
チオキサントン化合物及びこれを有する有機発光素子
【課題】発光効率が高く、駆動電圧の低い有機発光素子を提供する。
【解決手段】陽極と陰極と、前記陽極と前記陰極との間に配置される有機化合物層と、から構成される有機発光素子において、前記有機化合物層に、下記一般式[1]で示されるチオキサントン化合物が含まれることを特徴とする、有機発光素子。

【解決手段】陽極と陰極と、前記陽極と前記陰極との間に配置される有機化合物層と、から構成される有機発光素子において、前記有機化合物層に、下記一般式[1]で示されるチオキサントン化合物が含まれることを特徴とする、有機発光素子。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はチオキサントン化合物及びこれを有する有機発光素子に関する。
【背景技術】
【0002】
有機発光素子は、陽極と陰極と、これら両電極間に配置される有機化合物層とを有する電子素子である。各電極から注入させる正孔(ホール)及び電子が有機化合物層に含まれる発光層内で再結合することで生成される励起子が基底状態に戻る際に、有機発光素子は、光を放出する。有機発光素子の最近の進歩は著しく、その特徴として、低駆動電圧、発光波長の多様化、高速応答性、発光デバイスの薄型・軽量化が可能であることが挙げられる。
【0003】
ところで有機発光素子は、発光に関与する励起子の種類によって蛍光発光素子と燐光発光素子とに大別される。そのうち燐光発光素子は、有機発光素子を構成する有機化合物層、具体的には発光層中に燐光発光材料を有する電子素子である。ここで燐光発光材料は、正孔及び電子が再結合することによって生じるエネルギーを吸収して三重項励起子を生成する。このため燐光発光素子は、この三重項励起子由来の発光が得られる有機発光素子である。
【0004】
ところで、燐光の発光量子収率は理論上蛍光の量子収率の4倍にすることができるので、近年では燐光発光素子に注目が集まっている。しかし燐光発光素子において、発光効率についてはさらなる改善の余地がある。
【0005】
一方、燐光発光素子で使用される燐光発光材料について様々な提案がなされている。燐光発光材料として、例えば、下記に示される化合物H−1(チオキサンテン化合物、特許文献1参照。)及び化合物H−2(チオキナクリドン化合物、特許文献2参照。)が提案されている。
【0006】
【化1】
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開平03−177486号公報
【特許文献2】特開平11−54279号公報
【発明の概要】
【発明が解決しようとする課題】
【0008】
ところで特許文献1に開示されているチオキサンテン化合物は、ケトン骨格を有していないため、LUMO準位が浅くなる。また、特許文献2に開示されているチオキナクリドン化合物は、基本骨格のバンドギャップが狭く、それ故にT1エネルギーが低くなってしまう。
【0009】
一方で、有機発光素子においては、発光層に含まれる発光材料の他に、電子輸送層に含まれる電子輸送性材料の開発が求められている。具体的には、LUMO準位が2.7eV以上と深く、かつ化学的に安定な有機化合物が求められている。
【0010】
特に、発光層に燐光発光材料が含まれる有機発光素子においては、素子を構成する材料である発光材料や電子輸送材料についてT1エネルギーが高いことが求められている。
【0011】
本発明は、上述した課題を解決するためになされたものであり、その目的は、発光効率が高く、駆動電圧の低い有機発光素子を提供することである。
【課題を解決するための手段】
【0012】
本発明のチオキサントン化合物は、下記一般式[1]で示されることを特徴とする。
【0013】
【化2】
(式[1]において、R1乃至R8は、それぞれ水素原子、炭素数1乃至4のアルキル基又はフェニル基、ビフェニル基、ナフチル基、フェナンスレン基、フルオレニル基、トリフェニレニル基、ジベンゾフラニル基及びジベンゾチオフェニル基のいずれかから選択されるアリール基を表す。ただし、R1乃至R8のうち少なくともひとつが前記アリール基である。
【0014】
尚、前記アルキル基及び前記アリール基は、さらにアルキル基、芳香族炭化水素基又は芳香族複素環基を有してもよい。)
【発明の効果】
【0015】
本発明のチオキサントン化合物は、T1エネルギーが2.3eV以上と高く、かつLUMO準位が2.7eV以上と深い化合物である。このため本発明によれば、発光効率が高く、駆動電圧の低い有機発光素子を提供することができる。
【図面の簡単な説明】
【0016】
【図1】有機発光素子と、有機発光素子に接続するスイッチング素子とを示す断面模式図である。
【発明を実施するための形態】
【0017】
まず本発明のチオキサントン化合物について説明する。本発明のチオキサントン化合物は下記一般式[1]で示される化合物である。
【0018】
【化3】
【0019】
式[1]において、R1乃至R8は、それぞれ水素原子、炭素数1乃至4のアルキル基又はフェニル基、ビフェニル基、ナフチル基、フェナンスレン基、フルオレニル基、トリフェニレニル基、ジベンゾフラニル基及びジベンゾチオフェニル基のいずれかから選択されるアリール基を表す。
【0020】
R1乃至R8で表されるアルキル基として、メチル基、エチル基、n−プロピル基、iso−プロピル基、n−ブチル基、iso−ブチル基、sec−ブチル基、tert−ブチル基が挙げられる。
【0021】
ただし、R1乃至R8のうち少なくともひとつが前記アリール基である。
【0022】
尚、上記アルキル基及び上記アリール基は、さらにアルキル基、芳香族炭化水素基又は芳香族複素環基を有してもよい。
【0023】
上記アルキル基及び上記アリール基がさらに有してもよいアルキル基として、例えば、メチル基、エチル基、n−プロピル基、iso−プロピル基、n−ブチル基、iso−ブチル基、sec−ブチル基、tert−ブチル基等が挙げられる。
【0024】
上記アルキル基及び上記アリール基がさらに有してもよい芳香族炭化水素基として、フェニル基、ナフチル基、フェナントリル基、フルオレニル基等が挙げられる。
【0025】
上記アルキル基及び上記アリール基がさらに有してもよい芳香族複素環基として、チエニル基、ジベンゾフラニル基、ジベンゾチオフェニル基、ピロリル基、ピリジル基等の複素芳香環基が挙げられる。
【0026】
尚、上記アルキル基及び上記アリール基は、メトキシ基、エトキシ基等のアルコキシ基、フェノキシ基、ナフトキシ基等のアリールオキシ基、フッ素、塩素、臭素、ヨウ素等のハロゲン原子をさらに有してもよい。
【0027】
本発明のチオキサントン(チオキサンテン−9−オン)化合物は、チオキサントン自身でT1エネルギーが3.0eVと高く、かつLUMO準位が2.7eV以上と深い。このため、有機発光素子の高効率化や安定性の向上をもたらすと考えられる。
【0028】
次に、本発明のチオキサントン化合物の合成方法について説明する。本発明のチオキサントン化合物は、例えば、下記に示される合成スキームを利用して合成される。尚、下記合成スキームにおいて、Arは、フェニル基、ナフチル基、フェナンスレン基、フルオレニル基、トリフェニレニル基、ジベンゾフラニル基又はジベンゾチオフェニル基を表す。
【0029】
【化4】
【0030】
上記合成スキームに示されるように、本発明のチオキサントン化合物は、Pd触媒を用いたチオキサントンのハロゲン体(X)とアリールボロン酸もしくはアリールボロン酸エステルとのカップリング反応で合成することができる。尚、原料となるチオキサントンのハロゲン体は、市販品を利用することができる。
【0031】
ここで上記反応スキームを利用して本発明のチオキサントン化合物を合成する場合、Arを適宜選択することにより、所望のチオキサントン化合物を合成することができる。
【0032】
また本発明のチオキサントン化合物を有機発光素子の構成材料として使用する場合には、使用する直前に昇華精製を行うのが好ましい。なぜなら有機化合物の高純度化の観点からして昇華精製は精製効果(高純度化効果)が大きいからである。ただし昇華精製を行って有機化合物を精製する際には、一般に分子量が大きい化合物ほど高温が必要とされる。また化合物によっては高温による熱分解等を起こしやすくなる。従って、有機発光素子の構成材料として使用される有機化合物は、過大な加熱なく昇華精製を行うことができるように、分子量が1000以下であることが好ましい。
【0033】
本発明のチオキサントン化合物において、主骨格たるチオキサントン骨格は、カルボニル基を有しているため電子親和性が高い。このため発明者は、カルボニル基に由来する電子輸送能を考慮して、このチオキサントン骨格が電子輸送材料となる化合物として適した骨格であると考察した。
【0034】
また本発明のチオキサントン化合物は、チオキサントン骨格中の環構造の一部としてカルボニル基の炭素原子を含めているため、チオキサントン骨格の全体が平面構造になる。この構造を有するため、本発明のチオキサントン化合物は、分子同士の重なり合いが起こりやすくなり、固体状態における分子間電子移動が効率的に起こりやすくなる。これらの構造的性質から、本発明のチオキサントン化合物は、有機発光素子の構成材料、具体的には、陰極や隣接する層からの電子を注入・輸送する機能を担う材料として適しているといえる。即ち、本発明のチオキサントン化合物は、電子注入・輸送層に含まれる電子注入・輸送性材料や発光層のホストとして好適に用いることができる。
【0035】
またチオキサントン骨格が示す他の特長として、高いT1エネルギーがある。実際に無置換のチオキサントン(式[1]においてR1乃至R8が全て水素原子の化合物)の希薄トルエン溶液を77Kにおいて燐光スペクトル測定して得られる0−0バンドからT1エネルギーを求めることができる。その結果無置換のチオキサントンのT1エネルギーは2.74eV(451nm)であり、この値は青領域の一部(455nm〜480nm)や緑色領域(490nm〜530nm)よりも十分エネルギーが高い。従って、本発明のチオキサントン化合物は、発光色が幅広い燐光発光素子において電子輸送層に含まれる電子輸送材料や発光層のホストとして本質的に適用できる。
【0036】
ところで、特許文献1に開示されている化合物H1は、チオキサントン骨格中のカルボニルがメチレンになったチオキサンテン化合物である。ここで化合物H1は、T1エネルギーが高い点(波長換算で361nm)では優れているが、カルボニル基が存在しないために、チオキサントン化合物の特徴である電子受容性、電子輸送性が大きく低下している。
【0037】
ここで本発明のチオキサントン化合物、並びに特許文献1及び2にそれぞれ開示されている化合物の主骨格(チオキサントン骨格、チオキサンテン骨格、チオキナクリドン骨格)のT1エネルギーについて測定及び計算を行った。
【0038】
ここで測定によってT1エネルギーを求める際には、77Kにおけるトルエン溶液状態での燐光スペクトルの測定結果から求めた。より具体的には、燐光スペクトルの測定を行った後、この測定で得られた0−0バンドからT1エネルギーを求めた。
【0039】
一方、計算によってT1エネルギーを求める際には、密度汎関数法(Density Functional Theory)を用いて、B3LYP/6−31G*レベルでの分子軌道計算を行った。上記計算及び測定によって得られたT1エネルギーの値を下記表1に示す。
【0040】
【表1】
【0041】
表1より、チオキサンテンのLUMOがチオキサントンよりも浅いことが示されている。またチオキナクリドンは、チオキサントン骨格を含む構造であるが、チオキサントン骨格と比較するとT1エネルギーが大幅に低くなる(波長換算で約570nm)。このためチオキナクリドンは、発光色が青や緑のりん光発光素子の構成材料として適さない化合物である。
【0042】
以上より、表1で示される化合物において、発光色が青や緑のりん光発光素子の構成材料としてふさわしいT1エネルギーを有し、電子受容性や電子輸送性に適した深いHOMOを有する化合物は、チオキサントン化合物のみである。
【0043】
ところで、特許文献1及び2にて開示されているチオキサンテン化合物及びチオキナクリドン化合物は、共にその用途は蛍光性の発光材料である。一方、本発明のチオキサントン化合物は、化合物中に含まれるカルボニル基に由来する電子輸送性に着目して使用されるものであり、その用途は、電子輸送層に含まれる電子輸送性材料や発光層のホストである。
【0044】
一方で、有機発光素子を構成する電子輸送層あるいは発光層に、発光材料以外の化合物として本発明のチオキサントン化合物を使用する場合、以下のことを考慮することが大切である。それはその有機発光素子が有する発光材料の発光色を考慮して最適なバンドキャップ及びより深いLUMOを、本発明のチオキサントン化合物が有することである。
【0045】
ここで本発明のチオキサントン化合物において、化合物のバンドギャップを狭くするためには、チオキサントン骨格と共役がつながる部位に、アリール基等の共役を有する置換基を導入する必要がある。チオキサントン骨格と共役がつながる部位となる置換位置とは、下記に示されるチオキサントン骨格の1位乃至8位の炭素である。よってチオキサントン骨格の1位乃至8位にアリール基を導入することが好ましい。
【0046】
【化5】
【0047】
また、化合物が有する共役を広げつつバンドギャップを狭く制御するためにはチオキサントン骨格に対して立体障害の小さい置換位置に置換基を設けることが好ましい。この点を考慮すると、チオキサントン骨格について置換基を導入する場合、置換基が導入される位置は、式[1]中のR2、R3、R6及びR7の中から1つ又は2つ選択するのが好ましい。R2及びR3から1つ選択しR6及びR7から1つ選択する組み合わせがより好ましい。そして、R2及びR7の組み合わせ又はR3及びR6の組み合わせが特に好ましい。
【0048】
一方で、式[1]中のR1、R4、R5及びR8は、いずれも水素原子であることが好ましい。また置換基を複数置換する場合、置換する置換基は何れも同じにすることが好ましい。
【0049】
他方で、チオキサントン骨格のように平面性が高い骨格にアルキル基や芳香環基を導入することで、溶媒に対する溶解性や真空蒸着時の昇華性、薄膜状態のアモルファス性を向上させることができる。ここで、アルキル基を導入する際には、炭素数が炭素数1乃至4のアルキル基を導入すると化合物の昇華性を損なわずにアモルファス性を向上することができる。
【0050】
ところで、本発明のチオキサントン化合物を、燐光発光素子の構成材料である電子輸送性材料や発光層のホストとして使用する際には、燐光発光材料より高いT1エネルギーと最適なHOMO−LUMO準位を有している必要がある。ここで、チオキサントン骨格にアリール基を導入することで化合物のT1エネルギーをある程度調整することができる。
【0051】
特に、発光材料である燐光発光材料を有する有機発光素子において、本発明のチオキサントン化合物を発光層あるいは発光層に隣接する電荷輸送層(電子輸送層)の構成材料として使用する際には、化合物が有するT1エネルギーが重要となる。つまり燐光発光材料の発光色が青乃至赤、即ち、発光波長のスペクトルの最大ピークが455nm以上620nm以下である場合において、その燐光発光材料の発光色に対応するように本発明のチオキサントン化合物のT1エネルギーを設定することが重要である。
【0052】
ここで発明者は、本発明のチオキサントン化合物のT1エネルギーを設定するにあたり、式[1]中のR1乃至R8のいずれかに導入される置換基(縮合環)のT1エネルギーに注目した。下記表2は、ベンゼン及び主な縮合環のT1エネルギー(波長換算値)を示す表である。
【0053】
【表2】
【0054】
表2を考慮すると、ベンゼン・縮合環の中でも、ベンゼン、ナフタレン、フェナンスレン、フルオレン、トリフェニレン、クリセン、ジベンゾフラン、ジベンゾチオフェン及びピレンが好ましいといえる。
【0055】
さらに燐光発光材料の発光色が青から緑である場合、本発明のチオキサントン化合物の主骨格に導入される置換基(ベンゼン・縮合環)として好ましい縮合環は、ベンゼン、ナフタレン、フェナンスレン、フルオレン、トリフェニレン、ジベンゾフラン又はジベンゾチオフェンである。尚、この場合における「青から緑」とは、波長にして455nm以上530nm以下の範囲であり、440nm付近の深青は含まれない。
【0056】
また本発明のチオキサントン化合物の主骨格に導入される置換基のその置換位置は、より電子受容性を高めるために、化合物自体のLUMOを深くする位置が好ましい。下記表3は、置換基(フェニル基)の置換位置とHOMOエネルギー及びLUMOエネルギーとの関係を示す表である。
【0057】
【表3】
【0058】
表3より、同じ置換基(フェニル基)を置換した場合であっても、2位置換又は3位置換に相当するR2、R3、R6、R7に置換基を置換する方が、チオキサントン化合物のLUMOはより深くなるといえる。これより、電子受容性の高さが要求される電子輸送層や発光層の材料として本発明のチオキサントン化合物を使用する際には、R2、R3、R6及びR7のいずれかにアリール基を置換することが好ましい。
【0059】
本発明の化合物は燐光発光する有機発光素子の電子輸送層や発光層の少なくともいずれか一方に好ましく用いることができる。これは本発明に係るチオキサントン化合物はそのT1エネルギーが燐光発光材料のそれより高いからである。そして本発明に係るチオキサントン化合物はそのような層に用いる場合にも適切な広さのバンドギャップを有しているので好ましい。
【0060】
本発明のチオキサントン化合物の具体例を以下に示す。ただし、本発明ハこれらに限定されるものではない。
【0061】
【化6】
【0062】
【化7】
【0063】
【化8】
【0064】
例示化合物のうちA群に示す化合物は、式[1]に示されるチオキサンテン骨格中のR2及びR7又はR3乃至R6に同一の置換基(アルキル基又はアリール基)が導入されている化合物である。A群に示す化合物は、中心骨格であるチオキサントン骨格に対して同一の置換基を所定の位置に2つ導入することで、対称軸を持った安定な骨格を有している。従って、A群の化合物は、化学的安定性が非常に高い上に電子輸送性を有する。よって、それらを電子輸送性材料又は発光層に含まれるホストあるいはアシスト材料として使用すると、素子の長寿命化が期待できる。
【0065】
例示化合物のうちB群に示す化合物は、式[1]に示されるチオキサンテン骨格中のR1乃至R8のいずれか(例えば、R2及びR7又はR3乃至R6)に、複素環基あるいは複素環を含む置換基が導入されている化合物である。B群に示す化合物に含まれるヘテロ原子を環状基の内部に有する複素環基は、芳香族炭化水素基ほどではないが、芳香族炭化水素基に準じた安定性を有する。このため、B群の化合物を電子輸送性材料又は発光層に含まれるホストあるいはアシスト材料として使用すると、素子の長寿命化が期待できる。
【0066】
例示化合物のうちC群に示す化合物は、式[1]に示されるチオキサンテン骨格中のR1乃至R8のいずれか1つに置換基(アルキル基、アリール基、複素環基あるいはこれらを組み合わせた複合置換基)が導入されている化合物である。C群に示す化合物は、化合物全体として対称性が無いために、HOMO−LUMOがCT性を有する場合がある。これを利用して発光材料に適したHOMO−LUMOに調整することができる。このため、C群の化合物を電子輸送性材料又は発光層に含まれるホストあるいはアシスト材料として使用すると、素子の長寿命化が期待できる。
【0067】
例示化合物のうちD群に示す化合物は、A群乃至C群の思想を組み合わせたものであり、対称性を低くすることで化合物自体の溶解性、電子移動度の制御をすることができる。
【0068】
次に、本発明の有機発光素子を説明する。本発明の有機発光素子は、互いに対向しあう一対の電極である陽極と陰極と、陽極と陰極との間に配置される有機化合物層と、から構成される。そして、この有機化合物層に本発明のチオキサントン化合物が含まれる。
【0069】
有機発光素子を構成する有機化合物層は、発光層又は発光機能を有する層が含まれていれば、単一の層であってもよいし、複数の層からなる積層体であってもよい。ここで有機化合物層が複数の層からなる積層体である場合、有機化合物層を構成する層としては、ホール注入層、ホール輸送層、発光層、ホールブロック層、電子輸送層、電子注入層、エキシトンブロック層等が挙げられる。上記群に示されている層の中から複数の層を適宜選択し、かつそれらを組み合わせて用いることができる。
【0070】
ただし本発明の有機発光素子の構成はこれらに限定されるものではない。例えば、電極と有機化合物層との界面に絶縁性層、接着層あるいは干渉層を設ける、電子輸送層もしくはホール輸送層がイオン化ポテンシャルの異なる二層から構成される等多様な層構成をとることができる。
【0071】
本発明の有機発光素子の素子形態としては、基板と反対側の電極から光を取り出すいわゆるトップエミッション方式でも、基板側の電極から光を取り出すいわゆるボトムエミッション方式でもよい。また基板や電極を光透過性の材料にして、両面から光を取り出す構成にしてもよい。
【0072】
本発明のチオキサントン化合物は、有機発光素子を構成する有機化合物層に含まれるが、好ましくは、電子輸送層、ホール・エキシトンブロッキング層、電子注入層又は発光層に含まれる。ここで本発明のチオキサントン化合物が電子輸送層又はホール・エキシトンブロッキング層に含まれる場合は、本発明のチオキサントン化合物は電子輸送材料として使用される。
【0073】
ところでホールブロッキング層は、陽極から注入されたホールをブロックする(陰極方向へ移動させない)層の意味として用いられることがあるが、本発明においては、発光層の陰極側で発光層と隣接する層をホールブロッキング層と呼ぶことにする。この理由として、本発明のチオキサントン化合物は、本来はホールをブロッキングすることを目的する材料としてではなく、電子を発光層へ輸送するための材料として使用されているからである。しかし、層を設ける位置が一般的なホールブロッキング層と同じ位置にあることと、配置位置に関して電子輸送層と混同しないために、配置位置に合わせてホールブロッキング層と記すことがある。
【0074】
本発明のチオキサントン化合物がホールブロッキング層に含まれる場合、発光層は複数種の成分から構成されていてよく、それらを主成分と副成分とに分類することができる。つまり、発光層を構成する全化合物の中で重量比が最大の化合物である主成分と、それ以外の副成分とに分類することができる。
【0075】
ここで主成分は、ホストと呼ばれている。一方、副成分は、材料自体の機能に応じてゲスト(ドーパント)、発光アシスト材料又は電荷注入材料と呼ばれている。尚、発光アシスト材料及び電荷注入材料は、同一構造の有機化合物であってもよいし、異なる構造の有機化合物であってもよい。また副成分であるものの、材料によっては、ゲスト等と区別する意味で第二ホストと呼ぶこともできる。
【0076】
本発明の有機発光素子において、ゲストとは、発光層内で主たる発光を担う化合物である。これに対してホストとは、発光層内でゲストの周囲にマトリックスとして存在する化合物であって、主にキャリアの輸送、及びゲスト材料への励起エネルギー供与を担う化合物である。ここでホストは、複数種の材料を有してもよい。
【0077】
発光層内に含まれるゲストの濃度は、発光層の構成材料の全体量を基準として、0.01重量%以上50重量%以下であり、好ましくは0.1重量%以上20重量%以下である。さらに好ましくは、濃度消光を防ぐためにゲストの濃度は0.01重量%以上10重量%以下であることが望ましい。またゲストはホストからなる層の全体に均一に含まれてもよいし、濃度勾配を有して含まれてもよい。また特定の領域に部分的に含ませてゲストを含まないホスト層の領域を部分的に設けてもよい。
【0078】
本発明のチオキサントン化合物は、ゲストたる燐光発光材料に対応する発光層のホストや発光層に含まれる電荷注入材料又は電子輸送層・ホールブロック層に含まれる電子輸送材料として使用される。このとき燐光発光材料の発光色は特に限定されないが、その最大発光ピーク波長が460nm以上530nm以下の範囲にある青色から緑色発光材料であることが好ましい。特に最大発光ピーク波長が490nm以上530nm以下の範囲にある緑色発光材料であることが好ましい
一般に燐光発光する有機発光素子では、ホスト材料のT1から非放射失活による発光効率の低下を防ぐために、ホスト材料のT1エネルギーはゲスト材料である燐光発光材料のT1エネルギーよりも高いことが必要とされている。
【0079】
本発明のチオキサントン化合物は、その中心となるチオキサントン骨格のT1エネルギーが451nmであるので、緑色燐光発光材料のT1エネルギーよりも高い。従ってこれを緑色発光する有機発光素子の発光層もしくはその周辺層に用いると発光効率の高い有機発光素子を得る事ができる。
【0080】
さらに本発明のチオキサントン化合物は、LUMO準位が深いので、これを電子注入材料、電子輸送材料又はホールブロッキング層の構成材料のみならず、発光層に含まれる第二ホストとして使用することができる。これらに示される用途で本発明のチオキサントン化合物を用いることで、素子の駆動電圧を低くすることができる。なぜならLUMO準位が深いと、発光層の陰極側に隣接する電子輸送層又はホールブロッキング層からの電子注入障壁が小さくなるからである。
【0081】
一方、本発明のチオキサントン化合物が発光層に含まれる場合、発光層は、本発明のチオキサントン化合物のみからなる層でもよいし、ホストとゲストとからなる層であってもよい。ここで発光層がホストとゲストとからなる層である場合、本発明のチオキサントン化合物は、ホストとして使用するのが好ましい。
【0082】
ここで本発明のチオキサントン化合物を発光層のホストとして使用する場合、対応するゲストとして、好ましくは、燐光発光材料である。ゲストとして使用される燐光発光材料は、具体的には、イリジウム錯体、白金錯体、レニウム錯体、銅錯体、ユーロピウム錯体、ルテニウム錯体等の金属錯体である。中でも燐光発光性の強いイリジウム錯体が好ましい。また、励起子やキャリアの伝達を補助することを目的として、発光層が複数の燐光発光材料を有していてもよい。
【0083】
以下にイリジウム錯体の具体例を示すが、本発明はこれらに限定されるものではない。
【0084】
【化9】
【0085】
また発光層に含まれるホストを本発明のチオキサントン化合物を含めて複数種にしてもよい。以下に本発明の有機発光素子の構成材料として使用されるホスト(本発明のチオキサントン化合物を除く。)の具体例を示すが、本発明はこれらに限定されるものではない。
【0086】
【化10】
【0087】
また本発明の有機発光素子においては、本発明の値オキサントン化合物以外にも、必要に応じて従来公知の低分子系及び高分子系の化合物を使用することができる。より具体的にはホール注入・輸送性材料、ホスト材料、発光性化合物あるいは電子注入・輸送性化合物等を一緒に使用することができる。以下にこれらの化合物例を挙げる。
【0088】
ホール注入・輸送性材料としては、陽極からのホールの注入が容易で、注入されたホールを発光層へと輸送することができるように、ホール移動度が高い材料が好ましい。ホール注入輸送性能を有する低分子及び高分子系材料としては、トリアリールアミン誘導体、フェニレンジアミン誘導体、スチルベン誘導体、フタロシアニン誘導体、ポルフィリン誘導体、ポリ(ビニルカルバゾール)、ポリ(チオフェン)、その他導電性高分子が挙げられる。
【0089】
主に発光機能に関わる発光材料としては、前述の燐光発光ゲスト材料、もしくはその誘導体以外に、縮環化合物(例えばフルオレン誘導体、ナフタレン誘導体、ピレン誘導体、ペリレン誘導体、テトラセン誘導体、アントラセン誘導体、ルブレン等)、キナクリドン誘導体、クマリン誘導体、スチルベン誘導体、トリス(8−キノリノラート)アルミニウム等の有機アルミニウム錯体、有機ベリリウム錯体、及びポリ(フェニレンビニレン)誘導体、ポリ(フルオレン)誘導体、ポリ(フェニレン)誘導体等の高分子誘導体が挙げられる。
【0090】
電子注入・輸送性材料としては、陰極からの電子の注入が容易で注入された電子を発光層へ輸送することができるものから任意に選ぶことができ、ホール注入輸送性材料のホール移動度とのバランス等を考慮して選択される。電子注入輸送性能を有する材料としては、オキサジアゾール誘導体、オキサゾール誘導体、ピラジン誘導体、トリアゾール誘導体、トリアジン誘導体、キノリン誘導体、キノキサリン誘導体、フェナントロリン誘導体、有機アルミニウム錯体等が挙げられる。
【0091】
陽極を構成する材料としては仕事関数がなるべく大きなものがよい。例えば金、白金、銀、銅、ニッケル、パラジウム、コバルト、セレン、バナジウム、タングステン等の金属単体あるいはこれらを複数種組み合わせた合金、酸化錫、酸化亜鉛、酸化インジウム、酸化錫インジウム(ITO)、酸化亜鉛インジウム等の金属酸化物が使用できる。またポリアニリン、ポリピロール、ポリチオフェン等の導電性ポリマーも使用できる。これらの電極物質は一種類を単独で使用してもよいし、二種類以上を併用して使用してもよい。また、陽極は一層で構成されていてもよく、複数の層で構成されていてもよい。
【0092】
一方、陰極を構成する材料としては仕事関数の小さなものがよい。例えばリチウム等のアルカリ金属、カルシウム等のアルカリ土類金属、アルミニウム、チタニウム、マンガン、銀、鉛、クロム等の金属単体が挙げられる。あるいはこれら金属単体を複数種組み合わせた合金も使用することができる。例えばマグネシウム−銀、アルミニウム−リチウム、アルミニウム−マグネシウム等が使用できる。酸化錫インジウム(ITO)等の金属酸化物の利用も可能である。これらの電極物質は一種類を単独で使用してもよいし、二種類以上を併用して使用してもよい。また陰極は一層構成でもよく、多層構成でもよい。
【0093】
本発明の有機発光素子において、本発明の有機化合物を含有する層及びその他の有機化合物からなる層は、以下に示す方法により形成される。一般には真空蒸着法、イオン化蒸着法、スパッタリング、プラズマあるいは、適当な溶媒に溶解させて公知の塗布法(例えば、スピンコーティング、ディッピング、キャスト法、LB法、インクジェット法等)により薄膜を形成する。ここで真空蒸着法や溶液塗布法等によって層を形成すると、結晶化等が起こりにくく経時安定性に優れる。また塗布法で成膜する場合は、適当なバインダー樹脂と組み合わせて膜を形成することもできる。
【0094】
上記バインダー樹脂としては、ポリビニルカルバゾール樹脂、ポリカーボネート樹脂、ポリエステル樹脂、ABS樹脂、アクリル樹脂、ポリイミド樹脂、フェノール樹脂、エポキシ樹脂、シリコン樹脂、尿素樹脂等が挙げられるが、これらに限定されるものではない。また、これらバインダー樹脂は、ホモポリマー又は共重合体として一種単独で使用してもよいし、二種以上を混合して使用してもよい。さらに必要に応じて、公知の可塑剤、酸化防止剤、紫外線吸収剤等の添加剤を併用してもよい。
【0095】
本発明に係る有機発光素子は、表示装置や照明装置に用いることができる。他にも電子写真方式の画像形成装置の露光光源や液晶表示装置のバックライト等がある。
【0096】
表示装置は本実施形態に係る有機発光素子を表示部に有する。この表示部は複数の画素を有する。この画素は本実施形態に係る有機発光素子と発光輝度を制御するためのスイッチング素子の一例としてTFT素子とを有し、この有機発光素子の陽極又は陰極とTFT素子のドレイン電極又はソース電極とが接続されている。表示装置はPC等の画像表示装置として用いることができる。
【0097】
表示装置は、エリアCCD、リニアCCD、メモリーカード等からの画像情報を入力する入力部を有し、入力された画像を表示部に出力する画像入力装置でもよい。また、撮像装置やインクジェットプリンタが有する表示部として、外部から入力された画像情報を表示する画像出力機能と操作パネルとして画像への加工情報を入力する入力機能との両方を有していてもよい。また表示装置はマルチファンクションプリンタの表示部に用いられてもよい。
【0098】
次に、本実施形態に係る有機発光素子を使用した表示装置について図1を用いて説明する。
【0099】
図1は、本発明の有機発光素子を使用した表示装置の例を示す断面模式図である。図1の表示装置1では、有機発光素子とTFT素子との組み合わせが2組図示されている。構造の詳細を以下に説明する。
【0100】
図1の表示装置1は、ガラス等からなる基板1と、その上部にTFT素子又は有機化合物層を保護するための防湿膜2が設けられている。また符号3は金属材料からなるゲート電極3である。一方、符号4はゲート絶縁膜4であり、符号5は半導体層5である。
【0101】
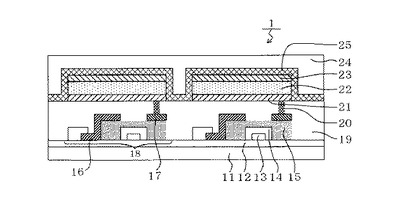
TFT素子8は、半導体層5と、ドレイン電極6と、ソース電極7と、を有している。そしてTFT素子8の上部には、絶縁膜9が設けられている。ここで図1の表示装置1においては、有機発光素子が有する陽極11と、ソース電極7と、がコンタクトホール10を介して接続されている。尚、本発明の表示装置は、図1の構成に限られず、陽極又は陰極のうちいずれか一方とTFT素子が有するソース電極又はドレイン電極のいずれか一方とが電気接続されていればよい。
【0102】
また図1の表示装置1において、有機化合物層22は、単一の層として表記されている。ただし、本発明において、有機化合物層22は単一の層に限定されるものではなく、複数の層からなる積層体であってもよい。また陰極23の上には有機発光素子の劣化を抑制するための第一の保護層24や第二の保護層25が設けられている。
【0103】
一方、図1の表示装置1において、スイッチング素子については特に制限はなく、単結晶シリコン基板やMIM素子、a−Si型の素子等を用いてもよい。
【実施例】
【0104】
[実施例1]例示化合物A−1の合成
【0105】
【化11】
【0106】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−2(フェニルボロン酸):1.5g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0107】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、290℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−1を1.45g得た(収率80%)。
【0108】
得られた化合物の同定は質量分析によって行った。
【0109】
[MALDI−TOF−MS(マトリックス支援イオン化−飛行時間型質量分析)]
実測値:m/z=364.55、計算値:C25H16OS=364.09
【0110】
また例示化合物A−1について、以下の方法でT1エネルギーの測定を行った。調製した例示化合物A−1のトルエン希薄溶液について、Ar雰囲気下、77K、励起波長350nmにおいて燐光スペクトルの測定を行った。得られた燐光スペクトルの第一発光ピークのピーク波長からT1エネルギーを求めた。その結果、T1エネルギーは、波長換算で475nmであった。
【0111】
次に、例示化合物A−1について、以下の方法でエネルギーギャップの測定を行った。ガラス基板上に例示化合物A−1を加熱蒸着して、膜厚20nmの蒸着薄膜を得た。この蒸着薄膜について、紫外可視分光光度計(日本分光株式会社製V−560)を用いて吸光スペクトルを測定した。得られた吸光スペクトルの吸収端から、例示化合物A−2のエネルギーギャップを求めた。その結果、エネルギーギャップは3.4eVであった。
【0112】
[実施例2]例示化合物A−3の合成
【0113】
【化12】
【0114】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−3(3−ビフェニルボロン酸):2.4g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0115】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、310℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−3を1.8g得た(収率70%)。
【0116】
[MALDI−TOF−MS]
実測値:m/z=515.98、計算値:516.15
また例示化合物A−3について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で474nmであった。さらに例示化合物A−3について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物A−3のエネルギーギャップは3.3eVであった。
【0117】
[実施例3]例示化合物A−5の合成
【0118】
【化13】
【0119】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−4:4.3g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0120】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をクロロベンゼンに加熱溶解してから熱時ろ過した後、クロロベンゼン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、380℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−5を1.8g得た(収率55%)。
【0121】
[MALDI−TOF−MS]
実測値:m/z=668.22、計算値:668.84
【0122】
また例示化合物A−5について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で475nmであった。さらに例示化合物A−5について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物A−5のエネルギーギャップは3.3eVであった。
【0123】
[実施例4]例示化合物A−8の合成
【0124】
【化14】
【0125】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−5:2.9g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0126】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、350℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−8を2.2g得た(収率75%)。
【0127】
[MALDI−TOF−MS]
実測値:m/z=596.45、計算値:596.22
【0128】
また例示化合物A−8について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で500nmであった。さらに例示化合物A−8について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物A−8のエネルギーギャップは2.7eVであった。
【0129】
[実施例5]例示化合物A−9の合成
【0130】
【化15】
【0131】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−6:2.9g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0132】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、340℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−9を2.1g得た(収率71%)。
【0133】
[MALDI−TOF−MS]
実測値:m/z=596.40、計算値:596.22
【0134】
また例示化合物A−9について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で482nmであった。
【0135】
[実施例6]例示化合物A−13の合成
【0136】
【化16】
【0137】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−7:4.3g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0138】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をクロロベンゼンに加熱溶解してから熱時ろ過した後、クロロベンゼン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を120℃で真空乾燥することにより、例示化合物A−13を1.7g得た(収率50%)。
【0139】
[MALDI−TOF−MS]
実測値:m/z=674.98、計算値:664.19
【0140】
また例示化合物A−13について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で503nmであった。
【0141】
[実施例7]例示化合物A−17の合成
【0142】
【化17】
【0143】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−8:5.3g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0144】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、380℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−17を3.0g得た(収率72%)。
【0145】
[MALDI−TOF−MS]
実測値:m/z=840.77、計算値:840.25
【0146】
また例示化合物A−17について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で500nmであった。さらに例示化合物A−17について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物A−17のエネルギーギャップは2.7eVであった。
【0147】
[実施例8]例示化合物A−21の合成
【0148】
【化18】
【0149】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.9g(5mmol)
F−9:2.0g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0150】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、310℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−21を1.7g得た(収率74%)。
【0151】
[MALDI−TOF−MS]
実測値:m/z=448.69、計算値:448.19
【0152】
また例示化合物A−21について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で463nmであった。
【0153】
[実施例9]例示化合物A−25の合成
【0154】
【化19】
【0155】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−10:1.85g(5mmol)
F−6:2.9g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0156】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、340℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−25を1.9g得た(収率64%)。
【0157】
[MALDI−TOF−MS]
実測値:m/z=596.89、計算値:596.22
【0158】
また例示化合物A−25について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で484nmであった。
【0159】
[実施例10]例示化合物B−1の合成
【0160】
【化20】
【0161】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−11:3.7g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0162】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、340℃の条件下で昇華精製を行うことにより、高純度の例示化合物B−1を1.7g得た(収率59%)。
【0163】
[MALDI−TOF−MS]
実測値:m/z=576.67、計算値:576.07
【0164】
また例示化合物C−1について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で456nmであった。さらに例示化合物B−1について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物B−1のエネルギーギャップは3.5eVであった。
【0165】
[実施例11]例示化合物B−2の合成
【0166】
【化21】
【0167】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−12:3.7g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0168】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、330℃の条件下で昇華精製を行うことにより、高純度の例示化合物B−2を1.9g得た(収率65%)。
【0169】
[MALDI−TOF−MS]
実測値:m/z=576.46、計算値:576.07
【0170】
また例示化合物B−2について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で455nmであった。さらに例示化合物B−2について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物B−2のエネルギーギャップは3.5eVであった。
【0171】
[実施例12]例示化合物B−3の合成
【0172】
【化22】
【0173】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−13:3.5g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0174】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、330℃の条件下で昇華精製を行うことにより、高純度の例示化合物B−3を1.5g得た(収率56%)。
【0175】
[MALDI−TOF−MS]
実測値:m/z=544.87、計算値:C37H20O3S=544.11
【0176】
また例示化合物B−3について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で454nmであった。さらに例示化合物B−3について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物B−3のエネルギーギャップは3.5eVであった。
【0177】
[実施例13]例示化合物C−2の合成
【0178】
【化23】
【0179】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−14:1.45g(5mmol)
F−15:3.0g(6mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0180】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、330℃の条件下で昇華精製を行うことにより、高純度の例示化合物C−2を2.2g得た(収率76%)。
【0181】
[MALDI−TOF−MS]
実測値:m/z=590.84、計算値:590.17
【0182】
また例示化合物C−2について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で490nmであった。さらに例示化合物C−2について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物C−2のエネルギーギャップは3.6eVであった。
【0183】
[実施例14]例示化合物C−7の合成
【0184】
【化24】
【0185】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−14:3.5g(12mmol)
F−16:2.0g(5mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0186】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、330℃の条件下で昇華精製を行うことにより、高純度の例示化合物C−7を2.2g得た(収率77%)。
【0187】
[MALDI−TOF−MS]
実測値:m/z=574.79、計算値:574.11
【0188】
また例示化合物C−7について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で470nmであった。さらに例示化合物C−7について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物C−7のエネルギーギャップは3.4eVであった。
【0189】
[実施例15]例示化合物C−9の合成
【0190】
【化25】
【0191】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−14:1.45g(5mmol)
F−17:2.3g(6mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0192】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、310℃の条件下で昇華精製を行うことにより、高純度の例示化合物C−9を1.5g得た(収率63%)。
【0193】
[MALDI−TOF−MS]
実測値:m/z=470.54、計算値:470.08
また例示化合物C−9について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で453nmであった。さらに例示化合物C−9について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物C−9のエネルギーギャップは3.5eVであった。
【0194】
[実施例16]例示化合物C−10の合成
【0195】
【化26】
【0196】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−14:1.5g(5mmol)
F−18:3.6g(6mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0197】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、320℃の条件下で昇華精製を行うことにより、高純度の例示化合物C−10を2.4g得た(収率70%)。
【0198】
[MALDI−TOF−MS]
実測値:m/z=678.87、計算値:678.20
【0199】
また例示化合物C−10について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で480nmであった。
【0200】
[実施例17]
本実施例では、基板上に順次陽極/ホール輸送層/発光層/ホールブロッキング層/電子輸送層/陰極が順次設けられている有機発光素子を、以下に示す方法で作製した。尚、本実施例で使用した化合物の一部を下記に示す。
【0201】
【化27】
【0202】
ガラス基板上に、スパッタ法により、ITOを成膜し陽極を形成した。このとき陽極の膜厚を120nmとした。このようにITO電極が形成されている基板を透明導電性支持基板(ITO基板)として後の工程で使用した。次に、このITO基板上に、下記表4に示される有機化合物層及び電極層を、1×10-5Paの真空チャンバー内における抵抗加熱による真空蒸着によって連続的に成膜した。このときITO電極に対向する電極(金属電極層)の面積を3mm2となるように作製した。
【0203】
【表4】
【0204】
次に、有機発光素子が水分の吸着によって素子劣化が起こらないように、乾燥空気雰囲気中で保護用ガラス板をかぶせアクリル樹脂系接着材で封止した。以上のようにして有機発光素子を得た。
【0205】
得られた有機発光素子について、ITO電極を正極、Al電極を負極にして、5.1Vの印加電圧をかけたところ、発光効率が47cd/Aで、輝度2000cd/m2の緑色発光が観測された。またこの素子においてCIE色度座標は、(x,y)=(0.30,0.63)であった。
【0206】
[実施例18〜29]
実施例17において、ホールブロック層(HB層)に含まれるホールブロック材料(HB材料)、並びに発光層に含まれる第一ホスト、第二ホスト及びゲストの組み合わせを下記表5に変更した他は、実施例17と同様の方法で素子を作製した。また得られた素子について実施例17と同様に評価を行った。結果を表5に示す。
【0207】
【表5】
【0208】
表5より、本発明のチオキサントン化合物は、燐光発光する有機発光素子において、電子輸送材料又は発光材料として用いることで良好な発光効率を得られることが分かった。
【0209】
[実施例30、31、比較例1]
実施例17において、ホールブロック層(HB層)に含まれるホールブロック材料(HB材料)、並びに発光層に含まれる第一ホスト、第二ホスト及びゲストの組み合わせを下記表6に変更した他は、実施例17と同様の方法で素子を作製した。ここで比較例1で使用した化合物H−1Aを以下に示す。
【0210】
【化28】
【0211】
得られた素子について素子の安定性を評価するために電流値40mA/cm2における有機発光素子の輝度半減寿命を測定した。結果を表6に示す。
【0212】
【表6】
【0213】
表6に示されるように、燐光発光する有機発光素子において、本発明のチオキサントン化合物は、比較例の化合物よりも、輝度半減寿命が長いことが示された。これは、カルボニル骨格によって適切なLUMO準位を有するために電荷のバランスが取れた安定な素子構造になるからである。このことによって有機発光素子において良好な素子寿命を得られることが分かった。
【産業上の利用可能性】
【0214】
以上のように、本発明のチオキサントン化合物は、T1エネルギーが高くLUMO準位が深い化合物である。このため有機発光素子の構成材料として使用すると、発光効率の高く、劣化しにくい安定な有機発光素子を得ることができる。
【符号の説明】
【0215】
1:表示装置、11:基板、12:防湿膜、13:ゲート電極、14:絶縁膜、15:半導体層、16:ドレイン電極、17:ソース電極、18:TFT素子、19:絶縁膜、20:コンタクトホール、21:陽極、22:有機化合物層、23:陰極、24:第一の保護層、25:第二の保護層
【技術分野】
【0001】
本発明はチオキサントン化合物及びこれを有する有機発光素子に関する。
【背景技術】
【0002】
有機発光素子は、陽極と陰極と、これら両電極間に配置される有機化合物層とを有する電子素子である。各電極から注入させる正孔(ホール)及び電子が有機化合物層に含まれる発光層内で再結合することで生成される励起子が基底状態に戻る際に、有機発光素子は、光を放出する。有機発光素子の最近の進歩は著しく、その特徴として、低駆動電圧、発光波長の多様化、高速応答性、発光デバイスの薄型・軽量化が可能であることが挙げられる。
【0003】
ところで有機発光素子は、発光に関与する励起子の種類によって蛍光発光素子と燐光発光素子とに大別される。そのうち燐光発光素子は、有機発光素子を構成する有機化合物層、具体的には発光層中に燐光発光材料を有する電子素子である。ここで燐光発光材料は、正孔及び電子が再結合することによって生じるエネルギーを吸収して三重項励起子を生成する。このため燐光発光素子は、この三重項励起子由来の発光が得られる有機発光素子である。
【0004】
ところで、燐光の発光量子収率は理論上蛍光の量子収率の4倍にすることができるので、近年では燐光発光素子に注目が集まっている。しかし燐光発光素子において、発光効率についてはさらなる改善の余地がある。
【0005】
一方、燐光発光素子で使用される燐光発光材料について様々な提案がなされている。燐光発光材料として、例えば、下記に示される化合物H−1(チオキサンテン化合物、特許文献1参照。)及び化合物H−2(チオキナクリドン化合物、特許文献2参照。)が提案されている。
【0006】
【化1】
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開平03−177486号公報
【特許文献2】特開平11−54279号公報
【発明の概要】
【発明が解決しようとする課題】
【0008】
ところで特許文献1に開示されているチオキサンテン化合物は、ケトン骨格を有していないため、LUMO準位が浅くなる。また、特許文献2に開示されているチオキナクリドン化合物は、基本骨格のバンドギャップが狭く、それ故にT1エネルギーが低くなってしまう。
【0009】
一方で、有機発光素子においては、発光層に含まれる発光材料の他に、電子輸送層に含まれる電子輸送性材料の開発が求められている。具体的には、LUMO準位が2.7eV以上と深く、かつ化学的に安定な有機化合物が求められている。
【0010】
特に、発光層に燐光発光材料が含まれる有機発光素子においては、素子を構成する材料である発光材料や電子輸送材料についてT1エネルギーが高いことが求められている。
【0011】
本発明は、上述した課題を解決するためになされたものであり、その目的は、発光効率が高く、駆動電圧の低い有機発光素子を提供することである。
【課題を解決するための手段】
【0012】
本発明のチオキサントン化合物は、下記一般式[1]で示されることを特徴とする。
【0013】
【化2】
(式[1]において、R1乃至R8は、それぞれ水素原子、炭素数1乃至4のアルキル基又はフェニル基、ビフェニル基、ナフチル基、フェナンスレン基、フルオレニル基、トリフェニレニル基、ジベンゾフラニル基及びジベンゾチオフェニル基のいずれかから選択されるアリール基を表す。ただし、R1乃至R8のうち少なくともひとつが前記アリール基である。
【0014】
尚、前記アルキル基及び前記アリール基は、さらにアルキル基、芳香族炭化水素基又は芳香族複素環基を有してもよい。)
【発明の効果】
【0015】
本発明のチオキサントン化合物は、T1エネルギーが2.3eV以上と高く、かつLUMO準位が2.7eV以上と深い化合物である。このため本発明によれば、発光効率が高く、駆動電圧の低い有機発光素子を提供することができる。
【図面の簡単な説明】
【0016】
【図1】有機発光素子と、有機発光素子に接続するスイッチング素子とを示す断面模式図である。
【発明を実施するための形態】
【0017】
まず本発明のチオキサントン化合物について説明する。本発明のチオキサントン化合物は下記一般式[1]で示される化合物である。
【0018】
【化3】
【0019】
式[1]において、R1乃至R8は、それぞれ水素原子、炭素数1乃至4のアルキル基又はフェニル基、ビフェニル基、ナフチル基、フェナンスレン基、フルオレニル基、トリフェニレニル基、ジベンゾフラニル基及びジベンゾチオフェニル基のいずれかから選択されるアリール基を表す。
【0020】
R1乃至R8で表されるアルキル基として、メチル基、エチル基、n−プロピル基、iso−プロピル基、n−ブチル基、iso−ブチル基、sec−ブチル基、tert−ブチル基が挙げられる。
【0021】
ただし、R1乃至R8のうち少なくともひとつが前記アリール基である。
【0022】
尚、上記アルキル基及び上記アリール基は、さらにアルキル基、芳香族炭化水素基又は芳香族複素環基を有してもよい。
【0023】
上記アルキル基及び上記アリール基がさらに有してもよいアルキル基として、例えば、メチル基、エチル基、n−プロピル基、iso−プロピル基、n−ブチル基、iso−ブチル基、sec−ブチル基、tert−ブチル基等が挙げられる。
【0024】
上記アルキル基及び上記アリール基がさらに有してもよい芳香族炭化水素基として、フェニル基、ナフチル基、フェナントリル基、フルオレニル基等が挙げられる。
【0025】
上記アルキル基及び上記アリール基がさらに有してもよい芳香族複素環基として、チエニル基、ジベンゾフラニル基、ジベンゾチオフェニル基、ピロリル基、ピリジル基等の複素芳香環基が挙げられる。
【0026】
尚、上記アルキル基及び上記アリール基は、メトキシ基、エトキシ基等のアルコキシ基、フェノキシ基、ナフトキシ基等のアリールオキシ基、フッ素、塩素、臭素、ヨウ素等のハロゲン原子をさらに有してもよい。
【0027】
本発明のチオキサントン(チオキサンテン−9−オン)化合物は、チオキサントン自身でT1エネルギーが3.0eVと高く、かつLUMO準位が2.7eV以上と深い。このため、有機発光素子の高効率化や安定性の向上をもたらすと考えられる。
【0028】
次に、本発明のチオキサントン化合物の合成方法について説明する。本発明のチオキサントン化合物は、例えば、下記に示される合成スキームを利用して合成される。尚、下記合成スキームにおいて、Arは、フェニル基、ナフチル基、フェナンスレン基、フルオレニル基、トリフェニレニル基、ジベンゾフラニル基又はジベンゾチオフェニル基を表す。
【0029】
【化4】
【0030】
上記合成スキームに示されるように、本発明のチオキサントン化合物は、Pd触媒を用いたチオキサントンのハロゲン体(X)とアリールボロン酸もしくはアリールボロン酸エステルとのカップリング反応で合成することができる。尚、原料となるチオキサントンのハロゲン体は、市販品を利用することができる。
【0031】
ここで上記反応スキームを利用して本発明のチオキサントン化合物を合成する場合、Arを適宜選択することにより、所望のチオキサントン化合物を合成することができる。
【0032】
また本発明のチオキサントン化合物を有機発光素子の構成材料として使用する場合には、使用する直前に昇華精製を行うのが好ましい。なぜなら有機化合物の高純度化の観点からして昇華精製は精製効果(高純度化効果)が大きいからである。ただし昇華精製を行って有機化合物を精製する際には、一般に分子量が大きい化合物ほど高温が必要とされる。また化合物によっては高温による熱分解等を起こしやすくなる。従って、有機発光素子の構成材料として使用される有機化合物は、過大な加熱なく昇華精製を行うことができるように、分子量が1000以下であることが好ましい。
【0033】
本発明のチオキサントン化合物において、主骨格たるチオキサントン骨格は、カルボニル基を有しているため電子親和性が高い。このため発明者は、カルボニル基に由来する電子輸送能を考慮して、このチオキサントン骨格が電子輸送材料となる化合物として適した骨格であると考察した。
【0034】
また本発明のチオキサントン化合物は、チオキサントン骨格中の環構造の一部としてカルボニル基の炭素原子を含めているため、チオキサントン骨格の全体が平面構造になる。この構造を有するため、本発明のチオキサントン化合物は、分子同士の重なり合いが起こりやすくなり、固体状態における分子間電子移動が効率的に起こりやすくなる。これらの構造的性質から、本発明のチオキサントン化合物は、有機発光素子の構成材料、具体的には、陰極や隣接する層からの電子を注入・輸送する機能を担う材料として適しているといえる。即ち、本発明のチオキサントン化合物は、電子注入・輸送層に含まれる電子注入・輸送性材料や発光層のホストとして好適に用いることができる。
【0035】
またチオキサントン骨格が示す他の特長として、高いT1エネルギーがある。実際に無置換のチオキサントン(式[1]においてR1乃至R8が全て水素原子の化合物)の希薄トルエン溶液を77Kにおいて燐光スペクトル測定して得られる0−0バンドからT1エネルギーを求めることができる。その結果無置換のチオキサントンのT1エネルギーは2.74eV(451nm)であり、この値は青領域の一部(455nm〜480nm)や緑色領域(490nm〜530nm)よりも十分エネルギーが高い。従って、本発明のチオキサントン化合物は、発光色が幅広い燐光発光素子において電子輸送層に含まれる電子輸送材料や発光層のホストとして本質的に適用できる。
【0036】
ところで、特許文献1に開示されている化合物H1は、チオキサントン骨格中のカルボニルがメチレンになったチオキサンテン化合物である。ここで化合物H1は、T1エネルギーが高い点(波長換算で361nm)では優れているが、カルボニル基が存在しないために、チオキサントン化合物の特徴である電子受容性、電子輸送性が大きく低下している。
【0037】
ここで本発明のチオキサントン化合物、並びに特許文献1及び2にそれぞれ開示されている化合物の主骨格(チオキサントン骨格、チオキサンテン骨格、チオキナクリドン骨格)のT1エネルギーについて測定及び計算を行った。
【0038】
ここで測定によってT1エネルギーを求める際には、77Kにおけるトルエン溶液状態での燐光スペクトルの測定結果から求めた。より具体的には、燐光スペクトルの測定を行った後、この測定で得られた0−0バンドからT1エネルギーを求めた。
【0039】
一方、計算によってT1エネルギーを求める際には、密度汎関数法(Density Functional Theory)を用いて、B3LYP/6−31G*レベルでの分子軌道計算を行った。上記計算及び測定によって得られたT1エネルギーの値を下記表1に示す。
【0040】
【表1】
【0041】
表1より、チオキサンテンのLUMOがチオキサントンよりも浅いことが示されている。またチオキナクリドンは、チオキサントン骨格を含む構造であるが、チオキサントン骨格と比較するとT1エネルギーが大幅に低くなる(波長換算で約570nm)。このためチオキナクリドンは、発光色が青や緑のりん光発光素子の構成材料として適さない化合物である。
【0042】
以上より、表1で示される化合物において、発光色が青や緑のりん光発光素子の構成材料としてふさわしいT1エネルギーを有し、電子受容性や電子輸送性に適した深いHOMOを有する化合物は、チオキサントン化合物のみである。
【0043】
ところで、特許文献1及び2にて開示されているチオキサンテン化合物及びチオキナクリドン化合物は、共にその用途は蛍光性の発光材料である。一方、本発明のチオキサントン化合物は、化合物中に含まれるカルボニル基に由来する電子輸送性に着目して使用されるものであり、その用途は、電子輸送層に含まれる電子輸送性材料や発光層のホストである。
【0044】
一方で、有機発光素子を構成する電子輸送層あるいは発光層に、発光材料以外の化合物として本発明のチオキサントン化合物を使用する場合、以下のことを考慮することが大切である。それはその有機発光素子が有する発光材料の発光色を考慮して最適なバンドキャップ及びより深いLUMOを、本発明のチオキサントン化合物が有することである。
【0045】
ここで本発明のチオキサントン化合物において、化合物のバンドギャップを狭くするためには、チオキサントン骨格と共役がつながる部位に、アリール基等の共役を有する置換基を導入する必要がある。チオキサントン骨格と共役がつながる部位となる置換位置とは、下記に示されるチオキサントン骨格の1位乃至8位の炭素である。よってチオキサントン骨格の1位乃至8位にアリール基を導入することが好ましい。
【0046】
【化5】
【0047】
また、化合物が有する共役を広げつつバンドギャップを狭く制御するためにはチオキサントン骨格に対して立体障害の小さい置換位置に置換基を設けることが好ましい。この点を考慮すると、チオキサントン骨格について置換基を導入する場合、置換基が導入される位置は、式[1]中のR2、R3、R6及びR7の中から1つ又は2つ選択するのが好ましい。R2及びR3から1つ選択しR6及びR7から1つ選択する組み合わせがより好ましい。そして、R2及びR7の組み合わせ又はR3及びR6の組み合わせが特に好ましい。
【0048】
一方で、式[1]中のR1、R4、R5及びR8は、いずれも水素原子であることが好ましい。また置換基を複数置換する場合、置換する置換基は何れも同じにすることが好ましい。
【0049】
他方で、チオキサントン骨格のように平面性が高い骨格にアルキル基や芳香環基を導入することで、溶媒に対する溶解性や真空蒸着時の昇華性、薄膜状態のアモルファス性を向上させることができる。ここで、アルキル基を導入する際には、炭素数が炭素数1乃至4のアルキル基を導入すると化合物の昇華性を損なわずにアモルファス性を向上することができる。
【0050】
ところで、本発明のチオキサントン化合物を、燐光発光素子の構成材料である電子輸送性材料や発光層のホストとして使用する際には、燐光発光材料より高いT1エネルギーと最適なHOMO−LUMO準位を有している必要がある。ここで、チオキサントン骨格にアリール基を導入することで化合物のT1エネルギーをある程度調整することができる。
【0051】
特に、発光材料である燐光発光材料を有する有機発光素子において、本発明のチオキサントン化合物を発光層あるいは発光層に隣接する電荷輸送層(電子輸送層)の構成材料として使用する際には、化合物が有するT1エネルギーが重要となる。つまり燐光発光材料の発光色が青乃至赤、即ち、発光波長のスペクトルの最大ピークが455nm以上620nm以下である場合において、その燐光発光材料の発光色に対応するように本発明のチオキサントン化合物のT1エネルギーを設定することが重要である。
【0052】
ここで発明者は、本発明のチオキサントン化合物のT1エネルギーを設定するにあたり、式[1]中のR1乃至R8のいずれかに導入される置換基(縮合環)のT1エネルギーに注目した。下記表2は、ベンゼン及び主な縮合環のT1エネルギー(波長換算値)を示す表である。
【0053】
【表2】
【0054】
表2を考慮すると、ベンゼン・縮合環の中でも、ベンゼン、ナフタレン、フェナンスレン、フルオレン、トリフェニレン、クリセン、ジベンゾフラン、ジベンゾチオフェン及びピレンが好ましいといえる。
【0055】
さらに燐光発光材料の発光色が青から緑である場合、本発明のチオキサントン化合物の主骨格に導入される置換基(ベンゼン・縮合環)として好ましい縮合環は、ベンゼン、ナフタレン、フェナンスレン、フルオレン、トリフェニレン、ジベンゾフラン又はジベンゾチオフェンである。尚、この場合における「青から緑」とは、波長にして455nm以上530nm以下の範囲であり、440nm付近の深青は含まれない。
【0056】
また本発明のチオキサントン化合物の主骨格に導入される置換基のその置換位置は、より電子受容性を高めるために、化合物自体のLUMOを深くする位置が好ましい。下記表3は、置換基(フェニル基)の置換位置とHOMOエネルギー及びLUMOエネルギーとの関係を示す表である。
【0057】
【表3】
【0058】
表3より、同じ置換基(フェニル基)を置換した場合であっても、2位置換又は3位置換に相当するR2、R3、R6、R7に置換基を置換する方が、チオキサントン化合物のLUMOはより深くなるといえる。これより、電子受容性の高さが要求される電子輸送層や発光層の材料として本発明のチオキサントン化合物を使用する際には、R2、R3、R6及びR7のいずれかにアリール基を置換することが好ましい。
【0059】
本発明の化合物は燐光発光する有機発光素子の電子輸送層や発光層の少なくともいずれか一方に好ましく用いることができる。これは本発明に係るチオキサントン化合物はそのT1エネルギーが燐光発光材料のそれより高いからである。そして本発明に係るチオキサントン化合物はそのような層に用いる場合にも適切な広さのバンドギャップを有しているので好ましい。
【0060】
本発明のチオキサントン化合物の具体例を以下に示す。ただし、本発明ハこれらに限定されるものではない。
【0061】
【化6】
【0062】
【化7】
【0063】
【化8】
【0064】
例示化合物のうちA群に示す化合物は、式[1]に示されるチオキサンテン骨格中のR2及びR7又はR3乃至R6に同一の置換基(アルキル基又はアリール基)が導入されている化合物である。A群に示す化合物は、中心骨格であるチオキサントン骨格に対して同一の置換基を所定の位置に2つ導入することで、対称軸を持った安定な骨格を有している。従って、A群の化合物は、化学的安定性が非常に高い上に電子輸送性を有する。よって、それらを電子輸送性材料又は発光層に含まれるホストあるいはアシスト材料として使用すると、素子の長寿命化が期待できる。
【0065】
例示化合物のうちB群に示す化合物は、式[1]に示されるチオキサンテン骨格中のR1乃至R8のいずれか(例えば、R2及びR7又はR3乃至R6)に、複素環基あるいは複素環を含む置換基が導入されている化合物である。B群に示す化合物に含まれるヘテロ原子を環状基の内部に有する複素環基は、芳香族炭化水素基ほどではないが、芳香族炭化水素基に準じた安定性を有する。このため、B群の化合物を電子輸送性材料又は発光層に含まれるホストあるいはアシスト材料として使用すると、素子の長寿命化が期待できる。
【0066】
例示化合物のうちC群に示す化合物は、式[1]に示されるチオキサンテン骨格中のR1乃至R8のいずれか1つに置換基(アルキル基、アリール基、複素環基あるいはこれらを組み合わせた複合置換基)が導入されている化合物である。C群に示す化合物は、化合物全体として対称性が無いために、HOMO−LUMOがCT性を有する場合がある。これを利用して発光材料に適したHOMO−LUMOに調整することができる。このため、C群の化合物を電子輸送性材料又は発光層に含まれるホストあるいはアシスト材料として使用すると、素子の長寿命化が期待できる。
【0067】
例示化合物のうちD群に示す化合物は、A群乃至C群の思想を組み合わせたものであり、対称性を低くすることで化合物自体の溶解性、電子移動度の制御をすることができる。
【0068】
次に、本発明の有機発光素子を説明する。本発明の有機発光素子は、互いに対向しあう一対の電極である陽極と陰極と、陽極と陰極との間に配置される有機化合物層と、から構成される。そして、この有機化合物層に本発明のチオキサントン化合物が含まれる。
【0069】
有機発光素子を構成する有機化合物層は、発光層又は発光機能を有する層が含まれていれば、単一の層であってもよいし、複数の層からなる積層体であってもよい。ここで有機化合物層が複数の層からなる積層体である場合、有機化合物層を構成する層としては、ホール注入層、ホール輸送層、発光層、ホールブロック層、電子輸送層、電子注入層、エキシトンブロック層等が挙げられる。上記群に示されている層の中から複数の層を適宜選択し、かつそれらを組み合わせて用いることができる。
【0070】
ただし本発明の有機発光素子の構成はこれらに限定されるものではない。例えば、電極と有機化合物層との界面に絶縁性層、接着層あるいは干渉層を設ける、電子輸送層もしくはホール輸送層がイオン化ポテンシャルの異なる二層から構成される等多様な層構成をとることができる。
【0071】
本発明の有機発光素子の素子形態としては、基板と反対側の電極から光を取り出すいわゆるトップエミッション方式でも、基板側の電極から光を取り出すいわゆるボトムエミッション方式でもよい。また基板や電極を光透過性の材料にして、両面から光を取り出す構成にしてもよい。
【0072】
本発明のチオキサントン化合物は、有機発光素子を構成する有機化合物層に含まれるが、好ましくは、電子輸送層、ホール・エキシトンブロッキング層、電子注入層又は発光層に含まれる。ここで本発明のチオキサントン化合物が電子輸送層又はホール・エキシトンブロッキング層に含まれる場合は、本発明のチオキサントン化合物は電子輸送材料として使用される。
【0073】
ところでホールブロッキング層は、陽極から注入されたホールをブロックする(陰極方向へ移動させない)層の意味として用いられることがあるが、本発明においては、発光層の陰極側で発光層と隣接する層をホールブロッキング層と呼ぶことにする。この理由として、本発明のチオキサントン化合物は、本来はホールをブロッキングすることを目的する材料としてではなく、電子を発光層へ輸送するための材料として使用されているからである。しかし、層を設ける位置が一般的なホールブロッキング層と同じ位置にあることと、配置位置に関して電子輸送層と混同しないために、配置位置に合わせてホールブロッキング層と記すことがある。
【0074】
本発明のチオキサントン化合物がホールブロッキング層に含まれる場合、発光層は複数種の成分から構成されていてよく、それらを主成分と副成分とに分類することができる。つまり、発光層を構成する全化合物の中で重量比が最大の化合物である主成分と、それ以外の副成分とに分類することができる。
【0075】
ここで主成分は、ホストと呼ばれている。一方、副成分は、材料自体の機能に応じてゲスト(ドーパント)、発光アシスト材料又は電荷注入材料と呼ばれている。尚、発光アシスト材料及び電荷注入材料は、同一構造の有機化合物であってもよいし、異なる構造の有機化合物であってもよい。また副成分であるものの、材料によっては、ゲスト等と区別する意味で第二ホストと呼ぶこともできる。
【0076】
本発明の有機発光素子において、ゲストとは、発光層内で主たる発光を担う化合物である。これに対してホストとは、発光層内でゲストの周囲にマトリックスとして存在する化合物であって、主にキャリアの輸送、及びゲスト材料への励起エネルギー供与を担う化合物である。ここでホストは、複数種の材料を有してもよい。
【0077】
発光層内に含まれるゲストの濃度は、発光層の構成材料の全体量を基準として、0.01重量%以上50重量%以下であり、好ましくは0.1重量%以上20重量%以下である。さらに好ましくは、濃度消光を防ぐためにゲストの濃度は0.01重量%以上10重量%以下であることが望ましい。またゲストはホストからなる層の全体に均一に含まれてもよいし、濃度勾配を有して含まれてもよい。また特定の領域に部分的に含ませてゲストを含まないホスト層の領域を部分的に設けてもよい。
【0078】
本発明のチオキサントン化合物は、ゲストたる燐光発光材料に対応する発光層のホストや発光層に含まれる電荷注入材料又は電子輸送層・ホールブロック層に含まれる電子輸送材料として使用される。このとき燐光発光材料の発光色は特に限定されないが、その最大発光ピーク波長が460nm以上530nm以下の範囲にある青色から緑色発光材料であることが好ましい。特に最大発光ピーク波長が490nm以上530nm以下の範囲にある緑色発光材料であることが好ましい
一般に燐光発光する有機発光素子では、ホスト材料のT1から非放射失活による発光効率の低下を防ぐために、ホスト材料のT1エネルギーはゲスト材料である燐光発光材料のT1エネルギーよりも高いことが必要とされている。
【0079】
本発明のチオキサントン化合物は、その中心となるチオキサントン骨格のT1エネルギーが451nmであるので、緑色燐光発光材料のT1エネルギーよりも高い。従ってこれを緑色発光する有機発光素子の発光層もしくはその周辺層に用いると発光効率の高い有機発光素子を得る事ができる。
【0080】
さらに本発明のチオキサントン化合物は、LUMO準位が深いので、これを電子注入材料、電子輸送材料又はホールブロッキング層の構成材料のみならず、発光層に含まれる第二ホストとして使用することができる。これらに示される用途で本発明のチオキサントン化合物を用いることで、素子の駆動電圧を低くすることができる。なぜならLUMO準位が深いと、発光層の陰極側に隣接する電子輸送層又はホールブロッキング層からの電子注入障壁が小さくなるからである。
【0081】
一方、本発明のチオキサントン化合物が発光層に含まれる場合、発光層は、本発明のチオキサントン化合物のみからなる層でもよいし、ホストとゲストとからなる層であってもよい。ここで発光層がホストとゲストとからなる層である場合、本発明のチオキサントン化合物は、ホストとして使用するのが好ましい。
【0082】
ここで本発明のチオキサントン化合物を発光層のホストとして使用する場合、対応するゲストとして、好ましくは、燐光発光材料である。ゲストとして使用される燐光発光材料は、具体的には、イリジウム錯体、白金錯体、レニウム錯体、銅錯体、ユーロピウム錯体、ルテニウム錯体等の金属錯体である。中でも燐光発光性の強いイリジウム錯体が好ましい。また、励起子やキャリアの伝達を補助することを目的として、発光層が複数の燐光発光材料を有していてもよい。
【0083】
以下にイリジウム錯体の具体例を示すが、本発明はこれらに限定されるものではない。
【0084】
【化9】
【0085】
また発光層に含まれるホストを本発明のチオキサントン化合物を含めて複数種にしてもよい。以下に本発明の有機発光素子の構成材料として使用されるホスト(本発明のチオキサントン化合物を除く。)の具体例を示すが、本発明はこれらに限定されるものではない。
【0086】
【化10】
【0087】
また本発明の有機発光素子においては、本発明の値オキサントン化合物以外にも、必要に応じて従来公知の低分子系及び高分子系の化合物を使用することができる。より具体的にはホール注入・輸送性材料、ホスト材料、発光性化合物あるいは電子注入・輸送性化合物等を一緒に使用することができる。以下にこれらの化合物例を挙げる。
【0088】
ホール注入・輸送性材料としては、陽極からのホールの注入が容易で、注入されたホールを発光層へと輸送することができるように、ホール移動度が高い材料が好ましい。ホール注入輸送性能を有する低分子及び高分子系材料としては、トリアリールアミン誘導体、フェニレンジアミン誘導体、スチルベン誘導体、フタロシアニン誘導体、ポルフィリン誘導体、ポリ(ビニルカルバゾール)、ポリ(チオフェン)、その他導電性高分子が挙げられる。
【0089】
主に発光機能に関わる発光材料としては、前述の燐光発光ゲスト材料、もしくはその誘導体以外に、縮環化合物(例えばフルオレン誘導体、ナフタレン誘導体、ピレン誘導体、ペリレン誘導体、テトラセン誘導体、アントラセン誘導体、ルブレン等)、キナクリドン誘導体、クマリン誘導体、スチルベン誘導体、トリス(8−キノリノラート)アルミニウム等の有機アルミニウム錯体、有機ベリリウム錯体、及びポリ(フェニレンビニレン)誘導体、ポリ(フルオレン)誘導体、ポリ(フェニレン)誘導体等の高分子誘導体が挙げられる。
【0090】
電子注入・輸送性材料としては、陰極からの電子の注入が容易で注入された電子を発光層へ輸送することができるものから任意に選ぶことができ、ホール注入輸送性材料のホール移動度とのバランス等を考慮して選択される。電子注入輸送性能を有する材料としては、オキサジアゾール誘導体、オキサゾール誘導体、ピラジン誘導体、トリアゾール誘導体、トリアジン誘導体、キノリン誘導体、キノキサリン誘導体、フェナントロリン誘導体、有機アルミニウム錯体等が挙げられる。
【0091】
陽極を構成する材料としては仕事関数がなるべく大きなものがよい。例えば金、白金、銀、銅、ニッケル、パラジウム、コバルト、セレン、バナジウム、タングステン等の金属単体あるいはこれらを複数種組み合わせた合金、酸化錫、酸化亜鉛、酸化インジウム、酸化錫インジウム(ITO)、酸化亜鉛インジウム等の金属酸化物が使用できる。またポリアニリン、ポリピロール、ポリチオフェン等の導電性ポリマーも使用できる。これらの電極物質は一種類を単独で使用してもよいし、二種類以上を併用して使用してもよい。また、陽極は一層で構成されていてもよく、複数の層で構成されていてもよい。
【0092】
一方、陰極を構成する材料としては仕事関数の小さなものがよい。例えばリチウム等のアルカリ金属、カルシウム等のアルカリ土類金属、アルミニウム、チタニウム、マンガン、銀、鉛、クロム等の金属単体が挙げられる。あるいはこれら金属単体を複数種組み合わせた合金も使用することができる。例えばマグネシウム−銀、アルミニウム−リチウム、アルミニウム−マグネシウム等が使用できる。酸化錫インジウム(ITO)等の金属酸化物の利用も可能である。これらの電極物質は一種類を単独で使用してもよいし、二種類以上を併用して使用してもよい。また陰極は一層構成でもよく、多層構成でもよい。
【0093】
本発明の有機発光素子において、本発明の有機化合物を含有する層及びその他の有機化合物からなる層は、以下に示す方法により形成される。一般には真空蒸着法、イオン化蒸着法、スパッタリング、プラズマあるいは、適当な溶媒に溶解させて公知の塗布法(例えば、スピンコーティング、ディッピング、キャスト法、LB法、インクジェット法等)により薄膜を形成する。ここで真空蒸着法や溶液塗布法等によって層を形成すると、結晶化等が起こりにくく経時安定性に優れる。また塗布法で成膜する場合は、適当なバインダー樹脂と組み合わせて膜を形成することもできる。
【0094】
上記バインダー樹脂としては、ポリビニルカルバゾール樹脂、ポリカーボネート樹脂、ポリエステル樹脂、ABS樹脂、アクリル樹脂、ポリイミド樹脂、フェノール樹脂、エポキシ樹脂、シリコン樹脂、尿素樹脂等が挙げられるが、これらに限定されるものではない。また、これらバインダー樹脂は、ホモポリマー又は共重合体として一種単独で使用してもよいし、二種以上を混合して使用してもよい。さらに必要に応じて、公知の可塑剤、酸化防止剤、紫外線吸収剤等の添加剤を併用してもよい。
【0095】
本発明に係る有機発光素子は、表示装置や照明装置に用いることができる。他にも電子写真方式の画像形成装置の露光光源や液晶表示装置のバックライト等がある。
【0096】
表示装置は本実施形態に係る有機発光素子を表示部に有する。この表示部は複数の画素を有する。この画素は本実施形態に係る有機発光素子と発光輝度を制御するためのスイッチング素子の一例としてTFT素子とを有し、この有機発光素子の陽極又は陰極とTFT素子のドレイン電極又はソース電極とが接続されている。表示装置はPC等の画像表示装置として用いることができる。
【0097】
表示装置は、エリアCCD、リニアCCD、メモリーカード等からの画像情報を入力する入力部を有し、入力された画像を表示部に出力する画像入力装置でもよい。また、撮像装置やインクジェットプリンタが有する表示部として、外部から入力された画像情報を表示する画像出力機能と操作パネルとして画像への加工情報を入力する入力機能との両方を有していてもよい。また表示装置はマルチファンクションプリンタの表示部に用いられてもよい。
【0098】
次に、本実施形態に係る有機発光素子を使用した表示装置について図1を用いて説明する。
【0099】
図1は、本発明の有機発光素子を使用した表示装置の例を示す断面模式図である。図1の表示装置1では、有機発光素子とTFT素子との組み合わせが2組図示されている。構造の詳細を以下に説明する。
【0100】
図1の表示装置1は、ガラス等からなる基板1と、その上部にTFT素子又は有機化合物層を保護するための防湿膜2が設けられている。また符号3は金属材料からなるゲート電極3である。一方、符号4はゲート絶縁膜4であり、符号5は半導体層5である。
【0101】
TFT素子8は、半導体層5と、ドレイン電極6と、ソース電極7と、を有している。そしてTFT素子8の上部には、絶縁膜9が設けられている。ここで図1の表示装置1においては、有機発光素子が有する陽極11と、ソース電極7と、がコンタクトホール10を介して接続されている。尚、本発明の表示装置は、図1の構成に限られず、陽極又は陰極のうちいずれか一方とTFT素子が有するソース電極又はドレイン電極のいずれか一方とが電気接続されていればよい。
【0102】
また図1の表示装置1において、有機化合物層22は、単一の層として表記されている。ただし、本発明において、有機化合物層22は単一の層に限定されるものではなく、複数の層からなる積層体であってもよい。また陰極23の上には有機発光素子の劣化を抑制するための第一の保護層24や第二の保護層25が設けられている。
【0103】
一方、図1の表示装置1において、スイッチング素子については特に制限はなく、単結晶シリコン基板やMIM素子、a−Si型の素子等を用いてもよい。
【実施例】
【0104】
[実施例1]例示化合物A−1の合成
【0105】
【化11】
【0106】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−2(フェニルボロン酸):1.5g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0107】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、290℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−1を1.45g得た(収率80%)。
【0108】
得られた化合物の同定は質量分析によって行った。
【0109】
[MALDI−TOF−MS(マトリックス支援イオン化−飛行時間型質量分析)]
実測値:m/z=364.55、計算値:C25H16OS=364.09
【0110】
また例示化合物A−1について、以下の方法でT1エネルギーの測定を行った。調製した例示化合物A−1のトルエン希薄溶液について、Ar雰囲気下、77K、励起波長350nmにおいて燐光スペクトルの測定を行った。得られた燐光スペクトルの第一発光ピークのピーク波長からT1エネルギーを求めた。その結果、T1エネルギーは、波長換算で475nmであった。
【0111】
次に、例示化合物A−1について、以下の方法でエネルギーギャップの測定を行った。ガラス基板上に例示化合物A−1を加熱蒸着して、膜厚20nmの蒸着薄膜を得た。この蒸着薄膜について、紫外可視分光光度計(日本分光株式会社製V−560)を用いて吸光スペクトルを測定した。得られた吸光スペクトルの吸収端から、例示化合物A−2のエネルギーギャップを求めた。その結果、エネルギーギャップは3.4eVであった。
【0112】
[実施例2]例示化合物A−3の合成
【0113】
【化12】
【0114】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−3(3−ビフェニルボロン酸):2.4g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0115】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、310℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−3を1.8g得た(収率70%)。
【0116】
[MALDI−TOF−MS]
実測値:m/z=515.98、計算値:516.15
また例示化合物A−3について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で474nmであった。さらに例示化合物A−3について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物A−3のエネルギーギャップは3.3eVであった。
【0117】
[実施例3]例示化合物A−5の合成
【0118】
【化13】
【0119】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−4:4.3g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0120】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をクロロベンゼンに加熱溶解してから熱時ろ過した後、クロロベンゼン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、380℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−5を1.8g得た(収率55%)。
【0121】
[MALDI−TOF−MS]
実測値:m/z=668.22、計算値:668.84
【0122】
また例示化合物A−5について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で475nmであった。さらに例示化合物A−5について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物A−5のエネルギーギャップは3.3eVであった。
【0123】
[実施例4]例示化合物A−8の合成
【0124】
【化14】
【0125】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−5:2.9g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0126】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、350℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−8を2.2g得た(収率75%)。
【0127】
[MALDI−TOF−MS]
実測値:m/z=596.45、計算値:596.22
【0128】
また例示化合物A−8について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で500nmであった。さらに例示化合物A−8について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物A−8のエネルギーギャップは2.7eVであった。
【0129】
[実施例5]例示化合物A−9の合成
【0130】
【化15】
【0131】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−6:2.9g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0132】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、340℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−9を2.1g得た(収率71%)。
【0133】
[MALDI−TOF−MS]
実測値:m/z=596.40、計算値:596.22
【0134】
また例示化合物A−9について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で482nmであった。
【0135】
[実施例6]例示化合物A−13の合成
【0136】
【化16】
【0137】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−7:4.3g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0138】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をクロロベンゼンに加熱溶解してから熱時ろ過した後、クロロベンゼン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を120℃で真空乾燥することにより、例示化合物A−13を1.7g得た(収率50%)。
【0139】
[MALDI−TOF−MS]
実測値:m/z=674.98、計算値:664.19
【0140】
また例示化合物A−13について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で503nmであった。
【0141】
[実施例7]例示化合物A−17の合成
【0142】
【化17】
【0143】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−8:5.3g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0144】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、380℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−17を3.0g得た(収率72%)。
【0145】
[MALDI−TOF−MS]
実測値:m/z=840.77、計算値:840.25
【0146】
また例示化合物A−17について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で500nmであった。さらに例示化合物A−17について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物A−17のエネルギーギャップは2.7eVであった。
【0147】
[実施例8]例示化合物A−21の合成
【0148】
【化18】
【0149】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.9g(5mmol)
F−9:2.0g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0150】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、310℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−21を1.7g得た(収率74%)。
【0151】
[MALDI−TOF−MS]
実測値:m/z=448.69、計算値:448.19
【0152】
また例示化合物A−21について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で463nmであった。
【0153】
[実施例9]例示化合物A−25の合成
【0154】
【化19】
【0155】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−10:1.85g(5mmol)
F−6:2.9g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0156】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、340℃の条件下で昇華精製を行うことにより、高純度の例示化合物A−25を1.9g得た(収率64%)。
【0157】
[MALDI−TOF−MS]
実測値:m/z=596.89、計算値:596.22
【0158】
また例示化合物A−25について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で484nmであった。
【0159】
[実施例10]例示化合物B−1の合成
【0160】
【化20】
【0161】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−11:3.7g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0162】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、340℃の条件下で昇華精製を行うことにより、高純度の例示化合物B−1を1.7g得た(収率59%)。
【0163】
[MALDI−TOF−MS]
実測値:m/z=576.67、計算値:576.07
【0164】
また例示化合物C−1について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で456nmであった。さらに例示化合物B−1について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物B−1のエネルギーギャップは3.5eVであった。
【0165】
[実施例11]例示化合物B−2の合成
【0166】
【化21】
【0167】
200mLナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−12:3.7g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0168】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、330℃の条件下で昇華精製を行うことにより、高純度の例示化合物B−2を1.9g得た(収率65%)。
【0169】
[MALDI−TOF−MS]
実測値:m/z=576.46、計算値:576.07
【0170】
また例示化合物B−2について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で455nmであった。さらに例示化合物B−2について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物B−2のエネルギーギャップは3.5eVであった。
【0171】
[実施例12]例示化合物B−3の合成
【0172】
【化22】
【0173】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−1:1.85g(5mmol)
F−13:3.5g(12mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0174】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、330℃の条件下で昇華精製を行うことにより、高純度の例示化合物B−3を1.5g得た(収率56%)。
【0175】
[MALDI−TOF−MS]
実測値:m/z=544.87、計算値:C37H20O3S=544.11
【0176】
また例示化合物B−3について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で454nmであった。さらに例示化合物B−3について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物B−3のエネルギーギャップは3.5eVであった。
【0177】
[実施例13]例示化合物C−2の合成
【0178】
【化23】
【0179】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−14:1.45g(5mmol)
F−15:3.0g(6mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0180】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、330℃の条件下で昇華精製を行うことにより、高純度の例示化合物C−2を2.2g得た(収率76%)。
【0181】
[MALDI−TOF−MS]
実測値:m/z=590.84、計算値:590.17
【0182】
また例示化合物C−2について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で490nmであった。さらに例示化合物C−2について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物C−2のエネルギーギャップは3.6eVであった。
【0183】
[実施例14]例示化合物C−7の合成
【0184】
【化24】
【0185】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−14:3.5g(12mmol)
F−16:2.0g(5mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0186】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、330℃の条件下で昇華精製を行うことにより、高純度の例示化合物C−7を2.2g得た(収率77%)。
【0187】
[MALDI−TOF−MS]
実測値:m/z=574.79、計算値:574.11
【0188】
また例示化合物C−7について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で470nmであった。さらに例示化合物C−7について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物C−7のエネルギーギャップは3.4eVであった。
【0189】
[実施例15]例示化合物C−9の合成
【0190】
【化25】
【0191】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−14:1.45g(5mmol)
F−17:2.3g(6mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0192】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、310℃の条件下で昇華精製を行うことにより、高純度の例示化合物C−9を1.5g得た(収率63%)。
【0193】
[MALDI−TOF−MS]
実測値:m/z=470.54、計算値:470.08
また例示化合物C−9について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で453nmであった。さらに例示化合物C−9について実施例1と同様の方法でエネルギーギャップの測定を行ったところ、例示化合物C−9のエネルギーギャップは3.5eVであった。
【0194】
[実施例16]例示化合物C−10の合成
【0195】
【化26】
【0196】
200mLのナスフラスコに、以下に示す試薬、溶媒を仕込んだ。
F−14:1.5g(5mmol)
F−18:3.6g(6mmol)
テトラキス(トリフェニルホスフィン)パラジウム(0):137mg(0.12mmol)
トルエン:50mL
エタノール:20mL
炭酸ナトリウム水溶液(濃度:30重量%):30mL
【0197】
次に、反応系内を窒素雰囲気にした後、反応溶液を還流させながら3時間撹拌した。反応終了後、反応溶液に水を加えてさらに撹拌し、析出した結晶をろ別した。次に、この結晶を水、エタノール、アセトンで順次洗浄することで粗生成物を得た。次に、この粗生成物をトルエンに加熱溶解してから熱時ろ過した後、トルエン溶媒を用いた再結晶を2回行い精製した結晶を得た。次に、得られた結晶を100℃で真空乾燥した後、1×10-4Pa、320℃の条件下で昇華精製を行うことにより、高純度の例示化合物C−10を2.4g得た(収率70%)。
【0198】
[MALDI−TOF−MS]
実測値:m/z=678.87、計算値:678.20
【0199】
また例示化合物C−10について、実施例1と同様の方法でT1エネルギーの測定を行ったところ、波長換算値で480nmであった。
【0200】
[実施例17]
本実施例では、基板上に順次陽極/ホール輸送層/発光層/ホールブロッキング層/電子輸送層/陰極が順次設けられている有機発光素子を、以下に示す方法で作製した。尚、本実施例で使用した化合物の一部を下記に示す。
【0201】
【化27】
【0202】
ガラス基板上に、スパッタ法により、ITOを成膜し陽極を形成した。このとき陽極の膜厚を120nmとした。このようにITO電極が形成されている基板を透明導電性支持基板(ITO基板)として後の工程で使用した。次に、このITO基板上に、下記表4に示される有機化合物層及び電極層を、1×10-5Paの真空チャンバー内における抵抗加熱による真空蒸着によって連続的に成膜した。このときITO電極に対向する電極(金属電極層)の面積を3mm2となるように作製した。
【0203】
【表4】
【0204】
次に、有機発光素子が水分の吸着によって素子劣化が起こらないように、乾燥空気雰囲気中で保護用ガラス板をかぶせアクリル樹脂系接着材で封止した。以上のようにして有機発光素子を得た。
【0205】
得られた有機発光素子について、ITO電極を正極、Al電極を負極にして、5.1Vの印加電圧をかけたところ、発光効率が47cd/Aで、輝度2000cd/m2の緑色発光が観測された。またこの素子においてCIE色度座標は、(x,y)=(0.30,0.63)であった。
【0206】
[実施例18〜29]
実施例17において、ホールブロック層(HB層)に含まれるホールブロック材料(HB材料)、並びに発光層に含まれる第一ホスト、第二ホスト及びゲストの組み合わせを下記表5に変更した他は、実施例17と同様の方法で素子を作製した。また得られた素子について実施例17と同様に評価を行った。結果を表5に示す。
【0207】
【表5】
【0208】
表5より、本発明のチオキサントン化合物は、燐光発光する有機発光素子において、電子輸送材料又は発光材料として用いることで良好な発光効率を得られることが分かった。
【0209】
[実施例30、31、比較例1]
実施例17において、ホールブロック層(HB層)に含まれるホールブロック材料(HB材料)、並びに発光層に含まれる第一ホスト、第二ホスト及びゲストの組み合わせを下記表6に変更した他は、実施例17と同様の方法で素子を作製した。ここで比較例1で使用した化合物H−1Aを以下に示す。
【0210】
【化28】
【0211】
得られた素子について素子の安定性を評価するために電流値40mA/cm2における有機発光素子の輝度半減寿命を測定した。結果を表6に示す。
【0212】
【表6】
【0213】
表6に示されるように、燐光発光する有機発光素子において、本発明のチオキサントン化合物は、比較例の化合物よりも、輝度半減寿命が長いことが示された。これは、カルボニル骨格によって適切なLUMO準位を有するために電荷のバランスが取れた安定な素子構造になるからである。このことによって有機発光素子において良好な素子寿命を得られることが分かった。
【産業上の利用可能性】
【0214】
以上のように、本発明のチオキサントン化合物は、T1エネルギーが高くLUMO準位が深い化合物である。このため有機発光素子の構成材料として使用すると、発光効率の高く、劣化しにくい安定な有機発光素子を得ることができる。
【符号の説明】
【0215】
1:表示装置、11:基板、12:防湿膜、13:ゲート電極、14:絶縁膜、15:半導体層、16:ドレイン電極、17:ソース電極、18:TFT素子、19:絶縁膜、20:コンタクトホール、21:陽極、22:有機化合物層、23:陰極、24:第一の保護層、25:第二の保護層
【特許請求の範囲】
【請求項1】
下記一般式[1]で示されることを特徴とする、チオキサントン化合物。
【化1】
(式[1]において、R1乃至R8は、それぞれ水素原子、炭素数1乃至4のアルキル基又はフェニル基、ビフェニル基、ナフチル基、フェナンスレン基、フルオレニル基、トリフェニレニル基、ジベンゾフラニル基及びジベンゾチオフェニル基のいずれかから選択されるアリール基を表す。ただし、R1乃至R8のうち少なくともひとつが前記アリール基である。
尚、前記アルキル基及び前記アリール基は、さらにアルキル基、芳香族炭化水素基又は芳香族複素環基を有してもよい。)
【請求項2】
前記R1、R4、R5及びR8が水素原子であることを特徴とする、請求項1に記載のチオキサントン化合物。
【請求項3】
陽極と陰極と、
前記陽極と前記陰極との間に配置される有機化合物層と、から構成される有機発光素子において、
前記有機化合物層に、請求項1又は2に記載のチオキサントン化合物が含まれることを特徴とする、有機発光素子。
【請求項4】
前記チオキサントン化合物が、ホールブロッキング層及び発光層のいずれかに含まれることを特徴とする、請求項3に記載の有機発光素子。
【請求項5】
前記チオキサントン化合物が前記発光層に含まれており、
前記発光層が、複数種のホストと、ゲストとからなり、
前記複数種のホストのうちの一種が前記チオキサントン化合物であることを特徴とする、請求項4に記載の有機発光素子。
【請求項6】
前記ゲストが燐光発光材料であることを特徴とする、請求項5に記載の有機発光素子。
【請求項7】
前記燐光発光材料がイリジウム錯体であることを特徴とする、請求項6に記載の有機発光素子。
【請求項8】
複数の画素を有し、
前記画素が、請求項3乃至7のいずれか一項に記載の有機発光素子と、前記有機発光素子に接続するスイッチング素子と、を有することを特徴とする表示装置。
【請求項9】
画像を表示するための表示部と、画像情報を入力するための入力部とを有し、
前記表示部が複数の画素を有し、
前記画素が、請求項3乃至7のいずれか一項に記載の有機発光素子と、前記有機発光素子に接続するスイッチング素子と、を有することを特徴とする画像入力装置。
【請求項1】
下記一般式[1]で示されることを特徴とする、チオキサントン化合物。
【化1】
(式[1]において、R1乃至R8は、それぞれ水素原子、炭素数1乃至4のアルキル基又はフェニル基、ビフェニル基、ナフチル基、フェナンスレン基、フルオレニル基、トリフェニレニル基、ジベンゾフラニル基及びジベンゾチオフェニル基のいずれかから選択されるアリール基を表す。ただし、R1乃至R8のうち少なくともひとつが前記アリール基である。
尚、前記アルキル基及び前記アリール基は、さらにアルキル基、芳香族炭化水素基又は芳香族複素環基を有してもよい。)
【請求項2】
前記R1、R4、R5及びR8が水素原子であることを特徴とする、請求項1に記載のチオキサントン化合物。
【請求項3】
陽極と陰極と、
前記陽極と前記陰極との間に配置される有機化合物層と、から構成される有機発光素子において、
前記有機化合物層に、請求項1又は2に記載のチオキサントン化合物が含まれることを特徴とする、有機発光素子。
【請求項4】
前記チオキサントン化合物が、ホールブロッキング層及び発光層のいずれかに含まれることを特徴とする、請求項3に記載の有機発光素子。
【請求項5】
前記チオキサントン化合物が前記発光層に含まれており、
前記発光層が、複数種のホストと、ゲストとからなり、
前記複数種のホストのうちの一種が前記チオキサントン化合物であることを特徴とする、請求項4に記載の有機発光素子。
【請求項6】
前記ゲストが燐光発光材料であることを特徴とする、請求項5に記載の有機発光素子。
【請求項7】
前記燐光発光材料がイリジウム錯体であることを特徴とする、請求項6に記載の有機発光素子。
【請求項8】
複数の画素を有し、
前記画素が、請求項3乃至7のいずれか一項に記載の有機発光素子と、前記有機発光素子に接続するスイッチング素子と、を有することを特徴とする表示装置。
【請求項9】
画像を表示するための表示部と、画像情報を入力するための入力部とを有し、
前記表示部が複数の画素を有し、
前記画素が、請求項3乃至7のいずれか一項に記載の有機発光素子と、前記有機発光素子に接続するスイッチング素子と、を有することを特徴とする画像入力装置。
【図1】

【公開番号】特開2012−102024(P2012−102024A)
【公開日】平成24年5月31日(2012.5.31)
【国際特許分類】
【出願番号】特願2010−249278(P2010−249278)
【出願日】平成22年11月8日(2010.11.8)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
【公開日】平成24年5月31日(2012.5.31)
【国際特許分類】
【出願日】平成22年11月8日(2010.11.8)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
[ Back to top ]