
タンパク質分子対、タンパク質分子対をコードする遺伝子および遺伝子導入ベクターとタンパク質分子対を産生する細胞
【課題】タンパク質同士の相互作用をアッセイする方法として知られているBiFC法では、目的とするタンパク質同士が相互作用をした際に蛍光タンパク質も再会合して発光することが必要である。しかし、タンパク質同士よりも蛍光タンパク質の断片同士のアフィニティが強いと、相互作用を行わないのに蛍光が観察されるという課題があった。
【解決手段】観察したいタンパク質に結合する蛍光タンパク質の断片のアミノ酸を部分的に他のアミノ酸に置き換え、蛍光タンパク質の断片同士のアフィニティを調節しタンパク質同士のアフィニティに合わせる。
【解決手段】観察したいタンパク質に結合する蛍光タンパク質の断片のアミノ酸を部分的に他のアミノ酸に置き換え、蛍光タンパク質の断片同士のアフィニティを調節しタンパク質同士のアフィニティに合わせる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、タンパク質同士の相互作用を観察するBiFC法において用いられる蛍光タンパク質分子対とその蛍光タンパク質分子対をコードする遺伝子、該遺伝子導入ベクターおよびその遺伝子導入ベクターが導入され、目的の蛍光タンパク質分子対を産生する細胞に関する。
【背景技術】
【0002】
タンパク質間相互作用を解析する技術の1つに、BiFC (Bimolecular Fluorescence Complementation : 二分子蛍光相補性)法がある。BiFCとは、蛍光タンパク質を2つの分割断片にすることで失われた蛍光が、相補的な分割断片の再会合により回復する現象である。この現象を利用して、生きた細胞内でのタンパク質間相互作用を可視化することができる。BiFC法を用いると、翻訳後修飾されたタンパク質の細胞内局在や機能解析など、タンパク質が機能している状態に近い条件でのタンパク質間の相互作用を調べることができる。
【0003】
蛍光タンパク質の分割断片を用いる手法は、2000年に初めて報告され、逆並行ロイシンジッパーと融合させたGFP(Green Fluorescent Protein)の分割断片が、大腸菌で、ロイシンジッパーの形成とそれに伴うGFP断片の会合によって蛍光回復することが明らかにされた(非特許文献1)。
【0004】
動物細胞内での蛍光回復は、GFPよりも蛍光強度が大きいYFP(Yellow Fluorescent Protein)で報告され(非特許文献2)、BiFC アッセイ(assay)という術語が初めて使用された。その後、GFP、YFP、CFP(Cyan Fluorescent Protein)などの複数の蛍光タンパク質を用いたマルチカラーBiFC(非特許文献3)が報告された。
【0005】
CFPを改良したCerulean(非特許文献4)、YFPを改良したCitrine(非特許文献5)とVenus(非特許文献6)は、生理的条件下(37℃)での発色団形成効率が高く、これらの蛍光タンパク質を用いたマルチカラーBiFC(非特許文献7)も報告されている。
【0006】
近年では、BiFC法で用いられる蛍光タンパク質も増え(非特許文献8、非特許文献9)、また、ユビキチン化されたタンパク質の検出(非特許文献10)、薬剤によるタンパク質間相互作用の誘導(非特許文献11)など、BiFC法の応用的な利用も進歩してきている。
【0007】
また、特許文献1では、時間経過に伴って発光色が変化する蛍光タンパク質をN末端側断片とC末端側断片にわけて、相互作用を調べたいそれぞれのタンパク質と融合させ、当該タンパク質間の相互作用の際に、分割された蛍光タンパク質が再会合して発色する技術が開示されている。ここでは、蛍光タンパク質としてKusabira−Orange(mKO)が選択され、特にN末端から70番目のアミノ酸であるブロリンが他のアミノ酸に置換された蛍光タンパク質であることを特徴としている。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2006−308568号公報
【非特許文献】
【0009】
【非特許文献1】インドラニール ゴーシュ(Indraneel Ghosh), アンドリュー D. ハミルトン(Andrew D. Hamilton)、とライニー レーガン(Lynne Regan)、「アンチパラレル ロイシン ジッパー−ディレクテッド プロテイン リアセンブリー:アプリケーション ツー ザ グリーン フローレセント プロテイン(Antiparallel Leucine Zipper−Direction Protein Reassembly: Application to the Green Fluorescent Protein」,アメリカン ケミカル ソサイアティ(American Chemical Society)、2000年、122、P.5658−5659
【非特許文献2】フュウ C.D.(Hu, C.D.), シネノフ Y.(Chinenov, Y.), とケルポラ T.K.(Kerppola, T.K.)「ビジュアリゼーション オブ インターラクション アマング ビーズィーアイピー アンド アールイーエル ファミリー プロテインズ イン リビング セルズ ユージング バイモレキュラー フローレセンス コンプリメンテーション( Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation.)」 モル セル(Mol. Cell) 9, 789−798 (2002).
【非特許文献3】フュウ C.D.(Hu, C.D.), シネノフ Y.(Chinenov, Y.), とケルポラ T.K.(Kerppola, T.K.)「シミュルテイニアス ビジュアリゼーション オブ マルチプル プロテイン インターラクションズ イン リビング セルズ ユージング マルチカラー フローレセンス コンプレメンテイション アナリシス(Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis.)ナット バイオテクノル(Nat. Biotechnol.)21, 539−545 (2003).
【非特許文献4】リッゾ M.A.(Rizzo, M.A.), スプリンガー G.H.(Springer, G.H.), グラナダ B.(Granada, B.), と ピストン D.W.(Piston, D.W.)「アン インプルーブド シアン フローレセント プロテイン バリアント ユースフル フォア フレッツ( An improved cyan fluorescent protein variant useful for FRET.)ナット バイオテクノル(Nat. Biotechnol.)22, 445−449 (2004).
【非特許文献5】グリエスベック O.(Griesbeck, O.), バイルド G.S.(Baird, G.S.), キャンベル R.E.(Campbell, R.E.), ザハリアスD.A.(Zacharias, D.A.), と ツジエン R.Y.(Tsien, R.Y.)「リデューシング ザ エンバイロメンタル センシティビィティ オブ イエロー フローレセント プロテイン(Reducing the environmental sensitivity of yellow fluorescent protein.)」J. Biol. Chem. 276, 29188−29194 (2001).
【非特許文献6】ナガイ T.(Nagai, T.), イバタ K.(Ibata, K.), パーク E.S.(Park, E.S.), クボタ M.(Kubota, M.), ミコシバ K.(Mikoshiba, K.), と ミヤワキ A.(Miyawaki, A.)「ア バリアント オブ イエロー フローレセント プロテイン ウイズ ファースト アンド エフィシェント マチュレーション フォア セル−バイオロジカル アプリケーション(A variant of yellow fluorescent protein with fast and efficient maturation for cell−biological applications.)」ナット バイオテクノ(Nat. Biotechnol.) 20, 87−90 (2002).
【非特許文献7】シュー Y.J.(Shyu, Y.J.), リー H.(Liu, H.),デング X.( Deng, X.), と フュウ C.D.(Hu, C.D.)「アイデンティフィケーション オブ ニュー フローレセント プロテイン フラグメンツ フォア バイモレキュラー フローレセンス コンプリメンテーション アナライシス アンダー フィシオロジカルコンディションズ( Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions.) バイオテクニークス( Biotechniques) 40, 61−66 (2006).
【非特許文献8】ファン J.Y.(Fan, J.Y.), キュイ Z.Q.(Cui, Z.Q.), ウェイ H.P.(Wei, H.P.), ザングZ.P.(Zhang, Z.P.), ズウ Y.F.(Zhou, Y.F.), ワング Y.P.(Wang, Y.P.), と ザング X.E.(Zhang, X.E.)「スプリット mチェリー アズ ア ニュー レッド バイモレキュラー フローレセンス コンプリメンテーション システム フォア ビジュアリジング プロテイン−プロテイン インターラクションズ イン リビング セルズ( Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein−protein interactions in living cells.)」 バイオケム バイオフィズ レス コミュ (Biochem. Biophys. Res. Commun.) 367, 47−53 (2007).
【非特許文献9】チュー J.(Chu, J.), ザング Z.(Zhang, Z.), ゼング Y.(Zheng, Y.),ヤング J.( Yang, J.), キン L.(Qin, L.), リュー J.(Lu, J.),フアング Z.L.( Huang, Z.L.), ゼング S.(Zeng, S.), と ルオ Q.(Luo, Q.)「 ア ノベル ファー−レッド バイモレキュラー フローレセンス コンプリメンテション システム ザット アローズ フォア エフィシェント ビジュアリザション オブ プロテイン インターラクションズ アンダー フィジオロジカル コンディションズ( A novel far−red bimolecular fluorescence complementation system that allows for efficient visualization of protein interactions under physiological conditions.)バイオセンス バイオエレクトロン (Biosens. Bioelectron.) 25, 234−239 (2009).
【非特許文献10】ファング D.(Fang, D.), と ケルポラ T.K.(Kerppola, T.K.)「アビクイチン−メディエーティッド フローレセンス コンプリメンテション リビールズ ザット ジュン アビクイティネイテッド バイ イッチ/エイアイピー4 イズ ローカライズド ツー リソソーム (Ubiquitin−mediated fluorescence complementation reveals that Jun ubiquitinated by Itch/AIP4 is localized to lysosomes.)」 Proc. Natl. Acad. Sci. U.S.A., 101, 14782−14787 (2004).
【非特許文献11】ロビダ M.A.(Robida, M.A,)と ケルポラT.K.(Kerppola, T.K.)「バイモレキュラー フローレセンス コンプリメンテーション アナライシス オブ インデューシブル プロテイン インターラクション:エフェクト オブ ファクターズ アフェクティング プロテイン フォールディング オン フローレセント プロテイン フラグメント アソシエーション(Bimolecular fluorescence complementation analysis of inducible protein interactions: effects of factors affecting protein folding on fluorescent protein fragment association.) J. Mol. Biol. 394, 391−409 (2009).
【発明の概要】
【発明が解決しようとする課題】
【0010】
BiFC法は、1つの蛍光タンパク質を分断し、それぞれの断片を2つの相互作用を調べたいタンパク質(目標タンパク質)にそれぞれ融合させておき、2つの目標タンパク質が相互作用を行った時に、蛍光タンパク質断片同士が会合し発光する現象を利用したタンパク質間相互作用を評価する方法の1つである。従って、2つの目標タンパク質が相互作用を行わない場合には、蛍光タンパク質断片同士は会合せず、発光しないという事が前提となっている。
【0011】
しかし、タンパク質は複雑な3次元構造を有しており、ペプチド主鎖の多彩な折れ曲がり構造と、アミノ酸側鎖の官能基が存在する。そして、これらペプチド主鎖の形状とアミノ酸側鎖の官能基間の相互作用によって、タンパク質間のアフィニティ(親和力)が決定されることが知られている。従って、2つの目標タンパク質間に相互作用がなくても、分断された蛍光タンパク質断片同士のアフィニティが強ければ、断片同士の会合が生じることが考えられる。その結果、目標タンパク質同士の相互作用がなくても発光してしまうという現象が生じる事が考えられる。この状態は、2つの目標タンパク質間の会合を示していることにならず、タンパク質間の相互作用を評価する方法として好ましくない状態であるといえる。本発明者は、事実このような状態を観測し、上記の課題を把握するに至った。
【課題を解決するための手段】
【0012】
本発明は、上記の課題に鑑みて想到されたものであり、分断された蛍光タンパク質間のアフィニティを調節する方法を提供するものである。より具体的には、
蛍光タンパク質のN末端側断片に接続された第1のタンパク質分子と、前記蛍光タンパク質のC末端側断片に接続された前記第2のタンパク質分子とからなる融合タンパク質分子対において、
前記N末端側断片と前記C末端側断片の少なくとも一方の断片で、少なくとも1箇所以上のアミノ酸が他のアミノ酸に置換され、
前記N末端側断片とC末端側断片同士のアフィニティは、アミノ酸の置換を受けない場合と比較し、前記アミノ酸の置換によって調節されたことを特徴とする融合タンパク質分子対を提供する。
【0013】
また、本発明の融合タンパク質分子対では、前記置換されるアミノ酸は、前記蛍光タンパク質のβ−シート上のアミノ酸である。
【0014】
また、本発明の融合タンパク質分子対では、前記変更されるアミノ酸は、β−シート上のロイシンであり、変更されたアミノ酸はイソロイシン、バリン、アラニンである。
【0015】
また、本発明の融合タンパク質分子対では、前記蛍光タンパク質は配列番号1の蛍光タンパク質である。
【0016】
また、本発明の融合タンパク質分子対では、前記変更されるアミノ酸は、C末端側断片のアミノ酸であって配列番号1のN末端側から201番目若しくは207番目のロイシンである。
【0017】
また、本発明は、上記の融合タンパク質分子対をコードする遺伝子を提供する。また、上記遺伝子のそれぞれを含む遺伝子導入/発現ベクターを提供する。さらに、その遺伝子導入/発現ベクターが導入され、上記の融合タンパク質分子対を産生する細胞を提供するものである。
【発明の効果】
【0018】
本発明は、蛍光タンパク質の分断された断片同士のアフィニティを調節するので、目標タンパク質同士が相互作用を行った時だけ、蛍光タンパク質が再会合し発光させることができる融合タンパク質分子対と、その融合タンパク質分子対を産生する細胞を提供することができる。これによって細胞内の活動はもとより、細胞外であっても、タンパク質同士の相互作用をアッセイするBiFC法を、より正確に行うことができる。
【図面の簡単な説明】
【0019】
【図1】本発明のタンパク質の相互作用の概念を示す図である。
【図2】蛍光タンパク質の構造を例示する図である。
【図3】本実施例に用いた蛍光タンパク質の構造とアミノ酸配列の関係を示す図である。
【図4】アミノ酸の置き換え点を求めるために相互作用を行うアミノ酸同士の距離を計算した結果を示す図である。
【図5】本実施例に用いたベクターの構成を示す図である。
【図6】メガ・プライマー法を説明する概念図である。
【図7】本実施例に用いたベクターを作製する工程を示す概念図である。
【図8】本実施例の結果(蛍光回復率)を示すグラフである。
【発明を実施するための形態】
【0020】
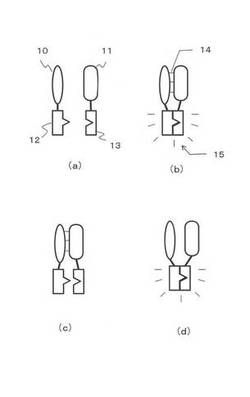
本発明の概要を図1を用いて説明する。図1(a)を参照して、目標タンパク質10、11のそれぞれには、蛍光タンパク質のN末端側断片12とC末端側断片13がそれぞれ結合されている。本発明では、これらタンパク質の対を融合タンパク質分子対と呼ぶ。目標タンパク質同士が接近して相互作用を及ぼしあうと、蛍光タンパク質のN末端側断片とC末端側断片も接近し会合する。図1(b)の符号14は、目標タンパク質が相互作用により会合していることを示す。この会合によって蛍光タンパク質断片同士12,13が会合し、蛍光タンパク質として発光機能を回復する(15)。これがBiFC法の原理である。従って、目標タンパク質同士が相互作用を行っても、蛍光タンパク質の断片同士が会合せず、発光を生じない場合は、タンパク質間の相互作用を評価する方法として利用することはできない。図1(c)は、この様子を模式的に表している。
【0021】
一方、目標タンパク質間の相互作用がないにも係らず、蛍光タンパク質の断片同士が会合し発光機能を回復する場合も、タンパク質間の相互作用を評価する方法として利用することはできない。図1(d)は、この様子を表している。すなわち、BiFC法においては、目標タンパク質同士のアフィニティと蛍光タンパク質の断片同士のアフィニティを適切に調節することが必要である。言い換えると、図1(c)または図1(d)で示した状態にならず、図1(b)に示したように目標タンパク質間の相互作用に応じて蛍光タンパク質の断片同士が会合して発光すること必要となる。本発明では、蛍光タンパク質断片の一部のアミノ酸を他のアミノ酸に置換し、上記のような目標タンパク質の相互作用に応じて発光するように蛍光タンパク質断片同士のアフィニティを調節する。ここで調節するとは、蛍光タンパク質断片同士のアフィニティを強くする若しくは弱くすることをいう。
【0022】
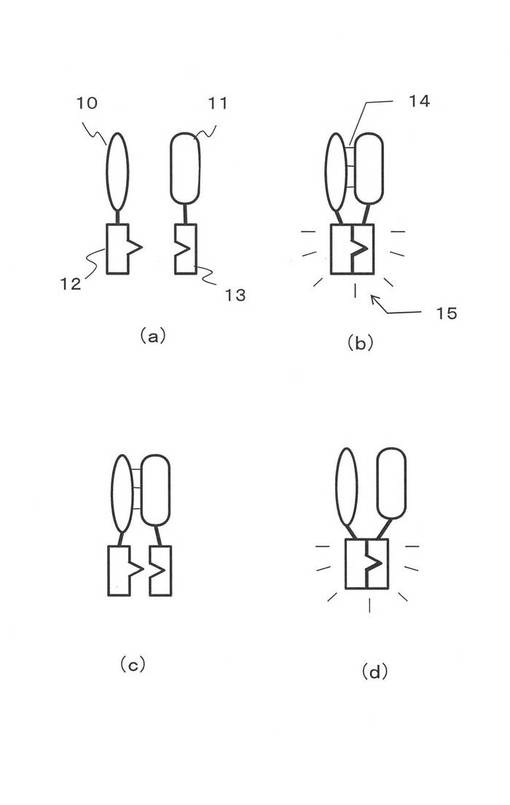
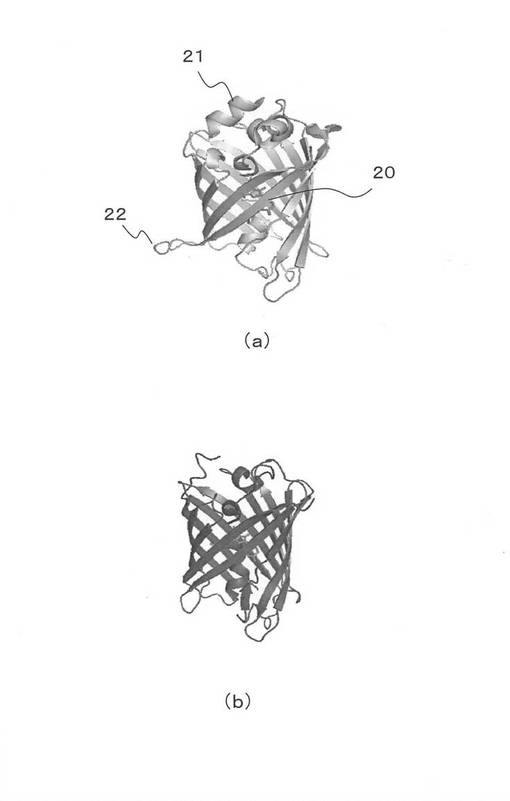
本発明で利用できる蛍光タンパク質は特に制限はない。CFP、GFPばかりでなく、近赤外領域や、近紫外域の光を発光する蛍光タンパク質でもよい。図2(a)には、後述する実施例で用いた黄色を発色する蛍光タンパク質Venusの立体構造モデルを示す。蛍光タンパク質には、β−シート(20)構造を形成する部分と、α−へリックス(21)およびループ(22)の部分が存在する。そして、全ての蛍光タンパク質には図2と同様なβ−シート構造の部分が存在する。例えば、図2(b)には、赤色蛍光を発色する蛍光タンパク質mCherryの構造を示す。図2(a)同様のβ−シート構造を有している。なお、図2の構造は、以下のURLで知られるプロテインデータベースでることができる。http://www.rcsb.org/pdb/explore/explore.do structureId=1MYW
蛍光タンパク質のN末端側断片とC末端側断片との切断点は特に限定するものではないが、β−シート以外の部分が望ましい。特にループの部分が望ましい。切断された蛍光タンパク質が再会合するには、疎水性相互作用が必要と考えられる。そして、β−シート部分での疎水性相互作用は、分割された切断片同士の会合に寄与すると考えられるので、β−シート部分は切断されずに残っているのが好ましいからである。
【0023】
本発明では、N末端側断片若しくはC末端側断片の少なくとも一方の断片で、少なくとも1つのアミノ酸を他のアミノ酸と置換する。置換するアミノ酸はβ−シート上のアミノ酸が望ましい。蛍光タンパク質は、図2に例示したように、並列したβ−シート構造を有している。分断された蛍光タンパク質断片同士が引きあって会合するには、このβ−シート部分の疎水性結合が最も有効に働くと考えられるからである。
【0024】
置換するアミノ酸は、ロイシンが望ましい。ロイシンは、3つの炭素原子が結合した直鎖構造を有する。そして、直鎖構造の炭素原子の数が3(イソロイシン)、2(バリン)、1(アラニン)であるアミノ酸への置換によって、両断片間のアフィニティを調節しやすいからである。ただし、ロイシン以外のアミノ酸の選択を棄却するものではない。また、置換後のアミノ酸がイソロイシン、バリン、アラニン以外のアミン酸である事を排除するものでもない。
【0025】
本発明で蛍光タンパク質の断片と融合される目標タンパク質は特に限定されるものではない。また、本発明を利用する温度やpH等の環境も、目標タンパク質やその断片が変性しない範囲であれば特に制限されない。上記条件を満たせば、例えば、in vitroでもよいし、in vivoであってもよい。
【0026】
蛍光タンパク質の断片と目標タンパク質の融合方法については、両タンパク質遺伝子のコドンフレームが一致(インフレーム)すれば特に制限はない。公知の組換え技術を用いてこの融合タンパク質遺伝子を作製できる。より具体的には、蛍光タンパク質の断片をコードするDNAと目標タンパク質をコードするDNAを、インフレームで直接もしくはリンカーを介して結合すれば、目標タンパク質と蛍光タンパク質の断片が融合したタンパク質をコードするDNAを得ることができる。
【0027】
同様にして、蛍光タンパク質の他方の断片ともう一方の目標タンパク質とが融合した融合タンパク質遺伝子を作製できる。さらに、この融合タンパク質遺伝子をこの遺伝子を導入する細胞に適したプラスミドに、発現ユニットと共に組み込んで、融合タンパク質遺伝子を該細胞で発現させるための組換えプラスミドを得ることができる。これら融合タンパク質遺伝子およびこれを組み込んだ組み換えプラスミドの作製は公知の方法、例えば、Maniates et al.,のMolecular cloningに記載の方法で作製できる。
【0028】
これらの組換えプラスミドを有する微生物、動物若しくは植物の細胞に導入・発現することで、蛍光タンパク質のいずれかの断片がそれぞれ融合された2つの目標タンパク質/蛍光タンパク質断片融合タンパク質を産生させることができる。また、組換えプラスミドを細胞に導入して、目的の目標タンパク質/蛍光タンパク質断片融合タンパク質を産生させることは、公知の方法を利用することができる。
【0029】
なお、プラスミドに組み込む蛍光タンパク質の断片をコードするDNAの少なくとも一方は、当該蛍光タンパク質断片中のβ−シート上のアミノ酸を置換した蛍光タンパク質断片をコードするDNAである。本発明は、蛍光タンパク質断片のアミノ酸を他のアミノ酸に置換することで、蛍光タンパク質の断片同士の会合のアフィニティを調節するからである。
【実施例】
【0030】
以下に本発明のタンパク質分子対について実施例を参考し、より詳細に説明する。実施例の概略は以下の通りである。
(1)目標タンパク質と蛍光タンパク質の断片が融合した融合タンパク質分子対をコードする遺伝子を含む組換えプラスミドを作製した。
(2)これらの組換えプラスミドで大腸菌形質転換し、形質転換した大腸菌を培養し組換えプラスミドDNAを抽出し精製した。
(3)精製したプラスミドDNA2種類を同時にマウス細胞に導入した。
(4)マウス細胞を固定した後、顕微鏡観察し、蛍光の有無を確認した。
【0031】
目標タンパク質同士のアフィニティと蛍光タンパク質同士のアフィニティの程度を知るために、本実施例では予め結合することが知られているタンパク質同士とそれらの一方の形を変えたミュータントを用意した。一方がミュータントである組では目標タンパク質同士は決して相互作用を行わない。また、蛍光タンパク質の断片の1つのアミノ酸も複数種類のアミノ酸に置換したものを用意した。アフィニティの程度を変えるためである。
【0032】
目標タンパク質としては、bFosとbJunを使用した。これらの2つのタンパク質は、ロイシンジッパーを構成することで相互作用を行うことが知られている。
【0033】
蛍光タンパク質は配列番号1で示すアミノ酸配列からなる蛍光タンパク質を用いた。これは、Venusと呼ばれる蛍光タンパク質であり、黄色に発光する。このN末端断片をbJunに融合させ、アミノ酸を置換したC末端断片をbFosに融合させた。アミノ酸の置き換えは、C末端側断片で行い、N末端側断片にはアミノ酸の置き換えを行わなかった。
【0034】
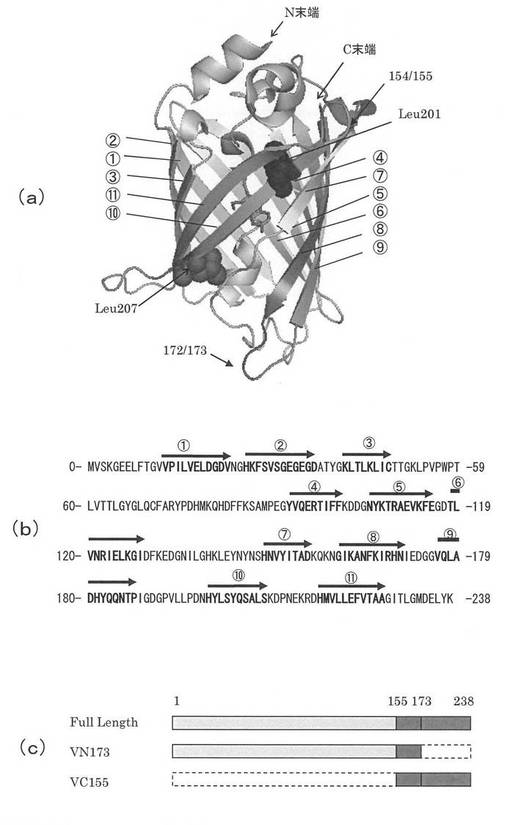
図3には、蛍光タンパク質の構造とアミノ酸配列の概念図を示す。蛍光タンパク質の構造は図2(a)と同じものである。図3(a)には、蛍光タンパク質の構造を、図3(b)にはN末端から番号をつけたアミノ酸配列を、そして図3(c)にはアミノ酸配列の切断を概念的に示す模式図を示した。図3(a)で示したβ−シート構造の番号と図3(b)で示したアミノ酸配列中の番号はそれぞれ対応する。
【0035】
この蛍光タンパク質の全長は238個のアミノ酸からなる。切断箇所は7番目のバレル構造の終端となる154番目のアミノ酸(アラニン)と155番目のアミノ酸(アスパラギン酸)の間とした。また、N末端側断片には155番目から173番目までの重複部分を有している。ここで、N末端側断片をVN173と呼び、C末端側断面をVC155と呼ぶ。
【0036】
蛍光タンパク質の断片同士のアフィニティの調節は配列のアミノ酸の置き換えで行う。置き換えを行うアミノ酸は以下のようにして決定した。図4には、蛍光タンパク質の構造中のアミノ酸同士の存在箇所と距離を計算で求めた表を示す。例えば、C末端側断片にある182番目のチロシン(Tyr)は9番目のβ−シート上にあり、V末端側断片の4番目のβ−シート上にある99番目のフェニルアラニン(Phe)と3.843オングストロームの距離関係にある。
【0037】
この結果からC末端側断片に存在する(N末端側から)201番目のロイシン(Leu)(符号30)と207番目のロイシン(Leu)(符号31)は、疎水性相互作用の相手になると考えられるN末端側断片のアミノ酸がそれぞれ4つ、3つと多い。そこで、201番目と207番目のロイシンを置き換えるアミノ酸とした。
【0038】
置き換えたアミノ酸は、イソロイシン、バリン、アラニンとした。これらは、それぞれ直鎖の炭素数が3、2、1と少なくなる。すなわち、疎水性相互作用は炭素数の減少に従って小さくなると考えた。疎水性相互作用の相手との距離が大きくなるからである。また、ロイシンはその直鎖の末端にメチル基が2つあるのに対して、イソロイシンの直鎖の末端にはメチル基は1つである。従って、イソロイシンはロイシン同様その直鎖の炭素数が3であるが、相互作用は若干小さくなると考えた。
【0039】
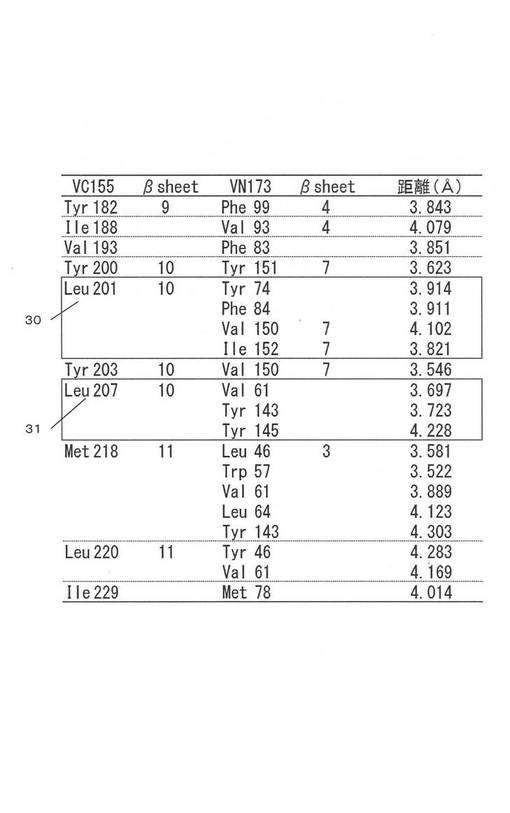
図5には作製したベクターの構造の概念図を示す。図5(a)を参照して、pHA−bFos−VC155(L201I)は、bFosと201番目のロイシンをイソロイシンに置換したVC155をコードする塩基配列が組み込まれたベクターである。なお、元々のpHA−bFos−VC155はパリュー大学由来である。サイトメガロウイルス由来のプロモータであるPCMV−IEと検出用のタンパク質であるHA−tagをコードする塩基配列の後、bFosと201番目のアミノ酸が置換されたVC155をコードする塩基配列が続く構造である。
【0040】
同様に、201番目のロイシンをバリン、アラニンに置換したものと、207番目のロイシンをイソロイシン、バリン、アラニンに置換したものを用意した。それぞれ、pHA−bFos−VC155(L201V)、pHA−bFos−VC155(L201A)、pHA−bFos−VC155(L207I)pHA−bFos−VC155(L207V)、pHA−bFos−VC155(L207A)のように表す。
【0041】
これらの相手方となる融合タンパク質分子は、bJunと蛍光タンパク質のN末端側断片である(図5(b)参照)。ベクターは、pFLAG−bJun−VN173であり、bJunと蛍光タンパク質のN末端断片をコードする塩基配列が組み込まれたベクターである。pHA−bFos−VC155(L201I)などと同様に、サイトメガロウイルス由来のプロモータであるPCMV−IEと検出用のタンパク質であるFLAG−tagをコードする塩基配列の後、bJunとVN173をコードする塩基配列が続く構造である。
【0042】
また、ネガティブコントロールとして、相互作用を起こさない目標タンパク質を用意した。これはbFosをミュータント化してbJunと相互作用を起こさないようにしたタンパク質である。これをbFos(m)とした。
【0043】
また、上記の組換えプラスミドDNAが導入されたことを確認するために、図2(b)で示した赤色の蛍光タンパク質をコードするプラスミドDNAも用意した。この蛍光タンパク質はmCherryと呼ばれる蛍光タンパク質で配列番号2番で示すアミノ酸配列を有する。
【0044】
<組換えプラスミドDNAの作製>
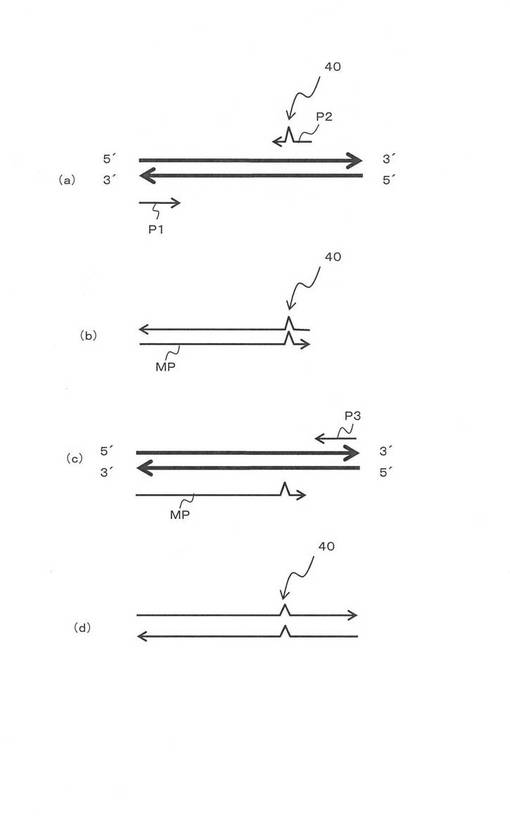
図6と図7を用いて、組換えプラスミドDNAの作製の工程を説明する。パリュー大学由来のpHA−bFos−VC155をAsp718とNotIで制限酵素処理し、VC155部分10を切り出し、塩基配列13を得た(図7(a、c))。一方、pHA−bFos−VC155由来のプラスミドのVC155部分の所定アミノ酸を図6で示したメガ・プライマー法で置き換えたプラスミド17を作製した(図7(b))。メガ・プライマー法で用いたプライマーを表1に示す。
【0045】
【表1】

【0046】
なお、上記のプライマー1は配列番号3、プイライマー2(L209I)は配列番号4、プイライマー2(L201V)は配列番号5、プイライマー2(L201A)は配列番号6、プイライマー2(L207I)は配列番号7、プイライマー2(L207VI)は配列番号8、プイライマー2(L207A)は配列番号9、プライマー3は配列番号10である。
【0047】
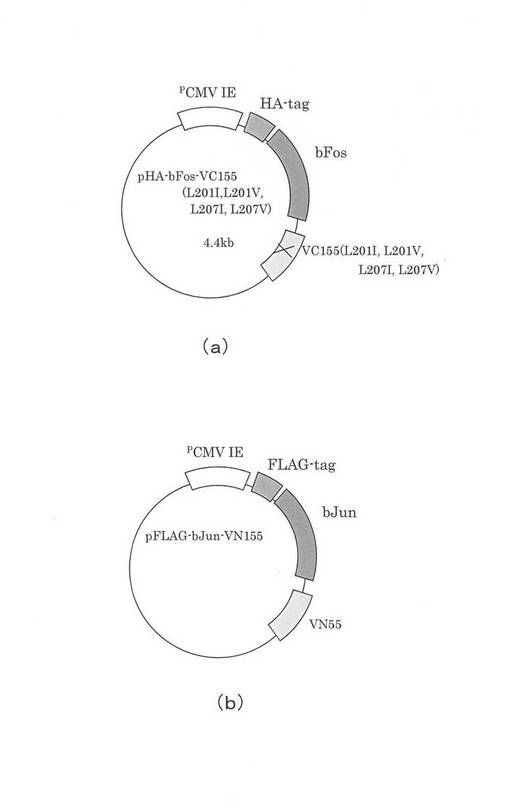
図6を参照して、メガ・プライマー法について概説しておく。図6(a)は、図7(b)で示すVC155部分のDNAを示す。プライマーP1は、この2本鎖DNAのうち、片方のDNA鎖の3´末端部分に対応するプライマーであり、プライマーP2は、もう片方のDNA鎖に対応し、置き換えたい塩基40を含み、その前後の塩基で構成されるプライマーである。
【0048】
VC155部分のDNAを鋳型とし、プライマーP1およびプライマーP2を用いてPCRを行うと、図6(b)で示すように、置換したい部分の塩基が、塩基40とそれに対応する塩基に置き換えられた2本鎖DNAが大量に得られる。これがメガ・プライマーMPである。
【0049】
そして、図6(c)で示すように、VC155部分のDNAを鋳型とし、このメガ・プライマーMPとプライマーP3を用いて再度PCRを行うと、図6(d)で示すような、VC155部分のDNAであって、所定の場所の塩基が、塩基40とそれに対応する塩基に置換された2本鎖DNAを得ることができる。従って、所定箇所のアミノ酸を置き換えるのは、そのアミノ酸に対応する箇所の塩基配列を変更したプライマーP2を用いる。以上の工程で、所定の箇所をイソロイシン(I)、バリン(V)やアラニン(A)に置き換えた。
【0050】
VC155の所定のアミノ酸を置き換えたプラスミドから置き換えられたVC155部分だけをAsp718とNotIで制限酵素処理し切り出し塩基配列17を得た(図7(b))。なお、図7(b)(d)でバツ印の部分は、アミノ酸が置き換えられた部位を示す。そして、塩基配列13と塩基配列17をライゲーションして、所定のプラスミド19を得た。
【0051】
なお、塩基配列13と17は読み枠が合うようにMCS(マルチクローニングサイト)内にある2種類の制限酵素で処理し、フェノール/クロロホルム/イソアミルアルコール溶液処理後、エタノール沈殿により回収した。さらに電気泳動を行った後、ゲルから切り出し、Mag ExtractorTM (TOYOBO社製)を用いて抽出し、以下のように反応液10μLを作製し、16℃で16時間ライゲーションを行った。
【0052】
< 反応液の組成 >
ベクター x μL
インサート y μL
T4 DNA ligase 0.4 μL
10×Ligation buffer 1 μL
ATP (5.0 mM) 1 μL
ベクターとインサートのモル比が1:3となるように加え、滅菌水で10μLにfill upした。
【0053】
<塩基配列の確認>
作製したベクターの塩基配列は、以下の手順によりシークエンシング反応によるキャピラリー電気泳動によって確認した。
【0054】
0.2mL PCRチューブに鋳型DNA(50−100pmoL/μLに調製)を4μL移し、サーマルサイクラー(Gene Amp PCR System 9700、 Applied Biosystems社製)を用いて96℃で1分間加熱後、直ちに氷上に置いた。これにCEQ DTCS Quick Start Master Mix (BECKMAN COULTER社製)を4μL、プライマーを1μMとなるように加え、反応液量を10μLとした。この反応液をサーマルサイクラーで以下の条件で反応を30サイクル行った。
【0055】
< 反応条件 >
Denaturation 96 ℃ 20sec
Annealing 50 ℃ 20sec ×30 cycle
Extension 60 ℃ 4 min
4 ℃ ∞
PCRで増幅させたベクターを次の手順でキャピラリー電気泳動を行った。1.5mLマイクロチューブにStop Solution(3M酢酸ナトリウム 1μL、20mg/mL グリコーゲン溶液0.5μLを入れておき、反応終了後のシークエンシング反応液を10μL加え、よく攪拌した。冷100%エタノールを30μLを加えて攪拌した後、直ちに4℃、14000rpmで15分間遠心分離した。上清を除き、冷70%エタノールを200μLを加えて、ペレットを洗浄した後、4℃、14000rpmで2分間遠心分離した。この洗浄操作をもう一度繰り返し、上清を除去した後、ペレットを真空乾燥し、30μLのSample Loading Solution に溶解した。
【0056】
サンプルを96−wellサンプルプレートに移し、Mineral Oil (BECKMAN COULTER社製)を1滴加えた。96−well バッファープレートに、CEQ Separation buffer (BECKMAN COULTER社製)をwellの8割程度加えた。これらのサンプルプレートとバッファープレート、そして、Separation Gel−LPA Iを、CEQTM2000XL DNA Analysis System (BECKMAN COULTER社製)にセットして、キャピラリー電気泳動を行った。塩基配列の結果はこのシステムの出力としてテキストデータで得ることができる。
【0057】
次に作製した組換えプラスミドを大腸菌を用いて増幅させた。これは、以下の手順によった。
(1)コンピテントセルを作製した。
(2)大腸菌(コンピテントセル)に組換えプラスミドを導入し培養させた。
以下にその手順を詳細に説明する。
【0058】
<コンピテントセルの作製>
大腸菌(DH10B)を、LB寒天培地に塗布し、37℃で一晩培養した。この培地から単一コロニーを選び、L字型試験管に入れたLB液体培地20mLに植菌して、37℃で一晩振盪培養した。一晩振盪培養したLB液体培地2mLを、坂口フラスコに入れたLB液体培地200mLに加えた。坂口フラスコ内のLB液体培地を37℃に保ち、波長660nmで測定した濁度(O.D.660)が0.6になるまで振盪培養した。その後坂口フラスコを氷上に20分間静置した。
【0059】
この振盪培養した大腸菌を洗浄した。具体的には、以下の手順を行った。フラスコ内のLB液体培地を遠心チューブに移し、0℃、5000rpmの条件で10分間遠心分離後、上清を取り除いた。上清を取り除いた遠心チューブに、オートクレーブ滅菌した氷冷超純水150mLを加えて懸濁した。懸濁した後、遠心チューブを、0℃、5000rpmの条件で10分間遠心分離後、上清を取り除いた。そして、再度遠心チューブに、オートクレーブ滅菌した氷冷超純水75mLを加えて懸濁した。懸濁した後、遠心チューブを、0℃、5000rpmでの条件で10分間遠心分離後、上清を取り除いた。以上のように、大腸菌を氷冷超純水で2回洗浄した。
【0060】
次に、遠心チューブにオートクレーブ滅菌した10%グリセロール12mLを加えて、懸濁した。グリセロールで懸濁した後、0℃、6000rpmの条件で10分間遠心分離後、10%グリセロールを取り除いた。そして再び、遠心チューブにオートクレーブ滅菌した10%グリセロール1.6mLを加え、懸濁した。
【0061】
以上の手順で、振盪培養された大腸菌を、DNAに対する膜透過性が増大した大腸菌であるコンピテントセルとした。保存するコンピテントセルは、遠心チューブ中の懸濁液を1.5mLマイクロチューブに40μLずつ分注し、液体窒素に入れて凍結させた。この保存するコンピテントセルは、−80℃で保存した。
【0062】
<エレクトロポレーション法による大腸菌の形質転換>
氷上で融解させたコンピテントセル40μLに、プラスミドDNA溶液1μLを加えて、混合し混合溶液とした。この混合溶液を氷上で冷やしておいたキュベット(電極間隔2.0mm)に移し、E.coli Pulser (BIO−RAD社製)を用いて、パルス(電圧2.1kV)をかけた。
【0063】
この混合溶液にSOC培地1mLを加えて懸濁し、1.5mLマイクロチューブに移した。これを適当な抗生物質を添加したLB寒天培地に塗布し、37℃で一晩培養した。
【0064】
<プラスミドDNAの抽出と精製>
次に増殖させた大腸菌からプラスミドDNAを抽出し精製した。プラスミドDNAを導入した大腸菌を培養したLB寒天培地から単一コロニーを選び、適当な抗生物質を添加したLB液体培地3mLに植菌して、37℃で一晩振盪培養した。
【0065】
培養液を1.5mLマイクロチューブに移し、4℃、10000rpmの条件で1分間遠心分離して、上清を取り除き集菌した。これにSolutionIを100μL加えて懸濁後、SolutionIIを200μL加えて穏やかに混合し、氷上に5分間置いた。SolutionIIを入れることで大腸菌は溶菌し、細胞内のプラスミドDNAが取り出せる。マイクロチューブ内の溶液を中和させるために、SolutionIIIを150μL加えて穏やかに混合し、氷上に5分間置いた。
【0066】
なお、SolutionIは、Tris緩衝液(25mM、pH8)にグルコースを50mMになるように加えた溶液である。また、SolutionIIは、0.2Nの水酸化ナトリウムに1%のSDS(ラウリル酸ナトリウム)を加えた溶液である。また、SolutionIIIは、3Mの酢酸カリウムと11.5%の酢酸を加えた溶液である。
【0067】
次に1.5mLマイクロチューブごと4℃、15000rpmの条件で10分間遠心分離後、ペレットを取り除いた。ペレットを取り除いた溶液に、フェノール/クロロホルム/イソアミルアルコール溶液を500μL加えて穏やかに混合し、プラスミドDNAを抽出した。
【0068】
20℃、15000rpmの条件で10分間遠心分離後、上清を別のマイクロチューブに移した。2/3倍量のイソプロパノールを加えて穏やかに混合し、室温で10分間置いた。
【0069】
さらに、20℃、15000rpmの条件で10分間遠心分離後、上清を取り除いた。上清を取り除いたペレットは、エタノール沈殿によって得たプラスミドDNAである。ペレットを冷70%エタノール400μLでリンスし、4℃、15000rpmの条件で3分間遠心分離後、上清を取り除き、15分間真空乾燥した。
【0070】
抽出したプラスミドDNA中のRNAを除去するために次のプロセスを行った。ペレットをTE buffer (pH8.0)50μLに溶解し、RNaseA溶液(0.5μg/mL)を1μL加えて、37℃で30分間インキュベートした。さらに、20%PEG6000/2.5M NaClを30μL加えて、よく混合し、氷上に1時間置いた。
【0071】
そして、4℃、15000rpmの条件で10分間遠心分離後、上清を取り除いた。ペレットを冷70%エタノール400μLでリンスし、4℃、15000rpmで3分間遠心分離した後、上清を取り除き、15分間真空乾燥した。ペレットをTE buffer (pH8.0)50μLに溶解した。以上の手順で精製したプラスミドDNAを得た。
【0072】
次に精製したプラスミドDNAをマウス細胞に導入した。具体的な手順は以下による。
(1)マウス細胞を培養した。
(2)リポフェクション法を用いてマウス細胞に遺伝子を導入した。
以下に詳細を説明する。
【0073】
まず、マウスの細胞は以下のようにして培養した。マウスC3H 10T1/2細胞を、D−MEM培地を8mL入れた100mmシャーレの中で、37℃、5%CO2存在下のCO2インキュベーターを用いて培養した。継代は、対数増殖期の細胞が、70−90%コンフルエントした時に行った。シャーレ内のD−MEM培地を取り除き、1×PBS(−)を4mL入れて洗浄し、これを取り除いた。トリプシン/EDTA溶液を1mL入れて、細胞をシャーレからはがし、球形になったことを確認してから、D−MEM培地を4mL入れて懸濁した。
【0074】
この単一細胞浮遊液0.5mLを、あらかじめD−MEM培地を8mL入れた別のシャーレに播種し、細胞をシャーレ全体に均一になるように分散させて、37℃、5% CO2存在下のCO2インキュベーターを用いて培養した。
【0075】
凍結ストック細胞の作製は、対数増殖期の細胞が、70−90%コンフルエントした時に、継代と同様にして細胞を100mmシャーレからはがした。細胞が球形になったことを確認してから、D−MEM培地を4mL入れて懸濁した。この単一細胞浮遊液を遠心チューブに移し、1000rpmで10分間遠心分離後、上清を取り除いた。無血清タイプ細胞凍結保存液バンバンカーTM(リンフォテック社製)を細胞凍結保存用チューブ(クライオチューブ)1本につき0.5mLとなるように加えて、懸濁した。この細胞懸濁液をクライオチューブに移し、−80℃で保存した。
【0076】
−80℃で凍結保存された細胞を培養する時は、まず、クライオチューブを37℃の恒温槽内で軽く揺すりながら細胞懸濁液を半解凍し、その後、室温で解凍を行った。この細胞懸濁液0.5mLを、あらかじめD−MEM培地を8mL入れた100mmシャーレに播種し、細胞をシャーレ全体に均一になるように分散させて、37℃、5%CO2存在下のCO2インキュベーターを用いて培養した。
【0077】
培養した細胞にプラスミドDNAをリポフェクション法で導入した。継代と同様にして細胞を100mmシャーレからはがし、細胞が球形になったことを確認してから、D−MEM培地を4mL入れて懸濁した。この単一細胞浮遊液をトランスフェクション時に50−80%コンフルエントになるように、12−wellプレートに1サンプルにつき(以下同様)、1μL播いた。Opti−MEMRIReduced Serum Medium(Invitrogen社製)に導入したいDNA1μgを加え、合計が200μLとなるようにOpti−MEMRIReduced Serum Mediumを加えて、穏やかに混合した。
【0078】
ここで導入したいDNAとは、先に作製したベクターである。より具体的には、BiFCを行わせる目標タンパク質と蛍光タンパク質の断片がコードされたプラスミドDNAと、赤色の蛍光タンパク質をコードするプラスミドDNAである。BiFCを行わせるプラスミドDNAは、赤色の蛍光タンパク質の3倍量となるように配合した。また、混合した溶液に3μLのLipofectamineTM LTX(Invitrogen社製)を加えて、穏やかに混合後、30分間室温で静置した。このDNA−LipofectamineTM LTX複合体200μLをwellに加えて、穏やかにプレートを振動させて混合した。
【0079】
次にマウス細胞を観察し、蛍光発光の有無を確認した。これは以下の手順によった。
(1)細胞の固定およびDAPIによる核の染色を行った。
(2)標本を作製した。
(3)顕微鏡による細胞観察を行った。
以下に具体的な手順を説明する。
【0080】
<細胞の固定およびDAPIによる核の染色>
培養細胞への遺伝子導入から24時間後に、固定と染色を行った。35mmシャーレ内のD−MEM培地を取り除き、4%パラホルムアルデヒドをカバーガラス全体に広がるように加えた。室温で15分間静置して固定後、4%パラホルムアルデヒドを取り除き、1×PBS(−)をカバーガラスがひたる程度に加えて、10分間振盪し洗浄した。1×PBS(−)を取り除き、0.1% Triton X−100を1mL加えて、室温で5分間静置した。
【0081】
0.1% Triton X−100を取り除き、1×PBS(−)をカバーガラスがひたる程度に加えて、10分間振盪して洗浄した。1×PBS(−)を取り除き、DAPI染色液をカバーガラス全体に広がるように加え、室温で5分間静置して染色した。DAPI染色液を取り除き、1×PBS(−)をカバーガラスがひたる程度に加えて、10分間振盪し洗浄した。
【0082】
<標本の作製>
マウント用グリセロールを10μLのせたスライドガラスの上に、細胞を固定・染色したカバーガラスを、気泡が入らないようにしてのせた。カバーガラスの上に濾紙を置き、垂直に押して余分な水分を取り除いた後、カバーガラスの周囲に黒いマニキュアを塗布し、遮光して乾燥させた。
【0083】
<蛍光顕微鏡による細胞の観察>
標本を、冷却型CCDカメラORCA−ER(浜松ホトニクス社製)を装備した蛍光顕微鏡Eclipse E600 (Nikon社製)を用いて観察した。フィルターブロックは、DAPI染色の観察用にUV−1Aフィルター(EX:360−370nm、DM:400nm、BM:400nm、Nicon社製)、蛍光タンパク質(Venus)の観察用にFITCフィルター(EX:465−495nm、DM:505nm、BM:515−555nm、Nicon社製)、蛍光タンパク質(mCherry)の観察用にTxRedフィルター(EX:540−580nm、DM:595nm、BM:600−660nm、Nicon社製)を用いた。画像の取り込みと解析は、画像解析ソフトウェア Lumina Vision for Mac(三谷商事社製)を用いて行った。
【0084】
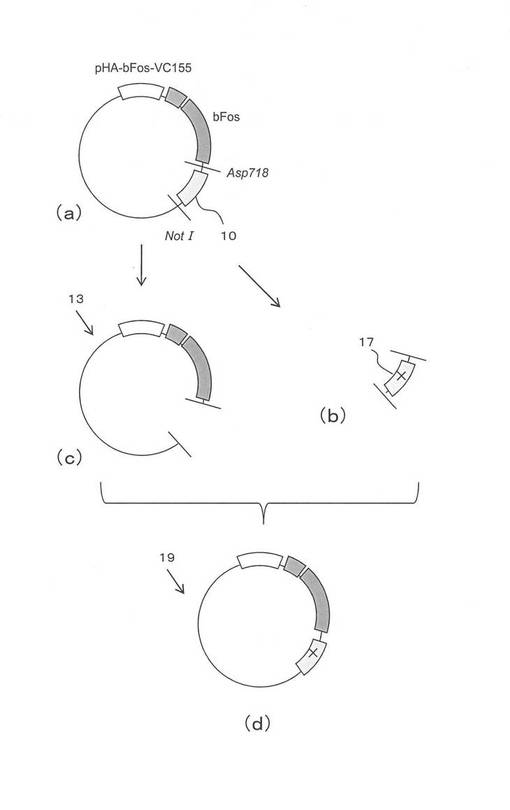
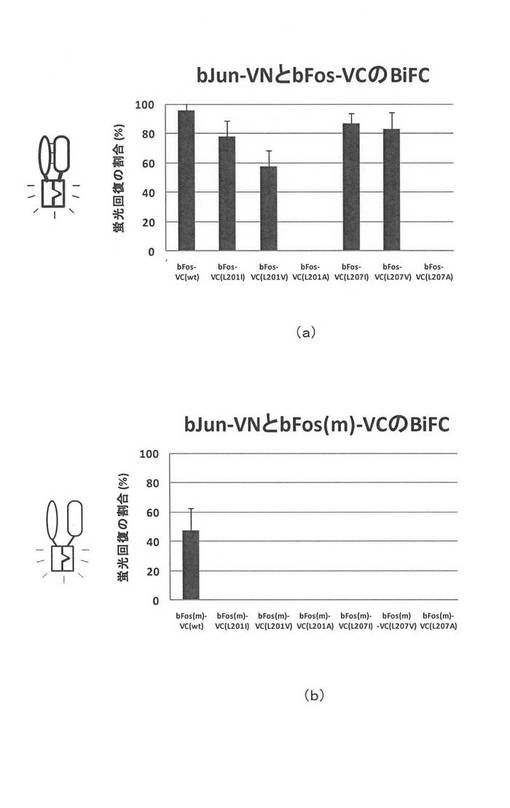
次に図8を参照して、観察結果を説明する。図8(a)は、横軸にサンプルの種類を示し、縦軸は蛍光回復の割合(%)を示す。蛍光回復率は、mCherry(赤色)の発光をする細胞の数に対して、それらの細胞の中で緑黄色の蛍光発光をしている細胞の数の割合で評価した。BiFCを行わせるための緑黄色の蛍光タンパク質(Venus)は、赤色の蛍光タンパク質(mCherry)の3倍量が導入されている。したがって、赤色の蛍光発光をする細胞の全てにBiFCを行わせるためのプラスミドDNAが導入されていると推定できる。言い換えると、赤色を発光していて緑黄色を発光していない細胞は、BiCFを行わせるためのプラスミドDNAは導入されているにもかかわらず、蛍光回復しなかったケースと判断できる。
【0085】
横軸のサンプルは、図8(a)、(b)ともに同じ順で表示されている。サンプルは横軸左から、bFosの野生種(bFosVC(wt))、201番目のアミノ酸を変更したもの(bFosVC(L201I)、bFosVC(L201V)、bFosVC(L201A))と207番目のアミノ酸を変更したもの(bFosVC(L207I)、bFosVC(L207V)、bFosVC(L207A))の7種類である。bFosの野生種は201番目および207番目のアミノ酸がロイシンである。
【0086】
図8(b)は、bFosをミュータントにした場合である。サンプルはbFosがミュータントになっている以外は、図7(a)と同じ蛍光タンパク質のC末端側断片が融合されたサンプルである。
【0087】
図8(a)を参照して、bFosVCの野生種では、目標タンパク質の相互作用によってほとんどの細胞内で蛍光発光が回復していることが観察された。一方、図8(b)を参照すると、目標タンパク質が相互作用を行わない場合であるにもかかわらず、およそ半分の細胞で蛍光回復が観察された。すなわち、目標タンパク質のアフィニティより蛍光タンパク質の断片同士のアフィニティが強いために、目標タンパク質の相互作用の有無に係らず、蛍光回復が行われた。
【0088】
再び図8(a)を参照して、本発明のタンパク質結合体である野生種以外のサンプルでは、置換したアミノ酸がイソロイシン、バリン、アラニンとアミノ酸の直鎖の炭素数が3、2、1と減るにしたがって、蛍光回復の割合が減少した。アラニンでは、全く蛍光が回復しなかった。これは、炭素数が減少するに従って、疎水性相互作用をする相手との距離が長くなるために、蛍光タンパク質の断片同士が再会合しにくくなるためと考えられた。すなわち、イソロイシン、バリン、アラニンへのアミノ酸置換によって蛍光タンパク質の断片同士のアフィニティが調節できた。
【0089】
再度図8(b)を参照すると、本発明のサンプルは目標タンパク質が相互作用を行わない時は、全く発光回復が認められなかった。従って、本発明のサンプルは、目標タンパク質の相互作用が行われた場合は蛍光発光を回復し、目標タンパク質が相互作用を行わない場合は蛍光発光を回復しなかった。これはBiFC法を用いてタンパク質同士の相互作用をアッセイするために必要不可欠な特性である。言い換えると、bFosとbJunを目標タンパク質としてBiFC法を用いる場合は、配列番号1の蛍光タンパク質の野生種を用いるのではなく、本発明のサンプルのように蛍光タンパク質の断片同士のアフィニティを調節しなければ、正しいアッセイができないことが示された。
【産業上の利用可能性】
【0090】
本発明は、たんぱく質の相互作用をアッセイするBiFC法に利用することができる。
【符号の説明】
【0091】
10、11 目標タンパク質
12、13 蛍光タンパク質の断片
14 目標タンパク質同士の相互作用
15 蛍光復活した蛍光タンパク質
20 蛍光タンパク質のβ−シート
21 蛍光タンパク質のα−へリックス
22 蛍光タンパク質のアミノ酸のループ部分
30 201番目のロイシン
31 207番目のロイシン
40 アミノ酸の修正点
【技術分野】
【0001】
本発明は、タンパク質同士の相互作用を観察するBiFC法において用いられる蛍光タンパク質分子対とその蛍光タンパク質分子対をコードする遺伝子、該遺伝子導入ベクターおよびその遺伝子導入ベクターが導入され、目的の蛍光タンパク質分子対を産生する細胞に関する。
【背景技術】
【0002】
タンパク質間相互作用を解析する技術の1つに、BiFC (Bimolecular Fluorescence Complementation : 二分子蛍光相補性)法がある。BiFCとは、蛍光タンパク質を2つの分割断片にすることで失われた蛍光が、相補的な分割断片の再会合により回復する現象である。この現象を利用して、生きた細胞内でのタンパク質間相互作用を可視化することができる。BiFC法を用いると、翻訳後修飾されたタンパク質の細胞内局在や機能解析など、タンパク質が機能している状態に近い条件でのタンパク質間の相互作用を調べることができる。
【0003】
蛍光タンパク質の分割断片を用いる手法は、2000年に初めて報告され、逆並行ロイシンジッパーと融合させたGFP(Green Fluorescent Protein)の分割断片が、大腸菌で、ロイシンジッパーの形成とそれに伴うGFP断片の会合によって蛍光回復することが明らかにされた(非特許文献1)。
【0004】
動物細胞内での蛍光回復は、GFPよりも蛍光強度が大きいYFP(Yellow Fluorescent Protein)で報告され(非特許文献2)、BiFC アッセイ(assay)という術語が初めて使用された。その後、GFP、YFP、CFP(Cyan Fluorescent Protein)などの複数の蛍光タンパク質を用いたマルチカラーBiFC(非特許文献3)が報告された。
【0005】
CFPを改良したCerulean(非特許文献4)、YFPを改良したCitrine(非特許文献5)とVenus(非特許文献6)は、生理的条件下(37℃)での発色団形成効率が高く、これらの蛍光タンパク質を用いたマルチカラーBiFC(非特許文献7)も報告されている。
【0006】
近年では、BiFC法で用いられる蛍光タンパク質も増え(非特許文献8、非特許文献9)、また、ユビキチン化されたタンパク質の検出(非特許文献10)、薬剤によるタンパク質間相互作用の誘導(非特許文献11)など、BiFC法の応用的な利用も進歩してきている。
【0007】
また、特許文献1では、時間経過に伴って発光色が変化する蛍光タンパク質をN末端側断片とC末端側断片にわけて、相互作用を調べたいそれぞれのタンパク質と融合させ、当該タンパク質間の相互作用の際に、分割された蛍光タンパク質が再会合して発色する技術が開示されている。ここでは、蛍光タンパク質としてKusabira−Orange(mKO)が選択され、特にN末端から70番目のアミノ酸であるブロリンが他のアミノ酸に置換された蛍光タンパク質であることを特徴としている。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2006−308568号公報
【非特許文献】
【0009】
【非特許文献1】インドラニール ゴーシュ(Indraneel Ghosh), アンドリュー D. ハミルトン(Andrew D. Hamilton)、とライニー レーガン(Lynne Regan)、「アンチパラレル ロイシン ジッパー−ディレクテッド プロテイン リアセンブリー:アプリケーション ツー ザ グリーン フローレセント プロテイン(Antiparallel Leucine Zipper−Direction Protein Reassembly: Application to the Green Fluorescent Protein」,アメリカン ケミカル ソサイアティ(American Chemical Society)、2000年、122、P.5658−5659
【非特許文献2】フュウ C.D.(Hu, C.D.), シネノフ Y.(Chinenov, Y.), とケルポラ T.K.(Kerppola, T.K.)「ビジュアリゼーション オブ インターラクション アマング ビーズィーアイピー アンド アールイーエル ファミリー プロテインズ イン リビング セルズ ユージング バイモレキュラー フローレセンス コンプリメンテーション( Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation.)」 モル セル(Mol. Cell) 9, 789−798 (2002).
【非特許文献3】フュウ C.D.(Hu, C.D.), シネノフ Y.(Chinenov, Y.), とケルポラ T.K.(Kerppola, T.K.)「シミュルテイニアス ビジュアリゼーション オブ マルチプル プロテイン インターラクションズ イン リビング セルズ ユージング マルチカラー フローレセンス コンプレメンテイション アナリシス(Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis.)ナット バイオテクノル(Nat. Biotechnol.)21, 539−545 (2003).
【非特許文献4】リッゾ M.A.(Rizzo, M.A.), スプリンガー G.H.(Springer, G.H.), グラナダ B.(Granada, B.), と ピストン D.W.(Piston, D.W.)「アン インプルーブド シアン フローレセント プロテイン バリアント ユースフル フォア フレッツ( An improved cyan fluorescent protein variant useful for FRET.)ナット バイオテクノル(Nat. Biotechnol.)22, 445−449 (2004).
【非特許文献5】グリエスベック O.(Griesbeck, O.), バイルド G.S.(Baird, G.S.), キャンベル R.E.(Campbell, R.E.), ザハリアスD.A.(Zacharias, D.A.), と ツジエン R.Y.(Tsien, R.Y.)「リデューシング ザ エンバイロメンタル センシティビィティ オブ イエロー フローレセント プロテイン(Reducing the environmental sensitivity of yellow fluorescent protein.)」J. Biol. Chem. 276, 29188−29194 (2001).
【非特許文献6】ナガイ T.(Nagai, T.), イバタ K.(Ibata, K.), パーク E.S.(Park, E.S.), クボタ M.(Kubota, M.), ミコシバ K.(Mikoshiba, K.), と ミヤワキ A.(Miyawaki, A.)「ア バリアント オブ イエロー フローレセント プロテイン ウイズ ファースト アンド エフィシェント マチュレーション フォア セル−バイオロジカル アプリケーション(A variant of yellow fluorescent protein with fast and efficient maturation for cell−biological applications.)」ナット バイオテクノ(Nat. Biotechnol.) 20, 87−90 (2002).
【非特許文献7】シュー Y.J.(Shyu, Y.J.), リー H.(Liu, H.),デング X.( Deng, X.), と フュウ C.D.(Hu, C.D.)「アイデンティフィケーション オブ ニュー フローレセント プロテイン フラグメンツ フォア バイモレキュラー フローレセンス コンプリメンテーション アナライシス アンダー フィシオロジカルコンディションズ( Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions.) バイオテクニークス( Biotechniques) 40, 61−66 (2006).
【非特許文献8】ファン J.Y.(Fan, J.Y.), キュイ Z.Q.(Cui, Z.Q.), ウェイ H.P.(Wei, H.P.), ザングZ.P.(Zhang, Z.P.), ズウ Y.F.(Zhou, Y.F.), ワング Y.P.(Wang, Y.P.), と ザング X.E.(Zhang, X.E.)「スプリット mチェリー アズ ア ニュー レッド バイモレキュラー フローレセンス コンプリメンテーション システム フォア ビジュアリジング プロテイン−プロテイン インターラクションズ イン リビング セルズ( Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein−protein interactions in living cells.)」 バイオケム バイオフィズ レス コミュ (Biochem. Biophys. Res. Commun.) 367, 47−53 (2007).
【非特許文献9】チュー J.(Chu, J.), ザング Z.(Zhang, Z.), ゼング Y.(Zheng, Y.),ヤング J.( Yang, J.), キン L.(Qin, L.), リュー J.(Lu, J.),フアング Z.L.( Huang, Z.L.), ゼング S.(Zeng, S.), と ルオ Q.(Luo, Q.)「 ア ノベル ファー−レッド バイモレキュラー フローレセンス コンプリメンテション システム ザット アローズ フォア エフィシェント ビジュアリザション オブ プロテイン インターラクションズ アンダー フィジオロジカル コンディションズ( A novel far−red bimolecular fluorescence complementation system that allows for efficient visualization of protein interactions under physiological conditions.)バイオセンス バイオエレクトロン (Biosens. Bioelectron.) 25, 234−239 (2009).
【非特許文献10】ファング D.(Fang, D.), と ケルポラ T.K.(Kerppola, T.K.)「アビクイチン−メディエーティッド フローレセンス コンプリメンテション リビールズ ザット ジュン アビクイティネイテッド バイ イッチ/エイアイピー4 イズ ローカライズド ツー リソソーム (Ubiquitin−mediated fluorescence complementation reveals that Jun ubiquitinated by Itch/AIP4 is localized to lysosomes.)」 Proc. Natl. Acad. Sci. U.S.A., 101, 14782−14787 (2004).
【非特許文献11】ロビダ M.A.(Robida, M.A,)と ケルポラT.K.(Kerppola, T.K.)「バイモレキュラー フローレセンス コンプリメンテーション アナライシス オブ インデューシブル プロテイン インターラクション:エフェクト オブ ファクターズ アフェクティング プロテイン フォールディング オン フローレセント プロテイン フラグメント アソシエーション(Bimolecular fluorescence complementation analysis of inducible protein interactions: effects of factors affecting protein folding on fluorescent protein fragment association.) J. Mol. Biol. 394, 391−409 (2009).
【発明の概要】
【発明が解決しようとする課題】
【0010】
BiFC法は、1つの蛍光タンパク質を分断し、それぞれの断片を2つの相互作用を調べたいタンパク質(目標タンパク質)にそれぞれ融合させておき、2つの目標タンパク質が相互作用を行った時に、蛍光タンパク質断片同士が会合し発光する現象を利用したタンパク質間相互作用を評価する方法の1つである。従って、2つの目標タンパク質が相互作用を行わない場合には、蛍光タンパク質断片同士は会合せず、発光しないという事が前提となっている。
【0011】
しかし、タンパク質は複雑な3次元構造を有しており、ペプチド主鎖の多彩な折れ曲がり構造と、アミノ酸側鎖の官能基が存在する。そして、これらペプチド主鎖の形状とアミノ酸側鎖の官能基間の相互作用によって、タンパク質間のアフィニティ(親和力)が決定されることが知られている。従って、2つの目標タンパク質間に相互作用がなくても、分断された蛍光タンパク質断片同士のアフィニティが強ければ、断片同士の会合が生じることが考えられる。その結果、目標タンパク質同士の相互作用がなくても発光してしまうという現象が生じる事が考えられる。この状態は、2つの目標タンパク質間の会合を示していることにならず、タンパク質間の相互作用を評価する方法として好ましくない状態であるといえる。本発明者は、事実このような状態を観測し、上記の課題を把握するに至った。
【課題を解決するための手段】
【0012】
本発明は、上記の課題に鑑みて想到されたものであり、分断された蛍光タンパク質間のアフィニティを調節する方法を提供するものである。より具体的には、
蛍光タンパク質のN末端側断片に接続された第1のタンパク質分子と、前記蛍光タンパク質のC末端側断片に接続された前記第2のタンパク質分子とからなる融合タンパク質分子対において、
前記N末端側断片と前記C末端側断片の少なくとも一方の断片で、少なくとも1箇所以上のアミノ酸が他のアミノ酸に置換され、
前記N末端側断片とC末端側断片同士のアフィニティは、アミノ酸の置換を受けない場合と比較し、前記アミノ酸の置換によって調節されたことを特徴とする融合タンパク質分子対を提供する。
【0013】
また、本発明の融合タンパク質分子対では、前記置換されるアミノ酸は、前記蛍光タンパク質のβ−シート上のアミノ酸である。
【0014】
また、本発明の融合タンパク質分子対では、前記変更されるアミノ酸は、β−シート上のロイシンであり、変更されたアミノ酸はイソロイシン、バリン、アラニンである。
【0015】
また、本発明の融合タンパク質分子対では、前記蛍光タンパク質は配列番号1の蛍光タンパク質である。
【0016】
また、本発明の融合タンパク質分子対では、前記変更されるアミノ酸は、C末端側断片のアミノ酸であって配列番号1のN末端側から201番目若しくは207番目のロイシンである。
【0017】
また、本発明は、上記の融合タンパク質分子対をコードする遺伝子を提供する。また、上記遺伝子のそれぞれを含む遺伝子導入/発現ベクターを提供する。さらに、その遺伝子導入/発現ベクターが導入され、上記の融合タンパク質分子対を産生する細胞を提供するものである。
【発明の効果】
【0018】
本発明は、蛍光タンパク質の分断された断片同士のアフィニティを調節するので、目標タンパク質同士が相互作用を行った時だけ、蛍光タンパク質が再会合し発光させることができる融合タンパク質分子対と、その融合タンパク質分子対を産生する細胞を提供することができる。これによって細胞内の活動はもとより、細胞外であっても、タンパク質同士の相互作用をアッセイするBiFC法を、より正確に行うことができる。
【図面の簡単な説明】
【0019】
【図1】本発明のタンパク質の相互作用の概念を示す図である。
【図2】蛍光タンパク質の構造を例示する図である。
【図3】本実施例に用いた蛍光タンパク質の構造とアミノ酸配列の関係を示す図である。
【図4】アミノ酸の置き換え点を求めるために相互作用を行うアミノ酸同士の距離を計算した結果を示す図である。
【図5】本実施例に用いたベクターの構成を示す図である。
【図6】メガ・プライマー法を説明する概念図である。
【図7】本実施例に用いたベクターを作製する工程を示す概念図である。
【図8】本実施例の結果(蛍光回復率)を示すグラフである。
【発明を実施するための形態】
【0020】
本発明の概要を図1を用いて説明する。図1(a)を参照して、目標タンパク質10、11のそれぞれには、蛍光タンパク質のN末端側断片12とC末端側断片13がそれぞれ結合されている。本発明では、これらタンパク質の対を融合タンパク質分子対と呼ぶ。目標タンパク質同士が接近して相互作用を及ぼしあうと、蛍光タンパク質のN末端側断片とC末端側断片も接近し会合する。図1(b)の符号14は、目標タンパク質が相互作用により会合していることを示す。この会合によって蛍光タンパク質断片同士12,13が会合し、蛍光タンパク質として発光機能を回復する(15)。これがBiFC法の原理である。従って、目標タンパク質同士が相互作用を行っても、蛍光タンパク質の断片同士が会合せず、発光を生じない場合は、タンパク質間の相互作用を評価する方法として利用することはできない。図1(c)は、この様子を模式的に表している。
【0021】
一方、目標タンパク質間の相互作用がないにも係らず、蛍光タンパク質の断片同士が会合し発光機能を回復する場合も、タンパク質間の相互作用を評価する方法として利用することはできない。図1(d)は、この様子を表している。すなわち、BiFC法においては、目標タンパク質同士のアフィニティと蛍光タンパク質の断片同士のアフィニティを適切に調節することが必要である。言い換えると、図1(c)または図1(d)で示した状態にならず、図1(b)に示したように目標タンパク質間の相互作用に応じて蛍光タンパク質の断片同士が会合して発光すること必要となる。本発明では、蛍光タンパク質断片の一部のアミノ酸を他のアミノ酸に置換し、上記のような目標タンパク質の相互作用に応じて発光するように蛍光タンパク質断片同士のアフィニティを調節する。ここで調節するとは、蛍光タンパク質断片同士のアフィニティを強くする若しくは弱くすることをいう。
【0022】
本発明で利用できる蛍光タンパク質は特に制限はない。CFP、GFPばかりでなく、近赤外領域や、近紫外域の光を発光する蛍光タンパク質でもよい。図2(a)には、後述する実施例で用いた黄色を発色する蛍光タンパク質Venusの立体構造モデルを示す。蛍光タンパク質には、β−シート(20)構造を形成する部分と、α−へリックス(21)およびループ(22)の部分が存在する。そして、全ての蛍光タンパク質には図2と同様なβ−シート構造の部分が存在する。例えば、図2(b)には、赤色蛍光を発色する蛍光タンパク質mCherryの構造を示す。図2(a)同様のβ−シート構造を有している。なお、図2の構造は、以下のURLで知られるプロテインデータベースでることができる。http://www.rcsb.org/pdb/explore/explore.do structureId=1MYW
蛍光タンパク質のN末端側断片とC末端側断片との切断点は特に限定するものではないが、β−シート以外の部分が望ましい。特にループの部分が望ましい。切断された蛍光タンパク質が再会合するには、疎水性相互作用が必要と考えられる。そして、β−シート部分での疎水性相互作用は、分割された切断片同士の会合に寄与すると考えられるので、β−シート部分は切断されずに残っているのが好ましいからである。
【0023】
本発明では、N末端側断片若しくはC末端側断片の少なくとも一方の断片で、少なくとも1つのアミノ酸を他のアミノ酸と置換する。置換するアミノ酸はβ−シート上のアミノ酸が望ましい。蛍光タンパク質は、図2に例示したように、並列したβ−シート構造を有している。分断された蛍光タンパク質断片同士が引きあって会合するには、このβ−シート部分の疎水性結合が最も有効に働くと考えられるからである。
【0024】
置換するアミノ酸は、ロイシンが望ましい。ロイシンは、3つの炭素原子が結合した直鎖構造を有する。そして、直鎖構造の炭素原子の数が3(イソロイシン)、2(バリン)、1(アラニン)であるアミノ酸への置換によって、両断片間のアフィニティを調節しやすいからである。ただし、ロイシン以外のアミノ酸の選択を棄却するものではない。また、置換後のアミノ酸がイソロイシン、バリン、アラニン以外のアミン酸である事を排除するものでもない。
【0025】
本発明で蛍光タンパク質の断片と融合される目標タンパク質は特に限定されるものではない。また、本発明を利用する温度やpH等の環境も、目標タンパク質やその断片が変性しない範囲であれば特に制限されない。上記条件を満たせば、例えば、in vitroでもよいし、in vivoであってもよい。
【0026】
蛍光タンパク質の断片と目標タンパク質の融合方法については、両タンパク質遺伝子のコドンフレームが一致(インフレーム)すれば特に制限はない。公知の組換え技術を用いてこの融合タンパク質遺伝子を作製できる。より具体的には、蛍光タンパク質の断片をコードするDNAと目標タンパク質をコードするDNAを、インフレームで直接もしくはリンカーを介して結合すれば、目標タンパク質と蛍光タンパク質の断片が融合したタンパク質をコードするDNAを得ることができる。
【0027】
同様にして、蛍光タンパク質の他方の断片ともう一方の目標タンパク質とが融合した融合タンパク質遺伝子を作製できる。さらに、この融合タンパク質遺伝子をこの遺伝子を導入する細胞に適したプラスミドに、発現ユニットと共に組み込んで、融合タンパク質遺伝子を該細胞で発現させるための組換えプラスミドを得ることができる。これら融合タンパク質遺伝子およびこれを組み込んだ組み換えプラスミドの作製は公知の方法、例えば、Maniates et al.,のMolecular cloningに記載の方法で作製できる。
【0028】
これらの組換えプラスミドを有する微生物、動物若しくは植物の細胞に導入・発現することで、蛍光タンパク質のいずれかの断片がそれぞれ融合された2つの目標タンパク質/蛍光タンパク質断片融合タンパク質を産生させることができる。また、組換えプラスミドを細胞に導入して、目的の目標タンパク質/蛍光タンパク質断片融合タンパク質を産生させることは、公知の方法を利用することができる。
【0029】
なお、プラスミドに組み込む蛍光タンパク質の断片をコードするDNAの少なくとも一方は、当該蛍光タンパク質断片中のβ−シート上のアミノ酸を置換した蛍光タンパク質断片をコードするDNAである。本発明は、蛍光タンパク質断片のアミノ酸を他のアミノ酸に置換することで、蛍光タンパク質の断片同士の会合のアフィニティを調節するからである。
【実施例】
【0030】
以下に本発明のタンパク質分子対について実施例を参考し、より詳細に説明する。実施例の概略は以下の通りである。
(1)目標タンパク質と蛍光タンパク質の断片が融合した融合タンパク質分子対をコードする遺伝子を含む組換えプラスミドを作製した。
(2)これらの組換えプラスミドで大腸菌形質転換し、形質転換した大腸菌を培養し組換えプラスミドDNAを抽出し精製した。
(3)精製したプラスミドDNA2種類を同時にマウス細胞に導入した。
(4)マウス細胞を固定した後、顕微鏡観察し、蛍光の有無を確認した。
【0031】
目標タンパク質同士のアフィニティと蛍光タンパク質同士のアフィニティの程度を知るために、本実施例では予め結合することが知られているタンパク質同士とそれらの一方の形を変えたミュータントを用意した。一方がミュータントである組では目標タンパク質同士は決して相互作用を行わない。また、蛍光タンパク質の断片の1つのアミノ酸も複数種類のアミノ酸に置換したものを用意した。アフィニティの程度を変えるためである。
【0032】
目標タンパク質としては、bFosとbJunを使用した。これらの2つのタンパク質は、ロイシンジッパーを構成することで相互作用を行うことが知られている。
【0033】
蛍光タンパク質は配列番号1で示すアミノ酸配列からなる蛍光タンパク質を用いた。これは、Venusと呼ばれる蛍光タンパク質であり、黄色に発光する。このN末端断片をbJunに融合させ、アミノ酸を置換したC末端断片をbFosに融合させた。アミノ酸の置き換えは、C末端側断片で行い、N末端側断片にはアミノ酸の置き換えを行わなかった。
【0034】
図3には、蛍光タンパク質の構造とアミノ酸配列の概念図を示す。蛍光タンパク質の構造は図2(a)と同じものである。図3(a)には、蛍光タンパク質の構造を、図3(b)にはN末端から番号をつけたアミノ酸配列を、そして図3(c)にはアミノ酸配列の切断を概念的に示す模式図を示した。図3(a)で示したβ−シート構造の番号と図3(b)で示したアミノ酸配列中の番号はそれぞれ対応する。
【0035】
この蛍光タンパク質の全長は238個のアミノ酸からなる。切断箇所は7番目のバレル構造の終端となる154番目のアミノ酸(アラニン)と155番目のアミノ酸(アスパラギン酸)の間とした。また、N末端側断片には155番目から173番目までの重複部分を有している。ここで、N末端側断片をVN173と呼び、C末端側断面をVC155と呼ぶ。
【0036】
蛍光タンパク質の断片同士のアフィニティの調節は配列のアミノ酸の置き換えで行う。置き換えを行うアミノ酸は以下のようにして決定した。図4には、蛍光タンパク質の構造中のアミノ酸同士の存在箇所と距離を計算で求めた表を示す。例えば、C末端側断片にある182番目のチロシン(Tyr)は9番目のβ−シート上にあり、V末端側断片の4番目のβ−シート上にある99番目のフェニルアラニン(Phe)と3.843オングストロームの距離関係にある。
【0037】
この結果からC末端側断片に存在する(N末端側から)201番目のロイシン(Leu)(符号30)と207番目のロイシン(Leu)(符号31)は、疎水性相互作用の相手になると考えられるN末端側断片のアミノ酸がそれぞれ4つ、3つと多い。そこで、201番目と207番目のロイシンを置き換えるアミノ酸とした。
【0038】
置き換えたアミノ酸は、イソロイシン、バリン、アラニンとした。これらは、それぞれ直鎖の炭素数が3、2、1と少なくなる。すなわち、疎水性相互作用は炭素数の減少に従って小さくなると考えた。疎水性相互作用の相手との距離が大きくなるからである。また、ロイシンはその直鎖の末端にメチル基が2つあるのに対して、イソロイシンの直鎖の末端にはメチル基は1つである。従って、イソロイシンはロイシン同様その直鎖の炭素数が3であるが、相互作用は若干小さくなると考えた。
【0039】
図5には作製したベクターの構造の概念図を示す。図5(a)を参照して、pHA−bFos−VC155(L201I)は、bFosと201番目のロイシンをイソロイシンに置換したVC155をコードする塩基配列が組み込まれたベクターである。なお、元々のpHA−bFos−VC155はパリュー大学由来である。サイトメガロウイルス由来のプロモータであるPCMV−IEと検出用のタンパク質であるHA−tagをコードする塩基配列の後、bFosと201番目のアミノ酸が置換されたVC155をコードする塩基配列が続く構造である。
【0040】
同様に、201番目のロイシンをバリン、アラニンに置換したものと、207番目のロイシンをイソロイシン、バリン、アラニンに置換したものを用意した。それぞれ、pHA−bFos−VC155(L201V)、pHA−bFos−VC155(L201A)、pHA−bFos−VC155(L207I)pHA−bFos−VC155(L207V)、pHA−bFos−VC155(L207A)のように表す。
【0041】
これらの相手方となる融合タンパク質分子は、bJunと蛍光タンパク質のN末端側断片である(図5(b)参照)。ベクターは、pFLAG−bJun−VN173であり、bJunと蛍光タンパク質のN末端断片をコードする塩基配列が組み込まれたベクターである。pHA−bFos−VC155(L201I)などと同様に、サイトメガロウイルス由来のプロモータであるPCMV−IEと検出用のタンパク質であるFLAG−tagをコードする塩基配列の後、bJunとVN173をコードする塩基配列が続く構造である。
【0042】
また、ネガティブコントロールとして、相互作用を起こさない目標タンパク質を用意した。これはbFosをミュータント化してbJunと相互作用を起こさないようにしたタンパク質である。これをbFos(m)とした。
【0043】
また、上記の組換えプラスミドDNAが導入されたことを確認するために、図2(b)で示した赤色の蛍光タンパク質をコードするプラスミドDNAも用意した。この蛍光タンパク質はmCherryと呼ばれる蛍光タンパク質で配列番号2番で示すアミノ酸配列を有する。
【0044】
<組換えプラスミドDNAの作製>
図6と図7を用いて、組換えプラスミドDNAの作製の工程を説明する。パリュー大学由来のpHA−bFos−VC155をAsp718とNotIで制限酵素処理し、VC155部分10を切り出し、塩基配列13を得た(図7(a、c))。一方、pHA−bFos−VC155由来のプラスミドのVC155部分の所定アミノ酸を図6で示したメガ・プライマー法で置き換えたプラスミド17を作製した(図7(b))。メガ・プライマー法で用いたプライマーを表1に示す。
【0045】
【表1】
【0046】
なお、上記のプライマー1は配列番号3、プイライマー2(L209I)は配列番号4、プイライマー2(L201V)は配列番号5、プイライマー2(L201A)は配列番号6、プイライマー2(L207I)は配列番号7、プイライマー2(L207VI)は配列番号8、プイライマー2(L207A)は配列番号9、プライマー3は配列番号10である。
【0047】
図6を参照して、メガ・プライマー法について概説しておく。図6(a)は、図7(b)で示すVC155部分のDNAを示す。プライマーP1は、この2本鎖DNAのうち、片方のDNA鎖の3´末端部分に対応するプライマーであり、プライマーP2は、もう片方のDNA鎖に対応し、置き換えたい塩基40を含み、その前後の塩基で構成されるプライマーである。
【0048】
VC155部分のDNAを鋳型とし、プライマーP1およびプライマーP2を用いてPCRを行うと、図6(b)で示すように、置換したい部分の塩基が、塩基40とそれに対応する塩基に置き換えられた2本鎖DNAが大量に得られる。これがメガ・プライマーMPである。
【0049】
そして、図6(c)で示すように、VC155部分のDNAを鋳型とし、このメガ・プライマーMPとプライマーP3を用いて再度PCRを行うと、図6(d)で示すような、VC155部分のDNAであって、所定の場所の塩基が、塩基40とそれに対応する塩基に置換された2本鎖DNAを得ることができる。従って、所定箇所のアミノ酸を置き換えるのは、そのアミノ酸に対応する箇所の塩基配列を変更したプライマーP2を用いる。以上の工程で、所定の箇所をイソロイシン(I)、バリン(V)やアラニン(A)に置き換えた。
【0050】
VC155の所定のアミノ酸を置き換えたプラスミドから置き換えられたVC155部分だけをAsp718とNotIで制限酵素処理し切り出し塩基配列17を得た(図7(b))。なお、図7(b)(d)でバツ印の部分は、アミノ酸が置き換えられた部位を示す。そして、塩基配列13と塩基配列17をライゲーションして、所定のプラスミド19を得た。
【0051】
なお、塩基配列13と17は読み枠が合うようにMCS(マルチクローニングサイト)内にある2種類の制限酵素で処理し、フェノール/クロロホルム/イソアミルアルコール溶液処理後、エタノール沈殿により回収した。さらに電気泳動を行った後、ゲルから切り出し、Mag ExtractorTM (TOYOBO社製)を用いて抽出し、以下のように反応液10μLを作製し、16℃で16時間ライゲーションを行った。
【0052】
< 反応液の組成 >
ベクター x μL
インサート y μL
T4 DNA ligase 0.4 μL
10×Ligation buffer 1 μL
ATP (5.0 mM) 1 μL
ベクターとインサートのモル比が1:3となるように加え、滅菌水で10μLにfill upした。
【0053】
<塩基配列の確認>
作製したベクターの塩基配列は、以下の手順によりシークエンシング反応によるキャピラリー電気泳動によって確認した。
【0054】
0.2mL PCRチューブに鋳型DNA(50−100pmoL/μLに調製)を4μL移し、サーマルサイクラー(Gene Amp PCR System 9700、 Applied Biosystems社製)を用いて96℃で1分間加熱後、直ちに氷上に置いた。これにCEQ DTCS Quick Start Master Mix (BECKMAN COULTER社製)を4μL、プライマーを1μMとなるように加え、反応液量を10μLとした。この反応液をサーマルサイクラーで以下の条件で反応を30サイクル行った。
【0055】
< 反応条件 >
Denaturation 96 ℃ 20sec
Annealing 50 ℃ 20sec ×30 cycle
Extension 60 ℃ 4 min
4 ℃ ∞
PCRで増幅させたベクターを次の手順でキャピラリー電気泳動を行った。1.5mLマイクロチューブにStop Solution(3M酢酸ナトリウム 1μL、20mg/mL グリコーゲン溶液0.5μLを入れておき、反応終了後のシークエンシング反応液を10μL加え、よく攪拌した。冷100%エタノールを30μLを加えて攪拌した後、直ちに4℃、14000rpmで15分間遠心分離した。上清を除き、冷70%エタノールを200μLを加えて、ペレットを洗浄した後、4℃、14000rpmで2分間遠心分離した。この洗浄操作をもう一度繰り返し、上清を除去した後、ペレットを真空乾燥し、30μLのSample Loading Solution に溶解した。
【0056】
サンプルを96−wellサンプルプレートに移し、Mineral Oil (BECKMAN COULTER社製)を1滴加えた。96−well バッファープレートに、CEQ Separation buffer (BECKMAN COULTER社製)をwellの8割程度加えた。これらのサンプルプレートとバッファープレート、そして、Separation Gel−LPA Iを、CEQTM2000XL DNA Analysis System (BECKMAN COULTER社製)にセットして、キャピラリー電気泳動を行った。塩基配列の結果はこのシステムの出力としてテキストデータで得ることができる。
【0057】
次に作製した組換えプラスミドを大腸菌を用いて増幅させた。これは、以下の手順によった。
(1)コンピテントセルを作製した。
(2)大腸菌(コンピテントセル)に組換えプラスミドを導入し培養させた。
以下にその手順を詳細に説明する。
【0058】
<コンピテントセルの作製>
大腸菌(DH10B)を、LB寒天培地に塗布し、37℃で一晩培養した。この培地から単一コロニーを選び、L字型試験管に入れたLB液体培地20mLに植菌して、37℃で一晩振盪培養した。一晩振盪培養したLB液体培地2mLを、坂口フラスコに入れたLB液体培地200mLに加えた。坂口フラスコ内のLB液体培地を37℃に保ち、波長660nmで測定した濁度(O.D.660)が0.6になるまで振盪培養した。その後坂口フラスコを氷上に20分間静置した。
【0059】
この振盪培養した大腸菌を洗浄した。具体的には、以下の手順を行った。フラスコ内のLB液体培地を遠心チューブに移し、0℃、5000rpmの条件で10分間遠心分離後、上清を取り除いた。上清を取り除いた遠心チューブに、オートクレーブ滅菌した氷冷超純水150mLを加えて懸濁した。懸濁した後、遠心チューブを、0℃、5000rpmの条件で10分間遠心分離後、上清を取り除いた。そして、再度遠心チューブに、オートクレーブ滅菌した氷冷超純水75mLを加えて懸濁した。懸濁した後、遠心チューブを、0℃、5000rpmでの条件で10分間遠心分離後、上清を取り除いた。以上のように、大腸菌を氷冷超純水で2回洗浄した。
【0060】
次に、遠心チューブにオートクレーブ滅菌した10%グリセロール12mLを加えて、懸濁した。グリセロールで懸濁した後、0℃、6000rpmの条件で10分間遠心分離後、10%グリセロールを取り除いた。そして再び、遠心チューブにオートクレーブ滅菌した10%グリセロール1.6mLを加え、懸濁した。
【0061】
以上の手順で、振盪培養された大腸菌を、DNAに対する膜透過性が増大した大腸菌であるコンピテントセルとした。保存するコンピテントセルは、遠心チューブ中の懸濁液を1.5mLマイクロチューブに40μLずつ分注し、液体窒素に入れて凍結させた。この保存するコンピテントセルは、−80℃で保存した。
【0062】
<エレクトロポレーション法による大腸菌の形質転換>
氷上で融解させたコンピテントセル40μLに、プラスミドDNA溶液1μLを加えて、混合し混合溶液とした。この混合溶液を氷上で冷やしておいたキュベット(電極間隔2.0mm)に移し、E.coli Pulser (BIO−RAD社製)を用いて、パルス(電圧2.1kV)をかけた。
【0063】
この混合溶液にSOC培地1mLを加えて懸濁し、1.5mLマイクロチューブに移した。これを適当な抗生物質を添加したLB寒天培地に塗布し、37℃で一晩培養した。
【0064】
<プラスミドDNAの抽出と精製>
次に増殖させた大腸菌からプラスミドDNAを抽出し精製した。プラスミドDNAを導入した大腸菌を培養したLB寒天培地から単一コロニーを選び、適当な抗生物質を添加したLB液体培地3mLに植菌して、37℃で一晩振盪培養した。
【0065】
培養液を1.5mLマイクロチューブに移し、4℃、10000rpmの条件で1分間遠心分離して、上清を取り除き集菌した。これにSolutionIを100μL加えて懸濁後、SolutionIIを200μL加えて穏やかに混合し、氷上に5分間置いた。SolutionIIを入れることで大腸菌は溶菌し、細胞内のプラスミドDNAが取り出せる。マイクロチューブ内の溶液を中和させるために、SolutionIIIを150μL加えて穏やかに混合し、氷上に5分間置いた。
【0066】
なお、SolutionIは、Tris緩衝液(25mM、pH8)にグルコースを50mMになるように加えた溶液である。また、SolutionIIは、0.2Nの水酸化ナトリウムに1%のSDS(ラウリル酸ナトリウム)を加えた溶液である。また、SolutionIIIは、3Mの酢酸カリウムと11.5%の酢酸を加えた溶液である。
【0067】
次に1.5mLマイクロチューブごと4℃、15000rpmの条件で10分間遠心分離後、ペレットを取り除いた。ペレットを取り除いた溶液に、フェノール/クロロホルム/イソアミルアルコール溶液を500μL加えて穏やかに混合し、プラスミドDNAを抽出した。
【0068】
20℃、15000rpmの条件で10分間遠心分離後、上清を別のマイクロチューブに移した。2/3倍量のイソプロパノールを加えて穏やかに混合し、室温で10分間置いた。
【0069】
さらに、20℃、15000rpmの条件で10分間遠心分離後、上清を取り除いた。上清を取り除いたペレットは、エタノール沈殿によって得たプラスミドDNAである。ペレットを冷70%エタノール400μLでリンスし、4℃、15000rpmの条件で3分間遠心分離後、上清を取り除き、15分間真空乾燥した。
【0070】
抽出したプラスミドDNA中のRNAを除去するために次のプロセスを行った。ペレットをTE buffer (pH8.0)50μLに溶解し、RNaseA溶液(0.5μg/mL)を1μL加えて、37℃で30分間インキュベートした。さらに、20%PEG6000/2.5M NaClを30μL加えて、よく混合し、氷上に1時間置いた。
【0071】
そして、4℃、15000rpmの条件で10分間遠心分離後、上清を取り除いた。ペレットを冷70%エタノール400μLでリンスし、4℃、15000rpmで3分間遠心分離した後、上清を取り除き、15分間真空乾燥した。ペレットをTE buffer (pH8.0)50μLに溶解した。以上の手順で精製したプラスミドDNAを得た。
【0072】
次に精製したプラスミドDNAをマウス細胞に導入した。具体的な手順は以下による。
(1)マウス細胞を培養した。
(2)リポフェクション法を用いてマウス細胞に遺伝子を導入した。
以下に詳細を説明する。
【0073】
まず、マウスの細胞は以下のようにして培養した。マウスC3H 10T1/2細胞を、D−MEM培地を8mL入れた100mmシャーレの中で、37℃、5%CO2存在下のCO2インキュベーターを用いて培養した。継代は、対数増殖期の細胞が、70−90%コンフルエントした時に行った。シャーレ内のD−MEM培地を取り除き、1×PBS(−)を4mL入れて洗浄し、これを取り除いた。トリプシン/EDTA溶液を1mL入れて、細胞をシャーレからはがし、球形になったことを確認してから、D−MEM培地を4mL入れて懸濁した。
【0074】
この単一細胞浮遊液0.5mLを、あらかじめD−MEM培地を8mL入れた別のシャーレに播種し、細胞をシャーレ全体に均一になるように分散させて、37℃、5% CO2存在下のCO2インキュベーターを用いて培養した。
【0075】
凍結ストック細胞の作製は、対数増殖期の細胞が、70−90%コンフルエントした時に、継代と同様にして細胞を100mmシャーレからはがした。細胞が球形になったことを確認してから、D−MEM培地を4mL入れて懸濁した。この単一細胞浮遊液を遠心チューブに移し、1000rpmで10分間遠心分離後、上清を取り除いた。無血清タイプ細胞凍結保存液バンバンカーTM(リンフォテック社製)を細胞凍結保存用チューブ(クライオチューブ)1本につき0.5mLとなるように加えて、懸濁した。この細胞懸濁液をクライオチューブに移し、−80℃で保存した。
【0076】
−80℃で凍結保存された細胞を培養する時は、まず、クライオチューブを37℃の恒温槽内で軽く揺すりながら細胞懸濁液を半解凍し、その後、室温で解凍を行った。この細胞懸濁液0.5mLを、あらかじめD−MEM培地を8mL入れた100mmシャーレに播種し、細胞をシャーレ全体に均一になるように分散させて、37℃、5%CO2存在下のCO2インキュベーターを用いて培養した。
【0077】
培養した細胞にプラスミドDNAをリポフェクション法で導入した。継代と同様にして細胞を100mmシャーレからはがし、細胞が球形になったことを確認してから、D−MEM培地を4mL入れて懸濁した。この単一細胞浮遊液をトランスフェクション時に50−80%コンフルエントになるように、12−wellプレートに1サンプルにつき(以下同様)、1μL播いた。Opti−MEMRIReduced Serum Medium(Invitrogen社製)に導入したいDNA1μgを加え、合計が200μLとなるようにOpti−MEMRIReduced Serum Mediumを加えて、穏やかに混合した。
【0078】
ここで導入したいDNAとは、先に作製したベクターである。より具体的には、BiFCを行わせる目標タンパク質と蛍光タンパク質の断片がコードされたプラスミドDNAと、赤色の蛍光タンパク質をコードするプラスミドDNAである。BiFCを行わせるプラスミドDNAは、赤色の蛍光タンパク質の3倍量となるように配合した。また、混合した溶液に3μLのLipofectamineTM LTX(Invitrogen社製)を加えて、穏やかに混合後、30分間室温で静置した。このDNA−LipofectamineTM LTX複合体200μLをwellに加えて、穏やかにプレートを振動させて混合した。
【0079】
次にマウス細胞を観察し、蛍光発光の有無を確認した。これは以下の手順によった。
(1)細胞の固定およびDAPIによる核の染色を行った。
(2)標本を作製した。
(3)顕微鏡による細胞観察を行った。
以下に具体的な手順を説明する。
【0080】
<細胞の固定およびDAPIによる核の染色>
培養細胞への遺伝子導入から24時間後に、固定と染色を行った。35mmシャーレ内のD−MEM培地を取り除き、4%パラホルムアルデヒドをカバーガラス全体に広がるように加えた。室温で15分間静置して固定後、4%パラホルムアルデヒドを取り除き、1×PBS(−)をカバーガラスがひたる程度に加えて、10分間振盪し洗浄した。1×PBS(−)を取り除き、0.1% Triton X−100を1mL加えて、室温で5分間静置した。
【0081】
0.1% Triton X−100を取り除き、1×PBS(−)をカバーガラスがひたる程度に加えて、10分間振盪して洗浄した。1×PBS(−)を取り除き、DAPI染色液をカバーガラス全体に広がるように加え、室温で5分間静置して染色した。DAPI染色液を取り除き、1×PBS(−)をカバーガラスがひたる程度に加えて、10分間振盪し洗浄した。
【0082】
<標本の作製>
マウント用グリセロールを10μLのせたスライドガラスの上に、細胞を固定・染色したカバーガラスを、気泡が入らないようにしてのせた。カバーガラスの上に濾紙を置き、垂直に押して余分な水分を取り除いた後、カバーガラスの周囲に黒いマニキュアを塗布し、遮光して乾燥させた。
【0083】
<蛍光顕微鏡による細胞の観察>
標本を、冷却型CCDカメラORCA−ER(浜松ホトニクス社製)を装備した蛍光顕微鏡Eclipse E600 (Nikon社製)を用いて観察した。フィルターブロックは、DAPI染色の観察用にUV−1Aフィルター(EX:360−370nm、DM:400nm、BM:400nm、Nicon社製)、蛍光タンパク質(Venus)の観察用にFITCフィルター(EX:465−495nm、DM:505nm、BM:515−555nm、Nicon社製)、蛍光タンパク質(mCherry)の観察用にTxRedフィルター(EX:540−580nm、DM:595nm、BM:600−660nm、Nicon社製)を用いた。画像の取り込みと解析は、画像解析ソフトウェア Lumina Vision for Mac(三谷商事社製)を用いて行った。
【0084】
次に図8を参照して、観察結果を説明する。図8(a)は、横軸にサンプルの種類を示し、縦軸は蛍光回復の割合(%)を示す。蛍光回復率は、mCherry(赤色)の発光をする細胞の数に対して、それらの細胞の中で緑黄色の蛍光発光をしている細胞の数の割合で評価した。BiFCを行わせるための緑黄色の蛍光タンパク質(Venus)は、赤色の蛍光タンパク質(mCherry)の3倍量が導入されている。したがって、赤色の蛍光発光をする細胞の全てにBiFCを行わせるためのプラスミドDNAが導入されていると推定できる。言い換えると、赤色を発光していて緑黄色を発光していない細胞は、BiCFを行わせるためのプラスミドDNAは導入されているにもかかわらず、蛍光回復しなかったケースと判断できる。
【0085】
横軸のサンプルは、図8(a)、(b)ともに同じ順で表示されている。サンプルは横軸左から、bFosの野生種(bFosVC(wt))、201番目のアミノ酸を変更したもの(bFosVC(L201I)、bFosVC(L201V)、bFosVC(L201A))と207番目のアミノ酸を変更したもの(bFosVC(L207I)、bFosVC(L207V)、bFosVC(L207A))の7種類である。bFosの野生種は201番目および207番目のアミノ酸がロイシンである。
【0086】
図8(b)は、bFosをミュータントにした場合である。サンプルはbFosがミュータントになっている以外は、図7(a)と同じ蛍光タンパク質のC末端側断片が融合されたサンプルである。
【0087】
図8(a)を参照して、bFosVCの野生種では、目標タンパク質の相互作用によってほとんどの細胞内で蛍光発光が回復していることが観察された。一方、図8(b)を参照すると、目標タンパク質が相互作用を行わない場合であるにもかかわらず、およそ半分の細胞で蛍光回復が観察された。すなわち、目標タンパク質のアフィニティより蛍光タンパク質の断片同士のアフィニティが強いために、目標タンパク質の相互作用の有無に係らず、蛍光回復が行われた。
【0088】
再び図8(a)を参照して、本発明のタンパク質結合体である野生種以外のサンプルでは、置換したアミノ酸がイソロイシン、バリン、アラニンとアミノ酸の直鎖の炭素数が3、2、1と減るにしたがって、蛍光回復の割合が減少した。アラニンでは、全く蛍光が回復しなかった。これは、炭素数が減少するに従って、疎水性相互作用をする相手との距離が長くなるために、蛍光タンパク質の断片同士が再会合しにくくなるためと考えられた。すなわち、イソロイシン、バリン、アラニンへのアミノ酸置換によって蛍光タンパク質の断片同士のアフィニティが調節できた。
【0089】
再度図8(b)を参照すると、本発明のサンプルは目標タンパク質が相互作用を行わない時は、全く発光回復が認められなかった。従って、本発明のサンプルは、目標タンパク質の相互作用が行われた場合は蛍光発光を回復し、目標タンパク質が相互作用を行わない場合は蛍光発光を回復しなかった。これはBiFC法を用いてタンパク質同士の相互作用をアッセイするために必要不可欠な特性である。言い換えると、bFosとbJunを目標タンパク質としてBiFC法を用いる場合は、配列番号1の蛍光タンパク質の野生種を用いるのではなく、本発明のサンプルのように蛍光タンパク質の断片同士のアフィニティを調節しなければ、正しいアッセイができないことが示された。
【産業上の利用可能性】
【0090】
本発明は、たんぱく質の相互作用をアッセイするBiFC法に利用することができる。
【符号の説明】
【0091】
10、11 目標タンパク質
12、13 蛍光タンパク質の断片
14 目標タンパク質同士の相互作用
15 蛍光復活した蛍光タンパク質
20 蛍光タンパク質のβ−シート
21 蛍光タンパク質のα−へリックス
22 蛍光タンパク質のアミノ酸のループ部分
30 201番目のロイシン
31 207番目のロイシン
40 アミノ酸の修正点
【特許請求の範囲】
【請求項1】
蛍光タンパク質のN末端側断片に接続された第1のタンパク質分子と、前記蛍光タンパク質のC末端側断片に接続された前記第2のタンパク質分子とからなる融合タンパク質分子対において、
前記N末端側断片と前記C末端側断片の少なくとも一方の断片で、少なくとも1箇所以上のアミノ酸が他のアミノ酸に置換され、
前記N末端側断片とC末端側断片同士のアフィニティは、アミノ酸の置換を受けない場合と比較し、前記アミノ酸の置換によって調節されたことを特徴とする融合タンパク質分子対。
【請求項2】
前記置換されるアミノ酸は、前記蛍光タンパク質のβ−シート上のアミノ酸である請求項1に記載された融合タンパク質分子対。
【請求項3】
前記変更されるアミノ酸は、ロイシンであり、変更されたアミノ酸はイソロイシン、バリン、アラニンである請求項1または2のいずれかの請求項に記載された融合タンパク質分子対。
【請求項4】
前記蛍光タンパク質は配列番号1の蛍光タンパク質である請求項1乃至3のいずれか1の請求項に記載された融合タンパク質分子対。
【請求項5】
前記変更されるアミノ酸は、C末端側断片のアミノ酸であって配列番号1のN末端側から201番目若しくは207番目のロイシンである請求項3に記載された融合タンパク質分子対。
【請求項6】
請求項1ないし請求項6のいずれか1項に記載された融合タンパク質分子対のそれぞれをコードする遺伝子。
【請求項7】
請求項6に記載の融合タンパク質分子対のそれぞれを含む遺伝子導入/発現ベクター。
【請求項8】
請求項7に記載の遺伝子導入/発現ベクターが導入された請求項1ないし請求項5のいずれか1項に記載された融合タンパク質分子対を産生する細胞。
【請求項1】
蛍光タンパク質のN末端側断片に接続された第1のタンパク質分子と、前記蛍光タンパク質のC末端側断片に接続された前記第2のタンパク質分子とからなる融合タンパク質分子対において、
前記N末端側断片と前記C末端側断片の少なくとも一方の断片で、少なくとも1箇所以上のアミノ酸が他のアミノ酸に置換され、
前記N末端側断片とC末端側断片同士のアフィニティは、アミノ酸の置換を受けない場合と比較し、前記アミノ酸の置換によって調節されたことを特徴とする融合タンパク質分子対。
【請求項2】
前記置換されるアミノ酸は、前記蛍光タンパク質のβ−シート上のアミノ酸である請求項1に記載された融合タンパク質分子対。
【請求項3】
前記変更されるアミノ酸は、ロイシンであり、変更されたアミノ酸はイソロイシン、バリン、アラニンである請求項1または2のいずれかの請求項に記載された融合タンパク質分子対。
【請求項4】
前記蛍光タンパク質は配列番号1の蛍光タンパク質である請求項1乃至3のいずれか1の請求項に記載された融合タンパク質分子対。
【請求項5】
前記変更されるアミノ酸は、C末端側断片のアミノ酸であって配列番号1のN末端側から201番目若しくは207番目のロイシンである請求項3に記載された融合タンパク質分子対。
【請求項6】
請求項1ないし請求項6のいずれか1項に記載された融合タンパク質分子対のそれぞれをコードする遺伝子。
【請求項7】
請求項6に記載の融合タンパク質分子対のそれぞれを含む遺伝子導入/発現ベクター。
【請求項8】
請求項7に記載の遺伝子導入/発現ベクターが導入された請求項1ないし請求項5のいずれか1項に記載された融合タンパク質分子対を産生する細胞。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2011−211983(P2011−211983A)
【公開日】平成23年10月27日(2011.10.27)
【国際特許分類】
【出願番号】特願2010−84306(P2010−84306)
【出願日】平成22年3月31日(2010.3.31)
【出願人】(505127721)公立大学法人大阪府立大学 (688)
【Fターム(参考)】
【公開日】平成23年10月27日(2011.10.27)
【国際特許分類】
【出願日】平成22年3月31日(2010.3.31)
【出願人】(505127721)公立大学法人大阪府立大学 (688)
【Fターム(参考)】
[ Back to top ]