
チロシンキナーゼ受容体由来の細胞外アロステリック阻害剤結合ドメイン
本発明は、1回膜貫通型チロシンキナーゼ受容体に由来する、アロステリック阻害剤に対する細胞外結合ドメインに関する。より具体的には、本発明は、線維芽細胞増殖因子受容体(FGFR)に由来する細胞外ドメインに関する。さらに、本発明は、他のチロシンキナーゼ受容体の細胞外部分における類似のドメインを同定するためのこのドメインの使用、および小分子化合物アロステリック阻害剤を同定するスクリーニング方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、1回膜貫通型(single membrane span)チロシンキナーゼ受容体に由来する、アロステリック阻害剤に対する細胞外結合ドメインに関する。より具体的には、本発明は、チロシンキナーゼ受容体、すなわち線維芽細胞増殖因子受容体(FGFR)、血管内皮増殖因子受容体(VEGFR)または血小板由来増殖因子受容体(PDGFR)に由来する細胞外ドメインに関する。さらに、本発明は、他のチロシンキナーゼ受容体の細胞外部分における類似のドメインを同定するためのこのドメインの使用、およびアロステリック阻害剤を同定するスクリーニング方法に関する。
【背景技術】
【0002】
細胞表面受容体は、全ての薬剤の大多数に標的を提示する(Overingtonら、2006年)。歴史的に、創薬プログラムは、オルソステリック部位で内因性リガンドと結合を競合するアンタゴニストを開発するための研究が多数派を占めていた。その一方、アロステリック部位、すなわち、オルソステリックリガンド(標的が受容体の場合)または基質(標的が酵素の場合)によって利用されるものとはトポグラフィー的に異なるドメインに結合し、タンパク質の活性を調節する薬剤は、同定するのがより困難であった。しかし、近年は、リガンド依存性イオンチャネルおよびGタンパク質共役受容体(GPCR)に対して同定されるアロステリック調節因子の数の増加をみている(Christopoulos、2002年;Kenakin、2010年。驚くべきことに、増殖因子受容体チロシンキナーゼ(RTK)に関しては、この受容体スーパーファミリーが極めて生物学的に重要であり医学的に意義があるという事実にもかかわらず、ならびにアロステリック剤が、より大きな安全性および/または選択性を含む、伝統的なオルソステリックリガンドに勝る明白な治療上の利点を提供することができるという事実にもかかわらず、アロステリック小分子化合物調節因子はこれまでに同定されていない。これまで、RTKを標的とするほとんどの治療が、増殖因子リガンドを認識するモノクローナル抗体、または受容体のチロシンキナーゼ活性を直接阻害する小分子化学化合物に焦点を合わせてきた。
【0003】
なかでも、より有効なおよび/または選択的なRTK小分子化合物阻害剤から実質的に利益を得られる一つの分野が、抗血管新生薬療法の分野である。VEGF標的化抗血管新生剤は癌患者の生存を延長させるが、全体の奏効は、内因的不応性、獲得耐性を介した逃避(escape)および少なくとも前臨床モデルでは転移の刺激により制限される。別の抗血管新生剤との併用療法は、これらの課題を克服するのに役立つ可能性があるとみなされてきた。最初に同定された血管新生因子である線維芽細胞増殖因子(FGF)−2は、魅力的な薬剤候補である。実際、FGFRシグナル伝達は、癌および炎症性疾患に関与しており(Shinら、2006年;Eswarakumarら、2005年;Malemudら、2007年;Carmeliet、2005年)、腫瘍血管新生スイッチに寄与し(Prestaら、2005年;Kuboら、2002年;Shineら、2006年;Lavineら、2006年)、腫瘍の血管新生およびVEGF阻害剤治療時の再発を助ける(Casanovasら、2005年)。それにもかかわらず、FGFファミリーは、抗血管新生剤開発に関し大きな注目は受けていない。この理由の一部に、18種類のリガンドおよび4種類のFGFRのこのスーパーファミリーのメンバー間の重複性がある(Eswarakumarら、2005年;BeenkenおよびMohammadi、2009年;Cenniら、2005年;Bossardら、2004年;Compagniら、2000年)。また、FGFRチロシンキナーゼの選択的阻害剤は、臨床使用が承認されていない(Dimitroffら、1999年;McDermottら、2005年)。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Overingtonら、Nat.Rev.Drug Discov.5,993−995(2006)
【非特許文献2】Christopoulos、Nat.Rev.Drug Discov.1,198−210(2002)
【非特許文献3】Kenakin、Pharmacol rev,62,265−304(2010)
【非特許文献4】Shinら、Mol Biol Cell 17,576−584(2006)
【非特許文献5】Eswarakumarら、Cytokine Growth Factor Rev 16,139−149(2005)
【非特許文献6】Malemudら、Clinica chimica acta;international journal of clinical chemistry 375,10−19(2007)
【非特許文献7】Carmeliet、Nature 438,932−936(2005)
【非特許文献8】Prestaら、Cytokine Growth Factor Rev 16,159−178(2005)
【非特許文献9】Kuboら、Proc Natl Acad Sci USA 99,8868−8873(2002)
【非特許文献10】Lavineら、Genes & development 20,1651−1666(2006)
【非特許文献11】Casanovasら、Cancer Cell 8,299−309(2005)
【非特許文献12】BeenkenおよびMohammadi、Nature reviews 8,235−253(2009)
【非特許文献13】Cenniら、Anticancer Res 25,1109−1113(2005)
【非特許文献14】Bossardら、Cancer Res 64,7507−7512(2004)
【非特許文献15】Compagniら、Cancer Res 60,7163−7169(2000)
【非特許文献16】Dimitroffら、Invest New Drugs 17,121−135(1999)
【非特許文献17】McDermottら、Bioorg Med Chem 13,4835−4841(2005)
【発明の概要】
【発明が解決しようとする課題】
【0005】
驚くべきことに本発明者らは、化学的最適化と組み合わされた高速スループットスクリーニングにより、RTK、すなわちFGFRの最初の経口的に活性な小分子化合物アロステリック阻害剤を同定できることを見出した。この化合物は、SSR128129(略して「SSR」)と呼ばれる(図3)。
【0006】
SSR活性に基づく詳細な研究により説明されるように、SSRは、同じファミリー(現在はFGFRファミリー)の全てのメンバーを阻害する能力を有する。以下の例で示されるように、SSRは、FGFR1活性(図4および6)、FGFR2活性(図8)、FGFR3活性(図9)およびFGFR4活性(図7)を阻害することができる。故に、このアロステリック阻害剤は、TKRの異なるメンバーにより共有される受容体の細胞外ドメインにある、進化的に保存されたFGFRアロステリック部位に結合する。この保存された部位は、FGFRのドメインIIIに位置する(図11)。結合部位へのSSRの結合は、「偏った拮抗作用(biased antagonism)」を誘導する。作用は、アロステリック結合部位へのSSR結合が受容体、特に判定されたフラストレートされた(frustrated)ドメインの立体構造変化をもたらすという事実により確認される。偏った拮抗作用のため、アロステリック阻害剤を同定する方法は、下記のようにリン酸シグナル伝達経路測定に基づくスクリーニングテストの使用により提供される。今後は、SSRは、RTKのアロステリック阻害剤の最初の例である。
【課題を解決するための手段】
【0007】
FGFR上のこのような部位の標的化ならびにVEGFR2およびPDGFRβのような他のRTKにおける類似の部位の標的化の検証は、重要な実用的意義を有しており、大きな治療効果をもたらす。
【図面の簡単な説明】
【0008】
【図1】AはHUVECでの定量的PCR実験がFGFR1およびFGFR4発現のみを示す図である。BはRT−PCR分析がアイソフォームFGFR1βおよびFGFR4を同定した図である。FGFR1がIIIcバリアントのフォーマット下にある図である(C)。
【図2】FGFR1βlllc−hMpIをトランスフェクトされたBaF/3細胞は、挿入されたFGFRが活性化される場合に増殖することができることを示す図である。FGF4(A)のみはFGFR1βlllcを誘導することができるが、FGF19は誘導することができない(B)。
【図3】SSR化合物表示を示す図である。
【図4−1】内皮細胞増殖に対するSSR活性に関する試験を示す図である。AはFGF2およびFGF4のみがHUVEC増殖を刺激し、それによりFGFR1がこれらの細胞で増殖を駆動することが示される。BはSSRがFGF2誘導HUVEC増殖を阻害し、それによりSSRによるFGFR1拮抗作用が示される。CはFGFR1をトランスフェクトされたラットファッドパッド(Fad Pad)内皮細胞(RFPEC)では、FGF2は、高用量でさえSSRにより部分的にのみ阻害されるFGFR1自己リン酸化を誘導した。Dはこの阻害が、PAECトランスフェクトFGFR1またはHUVECでのFGF2結合に対するSSRの競合的効果に起因しない。
【図4−2】内皮細胞増殖に対するSSR活性に関する試験を示す図である。AはFGF2およびFGF4のみがHUVEC増殖を刺激し、それによりFGFR1がこれらの細胞で増殖を駆動することが示される。BはSSRがFGF2誘導HUVEC増殖を阻害し、それによりSSRによるFGFR1拮抗作用が示される。CはFGFR1をトランスフェクトされたラットファッドパッド(Fad Pad)内皮細胞(RFPEC)では、FGF2は、高用量でさえSSRにより部分的にのみ阻害されるFGFR1自己リン酸化を誘導した。Dはこの阻害が、PAECトランスフェクトFGFR1またはHUVECでのFGF2結合に対するSSRの競合的効果に起因しない。
【図5】Aは異方性のパラメータとしての反転速度(tumbling speed)の測定による、Fc−タグ(FGFR2∂123)なしのFGFR2の精製ECDへの、蛍光lumioタグ付FGF1の結合(FGF1−lumio)を示す図である。SSRとFGF1−lumioの間の直接競合は観察されない。BはSSRが、FGFR2多量体化を阻害することができない、またはCはFGF2二量体化を阻害することができないことを示す図である。
【図6】内皮細胞走化性遊走に対するSSR活性に関する試験を示す図である。AはFGF2およびFGF4のみがHUVEC遊走を刺激し、それによりFGFR1がこれらの細胞で増殖を駆動することが示される。BはSSRが、FGFR1に対する拮抗アフェクト(antagonistic affect)に対応する、FGF2誘導HUVEC走化性遊走を阻害する。
【図7】内皮細胞インビトロ血管新生に対するSSR活性に関する試験を示す図である。AはFGF2およびFGF19のみがHUVEC血管新生を刺激し、それによりFGFR4がこれらの細胞におけるこの分化段階を制御することが示される。BはSSRが、FGFR4に対する拮抗アフェクトに対応する、FGF2誘導HUVECインビトロ血管新生を阻害する。
【図8】PANC02増殖および遊走に対するSSR活性に関する試験を示す図である。PANC02細胞は、VEGFの有無にかかわらずFGF7刺激下で増殖(A)または遊走(B)し、該系のFGFR2IIIb依存を示唆している。SSRは、FGF7誘導PANC02増殖およびFGF7+VEGF誘導細胞遊走を阻害し、FGFR2IIIbを阻害する能力を示している。
【図9】B9骨髄腫細胞増殖に対するSSR活性に関する試験を示す図である。AはFGF1がFGFR3を介してB9骨髄腫細胞増殖を誘導し、SSRがこの刺激を阻害し、それによりSSRがFGFR3をブロックできることが示される。Bはそれにもかかわらず、SSRが、自己活性FGFR3変異体(キナーゼドメインが構造的にリン酸化される。)をトランスフェクトされたB9細胞の増殖を阻害することはできない。これらの結果は、SSRの細胞外効果を示している。
【図10】SSRが、IGF、PDGF−BB、EGFまたはPIGFなどのさまざまな増殖因子により誘導された細胞遊走を大幅に阻害することができないHUVEC遊走アッセイを示す図である。SSRは、FGFRに特異的であり、およびFGF誘導HUVEC遊走のみをブロックする。
【図11−1】FGFRドメインIIIに結合するSSRを示すNMR試験を示す図である。AはFGFR1細胞外ドメインでのSSR結合の1D−およびSTD−NMR分析。飽和は対照TNFRIαでは観察されない。BはドメインIIIにおける相互作用部位を示唆する、異なるFGFR1ドメインに結合するSSRの1D−NMR試験は、FGFR1完全長およびFGFR1ドメインIIで得られたスペクトルが類似していることを実証する。CはSSRが、FGFR2およびFGFR3(D)細胞外ドメインに結合する能力を示す等温滴定熱量(Isothermal titration calorimetric)。
【図11−2】FGFRドメインIIIに結合するSSRを示すNMR試験を示す図である。AはFGFR1細胞外ドメインでのSSR結合の1D−およびSTD−NMR分析。飽和は対照TNFRIαでは観察されない。BはドメインIIIにおける相互作用部位を示唆する、異なるFGFR1ドメインに結合するSSRの1D−NMR試験は、FGFR1完全長およびFGFR1ドメインIIで得られたスペクトルが類似していることを実証する。CはSSRが、FGFR2およびFGFR3(D)細胞外ドメインに結合する能力を示す等温滴定熱量(Isothermal titration calorimetric)。
【図12−1】SSRがFGF1(A、B)およびFGF2(C)と相互作用できないことを実証するNMR(A、C)およびITC(B)試験を示す図である。Dはヘパリンミメティクスである八硫酸スクロース(SOS)の添加後にFGFR1に結合するSSRへの干渉は観察されず、SSRがFGFR1のヘパリン結合部位と相互作用しないことを確認する図である。
【図12−2】SSRがFGF1(A、B)およびFGF2(C)と相互作用できないことを実証するNMR(A、C)およびITC(B)試験を示す図である。Dはヘパリンミメティクスである八硫酸スクロース(SOS)の添加後にFGFR1に結合するSSRへの干渉は観察されず、SSRがFGFR1のヘパリン結合部位と相互作用しないことを確認する図である。
【図13】インシリコモデリングおよび変異誘発が、Y328アミノ酸近傍のSSRに対するアロステリック結合部を同定することを示す図である。WT FGFR2細胞外ドメイン(A)でのITC実験は、SSRとFGFR2の間の相互作用を示す。測定がY328D変異体(B)で実現される場合に失敗するこの結合は、Y328D変異がFGFR2をSSR結合に非感受性にすることを確認する。
【図14】100μM SSRなしまたは100μM SSRあり(それぞれ黒い線およびグレーの線)での精製された(A)FGFR2∂23−WT、(B)FGFR2−∂23−His、(C)FGFR2−∂23−Tyr、(D)FGFR2−∂23−H/T(H295L/Y328D二重変異体)のフーリエ変換赤外(FTIR)測定が、WTまたはHis295L変異体の両方で立体構造変化を同定したのに対し、変異Y328Dはこの変化に対しFGFR2を非感受性にすることを示す図である。
【図15】完全長FGFR2−WTまたは−Y328Dを有する安定にトランスフェクトされたHEK293細胞をFGF2で刺激(5分間で0.5ng/ml)した後の、活性化Erk1/2のウェスタンブロット分析を示す図である。濃度測定で確定された(densitometry defined)IC50値(平均±標準誤差;3つの独立した実験)は、FGFR2−Y328D変異体受容体(B)はFGFR2−WT(A)と比較してSSR阻害に対する感受性が約5倍低いことを示している。
【図16】AはFGF2で刺激された、FGFR2トランスフェクトHEK細胞に対する、FGFRチロシンキナーゼ阻害剤SU5402と比較したSSR効果のウェスタンブロット分析を示す図である。SU5402がPLCγ、FRS2およびErk1/2リン酸化を阻害するのに対し、SSRはPLCγ経路を阻害せず、SSRによる偏った拮抗作用を示している(B)。
【図17−1】HUVECにおけるFGF2誘導AKTリン酸化に対するSSR効果のウェスタンブロット分析(A)と対応する定量化グラフ(B)を示す図である。Cはこの効果が、ホスホ−AKT(Ser473)に対するオンセルELISA(on−cell ELISA)でも定量化できることを示す図である。Dはこの効果が、SSRがFGFRに結合するFGFと競合できないことと独立であることを示す図である。
【図17−2】HUVECにおけるFGF2誘導AKTリン酸化に対するSSR効果のウェスタンブロット分析(A)と対応する定量化グラフ(B)を示す図である。Cはこの効果が、ホスホ−AKT(Ser473)に対するオンセルELISA(on−cell ELISA)でも定量化できることを示す図である。Dはこの効果が、SSRがFGFRに結合するFGFと競合できないことと独立であることを示す図である。
【図18】ソフトウェアプログラムAGADIRおよび変異分析を用いた、VEGF−R2受容体における想定されるフラストレートされた帯域の同定を示す図である。(A)構造変化(例えばβシートからαヘリックスへの転移)を起こす傾向がある幾つかの領域が同定された。(B)2つのリジン(K609およびK648)が、膜貫通ドメインとの近さおよび変異後のヘリシティ特性に対する最も負の影響により、アスパラギン酸への変異に対して選択された。
【図19】ソフトウェアプログラムAGADIRおよび変異分析を用いた、PDGF−Rβ受容体における想定されるフラストレートされた帯域の同定を示す図である。アスパラギン酸への変異リジン387およびアスパラギン酸への変異ロイシン383は、ヘリシティ特性に対する最も負の影響を有するように思われる。
【図20】Erk1/2およびPLCγを通じたVEGF−R2およびPDGF−Rβ受容体シグナル伝達経路活性化の略図を示す図である。(A)VEGF刺激後のVEGFR2シグナル伝達および(B)PDGF刺激後のPDGF−Rβシグナル伝達。
【図21】ウェスタンブロットまたはシュアファイアアッセイ(surefire assay)を用いた、VEGF−R2またはPDGF−Rβ受容体の野生型または変異型を過剰発現したHEK293細胞におけるErk1/2リン酸化検出を示す図である。(A)VEGF−R2のマウス野生型およびK609DまたはK648Dで変異されたVEGF−R2は、HEK293細胞において安定にトランスフェクトされた。飢餓およびマウスVEGFなし(0)またはマウスVEGFあり(+)での刺激後、Erk1/2リン酸化はウェスタンブロットにより検出される。(B)タンパク質抽出物でのErk1/2(左の図式)またはPLCγ(右の図式)リン酸化を検出する確実なアッセイの略図。(C)10%FBS、1もしくは50ng/ml PDGF−BBで刺激または対照として刺激なし(0)後のPDGFRβトランスフェクトHEK293細胞における合計Erk1/2またはリン酸化Erk1/2のアルファスクリーンシュアファイア用量。
【発明を実施するための形態】
【0009】
本発明の種々の態様は、本発明の詳細な記載および以下の例において説明される。
【0010】
本発明の第1の態様は、チロシンキナーゼ受容体の細胞外ドメインに由来するアロステリック結合部位である。本明細書では、アロステリック結合部位とは、受容体のリガンド結合部位へのリガンドの結合の競合的阻害を引き起こすことなく、阻害剤、好ましくは小分子化合物が結合できる部位を意味する。本明細書では、由来するとは、アロステリック結合部位が細胞外ドメインの一部から成るが、完全な細胞外ドメインは含まないことを意味する。好ましくは、アロステリック結合部位は、10から200アミノ酸長の間、より好ましくは10から100アミノ酸の間、さらにより好ましくは20から50アミノ酸の間であり、前記アミノ酸は受容体の細胞外ドメインの部分である。
【0011】
本明細書では、小分子化合物は、好ましくは1000D未満、より好ましくは900D未満、より好ましくは800D未満、より好ましくは700D未満、より好ましくは600D未満、さらにより好ましくは500D未満の分子重量を有する非ポリマー性質の化合物である。
【0012】
チロシンキナーゼ受容体および受容体チロシンキナーゼ(RTK)は、本特許の範囲では、適用等価語である。「チロシンキナーゼ受容体」が受容体を示すのに使用されるのに対し、「受容体チロシンキナーゼ」はより具体的には受容体のキナーゼ活性を示すのに使用される。チロシンキナーゼ受容体は当業者に知られており、EGF、インスリン様増殖因子、PDGF、FGF、VEGF、HGF、Trk、AXL、LTK、TIE、ROR、DDR、PKT7、RYK、CCK4、EphおよびMuSK受容体ファミリーの受容体を含むが、これらに限定されない。好ましくは、前記アロステリック結合部位は、AXL、FGFR、MuSK、PDGFR、PTK7、ROR、TIEおよびVEGFRなどを含む、Igドメインを有するTKRの細胞外ドメインに由来する。さらにより好ましくは前記アロステリック結合部位は、細胞質ドメインにスプリットキナーゼドメインを有するTKRに由来する。本発明によるTKRの好ましい実施形態は、線維芽細胞増殖因子受容体(FGFR)、またはこのホモログ、オルソログもしくはパラログである。
【0013】
タンパク質の「ホモログ」は、当該非修飾タンパク質と比べてアミノ酸置換、欠失および/または挿入を有する、ならびに由来する非修飾タンパク質と類似の生物活性および機能活性を有するペプチド、オリゴペプチド、ポリペプチド、タンパク質および酵素を含む。「オルソログおよびパラログ」は、遺伝子の祖先関係を記述するのに使用される進化概念を含む。パラログは、遺伝子祖先遺伝子の複製を通じて生じた同じ種内の遺伝子である。オルソログは、種分化を通じて生じた異なる生物由来の、およびまた共通の祖先遺伝子に由来する遺伝子である。
【0014】
配列番号1を含むこのようなアロステリック結合は、FGFRファミリー、より具体的には、FGFR2に属する。好ましくは、前記アロステリック阻害剤部位は配列番号1を含み、さらにより好ましくは前記アロステリック阻害剤部位は配列番号1から成る。
【0015】
別の態様では、本発明は、アロステリック結合部位のホモログ、パラログまたはオルソログから成る。好ましくは、これらのホモログ、パラログまたはオルソログのポリペプチド配列は、配列番号1と少なくとも70%、80%、90%、95%以上の相同性を共有する。
【0016】
一例として、アロステリック結合部位のこのようなパラログは、FGFRファミリーに存在する。
【0017】
特に、本発明によるアロステリック結合部位は、FGFRのドメインIIIに位置する。
【0018】
同様に、VEGFR2に対するアロステリック結合部位は、リジン609およびリジン648を含む領域における、受容体のIgドメイン6に位置する。
【0019】
アロステリック結合部位はまた、PDGFRβにも存在し、および膜貫通領域近傍に位置する領域、特にIgドメイン3(ロイシン383およびリジン387を含む領域)に位置する。好ましくは、アロステリック結合部位に対するアロステリック阻害剤の結合により、偏った拮抗作用が誘導される。
【0020】
本明細書では、「偏った拮抗作用」とは、幾つかの下流経路を有する受容体に関して、アロステリック阻害剤結合部位にアロステリック阻害剤が結合した際に全ての経路が影響を受けるわけではないこと、または全ての経路が同程度に影響を受けるわけではないことを意味する。好ましい実施形態では、少なくとも1つの下流経路が阻害されるのに対し、少なくとももう1つの下流経路は影響を受けない。
【0021】
好ましくは、本発明によるアロステリック結合部位は、フラストレートされたドメインを含み、好ましくはフラストレートされたドメインから本質的に成り、さらにより好ましくはフラストレートされたドメインから成る。
【0022】
本明細書では、「フラストレートされたドメイン」とは、1つの構造的立体構造に明確に対するものではないタンパク質ドメインまたはこのフラグメントを意味する。フラストレートされたドメインは当業者に知られており、フラストレートされたドメインの存在は、1つのタンパク質二次構造予測プログラムでの曖昧な答え、または2つの異なるタンパク質二次構造予測プログラム間の予測における矛盾のどちらかにより検出される。優先的に、フラストレートされたドメインの存在は、タンパク質二次構造予測プログラムからの予測、ならびに結晶化およびX線回析などのタンパク質構造検出方法により判定されるような実構造における矛盾により検出される。非限定例として、矛盾は、1つの方法によるαヘリックスの表示、および別の方法によるβシートの表示であってもよい。タンパク質は、最小限にフラストレートされる。しかし、幾つかのドメインは、幾つかのフラストレーションを誘導しており(本明細書では「フラストレートされたドメイン」と呼ばれる)、これらのドメインは、タンパク質の立体構造変化を誘導する傾向がある。
【0023】
好ましい実施形態では、前記フラストレートされたドメインは配列番号2を含み、好ましくはこれは配列番号2から成る。このフラストレートされたドメインは、FGFRファミリー、特にFGFR2に属する。
【0024】
他のフラストレートされたドメインは、上記に示されたように同定することができる。
【0025】
本発明の別の態様は、アロステリック結合部位が位置するチロシンキナーゼ受容体の結合部位にリガンドが結合した際に、偏った拮抗作用を誘導するための本発明によるアロステリック結合部位の使用である。本発明のさらに別の態様は、前記部位に結合している、ランダムライブラリー由来の小分子化合物阻害剤をスクリーニングするための、本発明によるアロステリック結合部位の使用である。
【0026】
本発明のさらに別の態様は、チロシンキナーゼ受容体の細胞外ドメインにおけるフラストレートされたドメインの存在をスクリーニングすることを含む、前記細胞外ドメインにおけるアロステリック阻害剤結合部位の同定方法である。フラストレートされたドメインをスクリーニングする方法は当業者に知られており、このような方法の例は例8に記載されている。非限定例として、フラストレートされたドメインは、1つのタンパク質二次構造予測プログラムでの曖昧な答えにより、好ましくは2つの異なるタンパク質二次構造予測プログラム間の予測における矛盾により、さらにより好ましくはタンパク質二次構造予測プログラムからの予測、ならびに結晶化およびX線回析などのタンパク質構造検出方法により判定されるような実構造における矛盾により検出される。タンパク質二次構造予測のためのプログラムは当業者に知られている。非限定例として、このようなプログラムがRost(2003年)により記載されている。好ましくは、前記フラストレートされたドメインは、前記アロステリック結合部位の近傍にある。より好ましくはこれは結合部位の境界から20アミノ酸以内、さらにより好ましくは10アミノ酸以内に位置し、さらにより好ましくはこれは前記結合部位に隣接し、さらにより好ましくはこれは結合部位と重複し、最も好ましくはこれは結合部位に含まれる。考えられる阻害剤部位を同定した後、スクリーニングは、該部位に結合する、およびアロステリック阻害機能をテストできる小分子、小ペプチド、ペプチドミメティクス、抗体またはナノ抗体(nanobody)などの化合物を設計して、考えられる阻害剤部位の機能の確認により完了することができる。
【0027】
本発明の別の態様は、チロシンキナーゼ受容体の活性化に依存する2つの異なる下流経路により誘導される2つの異なるレポーターの比較を含む、本発明による前記チロシンキナーゼ受容体の細胞外ドメインにおけるアロステリック阻害剤部位に結合している小分子化合物アロステリック阻害剤の同定方法である。レポーターは、検出可能なシグナルをもたらす化合物の任意の遺伝子、タンパク質であり、非限定例として、抗生物質耐性遺伝子、細胞死をもたらす毒素遺伝子、GFPなどの蛍光タンパク質をコードする遺伝子、またはβガラクトシダーゼなどの酵素活性をコードする遺伝子、またはリン酸化もしくは脱リン酸化、アセチル化もしくは脱アセチル化されたまたは立体構造が変化しているタンパク質であってもよい。レポーター遺伝子の場合では、コード配列は、適切なプロモーター、すなわち受容体へのリガンドの結合およびレポーター経路の結果的な誘導により誘導されるプロモーターの制御下に置かれる。二重経路の場合では、2つの異なるプロモーターが必要とされる。非限定例として、アロステリック阻害剤の存在下または非存在下でタンパク質のリン酸化を比較することは、偏った拮抗作用によるリン酸化の違いを生み、リン酸化のこれらの違いはレポーターとして使用することができる。
【0028】
好ましい実施形態では、RTKのアロステリック阻害剤の同定は、
a)RTKのアロステリック結合部位をアロステリック阻害剤候補化合物と接触させる段階、
b)前記チロシンキナーゼ受容体の活性化/阻害に依存する少なくとも2つの下流経路の変化を測定する段階、
c)前記チロシンキナーゼ受容体の活性化/阻害に依存する少なくとも2つの異なる下流経路の各々に対する少なくとも1つのレポーターの状態の変化を比較する段階
を含み、
受容体のリガンド結合ドメインに結合しているリガンドの存在下、少なくとも1つの下流経路が阻害されるのに対し、少なくとももう1つの下流経路は影響を受けない場合に、アロステリック阻害剤が同定される、スクリーニングテストを実施して行うことができる。レポーターの状態の変化は使用されるレポーターに依存し、非限定例としてレポータータンパク質のリン酸化の変化、または遺伝子の非誘導から誘導への(または逆も同様)スイッチであってもよい。好ましくは、前記状態の変化はホスホイレーション(phosphoylation)状態の変化である。
【0029】
好ましくは、下流経路の変化は、ERK1/2 およびPLCγシグナル伝達経路を含む、リン酸シグナル伝達経路の変化の測定により実施される。
【0030】
別の実施形態では、FGF−Rのアロステリック調節因子は、下記に記載のようにSEC−LC/MSに基づく親和性スクリーニングを用いて同定することができる:
SEC−LC/MS方法論は、オンライン結合された2次元システム、分離のため高性能液体クロマトグラフィーに結合されたサイズ排除クロマトグラフィーと、これに続く検出のためのエレクトロスプレーイオン化−飛行時間型質量分析から成る、親和性スクリーニングに使用される分析技法である。
【0031】
方法は、可溶性ポリペプチド(ペプチド、タンパク質ドメイン、または完全長タンパク質を含む)と相互作用する幾つかの化合物の能力に基づく。目的とするペプチドと小分子化合物のプールを混合した後、ペプチド−リガンド複合体は、サイズ排除クロマトグラフィーによる非結合および結合小分子化合物の分離を可能にする質量シフトを誘導する。次いで、複合体は解離され、バインダーがペプチドから分離され、および正確な質量測定のため高分解能LC/ESI−TOFを用いて検出される(例えばWaters LCTプレミア質量分析計により)。データデコンボリューションアルゴリズムは、質量検出分析からの結合分子の同定を可能にする。
【0032】
FGFRの小分子化合物アロステリック調節因子を同定するには、この技術を天然のまたは変異された種々のFGF−Rの細胞外ドメインに適用することができる。天然型は、細胞外ドメインに対するバインダー全ての検出を可能にする。あるいはアロステリック調節因子は、SSR結合部位に近接するFGF−R2ヘリックスの「開口(open)」型を用いてスクリーニングすることができる。前記「開口」型は、アルファヘリックスを安定化し、これによりSSR結合への感作を可能にする変異Tyr328Arg−Ile329Lysにより得ることができる。変異FGF−R2は、次いでWT FGF−R2の代わりにスクリーニングに使用される。類似の戦略を、FGF−R2におけるTyr328およびIle329に対応するアミノ酸に変異を有するFGF−R1、−R3または−R4をスクリーニングするのに使用することができる。Tyr328Aspでの変異型(FGF−R2)または対応する位置で変異を有する他のFGF−Rを、対照として使用することができる。実際、SSRは、疎水ポケット近傍のTyr328Aspで変異されたFGF−R2には結合しない。したがってこの変異型は、FGF−R2における標的化ポケットと相互作用しない化合物の部分を廃棄するのに使用することができる。
【0033】
全てのこれらの場合において、この戦略は、目的とするペプチドの標的ポケットに結合することができる小分子化合物の同定をもたらす。第2の段階では、細胞におけるシグナル伝達に対する効果が評価されなければならない。R&D Systems社製のProteome Profiler(商標)Array「ヒトホスホ−キナーゼアレイキット」により同定されたリン酸シグナル伝達経路に基づき、プロテオームプロファイラーにより検出された影響を受けないキナーゼを阻害することなく、(PYK2、eNOS、p53、c−jun、AKT、CREB、Erk1/2において)リン酸化キナーゼレベルでHUVECに対するFGF−2効果を阻害する能力に関して、アロステリック調節因子を(細胞タンパク質抽出物でまたは細胞で直接)ELISAによりチェックすることができる。
【0034】
類似のアプローチを、他のRTKに対して踏むことができる。受容体の細胞外ドメインにおける1つ以上のフラストレートされたドメインを同定した後、前記フラストレートされたドメインをSEC−LC/MSアプローチで使用して、フラストレートされたドメインの領域におけるバインダーを同定することができる。シグナル伝達経路に対するバインダーの効果は、次いで上記のようなホスホマップ(phosphomap)アプローチ、または経路の任意の他のレポーター系を用いてテストすることができる。
【0035】
本発明のさらに別の態様は、本発明による「アロステリック阻害剤」とも呼ばれる、および/または本発明による方法により同定された、アロステリック結合部位に結合している小化合物化合物である。
【0036】
「化合物」とは、単純または複合有機および無機分子、ペプチド、ペプチドミメティクス、タンパク質、抗体、炭水化物、核酸またはこれらの誘導体を含む、任意の化学的または生物学的化合物を意味する。
【0037】
(実施例)
例に対する材料および方法
STD−NMR結合アッセイ
ヒトFGFR1遺伝子(P11362)の細胞外ドメイン(ECD;アミノ酸:39−358)をPCR増幅し、およびNdeIおよびBamHI制限部位を用いて、大腸菌(E. coli)ベクターpETTEV(N末端His−タグの後にTEVプロテアーゼ切断部位が続く)にクローン化した。タンパク質産生には、得られたプラスミド(pET FGFR1 D1D2D3)を大腸菌BL21(DE3)(Novagene社)に形質転換した。細胞はOD600が0.6に達するまで37℃で増殖させ、組換えタンパク質産生は1mM IPTG(イソプロピル−b−D−チオガラクトピラノシド)を添加して誘導した。4時間誘導後、細胞を回収し、使用まで−80℃で貯蔵した。細胞ペレット(1L培養)を解凍し、リゾチーム(2mg)、および40Uベンゾナーゼ(Merck社)を含有する50ml緩衝液1(20mM Tris/HCl、pH7.5、200mM NaCl)に再懸濁した。細胞を超音波処理により破壊し、封入体(IB)を遠心分離(15,000g、20分、4℃)により沈降させ、得られたペレットを緩衝液1で2回洗浄した。IBペレットは20ml変性緩衝液(6Mグアニジン−HCl、20mM Tris/HCl、pH8.0、200mM NaCl)に室温で40分間溶解した。可溶性デブリを遠心分離(30,000g、30分)により除去し、上清を、製造者の推奨に従って緩衝液Aで事前に平衡化したNi−NTAカラム(Qiagen社)に添加した。FGFR1 ECDは、500mMイミダゾールを含む変性緩衝液を用いてカラムから溶出した。ECDを含有する画分をプールし、50mM Tris/HCl、pH8.0、250mM NaCl、0.5M L−アルギニン、2mM EDTA、0.02%アジドへの可溶化タンパク質のフラッシュ希釈(flash−diluting)(希釈係数1:30)と、この後の24時間、4℃で穏やかな撹拌によるインキュベーションによりリフォールディングした。リフォールディング混合物を20分間、30,000gで遠心分離し、アミコン撹拌セル(Amicon stirred cell)でYM10膜を通じて濃縮し(最終タンパク質濃度1mg/ml)、25mMTris/HCl、pH8.0、2mM EDTA、0.02%アジドに対して透析し、HiTrapヘパリンHP 5ml(GE Healthcare社)に適用し、0から2M NaClへの直線勾配で溶出した。FGFR1 ECDの最終精製は、25mM Tris/HCl、pH8.0、200mM NaCl、25mM L−アルギニン、2mM EDTA、0.02%アジドで平衡化したHi Load26/60 75pGカラム(GE Healthcare社)を用いてサイズ排除クロマトグラフィーにより達成した。FGF1(アミノ酸:16−155)およびFGF2(アミノ酸9−155)およびTNF−Riαを発現させ、精製した。FGFR1 ECDの構造完全性は、ヘパリンカラム(上記参照)に結合する能力およびFGF1との複合体の形成により実証した。複合体形成は、サイズ排除クロマトグラフィーおよびこの後のSDS−PAGEでの分析により分析した。
【0038】
全てのSTD−および1D−NMR実験は、BRUKER3チャンネルDRX600およびBRUKER4チャンネルDRX800スペクトロメーターで298Kの標準温度で行い、および内部標準3−トリメチル−2,2,3,3−テトラジュウテロプロピオン酸−ナトリウム塩(TSP)を基準にした。典型的には、NMR試料は、25mM Tris/HCl、pH8.0、200mM NaCl、25mM L−アルギニン、2mM EDTA、0.02%アジド(95%H2O/5%D2O中)中に0.5mlのタンパク質(20−300mM)を含有した。タンパク質リガンド1D STD NMR測定のため、スペクトルを、別々のタンパク質メチル共鳴での弱い2秒RF照射により、1mMリガンドSSR128129E(100mM DMSO原液)および40mMタンパク質で記録した。水抑制は、標準Bruker WATERGATE3−9−19シーケンスを用いて行った。NMRデータは、Brukerプログラムxwin NMRソフトウェアを用いて処理した。
【0039】
等温滴定熱量測定(ITC)
熱量測定実験は全て、以前に記載されているように45、VP−ITC滴定熱量計(MicroCal Inc.社、ノーサンプトン、MA)により30℃で実施した。滴定は、1.407mLの10−20μM相互作用タンパク質(すなわちFGFR2∂123、FGFR2∂23ならびにこの記載された変異体およびサブドメイン、FGFR3∂123、FGF1、FGF2ならびにホリスタチン(陰性対照として)を含有する溶液細胞への、4分間隔、回転撹拌器−シリンジを介した1.25mM SSRの10μLアリコートの添加を伴った。300rpmの一定撹拌速度を維持し、データをMicroCal社により供給された標準非相互作用一部位モデル(nが1.0に固定される。)に適合させた。測定は全て、10mM HEPES pH7.2、150mM NaClで実施し、タンパク質を以前に記載されているように(Pellegriniら、2000年)精製した。変異誘発は「部位特異的変異誘発キット」(Stratagene社)を用いて実施した。
【0040】
フーリエ変換赤外線測定
フーリエ変換赤外線測定は、AquaSpecフローセルを備えたBruker Tensor 37 FT−IRスペクトロメーターを用いて実施した。試料コンパートメントは25℃に温度自動調節し、ノイズ比に対し良好なシグナルについて100スペクトルを平均化した。タンパク質は上記のように精製した。ゲル濾過直後、タンパク質を、SSRの存在下または非存在下、同じ調製の緩衝液(10mM Hepes pH7.2、150mM NaCl)で一晩透析した。透析緩衝液試料を使用してバックグラウンドシグナルを減算した。分析は、Brukerにより提供されたOPUSソフトウェアパッケージを用いて実施した。結果の解釈は記載されているように実施した{Barth、2002年 #60}。
【0041】
HEK293トランスフェクションならびにErk1/2、PLCγおよびFRS2リン酸化試験
HEK293細胞は、pcDNA3(Invitrogen社)にクローン化したhFGFR2IIIcaまたはhFGFR2IIIca−Y328Dを(FuGENE6、Roche社を用いて)一時的にまたは安定にトランスフェクトした。安定にトランスフェクト細胞はG418(400μg/ml)含有培地で増殖させた。刺激前、細胞を一晩、DMEM(0%血清)で飢餓状態にし、および必要とされる濃度でSSR128128Eと一緒にプレインキュベートした。細胞をこの後、1μMでのSSRまたはSU5402ありまたはなしにより37℃、5分間、FGF2(0.5−10ng/mlの間の濃度)で刺激した。ホスファターゼ阻害剤(Roche社)を含有する氷冷リン酸緩衝生理食塩水で洗浄後、細胞をRIPA緩衝液(製造者(Roche社)により記載されているようにホスファターゼおよびプロテアーゼ阻害剤を含有する、Tris 30mM HCl pH7.5、150mM NaCl、1mM EDTA、1%トリトン−X、0.5%w/vデオキシコール酸)で溶解した。細胞溶解物を10分間、12,000gで遠心分離し、上清を回収した。タンパク質をNovexポリアクリルアミドゲル(Invitrogen社、カールスバッド、CA)で分離し、この後Hybond ECLニトロセルロース膜(Amersham Pharmacia社)に移した。PBSで5%非脂肪乳粉と一緒にインキュベーションした後、膜を以下の抗体:ホスホ−ERK1/2(CST:9101)、ホスホ−FRS2(CST:3861)、ホスホ−PLCγ(CST:2821)およびFGFR2(F0300、Sigma社)と一緒に一晩、4℃でインキュベートした。
【0042】
FGFRトランスフェクトBaF/3細胞増殖:
この実験に使用されたBaF/3細胞の構築は、出願WO2007/080325に詳細に記載されている。
【0043】
定量的リアルタイムPCR
全RNAは、トリゾール試薬(Invitrogen社、USA)およびRNeasyキット(Qiagen社、ドイツ)を用いてHUVECから単離し、全RNAからこの後Quantitect Reverse Transcriptionキット(Qiagen社、ドイツ)を用いてcDNAを調製した。プライマーセットおよびFAM(商標)色素標識TaqMan(登録商標)MGBプローブ(Eurogentec社、ベルギー)を、ヒトFGFR1、FGFR2、FGFR3、FGFR4、およびTBP用に設計し、PCR反応は7500 FastリアルタイムPCRシステム(ABI社、ドイツ)で行った。各試料は、特定の標準対照および非テンプレート対照と一緒に三通りに分析した。増幅は、2×TaqMan(登録商標)Universal PCR Master Mix、20×Assays−on−demand(商標)Gene Expression Assay Mixを用いて行った。各試料における最初のmRNAコピー数の計算は、サイクル閾値(CT)方法により行った。FGFR1、FGFR2、FGFR3、FGFR4、mRNAのコピー数は、TBP mRNAレベルを用いて正規化した。
【0044】
FGFR1リン酸化測定
hFGFR1IIIcα−ヘマグルチニンを安定にトランスフェクトされたラット脂肪パッド内皮細胞を、80−90%培養密度に増殖させ、24時間血清飢餓状態にした(0.5%FBS)。刺激は、SSRまたはDMSO(対照として)と組み合わせて2ng/mlでのFGF2で5分間実施した。細胞溶解物を10分間、12,000gで遠心分離し、上清を回収した。HAタグ付タンパク質を、アガロース結合抗HA抗体の存在下、一晩、4℃で細胞培養物のインキュベーションにより免疫沈降させた。免疫複合体は1mlの溶解緩衝液で3回洗浄した。タンパク質は、50μlの2×SDS試料緩衝液と一緒のインキュベーションおよび煮沸を介して溶出した。タンパク質をNovexポリアクリルアミドゲル(Invitrogen社、カールスバッド、CA)で分離し、この後Hybond ECLニトロセルロース膜(Amersham Pharmacia社)に移した。PBSで5%非脂肪乳粉と一緒にインキュベーションした後、膜を以下の抗体:pFGFR(CST:3471)およびFGFR1(CST:3472)と一緒に一晩、4℃でインキュベートした。
【0045】
異方性測定
SSRが、結合ポケットへのFGF1の結合を阻害するかどうかを評価するため、本発明者らはFcタグなしのFGFR2の全細胞外ドメイン(FGFR2∂123)を精製し、およびSSR(1mM)なし(青)またはあり(赤)のさまざまな濃度のFGFR2∂123の存在下、蛍光lumioタグ付FGF1(FGF1−lumio;1μMの定濃度)の反転速度(異方性のパラメーターとして)を測定した。FGFR2∂123をFGF1−lumioに添加した場合、リガンド/受容体複合体の反転速度は、該複合体のより大きなサイズのためFGF−lumio単独の反転速度より遅かった。SSRの大きなモル過剰(1000倍)は複合体の反転速度を変化させず、SSRがFGFRからFGFを移動させないことを確認した。
【0046】
HUVEC増殖
コンフルエントなHUVEC細胞を回収し、0.5%FCS(Hyclone社、SH30070.03)、2mMグルタミン、MEM非必須アミノ酸 1×(Gibco社、11140−035)、MEMピルビン酸ナトリウム 1×(Gibco社、11360−039)を含む100μl RPMI 1640(Invitrogen社、32404−014)中の5 104細胞を、一晩、96ウェルコラーゲンIコーティングプレート(Beckton Dickinson社、354650)にウェルごとに播種する。次いで、培地を除去し、ならびに2×FGF2(R&D社、234−FSE−025)、FGF4(R&D社、235−F4−025)またはFGF−19(自家製造)および50μlの2×SSR(200または600nM)を含有する50μlの培地に置換する。細胞をCO2チャンバーで3日間、37℃でインキュベートし、増殖を100μlの「Cell Titer GIo Luminescent細胞生存能」キット(Promega社、G7571)でATP含量を定量化して評価する。
【0047】
HUVEC走化性遊走
コンフルエントなHUVEC細胞を回収し、およびFCS、2mMグルタミン、MEM非必須アミノ酸 1×(Gibco社、11140−035)、MEMピルビン酸ナトリウム 1×(Gibco社、11360−039)を含まないRPMI 1640(Invitrogen社、32404−014)に0.8 106細胞/mlで再懸濁する。250μlの細胞溶液を、内皮細胞遊走のための24ウェルBD Biocoat血管新生システム(BD Biocoat社、354144)の上のチャンバーに4×SSRで分配し、および750μlの培地を、FGF2(R&D社、234−FSE−025)、FGF4(R&D社、235−F4−025)またはFGF−19(自家製造)を含む下のチャンバーに67ng/mlで分配する。プレートをCO2チャンバーで22時間、37℃でインキュベートする。次いで、プレートインサートを除去し、および500μLのカルセイン(Molecular probes社、C−3100)を含有する新たな24ウェルプレート(Falcon社、353504)に90分間入れる。次いで遊走細胞が蛍光し、遊走を485nm励起および535nm発光後、ダウンステアズリーディング(downstairs reading)でルミノメーターにより測定する。
【0048】
HUVECインビトロ血管新生
コラーゲン/マトリゲルゲルは、チャンバースライド(Biocoat Cellwareコラーゲン、タイプI、8ウェル培養サイド(cultureside):Becton dickinson社 354630)の各ウェルに、コラーゲンI(ラット尾コラーゲン、タイプI:Becton dickinson社 354236)中160μlの1/6希釈マトリゲル(増殖因子低減マトリゲル:Becton dickinson社 356230)を分配して調製する。重合が1時間、37℃で生じる。次いで、15.103HUVECを、400μl EBM培地(Clonetics社 C3121)+2%FCS+hEGF 10μg/mlにウェルごとに添加する。内皮細胞を、10ng/mlのFGF2(R&D社、133−FB−025)、FGF4(R&D社、235−F4−025)またはFGF19(R&D社、969−FG−025)で、CO2チャンバーで24時間、37℃で刺激する。次いで、完全長の偽細管をバイオイメージングシステム(Imagenia Biocom社、Courtaboeuf、フランス)を用いて定量化する。
【0049】
HUVECにおけるAKTリン酸化のウェスタンブロット分析
HUVE細胞(Promocell社、C−12200)は、2%FBS(Clonetics社、CC−4101)、EGM singlequotsキット(Clonetics社、CC−4133)からの10μg/ml hEGF(Clonetics社、CC−4017)、1250ng/mlヘパリン(Sigma社、H3149)および375ng/ml ECGS(BD Biosciences社、356006)を含有する2mlのEBM培地(Clonetics社、CC−3121)中0.5.106細胞で、35mmコラーゲンIコーティングディスク(BD Biocoat、354456)に播種する。90%培養密度で、細胞を1.8mlのRPMI 1640(Invitrogen社、32404−014)、0.5%FCS、2mMグルタミン、1mM非必須アミノ酸(Invitrogen社、11140−050)、ピルビン酸ナトリウム(Invitrogen社、11360−070)で一晩飢餓状態にする。翌日、細胞を、10×SSR(3μM)ありまたはなしの10×FGF−4(30ng/ml;R&D社、235−F4−025)を含有する200μlの平衡化飢餓培地により10分間刺激する。次に、細胞を冷PBSですすぎ、細胞を、2.5mMオルトバナジウム酸およびプロテアーゼ阻害剤カクテル(Sigma社、P8340)を含有する75μl RIPAで溶解する。細胞溶解物を10分間、12,000gで遠心分離し、上清を回収した。タンパク質を4−20%Novex Tris−グリシンポリアクリルアミドゲル(Invitrogen社)で分離し、この後ニトロセルロース膜(Invitrogen社、IB3010−01)に移した。TBS−0.05%Tween80で5%非脂肪乳粉と一緒にインキュベーションした後、膜を、TBSで1000×希釈した抗ホスホAKT(Ser473、CST、4058)、tween、1%BSAと一緒に一晩、4℃でインキュベートした。各スポットのシグナルがSuperSignal(登録商標)West Dura Extended Duration Substrate(Thermo Scientific社、34076)による化学発光検出後に得られ、スポット密度をBiolmaging System Chemigenius2(Syngene社)を用いて定量化する。
【0050】
オンセルAKTリン酸化ELISA
コンフルエントなHUVEC細胞を回収し、0.5%FCS(Hyclone社、SH30070.03)、2mMグルタミン、MEM非必須アミノ酸 1×(Gibco社、11140−035)、MEMピルビン酸ナトリウム 1×(Gibco社、11360−039)を含む50μl RPMI 1640(Invitrogen社、32404−014)中の5 104細胞を、一晩、96ウェルコラーゲンIコーティングプレート(Beckton Dickinson社、354650)にウェルごとに播種する。細胞は、20ng/ml FGF4および600nMSSRを含有するFCSなしで100μl平衡化飢餓培地で5分間刺激する。次いで、50μlのPFA8%をPBS(Polysciences社、18814)に室温で15分間添加し、細胞を200μl PBSで2分間、3回洗浄する。非特異的部位は、PBS、トリトン0.3%、正常ヤギ血清0.1%(Zymed社、50−062Z)により室温で1時間ブロックし、ブロッキング緩衝液を作製し、およびPBSで1/500希釈した抗ホスホ−AKT(Ser473)抗体(CST、4058)、トリトン0.3%に一晩置換する。一次抗体を次いで除外し、および200μl PBSで2分間、3回洗浄した。HRP結合抗ウサギ二次抗体(CST、7074)を使用して、PBS、0.3%トリトンで2時間、室温で1/2000希釈した後、AKTリン酸化を検出する。次いで、細胞をPBSですすぎ、100μlのHRP基質(Uptima社、UP664781)を暗室で20分間添加する。酵素反応を100μlの停止緩衝液(Uptima社、UPS29590)で停止し、ODを450nmで測定した。
【0051】
FGFRトランスフェクト300−19細胞に結合するFGF2:
FGF2は、購入者の推奨に従ってAlexa Fluor488 C5−マレイミド(Invitrogen社、A10254)で標識した。
【0052】
このAF488−FGF2は、pEF6−V5/His Topoプラスミド(Invitrogen社)でFGFR1またはFGFR4構築物をトランスフェクトしたマウスプレ−B 300−19細胞での結合実験において10ng/mlで使用した。SSR(300nM最終)は、10%FCS(Hyclone社、SH30070.03)、2mMグルタミン、MEM非必須アミノ酸 1×(Gibco社、11140−035)、MEMピルビン酸ナトリウム 1×(Gibco社、11360−039)および150mMモノチオグリセロール(Sigma社、M6145)を含むRPMI 1640(Invitrogen社、32404−014)で、150rpm撹拌下、4℃で細胞と一緒に20分間プレインキュベートしていた。次いで、FGF2(10ng/ml最終)を30分間添加し、FACS Caliburフローサイトメーター(Beckton Dickinson社)を用いて結合を測定する。条件ごとに蛍光培地も分析する。
【0053】
さまざまな増殖因子による細胞遊走
細胞遊走は、ポリカーボネート膜(Costar、Corning Inc.社)を有する8μm孔径トランスウェル透過性支持体を含有する24ウェルインサートを用いて、改変ボイデンチャンバーアッセイにより評価した。指数関数的に増殖している細胞を0.2%FBS含有培地で16時間飢餓状態にし、および同じ低血清培地に5×105細胞/mlで再懸濁した。100μlの細胞懸濁液を上のチャンバーに播種したのに対し、化学誘引物質および/またはSSRは下のチャンバーに入れた。テストした化学誘引物質には、ヒトPDGF−BB、IGF−I、PIGF、EGF(SSR(1μM)の存在下または非存在下、全て100ng/ml)が含まれる。10%FBS含有培地は陽性対照として使用した。37℃で6時間インキュベーションした後、膜の上側の細胞は綿棒を用いて擦り取ったのに対し、下表面の遊走細胞はPBS中1%パラホルムアルデヒドで固定し、および蛍光マイクロソープ(microsope)を用いた定量化のためDAPIで核染色した。定量化は、10×の倍率で5枚のランダム画像を作製しおよび核の数をカウントして実施する。
【0054】
PANC02増殖および遊走:
細胞増殖は、0.2%FBS(Hyclone社、SH30070.03)、2mMグルタミン、MEM非必須アミノ酸 1×(Gibco社、11140−035)、MEMピルビン酸ナトリウム 1×(Gibco社、11360−039)を含む100μl RPMI 1640(Invitrogen社、32404−014)で16時間飢餓状態にしおよび96ウェルマイクロプレートに4,000細胞/ウェルで播種した、指数関数的に増殖している細胞で分析した。分裂促進因子および/またはSSRに72時間曝露した後、細胞増殖をCellTiter 96 AQueous One Solution Cell Proliferation Assay(Promega社、マジソン、ウィスコンシン、USA)を使用して製造者の指示に従ってアッセイした。細胞遊走は、ポリカーボネート膜(Costar、Corning Inc.社)を有する8μm孔径トランスウェル透過性支持体を含有する24ウェルインサートを用いて、改変ボイデンチャンバーアッセイにより評価した。指数関数的に増殖している細胞を0.2%FBS含有培地で16時間飢餓状態にし、および同じ低血清培地に5×105細胞/mlで再懸濁した。100μlの細胞懸濁液を上のチャンバーに播種したのに対し、化学誘引物質および/またはSSRは下のチャンバーに入れた。10%FBS含有培地は陽性対照として使用した。37℃で6時間インキュベーションした後、膜の上側の細胞は綿棒を用いて擦り取ったのに対し、下表面の遊走細胞は4%ホルムアルデヒドで固定し、および定量化のためDAPIで核染色した。
【0055】
B9骨髄腫細胞増殖:
細胞増殖は、IMDM(Invitrogen社、31980048)、0.2%FBS、2mMグルタミン含有培地で16時間飢餓状態にしおよび96ウェルマイクロプレートに4,000細胞/ウェルで播種した、指数関数的に増殖している細胞で分析した。分裂促進因子および/またはSSRに72時間曝露した後、細胞増殖をCellTiter 96 AQueous One Solution Cell Proliferation Assay(Promega社、マジソン、ウィスコンシン、USA)を使用して製造者の指示に従ってアッセイした。
【0056】
アルファスクリーンシュアファイアアッセイ
0日目:HEK293:mVEGFR2wt細胞またはHEK293:PDGFRβ細胞を10000細胞/ウェル(96ウェルプレート細胞結合Costar)でプレーティングし、一晩接触させる
1日目:細胞をDMEM(0%血清)で最低3時間飢餓状態にする:50ng/mlのVEGF164またはPDGF−BBの混合物をDMEM(0%血清)で調製し、5または15分間刺激する:シュアファイアアッセイ(Perkin Elmer社)からの溶解緩衝液で細胞を溶解する:50μlの緩衝液で細胞を溶解し、プレートをRTで10分間撹拌し、次いでさらなる使用まで−20℃で凍結する;タンパク質と溶解緩衝液の混合物を作製し、および製造者の指示に従ってpERK1/2、合計ERK1/2ならびにカスタムデザインされたpPLCγおよび合計PLCγで分析する。
【実施例1】
【0057】
アロステリック、マルチFGFR阻害剤としてのSSR128129Eの同定
この試験の目的は、FGFR細胞外ドメイン(ECD)に結合しおよびFGFRシグナル伝達を阻害する低分子量化学化合物を開発することであった。小分子化合物が、オルソステリック部位に対する単純な立体障害を介してはるかに大きなポリペプチド(すなわちFGF)とどのように相互作用できるかを構想することは困難であることから、複数のリガンド結合アッセイフォーマットを利用して、任意の同定された化合物がアロステリック機構を介してオルソステリックに作用しているかどうかを判定した。本発明者らは最初に、ハイスループットシンチレーション近接結合アッセイ(SPA)を開発して、Fc−フラグメントに結合した、3つのIg様ドメインD1−3(FGFR1∂123/Fc)から成る、FGFR1−ECDへの125I−FGF2の結合を阻害する化合物を同定した。>20,000化合物のスクリーニングおよび化学的最適化の後、1つの化合物、SSR128129E(以後「SSR」と略す)が、125I−FGF2結合を阻害した。追加のSPAアッセイでは、SSRは、さまざまなFGFRへの種々のFGFリガンドの結合をブロックする一方で、関連した構造相同性または全く異なる化学組成を有する>100の異なるリガンドと同種受容体との結合を阻害しない、マルチFGFR阻害剤として作用した。この知見は、競合的(オルソステリック)機構、または高い負の協同性(ChristopoulosおよびKenakin、2002年)を特徴とする他のアロステリック相互作用のどちらかを示唆した。
【0058】
アロステリック相互作用の1つの顕著な特徴は、「プローブ依存性(probe−dependence)」、すなわち、調節因子が相互作用しているオルソステリックリガンド−受容体複合体の性質に依存するアロステリック相互作用の大きさおよび方向の変動の現象である(Mayら、2007年)。SPAでの125I−FGF2結合に対するSSRの効果が、人工基質にプレーティングされた改変FGFR/Fc融合タンパク質の立体配置に依存したかどうかを判定するため、本発明者らは次に、異方性のパラメータとしての反転速度を測定して、SSRが、Fc−タグなしのFGFR2の精製ECD(FGFR2∂123)への、蛍光lumioタグ付FGF1(FGF1−lumio)の結合を阻害するかどうかを試験した。FGFR2∂123をFGF1−lumioに添加した場合、リガンド/受容体複合体の反転速度は、該複合体のより大きなサイズのためFGF−lumio単独の反転速度より遅かった。SSRがリガンド結合を阻害したなら、反転速度は再び増加するはずである。しかし、>1,000倍のモル過剰でさえ、SSRは複合体の反転速度を変化させなかった。これは、SSRとFGF1−lumioの間の直接競合の欠如を示唆している(図5A)。最後に、ヒト臍帯静脈内皮細胞(HUVEC)またはFGFR1を過剰発現するブタ大動脈内皮細胞(PAE−FGFR1)でのI125−FGF2による結合アッセイもまた、後者がPAE−FGFR1がインタクトな細胞においてより自然な立体構造で発現される場合、SSRが(高μM濃度でさえ)受容体へのI125−FGF2結合を阻害できないことを明らかにした。しかし、αFGF2抗体を中和することは効果があった。この後者の実験パラダイムでは、およびSPAとは対照的に、SSRは、他のFGFRへの別のFGFリガンド(すなわちFGFR2へのFGF2またはFGF4;FGFR4へのFGF2)の結合も拮抗しなかった。
【0059】
まとめると、これらの結果から、FGFリガンドの結合に対するSSRの阻害剤活性は、FGFRの立体構造に高度に依存しており、重複する結合ドメインの立体障害に依存する単純な競合的機構とは一致しなかったことが示される。FGFRについて本明細書で述べたように、アッセイ条件に応じてオルソステリックリガンドの結合に示差的に影響する小分子化合物アロステリック調節因子の能力は、GPCRの分野で以前に報告されている(Litschigら、1999年;Priceら、2005年)。恐らく、FGFR1∂123/Fcが、SSRの結合による125I−FGF2の親和性に対する負のアロステリック効果の伝達を可能にする立体構造に存在するのに対し、Fcタグ、すなわち天然の環境におけるインタクトな受容体全体の発現の欠如は存在しない。
【実施例2】
【0060】
SSRはアロステリックおよびマルチFGFR阻害剤である。
FGFR遺伝子発現(図1B)およびFGFR1バリアント(図1C)を検出する特定のプライマーを用いた定量的PCR(図1A)およびRT−PCRによるHUVEC細胞(C−12200、Promocell社)でのFGFR発現分析は、FGF−R4およびFGF−R1β3cの発現のみを実証したことから、本発明者らは、HUVE細胞を最初に用いて異なるFGFRでのSSRの拮抗活性を試験した。FGF19はFGFR4を特異的に刺激することが知られているのに対し、FGF4(FGF19ではなく)はFGFR1−hMpl融合タンパク質をトランスフェクトしたBaF/3細胞においてFGF−R1のみを活性化する(図2A)一方、FGF19は活性化できない(図2B)。したがって、HUVECにおいてFGFR1およびFGFR4は、それぞれFGF4およびFGF19で刺激することができる。
【0061】
HUVEC増殖は、FGF2およびFGF4で刺激されるが、FGF19では刺激されず(図4A)、HUVEC増殖がFGFR1の制御下にあることを示唆している。SSRは、FGF2誘導HUVEC増殖を阻害することができ、SSRがFGFR1β3c受容体を阻害することを示している(図4B)。ヒト臍帯静脈内皮細胞(HUVEC)またはFGFR1を過剰発現するブタ大動脈内皮細胞(PAE−FGFR1)でのI125−FGF2による結合アッセイは、SSRが(高μM濃度でさえ)受容体へのI125−FGF2結合を阻害できないことをさらに明らかにした。しかし、αFGF2抗体を中和することは効果があった(図4D)。本発明者らはまた、SSRが、FGFRシグナル伝達における重要な段階であるFGFRの自己リン酸化を阻害するかどうかも分析した。ラット脂肪パッド内皮細胞で発現されたFGFR1の免疫沈降とこの後のリン酸化FGFR1の免疫ブロットは、FGF2誘導FGFR1チロシンリン酸化が、ナノモル濃度範囲でSSRにより高度に低減されることを明らかにした(図4C)。特に、高用量でさえ、SSRはFGFR1チロシンリン酸化を完全には排除せず、低い残基レベルを残した(図4C)。FGFR2細胞外ドメインに結合するlumioタグ付FGF1に対するSSR効果が分析されており、SSRはFGF1/FGFR2相互作用を阻害しない(図5A)。同様に、SSRはFGFR2またはFGF2二量体化を阻害することができる(図5Bおよび5C)。
【0062】
HUVECの遊走も、FGF19ではなくFGF2−およびFGF4により刺激した(図6A)。この関連で、SSRは、FGFR1活性を阻害し、FGF2誘導HUVEC走化性遊走の低減をもたらすこともできる(図6B)。
【0063】
反対に、インビトロ血管新生は、FGF2およびFGF19により刺激されるのに対し、FGF4は不活性であり、FGFR4がこのアッセイではインビトロ血管新生を制御することを示唆している(図7A)。低いナノモル範囲では、SSRはFGF2誘導HUVEC血管新生をブロックし、SSRがFGFR4に制御される細胞プロセスを阻害できることを実証している(図7B)。
【0064】
FGFR2およびFGFR2−IIIbバリアントに対するSSR活性を評価するため、PANC02細胞の増殖および遊走が使用された。この理由は、これらの細胞反応が、FGFR2−IIIbの特異的リガンドである100ng/ml FGF7により刺激され得るためである(図8Aおよび8B)。VEGFの有無にかかわらず、FGF7誘導は100nM SSR添加によりブロックされ、SSRがFGFR2受容体および3bバリアントを阻害できることを示している(図8Aおよび8B)。
【0065】
FGFR3に対するSSR効果を試験するため、FGFR3WTまたはFGFR3TD(いずれかのリガンドの非存在下でさえ、K650E変異により誘導される構造的活性化FGFR3バリアント;Truedelら;blood 2006年)のどちらかを発現するB9−骨髄腫細胞の増殖を、FGF1(25ng/ml)による刺激によりアッセイした。B9−FGFR3WT細胞系が、FGF1により誘導され得および0.1μMのSSRにより阻害され得る(図9)のに対し、B9−FGFR3TD細胞系はSSRに非感受性であった(図9)。これは、SSRがFGFR3受容体を阻害できることを示しており、SSRがFGFRのキナーゼドメインで作用しないことを確認するものである。
【0066】
まとめると、これらの結果は、SSRが全てのFGFRアイソフォーム(FGFR1、R2、R3およびR4)およびFGFRバリアントを阻害できることを示している。
【実施例3】
【0067】
SSRは他の増殖因子により誘導される細胞反応を阻害できない。
SSRがFGF依存性シグナル伝達効果を示差的に阻害したことから、本発明者らは次に、SSRがインビトロでのFGF依存性細胞反応にも影響するかどうかを調査した。HUVECを用いると、SSRはFGF2の走化性効果を阻害した。
【0068】
SSRは、いずれもチロシンキナーゼ受容体ファミリーのメンバーを活性化することが知られているPIGF、EGF、PDGF−BBおよびIGFにより誘導された細胞反応に影響しなかった(図10)。
【実施例4】
【0069】
SSR128129EはFGFRの細胞外領域のIg様ドメインD3におけるアロステリック部位に結合する。
SSRはマルチFGFR阻害剤であったことから、本発明者らは、さまざまな(ヒト)FGFRサブタイプのポリペプチドフラグメントを使用した。SSRの飽和移動差NMR(STD−NMR)スペクトルは、SSRがFGFR1のECD(FGFR1∂123)に結合することを明らかにした(図11A)。これは、一次元(1D)−NMRプロファイルの分析により確認され、これがSSRの添加時のFGFR1∂123シグナルのピーク拡大を明らかにした(図11A)。この結合はTNF−R1細胞外タンパク質では観察されないため、この結合は特異的である(図11A)。本発明者らは次いで、FGFRのECDフラグメントを使用して3つのIgドメインの1つに対するSSRの結合部位をマッピングした。ドメインD3(FGFR1∂3)のみを含有するフラグメントの1D−NMR測定は、この膜近傍ドメインにおいてSSRの結合部位を同定した(図11B)。事実、FGFR1∂3およびFGFR1∂123は、本発明者らがこれらのタンパク質について同じ親和性を得たことを意味する同一のシグナル(太線)を与えるのに対し、FGFR1∂12およびFGFR1∂2は明確な線(図11B)をもたらす。2つのECDフラグメント、FGFR2∂23(ドメインD2および3から成る)およびFGFR3∂23を用いた等温滴定熱量測定(ITC)は、SSRがFGFR2およびFGFR3に結合することを明らかにした(図11Cおよび11D)。
【0070】
FGFRのドメインD3へのSSRの結合は特異的であった。この理由は、ITCまたはSTD−NMRにより分析した場合、該化合物がFGF−リガンド(FGF1およびFGF2;図12A、12Bおよび12C)に結合しなかったためである。さらに、ヘパリンはFGFRへのSSRの結合を妨げなかった。この理由は、STD−NMRが、ヘパリン類似体八硫酸スクロース(SOS、図12D)の存在下または非存在下、FGFRへのSSRの同等のシグナルを明らかにしたためである。
【実施例5】
【0071】
SSRのアロステリック結合はFGFRにおける立体構造変化を誘導する。
本発明者らは次いで、先行実験で同定された領域へのSSRの結合により媒介されたFGFRの立体構造変化の直接的証拠を、本発明者らが得ることができるかどうかを調べた。したがって、本発明者らは、ドメインドメインD2−3(FGFR2∂23)から成る、FGFR2のECDフラグメントのフーリエ変換赤外線(FTIR)分光測定を実施した。どちらかのバリアントへのSSRの添加は、全体的な立体構造変化と一致する、1,640cm−1周辺で最大となる、FTIRスペクトルのアミドIバンドの振幅の増加をもたらした(図12A)。
【0072】
本発明者らは次に、SSRが、D3におけるオルソステリック部位の部分を形成するアミノ酸残基または別のアロステリック部位に結合するかどうかを調べた。最初に、本発明者らは、ソフトウェアパッケージMOLEGRO、AutodockおよびYASARAならびに利用可能な結晶学的データの分子ドッキングアルゴリズムを使用した。両方の方法を用いたFGFR2∂3でのSSRのドッキングランは、2つの想定される結合部位(1つはHis293周辺に集中し、もう1つはTyr328周辺に集中していた。)を同定した。これらの想定される結合部位は、FGFリガンド結合部位と比べて、受容体の反対面に位置しおよびオルソステリック結合部位から25Åまでの疎水ポケットを形成する。特に、両方の残基は、オルソステリックFGF結合ポケットの残基と重複しない。両方の想定されるSSR結合部位の機能的関連性を評価するため、本発明者らは、分子力場FoIdXソフトウェア(Schymkowitzら、2005年)を使用して、アロステリックリガンド結合を低減または排除するが、構造の全体的な立体構造安定性を撹乱しない変異を設計した:(i)芳香族残基を負に帯電されたアスパラギン酸と置換してSSRとの疎水性相互作用を除去するFGFR2∂23−Y328D;(ii)SSRの他の想定される結合部位から必須残基を除去するFGFR2∂23−H293L;および(iii)FGFR2∂23−Y328D/H293L二重変異体(FGFR2∂23−YHと呼ばれる)。ITC結合実験は、SSRがFGFR2∂23−Y328Dに結合しないことを示している(図13Bおよび13C)。これらの知見は、SSRが、オルソステリックリガンド結合部位近傍の疎水ポケットにより形成されたアロステリック部位に結合するモデルで、および残基Tyr328はSSRとFGFR2の間の相互作用を媒介するのに必須と思われるモデルと一致する。本発明者らはまた、前述の変異FGFR2フラグメントのFTIRスペクトルも分析した。これらの単一または二重変異は、FTIRスペクトルにおける重大なシフトを誘導しなかった。これは、これらのFGFRバリアントの全体的な三次元立体配置が同等であることを示している。SSRは、FGFR2∂23−H293Lおよび天然のFGFR2∂23フラグメントのFTIRスペクトルの同等のシフトを誘導した(図14Aおよび14B)。これは、His293からLeu293への変異が十分に強烈でないこと、またはHis293がSSRとの相互作用にそれほど意味がないことを示唆している。その一方、SSRは、変異FGFR2∂23−Y328DまたはFGFR2∂23−YHフラグメントのFTIRスペクトルの変化を誘導しなかった(図14Cおよび14D)。これは、残基Tyr328が、SSRを結合する時のFGFR2のアロステリック立体構造変化の媒介に実際に必須であったことを示している。
【実施例6】
【0073】
アロステリックSSR結合部位の変異はFGFRシグナル伝達のSSR阻害を低減する。
FGFRシグナル伝達の調節におけるアロステリック部位の機能的重要性を評価するため、本発明者らは、機能的FGFR2WTまたはFGFR2∂23−Y328Dバリアントのどちらかを発現する安定なHEK293細胞系を作製し、SSRがこれらの細胞系でFGF2によるERK1/2の活性化を阻害するかどうかを分析した。免疫ブロッティングは、SSRによるFGFR2∂23−Y328D細胞でのFGF2誘導ERK1/2リン酸化の阻害が、FGFR2WT細胞での阻害能力(IC50値:28±12nM)と比べて低減する(IC50値:121±30nM)ことを明らかにした(図15Aおよび15B)。これは、このアロステリック部位が、インビトロでの精製FGFR2フラグメントへのSSR結合に関係がないだけでなく、インセルロ(in cellulo)での生理条件ではFGFR2シグナル伝達に対する阻害活性にも関係がないことを示している。Y328D変異が、SSRの阻害活性を完全には無効にしなかったという事実は、FGFR2が生理学的文脈で発現される場合に、Tyr328に加えて他の隣接残基もSSRの結合に寄与することを示唆している可能性がある。
【実施例7】
【0074】
SSRはFGFR依存性ホスホ−シグナル伝達の「偏った」阻害剤である。
FGFR制御リン光体−シグナル伝達に対するSSR効果を評価するため、HEK293細胞をFGFR2でトランスフェクトし、FGFR依存性FRS2およびPLCγカスケードを阻害することが記載された公表FGFRチロシンキナーゼ阻害剤SU5402(Zhenら、Oncogene 2007年)と比較して、FGFR自己リン酸化後の2つの主要経路、PLCγおよびFRS2をウェスタンブロットにより試験した。このような細胞では、FGFは、FRS2、Erk1/2およびPLCγリン酸化を誘導する。SU5402が全てのこれらの誘導をブロックするのに対し、SSRはFRS2経路を阻害するのみであり(図16Aおよび16B)、SSRにより得られた偏った拮抗作用を説明している。より一般的には、リン酸化のこれらの違いは、アロステリック調節因子を評価するためのレポーターとして使用することができる。
【実施例8】
【0075】
チロシンキナーゼ受容体の細胞外ドメインにおけるフラストレートされたドメインを探索するための方法論
例5からの記載された立体構造変化の考えられる分子機構の1つは、フラストレートされたドメイン(定義については上記参照)の存在を含む。AGADIR(ヘリックス安定性予測アルゴリズム;Munoz,V.&Serrano,L. 1994年)を用いてヒトFGFR2のドメインD2およびD3を分析した時、本発明者らは、βシートからαヘリックスへのシフトを起こす蛍光がある唯一の領域として、Tyr319からArg330の範囲の(故に必須残基Tyr328を含む)残基の配列を同定した。アルファヘリカリティ(helicality)を低減し故にドメインのフラストレーションを低減することがAGADIRにより予測されたアスパラギン酸によるTyr328の置換(FGFR2∂23−Y328D)は、このような理論モデルと一致して、FTIR分析により検出されたような観察された立体構造変化を実際に妨げた。他のTKRのうち、VEGFR1、−2および−3ならびにPDGFRβを含む別の増殖因子受容体の類似の配列分析は、比較的高いAGADIRスコアの領域を含有した。該領域はこの領域からアスパラギン酸に必須残基を変異させることで逆になり得る。本発明者らは、K609DおよびK648Dを変異させることが、VEGFによる刺激時にERK1/2シグナル伝達の低減をもたらした、VEGFR2の幾つかの予備データを持っている。
【実施例9】
【0076】
SEC−LC/MSによるFGF−Rアロステリック調節因子の親和性スクリーニングおよび同定された化合物の活性評価
SEC−LC/MS方法論は、オンライン結合された2次元システム、高性能液体クロマトグラフィーに結合されたサイズ排除クロマトグラフィーと、これに続く検出のためのエレクトロスプレーイオン化−飛行時間型質量分析に依存する、親和性スクリーニングに使用される分析技法である。
【0077】
可溶性ポリペプチド(ペプチド、タンパク質ドメイン、または完全長タンパク質)と相互作用する幾つかの化合物の能力に基づく。目的とするペプチドと小分子化合物のプールを混合した後、ペプチド−リガンド複合体は、サイズ排除クロマトグラフィーによる非結合と結合小分子化合物との分離を可能にする質量シフトを誘導する。次いで、複合体は解離され、バインダーが正確な質量測定のため高分解能LC/ESI−TOFを用いて検出される(例えばWaters LCTプレミア質量分析計により)。データデコンボリューションアルゴリズムは、質量検出分析からの結合分子の同定を可能にする。
【0078】
FGF−Rアロステリック調節因子を同定するには、この技術を天然のまたは変異された種々のFGF−Rの細胞外ドメインに適用することができる。天然型は、細胞外ドメインバインダー全ての検出を可能にする。アロステリック調節因子のスクリーニングを実現する別の方法は、アルファヘリックスを安定化し、SSR128129結合への感作を可能にする変異Tyr328Arg−Ile329Lysにより得られた、SSR128129結合部位に近接するFGF−R2ヘリックスの「開口」型を用いて行うことができる。この場合、この変異FGF−R2は、WT FGF−R2にとって代わることができる。類似の戦略を、FGF−R2におけるTyr328およびIle329に対応するアミノ酸に変異を有するFGF−R1、−R3または−R4のスクリーニングを行うのに使用することができる。Tyr328での変異型(FGF−R2)または他のFGF−Rにおける対応する変異アミノ酸は、カウンタースクリーニング(counterscreen)を行うのに使用することができる。SSR128129は、疎水ポケット近傍のTyr328Aspで変異されたFGF−R2に結合しないため、この変異型は、FGF−R2における標的化ポケットと相互作用しない化合物の部分を廃棄するのに使用することができる。
【0079】
全ての場合において、この戦略は、目的とするペプチドにおける標的ポケットに結合することができる分子の同定をもたらす。次の段階では、細胞におけるシグナル伝達に対する効果が評価されなければならない。第1に、選択された化合物は、ウェスタンブロット実験でSSRにより観察されたように(図17Aおよび17B)、HUVEC細胞におけるAKTリン酸化などのFGF誘導誘導経路を阻害しないければならない。HUVEC細胞におけるAKTのリン酸化状態は、オンセルELISA方法論により測定することができる。このアッセイフォーマットは、HUVECなどのさまざまな細胞においてAKTリン酸化を直接検出するため自家開発されており、およびFGF4刺激HUVECに対するSSR効果の検出を可能にする(図17C)。FGFRアロステリック調節因子の典型的な特徴は、FGF結合と競合する能力である。これを評価するため、FGFRをトランスフェクトされたマウスプレ−B 300−19細胞での結合アッセイが作成された。10ng/mlでのAlexaFluor488標識FGF2は、天然にはいずれのFGFRも発現しない300−19細胞で発現されたFGFR1またはFGFR4に結合し、300nMでのSSRは、フローサイトメトリー分析により、FGFR1またはFGFR4へのこの結合を競合することはできない(図17D)。
【実施例10】
【0080】
VEGF−R2受容体における想定されるフラストレートされた帯域の同定および想定されるフラストレートされた帯域の変異分析
FGF−R用に開発された戦略が、別の受容体TK、すなわちVEGF−R2またはKDRに適用される。想定されるアロステリック標的部位を持つことができる領域を同定する最初のアプローチとして、本発明者らは、マウスVEGF−R2受容体由来の利用可能な一次アミノ酸配列(EntrezアクセッションNP_034742.2)を用いて、構造変化(例えばβシートからαヘリックスへの転移)を起こす傾向がある領域を同定するソフトウェアプログラムAGADIR1を利用した。これは、高い螺旋傾向を有する幾つかの領域をもたらしたが、Igドメイン構造では主にβシート構造が期待されるはずである。Agadir分析の結果は図18Aに示されている。
【0081】
この後、VEGF−R2由来の各アミノ酸をアスパラギン酸残基(D)により連続的にインシリコ変異させた後、本発明者らは、AGADIRスコアを再び分析し、ならびに(i)螺旋傾向の変化が最大(最もネガティブ)であるおよび(ii)膜貫通ドメインに最も近傍に位置するようなIgドメインに位置するような変異を選択した。これらから、K609DおよびK648D(両方の残基ともmVEGF−R2のドメインlgD6にある(図18B))は螺旋傾向の最大の低下をもたらし、インセルロ分析にさらに使用された(さらなる例12参照)。
【実施例11】
【0082】
PDGF−Rβ受容体における想定されるフラストレートされた帯域の同定および想定されるフラストレートされた帯域の変異分析
想定されるアロステリック標的部位を持つことができる領域を同定する最初のアプローチとして、本発明者らは、ヒトPDGFRβ受容体由来の利用可能な一次アミノ酸配列(EntrezアクセッションNP_002600.1)を用いて、構造変化(例えばβシートからαヘリックスへの転移)を起こす傾向がある領域を同定するソフトウェアプログラムAGADIR1を利用した。これは、高い螺旋傾向を有する幾つかの領域をもたらしたが、Igドメイン構造では主にβシート構造が発現されるはずである(図19)。この後、PDGFRβ由来の各アミノ酸をアスパラギン酸残基(D)により連続的にインシリコ変異させた後、本発明者らは、AGADIRスコアを再び分析し、ならびに(i)螺旋傾向の変化が最大(最もネガティブ)であるおよび(ii)膜貫通ドメインに最も近傍に位置するようなIgドメインに位置するような変異を選択した。これらから、L383DおよびK387D(両方の残基ともhPDGFRβのドメインIgD3にある)は螺旋傾向の最大の低下をもたらした(図19)。
【実施例12】
【0083】
「偏った」シグナル伝達を誘導する化合物を同定するスクリーニング方法
VEGFまたはPDGF−BBの結合は、それぞれの同種受容体の二量体化を誘導し、これが今度は細胞内キナーゼドメインのリン酸化を誘導する。この後(FGF−Rの偏ったアンタゴニストSSRに一致する目的とする)2つの主要経路が、ERK1/2経路(図21Aおよび21C)およびPLCγ経路(図20に概略的に示されている)を含め活性化される。各経路の活性化の測定は、考えられる阻害剤の存在下または非存在下、シグナル伝達経路の1つのみの阻害により、「偏った」シグナル伝達を誘導する化合物の同定をもたらす。
【0084】
VEGF−R2については、実施例10で同定された2つの変異体VEGFR2受容体(VEGFR2K609DおよびVEGFR2K648D)およびVEGF−R2のウィルト型フォーム(wilt type form)を、HEK293細胞で安定に発現させた。VEGFR2WT受容体は、ERK1/2リン酸化の活性化により明らかに反応する。VEGFR2K609D変異体が、ERK1/2を通じたシグナル伝達能力を低減した(だがカウンタースクリーニングアッセイには十分である)のに対し、VEGFR2K648D変異体はこれを喪失した。結果は図21Aに要約されている。
【0085】
PDGF−Rβについては、hPDGFRβ細胞を過剰発現するHEK293細胞を、アルファスクリーンシュアファイア手順に従って添加剤なし(「0」)、10%ウシ胎仔血清(10%FBS)、50ng/ml PDGF−BB(50ng/ml)を含有する培地または1ng/ml PDGF−BB(1ng/ml)を含有する培地のどちらかで刺激した(図21B)。図21Cでは、左のパネルは、刺激時の活性化または合計ERK1/2タンパク質を検出する混合物の略図を再び表す。右のパネルは、アルファスクリーンシュアファイア検出方法により測定された細胞のPDGF−BB刺激後に検出された測定を表す。これは、細胞を10%FBSまたは50ng/mlのPDGF−BBのどちらかで刺激する場合、PDGF−BBによる1ng/ml刺激の有無にかかわらず、はるかに低いシグナルが存在する間は明らかなシグナルを検出できることを示している。PLCγの答えは、類似の方法で測定する。
【0086】
FGF−RにおけるSSRのように、VEGF−R2またはPDGF−Rβにおいて「偏った」シグナル伝達を誘導する化合物を同定するスクリーニング方法は、Erk1/2およびPLCγの答えに基づく。候補アロステリック阻害剤の存在下および非存在下におけるERK1/2およびPLCγ反応の比較は、偏った阻害剤として作用する、すなわち2つのシグナル伝達経路の1つのみを阻害する化合物の同定を可能にする。VEGF−R2またはPDGF−Rβの変異構築物は、同定されたアロステリック調節因子の作用の機構を検証するカウンタースクリーニングアッセイに役立つことができる。変異受容体では、化合物は受容体調節能力を喪失せざるを得ない。
【0087】
【表1】

【技術分野】
【0001】
本発明は、1回膜貫通型(single membrane span)チロシンキナーゼ受容体に由来する、アロステリック阻害剤に対する細胞外結合ドメインに関する。より具体的には、本発明は、チロシンキナーゼ受容体、すなわち線維芽細胞増殖因子受容体(FGFR)、血管内皮増殖因子受容体(VEGFR)または血小板由来増殖因子受容体(PDGFR)に由来する細胞外ドメインに関する。さらに、本発明は、他のチロシンキナーゼ受容体の細胞外部分における類似のドメインを同定するためのこのドメインの使用、およびアロステリック阻害剤を同定するスクリーニング方法に関する。
【背景技術】
【0002】
細胞表面受容体は、全ての薬剤の大多数に標的を提示する(Overingtonら、2006年)。歴史的に、創薬プログラムは、オルソステリック部位で内因性リガンドと結合を競合するアンタゴニストを開発するための研究が多数派を占めていた。その一方、アロステリック部位、すなわち、オルソステリックリガンド(標的が受容体の場合)または基質(標的が酵素の場合)によって利用されるものとはトポグラフィー的に異なるドメインに結合し、タンパク質の活性を調節する薬剤は、同定するのがより困難であった。しかし、近年は、リガンド依存性イオンチャネルおよびGタンパク質共役受容体(GPCR)に対して同定されるアロステリック調節因子の数の増加をみている(Christopoulos、2002年;Kenakin、2010年。驚くべきことに、増殖因子受容体チロシンキナーゼ(RTK)に関しては、この受容体スーパーファミリーが極めて生物学的に重要であり医学的に意義があるという事実にもかかわらず、ならびにアロステリック剤が、より大きな安全性および/または選択性を含む、伝統的なオルソステリックリガンドに勝る明白な治療上の利点を提供することができるという事実にもかかわらず、アロステリック小分子化合物調節因子はこれまでに同定されていない。これまで、RTKを標的とするほとんどの治療が、増殖因子リガンドを認識するモノクローナル抗体、または受容体のチロシンキナーゼ活性を直接阻害する小分子化学化合物に焦点を合わせてきた。
【0003】
なかでも、より有効なおよび/または選択的なRTK小分子化合物阻害剤から実質的に利益を得られる一つの分野が、抗血管新生薬療法の分野である。VEGF標的化抗血管新生剤は癌患者の生存を延長させるが、全体の奏効は、内因的不応性、獲得耐性を介した逃避(escape)および少なくとも前臨床モデルでは転移の刺激により制限される。別の抗血管新生剤との併用療法は、これらの課題を克服するのに役立つ可能性があるとみなされてきた。最初に同定された血管新生因子である線維芽細胞増殖因子(FGF)−2は、魅力的な薬剤候補である。実際、FGFRシグナル伝達は、癌および炎症性疾患に関与しており(Shinら、2006年;Eswarakumarら、2005年;Malemudら、2007年;Carmeliet、2005年)、腫瘍血管新生スイッチに寄与し(Prestaら、2005年;Kuboら、2002年;Shineら、2006年;Lavineら、2006年)、腫瘍の血管新生およびVEGF阻害剤治療時の再発を助ける(Casanovasら、2005年)。それにもかかわらず、FGFファミリーは、抗血管新生剤開発に関し大きな注目は受けていない。この理由の一部に、18種類のリガンドおよび4種類のFGFRのこのスーパーファミリーのメンバー間の重複性がある(Eswarakumarら、2005年;BeenkenおよびMohammadi、2009年;Cenniら、2005年;Bossardら、2004年;Compagniら、2000年)。また、FGFRチロシンキナーゼの選択的阻害剤は、臨床使用が承認されていない(Dimitroffら、1999年;McDermottら、2005年)。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Overingtonら、Nat.Rev.Drug Discov.5,993−995(2006)
【非特許文献2】Christopoulos、Nat.Rev.Drug Discov.1,198−210(2002)
【非特許文献3】Kenakin、Pharmacol rev,62,265−304(2010)
【非特許文献4】Shinら、Mol Biol Cell 17,576−584(2006)
【非特許文献5】Eswarakumarら、Cytokine Growth Factor Rev 16,139−149(2005)
【非特許文献6】Malemudら、Clinica chimica acta;international journal of clinical chemistry 375,10−19(2007)
【非特許文献7】Carmeliet、Nature 438,932−936(2005)
【非特許文献8】Prestaら、Cytokine Growth Factor Rev 16,159−178(2005)
【非特許文献9】Kuboら、Proc Natl Acad Sci USA 99,8868−8873(2002)
【非特許文献10】Lavineら、Genes & development 20,1651−1666(2006)
【非特許文献11】Casanovasら、Cancer Cell 8,299−309(2005)
【非特許文献12】BeenkenおよびMohammadi、Nature reviews 8,235−253(2009)
【非特許文献13】Cenniら、Anticancer Res 25,1109−1113(2005)
【非特許文献14】Bossardら、Cancer Res 64,7507−7512(2004)
【非特許文献15】Compagniら、Cancer Res 60,7163−7169(2000)
【非特許文献16】Dimitroffら、Invest New Drugs 17,121−135(1999)
【非特許文献17】McDermottら、Bioorg Med Chem 13,4835−4841(2005)
【発明の概要】
【発明が解決しようとする課題】
【0005】
驚くべきことに本発明者らは、化学的最適化と組み合わされた高速スループットスクリーニングにより、RTK、すなわちFGFRの最初の経口的に活性な小分子化合物アロステリック阻害剤を同定できることを見出した。この化合物は、SSR128129(略して「SSR」)と呼ばれる(図3)。
【0006】
SSR活性に基づく詳細な研究により説明されるように、SSRは、同じファミリー(現在はFGFRファミリー)の全てのメンバーを阻害する能力を有する。以下の例で示されるように、SSRは、FGFR1活性(図4および6)、FGFR2活性(図8)、FGFR3活性(図9)およびFGFR4活性(図7)を阻害することができる。故に、このアロステリック阻害剤は、TKRの異なるメンバーにより共有される受容体の細胞外ドメインにある、進化的に保存されたFGFRアロステリック部位に結合する。この保存された部位は、FGFRのドメインIIIに位置する(図11)。結合部位へのSSRの結合は、「偏った拮抗作用(biased antagonism)」を誘導する。作用は、アロステリック結合部位へのSSR結合が受容体、特に判定されたフラストレートされた(frustrated)ドメインの立体構造変化をもたらすという事実により確認される。偏った拮抗作用のため、アロステリック阻害剤を同定する方法は、下記のようにリン酸シグナル伝達経路測定に基づくスクリーニングテストの使用により提供される。今後は、SSRは、RTKのアロステリック阻害剤の最初の例である。
【課題を解決するための手段】
【0007】
FGFR上のこのような部位の標的化ならびにVEGFR2およびPDGFRβのような他のRTKにおける類似の部位の標的化の検証は、重要な実用的意義を有しており、大きな治療効果をもたらす。
【図面の簡単な説明】
【0008】
【図1】AはHUVECでの定量的PCR実験がFGFR1およびFGFR4発現のみを示す図である。BはRT−PCR分析がアイソフォームFGFR1βおよびFGFR4を同定した図である。FGFR1がIIIcバリアントのフォーマット下にある図である(C)。
【図2】FGFR1βlllc−hMpIをトランスフェクトされたBaF/3細胞は、挿入されたFGFRが活性化される場合に増殖することができることを示す図である。FGF4(A)のみはFGFR1βlllcを誘導することができるが、FGF19は誘導することができない(B)。
【図3】SSR化合物表示を示す図である。
【図4−1】内皮細胞増殖に対するSSR活性に関する試験を示す図である。AはFGF2およびFGF4のみがHUVEC増殖を刺激し、それによりFGFR1がこれらの細胞で増殖を駆動することが示される。BはSSRがFGF2誘導HUVEC増殖を阻害し、それによりSSRによるFGFR1拮抗作用が示される。CはFGFR1をトランスフェクトされたラットファッドパッド(Fad Pad)内皮細胞(RFPEC)では、FGF2は、高用量でさえSSRにより部分的にのみ阻害されるFGFR1自己リン酸化を誘導した。Dはこの阻害が、PAECトランスフェクトFGFR1またはHUVECでのFGF2結合に対するSSRの競合的効果に起因しない。
【図4−2】内皮細胞増殖に対するSSR活性に関する試験を示す図である。AはFGF2およびFGF4のみがHUVEC増殖を刺激し、それによりFGFR1がこれらの細胞で増殖を駆動することが示される。BはSSRがFGF2誘導HUVEC増殖を阻害し、それによりSSRによるFGFR1拮抗作用が示される。CはFGFR1をトランスフェクトされたラットファッドパッド(Fad Pad)内皮細胞(RFPEC)では、FGF2は、高用量でさえSSRにより部分的にのみ阻害されるFGFR1自己リン酸化を誘導した。Dはこの阻害が、PAECトランスフェクトFGFR1またはHUVECでのFGF2結合に対するSSRの競合的効果に起因しない。
【図5】Aは異方性のパラメータとしての反転速度(tumbling speed)の測定による、Fc−タグ(FGFR2∂123)なしのFGFR2の精製ECDへの、蛍光lumioタグ付FGF1の結合(FGF1−lumio)を示す図である。SSRとFGF1−lumioの間の直接競合は観察されない。BはSSRが、FGFR2多量体化を阻害することができない、またはCはFGF2二量体化を阻害することができないことを示す図である。
【図6】内皮細胞走化性遊走に対するSSR活性に関する試験を示す図である。AはFGF2およびFGF4のみがHUVEC遊走を刺激し、それによりFGFR1がこれらの細胞で増殖を駆動することが示される。BはSSRが、FGFR1に対する拮抗アフェクト(antagonistic affect)に対応する、FGF2誘導HUVEC走化性遊走を阻害する。
【図7】内皮細胞インビトロ血管新生に対するSSR活性に関する試験を示す図である。AはFGF2およびFGF19のみがHUVEC血管新生を刺激し、それによりFGFR4がこれらの細胞におけるこの分化段階を制御することが示される。BはSSRが、FGFR4に対する拮抗アフェクトに対応する、FGF2誘導HUVECインビトロ血管新生を阻害する。
【図8】PANC02増殖および遊走に対するSSR活性に関する試験を示す図である。PANC02細胞は、VEGFの有無にかかわらずFGF7刺激下で増殖(A)または遊走(B)し、該系のFGFR2IIIb依存を示唆している。SSRは、FGF7誘導PANC02増殖およびFGF7+VEGF誘導細胞遊走を阻害し、FGFR2IIIbを阻害する能力を示している。
【図9】B9骨髄腫細胞増殖に対するSSR活性に関する試験を示す図である。AはFGF1がFGFR3を介してB9骨髄腫細胞増殖を誘導し、SSRがこの刺激を阻害し、それによりSSRがFGFR3をブロックできることが示される。Bはそれにもかかわらず、SSRが、自己活性FGFR3変異体(キナーゼドメインが構造的にリン酸化される。)をトランスフェクトされたB9細胞の増殖を阻害することはできない。これらの結果は、SSRの細胞外効果を示している。
【図10】SSRが、IGF、PDGF−BB、EGFまたはPIGFなどのさまざまな増殖因子により誘導された細胞遊走を大幅に阻害することができないHUVEC遊走アッセイを示す図である。SSRは、FGFRに特異的であり、およびFGF誘導HUVEC遊走のみをブロックする。
【図11−1】FGFRドメインIIIに結合するSSRを示すNMR試験を示す図である。AはFGFR1細胞外ドメインでのSSR結合の1D−およびSTD−NMR分析。飽和は対照TNFRIαでは観察されない。BはドメインIIIにおける相互作用部位を示唆する、異なるFGFR1ドメインに結合するSSRの1D−NMR試験は、FGFR1完全長およびFGFR1ドメインIIで得られたスペクトルが類似していることを実証する。CはSSRが、FGFR2およびFGFR3(D)細胞外ドメインに結合する能力を示す等温滴定熱量(Isothermal titration calorimetric)。
【図11−2】FGFRドメインIIIに結合するSSRを示すNMR試験を示す図である。AはFGFR1細胞外ドメインでのSSR結合の1D−およびSTD−NMR分析。飽和は対照TNFRIαでは観察されない。BはドメインIIIにおける相互作用部位を示唆する、異なるFGFR1ドメインに結合するSSRの1D−NMR試験は、FGFR1完全長およびFGFR1ドメインIIで得られたスペクトルが類似していることを実証する。CはSSRが、FGFR2およびFGFR3(D)細胞外ドメインに結合する能力を示す等温滴定熱量(Isothermal titration calorimetric)。
【図12−1】SSRがFGF1(A、B)およびFGF2(C)と相互作用できないことを実証するNMR(A、C)およびITC(B)試験を示す図である。Dはヘパリンミメティクスである八硫酸スクロース(SOS)の添加後にFGFR1に結合するSSRへの干渉は観察されず、SSRがFGFR1のヘパリン結合部位と相互作用しないことを確認する図である。
【図12−2】SSRがFGF1(A、B)およびFGF2(C)と相互作用できないことを実証するNMR(A、C)およびITC(B)試験を示す図である。Dはヘパリンミメティクスである八硫酸スクロース(SOS)の添加後にFGFR1に結合するSSRへの干渉は観察されず、SSRがFGFR1のヘパリン結合部位と相互作用しないことを確認する図である。
【図13】インシリコモデリングおよび変異誘発が、Y328アミノ酸近傍のSSRに対するアロステリック結合部を同定することを示す図である。WT FGFR2細胞外ドメイン(A)でのITC実験は、SSRとFGFR2の間の相互作用を示す。測定がY328D変異体(B)で実現される場合に失敗するこの結合は、Y328D変異がFGFR2をSSR結合に非感受性にすることを確認する。
【図14】100μM SSRなしまたは100μM SSRあり(それぞれ黒い線およびグレーの線)での精製された(A)FGFR2∂23−WT、(B)FGFR2−∂23−His、(C)FGFR2−∂23−Tyr、(D)FGFR2−∂23−H/T(H295L/Y328D二重変異体)のフーリエ変換赤外(FTIR)測定が、WTまたはHis295L変異体の両方で立体構造変化を同定したのに対し、変異Y328Dはこの変化に対しFGFR2を非感受性にすることを示す図である。
【図15】完全長FGFR2−WTまたは−Y328Dを有する安定にトランスフェクトされたHEK293細胞をFGF2で刺激(5分間で0.5ng/ml)した後の、活性化Erk1/2のウェスタンブロット分析を示す図である。濃度測定で確定された(densitometry defined)IC50値(平均±標準誤差;3つの独立した実験)は、FGFR2−Y328D変異体受容体(B)はFGFR2−WT(A)と比較してSSR阻害に対する感受性が約5倍低いことを示している。
【図16】AはFGF2で刺激された、FGFR2トランスフェクトHEK細胞に対する、FGFRチロシンキナーゼ阻害剤SU5402と比較したSSR効果のウェスタンブロット分析を示す図である。SU5402がPLCγ、FRS2およびErk1/2リン酸化を阻害するのに対し、SSRはPLCγ経路を阻害せず、SSRによる偏った拮抗作用を示している(B)。
【図17−1】HUVECにおけるFGF2誘導AKTリン酸化に対するSSR効果のウェスタンブロット分析(A)と対応する定量化グラフ(B)を示す図である。Cはこの効果が、ホスホ−AKT(Ser473)に対するオンセルELISA(on−cell ELISA)でも定量化できることを示す図である。Dはこの効果が、SSRがFGFRに結合するFGFと競合できないことと独立であることを示す図である。
【図17−2】HUVECにおけるFGF2誘導AKTリン酸化に対するSSR効果のウェスタンブロット分析(A)と対応する定量化グラフ(B)を示す図である。Cはこの効果が、ホスホ−AKT(Ser473)に対するオンセルELISA(on−cell ELISA)でも定量化できることを示す図である。Dはこの効果が、SSRがFGFRに結合するFGFと競合できないことと独立であることを示す図である。
【図18】ソフトウェアプログラムAGADIRおよび変異分析を用いた、VEGF−R2受容体における想定されるフラストレートされた帯域の同定を示す図である。(A)構造変化(例えばβシートからαヘリックスへの転移)を起こす傾向がある幾つかの領域が同定された。(B)2つのリジン(K609およびK648)が、膜貫通ドメインとの近さおよび変異後のヘリシティ特性に対する最も負の影響により、アスパラギン酸への変異に対して選択された。
【図19】ソフトウェアプログラムAGADIRおよび変異分析を用いた、PDGF−Rβ受容体における想定されるフラストレートされた帯域の同定を示す図である。アスパラギン酸への変異リジン387およびアスパラギン酸への変異ロイシン383は、ヘリシティ特性に対する最も負の影響を有するように思われる。
【図20】Erk1/2およびPLCγを通じたVEGF−R2およびPDGF−Rβ受容体シグナル伝達経路活性化の略図を示す図である。(A)VEGF刺激後のVEGFR2シグナル伝達および(B)PDGF刺激後のPDGF−Rβシグナル伝達。
【図21】ウェスタンブロットまたはシュアファイアアッセイ(surefire assay)を用いた、VEGF−R2またはPDGF−Rβ受容体の野生型または変異型を過剰発現したHEK293細胞におけるErk1/2リン酸化検出を示す図である。(A)VEGF−R2のマウス野生型およびK609DまたはK648Dで変異されたVEGF−R2は、HEK293細胞において安定にトランスフェクトされた。飢餓およびマウスVEGFなし(0)またはマウスVEGFあり(+)での刺激後、Erk1/2リン酸化はウェスタンブロットにより検出される。(B)タンパク質抽出物でのErk1/2(左の図式)またはPLCγ(右の図式)リン酸化を検出する確実なアッセイの略図。(C)10%FBS、1もしくは50ng/ml PDGF−BBで刺激または対照として刺激なし(0)後のPDGFRβトランスフェクトHEK293細胞における合計Erk1/2またはリン酸化Erk1/2のアルファスクリーンシュアファイア用量。
【発明を実施するための形態】
【0009】
本発明の種々の態様は、本発明の詳細な記載および以下の例において説明される。
【0010】
本発明の第1の態様は、チロシンキナーゼ受容体の細胞外ドメインに由来するアロステリック結合部位である。本明細書では、アロステリック結合部位とは、受容体のリガンド結合部位へのリガンドの結合の競合的阻害を引き起こすことなく、阻害剤、好ましくは小分子化合物が結合できる部位を意味する。本明細書では、由来するとは、アロステリック結合部位が細胞外ドメインの一部から成るが、完全な細胞外ドメインは含まないことを意味する。好ましくは、アロステリック結合部位は、10から200アミノ酸長の間、より好ましくは10から100アミノ酸の間、さらにより好ましくは20から50アミノ酸の間であり、前記アミノ酸は受容体の細胞外ドメインの部分である。
【0011】
本明細書では、小分子化合物は、好ましくは1000D未満、より好ましくは900D未満、より好ましくは800D未満、より好ましくは700D未満、より好ましくは600D未満、さらにより好ましくは500D未満の分子重量を有する非ポリマー性質の化合物である。
【0012】
チロシンキナーゼ受容体および受容体チロシンキナーゼ(RTK)は、本特許の範囲では、適用等価語である。「チロシンキナーゼ受容体」が受容体を示すのに使用されるのに対し、「受容体チロシンキナーゼ」はより具体的には受容体のキナーゼ活性を示すのに使用される。チロシンキナーゼ受容体は当業者に知られており、EGF、インスリン様増殖因子、PDGF、FGF、VEGF、HGF、Trk、AXL、LTK、TIE、ROR、DDR、PKT7、RYK、CCK4、EphおよびMuSK受容体ファミリーの受容体を含むが、これらに限定されない。好ましくは、前記アロステリック結合部位は、AXL、FGFR、MuSK、PDGFR、PTK7、ROR、TIEおよびVEGFRなどを含む、Igドメインを有するTKRの細胞外ドメインに由来する。さらにより好ましくは前記アロステリック結合部位は、細胞質ドメインにスプリットキナーゼドメインを有するTKRに由来する。本発明によるTKRの好ましい実施形態は、線維芽細胞増殖因子受容体(FGFR)、またはこのホモログ、オルソログもしくはパラログである。
【0013】
タンパク質の「ホモログ」は、当該非修飾タンパク質と比べてアミノ酸置換、欠失および/または挿入を有する、ならびに由来する非修飾タンパク質と類似の生物活性および機能活性を有するペプチド、オリゴペプチド、ポリペプチド、タンパク質および酵素を含む。「オルソログおよびパラログ」は、遺伝子の祖先関係を記述するのに使用される進化概念を含む。パラログは、遺伝子祖先遺伝子の複製を通じて生じた同じ種内の遺伝子である。オルソログは、種分化を通じて生じた異なる生物由来の、およびまた共通の祖先遺伝子に由来する遺伝子である。
【0014】
配列番号1を含むこのようなアロステリック結合は、FGFRファミリー、より具体的には、FGFR2に属する。好ましくは、前記アロステリック阻害剤部位は配列番号1を含み、さらにより好ましくは前記アロステリック阻害剤部位は配列番号1から成る。
【0015】
別の態様では、本発明は、アロステリック結合部位のホモログ、パラログまたはオルソログから成る。好ましくは、これらのホモログ、パラログまたはオルソログのポリペプチド配列は、配列番号1と少なくとも70%、80%、90%、95%以上の相同性を共有する。
【0016】
一例として、アロステリック結合部位のこのようなパラログは、FGFRファミリーに存在する。
【0017】
特に、本発明によるアロステリック結合部位は、FGFRのドメインIIIに位置する。
【0018】
同様に、VEGFR2に対するアロステリック結合部位は、リジン609およびリジン648を含む領域における、受容体のIgドメイン6に位置する。
【0019】
アロステリック結合部位はまた、PDGFRβにも存在し、および膜貫通領域近傍に位置する領域、特にIgドメイン3(ロイシン383およびリジン387を含む領域)に位置する。好ましくは、アロステリック結合部位に対するアロステリック阻害剤の結合により、偏った拮抗作用が誘導される。
【0020】
本明細書では、「偏った拮抗作用」とは、幾つかの下流経路を有する受容体に関して、アロステリック阻害剤結合部位にアロステリック阻害剤が結合した際に全ての経路が影響を受けるわけではないこと、または全ての経路が同程度に影響を受けるわけではないことを意味する。好ましい実施形態では、少なくとも1つの下流経路が阻害されるのに対し、少なくとももう1つの下流経路は影響を受けない。
【0021】
好ましくは、本発明によるアロステリック結合部位は、フラストレートされたドメインを含み、好ましくはフラストレートされたドメインから本質的に成り、さらにより好ましくはフラストレートされたドメインから成る。
【0022】
本明細書では、「フラストレートされたドメイン」とは、1つの構造的立体構造に明確に対するものではないタンパク質ドメインまたはこのフラグメントを意味する。フラストレートされたドメインは当業者に知られており、フラストレートされたドメインの存在は、1つのタンパク質二次構造予測プログラムでの曖昧な答え、または2つの異なるタンパク質二次構造予測プログラム間の予測における矛盾のどちらかにより検出される。優先的に、フラストレートされたドメインの存在は、タンパク質二次構造予測プログラムからの予測、ならびに結晶化およびX線回析などのタンパク質構造検出方法により判定されるような実構造における矛盾により検出される。非限定例として、矛盾は、1つの方法によるαヘリックスの表示、および別の方法によるβシートの表示であってもよい。タンパク質は、最小限にフラストレートされる。しかし、幾つかのドメインは、幾つかのフラストレーションを誘導しており(本明細書では「フラストレートされたドメイン」と呼ばれる)、これらのドメインは、タンパク質の立体構造変化を誘導する傾向がある。
【0023】
好ましい実施形態では、前記フラストレートされたドメインは配列番号2を含み、好ましくはこれは配列番号2から成る。このフラストレートされたドメインは、FGFRファミリー、特にFGFR2に属する。
【0024】
他のフラストレートされたドメインは、上記に示されたように同定することができる。
【0025】
本発明の別の態様は、アロステリック結合部位が位置するチロシンキナーゼ受容体の結合部位にリガンドが結合した際に、偏った拮抗作用を誘導するための本発明によるアロステリック結合部位の使用である。本発明のさらに別の態様は、前記部位に結合している、ランダムライブラリー由来の小分子化合物阻害剤をスクリーニングするための、本発明によるアロステリック結合部位の使用である。
【0026】
本発明のさらに別の態様は、チロシンキナーゼ受容体の細胞外ドメインにおけるフラストレートされたドメインの存在をスクリーニングすることを含む、前記細胞外ドメインにおけるアロステリック阻害剤結合部位の同定方法である。フラストレートされたドメインをスクリーニングする方法は当業者に知られており、このような方法の例は例8に記載されている。非限定例として、フラストレートされたドメインは、1つのタンパク質二次構造予測プログラムでの曖昧な答えにより、好ましくは2つの異なるタンパク質二次構造予測プログラム間の予測における矛盾により、さらにより好ましくはタンパク質二次構造予測プログラムからの予測、ならびに結晶化およびX線回析などのタンパク質構造検出方法により判定されるような実構造における矛盾により検出される。タンパク質二次構造予測のためのプログラムは当業者に知られている。非限定例として、このようなプログラムがRost(2003年)により記載されている。好ましくは、前記フラストレートされたドメインは、前記アロステリック結合部位の近傍にある。より好ましくはこれは結合部位の境界から20アミノ酸以内、さらにより好ましくは10アミノ酸以内に位置し、さらにより好ましくはこれは前記結合部位に隣接し、さらにより好ましくはこれは結合部位と重複し、最も好ましくはこれは結合部位に含まれる。考えられる阻害剤部位を同定した後、スクリーニングは、該部位に結合する、およびアロステリック阻害機能をテストできる小分子、小ペプチド、ペプチドミメティクス、抗体またはナノ抗体(nanobody)などの化合物を設計して、考えられる阻害剤部位の機能の確認により完了することができる。
【0027】
本発明の別の態様は、チロシンキナーゼ受容体の活性化に依存する2つの異なる下流経路により誘導される2つの異なるレポーターの比較を含む、本発明による前記チロシンキナーゼ受容体の細胞外ドメインにおけるアロステリック阻害剤部位に結合している小分子化合物アロステリック阻害剤の同定方法である。レポーターは、検出可能なシグナルをもたらす化合物の任意の遺伝子、タンパク質であり、非限定例として、抗生物質耐性遺伝子、細胞死をもたらす毒素遺伝子、GFPなどの蛍光タンパク質をコードする遺伝子、またはβガラクトシダーゼなどの酵素活性をコードする遺伝子、またはリン酸化もしくは脱リン酸化、アセチル化もしくは脱アセチル化されたまたは立体構造が変化しているタンパク質であってもよい。レポーター遺伝子の場合では、コード配列は、適切なプロモーター、すなわち受容体へのリガンドの結合およびレポーター経路の結果的な誘導により誘導されるプロモーターの制御下に置かれる。二重経路の場合では、2つの異なるプロモーターが必要とされる。非限定例として、アロステリック阻害剤の存在下または非存在下でタンパク質のリン酸化を比較することは、偏った拮抗作用によるリン酸化の違いを生み、リン酸化のこれらの違いはレポーターとして使用することができる。
【0028】
好ましい実施形態では、RTKのアロステリック阻害剤の同定は、
a)RTKのアロステリック結合部位をアロステリック阻害剤候補化合物と接触させる段階、
b)前記チロシンキナーゼ受容体の活性化/阻害に依存する少なくとも2つの下流経路の変化を測定する段階、
c)前記チロシンキナーゼ受容体の活性化/阻害に依存する少なくとも2つの異なる下流経路の各々に対する少なくとも1つのレポーターの状態の変化を比較する段階
を含み、
受容体のリガンド結合ドメインに結合しているリガンドの存在下、少なくとも1つの下流経路が阻害されるのに対し、少なくとももう1つの下流経路は影響を受けない場合に、アロステリック阻害剤が同定される、スクリーニングテストを実施して行うことができる。レポーターの状態の変化は使用されるレポーターに依存し、非限定例としてレポータータンパク質のリン酸化の変化、または遺伝子の非誘導から誘導への(または逆も同様)スイッチであってもよい。好ましくは、前記状態の変化はホスホイレーション(phosphoylation)状態の変化である。
【0029】
好ましくは、下流経路の変化は、ERK1/2 およびPLCγシグナル伝達経路を含む、リン酸シグナル伝達経路の変化の測定により実施される。
【0030】
別の実施形態では、FGF−Rのアロステリック調節因子は、下記に記載のようにSEC−LC/MSに基づく親和性スクリーニングを用いて同定することができる:
SEC−LC/MS方法論は、オンライン結合された2次元システム、分離のため高性能液体クロマトグラフィーに結合されたサイズ排除クロマトグラフィーと、これに続く検出のためのエレクトロスプレーイオン化−飛行時間型質量分析から成る、親和性スクリーニングに使用される分析技法である。
【0031】
方法は、可溶性ポリペプチド(ペプチド、タンパク質ドメイン、または完全長タンパク質を含む)と相互作用する幾つかの化合物の能力に基づく。目的とするペプチドと小分子化合物のプールを混合した後、ペプチド−リガンド複合体は、サイズ排除クロマトグラフィーによる非結合および結合小分子化合物の分離を可能にする質量シフトを誘導する。次いで、複合体は解離され、バインダーがペプチドから分離され、および正確な質量測定のため高分解能LC/ESI−TOFを用いて検出される(例えばWaters LCTプレミア質量分析計により)。データデコンボリューションアルゴリズムは、質量検出分析からの結合分子の同定を可能にする。
【0032】
FGFRの小分子化合物アロステリック調節因子を同定するには、この技術を天然のまたは変異された種々のFGF−Rの細胞外ドメインに適用することができる。天然型は、細胞外ドメインに対するバインダー全ての検出を可能にする。あるいはアロステリック調節因子は、SSR結合部位に近接するFGF−R2ヘリックスの「開口(open)」型を用いてスクリーニングすることができる。前記「開口」型は、アルファヘリックスを安定化し、これによりSSR結合への感作を可能にする変異Tyr328Arg−Ile329Lysにより得ることができる。変異FGF−R2は、次いでWT FGF−R2の代わりにスクリーニングに使用される。類似の戦略を、FGF−R2におけるTyr328およびIle329に対応するアミノ酸に変異を有するFGF−R1、−R3または−R4をスクリーニングするのに使用することができる。Tyr328Aspでの変異型(FGF−R2)または対応する位置で変異を有する他のFGF−Rを、対照として使用することができる。実際、SSRは、疎水ポケット近傍のTyr328Aspで変異されたFGF−R2には結合しない。したがってこの変異型は、FGF−R2における標的化ポケットと相互作用しない化合物の部分を廃棄するのに使用することができる。
【0033】
全てのこれらの場合において、この戦略は、目的とするペプチドの標的ポケットに結合することができる小分子化合物の同定をもたらす。第2の段階では、細胞におけるシグナル伝達に対する効果が評価されなければならない。R&D Systems社製のProteome Profiler(商標)Array「ヒトホスホ−キナーゼアレイキット」により同定されたリン酸シグナル伝達経路に基づき、プロテオームプロファイラーにより検出された影響を受けないキナーゼを阻害することなく、(PYK2、eNOS、p53、c−jun、AKT、CREB、Erk1/2において)リン酸化キナーゼレベルでHUVECに対するFGF−2効果を阻害する能力に関して、アロステリック調節因子を(細胞タンパク質抽出物でまたは細胞で直接)ELISAによりチェックすることができる。
【0034】
類似のアプローチを、他のRTKに対して踏むことができる。受容体の細胞外ドメインにおける1つ以上のフラストレートされたドメインを同定した後、前記フラストレートされたドメインをSEC−LC/MSアプローチで使用して、フラストレートされたドメインの領域におけるバインダーを同定することができる。シグナル伝達経路に対するバインダーの効果は、次いで上記のようなホスホマップ(phosphomap)アプローチ、または経路の任意の他のレポーター系を用いてテストすることができる。
【0035】
本発明のさらに別の態様は、本発明による「アロステリック阻害剤」とも呼ばれる、および/または本発明による方法により同定された、アロステリック結合部位に結合している小化合物化合物である。
【0036】
「化合物」とは、単純または複合有機および無機分子、ペプチド、ペプチドミメティクス、タンパク質、抗体、炭水化物、核酸またはこれらの誘導体を含む、任意の化学的または生物学的化合物を意味する。
【0037】
(実施例)
例に対する材料および方法
STD−NMR結合アッセイ
ヒトFGFR1遺伝子(P11362)の細胞外ドメイン(ECD;アミノ酸:39−358)をPCR増幅し、およびNdeIおよびBamHI制限部位を用いて、大腸菌(E. coli)ベクターpETTEV(N末端His−タグの後にTEVプロテアーゼ切断部位が続く)にクローン化した。タンパク質産生には、得られたプラスミド(pET FGFR1 D1D2D3)を大腸菌BL21(DE3)(Novagene社)に形質転換した。細胞はOD600が0.6に達するまで37℃で増殖させ、組換えタンパク質産生は1mM IPTG(イソプロピル−b−D−チオガラクトピラノシド)を添加して誘導した。4時間誘導後、細胞を回収し、使用まで−80℃で貯蔵した。細胞ペレット(1L培養)を解凍し、リゾチーム(2mg)、および40Uベンゾナーゼ(Merck社)を含有する50ml緩衝液1(20mM Tris/HCl、pH7.5、200mM NaCl)に再懸濁した。細胞を超音波処理により破壊し、封入体(IB)を遠心分離(15,000g、20分、4℃)により沈降させ、得られたペレットを緩衝液1で2回洗浄した。IBペレットは20ml変性緩衝液(6Mグアニジン−HCl、20mM Tris/HCl、pH8.0、200mM NaCl)に室温で40分間溶解した。可溶性デブリを遠心分離(30,000g、30分)により除去し、上清を、製造者の推奨に従って緩衝液Aで事前に平衡化したNi−NTAカラム(Qiagen社)に添加した。FGFR1 ECDは、500mMイミダゾールを含む変性緩衝液を用いてカラムから溶出した。ECDを含有する画分をプールし、50mM Tris/HCl、pH8.0、250mM NaCl、0.5M L−アルギニン、2mM EDTA、0.02%アジドへの可溶化タンパク質のフラッシュ希釈(flash−diluting)(希釈係数1:30)と、この後の24時間、4℃で穏やかな撹拌によるインキュベーションによりリフォールディングした。リフォールディング混合物を20分間、30,000gで遠心分離し、アミコン撹拌セル(Amicon stirred cell)でYM10膜を通じて濃縮し(最終タンパク質濃度1mg/ml)、25mMTris/HCl、pH8.0、2mM EDTA、0.02%アジドに対して透析し、HiTrapヘパリンHP 5ml(GE Healthcare社)に適用し、0から2M NaClへの直線勾配で溶出した。FGFR1 ECDの最終精製は、25mM Tris/HCl、pH8.0、200mM NaCl、25mM L−アルギニン、2mM EDTA、0.02%アジドで平衡化したHi Load26/60 75pGカラム(GE Healthcare社)を用いてサイズ排除クロマトグラフィーにより達成した。FGF1(アミノ酸:16−155)およびFGF2(アミノ酸9−155)およびTNF−Riαを発現させ、精製した。FGFR1 ECDの構造完全性は、ヘパリンカラム(上記参照)に結合する能力およびFGF1との複合体の形成により実証した。複合体形成は、サイズ排除クロマトグラフィーおよびこの後のSDS−PAGEでの分析により分析した。
【0038】
全てのSTD−および1D−NMR実験は、BRUKER3チャンネルDRX600およびBRUKER4チャンネルDRX800スペクトロメーターで298Kの標準温度で行い、および内部標準3−トリメチル−2,2,3,3−テトラジュウテロプロピオン酸−ナトリウム塩(TSP)を基準にした。典型的には、NMR試料は、25mM Tris/HCl、pH8.0、200mM NaCl、25mM L−アルギニン、2mM EDTA、0.02%アジド(95%H2O/5%D2O中)中に0.5mlのタンパク質(20−300mM)を含有した。タンパク質リガンド1D STD NMR測定のため、スペクトルを、別々のタンパク質メチル共鳴での弱い2秒RF照射により、1mMリガンドSSR128129E(100mM DMSO原液)および40mMタンパク質で記録した。水抑制は、標準Bruker WATERGATE3−9−19シーケンスを用いて行った。NMRデータは、Brukerプログラムxwin NMRソフトウェアを用いて処理した。
【0039】
等温滴定熱量測定(ITC)
熱量測定実験は全て、以前に記載されているように45、VP−ITC滴定熱量計(MicroCal Inc.社、ノーサンプトン、MA)により30℃で実施した。滴定は、1.407mLの10−20μM相互作用タンパク質(すなわちFGFR2∂123、FGFR2∂23ならびにこの記載された変異体およびサブドメイン、FGFR3∂123、FGF1、FGF2ならびにホリスタチン(陰性対照として)を含有する溶液細胞への、4分間隔、回転撹拌器−シリンジを介した1.25mM SSRの10μLアリコートの添加を伴った。300rpmの一定撹拌速度を維持し、データをMicroCal社により供給された標準非相互作用一部位モデル(nが1.0に固定される。)に適合させた。測定は全て、10mM HEPES pH7.2、150mM NaClで実施し、タンパク質を以前に記載されているように(Pellegriniら、2000年)精製した。変異誘発は「部位特異的変異誘発キット」(Stratagene社)を用いて実施した。
【0040】
フーリエ変換赤外線測定
フーリエ変換赤外線測定は、AquaSpecフローセルを備えたBruker Tensor 37 FT−IRスペクトロメーターを用いて実施した。試料コンパートメントは25℃に温度自動調節し、ノイズ比に対し良好なシグナルについて100スペクトルを平均化した。タンパク質は上記のように精製した。ゲル濾過直後、タンパク質を、SSRの存在下または非存在下、同じ調製の緩衝液(10mM Hepes pH7.2、150mM NaCl)で一晩透析した。透析緩衝液試料を使用してバックグラウンドシグナルを減算した。分析は、Brukerにより提供されたOPUSソフトウェアパッケージを用いて実施した。結果の解釈は記載されているように実施した{Barth、2002年 #60}。
【0041】
HEK293トランスフェクションならびにErk1/2、PLCγおよびFRS2リン酸化試験
HEK293細胞は、pcDNA3(Invitrogen社)にクローン化したhFGFR2IIIcaまたはhFGFR2IIIca−Y328Dを(FuGENE6、Roche社を用いて)一時的にまたは安定にトランスフェクトした。安定にトランスフェクト細胞はG418(400μg/ml)含有培地で増殖させた。刺激前、細胞を一晩、DMEM(0%血清)で飢餓状態にし、および必要とされる濃度でSSR128128Eと一緒にプレインキュベートした。細胞をこの後、1μMでのSSRまたはSU5402ありまたはなしにより37℃、5分間、FGF2(0.5−10ng/mlの間の濃度)で刺激した。ホスファターゼ阻害剤(Roche社)を含有する氷冷リン酸緩衝生理食塩水で洗浄後、細胞をRIPA緩衝液(製造者(Roche社)により記載されているようにホスファターゼおよびプロテアーゼ阻害剤を含有する、Tris 30mM HCl pH7.5、150mM NaCl、1mM EDTA、1%トリトン−X、0.5%w/vデオキシコール酸)で溶解した。細胞溶解物を10分間、12,000gで遠心分離し、上清を回収した。タンパク質をNovexポリアクリルアミドゲル(Invitrogen社、カールスバッド、CA)で分離し、この後Hybond ECLニトロセルロース膜(Amersham Pharmacia社)に移した。PBSで5%非脂肪乳粉と一緒にインキュベーションした後、膜を以下の抗体:ホスホ−ERK1/2(CST:9101)、ホスホ−FRS2(CST:3861)、ホスホ−PLCγ(CST:2821)およびFGFR2(F0300、Sigma社)と一緒に一晩、4℃でインキュベートした。
【0042】
FGFRトランスフェクトBaF/3細胞増殖:
この実験に使用されたBaF/3細胞の構築は、出願WO2007/080325に詳細に記載されている。
【0043】
定量的リアルタイムPCR
全RNAは、トリゾール試薬(Invitrogen社、USA)およびRNeasyキット(Qiagen社、ドイツ)を用いてHUVECから単離し、全RNAからこの後Quantitect Reverse Transcriptionキット(Qiagen社、ドイツ)を用いてcDNAを調製した。プライマーセットおよびFAM(商標)色素標識TaqMan(登録商標)MGBプローブ(Eurogentec社、ベルギー)を、ヒトFGFR1、FGFR2、FGFR3、FGFR4、およびTBP用に設計し、PCR反応は7500 FastリアルタイムPCRシステム(ABI社、ドイツ)で行った。各試料は、特定の標準対照および非テンプレート対照と一緒に三通りに分析した。増幅は、2×TaqMan(登録商標)Universal PCR Master Mix、20×Assays−on−demand(商標)Gene Expression Assay Mixを用いて行った。各試料における最初のmRNAコピー数の計算は、サイクル閾値(CT)方法により行った。FGFR1、FGFR2、FGFR3、FGFR4、mRNAのコピー数は、TBP mRNAレベルを用いて正規化した。
【0044】
FGFR1リン酸化測定
hFGFR1IIIcα−ヘマグルチニンを安定にトランスフェクトされたラット脂肪パッド内皮細胞を、80−90%培養密度に増殖させ、24時間血清飢餓状態にした(0.5%FBS)。刺激は、SSRまたはDMSO(対照として)と組み合わせて2ng/mlでのFGF2で5分間実施した。細胞溶解物を10分間、12,000gで遠心分離し、上清を回収した。HAタグ付タンパク質を、アガロース結合抗HA抗体の存在下、一晩、4℃で細胞培養物のインキュベーションにより免疫沈降させた。免疫複合体は1mlの溶解緩衝液で3回洗浄した。タンパク質は、50μlの2×SDS試料緩衝液と一緒のインキュベーションおよび煮沸を介して溶出した。タンパク質をNovexポリアクリルアミドゲル(Invitrogen社、カールスバッド、CA)で分離し、この後Hybond ECLニトロセルロース膜(Amersham Pharmacia社)に移した。PBSで5%非脂肪乳粉と一緒にインキュベーションした後、膜を以下の抗体:pFGFR(CST:3471)およびFGFR1(CST:3472)と一緒に一晩、4℃でインキュベートした。
【0045】
異方性測定
SSRが、結合ポケットへのFGF1の結合を阻害するかどうかを評価するため、本発明者らはFcタグなしのFGFR2の全細胞外ドメイン(FGFR2∂123)を精製し、およびSSR(1mM)なし(青)またはあり(赤)のさまざまな濃度のFGFR2∂123の存在下、蛍光lumioタグ付FGF1(FGF1−lumio;1μMの定濃度)の反転速度(異方性のパラメーターとして)を測定した。FGFR2∂123をFGF1−lumioに添加した場合、リガンド/受容体複合体の反転速度は、該複合体のより大きなサイズのためFGF−lumio単独の反転速度より遅かった。SSRの大きなモル過剰(1000倍)は複合体の反転速度を変化させず、SSRがFGFRからFGFを移動させないことを確認した。
【0046】
HUVEC増殖
コンフルエントなHUVEC細胞を回収し、0.5%FCS(Hyclone社、SH30070.03)、2mMグルタミン、MEM非必須アミノ酸 1×(Gibco社、11140−035)、MEMピルビン酸ナトリウム 1×(Gibco社、11360−039)を含む100μl RPMI 1640(Invitrogen社、32404−014)中の5 104細胞を、一晩、96ウェルコラーゲンIコーティングプレート(Beckton Dickinson社、354650)にウェルごとに播種する。次いで、培地を除去し、ならびに2×FGF2(R&D社、234−FSE−025)、FGF4(R&D社、235−F4−025)またはFGF−19(自家製造)および50μlの2×SSR(200または600nM)を含有する50μlの培地に置換する。細胞をCO2チャンバーで3日間、37℃でインキュベートし、増殖を100μlの「Cell Titer GIo Luminescent細胞生存能」キット(Promega社、G7571)でATP含量を定量化して評価する。
【0047】
HUVEC走化性遊走
コンフルエントなHUVEC細胞を回収し、およびFCS、2mMグルタミン、MEM非必須アミノ酸 1×(Gibco社、11140−035)、MEMピルビン酸ナトリウム 1×(Gibco社、11360−039)を含まないRPMI 1640(Invitrogen社、32404−014)に0.8 106細胞/mlで再懸濁する。250μlの細胞溶液を、内皮細胞遊走のための24ウェルBD Biocoat血管新生システム(BD Biocoat社、354144)の上のチャンバーに4×SSRで分配し、および750μlの培地を、FGF2(R&D社、234−FSE−025)、FGF4(R&D社、235−F4−025)またはFGF−19(自家製造)を含む下のチャンバーに67ng/mlで分配する。プレートをCO2チャンバーで22時間、37℃でインキュベートする。次いで、プレートインサートを除去し、および500μLのカルセイン(Molecular probes社、C−3100)を含有する新たな24ウェルプレート(Falcon社、353504)に90分間入れる。次いで遊走細胞が蛍光し、遊走を485nm励起および535nm発光後、ダウンステアズリーディング(downstairs reading)でルミノメーターにより測定する。
【0048】
HUVECインビトロ血管新生
コラーゲン/マトリゲルゲルは、チャンバースライド(Biocoat Cellwareコラーゲン、タイプI、8ウェル培養サイド(cultureside):Becton dickinson社 354630)の各ウェルに、コラーゲンI(ラット尾コラーゲン、タイプI:Becton dickinson社 354236)中160μlの1/6希釈マトリゲル(増殖因子低減マトリゲル:Becton dickinson社 356230)を分配して調製する。重合が1時間、37℃で生じる。次いで、15.103HUVECを、400μl EBM培地(Clonetics社 C3121)+2%FCS+hEGF 10μg/mlにウェルごとに添加する。内皮細胞を、10ng/mlのFGF2(R&D社、133−FB−025)、FGF4(R&D社、235−F4−025)またはFGF19(R&D社、969−FG−025)で、CO2チャンバーで24時間、37℃で刺激する。次いで、完全長の偽細管をバイオイメージングシステム(Imagenia Biocom社、Courtaboeuf、フランス)を用いて定量化する。
【0049】
HUVECにおけるAKTリン酸化のウェスタンブロット分析
HUVE細胞(Promocell社、C−12200)は、2%FBS(Clonetics社、CC−4101)、EGM singlequotsキット(Clonetics社、CC−4133)からの10μg/ml hEGF(Clonetics社、CC−4017)、1250ng/mlヘパリン(Sigma社、H3149)および375ng/ml ECGS(BD Biosciences社、356006)を含有する2mlのEBM培地(Clonetics社、CC−3121)中0.5.106細胞で、35mmコラーゲンIコーティングディスク(BD Biocoat、354456)に播種する。90%培養密度で、細胞を1.8mlのRPMI 1640(Invitrogen社、32404−014)、0.5%FCS、2mMグルタミン、1mM非必須アミノ酸(Invitrogen社、11140−050)、ピルビン酸ナトリウム(Invitrogen社、11360−070)で一晩飢餓状態にする。翌日、細胞を、10×SSR(3μM)ありまたはなしの10×FGF−4(30ng/ml;R&D社、235−F4−025)を含有する200μlの平衡化飢餓培地により10分間刺激する。次に、細胞を冷PBSですすぎ、細胞を、2.5mMオルトバナジウム酸およびプロテアーゼ阻害剤カクテル(Sigma社、P8340)を含有する75μl RIPAで溶解する。細胞溶解物を10分間、12,000gで遠心分離し、上清を回収した。タンパク質を4−20%Novex Tris−グリシンポリアクリルアミドゲル(Invitrogen社)で分離し、この後ニトロセルロース膜(Invitrogen社、IB3010−01)に移した。TBS−0.05%Tween80で5%非脂肪乳粉と一緒にインキュベーションした後、膜を、TBSで1000×希釈した抗ホスホAKT(Ser473、CST、4058)、tween、1%BSAと一緒に一晩、4℃でインキュベートした。各スポットのシグナルがSuperSignal(登録商標)West Dura Extended Duration Substrate(Thermo Scientific社、34076)による化学発光検出後に得られ、スポット密度をBiolmaging System Chemigenius2(Syngene社)を用いて定量化する。
【0050】
オンセルAKTリン酸化ELISA
コンフルエントなHUVEC細胞を回収し、0.5%FCS(Hyclone社、SH30070.03)、2mMグルタミン、MEM非必須アミノ酸 1×(Gibco社、11140−035)、MEMピルビン酸ナトリウム 1×(Gibco社、11360−039)を含む50μl RPMI 1640(Invitrogen社、32404−014)中の5 104細胞を、一晩、96ウェルコラーゲンIコーティングプレート(Beckton Dickinson社、354650)にウェルごとに播種する。細胞は、20ng/ml FGF4および600nMSSRを含有するFCSなしで100μl平衡化飢餓培地で5分間刺激する。次いで、50μlのPFA8%をPBS(Polysciences社、18814)に室温で15分間添加し、細胞を200μl PBSで2分間、3回洗浄する。非特異的部位は、PBS、トリトン0.3%、正常ヤギ血清0.1%(Zymed社、50−062Z)により室温で1時間ブロックし、ブロッキング緩衝液を作製し、およびPBSで1/500希釈した抗ホスホ−AKT(Ser473)抗体(CST、4058)、トリトン0.3%に一晩置換する。一次抗体を次いで除外し、および200μl PBSで2分間、3回洗浄した。HRP結合抗ウサギ二次抗体(CST、7074)を使用して、PBS、0.3%トリトンで2時間、室温で1/2000希釈した後、AKTリン酸化を検出する。次いで、細胞をPBSですすぎ、100μlのHRP基質(Uptima社、UP664781)を暗室で20分間添加する。酵素反応を100μlの停止緩衝液(Uptima社、UPS29590)で停止し、ODを450nmで測定した。
【0051】
FGFRトランスフェクト300−19細胞に結合するFGF2:
FGF2は、購入者の推奨に従ってAlexa Fluor488 C5−マレイミド(Invitrogen社、A10254)で標識した。
【0052】
このAF488−FGF2は、pEF6−V5/His Topoプラスミド(Invitrogen社)でFGFR1またはFGFR4構築物をトランスフェクトしたマウスプレ−B 300−19細胞での結合実験において10ng/mlで使用した。SSR(300nM最終)は、10%FCS(Hyclone社、SH30070.03)、2mMグルタミン、MEM非必須アミノ酸 1×(Gibco社、11140−035)、MEMピルビン酸ナトリウム 1×(Gibco社、11360−039)および150mMモノチオグリセロール(Sigma社、M6145)を含むRPMI 1640(Invitrogen社、32404−014)で、150rpm撹拌下、4℃で細胞と一緒に20分間プレインキュベートしていた。次いで、FGF2(10ng/ml最終)を30分間添加し、FACS Caliburフローサイトメーター(Beckton Dickinson社)を用いて結合を測定する。条件ごとに蛍光培地も分析する。
【0053】
さまざまな増殖因子による細胞遊走
細胞遊走は、ポリカーボネート膜(Costar、Corning Inc.社)を有する8μm孔径トランスウェル透過性支持体を含有する24ウェルインサートを用いて、改変ボイデンチャンバーアッセイにより評価した。指数関数的に増殖している細胞を0.2%FBS含有培地で16時間飢餓状態にし、および同じ低血清培地に5×105細胞/mlで再懸濁した。100μlの細胞懸濁液を上のチャンバーに播種したのに対し、化学誘引物質および/またはSSRは下のチャンバーに入れた。テストした化学誘引物質には、ヒトPDGF−BB、IGF−I、PIGF、EGF(SSR(1μM)の存在下または非存在下、全て100ng/ml)が含まれる。10%FBS含有培地は陽性対照として使用した。37℃で6時間インキュベーションした後、膜の上側の細胞は綿棒を用いて擦り取ったのに対し、下表面の遊走細胞はPBS中1%パラホルムアルデヒドで固定し、および蛍光マイクロソープ(microsope)を用いた定量化のためDAPIで核染色した。定量化は、10×の倍率で5枚のランダム画像を作製しおよび核の数をカウントして実施する。
【0054】
PANC02増殖および遊走:
細胞増殖は、0.2%FBS(Hyclone社、SH30070.03)、2mMグルタミン、MEM非必須アミノ酸 1×(Gibco社、11140−035)、MEMピルビン酸ナトリウム 1×(Gibco社、11360−039)を含む100μl RPMI 1640(Invitrogen社、32404−014)で16時間飢餓状態にしおよび96ウェルマイクロプレートに4,000細胞/ウェルで播種した、指数関数的に増殖している細胞で分析した。分裂促進因子および/またはSSRに72時間曝露した後、細胞増殖をCellTiter 96 AQueous One Solution Cell Proliferation Assay(Promega社、マジソン、ウィスコンシン、USA)を使用して製造者の指示に従ってアッセイした。細胞遊走は、ポリカーボネート膜(Costar、Corning Inc.社)を有する8μm孔径トランスウェル透過性支持体を含有する24ウェルインサートを用いて、改変ボイデンチャンバーアッセイにより評価した。指数関数的に増殖している細胞を0.2%FBS含有培地で16時間飢餓状態にし、および同じ低血清培地に5×105細胞/mlで再懸濁した。100μlの細胞懸濁液を上のチャンバーに播種したのに対し、化学誘引物質および/またはSSRは下のチャンバーに入れた。10%FBS含有培地は陽性対照として使用した。37℃で6時間インキュベーションした後、膜の上側の細胞は綿棒を用いて擦り取ったのに対し、下表面の遊走細胞は4%ホルムアルデヒドで固定し、および定量化のためDAPIで核染色した。
【0055】
B9骨髄腫細胞増殖:
細胞増殖は、IMDM(Invitrogen社、31980048)、0.2%FBS、2mMグルタミン含有培地で16時間飢餓状態にしおよび96ウェルマイクロプレートに4,000細胞/ウェルで播種した、指数関数的に増殖している細胞で分析した。分裂促進因子および/またはSSRに72時間曝露した後、細胞増殖をCellTiter 96 AQueous One Solution Cell Proliferation Assay(Promega社、マジソン、ウィスコンシン、USA)を使用して製造者の指示に従ってアッセイした。
【0056】
アルファスクリーンシュアファイアアッセイ
0日目:HEK293:mVEGFR2wt細胞またはHEK293:PDGFRβ細胞を10000細胞/ウェル(96ウェルプレート細胞結合Costar)でプレーティングし、一晩接触させる
1日目:細胞をDMEM(0%血清)で最低3時間飢餓状態にする:50ng/mlのVEGF164またはPDGF−BBの混合物をDMEM(0%血清)で調製し、5または15分間刺激する:シュアファイアアッセイ(Perkin Elmer社)からの溶解緩衝液で細胞を溶解する:50μlの緩衝液で細胞を溶解し、プレートをRTで10分間撹拌し、次いでさらなる使用まで−20℃で凍結する;タンパク質と溶解緩衝液の混合物を作製し、および製造者の指示に従ってpERK1/2、合計ERK1/2ならびにカスタムデザインされたpPLCγおよび合計PLCγで分析する。
【実施例1】
【0057】
アロステリック、マルチFGFR阻害剤としてのSSR128129Eの同定
この試験の目的は、FGFR細胞外ドメイン(ECD)に結合しおよびFGFRシグナル伝達を阻害する低分子量化学化合物を開発することであった。小分子化合物が、オルソステリック部位に対する単純な立体障害を介してはるかに大きなポリペプチド(すなわちFGF)とどのように相互作用できるかを構想することは困難であることから、複数のリガンド結合アッセイフォーマットを利用して、任意の同定された化合物がアロステリック機構を介してオルソステリックに作用しているかどうかを判定した。本発明者らは最初に、ハイスループットシンチレーション近接結合アッセイ(SPA)を開発して、Fc−フラグメントに結合した、3つのIg様ドメインD1−3(FGFR1∂123/Fc)から成る、FGFR1−ECDへの125I−FGF2の結合を阻害する化合物を同定した。>20,000化合物のスクリーニングおよび化学的最適化の後、1つの化合物、SSR128129E(以後「SSR」と略す)が、125I−FGF2結合を阻害した。追加のSPAアッセイでは、SSRは、さまざまなFGFRへの種々のFGFリガンドの結合をブロックする一方で、関連した構造相同性または全く異なる化学組成を有する>100の異なるリガンドと同種受容体との結合を阻害しない、マルチFGFR阻害剤として作用した。この知見は、競合的(オルソステリック)機構、または高い負の協同性(ChristopoulosおよびKenakin、2002年)を特徴とする他のアロステリック相互作用のどちらかを示唆した。
【0058】
アロステリック相互作用の1つの顕著な特徴は、「プローブ依存性(probe−dependence)」、すなわち、調節因子が相互作用しているオルソステリックリガンド−受容体複合体の性質に依存するアロステリック相互作用の大きさおよび方向の変動の現象である(Mayら、2007年)。SPAでの125I−FGF2結合に対するSSRの効果が、人工基質にプレーティングされた改変FGFR/Fc融合タンパク質の立体配置に依存したかどうかを判定するため、本発明者らは次に、異方性のパラメータとしての反転速度を測定して、SSRが、Fc−タグなしのFGFR2の精製ECD(FGFR2∂123)への、蛍光lumioタグ付FGF1(FGF1−lumio)の結合を阻害するかどうかを試験した。FGFR2∂123をFGF1−lumioに添加した場合、リガンド/受容体複合体の反転速度は、該複合体のより大きなサイズのためFGF−lumio単独の反転速度より遅かった。SSRがリガンド結合を阻害したなら、反転速度は再び増加するはずである。しかし、>1,000倍のモル過剰でさえ、SSRは複合体の反転速度を変化させなかった。これは、SSRとFGF1−lumioの間の直接競合の欠如を示唆している(図5A)。最後に、ヒト臍帯静脈内皮細胞(HUVEC)またはFGFR1を過剰発現するブタ大動脈内皮細胞(PAE−FGFR1)でのI125−FGF2による結合アッセイもまた、後者がPAE−FGFR1がインタクトな細胞においてより自然な立体構造で発現される場合、SSRが(高μM濃度でさえ)受容体へのI125−FGF2結合を阻害できないことを明らかにした。しかし、αFGF2抗体を中和することは効果があった。この後者の実験パラダイムでは、およびSPAとは対照的に、SSRは、他のFGFRへの別のFGFリガンド(すなわちFGFR2へのFGF2またはFGF4;FGFR4へのFGF2)の結合も拮抗しなかった。
【0059】
まとめると、これらの結果から、FGFリガンドの結合に対するSSRの阻害剤活性は、FGFRの立体構造に高度に依存しており、重複する結合ドメインの立体障害に依存する単純な競合的機構とは一致しなかったことが示される。FGFRについて本明細書で述べたように、アッセイ条件に応じてオルソステリックリガンドの結合に示差的に影響する小分子化合物アロステリック調節因子の能力は、GPCRの分野で以前に報告されている(Litschigら、1999年;Priceら、2005年)。恐らく、FGFR1∂123/Fcが、SSRの結合による125I−FGF2の親和性に対する負のアロステリック効果の伝達を可能にする立体構造に存在するのに対し、Fcタグ、すなわち天然の環境におけるインタクトな受容体全体の発現の欠如は存在しない。
【実施例2】
【0060】
SSRはアロステリックおよびマルチFGFR阻害剤である。
FGFR遺伝子発現(図1B)およびFGFR1バリアント(図1C)を検出する特定のプライマーを用いた定量的PCR(図1A)およびRT−PCRによるHUVEC細胞(C−12200、Promocell社)でのFGFR発現分析は、FGF−R4およびFGF−R1β3cの発現のみを実証したことから、本発明者らは、HUVE細胞を最初に用いて異なるFGFRでのSSRの拮抗活性を試験した。FGF19はFGFR4を特異的に刺激することが知られているのに対し、FGF4(FGF19ではなく)はFGFR1−hMpl融合タンパク質をトランスフェクトしたBaF/3細胞においてFGF−R1のみを活性化する(図2A)一方、FGF19は活性化できない(図2B)。したがって、HUVECにおいてFGFR1およびFGFR4は、それぞれFGF4およびFGF19で刺激することができる。
【0061】
HUVEC増殖は、FGF2およびFGF4で刺激されるが、FGF19では刺激されず(図4A)、HUVEC増殖がFGFR1の制御下にあることを示唆している。SSRは、FGF2誘導HUVEC増殖を阻害することができ、SSRがFGFR1β3c受容体を阻害することを示している(図4B)。ヒト臍帯静脈内皮細胞(HUVEC)またはFGFR1を過剰発現するブタ大動脈内皮細胞(PAE−FGFR1)でのI125−FGF2による結合アッセイは、SSRが(高μM濃度でさえ)受容体へのI125−FGF2結合を阻害できないことをさらに明らかにした。しかし、αFGF2抗体を中和することは効果があった(図4D)。本発明者らはまた、SSRが、FGFRシグナル伝達における重要な段階であるFGFRの自己リン酸化を阻害するかどうかも分析した。ラット脂肪パッド内皮細胞で発現されたFGFR1の免疫沈降とこの後のリン酸化FGFR1の免疫ブロットは、FGF2誘導FGFR1チロシンリン酸化が、ナノモル濃度範囲でSSRにより高度に低減されることを明らかにした(図4C)。特に、高用量でさえ、SSRはFGFR1チロシンリン酸化を完全には排除せず、低い残基レベルを残した(図4C)。FGFR2細胞外ドメインに結合するlumioタグ付FGF1に対するSSR効果が分析されており、SSRはFGF1/FGFR2相互作用を阻害しない(図5A)。同様に、SSRはFGFR2またはFGF2二量体化を阻害することができる(図5Bおよび5C)。
【0062】
HUVECの遊走も、FGF19ではなくFGF2−およびFGF4により刺激した(図6A)。この関連で、SSRは、FGFR1活性を阻害し、FGF2誘導HUVEC走化性遊走の低減をもたらすこともできる(図6B)。
【0063】
反対に、インビトロ血管新生は、FGF2およびFGF19により刺激されるのに対し、FGF4は不活性であり、FGFR4がこのアッセイではインビトロ血管新生を制御することを示唆している(図7A)。低いナノモル範囲では、SSRはFGF2誘導HUVEC血管新生をブロックし、SSRがFGFR4に制御される細胞プロセスを阻害できることを実証している(図7B)。
【0064】
FGFR2およびFGFR2−IIIbバリアントに対するSSR活性を評価するため、PANC02細胞の増殖および遊走が使用された。この理由は、これらの細胞反応が、FGFR2−IIIbの特異的リガンドである100ng/ml FGF7により刺激され得るためである(図8Aおよび8B)。VEGFの有無にかかわらず、FGF7誘導は100nM SSR添加によりブロックされ、SSRがFGFR2受容体および3bバリアントを阻害できることを示している(図8Aおよび8B)。
【0065】
FGFR3に対するSSR効果を試験するため、FGFR3WTまたはFGFR3TD(いずれかのリガンドの非存在下でさえ、K650E変異により誘導される構造的活性化FGFR3バリアント;Truedelら;blood 2006年)のどちらかを発現するB9−骨髄腫細胞の増殖を、FGF1(25ng/ml)による刺激によりアッセイした。B9−FGFR3WT細胞系が、FGF1により誘導され得および0.1μMのSSRにより阻害され得る(図9)のに対し、B9−FGFR3TD細胞系はSSRに非感受性であった(図9)。これは、SSRがFGFR3受容体を阻害できることを示しており、SSRがFGFRのキナーゼドメインで作用しないことを確認するものである。
【0066】
まとめると、これらの結果は、SSRが全てのFGFRアイソフォーム(FGFR1、R2、R3およびR4)およびFGFRバリアントを阻害できることを示している。
【実施例3】
【0067】
SSRは他の増殖因子により誘導される細胞反応を阻害できない。
SSRがFGF依存性シグナル伝達効果を示差的に阻害したことから、本発明者らは次に、SSRがインビトロでのFGF依存性細胞反応にも影響するかどうかを調査した。HUVECを用いると、SSRはFGF2の走化性効果を阻害した。
【0068】
SSRは、いずれもチロシンキナーゼ受容体ファミリーのメンバーを活性化することが知られているPIGF、EGF、PDGF−BBおよびIGFにより誘導された細胞反応に影響しなかった(図10)。
【実施例4】
【0069】
SSR128129EはFGFRの細胞外領域のIg様ドメインD3におけるアロステリック部位に結合する。
SSRはマルチFGFR阻害剤であったことから、本発明者らは、さまざまな(ヒト)FGFRサブタイプのポリペプチドフラグメントを使用した。SSRの飽和移動差NMR(STD−NMR)スペクトルは、SSRがFGFR1のECD(FGFR1∂123)に結合することを明らかにした(図11A)。これは、一次元(1D)−NMRプロファイルの分析により確認され、これがSSRの添加時のFGFR1∂123シグナルのピーク拡大を明らかにした(図11A)。この結合はTNF−R1細胞外タンパク質では観察されないため、この結合は特異的である(図11A)。本発明者らは次いで、FGFRのECDフラグメントを使用して3つのIgドメインの1つに対するSSRの結合部位をマッピングした。ドメインD3(FGFR1∂3)のみを含有するフラグメントの1D−NMR測定は、この膜近傍ドメインにおいてSSRの結合部位を同定した(図11B)。事実、FGFR1∂3およびFGFR1∂123は、本発明者らがこれらのタンパク質について同じ親和性を得たことを意味する同一のシグナル(太線)を与えるのに対し、FGFR1∂12およびFGFR1∂2は明確な線(図11B)をもたらす。2つのECDフラグメント、FGFR2∂23(ドメインD2および3から成る)およびFGFR3∂23を用いた等温滴定熱量測定(ITC)は、SSRがFGFR2およびFGFR3に結合することを明らかにした(図11Cおよび11D)。
【0070】
FGFRのドメインD3へのSSRの結合は特異的であった。この理由は、ITCまたはSTD−NMRにより分析した場合、該化合物がFGF−リガンド(FGF1およびFGF2;図12A、12Bおよび12C)に結合しなかったためである。さらに、ヘパリンはFGFRへのSSRの結合を妨げなかった。この理由は、STD−NMRが、ヘパリン類似体八硫酸スクロース(SOS、図12D)の存在下または非存在下、FGFRへのSSRの同等のシグナルを明らかにしたためである。
【実施例5】
【0071】
SSRのアロステリック結合はFGFRにおける立体構造変化を誘導する。
本発明者らは次いで、先行実験で同定された領域へのSSRの結合により媒介されたFGFRの立体構造変化の直接的証拠を、本発明者らが得ることができるかどうかを調べた。したがって、本発明者らは、ドメインドメインD2−3(FGFR2∂23)から成る、FGFR2のECDフラグメントのフーリエ変換赤外線(FTIR)分光測定を実施した。どちらかのバリアントへのSSRの添加は、全体的な立体構造変化と一致する、1,640cm−1周辺で最大となる、FTIRスペクトルのアミドIバンドの振幅の増加をもたらした(図12A)。
【0072】
本発明者らは次に、SSRが、D3におけるオルソステリック部位の部分を形成するアミノ酸残基または別のアロステリック部位に結合するかどうかを調べた。最初に、本発明者らは、ソフトウェアパッケージMOLEGRO、AutodockおよびYASARAならびに利用可能な結晶学的データの分子ドッキングアルゴリズムを使用した。両方の方法を用いたFGFR2∂3でのSSRのドッキングランは、2つの想定される結合部位(1つはHis293周辺に集中し、もう1つはTyr328周辺に集中していた。)を同定した。これらの想定される結合部位は、FGFリガンド結合部位と比べて、受容体の反対面に位置しおよびオルソステリック結合部位から25Åまでの疎水ポケットを形成する。特に、両方の残基は、オルソステリックFGF結合ポケットの残基と重複しない。両方の想定されるSSR結合部位の機能的関連性を評価するため、本発明者らは、分子力場FoIdXソフトウェア(Schymkowitzら、2005年)を使用して、アロステリックリガンド結合を低減または排除するが、構造の全体的な立体構造安定性を撹乱しない変異を設計した:(i)芳香族残基を負に帯電されたアスパラギン酸と置換してSSRとの疎水性相互作用を除去するFGFR2∂23−Y328D;(ii)SSRの他の想定される結合部位から必須残基を除去するFGFR2∂23−H293L;および(iii)FGFR2∂23−Y328D/H293L二重変異体(FGFR2∂23−YHと呼ばれる)。ITC結合実験は、SSRがFGFR2∂23−Y328Dに結合しないことを示している(図13Bおよび13C)。これらの知見は、SSRが、オルソステリックリガンド結合部位近傍の疎水ポケットにより形成されたアロステリック部位に結合するモデルで、および残基Tyr328はSSRとFGFR2の間の相互作用を媒介するのに必須と思われるモデルと一致する。本発明者らはまた、前述の変異FGFR2フラグメントのFTIRスペクトルも分析した。これらの単一または二重変異は、FTIRスペクトルにおける重大なシフトを誘導しなかった。これは、これらのFGFRバリアントの全体的な三次元立体配置が同等であることを示している。SSRは、FGFR2∂23−H293Lおよび天然のFGFR2∂23フラグメントのFTIRスペクトルの同等のシフトを誘導した(図14Aおよび14B)。これは、His293からLeu293への変異が十分に強烈でないこと、またはHis293がSSRとの相互作用にそれほど意味がないことを示唆している。その一方、SSRは、変異FGFR2∂23−Y328DまたはFGFR2∂23−YHフラグメントのFTIRスペクトルの変化を誘導しなかった(図14Cおよび14D)。これは、残基Tyr328が、SSRを結合する時のFGFR2のアロステリック立体構造変化の媒介に実際に必須であったことを示している。
【実施例6】
【0073】
アロステリックSSR結合部位の変異はFGFRシグナル伝達のSSR阻害を低減する。
FGFRシグナル伝達の調節におけるアロステリック部位の機能的重要性を評価するため、本発明者らは、機能的FGFR2WTまたはFGFR2∂23−Y328Dバリアントのどちらかを発現する安定なHEK293細胞系を作製し、SSRがこれらの細胞系でFGF2によるERK1/2の活性化を阻害するかどうかを分析した。免疫ブロッティングは、SSRによるFGFR2∂23−Y328D細胞でのFGF2誘導ERK1/2リン酸化の阻害が、FGFR2WT細胞での阻害能力(IC50値:28±12nM)と比べて低減する(IC50値:121±30nM)ことを明らかにした(図15Aおよび15B)。これは、このアロステリック部位が、インビトロでの精製FGFR2フラグメントへのSSR結合に関係がないだけでなく、インセルロ(in cellulo)での生理条件ではFGFR2シグナル伝達に対する阻害活性にも関係がないことを示している。Y328D変異が、SSRの阻害活性を完全には無効にしなかったという事実は、FGFR2が生理学的文脈で発現される場合に、Tyr328に加えて他の隣接残基もSSRの結合に寄与することを示唆している可能性がある。
【実施例7】
【0074】
SSRはFGFR依存性ホスホ−シグナル伝達の「偏った」阻害剤である。
FGFR制御リン光体−シグナル伝達に対するSSR効果を評価するため、HEK293細胞をFGFR2でトランスフェクトし、FGFR依存性FRS2およびPLCγカスケードを阻害することが記載された公表FGFRチロシンキナーゼ阻害剤SU5402(Zhenら、Oncogene 2007年)と比較して、FGFR自己リン酸化後の2つの主要経路、PLCγおよびFRS2をウェスタンブロットにより試験した。このような細胞では、FGFは、FRS2、Erk1/2およびPLCγリン酸化を誘導する。SU5402が全てのこれらの誘導をブロックするのに対し、SSRはFRS2経路を阻害するのみであり(図16Aおよび16B)、SSRにより得られた偏った拮抗作用を説明している。より一般的には、リン酸化のこれらの違いは、アロステリック調節因子を評価するためのレポーターとして使用することができる。
【実施例8】
【0075】
チロシンキナーゼ受容体の細胞外ドメインにおけるフラストレートされたドメインを探索するための方法論
例5からの記載された立体構造変化の考えられる分子機構の1つは、フラストレートされたドメイン(定義については上記参照)の存在を含む。AGADIR(ヘリックス安定性予測アルゴリズム;Munoz,V.&Serrano,L. 1994年)を用いてヒトFGFR2のドメインD2およびD3を分析した時、本発明者らは、βシートからαヘリックスへのシフトを起こす蛍光がある唯一の領域として、Tyr319からArg330の範囲の(故に必須残基Tyr328を含む)残基の配列を同定した。アルファヘリカリティ(helicality)を低減し故にドメインのフラストレーションを低減することがAGADIRにより予測されたアスパラギン酸によるTyr328の置換(FGFR2∂23−Y328D)は、このような理論モデルと一致して、FTIR分析により検出されたような観察された立体構造変化を実際に妨げた。他のTKRのうち、VEGFR1、−2および−3ならびにPDGFRβを含む別の増殖因子受容体の類似の配列分析は、比較的高いAGADIRスコアの領域を含有した。該領域はこの領域からアスパラギン酸に必須残基を変異させることで逆になり得る。本発明者らは、K609DおよびK648Dを変異させることが、VEGFによる刺激時にERK1/2シグナル伝達の低減をもたらした、VEGFR2の幾つかの予備データを持っている。
【実施例9】
【0076】
SEC−LC/MSによるFGF−Rアロステリック調節因子の親和性スクリーニングおよび同定された化合物の活性評価
SEC−LC/MS方法論は、オンライン結合された2次元システム、高性能液体クロマトグラフィーに結合されたサイズ排除クロマトグラフィーと、これに続く検出のためのエレクトロスプレーイオン化−飛行時間型質量分析に依存する、親和性スクリーニングに使用される分析技法である。
【0077】
可溶性ポリペプチド(ペプチド、タンパク質ドメイン、または完全長タンパク質)と相互作用する幾つかの化合物の能力に基づく。目的とするペプチドと小分子化合物のプールを混合した後、ペプチド−リガンド複合体は、サイズ排除クロマトグラフィーによる非結合と結合小分子化合物との分離を可能にする質量シフトを誘導する。次いで、複合体は解離され、バインダーが正確な質量測定のため高分解能LC/ESI−TOFを用いて検出される(例えばWaters LCTプレミア質量分析計により)。データデコンボリューションアルゴリズムは、質量検出分析からの結合分子の同定を可能にする。
【0078】
FGF−Rアロステリック調節因子を同定するには、この技術を天然のまたは変異された種々のFGF−Rの細胞外ドメインに適用することができる。天然型は、細胞外ドメインバインダー全ての検出を可能にする。アロステリック調節因子のスクリーニングを実現する別の方法は、アルファヘリックスを安定化し、SSR128129結合への感作を可能にする変異Tyr328Arg−Ile329Lysにより得られた、SSR128129結合部位に近接するFGF−R2ヘリックスの「開口」型を用いて行うことができる。この場合、この変異FGF−R2は、WT FGF−R2にとって代わることができる。類似の戦略を、FGF−R2におけるTyr328およびIle329に対応するアミノ酸に変異を有するFGF−R1、−R3または−R4のスクリーニングを行うのに使用することができる。Tyr328での変異型(FGF−R2)または他のFGF−Rにおける対応する変異アミノ酸は、カウンタースクリーニング(counterscreen)を行うのに使用することができる。SSR128129は、疎水ポケット近傍のTyr328Aspで変異されたFGF−R2に結合しないため、この変異型は、FGF−R2における標的化ポケットと相互作用しない化合物の部分を廃棄するのに使用することができる。
【0079】
全ての場合において、この戦略は、目的とするペプチドにおける標的ポケットに結合することができる分子の同定をもたらす。次の段階では、細胞におけるシグナル伝達に対する効果が評価されなければならない。第1に、選択された化合物は、ウェスタンブロット実験でSSRにより観察されたように(図17Aおよび17B)、HUVEC細胞におけるAKTリン酸化などのFGF誘導誘導経路を阻害しないければならない。HUVEC細胞におけるAKTのリン酸化状態は、オンセルELISA方法論により測定することができる。このアッセイフォーマットは、HUVECなどのさまざまな細胞においてAKTリン酸化を直接検出するため自家開発されており、およびFGF4刺激HUVECに対するSSR効果の検出を可能にする(図17C)。FGFRアロステリック調節因子の典型的な特徴は、FGF結合と競合する能力である。これを評価するため、FGFRをトランスフェクトされたマウスプレ−B 300−19細胞での結合アッセイが作成された。10ng/mlでのAlexaFluor488標識FGF2は、天然にはいずれのFGFRも発現しない300−19細胞で発現されたFGFR1またはFGFR4に結合し、300nMでのSSRは、フローサイトメトリー分析により、FGFR1またはFGFR4へのこの結合を競合することはできない(図17D)。
【実施例10】
【0080】
VEGF−R2受容体における想定されるフラストレートされた帯域の同定および想定されるフラストレートされた帯域の変異分析
FGF−R用に開発された戦略が、別の受容体TK、すなわちVEGF−R2またはKDRに適用される。想定されるアロステリック標的部位を持つことができる領域を同定する最初のアプローチとして、本発明者らは、マウスVEGF−R2受容体由来の利用可能な一次アミノ酸配列(EntrezアクセッションNP_034742.2)を用いて、構造変化(例えばβシートからαヘリックスへの転移)を起こす傾向がある領域を同定するソフトウェアプログラムAGADIR1を利用した。これは、高い螺旋傾向を有する幾つかの領域をもたらしたが、Igドメイン構造では主にβシート構造が期待されるはずである。Agadir分析の結果は図18Aに示されている。
【0081】
この後、VEGF−R2由来の各アミノ酸をアスパラギン酸残基(D)により連続的にインシリコ変異させた後、本発明者らは、AGADIRスコアを再び分析し、ならびに(i)螺旋傾向の変化が最大(最もネガティブ)であるおよび(ii)膜貫通ドメインに最も近傍に位置するようなIgドメインに位置するような変異を選択した。これらから、K609DおよびK648D(両方の残基ともmVEGF−R2のドメインlgD6にある(図18B))は螺旋傾向の最大の低下をもたらし、インセルロ分析にさらに使用された(さらなる例12参照)。
【実施例11】
【0082】
PDGF−Rβ受容体における想定されるフラストレートされた帯域の同定および想定されるフラストレートされた帯域の変異分析
想定されるアロステリック標的部位を持つことができる領域を同定する最初のアプローチとして、本発明者らは、ヒトPDGFRβ受容体由来の利用可能な一次アミノ酸配列(EntrezアクセッションNP_002600.1)を用いて、構造変化(例えばβシートからαヘリックスへの転移)を起こす傾向がある領域を同定するソフトウェアプログラムAGADIR1を利用した。これは、高い螺旋傾向を有する幾つかの領域をもたらしたが、Igドメイン構造では主にβシート構造が発現されるはずである(図19)。この後、PDGFRβ由来の各アミノ酸をアスパラギン酸残基(D)により連続的にインシリコ変異させた後、本発明者らは、AGADIRスコアを再び分析し、ならびに(i)螺旋傾向の変化が最大(最もネガティブ)であるおよび(ii)膜貫通ドメインに最も近傍に位置するようなIgドメインに位置するような変異を選択した。これらから、L383DおよびK387D(両方の残基ともhPDGFRβのドメインIgD3にある)は螺旋傾向の最大の低下をもたらした(図19)。
【実施例12】
【0083】
「偏った」シグナル伝達を誘導する化合物を同定するスクリーニング方法
VEGFまたはPDGF−BBの結合は、それぞれの同種受容体の二量体化を誘導し、これが今度は細胞内キナーゼドメインのリン酸化を誘導する。この後(FGF−Rの偏ったアンタゴニストSSRに一致する目的とする)2つの主要経路が、ERK1/2経路(図21Aおよび21C)およびPLCγ経路(図20に概略的に示されている)を含め活性化される。各経路の活性化の測定は、考えられる阻害剤の存在下または非存在下、シグナル伝達経路の1つのみの阻害により、「偏った」シグナル伝達を誘導する化合物の同定をもたらす。
【0084】
VEGF−R2については、実施例10で同定された2つの変異体VEGFR2受容体(VEGFR2K609DおよびVEGFR2K648D)およびVEGF−R2のウィルト型フォーム(wilt type form)を、HEK293細胞で安定に発現させた。VEGFR2WT受容体は、ERK1/2リン酸化の活性化により明らかに反応する。VEGFR2K609D変異体が、ERK1/2を通じたシグナル伝達能力を低減した(だがカウンタースクリーニングアッセイには十分である)のに対し、VEGFR2K648D変異体はこれを喪失した。結果は図21Aに要約されている。
【0085】
PDGF−Rβについては、hPDGFRβ細胞を過剰発現するHEK293細胞を、アルファスクリーンシュアファイア手順に従って添加剤なし(「0」)、10%ウシ胎仔血清(10%FBS)、50ng/ml PDGF−BB(50ng/ml)を含有する培地または1ng/ml PDGF−BB(1ng/ml)を含有する培地のどちらかで刺激した(図21B)。図21Cでは、左のパネルは、刺激時の活性化または合計ERK1/2タンパク質を検出する混合物の略図を再び表す。右のパネルは、アルファスクリーンシュアファイア検出方法により測定された細胞のPDGF−BB刺激後に検出された測定を表す。これは、細胞を10%FBSまたは50ng/mlのPDGF−BBのどちらかで刺激する場合、PDGF−BBによる1ng/ml刺激の有無にかかわらず、はるかに低いシグナルが存在する間は明らかなシグナルを検出できることを示している。PLCγの答えは、類似の方法で測定する。
【0086】
FGF−RにおけるSSRのように、VEGF−R2またはPDGF−Rβにおいて「偏った」シグナル伝達を誘導する化合物を同定するスクリーニング方法は、Erk1/2およびPLCγの答えに基づく。候補アロステリック阻害剤の存在下および非存在下におけるERK1/2およびPLCγ反応の比較は、偏った阻害剤として作用する、すなわち2つのシグナル伝達経路の1つのみを阻害する化合物の同定を可能にする。VEGF−R2またはPDGF−Rβの変異構築物は、同定されたアロステリック調節因子の作用の機構を検証するカウンタースクリーニングアッセイに役立つことができる。変異受容体では、化合物は受容体調節能力を喪失せざるを得ない。
【0087】
【表1】
【特許請求の範囲】
【請求項1】
チロシンキナーゼ受容体の細胞外ドメインに由来するアロステリック阻害剤結合部位。
【請求項2】
前記結合部位がIgドメインを有するチロシンキナーゼ受容体に由来する、請求項1に記載のアロステリック阻害剤結合部位。
【請求項3】
前記結合部位が偏った拮抗作用を誘導する、請求項1または2に記載のアロステリック阻害剤結合部位。
【請求項4】
前記結合部位がフラストレートされたドメインを含む、請求項1から3のいずれかに記載のアロステリック阻害剤結合部位。
【請求項5】
前記チロシンキナーゼ受容体が、線維芽細胞増殖因子受容体、血管内皮増殖因子受容体もしくは血小板由来増殖因子受容体、またはこのホモログ、パラログもしくはオソログである、請求項1から4のいずれかに記載のアロステリック阻害剤結合部位。
【請求項6】
配列番号1:
His Val Glu Lys Asn Gly Ser Lys Tyr Gly Pro Asp Gly Leu Pro Tyr Leu Lys Val Leu Lys Ala Ala Gly Val Asn Thr Thr Asp Lys Glu Ile Glu Val Leu Tyr Ile Arg Asn
または配列番号1と少なくとも70%、80%、90%もしくは95%の相同性を示す配列にある、請求項1から5のいずれかに記載のアロステリック阻害剤結合部位。
【請求項7】
配列番号2
Leu Lys Ala Ala Gly Val Asn Thr Thr Asp Lys Glu Ile Glu Val Leu Tyr Ile Arg Asn
または配列番号2と少なくとも70%、80%、90%もしくは95%の相同性を示す配列にある、請求項4に記載のフラストレートされた結合部位。
【請求項8】
チロシンキナーゼ受容体経路において偏った拮抗作用を誘導するための、請求項1から6に記載のアロステリック阻害剤結合部位の使用。
【請求項9】
小分子化合物阻害剤をスクリーニングするための、請求項1から7に記載のアロステリック阻害剤結合部位の使用。
【請求項10】
チロシンキナーゼ受容体の細胞外ドメインにおけるフラストレートされたドメインのスクリーニングを含む、前記細胞外ドメインにおけるアロステリック阻害剤結合部位の同定方法。
【請求項11】
チロシンキナーゼ受容体の活性化/阻害に依存する2つの異なる下流経路により誘導される少なくとも2つの異なるレポーターの比較を含む、請求項1から7に記載のチロシンキナーゼ受容体の細胞外ドメインにおけるアロステリック阻害剤部位に結合する小分子化合物アロステリック阻害剤の同定方法。
【請求項12】
a)RTKのアロステリック結合部位を小分子化合物アロステリック阻害剤候補化合物と接触させる段階、
b)前記チロシンキナーゼ受容体の活性化/阻害に依存する少なくとも2つの下流経路の変化を測定する段階、
c)前記チロシンキナーゼ受容体の活性化/阻害に依存する少なくとも2つの異なる下流経路に対する少なくとも1つのレポーターの状態の変化を比較する段階
を含み、
受容体のリガンド結合ドメインに結合するリガンドの存在下、少なくとも1つの下流経路が阻害されるのに対し、少なくとももう1つの下流経路が影響を受けない場合に、アロステリック阻害剤が同定される、
RTKのアロステリック阻害剤の同定方法。
【請求項13】
前記2つの異なる下流経路が、ERK1/2シグナル伝達経路およびPLCγシグナル伝達経路に対応する、請求項12に記載のRTKのアロステリック阻害剤の同定方法。
【請求項14】
化合物が小分子である、請求項1から6のいずれかに記載のアロステリック結合部位に結合する前記化合物。
【請求項1】
チロシンキナーゼ受容体の細胞外ドメインに由来するアロステリック阻害剤結合部位。
【請求項2】
前記結合部位がIgドメインを有するチロシンキナーゼ受容体に由来する、請求項1に記載のアロステリック阻害剤結合部位。
【請求項3】
前記結合部位が偏った拮抗作用を誘導する、請求項1または2に記載のアロステリック阻害剤結合部位。
【請求項4】
前記結合部位がフラストレートされたドメインを含む、請求項1から3のいずれかに記載のアロステリック阻害剤結合部位。
【請求項5】
前記チロシンキナーゼ受容体が、線維芽細胞増殖因子受容体、血管内皮増殖因子受容体もしくは血小板由来増殖因子受容体、またはこのホモログ、パラログもしくはオソログである、請求項1から4のいずれかに記載のアロステリック阻害剤結合部位。
【請求項6】
配列番号1:
His Val Glu Lys Asn Gly Ser Lys Tyr Gly Pro Asp Gly Leu Pro Tyr Leu Lys Val Leu Lys Ala Ala Gly Val Asn Thr Thr Asp Lys Glu Ile Glu Val Leu Tyr Ile Arg Asn
または配列番号1と少なくとも70%、80%、90%もしくは95%の相同性を示す配列にある、請求項1から5のいずれかに記載のアロステリック阻害剤結合部位。
【請求項7】
配列番号2
Leu Lys Ala Ala Gly Val Asn Thr Thr Asp Lys Glu Ile Glu Val Leu Tyr Ile Arg Asn
または配列番号2と少なくとも70%、80%、90%もしくは95%の相同性を示す配列にある、請求項4に記載のフラストレートされた結合部位。
【請求項8】
チロシンキナーゼ受容体経路において偏った拮抗作用を誘導するための、請求項1から6に記載のアロステリック阻害剤結合部位の使用。
【請求項9】
小分子化合物阻害剤をスクリーニングするための、請求項1から7に記載のアロステリック阻害剤結合部位の使用。
【請求項10】
チロシンキナーゼ受容体の細胞外ドメインにおけるフラストレートされたドメインのスクリーニングを含む、前記細胞外ドメインにおけるアロステリック阻害剤結合部位の同定方法。
【請求項11】
チロシンキナーゼ受容体の活性化/阻害に依存する2つの異なる下流経路により誘導される少なくとも2つの異なるレポーターの比較を含む、請求項1から7に記載のチロシンキナーゼ受容体の細胞外ドメインにおけるアロステリック阻害剤部位に結合する小分子化合物アロステリック阻害剤の同定方法。
【請求項12】
a)RTKのアロステリック結合部位を小分子化合物アロステリック阻害剤候補化合物と接触させる段階、
b)前記チロシンキナーゼ受容体の活性化/阻害に依存する少なくとも2つの下流経路の変化を測定する段階、
c)前記チロシンキナーゼ受容体の活性化/阻害に依存する少なくとも2つの異なる下流経路に対する少なくとも1つのレポーターの状態の変化を比較する段階
を含み、
受容体のリガンド結合ドメインに結合するリガンドの存在下、少なくとも1つの下流経路が阻害されるのに対し、少なくとももう1つの下流経路が影響を受けない場合に、アロステリック阻害剤が同定される、
RTKのアロステリック阻害剤の同定方法。
【請求項13】
前記2つの異なる下流経路が、ERK1/2シグナル伝達経路およびPLCγシグナル伝達経路に対応する、請求項12に記載のRTKのアロステリック阻害剤の同定方法。
【請求項14】
化合物が小分子である、請求項1から6のいずれかに記載のアロステリック結合部位に結合する前記化合物。
【図1】

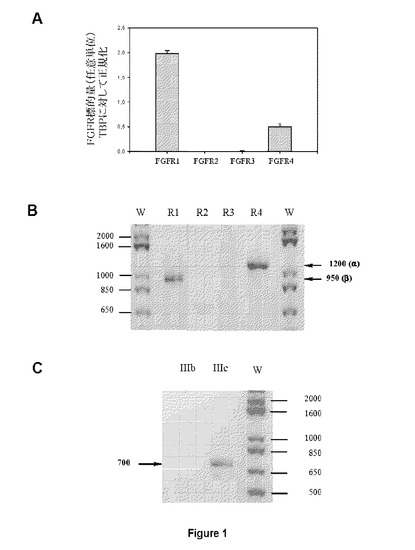
【図2】
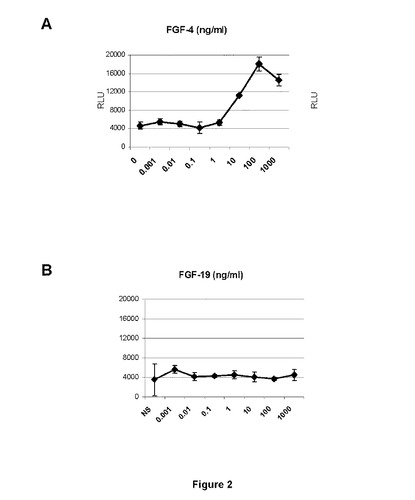
【図3】
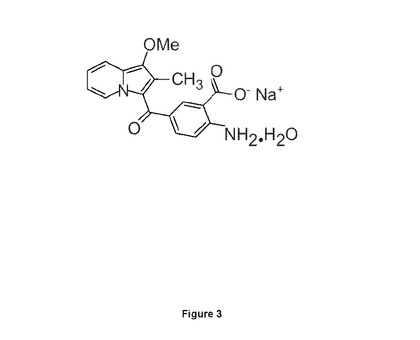
【図4−1】
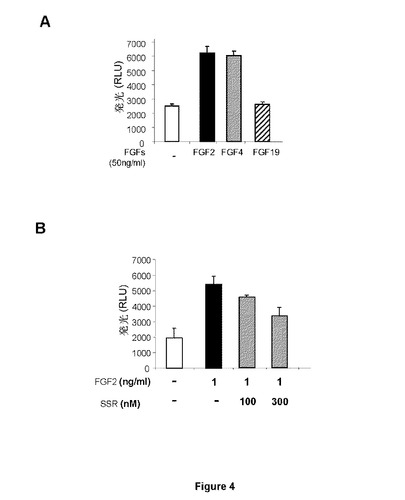
【図4−2】
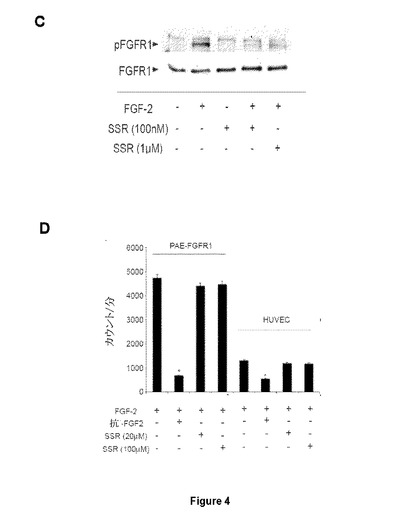
【図5】
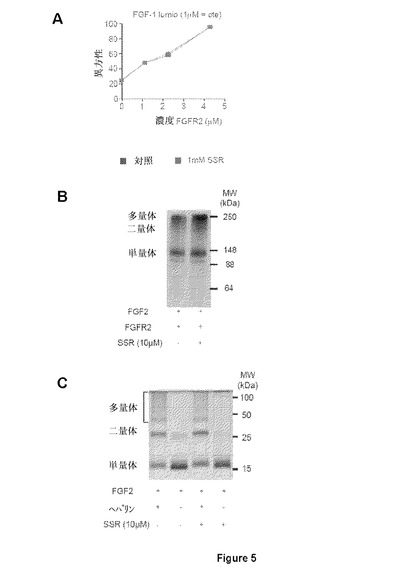
【図6】
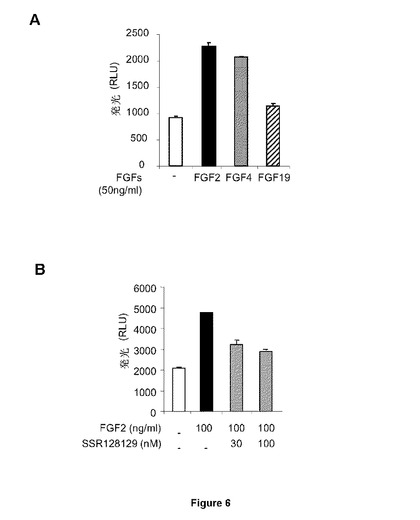
【図7】
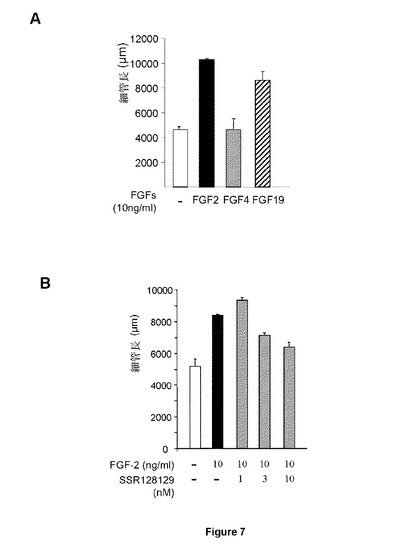
【図8】
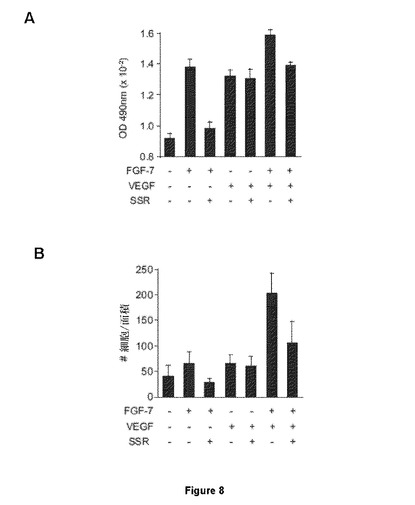
【図9】
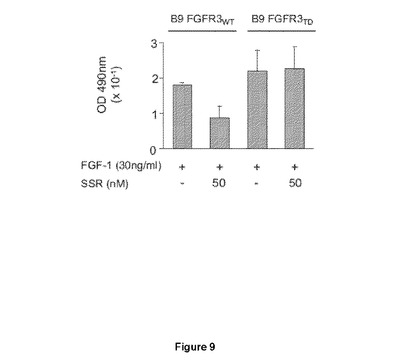
【図10】
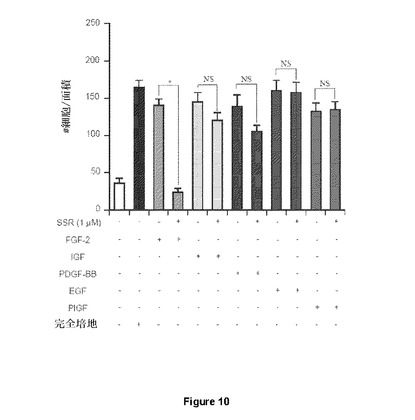
【図11−1】
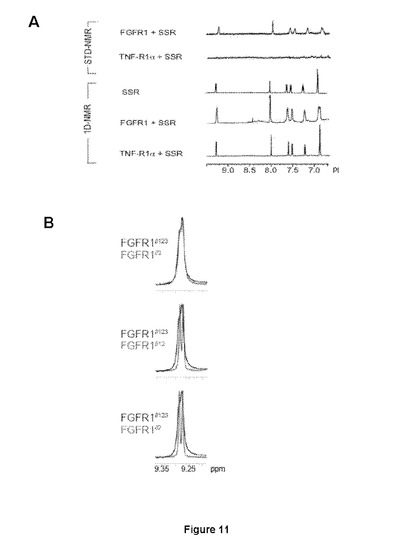
【図11−2】
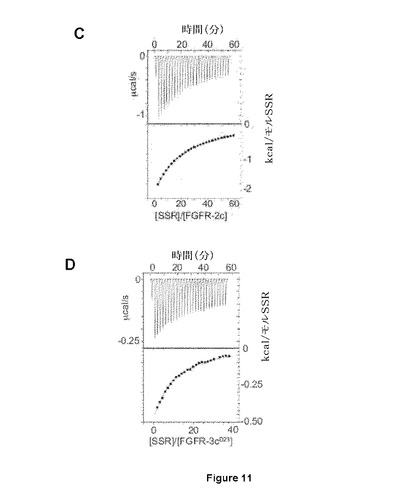
【図12−1】
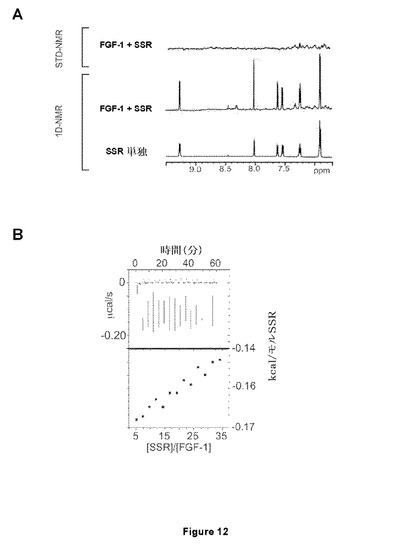
【図12−2】
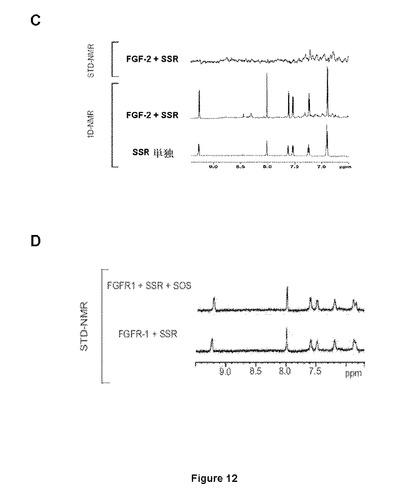
【図13】
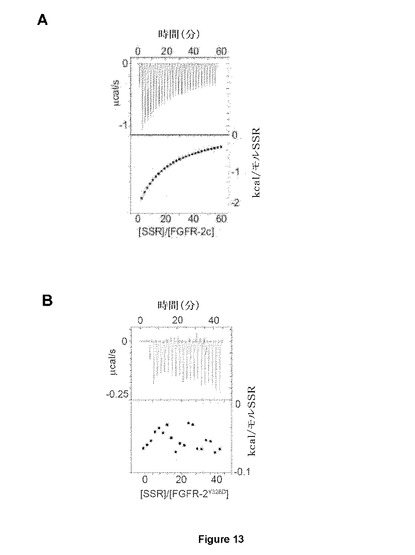
【図14】
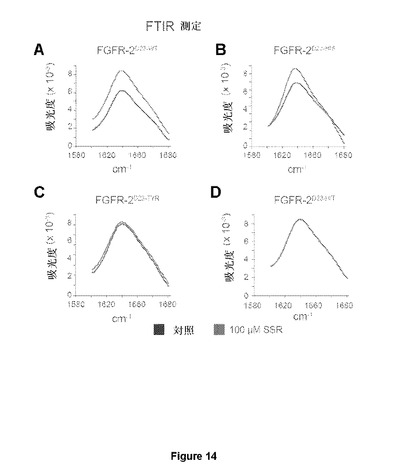
【図15】
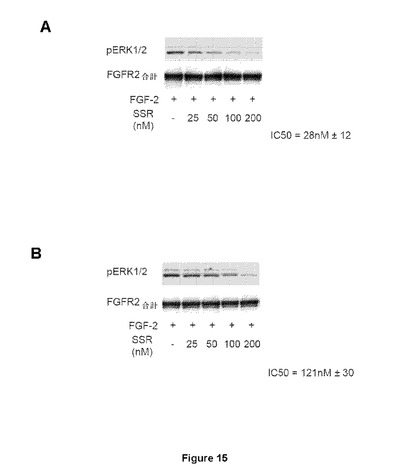
【図16】
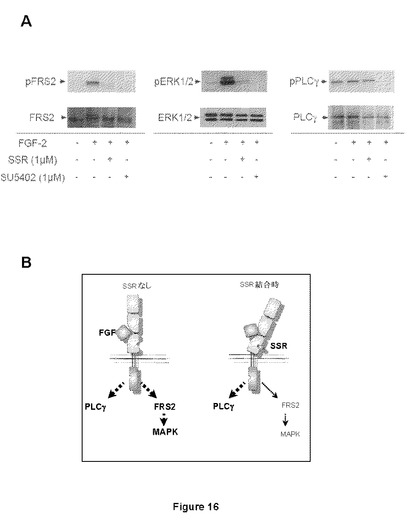
【図17−1】
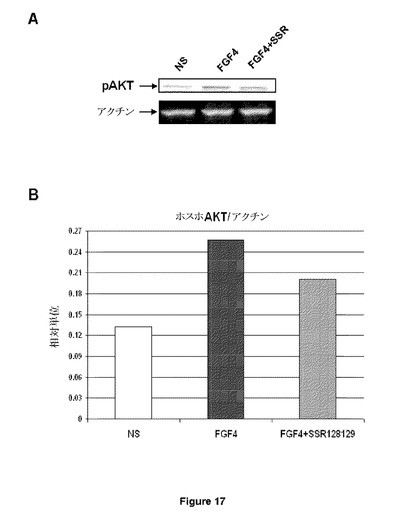
【図17−2】
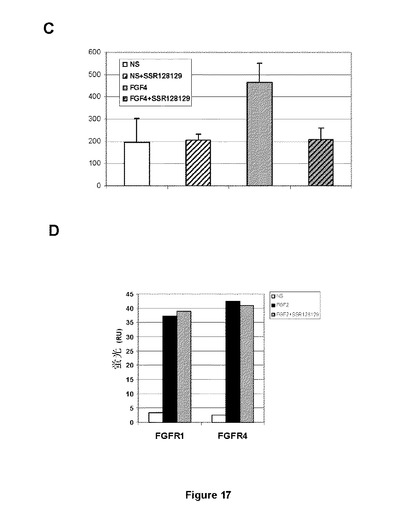
【図18】
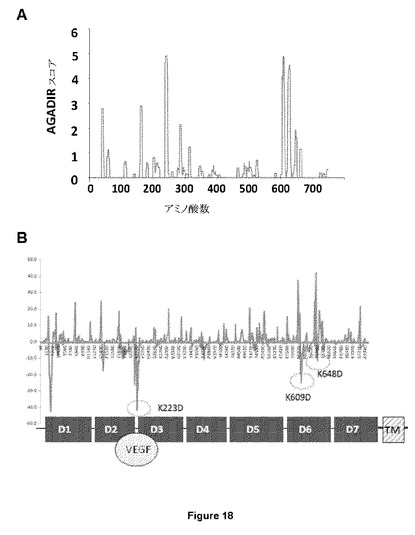
【図19】
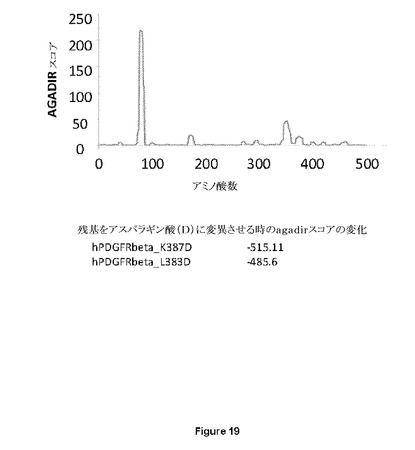
【図20】
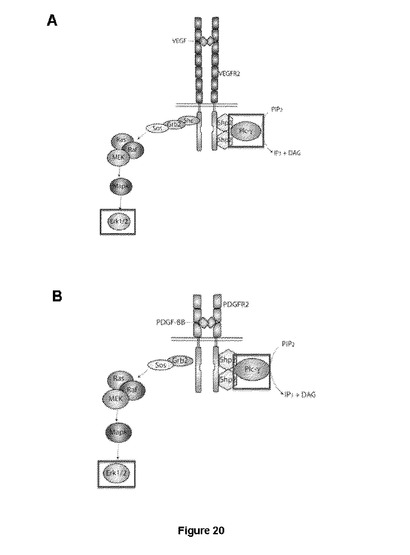
【図21】
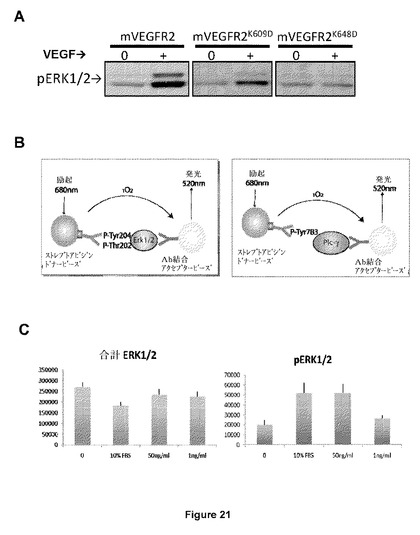
【図2】
【図3】
【図4−1】
【図4−2】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11−1】
【図11−2】
【図12−1】
【図12−2】
【図13】
【図14】
【図15】
【図16】
【図17−1】
【図17−2】
【図18】
【図19】
【図20】
【図21】
【公表番号】特表2012−532121(P2012−532121A)
【公表日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願番号】特願2012−518145(P2012−518145)
【出願日】平成22年7月2日(2010.7.2)
【国際出願番号】PCT/IB2010/053054
【国際公開番号】WO2011/001413
【国際公開日】平成23年1月6日(2011.1.6)
【出願人】(504456798)サノフイ (433)
【出願人】(509333933)
【出願人】(502014363)ライフ・サイエンシーズ・リサーチ・パートナーズ・フェレニゲング・ゾンデル・ウィンストーメルク (12)
【氏名又は名称原語表記】LIFE SCIENCES RESEARCH PARTNERS VZW
【出願人】(509333922)フリエ・ウニベルシテイト・ブリユツセル (2)
【Fターム(参考)】
【公表日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願日】平成22年7月2日(2010.7.2)
【国際出願番号】PCT/IB2010/053054
【国際公開番号】WO2011/001413
【国際公開日】平成23年1月6日(2011.1.6)
【出願人】(504456798)サノフイ (433)
【出願人】(509333933)
【出願人】(502014363)ライフ・サイエンシーズ・リサーチ・パートナーズ・フェレニゲング・ゾンデル・ウィンストーメルク (12)
【氏名又は名称原語表記】LIFE SCIENCES RESEARCH PARTNERS VZW
【出願人】(509333922)フリエ・ウニベルシテイト・ブリユツセル (2)
【Fターム(参考)】
[ Back to top ]