
タンパク質の機能部位またはエピトープの遮蔽方法
ポリペプチド内部の1つ以上の結合部位を部位特異的に遮蔽する方法が開示されている。方法では、該ポリペプチドに少なくとも1つの低分子量水溶性ポリマーを、結合部位が該ポリマーによって隠蔽されるように付着させる。開示した方法による結合部位(例えば、エピトープ)の遮蔽は、該結合部位とその対応受容体との相互作用によって誘導される生物応答を排除するかまたは実質的に減退させ、生物応答をポリペプチドの非隠蔽部分に集中させる。本文中の方法によって製造される医薬製品(例えば、ポリマー修飾抗原およびそれらを含むワクチン組成物)およびそれらの使用は、脊椎動物生体組織、好ましくはヒトまたはヒト以外の商業用もしくは飼育用の獣医学的に重要な哺乳動物のような哺乳類宿主に直接導入されたときに非隠蔽部分に対して特異的免疫応答を誘導し、該哺乳動物体内で選択的免疫防御を発生する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、結合部位がポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーを部位特異的にポリペプチドに付着させる処理を含むポリペプチド内部の1つ以上の結合部位の遮蔽方法に関する。1つの実施態様では、隠蔽された結合部位がポリペプチド抗原内部のエピトープである。開示された方法による結合部位(例えば、エピトープ)の遮蔽は、該結合部位とその対応受容体との相互作用によって誘導される生物応答を排除するかまたは実質的に減退させる効果を有しており、ポリペプチドの非隠蔽部分に生物応答が集中することを援助する。本発明はさらに、本文中に開示された方法によって製造された医薬製品(例えば、ポリマー修飾抗原およびそれらを含むワクチン組成物)およびそれらの使用に関する。これらは脊椎動物生体組織、好ましくはヒトまたは商業用もしくは飼育用の獣医学的に重要なヒト以外の哺乳動物のような哺乳類宿主に直接的に導入されると、ポリペプチドの非隠蔽部分に特異的免疫応答を誘導し、該哺乳動物体内で選択的免疫防御を生じる。
【背景技術】
【0002】
タンパク質内部の生物活性部位またはドメイン(例えば、免疫原性エピトープ)の選択的隠蔽または“遮蔽”の自然発生は、宿主免疫応答を破壊するために病原体が使用する戦略であると説明されてきた。例えば、インフルエンザウイルスの赤血球凝集素(HA)中で、モノクローナル抗体のエピトープはN−結合オリゴ糖鎖によって遮蔽されている(Wiley,D.C.ら,1981,Nature289:373−8)。Skehelおよび共同研究者らは、抗体エピトープの極めて近傍に付加N−結合オリゴ糖を含むHAをもつ変種に対して選択された抗体の存在下のウイルス増殖を証明した(1984,Proc.Natl.Acad.Sci.U.S.A.81:1779−83)。また、HIVのエンベロープ糖タンパク質のgp120サブユニットについては、宿主免疫系による中和を逃れるメカニズムであるグリコシル化によって重要なエピトープが掩蔽されることが詳細に記載されている(たとえば,Burton,D.R.ら,2004,Nat.Immunol.5:233−6;Johnson,W.E.and R.C.Desrosiers,2002,Annu.Rev.Med.53:499−518;Kwong,P.D.ら,1998,Nature 393:648−59;およびWei,X.ら,2003,Nature 422:307−12)。また、他のHIVエピトープについては構造的隠蔽によって同じ効果が達成されている(Kwong,P.D.ら,2002,Nature 420:678−82)。この“免疫回避”メカニズムはしばしば、デコイとして作用する免疫優性エピトープの慢性提示という結果を生み、ふつうなら最適防御を確保するより強力な防御エピトープ(“中和”エピトープ)に免疫系が集中することを妨害する。最終的にこのメカニズムはワクチン効果を低下させる一因となるであろう。
【0003】
糖質もまた非抗原性受容体結合部位を隠蔽できる。例えば、Goto,H.およびK.Kawaoka(1998,Proc.Natl.Acad.Sci.U.S.A.95:10224−8)は、インフルエンザウイルスのノイラミニダーゼタンパク質の特異的部化をオリゴ糖側鎖によって隠蔽し、該タンパク質へのプラスミノーゲンの結合を阻止することを提案した。空間的に近接したウイルスの赤血球凝集素を開裂し融合受容能を与えるためにはプラスミノーゲンが必要なので、糖質側鎖はウイルスの感染力を大きく変質させる。
【0004】
ポリマー遮蔽は、医薬タンパク質の免疫原性を減退させ、望ましくない免疫原性副作用を最小に抑制し、腎濾過経由のクリアランスを減少させることによって最終的に治療効果を改善するために広く使用されている方法である(総説:Schellekens,H.,2002,Clinical Therapeutics 24:1720−40参照)。広範囲に応用された治療用タンパク質遮蔽方法では、高分子量(“HMW”)の線状または分枝状ポリエチレングリコール(“PEG”)鎖を該タンパク質に付着させる(Harris,J.M.and R.B.Chess,2003,Nat.Rev.Drug Discov.2:214−21;米国特許番号4,179,337、Davisらに許諾)。所望効果はタンパク質の完全隠蔽なので、治療用タンパク質の修飾に使用するPEG部分は付着対象タンパク質と同等のサイズを有している。
【0005】
もっと最近には、選択エピトープ特異的にタンパク質を遮蔽する構想が提案され、“イムノフォーカシング”と命名された。この戦略は、タンパク質抗原または免疫原の内部の免疫優性の非中和性エピトープの選択的遮蔽であり、その結果として免疫応答がより強力な中和エピトープに指向および/または集中する(参照:Burton,D.R.ら,2004,Nat.Immunol.5:233−6;Delves,PJ.ら,1997,Mol. Med.Today 3:55−60;およびPantophlet,R.and D.R.Burton,2003,Trends Mol.Med.9:468−73)。この方法は、上記のような持続性病原体感染症に好都合に展開する複合的免疫回避戦略の対策として開発された。特定例として、Garrity,R.R.は、HIV gp120のV3超可変ループに付加N−結合グリコシリーション部位を組込んで、該タンパク質に対する抗体応答をかなりシフトさせた(1997,J.Immunol.159:279−89)。Pantophlet,R.らによって行われたもっと体系的な研究は、モノマーgp120内部に7つのN−グリコシレーションモチーフを付加すると、広範な非中和抗体パネルへの結合は完全に排除されるが、中和モノクローナル抗体mAB b12への結合は低効率で維持されることを示した(Pantophlet,R.and D.R.Burton,2003,前出 9:468−73;Pantophlet,R.ら,2003,J.Virol.77:5889−901)。しかしながら重要なことは、グリコシレーションがT細胞認識を必ずしも排除しないことである。実際、T細胞がグリコシル化ペプチドに結合することは多くの文献に記載されており(参照:たとえば,Cudic,M.ら,2002,Bioorg.Med.Chem.10:3859−70;Glithero, AJ.ら,1999,Immunity 10:63−74;Haurum,J.S.ら,1999,J.Exp.Med.190:145−50;Jackson,D.C.ら, 1994,Virology 199:422−30;およびRudd,P.M.ら,2001,Science 291:2370−6)、特に、糖質側鎖が短い場合もある(Cudicら,2002,前出;Jacksonら,1994,前出)。従って本発明は、SMW PEG部分を非限定例とする糖質側鎖以外の低分子量(“SMW”)ポリマー部分を結合部位の内部または近傍のペプチド/タンパク質に結合させること(“PEG付加”)によるペプチドまたはタンパク質内部の受容体の結合部位の選択的遮蔽に関する。
【0006】
1991年8月20日付けでGethingらに許諾された米国特許番号5,041,376は、オリゴヌクレオチド突然変異誘発を使用して補足N−結合オリゴ糖側鎖を部位特異的場所に組込むことによってタンパク質のエピトープを遮蔽する一般的方法を開示している。
【0007】
それぞれ1996年12月17日および1998年12月19日付けでGarrityらに許諾された米国特許番号5,585,250および5,853,724は、中和抗体応答がV3ループから逸れてタンパク質のより強力な中和部分を指向するようにしたHIV−1 gp120/160のV3ループのエピトープ鎮静方法を開示している。
【発明の開示】
【0008】
本発明は、結合部位がポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量(“SMW”)水溶性ポリマーを部位特異的にポリペプチドに付着させる処理を含むポリペプチド内部の1つ以上の結合部位の遮蔽方法に関する。本発明の1つの実施態様では、本文中の方法によって遮蔽される結合部位(すなわち、機能部位)はポリペプチド抗原内部の免疫原性または抗原性エピトープである。選択された結合部位(例えば、エピトープ)の隠蔽は、結合部位とそれらの対応受容体との相互作用によって誘導される望ましくない生物応答(例えば、免疫応答)を排除または減退させ、生物応答を該ポリマー修飾ポリペプチドの非隠蔽部分に集中させる。従って本発明は、少なくとも1つのSMW水溶性ポリマーを部位特異的にペプチド/ポリペプチドに付着させ、該結合部位/エピトープをポリマーによって選択的に隠蔽するペプチドまたはポリペプチドの結合部位またはエピトープの遮蔽方法に関する。この場合、ペプチド/ポリペプチドは1つ以上の結合部位/エピトープを内包している。SMW水溶性ポリマーは、遮蔽すべき結合部位/エピトープの内部または近傍の場所(例えば、アミノ酸)でポリペプチドに付着する。本発明の1つの実施態様では、ポリペプチドへの部位特異的付着によってポリペプチド内部の結合部位を選択的に遮蔽するために使用される低分子量ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から選択される。別の実施態様では、低分子量水溶性ポリマーがPEGである。
【0009】
本発明はさらに、より有益な応答(例えば、中和応答)をタンパク質/ペプチド内部の択一的エピトープに向けさせるために、ポリペプチド抗原内部に存在する免疫優性および/または有益でないエピトープを免疫減衰する方法に関する。この場合、免疫優性および/または有益でないエピトープは抗原に付着した少なくとも1つの低分子量水溶性ポリマーによって選択的に隠蔽される。従って本文中の方法は、免疫応答が免疫由来細胞またはタンパク質による認識がほぼ不可能になった隠蔽エピトープから逸れて、1つ以上の非隠蔽エピトープに集中するという結果を得るために使用される。抗原への部位特異的付着によって抗原内部の有益でないエピトープを選択的に遮蔽するために使用される低分子量ポリマーは、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から選択できる。別の実施態様では、低分子量水溶性ポリマーがPEGである。
【0010】
本発明の1つの特定実施態様は、エピトープがポリマーによって選択的に隠蔽されるように抗原内部の少なくとも1つのアミノ酸残基にSMW PEGを非限定例とする少なくとも1つのSMW水溶性ポリマーを部位特異的に付着させる処理を含むポリペプチド抗原のエピトープ遮蔽方法に関する。この場合、該エピトープは、エンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または少なくとも一部分を模倣する共有結合安定化三量体コイルドコイル構造のスカフォールドドメイン内部に局在している。該コイルドコイル構造は、ペプチド間の1つ以上の共有結合によって共有結合的に安定化された3つのキメラペプチドを含み、キメラペプチドのおのおのは、エンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでいる。本発明のこの部分の具体的実施態様で、エンベロープウイルス膜融合タンパク質はHIV gp41である。
【0011】
従って本発明の1つの実施態様は、エピトープがポリマーで選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーをコイルドコイル構造に部位特異的に付着させる処理を含む、HIV gp41の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造のエピトープの遮蔽方法に関する。別の実施態様で、隠蔽エピトープは、コイルドコイル構造のスカフォールドドメイン内部に局在し、該コイルドコイル構造は3つのキメラペプチドを含み、キメラペプチドのおのおのは、HIV gp41 NH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含む“スカフォールドドメイン”を有しており、該ペプチドはジスルフィド結合を非限定例とする1つ以上の共有結合または化学選択的結合反応から生じる共有結合のいずれかによって共有結合的に安定化されている。
【0012】
本発明はまた、本文中の方法によって製造されたポリマー修飾ペプチドまたはタンパク質、それらを含有する医薬/ワクチン組成物、および、哺乳動物体内で選択的免疫防御を誘導するための該医薬/ワクチン組成物による哺乳動物を非限定例とする動物の疾病状態に対する免疫方法に関する。
【0013】
従って本発明の1つの実施態様は、本文中の方法によってポリマー修飾されたキメラペプチドに関する。該ペプチドは、三量体コイルドコイル構造を生成するように共有結合的に安定化でき、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質(“N−ペプチド”部分)のNH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合した三量体コイルドコイル(“スカフォールド”部分)を形成できるα−ヘリカルスカフォールドタンパク質を含むアルファ(“α”)−ヘリカルドメインを含む。PEGを非限定例とする1つ以上の低分子量ポリマーをスカフォールドドメイン配列内部に局在する選択アミノ酸残基に付着させることによって部位特異的に遮蔽されるのは、少なくとも1つ以上のエピトープを含むこのスカフォールドドメインである。本発明はさらに、本文中に記載した方法によって製造したポリマー修飾キメラペプチドから成る共有結合安定化三量体コイルドコイル構造に関する。該ポリマー修飾コイルドコイルの非限定例は(CCIPN17)3、C(チオEPN17)3およびC(チオEP17GluN17)3である。
【0014】
本発明はさらに、特定の疾患または状態(非限定的にヒト免疫不全ウイルス−1感染/AIDSを含む)を特異的に認識する免疫応答を誘導するために、脊椎動物生体組織、好ましくはヒトまたは商業用または飼育用の獣医学的に重要なヒト以外の哺乳動物のような哺乳類宿主に直接的に導入できる医薬製品(例えば、ワクチン)、その製造および使用に関する。該医薬製品および/またはその抗原性接合体は、本文中の方法によって製造したポリマー修飾ポリペプチド抗原を含む。
【0015】
本文中に使用した“HIV”は、HIV−1、HIV−2またはHIV−1および/またはHIV−2を表す。
【0016】
本文中に使用した“中和性”はin vitro細胞/ウイルスベースアッセイでウイルス感染を防止する抗体の能力を表すために当業界で使用されている。中和活性は該特異的抗体に対するIC50値として定量的に測定できる。“中和抗体”または“HIV中和抗体”は、少なくとも1つのHIV単離物を中和するための当業界で承認された感染力アッセイで証明できる。
【0017】
本文中に使用した“PEG”は−ポリエチレングリコール−を表す。
【0018】
本文中に使用した“SMW”は低分子量を表し、“HMW”は高分子量を表し、“MW”は分子量を表す。
【0019】
本文中に使用した“SPPS”は固相ペプチド合成を表す。
【0020】
本文中に使用した“Michael受容体”は求核種(例えば、O−、C−またはS−)と反応できる分極した求電子性炭素炭素二重結合を含有する化学部分を表す。
【0021】
本発明は概括的には、特定受容体によって正常に認識されるペプチドまたはタンパク質内部の機能部位の選択的隠蔽または遮蔽方法を記載する。本発明の1つの実施態様では、開示された方法によって隠蔽すべき機能部位(すなわち、結合部位)は、免疫原性または抗原性エピトープ、例えばT細胞またはB細胞エピトープであり、該エピトープを認識することを阻害される受容体はT細胞の抗原受容体または免疫グロブリン分子である。選択されたエピトープの隠蔽は、ペプチド/タンパク質内部の択一的エピトープおよび一般にペプチド/タンパク質の残りの部分全部の免疫原性を損なうことなくペプチド/タンパク質の一部分に対する望ましくない免疫反応性を排除する。ペプチドまたはタンパク質の内部の1つ以上の結合部位(例えば、エピトープ)の選択的隠蔽は、本文中に記載したように、ペプチド/タンパク質内部の1つ以上の適正場所、すなわち、隠蔽すべき結合部位の内部または極めて近傍に1つ以上の低分子量(“SMW”)ポリマー(例えば、ポリエチレングリコール)を付着させることによって得られる。本発明の1つの実施態様では、SMWポリマーは、隠蔽すべき結合部位の内部または極めて近傍の1つ以上のアミノ酸残基に付着できる。該アミノ酸はコード化または非コード化残基でよい。従って本発明は、結合部位がポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーを部位特異的にポリペプチドに付着させる処理を含む、ポリペプチド内部の結合部位の遮蔽方法に関する。結合部位の非限定例はポリペプチド抗原のエピトープである。
【0022】
上述のように、本文中の方法は、ペプチドまたはタンパク質の内部に局在する受容体結合部位の選択的遮蔽に広く応用できるが、本文中では、免疫グロブリン分子またはT細胞受容体によって認識されるペプチドまたはタンパク質内部のエピトープ(すなわち、B−またはT−細胞エピトープ)の選択的遮蔽について本発明を詳細に記載し、その具体例を後述する。しかしながらこの記載は、記載したエピトープの選択的隠蔽方法に本発明を限定するものではない。当業者は、本文中に詳細に記載された方法が、対応受容体による結合部位の認識を防止する目的で対象ペプチドまたはポリペプチド内部の特異的結合部位を選択的に遮蔽するためにどのように修正できるかを理解されるであろう。従って、本文中に使用した“結合部位”は他のタンパク質(例えば、抗体または細胞性受容体)によって認識されるおよび/または相互作用するアミノ酸残基またはそれに付着した糖側鎖を含むペプチドまたはポリペプチドの領域を表す。従って、本発明の一部として記載した方法によって隠蔽された抗原性/免疫原性エピトープは、本文中に定義した“結合部位”を表す。タンパク質性結合部位は、(例えば、触媒活性部位でしばしば観察される)選択的非連続アミノ酸によって生成された適切に折り畳まれたペプチド/ポリペプチドの三次元コンホメーショナル領域または連続アミノ酸の線状エピトープ(例えば、T細胞エピトープ)を含み得る。
【0023】
本発明は、機能部位がポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーをペプチド/ポリペプチドに部位特異的に付着させる処理を含むペプチドまたはポリペプチドの機能部位(すなわち、結合部位)の遮蔽方法に関する。本発明の1つの実施態様で、機能部位はポリペプチド抗原内部のエピトープである。従って、本発明の1つの実施態様は、エピトープがポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーを抗原に部位特異的に付着させる処理を含むポリペプチド抗原のエピトープの遮蔽方法に関する。本文中に使用した“エピトープ”という用語は、従来の免疫原性(すなわち、体液性または細胞性応答を誘発する能力)および/または抗原性(すなわち、T細胞受容体の抗体と特異的に結合する能力)応答を誘発できるタンパク質決定基を包含する。従って、免疫原性エピトープは本発明の一部として記載された方法によって選択的に隠蔽できる多くの種類の結合部位のひとつにすぎない。エピトープが通常はアミノ酸または糖側鎖のような分子の化学的活性表面の集まりから成り、しばしば特異的三次元構造特性および特異的電荷特性を有していることは公知である。コンホメーショナルエピトープとノンコンホメーショナルエピトープとは、変性用溶媒の存在下で前者の結合は消失するが後者の結合は消失しないことによって識別される。
【0024】
ペプチド/ポリペプチドのエピトープの選択的遮蔽方法は、該ペプチド/ポリペプチドが2つ以上のエピトープを内包し、該エピトープの1つ以上に対する特異的免疫応答が望ましくそれ以外の免疫応答は排除したい場合に特に有用である。従って、本文中の方法は、免疫応答が免疫由来細胞による認識が不能になった隠蔽エピトープから逸れて、1つ以上の望ましい非隠蔽エピトープに集中するという結果を得るために使用される。ペプチド/ポリペプチド内部の適切な場所に低分子量ポリマー分子を付着させて隠蔽エピトープとその免疫細胞受容体(例えば、免疫グロブリン分子)との会合を阻止することによって、エピトープは物理的にも機能的にも有効に遮蔽される。例えば、ポリペプチド抗原(例えば、合成ペプチドまたは病原性タンパク質)は、宿主生物体内で免疫応答を選択的に誘発し該抗原内部の他のエピトープを実質的に排除するような1つ以上の免疫優性エピトープを含有し得る。このような免疫優性エピトープが有益でない免疫応答(例えば、非中和抗体または非保存中和抗体)を誘発し、同じペプチド/タンパク質内部に局在する免疫刺激性の低いエピトープに対する有益な免疫応答が失われるような場合、免疫刺激性の低いエピトープに免疫応答を集中させ、免疫優性エピトープから逸れるようにすることが重要である。従って、本発明の1つの実施態様は、より有益な免疫応答(例えば、中和応答または保存中和応答)がタンパク質/ペプチド内部の択一的エピトープを指向するように有益でない免疫優性エピトープを免疫減衰させることに関する。重要なことは、有益でない免疫応答を誘導するエピトープの遮蔽による無益な免疫応答の免疫減衰が望ましくない応答の完全な排除を必ずしも意味しないことである。
【0025】
本発明はさらに、本文中の方法によって作製したポリマー修飾ペプチドまたはタンパク質、それらを含有する医薬/ワクチン組成物、哺乳動物体内で選択的免疫防御を誘導するための医薬/ワクチン組成物による疾患状態に対する哺乳動物の免疫方法に関する。従って本発明は、免疫応答を1つのポリペプチドに集中させる方法に関する。この場合、ポリペプチドは免疫応答を誘導できる1つ以上のエピトープを含んでおり、方法は、少なくとも1つのエピトープがポリマーによって選択的に隠蔽され該ポリペプチドに対する免疫応答が該ポリペプチド内部の非遮蔽領域を指向するように、少なくとも1つの低分子量水溶性ポリマーを部位特異的にポリペプチドに付着させる処理を含む。従って本発明は、本文中の方法で少なくとも1つの低分子量水溶性ポリマーをポリペプチドに部位特異的に付着させることによって該ポリペプチド内部の1つ以上のエピトープを選択的に遮蔽することによってポリペプチドに対する免疫応答を変質させる方法に関する。
【0026】
後出の実施例の項のポリエチレングリコール(“PEG”)の使用によって証明されるように、本文中の方法で結合部位またはエピトープを選択的に隠蔽するために使用したポリマーは実質的に直鎖状のポリマーであり、実質的に非免疫原性、無毒性、高度に可溶性で生物活性を有していない。本文中の方法で結合部位またはエピトープを選択的に遮蔽するために使用したポリマーの好ましい実施態様としてPEGを選択した。しかしながらこのポリマーは限定でなく例示目的で使用しただけである。水溶性ポリマーの例は、メトキシポリエチレングリコールのようなポリアルキレングリコールおよびその誘導体、ポリエチレングリコールホモポリマー、ポリプロピレングリコールホモポリマーおよびエチレングリコール−プロピレングリコールコポリマー(ポリマーおよびコポリマーは未置換であるかまたは一端がアルキル基で置換されている)、ヘパリンおよびヘパリンフラグメント、ポリビニルアルコールおよびポリビニルエチルエーテル、ポリビニルピロリドン、アスパルタミド、デキストランおよびデキストリン誘導体、デキストリンおよびデキストリン誘導体でポリオキシエチル化されたポリオールを含む。具体的に記述した水溶性ポリマーの様々な誘導体も本文中の方法に使用できると考えてよい。上記のような水溶性ポリマー、特にPEGのようなポリアルキレンオキシド基材のポリマーは公知である(参照:たとえば,Poly(ethylene glycol)Chemistry:Biotechnical and Biomedical Applications,Ed.J.M.Harris,New York:Plenum Press,1992;Poly(ethylene glycol)Chemistry and Biological Applications,Eds.J.M. Harris and S.Zalipsky,ACS,1997;以下の公開番号を有する国際特許出願公開:WO 90/13540,WO 92/00748,WO 92/16555,WO 94/04193,WO94/14758,WO 94/17039,WO 94/18247,WO 94/28937,WO 95/11924,WO 96/00080,WO 96/23794,WO 98/07713,WO 98/41562,WO 98/48837,WO 99/30727,WO 99/32134,WO 99/33483,WO 99/53951,WO 01/26692,WO 95/13312,WO 96/21469,WO 97/03106,WO 99/45964;次の米国特許:4,179,337;5,075,046;5,089,261;5,100,992;5,134,192;5,166,309;5,171,264;5,213,891;5,219,564;5,275,838;5,281,698;5,298,643;5,312,808;5,321,095;5,324,844;5,349,001;5,352,756;5,405,877;5,455027;5,446,090;5,470,829;5,478,805;5,567,422;5,605,976;5,612,460;5,614,549;5,618,528;5,672,662;5,637,749;5,643,575;5,650,388;5,681,567;5,686,110;5,730,990;5,739,208;5,756,593;5,808,096;5,824,778;5,824,784;5,840,900;5,874,500;5,880,131;5,900,461;5,902,588;5,919,442;5,919,455;5,932,462;5,965,119;5,965,566;5,985,263;5,990,237;6,011,042;6,013,283;6,077,939;6,113,906;6,127,355;6,177,087;6,180,095;6,194,580;および6,214,966)。
【0027】
本発明の1つの実施態様で、ポリペプチド抗原のエピトープを非限定例とするポリペプチド内部の結合部位を該ポリペプチドへの部位特異的付着によって選択的に遮蔽するために使用した低分子量ポリマーは、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される。別の実施態様で、該低分子量水溶性ポリマーは、(CH2−CH2−O)−から成る水溶性反復単位をもつPEGである。PEGが有利な理由は、ポリエーテル主鎖が生物環境および大抵の化学反応条件下で不活性であり、修飾すべきペプチド/タンパク質の特定反応基に共有結合付着できる官能基を含むPEG誘導体(すなわち、“活性化”PEG)を生成するために必要な化学修飾を受入れ易い末端を有しているからである。様々な活性化PEGポリマーが当業界で公知であり、その官能基の非限定例は、カーボネート、活性エステル、アルデヒド、トレシラート、アクリレート、ケトン、アミノオキシ、アミン、カルボン酸、エステル、チオエステル、ハロゲン、チオール、シアノアセテート、ジパルミトイルホスファチジルエタノールアミン、ジステアロイルホスファチジルエタノールアミン、エポキシド、ヒドラジド、アジド、イソシアナート、マレイミド、メタクリレート、ニトロフェニル、オルトピリジルジスルフィド、シラン、スルフヒドリル、ビニルスルホン、スクシニミジルグルタレート、スクシニミジルスクシネートおよびコハク酸である。官能基は、修飾すべきポリペプチドの可用な反応基の種類に基づいて選択できる。さらに、1つのPEGポリマーが2つ以上のポリペプチドに付着して生じるポリペプチド架橋をできるだけ少なくしこれによって改善された均質性を与えるために誘導体化可能な末端を1つだけ有している一官能PEG誘導体(“mPEG”)を使用できる。ポリペプチドの最も普遍的な反応部位は、リシン残基のα−もしくはε−アミノ基または他のアミノ酸のNH2−末端アミノ基である。他の典型的な反応部位のリストは、システイン、ヒスチジン、アルギニン、アスパラギン酸、グルタミン酸、セリン、トレオニン、チロシンなどのアミノ酸およびアミノ酸のCOOH−末端カルボン酸を含む。あるいは、PEGを非限定例とする本発明のSMWポリマー分子をコード化または非コード化アミノ酸内部に存在する反応基以外の反応基との相互作用を介してターゲットポリペプチドまたはペプチドに部位特異的に付着させることもできる。例えば、糖質部分または合成リンカーがターゲットポリペプチド/ペプチド構造の一成分であるときにSMWポリマーは該糖質部分または合成リンカー内部の反応基と相互作用を介して付着できる。
【0028】
本文中の方法によってポリペプチドを選択的に修飾するために使用されるPEGを非限定例とする様々なポリマーは、低分子量ポリマーを包含する。一般に、本文中に使用した“低分子量”ポリマーは、約1000Da以下の分子量のポリマー、特にPEGポリマー(例えば、≦PEG1000)を表す。付着したポリマー部分の水溶性反復単位の数は様々な値を有し得る。しかしながら、例えば、付着PEG部分の場合、このような単位−(CH2−CH2−O)−)のより好ましい数は20単位までの範囲である。本発明に概説したペプチドまたはポリペプチド内部の選択結合部位またはエピトープの隠蔽に使用したPEGのような低分子量ポリマーは約1000Da以下の分子量を有すると予測されるが、ペプチド/タンパク質の修飾に使用できるポリマーのサイズ範囲が隠蔽すべき結合部位またはエピトープのサイズに大きく左右されることは当業者に理解されよう。このため、結合部位/エピトープの完全遮蔽または実質的遮蔽に高分子量ポリマーの付着が必要な場合には本発明は分子量約1000Da以下のポリマーの使用に限定されない。しかしながら、本文中の方法の最終目標は、隠蔽すべき結合部位/エピトープが含まれているペプチド/ポリペプチドの一部分だけを遮蔽することであって、タンパク質内部に存在するすべての結合部位/エピトープを全体的に遮蔽することではないと銘記するのが重要である。
【0029】
本発明は、1つ以上のエピトープ(例えば、望まない免疫応答を生じるエピトープ)または結合部位が選択的に隠蔽され、治療用物質の残部はより有益な択一的免疫応答または活性を誘導し易いように維持されているポリマー修飾した治療用ペプチドまたはポリペプチドに関する。従って、本発明の最終目標は、特定の結合部位/エピトープを隠蔽するために必要な程度にだけ治療用タンパク質を遮蔽することであるから、本発明方法の使用には低分子量非分枝状ポリマーがもっとも好ましい。ポリマー修飾タンパク質はこれまでにも記載されているが、本発明のポリマー修飾の性質、方法および目的はこのような従来の研究とは顕著に異なっている。従来技術で開示されたポリマー修飾生物活性接合体の大半は約1000Daを上回るサイズのPEG部分の付着によって修飾されている。例えば、食品医薬品局によって認可された6つのPEG付加製品(Adagen(R)、Oncaspar(R)、PEG−Intron(R)、Pegasys(R)、Somavert(R)およびNeulasta(R))のおのおのは、5,000−40,000Daの範囲の分子量をもつPEGポリマーによって修飾されている。本発明では、所望する以上のペプチド/ポリペプチドが隠蔽される可能性があり従って有益な結合部位/エピトープの認識が妨害される可能性があるので高分子量(“HMW”)ポリマーの使用は好ましくない。過去においては、治療用タンパク質または化学的化合物をHMWポリマーと結合させることに伴って治療薬が与える生物活性がある程度犠牲になることは容認されていた。付着したHMW PEGポリマーはしばしば治療薬の全体または実質部分を覆い隠す(例えば、遮蔽する)が、薬剤の活性部位があらわれると(例えば接近可能になると)PEG分子の高度な可撓性が所期の生物応答を実行するためのモーメントを提供できる。従って、修飾の結果として治療薬の比活性は低下するが、腎臓および細網内皮系(“RES”)のクリアランス減少およびタンパク質分解酵素からの遮蔽の双方によって治療薬の体内利用効率は向上する。最終的に、この体内利用効率の向上が治療薬の投与に対して全体的にはより有益な生物応答を提供する。いわば、本発明の意図はタンパク質/ペプチドの所与の活性に対する全体的に改善された応答を誘発することではない。むしろ、本文中に記載の修飾によってタンパク質/ポリペプチドの所与の活性を選択的に変質させることである。ポリエーテル主鎖の高度な可撓性およびその水分子結合能力が理由で、PEG分子は同等の分子量の可溶性タンパク質の5−10×の大きさであるかのように作用する。従って、本発明の発明者らは、エピトープまたは結合部位の選択的遮蔽がより小さい分子量(例えば、約1000Da未満の分子量)の水溶性ポリマーの部位特異的付着によって達成でき、タンパク質の残りの部分およびその内部の結合部位/エピトープは可用であり、制約されることなく対応受容体と相互作用して所望の応答を発生および/または促進すると推測した。
【0030】
従って、本発明の好ましい実施態様は、ペプチド/ポリペプチドが2つ以上のエピトープまたは結合部位を内包しており、1つのエピトープ/結合部位がポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーをペプチド/ポリペプチドに部位特異的に付着させるペプチドまたはポリペプチドのエピトープまたは結合部位の遮蔽方法に関する。ペプチド/ポリペプチドのポリマー修飾の結果として、生物に(例えば、ワクチンの形態で)投与されたときにペプチド/ポリペプチドによって誘導される生物応答はペプチド/ポリペプチドの非隠蔽エピトープまたは結合部位の活性の結果である。従って本発明は、低分子量水溶性ポリマーをペプチド/ポリペプチドに共有結合的に付着させることによってペプチドまたはポリペプチドを修飾し、生物に(例えば、ワクチンの形態)で投与したときに付着ポリマーによって遮蔽されないペプチド/ポリペプチドの1つ以上の領域に特異的な生物応答を誘導するための方法に関する。本発明の1つの実施態様では、対象ペプチドまたはポリペプチドが1つ以上のエピトープを、単独で含むかまたは免疫系に由来しないタンパク質もしくは化合物の1つ以上の結合部位と共に含む。本発明の1つの実施態様の目的は、アミノ酸配列の1つの部分または領域の免疫原性を減退させる(すなわち、別の免疫応答の利益となるように特定の免疫応答を減退させる)ために特定のエピトープを選択的に遮蔽することである。従って、ペプチド/ポリペプチドの生物活性は、タンパク質の異なる(すなわち、非隠蔽)部分に集中し、これが該ペプチド/タンパク質への特異的応答を変化させる。本発明の1つの実施態様では、選択的に隠蔽されたエピトープは、ペプチド間の1つ以上の共有結合によって共有結合的に安定化された3つのキメラペプチドを含むエンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造内部に局在する“スカフォールド”ドメインである。該コイルドコイルのキメラペプチドのおのおのは、該エンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメインの全部または一部分にアルファ−らせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでいる。本発明のこの部分の1つの実施態様では、該エンベロープウイルス膜融合タンパク質がHIV gp41である。
【0031】
従って本発明は、SMW PEGを非限定例とする少なくとも1つの低分子量水溶性ポリマーを抗原内部の少なくとも1つのアミノ酸残基に部位特異的に付着させるポリペプチド抗原のエピトープの遮蔽方法に関する。エピトープはポリマーによって選択的に隠蔽され、選択的に隠蔽されたエピトープは、ペプチド間の1つ以上の共有結合によって共有結合的に安定化された3つのキメラペプチドを含むエンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造内部のスカフォールドドメイン内部に局在する。キメラペプチドのおのおのは、該エンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでいる。本発明のこの部分の具体的な実施態様では、エンベロープウイルス膜融合タンパク質がHIV gp41である。
【0032】
本発明の代表的な用途では、発明者らは、HIV gp41の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造を含むワクチン構築物の一部分の選択的隠蔽が(暫定米国特許出願番号60/576,062,出願日2004.7.1、および、番号60/636,724,出願日2004.12.16に記載)、2つの低分子量PEG部分の付着によって実現し、この隠蔽は、免疫応答をペプチドミメティクスのHIV部分に選択的に集中させる(すなわち、gp41膜融合中間体のこのミメティクスの非HIV部分の免疫原性を減退させる)ために有効であることを知見した。これは後出の実施例1に詳細に記載する。コイルドコイル構造は、おのおのが非HIV由来スカフォールドドメインを含む3つのアルファ(“α”)−ヘリカルポリペプチドから成り、該ドメインはHIV N−ペプチドドメインにらせん相で融合したα−ヘリカル構造の安定化を助ける。共有結合的に安定化されたコイルドコイル構造内部に含まれた3つのポリペプチドのおのおののスカフォールドドメインのアミノ酸配列内部に局在する2つのリシンアミノ酸はPEG236部分の共有結合付着によって修飾されている(図3参照)。各ペプチドの修飾の結果として、共有結合付着した合計6個のSMW PEG分子をもつコイルドコイル構造が形成される。実施例1に示すように、該SMW PEG分子はスカフォールドドメインに対する抗体の産生を効果的に遮蔽し、免疫応答を構造のHIV部分に集中させる。この例は、対象ペプチド/タンパク質の1つの領域を選択的に遮蔽するためにSMW PEG分子、特にPEG236を使用することを初めて開示したものである。
【0033】
従って本発明の1つの実施態様は、エピトープがポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーをコイルドコイル構造に部位特異的に付着させる処理を含む、HIV gp41の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造のエピトープ遮蔽方法に関する。別の実施態様で、隠蔽されたエピトープは、コイルドコイル構造のスカフォールドドメインに局在し、該コイルドコイル構造は、HIV gp41 NH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含む“スカフォールド”ドメインをおのおのが有している3つのキメラペプチドを含み、該ペプチドは1つ以上の共有結合によって共有結合的に安定化されている。スカフォールドドメインは、該ドメイン内部の1つ以上のアミノ酸残基に1つ以上の低分子量水溶性ポリマーを付着させることによって修飾でき、該水溶性ポリマーは、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から選択され、より特定的にはPEGポリマー(例えば、PEG236)である。
【0034】
本文中の方法によって選択的に遮蔽できる三量体コイルドコイル構造は、暫定米国特許出願番号60/576,062,出願日2004.7.1、および番号60/636,724,出願日2004.12.16、に詳細に記載されている。双方の特許は参照によって本発明に含まれるものとする。これらの出願に記載された三量体コイルドコイル構造はエンベロープウイルス膜融合のミメティクスであり、特にHIV gp41を非限定例とするエンベロープウイルスの膜タンパク質の融合中間体のミメティクスを表す。多くのエンベロープウイルスによって使用される膜融合イベントの一例として、HIV gp41−媒介膜融合は、gp41糖タンパク質のエクトドメインに局在する3つの必須成分:NH2−末端融合ペプチド、NH2−末端ヘプタッドリピート(“N−ヘリックス”)およびCOOH−末端ヘプタッドリピート(“C−ヘリックス”)が関与する複合プロセスである。2つのヘプタッドリピート領域(NH2:HR1およびCOOH:HR2)は、糖タンパク質に周期的疎水性を付与し、多くのエンベロープウイルスの融合メカニズムに関与する共通構造モチーフである“ヘアピン三量体”と呼ばれるgp41の融合発生(すなわち融合活性)コンホメーションを形成するために互いに相互作用するα−ヘリカル構造を予測させる。ヘアピン三量体構造は6つのα−ヘリックスの束である。3つのgp41エクトドメインのC−ヘリックス領域によって形成された3つのα−ヘリックスが、3つのgp41エクトドメインのN−ヘリックス領域によって形成された中央の三重鎖コイルドコイルに逆平行にパックされている。融合プロセスは、N−ヘリックスコイルドコイルが露出したNH2−末端融合ペプチドをターゲット細胞膜の近傍/内部に配置する“プレヘアピン”コンホメーションの形成を経由して進行する。3つのC−ヘリックスが折り返されてこの中央N−ヘリックスコイルドコイルに会合し、ウイルスおよび宿主の細胞膜が膜融合の準備として近接状態に引っ張られるとヘアピン三量体が形成される。ウイルス/宿主細胞膜融合を防止する抗HIV薬は、プレヘアピンまたはヘアピン三量体複合体に結合する中和抗体を含む。中央N−ヘリックスコイルドコイルによって形成されたgp41エクトドメインのヘプタッドリピート1(HR1)領域内部の疎水性ポケットは、広い交差反応性のHIV中和抗体を誘導するための好ましいターゲットであると考えられている(参照:暫定米国特許出願番号60/576,062,出願日2004.7.1:この疎水性ポケット領域に結合する中和抗体を記載している)。“プレヘアピン”構造は多くのエンベロープウイルス膜融合タンパク質の共通特徴なので(参照:Singhら,1999,J.Mol.Biol.290:1031−1041)、ポリマー修飾された膜融合のミメティクスは、本文中の方法を使用し様々なエンベロープウイルスに特異的なワクチン構築物を製造するために作製できる。従って、本文中に例示した方法はHIVに特異的なワクチン構築物の製造を目標としているが、他のエンベロープウイルスをターゲットとするワクチン構築物を製造するためにこの同じ基本戦略を使用できることが当業者には理解されよう。
【0035】
従って、本発明の1つの実施態様は、3つのキメラペプチドを含むHIV gp41 N−ペプチドコイルドコイルの全部または一部分を模倣するポリマー修飾され共有結合的に安定化された三量体コイルドコイルに関する。キメラペプチドのおのおのは、HIV gp41 N−ペプチドの全部または一部分にらせん相に融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含み、該スカフォールドドメイン内部に局在するエピトープは該スカフォールドドメインの内部または極めて近傍の少なくとも1つのアミノ酸残基に部位特異的に付着した少なくとも1つの低分子量水溶性ポリマーによって選択的に隠蔽されている。該三量体コイルドコイルgp41ミメティクスは、例えばジスルフィド結合または化学選択的結合反応のいずれかから生じた共有結合のようなキメラペプチド間に形成された共有結合を介して安定化されたホモ三量体構造またはヘテロ三量体構造を示し得る。該コイルドコイル構造のスカフォールド部分の内部に局在するエピトープは、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択された分子量約1000Da未満のPEGポリマーを非限定例とするSMW水溶性ポリマーの付着によって遮蔽できる。
【0036】
従って、本発明の1つの実施態様では、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または一部分を模倣するポリマー修飾三量体コイルドコイル構造が、コイルドコイル構造内部に含まれた並列α−ヘリカルペプチドのシステイン残基間の共有ジスルフィド結合の形成を介して共有結合的に安定化されているコイルドコイルミメティクスを含み、該コイルドコイル構造の1つの領域(エピトープ含有)は該領域の内部または近傍の1つ以上のアミノ酸へのSMWポリマーの付着によって遮蔽されている。本発明のこの実施態様で、共有結合形成に参加するシステインアミノ酸残基は、個々のα−ヘリカルペプチドのα−ヘリカルドメインの外部に位置するように操作されている。従ってコイルドコイル構造に内包されている個別ペプチドのおのおのは、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメイン(“N−ペプチド”ドメイン)の全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態(“スカフォールド”ドメイン)またはその修飾形態を含むα−ヘリカルドメインと、該α−ヘリカルドメインの外部に局在する少なくとも2つのシステイン残基とを有している。複数の該可溶性キメラペプチドを三量体コイルドコイル構造が形成される濃度でインキュベートし次いで該構造を酸化させることによって、コイルドコイルの並列キメラペプチドのシステイン残基間に共有ジスルフィド結合が形成される。前述したように、本文中に開示の方法によって遮蔽すべき好ましいエピトープはスカフォールドドメイン内部に局在している。
【0037】
本発明の別の実施態様では、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または一部分を模倣するポリマー修飾三量体コイルドコイル構造が、化学選択的結合反応から生じたコイルドコイル構造に内包されたα−ヘリカルペプチド間の共有化学結合の形成を介して共有結合的に安定化されたコイルドコイルミメティクスを含み、コイルドコイル構造の1つの領域(少なくとも1つのエピトープ含有)は該領域の内部または近傍の1つ以上のアミノ酸へのSMWポリマーの付着によって遮蔽されている。この実施態様で、該コイルドコイル構造は、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメイン(“N−ペプチド”ドメイン)の全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態(“スカフォールド”ドメイン)またはその修飾形態を含むα−ヘリカルドメインをおのおのが有している3つのキメラペプチドを含む。3つのキメラペプチドは、チオエーテル結合の形成を非限定例とする化学選択的反応によって生じた共有化学結合の形成を介してコイルドコイル構造中で安定化されている。例えば、本発明に従ってポリマー修飾できるヘテロ三量体コイルドコイル構造は、該ペプチドのα−ヘリカルドメインの外部に局在する少なくとも2つのシステイン残基をさらに含む1つのキメラペプチドと、おのおのが求電子部分を組込むように誘導体化される2つのキメラペプチドとを含み、第一キメラペプチドの各システイン残基の求核スルフヒドリルが2つの誘導体化キメラペプチドのおのおのの求電子部分と共にチオエーテル結合を形成する。詳細は、暫定米国特許出願番号60/576,062(前出)に記載されている。
【0038】
コイルドコイル構造を安定化するために使用できる化学選択的結合反応の非限定例は、以下の化学種間の反応であり、これらの化学種は得られる三量体構造内部に内包された個々のキメラペプチド内部に組込まれる:(i)アルデヒド/ケトンとヒドラジドによるヒドラジンの形成;(ii)ケトンとアミノキシ基によるオキシムの形成;(iii)ケトンとチオセミカルバジドによるチオセミカルバゾンの形成;(iv)アルデヒドとβ−アミノチオールによるチアゾリジンの形成;(v)チオカルボキシレートとα−ハロカルボニルによるチオエステルの形成;(vi)チオエステルとN−末端ペプチドシステインによるアミドの形成;(vii)アルキルハライドとチオールによるチオエーテルの形成;および(viii)マレイミドとチオールによるチオエーテルの形成。
【0039】
従って本発明の1つの実施態様は、本文の方法によってポリマー修飾されたキメラペプチドに関する。該ペプチドは共有結合的に安定化されて三量体コイルドコイル構造を形成できる。該キメラペプチドは、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメイン(“N−ペプチド”部分)の全部または一部分にらせん相で融合した三量体コイルドコイルを形成できるα−ヘリカルスカフォールドタンパク質(“スカフォールド”部分)を含むアルファ(“α”)−ヘリカルドメインを含む。PEGを非限定例とする1つ以上の低分子量ポリマーがスカフォールドドメイン配列の内部または極めて近傍に局在する選択されたアミノ酸残基に付着することによって部位特異的に遮蔽される少なくとも1つまたは複数のエピトープはこのスカフォールドドメインに内包されている。前述のように、このポリマー修飾が免疫応答を三量体コイルドコイルのウイルスN−ペプチド部分に集中させる。
【0040】
暫定米国特許出願番号60/576,062および番号60/636,724(前出)に詳細に記載されているように、三量体コイルドコイルコンホメーションを獲得できるα−ヘリカルスカフォールドタンパク質に融合したHIV gp41のNH2−末端ヘプタッドリピートドメイン(N−ヘリックス)の全部または一部分を含むキメラペプチドは当業界で公知である(Eckert and Kim,2001,Proc.Natl.Acad.Sci.USA98:11187−11192)。この普遍構造を有しているペプチドを本文では“キメラN−ペプチド”と呼ぶが、これはHIV−由来ペプチドに限定されない。HIVによって使用される融合メカニズムと同様の融合メカニズム(すなわち、ヘアピン三量体)を利用することが判っているエンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメインを、本発明に使用するキメラN−ペプチドの作製に使用できる。本発明の1つの実施態様では、本文中の方法によってポリマー修飾できホモ三量体もしくはヘテロ三量体コイルドコイル構造内部で共有結合的に安定化される可溶性キメラペプチドはこれらのキメラN−ペプチドの誘導体を表す。本文中に記載のようにしてポリマー修飾できるこのようなキメラN−ペプチドの一例を“CC−キメラN−ペプチド”と呼ぶ。CC−キメラN−ペプチドは以下の成分を含む:(1)三量体コイルドコイルコンホメーションを獲得できるα−ヘリカルスカフォールドタンパク質(“スカフォールド”部分);(2)HIV gp41のN−ヘリックス領域の全部または一部分を非限定例とするエンベロープウイルス膜タンパク質(“N−ペプチド”部分)のNH2−末端ヘプタッドリピートドメイン(“N−ヘリックス”)の全部または一部分;および(3)可撓性を強化するために場合によってはリンカー/スペーサー領域によってコアキメラペプチド配列から隔てられたキメラペプチド配列のNH2−またはCOOH−末端に局在する少なくとも2つのシステイン残基(“システイン部分”)。スカフォールド部分は上記N−ペプチド部分にらせん相で融合し、システイン部分(場合によってはリンカー/スペーサー領域を含む)はキメラペプチドのこのα−ヘリカルコアの外部に局在する。該ペプチドは、可溶性三量体コイルドコイル構造を形成でき、この構造では、3つの等しい(すなわち、ホモ三量体コイルドコイルを形成する)または実質的に類似の(すなわち、ヘテロ三量体コイルドコイルを形成する)キメラペプチド鎖がパラレル配向で物理的に会合している。該領域の免疫原性を減衰させ三量体コイルドコイル構造のN−ペプチド部分に免疫応答を集中させるために、該ドメインの内部または近傍に局在する1つ以上のアミノ酸残基にPEGを非限定例とする1つ以上の低分子量ポリマー部分を付着させることによって選択的に遮蔽されるのはスカフォールドドメインおよびその内部に局在するエピトープである。
【0041】
従って本発明の1つの実施態様は、(a)コイルドコイルの可溶性三量体形態を含むスカフォールド部分と、(b)HIV gp41のN−ペプチドまたはその修飾形態の全部または一部分を含むN−ペプチド部分と、(c)少なくとも2つのシステイン残基を含むシステイン部分とを含むポリマー修飾された可溶性キメラペプチドに関する。(a)のスカフォールド部分は(b)のN−ペプチド部分にらせん相で融合してα−ヘリカルドメインを形成しており、(c)のシステイン部分はα−ヘリカルドメインの外部に局在しており、該スカフォールド部分の内部に局在している1つ以上のエピトープは該スカフォールド部分の内部の少なくとも1つのアミノ酸残基に部位特異的に付着した少なくとも1つの低分子量水溶性ポリマーによって選択的に隠蔽されている。
【0042】
“CCIZN17”は本発明の一部としてポリマー修飾できる(すなわち、スカフォールドドメイン内部のエピトープを選択的に遮蔽できる)1つのCC−キメラN−ペプチドである。CCIZN17のコアα−ヘリカルドメインは、Eckert and Kim,2001(前出)に開示されたキメラN−ペプチド“IZN17”である。IZN17(IKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL:配列1)はEckert and Kim, 2001(前出)によって、HIV gp41のN−ヘリックスドメインの一部分の非凝集性三量体コイルドコイルが形成できるように設計された。このキメラペプチドは、HIV gp 41(“N17”N−ペプチドドメイン;配列32;上記の下線部分)のN−ヘリックスの一部分のNH2−末端にらせん相で融合した三量体コイルドコイル(“IZ”スカフォールドドメイン;配列14;上記のイタリック体)を形成できる可溶性α−ヘリカルドメインから成り、長さ41アミノ酸の連続コイルドコイルを創製する。IZドメインはSuzukiら(1998,Protein Eng.11:1051−1055)によって記載された設計に基づく修飾されたイソロイシンジッパーであり、溶液中ではヘリカルで三量体である。N17ドメインは、gp41エクトドメインのタンパク質分析によって同定されたHIV gp41のN−ヘリックス領域に由来の36アミノ酸ペプチド(“N36”)の先端欠失部分である。暫定米国特許出願番号60/576,062および番号60/636,724(前出)に詳細に記載されているように、CCIZN17はIZN17のNH2−末端にアミノ酸配列Cys−Cys−Gly−Gly(配列:10)を付加することによって作製した。従ってCCIZN17は以下のアミノ酸配列から構成されている:CCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL(配列:2)。CCIZN17を作製するためにIZN17に付加したシステイン残基は、このCC−キメラN−ペプチドに共有結合安定化三量体コイルドコイル形成能力を与える(ジスルフィド結合安定化CCIZN17由来コイルドコイルについては図1参照;チオエーテル結合安定化CCIZN17−由来コイルドコイルについては図8参照)。CCIZN17中の付加グリシン残基はキメラIZN17ペプチドのα−ヘリカルコアからシステイン残基を隔てるスペーサー領域を表し、該システイン残基にジスルフィドまたは化学選択的(例えば、チオエーテル)結合を形成するためにコンホメーション的により大きい自由を与える。3つのCCIZN17キメラペプチド間の共有ジスルフィド結合の存在は共有結合的に安定化された三量体コイルドコイル(CCIZN17)3を生じる(図1参照)。このコイルドコイルではは、三量体の存続がモノマー鎖の濃度にもはや依存しないので出発IZN17三量体コイルドコイルよりも安定である(参照:暫定米国特許出願番号60/576,062および番号60/636,724(前出))。
【0043】
キメラN−ペプチド(例えばIZN17)はまた、共有結合的、化学選択的反応に参加できる求電子部分を組込むように誘導体化することもでき、“誘導体化キメラN−ペプチド”と呼ばれる。従って本発明の別の実施態様は、(例えば、スカフォールドドメイン内部のエピトープを選択的に遮蔽するために)本文の方法によってポリマー修飾されて三量体コイルドコイル構造内部で共有結合的に安定化できる誘導体化キメラN−ペプチドに関する。証明として、化学選択的反応を介して共有結合的に安定化されている三量体コイルドコイル構造は、単一のCC−キメラN−ペプチド(例えばCCCIZN17;配列:76)および2つの誘導体化キメラN−ペプチド(例えばブロモアセチル−GGGIZN17;配列:77)を含み、後者のおのおのは、等しいかまたは実質的に等しいα−コアドメイン(例えばIZN17;配列:1)を有しており、該キメラペプチド間で共有チオエーテル結合が生じる。これらの3つのキメラペプチドのおのおののスカフォールド部分内部のエピトープは、該スカフォールドドメイン内部の1つ以上のアミノ酸に低分子量ポリマー部分(例えばSMW PEG)を付着させることによって選択的に遮蔽できる。“NH2−C(Fm)(チオIZN17)3”と命名された共有結合安定化三量体コイルドコイルは、本発明の一部として記載した方法によってポリマー修飾され得るチオエーテル結合安定化コイルドコイルの一例を表す(図8、参照:暫定米国特許出願番号60/576,062および番号60/636,724(前出))。NH2−C(Fm)(チオIZN17)3は1つのCC−キメラN−ペプチドすなわちCCCIZN17ペプチド(配列:76)および2つの誘導体化キメラN−ペプチドすなわちブロモアセチル−GGGIZN17(配列:77)から成り、誘導体化キメラN−ペプチドのNH2−末端に局在するブロモアセチル部分のおのおのはCC−キメラN−ペプチドの“システイン部分”に局在するシステイン残基と共にチオエーテル結合を形成する。この特定のコイルドコイル中で、NH2−末端システイン残基は最初はFm基によって保護され、2つの内部システイン残基をブロモ部分とのチオエーテル結合形成に参加させる。誘導体化キメラN−ペプチドすなわちブロモアセチル−GGGIZN17(配列:77)で証明されるように、誘導体化キメラN−ペプチドの求電子部分は、キメラペプチドのコイルドコイル形成と得られたコイルドコイルを安定化する共有結合形成との間の干渉を最小にするために可撓性リンカー領域(たとえば、2つ以上の連続したグリシン残基)によってキメラN−ペプチドのα−ヘリカル部分から隔てられる。
【0044】
さらに、IQN17(RMKQIEDKIEEIESKQKKIENEIARIKKLLQLTVWGIKQLQARIL:配列:11)はIZN17と同様の設計図をもつキメラN−ペプチドである。しかしながらN17ドメイン(下線部分)は酵母転写アクチベーターGCN4に由来の代替的三量体コイルドコイルモチーフにらせん相で融合している(“IQ”ドメイン;配列:17;上記イタリック体)。コアIQN17ペプチドを含む上記同様の共有結合安定化三量体コイルドコイルを作製し本発明の一部としてポリマー修飾できる。
【0045】
本文中で“スカフォールド部分”と互換的に使用され本発明の一部として選択的に遮蔽されるCC−および誘導体化−キメラN−ペプチドの“スカフォールドドメイン”は、非−HIVアミノ酸残基を含んでおり、コイルドコイルの可溶性三量体形態を表す。スカフォールドドメインは上述のN−ペプチド領域のNH2−末端またはCOOH−末端に融合できるか、または、該ドメインがN−ペプチド領域のNH2−およびCOOH−末端の双方に局在するように分割できる。一部分がN−ペプチド部分の両端に局在するようにスカフォールドドメインが分割されているとき、この分割されたスカフォールドドメインと分割部分の一方または双方に内包されているエピトープとを適正に遮蔽するために、少なくとも1つのポリマー部分が分割スカフォールド領域の各部分(すなわち、NH2−末端部分とCOOH−末端部分)に存在するアミノ酸残基に付着するであろう。コイルドコイルタンパク質は文字“a”から“g”によって表されるアミノ酸の特徴的ヘプタッドリピートから構成され、ヘプタッドリピートの第一および第四の位置すなわち“a”および“d”の位置はそれぞれコイルドコイルの相互作用鎖の内部を形成し一般に疎水性である。本文に記載のポリマー修飾三量体コイルドコイル構造のスカフォールドドメインを表すコイルドコイルモチーフは、以下を非限定例とする様々なソースから単離できる:Suzukiらによって開示されたイソロイシンジッパーモチーフ(1998(前出);以後の本文中では“Suzuki−IZ”;YGGIEKKIEAIEKKIEAIEKKIEAIEKKIEA;配列:12)およびEckertらによって開示されたGCN4−pIQIコイルドコイルモチーフ、(1998,J.Mol.Biol.284:859−865および国際出願PCT/US01/29637,国際公開WO02/24735;RMKQIEDKIEEILSKQYHIENEIARIKKLIGER(配列:13))およびそれらの先端欠失および/または修飾形態。Suzuki−IZまたはGCN4−pIQIコイルドコイルモチーフは、1つのアミノ酸残基の付加、置換、修飾および/または欠失によって改変できる。例えば、該スカフォールドタンパク質内部のアミノ酸残基を該特定場所のSMWポリマーの付着を容易にするために修飾してもよい。スカフォールドドメインは短縮、修飾またはその双方が可能であるが、得られるCC−または誘導体化−キメラN−ペプチドがN−ペプチドコイルドコイル領域を適正に提示する能力を維持する(すなわち、N−ヘリックス三量体コイルドコイルの安定で忠実なミメティクスを生成する能力を維持する)ことが重要である。これは、暫定米国特許出願番号60/576,062および番号60/636,724(前出)に開示された実験によって判断できる。先端欠失および/または修飾されたSuzuki−IZ−様およびGCN4−pIQI−様スカフォールドドメインの様々な多数の実施例が、暫定米国特許出願番号60/576,062および番号60/636,724(前出)に開示されている。これらの文献のおのおのは参照によって本発明に含まれるものとする。Suzuki−IZ−様およびGCN4−pIQI−様スカフォールドタンパク質の例を表1に示す。しかしながらこの記述は表1に示したCC−または誘導体化−キメラN−ペプチドに内包されたスカフォールドタンパク質の選択的な遮蔽方法に本発明を限定するものではない。
【0046】
【表1】
【0047】
本発明の1つの実施態様では、本文中に記載のCC−および誘導体化−キメラN−ペプチドのスカフォールドドメインは、免疫原性担体またはアフィニティ樹脂へのそれらの接合および/または得られたコイルドコイル構造の接合を容易にするために特異的に修飾されてもよい。例えば、アミノ酸配列IEKKIEEIEKKIEEIEKKIEEIEK(配列:32;“a”位置は下線部分)から成る“EZ”スカフォールドは、暫定米国特許出願番号60/576,062および番号60/636,724(前出)に記載の共有結合安定化三量体コイルドコイルと免疫原性担体であるNeisseria meningitidis外膜プロテオソーム複合体(“OMPC”)粒子との接合を容易にするために設計した。担体へのペプチドの共有結合付加(すなわち、担体−ペプチド接合体)は接合体の不可逆的沈殿を生じることが多いが、OMPC−ペプチド接合体の場合も同様である。これを防止するために、1モルのOMPCに組込むペプチドの最大モル数(通常は、ペプチド“負荷”と呼ぶ)を慎重にコントロールしなければならない。何らかの配列のペプチドをOMPCに直接接合し、ペプチドエピトープに対して十分な免疫応答を得るために必要な閾値以上のペプチド負荷を確保できる戦略が最近開発された(参照:Bianchiら,暫定米国特許出願番号60/530,867“A Method to Make a Peptide−Carrier Conjugate with a High Immunogenicity,”出願日2003.12.18)。該方法はペプチド配列のpI(等電点)の操作を含み、その値を約3.5−5.0の範囲内で最適に調節する。pIがこの範囲内の値になるように、ペプチドを活性の免疫原性ドメインの外部で修飾し、ペプチドの免疫特性の維持を確保する。例えば、IZN17(配列:1)は10.7という極めて高いpIを有しているが、その理由は恐らくIZスカフォールド内部に多数の塩基性アミノ酸残基が存在するからであろう。EZスカフォールドドメインは、スカフォールドを含むキメラペプチドやそれらに内包されたコイルドコイル構造がOMPCとの接合を容易に行うことができるように、もっと酸性に設計した。IZスカフォールドを作製するために使用したSuzuki−IZスカフォールドはSuzukiら,1998(前出)によって設計された(IEKKIEA)n(配列:99)のヘプタッドリピート配列を有している。EZスカフォールド中では、Suzuki−IZのヘプタッドリピートの“c”のアラニン残基がグルタミン酸残基に突然変異することによってこのヘプタッドリピート配列が修飾され、(IEKKIEE)n(配列:100)となる。得られたキメラN−ペプチドEZN17(IEKKIEEIEKKIEEIEKKIEEIEKLLQLTVWGIKQLQARIL;配列:98)の5.2というpIは効率的なOMPC接合に最も有利な範囲に近い。
【0048】
前述のように、本発明の一部として記載した上記の方法によってポリマー修飾できる様々なキメラペプチドを作製するときには適正なヘリカル構造の維持が極めて重要である。例えば、N17よりも長い(例えば、“N23”または“N36”)HIV N−ペプチド配列セグメントを有しているIZN−キメラペプチドおよびそのCCIZN−誘導体は、IZと長いHIV N−ペプチドとを接合する融合イベント中にα−ヘリカルヘプタッドリピートが確実に維持されるように、1個から数個のアミノ酸で延長された修飾IZドメインを含み得る。IZN23およびIZN36はやはりEckert and Kim,2001(前出)に開示されたキメラN−ペプチドであり、本発明に記載したようにスカフォールドドメイン内部のエピトープを選択的に隠蔽するために三量体コイルドコイル内部で共有結合的に安定化されポリマー修飾され得る。IZN23およびIZN36はそれぞれ以下のアミノ酸配列を有している:IKKEIEAIKKEQEAIKKKIEAIEKEIEAQQHLLQLTVWGIKQLQARIL(配列:23)およびIKKEIEAIKKEQEAIKKKIEAIEKEISGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARIL(配列:24)。IZN23およびIZN36のスカフォールドドメインはイタリック体で示し、ペプチドの“a”位置は下線を付けて示す。IZN17およびそのCCIZN−誘導体であるCCIZN17の作製に使用したIZスカフォールドに比較すると、IZN23およびIZN36のスカフォールドドメインは、安定なα−ヘリカルコンホメーションの生成を容易にする適正な“a”−“g”スペーシングを維持するために1つ(IZN23)または2つの(IZN36)アミノ酸残基で延長されている。このようにしてスカフォールドドメインを延長するために選択されたアミノ酸は隣接ヘリックス間で静電相互作用し得る(Suzukiら,1998,(前出))。
【0049】
本発明は、既刊文献(参照:暫定米国特許出願番号60/576,062および番号60/636,724(前出))に詳細に記載された共有結合安定化三量体コイルドコイルのコア構造を有しており、タンパク質構造内部の1つ以上のエピトープを隠蔽しその結果として当該構築物に対して生じる免疫応答を非遮蔽領域に集中および/または集結させるために、SMW PEG部分を非限定例とする1つ以上の低分子量ポリマーの選択的付着によって修飾されたワクチン構築物に関する。上述のように、本発明の1つの実施態様では、該ワクチン構築物が3つのキメラペプチドを含み、おのおのはHIV gp41エクトドメインのN−ヘリックスヘプタッド領域の全部または一部分(“N−ペプチド”部分)にらせん相で融合した可溶性スカフォールドタンパク質を含む。HIV gp41エクトドメインは、約169アミノ酸残基(HIV−1 gp160エンベロープタンパク質中の位置に準拠して残基512−681)を表す。エクトドメインのNH2−およびCOOH−末端部分の内部に局在する2つの4−3ヘプタッドリピート領域はそれぞれN−およびC−ヘリックス領域と呼ばれるα−ヘリックスを形成すると予測される。N−ヘリックスおよびC−ヘリックスはそれぞれgp160のほぼ541−592位および623−663位のアミノ酸に局在する(参照:たとえば,Caffreyら,1998,EMBO J.17:4572−4584)。gp41エクトドメインのコア融合−活性構造(すなわち、“ヘアピン三量体”)は、それぞれN−ヘリックスおよびC−ヘリックス領域に局在する2つの相互作用性ペプチド、N36(SGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARIL;配列:29)およびC34(WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL;配列:30)の三量体から成る(参照:Chanら,1997,Cell 89:263−273;および、米国特許番号6,506,554)。N36は、HIV−1 gp160の位置に準拠してアミノ酸残基546−581(両端のアミノ酸を含む)を含む。他のグループは、Chanらによって開示されたものよりもやや大きい安定な6−ヘリックス束を形成するHIV−1 gp41の結晶化ヘリカルドメインを有している。例えば、Weissenhornら(1997,Nature 387:426−430)は、N36配列のNH2−末端の5つの追加アミノ酸残基とCOOH−末端の7つの追加アミノ酸残基とを含むN36よりも長いN−ペプチド領域を同定した:ARQLLSGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARILAVERYLK(配列:31)。従って、本発明によってポリマー修飾され得るCC−および誘導体化−キメラN−ペプチドのN−ペプチド領域は、上記に開示されたN36領域の全部または一部分に必ずしも限定されない。従って1つの実施態様では、そのスカフォールドドメインが本文中に記載のようにポリマー修飾されたCC−および誘導体化−キメラN−ペプチドは、N36のCOOH−末端側の半分に局在しHIV−1 gp160配列の残基565−581に対応する少なくとも17のアミノ酸(“N17”)(LLQLTVWGIKQLQARIL;配列:32)を含み得る。別の実施態様では、N17よりも長いまたは短いN−ペプチド領域のセグメントは上述のようなポリマー修飾スカフォールドドメインに融合し得る(例えば、“N−23”−IEAQQHLLQLTVWGIKQLQARIL;配列:33)。N−ペプチドドメインの延長は、N17内部で同定された推定免疫原性エッジ効果を克服するために有用であろう(参照:暫定米国特許出願番号60/576,062および番号60/636,724(前出))。ヘアピン三量体構造はまた、疎水性ポケットと呼ばれる深い空洞を備えている。1つのこのような空洞はN36α−ヘリックスの各溝の底部(すなわち、COOH−末端側の半分)に局在し、並列したC34ヘリックスのノブ状突起で充填され、ボール−ソケットアレンジメントを形成する(参照:Caoら,1993,J.Virol.67:2747−2755)。従って、CC−および誘導体化−キメラN−ペプチドは、N−ヘリックスα−ヘリカルドメインのCOOH−末端側の半分に存在する中央N−ヘリッスクコイルドコイルの疎水性ポケットまたは空洞を形成するアミノ酸残基を含むgp41 N−ヘリックスの一部分を含み得る。
【0050】
本文中に記載したワクチン構築物のHIV N−ペプチド領域はまた、野生型アミノ酸配列に比べて修飾されてもよい。例えば、N17のCOOH−末端内部に局在する免疫優性非中和エピトープはIZN17ペプチドのアラニン走査によって同定された(暫定米国特許出願番号60/576,062および番号60/636,724)。この望ましくない免疫優性応答は、非中和エピトープの原因であると考えられるN17の1つ以上のアミノ酸残基(ロイシン−581,アルギニン−579,グルタミン−577および/またはグルタミン−575)を突然変異させることによって克服できる。一例では、免疫優性エピトープの原因であると考えられる4つのアミノ酸残基のおのおのがアラニン残基で置換され、アミノ酸配列:LLQLTVWGIKALAAAI(配列:34)から成る“N17Ala4”が生じるようにN17を修飾し得る。あるいは、有益でない抗原応答を排除するために該非中和免疫優性領域の先端を欠失させるかまたは完全に除去してもよい(例えば“N13”−LLQLTVWGIKQLQ;配列:35)。本発明のキメラペプチドのN−ペプチド部分はまた、ペプチド全体がいっそう安定するように修飾することもできる。例えば、N−ペプチドドメインは、配列中により安定性のイソロイシン残基を組込むことによって修飾できる(例えば“N17Ile”−LIQLIVWGIKQIQARIL;配列:36)。
【0051】
上述のように、本文中の方法によってポリマー修飾された三量体コイルドコイルワクチン構築物の好ましい実施態様は、α−ヘリカル安定性スカフォールドドメインにらせん相で融合したHIV gp41 N−ヘリックス領域の一部分を有しているHIV融合中間体のミメティクスとなる。HIV gp41のこのN−ヘリックス部分は、HIV−1、HIV−2、別のHIV菌株または別のレンチウイルス種の菌株(例えば、サル免疫不全ウイルス(SIV)、ネコ免疫不全ウイルス(FTV)またはビスナウイルス)から単離できる。このために同様のHIV菌株および/または他の種の免疫不全ウイルス中の対応するN−ペプチド配列を同定することは容易であり、当業界で公知である。例えば、HIV−1 gp41のN36 N−ペプチド配列に位置合せする他のレンチウイルスNH2−末端α−ヘリカルドメインのアミノ酸配列を表2に示す。さらに、他のエンベロープウイルスの膜−融合タンパク質中のα−ヘリカルコイルドコイルドメインが同定された(参照:Singhら,1999(前出))。
【0052】
【表2】
【0053】
本文中で定義したキメラN−ペプチド(例えばIZN17)の作製については詳細に上述した。暫定米国特許出願番号60/576,062および番号60/636,724(前出)にはもっと詳細に記載されている。該キメラN−ペプチドは、安定なジスルフィドおよび/またはチオエーテル結合をそれぞれ形成する能力を該ペプチドに与えて共有結合安定化三量体コイルドコイル構造を得るために、コアキメラペプチド配列のNH2−またはCOOH−末端に少なくとも2つのシステイン残基(すなわち“CC−キメラN−ペプチド”)または求電子部分(すなわち“誘導体化−キメラN−ペプチド”)を付加することによって修飾できる。1つの実施態様では、少なくとも2つのシステインアミノ酸残基がキメラN−ペプチドのコアα−ヘリカルドメインの外側に付加されて、酸化性条件下で隣接のCC−キメラN−ペプチドと共にジスルフィドブリッドを形成できる“CC−キメラN−ペプチド”を生成する。付加システイン残基をヘリカル外側に配置するのは、該ペプチドのコイルドコイルコンホメーションと安定性ジスルフィド結合との干渉を最小にするためである。並列配向の3つのCC−キメラN−ペプチドが物理的に会合すると、個々のポリペプチドモノマーのα−ヘリカル構造の結果としてシステイン残基が緊密に接近する。酸化性条件下でこれらの並列システイン残基は3つの鎖間で自発的にジスルフィドブリッジを形成する。別の実施態様では、CC−キメラN−ペプチドの操作されたシステインアミノ酸残基に存在するチオール−反応性官能基が隣接の誘導体化N−ペプチドの求電子部分(たとえば、アルキルハライド部分またはMichael受容体)と共に共有結合的化学選択的結合を形成できる。該求電子部分は、キメラN−ペプチドの末端およびそのα−ヘリカルドメインの外側に付加される。これらのシステイン残基または求電子部分は、コアキメラN−ペプチドのNH2−末端に付加されてもCOOH−末端に付加されてもよい。CC−および誘導体化−キメラN−ペプチドの付加システイン残基または求電子部分はそれぞれ、リンカーまたはスペーサー領域によってキメラペプチドのα−ヘリカル部分から隔てられる。スペーサー領域はキメラN−ペプチドのヘリカル構造を破壊し、末端システイン残基または求電子部分が共有結合形成に参加するためにコンホメーション的により大きい自由を与える。スペーサー/リンカー領域は、α−ヘリカル二次構造を破壊する能力をもつことが知られた短いアミノ酸配列(例えば、グリシンまたはプロリン残基;または、単一D−アミノ酸、アミノ酸の右旋性異性体)を含み得る。α−ヘリカル構造の破壊に必要な最少数の残基をスペーサーとして使用するのが好ましい。あるいは、スペーサー/リンカー領域が、化学的または合成リンカー、例えば、アミノペンタン酸、アミノヘキサン酸、または、C−アルファ側鎖のない何らかのアミノ酸を含むこともできる。
【0054】
スカフォールドドメインの内部に局在する1つ以上のアミノ酸残基に1つ以上の低分子量ポリマー分子を付着させることによってポリマー修飾でき、該スカフォールドドメイン内部の1つ以上のエピトープを効果的に遮蔽し、免疫応答をキメラペプチドの非遮蔽部分(例えばHIV部分)に集中させる好ましい共有結合安定化三量体コイルドコイルは(CCIZN17)3(配列:2)3である。図1参照。このコイルドコイル構造は、以下のアミノ酸配列をもつ3つの等しいCCIZN17キメラペプチドから成る:CCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL(配列:2)。前述のようにCCIZN17は、コアキメラペプチド配列“IZN17”がN17(配列:32)のNH2−末端に融合したIZスカフォールドドメイン(上記配列の下線部分)から構成されているCC−キメラN−ペプチドを表す。CCIZN17はさらに、コアキメラペプチドのNH2−末端に付加された安定なCys−Cys−Gly−Gly(配列:10)を含む。2つの連続システイン残基は、ペプチドの酸化によって形成される緊密に会合したCC−キメラN−ペプチドの並列システイン残基と共にジスルフィド結合形成に参加する。2つの連続グリシン残基は、システイン残基をコアキメラペプチド配列のα−ヘリカルドメインから隔てるスペーサー領域を表す。グリシンスペーサー領域は、システイン残基がコアペプチド配列のヘリカル二次構造に巻き込まれないことを確保し、この遊離システイン残基がジスルフィド結合形成に参加することを援助する。
【0055】
従って、本発明の1つの実施態様は、(CCIZN17)3のコア構造を有している共有結合安定化三量体コイルドコイルである。該コイルドコイル構造のスカフォールドドメインおよび該ドメインに内包されたエピトープは、該ドメインの内部または近傍に局在する1つ以上のアミノ酸残基に1つ以上の低分子量水溶性ポリマーを共有結合付着させた結果として選択的に遮蔽されている。後出の実施例1に詳細に記載する代表的ワクチン構築物では、スカフォールドドメイン内部に局在する2つのリシンアミノ酸残基が、市販のPEG236部分によってPEG付加され、本文中で“CCIPN17”と呼ぶペプチドが作製される。CCIPN17はCCIZN17と同じアミノ酸配列(CCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL;配列:2)を有している。しかしながら上述のように、スカフォールドドメイン(イタリック体)の内部に局在する2つのリシン残基(下線部分)はPEG236によって共有結合的に修飾されている(図2参照)。従って、得られる(CCIPN17)3ワクチン構築物のスカフォールドドメインは、各ペプチドモノマーに2つずつ存在する6つのPEG236部分によって遮蔽されている。HIV gp41融合中間体のミメティクスを表す低分子量ポリマー修飾(PEG修飾を非限定例とする)されたコイルドコイル構造は、本文中に記載の種々のキメラペプチドおよび暫定米国特許出願番号60/576,062および番号60/636,724(前出)に記載のペプチドを使用して同様に作製し得る。
【0056】
ペプチド間に形成されたジスルフィド結合を介して共有結合安定化される3つのCC−キメラN−ペプチドをおのおのが含み本発明の一部として記載された方法によってポリマー修飾できる三量体コイルドコイル構造の例を表3に示す。表3の共有結合安定化三量体コイルドコイルの記述もまた、本発明方法がこれらの例に含まれたスカフォールドドメインの選択的遮蔽方法に限定されることを意味しない。
【0057】
【表3】
【0058】
上述のように、表3の共有結合安定化三量体コイルドコイルのおのおのはコアペプチド構造のα−ヘリカルドメイン外部に局在するシステイン残基間のジスルフィド結合の形成によって共有結合的に安定化されている3つのCC−キメラN−ペプチドから成る。あるいは、前述のように、本発明によってポリマー修飾できる三量体コイルドコイルワクチン構造は、化学選択的結合の形成を介して共有結合的に安定化できる。例えば、2つの誘導体化キメラN−ペプチドの求電子部分と1つのCC−キメラN−ペプチドの2つの付加システインアミノ酸残基のおのおののチオール官能基との間で2つの化学選択的結合が形成されて2つのチオエーテル共有結合が形成される。例えば、暫定米国特許出願番号60/576,062および番号60/636,724(前出)に詳細に記載されているNH2−C(Fm)(チオールIZN17)3と命名された三量体コイルドコイルミメティクスを本発明によってポリマー修飾できる。この三量体コイルドコイルは、以下のペプチドモノマーから成る:
1)NH2−C(Fm)Ttds−CCIZN17(表4のペプチド1):可撓性スペーサー(−GG−;他の可撓性スペーサーも好適)を介してスカフォールドドメインから隔てられた2つのシステイン残基と以後の接合用の保護チオール基(フルオロメトキシ(Fm)保護基を使用するが他の保護基も適当)をもつ第三のシステイン残基とを有するペプチド:
2)ブロモアセチル−GGGIZN17(表4のペプチド2):可撓性リンカー(−GGG−;2つの誘導体化キメラN−ペプチド鎖と1つのCC−キメラN−ペプチドとの間の2つのチオエーテル結合の形成に適合性であるならば他の可撓性スペーサーも適当である)を介してスカフォールドドメインから隔てられたブロモアセチル部分を有するペプチド。
【0059】
2つのペプチド前駆体NH2−C(Fm)Ttds−CCIZN17(表4のペプチド1)およびBr−アセチル−GGGIZN17(表4のペプチド2)をそれぞれ約1:2の濃度比でインキュベートして1つのCC−キメラN−ペプチド(ペプチド1)と2つの誘導体化N−ペプチド(ペプチド2)とを有する三量体コイルドコイルを作製できる。化学選択的結合反応は、2つのチオエーテル結合の形成を伴って進行し、この反応で2つのBr−アセチル−GGGIZN17ペプチドの各ブロモ誘導体がNH2−C(Fm)Ttds−CCIZN17ペプチドの2つの非保護システイン残基の1つと反応する(図8参照)。共有結合安定化三量体コイルドコイルの形成後、該三量体コイルドコイルの(例えば、精製樹脂またはOMPCへの)接合を容易にするために保護基を除去できる。
【0060】
本発明の方法によってポリマー修飾できる化学選択的結合の形成を介して共有結合安定化三量体コイルドコイルを作製するために使用し得る追加のペプチドモノマーを単なる例として表4に示す。例えば、“ac−C(チオIZN17)3”と命名された安定化コイルドコイルは、1つのac−C(Acm)Ttds−CCIZN17ペプチド(表4のペプチド3)と2つのBr−アセチル−GGGIZN17ペプチド(表4のペプチド2)とを含むように合成し得る。得られる三量体コイルドコイルはNH2−末端システイン残基の保護基としてAcm(アセタミドメチル基)を有しており、これは以後の接合を容易にするために除去できる。
【0061】
【表4】
【0062】
PEGおよび類縁ポリマーのようなポリマー部分をタンパク質に見出される反応性基に付着させるために様々な手段が使用されてきた。参照:たとえば,米国特許番号4,179,337,Davisらに許諾、および、米国特許番号4,002,531,Royerに許諾。総説についてはAbuchowskiら,Enzymes as Drugs,Eds.J.S.Holcerberg and J.Roberts,1998,pp.367−383およびZalipsky,S,1995,Bioconjugate Chem.6:150−165を参照するとよい。タンパク質を修飾するためのPEGおよび他のポリマーの使用はまた、以下の文献でも検討されている:Cheng,T.L.ら,1999,Bioconjugate Chem.10:520−528;Belcheva,N.ら,1999,Bioconjugate Chem.10:932−937;Bettinger,T.ら,Bioconjugate Chem.9:842−846;Huang,S.−Y.ら,1998,Bioconjugate Chem.9:612−617;Schwarz,J.B.ら,1999,J.Amer.Chem.Soc.121:2662−2673;Reuter,J.D.ら,1999,Bioconjugate Chem.10:271−278;およびChan,T.−H.ら,1997,J.Org.Chem.62:3500−3504。前述したように、タンパク質中の典型的なポリマー付着部位はリシン残基またはNH2−末端に存在する一次アミノ基、システイン側鎖に存在するチオール基、グルタメートもしくはアスパルテート残基、または、COOH−末端に存在するカルボキシル基、などを含む。(例えば、循環半減期を改善するため、タンパク質分解および免疫原性を抑制するために)PEGのような水溶性ポリマーを対象ポリペプチドに付着させる場合、ポリマー部分をユーザーの定めた精度でコントロール的に付着させるのが難しいならば、一様でない結果が得られることになる。過去には、付着の推計的特性および水溶性ポリマーのヘテロ分散的特性を理由としてポリマー−タンパク質コンジュゲートの精製および分析キャラクタリゼーションが困難であった。
【0063】
本文中に記載のようなSMWポリマー修飾ポリペプチドの好ましい作製方法では、1つ以上のSMWポリマーを対象タンパク質内部の部位特異的場所に付着させる。これは、本発明のように所期の生物応答(例えば、免疫応答)を誘導または促進できるように択一的部位を可用状態に維持しながらタンパク質内部の特定の(1つ以上の)生物活性部位を付着ポリマーによって遮蔽する予定であるときに特に重要である。ポリマーがタンパク質に部位特異的に結合した低分子量ポリマー誘導体化タンパク質(例えば、SMW PEG付加タンパク質)の調製は、付着鎖の個数および場所を完全にコントロールできる種々の方法によって達成される。例えば、ポリマーのランダムな付着を減少させる1つの試みでは、天然のリシンまたはシステインアミノ酸残基を除去し、同時に所望のポリマー付着部位にリシン/システインアミノ酸を再度付加したタンパク質を作製した(参照:たとえば,米国特許番号4,904,584)。あるいは、後出の実施例1に例示したワクチン構築物(CCIPN17)3を固相ペプチド合成(“SPPS”)によって作製した。従って、本発明方法によって修飾すべきペプチドおよび/またはポリペプチドの合成は、標準全自動ペプチドシンセサイザーを使用する段階的標準Bocおよび/またはFmoc固相ペプチド合成で行うか、または、販売業者に注文し購入した標準プロトコルに従って手動操作で行う。参照:たとえば,Synthetic Peptides,A User’s Guide,Ed.G.A.,New York.W.H.Freeman & Company,1992;Principles of Peptide Synthesis,2nd ed.,Ed.M.Bodanszky, Springer−Verlag,1993;The Practice of Peptide Synthesis,2nd ed.,Eds.M.Bodanszlky and A.Bodanszky,Springer−Verlag,1994;Protecting Groups,Ed.P.J.Kocienski,Stuttgart,Germany:Georg Thieme Verlag,1994;およびFmoc Solid Phase Peptide Synthesis,A Practical Approach,Eds.W.C.Chan and P.D.White,Oxford University Press,2000。
【0064】
天然型化学結合(Dawson,P.E.and S.B.Kent,2000,Ann.Rev.Biochem.69:923−60)のような化学選択的方法を使用すると、全化学合成によってもっと大きいポリマー修飾タンパク質も容易に得られる。その優れた代表例は、Kochendoerfer,G.G.ら(2003,Science 299:884−87)によって示されており、Kochendoerfer,G.G.ら,米国非暫定出願番号10/332,385,US公開番号US2004/0115774にいっそう詳細に記載されている。該文献では著者/発明者らが2つの部位特異的HWM分枝状PEG分子を含む166アミノ酸のタンパク質を累積収率25−40%で作製した。この文献に記載されたHMW分枝状PEGの付着に関する化学理論はSMW PEG部分の導入に容易に応用できる。しかしまた、利用可能な多くの他の選択肢も存在しており(Harris,J.M.and R.B.Chess,2003,Nat.Rev.Drug Discov.2:214−221)、それらは当業者に公知である。Kochendoerferら,2003(前出)で作製されたタンパク質SEPのサイズは、HIVエンベロープ糖タンパク質のgp120サブユニットのサイズと同等であり、糖残基の選択的付加に基づいて該タンパク質のエピトープを隠蔽する戦略が提案されている。従って、特定の免疫反応性を減退させるかまたは完全に除去して別の免疫反応性の利益になるようにSMW PEGで選択的に誘導体化されるgp120に基づくHIVワクチンが完全に化学的な合成方法で得られることは明らかである。
【0065】
本文中に記載したようなポリマー修飾タンパク質の代替的作製方法では、組換えDNA方法によって産生された適当な前駆体タンパク質にSMWポリマー(例えばPEG)を化学選択的に付着させる。より具体的に、Bertozziおよび共同研究者らは、組換えタンパク質内部の任意の位置にアジド基を組込む普遍的に応用できる戦略を開発した(Kiick,K.L.ら,2002,Proc.Natl.Acad.Sci.U.S.A.99:19−24)。該方法は、細胞の先天的翻訳装置によってプロセスされる非天然アミノ酸の利用から成る。アジドホモアラニン(メチオニン代用物)を大腸菌のメチオニル−tRNAシンテターゼによって活性化し、メチオニン欠失細菌培養物中で発現されたタンパク質のメチオニンを置換する。アジドはStaudinger結合と呼ばれる極めて化学選択的な反応にホスファンと共に参加して、最終的にタンパク質とホスファン付着部分とを接合する安定なアミド結合を形成する(参照:Kohn,M.and R.Breinbauer,2004,Angew Chem.Int.Ed.Engl.43:3106−16;Saxon,E.ら,2000,Org.Lett.2:2141−3;Saxon,E.and CR.Bertozzi,2000,Science 287:2007−10)。例えば、CCIPN17(実施例1参照)に使用したSMW PEGすなわちPEG236のStaudinger結合は、対応するホスフィノチオエステル1と共に行うことができ、後者は以下の反応スキーム1に従ってアジドホモアラニンに結合するであろう:
【0066】
【化1】
【0067】
もっと長鎖のPEGのホスフィノチオエステルの合成が記載されている(Soellner,M.B.ら,2003,J.Am.Chem.Soc.125:11790−1)。アジドホモアラニンを含有する組換えタンパク質を作製するためのタンパク質サイズの制限は先験的には存在しないので、一次配列内部のいずれかの選択的場所に付着したSMWポリマー鎖によって任意の長さのタンパク質を作製する方法は従来技術に存在している。
【0068】
本発明のポリマー修飾ポリペプチドを設計するとき、先ず、ターゲットタンパク質のアミノ酸配列を、典型的には遺伝子および/またはタンパク質のデータベースのようなデータベースを含む様々なソースのいずれか1つ以上から入手するかまたは例えば新しい対象タンパク質ターゲットのタンパク質同定によって新規に獲得する。このようなデータベースの非限定例は、GeneBank(Bensonら,1998,Nucleic Acids Res.26:1−7;USA National Center for Biotechnology Information,National Library of Medicine,National Institutes of Health,Bethesda,Md.,USA),TIGR Database(The Institute for Genomic Research,Rockville,Md.,USA),Protein Data Bank(Brookhaven National Laboratory,USA)およびExPASy and Swiss−Protein database(Swiss Institute of Bioinformatics,Geneve,Switzerland)を含む。設計戦略に使用されるポリペプチド主鎖は、ターゲット配列(例えば、ポリペプチド鎖の成熟形態)の一部分(例えば、ドメイン)または全部を表すことができる。次に、所与のターゲットタンパク質について所望のポリマー修飾ポリペプチド構造を設計する。この設計戦略は、ポリペプチドの修飾に使用する水溶性ポリマーの選択、ポリペプチド内部の(1つ以上の)ポリマー付着部位、使用する付着戦略を含む。本発明では特に、SMWポリマーの付着によってどの部位を特異的に遮蔽すべき/できるかを同定および査定するために、最初にポリペプチドアミノ酸配列について1つ以上の機能部位(例えば、グリコシレーション部位、免疫原性部位またはプロテアーゼ感受性部位)の存在を調べる。生物活性部位はしばしば、ヘリックスまたはベータ−シート二次構造;典型的にはループおよびターン領域が実質的に存在しない領域を表す。従って、本文中に記載したようにSMWポリマーの付着を介して隠蔽すべき機能部位は、モデリング方法を使用して同定できる。モデリングは、ターゲットタンパク質の所与の核酸またはアミノ酸配列の部分または全部について、対象ポリペプチドに関する二次元構造情報、対象タンパク質に関する三次元構造情報、またはそれらの組合せを比較できるホモロジーモデリングソフトウェアアルゴリズムまたはプログラムの使用のようなバイオコンピューター処理を含む。いくつかのソフトウェアプログラムおよびデータベースが公知でありこの目的に可用である。これらの方法を組込んだソフトウェアプログラムの一例は、“MAXVECTOR”(Oxford Molecular Group)であり、これは多重タンパク質分析ツールを内蔵し、二次構造(Chou−Fasman and Robson−Garnier methods);親水性(Hopp−Woods,Kyte−Doolittle,Goldman−Engelman−Steitz(GES) and von−Heijne methods);疎水性(Fauchere−Pliska,Janin,Manavalan,Sweet−Eisenberg methods);抗原性(Hopp−Woods,Parker,Protrusion Index(Thornton),Welling methods);表面確率(Karplus & Schultz’s methods);可撓性(Janin and Emini methods);両親媒性(Eisenberg method);PROSITE−に基づくサブ配列データベースおよびユーザーの定めたサブ配列;カスタム化配列セット;プロテアーゼ部位とタンパク質分解性消化の予測値、などを予測する。このようにして、ターゲット天然タンパク質またはミメティクスの三次元構造および化学的性質を考慮することはポリマー付着場所の選択に役立つであろう。
【0069】
例えば、タンパク質中の免疫原性部位を予測するために以下の方法を使用できる。タンパク質配列の二次構造を最初に予測し、無秩序領域、特にターンを同定する。この目的にはChou−Fasmanパラメーターが特に役立つ(Chouら,1978,Advanced Enzymology 47:45−148;Qianら,1988,J.Mol.Biol.202:865−884;およびGenetics Computer Group,Inc.の“PEPEPLOT”プログラム)。また、タンパク質配列の疎水性は、典型的には標準方法(たとえば,Fauchere−PliskaまたはHopp−Woodsの疎水性予測法;またはBull−Breese)による6残基の走行平均を使用して走査する。より親水性の領域はターンである領域に特に注意して同定する。同時に、N−結合部位およびO−結合部位を含むグリコシル化部位を同定する。タンパク質中の最も親水性の部位は、それがターンに存在しグリコシル化されていると予測されていないならば免疫原性の確率が高いのでポリマー修飾部位として使用できるであろう。親水性が順次に低くなる親水性領域に同じ基本法則を応用し二次的なポリマー修飾用部位を同定する。
【0070】
また、生物学的および化学的分析も本発明によるポリマー付着が効果的な特異的場所を同定するために使用し得る。例えば、アラニン走査は、免疫原性部位、プロテアーゼ感受性部位またはポリマー修飾に適した他の部位を同定するために使用でき、生物活性に必要な不変残基を同定するために特に有用である。アラニン走査はまた、上記のような他のタンパク質分析ツールと併用することもできる。別の方法は、修飾用水溶性ポリマー分子が各分子のポリペプチド鎖中の異なる部位に局在している合成した対象タンパク質のライブラリーを体系的に作製することである。ポリマー修飾ポリペプチド鎖のライブラリーのフォールデイング後、例えば、フォールドした分子をフォールドしない分子からまたは受容体結合分子を非結合分子から分離するために機能性選択/アッセイを実行できる。次いで機能性分子についてポリマー修飾部位の好ましい場所を決定できる。このような方法では、非干渉性ポリマー修飾部位の同定を容易にするために“protein signature analysis”の原則を利用できる(Muirら,1996,Chem.Biol.3:817−825)。上述のような様々な付着化学が存在するので、水溶性ポリマーとターゲットペプチドまたはポリペプチドとの間に形成される結合は、カーボネート、エステル、ウレタン、オルトエステル、アミド、アミン、オキシム、イミド、ウレア、チオウレア、チオエーテル、チオウレタン、チオエステル、エーテル、チアゾリジン、ヒドラゾン、オキサゾリジンなどから選択された結合を含むことができる。
【0071】
本発明の1つの目的は、特定の疾患または状態(非限定例としてHIV感染/AIDSを含む)に対する患者の免疫防御を誘発したりおよび/または患者体内でこのような状態の原因となる病理発生を抑制するためにヒトまたはヒト以外の哺乳類患者に投与できるワクチンに関する。一般に、本発明のワクチンは、真菌類、原生動物、細菌およびウイルス(例えばインフルエンザウイルス、特にHIV)を非限定例とする多くの病原性生物に対する免疫防御を与えることができる。本発明の1つの実施態様で、ワクチンは、本文中に記載しかつ暫定米国特許出願60/576,062および60/636,724(前出)により詳細に記載された共有結合安定化三量体コイルドコイル構造のポリマー修飾形態を含み、これはHIV gp41を非限定例とするエンベロープウイルスタンパク質の融合中間体のミメティクスを表す。本発明の1つの実施態様では、共有結合安定化コイルドコイルミメティクス内部に含まれた個々のキメラペプチドの1つ以上が、PEGを非限定例とする低分子量水溶性ポリマーによって修飾され、その結果としてコイルドコイルの安定性スカフォールドドメインの全部または一部分、従って該ドメインに内包されている1つ以上のエピトープが免疫由来受容体/細胞と相互作用しないようにポリマーによって遮蔽されている。従って本発明は、本文中に記載のポリマー修飾ワクチン構築物を治療有効量(後記に定義)で投与する哺乳動物の免疫方法または病原性生物に対する防御免疫応答の誘導方法に関する。方法は、単独で存在するかまたは非天然型ポリペプチド成分(例えば、前述のようなスカフォールドコイルドコイル)に結合して(例えば、融合ペプチド/タンパク質として)存在する天然型病原性抗原の全部または一部分のポリマー修飾形態(例えば、1つ以上のSME PEG部分によって修飾)を含むワクチンを哺乳動物に投与する段階を含む。ポリマーの付加は、該ポリマーの存在によって遮蔽された領域を免疫減衰し、免疫応答を抗原の非隠蔽部分に集中させる。従って、本発明の1つの実施態様は、脊椎動物の生体組織、好ましくはヒトまたはヒト以外の商業用もしくは飼育用の獣医学的に重要な哺乳動物のような哺乳類宿主に直接導入されるとヒト免疫不全ウイルス−1(HIV−1)を特異的に認識する細胞性免疫応答を誘導するHIV医薬製品、その製造および使用(本文中に記載)に関する。
【0072】
従って、本発明のポリマー修飾ポリペプチドの好ましい用途は、治療有効量のワクチン形態のポリペプチドを哺乳動物に投与する段階を含む、哺乳動物の疾患状態、例えばHIV感染の治療である。これは、ヒトまたはヒト以外の哺乳類の疾患個体のいかなる治療も包含し、例えば:(i)当該疾患への素因はあるけれども罹患していると診断されたことがない個体の発症予防;(ii)疾患の進行の阻害および/または停止;または、疾患の軽減、すなわち疾患の退行の惹起、を含む。本文中に使用した“治療有効量”という用語は、哺乳動物に投与したとき治療効果を生じるため、例えば哺乳動物のHIV−1感染を阻害するために十分な本発明のポリマー修飾ポリペプチドおよび/または該ポリペプチドを含有するワクチン組成物の量を意味する。“治療有効量”を構成する量は、ポリペプチド、状態または疾患の種類とその重篤度、治療される哺乳動物の体重、年齢、などに従って変更されるが、平均的な当業者は現状の知識およびこの開示に基づいてその量を容易に決定できるであろう。
【0073】
ペプチドのような低分子は免疫原性に乏しく、従って、該分子に対する実体的な免疫応答を誘発するためにより大きい巨大分子(担体)とのペプチド接合体を調製することがしばしば必要である。従って、本発明のポリマー修飾ポリペプチドはさらに免疫原性担体に接合し得る。本文中に記載したコイルドコイル構造の場合、個々のポリマー修飾α−ヘリカルキメラペプチド(例えば、CCIPN17)を最初に免疫原性担体に接合し、次いで、個別に接合した3つのペプチドから成る共有結合安定化ホモ三量体またはヘテロ三量体コイルドコイル構造の形成に導く条件を作用させる。あるいは、本発明のキメラペプチドから成る共有結合安定化ポリマー修飾ホモ三量体またはヘテロ三量体コイルドコイル構造を免疫原性担体にコンジュゲートさせてもよい。通常はヘテロロガスタンパク質から成る担体分子は、コイルドコイルの非隠蔽部分(例えば、HIV部分)に対する免疫応答の誘発および/または増進を援助する。ポリマー修飾ポリペプチドは、非特異的架橋剤、一属性(monogeneric)スペーサーまたは二属性(bigeneric)スペーサーによって免疫原性担体に連結できる。本発明のポリマー修飾ポリペプチドがコンジュゲートできる多くの免疫原性担体分子が当業界で公知である(参照:たとえば,Shodelら,1996,J.Biotechnol.44:91−96;Lang and Korhonen,1997,Behring Inst.Mitt.98:400−409;Brennanら,2001,Mol.Biotechnol.17:15−26;Pumpens and Grens,2201,Intervirology 44:98−114;およびSimpsonら, 1999,Cell Mol.Life Sci.56:47−61)。このような潜在能力をもつ免疫原性分子の非限定例は、Neisseria meningitidis OMPC粒子、キーホールリンペットヘモシアニン(KLH)、ウシ血清アルブミン(BSA)、オボアルブミン(OVA)、チログロブリン(TG)、HBV−コア抗原、HBV−表面抗原や、破傷風毒素、ジフテリア毒素またはロタウイルスVP6のような免疫原性タンパク質、および、p24含有HIVキャプシド粒子である。
【0074】
従って本発明の1つの実施態様では、本文中に記載のポリマー修飾HIV−特異的ペプチドミメティクスをHIV感染の防止または防御(例えば予防または治療)用ワクチンの成分として使用し、HIVに対して免疫活性のHIV−特異的中和抗体を生成させることができる。ワクチンはMPL−Aのような当業界で公知のアジュバントに配合し、ミョウバンまたはリン酸アルミニウムに吸着させる。抗原性接合体はまた、より強力なヘルパーT細胞応答を実現するために、合成の非天然pan HLA DR−結合エピトープ(PADRE)を非限定例とするT細胞ヘルパーエピトープを含み得る(参照:たとえば,Alexanderら,2000,J.Immunol.,164:1625−1633およびdel Guercioら,1997,Vaccine 15:441−448)。本文中に記載のポリマー修飾ポリペプチドの免疫原性接合体は、それらの構成成分パーツ(ポリマー修飾ポリペプチドと担体)を単離、合成および精製し、次いで2つの成分を接合することによって調製し得る。所望ならばその後にコンジュゲート混合物の精製を行ってもよい。ポリマー修飾ポリペプチドと適当な免疫原性担体との抗原性接合体は、構成成分パーツ(すなわち、ポリマー修飾ポリペプチドと担体)間に少なくとも1つの共有結合連結を有しており、典型的には2つ以上のポリマー修飾ポリペプチド分子が担体分子のおのおのに共有結合している。構成成分を別々に調製するならば、その後、非特異的架橋剤、一属性スペーサーまたは二属性スペーサーによって連結できる。非特異的架橋の方法は当業界で公知であり、その非限定例は、グルタルアルデヒドとの反応、スクシニル化担体添加または不添加で行うN−エチル−N’−(3−ジメチルアミノプロピル)カルボジイミドとの反応、グリコシル化置換基のペルヨージド酸化次いでホウ水素化ナトリウムまたはシアノホウ水素化ナトリウムの存在下で行うタンパク質担体の遊離アミノ基への結合、芳香族アミノ基のジアゾ化次いでタンパク質のチロシン側鎖残基への結合、イソシアナートとの反応、または、混合無水物との反応を含む。概要についてはBriandら,1985,J.Imm.Meth.78:59−69を参照するとよい。
【0075】
本文中に記載のキメラペプチドと例えば樹脂または担体との接合を容易にするために、本発明のポリマー修飾ポリペプチド、例えばCCIPN17をさらに修飾して、追加のシステイン残基を含有させてもよい。CCIZN17/CCIPN17の場合、これはペプチドのNH2−末端に1個の追加システインアミノ酸残基を組込むことである(たとえば,“CCCIZN17”−CCCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL;配列:76)。場合によっては、奇数番号の末端システイン残基を可撓性化学リンカー、例えば、Ttds(1−アミノ−4,7,10−トリオキサ−13−トリデカミンコハク酸;−NH−(CH2)3−O−(CH2)2−O−(CH2)2−O−(CH2)3−NH−CO−CH2)2−CO−を用いて残りの連続システイン残基から分離してもよい。暫定米国特許出願60/576,062および60/636,724(前出)参照。奇数番号のシステイン残基は、本文中に記載のようなポリマー修飾CC−および/または誘導体化キメラN−ペプチドを含む共有結合安定化三量体の形成後に、共有結合安定化三量体あたり少なくとも1つの反応性チオール基がマレイミジルのようなチオール−反応性基とさらに反応するために可用であることを確保する。リンカーは、三量体化ドメインとシステイン残基とのジスルフィドブリッジ間に接合/誘導体化に適した可撓性、可溶性および間隔を提供する。追加のシステイン残基は、該システイン残基がジスルフィド結合または化学選択的結合の形成(例えば、チオエーテル結合形成)および/またはポリマー付着に参加することを防止するために保護チオール基を有していてもよい。保護基(非限定例はフルオレニルメトキシ(“Fm”)またはアセタミドメチル(“Acm”)基を含む)は共有結合安定化三量体コイルドコイルの形成後に除去することができ、新しく露出したチオール基が以後の接合反応に可用である。
【0076】
本発明のワクチンは、宿主に投与するための医薬的に有効な配合物に配合し得る。このような配合物は例えば、リン酸塩緩衝生理食塩水(PBS)のような生理的塩類溶液でよい。本発明のワクチン構築物に長期間安定性も与えるような医薬的に許容される配合物の使用が有用であろう。様々な保存条件(例えば、pH、バッファの種類、塩濃度、光照射、高温度、低イオン強度)はペプチドワクチンの安定性を最適にするように配合物中でコントロールし得る。ワクチンは、それぞれのワクチン配合物にアジュバントを添加するかまたは不添加で投与し得る。従って本発明のペプチドワクチンはまた、本発明のワクチン構築物の免疫原性を増強する1種以上のアジュバントと配合し得る。数多くのこのようなアジュバントが当業界で公知である。本発明のワクチンに使用するためのアジュバントの2つの例は、1つ以上の形態のリン酸アルミニウム基剤のアジュバントおよびサポニンアジュバントQS21(サポニンと3D−モノホスホリル脂質A(MPL)の精製形態、リポ多糖(LPS)の無毒誘導体)であり、おのおのが多くの動物モデル体内でワクチンに対して有効な体液性および細胞性応答を示す。熟練の当業者は最適免疫応答を与えるようにペプチドワクチン対アジユバント比を調整し得る。別のアジュバントは、POE−POP−POEブロックコポリマーのようなポリオキシエチレン(POE)およびポリオキシプロピレン(POP)のブロックを含む非イオン性ブロックコポリマーである(Newmanら,1998,Critical Reviews in Therapeutic Drug Carrier Systems 15(2):89−142)。基本構造は、POE−POP−POEブロックコポリマーのようなポリオキシエチレン(POE)およびポリオキシプロピレン(POP)のブロックを含む。Newmanら(同書)は、ある種のPOE−POP−POEブロックコポリマーがインフルエンザタンパク質基剤のワクチンにアジュバントとして有用であろうと開示している。すなわち、約9000ドルトンから約20,000ドルトンの範囲の分子量を有している中央POPブロックとコポリマーの全分子量の約20%までを含むフランキングPOEブロックとを含む高分子量POE−POP−POEブロックコポリマーである(参照:米国再発行特許番号36,665,米国特許番号5,567,859,番号5,691,387,番号5,696,298および番号5,990,241、すべてEmanueleらに許諾、これらのPOE−POP−POEブロックコポリマー関連)。WO96/04932はさらに、界面活性剤特性を有しておりワクチンアジュバントとして生物効果を示す高分子量POE/POPブロックコポリマーを開示している。この文節に引用した上記文献はそれらの記載内容全部が参照によって本発明に含まれるものとする。従って、アジュバント不添加ワクチン投与に比べて本発明のワクチン構築物の免疫応答を増強するために有効なアジュバントの利用は当業者の認識範囲に入っている。
【0077】
本発明のワクチン構築物は、経腸的および非経口的経路のような当業界で公知の手段のいずれかによって投与できる。これらのデリバリー経路は非限定的に、筋肉内注射、腹腔内注射、静脈内注射、吸入または鼻腔内デリバリー、経口デリバリー、舌下投与、皮下投与、経皮投与(transdermal,transcutaneous,percutaneous)、または何らかの形態の粒子衝撃または何らかの可用な無針注入デバイスを含む。ワクチンレシピエントに導入すべきポリマー修飾ポリペプチド構築物の量は、ポリペプチドの免疫原性に従属するであろう。また、本発明のワクチンに対する全体的免疫応答を最適にするようにブースターワクチンを接種することも考察される。
【0078】
本文中で言及したすべての刊行物は本発明に関連して使用する方法および材料を記載および開示する目的で引用した。本発明が従来発明によるこれらの開示に先行する権利がないと是認したと解釈すべきではない。
【0079】
本発明の好ましい実施態様を添付図面と共に記載したが、本発明がこれらの具体的な実施態様に限定されないこと、特許請求の範囲に記載の本発明の範囲または要旨を逸脱することなく様々な変更および修正が当業者に可能であることを理解されたい。従って、以下の実施例は本発明の例示であるが本発明の限定ではない。
【実施例1】
【0080】
望ましくないエピトープを遮蔽するためのSMW PEGの使用:
SMW PEG−誘導体化HIVワクチンの設計
この実施例はHIVワクチンの分野に役立つ本発明の1つの用途を示す。HIV gp41のN−ヘリックスの三量体コイルドコイルのミメティクスを表すサブユニットワクチンとして使用するように操作したペプチドを予め設計した(参照:暫定米国特許出願番号60/576,062および60/636,724(前出))。これらのサブユニットワクチンは、設計した可溶性三量体コイルドコイル(“スカフォールド”領域)と、HIV gp41のN−ヘリックスコイルドコイルの一部分と、鎖間共有結合形成用共有結合安定化部分とを含むキメラペプチドである。このようなミメティクスの一例を図1Aに概略的に示す。これは“(CCIZN17)3”と命名した共有結合安定化三量体コイルドコイルを表す。(CCIZN17)3[配列:2]3内部の3つのキメラCCIZN17ペプチドを共有結合安定化するジスルフィド結合の化学構造図を図1Bに示す。3つのジスルフィド結合がNH2−末端システイン残基のチオール(−SH)基間に局在している。システイン残基のカルボキシ末端に局在する連続アミノ酸は一文字命名法の活字体で示す(図1B)。gp41のプレヘパリン複合体を模倣する三量体コイルドコイルのHIV部分(N−ペプチド部分;例えば(CCIZN17)3中の“N17”)の安定な提示には、スカフォールド領域および鎖間共有結合の双方が必須である。gp41のN17領域によって生成されるコンホメーションエピトープをターゲットする抗体は、N17を内包する三量体コイルドコイルすなわち疎水性ポケット内部に拘束されたとき、単一サイクル感染力アッセイでHIV−1に対する抗ウイルス活性を示す(参照:暫定米国特許出願、出願日2004.6.1)。HIV gp41の三量体コイルドコイルN−ペプチド部分を特異的にターゲットする抗体を増感するこのクラスのペプチドワクチンの使用を容易にするために、本発明の発明者らは、スカフォールドと鎖間共有結合とを含む部分を免疫認識から遮蔽しながらペプチドミメティクスのHIV部分に選択的に集中した免疫応答を誘導する戦略を計画した。
【0081】
CCIZN17(配列:2)の合成−
ペプチドCCIZN17(配列:2)は、Pioneerペプチドシンセサイザー(Applied Biosystems)でFmoc/t−Bu化学を使用して固相合成した。使用した樹脂は、Fmoc−Linker AM−Champion、1%架橋(Biosearch Technologies,Inc.)であり、これは、修飾Rinkリンカーp−[(R,S)−α−[9H−フルオレン−9−イル−メトキシホルムアミド]−2,4−ジメトキシベンジル]−フェノキシ酢酸(Rink,H.,1987,Tetrahedron Lett.28:3787−3789;Bernatowicz,M.S.ら,1989,Tetrahedron Lett.30:4645−4667)によって誘導体化したPEG−PS基材樹脂である。すべてのアシル化反応は樹脂遊離アミノ基の4倍過剰量の活性化アミノ酸で60−120分行った。アミノ酸は、等モル量のHBTU(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)および2倍モル過剰量のDIEA(N,N−ジイソプロピルエチルアミン)で活性化した。側鎖保護基は以下を使用した:グルタミン酸(Glu)およびトレオニン(Thr)にtert−ブチル;システイン(Cys)およびグルタミン(Gln)にトリチル;リシン(Lys)およびトリプトファン(Trp)にtert−ブトキシ−カルボニル;アルギニン(Arg)に2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニル。アセチル化反応は、ペプチド組立の終了後、10倍過剰量の無水酢酸とN,N−ジメチルホルムアミド(DMF)中で反応させることによって行った。合成の終了後、乾燥ペプチド−樹脂を、88%トリフルオロ酢酸(TFA)、5%フェノール、2%トリイソプロピルシランおよび5%水(Sole,N.A.and G.Barany,1992(前出))によって室温で1.5時間処理した。樹脂を濾過し、溶液を低温メチル−t−ブチルエーテルに加えてペプチドを沈殿させた。遠心後、ペプチドペレットを新しい低温メチル−t−ブチルエーテルで洗浄して有機スカベンジャーを除去した。このプロセスを2回繰返した。最終ペレットを乾燥し、H2O、20%アセトニトリルに再懸濁させ、凍結乾燥した。
【0082】
粗ペプチドCCIZN17(配列:2)をToyopearl HW−50S樹脂を詰めた700×26mmカラムで、30%アセトニトリルを含む水、0.1%TFAを溶出剤として使用し、ゲル透過クロマトグラフィー(GPC)によって精製した。典型的な処理では、300mgの粗ペプチドを12mLの溶出剤に溶解し、流速1mL/分でカラムに直接充填した。溶出画分の分析用HPLCは、Beckman HPLCで、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用い、溶出剤B(上記)の以下の勾配:40%−60%のB(20分)−80%(3分)、流速1mL/分を使用して行った。純度に基づいて画分をプールし、セミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用い、溶出剤として(A)水中の0.1%のTFA、(B)アセトニトリル中の0.1%のTFAを使用する逆相HPLCによってさらに精製した。溶出剤Bの以下の勾配を使用した:35%−50%,20分超,流速80mL/分。Micromass LCZ platform質量分析計でエレクトロスプレー質量分析法によって精製ペプチドのキャラクタリゼーションを行った。理論的平均分子量(MW))は5175Daであり、測定MWは5175Daである。
【0083】
CCIZN17から(CCIZN17)3への酸化−
精製ペプチド前駆体(12mg)、CCIZN17(配列:2)を0.1MのHepes,pH7.3(USB Corp.)に1mg/mLの濃度で溶解した。この条件下で、CCIZN17は空気によってゆっくりと酸化して共有結合三量体(CCIZN17)3([配列:2]3)になる。図1B参照。酸化反応は、Waters−Micromass LCZ Platformで、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用い、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用するLC−MS分析によってモニターした。溶出剤Bの以下の勾配を使用した:40%−60%B(20分)−80%(3分)、流速1mL/分。これらのクロマトグラフィー条件で、CCIZN17はtR=13.25’に溶出する。酸化反応は3時間円滑に進行しtR=15’に溶出する主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのCCIZN17ペプチド鎖([CCIZN17]3)を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液(12mL)に24μLのTFAを添加し、溶液をTSKgel Toyopearl HW−50S樹脂を詰めた700×26カラムに充填し、溶出剤としてH2O/アセトニトリル70/30、0.1%TFA、流速1mL/分を使用した。溶出分画をPhenomenex,Jupiter C4カラム(150×4.6mm,5μm)で、溶出剤として:(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用し、HPLCによって分析した。LC−MS Waters−Micromass LCZ Platformで溶出剤Bの以下の勾配を使用した:40%−60%B(20分)−80%(3分)、流速1mL/分。共有結合三量体に対応するプール画分を、ESI−QqToF(Applied Biosystems)、正モード(ES+)、ナノスプレー1μl注入、を用いる質量分析法によってさらに分析した。予想MWは15520.2Daであり、測定MWは15520.1Daである。
【0084】
CCIPN17の合成−
ペプチドCCIPN17(図2B参照)は上記のCCIZN17(配列:2)の場合と同じプロトコルに従って合成した。固相配列組立中に、IZ部分(図2Bおよび以下に示した配列の下線部分)の2つのリシン残基にアリルオキシカルボニル(aloc)のε−NH保護を導入した。他のリシン残基はすべて標準Fmoc/tBu化学に従ってBocで保護した:CCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL(配列:2;スカフォールドドメインはイタリック体)。CCIPN17のアミノ酸配列はCCIZN17のアミノ酸配列と同じであるが、PEG付加CCIZN17ペプチドを“CCIPN17”と表す。ペプチド組立の終了後、NH2−末端を10倍過剰量の無水酢酸とDHF中で反応させることによってアセチル化した。保護ペプチド樹脂を0.25当量のテトラキス(トリフェニルホスフィン)パラジウムPd(PPh3)4および24当量のフェニルシランで30分間処理して2つのリシン残基からAloc保護基を除去した。他の残基側鎖はすべて保護された状態に維持した。2つの脱保護リシン鎖をDIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−dPEG酸,Quanta BioDesign)によってさらに誘導体化した。
【0085】
粗ペプチドCCIPN17をCCIZN17(上記)の精製の場合と同様のゲル透過クロマトグラフィーによって精製した。GPCによって得られたプール画分(純度70%)を上記のCCIZN17場合と同様の逆相HPLCによってさらに>95%純度まで精製した。溶出剤Bの以下の勾配を使用した:40−55%、25分超、流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、CCIZN17の場合と同様に、Jupiter C4カラム(150×4.6mm,5μm,300A,Phenomenex)を用いた分析用HPLCによって行った。精製ペプチドのキャラクタリゼーションはMicromass LCZ platform ES+を用いた質量分析法によって行った。測定MWは5612.0Daであり計算した平均MWは5611.8Daである。
【0086】
CCIPN17から(CCIPN17)3への酸化−
精製ペプチド前駆体(26mg)、CCIPN17をバッファに溶解し、空気によってゆっくりと酸化して共有結合三量体(CCIPN17)3とした(CCIZN17の酸化の場合と同様)。酸化反応は、Waters−Micromass LCZ Platformを使用するLC−MS分析によって上記同様にモニターした。これらのクロマトグラフィー条件下で、CCIPN17はtR=15.6’に溶出する。酸化反応は円滑に進行し一夜でtR=18.7’に溶出する1つの主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのCCIPN17ペプチド鎖([CCIPN17]3)を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液に45μLのTFAを添加し、溶液を、TSKgel Toyopearl HW−50S樹脂を詰めた700×26mmカラムに直接充填し、溶出剤としてH2O/アセトニトリル40/60,0.1%TFA,流速1mL/分を使用した。溶出画分をPhenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用い、溶出剤として:(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用するHPLCによって分析した。LC−MS Waters−Micromass LCZ Platform、正イオンモード(ES+)で溶出剤Bの以下の勾配を使用した:35%−55%B(20分)−80%(3分),流速1mL/分。予想MWは16829.57Daであり、測定MWは16830Daである。
【0087】
ビオチン−CCIZ(ビオチン−配列:3)の合成−
ペプチド−CCIZ(ビオチン−GCCGGIKKEIEAIKKEQEAIKKKIEAIEKE;配列:3)はCCIZN17(配列:2)の場合と同じプロトコルで合成した。ビオチンとの反応は、ペプチド組立プロセスの終了後、等モル量のDIPC(N,N’−ジイソプロピルカルボジイミド)およびHOAt(7−アザ−1−ヒドロキシベンゾトリアゾール)によって活性化した4倍過剰量のビオチンと一夜反応させることによって行った。粗ペプチドビオチン−CCIZ(配列:3)を、セミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)、流速80mL/分を用い、溶出剤として(A)水中の0.1%のTFAと(B)アセトニトリル中の0.1%のTFAを使用する逆相HPLCによって精製した。溶出剤Bの以下の勾配を使用した:25%−35%,30分超,流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、Ultrasphere C18カラム(250×4.6mm,5μm,80A,Beckman),流速1mL/分を用い、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用する分析用HPLCによって行った。溶出剤Bの以下の勾配を使用した:25%−40%(20分超)−80%(3分超)。予想MWは3570.0Daであり、測定MWは3570.5Daである。
【0088】
ビオチン−CCIZから(ビオチン−CCIZ)3への酸化−
精製ペプチド前駆体(15mg)、ビオチン−CCIZ(ビオチン−配列:3)を0.1MのHepes,pH7.3(USB Corp.)に5mg/mLの濃度で溶解した。ビオチン−CCIZNは空気によってゆっくりと酸化して一夜で共有結合三量体(ビオチン−CCIZN)3となる。酸化反応は上記同様にWaters−Micromass LCZ Platformを使用するLC−MS分析によってモニターした。これらのクロマトグラフィー条件で、ビオチン−CCIZはtR=16.4’に溶出する。酸化反応は円滑に進行し一夜でtR=16.9’に溶出する主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのビオチン−CCIZペプチド鎖([ビオチン−CCIZ]3)を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液を水に透析し、次いで凍結乾燥した。生成物を、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用い、溶出剤として:(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用し、LC−MS Waters−Micromass LCZ Platform、正モード(ES+)で行うHPLCによって分析した。予想MWは10704.94Daであり、測定MWは10704.0Daである。
【0089】
ビオチン−CCIZN17(ビオチン−配列:82)の合成−
ビオチン−CCIZN17(ビオチン−GCCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL;配列:82)はCCIZN17(配列:2)の場合と同じプロトコルで合成した。ビオチンとの反応はペプチド組立プロセスの終了後、上記のビオチン−CCIZの場合と同様に行った。粗ペプチド、ビオチン−CCIZN17の精製は、上記のCCIZN17精製の場合と同様にゲル透過クロマトグラフィー(GPC)によって行った。GPCによって得られたプール画分(純度70%)を上記同様の逆相HPLCによってさらに>95%純度まで精製した。溶出剤Bの以下の勾配を使用した:35%−55%、25分超、流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、Jupiter C4カラム(150×4.6mm,5μm,300A,Phenomenex)、流速1mL/分を用い、溶出剤として:(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用する分析用HPLCによって行った。溶出剤Bの以下の勾配を使用した:40%−60%B(20分)−80%(3分)。精製ペプチドのキャラクタリゼーションはMicromass LCZ platform ES+で質量分析法によって行った。測定MWは5416.0Daであり計算した平均MWは5416Daである。
【0090】
ビオチン−CCIZN17から(ビオチン−CCIZN17)3への酸化−
精製ペプチド前駆体(20mg)、ビオチン−CCIZ17を0.1MのHepes,pH7.3(USB Corp.)に2mg/mLの濃度で溶解した。この条件下でビオチン−CCIZN17は空気によってゆっくりと酸化して共有結合三量体(ビオチン−CCIZN17)3となる。酸化反応は上記同様にWaters−Micromass LCZ Platformを使用するLC−MS分析によって、溶出剤Bの勾配:40%−60%B(20分)−80%(3分),流速1mL/分を使用してモニターした。これらのクロマトグラフィー条件下で、ビオチン−CCIZ17はtR=12.87’に溶出する。酸化反応は円滑に進行し一夜でtR=14.9’に溶出する1つの主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのビオチン−CCIZ17ペプチド鎖(ビオチン−CCIZN17)3を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液に45μLのTFAを添加し、TSKgel Toyopearl HW−50S樹脂を詰めた700×26mmカラムに溶液を直接充填し、溶出剤H2O/アセトニトリル40/60、0.1%TFA、流速1mL/分を使用した。プールした画分をセミ・プレパラティブPhenomenex C4カラム(10×250mm,15μm)、流速20mL/分を用い、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用する逆相HPLCによってさらに精製した。溶出剤Bの以下の勾配を使用した:35%−50%,20分超,流速80mL/分。溶出した画分を、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)で溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用するHPLCによって分析した。LC−MS Waters−Micromass LCZ Platform、正モード(ES+)で溶出剤Bの以下の勾配を使用した:40%−60%B(20分)−80%(3分)、流速1mL/分。予想MWは16244Daであり測定MWは16245Daである。
【0091】
ビオチン−CCIPN17の合成−
このペプチドは上記のビオチン−CCIZN17(配列:82)の場合と同じプロトコルに従って合成した。固相配列組立中に、IZ部分(下記配列の下線部分)の2つのリシン残基にアリルオキシカルボニル(aloc)のε−NH保護を導入し、他のリシンはすべて標準Fmoc/tBu化学に従ってBoc保護した:ビオチン−GCCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL(配列:82;スカフォールドドメインはイタリック体)。PEG付加ビオチン−CCIZN17ペプチドを“ビオチン−CCIPN17”と命名するが、ビオチン−CCIPN17のアミノ酸配列はビオチン−CCIZN17のアミノ酸配列と同じである。ペプチド組立の終了後、上記のビオチン−CCIZの場合と同様にビオチンと反応させた。保護ペプチド樹脂を0.25当量のテトラキス(トリフェニルホスフィン)パラジウムPd(PPh3)4および24当量のフェニルシランで30分間処理して2つのリシン残基からAloc保護基を除去し、他の残基側鎖はすべて保護された状態に維持した。2つの脱保護リシン鎖を、DIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−d PEG酸,Quanta BioDesign)によってさらに誘導体化した。
【0092】
粗ペプチドビオチン−CCIPN17を上記同様にTSKgel Toyopearl HW−50S樹脂(700×26mmカラム)上のゲル透過クロマトグラフィー(GPC)によって精製した。GPCによって得られたプール画分(純度70%)を、上記同様のセミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジを用いる逆相HPLCによって、溶出剤Bの勾配:35−55%、25分超、流速80mL/分を使用して、さらに>95%純度まで精製した。。粗ペプチド、溶出画分および精製プールの分析は、上記同様にJupiter C4カラム(150×4.6mm,5μm,300A,Phenomenex)を用いる分析用HPLCによって行った。精製ペプチドのキャラクタリゼーションはMicromass LCZ platform ES+の質量分析法によって行った。測定MWは5796Daであり、計算した平均MWは5796Daである。
【0093】
ビオチン−CCIP17から(ビオチン−CCIPN17)3への酸化−
精製したペプチド前駆体(20mg)、ビオチン−CCIPN17を0.1MのHepes,pH7.3(USB Corp.)に濃度2mg/mLで溶解した。この条件下で、ビオチン−CCIPN17は空気によってゆっくりと酸化して一夜で(ビオチン−CCIPN17)3となる。酸化反応は、Waters−Micromass LCZ PlatformをPhenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いるLC−MS分析によって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用してモニターした。溶出剤Bの以下の勾配を使用した:45%−60% B(20分)−80%(3分)、流速1mL/分。これらのクロマトグラフィー条件下で、ビオチン−CCIPN17はtR=11.9’に溶出する。酸化反応は円滑に進行し一夜でtR=17.8’に溶出する1つの主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのCビオチン−CIPN17ペプチド鎖([ビオチン−CCIPN17]3)を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液にグアニジニウムクロリドを最終濃度6Mまで添加し、溶液をTSKgel Toyopearl HW−50S樹脂を詰めた700×26mmカラムに直接充填し、溶出剤としてH2O/アセトニトリル40/60、0.1% TFA、流速1mL/分を使用した。溶出した画分をPhenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いるHPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して分析した。溶出剤Bを以下の勾配で使用した:45%−60% B(20分)−80%(3分)、流速1mL/分、LC−MS Waters−Micromass LCZ Platform、正モード(ES+)。予測MWは17383Daであり、計算MWは17384Daである。
【0094】
ビオチン−CCIPの合成−
このペプチドは上記のビオチン−CCIZ(配列:3)の場合と同じプロトコルに従って合成した。固相配列組立中、IZ部分(下記配列の下線部分)の2つのリシン残基にアリルオキシカルボニル(aloc)のε−NH保護を導入し、他のリシンはすべて標準Fmoc/tBu化学に従ってBocで保護した:ビオチン−GCCGGIKKEIEAIKKEQEAIKKKIEAIEK(配列:3;スカフォールドドメインはイタリック体)。PEG付加ビオチン−CCIZペプチドを“ビオチン−CCIP”と命名するが、ビオチン−CCIPのアミノ酸配列はビオチン−CCIZのアミノ酸配列と同じである。ペプチド組立の終了後、上記のビオチン−CCIZの場合と同様にビオチンと反応させた。保護されたペプチド−樹脂をさらに0.25当量のテトラキス(トリフェニルホスフィン)パラジウムPd(PPh3)4および24当量のフェニルシランで30分間処理して2つのリシン残基からAloc保護基を除去し、他の残基側鎖はすべて保護された状態に維持した。2つの脱保護リシン鎖を、DIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−d PEG酸,Quanta BioDesign)によってさらに誘導体化した。
【0095】
粗ペプチド、ビオチン−CCIPをTSKgel Toyopearl HW−50S樹脂(700×26mmカラム)上で溶出剤として水中の30%アセトニトリル、0.1%TFA、流速1mL/分を使用するゲル透過クロマトグラフィー(GPC)によって精製した。GPCによって得られたプール画分(純度70%)をセミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(4×200mm,15μm)を用いる逆相HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して>95%純度までさらに精製した。溶出剤Bの以下の勾配を使用した:20−35%、30分超、流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、Jupiter C4カラム(150×4.6mm,5μm,300A,Phenomenex)、流速1mL/分、を用いる分析用HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して行った。溶出剤Bの以下の勾配を使用した:20−40%(20分超)−80%(3分超)。予測MWは3821Daであり、測定MWは3821Daである。
【0096】
ビオチン−CCIPから(ビオチン−CCIPN)3への酸化−
精製したペプチド前駆体(20mg)、ビオチン−CCIPN17を0.1MのHepes,pH7.3(USB Corp.)に濃度2mg/mLで溶解した。この条件下で、ビオチン−CCIPは空気によってゆっくりと酸化して一夜で共有結合三量体(ビオチン−CCIP)3となる。酸化反応は、Waters−Micromass LCZ PlatformをPhenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いたLC−MS分析によって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用してモニターした。溶出剤Bの以下の勾配を使用した:25%−40% B(20分)−80%(3分)、流速1mL/分。これらのクロマトグラフィー条件下で、ビオチン−CCIPはtR=15.5’に溶出する。酸化反応は円滑に進行し一夜でtR=18.4’に溶出する1つの主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのビオチン−CCIPNペプチド鎖([ビオチン−CCIP]3)を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液にトリフルオロ酢酸を最終濃度0.2%までおよびグアニジニウムクロリドを最終濃度6Mまで添加し、溶液を、TSKgel Toyopearl HW−50S樹脂を詰めた700×26mmカラムに直接充填し、溶出剤としてH2O/アセトニトリル40/60、0.1% TFA、流速1mL/分を使用した。溶出した画分は、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いるHPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して分析した。溶出剤Bを以下の勾配で使用した:25%−40% B(20分)−80%(3分)、流速1 mL/分、LC−MS Waters−Micromass LCZ Platform、正モード(ES+)。予測MWは11456Daであり、測定MWは11456Daである。
【0097】
ワクチン設計−公称サイズ約15.6オングストロームのポリエチレングリコール部分をCCIZN17(配列2)のスカフォールドドメインに、該PEG部分が三量体コイルドコイル形成を少しは妨害するとしても妨害が最小になるように戦略的に付着させた。この小さいポリエチレングリコール部分m−dPEGTM酸(MW=236)(Quanta BIODESIGN,Powell,Ohio)は分子量約236Daであり、以下の構造:
【0098】
【化2】
を有している。
【0099】
最初に、三量体コイルドコイルのヘプタッドリピートのより外側の“f”位置に局在する3つのリシン残基のイプシロンアミンをIZスカフォールドへのポリエチレングリコール部分の付着に適した位置として同定した(図2A)。これらのリシン残基の2つをm−dPEGTM酸(MW=236)による誘導体化のために最終的に選択した(図2B参照、誘導体化リシン(“K”)残基は下線付き)。図2Bに示すように、このポリマー修飾CC−キメラN−ペプチドの名称は、CCIZN17からCCIPN17に変更されており、3つのCCIPN17キメラポリマー修飾ペプチドを含む共有結合安定化(CCIPN17)3三量体コイルドコイルを生成できる。このワクチン構築物(CCIPN17)3の概略モデルを図3に示す。三量体コイルドコイルを形成するヘリックスは、長さ約62オングストロームの円筒として示されている。各円筒の暗い灰色部分は長さ約36オングストロームのIZスカフォールドドメインを表し、明るい灰色部分は長さ約25オングストロームのHIV gp41のN17領域を表す。m−dPEGTM酸(MW=236)部分は長さ約15オングストロームの長方形として表されている。ポリマー修飾用に選択された2つの指定リシン残基の目的はIZスカフォールドだけを遮蔽することである。実際、コイルドコイルのN17ドメインに近いほうのリシン残基はN17コイルドコイル部分から約22オングストロームであり、15オングストロームのm−dPEGTM酸部分がこのドメインのいずれかの部分を遮蔽できないことを確保する。
【0100】
SMW−PEG付加ワクチンの免疫原性−
動物実験はPEGを付加しないワクチン(CCIZN17)3およびPEG付加ワクチン(CCIPN17)3によって誘発されたモルモットの抗体応答の比較によって行った。試験にはホモロガス方式(すなわち、抗原によるプライミング注射、次いで同じ抗原による2回のブースト注射)およびヘテロロガス方式(すなわち、PEGを付加しない抗原(CCIZN17)3によるプライミング注射、次いでPEG付加抗原(CCIPN17)3による2回のブースト注射)の双方を使用した(図4参照)。PEG付加抗原はQS21アジュバント(Antigenics;New York,NY)と共に動物に投与した。免疫応答の分析はELISAによって行った。ELISAプレートへの抗原の吸着能力の違いに起因するバイアスを除去するために、すべての捕獲抗原をビオチニル化し、捕獲ビオチニル化分子を添加する前にELISAプレートをストレプトアビジンで被覆した。このようにすると各抗原に対するELISA幾何平均力価(GMT)が、血清中に存在する対応特異性をもつ抗体の相対量を正確に反映する。免疫分子のビオチニル化形態(ビオチン−CCIZN17)3および(ビオチン−CCIPN17)3ならびにスカフォールド分子単独のビオチニル化形態(ビオチン−CCIZ)3および(ビオチン−CCIP)3をELISAアッセイの捕獲抗原として使用した。PEGを付加しないワクチン(CCIZN17)3で予防接種した動物の血清中には全抗原およびスカフォールド部分単独と同等の量の抗体が存在する。しかしながらPEG付加ワクチンによる免疫は、ホモロガス方式でもヘテロロガス方式でも、スカフォールド((CCIZ)3でもPEG付加(CCIP)3でもない)に対しては有意に減退した応答が生じるが、N−ペプチド部分に対しては極めて高い力価が誘発される。これは(CCIZN17)3および(CCIPN17)3の双方で等しく、従って、どちらの抗原に対して測定したときもGMT測定値が等しい。
【0101】
この実験は、タンパク質抗原の特定領域を選択的に隠蔽するSMW PEG部分の付着が、ペプチド/タンパク質の残りの部分の免疫原性を遮蔽することなく該領域の免疫反応性を除去できることを証明する。
【実施例2】
【0102】
アルツハイマー病の免疫療法
PEG236−Aβ1−42(配列:4;図5C参照)の合成−
ABI433シンセサイザー(Applied Biosystems)で標準Fmoc/t−Bu化学に従ってペプチドを固相合成した。使用した樹脂は予め誘導体化したFmoc−Ala−ALH−Champion,1%架橋(Biosearch Technologies,Inc.)であり、これはカルボキシル化アラニンを産生する誘導体化PEG−PS基材樹脂である。すべてのアシル化反応は8倍過剰量の活性化アミノ酸で樹脂の遊離アミノ基に60分間行った。アミノ酸は等モル量のHATU(O−(7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)および2倍モル過剰量のDIEA(N,N−ジイソプロピルエチルアミン)で活性化した。側鎖保護基は以下を用いた:グルタミン酸(E)、アスパラギン酸(A)、チロシン(Y)およびセリン(S)にtert−ブチル;ヒスチジン(H)、アスパラギン(N)およびグルタミン(Q)にトリチル;リシン(K)にtert−ブトキシ−カルボニル;アルギニン(R)に2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニル。Fmoc保護基を除去するためには、(8−42)残基アセンブリにDMF中の2%DBU(ジアザ−ビシクロ−ウンデセン)、(1−8)残基アセンブリにジメチルホルムアミド(DMF)中の20%ピペリジンを使用した。アミノ酸組立の終了後、DIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−dPEG酸,Quanta BioDesign)でNH2−末端をさらに誘導体化した。
【0103】
乾燥ペプチド樹脂を88%トリフルオロ酢酸(TFA)、5%フェノール、2%トリイソプロピルシランおよび5%水(Sole,N.A.and G.Barany,1992,(前出))によって室温で1.5時間処理した。樹脂を濾過し、溶液を低温メチル−t−ブチルエーテルに加えてペプチドを沈殿させた。遠心後、沈殿ペレットを新しい低温メチル−t−ブチルエーテルで洗浄して有機スカベンジャーを除去した。このプロセスを2回繰返した。最終ペレットを乾燥し、H2O、20%アセトニトリルに再懸濁させて凍結乾燥した。
【0104】
粗ペプチドPEG236−Aβ1−42
(DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA;配列:4)は、セミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用い、溶出剤として(A)水および(B)0.1%NH4OHを含むアセトニトリルを使用する逆相HPLCによって精製した。溶出剤Bの以下の勾配を使用した:10%−30%、20分超、流速80mL/分。溶出画分の分析用HPLCは、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いるBeckman HPLCで、溶出剤B(上記)の以下の勾配:15%−35% B(20分)−80%(3分)、流速1mL/分で行った。精製ペプチドのキャラクタリゼーションは、Micromass LCZ platformスペクトロメーターでHPLC−MSによって行った。理論的平均分子量(MW)は4731.0Daであり、測定MWは4731.0Daである。
【0105】
結果
本発明の1つの具体的用途は、アルツハイマー病(AD)の予防および治療処置のための改良ワクチンの開発である。最近の報告は、Elan/Wyeth AN−1792 Aβ 1−42ワクチン(図5B参照)の臨床試験の中断に導いた重篤な脳の炎症の問題が、T細胞応答に起因することを示唆している(Monsonego,A.and H.L.Weiner,2003,Science 302:834−8;Nicoll,J.A.ら,2003,Nat.Med.9:448−52)。この応答はAβペプチドのCOOH−末端を特異的に指向している(Monsogeo,A.ら,2003,J.Clin.Invest.112:415−22)。しかしながら注目すべきは、同じ臨床試験が、高いAβ抗体価をもつ患者でプラークの減少およびある程度限られた記憶の安定化を示したことであり、これはワクチンが治療効果を発揮していたことを表す。特に、Aβ1−42ワクチンを接種した患者の大半では病理形態のAβに特異的な抗体が誘発されたことが知見された(Hock,C.ら,2002,Nat.Med.8:1270−5)。これらの治療用抗体は、ペプチドのNH2−末端領域、すなわち残基4−10のエピトープを認識する(McLaurin,J.ら,2002,Nat.Med.8:1263−9;図5A参照)。これらの知見は疾病動物モデルでも確認された(Bard,F.ら,2003,Proc.Natl.Acad.Sci.U.S.A.100:2023−8)。そこでElan/Wyethらは、接合したN−末端(1−16)Aβペプチドに基づく第二世代のワクチンACC−001の臨床試験に進んでいる(図5B参照)。
【0106】
COOH−末端領域が欠失でなくSMWポリマー(例えばSMW PEG)によって遮蔽されている遊離形態またはコンジュゲート形態のAβペプチドの使用は、Elan/Wyethワクチンの有力な代替物である(図5C参照)。例えばSMW PEGはアミノ酸16−30に局在し上述の重篤な副作用の原因になると思われるAβのCOOH−末端T細胞エピトープの内部または極めて近傍で単一または多数コピーに付着できるが(Monsonego,A.ら,2003(前出))、NH2−末端領域のアミノ酸1−16またはもっと短い領域のアミノ酸4−10の免疫原性は元のまま維持される。双方の領域は有益なエピトープを内包すると考えられている(McLaurin,J.R.ら,2002(前出))。同じ分子が、SMW PEG付加Aβペプチドによる免疫後の従来のハイブリドーマテクノロジーによって、または、固定化SMW PEG付加Aβペプチドに関するファージディスプレイライブラリーをパニングすることによって受動免疫療法の抗体開発に使用できる。
【0107】
本発明の別の具体的用途は、Aβタンパク質の異なる凝集状態を識別する潜在能力を有している治療用抗体、例えば、Aβ誘発病理の原因と見做される小オリゴマーに特異的な抗体の開発である。(Cleary,J.P.ら,2005,Nat.Neurosci.8:79−84)。AβのNH2−末端領域は低分子量オリゴマー、高分子量オリゴマーおよびフィブリルへのAβの凝集には関与しないが、このNH2−末端は免疫優性であり、多様な形態に共通のエピトープに特異的なのでそれらの識別ができない抗体の優先的なin vivo形成を指令する。NH2−末端領域が欠失でなくSMWポリマー(例えばSMW PEG)で遮蔽されている遊離形態またはコンジュゲート形態のAβペプチドを使用することによって所望の特異性をもつ抗体を得ることができる。このペプチドは正常Aβの潜在的凝集能力は維持しているが、N−末端領域に対する免疫応答はPEG遮蔽によって抑制されている。
【実施例3】
【0108】
HIV−1の4E10/Trp−富化領域をターゲットする低分子量PEG付加ワクチン
中和抗体4E10(Salzwedel,K.ら,1999,J.Virol.73:2469−80)に結合するHIV−1 gp41内部のTrp−富化領域の構造は、脂質模擬環境でSchibli,DJ.らによって決定された(2001,Biochemistry 40:9570−8)。最近にはまた、4E10抗体に接触するアミノ酸残基が開示された(Cardoso,R.ら,2004(前出))。これらの接触残基は、構造内のペプチドの水に面する側を構成する(図6A参照)。図6Aで、4E10結合残基は下線部分で示し、672および680位のTrp残基を含むが、膜に面するTrp残基は二重下線部分で示し、666、670および678位に局在する。
【0109】
4E10エピトープの所望のα−ヘリカルコンホメーションは、4E10エピトープをGCN4ロイシンジッパーの表面にグラフトすることによって得ることができ(O’Shea,E.K.ら,1989,Science 245:646−8)、また、Sia,S.K.and P.S.Kim(2003,Proc.Natl.Acad.Sci.U.S.A.100:9756−61)に類似である。図6Aに示すTrp−富化領域の構造に基づいて、膜に面する残基のいくつかに突然変異を導入し(図6Bの“4E10コイル”の一部として下線を付けたアミノ酸)、GCN4ペプチド(“GCN4コイル”)に適合性のダイマー化表面をもつ4E10ペプチド(“4E10コイル”)を作製できる(図6B参照)。このコイルドコイルは、4E10領域を適正なヘリカル構造に拘束するであろう。GCN4/4E10コイルドコイルの安定性は、ジスルフィド結合の付加(Sia,S.K.and P.S.Kim,2003(前出))、および、GCN4と4E10との“a”および“d”位の残基をロイシンまたはイソロイシンに突然変異させることによってさらに強化できる(Lu,S.M.and R.S.Hodges,2002,J.Biol.Chem.277:23515−24)。該構造(すなわちGCN4コイル)のスカフォールド性部分に対する免疫応答を回避するために、GCN4コイルの表面に沿って適切なリシン残基(例えば、図7の二重下線を付けた残基)をPEG236で誘導体化できる(図7)。
【実施例4】
【0110】
OMPCに接合する酸性スカフォールドをもつPEG付加キメラN17ペプチド
Br−アセチル−GGGEP17GluN17(表5のペプチド1)の合成−
このペプチドは、Pioneerペプチドシンセサイザー(Applied Biosystems)で固相合成した。使用した樹脂はFmoc−Linker AM−Champion,1%架橋(Biosearch Technologies,Inc.)、修飾Rinkリンカーp−[(R,S)−α−[9H−フルオレン−9−イル−メトキシホルムアミド]−2,4−ジメトキシベンジル]−フェノキシ酢酸で誘導体化したPEG−PS基材樹脂であった(Rink,H.,1987,Tetrahedron Lett.28:3787−3789;Bernatowicz,M.S.ら,1989,Tetrahedron Lett.30:4645−4667)。すべてのアシル化反応は樹脂遊離アミノ基の4倍過剰量の活性化アミノ酸で90分間行った。アミノ酸は等モル量のHBTU(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)および2倍モル過剰量のDIEA(N,N−ジイソプロピルアミン)で活性化した。ペプチド配列:KIEEIEEKIEEIEKLLQLTVWGIKQLQARIL(配列:93;非−HIV残基はイタリック体)の最初の31残基部分については、標準Fmoc/t−Bu化学を使用した。側鎖保護基は以下を用いた:グルタミン酸(Glu)およびトレオニン(Thr)にtert−ブチル;システイン(Cys)およびグルタミン(Gln)にトリチル;リシン(Lys)およびトリプトファン(Trp)にtert−ブトキシ−カルボニル;アルギニン(Arg)に2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニル。最初の31残基の組立後、Dde−Lys(Fmoc)−OH,N−α−1−(4,4−ジメチル−2,6−ジオキソシクロヘキシ−1−イリデン)エチル−N−ε−Fmoc−L−リシンを32番目の残基としてアシル化した。ε−アミノ基のFmoc−保護基を20%ピペリジン含有ジメチルホルムアミド(DMF)溶液で処理することによって除去した。この条件下でDde保護基は完全に安定である。脱保護したリシン側鎖をさらにDIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−dPEG酸,Quanta BioDesign)でさらに誘導体化した。次にペプチド樹脂を1%ヒドラジン含有DMF溶液で処理してリシンのα−アミノ基からDde−基を除去した。ペプチド樹脂GGIEKKIEEIEEKIEEIEKLLQLTVWGIKQLQARIL(配列:94;非−HIV残基はイタリック体;PEG化リシン残基は下線部分)を得るために組立を継続した。次にペプチド樹脂を2つの部分に分割した。全体の2/3にあたる大きいほうの部分を使用して別のグリシン残基を組込むために組立を継続し、DMF中で10倍過剰量の無水ブロモ酢酸と反応させることによってさらにブロモアセチル化した。GGIEKKIEEIEEKIEEIEKLLQLTVWGIKQLQARIL(配列:94)−樹脂の残りの1/3を使用してAc−C(Acm)−TtdsCCEP17GluN17(下記参照)の合成を継続した。
【0111】
Br−アセチル−EP17GluN17の合成終了後、乾燥ペプチド−樹脂を88%トリフルオロ酢酸(TFA)、5%フェノール、2%トリイソプロピルシランおよび5%水(Sole,N.A.and G.Barany,1992(前出))によって室温で1.5時間処理した。樹脂を濾過し、溶液を低温メチル−t−ブチルエーテルに加えてペプチドを沈殿させた。遠心後、ペプチドペレットを新しい低温メチル−t−ブチルエーテルで洗浄して有機スカベンジャーを除去した。このプロセスを2回繰返した。最終ペレットを乾燥し、H2O、20%アセトニトリルに再懸濁させて凍結乾燥した。
【0112】
粗ペプチドBr−アセチル−GGGEP17GluN17(表5のペプチド1)の精製は、Toyopearl HW−50S樹脂を詰めた700×26mmカラム上で水中の60%のアセトニトリル、0.1%TFAを溶出剤として使用するゲル透過クロマトグラフィー(GPC)によって行った。典型的な処理では、900mgの粗ペプチドを25mLの溶出剤に溶解し、流速1mL/分でカラムに直接充填した。溶出画分の分析用HPLCは、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いたBeckman HPLCで溶出剤B(上記)の以下の勾配:45%−65% B(20分)−80%(3分)、流速1mL/分を使用した。純度に基づいて画分をプールし、セミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用いる逆相HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用してさらに精製した。溶出剤Bの以下の勾配を使用した:45%−60%、20分超、流速80mL/分。精製ペプチドのキャラクタリゼーションは、Micromass LCZ platformスペクトロメーターでエレクトロスプレー質量分析法によって行った。理論的平均分子量(MW)は4613.3Daであり、測定MWは4613.0Daである。
【0113】
Ac−C(Acm)−Ttds−CCEP17GluN17(表5のペプチド2)の合成−
上記アセンブリからGGIEKKIEEIEEKIEEIEKLLQLTVWGIKQLQARIL(配列:94−樹脂(上記同様に作製)の1/3を使用してAc−C(Acm)Ttds−CC−部分の合成を継続した。トリチル側鎖で保護された最初の(カルボキシ末端配列)の2つのシステインは上記のβ−アセチル−GGGEP17GluN17(表5のペプチド1)同様に標準Fmoc/tBu化学によって組立てた。Ttds(1−アミノ−4,7,10−トリオキサ−13−トリデカミンコハク酸(−NH−(CH2)3−O−(CH2)2−O−(CH2)2−O−(CH2)3−NH−CO−CH2)2−CO−)との反応は、等モル量のHBTUおよび2倍モル過剰量のDIEAで2時間活性化した4倍過剰量のTtdsとの反応によって行った。N−末端残基C(Acm)をFmoc−Cys(Acm)−OH(上記)と同じ条件下でアシル化し、次いでFmoc脱保護後にDMF中の10倍過剰量の無水酢酸と反応させることによってアセチル化した。先行実施例の記載と同様にして樹脂から分離後、粗ペプチドAc−C(Acm)−TtdsCCEP17GluN17(表5のペプチド2)を、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で、水中の60%アセトニトリル、0.1%TFA、流速1mL/分を使用するゲル透過クロマトグラフイー(GPC)によって精製した。GPCによって得られたプール画分(純度70%)をセミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用いた逆相HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して、>95%までさらに精製した。溶出剤Bの以下の勾配を使用した:45%−60%、20分超、流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、Jupiter C4カラム(150×4.6mm,5μm,5μm,300A,Phenomenex)、流速1mL/分を用いた分析用HPLCによって、溶出剤として(A)水中の0.1%TFA、および、(B)アセトニトリル中の0.1%TFAを使用して行った。溶出剤Bの以下の勾配を使用した:45−65%(20分超)−80%(3分超)。精製ペプチドのキャラクタリゼーションは、Micromass LCZ platformでエレクトロスプレー質量分析法によって行った。理論的平均分子量(MW)は5161.2Daであり、測定MWは5161.0Daである。
【0114】
Ac−C(Acm)−(チオEP17GluN17)3の合成−
チオエーテル結合の形成による共有結合的三量体化−
精製ペプチド前駆体(10.1mg)、Br−アセチル−GGGEP17GluN17(表5のペプチド1)を、6Mのグアニジウムクロリド、0.25MのTrisHCl,pH8.5、2mMのEDTAに5mg/mLの濃度で溶解した。この溶液に他のペプチド前駆体Ac−C(Acm)Ttds−CCEP17GluN17(表5のペプチド2)(5.3mg;1mg/mL)を添加した。2つのペプチド前駆体の実効モル比は2.2:1のBr−アセチル−GGGEP17GluN17:Ac−C(Acm)Ttds−CCEP17GluN17であった。三量体化反応のモニターは、Waters−Micromass LCZ Platformを使用するLC−MS分析によって、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)で溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して行った。溶出剤Bを以下の勾配で使用した:45%−65% B(20分)−80%(3分)、流速1mL/分。チオエーテル形成反応は円滑に進行した。2時間後、1つの生成物が形成され、その質量は、2つのチオエーテル結合を介して2つのBr−アセチル−GGGEP17GluN17ペプチド鎖に連結された1つのAc−C(Acm)Ttds−CCEP17GluN17ペプチド鎖に相当する。溶液に20μLのTFAを添加し、溶液を、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で水中の60%アセトニトリル、0.1%TFA、流量1mL/分の溶出剤を使用するゲル透過クロマトグラフィー(GPC)によって直接精製した。溶出画分の分析は、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)で溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用するHPLCによって行った。溶出剤Bを以下の勾配で使用した:45%−65% B(20分)−80%(3分)、流速1mL/分、LC−MS Waters−Micromass LCZ Platform、正モード(ES+)。予想MWは14225Daであり、測定MWは14226Daであった。
【0115】
Ac−C(Acm)(チオEP17GluN17)3のアセチル化システインの脱保護によるAc−C(チオEP17GluN17)3の形成−
精製したコイルドコイル前駆体ac−C(Acm)(チオEP17GluN17)3を20mg/mlの濃度でTFA、アニソール2%および50当量のAgOTf(トリフルオロメタンスルホン酸銀)に40℃で3時間溶解した。氷冷した無水エーテルを反応混合物に添加し、沈殿物を遠心によって単離した。沈殿物を氷冷した無水エーテルで2回洗浄した。ペプチドをH2O/アセトニトリル、0.1%TFAに再溶解し、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で溶出剤として水中の60%アセトニトリル、0.1%TFA、流速1mL/分を使用するゲル透過クロマトグラフィー(GPC)によって精製した。精製したペプチドのキャラクタリゼーションは、Micromass LCZ platformでエレクトロスプレー質量分析法によって行った。理論的平均MWは14157Daであり、測定MWは14157Daであった。
【0116】
Br−アセチル−GGGEPN17(表5のペプチド3)の合成−
このペプチドは、Pioneerペプチドシンセサイザー(Applied Biosystems)で固相合成した。使用した樹脂はFmoc−Linker AM−Champion,1%架橋(Biosearch Technologies,Inc.)、修飾Rinkリンカーp−[(R,S)−α−[9H−フルオレン−9−イル−メトキシホルムアミド]−2,4−ジメトキシベンジル]−フェノキシ酢酸で誘導体化したPEG−PS基材樹脂であった(Rink,H.,1987,Tetrahedron Lett.28:3787−3789;Bernatowicz,M.S.ら,1989,Tetrahedron Lett.30:4645−4667)。すべてのアシル化反応は樹脂遊離アミノ基の4倍過剰量の活性化アミノ酸で90分間行った。アミノ酸は等モル量のHBTU(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)および2倍モル過剰量のDIEA(N,N−ジイソプロピルアミン)で活性化した。ペプチド配列:KIEEIEKKIEEIEKLLQLTYWGIKQLQARIL(配列:95;非−HIV残基はイタリック体)の最初の31残基部分については、標準Fmoc/t−Bu化学を使用した。側鎖保護基は以下を用いた:グルタミン酸(Glu)およびトレオニン(Thr)にtert−ブチル;システイン(Cys)およびグルタミン(Gln)にトリチル;リシン(Lys)およびトリプトファン(Trp)にtert−ブトキシ−カルボニル;アルギニン(Arg)に2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニル。最初の31残基の組立後、Dde−Lys(Fmoc)−OH,N−α−1−(4,4−ジメチル−2,6−ジオキソシクロヘキシ−1−イリデン)エチル−N−ε−Fmoc−L−リシンを32番目の残基としてアシル化した。ε−アミノ基のFmoc−保護基を20%ピペリジン含有ジメチルホルムアミド(DMF)溶液で処理することによって除去した。この条件下でDde保護基は完全に安定である。脱保護したリシン側鎖をさらにDIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−dPEG酸,Quanta BioDesign)でさらに誘導体化した。次にペプチド樹脂を1%ヒドラジン含有DMF溶液で処理してリシンのα−アミノ基からDde−基を除去した。組立を継続してスカフォールドの他の6つの残基、非HIVドメインすなわち配列KIEEIE(配列:96)をペプチド樹脂に組込んだ。次いで、Dde−Lys(Fmoc)−OH,N−α−1−(4,4−ジメチル−2,6−ジオキソシクロヘキサ−1−イリデン)エチル−N−ε−Fmoc−L−リシンをアシル化した。ε−アミノ基のFmoc−保護基はジメチルホルムアミド(DMF)中の20%ピペリジン溶液によって選択的に除去した。この条件下でDde−保護基は完全に安定である。脱保護したリシン側鎖を、DIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−d PEG酸, Quanta BioDesign)によってさらに誘導体化した。ペプチド樹脂GGIEKKIEEIEKKIEEIEKKIEEIEKLLQLTVWGIKQLQARIL(配列:97;非−HIV残基はイタリック体;PEG化リシン残基は下線部分)を得るために組立を継続した。次にペプチド樹脂を2つの部分に分割した。全体の2/3にあたる大きいほうの部分を使用して別のグリシン残基を組込むために組立を継続し、DMF中で10倍過剰量の無水ブロモ酢酸と反応させることによってさらにブロモアセチル化した。ペプチド−樹脂の残りの1/3を使用してAc−C(Acm)−TtdsCCEPN17(下記参照)の合成を継続した。
【0117】
Br−アセチル−EPN17の合成終了後、乾燥ペプチド−樹脂を88%トリフルオロ酢酸(TFA)、5%フェノール、2%トリイソプロピルシランおよび5%水(Sole,N.A.and G.Barany,1992(前出))によって室温で1.5時間処理した。樹脂を濾過し、溶液を低温メチル−t−ブチルエーテルに加えてペプチドを沈殿させた。遠心後、ペプチドペレットを新しい低温メチル−t−ブチルエーテルで洗浄して有機スカベンジャーを除去した。このプロセスを2回繰返した。最終ペレットを乾燥し、H2O、20%アセトニトリルに再懸濁させて凍結乾燥した。
【0118】
粗ペプチドBr−アセチル−GGGEPN17(表5のペプチド3)の精製は、Toyopearl HW−50S樹脂を詰めた700×26mmカラム上で水中の60%のアセトニトリル、0.1%TFAを溶出剤として使用するゲル透過クロマトグラフィー(GPC)によって行った。典型的な処理では、900mgの粗ペプチドを25mLの溶出剤に溶解し、流速1mL/分でカラムに直接充填した。溶出画分の分析用HPLCは、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いたBeckman HPLCで溶出剤B(上記)の以下の勾配:45%−65% B(20分)−80%(3分)、流速1mL/分を使用した。純度に基づいて画分をプールし、セミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用いる逆相HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用してさらに精製した。溶出剤Bの以下の勾配を使用した:45%−60%、20分超、流速80mL/分。精製ペプチドのキャラクタリゼーションは、Micromass LCZ platformスペクトロメーターでエレクトロスプレー質量分析法によって行った。理論的平均分子量(MW)は5699Daであり、測定MWは5700Daである。
【0119】
Ac−C(Acm)−Ttds−CCEPN17(表5のペプチド4)の合成−
上記アセンブリからGGIEKKIEEIEKKIEEIEKKIEEIEKLLQLTVWGIKQLQARIL(配列:97)−樹脂(詳細は上記参照)の1/3を使用してAc−C(Acm)Ttds−CC−部分の合成を継続した。トリチル側鎖で保護された最初の(カルボキシ末端配列)の2つのシステインは標準Fmoc/tBu化学によって組立てた。Ttds(1−アミノ−4,7,10−トリオキサ−13−トリデカミンコハク酸(−NH−(CH2)3−O−(CH2)2−O−(CH2)2−O−(CH2)3−NH−CO−CH2)2−CO−)との反応は、等モル量のHBTUおよび2倍モル過剰量のDIEAで2時間活性化した4倍過剰量のTtdsとの反応によって行った。N−末端残基C(Acm)をFmoc−Cys(Acm)−OHと同じ条件下でアシル化し、次いでFmoc脱保護後にDMF中の10倍過剰量の無水酢酸との反応によってアセチル化した。
【0120】
先行実施例の記載と同様にして樹脂から分離後、粗ペプチド、アセチル−C(Acm)−TtdsCCEPN17を、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で、溶出剤として水中の60%アセトニトリル、0.1%TFA、流速1mL/分を使用するゲル透過クロマトグラフイー(GPC)によって精製した。GPCによって得られたプール画分(純度70%)をセミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用いた逆相HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して、>95%純度までさらに精製した。溶出剤Bの以下の勾配を使用した:45%−60%、20分超、流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、Jupiter C4カラム(150×4.6mm,5μm,300A,Phenomenex)、流速1mL/分を用いた分析用HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して行った。溶出剤Bの以下の勾配を使用した:45−65%(20分超)−80%(3分超)。精製ペプチドのキャラクタリゼーションは、Micromass LCZ platformでエレクトロスプレー質量分析法によって行った。理論的平均分子量(MW)は6248Daであり、測定MWは6247Daであった。
【0121】
アセチル−C(Acm)(チオEPN17)3の合成−
チオエーテル結合の形成による共有結合的三量体化−
精製ペプチド前駆体(12mg)、Br−アセチル−GGGEPN17(表5のペプチド3)を、6Mのグアニジウムクロリド、0.25MのTrisHCl,pH8.5、2mMのEDTAに5mg/mLの濃度で溶解した。この溶液に他のペプチド前駆体アセチル−C(Acm)Ttds−CCEPN17(表5のペプチド4)(6mg;1mg/mL)を添加した。2つのペプチド前駆体の実効モル比は2.2:1のBr−アセチル−GGGEP1N17:Ac−C(Acm)Ttds−CCEPN17であった。三量体化反応のモニターは、Waters−Micromass LCZ Platformを使用するLC−MS分析によって、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)で溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して行った。溶出剤Bを以下の勾配で使用した:35%−70% B(20分)−80%(3分)、流速1mL/分。チオエーテル形成反応は円滑に進行した。2時間後、1つの生成物が形成され、その質量は、2つのチオエーテル結合を介して2つのBr−アセチル−GGGEP1N17ペプチド鎖に連結された1つのAc−C(Acm)Ttds−CCEPN17ペプチド鎖に相当する。溶液に20μLのTFAを添加し、溶液を、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で水中の60%アセトニトリル、0.1%TFA、流量1mL/分の溶出剤を使用するゲル透過クロマトグラフィー(GPC)によって直接精製した。溶出画分の分析は、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)で溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用するHPLCによって行った。溶出剤Bを以下の勾配で使用した:45%−65% B(20分)−80%(3分)、流速1mL/分、LC−MS Waters−Micromass LCZ Platform、正モード(ES+)。予想MWは17489Daであり、測定MWは17489Daであった。
【0122】
Ac−C(Acm)(チオEPN17)3のアセチル化システインの脱保護によるAc−C(チオEPN17)3の形成−
精製したコイルドコイル前駆体ac−C(Acm)(チオEPN17)3を20mg/mlの濃度でTFA、アニソール2%および50当量のAgOTf(トリフルオロメタンスルホン酸銀)に40℃で3時間溶解した。氷冷した無水エーテルを反応混合物に添加し、沈殿物を遠心によって単離した。沈殿物を氷冷した無水エーテルで2回洗浄した。ペプチドをH2O/アセトニトリル、0.1%TFAに再溶解し、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で溶出剤として水中の60%アセトニトリル、0.1%TFA、流速1mL/分を使用するゲル透過クロマトグラフィー(GPC)によって精製した。精製したペプチドのキャラクタリゼーションは、Micromass LCZ platformでエレクトロスプレー質量分析法によって行った。理論的平均MWは17415Daであり、測定MWは17416Daであった。
【0123】
ac−C(チオEP17GluN17)3およびac−C(チオEPN17)3とOMPCの接合−
グラム陰性細菌からOMPCを精製する様々な方法が計画されている(Fraschら,1997,J Exp.Med.140:87;Fraschら,1978,J Exp.Med.147:629;U.S.Pat.番号4,707,543;Heltingら,1981.Acta Path.Microbiol.Scand.Sect.C.89:69;およびU.S.Pat.番号4,271,147)。N.meningitidis Bの改良外膜タンパク質複合体(improved Outer Membrane Protein Complex(iOMPC))は、Fuが米国特許番号5,494,808に記載したような当業界で公知の技術を使用して得られる。
【0124】
1.36mLのNeisseria meningitidisの改良外膜タンパク質複合体(iOMPC)溶液(7.13mg/ml)に、0.5MのNaHCO3(0.151mL)を最終濃度50mM,pH8.5まで添加した。この溶液に、0.407mLのヘテロ二官能性架橋剤スルホスクシニミジル4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシレート(sSMCC,Pierce Chemical Co.)の20μM溶液を2倍過剰量で滴下した(OMPCのリシン残基に対して、0.42μmolのリシン/mgのOMPCタンパク質)。0.045mLの0.5MのNaHCO3をさらに添加することによってpHを再調整した。溶液を暗中、4℃で1時間熟成後、1MのNaH2PO4溶液(16μl)を添加してpHを中性まで低下させた。溶液を4℃で300K MWCO DispoDialyser(Spectrum Laboratories Inc.,CA)を使用し、バッファを6回交換し(2時間毎)、2Lの20mMのHEPES,pH7.3、(4−(2−ヒドロキシエチル)ピペラジン−1−エタンスルホン酸)、2mMのEDTA(エチレンジアミン四酢酸)に透析して余剰の試薬を除去した。透析後に合計1.9mLの活性化OMPC(aOMPC)を回収した。
【0125】
2つのCys−含有ペプチド、ac−C(チオEP17GluN17)3およびac−C(チオEPN17)3の0.5mg/mlの予製液を0.1MのHEPES、2mMのEDTA,pH7.3の脱ガス溶液中で調製した。沈殿を生じることなくaOMPCに安全に組込めるペプチドリガンドの最大量を確定するために、先ずaOMPCを漸増量のペプチドリガンドとインキュベートする小規模試験で接合反応を追跡した。OMPCに組込めるマレイミド基の最大数は1mgのOMPCあたりのリシン残基総数によって限定され、0.42μMのリシン/mgのOMPCである。OMPCの平均MWが40×106であると考えると、これは16,000モルのリシン/1モルのOMPCに相当する。これらのうちの一部分だけがsSMCCによって活性化され(35%以下)、これは達成可能な最大ペプチド負荷約5000モルに対応する。この場合にはaOMPCをOMPC1モルあたり1000、2000および3000モル過剰量のペプチドリガンドとインキュベートした。1時間後、サンプルをaOMPCサンプルに比較して沈殿の存在または濁りの増加を点検した。ac−C(チオEP17GluN17)3の場合には、3000モルのリガンド/1モルのOMPCという最大モル過剰量の使用まで沈殿または濁りの増加は全く観察されなかったが、ac−C(チオEPN17)3の場合には、3000モル/OMPCモルで多少の沈殿が観察され、2500モル/OMPCモルでは完全溶解であった。
【0126】
これらの観察に基づいて大規模反応を行った。20mMのHEPES、2mMのEDTA,pH7.3中の0.75mL(3.8mg)のaOMPCに7mLのac−C(チオEP17GluN17)3ペプチド予製液を、緩やかに渦流させながら滴下した。これは3000モル過剰量のペプチドモル数/OMPCモルに対応する。ac−C(チオEPN17)3の場合は、20mMのHEPES、2mMのEDTA,pH7.3中の0.45mL(2.3mg)のaOMPCに9mLのac−C(チオEPN17)3ペプチド予製液を緩やかに渦流させながら滴下した。これは2500モル過剰量のペプチドモル数/OMPCモルに対応する。最終コンジュゲートのペプチド負荷を定量するためにマレイミド活性化OMPCのサンプルをブランクとして維持した。接合反応混合物を暗中、4℃で17時間熟成した。次いで、最終濃度15mMまでのμ−メルカプトエタノール(総添加量10−12μL)を、暗中、4℃で1時間使用してOMPCの残留マレイミド基を反応停止させた。溶液を300K MWCO DispoDialyserで1Lの20mMのHEPES,pH7.3を4℃で4時間毎に4回交換して透析し、接合しなかったペプチドおよびβ−メルカプトエタノールを除去した。Lowryアッセイ(Lowryら,1951,J.Biol.Chem.193:265)によって濃度を測定すると、0.41mg/mLのOMPC−(チオEP17GluN17)3および0.32mg/mLのOMPC−(チオEPN17)3が存在した。コンジュゲートおよびaOMPCサンプルを、排気した密封ガラス管に入れ共沸HClと共に110℃で70時間加水分解した。アミノ酸解析によってアミノ酸組成を決定した。OMPCタンパク質へのペプチド接合負荷は、コンジュゲートアミノ酸組成をOMPC担体およびペプチドリガンドの双方の組成に比較し、データの重回帰最小二乗分析によって決定した(Shulerら,1992,J.Immunol.Methods 156:137−149)。コンジュゲートOMPC−(チオEP17GluN17)3では得られたペプチド対OMPCモル比は541であり、OMPC−(チオEPN17)3では得られたペプチド対OMPCモル比は443であった。
【0127】
結果−
この実施例は、CCEZN17配列(配列:44)に基づく2つのチオエーテル結合安定化キメラペプチドの設計を記載している。第一の三量体コイルドコイルAc−C(Acm)−(チオEP17GluN17)3は17アミノ酸のEZ−様スカフォールドに基づき、第二のコイルドコイルAc−C(Acm)−(チオEPN17)3は24アミノ酸のEZスカフォールドに基づく。2つの構築物は詳細に前述したように個々のキメラペプチド間のチオエーテル結合の形成によって共有結合的に安定化されている。設計したコイルドコイルのスカフォールド配列内部の様々な場所に、変化すなわち突然変異または側鎖の誘導体化を導入し、表5に“下線を付けて”示す。
【0128】
【表5】
【0129】
C(Acm)−(チオEP17GluN17)3は1つのCC−キメラN−ペプチドすなわちCCCE17N17(配列:88;表5のペプチド2)および2つの誘導体化キメラN−ペプチドすなわちブロモアセチル−GGGE17GluN17(配列:87;表5のペプチド1)から成る。CCCE17GluN17のNH2−末端システイン残基は、Acm(アセタミドメチル)基で保護され、残りの非保護システイン残基のおのおのは各誘導体化キメラN−ペプチドのブロモ−アセチル部分と共に化学選択的チオエーテル結合を形成する。C(Acm)−(チオEP17GluN17)3に内包された各キメラペプチドのスカフォールドドメインは、EZスカフォールド(配列:15)の24アミノ酸の代わりに17アミノ酸およびリシンからグルタミン酸への突然変異を含む短縮EZコイルドコイル(“E17Glu”スカフォールド;配列:84)から成る。C(Acm)−(チオEP17GluN17)3のスカフォールドドメインは、三量体コイルドコイルに内包された個別ペプチドのおのおののスカフォールド領域内部の1つのリシン残基へのPEG236部分の付着によって選択的に遮蔽されている(表5のペプチド1および2の下線部分)。各ペプチドのPEG付加リシン残基は、スカフォールドヘリックスのヘプタッドリピートの“f”位置に局在し、該残基のε−アミノ基を介してPEG部分が付着している。24アミノ酸から17アミノ酸へのスカフォールドの短縮は、得られるコイルドコイルC(Acm)−(チオEP17GluN17)3とOMPC担体との接合を促進し、また、接合に使用した水性バッファ中のペプチドの可溶化を、コイルドコイルの等電点(“pI”)を下げることによって促進する。PEG236によるリシン残基のεアミノ基の誘導体化に伴う遊離アミノ基の除去、および、LysからGluへの突然変異はいずれも、得られるコイルドコイルのpIをいっそう低下させる。この実施例に記載したHIV gp41融合中間体の種々のミメティクスの等電点の比較については表6を参照するとよい。マレイミド活性化OMPC担体へのC(Acm)−(チオEP17GluN17)3の接合は、C−(チオEP17GluN17)3を形成するチオール化ペプチドを得るために脱保護されたNH2−末端システイン残基を介して惹起される。
【0130】
C(Acm)−(チオEPN17)3は1つのCC−キメラN−ペプチドすなわちCCCEZN17(配列:90;表5のペプチド4)と2つの誘導体化キメラN−ペプチドすなわちブロモアセチル−GGGEZN17(配列:89;表5のペプチド3)とから成る。CCCEZN17のNH2−末端システイン残基はAcm(アセタミドメチル)基によって保護され、残りの非保護システイン残基のおのおのは各誘導体化キメラN−ペプチドのブロモ−アセチル部分と共に化学選択的チオエーテル結合を形成する。C(Acm)−(チオEPN17)3に内包された各キメラペプチドのスカフォールドドメインは、EZスカフォールド(配列:15)の24アミノ酸から成る。C(Acm)−(チオEPN17)3のスカフォールドドメインは、三量体コイルドコイルに内包された個別ペプチドのおのおののスカフォールド領域内部の2つのリシン残基へのPEG236部分の付着によって選択的に遮蔽されている(表5のペプチド3および4の下線部分)。各ペプチドのPEG付加リシン残基は、スカフォールドヘリックスのヘプタッドリピートの“f”位置に局在し、該残基のε−アミノ基を介してPEG部分が付着している。PEG236によるリシン残基のεアミノ基の誘導体化は遊離アミノ基を除去し、得られるコイルドコイルのpI低下を助ける(表6)。マレイミド活性化OMPC担体へのC(Acm)−(チオEPN17)3の接合は、C−(チオEPN17)3を形成するチオール化ペプチドを得るために脱保護されたNH2−末端システイン残基を介して惹起される。
【0131】
【表6】
【図面の簡単な説明】
【0132】
【図1】図1Aは、共有結合安定化ホモ三量体コイルドコイル(CCIZN17)3を生じる酸化反応の概略図である。三量体コイルドコイルを安定化する3つのジスルフィド結合が各ペプチド鎖間に1つずつ存在する。図1Bは、(CCIZN17)3([配列2]3)を安定化するジスルフィド結合の化学構造図である。3つのジスルフィド結合がNH2−末端システインアミノ酸残基のチオール(−SH)基の間に局在しており、その化学構造が図示されている。2つのシステイン残基の下流に局在するアミノ酸は大文字の一文字記号で示されている。
【図2】図2Aは三量体“IZ”スカフォールドのヘリカルホイール表示である。ヘプタッドリピートの“f”位に局在する3つのリシンアミノ酸(“K”)がコイルドコイルの外側に近い位置に存在する状態で示されている。図2Bの(CCIPN17)3の配列表示からわかるように、これらのリシン残基の2つが、側鎖のε−アミノ基をm−dPEGTM酸(MW=236)で誘導体化して選択的に遮蔽された三量体コイルドコイル(CCIPN17)3を生成するために選択された。IZスカフォールドドメイン(イタリック体)のPEG付加リシン残基は下線を付けて示す。HIV N17ドメインは大文字で示す。
【図3】(CCIPN17)3ペプチドワクチンの概略モデルである。個々のキメラペプチドCCIPN17は長さ約62オングストロームの円筒として示されている。暗い灰色部分は長さ約36オングストロームのIZスカフォールドドメインを表し、明るい灰色部分は長さ約25オングストロームのHIV gp41 N−ペプチドのN17領域を表す。m−dPEGTM酸(MW=236)部分は長さ約15オングストロームの長方形として示されている。
【図4】(CCIPN17)3によるホモロガスおよびヘテロロガスプライム/ブースト免疫方式で行ったモルモット試験のELISA結果を示す。プライム/ブーストの組合せは各棒グラフ群の下部に示す。各抗原に対するELISA幾何平均力価(GMT)は血清中に存在する対応特異性所有抗体の相対量を正確に反映する。
【図5】図5Aは、アミロイドβ−ペプチド(Aβ)1−42ペプチドのアミノ酸配列を示す。免疫優性T細胞エピトープ(Monsogeo,A.ら,2003,J.Clin.Invest.112:415−22)は一本の下線で示し、防御B細胞エピトープ(McLaurin,J.ら,2002,Nat.Med.8:1263−9)は二重下線で示す。図5Bは、Elan/Wyeth,AN−1792(Aβ1−42)およびACC−001((Aβ1−16)の2つのAβペプチドワクチンのアミノ酸配列を示す。図5Cは、本発明で提案したAβ1−42ペプチドワクチンの概略図であり、2つのリシンアミノ酸残基(下線部分)をm−dPEGTM酸(MW=236)部分で誘導体化して免疫優性T細胞エピトープを選択的に遮蔽し、ペプチドのNH2−末端部分の防御B細胞エピトープを認識し易くしている。
【図6】図6Aは、HIV−1 gp41エクトドメイン(配列6)中のトリプトファン(Trp)−富化モチーフのアミノ酸配列、および、Schibli,DJ.ら(2001,Biochemistry 40:9570−8)によって同定されたその二次構造の概略図を示す。一本下線で示したアミノ酸残基(W672、F673、1675、T676およびW680)がHIV−中和抗体4E10と相互作用することは最近になって確認された(Cardoso,R.M.F.ら,2005,Immunity vol.22:163−173)。二重下線で示したアミノ酸残基(W666、W370およびW678)は膜に面したTrp残基である。図6Bは、ロイシンジッパーGCN4に基づく提案4E10ターゲット化ワクチンの概略図を示す。下線を付けたアミノ酸残基は、“GCN4コイル”ペプチド(配列8)に適合性のダイマー化表面を有しているペプチド“4E10コイル”(配列7)を作製するために、4E10結合性アミノ酸を含有するTrp富化領域のいくつかの膜対面残基に導入した突然変異を表す。ジスルフィド結合がGCN4/4E10コイルドコイルをさらに強化する。
【図7】リシン残基(二重下線)にm−dPEGTM酸(MW=236)部分を付着させることによってPEG修飾した図6Bの提案4E10ターゲット化ワクチンの概略図である。
【図8】チオエーテル化学選択的結合によって共有結合的に安定化され本発明によってポリマー修飾できる三量体コイルドコイル構造NH2−C(Fm)(チオIZN17)3の化学構造図を表す。三量体コイルドコイルは、1つのCCキメラN−ペプチドすなわちNH2−C(Fm)Ttds−CCIZN17(配列76)の2つのシステインアミノ酸残基のおのおののチオール(−SH)基間に局在する2つのチオエーテル結合と、2つの誘導体化キメラペプチドすなわちBr−アセチル−GGGIZN17(配列77)のおのおのの求電子性ブロモ部分とによって形成される。結合システイン残基のCOOH−末端に局在するアミノ酸残基は一文字記号で示している。このコイルドコイル構造は、CC−キメラN−ペプチドのNH2−末端システイン残基のチオールを遮蔽するフルオレニルメトキシ(“Fm”)保護基およびNH2−末端の遊離アミノ基を有している。
【技術分野】
【0001】
本発明は、結合部位がポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーを部位特異的にポリペプチドに付着させる処理を含むポリペプチド内部の1つ以上の結合部位の遮蔽方法に関する。1つの実施態様では、隠蔽された結合部位がポリペプチド抗原内部のエピトープである。開示された方法による結合部位(例えば、エピトープ)の遮蔽は、該結合部位とその対応受容体との相互作用によって誘導される生物応答を排除するかまたは実質的に減退させる効果を有しており、ポリペプチドの非隠蔽部分に生物応答が集中することを援助する。本発明はさらに、本文中に開示された方法によって製造された医薬製品(例えば、ポリマー修飾抗原およびそれらを含むワクチン組成物)およびそれらの使用に関する。これらは脊椎動物生体組織、好ましくはヒトまたは商業用もしくは飼育用の獣医学的に重要なヒト以外の哺乳動物のような哺乳類宿主に直接的に導入されると、ポリペプチドの非隠蔽部分に特異的免疫応答を誘導し、該哺乳動物体内で選択的免疫防御を生じる。
【背景技術】
【0002】
タンパク質内部の生物活性部位またはドメイン(例えば、免疫原性エピトープ)の選択的隠蔽または“遮蔽”の自然発生は、宿主免疫応答を破壊するために病原体が使用する戦略であると説明されてきた。例えば、インフルエンザウイルスの赤血球凝集素(HA)中で、モノクローナル抗体のエピトープはN−結合オリゴ糖鎖によって遮蔽されている(Wiley,D.C.ら,1981,Nature289:373−8)。Skehelおよび共同研究者らは、抗体エピトープの極めて近傍に付加N−結合オリゴ糖を含むHAをもつ変種に対して選択された抗体の存在下のウイルス増殖を証明した(1984,Proc.Natl.Acad.Sci.U.S.A.81:1779−83)。また、HIVのエンベロープ糖タンパク質のgp120サブユニットについては、宿主免疫系による中和を逃れるメカニズムであるグリコシル化によって重要なエピトープが掩蔽されることが詳細に記載されている(たとえば,Burton,D.R.ら,2004,Nat.Immunol.5:233−6;Johnson,W.E.and R.C.Desrosiers,2002,Annu.Rev.Med.53:499−518;Kwong,P.D.ら,1998,Nature 393:648−59;およびWei,X.ら,2003,Nature 422:307−12)。また、他のHIVエピトープについては構造的隠蔽によって同じ効果が達成されている(Kwong,P.D.ら,2002,Nature 420:678−82)。この“免疫回避”メカニズムはしばしば、デコイとして作用する免疫優性エピトープの慢性提示という結果を生み、ふつうなら最適防御を確保するより強力な防御エピトープ(“中和”エピトープ)に免疫系が集中することを妨害する。最終的にこのメカニズムはワクチン効果を低下させる一因となるであろう。
【0003】
糖質もまた非抗原性受容体結合部位を隠蔽できる。例えば、Goto,H.およびK.Kawaoka(1998,Proc.Natl.Acad.Sci.U.S.A.95:10224−8)は、インフルエンザウイルスのノイラミニダーゼタンパク質の特異的部化をオリゴ糖側鎖によって隠蔽し、該タンパク質へのプラスミノーゲンの結合を阻止することを提案した。空間的に近接したウイルスの赤血球凝集素を開裂し融合受容能を与えるためにはプラスミノーゲンが必要なので、糖質側鎖はウイルスの感染力を大きく変質させる。
【0004】
ポリマー遮蔽は、医薬タンパク質の免疫原性を減退させ、望ましくない免疫原性副作用を最小に抑制し、腎濾過経由のクリアランスを減少させることによって最終的に治療効果を改善するために広く使用されている方法である(総説:Schellekens,H.,2002,Clinical Therapeutics 24:1720−40参照)。広範囲に応用された治療用タンパク質遮蔽方法では、高分子量(“HMW”)の線状または分枝状ポリエチレングリコール(“PEG”)鎖を該タンパク質に付着させる(Harris,J.M.and R.B.Chess,2003,Nat.Rev.Drug Discov.2:214−21;米国特許番号4,179,337、Davisらに許諾)。所望効果はタンパク質の完全隠蔽なので、治療用タンパク質の修飾に使用するPEG部分は付着対象タンパク質と同等のサイズを有している。
【0005】
もっと最近には、選択エピトープ特異的にタンパク質を遮蔽する構想が提案され、“イムノフォーカシング”と命名された。この戦略は、タンパク質抗原または免疫原の内部の免疫優性の非中和性エピトープの選択的遮蔽であり、その結果として免疫応答がより強力な中和エピトープに指向および/または集中する(参照:Burton,D.R.ら,2004,Nat.Immunol.5:233−6;Delves,PJ.ら,1997,Mol. Med.Today 3:55−60;およびPantophlet,R.and D.R.Burton,2003,Trends Mol.Med.9:468−73)。この方法は、上記のような持続性病原体感染症に好都合に展開する複合的免疫回避戦略の対策として開発された。特定例として、Garrity,R.R.は、HIV gp120のV3超可変ループに付加N−結合グリコシリーション部位を組込んで、該タンパク質に対する抗体応答をかなりシフトさせた(1997,J.Immunol.159:279−89)。Pantophlet,R.らによって行われたもっと体系的な研究は、モノマーgp120内部に7つのN−グリコシレーションモチーフを付加すると、広範な非中和抗体パネルへの結合は完全に排除されるが、中和モノクローナル抗体mAB b12への結合は低効率で維持されることを示した(Pantophlet,R.and D.R.Burton,2003,前出 9:468−73;Pantophlet,R.ら,2003,J.Virol.77:5889−901)。しかしながら重要なことは、グリコシレーションがT細胞認識を必ずしも排除しないことである。実際、T細胞がグリコシル化ペプチドに結合することは多くの文献に記載されており(参照:たとえば,Cudic,M.ら,2002,Bioorg.Med.Chem.10:3859−70;Glithero, AJ.ら,1999,Immunity 10:63−74;Haurum,J.S.ら,1999,J.Exp.Med.190:145−50;Jackson,D.C.ら, 1994,Virology 199:422−30;およびRudd,P.M.ら,2001,Science 291:2370−6)、特に、糖質側鎖が短い場合もある(Cudicら,2002,前出;Jacksonら,1994,前出)。従って本発明は、SMW PEG部分を非限定例とする糖質側鎖以外の低分子量(“SMW”)ポリマー部分を結合部位の内部または近傍のペプチド/タンパク質に結合させること(“PEG付加”)によるペプチドまたはタンパク質内部の受容体の結合部位の選択的遮蔽に関する。
【0006】
1991年8月20日付けでGethingらに許諾された米国特許番号5,041,376は、オリゴヌクレオチド突然変異誘発を使用して補足N−結合オリゴ糖側鎖を部位特異的場所に組込むことによってタンパク質のエピトープを遮蔽する一般的方法を開示している。
【0007】
それぞれ1996年12月17日および1998年12月19日付けでGarrityらに許諾された米国特許番号5,585,250および5,853,724は、中和抗体応答がV3ループから逸れてタンパク質のより強力な中和部分を指向するようにしたHIV−1 gp120/160のV3ループのエピトープ鎮静方法を開示している。
【発明の開示】
【0008】
本発明は、結合部位がポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量(“SMW”)水溶性ポリマーを部位特異的にポリペプチドに付着させる処理を含むポリペプチド内部の1つ以上の結合部位の遮蔽方法に関する。本発明の1つの実施態様では、本文中の方法によって遮蔽される結合部位(すなわち、機能部位)はポリペプチド抗原内部の免疫原性または抗原性エピトープである。選択された結合部位(例えば、エピトープ)の隠蔽は、結合部位とそれらの対応受容体との相互作用によって誘導される望ましくない生物応答(例えば、免疫応答)を排除または減退させ、生物応答を該ポリマー修飾ポリペプチドの非隠蔽部分に集中させる。従って本発明は、少なくとも1つのSMW水溶性ポリマーを部位特異的にペプチド/ポリペプチドに付着させ、該結合部位/エピトープをポリマーによって選択的に隠蔽するペプチドまたはポリペプチドの結合部位またはエピトープの遮蔽方法に関する。この場合、ペプチド/ポリペプチドは1つ以上の結合部位/エピトープを内包している。SMW水溶性ポリマーは、遮蔽すべき結合部位/エピトープの内部または近傍の場所(例えば、アミノ酸)でポリペプチドに付着する。本発明の1つの実施態様では、ポリペプチドへの部位特異的付着によってポリペプチド内部の結合部位を選択的に遮蔽するために使用される低分子量ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から選択される。別の実施態様では、低分子量水溶性ポリマーがPEGである。
【0009】
本発明はさらに、より有益な応答(例えば、中和応答)をタンパク質/ペプチド内部の択一的エピトープに向けさせるために、ポリペプチド抗原内部に存在する免疫優性および/または有益でないエピトープを免疫減衰する方法に関する。この場合、免疫優性および/または有益でないエピトープは抗原に付着した少なくとも1つの低分子量水溶性ポリマーによって選択的に隠蔽される。従って本文中の方法は、免疫応答が免疫由来細胞またはタンパク質による認識がほぼ不可能になった隠蔽エピトープから逸れて、1つ以上の非隠蔽エピトープに集中するという結果を得るために使用される。抗原への部位特異的付着によって抗原内部の有益でないエピトープを選択的に遮蔽するために使用される低分子量ポリマーは、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から選択できる。別の実施態様では、低分子量水溶性ポリマーがPEGである。
【0010】
本発明の1つの特定実施態様は、エピトープがポリマーによって選択的に隠蔽されるように抗原内部の少なくとも1つのアミノ酸残基にSMW PEGを非限定例とする少なくとも1つのSMW水溶性ポリマーを部位特異的に付着させる処理を含むポリペプチド抗原のエピトープ遮蔽方法に関する。この場合、該エピトープは、エンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または少なくとも一部分を模倣する共有結合安定化三量体コイルドコイル構造のスカフォールドドメイン内部に局在している。該コイルドコイル構造は、ペプチド間の1つ以上の共有結合によって共有結合的に安定化された3つのキメラペプチドを含み、キメラペプチドのおのおのは、エンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでいる。本発明のこの部分の具体的実施態様で、エンベロープウイルス膜融合タンパク質はHIV gp41である。
【0011】
従って本発明の1つの実施態様は、エピトープがポリマーで選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーをコイルドコイル構造に部位特異的に付着させる処理を含む、HIV gp41の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造のエピトープの遮蔽方法に関する。別の実施態様で、隠蔽エピトープは、コイルドコイル構造のスカフォールドドメイン内部に局在し、該コイルドコイル構造は3つのキメラペプチドを含み、キメラペプチドのおのおのは、HIV gp41 NH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含む“スカフォールドドメイン”を有しており、該ペプチドはジスルフィド結合を非限定例とする1つ以上の共有結合または化学選択的結合反応から生じる共有結合のいずれかによって共有結合的に安定化されている。
【0012】
本発明はまた、本文中の方法によって製造されたポリマー修飾ペプチドまたはタンパク質、それらを含有する医薬/ワクチン組成物、および、哺乳動物体内で選択的免疫防御を誘導するための該医薬/ワクチン組成物による哺乳動物を非限定例とする動物の疾病状態に対する免疫方法に関する。
【0013】
従って本発明の1つの実施態様は、本文中の方法によってポリマー修飾されたキメラペプチドに関する。該ペプチドは、三量体コイルドコイル構造を生成するように共有結合的に安定化でき、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質(“N−ペプチド”部分)のNH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合した三量体コイルドコイル(“スカフォールド”部分)を形成できるα−ヘリカルスカフォールドタンパク質を含むアルファ(“α”)−ヘリカルドメインを含む。PEGを非限定例とする1つ以上の低分子量ポリマーをスカフォールドドメイン配列内部に局在する選択アミノ酸残基に付着させることによって部位特異的に遮蔽されるのは、少なくとも1つ以上のエピトープを含むこのスカフォールドドメインである。本発明はさらに、本文中に記載した方法によって製造したポリマー修飾キメラペプチドから成る共有結合安定化三量体コイルドコイル構造に関する。該ポリマー修飾コイルドコイルの非限定例は(CCIPN17)3、C(チオEPN17)3およびC(チオEP17GluN17)3である。
【0014】
本発明はさらに、特定の疾患または状態(非限定的にヒト免疫不全ウイルス−1感染/AIDSを含む)を特異的に認識する免疫応答を誘導するために、脊椎動物生体組織、好ましくはヒトまたは商業用または飼育用の獣医学的に重要なヒト以外の哺乳動物のような哺乳類宿主に直接的に導入できる医薬製品(例えば、ワクチン)、その製造および使用に関する。該医薬製品および/またはその抗原性接合体は、本文中の方法によって製造したポリマー修飾ポリペプチド抗原を含む。
【0015】
本文中に使用した“HIV”は、HIV−1、HIV−2またはHIV−1および/またはHIV−2を表す。
【0016】
本文中に使用した“中和性”はin vitro細胞/ウイルスベースアッセイでウイルス感染を防止する抗体の能力を表すために当業界で使用されている。中和活性は該特異的抗体に対するIC50値として定量的に測定できる。“中和抗体”または“HIV中和抗体”は、少なくとも1つのHIV単離物を中和するための当業界で承認された感染力アッセイで証明できる。
【0017】
本文中に使用した“PEG”は−ポリエチレングリコール−を表す。
【0018】
本文中に使用した“SMW”は低分子量を表し、“HMW”は高分子量を表し、“MW”は分子量を表す。
【0019】
本文中に使用した“SPPS”は固相ペプチド合成を表す。
【0020】
本文中に使用した“Michael受容体”は求核種(例えば、O−、C−またはS−)と反応できる分極した求電子性炭素炭素二重結合を含有する化学部分を表す。
【0021】
本発明は概括的には、特定受容体によって正常に認識されるペプチドまたはタンパク質内部の機能部位の選択的隠蔽または遮蔽方法を記載する。本発明の1つの実施態様では、開示された方法によって隠蔽すべき機能部位(すなわち、結合部位)は、免疫原性または抗原性エピトープ、例えばT細胞またはB細胞エピトープであり、該エピトープを認識することを阻害される受容体はT細胞の抗原受容体または免疫グロブリン分子である。選択されたエピトープの隠蔽は、ペプチド/タンパク質内部の択一的エピトープおよび一般にペプチド/タンパク質の残りの部分全部の免疫原性を損なうことなくペプチド/タンパク質の一部分に対する望ましくない免疫反応性を排除する。ペプチドまたはタンパク質の内部の1つ以上の結合部位(例えば、エピトープ)の選択的隠蔽は、本文中に記載したように、ペプチド/タンパク質内部の1つ以上の適正場所、すなわち、隠蔽すべき結合部位の内部または極めて近傍に1つ以上の低分子量(“SMW”)ポリマー(例えば、ポリエチレングリコール)を付着させることによって得られる。本発明の1つの実施態様では、SMWポリマーは、隠蔽すべき結合部位の内部または極めて近傍の1つ以上のアミノ酸残基に付着できる。該アミノ酸はコード化または非コード化残基でよい。従って本発明は、結合部位がポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーを部位特異的にポリペプチドに付着させる処理を含む、ポリペプチド内部の結合部位の遮蔽方法に関する。結合部位の非限定例はポリペプチド抗原のエピトープである。
【0022】
上述のように、本文中の方法は、ペプチドまたはタンパク質の内部に局在する受容体結合部位の選択的遮蔽に広く応用できるが、本文中では、免疫グロブリン分子またはT細胞受容体によって認識されるペプチドまたはタンパク質内部のエピトープ(すなわち、B−またはT−細胞エピトープ)の選択的遮蔽について本発明を詳細に記載し、その具体例を後述する。しかしながらこの記載は、記載したエピトープの選択的隠蔽方法に本発明を限定するものではない。当業者は、本文中に詳細に記載された方法が、対応受容体による結合部位の認識を防止する目的で対象ペプチドまたはポリペプチド内部の特異的結合部位を選択的に遮蔽するためにどのように修正できるかを理解されるであろう。従って、本文中に使用した“結合部位”は他のタンパク質(例えば、抗体または細胞性受容体)によって認識されるおよび/または相互作用するアミノ酸残基またはそれに付着した糖側鎖を含むペプチドまたはポリペプチドの領域を表す。従って、本発明の一部として記載した方法によって隠蔽された抗原性/免疫原性エピトープは、本文中に定義した“結合部位”を表す。タンパク質性結合部位は、(例えば、触媒活性部位でしばしば観察される)選択的非連続アミノ酸によって生成された適切に折り畳まれたペプチド/ポリペプチドの三次元コンホメーショナル領域または連続アミノ酸の線状エピトープ(例えば、T細胞エピトープ)を含み得る。
【0023】
本発明は、機能部位がポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーをペプチド/ポリペプチドに部位特異的に付着させる処理を含むペプチドまたはポリペプチドの機能部位(すなわち、結合部位)の遮蔽方法に関する。本発明の1つの実施態様で、機能部位はポリペプチド抗原内部のエピトープである。従って、本発明の1つの実施態様は、エピトープがポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーを抗原に部位特異的に付着させる処理を含むポリペプチド抗原のエピトープの遮蔽方法に関する。本文中に使用した“エピトープ”という用語は、従来の免疫原性(すなわち、体液性または細胞性応答を誘発する能力)および/または抗原性(すなわち、T細胞受容体の抗体と特異的に結合する能力)応答を誘発できるタンパク質決定基を包含する。従って、免疫原性エピトープは本発明の一部として記載された方法によって選択的に隠蔽できる多くの種類の結合部位のひとつにすぎない。エピトープが通常はアミノ酸または糖側鎖のような分子の化学的活性表面の集まりから成り、しばしば特異的三次元構造特性および特異的電荷特性を有していることは公知である。コンホメーショナルエピトープとノンコンホメーショナルエピトープとは、変性用溶媒の存在下で前者の結合は消失するが後者の結合は消失しないことによって識別される。
【0024】
ペプチド/ポリペプチドのエピトープの選択的遮蔽方法は、該ペプチド/ポリペプチドが2つ以上のエピトープを内包し、該エピトープの1つ以上に対する特異的免疫応答が望ましくそれ以外の免疫応答は排除したい場合に特に有用である。従って、本文中の方法は、免疫応答が免疫由来細胞による認識が不能になった隠蔽エピトープから逸れて、1つ以上の望ましい非隠蔽エピトープに集中するという結果を得るために使用される。ペプチド/ポリペプチド内部の適切な場所に低分子量ポリマー分子を付着させて隠蔽エピトープとその免疫細胞受容体(例えば、免疫グロブリン分子)との会合を阻止することによって、エピトープは物理的にも機能的にも有効に遮蔽される。例えば、ポリペプチド抗原(例えば、合成ペプチドまたは病原性タンパク質)は、宿主生物体内で免疫応答を選択的に誘発し該抗原内部の他のエピトープを実質的に排除するような1つ以上の免疫優性エピトープを含有し得る。このような免疫優性エピトープが有益でない免疫応答(例えば、非中和抗体または非保存中和抗体)を誘発し、同じペプチド/タンパク質内部に局在する免疫刺激性の低いエピトープに対する有益な免疫応答が失われるような場合、免疫刺激性の低いエピトープに免疫応答を集中させ、免疫優性エピトープから逸れるようにすることが重要である。従って、本発明の1つの実施態様は、より有益な免疫応答(例えば、中和応答または保存中和応答)がタンパク質/ペプチド内部の択一的エピトープを指向するように有益でない免疫優性エピトープを免疫減衰させることに関する。重要なことは、有益でない免疫応答を誘導するエピトープの遮蔽による無益な免疫応答の免疫減衰が望ましくない応答の完全な排除を必ずしも意味しないことである。
【0025】
本発明はさらに、本文中の方法によって作製したポリマー修飾ペプチドまたはタンパク質、それらを含有する医薬/ワクチン組成物、哺乳動物体内で選択的免疫防御を誘導するための医薬/ワクチン組成物による疾患状態に対する哺乳動物の免疫方法に関する。従って本発明は、免疫応答を1つのポリペプチドに集中させる方法に関する。この場合、ポリペプチドは免疫応答を誘導できる1つ以上のエピトープを含んでおり、方法は、少なくとも1つのエピトープがポリマーによって選択的に隠蔽され該ポリペプチドに対する免疫応答が該ポリペプチド内部の非遮蔽領域を指向するように、少なくとも1つの低分子量水溶性ポリマーを部位特異的にポリペプチドに付着させる処理を含む。従って本発明は、本文中の方法で少なくとも1つの低分子量水溶性ポリマーをポリペプチドに部位特異的に付着させることによって該ポリペプチド内部の1つ以上のエピトープを選択的に遮蔽することによってポリペプチドに対する免疫応答を変質させる方法に関する。
【0026】
後出の実施例の項のポリエチレングリコール(“PEG”)の使用によって証明されるように、本文中の方法で結合部位またはエピトープを選択的に隠蔽するために使用したポリマーは実質的に直鎖状のポリマーであり、実質的に非免疫原性、無毒性、高度に可溶性で生物活性を有していない。本文中の方法で結合部位またはエピトープを選択的に遮蔽するために使用したポリマーの好ましい実施態様としてPEGを選択した。しかしながらこのポリマーは限定でなく例示目的で使用しただけである。水溶性ポリマーの例は、メトキシポリエチレングリコールのようなポリアルキレングリコールおよびその誘導体、ポリエチレングリコールホモポリマー、ポリプロピレングリコールホモポリマーおよびエチレングリコール−プロピレングリコールコポリマー(ポリマーおよびコポリマーは未置換であるかまたは一端がアルキル基で置換されている)、ヘパリンおよびヘパリンフラグメント、ポリビニルアルコールおよびポリビニルエチルエーテル、ポリビニルピロリドン、アスパルタミド、デキストランおよびデキストリン誘導体、デキストリンおよびデキストリン誘導体でポリオキシエチル化されたポリオールを含む。具体的に記述した水溶性ポリマーの様々な誘導体も本文中の方法に使用できると考えてよい。上記のような水溶性ポリマー、特にPEGのようなポリアルキレンオキシド基材のポリマーは公知である(参照:たとえば,Poly(ethylene glycol)Chemistry:Biotechnical and Biomedical Applications,Ed.J.M.Harris,New York:Plenum Press,1992;Poly(ethylene glycol)Chemistry and Biological Applications,Eds.J.M. Harris and S.Zalipsky,ACS,1997;以下の公開番号を有する国際特許出願公開:WO 90/13540,WO 92/00748,WO 92/16555,WO 94/04193,WO94/14758,WO 94/17039,WO 94/18247,WO 94/28937,WO 95/11924,WO 96/00080,WO 96/23794,WO 98/07713,WO 98/41562,WO 98/48837,WO 99/30727,WO 99/32134,WO 99/33483,WO 99/53951,WO 01/26692,WO 95/13312,WO 96/21469,WO 97/03106,WO 99/45964;次の米国特許:4,179,337;5,075,046;5,089,261;5,100,992;5,134,192;5,166,309;5,171,264;5,213,891;5,219,564;5,275,838;5,281,698;5,298,643;5,312,808;5,321,095;5,324,844;5,349,001;5,352,756;5,405,877;5,455027;5,446,090;5,470,829;5,478,805;5,567,422;5,605,976;5,612,460;5,614,549;5,618,528;5,672,662;5,637,749;5,643,575;5,650,388;5,681,567;5,686,110;5,730,990;5,739,208;5,756,593;5,808,096;5,824,778;5,824,784;5,840,900;5,874,500;5,880,131;5,900,461;5,902,588;5,919,442;5,919,455;5,932,462;5,965,119;5,965,566;5,985,263;5,990,237;6,011,042;6,013,283;6,077,939;6,113,906;6,127,355;6,177,087;6,180,095;6,194,580;および6,214,966)。
【0027】
本発明の1つの実施態様で、ポリペプチド抗原のエピトープを非限定例とするポリペプチド内部の結合部位を該ポリペプチドへの部位特異的付着によって選択的に遮蔽するために使用した低分子量ポリマーは、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される。別の実施態様で、該低分子量水溶性ポリマーは、(CH2−CH2−O)−から成る水溶性反復単位をもつPEGである。PEGが有利な理由は、ポリエーテル主鎖が生物環境および大抵の化学反応条件下で不活性であり、修飾すべきペプチド/タンパク質の特定反応基に共有結合付着できる官能基を含むPEG誘導体(すなわち、“活性化”PEG)を生成するために必要な化学修飾を受入れ易い末端を有しているからである。様々な活性化PEGポリマーが当業界で公知であり、その官能基の非限定例は、カーボネート、活性エステル、アルデヒド、トレシラート、アクリレート、ケトン、アミノオキシ、アミン、カルボン酸、エステル、チオエステル、ハロゲン、チオール、シアノアセテート、ジパルミトイルホスファチジルエタノールアミン、ジステアロイルホスファチジルエタノールアミン、エポキシド、ヒドラジド、アジド、イソシアナート、マレイミド、メタクリレート、ニトロフェニル、オルトピリジルジスルフィド、シラン、スルフヒドリル、ビニルスルホン、スクシニミジルグルタレート、スクシニミジルスクシネートおよびコハク酸である。官能基は、修飾すべきポリペプチドの可用な反応基の種類に基づいて選択できる。さらに、1つのPEGポリマーが2つ以上のポリペプチドに付着して生じるポリペプチド架橋をできるだけ少なくしこれによって改善された均質性を与えるために誘導体化可能な末端を1つだけ有している一官能PEG誘導体(“mPEG”)を使用できる。ポリペプチドの最も普遍的な反応部位は、リシン残基のα−もしくはε−アミノ基または他のアミノ酸のNH2−末端アミノ基である。他の典型的な反応部位のリストは、システイン、ヒスチジン、アルギニン、アスパラギン酸、グルタミン酸、セリン、トレオニン、チロシンなどのアミノ酸およびアミノ酸のCOOH−末端カルボン酸を含む。あるいは、PEGを非限定例とする本発明のSMWポリマー分子をコード化または非コード化アミノ酸内部に存在する反応基以外の反応基との相互作用を介してターゲットポリペプチドまたはペプチドに部位特異的に付着させることもできる。例えば、糖質部分または合成リンカーがターゲットポリペプチド/ペプチド構造の一成分であるときにSMWポリマーは該糖質部分または合成リンカー内部の反応基と相互作用を介して付着できる。
【0028】
本文中の方法によってポリペプチドを選択的に修飾するために使用されるPEGを非限定例とする様々なポリマーは、低分子量ポリマーを包含する。一般に、本文中に使用した“低分子量”ポリマーは、約1000Da以下の分子量のポリマー、特にPEGポリマー(例えば、≦PEG1000)を表す。付着したポリマー部分の水溶性反復単位の数は様々な値を有し得る。しかしながら、例えば、付着PEG部分の場合、このような単位−(CH2−CH2−O)−)のより好ましい数は20単位までの範囲である。本発明に概説したペプチドまたはポリペプチド内部の選択結合部位またはエピトープの隠蔽に使用したPEGのような低分子量ポリマーは約1000Da以下の分子量を有すると予測されるが、ペプチド/タンパク質の修飾に使用できるポリマーのサイズ範囲が隠蔽すべき結合部位またはエピトープのサイズに大きく左右されることは当業者に理解されよう。このため、結合部位/エピトープの完全遮蔽または実質的遮蔽に高分子量ポリマーの付着が必要な場合には本発明は分子量約1000Da以下のポリマーの使用に限定されない。しかしながら、本文中の方法の最終目標は、隠蔽すべき結合部位/エピトープが含まれているペプチド/ポリペプチドの一部分だけを遮蔽することであって、タンパク質内部に存在するすべての結合部位/エピトープを全体的に遮蔽することではないと銘記するのが重要である。
【0029】
本発明は、1つ以上のエピトープ(例えば、望まない免疫応答を生じるエピトープ)または結合部位が選択的に隠蔽され、治療用物質の残部はより有益な択一的免疫応答または活性を誘導し易いように維持されているポリマー修飾した治療用ペプチドまたはポリペプチドに関する。従って、本発明の最終目標は、特定の結合部位/エピトープを隠蔽するために必要な程度にだけ治療用タンパク質を遮蔽することであるから、本発明方法の使用には低分子量非分枝状ポリマーがもっとも好ましい。ポリマー修飾タンパク質はこれまでにも記載されているが、本発明のポリマー修飾の性質、方法および目的はこのような従来の研究とは顕著に異なっている。従来技術で開示されたポリマー修飾生物活性接合体の大半は約1000Daを上回るサイズのPEG部分の付着によって修飾されている。例えば、食品医薬品局によって認可された6つのPEG付加製品(Adagen(R)、Oncaspar(R)、PEG−Intron(R)、Pegasys(R)、Somavert(R)およびNeulasta(R))のおのおのは、5,000−40,000Daの範囲の分子量をもつPEGポリマーによって修飾されている。本発明では、所望する以上のペプチド/ポリペプチドが隠蔽される可能性があり従って有益な結合部位/エピトープの認識が妨害される可能性があるので高分子量(“HMW”)ポリマーの使用は好ましくない。過去においては、治療用タンパク質または化学的化合物をHMWポリマーと結合させることに伴って治療薬が与える生物活性がある程度犠牲になることは容認されていた。付着したHMW PEGポリマーはしばしば治療薬の全体または実質部分を覆い隠す(例えば、遮蔽する)が、薬剤の活性部位があらわれると(例えば接近可能になると)PEG分子の高度な可撓性が所期の生物応答を実行するためのモーメントを提供できる。従って、修飾の結果として治療薬の比活性は低下するが、腎臓および細網内皮系(“RES”)のクリアランス減少およびタンパク質分解酵素からの遮蔽の双方によって治療薬の体内利用効率は向上する。最終的に、この体内利用効率の向上が治療薬の投与に対して全体的にはより有益な生物応答を提供する。いわば、本発明の意図はタンパク質/ペプチドの所与の活性に対する全体的に改善された応答を誘発することではない。むしろ、本文中に記載の修飾によってタンパク質/ポリペプチドの所与の活性を選択的に変質させることである。ポリエーテル主鎖の高度な可撓性およびその水分子結合能力が理由で、PEG分子は同等の分子量の可溶性タンパク質の5−10×の大きさであるかのように作用する。従って、本発明の発明者らは、エピトープまたは結合部位の選択的遮蔽がより小さい分子量(例えば、約1000Da未満の分子量)の水溶性ポリマーの部位特異的付着によって達成でき、タンパク質の残りの部分およびその内部の結合部位/エピトープは可用であり、制約されることなく対応受容体と相互作用して所望の応答を発生および/または促進すると推測した。
【0030】
従って、本発明の好ましい実施態様は、ペプチド/ポリペプチドが2つ以上のエピトープまたは結合部位を内包しており、1つのエピトープ/結合部位がポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーをペプチド/ポリペプチドに部位特異的に付着させるペプチドまたはポリペプチドのエピトープまたは結合部位の遮蔽方法に関する。ペプチド/ポリペプチドのポリマー修飾の結果として、生物に(例えば、ワクチンの形態で)投与されたときにペプチド/ポリペプチドによって誘導される生物応答はペプチド/ポリペプチドの非隠蔽エピトープまたは結合部位の活性の結果である。従って本発明は、低分子量水溶性ポリマーをペプチド/ポリペプチドに共有結合的に付着させることによってペプチドまたはポリペプチドを修飾し、生物に(例えば、ワクチンの形態)で投与したときに付着ポリマーによって遮蔽されないペプチド/ポリペプチドの1つ以上の領域に特異的な生物応答を誘導するための方法に関する。本発明の1つの実施態様では、対象ペプチドまたはポリペプチドが1つ以上のエピトープを、単独で含むかまたは免疫系に由来しないタンパク質もしくは化合物の1つ以上の結合部位と共に含む。本発明の1つの実施態様の目的は、アミノ酸配列の1つの部分または領域の免疫原性を減退させる(すなわち、別の免疫応答の利益となるように特定の免疫応答を減退させる)ために特定のエピトープを選択的に遮蔽することである。従って、ペプチド/ポリペプチドの生物活性は、タンパク質の異なる(すなわち、非隠蔽)部分に集中し、これが該ペプチド/タンパク質への特異的応答を変化させる。本発明の1つの実施態様では、選択的に隠蔽されたエピトープは、ペプチド間の1つ以上の共有結合によって共有結合的に安定化された3つのキメラペプチドを含むエンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造内部に局在する“スカフォールド”ドメインである。該コイルドコイルのキメラペプチドのおのおのは、該エンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメインの全部または一部分にアルファ−らせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでいる。本発明のこの部分の1つの実施態様では、該エンベロープウイルス膜融合タンパク質がHIV gp41である。
【0031】
従って本発明は、SMW PEGを非限定例とする少なくとも1つの低分子量水溶性ポリマーを抗原内部の少なくとも1つのアミノ酸残基に部位特異的に付着させるポリペプチド抗原のエピトープの遮蔽方法に関する。エピトープはポリマーによって選択的に隠蔽され、選択的に隠蔽されたエピトープは、ペプチド間の1つ以上の共有結合によって共有結合的に安定化された3つのキメラペプチドを含むエンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造内部のスカフォールドドメイン内部に局在する。キメラペプチドのおのおのは、該エンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでいる。本発明のこの部分の具体的な実施態様では、エンベロープウイルス膜融合タンパク質がHIV gp41である。
【0032】
本発明の代表的な用途では、発明者らは、HIV gp41の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造を含むワクチン構築物の一部分の選択的隠蔽が(暫定米国特許出願番号60/576,062,出願日2004.7.1、および、番号60/636,724,出願日2004.12.16に記載)、2つの低分子量PEG部分の付着によって実現し、この隠蔽は、免疫応答をペプチドミメティクスのHIV部分に選択的に集中させる(すなわち、gp41膜融合中間体のこのミメティクスの非HIV部分の免疫原性を減退させる)ために有効であることを知見した。これは後出の実施例1に詳細に記載する。コイルドコイル構造は、おのおのが非HIV由来スカフォールドドメインを含む3つのアルファ(“α”)−ヘリカルポリペプチドから成り、該ドメインはHIV N−ペプチドドメインにらせん相で融合したα−ヘリカル構造の安定化を助ける。共有結合的に安定化されたコイルドコイル構造内部に含まれた3つのポリペプチドのおのおののスカフォールドドメインのアミノ酸配列内部に局在する2つのリシンアミノ酸はPEG236部分の共有結合付着によって修飾されている(図3参照)。各ペプチドの修飾の結果として、共有結合付着した合計6個のSMW PEG分子をもつコイルドコイル構造が形成される。実施例1に示すように、該SMW PEG分子はスカフォールドドメインに対する抗体の産生を効果的に遮蔽し、免疫応答を構造のHIV部分に集中させる。この例は、対象ペプチド/タンパク質の1つの領域を選択的に遮蔽するためにSMW PEG分子、特にPEG236を使用することを初めて開示したものである。
【0033】
従って本発明の1つの実施態様は、エピトープがポリマーによって選択的に隠蔽されるように少なくとも1つの低分子量水溶性ポリマーをコイルドコイル構造に部位特異的に付着させる処理を含む、HIV gp41の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造のエピトープ遮蔽方法に関する。別の実施態様で、隠蔽されたエピトープは、コイルドコイル構造のスカフォールドドメインに局在し、該コイルドコイル構造は、HIV gp41 NH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含む“スカフォールド”ドメインをおのおのが有している3つのキメラペプチドを含み、該ペプチドは1つ以上の共有結合によって共有結合的に安定化されている。スカフォールドドメインは、該ドメイン内部の1つ以上のアミノ酸残基に1つ以上の低分子量水溶性ポリマーを付着させることによって修飾でき、該水溶性ポリマーは、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から選択され、より特定的にはPEGポリマー(例えば、PEG236)である。
【0034】
本文中の方法によって選択的に遮蔽できる三量体コイルドコイル構造は、暫定米国特許出願番号60/576,062,出願日2004.7.1、および番号60/636,724,出願日2004.12.16、に詳細に記載されている。双方の特許は参照によって本発明に含まれるものとする。これらの出願に記載された三量体コイルドコイル構造はエンベロープウイルス膜融合のミメティクスであり、特にHIV gp41を非限定例とするエンベロープウイルスの膜タンパク質の融合中間体のミメティクスを表す。多くのエンベロープウイルスによって使用される膜融合イベントの一例として、HIV gp41−媒介膜融合は、gp41糖タンパク質のエクトドメインに局在する3つの必須成分:NH2−末端融合ペプチド、NH2−末端ヘプタッドリピート(“N−ヘリックス”)およびCOOH−末端ヘプタッドリピート(“C−ヘリックス”)が関与する複合プロセスである。2つのヘプタッドリピート領域(NH2:HR1およびCOOH:HR2)は、糖タンパク質に周期的疎水性を付与し、多くのエンベロープウイルスの融合メカニズムに関与する共通構造モチーフである“ヘアピン三量体”と呼ばれるgp41の融合発生(すなわち融合活性)コンホメーションを形成するために互いに相互作用するα−ヘリカル構造を予測させる。ヘアピン三量体構造は6つのα−ヘリックスの束である。3つのgp41エクトドメインのC−ヘリックス領域によって形成された3つのα−ヘリックスが、3つのgp41エクトドメインのN−ヘリックス領域によって形成された中央の三重鎖コイルドコイルに逆平行にパックされている。融合プロセスは、N−ヘリックスコイルドコイルが露出したNH2−末端融合ペプチドをターゲット細胞膜の近傍/内部に配置する“プレヘアピン”コンホメーションの形成を経由して進行する。3つのC−ヘリックスが折り返されてこの中央N−ヘリックスコイルドコイルに会合し、ウイルスおよび宿主の細胞膜が膜融合の準備として近接状態に引っ張られるとヘアピン三量体が形成される。ウイルス/宿主細胞膜融合を防止する抗HIV薬は、プレヘアピンまたはヘアピン三量体複合体に結合する中和抗体を含む。中央N−ヘリックスコイルドコイルによって形成されたgp41エクトドメインのヘプタッドリピート1(HR1)領域内部の疎水性ポケットは、広い交差反応性のHIV中和抗体を誘導するための好ましいターゲットであると考えられている(参照:暫定米国特許出願番号60/576,062,出願日2004.7.1:この疎水性ポケット領域に結合する中和抗体を記載している)。“プレヘアピン”構造は多くのエンベロープウイルス膜融合タンパク質の共通特徴なので(参照:Singhら,1999,J.Mol.Biol.290:1031−1041)、ポリマー修飾された膜融合のミメティクスは、本文中の方法を使用し様々なエンベロープウイルスに特異的なワクチン構築物を製造するために作製できる。従って、本文中に例示した方法はHIVに特異的なワクチン構築物の製造を目標としているが、他のエンベロープウイルスをターゲットとするワクチン構築物を製造するためにこの同じ基本戦略を使用できることが当業者には理解されよう。
【0035】
従って、本発明の1つの実施態様は、3つのキメラペプチドを含むHIV gp41 N−ペプチドコイルドコイルの全部または一部分を模倣するポリマー修飾され共有結合的に安定化された三量体コイルドコイルに関する。キメラペプチドのおのおのは、HIV gp41 N−ペプチドの全部または一部分にらせん相に融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含み、該スカフォールドドメイン内部に局在するエピトープは該スカフォールドドメインの内部または極めて近傍の少なくとも1つのアミノ酸残基に部位特異的に付着した少なくとも1つの低分子量水溶性ポリマーによって選択的に隠蔽されている。該三量体コイルドコイルgp41ミメティクスは、例えばジスルフィド結合または化学選択的結合反応のいずれかから生じた共有結合のようなキメラペプチド間に形成された共有結合を介して安定化されたホモ三量体構造またはヘテロ三量体構造を示し得る。該コイルドコイル構造のスカフォールド部分の内部に局在するエピトープは、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択された分子量約1000Da未満のPEGポリマーを非限定例とするSMW水溶性ポリマーの付着によって遮蔽できる。
【0036】
従って、本発明の1つの実施態様では、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または一部分を模倣するポリマー修飾三量体コイルドコイル構造が、コイルドコイル構造内部に含まれた並列α−ヘリカルペプチドのシステイン残基間の共有ジスルフィド結合の形成を介して共有結合的に安定化されているコイルドコイルミメティクスを含み、該コイルドコイル構造の1つの領域(エピトープ含有)は該領域の内部または近傍の1つ以上のアミノ酸へのSMWポリマーの付着によって遮蔽されている。本発明のこの実施態様で、共有結合形成に参加するシステインアミノ酸残基は、個々のα−ヘリカルペプチドのα−ヘリカルドメインの外部に位置するように操作されている。従ってコイルドコイル構造に内包されている個別ペプチドのおのおのは、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメイン(“N−ペプチド”ドメイン)の全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態(“スカフォールド”ドメイン)またはその修飾形態を含むα−ヘリカルドメインと、該α−ヘリカルドメインの外部に局在する少なくとも2つのシステイン残基とを有している。複数の該可溶性キメラペプチドを三量体コイルドコイル構造が形成される濃度でインキュベートし次いで該構造を酸化させることによって、コイルドコイルの並列キメラペプチドのシステイン残基間に共有ジスルフィド結合が形成される。前述したように、本文中に開示の方法によって遮蔽すべき好ましいエピトープはスカフォールドドメイン内部に局在している。
【0037】
本発明の別の実施態様では、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または一部分を模倣するポリマー修飾三量体コイルドコイル構造が、化学選択的結合反応から生じたコイルドコイル構造に内包されたα−ヘリカルペプチド間の共有化学結合の形成を介して共有結合的に安定化されたコイルドコイルミメティクスを含み、コイルドコイル構造の1つの領域(少なくとも1つのエピトープ含有)は該領域の内部または近傍の1つ以上のアミノ酸へのSMWポリマーの付着によって遮蔽されている。この実施態様で、該コイルドコイル構造は、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメイン(“N−ペプチド”ドメイン)の全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態(“スカフォールド”ドメイン)またはその修飾形態を含むα−ヘリカルドメインをおのおのが有している3つのキメラペプチドを含む。3つのキメラペプチドは、チオエーテル結合の形成を非限定例とする化学選択的反応によって生じた共有化学結合の形成を介してコイルドコイル構造中で安定化されている。例えば、本発明に従ってポリマー修飾できるヘテロ三量体コイルドコイル構造は、該ペプチドのα−ヘリカルドメインの外部に局在する少なくとも2つのシステイン残基をさらに含む1つのキメラペプチドと、おのおのが求電子部分を組込むように誘導体化される2つのキメラペプチドとを含み、第一キメラペプチドの各システイン残基の求核スルフヒドリルが2つの誘導体化キメラペプチドのおのおのの求電子部分と共にチオエーテル結合を形成する。詳細は、暫定米国特許出願番号60/576,062(前出)に記載されている。
【0038】
コイルドコイル構造を安定化するために使用できる化学選択的結合反応の非限定例は、以下の化学種間の反応であり、これらの化学種は得られる三量体構造内部に内包された個々のキメラペプチド内部に組込まれる:(i)アルデヒド/ケトンとヒドラジドによるヒドラジンの形成;(ii)ケトンとアミノキシ基によるオキシムの形成;(iii)ケトンとチオセミカルバジドによるチオセミカルバゾンの形成;(iv)アルデヒドとβ−アミノチオールによるチアゾリジンの形成;(v)チオカルボキシレートとα−ハロカルボニルによるチオエステルの形成;(vi)チオエステルとN−末端ペプチドシステインによるアミドの形成;(vii)アルキルハライドとチオールによるチオエーテルの形成;および(viii)マレイミドとチオールによるチオエーテルの形成。
【0039】
従って本発明の1つの実施態様は、本文の方法によってポリマー修飾されたキメラペプチドに関する。該ペプチドは共有結合的に安定化されて三量体コイルドコイル構造を形成できる。該キメラペプチドは、HIV gp41を非限定例とするエンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメイン(“N−ペプチド”部分)の全部または一部分にらせん相で融合した三量体コイルドコイルを形成できるα−ヘリカルスカフォールドタンパク質(“スカフォールド”部分)を含むアルファ(“α”)−ヘリカルドメインを含む。PEGを非限定例とする1つ以上の低分子量ポリマーがスカフォールドドメイン配列の内部または極めて近傍に局在する選択されたアミノ酸残基に付着することによって部位特異的に遮蔽される少なくとも1つまたは複数のエピトープはこのスカフォールドドメインに内包されている。前述のように、このポリマー修飾が免疫応答を三量体コイルドコイルのウイルスN−ペプチド部分に集中させる。
【0040】
暫定米国特許出願番号60/576,062および番号60/636,724(前出)に詳細に記載されているように、三量体コイルドコイルコンホメーションを獲得できるα−ヘリカルスカフォールドタンパク質に融合したHIV gp41のNH2−末端ヘプタッドリピートドメイン(N−ヘリックス)の全部または一部分を含むキメラペプチドは当業界で公知である(Eckert and Kim,2001,Proc.Natl.Acad.Sci.USA98:11187−11192)。この普遍構造を有しているペプチドを本文では“キメラN−ペプチド”と呼ぶが、これはHIV−由来ペプチドに限定されない。HIVによって使用される融合メカニズムと同様の融合メカニズム(すなわち、ヘアピン三量体)を利用することが判っているエンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメインを、本発明に使用するキメラN−ペプチドの作製に使用できる。本発明の1つの実施態様では、本文中の方法によってポリマー修飾できホモ三量体もしくはヘテロ三量体コイルドコイル構造内部で共有結合的に安定化される可溶性キメラペプチドはこれらのキメラN−ペプチドの誘導体を表す。本文中に記載のようにしてポリマー修飾できるこのようなキメラN−ペプチドの一例を“CC−キメラN−ペプチド”と呼ぶ。CC−キメラN−ペプチドは以下の成分を含む:(1)三量体コイルドコイルコンホメーションを獲得できるα−ヘリカルスカフォールドタンパク質(“スカフォールド”部分);(2)HIV gp41のN−ヘリックス領域の全部または一部分を非限定例とするエンベロープウイルス膜タンパク質(“N−ペプチド”部分)のNH2−末端ヘプタッドリピートドメイン(“N−ヘリックス”)の全部または一部分;および(3)可撓性を強化するために場合によってはリンカー/スペーサー領域によってコアキメラペプチド配列から隔てられたキメラペプチド配列のNH2−またはCOOH−末端に局在する少なくとも2つのシステイン残基(“システイン部分”)。スカフォールド部分は上記N−ペプチド部分にらせん相で融合し、システイン部分(場合によってはリンカー/スペーサー領域を含む)はキメラペプチドのこのα−ヘリカルコアの外部に局在する。該ペプチドは、可溶性三量体コイルドコイル構造を形成でき、この構造では、3つの等しい(すなわち、ホモ三量体コイルドコイルを形成する)または実質的に類似の(すなわち、ヘテロ三量体コイルドコイルを形成する)キメラペプチド鎖がパラレル配向で物理的に会合している。該領域の免疫原性を減衰させ三量体コイルドコイル構造のN−ペプチド部分に免疫応答を集中させるために、該ドメインの内部または近傍に局在する1つ以上のアミノ酸残基にPEGを非限定例とする1つ以上の低分子量ポリマー部分を付着させることによって選択的に遮蔽されるのはスカフォールドドメインおよびその内部に局在するエピトープである。
【0041】
従って本発明の1つの実施態様は、(a)コイルドコイルの可溶性三量体形態を含むスカフォールド部分と、(b)HIV gp41のN−ペプチドまたはその修飾形態の全部または一部分を含むN−ペプチド部分と、(c)少なくとも2つのシステイン残基を含むシステイン部分とを含むポリマー修飾された可溶性キメラペプチドに関する。(a)のスカフォールド部分は(b)のN−ペプチド部分にらせん相で融合してα−ヘリカルドメインを形成しており、(c)のシステイン部分はα−ヘリカルドメインの外部に局在しており、該スカフォールド部分の内部に局在している1つ以上のエピトープは該スカフォールド部分の内部の少なくとも1つのアミノ酸残基に部位特異的に付着した少なくとも1つの低分子量水溶性ポリマーによって選択的に隠蔽されている。
【0042】
“CCIZN17”は本発明の一部としてポリマー修飾できる(すなわち、スカフォールドドメイン内部のエピトープを選択的に遮蔽できる)1つのCC−キメラN−ペプチドである。CCIZN17のコアα−ヘリカルドメインは、Eckert and Kim,2001(前出)に開示されたキメラN−ペプチド“IZN17”である。IZN17(IKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL:配列1)はEckert and Kim, 2001(前出)によって、HIV gp41のN−ヘリックスドメインの一部分の非凝集性三量体コイルドコイルが形成できるように設計された。このキメラペプチドは、HIV gp 41(“N17”N−ペプチドドメイン;配列32;上記の下線部分)のN−ヘリックスの一部分のNH2−末端にらせん相で融合した三量体コイルドコイル(“IZ”スカフォールドドメイン;配列14;上記のイタリック体)を形成できる可溶性α−ヘリカルドメインから成り、長さ41アミノ酸の連続コイルドコイルを創製する。IZドメインはSuzukiら(1998,Protein Eng.11:1051−1055)によって記載された設計に基づく修飾されたイソロイシンジッパーであり、溶液中ではヘリカルで三量体である。N17ドメインは、gp41エクトドメインのタンパク質分析によって同定されたHIV gp41のN−ヘリックス領域に由来の36アミノ酸ペプチド(“N36”)の先端欠失部分である。暫定米国特許出願番号60/576,062および番号60/636,724(前出)に詳細に記載されているように、CCIZN17はIZN17のNH2−末端にアミノ酸配列Cys−Cys−Gly−Gly(配列:10)を付加することによって作製した。従ってCCIZN17は以下のアミノ酸配列から構成されている:CCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL(配列:2)。CCIZN17を作製するためにIZN17に付加したシステイン残基は、このCC−キメラN−ペプチドに共有結合安定化三量体コイルドコイル形成能力を与える(ジスルフィド結合安定化CCIZN17由来コイルドコイルについては図1参照;チオエーテル結合安定化CCIZN17−由来コイルドコイルについては図8参照)。CCIZN17中の付加グリシン残基はキメラIZN17ペプチドのα−ヘリカルコアからシステイン残基を隔てるスペーサー領域を表し、該システイン残基にジスルフィドまたは化学選択的(例えば、チオエーテル)結合を形成するためにコンホメーション的により大きい自由を与える。3つのCCIZN17キメラペプチド間の共有ジスルフィド結合の存在は共有結合的に安定化された三量体コイルドコイル(CCIZN17)3を生じる(図1参照)。このコイルドコイルではは、三量体の存続がモノマー鎖の濃度にもはや依存しないので出発IZN17三量体コイルドコイルよりも安定である(参照:暫定米国特許出願番号60/576,062および番号60/636,724(前出))。
【0043】
キメラN−ペプチド(例えばIZN17)はまた、共有結合的、化学選択的反応に参加できる求電子部分を組込むように誘導体化することもでき、“誘導体化キメラN−ペプチド”と呼ばれる。従って本発明の別の実施態様は、(例えば、スカフォールドドメイン内部のエピトープを選択的に遮蔽するために)本文の方法によってポリマー修飾されて三量体コイルドコイル構造内部で共有結合的に安定化できる誘導体化キメラN−ペプチドに関する。証明として、化学選択的反応を介して共有結合的に安定化されている三量体コイルドコイル構造は、単一のCC−キメラN−ペプチド(例えばCCCIZN17;配列:76)および2つの誘導体化キメラN−ペプチド(例えばブロモアセチル−GGGIZN17;配列:77)を含み、後者のおのおのは、等しいかまたは実質的に等しいα−コアドメイン(例えばIZN17;配列:1)を有しており、該キメラペプチド間で共有チオエーテル結合が生じる。これらの3つのキメラペプチドのおのおののスカフォールド部分内部のエピトープは、該スカフォールドドメイン内部の1つ以上のアミノ酸に低分子量ポリマー部分(例えばSMW PEG)を付着させることによって選択的に遮蔽できる。“NH2−C(Fm)(チオIZN17)3”と命名された共有結合安定化三量体コイルドコイルは、本発明の一部として記載した方法によってポリマー修飾され得るチオエーテル結合安定化コイルドコイルの一例を表す(図8、参照:暫定米国特許出願番号60/576,062および番号60/636,724(前出))。NH2−C(Fm)(チオIZN17)3は1つのCC−キメラN−ペプチドすなわちCCCIZN17ペプチド(配列:76)および2つの誘導体化キメラN−ペプチドすなわちブロモアセチル−GGGIZN17(配列:77)から成り、誘導体化キメラN−ペプチドのNH2−末端に局在するブロモアセチル部分のおのおのはCC−キメラN−ペプチドの“システイン部分”に局在するシステイン残基と共にチオエーテル結合を形成する。この特定のコイルドコイル中で、NH2−末端システイン残基は最初はFm基によって保護され、2つの内部システイン残基をブロモ部分とのチオエーテル結合形成に参加させる。誘導体化キメラN−ペプチドすなわちブロモアセチル−GGGIZN17(配列:77)で証明されるように、誘導体化キメラN−ペプチドの求電子部分は、キメラペプチドのコイルドコイル形成と得られたコイルドコイルを安定化する共有結合形成との間の干渉を最小にするために可撓性リンカー領域(たとえば、2つ以上の連続したグリシン残基)によってキメラN−ペプチドのα−ヘリカル部分から隔てられる。
【0044】
さらに、IQN17(RMKQIEDKIEEIESKQKKIENEIARIKKLLQLTVWGIKQLQARIL:配列:11)はIZN17と同様の設計図をもつキメラN−ペプチドである。しかしながらN17ドメイン(下線部分)は酵母転写アクチベーターGCN4に由来の代替的三量体コイルドコイルモチーフにらせん相で融合している(“IQ”ドメイン;配列:17;上記イタリック体)。コアIQN17ペプチドを含む上記同様の共有結合安定化三量体コイルドコイルを作製し本発明の一部としてポリマー修飾できる。
【0045】
本文中で“スカフォールド部分”と互換的に使用され本発明の一部として選択的に遮蔽されるCC−および誘導体化−キメラN−ペプチドの“スカフォールドドメイン”は、非−HIVアミノ酸残基を含んでおり、コイルドコイルの可溶性三量体形態を表す。スカフォールドドメインは上述のN−ペプチド領域のNH2−末端またはCOOH−末端に融合できるか、または、該ドメインがN−ペプチド領域のNH2−およびCOOH−末端の双方に局在するように分割できる。一部分がN−ペプチド部分の両端に局在するようにスカフォールドドメインが分割されているとき、この分割されたスカフォールドドメインと分割部分の一方または双方に内包されているエピトープとを適正に遮蔽するために、少なくとも1つのポリマー部分が分割スカフォールド領域の各部分(すなわち、NH2−末端部分とCOOH−末端部分)に存在するアミノ酸残基に付着するであろう。コイルドコイルタンパク質は文字“a”から“g”によって表されるアミノ酸の特徴的ヘプタッドリピートから構成され、ヘプタッドリピートの第一および第四の位置すなわち“a”および“d”の位置はそれぞれコイルドコイルの相互作用鎖の内部を形成し一般に疎水性である。本文に記載のポリマー修飾三量体コイルドコイル構造のスカフォールドドメインを表すコイルドコイルモチーフは、以下を非限定例とする様々なソースから単離できる:Suzukiらによって開示されたイソロイシンジッパーモチーフ(1998(前出);以後の本文中では“Suzuki−IZ”;YGGIEKKIEAIEKKIEAIEKKIEAIEKKIEA;配列:12)およびEckertらによって開示されたGCN4−pIQIコイルドコイルモチーフ、(1998,J.Mol.Biol.284:859−865および国際出願PCT/US01/29637,国際公開WO02/24735;RMKQIEDKIEEILSKQYHIENEIARIKKLIGER(配列:13))およびそれらの先端欠失および/または修飾形態。Suzuki−IZまたはGCN4−pIQIコイルドコイルモチーフは、1つのアミノ酸残基の付加、置換、修飾および/または欠失によって改変できる。例えば、該スカフォールドタンパク質内部のアミノ酸残基を該特定場所のSMWポリマーの付着を容易にするために修飾してもよい。スカフォールドドメインは短縮、修飾またはその双方が可能であるが、得られるCC−または誘導体化−キメラN−ペプチドがN−ペプチドコイルドコイル領域を適正に提示する能力を維持する(すなわち、N−ヘリックス三量体コイルドコイルの安定で忠実なミメティクスを生成する能力を維持する)ことが重要である。これは、暫定米国特許出願番号60/576,062および番号60/636,724(前出)に開示された実験によって判断できる。先端欠失および/または修飾されたSuzuki−IZ−様およびGCN4−pIQI−様スカフォールドドメインの様々な多数の実施例が、暫定米国特許出願番号60/576,062および番号60/636,724(前出)に開示されている。これらの文献のおのおのは参照によって本発明に含まれるものとする。Suzuki−IZ−様およびGCN4−pIQI−様スカフォールドタンパク質の例を表1に示す。しかしながらこの記述は表1に示したCC−または誘導体化−キメラN−ペプチドに内包されたスカフォールドタンパク質の選択的な遮蔽方法に本発明を限定するものではない。
【0046】
【表1】
【0047】
本発明の1つの実施態様では、本文中に記載のCC−および誘導体化−キメラN−ペプチドのスカフォールドドメインは、免疫原性担体またはアフィニティ樹脂へのそれらの接合および/または得られたコイルドコイル構造の接合を容易にするために特異的に修飾されてもよい。例えば、アミノ酸配列IEKKIEEIEKKIEEIEKKIEEIEK(配列:32;“a”位置は下線部分)から成る“EZ”スカフォールドは、暫定米国特許出願番号60/576,062および番号60/636,724(前出)に記載の共有結合安定化三量体コイルドコイルと免疫原性担体であるNeisseria meningitidis外膜プロテオソーム複合体(“OMPC”)粒子との接合を容易にするために設計した。担体へのペプチドの共有結合付加(すなわち、担体−ペプチド接合体)は接合体の不可逆的沈殿を生じることが多いが、OMPC−ペプチド接合体の場合も同様である。これを防止するために、1モルのOMPCに組込むペプチドの最大モル数(通常は、ペプチド“負荷”と呼ぶ)を慎重にコントロールしなければならない。何らかの配列のペプチドをOMPCに直接接合し、ペプチドエピトープに対して十分な免疫応答を得るために必要な閾値以上のペプチド負荷を確保できる戦略が最近開発された(参照:Bianchiら,暫定米国特許出願番号60/530,867“A Method to Make a Peptide−Carrier Conjugate with a High Immunogenicity,”出願日2003.12.18)。該方法はペプチド配列のpI(等電点)の操作を含み、その値を約3.5−5.0の範囲内で最適に調節する。pIがこの範囲内の値になるように、ペプチドを活性の免疫原性ドメインの外部で修飾し、ペプチドの免疫特性の維持を確保する。例えば、IZN17(配列:1)は10.7という極めて高いpIを有しているが、その理由は恐らくIZスカフォールド内部に多数の塩基性アミノ酸残基が存在するからであろう。EZスカフォールドドメインは、スカフォールドを含むキメラペプチドやそれらに内包されたコイルドコイル構造がOMPCとの接合を容易に行うことができるように、もっと酸性に設計した。IZスカフォールドを作製するために使用したSuzuki−IZスカフォールドはSuzukiら,1998(前出)によって設計された(IEKKIEA)n(配列:99)のヘプタッドリピート配列を有している。EZスカフォールド中では、Suzuki−IZのヘプタッドリピートの“c”のアラニン残基がグルタミン酸残基に突然変異することによってこのヘプタッドリピート配列が修飾され、(IEKKIEE)n(配列:100)となる。得られたキメラN−ペプチドEZN17(IEKKIEEIEKKIEEIEKKIEEIEKLLQLTVWGIKQLQARIL;配列:98)の5.2というpIは効率的なOMPC接合に最も有利な範囲に近い。
【0048】
前述のように、本発明の一部として記載した上記の方法によってポリマー修飾できる様々なキメラペプチドを作製するときには適正なヘリカル構造の維持が極めて重要である。例えば、N17よりも長い(例えば、“N23”または“N36”)HIV N−ペプチド配列セグメントを有しているIZN−キメラペプチドおよびそのCCIZN−誘導体は、IZと長いHIV N−ペプチドとを接合する融合イベント中にα−ヘリカルヘプタッドリピートが確実に維持されるように、1個から数個のアミノ酸で延長された修飾IZドメインを含み得る。IZN23およびIZN36はやはりEckert and Kim,2001(前出)に開示されたキメラN−ペプチドであり、本発明に記載したようにスカフォールドドメイン内部のエピトープを選択的に隠蔽するために三量体コイルドコイル内部で共有結合的に安定化されポリマー修飾され得る。IZN23およびIZN36はそれぞれ以下のアミノ酸配列を有している:IKKEIEAIKKEQEAIKKKIEAIEKEIEAQQHLLQLTVWGIKQLQARIL(配列:23)およびIKKEIEAIKKEQEAIKKKIEAIEKEISGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARIL(配列:24)。IZN23およびIZN36のスカフォールドドメインはイタリック体で示し、ペプチドの“a”位置は下線を付けて示す。IZN17およびそのCCIZN−誘導体であるCCIZN17の作製に使用したIZスカフォールドに比較すると、IZN23およびIZN36のスカフォールドドメインは、安定なα−ヘリカルコンホメーションの生成を容易にする適正な“a”−“g”スペーシングを維持するために1つ(IZN23)または2つの(IZN36)アミノ酸残基で延長されている。このようにしてスカフォールドドメインを延長するために選択されたアミノ酸は隣接ヘリックス間で静電相互作用し得る(Suzukiら,1998,(前出))。
【0049】
本発明は、既刊文献(参照:暫定米国特許出願番号60/576,062および番号60/636,724(前出))に詳細に記載された共有結合安定化三量体コイルドコイルのコア構造を有しており、タンパク質構造内部の1つ以上のエピトープを隠蔽しその結果として当該構築物に対して生じる免疫応答を非遮蔽領域に集中および/または集結させるために、SMW PEG部分を非限定例とする1つ以上の低分子量ポリマーの選択的付着によって修飾されたワクチン構築物に関する。上述のように、本発明の1つの実施態様では、該ワクチン構築物が3つのキメラペプチドを含み、おのおのはHIV gp41エクトドメインのN−ヘリックスヘプタッド領域の全部または一部分(“N−ペプチド”部分)にらせん相で融合した可溶性スカフォールドタンパク質を含む。HIV gp41エクトドメインは、約169アミノ酸残基(HIV−1 gp160エンベロープタンパク質中の位置に準拠して残基512−681)を表す。エクトドメインのNH2−およびCOOH−末端部分の内部に局在する2つの4−3ヘプタッドリピート領域はそれぞれN−およびC−ヘリックス領域と呼ばれるα−ヘリックスを形成すると予測される。N−ヘリックスおよびC−ヘリックスはそれぞれgp160のほぼ541−592位および623−663位のアミノ酸に局在する(参照:たとえば,Caffreyら,1998,EMBO J.17:4572−4584)。gp41エクトドメインのコア融合−活性構造(すなわち、“ヘアピン三量体”)は、それぞれN−ヘリックスおよびC−ヘリックス領域に局在する2つの相互作用性ペプチド、N36(SGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARIL;配列:29)およびC34(WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL;配列:30)の三量体から成る(参照:Chanら,1997,Cell 89:263−273;および、米国特許番号6,506,554)。N36は、HIV−1 gp160の位置に準拠してアミノ酸残基546−581(両端のアミノ酸を含む)を含む。他のグループは、Chanらによって開示されたものよりもやや大きい安定な6−ヘリックス束を形成するHIV−1 gp41の結晶化ヘリカルドメインを有している。例えば、Weissenhornら(1997,Nature 387:426−430)は、N36配列のNH2−末端の5つの追加アミノ酸残基とCOOH−末端の7つの追加アミノ酸残基とを含むN36よりも長いN−ペプチド領域を同定した:ARQLLSGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARILAVERYLK(配列:31)。従って、本発明によってポリマー修飾され得るCC−および誘導体化−キメラN−ペプチドのN−ペプチド領域は、上記に開示されたN36領域の全部または一部分に必ずしも限定されない。従って1つの実施態様では、そのスカフォールドドメインが本文中に記載のようにポリマー修飾されたCC−および誘導体化−キメラN−ペプチドは、N36のCOOH−末端側の半分に局在しHIV−1 gp160配列の残基565−581に対応する少なくとも17のアミノ酸(“N17”)(LLQLTVWGIKQLQARIL;配列:32)を含み得る。別の実施態様では、N17よりも長いまたは短いN−ペプチド領域のセグメントは上述のようなポリマー修飾スカフォールドドメインに融合し得る(例えば、“N−23”−IEAQQHLLQLTVWGIKQLQARIL;配列:33)。N−ペプチドドメインの延長は、N17内部で同定された推定免疫原性エッジ効果を克服するために有用であろう(参照:暫定米国特許出願番号60/576,062および番号60/636,724(前出))。ヘアピン三量体構造はまた、疎水性ポケットと呼ばれる深い空洞を備えている。1つのこのような空洞はN36α−ヘリックスの各溝の底部(すなわち、COOH−末端側の半分)に局在し、並列したC34ヘリックスのノブ状突起で充填され、ボール−ソケットアレンジメントを形成する(参照:Caoら,1993,J.Virol.67:2747−2755)。従って、CC−および誘導体化−キメラN−ペプチドは、N−ヘリックスα−ヘリカルドメインのCOOH−末端側の半分に存在する中央N−ヘリッスクコイルドコイルの疎水性ポケットまたは空洞を形成するアミノ酸残基を含むgp41 N−ヘリックスの一部分を含み得る。
【0050】
本文中に記載したワクチン構築物のHIV N−ペプチド領域はまた、野生型アミノ酸配列に比べて修飾されてもよい。例えば、N17のCOOH−末端内部に局在する免疫優性非中和エピトープはIZN17ペプチドのアラニン走査によって同定された(暫定米国特許出願番号60/576,062および番号60/636,724)。この望ましくない免疫優性応答は、非中和エピトープの原因であると考えられるN17の1つ以上のアミノ酸残基(ロイシン−581,アルギニン−579,グルタミン−577および/またはグルタミン−575)を突然変異させることによって克服できる。一例では、免疫優性エピトープの原因であると考えられる4つのアミノ酸残基のおのおのがアラニン残基で置換され、アミノ酸配列:LLQLTVWGIKALAAAI(配列:34)から成る“N17Ala4”が生じるようにN17を修飾し得る。あるいは、有益でない抗原応答を排除するために該非中和免疫優性領域の先端を欠失させるかまたは完全に除去してもよい(例えば“N13”−LLQLTVWGIKQLQ;配列:35)。本発明のキメラペプチドのN−ペプチド部分はまた、ペプチド全体がいっそう安定するように修飾することもできる。例えば、N−ペプチドドメインは、配列中により安定性のイソロイシン残基を組込むことによって修飾できる(例えば“N17Ile”−LIQLIVWGIKQIQARIL;配列:36)。
【0051】
上述のように、本文中の方法によってポリマー修飾された三量体コイルドコイルワクチン構築物の好ましい実施態様は、α−ヘリカル安定性スカフォールドドメインにらせん相で融合したHIV gp41 N−ヘリックス領域の一部分を有しているHIV融合中間体のミメティクスとなる。HIV gp41のこのN−ヘリックス部分は、HIV−1、HIV−2、別のHIV菌株または別のレンチウイルス種の菌株(例えば、サル免疫不全ウイルス(SIV)、ネコ免疫不全ウイルス(FTV)またはビスナウイルス)から単離できる。このために同様のHIV菌株および/または他の種の免疫不全ウイルス中の対応するN−ペプチド配列を同定することは容易であり、当業界で公知である。例えば、HIV−1 gp41のN36 N−ペプチド配列に位置合せする他のレンチウイルスNH2−末端α−ヘリカルドメインのアミノ酸配列を表2に示す。さらに、他のエンベロープウイルスの膜−融合タンパク質中のα−ヘリカルコイルドコイルドメインが同定された(参照:Singhら,1999(前出))。
【0052】
【表2】
【0053】
本文中で定義したキメラN−ペプチド(例えばIZN17)の作製については詳細に上述した。暫定米国特許出願番号60/576,062および番号60/636,724(前出)にはもっと詳細に記載されている。該キメラN−ペプチドは、安定なジスルフィドおよび/またはチオエーテル結合をそれぞれ形成する能力を該ペプチドに与えて共有結合安定化三量体コイルドコイル構造を得るために、コアキメラペプチド配列のNH2−またはCOOH−末端に少なくとも2つのシステイン残基(すなわち“CC−キメラN−ペプチド”)または求電子部分(すなわち“誘導体化−キメラN−ペプチド”)を付加することによって修飾できる。1つの実施態様では、少なくとも2つのシステインアミノ酸残基がキメラN−ペプチドのコアα−ヘリカルドメインの外側に付加されて、酸化性条件下で隣接のCC−キメラN−ペプチドと共にジスルフィドブリッドを形成できる“CC−キメラN−ペプチド”を生成する。付加システイン残基をヘリカル外側に配置するのは、該ペプチドのコイルドコイルコンホメーションと安定性ジスルフィド結合との干渉を最小にするためである。並列配向の3つのCC−キメラN−ペプチドが物理的に会合すると、個々のポリペプチドモノマーのα−ヘリカル構造の結果としてシステイン残基が緊密に接近する。酸化性条件下でこれらの並列システイン残基は3つの鎖間で自発的にジスルフィドブリッジを形成する。別の実施態様では、CC−キメラN−ペプチドの操作されたシステインアミノ酸残基に存在するチオール−反応性官能基が隣接の誘導体化N−ペプチドの求電子部分(たとえば、アルキルハライド部分またはMichael受容体)と共に共有結合的化学選択的結合を形成できる。該求電子部分は、キメラN−ペプチドの末端およびそのα−ヘリカルドメインの外側に付加される。これらのシステイン残基または求電子部分は、コアキメラN−ペプチドのNH2−末端に付加されてもCOOH−末端に付加されてもよい。CC−および誘導体化−キメラN−ペプチドの付加システイン残基または求電子部分はそれぞれ、リンカーまたはスペーサー領域によってキメラペプチドのα−ヘリカル部分から隔てられる。スペーサー領域はキメラN−ペプチドのヘリカル構造を破壊し、末端システイン残基または求電子部分が共有結合形成に参加するためにコンホメーション的により大きい自由を与える。スペーサー/リンカー領域は、α−ヘリカル二次構造を破壊する能力をもつことが知られた短いアミノ酸配列(例えば、グリシンまたはプロリン残基;または、単一D−アミノ酸、アミノ酸の右旋性異性体)を含み得る。α−ヘリカル構造の破壊に必要な最少数の残基をスペーサーとして使用するのが好ましい。あるいは、スペーサー/リンカー領域が、化学的または合成リンカー、例えば、アミノペンタン酸、アミノヘキサン酸、または、C−アルファ側鎖のない何らかのアミノ酸を含むこともできる。
【0054】
スカフォールドドメインの内部に局在する1つ以上のアミノ酸残基に1つ以上の低分子量ポリマー分子を付着させることによってポリマー修飾でき、該スカフォールドドメイン内部の1つ以上のエピトープを効果的に遮蔽し、免疫応答をキメラペプチドの非遮蔽部分(例えばHIV部分)に集中させる好ましい共有結合安定化三量体コイルドコイルは(CCIZN17)3(配列:2)3である。図1参照。このコイルドコイル構造は、以下のアミノ酸配列をもつ3つの等しいCCIZN17キメラペプチドから成る:CCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL(配列:2)。前述のようにCCIZN17は、コアキメラペプチド配列“IZN17”がN17(配列:32)のNH2−末端に融合したIZスカフォールドドメイン(上記配列の下線部分)から構成されているCC−キメラN−ペプチドを表す。CCIZN17はさらに、コアキメラペプチドのNH2−末端に付加された安定なCys−Cys−Gly−Gly(配列:10)を含む。2つの連続システイン残基は、ペプチドの酸化によって形成される緊密に会合したCC−キメラN−ペプチドの並列システイン残基と共にジスルフィド結合形成に参加する。2つの連続グリシン残基は、システイン残基をコアキメラペプチド配列のα−ヘリカルドメインから隔てるスペーサー領域を表す。グリシンスペーサー領域は、システイン残基がコアペプチド配列のヘリカル二次構造に巻き込まれないことを確保し、この遊離システイン残基がジスルフィド結合形成に参加することを援助する。
【0055】
従って、本発明の1つの実施態様は、(CCIZN17)3のコア構造を有している共有結合安定化三量体コイルドコイルである。該コイルドコイル構造のスカフォールドドメインおよび該ドメインに内包されたエピトープは、該ドメインの内部または近傍に局在する1つ以上のアミノ酸残基に1つ以上の低分子量水溶性ポリマーを共有結合付着させた結果として選択的に遮蔽されている。後出の実施例1に詳細に記載する代表的ワクチン構築物では、スカフォールドドメイン内部に局在する2つのリシンアミノ酸残基が、市販のPEG236部分によってPEG付加され、本文中で“CCIPN17”と呼ぶペプチドが作製される。CCIPN17はCCIZN17と同じアミノ酸配列(CCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL;配列:2)を有している。しかしながら上述のように、スカフォールドドメイン(イタリック体)の内部に局在する2つのリシン残基(下線部分)はPEG236によって共有結合的に修飾されている(図2参照)。従って、得られる(CCIPN17)3ワクチン構築物のスカフォールドドメインは、各ペプチドモノマーに2つずつ存在する6つのPEG236部分によって遮蔽されている。HIV gp41融合中間体のミメティクスを表す低分子量ポリマー修飾(PEG修飾を非限定例とする)されたコイルドコイル構造は、本文中に記載の種々のキメラペプチドおよび暫定米国特許出願番号60/576,062および番号60/636,724(前出)に記載のペプチドを使用して同様に作製し得る。
【0056】
ペプチド間に形成されたジスルフィド結合を介して共有結合安定化される3つのCC−キメラN−ペプチドをおのおのが含み本発明の一部として記載された方法によってポリマー修飾できる三量体コイルドコイル構造の例を表3に示す。表3の共有結合安定化三量体コイルドコイルの記述もまた、本発明方法がこれらの例に含まれたスカフォールドドメインの選択的遮蔽方法に限定されることを意味しない。
【0057】
【表3】
【0058】
上述のように、表3の共有結合安定化三量体コイルドコイルのおのおのはコアペプチド構造のα−ヘリカルドメイン外部に局在するシステイン残基間のジスルフィド結合の形成によって共有結合的に安定化されている3つのCC−キメラN−ペプチドから成る。あるいは、前述のように、本発明によってポリマー修飾できる三量体コイルドコイルワクチン構造は、化学選択的結合の形成を介して共有結合的に安定化できる。例えば、2つの誘導体化キメラN−ペプチドの求電子部分と1つのCC−キメラN−ペプチドの2つの付加システインアミノ酸残基のおのおののチオール官能基との間で2つの化学選択的結合が形成されて2つのチオエーテル共有結合が形成される。例えば、暫定米国特許出願番号60/576,062および番号60/636,724(前出)に詳細に記載されているNH2−C(Fm)(チオールIZN17)3と命名された三量体コイルドコイルミメティクスを本発明によってポリマー修飾できる。この三量体コイルドコイルは、以下のペプチドモノマーから成る:
1)NH2−C(Fm)Ttds−CCIZN17(表4のペプチド1):可撓性スペーサー(−GG−;他の可撓性スペーサーも好適)を介してスカフォールドドメインから隔てられた2つのシステイン残基と以後の接合用の保護チオール基(フルオロメトキシ(Fm)保護基を使用するが他の保護基も適当)をもつ第三のシステイン残基とを有するペプチド:
2)ブロモアセチル−GGGIZN17(表4のペプチド2):可撓性リンカー(−GGG−;2つの誘導体化キメラN−ペプチド鎖と1つのCC−キメラN−ペプチドとの間の2つのチオエーテル結合の形成に適合性であるならば他の可撓性スペーサーも適当である)を介してスカフォールドドメインから隔てられたブロモアセチル部分を有するペプチド。
【0059】
2つのペプチド前駆体NH2−C(Fm)Ttds−CCIZN17(表4のペプチド1)およびBr−アセチル−GGGIZN17(表4のペプチド2)をそれぞれ約1:2の濃度比でインキュベートして1つのCC−キメラN−ペプチド(ペプチド1)と2つの誘導体化N−ペプチド(ペプチド2)とを有する三量体コイルドコイルを作製できる。化学選択的結合反応は、2つのチオエーテル結合の形成を伴って進行し、この反応で2つのBr−アセチル−GGGIZN17ペプチドの各ブロモ誘導体がNH2−C(Fm)Ttds−CCIZN17ペプチドの2つの非保護システイン残基の1つと反応する(図8参照)。共有結合安定化三量体コイルドコイルの形成後、該三量体コイルドコイルの(例えば、精製樹脂またはOMPCへの)接合を容易にするために保護基を除去できる。
【0060】
本発明の方法によってポリマー修飾できる化学選択的結合の形成を介して共有結合安定化三量体コイルドコイルを作製するために使用し得る追加のペプチドモノマーを単なる例として表4に示す。例えば、“ac−C(チオIZN17)3”と命名された安定化コイルドコイルは、1つのac−C(Acm)Ttds−CCIZN17ペプチド(表4のペプチド3)と2つのBr−アセチル−GGGIZN17ペプチド(表4のペプチド2)とを含むように合成し得る。得られる三量体コイルドコイルはNH2−末端システイン残基の保護基としてAcm(アセタミドメチル基)を有しており、これは以後の接合を容易にするために除去できる。
【0061】
【表4】
【0062】
PEGおよび類縁ポリマーのようなポリマー部分をタンパク質に見出される反応性基に付着させるために様々な手段が使用されてきた。参照:たとえば,米国特許番号4,179,337,Davisらに許諾、および、米国特許番号4,002,531,Royerに許諾。総説についてはAbuchowskiら,Enzymes as Drugs,Eds.J.S.Holcerberg and J.Roberts,1998,pp.367−383およびZalipsky,S,1995,Bioconjugate Chem.6:150−165を参照するとよい。タンパク質を修飾するためのPEGおよび他のポリマーの使用はまた、以下の文献でも検討されている:Cheng,T.L.ら,1999,Bioconjugate Chem.10:520−528;Belcheva,N.ら,1999,Bioconjugate Chem.10:932−937;Bettinger,T.ら,Bioconjugate Chem.9:842−846;Huang,S.−Y.ら,1998,Bioconjugate Chem.9:612−617;Schwarz,J.B.ら,1999,J.Amer.Chem.Soc.121:2662−2673;Reuter,J.D.ら,1999,Bioconjugate Chem.10:271−278;およびChan,T.−H.ら,1997,J.Org.Chem.62:3500−3504。前述したように、タンパク質中の典型的なポリマー付着部位はリシン残基またはNH2−末端に存在する一次アミノ基、システイン側鎖に存在するチオール基、グルタメートもしくはアスパルテート残基、または、COOH−末端に存在するカルボキシル基、などを含む。(例えば、循環半減期を改善するため、タンパク質分解および免疫原性を抑制するために)PEGのような水溶性ポリマーを対象ポリペプチドに付着させる場合、ポリマー部分をユーザーの定めた精度でコントロール的に付着させるのが難しいならば、一様でない結果が得られることになる。過去には、付着の推計的特性および水溶性ポリマーのヘテロ分散的特性を理由としてポリマー−タンパク質コンジュゲートの精製および分析キャラクタリゼーションが困難であった。
【0063】
本文中に記載のようなSMWポリマー修飾ポリペプチドの好ましい作製方法では、1つ以上のSMWポリマーを対象タンパク質内部の部位特異的場所に付着させる。これは、本発明のように所期の生物応答(例えば、免疫応答)を誘導または促進できるように択一的部位を可用状態に維持しながらタンパク質内部の特定の(1つ以上の)生物活性部位を付着ポリマーによって遮蔽する予定であるときに特に重要である。ポリマーがタンパク質に部位特異的に結合した低分子量ポリマー誘導体化タンパク質(例えば、SMW PEG付加タンパク質)の調製は、付着鎖の個数および場所を完全にコントロールできる種々の方法によって達成される。例えば、ポリマーのランダムな付着を減少させる1つの試みでは、天然のリシンまたはシステインアミノ酸残基を除去し、同時に所望のポリマー付着部位にリシン/システインアミノ酸を再度付加したタンパク質を作製した(参照:たとえば,米国特許番号4,904,584)。あるいは、後出の実施例1に例示したワクチン構築物(CCIPN17)3を固相ペプチド合成(“SPPS”)によって作製した。従って、本発明方法によって修飾すべきペプチドおよび/またはポリペプチドの合成は、標準全自動ペプチドシンセサイザーを使用する段階的標準Bocおよび/またはFmoc固相ペプチド合成で行うか、または、販売業者に注文し購入した標準プロトコルに従って手動操作で行う。参照:たとえば,Synthetic Peptides,A User’s Guide,Ed.G.A.,New York.W.H.Freeman & Company,1992;Principles of Peptide Synthesis,2nd ed.,Ed.M.Bodanszky, Springer−Verlag,1993;The Practice of Peptide Synthesis,2nd ed.,Eds.M.Bodanszlky and A.Bodanszky,Springer−Verlag,1994;Protecting Groups,Ed.P.J.Kocienski,Stuttgart,Germany:Georg Thieme Verlag,1994;およびFmoc Solid Phase Peptide Synthesis,A Practical Approach,Eds.W.C.Chan and P.D.White,Oxford University Press,2000。
【0064】
天然型化学結合(Dawson,P.E.and S.B.Kent,2000,Ann.Rev.Biochem.69:923−60)のような化学選択的方法を使用すると、全化学合成によってもっと大きいポリマー修飾タンパク質も容易に得られる。その優れた代表例は、Kochendoerfer,G.G.ら(2003,Science 299:884−87)によって示されており、Kochendoerfer,G.G.ら,米国非暫定出願番号10/332,385,US公開番号US2004/0115774にいっそう詳細に記載されている。該文献では著者/発明者らが2つの部位特異的HWM分枝状PEG分子を含む166アミノ酸のタンパク質を累積収率25−40%で作製した。この文献に記載されたHMW分枝状PEGの付着に関する化学理論はSMW PEG部分の導入に容易に応用できる。しかしまた、利用可能な多くの他の選択肢も存在しており(Harris,J.M.and R.B.Chess,2003,Nat.Rev.Drug Discov.2:214−221)、それらは当業者に公知である。Kochendoerferら,2003(前出)で作製されたタンパク質SEPのサイズは、HIVエンベロープ糖タンパク質のgp120サブユニットのサイズと同等であり、糖残基の選択的付加に基づいて該タンパク質のエピトープを隠蔽する戦略が提案されている。従って、特定の免疫反応性を減退させるかまたは完全に除去して別の免疫反応性の利益になるようにSMW PEGで選択的に誘導体化されるgp120に基づくHIVワクチンが完全に化学的な合成方法で得られることは明らかである。
【0065】
本文中に記載したようなポリマー修飾タンパク質の代替的作製方法では、組換えDNA方法によって産生された適当な前駆体タンパク質にSMWポリマー(例えばPEG)を化学選択的に付着させる。より具体的に、Bertozziおよび共同研究者らは、組換えタンパク質内部の任意の位置にアジド基を組込む普遍的に応用できる戦略を開発した(Kiick,K.L.ら,2002,Proc.Natl.Acad.Sci.U.S.A.99:19−24)。該方法は、細胞の先天的翻訳装置によってプロセスされる非天然アミノ酸の利用から成る。アジドホモアラニン(メチオニン代用物)を大腸菌のメチオニル−tRNAシンテターゼによって活性化し、メチオニン欠失細菌培養物中で発現されたタンパク質のメチオニンを置換する。アジドはStaudinger結合と呼ばれる極めて化学選択的な反応にホスファンと共に参加して、最終的にタンパク質とホスファン付着部分とを接合する安定なアミド結合を形成する(参照:Kohn,M.and R.Breinbauer,2004,Angew Chem.Int.Ed.Engl.43:3106−16;Saxon,E.ら,2000,Org.Lett.2:2141−3;Saxon,E.and CR.Bertozzi,2000,Science 287:2007−10)。例えば、CCIPN17(実施例1参照)に使用したSMW PEGすなわちPEG236のStaudinger結合は、対応するホスフィノチオエステル1と共に行うことができ、後者は以下の反応スキーム1に従ってアジドホモアラニンに結合するであろう:
【0066】
【化1】
【0067】
もっと長鎖のPEGのホスフィノチオエステルの合成が記載されている(Soellner,M.B.ら,2003,J.Am.Chem.Soc.125:11790−1)。アジドホモアラニンを含有する組換えタンパク質を作製するためのタンパク質サイズの制限は先験的には存在しないので、一次配列内部のいずれかの選択的場所に付着したSMWポリマー鎖によって任意の長さのタンパク質を作製する方法は従来技術に存在している。
【0068】
本発明のポリマー修飾ポリペプチドを設計するとき、先ず、ターゲットタンパク質のアミノ酸配列を、典型的には遺伝子および/またはタンパク質のデータベースのようなデータベースを含む様々なソースのいずれか1つ以上から入手するかまたは例えば新しい対象タンパク質ターゲットのタンパク質同定によって新規に獲得する。このようなデータベースの非限定例は、GeneBank(Bensonら,1998,Nucleic Acids Res.26:1−7;USA National Center for Biotechnology Information,National Library of Medicine,National Institutes of Health,Bethesda,Md.,USA),TIGR Database(The Institute for Genomic Research,Rockville,Md.,USA),Protein Data Bank(Brookhaven National Laboratory,USA)およびExPASy and Swiss−Protein database(Swiss Institute of Bioinformatics,Geneve,Switzerland)を含む。設計戦略に使用されるポリペプチド主鎖は、ターゲット配列(例えば、ポリペプチド鎖の成熟形態)の一部分(例えば、ドメイン)または全部を表すことができる。次に、所与のターゲットタンパク質について所望のポリマー修飾ポリペプチド構造を設計する。この設計戦略は、ポリペプチドの修飾に使用する水溶性ポリマーの選択、ポリペプチド内部の(1つ以上の)ポリマー付着部位、使用する付着戦略を含む。本発明では特に、SMWポリマーの付着によってどの部位を特異的に遮蔽すべき/できるかを同定および査定するために、最初にポリペプチドアミノ酸配列について1つ以上の機能部位(例えば、グリコシレーション部位、免疫原性部位またはプロテアーゼ感受性部位)の存在を調べる。生物活性部位はしばしば、ヘリックスまたはベータ−シート二次構造;典型的にはループおよびターン領域が実質的に存在しない領域を表す。従って、本文中に記載したようにSMWポリマーの付着を介して隠蔽すべき機能部位は、モデリング方法を使用して同定できる。モデリングは、ターゲットタンパク質の所与の核酸またはアミノ酸配列の部分または全部について、対象ポリペプチドに関する二次元構造情報、対象タンパク質に関する三次元構造情報、またはそれらの組合せを比較できるホモロジーモデリングソフトウェアアルゴリズムまたはプログラムの使用のようなバイオコンピューター処理を含む。いくつかのソフトウェアプログラムおよびデータベースが公知でありこの目的に可用である。これらの方法を組込んだソフトウェアプログラムの一例は、“MAXVECTOR”(Oxford Molecular Group)であり、これは多重タンパク質分析ツールを内蔵し、二次構造(Chou−Fasman and Robson−Garnier methods);親水性(Hopp−Woods,Kyte−Doolittle,Goldman−Engelman−Steitz(GES) and von−Heijne methods);疎水性(Fauchere−Pliska,Janin,Manavalan,Sweet−Eisenberg methods);抗原性(Hopp−Woods,Parker,Protrusion Index(Thornton),Welling methods);表面確率(Karplus & Schultz’s methods);可撓性(Janin and Emini methods);両親媒性(Eisenberg method);PROSITE−に基づくサブ配列データベースおよびユーザーの定めたサブ配列;カスタム化配列セット;プロテアーゼ部位とタンパク質分解性消化の予測値、などを予測する。このようにして、ターゲット天然タンパク質またはミメティクスの三次元構造および化学的性質を考慮することはポリマー付着場所の選択に役立つであろう。
【0069】
例えば、タンパク質中の免疫原性部位を予測するために以下の方法を使用できる。タンパク質配列の二次構造を最初に予測し、無秩序領域、特にターンを同定する。この目的にはChou−Fasmanパラメーターが特に役立つ(Chouら,1978,Advanced Enzymology 47:45−148;Qianら,1988,J.Mol.Biol.202:865−884;およびGenetics Computer Group,Inc.の“PEPEPLOT”プログラム)。また、タンパク質配列の疎水性は、典型的には標準方法(たとえば,Fauchere−PliskaまたはHopp−Woodsの疎水性予測法;またはBull−Breese)による6残基の走行平均を使用して走査する。より親水性の領域はターンである領域に特に注意して同定する。同時に、N−結合部位およびO−結合部位を含むグリコシル化部位を同定する。タンパク質中の最も親水性の部位は、それがターンに存在しグリコシル化されていると予測されていないならば免疫原性の確率が高いのでポリマー修飾部位として使用できるであろう。親水性が順次に低くなる親水性領域に同じ基本法則を応用し二次的なポリマー修飾用部位を同定する。
【0070】
また、生物学的および化学的分析も本発明によるポリマー付着が効果的な特異的場所を同定するために使用し得る。例えば、アラニン走査は、免疫原性部位、プロテアーゼ感受性部位またはポリマー修飾に適した他の部位を同定するために使用でき、生物活性に必要な不変残基を同定するために特に有用である。アラニン走査はまた、上記のような他のタンパク質分析ツールと併用することもできる。別の方法は、修飾用水溶性ポリマー分子が各分子のポリペプチド鎖中の異なる部位に局在している合成した対象タンパク質のライブラリーを体系的に作製することである。ポリマー修飾ポリペプチド鎖のライブラリーのフォールデイング後、例えば、フォールドした分子をフォールドしない分子からまたは受容体結合分子を非結合分子から分離するために機能性選択/アッセイを実行できる。次いで機能性分子についてポリマー修飾部位の好ましい場所を決定できる。このような方法では、非干渉性ポリマー修飾部位の同定を容易にするために“protein signature analysis”の原則を利用できる(Muirら,1996,Chem.Biol.3:817−825)。上述のような様々な付着化学が存在するので、水溶性ポリマーとターゲットペプチドまたはポリペプチドとの間に形成される結合は、カーボネート、エステル、ウレタン、オルトエステル、アミド、アミン、オキシム、イミド、ウレア、チオウレア、チオエーテル、チオウレタン、チオエステル、エーテル、チアゾリジン、ヒドラゾン、オキサゾリジンなどから選択された結合を含むことができる。
【0071】
本発明の1つの目的は、特定の疾患または状態(非限定例としてHIV感染/AIDSを含む)に対する患者の免疫防御を誘発したりおよび/または患者体内でこのような状態の原因となる病理発生を抑制するためにヒトまたはヒト以外の哺乳類患者に投与できるワクチンに関する。一般に、本発明のワクチンは、真菌類、原生動物、細菌およびウイルス(例えばインフルエンザウイルス、特にHIV)を非限定例とする多くの病原性生物に対する免疫防御を与えることができる。本発明の1つの実施態様で、ワクチンは、本文中に記載しかつ暫定米国特許出願60/576,062および60/636,724(前出)により詳細に記載された共有結合安定化三量体コイルドコイル構造のポリマー修飾形態を含み、これはHIV gp41を非限定例とするエンベロープウイルスタンパク質の融合中間体のミメティクスを表す。本発明の1つの実施態様では、共有結合安定化コイルドコイルミメティクス内部に含まれた個々のキメラペプチドの1つ以上が、PEGを非限定例とする低分子量水溶性ポリマーによって修飾され、その結果としてコイルドコイルの安定性スカフォールドドメインの全部または一部分、従って該ドメインに内包されている1つ以上のエピトープが免疫由来受容体/細胞と相互作用しないようにポリマーによって遮蔽されている。従って本発明は、本文中に記載のポリマー修飾ワクチン構築物を治療有効量(後記に定義)で投与する哺乳動物の免疫方法または病原性生物に対する防御免疫応答の誘導方法に関する。方法は、単独で存在するかまたは非天然型ポリペプチド成分(例えば、前述のようなスカフォールドコイルドコイル)に結合して(例えば、融合ペプチド/タンパク質として)存在する天然型病原性抗原の全部または一部分のポリマー修飾形態(例えば、1つ以上のSME PEG部分によって修飾)を含むワクチンを哺乳動物に投与する段階を含む。ポリマーの付加は、該ポリマーの存在によって遮蔽された領域を免疫減衰し、免疫応答を抗原の非隠蔽部分に集中させる。従って、本発明の1つの実施態様は、脊椎動物の生体組織、好ましくはヒトまたはヒト以外の商業用もしくは飼育用の獣医学的に重要な哺乳動物のような哺乳類宿主に直接導入されるとヒト免疫不全ウイルス−1(HIV−1)を特異的に認識する細胞性免疫応答を誘導するHIV医薬製品、その製造および使用(本文中に記載)に関する。
【0072】
従って、本発明のポリマー修飾ポリペプチドの好ましい用途は、治療有効量のワクチン形態のポリペプチドを哺乳動物に投与する段階を含む、哺乳動物の疾患状態、例えばHIV感染の治療である。これは、ヒトまたはヒト以外の哺乳類の疾患個体のいかなる治療も包含し、例えば:(i)当該疾患への素因はあるけれども罹患していると診断されたことがない個体の発症予防;(ii)疾患の進行の阻害および/または停止;または、疾患の軽減、すなわち疾患の退行の惹起、を含む。本文中に使用した“治療有効量”という用語は、哺乳動物に投与したとき治療効果を生じるため、例えば哺乳動物のHIV−1感染を阻害するために十分な本発明のポリマー修飾ポリペプチドおよび/または該ポリペプチドを含有するワクチン組成物の量を意味する。“治療有効量”を構成する量は、ポリペプチド、状態または疾患の種類とその重篤度、治療される哺乳動物の体重、年齢、などに従って変更されるが、平均的な当業者は現状の知識およびこの開示に基づいてその量を容易に決定できるであろう。
【0073】
ペプチドのような低分子は免疫原性に乏しく、従って、該分子に対する実体的な免疫応答を誘発するためにより大きい巨大分子(担体)とのペプチド接合体を調製することがしばしば必要である。従って、本発明のポリマー修飾ポリペプチドはさらに免疫原性担体に接合し得る。本文中に記載したコイルドコイル構造の場合、個々のポリマー修飾α−ヘリカルキメラペプチド(例えば、CCIPN17)を最初に免疫原性担体に接合し、次いで、個別に接合した3つのペプチドから成る共有結合安定化ホモ三量体またはヘテロ三量体コイルドコイル構造の形成に導く条件を作用させる。あるいは、本発明のキメラペプチドから成る共有結合安定化ポリマー修飾ホモ三量体またはヘテロ三量体コイルドコイル構造を免疫原性担体にコンジュゲートさせてもよい。通常はヘテロロガスタンパク質から成る担体分子は、コイルドコイルの非隠蔽部分(例えば、HIV部分)に対する免疫応答の誘発および/または増進を援助する。ポリマー修飾ポリペプチドは、非特異的架橋剤、一属性(monogeneric)スペーサーまたは二属性(bigeneric)スペーサーによって免疫原性担体に連結できる。本発明のポリマー修飾ポリペプチドがコンジュゲートできる多くの免疫原性担体分子が当業界で公知である(参照:たとえば,Shodelら,1996,J.Biotechnol.44:91−96;Lang and Korhonen,1997,Behring Inst.Mitt.98:400−409;Brennanら,2001,Mol.Biotechnol.17:15−26;Pumpens and Grens,2201,Intervirology 44:98−114;およびSimpsonら, 1999,Cell Mol.Life Sci.56:47−61)。このような潜在能力をもつ免疫原性分子の非限定例は、Neisseria meningitidis OMPC粒子、キーホールリンペットヘモシアニン(KLH)、ウシ血清アルブミン(BSA)、オボアルブミン(OVA)、チログロブリン(TG)、HBV−コア抗原、HBV−表面抗原や、破傷風毒素、ジフテリア毒素またはロタウイルスVP6のような免疫原性タンパク質、および、p24含有HIVキャプシド粒子である。
【0074】
従って本発明の1つの実施態様では、本文中に記載のポリマー修飾HIV−特異的ペプチドミメティクスをHIV感染の防止または防御(例えば予防または治療)用ワクチンの成分として使用し、HIVに対して免疫活性のHIV−特異的中和抗体を生成させることができる。ワクチンはMPL−Aのような当業界で公知のアジュバントに配合し、ミョウバンまたはリン酸アルミニウムに吸着させる。抗原性接合体はまた、より強力なヘルパーT細胞応答を実現するために、合成の非天然pan HLA DR−結合エピトープ(PADRE)を非限定例とするT細胞ヘルパーエピトープを含み得る(参照:たとえば,Alexanderら,2000,J.Immunol.,164:1625−1633およびdel Guercioら,1997,Vaccine 15:441−448)。本文中に記載のポリマー修飾ポリペプチドの免疫原性接合体は、それらの構成成分パーツ(ポリマー修飾ポリペプチドと担体)を単離、合成および精製し、次いで2つの成分を接合することによって調製し得る。所望ならばその後にコンジュゲート混合物の精製を行ってもよい。ポリマー修飾ポリペプチドと適当な免疫原性担体との抗原性接合体は、構成成分パーツ(すなわち、ポリマー修飾ポリペプチドと担体)間に少なくとも1つの共有結合連結を有しており、典型的には2つ以上のポリマー修飾ポリペプチド分子が担体分子のおのおのに共有結合している。構成成分を別々に調製するならば、その後、非特異的架橋剤、一属性スペーサーまたは二属性スペーサーによって連結できる。非特異的架橋の方法は当業界で公知であり、その非限定例は、グルタルアルデヒドとの反応、スクシニル化担体添加または不添加で行うN−エチル−N’−(3−ジメチルアミノプロピル)カルボジイミドとの反応、グリコシル化置換基のペルヨージド酸化次いでホウ水素化ナトリウムまたはシアノホウ水素化ナトリウムの存在下で行うタンパク質担体の遊離アミノ基への結合、芳香族アミノ基のジアゾ化次いでタンパク質のチロシン側鎖残基への結合、イソシアナートとの反応、または、混合無水物との反応を含む。概要についてはBriandら,1985,J.Imm.Meth.78:59−69を参照するとよい。
【0075】
本文中に記載のキメラペプチドと例えば樹脂または担体との接合を容易にするために、本発明のポリマー修飾ポリペプチド、例えばCCIPN17をさらに修飾して、追加のシステイン残基を含有させてもよい。CCIZN17/CCIPN17の場合、これはペプチドのNH2−末端に1個の追加システインアミノ酸残基を組込むことである(たとえば,“CCCIZN17”−CCCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL;配列:76)。場合によっては、奇数番号の末端システイン残基を可撓性化学リンカー、例えば、Ttds(1−アミノ−4,7,10−トリオキサ−13−トリデカミンコハク酸;−NH−(CH2)3−O−(CH2)2−O−(CH2)2−O−(CH2)3−NH−CO−CH2)2−CO−を用いて残りの連続システイン残基から分離してもよい。暫定米国特許出願60/576,062および60/636,724(前出)参照。奇数番号のシステイン残基は、本文中に記載のようなポリマー修飾CC−および/または誘導体化キメラN−ペプチドを含む共有結合安定化三量体の形成後に、共有結合安定化三量体あたり少なくとも1つの反応性チオール基がマレイミジルのようなチオール−反応性基とさらに反応するために可用であることを確保する。リンカーは、三量体化ドメインとシステイン残基とのジスルフィドブリッジ間に接合/誘導体化に適した可撓性、可溶性および間隔を提供する。追加のシステイン残基は、該システイン残基がジスルフィド結合または化学選択的結合の形成(例えば、チオエーテル結合形成)および/またはポリマー付着に参加することを防止するために保護チオール基を有していてもよい。保護基(非限定例はフルオレニルメトキシ(“Fm”)またはアセタミドメチル(“Acm”)基を含む)は共有結合安定化三量体コイルドコイルの形成後に除去することができ、新しく露出したチオール基が以後の接合反応に可用である。
【0076】
本発明のワクチンは、宿主に投与するための医薬的に有効な配合物に配合し得る。このような配合物は例えば、リン酸塩緩衝生理食塩水(PBS)のような生理的塩類溶液でよい。本発明のワクチン構築物に長期間安定性も与えるような医薬的に許容される配合物の使用が有用であろう。様々な保存条件(例えば、pH、バッファの種類、塩濃度、光照射、高温度、低イオン強度)はペプチドワクチンの安定性を最適にするように配合物中でコントロールし得る。ワクチンは、それぞれのワクチン配合物にアジュバントを添加するかまたは不添加で投与し得る。従って本発明のペプチドワクチンはまた、本発明のワクチン構築物の免疫原性を増強する1種以上のアジュバントと配合し得る。数多くのこのようなアジュバントが当業界で公知である。本発明のワクチンに使用するためのアジュバントの2つの例は、1つ以上の形態のリン酸アルミニウム基剤のアジュバントおよびサポニンアジュバントQS21(サポニンと3D−モノホスホリル脂質A(MPL)の精製形態、リポ多糖(LPS)の無毒誘導体)であり、おのおのが多くの動物モデル体内でワクチンに対して有効な体液性および細胞性応答を示す。熟練の当業者は最適免疫応答を与えるようにペプチドワクチン対アジユバント比を調整し得る。別のアジュバントは、POE−POP−POEブロックコポリマーのようなポリオキシエチレン(POE)およびポリオキシプロピレン(POP)のブロックを含む非イオン性ブロックコポリマーである(Newmanら,1998,Critical Reviews in Therapeutic Drug Carrier Systems 15(2):89−142)。基本構造は、POE−POP−POEブロックコポリマーのようなポリオキシエチレン(POE)およびポリオキシプロピレン(POP)のブロックを含む。Newmanら(同書)は、ある種のPOE−POP−POEブロックコポリマーがインフルエンザタンパク質基剤のワクチンにアジュバントとして有用であろうと開示している。すなわち、約9000ドルトンから約20,000ドルトンの範囲の分子量を有している中央POPブロックとコポリマーの全分子量の約20%までを含むフランキングPOEブロックとを含む高分子量POE−POP−POEブロックコポリマーである(参照:米国再発行特許番号36,665,米国特許番号5,567,859,番号5,691,387,番号5,696,298および番号5,990,241、すべてEmanueleらに許諾、これらのPOE−POP−POEブロックコポリマー関連)。WO96/04932はさらに、界面活性剤特性を有しておりワクチンアジュバントとして生物効果を示す高分子量POE/POPブロックコポリマーを開示している。この文節に引用した上記文献はそれらの記載内容全部が参照によって本発明に含まれるものとする。従って、アジュバント不添加ワクチン投与に比べて本発明のワクチン構築物の免疫応答を増強するために有効なアジュバントの利用は当業者の認識範囲に入っている。
【0077】
本発明のワクチン構築物は、経腸的および非経口的経路のような当業界で公知の手段のいずれかによって投与できる。これらのデリバリー経路は非限定的に、筋肉内注射、腹腔内注射、静脈内注射、吸入または鼻腔内デリバリー、経口デリバリー、舌下投与、皮下投与、経皮投与(transdermal,transcutaneous,percutaneous)、または何らかの形態の粒子衝撃または何らかの可用な無針注入デバイスを含む。ワクチンレシピエントに導入すべきポリマー修飾ポリペプチド構築物の量は、ポリペプチドの免疫原性に従属するであろう。また、本発明のワクチンに対する全体的免疫応答を最適にするようにブースターワクチンを接種することも考察される。
【0078】
本文中で言及したすべての刊行物は本発明に関連して使用する方法および材料を記載および開示する目的で引用した。本発明が従来発明によるこれらの開示に先行する権利がないと是認したと解釈すべきではない。
【0079】
本発明の好ましい実施態様を添付図面と共に記載したが、本発明がこれらの具体的な実施態様に限定されないこと、特許請求の範囲に記載の本発明の範囲または要旨を逸脱することなく様々な変更および修正が当業者に可能であることを理解されたい。従って、以下の実施例は本発明の例示であるが本発明の限定ではない。
【実施例1】
【0080】
望ましくないエピトープを遮蔽するためのSMW PEGの使用:
SMW PEG−誘導体化HIVワクチンの設計
この実施例はHIVワクチンの分野に役立つ本発明の1つの用途を示す。HIV gp41のN−ヘリックスの三量体コイルドコイルのミメティクスを表すサブユニットワクチンとして使用するように操作したペプチドを予め設計した(参照:暫定米国特許出願番号60/576,062および60/636,724(前出))。これらのサブユニットワクチンは、設計した可溶性三量体コイルドコイル(“スカフォールド”領域)と、HIV gp41のN−ヘリックスコイルドコイルの一部分と、鎖間共有結合形成用共有結合安定化部分とを含むキメラペプチドである。このようなミメティクスの一例を図1Aに概略的に示す。これは“(CCIZN17)3”と命名した共有結合安定化三量体コイルドコイルを表す。(CCIZN17)3[配列:2]3内部の3つのキメラCCIZN17ペプチドを共有結合安定化するジスルフィド結合の化学構造図を図1Bに示す。3つのジスルフィド結合がNH2−末端システイン残基のチオール(−SH)基間に局在している。システイン残基のカルボキシ末端に局在する連続アミノ酸は一文字命名法の活字体で示す(図1B)。gp41のプレヘパリン複合体を模倣する三量体コイルドコイルのHIV部分(N−ペプチド部分;例えば(CCIZN17)3中の“N17”)の安定な提示には、スカフォールド領域および鎖間共有結合の双方が必須である。gp41のN17領域によって生成されるコンホメーションエピトープをターゲットする抗体は、N17を内包する三量体コイルドコイルすなわち疎水性ポケット内部に拘束されたとき、単一サイクル感染力アッセイでHIV−1に対する抗ウイルス活性を示す(参照:暫定米国特許出願、出願日2004.6.1)。HIV gp41の三量体コイルドコイルN−ペプチド部分を特異的にターゲットする抗体を増感するこのクラスのペプチドワクチンの使用を容易にするために、本発明の発明者らは、スカフォールドと鎖間共有結合とを含む部分を免疫認識から遮蔽しながらペプチドミメティクスのHIV部分に選択的に集中した免疫応答を誘導する戦略を計画した。
【0081】
CCIZN17(配列:2)の合成−
ペプチドCCIZN17(配列:2)は、Pioneerペプチドシンセサイザー(Applied Biosystems)でFmoc/t−Bu化学を使用して固相合成した。使用した樹脂は、Fmoc−Linker AM−Champion、1%架橋(Biosearch Technologies,Inc.)であり、これは、修飾Rinkリンカーp−[(R,S)−α−[9H−フルオレン−9−イル−メトキシホルムアミド]−2,4−ジメトキシベンジル]−フェノキシ酢酸(Rink,H.,1987,Tetrahedron Lett.28:3787−3789;Bernatowicz,M.S.ら,1989,Tetrahedron Lett.30:4645−4667)によって誘導体化したPEG−PS基材樹脂である。すべてのアシル化反応は樹脂遊離アミノ基の4倍過剰量の活性化アミノ酸で60−120分行った。アミノ酸は、等モル量のHBTU(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)および2倍モル過剰量のDIEA(N,N−ジイソプロピルエチルアミン)で活性化した。側鎖保護基は以下を使用した:グルタミン酸(Glu)およびトレオニン(Thr)にtert−ブチル;システイン(Cys)およびグルタミン(Gln)にトリチル;リシン(Lys)およびトリプトファン(Trp)にtert−ブトキシ−カルボニル;アルギニン(Arg)に2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニル。アセチル化反応は、ペプチド組立の終了後、10倍過剰量の無水酢酸とN,N−ジメチルホルムアミド(DMF)中で反応させることによって行った。合成の終了後、乾燥ペプチド−樹脂を、88%トリフルオロ酢酸(TFA)、5%フェノール、2%トリイソプロピルシランおよび5%水(Sole,N.A.and G.Barany,1992(前出))によって室温で1.5時間処理した。樹脂を濾過し、溶液を低温メチル−t−ブチルエーテルに加えてペプチドを沈殿させた。遠心後、ペプチドペレットを新しい低温メチル−t−ブチルエーテルで洗浄して有機スカベンジャーを除去した。このプロセスを2回繰返した。最終ペレットを乾燥し、H2O、20%アセトニトリルに再懸濁させ、凍結乾燥した。
【0082】
粗ペプチドCCIZN17(配列:2)をToyopearl HW−50S樹脂を詰めた700×26mmカラムで、30%アセトニトリルを含む水、0.1%TFAを溶出剤として使用し、ゲル透過クロマトグラフィー(GPC)によって精製した。典型的な処理では、300mgの粗ペプチドを12mLの溶出剤に溶解し、流速1mL/分でカラムに直接充填した。溶出画分の分析用HPLCは、Beckman HPLCで、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用い、溶出剤B(上記)の以下の勾配:40%−60%のB(20分)−80%(3分)、流速1mL/分を使用して行った。純度に基づいて画分をプールし、セミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用い、溶出剤として(A)水中の0.1%のTFA、(B)アセトニトリル中の0.1%のTFAを使用する逆相HPLCによってさらに精製した。溶出剤Bの以下の勾配を使用した:35%−50%,20分超,流速80mL/分。Micromass LCZ platform質量分析計でエレクトロスプレー質量分析法によって精製ペプチドのキャラクタリゼーションを行った。理論的平均分子量(MW))は5175Daであり、測定MWは5175Daである。
【0083】
CCIZN17から(CCIZN17)3への酸化−
精製ペプチド前駆体(12mg)、CCIZN17(配列:2)を0.1MのHepes,pH7.3(USB Corp.)に1mg/mLの濃度で溶解した。この条件下で、CCIZN17は空気によってゆっくりと酸化して共有結合三量体(CCIZN17)3([配列:2]3)になる。図1B参照。酸化反応は、Waters−Micromass LCZ Platformで、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用い、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用するLC−MS分析によってモニターした。溶出剤Bの以下の勾配を使用した:40%−60%B(20分)−80%(3分)、流速1mL/分。これらのクロマトグラフィー条件で、CCIZN17はtR=13.25’に溶出する。酸化反応は3時間円滑に進行しtR=15’に溶出する主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのCCIZN17ペプチド鎖([CCIZN17]3)を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液(12mL)に24μLのTFAを添加し、溶液をTSKgel Toyopearl HW−50S樹脂を詰めた700×26カラムに充填し、溶出剤としてH2O/アセトニトリル70/30、0.1%TFA、流速1mL/分を使用した。溶出分画をPhenomenex,Jupiter C4カラム(150×4.6mm,5μm)で、溶出剤として:(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用し、HPLCによって分析した。LC−MS Waters−Micromass LCZ Platformで溶出剤Bの以下の勾配を使用した:40%−60%B(20分)−80%(3分)、流速1mL/分。共有結合三量体に対応するプール画分を、ESI−QqToF(Applied Biosystems)、正モード(ES+)、ナノスプレー1μl注入、を用いる質量分析法によってさらに分析した。予想MWは15520.2Daであり、測定MWは15520.1Daである。
【0084】
CCIPN17の合成−
ペプチドCCIPN17(図2B参照)は上記のCCIZN17(配列:2)の場合と同じプロトコルに従って合成した。固相配列組立中に、IZ部分(図2Bおよび以下に示した配列の下線部分)の2つのリシン残基にアリルオキシカルボニル(aloc)のε−NH保護を導入した。他のリシン残基はすべて標準Fmoc/tBu化学に従ってBocで保護した:CCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL(配列:2;スカフォールドドメインはイタリック体)。CCIPN17のアミノ酸配列はCCIZN17のアミノ酸配列と同じであるが、PEG付加CCIZN17ペプチドを“CCIPN17”と表す。ペプチド組立の終了後、NH2−末端を10倍過剰量の無水酢酸とDHF中で反応させることによってアセチル化した。保護ペプチド樹脂を0.25当量のテトラキス(トリフェニルホスフィン)パラジウムPd(PPh3)4および24当量のフェニルシランで30分間処理して2つのリシン残基からAloc保護基を除去した。他の残基側鎖はすべて保護された状態に維持した。2つの脱保護リシン鎖をDIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−dPEG酸,Quanta BioDesign)によってさらに誘導体化した。
【0085】
粗ペプチドCCIPN17をCCIZN17(上記)の精製の場合と同様のゲル透過クロマトグラフィーによって精製した。GPCによって得られたプール画分(純度70%)を上記のCCIZN17場合と同様の逆相HPLCによってさらに>95%純度まで精製した。溶出剤Bの以下の勾配を使用した:40−55%、25分超、流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、CCIZN17の場合と同様に、Jupiter C4カラム(150×4.6mm,5μm,300A,Phenomenex)を用いた分析用HPLCによって行った。精製ペプチドのキャラクタリゼーションはMicromass LCZ platform ES+を用いた質量分析法によって行った。測定MWは5612.0Daであり計算した平均MWは5611.8Daである。
【0086】
CCIPN17から(CCIPN17)3への酸化−
精製ペプチド前駆体(26mg)、CCIPN17をバッファに溶解し、空気によってゆっくりと酸化して共有結合三量体(CCIPN17)3とした(CCIZN17の酸化の場合と同様)。酸化反応は、Waters−Micromass LCZ Platformを使用するLC−MS分析によって上記同様にモニターした。これらのクロマトグラフィー条件下で、CCIPN17はtR=15.6’に溶出する。酸化反応は円滑に進行し一夜でtR=18.7’に溶出する1つの主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのCCIPN17ペプチド鎖([CCIPN17]3)を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液に45μLのTFAを添加し、溶液を、TSKgel Toyopearl HW−50S樹脂を詰めた700×26mmカラムに直接充填し、溶出剤としてH2O/アセトニトリル40/60,0.1%TFA,流速1mL/分を使用した。溶出画分をPhenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用い、溶出剤として:(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用するHPLCによって分析した。LC−MS Waters−Micromass LCZ Platform、正イオンモード(ES+)で溶出剤Bの以下の勾配を使用した:35%−55%B(20分)−80%(3分),流速1mL/分。予想MWは16829.57Daであり、測定MWは16830Daである。
【0087】
ビオチン−CCIZ(ビオチン−配列:3)の合成−
ペプチド−CCIZ(ビオチン−GCCGGIKKEIEAIKKEQEAIKKKIEAIEKE;配列:3)はCCIZN17(配列:2)の場合と同じプロトコルで合成した。ビオチンとの反応は、ペプチド組立プロセスの終了後、等モル量のDIPC(N,N’−ジイソプロピルカルボジイミド)およびHOAt(7−アザ−1−ヒドロキシベンゾトリアゾール)によって活性化した4倍過剰量のビオチンと一夜反応させることによって行った。粗ペプチドビオチン−CCIZ(配列:3)を、セミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)、流速80mL/分を用い、溶出剤として(A)水中の0.1%のTFAと(B)アセトニトリル中の0.1%のTFAを使用する逆相HPLCによって精製した。溶出剤Bの以下の勾配を使用した:25%−35%,30分超,流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、Ultrasphere C18カラム(250×4.6mm,5μm,80A,Beckman),流速1mL/分を用い、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用する分析用HPLCによって行った。溶出剤Bの以下の勾配を使用した:25%−40%(20分超)−80%(3分超)。予想MWは3570.0Daであり、測定MWは3570.5Daである。
【0088】
ビオチン−CCIZから(ビオチン−CCIZ)3への酸化−
精製ペプチド前駆体(15mg)、ビオチン−CCIZ(ビオチン−配列:3)を0.1MのHepes,pH7.3(USB Corp.)に5mg/mLの濃度で溶解した。ビオチン−CCIZNは空気によってゆっくりと酸化して一夜で共有結合三量体(ビオチン−CCIZN)3となる。酸化反応は上記同様にWaters−Micromass LCZ Platformを使用するLC−MS分析によってモニターした。これらのクロマトグラフィー条件で、ビオチン−CCIZはtR=16.4’に溶出する。酸化反応は円滑に進行し一夜でtR=16.9’に溶出する主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのビオチン−CCIZペプチド鎖([ビオチン−CCIZ]3)を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液を水に透析し、次いで凍結乾燥した。生成物を、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用い、溶出剤として:(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用し、LC−MS Waters−Micromass LCZ Platform、正モード(ES+)で行うHPLCによって分析した。予想MWは10704.94Daであり、測定MWは10704.0Daである。
【0089】
ビオチン−CCIZN17(ビオチン−配列:82)の合成−
ビオチン−CCIZN17(ビオチン−GCCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL;配列:82)はCCIZN17(配列:2)の場合と同じプロトコルで合成した。ビオチンとの反応はペプチド組立プロセスの終了後、上記のビオチン−CCIZの場合と同様に行った。粗ペプチド、ビオチン−CCIZN17の精製は、上記のCCIZN17精製の場合と同様にゲル透過クロマトグラフィー(GPC)によって行った。GPCによって得られたプール画分(純度70%)を上記同様の逆相HPLCによってさらに>95%純度まで精製した。溶出剤Bの以下の勾配を使用した:35%−55%、25分超、流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、Jupiter C4カラム(150×4.6mm,5μm,300A,Phenomenex)、流速1mL/分を用い、溶出剤として:(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用する分析用HPLCによって行った。溶出剤Bの以下の勾配を使用した:40%−60%B(20分)−80%(3分)。精製ペプチドのキャラクタリゼーションはMicromass LCZ platform ES+で質量分析法によって行った。測定MWは5416.0Daであり計算した平均MWは5416Daである。
【0090】
ビオチン−CCIZN17から(ビオチン−CCIZN17)3への酸化−
精製ペプチド前駆体(20mg)、ビオチン−CCIZ17を0.1MのHepes,pH7.3(USB Corp.)に2mg/mLの濃度で溶解した。この条件下でビオチン−CCIZN17は空気によってゆっくりと酸化して共有結合三量体(ビオチン−CCIZN17)3となる。酸化反応は上記同様にWaters−Micromass LCZ Platformを使用するLC−MS分析によって、溶出剤Bの勾配:40%−60%B(20分)−80%(3分),流速1mL/分を使用してモニターした。これらのクロマトグラフィー条件下で、ビオチン−CCIZ17はtR=12.87’に溶出する。酸化反応は円滑に進行し一夜でtR=14.9’に溶出する1つの主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのビオチン−CCIZ17ペプチド鎖(ビオチン−CCIZN17)3を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液に45μLのTFAを添加し、TSKgel Toyopearl HW−50S樹脂を詰めた700×26mmカラムに溶液を直接充填し、溶出剤H2O/アセトニトリル40/60、0.1%TFA、流速1mL/分を使用した。プールした画分をセミ・プレパラティブPhenomenex C4カラム(10×250mm,15μm)、流速20mL/分を用い、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用する逆相HPLCによってさらに精製した。溶出剤Bの以下の勾配を使用した:35%−50%,20分超,流速80mL/分。溶出した画分を、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)で溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用するHPLCによって分析した。LC−MS Waters−Micromass LCZ Platform、正モード(ES+)で溶出剤Bの以下の勾配を使用した:40%−60%B(20分)−80%(3分)、流速1mL/分。予想MWは16244Daであり測定MWは16245Daである。
【0091】
ビオチン−CCIPN17の合成−
このペプチドは上記のビオチン−CCIZN17(配列:82)の場合と同じプロトコルに従って合成した。固相配列組立中に、IZ部分(下記配列の下線部分)の2つのリシン残基にアリルオキシカルボニル(aloc)のε−NH保護を導入し、他のリシンはすべて標準Fmoc/tBu化学に従ってBoc保護した:ビオチン−GCCGGIKKEIEAIKKEQEAIKKKIEAIEKLLQLTVWGIKQLQARIL(配列:82;スカフォールドドメインはイタリック体)。PEG付加ビオチン−CCIZN17ペプチドを“ビオチン−CCIPN17”と命名するが、ビオチン−CCIPN17のアミノ酸配列はビオチン−CCIZN17のアミノ酸配列と同じである。ペプチド組立の終了後、上記のビオチン−CCIZの場合と同様にビオチンと反応させた。保護ペプチド樹脂を0.25当量のテトラキス(トリフェニルホスフィン)パラジウムPd(PPh3)4および24当量のフェニルシランで30分間処理して2つのリシン残基からAloc保護基を除去し、他の残基側鎖はすべて保護された状態に維持した。2つの脱保護リシン鎖を、DIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−d PEG酸,Quanta BioDesign)によってさらに誘導体化した。
【0092】
粗ペプチドビオチン−CCIPN17を上記同様にTSKgel Toyopearl HW−50S樹脂(700×26mmカラム)上のゲル透過クロマトグラフィー(GPC)によって精製した。GPCによって得られたプール画分(純度70%)を、上記同様のセミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジを用いる逆相HPLCによって、溶出剤Bの勾配:35−55%、25分超、流速80mL/分を使用して、さらに>95%純度まで精製した。。粗ペプチド、溶出画分および精製プールの分析は、上記同様にJupiter C4カラム(150×4.6mm,5μm,300A,Phenomenex)を用いる分析用HPLCによって行った。精製ペプチドのキャラクタリゼーションはMicromass LCZ platform ES+の質量分析法によって行った。測定MWは5796Daであり、計算した平均MWは5796Daである。
【0093】
ビオチン−CCIP17から(ビオチン−CCIPN17)3への酸化−
精製したペプチド前駆体(20mg)、ビオチン−CCIPN17を0.1MのHepes,pH7.3(USB Corp.)に濃度2mg/mLで溶解した。この条件下で、ビオチン−CCIPN17は空気によってゆっくりと酸化して一夜で(ビオチン−CCIPN17)3となる。酸化反応は、Waters−Micromass LCZ PlatformをPhenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いるLC−MS分析によって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用してモニターした。溶出剤Bの以下の勾配を使用した:45%−60% B(20分)−80%(3分)、流速1mL/分。これらのクロマトグラフィー条件下で、ビオチン−CCIPN17はtR=11.9’に溶出する。酸化反応は円滑に進行し一夜でtR=17.8’に溶出する1つの主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのCビオチン−CIPN17ペプチド鎖([ビオチン−CCIPN17]3)を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液にグアニジニウムクロリドを最終濃度6Mまで添加し、溶液をTSKgel Toyopearl HW−50S樹脂を詰めた700×26mmカラムに直接充填し、溶出剤としてH2O/アセトニトリル40/60、0.1% TFA、流速1mL/分を使用した。溶出した画分をPhenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いるHPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して分析した。溶出剤Bを以下の勾配で使用した:45%−60% B(20分)−80%(3分)、流速1mL/分、LC−MS Waters−Micromass LCZ Platform、正モード(ES+)。予測MWは17383Daであり、計算MWは17384Daである。
【0094】
ビオチン−CCIPの合成−
このペプチドは上記のビオチン−CCIZ(配列:3)の場合と同じプロトコルに従って合成した。固相配列組立中、IZ部分(下記配列の下線部分)の2つのリシン残基にアリルオキシカルボニル(aloc)のε−NH保護を導入し、他のリシンはすべて標準Fmoc/tBu化学に従ってBocで保護した:ビオチン−GCCGGIKKEIEAIKKEQEAIKKKIEAIEK(配列:3;スカフォールドドメインはイタリック体)。PEG付加ビオチン−CCIZペプチドを“ビオチン−CCIP”と命名するが、ビオチン−CCIPのアミノ酸配列はビオチン−CCIZのアミノ酸配列と同じである。ペプチド組立の終了後、上記のビオチン−CCIZの場合と同様にビオチンと反応させた。保護されたペプチド−樹脂をさらに0.25当量のテトラキス(トリフェニルホスフィン)パラジウムPd(PPh3)4および24当量のフェニルシランで30分間処理して2つのリシン残基からAloc保護基を除去し、他の残基側鎖はすべて保護された状態に維持した。2つの脱保護リシン鎖を、DIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−d PEG酸,Quanta BioDesign)によってさらに誘導体化した。
【0095】
粗ペプチド、ビオチン−CCIPをTSKgel Toyopearl HW−50S樹脂(700×26mmカラム)上で溶出剤として水中の30%アセトニトリル、0.1%TFA、流速1mL/分を使用するゲル透過クロマトグラフィー(GPC)によって精製した。GPCによって得られたプール画分(純度70%)をセミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(4×200mm,15μm)を用いる逆相HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して>95%純度までさらに精製した。溶出剤Bの以下の勾配を使用した:20−35%、30分超、流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、Jupiter C4カラム(150×4.6mm,5μm,300A,Phenomenex)、流速1mL/分、を用いる分析用HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して行った。溶出剤Bの以下の勾配を使用した:20−40%(20分超)−80%(3分超)。予測MWは3821Daであり、測定MWは3821Daである。
【0096】
ビオチン−CCIPから(ビオチン−CCIPN)3への酸化−
精製したペプチド前駆体(20mg)、ビオチン−CCIPN17を0.1MのHepes,pH7.3(USB Corp.)に濃度2mg/mLで溶解した。この条件下で、ビオチン−CCIPは空気によってゆっくりと酸化して一夜で共有結合三量体(ビオチン−CCIP)3となる。酸化反応は、Waters−Micromass LCZ PlatformをPhenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いたLC−MS分析によって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用してモニターした。溶出剤Bの以下の勾配を使用した:25%−40% B(20分)−80%(3分)、流速1mL/分。これらのクロマトグラフィー条件下で、ビオチン−CCIPはtR=15.5’に溶出する。酸化反応は円滑に進行し一夜でtR=18.4’に溶出する1つの主生成物が形成される。生成物の質量は、3つのジスルフィドブリッジの形成に合致する質量減を有する3つのビオチン−CCIPNペプチド鎖([ビオチン−CCIP]3)を含む1分子の質量に対応する。酸化反応の総収率は80%超である。溶液にトリフルオロ酢酸を最終濃度0.2%までおよびグアニジニウムクロリドを最終濃度6Mまで添加し、溶液を、TSKgel Toyopearl HW−50S樹脂を詰めた700×26mmカラムに直接充填し、溶出剤としてH2O/アセトニトリル40/60、0.1% TFA、流速1mL/分を使用した。溶出した画分は、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いるHPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して分析した。溶出剤Bを以下の勾配で使用した:25%−40% B(20分)−80%(3分)、流速1 mL/分、LC−MS Waters−Micromass LCZ Platform、正モード(ES+)。予測MWは11456Daであり、測定MWは11456Daである。
【0097】
ワクチン設計−公称サイズ約15.6オングストロームのポリエチレングリコール部分をCCIZN17(配列2)のスカフォールドドメインに、該PEG部分が三量体コイルドコイル形成を少しは妨害するとしても妨害が最小になるように戦略的に付着させた。この小さいポリエチレングリコール部分m−dPEGTM酸(MW=236)(Quanta BIODESIGN,Powell,Ohio)は分子量約236Daであり、以下の構造:
【0098】
【化2】
を有している。
【0099】
最初に、三量体コイルドコイルのヘプタッドリピートのより外側の“f”位置に局在する3つのリシン残基のイプシロンアミンをIZスカフォールドへのポリエチレングリコール部分の付着に適した位置として同定した(図2A)。これらのリシン残基の2つをm−dPEGTM酸(MW=236)による誘導体化のために最終的に選択した(図2B参照、誘導体化リシン(“K”)残基は下線付き)。図2Bに示すように、このポリマー修飾CC−キメラN−ペプチドの名称は、CCIZN17からCCIPN17に変更されており、3つのCCIPN17キメラポリマー修飾ペプチドを含む共有結合安定化(CCIPN17)3三量体コイルドコイルを生成できる。このワクチン構築物(CCIPN17)3の概略モデルを図3に示す。三量体コイルドコイルを形成するヘリックスは、長さ約62オングストロームの円筒として示されている。各円筒の暗い灰色部分は長さ約36オングストロームのIZスカフォールドドメインを表し、明るい灰色部分は長さ約25オングストロームのHIV gp41のN17領域を表す。m−dPEGTM酸(MW=236)部分は長さ約15オングストロームの長方形として表されている。ポリマー修飾用に選択された2つの指定リシン残基の目的はIZスカフォールドだけを遮蔽することである。実際、コイルドコイルのN17ドメインに近いほうのリシン残基はN17コイルドコイル部分から約22オングストロームであり、15オングストロームのm−dPEGTM酸部分がこのドメインのいずれかの部分を遮蔽できないことを確保する。
【0100】
SMW−PEG付加ワクチンの免疫原性−
動物実験はPEGを付加しないワクチン(CCIZN17)3およびPEG付加ワクチン(CCIPN17)3によって誘発されたモルモットの抗体応答の比較によって行った。試験にはホモロガス方式(すなわち、抗原によるプライミング注射、次いで同じ抗原による2回のブースト注射)およびヘテロロガス方式(すなわち、PEGを付加しない抗原(CCIZN17)3によるプライミング注射、次いでPEG付加抗原(CCIPN17)3による2回のブースト注射)の双方を使用した(図4参照)。PEG付加抗原はQS21アジュバント(Antigenics;New York,NY)と共に動物に投与した。免疫応答の分析はELISAによって行った。ELISAプレートへの抗原の吸着能力の違いに起因するバイアスを除去するために、すべての捕獲抗原をビオチニル化し、捕獲ビオチニル化分子を添加する前にELISAプレートをストレプトアビジンで被覆した。このようにすると各抗原に対するELISA幾何平均力価(GMT)が、血清中に存在する対応特異性をもつ抗体の相対量を正確に反映する。免疫分子のビオチニル化形態(ビオチン−CCIZN17)3および(ビオチン−CCIPN17)3ならびにスカフォールド分子単独のビオチニル化形態(ビオチン−CCIZ)3および(ビオチン−CCIP)3をELISAアッセイの捕獲抗原として使用した。PEGを付加しないワクチン(CCIZN17)3で予防接種した動物の血清中には全抗原およびスカフォールド部分単独と同等の量の抗体が存在する。しかしながらPEG付加ワクチンによる免疫は、ホモロガス方式でもヘテロロガス方式でも、スカフォールド((CCIZ)3でもPEG付加(CCIP)3でもない)に対しては有意に減退した応答が生じるが、N−ペプチド部分に対しては極めて高い力価が誘発される。これは(CCIZN17)3および(CCIPN17)3の双方で等しく、従って、どちらの抗原に対して測定したときもGMT測定値が等しい。
【0101】
この実験は、タンパク質抗原の特定領域を選択的に隠蔽するSMW PEG部分の付着が、ペプチド/タンパク質の残りの部分の免疫原性を遮蔽することなく該領域の免疫反応性を除去できることを証明する。
【実施例2】
【0102】
アルツハイマー病の免疫療法
PEG236−Aβ1−42(配列:4;図5C参照)の合成−
ABI433シンセサイザー(Applied Biosystems)で標準Fmoc/t−Bu化学に従ってペプチドを固相合成した。使用した樹脂は予め誘導体化したFmoc−Ala−ALH−Champion,1%架橋(Biosearch Technologies,Inc.)であり、これはカルボキシル化アラニンを産生する誘導体化PEG−PS基材樹脂である。すべてのアシル化反応は8倍過剰量の活性化アミノ酸で樹脂の遊離アミノ基に60分間行った。アミノ酸は等モル量のHATU(O−(7−アザベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)および2倍モル過剰量のDIEA(N,N−ジイソプロピルエチルアミン)で活性化した。側鎖保護基は以下を用いた:グルタミン酸(E)、アスパラギン酸(A)、チロシン(Y)およびセリン(S)にtert−ブチル;ヒスチジン(H)、アスパラギン(N)およびグルタミン(Q)にトリチル;リシン(K)にtert−ブトキシ−カルボニル;アルギニン(R)に2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニル。Fmoc保護基を除去するためには、(8−42)残基アセンブリにDMF中の2%DBU(ジアザ−ビシクロ−ウンデセン)、(1−8)残基アセンブリにジメチルホルムアミド(DMF)中の20%ピペリジンを使用した。アミノ酸組立の終了後、DIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−dPEG酸,Quanta BioDesign)でNH2−末端をさらに誘導体化した。
【0103】
乾燥ペプチド樹脂を88%トリフルオロ酢酸(TFA)、5%フェノール、2%トリイソプロピルシランおよび5%水(Sole,N.A.and G.Barany,1992,(前出))によって室温で1.5時間処理した。樹脂を濾過し、溶液を低温メチル−t−ブチルエーテルに加えてペプチドを沈殿させた。遠心後、沈殿ペレットを新しい低温メチル−t−ブチルエーテルで洗浄して有機スカベンジャーを除去した。このプロセスを2回繰返した。最終ペレットを乾燥し、H2O、20%アセトニトリルに再懸濁させて凍結乾燥した。
【0104】
粗ペプチドPEG236−Aβ1−42
(DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA;配列:4)は、セミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用い、溶出剤として(A)水および(B)0.1%NH4OHを含むアセトニトリルを使用する逆相HPLCによって精製した。溶出剤Bの以下の勾配を使用した:10%−30%、20分超、流速80mL/分。溶出画分の分析用HPLCは、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いるBeckman HPLCで、溶出剤B(上記)の以下の勾配:15%−35% B(20分)−80%(3分)、流速1mL/分で行った。精製ペプチドのキャラクタリゼーションは、Micromass LCZ platformスペクトロメーターでHPLC−MSによって行った。理論的平均分子量(MW)は4731.0Daであり、測定MWは4731.0Daである。
【0105】
結果
本発明の1つの具体的用途は、アルツハイマー病(AD)の予防および治療処置のための改良ワクチンの開発である。最近の報告は、Elan/Wyeth AN−1792 Aβ 1−42ワクチン(図5B参照)の臨床試験の中断に導いた重篤な脳の炎症の問題が、T細胞応答に起因することを示唆している(Monsonego,A.and H.L.Weiner,2003,Science 302:834−8;Nicoll,J.A.ら,2003,Nat.Med.9:448−52)。この応答はAβペプチドのCOOH−末端を特異的に指向している(Monsogeo,A.ら,2003,J.Clin.Invest.112:415−22)。しかしながら注目すべきは、同じ臨床試験が、高いAβ抗体価をもつ患者でプラークの減少およびある程度限られた記憶の安定化を示したことであり、これはワクチンが治療効果を発揮していたことを表す。特に、Aβ1−42ワクチンを接種した患者の大半では病理形態のAβに特異的な抗体が誘発されたことが知見された(Hock,C.ら,2002,Nat.Med.8:1270−5)。これらの治療用抗体は、ペプチドのNH2−末端領域、すなわち残基4−10のエピトープを認識する(McLaurin,J.ら,2002,Nat.Med.8:1263−9;図5A参照)。これらの知見は疾病動物モデルでも確認された(Bard,F.ら,2003,Proc.Natl.Acad.Sci.U.S.A.100:2023−8)。そこでElan/Wyethらは、接合したN−末端(1−16)Aβペプチドに基づく第二世代のワクチンACC−001の臨床試験に進んでいる(図5B参照)。
【0106】
COOH−末端領域が欠失でなくSMWポリマー(例えばSMW PEG)によって遮蔽されている遊離形態またはコンジュゲート形態のAβペプチドの使用は、Elan/Wyethワクチンの有力な代替物である(図5C参照)。例えばSMW PEGはアミノ酸16−30に局在し上述の重篤な副作用の原因になると思われるAβのCOOH−末端T細胞エピトープの内部または極めて近傍で単一または多数コピーに付着できるが(Monsonego,A.ら,2003(前出))、NH2−末端領域のアミノ酸1−16またはもっと短い領域のアミノ酸4−10の免疫原性は元のまま維持される。双方の領域は有益なエピトープを内包すると考えられている(McLaurin,J.R.ら,2002(前出))。同じ分子が、SMW PEG付加Aβペプチドによる免疫後の従来のハイブリドーマテクノロジーによって、または、固定化SMW PEG付加Aβペプチドに関するファージディスプレイライブラリーをパニングすることによって受動免疫療法の抗体開発に使用できる。
【0107】
本発明の別の具体的用途は、Aβタンパク質の異なる凝集状態を識別する潜在能力を有している治療用抗体、例えば、Aβ誘発病理の原因と見做される小オリゴマーに特異的な抗体の開発である。(Cleary,J.P.ら,2005,Nat.Neurosci.8:79−84)。AβのNH2−末端領域は低分子量オリゴマー、高分子量オリゴマーおよびフィブリルへのAβの凝集には関与しないが、このNH2−末端は免疫優性であり、多様な形態に共通のエピトープに特異的なのでそれらの識別ができない抗体の優先的なin vivo形成を指令する。NH2−末端領域が欠失でなくSMWポリマー(例えばSMW PEG)で遮蔽されている遊離形態またはコンジュゲート形態のAβペプチドを使用することによって所望の特異性をもつ抗体を得ることができる。このペプチドは正常Aβの潜在的凝集能力は維持しているが、N−末端領域に対する免疫応答はPEG遮蔽によって抑制されている。
【実施例3】
【0108】
HIV−1の4E10/Trp−富化領域をターゲットする低分子量PEG付加ワクチン
中和抗体4E10(Salzwedel,K.ら,1999,J.Virol.73:2469−80)に結合するHIV−1 gp41内部のTrp−富化領域の構造は、脂質模擬環境でSchibli,DJ.らによって決定された(2001,Biochemistry 40:9570−8)。最近にはまた、4E10抗体に接触するアミノ酸残基が開示された(Cardoso,R.ら,2004(前出))。これらの接触残基は、構造内のペプチドの水に面する側を構成する(図6A参照)。図6Aで、4E10結合残基は下線部分で示し、672および680位のTrp残基を含むが、膜に面するTrp残基は二重下線部分で示し、666、670および678位に局在する。
【0109】
4E10エピトープの所望のα−ヘリカルコンホメーションは、4E10エピトープをGCN4ロイシンジッパーの表面にグラフトすることによって得ることができ(O’Shea,E.K.ら,1989,Science 245:646−8)、また、Sia,S.K.and P.S.Kim(2003,Proc.Natl.Acad.Sci.U.S.A.100:9756−61)に類似である。図6Aに示すTrp−富化領域の構造に基づいて、膜に面する残基のいくつかに突然変異を導入し(図6Bの“4E10コイル”の一部として下線を付けたアミノ酸)、GCN4ペプチド(“GCN4コイル”)に適合性のダイマー化表面をもつ4E10ペプチド(“4E10コイル”)を作製できる(図6B参照)。このコイルドコイルは、4E10領域を適正なヘリカル構造に拘束するであろう。GCN4/4E10コイルドコイルの安定性は、ジスルフィド結合の付加(Sia,S.K.and P.S.Kim,2003(前出))、および、GCN4と4E10との“a”および“d”位の残基をロイシンまたはイソロイシンに突然変異させることによってさらに強化できる(Lu,S.M.and R.S.Hodges,2002,J.Biol.Chem.277:23515−24)。該構造(すなわちGCN4コイル)のスカフォールド性部分に対する免疫応答を回避するために、GCN4コイルの表面に沿って適切なリシン残基(例えば、図7の二重下線を付けた残基)をPEG236で誘導体化できる(図7)。
【実施例4】
【0110】
OMPCに接合する酸性スカフォールドをもつPEG付加キメラN17ペプチド
Br−アセチル−GGGEP17GluN17(表5のペプチド1)の合成−
このペプチドは、Pioneerペプチドシンセサイザー(Applied Biosystems)で固相合成した。使用した樹脂はFmoc−Linker AM−Champion,1%架橋(Biosearch Technologies,Inc.)、修飾Rinkリンカーp−[(R,S)−α−[9H−フルオレン−9−イル−メトキシホルムアミド]−2,4−ジメトキシベンジル]−フェノキシ酢酸で誘導体化したPEG−PS基材樹脂であった(Rink,H.,1987,Tetrahedron Lett.28:3787−3789;Bernatowicz,M.S.ら,1989,Tetrahedron Lett.30:4645−4667)。すべてのアシル化反応は樹脂遊離アミノ基の4倍過剰量の活性化アミノ酸で90分間行った。アミノ酸は等モル量のHBTU(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)および2倍モル過剰量のDIEA(N,N−ジイソプロピルアミン)で活性化した。ペプチド配列:KIEEIEEKIEEIEKLLQLTVWGIKQLQARIL(配列:93;非−HIV残基はイタリック体)の最初の31残基部分については、標準Fmoc/t−Bu化学を使用した。側鎖保護基は以下を用いた:グルタミン酸(Glu)およびトレオニン(Thr)にtert−ブチル;システイン(Cys)およびグルタミン(Gln)にトリチル;リシン(Lys)およびトリプトファン(Trp)にtert−ブトキシ−カルボニル;アルギニン(Arg)に2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニル。最初の31残基の組立後、Dde−Lys(Fmoc)−OH,N−α−1−(4,4−ジメチル−2,6−ジオキソシクロヘキシ−1−イリデン)エチル−N−ε−Fmoc−L−リシンを32番目の残基としてアシル化した。ε−アミノ基のFmoc−保護基を20%ピペリジン含有ジメチルホルムアミド(DMF)溶液で処理することによって除去した。この条件下でDde保護基は完全に安定である。脱保護したリシン側鎖をさらにDIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−dPEG酸,Quanta BioDesign)でさらに誘導体化した。次にペプチド樹脂を1%ヒドラジン含有DMF溶液で処理してリシンのα−アミノ基からDde−基を除去した。ペプチド樹脂GGIEKKIEEIEEKIEEIEKLLQLTVWGIKQLQARIL(配列:94;非−HIV残基はイタリック体;PEG化リシン残基は下線部分)を得るために組立を継続した。次にペプチド樹脂を2つの部分に分割した。全体の2/3にあたる大きいほうの部分を使用して別のグリシン残基を組込むために組立を継続し、DMF中で10倍過剰量の無水ブロモ酢酸と反応させることによってさらにブロモアセチル化した。GGIEKKIEEIEEKIEEIEKLLQLTVWGIKQLQARIL(配列:94)−樹脂の残りの1/3を使用してAc−C(Acm)−TtdsCCEP17GluN17(下記参照)の合成を継続した。
【0111】
Br−アセチル−EP17GluN17の合成終了後、乾燥ペプチド−樹脂を88%トリフルオロ酢酸(TFA)、5%フェノール、2%トリイソプロピルシランおよび5%水(Sole,N.A.and G.Barany,1992(前出))によって室温で1.5時間処理した。樹脂を濾過し、溶液を低温メチル−t−ブチルエーテルに加えてペプチドを沈殿させた。遠心後、ペプチドペレットを新しい低温メチル−t−ブチルエーテルで洗浄して有機スカベンジャーを除去した。このプロセスを2回繰返した。最終ペレットを乾燥し、H2O、20%アセトニトリルに再懸濁させて凍結乾燥した。
【0112】
粗ペプチドBr−アセチル−GGGEP17GluN17(表5のペプチド1)の精製は、Toyopearl HW−50S樹脂を詰めた700×26mmカラム上で水中の60%のアセトニトリル、0.1%TFAを溶出剤として使用するゲル透過クロマトグラフィー(GPC)によって行った。典型的な処理では、900mgの粗ペプチドを25mLの溶出剤に溶解し、流速1mL/分でカラムに直接充填した。溶出画分の分析用HPLCは、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いたBeckman HPLCで溶出剤B(上記)の以下の勾配:45%−65% B(20分)−80%(3分)、流速1mL/分を使用した。純度に基づいて画分をプールし、セミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用いる逆相HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用してさらに精製した。溶出剤Bの以下の勾配を使用した:45%−60%、20分超、流速80mL/分。精製ペプチドのキャラクタリゼーションは、Micromass LCZ platformスペクトロメーターでエレクトロスプレー質量分析法によって行った。理論的平均分子量(MW)は4613.3Daであり、測定MWは4613.0Daである。
【0113】
Ac−C(Acm)−Ttds−CCEP17GluN17(表5のペプチド2)の合成−
上記アセンブリからGGIEKKIEEIEEKIEEIEKLLQLTVWGIKQLQARIL(配列:94−樹脂(上記同様に作製)の1/3を使用してAc−C(Acm)Ttds−CC−部分の合成を継続した。トリチル側鎖で保護された最初の(カルボキシ末端配列)の2つのシステインは上記のβ−アセチル−GGGEP17GluN17(表5のペプチド1)同様に標準Fmoc/tBu化学によって組立てた。Ttds(1−アミノ−4,7,10−トリオキサ−13−トリデカミンコハク酸(−NH−(CH2)3−O−(CH2)2−O−(CH2)2−O−(CH2)3−NH−CO−CH2)2−CO−)との反応は、等モル量のHBTUおよび2倍モル過剰量のDIEAで2時間活性化した4倍過剰量のTtdsとの反応によって行った。N−末端残基C(Acm)をFmoc−Cys(Acm)−OH(上記)と同じ条件下でアシル化し、次いでFmoc脱保護後にDMF中の10倍過剰量の無水酢酸と反応させることによってアセチル化した。先行実施例の記載と同様にして樹脂から分離後、粗ペプチドAc−C(Acm)−TtdsCCEP17GluN17(表5のペプチド2)を、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で、水中の60%アセトニトリル、0.1%TFA、流速1mL/分を使用するゲル透過クロマトグラフイー(GPC)によって精製した。GPCによって得られたプール画分(純度70%)をセミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用いた逆相HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して、>95%までさらに精製した。溶出剤Bの以下の勾配を使用した:45%−60%、20分超、流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、Jupiter C4カラム(150×4.6mm,5μm,5μm,300A,Phenomenex)、流速1mL/分を用いた分析用HPLCによって、溶出剤として(A)水中の0.1%TFA、および、(B)アセトニトリル中の0.1%TFAを使用して行った。溶出剤Bの以下の勾配を使用した:45−65%(20分超)−80%(3分超)。精製ペプチドのキャラクタリゼーションは、Micromass LCZ platformでエレクトロスプレー質量分析法によって行った。理論的平均分子量(MW)は5161.2Daであり、測定MWは5161.0Daである。
【0114】
Ac−C(Acm)−(チオEP17GluN17)3の合成−
チオエーテル結合の形成による共有結合的三量体化−
精製ペプチド前駆体(10.1mg)、Br−アセチル−GGGEP17GluN17(表5のペプチド1)を、6Mのグアニジウムクロリド、0.25MのTrisHCl,pH8.5、2mMのEDTAに5mg/mLの濃度で溶解した。この溶液に他のペプチド前駆体Ac−C(Acm)Ttds−CCEP17GluN17(表5のペプチド2)(5.3mg;1mg/mL)を添加した。2つのペプチド前駆体の実効モル比は2.2:1のBr−アセチル−GGGEP17GluN17:Ac−C(Acm)Ttds−CCEP17GluN17であった。三量体化反応のモニターは、Waters−Micromass LCZ Platformを使用するLC−MS分析によって、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)で溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して行った。溶出剤Bを以下の勾配で使用した:45%−65% B(20分)−80%(3分)、流速1mL/分。チオエーテル形成反応は円滑に進行した。2時間後、1つの生成物が形成され、その質量は、2つのチオエーテル結合を介して2つのBr−アセチル−GGGEP17GluN17ペプチド鎖に連結された1つのAc−C(Acm)Ttds−CCEP17GluN17ペプチド鎖に相当する。溶液に20μLのTFAを添加し、溶液を、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で水中の60%アセトニトリル、0.1%TFA、流量1mL/分の溶出剤を使用するゲル透過クロマトグラフィー(GPC)によって直接精製した。溶出画分の分析は、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)で溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用するHPLCによって行った。溶出剤Bを以下の勾配で使用した:45%−65% B(20分)−80%(3分)、流速1mL/分、LC−MS Waters−Micromass LCZ Platform、正モード(ES+)。予想MWは14225Daであり、測定MWは14226Daであった。
【0115】
Ac−C(Acm)(チオEP17GluN17)3のアセチル化システインの脱保護によるAc−C(チオEP17GluN17)3の形成−
精製したコイルドコイル前駆体ac−C(Acm)(チオEP17GluN17)3を20mg/mlの濃度でTFA、アニソール2%および50当量のAgOTf(トリフルオロメタンスルホン酸銀)に40℃で3時間溶解した。氷冷した無水エーテルを反応混合物に添加し、沈殿物を遠心によって単離した。沈殿物を氷冷した無水エーテルで2回洗浄した。ペプチドをH2O/アセトニトリル、0.1%TFAに再溶解し、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で溶出剤として水中の60%アセトニトリル、0.1%TFA、流速1mL/分を使用するゲル透過クロマトグラフィー(GPC)によって精製した。精製したペプチドのキャラクタリゼーションは、Micromass LCZ platformでエレクトロスプレー質量分析法によって行った。理論的平均MWは14157Daであり、測定MWは14157Daであった。
【0116】
Br−アセチル−GGGEPN17(表5のペプチド3)の合成−
このペプチドは、Pioneerペプチドシンセサイザー(Applied Biosystems)で固相合成した。使用した樹脂はFmoc−Linker AM−Champion,1%架橋(Biosearch Technologies,Inc.)、修飾Rinkリンカーp−[(R,S)−α−[9H−フルオレン−9−イル−メトキシホルムアミド]−2,4−ジメトキシベンジル]−フェノキシ酢酸で誘導体化したPEG−PS基材樹脂であった(Rink,H.,1987,Tetrahedron Lett.28:3787−3789;Bernatowicz,M.S.ら,1989,Tetrahedron Lett.30:4645−4667)。すべてのアシル化反応は樹脂遊離アミノ基の4倍過剰量の活性化アミノ酸で90分間行った。アミノ酸は等モル量のHBTU(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)および2倍モル過剰量のDIEA(N,N−ジイソプロピルアミン)で活性化した。ペプチド配列:KIEEIEKKIEEIEKLLQLTYWGIKQLQARIL(配列:95;非−HIV残基はイタリック体)の最初の31残基部分については、標準Fmoc/t−Bu化学を使用した。側鎖保護基は以下を用いた:グルタミン酸(Glu)およびトレオニン(Thr)にtert−ブチル;システイン(Cys)およびグルタミン(Gln)にトリチル;リシン(Lys)およびトリプトファン(Trp)にtert−ブトキシ−カルボニル;アルギニン(Arg)に2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニル。最初の31残基の組立後、Dde−Lys(Fmoc)−OH,N−α−1−(4,4−ジメチル−2,6−ジオキソシクロヘキシ−1−イリデン)エチル−N−ε−Fmoc−L−リシンを32番目の残基としてアシル化した。ε−アミノ基のFmoc−保護基を20%ピペリジン含有ジメチルホルムアミド(DMF)溶液で処理することによって除去した。この条件下でDde保護基は完全に安定である。脱保護したリシン側鎖をさらにDIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−dPEG酸,Quanta BioDesign)でさらに誘導体化した。次にペプチド樹脂を1%ヒドラジン含有DMF溶液で処理してリシンのα−アミノ基からDde−基を除去した。組立を継続してスカフォールドの他の6つの残基、非HIVドメインすなわち配列KIEEIE(配列:96)をペプチド樹脂に組込んだ。次いで、Dde−Lys(Fmoc)−OH,N−α−1−(4,4−ジメチル−2,6−ジオキソシクロヘキサ−1−イリデン)エチル−N−ε−Fmoc−L−リシンをアシル化した。ε−アミノ基のFmoc−保護基はジメチルホルムアミド(DMF)中の20%ピペリジン溶液によって選択的に除去した。この条件下でDde−保護基は完全に安定である。脱保護したリシン側鎖を、DIPC(ジイソプロピルカルボジイミド)およびHOBt(ヒドロキシベンゾトリアゾール)と3時間反応させたMW236Daのポリエチレングリコール酸誘導体(m−d PEG酸, Quanta BioDesign)によってさらに誘導体化した。ペプチド樹脂GGIEKKIEEIEKKIEEIEKKIEEIEKLLQLTVWGIKQLQARIL(配列:97;非−HIV残基はイタリック体;PEG化リシン残基は下線部分)を得るために組立を継続した。次にペプチド樹脂を2つの部分に分割した。全体の2/3にあたる大きいほうの部分を使用して別のグリシン残基を組込むために組立を継続し、DMF中で10倍過剰量の無水ブロモ酢酸と反応させることによってさらにブロモアセチル化した。ペプチド−樹脂の残りの1/3を使用してAc−C(Acm)−TtdsCCEPN17(下記参照)の合成を継続した。
【0117】
Br−アセチル−EPN17の合成終了後、乾燥ペプチド−樹脂を88%トリフルオロ酢酸(TFA)、5%フェノール、2%トリイソプロピルシランおよび5%水(Sole,N.A.and G.Barany,1992(前出))によって室温で1.5時間処理した。樹脂を濾過し、溶液を低温メチル−t−ブチルエーテルに加えてペプチドを沈殿させた。遠心後、ペプチドペレットを新しい低温メチル−t−ブチルエーテルで洗浄して有機スカベンジャーを除去した。このプロセスを2回繰返した。最終ペレットを乾燥し、H2O、20%アセトニトリルに再懸濁させて凍結乾燥した。
【0118】
粗ペプチドBr−アセチル−GGGEPN17(表5のペプチド3)の精製は、Toyopearl HW−50S樹脂を詰めた700×26mmカラム上で水中の60%のアセトニトリル、0.1%TFAを溶出剤として使用するゲル透過クロマトグラフィー(GPC)によって行った。典型的な処理では、900mgの粗ペプチドを25mLの溶出剤に溶解し、流速1mL/分でカラムに直接充填した。溶出画分の分析用HPLCは、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)を用いたBeckman HPLCで溶出剤B(上記)の以下の勾配:45%−65% B(20分)−80%(3分)、流速1mL/分を使用した。純度に基づいて画分をプールし、セミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用いる逆相HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用してさらに精製した。溶出剤Bの以下の勾配を使用した:45%−60%、20分超、流速80mL/分。精製ペプチドのキャラクタリゼーションは、Micromass LCZ platformスペクトロメーターでエレクトロスプレー質量分析法によって行った。理論的平均分子量(MW)は5699Daであり、測定MWは5700Daである。
【0119】
Ac−C(Acm)−Ttds−CCEPN17(表5のペプチド4)の合成−
上記アセンブリからGGIEKKIEEIEKKIEEIEKKIEEIEKLLQLTVWGIKQLQARIL(配列:97)−樹脂(詳細は上記参照)の1/3を使用してAc−C(Acm)Ttds−CC−部分の合成を継続した。トリチル側鎖で保護された最初の(カルボキシ末端配列)の2つのシステインは標準Fmoc/tBu化学によって組立てた。Ttds(1−アミノ−4,7,10−トリオキサ−13−トリデカミンコハク酸(−NH−(CH2)3−O−(CH2)2−O−(CH2)2−O−(CH2)3−NH−CO−CH2)2−CO−)との反応は、等モル量のHBTUおよび2倍モル過剰量のDIEAで2時間活性化した4倍過剰量のTtdsとの反応によって行った。N−末端残基C(Acm)をFmoc−Cys(Acm)−OHと同じ条件下でアシル化し、次いでFmoc脱保護後にDMF中の10倍過剰量の無水酢酸との反応によってアセチル化した。
【0120】
先行実施例の記載と同様にして樹脂から分離後、粗ペプチド、アセチル−C(Acm)−TtdsCCEPN17を、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で、溶出剤として水中の60%アセトニトリル、0.1%TFA、流速1mL/分を使用するゲル透過クロマトグラフイー(GPC)によって精製した。GPCによって得られたプール画分(純度70%)をセミ・プレパラティブWaters RCM Delta−PakTM C4カートリッジ(40×200mm,15μm)を用いた逆相HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して、>95%純度までさらに精製した。溶出剤Bの以下の勾配を使用した:45%−60%、20分超、流速80mL/分。粗ペプチド、溶出画分および精製プールの分析は、Jupiter C4カラム(150×4.6mm,5μm,300A,Phenomenex)、流速1mL/分を用いた分析用HPLCによって、溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して行った。溶出剤Bの以下の勾配を使用した:45−65%(20分超)−80%(3分超)。精製ペプチドのキャラクタリゼーションは、Micromass LCZ platformでエレクトロスプレー質量分析法によって行った。理論的平均分子量(MW)は6248Daであり、測定MWは6247Daであった。
【0121】
アセチル−C(Acm)(チオEPN17)3の合成−
チオエーテル結合の形成による共有結合的三量体化−
精製ペプチド前駆体(12mg)、Br−アセチル−GGGEPN17(表5のペプチド3)を、6Mのグアニジウムクロリド、0.25MのTrisHCl,pH8.5、2mMのEDTAに5mg/mLの濃度で溶解した。この溶液に他のペプチド前駆体アセチル−C(Acm)Ttds−CCEPN17(表5のペプチド4)(6mg;1mg/mL)を添加した。2つのペプチド前駆体の実効モル比は2.2:1のBr−アセチル−GGGEP1N17:Ac−C(Acm)Ttds−CCEPN17であった。三量体化反応のモニターは、Waters−Micromass LCZ Platformを使用するLC−MS分析によって、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)で溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用して行った。溶出剤Bを以下の勾配で使用した:35%−70% B(20分)−80%(3分)、流速1mL/分。チオエーテル形成反応は円滑に進行した。2時間後、1つの生成物が形成され、その質量は、2つのチオエーテル結合を介して2つのBr−アセチル−GGGEP1N17ペプチド鎖に連結された1つのAc−C(Acm)Ttds−CCEPN17ペプチド鎖に相当する。溶液に20μLのTFAを添加し、溶液を、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で水中の60%アセトニトリル、0.1%TFA、流量1mL/分の溶出剤を使用するゲル透過クロマトグラフィー(GPC)によって直接精製した。溶出画分の分析は、Phenomenex,Jupiter C4カラム(150×4.6mm,5μm)で溶出剤として(A)水中の0.1%TFAおよび(B)アセトニトリル中の0.1%TFAを使用するHPLCによって行った。溶出剤Bを以下の勾配で使用した:45%−65% B(20分)−80%(3分)、流速1mL/分、LC−MS Waters−Micromass LCZ Platform、正モード(ES+)。予想MWは17489Daであり、測定MWは17489Daであった。
【0122】
Ac−C(Acm)(チオEPN17)3のアセチル化システインの脱保護によるAc−C(チオEPN17)3の形成−
精製したコイルドコイル前駆体ac−C(Acm)(チオEPN17)3を20mg/mlの濃度でTFA、アニソール2%および50当量のAgOTf(トリフルオロメタンスルホン酸銀)に40℃で3時間溶解した。氷冷した無水エーテルを反応混合物に添加し、沈殿物を遠心によって単離した。沈殿物を氷冷した無水エーテルで2回洗浄した。ペプチドをH2O/アセトニトリル、0.1%TFAに再溶解し、TSK−gel Toyopearl HW−50S樹脂(700×26mmカラム)で溶出剤として水中の60%アセトニトリル、0.1%TFA、流速1mL/分を使用するゲル透過クロマトグラフィー(GPC)によって精製した。精製したペプチドのキャラクタリゼーションは、Micromass LCZ platformでエレクトロスプレー質量分析法によって行った。理論的平均MWは17415Daであり、測定MWは17416Daであった。
【0123】
ac−C(チオEP17GluN17)3およびac−C(チオEPN17)3とOMPCの接合−
グラム陰性細菌からOMPCを精製する様々な方法が計画されている(Fraschら,1997,J Exp.Med.140:87;Fraschら,1978,J Exp.Med.147:629;U.S.Pat.番号4,707,543;Heltingら,1981.Acta Path.Microbiol.Scand.Sect.C.89:69;およびU.S.Pat.番号4,271,147)。N.meningitidis Bの改良外膜タンパク質複合体(improved Outer Membrane Protein Complex(iOMPC))は、Fuが米国特許番号5,494,808に記載したような当業界で公知の技術を使用して得られる。
【0124】
1.36mLのNeisseria meningitidisの改良外膜タンパク質複合体(iOMPC)溶液(7.13mg/ml)に、0.5MのNaHCO3(0.151mL)を最終濃度50mM,pH8.5まで添加した。この溶液に、0.407mLのヘテロ二官能性架橋剤スルホスクシニミジル4−(N−マレイミドメチル)シクロヘキサン−1−カルボキシレート(sSMCC,Pierce Chemical Co.)の20μM溶液を2倍過剰量で滴下した(OMPCのリシン残基に対して、0.42μmolのリシン/mgのOMPCタンパク質)。0.045mLの0.5MのNaHCO3をさらに添加することによってpHを再調整した。溶液を暗中、4℃で1時間熟成後、1MのNaH2PO4溶液(16μl)を添加してpHを中性まで低下させた。溶液を4℃で300K MWCO DispoDialyser(Spectrum Laboratories Inc.,CA)を使用し、バッファを6回交換し(2時間毎)、2Lの20mMのHEPES,pH7.3、(4−(2−ヒドロキシエチル)ピペラジン−1−エタンスルホン酸)、2mMのEDTA(エチレンジアミン四酢酸)に透析して余剰の試薬を除去した。透析後に合計1.9mLの活性化OMPC(aOMPC)を回収した。
【0125】
2つのCys−含有ペプチド、ac−C(チオEP17GluN17)3およびac−C(チオEPN17)3の0.5mg/mlの予製液を0.1MのHEPES、2mMのEDTA,pH7.3の脱ガス溶液中で調製した。沈殿を生じることなくaOMPCに安全に組込めるペプチドリガンドの最大量を確定するために、先ずaOMPCを漸増量のペプチドリガンドとインキュベートする小規模試験で接合反応を追跡した。OMPCに組込めるマレイミド基の最大数は1mgのOMPCあたりのリシン残基総数によって限定され、0.42μMのリシン/mgのOMPCである。OMPCの平均MWが40×106であると考えると、これは16,000モルのリシン/1モルのOMPCに相当する。これらのうちの一部分だけがsSMCCによって活性化され(35%以下)、これは達成可能な最大ペプチド負荷約5000モルに対応する。この場合にはaOMPCをOMPC1モルあたり1000、2000および3000モル過剰量のペプチドリガンドとインキュベートした。1時間後、サンプルをaOMPCサンプルに比較して沈殿の存在または濁りの増加を点検した。ac−C(チオEP17GluN17)3の場合には、3000モルのリガンド/1モルのOMPCという最大モル過剰量の使用まで沈殿または濁りの増加は全く観察されなかったが、ac−C(チオEPN17)3の場合には、3000モル/OMPCモルで多少の沈殿が観察され、2500モル/OMPCモルでは完全溶解であった。
【0126】
これらの観察に基づいて大規模反応を行った。20mMのHEPES、2mMのEDTA,pH7.3中の0.75mL(3.8mg)のaOMPCに7mLのac−C(チオEP17GluN17)3ペプチド予製液を、緩やかに渦流させながら滴下した。これは3000モル過剰量のペプチドモル数/OMPCモルに対応する。ac−C(チオEPN17)3の場合は、20mMのHEPES、2mMのEDTA,pH7.3中の0.45mL(2.3mg)のaOMPCに9mLのac−C(チオEPN17)3ペプチド予製液を緩やかに渦流させながら滴下した。これは2500モル過剰量のペプチドモル数/OMPCモルに対応する。最終コンジュゲートのペプチド負荷を定量するためにマレイミド活性化OMPCのサンプルをブランクとして維持した。接合反応混合物を暗中、4℃で17時間熟成した。次いで、最終濃度15mMまでのμ−メルカプトエタノール(総添加量10−12μL)を、暗中、4℃で1時間使用してOMPCの残留マレイミド基を反応停止させた。溶液を300K MWCO DispoDialyserで1Lの20mMのHEPES,pH7.3を4℃で4時間毎に4回交換して透析し、接合しなかったペプチドおよびβ−メルカプトエタノールを除去した。Lowryアッセイ(Lowryら,1951,J.Biol.Chem.193:265)によって濃度を測定すると、0.41mg/mLのOMPC−(チオEP17GluN17)3および0.32mg/mLのOMPC−(チオEPN17)3が存在した。コンジュゲートおよびaOMPCサンプルを、排気した密封ガラス管に入れ共沸HClと共に110℃で70時間加水分解した。アミノ酸解析によってアミノ酸組成を決定した。OMPCタンパク質へのペプチド接合負荷は、コンジュゲートアミノ酸組成をOMPC担体およびペプチドリガンドの双方の組成に比較し、データの重回帰最小二乗分析によって決定した(Shulerら,1992,J.Immunol.Methods 156:137−149)。コンジュゲートOMPC−(チオEP17GluN17)3では得られたペプチド対OMPCモル比は541であり、OMPC−(チオEPN17)3では得られたペプチド対OMPCモル比は443であった。
【0127】
結果−
この実施例は、CCEZN17配列(配列:44)に基づく2つのチオエーテル結合安定化キメラペプチドの設計を記載している。第一の三量体コイルドコイルAc−C(Acm)−(チオEP17GluN17)3は17アミノ酸のEZ−様スカフォールドに基づき、第二のコイルドコイルAc−C(Acm)−(チオEPN17)3は24アミノ酸のEZスカフォールドに基づく。2つの構築物は詳細に前述したように個々のキメラペプチド間のチオエーテル結合の形成によって共有結合的に安定化されている。設計したコイルドコイルのスカフォールド配列内部の様々な場所に、変化すなわち突然変異または側鎖の誘導体化を導入し、表5に“下線を付けて”示す。
【0128】
【表5】
【0129】
C(Acm)−(チオEP17GluN17)3は1つのCC−キメラN−ペプチドすなわちCCCE17N17(配列:88;表5のペプチド2)および2つの誘導体化キメラN−ペプチドすなわちブロモアセチル−GGGE17GluN17(配列:87;表5のペプチド1)から成る。CCCE17GluN17のNH2−末端システイン残基は、Acm(アセタミドメチル)基で保護され、残りの非保護システイン残基のおのおのは各誘導体化キメラN−ペプチドのブロモ−アセチル部分と共に化学選択的チオエーテル結合を形成する。C(Acm)−(チオEP17GluN17)3に内包された各キメラペプチドのスカフォールドドメインは、EZスカフォールド(配列:15)の24アミノ酸の代わりに17アミノ酸およびリシンからグルタミン酸への突然変異を含む短縮EZコイルドコイル(“E17Glu”スカフォールド;配列:84)から成る。C(Acm)−(チオEP17GluN17)3のスカフォールドドメインは、三量体コイルドコイルに内包された個別ペプチドのおのおののスカフォールド領域内部の1つのリシン残基へのPEG236部分の付着によって選択的に遮蔽されている(表5のペプチド1および2の下線部分)。各ペプチドのPEG付加リシン残基は、スカフォールドヘリックスのヘプタッドリピートの“f”位置に局在し、該残基のε−アミノ基を介してPEG部分が付着している。24アミノ酸から17アミノ酸へのスカフォールドの短縮は、得られるコイルドコイルC(Acm)−(チオEP17GluN17)3とOMPC担体との接合を促進し、また、接合に使用した水性バッファ中のペプチドの可溶化を、コイルドコイルの等電点(“pI”)を下げることによって促進する。PEG236によるリシン残基のεアミノ基の誘導体化に伴う遊離アミノ基の除去、および、LysからGluへの突然変異はいずれも、得られるコイルドコイルのpIをいっそう低下させる。この実施例に記載したHIV gp41融合中間体の種々のミメティクスの等電点の比較については表6を参照するとよい。マレイミド活性化OMPC担体へのC(Acm)−(チオEP17GluN17)3の接合は、C−(チオEP17GluN17)3を形成するチオール化ペプチドを得るために脱保護されたNH2−末端システイン残基を介して惹起される。
【0130】
C(Acm)−(チオEPN17)3は1つのCC−キメラN−ペプチドすなわちCCCEZN17(配列:90;表5のペプチド4)と2つの誘導体化キメラN−ペプチドすなわちブロモアセチル−GGGEZN17(配列:89;表5のペプチド3)とから成る。CCCEZN17のNH2−末端システイン残基はAcm(アセタミドメチル)基によって保護され、残りの非保護システイン残基のおのおのは各誘導体化キメラN−ペプチドのブロモ−アセチル部分と共に化学選択的チオエーテル結合を形成する。C(Acm)−(チオEPN17)3に内包された各キメラペプチドのスカフォールドドメインは、EZスカフォールド(配列:15)の24アミノ酸から成る。C(Acm)−(チオEPN17)3のスカフォールドドメインは、三量体コイルドコイルに内包された個別ペプチドのおのおののスカフォールド領域内部の2つのリシン残基へのPEG236部分の付着によって選択的に遮蔽されている(表5のペプチド3および4の下線部分)。各ペプチドのPEG付加リシン残基は、スカフォールドヘリックスのヘプタッドリピートの“f”位置に局在し、該残基のε−アミノ基を介してPEG部分が付着している。PEG236によるリシン残基のεアミノ基の誘導体化は遊離アミノ基を除去し、得られるコイルドコイルのpI低下を助ける(表6)。マレイミド活性化OMPC担体へのC(Acm)−(チオEPN17)3の接合は、C−(チオEPN17)3を形成するチオール化ペプチドを得るために脱保護されたNH2−末端システイン残基を介して惹起される。
【0131】
【表6】
【図面の簡単な説明】
【0132】
【図1】図1Aは、共有結合安定化ホモ三量体コイルドコイル(CCIZN17)3を生じる酸化反応の概略図である。三量体コイルドコイルを安定化する3つのジスルフィド結合が各ペプチド鎖間に1つずつ存在する。図1Bは、(CCIZN17)3([配列2]3)を安定化するジスルフィド結合の化学構造図である。3つのジスルフィド結合がNH2−末端システインアミノ酸残基のチオール(−SH)基の間に局在しており、その化学構造が図示されている。2つのシステイン残基の下流に局在するアミノ酸は大文字の一文字記号で示されている。
【図2】図2Aは三量体“IZ”スカフォールドのヘリカルホイール表示である。ヘプタッドリピートの“f”位に局在する3つのリシンアミノ酸(“K”)がコイルドコイルの外側に近い位置に存在する状態で示されている。図2Bの(CCIPN17)3の配列表示からわかるように、これらのリシン残基の2つが、側鎖のε−アミノ基をm−dPEGTM酸(MW=236)で誘導体化して選択的に遮蔽された三量体コイルドコイル(CCIPN17)3を生成するために選択された。IZスカフォールドドメイン(イタリック体)のPEG付加リシン残基は下線を付けて示す。HIV N17ドメインは大文字で示す。
【図3】(CCIPN17)3ペプチドワクチンの概略モデルである。個々のキメラペプチドCCIPN17は長さ約62オングストロームの円筒として示されている。暗い灰色部分は長さ約36オングストロームのIZスカフォールドドメインを表し、明るい灰色部分は長さ約25オングストロームのHIV gp41 N−ペプチドのN17領域を表す。m−dPEGTM酸(MW=236)部分は長さ約15オングストロームの長方形として示されている。
【図4】(CCIPN17)3によるホモロガスおよびヘテロロガスプライム/ブースト免疫方式で行ったモルモット試験のELISA結果を示す。プライム/ブーストの組合せは各棒グラフ群の下部に示す。各抗原に対するELISA幾何平均力価(GMT)は血清中に存在する対応特異性所有抗体の相対量を正確に反映する。
【図5】図5Aは、アミロイドβ−ペプチド(Aβ)1−42ペプチドのアミノ酸配列を示す。免疫優性T細胞エピトープ(Monsogeo,A.ら,2003,J.Clin.Invest.112:415−22)は一本の下線で示し、防御B細胞エピトープ(McLaurin,J.ら,2002,Nat.Med.8:1263−9)は二重下線で示す。図5Bは、Elan/Wyeth,AN−1792(Aβ1−42)およびACC−001((Aβ1−16)の2つのAβペプチドワクチンのアミノ酸配列を示す。図5Cは、本発明で提案したAβ1−42ペプチドワクチンの概略図であり、2つのリシンアミノ酸残基(下線部分)をm−dPEGTM酸(MW=236)部分で誘導体化して免疫優性T細胞エピトープを選択的に遮蔽し、ペプチドのNH2−末端部分の防御B細胞エピトープを認識し易くしている。
【図6】図6Aは、HIV−1 gp41エクトドメイン(配列6)中のトリプトファン(Trp)−富化モチーフのアミノ酸配列、および、Schibli,DJ.ら(2001,Biochemistry 40:9570−8)によって同定されたその二次構造の概略図を示す。一本下線で示したアミノ酸残基(W672、F673、1675、T676およびW680)がHIV−中和抗体4E10と相互作用することは最近になって確認された(Cardoso,R.M.F.ら,2005,Immunity vol.22:163−173)。二重下線で示したアミノ酸残基(W666、W370およびW678)は膜に面したTrp残基である。図6Bは、ロイシンジッパーGCN4に基づく提案4E10ターゲット化ワクチンの概略図を示す。下線を付けたアミノ酸残基は、“GCN4コイル”ペプチド(配列8)に適合性のダイマー化表面を有しているペプチド“4E10コイル”(配列7)を作製するために、4E10結合性アミノ酸を含有するTrp富化領域のいくつかの膜対面残基に導入した突然変異を表す。ジスルフィド結合がGCN4/4E10コイルドコイルをさらに強化する。
【図7】リシン残基(二重下線)にm−dPEGTM酸(MW=236)部分を付着させることによってPEG修飾した図6Bの提案4E10ターゲット化ワクチンの概略図である。
【図8】チオエーテル化学選択的結合によって共有結合的に安定化され本発明によってポリマー修飾できる三量体コイルドコイル構造NH2−C(Fm)(チオIZN17)3の化学構造図を表す。三量体コイルドコイルは、1つのCCキメラN−ペプチドすなわちNH2−C(Fm)Ttds−CCIZN17(配列76)の2つのシステインアミノ酸残基のおのおののチオール(−SH)基間に局在する2つのチオエーテル結合と、2つの誘導体化キメラペプチドすなわちBr−アセチル−GGGIZN17(配列77)のおのおのの求電子性ブロモ部分とによって形成される。結合システイン残基のCOOH−末端に局在するアミノ酸残基は一文字記号で示している。このコイルドコイル構造は、CC−キメラN−ペプチドのNH2−末端システイン残基のチオールを遮蔽するフルオレニルメトキシ(“Fm”)保護基およびNH2−末端の遊離アミノ基を有している。
【特許請求の範囲】
【請求項1】
ポリペプチド内部の結合部位の遮蔽方法であり、少なくとも1つの低分子量水溶性ポリマーを結合部位が前記ポリマーによって選択的に隠蔽されるように部位特異的に前記ポリペプチドに付着させる、遮蔽方法。
【請求項2】
前記結合部位がポリペプチド抗原のエピトープである請求項1の方法。
【請求項3】
前記低分子量水溶性ポリマーが前記ポリペプチドの1つのアミノ酸残基に部位特異的に付着する請求項2の方法。
【請求項4】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項3の方法。
【請求項5】
前記低分子量水溶性ポリマーがポリエチレングリコール(PEG)である請求項4の方法。
【請求項6】
前記PEGポリマーが約1000Da未満の分子量を有している請求項5の方法。
【請求項7】
前記エピトープが、前記ペプチド間の1つ以上の共有結合によって共有結合的に安定化された3つのキメラペプチドを含むエンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造のスカフォールドドメイン内部に局在しており、各キメラペプチドが前記エンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでいる請求項6の方法。
【請求項8】
前記エンベロープウイルス膜融合タンパク質がHIV gp41である請求項7の方法。
【請求項9】
ポリペプチド抗原のエピトープの遮蔽方法であり、少なくとも1つの低分子量水溶性ポリマーをエピトープが前記ポリマーによって選択的に隠蔽されるように前記ポリペプチド内部の少なくとも1つのアミノ酸残基に部位特異的に付着させる、遮蔽方法。
【請求項10】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項9の方法。
【請求項11】
前記低分子量水溶性ポリマーが約1000Da未満の分子量をもつポリエチレングリコール(PEG)である請求項10の方法。
【請求項12】
HIV gp41の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造のエピトープを遮蔽する方法であって、少なくとも1つの低分子量水溶性ポリマーを前記エピトープが前記ポリマーによって選択的に隠蔽されるようにコイルドコイル構造の少なくとも1つのアミノ酸残基に部位特異的に付着させることを含み、前記コイルドコイル構造が3つのキメラペプチド(前記ペプチド間の1つ以上の共有結合によって共有結合的に安定化される)を含み、各キメラペプチドがHIV gp41のN−ペプチドの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでおり、ならびに前記選択的に隠蔽されたエピトープが前記キメラペプチドのスカフォールドドメインに局在している方法。
【請求項13】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項12の方法。
【請求項14】
前記低分子量水溶性ポリマーが約1000Da未満の分子量を有しているポリエチレングリコール(PEG)である請求項13の方法。
【請求項15】
3つのキメラペプチドを含むHIV gp41 N−ペプチドのコイルドコイルの全部または一部分を模倣するポリマー修飾された共有結合安定化三量体コイルドコイルであって、各キメラペプチドが、HIV gp41 N−ペプチドの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでおり、スカフォールドドメイン内部に局在するエピトープが前記スカフォールドドメイン内部の少なくとも1つのアミノ酸残基に部位特異的に付着した少なくとも1つの低分子量水溶性ポリマーによって隠蔽されている前記三量体コイルドコイル。
【請求項16】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項15に記載のコイルドコイル。
【請求項17】
前記低分子量水溶性ポリマーが約1000Da未満の分子量を有しているポリエチレングリコール(PEG)である請求項16に記載のコイルドコイル。
【請求項18】
前記コイルドコイル内部の3つのキメラペプチドのおのおのがさらに、前記ペプチドのヘリカルドメインの外部に局在する少なくとも2つのシステイン残基を含み、および並列したペプチド上の前記システイン残基間のジスルフィド結合によってコイルドコイルが共有結合的に安定化されている請求項17に記載のコイルドコイル。
【請求項19】
前記コイルドコイル内部の3つのキメラペプチドのうちの第一キメラペプチドがさらに、前記ペプチドのヘリカルドメインの外部に局在する少なくとも2つのシステイン残基を含み、ならびに前記コイルドコイル内部の3つのキメラペプチドのうちの第二および第三のキメラペプチドがさらに、前記ペプチドのヘリカルドメインの外部に局在しチオエーテル結合を形成し得る求電子部分を含み、ならびにコイルドコイルが第一キメラペプチドの各システイン残基の求核性スルフヒドリルと各第二および第三キメラペプチドの求電子部分との間のチオエーテル結合を介して共有結合的に安定化されている請求項17に記載のコイルドコイル。
【請求項20】
(CCIZN17)3、C(チオEZN17)3およびC(チオE17GluN17)3から成るグループから選択されたHIV gp41 N−ペプチドコイルドコイルの全部または一部分を模倣するコイルドコイルであって、前記コイルドコイルのスカフォールドドメイン内部に局在するエピトープが前記スカフォールドドメイン内部の少なくとも1つのアミノ酸残基に部位特異的に付着した少なくとも1つの低分子量水溶性ポリマーによって選択的に隠蔽されているポリマー修飾された共有結合安定化三量体コイルドコイル。
【請求項21】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項20に記載のコイルドコイル。
【請求項22】
前記低分子量水溶性ポリマーが約1000Da未満の分子量を有しているポリエチレングリコール(PEG)である請求項21に記載のコイルドコイル。
【請求項23】
(a)コイルドコイルの可溶性三量体形態を含むスカフォールド部分と、
(b)HIV gp41 N−ペプチドの全部または一部分を含むN−ペプチド部分またはその修飾形態と、
(c)少なくとも2つのシステイン残基を含むシステイン部分と、
を含み、(a)の前記スカフォールド部分が(b)の前記N−ペプチド部分にらせん相で融合してα−ヘリカルドメインを形成し、(c)の前記システイン部分が前記α−ヘリカルドメインの外部に局在し、および前記スカフォールド部分の内部に局在するエピトープが前記スカフォールド部分内部の少なくとも1つのアミノ酸残基に部位特異的に付着した少なくとも1つの低分子量水溶性ポリマーによって選択的に隠蔽されているポリマー修飾された可溶性キメラペプチド。
【請求項24】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項23に記載のキメラペプチド。
【請求項25】
前記低分子量水溶性ポリマーが約1000Da未満の分子量を有しているポリエチレングリコール(PEG)である請求項24に記載のキメラペプチド。
【請求項26】
前記N−ペプチド部分がN−ヘリックス疎水性ポケットを形成するアミノ酸残基を内包するのに十分なHIV gp41 N−ペプチドの部分を含む請求項25に記載のキメラペプチド。
【請求項27】
CCIPN17であるポリマー修飾キメラペプチド。
【請求項28】
免疫原性担体分子に共有結合的に連結されている請求項23に記載の可溶性キメラペプチドの抗原性接合体。
【請求項29】
前記担体分子がOMPCである請求項28に記載の抗原性接合体。
【請求項30】
免疫原性担体分子に共有結合的に連結されている請求項15に記載の共有結合安定化三量体コイルドコイルの抗原性接合体。
【請求項31】
前記担体分子がOMPCである請求項30に記載の抗原性接合体。
【請求項32】
生物学的に有効なアジュバント、タンパク質、または、免疫応答を増進し得る他の成分と混合した請求項30に記載の抗原性接合体を有効量で含み、哺乳動物体内でHIV特異的中和抗体を誘発し得る免疫原として有用な医薬組成物。
【請求項1】
ポリペプチド内部の結合部位の遮蔽方法であり、少なくとも1つの低分子量水溶性ポリマーを結合部位が前記ポリマーによって選択的に隠蔽されるように部位特異的に前記ポリペプチドに付着させる、遮蔽方法。
【請求項2】
前記結合部位がポリペプチド抗原のエピトープである請求項1の方法。
【請求項3】
前記低分子量水溶性ポリマーが前記ポリペプチドの1つのアミノ酸残基に部位特異的に付着する請求項2の方法。
【請求項4】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項3の方法。
【請求項5】
前記低分子量水溶性ポリマーがポリエチレングリコール(PEG)である請求項4の方法。
【請求項6】
前記PEGポリマーが約1000Da未満の分子量を有している請求項5の方法。
【請求項7】
前記エピトープが、前記ペプチド間の1つ以上の共有結合によって共有結合的に安定化された3つのキメラペプチドを含むエンベロープウイルス膜融合タンパク質の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造のスカフォールドドメイン内部に局在しており、各キメラペプチドが前記エンベロープウイルス膜融合タンパク質のNH2−末端ヘプタッドリピートドメインの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでいる請求項6の方法。
【請求項8】
前記エンベロープウイルス膜融合タンパク質がHIV gp41である請求項7の方法。
【請求項9】
ポリペプチド抗原のエピトープの遮蔽方法であり、少なくとも1つの低分子量水溶性ポリマーをエピトープが前記ポリマーによって選択的に隠蔽されるように前記ポリペプチド内部の少なくとも1つのアミノ酸残基に部位特異的に付着させる、遮蔽方法。
【請求項10】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項9の方法。
【請求項11】
前記低分子量水溶性ポリマーが約1000Da未満の分子量をもつポリエチレングリコール(PEG)である請求項10の方法。
【請求項12】
HIV gp41の内部三量体コイルドコイルモチーフの全部または一部分を模倣する共有結合安定化三量体コイルドコイル構造のエピトープを遮蔽する方法であって、少なくとも1つの低分子量水溶性ポリマーを前記エピトープが前記ポリマーによって選択的に隠蔽されるようにコイルドコイル構造の少なくとも1つのアミノ酸残基に部位特異的に付着させることを含み、前記コイルドコイル構造が3つのキメラペプチド(前記ペプチド間の1つ以上の共有結合によって共有結合的に安定化される)を含み、各キメラペプチドがHIV gp41のN−ペプチドの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでおり、ならびに前記選択的に隠蔽されたエピトープが前記キメラペプチドのスカフォールドドメインに局在している方法。
【請求項13】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項12の方法。
【請求項14】
前記低分子量水溶性ポリマーが約1000Da未満の分子量を有しているポリエチレングリコール(PEG)である請求項13の方法。
【請求項15】
3つのキメラペプチドを含むHIV gp41 N−ペプチドのコイルドコイルの全部または一部分を模倣するポリマー修飾された共有結合安定化三量体コイルドコイルであって、各キメラペプチドが、HIV gp41 N−ペプチドの全部または一部分にらせん相で融合したコイルドコイルの可溶性三量体形態またはその修飾形態を含むスカフォールドドメインを含んでおり、スカフォールドドメイン内部に局在するエピトープが前記スカフォールドドメイン内部の少なくとも1つのアミノ酸残基に部位特異的に付着した少なくとも1つの低分子量水溶性ポリマーによって隠蔽されている前記三量体コイルドコイル。
【請求項16】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項15に記載のコイルドコイル。
【請求項17】
前記低分子量水溶性ポリマーが約1000Da未満の分子量を有しているポリエチレングリコール(PEG)である請求項16に記載のコイルドコイル。
【請求項18】
前記コイルドコイル内部の3つのキメラペプチドのおのおのがさらに、前記ペプチドのヘリカルドメインの外部に局在する少なくとも2つのシステイン残基を含み、および並列したペプチド上の前記システイン残基間のジスルフィド結合によってコイルドコイルが共有結合的に安定化されている請求項17に記載のコイルドコイル。
【請求項19】
前記コイルドコイル内部の3つのキメラペプチドのうちの第一キメラペプチドがさらに、前記ペプチドのヘリカルドメインの外部に局在する少なくとも2つのシステイン残基を含み、ならびに前記コイルドコイル内部の3つのキメラペプチドのうちの第二および第三のキメラペプチドがさらに、前記ペプチドのヘリカルドメインの外部に局在しチオエーテル結合を形成し得る求電子部分を含み、ならびにコイルドコイルが第一キメラペプチドの各システイン残基の求核性スルフヒドリルと各第二および第三キメラペプチドの求電子部分との間のチオエーテル結合を介して共有結合的に安定化されている請求項17に記載のコイルドコイル。
【請求項20】
(CCIZN17)3、C(チオEZN17)3およびC(チオE17GluN17)3から成るグループから選択されたHIV gp41 N−ペプチドコイルドコイルの全部または一部分を模倣するコイルドコイルであって、前記コイルドコイルのスカフォールドドメイン内部に局在するエピトープが前記スカフォールドドメイン内部の少なくとも1つのアミノ酸残基に部位特異的に付着した少なくとも1つの低分子量水溶性ポリマーによって選択的に隠蔽されているポリマー修飾された共有結合安定化三量体コイルドコイル。
【請求項21】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項20に記載のコイルドコイル。
【請求項22】
前記低分子量水溶性ポリマーが約1000Da未満の分子量を有しているポリエチレングリコール(PEG)である請求項21に記載のコイルドコイル。
【請求項23】
(a)コイルドコイルの可溶性三量体形態を含むスカフォールド部分と、
(b)HIV gp41 N−ペプチドの全部または一部分を含むN−ペプチド部分またはその修飾形態と、
(c)少なくとも2つのシステイン残基を含むシステイン部分と、
を含み、(a)の前記スカフォールド部分が(b)の前記N−ペプチド部分にらせん相で融合してα−ヘリカルドメインを形成し、(c)の前記システイン部分が前記α−ヘリカルドメインの外部に局在し、および前記スカフォールド部分の内部に局在するエピトープが前記スカフォールド部分内部の少なくとも1つのアミノ酸残基に部位特異的に付着した少なくとも1つの低分子量水溶性ポリマーによって選択的に隠蔽されているポリマー修飾された可溶性キメラペプチド。
【請求項24】
前記低分子量水溶性ポリマーが、ポリアルキレンオキシド、ポリアミドアルキレンオキシドおよびそれらの誘導体から成るグループから選択される請求項23に記載のキメラペプチド。
【請求項25】
前記低分子量水溶性ポリマーが約1000Da未満の分子量を有しているポリエチレングリコール(PEG)である請求項24に記載のキメラペプチド。
【請求項26】
前記N−ペプチド部分がN−ヘリックス疎水性ポケットを形成するアミノ酸残基を内包するのに十分なHIV gp41 N−ペプチドの部分を含む請求項25に記載のキメラペプチド。
【請求項27】
CCIPN17であるポリマー修飾キメラペプチド。
【請求項28】
免疫原性担体分子に共有結合的に連結されている請求項23に記載の可溶性キメラペプチドの抗原性接合体。
【請求項29】
前記担体分子がOMPCである請求項28に記載の抗原性接合体。
【請求項30】
免疫原性担体分子に共有結合的に連結されている請求項15に記載の共有結合安定化三量体コイルドコイルの抗原性接合体。
【請求項31】
前記担体分子がOMPCである請求項30に記載の抗原性接合体。
【請求項32】
生物学的に有効なアジュバント、タンパク質、または、免疫応答を増進し得る他の成分と混合した請求項30に記載の抗原性接合体を有効量で含み、哺乳動物体内でHIV特異的中和抗体を誘発し得る免疫原として有用な医薬組成物。
【図1】

【図2】
【図3】
【図4】
【図5】

【図6】
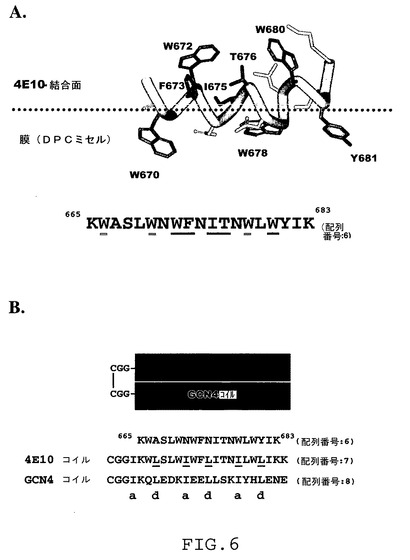
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2008−534640(P2008−534640A)
【公表日】平成20年8月28日(2008.8.28)
【国際特許分類】
【出願番号】特願2008−504703(P2008−504703)
【出願日】平成18年3月31日(2006.3.31)
【国際出願番号】PCT/EP2006/003361
【国際公開番号】WO2006/105993
【国際公開日】平成18年10月12日(2006.10.12)
【出願人】(501209427)イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー (90)
【Fターム(参考)】
【公表日】平成20年8月28日(2008.8.28)
【国際特許分類】
【出願日】平成18年3月31日(2006.3.31)
【国際出願番号】PCT/EP2006/003361
【国際公開番号】WO2006/105993
【国際公開日】平成18年10月12日(2006.10.12)
【出願人】(501209427)イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー (90)
【Fターム(参考)】
[ Back to top ]