
チオアミド化合物、チオアミド化合物の製造方法、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法、及びアトルバスタチンの製造方法
【課題】[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を、効率よく且つ銅触媒などの安価に調製可能な触媒を用いて製造可能なチオアミド化合物、該チオアミド化合物の製造方法、前記チオアミド化合物を用いた[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法などの提供。
【解決手段】下記一般式(1)で表されるチオアミド化合物。

ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
【解決手段】下記一般式(1)で表されるチオアミド化合物。
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アトルバスタチンの合成に有用なチオアミド化合物、該チオアミド化合物の製造方法、前記チオアミド化合物を用いた[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法、及び前記チオアミド化合物を用いたアトルバスタチンの製造方法に関する。
【背景技術】
【0002】
3−ヒドロキシ−3−メチルグルタリル−補酵素A(HMG−CoA)のメバロン酸塩への変換は、コレステロール生合成経路における初期の律速工程である。この工程は、酵素HMG−CoAレダクターゼによって触媒される。スタチンは、HMG−CoAレダクターゼがこの変換を触媒するのを阻害する。そのようなことから、スタチンは、総体的に強力な脂質低減作用物質である。
【0003】
現在、アトルバスタチンカルシウム水和物はLipitor(登録商標)として販売されており、下記式を有する(例えば、特許文献1参照)。
【化1】
【0004】
アトルバスタチンカルシウムは、HMG−CoAの選択的、競合的阻害剤である。そのようなことから、アトルバスタチンは強力な脂質低減作用物質であり、このため脂質低下剤及び/又はコレステロール低下剤として有用である。また、骨粗しょう症、良性の前立腺肥大(BPH)、及びアルツハイマー病の治療にも有用である。
【0005】
アトルバスタチンカルシウムの合成方法としては、図1に記載の合成方法が知られている(例えば、非特許文献1参照)。
しかし、図1に記載の合成方法は、HBr、及びNaCNの使用や、極低温工程を必要とすることから、工業生産には不適であるという問題がある。
【0006】
そこで、アトルバスタチンの新規合成方法の探索を目的として、種々の検討がされている。
例えば、図1に記載のアトルバスタチンの合成方法における合成中間体である、(4R−シス)−1,1−ジメチルエチル 6−シアノエチル−2,2−ジメチル−1,3−ジオキサン−4−アセテートの合成方法が提案されている(例えば、非特許文献2参照)。
しかし、この提案の技術では、ベンゼンスルホニル誘導体を経由して(4R−シス)−1,1−ジメチルエチル 6−シアノエチル−2,2−ジメチル−1,3−ジオキサン−4−アセテートが合成されているが、前記ベンゼンスルホニル誘導体は、次のステップへの反応性が十分ではないという問題がある。
【0007】
また、前記(4R−シス)−1,1−ジメチルエチル 6−シアノエチル−2,2−ジメチル−1,3−ジオキサン−4−アセテートを酵素法により合成する方法が提案されている(例えば、特許文献2参照)。
この提案の技術は、図2に示すような反応経路であるが、ケトレダクターゼ(KRED)、グルコースデヒドロゲナーゼ(GDH)、ハロヒドリンデハロゲナーゼ(HHDH)の3種の酵素を用い、3度に渡る煩雑な酵素反応が必要であるという問題がある。
【0008】
また、他の方法として、図1に記載のアトルバスタチンの合成方法における中間体である、tert−ブチル[(4R,6R)−6−アミノエチル−2,2−ジメチル−1,3−ジオキサン−4−イル]アセテートの合成方法が提案されている(例えば、非特許文献3参照)。
しかし、この提案の技術では、窒素官能基の導入に一級アルコールの酸化、ニトロアルドール反応、及び生じる二級アルコールの脱離反応を必要とし、煩雑な工程を要するという問題がある。
【0009】
したがって、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を、効率よく且つ銅触媒などの安価に調製可能な触媒を用いて製造可能な化合物、該化合物の製造方法、前記化合物を用いた[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法、及び前記化合物を用いたアトルバスタチンの製造方法の提供が求められているのが現状である。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】米国特許第5,273,995号公報
【特許文献2】国際公開第2004/015132号パンフレット
【非特許文献】
【0011】
【非特許文献1】Roth B.D.,et al.,Progress in Medicinal Chemistry,2002,40,1−22
【非特許文献2】Philip L.Brower,et al.,Tetrahedron Letters,Vol.33,No.17,pp.2279−2282,1992
【非特許文献3】Stanislav Radl,SYNTHETIC COMMUNICATIONS,Vol.33,No.13,pp.2275−2283,2003
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明は、従来における前記諸問題を解決し、以下の目的を達成することを課題とする。即ち、本発明は、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を、効率よく且つ銅触媒などの安価に調製可能な触媒を用いて製造可能なチオアミド化合物、該チオアミド化合物の製造方法、前記チオアミド化合物を用いた[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法、及び前記チオアミド化合物を用いたアトルバスタチンの製造方法を提供することを目的とする。
【課題を解決するための手段】
【0013】
前記課題を解決するための手段としては、以下の通りである。即ち、
<1> 下記一般式(1)で表されることを特徴とするチオアミド化合物である。
【化2】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
<2> 前記<1>に記載のチオアミド化合物の製造方法であって、
下記一般式(2)で表される化合物と、下記一般式(3)で表される化合物とを反応させる反応工程を含むことを特徴とするチオアミド化合物の製造方法である。
【化3】
【化4】
ただし、前記一般式(2)中、R1は、−OR11、及び−NR12R13のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、水素原子及びアミノ基の保護基のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。前記一般式(3)中、R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。
<3> 反応工程が、銅錯体を用いて行われる前記<2>に記載のチオアミド化合物の製造方法である。
<4> 銅錯体が、銅−光学活性ホスフィン錯体である前記<3>に記載のチオアミド化合物の製造方法である。
<5> 下記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程を含むことを特徴とする[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法である。
【化5】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
【化6】
ただし、前記一般式(4)中、R4は、カルボキシル基の保護基及び水素原子のいずれかを表す。R5及びR6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
<6> 下記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程を含むことを特徴とするアトルバスタチンの製造方法である。
【化7】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい。)。
【化8】
ただし、前記一般式(4)中、R4は、カルボキシル基の保護基及び水素原子のいずれかを表す。R5及びR6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
【発明の効果】
【0014】
本発明によれば、従来における前記諸問題を解決し、前記目的を達成することができ、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を、効率よく且つ銅触媒などの安価に調製可能な触媒を用いて製造可能なチオアミド化合物、該チオアミド化合物の製造方法、前記チオアミド化合物を用いた[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法、及び前記チオアミド化合物を用いたアトルバスタチンの製造方法を提供することができる。
【図面の簡単な説明】
【0015】
【図1】図1は、従来のアトルバスタチンの合成方法の一例を示す合成スキームである。
【図2】図2は、酵素法による(4R−シス)−1,1−ジメチルエチル 6−シアノエチル−2,2−ジメチル−1,3−ジオキサン−4−アセテートの合成方法を示す合成スキームである。
【発明を実施するための形態】
【0016】
本明細書、及び特許請求の範囲に記載された化学式及び一般式における立体配置は特に言及しない場合には、絶対配置を表す。
【0017】
(チオアミド化合物)
本発明のチオアミド化合物は、下記一般式(1)で表される。
【化9】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
【0018】
前記R11における水酸基の保護基としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、Greenら、Protective Groups in Organic Synthesis, 3rd Edition,1999,John Wiley & Sons, Inc.などの成書を参照することができる。
前記水酸基の保護基としては、例えば、アラルキル基、トリアルキルシリル基、アルコキシアルキル基、アルカノイル基、アリールカルボニル基などが挙げられる。保護基におけるアリール環(ベンゼン環など)が置換基を有する場合には、置換基としてハロゲン原子やアルコキシ基などが挙げられる。
前記アラルキル基としては、例えば、ベンジル基、p−メトキシベンジル基、p−アミノベンジル基などが挙げられる。
前記トリアルキルシリル基としては、例えば、トリメチルシリル基、トリエチルシリル基、tert−ブチルジメチルシリル基などが挙げられる。
前記アルコキシアルキル基としては、例えば、メトキシメチル基、エトキシメチル基などが挙げられる。
前記アルカノイル基としては、例えば、アセチル基、トリフルオロアセチル基などが挙げられる。
前記アリールカルボニル基としては、例えば、ベンゾイル基、置換フェニルカルボニル基などが挙げられる。
これらの中でも、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を合成する際の脱保護の容易性の点から、アラルキル基が好ましく、ベンジル基がより好ましい。
【0019】
前記R12及び前記R13におけるアミノ基の保護基としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、メトキシカルボニル基、t−ブトキシカルボニル基、ベンジルオキシカルボニル基、アリルオキシカルボニル基、ホルミル基、アセチル基、ベンゾイル基、メチル基、エチル基、アリル基、ベンゼンスルホニル基などが挙げられる。また、前記R12及び前記R13が、一緒になって環構造の保護基を形成している場合は、例えば、フタロイル基(Phth基)が挙げられる。これらの中でも、温和な反応条件で脱保護が可能な点で、フタロイル基が好ましい。
【0020】
前記R2及び前記R3におけるアミド基の保護基としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、トシル基、メトキシメチル基、ベンジルオキシメチル基、アリル基、トリイソプロピルシリル基、ベンジル基、メトキシカルボニル基などが挙げられる。
これらの中でも、アルドール反応における反応成績に優れる点からアリル基が好ましい。
【0021】
本発明のチオアミド化合物は、立体異性体の混合物として得られる場合もあるが、この場合も本発明の範囲に包含される。
【0022】
前記一般式(1)で表されるチオアミド化合物の製造方法としては、特に制限はなく、目的に応じて適宜選択することができるが、下記本発明のチオアミド化合物の製造方法が好ましい。
【0023】
本発明のチオアミド化合物は、アトルバスタチン合成の有用な中間体である、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を合成するための物質、更にはアトルバスタチンを合成するための物質として有用である。
【0024】
(チオアミド化合物の製造方法)
本発明のチオアミド化合物の製造方法は、本発明の前記一般式(1)で表されるチオアミド化合物の製造方法であって、下記一般式(2)で表される化合物と、下記一般式(3)で表される化合物とを反応させる反応工程を含み、更に必要に応じて、その他の工程を含む。
【化10】
【化11】
ただし、前記一般式(2)中、R1は、−OR11、及び−NR12R13のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、水素原子及びアミノ基の保護基のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。前記一般式(3)中、R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。
前記一般式(2)中のR1は、前記一般式(1)中のR1と同じである。
前記一般式(3)中のR2及びR3は、前記一般式(1)中のR2及びR3とそれぞれ同じである。
【0025】
<反応工程>
前記反応工程としては、前記一般式(2)で表される化合物と、前記一般式(3)で表される化合物とを反応させる工程であれば、特に制限はなく、目的に応じて適宜選択することができるが、銅錯体を用いることが好ましい。前記反応工程において、前記銅錯体を触媒として用いることにより、安価な銅を触媒源に用いて前記一般式(1)で表されるチオアミド化合物を製造することができる。
【0026】
前記銅錯体としては、特に制限はなく、目的に応じて適宜選択することができるが、銅−光学活性ホスフィン錯体であることが、エナンチオ選択性が優れる点で好ましい。
前記銅−光学活性ホスフィン錯体は、銅と光学活性ホスフィン配位子の錯体である。前記光学活性ホスフィン配位子としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、シクロヘキシルアニシルメチルホスフィン(CAMP)、1,2−ビス(アニシルフェニルホスフィノ)エタン(DIPAMP)、1,2−ビス(アルキルメチルホスフィノ)エタン(BisP*)、2,3−ビス(ジフェニルホスフィノ)ブタン(CHIRAPHOS)、1,2−ビス(ジフェニルホスフィノ)プロパン(PROPHOS)、2,3−ビス(ジフェニルホスフィノ)−5−ノルボルネン(NORPHOS)、2,3−O−イソプロピリデン−2,3−ジヒドロキシ−1,4−ビス(ジフェニルホスフィノ)ブタン(DIOP)、1−シクロヘキシル−1,2−ビス(ジフェニルホスフィノ)エタン(CYCPHOS)、1−置換−3,4−ビス(ジフェニルホスフィノ)ピロリジン(DEGPHOS)、2,4−ビス−(ジフェニルホスフィノ)ペンタン(SKEWPHOS)、1,2−ビス(置換ホスホラノ)ベンゼン(DuPHOS)、1,2−ビス(置換ホスホラノ)エタン(BPE)、1−((置換ホスホラノ)−2−(ジフェニルホスフィノ)ベンゼン(UCAP−Ph)、1−(ビス(3,5−ジメチルフェニル)ホスフィノ)−2−(置換ホスホラノ)ベンゼン(UCAP−DM)、1−((置換ホスホラノ)−2−(ビス(3,5−ジ(t−ブチル)−4−メトキシフェニル)ホスフィノ)ベンゼン(UCAP−DTBM)、1−((置換ホスホラノ)−2−(ジ−ナフタレン−1−イル−ホスフィノ)ベンゼン(UCAP−(1−Nap))、1−[1’,2−ビス(ジフェニルホスフィノ)フェロセニル]エチルアミン(BPPFA)、1−[1’,2−ビス(ジフェニルホスフィノ)フェロセニル]エチルアルコール(BPPFOH)、2,2’−ビス(ジフェニルホスフィノ)−1,1’−ジシクロペンタン(BICP)、2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル(BINAP)、2,2’−ビス(ジフェニルホスフィノ)−1,1’−(5,5’,6,6’,7,7’,8,8’,−オクタヒドロビナフチル)(H8−BINAP)、2,2’−ビス(ジ−p−トリルホスフィノ)−1,1’−ビナフチル(TOL−BINAP)、2,2’−ビス(ジ(3,5−ジメチルフェニル)ホスフィノ)−1,1’−ビナフチル(DM−BINAP)、2,2’−ビス(ジフェニルホスフィノ)−6,6’−ジメチル−1,1’−ビフェニル(BICHEP)、((5,6),(5’,6’)−ビス(メチレンジオキシ)ビフェニル−2,2’−ジイル)(ビスジフェニルホスフィン)(SEGPHOS)、((5,6),(5’,6’)−ビス(メチレンジオキシ)ビフェニル−2,2’−ジイル)(ビス(3,5−ジメチルフェニル)ホスフィン)(DM−SEGPHOS)、((5,6),(5’,6’)−ビス(メチレンジオキシ)ビフェニル−2,2’−ジイル)(ビス(3,5−ジ(tert−ブチル)−4−メトキシフェニル)ホスフィン)(DTBM−SEGPHOS)などが挙げられる。
これらの中でも、エナンチオ選択性がより優れる点から、(S,S)−2,5置換−BPEが好ましく、(S,S)−Ph−BPEがより好ましい。
なお、(S,S)−Ph−BPEは、以下の構造式で表される化合物である。
【化12】
ただし、「Ph」はフェニル基を表す。
【0027】
なお、チオアミドとアルデヒドとのアルドール反応においては、アルデヒドのα位に酸性の水素原子がある場合には、通常の触媒においては、量論的にチオアミドを活性化するため、多量の触媒が必要である。
しかし、前記銅−光学活性ホスフィン錯体を用いることにより、チオアミド(前記一般式(3)で表される化合物)を用いるアルドール反応において、アルデヒドのα位に酸性の水素原子がある場合にも、触媒的にチオアミドを活性化し(言い換えれば、量論的にチオアミドを活性化せず)、前記一般式(1)で表されるチオアミド化合物を製造することができる。
そのため、前記銅−光学活性ホスフィン錯体は、触媒量の使用でよい。前記触媒量としては、前記一般式(2)で表される化合物に対して、1mol%〜9mol%が好ましい。
【0028】
前記反応工程における前記一般式(2)で表される化合物と前記一般式(3)で表される化合物との割合としては、特に制限はなく、目的に応じて適宜選択することができるが、前記一般式(2)で表される化合物に対して、前記一般式(3)で表される化合物が1.0当量〜1.5当量であることが好ましい。
【0029】
前記反応工程において使用される溶媒としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、THF(テトラヒドロフラン)、DMF(N,N−ジメチルホルムアミド)などが挙げられる。
【0030】
前記反応工程における反応温度としては、特に制限はなく、目的に応じて適宜選択することができるが、−60℃〜−40℃が好ましい。
前記反応工程における反応時間としては、特に制限はなく、目的に応じて適宜選択することができるが、5時間〜36時間が好ましい。
【0031】
本発明のチオアミド化合物の製造方法により得られるチオアミド化合物は、立体異性体の混合物として得られる場合もあるが、この場合も本発明の範囲に包含される。
【0032】
本発明の製造方法により得られる前記一般式(1)で表されるチオアミド化合物は、アトルバスタチン合成の有用な中間体である、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を合成するための物質、更にはアトルバスタチンを合成するための物質として有用である。
【0033】
([(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法)
本発明の[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法は、前記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程を含み、更に必要に応じて、その他の工程を含む。
【0034】
<転化する工程>
前記転化する工程は、前記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができる。
【化13】
ただし、前記一般式(4)中、R4は、カルボキシル基の保護基及び水素原子のいずれかを表す。R5及びR6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
【0035】
前記R4におけるカルボキシル基の保護基としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、Greenら、Protective Groups in Organic Synthesis, 3rd Edition,1999,John Wiley & Sons, Inc.などの成書を参照することができる。
前記カルボキシル基の保護基としては、例えば、アルキル基、トリアルキルシリル基などを挙げることができる。
前記アルキル基としては、メチル基、エチル基、tert−ブチル基などが挙げられる。
前記トリアルキルシリル基としては、例えば、トリメチルシリル基、トリエチルシリル基などが挙げられる。
これらの中でも、酸性条件でも脱保護が可能である点からtert−ブチル基が好ましい。
【0036】
前記R5及び前記R6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
炭素数1〜6の炭化水素基としては、例えば、メチル基、エチル基、n−プロピル基、n−ブチル基などが挙げられる。
前記R5及び前記R6が一緒になって形成される環構造としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、脂肪族炭化水素環が挙げられる。前記脂肪族炭化水素環としては、例えば、シクロペンタン環、シクロヘキサン環、シクロヘプタン環などが挙げられる。
【0037】
前記転化する工程について、前記一般式(1)におけるR1が−OR11の場合と、−NR12R13の場合に分けて、その一例を説明する。
【0038】
−転化する工程A(前記一般式(1)におけるR1が−OR11の場合)−
前記転化する工程Aとしては、例えば、下記一般式(1−1)で表されるチオアミド化合物を下記一般式(5)で表される化合物へ転化する工程(工程(A−I))、下記一般式(5)で表される化合物を下記一般式(6)で表される化合物へ転化する工程(工程(A−II))、下記一般式(6)で表される化合物を下記一般式(9)で表される化合物へ転化する工程(工程(A−III))、下記一般式(9)で表される化合物を下記一般式(11)で表される化合物へ転化する工程(工程(A−IV))、及び下記一般式(11)で表される化合物を前記一般式(4)で表されるアセテート誘導体へ転化する工程(工程(A−V))をこの順で行うことが挙げられる。
【0039】
前記転化する工程Aを前記工程(A−I)〜工程(A−V)で行う場合には、前記工程(A−I)〜工程(A−V)をこの順で行う際に、他の工程を加えてもよい。
また、前記工程(A−I)〜工程(A−V)のいずれかの工程を、これら以外のその他の工程に代えて行ってもよい。
【0040】
−−工程(A−I)−−
前記工程(A−I)は、下記一般式(1−1)で表されるチオアミド化合物を下記一般式(5)で表される化合物へ転化する工程である。
【化14】
ただし、前記一般式(1−1)中、R2、R3及びR11は、前記一般式(1)中のR2、R3及びR11とそれぞれ同じである。前記一般式(5)中、R2、R3及びR11は、前記一般式(1−1)中のR2、R3及びR11とそれぞれ同じである。前記R7は、水酸基の保護基を表す。
前記R7としては、水酸基の保護基であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記一般式(1)における前記R11の説明で例示した保護基が挙げられる。
前記R11と前記R7の水酸基の保護基の組合せとしては、特に制限はなく、目的に応じて適宜選択することができるが、前記R7の基が前記R11の基よりも脱保護し易いことが好ましい。そのような組合せとしては、前記R11がベンジル基であり前記R7がtert−ブチルジメチルシリル(TBS)基であることが、選択的に前記R7を脱保護できる点で好ましい。
【0041】
前記工程(A−I)としては、前記一般式(1−1)中の水酸基の水素原子を前記R7基に変換できる限り、特に制限はなく、目的に応じて適宜選択することができ、例えば、水酸基を保護する公知の方法に従って行うことができる。
【0042】
前記工程(A−I)おいて、前記一般式(1−1)中の水酸基の水素原子をtert−ブチルジメチルシリル(TBS)基に置換する場合には、塩基存在下で反応を行うことが好ましい。前記塩基としては、特に制限はなく、目的に応じて適宜選択することができるが、求核性が低い塩基が好ましく、2,6−ルチジンがより好ましい。
【0043】
−−工程(A−II)−−
前記工程(A−II)は、前記一般式(5)で表される化合物を下記一般式(6)で表される化合物へ転化する工程である。
【化15】
ただし、前記一般式(6)中、R7及びR11は、前記一般式(5)中のR7及びR11とそれぞれ同じである。前記一般式(6)中、R4は、前記一般式(4)中のR4と同じである。
【0044】
前記工程(A−II)としては、チオアミドをβケトエステルに変換できる限り、特に制限はなく、目的に応じて適宜選択することができ、例えば、Sメチル化に続く、相当するリチウムエノラートの付加反応を用いて行うことができる。また、前記工程(A−II)は、Yuichiro Mutoh, Toshiaki Murai,Organic Letters 2003,Vol5,No.8,1361−1364.を参考にして行うことができる。
【0045】
−−工程(A−III)−−
前記工程(A−III)としては、前記一般式(6)で表される化合物を下記一般式(9)で表される化合物へ転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記R7の脱保護の後にジアステレオ選択的還元を行い、更にsyn−1,3−ジオールに対して保護基を導入し1,3−ジオキサン環を形成する方法などが挙げられる。
【化16】
ただし、前記一般式(9)中、R4及びR11は、前記一般式(6)中のR4及びR11とそれぞれ同じである。前記一般式(9)中、R5及びR6は、前記一般式(4)中のR5及びR6とそれぞれと同じである。
【0046】
前記R7の脱保護の後にジアステレオ選択的還元を行い、更にsyn−1,3−ジオールに対して保護基を導入し1,3−ジオキサン環を形成する方法としては、例えば、前記一般式(6)で表される化合物を下記一般式(7)で表される化合物に変換する工程(工程(A−III−1))、下記一般式(7)で表される化合物を下記一般式(8)で表される化合物に変換する工程(工程(A−III−2))、及び下記一般式(8)で表される化合物を前記一般式(9)で表される化合物に変換する工程(工程(A−III−3))をこの順で行う方法が挙げられる。
【化17】
ただし、前記一般式(7)、及び一般式(8)中、R4及びR11は、前記一般式(6)中のR4及びR11とそれぞれ同じである。
【0047】
前記工程(A−III−1)としては、前記一般式(6)中の前記R7を水素原子に置換する、即ち、水酸基の保護基である前記R7を脱保護する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記R7がトリアルキルシリル基(例えば、tert−ブチルジメチルシリル(TBS)基)の場合には、酸性条件、又はフッ化物イオンによって脱保護する方法が挙げられる。前記フッ化物イオンによって脱保護する場合には、例えば、テトラブチルアンモニウムフルオリド(TBAF)、フッ化水素酸(HF)、フッ化セシウム(CsF)などを用いることができる。
【0048】
前記工程(A−III−2)としては、前記一般式(7)中のカルボニル基を水酸基に還元することができる工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、LiAlH4、NaBH4などの還元剤を用いる工程が挙げられる。前記NaBH4を用いる場合には、通常、メタノール、エタノールなどのアルコール存在下で還元が行われる。なお、前記工程(A−III−2)においては、コンフォメーションを制御することでsyn選択性を発現することができる。コンフォメーションを制御する方法としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、トリエチルボラン、ジエチルメトキシボランなどによるアルコールとケトン部位のキレート形成によりコンフォメーションを制御することが挙げられる。
【0049】
前記工程(A−III−3)としては、前記一般式(8)中の光学活性syn−1,3ジオールを、前記一般式(9)中の1,3−ジオキサン環に変換する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、2,2−ジメトキシプロパン、2−メトキシ−2−プロペン、アセトン、3,3−ジメトキシペンタン、シクロヘキサノン、シクロペンタノンなどを用いる方法が挙げられる。
【0050】
前記工程(A−III)において、前記工程(A−III−1)及び工程(A−III−2)は、精製をすることなく行い、前記工程(A−III−3)で精製を行い、前記一般式(9)で表される化合物を得てもよい。
【0051】
−−工程(A−IV)−−
前記工程(A−IV)としては、前記一般式(9)で表される化合物を下記一般式(11)で表される化合物へ転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができる。
【化18】
ただし、前記一般式(11)中、R4、R5及びR6は、前記一般式(9)中のR4、R5及びR6とそれぞれ同じである。R8は、アジ基に変換可能な基を表す。
【0052】
前記R8としては、アジ基に変換可能な基であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、−OR10(R10は、Ts基(トシル基)、Ms基(メシル基)、Tf基(トリフルオロメタンスルホニル基)等)、ヨウ素原子などが挙げられる。
【0053】
前記工程(A−IV)としては、例えば、前記一般式(9)で表される化合物を下記一般式(10)で表される化合物に変換する工程(工程(A−IV−1))の後に、続いて、下記一般式(10)で表される化合物を前記一般式(11)で表される化合物に変換する工程(工程(A−IV−2))などが挙げられる。
【化19】
ただし、前記一般式(10)中、R4、R5及びR6は、前記一般式(9)中のR4、R5及びR6とそれぞれ同じである。
【0054】
前記工程(A−IV−1)としては、前記一般式(9)中の前記R11を水素原子に置換する、即ち、水酸基の保護基R11を脱保護する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、従来公知の水酸基の脱保護の方法により行うことができる。前記R11が、ベンジル基の場合には、前記工程(A−IV−1)としては、例えば、水酸化パラジウム/炭素(パールマン触媒)などのパラジウム炭素(Pd/C)を用いてベンジル基を脱保護する方法が挙げられる。
【0055】
前記工程(A−IV−2)としては、前記一般式(10)中の水酸基をアジ基に変換可能な基(例えば、−OR10(R10はトシル基))に置換する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、パラトルエンスルホニルクロリドを用いて水酸基をトシル化する方法などが挙げられる。
【0056】
前記工程(A−IV)において、前記工程(A−IV−1)は、精製をすることなく行い、前記工程(A−IV−2)で精製を行い、前記一般式(11)で表される化合物を得てもよい。
【0057】
−−工程(A−V)−−
前記工程(A−V)は、前記一般式(11)で表される化合物を下記一般式(4)で表されるアセテート誘導体へ転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができる。
【化20】
【0058】
前記工程(A−V)としては、例えば、前記一般式(11)で表される化合物を下記一般式(12)で表される化合物に変換する工程(工程(A−V−1))の後に、続いて、下記一般式(12)で表される化合物を前記一般式(4)で表されるアセテート誘導体に変換する工程(工程(A−V−2))が挙げられる。
【化21】
ただし、前記一般式(12)中、R4、R5及びR6は、前記一般式(11)中のR4、R5及びR6とそれぞれ同じである。
【0059】
前記工程(A−V−1)としては、前記一般式(11)中のR8(アジ基に変換可能な基)をアジ基(N3−)に置換する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、−OR10(R10はトシル基)をアジ基に置換する場合、求核剤としてのアジ化ナトリウムを反応させ、トシラートアニオン(TsO−)を脱離する求核置換反応を起こすことにより、アジ基を生成させる方法などが挙げられる。
【0060】
前記工程(A−V−2)としては、前記一般式(12)中のアジ基(N3−)を1級アミノ基に置換する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、トリフェニルホスフィンを用いたシュタウディンガー還元を用いる方法が挙げられる。
【0061】
前記工程(A−V)において、前記工程(A−V−1)は、精製をすることなく行い、前記工程(A−V−2)で精製を行い、前記一般式(4)で表されるアセテート誘導体を得てもよい。
【0062】
−転化する工程B(前記一般式(1)におけるR1が−NR12R13の場合)−
前記転化する工程Bとしては、例えば、下記一般式(1−2)で表されるチオアミド化合物を下記一般式(13)で表される化合物へ転化する工程(工程(B−I))、下記一般式(13)で表される化合物を下記一般式(14)で表される化合物へ転化する工程(工程(B−II))、下記一般式(14)で表される化合物を下記一般式(17)で表される化合物へ転化する工程(工程(B−III))、及び下記一般式(17)で表される化合物を前記一般式(4)で表されるアセテート誘導体へ転化する工程(工程(B−IV))をこの順で行うことが挙げられる。
【0063】
前記転化する工程Bを前記工程(B−I)〜工程(B−IV)で行う場合には、前記工程(B−I)〜工程(B−IV)を順で行う際に、他の工程を加えてもよい。
また、前記工程(B−I)〜工程(B−IV)のいずれかの工程を、これら以外のその他の工程に代えて行ってもよい。
【0064】
−−工程(B−I)−−
前記工程(B−I)は、下記一般式(1−2)で表されるチオアミド化合物を下記一般式(5)で表される化合物へ転化する工程である。
【化22】
ただし、前記一般式(1−2)中、R2、R3、R12及びR13は、前記一般式(1)中のR2、R3、R12及びR13とそれぞれ同じである。前記一般式(13)中、R2、R3、R12及びR13は、前記一般式(1−2)中のR2、R3、R12及びR13とそれぞれ同じである。前記R9は、水酸基の保護基を表す。
【0065】
前記R9としては、水酸基の保護基であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記一般式(1)における前記R11の説明で例示した保護基などが挙げられる。
【0066】
前記工程(B−I)としては、前記一般式(1−2)中の水酸基の水素原子を前記R9基に変換できる限り、特に制限はなく、目的に応じて適宜選択することができ、例えば、水酸基を保護する公知の方法に従って行うことができる。
【0067】
前記工程(B−I)おいて、前記一般式(1−2)中の水酸基の水素原子をtert−ブチルジメチルシリル(TBS)基に置換する場合には、塩基存在下で、反応を行うことが好ましい。前記塩基としては、特に制限はなく、目的に応じて適宜選択することができるが、求核性が低い塩基が好ましく、2,6−ルチジンがより好ましい。
【0068】
−−工程(B−II)−−
前記工程(B−II)は、下記一般式(13)で表される化合物を下記一般式(14)で表される化合物へ転化する工程である。
【化23】
ただし、前記一般式(14)中、R9、R12及びR13は、前記一般式(13)中のR9、R12及びR13とそれぞれ同じである。前記一般式(14)中、R4は、前記一般式(4)中のR4と同じである。
【0069】
前記工程(B−II)としては、チオアミドをβケトエステルに変換できる限り、特に制限はなく、目的に応じて適宜選択することができ、例えば、Sメチル化に続く、相当するリチウムエノラートの付加反応を用いて行うことができる。また、前記工程(B−II)は、Yuichiro Mutoh, Toshiaki Murai,Organic Letters 2003,Vol5,No.8,1361−1364.を参考にして行うことができる。
【0070】
−−工程(B−III)−−
前記工程(B−III)としては、前記一般式(14)で表される化合物を下記一般式(17)で表される化合物へ転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記R9の脱保護の後にジアステレオ選択的還元を行い、更にsyn−1,3−ジオールに対して保護基を導入し1,3−ジオキサン環を形成する方法などが挙げられる。
【化24】
ただし、前記一般式(17)中、R4、R12及びR13は、前記一般式(6)中のR4、R12及びR13とそれぞれ同じである。前記一般式(17)中、R5及びR6は、前記一般式(4)中のR5及びR6とそれぞれと同じである。
【0071】
前記R9の脱保護の後にジアステレオ選択的還元を行い、更にsyn−1,3−ジオールに対して保護基を導入し1,3−ジオキサン環を形成する方法としては、例えば、前記一般式(14)で表される化合物を下記一般式(15)で表される化合物に変換する工程(工程(B−III−1))、下記一般式(15)で表される化合物を下記一般式(16)で表される化合物に変換する工程(工程(B−III−2))、及び下記一般式(16)で表される化合物を下記一般式(17)で表される化合物に変換する工程(工程(B−III−3))をこの順で行う方法などが挙げられる。
【化25】
ただし、前記一般式(15)、及び一般式(16)中、R4、R12及びR13は、前記一般式(14)中のR4、R12及びR13とそれぞれ同じである。
【0072】
前記工程(B−III−1)としては、前記一般式(14)中の前記R9を水素原子に置換する、即ち、水酸基の保護基である前記R9を脱保護する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記R9がトリアルキルシリル基(例えば、tert−ブチルジメチルシリル(TBS)基)の場合には、酸性条件、又はフッ化物イオンによって脱保護する方法が挙げられる。前記フッ化物イオンによって脱保護する場合には、例えば、テトラブチルアンモニウムフルオリド(TBAF)、フッ化水素酸(HF)、フッ化セシウム(CsF)などを用いることができる。
【0073】
前記工程(B−III−2)としては、前記一般式(15)中のカルボニル基を水酸基に還元することができる工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、LiAlH4、NaBH4などの還元剤を用いる工程が挙げられる。前記NaBH4を用いる場合には、通常、メタノール、エタノールなどのアルコール存在下で還元が行われる。なお、前記工程(B−III−2)においては、コンフォメーションを制御することでsyn選択性を発現することができる。コンフォメーションを制御する方法としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、トリエチルボラン、ジエチルメトキシボランなどによるアルコールとケトン部位のキレート形成によりコンフォメーションを制御することが挙げられる。
【0074】
前記工程(B−III−3)としては、前記一般式(16)中の光学活性syn−1,3ジオールを、前記一般式(17)中の1,3−ジオキサン環に変換する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、2,2−ジメトキシプロパン、2−メトキシ−2−プロペン、アセトン、3,3−ジメトキシペンタン、シクロヘキサノン、シクロペンタノンなどを用いる方法が挙げられる。
【0075】
前記工程(B−III)において、前記工程(B−III−1)及び工程(B−III−2)は、精製をすることなく行い、前記工程(B−III−3)で精製を行い、前記一般式(17)で表される化合物を得てもよい。
【0076】
−−工程(B−IV)−−
前記工程(B−IV)としては、前記一般式(17)で表される化合物を前記一般式(4)で表されるアセテート誘導体へ転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができる。
【化26】
【0077】
前記工程(B−IV)において、前記NR12R13をNH2に変換する、即ち、アミノ基の保護基を脱保護する方法としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記R12及び前記R13が、一緒になってフタロイル基を形成している場合には、メチルアミン、ヒドラジン等を作用させることで脱保護する方法などが挙げられる。
【0078】
本発明の[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法は、アトルバスタチンの製造に有用な[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を効率よく安価に製造することができる。
【0079】
(アトルバスタチンの製造方法)
前記アトルバスタチンの製造方法は、前記一般式(1)で表されるチオアミド化合物を前記一般式(4)で表されるアセテート誘導体に転化する工程を少なくとも含み、更に必要に応じて、その他の工程を含む。
【0080】
本発明においてアトルバスタチンとは、アトルバスタチン((3R,5R)−7−[2−(4−フルオロフェニル)−5−イソプロピル−3−フェニル−4−フェニルカルバモイル−1H−ピロル−1−イル]−3,5−ジヒドロキシヘプタン酸)に加え、その製薬学的に許容される塩を包含する。
【0081】
前記塩としては、例えば、ナトリウム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アンモニウム塩;トリアルキルアミン塩等の有機塩基塩などが挙げられる。
前記アトルバスタチンとしては、アトルバスタチンのアルカリ土類金属塩が好ましく、アトルバスタチンカルシウムがより好ましい。
【0082】
<転化する工程>
前記転化する工程としては、本発明の前記[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法に記載した転化する工程が挙げられる。
【0083】
<その他の工程>
前記その他の工程としては、例えば、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体から、アトルバスタチン(特にアトルバスタチンカルシウム)を製造する工程が挙げられる。
【0084】
【化27】
【0085】
前記[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体、即ち、前記一般式(4)で表されるアセテート誘導体から、アトルバスタチンカルシウムを製造する工程としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、Kelvin L.Baumann,et al.,Tetrahedron Letters,Vol.33,No.17,pp.2283−2284,1992に記載の方法などが挙げられる。
【0086】
本発明のアトルバスタチンの製造方法は、アトルバスタチンを効率よく且つ安価に製造することができる。
【実施例】
【0087】
以下に本発明の実施例を挙げて本発明を具体的に説明するが、本発明はこれらの実施例に何ら限定されるものではない。
なお、以下の実施例において、「THF」は「テトラヒドロフラン」を表す。「DMF」は「N,N−ジメチルホルムアミド」を表す。「Bn」は「ベンジル基」を表す。「TBS」は「tert−ブチルジメチルシリル基」を表す。「Ts」は「トシル基」を表す。
【0088】
(実施例1)
<チオアミド化合物A−1の合成1>
<<化合物X(N,N−ジアリルチオアセトアミド)の合成>>
300mLナスフラスコにN,N−ジアリルアセトアミド(5.0g、36mmol、1当量、Stanislaw Krompiec, et al.,Journal of Molecular Catalysis A: Chemical 2005,Vil 225,No.1,91−101.に従って合成)、ローソン試薬(7.3g、18mmol、0.5当量)、及び乾燥THF(180mL)を順次加えて12時間加熱還流した後、室温に戻して1規定塩酸(3mL)加えた。得られた二相混合物を酢酸エチル(20mL)で抽出し、有機層を飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られた残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=10/1(体積比))で精製し、下記化合物X(N,N−ジアリルチオアセトアミド)を淡黄色油状物として得た。収量4.8g(収率86%)。
【化28】
【0089】
<<2,2,5,7,8−ペンタメチルクロマノールリチウム塩の合成>>
加熱真空乾燥した5mLナスフラスコに2,2,5,7,8−ペンタメチルクロマノール(88.1mg、0.40mmol)を加え1時間減圧乾燥した後、フラスコ内をアルゴン雰囲気にして乾燥テトラヒドロフラン(THF)(2mL)を加えた。フラスコをアセトン−ドライアイス浴で−78℃に冷却し、n−ブチルリチウム(247μL、0.40mmol、1.62Mへキサン溶液)をゆっくりと加え、同温度で1時間撹拌し、2,2,5,7,8−ペンタメチルクロマノールリチウム塩の0.2M THF溶液を得た。
【0090】
<<Cu/(S,S)−Ph−BPEの合成>>
加熱真空乾燥した5mLナスフラスコに[Cu(CH3CN)4]PF6(Strem社製、149.0mg、0.40mmol)と(S,S)−Ph−BPE(Strem社製、202.5mg、0.40mmol)をグローブボックス内で加えた後、アルゴン雰囲気下にてTHF(4mL)を加えてCu/(S,S)−Ph−BPE(銅−光学活性ホスフィン錯体)の0.1M THF溶液を得た。
【0091】
<<チオアミド化合物A−1の合成>>
加熱真空乾燥した100mLナスフラスコにN,N−ジメチルホルムアミド(DMF)(60mL)、3−(ベンジルオキシ)プロパナール(1.0g、6.09mmol、1当量、Amanda M.Heapy, Margaret A. Brimble, Tetrahedron,Vol.66,No.29,5424−5431.に従って合成)、N,N−ジアリルチオアセトアミド(上記化合物X、1.13g、7.30mmol、1.2当量)、上記で調製したCu/(S,S)−Ph−BPE溶液(3.65mL、0.365mmol、0.06当量)をアルゴン雰囲気下加えた。ナスフラスコを−40℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩のTHF溶液(1.83mL、0.365mmol、0.06当量)を加え、−40℃で36時間撹拌した後、飽和塩化アンモニウム水溶液(30mL)と2,2−ビピリジン(57mg、0.365mmol、0.06当量)を加え、その混合溶液を酢酸エチル(20mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=5/1〜2/1(体積比))で精製し、下記チオアミド化合物A−1を黄色油状物として得た。収量1.64g(収率84%、92%ee)。
【化29】
【0092】
得られたチオアミド化合物A−1のIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν3413, 3085, 2919, 2861, 1643, 1496, 1411cm−1
1H NMR(CDCl3) δ7.34−7.29(m,5H), 5.86(dddd,J=5.7, 5.7, 10.3, 17.2Hz,1H), 5.73(dddd,J=5.0, 5.0, 10.3, 17.2Hz,1H), 5.26−5.09(m,4H), 4.75(dd,J=5.7, 14.7Hz,1H), 4.54−4.50(m,1H), 4.51(s,2H), 4.41−4.35(m,1H), 4.32−4.25(m,1H), 4.10−4.04(m,1H), 3.71−3.68(m,2H), 2.84(dd,J=2.8, 15.4Hz,1H), 2.76(dd,J=8.7, 15.4Hz,1H), 1.91−1.79(m,2H)
13CNMR(CDCl3) δ202.5, 138.1, 130.5, 130.5,130.5, 128.3, 127.6, 127.5, 118.4, 117.7, 73.1, 69.0, 67.9, 55.5, 52.8, 48.3, 36.3
[α]D22−33.9(c0.33,CHCl3,92%eesample)
ESI−MS m/z 342.2 [M+Na]+
HRMS(ESI) Anal.calcd. for C18H25NNaO2S m/z 342.1498 [M+Na]+,found;342.1500.
【0093】
(実施例2)
<チオアミド化合物A−1の合成2>
触媒の種類を変えて、チオアミド化合物A−1を合成した。具体的には以下のとおりである。
加熱真空乾燥した20mLナスフラスコに[Cu(CH3CN)4]PF6(分子量372.72)(6.7mg、0.018mmol、0.09当量)と(S)−BINAP(Strem社製、分子量622.67、11.2mg、0.018mmol、0.09当量)をグローブボックス内で加え、アルゴン雰囲気下THF(180μL)を加えて5分間撹拌した後、DMF(2mL)、3−(ベンジルオキシ)プロパナール(32.8μL、0.20mmol、1当量)、及びN,N−ジアリルチオアセトアミド(上記化合物X、38.2μL、0.24mmol、1.2当量)を加えた。試験管を−60℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩の0.2M THF溶液(90μL、0.018mmol、0.09当量)を加え、−60℃で60時間撹拌した後、THF/酢酸混合溶液(10:1(体積比))(2mL)と2,2−ビピリジン(2.8mg、0.018mmol、0.09当量)を加えて反応を停止した。蒸留水(2mL)を加えた後、混合溶液を酢酸エチル(2mL)で3回抽出した。得られた有機層を飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥し、チオアミド化合物A−1(NMR収率25%、−10%ee)を得た。
【0094】
(実施例3)
<チオアミド化合物A−1の合成3>
触媒の種類を変えて、チオアミドチオアミド化合物A−1を合成した。具体的には以下のとおりである。
加熱真空乾燥した20mLナスフラスコに[Cu(CH3CN)4]PF6(分子量372.72)(6.7mg、0.018mmol、0.09当量)と(R,R)−iPr−DuPhos(分子量418.58)(7.5mg、0.018mmol、0.09当量)をグローブボックス内で加え、アルゴン雰囲気下THF(180μL)を加えて5分間撹拌した後、DMF(2mL)、3−(ベンジルオキシ)プロパナール(32.8μL、0.20mmol、1当量)、及びN,N−ジアリルチオアセトアミド(上記化合物X、38.2μL、0.24mmol、1.2当量)を加えた。試験管を−60℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩の0.2M THF溶液(90μL、0.018mmol、0.09当量)を加え、−60℃で60時間撹拌した後、THF/酢酸混合溶液(10:1(体積比))(2mL)と2,2−ビピリジン(2.8mg、0.018mmol、0.09当量)を加えて反応を停止した。蒸留水(2mL)を加えた後、混合溶液を酢酸エチル(2mL)で3回抽出した。得られた有機層を飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥し、チオアミド化合物A−1(NMR収率52%,−46%ee)を得た。
【0095】
(実施例4)
<チオアミド化合物A−2の合成>
チオアミド化合物A−1とは保護基の種類が異なるチオアミド化合物A−2を合成した。具体的に以下のとおりである。
加熱真空乾燥した20mL試験管にDMF(2mL)、3−(ベンジルオキシ)プロパナール(33μL、0.20mmol、1当量、Amanda M.Heapy, Margaret A. Brimble, Tetrahedron,Vol.66,No.29,5424−5431.に従って合成)、N,N−ジメチルチオアセトアミド(Alfa Aesar社製、24.5g、0.24mmol、1.2当量)、上記で調製したCu/(S,S)−Ph−BPE溶液(120μL、0.012mmol、0.06当量)をアルゴン雰囲気下加えた。ナスフラスコを−40℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩のTHF溶液(60μL、0.012mmol、0.06当量)を加え、−40℃で40時間撹拌した後、飽和塩化アンモニウム水溶液(2mL)と2,2−ビピリジン(1.8mg、0.012mmol、0.06当量)を加え、その混合溶液を酢酸エチル(3mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=5/1〜1/1(体積比))で精製し、下記チオアミド化合物A−2を黄色油状物として得た。収量12.8mg(収率24%、90%ee)。
【化30】
【0096】
得られたチオアミド化合物A−2のIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν3406, 2960, 2871, 1523cm−1
1H NMR(CDCl3) δ7.36−7.27(m,5H), 4.52(s,2H), 4.37−4.34(m,1H), 3.71−3.69(m,2H), 3.46(s,3H), 3.28(s,3H), 2.79(dd,J=2.5, 15.8Hz,1H), 2.72(dd,J=8.9, 15.8Hz,1H), 1.91−1.84(m,2H)
13C NMR(CDCl3) δ202.5, 138.5, 128.3, 127.6, 127.5, 73.1, 69.0, 67.8, 52.9, 44.6, 44.5, 41.9
[α]D22−34.8(c0.13,CHCl3,90%ee)
ESI−MS m/z 280.2 [M+Na]+
HRMS (ESI) Anal.calcd. for C14H21NNaO2S m/z 290.1185 [M+Na]+,found;290.1186.
【0097】
(実施例5)
<チオアミド化合物A−3の合成>
チオアミド化合物A−1とは保護基の種類が異なるチオアミド化合物A−3を合成した。具体的に以下のとおりである。
加熱真空乾燥した20mL試験管にDMF(2mL)、3−(tert−ブチルジメチルシリルオキシ)プロパナール(37μL、0.20mmol、1当量、Christian U. Gruenanger, Bernhard Breit, Angewandte Chemie, International Edition, 2010, Vol 49, No.5, 967−970.に従って合成)、上記で調製したN,N−ジアリルチオアセトアミド(38μL、0.24mmol、1.2当量)、上記で調製したCu/(S,S)−Ph−BPE溶液(180μL、0.018mmol、0.09当量)をアルゴン雰囲気下加えた。ナスフラスコを−40℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩のTHF溶液(90μL、0.018mmol、0.09当量)を加え、−40℃で40時間撹拌した後、飽和塩化アンモニウム水溶液(2mL)と2,2−ビピリジン(2.8mg、0.018mmol、0.06当量)を加え、その混合溶液を酢酸エチル(3mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=5/1〜2/1(体積比))で精製し、下記チオアミド化合物A−3を黄色油状物として得た。収量35.7mg(収率52%、45%ee)。
【化31】
【0098】
得られたチオアミド化合物A−3のIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν3421, 2927, 2858, 1643, 1492, 1411, 1250cm−1
1H NMR (CDCl3) δ5.92(dddd,J=5.7, 5.7, 10.3, 17.2Hz,1H), 5.82(dddd,J=5.5, 5.5, 10.3, 17.2Hz,1H), 5.29−5.15(m,4H), 4.77(dd,J=5.5, 14.9Hz,1H), 4.47(dd,J= 6.0, 14.9Hz,1H), 4.37−4.32(m,2H), 4.14−4.08(m,1H), 3.87−3.78(m,2H), 2.86−2.81(m,2H), 1.81−1.68(m,2H), 0.84(s,9H), 0.04(s,6H)
13C NMR(CDCl3) δ202.7, 130.7, 118.4, 117.8, 69.5, 61.2, 55.7, 53.4, 48.7, 32.2, 25.9, 18.2, −5.5
[α]D22−17.2(c0.17,CHCl3,45%ee)
ESI−MS m/z 366.2 [M+Na]+
HRMS(ESI) Anal.calcd. for C17H33NNaO2SSi m/z 366.1893 [M+Na]+,found;366.1892.
【0099】
(実施例6)
<チオアミド化合物A−4の合成>
チオアミド化合物A−4の合成を行った。
加熱真空乾燥した50mLナスフラスコにDMF(13mL)、3−フタルイミジルプロパナール(Aurora Fine Chemicals社製、266.0mg、1.13mmol、1当量)、N,N−ジアリルチオアセトアミド(244μL、1.35mmol、1.2当量)、上記で調製したCu/(S,S)−Ph−BPE溶液(100μL、0.10mmol、0.09当量)をアルゴン雰囲気下加えた。ナスフラスコを−40℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩のTHF溶液(50μL、0.10mmol、0.09当量)を加え、−40℃で24時間撹拌した後、飽和塩化アンモニウム水溶液(5mL)と2,2−ビピリジン(15.6mg、0.10mmol、0.09当量)を加え、その混合溶液を酢酸エチル(5mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=7/1〜2/1(体積比))で精製し、下記チオアミド化合物A−4を黄色油状物として得た。収量251.1mg(収率62%、75%ee)。
【化32】
【0100】
得られたチオアミド化合物A−4のIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν 3463, 3086, 2923, 2865, 1770, 1712, 1612, 1492, 1396 cm−1
1H NMR(CDCl3) δ7.85−7.83 (m,2H), 7.72−7.71 (m,2H), 5.86(dddd,J=6.4, 6.4, 10.7, 17.2Hz,1H), 5.78(dddd,J=4.6 , 4.6, 10.7, 17.2Hz,1H), 5.30−5.12(m,4H), 4.78(dd,J=4.8, 14.9Hz,1H), 4.47(dd,J=5.7, 14.9Hz,1H), 4.36−4.30(m,1H), 4.27−4.08(m,3H), 3.91−3.84(m,2H), 2.78−2.76(m,2H), 1.90−1.85(m,2H)
13C NMR(CDCl3) δ202.1, 168.5, 133.9, 132.0, 130.6, 130.5, 123.2, 118.5, 118.0, 68.0, 55.7, 53.0, 47.8, 35.2, 34.7
[α]D22−11.4(c0.55,CHCl3,75%ee sample)
ESI−MS m/z 381.4 [M+Na]+
HRMS (ESI) Anal.calcd. for C19H22N2NaO3S m/z 381.1243 [M+Na]+,found;381.1243.
【0101】
(実施例7)
<tert−ブチル[(4R,6R)−6−アミノエチル−2,2−ジメチル−1,3−ジオキサン−4−イル]アセテートの合成1>
<<化合物Bの合成>>
加熱真空乾燥した100mLナスフラスコにアルゴン雰囲気下で、実施例1で合成したチオアミド化合物A−1(800mg、2.5mmol、1当量)、塩化メチレン(30mL)を加え、氷浴で0℃に冷却した。2,6−ルチジン(575μL、5.0mmol、2当量)、tert−ブチルジメチルシリル(TBS)トリフラート(860μL、3.75mmol、1.5当量)を加え、室温にて3時間撹拌後、飽和塩化アンモニウム水溶液(20mL)を加えて反応を停止し、その混合溶液を塩化メチレン(10mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、化合物Bを淡黄色油状物として得た。収量1.04g(収率96%)。
【化33】
【0102】
得られた化合物BのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2954, 2858, 1643, 1493, 1257, 1115cm−1
1H NMR(CDCl3) δ7.33−7.29(m,5H), 5.92(dddd,J=5.8, 5.8, 10.8, 16.7Hz,1H), 5.75(dddd,J=4.8, 4.8, 10.3, 17.2Hz,1H), 5.25−5.08(m,4H), 4.83(dd,J=5.7, 14.2Hz,1H), 4.61−4.53(m,2H), 4.48(dd,J=3.0, 15.1Hz,1H), 4.34(dd,J=6.9, 14.2Hz,1H), 4.00−3.95(m,1H), 3.64−3.51(m,2H), 3.10(dd,J=8.2, 13.8Hz,1H), 2.83(dd,J=4.1, 13.8Hz,1H), 1.90−1.86(m,2H), 0.84(s,9H), 0.06(s,3H), 0.02(s,3H)
13C NMR(CDCl3) δ203.0, 138.6, 131.5, 131.3, 128.3, 127.6, 127.5, 119.3, 117.6, 73.0, 72.1, 66.5, 56.4, 53.1, 50.2, 37.4, 25.9, 25.7, 25.6, 17.9, −4.3, −4.8
[α]D22+6.8(c1.0,CHCl3)
ESI−MS m/z 456.3 [M+Na]+
HRMS(ESI) Anal.calcd. for C24H39NNaO2SSi m/z 456.2363 m/z [M+Na]+,found;456.2358.
【0103】
<<化合物Cの合成>>
−酢酸tert−ブチルエステルリチウムエノラートの調製−
加熱真空乾燥した20mLナスフラスコにアルゴン雰囲気下で酢酸tert−ブチルエステル(1.0mL、7.46mmol、1当量)、リチウムヘキサメチルジシラジド(1.25g、7.46mmol、1当量)を加えてアセトン−ドライアイス浴で−78℃に冷却した。−78℃に冷却した乾燥THF(7.46mL)をゆっくりと滴下後、同温度にて1時間撹拌し、酢酸tert−ブチルエステルリチウムエノラートの1.0M THF溶液を得た。
【0104】
−化合物Cの合成−
加熱真空乾燥した100mLナスフラスコにアルゴン雰囲気下で化合物B(920mg、2.12mmol、1当量)とジエチルエーテル(23mL)を加え、氷浴で0℃に冷却した。メチルトリフラート(467μL、4.24mmol、2当量)を加えて0℃にて5分間、室温にて4時間半撹拌後、フラスコをアセトン−ドライアイス浴で−78℃に冷却し、上記で調製した酢酸tert−ブチルエステルリチウムエノラート(64μL、6.40mmol、3当量)をゆっくりと滴下して加えた。3時間撹拌後、塩化メチレン(7mL)、シリカゲル(5g)を加え、室温にて1時間半撹拌した。混合物をシリカゲルショートパッドカラムに通し、濾液を濃縮して得られた残渣にTHF(20mL)、1規定塩酸(2mL)を加え室温にて2時間撹拌後、混合溶液を酢酸エチル(20mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られた残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=20/1(体積比))で精製し、下記化合物Cを無色油状物として得た。収量662mg(収率72%)。
【化34】
【0105】
得られた化合物CのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2954, 2931, 2858, 1739, 1716, 1647, 1458, 1369, 1254, 1146, 1115cm−1
1H NMR(CDCl3);
keto form;δ7.36−7.28(m,5H), 4.50(s,2H), 4.50−4.43(m,1H), 3.56−3.48(m,2H), 3.34(s,2H),2.69(d,J=6.4Hz,2H), 1.81−1.77(m,2H), 1.45(s,9H), 0.85(s,9H), 0.05(s,3H), 0.03(s,3H);
enol form;δ12.15(brs,1H), 7.36−7.28(m,5H),4.90(s,1H), 4.33(s,2H), 4.35−4.31(m,1H), 3.56−3.48(m,2H), 2.29(d,J=6.6Hz,2H), 1.77−1.73(m,2H), 1.47(s,9H), 0.87(s,9H), 0.05(s,3H), 0.02(s,3H)
13C NMR(CDCl3) keto−enol mixture;δ202.1, 166.3, 138.4, 128.3, 127.6, 127.5, 113.9, 92.8, 81.8, 80.6, 73.0, 73.0, 72.9, 67.2, 66.6, 66.4, 52.0, 50.3, 43.8, 37.3, 37.2, 28.6, 28.3, 28.0, 25.8, 17.9, −4.7, −4.8, −4.9
[α]D22−7.6(c0.5,CHCl3)
ESI−MS m/z 459.3 [M+Na]+
HRMS(ESI) Anal.calcd. for C24H40NaO5Si m/z 459.2537 [M+Na]+,found;459.2534.
【0106】
<<化合物D、E、及びFの合成>>
30mLナスフラスコにアルゴン雰囲気下、化合物C(400mg、0.916mmol、1当量)と乾燥THF(2.5mL)を入れ、氷浴で0℃に冷却した。続いてテトラブチルアンモニウムフルオリド(1.3mL、1.30mmol、1.4当量、1.0M THF溶液)をゆっくりと滴下し、0℃で30分間、室温にて3時間撹拌後、蒸留水(2mL)を加え、混合液を酢酸エチル(5mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Dの粗生成物を淡黄色油状物質として得た。
続いて、50mLナスフラスコに上記で得られた化合物D(292mg、0.906mmol、1当量)、乾燥THF(8.5mL)、メタノール(2.4mL)を入れ、ナスフラスコを−80℃の恒温槽に移した後、ジエチルメトキシボラン(1.0mL、1.0mmol、1.1当量、1.0M THF溶液)を加えて30分間撹拌した。水素化ホウ素ナトリウム(37.8mg、1.0mmol、1.1当量)を加えて−80℃にて10時間撹拌後、酢酸(2mL)を加え、混合液を酢酸エチル(10mL)で3回抽出した。得られた有機層を飽和重曹水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Eの粗生成物を淡黄色油状物として得た。
上記で得られた化合物E(273mg、0.84mmol、1当量)、アセトン(2.6mL)、パラトルエンスルホン酸一水和物(16.0mg)を30mLナスフラスコに移し、ジメトキシプロパン(205μL、1.68mmol、2当量)を加えて室温で4時間撹拌後、飽和重曹水を加えてpHを約7とした後、混合溶液をエーテル(5mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Fを淡黄色油状物質として得た。収量294mg(3段階収率88%)。
【化35】
【0107】
得られた化合物FのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2981, 2938, 1724, 1454, 1369, 1277, 1153cm−1
1H NMR(CDCl3) δ7.38−7.27(m,5H), 4.50(dd,J=4.8, 12.1Hz,2H), 4.28−4.21(m,1H), 4.09−4.02(m,1H), 3.61−3.50(m,2H), 2.42(dd,J=7.1, 15.1Hz,1H), 2.29(dd,J=6.2, 15.1Hz,1H), 1.81−1.68(m,2H), 1.56(dt,J=2.3, 12.6Hz,1H), 1.44(s,9H), 1.44(s,3H), 1.35(s,3H), 1.25−1.15(m,1H)
13CNMR(CDCl3) δ170.3, 138.5, 128.4, 127.6, 127.5, 98.7, 80.5, 73.0, 66.3, 66.2, 66.0, 42.8, 36.6, 36.5, 30.1, 28.1, 19.6
[α]D22+21.9(c0.26,CHCl3)
ESI−MS m/z 387.2 [M+Na]+
HRMS(ESI)Anal.calcd. for C21H32NaO5 m/z 387.2142 [M+Na]+,found;387.2140.
【0108】
<<化合部G、及びHの合成>>
化合物F(153mg、0.419mmol、1当量)、酢酸エチル(1.5mL)、水酸化パラジウム/炭素(25mg、20%w/w)を20mLナスフラスコに加えて1気圧の水素雰囲気下、60℃にて24時間撹拌した後、不要物をセライト濾過し、濾液を減圧濃縮して下記化合物Gを淡黄色油状物として得た。
続いて、上記で得られた化合物G(120.5mg、0.417mmol、1当量)、4−ジメチルアミノピリジン(15.3mg、0.125mmol、0.3当量)、塩化メチレン(2.5mL)を20mLナスフラスコに入れ、氷浴で0℃に冷却した。トリエチルアミン(17μL、1.25mmol、3当量)を加えて5分間撹拌し、パラトルエンスルホニルクロリド(159.3mg、0.834mmol、2当量)を加え、室温で4時間撹拌した後、塩化メチレン(10mL)、蒸留水(10mL)を加えた。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=10/1〜5/1(体積比))で精製し、化合物Hを無色油状物質として得た。収量165.1mg(2段階収率91%)。
【化36】
【0109】
得られた化合物HのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2981, 1728, 1365, 1176cm−1
1H NMR (CDCl3) δ7.91(d,J=8.2Hz,2H), 7.79(d,4H), 7.34(d,J=8.2Hz,2H), 4.26−4.18(m,1H), 4.18−4.13(m,1H), 4.11−4.06(m,1H), 3.99−3.91(m,1H), 2.45(s,3H), 2.39(dd,J=7.1, 15.1Hz,1H), 2.26(dd,J=6.2, 15.1Hz,1H), 1.89−1.67(m,2H), 1.51−1.45(m,2H), 1.44(s,9H), 1.34(s,3H), 1.27(s,3H)
13C NMR(CDCl3) δ170.1, 144.7, 133.0, 129.8, 127.9, 98.8, 80.6, 66.7, 66.0, 64.7, 42.6, 36.2, 35.4, 29.9, 28.1, 21.6, 19.5
[α]D22+12.9(c0.9,CHCl3)
ESI−MS m/z 451.2 [M+Na]+
HRMS(ESI) Anal.calcd. for C21H32NaO7S m/z 451.1761 [M+Na]+,found;451.1756.
【0110】
<<化合物I、及びJの合成>>
20mLナスフラスコに化合物H(120.0mg、0.277mmol、1当量)、アジ化ナトリウム(36.0mg、0.555mmol、2当量)、N,N−ジメチルホルムアミド(1.5mL)を加えて室温にて6時間撹拌した後、蒸留水(1mL)を加えた。混合液を酢酸エチル(3mL)で抽出後、得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。シリカゲルショートパッドカラムを通して濾過し、濾液を濃縮して下記化合物Iを得た。収量67.8mg(収率82%)。
10mL試験管に化合物I(52.1mg、0.174mmol、1当量)、THF(1.0mL)、蒸留水(0.10mL)、トリフェニルホスフィン(91.3mg、0.348mmol、2当量)を入れて50℃にて2時間撹拌した後、THFを減圧留去した。残渣にトルエン(5mL)を加えて混合液を減圧濃縮し、残渣に含まれる水を共沸混合物として留去した。得られた残渣に対して再びトルエン(5mL)を加え混合液を減圧濃縮し、得られた残渣をフラッシュカラムクロマトグラフィー(塩化メチレン/メタノール/トリエチルアミン=95/4/1(体積比))で精製し、化合物J(tert−ブチル[(4R,6R)−6−アミノエチル−2,2−ジメチル−1,3−ジオキサン−4−イル]アセテート)を無色油状物質として得た。収量41.2mg(収率87%)。
【化37】
【0111】
得られた化合物IのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2978, 2939, 2877, 2098, 1732cm−1
1H NMR(CDCl3) δ4.37−4.30(m,1H), 4.12−4.06(m,1H), 3.48−3.41(m,2H), 2.37(dd,J=5.5, 14.9Hz,1H), 2.33(dd,J=7.6, 14.9Hz,1H), 1.80−1.65(m,2H), 1.66(dt,2.5, 12.6Hz,1H), 1.48(s,3H), 1.46(s,9H), 1.32(s,3H), 1.22−1.13(m,1H)
13C NMR(CDCl3)δ170.1, 98.8, 80.6, 66.1, 65.8, 47.5, 42.6, 36.4, 35.6, 30.0, 28.1, 19.6
[α]D22+17.9(c0.21,CHCl3)
ESI−MS m/z 322.2 [M+Na]+
HRMS(ESI) Anal.calcd. for C14H25N3NaO4 m/z 322.1737 [M+Na]+,found;322.1737.
【0112】
得られた化合物JのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν3374, 2981, 2938, 2873, 1731, 1176cm−1
1H NMR(CDCl3) δ4.25−4.19(m,1H), 3.98−3.92(m,1H), 2.80−2.77(m,2H), 2.39(dd,J=6.9, 15.1,1H),2.26(dd,J=6.2, 15.1,1H), 1.95(brs,2H), 1.64−1.50(m,3H), 1.42(s,9H), 1.42(s,3H), 1.33(s,3H), 1.30−1.15(m,1H)
13C NMR(CDCl3)δ170.2, 98.6, 80.5, 67.4, 66.2, 42.6, 39.5, 38.4, 36.5, 30.1, 28.0, 19.7
[α]D22+11.5(c0.28,CHCl3)
ESI−MS m/z 274.2 [M+H]+
HRMS(ESI) Anal.calcd. for C14H28NO4 m/z 274.2013 [M+H]+,found;274.2015.
【0113】
(実施例8)
<tert−ブチル[(4R,6R)−6−アミノエチル−2,2−ジメチル−1,3−ジオキサン−4−イル]アセテートの合成2>
<<化合物Kの合成>>
加熱真空乾燥した30mLナスフラスコにアルゴン雰囲気下で、実施例6で合成したチオアミド化合物A−4(100.2mg、0.28mmol、1当量)、及び塩化メチレン(3.5mL)を加え、氷浴で0℃に冷却した。2,6−ルチジン(64.0μL、0.56mmol、2当量)、tert−ブチルジメチルシリル(TBS)トリフラート(96.5μL、0.42mmol、1.5当量)を加え、室温にて3時間撹拌後、飽和塩化アンモニウム水溶液(3mL)を加えて反応を停止し、その混合溶液を塩化メチレン(5mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Kを淡黄色油状物として得た。収量124.4mg(収率94%)。
【化38】
【0114】
得られた化合物KのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2954, 2857, 1774, 1712, 1593, 1496, 1400cm−1
1H NMR(CDCl3) δ7.82−7.80(m,2H), 7.71−7.68(m,2H), 5.92(dddd,J=5.7, 5.7, 10.3, 17.2Hz,1H), 5.82(dddd,J=5.5 , 5.5, 10.3, 17.2Hz,1H), 5.27−5.11(m,4H), 4.85(dd,J=7.2, 14.2Hz,1H), 4.63−4.53(m,2H), 4.31(dd,J=7.3, 14.2Hz,1H), 4.00(dd,J=4.6, 17.2Hz,1H), 3.82−3.72(m,2H), 3.16(dd,J=8.5, 13.5Hz,1H), 2.81(dd,J=4.1, 13.5Hz,1H), 2.03−1.86(m,2H), 0.85(s,9H), 0.10(s,3H), 0.03(s,3H)
13C NMR(CDCl3) δ202.5, 168.2, 133.8, 132.2, 131.3, 131.2, 123.1, 119.4, 117.6, 72.2, 56.5, 53.1, 49.1, 35.7, 34.0, 25.8, 17.8, −4.4, −4.7
[α]D23+10.1(c0.19,CHCl3)
ESI−MS m/z 495.2 [M+Na]+
HRMS (ESI) Anal.calcd. for C25H36N2NaO3SSi m/z 495.2108 m/z [M+Na]+,found;495.2111
【0115】
<<化合物L、M、N、及びOの合成>>
加熱真空乾燥した20mL試験管にアルゴン雰囲気下で化合物K(83.5mg、0.177mmol、1当量)とジエチルエーテル(1.5mL)を加え、氷浴で0℃に冷却した。メチルトリフラート(39μmL、0.353mmol、2当量)を加えて0℃にて5分間、室温にて4時間半撹拌後、フラスコをアセトン−ドライアイス浴で−78℃に冷却し、上記で調製した酢酸tert−ブチルエステルリチウムエノラート(177μL、0.177mmol、3当量)をゆっくりと滴下して加えた。3時間撹拌後、塩化メチレン(3mL)、及びシリカゲル(2g)を加え、室温にて1時間半撹拌した。混合物をシリカゲルショートパッドカラムに通し、濾液を濃縮して得られた残渣にTHF(10mL)、1規定塩酸(1mL)を加え室温にて2時間撹拌後、混合溶液を酢酸エチル(5mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Lの粗生成物を淡黄色油状物として得た。
続いて、アルゴン雰囲気下、化合物L(60mg、0.126mmol、1当量)と乾燥THF(800μL)を20mL試験管に移し、氷浴で0℃に冷却した。続いてテトラブチルアンモニウムフルオリド(176μL、0.176mmol、1.4当量、1.0M THF溶液)をゆっくりと滴下し、0℃で30分間、室温にて3時間撹拌後、蒸留水(0.5mL)を加え、混合液を酢酸エチル(3mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Mの粗生成物を淡黄色油状物質として得た。
続いて、20mLナスフラスコに化合物M(45mg、0.124mmol、1当量)、乾燥THF(1.7mL)、及びメタノール(0.4mL)を入れ、ナスフラスコを−80℃の恒温槽に移した後、ジエチルメトキシボラン(136μL、0.136mmol、1.1当量、1.0M THF溶液)を加えて30分間撹拌した。水素化ホウ素ナトリウム(5.2mg、0.136mmol、1.1当量)を加えて−80℃にて10時間撹拌後、酢酸(0.2mL)を加え、混合液を酢酸エチル(3mL)で3回抽出した。得られた有機層を飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Nの粗生成物を淡黄色油状物として得た。
続いて、得られた化合物N(43.0mg、0.12mmol、1当量)、アセトン(0.5mL)、及びパラトルエンスルホン酸一水和物(2.3mg、0.012mmol、0.1当量)を20mL試験管に移し、ジメトキシプロパン(29.5μL、0.24mmol、2当量)を加えて室温で4時間撹拌後、飽和重曹水を加えてpHを約7とした後、混合溶液をエーテル(2mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、化合物Oを淡黄色油状物質して得た。収量47mg(4段階収率66%)。
【化39】
【0116】
得られた化合物OのIRスペクトル、1H NMRスペクトル、及び13C NMRスペクトルデータを示す。
IR(neat) ν2923, 2865, 1770, 1725, 1712, 1612, 1492, 1396cm−1
1H NMR(CDCl3) δ7.86−7.83(m,2H), 7.74−7.71(m,2H), 4.35−4.27(m,1H), 4.12−4.04(m,1H), 3.65−3.55(m,2H), 2.44(dd,J=7.1, 15.1Hz,1H), 2.32(dd,J=6.2, 15.1Hz,1H), 1.82−1.69(m,2H), 1.57(dt,J=2.3, 12.6Hz,1H), 1.44(s,9H), 1.44(s,3H), 1.35(s,3H), 1.25−1.15(m,1H)
13C NMR(CDCl3) δ170.5, 168.6, 133.9, 132.0, 130.6, 80.5, 73.0, 67.9, 66.2, 42.8, 36.7, 36.6, 33.1, 30.1, 28.1, 19.6
【0117】
<<化合物Jの合成>>
エタノール(2.5mL)に化合物O(45mg、0.11mmol、1当量)を溶解し、ヒドラジン一水和物(70mL、2.25mmol、20当量)を加えた後、60℃にて1時間撹拌した。反応溶液を室温に戻した後、析出した固体を濾別し、濾液を減圧濃縮した。残渣に塩化メチレンと飽和食塩水を加え、水層を塩化メチレンで抽出し、得られた有機層を無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られた残渣をフラッシュカラムクロマトグラフィー(塩化メチレン/メタノール/トリエチルアミン=95/4/1(体積比))で精製し、化合物Jを無色油状物質して得た。収量25.5mg(収率85%)。
【化40】
【産業上の利用可能性】
【0118】
本発明のチオアミド化合物は、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を、効率よく安価に製造可能であることから、アトルバスタチンの合成に好適に用いることができる。
【技術分野】
【0001】
本発明は、アトルバスタチンの合成に有用なチオアミド化合物、該チオアミド化合物の製造方法、前記チオアミド化合物を用いた[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法、及び前記チオアミド化合物を用いたアトルバスタチンの製造方法に関する。
【背景技術】
【0002】
3−ヒドロキシ−3−メチルグルタリル−補酵素A(HMG−CoA)のメバロン酸塩への変換は、コレステロール生合成経路における初期の律速工程である。この工程は、酵素HMG−CoAレダクターゼによって触媒される。スタチンは、HMG−CoAレダクターゼがこの変換を触媒するのを阻害する。そのようなことから、スタチンは、総体的に強力な脂質低減作用物質である。
【0003】
現在、アトルバスタチンカルシウム水和物はLipitor(登録商標)として販売されており、下記式を有する(例えば、特許文献1参照)。
【化1】
【0004】
アトルバスタチンカルシウムは、HMG−CoAの選択的、競合的阻害剤である。そのようなことから、アトルバスタチンは強力な脂質低減作用物質であり、このため脂質低下剤及び/又はコレステロール低下剤として有用である。また、骨粗しょう症、良性の前立腺肥大(BPH)、及びアルツハイマー病の治療にも有用である。
【0005】
アトルバスタチンカルシウムの合成方法としては、図1に記載の合成方法が知られている(例えば、非特許文献1参照)。
しかし、図1に記載の合成方法は、HBr、及びNaCNの使用や、極低温工程を必要とすることから、工業生産には不適であるという問題がある。
【0006】
そこで、アトルバスタチンの新規合成方法の探索を目的として、種々の検討がされている。
例えば、図1に記載のアトルバスタチンの合成方法における合成中間体である、(4R−シス)−1,1−ジメチルエチル 6−シアノエチル−2,2−ジメチル−1,3−ジオキサン−4−アセテートの合成方法が提案されている(例えば、非特許文献2参照)。
しかし、この提案の技術では、ベンゼンスルホニル誘導体を経由して(4R−シス)−1,1−ジメチルエチル 6−シアノエチル−2,2−ジメチル−1,3−ジオキサン−4−アセテートが合成されているが、前記ベンゼンスルホニル誘導体は、次のステップへの反応性が十分ではないという問題がある。
【0007】
また、前記(4R−シス)−1,1−ジメチルエチル 6−シアノエチル−2,2−ジメチル−1,3−ジオキサン−4−アセテートを酵素法により合成する方法が提案されている(例えば、特許文献2参照)。
この提案の技術は、図2に示すような反応経路であるが、ケトレダクターゼ(KRED)、グルコースデヒドロゲナーゼ(GDH)、ハロヒドリンデハロゲナーゼ(HHDH)の3種の酵素を用い、3度に渡る煩雑な酵素反応が必要であるという問題がある。
【0008】
また、他の方法として、図1に記載のアトルバスタチンの合成方法における中間体である、tert−ブチル[(4R,6R)−6−アミノエチル−2,2−ジメチル−1,3−ジオキサン−4−イル]アセテートの合成方法が提案されている(例えば、非特許文献3参照)。
しかし、この提案の技術では、窒素官能基の導入に一級アルコールの酸化、ニトロアルドール反応、及び生じる二級アルコールの脱離反応を必要とし、煩雑な工程を要するという問題がある。
【0009】
したがって、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を、効率よく且つ銅触媒などの安価に調製可能な触媒を用いて製造可能な化合物、該化合物の製造方法、前記化合物を用いた[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法、及び前記化合物を用いたアトルバスタチンの製造方法の提供が求められているのが現状である。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】米国特許第5,273,995号公報
【特許文献2】国際公開第2004/015132号パンフレット
【非特許文献】
【0011】
【非特許文献1】Roth B.D.,et al.,Progress in Medicinal Chemistry,2002,40,1−22
【非特許文献2】Philip L.Brower,et al.,Tetrahedron Letters,Vol.33,No.17,pp.2279−2282,1992
【非特許文献3】Stanislav Radl,SYNTHETIC COMMUNICATIONS,Vol.33,No.13,pp.2275−2283,2003
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明は、従来における前記諸問題を解決し、以下の目的を達成することを課題とする。即ち、本発明は、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を、効率よく且つ銅触媒などの安価に調製可能な触媒を用いて製造可能なチオアミド化合物、該チオアミド化合物の製造方法、前記チオアミド化合物を用いた[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法、及び前記チオアミド化合物を用いたアトルバスタチンの製造方法を提供することを目的とする。
【課題を解決するための手段】
【0013】
前記課題を解決するための手段としては、以下の通りである。即ち、
<1> 下記一般式(1)で表されることを特徴とするチオアミド化合物である。
【化2】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
<2> 前記<1>に記載のチオアミド化合物の製造方法であって、
下記一般式(2)で表される化合物と、下記一般式(3)で表される化合物とを反応させる反応工程を含むことを特徴とするチオアミド化合物の製造方法である。
【化3】
【化4】
ただし、前記一般式(2)中、R1は、−OR11、及び−NR12R13のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、水素原子及びアミノ基の保護基のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。前記一般式(3)中、R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。
<3> 反応工程が、銅錯体を用いて行われる前記<2>に記載のチオアミド化合物の製造方法である。
<4> 銅錯体が、銅−光学活性ホスフィン錯体である前記<3>に記載のチオアミド化合物の製造方法である。
<5> 下記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程を含むことを特徴とする[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法である。
【化5】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
【化6】
ただし、前記一般式(4)中、R4は、カルボキシル基の保護基及び水素原子のいずれかを表す。R5及びR6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
<6> 下記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程を含むことを特徴とするアトルバスタチンの製造方法である。
【化7】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい。)。
【化8】
ただし、前記一般式(4)中、R4は、カルボキシル基の保護基及び水素原子のいずれかを表す。R5及びR6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
【発明の効果】
【0014】
本発明によれば、従来における前記諸問題を解決し、前記目的を達成することができ、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を、効率よく且つ銅触媒などの安価に調製可能な触媒を用いて製造可能なチオアミド化合物、該チオアミド化合物の製造方法、前記チオアミド化合物を用いた[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法、及び前記チオアミド化合物を用いたアトルバスタチンの製造方法を提供することができる。
【図面の簡単な説明】
【0015】
【図1】図1は、従来のアトルバスタチンの合成方法の一例を示す合成スキームである。
【図2】図2は、酵素法による(4R−シス)−1,1−ジメチルエチル 6−シアノエチル−2,2−ジメチル−1,3−ジオキサン−4−アセテートの合成方法を示す合成スキームである。
【発明を実施するための形態】
【0016】
本明細書、及び特許請求の範囲に記載された化学式及び一般式における立体配置は特に言及しない場合には、絶対配置を表す。
【0017】
(チオアミド化合物)
本発明のチオアミド化合物は、下記一般式(1)で表される。
【化9】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
【0018】
前記R11における水酸基の保護基としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、Greenら、Protective Groups in Organic Synthesis, 3rd Edition,1999,John Wiley & Sons, Inc.などの成書を参照することができる。
前記水酸基の保護基としては、例えば、アラルキル基、トリアルキルシリル基、アルコキシアルキル基、アルカノイル基、アリールカルボニル基などが挙げられる。保護基におけるアリール環(ベンゼン環など)が置換基を有する場合には、置換基としてハロゲン原子やアルコキシ基などが挙げられる。
前記アラルキル基としては、例えば、ベンジル基、p−メトキシベンジル基、p−アミノベンジル基などが挙げられる。
前記トリアルキルシリル基としては、例えば、トリメチルシリル基、トリエチルシリル基、tert−ブチルジメチルシリル基などが挙げられる。
前記アルコキシアルキル基としては、例えば、メトキシメチル基、エトキシメチル基などが挙げられる。
前記アルカノイル基としては、例えば、アセチル基、トリフルオロアセチル基などが挙げられる。
前記アリールカルボニル基としては、例えば、ベンゾイル基、置換フェニルカルボニル基などが挙げられる。
これらの中でも、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を合成する際の脱保護の容易性の点から、アラルキル基が好ましく、ベンジル基がより好ましい。
【0019】
前記R12及び前記R13におけるアミノ基の保護基としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、メトキシカルボニル基、t−ブトキシカルボニル基、ベンジルオキシカルボニル基、アリルオキシカルボニル基、ホルミル基、アセチル基、ベンゾイル基、メチル基、エチル基、アリル基、ベンゼンスルホニル基などが挙げられる。また、前記R12及び前記R13が、一緒になって環構造の保護基を形成している場合は、例えば、フタロイル基(Phth基)が挙げられる。これらの中でも、温和な反応条件で脱保護が可能な点で、フタロイル基が好ましい。
【0020】
前記R2及び前記R3におけるアミド基の保護基としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、トシル基、メトキシメチル基、ベンジルオキシメチル基、アリル基、トリイソプロピルシリル基、ベンジル基、メトキシカルボニル基などが挙げられる。
これらの中でも、アルドール反応における反応成績に優れる点からアリル基が好ましい。
【0021】
本発明のチオアミド化合物は、立体異性体の混合物として得られる場合もあるが、この場合も本発明の範囲に包含される。
【0022】
前記一般式(1)で表されるチオアミド化合物の製造方法としては、特に制限はなく、目的に応じて適宜選択することができるが、下記本発明のチオアミド化合物の製造方法が好ましい。
【0023】
本発明のチオアミド化合物は、アトルバスタチン合成の有用な中間体である、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を合成するための物質、更にはアトルバスタチンを合成するための物質として有用である。
【0024】
(チオアミド化合物の製造方法)
本発明のチオアミド化合物の製造方法は、本発明の前記一般式(1)で表されるチオアミド化合物の製造方法であって、下記一般式(2)で表される化合物と、下記一般式(3)で表される化合物とを反応させる反応工程を含み、更に必要に応じて、その他の工程を含む。
【化10】
【化11】
ただし、前記一般式(2)中、R1は、−OR11、及び−NR12R13のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、水素原子及びアミノ基の保護基のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。前記一般式(3)中、R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。
前記一般式(2)中のR1は、前記一般式(1)中のR1と同じである。
前記一般式(3)中のR2及びR3は、前記一般式(1)中のR2及びR3とそれぞれ同じである。
【0025】
<反応工程>
前記反応工程としては、前記一般式(2)で表される化合物と、前記一般式(3)で表される化合物とを反応させる工程であれば、特に制限はなく、目的に応じて適宜選択することができるが、銅錯体を用いることが好ましい。前記反応工程において、前記銅錯体を触媒として用いることにより、安価な銅を触媒源に用いて前記一般式(1)で表されるチオアミド化合物を製造することができる。
【0026】
前記銅錯体としては、特に制限はなく、目的に応じて適宜選択することができるが、銅−光学活性ホスフィン錯体であることが、エナンチオ選択性が優れる点で好ましい。
前記銅−光学活性ホスフィン錯体は、銅と光学活性ホスフィン配位子の錯体である。前記光学活性ホスフィン配位子としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、シクロヘキシルアニシルメチルホスフィン(CAMP)、1,2−ビス(アニシルフェニルホスフィノ)エタン(DIPAMP)、1,2−ビス(アルキルメチルホスフィノ)エタン(BisP*)、2,3−ビス(ジフェニルホスフィノ)ブタン(CHIRAPHOS)、1,2−ビス(ジフェニルホスフィノ)プロパン(PROPHOS)、2,3−ビス(ジフェニルホスフィノ)−5−ノルボルネン(NORPHOS)、2,3−O−イソプロピリデン−2,3−ジヒドロキシ−1,4−ビス(ジフェニルホスフィノ)ブタン(DIOP)、1−シクロヘキシル−1,2−ビス(ジフェニルホスフィノ)エタン(CYCPHOS)、1−置換−3,4−ビス(ジフェニルホスフィノ)ピロリジン(DEGPHOS)、2,4−ビス−(ジフェニルホスフィノ)ペンタン(SKEWPHOS)、1,2−ビス(置換ホスホラノ)ベンゼン(DuPHOS)、1,2−ビス(置換ホスホラノ)エタン(BPE)、1−((置換ホスホラノ)−2−(ジフェニルホスフィノ)ベンゼン(UCAP−Ph)、1−(ビス(3,5−ジメチルフェニル)ホスフィノ)−2−(置換ホスホラノ)ベンゼン(UCAP−DM)、1−((置換ホスホラノ)−2−(ビス(3,5−ジ(t−ブチル)−4−メトキシフェニル)ホスフィノ)ベンゼン(UCAP−DTBM)、1−((置換ホスホラノ)−2−(ジ−ナフタレン−1−イル−ホスフィノ)ベンゼン(UCAP−(1−Nap))、1−[1’,2−ビス(ジフェニルホスフィノ)フェロセニル]エチルアミン(BPPFA)、1−[1’,2−ビス(ジフェニルホスフィノ)フェロセニル]エチルアルコール(BPPFOH)、2,2’−ビス(ジフェニルホスフィノ)−1,1’−ジシクロペンタン(BICP)、2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル(BINAP)、2,2’−ビス(ジフェニルホスフィノ)−1,1’−(5,5’,6,6’,7,7’,8,8’,−オクタヒドロビナフチル)(H8−BINAP)、2,2’−ビス(ジ−p−トリルホスフィノ)−1,1’−ビナフチル(TOL−BINAP)、2,2’−ビス(ジ(3,5−ジメチルフェニル)ホスフィノ)−1,1’−ビナフチル(DM−BINAP)、2,2’−ビス(ジフェニルホスフィノ)−6,6’−ジメチル−1,1’−ビフェニル(BICHEP)、((5,6),(5’,6’)−ビス(メチレンジオキシ)ビフェニル−2,2’−ジイル)(ビスジフェニルホスフィン)(SEGPHOS)、((5,6),(5’,6’)−ビス(メチレンジオキシ)ビフェニル−2,2’−ジイル)(ビス(3,5−ジメチルフェニル)ホスフィン)(DM−SEGPHOS)、((5,6),(5’,6’)−ビス(メチレンジオキシ)ビフェニル−2,2’−ジイル)(ビス(3,5−ジ(tert−ブチル)−4−メトキシフェニル)ホスフィン)(DTBM−SEGPHOS)などが挙げられる。
これらの中でも、エナンチオ選択性がより優れる点から、(S,S)−2,5置換−BPEが好ましく、(S,S)−Ph−BPEがより好ましい。
なお、(S,S)−Ph−BPEは、以下の構造式で表される化合物である。
【化12】
ただし、「Ph」はフェニル基を表す。
【0027】
なお、チオアミドとアルデヒドとのアルドール反応においては、アルデヒドのα位に酸性の水素原子がある場合には、通常の触媒においては、量論的にチオアミドを活性化するため、多量の触媒が必要である。
しかし、前記銅−光学活性ホスフィン錯体を用いることにより、チオアミド(前記一般式(3)で表される化合物)を用いるアルドール反応において、アルデヒドのα位に酸性の水素原子がある場合にも、触媒的にチオアミドを活性化し(言い換えれば、量論的にチオアミドを活性化せず)、前記一般式(1)で表されるチオアミド化合物を製造することができる。
そのため、前記銅−光学活性ホスフィン錯体は、触媒量の使用でよい。前記触媒量としては、前記一般式(2)で表される化合物に対して、1mol%〜9mol%が好ましい。
【0028】
前記反応工程における前記一般式(2)で表される化合物と前記一般式(3)で表される化合物との割合としては、特に制限はなく、目的に応じて適宜選択することができるが、前記一般式(2)で表される化合物に対して、前記一般式(3)で表される化合物が1.0当量〜1.5当量であることが好ましい。
【0029】
前記反応工程において使用される溶媒としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、THF(テトラヒドロフラン)、DMF(N,N−ジメチルホルムアミド)などが挙げられる。
【0030】
前記反応工程における反応温度としては、特に制限はなく、目的に応じて適宜選択することができるが、−60℃〜−40℃が好ましい。
前記反応工程における反応時間としては、特に制限はなく、目的に応じて適宜選択することができるが、5時間〜36時間が好ましい。
【0031】
本発明のチオアミド化合物の製造方法により得られるチオアミド化合物は、立体異性体の混合物として得られる場合もあるが、この場合も本発明の範囲に包含される。
【0032】
本発明の製造方法により得られる前記一般式(1)で表されるチオアミド化合物は、アトルバスタチン合成の有用な中間体である、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を合成するための物質、更にはアトルバスタチンを合成するための物質として有用である。
【0033】
([(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法)
本発明の[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法は、前記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程を含み、更に必要に応じて、その他の工程を含む。
【0034】
<転化する工程>
前記転化する工程は、前記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができる。
【化13】
ただし、前記一般式(4)中、R4は、カルボキシル基の保護基及び水素原子のいずれかを表す。R5及びR6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
【0035】
前記R4におけるカルボキシル基の保護基としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、Greenら、Protective Groups in Organic Synthesis, 3rd Edition,1999,John Wiley & Sons, Inc.などの成書を参照することができる。
前記カルボキシル基の保護基としては、例えば、アルキル基、トリアルキルシリル基などを挙げることができる。
前記アルキル基としては、メチル基、エチル基、tert−ブチル基などが挙げられる。
前記トリアルキルシリル基としては、例えば、トリメチルシリル基、トリエチルシリル基などが挙げられる。
これらの中でも、酸性条件でも脱保護が可能である点からtert−ブチル基が好ましい。
【0036】
前記R5及び前記R6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
炭素数1〜6の炭化水素基としては、例えば、メチル基、エチル基、n−プロピル基、n−ブチル基などが挙げられる。
前記R5及び前記R6が一緒になって形成される環構造としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、脂肪族炭化水素環が挙げられる。前記脂肪族炭化水素環としては、例えば、シクロペンタン環、シクロヘキサン環、シクロヘプタン環などが挙げられる。
【0037】
前記転化する工程について、前記一般式(1)におけるR1が−OR11の場合と、−NR12R13の場合に分けて、その一例を説明する。
【0038】
−転化する工程A(前記一般式(1)におけるR1が−OR11の場合)−
前記転化する工程Aとしては、例えば、下記一般式(1−1)で表されるチオアミド化合物を下記一般式(5)で表される化合物へ転化する工程(工程(A−I))、下記一般式(5)で表される化合物を下記一般式(6)で表される化合物へ転化する工程(工程(A−II))、下記一般式(6)で表される化合物を下記一般式(9)で表される化合物へ転化する工程(工程(A−III))、下記一般式(9)で表される化合物を下記一般式(11)で表される化合物へ転化する工程(工程(A−IV))、及び下記一般式(11)で表される化合物を前記一般式(4)で表されるアセテート誘導体へ転化する工程(工程(A−V))をこの順で行うことが挙げられる。
【0039】
前記転化する工程Aを前記工程(A−I)〜工程(A−V)で行う場合には、前記工程(A−I)〜工程(A−V)をこの順で行う際に、他の工程を加えてもよい。
また、前記工程(A−I)〜工程(A−V)のいずれかの工程を、これら以外のその他の工程に代えて行ってもよい。
【0040】
−−工程(A−I)−−
前記工程(A−I)は、下記一般式(1−1)で表されるチオアミド化合物を下記一般式(5)で表される化合物へ転化する工程である。
【化14】
ただし、前記一般式(1−1)中、R2、R3及びR11は、前記一般式(1)中のR2、R3及びR11とそれぞれ同じである。前記一般式(5)中、R2、R3及びR11は、前記一般式(1−1)中のR2、R3及びR11とそれぞれ同じである。前記R7は、水酸基の保護基を表す。
前記R7としては、水酸基の保護基であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記一般式(1)における前記R11の説明で例示した保護基が挙げられる。
前記R11と前記R7の水酸基の保護基の組合せとしては、特に制限はなく、目的に応じて適宜選択することができるが、前記R7の基が前記R11の基よりも脱保護し易いことが好ましい。そのような組合せとしては、前記R11がベンジル基であり前記R7がtert−ブチルジメチルシリル(TBS)基であることが、選択的に前記R7を脱保護できる点で好ましい。
【0041】
前記工程(A−I)としては、前記一般式(1−1)中の水酸基の水素原子を前記R7基に変換できる限り、特に制限はなく、目的に応じて適宜選択することができ、例えば、水酸基を保護する公知の方法に従って行うことができる。
【0042】
前記工程(A−I)おいて、前記一般式(1−1)中の水酸基の水素原子をtert−ブチルジメチルシリル(TBS)基に置換する場合には、塩基存在下で反応を行うことが好ましい。前記塩基としては、特に制限はなく、目的に応じて適宜選択することができるが、求核性が低い塩基が好ましく、2,6−ルチジンがより好ましい。
【0043】
−−工程(A−II)−−
前記工程(A−II)は、前記一般式(5)で表される化合物を下記一般式(6)で表される化合物へ転化する工程である。
【化15】
ただし、前記一般式(6)中、R7及びR11は、前記一般式(5)中のR7及びR11とそれぞれ同じである。前記一般式(6)中、R4は、前記一般式(4)中のR4と同じである。
【0044】
前記工程(A−II)としては、チオアミドをβケトエステルに変換できる限り、特に制限はなく、目的に応じて適宜選択することができ、例えば、Sメチル化に続く、相当するリチウムエノラートの付加反応を用いて行うことができる。また、前記工程(A−II)は、Yuichiro Mutoh, Toshiaki Murai,Organic Letters 2003,Vol5,No.8,1361−1364.を参考にして行うことができる。
【0045】
−−工程(A−III)−−
前記工程(A−III)としては、前記一般式(6)で表される化合物を下記一般式(9)で表される化合物へ転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記R7の脱保護の後にジアステレオ選択的還元を行い、更にsyn−1,3−ジオールに対して保護基を導入し1,3−ジオキサン環を形成する方法などが挙げられる。
【化16】
ただし、前記一般式(9)中、R4及びR11は、前記一般式(6)中のR4及びR11とそれぞれ同じである。前記一般式(9)中、R5及びR6は、前記一般式(4)中のR5及びR6とそれぞれと同じである。
【0046】
前記R7の脱保護の後にジアステレオ選択的還元を行い、更にsyn−1,3−ジオールに対して保護基を導入し1,3−ジオキサン環を形成する方法としては、例えば、前記一般式(6)で表される化合物を下記一般式(7)で表される化合物に変換する工程(工程(A−III−1))、下記一般式(7)で表される化合物を下記一般式(8)で表される化合物に変換する工程(工程(A−III−2))、及び下記一般式(8)で表される化合物を前記一般式(9)で表される化合物に変換する工程(工程(A−III−3))をこの順で行う方法が挙げられる。
【化17】
ただし、前記一般式(7)、及び一般式(8)中、R4及びR11は、前記一般式(6)中のR4及びR11とそれぞれ同じである。
【0047】
前記工程(A−III−1)としては、前記一般式(6)中の前記R7を水素原子に置換する、即ち、水酸基の保護基である前記R7を脱保護する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記R7がトリアルキルシリル基(例えば、tert−ブチルジメチルシリル(TBS)基)の場合には、酸性条件、又はフッ化物イオンによって脱保護する方法が挙げられる。前記フッ化物イオンによって脱保護する場合には、例えば、テトラブチルアンモニウムフルオリド(TBAF)、フッ化水素酸(HF)、フッ化セシウム(CsF)などを用いることができる。
【0048】
前記工程(A−III−2)としては、前記一般式(7)中のカルボニル基を水酸基に還元することができる工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、LiAlH4、NaBH4などの還元剤を用いる工程が挙げられる。前記NaBH4を用いる場合には、通常、メタノール、エタノールなどのアルコール存在下で還元が行われる。なお、前記工程(A−III−2)においては、コンフォメーションを制御することでsyn選択性を発現することができる。コンフォメーションを制御する方法としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、トリエチルボラン、ジエチルメトキシボランなどによるアルコールとケトン部位のキレート形成によりコンフォメーションを制御することが挙げられる。
【0049】
前記工程(A−III−3)としては、前記一般式(8)中の光学活性syn−1,3ジオールを、前記一般式(9)中の1,3−ジオキサン環に変換する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、2,2−ジメトキシプロパン、2−メトキシ−2−プロペン、アセトン、3,3−ジメトキシペンタン、シクロヘキサノン、シクロペンタノンなどを用いる方法が挙げられる。
【0050】
前記工程(A−III)において、前記工程(A−III−1)及び工程(A−III−2)は、精製をすることなく行い、前記工程(A−III−3)で精製を行い、前記一般式(9)で表される化合物を得てもよい。
【0051】
−−工程(A−IV)−−
前記工程(A−IV)としては、前記一般式(9)で表される化合物を下記一般式(11)で表される化合物へ転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができる。
【化18】
ただし、前記一般式(11)中、R4、R5及びR6は、前記一般式(9)中のR4、R5及びR6とそれぞれ同じである。R8は、アジ基に変換可能な基を表す。
【0052】
前記R8としては、アジ基に変換可能な基であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、−OR10(R10は、Ts基(トシル基)、Ms基(メシル基)、Tf基(トリフルオロメタンスルホニル基)等)、ヨウ素原子などが挙げられる。
【0053】
前記工程(A−IV)としては、例えば、前記一般式(9)で表される化合物を下記一般式(10)で表される化合物に変換する工程(工程(A−IV−1))の後に、続いて、下記一般式(10)で表される化合物を前記一般式(11)で表される化合物に変換する工程(工程(A−IV−2))などが挙げられる。
【化19】
ただし、前記一般式(10)中、R4、R5及びR6は、前記一般式(9)中のR4、R5及びR6とそれぞれ同じである。
【0054】
前記工程(A−IV−1)としては、前記一般式(9)中の前記R11を水素原子に置換する、即ち、水酸基の保護基R11を脱保護する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、従来公知の水酸基の脱保護の方法により行うことができる。前記R11が、ベンジル基の場合には、前記工程(A−IV−1)としては、例えば、水酸化パラジウム/炭素(パールマン触媒)などのパラジウム炭素(Pd/C)を用いてベンジル基を脱保護する方法が挙げられる。
【0055】
前記工程(A−IV−2)としては、前記一般式(10)中の水酸基をアジ基に変換可能な基(例えば、−OR10(R10はトシル基))に置換する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、パラトルエンスルホニルクロリドを用いて水酸基をトシル化する方法などが挙げられる。
【0056】
前記工程(A−IV)において、前記工程(A−IV−1)は、精製をすることなく行い、前記工程(A−IV−2)で精製を行い、前記一般式(11)で表される化合物を得てもよい。
【0057】
−−工程(A−V)−−
前記工程(A−V)は、前記一般式(11)で表される化合物を下記一般式(4)で表されるアセテート誘導体へ転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができる。
【化20】
【0058】
前記工程(A−V)としては、例えば、前記一般式(11)で表される化合物を下記一般式(12)で表される化合物に変換する工程(工程(A−V−1))の後に、続いて、下記一般式(12)で表される化合物を前記一般式(4)で表されるアセテート誘導体に変換する工程(工程(A−V−2))が挙げられる。
【化21】
ただし、前記一般式(12)中、R4、R5及びR6は、前記一般式(11)中のR4、R5及びR6とそれぞれ同じである。
【0059】
前記工程(A−V−1)としては、前記一般式(11)中のR8(アジ基に変換可能な基)をアジ基(N3−)に置換する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、−OR10(R10はトシル基)をアジ基に置換する場合、求核剤としてのアジ化ナトリウムを反応させ、トシラートアニオン(TsO−)を脱離する求核置換反応を起こすことにより、アジ基を生成させる方法などが挙げられる。
【0060】
前記工程(A−V−2)としては、前記一般式(12)中のアジ基(N3−)を1級アミノ基に置換する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、トリフェニルホスフィンを用いたシュタウディンガー還元を用いる方法が挙げられる。
【0061】
前記工程(A−V)において、前記工程(A−V−1)は、精製をすることなく行い、前記工程(A−V−2)で精製を行い、前記一般式(4)で表されるアセテート誘導体を得てもよい。
【0062】
−転化する工程B(前記一般式(1)におけるR1が−NR12R13の場合)−
前記転化する工程Bとしては、例えば、下記一般式(1−2)で表されるチオアミド化合物を下記一般式(13)で表される化合物へ転化する工程(工程(B−I))、下記一般式(13)で表される化合物を下記一般式(14)で表される化合物へ転化する工程(工程(B−II))、下記一般式(14)で表される化合物を下記一般式(17)で表される化合物へ転化する工程(工程(B−III))、及び下記一般式(17)で表される化合物を前記一般式(4)で表されるアセテート誘導体へ転化する工程(工程(B−IV))をこの順で行うことが挙げられる。
【0063】
前記転化する工程Bを前記工程(B−I)〜工程(B−IV)で行う場合には、前記工程(B−I)〜工程(B−IV)を順で行う際に、他の工程を加えてもよい。
また、前記工程(B−I)〜工程(B−IV)のいずれかの工程を、これら以外のその他の工程に代えて行ってもよい。
【0064】
−−工程(B−I)−−
前記工程(B−I)は、下記一般式(1−2)で表されるチオアミド化合物を下記一般式(5)で表される化合物へ転化する工程である。
【化22】
ただし、前記一般式(1−2)中、R2、R3、R12及びR13は、前記一般式(1)中のR2、R3、R12及びR13とそれぞれ同じである。前記一般式(13)中、R2、R3、R12及びR13は、前記一般式(1−2)中のR2、R3、R12及びR13とそれぞれ同じである。前記R9は、水酸基の保護基を表す。
【0065】
前記R9としては、水酸基の保護基であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記一般式(1)における前記R11の説明で例示した保護基などが挙げられる。
【0066】
前記工程(B−I)としては、前記一般式(1−2)中の水酸基の水素原子を前記R9基に変換できる限り、特に制限はなく、目的に応じて適宜選択することができ、例えば、水酸基を保護する公知の方法に従って行うことができる。
【0067】
前記工程(B−I)おいて、前記一般式(1−2)中の水酸基の水素原子をtert−ブチルジメチルシリル(TBS)基に置換する場合には、塩基存在下で、反応を行うことが好ましい。前記塩基としては、特に制限はなく、目的に応じて適宜選択することができるが、求核性が低い塩基が好ましく、2,6−ルチジンがより好ましい。
【0068】
−−工程(B−II)−−
前記工程(B−II)は、下記一般式(13)で表される化合物を下記一般式(14)で表される化合物へ転化する工程である。
【化23】
ただし、前記一般式(14)中、R9、R12及びR13は、前記一般式(13)中のR9、R12及びR13とそれぞれ同じである。前記一般式(14)中、R4は、前記一般式(4)中のR4と同じである。
【0069】
前記工程(B−II)としては、チオアミドをβケトエステルに変換できる限り、特に制限はなく、目的に応じて適宜選択することができ、例えば、Sメチル化に続く、相当するリチウムエノラートの付加反応を用いて行うことができる。また、前記工程(B−II)は、Yuichiro Mutoh, Toshiaki Murai,Organic Letters 2003,Vol5,No.8,1361−1364.を参考にして行うことができる。
【0070】
−−工程(B−III)−−
前記工程(B−III)としては、前記一般式(14)で表される化合物を下記一般式(17)で表される化合物へ転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記R9の脱保護の後にジアステレオ選択的還元を行い、更にsyn−1,3−ジオールに対して保護基を導入し1,3−ジオキサン環を形成する方法などが挙げられる。
【化24】
ただし、前記一般式(17)中、R4、R12及びR13は、前記一般式(6)中のR4、R12及びR13とそれぞれ同じである。前記一般式(17)中、R5及びR6は、前記一般式(4)中のR5及びR6とそれぞれと同じである。
【0071】
前記R9の脱保護の後にジアステレオ選択的還元を行い、更にsyn−1,3−ジオールに対して保護基を導入し1,3−ジオキサン環を形成する方法としては、例えば、前記一般式(14)で表される化合物を下記一般式(15)で表される化合物に変換する工程(工程(B−III−1))、下記一般式(15)で表される化合物を下記一般式(16)で表される化合物に変換する工程(工程(B−III−2))、及び下記一般式(16)で表される化合物を下記一般式(17)で表される化合物に変換する工程(工程(B−III−3))をこの順で行う方法などが挙げられる。
【化25】
ただし、前記一般式(15)、及び一般式(16)中、R4、R12及びR13は、前記一般式(14)中のR4、R12及びR13とそれぞれ同じである。
【0072】
前記工程(B−III−1)としては、前記一般式(14)中の前記R9を水素原子に置換する、即ち、水酸基の保護基である前記R9を脱保護する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記R9がトリアルキルシリル基(例えば、tert−ブチルジメチルシリル(TBS)基)の場合には、酸性条件、又はフッ化物イオンによって脱保護する方法が挙げられる。前記フッ化物イオンによって脱保護する場合には、例えば、テトラブチルアンモニウムフルオリド(TBAF)、フッ化水素酸(HF)、フッ化セシウム(CsF)などを用いることができる。
【0073】
前記工程(B−III−2)としては、前記一般式(15)中のカルボニル基を水酸基に還元することができる工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、LiAlH4、NaBH4などの還元剤を用いる工程が挙げられる。前記NaBH4を用いる場合には、通常、メタノール、エタノールなどのアルコール存在下で還元が行われる。なお、前記工程(B−III−2)においては、コンフォメーションを制御することでsyn選択性を発現することができる。コンフォメーションを制御する方法としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、トリエチルボラン、ジエチルメトキシボランなどによるアルコールとケトン部位のキレート形成によりコンフォメーションを制御することが挙げられる。
【0074】
前記工程(B−III−3)としては、前記一般式(16)中の光学活性syn−1,3ジオールを、前記一般式(17)中の1,3−ジオキサン環に変換する工程であれば、特に制限はなく、目的に応じて適宜選択することができ、例えば、2,2−ジメトキシプロパン、2−メトキシ−2−プロペン、アセトン、3,3−ジメトキシペンタン、シクロヘキサノン、シクロペンタノンなどを用いる方法が挙げられる。
【0075】
前記工程(B−III)において、前記工程(B−III−1)及び工程(B−III−2)は、精製をすることなく行い、前記工程(B−III−3)で精製を行い、前記一般式(17)で表される化合物を得てもよい。
【0076】
−−工程(B−IV)−−
前記工程(B−IV)としては、前記一般式(17)で表される化合物を前記一般式(4)で表されるアセテート誘導体へ転化する工程であれば、特に制限はなく、目的に応じて適宜選択することができる。
【化26】
【0077】
前記工程(B−IV)において、前記NR12R13をNH2に変換する、即ち、アミノ基の保護基を脱保護する方法としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、前記R12及び前記R13が、一緒になってフタロイル基を形成している場合には、メチルアミン、ヒドラジン等を作用させることで脱保護する方法などが挙げられる。
【0078】
本発明の[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法は、アトルバスタチンの製造に有用な[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を効率よく安価に製造することができる。
【0079】
(アトルバスタチンの製造方法)
前記アトルバスタチンの製造方法は、前記一般式(1)で表されるチオアミド化合物を前記一般式(4)で表されるアセテート誘導体に転化する工程を少なくとも含み、更に必要に応じて、その他の工程を含む。
【0080】
本発明においてアトルバスタチンとは、アトルバスタチン((3R,5R)−7−[2−(4−フルオロフェニル)−5−イソプロピル−3−フェニル−4−フェニルカルバモイル−1H−ピロル−1−イル]−3,5−ジヒドロキシヘプタン酸)に加え、その製薬学的に許容される塩を包含する。
【0081】
前記塩としては、例えば、ナトリウム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アンモニウム塩;トリアルキルアミン塩等の有機塩基塩などが挙げられる。
前記アトルバスタチンとしては、アトルバスタチンのアルカリ土類金属塩が好ましく、アトルバスタチンカルシウムがより好ましい。
【0082】
<転化する工程>
前記転化する工程としては、本発明の前記[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法に記載した転化する工程が挙げられる。
【0083】
<その他の工程>
前記その他の工程としては、例えば、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体から、アトルバスタチン(特にアトルバスタチンカルシウム)を製造する工程が挙げられる。
【0084】
【化27】
【0085】
前記[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体、即ち、前記一般式(4)で表されるアセテート誘導体から、アトルバスタチンカルシウムを製造する工程としては、特に制限はなく、目的に応じて適宜選択することができ、例えば、Kelvin L.Baumann,et al.,Tetrahedron Letters,Vol.33,No.17,pp.2283−2284,1992に記載の方法などが挙げられる。
【0086】
本発明のアトルバスタチンの製造方法は、アトルバスタチンを効率よく且つ安価に製造することができる。
【実施例】
【0087】
以下に本発明の実施例を挙げて本発明を具体的に説明するが、本発明はこれらの実施例に何ら限定されるものではない。
なお、以下の実施例において、「THF」は「テトラヒドロフラン」を表す。「DMF」は「N,N−ジメチルホルムアミド」を表す。「Bn」は「ベンジル基」を表す。「TBS」は「tert−ブチルジメチルシリル基」を表す。「Ts」は「トシル基」を表す。
【0088】
(実施例1)
<チオアミド化合物A−1の合成1>
<<化合物X(N,N−ジアリルチオアセトアミド)の合成>>
300mLナスフラスコにN,N−ジアリルアセトアミド(5.0g、36mmol、1当量、Stanislaw Krompiec, et al.,Journal of Molecular Catalysis A: Chemical 2005,Vil 225,No.1,91−101.に従って合成)、ローソン試薬(7.3g、18mmol、0.5当量)、及び乾燥THF(180mL)を順次加えて12時間加熱還流した後、室温に戻して1規定塩酸(3mL)加えた。得られた二相混合物を酢酸エチル(20mL)で抽出し、有機層を飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られた残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=10/1(体積比))で精製し、下記化合物X(N,N−ジアリルチオアセトアミド)を淡黄色油状物として得た。収量4.8g(収率86%)。
【化28】
【0089】
<<2,2,5,7,8−ペンタメチルクロマノールリチウム塩の合成>>
加熱真空乾燥した5mLナスフラスコに2,2,5,7,8−ペンタメチルクロマノール(88.1mg、0.40mmol)を加え1時間減圧乾燥した後、フラスコ内をアルゴン雰囲気にして乾燥テトラヒドロフラン(THF)(2mL)を加えた。フラスコをアセトン−ドライアイス浴で−78℃に冷却し、n−ブチルリチウム(247μL、0.40mmol、1.62Mへキサン溶液)をゆっくりと加え、同温度で1時間撹拌し、2,2,5,7,8−ペンタメチルクロマノールリチウム塩の0.2M THF溶液を得た。
【0090】
<<Cu/(S,S)−Ph−BPEの合成>>
加熱真空乾燥した5mLナスフラスコに[Cu(CH3CN)4]PF6(Strem社製、149.0mg、0.40mmol)と(S,S)−Ph−BPE(Strem社製、202.5mg、0.40mmol)をグローブボックス内で加えた後、アルゴン雰囲気下にてTHF(4mL)を加えてCu/(S,S)−Ph−BPE(銅−光学活性ホスフィン錯体)の0.1M THF溶液を得た。
【0091】
<<チオアミド化合物A−1の合成>>
加熱真空乾燥した100mLナスフラスコにN,N−ジメチルホルムアミド(DMF)(60mL)、3−(ベンジルオキシ)プロパナール(1.0g、6.09mmol、1当量、Amanda M.Heapy, Margaret A. Brimble, Tetrahedron,Vol.66,No.29,5424−5431.に従って合成)、N,N−ジアリルチオアセトアミド(上記化合物X、1.13g、7.30mmol、1.2当量)、上記で調製したCu/(S,S)−Ph−BPE溶液(3.65mL、0.365mmol、0.06当量)をアルゴン雰囲気下加えた。ナスフラスコを−40℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩のTHF溶液(1.83mL、0.365mmol、0.06当量)を加え、−40℃で36時間撹拌した後、飽和塩化アンモニウム水溶液(30mL)と2,2−ビピリジン(57mg、0.365mmol、0.06当量)を加え、その混合溶液を酢酸エチル(20mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=5/1〜2/1(体積比))で精製し、下記チオアミド化合物A−1を黄色油状物として得た。収量1.64g(収率84%、92%ee)。
【化29】
【0092】
得られたチオアミド化合物A−1のIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν3413, 3085, 2919, 2861, 1643, 1496, 1411cm−1
1H NMR(CDCl3) δ7.34−7.29(m,5H), 5.86(dddd,J=5.7, 5.7, 10.3, 17.2Hz,1H), 5.73(dddd,J=5.0, 5.0, 10.3, 17.2Hz,1H), 5.26−5.09(m,4H), 4.75(dd,J=5.7, 14.7Hz,1H), 4.54−4.50(m,1H), 4.51(s,2H), 4.41−4.35(m,1H), 4.32−4.25(m,1H), 4.10−4.04(m,1H), 3.71−3.68(m,2H), 2.84(dd,J=2.8, 15.4Hz,1H), 2.76(dd,J=8.7, 15.4Hz,1H), 1.91−1.79(m,2H)
13CNMR(CDCl3) δ202.5, 138.1, 130.5, 130.5,130.5, 128.3, 127.6, 127.5, 118.4, 117.7, 73.1, 69.0, 67.9, 55.5, 52.8, 48.3, 36.3
[α]D22−33.9(c0.33,CHCl3,92%eesample)
ESI−MS m/z 342.2 [M+Na]+
HRMS(ESI) Anal.calcd. for C18H25NNaO2S m/z 342.1498 [M+Na]+,found;342.1500.
【0093】
(実施例2)
<チオアミド化合物A−1の合成2>
触媒の種類を変えて、チオアミド化合物A−1を合成した。具体的には以下のとおりである。
加熱真空乾燥した20mLナスフラスコに[Cu(CH3CN)4]PF6(分子量372.72)(6.7mg、0.018mmol、0.09当量)と(S)−BINAP(Strem社製、分子量622.67、11.2mg、0.018mmol、0.09当量)をグローブボックス内で加え、アルゴン雰囲気下THF(180μL)を加えて5分間撹拌した後、DMF(2mL)、3−(ベンジルオキシ)プロパナール(32.8μL、0.20mmol、1当量)、及びN,N−ジアリルチオアセトアミド(上記化合物X、38.2μL、0.24mmol、1.2当量)を加えた。試験管を−60℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩の0.2M THF溶液(90μL、0.018mmol、0.09当量)を加え、−60℃で60時間撹拌した後、THF/酢酸混合溶液(10:1(体積比))(2mL)と2,2−ビピリジン(2.8mg、0.018mmol、0.09当量)を加えて反応を停止した。蒸留水(2mL)を加えた後、混合溶液を酢酸エチル(2mL)で3回抽出した。得られた有機層を飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥し、チオアミド化合物A−1(NMR収率25%、−10%ee)を得た。
【0094】
(実施例3)
<チオアミド化合物A−1の合成3>
触媒の種類を変えて、チオアミドチオアミド化合物A−1を合成した。具体的には以下のとおりである。
加熱真空乾燥した20mLナスフラスコに[Cu(CH3CN)4]PF6(分子量372.72)(6.7mg、0.018mmol、0.09当量)と(R,R)−iPr−DuPhos(分子量418.58)(7.5mg、0.018mmol、0.09当量)をグローブボックス内で加え、アルゴン雰囲気下THF(180μL)を加えて5分間撹拌した後、DMF(2mL)、3−(ベンジルオキシ)プロパナール(32.8μL、0.20mmol、1当量)、及びN,N−ジアリルチオアセトアミド(上記化合物X、38.2μL、0.24mmol、1.2当量)を加えた。試験管を−60℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩の0.2M THF溶液(90μL、0.018mmol、0.09当量)を加え、−60℃で60時間撹拌した後、THF/酢酸混合溶液(10:1(体積比))(2mL)と2,2−ビピリジン(2.8mg、0.018mmol、0.09当量)を加えて反応を停止した。蒸留水(2mL)を加えた後、混合溶液を酢酸エチル(2mL)で3回抽出した。得られた有機層を飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥し、チオアミド化合物A−1(NMR収率52%,−46%ee)を得た。
【0095】
(実施例4)
<チオアミド化合物A−2の合成>
チオアミド化合物A−1とは保護基の種類が異なるチオアミド化合物A−2を合成した。具体的に以下のとおりである。
加熱真空乾燥した20mL試験管にDMF(2mL)、3−(ベンジルオキシ)プロパナール(33μL、0.20mmol、1当量、Amanda M.Heapy, Margaret A. Brimble, Tetrahedron,Vol.66,No.29,5424−5431.に従って合成)、N,N−ジメチルチオアセトアミド(Alfa Aesar社製、24.5g、0.24mmol、1.2当量)、上記で調製したCu/(S,S)−Ph−BPE溶液(120μL、0.012mmol、0.06当量)をアルゴン雰囲気下加えた。ナスフラスコを−40℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩のTHF溶液(60μL、0.012mmol、0.06当量)を加え、−40℃で40時間撹拌した後、飽和塩化アンモニウム水溶液(2mL)と2,2−ビピリジン(1.8mg、0.012mmol、0.06当量)を加え、その混合溶液を酢酸エチル(3mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=5/1〜1/1(体積比))で精製し、下記チオアミド化合物A−2を黄色油状物として得た。収量12.8mg(収率24%、90%ee)。
【化30】
【0096】
得られたチオアミド化合物A−2のIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν3406, 2960, 2871, 1523cm−1
1H NMR(CDCl3) δ7.36−7.27(m,5H), 4.52(s,2H), 4.37−4.34(m,1H), 3.71−3.69(m,2H), 3.46(s,3H), 3.28(s,3H), 2.79(dd,J=2.5, 15.8Hz,1H), 2.72(dd,J=8.9, 15.8Hz,1H), 1.91−1.84(m,2H)
13C NMR(CDCl3) δ202.5, 138.5, 128.3, 127.6, 127.5, 73.1, 69.0, 67.8, 52.9, 44.6, 44.5, 41.9
[α]D22−34.8(c0.13,CHCl3,90%ee)
ESI−MS m/z 280.2 [M+Na]+
HRMS (ESI) Anal.calcd. for C14H21NNaO2S m/z 290.1185 [M+Na]+,found;290.1186.
【0097】
(実施例5)
<チオアミド化合物A−3の合成>
チオアミド化合物A−1とは保護基の種類が異なるチオアミド化合物A−3を合成した。具体的に以下のとおりである。
加熱真空乾燥した20mL試験管にDMF(2mL)、3−(tert−ブチルジメチルシリルオキシ)プロパナール(37μL、0.20mmol、1当量、Christian U. Gruenanger, Bernhard Breit, Angewandte Chemie, International Edition, 2010, Vol 49, No.5, 967−970.に従って合成)、上記で調製したN,N−ジアリルチオアセトアミド(38μL、0.24mmol、1.2当量)、上記で調製したCu/(S,S)−Ph−BPE溶液(180μL、0.018mmol、0.09当量)をアルゴン雰囲気下加えた。ナスフラスコを−40℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩のTHF溶液(90μL、0.018mmol、0.09当量)を加え、−40℃で40時間撹拌した後、飽和塩化アンモニウム水溶液(2mL)と2,2−ビピリジン(2.8mg、0.018mmol、0.06当量)を加え、その混合溶液を酢酸エチル(3mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=5/1〜2/1(体積比))で精製し、下記チオアミド化合物A−3を黄色油状物として得た。収量35.7mg(収率52%、45%ee)。
【化31】
【0098】
得られたチオアミド化合物A−3のIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν3421, 2927, 2858, 1643, 1492, 1411, 1250cm−1
1H NMR (CDCl3) δ5.92(dddd,J=5.7, 5.7, 10.3, 17.2Hz,1H), 5.82(dddd,J=5.5, 5.5, 10.3, 17.2Hz,1H), 5.29−5.15(m,4H), 4.77(dd,J=5.5, 14.9Hz,1H), 4.47(dd,J= 6.0, 14.9Hz,1H), 4.37−4.32(m,2H), 4.14−4.08(m,1H), 3.87−3.78(m,2H), 2.86−2.81(m,2H), 1.81−1.68(m,2H), 0.84(s,9H), 0.04(s,6H)
13C NMR(CDCl3) δ202.7, 130.7, 118.4, 117.8, 69.5, 61.2, 55.7, 53.4, 48.7, 32.2, 25.9, 18.2, −5.5
[α]D22−17.2(c0.17,CHCl3,45%ee)
ESI−MS m/z 366.2 [M+Na]+
HRMS(ESI) Anal.calcd. for C17H33NNaO2SSi m/z 366.1893 [M+Na]+,found;366.1892.
【0099】
(実施例6)
<チオアミド化合物A−4の合成>
チオアミド化合物A−4の合成を行った。
加熱真空乾燥した50mLナスフラスコにDMF(13mL)、3−フタルイミジルプロパナール(Aurora Fine Chemicals社製、266.0mg、1.13mmol、1当量)、N,N−ジアリルチオアセトアミド(244μL、1.35mmol、1.2当量)、上記で調製したCu/(S,S)−Ph−BPE溶液(100μL、0.10mmol、0.09当量)をアルゴン雰囲気下加えた。ナスフラスコを−40℃の恒温槽へ移し、上記で調製した2,2,5,7,8−ペンタメチルクロマノールリチウム塩のTHF溶液(50μL、0.10mmol、0.09当量)を加え、−40℃で24時間撹拌した後、飽和塩化アンモニウム水溶液(5mL)と2,2−ビピリジン(15.6mg、0.10mmol、0.09当量)を加え、その混合溶液を酢酸エチル(5mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=7/1〜2/1(体積比))で精製し、下記チオアミド化合物A−4を黄色油状物として得た。収量251.1mg(収率62%、75%ee)。
【化32】
【0100】
得られたチオアミド化合物A−4のIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν 3463, 3086, 2923, 2865, 1770, 1712, 1612, 1492, 1396 cm−1
1H NMR(CDCl3) δ7.85−7.83 (m,2H), 7.72−7.71 (m,2H), 5.86(dddd,J=6.4, 6.4, 10.7, 17.2Hz,1H), 5.78(dddd,J=4.6 , 4.6, 10.7, 17.2Hz,1H), 5.30−5.12(m,4H), 4.78(dd,J=4.8, 14.9Hz,1H), 4.47(dd,J=5.7, 14.9Hz,1H), 4.36−4.30(m,1H), 4.27−4.08(m,3H), 3.91−3.84(m,2H), 2.78−2.76(m,2H), 1.90−1.85(m,2H)
13C NMR(CDCl3) δ202.1, 168.5, 133.9, 132.0, 130.6, 130.5, 123.2, 118.5, 118.0, 68.0, 55.7, 53.0, 47.8, 35.2, 34.7
[α]D22−11.4(c0.55,CHCl3,75%ee sample)
ESI−MS m/z 381.4 [M+Na]+
HRMS (ESI) Anal.calcd. for C19H22N2NaO3S m/z 381.1243 [M+Na]+,found;381.1243.
【0101】
(実施例7)
<tert−ブチル[(4R,6R)−6−アミノエチル−2,2−ジメチル−1,3−ジオキサン−4−イル]アセテートの合成1>
<<化合物Bの合成>>
加熱真空乾燥した100mLナスフラスコにアルゴン雰囲気下で、実施例1で合成したチオアミド化合物A−1(800mg、2.5mmol、1当量)、塩化メチレン(30mL)を加え、氷浴で0℃に冷却した。2,6−ルチジン(575μL、5.0mmol、2当量)、tert−ブチルジメチルシリル(TBS)トリフラート(860μL、3.75mmol、1.5当量)を加え、室温にて3時間撹拌後、飽和塩化アンモニウム水溶液(20mL)を加えて反応を停止し、その混合溶液を塩化メチレン(10mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、化合物Bを淡黄色油状物として得た。収量1.04g(収率96%)。
【化33】
【0102】
得られた化合物BのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2954, 2858, 1643, 1493, 1257, 1115cm−1
1H NMR(CDCl3) δ7.33−7.29(m,5H), 5.92(dddd,J=5.8, 5.8, 10.8, 16.7Hz,1H), 5.75(dddd,J=4.8, 4.8, 10.3, 17.2Hz,1H), 5.25−5.08(m,4H), 4.83(dd,J=5.7, 14.2Hz,1H), 4.61−4.53(m,2H), 4.48(dd,J=3.0, 15.1Hz,1H), 4.34(dd,J=6.9, 14.2Hz,1H), 4.00−3.95(m,1H), 3.64−3.51(m,2H), 3.10(dd,J=8.2, 13.8Hz,1H), 2.83(dd,J=4.1, 13.8Hz,1H), 1.90−1.86(m,2H), 0.84(s,9H), 0.06(s,3H), 0.02(s,3H)
13C NMR(CDCl3) δ203.0, 138.6, 131.5, 131.3, 128.3, 127.6, 127.5, 119.3, 117.6, 73.0, 72.1, 66.5, 56.4, 53.1, 50.2, 37.4, 25.9, 25.7, 25.6, 17.9, −4.3, −4.8
[α]D22+6.8(c1.0,CHCl3)
ESI−MS m/z 456.3 [M+Na]+
HRMS(ESI) Anal.calcd. for C24H39NNaO2SSi m/z 456.2363 m/z [M+Na]+,found;456.2358.
【0103】
<<化合物Cの合成>>
−酢酸tert−ブチルエステルリチウムエノラートの調製−
加熱真空乾燥した20mLナスフラスコにアルゴン雰囲気下で酢酸tert−ブチルエステル(1.0mL、7.46mmol、1当量)、リチウムヘキサメチルジシラジド(1.25g、7.46mmol、1当量)を加えてアセトン−ドライアイス浴で−78℃に冷却した。−78℃に冷却した乾燥THF(7.46mL)をゆっくりと滴下後、同温度にて1時間撹拌し、酢酸tert−ブチルエステルリチウムエノラートの1.0M THF溶液を得た。
【0104】
−化合物Cの合成−
加熱真空乾燥した100mLナスフラスコにアルゴン雰囲気下で化合物B(920mg、2.12mmol、1当量)とジエチルエーテル(23mL)を加え、氷浴で0℃に冷却した。メチルトリフラート(467μL、4.24mmol、2当量)を加えて0℃にて5分間、室温にて4時間半撹拌後、フラスコをアセトン−ドライアイス浴で−78℃に冷却し、上記で調製した酢酸tert−ブチルエステルリチウムエノラート(64μL、6.40mmol、3当量)をゆっくりと滴下して加えた。3時間撹拌後、塩化メチレン(7mL)、シリカゲル(5g)を加え、室温にて1時間半撹拌した。混合物をシリカゲルショートパッドカラムに通し、濾液を濃縮して得られた残渣にTHF(20mL)、1規定塩酸(2mL)を加え室温にて2時間撹拌後、混合溶液を酢酸エチル(20mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られた残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=20/1(体積比))で精製し、下記化合物Cを無色油状物として得た。収量662mg(収率72%)。
【化34】
【0105】
得られた化合物CのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2954, 2931, 2858, 1739, 1716, 1647, 1458, 1369, 1254, 1146, 1115cm−1
1H NMR(CDCl3);
keto form;δ7.36−7.28(m,5H), 4.50(s,2H), 4.50−4.43(m,1H), 3.56−3.48(m,2H), 3.34(s,2H),2.69(d,J=6.4Hz,2H), 1.81−1.77(m,2H), 1.45(s,9H), 0.85(s,9H), 0.05(s,3H), 0.03(s,3H);
enol form;δ12.15(brs,1H), 7.36−7.28(m,5H),4.90(s,1H), 4.33(s,2H), 4.35−4.31(m,1H), 3.56−3.48(m,2H), 2.29(d,J=6.6Hz,2H), 1.77−1.73(m,2H), 1.47(s,9H), 0.87(s,9H), 0.05(s,3H), 0.02(s,3H)
13C NMR(CDCl3) keto−enol mixture;δ202.1, 166.3, 138.4, 128.3, 127.6, 127.5, 113.9, 92.8, 81.8, 80.6, 73.0, 73.0, 72.9, 67.2, 66.6, 66.4, 52.0, 50.3, 43.8, 37.3, 37.2, 28.6, 28.3, 28.0, 25.8, 17.9, −4.7, −4.8, −4.9
[α]D22−7.6(c0.5,CHCl3)
ESI−MS m/z 459.3 [M+Na]+
HRMS(ESI) Anal.calcd. for C24H40NaO5Si m/z 459.2537 [M+Na]+,found;459.2534.
【0106】
<<化合物D、E、及びFの合成>>
30mLナスフラスコにアルゴン雰囲気下、化合物C(400mg、0.916mmol、1当量)と乾燥THF(2.5mL)を入れ、氷浴で0℃に冷却した。続いてテトラブチルアンモニウムフルオリド(1.3mL、1.30mmol、1.4当量、1.0M THF溶液)をゆっくりと滴下し、0℃で30分間、室温にて3時間撹拌後、蒸留水(2mL)を加え、混合液を酢酸エチル(5mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Dの粗生成物を淡黄色油状物質として得た。
続いて、50mLナスフラスコに上記で得られた化合物D(292mg、0.906mmol、1当量)、乾燥THF(8.5mL)、メタノール(2.4mL)を入れ、ナスフラスコを−80℃の恒温槽に移した後、ジエチルメトキシボラン(1.0mL、1.0mmol、1.1当量、1.0M THF溶液)を加えて30分間撹拌した。水素化ホウ素ナトリウム(37.8mg、1.0mmol、1.1当量)を加えて−80℃にて10時間撹拌後、酢酸(2mL)を加え、混合液を酢酸エチル(10mL)で3回抽出した。得られた有機層を飽和重曹水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Eの粗生成物を淡黄色油状物として得た。
上記で得られた化合物E(273mg、0.84mmol、1当量)、アセトン(2.6mL)、パラトルエンスルホン酸一水和物(16.0mg)を30mLナスフラスコに移し、ジメトキシプロパン(205μL、1.68mmol、2当量)を加えて室温で4時間撹拌後、飽和重曹水を加えてpHを約7とした後、混合溶液をエーテル(5mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Fを淡黄色油状物質として得た。収量294mg(3段階収率88%)。
【化35】
【0107】
得られた化合物FのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2981, 2938, 1724, 1454, 1369, 1277, 1153cm−1
1H NMR(CDCl3) δ7.38−7.27(m,5H), 4.50(dd,J=4.8, 12.1Hz,2H), 4.28−4.21(m,1H), 4.09−4.02(m,1H), 3.61−3.50(m,2H), 2.42(dd,J=7.1, 15.1Hz,1H), 2.29(dd,J=6.2, 15.1Hz,1H), 1.81−1.68(m,2H), 1.56(dt,J=2.3, 12.6Hz,1H), 1.44(s,9H), 1.44(s,3H), 1.35(s,3H), 1.25−1.15(m,1H)
13CNMR(CDCl3) δ170.3, 138.5, 128.4, 127.6, 127.5, 98.7, 80.5, 73.0, 66.3, 66.2, 66.0, 42.8, 36.6, 36.5, 30.1, 28.1, 19.6
[α]D22+21.9(c0.26,CHCl3)
ESI−MS m/z 387.2 [M+Na]+
HRMS(ESI)Anal.calcd. for C21H32NaO5 m/z 387.2142 [M+Na]+,found;387.2140.
【0108】
<<化合部G、及びHの合成>>
化合物F(153mg、0.419mmol、1当量)、酢酸エチル(1.5mL)、水酸化パラジウム/炭素(25mg、20%w/w)を20mLナスフラスコに加えて1気圧の水素雰囲気下、60℃にて24時間撹拌した後、不要物をセライト濾過し、濾液を減圧濃縮して下記化合物Gを淡黄色油状物として得た。
続いて、上記で得られた化合物G(120.5mg、0.417mmol、1当量)、4−ジメチルアミノピリジン(15.3mg、0.125mmol、0.3当量)、塩化メチレン(2.5mL)を20mLナスフラスコに入れ、氷浴で0℃に冷却した。トリエチルアミン(17μL、1.25mmol、3当量)を加えて5分間撹拌し、パラトルエンスルホニルクロリド(159.3mg、0.834mmol、2当量)を加え、室温で4時間撹拌した後、塩化メチレン(10mL)、蒸留水(10mL)を加えた。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=10/1〜5/1(体積比))で精製し、化合物Hを無色油状物質として得た。収量165.1mg(2段階収率91%)。
【化36】
【0109】
得られた化合物HのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2981, 1728, 1365, 1176cm−1
1H NMR (CDCl3) δ7.91(d,J=8.2Hz,2H), 7.79(d,4H), 7.34(d,J=8.2Hz,2H), 4.26−4.18(m,1H), 4.18−4.13(m,1H), 4.11−4.06(m,1H), 3.99−3.91(m,1H), 2.45(s,3H), 2.39(dd,J=7.1, 15.1Hz,1H), 2.26(dd,J=6.2, 15.1Hz,1H), 1.89−1.67(m,2H), 1.51−1.45(m,2H), 1.44(s,9H), 1.34(s,3H), 1.27(s,3H)
13C NMR(CDCl3) δ170.1, 144.7, 133.0, 129.8, 127.9, 98.8, 80.6, 66.7, 66.0, 64.7, 42.6, 36.2, 35.4, 29.9, 28.1, 21.6, 19.5
[α]D22+12.9(c0.9,CHCl3)
ESI−MS m/z 451.2 [M+Na]+
HRMS(ESI) Anal.calcd. for C21H32NaO7S m/z 451.1761 [M+Na]+,found;451.1756.
【0110】
<<化合物I、及びJの合成>>
20mLナスフラスコに化合物H(120.0mg、0.277mmol、1当量)、アジ化ナトリウム(36.0mg、0.555mmol、2当量)、N,N−ジメチルホルムアミド(1.5mL)を加えて室温にて6時間撹拌した後、蒸留水(1mL)を加えた。混合液を酢酸エチル(3mL)で抽出後、得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。シリカゲルショートパッドカラムを通して濾過し、濾液を濃縮して下記化合物Iを得た。収量67.8mg(収率82%)。
10mL試験管に化合物I(52.1mg、0.174mmol、1当量)、THF(1.0mL)、蒸留水(0.10mL)、トリフェニルホスフィン(91.3mg、0.348mmol、2当量)を入れて50℃にて2時間撹拌した後、THFを減圧留去した。残渣にトルエン(5mL)を加えて混合液を減圧濃縮し、残渣に含まれる水を共沸混合物として留去した。得られた残渣に対して再びトルエン(5mL)を加え混合液を減圧濃縮し、得られた残渣をフラッシュカラムクロマトグラフィー(塩化メチレン/メタノール/トリエチルアミン=95/4/1(体積比))で精製し、化合物J(tert−ブチル[(4R,6R)−6−アミノエチル−2,2−ジメチル−1,3−ジオキサン−4−イル]アセテート)を無色油状物質として得た。収量41.2mg(収率87%)。
【化37】
【0111】
得られた化合物IのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2978, 2939, 2877, 2098, 1732cm−1
1H NMR(CDCl3) δ4.37−4.30(m,1H), 4.12−4.06(m,1H), 3.48−3.41(m,2H), 2.37(dd,J=5.5, 14.9Hz,1H), 2.33(dd,J=7.6, 14.9Hz,1H), 1.80−1.65(m,2H), 1.66(dt,2.5, 12.6Hz,1H), 1.48(s,3H), 1.46(s,9H), 1.32(s,3H), 1.22−1.13(m,1H)
13C NMR(CDCl3)δ170.1, 98.8, 80.6, 66.1, 65.8, 47.5, 42.6, 36.4, 35.6, 30.0, 28.1, 19.6
[α]D22+17.9(c0.21,CHCl3)
ESI−MS m/z 322.2 [M+Na]+
HRMS(ESI) Anal.calcd. for C14H25N3NaO4 m/z 322.1737 [M+Na]+,found;322.1737.
【0112】
得られた化合物JのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν3374, 2981, 2938, 2873, 1731, 1176cm−1
1H NMR(CDCl3) δ4.25−4.19(m,1H), 3.98−3.92(m,1H), 2.80−2.77(m,2H), 2.39(dd,J=6.9, 15.1,1H),2.26(dd,J=6.2, 15.1,1H), 1.95(brs,2H), 1.64−1.50(m,3H), 1.42(s,9H), 1.42(s,3H), 1.33(s,3H), 1.30−1.15(m,1H)
13C NMR(CDCl3)δ170.2, 98.6, 80.5, 67.4, 66.2, 42.6, 39.5, 38.4, 36.5, 30.1, 28.0, 19.7
[α]D22+11.5(c0.28,CHCl3)
ESI−MS m/z 274.2 [M+H]+
HRMS(ESI) Anal.calcd. for C14H28NO4 m/z 274.2013 [M+H]+,found;274.2015.
【0113】
(実施例8)
<tert−ブチル[(4R,6R)−6−アミノエチル−2,2−ジメチル−1,3−ジオキサン−4−イル]アセテートの合成2>
<<化合物Kの合成>>
加熱真空乾燥した30mLナスフラスコにアルゴン雰囲気下で、実施例6で合成したチオアミド化合物A−4(100.2mg、0.28mmol、1当量)、及び塩化メチレン(3.5mL)を加え、氷浴で0℃に冷却した。2,6−ルチジン(64.0μL、0.56mmol、2当量)、tert−ブチルジメチルシリル(TBS)トリフラート(96.5μL、0.42mmol、1.5当量)を加え、室温にて3時間撹拌後、飽和塩化アンモニウム水溶液(3mL)を加えて反応を停止し、その混合溶液を塩化メチレン(5mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Kを淡黄色油状物として得た。収量124.4mg(収率94%)。
【化38】
【0114】
得られた化合物KのIRスペクトル、1H NMRスペクトル、13C NMRスペクトル、比旋光度、ESI−MSスペクトル、及びHRMSスペクトルデータを示す。
IR(neat) ν2954, 2857, 1774, 1712, 1593, 1496, 1400cm−1
1H NMR(CDCl3) δ7.82−7.80(m,2H), 7.71−7.68(m,2H), 5.92(dddd,J=5.7, 5.7, 10.3, 17.2Hz,1H), 5.82(dddd,J=5.5 , 5.5, 10.3, 17.2Hz,1H), 5.27−5.11(m,4H), 4.85(dd,J=7.2, 14.2Hz,1H), 4.63−4.53(m,2H), 4.31(dd,J=7.3, 14.2Hz,1H), 4.00(dd,J=4.6, 17.2Hz,1H), 3.82−3.72(m,2H), 3.16(dd,J=8.5, 13.5Hz,1H), 2.81(dd,J=4.1, 13.5Hz,1H), 2.03−1.86(m,2H), 0.85(s,9H), 0.10(s,3H), 0.03(s,3H)
13C NMR(CDCl3) δ202.5, 168.2, 133.8, 132.2, 131.3, 131.2, 123.1, 119.4, 117.6, 72.2, 56.5, 53.1, 49.1, 35.7, 34.0, 25.8, 17.8, −4.4, −4.7
[α]D23+10.1(c0.19,CHCl3)
ESI−MS m/z 495.2 [M+Na]+
HRMS (ESI) Anal.calcd. for C25H36N2NaO3SSi m/z 495.2108 m/z [M+Na]+,found;495.2111
【0115】
<<化合物L、M、N、及びOの合成>>
加熱真空乾燥した20mL試験管にアルゴン雰囲気下で化合物K(83.5mg、0.177mmol、1当量)とジエチルエーテル(1.5mL)を加え、氷浴で0℃に冷却した。メチルトリフラート(39μmL、0.353mmol、2当量)を加えて0℃にて5分間、室温にて4時間半撹拌後、フラスコをアセトン−ドライアイス浴で−78℃に冷却し、上記で調製した酢酸tert−ブチルエステルリチウムエノラート(177μL、0.177mmol、3当量)をゆっくりと滴下して加えた。3時間撹拌後、塩化メチレン(3mL)、及びシリカゲル(2g)を加え、室温にて1時間半撹拌した。混合物をシリカゲルショートパッドカラムに通し、濾液を濃縮して得られた残渣にTHF(10mL)、1規定塩酸(1mL)を加え室温にて2時間撹拌後、混合溶液を酢酸エチル(5mL)で3回抽出した。得られた有機層を蒸留水、飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Lの粗生成物を淡黄色油状物として得た。
続いて、アルゴン雰囲気下、化合物L(60mg、0.126mmol、1当量)と乾燥THF(800μL)を20mL試験管に移し、氷浴で0℃に冷却した。続いてテトラブチルアンモニウムフルオリド(176μL、0.176mmol、1.4当量、1.0M THF溶液)をゆっくりと滴下し、0℃で30分間、室温にて3時間撹拌後、蒸留水(0.5mL)を加え、混合液を酢酸エチル(3mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Mの粗生成物を淡黄色油状物質として得た。
続いて、20mLナスフラスコに化合物M(45mg、0.124mmol、1当量)、乾燥THF(1.7mL)、及びメタノール(0.4mL)を入れ、ナスフラスコを−80℃の恒温槽に移した後、ジエチルメトキシボラン(136μL、0.136mmol、1.1当量、1.0M THF溶液)を加えて30分間撹拌した。水素化ホウ素ナトリウム(5.2mg、0.136mmol、1.1当量)を加えて−80℃にて10時間撹拌後、酢酸(0.2mL)を加え、混合液を酢酸エチル(3mL)で3回抽出した。得られた有機層を飽和重曹水、及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、下記化合物Nの粗生成物を淡黄色油状物として得た。
続いて、得られた化合物N(43.0mg、0.12mmol、1当量)、アセトン(0.5mL)、及びパラトルエンスルホン酸一水和物(2.3mg、0.012mmol、0.1当量)を20mL試験管に移し、ジメトキシプロパン(29.5μL、0.24mmol、2当量)を加えて室温で4時間撹拌後、飽和重曹水を加えてpHを約7とした後、混合溶液をエーテル(2mL)で3回抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過及び濃縮し、化合物Oを淡黄色油状物質して得た。収量47mg(4段階収率66%)。
【化39】
【0116】
得られた化合物OのIRスペクトル、1H NMRスペクトル、及び13C NMRスペクトルデータを示す。
IR(neat) ν2923, 2865, 1770, 1725, 1712, 1612, 1492, 1396cm−1
1H NMR(CDCl3) δ7.86−7.83(m,2H), 7.74−7.71(m,2H), 4.35−4.27(m,1H), 4.12−4.04(m,1H), 3.65−3.55(m,2H), 2.44(dd,J=7.1, 15.1Hz,1H), 2.32(dd,J=6.2, 15.1Hz,1H), 1.82−1.69(m,2H), 1.57(dt,J=2.3, 12.6Hz,1H), 1.44(s,9H), 1.44(s,3H), 1.35(s,3H), 1.25−1.15(m,1H)
13C NMR(CDCl3) δ170.5, 168.6, 133.9, 132.0, 130.6, 80.5, 73.0, 67.9, 66.2, 42.8, 36.7, 36.6, 33.1, 30.1, 28.1, 19.6
【0117】
<<化合物Jの合成>>
エタノール(2.5mL)に化合物O(45mg、0.11mmol、1当量)を溶解し、ヒドラジン一水和物(70mL、2.25mmol、20当量)を加えた後、60℃にて1時間撹拌した。反応溶液を室温に戻した後、析出した固体を濾別し、濾液を減圧濃縮した。残渣に塩化メチレンと飽和食塩水を加え、水層を塩化メチレンで抽出し、得られた有機層を無水硫酸ナトリウムで乾燥した。濾過及び濃縮後に得られた残渣をフラッシュカラムクロマトグラフィー(塩化メチレン/メタノール/トリエチルアミン=95/4/1(体積比))で精製し、化合物Jを無色油状物質して得た。収量25.5mg(収率85%)。
【化40】
【産業上の利用可能性】
【0118】
本発明のチオアミド化合物は、[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体を、効率よく安価に製造可能であることから、アトルバスタチンの合成に好適に用いることができる。
【特許請求の範囲】
【請求項1】
下記一般式(1)で表されることを特徴とするチオアミド化合物。
【化1】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
【請求項2】
請求項1に記載のチオアミド化合物の製造方法であって、
下記一般式(2)で表される化合物と、下記一般式(3)で表される化合物とを反応させる反応工程を含むことを特徴とするチオアミド化合物の製造方法。
【化2】
【化3】
ただし、前記一般式(2)中、R1は、−OR11、及び−NR12R13のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、水素原子及びアミノ基の保護基のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。前記一般式(3)中、R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。
【請求項3】
反応工程が、銅錯体を用いて行われる請求項2に記載のチオアミド化合物の製造方法。
【請求項4】
銅錯体が、銅−光学活性ホスフィン錯体である請求項3に記載のチオアミド化合物の製造方法。
【請求項5】
下記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程を含むことを特徴とする[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法。
【化4】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
【化5】
ただし、前記一般式(4)中、R4は、カルボキシル基の保護基及び水素原子のいずれかを表す。R5及びR6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
【請求項6】
下記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程を含むことを特徴とするアトルバスタチンの製造方法。
【化6】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい。)。
【化7】
ただし、前記一般式(4)中、R4は、カルボキシル基の保護基及び水素原子のいずれかを表す。R5及びR6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
【請求項1】
下記一般式(1)で表されることを特徴とするチオアミド化合物。
【化1】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
【請求項2】
請求項1に記載のチオアミド化合物の製造方法であって、
下記一般式(2)で表される化合物と、下記一般式(3)で表される化合物とを反応させる反応工程を含むことを特徴とするチオアミド化合物の製造方法。
【化2】
【化3】
ただし、前記一般式(2)中、R1は、−OR11、及び−NR12R13のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、水素原子及びアミノ基の保護基のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。前記一般式(3)中、R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。
【請求項3】
反応工程が、銅錯体を用いて行われる請求項2に記載のチオアミド化合物の製造方法。
【請求項4】
銅錯体が、銅−光学活性ホスフィン錯体である請求項3に記載のチオアミド化合物の製造方法。
【請求項5】
下記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程を含むことを特徴とする[(4R,6R)−6−アミノエチル−1,3−ジオキサン−4−イル]アセテート誘導体の製造方法。
【化4】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい)。
【化5】
ただし、前記一般式(4)中、R4は、カルボキシル基の保護基及び水素原子のいずれかを表す。R5及びR6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
【請求項6】
下記一般式(1)で表されるチオアミド化合物を下記一般式(4)で表されるアセテート誘導体に転化する工程を含むことを特徴とするアトルバスタチンの製造方法。
【化6】
ただし、前記一般式(1)中、R1は、−OR11、及び−NR12R13のいずれかを表す。R2及びR3は、それぞれ独立に、アミド基の保護基及び水素原子のいずれかを表す。前記R11は、水酸基の保護基及び水素原子のいずれかを表す。前記R12及び前記R13は、それぞれ独立に、アミノ基の保護基及び水素原子のいずれかを表す(なお、前記R12及び前記R13は、一緒になって環構造の保護基を形成していてもよい。)。
【化7】
ただし、前記一般式(4)中、R4は、カルボキシル基の保護基及び水素原子のいずれかを表す。R5及びR6は、それぞれ独立に、炭素数1〜6の炭化水素基及び水素原子のいずれかを表す(なお、前記R5及び前記R6は、一緒になって環構造を形成していてもよい)。
【図1】

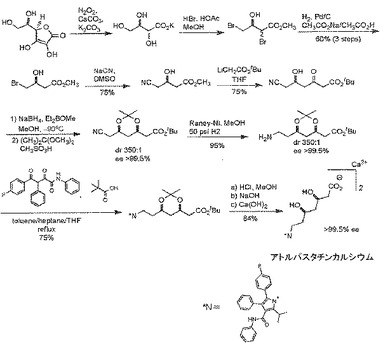
【図2】
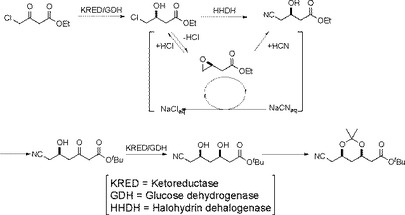
【図2】
【公開番号】特開2012−171904(P2012−171904A)
【公開日】平成24年9月10日(2012.9.10)
【国際特許分類】
【出願番号】特願2011−35006(P2011−35006)
【出願日】平成23年2月21日(2011.2.21)
【出願人】(000173913)公益財団法人微生物化学研究会 (29)
【Fターム(参考)】
【公開日】平成24年9月10日(2012.9.10)
【国際特許分類】
【出願日】平成23年2月21日(2011.2.21)
【出願人】(000173913)公益財団法人微生物化学研究会 (29)
【Fターム(参考)】
[ Back to top ]