
テトラサイクリンの1日1回製剤
【課題】急性または慢性疾患、例えば炎症性コンポーネントを備えるものの治療に使用し得るドキシサイクリンの薬学的組成物の提供
【解決手段】急性及び慢性炎症性疾患状態(酒さ及び関節炎のような)の治療に有用である、即効型(IR)製剤にテトラサイクリンを含有するコンポーネントと遅延放出型(DR)製剤にテトラサイクリンを含有する第二コンポーネントの組み合わせであり、ここで、IRとDRの割合が約99:1〜約70:30である組成物。
【解決手段】急性及び慢性炎症性疾患状態(酒さ及び関節炎のような)の治療に有用である、即効型(IR)製剤にテトラサイクリンを含有するコンポーネントと遅延放出型(DR)製剤にテトラサイクリンを含有する第二コンポーネントの組み合わせであり、ここで、IRとDRの割合が約99:1〜約70:30である組成物。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明はテトラサイクリンの1日1回組成物(once−daily compositions)に関し、これは急性または慢性疾患、例えば炎症性コンポーネントを備えるものの治療に使用し得る。より詳細には、本発明は、炎症のようなものに関わるコラーゲン破壊酵素または分子が要因である疾患または状態の治療のためのドキシサイクリンの薬学的組成物に向けられ、そして、これは1日1回製剤である。該組成物は、特に歯周病、酒さ(rosacea)、ドライアイ、アクネ(acne)及び慢性関節リウマチのような一般的な疾患状態の処置に有用である。
【背景技術】
【0002】
発明の背景
通常、ドキシサイクリン及び関連するテトラサイクリンは、様々な細菌感染症を治療するための広域スペクトル抗生物質として使用される。テトラサイクリンはアミノアシル−tRNAのリボゾームへの結合を阻止することにより、グラム陽性及びグラム陰性細菌のタンパク合成を阻害する。これらの作用は殺菌(殺菌性)よりむしろ静菌(細菌の増殖阻止)である。抗生物質効果を達成するために一般的に使用されるドキシサイクリンの用量は、100mg及び50mgである。
【0003】
ドキシサイクリンはまた、他のテトラサイクリンと同様に、その抗生物質特性に加えて他の治療用途も有する。例えば、ドキシサイクリンは、コラゲナーゼ、ゼラチナーゼ及びエラスターゼのようなコラーゲン破壊酵素の作用を阻害することが知られている。そのコラゲナーゼ阻害活性は歯周病を治療するために使用されている。他の例では、ドキシサイクリンは、細菌P.acnesにより産生されるリパーゼを阻害し、そして、炎症に関係する遊離脂肪酸の利用能を低下し得る。ドキシサイクリンはまた、サイトカインレベルを低下することにより炎症を減少し得、そして、濾胞壁の完全性を保持する。このように、ドキシサイクリンはまた、アクネのような皮膚病を治療する潜在能力も有する。
【0004】
効果に関するメカニズムは完全には明らかではないが、研究者はテトラサイクリンの抗微生物用量以下で、様々な病気(ailments)の治療に有用であることを発見している。例えば、米国特許第6,455,583号では、患者に非−抗菌量のテトラサイクリンを経
口投与することにより、瞼板腺炎を治療することを開示している。米国特許第6,100,248
号は、これらの抗菌活性を弱めるまたは消すために化学的に修飾されたテトラサイクリンを投与することにより、癌増殖を抑制する方法を教示している。抗菌治療に使用される通常の量より一般的に少量のテトラサイクリン量を投与することにより、コラーゲン分解酵素を減少させる方法が、米国特許第4,666,897号に開示されている。この段落で引用した
特許は、これより、本明細書において参考文献として組み込まれる。
【0005】
市場において、歯周病の治療において部位特異的に使用するための2種のはめ込み型製品がある。PerioChip(登録商標)は小さく、オレンジ−褐色チップであり、これは歯周ポケットに挿入される。それぞれのPerioChip(登録商標)は、生分解性で、再吸収可能なマトリックスに、2.5mgのグルコン酸クロルヘキシジンを含有する。PerioChip(登録商標)治療は、5mmまたはより深く残存するポケットに3ヶ月毎に1回投与することが推奨されている。第2の製品、Atridox(登録商標)は、注射可能で、再吸収可能なゲルであり、約一週間、42.5mgのドキシサイクリンの歯肉下への制御放出を提供する。さらに、現在、Periostat(登録商標)と呼ばれる新しい経口薬剤を利用することが可能であり、これは、成人の歯周病患者に使用
されるコラゲナーゼ阻害剤として、20mgのドキシサイクリンを全身に送達する。多くの人々は、インプラントよりもピルを好む。しかし、Periostat(登録商標)は1日2回の投与が必要であり、そして、患者の薬剤服用順守に対する懸念がある。このように、ドキシサイクリンの1日1回の製剤を開発することは非常に利益をもたらす。
【0006】
ドキシサイクリンは感染症を治療する上で一般的に効果的であるが、ドキシサイクリンの使用は望ましくない副作用も導き得る。例えば、抗生物質のドキシサイクリンの長期間投与は、健全な生物フローラ(腸内細菌叢のような)を減少または排除し得、そして、抗生物質耐性生物の産生または酵母及びカビの過剰増殖を導き得る。抗生物質特性の好ましくない副作用のために、テトラサイクリン(特にドキシサイクリン)の抗コラーゲン破壊酵素または他の有益な効果は獲得するが、抗菌効果は回避するようなドキシサイクリン送達のための独特の用量及び改良された製剤が必要である。
【発明の概要】
【課題を解決するための手段】
【0007】
本発明の要約
本発明は、その最も広い意味において、定常状態で、疾患または状態の処置において有益な効果を有することには十分有効であるが、抗菌効果を発揮するほど高くない有効成分のin vivoレベルの徐放性プロフィールを提供するために設計されたテトラサイクリンの薬学的組成物に向けられている。このような薬学的組成物は、1日1回摂取され得る剤形で処方される。
【0008】
本発明の一つの目的は、ドキシサイクリンの薬学的組成物を提供することであり、これは、長期間の抗菌活性の望ましくない影響を伴うことなく、コラゲナーゼの過剰産生(歯周病のような)またはドキシサイクリンで調節され得る特定の疾患状態(炎症を伴う状態のような)に関係する他の生化学剤が原因で生じる疾患または状態の治療または予防に必要とされる定常状態の血中レベルをもたらすにもかかわらず、1日1回で与えられ得る。
【0009】
本発明の一つの目的は、最小約0.1μg/ml及び最大約1.0μg/mlのドキシサイクリンの定常状態の血中レベルを与えるドキシサイクリンを含有する1日1回の薬学的組成物を提供することである。
【0010】
本発明の一つの側面は、ドキシサイクリンベースで、50mg未満であるが25mgより多く、好ましくは約40mgを含有するドキシサイクリンの即効型(immediate release)製剤である。
【0011】
他の側面において、本発明は、薬剤の即効型(IR)コンポーネント及び薬剤の遅延放出型(delayed release(DR))コンポーネントを含むドキシサイクリンの薬学的組成物に向けられ、これらは1日1回投与のための一つの単位用量に合わせられる。該コンポーネントは様々な割合で存在し得るが、好ましくはIR:DRが約70:30〜約80:20、最も好ましくは75:25の割合であるが、ドキシサイクリンの全投与量は約50mg未満、そして、好ましくは約40mgである。該割合はIRとDRの用量の内訳を示しており、例えば、80:20は、40mgの80%がIR部分からであり、40mgの20%がDR部分からであることを意味する。
【0012】
さらに、本発明の他の目的は、ドキシサイクリンの1日1回用量を投与することにより、歯周病、慢性関節リウマチ、副甲状腺機能亢進症、糖尿病及びアクネのような組織の病理学的破壊を生じるコラゲナーゼが過剰量産生される疾患または状態を治療する方法を提供することである。例えば、Golubの米国特許第4,666,897号を参照。
【0013】
本発明の他の目的は、本発明に従うドキシサイクリンの1日1回用量を投与することにより、酒さ、ヒトの皮膚科学的状態の全身治療方法を提供することである。
【0014】
本発明の他の目的は、本発明の1日1回組成物を調製するためのプロセスを提供することである。
【0015】
本発明の詳細な説明
次の記載は主としてドキシサイクリンに向けられるが、本発明は他のテトラサイクリン類、特にドキシサイクリンと類似のin vivo吸収プロフィールを有する他のテトラサイクリン類、より具体的にはドキシサイクリンと同様の消化管内での吸収域を有するテトラサイクリン類に適用できるということが予想される。様々な種類のテトラサイクリン類及び様々な疾患状態に対する有益な効果は、例えばWO02/083106及び米国特許第6,638,922号に開示され、これらのそれぞれは、これら全体を本明細書において参考として援用する。
【0016】
本発明は、例として、哺乳動物、好ましくはヒトが、1日に1回のみの用量を行う必要のある製剤ではあるが、薬剤の特定の定常状態の血中レベルを提供するために設計されたドキシサイクリンの経口投与組成物を提供することにより成し遂げられ得る。本発明の組成物は、同じ効果を達成する組成物の1日に複数回の投与(1日2回の投与のような)に代わって、有用であることが意図される。ドキシサイクリン(または比較可能な生理学的及び吸収特性の他のテトラサイクリン類)の好ましい血中レベルは、定常状態で約0.1〜約1.0μg/mlである。好ましくは、血中レベルは、治療の全体を通して、日々の投与を伴う、好ましい血中レベル内にある。さらに好ましくは、血中レベルは約0.3μg/ml〜約0.8μg/mlである。
【0017】
上記の血清レベルは、望ましくない抗生物質活性を伴うことなく、ドキシサイクリンの望ましい抗−コラゲナーゼ及び抗−炎症活性を可能にする。これらのレベルは、ドキシサイクリンベースで50mgより低いが25mgより多い、好ましくは約40mgを含有する即効型製剤の単一の1日1回の投与で成し遂げ得るという驚くべき発見であった。
【0018】
「約」は、活性のある薬学的成分の量に関して、米国薬局方(USP−NF 21)、2003 Annual Editionにみられる、または、www.usp.orgで利用できる薬学的に許容される制限内であることを意味する。血中レベルの側面からみると、「約」はFDAの許容するガイドライン内であることを意味する。
【0019】
「即効型」製剤は、促進、遅延、または、徐放性効果を伴うことなく投与時に、全ての活性成分を実質的に放出することを意図する剤形を意味する。
ドキシサイクリンのこのような組成物は、液状懸濁液または溶液の形態で、あるいは、錠剤、ペレット(本明細書ではビード(bead)とビードレット(beadlet)を区別なく使用する)、粒子(particle)、カプセルまたはゲルのような固形物の形態であり得る。本発明において好ましくは、錠剤またはカプセル中のビードレットである。
【0020】
薬学的な活性成分に関して、本発明の要求される血清レベルに従うならば、テトラサイクリン化合物のいかなる形態でも使用し得る。例えばドキシサイクリンは、2つの化学的な形態:モノハイドレート形態及びヒクレート(hyclate)形態、にある薬学的製剤で一般
的に使用される。モノハイドレートは1分子の水で水和された塩基分子であり、カプセル及び、幾つかのマーケットにおいては、粉末の経口懸濁液(水を加えて再構成される)の製剤で使用される。ヒクレートは、水及びエタノールで溶媒和された塩酸塩であり、そしてカプセルまたは錠剤の製剤で概して使用される。本発明の組成物にあるドキシサイクリ
ンの量は、ドキシサイクリンベースを意味する。また、本発明の組成物において、一つ以上の活性成分があり得る。すなわち、ドキシサイクリンは、剤形において、他の治療に役立つまたは栄養性の物質と混合され得る。
【0021】
即効型剤形
このような即効型剤形を処方するための当業者に公知の多くの方法がある。例えば、即効型錠剤は、ドキシサイクリンと、微結晶性セルロース、例えばAVICEL(登録商標)(FMC Corp.)またはEMCOCEL(登録商標)(Mendell Inc.);リン酸ジカルシウム、例えばEMCOMPRESS(登録商標)(Mendell
Inc.);硫酸カルシウム、例えばCOMPACTROL(登録商標)(Mendell Inc.);及びスターチ、例えばSTARCH 1500のようなバルキング剤(bulking agent)を混合することにより調製され得る。さらに、微結晶性セルロース、スターチ、クロスポビドン、例えばPOLYPLASDONE XL(登録商標)(International Specialty Products);ソディウムスターチグリコレート、例えばEXPLOTAB(登録商標)(Mendell Inc.);及びクロスカルメロースナトリウム(croscarmellose sodium)、例えばAC−DI−SOL(登録商標)(FMC Corp.)のような崩壊剤を添加し得る。本明細書で使用する抗粘着剤(Antiadherant)及びグリダント(glidant)はタルク、コーンスターチ、二酸化珪素、ラウリル硫酸ナトリウム、コロイド状二酸化珪素(silica dioxide)及びメタリックステアラート(metallic stearate)を挙げることができる。
【0022】
滑沢剤(lubricants)は、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸ナトリウム、ステアリン酸、フマル酸ステアリルナトリウム、ステロテックス(sterotex)、タルク、ワックスなどが使用され得る。結合剤(binding agents)は、ポリビニルピロリドン、スターチ、メチルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースなどが使用され得る。
【0023】
本発明は、当業者に公知の方法(湿式造粒法及び直接圧縮法を含む)を使用して調製した錠剤に好ましくは処方される。経口錠剤は当業者に公知のいずれかの適したプロセスを使用して調製される。例えば、Remington’s Pharmaceutical Sciences, 18th Edition, A. Gennaro, Ed., Mack Pub. Co. (Easton, PA 1990) Chapters 88-91を参照し、この全
体はこれより参考として援用される。概して、活性成分、ドキシサイクリンは、薬学的に許容される賦形剤(例えば、上に列挙した結合剤、滑沢剤など)と混合し、そして、錠剤に圧縮する。好ましくは、剤形は湿式造粒技術または直接圧縮法により調製され、そして、均一な顆粒を形成する。あるいは、活性成分は、顆粒が調製された後に顆粒と混合し得る。そして、湿った顆粒マスを乾燥させ、そして適したふるいわけ装置(screening device)を使用して大きさをそろえ、そして粉末を提供し、そして、これは、望ましいように、カプセルに充填あるいはマトリックス錠剤またはキャプレッツ(caplets)に圧縮し得る。
【0024】
好ましい実施形態において、錠剤は直接圧縮法を使用して調製される。直接圧縮法は、特に比較的簡単な製造に関して、湿式造粒法以上に数多くの潜在的な利点を提供する。直接圧縮法において、少なくとも一つの薬学的活性剤及び賦形剤または他の成分が、ステンレススチールふるい(40メッシュスチールふるいのような)を通してふるいにかけられる。そして、ふるいにかけた材料を適切な混合機に移し、そして、3分間増圧バーとともに10分間混合する。そして、混合物は適した道具を使用して回転プレス上で錠剤に圧縮する。
【0025】
上述したように、即効型組成物の他の好ましい剤形は、即効型ビードレットまたはペレ
ットを含有するカプセルである。このようなペレットを製造する方法は、以下の段落(例えば、IRペレット)に記載する。該ペレットを従来技術によりカプセルに、例えばゼラチンカプセルに、充填する。
【0026】
組み合わせIR/DR剤形
本発明の他の実施形態は、1単位用量において、ドキシサイクリンの実質的な即効投与を有する組成物に、あらかじめ決められた時間少なくとも一つのさらなる投与が続く。組成物の第1の即効型用量は散剤、顆粒剤、ビードレットまたは錠剤の形態であり得、第2の遅延放出部分は被覆された顆粒、被覆されたビードレット、被覆された錠剤、または被覆されていないマトリックス錠剤であり得る。即効部分またはコンポーネントと遅延放出部分またはコンポーネントの割合は、in vitroでの薬剤放出プロフィール及びin vivoでの血中濃度プロフィールを調整するために利用され得る。このような薬剤放出プロフィールを提供することにより、組成物はその日の2回目の投与の必要性を排除する。さらに、ドキシサイクリンの全投与量は、その抗生物質特性由来の望ましくない副作用を回避するために、50mg未満であるが、抗−コラゲナーゼ及び/または抗−炎症効果を達成するために25mgより多い。
【0027】
幾つかの剤形バリエーションは、これらの寄与を備えた製品を達成するために使用され得る。例えば、即効粉末混合物は、遅延放出錠剤または遅延放出ペレットと共にカプセル化され得る。さらなる例は、別々に調製され、そして、適切な大きさのカプセルシェルにカプセル化される即効型錠剤及び遅延放出型錠剤である。または、例えば、遅延放出型錠剤がコアとして使用され得、そして即効部分がプレスコーターを使用して外層として圧縮され得、または、薬剤積層技術を使用してオーバーコートされ得る(両方の技術は、H.A.Liberman, L.Lachman及びJ.B.Schwartzにより編集された, Maecel Dekker, Inc. New York and Basel による1990年のPharmaceutical Dosage Forms:Tablets, Second Edition, Volume 1,のGunsel and Dusel, Chapter 5,「Compression-coated and layer tablets」に見られ得る)。
【0028】
複合粒子カプセル
好ましい実施形態として、ドキシサイクリンのIR/DR組成物は、ビードレットを含有するカプセルの形態である。現在、単一形態の複合単位剤形において、2つの異なるタイプの単位を有することが好ましい。第1ユニットは即効剤形であり、好ましくはペレット形態にある。このコンポーネントはまた、望ましいまたは必要ならば、粉末であり得る。いずれの場合でも、剤形はラウリル硫酸ナトリウム、ソディウムモノグリセラート、ソルビタンモノオレエート、ポリオキシエチレンソルビタンモノオレエート、グリセリルモノステアレート、グリセリルモノオレエート、グリセリルモノブチレート、界面活性ポリマーのプルロニック(Pluronic)系列のいずれか一つ、あるいは、界面活性特性を有する任意の他の適した物質または上記の任意の組み合わせのような界面活性剤を有し得る。好ましくは、界面活性剤はソディウムモノグリセラートとラウリル硫酸ナトリウムの組み合わせである。このコンポーネントのこれらの物質の濃度は約0.05〜約10.0%(W/W)の範囲であり得る。
【0029】
薬剤含有ペレットを製造する上で使用され得る他の賦形剤物質は、薬剤学において一般的に使用されるそれらのいずれかであり、そして、活性薬剤とペレットの物理化学的特性の適合性を基準にして選択すべきである。これらは、例えば、メチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、ポリビニルピロリドン/ビニルアセテートコポリマーなどのようなセルロース誘導体等の結合剤(binder);コーンスターチ、アルファ化デンプン、架橋カルボキシメチルセルロース(AC−DI−SOL(登録商標))、ソディウムスターチグリコレート(EXPLOTAB(登録商標))、架橋ポリビニルピロリ
ドン(PLASDONE(登録商標)XL)のような崩壊剤、及び錠剤調製で使用される任意の崩壊剤(これらは一般的に、本発明の一つのような即効用量において使用される);ラクトース、炭酸カルシウム、リン酸カルシウム、硫酸カルシウム、微結晶性セルロース、デキストラン、スターチ、スクロース、キシリトール、ラクチトール、マンニトール、ソルビトール、塩化ナトリウム、ポリエチレングリコールなどのような充填剤;ラウリル硫酸ナトリウム、ソルビタンモノオレエート、ポリオキシエチレンソルビタンモノオレエート、胆汁酸塩、グリセリルモノステアレート、PLURONIC(登録商標)系列(BASF)などのような界面活性剤;クエン酸、コハク酸、フマル酸、リンゴ酸、酒石酸、マレイン酸、グルタル酸 重炭酸ナトリウム及び炭酸ナトリウムなどのような可溶化剤(solubilizers);抗酸化剤、緩衝液、酸などのような安定剤が挙げられ、これらもまた利用し得る。
【0030】
ペレットは、例えば、単純な顆粒化およびそれに続くふるい分け;押し出し及びマルメライゼーション(marumerization);回転造粒(rotogranulation);または、妥当な大きさ及びローバスト性(robustness)のペレットを生じる任意の凝集プロセスにより作成され得る。押し出し及びマルメライゼーションに関して、薬剤及び他の添加剤は、結合剤溶液を添加することにより粒状化される。湿ったマスを特定のサイズのふるいを備えた押し出し器に通し、そして、押し出されたものはマルメライザーで球状にする。得られたペレットは乾燥させ、そしてさらなる適用のためにふるいにかける。高剪断造粒機(high−shear granulation)を使用し得、ここで、薬剤及び他の添加剤は乾燥混合され、そして、高剪断造粒機/混合機に結合剤溶液を添加することにより混合物を湿らせる。混合及びミリングの複合作用により湿らせた後に、粒状物を混練する。結果として生じた顆粒またはペレットは乾燥させ、そしてさらなる適用のためにふるいにかける。
【0031】
以前に記載したように、混合及び混合しない場合(consideration)において好ましい形態がペレットであっても、この即効型コンポーネントを粉末として含有することもまた可能である。
【0032】
あるいは、組成物の即効ビードレットまたはペレットは溶液または懸濁液積層により調製され得、それによって、薬剤溶液またはディスパージョンが、結合剤を伴っても伴わなくても、流動床プロセッサーまたは他の適した装置において、コアまたは最初シード(調製されたものか市販の製品のいずれか)上に噴霧される。コアまたは最初のシードは、例えば、シュガー球体(sugar sphere)または微結晶性セルロースからなる球体であり得る。このように薬剤は最初のシードの表面に被覆される。薬剤を詰めたペレットは、さらなる適用のために乾燥する。
【0033】
第2ユニットは遅延放出(DR)プロフィールを有するべきであり、そして、GI管のpH変化ならびにドキシサイクリンまたは他のテトラサイクリンの吸収に対するその影響に対応できる必要がある。このペレットは、ペレットの微小環境のpHを低下するために有用である必要に応じた何らかの有機酸と同様に、第1ユニットのペレットに関して記載したように全ての成分を含有し得、そして、溶解を促進する。これらの物質は、制限されないが、クエン酸、乳酸、酒石酸、または他の適した有機酸である。これらの物質は、約0〜約15.0パーセント(W/W)の濃度で存在すべきであり;好ましくは、これらの物質は約5.0〜約10.0パーセント(W/W)の濃度で存在する。これらのペレットを製造するためのプロセスは、第1ユニットのペレットに関して上述したプロセスに一致する。
【0034】
第1ユニットのペレットと異なり、第2ユニットの遅延放出型コンポーネントは、ペレットからの薬剤の放出が遅らせるようなペレットの表面に適用される制御コートを有する
。これは、腸溶性物質の被覆を適用することにより達成される。「腸溶性物質」は、胃の酸性環境で実質的に不溶性であるが、特定のpHの腸液に主に溶解するポリマーである。腸溶性物質は毒性がなく、薬学的に許容されるポリマーであり、そして、例えば、セルロースアセテートフタレート(CAP)、ヒドロキシプロピルメチルセルロースフタレート(HPMCP)、ポリビニルアセテートフタレート(PVAP)、ヒドロキシプロピルメチルセルロースアセテートスクシナート(HPMCAS)、セルロースアセテートトリメリテート、ヒドロキシプロピルメチルセルローススクシナート、セルロースアセテートスクシナート、セルロースアセテートヘキサヒドロフタレート、セルロースプロピオナートフタレート、メチルメタクリル酸及びメチルメタクリレートのコポリマー、メチルアクリレート、メチルメタクリレート及びメタクリル酸のコポリマー、メチルビニルエーテル及び無水マレイン酸のコポリマー(Gantrez ES series)、エチルメチルアクリレート−メチルメタクリレート−クロロトリメチルアンモニウムエチルアクリレートコポリマー、ゼイン、シェラック及びコパールコロホリウム(copal collophorium)のような天然樹脂及び幾つかの市販の入手可能な腸ディスパージョンシステム(例えば、EUDRAGIT(登録商標)L30D55、EUDRAGIT(登録商標)FS30D、EUDRAGIT(登録商標)L100、KOLLICOAT(登録商標)EMM30D、ESTACRYL(登録商標)30D、COATERIC(登録商標)及びAQUATERIC(登録商標))を含有する。上記は、可能な物質のリストであるが、当業者は、それは幅広くなく、そして、遅延放出プロフィールを提供する本発明の目的に合致する他の腸溶性物質があるということを認識する。これらの被覆物質は、ペレット組成物の約1.0%(W/W)〜約50%(W/W)の範囲で表面を被覆することに使用され得る。好ましくは、これらの被覆物質は、約20〜約40パーセント(W/W)の範囲であるべきである。該ペレットは、例えば流動床装置またはパンコーティング(pan coating)において被覆され得る。
【0035】
腸溶性被覆ペレットに関して、pH4.5未満程度の酸性胃環境においてドキシサイクリンの実質的な放出はない。pH感受性層が小腸よりも高いpHに溶解した時;特定の遅延期間の後;またはユニットが胃を通過した後に、ドキシサイクリンは利用可能になる。好ましい遅延時間は2〜6時間の範囲にある。
【0036】
この実施形態のバリエーションに関して、DRペレットは、保護層そして最終的の腸溶性被膜により分離される、ドキシサイクリンの層を含有し、結果として「繰り返し作用(repeat−action)」用量デリバリーとなる。仮に全ての層におけるドキシサイクリン、または他のテトラサイクリンの放出が薬剤の吸収ウィンドウ内にある場合、このような剤形は、本発明の放出プロフィールの血液レベル要求に見合い得る。
【0037】
オーバーコート層はさらに、必要に応じて、本発明のIR/DRペレットに適用し得る。OPADRY(登録商標)、OPADRY II(登録商標)(Colorcon)及びColorconからの対応するカラー及びカラーレスグレードは、ペレットが粘着性になることから保護するために及び産物に色を提供するために使用し得る。保護的な及びカラーコーティングの推奨されているレベルは、1〜6%、好ましくは2−3%(W/W)である。
【0038】
多くの成分は、例えばコーティングプロセス及び製品特性を改良するために、可塑剤:アセチルトリエチルシトレート、トリエチルシトレート、アセチルトリブチルシトレート、セバシン酸ジブチル、トリアセチン、ポリエチレングリコール、プロピレングリコール及び他のもの;滑沢剤:タルク、コロイド状二酸化珪素(silica dioxide)、ステアリン酸マグネシウム、ステアリン酸カルシウム、二酸化チタニウム、珪酸マグネシウムなどのような、オーバーコーティング配合物(overcoating formula)に取り込まれ得る。
【0039】
遅延放出及び即効ユニットはあらかじめ決定した割合、好ましくは約70:30〜約80:20、より好ましくは75:25(IR/DR)で剤形(この例において、異種のペレットがカプセルに詰められる)に配合され、これは、1日1回のみの投与で望ましい定常状態の血清レベルを達成する。
【0040】
該組成物は、好ましくはビードレット形態で、硬ゼラチンカプセル中に組み込まれ得、さらなる賦形剤を備えてもまたは単独でもよい。カプセル製剤に添加される典型的な賦形剤は、制限されることはないが:微結晶セルロース、ソイポリサッカライド、リン酸カルシウム二水和物、硫酸カルシウム、ラクトース、スクロース、ソルビトールのようなフィラー(fillers)、または任意の他の不活化フィラーが挙げられる。さらに、フュームド(fumed)二酸化珪素、シリカゲル、ステアリン酸マグネシウム、ステアリン酸カルシウムのような流動補助剤または粉末に流動を与える任意の別の物質であり得る。仮に必要である場合には、ポリエチレングリコール、ロイシン、グリセリルベヘネート(glyceryl behenate)、ステアリン酸マグネシウムまたはステアリン酸カルシウムを使用することにより、滑沢剤をさらに添加し得る。
【0041】
該組成物はまた、特に錠剤マトリックスへの組み込みにより、錠剤に組み込まれ得、これは摂取の後、粒子をすばやく分散させる。これらの粒子をこのような錠剤に組み込むために、粒子を受け入れ得る錠剤にフィラー/結合剤を添加しなければならないが、錠剤形成プロセス中、これらを破壊させてはいけない。この目的に適する物質は、制限されないが、微結晶セルロース(AVICEL(登録商標))、ソイポリサッカライド(EMCOSOY(登録商標))、α化スターチ(STARCH(登録商標)1500、NATIONAL(登録商標)1551)、及びポリエチレングリコール(CARBOWAX(登録商標))を含む。物質は5−75%(W/W)の範囲で存在すべきであり、好ましい範囲は25−50%(W/W)である。
【0042】
さらに、いったん錠剤を摂取したときに、ビーズ(beads)を分散するために崩壊剤を添加する。適した崩壊剤は、制限されないが:架橋ソディウムカルボキシメチルセルロース(AC−DI−SOL(登録商標))、ソディウムスターチグリコレート(EXPLOTAB(登録商標)、PRIMOJEL(登録商標))、及び架橋ポリビニルポリピロリドン(Plasone−XL)を含む。これらの物質は3−15%(W/W)の割合で存在すべきであり、好ましい範囲は5−10%(W/W)である。
【0043】
滑沢剤もまた、適当な錠剤化を行うために添加され、そして、これらは制限されないが:ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、ポリエチレングリコール、ロイシン、グリセリルベヘネート、及び水素添加植物性油を含む。これらの滑沢剤は0.1−10%(W/W)の量で存在すべきであり、好ましい範囲は0.3−3.0%(W/W)である。
【0044】
錠剤は例えば次のように形成される。粒子をAVICEL(登録商標)、崩壊剤及び滑沢剤と共にブレンダーに導入し、設定した数の時間(分)で混合し、均一ブレンドを提供し、次いで、これは錠剤が圧縮される錠剤プレスのホッパーに入れられる。使用される圧縮力は錠剤を形成するために十分である;しかし、ビーズまたはコーティングを砕くには十分ではない。
【0045】
本発明の複合投薬形態は、1日1回の経口投与で約24時間にわたり定常状態でレシピエント中で望ましい薬物レベルを達成するための薬学的に活性なドキシサイクリン、または他のテトラサイクリンの用量を送達することが理解される。
【0046】
本発明はまた、哺乳動物をドキシサイクリンまたは他のテトラサイクリンで治療するための方法を提供する。該方法は、本発明に従うドキシサイクリン、または他のテトラサイクリン組成物を、実質的に抗生物質活性を伴うことのない、ドキシサイクリンまたは他のテトラサイクリンの抗−コラゲナーゼまたは抗−炎症活性を必要とする哺乳動物、好ましくはヒトに投与することを含む。全身投与が好ましく、そして経口投与が最も好ましい。
【0047】
本発明の組成物を使用すると、炎症状態と同様に、歯周病、皮膚病などの増大したコラゲナーゼ産生を伴う疾患を治療するために、最小約0.1μg/ml、好ましくは約0.3μg/ml及び最大約1.0μg/ml、より好ましくは約0.8μg/mlのドキシサイクリンまたは他のテトラサイクリンの定常状態の血中レベル(blood level)が達成さ
れ得る。実際、毎日のマルチ投薬において与えられるテトラサイクリンの抗微生物血中レベル以下で治療可能な任意の疾患状態もまた、本発明の対応する1日1回製剤を使用して治療し得る。
【0048】
本発明は、ここで、次の実施例により説明するが、これは制限として理解されない。
【実施例】
【0049】
実施例
実施例1:ドキシサイクリンモノハイドレートを含有する層状IRペレットの調製
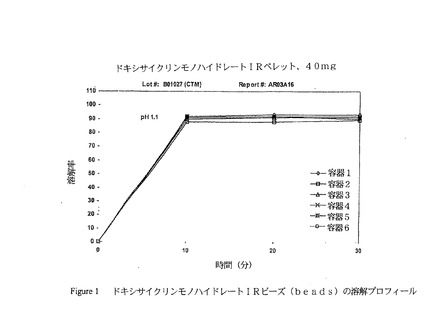
ドキシサイクリンモノハイドレートのディスパージョンは次のように調製した:5.725キログラムの脱イオン水を0.113キログラムのヒドロキシプロピルメチルセルロース及び1.5キログラムのドキシサイクリンモノハイドレートに添加し、続いて撹拌パドルを使用して30分間ゆっくり混合した。薬物ディスパージョンはGPCG−15流動床プロセッサーの9”Wurster Columnにおいてシュガーシード(sugar seeds)(30/35メッシュ)上にスプレーした。全ディスパージョンが適用されるまで、ペレットはカラムにおいて5分間乾燥させた。薬剤を詰めたペレットは、Wurster Columnから出し、そして、20メッシュのふるいを通過させた。保護コート(例えば、OPADRY(登録商標)ベージュ)もまた、色彩及び物理的な保護を提供するためにIRビーズ上に適用し得る。図1は、ドキシサイクリンモノハイドレート即効型ビーズに関する典型的な溶解プロフィールを示す。
【0050】
実施例2:ドキシサイクリンモノハイドレートを含有する腸溶性被覆ペレットの調製
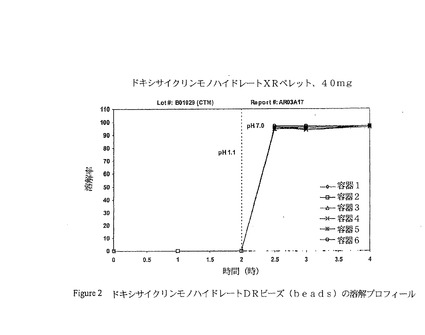
0.127キログラムのクエン酸トリエチルを3.538キログラムのEUDRAGIT(登録商標)L30D55(固形含有量:1.061キログラム)に添加し、そして少なくとも30分間撹拌することにより、EUDRAGIT(登録商標)L30D55コーティングディスパージョンを調製した。タルク0.315キログラムを2.939キログラムの脱イオン水に分散させた。可塑化EUDRAGIT(登録商標)L30D55をタルクディスパージョンと合わせ、60メッシュのふるいを通してふるいにかけた。生じた混合ディスパージョンを、GPCG−15流動床プロセッサーの9”Wurster Columnにおいて実施例1に従い調製した薬剤を詰めたペレット(3.5キログラム)にスプレーした。保護コート(例えば、OPADRY(登録商標)ベージュ)を、色彩及び物理的な保護を提供するためにDRビーズ上に適用し得る。図2はドキシサイクリンモノハイドレートの遅延放出ビーズに関する典型的な溶解プロフィールを示す。
【0051】
実施例3:薬剤を詰めたペレット及び腸溶性被覆ペレットのカプセル化
薬剤を詰めたペレット及び腸溶性被覆ペレットのそれぞれを適当なサイズのカプセルシェルに充填することにより、カプセルを調製し得る。薬剤を詰めたペレットと腸溶性被覆ペレットの割合は、100:0〜70:30であり得る。例えば、75:25の割合において、薬剤を詰めたペレットの充填重量は、薬剤を詰めたペレットの実際の有効性に基づいて、30mgのドキシサイクリンを送達するように算出し得;腸溶性被覆ペレットの充
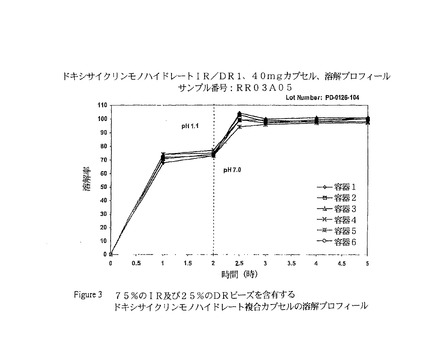
填重量は、腸溶性被覆ペレットの実際の有効性に基づいて、10mgのドキシサイクリンを送達するように算出し得る。Romoco CD5またはMG−2ペレット充填機は、ペレットを望ましいカプセルシェルに正確に充填するために使用し得る。図3は、即効ビーズ75%及び遅延放出ビーズ25%の複合カプセルに関する典型的な溶解プロフィールを示す。
【0052】
実施例4:ドキシサイクリンモノハイドレートを含有する遅延錠剤の調製
ドキシサイクリンモノハイドレート0.5625キログラムを、V型ブレンダー中で15分間、3.15キログラムの微結晶セルロースと混合し、粉末ブレンドをステアリン酸マグネシウム(0.0375キログラム)で、さらに5分間、滑らかにした。造粒流体としてイソプロピルアルコールを使用して、ドキシサイクリンモノハイドレート(0.2キログラム)をEUDRAGIT(登録商標)L100粉末(1.280キログラム)及び微結晶セルロース粉末(0.5キログラム)と共に粒状にした。湿った粒を流動床ドライヤー内で乾燥させ、そして、乾燥した粒を、V型ブレンダー中で5分間、ステアリン酸マグネシウム(0.020キログラム)とブレンドした。ドキシサイクリン粉末ブレンド及び粒を遅延錠剤プレス(belayed tablet press)に置き、粉末ブレンド及び粒状層のそれぞれが200mg及び100mgである目的の重量を備えた遅延錠剤(belayed tablet)に圧縮した。
【0053】
実施例5:ドキシサイクリンモノハイドレートを含有する即効型錠剤の調製
ドキシサイクリンモノハイドレート1.0キログラムを、V型ブレンダー中で5分間、2.225キログラムの微結晶セルロース(AVICEL(登録商標)PH102)と混合した。そして、残りの微結晶セルロース(1.75キログラムのAVICEL(登録商標)PH202)を、V型ブレンダー中の粉末ブレンドに添加し、さらに30分間混合した。そして、該粉末ブレンドをステアリン酸マグネシウム(0.025キログラム)で、5分間、滑らかにした。滑らかになった粉末混合物は200mgの目的の重量を備えた錠剤に圧縮した。該錠剤はさらに重合保護層で被覆し得る。
【0054】
実施例6
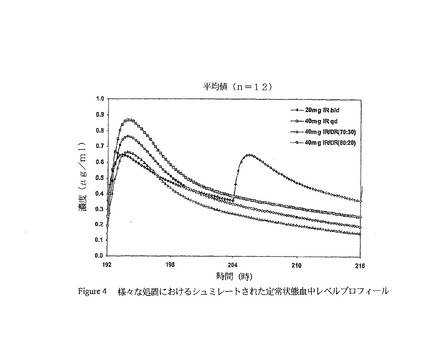
様々な治療(例えば、40mgの1日1回のIR処方、70:30及び80:20の割合の40mgの1日1回のIR及びDR組み合わせ、及び、1日2回の20mgドキシサイクリン治療)に関する定常状態のシミュレートされた血中レベル−時間プロフィールをin silicoモデリングにより決定し、そして図4に示す。独特の投与量(すなわち、<50mg、好ましくは40mg)及び組成物(IRビーズまたはIR/DR組み合わせ)を使用すると、歯周病及び皮膚病のような状態を処置するために、最小約0.1μg/ml、好ましくは約0.3μg/ml及び最大約1.0μg/ml、さらに好ましくは約0.8μg/mlのドキシサイクリンの定常状態の血中レベルが達成され得る。
【0055】
実施例7
薬剤を詰めたIRペレット及び腸溶性被覆DRペレットを75:25の割合で含有するサイズ0カプセルは、次のように調製する。IR及びDRペレットは、実施例1及び2に記載するように調製した。ペレットを製造するために使用されるドキシサイクリンのアッセイ値から、41.26mgのカプセルの有効性は40mgのドキシサイクリンの実際の濃度と一致しているということを決定した。IRペレットの有効性は、ペレットのグラムあたり194mgドキシサイクリンであり(mg/g)、DRペレットに関しては133mg/gであった。従って、各カプセルにおいて、IRビーズの充填質量は159.5mgであり、そしてDRビーズは77.6mgである(40mgカプセルのIR:DRで75:25に一致している)ことを算出した。
【0056】
実施例8
薬物動力学的な(PK)研究を、1日1回経口投与される徐放性ドキシサイクリンカプセルを摂取する第1群(実施例7を参照)(75/25 IR/DR 40mg)と1日2回、12時間間隔で、経口投与されるPeriostat(登録商標)錠剤(20mg)を摂取する第2群を比較するために、ヒト被験者で、実施した。
【0057】
薬物動力学的な採血は、第1群及び第2群についてはノミナルスタディー(Nominal Study)1日目に、そして、第1群については7日目に、次のように採集した;モーニング投薬後0(投与前)、0.5、1、1.5、2、2.5、3、4、6、8、12(適用される場合、ポスト−モーニング投薬(post-morning dose)前)、12.5、
13、13.5、14、14.5、15、16、18、20及び24時間。
【0058】
この実験のデータを次の表1に示す。
【0059】
【表1】

【0060】
1日目の75/25 IR/DR 40mgカプセルの平均Cmaxは、Periostat(登録商標)錠剤のものと匹敵し、そして、潜在的な抗生物質の効果濃度(1000ng/ml)を十分下回った。平均Cmin(24時間の時点で177ng/ml)は、最小の有効血漿濃度(100ng/ml)を十分に上回った。75/25 IR/DR
40mgカプセル及びPeriostat(登録商標)20mg錠剤の両方からのそれぞれの薬物動力学的データは、75/25 IR/DR 40mgカプセルが、
それぞれの投薬終了時に、高ピークの血漿濃度(>1000ng/ml)と低血漿濃度(<100ng/ml)の頻度がより少ない点で、より一貫したin vivo性能を提供することが分かる。
【0061】
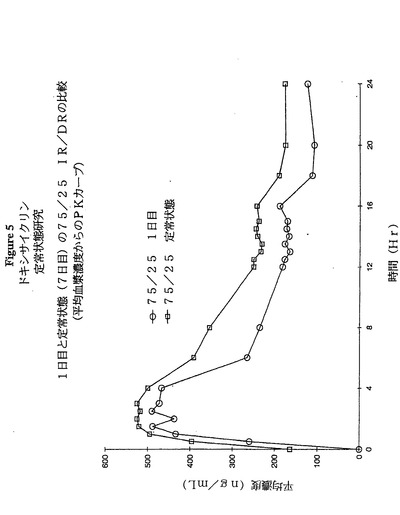
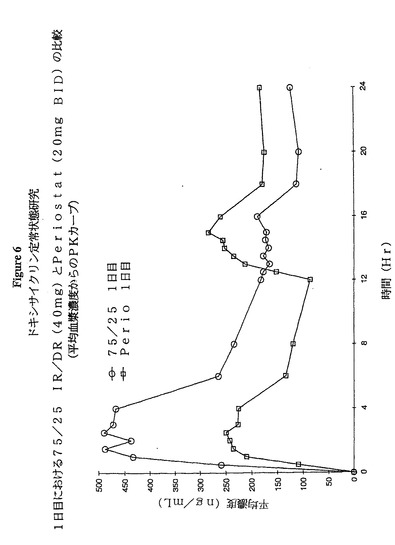
図5及び6は、本実験から得られた結果の二つの側面である。図5は、1日目及びまた7日目(定常状態)について24時間にわたる75:25 IR:DR 40mgドキシサイクリン製剤のPKプロフィールを比較する。図6は、75:25 40mgの1日1回剤形とPeriostat(登録商標)20mg(1日2回)剤形のPKプロフィールを比較する。
【図面の簡単な説明】
【0062】
【図1】図1は、本発明の範囲内のドキシサイクリンモノハイドレート即効型ビーズの溶解プロフィールを示し、これは、in vivoでのヒト血漿データからin vivoでの放出プロフィールをデコンボリュート(deconvolute)するためのコンパートメント吸収および通過モデルに基づくコンピューターアルゴリズムを利用することにより決定した。in silicoによるモデルは、即効剤形からのヒト血漿データを使用して最初に確認し、次いで、試験した。
【図2】図2は、ドキシサイクリンモノハイドレート遅延放出型ビーズのin silico溶解プロフィールを示す。
【図3】図3は、75%の即効ビーズ及び25%の遅延放出ビーズを備えた複合カプセルのin silico溶解プロフィールを示す。
【図4】図4は、様々な処置における定常状態での時間に対する予測血中レベルプロフィールを示す(すなわち、40mgの1日1回のIR処方、40mgの1日1回の70:30及び80:20の割合でのIR及びDRの組み合わせ、ならびに1日2回の20mgのドキシサイクリン処置)。
【図5】図5は、ヒトにおいて1日目及び7日目(定常状態)の75:25 IR:DR(40mg.)製剤の薬物動力学的プロフィールを示す。
【図6】図6は、75:25 IR:DR(40mg.)製剤とPeriostat(登録商標)20mgの1日2回剤形の薬物動力学的曲線を比較する。
【技術分野】
【0001】
発明の分野
本発明はテトラサイクリンの1日1回組成物(once−daily compositions)に関し、これは急性または慢性疾患、例えば炎症性コンポーネントを備えるものの治療に使用し得る。より詳細には、本発明は、炎症のようなものに関わるコラーゲン破壊酵素または分子が要因である疾患または状態の治療のためのドキシサイクリンの薬学的組成物に向けられ、そして、これは1日1回製剤である。該組成物は、特に歯周病、酒さ(rosacea)、ドライアイ、アクネ(acne)及び慢性関節リウマチのような一般的な疾患状態の処置に有用である。
【背景技術】
【0002】
発明の背景
通常、ドキシサイクリン及び関連するテトラサイクリンは、様々な細菌感染症を治療するための広域スペクトル抗生物質として使用される。テトラサイクリンはアミノアシル−tRNAのリボゾームへの結合を阻止することにより、グラム陽性及びグラム陰性細菌のタンパク合成を阻害する。これらの作用は殺菌(殺菌性)よりむしろ静菌(細菌の増殖阻止)である。抗生物質効果を達成するために一般的に使用されるドキシサイクリンの用量は、100mg及び50mgである。
【0003】
ドキシサイクリンはまた、他のテトラサイクリンと同様に、その抗生物質特性に加えて他の治療用途も有する。例えば、ドキシサイクリンは、コラゲナーゼ、ゼラチナーゼ及びエラスターゼのようなコラーゲン破壊酵素の作用を阻害することが知られている。そのコラゲナーゼ阻害活性は歯周病を治療するために使用されている。他の例では、ドキシサイクリンは、細菌P.acnesにより産生されるリパーゼを阻害し、そして、炎症に関係する遊離脂肪酸の利用能を低下し得る。ドキシサイクリンはまた、サイトカインレベルを低下することにより炎症を減少し得、そして、濾胞壁の完全性を保持する。このように、ドキシサイクリンはまた、アクネのような皮膚病を治療する潜在能力も有する。
【0004】
効果に関するメカニズムは完全には明らかではないが、研究者はテトラサイクリンの抗微生物用量以下で、様々な病気(ailments)の治療に有用であることを発見している。例えば、米国特許第6,455,583号では、患者に非−抗菌量のテトラサイクリンを経
口投与することにより、瞼板腺炎を治療することを開示している。米国特許第6,100,248
号は、これらの抗菌活性を弱めるまたは消すために化学的に修飾されたテトラサイクリンを投与することにより、癌増殖を抑制する方法を教示している。抗菌治療に使用される通常の量より一般的に少量のテトラサイクリン量を投与することにより、コラーゲン分解酵素を減少させる方法が、米国特許第4,666,897号に開示されている。この段落で引用した
特許は、これより、本明細書において参考文献として組み込まれる。
【0005】
市場において、歯周病の治療において部位特異的に使用するための2種のはめ込み型製品がある。PerioChip(登録商標)は小さく、オレンジ−褐色チップであり、これは歯周ポケットに挿入される。それぞれのPerioChip(登録商標)は、生分解性で、再吸収可能なマトリックスに、2.5mgのグルコン酸クロルヘキシジンを含有する。PerioChip(登録商標)治療は、5mmまたはより深く残存するポケットに3ヶ月毎に1回投与することが推奨されている。第2の製品、Atridox(登録商標)は、注射可能で、再吸収可能なゲルであり、約一週間、42.5mgのドキシサイクリンの歯肉下への制御放出を提供する。さらに、現在、Periostat(登録商標)と呼ばれる新しい経口薬剤を利用することが可能であり、これは、成人の歯周病患者に使用
されるコラゲナーゼ阻害剤として、20mgのドキシサイクリンを全身に送達する。多くの人々は、インプラントよりもピルを好む。しかし、Periostat(登録商標)は1日2回の投与が必要であり、そして、患者の薬剤服用順守に対する懸念がある。このように、ドキシサイクリンの1日1回の製剤を開発することは非常に利益をもたらす。
【0006】
ドキシサイクリンは感染症を治療する上で一般的に効果的であるが、ドキシサイクリンの使用は望ましくない副作用も導き得る。例えば、抗生物質のドキシサイクリンの長期間投与は、健全な生物フローラ(腸内細菌叢のような)を減少または排除し得、そして、抗生物質耐性生物の産生または酵母及びカビの過剰増殖を導き得る。抗生物質特性の好ましくない副作用のために、テトラサイクリン(特にドキシサイクリン)の抗コラーゲン破壊酵素または他の有益な効果は獲得するが、抗菌効果は回避するようなドキシサイクリン送達のための独特の用量及び改良された製剤が必要である。
【発明の概要】
【課題を解決するための手段】
【0007】
本発明の要約
本発明は、その最も広い意味において、定常状態で、疾患または状態の処置において有益な効果を有することには十分有効であるが、抗菌効果を発揮するほど高くない有効成分のin vivoレベルの徐放性プロフィールを提供するために設計されたテトラサイクリンの薬学的組成物に向けられている。このような薬学的組成物は、1日1回摂取され得る剤形で処方される。
【0008】
本発明の一つの目的は、ドキシサイクリンの薬学的組成物を提供することであり、これは、長期間の抗菌活性の望ましくない影響を伴うことなく、コラゲナーゼの過剰産生(歯周病のような)またはドキシサイクリンで調節され得る特定の疾患状態(炎症を伴う状態のような)に関係する他の生化学剤が原因で生じる疾患または状態の治療または予防に必要とされる定常状態の血中レベルをもたらすにもかかわらず、1日1回で与えられ得る。
【0009】
本発明の一つの目的は、最小約0.1μg/ml及び最大約1.0μg/mlのドキシサイクリンの定常状態の血中レベルを与えるドキシサイクリンを含有する1日1回の薬学的組成物を提供することである。
【0010】
本発明の一つの側面は、ドキシサイクリンベースで、50mg未満であるが25mgより多く、好ましくは約40mgを含有するドキシサイクリンの即効型(immediate release)製剤である。
【0011】
他の側面において、本発明は、薬剤の即効型(IR)コンポーネント及び薬剤の遅延放出型(delayed release(DR))コンポーネントを含むドキシサイクリンの薬学的組成物に向けられ、これらは1日1回投与のための一つの単位用量に合わせられる。該コンポーネントは様々な割合で存在し得るが、好ましくはIR:DRが約70:30〜約80:20、最も好ましくは75:25の割合であるが、ドキシサイクリンの全投与量は約50mg未満、そして、好ましくは約40mgである。該割合はIRとDRの用量の内訳を示しており、例えば、80:20は、40mgの80%がIR部分からであり、40mgの20%がDR部分からであることを意味する。
【0012】
さらに、本発明の他の目的は、ドキシサイクリンの1日1回用量を投与することにより、歯周病、慢性関節リウマチ、副甲状腺機能亢進症、糖尿病及びアクネのような組織の病理学的破壊を生じるコラゲナーゼが過剰量産生される疾患または状態を治療する方法を提供することである。例えば、Golubの米国特許第4,666,897号を参照。
【0013】
本発明の他の目的は、本発明に従うドキシサイクリンの1日1回用量を投与することにより、酒さ、ヒトの皮膚科学的状態の全身治療方法を提供することである。
【0014】
本発明の他の目的は、本発明の1日1回組成物を調製するためのプロセスを提供することである。
【0015】
本発明の詳細な説明
次の記載は主としてドキシサイクリンに向けられるが、本発明は他のテトラサイクリン類、特にドキシサイクリンと類似のin vivo吸収プロフィールを有する他のテトラサイクリン類、より具体的にはドキシサイクリンと同様の消化管内での吸収域を有するテトラサイクリン類に適用できるということが予想される。様々な種類のテトラサイクリン類及び様々な疾患状態に対する有益な効果は、例えばWO02/083106及び米国特許第6,638,922号に開示され、これらのそれぞれは、これら全体を本明細書において参考として援用する。
【0016】
本発明は、例として、哺乳動物、好ましくはヒトが、1日に1回のみの用量を行う必要のある製剤ではあるが、薬剤の特定の定常状態の血中レベルを提供するために設計されたドキシサイクリンの経口投与組成物を提供することにより成し遂げられ得る。本発明の組成物は、同じ効果を達成する組成物の1日に複数回の投与(1日2回の投与のような)に代わって、有用であることが意図される。ドキシサイクリン(または比較可能な生理学的及び吸収特性の他のテトラサイクリン類)の好ましい血中レベルは、定常状態で約0.1〜約1.0μg/mlである。好ましくは、血中レベルは、治療の全体を通して、日々の投与を伴う、好ましい血中レベル内にある。さらに好ましくは、血中レベルは約0.3μg/ml〜約0.8μg/mlである。
【0017】
上記の血清レベルは、望ましくない抗生物質活性を伴うことなく、ドキシサイクリンの望ましい抗−コラゲナーゼ及び抗−炎症活性を可能にする。これらのレベルは、ドキシサイクリンベースで50mgより低いが25mgより多い、好ましくは約40mgを含有する即効型製剤の単一の1日1回の投与で成し遂げ得るという驚くべき発見であった。
【0018】
「約」は、活性のある薬学的成分の量に関して、米国薬局方(USP−NF 21)、2003 Annual Editionにみられる、または、www.usp.orgで利用できる薬学的に許容される制限内であることを意味する。血中レベルの側面からみると、「約」はFDAの許容するガイドライン内であることを意味する。
【0019】
「即効型」製剤は、促進、遅延、または、徐放性効果を伴うことなく投与時に、全ての活性成分を実質的に放出することを意図する剤形を意味する。
ドキシサイクリンのこのような組成物は、液状懸濁液または溶液の形態で、あるいは、錠剤、ペレット(本明細書ではビード(bead)とビードレット(beadlet)を区別なく使用する)、粒子(particle)、カプセルまたはゲルのような固形物の形態であり得る。本発明において好ましくは、錠剤またはカプセル中のビードレットである。
【0020】
薬学的な活性成分に関して、本発明の要求される血清レベルに従うならば、テトラサイクリン化合物のいかなる形態でも使用し得る。例えばドキシサイクリンは、2つの化学的な形態:モノハイドレート形態及びヒクレート(hyclate)形態、にある薬学的製剤で一般
的に使用される。モノハイドレートは1分子の水で水和された塩基分子であり、カプセル及び、幾つかのマーケットにおいては、粉末の経口懸濁液(水を加えて再構成される)の製剤で使用される。ヒクレートは、水及びエタノールで溶媒和された塩酸塩であり、そしてカプセルまたは錠剤の製剤で概して使用される。本発明の組成物にあるドキシサイクリ
ンの量は、ドキシサイクリンベースを意味する。また、本発明の組成物において、一つ以上の活性成分があり得る。すなわち、ドキシサイクリンは、剤形において、他の治療に役立つまたは栄養性の物質と混合され得る。
【0021】
即効型剤形
このような即効型剤形を処方するための当業者に公知の多くの方法がある。例えば、即効型錠剤は、ドキシサイクリンと、微結晶性セルロース、例えばAVICEL(登録商標)(FMC Corp.)またはEMCOCEL(登録商標)(Mendell Inc.);リン酸ジカルシウム、例えばEMCOMPRESS(登録商標)(Mendell
Inc.);硫酸カルシウム、例えばCOMPACTROL(登録商標)(Mendell Inc.);及びスターチ、例えばSTARCH 1500のようなバルキング剤(bulking agent)を混合することにより調製され得る。さらに、微結晶性セルロース、スターチ、クロスポビドン、例えばPOLYPLASDONE XL(登録商標)(International Specialty Products);ソディウムスターチグリコレート、例えばEXPLOTAB(登録商標)(Mendell Inc.);及びクロスカルメロースナトリウム(croscarmellose sodium)、例えばAC−DI−SOL(登録商標)(FMC Corp.)のような崩壊剤を添加し得る。本明細書で使用する抗粘着剤(Antiadherant)及びグリダント(glidant)はタルク、コーンスターチ、二酸化珪素、ラウリル硫酸ナトリウム、コロイド状二酸化珪素(silica dioxide)及びメタリックステアラート(metallic stearate)を挙げることができる。
【0022】
滑沢剤(lubricants)は、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸ナトリウム、ステアリン酸、フマル酸ステアリルナトリウム、ステロテックス(sterotex)、タルク、ワックスなどが使用され得る。結合剤(binding agents)は、ポリビニルピロリドン、スターチ、メチルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースなどが使用され得る。
【0023】
本発明は、当業者に公知の方法(湿式造粒法及び直接圧縮法を含む)を使用して調製した錠剤に好ましくは処方される。経口錠剤は当業者に公知のいずれかの適したプロセスを使用して調製される。例えば、Remington’s Pharmaceutical Sciences, 18th Edition, A. Gennaro, Ed., Mack Pub. Co. (Easton, PA 1990) Chapters 88-91を参照し、この全
体はこれより参考として援用される。概して、活性成分、ドキシサイクリンは、薬学的に許容される賦形剤(例えば、上に列挙した結合剤、滑沢剤など)と混合し、そして、錠剤に圧縮する。好ましくは、剤形は湿式造粒技術または直接圧縮法により調製され、そして、均一な顆粒を形成する。あるいは、活性成分は、顆粒が調製された後に顆粒と混合し得る。そして、湿った顆粒マスを乾燥させ、そして適したふるいわけ装置(screening device)を使用して大きさをそろえ、そして粉末を提供し、そして、これは、望ましいように、カプセルに充填あるいはマトリックス錠剤またはキャプレッツ(caplets)に圧縮し得る。
【0024】
好ましい実施形態において、錠剤は直接圧縮法を使用して調製される。直接圧縮法は、特に比較的簡単な製造に関して、湿式造粒法以上に数多くの潜在的な利点を提供する。直接圧縮法において、少なくとも一つの薬学的活性剤及び賦形剤または他の成分が、ステンレススチールふるい(40メッシュスチールふるいのような)を通してふるいにかけられる。そして、ふるいにかけた材料を適切な混合機に移し、そして、3分間増圧バーとともに10分間混合する。そして、混合物は適した道具を使用して回転プレス上で錠剤に圧縮する。
【0025】
上述したように、即効型組成物の他の好ましい剤形は、即効型ビードレットまたはペレ
ットを含有するカプセルである。このようなペレットを製造する方法は、以下の段落(例えば、IRペレット)に記載する。該ペレットを従来技術によりカプセルに、例えばゼラチンカプセルに、充填する。
【0026】
組み合わせIR/DR剤形
本発明の他の実施形態は、1単位用量において、ドキシサイクリンの実質的な即効投与を有する組成物に、あらかじめ決められた時間少なくとも一つのさらなる投与が続く。組成物の第1の即効型用量は散剤、顆粒剤、ビードレットまたは錠剤の形態であり得、第2の遅延放出部分は被覆された顆粒、被覆されたビードレット、被覆された錠剤、または被覆されていないマトリックス錠剤であり得る。即効部分またはコンポーネントと遅延放出部分またはコンポーネントの割合は、in vitroでの薬剤放出プロフィール及びin vivoでの血中濃度プロフィールを調整するために利用され得る。このような薬剤放出プロフィールを提供することにより、組成物はその日の2回目の投与の必要性を排除する。さらに、ドキシサイクリンの全投与量は、その抗生物質特性由来の望ましくない副作用を回避するために、50mg未満であるが、抗−コラゲナーゼ及び/または抗−炎症効果を達成するために25mgより多い。
【0027】
幾つかの剤形バリエーションは、これらの寄与を備えた製品を達成するために使用され得る。例えば、即効粉末混合物は、遅延放出錠剤または遅延放出ペレットと共にカプセル化され得る。さらなる例は、別々に調製され、そして、適切な大きさのカプセルシェルにカプセル化される即効型錠剤及び遅延放出型錠剤である。または、例えば、遅延放出型錠剤がコアとして使用され得、そして即効部分がプレスコーターを使用して外層として圧縮され得、または、薬剤積層技術を使用してオーバーコートされ得る(両方の技術は、H.A.Liberman, L.Lachman及びJ.B.Schwartzにより編集された, Maecel Dekker, Inc. New York and Basel による1990年のPharmaceutical Dosage Forms:Tablets, Second Edition, Volume 1,のGunsel and Dusel, Chapter 5,「Compression-coated and layer tablets」に見られ得る)。
【0028】
複合粒子カプセル
好ましい実施形態として、ドキシサイクリンのIR/DR組成物は、ビードレットを含有するカプセルの形態である。現在、単一形態の複合単位剤形において、2つの異なるタイプの単位を有することが好ましい。第1ユニットは即効剤形であり、好ましくはペレット形態にある。このコンポーネントはまた、望ましいまたは必要ならば、粉末であり得る。いずれの場合でも、剤形はラウリル硫酸ナトリウム、ソディウムモノグリセラート、ソルビタンモノオレエート、ポリオキシエチレンソルビタンモノオレエート、グリセリルモノステアレート、グリセリルモノオレエート、グリセリルモノブチレート、界面活性ポリマーのプルロニック(Pluronic)系列のいずれか一つ、あるいは、界面活性特性を有する任意の他の適した物質または上記の任意の組み合わせのような界面活性剤を有し得る。好ましくは、界面活性剤はソディウムモノグリセラートとラウリル硫酸ナトリウムの組み合わせである。このコンポーネントのこれらの物質の濃度は約0.05〜約10.0%(W/W)の範囲であり得る。
【0029】
薬剤含有ペレットを製造する上で使用され得る他の賦形剤物質は、薬剤学において一般的に使用されるそれらのいずれかであり、そして、活性薬剤とペレットの物理化学的特性の適合性を基準にして選択すべきである。これらは、例えば、メチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、ポリビニルピロリドン/ビニルアセテートコポリマーなどのようなセルロース誘導体等の結合剤(binder);コーンスターチ、アルファ化デンプン、架橋カルボキシメチルセルロース(AC−DI−SOL(登録商標))、ソディウムスターチグリコレート(EXPLOTAB(登録商標))、架橋ポリビニルピロリ
ドン(PLASDONE(登録商標)XL)のような崩壊剤、及び錠剤調製で使用される任意の崩壊剤(これらは一般的に、本発明の一つのような即効用量において使用される);ラクトース、炭酸カルシウム、リン酸カルシウム、硫酸カルシウム、微結晶性セルロース、デキストラン、スターチ、スクロース、キシリトール、ラクチトール、マンニトール、ソルビトール、塩化ナトリウム、ポリエチレングリコールなどのような充填剤;ラウリル硫酸ナトリウム、ソルビタンモノオレエート、ポリオキシエチレンソルビタンモノオレエート、胆汁酸塩、グリセリルモノステアレート、PLURONIC(登録商標)系列(BASF)などのような界面活性剤;クエン酸、コハク酸、フマル酸、リンゴ酸、酒石酸、マレイン酸、グルタル酸 重炭酸ナトリウム及び炭酸ナトリウムなどのような可溶化剤(solubilizers);抗酸化剤、緩衝液、酸などのような安定剤が挙げられ、これらもまた利用し得る。
【0030】
ペレットは、例えば、単純な顆粒化およびそれに続くふるい分け;押し出し及びマルメライゼーション(marumerization);回転造粒(rotogranulation);または、妥当な大きさ及びローバスト性(robustness)のペレットを生じる任意の凝集プロセスにより作成され得る。押し出し及びマルメライゼーションに関して、薬剤及び他の添加剤は、結合剤溶液を添加することにより粒状化される。湿ったマスを特定のサイズのふるいを備えた押し出し器に通し、そして、押し出されたものはマルメライザーで球状にする。得られたペレットは乾燥させ、そしてさらなる適用のためにふるいにかける。高剪断造粒機(high−shear granulation)を使用し得、ここで、薬剤及び他の添加剤は乾燥混合され、そして、高剪断造粒機/混合機に結合剤溶液を添加することにより混合物を湿らせる。混合及びミリングの複合作用により湿らせた後に、粒状物を混練する。結果として生じた顆粒またはペレットは乾燥させ、そしてさらなる適用のためにふるいにかける。
【0031】
以前に記載したように、混合及び混合しない場合(consideration)において好ましい形態がペレットであっても、この即効型コンポーネントを粉末として含有することもまた可能である。
【0032】
あるいは、組成物の即効ビードレットまたはペレットは溶液または懸濁液積層により調製され得、それによって、薬剤溶液またはディスパージョンが、結合剤を伴っても伴わなくても、流動床プロセッサーまたは他の適した装置において、コアまたは最初シード(調製されたものか市販の製品のいずれか)上に噴霧される。コアまたは最初のシードは、例えば、シュガー球体(sugar sphere)または微結晶性セルロースからなる球体であり得る。このように薬剤は最初のシードの表面に被覆される。薬剤を詰めたペレットは、さらなる適用のために乾燥する。
【0033】
第2ユニットは遅延放出(DR)プロフィールを有するべきであり、そして、GI管のpH変化ならびにドキシサイクリンまたは他のテトラサイクリンの吸収に対するその影響に対応できる必要がある。このペレットは、ペレットの微小環境のpHを低下するために有用である必要に応じた何らかの有機酸と同様に、第1ユニットのペレットに関して記載したように全ての成分を含有し得、そして、溶解を促進する。これらの物質は、制限されないが、クエン酸、乳酸、酒石酸、または他の適した有機酸である。これらの物質は、約0〜約15.0パーセント(W/W)の濃度で存在すべきであり;好ましくは、これらの物質は約5.0〜約10.0パーセント(W/W)の濃度で存在する。これらのペレットを製造するためのプロセスは、第1ユニットのペレットに関して上述したプロセスに一致する。
【0034】
第1ユニットのペレットと異なり、第2ユニットの遅延放出型コンポーネントは、ペレットからの薬剤の放出が遅らせるようなペレットの表面に適用される制御コートを有する
。これは、腸溶性物質の被覆を適用することにより達成される。「腸溶性物質」は、胃の酸性環境で実質的に不溶性であるが、特定のpHの腸液に主に溶解するポリマーである。腸溶性物質は毒性がなく、薬学的に許容されるポリマーであり、そして、例えば、セルロースアセテートフタレート(CAP)、ヒドロキシプロピルメチルセルロースフタレート(HPMCP)、ポリビニルアセテートフタレート(PVAP)、ヒドロキシプロピルメチルセルロースアセテートスクシナート(HPMCAS)、セルロースアセテートトリメリテート、ヒドロキシプロピルメチルセルローススクシナート、セルロースアセテートスクシナート、セルロースアセテートヘキサヒドロフタレート、セルロースプロピオナートフタレート、メチルメタクリル酸及びメチルメタクリレートのコポリマー、メチルアクリレート、メチルメタクリレート及びメタクリル酸のコポリマー、メチルビニルエーテル及び無水マレイン酸のコポリマー(Gantrez ES series)、エチルメチルアクリレート−メチルメタクリレート−クロロトリメチルアンモニウムエチルアクリレートコポリマー、ゼイン、シェラック及びコパールコロホリウム(copal collophorium)のような天然樹脂及び幾つかの市販の入手可能な腸ディスパージョンシステム(例えば、EUDRAGIT(登録商標)L30D55、EUDRAGIT(登録商標)FS30D、EUDRAGIT(登録商標)L100、KOLLICOAT(登録商標)EMM30D、ESTACRYL(登録商標)30D、COATERIC(登録商標)及びAQUATERIC(登録商標))を含有する。上記は、可能な物質のリストであるが、当業者は、それは幅広くなく、そして、遅延放出プロフィールを提供する本発明の目的に合致する他の腸溶性物質があるということを認識する。これらの被覆物質は、ペレット組成物の約1.0%(W/W)〜約50%(W/W)の範囲で表面を被覆することに使用され得る。好ましくは、これらの被覆物質は、約20〜約40パーセント(W/W)の範囲であるべきである。該ペレットは、例えば流動床装置またはパンコーティング(pan coating)において被覆され得る。
【0035】
腸溶性被覆ペレットに関して、pH4.5未満程度の酸性胃環境においてドキシサイクリンの実質的な放出はない。pH感受性層が小腸よりも高いpHに溶解した時;特定の遅延期間の後;またはユニットが胃を通過した後に、ドキシサイクリンは利用可能になる。好ましい遅延時間は2〜6時間の範囲にある。
【0036】
この実施形態のバリエーションに関して、DRペレットは、保護層そして最終的の腸溶性被膜により分離される、ドキシサイクリンの層を含有し、結果として「繰り返し作用(repeat−action)」用量デリバリーとなる。仮に全ての層におけるドキシサイクリン、または他のテトラサイクリンの放出が薬剤の吸収ウィンドウ内にある場合、このような剤形は、本発明の放出プロフィールの血液レベル要求に見合い得る。
【0037】
オーバーコート層はさらに、必要に応じて、本発明のIR/DRペレットに適用し得る。OPADRY(登録商標)、OPADRY II(登録商標)(Colorcon)及びColorconからの対応するカラー及びカラーレスグレードは、ペレットが粘着性になることから保護するために及び産物に色を提供するために使用し得る。保護的な及びカラーコーティングの推奨されているレベルは、1〜6%、好ましくは2−3%(W/W)である。
【0038】
多くの成分は、例えばコーティングプロセス及び製品特性を改良するために、可塑剤:アセチルトリエチルシトレート、トリエチルシトレート、アセチルトリブチルシトレート、セバシン酸ジブチル、トリアセチン、ポリエチレングリコール、プロピレングリコール及び他のもの;滑沢剤:タルク、コロイド状二酸化珪素(silica dioxide)、ステアリン酸マグネシウム、ステアリン酸カルシウム、二酸化チタニウム、珪酸マグネシウムなどのような、オーバーコーティング配合物(overcoating formula)に取り込まれ得る。
【0039】
遅延放出及び即効ユニットはあらかじめ決定した割合、好ましくは約70:30〜約80:20、より好ましくは75:25(IR/DR)で剤形(この例において、異種のペレットがカプセルに詰められる)に配合され、これは、1日1回のみの投与で望ましい定常状態の血清レベルを達成する。
【0040】
該組成物は、好ましくはビードレット形態で、硬ゼラチンカプセル中に組み込まれ得、さらなる賦形剤を備えてもまたは単独でもよい。カプセル製剤に添加される典型的な賦形剤は、制限されることはないが:微結晶セルロース、ソイポリサッカライド、リン酸カルシウム二水和物、硫酸カルシウム、ラクトース、スクロース、ソルビトールのようなフィラー(fillers)、または任意の他の不活化フィラーが挙げられる。さらに、フュームド(fumed)二酸化珪素、シリカゲル、ステアリン酸マグネシウム、ステアリン酸カルシウムのような流動補助剤または粉末に流動を与える任意の別の物質であり得る。仮に必要である場合には、ポリエチレングリコール、ロイシン、グリセリルベヘネート(glyceryl behenate)、ステアリン酸マグネシウムまたはステアリン酸カルシウムを使用することにより、滑沢剤をさらに添加し得る。
【0041】
該組成物はまた、特に錠剤マトリックスへの組み込みにより、錠剤に組み込まれ得、これは摂取の後、粒子をすばやく分散させる。これらの粒子をこのような錠剤に組み込むために、粒子を受け入れ得る錠剤にフィラー/結合剤を添加しなければならないが、錠剤形成プロセス中、これらを破壊させてはいけない。この目的に適する物質は、制限されないが、微結晶セルロース(AVICEL(登録商標))、ソイポリサッカライド(EMCOSOY(登録商標))、α化スターチ(STARCH(登録商標)1500、NATIONAL(登録商標)1551)、及びポリエチレングリコール(CARBOWAX(登録商標))を含む。物質は5−75%(W/W)の範囲で存在すべきであり、好ましい範囲は25−50%(W/W)である。
【0042】
さらに、いったん錠剤を摂取したときに、ビーズ(beads)を分散するために崩壊剤を添加する。適した崩壊剤は、制限されないが:架橋ソディウムカルボキシメチルセルロース(AC−DI−SOL(登録商標))、ソディウムスターチグリコレート(EXPLOTAB(登録商標)、PRIMOJEL(登録商標))、及び架橋ポリビニルポリピロリドン(Plasone−XL)を含む。これらの物質は3−15%(W/W)の割合で存在すべきであり、好ましい範囲は5−10%(W/W)である。
【0043】
滑沢剤もまた、適当な錠剤化を行うために添加され、そして、これらは制限されないが:ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、ポリエチレングリコール、ロイシン、グリセリルベヘネート、及び水素添加植物性油を含む。これらの滑沢剤は0.1−10%(W/W)の量で存在すべきであり、好ましい範囲は0.3−3.0%(W/W)である。
【0044】
錠剤は例えば次のように形成される。粒子をAVICEL(登録商標)、崩壊剤及び滑沢剤と共にブレンダーに導入し、設定した数の時間(分)で混合し、均一ブレンドを提供し、次いで、これは錠剤が圧縮される錠剤プレスのホッパーに入れられる。使用される圧縮力は錠剤を形成するために十分である;しかし、ビーズまたはコーティングを砕くには十分ではない。
【0045】
本発明の複合投薬形態は、1日1回の経口投与で約24時間にわたり定常状態でレシピエント中で望ましい薬物レベルを達成するための薬学的に活性なドキシサイクリン、または他のテトラサイクリンの用量を送達することが理解される。
【0046】
本発明はまた、哺乳動物をドキシサイクリンまたは他のテトラサイクリンで治療するための方法を提供する。該方法は、本発明に従うドキシサイクリン、または他のテトラサイクリン組成物を、実質的に抗生物質活性を伴うことのない、ドキシサイクリンまたは他のテトラサイクリンの抗−コラゲナーゼまたは抗−炎症活性を必要とする哺乳動物、好ましくはヒトに投与することを含む。全身投与が好ましく、そして経口投与が最も好ましい。
【0047】
本発明の組成物を使用すると、炎症状態と同様に、歯周病、皮膚病などの増大したコラゲナーゼ産生を伴う疾患を治療するために、最小約0.1μg/ml、好ましくは約0.3μg/ml及び最大約1.0μg/ml、より好ましくは約0.8μg/mlのドキシサイクリンまたは他のテトラサイクリンの定常状態の血中レベル(blood level)が達成さ
れ得る。実際、毎日のマルチ投薬において与えられるテトラサイクリンの抗微生物血中レベル以下で治療可能な任意の疾患状態もまた、本発明の対応する1日1回製剤を使用して治療し得る。
【0048】
本発明は、ここで、次の実施例により説明するが、これは制限として理解されない。
【実施例】
【0049】
実施例
実施例1:ドキシサイクリンモノハイドレートを含有する層状IRペレットの調製
ドキシサイクリンモノハイドレートのディスパージョンは次のように調製した:5.725キログラムの脱イオン水を0.113キログラムのヒドロキシプロピルメチルセルロース及び1.5キログラムのドキシサイクリンモノハイドレートに添加し、続いて撹拌パドルを使用して30分間ゆっくり混合した。薬物ディスパージョンはGPCG−15流動床プロセッサーの9”Wurster Columnにおいてシュガーシード(sugar seeds)(30/35メッシュ)上にスプレーした。全ディスパージョンが適用されるまで、ペレットはカラムにおいて5分間乾燥させた。薬剤を詰めたペレットは、Wurster Columnから出し、そして、20メッシュのふるいを通過させた。保護コート(例えば、OPADRY(登録商標)ベージュ)もまた、色彩及び物理的な保護を提供するためにIRビーズ上に適用し得る。図1は、ドキシサイクリンモノハイドレート即効型ビーズに関する典型的な溶解プロフィールを示す。
【0050】
実施例2:ドキシサイクリンモノハイドレートを含有する腸溶性被覆ペレットの調製
0.127キログラムのクエン酸トリエチルを3.538キログラムのEUDRAGIT(登録商標)L30D55(固形含有量:1.061キログラム)に添加し、そして少なくとも30分間撹拌することにより、EUDRAGIT(登録商標)L30D55コーティングディスパージョンを調製した。タルク0.315キログラムを2.939キログラムの脱イオン水に分散させた。可塑化EUDRAGIT(登録商標)L30D55をタルクディスパージョンと合わせ、60メッシュのふるいを通してふるいにかけた。生じた混合ディスパージョンを、GPCG−15流動床プロセッサーの9”Wurster Columnにおいて実施例1に従い調製した薬剤を詰めたペレット(3.5キログラム)にスプレーした。保護コート(例えば、OPADRY(登録商標)ベージュ)を、色彩及び物理的な保護を提供するためにDRビーズ上に適用し得る。図2はドキシサイクリンモノハイドレートの遅延放出ビーズに関する典型的な溶解プロフィールを示す。
【0051】
実施例3:薬剤を詰めたペレット及び腸溶性被覆ペレットのカプセル化
薬剤を詰めたペレット及び腸溶性被覆ペレットのそれぞれを適当なサイズのカプセルシェルに充填することにより、カプセルを調製し得る。薬剤を詰めたペレットと腸溶性被覆ペレットの割合は、100:0〜70:30であり得る。例えば、75:25の割合において、薬剤を詰めたペレットの充填重量は、薬剤を詰めたペレットの実際の有効性に基づいて、30mgのドキシサイクリンを送達するように算出し得;腸溶性被覆ペレットの充
填重量は、腸溶性被覆ペレットの実際の有効性に基づいて、10mgのドキシサイクリンを送達するように算出し得る。Romoco CD5またはMG−2ペレット充填機は、ペレットを望ましいカプセルシェルに正確に充填するために使用し得る。図3は、即効ビーズ75%及び遅延放出ビーズ25%の複合カプセルに関する典型的な溶解プロフィールを示す。
【0052】
実施例4:ドキシサイクリンモノハイドレートを含有する遅延錠剤の調製
ドキシサイクリンモノハイドレート0.5625キログラムを、V型ブレンダー中で15分間、3.15キログラムの微結晶セルロースと混合し、粉末ブレンドをステアリン酸マグネシウム(0.0375キログラム)で、さらに5分間、滑らかにした。造粒流体としてイソプロピルアルコールを使用して、ドキシサイクリンモノハイドレート(0.2キログラム)をEUDRAGIT(登録商標)L100粉末(1.280キログラム)及び微結晶セルロース粉末(0.5キログラム)と共に粒状にした。湿った粒を流動床ドライヤー内で乾燥させ、そして、乾燥した粒を、V型ブレンダー中で5分間、ステアリン酸マグネシウム(0.020キログラム)とブレンドした。ドキシサイクリン粉末ブレンド及び粒を遅延錠剤プレス(belayed tablet press)に置き、粉末ブレンド及び粒状層のそれぞれが200mg及び100mgである目的の重量を備えた遅延錠剤(belayed tablet)に圧縮した。
【0053】
実施例5:ドキシサイクリンモノハイドレートを含有する即効型錠剤の調製
ドキシサイクリンモノハイドレート1.0キログラムを、V型ブレンダー中で5分間、2.225キログラムの微結晶セルロース(AVICEL(登録商標)PH102)と混合した。そして、残りの微結晶セルロース(1.75キログラムのAVICEL(登録商標)PH202)を、V型ブレンダー中の粉末ブレンドに添加し、さらに30分間混合した。そして、該粉末ブレンドをステアリン酸マグネシウム(0.025キログラム)で、5分間、滑らかにした。滑らかになった粉末混合物は200mgの目的の重量を備えた錠剤に圧縮した。該錠剤はさらに重合保護層で被覆し得る。
【0054】
実施例6
様々な治療(例えば、40mgの1日1回のIR処方、70:30及び80:20の割合の40mgの1日1回のIR及びDR組み合わせ、及び、1日2回の20mgドキシサイクリン治療)に関する定常状態のシミュレートされた血中レベル−時間プロフィールをin silicoモデリングにより決定し、そして図4に示す。独特の投与量(すなわち、<50mg、好ましくは40mg)及び組成物(IRビーズまたはIR/DR組み合わせ)を使用すると、歯周病及び皮膚病のような状態を処置するために、最小約0.1μg/ml、好ましくは約0.3μg/ml及び最大約1.0μg/ml、さらに好ましくは約0.8μg/mlのドキシサイクリンの定常状態の血中レベルが達成され得る。
【0055】
実施例7
薬剤を詰めたIRペレット及び腸溶性被覆DRペレットを75:25の割合で含有するサイズ0カプセルは、次のように調製する。IR及びDRペレットは、実施例1及び2に記載するように調製した。ペレットを製造するために使用されるドキシサイクリンのアッセイ値から、41.26mgのカプセルの有効性は40mgのドキシサイクリンの実際の濃度と一致しているということを決定した。IRペレットの有効性は、ペレットのグラムあたり194mgドキシサイクリンであり(mg/g)、DRペレットに関しては133mg/gであった。従って、各カプセルにおいて、IRビーズの充填質量は159.5mgであり、そしてDRビーズは77.6mgである(40mgカプセルのIR:DRで75:25に一致している)ことを算出した。
【0056】
実施例8
薬物動力学的な(PK)研究を、1日1回経口投与される徐放性ドキシサイクリンカプセルを摂取する第1群(実施例7を参照)(75/25 IR/DR 40mg)と1日2回、12時間間隔で、経口投与されるPeriostat(登録商標)錠剤(20mg)を摂取する第2群を比較するために、ヒト被験者で、実施した。
【0057】
薬物動力学的な採血は、第1群及び第2群についてはノミナルスタディー(Nominal Study)1日目に、そして、第1群については7日目に、次のように採集した;モーニング投薬後0(投与前)、0.5、1、1.5、2、2.5、3、4、6、8、12(適用される場合、ポスト−モーニング投薬(post-morning dose)前)、12.5、
13、13.5、14、14.5、15、16、18、20及び24時間。
【0058】
この実験のデータを次の表1に示す。
【0059】
【表1】
【0060】
1日目の75/25 IR/DR 40mgカプセルの平均Cmaxは、Periostat(登録商標)錠剤のものと匹敵し、そして、潜在的な抗生物質の効果濃度(1000ng/ml)を十分下回った。平均Cmin(24時間の時点で177ng/ml)は、最小の有効血漿濃度(100ng/ml)を十分に上回った。75/25 IR/DR
40mgカプセル及びPeriostat(登録商標)20mg錠剤の両方からのそれぞれの薬物動力学的データは、75/25 IR/DR 40mgカプセルが、
それぞれの投薬終了時に、高ピークの血漿濃度(>1000ng/ml)と低血漿濃度(<100ng/ml)の頻度がより少ない点で、より一貫したin vivo性能を提供することが分かる。
【0061】
図5及び6は、本実験から得られた結果の二つの側面である。図5は、1日目及びまた7日目(定常状態)について24時間にわたる75:25 IR:DR 40mgドキシサイクリン製剤のPKプロフィールを比較する。図6は、75:25 40mgの1日1回剤形とPeriostat(登録商標)20mg(1日2回)剤形のPKプロフィールを比較する。
【図面の簡単な説明】
【0062】
【図1】図1は、本発明の範囲内のドキシサイクリンモノハイドレート即効型ビーズの溶解プロフィールを示し、これは、in vivoでのヒト血漿データからin vivoでの放出プロフィールをデコンボリュート(deconvolute)するためのコンパートメント吸収および通過モデルに基づくコンピューターアルゴリズムを利用することにより決定した。in silicoによるモデルは、即効剤形からのヒト血漿データを使用して最初に確認し、次いで、試験した。
【図2】図2は、ドキシサイクリンモノハイドレート遅延放出型ビーズのin silico溶解プロフィールを示す。
【図3】図3は、75%の即効ビーズ及び25%の遅延放出ビーズを備えた複合カプセルのin silico溶解プロフィールを示す。
【図4】図4は、様々な処置における定常状態での時間に対する予測血中レベルプロフィールを示す(すなわち、40mgの1日1回のIR処方、40mgの1日1回の70:30及び80:20の割合でのIR及びDRの組み合わせ、ならびに1日2回の20mgのドキシサイクリン処置)。
【図5】図5は、ヒトにおいて1日目及び7日目(定常状態)の75:25 IR:DR(40mg.)製剤の薬物動力学的プロフィールを示す。
【図6】図6は、75:25 IR:DR(40mg.)製剤とPeriostat(登録商標)20mgの1日2回剤形の薬物動力学的曲線を比較する。
【特許請求の範囲】
【請求項1】
1日1回の投与で、最小約0.1μg/ml及び最大約1.0μg/mlのテトラサイクリンの定常状態の血中レベルを与える、テトラサイクリンを含有する経口薬学的組成物。
【請求項2】
1日1回の用量が、約0.3μg/ml〜約0.8μg/mlの間のテトラサイクリンの定常状態の血中レベルを与える、請求項1の組成物。
【請求項3】
テトラサイクリンがドキシサイクリンである請求項1の組成物。
【請求項4】
50mgと25mgの間のドキシサイクリンが存在する請求項3の組成物。
【請求項5】
約40mgのドキシサイクリンが存在する請求項3の組成物。
【請求項6】
即効型製剤である請求項4の組成物。
【請求項7】
即効型製剤である請求項5の組成物。
【請求項8】
即効型(IR)製剤にテトラサイクリンを含有するコンポーネントと遅延放出型(DR)製剤にテトラサイクリンを含有する第二コンポーネントの組み合わせであり、ここで、IRとDRの割合が約99:1〜約70:30である、請求項1の組成物。
【請求項9】
テトラサイクリンがドキシサイクリンである請求項8の組成物。
【請求項10】
即効型コンポーネントと遅延放出型コンポーネントの割合が約80:20〜約70:30である請求項9の組成物。
【請求項11】
即効型コンポーネントと遅延放出型コンポーネントの割合が75:25である請求項9の組成物。
【請求項12】
顆粒剤、錠剤、ペレット剤、散剤、サシェ、カプセル剤、ゲル剤、分散剤または懸濁剤の形態である請求項1の組成物。
【請求項13】
ペレットの組み合わせの剤形であり、ここで、ペレットの一部は即効型製剤であり、そして、ペレットの残りが遅延放出型製剤である、請求項12の組成物。
【請求項14】
ペレットがカプセル中に含まれる請求項13の組成物。
【請求項15】
IR及びDRコンポーネントが錠剤に圧縮される請求項9の組成物。
【請求項16】
層状錠剤であり、ここで、一つの層はDRコンポーネントを含有し、そして、もう一方の層はIRコンポーネントを含有する請求項15の組成物。
【請求項17】
層状ペレットであり、DR部分がペレットのコアを構成し、そして、IR部分がコアを取り囲む請求項12の組成物。
【請求項18】
25と50mgの間のテトラサイクリンを含有する経口薬学的剤形。
【請求項19】
テトラサイクリンがドキシサイクリンである請求項18の剤形。
【請求項20】
約40mgのドキシサイクリンを含有する請求項19の剤形。
【請求項21】
即効製剤である請求項18の剤形。
【請求項22】
顆粒剤、錠剤、ペレット剤、散剤、サシェ、カプセル剤、ゲル剤、分散剤または懸濁剤である請求項18の剤形。
【請求項23】
薬学的に許容される賦形剤とドキシサイクリンを含有する錠剤である請求項19の剤形。
【請求項24】
薬学的に許容される賦形剤とドキシサイクリンのペレットを含有するカプセル剤である請求項19の剤形。
【請求項25】
その一部が遅延放出型製剤であり、そして、もう一方の部分が即効型製剤である請求項19の剤形。
【請求項26】
哺乳動物に請求項1に従うテトラサイクリン組成物の日用量を投与することを包含し、疾患または状態を改善するために少なくとも1回で十分である、哺乳動物のコラゲナーゼ依存疾患または状態、及び/又は、急性もしくは慢性炎症状態を治療するための方法。
【請求項27】
哺乳動物がヒトである請求項26の方法。
【請求項28】
疾患または状態が、歯周病、酒さ(rosacea)、アクネ、ドライアイ、慢性関節リウマチ、副甲状腺機能亢進症及び糖尿病から選択される請求項26の方法。
【請求項29】
疾患が歯周病である請求項28の方法。
【請求項30】
テトラサイクリンがドキシサイクリンである請求項29の方法。
【請求項31】
状態が酒さである請求項28の方法。
【請求項32】
テトラサイクリンがドキシサイクリンである請求項31の方法。
【請求項33】
状態がドライアイである請求項28の方法。
【請求項34】
テトラサイクリンがドキシサイクリンである請求項33の方法。
【請求項35】
50mgと25mgの間のドキシサイクリンが該組成物中に存在する請求項30の方法。
【請求項36】
約40mgのドキシサイクリンが該組成物中に存在する請求項35の方法。
【請求項37】
即効型組成物である請求項30の方法。
【請求項38】
該組成物が、即効部分と遅延放出部分を含有する単位用量の形態である請求項30の方法。
【請求項39】
50mgと25mgの間のドキシサイクリンが該組成物中に存在する請求項32の方法。
【請求項40】
約40mgのドキシサイクリンが該組成物中に存在する請求項39の方法。
【請求項41】
即効型組成物である請求項32の方法。
【請求項42】
該組成物が、即効部分と遅延放出部分を含有する単位用量の形態にある請求項32の方法。
【請求項43】
50mgと25mgの間のドキシサイクリンが該組成物中に存在する請求項34の方法。
【請求項44】
約40mgのドキシサイクリンが該組成物中に存在する請求項43の方法。
【請求項45】
即効型組成物である請求項34の方法。
【請求項46】
組成物が、即効部分と遅延放出部分を含有する単位用量の形態にある請求項34の方法。
【請求項47】
最小約0.1μg/ml及び最大約1.0μg/mlのテトラサイクリンの定常状態の血中レベルを与え、50mgと25mgの間のドキシサイクリンと経口で薬学的に許容される賦形剤を組み合わせて含有する、テトラサイクリンを含む1日1回の経口薬学的組成物を調製するためのプロセス。
【請求項48】
テトラサイクリンがドキシサイクリンである請求項41のプロセス。
【請求項1】
1日1回の投与で、最小約0.1μg/ml及び最大約1.0μg/mlのテトラサイクリンの定常状態の血中レベルを与える、テトラサイクリンを含有する経口薬学的組成物。
【請求項2】
1日1回の用量が、約0.3μg/ml〜約0.8μg/mlの間のテトラサイクリンの定常状態の血中レベルを与える、請求項1の組成物。
【請求項3】
テトラサイクリンがドキシサイクリンである請求項1の組成物。
【請求項4】
50mgと25mgの間のドキシサイクリンが存在する請求項3の組成物。
【請求項5】
約40mgのドキシサイクリンが存在する請求項3の組成物。
【請求項6】
即効型製剤である請求項4の組成物。
【請求項7】
即効型製剤である請求項5の組成物。
【請求項8】
即効型(IR)製剤にテトラサイクリンを含有するコンポーネントと遅延放出型(DR)製剤にテトラサイクリンを含有する第二コンポーネントの組み合わせであり、ここで、IRとDRの割合が約99:1〜約70:30である、請求項1の組成物。
【請求項9】
テトラサイクリンがドキシサイクリンである請求項8の組成物。
【請求項10】
即効型コンポーネントと遅延放出型コンポーネントの割合が約80:20〜約70:30である請求項9の組成物。
【請求項11】
即効型コンポーネントと遅延放出型コンポーネントの割合が75:25である請求項9の組成物。
【請求項12】
顆粒剤、錠剤、ペレット剤、散剤、サシェ、カプセル剤、ゲル剤、分散剤または懸濁剤の形態である請求項1の組成物。
【請求項13】
ペレットの組み合わせの剤形であり、ここで、ペレットの一部は即効型製剤であり、そして、ペレットの残りが遅延放出型製剤である、請求項12の組成物。
【請求項14】
ペレットがカプセル中に含まれる請求項13の組成物。
【請求項15】
IR及びDRコンポーネントが錠剤に圧縮される請求項9の組成物。
【請求項16】
層状錠剤であり、ここで、一つの層はDRコンポーネントを含有し、そして、もう一方の層はIRコンポーネントを含有する請求項15の組成物。
【請求項17】
層状ペレットであり、DR部分がペレットのコアを構成し、そして、IR部分がコアを取り囲む請求項12の組成物。
【請求項18】
25と50mgの間のテトラサイクリンを含有する経口薬学的剤形。
【請求項19】
テトラサイクリンがドキシサイクリンである請求項18の剤形。
【請求項20】
約40mgのドキシサイクリンを含有する請求項19の剤形。
【請求項21】
即効製剤である請求項18の剤形。
【請求項22】
顆粒剤、錠剤、ペレット剤、散剤、サシェ、カプセル剤、ゲル剤、分散剤または懸濁剤である請求項18の剤形。
【請求項23】
薬学的に許容される賦形剤とドキシサイクリンを含有する錠剤である請求項19の剤形。
【請求項24】
薬学的に許容される賦形剤とドキシサイクリンのペレットを含有するカプセル剤である請求項19の剤形。
【請求項25】
その一部が遅延放出型製剤であり、そして、もう一方の部分が即効型製剤である請求項19の剤形。
【請求項26】
哺乳動物に請求項1に従うテトラサイクリン組成物の日用量を投与することを包含し、疾患または状態を改善するために少なくとも1回で十分である、哺乳動物のコラゲナーゼ依存疾患または状態、及び/又は、急性もしくは慢性炎症状態を治療するための方法。
【請求項27】
哺乳動物がヒトである請求項26の方法。
【請求項28】
疾患または状態が、歯周病、酒さ(rosacea)、アクネ、ドライアイ、慢性関節リウマチ、副甲状腺機能亢進症及び糖尿病から選択される請求項26の方法。
【請求項29】
疾患が歯周病である請求項28の方法。
【請求項30】
テトラサイクリンがドキシサイクリンである請求項29の方法。
【請求項31】
状態が酒さである請求項28の方法。
【請求項32】
テトラサイクリンがドキシサイクリンである請求項31の方法。
【請求項33】
状態がドライアイである請求項28の方法。
【請求項34】
テトラサイクリンがドキシサイクリンである請求項33の方法。
【請求項35】
50mgと25mgの間のドキシサイクリンが該組成物中に存在する請求項30の方法。
【請求項36】
約40mgのドキシサイクリンが該組成物中に存在する請求項35の方法。
【請求項37】
即効型組成物である請求項30の方法。
【請求項38】
該組成物が、即効部分と遅延放出部分を含有する単位用量の形態である請求項30の方法。
【請求項39】
50mgと25mgの間のドキシサイクリンが該組成物中に存在する請求項32の方法。
【請求項40】
約40mgのドキシサイクリンが該組成物中に存在する請求項39の方法。
【請求項41】
即効型組成物である請求項32の方法。
【請求項42】
該組成物が、即効部分と遅延放出部分を含有する単位用量の形態にある請求項32の方法。
【請求項43】
50mgと25mgの間のドキシサイクリンが該組成物中に存在する請求項34の方法。
【請求項44】
約40mgのドキシサイクリンが該組成物中に存在する請求項43の方法。
【請求項45】
即効型組成物である請求項34の方法。
【請求項46】
組成物が、即効部分と遅延放出部分を含有する単位用量の形態にある請求項34の方法。
【請求項47】
最小約0.1μg/ml及び最大約1.0μg/mlのテトラサイクリンの定常状態の血中レベルを与え、50mgと25mgの間のドキシサイクリンと経口で薬学的に許容される賦形剤を組み合わせて含有する、テトラサイクリンを含む1日1回の経口薬学的組成物を調製するためのプロセス。
【請求項48】
テトラサイクリンがドキシサイクリンである請求項41のプロセス。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2012−144554(P2012−144554A)
【公開日】平成24年8月2日(2012.8.2)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−59667(P2012−59667)
【出願日】平成24年3月16日(2012.3.16)
【分割の表示】特願2006−509767(P2006−509767)の分割
【原出願日】平成16年4月7日(2004.4.7)
【出願人】(506339316)スパーナス ファーマシューティカルズ インコーポレイテッド (23)
【Fターム(参考)】
【公開日】平成24年8月2日(2012.8.2)
【国際特許分類】
【出願番号】特願2012−59667(P2012−59667)
【出願日】平成24年3月16日(2012.3.16)
【分割の表示】特願2006−509767(P2006−509767)の分割
【原出願日】平成16年4月7日(2004.4.7)
【出願人】(506339316)スパーナス ファーマシューティカルズ インコーポレイテッド (23)
【Fターム(参考)】
[ Back to top ]