
テトラサイクリンのニトロ化
本発明は、一実施形態において、式(1)の化合物または薬学的に許容できるその塩[式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは、−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]の調製方法であって、(a)C1〜C12硝酸アルキルを、式(2)の化合物またはその塩と、酸重量/溶液重量が70%を超える濃度の酸の存在下で反応させて、式(3)の化合物またはその塩を含有する反応混合物を生成するステップであって、前記酸が、硫酸およびR5−SO3H[式中、R5は、1個または複数のハロゲンで置換されていてもよいC1〜C4アルキルであるか、またはR5は、1個または複数のC1〜C4アルキルもしくはハロゲンで置換されていてもよいC6〜C10アリールである]からなる群から選択される、ステップと、(b)式(3)の化合物またはその塩を還元して、式(4)の化合物またはその塩を形成するステップと、(c)式(4)の化合物をアシル化して、式(1)の化合物を形成するステップと、(d)場合により、式(1)の化合物の薬学的に許容できるその塩を任意選択により形成するステップとを含む方法を対象とする。
【化1】

【化2】
【化3】
【化4】
【図1】
【化1】
【化2】
【化3】
【化4】
【図1】
【発明の詳細な説明】
【技術分野】
【0001】
【背景技術】
【0002】
チゲサイクリンは、抗生物質への耐性出現という世界的な脅威に呼応して開発された。チゲサイクリンは、in vitroおよびin vivoの両方において拡大された広域スペクトルの抗菌活性を有する。グリシルサイクリン系抗生物質は、テトラサイクリン系抗生物質のように、細菌のタンパク質翻訳を阻害することにより作用する。
【0003】
チゲサイクリンは、テトラサイクリンファミリーの既知の抗生物質であり、ミノサイクリンの化学的類似体である。チゲサイクリンは、薬剤耐性細菌に対する処置として使用することができ、他の抗生物質が効かなかった場合に効果を示すことがわかっている。チゲサイクリンは、グラム陰性およびグラム陽性病原体、嫌気性菌、メチシリン感受性およびメチシリン耐性の両系統の黄色ブドウ球菌(MSSAおよびMRSA)によって引き起こされることのある、複雑性腹腔内感染症(cIAI)、複雑性皮膚・皮膚組織感染症(cSSSI)、市中肺炎(CAP)、院内肺炎(HAP)徴候などの多くの細菌感染の治療において使用することができる。さらに、チゲサイクリンを使用して、TetMおよびTetK耐性決定因子を有する細菌によって引き起こされる温血動物の細菌感染を治療またはコントロールすることもできる。チゲサイクリンはまた、骨・関節感染症、カテーテルに関連した好中球減少症、産婦人科感染症の治療、またはVRE、ESBL、腸内細菌、急速増殖性マイコバクテリア(rapid growing mycobacteria)などの他の耐性病原体の処置にも使用することができる。
【0004】
チゲサイクリンは、エピマー化によって分解することがあるという点で、いくつかの不都合な欠点をもつ。エピマー化は、テトラサイクリンの種類に応じて分解速度は様々に異なり得るものの、一般に、テトラサイクリンの知られている分解経路である。比較すると、チゲサイクリンのエピマー化速度は、たとえば弱酸性条件下かつ/またはやや高温であっても、速いことがある。テトラサイクリンの文献には、科学者らがテトラサイクリンのエピマー形成に対処し、これを最小限に抑えるのに使用してきたいくつかの方法が報告されている。一部の方法では、非水性溶液中にて塩基性pHで実施する場合、テトラサイクリンのカルシウム、マグネシウム、亜鉛、またはアルミニウム金属塩の形成により、エピマー形成が制限される(Gordon,P.N、Stephens Jr,C.R.、Noseworthy,M.M.、Teare,F.W.、英国特許第901,107号)。他の方法(Tobkes、米国特許第4,038,315号)では、金属錯体の形成を酸性pHで実施し、引き続いて安定な固体状の薬物を調製している。
【0005】
チゲサイクリンは、そのエピマーと構造が1箇所だけ異なる。チゲサイクリンでは、式Iに示すように、4位炭素のN−ジメチル基が、近接する水素に対してシスであるのに対し、
【0006】
【化1】
エピマー(すなわち、C4−エピマー)では、これらは、以下の構造に示されるように互いにトランスである。
【0007】
【化2】
【0008】
チゲサイクリンエピマーは、非毒性であると考えられているものの、ある種の条件下では、チゲサイクリンの抗菌効果を欠く場合もあり、したがって、望ましくない分解生成物となり得る。その上、チゲサイクリンを大規模で合成する場合には、エピマー化の量が増大する可能性がある。
【0009】
エピマー形成を低減する他の方法として、処理中のpHを約6.0より高く保つこと、ギ酸、酢酸、リン酸、ボロン酸などの弱酸のコンジュゲートとの接触を回避すること、および水ベースの溶液を含めた水分との接触を回避することが挙げられる。水分からの保護に関しては、NoseworthyおよびSpiegel(米国特許第3,026,248号)ならびにNashおよびHaeger(米国特許第3,219,529号)が、非水性媒体中にテトラサイクリン類似体を配合して、薬物の安定性を向上させることを提案している。しかし、これらの開示に含まれる媒体の大部分は、非経口よりも局所での使用に相応しいものである。テトラサイクリンエピマー化は、温度依存的であることも知られているため、テトラサイクリンを低温で製造および保管しても、エピマー形成速度を遅くすることができる(Yuen,P.H.、Sokoloski,T.D.、J.Pharm.Sci.66:1648〜1650、1977;Pawelczyk,E.、Matlak,B、Pol.J.Pharmacol.Pharm.34:409〜421、1982)。これらの方法のいくつかがチゲサイクリンで試みられてきたが、追加の分解剤を導入せずに、エピマー形成および酸化分解の両方を低減することに成功しているものはないようである。たとえば、金属錯化は、一般に塩基性pHではエピマー形成または分解にほとんど効果を示さないことがわかった。
【0010】
リン酸、酢酸、およびクエン酸緩衝剤の使用は、溶液状態の安定性を向上させるものの、凍結乾燥状態のチゲサイクリンの分解を促進するようである。しかし、緩衝剤なしでも、エピマー化は、チゲサイクリンではミノサイクリンなどの他のテトラサイクリンよりも深刻な問題である。
【0011】
C4−エピマーに加えて、他の不純物として、酸化副生成物が挙げられる。こうした副生成物の一部は、分子のD環が酸化されて得られる、アミノフェノールである。式3の化合物(本明細書のスキーム1を参照されたい)は、C−11位およびC−12a位が容易に酸化され得る。非溶媒を用いた沈殿による式3の化合物の単離は、酸化副生成物および金属塩が生成物と同時に沈殿し、純度が非常に低くなるという問題を伴う。式3の化合物の核の酸化および分解は、塩基性反応条件下ではより顕著になる可能性があり、大規模な工程においては、経過時間が通常はより長く、化合物がより長い時間塩基と接触するため、なおさらである。
【0012】
さらに、分解生成物は、1つのスキームの異なる合成ステップそれぞれの間に得られる場合もあり、こうした分解生成物から必要な化合物を分離することは、面倒な作業となる可能性がある。たとえば、シリカゲルクロマトグラフィーや分取HPLCなどの従来の精製技術を使用しても、こうした化合物は、そのキレート化特性のために簡単には精製できない。一部のテトラサイクリンは、EDTAのような金属イオン封鎖剤を含有する、緩衝化した固定相で飽和させた珪藻土でできたカラムを使用する分配クロマトグラフィーによって精製されているが、こうした技術は、分離能、再現精度、および処理能力が非常に低いという欠点をもつ場合がある。これらの不利点は、大規模合成の妨げとなる可能性がある。HPLCも精製に使用されているが、HPLCカラムで種々の成分を十分に分離するには、移動相中にイオン対形成剤が存在する必要がある。移動相中の金属イオン封鎖剤およびイオン対形成剤から最終生成物を分離することは、困難である可能性がある。
【0013】
小規模では、沈殿によって得られる不純な化合物を分取逆相HPLCによって精製してもよいが、逆相液体クロマトグラフィーによる精製は、キログラム分量の材料を扱う場合は非効率でコスト高になりかねない。
【発明の概要】
【発明が解決しようとする課題】
【0014】
チゲサイクリン、式1の化合物、構造的に関連する化合物などの化合物の形成のためのプロセスは、米国特許公開第2007−0026080A1号、第2007−0049560A1号、第2007−0049563A1号、第2007−0049562A1号、第2007−0049561A1号、および米国特許出願第12/251,488号に記載されており、これら文献をそれぞれ、全体として参照により本明細書に援用する。しかし、式1の1種類の化合物を、これまでに実現されているものより精製された形で得ることが依然として求められている。また、精製のためのクロマトグラフィーの使用を最小限にする新しい合成も依然として求められている。
【0015】
一例として、米国特許公開第2007−0049560号で開示されているチゲサイクリンの調製では、硝酸によるミノサイクリンのニトロ化が、プロセスのステップの1つである。テトラサイクリンの硝酸によるニトロ化も、US2007−0244335に記載されている。他の化学化合物の硝酸アルキルによるニトロ化が、WO2003/011810、米国特許第3,694,513号、Chem.Rev.1955、55、485〜510、J.Am.Chem.Soc.1974、96、2892〜98、米国特許第2,4169,74号、および米国特許第7,005,553号で報告されている。しかし、ミノサイクリンのニトロ化のための硝酸アルキルの使用は報告されていない。
【課題を解決するための手段】
【0016】
一実施形態では、本発明は、式1の化合物
【0017】
【化3】
または薬学的に許容できるその塩[式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは、−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]の調製方法であって、
(a)C1〜C12硝酸アルキルを、式2の化合物
【0018】
【化4】
またはその塩と、酸重量/溶液重量が70%を超える濃度の酸の存在下で反応させて、式3の化合物またはその塩
【0019】
【化5】
を含有する反応混合物を生成するステップであって、前記塩が、硫酸およびR5−SO3H[式中、R5は、1個または複数のハロゲンで置換されていてもよいC1〜C4アルキルであるか、またはR5は、1個または複数のC1〜C4アルキルまたはハロゲンで置換されていてもよいC6〜C10アリールである]からなる群から選択される、ステップと、
(b)式3の化合物またはその塩を還元して、式4の化合物またはその塩
【0020】
【化6】
を形成するステップと、
(c)式4の化合物をアシル化して、式1の化合物を形成するステップと、
(d)場合により、式1の化合物の薬学的に許容できるその塩を形成するステップと
を含む方法を対象とする。
【0021】
本発明は、別の実施形態では、式3の化合物
【0022】
【化7】
またはその塩[式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは、−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]の調製方法であって、C1〜C12硝酸アルキルを、式2の化合物
【0023】
【化8】
またはその塩と、酸重量/溶液重量が70%を超えるに等しい濃度の酸の存在下で反応させて、式3の化合物または式3の化合物の塩を生成するステップであって、前記酸が、硫酸およびR5−SO3H[式中、R5は、1個または複数のハロゲンで置換されていてもよいC1〜C4アルキルであるか、またはR5は、1個または複数のC1〜C4アルキルもしくはハロゲンで置換されていてもよいC6〜C10アリールである]からなる群から選択される、ステップを含む方法を対象とする。
【0024】
本明細書で開示する方法では、エピマー形成、出発試薬の存在、酸化副生成物などの、最終生成物中に存在する少なくとも1種の不純物の量を低減しながら、所望の生成物を形成することができる。不純物のそうした低減は、合成の少なくとも1段階の間、すなわち、ニトロ化、還元、およびアシル化反応のいずれか1つの間に実現することができる。本明細書で開示する方法は、最終生成物の純度が適切な状態で大規模合成を容易にすることもできる。
【図面の簡単な説明】
【0025】
【技術分野】
【0001】
【背景技術】
【0002】
チゲサイクリンは、抗生物質への耐性出現という世界的な脅威に呼応して開発された。チゲサイクリンは、in vitroおよびin vivoの両方において拡大された広域スペクトルの抗菌活性を有する。グリシルサイクリン系抗生物質は、テトラサイクリン系抗生物質のように、細菌のタンパク質翻訳を阻害することにより作用する。
【0003】
チゲサイクリンは、テトラサイクリンファミリーの既知の抗生物質であり、ミノサイクリンの化学的類似体である。チゲサイクリンは、薬剤耐性細菌に対する処置として使用することができ、他の抗生物質が効かなかった場合に効果を示すことがわかっている。チゲサイクリンは、グラム陰性およびグラム陽性病原体、嫌気性菌、メチシリン感受性およびメチシリン耐性の両系統の黄色ブドウ球菌(MSSAおよびMRSA)によって引き起こされることのある、複雑性腹腔内感染症(cIAI)、複雑性皮膚・皮膚組織感染症(cSSSI)、市中肺炎(CAP)、院内肺炎(HAP)徴候などの多くの細菌感染の治療において使用することができる。さらに、チゲサイクリンを使用して、TetMおよびTetK耐性決定因子を有する細菌によって引き起こされる温血動物の細菌感染を治療またはコントロールすることもできる。チゲサイクリンはまた、骨・関節感染症、カテーテルに関連した好中球減少症、産婦人科感染症の治療、またはVRE、ESBL、腸内細菌、急速増殖性マイコバクテリア(rapid growing mycobacteria)などの他の耐性病原体の処置にも使用することができる。
【0004】
チゲサイクリンは、エピマー化によって分解することがあるという点で、いくつかの不都合な欠点をもつ。エピマー化は、テトラサイクリンの種類に応じて分解速度は様々に異なり得るものの、一般に、テトラサイクリンの知られている分解経路である。比較すると、チゲサイクリンのエピマー化速度は、たとえば弱酸性条件下かつ/またはやや高温であっても、速いことがある。テトラサイクリンの文献には、科学者らがテトラサイクリンのエピマー形成に対処し、これを最小限に抑えるのに使用してきたいくつかの方法が報告されている。一部の方法では、非水性溶液中にて塩基性pHで実施する場合、テトラサイクリンのカルシウム、マグネシウム、亜鉛、またはアルミニウム金属塩の形成により、エピマー形成が制限される(Gordon,P.N、Stephens Jr,C.R.、Noseworthy,M.M.、Teare,F.W.、英国特許第901,107号)。他の方法(Tobkes、米国特許第4,038,315号)では、金属錯体の形成を酸性pHで実施し、引き続いて安定な固体状の薬物を調製している。
【0005】
チゲサイクリンは、そのエピマーと構造が1箇所だけ異なる。チゲサイクリンでは、式Iに示すように、4位炭素のN−ジメチル基が、近接する水素に対してシスであるのに対し、
【0006】
【化1】
エピマー(すなわち、C4−エピマー)では、これらは、以下の構造に示されるように互いにトランスである。
【0007】
【化2】
【0008】
チゲサイクリンエピマーは、非毒性であると考えられているものの、ある種の条件下では、チゲサイクリンの抗菌効果を欠く場合もあり、したがって、望ましくない分解生成物となり得る。その上、チゲサイクリンを大規模で合成する場合には、エピマー化の量が増大する可能性がある。
【0009】
エピマー形成を低減する他の方法として、処理中のpHを約6.0より高く保つこと、ギ酸、酢酸、リン酸、ボロン酸などの弱酸のコンジュゲートとの接触を回避すること、および水ベースの溶液を含めた水分との接触を回避することが挙げられる。水分からの保護に関しては、NoseworthyおよびSpiegel(米国特許第3,026,248号)ならびにNashおよびHaeger(米国特許第3,219,529号)が、非水性媒体中にテトラサイクリン類似体を配合して、薬物の安定性を向上させることを提案している。しかし、これらの開示に含まれる媒体の大部分は、非経口よりも局所での使用に相応しいものである。テトラサイクリンエピマー化は、温度依存的であることも知られているため、テトラサイクリンを低温で製造および保管しても、エピマー形成速度を遅くすることができる(Yuen,P.H.、Sokoloski,T.D.、J.Pharm.Sci.66:1648〜1650、1977;Pawelczyk,E.、Matlak,B、Pol.J.Pharmacol.Pharm.34:409〜421、1982)。これらの方法のいくつかがチゲサイクリンで試みられてきたが、追加の分解剤を導入せずに、エピマー形成および酸化分解の両方を低減することに成功しているものはないようである。たとえば、金属錯化は、一般に塩基性pHではエピマー形成または分解にほとんど効果を示さないことがわかった。
【0010】
リン酸、酢酸、およびクエン酸緩衝剤の使用は、溶液状態の安定性を向上させるものの、凍結乾燥状態のチゲサイクリンの分解を促進するようである。しかし、緩衝剤なしでも、エピマー化は、チゲサイクリンではミノサイクリンなどの他のテトラサイクリンよりも深刻な問題である。
【0011】
C4−エピマーに加えて、他の不純物として、酸化副生成物が挙げられる。こうした副生成物の一部は、分子のD環が酸化されて得られる、アミノフェノールである。式3の化合物(本明細書のスキーム1を参照されたい)は、C−11位およびC−12a位が容易に酸化され得る。非溶媒を用いた沈殿による式3の化合物の単離は、酸化副生成物および金属塩が生成物と同時に沈殿し、純度が非常に低くなるという問題を伴う。式3の化合物の核の酸化および分解は、塩基性反応条件下ではより顕著になる可能性があり、大規模な工程においては、経過時間が通常はより長く、化合物がより長い時間塩基と接触するため、なおさらである。
【0012】
さらに、分解生成物は、1つのスキームの異なる合成ステップそれぞれの間に得られる場合もあり、こうした分解生成物から必要な化合物を分離することは、面倒な作業となる可能性がある。たとえば、シリカゲルクロマトグラフィーや分取HPLCなどの従来の精製技術を使用しても、こうした化合物は、そのキレート化特性のために簡単には精製できない。一部のテトラサイクリンは、EDTAのような金属イオン封鎖剤を含有する、緩衝化した固定相で飽和させた珪藻土でできたカラムを使用する分配クロマトグラフィーによって精製されているが、こうした技術は、分離能、再現精度、および処理能力が非常に低いという欠点をもつ場合がある。これらの不利点は、大規模合成の妨げとなる可能性がある。HPLCも精製に使用されているが、HPLCカラムで種々の成分を十分に分離するには、移動相中にイオン対形成剤が存在する必要がある。移動相中の金属イオン封鎖剤およびイオン対形成剤から最終生成物を分離することは、困難である可能性がある。
【0013】
小規模では、沈殿によって得られる不純な化合物を分取逆相HPLCによって精製してもよいが、逆相液体クロマトグラフィーによる精製は、キログラム分量の材料を扱う場合は非効率でコスト高になりかねない。
【発明の概要】
【発明が解決しようとする課題】
【0014】
チゲサイクリン、式1の化合物、構造的に関連する化合物などの化合物の形成のためのプロセスは、米国特許公開第2007−0026080A1号、第2007−0049560A1号、第2007−0049563A1号、第2007−0049562A1号、第2007−0049561A1号、および米国特許出願第12/251,488号に記載されており、これら文献をそれぞれ、全体として参照により本明細書に援用する。しかし、式1の1種類の化合物を、これまでに実現されているものより精製された形で得ることが依然として求められている。また、精製のためのクロマトグラフィーの使用を最小限にする新しい合成も依然として求められている。
【0015】
一例として、米国特許公開第2007−0049560号で開示されているチゲサイクリンの調製では、硝酸によるミノサイクリンのニトロ化が、プロセスのステップの1つである。テトラサイクリンの硝酸によるニトロ化も、US2007−0244335に記載されている。他の化学化合物の硝酸アルキルによるニトロ化が、WO2003/011810、米国特許第3,694,513号、Chem.Rev.1955、55、485〜510、J.Am.Chem.Soc.1974、96、2892〜98、米国特許第2,4169,74号、および米国特許第7,005,553号で報告されている。しかし、ミノサイクリンのニトロ化のための硝酸アルキルの使用は報告されていない。
【課題を解決するための手段】
【0016】
一実施形態では、本発明は、式1の化合物
【0017】
【化3】
または薬学的に許容できるその塩[式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは、−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]の調製方法であって、
(a)C1〜C12硝酸アルキルを、式2の化合物
【0018】
【化4】
またはその塩と、酸重量/溶液重量が70%を超える濃度の酸の存在下で反応させて、式3の化合物またはその塩
【0019】
【化5】
を含有する反応混合物を生成するステップであって、前記塩が、硫酸およびR5−SO3H[式中、R5は、1個または複数のハロゲンで置換されていてもよいC1〜C4アルキルであるか、またはR5は、1個または複数のC1〜C4アルキルまたはハロゲンで置換されていてもよいC6〜C10アリールである]からなる群から選択される、ステップと、
(b)式3の化合物またはその塩を還元して、式4の化合物またはその塩
【0020】
【化6】
を形成するステップと、
(c)式4の化合物をアシル化して、式1の化合物を形成するステップと、
(d)場合により、式1の化合物の薬学的に許容できるその塩を形成するステップと
を含む方法を対象とする。
【0021】
本発明は、別の実施形態では、式3の化合物
【0022】
【化7】
またはその塩[式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは、−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]の調製方法であって、C1〜C12硝酸アルキルを、式2の化合物
【0023】
【化8】
またはその塩と、酸重量/溶液重量が70%を超えるに等しい濃度の酸の存在下で反応させて、式3の化合物または式3の化合物の塩を生成するステップであって、前記酸が、硫酸およびR5−SO3H[式中、R5は、1個または複数のハロゲンで置換されていてもよいC1〜C4アルキルであるか、またはR5は、1個または複数のC1〜C4アルキルもしくはハロゲンで置換されていてもよいC6〜C10アリールである]からなる群から選択される、ステップを含む方法を対象とする。
【0024】
本明細書で開示する方法では、エピマー形成、出発試薬の存在、酸化副生成物などの、最終生成物中に存在する少なくとも1種の不純物の量を低減しながら、所望の生成物を形成することができる。不純物のそうした低減は、合成の少なくとも1段階の間、すなわち、ニトロ化、還元、およびアシル化反応のいずれか1つの間に実現することができる。本明細書で開示する方法は、最終生成物の純度が適切な状態で大規模合成を容易にすることもできる。
【図面の簡単な説明】
【0025】
【特許請求の範囲】
【請求項1】
式1の化合物
【化1】
または薬学的に許容できるその塩[式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは、−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]の調製方法であって、(a)C1〜C12硝酸アルキルを、式2の化合物
【化2】
またはその塩と、酸重量/溶液重量が70%を超える濃度の酸の存在下で反応させて、式3の化合物またはその塩
【化3】
を含有する反応混合物を生成するステップであって、前記酸が、硫酸およびR5−SO3H[式中、R5は、1個または複数のハロゲンで置換されていてもよいC1〜C4アルキルであるか、またはR5は、1個または複数のC1〜C4アルキルまたはハロゲンで置換されていてもよいC6〜C10アリールである]からなる群から選択される、ステップと、
(b)式3の化合物またはその塩を還元して、式4の化合物またはその塩
【化4】
を形成するステップと、
(c)式4の化合物をアシル化して、式1の化合物を形成するステップと、
(d)場合により、式1の化合物の薬学的に許容できるその塩を形成するステップと
を含む方法。
【請求項2】
式3の化合物が、反応混合物から単離される、請求項1に記載の方法。
【請求項3】
式3の化合物が、ステップ(b)より前に反応混合物から単離されない、請求項1に記載の方法。
【請求項4】
酸が硫酸である、請求項1、2または3に記載の方法。
【請求項5】
硫酸の濃度が少なくとも95%である、請求項1、2、3または4に記載の方法。
【請求項6】
硝酸アルキルが、式2の化合物に対してモル過剰で存在する、請求項1に記載の方法。
【請求項7】
モル過剰が少なくとも1.05当量である、請求項6に記載の方法。
【請求項8】
(a)の反応ステップが、10℃〜30℃の範囲の温度の反応である、請求項1に記載の方法。
【請求項9】
(a)の反応ステップが、式2の化合物の塩との反応である、請求項1に記載の方法。
【請求項10】
式2の化合物の塩が、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩、硝酸塩、硫酸塩、酢酸塩、安息香酸塩、クエン酸塩、システイン塩、フマル酸塩、グリコール酸塩、マレイン酸塩、コハク酸塩、酒石酸塩、硫酸塩、およびクロロベンゼンスルホン酸塩からなる群から選択される、請求項9に記載の方法。
【請求項11】
式2の化合物の塩が、塩酸塩または硫酸塩である、請求項9に記載の方法。
【請求項12】
高速液体クロマトグラフィーによって求めたとき、反応混合物が、式3の化合物の2.5%以下の量の式3のC4−エピマーを含む、請求項1に記載の方法。
【請求項13】
R1が水素であり、R2がt−ブチルであり、R3がメチルであり、R4がメチルであり、nが1である、前記請求項のいずれかに記載の方法。
【請求項14】
式1の化合物がチゲサイクリンである、請求項1に記載の方法。
【請求項15】
式1の化合物がチゲサイクリンHClである、請求項1に記載の方法。
【請求項16】
C1〜C12硝酸アルキルがC1〜C8硝酸アルキルである、請求項1に記載の方法。
【請求項17】
C1〜C8硝酸アルキルが硝酸2−エチルヘキシルである、請求項16に記載の方法。
【請求項18】
C1〜C8硝酸アルキルがC3〜C6硝酸アルキルである、請求項16に記載の方法。
【請求項19】
C3〜C6硝酸アルキルが硝酸イソプロピルである、請求項17に記載の方法。
【請求項20】
式3の化合物
【化5】
またはその塩[式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは、−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]の調製方法であって、C1〜C12硝酸アルキルを、式2の化合物
【化6】
またはその塩と、酸重量/溶液重量が70%を超える濃度の酸の存在下で反応させて、式3の化合物または式3の化合物の塩を形成するステップであって、前記酸が、硫酸およびR5−SO3H[式中、R5は、1個または複数のハロゲンで置換されていてもよいC1〜C4アルキルであるか、またはR5は、1個または複数のC1〜C4アルキルまたはハロゲンで置換されていてもよいC6〜C10アリールである]からなる群から選択される、ステップを含む方法。
【請求項21】
式3の化合物が、9−ニトロミノサイクリンまたは薬学的に許容できるその塩である、請求項20に記載の方法。
【請求項22】
式3の化合物が、9−ニトロミノサイクリンHClである、請求項20に記載の方法。
【請求項23】
C1〜C12硝酸アルキルがC1〜C8硝酸アルキルである、請求項20に記載の方法。
【請求項24】
C1〜C8硝酸アルキルが硝酸2−エチルヘキシルである、請求項23に記載の方法。
【請求項25】
C1〜C8硝酸アルキルがC3〜C6硝酸アルキルである、請求項23に記載の方法。
【請求項26】
C3〜C6硝酸アルキルが硝酸イソプロピルである、請求項25に記載の方法。
【請求項1】
式1の化合物
【化1】
または薬学的に許容できるその塩[式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは、−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]の調製方法であって、(a)C1〜C12硝酸アルキルを、式2の化合物
【化2】
またはその塩と、酸重量/溶液重量が70%を超える濃度の酸の存在下で反応させて、式3の化合物またはその塩
【化3】
を含有する反応混合物を生成するステップであって、前記酸が、硫酸およびR5−SO3H[式中、R5は、1個または複数のハロゲンで置換されていてもよいC1〜C4アルキルであるか、またはR5は、1個または複数のC1〜C4アルキルまたはハロゲンで置換されていてもよいC6〜C10アリールである]からなる群から選択される、ステップと、
(b)式3の化合物またはその塩を還元して、式4の化合物またはその塩
【化4】
を形成するステップと、
(c)式4の化合物をアシル化して、式1の化合物を形成するステップと、
(d)場合により、式1の化合物の薬学的に許容できるその塩を形成するステップと
を含む方法。
【請求項2】
式3の化合物が、反応混合物から単離される、請求項1に記載の方法。
【請求項3】
式3の化合物が、ステップ(b)より前に反応混合物から単離されない、請求項1に記載の方法。
【請求項4】
酸が硫酸である、請求項1、2または3に記載の方法。
【請求項5】
硫酸の濃度が少なくとも95%である、請求項1、2、3または4に記載の方法。
【請求項6】
硝酸アルキルが、式2の化合物に対してモル過剰で存在する、請求項1に記載の方法。
【請求項7】
モル過剰が少なくとも1.05当量である、請求項6に記載の方法。
【請求項8】
(a)の反応ステップが、10℃〜30℃の範囲の温度の反応である、請求項1に記載の方法。
【請求項9】
(a)の反応ステップが、式2の化合物の塩との反応である、請求項1に記載の方法。
【請求項10】
式2の化合物の塩が、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩、硝酸塩、硫酸塩、酢酸塩、安息香酸塩、クエン酸塩、システイン塩、フマル酸塩、グリコール酸塩、マレイン酸塩、コハク酸塩、酒石酸塩、硫酸塩、およびクロロベンゼンスルホン酸塩からなる群から選択される、請求項9に記載の方法。
【請求項11】
式2の化合物の塩が、塩酸塩または硫酸塩である、請求項9に記載の方法。
【請求項12】
高速液体クロマトグラフィーによって求めたとき、反応混合物が、式3の化合物の2.5%以下の量の式3のC4−エピマーを含む、請求項1に記載の方法。
【請求項13】
R1が水素であり、R2がt−ブチルであり、R3がメチルであり、R4がメチルであり、nが1である、前記請求項のいずれかに記載の方法。
【請求項14】
式1の化合物がチゲサイクリンである、請求項1に記載の方法。
【請求項15】
式1の化合物がチゲサイクリンHClである、請求項1に記載の方法。
【請求項16】
C1〜C12硝酸アルキルがC1〜C8硝酸アルキルである、請求項1に記載の方法。
【請求項17】
C1〜C8硝酸アルキルが硝酸2−エチルヘキシルである、請求項16に記載の方法。
【請求項18】
C1〜C8硝酸アルキルがC3〜C6硝酸アルキルである、請求項16に記載の方法。
【請求項19】
C3〜C6硝酸アルキルが硝酸イソプロピルである、請求項17に記載の方法。
【請求項20】
式3の化合物
【化5】
またはその塩[式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは、−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]の調製方法であって、C1〜C12硝酸アルキルを、式2の化合物
【化6】
またはその塩と、酸重量/溶液重量が70%を超える濃度の酸の存在下で反応させて、式3の化合物または式3の化合物の塩を形成するステップであって、前記酸が、硫酸およびR5−SO3H[式中、R5は、1個または複数のハロゲンで置換されていてもよいC1〜C4アルキルであるか、またはR5は、1個または複数のC1〜C4アルキルまたはハロゲンで置換されていてもよいC6〜C10アリールである]からなる群から選択される、ステップを含む方法。
【請求項21】
式3の化合物が、9−ニトロミノサイクリンまたは薬学的に許容できるその塩である、請求項20に記載の方法。
【請求項22】
式3の化合物が、9−ニトロミノサイクリンHClである、請求項20に記載の方法。
【請求項23】
C1〜C12硝酸アルキルがC1〜C8硝酸アルキルである、請求項20に記載の方法。
【請求項24】
C1〜C8硝酸アルキルが硝酸2−エチルヘキシルである、請求項23に記載の方法。
【請求項25】
C1〜C8硝酸アルキルがC3〜C6硝酸アルキルである、請求項23に記載の方法。
【請求項26】
C3〜C6硝酸アルキルが硝酸イソプロピルである、請求項25に記載の方法。
【図1】(a)硝酸イソプロピルおよび硫酸、ならびに(b)硝酸および硫酸によってミノサイクリンをニトロ化した生成物のHPLCの比較を示す図である。
【発明を実施するための形態】
【0026】
定義
本明細書および付属の特許請求の範囲で使用するとき、単数形の「1つの(a)」、「1つの(an)」および「その(the)」は、内容が明らかに別途規定していない限り、複数の指示物を包含することを留意されたい。したがって、たとえば、「1種の化合物(a compound)」を含有する組成物への言及は、2種以上の化合物の混合物を包含する。また用語「または(or)」は、内容が明らかに別途規定していない限り、「および/または(and/or)」を含めた意味で一般に用いることを留意されたい。
【0027】
「チゲサイクリン」は、本明細書では、任意の薬学的に許容できる塩、鏡像異性体、エピマーなどの、遊離塩基形態および塩形態のチゲサイクリンを包含する。チゲサイクリンは、本明細書では、当業界で知られている方法に従って製剤することができる。同様に、「ミノサイクリン」も、本明細書では、任意の薬学的に許容できる塩、鏡像異性体、エピマーなどの、遊離塩基形態および塩形態のミノサイクリンを包含する。
【0028】
「化合物」とは、本明細書では、中性化合物(たとえば、遊離塩基)およびその塩形態(薬学的に許容できる塩など)を指す。化合物は、無水形態で、または水和物として、または溶媒和物として存在する場合がある。化合物は、立体異性体(たとえば、鏡像異性体およびジアステレオ異性体)として存在することもあり、鏡像異性体、ラセミ混合物、ジアステレオ異性体、およびその混合物として単離することができる。固体形態の化合物は、種々の結晶形態および非晶質形態で存在する場合がある。
【0029】
「薬学的に許容できる」とは、本明細書では、信頼できる医学的判断の範囲内で、妥当なリスク/利益比で、過度の毒性、刺激、アレルギー反応、または他の問題もしくは合併症なしに、患者の組織と接触させて使用するのに適する化合物、材料、組成物、および/または剤形を指す。
【0030】
用語「ハロゲン」とは、本明細書では、フルオロ、クロロ、ブロモ、および/またはヨードを指す。
【0031】
用語「アルキル」とは、本明細書では、1〜12個の炭素原子、たとえば、1〜8個、1〜6個、または1〜4個の炭素原子を有する、直鎖または分枝鎖の飽和脂肪族炭化水素基を指す。
【0032】
アルキル基の例として、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、イソペンチル、ネオペンチル、ヘキシル、ヘプチル、オクチルなどが挙げられる。
【0033】
用語「アルキレン」とは、本明細書では、1〜6個の炭素原子、たとえば、1〜4個、1〜3個、または1〜2個の炭素原子を有する、ジラジカルの直鎖または分枝鎖飽和脂肪族炭化水素基を指す。
【0034】
アルキレン基の例として、メチレン、エチレン、トリメチレン(1,3−プロパンジイル)、プロピレン(1,2−プロパンジイル)、テトラメチレン(1,4−ブタンジイル)、ブチレン(1,2−ブタンジイル)、1,3−ブタンジイル、2−メチル−1,3−プロパンジイル、ペンタメチレン(1,5−ペンタンジイル)、ペンチレン(1,2−ペンタンジイル)、ヘキサメチレン(1,6−ヘキサンジイル)、ヘキシレン(1,2−ヘキサンジイル)、2,3−ジメチル−1,4−ブタンジイルなどが挙げられる。
【0035】
用語「ヒドロキシ」または「ヒドロキシル」とは、本明細書では、−OH基を指す。
【0036】
用語「ニトロ」とは、本明細書では、基−NO2を指す。
【0037】
用語「アミノ」とは、本明細書では、−NH2基を指す。
【0038】
用語「アリール」とは、本明細書では、6〜14個の環炭素原子を含んでいる芳香族炭化水素基を指す。「C6〜C14アリール」とは、フェニル、ナフチル、ビフェニル、アントリル、テトラヒドロナフチル、フルオレニル、インダニル、ビフェニレニル、およびアセナフテニル基を指す。C6〜C14アリール基の例として、限定はしないが、フェニル、1−ナフチル、2−ナフチル、および3−ビフェン−1−イルが挙げられる。
【0039】
用語「シクロアルキル」とは、本明細書では、3〜8個の炭素原子を含んでいる非芳香族の単環式飽和炭化水素環を指す。C3〜C8シクロアルキルの代表例として、限定はしないが、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、およびシクロオクチルが挙げられる。炭素環の同じ炭素原子上にある任意の2個の水素原子のそれぞれが酸素原子で置き換えられて、オキソ(=O)置換基を形成していてもよく、または、アルキレンジオキシ基が、結合している炭素原子と一緒になったとき、2個の酸素原子を含んでいる5〜7員ヘテロ環を形成するように、その2個の水素原子がアルキレンジオキシ基で置き換えられていてもよい。
【0040】
別段指摘しない限り、本明細書で明確に規定しない置換基の命名は、官能基の末端部分に続いて、近接する官能基へと、結合点に向かって命名することによりなされる。たとえば、置換基「アリールアルキルオキシカルボニル」とは、基(アリール)−(アルキル)−O−C(O)−を指す。
【0041】
連続反応における各化合物は、遊離塩基の形でも塩の形でもよい。「塩」は、本明細書では、遊離塩基を適切な酸と反応させることにより、その場で、または別途調製することができる。例となる塩として、限定はしないが、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩、硝酸塩、硫酸塩、酢酸塩、安息香酸塩、クエン酸塩、システイン塩、フマル酸塩、グリコール酸塩、マレイン酸塩、コハク酸塩、酒石酸塩、硫酸塩、およびクロロベンゼンスルホン酸塩が挙げられる。別の実施形態では、塩は、アルキルスルホン酸塩およびアリールスルホン酸塩から選択することができる。一実施形態では、式2の化合物は、塩酸塩などの、上で開示した塩のいずれかであってよい塩として、または硫酸塩として提供される。
【0042】
「中間体」とは、本明細書では、出発材料と最終生成物の中間の生成物として生成される化合物を指す。一実施形態では、中間体は、式2の化合物をニトロ化した生成物である。たとえば、中間体は、式3の化合物またはその塩であってよい。
【0043】
中間体は、遊離塩基として存在しても、または本明細書で開示する塩のいずれかのような塩として存在してもよい。一実施形態では、中間体は硫酸塩である。
【0044】
一実施形態では、中間体を反応混合物から単離しない。「反応混合物」とは、本明細書では、試薬同士の化学反応の生成物だけでなく、副生成物、たとえば、不純物(所望でない立体化学の化合物を含める)、溶媒、および残存する任意の試薬、たとえば出発材料も含む、溶液またはスラリーを指す。一実施形態では、中間体は、式3の化合物であり、出発試薬(ニトロ化剤および/または式2の化合物など)、副生成物(式2または式3いずれかのC4−エピマーなど)も含有し得る反応混合物中に存在する。一実施形態では、反応混合物は、スラリーであり、スラリーは、少なくとも1種の固体と、少なくとも1種の液体(水、酸、溶媒など)とを含む組成物、たとえば、固体の懸濁液または分散液であってよい。
【0045】
別の実施形態では、中間体を反応混合物から単離する。一実施形態では、単離する中間体は、式3の化合物である。
【0046】
式3の化合物の調製方法の一実施形態では、調製方法は、式3の化合物を還元剤と反応させて、式4の化合物を形成するステップと、(c)場合により、式4の化合物の塩を形成するステップとをさらに含む。調製方法は、式4の化合物をアシル化して、式1の化合物を形成するステップと、場合により、式1の化合物の薬学的に許容できる塩を形成するステップとをさらに含んでもよい。
【0047】
式3の化合物の調製方法の一実施形態では、化合物は、反応混合物中で生成され、調製方法は、式3の化合物を含む反応混合物を還元剤と接触させて、式4の化合物を形成するステップと、(c)場合により、式4の化合物の薬学的に許容できる塩を形成するステップとをさらに含む。調製方法はさらに、式4の化合物をアシル化して、式1の化合物を形成するステップと、場合により、式1の化合物の薬学的に許容できる塩を形成するステップとを含んでもよい。
【0048】
一実施形態では、本発明の任意のプロセスにおけるC1〜C12硝酸アルキルは、C3〜C6硝酸アルキルなどのC1〜C8硝酸アルキルである。C1〜C8硝酸アルキルは、たとえば、硝酸2−エチルヘキシルであってよい。C3〜C6硝酸アルキルは、たとえば、硝酸イソプロピルであってよい。
【0049】
一実施形態では、式3、4および1の化合物をスキーム1に示すとおりに調製する。式2の化合物をC1〜C12硝酸アルキルと反応させると、−NO2置換基が挿入されて、式3の化合物が生成する。式3の−NO2置換基を、引き続いて水素化などによりアミノに還元すると、式4の化合物を形成することができる。最後に、式4の化合物をアシル化すると、式1の化合物が生じる。スキーム1では、R、R1、R2およびnは、本明細書でさらに規定するとおりである。場合により、スキーム1は、式2、3、4および1の化合物のいずれか1つまたは複数の塩の生成、ならびに式2、3、および4の化合物のそれぞれの塩のニトロ化、還元、およびアシル化を含んでもよい。
【0050】
【化9】

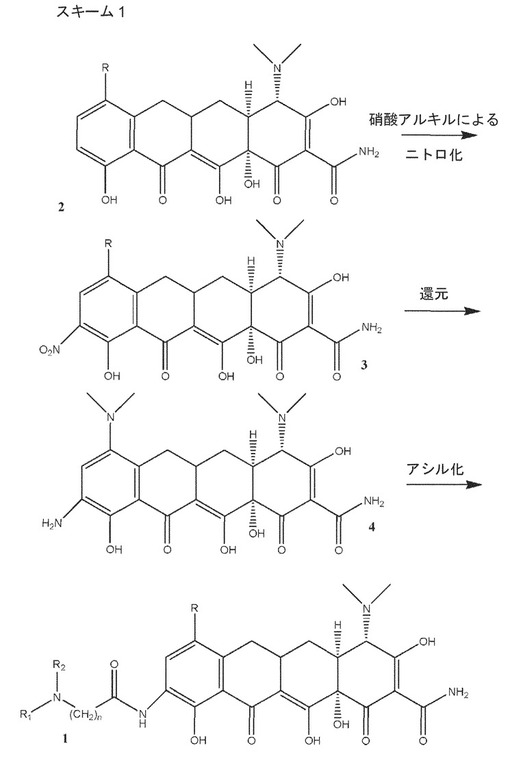
【0051】
一実施形態は、R1およびR2が、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rが−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nが1〜4の範囲をとる、式1の化合物または薬学的に許容できるその塩の調製方法を開示する。
【0052】
ニトロ化
一実施形態は、式3の化合物を単離しないニトロ化反応を開示する。別の実施形態は、式3の化合物を単離するニトロ化反応を開示する。
【0053】
C1〜C12硝酸アルキルは、当業者によって適切であるとされる任意の溶媒中で、式2の化合物またはその塩と反応させることができる。一実施形態では、使用する酸は、70%を超える濃度の硫酸である。硫酸の濃度を70%とした反応は、より高い濃度にするより緩慢になることがわかっている。したがって、濃度は、少なくとも75%、少なくとも80%、少なくとも85%、少なくとも90%、または少なくとも95%などの、70%を超える濃度とする。たとえば、濃度は98%以上でもよい。
【0054】
一実施形態では、C1〜C12硝酸アルキルを、式2の化合物に対してモル過剰で用意する。適切なモル過剰量は、当業者によって決定することができ、限定はしないが、少なくとも1.05、たとえば、1.05〜2.5当量の範囲のモル過剰などの値、たとえば、1.05〜2.0、1.05〜1.75、1.05〜1.5、1.05〜1.25、または1.05〜1.1当量の範囲のモル過剰などを挙げることができる。
【0055】
一実施形態では、C1〜C12硝酸アルキルを一定の時間をかけて加えることにより、C1〜C12硝酸アルキルを式2の化合物と反応させる。当業者なら、C1〜C12硝酸アルキルの総量を加える期間を、反応条件が最適化されるように決定することができる。たとえば、C1〜C12硝酸アルキルの添加をたとえばHPLCによってモニターして、使用するC1〜C12硝酸アルキルの量を制御することができる。一実施形態では、C1〜C12硝酸アルキルの総量を、少なくとも2時間、少なくとも3時間、少なくとも5時間、少なくとも10時間、少なくとも24時間、または1時間〜1週間の範囲、1時間〜48時間の範囲、1時間〜24時間の範囲、もしくは1時間〜12時間の範囲の時間などの、少なくとも1時間の時間をかけて加える。
【0056】
一実施形態では、C1〜C12硝酸アルキルを継続的に加えることができる。
【0057】
一実施形態では、C1〜C12硝酸アルキルと式2の化合物とを、0〜65℃の範囲の温度、たとえば、10℃〜30℃の範囲の温度で反応させることができる。
【0058】
驚くことに、また思いがけなく、式1の化合物のニトロ化に硝酸アルキルを使用することは、他のニトロ化剤を使用するよりも有利であることがわかった。以下でさらに論じるとおり、有利点として、他のニトロ化剤で通常必要となる条件より穏やかな条件を使用しても収率がより高いことに加え、他のニトロ化剤との反応から得られる生成物と比較して、生成物の外観がより良好であることが挙げられる。たとえば、ニトロ化剤として一般的に使用される硫酸と硝酸の混合物は、腐食性が強いことが知られており、その使用にはいくつかの安全対策が必要となる。ニトロ化に硝酸アルキルを使用することの追加の有利点として、(a)硝酸イソプロピルなどの特定の硝酸アルキルが市販されていること、(b)硝酸イソプロピルなどの特定の硝酸アルキルの保管および輸送において安全性の問題がないこと、および(c)硝酸とは異なり、硝酸イソプロピルなどの特定の硝酸アルキルは、塩酸などの特定の酸と反応して(塩素ガスを生成する)という欠点をもたず、したがって塩酸などの特定の酸と共に使用してもよいこと、が挙げられる。
【0059】
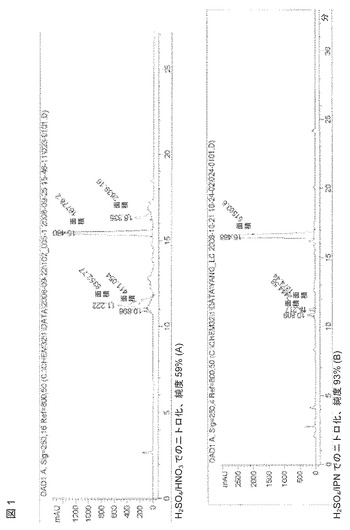
図1は、ミノサイクリンを(a)硝酸イソプロピルと硫酸、および(b)硝酸と硫酸でニトロ化した生成物のHPLCの比較を示すものである。(a)のHPLCプロットでは、(b)のHPLCプロットと比較して、生成物のピーク以外のピークの数がより少ないこと、および生成物のピーク以外のピークのそれぞれの大きさがより小さいことが示すように、(a)の生成物の方が純度が高いことが容易にわかる。(a)の反応混合物は、HPLCによる純度が93%を超えており、それに比較して(b)の純度は59%である。
【0060】
本発明のプロセスの例として、ミノサイクリンを硝酸イソプロピルと硫酸によってニトロ化して、9−ニトロミノサイクリンを得た後、メタノール中にてPd/Cで還元することにより、9−アミノミノサイクリン硫酸塩を得ることができる(スキーム2)。全収率は、9−ニトロミノサイクリンの単離なしで87%、9−ニトロミノサイクリンを単離して76%である。比較すると、ミノサイクリンの9−アミノミノサイクリンへの変換の全収率は、ミノサイクリンのニトロ化を硝酸イソプロピルと硫酸でなく硝酸と硫酸で実施した場合はわずか50%である。
【0061】
一実施形態では、ニトロ化反応によって、対応するC4−エピマーが、高速液体クロマトグラフィー(HPLC)によって求めたとき、式3の化合物の2.5%以下の量しか生成せずに、式3の中間体が生成される。
【0062】
各ステップ、すなわち、ニトロ化、還元、およびアシル化についてのHPLCパラメータは、実施例の部で提供する。
【0063】
一実施形態では、出発材料、たとえば式2の化合物の量が少なくなるようにニトロ化を実施する。一実施形態では、式2の化合物は、HPLCによって求めたとき、ニトロ化生成物中に10%未満、5%未満、3%未満、2%未満、1%未満、または0.5%未満の量でしか存在しない。
【0064】
一実施形態では、ニトロ化を大規模で実施することができる。一実施形態では、「大規模」とは、式2による化合物を少なくとも1グラム使用すること、たとえば、少なくとも2グラム、少なくとも5グラム、または少なくとも10グラム使用することをいう。
【0065】
一実施形態では、R1は水素であり、R2はt−ブチルであり、Rは−NR3R4であり、R3およびR4は、それぞれメチルであり、nは1である。別の実施形態では、R1およびR2は、これらが結合しているNと一緒になって、ピロリジニル基を形成しており、R3およびR4は、それぞれメチルであり、nは1である。
【0066】
一実施形態では、式1の化合物は、チゲサイクリンであり、遊離塩基の形でも、塩酸塩としてのものでもよい。この実施形態では、式3の化合物は、9−ニトロミノサイクリンであり、遊離塩基の形でも、塩酸塩としてのものでもよい。
【0067】
還元
一実施形態では、本発明のプロセスは、ニトロ化ステップで生成する中間体を還元するステップをさらに含む。還元された中間体は、式4の化合物でもその塩でもよい。
【0068】
還元は、水素などの還元剤、または化合物に水素を付加する他の任意の化学薬品を使用して実施することができる。たとえば、還元は、当業者によって決定される適切な圧力の水素雰囲気中にて実施することができる。一実施形態では、水素は、14〜100psiの範囲の圧力、たとえば、14〜45psiの範囲の圧力、たとえば、45psiで供給する。
【0069】
別の実施形態では、少なくとも1種の触媒の存在下で還元を実施する。例となる触媒として、限定はしないが、希土類金属酸化物、VIII族金属含有触媒、およびVIII族金属含有触媒の塩が挙げられる。VIII族金属含有触媒の例は、パラジウム担持炭素などのパラジウムである。
【0070】
触媒がパラジウム担持炭素である場合、一実施形態では、触媒は、C1〜C12硝酸アルキルとの反応より前に存在する式2の化合物の量に対して、0.1部〜1部の範囲の量で存在する。
【0071】
一実施形態では、中間体は、式3の化合物である。一実施形態では、式3の化合物において、R1は水素であり、R2はt−ブチルであり、R3はメチルであり、R4はメチルであり、nは1である。
【0072】
当業者なら、還元反応に適する溶媒を決定することができる。一実施形態では、還元より前に、反応混合物を、少なくとも1種の(C1〜C8)アルコールおよび/または少なくとも1種の(C1〜C8)ジオールを含む溶媒と合わせて、次の反応混合物を生成する。少なくとも1種の(C1〜C8)アルコールは、たとえば、メタノールおよびエタノールから選択されるものであってよい。少なくとも1種の(C1〜C8)ジオールは、たとえば、エチレングリコールであってよい。
【0073】
当業者なら、還元反応に適する温度を決定することができる。一実施形態では、0℃〜100℃の範囲の温度、たとえば、20℃〜80℃、25℃〜50℃、または26℃〜28℃の範囲の温度で還元を実施する。
【0074】
一実施形態では、還元の後、得られる反応混合物を、エーテル、ハロゲン化炭化水素、(C1〜C8)分枝鎖アルコール、(C1〜C8)炭化水素、またはこれらの組合せからなる群から選択される溶媒を含む溶媒系に加えるか、またはそれと合わせる。一実施形態では、エーテルは、MTBE、THF、またはジブチルエーテルである。一実施形態では、ハロゲン化炭化水素は、DCMまたはDCEである。一実施形態では、(C1〜C8)分枝鎖アルコールは、イソプロパノールである。一実施形態では、(C1〜C8)炭化水素は、ヘキサン、ヘプタン、またはオクタンである。
【0075】
一実施形態では、還元の後、得られる反応混合物を、−10℃〜50℃の範囲の温度、たとえば、0℃〜10℃の範囲の温度で溶媒系に加える。
【0076】
一実施形態では、本方法は、式4の化合物を固体または固体組成物として単離するステップをさらに含む。一実施形態では、式4の化合物を、本明細書に記載の塩のいずれかなどの塩として沈殿させるか、または単離する。
【0077】
一実施形態では、固体組成物は、高速液体クロマトグラフィーによって求めたとき、式4のC4−エピマーを10%未満の量しか含まない。別の実施形態では、C4−エピマーは、5%未満、3%未満、2%未満、1%未満、または0.5%未満の量しか存在しない。
【0078】
一実施形態では、固体組成物は、高速液体クロマトグラフィーによって求めたとき、式2を、2%未満の量、たとえば、1%未満または0.5%未満の量しか含まない。
【0079】
一実施形態では、還元を大規模で実施することができる。一実施形態では、「大規模」とは、式2による化合物を少なくとも1グラム使用すること、たとえば、少なくとも2グラム、少なくとも5グラム、少なくとも10グラム、少なくとも25グラム、少なくとも50グラム、少なくとも100グラム、少なくとも500g、少なくとも1kg、少なくとも5kg、少なくとも10kg、少なくとも25kg、少なくとも50kg、または少なくとも100kg使用することをいう。
【0080】
一実施形態では、アミノアシル化合物をアシル化剤として用いてアシル化を実施する。一実施形態では、アシル化は、水性媒質から選択されるものであってよい反応媒質、および試薬塩基なしの少なくとも1種の塩基性溶媒中で実施する。
【0081】
一実施形態では、式Iの化合物の調製方法は、チゲサイクリンまたは薬学的に許容できるその塩の調製方法である。
【0082】
式4の化合物の塩は、塩酸塩などのハロゲン化塩であってよい。
【0083】
アシル化反応媒質は、極性非プロトン性溶媒またはその溶媒混合物から選択される溶媒であってよい。一実施形態では、極性非プロトン性溶媒は、アセトニトリル、1,2−ジメトキシエタン、ジメチルアセトアミド、ジメチルホルムアミド、ヘキサメチルホスホルアミド、N,N’−ジメチルエチレン尿素、N,N’−ジメチルプロピレン尿素、塩化メチレン、N−メチルピロリジノン、テトラヒドロフラン、およびこれらの混合物から選択する。別の実施形態では、極性非プロトン性溶媒は、アセトニトリル、ジメチルホルムアミド、N,N’−ジメチルプロピレン尿素、N−メチルピロリジノン、テトラヒドロフラン、およびこれらの混合物から選択する。少なくとも1種の塩基性溶媒は、アセトニトリルとN,N’−ジメチルプロピレン尿素の混合物であってよい。別の実施形態では、少なくとも1種の塩基性溶媒は、水とN,N’−ジメチルプロピレン尿素の混合物であってよい。さらなる実施形態では、少なくとも1種の塩基性溶媒は、N,N’−ジメチルプロピレン尿素である。
【0084】
反応媒質は、水性媒質でもよい。さらなる実施形態では、塩基なしの少なくとも1種の塩基性溶媒は、塩基なしの水である。別の実施形態では、反応媒質は、試薬塩基なしの少なくとも1種の塩基性溶媒であってよい。塩基性溶媒とは、プロトンを部分的または完全に受容することのできる溶媒である。試薬塩基とは、反応の開始時に、式4の化合物およびアミノアシル化合物と同時にまたはそれに引き続いて加えられ、プロトンを部分的または完全に受容することのできる塩基をいう。試薬塩基は、反応の最中に加えられる塩基も指す。
【0085】
アミノアシル化合物は、ハロゲン化アミノアシル、アミノアシル無水物、および混合アミノアシル無水物から選択されるものであってよい。一実施形態では、アミノアシル化合物は、式6のハロゲン化アミノアシル
【0086】
【化10】
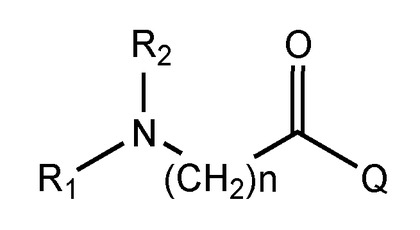
またはその塩であり、
式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Qは、フッ化物、臭化物、塩化物、およびヨウ化物から選択されるハロゲンである。
【0087】
さらなる実施形態では、Qは塩化物である。式6の化合物の塩は、ハロゲン化塩から選択されるものであってよい。ハロゲン化塩とは、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩などの、ハロゲンアニオンとの相互作用から生成する任意の塩を指す。一実施形態では、ハロゲン化塩は、塩酸塩である。
【0088】
別の実施形態では、nは1である。さらなる実施形態では、Xは臭化物である。
【0089】
式4の化合物とアミノアシル化合物の反応は、0℃〜30℃、たとえば20℃〜25℃、たとえば10℃〜17℃、たとえば0℃〜6℃、さらにはたとえば2℃〜8℃の範囲の温度で実施することができる。反応させる時間は、1時間〜24時間、たとえば0.5時間〜4時間、さらにはたとえば2時間〜8時間の範囲であってよい。式4の化合物の量に対して過剰のアミノアシル化合物を反応において使用することができる。一実施形態では、過剰は、1当量の式4の化合物に対して3当量のアミノアシル化合物であってよい。別の実施形態では、水性媒質対式4の化合物の比を、6:1w/wまたは5:1体積にすることができる。一実施形態では、アミノアシル化合物は、式4の化合物を水性媒質に溶かした溶液に加えるかそれと合わせる。
【0090】
一実施形態では、反応媒質が水性媒質である場合、水性媒質のpHは、4〜9、たとえば5〜7.5、たとえば6.3〜6.7、たとえば7.0〜7.5の範囲のpH、さらにはたとえば6.5、さらにまたたとえば7.2に調整することができる。pHを調整する前に水を加えてもよい。pHの調整は、限定はしないが水酸化アンモニウムを含めた、塩基を加えるものであってよい。水酸化アンモニウムの濃度は、25%〜30%の範囲であってよい。別の実施形態では、塩酸などの酸を使用してpHを調整してもよい。pH調整の際の反応媒質は、−5℃〜℃、たとえば5℃〜8℃、さらにはたとえば0℃〜5℃の範囲の温度にすることができる。
【0091】
pHの調整に続いて、水性媒質に少なくとも1種の有機溶媒または溶媒混合物を加えることができる。一実施形態では、少なくとも1種の有機溶媒混合物は、メタノールと塩化メチレンを含むものであってよい。メタノールの濃度は、限定はしないが20%や30%などの、5%〜30%の範囲であってよい。別の実施形態では、少なくとも1種の有機溶媒または溶媒混合物は、テトラヒドロフランを含むものであってよい。混合物の温度は、15℃〜25℃の範囲であってよい。
【0092】
一実施形態では、水性媒質を、少なくとも1種の極性プロトン性溶媒と少なくとも1種の極性非プロトン性溶媒の混合物で抽出してもよい。一実施形態では、少なくとも1種の極性非プロトン性溶媒は、塩化メチレンを含み、少なくとも1種の極性プロトン性溶媒は、メタノールを含む。別の実施形態では、水性媒質を、塩化メチレンなどの少なくとも1種の極性非プロトン性溶媒で抽出する。抽出は、−5℃〜25℃、さらにはたとえば0℃〜5℃の範囲の温度で実施することができる。さらなる実施形態では、各抽出の後、水性媒質のpHを7.0〜7.5の範囲、たとえば7.2に調整する。抽出プロセスは、たとえば10回まで繰り返してもよい。
【0093】
一実施形態では、合わせた有機抽出物を、硫酸ナトリウムなどの乾燥剤で処理してもよい。有機抽出物は、Norit CA−1などの炭で処理してもよい。濾過によって固体を除去して溶液を得る。一実施形態では、溶液を濃縮して式1の化合物を得てもよい。
【0094】
反応から得られた式1の化合物は、少なくとも1種の有機溶媒または溶媒混合物中に結晶化させることができる。一実施形態では、有機溶媒混合物は、メタノールと塩化メチレンを含む。結晶化は、たとえば、−15℃〜155℃、たとえば0℃〜15℃、さらにはたとえば2℃〜5℃の範囲の温度で行うことができる。
【0095】
別の実施形態では、抽出に続いて、得られる、少なくとも1種の極性プロトン性溶媒と少なくとも1種の極性非プロトン性溶媒の有機混合物を濃縮してスラリーを得、濾過して式1の化合物を得てもよい。濃縮および濾過は、たとえば0℃〜5℃で行うことができる。
【0096】
式1の化合物の調製方法は、5グラム超、たとえば10グラム超、たとえば50グラム超、たとえば100グラム超、たとえば500グラム超、たとえば1キログラム超、さらにはたとえば10キログラム超の式4のアミンを使用して実施することができる。
【0097】
一実施形態では、本明細書に記載の方法のいずれかによって調製した式1の化合物は、高速液体クロマトグラフィーによって求めたとき、10.0%未満の不純物、たとえば5%未満の不純物、たとえば2%未満の不純物、さらにはたとえば1〜1.4%の不純物を含有する。さらなる実施形態では、式1の化合物は、高速液体クロマトグラフィーによって求めたとき、0.5%未満のC4−エピマー、さらには0.2%未満のC4−エピマーなどの、1.0%未満の量のC4−エピマーを含有する。一実施形態では、式1の化合物は、高速液体クロマトグラフィーによって求めたとき、1%未満のミノサイクリン、たとえば0.6%未満のミノサイクリンを含有する。別の実施形態では、式1の化合物は、5%未満のジクロロメタン、たとえば2〜3%未満のジクロロメタンを含有する。
【0098】
精製
本発明の一実施形態では、本発明のプロセスは、式1の化合物または薬学的に許容できるその塩、
または薬学的に許容できるその塩[式中、R1およびR2は、水素、直鎖および分枝鎖(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは−NR3R4であり、R3およびR4は、水素ならびに直鎖および分枝鎖(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]を精製するステップをさらに含んでもよく、
精製は、
A)式1の化合物を、少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせて、第一の混合物を得るステップと、
B)第一の混合物を、0℃〜40℃の範囲の温度で15分〜2時間などの少なくとも1回の期間、混合するステップと、
C)式1の化合物を取得するステップとを含む。
【0099】
ここでは、用語「取得する」とは、限定はしないが、90%、95%、96%、97%、98%および99%より高いレベルを含めた有用なレベルの純度で、化合物を単離することを指す。純度のレベルは、高圧液体クロマトグラフィーによって求めることができる。
【0100】
一実施形態では、式1の化合物の精製は、
A)式1の化合物を、少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせて、第一の混合物を得るステップと、
B)第一の混合物を30℃〜40℃の範囲の温度で一定期間混合するステップと、
C)その第一の混合物を15℃〜25℃の範囲の温度に冷却し、混合物を混合せずに第二の期間静置するステップと、
D)その第一の混合物を0℃〜6℃の範囲の温度に冷却し、混合物を混合せずに第三の期間静置するステップと、
E)式1の化合物を取得するステップとを含む。
【0101】
一実施形態では、この方法は、nが1であり、R1が水素であり、R2がt−ブチルであり、R3およびR4がそれぞれメチルである式1の化合物を使用するものでもよい。少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせる式1の化合物は、固体、スラリー、懸濁液、および溶液から選択される形で用意することができる。
【0102】
一実施形態では、少なくとも1種の極性非プロトン性溶媒は、アセトン、1,2−ジクロロエタン、酢酸メチル、メチルエチルケトン、メチルイソブチルケトン、塩化メチレン、および酢酸エチルから選択されるものであってよい。さらなる実施形態では、少なくとも1種の極性非プロトン性溶媒は、アセトンおよび塩化メチレンから選択されるものであってよい。別の実施形態では、少なくとも1種の極性プロトン性溶媒は、メタノール、エタノール、イソプロパノール、およびt−ブタノールから選択されるものであってよい。さらなる実施形態では、少なくとも1種の極性プロトン性溶媒は、メタノールであってよい。
【0103】
少なくとも1種の極性非プロトン性溶媒と少なくとも1種の極性プロトン性溶媒の組合せとして、アセトンとメタノールを挙げることができる。別の実施形態では、少なくとも1種の極性非プロトン性溶媒の塩化メチレンと、少なくとも1種の極性プロトン性溶媒のメタノールの組合せを用意する。さらなる実施形態では、少なくとも1種の極性非プロトン性溶媒と少なくとも1種の極性プロトン性溶媒の組合せとして、酢酸メチルとメタノールを挙げることができる。式1の化合物は、たとえば、等体積の少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせることができる。
【0104】
一実施形態では、第一の混合物は、たとえば、温度が15℃〜25℃の範囲である、30分〜2時間の範囲の第一の期間、次いで温度が0℃〜2℃の範囲である、30分〜2時間の範囲の第二の期間の間、混合する。一実施形態では、第一の期間および第二の期間は、それぞれ1時間である。別の実施形態では、この方法は、第一の混合物を、15℃〜25℃の範囲の温度で、30分〜2時間の範囲の少なくとも1回の期間混合し、次いでその第一の混合物を濾過して固体を得るステップを含んでもよい。この方法は、その固体を、15℃〜25℃の範囲の温度で30分〜2時間の範囲の第一の期間をかけて、たとえば等体積の少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせ、濾過して第二の固体を得るステップをさらに含んでもよい。さらなる実施形態では、合わせ、濾過するこれらのステップを2回から15回繰り返してもよい。
【0105】
式1の化合物の精製は、第一の混合物から固体を得、その固体を少なくとも1種の極性プロトン性溶媒および少なくとも1種の極性非プロトン性溶媒と合わせて第二の混合物を得るステップをさらに含んでもよい。第二の混合物は、たとえば、メタノールおよび塩化メチレンを、1:5〜1:15のメタノール:塩化メチレンの範囲の体積比で含むものであってよい。一実施形態では、第二の混合物を30℃〜36℃の範囲の温度で混合し、次いで濾過して溶液を得てもよい。さらなる実施形態では、溶液中の極性プロトン性溶媒の濃度を5%未満のレベルに減らしてもよく、溶液を、たとえば0℃〜6℃の範囲の温度で、たとえば30分〜2時間の範囲の期間混合してから濾過してもよい。
【0106】
一実施形態では、第一の混合物を混合するステップは、10〜20分の範囲の期間、たとえば15分間行うものであってよい。一実施形態では、第一の混合物を15℃〜25℃の範囲の温度に冷却し、混合物を混合せずに静置するステップは、30分〜3時間、たとえば1時間〜2時間の範囲の第二の期間行うものであってよい。第一の混合物は、0℃〜6℃の範囲の温度にさらに冷却し、混合せずに30分〜2時間の範囲の第三の期間、たとえば1時間静置してもよい。
【0107】
式1の化合物を取得するステップは、本明細書に記載の任意の混合物を、発熱物質低減用フィルターおよび清澄化用フィルターから選択される少なくとも1種類のフィルターで濾過するステップを包含するものであってよい。本明細書で開示するとおり、混合は、機械式混合装置、たとえば撹拌子または撹拌機を使用して実施することができる。混合は、式1を有する化合物の溶媒系への溶解性によって実現することもできる。温度を上昇させると、溶解性を増大させることができる。
【0108】
一実施形態では、式1の化合物を少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせることにする場合、式1の化合物は、薬学的に許容できるその塩の形で使用してもよい。式1の化合物を本発明の方法の生成物として取得する場合、式1の化合物を薬学的に許容できるその塩の形で回収してもよい。
【0109】
本発明による方法によって式1の化合物を取得する別の実施形態では、酸を加えることにより、化合物を薬学的に許容できるその塩に変換してもよい。
【0110】
一実施形態では、精製された式1の化合物は、チゲサイクリンである。
【0111】
チゲサイクリンを少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせたものは、固体、スラリー、懸濁液、および溶液から選択される形で用意することができる。一実施形態では、本方法から取得したチゲサイクリンは、高圧液体クロマトグラフィー(HPLC)によって求めたとき、チゲサイクリンのC−4エピマーまたは薬学的に許容できるその塩を1%未満含有してもよい。
【0112】
本方法から取得した式1の化合物は、HPLCによって求めたとき、3.0%未満の不純物、たとえば1.0%未満の不純物、たとえば0.7%未満の不純物を含有してもよい。別の実施形態では、式1の化合物は、HPLCによって求めたとき、式1の化合物のC4エピマーまたは薬学的に許容できるその塩を2%未満、たとえばC4エピマーを1%未満、たとえばC4エピマーを0.5%未満含有してもよい。
【0113】
本方法は、5グラム超、たとえば50グラム超、たとえば100グラム超、たとえば500グラム超、たとえば1キログラム超、さらにはたとえば10キログラム超の式1の化合物で実施することができる。
【0114】
本開示の広い範囲を述べる、数字による範囲およびパラメータは、概数であるにもかかわらず、詳細な実施例で述べるその数値は、できる限り正確に報告する。しかし、どんな数値も、そのそれぞれの試験測定で見出される標準偏差から必然的に得られる一定の誤差を本質的に含んでいる。
【0115】
以下の実施例は、限定しない形で本発明を例示するものである。
【実施例】
【0116】
HPLC分析は、以下の条件下で実施した。
カラム:Waters Symmetratry RP8 15×0.46cm
移動相:A;0.03MのKH2PO4、H3PO4によりpH2、B;9:1のアセトニトリル:水
流量:0.8ml/分
検出波長:250nm
カラムオーブン温度 35℃
定組成プログラム:時間(分) A(%) B(%)
0 90 10
2 90 10
30 45 55
32 90 10
【0117】
ニトロ化
式2の化合物の一例であるミノサイクリンを、米国特許第3,226,436号に記載の方法に従って調製した。次いで、ミノサイクリン塩化物を以下の手順に従ってニトロ化した。
【0118】
15gの98%硫酸に5.3gのミノサイクリン塩化物を10〜30℃で数回に分けて加えた。懸濁液をN2中にて20℃で4時間撹拌し、均質な溶液を生成した。この溶液に2.2mLの硝酸イソプロピルをゆっくりと加えて、反応温度を30℃より低く保った。加えた後、反応液をもう2時間ねかせた。反応混合物をIPA/Hep溶液(125mL/25mL)に10℃未満でゆっくりと加え、次いで溶液をこの温度でもう1時間撹拌した。生成した固体を濾過し、予め冷却してある30mLのIPAで洗浄した。固体を真空オーブンにて40℃で20時間乾燥させた。黄色の固体(7.3g、純度91.3重量%、収率92.6%)が得られ、これ以上精製せずに次の反応へ進めた。
【0119】
ニトロ化反応から生成した中間体を単離した後の還元
加圧ガラス瓶に、50mLのメタノールおよび10mLの硫酸を加えた。予め調製しておいた9−ニトロミノサイクリン硫酸塩5gを5℃で一度に加えた。溶液に0.4gの10%Pd/Cを加え、次いで懸濁液をN2で3回、H2で1回パージした。Parr振盪機において、懸濁液を圧力45PSIのH2中にて20℃で3時間維持した。単純濾過によって触媒を取り除いた後、反応溶液をIPA/Hep溶液(100mL/30mL)に5〜10℃で加えた。固体を濾過し、予め冷却しておいた20mLのIPAで洗浄した。固体を真空オーブン中にて40℃で20時間乾燥させ、黄色の固体(4.3g、純度74重量%)を収率76%で得た。
【0120】
ニトロ化反応から生成した中間体を単離せずに行う還元
15gの98%硫酸に5.3gのミノサイクリン塩化物を10〜30℃で数回に分けて加えた。懸濁液をN2中にて20℃で4時間撹拌し、均質な溶液を生成した。この溶液に2.2mLの硝酸イソプロピルをゆっくりと加えて、反応温度を30℃より低く保った。加えた後、反応液をもう2時間ねかせた。反応混合物を5〜10℃にて50mLのメタノールで希釈し、次いで1gの10%Pd/Cを加えた。懸濁液をN2で3回、H2で1回パージし、次いでParr振盪機において圧力45PSIのH2中で3時間維持した。単純濾過によって触媒を取り除いた後、反応溶液をIPA/Hep溶液(250mL/50mL)に5〜10℃で加えた。固体を濾過し、予め冷却しておいた40mLのIPAで洗浄した。固体を真空オーブン中にて40℃で20時間乾燥させ、黄色の固体(7.4g、純度72重量%)を収率87%で得た。
【0121】
アシル化
式4の化合物のアシル化は、上で開示したとおりに実施することができる。アシル化は、参照により本明細書に援用される米国特許公開第2007−0049560A1号の[0143]〜[0213]段落でも開示されている。
【0122】
精製
式1の化合物の精製は、上記で開示したとおりに実施することができる。精製は、参照により本明細書に援用される米国特許公開第2007−0049560 A1号の[0214]〜[0283]段落でも開示されている。
【0123】
本発明について、本発明の実施形態およびその非限定的な実施例の論述によって説明してきたが、当業者なら、本明細書および特許請求の範囲を読めば、同じく本発明の企図される範囲内にある他の実施形態および変形形態を想定することができ、したがって本発明の範囲は、付属の特許請求の範囲によってのみ解釈され規定されるものとする。
【図1】
【発明を実施するための形態】
【0026】
定義
本明細書および付属の特許請求の範囲で使用するとき、単数形の「1つの(a)」、「1つの(an)」および「その(the)」は、内容が明らかに別途規定していない限り、複数の指示物を包含することを留意されたい。したがって、たとえば、「1種の化合物(a compound)」を含有する組成物への言及は、2種以上の化合物の混合物を包含する。また用語「または(or)」は、内容が明らかに別途規定していない限り、「および/または(and/or)」を含めた意味で一般に用いることを留意されたい。
【0027】
「チゲサイクリン」は、本明細書では、任意の薬学的に許容できる塩、鏡像異性体、エピマーなどの、遊離塩基形態および塩形態のチゲサイクリンを包含する。チゲサイクリンは、本明細書では、当業界で知られている方法に従って製剤することができる。同様に、「ミノサイクリン」も、本明細書では、任意の薬学的に許容できる塩、鏡像異性体、エピマーなどの、遊離塩基形態および塩形態のミノサイクリンを包含する。
【0028】
「化合物」とは、本明細書では、中性化合物(たとえば、遊離塩基)およびその塩形態(薬学的に許容できる塩など)を指す。化合物は、無水形態で、または水和物として、または溶媒和物として存在する場合がある。化合物は、立体異性体(たとえば、鏡像異性体およびジアステレオ異性体)として存在することもあり、鏡像異性体、ラセミ混合物、ジアステレオ異性体、およびその混合物として単離することができる。固体形態の化合物は、種々の結晶形態および非晶質形態で存在する場合がある。
【0029】
「薬学的に許容できる」とは、本明細書では、信頼できる医学的判断の範囲内で、妥当なリスク/利益比で、過度の毒性、刺激、アレルギー反応、または他の問題もしくは合併症なしに、患者の組織と接触させて使用するのに適する化合物、材料、組成物、および/または剤形を指す。
【0030】
用語「ハロゲン」とは、本明細書では、フルオロ、クロロ、ブロモ、および/またはヨードを指す。
【0031】
用語「アルキル」とは、本明細書では、1〜12個の炭素原子、たとえば、1〜8個、1〜6個、または1〜4個の炭素原子を有する、直鎖または分枝鎖の飽和脂肪族炭化水素基を指す。
【0032】
アルキル基の例として、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、イソペンチル、ネオペンチル、ヘキシル、ヘプチル、オクチルなどが挙げられる。
【0033】
用語「アルキレン」とは、本明細書では、1〜6個の炭素原子、たとえば、1〜4個、1〜3個、または1〜2個の炭素原子を有する、ジラジカルの直鎖または分枝鎖飽和脂肪族炭化水素基を指す。
【0034】
アルキレン基の例として、メチレン、エチレン、トリメチレン(1,3−プロパンジイル)、プロピレン(1,2−プロパンジイル)、テトラメチレン(1,4−ブタンジイル)、ブチレン(1,2−ブタンジイル)、1,3−ブタンジイル、2−メチル−1,3−プロパンジイル、ペンタメチレン(1,5−ペンタンジイル)、ペンチレン(1,2−ペンタンジイル)、ヘキサメチレン(1,6−ヘキサンジイル)、ヘキシレン(1,2−ヘキサンジイル)、2,3−ジメチル−1,4−ブタンジイルなどが挙げられる。
【0035】
用語「ヒドロキシ」または「ヒドロキシル」とは、本明細書では、−OH基を指す。
【0036】
用語「ニトロ」とは、本明細書では、基−NO2を指す。
【0037】
用語「アミノ」とは、本明細書では、−NH2基を指す。
【0038】
用語「アリール」とは、本明細書では、6〜14個の環炭素原子を含んでいる芳香族炭化水素基を指す。「C6〜C14アリール」とは、フェニル、ナフチル、ビフェニル、アントリル、テトラヒドロナフチル、フルオレニル、インダニル、ビフェニレニル、およびアセナフテニル基を指す。C6〜C14アリール基の例として、限定はしないが、フェニル、1−ナフチル、2−ナフチル、および3−ビフェン−1−イルが挙げられる。
【0039】
用語「シクロアルキル」とは、本明細書では、3〜8個の炭素原子を含んでいる非芳香族の単環式飽和炭化水素環を指す。C3〜C8シクロアルキルの代表例として、限定はしないが、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、およびシクロオクチルが挙げられる。炭素環の同じ炭素原子上にある任意の2個の水素原子のそれぞれが酸素原子で置き換えられて、オキソ(=O)置換基を形成していてもよく、または、アルキレンジオキシ基が、結合している炭素原子と一緒になったとき、2個の酸素原子を含んでいる5〜7員ヘテロ環を形成するように、その2個の水素原子がアルキレンジオキシ基で置き換えられていてもよい。
【0040】
別段指摘しない限り、本明細書で明確に規定しない置換基の命名は、官能基の末端部分に続いて、近接する官能基へと、結合点に向かって命名することによりなされる。たとえば、置換基「アリールアルキルオキシカルボニル」とは、基(アリール)−(アルキル)−O−C(O)−を指す。
【0041】
連続反応における各化合物は、遊離塩基の形でも塩の形でもよい。「塩」は、本明細書では、遊離塩基を適切な酸と反応させることにより、その場で、または別途調製することができる。例となる塩として、限定はしないが、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩、硝酸塩、硫酸塩、酢酸塩、安息香酸塩、クエン酸塩、システイン塩、フマル酸塩、グリコール酸塩、マレイン酸塩、コハク酸塩、酒石酸塩、硫酸塩、およびクロロベンゼンスルホン酸塩が挙げられる。別の実施形態では、塩は、アルキルスルホン酸塩およびアリールスルホン酸塩から選択することができる。一実施形態では、式2の化合物は、塩酸塩などの、上で開示した塩のいずれかであってよい塩として、または硫酸塩として提供される。
【0042】
「中間体」とは、本明細書では、出発材料と最終生成物の中間の生成物として生成される化合物を指す。一実施形態では、中間体は、式2の化合物をニトロ化した生成物である。たとえば、中間体は、式3の化合物またはその塩であってよい。
【0043】
中間体は、遊離塩基として存在しても、または本明細書で開示する塩のいずれかのような塩として存在してもよい。一実施形態では、中間体は硫酸塩である。
【0044】
一実施形態では、中間体を反応混合物から単離しない。「反応混合物」とは、本明細書では、試薬同士の化学反応の生成物だけでなく、副生成物、たとえば、不純物(所望でない立体化学の化合物を含める)、溶媒、および残存する任意の試薬、たとえば出発材料も含む、溶液またはスラリーを指す。一実施形態では、中間体は、式3の化合物であり、出発試薬(ニトロ化剤および/または式2の化合物など)、副生成物(式2または式3いずれかのC4−エピマーなど)も含有し得る反応混合物中に存在する。一実施形態では、反応混合物は、スラリーであり、スラリーは、少なくとも1種の固体と、少なくとも1種の液体(水、酸、溶媒など)とを含む組成物、たとえば、固体の懸濁液または分散液であってよい。
【0045】
別の実施形態では、中間体を反応混合物から単離する。一実施形態では、単離する中間体は、式3の化合物である。
【0046】
式3の化合物の調製方法の一実施形態では、調製方法は、式3の化合物を還元剤と反応させて、式4の化合物を形成するステップと、(c)場合により、式4の化合物の塩を形成するステップとをさらに含む。調製方法は、式4の化合物をアシル化して、式1の化合物を形成するステップと、場合により、式1の化合物の薬学的に許容できる塩を形成するステップとをさらに含んでもよい。
【0047】
式3の化合物の調製方法の一実施形態では、化合物は、反応混合物中で生成され、調製方法は、式3の化合物を含む反応混合物を還元剤と接触させて、式4の化合物を形成するステップと、(c)場合により、式4の化合物の薬学的に許容できる塩を形成するステップとをさらに含む。調製方法はさらに、式4の化合物をアシル化して、式1の化合物を形成するステップと、場合により、式1の化合物の薬学的に許容できる塩を形成するステップとを含んでもよい。
【0048】
一実施形態では、本発明の任意のプロセスにおけるC1〜C12硝酸アルキルは、C3〜C6硝酸アルキルなどのC1〜C8硝酸アルキルである。C1〜C8硝酸アルキルは、たとえば、硝酸2−エチルヘキシルであってよい。C3〜C6硝酸アルキルは、たとえば、硝酸イソプロピルであってよい。
【0049】
一実施形態では、式3、4および1の化合物をスキーム1に示すとおりに調製する。式2の化合物をC1〜C12硝酸アルキルと反応させると、−NO2置換基が挿入されて、式3の化合物が生成する。式3の−NO2置換基を、引き続いて水素化などによりアミノに還元すると、式4の化合物を形成することができる。最後に、式4の化合物をアシル化すると、式1の化合物が生じる。スキーム1では、R、R1、R2およびnは、本明細書でさらに規定するとおりである。場合により、スキーム1は、式2、3、4および1の化合物のいずれか1つまたは複数の塩の生成、ならびに式2、3、および4の化合物のそれぞれの塩のニトロ化、還元、およびアシル化を含んでもよい。
【0050】
【化9】
【0051】
一実施形態は、R1およびR2が、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rが−NR3R4であり、R3およびR4は、水素および(C1〜C4)アルキルからそれぞれ独立に選択され、nが1〜4の範囲をとる、式1の化合物または薬学的に許容できるその塩の調製方法を開示する。
【0052】
ニトロ化
一実施形態は、式3の化合物を単離しないニトロ化反応を開示する。別の実施形態は、式3の化合物を単離するニトロ化反応を開示する。
【0053】
C1〜C12硝酸アルキルは、当業者によって適切であるとされる任意の溶媒中で、式2の化合物またはその塩と反応させることができる。一実施形態では、使用する酸は、70%を超える濃度の硫酸である。硫酸の濃度を70%とした反応は、より高い濃度にするより緩慢になることがわかっている。したがって、濃度は、少なくとも75%、少なくとも80%、少なくとも85%、少なくとも90%、または少なくとも95%などの、70%を超える濃度とする。たとえば、濃度は98%以上でもよい。
【0054】
一実施形態では、C1〜C12硝酸アルキルを、式2の化合物に対してモル過剰で用意する。適切なモル過剰量は、当業者によって決定することができ、限定はしないが、少なくとも1.05、たとえば、1.05〜2.5当量の範囲のモル過剰などの値、たとえば、1.05〜2.0、1.05〜1.75、1.05〜1.5、1.05〜1.25、または1.05〜1.1当量の範囲のモル過剰などを挙げることができる。
【0055】
一実施形態では、C1〜C12硝酸アルキルを一定の時間をかけて加えることにより、C1〜C12硝酸アルキルを式2の化合物と反応させる。当業者なら、C1〜C12硝酸アルキルの総量を加える期間を、反応条件が最適化されるように決定することができる。たとえば、C1〜C12硝酸アルキルの添加をたとえばHPLCによってモニターして、使用するC1〜C12硝酸アルキルの量を制御することができる。一実施形態では、C1〜C12硝酸アルキルの総量を、少なくとも2時間、少なくとも3時間、少なくとも5時間、少なくとも10時間、少なくとも24時間、または1時間〜1週間の範囲、1時間〜48時間の範囲、1時間〜24時間の範囲、もしくは1時間〜12時間の範囲の時間などの、少なくとも1時間の時間をかけて加える。
【0056】
一実施形態では、C1〜C12硝酸アルキルを継続的に加えることができる。
【0057】
一実施形態では、C1〜C12硝酸アルキルと式2の化合物とを、0〜65℃の範囲の温度、たとえば、10℃〜30℃の範囲の温度で反応させることができる。
【0058】
驚くことに、また思いがけなく、式1の化合物のニトロ化に硝酸アルキルを使用することは、他のニトロ化剤を使用するよりも有利であることがわかった。以下でさらに論じるとおり、有利点として、他のニトロ化剤で通常必要となる条件より穏やかな条件を使用しても収率がより高いことに加え、他のニトロ化剤との反応から得られる生成物と比較して、生成物の外観がより良好であることが挙げられる。たとえば、ニトロ化剤として一般的に使用される硫酸と硝酸の混合物は、腐食性が強いことが知られており、その使用にはいくつかの安全対策が必要となる。ニトロ化に硝酸アルキルを使用することの追加の有利点として、(a)硝酸イソプロピルなどの特定の硝酸アルキルが市販されていること、(b)硝酸イソプロピルなどの特定の硝酸アルキルの保管および輸送において安全性の問題がないこと、および(c)硝酸とは異なり、硝酸イソプロピルなどの特定の硝酸アルキルは、塩酸などの特定の酸と反応して(塩素ガスを生成する)という欠点をもたず、したがって塩酸などの特定の酸と共に使用してもよいこと、が挙げられる。
【0059】
図1は、ミノサイクリンを(a)硝酸イソプロピルと硫酸、および(b)硝酸と硫酸でニトロ化した生成物のHPLCの比較を示すものである。(a)のHPLCプロットでは、(b)のHPLCプロットと比較して、生成物のピーク以外のピークの数がより少ないこと、および生成物のピーク以外のピークのそれぞれの大きさがより小さいことが示すように、(a)の生成物の方が純度が高いことが容易にわかる。(a)の反応混合物は、HPLCによる純度が93%を超えており、それに比較して(b)の純度は59%である。
【0060】
本発明のプロセスの例として、ミノサイクリンを硝酸イソプロピルと硫酸によってニトロ化して、9−ニトロミノサイクリンを得た後、メタノール中にてPd/Cで還元することにより、9−アミノミノサイクリン硫酸塩を得ることができる(スキーム2)。全収率は、9−ニトロミノサイクリンの単離なしで87%、9−ニトロミノサイクリンを単離して76%である。比較すると、ミノサイクリンの9−アミノミノサイクリンへの変換の全収率は、ミノサイクリンのニトロ化を硝酸イソプロピルと硫酸でなく硝酸と硫酸で実施した場合はわずか50%である。
【0061】
一実施形態では、ニトロ化反応によって、対応するC4−エピマーが、高速液体クロマトグラフィー(HPLC)によって求めたとき、式3の化合物の2.5%以下の量しか生成せずに、式3の中間体が生成される。
【0062】
各ステップ、すなわち、ニトロ化、還元、およびアシル化についてのHPLCパラメータは、実施例の部で提供する。
【0063】
一実施形態では、出発材料、たとえば式2の化合物の量が少なくなるようにニトロ化を実施する。一実施形態では、式2の化合物は、HPLCによって求めたとき、ニトロ化生成物中に10%未満、5%未満、3%未満、2%未満、1%未満、または0.5%未満の量でしか存在しない。
【0064】
一実施形態では、ニトロ化を大規模で実施することができる。一実施形態では、「大規模」とは、式2による化合物を少なくとも1グラム使用すること、たとえば、少なくとも2グラム、少なくとも5グラム、または少なくとも10グラム使用することをいう。
【0065】
一実施形態では、R1は水素であり、R2はt−ブチルであり、Rは−NR3R4であり、R3およびR4は、それぞれメチルであり、nは1である。別の実施形態では、R1およびR2は、これらが結合しているNと一緒になって、ピロリジニル基を形成しており、R3およびR4は、それぞれメチルであり、nは1である。
【0066】
一実施形態では、式1の化合物は、チゲサイクリンであり、遊離塩基の形でも、塩酸塩としてのものでもよい。この実施形態では、式3の化合物は、9−ニトロミノサイクリンであり、遊離塩基の形でも、塩酸塩としてのものでもよい。
【0067】
還元
一実施形態では、本発明のプロセスは、ニトロ化ステップで生成する中間体を還元するステップをさらに含む。還元された中間体は、式4の化合物でもその塩でもよい。
【0068】
還元は、水素などの還元剤、または化合物に水素を付加する他の任意の化学薬品を使用して実施することができる。たとえば、還元は、当業者によって決定される適切な圧力の水素雰囲気中にて実施することができる。一実施形態では、水素は、14〜100psiの範囲の圧力、たとえば、14〜45psiの範囲の圧力、たとえば、45psiで供給する。
【0069】
別の実施形態では、少なくとも1種の触媒の存在下で還元を実施する。例となる触媒として、限定はしないが、希土類金属酸化物、VIII族金属含有触媒、およびVIII族金属含有触媒の塩が挙げられる。VIII族金属含有触媒の例は、パラジウム担持炭素などのパラジウムである。
【0070】
触媒がパラジウム担持炭素である場合、一実施形態では、触媒は、C1〜C12硝酸アルキルとの反応より前に存在する式2の化合物の量に対して、0.1部〜1部の範囲の量で存在する。
【0071】
一実施形態では、中間体は、式3の化合物である。一実施形態では、式3の化合物において、R1は水素であり、R2はt−ブチルであり、R3はメチルであり、R4はメチルであり、nは1である。
【0072】
当業者なら、還元反応に適する溶媒を決定することができる。一実施形態では、還元より前に、反応混合物を、少なくとも1種の(C1〜C8)アルコールおよび/または少なくとも1種の(C1〜C8)ジオールを含む溶媒と合わせて、次の反応混合物を生成する。少なくとも1種の(C1〜C8)アルコールは、たとえば、メタノールおよびエタノールから選択されるものであってよい。少なくとも1種の(C1〜C8)ジオールは、たとえば、エチレングリコールであってよい。
【0073】
当業者なら、還元反応に適する温度を決定することができる。一実施形態では、0℃〜100℃の範囲の温度、たとえば、20℃〜80℃、25℃〜50℃、または26℃〜28℃の範囲の温度で還元を実施する。
【0074】
一実施形態では、還元の後、得られる反応混合物を、エーテル、ハロゲン化炭化水素、(C1〜C8)分枝鎖アルコール、(C1〜C8)炭化水素、またはこれらの組合せからなる群から選択される溶媒を含む溶媒系に加えるか、またはそれと合わせる。一実施形態では、エーテルは、MTBE、THF、またはジブチルエーテルである。一実施形態では、ハロゲン化炭化水素は、DCMまたはDCEである。一実施形態では、(C1〜C8)分枝鎖アルコールは、イソプロパノールである。一実施形態では、(C1〜C8)炭化水素は、ヘキサン、ヘプタン、またはオクタンである。
【0075】
一実施形態では、還元の後、得られる反応混合物を、−10℃〜50℃の範囲の温度、たとえば、0℃〜10℃の範囲の温度で溶媒系に加える。
【0076】
一実施形態では、本方法は、式4の化合物を固体または固体組成物として単離するステップをさらに含む。一実施形態では、式4の化合物を、本明細書に記載の塩のいずれかなどの塩として沈殿させるか、または単離する。
【0077】
一実施形態では、固体組成物は、高速液体クロマトグラフィーによって求めたとき、式4のC4−エピマーを10%未満の量しか含まない。別の実施形態では、C4−エピマーは、5%未満、3%未満、2%未満、1%未満、または0.5%未満の量しか存在しない。
【0078】
一実施形態では、固体組成物は、高速液体クロマトグラフィーによって求めたとき、式2を、2%未満の量、たとえば、1%未満または0.5%未満の量しか含まない。
【0079】
一実施形態では、還元を大規模で実施することができる。一実施形態では、「大規模」とは、式2による化合物を少なくとも1グラム使用すること、たとえば、少なくとも2グラム、少なくとも5グラム、少なくとも10グラム、少なくとも25グラム、少なくとも50グラム、少なくとも100グラム、少なくとも500g、少なくとも1kg、少なくとも5kg、少なくとも10kg、少なくとも25kg、少なくとも50kg、または少なくとも100kg使用することをいう。
【0080】
一実施形態では、アミノアシル化合物をアシル化剤として用いてアシル化を実施する。一実施形態では、アシル化は、水性媒質から選択されるものであってよい反応媒質、および試薬塩基なしの少なくとも1種の塩基性溶媒中で実施する。
【0081】
一実施形態では、式Iの化合物の調製方法は、チゲサイクリンまたは薬学的に許容できるその塩の調製方法である。
【0082】
式4の化合物の塩は、塩酸塩などのハロゲン化塩であってよい。
【0083】
アシル化反応媒質は、極性非プロトン性溶媒またはその溶媒混合物から選択される溶媒であってよい。一実施形態では、極性非プロトン性溶媒は、アセトニトリル、1,2−ジメトキシエタン、ジメチルアセトアミド、ジメチルホルムアミド、ヘキサメチルホスホルアミド、N,N’−ジメチルエチレン尿素、N,N’−ジメチルプロピレン尿素、塩化メチレン、N−メチルピロリジノン、テトラヒドロフラン、およびこれらの混合物から選択する。別の実施形態では、極性非プロトン性溶媒は、アセトニトリル、ジメチルホルムアミド、N,N’−ジメチルプロピレン尿素、N−メチルピロリジノン、テトラヒドロフラン、およびこれらの混合物から選択する。少なくとも1種の塩基性溶媒は、アセトニトリルとN,N’−ジメチルプロピレン尿素の混合物であってよい。別の実施形態では、少なくとも1種の塩基性溶媒は、水とN,N’−ジメチルプロピレン尿素の混合物であってよい。さらなる実施形態では、少なくとも1種の塩基性溶媒は、N,N’−ジメチルプロピレン尿素である。
【0084】
反応媒質は、水性媒質でもよい。さらなる実施形態では、塩基なしの少なくとも1種の塩基性溶媒は、塩基なしの水である。別の実施形態では、反応媒質は、試薬塩基なしの少なくとも1種の塩基性溶媒であってよい。塩基性溶媒とは、プロトンを部分的または完全に受容することのできる溶媒である。試薬塩基とは、反応の開始時に、式4の化合物およびアミノアシル化合物と同時にまたはそれに引き続いて加えられ、プロトンを部分的または完全に受容することのできる塩基をいう。試薬塩基は、反応の最中に加えられる塩基も指す。
【0085】
アミノアシル化合物は、ハロゲン化アミノアシル、アミノアシル無水物、および混合アミノアシル無水物から選択されるものであってよい。一実施形態では、アミノアシル化合物は、式6のハロゲン化アミノアシル
【0086】
【化10】
またはその塩であり、
式中、R1およびR2は、水素、(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Qは、フッ化物、臭化物、塩化物、およびヨウ化物から選択されるハロゲンである。
【0087】
さらなる実施形態では、Qは塩化物である。式6の化合物の塩は、ハロゲン化塩から選択されるものであってよい。ハロゲン化塩とは、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩などの、ハロゲンアニオンとの相互作用から生成する任意の塩を指す。一実施形態では、ハロゲン化塩は、塩酸塩である。
【0088】
別の実施形態では、nは1である。さらなる実施形態では、Xは臭化物である。
【0089】
式4の化合物とアミノアシル化合物の反応は、0℃〜30℃、たとえば20℃〜25℃、たとえば10℃〜17℃、たとえば0℃〜6℃、さらにはたとえば2℃〜8℃の範囲の温度で実施することができる。反応させる時間は、1時間〜24時間、たとえば0.5時間〜4時間、さらにはたとえば2時間〜8時間の範囲であってよい。式4の化合物の量に対して過剰のアミノアシル化合物を反応において使用することができる。一実施形態では、過剰は、1当量の式4の化合物に対して3当量のアミノアシル化合物であってよい。別の実施形態では、水性媒質対式4の化合物の比を、6:1w/wまたは5:1体積にすることができる。一実施形態では、アミノアシル化合物は、式4の化合物を水性媒質に溶かした溶液に加えるかそれと合わせる。
【0090】
一実施形態では、反応媒質が水性媒質である場合、水性媒質のpHは、4〜9、たとえば5〜7.5、たとえば6.3〜6.7、たとえば7.0〜7.5の範囲のpH、さらにはたとえば6.5、さらにまたたとえば7.2に調整することができる。pHを調整する前に水を加えてもよい。pHの調整は、限定はしないが水酸化アンモニウムを含めた、塩基を加えるものであってよい。水酸化アンモニウムの濃度は、25%〜30%の範囲であってよい。別の実施形態では、塩酸などの酸を使用してpHを調整してもよい。pH調整の際の反応媒質は、−5℃〜℃、たとえば5℃〜8℃、さらにはたとえば0℃〜5℃の範囲の温度にすることができる。
【0091】
pHの調整に続いて、水性媒質に少なくとも1種の有機溶媒または溶媒混合物を加えることができる。一実施形態では、少なくとも1種の有機溶媒混合物は、メタノールと塩化メチレンを含むものであってよい。メタノールの濃度は、限定はしないが20%や30%などの、5%〜30%の範囲であってよい。別の実施形態では、少なくとも1種の有機溶媒または溶媒混合物は、テトラヒドロフランを含むものであってよい。混合物の温度は、15℃〜25℃の範囲であってよい。
【0092】
一実施形態では、水性媒質を、少なくとも1種の極性プロトン性溶媒と少なくとも1種の極性非プロトン性溶媒の混合物で抽出してもよい。一実施形態では、少なくとも1種の極性非プロトン性溶媒は、塩化メチレンを含み、少なくとも1種の極性プロトン性溶媒は、メタノールを含む。別の実施形態では、水性媒質を、塩化メチレンなどの少なくとも1種の極性非プロトン性溶媒で抽出する。抽出は、−5℃〜25℃、さらにはたとえば0℃〜5℃の範囲の温度で実施することができる。さらなる実施形態では、各抽出の後、水性媒質のpHを7.0〜7.5の範囲、たとえば7.2に調整する。抽出プロセスは、たとえば10回まで繰り返してもよい。
【0093】
一実施形態では、合わせた有機抽出物を、硫酸ナトリウムなどの乾燥剤で処理してもよい。有機抽出物は、Norit CA−1などの炭で処理してもよい。濾過によって固体を除去して溶液を得る。一実施形態では、溶液を濃縮して式1の化合物を得てもよい。
【0094】
反応から得られた式1の化合物は、少なくとも1種の有機溶媒または溶媒混合物中に結晶化させることができる。一実施形態では、有機溶媒混合物は、メタノールと塩化メチレンを含む。結晶化は、たとえば、−15℃〜155℃、たとえば0℃〜15℃、さらにはたとえば2℃〜5℃の範囲の温度で行うことができる。
【0095】
別の実施形態では、抽出に続いて、得られる、少なくとも1種の極性プロトン性溶媒と少なくとも1種の極性非プロトン性溶媒の有機混合物を濃縮してスラリーを得、濾過して式1の化合物を得てもよい。濃縮および濾過は、たとえば0℃〜5℃で行うことができる。
【0096】
式1の化合物の調製方法は、5グラム超、たとえば10グラム超、たとえば50グラム超、たとえば100グラム超、たとえば500グラム超、たとえば1キログラム超、さらにはたとえば10キログラム超の式4のアミンを使用して実施することができる。
【0097】
一実施形態では、本明細書に記載の方法のいずれかによって調製した式1の化合物は、高速液体クロマトグラフィーによって求めたとき、10.0%未満の不純物、たとえば5%未満の不純物、たとえば2%未満の不純物、さらにはたとえば1〜1.4%の不純物を含有する。さらなる実施形態では、式1の化合物は、高速液体クロマトグラフィーによって求めたとき、0.5%未満のC4−エピマー、さらには0.2%未満のC4−エピマーなどの、1.0%未満の量のC4−エピマーを含有する。一実施形態では、式1の化合物は、高速液体クロマトグラフィーによって求めたとき、1%未満のミノサイクリン、たとえば0.6%未満のミノサイクリンを含有する。別の実施形態では、式1の化合物は、5%未満のジクロロメタン、たとえば2〜3%未満のジクロロメタンを含有する。
【0098】
精製
本発明の一実施形態では、本発明のプロセスは、式1の化合物または薬学的に許容できるその塩、
または薬学的に許容できるその塩[式中、R1およびR2は、水素、直鎖および分枝鎖(C1〜C6)アルキル、およびシクロアルキルからそれぞれ独立に選択され、Rは−NR3R4であり、R3およびR4は、水素ならびに直鎖および分枝鎖(C1〜C4)アルキルからそれぞれ独立に選択され、nは、1〜4の範囲をとる]を精製するステップをさらに含んでもよく、
精製は、
A)式1の化合物を、少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせて、第一の混合物を得るステップと、
B)第一の混合物を、0℃〜40℃の範囲の温度で15分〜2時間などの少なくとも1回の期間、混合するステップと、
C)式1の化合物を取得するステップとを含む。
【0099】
ここでは、用語「取得する」とは、限定はしないが、90%、95%、96%、97%、98%および99%より高いレベルを含めた有用なレベルの純度で、化合物を単離することを指す。純度のレベルは、高圧液体クロマトグラフィーによって求めることができる。
【0100】
一実施形態では、式1の化合物の精製は、
A)式1の化合物を、少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせて、第一の混合物を得るステップと、
B)第一の混合物を30℃〜40℃の範囲の温度で一定期間混合するステップと、
C)その第一の混合物を15℃〜25℃の範囲の温度に冷却し、混合物を混合せずに第二の期間静置するステップと、
D)その第一の混合物を0℃〜6℃の範囲の温度に冷却し、混合物を混合せずに第三の期間静置するステップと、
E)式1の化合物を取得するステップとを含む。
【0101】
一実施形態では、この方法は、nが1であり、R1が水素であり、R2がt−ブチルであり、R3およびR4がそれぞれメチルである式1の化合物を使用するものでもよい。少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせる式1の化合物は、固体、スラリー、懸濁液、および溶液から選択される形で用意することができる。
【0102】
一実施形態では、少なくとも1種の極性非プロトン性溶媒は、アセトン、1,2−ジクロロエタン、酢酸メチル、メチルエチルケトン、メチルイソブチルケトン、塩化メチレン、および酢酸エチルから選択されるものであってよい。さらなる実施形態では、少なくとも1種の極性非プロトン性溶媒は、アセトンおよび塩化メチレンから選択されるものであってよい。別の実施形態では、少なくとも1種の極性プロトン性溶媒は、メタノール、エタノール、イソプロパノール、およびt−ブタノールから選択されるものであってよい。さらなる実施形態では、少なくとも1種の極性プロトン性溶媒は、メタノールであってよい。
【0103】
少なくとも1種の極性非プロトン性溶媒と少なくとも1種の極性プロトン性溶媒の組合せとして、アセトンとメタノールを挙げることができる。別の実施形態では、少なくとも1種の極性非プロトン性溶媒の塩化メチレンと、少なくとも1種の極性プロトン性溶媒のメタノールの組合せを用意する。さらなる実施形態では、少なくとも1種の極性非プロトン性溶媒と少なくとも1種の極性プロトン性溶媒の組合せとして、酢酸メチルとメタノールを挙げることができる。式1の化合物は、たとえば、等体積の少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせることができる。
【0104】
一実施形態では、第一の混合物は、たとえば、温度が15℃〜25℃の範囲である、30分〜2時間の範囲の第一の期間、次いで温度が0℃〜2℃の範囲である、30分〜2時間の範囲の第二の期間の間、混合する。一実施形態では、第一の期間および第二の期間は、それぞれ1時間である。別の実施形態では、この方法は、第一の混合物を、15℃〜25℃の範囲の温度で、30分〜2時間の範囲の少なくとも1回の期間混合し、次いでその第一の混合物を濾過して固体を得るステップを含んでもよい。この方法は、その固体を、15℃〜25℃の範囲の温度で30分〜2時間の範囲の第一の期間をかけて、たとえば等体積の少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせ、濾過して第二の固体を得るステップをさらに含んでもよい。さらなる実施形態では、合わせ、濾過するこれらのステップを2回から15回繰り返してもよい。
【0105】
式1の化合物の精製は、第一の混合物から固体を得、その固体を少なくとも1種の極性プロトン性溶媒および少なくとも1種の極性非プロトン性溶媒と合わせて第二の混合物を得るステップをさらに含んでもよい。第二の混合物は、たとえば、メタノールおよび塩化メチレンを、1:5〜1:15のメタノール:塩化メチレンの範囲の体積比で含むものであってよい。一実施形態では、第二の混合物を30℃〜36℃の範囲の温度で混合し、次いで濾過して溶液を得てもよい。さらなる実施形態では、溶液中の極性プロトン性溶媒の濃度を5%未満のレベルに減らしてもよく、溶液を、たとえば0℃〜6℃の範囲の温度で、たとえば30分〜2時間の範囲の期間混合してから濾過してもよい。
【0106】
一実施形態では、第一の混合物を混合するステップは、10〜20分の範囲の期間、たとえば15分間行うものであってよい。一実施形態では、第一の混合物を15℃〜25℃の範囲の温度に冷却し、混合物を混合せずに静置するステップは、30分〜3時間、たとえば1時間〜2時間の範囲の第二の期間行うものであってよい。第一の混合物は、0℃〜6℃の範囲の温度にさらに冷却し、混合せずに30分〜2時間の範囲の第三の期間、たとえば1時間静置してもよい。
【0107】
式1の化合物を取得するステップは、本明細書に記載の任意の混合物を、発熱物質低減用フィルターおよび清澄化用フィルターから選択される少なくとも1種類のフィルターで濾過するステップを包含するものであってよい。本明細書で開示するとおり、混合は、機械式混合装置、たとえば撹拌子または撹拌機を使用して実施することができる。混合は、式1を有する化合物の溶媒系への溶解性によって実現することもできる。温度を上昇させると、溶解性を増大させることができる。
【0108】
一実施形態では、式1の化合物を少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせることにする場合、式1の化合物は、薬学的に許容できるその塩の形で使用してもよい。式1の化合物を本発明の方法の生成物として取得する場合、式1の化合物を薬学的に許容できるその塩の形で回収してもよい。
【0109】
本発明による方法によって式1の化合物を取得する別の実施形態では、酸を加えることにより、化合物を薬学的に許容できるその塩に変換してもよい。
【0110】
一実施形態では、精製された式1の化合物は、チゲサイクリンである。
【0111】
チゲサイクリンを少なくとも1種の極性非プロトン性溶媒および少なくとも1種の極性プロトン性溶媒と合わせたものは、固体、スラリー、懸濁液、および溶液から選択される形で用意することができる。一実施形態では、本方法から取得したチゲサイクリンは、高圧液体クロマトグラフィー(HPLC)によって求めたとき、チゲサイクリンのC−4エピマーまたは薬学的に許容できるその塩を1%未満含有してもよい。
【0112】
本方法から取得した式1の化合物は、HPLCによって求めたとき、3.0%未満の不純物、たとえば1.0%未満の不純物、たとえば0.7%未満の不純物を含有してもよい。別の実施形態では、式1の化合物は、HPLCによって求めたとき、式1の化合物のC4エピマーまたは薬学的に許容できるその塩を2%未満、たとえばC4エピマーを1%未満、たとえばC4エピマーを0.5%未満含有してもよい。
【0113】
本方法は、5グラム超、たとえば50グラム超、たとえば100グラム超、たとえば500グラム超、たとえば1キログラム超、さらにはたとえば10キログラム超の式1の化合物で実施することができる。
【0114】
本開示の広い範囲を述べる、数字による範囲およびパラメータは、概数であるにもかかわらず、詳細な実施例で述べるその数値は、できる限り正確に報告する。しかし、どんな数値も、そのそれぞれの試験測定で見出される標準偏差から必然的に得られる一定の誤差を本質的に含んでいる。
【0115】
以下の実施例は、限定しない形で本発明を例示するものである。
【実施例】
【0116】
HPLC分析は、以下の条件下で実施した。
カラム:Waters Symmetratry RP8 15×0.46cm
移動相:A;0.03MのKH2PO4、H3PO4によりpH2、B;9:1のアセトニトリル:水
流量:0.8ml/分
検出波長:250nm
カラムオーブン温度 35℃
定組成プログラム:時間(分) A(%) B(%)
0 90 10
2 90 10
30 45 55
32 90 10
【0117】
ニトロ化
式2の化合物の一例であるミノサイクリンを、米国特許第3,226,436号に記載の方法に従って調製した。次いで、ミノサイクリン塩化物を以下の手順に従ってニトロ化した。
【0118】
15gの98%硫酸に5.3gのミノサイクリン塩化物を10〜30℃で数回に分けて加えた。懸濁液をN2中にて20℃で4時間撹拌し、均質な溶液を生成した。この溶液に2.2mLの硝酸イソプロピルをゆっくりと加えて、反応温度を30℃より低く保った。加えた後、反応液をもう2時間ねかせた。反応混合物をIPA/Hep溶液(125mL/25mL)に10℃未満でゆっくりと加え、次いで溶液をこの温度でもう1時間撹拌した。生成した固体を濾過し、予め冷却してある30mLのIPAで洗浄した。固体を真空オーブンにて40℃で20時間乾燥させた。黄色の固体(7.3g、純度91.3重量%、収率92.6%)が得られ、これ以上精製せずに次の反応へ進めた。
【0119】
ニトロ化反応から生成した中間体を単離した後の還元
加圧ガラス瓶に、50mLのメタノールおよび10mLの硫酸を加えた。予め調製しておいた9−ニトロミノサイクリン硫酸塩5gを5℃で一度に加えた。溶液に0.4gの10%Pd/Cを加え、次いで懸濁液をN2で3回、H2で1回パージした。Parr振盪機において、懸濁液を圧力45PSIのH2中にて20℃で3時間維持した。単純濾過によって触媒を取り除いた後、反応溶液をIPA/Hep溶液(100mL/30mL)に5〜10℃で加えた。固体を濾過し、予め冷却しておいた20mLのIPAで洗浄した。固体を真空オーブン中にて40℃で20時間乾燥させ、黄色の固体(4.3g、純度74重量%)を収率76%で得た。
【0120】
ニトロ化反応から生成した中間体を単離せずに行う還元
15gの98%硫酸に5.3gのミノサイクリン塩化物を10〜30℃で数回に分けて加えた。懸濁液をN2中にて20℃で4時間撹拌し、均質な溶液を生成した。この溶液に2.2mLの硝酸イソプロピルをゆっくりと加えて、反応温度を30℃より低く保った。加えた後、反応液をもう2時間ねかせた。反応混合物を5〜10℃にて50mLのメタノールで希釈し、次いで1gの10%Pd/Cを加えた。懸濁液をN2で3回、H2で1回パージし、次いでParr振盪機において圧力45PSIのH2中で3時間維持した。単純濾過によって触媒を取り除いた後、反応溶液をIPA/Hep溶液(250mL/50mL)に5〜10℃で加えた。固体を濾過し、予め冷却しておいた40mLのIPAで洗浄した。固体を真空オーブン中にて40℃で20時間乾燥させ、黄色の固体(7.4g、純度72重量%)を収率87%で得た。
【0121】
アシル化
式4の化合物のアシル化は、上で開示したとおりに実施することができる。アシル化は、参照により本明細書に援用される米国特許公開第2007−0049560A1号の[0143]〜[0213]段落でも開示されている。
【0122】
精製
式1の化合物の精製は、上記で開示したとおりに実施することができる。精製は、参照により本明細書に援用される米国特許公開第2007−0049560 A1号の[0214]〜[0283]段落でも開示されている。
【0123】
本発明について、本発明の実施形態およびその非限定的な実施例の論述によって説明してきたが、当業者なら、本明細書および特許請求の範囲を読めば、同じく本発明の企図される範囲内にある他の実施形態および変形形態を想定することができ、したがって本発明の範囲は、付属の特許請求の範囲によってのみ解釈され規定されるものとする。
【図1】
【公表番号】特表2012−520305(P2012−520305A)
【公表日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願番号】特願2011−554114(P2011−554114)
【出願日】平成22年3月9日(2010.3.9)
【国際出願番号】PCT/US2010/026630
【国際公開番号】WO2010/114680
【国際公開日】平成22年10月7日(2010.10.7)
【出願人】(309040701)ワイス・エルエルシー (181)
【Fターム(参考)】
【公表日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願日】平成22年3月9日(2010.3.9)
【国際出願番号】PCT/US2010/026630
【国際公開番号】WO2010/114680
【国際公開日】平成22年10月7日(2010.10.7)
【出願人】(309040701)ワイス・エルエルシー (181)
【Fターム(参考)】
[ Back to top ]