
テトラアルキルアンモニウムアルキルホスフェートを含むセルロース溶液およびそれから製造される製品
本発明は、セルロースおよび少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートを含むセルロース溶液、ならびに該セルロース溶液の製造方法に関する。本発明の別の側面は、セルロースおよび少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートを含むセルロース溶液から製造される成形品に関する。本発明の別の態様は、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートを含むセルロース溶液から製造されるセルロース誘導体を含む組成物に関する。本発明の別の態様は、セルロースおよび少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートを含むセルロース溶液から製造される、位置選択的に置換されたセルロースエステルを含む組成物に関する。本発明の別の態様において、本発明のセルロースエステルは、液晶ディスプレイ用の保護フィルムおよび補償フィルムとして使用される。

【発明の詳細な説明】
【技術分野】
【0001】
関連出願
この元の出願は、米国仮出願整理番号61/169,560、発明の名称“Tetraalkylammonium Alkylphosphates”(2009年4月15日出願)(参照により本明細書に組入れる)の利益を主張する。
【0002】
発明の分野
本発明はセルロース化学、詳細にはセルロース溶液、セルロースエステルの製造、およびそれから製造される製品の分野に関する。
【背景技術】
【0003】
発明の背景
セルロースは無水グルコースのβ−1,4−結合ポリマーである。セルロースは、典型的には高分子量の多分散ポリマーであり、水および実質的に全ての一般的な有機溶媒に不溶である。木材またはコットンの製品,例えばハウジングまたは布帛における非変性セルロースの使用は周知である。非変性セルロースはまた、種々の他の用途において、通常はフィルム(例えばセロハン(登録商標))として、繊維(例えばビスコースレーヨン)として、または粉末(例えば微結晶セルロース)として(医薬用途において使用される)、利用される。変性セルロース(例えばセルロースエステル)もまた広範な商業用途において広く利用されている[Prog.Polym.Sci.2001,26,1605−1688]。セルロースエステルは、一般的に、まずセルロースをセルローストリエステルに転化した後、セルローストリエステルを酸性水性媒体中で所望の置換度(DS,無水グルコースモノマー当たりの置換基の数)まで加水分解することによって調製する。これらの条件下でのセルローストリアセテートの加水分解により、ランダムコポリマーが得られ、これは、最終DSに応じて8つの異なるモノマーからなることができる[Macromolecules 1991,24,3050]。
【0004】
イオン液体中でのセルロースの溶解は公知である。当業者は、特定のイオン液体中、任意の既知温度にて溶解するセルロースの最大量は、初期セルロースの重合度(DP)および溶解プロセス中のセルロースの分解程度に左右されることを理解するであろう。
【0005】
最も広い意味において、イオン液体(IL)は単純にイオンのみを含有する任意の液体である。よって、溶融塩,例えばNaCl(温度800℃超で溶融する)はイオン液体と分類できる。実際的な観点から、イオン液体という用語は、ここでは、約100℃未満で溶融する有機塩に対して用いる。
【0006】
イオン液体のカチオンは構造的に多様であるが、これらは一般的には窒素またはリン(これらは4級アンモニウムまたはホスホニウムに転化されることができる)を含有する。一般的に、最も有用なイオン液体は、環構造の一部である窒素を含有する。これらのカチオンの例としては、ピリジニウム、ピリダジニウム、ピリミジニウム、ピラジニウム、イミダゾリウム、ピラゾリウム、オキサゾリウム、トリアゾリウム、チアゾリウム、ピペリジニウム、ピロリジニウム、キノリニウム、およびイソキノリニウムが挙げられる。
【0007】
【化1】
【0008】
イオン液体のアニオンもまた構造的に多様であることができる。アニオンは無機または有機のいずれであることもでき、そしてこれらは種々の媒体中でのイオン液体の溶解性に対する顕著な影響を有する。例えば、疎水性アニオン,例えばヘキサフルオロホスフェートまたはトリフルイミドを含有するイオン液体は、水中の極めて低い溶解性を有する。一方、親水性アニオン,例えばクロリドまたはアセテートを含むイオン液体は水中で完全に混和性である。
【0009】
イオン液体の名称は一般的には省略される。アルキルカチオンはしばしばアルキル置換基およびカチオン(1組の括弧内に与えられる)の記号、続いてアニオンの略号で命名される。表記しないが、カチオンは正電荷を有し、アニオンは負電荷を有することが理解される。例えば、[BMIm]OAcは、1−ブチル−3−メチルイミダゾリウムアセテートを意味し、そして[AMIm]Clは1−アリル−3−メチルイミダゾリウムクロリドを意味する。
【0010】
窒素が環状カチオンの一部である(例えばイミダゾリウム)場合、これらのイオン液体は典型的には、対応するアンモニウムまたはホスホニウムを含有するイオン液体と比べ、より低い融点を有し、より含水性が低く、そしてより安定である。セルロースの溶解およびエステル化の点で、イミダゾリウムをカチオンとして含有するイオン液体は一般的に好ましい。これらのイオン液体では、濃厚セルロース溶液(約20質量%,[EMIm]OAc中)を実現可能であり、これから種々のセルロースエステルを調製できる(第US6,824,599号、米国出願第20080194807号、米国出願第20080194808号、および米国出願第20080194834号)。
【0011】
最も一般的に利用される非環式アンモニウム系イオン液体は、テトラアルキルアンモニウムハライドである。イオン液体の分類は、セルロースの溶解およびエステル化において制限された利用性を有する。2つのテトラアルキルアンモニウムハライド,テトラエチルアンモニウムクロリド(第US4,597,798号)およびテトラブチルアンモニウムフルオリド水和物(Macromol.Biosci.2007,7,307−314)のみが、セルロースを顕著レベル(約6質量%セルロース)で可溶化させることが示されている。両者の場合で、かなりの濃度の共溶媒(30〜90質量%),例えばDMSOまたはDMFがセルロース可溶化に必要であった。イオン液体中の低濃度のセルロース、これらのイオン液体から水を除去することの困難性、これらのイオン液体の不安定性、更にこれらのイオン液体の腐食性および有毒性に起因して、これらのテトラアルキルアンモニウムハライドはセルロース誘導体の製造のための実際的な媒体ではない。
【発明の概要】
【課題を解決するための手段】
【0012】
発明の要約
本発明の一態様は、1種以上のテトラアルキルアンモニウムアルキルホスフェート中でのセルロースの溶解に関する。本発明は、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上の非プロトン性溶媒を含む混合物中、セルロースを溶解させるのに十分な接触時間および接触温度での、セルロースの溶解に更に関する。本発明は更に、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上のイオン液体であってカチオンがイミダゾリウムであるものを含む混合物中での、セルロースの溶解に関する。本発明は更に、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上の酸(ここで酸はセルロースと組合せない)を含む混合物中、セルロースを溶解させるのに十分な接触時間および接触温度でのセルロースの溶解に関する。本発明の別の態様は、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートまたはその混合物を含むセルロース溶液から製造される成形品に関する。本発明の別の態様は、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートまたはその混合物を含むセルロース溶液から製造されるセルロースの誘導体を含む組成物に関する。本発明の別の態様は、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートまたはその混合物を含むセルロース溶液から製造される、位置選択的に置換されたセルロースエステルを含む組成物に関する。本発明の更に別の態様において、本発明のセルロースエステルは、液晶ディスプレイ用の保護フィルムおよび補償フィルムとして使用される。
【図面の簡単な説明】
【0013】
【図1】図1は、トリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)中、12.5質量%のセルロースの溶解を示す。セルロース吸収は1000cm-1で測定した。
【図2】図2は、モデルでのセルロースwt%および実験の吸収値([TBMA]DMP中に溶解した10wt%セルロースについて)を示す。
【図3】図3は、[TBMA]DMP中に溶解したセルロースの、1825cm-1(無水酢酸)および1220cm-1(酢酸エステルおよび酢酸)での赤外バンドの吸収 対 エステル化(3eq無水酢酸)中の接触時間、のプロットを示す。
【図4】図4は、[BMIm]DMP中に溶解したセルロースの、1825cm-1(無水酢酸)および1220cm-1(酢酸エステルおよび酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを示す。
【図5】図5は、[BMIm]DMP中に溶解したセルロースの、1825cm-1(無水酢酸)および1220cm-1(酢酸エステルおよび酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを、MSAの存在下および不存在下で比較する。
【図6】図6は、[TBMA]DMP中に溶解したセルロースの、1825cm-1(無水酢酸)および1724cm-1(酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを示す。
【図7】図7は、0.5eqのAc2Oの100℃での添加を含む接触時間を示す。x軸は、各反応が同じ無水物添加時点(15分)で始まるようにシフトさせた。
【図8】図8は、2.5eqのAc2Oの80℃での添加を含む接触時間を示す。x軸は、各反応が同じ無水物添加時点(72分)で始まるようにシフトさせた。
【図9】図9は、DS 対 2.5eqのAc2Oの添加を含む時間(酸の不存在下および存在下、80℃にて)、のプロットを示す。
【図10】図10は、[TBMA]DMP中に溶解したセルロースの、1815cm-1(無水物)、1732cm-1(酸)、および1226cm-1(エステル+酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを示す。
【図11】図11は、[TBMA]DMP中に溶解したセルロースの、1815cm-1および1732cm-1での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを、Ac2O/Pr2OまたはAc2O/Pr2O+MSAを100℃にて添加する場合で比較する。
【図12】図12は、[TBMA]DMP中に溶解したセルロースの、1815cm-1および1732cm-1での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを、Ac2O/Pr2OまたはAc2O/Pr2O+MSAを60℃にて添加する場合で比較する。
【図13】図13は、DS 対 1.0eqのAc2Oおよび1.0eqのPr2O(MSAの不存在下および存在下、60℃にて)の添加を含む時間、のプロットを示す。
【図14】図14は、75/25wt/wt[TBMA]DMP/DMF混合物中に溶解したセルロースの、1825cm-1(無水酢酸)および1724cm-1(酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを示す。
【図15】図15は、本発明の位置選択的に置換されたセルロースエステルについてのRthと総置換度との関係を示す。
【発明を実施するための形態】
【0014】
発明の詳細な説明
本発明は、本発明の以下の詳細な説明およびその中で与えられる例を参照して、より容易に理解できる。本発明は、記載される特定の方法、配合、および条件に限定されず、従って変更できることを理解すべきである。本明細書で用いる述語は本発明の特定の側面の説明の目的のみであり、限定を意図しないこともまた理解すべきである。
【0015】
本明細書および特許請求の範囲において、多くの用語を参照し、これらは以下の意味を有すると定義するものとする。
【0016】
単数形“a”,“an”および“the”は明示の特記がない限り複数の指示対象を包含する。
【0017】
値は「約」または「ほぼ」与えられる数値、とし表すことができる。同様に、範囲は本明細書で「約」1つの特定値から、および/または「約」もしくは別の特定値までとして表すことができる。このような範囲を表す場合、別の側面は、1つの特定値から、および/または他の特定値までを包含する。同様に、値が近似として表される場合、先行詞「約」の使用により、特定値が別の側面を形成することが理解されよう。
【0018】
本発明の一態様は、1種以上のテトラアルキルアンモニウムアルキルホスフェート中、セルロースを溶解させるのに十分な接触時間および接触温度での、セルロースの溶解に関する。本発明は、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上の非プロトン性溶媒を含む混合物中、セルロースを溶解させるのに十分な接触時間および接触温度での、セルロースの溶解に更に関する。本発明は、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上のイオン液体(カチオンがイミダゾリウムであるもの)を含む混合物中、セルロースを溶解させるのに十分な接触時間および接触温度での、セルロースの溶解に更に関する。本発明は、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上の酸(酸はセルロースと組合せない)を含む混合物中、セルロースを溶解させるのに十分な接触時間および接触温度での、セルロースの溶解に更に関する。本発明の別の態様は、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートを含むセルロース溶液から製造される成形品に関する。
【0019】
本発明に好適なセルロースは、セルロースエステルの製造に好適であることが当該分野で公知の任意のセルロースであることができる。一態様において、セルロースは、針葉樹または広葉樹から木材パルプとして、または一年生植物(例えばコットンまたはコーン)から得ることができる。セルロースは、複数の無水グルコースモノマー単位を含むβ−1,4−結合ポリマーであることができる。本発明において用いるのに好適なセルロースは、一般的には、以下の構造:
【0020】
【化2】
【0021】
を含むことができる。
【0022】
セルロースは、α−セルロース量少なくとも90%、およびより好ましくはα−セルロース量少なくとも95%を有することができる。セルロースは、重合度(DP)少なくとも10、少なくとも250、少なくとも1,000、または少なくとも5,000を有することができる。本明細書で用いる用語「重合度」は、セルロースおよび/またはセルロースエステルを意味する場合、セルロースポリマー鎖当たりの無水グルコースモノマー単位の平均数を意味するものとする。更に、セルロースの重量平均分子量は、約1,500〜約850,000の範囲、約40,000〜約200,000の範囲、または55,000〜約160,000の範囲であることができる。加えて、本発明において使用するために好適なセルロースは、シート、ハンマーミルされたシート、繊維、または粉末の形状であることができる。一態様において、セルロースは、平均粒子サイズ約500マイクロメートル(「μm」)未満、約400μm未満、または300μm未満の粉末であることができる。
【0023】
本発明のために好適なテトラアルキルアンモニウムアルキルホスフェートは、構造:
【0024】
【化3】
【0025】
(式中、R1、R2、R3およびR4は独立にC1−C5の直鎖もしくは分岐鎖のアルキル基またはC2−C20のアルコキシ基であり、そしてR5およびR6は独立にヒドリド、C1−C5の直鎖もしくは分岐鎖のアルキル基、またはC2−C20のアルコキシ基である)
に対応する。本発明の一態様においては、R1がメチルまたはエチルであり;R2、R3およびR4が独立にメチル、エチル、プロピル、ブチル、イソブチル、ペンチル、エタノール、エトキシエタノールであり;R1、R2、R3およびR4が同一ではなく;そしてR5およびR6がメチル、エチル、プロピルまたはブチルである。別の態様においては、R1がメチルであり;R2、R3およびR4がプロピルまたはブチルであり;そしてR5およびR6がメチルまたはエチルである。好適なテトラアルキルアンモニウムアルキルホスフェートの例としては、これらに限定するものではないが、トリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)、トリブチルエチルアンモニウムジエチルホスフェート([TBEA]DEP)、トリプロピルメチルアンモニウムジメチルホスフェート([TPMA]DMP)、トリプロピルエチルアンモニウムジエチルホスフェート([TPEA]DEP)が挙げられる。潤滑剤および帯電防止剤として使用するためのこれらのテトラアルキルアンモニウムアルキルホスフェートの調製は、第US2,563,506号(1951)(本明細書の記載と矛盾しない限りにおいて参照により本明細書に組入れる)に開示されている。
【0026】
一態様において、テトラアルキルアンモニウムアルキルホスフェートのカチオン:アニオンの比は、約1:1〜約1:10である。別の態様において、カチオン:アニオンの比は約1:1である。更に別の態様において、カチオン:アニオンの比は約1:2である。
【0027】
テトラアルキルアンモニウムアルキルホスフェートと混合する1種以上の共溶媒は、セルロースの溶液の調製において有用であることができる。共溶媒としては、これらに限定するものではないが、非プロトン性溶媒、プロトン性溶媒、酸、およびテトラアルキルアンモニウムアルキルホスフェート以外のイオン液体が挙げられる。例えば、非プロトン性溶媒は本発明において有用である。本発明において、非プロトン性溶媒は、酸素、窒素、または硫黄に結合する水素を含有しないものである。これは解離して誘電率約30超を有する可能性がある。更に、好適な非プロトン性溶媒は、セルロースの溶解中またはエステル化の間に塩基によって外れる可能性がある酸性プロトンを有さない。非プロトン性溶媒をテトラアルキルアンモニウムアルキルホスフェートとともに用いる場合、より高いセルロース濃度をより低いセルロース溶液粘度とともに実現でき、これは接触温度を低くできる。更に、セルロースエステル生成物は、しばしば、非プロトン性溶媒を用いる場合、テトラアルキルアンモニウムアルキルホスフェート単独においてよりも良好な溶解性を与える。これはしばしば、選択されたセルロースエステル組成物を製造する場合に重要である。好適な非プロトン性溶媒の例としては、ヘキサメチルホスホルアミド、N−メチルピロリドン、ニトロメタン、ジメチルホルムアミド、ジメチルアセトアミド、アセトニトリル、スルホラン、ジメチルスルホキシド等が挙げられる。一態様において、非プロトン性溶媒としては、これらに限定するものではないが、N−メチルピロリドン(NMP)およびジメチルホルムアミド(DMF)が挙げられる。非プロトン性溶媒は、共溶媒およびテトラアルキルアンモニウムアルキルホスフェートの総質量基準で約0.1〜約99質量%の範囲の量で存在できる。本発明の別の態様において、共溶媒の量は、共溶媒およびテトラアルキルアンモニウムアルキルホスフェートの総質量基準で約5〜約90質量%、または約10〜約25質量%である。
【0028】
特定のプロトン性溶媒もまた本発明において共溶媒として有用である。本発明の目的のために、プロトン性溶媒は、酸素、窒素、または硫黄に結合した水素を含有するものである。これは解離して誘電率約10未満を有する可能性がある。好適なプロトン性溶媒の例としては、脂肪族カルボン酸(例えば酢酸、プロピオン酸、酪酸、イソ酪酸)、およびアミン(例えばジエチルアミン、ブチルアミン、ジブチルアミン、プロピルアミン、ジプロピルアミン)が挙げられる。本発明の別の態様において、プロトン性溶媒としては、酢酸、プロピオン酸、酪酸およびこれらの組合せが挙げられる。プロトン性溶媒は、共溶媒およびテトラアルキルアンモニウムアルキルホスフェートの総質量基準で約0.1〜約25質量%の量で存在できる。別の態様において、共溶媒の量は、共溶媒およびテトラアルキルアンモニウムアルキルホスフェートの総質量基準で約1〜約15質量%、または約3〜約10質量%である。
【0029】
本発明の別の態様において、テトラアルキルアンモニウムアルキルホスフェート以外のイオン液体を共溶媒として用いることができる。イオン液体は、テトラアルキルアンモニウムアルキルホスフェート中でのセルロースの溶解を助ける能力を有する当該分野で公知の任意のものであることができる。このようなイオン液体の好適例は、米国特許出願、発明の名称“Cellulose Esters and Their Production In Carboxylated Ionic Liquids”(2008年2月13日出願、整理番号12/030387)、および米国特許出願、発明の名称“Cellulose Esters and Their Production in Halogenated Ionic Liquids”(2008年8月11日出願、整理番号12/189415)(これらの両者は、本明細書の記載と矛盾しない限りにおいて参照により本明細書に組入れる)に開示されている。
【0030】
本発明の一態様において、本発明における共溶媒として有用なイオン液体は、構造:
【0031】
【化4】
【0032】
に対応するアルキルまたはアルケニル置換されたイミダゾリウム塩である。式中、R1およびR3は独立にC1−C8アルキル基、C2−C8アルケニル基、またはC1−C8アルコキシアルキル基であり、そしてR2、R4およびR5は独立にヒドリド、C1−C8アルキル基、C1−C8アルケニル基、C1−C8アルコキシアルキル基、またはC1−C8アルコキシ基である。アニオン(X-)はクロリド、C1−C20の直鎖もしくは分岐鎖のカルボキシレートもしくは置換カルボキシレート、またはアルキルホスフェートである。カルボキシレートアニオンの例としては、ホルメート、アセテート、プロピオネート、ブチレート、バレレート、ヘキサノエート、ラクテート、オキサレート、またはクロロ−、ブロモ−、フルオロ−置換されたアセテート、プロピオネート、またはブチレート等が挙げられる。ジアルキルホスフェートの例としては、ジメチルホスフェート、ジエチルホスフェート、ジプロピルホスフェート、またはジブチルホスフェート等が挙げられる。セルロース溶解のためのこれらの種類のイミダゾリウム塩の例は、米国出願第20080194807号、第20080194808号、第20080194834号;Green Chemistry 2008,10,44−46;Green Chemistry 2007,9,233−242に見出すことができる。イミダゾリウム系イオン液体は、セルロースを溶解するのに用いる液体成分の総質量基準で約99質量%〜約1質量%の量で存在できる。別の態様において、イミダゾリウム系イオン液体の量は、セルロースを溶解するのに用いる液体の総質量基準で約75質量%〜約2質量%、または約20質量%〜約5質量%である。
【0033】
共溶媒としての1種以上のテトラアルキルアンモニウムアルキルホスフェートと混合できる酸は、セルロースの溶解中またはエステル化の間にセルロースと組合せないものである。好適な酸の例としては、これらに限定するものではないが、アルキルスルホン酸,例えばメタンスルホン酸およびアリールスルホン酸,例えばp−トルエンスルホン酸が挙げられる。酸は、酸およびテトラアルキルアンモニウムアルキルホスフェートならびに任意に他の混合物成分の総質量基準で約0.01〜約10質量%の量で存在できる。本発明の別の態様において、酸の量は、酸およびテトラアルキルアンモニウムアルキルホスフェートならびに任意に他の混合物成分の総質量基準で約0.1〜約7質量%、または約1〜約5質量%である。本態様の一側面において、酸はセルロースの分子量を顕著に変えない。
【0034】
本態様の別の側面において、少なくとも1種の酸を、1種以上のテトラアルキルアンモニウムアルキルホスフェート中にセルロースを溶解させた後に混合できる。本態様の別の側面において、セルロース溶解前に酸をテトラアルキルアンモニウムアルキルホスフェートと混合できる。別の態様において、酸を、アシル化試薬の一部とともに、混合物として添加できる。驚くべきことに、酸が反応速度を遅化でき、そして顕著な分子量低下を招来しないことを見出した。更に、セルロースエステル生成物の色が、酸を用いない場合と比べて改善される。従って、本態様の一側面において、酸は、酸が存在しない場合と比べてより遅い反応速度を招来できる。別の側面において、酸はセルロースの分子量を顕著に変えない。更に別の側面において、酸は、酸が存在しない場合と比べて改善されたセルロースエステル色を与える。厳密な反応速度変化、製品分子量、および製品色の改善は、多くの要因,例えば酸の選択、酸濃度、接触温度、および接触時間に左右される。
【0035】
本発明においてセルロースを溶解させてセルロース溶液を生成する際、接触温度および接触時間は、テトラアルキルアンモニウムアルキルホスフェート中でのセルロースの均一な混合物を得るのに十分なものである。本発明の一態様において、接触温度は約20℃〜約150℃、または約50℃〜約120℃である。本発明の一態様において、接触時間は約5分間〜約24時間、または約30分間〜約3時間である。当業者は、溶解の速度が、温度、およびどの程度良好にセルロースがテトラアルキルアンモニウムアルキルホスフェート中に分散するかに左右されることを理解するであろう。本発明のテトラアルキルアンモニウムアルキルホスフェート中に溶解できるセルロースの量は、セルロースを溶解させるのに用いる特定のテトラアルキルアンモニウムアルキルホスフェート、およびセルロースのDPに左右される。一態様において、セルロース溶液中のセルロースの濃度は、セルロース溶液の総質量基準で約1質量%〜約40質量%、または約7質量%〜約20質量%である。
【0036】
本発明の一態様において、少なくとも1つの成分がテトラアルキルアンモニウムアルキルホスフェートである液体が、H2O、窒素含有塩基、またはアルコール酸をセルロース溶解前に含まないことは必須ではない。従って、セルロース溶液は、セルロースおよびテトラアルキルアンモニウムアルキルホスフェート、ならびに水、窒素含有塩基およびアルコールからなる群から選択される少なくとも1種の成分を含むことができる。一態様において、テトラアルキルアンモニウムアルキルホスフェートは、セルロース溶液中に含有される液体の総質量基準で約20質量%未満の水、窒素含有塩基、および/またはアルコールを含有する。別の態様において、テトラアルキルアンモニウムアルキルホスフェートは、セルロース溶液中に含有される液体の総質量基準で約5質量%未満の水、塩基、および/もしくはアルコール、または約2質量%未満の水、塩基、および/もしくはアルコールを含有する。
【0037】
本発明の別の態様において、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートまたはその混合物を含むセルロース溶液から種々の成形品を製造できる。これらの成形物としては、粉末、フィルム、繊維、チューブ等が挙げられる。典型的には、セルロース溶液は、所望の形状に形成し、次いで直ちに非溶媒に接触させる。これにより、セルロースは再生または沈殿するが、テトラアルキルアンモニウムアルキルホスフェートと混和できる。このような非溶媒の例としては、これらに限定するものではないが、水およびアルコール,例えばメタノール、エタノール、n−プロパノール、イソプロパノール等が挙げられる。このプロセスの例として、セルロース溶液をダイ(これは特定形状を与える)経由で押出し、次いでセルロース溶液を非溶媒に接触させることによって繊維を形成することができる。電界紡糸の場合には、ナノファイバーを形成可能である。別の例は、表面上へ薄いフィルムをキャストし、次いで非溶媒に接触させることによって固体フィルムを形成することである。
【0038】
本発明の別の態様は、セルロースおよび少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートまたはその混合物を含むセルロース溶液から製造されるセルロース誘導体を含む組成物に関する。セルロース誘導体は、セルロースエステル、セルロースエーテル、または混合セルロースエステル−エーテルであることができる。
【0039】
セルロースエステルは、所望の置換度(DS)および重合度(DP)を有するセルロースエステルを生成するのに十分な接触温度および接触時間で、セルロース溶液を1種以上のC1−C20アシル化試薬に接触させることによって製造できる。よって、製造されるセルロースエステルは、一般的には以下の構造:
【0040】
【化5】
【0041】
(式中、R2、R3、R6は水素(但しR2、R3、R6は同時に水素ではない)、またはC1−C20の直鎖もしくは分岐鎖のアルキル基もしくはアリール基であってエステル結合を介してセルロースに結合しているものである)
を含む。
【0042】
本発明の方法によって製造されるセルロースエステルは、DS約0.1〜約3.5、約0.1〜約3.08、約0.1〜約3.0、約1.8〜約2.9、または約2.0〜約2.6を有する。本発明の方法によって製造されるセルロースエステルのDPは、少なくとも5、または少なくとも10となる。別の態様において、セルロースエステルのDPは、少なくとも50、または少なくとも100、または少なくとも250である。更に別の態様において、セルロースエステルのDPは、約5〜約1000、約10〜約250、または約10〜約50である。
【0043】
アシル化剤は、セルロースをアシル化してセルロースエステルを生成するための当該分野で公知の任意のものであることができる。本発明の一態様において、アシル化試薬は、1種以上のC1−C20の直鎖または分岐鎖のアルキルまたはアリールカルボン酸無水物、カルボン酸ハロゲン化物、ジケテン、またはアセト酢酸エステルである。カルボン酸無水物の例としては、これらに限定するものではないが、無水酢酸、プロピオン酸無水物、酪酸無水物、イソ酪酸無水物、吉草酸無水物、ヘキサン酸無水物、2−エチルヘキサン酸無水物、ノナン酸無水物、ラウリン酸無水物、パルミチン酸無水物、ステアリン酸無水物、安息香酸無水物、置換安息香酸無水物、無水フタル酸、およびイソフタル酸無水物が挙げられる。カルボン酸ハロゲン化物の例としては、これらに限定するものではないが、アセチルハライド、プロピオニルハライド、ブチリルハライド、ヘキサノイルハライド、2−エチルヘキサノイルハライド、ラウロイルハライド、パルミトイルハライド、ベンゾイルハライド、置換ベンゾイルハライド、およびステアロイルハライドが挙げられる。アセト酢酸エステルの例としては、これらに限定するものではないが、メチルアセトアセテート、エチルアセトアセテート、プロピルアセトアセテート、ブチルアセトアセテート、およびtert−ブチルアセトアセテートが挙げられる。本発明の一態様において、アシル化試薬は、無水酢酸、プロピオン酸無水物、酪酸無水物、2−エチルヘキサン酸無水物、ノナン酸無水物およびステアリン酸無水物からなる群から選択される、少なくとも1種のC2−C9の直鎖または分岐鎖のアルキルカルボン酸無水物であることができる。アシル化試薬は、セルロースがテトラアルキルアンモニウムアルキルホスフェート中に溶解した後に添加できる。所望される場合には、テトラアルキルアンモニウムアルキルホスフェート中にセルロースを溶解させる前に、アシル化試薬をテトラアルキルアンモニウムアルキルホスフェートに添加できる。別の態様において、テトラアルキルアンモニウムアルキルホスフェートおよびアシル化試薬を同時にセルロースに添加してセルロース溶液を生成できる。
【0044】
テトラアルキルアンモニウムアルキルホスフェート中に溶解したセルロースのエステル化において、接触温度は、所望のセルロースエステルを生成するのに十分なものである。一態様において、接触温度は、約20℃〜約140℃である。別の態様において、接触温度は約50℃〜約100℃、または約60℃〜約80℃である。
【0045】
テトラアルキルアンモニウムアルキルホスフェート中に溶解したセルロースのエステル化において、接触時間は所望のセルロースエステルを生成するのに十分なものである。本発明の一態様において、接触時間は約1分間〜約48時間である。別の態様において、接触時間は約10分間〜約24時間、または約30分間〜約5時間である。
【0046】
本発明においては、セルロースエステル化の間にセルロースホスフェートエステルが殆どまたは全く形成されないことを理解することが重要である。すなわち、テトラアルキルアンモニウムアルキルホスフェートはホスフェート供与体としては作用しない。これは、イオン液体,例えば1,3−ジアルキルイミダゾリウムカルボキシレート(セルロースエステルのアシル基の少なくとも1つがイオン液体によって供与される)とは対照的である。これは米国特許出願、発明の名称“Cellulose Esters and Their Production In Carboxylated Ionic Liquids”(2008年2月13日出願、および整理番号12/030387を有し、先に本明細書の記載と矛盾しない限りにおいて参照により本明細書に組入れている)に記載されている。すなわち、イオン液体はアシル化試薬として作用する。本発明のセルロースエステルは:
a)セルロースを少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートと接触させてセルロース溶液を形成すること;
b)少なくとも1種のセルロースエステルを含むアシル化セルロース溶液を生成するのに十分な接触温度および接触時間で、該セルロース溶液を少なくとも1種のアシル化試薬と接触させて物品を生成すること;
c)該アシル化セルロース溶液を少なくとも1種の非溶媒と接触させることによって該セルロースエステルを沈殿させて、沈殿したセルロースエステルとテトラアルキルアンモニウムアルキルホスフェートとを含むセルロースエステルスラリーを生成すること;
d)該沈殿したセルロースエステルの少なくとも一部をセルロースエステルスラリーから分離して、回収されたセルロースエステル、およびテトラアルキルアンモニウムアルキルホスフェートを含む沈殿液体を生成すること;
を含む方法によって製造できる。
【0047】
本発明の別の態様において、セルロースエステルの製造方法は、回収されたセルロースエステルを洗浄液体で洗浄して、洗浄されたセルロースエステルを生成することを更に含む。
【0048】
本発明の別の態様において、セルロースエステルの製造方法は、洗浄されたセルロースエステルを乾燥させて、乾燥されたセルロースエステル製品を生成することを更に含む。
【0049】
本発明の別の態様において、セルロースエステルの製造方法は、テトラアルキルアンモニウムアルキルホスフェートを沈殿液体から分離して、回収されたテトラアルキルアンモニウムアルキルホスフェートを生成することを更に含む。
【0050】
本発明の別の態様において、セルロースエステルの製造方法は、回収されたテトラアルキルアンモニウムアルキルホスフェートを再循環させてセルロースを溶解させてセルロース溶液を生成することを更に含む。
【0051】
本発明の方法において、1種以上のアシル化試薬をセルロース溶液と接触させる場合、添加するアシル化試薬の量およびアシル化試薬を添加する順序は、溶液粘度、製品品質、および相対置換度(RDS)等の因子に大きく影響する可能性がある。
【0052】
本発明の一側面において、1種以上のアシル化試薬は単独添加で添加する。この側面において、約0.1当量〜約20当量のアシル化試薬を、1つの添加時期の間に添加する。ここで当量は、無水グルコース1モル当たりのアシル化試薬のモル数である。別の態様において、約0.5当量〜約5当量のアシル化試薬を、1つの添加時期の間に添加する。最も好ましいのは、約1当量〜約3当量のアシル化試薬を1つの添加時期の間に添加することである。
【0053】
本発明の別の態様において、アシル化試薬の添加は段階的であり、これはアシル化試薬を引き続いて添加することを意味する。この側面において、合計で0.5当量〜約20当量のアシル化試薬をセルロース溶液に対して添加し、または合計で約1.5当量〜約5当量のアシル化試薬を添加する。しかし、段階的な添加において、約0.1当量〜約2当量のアシル化試薬を、1つの添加時期の間に添加し、残りのアシル化試薬を1つ以上の異なる添加時期に添加する。別の態様において、約0.5当量〜約1当量のアシル化試薬を1つの添加時期の間に添加し、残りのアシル化試薬を1つ以上の異なる添加時期に添加する。本発明のこの側面の1つの利点は、初期のアシル化試薬添加が接触混合物の溶液粘度の低減を招来することである。溶液粘度の低減により、1つの容器から別の容器へのより容易な移動が可能になり、また後続のアシル化試薬添加の間の接触温度の低減が可能になる。これは製品品質に影響する可能性がある。本発明のこの側面の別の利点は、2つ以上のアシル化試薬を、段階的な添加でいつ添加するかに関する。この側面において、1つのアシル化試薬を添加して第1段階の間の反応を可能にでき、次いで第2のアシル化試薬を添加して第2段階の間の反応を可能にでき、これにより、特異な置換パターンを有する新規なセルロースエステルが得られる。
【0054】
本発明において、1種以上のアシル化試薬を添加する場合、セルロースのC6位がC2およびC3よりも大幅に速くアシル化されたことを見出した。よって、C6/C3およびC6/C2のRDS比は、1よりも大きく、これは位置選択的に置換されたセルロースエステルの特徴である。位置選択性の程度は、以下の要因:アシル置換基の種類、接触温度、イオン液体相互作用、アシル化試薬の当量、添加の順序等の少なくとも1つに左右される。典型的には、アシル置換基における炭素原子数が大きくなると、セルロースのC6位はC2位およびC3位よりも優先的にアシル化される。加えて、接触温度をエステル化において低くするに従って、セルロースのC6位をC2位またはC3位よりも優先的にアシル化できる。前記したように、イオン液体の種類およびプロセスにおけるそのセルロースとの相互作用は、セルロースエステルの位置選択性に影響する可能性がある。例えば、カルボキシル化イオン液体を用いる場合、位置選択的に置換されたセルロースエステルは、RDSがC6>C2>C3の場合に生成する。本発明のテトラアルキルアンモニウムジアルキルホスフェートを用いる場合、位置選択的に置換されたセルロースエステルは、RDSがC6>C3>C2の場合に生成する。これは、セルロースエステルにおける置換基の位置選択的な配置が、従来のセルロースエステルとは異なる物理的特性を有する位置選択的に置換されたセルロースエステルをもたらすという点で顕著である。
【0055】
本発明の一態様において、セルロースとアシル化試薬との反応を防止する保護基を用いない。
【0056】
本発明の一態様において、C6/C3またはC6/C2についての環RDS比は少なくとも1.05である。別の態様において、C6/C3またはC6/C2についての環RDS比は少なくとも1.1である。本発明の別の態様は、C6/C3またはC6/C2についての環RDS比が少なくとも1.3である場合である。
【0057】
本発明の別の態様において、C6/C3またはC6/C2についての環RDS比に総DS[(C6/C3)*DSまたは(C6/C2)*DS]を乗じた積は少なくとも2.9である。別の態様において、C6/C3またはC6/C2についての環RDS比に総DSを乗じた積は少なくとも3.0である。別の態様において、C6/C3またはC6/C2についての環RDS比に総DSを乗じた積は少なくとも3.2である。
【0058】
本発明の別の態様において、C6/C3またはC6/C2についての環RDS比は少なくとも1.05であり、そしてC6/C3またはC6/C2についての環RDS比に総DSを乗じた積は少なくとも2.9である。別の態様において、C6/C3またはC6/C2についての環RDS比は少なくとも1.1であり、そしてC6/C3またはC6/C2についての環RDS比に総DSを乗じた積は少なくとも3.0である。更に別の態様において、C6/C3またはC6/C2についての環RDS比は少なくとも1.3であり、そしてC6/C3またはC6/C2についての環RDS比に総DSを乗じた積は少なくとも3.2である。
【0059】
前記したように、2以上のアシル置換基がより等量で存在する場合、各置換基のRDSを独立に評価するためにカルボニル炭素を纏めることが望ましい場合がある。よって、本発明の一態様において、C6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比は少なくとも1.3である。別の態様において、C6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比は少なくとも1.5である。別の態様において、C6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比は少なくとも1.7である。
【0060】
本発明の別の態様において、C6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比にアシル置換基のDS[(C6/C3)*DSアシルまたは(C6/C2)*DSアシル]を乗じた積は少なくとも2.3である。別の態様において、C6/C3またはC6/C2についてのカルボニルRDS比にアシル置換基のDSを乗じた積は少なくとも2.5である。別の態様において、C6/C3またはC6/C2についてのカルボニルRDS比にアシル置換基のDSを乗じた積は少なくとも2.7である。本発明の別の態様において、C6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比は少なくとも1.3であり、そしてC6/C3またはC6/C2についてのカルボニルRDS比にアシルDSを乗じた積は少なくとも2.3である。別の態様において、C6/C3またはC6/C2についてのカルボニルRDS比は少なくとも1.5であり、そしてC6/C3またはC6/C2についてのカルボニルRDS比にアシルDSを乗じた積は少なくとも2.5である。更に別の態様において、C6/C3またはC6/C2についてのカルボニルRDS比は少なくとも1.7であり、そしてC6/C3またはC6/C2についてのカルボニルRDS比にアシルDSを乗じた積は少なくとも2.7である。
【0061】
驚くべきことに、本発明の一態様において、アシル化試薬の段階的な添加は、アシル化試薬の混合添加において得られるものとは異なる相対置換度(RDS)を与えた。更に、本発明の段階的および混合の添加は、一般的にRDSがC6、C3およびC2で約1:1:1であるセルロースエステルを与える混合セルロースエステルを形成するための当該分野において公知の他の手段とは異なるRDSを与える。幾つかの場合、先行技術の方法は、RDSがC6でC2およびC3のものよりも小さいようなRDSを与える。
【0062】
本発明の方法によって製造されるセルロースエステルは、セルロースエステル溶液を非溶媒と接触させることによってセルロースエステルの少なくとも一部を沈殿させることによって沈殿させ、そしてこれにより、沈殿したセルロースエステルと沈殿液体とを含むスラリーを生成する。非溶媒の例としては、これらに限定するものではないが、C1−C8アルコール、水、またはこれらの混合物が挙げられる。一態様において、セルロースエステルの沈殿のためにメタノールを用いる。非溶媒の量は、セルロースエステルの少なくとも一部を沈殿させるのに十分な任意の量であることができる。一態様において、非溶媒の量は、アシル化セルロース溶液の総体積基準で少なくとも約10体積、少なくとも約5体積、または少なくとも0.5体積であることができる。セルロースエステルの沈殿のために必要な接触時間および接触温度は、所望レベルの沈殿を実現するのに必要な任意の時間または温度であることができる。本発明の態様において、沈殿のための接触時間は、約1〜約300分間、約10〜約200分間、または20〜100分間である。沈殿のための接触温度は、約0〜約120℃、約20〜約100℃、または25〜約50℃の範囲であることができる。
【0063】
沈殿に続き、本発明の方法によって製造されるセルロースエステルは、セルロースエスラリーから分離して、回収されたセルロースエステルおよび沈殿液体を生成できる。液体の少なくとも一部を固体のスラリーから分離するための当該分野において公知の任意の固/液分離方法を用いることができる。本発明において用いるのに好適な、好適な固/液分離方法の例としては、これらに限定するものではないが、遠心分離、濾過等が挙げられる。一態様において、セルロースエステルスラリーの沈殿液体の少なくとも50質量%、少なくとも70質量%、または少なくとも90質量%を除去できる。
【0064】
分離に続き、本発明の方法によって製造される、回収されたセルロースエステル、またはセルロースエステルの湿潤ケークを、湿潤ケークを洗浄するのに好適な当該分野で公知の任意の方法を用いて洗浄液体で洗浄できる。本発明において用いるのに好適な洗浄方法の例としては、これらに限定するものではないが、多段階向流洗浄が挙げられる。洗浄液体は、任意のセルロースエステル非溶媒であることができる。非溶媒は本開示において前記した。好適な非溶媒の例としては、これらに限定するものではないが、C1−C8アルコール、水、またはこれらの混合物が挙げられる。
【0065】
一態様において、回収されたセルロースエステルの洗浄は、任意の不所望の副生成物および/または着色体の少なくとも一部を、回収されたセルロースエステルから除去して、洗浄されたセルロースエステルを生成するような様式で実施できる。一態様において、洗浄液体として用いる非溶媒は、漂白剤を、洗浄液体の総質量基準で約0.001〜約50質量%の範囲、または0.01〜5質量%の範囲で含有できる。本発明において用いるのに好適な漂白剤の例としては、これらに限定するものではないが、亜塩素酸塩,例えば亜塩素酸ナトリウム(NaClO2);次亜ハロゲン化物,例えばNaOCl、NaOBr等;過酸化物,例えば過酸化水素等;過酸,例えば過酢酸等;金属,例えばFe、Mn、Cu、Cr等;亜硫酸ナトリウム類,例えば亜硫酸ナトリウム(Na2SO3)、メタ重亜硫酸ナトリウム(Na2S2O5)、亜硫酸水素ナトリウム(NaHSO3)等;過ホウ酸塩,例えば過ホウ酸ナトリウム(NaBO3・nH2O,n=1または4);二酸化塩素(ClO2);酸素;およびオゾンが挙げられる。一態様において、本発明において用いる漂白剤としては、過酸化水素、NaOCl、亜塩素酸ナトリウムおよび/または亜硫酸ナトリウムを挙げることができる。一態様において、副生成物および/または着色体の総量の少なくとも70%、または少なくとも90%を、回収されたセルロースエステルから除去する。
【0066】
洗浄に続き、本発明の方法によって洗浄されたセルロースエステルを、当該分野で公知の任意の乾燥方法によって乾燥して、洗浄されたセルロースエステル生成物の液体分の少なくとも一部を除去できる。乾燥設備の例としては、これらに限定するものではないが、ロータリードライヤー、スクリュー型ドライヤー、パドルドライヤー、および/またはジャケット付ドライヤーが挙げられる。一態様において、乾燥されたセルロースエステル生成物は、5質量%未満、3質量%未満、または1質量%未満の液体を含む。
【0067】
本発明の一態様において、本発明の方法によって調製したセルロースエステルスラリーから沈殿させたセルロースエステルの分離に続き、テトラアルキルアンモニウムアルキルホスフェートの少なくとも一部を、上記の沈殿液体から回収して、セルロース溶解において可能な再使用のための再循環されたテトラアルキルアンモニウムアルキルホスフェートを生成する。一態様において、1種以上のアルコール、水、および/または残留カルボン酸ならびに任意の共溶媒を、少なくとも1種の液/液分離方法で沈殿液体を処理することによって実質的に除去する。このような分離方法は、当該分野で公知の任意の液/液分離方法,例えば、フラッシュ蒸発および/または蒸留を含むことができる。一態様において、1種以上のアルコール、水、および/または残留カルボン酸ならびに任意の共溶媒の少なくとも80質量%、少なくとも90質量%、または少なくとも95質量%を沈殿液体から除去でき、これにより再循環されたテトラアルキルアンモニウムアルキルホスフェートを生成できる。
【0068】
別の態様において、セルロースエステル非溶媒がC1−C8アルコールまたはその混合物,例えばMeOHである場合、アルコールの一部のみを、液/液分離方法によって沈殿液体から除去して、分画された沈殿液体を生成する。一部分離後、分画された沈殿液体中のアルコールの総量は、約0.1〜約60質量%の範囲、約5〜約55質量%の範囲、または15〜50質量%の範囲である。分画された沈殿液体は、次いで、沈殿液体中に含有されるカルボン酸の少なくとも一部をアルキルエステル,例えばメチルエステルに転化するのに十分な温度、圧力および時間で、カルボン酸を分画された沈殿液体中に存在するアルコールと反応させることによって、処理できる。エステル化は、100℃〜180℃の範囲、または130℃〜160℃の範囲の温度で実施できる。加えて、エステル化の間の圧力は、約10〜約1,000ポンド毎平方インチゲージ(「psig」)の範囲、または100〜300psigの範囲であることができる。分画された沈殿液体は、エステル化の間の滞留時間約10〜約1,000分間の範囲、または120〜600分間の範囲を有することができる。一態様において、沈殿液体中のカルボン酸の少なくとも5モル%、少なくとも20モル%、または少なくとも50モル%を、上記エステル化の間にエステル化して、改質された分画された沈殿液体を生成できる。上記エステル化に続き、カルボキシレートエステル、1種以上のアルコール、および水(エステル化の間に生成するもの)を、改質された分画された沈殿液体を少なくとも1種の液/液分離方法で処理することによって実質的に除去できる。カルボン酸のアルキルエステルへの転化は、沈殿液体からのその除去を促進することを見出した。この態様において、1種以上のアルコール、カルボキシレートエステル、水、および/または残留カルボン酸ならびに任意に共溶媒の少なくとも80質量%、少なくとも90質量%、または少なくとも95質量%を沈殿混合物から除去でき、これにより、再循環するテトラアルキルアンモニウムアルキルホスフェート流を生成できる。この方法で除去されるカルボキシレートエステルの少なくとも一部を、CO挿入によって無水物に転化できる。
【0069】
本発明の方法によって製造されるセルロースエステルは種々の用途において有用である。当業者は、具体的な用途はセルロースエステルの具体的な種類(例えばアシル置換基の種類、DS、Mw、およびセルロースエステルコポリマーの種類等の要因として)がセルロースエステルの物理特性に顕著に影響することを理解するであろう[Prog.Polym.Sci.2001,26,1605−1688]。
【0070】
本発明の一態様において、セルロースエステルは熱可塑性用途において用いる。ここでセルロースエステルはフィルムまたは成形品を形成するために用いる。熱可塑性用途において用いるための好ましいセルロースエステルの例としては、セルロースアセテート、セルロースプロピオネート、セルロースブチレート、セルロースアセテートプロピオネート、セルロースアセテートブチレートまたはこれらの混合物が挙げられる。
【0071】
本発明の更に別の態様において、セルロースエステルはコーティング用途において用いる。コーティング用途の例としては、これらに限定するものではないが、自動車、木材、プラスチック、または金属コーティングが挙げられる。コーティング用途において用いるための好ましいセルロースエステルの例としては、セルロースアセテート、セルロースプロピオネート、セルロースブチレート、セルロースアセテートプロピオネート、セルロースアセテートブチレートまたはこれらの混合物が挙げられる。
【0072】
本発明の更に別の態様において、セルロースエステルはパーソナルケア用途において用いる。パーソナルケア用途において、セルロースエステルは一般的には適切な溶媒中に溶解または懸濁させる。そして、セルロースエステルは、皮膚または毛髪に適用される際に、構造化剤、送達剤、およびフィルム形成剤として働く。パーソナル用途において用いるための好ましいセルロースエステルの例としては、セルロースアセテート、セルロースプロピオネート、セルロースブチレート、セルロースアセテートプロピオネート、セルロースアセテートブチレート、セルロースヘキサノエート、セルロース2−エチルヘキサネート、セルロースラウレート、セルロースパルミテート、セルロースステアレートまたはこれらの混合物が挙げられる。
【0073】
本発明の更に別の態様において、セルロースエステルは、薬物送達用途において用いる。薬物送達用途において、セルロースエステルはフィルム形成剤として、例えばタブレットまたは粒子のコーティングにおいて働くことができる。セルロースエステルはまた、溶解性が乏しい薬物のアモルファス混合物を形成することによって薬物の溶解性および生物学的利用性を改善するのに用いることができる。セルロースエステルは、制御薬物送達において用いることができる。ここで薬物は、外部刺激,例えばpH変化に応答してセルロースエステルマトリクスから放出される。薬物送達用途において用いるための好ましいセルロースエステルの例としては、セルロースアセテート、セルロースプロピオネート、セルロースブチレート、セルロースアセテートプロピオネート、セルロースアセテートブチレート、セルロースアセテートフタレートまたはこれらの混合物が挙げられる。
【0074】
本発明の更に別の態様において、セルロースエステルは、フィルムの溶媒キャストを含む用途において用いる。これらの用途の例としては、写真用フィルムならびに液晶ディスプレイ用の保護フィルムおよび補償フィルムが挙げられる。溶媒キャストフィルム用途において用いるための好ましいセルロースエステルの例としては、セルローストリアセテート、セルロースアセテート、セルロースプロピオネート、およびセルロースアセテートプロピオネートが挙げられる。
【0075】
本発明の更に別の態様において、本発明のセルロースエステルは、フィルムの溶媒キャストを含む用途で使用できる。このような用途の例としては、写真用フィルム、液晶ディスプレイ用の保護フィルムおよび補償フィルムが挙げられる。溶媒キャストフィルム用途において用いるために好適なセルロースエステルの例としては、これらに限定するものではないが、セルローストリアセテート、セルロースアセテート、セルロースプロピオネート、およびセルロースアセテートプロピオネートが挙げられる。
【0076】
本発明の態様において、フィルムは、本発明のセルロースエステルを含んで製造され、液晶ディスプレイ(LCD)用の保護フィルムおよび補償フィルムとして用いる。これらのフィルムは、米国出願第2009/0096962号に記載されるような溶媒キャストによって、または米国出願第2009/0050842号に記載されるような溶融押出しによって、作製できる(これらの両者は、本明細書の記載と矛盾しない限りにおいて参照によりその全部を本明細書に組入れる)。
【0077】
保護フィルムとして用いる場合、フィルムは、典型的には、配向したヨウ素化ポリビニルアルコール(PVOH)偏光フィルムのいずれかの側にラミネートして、PVOH層をスクラッチまたは水分から保護する一方、構造的な剛直性もまた増大させる。補償フィルム(または板)として用いる場合、これらは偏光子積層体とラミネートされるか、そうでなければ偏光子と液晶層との間に含まれることができる。これらの補償フィルムは、LCDのコントラスト比、広視野角、および色シフト性能を改善できる。この重要な機能の理由は、LCDにおいて用いる典型的な組の交差した偏光子について、対角線に沿って顕著な光漏失(これは悪いコントラスト比を招来する)が、特に視野角が増大するに従って、存在することである。光学フィルムの種々の組合せを使用してこの光漏失を較正または「補償」できることが公知である。これらの補償フィルムは、特定の明確に定義されたレタデーション(または複屈折)値を有さなければならない。これは液晶セルの種類または用いるモードに応じて変わる。液晶セル自体もまた特定の程度の不所望の較正すべき光学レタデーションを与えるからである。
【0078】
補償フィルムは、一般的に複屈折にて定量する。これはすなわち屈折率nに関連する。セルロースエステルについて、屈折率は約1.46〜1.50である。非配向の等方性物質について、屈折率は、入射光波の偏光状態に関わらず同じになる。物質が配向するに従い、または異方性になるに従い、屈折率は物質の方向に依存するようになる。本発明の目的のために、nx、nyおよびnzという重要な3つの屈折率が存在し、これらはそれぞれ機械方向(MD)、横方向(TD)および厚み方向に対応する。物質がより異方性になる(例えば延伸によって)に従って、任意の2つの屈折率の間の差は増大する。屈折率のこの相違は、屈折率のその特定の組合せについての物質の複屈折という。選択する物質方向の多くの組合せが存在するため、対応して異なる値の複屈折が存在する。2つの最も一般的な複屈折パラメータは、平面複屈折(Δe=nx−ny と定義される)および厚み複屈折(Δth)(Δth=nz−(nx+ny)/2 と定義される)である。複屈折Δeは、MDとTDとの間の相対的な面内配向の指標であり、無次元である。これに対し、Δthは、平均面配向に対する厚み方向の配向の指標を与える。
【0079】
光学レタデーション(R)は、フィルムの厚み(d)による複屈折に関する:Re=Δed=(nx−ny)d; Rth=Δthd=[nz−(nx+ny)/2]。レタデーションは、2つの直交する光波の間の相対的な位相シフトの直接の指標であり、そして典型的にはナノメートル(nm)単位で報告する。Rthの定義は、何人かの著者によって、特に符号(±)に関して異なることに留意されたい。
【0080】
補償フィルムまたは補償板は、LCDディスプレイ装置を操作するモードに応じて多くの形状をとることができる。例えば、C−板補償フィルムはx−y平面において等方性であり、そして板は正(+C)または負(−C)であることができる。+C板の場合、nx=ny<nzである。−C板の場合、nx=ny>nzである。別の例は、A−板補償フィルムであり、これはy-z方向において等方性であり、そして繰り返すが、板は正(+A)または負(−A)であることができる。+A板の場合、nx>ny=nzである。−A板の場合、nx<ny=nzである。
【0081】
一般的に、脂肪族セルロースエステルは、Rthの値約0〜約−350nmを、フィルム厚み60μmで与える。観察されるRthに影響する最も重要な因子は、置換基の種類およヒドロキシルの置換度(DSOH)である。極低DSOHを有するセルロース混合エステルを用いて製造されるフィルム(Shelbyら,第US2009/0050842号における)は、Rth値約0〜約−50nmを有していた。セルロース混合エステルのDSOHを顕著に増大させることによって、Sheltonら(第US2009/0096962号)は、より大きい絶対値のRthである約−100〜約−350nmが得られることを示した。セルロースアセテートは、典型的には、Rth値約−40〜約−90nm(DSOHに応じて)を与える。
【0082】
本発明の一側面は、位置選択的に置換されたセルロースエステルを含む補償フィルムに関し、該補償フィルムは、Rth約−400〜約+100nmを有する。本発明の別の態様において、補償フィルムは、総DS約1.5〜約2.95の単一アシル置換基(第2のアシル置換基のDS≦0.2)を有する位置選択的に置換されたセルロースエステルを含んで製造され、そして補償フィルムはRth値約−400〜約+100nmを有する。
【0083】
本発明の一態様において、フィルムを製造するために用いる位置選択的に置換されたセルロースエステルは、セルロースアセテート、セルロースプロピオネート、およびセルロースブチレートからなる群から選択され、位置選択的に置換されたセルロースエステルは、総DS約1.6〜約2.9を有する。本発明の別の態様において、補償フィルムはRth値約−380〜約−110nmを有し、そして総DSが約1.7〜約2.5である位置選択的に置換されたセルロースプロピオネートで構成される。更に別の態様において、補償フィルムはRth値約−380〜約−110nmを有し、そして総DSが約1.7〜約2.5およびC6/C3またはC6/C2についての環RDS比が少なくとも1.05である位置選択的に置換されたセルロースプロピオネートで構成される。別の態様において、補償フィルムはRth値約−60〜約+100nmを有し、そして総DSが約2.6〜約2.9である位置選択的に置換されたセルロースプロピオネートで構成される。更に別の態様において、補償フィルムはRth値約−60〜約+100nmを有し、そして総DSが約2.6〜約2.9およびC6/C3またはC6/C2についての環RDS比が少なくとも1.05である位置選択的に置換されたセルロースプロピオネートで構成される。別の態様において、補償フィルムはRth値約0〜約+100nmを有し、そして総DSが約2.75〜約2.9である位置選択的に置換されたセルロースプロピオネートで構成される。更に別の態様において、補償フィルムはRth値約0〜約+100nmを有し、そして総DSが約2.75〜約2.9およびC6/C3またはC6/C2についての環RDS比が少なくとも1.05である位置選択的に置換されたセルロースプロピオネートで構成される。
【0084】
本発明の別の側面は、Rth範囲約−160〜約+270nmを有する補償フィルムであって、複数の2以上のアシル置換基の総DSが約1.5〜約3.0である位置選択的に置換されたセルロースエステルで構成された、補償フィルムに関する。本発明の一態様において、セルロースエステルは、セルロースアセテートプロピオネート、セルロースアセテートブチレート、セルロースベンゾエートプロピオネート、およびセルロースベンゾエートブチレートからなる群から選択でき;ここで位置選択的に置換されたセルロースエステルは総DS約2.0〜約3.0を有する。別の態様において、補償フィルムはRth値約−160〜約0nmを有し、そして総DSが約2.0〜約3.0、C6/C3またはC6/C2についての環RDS比が少なくとも1.05、およびC6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比が少なくとも約1.3である位置選択的に置換されたセルロースアセテートプロピオネートで構成される。別の態様において、補償フィルムはRth値約+100〜約+270nmを有し、そして総DSが約2.0〜約3.0、C6/C3またはC6/C2についての環RDS比が少なくとも1.05、およびC6/C3またはC6/C2についての少なくとも1つのアシル置換基についてのカルボニルRDS比が少なくとも約1.3である位置選択的に置換されたセルロースベンゾエートプロピオネートで構成される。別の態様において、補償フィルムはRth値約+100〜約+270nmを有し、そして総DSが約2.0〜約2.85、ベンゾエートDSが約0.75〜約0.90、C6/C3またはC6/C2についての環RDS比が少なくとも1.05であり、C6/C3またはC6/C2についてのプロピオネートについてのカルボニルRDS比が少なくとも約1.3である、位置選択的に置換されたセルロースベンゾエートプロピオネートで構成される。
【0085】
イオン液体、セルロースエステルおよびセルロース誘導体の製造におけるこれらの使用、セルロースエステルおよびセルロース誘導体を製造する方法においてイオン液体とともに共溶媒を使用すること、ならびにセルロースエステルの処理に関する更なる情報は、米国特許出願、発明の名称”Cellulose Esters and Their Production In Carboxylated Ionic Liquids”(2008年2月13日出願、および整理番号12/030387を有する)、およびその一部継続出願、発明の名称“Regioselectively Substituted Cellulose Esters Produced In A Carboxylated Ionic Liquid Process and Products Produced Therefrom”(2009年9月12日出願;米国特許出願、発明の名称”Cellulose Esters and Their Production in Halogenated Ionic Liquids”(2008年8月11日出願、および整理番号12/189415を有する)、およびその一部継続出願、発明の名称”Regioselectively Substituted Cellulose Esters Produced In A Halogenated Ionic Liquid Process and Products Produced Therefrom”(2009年9月12日出願);米国特許出願”Production of Ionic Liquids”(2008年2月13日出願、整理番号12/030,425を有する);および米国特許出願、発明の名称”Reformation of Ionic Liquids”(2008年2月13日出願、整理番号12/030,424を有する);米国特許出願、発明の名称”Treatment of Cellulose Esters”(2008年8月11日出願、整理番号12/189,421を有する);米国特許出願、発明の名称”Production of Cellulose Esters In the Presence of A Cosolvent”(2008年8月11日出願、整理番号12/189,753を有する);および米国仮出願、発明の名称”Regioselectively Substituted Cellulose Esters and Their Production in Ionic Liquids”(2008年8月13日出願、整理番号61/088,423を有する);に記載されており、これらの全ては本明細書の記載と矛盾しない範囲で参照により本明細書に組入れる。
【0086】
本発明は、その態様の以下の例によって更に説明できるが、これらの例は例示の目的のみで包含され、発明の範囲の限定を意図しないことが理解されよう。
【0087】
例
材料および方法
実験のテトラアルキルアンモニウムアルキルホスフェートは例において記載するように調製した。セルロースの重合度は、銅エチレンジアミン(Cuen)を溶媒として用いてキャピラリー粘度測定により評価した。溶解前に、セルロースを典型的には14〜18時間、50℃で5mmHgにて乾燥させた。
【0088】
本発明のセルロースエステルにおけるC6、C3およびC2での相対置換度(RDS)は、カーボン13NMRで、”Cellulose Derivatives”,ACS Symposium Series 688,1998,T.J.Heinze and W.G.Glasser,Editors(本明細書の記載と矛盾しない範囲で参照により本明細書に組入れる)に記載される一般的な方法に従って評価した。簡単に、カーボン13NMRデータは、100MHzで運転するJEOL NMRスペクトロメータまたは125MHzで運転するBruker NMRスペクトロメータを用いて得た。サンプル濃度は、100mg/mLのDMSO−d6であった。5mgのCr(OAcAc)3(100mgのサンプル当たり)を緩和剤として添加した。スペクトルを80℃にてパルス遅延1秒を用いて収集した。通常、15,000スキャンを各実験において収集した。ヒドロキシルのエステルへの転換は、ヒドロキシルを伴うカーボンの低磁場シフトおよびカルボニル官能基に対するカーボンガンマの高磁場シフトをもたらす。よって、C2およびC6の環炭素のRDSは、置換および非置換のC1炭素およびC6炭素の直接積分によって評価した。C3でのRDSは、C6およびC2のRDSの合計を総DSから減算することによって評価した。カルボニルRDSは、Macromolecules,1991,24,3050−3059(本明細書の記載と矛盾しない範囲で参照により本明細書に組入れる)に記載される一般的な配置を用いたカルボニル炭素の積算によって評価した。複数のアシル基を含有するセルロース混合エステルの場合、セルロースエステルを、完全に置換されたセルロース混合p−ニトロベンゾエートエステルにまず転化した。p−ニトロベンゾエートエステルの位置は、セルロース混合エステル中のヒドロキシルの位置を示す。
【0089】
色測定は、ASTM D1925の以下の一般的な手順に従って行った。色測定用のサンプルは、1.7gのセルロースエステルを41.1gのn−メチルピロリドン(NMP)中に溶解させることによって準備した。HunterLab ColorQuest XE測色計(透過モードで操作する20mm路長セルを有する)を測定用に用いた。測色計は、標準的なコンピュータ(EasyMatch QC Software(HunterLab)が動作するもの)にインターフェース接続した。値(L*;白色から黒色、a*;+赤から−緑;b*;+黄から−青)を、NMP(セルロースエステルなし)およびセルロースエステル/NMP溶液について得た。NMP溶媒とサンプル溶液との間の色差(E*)を次いで算出した(E*=[(Δa*)2+[(Δb*)2+[(ΔL*)2]0.5(式中、Δはサンプル溶液についての値から溶媒についての値を減じたものである))。値E*がゼロに近づくほど色が良好である。
【0090】
セルロースエステル中の硫黄およびリンの濃度は、誘導結合プラズマ発光分析(ICP)で評価した。サンプルは、濃HNO3中での消化、続いて超純水による希釈(内部標準添加後)によって調製した。最終マトリクスは、水中5質量%NHO3であった。次いでサンプルのリン(177.434nm)および硫黄(180.669nm)量を、Perkin Elmer 2100DV誘導結合プラズマ発光分析計を用いて分析した。これはNISTトレーサブル標準を用いて較正した。
【0091】
フィルムの溶媒キャストを、以下の一般的な手順に従って実施した:乾燥させたセルロースエステルおよび10wt%可塑剤を、CH2Cl2/メタノール(またはエタノール)の90/10wt%溶媒混合物に添加して、最終濃度5〜30wt%(セルロースエステル+可塑剤基準)を与えてセルロースエステル/溶媒混合物を生成した。セルロースエステル/溶媒混合物をシールし、ローラ上に置き、そして24時間混合して均一セルロースエステル溶液を得た。混合後、セルロースエステル溶液はガラス板上にドクターブレードを用いてキャストして、所望厚みを有するフィルムを得た。キャストは、相対湿度を50%に制御したドラフト中で行った。キャスト後、フィルムおよびガラスを1時間カバーパン下(溶媒蒸発の程度を最小化するため)で乾燥させた。この初期乾燥の後、フィルムをガラスから剥離し、強制換気オーブン中で10分間100℃にてアニールした。100℃でのアニール後、フィルムをより高温(120℃)で更に10分間アニールした。
【0092】
フィルム光学レタデーション測定は、J.A.Woollam M−2000V Spectroscopic Ellipsometerを用いて370〜1000nmのスペクトル範囲で行った。RetMeas(Retardation Measurement)プログラム(J.A.Woollam Co.,Inc.より)を用いて光学フィルムの面内(Re)および面外(Rth)のレタデーションを得た。値を589nmまたは633nmでフィルム厚み60μmにて報告する。
【0093】
例1. トリブチルメチルアンモニウムジメチルホスフェート[TBMA]DMPの調製
1Lの三口丸底フラスコ(機械撹拌を備える)に、400gの新しく蒸留したトリブチルアミンおよび302gのトリメチルホスフェート(1eq)を添加した。これは、2つの明澄な相(上部相がトリブチルアミンである)を与えた。次いでフラスコを油浴中に入れ、反応混合物をN2で空にした。油浴を120℃まで加熱し、接触混合物を49時間撹拌して、淡黄色の均一な混合物を得た。室温まで冷却した後、サンプルは固化して白色固体が得られた(収率>99%)。
【0094】
プロトンNMRによる分析は、出発物質のトリブチルメチルアンモニウムジメチルホスフェートへの>99%の転化を示した。熱重量分析(TGA)は、[TBMA]DMPの熱分解の開始が約240℃であることを示した。示差走査熱量計(DSC)による分析は、溶融(Tm)が、100℃までの20℃/分での1回目の加熱スキャンの間に55℃を中心とすることを示した。溶融から20℃/分で−100℃まで冷却し、再度100℃まで20℃/分で加熱した後、Tg(ガラス転移温度)はー52℃で観測され、2つのTc(結晶化温度)は14℃および23℃で観測され、2つのTmは54℃および66℃で観測された。
【0095】
この例は、融点100℃未満のテトラアルキルアンモニウムアルキルホスフェートの調製を示す。
【0096】
例2. トリブチルメチルアンモニウムジメチルホスフェート([TMBA]DMP)中のセルロースの溶解
セルロース溶解前に、52.26gのトリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)を三口100mL丸底フラスコ(機械撹拌およびN2/減圧入口を備える)に添加した。フラスコを80℃の油浴中に入れ、[TBMA]DMPを17時間、約0.9mmHgで撹拌した。[TBMA]DMPを70℃まで冷却し、iC10ダイヤモンド付赤外プローブ(Mettler−Toledo AutoChem,Inc.,Columbia,MD,USA)を挿入して吸収を測定した。
【0097】
激しく撹拌しながら、[TBMA]DMPに、7.46g(12.5wt%)のセルロース(DP約335)を添加した(5分添加)。セルロースは容易かつ迅速に[TBMA]DMP中に分散してセルロース溶液を生成した。ブリード弁の補助で減圧を適用し(約1mmHg)、油浴を100℃まで加熱した。セルロースおよび[TBMA]DMPが100℃に到達するまで(約60分間)に、混合物は粘稠、半透明のセルロース溶液であり、視認できる粒子はなかった。セルロース溶液を更に65分間100℃にて撹拌し、この時点でこれは明澄なセルロース溶液であった。
【0098】
図1は、12.5wt%セルロースの[TBMA]DMP中での溶解を示す。セルロース吸収は1000cm-1で測定した。セルロースの分散を確保してクランピングを回避するために70℃まで加熱しながら、セルロースを[TBMA]DMPに添加した。全てのセルロースが添加されるまでに、セルロースは溶解し始めた。次いで接触温度を100℃まで上げた。接触温度が増大するに従い、セルロース溶解の程度は顕著に増大し、セルロースおよび[TBMA]DMPが100℃に到達するまでに、セルロースは[TBMA]DMP中に本質的に溶解して均一なセルロース溶液を生成した。
【0099】
この例は、セルロースが迅速かつ容易に[TBMA]DMP中に相当濃度で溶解できることを示す。
【0100】
例3. テトラアルキルアンモニウムアルキルホスフェート中に溶解したセルロースの量の分析のためのモデルの開発
例2の一般的な手順に従い、種々の濃度のセルロース(2.5,5.0,7.5,10.0,12.5wt%)をトリブチルメチルアンモニウムジメチルホスフェート中に溶解させた。初期および最終の濃度を用い、種々の濃度での吸収を、部分最小自乗法を用いて、スペクトル領域860〜1320cm-1に亘って適合させた。加えて、1157cm-1におけるセルロース吸収バンドの二次導関数に対する線形最小自乗適合を行った。このバンドは、このスペクトル領域においてテトラアルキルアンモニウムアルキルホスフェートからの干渉が殆どまたは全く存在しないことに基づいて選択した。図2は、モデルwt%セルロース、およびトリブチルメチルアンモニウムジメチルホスフェート中に溶解した10wt%セルロースについての実験の吸収値を示す。示されるように、モデルwt%セルロースおよび実験の吸収は優れた一致を示した。
【0101】
この例は、in situでの赤外分光分析に基づくモデルを用いてテトラアルキルアンモニウムアルキルホスフェート中のセルロースの濃度を評価できることを示す。この種の分析は、セルロースの一部のみが特定のテトラアルキルアンモニウムアルキルホスフェート中に既定濃度で可溶である場合に特に有用である。
【0102】
例4. 種々のテトラアルキルアンモニウムアルキルホスフェート中でのセルロースの溶解の比較
以下の更なるテトラアルキルアンモニウムアルキルホスフェートを、例1の一般的な方法によって調製した:トリブチルメチルアンモニウムジメチルホスフェート[TBMA]DMP、トリペンチルメチルアンモニウムジメチルホスフェート([TPMA]DMP)、トリオクチルメチルアンモニウムジメチルホスフェート[TOMA]DMP、トリメチルエタノールアンモニウムジメチルホスフェート[TMEA]DMP、トリメチルエトキシエタノールアンモニウムジメチルホスフェート[TMEEA]DMP、およびトリメチルエチルアセテートアンモニウムジメチルホスフェート[TMEAA]DMP。例2の一般的な方法に従い、固定濃度のセルロース(7.5wt%、DP335)を用い、これらのテトラアルキルアンモニウムアルキルホスフェートのそれぞれにおけるセルロースの溶解性を評価した。溶解したセルロースの総量を、例3に記載する線形モデルを用いて評価した。結果を表1に纏める。
【0103】
【表1】
【0104】
この例は、アンモニウムカチオンに結合したアルキル基(同じアルキルホスフェートアニオンを有する)が、イオン液体中のセルロースの溶解性に対する極めて顕著な効果を有することを示す。例えば、メチルアンモニウムの場合、他の3つのアルキル基が、C4(トリブチル)、C5(トリペンチル)、C8(トリオクチル)の順で変化するに従い、溶解するセルロースの質量%はそれぞれ100,28,および0wt%である。トリメチルアンモニウムでは、エタノールからエトキシエタノールへの移動がセルロースの溶解性を損なう。エタノールのアセチル化の際、セルロースの溶解性は顕著に低下する。明確に、セルロースは特定のテトラアルキルアンモニウムアルキルホスフェート中での優れた溶解性を有する。注意深い実験によってのみ、セルロース溶解における特定のテトラアルキルアンモニウムアルキルホスフェートの有用性を評価できる。
【0105】
例5. トリブチルメチルアンモニウムジメチルホスフェートおよびジメチルスルホキシドの混合物中でのセルロースの溶解
三口100mL丸底フラスコ(機械撹拌を備え、N2/減圧入口およびiC10ダイヤモンド付赤外プローブ(Mettler−Toledo AutoChem,Inc.,Columbia,MD,USA)を有する)に、51.31gの[TBMA]DMPおよび17.10gのジメチルスルホキシド(DMSO)(25wt%)の混合物を添加した。室温で急速に撹拌しながら、7.60gのセルロース(10wt%、DP約335)を[TBMA]DMPおよびDMSOの溶液に添加した(6分間添加)。セルロース/[TBMA]DMP/DMSO混合物を更に4分間撹拌して、予熱した80℃油浴をフラスコまで上げる前にセルロースの良好な分散を確保した。油浴を上げてから10分後、全てのセルロースが溶解し、淡黄色、低粘度のセルロース溶液が得られた。
【0106】
この例は、より低いセルロース溶液粘度のための手段として、テトラアルキルアンモニウムアルキルホスフェート中にセルロースを溶解させる場合に共溶媒を使用できることを示す。
【0107】
例6. トリブチルメチルアンモニウムジメチルホスフェート中に溶解したセルロースの、触媒不存在下でのエステル化
例2の一般的な方法に従い、7.5wt%のセルロース溶液を調製した。セルロース溶液の温度を80℃に調整した後、3当量の無水酢酸(Ac2O)を滴下(6分間)してアシル化セルロース溶液を生成した。アシル化セルロース溶液のサンプルは、反応の過程で取り出し、セルロースエステルはメタノール中で沈殿させることによって単離してセルロースエステルスラリーを回収した。セルロースエステルスラリーを濾過して、回収されたセルロースエステルを生成した。各々の回収したセルロースエステルサンプルを、4つの200mL分量のメタノールで洗浄して、洗浄されたセルロースを生成し、次いで50℃、10mmHgで乾燥させ、乾燥されたセルロースエステル生成物を得た。
【0108】
図3は、1825cm-1(無水酢酸)および1220cm-1(酢酸エステルおよび酢酸)での赤外バンドの吸収 対 [TBMA]DMP中に溶解したセルロースのエステル化(3eq無水酢酸)中の接触時間のプロットを示す。図3に示すDS値は、プロトンNMR分光法によって評価し、接触時間の間に取り出したアシル化セルロース溶液サンプルに対応する。図3が示すように、反応の速度は極めて速かった。無水物添加の開始後、DSが2.52に到達するまで、23分間が必要であるのみであった。
【0109】
この例は、テトラアルキルアンモニウムアルキルホスフェート中に溶解したセルロースのエステル化において、触媒を添加して反応を促進させる必要が無いことを示す。
【0110】
例7. 1−ブチル−3−メチルイミダゾリウムジメチルホスフェート中に溶解したセルロースのエステル化(比較)
三口100mL丸底フラスコは、機械撹拌を備え、iC10ダイヤモンド付IRプローブ(Mettler−Toledo AutoChem,Inc.,Columbia,MD,USA)を有し、そしてN2/減圧入口を有していた。フラスコに、50.65gの1−ブチル−3−メチルイミダゾリウムジメチルホスフェート([BMIm]DMP)を添加した。室温で撹拌しながら、4.11g(7.5wt%)のセルロース(DP約335、6分間添加)を添加した。次いで、ブリード弁の補助で減圧(2mmHg)を適用し、80℃に予熱した油浴をフラスコまで上げた。油浴を上げてから6分後に、明澄なセルロース溶液が生成された。セルロース溶液を更に11分間撹拌した後、油浴を下ろし、そしてセルロース溶液を室温まで放冷した。
【0111】
9.57g(3.7eq)の無水酢酸を、10分間に亘って滴下添加して反応混合物を生成した。添加が完了した後、反応混合物を33分間撹拌した後、サンプルを取り出し、そしてセルロースエステルをメタノール中に沈殿させた(サンプル1)。この時点で、反応混合物中には色は形成されなかった。予熱した50℃の油浴をフラスコまで上げた。反応混合物を34分間撹拌した後サンプルを取り出し、セルロースエステルをメタノール中で沈殿させた(サンプル2)。反応混合物の色の変化は殆どなかった。油浴温度設定を80℃まで上げた。13分以内に、反応混合物の色が濃琥珀色になり、粘度が増大し始めた。更に8分後、反応混合物がゲル化した。撹拌を更に30分間継続した後、油浴を下ろした。反応混合物は黒色ゲルであった。ゲルを固化させるために、メタノールをフラスコに直接添加した(サンプル3)。濾過後、各サンプルを4つの200mL分量のメタノールで洗浄し、セルロースエステル固体を1晩50℃、5mmHgで乾燥させた。サンプル1(白色固体)およびサンプル2(褐色固体)はDMSOおよびNMPのような溶媒中で可溶であった。サンプル3(黒色固体)は評価した全溶媒中で不溶であった。サンプル2(DS=2.48)がアセトン中100mg/mLで可溶でなかったことに留意することが重要であり;ゲルが形成された。本発明においては、テトラアルキルアンモニウムアルキルホスフェート中に溶解したセルロースから調製されたセルロースアセテートは、これらがDS約2.45〜約2.55を有する場合、アセトン中100mg/mLで完全に可溶であることを見出した。
【0112】
図4は、[BMIm]DMP中に溶解したセルロースの、1825cm-1(無水酢酸)および1220cm-1(酢酸エステルおよび酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを示す。
【0113】
別の反応を、0.1eqMSAを触媒として用いたことを除いて先の反応と同一の方法で行った。図5に示すように、メチルスルホン酸(MSA)を含むことは、MSAが存在しない場合と比べたときの反応速度の変化をさせなかった。MSAを含まない反応の場合でのように、反応温度を80℃に上げた後、反応混合物粘度の増大が観察された。サンプルを迅速に取り出してMeOH中に沈殿させた。サンプリングの数分以内に、反応混合物はまたゲル化した。すなわち、MSAの存在はゲル化を抑止しなかった。
【0114】
これらの例は多くの重要な点を示す。[BMIm]DMP中でのセルロースの溶解は公知である(Green Chemistry 2008,10,44−46;Green Chemistry 2007,9,233−242)。しかし、この例は、[BMIm]DMP中に溶解したセルロースのエステル化が成功でないことを示す。[TBMA]DMP中に溶解したセルロースの80℃でのエステル化はゲル化を招来しない(例6)。得られるセルロースエステル生成物は、典型的には、DS範囲2.45〜2.55を有する白色固体であり、アセトン中で完全に可溶である。これに反し、[BMIm]DMP中に溶解したセルロースの80℃でのエステル化は、急速なゲル化を招来する。セルロースエステル生成物は常に高度に着色し、全ての溶媒中で不溶である。MSAを含むことは反応速度を変化させず、ゲル化の防止もしない。これは、MSAを含むことが反応速度を加速しゲル化を防止するという[BMIm]Cl中に溶解したセルロースのエステル化で観測されるもの(米国特許出願、発明の名称“Cellulose Esters and Their Production in Halogenated Ionic Liquids”(2008年8月11日出願、整理番号第12/189415号を有する)に記載される通り)に反する。
【0115】
例8. テトラアルキルアンモニウムアルキルホスフェート−強酸混合物中に溶解したセルロースのエステル化
セルロース溶解前に、53.46gのトリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)を三口100mL丸底フラスコ(機械撹拌およびN2/減圧入口を備える)に添加した。フラスコを80℃の油浴中に入れ、溶液を1晩減圧(16.5時間、約0.5mmHg)にて撹拌した。[TBMA]DMPを70℃まで冷却し、IRプローブ(Mettler−Toledo AutoChem,Inc.,Columbia,MD,USA)を挿入して吸収データを得た。
【0116】
激しく撹拌しながら、[TBMA]DMPに、5.94g(10wt%)の微結晶セルロースを添加した(5分添加)。セルロースは容易かつ迅速に[TBMA]DMP中に分散した。ブリード弁の補助で減圧を適用し(約1mmHg)、油浴を100℃まで加熱した。セルロースおよび[TBMA]DMPが98℃に到達するまで(約20分間)に、ほぼ全てのセルロースが溶解した。セルロースおよび[TBMA]DMPを更に100分間100℃にて撹拌し、この時点で混合物は明澄な淡黄色のセルロース溶液であった。
【0117】
セルロース溶液に、1.87g(0.5eq)の無水酢酸(Ac2O)を3分間かけて滴下添加した。セルロース溶液粘度は顕著に低下し、セルロース溶液色は若干濃くなった。この時点で、セルロース溶液を80℃まで冷却した後、9.35g(2.5eq)のAc2Oを9分間かけて滴下添加し、アシル化セルロース溶液を生成した。アシル化セルロース溶液のサンプルを種々の時間間隔で取り出し、セルロースエステルを200mLメタノール中での沈殿によって単離してセルロースエステルスラリーを生成した。沈殿したセルロースエステルを濾過によって単離して、回収されたセルロースエステルを生成した。各々の回収したセルロースエステルサンプルを4つの200mL分量のメタノールで洗浄して、洗浄されたセルロースエステルを生成し、次いで減圧下(50℃、約10mmHg)で乾燥させた。in situでのIRに基づき、反応は、2回目の無水物添加の終了から11分後にほぼ完了した。アシル化セルロース溶液の色がどのように変化するかを見るために、接触時間を更に延長した。反応が進行するに従い、アシル化セルロース溶液はより濃い琥珀色になり、反応の終了時にはほぼ茶色になった。
【0118】
図6は、1825cm-1(無水酢酸)および1724cm-1([TBMA]DMPとの相互作用によって高波数側にシフトした酢酸)での赤外バンドの吸収 対 [TBMA]DMP中に溶解したセルロースのエステル化の間の接触時間のプロットを示す。図6に示すDS値は、プロトンNMR分光法によって評価し、接触時間の間に取り出したサンプルに対応する。図6が示すように、100℃での反応の速度は極めて速い。Ac2Oが極めて急速に消費されたためAc2Oは添加中に観察されなかった。80℃にて、反応の速度は若干遅くなったが、なお極めて急速であった。2回目の無水物添加の開始後、DSが2.48に到達するまで、21分間が必要であるのみであった。
【0119】
沈殿、洗浄、および乾燥の後、サンプル1(DS=2.04)は白色固体であった。サンプル2(DS=2.48)は白色固体であった。サンプル3(DS=2.61)はオフホワイト固体であった。サンプル4(DS=2.71)は淡褐色固体であった。サンプル2(DS=2.48)はアセトン中、100mg/mLにて完全に可溶であった。
【0120】
沈殿液体を減圧中で濃縮し、8.3wt%の残存酢酸(HOAc)を含有する[TBMA]DMPが得られた。[TBMA]DMPのカチオン:アニオン比は1:1であり、アニオン交換がないことを示す。
【0121】
2つの追加の反応を、1wt%過塩素酸(HClO4)([TBMA]DMP基準で)または1wt%メタンスルホン酸(MSA)([TBMA]DMP基準で)を0.5eqAc2Oとともに混合物として添加したことを除いて上記の全く同じ手順に従って行なった。図7および8は、Ac2O、Ac2O+HClO4、またはAc2O+MSAを添加する場合の、[TBMA]DMP中に溶解したセルロースの、1724cm-1での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを比較する。図8に示すDS値は、プロトンNMR分光法によって評価し、接触時間の間に取り出したサンプルに対応する。図7は、0.5eqAc2Oの100℃での添加を含む接触時間を示す。各反応が無水物添加(15分)の同じ時点で開始するようにx軸をシフトさせた。全3つの場合で、100℃での反応速度は極めて速かった。Ac2Oが極めて急速に消費されたためAc2Oは添加中に観測されなかった。図8は、80℃での2.5eqAc2Oの添加を含む接触時間を示す。各反応が無水物添加(72分)の同じ時点で開始するようにx軸をシフトさせた。全3つの場合で、反応速度は、温度低下に起因して若干低下したが、なお極めて急速であった。すなわち、これらの酸を含むことは反応速度を加速しなかった。実際、各反応について得られるプロットの初期スロープ(72〜90分)およびDS値の比較は、MSAを含むことが反応速度を顕著に遅くすることを示した。これは図9にてより容易に見られ、DS 対 2.5eqAc2Oの添加(酸の不存在下および存在下、80℃にて)を含む時間のプロットを示す。更に、これらのサンプルのゲルパーミエーションクロマトグラフィ(GPC、表2)による分析は、これらの強酸を高濃度で含むことがセルロースエステル生成物の分子量に対して有した影響は無視できることを示した。典型的には、これらの酸をこれらの濃度で含むことは、セルロースエステル生成物の分子量を顕著に低下させる。
【0122】
【表2】
【0123】
極めて驚くべきことに、表2に記す通り、MSAを含むことは、反応溶液およびセルロースエステル生成物の色を顕著に改善した。HClO4(強い酸化剤)の場合、セルロースエステル生成物の色は、Ac2Oのみを含む(酸なし)反応について上記したものと極めて類似した。MSAの場合、アシル化セルロース溶液から取り出した全ての4つのサンプルは白色であった。Ac2O+MSAアシル化セルロース溶液から単離したサンプル4のセルロースエステルはNMP中に溶解し、セルロースエステル/NMP溶液はE*17.8を有し、一方、Ac2Oアシル化セルロース溶液から単離したサンプル4のセルロースエステルは、NMP中に溶解した際に、E*31.4を有した。すなわち、同一の接触条件下では、アシル化セルロース溶液中にMSAを含むことは、溶液色を約45%低下させた。
【0124】
Ac2Oアシル化セルロース溶液から単離したサンプル2、およびAc2O+MSAアシル化セルロース溶液からのサンプル3は、残存する硫黄およびリンについてICPで分析した。Ac2Oアシル化セルロース溶液から単離したサンプル2は、14.8ppmのSおよび12.9ppmのリンを含有することを見出した。Ac2O+MSAアシル化セルロース溶液からのサンプル3は、15.3ppmのSおよび24.5ppmのリンを含有することを見出した。このデータは、セルロースエステル生成物が、スルフェートまたはホスフェートの無機エステルを殆どまたは全く含有しないことを示す。
【0125】
この例は、本発明の多くの驚くべきそして重要な特徴を示す。テトラアルキルアンモニウムアルキルホスフェートをセルロースのための、および後続のセルロースのエステル化の間の溶媒として用いる場合、強酸を用いて反応の結果を変えることができる。これらの酸を含むことは、分子量に殆どまたは全く影響しないし、反応速度を加速させることもない。全く驚くべきことに、十分な濃度では、反応速度が低下する。テトラアルキルアンモニウムアルキルホスフェート中にこれらの酸を含むことは、セルロースのエステル化の間に得られる生成物の色を変化させまたは改善することができる。更に、少量の無水物をテトラアルキルアンモニウムアルキルホスフェート−セルロース溶液に添加することは、溶液粘度の顕著な低減を招来し、接触温度の低減を可能することに留意すべきである。
【0126】
例9. テトラアルキルアンモニウムアルキルホスフェート−強酸混合物中に溶解したセルロースからのセルロース混合エステルの調製
セルロース溶解前に、61.11gのトリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)を三口100mL丸底フラスコ(機械撹拌およびN2/減圧入口を備える)に添加した。フラスコを80℃の油浴中に入れ、[TBMA]DMPを1晩減圧(約16.5時間、約0.5mmHg)にて撹拌した。液体を70℃まで冷却し、IRプローブ(Mettler−Toledo AutoChem,Inc.,Columbia,MD,USA)を挿入した。
【0127】
激しく撹拌しながら、[TBMA]DMPに6.79g(10wt%)の微結晶セルロースを添加した(5分添加)。セルロースは容易かつ迅速に[TBMA]DMP中に分散した。油浴を100℃まで加熱した。セルロースおよび[TBMA]DMPが97℃に到達するまで(約35分間)に、ほぼ全てのセルロースが溶解した。セルロースおよび[TBMA]DMPを更に75分間100℃にて撹拌し、この時点で混合物は明澄な淡黄色のセルロース溶液であった。
【0128】
セルロース溶液に、2.14g(0.5eq)の無水酢酸(Ac2O)および2.73gのプロピオン酸無水物(Pr2O)の冷却された混合物を、3分間かけて滴下添加してアシル化セルロース溶液を生成した。アシル化セルロース溶液の粘度は顕著に低下し、アシル化セルロース溶液の色は若干濃くなった。この時点で、アシル化セルロース溶液を60℃まで冷却した後、4.28g(1eq)のAc2Oおよび5.45gのPr2Oの冷却された混合物を7分間かけて滴下添加した。アシル化セルロース溶液のサンプルを種々の時間間隔で取り出し、セルロースエステルを、200mLの75/25MeOH/H2O中での沈殿によって単離してセルロースエステルスラリーを生成した。セルロースエステルスラリーを濾過して、回収されたセルロースエステルおよび沈殿液体を生成した。各々の回収されたセルロースエステルサンプルを4つの200mL分量の75/25MeOH/H2O中で洗浄して、洗浄されたセルロースエステルを生成し、次いで減圧(50℃、約10mmHg)下で乾燥させて、乾燥されたセルロースエステル生成物を生成した。
【0129】
図10は、1815cm-1(無水物)、1732cm-1([TBMA]DMPとの相互作用によって高波数側にシフトした酸)および1226cm-1(エステル+酸)赤外バンドの吸収 対 [TBMA]DMP中に溶解したセルロースのエステル化の間の接触時間、のプロットを示す。図10に示すDS値は、プロトンNMR分光法によって評価し、接触時間の間に取り出したサンプルに対応した。図10が示すように、100℃での反応速度は十分速く、添加中に無水物は観測されなかった。60℃にて、反応速度は遅くなった。2回目の無水物添加の開始後、DS2.42に到達するのに142分間が必要であった。例8と比べ、2回目添加後の反応速度は、主に接触温度の相違に起因して、より遅かった。
【0130】
沈殿、洗浄、および乾燥の後、サンプル1(DS=1.36)、2(DS=1.78)および3(DS=2.08)は白色固体であった。サンプル4(DS=2.42)は淡黄色固体であった。
【0131】
追加の反応は、1wt%メタンスルホン酸(MSA)を0.5eqAc2O+0.5eqPr2Oとともに混合物として添加したことを除いて上記の全く同じ手順で行った。図11および12は、Ac2O/Pr2OまたはAc2O/Pr2O+MSAを添加した場合の、[TBMA]DMP中に溶解したセルロースの、1815cm-1および1732cm-1での赤外バンドについての吸収 対 エステル化の間の接触時間、のプロットを比較する。図11は、0.5eqAc2Oおよび0.5eqPr2Oの100℃での添加を含む接触時間を示す。各反応が無水物添加の同じ時点(30分)で開始するようにx軸をシフトさせた。両者の場合で、100℃での反応速度は速かった。Ac2O/Pr2O(MSAなし)の場合、無水物が極めて急速に消費されたため添加中に何も観測されなかった。しかし、Ac2O/Pr2O+MSAでは、小さい濃度の無水物が添加時間中に観測された。図12は、1eqAc2Oおよび1eqPr2Oの、MSAありおよびなしでの、60℃での添加を含む接触時間を示す。各反応が無水物添加の同じ時点(113分)で開始するようにx軸をシフトさせた。両者の場合で、反応速度は、より低い接触温度に起因して遅くなった。図12に基づき、MSAが接触混合物中に存在する場合、無水物消費速度がより低かったことが明らかである。更に、1732cm-1吸収の初期スロープ(115〜175分)の比較もまた、MSAを含むことが反応速度を遅くしたことを示す。これは、1.0eqAc2Oおよび1.0eqPr2Oの酸の不存在下および存在下での60℃での添加を含む時間の、DS 対 時間のプロットを示す図14から容易に分かる。更に、ゲルパーミエーションクロマトグラフィによるこれらのサンプルの分析(表3)は、これらの強酸を高濃度で含むことが生成物の分子量に与える影響は無視できることを示した。典型的な溶媒を用いるセルロースのエステル化において、これらの酸をこれらの濃度で含むことは、セルロースエステル生成物の分子量を顕著に低減する。
【0132】
【表3】
【0133】
加えて、MSAは最終セルロースエステル生成物の色を、MSAが存在しない場合と比べて改善した。Ac2O+MSAアシル化セルロース溶液から単離したサンプル5のセルロースエステルをNMP中に溶解させ、セルロースエステル/NMP溶液はE*14.3を有した。Ac2Oアシル化セルロース溶液から単離したサンプル4のセルロースエステルもまたNMP中に溶解させ、セルロースエステル/NMP溶液はE*17.8を有した。すなわち、より長い接触時間である135分間にも関わらず、アシル化セルロース溶液中にMSAが含まれることは、MSAが不存在である場合と比べてアシル化セルロース溶液における色を低減した。
【0134】
Ac2O/Pr2Oアシル化セルロース溶液から単離したサンプル4のセルロースエステル、およびAc2O/Pr2O+MSAアシル化セルロース溶液からのサンプル5のセルロースエステルは、残存する硫黄およびリンについてICPによって分析した。Ac2O/Pr2Oアシル化セルロース溶液から単離したサンプル4は、9.1ppmのSおよび121.9ppmのリンを含有することを見出した。Ac2O/Pr2O+MSAアシル化セルロース溶液からのサンプル5は、9.4ppmのSおよび67.8ppmのリンを含有することを見出した。これらのデータは、セルロースエステル生成物がスルフェートまたはホスフェートの無機エステルを殆どまたは全く含有しないことを示す。
【0135】
この例は、本発明の多くの驚くべきそして重要な特徴を示す。セルロースのための、および後続のセルロースのエステル化の間の溶媒としてテトラアルキルアンモニウムアルキルホスフェートを用いる場合、強酸を用いてアシル化セルロース溶液の結果を変えることができる。これらの酸を含むことは、分子量に殆どまたは全く影響しないし、反応速度を低下させる可能性もない。テトラアルキルアンモニウムアルキルホスフェート中にこれらの酸を含むことは、セルロースのエステル化の間に得られる生成物の色を変化させまたは改善することができる。更に、少量の無水物をセルロース溶液に添加することは、セルロース溶液粘度の顕著な低減を招来し、接触温度の低減を可能することに留意すべきである。最後に、この例で示すような混合エステルの調製において、アシル基の分布(位置選択性)は、アシル化試薬を添加する順序およびアシル化試薬を添加する際の濃度によって制御できることに留意すべきである。
【0136】
例10. テトラアルキルアンモニウムアルキルホスフェート−非プロトン性溶媒混合物中に溶解したセルロースからのセルロースエステルの調製
例5の一般的な手順に従って、セルロース(10wt%、DP約335)を、質量基準で75/25の[TBMA]DMP/ジメチルホルムアミドの混合物中に溶解させた。これにより薄黄色溶液が得られた。セルロース溶液の温度を50℃に調整した後、3当量のAc2Oを16分間かけて滴下添加して、アシル化セルロース溶液を生成した。Ac2Oの添加後、アシル化セルロース溶液を64分間50℃にて撹拌した後、接触温度を80℃まで上げた。アシル化セルロース溶液の色は接触時間を通じて薄黄色であった。アシル化セルロース溶液のサンプルを反応の過程で取り出し、メタノール中での沈殿によってセルロースエステルを単離してセルロースエステルスラリーを生成した。セルロースエステルスラリーを濾過して白色の回収されたセルロースエステルおよび沈殿液体を生成した。回収したセルロースエステルの各サンプルを、3つの200mL分量で洗浄して、洗浄されたセルロースエステルを生成し、次いで50℃、10mmHgにて乾燥させて、乾燥されたセルロースエステル生成物を生成した。
【0137】
図14は、[TBMA]DMP/DMF中に溶解したセルロースの、1825cm-1(無水酢酸)および1724cm-1(酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間のプロットを示す。図14に示すDS値は、プロトンNMR分光法によって評価し、接触時間の間に取り出したアシル化セルロース溶液サンプルに対応する。図14が示すように、50℃でも、反応速度は適切であった。無水物添加の開始後、DSが0.85に到達するまで、23分間が必要であるのみであった。接触温度を80℃まで上げる場合、反応速度が増大し、反応が終了したときにDS2.48をもたらした。アシル化セルロース溶液の色は、接触時間を通じて維持されたこと、およびアシル化セルロース溶液のサンプリングによって得られるセルロースエステルが白色であったことに留意することが重要である。
【0138】
2回目の反応は、例5で調製したセルロース溶液(10wt%セルロース,75/25[TBMA]DMP/DMSO中)を用いて同一の方法で行った。この場合において、50℃であっても、アシル化セルロース溶液の色は急速に濃くなり、そしてアシル化セルロース溶液の粘度は顕著に低下した。接触温度を80℃まで上げた場合、アシル化セルロース溶液の色は黒色になり、そして溶液粘度は極めて低くなった。セルロースエステルは、メタノールで沈殿させることによって単離してセルロースエステルスラリーを形成し、次いで濾過して、回収されたセルロースエステルを生成した。サンプル1および2のセルロースエステルは白色;サンプル3は濃茶色;そしてサンプル4は灰−黒の外観であった。表4は、[TBMA]DMP/DMSOアシル化セルロース溶液から得られるセルロースエステル生成物 対 [TBMA]DMP/DMFアシル化セルロース溶液からのセルロースエステル生成物、の分子量を比較する(表4)。[TBMA]DMP/DMSOアシル化セルロース溶液の場合、セルロースアセテートについてのMnおよびMwの両者は、[TBMA]DMP/DMFアシル化セルロース溶液からのセルロースアセテートと比べて顕著に低かった。理論に拘束されることを望まないが、変色およびセルロースエステル生成物の分解は、比較的酸性のプロトンのアルファからSO二重結合(DMFの場合は不存在)によって生じると考えられる。
【0139】
【表4】
【0140】
この例は、多様な非プロトン性溶媒を共溶媒としてテトラアルキルアンモニウムアルキルホスフェートとともにセルロースを溶解させるために使用できるが、選択された非プロトン性溶媒,例えばN,N−ジメチルホルムアミドまたはN−メチルピロリドンのみがセルロースエステル化の間の適切な共溶媒であることを示す。一般的に、副反応を招来する酸性プロトンを有さない誘電率約30超の任意の非プロトン性溶媒は、テトラアルキルアンモニウムアルキルホスフェートとの組合せで用いる場合、セルロースの溶解およびエステル化において用いるのに好適である。
【0141】
例11. 75/25トリブチルメチルアンモニウムジメチルホスフェート/N−メチルピロリドン中に溶解したセルロースからのセルロースアセテートプロピオネートの調製
例5の一般的な手順に従って、セルロース(10wt%、DP約335)を、質量基準で75/25の[TBMA]DMP/NMPの混合物中に、100℃にて10分間で、溶解させた。これにより薄黄色セルロース溶液が得られた。セルロース溶液に、100℃にて、3.3当量のPr2O(微量のAc2Oを含有した)を13分間かけて滴下添加して、アシル化セルロース溶液を生成した。添加終了から15分で、IR吸収値が平坦になり始め、そして吸収値は、DSが所望値近傍であることを示した。アシル化セルロース溶液からIRプローブを取り出し、アシル化セルロース溶液を直ちに300mLの75/25メタノール/H2O中に注ぎ入れる一方、Heidolphホモジナイザーで混合して、沈殿したセルロースエステルおよび沈殿液体を含むセルロースエステルスラリーを生成した。沈殿したセルロースエステルを濾過し、3×で200mL分量の75/25MeOH/H2Oで洗浄することによって単離して、洗浄されたセルロースエステルを生成した後、1晩10mmHg、50℃にて乾燥させ、これは11.9gの雪白色の乾燥されたセルロースエステル固体を与えた。1H NMRによる分析から、セルロースエステルはDS2.34(DSPr=2.30,DSAc=0.04)を有したことが明らかになった。定量カーボン13NMRによる分析は、乾燥されたセルロースエステル生成物が環RDSの:RDS C6=0.97、RDS C3=0.64、RDS C2=0.73を有して位置選択的に置換されたことを示した。セルロースエステル生成物は、DMSO、NMP、アセトン、および90/10 CH2Cl2/MeOH等の多様な溶媒中、100mg/1mLで完全に可溶であった。
【0142】
この例は、セルロースがテトラアルキルアンモニウム−非プロトン性溶媒混合物中に迅速に溶解でき、次いで高温でエステル化されて高品質のセルロースエステルを得ることができることを示す。
【0143】
例12. フィルムのキャストおよびフィルム光学測定
一連の本質的に6,3−および6,2−で位置選択的に置換されたセルロースエステル(1−3)を、例11の一般的な手順に従って調製した(高C6RDS)。市販(比較例4および6)セルロースエステル(Eastman Chemical Companyから入手可能)を、米国特許出願公開第2009/0096962号および米国特許出願公開第2009/0050842号に記載される一般的な手順によって調製した。比較例5は、米国特許出願公開第2005/0192434号に記載されるようにして調製した。例5におけるセルロースエステルは、本質的に2,3−で位置選択的に置換され、そしてこれがC6にて低RDSを有する一方、本発明のセルロースエステルがC6にて高RDSを有する点で、本発明の例とは異なっていた。環RDSはフィルムをキャストする前に各サンプルについて評価し、そしてフィルム光学特性を評価した。結果を表5に纏める。
【0144】
【表5】
【0145】
6,3−、6,2−セルロースプロピオネート(例3、DS=2.10、C6/C3=1.58、C6/C2=1.73、DS*C6/C3=3.3、DS*C6/C2=3.6)についてのRthの値を、2,3−セルロースプロピオネート(例5、DS=1.99、C6/C3=0.45、C6/C2=0.43、DS*C6/C3=0.90、DS*C6/C2=0.86)に対して比較し、6,3−、6,2−セルロースプロピオネートは大幅により負のRth値(−382nm 対 −80nm)を与えた(2つのセルロースエステルは同様のDS値を有していても)ことが明らかであった。同様に、Rth値は、例3について、例6(DS=1.93、C6/C3=0.79、C6/C2=0.85、DS*C6/C3=1.5、DS*C6/C2=1.6)に対して顕著により負であった。位置選択的に置換された6,3−、6,2−セルロースプロピオネートの総DSが増大した場合(例1および2)、Rthの値は急速かつ顕著に増大した。例えば、DS2.45(例2)において、Rthは−140nmに増大した。DSが若干、DS2.49(例2)まで増大する場合、Rthは更に−119nmまで増大した。図15が示すように、総DSが更に増大すると、Rthは正になった。この挙動は、市販のセルロースエステルで観測されるものと顕著に異なった(例4と比較)。
【0146】
この例は、セルロースエステルの置換パターンが面外レタデーションに顕著に影響する可能性があることを示す。具体的には、本発明の位置選択的に置換されたセルロースエステルは、異なる置換パターンを有するセルロースエステルを用いては到達できないレタデーション値を与えることができる。より低い総DSで、本発明のセルロースプロピオネートについてのRthは、従来のセルロースプロピオネートに対して大幅により負であった。より高い総DSで、本発明のセルロースプロピオネートについてのRthは、従来のセルロースプロピオネートに対してより小さく負またはより大きく正であった。
【0147】
例13. 位置選択的に置換されたセルロースアセテートプロピオネートからのセルロースアセテートプロピオネートベンゾエートの調製
例11で調製されたセルロースアセテートプロピオネート(5g)およびピリジン(50g)を300mL丸底フラスコ(機械撹拌および水凝縮器を備える)に入れた。混合物を室温で窒素雰囲気下で撹拌して明澄溶液を得た。次いで塩化ベンゾイル(12.8g)を添加漏斗経由で滴下添加した。添加の完了後(約1時間)、温度を70℃まで上げた。混合物を更に5時間撹拌し、次いで室温まで放冷した。得られた溶液をメタノール(800g)中に激しく撹拌しながら沈殿させ、濾過し、メタノールで繰り返し洗浄し、そして減圧下で乾燥させて繊維状生成物(4.7g)を得た。1HNMRによる分析から、DSPr=2.29、DSAc=0.04、DSBz=0.70が明らかになった。13CNMRによる分析から、初期サンプルの位置選択性が反応条件下で保存されたことが明らかになった。
【0148】
フィルムキャストおよびフィルム光学特性の測定のための一般的な手順に従い、セルロースアセテートプロピオネートベンゾエートが、面外レタデーション(Rth、633)+156nmをフィルム厚み43μmで(+218を60μmで)有することを見出した。
【0149】
この例は、脂肪族アシル基の位置選択的な配置が高いC6/C3比またはC6/C2比を有するセルロースエステルをもたらし、続いて後続の芳香族基のC2およびC3での配置が、大きい正の値のRthを有するセルロースエステルをもたらしたことを示した。この例は、これらの種類のセルロースエステルが、2段階プロセスによって、第1段階で達成した置換パターンを乱すことなく調製できることを示した。
【0150】
例14. 段階的な無水物添加を用いた、70/30トリブチルメチルアンモニウムジメチルホスフェート/N−メチルピロリドン中に溶解したセルロースからのセルロースベンゾエートプロピオネートの調製
例5の一般的な手順に従って、セルロース(10wt%、DP約335)を、質量基準で70/30の[TBMA]DMP/NMPの混合物中に100℃で溶解させた。これにより薄黄色セルロース溶液が得られた。セルロース溶液に、100℃にて、2.5当量のPr2Oを3分間かけて滴下添加して、アシル化セルロース溶液を生成した。Pr2O添加終了から10分後に、安息香酸無水物(4eq)を一度に溶融物として添加した(85℃で溶融した)。アシル化セルロース溶液を更に35分間撹拌し、この時点でIR吸収値は、DSが所望値近傍であることを示した。アシル化セルロース溶液からIRプローブを取り出し、アシル化セルロース溶液を直ちに300mLのメタノールに注ぎ入れる一方、Heidolphホモジナイザーで混合して、セルロースベンゾエートプロピオネートスラリーを生成した。セルロースベンゾエートプロピオネートを濾過し、8×で200mL分量のメタノールで洗浄することによって単離した後、1晩10mmHg、50℃にて乾燥させた。1H NMRによる分析から、セルロースベンゾエートプロピオネートはDS2.91(DSPr=2.58,DSBz=0.33)を有したことが明らかになった。定量カーボン13NMRによる分析は、生成物が環RDSの:RDS C6=1.00、RDS C3=0.91、RDS C2=1.00、およびベンゾエートカルボニルRDSの:RDS C6=0.04、RDS C3=0.12、RDS C2=0.17を有して位置選択的に置換されたことを示した。生成物は、DMSO、NMP、およびCH2Cl2等の多様な溶媒中で可溶であった。
【0151】
フィルムキャストおよびフィルム光学特性の測定のための一般的な基準に従って、セルロースベンゾエートプロピオネートは、面外レタデーション(Rth、589)+50.5nmをフィルム厚み27μmで(+112.2を60μmで)有することを見出した。
【0152】
この例は、脂肪族反応剤をまず添加し、続いて芳香族反応剤を添加する段階的な添加を採用することによって、脂肪族アシル基の位置選択的な配置(高いC6/C3比またはC6/C2比)および芳香族基のC2およびC3での導入がもたらされることを示した。これらの位置選択的に置換されたセルロースエステルから製造される補償フィルムは大きい正の値のRthを示した。
【技術分野】
【0001】
関連出願
この元の出願は、米国仮出願整理番号61/169,560、発明の名称“Tetraalkylammonium Alkylphosphates”(2009年4月15日出願)(参照により本明細書に組入れる)の利益を主張する。
【0002】
発明の分野
本発明はセルロース化学、詳細にはセルロース溶液、セルロースエステルの製造、およびそれから製造される製品の分野に関する。
【背景技術】
【0003】
発明の背景
セルロースは無水グルコースのβ−1,4−結合ポリマーである。セルロースは、典型的には高分子量の多分散ポリマーであり、水および実質的に全ての一般的な有機溶媒に不溶である。木材またはコットンの製品,例えばハウジングまたは布帛における非変性セルロースの使用は周知である。非変性セルロースはまた、種々の他の用途において、通常はフィルム(例えばセロハン(登録商標))として、繊維(例えばビスコースレーヨン)として、または粉末(例えば微結晶セルロース)として(医薬用途において使用される)、利用される。変性セルロース(例えばセルロースエステル)もまた広範な商業用途において広く利用されている[Prog.Polym.Sci.2001,26,1605−1688]。セルロースエステルは、一般的に、まずセルロースをセルローストリエステルに転化した後、セルローストリエステルを酸性水性媒体中で所望の置換度(DS,無水グルコースモノマー当たりの置換基の数)まで加水分解することによって調製する。これらの条件下でのセルローストリアセテートの加水分解により、ランダムコポリマーが得られ、これは、最終DSに応じて8つの異なるモノマーからなることができる[Macromolecules 1991,24,3050]。
【0004】
イオン液体中でのセルロースの溶解は公知である。当業者は、特定のイオン液体中、任意の既知温度にて溶解するセルロースの最大量は、初期セルロースの重合度(DP)および溶解プロセス中のセルロースの分解程度に左右されることを理解するであろう。
【0005】
最も広い意味において、イオン液体(IL)は単純にイオンのみを含有する任意の液体である。よって、溶融塩,例えばNaCl(温度800℃超で溶融する)はイオン液体と分類できる。実際的な観点から、イオン液体という用語は、ここでは、約100℃未満で溶融する有機塩に対して用いる。
【0006】
イオン液体のカチオンは構造的に多様であるが、これらは一般的には窒素またはリン(これらは4級アンモニウムまたはホスホニウムに転化されることができる)を含有する。一般的に、最も有用なイオン液体は、環構造の一部である窒素を含有する。これらのカチオンの例としては、ピリジニウム、ピリダジニウム、ピリミジニウム、ピラジニウム、イミダゾリウム、ピラゾリウム、オキサゾリウム、トリアゾリウム、チアゾリウム、ピペリジニウム、ピロリジニウム、キノリニウム、およびイソキノリニウムが挙げられる。
【0007】
【化1】
【0008】
イオン液体のアニオンもまた構造的に多様であることができる。アニオンは無機または有機のいずれであることもでき、そしてこれらは種々の媒体中でのイオン液体の溶解性に対する顕著な影響を有する。例えば、疎水性アニオン,例えばヘキサフルオロホスフェートまたはトリフルイミドを含有するイオン液体は、水中の極めて低い溶解性を有する。一方、親水性アニオン,例えばクロリドまたはアセテートを含むイオン液体は水中で完全に混和性である。
【0009】
イオン液体の名称は一般的には省略される。アルキルカチオンはしばしばアルキル置換基およびカチオン(1組の括弧内に与えられる)の記号、続いてアニオンの略号で命名される。表記しないが、カチオンは正電荷を有し、アニオンは負電荷を有することが理解される。例えば、[BMIm]OAcは、1−ブチル−3−メチルイミダゾリウムアセテートを意味し、そして[AMIm]Clは1−アリル−3−メチルイミダゾリウムクロリドを意味する。
【0010】
窒素が環状カチオンの一部である(例えばイミダゾリウム)場合、これらのイオン液体は典型的には、対応するアンモニウムまたはホスホニウムを含有するイオン液体と比べ、より低い融点を有し、より含水性が低く、そしてより安定である。セルロースの溶解およびエステル化の点で、イミダゾリウムをカチオンとして含有するイオン液体は一般的に好ましい。これらのイオン液体では、濃厚セルロース溶液(約20質量%,[EMIm]OAc中)を実現可能であり、これから種々のセルロースエステルを調製できる(第US6,824,599号、米国出願第20080194807号、米国出願第20080194808号、および米国出願第20080194834号)。
【0011】
最も一般的に利用される非環式アンモニウム系イオン液体は、テトラアルキルアンモニウムハライドである。イオン液体の分類は、セルロースの溶解およびエステル化において制限された利用性を有する。2つのテトラアルキルアンモニウムハライド,テトラエチルアンモニウムクロリド(第US4,597,798号)およびテトラブチルアンモニウムフルオリド水和物(Macromol.Biosci.2007,7,307−314)のみが、セルロースを顕著レベル(約6質量%セルロース)で可溶化させることが示されている。両者の場合で、かなりの濃度の共溶媒(30〜90質量%),例えばDMSOまたはDMFがセルロース可溶化に必要であった。イオン液体中の低濃度のセルロース、これらのイオン液体から水を除去することの困難性、これらのイオン液体の不安定性、更にこれらのイオン液体の腐食性および有毒性に起因して、これらのテトラアルキルアンモニウムハライドはセルロース誘導体の製造のための実際的な媒体ではない。
【発明の概要】
【課題を解決するための手段】
【0012】
発明の要約
本発明の一態様は、1種以上のテトラアルキルアンモニウムアルキルホスフェート中でのセルロースの溶解に関する。本発明は、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上の非プロトン性溶媒を含む混合物中、セルロースを溶解させるのに十分な接触時間および接触温度での、セルロースの溶解に更に関する。本発明は更に、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上のイオン液体であってカチオンがイミダゾリウムであるものを含む混合物中での、セルロースの溶解に関する。本発明は更に、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上の酸(ここで酸はセルロースと組合せない)を含む混合物中、セルロースを溶解させるのに十分な接触時間および接触温度でのセルロースの溶解に関する。本発明の別の態様は、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートまたはその混合物を含むセルロース溶液から製造される成形品に関する。本発明の別の態様は、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートまたはその混合物を含むセルロース溶液から製造されるセルロースの誘導体を含む組成物に関する。本発明の別の態様は、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートまたはその混合物を含むセルロース溶液から製造される、位置選択的に置換されたセルロースエステルを含む組成物に関する。本発明の更に別の態様において、本発明のセルロースエステルは、液晶ディスプレイ用の保護フィルムおよび補償フィルムとして使用される。
【図面の簡単な説明】
【0013】
【図1】図1は、トリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)中、12.5質量%のセルロースの溶解を示す。セルロース吸収は1000cm-1で測定した。
【図2】図2は、モデルでのセルロースwt%および実験の吸収値([TBMA]DMP中に溶解した10wt%セルロースについて)を示す。
【図3】図3は、[TBMA]DMP中に溶解したセルロースの、1825cm-1(無水酢酸)および1220cm-1(酢酸エステルおよび酢酸)での赤外バンドの吸収 対 エステル化(3eq無水酢酸)中の接触時間、のプロットを示す。
【図4】図4は、[BMIm]DMP中に溶解したセルロースの、1825cm-1(無水酢酸)および1220cm-1(酢酸エステルおよび酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを示す。
【図5】図5は、[BMIm]DMP中に溶解したセルロースの、1825cm-1(無水酢酸)および1220cm-1(酢酸エステルおよび酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを、MSAの存在下および不存在下で比較する。
【図6】図6は、[TBMA]DMP中に溶解したセルロースの、1825cm-1(無水酢酸)および1724cm-1(酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを示す。
【図7】図7は、0.5eqのAc2Oの100℃での添加を含む接触時間を示す。x軸は、各反応が同じ無水物添加時点(15分)で始まるようにシフトさせた。
【図8】図8は、2.5eqのAc2Oの80℃での添加を含む接触時間を示す。x軸は、各反応が同じ無水物添加時点(72分)で始まるようにシフトさせた。
【図9】図9は、DS 対 2.5eqのAc2Oの添加を含む時間(酸の不存在下および存在下、80℃にて)、のプロットを示す。
【図10】図10は、[TBMA]DMP中に溶解したセルロースの、1815cm-1(無水物)、1732cm-1(酸)、および1226cm-1(エステル+酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを示す。
【図11】図11は、[TBMA]DMP中に溶解したセルロースの、1815cm-1および1732cm-1での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを、Ac2O/Pr2OまたはAc2O/Pr2O+MSAを100℃にて添加する場合で比較する。
【図12】図12は、[TBMA]DMP中に溶解したセルロースの、1815cm-1および1732cm-1での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを、Ac2O/Pr2OまたはAc2O/Pr2O+MSAを60℃にて添加する場合で比較する。
【図13】図13は、DS 対 1.0eqのAc2Oおよび1.0eqのPr2O(MSAの不存在下および存在下、60℃にて)の添加を含む時間、のプロットを示す。
【図14】図14は、75/25wt/wt[TBMA]DMP/DMF混合物中に溶解したセルロースの、1825cm-1(無水酢酸)および1724cm-1(酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを示す。
【図15】図15は、本発明の位置選択的に置換されたセルロースエステルについてのRthと総置換度との関係を示す。
【発明を実施するための形態】
【0014】
発明の詳細な説明
本発明は、本発明の以下の詳細な説明およびその中で与えられる例を参照して、より容易に理解できる。本発明は、記載される特定の方法、配合、および条件に限定されず、従って変更できることを理解すべきである。本明細書で用いる述語は本発明の特定の側面の説明の目的のみであり、限定を意図しないこともまた理解すべきである。
【0015】
本明細書および特許請求の範囲において、多くの用語を参照し、これらは以下の意味を有すると定義するものとする。
【0016】
単数形“a”,“an”および“the”は明示の特記がない限り複数の指示対象を包含する。
【0017】
値は「約」または「ほぼ」与えられる数値、とし表すことができる。同様に、範囲は本明細書で「約」1つの特定値から、および/または「約」もしくは別の特定値までとして表すことができる。このような範囲を表す場合、別の側面は、1つの特定値から、および/または他の特定値までを包含する。同様に、値が近似として表される場合、先行詞「約」の使用により、特定値が別の側面を形成することが理解されよう。
【0018】
本発明の一態様は、1種以上のテトラアルキルアンモニウムアルキルホスフェート中、セルロースを溶解させるのに十分な接触時間および接触温度での、セルロースの溶解に関する。本発明は、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上の非プロトン性溶媒を含む混合物中、セルロースを溶解させるのに十分な接触時間および接触温度での、セルロースの溶解に更に関する。本発明は、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上のイオン液体(カチオンがイミダゾリウムであるもの)を含む混合物中、セルロースを溶解させるのに十分な接触時間および接触温度での、セルロースの溶解に更に関する。本発明は、1種以上のテトラアルキルアンモニウムアルキルホスフェートおよび1種以上の酸(酸はセルロースと組合せない)を含む混合物中、セルロースを溶解させるのに十分な接触時間および接触温度での、セルロースの溶解に更に関する。本発明の別の態様は、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートを含むセルロース溶液から製造される成形品に関する。
【0019】
本発明に好適なセルロースは、セルロースエステルの製造に好適であることが当該分野で公知の任意のセルロースであることができる。一態様において、セルロースは、針葉樹または広葉樹から木材パルプとして、または一年生植物(例えばコットンまたはコーン)から得ることができる。セルロースは、複数の無水グルコースモノマー単位を含むβ−1,4−結合ポリマーであることができる。本発明において用いるのに好適なセルロースは、一般的には、以下の構造:
【0020】
【化2】
【0021】
を含むことができる。
【0022】
セルロースは、α−セルロース量少なくとも90%、およびより好ましくはα−セルロース量少なくとも95%を有することができる。セルロースは、重合度(DP)少なくとも10、少なくとも250、少なくとも1,000、または少なくとも5,000を有することができる。本明細書で用いる用語「重合度」は、セルロースおよび/またはセルロースエステルを意味する場合、セルロースポリマー鎖当たりの無水グルコースモノマー単位の平均数を意味するものとする。更に、セルロースの重量平均分子量は、約1,500〜約850,000の範囲、約40,000〜約200,000の範囲、または55,000〜約160,000の範囲であることができる。加えて、本発明において使用するために好適なセルロースは、シート、ハンマーミルされたシート、繊維、または粉末の形状であることができる。一態様において、セルロースは、平均粒子サイズ約500マイクロメートル(「μm」)未満、約400μm未満、または300μm未満の粉末であることができる。
【0023】
本発明のために好適なテトラアルキルアンモニウムアルキルホスフェートは、構造:
【0024】
【化3】
【0025】
(式中、R1、R2、R3およびR4は独立にC1−C5の直鎖もしくは分岐鎖のアルキル基またはC2−C20のアルコキシ基であり、そしてR5およびR6は独立にヒドリド、C1−C5の直鎖もしくは分岐鎖のアルキル基、またはC2−C20のアルコキシ基である)
に対応する。本発明の一態様においては、R1がメチルまたはエチルであり;R2、R3およびR4が独立にメチル、エチル、プロピル、ブチル、イソブチル、ペンチル、エタノール、エトキシエタノールであり;R1、R2、R3およびR4が同一ではなく;そしてR5およびR6がメチル、エチル、プロピルまたはブチルである。別の態様においては、R1がメチルであり;R2、R3およびR4がプロピルまたはブチルであり;そしてR5およびR6がメチルまたはエチルである。好適なテトラアルキルアンモニウムアルキルホスフェートの例としては、これらに限定するものではないが、トリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)、トリブチルエチルアンモニウムジエチルホスフェート([TBEA]DEP)、トリプロピルメチルアンモニウムジメチルホスフェート([TPMA]DMP)、トリプロピルエチルアンモニウムジエチルホスフェート([TPEA]DEP)が挙げられる。潤滑剤および帯電防止剤として使用するためのこれらのテトラアルキルアンモニウムアルキルホスフェートの調製は、第US2,563,506号(1951)(本明細書の記載と矛盾しない限りにおいて参照により本明細書に組入れる)に開示されている。
【0026】
一態様において、テトラアルキルアンモニウムアルキルホスフェートのカチオン:アニオンの比は、約1:1〜約1:10である。別の態様において、カチオン:アニオンの比は約1:1である。更に別の態様において、カチオン:アニオンの比は約1:2である。
【0027】
テトラアルキルアンモニウムアルキルホスフェートと混合する1種以上の共溶媒は、セルロースの溶液の調製において有用であることができる。共溶媒としては、これらに限定するものではないが、非プロトン性溶媒、プロトン性溶媒、酸、およびテトラアルキルアンモニウムアルキルホスフェート以外のイオン液体が挙げられる。例えば、非プロトン性溶媒は本発明において有用である。本発明において、非プロトン性溶媒は、酸素、窒素、または硫黄に結合する水素を含有しないものである。これは解離して誘電率約30超を有する可能性がある。更に、好適な非プロトン性溶媒は、セルロースの溶解中またはエステル化の間に塩基によって外れる可能性がある酸性プロトンを有さない。非プロトン性溶媒をテトラアルキルアンモニウムアルキルホスフェートとともに用いる場合、より高いセルロース濃度をより低いセルロース溶液粘度とともに実現でき、これは接触温度を低くできる。更に、セルロースエステル生成物は、しばしば、非プロトン性溶媒を用いる場合、テトラアルキルアンモニウムアルキルホスフェート単独においてよりも良好な溶解性を与える。これはしばしば、選択されたセルロースエステル組成物を製造する場合に重要である。好適な非プロトン性溶媒の例としては、ヘキサメチルホスホルアミド、N−メチルピロリドン、ニトロメタン、ジメチルホルムアミド、ジメチルアセトアミド、アセトニトリル、スルホラン、ジメチルスルホキシド等が挙げられる。一態様において、非プロトン性溶媒としては、これらに限定するものではないが、N−メチルピロリドン(NMP)およびジメチルホルムアミド(DMF)が挙げられる。非プロトン性溶媒は、共溶媒およびテトラアルキルアンモニウムアルキルホスフェートの総質量基準で約0.1〜約99質量%の範囲の量で存在できる。本発明の別の態様において、共溶媒の量は、共溶媒およびテトラアルキルアンモニウムアルキルホスフェートの総質量基準で約5〜約90質量%、または約10〜約25質量%である。
【0028】
特定のプロトン性溶媒もまた本発明において共溶媒として有用である。本発明の目的のために、プロトン性溶媒は、酸素、窒素、または硫黄に結合した水素を含有するものである。これは解離して誘電率約10未満を有する可能性がある。好適なプロトン性溶媒の例としては、脂肪族カルボン酸(例えば酢酸、プロピオン酸、酪酸、イソ酪酸)、およびアミン(例えばジエチルアミン、ブチルアミン、ジブチルアミン、プロピルアミン、ジプロピルアミン)が挙げられる。本発明の別の態様において、プロトン性溶媒としては、酢酸、プロピオン酸、酪酸およびこれらの組合せが挙げられる。プロトン性溶媒は、共溶媒およびテトラアルキルアンモニウムアルキルホスフェートの総質量基準で約0.1〜約25質量%の量で存在できる。別の態様において、共溶媒の量は、共溶媒およびテトラアルキルアンモニウムアルキルホスフェートの総質量基準で約1〜約15質量%、または約3〜約10質量%である。
【0029】
本発明の別の態様において、テトラアルキルアンモニウムアルキルホスフェート以外のイオン液体を共溶媒として用いることができる。イオン液体は、テトラアルキルアンモニウムアルキルホスフェート中でのセルロースの溶解を助ける能力を有する当該分野で公知の任意のものであることができる。このようなイオン液体の好適例は、米国特許出願、発明の名称“Cellulose Esters and Their Production In Carboxylated Ionic Liquids”(2008年2月13日出願、整理番号12/030387)、および米国特許出願、発明の名称“Cellulose Esters and Their Production in Halogenated Ionic Liquids”(2008年8月11日出願、整理番号12/189415)(これらの両者は、本明細書の記載と矛盾しない限りにおいて参照により本明細書に組入れる)に開示されている。
【0030】
本発明の一態様において、本発明における共溶媒として有用なイオン液体は、構造:
【0031】
【化4】
【0032】
に対応するアルキルまたはアルケニル置換されたイミダゾリウム塩である。式中、R1およびR3は独立にC1−C8アルキル基、C2−C8アルケニル基、またはC1−C8アルコキシアルキル基であり、そしてR2、R4およびR5は独立にヒドリド、C1−C8アルキル基、C1−C8アルケニル基、C1−C8アルコキシアルキル基、またはC1−C8アルコキシ基である。アニオン(X-)はクロリド、C1−C20の直鎖もしくは分岐鎖のカルボキシレートもしくは置換カルボキシレート、またはアルキルホスフェートである。カルボキシレートアニオンの例としては、ホルメート、アセテート、プロピオネート、ブチレート、バレレート、ヘキサノエート、ラクテート、オキサレート、またはクロロ−、ブロモ−、フルオロ−置換されたアセテート、プロピオネート、またはブチレート等が挙げられる。ジアルキルホスフェートの例としては、ジメチルホスフェート、ジエチルホスフェート、ジプロピルホスフェート、またはジブチルホスフェート等が挙げられる。セルロース溶解のためのこれらの種類のイミダゾリウム塩の例は、米国出願第20080194807号、第20080194808号、第20080194834号;Green Chemistry 2008,10,44−46;Green Chemistry 2007,9,233−242に見出すことができる。イミダゾリウム系イオン液体は、セルロースを溶解するのに用いる液体成分の総質量基準で約99質量%〜約1質量%の量で存在できる。別の態様において、イミダゾリウム系イオン液体の量は、セルロースを溶解するのに用いる液体の総質量基準で約75質量%〜約2質量%、または約20質量%〜約5質量%である。
【0033】
共溶媒としての1種以上のテトラアルキルアンモニウムアルキルホスフェートと混合できる酸は、セルロースの溶解中またはエステル化の間にセルロースと組合せないものである。好適な酸の例としては、これらに限定するものではないが、アルキルスルホン酸,例えばメタンスルホン酸およびアリールスルホン酸,例えばp−トルエンスルホン酸が挙げられる。酸は、酸およびテトラアルキルアンモニウムアルキルホスフェートならびに任意に他の混合物成分の総質量基準で約0.01〜約10質量%の量で存在できる。本発明の別の態様において、酸の量は、酸およびテトラアルキルアンモニウムアルキルホスフェートならびに任意に他の混合物成分の総質量基準で約0.1〜約7質量%、または約1〜約5質量%である。本態様の一側面において、酸はセルロースの分子量を顕著に変えない。
【0034】
本態様の別の側面において、少なくとも1種の酸を、1種以上のテトラアルキルアンモニウムアルキルホスフェート中にセルロースを溶解させた後に混合できる。本態様の別の側面において、セルロース溶解前に酸をテトラアルキルアンモニウムアルキルホスフェートと混合できる。別の態様において、酸を、アシル化試薬の一部とともに、混合物として添加できる。驚くべきことに、酸が反応速度を遅化でき、そして顕著な分子量低下を招来しないことを見出した。更に、セルロースエステル生成物の色が、酸を用いない場合と比べて改善される。従って、本態様の一側面において、酸は、酸が存在しない場合と比べてより遅い反応速度を招来できる。別の側面において、酸はセルロースの分子量を顕著に変えない。更に別の側面において、酸は、酸が存在しない場合と比べて改善されたセルロースエステル色を与える。厳密な反応速度変化、製品分子量、および製品色の改善は、多くの要因,例えば酸の選択、酸濃度、接触温度、および接触時間に左右される。
【0035】
本発明においてセルロースを溶解させてセルロース溶液を生成する際、接触温度および接触時間は、テトラアルキルアンモニウムアルキルホスフェート中でのセルロースの均一な混合物を得るのに十分なものである。本発明の一態様において、接触温度は約20℃〜約150℃、または約50℃〜約120℃である。本発明の一態様において、接触時間は約5分間〜約24時間、または約30分間〜約3時間である。当業者は、溶解の速度が、温度、およびどの程度良好にセルロースがテトラアルキルアンモニウムアルキルホスフェート中に分散するかに左右されることを理解するであろう。本発明のテトラアルキルアンモニウムアルキルホスフェート中に溶解できるセルロースの量は、セルロースを溶解させるのに用いる特定のテトラアルキルアンモニウムアルキルホスフェート、およびセルロースのDPに左右される。一態様において、セルロース溶液中のセルロースの濃度は、セルロース溶液の総質量基準で約1質量%〜約40質量%、または約7質量%〜約20質量%である。
【0036】
本発明の一態様において、少なくとも1つの成分がテトラアルキルアンモニウムアルキルホスフェートである液体が、H2O、窒素含有塩基、またはアルコール酸をセルロース溶解前に含まないことは必須ではない。従って、セルロース溶液は、セルロースおよびテトラアルキルアンモニウムアルキルホスフェート、ならびに水、窒素含有塩基およびアルコールからなる群から選択される少なくとも1種の成分を含むことができる。一態様において、テトラアルキルアンモニウムアルキルホスフェートは、セルロース溶液中に含有される液体の総質量基準で約20質量%未満の水、窒素含有塩基、および/またはアルコールを含有する。別の態様において、テトラアルキルアンモニウムアルキルホスフェートは、セルロース溶液中に含有される液体の総質量基準で約5質量%未満の水、塩基、および/もしくはアルコール、または約2質量%未満の水、塩基、および/もしくはアルコールを含有する。
【0037】
本発明の別の態様において、少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートまたはその混合物を含むセルロース溶液から種々の成形品を製造できる。これらの成形物としては、粉末、フィルム、繊維、チューブ等が挙げられる。典型的には、セルロース溶液は、所望の形状に形成し、次いで直ちに非溶媒に接触させる。これにより、セルロースは再生または沈殿するが、テトラアルキルアンモニウムアルキルホスフェートと混和できる。このような非溶媒の例としては、これらに限定するものではないが、水およびアルコール,例えばメタノール、エタノール、n−プロパノール、イソプロパノール等が挙げられる。このプロセスの例として、セルロース溶液をダイ(これは特定形状を与える)経由で押出し、次いでセルロース溶液を非溶媒に接触させることによって繊維を形成することができる。電界紡糸の場合には、ナノファイバーを形成可能である。別の例は、表面上へ薄いフィルムをキャストし、次いで非溶媒に接触させることによって固体フィルムを形成することである。
【0038】
本発明の別の態様は、セルロースおよび少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートまたはその混合物を含むセルロース溶液から製造されるセルロース誘導体を含む組成物に関する。セルロース誘導体は、セルロースエステル、セルロースエーテル、または混合セルロースエステル−エーテルであることができる。
【0039】
セルロースエステルは、所望の置換度(DS)および重合度(DP)を有するセルロースエステルを生成するのに十分な接触温度および接触時間で、セルロース溶液を1種以上のC1−C20アシル化試薬に接触させることによって製造できる。よって、製造されるセルロースエステルは、一般的には以下の構造:
【0040】
【化5】
【0041】
(式中、R2、R3、R6は水素(但しR2、R3、R6は同時に水素ではない)、またはC1−C20の直鎖もしくは分岐鎖のアルキル基もしくはアリール基であってエステル結合を介してセルロースに結合しているものである)
を含む。
【0042】
本発明の方法によって製造されるセルロースエステルは、DS約0.1〜約3.5、約0.1〜約3.08、約0.1〜約3.0、約1.8〜約2.9、または約2.0〜約2.6を有する。本発明の方法によって製造されるセルロースエステルのDPは、少なくとも5、または少なくとも10となる。別の態様において、セルロースエステルのDPは、少なくとも50、または少なくとも100、または少なくとも250である。更に別の態様において、セルロースエステルのDPは、約5〜約1000、約10〜約250、または約10〜約50である。
【0043】
アシル化剤は、セルロースをアシル化してセルロースエステルを生成するための当該分野で公知の任意のものであることができる。本発明の一態様において、アシル化試薬は、1種以上のC1−C20の直鎖または分岐鎖のアルキルまたはアリールカルボン酸無水物、カルボン酸ハロゲン化物、ジケテン、またはアセト酢酸エステルである。カルボン酸無水物の例としては、これらに限定するものではないが、無水酢酸、プロピオン酸無水物、酪酸無水物、イソ酪酸無水物、吉草酸無水物、ヘキサン酸無水物、2−エチルヘキサン酸無水物、ノナン酸無水物、ラウリン酸無水物、パルミチン酸無水物、ステアリン酸無水物、安息香酸無水物、置換安息香酸無水物、無水フタル酸、およびイソフタル酸無水物が挙げられる。カルボン酸ハロゲン化物の例としては、これらに限定するものではないが、アセチルハライド、プロピオニルハライド、ブチリルハライド、ヘキサノイルハライド、2−エチルヘキサノイルハライド、ラウロイルハライド、パルミトイルハライド、ベンゾイルハライド、置換ベンゾイルハライド、およびステアロイルハライドが挙げられる。アセト酢酸エステルの例としては、これらに限定するものではないが、メチルアセトアセテート、エチルアセトアセテート、プロピルアセトアセテート、ブチルアセトアセテート、およびtert−ブチルアセトアセテートが挙げられる。本発明の一態様において、アシル化試薬は、無水酢酸、プロピオン酸無水物、酪酸無水物、2−エチルヘキサン酸無水物、ノナン酸無水物およびステアリン酸無水物からなる群から選択される、少なくとも1種のC2−C9の直鎖または分岐鎖のアルキルカルボン酸無水物であることができる。アシル化試薬は、セルロースがテトラアルキルアンモニウムアルキルホスフェート中に溶解した後に添加できる。所望される場合には、テトラアルキルアンモニウムアルキルホスフェート中にセルロースを溶解させる前に、アシル化試薬をテトラアルキルアンモニウムアルキルホスフェートに添加できる。別の態様において、テトラアルキルアンモニウムアルキルホスフェートおよびアシル化試薬を同時にセルロースに添加してセルロース溶液を生成できる。
【0044】
テトラアルキルアンモニウムアルキルホスフェート中に溶解したセルロースのエステル化において、接触温度は、所望のセルロースエステルを生成するのに十分なものである。一態様において、接触温度は、約20℃〜約140℃である。別の態様において、接触温度は約50℃〜約100℃、または約60℃〜約80℃である。
【0045】
テトラアルキルアンモニウムアルキルホスフェート中に溶解したセルロースのエステル化において、接触時間は所望のセルロースエステルを生成するのに十分なものである。本発明の一態様において、接触時間は約1分間〜約48時間である。別の態様において、接触時間は約10分間〜約24時間、または約30分間〜約5時間である。
【0046】
本発明においては、セルロースエステル化の間にセルロースホスフェートエステルが殆どまたは全く形成されないことを理解することが重要である。すなわち、テトラアルキルアンモニウムアルキルホスフェートはホスフェート供与体としては作用しない。これは、イオン液体,例えば1,3−ジアルキルイミダゾリウムカルボキシレート(セルロースエステルのアシル基の少なくとも1つがイオン液体によって供与される)とは対照的である。これは米国特許出願、発明の名称“Cellulose Esters and Their Production In Carboxylated Ionic Liquids”(2008年2月13日出願、および整理番号12/030387を有し、先に本明細書の記載と矛盾しない限りにおいて参照により本明細書に組入れている)に記載されている。すなわち、イオン液体はアシル化試薬として作用する。本発明のセルロースエステルは:
a)セルロースを少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートと接触させてセルロース溶液を形成すること;
b)少なくとも1種のセルロースエステルを含むアシル化セルロース溶液を生成するのに十分な接触温度および接触時間で、該セルロース溶液を少なくとも1種のアシル化試薬と接触させて物品を生成すること;
c)該アシル化セルロース溶液を少なくとも1種の非溶媒と接触させることによって該セルロースエステルを沈殿させて、沈殿したセルロースエステルとテトラアルキルアンモニウムアルキルホスフェートとを含むセルロースエステルスラリーを生成すること;
d)該沈殿したセルロースエステルの少なくとも一部をセルロースエステルスラリーから分離して、回収されたセルロースエステル、およびテトラアルキルアンモニウムアルキルホスフェートを含む沈殿液体を生成すること;
を含む方法によって製造できる。
【0047】
本発明の別の態様において、セルロースエステルの製造方法は、回収されたセルロースエステルを洗浄液体で洗浄して、洗浄されたセルロースエステルを生成することを更に含む。
【0048】
本発明の別の態様において、セルロースエステルの製造方法は、洗浄されたセルロースエステルを乾燥させて、乾燥されたセルロースエステル製品を生成することを更に含む。
【0049】
本発明の別の態様において、セルロースエステルの製造方法は、テトラアルキルアンモニウムアルキルホスフェートを沈殿液体から分離して、回収されたテトラアルキルアンモニウムアルキルホスフェートを生成することを更に含む。
【0050】
本発明の別の態様において、セルロースエステルの製造方法は、回収されたテトラアルキルアンモニウムアルキルホスフェートを再循環させてセルロースを溶解させてセルロース溶液を生成することを更に含む。
【0051】
本発明の方法において、1種以上のアシル化試薬をセルロース溶液と接触させる場合、添加するアシル化試薬の量およびアシル化試薬を添加する順序は、溶液粘度、製品品質、および相対置換度(RDS)等の因子に大きく影響する可能性がある。
【0052】
本発明の一側面において、1種以上のアシル化試薬は単独添加で添加する。この側面において、約0.1当量〜約20当量のアシル化試薬を、1つの添加時期の間に添加する。ここで当量は、無水グルコース1モル当たりのアシル化試薬のモル数である。別の態様において、約0.5当量〜約5当量のアシル化試薬を、1つの添加時期の間に添加する。最も好ましいのは、約1当量〜約3当量のアシル化試薬を1つの添加時期の間に添加することである。
【0053】
本発明の別の態様において、アシル化試薬の添加は段階的であり、これはアシル化試薬を引き続いて添加することを意味する。この側面において、合計で0.5当量〜約20当量のアシル化試薬をセルロース溶液に対して添加し、または合計で約1.5当量〜約5当量のアシル化試薬を添加する。しかし、段階的な添加において、約0.1当量〜約2当量のアシル化試薬を、1つの添加時期の間に添加し、残りのアシル化試薬を1つ以上の異なる添加時期に添加する。別の態様において、約0.5当量〜約1当量のアシル化試薬を1つの添加時期の間に添加し、残りのアシル化試薬を1つ以上の異なる添加時期に添加する。本発明のこの側面の1つの利点は、初期のアシル化試薬添加が接触混合物の溶液粘度の低減を招来することである。溶液粘度の低減により、1つの容器から別の容器へのより容易な移動が可能になり、また後続のアシル化試薬添加の間の接触温度の低減が可能になる。これは製品品質に影響する可能性がある。本発明のこの側面の別の利点は、2つ以上のアシル化試薬を、段階的な添加でいつ添加するかに関する。この側面において、1つのアシル化試薬を添加して第1段階の間の反応を可能にでき、次いで第2のアシル化試薬を添加して第2段階の間の反応を可能にでき、これにより、特異な置換パターンを有する新規なセルロースエステルが得られる。
【0054】
本発明において、1種以上のアシル化試薬を添加する場合、セルロースのC6位がC2およびC3よりも大幅に速くアシル化されたことを見出した。よって、C6/C3およびC6/C2のRDS比は、1よりも大きく、これは位置選択的に置換されたセルロースエステルの特徴である。位置選択性の程度は、以下の要因:アシル置換基の種類、接触温度、イオン液体相互作用、アシル化試薬の当量、添加の順序等の少なくとも1つに左右される。典型的には、アシル置換基における炭素原子数が大きくなると、セルロースのC6位はC2位およびC3位よりも優先的にアシル化される。加えて、接触温度をエステル化において低くするに従って、セルロースのC6位をC2位またはC3位よりも優先的にアシル化できる。前記したように、イオン液体の種類およびプロセスにおけるそのセルロースとの相互作用は、セルロースエステルの位置選択性に影響する可能性がある。例えば、カルボキシル化イオン液体を用いる場合、位置選択的に置換されたセルロースエステルは、RDSがC6>C2>C3の場合に生成する。本発明のテトラアルキルアンモニウムジアルキルホスフェートを用いる場合、位置選択的に置換されたセルロースエステルは、RDSがC6>C3>C2の場合に生成する。これは、セルロースエステルにおける置換基の位置選択的な配置が、従来のセルロースエステルとは異なる物理的特性を有する位置選択的に置換されたセルロースエステルをもたらすという点で顕著である。
【0055】
本発明の一態様において、セルロースとアシル化試薬との反応を防止する保護基を用いない。
【0056】
本発明の一態様において、C6/C3またはC6/C2についての環RDS比は少なくとも1.05である。別の態様において、C6/C3またはC6/C2についての環RDS比は少なくとも1.1である。本発明の別の態様は、C6/C3またはC6/C2についての環RDS比が少なくとも1.3である場合である。
【0057】
本発明の別の態様において、C6/C3またはC6/C2についての環RDS比に総DS[(C6/C3)*DSまたは(C6/C2)*DS]を乗じた積は少なくとも2.9である。別の態様において、C6/C3またはC6/C2についての環RDS比に総DSを乗じた積は少なくとも3.0である。別の態様において、C6/C3またはC6/C2についての環RDS比に総DSを乗じた積は少なくとも3.2である。
【0058】
本発明の別の態様において、C6/C3またはC6/C2についての環RDS比は少なくとも1.05であり、そしてC6/C3またはC6/C2についての環RDS比に総DSを乗じた積は少なくとも2.9である。別の態様において、C6/C3またはC6/C2についての環RDS比は少なくとも1.1であり、そしてC6/C3またはC6/C2についての環RDS比に総DSを乗じた積は少なくとも3.0である。更に別の態様において、C6/C3またはC6/C2についての環RDS比は少なくとも1.3であり、そしてC6/C3またはC6/C2についての環RDS比に総DSを乗じた積は少なくとも3.2である。
【0059】
前記したように、2以上のアシル置換基がより等量で存在する場合、各置換基のRDSを独立に評価するためにカルボニル炭素を纏めることが望ましい場合がある。よって、本発明の一態様において、C6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比は少なくとも1.3である。別の態様において、C6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比は少なくとも1.5である。別の態様において、C6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比は少なくとも1.7である。
【0060】
本発明の別の態様において、C6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比にアシル置換基のDS[(C6/C3)*DSアシルまたは(C6/C2)*DSアシル]を乗じた積は少なくとも2.3である。別の態様において、C6/C3またはC6/C2についてのカルボニルRDS比にアシル置換基のDSを乗じた積は少なくとも2.5である。別の態様において、C6/C3またはC6/C2についてのカルボニルRDS比にアシル置換基のDSを乗じた積は少なくとも2.7である。本発明の別の態様において、C6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比は少なくとも1.3であり、そしてC6/C3またはC6/C2についてのカルボニルRDS比にアシルDSを乗じた積は少なくとも2.3である。別の態様において、C6/C3またはC6/C2についてのカルボニルRDS比は少なくとも1.5であり、そしてC6/C3またはC6/C2についてのカルボニルRDS比にアシルDSを乗じた積は少なくとも2.5である。更に別の態様において、C6/C3またはC6/C2についてのカルボニルRDS比は少なくとも1.7であり、そしてC6/C3またはC6/C2についてのカルボニルRDS比にアシルDSを乗じた積は少なくとも2.7である。
【0061】
驚くべきことに、本発明の一態様において、アシル化試薬の段階的な添加は、アシル化試薬の混合添加において得られるものとは異なる相対置換度(RDS)を与えた。更に、本発明の段階的および混合の添加は、一般的にRDSがC6、C3およびC2で約1:1:1であるセルロースエステルを与える混合セルロースエステルを形成するための当該分野において公知の他の手段とは異なるRDSを与える。幾つかの場合、先行技術の方法は、RDSがC6でC2およびC3のものよりも小さいようなRDSを与える。
【0062】
本発明の方法によって製造されるセルロースエステルは、セルロースエステル溶液を非溶媒と接触させることによってセルロースエステルの少なくとも一部を沈殿させることによって沈殿させ、そしてこれにより、沈殿したセルロースエステルと沈殿液体とを含むスラリーを生成する。非溶媒の例としては、これらに限定するものではないが、C1−C8アルコール、水、またはこれらの混合物が挙げられる。一態様において、セルロースエステルの沈殿のためにメタノールを用いる。非溶媒の量は、セルロースエステルの少なくとも一部を沈殿させるのに十分な任意の量であることができる。一態様において、非溶媒の量は、アシル化セルロース溶液の総体積基準で少なくとも約10体積、少なくとも約5体積、または少なくとも0.5体積であることができる。セルロースエステルの沈殿のために必要な接触時間および接触温度は、所望レベルの沈殿を実現するのに必要な任意の時間または温度であることができる。本発明の態様において、沈殿のための接触時間は、約1〜約300分間、約10〜約200分間、または20〜100分間である。沈殿のための接触温度は、約0〜約120℃、約20〜約100℃、または25〜約50℃の範囲であることができる。
【0063】
沈殿に続き、本発明の方法によって製造されるセルロースエステルは、セルロースエスラリーから分離して、回収されたセルロースエステルおよび沈殿液体を生成できる。液体の少なくとも一部を固体のスラリーから分離するための当該分野において公知の任意の固/液分離方法を用いることができる。本発明において用いるのに好適な、好適な固/液分離方法の例としては、これらに限定するものではないが、遠心分離、濾過等が挙げられる。一態様において、セルロースエステルスラリーの沈殿液体の少なくとも50質量%、少なくとも70質量%、または少なくとも90質量%を除去できる。
【0064】
分離に続き、本発明の方法によって製造される、回収されたセルロースエステル、またはセルロースエステルの湿潤ケークを、湿潤ケークを洗浄するのに好適な当該分野で公知の任意の方法を用いて洗浄液体で洗浄できる。本発明において用いるのに好適な洗浄方法の例としては、これらに限定するものではないが、多段階向流洗浄が挙げられる。洗浄液体は、任意のセルロースエステル非溶媒であることができる。非溶媒は本開示において前記した。好適な非溶媒の例としては、これらに限定するものではないが、C1−C8アルコール、水、またはこれらの混合物が挙げられる。
【0065】
一態様において、回収されたセルロースエステルの洗浄は、任意の不所望の副生成物および/または着色体の少なくとも一部を、回収されたセルロースエステルから除去して、洗浄されたセルロースエステルを生成するような様式で実施できる。一態様において、洗浄液体として用いる非溶媒は、漂白剤を、洗浄液体の総質量基準で約0.001〜約50質量%の範囲、または0.01〜5質量%の範囲で含有できる。本発明において用いるのに好適な漂白剤の例としては、これらに限定するものではないが、亜塩素酸塩,例えば亜塩素酸ナトリウム(NaClO2);次亜ハロゲン化物,例えばNaOCl、NaOBr等;過酸化物,例えば過酸化水素等;過酸,例えば過酢酸等;金属,例えばFe、Mn、Cu、Cr等;亜硫酸ナトリウム類,例えば亜硫酸ナトリウム(Na2SO3)、メタ重亜硫酸ナトリウム(Na2S2O5)、亜硫酸水素ナトリウム(NaHSO3)等;過ホウ酸塩,例えば過ホウ酸ナトリウム(NaBO3・nH2O,n=1または4);二酸化塩素(ClO2);酸素;およびオゾンが挙げられる。一態様において、本発明において用いる漂白剤としては、過酸化水素、NaOCl、亜塩素酸ナトリウムおよび/または亜硫酸ナトリウムを挙げることができる。一態様において、副生成物および/または着色体の総量の少なくとも70%、または少なくとも90%を、回収されたセルロースエステルから除去する。
【0066】
洗浄に続き、本発明の方法によって洗浄されたセルロースエステルを、当該分野で公知の任意の乾燥方法によって乾燥して、洗浄されたセルロースエステル生成物の液体分の少なくとも一部を除去できる。乾燥設備の例としては、これらに限定するものではないが、ロータリードライヤー、スクリュー型ドライヤー、パドルドライヤー、および/またはジャケット付ドライヤーが挙げられる。一態様において、乾燥されたセルロースエステル生成物は、5質量%未満、3質量%未満、または1質量%未満の液体を含む。
【0067】
本発明の一態様において、本発明の方法によって調製したセルロースエステルスラリーから沈殿させたセルロースエステルの分離に続き、テトラアルキルアンモニウムアルキルホスフェートの少なくとも一部を、上記の沈殿液体から回収して、セルロース溶解において可能な再使用のための再循環されたテトラアルキルアンモニウムアルキルホスフェートを生成する。一態様において、1種以上のアルコール、水、および/または残留カルボン酸ならびに任意の共溶媒を、少なくとも1種の液/液分離方法で沈殿液体を処理することによって実質的に除去する。このような分離方法は、当該分野で公知の任意の液/液分離方法,例えば、フラッシュ蒸発および/または蒸留を含むことができる。一態様において、1種以上のアルコール、水、および/または残留カルボン酸ならびに任意の共溶媒の少なくとも80質量%、少なくとも90質量%、または少なくとも95質量%を沈殿液体から除去でき、これにより再循環されたテトラアルキルアンモニウムアルキルホスフェートを生成できる。
【0068】
別の態様において、セルロースエステル非溶媒がC1−C8アルコールまたはその混合物,例えばMeOHである場合、アルコールの一部のみを、液/液分離方法によって沈殿液体から除去して、分画された沈殿液体を生成する。一部分離後、分画された沈殿液体中のアルコールの総量は、約0.1〜約60質量%の範囲、約5〜約55質量%の範囲、または15〜50質量%の範囲である。分画された沈殿液体は、次いで、沈殿液体中に含有されるカルボン酸の少なくとも一部をアルキルエステル,例えばメチルエステルに転化するのに十分な温度、圧力および時間で、カルボン酸を分画された沈殿液体中に存在するアルコールと反応させることによって、処理できる。エステル化は、100℃〜180℃の範囲、または130℃〜160℃の範囲の温度で実施できる。加えて、エステル化の間の圧力は、約10〜約1,000ポンド毎平方インチゲージ(「psig」)の範囲、または100〜300psigの範囲であることができる。分画された沈殿液体は、エステル化の間の滞留時間約10〜約1,000分間の範囲、または120〜600分間の範囲を有することができる。一態様において、沈殿液体中のカルボン酸の少なくとも5モル%、少なくとも20モル%、または少なくとも50モル%を、上記エステル化の間にエステル化して、改質された分画された沈殿液体を生成できる。上記エステル化に続き、カルボキシレートエステル、1種以上のアルコール、および水(エステル化の間に生成するもの)を、改質された分画された沈殿液体を少なくとも1種の液/液分離方法で処理することによって実質的に除去できる。カルボン酸のアルキルエステルへの転化は、沈殿液体からのその除去を促進することを見出した。この態様において、1種以上のアルコール、カルボキシレートエステル、水、および/または残留カルボン酸ならびに任意に共溶媒の少なくとも80質量%、少なくとも90質量%、または少なくとも95質量%を沈殿混合物から除去でき、これにより、再循環するテトラアルキルアンモニウムアルキルホスフェート流を生成できる。この方法で除去されるカルボキシレートエステルの少なくとも一部を、CO挿入によって無水物に転化できる。
【0069】
本発明の方法によって製造されるセルロースエステルは種々の用途において有用である。当業者は、具体的な用途はセルロースエステルの具体的な種類(例えばアシル置換基の種類、DS、Mw、およびセルロースエステルコポリマーの種類等の要因として)がセルロースエステルの物理特性に顕著に影響することを理解するであろう[Prog.Polym.Sci.2001,26,1605−1688]。
【0070】
本発明の一態様において、セルロースエステルは熱可塑性用途において用いる。ここでセルロースエステルはフィルムまたは成形品を形成するために用いる。熱可塑性用途において用いるための好ましいセルロースエステルの例としては、セルロースアセテート、セルロースプロピオネート、セルロースブチレート、セルロースアセテートプロピオネート、セルロースアセテートブチレートまたはこれらの混合物が挙げられる。
【0071】
本発明の更に別の態様において、セルロースエステルはコーティング用途において用いる。コーティング用途の例としては、これらに限定するものではないが、自動車、木材、プラスチック、または金属コーティングが挙げられる。コーティング用途において用いるための好ましいセルロースエステルの例としては、セルロースアセテート、セルロースプロピオネート、セルロースブチレート、セルロースアセテートプロピオネート、セルロースアセテートブチレートまたはこれらの混合物が挙げられる。
【0072】
本発明の更に別の態様において、セルロースエステルはパーソナルケア用途において用いる。パーソナルケア用途において、セルロースエステルは一般的には適切な溶媒中に溶解または懸濁させる。そして、セルロースエステルは、皮膚または毛髪に適用される際に、構造化剤、送達剤、およびフィルム形成剤として働く。パーソナル用途において用いるための好ましいセルロースエステルの例としては、セルロースアセテート、セルロースプロピオネート、セルロースブチレート、セルロースアセテートプロピオネート、セルロースアセテートブチレート、セルロースヘキサノエート、セルロース2−エチルヘキサネート、セルロースラウレート、セルロースパルミテート、セルロースステアレートまたはこれらの混合物が挙げられる。
【0073】
本発明の更に別の態様において、セルロースエステルは、薬物送達用途において用いる。薬物送達用途において、セルロースエステルはフィルム形成剤として、例えばタブレットまたは粒子のコーティングにおいて働くことができる。セルロースエステルはまた、溶解性が乏しい薬物のアモルファス混合物を形成することによって薬物の溶解性および生物学的利用性を改善するのに用いることができる。セルロースエステルは、制御薬物送達において用いることができる。ここで薬物は、外部刺激,例えばpH変化に応答してセルロースエステルマトリクスから放出される。薬物送達用途において用いるための好ましいセルロースエステルの例としては、セルロースアセテート、セルロースプロピオネート、セルロースブチレート、セルロースアセテートプロピオネート、セルロースアセテートブチレート、セルロースアセテートフタレートまたはこれらの混合物が挙げられる。
【0074】
本発明の更に別の態様において、セルロースエステルは、フィルムの溶媒キャストを含む用途において用いる。これらの用途の例としては、写真用フィルムならびに液晶ディスプレイ用の保護フィルムおよび補償フィルムが挙げられる。溶媒キャストフィルム用途において用いるための好ましいセルロースエステルの例としては、セルローストリアセテート、セルロースアセテート、セルロースプロピオネート、およびセルロースアセテートプロピオネートが挙げられる。
【0075】
本発明の更に別の態様において、本発明のセルロースエステルは、フィルムの溶媒キャストを含む用途で使用できる。このような用途の例としては、写真用フィルム、液晶ディスプレイ用の保護フィルムおよび補償フィルムが挙げられる。溶媒キャストフィルム用途において用いるために好適なセルロースエステルの例としては、これらに限定するものではないが、セルローストリアセテート、セルロースアセテート、セルロースプロピオネート、およびセルロースアセテートプロピオネートが挙げられる。
【0076】
本発明の態様において、フィルムは、本発明のセルロースエステルを含んで製造され、液晶ディスプレイ(LCD)用の保護フィルムおよび補償フィルムとして用いる。これらのフィルムは、米国出願第2009/0096962号に記載されるような溶媒キャストによって、または米国出願第2009/0050842号に記載されるような溶融押出しによって、作製できる(これらの両者は、本明細書の記載と矛盾しない限りにおいて参照によりその全部を本明細書に組入れる)。
【0077】
保護フィルムとして用いる場合、フィルムは、典型的には、配向したヨウ素化ポリビニルアルコール(PVOH)偏光フィルムのいずれかの側にラミネートして、PVOH層をスクラッチまたは水分から保護する一方、構造的な剛直性もまた増大させる。補償フィルム(または板)として用いる場合、これらは偏光子積層体とラミネートされるか、そうでなければ偏光子と液晶層との間に含まれることができる。これらの補償フィルムは、LCDのコントラスト比、広視野角、および色シフト性能を改善できる。この重要な機能の理由は、LCDにおいて用いる典型的な組の交差した偏光子について、対角線に沿って顕著な光漏失(これは悪いコントラスト比を招来する)が、特に視野角が増大するに従って、存在することである。光学フィルムの種々の組合せを使用してこの光漏失を較正または「補償」できることが公知である。これらの補償フィルムは、特定の明確に定義されたレタデーション(または複屈折)値を有さなければならない。これは液晶セルの種類または用いるモードに応じて変わる。液晶セル自体もまた特定の程度の不所望の較正すべき光学レタデーションを与えるからである。
【0078】
補償フィルムは、一般的に複屈折にて定量する。これはすなわち屈折率nに関連する。セルロースエステルについて、屈折率は約1.46〜1.50である。非配向の等方性物質について、屈折率は、入射光波の偏光状態に関わらず同じになる。物質が配向するに従い、または異方性になるに従い、屈折率は物質の方向に依存するようになる。本発明の目的のために、nx、nyおよびnzという重要な3つの屈折率が存在し、これらはそれぞれ機械方向(MD)、横方向(TD)および厚み方向に対応する。物質がより異方性になる(例えば延伸によって)に従って、任意の2つの屈折率の間の差は増大する。屈折率のこの相違は、屈折率のその特定の組合せについての物質の複屈折という。選択する物質方向の多くの組合せが存在するため、対応して異なる値の複屈折が存在する。2つの最も一般的な複屈折パラメータは、平面複屈折(Δe=nx−ny と定義される)および厚み複屈折(Δth)(Δth=nz−(nx+ny)/2 と定義される)である。複屈折Δeは、MDとTDとの間の相対的な面内配向の指標であり、無次元である。これに対し、Δthは、平均面配向に対する厚み方向の配向の指標を与える。
【0079】
光学レタデーション(R)は、フィルムの厚み(d)による複屈折に関する:Re=Δed=(nx−ny)d; Rth=Δthd=[nz−(nx+ny)/2]。レタデーションは、2つの直交する光波の間の相対的な位相シフトの直接の指標であり、そして典型的にはナノメートル(nm)単位で報告する。Rthの定義は、何人かの著者によって、特に符号(±)に関して異なることに留意されたい。
【0080】
補償フィルムまたは補償板は、LCDディスプレイ装置を操作するモードに応じて多くの形状をとることができる。例えば、C−板補償フィルムはx−y平面において等方性であり、そして板は正(+C)または負(−C)であることができる。+C板の場合、nx=ny<nzである。−C板の場合、nx=ny>nzである。別の例は、A−板補償フィルムであり、これはy-z方向において等方性であり、そして繰り返すが、板は正(+A)または負(−A)であることができる。+A板の場合、nx>ny=nzである。−A板の場合、nx<ny=nzである。
【0081】
一般的に、脂肪族セルロースエステルは、Rthの値約0〜約−350nmを、フィルム厚み60μmで与える。観察されるRthに影響する最も重要な因子は、置換基の種類およヒドロキシルの置換度(DSOH)である。極低DSOHを有するセルロース混合エステルを用いて製造されるフィルム(Shelbyら,第US2009/0050842号における)は、Rth値約0〜約−50nmを有していた。セルロース混合エステルのDSOHを顕著に増大させることによって、Sheltonら(第US2009/0096962号)は、より大きい絶対値のRthである約−100〜約−350nmが得られることを示した。セルロースアセテートは、典型的には、Rth値約−40〜約−90nm(DSOHに応じて)を与える。
【0082】
本発明の一側面は、位置選択的に置換されたセルロースエステルを含む補償フィルムに関し、該補償フィルムは、Rth約−400〜約+100nmを有する。本発明の別の態様において、補償フィルムは、総DS約1.5〜約2.95の単一アシル置換基(第2のアシル置換基のDS≦0.2)を有する位置選択的に置換されたセルロースエステルを含んで製造され、そして補償フィルムはRth値約−400〜約+100nmを有する。
【0083】
本発明の一態様において、フィルムを製造するために用いる位置選択的に置換されたセルロースエステルは、セルロースアセテート、セルロースプロピオネート、およびセルロースブチレートからなる群から選択され、位置選択的に置換されたセルロースエステルは、総DS約1.6〜約2.9を有する。本発明の別の態様において、補償フィルムはRth値約−380〜約−110nmを有し、そして総DSが約1.7〜約2.5である位置選択的に置換されたセルロースプロピオネートで構成される。更に別の態様において、補償フィルムはRth値約−380〜約−110nmを有し、そして総DSが約1.7〜約2.5およびC6/C3またはC6/C2についての環RDS比が少なくとも1.05である位置選択的に置換されたセルロースプロピオネートで構成される。別の態様において、補償フィルムはRth値約−60〜約+100nmを有し、そして総DSが約2.6〜約2.9である位置選択的に置換されたセルロースプロピオネートで構成される。更に別の態様において、補償フィルムはRth値約−60〜約+100nmを有し、そして総DSが約2.6〜約2.9およびC6/C3またはC6/C2についての環RDS比が少なくとも1.05である位置選択的に置換されたセルロースプロピオネートで構成される。別の態様において、補償フィルムはRth値約0〜約+100nmを有し、そして総DSが約2.75〜約2.9である位置選択的に置換されたセルロースプロピオネートで構成される。更に別の態様において、補償フィルムはRth値約0〜約+100nmを有し、そして総DSが約2.75〜約2.9およびC6/C3またはC6/C2についての環RDS比が少なくとも1.05である位置選択的に置換されたセルロースプロピオネートで構成される。
【0084】
本発明の別の側面は、Rth範囲約−160〜約+270nmを有する補償フィルムであって、複数の2以上のアシル置換基の総DSが約1.5〜約3.0である位置選択的に置換されたセルロースエステルで構成された、補償フィルムに関する。本発明の一態様において、セルロースエステルは、セルロースアセテートプロピオネート、セルロースアセテートブチレート、セルロースベンゾエートプロピオネート、およびセルロースベンゾエートブチレートからなる群から選択でき;ここで位置選択的に置換されたセルロースエステルは総DS約2.0〜約3.0を有する。別の態様において、補償フィルムはRth値約−160〜約0nmを有し、そして総DSが約2.0〜約3.0、C6/C3またはC6/C2についての環RDS比が少なくとも1.05、およびC6/C3またはC6/C2についての少なくとも1つのアシル置換基のカルボニルRDS比が少なくとも約1.3である位置選択的に置換されたセルロースアセテートプロピオネートで構成される。別の態様において、補償フィルムはRth値約+100〜約+270nmを有し、そして総DSが約2.0〜約3.0、C6/C3またはC6/C2についての環RDS比が少なくとも1.05、およびC6/C3またはC6/C2についての少なくとも1つのアシル置換基についてのカルボニルRDS比が少なくとも約1.3である位置選択的に置換されたセルロースベンゾエートプロピオネートで構成される。別の態様において、補償フィルムはRth値約+100〜約+270nmを有し、そして総DSが約2.0〜約2.85、ベンゾエートDSが約0.75〜約0.90、C6/C3またはC6/C2についての環RDS比が少なくとも1.05であり、C6/C3またはC6/C2についてのプロピオネートについてのカルボニルRDS比が少なくとも約1.3である、位置選択的に置換されたセルロースベンゾエートプロピオネートで構成される。
【0085】
イオン液体、セルロースエステルおよびセルロース誘導体の製造におけるこれらの使用、セルロースエステルおよびセルロース誘導体を製造する方法においてイオン液体とともに共溶媒を使用すること、ならびにセルロースエステルの処理に関する更なる情報は、米国特許出願、発明の名称”Cellulose Esters and Their Production In Carboxylated Ionic Liquids”(2008年2月13日出願、および整理番号12/030387を有する)、およびその一部継続出願、発明の名称“Regioselectively Substituted Cellulose Esters Produced In A Carboxylated Ionic Liquid Process and Products Produced Therefrom”(2009年9月12日出願;米国特許出願、発明の名称”Cellulose Esters and Their Production in Halogenated Ionic Liquids”(2008年8月11日出願、および整理番号12/189415を有する)、およびその一部継続出願、発明の名称”Regioselectively Substituted Cellulose Esters Produced In A Halogenated Ionic Liquid Process and Products Produced Therefrom”(2009年9月12日出願);米国特許出願”Production of Ionic Liquids”(2008年2月13日出願、整理番号12/030,425を有する);および米国特許出願、発明の名称”Reformation of Ionic Liquids”(2008年2月13日出願、整理番号12/030,424を有する);米国特許出願、発明の名称”Treatment of Cellulose Esters”(2008年8月11日出願、整理番号12/189,421を有する);米国特許出願、発明の名称”Production of Cellulose Esters In the Presence of A Cosolvent”(2008年8月11日出願、整理番号12/189,753を有する);および米国仮出願、発明の名称”Regioselectively Substituted Cellulose Esters and Their Production in Ionic Liquids”(2008年8月13日出願、整理番号61/088,423を有する);に記載されており、これらの全ては本明細書の記載と矛盾しない範囲で参照により本明細書に組入れる。
【0086】
本発明は、その態様の以下の例によって更に説明できるが、これらの例は例示の目的のみで包含され、発明の範囲の限定を意図しないことが理解されよう。
【0087】
例
材料および方法
実験のテトラアルキルアンモニウムアルキルホスフェートは例において記載するように調製した。セルロースの重合度は、銅エチレンジアミン(Cuen)を溶媒として用いてキャピラリー粘度測定により評価した。溶解前に、セルロースを典型的には14〜18時間、50℃で5mmHgにて乾燥させた。
【0088】
本発明のセルロースエステルにおけるC6、C3およびC2での相対置換度(RDS)は、カーボン13NMRで、”Cellulose Derivatives”,ACS Symposium Series 688,1998,T.J.Heinze and W.G.Glasser,Editors(本明細書の記載と矛盾しない範囲で参照により本明細書に組入れる)に記載される一般的な方法に従って評価した。簡単に、カーボン13NMRデータは、100MHzで運転するJEOL NMRスペクトロメータまたは125MHzで運転するBruker NMRスペクトロメータを用いて得た。サンプル濃度は、100mg/mLのDMSO−d6であった。5mgのCr(OAcAc)3(100mgのサンプル当たり)を緩和剤として添加した。スペクトルを80℃にてパルス遅延1秒を用いて収集した。通常、15,000スキャンを各実験において収集した。ヒドロキシルのエステルへの転換は、ヒドロキシルを伴うカーボンの低磁場シフトおよびカルボニル官能基に対するカーボンガンマの高磁場シフトをもたらす。よって、C2およびC6の環炭素のRDSは、置換および非置換のC1炭素およびC6炭素の直接積分によって評価した。C3でのRDSは、C6およびC2のRDSの合計を総DSから減算することによって評価した。カルボニルRDSは、Macromolecules,1991,24,3050−3059(本明細書の記載と矛盾しない範囲で参照により本明細書に組入れる)に記載される一般的な配置を用いたカルボニル炭素の積算によって評価した。複数のアシル基を含有するセルロース混合エステルの場合、セルロースエステルを、完全に置換されたセルロース混合p−ニトロベンゾエートエステルにまず転化した。p−ニトロベンゾエートエステルの位置は、セルロース混合エステル中のヒドロキシルの位置を示す。
【0089】
色測定は、ASTM D1925の以下の一般的な手順に従って行った。色測定用のサンプルは、1.7gのセルロースエステルを41.1gのn−メチルピロリドン(NMP)中に溶解させることによって準備した。HunterLab ColorQuest XE測色計(透過モードで操作する20mm路長セルを有する)を測定用に用いた。測色計は、標準的なコンピュータ(EasyMatch QC Software(HunterLab)が動作するもの)にインターフェース接続した。値(L*;白色から黒色、a*;+赤から−緑;b*;+黄から−青)を、NMP(セルロースエステルなし)およびセルロースエステル/NMP溶液について得た。NMP溶媒とサンプル溶液との間の色差(E*)を次いで算出した(E*=[(Δa*)2+[(Δb*)2+[(ΔL*)2]0.5(式中、Δはサンプル溶液についての値から溶媒についての値を減じたものである))。値E*がゼロに近づくほど色が良好である。
【0090】
セルロースエステル中の硫黄およびリンの濃度は、誘導結合プラズマ発光分析(ICP)で評価した。サンプルは、濃HNO3中での消化、続いて超純水による希釈(内部標準添加後)によって調製した。最終マトリクスは、水中5質量%NHO3であった。次いでサンプルのリン(177.434nm)および硫黄(180.669nm)量を、Perkin Elmer 2100DV誘導結合プラズマ発光分析計を用いて分析した。これはNISTトレーサブル標準を用いて較正した。
【0091】
フィルムの溶媒キャストを、以下の一般的な手順に従って実施した:乾燥させたセルロースエステルおよび10wt%可塑剤を、CH2Cl2/メタノール(またはエタノール)の90/10wt%溶媒混合物に添加して、最終濃度5〜30wt%(セルロースエステル+可塑剤基準)を与えてセルロースエステル/溶媒混合物を生成した。セルロースエステル/溶媒混合物をシールし、ローラ上に置き、そして24時間混合して均一セルロースエステル溶液を得た。混合後、セルロースエステル溶液はガラス板上にドクターブレードを用いてキャストして、所望厚みを有するフィルムを得た。キャストは、相対湿度を50%に制御したドラフト中で行った。キャスト後、フィルムおよびガラスを1時間カバーパン下(溶媒蒸発の程度を最小化するため)で乾燥させた。この初期乾燥の後、フィルムをガラスから剥離し、強制換気オーブン中で10分間100℃にてアニールした。100℃でのアニール後、フィルムをより高温(120℃)で更に10分間アニールした。
【0092】
フィルム光学レタデーション測定は、J.A.Woollam M−2000V Spectroscopic Ellipsometerを用いて370〜1000nmのスペクトル範囲で行った。RetMeas(Retardation Measurement)プログラム(J.A.Woollam Co.,Inc.より)を用いて光学フィルムの面内(Re)および面外(Rth)のレタデーションを得た。値を589nmまたは633nmでフィルム厚み60μmにて報告する。
【0093】
例1. トリブチルメチルアンモニウムジメチルホスフェート[TBMA]DMPの調製
1Lの三口丸底フラスコ(機械撹拌を備える)に、400gの新しく蒸留したトリブチルアミンおよび302gのトリメチルホスフェート(1eq)を添加した。これは、2つの明澄な相(上部相がトリブチルアミンである)を与えた。次いでフラスコを油浴中に入れ、反応混合物をN2で空にした。油浴を120℃まで加熱し、接触混合物を49時間撹拌して、淡黄色の均一な混合物を得た。室温まで冷却した後、サンプルは固化して白色固体が得られた(収率>99%)。
【0094】
プロトンNMRによる分析は、出発物質のトリブチルメチルアンモニウムジメチルホスフェートへの>99%の転化を示した。熱重量分析(TGA)は、[TBMA]DMPの熱分解の開始が約240℃であることを示した。示差走査熱量計(DSC)による分析は、溶融(Tm)が、100℃までの20℃/分での1回目の加熱スキャンの間に55℃を中心とすることを示した。溶融から20℃/分で−100℃まで冷却し、再度100℃まで20℃/分で加熱した後、Tg(ガラス転移温度)はー52℃で観測され、2つのTc(結晶化温度)は14℃および23℃で観測され、2つのTmは54℃および66℃で観測された。
【0095】
この例は、融点100℃未満のテトラアルキルアンモニウムアルキルホスフェートの調製を示す。
【0096】
例2. トリブチルメチルアンモニウムジメチルホスフェート([TMBA]DMP)中のセルロースの溶解
セルロース溶解前に、52.26gのトリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)を三口100mL丸底フラスコ(機械撹拌およびN2/減圧入口を備える)に添加した。フラスコを80℃の油浴中に入れ、[TBMA]DMPを17時間、約0.9mmHgで撹拌した。[TBMA]DMPを70℃まで冷却し、iC10ダイヤモンド付赤外プローブ(Mettler−Toledo AutoChem,Inc.,Columbia,MD,USA)を挿入して吸収を測定した。
【0097】
激しく撹拌しながら、[TBMA]DMPに、7.46g(12.5wt%)のセルロース(DP約335)を添加した(5分添加)。セルロースは容易かつ迅速に[TBMA]DMP中に分散してセルロース溶液を生成した。ブリード弁の補助で減圧を適用し(約1mmHg)、油浴を100℃まで加熱した。セルロースおよび[TBMA]DMPが100℃に到達するまで(約60分間)に、混合物は粘稠、半透明のセルロース溶液であり、視認できる粒子はなかった。セルロース溶液を更に65分間100℃にて撹拌し、この時点でこれは明澄なセルロース溶液であった。
【0098】
図1は、12.5wt%セルロースの[TBMA]DMP中での溶解を示す。セルロース吸収は1000cm-1で測定した。セルロースの分散を確保してクランピングを回避するために70℃まで加熱しながら、セルロースを[TBMA]DMPに添加した。全てのセルロースが添加されるまでに、セルロースは溶解し始めた。次いで接触温度を100℃まで上げた。接触温度が増大するに従い、セルロース溶解の程度は顕著に増大し、セルロースおよび[TBMA]DMPが100℃に到達するまでに、セルロースは[TBMA]DMP中に本質的に溶解して均一なセルロース溶液を生成した。
【0099】
この例は、セルロースが迅速かつ容易に[TBMA]DMP中に相当濃度で溶解できることを示す。
【0100】
例3. テトラアルキルアンモニウムアルキルホスフェート中に溶解したセルロースの量の分析のためのモデルの開発
例2の一般的な手順に従い、種々の濃度のセルロース(2.5,5.0,7.5,10.0,12.5wt%)をトリブチルメチルアンモニウムジメチルホスフェート中に溶解させた。初期および最終の濃度を用い、種々の濃度での吸収を、部分最小自乗法を用いて、スペクトル領域860〜1320cm-1に亘って適合させた。加えて、1157cm-1におけるセルロース吸収バンドの二次導関数に対する線形最小自乗適合を行った。このバンドは、このスペクトル領域においてテトラアルキルアンモニウムアルキルホスフェートからの干渉が殆どまたは全く存在しないことに基づいて選択した。図2は、モデルwt%セルロース、およびトリブチルメチルアンモニウムジメチルホスフェート中に溶解した10wt%セルロースについての実験の吸収値を示す。示されるように、モデルwt%セルロースおよび実験の吸収は優れた一致を示した。
【0101】
この例は、in situでの赤外分光分析に基づくモデルを用いてテトラアルキルアンモニウムアルキルホスフェート中のセルロースの濃度を評価できることを示す。この種の分析は、セルロースの一部のみが特定のテトラアルキルアンモニウムアルキルホスフェート中に既定濃度で可溶である場合に特に有用である。
【0102】
例4. 種々のテトラアルキルアンモニウムアルキルホスフェート中でのセルロースの溶解の比較
以下の更なるテトラアルキルアンモニウムアルキルホスフェートを、例1の一般的な方法によって調製した:トリブチルメチルアンモニウムジメチルホスフェート[TBMA]DMP、トリペンチルメチルアンモニウムジメチルホスフェート([TPMA]DMP)、トリオクチルメチルアンモニウムジメチルホスフェート[TOMA]DMP、トリメチルエタノールアンモニウムジメチルホスフェート[TMEA]DMP、トリメチルエトキシエタノールアンモニウムジメチルホスフェート[TMEEA]DMP、およびトリメチルエチルアセテートアンモニウムジメチルホスフェート[TMEAA]DMP。例2の一般的な方法に従い、固定濃度のセルロース(7.5wt%、DP335)を用い、これらのテトラアルキルアンモニウムアルキルホスフェートのそれぞれにおけるセルロースの溶解性を評価した。溶解したセルロースの総量を、例3に記載する線形モデルを用いて評価した。結果を表1に纏める。
【0103】
【表1】
【0104】
この例は、アンモニウムカチオンに結合したアルキル基(同じアルキルホスフェートアニオンを有する)が、イオン液体中のセルロースの溶解性に対する極めて顕著な効果を有することを示す。例えば、メチルアンモニウムの場合、他の3つのアルキル基が、C4(トリブチル)、C5(トリペンチル)、C8(トリオクチル)の順で変化するに従い、溶解するセルロースの質量%はそれぞれ100,28,および0wt%である。トリメチルアンモニウムでは、エタノールからエトキシエタノールへの移動がセルロースの溶解性を損なう。エタノールのアセチル化の際、セルロースの溶解性は顕著に低下する。明確に、セルロースは特定のテトラアルキルアンモニウムアルキルホスフェート中での優れた溶解性を有する。注意深い実験によってのみ、セルロース溶解における特定のテトラアルキルアンモニウムアルキルホスフェートの有用性を評価できる。
【0105】
例5. トリブチルメチルアンモニウムジメチルホスフェートおよびジメチルスルホキシドの混合物中でのセルロースの溶解
三口100mL丸底フラスコ(機械撹拌を備え、N2/減圧入口およびiC10ダイヤモンド付赤外プローブ(Mettler−Toledo AutoChem,Inc.,Columbia,MD,USA)を有する)に、51.31gの[TBMA]DMPおよび17.10gのジメチルスルホキシド(DMSO)(25wt%)の混合物を添加した。室温で急速に撹拌しながら、7.60gのセルロース(10wt%、DP約335)を[TBMA]DMPおよびDMSOの溶液に添加した(6分間添加)。セルロース/[TBMA]DMP/DMSO混合物を更に4分間撹拌して、予熱した80℃油浴をフラスコまで上げる前にセルロースの良好な分散を確保した。油浴を上げてから10分後、全てのセルロースが溶解し、淡黄色、低粘度のセルロース溶液が得られた。
【0106】
この例は、より低いセルロース溶液粘度のための手段として、テトラアルキルアンモニウムアルキルホスフェート中にセルロースを溶解させる場合に共溶媒を使用できることを示す。
【0107】
例6. トリブチルメチルアンモニウムジメチルホスフェート中に溶解したセルロースの、触媒不存在下でのエステル化
例2の一般的な方法に従い、7.5wt%のセルロース溶液を調製した。セルロース溶液の温度を80℃に調整した後、3当量の無水酢酸(Ac2O)を滴下(6分間)してアシル化セルロース溶液を生成した。アシル化セルロース溶液のサンプルは、反応の過程で取り出し、セルロースエステルはメタノール中で沈殿させることによって単離してセルロースエステルスラリーを回収した。セルロースエステルスラリーを濾過して、回収されたセルロースエステルを生成した。各々の回収したセルロースエステルサンプルを、4つの200mL分量のメタノールで洗浄して、洗浄されたセルロースを生成し、次いで50℃、10mmHgで乾燥させ、乾燥されたセルロースエステル生成物を得た。
【0108】
図3は、1825cm-1(無水酢酸)および1220cm-1(酢酸エステルおよび酢酸)での赤外バンドの吸収 対 [TBMA]DMP中に溶解したセルロースのエステル化(3eq無水酢酸)中の接触時間のプロットを示す。図3に示すDS値は、プロトンNMR分光法によって評価し、接触時間の間に取り出したアシル化セルロース溶液サンプルに対応する。図3が示すように、反応の速度は極めて速かった。無水物添加の開始後、DSが2.52に到達するまで、23分間が必要であるのみであった。
【0109】
この例は、テトラアルキルアンモニウムアルキルホスフェート中に溶解したセルロースのエステル化において、触媒を添加して反応を促進させる必要が無いことを示す。
【0110】
例7. 1−ブチル−3−メチルイミダゾリウムジメチルホスフェート中に溶解したセルロースのエステル化(比較)
三口100mL丸底フラスコは、機械撹拌を備え、iC10ダイヤモンド付IRプローブ(Mettler−Toledo AutoChem,Inc.,Columbia,MD,USA)を有し、そしてN2/減圧入口を有していた。フラスコに、50.65gの1−ブチル−3−メチルイミダゾリウムジメチルホスフェート([BMIm]DMP)を添加した。室温で撹拌しながら、4.11g(7.5wt%)のセルロース(DP約335、6分間添加)を添加した。次いで、ブリード弁の補助で減圧(2mmHg)を適用し、80℃に予熱した油浴をフラスコまで上げた。油浴を上げてから6分後に、明澄なセルロース溶液が生成された。セルロース溶液を更に11分間撹拌した後、油浴を下ろし、そしてセルロース溶液を室温まで放冷した。
【0111】
9.57g(3.7eq)の無水酢酸を、10分間に亘って滴下添加して反応混合物を生成した。添加が完了した後、反応混合物を33分間撹拌した後、サンプルを取り出し、そしてセルロースエステルをメタノール中に沈殿させた(サンプル1)。この時点で、反応混合物中には色は形成されなかった。予熱した50℃の油浴をフラスコまで上げた。反応混合物を34分間撹拌した後サンプルを取り出し、セルロースエステルをメタノール中で沈殿させた(サンプル2)。反応混合物の色の変化は殆どなかった。油浴温度設定を80℃まで上げた。13分以内に、反応混合物の色が濃琥珀色になり、粘度が増大し始めた。更に8分後、反応混合物がゲル化した。撹拌を更に30分間継続した後、油浴を下ろした。反応混合物は黒色ゲルであった。ゲルを固化させるために、メタノールをフラスコに直接添加した(サンプル3)。濾過後、各サンプルを4つの200mL分量のメタノールで洗浄し、セルロースエステル固体を1晩50℃、5mmHgで乾燥させた。サンプル1(白色固体)およびサンプル2(褐色固体)はDMSOおよびNMPのような溶媒中で可溶であった。サンプル3(黒色固体)は評価した全溶媒中で不溶であった。サンプル2(DS=2.48)がアセトン中100mg/mLで可溶でなかったことに留意することが重要であり;ゲルが形成された。本発明においては、テトラアルキルアンモニウムアルキルホスフェート中に溶解したセルロースから調製されたセルロースアセテートは、これらがDS約2.45〜約2.55を有する場合、アセトン中100mg/mLで完全に可溶であることを見出した。
【0112】
図4は、[BMIm]DMP中に溶解したセルロースの、1825cm-1(無水酢酸)および1220cm-1(酢酸エステルおよび酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを示す。
【0113】
別の反応を、0.1eqMSAを触媒として用いたことを除いて先の反応と同一の方法で行った。図5に示すように、メチルスルホン酸(MSA)を含むことは、MSAが存在しない場合と比べたときの反応速度の変化をさせなかった。MSAを含まない反応の場合でのように、反応温度を80℃に上げた後、反応混合物粘度の増大が観察された。サンプルを迅速に取り出してMeOH中に沈殿させた。サンプリングの数分以内に、反応混合物はまたゲル化した。すなわち、MSAの存在はゲル化を抑止しなかった。
【0114】
これらの例は多くの重要な点を示す。[BMIm]DMP中でのセルロースの溶解は公知である(Green Chemistry 2008,10,44−46;Green Chemistry 2007,9,233−242)。しかし、この例は、[BMIm]DMP中に溶解したセルロースのエステル化が成功でないことを示す。[TBMA]DMP中に溶解したセルロースの80℃でのエステル化はゲル化を招来しない(例6)。得られるセルロースエステル生成物は、典型的には、DS範囲2.45〜2.55を有する白色固体であり、アセトン中で完全に可溶である。これに反し、[BMIm]DMP中に溶解したセルロースの80℃でのエステル化は、急速なゲル化を招来する。セルロースエステル生成物は常に高度に着色し、全ての溶媒中で不溶である。MSAを含むことは反応速度を変化させず、ゲル化の防止もしない。これは、MSAを含むことが反応速度を加速しゲル化を防止するという[BMIm]Cl中に溶解したセルロースのエステル化で観測されるもの(米国特許出願、発明の名称“Cellulose Esters and Their Production in Halogenated Ionic Liquids”(2008年8月11日出願、整理番号第12/189415号を有する)に記載される通り)に反する。
【0115】
例8. テトラアルキルアンモニウムアルキルホスフェート−強酸混合物中に溶解したセルロースのエステル化
セルロース溶解前に、53.46gのトリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)を三口100mL丸底フラスコ(機械撹拌およびN2/減圧入口を備える)に添加した。フラスコを80℃の油浴中に入れ、溶液を1晩減圧(16.5時間、約0.5mmHg)にて撹拌した。[TBMA]DMPを70℃まで冷却し、IRプローブ(Mettler−Toledo AutoChem,Inc.,Columbia,MD,USA)を挿入して吸収データを得た。
【0116】
激しく撹拌しながら、[TBMA]DMPに、5.94g(10wt%)の微結晶セルロースを添加した(5分添加)。セルロースは容易かつ迅速に[TBMA]DMP中に分散した。ブリード弁の補助で減圧を適用し(約1mmHg)、油浴を100℃まで加熱した。セルロースおよび[TBMA]DMPが98℃に到達するまで(約20分間)に、ほぼ全てのセルロースが溶解した。セルロースおよび[TBMA]DMPを更に100分間100℃にて撹拌し、この時点で混合物は明澄な淡黄色のセルロース溶液であった。
【0117】
セルロース溶液に、1.87g(0.5eq)の無水酢酸(Ac2O)を3分間かけて滴下添加した。セルロース溶液粘度は顕著に低下し、セルロース溶液色は若干濃くなった。この時点で、セルロース溶液を80℃まで冷却した後、9.35g(2.5eq)のAc2Oを9分間かけて滴下添加し、アシル化セルロース溶液を生成した。アシル化セルロース溶液のサンプルを種々の時間間隔で取り出し、セルロースエステルを200mLメタノール中での沈殿によって単離してセルロースエステルスラリーを生成した。沈殿したセルロースエステルを濾過によって単離して、回収されたセルロースエステルを生成した。各々の回収したセルロースエステルサンプルを4つの200mL分量のメタノールで洗浄して、洗浄されたセルロースエステルを生成し、次いで減圧下(50℃、約10mmHg)で乾燥させた。in situでのIRに基づき、反応は、2回目の無水物添加の終了から11分後にほぼ完了した。アシル化セルロース溶液の色がどのように変化するかを見るために、接触時間を更に延長した。反応が進行するに従い、アシル化セルロース溶液はより濃い琥珀色になり、反応の終了時にはほぼ茶色になった。
【0118】
図6は、1825cm-1(無水酢酸)および1724cm-1([TBMA]DMPとの相互作用によって高波数側にシフトした酢酸)での赤外バンドの吸収 対 [TBMA]DMP中に溶解したセルロースのエステル化の間の接触時間のプロットを示す。図6に示すDS値は、プロトンNMR分光法によって評価し、接触時間の間に取り出したサンプルに対応する。図6が示すように、100℃での反応の速度は極めて速い。Ac2Oが極めて急速に消費されたためAc2Oは添加中に観察されなかった。80℃にて、反応の速度は若干遅くなったが、なお極めて急速であった。2回目の無水物添加の開始後、DSが2.48に到達するまで、21分間が必要であるのみであった。
【0119】
沈殿、洗浄、および乾燥の後、サンプル1(DS=2.04)は白色固体であった。サンプル2(DS=2.48)は白色固体であった。サンプル3(DS=2.61)はオフホワイト固体であった。サンプル4(DS=2.71)は淡褐色固体であった。サンプル2(DS=2.48)はアセトン中、100mg/mLにて完全に可溶であった。
【0120】
沈殿液体を減圧中で濃縮し、8.3wt%の残存酢酸(HOAc)を含有する[TBMA]DMPが得られた。[TBMA]DMPのカチオン:アニオン比は1:1であり、アニオン交換がないことを示す。
【0121】
2つの追加の反応を、1wt%過塩素酸(HClO4)([TBMA]DMP基準で)または1wt%メタンスルホン酸(MSA)([TBMA]DMP基準で)を0.5eqAc2Oとともに混合物として添加したことを除いて上記の全く同じ手順に従って行なった。図7および8は、Ac2O、Ac2O+HClO4、またはAc2O+MSAを添加する場合の、[TBMA]DMP中に溶解したセルロースの、1724cm-1での赤外バンドの吸収 対 エステル化の間の接触時間、のプロットを比較する。図8に示すDS値は、プロトンNMR分光法によって評価し、接触時間の間に取り出したサンプルに対応する。図7は、0.5eqAc2Oの100℃での添加を含む接触時間を示す。各反応が無水物添加(15分)の同じ時点で開始するようにx軸をシフトさせた。全3つの場合で、100℃での反応速度は極めて速かった。Ac2Oが極めて急速に消費されたためAc2Oは添加中に観測されなかった。図8は、80℃での2.5eqAc2Oの添加を含む接触時間を示す。各反応が無水物添加(72分)の同じ時点で開始するようにx軸をシフトさせた。全3つの場合で、反応速度は、温度低下に起因して若干低下したが、なお極めて急速であった。すなわち、これらの酸を含むことは反応速度を加速しなかった。実際、各反応について得られるプロットの初期スロープ(72〜90分)およびDS値の比較は、MSAを含むことが反応速度を顕著に遅くすることを示した。これは図9にてより容易に見られ、DS 対 2.5eqAc2Oの添加(酸の不存在下および存在下、80℃にて)を含む時間のプロットを示す。更に、これらのサンプルのゲルパーミエーションクロマトグラフィ(GPC、表2)による分析は、これらの強酸を高濃度で含むことがセルロースエステル生成物の分子量に対して有した影響は無視できることを示した。典型的には、これらの酸をこれらの濃度で含むことは、セルロースエステル生成物の分子量を顕著に低下させる。
【0122】
【表2】
【0123】
極めて驚くべきことに、表2に記す通り、MSAを含むことは、反応溶液およびセルロースエステル生成物の色を顕著に改善した。HClO4(強い酸化剤)の場合、セルロースエステル生成物の色は、Ac2Oのみを含む(酸なし)反応について上記したものと極めて類似した。MSAの場合、アシル化セルロース溶液から取り出した全ての4つのサンプルは白色であった。Ac2O+MSAアシル化セルロース溶液から単離したサンプル4のセルロースエステルはNMP中に溶解し、セルロースエステル/NMP溶液はE*17.8を有し、一方、Ac2Oアシル化セルロース溶液から単離したサンプル4のセルロースエステルは、NMP中に溶解した際に、E*31.4を有した。すなわち、同一の接触条件下では、アシル化セルロース溶液中にMSAを含むことは、溶液色を約45%低下させた。
【0124】
Ac2Oアシル化セルロース溶液から単離したサンプル2、およびAc2O+MSAアシル化セルロース溶液からのサンプル3は、残存する硫黄およびリンについてICPで分析した。Ac2Oアシル化セルロース溶液から単離したサンプル2は、14.8ppmのSおよび12.9ppmのリンを含有することを見出した。Ac2O+MSAアシル化セルロース溶液からのサンプル3は、15.3ppmのSおよび24.5ppmのリンを含有することを見出した。このデータは、セルロースエステル生成物が、スルフェートまたはホスフェートの無機エステルを殆どまたは全く含有しないことを示す。
【0125】
この例は、本発明の多くの驚くべきそして重要な特徴を示す。テトラアルキルアンモニウムアルキルホスフェートをセルロースのための、および後続のセルロースのエステル化の間の溶媒として用いる場合、強酸を用いて反応の結果を変えることができる。これらの酸を含むことは、分子量に殆どまたは全く影響しないし、反応速度を加速させることもない。全く驚くべきことに、十分な濃度では、反応速度が低下する。テトラアルキルアンモニウムアルキルホスフェート中にこれらの酸を含むことは、セルロースのエステル化の間に得られる生成物の色を変化させまたは改善することができる。更に、少量の無水物をテトラアルキルアンモニウムアルキルホスフェート−セルロース溶液に添加することは、溶液粘度の顕著な低減を招来し、接触温度の低減を可能することに留意すべきである。
【0126】
例9. テトラアルキルアンモニウムアルキルホスフェート−強酸混合物中に溶解したセルロースからのセルロース混合エステルの調製
セルロース溶解前に、61.11gのトリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)を三口100mL丸底フラスコ(機械撹拌およびN2/減圧入口を備える)に添加した。フラスコを80℃の油浴中に入れ、[TBMA]DMPを1晩減圧(約16.5時間、約0.5mmHg)にて撹拌した。液体を70℃まで冷却し、IRプローブ(Mettler−Toledo AutoChem,Inc.,Columbia,MD,USA)を挿入した。
【0127】
激しく撹拌しながら、[TBMA]DMPに6.79g(10wt%)の微結晶セルロースを添加した(5分添加)。セルロースは容易かつ迅速に[TBMA]DMP中に分散した。油浴を100℃まで加熱した。セルロースおよび[TBMA]DMPが97℃に到達するまで(約35分間)に、ほぼ全てのセルロースが溶解した。セルロースおよび[TBMA]DMPを更に75分間100℃にて撹拌し、この時点で混合物は明澄な淡黄色のセルロース溶液であった。
【0128】
セルロース溶液に、2.14g(0.5eq)の無水酢酸(Ac2O)および2.73gのプロピオン酸無水物(Pr2O)の冷却された混合物を、3分間かけて滴下添加してアシル化セルロース溶液を生成した。アシル化セルロース溶液の粘度は顕著に低下し、アシル化セルロース溶液の色は若干濃くなった。この時点で、アシル化セルロース溶液を60℃まで冷却した後、4.28g(1eq)のAc2Oおよび5.45gのPr2Oの冷却された混合物を7分間かけて滴下添加した。アシル化セルロース溶液のサンプルを種々の時間間隔で取り出し、セルロースエステルを、200mLの75/25MeOH/H2O中での沈殿によって単離してセルロースエステルスラリーを生成した。セルロースエステルスラリーを濾過して、回収されたセルロースエステルおよび沈殿液体を生成した。各々の回収されたセルロースエステルサンプルを4つの200mL分量の75/25MeOH/H2O中で洗浄して、洗浄されたセルロースエステルを生成し、次いで減圧(50℃、約10mmHg)下で乾燥させて、乾燥されたセルロースエステル生成物を生成した。
【0129】
図10は、1815cm-1(無水物)、1732cm-1([TBMA]DMPとの相互作用によって高波数側にシフトした酸)および1226cm-1(エステル+酸)赤外バンドの吸収 対 [TBMA]DMP中に溶解したセルロースのエステル化の間の接触時間、のプロットを示す。図10に示すDS値は、プロトンNMR分光法によって評価し、接触時間の間に取り出したサンプルに対応した。図10が示すように、100℃での反応速度は十分速く、添加中に無水物は観測されなかった。60℃にて、反応速度は遅くなった。2回目の無水物添加の開始後、DS2.42に到達するのに142分間が必要であった。例8と比べ、2回目添加後の反応速度は、主に接触温度の相違に起因して、より遅かった。
【0130】
沈殿、洗浄、および乾燥の後、サンプル1(DS=1.36)、2(DS=1.78)および3(DS=2.08)は白色固体であった。サンプル4(DS=2.42)は淡黄色固体であった。
【0131】
追加の反応は、1wt%メタンスルホン酸(MSA)を0.5eqAc2O+0.5eqPr2Oとともに混合物として添加したことを除いて上記の全く同じ手順で行った。図11および12は、Ac2O/Pr2OまたはAc2O/Pr2O+MSAを添加した場合の、[TBMA]DMP中に溶解したセルロースの、1815cm-1および1732cm-1での赤外バンドについての吸収 対 エステル化の間の接触時間、のプロットを比較する。図11は、0.5eqAc2Oおよび0.5eqPr2Oの100℃での添加を含む接触時間を示す。各反応が無水物添加の同じ時点(30分)で開始するようにx軸をシフトさせた。両者の場合で、100℃での反応速度は速かった。Ac2O/Pr2O(MSAなし)の場合、無水物が極めて急速に消費されたため添加中に何も観測されなかった。しかし、Ac2O/Pr2O+MSAでは、小さい濃度の無水物が添加時間中に観測された。図12は、1eqAc2Oおよび1eqPr2Oの、MSAありおよびなしでの、60℃での添加を含む接触時間を示す。各反応が無水物添加の同じ時点(113分)で開始するようにx軸をシフトさせた。両者の場合で、反応速度は、より低い接触温度に起因して遅くなった。図12に基づき、MSAが接触混合物中に存在する場合、無水物消費速度がより低かったことが明らかである。更に、1732cm-1吸収の初期スロープ(115〜175分)の比較もまた、MSAを含むことが反応速度を遅くしたことを示す。これは、1.0eqAc2Oおよび1.0eqPr2Oの酸の不存在下および存在下での60℃での添加を含む時間の、DS 対 時間のプロットを示す図14から容易に分かる。更に、ゲルパーミエーションクロマトグラフィによるこれらのサンプルの分析(表3)は、これらの強酸を高濃度で含むことが生成物の分子量に与える影響は無視できることを示した。典型的な溶媒を用いるセルロースのエステル化において、これらの酸をこれらの濃度で含むことは、セルロースエステル生成物の分子量を顕著に低減する。
【0132】
【表3】
【0133】
加えて、MSAは最終セルロースエステル生成物の色を、MSAが存在しない場合と比べて改善した。Ac2O+MSAアシル化セルロース溶液から単離したサンプル5のセルロースエステルをNMP中に溶解させ、セルロースエステル/NMP溶液はE*14.3を有した。Ac2Oアシル化セルロース溶液から単離したサンプル4のセルロースエステルもまたNMP中に溶解させ、セルロースエステル/NMP溶液はE*17.8を有した。すなわち、より長い接触時間である135分間にも関わらず、アシル化セルロース溶液中にMSAが含まれることは、MSAが不存在である場合と比べてアシル化セルロース溶液における色を低減した。
【0134】
Ac2O/Pr2Oアシル化セルロース溶液から単離したサンプル4のセルロースエステル、およびAc2O/Pr2O+MSAアシル化セルロース溶液からのサンプル5のセルロースエステルは、残存する硫黄およびリンについてICPによって分析した。Ac2O/Pr2Oアシル化セルロース溶液から単離したサンプル4は、9.1ppmのSおよび121.9ppmのリンを含有することを見出した。Ac2O/Pr2O+MSAアシル化セルロース溶液からのサンプル5は、9.4ppmのSおよび67.8ppmのリンを含有することを見出した。これらのデータは、セルロースエステル生成物がスルフェートまたはホスフェートの無機エステルを殆どまたは全く含有しないことを示す。
【0135】
この例は、本発明の多くの驚くべきそして重要な特徴を示す。セルロースのための、および後続のセルロースのエステル化の間の溶媒としてテトラアルキルアンモニウムアルキルホスフェートを用いる場合、強酸を用いてアシル化セルロース溶液の結果を変えることができる。これらの酸を含むことは、分子量に殆どまたは全く影響しないし、反応速度を低下させる可能性もない。テトラアルキルアンモニウムアルキルホスフェート中にこれらの酸を含むことは、セルロースのエステル化の間に得られる生成物の色を変化させまたは改善することができる。更に、少量の無水物をセルロース溶液に添加することは、セルロース溶液粘度の顕著な低減を招来し、接触温度の低減を可能することに留意すべきである。最後に、この例で示すような混合エステルの調製において、アシル基の分布(位置選択性)は、アシル化試薬を添加する順序およびアシル化試薬を添加する際の濃度によって制御できることに留意すべきである。
【0136】
例10. テトラアルキルアンモニウムアルキルホスフェート−非プロトン性溶媒混合物中に溶解したセルロースからのセルロースエステルの調製
例5の一般的な手順に従って、セルロース(10wt%、DP約335)を、質量基準で75/25の[TBMA]DMP/ジメチルホルムアミドの混合物中に溶解させた。これにより薄黄色溶液が得られた。セルロース溶液の温度を50℃に調整した後、3当量のAc2Oを16分間かけて滴下添加して、アシル化セルロース溶液を生成した。Ac2Oの添加後、アシル化セルロース溶液を64分間50℃にて撹拌した後、接触温度を80℃まで上げた。アシル化セルロース溶液の色は接触時間を通じて薄黄色であった。アシル化セルロース溶液のサンプルを反応の過程で取り出し、メタノール中での沈殿によってセルロースエステルを単離してセルロースエステルスラリーを生成した。セルロースエステルスラリーを濾過して白色の回収されたセルロースエステルおよび沈殿液体を生成した。回収したセルロースエステルの各サンプルを、3つの200mL分量で洗浄して、洗浄されたセルロースエステルを生成し、次いで50℃、10mmHgにて乾燥させて、乾燥されたセルロースエステル生成物を生成した。
【0137】
図14は、[TBMA]DMP/DMF中に溶解したセルロースの、1825cm-1(無水酢酸)および1724cm-1(酢酸)での赤外バンドの吸収 対 エステル化の間の接触時間のプロットを示す。図14に示すDS値は、プロトンNMR分光法によって評価し、接触時間の間に取り出したアシル化セルロース溶液サンプルに対応する。図14が示すように、50℃でも、反応速度は適切であった。無水物添加の開始後、DSが0.85に到達するまで、23分間が必要であるのみであった。接触温度を80℃まで上げる場合、反応速度が増大し、反応が終了したときにDS2.48をもたらした。アシル化セルロース溶液の色は、接触時間を通じて維持されたこと、およびアシル化セルロース溶液のサンプリングによって得られるセルロースエステルが白色であったことに留意することが重要である。
【0138】
2回目の反応は、例5で調製したセルロース溶液(10wt%セルロース,75/25[TBMA]DMP/DMSO中)を用いて同一の方法で行った。この場合において、50℃であっても、アシル化セルロース溶液の色は急速に濃くなり、そしてアシル化セルロース溶液の粘度は顕著に低下した。接触温度を80℃まで上げた場合、アシル化セルロース溶液の色は黒色になり、そして溶液粘度は極めて低くなった。セルロースエステルは、メタノールで沈殿させることによって単離してセルロースエステルスラリーを形成し、次いで濾過して、回収されたセルロースエステルを生成した。サンプル1および2のセルロースエステルは白色;サンプル3は濃茶色;そしてサンプル4は灰−黒の外観であった。表4は、[TBMA]DMP/DMSOアシル化セルロース溶液から得られるセルロースエステル生成物 対 [TBMA]DMP/DMFアシル化セルロース溶液からのセルロースエステル生成物、の分子量を比較する(表4)。[TBMA]DMP/DMSOアシル化セルロース溶液の場合、セルロースアセテートについてのMnおよびMwの両者は、[TBMA]DMP/DMFアシル化セルロース溶液からのセルロースアセテートと比べて顕著に低かった。理論に拘束されることを望まないが、変色およびセルロースエステル生成物の分解は、比較的酸性のプロトンのアルファからSO二重結合(DMFの場合は不存在)によって生じると考えられる。
【0139】
【表4】
【0140】
この例は、多様な非プロトン性溶媒を共溶媒としてテトラアルキルアンモニウムアルキルホスフェートとともにセルロースを溶解させるために使用できるが、選択された非プロトン性溶媒,例えばN,N−ジメチルホルムアミドまたはN−メチルピロリドンのみがセルロースエステル化の間の適切な共溶媒であることを示す。一般的に、副反応を招来する酸性プロトンを有さない誘電率約30超の任意の非プロトン性溶媒は、テトラアルキルアンモニウムアルキルホスフェートとの組合せで用いる場合、セルロースの溶解およびエステル化において用いるのに好適である。
【0141】
例11. 75/25トリブチルメチルアンモニウムジメチルホスフェート/N−メチルピロリドン中に溶解したセルロースからのセルロースアセテートプロピオネートの調製
例5の一般的な手順に従って、セルロース(10wt%、DP約335)を、質量基準で75/25の[TBMA]DMP/NMPの混合物中に、100℃にて10分間で、溶解させた。これにより薄黄色セルロース溶液が得られた。セルロース溶液に、100℃にて、3.3当量のPr2O(微量のAc2Oを含有した)を13分間かけて滴下添加して、アシル化セルロース溶液を生成した。添加終了から15分で、IR吸収値が平坦になり始め、そして吸収値は、DSが所望値近傍であることを示した。アシル化セルロース溶液からIRプローブを取り出し、アシル化セルロース溶液を直ちに300mLの75/25メタノール/H2O中に注ぎ入れる一方、Heidolphホモジナイザーで混合して、沈殿したセルロースエステルおよび沈殿液体を含むセルロースエステルスラリーを生成した。沈殿したセルロースエステルを濾過し、3×で200mL分量の75/25MeOH/H2Oで洗浄することによって単離して、洗浄されたセルロースエステルを生成した後、1晩10mmHg、50℃にて乾燥させ、これは11.9gの雪白色の乾燥されたセルロースエステル固体を与えた。1H NMRによる分析から、セルロースエステルはDS2.34(DSPr=2.30,DSAc=0.04)を有したことが明らかになった。定量カーボン13NMRによる分析は、乾燥されたセルロースエステル生成物が環RDSの:RDS C6=0.97、RDS C3=0.64、RDS C2=0.73を有して位置選択的に置換されたことを示した。セルロースエステル生成物は、DMSO、NMP、アセトン、および90/10 CH2Cl2/MeOH等の多様な溶媒中、100mg/1mLで完全に可溶であった。
【0142】
この例は、セルロースがテトラアルキルアンモニウム−非プロトン性溶媒混合物中に迅速に溶解でき、次いで高温でエステル化されて高品質のセルロースエステルを得ることができることを示す。
【0143】
例12. フィルムのキャストおよびフィルム光学測定
一連の本質的に6,3−および6,2−で位置選択的に置換されたセルロースエステル(1−3)を、例11の一般的な手順に従って調製した(高C6RDS)。市販(比較例4および6)セルロースエステル(Eastman Chemical Companyから入手可能)を、米国特許出願公開第2009/0096962号および米国特許出願公開第2009/0050842号に記載される一般的な手順によって調製した。比較例5は、米国特許出願公開第2005/0192434号に記載されるようにして調製した。例5におけるセルロースエステルは、本質的に2,3−で位置選択的に置換され、そしてこれがC6にて低RDSを有する一方、本発明のセルロースエステルがC6にて高RDSを有する点で、本発明の例とは異なっていた。環RDSはフィルムをキャストする前に各サンプルについて評価し、そしてフィルム光学特性を評価した。結果を表5に纏める。
【0144】
【表5】
【0145】
6,3−、6,2−セルロースプロピオネート(例3、DS=2.10、C6/C3=1.58、C6/C2=1.73、DS*C6/C3=3.3、DS*C6/C2=3.6)についてのRthの値を、2,3−セルロースプロピオネート(例5、DS=1.99、C6/C3=0.45、C6/C2=0.43、DS*C6/C3=0.90、DS*C6/C2=0.86)に対して比較し、6,3−、6,2−セルロースプロピオネートは大幅により負のRth値(−382nm 対 −80nm)を与えた(2つのセルロースエステルは同様のDS値を有していても)ことが明らかであった。同様に、Rth値は、例3について、例6(DS=1.93、C6/C3=0.79、C6/C2=0.85、DS*C6/C3=1.5、DS*C6/C2=1.6)に対して顕著により負であった。位置選択的に置換された6,3−、6,2−セルロースプロピオネートの総DSが増大した場合(例1および2)、Rthの値は急速かつ顕著に増大した。例えば、DS2.45(例2)において、Rthは−140nmに増大した。DSが若干、DS2.49(例2)まで増大する場合、Rthは更に−119nmまで増大した。図15が示すように、総DSが更に増大すると、Rthは正になった。この挙動は、市販のセルロースエステルで観測されるものと顕著に異なった(例4と比較)。
【0146】
この例は、セルロースエステルの置換パターンが面外レタデーションに顕著に影響する可能性があることを示す。具体的には、本発明の位置選択的に置換されたセルロースエステルは、異なる置換パターンを有するセルロースエステルを用いては到達できないレタデーション値を与えることができる。より低い総DSで、本発明のセルロースプロピオネートについてのRthは、従来のセルロースプロピオネートに対して大幅により負であった。より高い総DSで、本発明のセルロースプロピオネートについてのRthは、従来のセルロースプロピオネートに対してより小さく負またはより大きく正であった。
【0147】
例13. 位置選択的に置換されたセルロースアセテートプロピオネートからのセルロースアセテートプロピオネートベンゾエートの調製
例11で調製されたセルロースアセテートプロピオネート(5g)およびピリジン(50g)を300mL丸底フラスコ(機械撹拌および水凝縮器を備える)に入れた。混合物を室温で窒素雰囲気下で撹拌して明澄溶液を得た。次いで塩化ベンゾイル(12.8g)を添加漏斗経由で滴下添加した。添加の完了後(約1時間)、温度を70℃まで上げた。混合物を更に5時間撹拌し、次いで室温まで放冷した。得られた溶液をメタノール(800g)中に激しく撹拌しながら沈殿させ、濾過し、メタノールで繰り返し洗浄し、そして減圧下で乾燥させて繊維状生成物(4.7g)を得た。1HNMRによる分析から、DSPr=2.29、DSAc=0.04、DSBz=0.70が明らかになった。13CNMRによる分析から、初期サンプルの位置選択性が反応条件下で保存されたことが明らかになった。
【0148】
フィルムキャストおよびフィルム光学特性の測定のための一般的な手順に従い、セルロースアセテートプロピオネートベンゾエートが、面外レタデーション(Rth、633)+156nmをフィルム厚み43μmで(+218を60μmで)有することを見出した。
【0149】
この例は、脂肪族アシル基の位置選択的な配置が高いC6/C3比またはC6/C2比を有するセルロースエステルをもたらし、続いて後続の芳香族基のC2およびC3での配置が、大きい正の値のRthを有するセルロースエステルをもたらしたことを示した。この例は、これらの種類のセルロースエステルが、2段階プロセスによって、第1段階で達成した置換パターンを乱すことなく調製できることを示した。
【0150】
例14. 段階的な無水物添加を用いた、70/30トリブチルメチルアンモニウムジメチルホスフェート/N−メチルピロリドン中に溶解したセルロースからのセルロースベンゾエートプロピオネートの調製
例5の一般的な手順に従って、セルロース(10wt%、DP約335)を、質量基準で70/30の[TBMA]DMP/NMPの混合物中に100℃で溶解させた。これにより薄黄色セルロース溶液が得られた。セルロース溶液に、100℃にて、2.5当量のPr2Oを3分間かけて滴下添加して、アシル化セルロース溶液を生成した。Pr2O添加終了から10分後に、安息香酸無水物(4eq)を一度に溶融物として添加した(85℃で溶融した)。アシル化セルロース溶液を更に35分間撹拌し、この時点でIR吸収値は、DSが所望値近傍であることを示した。アシル化セルロース溶液からIRプローブを取り出し、アシル化セルロース溶液を直ちに300mLのメタノールに注ぎ入れる一方、Heidolphホモジナイザーで混合して、セルロースベンゾエートプロピオネートスラリーを生成した。セルロースベンゾエートプロピオネートを濾過し、8×で200mL分量のメタノールで洗浄することによって単離した後、1晩10mmHg、50℃にて乾燥させた。1H NMRによる分析から、セルロースベンゾエートプロピオネートはDS2.91(DSPr=2.58,DSBz=0.33)を有したことが明らかになった。定量カーボン13NMRによる分析は、生成物が環RDSの:RDS C6=1.00、RDS C3=0.91、RDS C2=1.00、およびベンゾエートカルボニルRDSの:RDS C6=0.04、RDS C3=0.12、RDS C2=0.17を有して位置選択的に置換されたことを示した。生成物は、DMSO、NMP、およびCH2Cl2等の多様な溶媒中で可溶であった。
【0151】
フィルムキャストおよびフィルム光学特性の測定のための一般的な基準に従って、セルロースベンゾエートプロピオネートは、面外レタデーション(Rth、589)+50.5nmをフィルム厚み27μmで(+112.2を60μmで)有することを見出した。
【0152】
この例は、脂肪族反応剤をまず添加し、続いて芳香族反応剤を添加する段階的な添加を採用することによって、脂肪族アシル基の位置選択的な配置(高いC6/C3比またはC6/C2比)および芳香族基のC2およびC3での導入がもたらされることを示した。これらの位置選択的に置換されたセルロースエステルから製造される補償フィルムは大きい正の値のRthを示した。
【特許請求の範囲】
【請求項1】
セルロースおよび少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートを含む、セルロース溶液。
【請求項2】
該テトラアルキルアンモニウムアルキルホスフェートが、式:
【化1】
(式中、R1、R2、R3およびR4は独立にC1−C5の直鎖もしくは分岐鎖のアルキル基またはC2−C20のアルコキシ基であり、そしてR5およびR6は独立にヒドリド、C1−C5の直鎖もしくは分岐鎖のアルキル基、またはC2−C20のアルコキシ基である)
を有する、請求項1に記載のセルロース溶液。
【請求項3】
R1がメチルまたはエチルであり;R2、R3およびR4が独立にメチル、エチル、プロピル、ブチル、イソブチル、ペンチル、エタノール、エトキシエタノールであり;R1、R2、R3およびR4が同一ではなく;そしてR5およびR6がメチル、エチル、プロピルまたはブチルである、請求項1に記載のセルロース溶液。
【請求項4】
R1がメチルであり;R2、R3およびR4がプロピルまたはブチルであり;そしてR5およびR6がメチルまたはエチルである、請求項5に記載のセルロース溶液。
【請求項5】
該テトラアルキルアンモニウムアルキルホスフェートが、トリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)、トリブチルエチルアンモニウムジエチルホスフェート([TBEA]DEP)、トリプロピルメチルアンモニウムジメチルホスフェート([TPMA]DMP)、およびトリプロピルエチルアンモニウムジエチルホスフェート([TPEA]DEP)からなる群から選択される少なくとも1種である、請求項4に記載のセルロース溶液。
【請求項6】
少なくとも1種の共溶媒を更に含む、請求項1に記載のセルロース溶液。
【請求項7】
該共溶媒が、非プロトン性溶媒、プロトン性溶媒、酸、およびテトラアルキルアンモニウムアルキルホスフェート以外のイオン液体からなる群から選択される少なくとも1種である、請求項6に記載のセルロース溶液。
【請求項8】
該非プロトン性溶媒が、ヘキサメチルホスホルアミド、N−メチルピロリドン、ニトロメタン、ジメチルホルムアミド、ジメチルアセトアミド、アセトニトリル、スルホラン、およびジメチルスルホキシドからなる群から選択される少なくとも1種である、請求項7に記載のセルロース溶液。
【請求項9】
該プロトン性溶媒が、脂肪族カルボン酸およびアミンからなる群から選択される少なくとも1種である、請求項7に記載のセルロース溶液。
【請求項10】
該脂肪族カルボン酸が、酢酸、プロピオン酸、酪酸、およびイソ酪酸からなる群から選択される少なくとも1種である、請求項9に記載のセルロース溶液。
【請求項11】
該アミンが、ジエチルアミン、ブチルアミン、ジブチルアミン、プロピルアミン、およびジプロピルアミンからなる群から選択される少なくとも1種である、請求項9に記載のセルロース溶液。
【請求項12】
該テトラアルキルアンモニウムアルキルホスフェート以外のイオン液体が、少なくとも1種のカルボキシル化イオン液体または少なくとも1種のハロゲン化イオン液体である、請求項6に記載のセルロース溶液。
【請求項13】
該イオン液体が、構造:
【化2】
(式中、R1およびR3は独立にC1−C8アルキル基、C2−C8アルケニル基、またはC1−C8アルコキシアルキル基であり、そしてR2、R4およびR5は独立にヒドリド、C1−C8アルキル基、C2−C8アルケニル基、C1−C8アルコキシアルキル基、またはC1−C8アルコキシ基であり;そして(X-)はクロリド、C1−C20の直鎖もしくは分岐鎖のカルボキシレートもしくは置換カルボキシレート、またはアルキルホスフェートである)
に対応する少なくとも1種のアルキルまたはアルケニル置換されたイミダゾリウム塩である、請求項12に記載のセルロース溶液。
【請求項14】
該酸が、アルキルスルホン酸およびアリールスルホン酸からなる群から選択される少なくとも1種である、請求項6に記載のセルロース溶液。
【請求項15】
該酸の量が、酸およびテトラアルキルアンモニウムアルキルホスフェートの総質量基準で約0.01〜約10質量%の範囲である、請求項14に記載のセルロース溶液。
【請求項16】
セルロースを少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートと接触させることを含む、セルロース溶液の製造方法。
【請求項17】
接触温度が約20℃〜約150℃である、請求項16に記載のセルロース溶液の製造方法。
【請求項18】
テトラアルキルアンモニウムアルキルホスフェート中に溶解できるセルロースの量が、セルロース溶液の総質量基準で約1質量%〜約40質量%の範囲である、請求項16に記載のセルロース溶液の製造方法。
【請求項19】
請求項1に記載のセルロース溶液から製造される、物品。
【請求項20】
a)セルロースおよび少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートを含むセルロース溶液を成形物に形成すること;ならびに
b)該成形物を少なくとも1種の非溶媒と接触させて物品を生成すること;
を含む、セルロース物品の製造方法。
【請求項1】
セルロースおよび少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートを含む、セルロース溶液。
【請求項2】
該テトラアルキルアンモニウムアルキルホスフェートが、式:
【化1】
(式中、R1、R2、R3およびR4は独立にC1−C5の直鎖もしくは分岐鎖のアルキル基またはC2−C20のアルコキシ基であり、そしてR5およびR6は独立にヒドリド、C1−C5の直鎖もしくは分岐鎖のアルキル基、またはC2−C20のアルコキシ基である)
を有する、請求項1に記載のセルロース溶液。
【請求項3】
R1がメチルまたはエチルであり;R2、R3およびR4が独立にメチル、エチル、プロピル、ブチル、イソブチル、ペンチル、エタノール、エトキシエタノールであり;R1、R2、R3およびR4が同一ではなく;そしてR5およびR6がメチル、エチル、プロピルまたはブチルである、請求項1に記載のセルロース溶液。
【請求項4】
R1がメチルであり;R2、R3およびR4がプロピルまたはブチルであり;そしてR5およびR6がメチルまたはエチルである、請求項5に記載のセルロース溶液。
【請求項5】
該テトラアルキルアンモニウムアルキルホスフェートが、トリブチルメチルアンモニウムジメチルホスフェート([TBMA]DMP)、トリブチルエチルアンモニウムジエチルホスフェート([TBEA]DEP)、トリプロピルメチルアンモニウムジメチルホスフェート([TPMA]DMP)、およびトリプロピルエチルアンモニウムジエチルホスフェート([TPEA]DEP)からなる群から選択される少なくとも1種である、請求項4に記載のセルロース溶液。
【請求項6】
少なくとも1種の共溶媒を更に含む、請求項1に記載のセルロース溶液。
【請求項7】
該共溶媒が、非プロトン性溶媒、プロトン性溶媒、酸、およびテトラアルキルアンモニウムアルキルホスフェート以外のイオン液体からなる群から選択される少なくとも1種である、請求項6に記載のセルロース溶液。
【請求項8】
該非プロトン性溶媒が、ヘキサメチルホスホルアミド、N−メチルピロリドン、ニトロメタン、ジメチルホルムアミド、ジメチルアセトアミド、アセトニトリル、スルホラン、およびジメチルスルホキシドからなる群から選択される少なくとも1種である、請求項7に記載のセルロース溶液。
【請求項9】
該プロトン性溶媒が、脂肪族カルボン酸およびアミンからなる群から選択される少なくとも1種である、請求項7に記載のセルロース溶液。
【請求項10】
該脂肪族カルボン酸が、酢酸、プロピオン酸、酪酸、およびイソ酪酸からなる群から選択される少なくとも1種である、請求項9に記載のセルロース溶液。
【請求項11】
該アミンが、ジエチルアミン、ブチルアミン、ジブチルアミン、プロピルアミン、およびジプロピルアミンからなる群から選択される少なくとも1種である、請求項9に記載のセルロース溶液。
【請求項12】
該テトラアルキルアンモニウムアルキルホスフェート以外のイオン液体が、少なくとも1種のカルボキシル化イオン液体または少なくとも1種のハロゲン化イオン液体である、請求項6に記載のセルロース溶液。
【請求項13】
該イオン液体が、構造:
【化2】
(式中、R1およびR3は独立にC1−C8アルキル基、C2−C8アルケニル基、またはC1−C8アルコキシアルキル基であり、そしてR2、R4およびR5は独立にヒドリド、C1−C8アルキル基、C2−C8アルケニル基、C1−C8アルコキシアルキル基、またはC1−C8アルコキシ基であり;そして(X-)はクロリド、C1−C20の直鎖もしくは分岐鎖のカルボキシレートもしくは置換カルボキシレート、またはアルキルホスフェートである)
に対応する少なくとも1種のアルキルまたはアルケニル置換されたイミダゾリウム塩である、請求項12に記載のセルロース溶液。
【請求項14】
該酸が、アルキルスルホン酸およびアリールスルホン酸からなる群から選択される少なくとも1種である、請求項6に記載のセルロース溶液。
【請求項15】
該酸の量が、酸およびテトラアルキルアンモニウムアルキルホスフェートの総質量基準で約0.01〜約10質量%の範囲である、請求項14に記載のセルロース溶液。
【請求項16】
セルロースを少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートと接触させることを含む、セルロース溶液の製造方法。
【請求項17】
接触温度が約20℃〜約150℃である、請求項16に記載のセルロース溶液の製造方法。
【請求項18】
テトラアルキルアンモニウムアルキルホスフェート中に溶解できるセルロースの量が、セルロース溶液の総質量基準で約1質量%〜約40質量%の範囲である、請求項16に記載のセルロース溶液の製造方法。
【請求項19】
請求項1に記載のセルロース溶液から製造される、物品。
【請求項20】
a)セルロースおよび少なくとも1種のテトラアルキルアンモニウムアルキルホスフェートを含むセルロース溶液を成形物に形成すること;ならびに
b)該成形物を少なくとも1種の非溶媒と接触させて物品を生成すること;
を含む、セルロース物品の製造方法。
【図1】

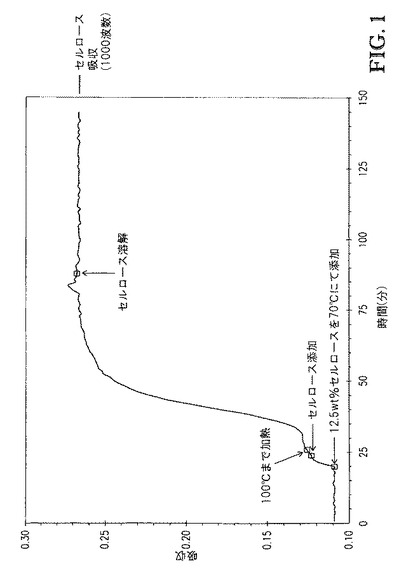
【図2】
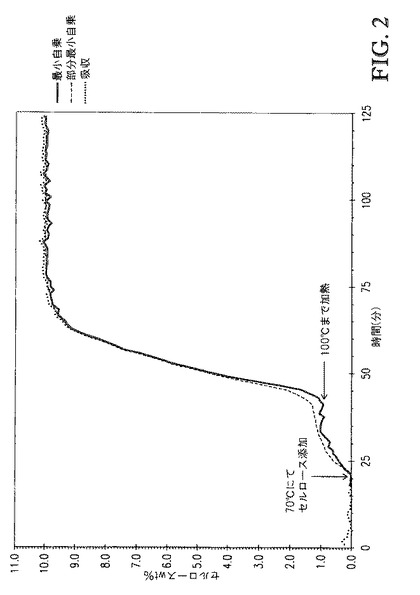
【図3】
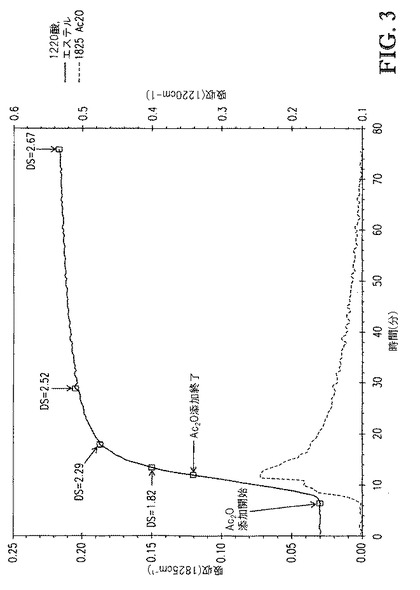
【図4】
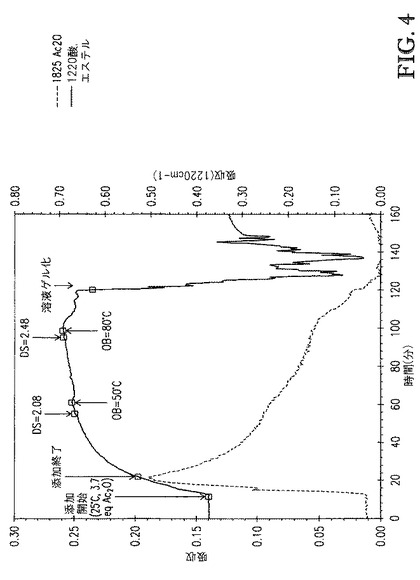
【図5】
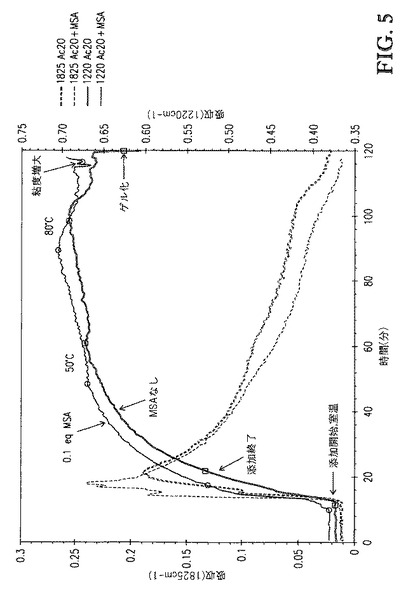
【図6】
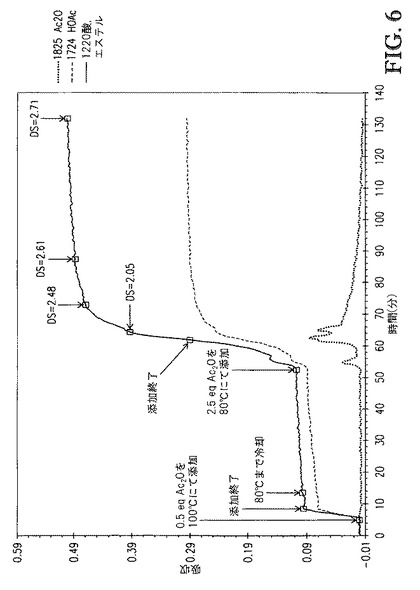
【図7】
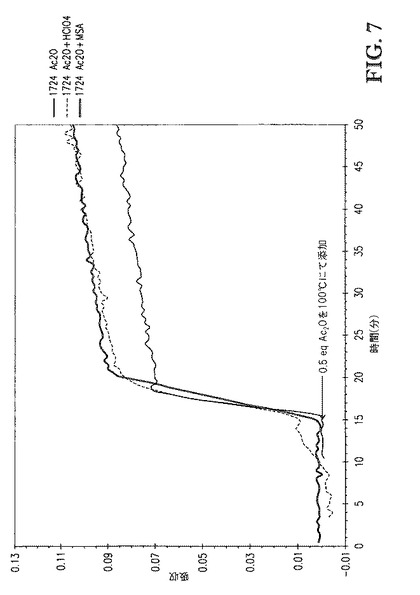
【図8】
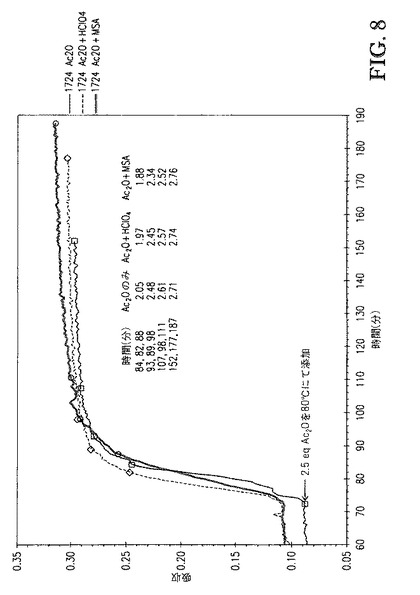
【図9】
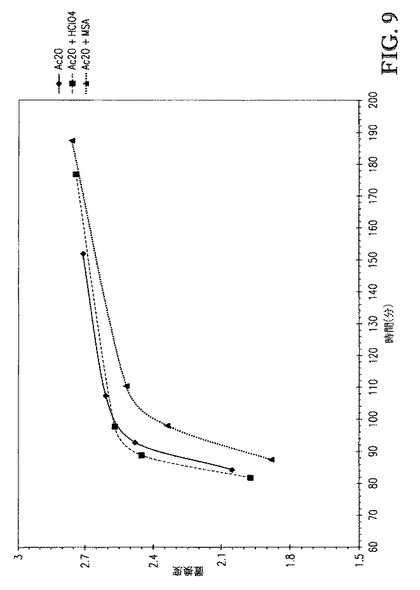
【図10】
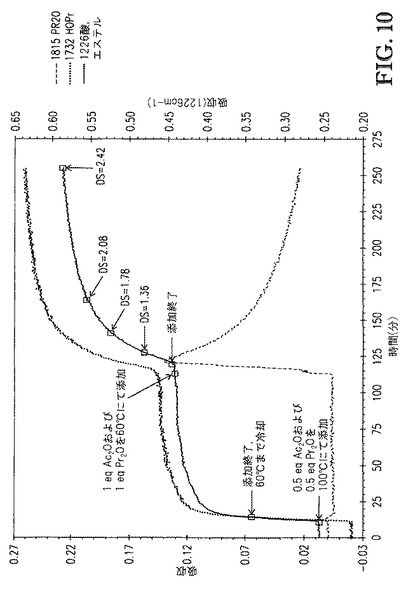
【図11】
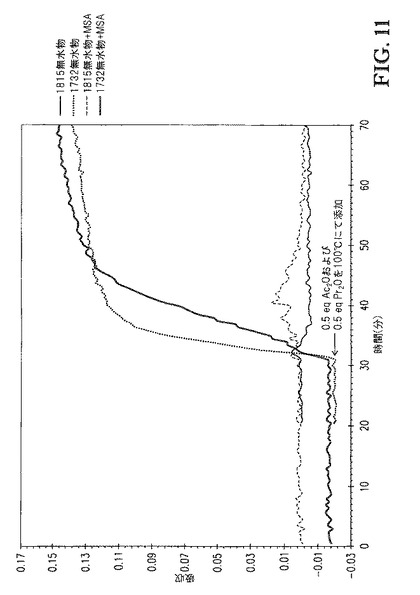
【図12】
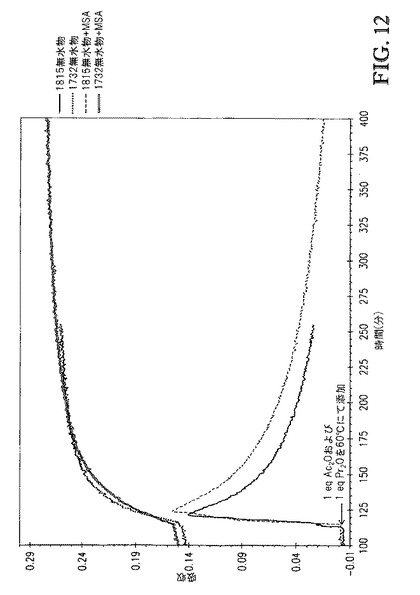
【図13】
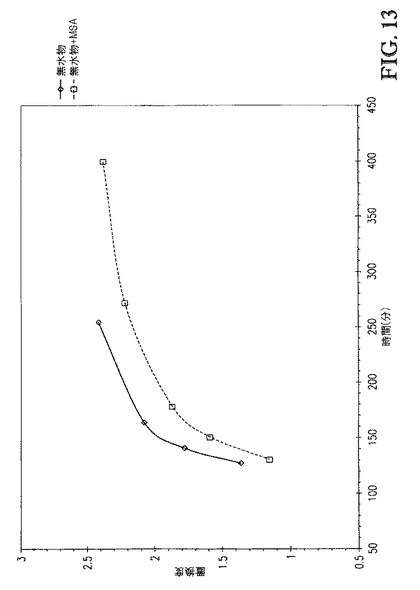
【図14】
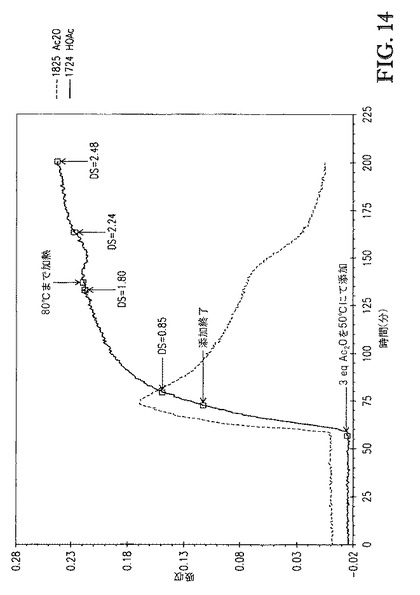
【図15】
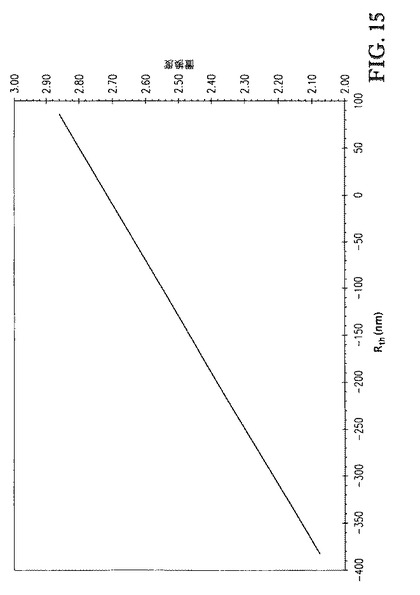
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公表番号】特表2012−524145(P2012−524145A)
【公表日】平成24年10月11日(2012.10.11)
【国際特許分類】
【出願番号】特願2012−505861(P2012−505861)
【出願日】平成21年8月13日(2009.8.13)
【国際出願番号】PCT/US2009/004626
【国際公開番号】WO2010/120269
【国際公開日】平成22年10月21日(2010.10.21)
【出願人】(594055158)イーストマン ケミカル カンパニー (391)
【Fターム(参考)】
【公表日】平成24年10月11日(2012.10.11)
【国際特許分類】
【出願日】平成21年8月13日(2009.8.13)
【国際出願番号】PCT/US2009/004626
【国際公開番号】WO2010/120269
【国際公開日】平成22年10月21日(2010.10.21)
【出願人】(594055158)イーストマン ケミカル カンパニー (391)
【Fターム(参考)】
[ Back to top ]