
タンパク質ディスプレイ
本発明は、標的分子に対する所望の活性に関してポリペプチドをスクリーニングするための方法に関する。特に、本発明は、細菌細胞においてポリペプチドを発現し、細胞を透過化することによって、標的分子に対する所望の活性に関してポリペプチドをスクリーニングする方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ポリペプチドを標的分子に対する所望の活性に関してスクリーニングするための方法に関する。特に、本発明は、細菌細胞においてポリペプチドを発現し、前記細胞を透過化することにより、標的分子に対する所望の活性に関してポリペプチドをスクリーニングするための方法に関する。
【背景技術】
【0002】
タンパク質ディスプレイの最も初期の方法は、ファージディスプレイ(非特許文献1)であり、この方法では、関心対象のタンパク質を、タンパク質の野生型コピーとともに存在し得るファージの外皮タンパク質の1つと融合させる。たとえば、pIIIタンパク質に対する融合を使用したM13繊維状ファージを基礎とするディスプレイプラットフォーム。
【0003】
他のディスプレイ法には、「インビトロ」ディスプレイ法が含まれ、この方法では、タンパク質は細胞翻訳抽出物を用いて発現され、タンパク質とコーディング核酸との間のカップリングは、物理的結合(例えば、リボソームディスプレイ、mRNAディスプレイ)によるか、または通常のスカフォールドへの付着への付着もしくは膜内への封入により、例えばインビトロ区画化(IVC)で達成され、IVCでは、mRNAは、mRNAおよびタンパク質両方のマイクロビーズ(磁気またはセファロース)キャプチャシステムも含み得るミセル懸濁液内で翻訳される。
【0004】
タンパク質ディスプレイの別の方法は微生物表面ディスプレイであり、これは、グラム陰性、グラム陽性真正細菌または酵母のいずれかの微生物細胞外側に対する発現されたタンパク質の標的配置を含む。タンパク質を、細胞表面にそれらを付着させるアンカードメインと融合させる。アンカードメインは、脂質化もしくは細胞壁への共有結合を要求するモチーフを有し得るか、または露出したループ領域内の内在性膜タンパク質に対する融合物であり得る。産生の拡張性のために、微生物表面ディスプレイは、多様なライブラリーから改善されたタンパク質変異体についてスクリーニングするために用いられるだけでなく、ワクチン接種用の抗原を提示するためもしくは工業バイオテクノロジーの酵素用細胞スカフォールドとして用いることもできる。
【0005】
タンパク質ディスプレイ方法は、通常、抗体などの親和性タンパク質の進化に適用される。単一分子ディスプレイ法は歴史上最も普及しているが、高いバックグラウンドおよび親和性スケール間の低い分解能の問題を抱えている。周辺質収量が細胞質での発現に比べて非常に低いことが多い場合でも、酵母における表面ディスプレイによるかまたはファージ系により同定されるタンパク質は、通常、大腸菌(E. coli)周辺質における発現のために再編成される。しかし、抗体が細胞質において高収率で発現される場合、ほぼすべての場合に、それらは不溶性封入体を形成し、これは難儀してリフォールディングし、活性に関して試験しなければならない。
【0006】
このように、特に、親和性タンパク質ディスプレイライブラリおよび酵素ライブラリーをスクリーニングするためのタンパク質ディスプレイの方法が依然として必要とされている。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Smith (1985) Science, 228:1315-1317
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明者らは、透過化された細菌細胞において標的分子に対する所望の活性に関してポリペプチドをスクリーニングすることを可能にするタンパク質ディスプレイの方法を開発した。ポリペプチドは、透過化された細菌細胞内に保持されているか、または透過化された細菌細胞に結合しているかのいずれかである。
【0009】
したがって、本発明は、標的分子に対する所望の活性に関してポリペプチドをスクリーニングする方法であって:
a)ポリペプチドをコード化するポリヌクレオチドを含む細菌細胞を培養して、ポリペプチドを産生し、
b)細菌細胞を透過化し、ここで、ポリペプチドおよびポリペプチドをコード化するポリヌクレオチドは透過化された細菌細胞の内部に保持され、
c)透過化された細菌細胞を標的分子と接触させて、該標的分子を透過化された細菌細胞中に拡散させ、そして
d)ポリペプチドを所望の活性に関してスクリーニングする
ことを含む方法を提供する。
【0010】
本発明はさらに、標的分子に対する所望の活性に関してポリペプチドをスクリーニングする方法であって:
a)ポリペプチドをコード化するポリヌクレオチドを含む細菌細胞を培養して、ポリペプチドを産生し、細菌細胞壁に付着させ、
b)細菌細胞を透過化し、ここで、ポリペプチドをコード化するポリヌクレオチドは、透過化された細菌細胞内部に保持される、
c)透過化された細菌細胞を標的分子と接触させ、そして
d)ポリペプチドを所望の活性に関してスクリーニングする
ことを含む方法を提供する。
【0011】
1つの実施形態において、ステップd)は:
i)ポリペプチドが標的分子に結合するかどうか、および/もしくは標的分子に結合する程度を決定すること、ならびに/または
ii)ポリペプチドが標的分子を酵素的に修飾するかどうか、および/もしくは標的分子の酵素修飾率を決定する
ことを含む。
【0012】
細菌細胞は、細胞膜を可溶化するが細菌細胞壁の完全性を保持する任意の薬剤で透過化することができる。そのような薬剤としては、界面活性剤および有機溶媒が挙げられる。1つの実施形態において、細菌細胞を界面活性剤、たとえば非イオン性界面活性剤で透過化する。
【0013】
本発明の方法は、任意の好適なグラム陰性またはグラム陽性細菌細胞において実施することができるが、好ましくは、細菌細胞はグラム陰性細菌細胞である。
【0014】
1つの実施形態において、ポリペプチドは、少なくとも第2ポリペプチドと結合して、透過化された細菌細胞の内部に保持された、および/または細菌細胞壁に付着した、タンパク質複合体を形成する。ポリペプチドは、たとえば非共有もしくは共有結合によるなどで第2ポリペプチドと間接的に結合することができるか、またはポリペプチドは、第2ポリペプチドと直接的に結合することができ、たとえば融合タンパク質であり得る。
【0015】
したがって、1つの実施形態において、ポリペプチドを、第2ポリペプチド、またはそのサブユニットと融合させる。
【0016】
本発明の方法において、第2ポリペプチドは:
i)タンパク質複合体が透過化された細菌細胞壁の内部に保持されるような分子サイズを有するポリペプチド;
ii)DNA結合タンパク質;および/または
iii)細菌細胞壁結合タンパク質
から選択することができる。
【0017】
1つの実施形態において、タンパク質複合体の分子量は、少なくとも約120kDaである。
【0018】
別の実施形態において、第2ポリペプチドは、透過化された細菌細胞の内部でマルチマーを形成する。マルチマーは、たとえば、ダイマー、トリマー、テトラマー、ペンタマー、ヘキサマーまたはさらに高次のマルチマーであり得る。1つの実施形態において、マルチマーはテトラマーである。
【0019】
特定の1つの実施形態において、第2ポリペプチドは、RhnA、β−ガラクトシダーゼ、BetB、G5K、GshB、およびYdcWから選択される。
【0020】
所望の活性に関してスクリーニングされるポリペプチドを細菌宿主細胞DNAと結合させるために、任意のDNA結合タンパク質を本発明の方法で用いることができる。1つの実施形態において、DNA結合タンパク質はComEである。
【0021】
代替的または付加的に、ポリペプチドを細菌細胞壁結合タンパク質と結合させることができ、この場合、細菌細胞壁結合タンパク質は、ペプチドグリカン結合タンパク質、および細胞壁と結合できるリポタンパク質またはそのフラグメントから選択される。
【0022】
1つの実施形態において、細菌細胞壁結合タンパク質は、KzPG、PAL、OmpA、YiaD、YfiBおよびMotBから選択されるペプチドグリカン結合タンパク質である。
【0023】
ポリペプチドは細菌細胞壁と非共有的または共有的のいずれかで結合し得るが、1つの実施形態では、ポリペプチドは細菌細胞壁と共有結合する。
【0024】
別の実施形態において、細胞壁と結合できるリポタンパク質は、外膜付着に必要な機能的N末端シグナル配列が欠如したリポタンパク質である。
【0025】
特定の1つの実施形態において、リポタンパク質は大腸菌LPPである。
【0026】
本発明の方法の1つの実施形態において、非イオン性界面活性剤は、デカノイル−N−メチルグルカミド(Mega10)、ジメチルオクチルホスフィンオキシド(Apo8)、n−オクチル−β−D−チオグルコピラノシド(8TGP)、ならびにデカノイル−N−メチルグルカミド(Mega10)およびジメチルオクチルホスフィンオキシド(Apo8)の混合物から選択される。
【0027】
特定の1つの実施形態において、細菌細胞の透過化は:
i)LB中約0.5%のn−オクチル−β−D−チオグルコピラノシド(8TGP)、および
ii)LB中約0.5%のデカノイル−N−メチルグルカミド(Mega10)および約0.5%のジメチルオクチルホスフィンオキシド(Apo8)
から選択される溶液中で実施される。
【0028】
本発明の方法の別の実施形態において、ステップb)は、細菌細胞を選択的に透過化することを含み、それにより、細菌細胞の外膜は細菌細胞の内膜よりもさらに透過化される。
【0029】
1つの実施形態において、細菌細胞は、ジメチルオクチルホスフィンオキシド(Apo8)および/またはポリソルベート20(Tween20)から選択される界面活性剤で選択的に透過化される。
【0030】
1つの実施形態において、細菌細胞の選択的透過化は、界面活性剤を約0.2%で含む溶液中で実施される。
【0031】
別の実施形態において、細菌細胞の選択的透過化は、EDTAまたはCa2+を含む溶液中で実施される。
【0032】
別の実施形態において、方法は、ポリペプチドをコード化するポリヌクレオチドを含むDNAを透過化された細菌細胞から単離することをさらに含む。
【0033】
細菌細胞から単離されるDNAは、ゲノムDNAおよび/またはエピソームDNAであり得る。エピソームDNAは、たとえば、プラスミドまたはコスミドであり得る。
【0034】
1つの実施形態において、ポリヌクレオチドは外因性ポリヌクレオチドである。
【0035】
別の実施形態において、標的分子の分子量は約120kDa未満である。
【0036】
本発明はさらに、
所望の活性を有するポリペプチドを同定するための方法であって:
a)本発明の方法を用いてポリペプチドのライブラリーをスクリーニングすること;および
b)所望の活性を有する1以上のポリペプチドを選択すること
を含む方法を提供する。
【0037】
1つの実施形態において、方法はさらに、c)ポリペプチドをコード化するポリヌクレオチドを含むDNAを細菌細胞から単離することを含む。
【0038】
別の実施形態において、方法は、d)ポリペプチドをコード化するポリヌクレオチドの配列を決定することをさらに含む。
【0039】
1つの実施形態において、ポリペプチドのライブラリーは、細胞、組織、器官または生物から得られるポリヌクレオチドによりコード化される。
【0040】
別の実施形態において、ポリペプチドのライブラリーは、1以上の親ポリヌクレオチドを突然変異させることによって得られるポリヌクレオチドによりコード化される。
【0041】
1つの実施形態において、ポリペプチドは抗体または酵素である。
【0042】
特定の1つの実施形態において、抗体は単鎖可変フラグメント(scFV)である。
【0043】
別の実施形態において、ポリペプチドは酵素であり、標的分子を透過化された細菌細胞に結合させる。標的分子を透過化された細菌細胞に結合させるために、標的分子を細菌細胞に直接的または間接的のいずれかで結合させることができる。標的分子を透過化された細菌細胞に直接的に結合させるために、標的分子をたとえば、細菌細胞壁結合タンパク質と結合させることができる。
【0044】
別の実施形態において、ポリペプチドは、抗体以外の結合タンパク質である。たとえば、ポリペプチドは、これらに限定されるものではないが、リポカリン、フィブロネクチンIII型ドメイン(FN3)、ユビキチン、またはγ−B−クリスタリンをはじめとする結合タンパク質であり得る。
【0045】
本発明の方法の1つの実施形態において、ポリペプチドは、I27、RL6、KzPG、SNAP,および/またはDBPのいずれか1つから選択されるドメインを含む。特定の1つの実施形態において、ポリペプチドは、ドメインI27、RL6、KzPG、SNAP、およびDBPを含む。
【0046】
本発明の方法の別の実施形態において、ポリペプチドは、配列番号13と少なくとも80%、好ましくは少なくとも90%、さらに好ましくは少なくとも95%、さらに好ましくは100%同一のアミノ酸配列を含む。
【0047】
本発明はさらに、第2ポリペプチドと結合して、透過化された細菌細胞の内部に保持されるタンパク質複合体を形成する外因性タンパク質を含む透過化された細菌細胞を提供する。
【0048】
本発明はさらに、細菌細胞壁に付着した外因性ポリペプチドを含む透過化された細菌細胞を提供する。
【0049】
本発明はさらに:
a)i)第1ポリペプチドをコード化するポリヌクレオチドをベクターに挿入するための部位、および
ii)第1ポリペプチドと結合して、透過化された細菌細胞の内部に保持されるタンパク質複合体を形成する、第2ポリペプチドをコード化するオープンリーディングフレーム
を含むベクター、ならびに
b)細菌細胞を透過化することができる薬剤
を含むキットを提供する。
【0050】
本発明はさらに:
a)i)第1ポリペプチドをコード化するポリヌクレオチドをベクターに挿入するための部位、および
ii)第1ポリペプチドと結合して、細菌細胞壁に付着したタンパク質複合体を形成する第2ポリペプチドをコード化するオープンリーディングフレーム
を含むベクター、ならびに
b)細菌細胞を透過化することができる薬剤
を含むキットを提供する。
【0051】
1つの実施形態において、部位およびオープンリーディングフレームは、第1ポリペプチドおよび第2ポリペプチド、またはそのサブユニットが融合タンパク質として発現されるような位置にある。
【0052】
別の実施形態において、細菌細胞を透過化することができる薬剤は界面活性剤である。
【0053】
さらに別の実施形態において、界面活性剤は、デカノイル−N−メチルグルカミド(Mega10)、ジメチルオクチルホスフィンオキシド(Apo8)、n−オクチル−β−D−チオグルコピラノシド(8TGP)、ならびにデカノイル−N−メチルグルカミド(Mega10)およびジメチルオクチルホスフィンオキシド(Apo8)の混合物から選択される非イオン性界面活性剤である。
【0054】
1つの実施形態において、キットは細菌細胞をさらに含む。
【0055】
好ましくは、細菌細胞はグラム陰性である。たとえば、細菌細胞は大腸菌であり得る。
【0056】
本発明はさらに、配列番号13と少なくとも80%、好ましくは少なくとも90%、さらに好ましくは少なくとも95%、さらに好ましくは100%同一であるアミノ酸配列を提供する。
【0057】
本発明はさらに、配列番号14または配列番号15と少なくとも80%、好ましくは少なくとも90%、さらに好ましくは少なくとも95%、さらに好ましくは100%同一であるヌクレオチド配列を含むポリヌクレオチドを提供する。
【0058】
本発明はさらに、本発明のポリヌクレオチド配列を含むベクターを提供する。
【0059】
本発明はさらに、配列番号6〜12または16のいずれか1つと少なくとも90%、さらに好ましくは100%同一であるアミノ酸配列を含むポリペプチドスペーサーを提供する。
【0060】
明らかなように、本発明の1つの態様の好ましい特徴および特性は、本発明の多くの他の態様に適用可能である。
【0061】
本明細書全体を通して、「comprise(含む)」という語、または「comprises」もしくは「comprising」などの変形は、記載された要素、整数もしくはステップ、または要素、整数もしくはステップの群を包含するが、任意の他の要素、整数若しくはステップ、または要素、整数もしくはステップの群を除外しないことを意味すると理解される。
【0062】
本発明を、以下の非限定的例により、添付の図面を参照して後述する。
【図面の簡単な説明】
【0063】
【図1】大腸菌細胞の界面活性剤透過化。GFPを発現する大腸菌細胞を界面活性剤で処理して、膜透過化の有効性を測定した。細胞を明視野(第1列)または蛍光顕微鏡法(第2列および第3列)のいずれかにより観察した。透過化は、膜不透過性DNA結合色素Gel Red(第3列)の吸収と同時にGFP(緑色、第2列)が、細胞から放出される場合に有効であった。界面活性剤8TGP(0.5%)および0.5%のMega10/0.5%のApo8(「Agent86」)は大腸菌細胞の透過化において最も有効であることが判明した。
【図2】界面活性剤上清のSDS−PAGE。図1で示される大腸菌細胞の界面活性剤透過化の上清を、9%のSDS−PAGE上にロードして、界面活性剤によるタンパク質放出を定性的に評価した(第1レーン)。界面活性剤透過化細胞における細胞壁カプセルによる細胞タンパク質のサブセットの保持を示すために、界面活性剤透過化細胞の試料をリゾチーム(2mg/mL)で処理して、細胞壁(第2レーン)を加水分解した。
【図3】テトラマー融合タンパク質発現。(A)His6::SNAP::テトラマー融合タンパク質の大腸菌における発現を、全細胞タンパク質に対してプローブされたαHis抗体を用いてウェスタンブロットにより調べた。予想される分子量のバンドに加えて、>250kDの高分子量バンドがRhnAテトラマー融合物(レーン4)で観察され、これはテトラマーとして移動した複合体の推定されるSDS耐性形態である。(B)BetBテトラマー融合タンパク質抽出物を界面活性剤可溶性および界面活性剤不溶性(細胞カプセルペレット)抽出物に分離し、そしてSDS−PAGEにより調べた。
【図4】テトラマー融合タンパク質のSNAP標識。例1で記載されているようにして、His6::SNAP::テトラマー融合タンパク質を大腸菌において発現させ、細胞を8TGPで透過化した。例3で記載されているようにして、融合タンパク質の発現を、膜不透過性SNAPリガンドBG−547で標識された透過化細胞の蛍光顕微鏡検査(第2列)により検出した。細胞DNAを膜透過性色素、Sytox Greenで標識した(第1列)。SNAPおよびSytox Greenシグナルのオーバーレイを第3列に示す。
【図5】透過化細胞におけるHis6::SNAP::BetBテトラマーのαHis抗体標識。例3で記載されるような、αHis抗体でプローブされたHis6::SNAP::BetBテトラマーを発現する透過化細胞の蛍光顕微鏡検査(第1パネル)。細胞をSNAPリガンドBG−547で標識した(第2パネル)。αHisおよびSNAP両方の共存(第3パネル)は、αHis抗体が透過化細胞の細胞壁を透過することを示す。
【図6】HALOおよびSNAP発現レポーターとのBetB、RhnAおよびYdcWテトラマー融合物。BetB、RhnAおよびYdcWテトラマーを別々に発現レポーターHALOおよびSNAPと融合させた。融合タンパク質を発現する細胞を透過化し、宿主DNAをGel Redで標識し、HALO(G1001)およびSNAP(BG−488)の蛍光リガンドを用いて融合タンパク質を検出した。
【図7】GFP5::DNA結合タンパク質(DBP)融合物の発現。淋菌(N. gonorrhoeae)由来の非特異的高親和性DNA結合タンパク質であるComEをGFP5のC末端に融合させ、大腸菌で発現させた。細胞を透過化し、GFP(第1パネル)およびGel Red(第2パネル)に関して蛍光顕微鏡法により観察した。蛍光の共存(第3パネル)は、融合タンパク質および宿主DNAの両方が透過化細胞カプセル内に保持されたことを示す。
【図8】透過化細胞におけるDNAの保持。GFP5::DBP融合物、またはHis6::eGFP融合物を発現する大腸菌細胞を、未処理のまま放置するか(1および4行)または例1で記載されるように透過化させるか(2、3、5および6行)のいずれかであった。透過化細胞を4℃で一晩保存するか、またはTBS中に再懸濁させ、37℃で一晩振盪した後、GFP(第1列)またはGel Red(第2列)に関して蛍光顕微鏡法により観察した。GFPおよびGel Redの共存を第3列に示す。
【図9】透過化細胞からのDNA抽出。(A)GFP5::DBP融合タンパク質または(B)His6::eGFP融合タンパク質を発現する大腸菌細胞を例1で記載されるようにして透過化した。透過化細胞を一晩4℃で保存するか、またはTBS中に再懸濁させ、37℃で一晩振盪した後、プラスミドDNAを抽出し、エチジウムブロミド染色された1%アガロースゲル上、TAE緩衝液で電気泳動させた。レーン1は未処理細胞中の全プラスミドDNAである。レーン2および4は、それぞれ4℃で一晩保存し、37℃で振盪した細胞カプセルの透過化ステップからの上清であり、レーン3および5は、それぞれ4℃で一晩保存し、37℃で振盪した細胞カプセルからのプラスミド調製物である。
【図10】OmpF::SNAP::LPP融合タンパク質のSNAP標識。OmpF::SNAP::LPP融合タンパク質を発現する大腸菌細胞を例1で記載されるようにして透過化した。融合タンパク質局在化を、例3で記載されるようにSNAPリガンドBG−488での標識により検出した。標識された細胞を明視野顕微鏡検査(第1パネル)および蛍光顕微鏡検査(第2パネル)により観察した。第3パネルは明視野図および蛍光図両方のオーバーレイである。
【図11】αGFP::HALO::RhnA融合タンパク質によるeGFPの結合。αGFP::HALO::RhnA融合タンパク質を発現する大腸菌細胞を例1で記載されるようにして透過化した。精製されたeGFPタンパク質を例8で記載されるように細胞カプセルと結合させ、eGFPを蛍光顕微鏡検査により視覚化した。第1パネル、明視野図;第2パネル、eGFP蛍光;第3パネル、明視野および蛍光のオーバーレイ。
【図12】αGFP::KzPG::SNAP::DBP融合タンパク質によるeGFPの結合。αGFP::KzPG::SNAP::DBP融合タンパク質を発現する大腸菌細胞を例1で記載されるようにして透過化した。精製されたeGFPタンパク質を例8で記載されるようにして細胞カプセルと結合させ、例3で記載されているようにして、eGFPを2つの方法、ウェットマウントおよびドライマウントによる蛍光顕微鏡法で視覚化した。(A)ウェットマウント細胞カプセルに結合させたeGFP。挿入パネル(i)および(ii)は、αGFP::KzPG::SNAP::DBP融合タンパク質により結合したeGFPの細胞壁局在化を示す。(B)および挿入パネル(Aiii)は、DABCO/グリセロール中でドライマウントによる顕微鏡検査のために調製された同じ細胞を示す。
【図13】OmpF::αGFP::SNAP::LPP融合タンパク質によるeGFPの結合。OmpF::SNAP::LPPまたはOmpF::αGFP::SNAP::LPP融合タンパク質を発現する大腸菌細胞を例1で記載されているようにして透過化した。精製されたeGFPタンパク質を例8で記載されているようにして細胞カプセルと結合させ、例3で記載されているようにして、eGFPをドライマウントによる蛍光顕微鏡法によって視覚化した。(A)OmpF::SNAP::LPP融合タンパク質を発現する細胞は、OmpF::αGFP::SNAP::LPP融合タンパク質を発現する細胞(第2パネル、下行)と異なり、eGFP蛍光がない(第2パネル、上行)。
【図14】LPP融合タンパク質による細胞壁に対する共有結合の証明。OmpF::αGFP::SNAP::LPP融合タンパク質を発現する大腸菌細胞を例1で記載されているようにして透過化した。融合タンパク質局在化を例3で記載されているようにしてSNAPリガンドBG−488で標識することにより検出し、DNAをGel Redで染色した。試料を5分間22℃(A)または95℃(B)で加熱した後、ドライマウントし、蛍光顕微鏡検査により観察した。
【図15】界面活性剤/Ca2+緩衝液を用いた外膜透過化。OmpF::αGFP::SNAP::LPP融合タンパク質(外部αGFPまたはαGFP::HALO::FLAG::RhnA融合タンパク質(内部αGFP)を発現する大腸菌細胞を例10で記載されているようにして透過化した。大きなリガンドに対する外膜の透過化は、細胞壁に付着したαGFPドメインにeGFPを結合させることにより評価した。内膜の透過化は、大きなリガンド(eGFP)および小さなリガンド(Gel Red)を用いて評価した。Ca2+緩衝液中の界面活性剤Apo8(A)およびTween20(B)はどちらも、大きなリガンドに対する外膜の選択的透過性を示した。
【図16】界面活性剤/EDTA緩衝液を用いた外膜透過化。OmpF::αGFP::SNAP::LPP融合タンパク質(外部αGFP)またはαGFP::HALO::FLAG::RhnA融合タンパク質(内部αGFP)を発現する大腸菌細胞を例10で記載されるようにして透過化した。大きなリガンドに対する外膜の透過化を、eGFPを細胞壁に付着したαGFPドメインに結合させることにより評価した。内膜の透過化は、大きなリガンド(eGFP)および小さなリガンド(Gel Red)を用いて評価した。EDTA緩衝液中の界面活性剤Apo8(A)およびTween20(B)はどちらも大きなリガンドに対する外膜の選択的透過性を示した。
【図17】混合eGFP、およびSNAP標識された細胞のFACS分析。eGFP(#1矢印);SNAPリガンドBG−488で標識されたαGFP::KzPG::SNAP::DBP融合タンパク質(#2矢印);およびSNAPリガンドBG−547で標識されたHis6::SNAP::BetB(#3矢印)を発現する大腸菌細胞の3つの集団を、FACSにより分類した。分類された集団を初回分類の60分後に純度および細胞完全性について再分析した。
【図18】αGFPおよびKzPGドメイン間のペプチドリンカーは、αGFP::KzPG::SNAP::DBP融合タンパク質を発現する大腸菌細胞のセファロース支持体に対する結合を可能にする。12−merリンカー領域RL6をαGFPおよびKzPGドメイン間に有するαGFP::KzPG::SNAP::DBP融合タンパク質を発現する細胞を、His6::eGFP中間体によりCo2+−セファロース支持体Co2+−セファロース支持体と結合させた。GFP結合を左のパネルに示し(緑);融合タンパク質のSNAPリガンド(赤)結合を中央のパネルに示し;それぞれのオーバーレイを右に示す。
【図19】αGFP::RL6::KzPG::SNAP::DBP融合タンパク質を発現する大腸菌細胞のストレプトアビジン標識された磁気ビーズに対する結合。(A)ビオチン標識されたeGFP(中央および右パネル)を、αGFP::RL6::KzPG::SNAP::DBP融合タンパク質を発現する細胞と結合させ、これを次にストレプトアビジン標識された磁気粒子と結合させた。(B)最初にビオチニル化eGFPで標識されたストレプトアビジン標識磁気粒子に対するαGFP::RL6::KzPG::SNAP::DBP融合タンパク質を発現する細胞の逆の結合。この例では、ビーズは緑色に標識され(GFPパネル)、細胞はBG−547SNAPリガンドで標識された(赤、SNAP redパネル)。(C)ドメインリンカー(ヒトタイチンの27番目のIgドメイン)も結合スペーサーとして有効であった。αGFP::I27::RL6::KzPG::SNAP::DBP融合タンパク質を発現する大腸菌細胞をまずビオチニル化eGFPと結合させ(緑、GFPパネル)、BG−547SNAPリガンドで標識(赤、SNAP redパネル)した後、ストレプトアビジン標識磁気粒子と結合させる。
【図20】大腸菌細胞質におけるscFv::I27::RL6::KzPG::SNAP::DBP融合タンパク質としてのマウスscFv遺伝子の発現。マウスscFvライブラリーを構築し、本発明の方法にしたがって大腸菌細胞質でディスプレイした。検出可能な発現を有するクローンをSNAPリガンド結合により検出し、ミスフォールド(左パネル)、わずかに発現されるが可溶性(中央のパネル)または高度に発現され可溶性(右パネル)に分類した。
【図21】可溶性および不溶性scFv発現の大腸菌細胞質における検出。マウスscFv発現ライブラリーからの限定されたスクリーンにおいて高度に発現され可溶性であることが判明した、選択されたクローンを、scFv::I27::RL6::FLAGおよびscFv::RL6::FLAG融合タンパク質として発現構築物中にサブクローンした。タンパク質フラクションを可溶性または不溶性のいずれかとしてSDS−PAGEゲル上にロードし、ニトロセルロース膜に移し、αFLAG抗体を用いて検出した。可溶性(S)または不溶性(P)フラクションについて、試料、ならびに発現される各クローンをI27::RL6(I)またはRL6(R)リンカーと対にする。ライブラリスクリーンから単離されるI27::RL6::KzPG::SNAP::DBPディスプレイ構築物におけるオリジナルのscFvクローンの蛍光顕微鏡画像も下のパネルに示す。
【0064】
配列表の凡例
配列番号1 pAra3::His6::SNAPアラビノースベクターのヌクレオチド配列
配列番号2 pAra3::His6::KzPG::SNAP::DBPベクターのヌクレオチド配列
配列番号3 pAra3::OmpF::SNAP::LPPベクターのヌクレオチド配列
配列番号4 pAra3::αGFP(R35)::HALO::FLAG::RhnAベクターのヌクレオチド配列
配列番号5 ランダム化ペプチドスペーサードメイン
配列番号6〜12 ペプチドリンカースペーサー
配列番号13 I27::RL6::KzPG::SNAP::DBP
配列番号14 I27::RL6::KzPG::SNAP::DBPコーディング配列
配列番号15 ライブラリスカフォールドベクター
配列番号16 I27スペーサー
【発明を実施するための形態】
【0065】
一般的技術および定義
別段の明確な規定がない限り、本明細書中で用いられる全ての専門用語および科学用語は、(たとえば、タンパク質化学、生化学,細胞培養、分子遺伝学、微生物学、および免疫学の)当業者により通常理解されるのと同じ意味を有すると解釈されるべきである。
【0066】
特に明記しない限り、本発明で利用される組換えタンパク質、細胞培養、および免疫手法は、当業者に周知の標準的手順である。そのような技術は、J. Perbal, A Practical Guide to Molecular Cloning, John Wiley and Sons (1984)、J. Sambrook et al., Molecular Cloning: A Laboratory Manual, 3rd edn, Cold Spring Harbour Laboratory Press (2001)、R. Scopes, Protein Purification - Principals and Practice, 3rd edn, Springer (1994)、T.A. Brown (editor), Essential Molecular Biology: A Practical Approach, Volumes 1 and 2, IRL Press (1991)、D.M. Glover and B.D. Hames (editors), DNA Cloning: A Practical Approach, Volumes 1-4, IRL Press (1995 and 1996)、およびF.M. Ausubel et al. (editors), Current Protocols in Molecular Biology, Greene Pub. Associates and Wiley-Interscience (1988、including all updates until present), Ed Harlow and David Lane (editors) Antibodies: A Laboratory Manual, Cold Spring Harbour Laboratory, (1988)、およびJ.E. Coligan et al. (editors) Current Protocols in Immunology, John Wiley & Sons (including all updates until present)などの出典で文献全体にわたって記載され、説明されている。
【0067】
「ポリペプチド」、「タンパク質」および「ペプチド」という用語は、本明細書中では一般的に交換可能に用いられる。本明細書中で用いられる場合、「外因性ポリペプチド」という用語は、外因性ポリヌクレオチドによりコード化されるポリペプチドを指す。「外因性ポリヌクレオチド」という用語は、本明細書中で用いられる場合、それが導入される細胞と異なるポリヌクレオチド、またはポリペプチドが通常見いだされない宿主細胞核酸内の位置以外は導入される細胞における配列と配列が相同性であることを意味する。
【0068】
「抗体」という用語は、本発明で用いられる場合、ポリクローナル抗体、モノクローナル抗体、二重特異性抗体、二特異性抗体、三特異性抗体、多特異性抗体、ヘテロ結合抗体、キメラ抗体、例えばインタクト分子ならびにそのフラグメント、例えばFab、F(ab’)2、FvおよびscFvならびに他の抗体様分子が挙げられる。
【0069】
「約」という用語は、本明細書中で用いられる場合、所定値の+/−5%の範囲を指す。
【0070】
透過化
本発明の方法では、細菌細胞を透過化し、かくして可溶性細胞成分の少なくとも一部を、細胞壁を通して拡散させる。所望の活性に関してスクリーニングされるポリペプチドは、細菌細胞壁内に保持されるか、または細菌細胞壁に付着する。本明細書中で用いられる場合、「透過化された細菌細胞」とは、1以上細胞膜中に孔を生成させるか、または細胞膜を可溶化し、一方、ペプチドグリカン間の結合を加水分解せず、それにより細胞壁をインタクトのままで保持する透過化剤の使用を指す。細菌細胞を透過化する薬剤の非限定的例としては、界面活性剤および有機溶媒が挙げられる。透過化は、有利には、小〜中程度のサイズのタンパク質、たとえば120kDaまで、または同等以下のサイズの他の分子がインタクトなままの細胞カプセル中に侵入させる。さらに、細菌細胞壁の完全性を維持することにより、透過化された細菌細胞は、従来法で、たとえば細菌細胞をTris−EDTA−リゾチームで処理することにより産生されるスフェロプラストほど脆弱ではなく、スフェロプラストでは、細菌細胞壁が少なくとも部分的に加水分解されている。本発明の方法で産生される透過化された細菌細胞は、蛍光活性化細胞分類(FACS)などの技術によく適しており、一方、スフェロプラストは高剪断フローサイトメトリー環境により損なわれ、制御された浸透圧条件を必要とし、したがってそれらの潜在的使用が限定される。
【0071】
好ましくは、透過化処理は、細胞タンパク質をそれらの自然な状態および相互関係で保存する。非イオン性界面活性剤は、タンパク質フォールディングおよびタンパク質複合体に対してイオン性界面活性剤ほど破壊的でない。したがって、好ましい実施形態では、非イオン性界面活性剤を用いて細菌細胞壁を透過化する。非イオン性界面活性剤の非限定的例としては、Triton X−100、Triton X−114、Brij 35、Brij 58、Tween 20、Tween 80、Nonidet P−40 Substitute、Octyl β Glucoside、Mega 8、Mega 9、Mega 10、BigCHAP、Deoxy BigCHAP、Apo8、および8TGPが挙げられる。
【0072】
界面活性剤の混合物を用いて細菌細胞を透過化することができる。たとえば、界面活性剤は、2以上の非イオン性界面活性剤の混合物であり得る。1つの実施形態において、界面活性剤はMega 10およびApo8の混合物である。
【0073】
所望の活性に関してスクリーニングされるポリペプチドが細菌細胞壁に付着しているか、または内側細胞膜と一体化しているかもしくは付着している場合、必ずしも細菌細胞の内膜を透過化する必要はない可能性があることを当業者は理解するであろう。したがって、1つの実施形態では、細菌細胞は選択的に透過化される。「選択的に透過化される」により、透過化された細菌細胞の外膜が内膜よりもさらに透過化され、それによって、内膜および外膜の両方が、0.5%のMega 10および0.5%のApo8を含む溶液を用いることによるなどで透過化された透過化細胞と比較した場合、膜不透過性物質、たとえば膜不透過性DNA結合リガンドGel Redの50%以下、もしくは更に好ましくは40%、30%、20%、10%、5%、4%、3%、2%、1%以下が選択的に透過化された細胞の内膜を透過するか、または透過しないことを意味する。
【0074】
当業者は、本発明の方法にしたがって細菌細胞を選択的に透過化するために好適な条件を決定することができるが、1つの実施形態では、細菌細胞は、非イオン性界面活性剤で選択的に透過化される。たとえば、非イオン性界面活性剤は、Apo8およびTween20から選択することができる。1つの実施形態において、細菌細胞を選択的に透過化するための溶液は、界面活性剤を約0.2%〜約0.4%、または約0.2%〜約0.3%、または約0.2%の濃度で含む。好ましくは、細菌細胞を選択的に透過化するための溶液は、Ca2+またはEDTAを含む緩衝液中界面活性剤を含む。細菌細胞を選択的に透過化するために好適な例示的緩衝液としては、25mMのTris中0.2〜0.4%のApo8もしくはTween20、1mMのEDTA(pH8.0)、または25mMのTris、2mMのCa2+(pH8.0)が挙げられる。1つの実施形態において、細菌細胞の選択的透過は、細胞を好適な緩衝液中、約25℃にて約10分間インキュベートすることによって達成することができる。
【0075】
ポリペプチド発現
所望の活性に関してスクリーニングされるポリペプチドを、細菌細胞における発現の好適なベクター中にクローンすることができる。「ベクター」は本明細書中で用いられる場合、細菌細胞を形質転換するために好適であることが当該技術分野で知られている任意のベクターを指す。好ましくは、ベクターはまた、宿主のゲノムから独立して、細菌細胞内で複製することができる。ベクターとしては、プラスミド、ウイルスおよびコスミドならびに線状DNAエレメント、例えば大腸菌の線状ファージN15、および/または細菌細胞ゲノムから独立して複製する染色体外DNAが挙げられる。好ましくは、ベクターは発現ベクターである。
【0076】
本明細書中で用いられる場合、「発現ベクター」は、細菌細胞において特定のポリヌクレオチド分子を発現させることができるベクターである。好ましくは、発現ベクターは、細菌細胞内で複製することもできる。好適な発現ベクターは、典型的には、調節配列、たとえば転写制御配列、翻訳制御配列、複製起点、および組換え細菌細胞と適合性であり、ポリペプチドをコード化するポリヌクレオチド分子の発現を制御する他の調節配列を含む。転写制御配列は、転写の開始、伸長、および終止を制御する配列である。特に重要な転写制御配列は、転写開始を制御するもの、たとえば、プロモーター、エンハンサー、オペレーターおよびリプレッサー配列である。好適な転写制御配列は、細菌細胞において機能することができる任意の転写制御配列を含む。種々のそのような転写制御配列は、当業者に公知である。
【0077】
発現ベクターの細菌細胞への形質転換は、ポリヌクレオチド分子を細胞中に挿入できる任意の好適な方法により達成することができる。形質転換技術としては、これらに限定されるものではないが、エレクトロポレーションおよび化学変換が挙げられる。形質転換されたポリヌクレオチド分子は、染色体外にとどまる可能性があるか、またはそれらの発現される能力が保持されるような方法で形質転換された(すなわち、組換え)細胞の染色体内の1以上の部位に組み込まれる可能性がある。
【0078】
組換えDNA技術を用いて、たとえば、宿主細胞内のポリヌクレオチド分子のコピー数、それらのポリヌクレオチド分子が転写される効率、結果として得られる転写物が翻訳される効率、および翻訳後修飾の効率を操作することによって、形質転換されたポリヌクレオチド分子の発現を改善することができる。ポリヌクレオチド分子の発現を増大させるために有用な組換え技術としては、これらに限定されるものではないが、ポリヌクレオチド分子を高コピー数のプラスミドに機能的に連結すること、ベクター安定性配列のプラスミドへの添加、転写制御シグナル(たとえば、プロモーター、オペレーター、エンハンサー)の置換もしくは修飾、翻訳制御シグナル(たとえば、リボソーム結合部位、Shine−Dalgarno配列)の置換もしくは修飾、宿主細胞のコドン使用に対応させるためのポリヌクレオチド分子の修飾、および転写物を不安定化する配列の欠失が挙げられる。
【0079】
当業者は、本発明の方法でポリペプチドを発現するために好適な細菌株を容易に決定することができるであろう。当業者は、本発明の方法での使用に好適なグラム陰性菌には、サルモネラ属(Salmonella)、大腸菌、赤痢菌(Shigella)、カンピロバクター属(Campylobacter)、フゾバクテリウム属(Fusobacterium)、ボルデテラ属(Bordetella)、パスツレラ属(Pasteurella)、アクチノバチルス属(アクチンobacillus)、ヘモフィルス属(Haemophilus)およびヒストフィルス属(Histophilus)が含まれることを理解するであろう。好ましい実施形態において、グラム陰性菌は大腸菌である。
【0080】
タンパク質複合体
所望の活性に関してスクリーニングされるポリペプチドを少なくとも第2ポリペプチドと結合させて、タンパク質複合体が透過化された細菌細胞の内部に保持されるような分子サイズを有するタンパク質複合体を形成することができる。ポリペプチドをたとえば、ジスルフィド架橋などの共有結合によるか、または非共有結合により、第2ポリペプチドと結合させることができる。「非共有結合」とは、原子間結合が関与しない分子相互作用を指す。たとえば、非共有相互作用は、イオン結合、水素結合、疎水性相互作用、およびファンデルワールス力が関与する。非共有結合力を用いて、別個のポリペプチド鎖をタンパク質またはタンパク質複合体中に一緒に保持することができる。したがって、ポリペプチドおよび第2ポリペプチドを同じかもしくは異なるベクターのいずれかから別のポリペプチドとして発現することができるか、またはポリペプチドの一方もしくは両方を、細菌細胞ゲノム中に組み入れられたポリペプチドをコード化するDNAから発現することができる。
【0081】
あるいは、タンパク質複合体と結合するポリペプチドおよび第2ポリペプチドは融合タンパク質であり得る。本明細書中で用いられる場合、「融合タンパク質」とは、2つのポリペプチドのそれぞれの少なくとも一部をコード化するポリヌクレオチドの発現から生じる2以上のポリペプチド、またはそれらのフラグメントからなるハイブリッドタンパク質を指す。
【0082】
分子サイズにより透過化された細菌細胞中に保持されるタンパク質複合体
第2ポリペプチドは、所望の活性に関してスクリーニングされるポリペプチドで形成された複合体の少なくともいくつかが、透過化された細菌細胞から拡散することができないような十分な分子サイズ、すなわち十分な分子量または分子半径を有する任意のポリペプチドであり得る。したがって、タンパク質複合体は、細胞の透過化後に細菌細胞内に保持される。当業者は、その分子量および球状または桿状(糸状)タンパク質であるかどうかをはじめとする第2ポリペプチドの性質が、細菌細胞壁を通るタンパク質複合体の拡散を防止または阻害するその能力を決定することを理解するであろう。1つの実施形態において、第2ポリペプチドの分子量は、少なくとも約30kDa、または少なくとも約40、50、60、70、80、90、100、120、130、140、150kDaもしくはそれ以上である。1つの実施形態において、第2ポリペプチドは少なくとも約120kDaである。
【0083】
1つの実施形態において、第2ポリペプチドは、透過化された細菌細胞の孔排除サイズよりも大きな分子サイズを有するマルチマーを形成する。本明細書中で用いられる場合、「マルチマー」という用語およびその文法的変異は、2以上の異なる分子間のマルチマー複合体の形成を指す。マルチマーは、たとえば、同じタンパク質の2以上の分子(すなわち、ホモマルチマー)または2以上の異なるか、もしくは非同一タンパク質(すなわち、ヘテロマルチマー)の混合物を含み得る。本発明の方法における使用に好適なマルチマーを形成するタンパク質としては、ダイマー、トリマー、テトラマー、ペンタマー、ヘキサマー、および7以上のサブユニットを含むさらに高次のマルチマーが挙げられる。
【0084】
マルチマータンパク質には、ホモダイマー、たとえば、PDGF受容体α、およびβイソ型、エリスロポエチン受容体、MPL、ならびに−CSF受容体、それらのサブユニットがそれぞれリガンド結合およびエフェクタードメインを有するヘテロダイマー、たとえば、PDGF受容体αβイソ型、ならびに異なる機能を有する成分サブユニットを有するマルチマー、たとえば、IL−2、IL−3、IL−4、IL−5、IL−6、IL−7、およびGM−CSF受容体が含まれる。本発明の方法で用いることができる他のマルチマータンパク質の非限定的例としては、DNAの合成または複製に関与する因子、例えばTFIIDおよびTFIIHなどのmRNAの産生に関与するDNAポリメラーゼタンパク質;細胞、核および他の膜関連タンパク質、例えばホルモンおよび他のシグナル伝達受容体、能動輸送タンパク質およびイオンチャンネル、ヘモグロビン、フィブリノゲンおよびフォンウィルブランド因子をはじめとする血液中のマルチマータンパク質;細胞内の構造を形成するタンパク質、例えばアクチン、ミオシン、およびチューブリンならびに他の細胞骨格タンパク質;細胞外環境で構造を形成するタンパク質、例えばコラーゲン、エラスチンおよびフィブロネクチン;細胞内および細胞外輸送に関与するタンパク質、例えばキネシンおよびダイニン、タンパク質のSNAREファミリー(可溶性NSF付着タンパク質受容体)およびクラスリン;クロマチン構造の調節に役立つタンパク質、例えばヒストンおよびプロタミン、Swi3p、Rsc8pおよびモイラ;マルチマー転写因子、例えばFos、JunおよびCBTF(CCAATボックス転写因子);マルチマー酵素、例えばアセチルコリンエステラーゼおよびアルコールデヒドロゲナーゼ;シャペロンタンパク質、例えばGroE、GroEL(シャペロニン60)およびGroES(シャペロニン10);抗毒素、たとえばヘビ毒、ボツリヌス毒素、ストレプトコッカス(Streptococcus)超抗原;リシン(バクテリオファージおよびウイルス由来の酵素);ならびにほとんどのアロステリックタンパク質が挙げられる。1つの実施形態において、マルチマータンパク質は大腸菌タンパク質である。マルチマーを形成する大腸菌タンパク質の非限定的例としては、L−ラムノースイソメラーゼ(RhnA;たとえばNCBI受入CAA43002)、β−ガラクトシダーゼ(β−gal;たとえばNCBI受入YP001461520)、ベタインアルデヒドデヒドロゲナーゼ(BetB;たとえばNCBI受入AAA23506)、グルタメート−5−キナーゼ(G5K;たとえばNCBI受入AAB08662)、グルタチオンシンターゼ(GshB;たとえばNCBI受入AP_003504)、および中鎖アルデヒドデヒドロゲナーゼ(YdcW;たとえばNCBI受入AP_002067)が挙げられる。
【0085】
1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドは、細菌細胞壁内にポリペプチドを保持するために十分な分子サイズを有する。したがって、当業者は、透過化された細菌細胞内にポリペプチドを保持するために、そのようなポリペプチドを第2ポリペプチドと必ずしも結合させる必要はないことを理解するであろう。
【0086】
DNA結合タンパク質
本発明者は、透過化後の細菌細胞内にDNAが保持されることを見出した。したがって、1つの実施形態において、ポリペプチドをDNA結合タンパク質と結合させて、タンパク質複合体を形成し、これはDNAと結合し、細菌細胞の内部に保持される。本明細書中で用いられる場合、「DNA結合タンパク質」とは、2本鎖または1本鎖DNAを認識する少なくとも1つのモチーフを含むDNA結合ドメインを含む任意のタンパク質を指す。当業者には知られているように、DNA結合ドメインは、ヘリックス・ターン・ヘリックス、亜鉛フィンガー、ロイシンジッパー、翼状ヘリックス、翼状ヘリックス・ターン・ヘリックス、ヘリックス・ループ・ヘリックス、DNAを認識する免疫グロブリンフォールド、またはB3ドメインを含む。ポリペプチドをDNA結合タンパク質と結合することによって、有利には、本発明のスクリーニング法においてポリペプチドをコード化するプラスミドなどのDNAの回収が増大する。
【0087】
DNA結合タンパク質の例としては、細菌コンピテンスタンパク質、例えばこれらに限定されるものではないが、大腸菌DNA結合タンパク質、淋菌(Neisseria gonorhoeae)DNA結合タンパク質、たとえばComE、アデノウイルスE2タンパク質、AraC転写因子、塩基性ヘリックス・ループ・ヘリックス転写因子、塩基性ロイシンジッパー転写因子、ブチラート反応因子、セントロメアタンパク質B、COUP転写因子、初期増殖応答転写因子、Gボックス結合因子、GATA転写因子、HMGAタンパク質、ホメオドメインタンパク質、I−カッパBタンパク質、組み込み宿主因子、インターフェロン調節因子、インターフェロン−刺激遺伝子因子3、Kruppel様転写因子、ロイシン応答調節タンパク質、マトリックス付着領域結合タンパク質、メチル−CpG結合タンパク質、MutSホモログ2タンパク質、骨髄性−リンパ性白血病タンパク質、NF−カッパB、NF1転写因子、核呼吸因子、ガン遺伝子タンパク質p55、起点認識複合体、対合ボックス転写因子、POUドメイン因子、プロトオンコジーン因子、Rad51リコンビナーゼ、Rad52DNA修復および組換えタンパク質、複製タンパク質A、複製タンパク質C、網膜芽細胞種タンパク質、Smadタンパク質、SOX転写因子、Tボックスドメインタンパク質、TCF転写因子、テロメア結合タンパク質、Toll様受容体9、トランス活性化因子、および翼状ヘリックス転写因子が挙げられる。1つの実施形態において、DNA結合タンパク質は大腸菌DNA結合タンパク質である。別の実施形態において、DNA結合タンパク質は淋菌タンパク質、たとえばComEである。
【0088】
細胞壁結合タンパク質
所望の活性に関してスクリーニングされるポリペプチドを、細菌細胞壁結合タンパク質と結合させることができる。当業者は、細胞壁結合タンパク質の選択が宿主細胞種に依存することを理解するであろう。なぜなら、異なる細菌は異なる細胞壁組成を有するからである。細菌はペプチドグリカン(PG)から構成される細胞壁を有するが、種間の化学修飾は、異種間結合に影響を及ぼし得る。当業者は、特定の細菌種における使用に好適な細胞壁結合タンパク質を容易に決定することができるであろう。
【0089】
細菌細胞壁結合タンパク質は、それによって天然の構造中のポリペプチド鎖が細菌細胞壁上の特定の分子または分子構造を認識でき、結合できるドメイン構造を有することが知られているタンパク質を含む。したがって、「細菌細胞壁結合タンパク質」という用語は、細菌細胞壁に特異的に結合するタンパク質の部分であるタンパク質ドメインを包含する。細菌細胞壁結合タンパク質の例としては、バクテリオファージによりコードされるような細胞壁ヒドロラーゼ、細菌の細胞壁ヒドロラーゼおよび異なる自己溶菌酵素が挙げられる。さらに含まれるのは、バクテリオファージおよび他のウイルスのDNAによりコードされる受容体分子である。細菌細胞壁結合タンパク質が、細菌に特異的に結合できるバクテリオファージ起源の加水分解酵素由来である場合、細胞壁結合タンパク質はそれらの結合能力を保持するが、好ましくは著しい加水分解活性を有さない。
【0090】
1つの実施形態において、細胞壁結合タンパク質は大腸菌の細胞壁に共有結合する。たとえば、大腸菌宿主細胞について、PAL、OmpA、YiaD、YfiB、およびMotBに存在する、保存された約100のアミノ酸PG結合ドメインを有する内因性PG結合タンパク質がある(Parsons et al., 2006)。しかし、たとえばシュードモナス属(Pseudomonas)φKZファージ(KzPG)由来の約70のアミノ酸PG結合ドメインなどの他の生物由来のタンパク質は、大腸菌で十分に発現され、高親和性で細胞壁に結合することが示されている(Briers et al., 2009)。したがって、PGと結合するタンパク質由来のPG結合ドメインを本発明の方法で細菌細胞壁結合タンパク質として用いることができる。
【0091】
例示的実施形態において、所望の活性に関してスクリーニングされ、細菌細胞のサイトゾルで発現されるポリペプチドに、PG結合ドメインを融合させることができる。膜透過化により、PG結合ドメインは細胞壁と結合し、その結果、透過化細胞内で関心対象のポリペプチドが保持される。細胞内での関心対象のポリペプチドの保持を潜在的にさらに増強するために、当業者は、ポリペプチドが細菌細胞壁結合タンパク質に加えてDNA結合タンパク質と結合できることを理解するであろう。
【0092】
あるいは、関心対象のポリペプチドは、細菌細胞壁に共有結合することができるタンパク質と結合することができる。好ましくは、タンパク質は周辺質を標的とするシグナルを含む。したがって、ポリペプチドは細菌細胞のサイトゾル中で発現されるが、周辺質を標的とし、ここで膜透過化前に細胞壁に結合する。
【0093】
非限定的例として、細胞壁と共有結合する細菌細胞壁結合タンパク質は、細胞壁と結合することができ、外膜付着に必要な機能的N末端シグナル配列が欠如したリポタンパク質であり得る。たとえば、リポタンパク質は大腸菌LPPであり得る。LPPは、トリマーコイルドコイルを形成する多量の大腸菌タンパク質である。その天然の形態において、一端は脂質化により外膜に結合し、多端はC−末端リシンにより細胞壁に共有結合する。リポタンパク質は、リポタンパク質に周辺質を標的とさせる配列、たとえばOmpF周辺質ターゲティング配列をさらに含み得る。1つの実施形態において、リポタンパク質は、外膜付着に必要な機能的N末端シグナル配列が欠如した大腸菌リポタンパク質である。
【0094】
本明細書の教示を考慮して、当業者は、細菌細胞壁と共有結合し、本発明の方法での使用に好適なタンパク質を同定または設計できるであろう。
【0095】
本発明の1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドは、KzPGドメインならびにスペーサー、SNAPおよび/またはDBPから選択される1以上の他のドメインを含む融合ポリペプチドである。特定の1つの実施形態において、融合ポリペプチドは、1以上スペーサーならびにKzPG、SNAPおよびDBPドメインを含む。
【0096】
スペーサー
1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドを、1以上スペーサーを含む融合ポリペプチドとして発現することができる。「スペーサー」は、本明細書中で用いられる場合、融合ポリペプチド中に含まれて、細菌細胞におけるポリペプチドの発現を増強するか、または立体障害を減少させて、所望の活性に関してスクリーニングされるポリペプチドがその所望の三次構造をとることができ、および/またはその標的分子と適切に相互作用することができるペプチドまたはポリペプチドを指す。したがって、融合タンパク質は、融合ポリペプチド中の1以上のポリペプチドドメインの前、後、または間に1以上スペーサーを含み得る。スペーサーおよび所望のスペーサーの同定方法については、たとえば、George, et al. (2003)を参照のこと。
【0097】
1つの実施形態において、スペーサーは、1〜50のアミノ酸残基の長さ、または約1〜25残基、または約5〜15残基の長さである1以上のアミノ酸配列を含む。たとえば、スペーサーは、I27、RL1、RL2、RL3、RL4、RL5および/またはRL6の1以上から選択することができる。当業者は、限定された数のアミノ酸置換、たとえば、1、2、3、4または5のアミノ酸置換を、スペーサーとして機能するその能力に影響を及ぼすことなくスペーサーに導入することができることを理解するであろう。特定の1つの実施形態において、1以上スペーサーは、配列番号6〜12または16のいずれか1つから選択される。したがって、1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドは、I27、RL6、KzPG、SNAPおよびDBPを含む融合ポリペプチドである。
【0098】
スクリーニング法およびタンパク質進化
本発明は、標的分子に対する所望の活性に関してポリペプチドをスクリーニングするための方法を提供する。本明細書中で用いられる場合、「所望の活性」という用語は、ポリペプチドの任意の潜在的に有用な活性を指し、これらに限定されるものではないが、結合、酵素修飾、フォールディング安定性および/または熱安定性を包含する。
【0099】
「標的分子」という用語は、ポリペプチドと結合し、および/またはポリペプチドにより修飾される分子を指し、たとえば抗体、受容体、抗原、酵素などであり得る。したがって、「標的分子」は、酵素基質または結合に関して評価される分子(たとえば、リガンド、エピトープ、抗原、マルチマー化パートナー、例えばホモもしくはヘテロダイマーパートナーなど、またはそれらの任意の組み合わせ)について言及する際に使用することができる。
【0100】
ポリペプチド活性は、単一のポリペプチドを発現する単一種の細胞の関連で、またはそれぞれが異なるポリペプチドもしくはポリペプチド変異体を発現する細胞のライブラリーの関連で、スクリーニングあるいは選択することができると理解される。したがって、本発明の方法は、インビトロタンパク質進化について用いることができる。インビトロタンパク質進化により、多数のタンパク質機能および特性を調査することが可能になり、典型的には2つの主なステップ、すなわち多様化および選択を含む。多様化は、ポリペプチドをコードする核酸の多様なライブラリーを生成する能力に依存する。選択は、所望の活性に関してライブラリーをスクリーニングし、活性を遺伝子型と結びつけることにより、たとえば、観察された活性の原因となる遺伝子型を含むライブラリーの数を特定することにより達成することができる。
【0101】
DNAライブラリーは、ポリペプチドをコード化するDNA挿入物(DNAフラグメント)を含む組換えベクターのコレクションである。DNA挿入物の起源は、ゲノム、DNA、合成または半合成であり得る。ポリペプチドは、所望の活性を有し得、たとえば関心対象のポリペプチドは、結合タンパク質、たとえば抗体、または酵素、たとえば、ポリメラーゼ、リガーゼ、制限酵素、トポイソメラーゼ、キナーゼ、ホスファターゼ、代謝性酵素、触媒酵素、または成長因子ホルモン、抗菌ペプチド、抗原、受容体、レポータータンパク質、免疫調節タンパク質、神経伝達物質、構造タンパク質、転写因子またはトランスポーターであり得る。1つの実施形態において、ポリペプチドは抗体または酵素である。したがって、本発明の方法を、所望の活性を有するポリペプチドの変異体に関してスクリーニングするために用いることができる。
【0102】
たとえば、結合タンパク質または酵素のDNAライブラリーのクローニングおよび構築は、当該技術分野で公知の方法を用いて実施することができる。たとえば、Lutz and Patrick (2004)は、ライブラリー多様性を生成させる方法およびタンパク質工学で使用するための遺伝子組換えの方策を概説している。ディスプレイされたポリペプチド変異体のスクリーニングのために、表面ディスプレイされたライブラリーに用いられる方策を、本発明の方法に採用し、適応させることができる(Becker et al., 2004; Kenrick et al., 2007; Miller et al., 2006; Daugherty et al., 2000)。
【0103】
核酸のライブラリーを複数の細菌細胞に導入し、その結果、細菌細胞のそれぞれにおいてライブラリーのメンバーが発現される。発現されることに加えて、ポリペプチドを、それらの機能または特性を評価するために、透過化された細菌細胞内で保持するか、または細胞壁に付着させる。ポリペプチド、たとえば抗体などの結合タンパク質の核酸ライブラリー、または酵素の核酸ライブラリーを、種々の方法により、例えば点突然変異などの突然変異、欠失、および挿入の導入によるか、または組換え事象により、生成させることができる。変異体のライブラリーを生成させる方法は、当該技術分野で公知であり、エラープローンPCR、DNA修復不全細菌におけるDNAの合成およびDNAの化学修飾を含む。組換えによりライブラリーを生成させる方法は、当該技術分野で公知であり、遺伝子シャッフリング、高組換え誘導細菌におけるDNAのアセンブリ、合成核酸ライブラリアセンブリなど、またはそれらの任意の組み合わせが挙げられる。このように、ポリペプチドをコード化するポリヌクレオチドのライブラリーを複数の細菌細胞に導入することができ、その結果、細菌細胞のそれぞれにおいてライブラリーの1つまたはメンバーが発現される。
【0104】
いくつかの実施形態において、ライブラリーは、ポリペプチドの2以上の変異体を含み、この場合、それぞれの変異体は、アミノ酸配列において若干の変化を有する独特のポリペプチドを含む。他の実施形態において、ライブラリーは、2以上の無関係な配列を含む。たとえば、酵素を阻害できる候補ポリペプチドを同定するために、ランダム配列または所定の配列のライブラリーを調べることができる。ライブラリーは、少なくとも2、少なくとも5、少なくとも10、少なくとも50、少なくとも100、少なくとも1000、少なくとも10,000、少なくとも100,000、少なくとも1,000,000、少なくとも107またはそれ以上のメンバーを有し得る。
【0105】
結合タンパク質ディスプレイ
1つの実施形態において、本発明の方法を、たとえば抗体などの結合タンパク質の発生に適用することができる。したがって、1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドは結合タンパク質であり、標的分子は、結合タンパク質が結合することができる任意の分子であり、所望の活性は、標的分子に対する結合、および/または結合の程度である。本発明の方法は、たとえば、結合タンパク質をコード化するポリヌクレオチドを含む細菌細胞を培養して、細胞においてタンパク質を産生することを含み得る。細胞を続いて透過化し、透過化された細胞を標的分子と接触させる。当該技術分野における任意の好適な方法を、ポリペプチドが標的分子と結合するかどうか、および/または標的分子と結合する程度を決定するために用いることができる。
【0106】
本発明の方法は、結合タンパク質ディスプレイライブラリのスクリーニングに特に適している。タンパク質の細胞外空間に対するターゲティングを絶対的に必要とするインビボ表面ディスプレイの他の方法と異なり、細胞膜はディスプレイタンパク質に対して提示された標識された標的との相互作用を防止するので、本発明の方法は、宿主細胞の細胞質において親和性タンパク質を発現でき、フォールディングすることができる。したがって、スクリーニングパラメータは、細菌の細胞質における親和性変異体タンパク質の高収率および生産的フォールディングを含み得る。
【0107】
さらに、細胞質タンパク質発現およびフォールディングが還元環境にある場合、本発明の方法を適用して、抗体の変異体、またはそれらの自然の形態でジスルフィド結合を有し還元環境において生産的フォールディングが可能である他のタンパク質を選択することができる。選択された変異体は、フォールディング安定性に関してドメイン内またはドメイン間ジスルフィド結合に依存しないので、更に安定であることが予想される。このアプローチは、中和または標識のいずれかのために標的の細胞内結合に使用できる抗体を開発するために応用される。
【0108】
本発明の方法は、したがって、当該技術分野で公知のスカフォールド、例えば1本鎖抗体(scFv)、ドメイン抗体、Fab、および非抗体スカフォールド、例えばリポカリン、FN3、ユビキチン、γ−B−クリスタリンなどをはじめとする種々の結合タンパク質のディスプレイおよび選択のためのプラットフォームとして用いることができる。
【0109】
酵素ディスプレイ
本発明の方法は、酵素および酵素ライブラリーのディスプレイのため、そして酵素特性の発生のために用いることができる。したがって、1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドは酵素であり、標的分子は酵素の基質であり、所望の活性は標的分子に対する結合および/または標的分子の酵素修飾である。当業者は、他の表面ディスプレイ技術を用いる酵素活性に関してのアッセイを開発するための方法を、本発明の方法に対するアッセイと同等に適用することができることを理解するであろう。
【0110】
本発明の方法はまた、透過化され、次いで水中油中水(w/o/w)エマルジョンとして懸濁された宿主細胞において発現された酵素ライブラリーの使用によく適している。Aharoni et al. (2005)は、パラキソナーゼの改善のためにFACSによりw/o/wエマルジョン中で表面ディスプレイされた酵素ライブラリをを使用することの有用性を示した。非透過性油膜中に封入することの利点は、拡散性基質および生成物を酵素活性およびコーディング核酸配列の近くに保持できることである。しかし、Aharoni et al. (2005)により記載されているスクリーンは、酵素が宿主細胞の外部でディスプレイされることを必要とする。本発明の方法を使用して、酵素ライブラリーの細胞内発現およびフォールディングを酵素機能における改善のために用いることができた。
【0111】
本発明の方法において、酵素を産生するために、酵素をコード化するポリヌクレオチドを含む細菌細胞を培養する。細菌細胞の透過化後に、細胞を酵素の基質と接触させ、公知方法を用いて、酵素が基質を修飾するかどうか、および/または基質の酵素修飾率を測定することができる。
【0112】
数例において、標的分子(たとえば酵素基質)を細菌細胞と結合させることが望ましい場合がある。当業者は、標的分子を透過化された細菌細胞の任意の成分に、直接的または間接的のいずれかで結合させることができることを理解するであろう。直接結合は、非限定的例として、標的分子を細菌細胞壁と結合させることにより達成することができる。標的分子の間接的結合は、標的分子を、所望の活性に関してスクリーニングされるポリペプチドと結合している第2ポリペプチドと結合してタンパク質複合体を形成することによって達成することができる。たとえば、標的分子を、透過化された細菌細胞の内部にタンパク質複合体を保持するために十分な分子サイズを有するポリペプチドと結合させることができるか、またはDNA結合タンパク質と、もしくは本発明の方法で用いられるような細菌細胞壁結合タンパク質と結合させることができる。標的分子を細菌細胞と結合させることにより、有利には、たとえばFACSまたは磁気ビーズ選択によるなどの技術を使用して、活性酵素を提示する細菌細胞の単離が可能になる。
【0113】
当業者は、標的分子を細菌細胞と結合させるために好適なカップリング化学を容易に決定することができるであろう。好適なカップリング化学としては、アクリダイトおよびマレイミドなどのチオールカップリング試薬でのシステイン標識、アミン標識、ならびにカルボキシル標識が挙げられ、これらはPierce Protein Research ProductsおよびInvitrogenをはじめとする供給元から市販されている。
【0114】
フローサイトメトリー分析
本発明の細胞ディスプレイ技術は、関心対象のポリペプチドの数千もの分子を一度に提示することができ、リボソーム/mRNAディスプレイまたはファージディスプレイなどの分子ディスプレイ技術と異なり、フローサイトメトリー技術を用いて、たとえば蛍光活性化細胞分類(FACS)機械を用いてスクリーニングすることができる。ライブラリーにおける陽性事象をとらえることができるだけでなく、酵素活性または親和性などのパラメーターを各陽性メンバーについて同時に定義することができ、これによりスクリーンのアウトプットを改善することができる。フローサイトメトリーを実施するための機器は当該技術分野で公知であり、FACS Star Plus、FACScanおよびFACSort(Becton Dickinson)、Epics C、ならびにMoFloが挙げられる。フローサイトメトリー技術は、一般的に、液体試料中の細胞の分離を含む。典型的には、FACSの目的は、1以上の特性、たとえば、標的分子の存在について細胞を分析することである。フローサイトメトリー分析を実施するための方法は、当該技術分野で周知である。たとえば、酵素活性を分析するためのFACSの使用法の概説は、Farinas(2006)により記載されている。
【0115】
本発明に関して、フローサイトメトリーは、連続して実施できる複数回のスクリーニングに有用である。細胞を初回の分類から単離することができ、直ちにフローサイトメーターに再導入し、再度スクリーニングして、スクリーンのストリンジェンシーを改善することができる。フローサイトメトリーは本質的に粒子分類技術であるので、細胞を培養する能力は必要ではない。無生育性細胞から核酸を回収するための技術は当該技術分野で周知であり、たとえば、PCRをはじめとするテンプレート依存性増幅技術を挙げることができる。
【0116】
所望の活性を有するポリペプチドを生成する細菌細胞が同定された後、ポリペプチドをコード化するポリヌクレオチドを含むDNAを、任意の好適な既知技術を用いて細菌細胞から単離することができる。したがって、通常の手順を使用して、ポリペプチドをコード化するDNAを単離し、配列決定することができる。必要に応じて、ポリヌクレオチドは、所望の活性に関してスクリーニングされる変異体の別のライブラリーを生成させるために、もう1ラウンドの多様化をおこなうことができる。このようにして、反復プロセスを使用して、ポリペプチドの所望の活性を最適化することが可能である。
【0117】
キット
本発明の方法の実施に必要な成分は、キットの形態で都合良く提供することができる。当業者には理解されるように、キット中の種々の成分は、個々の容器中もしくはアリコートで供給することができるか、または溶液成分を異なる組み合わせ、異なる濃度で組み合わせて、本発明の方法の最適な実施を達成することができる。使用するまで成分が安定な形態で維持されるように、キットのどの成分を組み合わせることができるかを決定することは、当業者の知識の範囲内である。
【0118】
本発明のキットは、典型的には、最低でも、第1ポリペプチドをコード化するポリヌクレオチドをベクター中に挿入するための部位、および第1ポリペプチドと結合して、透過化された細菌細胞の細胞壁の内部に保持されるかまたは細胞壁に付着したタンパク質複合体を形成する第2ポリペプチドをコード化するオープンリーディングフレームを含むベクターを含むであろう。好ましくは、キットはまた、細菌細胞を透過化するための薬剤も含む。1つの実施形態において、キットは、細菌細胞、好ましくはグラム陰性細菌細胞をさらに含む。他のさらなる成分がキットとともに含まれていてもよいか、または所望により他の成分を最終使用者が提供してもよい。
【実施例】
【0119】
実施例1 大腸菌を透過性にする界面活性剤のスクリーニング
大腸菌細胞を透過性にするであろう、界面活性剤を同定するため、イオン性(n−ドデシル−β−イミノジプロピオン酸;デシルトリメチルアンモニウムクロライド;ドデカノイルサルコシンナトリウム;アンゼルゲント3−10)、および非イオン性(ジメチルオクチルホスフィンオキシド[Apo8];ジメチルデシルホスフィンオキシド;n−オクチル−β−D−チオグルコピラノシド[8TGP];スクロースモノドデカノアート;メガ(Mega)10;トウィーン(Tween)80;トリトン(Triton)X100;トリトン(Triton)X114)の両方の多くの界面活性剤を、膜−不透過性色素、ゲルレッド(Gel Red)(Biotium社、カタログ番号41002)の取り込み、およびGFPの遊離の両方によってスクリーニングした。透過処理のテストを行った界面活性剤は、Anatraceから購入した。
ここに述べる実験に用いた大腸菌宿主株は、全てK12由来のArgentum細胞株(ΔmcrAΔ(mrr−hsdRMS−mcrBC)ΔendA lacZΔM15)(Alchemy Biosciences社)であった。しかしながら、本発明の方法は、B株由来のBL21(F−dcm ompT hsdS(rB−mB−)gal)、およびK12クローン株DH5α(F−endA1 glnV44 thi−1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ(lacZYA−argF)U169,hsdR17(rK−mK+),λ−)を用いてもテストを行い、同様な結果を得た。
GFPを、アラビノース誘導性の高コピー数ベクター(pAra1::GFP5)にクローン化した。発現は、新たに画線したコロニーを含むシャーレから重度に播種した培養液より行った。培養液は、37°Cで、アラビノースを最終濃度0.2%になるまで添加して発現を誘導するときに、OD600が約0.3となるまで培養した。誘導された培養液は、収穫まで25°Cで2時間振盪した。
1mLの誘導培養液を遠心分離して細胞を沈殿させ、300μLの0.5%界面活性剤含有LB中に懸濁して透過性にした後、25°Cで10分間培養した。透過性にした細胞は、沈澱させ、再懸濁し1xゲルレッド(Gel Red)を含む水に再懸濁し2分間置き、その後沈澱させ300μLのTBSで1度洗浄した。細胞は、300μLのTBSに懸濁し、蛍光顕微鏡検査のために、DABCO/グリセリン(0.0325gのDABCOを900μLのグリセリン+100μLのPBS中に溶解)を加えて処理した。
サンプルは、スライド式カメラ(SPOT RT 2.3.0ソフトウェアv4.6)を用いたオリンパスProvis AX70光学顕微鏡、またはLeica TCS SP2共焦点レーザ走査顕微鏡/Leica DM IRE2倒立顕微鏡(Leica共焦点ソフトウェアv2.0)のいずれかにより可視化した。
図1は、界面活性剤によるGFP発現大腸菌の透過処理の結果を示す。未処理細胞は緑色(GFP)であるが、透過性にされた細胞は、内部のGFPを失い、DNA結合性ゲルレッド(Gel Red)色素を取り込み赤色に染色される。また、ノニデット−40もいくらか透過性化を示すが、Apo8とMega10は、より高い割合の透過化された細胞を示す。これら2種の界面活性剤の0.5%ずつの混合物は、剤86と呼ばれ、別の界面活性剤n-オクチル−β−D-チオグルコピラノシド(8TGP)と同様に、ほとんど完全に透過処理することを示した。Mega10、Apo8および8TGPは、すべて非イオン性界面活性剤で、イオン性界面活性剤と比べ、タンパク質の折りたたみや機能に対して、破壊の程度が低い。
細胞壁は、透過処理によっても無傷であったので、上述した界面活性剤での透過処理による上澄みの可溶性タンパク質の抽出物をSDS−PAGEにより分析した。透過処理した細胞に、ニワトリ卵白リゾチーム(Boehringer Mannheim社;837 059)を最終濃度2mg/mLまで、透過処理している細胞のサンプルに加え、細胞壁を除去し、全細胞タンパク質を遊離させた。β−メルカプトエタノールを含むSDS−PAGEローディング色素を、95°Cで2分間変性したサンプルに加えた。20μLのサンプルを9%SDS−PAGEにロードし、クーマシー・ブリリアント・ブルー/メタノール/酢酸で染色/固定した。
図2は、可溶性タンパク質の遊離は、顕微鏡検査で見られたGFPの遊離およびGel Redの取り込みと直接的に関連していることを示す。無傷な細胞壁を有する細胞からのタンパク質の遊離と、リゾチームを用いて細胞壁を除去した細胞からのそれとの間には、明白に差があったが、細胞壁に被包された細胞が遊離する可溶性タンパク質は、大きさが約120kDまでであった。これは、球状タンパク質が、グラム陰性真正細菌の細胞壁を構成するペプチドグリカン格子の細孔を通って、細胞から出ることができなくなる限界寸法であると思われる。
【0120】
実施例2. 宿主DNAを保持する透過処理溶液のスクリーニング
本発明の方法を、改良された特性のタンパク質変異体を求めて遺伝子ライブラリーをスクリーニングするのに使用するなら、発現されるタンパク質と、それをコードする核酸との間に関連がなければならない。膜の透過処理工程で、DNAが、細胞壁を通って失われるのを防ぐ障壁が除去されるので、宿主DNAの損失を減少し、あるいは防止しうる、透過処理の条件を調べた。
細胞の透過処理を、0.5%8TGPを用い異なる培地中で行い、DNAの損失を、DNA結合色素、Gel Redを用い、蛍光顕微鏡検査法により調べた。
【0121】
テストした透過処理培地(全培地に0.5%8TGPを含む)の組成
LB培地(10gトリプトン、5g酵母抽出物、10g/LのNaCl)
LB(塩)培地(10gトリプトン、5g/Lの酵母抽出物)
50mM−Tris、pH7.5
50mM−HEPES、pH7.0
170mM−NaCl
250mM−NaCl
25mM−Tris、pH7.5+1.5%PEG6000(w/v)
50mM−Tris、pH7.5+3%PEG6000(w/v)
50mM−Tris、pH7.5+170mM−NaCl
50mM−Tris、pH7.5+250mM−NaCl
好適な透過処理培地は、LB細菌培地と確認された。したがって、以降、透過処理は、0.5%8TGPを含むLBまたは剤86を含むLB(0.5%Mega10および0.5%Apo8を含むLB)のいずれかを用いて行った。
【0122】
実施例3. タンパク質のテトラマー骨格への融合
例1に記述した実験で見られたとおり、大きさが約120kDより大きいタンパク質は、細胞壁により、透過性にした大腸菌細胞の内部に保持された。したがって、120kDより小さい目的タンパク質も、タンパク質パ−トナーと融合することで合計寸法が120kDを超えるようにできれば、細胞壁カプセル中に保持されるだろうと考えられた。
このため、融合パートナーとして用いるため、大腸菌から6種の異なる四量体タンパク質をクローン化した。すなわち、β−gal、BetB、G5K、GshB、RhnA、およびYdcWで、それぞれのモノマー寸法は116kD、52kD、39kD、35kD、47kDおよび50kDであった。
アラビノース誘導性高コピー数ベクターを、四量体発現のために構築した。蛍光性基質と共有結合で結合する20kD領域であるSNAPタグ(NEB社/Covalys)が、テトラマー遺伝子の上流にクローン化され、発現レポーターとして使用された。精製と検知を容易にするため、6xHisエピトープも、融合タンパク質のN末端に挿入された。
前記アラビノースベクターpAra3::His6::SNAPの配列が、SEQ ID NO:1(配列番号)として示される。
融合タンパク質の発現は、0.2%アラビノースを加えて誘導し、培養液を25°Cで2時間培養した。
本発明の方法により、蛋白質ディスプレイのために細胞を透過性にするプロトコルは、以下のとおりであった:
1. 遠心分離により細胞1mLを沈澱させる。
2. 300μLの0.5%8TGP/LBに細胞を再懸濁する。
3. 25°Cで10分間培養する。
4. 遠心分離により細胞を沈澱させる。
5. 200μLのTBSまたはLBに細胞を再懸濁する。
【0123】
SNAP発現レポーター領域を、膜不透過性SNAP色素(Covalys、New England Biolabs社)で標識付けするプロトコルは、以下のとおりであった:
1. 20nmolのBG−488(緑色色素)またはBG−547(赤色色素)を、300μLのDMSOに200x原液として溶解する。
2. 1μLの200x原液を200μLの透過性にした細胞を懸濁したTBSまたはLBに加える。
3. 25°Cで15分間培養する。
4. 遠心分離により沈澱させ、また300μLのTBSに再懸濁させて、細胞を2度洗浄する。
【0124】
四量体融合タンパク質が、透過性にした細胞カプセル中に保持されるのを蛍光顕微鏡検査法により観察するためのプロトコルは、以下のとおりであった:
1. 20μLの細胞懸濁液を顕微鏡用スライドガラス上に滴下し、カバーガラスで覆い、端部をマニキュア液でシールする(湿式プレパラート);あるいは細胞の液滴をほぼ乾燥させて、その上に20μLのDABCO/グリセリンを滴下し、カバーガラスで覆い、端部をマニキュア液でシールする(乾式プレパラート)。
2. オリンパスまたはLeica蛍光顕微鏡のいずれかを用いて可視化する。
【0125】
完全長融合タンパク質の発現は、SDS−PAGEゲルに掛けてから、タンパク質抽出物のウェスタンブロット法でα−His6抗体をプローブに用い確認した。全ての四量体コンストラクトは、大腸菌中で検知可能なレベルで発現した(図3A)。
大腸菌内で発現した四量体融合タンパク質の蛍光顕微鏡検査によれば、β−galおよびG5Kは、顕著な封入体を含み、恐らく前記融合タンパク質の折りたたみが困難であるために、蛍光レベルが低いことが分かった。しかしながら、図4によれば、SNAP蛍光により判定されるとおり、前記融合タンパク質の発現は、GshBでは良好、RhnA、BetBおよびYdcWでは、極めて良好であった。透過性にした宿主細胞中の融合タンパク質の分布は、明視野顕微鏡検査および蛍光の両方から明らかなように複数のフォーカスを有し、均一でないことが注目された。しかし、蛍光性SNAP基質は、ミスフォールドされた領域には結合されていないであろうから、かつ信号が非常に強いことから、これらの物体は恐らく折り畳まれたタンパク質の凝集体であって、大腸菌中で過剰発現しているタンパク質においてしばしばみられる、折り畳まれていないタンパク質の封入体ではないと考えられる。
SNAP::テトラマー融合体は、His6-N末端エピトープをまた有していた。抗体といった大きな分子が、大腸菌細胞壁の格子構造を通り抜けられるかを調べるため、透過性にした細胞にSNAP::テトラマー融合体を検出するαHis抗体をプローブとして設けた。
1. 前記His6::SNAP::BetB骨格融合体の発現および透過処理を上述のように実施した。
2. BG−547 SNAPリガンドによる標識化を上述のように実施した。
3. 200μLの透過性にしたSNAP−標識化細胞をLB中で3回洗浄し、ポリエチレンイミン(PEI)コートしたカバーガラス上に沈降させた。過剰な細胞培地を、吸引して除去し、前記スライドガラスを風乾させた。
4. 細胞を、ブロッキング緩衝液(1%BSA、1%冷水魚ゼラチン(Sigma社、G7765)、アジドの0.02%PBS−Tween20溶液)中で、1時間ブロックした。
5. 細胞を、ブロッキング緩衝液で1:10に希釈したαHis一次抗体(Abcam社、AB9136−100)中で、一晩25°Cで培養した。
6. 細胞を3回PBS−Tween20中で洗浄した(各回10分間)。
7. 細胞を、ブロッキング緩衝液で1:2,000に希釈した二次抗体(Molecular Probes、A11015)中、室温1時間培養した。
8. 細胞を3回PBS−Tween20中で洗浄した。
9. DABCO/グリセリンを封入剤に用い、共焦点オリンパス顕微鏡で観察した。
【0126】
図5は、αHis抗体が細胞壁カプセルの内部でSNAP蛍光性リガンドと共存しており、細胞壁の細孔が、比較的大きなタンパク質が、カプセル内部空間中に拡散を許すのに十分な大きさであることを表わしている。このように、非常に大きなタンパク質リガンドも、本発明の方法により、細胞質中で発現する親和性タンパク質のための親和性基質として使用できる。
細胞下物体の生成が変化するか否かを見るためにSNAP融合パートナーおよび発現レポーターをHALOタンパク質(Promega社)と比較した。前記HALOタンパク質は、SNAPと同様に膜不透過性蛍光性基質(Alexa fluor 488;G1001、Promega社)と共有結合で結合する。HALOレポーター遺伝子は、四量体発現コンストラクト中のSNAP遺伝子の位置にインフレームに直接クローン化した。HALO::四量体骨格タンパク質の発現を、SNAP変異体と比較した。透過性にしたHALO細胞の標識付けは、実質的にSNAPに対して記述したように、またメーカーの説明書に従い実施した。図6は、HALO::テトラマーおよびSNAP::テトラマーの発現パターンは、同様であったが、例外的にHALO::RhnA融合タンパク質は、部分的にSNAP::RhnA融合体より溶解性で、複数の蛍光フォーカスを有する細胞がより少数であったことを示す。
したがって、タンパク質を四量体骨格(ここでは、SNAPまたはHALO)への融合体として発現させ、つづいて大腸菌宿主細胞を適当な界面活性剤で透過性にすると、目的のタンパク質が、細胞壁の内側に保持できる。
【0127】
実施例4. 細胞骨格としてのDNA結合タンパク質
表現型を遺伝子型に結合するには、宿主細胞は、透過処理の後、また機能スクリーニングを通して、少なくともいくらかのエピソームDNAを保持しなければならない。宿主ゲノムのDNAならびにプラスミドDNAを保持する透過処理条件を見出したことで、発現した目的のタンパク質の保持骨格としてDNAが使用できると結論付けた。
このため、小型(80aa)の高親和性らせん−ヘアピン−らせんDNA結合タンパク質(DBP)を、Neisseria gonorrhoeae ComE遺伝子(ChenおよびGotschlich、2001)からクローン化し、これをアラビノース誘導性コンストラクト(pAra3::GFP::DBP;seq2)中で、GFPのC末端に融合した。
アラビノース誘導による発現を例1に記述したように実施した。細胞を透過性にし、例1および3に記述したように蛍光顕微鏡検査のために調製した。
図7は、GFP::DBP融合体(緑色)が透過性にした細胞中に保持され、DNA結合色素、Gel Red(赤色)と共存したことを示す。
したがって、タンパク質を、高親和性、非特異的DNA結合タンパク質への融合体として発現させ、つづいて大腸菌宿主細胞を適当な界面活性剤で透過性にすると、目的のタンパク質が、細胞カプセルの内側に保持できる。
【0128】
実施例5. 透過性にした細胞中へのDNA保持
DNA、ゲノムおよびエピソーム両方のプラスミドの、透過性にした細胞カプセル中への保持を実証するため、蛍光顕微鏡検査およびプラスミドDNA抽出のためにGFP5::DBPおよびHis6::eGFPを発現する細胞を調製した。
誘導後、細胞を透過性にし、つづいて凍結するか、TBS中で37°Cにおいて一晩振盪した。次の日、全サンプルを、蛍光顕微鏡検査でGFP、およびDNA結合色素Gel RedでカプセルDNA含量を見るために、または、プラスミドDNA調製のために処理した。
蛍光顕微鏡検査を例3に対して記述したように実施した。図8は、宿主細胞DNA(赤色)およびGFP5::DBP(緑色)双方とも、細胞カプセル中に、透過処理直後も、一晩37°Cで培養しても目立った減少もなく、保持されたことを示す。His6::GFP タンパク質は、透過処理直後に失われたが、宿主細胞DNA(赤色)は、透過処理直後も、一晩後も、同じく目立った減少もなく、保持された。
宿主ゲノムだけでなくプラスミドDNAも透過性にした細胞内に保持されることを確認するため、同じように調製されたサンプルに対してプラスミド少量調製を行った。
1mLの界面活性剤処理または無処理細胞から、プラスミド少量調製アルカリ溶解プロトコルによりプラスミドDNAを調製した。界面活性剤抽出による上澄み中に遊離されたプラスミドDNAを、Perfectprep Gel Cleanup(Eppendorf社、955152051)コレムおよび溶液を、メーカーのプロトコルに従って用いて、抽出した。
各サンプルの全量を1%アガロースゲルにロードし、富士フィルム社LAS−3000インテリジェントダークボックス(Intelligent Darkbox)でイメージリーダー(Image Reader)LAS−3000ソフトウェアおよびMulti Gauge v3.0ソフトウェアを使用して画像化した。
図9は、両細胞株からのプラスミドDNAをサンプルとした、TAE緩衝液を用いた臭化エチジウム染色1%アガロースゲルを示す。
図9のレーン1は、無処理細胞中の全プラスミドDNAである。レーン2は、透過処理工程の上澄み、レーン3は、透過処理後に細胞カプセル中に保持されたプラスミドである。図8で観察された可溶性His6::GFP タンパク質の完全な消失に拘わらず、透過処理による上澄み中へのプラスミド遊離が殆どないことが観測された。したがって、プラスミドDNAは、ほとんど完全に細胞壁により保持され、本発明の方法において改良されたタンパク質変異体の選別のために、遺伝子型を表現型に結合するために使用できる。
顕微鏡検査のデータを確認するように、TBSに懸濁した透過性にした細胞の37°Cでの一晩の培養後も、この培養でのプラスミドDNAの消失は全く見られなかった(レーン5)。
【0129】
実施例6. ペプチドグリカン結合骨格
膜透過処理後も保持されるいま一つの細胞構造は、ペプチドグリカン(PG)の格子状ポリマーでなる細胞壁である。
PGを非共有結合で結合するために、大腸菌内で良好に発現し、細胞壁に高親和性(K=3x107M−1)をもって結合することが既に知られている(Briersら、2009)、シュードモナスφKZファージ(KzPG)からの70aaPG結合領域をクローン化した。親和性タンパク質の選別用に、PGに対するKzPG−結合領域の親和性より、より高い親和性を標的に対して有する変異体を見つけるため、骨格結合タンパク質の親和性を増す必要がある。骨格結合部分の親和性を増すため、ComE DNA結合領域(DBD)およびPG結合領域の両方を同一の融合タンパク質内で結合した。したがって、両骨格(PGまたはDNA)からの融合タンパク質の最終解離定数は、それぞれの速度定数の倍数の近似値となる。
このため、発現ベクターpAra3::His6::KzPG::SNAP::DBP(SEQ ID NO:2(配列番号))を構築した。例1に記述したように発現を誘導し、細胞を例3に記述したように蛍光顕微鏡検査のために調製した。融合タンパク質の発現および分布は、例3に記述したようにSNAP標識化により観察した。
蛍光は、細胞の辺縁で、細胞壁の領域において認められ、より軽度には、カプセルの細胞壁で囲まれる空間内の広汎な領域において認められた。
本発明のさらなる態様では、目的のタンパク質を共有結合で、透過処理前の細胞骨格に付着させる。これを実現するため、周辺質内にコイルドコイル三量体を形成する、大腸菌に豊富なタンパク質LPPへのタンパク質融合体を用いた。その天然型においては、1端が脂質化により外膜と連結し、他端はC末端リジンにより共有結合で細胞壁に結合している。
OmpF周辺質標的シグナル配列をSNAP発現レポーターに融合し、さらにN末端シグナル配列を欠く57aa大腸菌LPP配列、外膜付着に必要なシステインが続く発現コンストラクトを構築した。発現ベクター、pAra3::OmpF::SNAP::LPP(SEQ ID NO:3(配列番号))は、例1に記述されるようにアラビノースで誘導され、細胞を蛍光顕微鏡検査のため、例3に記述されるように、調製した。融合タンパク質の発現および分布は、例3に記述したようにSNAP標識化により観察した。
図10が示すように、LPP融合タンパク質の分布は、細胞壁の表面にわたって不均一で、強い蛍光の場所と、蛍光のない場所があった。しかし、ほとんどのケースで細胞の極が、標識化された。
【0130】
実施例7. 四量体タンパク質骨格を用いたαGFP親和性タンパク質のディスプレイ
親和性タンパク質に適用する本発明の方法を実証するため、eGFPに対して免疫したラマから生成した単一領域抗体を細胞骨格ベクター中にクローン化した。ここに、αGFP抗体に対する特許出願に(WO2007/068313)挙げられている2配列の内、R35変異体のみが機能することを指摘したい(αGFP−R35;タンパク質データベースID 3K1K)。したがって、この配列を全実験において使用した。
前記αGFP−R35遺伝子を、pAra3::HALO::FLAG::RhnA四量体骨格のN末端融合体として、pAra3::αGFP(R35)::HALO::FLAG::RhnAベクター(SEQ ID NO:4(配列番号))を製作するためクローン化した。
前記抗体の標的基質としてHis6::eGFP融合タンパク質を製造するため、pAra3::His6::eGFPベクターも、構築した。例1に記述されるように、His6::eGFPタンパク質を誘導した。可溶性タンパク質が、0.5%8TGPを用いて細胞から遊離され、Ni−NTAアガロース樹脂(Qiagen社;30230)を用いてIMACにより精製された。His6::eGFPは、Ni−NTA樹脂からNTTW緩衝液+イミダゾール(500mM NaCl、50mM Tris−HCl、pH7.5、0.1%Tween20+200mMイミダゾール)に溶出された。
抗体::四量体融合タンパク質の発現および宿主細胞の透過処理は、例1および3に記述されるように実施した。
透過性にした細胞カプセル内で、αGFPをeGFPに結合するため、カプセル沈殿物を300μLのeGFP中に懸濁し、25°Cで20分間平衡化させてから、カプセルを遠心分離で沈澱させ、300μLのTBS中で1度洗浄した後、TBS中に再懸濁させた。αGFP/eGFPカプセルに対する蛍光顕微鏡検査を、例3に記述されるように、実施した。
図11は、HALOリガンド標識化した図5で観察されるフォーカスと関係があるかもしれない、より強く染色された複数のフォーカスが生じているが、透過性にしたαGFP::HALO::RhnA融合タンパク質を発現しているカプセルが、細胞の全域でeGFPに結合したのを示している。
したがって、ラマαGFP抗体の機能は細胞質内で発現し、さらに界面活性剤透過処理後も、カプセル内に保持される。
例3で記述され、また図5で観察されたαHis抗体の標識化は、すでに約150kDと大きめなタンパク質が、透過性にした細胞壁を拡散して通り、カプセルの内部に入ることができることを実証した。しかし、未変性の抗体は、柔軟なヒンジ領域で分けられた、3つのほぼ同じ大きさの領域を有する変則的な形状のタンパク質である。したがって、これらのタンパク質の示す有効半径は、はるかに小さな球状タンパク質に相応すると思われる。しかし、βバレル構造と約27kDの分子の大きさを有するGFPは、半径がその大きさに比例する、対称性のタンパク質であり、内部のαGFP抗体と結合するために、透過性にしたカプセルの細胞壁を通過することができた。
したがって、本発明の方法は、大腸菌細胞質中で親和性タンパク質を発現して、少なくとも30kDの対称性の標的に結合する親和性ライブラリーのディスプレイに使用できる。
【0131】
実施例8. PG−およびDNA結合タンパク質骨格を用いるαGFP親和性タンパク質のディスプレイ
本発明の方法は、PG−およびDNA結合領域と融合したαGFPラクダ抗体を用いてさらに実証された。
抗体::KzPG::SNAP::DBP融合タンパク質の発現、宿主細胞の透過処理、およびHis6::eGFPによる標識付けは、例6に記述されるように実施した。
αGFP融合タンパク質によるeGFPの結合の画像化には、湿式および乾式プレパラートの両方が用いられた。図12は、GFP蛍光において、両方の画像化法で有意な差があったことを示している。乾式プレーパラート(DABCO/グリセリン)にされた細胞は、ほとんどが内部蛍光で、明視野とeGFP標識の間の混合があり、細胞壁の周囲の領域は内部空間より光が強くない(図12B)。しかしながら、TBS中で直接プレパラートにされた細胞は、細胞壁に結合したeGFPと思われる強い蛍光の外部境界のはっきりした形状と、より弱い内部信号を有していた(図12A)。理論に縛られることなく、推測するならば、DABCO/グリセリン溶媒環境は、粘度が高くまた水性でないので、KzPG領域とペプチドグリカン細胞壁の間の相互作用を抑止したが、αGFPのeGFPとの、あるいはDBPのDNAとの結合は抑止しなかった。
しかしながら、親和性タンパク質または酵素のスクリーニング操作は、通常は水系環境で行われるので、親和性融合タンパク質の分布は、図12Aに観測された細胞壁結合湿式プレパラートに近いであろう。
【0132】
実施例9. 細胞壁への共有結合性付着によるαGFP親和性タンパク質のディスプレイ
本発明の方法は、αGFP抗体の共有結合による細胞壁への結合によりさらに実証された。
αGFP抗体は、アラビノース誘導性融合体として、OmpFシグナル配列の下流、かつSNAPおよびLPP配列の上流にクローン化された。
アラビノースで発現を誘導した後、OmpFシグナル配列は、新生タンパク質を、内部細胞膜を通して周辺質に送り、これが膜細孔を通るときに分裂される。
周辺質において、LPP領域は、2つの別のパートナー、野生型LPPまたは他のαGFP融合タンパク質とともに、三量体コイルドコイルを形成すると思われる。LPP領域のC末端の残基はリジンで、共有結合により、εアミン基を介して、恐らくはYbiS L,D−トランスペプチダーゼにより(Magnetら、2007)、大腸菌細胞壁に結合する。
OmpF::αGFP::SNAP::LPP融合タンパク質の発現、細胞透過処理、およびeGFP標識付けは、例8に記述のように実施した。
図13は、eGFPは、細胞壁の周りに不均一に、しかし強力に結合したことを示す(図13B)。eGFPは、OmpF::SNAP::LPP融合体をαGFP領域無しに発現している細胞に結合されなかった(図13A)。
OmpF::SNAP::LPP融合体の細胞壁への共有結合性付着は、最初に、前記融合タンパク質を発現している透過性にした細胞をSNAPリガンドで標識化し、その後標識化細胞カプセルのサンプルを95°Cで5分間加熱して実証した。図14は、細胞壁を標識化するSNAPリガンドからの蛍光は、加熱処理されたサンプルと、加熱されていないコントロールとの間で差が生じなかったことを示す。Gel Red染色でも、加熱処理されたサンプルにおいても、ゲノムDNAは、細胞中に保持され続けることを実証した。
【0133】
実施例10. 外膜透過処理実験
本発明のさらなる態様においては、外膜は、リガンド標的、例えば酵素基質、あるいはポリペプチド等に対して選択的に透過性にしてもよいが、このとき、スクリーンされた前記ポリペプチドは、細胞壁の内部に、あるいはこれに付着して保持される。
外膜を選択的に透過性にする条件を見出すため、一連の界面活性剤および緩衝液を選別した。大型リガンド(eGFP)および小型リガンド(Gel Red)の両方を用いて、外/内膜の透過処理で大きな、または小さな膜細孔が生成したかを調べた。
アラビノース誘導性OmpF::αGFP::SNAP::LPP(細胞壁に付着)またはαGFP::HALO::FLAG::RhnA(細胞質内性)を発現する大腸菌株を培養し、例1に記述されるように誘導した。
誘導された培養液1mLを、50mMのTris(pH8)で1回洗浄し、25mM Tris+1mM EDTA(pH8)または25mM Tris+2mM Ca2+(pH8)のいずれかの中に0.2〜0.4%の界面活性剤をいれた透過処理緩衝液変種中に懸濁し、25℃で10分間培養した。
透過性にした細胞は、適当な緩衝液で1回洗浄してから、Gel Red(1x水中)で染色し、TBSで洗浄した。これを、精製したHis6::eGFPとともに25℃で1時間培養し、遠心分離で沈殿させ、TBS中に再懸濁させ、湿式プレパラートとして蛍光顕微鏡検査法により観察した。
図15及び16は、Tris/Ca2+またはTris/EDTA緩衝液中の0.2%のApo8(A)またはTween20(B)が、外膜を選択的に透過性にし、大きなリガンド(eGFP)に、外膜を通させるが、内膜は通させないことを実証している。より小さな、膜不透過性のDNA結合リガンドGel Redは、大部分のサンプルで、細胞質まで部分的に透過性であって、いくつかの細胞において内膜にある程度の細孔形成が起きたことを示している。しかしながら、0.5%の8TGPまたは剤86である界面活性剤で処理され、外膜、内膜ともにeGFPに対して完全に透過性であるサンプルに比べて、Gel Red結合の程度は著しく減少した。
【0134】
実施例11. 蛍光選別および被包性ディスプレイの解析
細胞ディスプレイプラットフォームとして、リガンド結合クローンを識別するのに、本発明の方法は、蛍光標示式細胞選別(FACS)に非常に向いている。透過性にした大腸菌細胞のFACSによる選別での安定性を試験するため、3集団:i)eGFP、ii)αGFP::KzPG::SNAP::DBP、およびiii)His6::SNAP::BetBを誘導して発現させた。
eGFP発現細胞は、透過性にせず、無傷の大腸菌細胞での蛍光のポジティブコントロールとした。αGFP::KzPG::SNAP::DBP発現細胞は、本発明の方法により透過性にし、SNAP BG−488リガンド(緑色)で標識付けした。His6::SNAP::BetB発現細胞は、本発明の方法により透過性にし、SNAP BG−547リガンド(赤色)で標識付けした。
細胞はPBS中に懸濁し、混合集団の選別のために、ほぼ同数を混合し、あるいは信号の較正のため、別々に選別した。細胞選別は、Becton Dickson Influx FACSにより実施した。データ解析は、FlowJoソフトウェアにより実施した。大腸菌選別のためのパラメーターは、操作者が決定した。
図17は、蛍光により3集団が識別できたことを示している。選別された集団の再分析は、前記選別がそれぞれの比較的純粋な集団をもたらすことを示した。グラフの低蛍光領域にある信号は、信号の内部雑音であることが後で判明したので、後に操作者により装置の修正により除去した。
【0135】
実施例12. 固体サポート結合のためのスペーサー領域選定
αGFP::KzPG::SNAP::DBP融合タンパク質を発現している細胞を、8TGP培地を用いて透過性にし、中間体His6−タグ化eGFPを経由してHisPur Co2+セファロース・ビーズ(Thermo Scientific社)に結合した。細胞またはビーズは、最初に過剰のHis6−eGFPとともに培養し、TBS中で洗浄し、一緒に25℃で30分間培養した。結合しなかった細胞は、ビーズから洗い流し、つぎに蛍光顕微鏡検査法でビーズの結合の程度を評価した。
最初は、αGFP::KzPG::SNAP::DBP融合タンパク質のセファロース・ビーズへの結合は検知されなかった。αGFP結合領域が、細胞壁に近すぎて、セファロース樹脂上のコバルト錯化eGFPに到達できないためと理論を立てた。このため、ランダムコドンによる12残基ペプチド・スペーサー領域を、αGFP結合領域およびkzPGペプチドグリカン結合領域の間にクローン化した(GGT ACC gcy gcy gkk wtb gck wtb gkk gkk gck gkk gcy gcy GGT CTG(SEQ ID NO:5(配列番号)))。
スペーサー変異体の小型ライブラリー(約2,000要素)を発現し、上述のようにCo2+セファロースに結合した。前記ライブラリーの一部は、ビーズに結合することが観測された。これらのクローンはPCRで増幅し、再クローン化し、十余のクローンの個別の結合性を試験し、配列を調べた。種々のペプチド・スペーサーが、タンパク分解に耐性があり(αGFPを高度に融合タンパク質中に保持する)、また、図18に示されているように、界面活性剤処理細胞をセファロース・ビーズに結合できることが判明した。サポート結合に機能することが判明したスペーサー配列を表1に挙げる。
【表1】

スペーサー配列の一つRL6を選んで、後の結合の検討に用いた。固体サポート・マトリックスへの強い結合に寄与する他の要素を調べた。細胞のマトリックスとの培養時間、および結合溶液の塩(NaCl)の濃度は、両方とも結合に対してポジティブな効果を有することが分かった。培養時間30分と、NaCl濃度の範囲約200mM〜500mMが、効果的であることが判明したが、300mMが最適と考えられる。結合は、Tris、リン酸塩、MOPSの300mMの塩含有緩衝溶液を含む一連の緩衝液中で効率的である。
αGFP::RL6::KzPG::SNAP::DBP融合タンパク質を発現している細胞を、ビオチン化eGFPを介してストレプトアビジン磁性ナノ粒子(MagneSphere、Roche Diagnostics社)に結合する条件は、図19に明示されるセファロース・ビーズ結合に対して求められた範囲内にあることも判明した。
12残基スペーサーに加えて、タンパク質領域をも、スペーサー領域として用いることが考えられた。ヒトのタイチン遺伝子(I27)からの小型で安定な高度に発現した27番目のイムノグロブリン領域をRL6スペーサーの上流にクローン化した。この領域も、N末端のαGFP領域の高度かつ安定的な発現と、同時に非常に良好な固定マトリックス結合をも可能にすることが分かった(図19)。
【0136】
実施例13. 被包性ディスプレイ用のマウスscFvライブラリーの構築
一本鎖抗体(scFv)ライブラリーの細胞内ディスプレイ用の最後の領域構造は、scFv::I27::RL6::KzPG::SNAP::DBPであった。scFv領域を除いた、融合タンパク質のタンパク質およびDNAの配列は、SEQ ID NO:13(配列番号)およびSEQ ID NO:14(配列番号)に与えられている。このタンパク質融合体は、N末端にscFv、つづいて2つのスペーサー領域であるI27およびRL6、つぎにペプチドグリカン結合領域のKzPG、SNAPレポーター領域、最後にDNA結合領域(DBP)を有する。
ランダムプライマーを用いたcDNAを、酵素Superscript III(Invitrogen社)を用いて、マウス脾臓全RNAから製造した。このcDNAから、シェーファー(Schaefer)ら(2010)が記述したように、Vent DNAポリメラーゼ(New England Biolabs社)、およびマウス抗体ファミリー配列用変性オリゴヌクレオチド・プライマーを用いて、scFv軽鎖(VL)および重鎖(VH)可変領域を増幅した。ライブラリー・クローン化に用いたオリゴヌクレオチド・プライマーは、Schaeferらの記述したものと、Bsm BIを介して我々のライブラリー骨格ベクター(SEQ ID NO:15(配列番号))内にクローン化するのに適当な末端を有する点で相違している。VLおよびVH領域は、オーバーラップ伸長PCRを用いて加えられる。最終的scFvバンドは、合計60回のPCR増幅サイクル(1回目30回、2回目30回)にかけられた。
ライブラリー・クローン化のために、ディスプレイ・コンストラクト900ngを、BsmBIで切断し、メーカーの説明書にしたがいSureclean(Bioline社)を用い沈澱させ、T4 DNAリガーゼを用い、同様に処理されたscFv生成物400ngに連結した。リガーゼを65°C、10分間の培養で失活させ、連結体をE. Coli Argentum株(Alchemy Biosciences社)中に電気穿孔で入れた。電気穿孔された細胞は、SOC培地中に回収し、37°Cで1時間培養し、プール後、75μg/mLアンピシリンを含む20x150mmLB寒天シャーレに拡げた。このシャーレを一晩30°Cで培養した。ライブラリーの大きさは4×105独立クローン程度と推定された。20クローンの内、20クローンに予想された大きさの挿入断片が含まれていることが判明した。
【0137】
実施例14. 被包性ディスプレイ・マウスscFvライブラリーのスクリーニング
ファージディスプレイ・ライブラリーから単離される一本鎖抗体は、多くの場合大腸菌中での発現が困難で、周辺質での発現レベルが低く、あるいはβシートとIg群の間にジスルフィド結合形成を欠くために、細胞質中で完全に不溶性である。大腸菌細胞質中で可溶性のマウスscFv骨格を選別するのに被包性ディスプレイを用いることができるか否かを決定するには、scFvの溶解性が融合タンパク質の性質と相関しているか否かを決定する必要があった。
有用な可溶性scFvは、凝集のレベルが低く、発現レベルが少なくとも中程度であることが予想された。これは、透過性にした細胞中のKzPG領域を、細胞壁に結合させる(したがって、細胞中の封入体に局在しない)、かつSNAPレポーター領域の少なくとも中程度の発現を示すクローンとして、視覚的に判断できた。
これらのパラメーターを選別するため、単一のコロニーを採取し、すでに例1に記述したようにアラビノースを用いて融合タンパク質の発現を誘導した。透過処理後、これらをSNAPリガンドで標識付けし蛍光顕微鏡検査法をもちいて観察した。前記ライブラリー・クローンを、発現、および、図20に例が示されるような、SNAPレポーターの細胞での分布に基づき、4類に分類した。
1. SNAPの発現なし。
2. 凝集した封入体中でのSNAPの中〜高程度の発現(図20、左図)
3. 細胞壁に局在してのSNAPの弱程度の発現(図20、中央図)
4. 細胞壁に局在してのSNAPの高程度の発現(図20、右図)
【0138】
発現と溶解性の両方が高いクローンのみを、さらに分析した。しかし前記SNAPレポーターの発現の弱かったのは、大腸菌発現に最適化されなかったタンパク質の非効率な発現のためでありうるので、こうしたクローンのうちのある部分は、コドンの使用が適性化されたならば、可溶性細胞質ライブラリー・ディスプレイに対して非常に適していると判明することがあり得る。
SNAPレポーターの発現レベルが高く、透過性にした細胞の細胞壁の周りに均一に分布したクローンの配列を調べ、残りの融合タンパク質に対して正しい翻訳枠にあるscFv挿入断片の存在を確認した。分析した21クローンの全てで、scFv挿入断片は、全長で、正しい長さのグリシン/セリン・リンカー領域を有し、全融合タンパク質の翻訳のために正しい読み枠中にあることが判明した。このことは、本発明のスクリーニング方法は、大腸菌細胞の細胞質中で、溶解状態で発現したマウスscFv遺伝子を正しく識別していることを示唆した。前記ライブラリーから単離されたscFvタンパク質が、大腸菌細胞質中で可溶であることを確認するため、これを、ライブラリー・コンストラクトから、介在性のスペーサー領域I27−RL6またはRL6のいずれかを含むC末端のFLAGエピトープを有するアラビノース誘導性発現ベクターにシャトルした。
アラビノースによるタンパク質発現の誘導後、可溶性のscFv::I27::RL6::FLAGまたはscFv::RL6::FLAG融合タンパク質を、0.5%8TGPで抽出した。不溶性の細胞物質は沈澱させ、β-メルカプトエタノールを含むSDS−PAGEローディング緩衝液中に、サンプルに超音波をかけ95℃で5分間加熱して、再懸濁した。各画分の等量ずつを10%SDS−PAGEゲルにかけ、電気泳動した。分離されたタンパク質は、ニトロセルロース膜に移し、5%脱脂粉乳でブロックした。組換えタンパク質発現体に、1:1000に希釈したヒツジαFLAG抗体(Sigma社)、つづいて、抗マウスHRP抱合二次抗体でプローブ付けを行った。化学発光を用いて検出した。
図21は、本発明の方法は、ほとんどが溶解した状態で、細菌細胞質内で発現するscFv遺伝子を識別することができることを明らかにしている。ウェスタンブロット法による発現プロフィールは、各サンプルで、scFv::I27::RL6::FLAGコンストラクトについてSNAPリガンドで検知した蛍光顕微鏡検査法と一致している。
【0139】
広く記述された本発明の範囲を逸脱することなく、特定の態様において示したように、多くの変化形および/または改良が可能であることを当業者は理解するであろう。本実施態様は、したがって、いかなる点においても説明のためであって、制限を課すものと考えてはならない。
本明細書において、議論され、および/または参照された公刊物は、その全体を参照のため本明細書に組み入れる。
本出願は、AU2009906310による優先権を主張するものであり、その開示全体は、参照により本願に組み込まれている。
本明細書に含まれている文書、行為、材料、装置、物品等々に関するいかなる議論もひとえに本発明の状況を提供することを目的としている。これらの事項のいずれか、または全てが、当出願の各請求項の優先日より以前に存在していたことを理由として、それが先行技術の基盤の一部を形成すること、あるいは本発明に関連する分野において普通の一般的な知識であったことを是認するとして取られるべきでない。
【0140】
参考文献
Aharoni et al. (2005) Chem Biol, 12:1281-1289
Becker et al. (2004) Curr Opin Biot, 15:323-329
Briers et al. (2009) Biochem Biophys Res Comm, 383:187-191
Chen and Gotschlich (2001) J Bact, 183: 3160-3168
Daugherty et al. (2000) J Immunol Methods, 243:211-227
Farinas (2006) Comb Chem High Thro Screen, 9:321-328
George, et al. (2003) Protein Engineering, 15:871-879
Kenrick et al. (2007) Curr Prot Cyt, 4.6.1-4.6.27
Lutz and Patrick (2004) Curr Opin Biot, 15:291-297
Magnet et al. (2007) J Bact 189:3927-3931
Miller et al. (2006) Nat Meth, 3:561-570
Parsons et al. (2006) Biochem 45:2122-2128
Schaefer et al. (2010) Antibody Eng, 1:21-44
Smith (1985) Science, 228:1315-1317
【技術分野】
【0001】
本発明は、ポリペプチドを標的分子に対する所望の活性に関してスクリーニングするための方法に関する。特に、本発明は、細菌細胞においてポリペプチドを発現し、前記細胞を透過化することにより、標的分子に対する所望の活性に関してポリペプチドをスクリーニングするための方法に関する。
【背景技術】
【0002】
タンパク質ディスプレイの最も初期の方法は、ファージディスプレイ(非特許文献1)であり、この方法では、関心対象のタンパク質を、タンパク質の野生型コピーとともに存在し得るファージの外皮タンパク質の1つと融合させる。たとえば、pIIIタンパク質に対する融合を使用したM13繊維状ファージを基礎とするディスプレイプラットフォーム。
【0003】
他のディスプレイ法には、「インビトロ」ディスプレイ法が含まれ、この方法では、タンパク質は細胞翻訳抽出物を用いて発現され、タンパク質とコーディング核酸との間のカップリングは、物理的結合(例えば、リボソームディスプレイ、mRNAディスプレイ)によるか、または通常のスカフォールドへの付着への付着もしくは膜内への封入により、例えばインビトロ区画化(IVC)で達成され、IVCでは、mRNAは、mRNAおよびタンパク質両方のマイクロビーズ(磁気またはセファロース)キャプチャシステムも含み得るミセル懸濁液内で翻訳される。
【0004】
タンパク質ディスプレイの別の方法は微生物表面ディスプレイであり、これは、グラム陰性、グラム陽性真正細菌または酵母のいずれかの微生物細胞外側に対する発現されたタンパク質の標的配置を含む。タンパク質を、細胞表面にそれらを付着させるアンカードメインと融合させる。アンカードメインは、脂質化もしくは細胞壁への共有結合を要求するモチーフを有し得るか、または露出したループ領域内の内在性膜タンパク質に対する融合物であり得る。産生の拡張性のために、微生物表面ディスプレイは、多様なライブラリーから改善されたタンパク質変異体についてスクリーニングするために用いられるだけでなく、ワクチン接種用の抗原を提示するためもしくは工業バイオテクノロジーの酵素用細胞スカフォールドとして用いることもできる。
【0005】
タンパク質ディスプレイ方法は、通常、抗体などの親和性タンパク質の進化に適用される。単一分子ディスプレイ法は歴史上最も普及しているが、高いバックグラウンドおよび親和性スケール間の低い分解能の問題を抱えている。周辺質収量が細胞質での発現に比べて非常に低いことが多い場合でも、酵母における表面ディスプレイによるかまたはファージ系により同定されるタンパク質は、通常、大腸菌(E. coli)周辺質における発現のために再編成される。しかし、抗体が細胞質において高収率で発現される場合、ほぼすべての場合に、それらは不溶性封入体を形成し、これは難儀してリフォールディングし、活性に関して試験しなければならない。
【0006】
このように、特に、親和性タンパク質ディスプレイライブラリおよび酵素ライブラリーをスクリーニングするためのタンパク質ディスプレイの方法が依然として必要とされている。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Smith (1985) Science, 228:1315-1317
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明者らは、透過化された細菌細胞において標的分子に対する所望の活性に関してポリペプチドをスクリーニングすることを可能にするタンパク質ディスプレイの方法を開発した。ポリペプチドは、透過化された細菌細胞内に保持されているか、または透過化された細菌細胞に結合しているかのいずれかである。
【0009】
したがって、本発明は、標的分子に対する所望の活性に関してポリペプチドをスクリーニングする方法であって:
a)ポリペプチドをコード化するポリヌクレオチドを含む細菌細胞を培養して、ポリペプチドを産生し、
b)細菌細胞を透過化し、ここで、ポリペプチドおよびポリペプチドをコード化するポリヌクレオチドは透過化された細菌細胞の内部に保持され、
c)透過化された細菌細胞を標的分子と接触させて、該標的分子を透過化された細菌細胞中に拡散させ、そして
d)ポリペプチドを所望の活性に関してスクリーニングする
ことを含む方法を提供する。
【0010】
本発明はさらに、標的分子に対する所望の活性に関してポリペプチドをスクリーニングする方法であって:
a)ポリペプチドをコード化するポリヌクレオチドを含む細菌細胞を培養して、ポリペプチドを産生し、細菌細胞壁に付着させ、
b)細菌細胞を透過化し、ここで、ポリペプチドをコード化するポリヌクレオチドは、透過化された細菌細胞内部に保持される、
c)透過化された細菌細胞を標的分子と接触させ、そして
d)ポリペプチドを所望の活性に関してスクリーニングする
ことを含む方法を提供する。
【0011】
1つの実施形態において、ステップd)は:
i)ポリペプチドが標的分子に結合するかどうか、および/もしくは標的分子に結合する程度を決定すること、ならびに/または
ii)ポリペプチドが標的分子を酵素的に修飾するかどうか、および/もしくは標的分子の酵素修飾率を決定する
ことを含む。
【0012】
細菌細胞は、細胞膜を可溶化するが細菌細胞壁の完全性を保持する任意の薬剤で透過化することができる。そのような薬剤としては、界面活性剤および有機溶媒が挙げられる。1つの実施形態において、細菌細胞を界面活性剤、たとえば非イオン性界面活性剤で透過化する。
【0013】
本発明の方法は、任意の好適なグラム陰性またはグラム陽性細菌細胞において実施することができるが、好ましくは、細菌細胞はグラム陰性細菌細胞である。
【0014】
1つの実施形態において、ポリペプチドは、少なくとも第2ポリペプチドと結合して、透過化された細菌細胞の内部に保持された、および/または細菌細胞壁に付着した、タンパク質複合体を形成する。ポリペプチドは、たとえば非共有もしくは共有結合によるなどで第2ポリペプチドと間接的に結合することができるか、またはポリペプチドは、第2ポリペプチドと直接的に結合することができ、たとえば融合タンパク質であり得る。
【0015】
したがって、1つの実施形態において、ポリペプチドを、第2ポリペプチド、またはそのサブユニットと融合させる。
【0016】
本発明の方法において、第2ポリペプチドは:
i)タンパク質複合体が透過化された細菌細胞壁の内部に保持されるような分子サイズを有するポリペプチド;
ii)DNA結合タンパク質;および/または
iii)細菌細胞壁結合タンパク質
から選択することができる。
【0017】
1つの実施形態において、タンパク質複合体の分子量は、少なくとも約120kDaである。
【0018】
別の実施形態において、第2ポリペプチドは、透過化された細菌細胞の内部でマルチマーを形成する。マルチマーは、たとえば、ダイマー、トリマー、テトラマー、ペンタマー、ヘキサマーまたはさらに高次のマルチマーであり得る。1つの実施形態において、マルチマーはテトラマーである。
【0019】
特定の1つの実施形態において、第2ポリペプチドは、RhnA、β−ガラクトシダーゼ、BetB、G5K、GshB、およびYdcWから選択される。
【0020】
所望の活性に関してスクリーニングされるポリペプチドを細菌宿主細胞DNAと結合させるために、任意のDNA結合タンパク質を本発明の方法で用いることができる。1つの実施形態において、DNA結合タンパク質はComEである。
【0021】
代替的または付加的に、ポリペプチドを細菌細胞壁結合タンパク質と結合させることができ、この場合、細菌細胞壁結合タンパク質は、ペプチドグリカン結合タンパク質、および細胞壁と結合できるリポタンパク質またはそのフラグメントから選択される。
【0022】
1つの実施形態において、細菌細胞壁結合タンパク質は、KzPG、PAL、OmpA、YiaD、YfiBおよびMotBから選択されるペプチドグリカン結合タンパク質である。
【0023】
ポリペプチドは細菌細胞壁と非共有的または共有的のいずれかで結合し得るが、1つの実施形態では、ポリペプチドは細菌細胞壁と共有結合する。
【0024】
別の実施形態において、細胞壁と結合できるリポタンパク質は、外膜付着に必要な機能的N末端シグナル配列が欠如したリポタンパク質である。
【0025】
特定の1つの実施形態において、リポタンパク質は大腸菌LPPである。
【0026】
本発明の方法の1つの実施形態において、非イオン性界面活性剤は、デカノイル−N−メチルグルカミド(Mega10)、ジメチルオクチルホスフィンオキシド(Apo8)、n−オクチル−β−D−チオグルコピラノシド(8TGP)、ならびにデカノイル−N−メチルグルカミド(Mega10)およびジメチルオクチルホスフィンオキシド(Apo8)の混合物から選択される。
【0027】
特定の1つの実施形態において、細菌細胞の透過化は:
i)LB中約0.5%のn−オクチル−β−D−チオグルコピラノシド(8TGP)、および
ii)LB中約0.5%のデカノイル−N−メチルグルカミド(Mega10)および約0.5%のジメチルオクチルホスフィンオキシド(Apo8)
から選択される溶液中で実施される。
【0028】
本発明の方法の別の実施形態において、ステップb)は、細菌細胞を選択的に透過化することを含み、それにより、細菌細胞の外膜は細菌細胞の内膜よりもさらに透過化される。
【0029】
1つの実施形態において、細菌細胞は、ジメチルオクチルホスフィンオキシド(Apo8)および/またはポリソルベート20(Tween20)から選択される界面活性剤で選択的に透過化される。
【0030】
1つの実施形態において、細菌細胞の選択的透過化は、界面活性剤を約0.2%で含む溶液中で実施される。
【0031】
別の実施形態において、細菌細胞の選択的透過化は、EDTAまたはCa2+を含む溶液中で実施される。
【0032】
別の実施形態において、方法は、ポリペプチドをコード化するポリヌクレオチドを含むDNAを透過化された細菌細胞から単離することをさらに含む。
【0033】
細菌細胞から単離されるDNAは、ゲノムDNAおよび/またはエピソームDNAであり得る。エピソームDNAは、たとえば、プラスミドまたはコスミドであり得る。
【0034】
1つの実施形態において、ポリヌクレオチドは外因性ポリヌクレオチドである。
【0035】
別の実施形態において、標的分子の分子量は約120kDa未満である。
【0036】
本発明はさらに、
所望の活性を有するポリペプチドを同定するための方法であって:
a)本発明の方法を用いてポリペプチドのライブラリーをスクリーニングすること;および
b)所望の活性を有する1以上のポリペプチドを選択すること
を含む方法を提供する。
【0037】
1つの実施形態において、方法はさらに、c)ポリペプチドをコード化するポリヌクレオチドを含むDNAを細菌細胞から単離することを含む。
【0038】
別の実施形態において、方法は、d)ポリペプチドをコード化するポリヌクレオチドの配列を決定することをさらに含む。
【0039】
1つの実施形態において、ポリペプチドのライブラリーは、細胞、組織、器官または生物から得られるポリヌクレオチドによりコード化される。
【0040】
別の実施形態において、ポリペプチドのライブラリーは、1以上の親ポリヌクレオチドを突然変異させることによって得られるポリヌクレオチドによりコード化される。
【0041】
1つの実施形態において、ポリペプチドは抗体または酵素である。
【0042】
特定の1つの実施形態において、抗体は単鎖可変フラグメント(scFV)である。
【0043】
別の実施形態において、ポリペプチドは酵素であり、標的分子を透過化された細菌細胞に結合させる。標的分子を透過化された細菌細胞に結合させるために、標的分子を細菌細胞に直接的または間接的のいずれかで結合させることができる。標的分子を透過化された細菌細胞に直接的に結合させるために、標的分子をたとえば、細菌細胞壁結合タンパク質と結合させることができる。
【0044】
別の実施形態において、ポリペプチドは、抗体以外の結合タンパク質である。たとえば、ポリペプチドは、これらに限定されるものではないが、リポカリン、フィブロネクチンIII型ドメイン(FN3)、ユビキチン、またはγ−B−クリスタリンをはじめとする結合タンパク質であり得る。
【0045】
本発明の方法の1つの実施形態において、ポリペプチドは、I27、RL6、KzPG、SNAP,および/またはDBPのいずれか1つから選択されるドメインを含む。特定の1つの実施形態において、ポリペプチドは、ドメインI27、RL6、KzPG、SNAP、およびDBPを含む。
【0046】
本発明の方法の別の実施形態において、ポリペプチドは、配列番号13と少なくとも80%、好ましくは少なくとも90%、さらに好ましくは少なくとも95%、さらに好ましくは100%同一のアミノ酸配列を含む。
【0047】
本発明はさらに、第2ポリペプチドと結合して、透過化された細菌細胞の内部に保持されるタンパク質複合体を形成する外因性タンパク質を含む透過化された細菌細胞を提供する。
【0048】
本発明はさらに、細菌細胞壁に付着した外因性ポリペプチドを含む透過化された細菌細胞を提供する。
【0049】
本発明はさらに:
a)i)第1ポリペプチドをコード化するポリヌクレオチドをベクターに挿入するための部位、および
ii)第1ポリペプチドと結合して、透過化された細菌細胞の内部に保持されるタンパク質複合体を形成する、第2ポリペプチドをコード化するオープンリーディングフレーム
を含むベクター、ならびに
b)細菌細胞を透過化することができる薬剤
を含むキットを提供する。
【0050】
本発明はさらに:
a)i)第1ポリペプチドをコード化するポリヌクレオチドをベクターに挿入するための部位、および
ii)第1ポリペプチドと結合して、細菌細胞壁に付着したタンパク質複合体を形成する第2ポリペプチドをコード化するオープンリーディングフレーム
を含むベクター、ならびに
b)細菌細胞を透過化することができる薬剤
を含むキットを提供する。
【0051】
1つの実施形態において、部位およびオープンリーディングフレームは、第1ポリペプチドおよび第2ポリペプチド、またはそのサブユニットが融合タンパク質として発現されるような位置にある。
【0052】
別の実施形態において、細菌細胞を透過化することができる薬剤は界面活性剤である。
【0053】
さらに別の実施形態において、界面活性剤は、デカノイル−N−メチルグルカミド(Mega10)、ジメチルオクチルホスフィンオキシド(Apo8)、n−オクチル−β−D−チオグルコピラノシド(8TGP)、ならびにデカノイル−N−メチルグルカミド(Mega10)およびジメチルオクチルホスフィンオキシド(Apo8)の混合物から選択される非イオン性界面活性剤である。
【0054】
1つの実施形態において、キットは細菌細胞をさらに含む。
【0055】
好ましくは、細菌細胞はグラム陰性である。たとえば、細菌細胞は大腸菌であり得る。
【0056】
本発明はさらに、配列番号13と少なくとも80%、好ましくは少なくとも90%、さらに好ましくは少なくとも95%、さらに好ましくは100%同一であるアミノ酸配列を提供する。
【0057】
本発明はさらに、配列番号14または配列番号15と少なくとも80%、好ましくは少なくとも90%、さらに好ましくは少なくとも95%、さらに好ましくは100%同一であるヌクレオチド配列を含むポリヌクレオチドを提供する。
【0058】
本発明はさらに、本発明のポリヌクレオチド配列を含むベクターを提供する。
【0059】
本発明はさらに、配列番号6〜12または16のいずれか1つと少なくとも90%、さらに好ましくは100%同一であるアミノ酸配列を含むポリペプチドスペーサーを提供する。
【0060】
明らかなように、本発明の1つの態様の好ましい特徴および特性は、本発明の多くの他の態様に適用可能である。
【0061】
本明細書全体を通して、「comprise(含む)」という語、または「comprises」もしくは「comprising」などの変形は、記載された要素、整数もしくはステップ、または要素、整数もしくはステップの群を包含するが、任意の他の要素、整数若しくはステップ、または要素、整数もしくはステップの群を除外しないことを意味すると理解される。
【0062】
本発明を、以下の非限定的例により、添付の図面を参照して後述する。
【図面の簡単な説明】
【0063】
【図1】大腸菌細胞の界面活性剤透過化。GFPを発現する大腸菌細胞を界面活性剤で処理して、膜透過化の有効性を測定した。細胞を明視野(第1列)または蛍光顕微鏡法(第2列および第3列)のいずれかにより観察した。透過化は、膜不透過性DNA結合色素Gel Red(第3列)の吸収と同時にGFP(緑色、第2列)が、細胞から放出される場合に有効であった。界面活性剤8TGP(0.5%)および0.5%のMega10/0.5%のApo8(「Agent86」)は大腸菌細胞の透過化において最も有効であることが判明した。
【図2】界面活性剤上清のSDS−PAGE。図1で示される大腸菌細胞の界面活性剤透過化の上清を、9%のSDS−PAGE上にロードして、界面活性剤によるタンパク質放出を定性的に評価した(第1レーン)。界面活性剤透過化細胞における細胞壁カプセルによる細胞タンパク質のサブセットの保持を示すために、界面活性剤透過化細胞の試料をリゾチーム(2mg/mL)で処理して、細胞壁(第2レーン)を加水分解した。
【図3】テトラマー融合タンパク質発現。(A)His6::SNAP::テトラマー融合タンパク質の大腸菌における発現を、全細胞タンパク質に対してプローブされたαHis抗体を用いてウェスタンブロットにより調べた。予想される分子量のバンドに加えて、>250kDの高分子量バンドがRhnAテトラマー融合物(レーン4)で観察され、これはテトラマーとして移動した複合体の推定されるSDS耐性形態である。(B)BetBテトラマー融合タンパク質抽出物を界面活性剤可溶性および界面活性剤不溶性(細胞カプセルペレット)抽出物に分離し、そしてSDS−PAGEにより調べた。
【図4】テトラマー融合タンパク質のSNAP標識。例1で記載されているようにして、His6::SNAP::テトラマー融合タンパク質を大腸菌において発現させ、細胞を8TGPで透過化した。例3で記載されているようにして、融合タンパク質の発現を、膜不透過性SNAPリガンドBG−547で標識された透過化細胞の蛍光顕微鏡検査(第2列)により検出した。細胞DNAを膜透過性色素、Sytox Greenで標識した(第1列)。SNAPおよびSytox Greenシグナルのオーバーレイを第3列に示す。
【図5】透過化細胞におけるHis6::SNAP::BetBテトラマーのαHis抗体標識。例3で記載されるような、αHis抗体でプローブされたHis6::SNAP::BetBテトラマーを発現する透過化細胞の蛍光顕微鏡検査(第1パネル)。細胞をSNAPリガンドBG−547で標識した(第2パネル)。αHisおよびSNAP両方の共存(第3パネル)は、αHis抗体が透過化細胞の細胞壁を透過することを示す。
【図6】HALOおよびSNAP発現レポーターとのBetB、RhnAおよびYdcWテトラマー融合物。BetB、RhnAおよびYdcWテトラマーを別々に発現レポーターHALOおよびSNAPと融合させた。融合タンパク質を発現する細胞を透過化し、宿主DNAをGel Redで標識し、HALO(G1001)およびSNAP(BG−488)の蛍光リガンドを用いて融合タンパク質を検出した。
【図7】GFP5::DNA結合タンパク質(DBP)融合物の発現。淋菌(N. gonorrhoeae)由来の非特異的高親和性DNA結合タンパク質であるComEをGFP5のC末端に融合させ、大腸菌で発現させた。細胞を透過化し、GFP(第1パネル)およびGel Red(第2パネル)に関して蛍光顕微鏡法により観察した。蛍光の共存(第3パネル)は、融合タンパク質および宿主DNAの両方が透過化細胞カプセル内に保持されたことを示す。
【図8】透過化細胞におけるDNAの保持。GFP5::DBP融合物、またはHis6::eGFP融合物を発現する大腸菌細胞を、未処理のまま放置するか(1および4行)または例1で記載されるように透過化させるか(2、3、5および6行)のいずれかであった。透過化細胞を4℃で一晩保存するか、またはTBS中に再懸濁させ、37℃で一晩振盪した後、GFP(第1列)またはGel Red(第2列)に関して蛍光顕微鏡法により観察した。GFPおよびGel Redの共存を第3列に示す。
【図9】透過化細胞からのDNA抽出。(A)GFP5::DBP融合タンパク質または(B)His6::eGFP融合タンパク質を発現する大腸菌細胞を例1で記載されるようにして透過化した。透過化細胞を一晩4℃で保存するか、またはTBS中に再懸濁させ、37℃で一晩振盪した後、プラスミドDNAを抽出し、エチジウムブロミド染色された1%アガロースゲル上、TAE緩衝液で電気泳動させた。レーン1は未処理細胞中の全プラスミドDNAである。レーン2および4は、それぞれ4℃で一晩保存し、37℃で振盪した細胞カプセルの透過化ステップからの上清であり、レーン3および5は、それぞれ4℃で一晩保存し、37℃で振盪した細胞カプセルからのプラスミド調製物である。
【図10】OmpF::SNAP::LPP融合タンパク質のSNAP標識。OmpF::SNAP::LPP融合タンパク質を発現する大腸菌細胞を例1で記載されるようにして透過化した。融合タンパク質局在化を、例3で記載されるようにSNAPリガンドBG−488での標識により検出した。標識された細胞を明視野顕微鏡検査(第1パネル)および蛍光顕微鏡検査(第2パネル)により観察した。第3パネルは明視野図および蛍光図両方のオーバーレイである。
【図11】αGFP::HALO::RhnA融合タンパク質によるeGFPの結合。αGFP::HALO::RhnA融合タンパク質を発現する大腸菌細胞を例1で記載されるようにして透過化した。精製されたeGFPタンパク質を例8で記載されるように細胞カプセルと結合させ、eGFPを蛍光顕微鏡検査により視覚化した。第1パネル、明視野図;第2パネル、eGFP蛍光;第3パネル、明視野および蛍光のオーバーレイ。
【図12】αGFP::KzPG::SNAP::DBP融合タンパク質によるeGFPの結合。αGFP::KzPG::SNAP::DBP融合タンパク質を発現する大腸菌細胞を例1で記載されるようにして透過化した。精製されたeGFPタンパク質を例8で記載されるようにして細胞カプセルと結合させ、例3で記載されているようにして、eGFPを2つの方法、ウェットマウントおよびドライマウントによる蛍光顕微鏡法で視覚化した。(A)ウェットマウント細胞カプセルに結合させたeGFP。挿入パネル(i)および(ii)は、αGFP::KzPG::SNAP::DBP融合タンパク質により結合したeGFPの細胞壁局在化を示す。(B)および挿入パネル(Aiii)は、DABCO/グリセロール中でドライマウントによる顕微鏡検査のために調製された同じ細胞を示す。
【図13】OmpF::αGFP::SNAP::LPP融合タンパク質によるeGFPの結合。OmpF::SNAP::LPPまたはOmpF::αGFP::SNAP::LPP融合タンパク質を発現する大腸菌細胞を例1で記載されているようにして透過化した。精製されたeGFPタンパク質を例8で記載されているようにして細胞カプセルと結合させ、例3で記載されているようにして、eGFPをドライマウントによる蛍光顕微鏡法によって視覚化した。(A)OmpF::SNAP::LPP融合タンパク質を発現する細胞は、OmpF::αGFP::SNAP::LPP融合タンパク質を発現する細胞(第2パネル、下行)と異なり、eGFP蛍光がない(第2パネル、上行)。
【図14】LPP融合タンパク質による細胞壁に対する共有結合の証明。OmpF::αGFP::SNAP::LPP融合タンパク質を発現する大腸菌細胞を例1で記載されているようにして透過化した。融合タンパク質局在化を例3で記載されているようにしてSNAPリガンドBG−488で標識することにより検出し、DNAをGel Redで染色した。試料を5分間22℃(A)または95℃(B)で加熱した後、ドライマウントし、蛍光顕微鏡検査により観察した。
【図15】界面活性剤/Ca2+緩衝液を用いた外膜透過化。OmpF::αGFP::SNAP::LPP融合タンパク質(外部αGFPまたはαGFP::HALO::FLAG::RhnA融合タンパク質(内部αGFP)を発現する大腸菌細胞を例10で記載されているようにして透過化した。大きなリガンドに対する外膜の透過化は、細胞壁に付着したαGFPドメインにeGFPを結合させることにより評価した。内膜の透過化は、大きなリガンド(eGFP)および小さなリガンド(Gel Red)を用いて評価した。Ca2+緩衝液中の界面活性剤Apo8(A)およびTween20(B)はどちらも、大きなリガンドに対する外膜の選択的透過性を示した。
【図16】界面活性剤/EDTA緩衝液を用いた外膜透過化。OmpF::αGFP::SNAP::LPP融合タンパク質(外部αGFP)またはαGFP::HALO::FLAG::RhnA融合タンパク質(内部αGFP)を発現する大腸菌細胞を例10で記載されるようにして透過化した。大きなリガンドに対する外膜の透過化を、eGFPを細胞壁に付着したαGFPドメインに結合させることにより評価した。内膜の透過化は、大きなリガンド(eGFP)および小さなリガンド(Gel Red)を用いて評価した。EDTA緩衝液中の界面活性剤Apo8(A)およびTween20(B)はどちらも大きなリガンドに対する外膜の選択的透過性を示した。
【図17】混合eGFP、およびSNAP標識された細胞のFACS分析。eGFP(#1矢印);SNAPリガンドBG−488で標識されたαGFP::KzPG::SNAP::DBP融合タンパク質(#2矢印);およびSNAPリガンドBG−547で標識されたHis6::SNAP::BetB(#3矢印)を発現する大腸菌細胞の3つの集団を、FACSにより分類した。分類された集団を初回分類の60分後に純度および細胞完全性について再分析した。
【図18】αGFPおよびKzPGドメイン間のペプチドリンカーは、αGFP::KzPG::SNAP::DBP融合タンパク質を発現する大腸菌細胞のセファロース支持体に対する結合を可能にする。12−merリンカー領域RL6をαGFPおよびKzPGドメイン間に有するαGFP::KzPG::SNAP::DBP融合タンパク質を発現する細胞を、His6::eGFP中間体によりCo2+−セファロース支持体Co2+−セファロース支持体と結合させた。GFP結合を左のパネルに示し(緑);融合タンパク質のSNAPリガンド(赤)結合を中央のパネルに示し;それぞれのオーバーレイを右に示す。
【図19】αGFP::RL6::KzPG::SNAP::DBP融合タンパク質を発現する大腸菌細胞のストレプトアビジン標識された磁気ビーズに対する結合。(A)ビオチン標識されたeGFP(中央および右パネル)を、αGFP::RL6::KzPG::SNAP::DBP融合タンパク質を発現する細胞と結合させ、これを次にストレプトアビジン標識された磁気粒子と結合させた。(B)最初にビオチニル化eGFPで標識されたストレプトアビジン標識磁気粒子に対するαGFP::RL6::KzPG::SNAP::DBP融合タンパク質を発現する細胞の逆の結合。この例では、ビーズは緑色に標識され(GFPパネル)、細胞はBG−547SNAPリガンドで標識された(赤、SNAP redパネル)。(C)ドメインリンカー(ヒトタイチンの27番目のIgドメイン)も結合スペーサーとして有効であった。αGFP::I27::RL6::KzPG::SNAP::DBP融合タンパク質を発現する大腸菌細胞をまずビオチニル化eGFPと結合させ(緑、GFPパネル)、BG−547SNAPリガンドで標識(赤、SNAP redパネル)した後、ストレプトアビジン標識磁気粒子と結合させる。
【図20】大腸菌細胞質におけるscFv::I27::RL6::KzPG::SNAP::DBP融合タンパク質としてのマウスscFv遺伝子の発現。マウスscFvライブラリーを構築し、本発明の方法にしたがって大腸菌細胞質でディスプレイした。検出可能な発現を有するクローンをSNAPリガンド結合により検出し、ミスフォールド(左パネル)、わずかに発現されるが可溶性(中央のパネル)または高度に発現され可溶性(右パネル)に分類した。
【図21】可溶性および不溶性scFv発現の大腸菌細胞質における検出。マウスscFv発現ライブラリーからの限定されたスクリーンにおいて高度に発現され可溶性であることが判明した、選択されたクローンを、scFv::I27::RL6::FLAGおよびscFv::RL6::FLAG融合タンパク質として発現構築物中にサブクローンした。タンパク質フラクションを可溶性または不溶性のいずれかとしてSDS−PAGEゲル上にロードし、ニトロセルロース膜に移し、αFLAG抗体を用いて検出した。可溶性(S)または不溶性(P)フラクションについて、試料、ならびに発現される各クローンをI27::RL6(I)またはRL6(R)リンカーと対にする。ライブラリスクリーンから単離されるI27::RL6::KzPG::SNAP::DBPディスプレイ構築物におけるオリジナルのscFvクローンの蛍光顕微鏡画像も下のパネルに示す。
【0064】
配列表の凡例
配列番号1 pAra3::His6::SNAPアラビノースベクターのヌクレオチド配列
配列番号2 pAra3::His6::KzPG::SNAP::DBPベクターのヌクレオチド配列
配列番号3 pAra3::OmpF::SNAP::LPPベクターのヌクレオチド配列
配列番号4 pAra3::αGFP(R35)::HALO::FLAG::RhnAベクターのヌクレオチド配列
配列番号5 ランダム化ペプチドスペーサードメイン
配列番号6〜12 ペプチドリンカースペーサー
配列番号13 I27::RL6::KzPG::SNAP::DBP
配列番号14 I27::RL6::KzPG::SNAP::DBPコーディング配列
配列番号15 ライブラリスカフォールドベクター
配列番号16 I27スペーサー
【発明を実施するための形態】
【0065】
一般的技術および定義
別段の明確な規定がない限り、本明細書中で用いられる全ての専門用語および科学用語は、(たとえば、タンパク質化学、生化学,細胞培養、分子遺伝学、微生物学、および免疫学の)当業者により通常理解されるのと同じ意味を有すると解釈されるべきである。
【0066】
特に明記しない限り、本発明で利用される組換えタンパク質、細胞培養、および免疫手法は、当業者に周知の標準的手順である。そのような技術は、J. Perbal, A Practical Guide to Molecular Cloning, John Wiley and Sons (1984)、J. Sambrook et al., Molecular Cloning: A Laboratory Manual, 3rd edn, Cold Spring Harbour Laboratory Press (2001)、R. Scopes, Protein Purification - Principals and Practice, 3rd edn, Springer (1994)、T.A. Brown (editor), Essential Molecular Biology: A Practical Approach, Volumes 1 and 2, IRL Press (1991)、D.M. Glover and B.D. Hames (editors), DNA Cloning: A Practical Approach, Volumes 1-4, IRL Press (1995 and 1996)、およびF.M. Ausubel et al. (editors), Current Protocols in Molecular Biology, Greene Pub. Associates and Wiley-Interscience (1988、including all updates until present), Ed Harlow and David Lane (editors) Antibodies: A Laboratory Manual, Cold Spring Harbour Laboratory, (1988)、およびJ.E. Coligan et al. (editors) Current Protocols in Immunology, John Wiley & Sons (including all updates until present)などの出典で文献全体にわたって記載され、説明されている。
【0067】
「ポリペプチド」、「タンパク質」および「ペプチド」という用語は、本明細書中では一般的に交換可能に用いられる。本明細書中で用いられる場合、「外因性ポリペプチド」という用語は、外因性ポリヌクレオチドによりコード化されるポリペプチドを指す。「外因性ポリヌクレオチド」という用語は、本明細書中で用いられる場合、それが導入される細胞と異なるポリヌクレオチド、またはポリペプチドが通常見いだされない宿主細胞核酸内の位置以外は導入される細胞における配列と配列が相同性であることを意味する。
【0068】
「抗体」という用語は、本発明で用いられる場合、ポリクローナル抗体、モノクローナル抗体、二重特異性抗体、二特異性抗体、三特異性抗体、多特異性抗体、ヘテロ結合抗体、キメラ抗体、例えばインタクト分子ならびにそのフラグメント、例えばFab、F(ab’)2、FvおよびscFvならびに他の抗体様分子が挙げられる。
【0069】
「約」という用語は、本明細書中で用いられる場合、所定値の+/−5%の範囲を指す。
【0070】
透過化
本発明の方法では、細菌細胞を透過化し、かくして可溶性細胞成分の少なくとも一部を、細胞壁を通して拡散させる。所望の活性に関してスクリーニングされるポリペプチドは、細菌細胞壁内に保持されるか、または細菌細胞壁に付着する。本明細書中で用いられる場合、「透過化された細菌細胞」とは、1以上細胞膜中に孔を生成させるか、または細胞膜を可溶化し、一方、ペプチドグリカン間の結合を加水分解せず、それにより細胞壁をインタクトのままで保持する透過化剤の使用を指す。細菌細胞を透過化する薬剤の非限定的例としては、界面活性剤および有機溶媒が挙げられる。透過化は、有利には、小〜中程度のサイズのタンパク質、たとえば120kDaまで、または同等以下のサイズの他の分子がインタクトなままの細胞カプセル中に侵入させる。さらに、細菌細胞壁の完全性を維持することにより、透過化された細菌細胞は、従来法で、たとえば細菌細胞をTris−EDTA−リゾチームで処理することにより産生されるスフェロプラストほど脆弱ではなく、スフェロプラストでは、細菌細胞壁が少なくとも部分的に加水分解されている。本発明の方法で産生される透過化された細菌細胞は、蛍光活性化細胞分類(FACS)などの技術によく適しており、一方、スフェロプラストは高剪断フローサイトメトリー環境により損なわれ、制御された浸透圧条件を必要とし、したがってそれらの潜在的使用が限定される。
【0071】
好ましくは、透過化処理は、細胞タンパク質をそれらの自然な状態および相互関係で保存する。非イオン性界面活性剤は、タンパク質フォールディングおよびタンパク質複合体に対してイオン性界面活性剤ほど破壊的でない。したがって、好ましい実施形態では、非イオン性界面活性剤を用いて細菌細胞壁を透過化する。非イオン性界面活性剤の非限定的例としては、Triton X−100、Triton X−114、Brij 35、Brij 58、Tween 20、Tween 80、Nonidet P−40 Substitute、Octyl β Glucoside、Mega 8、Mega 9、Mega 10、BigCHAP、Deoxy BigCHAP、Apo8、および8TGPが挙げられる。
【0072】
界面活性剤の混合物を用いて細菌細胞を透過化することができる。たとえば、界面活性剤は、2以上の非イオン性界面活性剤の混合物であり得る。1つの実施形態において、界面活性剤はMega 10およびApo8の混合物である。
【0073】
所望の活性に関してスクリーニングされるポリペプチドが細菌細胞壁に付着しているか、または内側細胞膜と一体化しているかもしくは付着している場合、必ずしも細菌細胞の内膜を透過化する必要はない可能性があることを当業者は理解するであろう。したがって、1つの実施形態では、細菌細胞は選択的に透過化される。「選択的に透過化される」により、透過化された細菌細胞の外膜が内膜よりもさらに透過化され、それによって、内膜および外膜の両方が、0.5%のMega 10および0.5%のApo8を含む溶液を用いることによるなどで透過化された透過化細胞と比較した場合、膜不透過性物質、たとえば膜不透過性DNA結合リガンドGel Redの50%以下、もしくは更に好ましくは40%、30%、20%、10%、5%、4%、3%、2%、1%以下が選択的に透過化された細胞の内膜を透過するか、または透過しないことを意味する。
【0074】
当業者は、本発明の方法にしたがって細菌細胞を選択的に透過化するために好適な条件を決定することができるが、1つの実施形態では、細菌細胞は、非イオン性界面活性剤で選択的に透過化される。たとえば、非イオン性界面活性剤は、Apo8およびTween20から選択することができる。1つの実施形態において、細菌細胞を選択的に透過化するための溶液は、界面活性剤を約0.2%〜約0.4%、または約0.2%〜約0.3%、または約0.2%の濃度で含む。好ましくは、細菌細胞を選択的に透過化するための溶液は、Ca2+またはEDTAを含む緩衝液中界面活性剤を含む。細菌細胞を選択的に透過化するために好適な例示的緩衝液としては、25mMのTris中0.2〜0.4%のApo8もしくはTween20、1mMのEDTA(pH8.0)、または25mMのTris、2mMのCa2+(pH8.0)が挙げられる。1つの実施形態において、細菌細胞の選択的透過は、細胞を好適な緩衝液中、約25℃にて約10分間インキュベートすることによって達成することができる。
【0075】
ポリペプチド発現
所望の活性に関してスクリーニングされるポリペプチドを、細菌細胞における発現の好適なベクター中にクローンすることができる。「ベクター」は本明細書中で用いられる場合、細菌細胞を形質転換するために好適であることが当該技術分野で知られている任意のベクターを指す。好ましくは、ベクターはまた、宿主のゲノムから独立して、細菌細胞内で複製することができる。ベクターとしては、プラスミド、ウイルスおよびコスミドならびに線状DNAエレメント、例えば大腸菌の線状ファージN15、および/または細菌細胞ゲノムから独立して複製する染色体外DNAが挙げられる。好ましくは、ベクターは発現ベクターである。
【0076】
本明細書中で用いられる場合、「発現ベクター」は、細菌細胞において特定のポリヌクレオチド分子を発現させることができるベクターである。好ましくは、発現ベクターは、細菌細胞内で複製することもできる。好適な発現ベクターは、典型的には、調節配列、たとえば転写制御配列、翻訳制御配列、複製起点、および組換え細菌細胞と適合性であり、ポリペプチドをコード化するポリヌクレオチド分子の発現を制御する他の調節配列を含む。転写制御配列は、転写の開始、伸長、および終止を制御する配列である。特に重要な転写制御配列は、転写開始を制御するもの、たとえば、プロモーター、エンハンサー、オペレーターおよびリプレッサー配列である。好適な転写制御配列は、細菌細胞において機能することができる任意の転写制御配列を含む。種々のそのような転写制御配列は、当業者に公知である。
【0077】
発現ベクターの細菌細胞への形質転換は、ポリヌクレオチド分子を細胞中に挿入できる任意の好適な方法により達成することができる。形質転換技術としては、これらに限定されるものではないが、エレクトロポレーションおよび化学変換が挙げられる。形質転換されたポリヌクレオチド分子は、染色体外にとどまる可能性があるか、またはそれらの発現される能力が保持されるような方法で形質転換された(すなわち、組換え)細胞の染色体内の1以上の部位に組み込まれる可能性がある。
【0078】
組換えDNA技術を用いて、たとえば、宿主細胞内のポリヌクレオチド分子のコピー数、それらのポリヌクレオチド分子が転写される効率、結果として得られる転写物が翻訳される効率、および翻訳後修飾の効率を操作することによって、形質転換されたポリヌクレオチド分子の発現を改善することができる。ポリヌクレオチド分子の発現を増大させるために有用な組換え技術としては、これらに限定されるものではないが、ポリヌクレオチド分子を高コピー数のプラスミドに機能的に連結すること、ベクター安定性配列のプラスミドへの添加、転写制御シグナル(たとえば、プロモーター、オペレーター、エンハンサー)の置換もしくは修飾、翻訳制御シグナル(たとえば、リボソーム結合部位、Shine−Dalgarno配列)の置換もしくは修飾、宿主細胞のコドン使用に対応させるためのポリヌクレオチド分子の修飾、および転写物を不安定化する配列の欠失が挙げられる。
【0079】
当業者は、本発明の方法でポリペプチドを発現するために好適な細菌株を容易に決定することができるであろう。当業者は、本発明の方法での使用に好適なグラム陰性菌には、サルモネラ属(Salmonella)、大腸菌、赤痢菌(Shigella)、カンピロバクター属(Campylobacter)、フゾバクテリウム属(Fusobacterium)、ボルデテラ属(Bordetella)、パスツレラ属(Pasteurella)、アクチノバチルス属(アクチンobacillus)、ヘモフィルス属(Haemophilus)およびヒストフィルス属(Histophilus)が含まれることを理解するであろう。好ましい実施形態において、グラム陰性菌は大腸菌である。
【0080】
タンパク質複合体
所望の活性に関してスクリーニングされるポリペプチドを少なくとも第2ポリペプチドと結合させて、タンパク質複合体が透過化された細菌細胞の内部に保持されるような分子サイズを有するタンパク質複合体を形成することができる。ポリペプチドをたとえば、ジスルフィド架橋などの共有結合によるか、または非共有結合により、第2ポリペプチドと結合させることができる。「非共有結合」とは、原子間結合が関与しない分子相互作用を指す。たとえば、非共有相互作用は、イオン結合、水素結合、疎水性相互作用、およびファンデルワールス力が関与する。非共有結合力を用いて、別個のポリペプチド鎖をタンパク質またはタンパク質複合体中に一緒に保持することができる。したがって、ポリペプチドおよび第2ポリペプチドを同じかもしくは異なるベクターのいずれかから別のポリペプチドとして発現することができるか、またはポリペプチドの一方もしくは両方を、細菌細胞ゲノム中に組み入れられたポリペプチドをコード化するDNAから発現することができる。
【0081】
あるいは、タンパク質複合体と結合するポリペプチドおよび第2ポリペプチドは融合タンパク質であり得る。本明細書中で用いられる場合、「融合タンパク質」とは、2つのポリペプチドのそれぞれの少なくとも一部をコード化するポリヌクレオチドの発現から生じる2以上のポリペプチド、またはそれらのフラグメントからなるハイブリッドタンパク質を指す。
【0082】
分子サイズにより透過化された細菌細胞中に保持されるタンパク質複合体
第2ポリペプチドは、所望の活性に関してスクリーニングされるポリペプチドで形成された複合体の少なくともいくつかが、透過化された細菌細胞から拡散することができないような十分な分子サイズ、すなわち十分な分子量または分子半径を有する任意のポリペプチドであり得る。したがって、タンパク質複合体は、細胞の透過化後に細菌細胞内に保持される。当業者は、その分子量および球状または桿状(糸状)タンパク質であるかどうかをはじめとする第2ポリペプチドの性質が、細菌細胞壁を通るタンパク質複合体の拡散を防止または阻害するその能力を決定することを理解するであろう。1つの実施形態において、第2ポリペプチドの分子量は、少なくとも約30kDa、または少なくとも約40、50、60、70、80、90、100、120、130、140、150kDaもしくはそれ以上である。1つの実施形態において、第2ポリペプチドは少なくとも約120kDaである。
【0083】
1つの実施形態において、第2ポリペプチドは、透過化された細菌細胞の孔排除サイズよりも大きな分子サイズを有するマルチマーを形成する。本明細書中で用いられる場合、「マルチマー」という用語およびその文法的変異は、2以上の異なる分子間のマルチマー複合体の形成を指す。マルチマーは、たとえば、同じタンパク質の2以上の分子(すなわち、ホモマルチマー)または2以上の異なるか、もしくは非同一タンパク質(すなわち、ヘテロマルチマー)の混合物を含み得る。本発明の方法における使用に好適なマルチマーを形成するタンパク質としては、ダイマー、トリマー、テトラマー、ペンタマー、ヘキサマー、および7以上のサブユニットを含むさらに高次のマルチマーが挙げられる。
【0084】
マルチマータンパク質には、ホモダイマー、たとえば、PDGF受容体α、およびβイソ型、エリスロポエチン受容体、MPL、ならびに−CSF受容体、それらのサブユニットがそれぞれリガンド結合およびエフェクタードメインを有するヘテロダイマー、たとえば、PDGF受容体αβイソ型、ならびに異なる機能を有する成分サブユニットを有するマルチマー、たとえば、IL−2、IL−3、IL−4、IL−5、IL−6、IL−7、およびGM−CSF受容体が含まれる。本発明の方法で用いることができる他のマルチマータンパク質の非限定的例としては、DNAの合成または複製に関与する因子、例えばTFIIDおよびTFIIHなどのmRNAの産生に関与するDNAポリメラーゼタンパク質;細胞、核および他の膜関連タンパク質、例えばホルモンおよび他のシグナル伝達受容体、能動輸送タンパク質およびイオンチャンネル、ヘモグロビン、フィブリノゲンおよびフォンウィルブランド因子をはじめとする血液中のマルチマータンパク質;細胞内の構造を形成するタンパク質、例えばアクチン、ミオシン、およびチューブリンならびに他の細胞骨格タンパク質;細胞外環境で構造を形成するタンパク質、例えばコラーゲン、エラスチンおよびフィブロネクチン;細胞内および細胞外輸送に関与するタンパク質、例えばキネシンおよびダイニン、タンパク質のSNAREファミリー(可溶性NSF付着タンパク質受容体)およびクラスリン;クロマチン構造の調節に役立つタンパク質、例えばヒストンおよびプロタミン、Swi3p、Rsc8pおよびモイラ;マルチマー転写因子、例えばFos、JunおよびCBTF(CCAATボックス転写因子);マルチマー酵素、例えばアセチルコリンエステラーゼおよびアルコールデヒドロゲナーゼ;シャペロンタンパク質、例えばGroE、GroEL(シャペロニン60)およびGroES(シャペロニン10);抗毒素、たとえばヘビ毒、ボツリヌス毒素、ストレプトコッカス(Streptococcus)超抗原;リシン(バクテリオファージおよびウイルス由来の酵素);ならびにほとんどのアロステリックタンパク質が挙げられる。1つの実施形態において、マルチマータンパク質は大腸菌タンパク質である。マルチマーを形成する大腸菌タンパク質の非限定的例としては、L−ラムノースイソメラーゼ(RhnA;たとえばNCBI受入CAA43002)、β−ガラクトシダーゼ(β−gal;たとえばNCBI受入YP001461520)、ベタインアルデヒドデヒドロゲナーゼ(BetB;たとえばNCBI受入AAA23506)、グルタメート−5−キナーゼ(G5K;たとえばNCBI受入AAB08662)、グルタチオンシンターゼ(GshB;たとえばNCBI受入AP_003504)、および中鎖アルデヒドデヒドロゲナーゼ(YdcW;たとえばNCBI受入AP_002067)が挙げられる。
【0085】
1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドは、細菌細胞壁内にポリペプチドを保持するために十分な分子サイズを有する。したがって、当業者は、透過化された細菌細胞内にポリペプチドを保持するために、そのようなポリペプチドを第2ポリペプチドと必ずしも結合させる必要はないことを理解するであろう。
【0086】
DNA結合タンパク質
本発明者は、透過化後の細菌細胞内にDNAが保持されることを見出した。したがって、1つの実施形態において、ポリペプチドをDNA結合タンパク質と結合させて、タンパク質複合体を形成し、これはDNAと結合し、細菌細胞の内部に保持される。本明細書中で用いられる場合、「DNA結合タンパク質」とは、2本鎖または1本鎖DNAを認識する少なくとも1つのモチーフを含むDNA結合ドメインを含む任意のタンパク質を指す。当業者には知られているように、DNA結合ドメインは、ヘリックス・ターン・ヘリックス、亜鉛フィンガー、ロイシンジッパー、翼状ヘリックス、翼状ヘリックス・ターン・ヘリックス、ヘリックス・ループ・ヘリックス、DNAを認識する免疫グロブリンフォールド、またはB3ドメインを含む。ポリペプチドをDNA結合タンパク質と結合することによって、有利には、本発明のスクリーニング法においてポリペプチドをコード化するプラスミドなどのDNAの回収が増大する。
【0087】
DNA結合タンパク質の例としては、細菌コンピテンスタンパク質、例えばこれらに限定されるものではないが、大腸菌DNA結合タンパク質、淋菌(Neisseria gonorhoeae)DNA結合タンパク質、たとえばComE、アデノウイルスE2タンパク質、AraC転写因子、塩基性ヘリックス・ループ・ヘリックス転写因子、塩基性ロイシンジッパー転写因子、ブチラート反応因子、セントロメアタンパク質B、COUP転写因子、初期増殖応答転写因子、Gボックス結合因子、GATA転写因子、HMGAタンパク質、ホメオドメインタンパク質、I−カッパBタンパク質、組み込み宿主因子、インターフェロン調節因子、インターフェロン−刺激遺伝子因子3、Kruppel様転写因子、ロイシン応答調節タンパク質、マトリックス付着領域結合タンパク質、メチル−CpG結合タンパク質、MutSホモログ2タンパク質、骨髄性−リンパ性白血病タンパク質、NF−カッパB、NF1転写因子、核呼吸因子、ガン遺伝子タンパク質p55、起点認識複合体、対合ボックス転写因子、POUドメイン因子、プロトオンコジーン因子、Rad51リコンビナーゼ、Rad52DNA修復および組換えタンパク質、複製タンパク質A、複製タンパク質C、網膜芽細胞種タンパク質、Smadタンパク質、SOX転写因子、Tボックスドメインタンパク質、TCF転写因子、テロメア結合タンパク質、Toll様受容体9、トランス活性化因子、および翼状ヘリックス転写因子が挙げられる。1つの実施形態において、DNA結合タンパク質は大腸菌DNA結合タンパク質である。別の実施形態において、DNA結合タンパク質は淋菌タンパク質、たとえばComEである。
【0088】
細胞壁結合タンパク質
所望の活性に関してスクリーニングされるポリペプチドを、細菌細胞壁結合タンパク質と結合させることができる。当業者は、細胞壁結合タンパク質の選択が宿主細胞種に依存することを理解するであろう。なぜなら、異なる細菌は異なる細胞壁組成を有するからである。細菌はペプチドグリカン(PG)から構成される細胞壁を有するが、種間の化学修飾は、異種間結合に影響を及ぼし得る。当業者は、特定の細菌種における使用に好適な細胞壁結合タンパク質を容易に決定することができるであろう。
【0089】
細菌細胞壁結合タンパク質は、それによって天然の構造中のポリペプチド鎖が細菌細胞壁上の特定の分子または分子構造を認識でき、結合できるドメイン構造を有することが知られているタンパク質を含む。したがって、「細菌細胞壁結合タンパク質」という用語は、細菌細胞壁に特異的に結合するタンパク質の部分であるタンパク質ドメインを包含する。細菌細胞壁結合タンパク質の例としては、バクテリオファージによりコードされるような細胞壁ヒドロラーゼ、細菌の細胞壁ヒドロラーゼおよび異なる自己溶菌酵素が挙げられる。さらに含まれるのは、バクテリオファージおよび他のウイルスのDNAによりコードされる受容体分子である。細菌細胞壁結合タンパク質が、細菌に特異的に結合できるバクテリオファージ起源の加水分解酵素由来である場合、細胞壁結合タンパク質はそれらの結合能力を保持するが、好ましくは著しい加水分解活性を有さない。
【0090】
1つの実施形態において、細胞壁結合タンパク質は大腸菌の細胞壁に共有結合する。たとえば、大腸菌宿主細胞について、PAL、OmpA、YiaD、YfiB、およびMotBに存在する、保存された約100のアミノ酸PG結合ドメインを有する内因性PG結合タンパク質がある(Parsons et al., 2006)。しかし、たとえばシュードモナス属(Pseudomonas)φKZファージ(KzPG)由来の約70のアミノ酸PG結合ドメインなどの他の生物由来のタンパク質は、大腸菌で十分に発現され、高親和性で細胞壁に結合することが示されている(Briers et al., 2009)。したがって、PGと結合するタンパク質由来のPG結合ドメインを本発明の方法で細菌細胞壁結合タンパク質として用いることができる。
【0091】
例示的実施形態において、所望の活性に関してスクリーニングされ、細菌細胞のサイトゾルで発現されるポリペプチドに、PG結合ドメインを融合させることができる。膜透過化により、PG結合ドメインは細胞壁と結合し、その結果、透過化細胞内で関心対象のポリペプチドが保持される。細胞内での関心対象のポリペプチドの保持を潜在的にさらに増強するために、当業者は、ポリペプチドが細菌細胞壁結合タンパク質に加えてDNA結合タンパク質と結合できることを理解するであろう。
【0092】
あるいは、関心対象のポリペプチドは、細菌細胞壁に共有結合することができるタンパク質と結合することができる。好ましくは、タンパク質は周辺質を標的とするシグナルを含む。したがって、ポリペプチドは細菌細胞のサイトゾル中で発現されるが、周辺質を標的とし、ここで膜透過化前に細胞壁に結合する。
【0093】
非限定的例として、細胞壁と共有結合する細菌細胞壁結合タンパク質は、細胞壁と結合することができ、外膜付着に必要な機能的N末端シグナル配列が欠如したリポタンパク質であり得る。たとえば、リポタンパク質は大腸菌LPPであり得る。LPPは、トリマーコイルドコイルを形成する多量の大腸菌タンパク質である。その天然の形態において、一端は脂質化により外膜に結合し、多端はC−末端リシンにより細胞壁に共有結合する。リポタンパク質は、リポタンパク質に周辺質を標的とさせる配列、たとえばOmpF周辺質ターゲティング配列をさらに含み得る。1つの実施形態において、リポタンパク質は、外膜付着に必要な機能的N末端シグナル配列が欠如した大腸菌リポタンパク質である。
【0094】
本明細書の教示を考慮して、当業者は、細菌細胞壁と共有結合し、本発明の方法での使用に好適なタンパク質を同定または設計できるであろう。
【0095】
本発明の1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドは、KzPGドメインならびにスペーサー、SNAPおよび/またはDBPから選択される1以上の他のドメインを含む融合ポリペプチドである。特定の1つの実施形態において、融合ポリペプチドは、1以上スペーサーならびにKzPG、SNAPおよびDBPドメインを含む。
【0096】
スペーサー
1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドを、1以上スペーサーを含む融合ポリペプチドとして発現することができる。「スペーサー」は、本明細書中で用いられる場合、融合ポリペプチド中に含まれて、細菌細胞におけるポリペプチドの発現を増強するか、または立体障害を減少させて、所望の活性に関してスクリーニングされるポリペプチドがその所望の三次構造をとることができ、および/またはその標的分子と適切に相互作用することができるペプチドまたはポリペプチドを指す。したがって、融合タンパク質は、融合ポリペプチド中の1以上のポリペプチドドメインの前、後、または間に1以上スペーサーを含み得る。スペーサーおよび所望のスペーサーの同定方法については、たとえば、George, et al. (2003)を参照のこと。
【0097】
1つの実施形態において、スペーサーは、1〜50のアミノ酸残基の長さ、または約1〜25残基、または約5〜15残基の長さである1以上のアミノ酸配列を含む。たとえば、スペーサーは、I27、RL1、RL2、RL3、RL4、RL5および/またはRL6の1以上から選択することができる。当業者は、限定された数のアミノ酸置換、たとえば、1、2、3、4または5のアミノ酸置換を、スペーサーとして機能するその能力に影響を及ぼすことなくスペーサーに導入することができることを理解するであろう。特定の1つの実施形態において、1以上スペーサーは、配列番号6〜12または16のいずれか1つから選択される。したがって、1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドは、I27、RL6、KzPG、SNAPおよびDBPを含む融合ポリペプチドである。
【0098】
スクリーニング法およびタンパク質進化
本発明は、標的分子に対する所望の活性に関してポリペプチドをスクリーニングするための方法を提供する。本明細書中で用いられる場合、「所望の活性」という用語は、ポリペプチドの任意の潜在的に有用な活性を指し、これらに限定されるものではないが、結合、酵素修飾、フォールディング安定性および/または熱安定性を包含する。
【0099】
「標的分子」という用語は、ポリペプチドと結合し、および/またはポリペプチドにより修飾される分子を指し、たとえば抗体、受容体、抗原、酵素などであり得る。したがって、「標的分子」は、酵素基質または結合に関して評価される分子(たとえば、リガンド、エピトープ、抗原、マルチマー化パートナー、例えばホモもしくはヘテロダイマーパートナーなど、またはそれらの任意の組み合わせ)について言及する際に使用することができる。
【0100】
ポリペプチド活性は、単一のポリペプチドを発現する単一種の細胞の関連で、またはそれぞれが異なるポリペプチドもしくはポリペプチド変異体を発現する細胞のライブラリーの関連で、スクリーニングあるいは選択することができると理解される。したがって、本発明の方法は、インビトロタンパク質進化について用いることができる。インビトロタンパク質進化により、多数のタンパク質機能および特性を調査することが可能になり、典型的には2つの主なステップ、すなわち多様化および選択を含む。多様化は、ポリペプチドをコードする核酸の多様なライブラリーを生成する能力に依存する。選択は、所望の活性に関してライブラリーをスクリーニングし、活性を遺伝子型と結びつけることにより、たとえば、観察された活性の原因となる遺伝子型を含むライブラリーの数を特定することにより達成することができる。
【0101】
DNAライブラリーは、ポリペプチドをコード化するDNA挿入物(DNAフラグメント)を含む組換えベクターのコレクションである。DNA挿入物の起源は、ゲノム、DNA、合成または半合成であり得る。ポリペプチドは、所望の活性を有し得、たとえば関心対象のポリペプチドは、結合タンパク質、たとえば抗体、または酵素、たとえば、ポリメラーゼ、リガーゼ、制限酵素、トポイソメラーゼ、キナーゼ、ホスファターゼ、代謝性酵素、触媒酵素、または成長因子ホルモン、抗菌ペプチド、抗原、受容体、レポータータンパク質、免疫調節タンパク質、神経伝達物質、構造タンパク質、転写因子またはトランスポーターであり得る。1つの実施形態において、ポリペプチドは抗体または酵素である。したがって、本発明の方法を、所望の活性を有するポリペプチドの変異体に関してスクリーニングするために用いることができる。
【0102】
たとえば、結合タンパク質または酵素のDNAライブラリーのクローニングおよび構築は、当該技術分野で公知の方法を用いて実施することができる。たとえば、Lutz and Patrick (2004)は、ライブラリー多様性を生成させる方法およびタンパク質工学で使用するための遺伝子組換えの方策を概説している。ディスプレイされたポリペプチド変異体のスクリーニングのために、表面ディスプレイされたライブラリーに用いられる方策を、本発明の方法に採用し、適応させることができる(Becker et al., 2004; Kenrick et al., 2007; Miller et al., 2006; Daugherty et al., 2000)。
【0103】
核酸のライブラリーを複数の細菌細胞に導入し、その結果、細菌細胞のそれぞれにおいてライブラリーのメンバーが発現される。発現されることに加えて、ポリペプチドを、それらの機能または特性を評価するために、透過化された細菌細胞内で保持するか、または細胞壁に付着させる。ポリペプチド、たとえば抗体などの結合タンパク質の核酸ライブラリー、または酵素の核酸ライブラリーを、種々の方法により、例えば点突然変異などの突然変異、欠失、および挿入の導入によるか、または組換え事象により、生成させることができる。変異体のライブラリーを生成させる方法は、当該技術分野で公知であり、エラープローンPCR、DNA修復不全細菌におけるDNAの合成およびDNAの化学修飾を含む。組換えによりライブラリーを生成させる方法は、当該技術分野で公知であり、遺伝子シャッフリング、高組換え誘導細菌におけるDNAのアセンブリ、合成核酸ライブラリアセンブリなど、またはそれらの任意の組み合わせが挙げられる。このように、ポリペプチドをコード化するポリヌクレオチドのライブラリーを複数の細菌細胞に導入することができ、その結果、細菌細胞のそれぞれにおいてライブラリーの1つまたはメンバーが発現される。
【0104】
いくつかの実施形態において、ライブラリーは、ポリペプチドの2以上の変異体を含み、この場合、それぞれの変異体は、アミノ酸配列において若干の変化を有する独特のポリペプチドを含む。他の実施形態において、ライブラリーは、2以上の無関係な配列を含む。たとえば、酵素を阻害できる候補ポリペプチドを同定するために、ランダム配列または所定の配列のライブラリーを調べることができる。ライブラリーは、少なくとも2、少なくとも5、少なくとも10、少なくとも50、少なくとも100、少なくとも1000、少なくとも10,000、少なくとも100,000、少なくとも1,000,000、少なくとも107またはそれ以上のメンバーを有し得る。
【0105】
結合タンパク質ディスプレイ
1つの実施形態において、本発明の方法を、たとえば抗体などの結合タンパク質の発生に適用することができる。したがって、1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドは結合タンパク質であり、標的分子は、結合タンパク質が結合することができる任意の分子であり、所望の活性は、標的分子に対する結合、および/または結合の程度である。本発明の方法は、たとえば、結合タンパク質をコード化するポリヌクレオチドを含む細菌細胞を培養して、細胞においてタンパク質を産生することを含み得る。細胞を続いて透過化し、透過化された細胞を標的分子と接触させる。当該技術分野における任意の好適な方法を、ポリペプチドが標的分子と結合するかどうか、および/または標的分子と結合する程度を決定するために用いることができる。
【0106】
本発明の方法は、結合タンパク質ディスプレイライブラリのスクリーニングに特に適している。タンパク質の細胞外空間に対するターゲティングを絶対的に必要とするインビボ表面ディスプレイの他の方法と異なり、細胞膜はディスプレイタンパク質に対して提示された標識された標的との相互作用を防止するので、本発明の方法は、宿主細胞の細胞質において親和性タンパク質を発現でき、フォールディングすることができる。したがって、スクリーニングパラメータは、細菌の細胞質における親和性変異体タンパク質の高収率および生産的フォールディングを含み得る。
【0107】
さらに、細胞質タンパク質発現およびフォールディングが還元環境にある場合、本発明の方法を適用して、抗体の変異体、またはそれらの自然の形態でジスルフィド結合を有し還元環境において生産的フォールディングが可能である他のタンパク質を選択することができる。選択された変異体は、フォールディング安定性に関してドメイン内またはドメイン間ジスルフィド結合に依存しないので、更に安定であることが予想される。このアプローチは、中和または標識のいずれかのために標的の細胞内結合に使用できる抗体を開発するために応用される。
【0108】
本発明の方法は、したがって、当該技術分野で公知のスカフォールド、例えば1本鎖抗体(scFv)、ドメイン抗体、Fab、および非抗体スカフォールド、例えばリポカリン、FN3、ユビキチン、γ−B−クリスタリンなどをはじめとする種々の結合タンパク質のディスプレイおよび選択のためのプラットフォームとして用いることができる。
【0109】
酵素ディスプレイ
本発明の方法は、酵素および酵素ライブラリーのディスプレイのため、そして酵素特性の発生のために用いることができる。したがって、1つの実施形態において、所望の活性に関してスクリーニングされるポリペプチドは酵素であり、標的分子は酵素の基質であり、所望の活性は標的分子に対する結合および/または標的分子の酵素修飾である。当業者は、他の表面ディスプレイ技術を用いる酵素活性に関してのアッセイを開発するための方法を、本発明の方法に対するアッセイと同等に適用することができることを理解するであろう。
【0110】
本発明の方法はまた、透過化され、次いで水中油中水(w/o/w)エマルジョンとして懸濁された宿主細胞において発現された酵素ライブラリーの使用によく適している。Aharoni et al. (2005)は、パラキソナーゼの改善のためにFACSによりw/o/wエマルジョン中で表面ディスプレイされた酵素ライブラリをを使用することの有用性を示した。非透過性油膜中に封入することの利点は、拡散性基質および生成物を酵素活性およびコーディング核酸配列の近くに保持できることである。しかし、Aharoni et al. (2005)により記載されているスクリーンは、酵素が宿主細胞の外部でディスプレイされることを必要とする。本発明の方法を使用して、酵素ライブラリーの細胞内発現およびフォールディングを酵素機能における改善のために用いることができた。
【0111】
本発明の方法において、酵素を産生するために、酵素をコード化するポリヌクレオチドを含む細菌細胞を培養する。細菌細胞の透過化後に、細胞を酵素の基質と接触させ、公知方法を用いて、酵素が基質を修飾するかどうか、および/または基質の酵素修飾率を測定することができる。
【0112】
数例において、標的分子(たとえば酵素基質)を細菌細胞と結合させることが望ましい場合がある。当業者は、標的分子を透過化された細菌細胞の任意の成分に、直接的または間接的のいずれかで結合させることができることを理解するであろう。直接結合は、非限定的例として、標的分子を細菌細胞壁と結合させることにより達成することができる。標的分子の間接的結合は、標的分子を、所望の活性に関してスクリーニングされるポリペプチドと結合している第2ポリペプチドと結合してタンパク質複合体を形成することによって達成することができる。たとえば、標的分子を、透過化された細菌細胞の内部にタンパク質複合体を保持するために十分な分子サイズを有するポリペプチドと結合させることができるか、またはDNA結合タンパク質と、もしくは本発明の方法で用いられるような細菌細胞壁結合タンパク質と結合させることができる。標的分子を細菌細胞と結合させることにより、有利には、たとえばFACSまたは磁気ビーズ選択によるなどの技術を使用して、活性酵素を提示する細菌細胞の単離が可能になる。
【0113】
当業者は、標的分子を細菌細胞と結合させるために好適なカップリング化学を容易に決定することができるであろう。好適なカップリング化学としては、アクリダイトおよびマレイミドなどのチオールカップリング試薬でのシステイン標識、アミン標識、ならびにカルボキシル標識が挙げられ、これらはPierce Protein Research ProductsおよびInvitrogenをはじめとする供給元から市販されている。
【0114】
フローサイトメトリー分析
本発明の細胞ディスプレイ技術は、関心対象のポリペプチドの数千もの分子を一度に提示することができ、リボソーム/mRNAディスプレイまたはファージディスプレイなどの分子ディスプレイ技術と異なり、フローサイトメトリー技術を用いて、たとえば蛍光活性化細胞分類(FACS)機械を用いてスクリーニングすることができる。ライブラリーにおける陽性事象をとらえることができるだけでなく、酵素活性または親和性などのパラメーターを各陽性メンバーについて同時に定義することができ、これによりスクリーンのアウトプットを改善することができる。フローサイトメトリーを実施するための機器は当該技術分野で公知であり、FACS Star Plus、FACScanおよびFACSort(Becton Dickinson)、Epics C、ならびにMoFloが挙げられる。フローサイトメトリー技術は、一般的に、液体試料中の細胞の分離を含む。典型的には、FACSの目的は、1以上の特性、たとえば、標的分子の存在について細胞を分析することである。フローサイトメトリー分析を実施するための方法は、当該技術分野で周知である。たとえば、酵素活性を分析するためのFACSの使用法の概説は、Farinas(2006)により記載されている。
【0115】
本発明に関して、フローサイトメトリーは、連続して実施できる複数回のスクリーニングに有用である。細胞を初回の分類から単離することができ、直ちにフローサイトメーターに再導入し、再度スクリーニングして、スクリーンのストリンジェンシーを改善することができる。フローサイトメトリーは本質的に粒子分類技術であるので、細胞を培養する能力は必要ではない。無生育性細胞から核酸を回収するための技術は当該技術分野で周知であり、たとえば、PCRをはじめとするテンプレート依存性増幅技術を挙げることができる。
【0116】
所望の活性を有するポリペプチドを生成する細菌細胞が同定された後、ポリペプチドをコード化するポリヌクレオチドを含むDNAを、任意の好適な既知技術を用いて細菌細胞から単離することができる。したがって、通常の手順を使用して、ポリペプチドをコード化するDNAを単離し、配列決定することができる。必要に応じて、ポリヌクレオチドは、所望の活性に関してスクリーニングされる変異体の別のライブラリーを生成させるために、もう1ラウンドの多様化をおこなうことができる。このようにして、反復プロセスを使用して、ポリペプチドの所望の活性を最適化することが可能である。
【0117】
キット
本発明の方法の実施に必要な成分は、キットの形態で都合良く提供することができる。当業者には理解されるように、キット中の種々の成分は、個々の容器中もしくはアリコートで供給することができるか、または溶液成分を異なる組み合わせ、異なる濃度で組み合わせて、本発明の方法の最適な実施を達成することができる。使用するまで成分が安定な形態で維持されるように、キットのどの成分を組み合わせることができるかを決定することは、当業者の知識の範囲内である。
【0118】
本発明のキットは、典型的には、最低でも、第1ポリペプチドをコード化するポリヌクレオチドをベクター中に挿入するための部位、および第1ポリペプチドと結合して、透過化された細菌細胞の細胞壁の内部に保持されるかまたは細胞壁に付着したタンパク質複合体を形成する第2ポリペプチドをコード化するオープンリーディングフレームを含むベクターを含むであろう。好ましくは、キットはまた、細菌細胞を透過化するための薬剤も含む。1つの実施形態において、キットは、細菌細胞、好ましくはグラム陰性細菌細胞をさらに含む。他のさらなる成分がキットとともに含まれていてもよいか、または所望により他の成分を最終使用者が提供してもよい。
【実施例】
【0119】
実施例1 大腸菌を透過性にする界面活性剤のスクリーニング
大腸菌細胞を透過性にするであろう、界面活性剤を同定するため、イオン性(n−ドデシル−β−イミノジプロピオン酸;デシルトリメチルアンモニウムクロライド;ドデカノイルサルコシンナトリウム;アンゼルゲント3−10)、および非イオン性(ジメチルオクチルホスフィンオキシド[Apo8];ジメチルデシルホスフィンオキシド;n−オクチル−β−D−チオグルコピラノシド[8TGP];スクロースモノドデカノアート;メガ(Mega)10;トウィーン(Tween)80;トリトン(Triton)X100;トリトン(Triton)X114)の両方の多くの界面活性剤を、膜−不透過性色素、ゲルレッド(Gel Red)(Biotium社、カタログ番号41002)の取り込み、およびGFPの遊離の両方によってスクリーニングした。透過処理のテストを行った界面活性剤は、Anatraceから購入した。
ここに述べる実験に用いた大腸菌宿主株は、全てK12由来のArgentum細胞株(ΔmcrAΔ(mrr−hsdRMS−mcrBC)ΔendA lacZΔM15)(Alchemy Biosciences社)であった。しかしながら、本発明の方法は、B株由来のBL21(F−dcm ompT hsdS(rB−mB−)gal)、およびK12クローン株DH5α(F−endA1 glnV44 thi−1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ(lacZYA−argF)U169,hsdR17(rK−mK+),λ−)を用いてもテストを行い、同様な結果を得た。
GFPを、アラビノース誘導性の高コピー数ベクター(pAra1::GFP5)にクローン化した。発現は、新たに画線したコロニーを含むシャーレから重度に播種した培養液より行った。培養液は、37°Cで、アラビノースを最終濃度0.2%になるまで添加して発現を誘導するときに、OD600が約0.3となるまで培養した。誘導された培養液は、収穫まで25°Cで2時間振盪した。
1mLの誘導培養液を遠心分離して細胞を沈殿させ、300μLの0.5%界面活性剤含有LB中に懸濁して透過性にした後、25°Cで10分間培養した。透過性にした細胞は、沈澱させ、再懸濁し1xゲルレッド(Gel Red)を含む水に再懸濁し2分間置き、その後沈澱させ300μLのTBSで1度洗浄した。細胞は、300μLのTBSに懸濁し、蛍光顕微鏡検査のために、DABCO/グリセリン(0.0325gのDABCOを900μLのグリセリン+100μLのPBS中に溶解)を加えて処理した。
サンプルは、スライド式カメラ(SPOT RT 2.3.0ソフトウェアv4.6)を用いたオリンパスProvis AX70光学顕微鏡、またはLeica TCS SP2共焦点レーザ走査顕微鏡/Leica DM IRE2倒立顕微鏡(Leica共焦点ソフトウェアv2.0)のいずれかにより可視化した。
図1は、界面活性剤によるGFP発現大腸菌の透過処理の結果を示す。未処理細胞は緑色(GFP)であるが、透過性にされた細胞は、内部のGFPを失い、DNA結合性ゲルレッド(Gel Red)色素を取り込み赤色に染色される。また、ノニデット−40もいくらか透過性化を示すが、Apo8とMega10は、より高い割合の透過化された細胞を示す。これら2種の界面活性剤の0.5%ずつの混合物は、剤86と呼ばれ、別の界面活性剤n-オクチル−β−D-チオグルコピラノシド(8TGP)と同様に、ほとんど完全に透過処理することを示した。Mega10、Apo8および8TGPは、すべて非イオン性界面活性剤で、イオン性界面活性剤と比べ、タンパク質の折りたたみや機能に対して、破壊の程度が低い。
細胞壁は、透過処理によっても無傷であったので、上述した界面活性剤での透過処理による上澄みの可溶性タンパク質の抽出物をSDS−PAGEにより分析した。透過処理した細胞に、ニワトリ卵白リゾチーム(Boehringer Mannheim社;837 059)を最終濃度2mg/mLまで、透過処理している細胞のサンプルに加え、細胞壁を除去し、全細胞タンパク質を遊離させた。β−メルカプトエタノールを含むSDS−PAGEローディング色素を、95°Cで2分間変性したサンプルに加えた。20μLのサンプルを9%SDS−PAGEにロードし、クーマシー・ブリリアント・ブルー/メタノール/酢酸で染色/固定した。
図2は、可溶性タンパク質の遊離は、顕微鏡検査で見られたGFPの遊離およびGel Redの取り込みと直接的に関連していることを示す。無傷な細胞壁を有する細胞からのタンパク質の遊離と、リゾチームを用いて細胞壁を除去した細胞からのそれとの間には、明白に差があったが、細胞壁に被包された細胞が遊離する可溶性タンパク質は、大きさが約120kDまでであった。これは、球状タンパク質が、グラム陰性真正細菌の細胞壁を構成するペプチドグリカン格子の細孔を通って、細胞から出ることができなくなる限界寸法であると思われる。
【0120】
実施例2. 宿主DNAを保持する透過処理溶液のスクリーニング
本発明の方法を、改良された特性のタンパク質変異体を求めて遺伝子ライブラリーをスクリーニングするのに使用するなら、発現されるタンパク質と、それをコードする核酸との間に関連がなければならない。膜の透過処理工程で、DNAが、細胞壁を通って失われるのを防ぐ障壁が除去されるので、宿主DNAの損失を減少し、あるいは防止しうる、透過処理の条件を調べた。
細胞の透過処理を、0.5%8TGPを用い異なる培地中で行い、DNAの損失を、DNA結合色素、Gel Redを用い、蛍光顕微鏡検査法により調べた。
【0121】
テストした透過処理培地(全培地に0.5%8TGPを含む)の組成
LB培地(10gトリプトン、5g酵母抽出物、10g/LのNaCl)
LB(塩)培地(10gトリプトン、5g/Lの酵母抽出物)
50mM−Tris、pH7.5
50mM−HEPES、pH7.0
170mM−NaCl
250mM−NaCl
25mM−Tris、pH7.5+1.5%PEG6000(w/v)
50mM−Tris、pH7.5+3%PEG6000(w/v)
50mM−Tris、pH7.5+170mM−NaCl
50mM−Tris、pH7.5+250mM−NaCl
好適な透過処理培地は、LB細菌培地と確認された。したがって、以降、透過処理は、0.5%8TGPを含むLBまたは剤86を含むLB(0.5%Mega10および0.5%Apo8を含むLB)のいずれかを用いて行った。
【0122】
実施例3. タンパク質のテトラマー骨格への融合
例1に記述した実験で見られたとおり、大きさが約120kDより大きいタンパク質は、細胞壁により、透過性にした大腸菌細胞の内部に保持された。したがって、120kDより小さい目的タンパク質も、タンパク質パ−トナーと融合することで合計寸法が120kDを超えるようにできれば、細胞壁カプセル中に保持されるだろうと考えられた。
このため、融合パートナーとして用いるため、大腸菌から6種の異なる四量体タンパク質をクローン化した。すなわち、β−gal、BetB、G5K、GshB、RhnA、およびYdcWで、それぞれのモノマー寸法は116kD、52kD、39kD、35kD、47kDおよび50kDであった。
アラビノース誘導性高コピー数ベクターを、四量体発現のために構築した。蛍光性基質と共有結合で結合する20kD領域であるSNAPタグ(NEB社/Covalys)が、テトラマー遺伝子の上流にクローン化され、発現レポーターとして使用された。精製と検知を容易にするため、6xHisエピトープも、融合タンパク質のN末端に挿入された。
前記アラビノースベクターpAra3::His6::SNAPの配列が、SEQ ID NO:1(配列番号)として示される。
融合タンパク質の発現は、0.2%アラビノースを加えて誘導し、培養液を25°Cで2時間培養した。
本発明の方法により、蛋白質ディスプレイのために細胞を透過性にするプロトコルは、以下のとおりであった:
1. 遠心分離により細胞1mLを沈澱させる。
2. 300μLの0.5%8TGP/LBに細胞を再懸濁する。
3. 25°Cで10分間培養する。
4. 遠心分離により細胞を沈澱させる。
5. 200μLのTBSまたはLBに細胞を再懸濁する。
【0123】
SNAP発現レポーター領域を、膜不透過性SNAP色素(Covalys、New England Biolabs社)で標識付けするプロトコルは、以下のとおりであった:
1. 20nmolのBG−488(緑色色素)またはBG−547(赤色色素)を、300μLのDMSOに200x原液として溶解する。
2. 1μLの200x原液を200μLの透過性にした細胞を懸濁したTBSまたはLBに加える。
3. 25°Cで15分間培養する。
4. 遠心分離により沈澱させ、また300μLのTBSに再懸濁させて、細胞を2度洗浄する。
【0124】
四量体融合タンパク質が、透過性にした細胞カプセル中に保持されるのを蛍光顕微鏡検査法により観察するためのプロトコルは、以下のとおりであった:
1. 20μLの細胞懸濁液を顕微鏡用スライドガラス上に滴下し、カバーガラスで覆い、端部をマニキュア液でシールする(湿式プレパラート);あるいは細胞の液滴をほぼ乾燥させて、その上に20μLのDABCO/グリセリンを滴下し、カバーガラスで覆い、端部をマニキュア液でシールする(乾式プレパラート)。
2. オリンパスまたはLeica蛍光顕微鏡のいずれかを用いて可視化する。
【0125】
完全長融合タンパク質の発現は、SDS−PAGEゲルに掛けてから、タンパク質抽出物のウェスタンブロット法でα−His6抗体をプローブに用い確認した。全ての四量体コンストラクトは、大腸菌中で検知可能なレベルで発現した(図3A)。
大腸菌内で発現した四量体融合タンパク質の蛍光顕微鏡検査によれば、β−galおよびG5Kは、顕著な封入体を含み、恐らく前記融合タンパク質の折りたたみが困難であるために、蛍光レベルが低いことが分かった。しかしながら、図4によれば、SNAP蛍光により判定されるとおり、前記融合タンパク質の発現は、GshBでは良好、RhnA、BetBおよびYdcWでは、極めて良好であった。透過性にした宿主細胞中の融合タンパク質の分布は、明視野顕微鏡検査および蛍光の両方から明らかなように複数のフォーカスを有し、均一でないことが注目された。しかし、蛍光性SNAP基質は、ミスフォールドされた領域には結合されていないであろうから、かつ信号が非常に強いことから、これらの物体は恐らく折り畳まれたタンパク質の凝集体であって、大腸菌中で過剰発現しているタンパク質においてしばしばみられる、折り畳まれていないタンパク質の封入体ではないと考えられる。
SNAP::テトラマー融合体は、His6-N末端エピトープをまた有していた。抗体といった大きな分子が、大腸菌細胞壁の格子構造を通り抜けられるかを調べるため、透過性にした細胞にSNAP::テトラマー融合体を検出するαHis抗体をプローブとして設けた。
1. 前記His6::SNAP::BetB骨格融合体の発現および透過処理を上述のように実施した。
2. BG−547 SNAPリガンドによる標識化を上述のように実施した。
3. 200μLの透過性にしたSNAP−標識化細胞をLB中で3回洗浄し、ポリエチレンイミン(PEI)コートしたカバーガラス上に沈降させた。過剰な細胞培地を、吸引して除去し、前記スライドガラスを風乾させた。
4. 細胞を、ブロッキング緩衝液(1%BSA、1%冷水魚ゼラチン(Sigma社、G7765)、アジドの0.02%PBS−Tween20溶液)中で、1時間ブロックした。
5. 細胞を、ブロッキング緩衝液で1:10に希釈したαHis一次抗体(Abcam社、AB9136−100)中で、一晩25°Cで培養した。
6. 細胞を3回PBS−Tween20中で洗浄した(各回10分間)。
7. 細胞を、ブロッキング緩衝液で1:2,000に希釈した二次抗体(Molecular Probes、A11015)中、室温1時間培養した。
8. 細胞を3回PBS−Tween20中で洗浄した。
9. DABCO/グリセリンを封入剤に用い、共焦点オリンパス顕微鏡で観察した。
【0126】
図5は、αHis抗体が細胞壁カプセルの内部でSNAP蛍光性リガンドと共存しており、細胞壁の細孔が、比較的大きなタンパク質が、カプセル内部空間中に拡散を許すのに十分な大きさであることを表わしている。このように、非常に大きなタンパク質リガンドも、本発明の方法により、細胞質中で発現する親和性タンパク質のための親和性基質として使用できる。
細胞下物体の生成が変化するか否かを見るためにSNAP融合パートナーおよび発現レポーターをHALOタンパク質(Promega社)と比較した。前記HALOタンパク質は、SNAPと同様に膜不透過性蛍光性基質(Alexa fluor 488;G1001、Promega社)と共有結合で結合する。HALOレポーター遺伝子は、四量体発現コンストラクト中のSNAP遺伝子の位置にインフレームに直接クローン化した。HALO::四量体骨格タンパク質の発現を、SNAP変異体と比較した。透過性にしたHALO細胞の標識付けは、実質的にSNAPに対して記述したように、またメーカーの説明書に従い実施した。図6は、HALO::テトラマーおよびSNAP::テトラマーの発現パターンは、同様であったが、例外的にHALO::RhnA融合タンパク質は、部分的にSNAP::RhnA融合体より溶解性で、複数の蛍光フォーカスを有する細胞がより少数であったことを示す。
したがって、タンパク質を四量体骨格(ここでは、SNAPまたはHALO)への融合体として発現させ、つづいて大腸菌宿主細胞を適当な界面活性剤で透過性にすると、目的のタンパク質が、細胞壁の内側に保持できる。
【0127】
実施例4. 細胞骨格としてのDNA結合タンパク質
表現型を遺伝子型に結合するには、宿主細胞は、透過処理の後、また機能スクリーニングを通して、少なくともいくらかのエピソームDNAを保持しなければならない。宿主ゲノムのDNAならびにプラスミドDNAを保持する透過処理条件を見出したことで、発現した目的のタンパク質の保持骨格としてDNAが使用できると結論付けた。
このため、小型(80aa)の高親和性らせん−ヘアピン−らせんDNA結合タンパク質(DBP)を、Neisseria gonorrhoeae ComE遺伝子(ChenおよびGotschlich、2001)からクローン化し、これをアラビノース誘導性コンストラクト(pAra3::GFP::DBP;seq2)中で、GFPのC末端に融合した。
アラビノース誘導による発現を例1に記述したように実施した。細胞を透過性にし、例1および3に記述したように蛍光顕微鏡検査のために調製した。
図7は、GFP::DBP融合体(緑色)が透過性にした細胞中に保持され、DNA結合色素、Gel Red(赤色)と共存したことを示す。
したがって、タンパク質を、高親和性、非特異的DNA結合タンパク質への融合体として発現させ、つづいて大腸菌宿主細胞を適当な界面活性剤で透過性にすると、目的のタンパク質が、細胞カプセルの内側に保持できる。
【0128】
実施例5. 透過性にした細胞中へのDNA保持
DNA、ゲノムおよびエピソーム両方のプラスミドの、透過性にした細胞カプセル中への保持を実証するため、蛍光顕微鏡検査およびプラスミドDNA抽出のためにGFP5::DBPおよびHis6::eGFPを発現する細胞を調製した。
誘導後、細胞を透過性にし、つづいて凍結するか、TBS中で37°Cにおいて一晩振盪した。次の日、全サンプルを、蛍光顕微鏡検査でGFP、およびDNA結合色素Gel RedでカプセルDNA含量を見るために、または、プラスミドDNA調製のために処理した。
蛍光顕微鏡検査を例3に対して記述したように実施した。図8は、宿主細胞DNA(赤色)およびGFP5::DBP(緑色)双方とも、細胞カプセル中に、透過処理直後も、一晩37°Cで培養しても目立った減少もなく、保持されたことを示す。His6::GFP タンパク質は、透過処理直後に失われたが、宿主細胞DNA(赤色)は、透過処理直後も、一晩後も、同じく目立った減少もなく、保持された。
宿主ゲノムだけでなくプラスミドDNAも透過性にした細胞内に保持されることを確認するため、同じように調製されたサンプルに対してプラスミド少量調製を行った。
1mLの界面活性剤処理または無処理細胞から、プラスミド少量調製アルカリ溶解プロトコルによりプラスミドDNAを調製した。界面活性剤抽出による上澄み中に遊離されたプラスミドDNAを、Perfectprep Gel Cleanup(Eppendorf社、955152051)コレムおよび溶液を、メーカーのプロトコルに従って用いて、抽出した。
各サンプルの全量を1%アガロースゲルにロードし、富士フィルム社LAS−3000インテリジェントダークボックス(Intelligent Darkbox)でイメージリーダー(Image Reader)LAS−3000ソフトウェアおよびMulti Gauge v3.0ソフトウェアを使用して画像化した。
図9は、両細胞株からのプラスミドDNAをサンプルとした、TAE緩衝液を用いた臭化エチジウム染色1%アガロースゲルを示す。
図9のレーン1は、無処理細胞中の全プラスミドDNAである。レーン2は、透過処理工程の上澄み、レーン3は、透過処理後に細胞カプセル中に保持されたプラスミドである。図8で観察された可溶性His6::GFP タンパク質の完全な消失に拘わらず、透過処理による上澄み中へのプラスミド遊離が殆どないことが観測された。したがって、プラスミドDNAは、ほとんど完全に細胞壁により保持され、本発明の方法において改良されたタンパク質変異体の選別のために、遺伝子型を表現型に結合するために使用できる。
顕微鏡検査のデータを確認するように、TBSに懸濁した透過性にした細胞の37°Cでの一晩の培養後も、この培養でのプラスミドDNAの消失は全く見られなかった(レーン5)。
【0129】
実施例6. ペプチドグリカン結合骨格
膜透過処理後も保持されるいま一つの細胞構造は、ペプチドグリカン(PG)の格子状ポリマーでなる細胞壁である。
PGを非共有結合で結合するために、大腸菌内で良好に発現し、細胞壁に高親和性(K=3x107M−1)をもって結合することが既に知られている(Briersら、2009)、シュードモナスφKZファージ(KzPG)からの70aaPG結合領域をクローン化した。親和性タンパク質の選別用に、PGに対するKzPG−結合領域の親和性より、より高い親和性を標的に対して有する変異体を見つけるため、骨格結合タンパク質の親和性を増す必要がある。骨格結合部分の親和性を増すため、ComE DNA結合領域(DBD)およびPG結合領域の両方を同一の融合タンパク質内で結合した。したがって、両骨格(PGまたはDNA)からの融合タンパク質の最終解離定数は、それぞれの速度定数の倍数の近似値となる。
このため、発現ベクターpAra3::His6::KzPG::SNAP::DBP(SEQ ID NO:2(配列番号))を構築した。例1に記述したように発現を誘導し、細胞を例3に記述したように蛍光顕微鏡検査のために調製した。融合タンパク質の発現および分布は、例3に記述したようにSNAP標識化により観察した。
蛍光は、細胞の辺縁で、細胞壁の領域において認められ、より軽度には、カプセルの細胞壁で囲まれる空間内の広汎な領域において認められた。
本発明のさらなる態様では、目的のタンパク質を共有結合で、透過処理前の細胞骨格に付着させる。これを実現するため、周辺質内にコイルドコイル三量体を形成する、大腸菌に豊富なタンパク質LPPへのタンパク質融合体を用いた。その天然型においては、1端が脂質化により外膜と連結し、他端はC末端リジンにより共有結合で細胞壁に結合している。
OmpF周辺質標的シグナル配列をSNAP発現レポーターに融合し、さらにN末端シグナル配列を欠く57aa大腸菌LPP配列、外膜付着に必要なシステインが続く発現コンストラクトを構築した。発現ベクター、pAra3::OmpF::SNAP::LPP(SEQ ID NO:3(配列番号))は、例1に記述されるようにアラビノースで誘導され、細胞を蛍光顕微鏡検査のため、例3に記述されるように、調製した。融合タンパク質の発現および分布は、例3に記述したようにSNAP標識化により観察した。
図10が示すように、LPP融合タンパク質の分布は、細胞壁の表面にわたって不均一で、強い蛍光の場所と、蛍光のない場所があった。しかし、ほとんどのケースで細胞の極が、標識化された。
【0130】
実施例7. 四量体タンパク質骨格を用いたαGFP親和性タンパク質のディスプレイ
親和性タンパク質に適用する本発明の方法を実証するため、eGFPに対して免疫したラマから生成した単一領域抗体を細胞骨格ベクター中にクローン化した。ここに、αGFP抗体に対する特許出願に(WO2007/068313)挙げられている2配列の内、R35変異体のみが機能することを指摘したい(αGFP−R35;タンパク質データベースID 3K1K)。したがって、この配列を全実験において使用した。
前記αGFP−R35遺伝子を、pAra3::HALO::FLAG::RhnA四量体骨格のN末端融合体として、pAra3::αGFP(R35)::HALO::FLAG::RhnAベクター(SEQ ID NO:4(配列番号))を製作するためクローン化した。
前記抗体の標的基質としてHis6::eGFP融合タンパク質を製造するため、pAra3::His6::eGFPベクターも、構築した。例1に記述されるように、His6::eGFPタンパク質を誘導した。可溶性タンパク質が、0.5%8TGPを用いて細胞から遊離され、Ni−NTAアガロース樹脂(Qiagen社;30230)を用いてIMACにより精製された。His6::eGFPは、Ni−NTA樹脂からNTTW緩衝液+イミダゾール(500mM NaCl、50mM Tris−HCl、pH7.5、0.1%Tween20+200mMイミダゾール)に溶出された。
抗体::四量体融合タンパク質の発現および宿主細胞の透過処理は、例1および3に記述されるように実施した。
透過性にした細胞カプセル内で、αGFPをeGFPに結合するため、カプセル沈殿物を300μLのeGFP中に懸濁し、25°Cで20分間平衡化させてから、カプセルを遠心分離で沈澱させ、300μLのTBS中で1度洗浄した後、TBS中に再懸濁させた。αGFP/eGFPカプセルに対する蛍光顕微鏡検査を、例3に記述されるように、実施した。
図11は、HALOリガンド標識化した図5で観察されるフォーカスと関係があるかもしれない、より強く染色された複数のフォーカスが生じているが、透過性にしたαGFP::HALO::RhnA融合タンパク質を発現しているカプセルが、細胞の全域でeGFPに結合したのを示している。
したがって、ラマαGFP抗体の機能は細胞質内で発現し、さらに界面活性剤透過処理後も、カプセル内に保持される。
例3で記述され、また図5で観察されたαHis抗体の標識化は、すでに約150kDと大きめなタンパク質が、透過性にした細胞壁を拡散して通り、カプセルの内部に入ることができることを実証した。しかし、未変性の抗体は、柔軟なヒンジ領域で分けられた、3つのほぼ同じ大きさの領域を有する変則的な形状のタンパク質である。したがって、これらのタンパク質の示す有効半径は、はるかに小さな球状タンパク質に相応すると思われる。しかし、βバレル構造と約27kDの分子の大きさを有するGFPは、半径がその大きさに比例する、対称性のタンパク質であり、内部のαGFP抗体と結合するために、透過性にしたカプセルの細胞壁を通過することができた。
したがって、本発明の方法は、大腸菌細胞質中で親和性タンパク質を発現して、少なくとも30kDの対称性の標的に結合する親和性ライブラリーのディスプレイに使用できる。
【0131】
実施例8. PG−およびDNA結合タンパク質骨格を用いるαGFP親和性タンパク質のディスプレイ
本発明の方法は、PG−およびDNA結合領域と融合したαGFPラクダ抗体を用いてさらに実証された。
抗体::KzPG::SNAP::DBP融合タンパク質の発現、宿主細胞の透過処理、およびHis6::eGFPによる標識付けは、例6に記述されるように実施した。
αGFP融合タンパク質によるeGFPの結合の画像化には、湿式および乾式プレパラートの両方が用いられた。図12は、GFP蛍光において、両方の画像化法で有意な差があったことを示している。乾式プレーパラート(DABCO/グリセリン)にされた細胞は、ほとんどが内部蛍光で、明視野とeGFP標識の間の混合があり、細胞壁の周囲の領域は内部空間より光が強くない(図12B)。しかしながら、TBS中で直接プレパラートにされた細胞は、細胞壁に結合したeGFPと思われる強い蛍光の外部境界のはっきりした形状と、より弱い内部信号を有していた(図12A)。理論に縛られることなく、推測するならば、DABCO/グリセリン溶媒環境は、粘度が高くまた水性でないので、KzPG領域とペプチドグリカン細胞壁の間の相互作用を抑止したが、αGFPのeGFPとの、あるいはDBPのDNAとの結合は抑止しなかった。
しかしながら、親和性タンパク質または酵素のスクリーニング操作は、通常は水系環境で行われるので、親和性融合タンパク質の分布は、図12Aに観測された細胞壁結合湿式プレパラートに近いであろう。
【0132】
実施例9. 細胞壁への共有結合性付着によるαGFP親和性タンパク質のディスプレイ
本発明の方法は、αGFP抗体の共有結合による細胞壁への結合によりさらに実証された。
αGFP抗体は、アラビノース誘導性融合体として、OmpFシグナル配列の下流、かつSNAPおよびLPP配列の上流にクローン化された。
アラビノースで発現を誘導した後、OmpFシグナル配列は、新生タンパク質を、内部細胞膜を通して周辺質に送り、これが膜細孔を通るときに分裂される。
周辺質において、LPP領域は、2つの別のパートナー、野生型LPPまたは他のαGFP融合タンパク質とともに、三量体コイルドコイルを形成すると思われる。LPP領域のC末端の残基はリジンで、共有結合により、εアミン基を介して、恐らくはYbiS L,D−トランスペプチダーゼにより(Magnetら、2007)、大腸菌細胞壁に結合する。
OmpF::αGFP::SNAP::LPP融合タンパク質の発現、細胞透過処理、およびeGFP標識付けは、例8に記述のように実施した。
図13は、eGFPは、細胞壁の周りに不均一に、しかし強力に結合したことを示す(図13B)。eGFPは、OmpF::SNAP::LPP融合体をαGFP領域無しに発現している細胞に結合されなかった(図13A)。
OmpF::SNAP::LPP融合体の細胞壁への共有結合性付着は、最初に、前記融合タンパク質を発現している透過性にした細胞をSNAPリガンドで標識化し、その後標識化細胞カプセルのサンプルを95°Cで5分間加熱して実証した。図14は、細胞壁を標識化するSNAPリガンドからの蛍光は、加熱処理されたサンプルと、加熱されていないコントロールとの間で差が生じなかったことを示す。Gel Red染色でも、加熱処理されたサンプルにおいても、ゲノムDNAは、細胞中に保持され続けることを実証した。
【0133】
実施例10. 外膜透過処理実験
本発明のさらなる態様においては、外膜は、リガンド標的、例えば酵素基質、あるいはポリペプチド等に対して選択的に透過性にしてもよいが、このとき、スクリーンされた前記ポリペプチドは、細胞壁の内部に、あるいはこれに付着して保持される。
外膜を選択的に透過性にする条件を見出すため、一連の界面活性剤および緩衝液を選別した。大型リガンド(eGFP)および小型リガンド(Gel Red)の両方を用いて、外/内膜の透過処理で大きな、または小さな膜細孔が生成したかを調べた。
アラビノース誘導性OmpF::αGFP::SNAP::LPP(細胞壁に付着)またはαGFP::HALO::FLAG::RhnA(細胞質内性)を発現する大腸菌株を培養し、例1に記述されるように誘導した。
誘導された培養液1mLを、50mMのTris(pH8)で1回洗浄し、25mM Tris+1mM EDTA(pH8)または25mM Tris+2mM Ca2+(pH8)のいずれかの中に0.2〜0.4%の界面活性剤をいれた透過処理緩衝液変種中に懸濁し、25℃で10分間培養した。
透過性にした細胞は、適当な緩衝液で1回洗浄してから、Gel Red(1x水中)で染色し、TBSで洗浄した。これを、精製したHis6::eGFPとともに25℃で1時間培養し、遠心分離で沈殿させ、TBS中に再懸濁させ、湿式プレパラートとして蛍光顕微鏡検査法により観察した。
図15及び16は、Tris/Ca2+またはTris/EDTA緩衝液中の0.2%のApo8(A)またはTween20(B)が、外膜を選択的に透過性にし、大きなリガンド(eGFP)に、外膜を通させるが、内膜は通させないことを実証している。より小さな、膜不透過性のDNA結合リガンドGel Redは、大部分のサンプルで、細胞質まで部分的に透過性であって、いくつかの細胞において内膜にある程度の細孔形成が起きたことを示している。しかしながら、0.5%の8TGPまたは剤86である界面活性剤で処理され、外膜、内膜ともにeGFPに対して完全に透過性であるサンプルに比べて、Gel Red結合の程度は著しく減少した。
【0134】
実施例11. 蛍光選別および被包性ディスプレイの解析
細胞ディスプレイプラットフォームとして、リガンド結合クローンを識別するのに、本発明の方法は、蛍光標示式細胞選別(FACS)に非常に向いている。透過性にした大腸菌細胞のFACSによる選別での安定性を試験するため、3集団:i)eGFP、ii)αGFP::KzPG::SNAP::DBP、およびiii)His6::SNAP::BetBを誘導して発現させた。
eGFP発現細胞は、透過性にせず、無傷の大腸菌細胞での蛍光のポジティブコントロールとした。αGFP::KzPG::SNAP::DBP発現細胞は、本発明の方法により透過性にし、SNAP BG−488リガンド(緑色)で標識付けした。His6::SNAP::BetB発現細胞は、本発明の方法により透過性にし、SNAP BG−547リガンド(赤色)で標識付けした。
細胞はPBS中に懸濁し、混合集団の選別のために、ほぼ同数を混合し、あるいは信号の較正のため、別々に選別した。細胞選別は、Becton Dickson Influx FACSにより実施した。データ解析は、FlowJoソフトウェアにより実施した。大腸菌選別のためのパラメーターは、操作者が決定した。
図17は、蛍光により3集団が識別できたことを示している。選別された集団の再分析は、前記選別がそれぞれの比較的純粋な集団をもたらすことを示した。グラフの低蛍光領域にある信号は、信号の内部雑音であることが後で判明したので、後に操作者により装置の修正により除去した。
【0135】
実施例12. 固体サポート結合のためのスペーサー領域選定
αGFP::KzPG::SNAP::DBP融合タンパク質を発現している細胞を、8TGP培地を用いて透過性にし、中間体His6−タグ化eGFPを経由してHisPur Co2+セファロース・ビーズ(Thermo Scientific社)に結合した。細胞またはビーズは、最初に過剰のHis6−eGFPとともに培養し、TBS中で洗浄し、一緒に25℃で30分間培養した。結合しなかった細胞は、ビーズから洗い流し、つぎに蛍光顕微鏡検査法でビーズの結合の程度を評価した。
最初は、αGFP::KzPG::SNAP::DBP融合タンパク質のセファロース・ビーズへの結合は検知されなかった。αGFP結合領域が、細胞壁に近すぎて、セファロース樹脂上のコバルト錯化eGFPに到達できないためと理論を立てた。このため、ランダムコドンによる12残基ペプチド・スペーサー領域を、αGFP結合領域およびkzPGペプチドグリカン結合領域の間にクローン化した(GGT ACC gcy gcy gkk wtb gck wtb gkk gkk gck gkk gcy gcy GGT CTG(SEQ ID NO:5(配列番号)))。
スペーサー変異体の小型ライブラリー(約2,000要素)を発現し、上述のようにCo2+セファロースに結合した。前記ライブラリーの一部は、ビーズに結合することが観測された。これらのクローンはPCRで増幅し、再クローン化し、十余のクローンの個別の結合性を試験し、配列を調べた。種々のペプチド・スペーサーが、タンパク分解に耐性があり(αGFPを高度に融合タンパク質中に保持する)、また、図18に示されているように、界面活性剤処理細胞をセファロース・ビーズに結合できることが判明した。サポート結合に機能することが判明したスペーサー配列を表1に挙げる。
【表1】
スペーサー配列の一つRL6を選んで、後の結合の検討に用いた。固体サポート・マトリックスへの強い結合に寄与する他の要素を調べた。細胞のマトリックスとの培養時間、および結合溶液の塩(NaCl)の濃度は、両方とも結合に対してポジティブな効果を有することが分かった。培養時間30分と、NaCl濃度の範囲約200mM〜500mMが、効果的であることが判明したが、300mMが最適と考えられる。結合は、Tris、リン酸塩、MOPSの300mMの塩含有緩衝溶液を含む一連の緩衝液中で効率的である。
αGFP::RL6::KzPG::SNAP::DBP融合タンパク質を発現している細胞を、ビオチン化eGFPを介してストレプトアビジン磁性ナノ粒子(MagneSphere、Roche Diagnostics社)に結合する条件は、図19に明示されるセファロース・ビーズ結合に対して求められた範囲内にあることも判明した。
12残基スペーサーに加えて、タンパク質領域をも、スペーサー領域として用いることが考えられた。ヒトのタイチン遺伝子(I27)からの小型で安定な高度に発現した27番目のイムノグロブリン領域をRL6スペーサーの上流にクローン化した。この領域も、N末端のαGFP領域の高度かつ安定的な発現と、同時に非常に良好な固定マトリックス結合をも可能にすることが分かった(図19)。
【0136】
実施例13. 被包性ディスプレイ用のマウスscFvライブラリーの構築
一本鎖抗体(scFv)ライブラリーの細胞内ディスプレイ用の最後の領域構造は、scFv::I27::RL6::KzPG::SNAP::DBPであった。scFv領域を除いた、融合タンパク質のタンパク質およびDNAの配列は、SEQ ID NO:13(配列番号)およびSEQ ID NO:14(配列番号)に与えられている。このタンパク質融合体は、N末端にscFv、つづいて2つのスペーサー領域であるI27およびRL6、つぎにペプチドグリカン結合領域のKzPG、SNAPレポーター領域、最後にDNA結合領域(DBP)を有する。
ランダムプライマーを用いたcDNAを、酵素Superscript III(Invitrogen社)を用いて、マウス脾臓全RNAから製造した。このcDNAから、シェーファー(Schaefer)ら(2010)が記述したように、Vent DNAポリメラーゼ(New England Biolabs社)、およびマウス抗体ファミリー配列用変性オリゴヌクレオチド・プライマーを用いて、scFv軽鎖(VL)および重鎖(VH)可変領域を増幅した。ライブラリー・クローン化に用いたオリゴヌクレオチド・プライマーは、Schaeferらの記述したものと、Bsm BIを介して我々のライブラリー骨格ベクター(SEQ ID NO:15(配列番号))内にクローン化するのに適当な末端を有する点で相違している。VLおよびVH領域は、オーバーラップ伸長PCRを用いて加えられる。最終的scFvバンドは、合計60回のPCR増幅サイクル(1回目30回、2回目30回)にかけられた。
ライブラリー・クローン化のために、ディスプレイ・コンストラクト900ngを、BsmBIで切断し、メーカーの説明書にしたがいSureclean(Bioline社)を用い沈澱させ、T4 DNAリガーゼを用い、同様に処理されたscFv生成物400ngに連結した。リガーゼを65°C、10分間の培養で失活させ、連結体をE. Coli Argentum株(Alchemy Biosciences社)中に電気穿孔で入れた。電気穿孔された細胞は、SOC培地中に回収し、37°Cで1時間培養し、プール後、75μg/mLアンピシリンを含む20x150mmLB寒天シャーレに拡げた。このシャーレを一晩30°Cで培養した。ライブラリーの大きさは4×105独立クローン程度と推定された。20クローンの内、20クローンに予想された大きさの挿入断片が含まれていることが判明した。
【0137】
実施例14. 被包性ディスプレイ・マウスscFvライブラリーのスクリーニング
ファージディスプレイ・ライブラリーから単離される一本鎖抗体は、多くの場合大腸菌中での発現が困難で、周辺質での発現レベルが低く、あるいはβシートとIg群の間にジスルフィド結合形成を欠くために、細胞質中で完全に不溶性である。大腸菌細胞質中で可溶性のマウスscFv骨格を選別するのに被包性ディスプレイを用いることができるか否かを決定するには、scFvの溶解性が融合タンパク質の性質と相関しているか否かを決定する必要があった。
有用な可溶性scFvは、凝集のレベルが低く、発現レベルが少なくとも中程度であることが予想された。これは、透過性にした細胞中のKzPG領域を、細胞壁に結合させる(したがって、細胞中の封入体に局在しない)、かつSNAPレポーター領域の少なくとも中程度の発現を示すクローンとして、視覚的に判断できた。
これらのパラメーターを選別するため、単一のコロニーを採取し、すでに例1に記述したようにアラビノースを用いて融合タンパク質の発現を誘導した。透過処理後、これらをSNAPリガンドで標識付けし蛍光顕微鏡検査法をもちいて観察した。前記ライブラリー・クローンを、発現、および、図20に例が示されるような、SNAPレポーターの細胞での分布に基づき、4類に分類した。
1. SNAPの発現なし。
2. 凝集した封入体中でのSNAPの中〜高程度の発現(図20、左図)
3. 細胞壁に局在してのSNAPの弱程度の発現(図20、中央図)
4. 細胞壁に局在してのSNAPの高程度の発現(図20、右図)
【0138】
発現と溶解性の両方が高いクローンのみを、さらに分析した。しかし前記SNAPレポーターの発現の弱かったのは、大腸菌発現に最適化されなかったタンパク質の非効率な発現のためでありうるので、こうしたクローンのうちのある部分は、コドンの使用が適性化されたならば、可溶性細胞質ライブラリー・ディスプレイに対して非常に適していると判明することがあり得る。
SNAPレポーターの発現レベルが高く、透過性にした細胞の細胞壁の周りに均一に分布したクローンの配列を調べ、残りの融合タンパク質に対して正しい翻訳枠にあるscFv挿入断片の存在を確認した。分析した21クローンの全てで、scFv挿入断片は、全長で、正しい長さのグリシン/セリン・リンカー領域を有し、全融合タンパク質の翻訳のために正しい読み枠中にあることが判明した。このことは、本発明のスクリーニング方法は、大腸菌細胞の細胞質中で、溶解状態で発現したマウスscFv遺伝子を正しく識別していることを示唆した。前記ライブラリーから単離されたscFvタンパク質が、大腸菌細胞質中で可溶であることを確認するため、これを、ライブラリー・コンストラクトから、介在性のスペーサー領域I27−RL6またはRL6のいずれかを含むC末端のFLAGエピトープを有するアラビノース誘導性発現ベクターにシャトルした。
アラビノースによるタンパク質発現の誘導後、可溶性のscFv::I27::RL6::FLAGまたはscFv::RL6::FLAG融合タンパク質を、0.5%8TGPで抽出した。不溶性の細胞物質は沈澱させ、β-メルカプトエタノールを含むSDS−PAGEローディング緩衝液中に、サンプルに超音波をかけ95℃で5分間加熱して、再懸濁した。各画分の等量ずつを10%SDS−PAGEゲルにかけ、電気泳動した。分離されたタンパク質は、ニトロセルロース膜に移し、5%脱脂粉乳でブロックした。組換えタンパク質発現体に、1:1000に希釈したヒツジαFLAG抗体(Sigma社)、つづいて、抗マウスHRP抱合二次抗体でプローブ付けを行った。化学発光を用いて検出した。
図21は、本発明の方法は、ほとんどが溶解した状態で、細菌細胞質内で発現するscFv遺伝子を識別することができることを明らかにしている。ウェスタンブロット法による発現プロフィールは、各サンプルで、scFv::I27::RL6::FLAGコンストラクトについてSNAPリガンドで検知した蛍光顕微鏡検査法と一致している。
【0139】
広く記述された本発明の範囲を逸脱することなく、特定の態様において示したように、多くの変化形および/または改良が可能であることを当業者は理解するであろう。本実施態様は、したがって、いかなる点においても説明のためであって、制限を課すものと考えてはならない。
本明細書において、議論され、および/または参照された公刊物は、その全体を参照のため本明細書に組み入れる。
本出願は、AU2009906310による優先権を主張するものであり、その開示全体は、参照により本願に組み込まれている。
本明細書に含まれている文書、行為、材料、装置、物品等々に関するいかなる議論もひとえに本発明の状況を提供することを目的としている。これらの事項のいずれか、または全てが、当出願の各請求項の優先日より以前に存在していたことを理由として、それが先行技術の基盤の一部を形成すること、あるいは本発明に関連する分野において普通の一般的な知識であったことを是認するとして取られるべきでない。
【0140】
参考文献
Aharoni et al. (2005) Chem Biol, 12:1281-1289
Becker et al. (2004) Curr Opin Biot, 15:323-329
Briers et al. (2009) Biochem Biophys Res Comm, 383:187-191
Chen and Gotschlich (2001) J Bact, 183: 3160-3168
Daugherty et al. (2000) J Immunol Methods, 243:211-227
Farinas (2006) Comb Chem High Thro Screen, 9:321-328
George, et al. (2003) Protein Engineering, 15:871-879
Kenrick et al. (2007) Curr Prot Cyt, 4.6.1-4.6.27
Lutz and Patrick (2004) Curr Opin Biot, 15:291-297
Magnet et al. (2007) J Bact 189:3927-3931
Miller et al. (2006) Nat Meth, 3:561-570
Parsons et al. (2006) Biochem 45:2122-2128
Schaefer et al. (2010) Antibody Eng, 1:21-44
Smith (1985) Science, 228:1315-1317
【特許請求の範囲】
【請求項1】
標的分子に対する所望の活性に関してポリペプチドをスクリーニングする方法であって:
a)ポリペプチドをコード化するポリヌクレオチドを含む細菌細胞を培養して、ポリペプチドを産生し、
b)細菌細胞を透過化し、ここで、ポリペプチドおよびポリペプチドをコード化するポリヌクレオチドは、透過化された細菌細胞内部に保持され、
c)透過化された細菌細胞を標的分子と接触させて、該標的分子を透過化された細菌細胞中に拡散させ、そして
d)ポリペプチドを所望の活性に関してスクリーニングする
ことを含む、方法。
【請求項2】
標的分子に対する所望の活性に関してポリペプチドをスクリーニングする方法であって:
a)ポリペプチドをコード化するポリヌクレオチドを含む細菌細胞を培養して、ポリペプチドを産生し、細菌細胞壁に付着させ、
b)細菌細胞を透過化し、ここで、ポリペプチドをコード化するポリヌクレオチドは、透過化された細菌細胞内部に保持され、
c)透過化された細菌細胞を標的分子と接触させ、そして
d)ポリペプチドを所望の活性に関してスクリーニングする
ことを含む、方法。
【請求項3】
ステップd)が:
i)ポリペプチドが標的分子と結合するかどうか、および/または標的分子と結合する程度を決定すること、および/または
ii)ポリペプチドが標的分子を酵素的に修飾するかどうか、および/または標的分子の酵素修飾率を決定すること
を含む、請求項1または2のいずれかに記載の方法。
【請求項4】
細菌細胞が界面活性剤で透過化される、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
界面活性剤が非イオン性界面活性剤である、請求項4記載の方法。
【請求項6】
細菌細胞がグラム陰性細菌細胞である、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
ポリペプチドが、少なくとも第2ポリペプチドと結合して、透過化された細菌細胞の内部に保持され、および/または細菌細胞壁に結合するタンパク質複合体を形成する、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
ポリペプチドが第2ポリペプチド、またはそのサブユニットに融合している、請求項7記載の方法。
【請求項9】
第2ポリペプチドが:
i)タンパク質複合体が透過化された細菌細胞壁内部に保持されるような分子サイズを有するポリペプチド;
ii)DNA結合タンパク質;および/または
iii)細菌細胞壁結合タンパク質
から選択される、請求項7または請求項8記載の方法。
【請求項10】
タンパク質複合体の分子量が少なくとも約120kDaである、請求項9記載の方法。
【請求項11】
第2ポリペプチドが、透過化された細菌細胞の内部でマルチマーを形成する、請求項9または請求項10記載の方法。
【請求項12】
マルチマーがテトラマーである、請求項11記載の方法。
【請求項13】
第2ポリペプチドが、RhnA、β−ガラクトシダーゼ、BetB、G5K、GshBおよびYdcWから選択される、請求項12記載の方法。
【請求項14】
DNA結合タンパク質がComEである、請求項9記載の方法。
【請求項15】
細菌細胞壁結合タンパク質が、ペプチドグリカン結合タンパク質、および細胞壁と結合できるリポタンパク質またはそのフラグメントから選択される、請求項9記載の方法。
【請求項16】
細菌細胞壁結合タンパク質が、KzPG、PAL、OmpA、YiaD、YfiBおよびMotBから選択されるペプチドグリカン結合タンパク質である、請求項15記載の方法。
【請求項17】
タンパク質複合体が細菌細胞壁に共有結合している、請求項15記載の方法。
【請求項18】
細胞壁と結合できるリポタンパク質またはそのフラグメントが、外膜付着に必要な機能的N末端シグナル配列が欠如したリポタンパク質である、請求項17記載の方法。
【請求項19】
リポタンパク質が大腸菌(E. coli)LPPである、請求項18記載の方法。
【請求項20】
非イオン性界面活性剤が、デカノイル−N−メチルグルカミド(Mega10)、ジメチルオクチルホスフィンオキシド(Apo8)、n−オクチル−β−D−チオグルコピラノシド(8TGP)、ならびにデカノイル−N−メチルグルカミド(Mega10)およびジメチルオクチルホスフィンオキシド(Apo8)の混合物から選択される、請求項5〜19のいずれか1項に記載の方法。
【請求項21】
細菌細胞の透過化が:
i)Luriaブロス(LB)中約0.5%のn−オクチル−β−D−チオグルコピラノシド(8TGP)、ならびに
ii)Luriaブロス(LB)中約0.5%のデカノイル−N−メチルグルカミド(Mega10)および約0.5%のジメチルオクチルホスフィンオキシド(Apo8)
から選択される溶液中で実施される、請求項20記載の方法。
【請求項22】
ステップb)が細菌細胞を選択的に透過化することを含み、それにより、細菌細胞の外膜が細菌細胞の内膜よりもさらに透過化される、請求項15〜19のいずれか1項に記載の方法。
【請求項23】
細菌細胞が、ジメチルオクチルホスフィンオキシド(Apo8)および/またはポリソルベート20(Tween20)から選択される界面活性剤で選択的に透過化される、請求項22記載の方法。
【請求項24】
細菌細胞の選択的透過化を、EDTAまたはCa2+を含む溶液中で実施する、請求項22または23記載の方法。
【請求項25】
e)ポリペプチドをコード化するポリヌクレオチドを含むDNAを、透過化された細菌細胞から単離することを更に含む、請求項1〜24のいずれか1項に記載の方法。
【請求項26】
DNAがゲノムDNAおよび/またはエピソームDNAである、請求項1〜25のいずれか1項に記載の方法。
【請求項27】
エピソームDNAがプラスミドまたはコスミドである、請求項26記載の方法。
【請求項28】
ポリヌクレオチドが外因性ポリヌクレオチドである、請求項1〜27のいずれか1項に記載の方法。
【請求項29】
標的分子の分子量が約120kDa未満である、請求項1〜28のいずれか1項に記載の方法。
【請求項30】
所望の活性を有するポリペプチドを同定する方法であって:
a)請求項1〜29のいずれか1項に記載の方法を用いてポリペプチドのライブラリーをスクリーニングし;そして
b)所望の活性を有する1以上のポリペプチドを選択することを含む、方法。
【請求項31】
c)細菌細胞からポリペプチドをコード化するポリヌクレオチドを含むDNAを単離することを更に含む、請求項30記載の方法。
【請求項32】
d)ポリペプチドをコード化するポリヌクレオチドの配列を決定することを更に含む、請求項31記載の方法。
【請求項33】
ポリペプチドのライブラリーが、細胞、組織、器官または生物から得られるポリヌクレオチドによってコード化される、請求項30〜32のいずれか1項に記載の方法。
【請求項34】
ポリペプチドのライブラリーが、1以上の親ポリヌクレオチドを突然変異させることによって得られるポリヌクレオチドによりコード化される、請求項30〜32のいずれか1項に記載の方法。
【請求項35】
ポリペプチドが抗体または酵素である、請求項1〜34のいずれか1項に記載の方法。
【請求項36】
抗体が単鎖可変フラグメント(scFV)である、請求項35記載の方法。
【請求項37】
ポリペプチドが酵素であり、標的分子が透過化された細菌細胞に連結される、請求項35記載の方法。
【請求項38】
標的分子が細菌細胞壁結合タンパク質に連結される、請求項37記載の方法。
【請求項39】
第2ポリペプチドと結合して、透過化された細菌細胞の内部に保持されるタンパク質複合体を形成する外因性ポリペプチドを含む、透過化された細菌細胞。
【請求項40】
細菌細胞壁に付着した外因性ポリペプチドを含む、透過化された細菌細胞。
【請求項41】
a)i)第1ポリペプチドをコード化するポリヌクレオチドをベクター中に挿入するための部位、および
ii)第1ポリペプチドと結合して、透過化された細菌細胞の内部に保持されるタンパク質複合体を形成する、第2ポリペプチドをコード化するオープンリーディングフレーム
を含むベクター、ならびに
b)細菌細胞を透過化することができる薬剤
を含む、キット。
【請求項42】
a)i)第1ポリペプチドをコード化するポリヌクレオチドをベクター中に挿入するための部位、および
ii)第1ポリペプチドと結合して、細菌細胞壁に付着するタンパク質複合体を形成する、第2ポリペプチドをコード化するオープンリーディングフレーム
を含むベクター、ならびに
b)細菌細胞を透過化することができる薬剤
を含む、キット。
【請求項43】
部位およびオープンリーディングフレームが、第1ポリペプチドおよび第2ポリペプチドまたはそのサブユニットが融合タンパク質として発現されるような位置にある、請求項41または42記載のキット。
【請求項44】
細菌細胞を透過化することができる薬剤が界面活性剤である、請求項41〜43のいずれか1項に記載のキット。
【請求項45】
界面活性剤が、デカノイル−N−メチルグルカミド(Mega10)、ジメチルオクチルホスフィンオキシド(Apo8)、n−オクチル−β−D−チオグルコピラノシド(8TGP)、ポリソルベート20(Tween20)、ならびにデカノイル−N−メチルグルカミド(Mega10)およびジメチルオクチルホスフィンオキシド(Apo8)の混合物からなる群から選択される非イオン性界面活性剤である、請求項44記載のキット。
【請求項46】
キットが細菌細胞を更に含む、請求項41〜45のいずれか1項に記載のキット。
【請求項47】
細菌細胞がグラム陰性である、請求項46記載の方法。
【請求項48】
細菌細胞が大腸菌である、請求項47記載のキット。
【請求項1】
標的分子に対する所望の活性に関してポリペプチドをスクリーニングする方法であって:
a)ポリペプチドをコード化するポリヌクレオチドを含む細菌細胞を培養して、ポリペプチドを産生し、
b)細菌細胞を透過化し、ここで、ポリペプチドおよびポリペプチドをコード化するポリヌクレオチドは、透過化された細菌細胞内部に保持され、
c)透過化された細菌細胞を標的分子と接触させて、該標的分子を透過化された細菌細胞中に拡散させ、そして
d)ポリペプチドを所望の活性に関してスクリーニングする
ことを含む、方法。
【請求項2】
標的分子に対する所望の活性に関してポリペプチドをスクリーニングする方法であって:
a)ポリペプチドをコード化するポリヌクレオチドを含む細菌細胞を培養して、ポリペプチドを産生し、細菌細胞壁に付着させ、
b)細菌細胞を透過化し、ここで、ポリペプチドをコード化するポリヌクレオチドは、透過化された細菌細胞内部に保持され、
c)透過化された細菌細胞を標的分子と接触させ、そして
d)ポリペプチドを所望の活性に関してスクリーニングする
ことを含む、方法。
【請求項3】
ステップd)が:
i)ポリペプチドが標的分子と結合するかどうか、および/または標的分子と結合する程度を決定すること、および/または
ii)ポリペプチドが標的分子を酵素的に修飾するかどうか、および/または標的分子の酵素修飾率を決定すること
を含む、請求項1または2のいずれかに記載の方法。
【請求項4】
細菌細胞が界面活性剤で透過化される、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
界面活性剤が非イオン性界面活性剤である、請求項4記載の方法。
【請求項6】
細菌細胞がグラム陰性細菌細胞である、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
ポリペプチドが、少なくとも第2ポリペプチドと結合して、透過化された細菌細胞の内部に保持され、および/または細菌細胞壁に結合するタンパク質複合体を形成する、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
ポリペプチドが第2ポリペプチド、またはそのサブユニットに融合している、請求項7記載の方法。
【請求項9】
第2ポリペプチドが:
i)タンパク質複合体が透過化された細菌細胞壁内部に保持されるような分子サイズを有するポリペプチド;
ii)DNA結合タンパク質;および/または
iii)細菌細胞壁結合タンパク質
から選択される、請求項7または請求項8記載の方法。
【請求項10】
タンパク質複合体の分子量が少なくとも約120kDaである、請求項9記載の方法。
【請求項11】
第2ポリペプチドが、透過化された細菌細胞の内部でマルチマーを形成する、請求項9または請求項10記載の方法。
【請求項12】
マルチマーがテトラマーである、請求項11記載の方法。
【請求項13】
第2ポリペプチドが、RhnA、β−ガラクトシダーゼ、BetB、G5K、GshBおよびYdcWから選択される、請求項12記載の方法。
【請求項14】
DNA結合タンパク質がComEである、請求項9記載の方法。
【請求項15】
細菌細胞壁結合タンパク質が、ペプチドグリカン結合タンパク質、および細胞壁と結合できるリポタンパク質またはそのフラグメントから選択される、請求項9記載の方法。
【請求項16】
細菌細胞壁結合タンパク質が、KzPG、PAL、OmpA、YiaD、YfiBおよびMotBから選択されるペプチドグリカン結合タンパク質である、請求項15記載の方法。
【請求項17】
タンパク質複合体が細菌細胞壁に共有結合している、請求項15記載の方法。
【請求項18】
細胞壁と結合できるリポタンパク質またはそのフラグメントが、外膜付着に必要な機能的N末端シグナル配列が欠如したリポタンパク質である、請求項17記載の方法。
【請求項19】
リポタンパク質が大腸菌(E. coli)LPPである、請求項18記載の方法。
【請求項20】
非イオン性界面活性剤が、デカノイル−N−メチルグルカミド(Mega10)、ジメチルオクチルホスフィンオキシド(Apo8)、n−オクチル−β−D−チオグルコピラノシド(8TGP)、ならびにデカノイル−N−メチルグルカミド(Mega10)およびジメチルオクチルホスフィンオキシド(Apo8)の混合物から選択される、請求項5〜19のいずれか1項に記載の方法。
【請求項21】
細菌細胞の透過化が:
i)Luriaブロス(LB)中約0.5%のn−オクチル−β−D−チオグルコピラノシド(8TGP)、ならびに
ii)Luriaブロス(LB)中約0.5%のデカノイル−N−メチルグルカミド(Mega10)および約0.5%のジメチルオクチルホスフィンオキシド(Apo8)
から選択される溶液中で実施される、請求項20記載の方法。
【請求項22】
ステップb)が細菌細胞を選択的に透過化することを含み、それにより、細菌細胞の外膜が細菌細胞の内膜よりもさらに透過化される、請求項15〜19のいずれか1項に記載の方法。
【請求項23】
細菌細胞が、ジメチルオクチルホスフィンオキシド(Apo8)および/またはポリソルベート20(Tween20)から選択される界面活性剤で選択的に透過化される、請求項22記載の方法。
【請求項24】
細菌細胞の選択的透過化を、EDTAまたはCa2+を含む溶液中で実施する、請求項22または23記載の方法。
【請求項25】
e)ポリペプチドをコード化するポリヌクレオチドを含むDNAを、透過化された細菌細胞から単離することを更に含む、請求項1〜24のいずれか1項に記載の方法。
【請求項26】
DNAがゲノムDNAおよび/またはエピソームDNAである、請求項1〜25のいずれか1項に記載の方法。
【請求項27】
エピソームDNAがプラスミドまたはコスミドである、請求項26記載の方法。
【請求項28】
ポリヌクレオチドが外因性ポリヌクレオチドである、請求項1〜27のいずれか1項に記載の方法。
【請求項29】
標的分子の分子量が約120kDa未満である、請求項1〜28のいずれか1項に記載の方法。
【請求項30】
所望の活性を有するポリペプチドを同定する方法であって:
a)請求項1〜29のいずれか1項に記載の方法を用いてポリペプチドのライブラリーをスクリーニングし;そして
b)所望の活性を有する1以上のポリペプチドを選択することを含む、方法。
【請求項31】
c)細菌細胞からポリペプチドをコード化するポリヌクレオチドを含むDNAを単離することを更に含む、請求項30記載の方法。
【請求項32】
d)ポリペプチドをコード化するポリヌクレオチドの配列を決定することを更に含む、請求項31記載の方法。
【請求項33】
ポリペプチドのライブラリーが、細胞、組織、器官または生物から得られるポリヌクレオチドによってコード化される、請求項30〜32のいずれか1項に記載の方法。
【請求項34】
ポリペプチドのライブラリーが、1以上の親ポリヌクレオチドを突然変異させることによって得られるポリヌクレオチドによりコード化される、請求項30〜32のいずれか1項に記載の方法。
【請求項35】
ポリペプチドが抗体または酵素である、請求項1〜34のいずれか1項に記載の方法。
【請求項36】
抗体が単鎖可変フラグメント(scFV)である、請求項35記載の方法。
【請求項37】
ポリペプチドが酵素であり、標的分子が透過化された細菌細胞に連結される、請求項35記載の方法。
【請求項38】
標的分子が細菌細胞壁結合タンパク質に連結される、請求項37記載の方法。
【請求項39】
第2ポリペプチドと結合して、透過化された細菌細胞の内部に保持されるタンパク質複合体を形成する外因性ポリペプチドを含む、透過化された細菌細胞。
【請求項40】
細菌細胞壁に付着した外因性ポリペプチドを含む、透過化された細菌細胞。
【請求項41】
a)i)第1ポリペプチドをコード化するポリヌクレオチドをベクター中に挿入するための部位、および
ii)第1ポリペプチドと結合して、透過化された細菌細胞の内部に保持されるタンパク質複合体を形成する、第2ポリペプチドをコード化するオープンリーディングフレーム
を含むベクター、ならびに
b)細菌細胞を透過化することができる薬剤
を含む、キット。
【請求項42】
a)i)第1ポリペプチドをコード化するポリヌクレオチドをベクター中に挿入するための部位、および
ii)第1ポリペプチドと結合して、細菌細胞壁に付着するタンパク質複合体を形成する、第2ポリペプチドをコード化するオープンリーディングフレーム
を含むベクター、ならびに
b)細菌細胞を透過化することができる薬剤
を含む、キット。
【請求項43】
部位およびオープンリーディングフレームが、第1ポリペプチドおよび第2ポリペプチドまたはそのサブユニットが融合タンパク質として発現されるような位置にある、請求項41または42記載のキット。
【請求項44】
細菌細胞を透過化することができる薬剤が界面活性剤である、請求項41〜43のいずれか1項に記載のキット。
【請求項45】
界面活性剤が、デカノイル−N−メチルグルカミド(Mega10)、ジメチルオクチルホスフィンオキシド(Apo8)、n−オクチル−β−D−チオグルコピラノシド(8TGP)、ポリソルベート20(Tween20)、ならびにデカノイル−N−メチルグルカミド(Mega10)およびジメチルオクチルホスフィンオキシド(Apo8)の混合物からなる群から選択される非イオン性界面活性剤である、請求項44記載のキット。
【請求項46】
キットが細菌細胞を更に含む、請求項41〜45のいずれか1項に記載のキット。
【請求項47】
細菌細胞がグラム陰性である、請求項46記載の方法。
【請求項48】
細菌細胞が大腸菌である、請求項47記載のキット。
【図1】

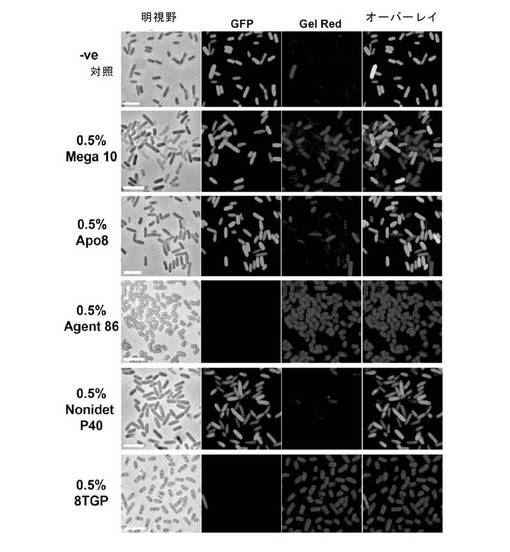
【図2】
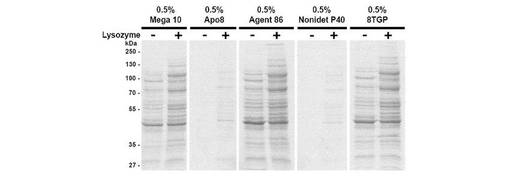
【図3】
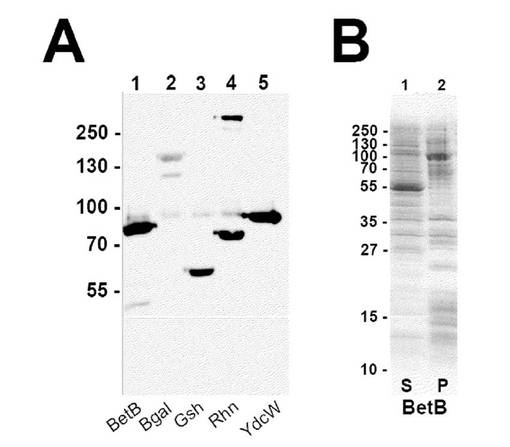
【図4】
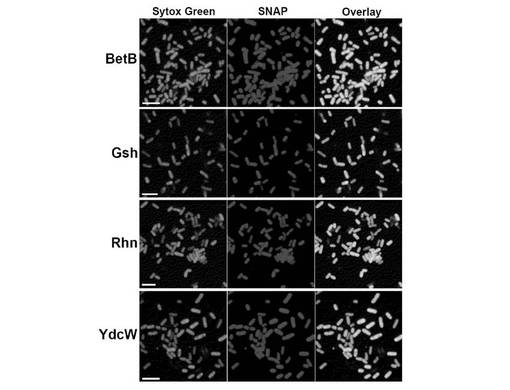
【図5】
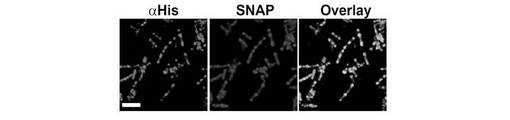
【図6】
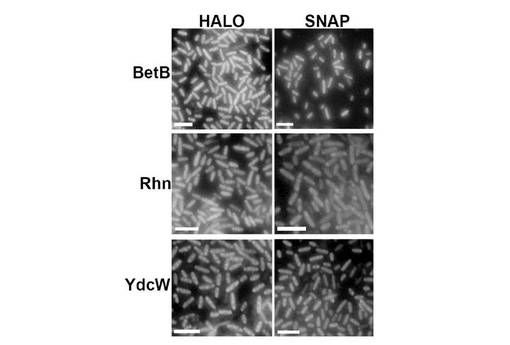
【図7】
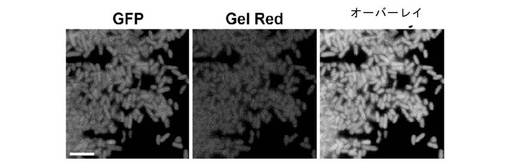
【図8】
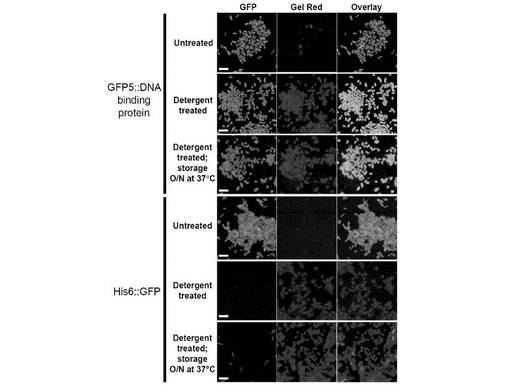
【図9】
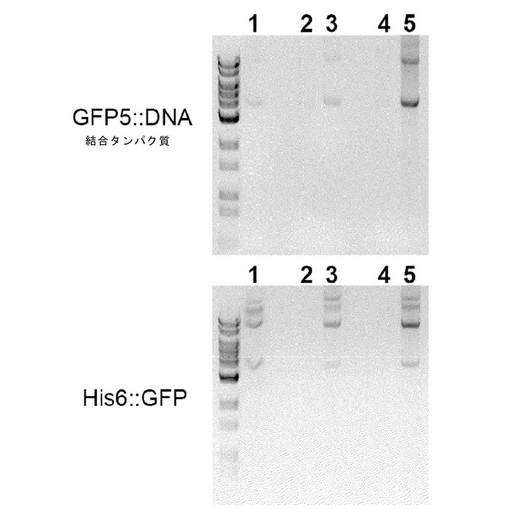
【図10】
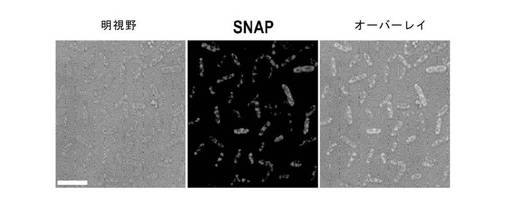
【図11】
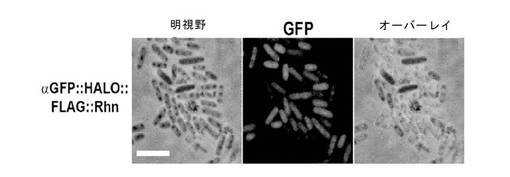
【図12】
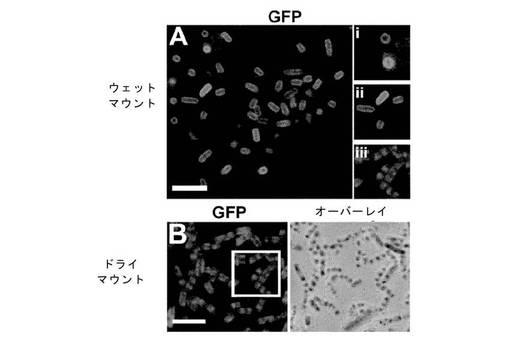
【図13】
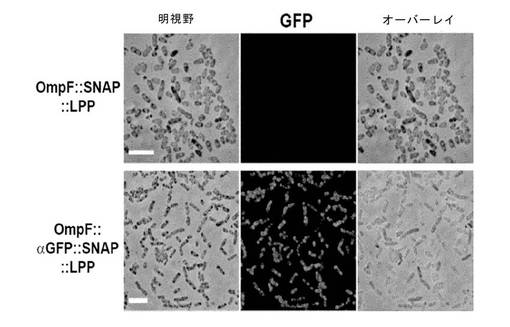
【図14】
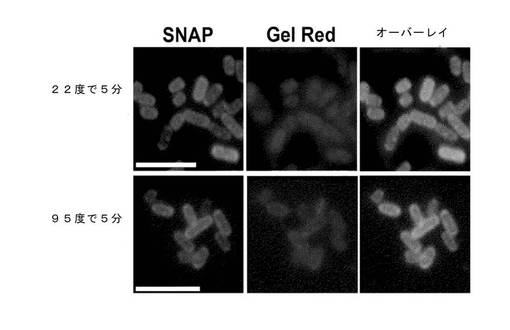
【図15】
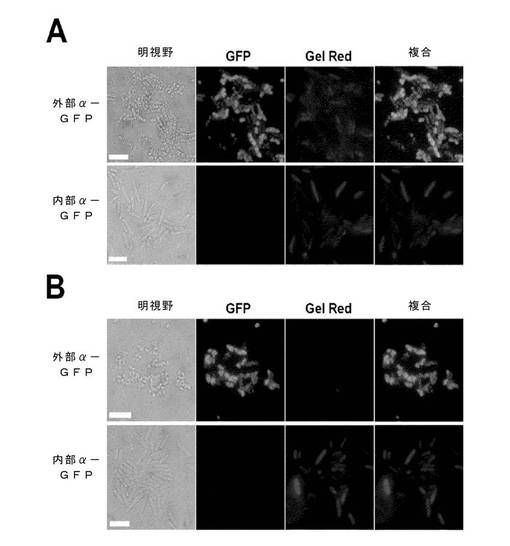
【図16】
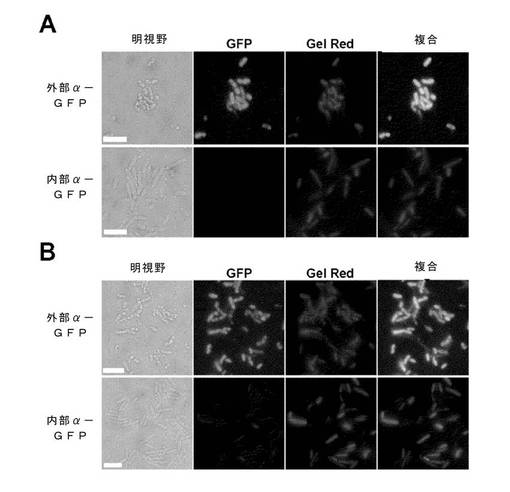
【図17】
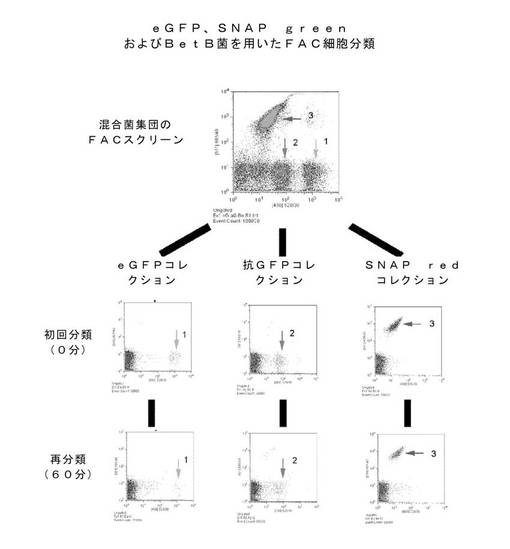
【図18】
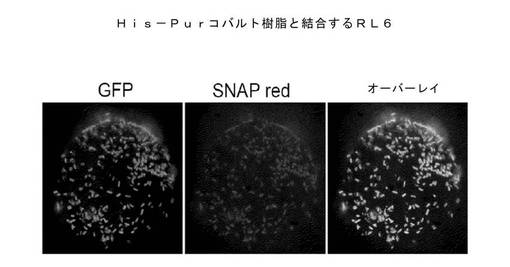
【図19】
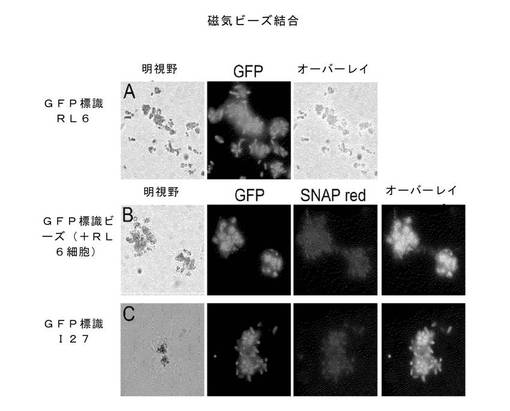
【図20】
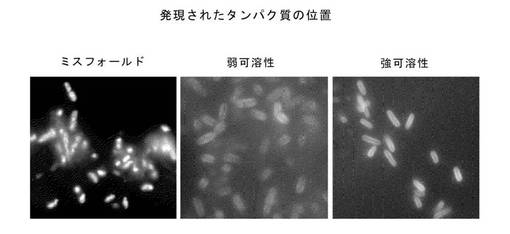
【図21】
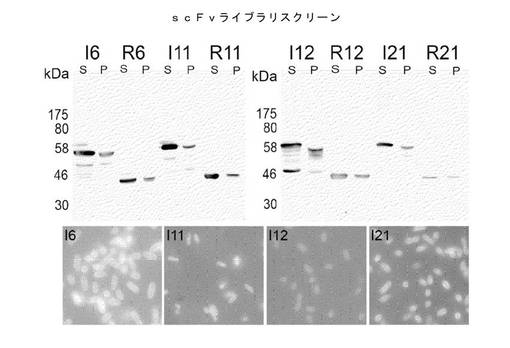
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公表番号】特表2013−514787(P2013−514787A)
【公表日】平成25年5月2日(2013.5.2)
【国際特許分類】
【出願番号】特願2012−545011(P2012−545011)
【出願日】平成22年12月20日(2010.12.20)
【国際出願番号】PCT/AU2010/001702
【国際公開番号】WO2011/075761
【国際公開日】平成23年6月30日(2011.6.30)
【出願人】(512165134)アフィニティ バイオサイエンス ピーティーワイ リミテッド (1)
【Fターム(参考)】
【公表日】平成25年5月2日(2013.5.2)
【国際特許分類】
【出願日】平成22年12月20日(2010.12.20)
【国際出願番号】PCT/AU2010/001702
【国際公開番号】WO2011/075761
【国際公開日】平成23年6月30日(2011.6.30)
【出願人】(512165134)アフィニティ バイオサイエンス ピーティーワイ リミテッド (1)
【Fターム(参考)】
[ Back to top ]