドメインが一つでもあれば、SGT酵素活性があることが分かった。
【0054】
〔実施例13〕SGTの抑制法
現在、ゲノムDNA配列情報が解読され、公開されている生物種については、SGT1遺伝子配列情報をホモロジー検索により容易に取得することができるが、今後明らかにされる生物種においても、同様の方法によりその生物種のSGT遺伝子の取得が可能である。それらのSGT遺伝子情報に基づいて、SGTを抑制することができる。1)SGT遺伝子DNA配列情報を元にして、相同性組み換え法による遺伝子破壊法(ノックアウト)、相補的核酸配列を発現させるアンチRNA法、RNAi法、トランスポゾン挿入法、T-DNA変異株作製法など、既知の方法でSGT遺伝子の破壊あるいは機能抑制を行うことができる。一例として、シロイヌナズナのT-DNA変異株が多数作製されており、そのうちAtSGT1に関するものは、SALK_054682、SALK_059879C、WiscDsLox451B08であり、これらは、AtSGT遺伝子にT-DNAの挿入、あるいはトランスポゾンの挿入によりORFが破壊されており、SGT活性が損なわれている。これらAtSGT1遺伝子(At3g01720)の変異を持つシロイヌナズナの種子をThe Arabidopsis Information Resource (TAIR) (http://www.arabidopsis.org/)から購入し、発芽させ、植物体を成育させ、あるいは、植物体よりカルスを誘導して、細胞培養を行った。FLAGタグを融合したエクステンシン繰り返し配列(VYKSOOOOV)を含むペプチドをコードする遺伝子を、植物体あるいはカルスに形質転換し、FLAGタグを利用して、エクステンシン配列を抗体で精製して、セリン残基へのO−ガラクトース付加が抑制されていることを検証している。
前述したように、コケ以上の陸上植物では、SGT遺伝子はゲノムあたり1遺伝子しかないことが分かっており、上記の方法で同定したSGT遺伝子を破壊あるいは抑制することにより、ペプチドのセリン残基へのO−ガラクトースの付加を抑制することができる。
【0055】
〔実施例14〕SGT1変異株における酵素活性の減少
シロイヌナズナにおいては、T-DNA挿入変異株のシリーズが作成されており、各ORFの変異株を購入することができる。SGT1(At3g01720)遺伝子に対応する位置のORFにT-DNAが挿入された変異株(Δsgtl1〜3)をオハイオ大学のアラビドプシスセンターから入手した。正常株(コロンビア)及び上記SGT1変異植物株の種子を発芽させ、茎を寒天培地上で培養してカルスを誘導し、細胞培養した。
約1gの正常株及び変異株のカルス細胞を集めて、乳鉢で破砕し、膜画分を調製した。実施例11と同様のHPLC解析によりSGT酵素活性を測定した結果、正常株ではSGT酵素活性が確認できたが、SGT1変異株では検出されなかった。(図11)
このことから、SGT1遺伝子のT-DNA挿入変異株では、SGT酵素活性が消失していることが示された。
【0056】
〔実施例15〕SGT1変異株における糖鎖付加の減少
正常株及び変異株のカルス細胞を破砕し、細胞壁画分を調製した。具体的には、カルス細胞1gを乳鉢で破砕し、1,000×gで遠心した沈殿を、クエン酸緩衝液(20mM,pH7.0)に懸濁してオートクレーブに2時間かけ、1,000×gで遠心した沈殿を細胞壁画分とした。SGT酵素によりペプチドにα結合で付加されたガラクトースはαガラクトシダーゼで遊離することが分かっており、一方で、ガラクタンおよびアラビノガラクタンに含まれるガラクトースはβ結合で、ガラクトシダーゼにより分離されるから、細胞壁画分0.1ODを、100μlの20mM酢酸アンモニウム(pH4.5)、αガラクトシダーゼ(Guar,5U,40℃)あるいはβガラクトシダーゼ(Oryzae,1U,30℃)処理を行って、ガラクトースを遊離させた。反応液をエタノール沈殿し、上清を乾燥させた。遊離した単糖をABEE標識後、HPLCで解析した。
その結果、正常株の細胞壁から遊離されるαガラクトースの量を100としたときに、変異株から遊離されるαガラクトースの量が約60に有意に減少していた。一方、βガラクトシダーゼにより遊離されたガラクトースの量は、正常株と変異株で差はなかった。
このことから、SGT1遺伝子のT-DNA挿入変異株では、糖タンパク質あるいはペプチドのセリン残基へのO−結合型ガラクトース付加が減少していることが示された。(図12)
【0057】
〔実施例16〕タバコからのSGT1遺伝子クローニング
(16−1)タバコのESTデータベースの検索
ゲノム配列が非公開の実用植物であるタバコから、PCRによりSGT1遺伝子に対応するcDNAをクローニングした。シロイヌナズナのSGT1配列(AtSGT1)をクエリーにして、公共データベース(http://blast.ncbi.nlm.nih.gov/Blast.cgi)にあるタバコのESTを検索したところ、FG201805とFG200221、FG146884という3つのEST配列を見いだした。遺伝情報処理ソフトウェア GENETYX-MAC Ver12((株)ゼネティックス)を用いて、これらの3つのEST配列を繋げて、シグナル配列、膜貫通領域、塩基性アミノ酸に富む領域を欠く2074bpからなるタバコSGT1遺伝子(NtSGT1)の部分配列を見出した。この配列をもとに遺伝子特異的プライマーNtSGTFw-1 (5’-TTGCACTGATGAAGAGAGGAA-3’;配列番号24)及びNtSGTRv-2 (5’- ATCATTGGCTCTAGTTTCAG-3’;配列番号25)をそれぞれフォーワードプライマー、リバースプライマーとして設計して合成した。
【0058】
(16−2)タバコSGT1遺伝子(NtSGT1)の全長クローニング
培養4日目のタバコ培養細胞BY-2からRNeasy Plant Mini Kit ((株)キアゲン)を用いてTotal RNAを抽出した。さらにGenElute mRNA Miniprep Kit (SIGMA)を用いてTotal RNAからmRNAを精製した。
得られたmRNA 500ngを鋳型として、TaKaRa RNA PCR Kit (AMV) ver. 3.0(タカラバイオ(株))を用いてRT-PCRを行った。逆転写はTaKaRa RNA PCR Kit (AMV) ver. 3.0に付属のOligo dT-Adaptor Primer 1.25 pmolを用いて42℃、1時間、次いで95℃、5分反応した。PCRはNtSGTFw-1とNtSGTRv-2を各10 pmol 用いて、94℃、30秒、53℃、30秒、72℃、2分10秒を1サイクルとする、計30サイクルを行った。このPCR産物についてアガロース電気泳動を行ったところ、予測される増幅DNAに相当する約2-kbのPCR産物が確認できたため、この部分を切り出してDNAを精製し、プラスミドpUC19に導入し、大腸菌DH5αを形質転換した。得られたクローンを鋳型とし、DNA配列(NtSGT1部分配列)を決定した。
【0059】
次に3’末端配列をクローニングするため、TaKaRa RNA PCR Kit (AMV) ver. 3.0 (タカラバイオ(株))を用いた3’ RACEを行った。上記で決定したNtSGT1部分配列をもとに遺伝子特異的プライマーNtSGTFw-19 (5’-TCCTCCTGATCCATCGTCACTTG-3’;配列番号26)を3’RACE用アンチセンスプライマーとして設計して合成した。逆転写はTaKaRa RNA PCR Kit (AMV) ver. 3.0に付属のOligo dT-Adaptor Primer 1.25 pmolを用いて42℃、1時間、次いで95℃、5分反応した。PCRはNtSGTFw-19とNtSGTRv-2を各10 pmol 用いて、94℃、30秒、50℃、30秒、72℃、2分を1サイクルとする、計30サイクルを行った。このPCR産物についてアガロース電気泳動を行ったところ、約1.2-kbのPCR産物が確認できたため、この部分を切り出してDNAを精製し、プラスミドpUC19に導入し、大腸菌DH5αを形質転換した。得られたクローンを鋳型とし、DNA塩基配列を決定した。
【0060】
次いで、5’末端配列をクローニングするため、5'-Full RACE Core Set (タカラバイオ(株))を用いた5’ RACEを行った。
上記で決定したNtSGT1部分配列をもとに5’末端リン酸化逆転写反応用プライマーNtSGT1-5phos (5’- [5’リン酸化]AACACCCTCTCGTGG-3’:配列番号39)、1st PCR用センスプライマーNtSGT1-S1 (5’-ATAAACAAACCAGCTGGAGT-3’:配列番号40)、1st PCR用アンチセンスプライマーNtSGT1-A1 (5’-CCAGTTTTGGGATGTCTGCT-3’:配列番号41)、2nd PCR用センスプライマーNtSGT1-S2 (5’-ACAGCAAAGAGGCACAAAAT-3’:配列番号42)、2nd PCR用アンチセンスプライマーNtSGT1-A2 (5’-AAAGTGGGAGCCAACTCCAT-3’:配列番号43)を設計し、合成した。上記で単離したタバコ培養細胞BY-2のmRNA 1μgを鋳型とし、NtSGT1-5phos 500 pmolを用いて30℃、10分、42℃、1時間、次いで80℃、2分反応した。得られたRNA/DNAハイブリッド中のRNAを5'-Full RACE Core Set 付属のRNaseHで分解し、得られたシングルストランドcDNAをエタノール沈殿後、付属のT4 RNA ligase でライゲーションした。このライゲーション液1μlを鋳型としてNtSGT1-S1 とNtSGT1-A1を各15 pmol用いてPrimeSTAR GXL DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、50℃、15秒、68℃、2分30秒を1サイクルとする、計30サイクルを行った。次にこのPCR反応液1μlを鋳型としてNtSGT1-S2 とNtSGT1-A2を各15 pmol用いてPrimeSTAR GXL DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、50℃、15秒、68℃、2分30秒を1サイクルとする、計30サイクルを行った。このPCR産物についてアガロース電気泳動を行ったところ、約700-bpのPCR産物が確認できたため、この部分を切り出してDNAを精製し、プラスミドpUC19に導入し、大腸菌DH5αを形質転換した。得られたクローンを鋳型とし、DNA配列を決定した。
【0061】
3’RACEと5’RACEにより得られた塩基配列をもとに、NtSGT1の全長を増幅するようにNtSGTFw-5term (5’-GGACACACAAACCTTTGACT-3’:配列番号44)とNtSGTRv-3term (TGGAGCTCTTTTTTTTTTTTTTTTTTTTTTTTVN-3’:配列番号45)を設計し、合成した。mRNA 500ngを鋳型として、TaKaRa RNA PCR Kit (AMV) ver. 3.0(タカラバイオ(株))を用いてRT-PCRを行った。逆転写はTaKaRa RNA PCR Kit (AMV) ver. 3.0に付属のOligo dT-Adaptor Primer 1.25 pmolを用いて42℃、1時間、次いで95℃、5分反応した。PCRはNtSGTFw-5termとNtSGTRv-3termを各10 pmol 用いてPrimeSTAR GXL DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、52℃、15秒、68℃、3分30秒を1サイクルとする、計30サイクルを行った。このPCR産物についてアガロース電気泳動を行ったところ、予測される増幅DNAに相当する約3.3-kbのPCR産物が確認できたため、この部分を切り出してDNAを精製し、プラスミドpUC19に導入し、大腸菌DH5αを形質転換した。得られたクローンを鋳型とし、DNA配列を決定した。
得られた完全長クローン(配列番号20)は3,336bpであり、899aa(配列番号21)をコードし、シロイヌナズナSGT1と80%以上のアミノ酸配列同一性を示していた。
タバコのSGT活性ドメイン1及び2のアミノ酸配列は、それぞれ配列番号22及び23で表すことができる。ドメイン1は、34aa〜337aa、ドメイン2は、398aa〜697aaである。
【0062】
〔実施例17〕タバコSGT1遺伝子を用いた形質転換酵母によるSGT酵素活性の発現
上記〔実施例11〕と同様に、タバコSGT1のシグナル配列および膜貫通領域を除いた主要部分(配列番号21に示されたNtSGT1の400〜720番目の領域)を、HAタグを付けた酵母PIR細胞壁タンパク質に結合して酵母細胞壁に発現させ、SGT酵素活性を測定したところ、HPLCでプロダクト#1と同じ溶出ピークが得られた。すなわち、タバコSGT1についても、FITC-VYKSOOOO基質のセリンに対してガラクトースを転位させることができることを確認した(図11)。
【0063】
〔実施例18〕RNAiを用いたタバコSGT1遺伝子発現の抑制
〔18−1〕dsRNAの設計及び発現プラスミドの構築
ヘアピン型二本鎖RNA(dsRNA)発現プラスミドを用いたRNAiにより、NtSGT1の制御を行うために、まずNtSGT1をターゲットとして549-bpのdsRNAの発現プラスミドを次の手順で構築した。
(1)ターゲット領域(センス配列およびアンチセンス配列)とリンカー配列のPCR増幅
ターゲット領域としてNtSGT1の549-bp断片(296番目のTから844番目のTまで)を選択した。PCRによりターゲット領域のセンス配列とアンチセンス配列を増幅した。
センス配列の増幅はプライマーSGTsenseFw (5’-AGATTGTCGACTTGCACTGATGAAGAGAGGAAG-3’ :配列番号46、下線部はSalIサイト)およびSGTsenseRv (5’-GAATAGCATGC AGATCTATCATCAAGTTATCATTAATC-3’:配列番号47、1つ目の下線部はSphIサイト、2つ目の下線部はBglIIサイト)を用いた。これらのプライマーを各々10 pmol用いてPrimeSTAR HS DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、53℃、5秒、72℃、40秒を1サイクルとする計5サイクルを行い、続いて98℃、10秒、59℃、5秒、72℃、40秒を1サイクルとする計25サイクルを行った。
アンチセンス配列の増幅はプライマーSGTantiFw (5’-AGATTGGTACCTTGCACT
GATGAAGAGAGGAAG-3’:配列番号48、下線部はKpnIサイト)およびSGTantiRv (5’-GAATAGAATTC AAGCTTATCATCAAGTTATCATTAATC:配列番号49、1つ目の下線部はEcoRIサイト、2つ目の下線部はHindIIIサイト)を用いた。これらのプライマーを各々10 pmol用いてPrimeSTAR HS DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、51℃、5秒、72℃、40秒を1サイクルとする計5サイクルを行い、続いて98℃、10秒、57℃、5秒、72℃、40秒を1サイクルとする計25サイクルを行った。
また、pIG221(Ohta et al., Plant Cell Physiol. 31: 805-831, 1990)からcastor bean catalase 遺伝子(CAT-1)の第一イントロン遺伝子201-bpをRNAiコンストラクトのリンカー配列として増幅した。プライマーIntron1Fw (5’- GGCGGGGATCCCTACAGG
GTAAATTTCT-3’:配列番号50、下線部はBamHIサイト)およびIntron1Rv (5’- GGCGGGGTACCGGTTCTGTAAC-3’:配列番号51、下線部はKpnIサイト) を用いた。これらのプライマーを各々10 pmol用いてPrimeSTAR HS DNA polymerase(タカラバイオ(株))によりPCRを行った。 PCR反応条件は98℃、10秒、51℃、5秒、72℃、40秒を1サイクルとする計5サイクルを行い、続いて98℃、10秒、56℃、5秒、72℃、30秒を1サイクルとする計25サイクルを行った。
【0064】
(2)dsRNA発現ベクターの構築
リンカー配列を制限酵素(BamHIおよびKpnI)で切断した後、プラスミドpUC19上のBamHIおよびKpnIサイトに導入し、プラスミドpUC19-Intを得た。次にターゲット領域のアンチセンス配列を制限酵素(SphIおよびSalI)で切断した後、プラスミドpUC19-Int上のSphIおよびSalIサイトに導入し、プラスミドpUC19-SGTanti-Intを得た。次にターゲット領域のセンス配列を制限酵素(KpnIおよびEcoRI)で切断した後、プラスミドpUC19-SGTanti-Int上のKpnIおよびEcoRIに導入し、プラスミドpUC19-SGT RNAi296を得た。
植物導入用の遺伝子発現ベクターには様々な種類があり、それらを適宜用いることができるが、本実施例では pMAT037 (Matsuoka and Nakamura, Proc. Natl. Acad. Sci. USA. 88:834-838, 1991)を用いた。pUC19-SGT RNAi296を制限酵素(BglIIおよびHindIII)で切断し、アンチセンス配列とリンカー配列およびセンス配列を含む約1.2-kbの断片を切り出して、pMAT037のCaMV35Sプロモーター後のマルチクローニングサイト中のBglIIおよびHindIIIサイトに導入し、プラスミドpMAT- SGT RNAi296を得た。
【0065】
〔18−2〕タバコ培養細胞BY-2へのアグロバクテリウム属細菌による遺伝子導入
タバコ培養細胞BY-2への遺伝子導入に先立ち、プラスミドpMAT- SGT RNAi296をエレクトロポーレーション法によりアグロバクテリウム細菌EHA101に組み込んだ。プラスミドを組み込んだアグロバクテリウム細菌を抗生物質であるカナマイシン(12.5 mg/l)およびテトラサイクリン(5 mg/l)を含む培地で増殖させ、600 nmにおける光学濃度(O.D.)値約1のアグロバクテリウム懸濁液を調製した。この懸濁液と培養3日目のBY-2細胞をシャーレに加え、シャーレを軽く旋回することで混合すると同時に培養液をシャーレ全面に広げる。野菜把捉テープでシャーレをシールし26℃、暗所で72時間静置培養した。ピペットを用いてシャーレ上のBY-2を15 mlの滅菌済みチューブに移し、5 mlのBY-2細胞用液体培地を加え、10回程度ピペッティングを行った。これを800 rpm、1分遠心しBY-2細胞を沈殿させる。アグロバクテリウムで濁った培地を除去し、新しい培地を5 ml加えBY-2細胞と上記の方法で混合し遠心によりBY-2細胞を回収した。この操作をさらに4回繰り返した後、BY-2の沈殿を得た。BY-2細胞の体積に対して4倍量または9倍量の新しい培地を加え、よく混合し、各々1 mlを形質転換用の固形培地に広げた。シャーレを傾けて液体培地を一方の隅に集めピペットで吸い取った後、野菜把捉テープでシャーレをシールし28℃、暗所で2週間静置培養し、培地上に出現した形質転換体を得た。
【0066】
〔18−3〕RNAi によるSGT転写抑制効果(RT-PCRによる転写産物量の測定)
RNAiコンストラクトを導入したタバコ培養細胞BY-2と、コントロールとして形質転換を行っていないタバコ培養細胞BY-2を8日間培養した後、培養液を2号濾紙(2枚重ねる)で濾過してBY-2細胞を回収した。このBY-2細胞100 mgを液体窒素で凍結させながら乳鉢で粉砕した。この粉砕したBY-2細胞からRNeasy Plant Mini Kit ((株)キアゲン)を用いてTotal RNAを抽出した。また抽出操作中に ((株)キアゲン)を用いてDNase処理を行った。得られた Total RNA 500ngを鋳型として、TaKaRa RNA PCR Kit (AMV) ver. 3.0(タカラバイオ(株))を用いてRT-PCRを行った。逆転写はTaKaRa RNA PCR Kit (AMV) ver. 3.0に付属のOligo dT-Adaptor Primer 1.25 pmolを用いて42℃、1時間、次いで95℃、5分反応した。PCRに用いた配列と反応条件は以下のとおりである。NtSGT1 mRNAの増幅にはプライマーNtSGTFw-3(5’-GGACACACAAACCTTTGACT-3’:配列番号52)、NtSGTRv-4(5’-CCAGTTTTGGGATGTCTGCT-3’:配列番号53)を各10 pmol用いて、94℃、30秒、53℃、30秒、72℃、30秒を1サイクルとする、計30サイクルを行った。またRT-PCRの内部標準として用いたactin mRNAの増幅にはBY-2 actin Fw(5’-CAATTCTTCGGTTGGATCTTG-3’:配列番号54)、BY-2 actin Rv(5’-CTTCATGCTGCTGGGAG-3’配列番号55)を各10 pmol用いて、94℃、30秒、53℃、30秒、72℃、30秒を1サイクルとする、計20サイクルを行った。RT-PCRは4回繰り返し実験を行い、得られたRT-PCR産物はDNAの蛍光検出剤であるSYBR Safe DNA gel stain(Invitrogen)を添加したアガロースゲルを用いて電気泳動に供し、スキャン型イメージャーであるTyphoon 9400(GEヘルスケア)で検出した。予測される増幅DNAの長さはNtSGT1においては約370-bp、actinにおいては約460-bpであるが、これらに相当するPCR産物が確認できたため、ソフトウェアImage Quant TL(GEヘルスケア・ジャパン)を用いてバンド強度を測定した。図12にRNAi によるSGT転写抑制効果を示した。図12AはRT-PCRの電気泳動結果。図12Bは、図12Aで検出したPCR産物のシグナル強度をグラフ化したものである。RNAiコンストラクト導入BY-2(RNAi1、 RNAi2、RNAi3)では、転写産物量が非形質転換体の50%未満に減少しており、RNAiによるSGT転写抑制効果が認められた。student testを行い、RNAiコンストラクト導入BY-2は全ての非形質転換体に対して p<0.01で有意であった。
【0067】
〔18−4〕RNAi によるSGT転写抑制効果(SGT活性の測定)
(1)マイクロソームの調製
前記実施例1でも用いたタバコ細胞抽出液マイクロソームと同様の調製を行った。具体的には、7日間培養したタバコ培養細胞BY-2の培養液を2号濾紙(2枚重ねる)で濾過してBY-2細胞を回収した。BY-2細胞1 gあたり破砕バッファー1 mlを添加して、ホモジナイザーを用いて、3,700 rpmから4,000 rpmの回転数で4回上下させ、破砕した。破砕バッファーの組成は、40 mM HEPES-KOH (pH7.4)、400 mMソルビトール、2 mM ジチオスレイトールである。この破砕液を1,000×g、10分、4℃で遠心し、上清を回収した。この上清を10,000×g、10分、4℃で遠心し、上清を回収した。この上清を100,000×g、10分、4℃で遠心し、沈殿を回収し、マイクロソーム画分とした。
【0068】
(2)SGT活性測定
上記で得たマイクロソーム画分を用いてSGT活性を測定した。活性測定に用いた組成は、50 μgマイクロソーム画分、0.1 M MES (pH6.0)、12.5 μM VYKSO4-Ac1 peptide、5 mM UDP-Galactose、5 mM MnCl2、2 mM EDTA、5×Protease inhibitor cocktail、0.10 % TritonX-100であり、総量6.25μlとした。反応温度は30℃であった。
測定結果を図12Cに示した。RNAiコンストラクト導入BY-2(RNAi1、 RNAi2、RNAi3)では、SGT活性が非形質転換体の50%未満に減少しており、RNAiによるSGT転写抑制効果が認められた。SGT転写産物量とSGT活性の両方においてRNAiによるSGT転写抑制効果が認められた。
【配列表フリーテキスト】
【0069】
1.クラミドモナス SGT遺伝子配列 Chla(g)
2.クラミドモナス SGTアミノ酸配列 Chla(a)
3.クラミドモナス SGT活性ドメインのアミノ酸配列 Chla-1
4.シロイヌナズナ SGT遺伝子配列 Arab(g)
5.シロイヌナズナ SGTアミノ酸配列 Arab(a)
6.シロイヌナズナ SGT活性ドメイン1のアミノ酸配列 Arab-1
7.シロイヌナズナ SGT活性ドメイン2のアミノ酸配列 Arab-2
8.イネ SGT遺伝子配列 Oryz(g)
9.イネ SGTのアミノ酸配列 Oryz(a)
10.イネ SGT活性ドメイン1のアミノ酸配列 Oryz-1
11.イネ SGT活性ドメイン2のアミノ酸配列 Oryz-2
12.ニセツリガネゴケ SGTのアミノ酸配列 Phys(a)
13.ニセツリガネゴケ SGT活性ドメイン1のアミノ酸配列 Phys-1
14.ニセツリガネゴケ SGT活性ドメイン2のアミノ酸配列 Phys-2
15.ブラシノ藻 SGTアミノ酸配列 Ostreo (a)
16.ブラシノ藻 SGT活性ドメインのアミノ酸配列 Ostreo -1
17.クラミドモナス SGT活性ドメイン1を含む活性化領域 Chla(act)1
18.シロイヌナズナ SGT活性ドメイン2を含む活性化領域 Arab(act)2
19.SGT活性測定用受容体基質ペプチド R-peptide
20.タバコ(nicotiana) SGT遺伝子配列
21.タバコ(nicotiana) SGTアミノ酸配列
22.タバコSGT CD1:
23.タバコSGT CD2:
24.タバコSGT 3’-RACE Forward primer(NtSGTFw-1)
25.タバコSGT 3’-RACE Reverse primer(NtSGTRv-2)
26.タバコSGT 3’-RACE antisense primer(NtSGTFw-19)
27.スポラミン配列(Sporamin sequence)
28.エクステンシン配列(extension sequence)
29.改変エクステンシン配列-1(Extension sequence(mod-1))
30.プロリンが水酸化されたエクステンシン配列(Hydroxyl-proline-extension sequence)
31.プロリンが水酸化された改変エクステンシン配列-1(Hydroxyl-prolin-extension sequence(mod-1))
32.プロリンが水酸化された改変エクステンシン配列-2(Hydroxyl-proline-extension sequence(mod-2))
33.プロリンが水酸化された改変エクステンシン配列-3(Hydroxyl-proline-extension sequence(mod-3))
34.プロリンが水酸化されたスポラミン配列(Hydroxyl-proline-sporamin sequence)
35.プロリンが水酸化されたAGP14配列(Hydroxyl-proline arabinogalactan protein14 (AGP14) sequence)
36.プロリンが水酸化された改変AGP14配列(Hydroxyl-proline AGP14 sequence(mod))
37.Ser繰り返し配列(Ser repetitive sequence)
38.タバコSGT mRNA(Nicotiana tabacum SGTmRNA)
39.5’末端リン酸化逆転写反応用プライマー(phosphorylation RT-PCR primer)(NtSGT1-5phos)
40.1st PCR用センスプライマーNtSGT1-S1(forward primer-1)
41.1st PCR用アンチセンスプライマーNtSGT1-A1(reverse primer-1)
42.2nd PCR用センスプライマーNtSGT1-S2(forward primer-2)
43.2nd PCR用アンチセンスプライマーNtSGT1-A2(reverse primer-2)
44.mRNA全長用NtSGTFw-5term(NtSGT mRNA forward primer)
5’-GGACACACAAACCTTTGACT-3’
45.mRNA全長用NtSGTRv-3term(NtSGT mRNA reverse primer)
TGGAGCTCTTTTTTTTTTTTTTTTTTTTTTTTVN-3’
46.NtSGT1ターゲット領域センス配列増幅用プライマーSGTsenseFw
5’-AGATTGTCGACTTGCACTGATGAAGAGAGGAAG-3’
47.NtSGT1ターゲット領域センス配列増幅用プライマーSGTsenseRv
5’-GAATAGCATGCAGATCTATCATCAAGTTATCATTAATC-3’
48.NtSGT1ターゲット領域アンチセンス配列増幅用プライマーSGTantiFw
5’-AGATTGGTACCTTGCACT
49.NtSGT1ターゲット領域アンチセンス配列増幅用プライマーSGTantiRv
5’-GAATAGAATTCAAGCTTATCATCAAGTTATCATTAATC
50.RNAiコンストラクトリンカー配列増幅用プライマーIntron1Fw
5’- GGCGGGGATCCCTACAGGGTAAATTTCT-3’
51.RNAiコンストラクトリンカー配列増幅用プライマーIntron1Rv
5’- GGCGGGGTACCGGTTCTGTAAC-3’
52.NtSGT1 mRNA増幅用プライマーNtSGTFw-3=配列番号44
5’-GGACACACAAACCTTTGACT-3’
53.NtSGT1 mRNA増幅用プライマーNtSGTRv-4
5’-CCAGTTTTGGGATGTCTGCT-3’
54.RT-PCRの内部標準actin mRNA増幅用プライマーBY-2 actin Fw
5’-CAATTCTTCGGTTGGATCTTG-3’
55.RT-PCRの内部標準actin mRNA増幅用プライマーBY-2 actin Rv
5’-CTTCATGCTGCTGGGAG-3’
【特許請求の範囲】
【請求項1】
下記の(1)〜(4)のいずれかのアミノ酸配列を含む、セリンO−結合型ガラクトース転移酵素活性(SGT酵素活性)を有するポリペプチド;
(1)配列番号21に示されるアミノ酸配列、
(2)配列番号21に示されるアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列。
(3)配列番号21と80%以上の同一性を有しているアミノ酸配列からなり、かつそのSGT酵素活性ドメインとして、DXDモチーフを有し、配列番号22又は23と90%以上の同一性を有するアミノ酸配列からなる部分配列を有しているアミノ酸配列、
(4)植物のゲノム中の塩基配列によりコードされるアミノ酸配列であって、配列番号21に示されるアミノ酸配列と80%以上の同一性を有するアミノ酸配列からなり、かつDXDモチーフを含むアミノ酸配列。
【請求項2】
下記(5)〜(7)に記載のアミノ酸配列を含む、セリンO−結合型ガラクトース転移酵素活性(SGT酵素活性)を有するSGT酵素活性ドメインポリペプチド;
(5)配列番号22又は23に示されるアミノ酸配列、
(6)配列番号22又は23に示されるアミノ酸配列のいずれかにおいて1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列、
(7)配列番号22又は23に示されるアミノ酸配列と少なくとも90%以上の同一性を有するアミノ酸配列であって、かつDXDモチーフを含むアミノ酸配列。
【請求項3】
請求項1又は2に記載のポリペプチドをコードする塩基配列からなる核酸。
【請求項4】
セリンO−結合型ガラクトース転移酵素活性を有するポリペプチドをコードする核酸であって、配列番号20に示される塩基配列、又は当該塩基配列の相補配列とストリンジェントな条件下でハイブリダイズする塩基配列からなる核酸。
【請求項5】
請求項3又は4に記載の核酸を組み込んだ発現ベクター。
【請求項6】
請求項3又は4に記載の核酸が発現可能に組みこまれた形質転換体。
【請求項7】
請求項6に記載の形質転換体を用いたO−結合型糖鎖を有する組換え糖タンパク質の製造方法であって、当該形質転換体が外来性糖タンパク質をコードする核酸により形質転換されていることを特徴とする、セリンO−結合型ガラクトースを含む糖鎖を有する組換え糖タンパク質の製造方法。
【請求項8】
請求項2に記載された、SGT酵素活性を有する活性ドメインポリペプチドに結合する阻害物質により当該ポリペプチドのSGT酵素活性を阻害することを特徴とする、形質転換植物細胞または形質転換植物における外来タンパク質のセリン残基へのO−結合型ガラクトース付加抑制方法。
【請求項9】
形質転換植物細胞または形質転換植物により外来タンパク質を産生する際に、外来タンパク質に含まれるセリン残基へのO−結合型ガラクトース付加を抑制する方法であって、形質転換宿主となる植物細胞が本来有しているSGT遺伝子である、請求項3又は4に記載されたSGTをコードする核酸の発現を抑制することを特徴とする、形質転換植物細胞または形質転換植物における外来タンパク質のセリン残基へのO−結合型ガラクトース付加抑制方法。
【請求項10】
請求項8又は9に記載の方法により外来タンパク質のセリン残基へのO−結合型ガラクトース付加が抑制された、形質転換植物細胞または形質転換植物。
【請求項11】
形質転換植物細胞または形質転換植物がタバコである請求項10に記載の形質転換植物細胞または形質転換植物。
【請求項1】
下記の(1)〜(4)のいずれかのアミノ酸配列を含む、セリンO−結合型ガラクトース転移酵素活性(SGT酵素活性)を有するポリペプチド;
(1)配列番号21に示されるアミノ酸配列、
(2)配列番号21に示されるアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列。
(3)配列番号21と80%以上の同一性を有しているアミノ酸配列からなり、かつそのSGT酵素活性ドメインとして、DXDモチーフを有し、配列番号22又は23と90%以上の同一性を有するアミノ酸配列からなる部分配列を有しているアミノ酸配列、
(4)植物のゲノム中の塩基配列によりコードされるアミノ酸配列であって、配列番号21に示されるアミノ酸配列と80%以上の同一性を有するアミノ酸配列からなり、かつDXDモチーフを含むアミノ酸配列。
【請求項2】
下記(5)〜(7)に記載のアミノ酸配列を含む、セリンO−結合型ガラクトース転移酵素活性(SGT酵素活性)を有するSGT酵素活性ドメインポリペプチド;
(5)配列番号22又は23に示されるアミノ酸配列、
(6)配列番号22又は23に示されるアミノ酸配列のいずれかにおいて1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列、
(7)配列番号22又は23に示されるアミノ酸配列と少なくとも90%以上の同一性を有するアミノ酸配列であって、かつDXDモチーフを含むアミノ酸配列。
【請求項3】
請求項1又は2に記載のポリペプチドをコードする塩基配列からなる核酸。
【請求項4】
セリンO−結合型ガラクトース転移酵素活性を有するポリペプチドをコードする核酸であって、配列番号20に示される塩基配列、又は当該塩基配列の相補配列とストリンジェントな条件下でハイブリダイズする塩基配列からなる核酸。
【請求項5】
請求項3又は4に記載の核酸を組み込んだ発現ベクター。
【請求項6】
請求項3又は4に記載の核酸が発現可能に組みこまれた形質転換体。
【請求項7】
請求項6に記載の形質転換体を用いたO−結合型糖鎖を有する組換え糖タンパク質の製造方法であって、当該形質転換体が外来性糖タンパク質をコードする核酸により形質転換されていることを特徴とする、セリンO−結合型ガラクトースを含む糖鎖を有する組換え糖タンパク質の製造方法。
【請求項8】
請求項2に記載された、SGT酵素活性を有する活性ドメインポリペプチドに結合する阻害物質により当該ポリペプチドのSGT酵素活性を阻害することを特徴とする、形質転換植物細胞または形質転換植物における外来タンパク質のセリン残基へのO−結合型ガラクトース付加抑制方法。
【請求項9】
形質転換植物細胞または形質転換植物により外来タンパク質を産生する際に、外来タンパク質に含まれるセリン残基へのO−結合型ガラクトース付加を抑制する方法であって、形質転換宿主となる植物細胞が本来有しているSGT遺伝子である、請求項3又は4に記載されたSGTをコードする核酸の発現を抑制することを特徴とする、形質転換植物細胞または形質転換植物における外来タンパク質のセリン残基へのO−結合型ガラクトース付加抑制方法。
【請求項10】
請求項8又は9に記載の方法により外来タンパク質のセリン残基へのO−結合型ガラクトース付加が抑制された、形質転換植物細胞または形質転換植物。
【請求項11】
形質転換植物細胞または形質転換植物がタバコである請求項10に記載の形質転換植物細胞または形質転換植物。
【図1】

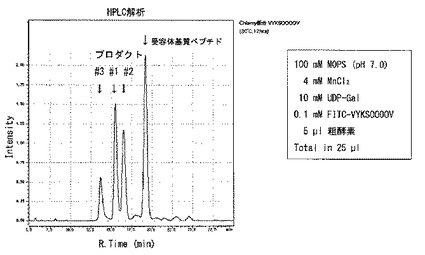
【図2】
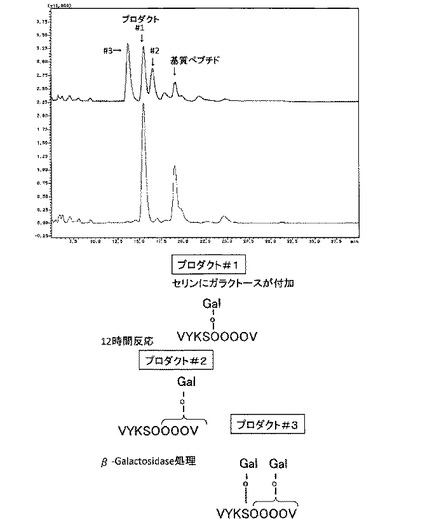
【図3AB】
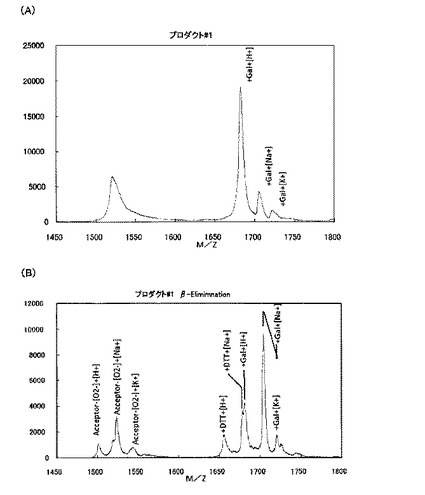
【図3CD】
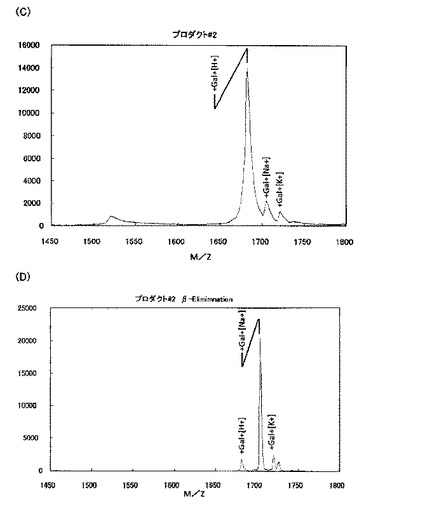
【図6B】
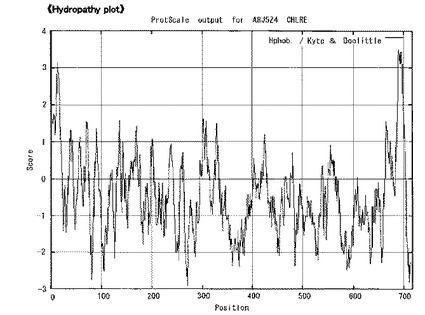
【図7B】
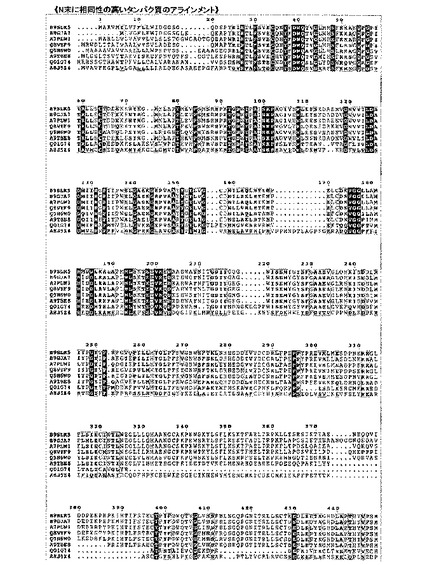
【図7C】

【図9】
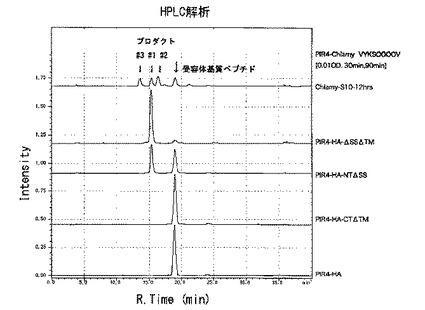
【図10】
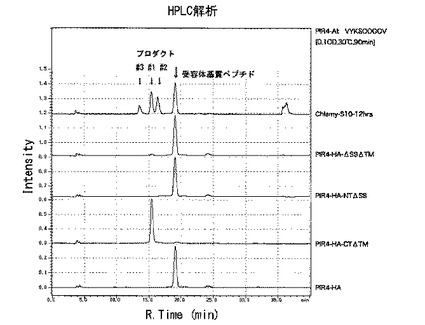
【図4】
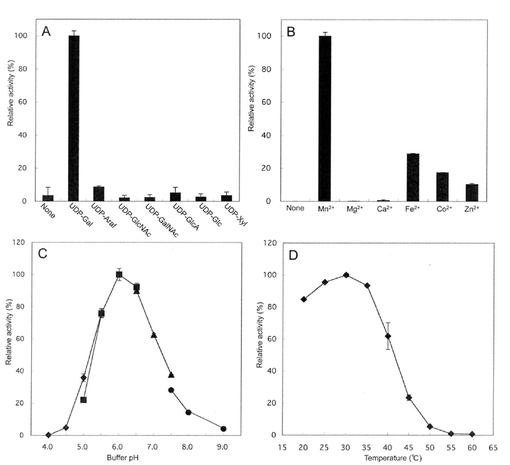
【図5A】
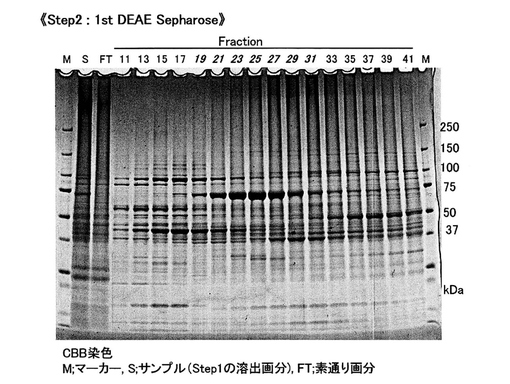
【図5B】

【図5C】
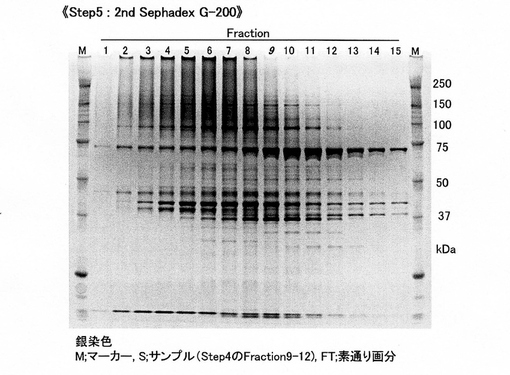
【図6A】
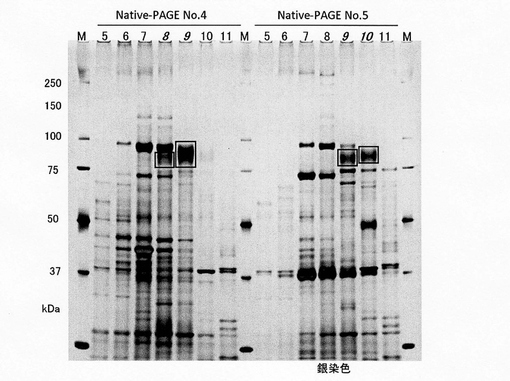
【図7A】

【図7D】

【図7E】
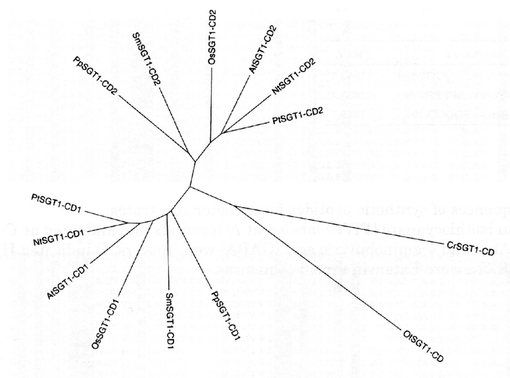
【図8】
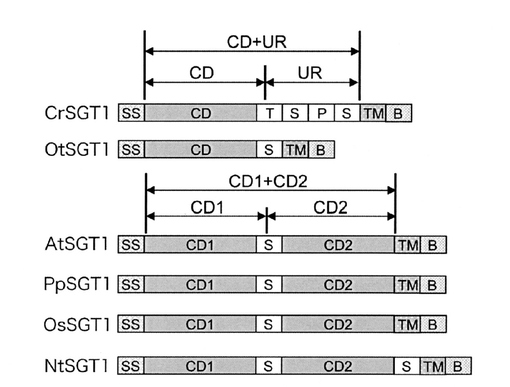
【図11】
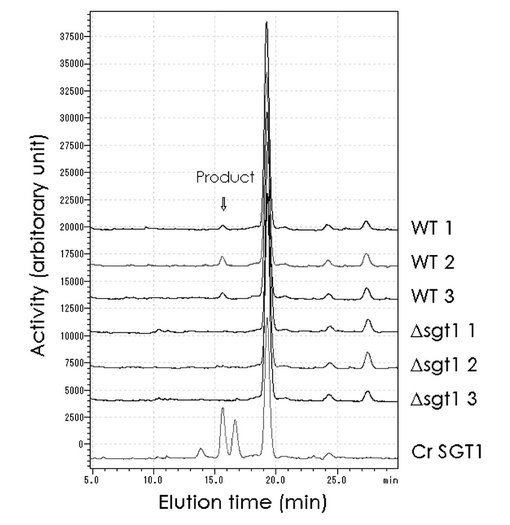
【図12】
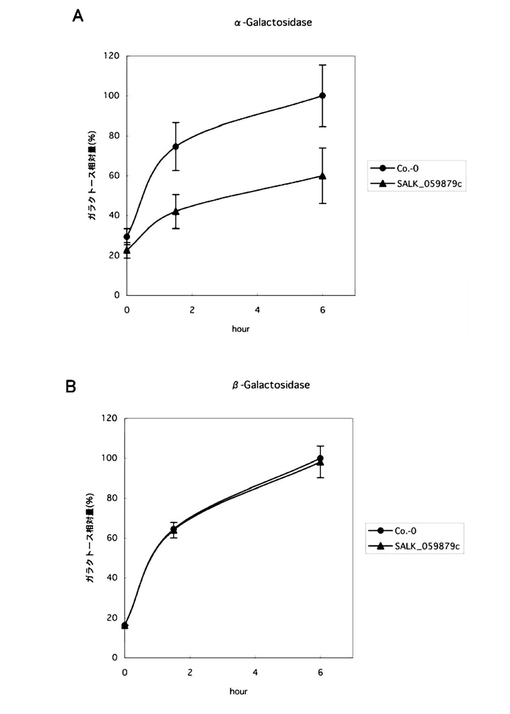
【図13】
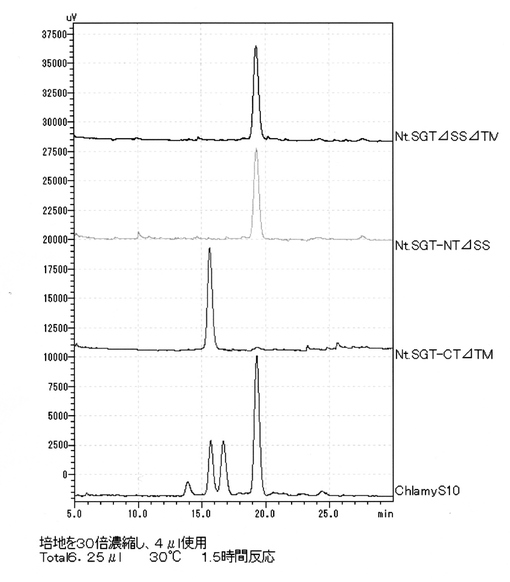
【図14】
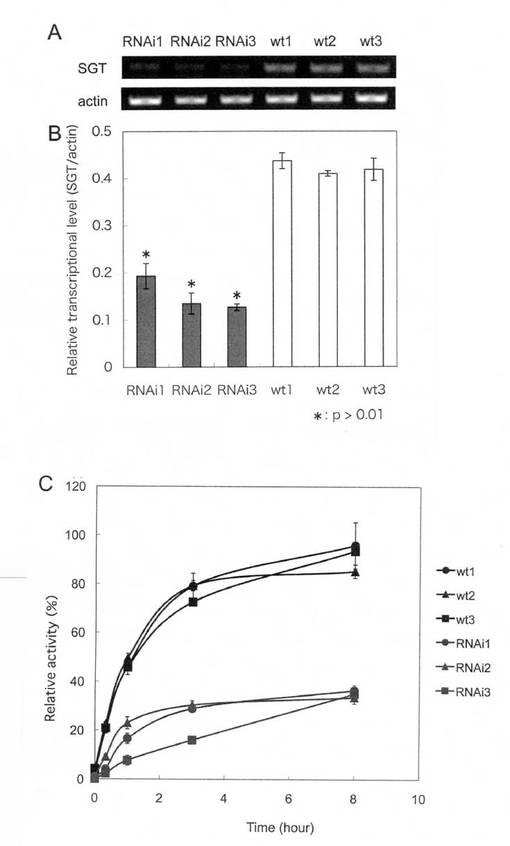
【図2】
【図3AB】
【図3CD】
【図6B】
【図7B】
【図7C】
【図9】
【図10】
【図4】
【図5A】
【図5B】
【図5C】
【図6A】
【図7A】
【図7D】
【図7E】
【図8】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2012−183022(P2012−183022A)
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願番号】特願2011−48303(P2011−48303)
【出願日】平成23年3月4日(2011.3.4)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【出願人】(504145342)国立大学法人九州大学 (960)
【Fターム(参考)】
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願日】平成23年3月4日(2011.3.4)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【出願人】(504145342)国立大学法人九州大学 (960)
【Fターム(参考)】
[ Back to top ]