
テトラヒドロビオプテリンの安定性錠剤処方物
本発明は、テトラヒドロビオプテリンの安定な固体処方物、それらを生産するためのプロセスおよびそのような処方物を使用する処置方法に関連する。本発明はまた、テトラヒドロビオプテリン、あるいはその前駆体、誘導体またはアナログの安定な固体処方物を提供し、これは延長した期間のための安定性を維持する。本発明の組成物は、室温で8時間より長く安定なBH4の安定な結晶形態および薬学的に受容可能なキャリア、希釈剤または賦形剤を含有し得る。本発明の例示的な安定な錠剤は、乾燥錠剤化プロセスを使用して調製され、室温で少なくとも6〜9ヶ月の貯蔵寿命を有することが示されている。
【発明の詳細な説明】
【技術分野】
【0001】
(背景)
(分野)
本発明は、一般にヒトを処置するためのテトラヒドロビオプテリンあるいはそれらの前駆体、誘導体またはアナログの安定性錠剤処方物に関する。
【背景技術】
【0002】
(関連技術の背景)
テトラヒドロビオプテリン(時々BH4と呼ばれる)は、天然に存在するプテリンファミリーの生体アミンであり、フェニルアラニンヒドロキシラーゼ(PAH)、チロシンヒドロキシラーゼ、トリプトファンヒドロキシラーゼおよび一酸化窒素合成酵素を含む多くの異なる酵素に対する補因子である。プテリンは、還元型および酸化型で生理学的体液および組織中に存在するが、5,6,7,8,テトラヒドロビオプテリンのみが生物学的に活性である。これはキラル分子であり、この補因子の6Rエナンチオマーが、生物学的に活性なエナンチオマーであることが公知である。BH4に関する合成および疾患の詳細な概説は、非特許文献1を参照のこと。
【0003】
PAH酵素の欠失または変異したPAH酵素、あるいはPAHの補因子であるBH4の欠損によるPAHの活性の欠損は、アミノ酸のフェニルアラニン(Phe)の過剰として現れ、これは極めて軽度の形態の場合には高フェニルアラニン血症(HPA)、中程度または重篤な形態の場合にはフェニルケトン尿症(PKU)として公知である。PAHの欠損はまた、神経伝達物質の合成のための前駆体であるアミノ酸のチロシンの欠損を生じる。チロシンヒドロキシラーゼ活性またはトリプトファンヒドロキシラーゼ活性の欠損は、神経伝達物質の産生の欠損として現れ得る。
【0004】
フェニルケトン尿症におけるBH4の欠損の役割の解明にもかかわらず、BH4を用いる処置は提案されていない。なぜなら、フェニルアラニンを制限した食事療法が青年または成人において1年あたり6,000ドルであるのに対して、BH4を用いる処置は青年または成人において1年あたり30,000ドルと極めて高価なためである(非特許文献2)。BH4に関する別の重要な問題としては、本化合物は不安定であり、容易に室温で好気的酸化を受け(非特許文献3;特許文献1)、室温で8時間未満の貯蔵寿命を有する(非特許文献4)ことである。
【0005】
市場において入手可能な他のテトラヒドロビオプテリン製品は、特別にパッケージ化されるかまたは凍結して保存される必要がある。例えばSchirck’s Laboratoryにより販売される錠剤のラベリングは、この錠剤が凍結して保存されるべきであることを指定し、この製品が室温でたった2ヶ月の貯蔵寿命しか有しないことを提示する。BIOPTEN(テトラヒドロビオプテリンの顆粒)は、室温での安定性を維持するために、高価で、密封したホイルパッケージ化(hermetically−sealed foil packaging)を必要とする。そのようなBH4組成物の不安定性は、商業的に望ましくなく、不適当な貯蔵による有意な分解は患者の治療を妨げ得る。
【0006】
薬剤物質の多形相(polymorphic form)は、吸湿性、粒子の形状、密度、流動性および成形性を含む異なる物理的特性および機械的特性を示し得、このことは今度は、薬剤物質の加工および/また製剤の製造に影響を与え得る。薬学的加工における多形性の影響はまた、処方および製造プロセスに依存している。薬剤物質の多形相は、乾燥、製粉、微粉末化、湿式造粒、噴霧乾燥および圧縮のような一連の製造プロセスに曝露されると相変化を起こし得る。湿気および気温のような環境条件への曝露はまた、多形変化を誘導し得る。変化の程度は、一般的に多形の相対的な安定性、相変化の動的な障壁および加えた圧力に依存する。FDA Center for Drug Evaluation and Research(CDER)Draft Guidance for Industry ANDAs:Pharmaceutical Solid Polymorphism Chemistry, Manufacturing, and Controls Information,2004年12月を参照のこと。
【特許文献1】米国特許第4,701,455号明細書
【非特許文献1】Blauら、「Disorders of tetrahydrobiopterin and related biogenic amines.」:Scriver CR、Beaudet AL、Sly WS、Valle D、Childs B、Vogelstein B編。The Metabolic and Molecular Bases of Inherited Disease 第8版 New York:McGraw−Hill、2001年:1275〜1776
【非特許文献2】Hanley、N.Engl.J.Med、2003年 348(17):1723
【非特許文献3】Davisら、1988年、Eur.J.Biochem. Vol 173、345〜351
【非特許文献4】BerneggarおよびBlau、2002年、Mol.Genet.Metabol.77:304〜313
【発明の開示】
【発明が解決しようとする課題】
【0007】
従って、テトラヒドロビオプテリンの安定な固体処方物、およびそのような安定な処方物を製造するためのプロセスに対する必要性が残っている。本発明は、そのような必要性に取り込むことに関する。
【課題を解決するための手段】
【0008】
(発明の要旨)
本発明は、テトラヒドロビオプテリンの安定な固体処方物(特に安定な錠剤)、そのような処方物を生産するためのプロセスおよびそのような処方物を使用する処置方法に関する。
【0009】
本発明は、テトラヒドロビオプテリン、あるいはその前駆体、誘導体またはアナログの安定な固体処方物を提供し、これは延長した期間のための安定性を維持する。本発明の組成物は、室温で8時間より長く安定なBH4の安定な結晶形態および薬学的に受容可能なキャリア、希釈剤または賦形剤を含有し得る。本発明の例示的な安定な錠剤は、乾燥錠剤化プロセスを使用して調製され、室温で少なくとも6〜9ヶ月の貯蔵寿命を有することが示されている。
【0010】
本発明の別の局面は、安定な固体処方物を調製するための乾燥処方プロセスを提供し、このプロセスは、水を添加せずに、テトラヒドロビオプテリン、あるいはその前駆体、誘導体またはアナログと別の薬学的キャリア、希釈剤または賦形剤とを混合する工程を包含する。
【0011】
例示的な実施形態において、この活性な薬学的成分および賦形剤は乾燥混和され、圧縮される。この錠剤は、湿度が、約65%(±5%)以下に維持される湿度制御された部屋において加工される。一度加工されると、この錠剤は、プラスチックバッグの外側2層の間に満たされた乾燥ピロー(desiccant pillow)を有するプラスチックで三重に裏打ちされた耐水性容器に貯蔵される。従って、本発明は、初期量の(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形(好ましくはpolymorph B)と1つ以上の薬学的に受容可能な賦形剤とを混合する工程、およびこの混合物から錠剤を形成する工程を包含する乾燥処方方法を含み、ここでこの工程は、液体の水を加える工程を包含しない。例示的な粒子サイズとしては、例えば約0.2μm〜約500μm、約1μm〜約250μmまたは約2μm〜約200μm、あるいは約500μmより大きい、約600μmより大きい、約700μmより大きい、または約850μmより小さい粒子サイズが挙げられる。
【0012】
例示的な実施形態において、この錠剤は、以下に「polymorph B」として記載される(6R)−5,6,7,8−テトラヒドロビオプテリンの安定な結晶型を使用して最初に製造され、そして少なくとも約95%の活性薬学的成分(API)が、室温で3ヶ月後、6ヶ月後、または9ヶ月後、あるいは好ましくは12ヶ月以上(例えば、15ヶ月、18ヶ月、21ヶ月、2年、2.5年、3年またはそれ以上)後に保持される。好ましくは、この錠剤は、そのような期間、室温で貯蔵後、少なくとも約90%、91%、92%、93%、94%、95%、96%、97%、98%、99%または99.5%のAPIを保持している。この錠剤はまた、好ましくは、そのような期間後、2%以下、1.5%以下、1%以下、0.9%以下、0.8%以下、0.7%以下、0.6%以下の乾燥による損失を示す。例示的な錠剤は、テトラヒドロプテリンの活性薬学的成分の初期量が、約25mg、約50mg、約75mg、約100mg、約125mg、約150mg、約175mg、約200mg、約250mg、約300mg、約350mg、約400mg、約450mg、約500mg、約550mg、約600mg、約650mg、約700mg、約750mg、約800mg、約850mg、約900mgまたはそれ以上の用量で製造され得る。好ましい錠剤はまた、投与時にて迅速な崩壊(例えば、3分以下)を示し、投与の容易さを改善する。
【0013】
従って、本発明は、初期量のpolymorph Bとの名称の(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形および薬学的に受容可能な賦形剤を含有する安定性錠剤処方物を提供し、ここで室温および約60%の湿度で6ヶ月後、この安定性錠剤処方物は、(6R)−L−エリスロ−テトラヒドロビオプテリンの初期量の少なくとも約95%を保持し、ここでこの結晶多形は、塩酸塩として、d値(A)で表される以下の特性ピーク:8.7(vs)、5.63(m)、4.76(m)、4.40(m)、4.00(s)、3.23(s)、3.11(vs)、好ましくは8.7(vs)、6.9(w)、5.90(vw)、5.63(m)、5.07(m)、4.76(m)、4.40(m)、4.15(w)、4.00(s)、3.95(m)、3.52(m)、3.44(w)、3.32(m)、3.23 (s)、3.17(w)、3.11(vs)、3.06(w)、2.99(w)、2.96 (w)、2.94(m)、2.87(w)、2.84(s)、2.82(m)、2.69 (w)、2.59(w)および2.44(w)、を有するX線粉末回折パターンを示す。好ましくは、この錠剤は、(6R)−L−エリスロ−テトラヒドロビオプテリンの初期量の少なくとも約90%、91%、92%、93%、94%、95%、96%、97%、98%、99%または99.5%を保持する。
【0014】
安定な固体処方物は、好ましくは処方物の安定性または他の特性を改善する以下の付加的な成分:結合剤、崩壊剤、酸性抗酸化剤または滑沢剤、あるいはそれらの組み合わせ、を1つ以上含有する。1つの例示的な好ましい組成物は、無水二塩基リン酸カルシウム、クロスポビドン、アスコルビン酸およびフマル酸ステアリルを、必要に応じてマンニトールおよびリボフラビンとともに含有する。安定な固体処方物は、必要に応じて処置される状態に適した他の治療薬、例えば、葉酸塩(葉酸前駆体、葉酸または葉酸誘導体を含む);および/またはビタミン(ビタミンCおよび/またはビタミンB12のような);および/または神経伝達物質前駆体(L−ドパまたはカルビドパのような);および/または5−ヒドロキシトリプトファン;および/またはアルギニンを含有し得る。テトラヒドロビオプテリン(または前駆体または誘導体またはアナログ)および葉酸塩を含有する組成物、および必要に応じてさらにアルギニンを含有する組成物が、特に意図される。
【0015】
さらに本発明は、経口投与のための他の安定な固体処方物、例えば、同様に安定な特性を有するカプセル、丸剤またはトローチを意図する。
【0016】
さらに本発明の別の局面は、そのような安定な固体処方物を使用する処置方法を提供する。本発明は、そのような本発明の処方物が、代謝障害、特にアミノ酸代謝に関わる代謝障害における介入に有用であることを意図する。さらに詳しくは、この安定性処方物は、上昇したフェニルアラニンレベルまたは減少したチロシンレベルを示す被験体(例えば、高フェニルアラニン血症、軽度のフェニルケトン尿症または古典的に重篤なフェニルケトン尿症を患っている被験体);および一酸化窒素合成酵素活性の増強の恩恵を受ける状態(血管疾患、虚血性疾患、炎症性疾患、糖尿病またはインスリン抵抗性を含む)を患っている被験体の処置のために使用され得る。各処置に必要とされる総用量は、複数回用量または単回用量で投与され得る。この安定性処方物は、毎日またはある他の間隔(例えば、1日おき毎またはさらには1週毎に)で投与され得る。
【0017】
この安定性処方物は、単独で使用され得るかまたは、処置される障害(内在する疾患または臨床症状を含む)に適した他の治療との組み合わせで使用され得る。例えば、HPAに対して、本発明の安定性処方物は、タンパク質を制限した食事(例えば、被験体は、1日のタンパク質が約600mg以下または約300mg以下に制限される)との組み合わせで、必要に応じて、チロシン、バリン、イソロイシンおよびロイシンのような補充アミノ酸とともに投与され得る。この安定性処方物はまた、葉酸塩、アルギニン、ビタミンまたは神経伝達物質前駆体との組み合わせで投与され得る。別の例として、血管疾患、糖尿病またはインスリン抵抗性に対して、本発明の安定性処方物は、抗高血圧薬、抗血小板薬、コレステロール降下薬、インスリンまたは経口血糖降下薬のような他の治療薬との組み合わせで投与され得る。
【0018】
本発明の他の特徴および利点は、以下の詳細な説明により明らかになる。しかしながら、詳細な説明および特定の実施例は、本発明の好ましい実施形態を示すが、例証のみで与えられることが理解されるべきである。なぜなら本発明の精神および範囲内における種々の変更および改変は、この詳細な説明より当業者に明らかになるからである。
【発明を実施するための最良の形態】
【0019】
(好ましい実施形態の説明)
本発明は、活性成分の安定な結晶多形を維持する安定性処方物を提供する。以下にpolymorph Bとして記載される、室温で空気中の酸素および通常の湿度に対して安定である(6R)−5,6,7,8−テトラヒドロビオプテリンジヒドロクロライドの無水多形が同定された。しかしながら、相対湿度のパーセントが80%に近づくと、polymorph Bはさらに多くの水を吸収するようであり、その結晶形態を失い、酸化を起こしやすくなる。
【0020】
乾燥処方プロセスを使用することにより、この多形の安定な結晶構造は、最終生成物において維持される。対照的に、テトラヒドロビオプテリン組成物を調製するための他のプロセスは、本発明の生成物に比較して、安定性を欠いた生成物を生じる。
【0021】
本発明の安定性錠剤処方物は、乾燥処方プロセスにおいてpolymorph Bを使用して作られ、通常の室温および通常の湿度、ならびに加速試験条件下で、少なくとも6ヶ月間または9ヶ月間、最初の(6R)−5,6,7,8−テトラヒドロビオプテリンのうち、99%以上を保持することが示された。加速試験条件下(すなわち、高温および高湿度)で観察された安定性は、この錠剤処方物が通常の室温および通常の湿度で6ヶ月間または9ヶ月間よりはるかに長く安定であることを示す。
【0022】
本明細書中で用いられる場合、「貯蔵寿命」は、薬学的処方物が、特定の貯蔵条件下(例えば、通常の湿度で室温)で貯蔵される場合、薬学的処方物中の活性薬学的成分(API)が、最小の分解(例えば、約5%以下の分解)を有する間の貯蔵期間を意味する。
【0023】
本発明の安定性処方物の貯蔵寿命は以下のように測定され得る。試験される処方物は、1つ以上の異なるバッチに分けられ、代表的な貯蔵条件下(例えば、4℃(冷蔵庫)、または25℃(室温))で貯蔵され得る。薬学的処方物におけるAPIの分解もまた、薬剤物質の分解速度を増加させることを意図した過度の貯蔵条件下で、加速試験を使用して検出され得る。例えば、1つのバッチは、「負荷を加えられる(stressed)」(45℃の温度および75%湿度を維持するチャンバーに置かれる)であり得る。次いで処方物の各バッチのサンプルは、処方物において依然として存在するAPIの量を、異なる時間点(例えば、時間0、2週、1ヶ月、3ヶ月、6ヶ月、9ヶ月、1年、1.5年、2年、2.5年、3年またはより長い)で分析される。処方物中のAPIの分析は、高速液体クロマトグラフィー、結晶または粉末のX線回折、赤外線またはラーマンスペクトルの研究、顕微鏡、示差走査熱量測定、熱性重量分析、高温ステージ顕微鏡(hot−stage microscopy)および固体核磁気共鳴を含む種々の検出方法により実施され得る。ある特定の多形形態の維持は、例えば、粉末または結晶のX線回折研究または最初に多形を分析するために使用される任意の同じ手法を実施することにより決定され得る。
【0024】
(I.テトラヒドロビオプテリン、前駆体、誘導体およびアナログの合成)
テトラヒドロビオプテリン、前駆体、誘導体およびアナログを合成するための種々の方法が当該分野において公知である。米国特許第5,698,408号;同第2,601,215号;同第3505329号;同第4,540,783号;同第4,550,109号;同第4,587,340号;同第4,595,752号;同第4,649,197号;同第4,665,182号;同第4,701,455号;同第4,713,454号;同第4,937,342号;同第5,037,981号;同第5,198,547号;同第5,350,851号;同第5,401,844号;同第5,698,408号、カナダ出願CA2420374、欧州出願EP079 574、EP191 335およびサントリーの日本特許公開JP4−082888、JP59−021685およびJP9−157270およびSugimotoおよびMatsuura、Bull.Chem.Soc.Japan、48(12):3767〜3768(1975)、SugimotoおよびMatsuura、Bull.Chem.Soc.Japan、52(1):181〜183(1979)、Matsuuraら.、Chem.Lett.(日本)、735〜738(1984)、Matsuuraら.、Heterocycles、Vol.23、No.12、3115〜3120、1985およびWhiteleyら.、Anal Biochem.137(2):394〜6(1984)(それぞれは本明細書中に参考として援用される)はそれぞれ本発明の組成物として使用され得るジヒドロビオプテリン、BH4およびそれらの誘導体の作製方法を記載する。
【0025】
その全体が参考として本明細書中に援用される国際公開第2005049614号、米国特許第4,540,783号、特許第59−021685号、Schircksら.、Helv.Chim.Acta、60:211(1977)、Sugimotoら.、Bull.Chem.Soc.Jp、52(1):181(1979)、Sugimotoら.、Bull.Chem.Soc.Jp、48(12):3767(1975)、Visontiniら.、Helv.Chim.Acta、52:1225(1969)およびMatsuuraら.、Chem.Lett.、p735(1984)はBH4を合成する方法を記載する。
【0026】
明細書中に記載される組成物および方法における使用のためのアナログの非限定的な例としては、プテリジン、プテリン、ネオプテリン、ビオプテリン、7,8−ジヒドロビオプテリン、6−メチルテトラヒドロプテリンならびに他の6−置換したテトラヒドロプテリンおよび他の6−置換したテトラヒドロプテリン類、セピアプテリン、6,7−ジメチルテトラヒドロプテリン、6−メチルビオプテリンおよび他の6−置換したビオプテリン類、ならびに当該分野において記載される他のアナログが挙げられる。本明細書中に記載される組成物および方法の使用のための誘導体の非限定的な例としては、米国特許第4,758,571号;同第4,774,244号;同第6,162,806号;同第5,902,810号;同第2,955,110号;同第2,541,717号;同第2,603,643号;および同第4,371,514号に記載される誘導体が挙げられ、それらの開示が本明細書中に援用される。
【0027】
当該分野において公知の任意のそのような方法または他の方法が、本発明の安定性処方物および治療方法における使用のためにBH4あるいは前駆体、誘導体またはアナログの生成のために使用され得る。
【0028】
(II.6R−テトラヒドロビオプテリン塩酸塩の結晶多形)
BH4、および特にBH4のジヒドロクロライドは、結晶多形性を示すことが見出されている。BH4の構造は以下:
【0029】
【化1】

に示される。
【0030】
BH4のこの(6R)の形態は、生物学的に活性型として公知であるが、BH4は、周囲温度に不安定であることが公知である。
【0031】
BH4は、取り扱いが困難である。それゆえ、吸湿特性および酸化に対する感受性による物質の分解を防止するために窒素下で封入されたアンプル内で、BH4のジヒドロクロライド(Schircks Laboratories、Jona、Switzerland)として生産され提供された。米国特許第4,649,197号は、(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドおよび6(S)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドをそのジアステレオマーに分離することは、6(R,S)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの乏しい結晶性のため困難であることを開示する。欧州特許番号第0 079 574号は、テトラヒドロビオプテリンの調製を記載し、ここで固体のテトラヒドロビオプテリンジヒドロクロライドは、中間体として得られる。S.Matsuuraらは、Chemistry Letters 1984年、735〜738頁およびHeterocycles、Vol.23、No.12、1985年、3115〜3120頁に、無色針状結晶の形態の結晶性固体として、6(R)−テトラヒドロビオプテリンジヒドロクロライドを記載し、これはJ.Biochem.98、1341〜1348(1985)に開示されるX線解析により特徴付けられる。旋光度6.81°は結晶生成物であることが見出され、これはEP−A2−0 191 335の実施例6における白色結晶の形態の結晶性固体について報告される旋光度6.51°に極めて類似している。
【0032】
(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの開発中に得られた結果は、この化合物が、多形形態および溶媒和物を含む異なる結晶形態で存在し得ることを示した。1つのBH4の結晶多形がより安定であり、周囲条件下で分解に安定であることが見出された。
【0033】
(多形形態B)
最も安定であることが見出された結晶多形は、本明細書中で「形態B(form B)」あるいは「polymorph B」と呼ばれる。
【0034】
Polymorph Bは、約20℃より上で最も高い熱力学的安定性を有するわずかに吸湿性の無水物である。さらに、形態Bは、その熱安定性、標的とした条件による調製の可能性、その適切な形態および粒子サイズのため、容易に加工され取り扱われ得る。融点は260℃付近(ΔHf>140 J/g)であるが、融解前および融解中の分解のため、明確な融点は検出され得ない。これらの顕著な特性は、特に薬学的適用に適した多形形態Bを与え、これはしばしば上昇した温度で調製される。Polymorph Bは、0.2μm〜500μmの範囲であり得る粒子サイズを有する微細粉末として得られ得る。
【0035】
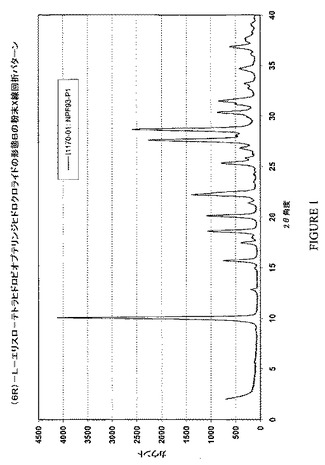
形態Bは、d値(A):8.7(vs)、6.9(w)、5.90(vw)、5.63(m)、5.07(m)、4.76(m)、4.40(m)、4.15(w)、4.00(s)、3.95(m)、3.52(m)、3.44(w)、3.32(m)、3.23(s)、3.17(w)、3.11(vs)、3.06(w)、2.99(w)、2.96(w)、2.94(m)、2.87(w)、2.84(s)、2.82(m)、2.69(w)、2.59(w)、2.44(w)で表されるX線粉末回折パターンを示す。図1は、(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの形態Bにより示される、特徴的なX線回折パターンのグラフである。本明細書中に使用される、以下の括弧内の略語は:(vs)=極めて強い強度;(s)=強い強度;(m)=中程度の強度;(w)=弱い強度;および(vw)=極めて弱い強度、を意味する。
【0036】
形態Bは、極めて大量(例えば、100キロスケール)に調製され得、延長した期間にわたり貯蔵され得る。
【0037】
結晶形態Bを含むすべての結晶形態(多形、水和物および溶媒和物)は、最も安定なpolymorph Bの調製のために使用され得る。Polymorph Bは、適した極性の非水溶性溶媒中において、非結晶性または他の形態の懸濁液の相平衡により得られ得る。
【0038】
BH4の他の形態は、BH4の他の形態を室温で溶媒中に分散し、多形形態Bを生成するのに十分な時間、この懸濁液を周囲温度で攪拌し、その後結晶形態Bを単離し、そして単離した形態Bから溶媒を除去することにより、形態Bに変換され得る。本明細書中に使用される周囲温度は、0℃〜60℃、好ましくは15℃〜40℃の範囲の温度を意味する。適用される温度は、処理中および攪拌中に段階的または継続的に温度を低下させることにより変更され得る。他の形態の形態Bへの変換のために適した溶媒は限定されないが、メタノール、エタノール、イソプロパノール、他のC3−アルコールおよびC4−アルコール、酢酸、アセトニトリル、テトラヒドロフラン、メチル(methy)−t−ブチルエーテル、1,4−ジオキサン、酢酸エチル、酢酸イソプロピル、他のC3−C6−酢酸塩、メチルエチルケトンおよび他のメチル−C3−C5アルキル−ケトンが挙げられる。完全な相平衡に至るまでの時間は、30時間までおよび好ましくは20時間以下までであり得る。
【0039】
Polymorph Bは、約5%まで水を含有する溶媒混合物(具体的にはエタノール、酢酸および水の混合物からなる)からの結晶化により得られ得る。(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドのPolymorph Bは、必要に応じて上昇した温度で、好ましくは形態Bより低いエネルギーの固体形態または(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの形態Bをエタノール、酢酸および水を含有する溶媒混合物中に溶解し、この溶液にシードを添加し、得られた懸濁液を冷却し、形成された結晶を単離することにより調製され得る。溶解は、室温または70℃までで、好ましくは50℃までで実施され得る。溶解のために最終溶媒混合物が使用され得るか、または出発物質が最初に水中に溶解され得、そして他の溶媒が両方または交互に溶媒に加えられ得るかである。この溶媒混合物の組成物は、水:酢酸:テトラヒドロフランを容量比1:3:2〜1:9:4および好ましくは1:5:4で含有し得る。この溶液は、好ましくは攪拌される。冷却は、−40℃〜0℃まで、好ましくは10℃〜30℃までの温度の低下を意味する。適したシードは、別のバッチ由来の多形形態Bあるいは同様なまたは同一の形態を有する結晶である。単離後、この結晶形態Bは、アセトンまたはテトラヒドロフランのような非溶媒を用いて洗浄され得、常法で乾燥され得る。
【0040】
Polymorph Bはまた、メタノール、エタノールおよび酢酸のような非溶媒の添加を通して水溶液からの結晶化により得られ得る。この結晶化手順および単離手順は、溶液を冷却することなしに室温で有利に実施され得る。そのためこのプロセスは、産業規模での実施に極めて適している。
【0041】
本明細書に記載される組成物および方法の1つの実施形態において、(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの多形形態Bを含む組成物は、周囲温度で水中において形態B以外の固体形態または(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの形態Bを溶解し、懸濁液を形成するのに十分な量の非溶媒の添加し、必要に応じてこの懸濁液をある時間攪拌し、そしてその後形成された結晶を単離することにより調製される。この組成物は、以下に記載されるように、さらに薬学的組成物に改変される。
【0042】
水溶液中における(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの濃度は、この溶液を参照すると10〜80重量%、より好ましくは20〜60重量%であり得る。好ましい非溶媒(すなわち、BH4の懸濁液を調製するのに有用な溶媒)は、メタノール、エタノールおよび酢酸である。この非溶媒は、水溶液に加えられ得る。さらに好ましくは、この水溶液は非溶媒に加えられる。懸濁液形成後の攪拌時間は30時間まで、好ましくは20時間以下までであり得る。濾過および乾燥による単離は上に記載されるように公知の様式で実施される。
【0043】
多形形態Bは極めて安定な結晶形態であり、容易に濾過、乾燥され、薬学的処方物に望ましい粒子サイズに粉砕される。これらのすぐれた特性は、特に薬学的適用に適した多形形態Bを与える。
【0044】
(III 安定な薬学的処方物)
薬学的処方物は、初めに薬学的に受容可能なキャリアとともにテトラヒドロビオプテリンあるいはその前駆体または誘導体またはアナログの安定な結晶形態を含有する。本発明の安定性処方物は、好ましくは1つ以上の以下のさらなる成分:結合剤、崩壊剤、酸性抗酸化剤または滑沢剤あるいはそれらの組み合わせを含有し、これらは処方物の安定性または他の特性を改善する。好ましくは、安定性錠剤処方物は、結合剤および崩壊剤を必要に応じて酸性抗酸化剤とともに含有し、そして必要に応じてさらに滑沢剤を含有する。
【0045】
処方物を調製するために使用される(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形の初期量は、例えば、処方物の約30wt%〜約40wt%の範囲または約32wt%〜約35wt%の範囲あるいは約33wt%であり得る。
【0046】
結合剤は、錠剤処方物を維持するのを助ける。ある場合には、無水の結合剤が無水状態のpolymorph Bを貯蔵するために使用される。ある場合には、結合剤が乾燥剤として作用し得る。例示的な結合剤としては、無水二塩基リン酸カルシウムおよびその一水和物が挙げられる。
【0047】
本発明の安定性錠剤処方物における結合剤の例示的な濃度は、約1wt%〜約5wt%の間である。特に意図した濃度は、約1.5wt%〜3wt%の間である。また少なくとも約1.6wt%、1.7wt%、1.8wt%、1.9wt%、2.0wt%、2.1wt%、2.2wt%、2.3wt%、2.4wt%、2.5wt%、2.6wt%、2.7wt%、2.8wt%、2.9wt%および3.0wt%の結合剤の濃度が意図されるかまたは約3.1wt%、3.2wt%、3.3wt%、3.4wt%、3.5wt%、3.6wt%、3.7wt%、3.8wt%、3.9wt%、4.0wt%、4.1wt%、4.2wt%、4.3wt%、4.4wt%、4.5wt%、4.6wt%、4.7wt%、4.8wt%、4.9wt%および5.0wt%までの濃度が意図される。本発明の安定性錠剤処方物中の結合剤のテトラヒドロビオプテリンに対する重量比は、例えば約1:10から約1:20の範囲である。また、約1:10.25、1:10.5、1:10.75、1:11、1:11.25、1:11.5、1:11.75、1:12、1:12.25、1:12.5、1:12.75、1:13、1:13.25、1:13.5、1:13.75、1:14、1:14.25、1:14.5、1:14.75、1:15、1:15.25、1:15.5、1:15.75、1:16、1:16.25、1:16.5、1:16.75、1:17、1:17.25、1:17.5、1:17.75、1:18、1:18.25、1:18.5、1:18.75、1:19、1:19.25、1:19.5および1:19.75の重量比が意図される。
【0048】
崩壊剤は、水を吸収し膨張することにより固体処方物の迅速な崩壊を助ける。例示的な崩壊剤としては、ポリビニルピロリドン(PVP、例えばPOVIDONEという名で販売されている)、ポビドンの架橋した形態(CPVP、例えばCROSPOVIDONEという名で販売されている)、カルボキシメチルセルロースナトリウムの架橋した形態(NaCMC、例えばAC−DI−SOLという名で販売されている)、他の改変したセルロースおよび改変したデンプンが挙げられる。CPVPを用いて処方された錠剤は、PVPを用いて処方された錠剤より極めて迅速な崩壊を示した。
【0049】
本発明の安定性錠剤処方物中の崩壊剤の例示的濃度は、約1wt%〜約20wt%の間である。特に意図した濃度は、約3wt%と約10wt%との間である。また少なくとも約1.1wt%、1.2wt%、1.3wt%、1.4wt%、1.5wt%、1.6wt%、1.7wt%、1.8wt%、1.9wt%、2.0wt%、2.1wt%、2.2wt%、2.3wt%、2.4wt%、2.5wt%、2.6wt%、2.7wt%、2.8wt%、2.9wt%および3.0wt%の崩壊剤の濃度あるいは約4.0wt%、4.1wt%、4.2wt%、4.3wt%、4.4wt%、4.5wt%、4.6wt%、4.7wt%、4.8wt%、4.9wt%、5.0wt%、5.2wt%、5.4wt%、5.6wt%、5.7wt%、5.8wt%、6.0wt%、6.25wt%、6.5wt%、6.75wt%、8.0wt%、8.25wt%、8.5wt%、8.75wt%、9.0wt%、9.25wt%、9.5wt%、9.75wt%および10.0wt%までの崩壊剤の濃度が意図される。本発明の安定性錠剤処方物中の崩壊剤対テトラヒドロビオプテリンの重量比は、例えば約1:5〜約1:10の範囲である。また、約1:5.25、1:5.5、1:5.75、1:6.0、1:6.25、1:6.5、1:6.75、1:7.0、1:7.25、1:7.5、1:7.75、1:8.0、1:8.25、1:8.5、1:8.75、1:9.0、1:9.25、1:9.5および1:9.75の重量比が意図される。
【0050】
抗酸化剤が含まれ得、特に溶解後のテトラヒドロビオプテリン生成物を安定化させるのを助け得る。APIの低pH水溶液は、高pH溶液より安定である。例示的な酸性抗酸化剤としては、アスコルビン酸、アスコルビン酸パルミテートおよびアスコルビン酸ステアレートのようなアスコルビン酸の脂肪酸エステルおよびアスコルビン酸ナトリウム、アスコルビン酸カルシウムまたはアスコルビン酸カリウムのようなアスコルビン酸の塩が挙げられる。非酸性抗酸化剤もまた、安定性錠剤処方物に使用され得る。非酸性抗酸化剤の非限定的な例として、β−カロテン、α−トコフェロールが挙げられる。酸性添加物は、錠剤処方物の安定性を増強するために加えられ得、これらとしてはクエン酸またはリンゴ酸が挙げられる。
【0051】
本発明の安定性錠剤処方物中の抗酸化剤の例示的濃度は、約1wt%と約3wt%との間である。特に意図した濃度は、少なくとも約1.1wt%、1.2wt%、1.3wt%、1.4wt%、1.5wt%、1.6wt%、1.7wt%、1.8wt%、1.9wt%および2.0wt%であるかまたは約2.1wt%、2.2wt%、2.3wt%、2.4wt%、2.5wt%、2.6wt%、2.7wt%、2.8wt%、2.9wt%および3.0wt%までの濃度である。本発明の安定性錠剤処方物中の抗酸化剤対テトラヒドロビオプテリンの重量比は、例えば約1:5〜1:30の範囲である。また約1:5.5、1:6、1:6.5、1:7、1:7.5、1:8、1:8.5、1:9、1:9.5、1:10、1:10.5、1:11、1:11.5、1:12、1:12.5、1:13、1:13.5、1:14、1:14.5、1:15、1:15.5、1:16、1:16.5、1:17、1:17.5、1:18、1:18.5、1:19、1:19.5、1:20、1:20.5、1:21、1:21.5、1:22、1:22.5、1:23、1:23.5、1:24、1:24.5、1:25、1:25.5、1:26、1:26.5、1:27、1:27.5、1:28、1:28.5、1:29および1:29.5の重量比が意図される。
【0052】
Schirk’s Laboratoryの錠剤において、アスコルビン酸は、BH4に対して1:1の比で存在する。対照的に、本発明の安定性処方物中のアスコルビン酸の濃度ははるかに低く、例えばアスコルビン酸対BH4が1:20の重量比(mg/mg)である。従って本発明はまた、1:1未満(例えば、1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14、1:15、1:16、1:17、1:18または1:19であり、好ましくは1:10未満の比)のBH4に対する比でアスコルビン酸を含有する処方物を意図する。
【0053】
滑沢剤は固体処方物の安定性、硬度および均一性を改善する。例示的な滑沢剤としては、フマル酸ステアリルおよびステアリン酸マグネシウムが挙げられる。
【0054】
本発明の安定性錠剤処方物中の滑沢剤の例示的な濃度は、約0.1wt%と約2wt%との間である。特に意図した濃度は、約0.5wt%と1wt%との間である。また、少なくとも約0.1wt%、0.2wt%、0.3wt%、0.4wt%、0.5wt%、0.6wt%、0.7wt%、0.8wt%、0.9wt%および1.0wt%の滑沢剤の濃度、あるいは約1.1wt%、1.2wt%、1.3wt%、1.4wt%、1.5wt%、1.6wt%、1.7wt%、1.8wt%、1.9wt%および2.0wt%までの滑沢剤の濃度が意図される。本発明の安定性錠剤処方物中の滑沢剤対テトラヒドロビオプテリンの重量比は、例えば、約1:25〜1:65の範囲である。また、約1:26、1:27、1:28、1:29、1:30、1:31、1:32、1:33、1:34、1:35、1:36、1:37、1:38、1:39、1:40、1:41、1:42、1:43、1:44、1:45、1:46、1:47、1:48、1:49、1:50、1:51、1:52、1:53、1:54、1:55、1:56、1:57、1:58、1:59、1:60、1:61、1:62、1:63、1:64および1:65の重量比が意図される。
【0055】
安定な固体処方物は、必要に応じて処置される状態に適した他の治療薬、例えば、葉酸前駆体、葉酸または葉酸誘導体を含む葉酸塩;および/またはアルギニン;および/またはビタミンCおよび/またはビタミンB2(リボフラビン)および/またはビタミンB12のようなビタミン;および/またはL−ドパまたはカルビドパのような神経伝達物質前駆体;および/または5−ヒドロキシトリプトファンを含有し得る。
【0056】
葉酸前駆体、葉酸または葉酸誘導体を含む例示的な葉酸塩は、米国特許第6,011,040号および同第6,544,994号に開示され、これら両方は参考として本明細書中に援用され、葉酸(プテリルモノグルタミン酸塩)、ジヒドロ葉酸、テトラヒドロ葉酸、5−メチルテトラヒドロ葉酸、5,10−メチレンテトラヒドロ葉酸、5,10−メテニルテトラヒドロ葉酸、5,10−ホルムイミノテトラヒドロ葉酸、5−ホルミルテトラヒドロ葉酸(ロイコボリン)、10−ホルミルテトラヒドロ葉酸、10−メチルテトラヒドロ葉酸、1つ以上のホリルポリグルタミン酸塩、ジヒドロ葉酸塩またはテトラヒドロ葉酸塩を与えるために葉酸のプテリン部分のピラジン環またはホリルポリグルタミン酸塩のピラジン環が減少された化合物、あるいは種々の酸化レベルでN−5またはN−10部分が1つの炭素単位を保有する前述のすべての化合物の誘導体、あるいはそれらの薬学的に適合性の塩、あるいはそれらの2つ以上の組み合わせが挙げられる。例示的なテトラヒドロ葉酸塩としては、5−ホルミル−(6S)−テトラヒドロ葉酸、5−メチル−(6S)−テトラヒドロ葉酸、5,10−メチレン−(6R)−テトラヒドロ葉酸、5,10−メテニル−(6R)−テトラヒドロ葉酸、10−ホルミル−(6R)−テトラヒドロ葉酸、5−ホルムイミノ−(6S)−テトラヒドロ葉酸または(6S)−テトラヒドロ葉酸およびそれらの薬学的に受容可能な塩が挙げられる。例示的な塩としては、ナトリウム塩、カリウム塩、カルシウム塩またはアンモニウム塩が挙げられる。
【0057】
例示的なBH4対葉酸塩対アルギニンの相対重量比は、約1:10:10〜約10:1:1であり得る。
【0058】
必要に応じて本発明の安定性処方物はまた、マンニトール、ヒドロキシルプロピルセルロース、微結晶性セルロースまたはスクロース、トレハロース、メレジトース(melezitose)、プランテオース(planteose)およびラフィノースのような他の非還元糖も含み得る。還元糖は、BH4と反応し得る。
【0059】
経口投与の固体処方物を製造するための薬学的に受容可能な成分は、例えば、成分を混合し、必要に応じてそれらを微細に分割し、次いでカプセル(例えば硬ゼラチンまたは軟ゼラチンからなるカプセル)を充填するか、錠剤、丸剤またはトローチを圧縮することにより組み込まれ得る。コーティングは、丸剤を形成するために圧縮後適用され得る。
【0060】
薬学的に受容可能な成分は、種々のタイプの処方において周知であり、例えば天然ポリマーまたは合成ポリマーのような結合剤、賦形剤、滑沢剤、界面活性剤、甘味剤および香料、コーティング剤、保存料、色素、増粘剤、アジュバント、抗菌剤、抗酸化剤および種々の処方タイプのためのキャリアであり得る。本明細書に記載される組成物に有用な結合剤の非限定的な例としては、例えば、トラガカントゴム(gum tragacanth)、アカシア、デンプン、ゼラチン、ならびにジカルボン酸、アルキレングリコール、ポリアルキレングリコールおよび/または脂肪性のヒドロキシルカルボン酸のホモポリエステルまたはコポリエステル;ジカルボン酸、アルキレンジアミンおよび/または脂肪性のアミノカルボン酸のホモポリアミドまたはコポリアミド;関連したポリエステルポリアミドコポリマー、ポリ無水物、ポリオルトエステル、ポリホスファゼンおよびポリカーボネートのような生物学的に分解可能なポリマーが挙げられる。生物学的に分解可能なポリマーは線状であるか、分枝されるかまたは架橋され得る。具体的な例は、ポリグリコール酸、ポリ乳酸およびポリd,l−ラクチド/グリコリドである。ポリマーの他の例はポリオキサアルキレン(ポリオキサエチレン、ポリオキサプロピレンおよびそれらの混合ポリマー、ポリアクリルアミドおよびヒドロキシルアルキル化ポリアクリルアミド、ポリマレイン酸およびそれらのポリエステルまたはポリアミド、ポリアクリル酸およびそれらのポリエステルまたはポリアミド、ポリビニルアルコールおよびそれらのポリエステルまたはポリエーテル、ポリビニルイミダゾール、ポリビニルピロリドンおよびキトサンのような天然ポリマーのような水溶解性ポリマーである。
【0061】
本明細書中に記載される組成物に有用な賦形剤の非限定的な例としては、リン酸二カルシウムのようなリン酸塩が挙げられる。本明細書中に記載される組成物に有用な滑沢剤の非限定的な例としては、天然油または合成油、脂質、ろう、ステアリン酸マグネシウムのような脂肪酸塩が挙げられる。
【0062】
本明細書中に記載される組成物に有用な界面活性剤は、陰イオン性、陰イオン性、両性または中性であり得る。本明細書中に記載される組成物に有用な界面活性剤の非限定的な例としては、レシチン、リン脂質、硫酸オクチル、硫酸デシル、硫酸ドデシル、硫酸テトラデシル、硫酸ヘキサデシルおよび硫酸オクタデシル、オレイン酸ナトリウムまたはカプリン酸ナトリウム、1−オクタノイルアミノエタン−2−スルホン酸、1−デカノイルアミノエタン−2−スルホン酸、1−ドデカノイルアミノエタン−2−スルホン酸、1−テトラデカノイルアミノエタン−2−スルホン酸、1−ヘキサデカノイルアミノエタン−2−スルホン酸および1−オクタデカノイルアミノエタン−2−スルホン酸のような1−アシルアミノエタン−2−スルホン酸ならびにタウロコール酸およびタウロデオキシコール酸、胆汁酸および、コール酸、デオキシコール酸およびグリココール酸ナトリウムのようなそれらの塩、カプリン酸ナトリウムまたはラウリン酸ナトリウム、オレイン酸ナトリウム、ラウリル硫酸ナトリウム、セチル硫酸ナトリウム、硫酸化ヒマシ油およびジオクチルスルホコハク酸ナトリウム、コカミドプロピルベタインおよびラウリルベタイン、脂肪アルコール、コレステロール、モノステアリン酸グリセロールまたはジステアリン酸グリセロール、モノオレイン酸グリセロールまたはジオレイン酸グリセロールおよびモノパルミチン酸グリセロールまたはジパルミチン酸グリセロールおよびステアリン酸ポリオキシエチレンが挙げられる。
【0063】
本明細書中に記載される組成物に有用な甘味剤の非限定的な例としては、スクロース、フルクトース、ラクトースまたはアスパルテームが挙げられる。本明細書中に記載される組成物に有用な香料の非限定的な例としては、ペパーミント、ウインターグリーンオイルまたはチェリーの香料またはオレンジの香料のような果実の香料が挙げられる。本明細書中に記載される組成物に有用なコーティング剤の非限定的な例としては、ゼラチン、ろう、シェラック、糖または他の生物学的に分解可能なポリマーが挙げられる。本明細書中に記載される組成物に有用な保存料の非限定的な例としては、メチルパラベンまたはプロピルパラベン、ソルビン酸、クロロブタノール、フェノールおよびチメロサールが挙げられる。
【0064】
本明細書に記載される多形はまた、発泡性錠剤または粉末として処方され得、水性環境において崩壊し飲用溶液を提供する。徐放性処方物はまた、消化管中の体液と接触して活性物質の制御放出を達成するために、本明細書に記載される多形から調製され得、実質的に一定かつ有効な血漿中の活性物質レベルを提供する。結晶形態は本目的のために、生物学的に分解可能なポリマーのポリマーマトリックス、水溶性ポリマーのポリマーマトリックスまたは両方の混合物のポリマーマトリックスおよび必要に応じて適した界面活性剤に埋め込まれ得る。この文脈において埋め込みは、ポリマーのマトリックス中に微粒子を組み込むことを意味し得る。制御放出処方物はまた、公知の分散技術またはエマルジョンコーティング技術を介して分散した微粒子または乳化した微小な液滴(micro−droplet)のカプセル化により得られる。
【0065】
本明細書に記載される組成物に使用されるBH4は、好ましくは、ジヒドロクロライドとして処方されるが、BH4の他の塩の形態は望ましい生物学的活性を有し、結果としてBH4の他の塩の形態が使用され得ることが意図される。具体的には、無機酸または有機酸のBH4の塩が好ましい。代替のBH4塩の形態の非限定的な例としては、酢酸、クエン酸、シュウ酸、酒石酸、フマル酸およびマンデル酸のBH4の塩が挙げられる。
【0066】
薬学的に受容可能な塩基性付加塩は、アルカリ金属およびアルカリ土類金属または有機アミンのような金属またはアミンを用いて形成され得る。化合物の薬学的に受容可能な塩はまた、薬学的に受容可能な陽イオンを用いて調製され得る。適した薬学的に受容可能な陽イオンは、当業者において公知であり、それらとしてはアルカリ金属、アルカリ土類金属、アンモニウムおよび4級アンモニウムの陽イオンが挙げられる。炭酸塩または炭酸水素塩もまた可能である。陽イオンとして使用される金属の例は、ナトリウム、カリウム、マグネシウム、アンモニウム、カルシウムまたは第二鉄などである。適したアミンの例としては、イソプロピルアミン、トリメチルアミン、ヒスチジン、N,N’ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、ジシクロヘキシルアミン、エチレンジアミン、Nメチルグルカミンおよびプロカインが挙げられる。
【0067】
薬学的に受容可能な酸性付加塩としては、無機酸塩または有機酸塩が挙げられる。適した酸性塩としては、塩酸塩、酢酸塩、クエン酸塩、サリチル酸塩、硝酸塩、リン酸塩が挙げられる。他の適した薬学的に受容可能な塩は当業者において周知であり、例えば、酢酸、クエン酸、シュウ酸、酒石酸またはマンデル酸、塩酸、臭化水素酸、硫酸またはリン酸;有機カルボン酸、有機スルホン酸、有機スルホ酸(sulfo acid)または有機リン酸(phospho acid)またはN置換したスルファミン酸(例えば酢酸、プロピオン酸、グリコール酸、コハク酸、マレイン酸、ヒドロキシルマレイン酸、メチルマレイン酸、フマル酸、リンゴ酸、酒石酸、乳酸、シュウ酸、 グルコン酸、グルカル酸、グルクロン酸、クエン酸、安息香酸、ケイ皮酸、マンデル酸、サリチル酸、4アミノサリチル酸、2フェノキシ安息香酸、2アセトキシ安息香酸、パモ酸(embonic acid)、ニコチン酸またはイソニコチン酸)との塩;および天然においてタンパク質の合成に関与する20のαアミノ酸のようなアミノ酸(例えばグルタミン酸またはアスパラギン酸)との塩およびまたフェニル酢酸、メタンスルホン酸、エタンスルホン酸、2ヒドロキシエタンスルホン酸、エタン1,2ジスルホン酸、ベンゼンスルホン酸、4メチルベンゼンスルホン酸、ナフタレン2スルホン酸、ナフタレン1,5ジスルホン酸、2または3ホスホグリセリン酸、グルコース6リン酸、Nシクロヘキシルスルファミン酸(シクラメートの形成を伴う)、あるいはアスコルビン酸のような他の酸性有機化合物との塩が挙げられる。
【0068】
本発明の安定性処方物は、商業的に望まれる場合、例えば乾燥カプセルまたは乾燥ポーチを用いて提供されるHDPEボトル中;あるいはホイルの上にホイルを重ねた(foil−on−foil)ブリスター包装中、またはシースルーポリマーフィルムを含むブリスター包装中に錠剤または丸剤あるいはカプセルとして提供され得る。
【0069】
句「薬学的に受容可能または薬理学的に受容可能」は、動物またはヒトに投与した場合、副反応、アレルギー反応または他の有害反応を生じない分子の実体および組成を言及する。本明細書中に使用される場合、「薬学的に受容可能なキャリア」は任意かつすべての溶媒、分散媒、コーティング、抗菌剤および抗真菌剤、等張性物質および吸収遅延物質などを含む。薬学的に活性な物質のためにそのような溶媒や薬剤を使用することは、当業者において周知である。任意の従来の媒体または薬剤が治療組成物と配合禁忌である場合を除いて、治療組成物において溶媒や薬剤を使用することが意図される。補充的な活性成分はまた、組成物に組み込まれ得る。
【0070】
本明細書中に使用される場合、「薬学的に受容可能なキャリア」は任意のかつすべての溶媒、分散媒、コーティング、抗菌剤および抗真菌剤、等張性物質および吸収遅延物質などを含む。薬学的に活性な物質のためにそのような溶媒や薬剤を使用することは、当業者において周知である。任意の従来のよい媒体または薬剤が治療組成物と配合禁忌である場合を除いて、治療組成物において溶媒や薬剤を使用することが意図される。補充的な活性成分はまた、組成物に組み込まれ得る。
【0071】
本発明の薬学的組成物および処置方法は、ヒト医薬品および獣医医薬品の分野において有用であり得ると認識される。従って、処置される被験体は哺乳動物、好ましくはヒトまたは他の動物であり得る。獣医学的目的のために、被験体として、例えばウシ、ヒツジ、ブタ、ウマおよびヤギを含む家畜、イヌおよびネコのようなコンパニオン動物、外来動物および/または動物園の動物、マウス、ラット、ウサギ、モルモットおよびハムスターを含む実験動物;およびニワトリ、シチメンショウ、アヒルおよびガチョウのような家禽が挙げられる。
【0072】
(IV.安定性処方物を使用する処置方法)
本発明の安定性処方物は、例えば減少したフェニルアラニンヒドロキシラーゼ活性、チロシンヒドロキシラーゼ活性またはトリプトファンヒドロキシラーゼ活性により引き起こされ得る上昇したフェニルアラニンレベル、あるいは減少したチロシンレベルまたはトリプトファンレベルと関連する状態の処置に使用され得る。上昇したフェニルアラニンレベルと関連する状態としては具体的に、本明細書に記載されるように軽度および古典的両方のフェニルケトン尿症、高フェニルアラニン血症が挙げられ、例示的な患者集団として、本明細書に記載される患者のサブグループおよび正常より高いフェニルアラニンレベルを示す任意の他の患者が挙げられる。減少したチロシンレベルまたはトリプトファンレベルと関連する状態としては、神経伝達物質の欠損、パーキンソン病のような神経性障害および精神医学的障害、ジストニー、脊髄小脳の変性、痛み、疲労、うつ病、他の情動障害および統合失調症が挙げられる。
【0073】
安定性処方物はまたBH4欠損(例えば限定されないが、ドパ反応性失調症(DRD)、セピアプテリンレダクターゼ(SR)欠乏症またはジヒドロプテリジンレダクターゼ(DHPR)欠乏症を含むBH4合成のための経路を欠損していることによる)を患っている患者を処置するために使用され得る。
【0074】
本発明の安定性処方物を用いる処置に適した被験体として、治療なしで上昇した血漿Phe濃度(例えば、1800mM/Lより高い、または1600mMより高い、1400mMより高い、1200mMより高い、1000mMより高い、800mMより高い、または600mMより高い、420mMより高い、300mMより高い、200μMより高い、または180mMより高い)を有する被験体が挙げられる。一般的に、軽度のPKUは600μM/Lまでの血漿Phe濃度として、中程度のPKUは600μM/L〜約1200μM/Lの間の血漿Phe濃度として、および古典的または重篤なPKUは1200μM/Lより高い血漿Phe濃度として分類される。好ましくは、安定性処方物のみを用いる処置またはタンパク質を制限した食事とともに安定性処方物を用いる処置は、被験体の血漿フェニルアラニン濃度を600mM未満、または500mM未満、または360mM±15mM未満、または200mM未満、または100mM未満に減少させる。他の適した被験体として、減少したフェニルアラニンヒドロキシラーゼ(PAH)活性を有すると診断された被験体が挙げられる。減少したPAH活性は、PAH酵素の変異(例えばPAHの触媒ドメインの変異またはF39L、L48S、I65T、R68S、A104D、S110C、D129G、E178G、V190A、P211T、R241C、R261Q、A300S、L308F、A313T、K320N、A373T、V388M E390G、A395P、P407SおよびY414Cからなる群より選択される1つ以上の変異);あるいは妊娠した女性、妊娠する可能性のある子供を出産する(child−bearing)年齢の女性、あるいは0歳と3歳との間、または0〜2歳、0〜1.5歳または0〜1歳の幼児である被験体;または単回用量BH4負荷試験または4用量負荷試験または7日負荷試験のような複数回用量負荷試験に対して24時間以内に応答しないと診断された被験体に起因し得る。例示的な患者集団および例示的なBH4負荷試験は、国際公開第2005/049000号に開示され、その全体が参考として援用される。
【0075】
米国特許第4,752,573号;同第4,758,571号;同第4,774,244号;同第4,920,122号;同第5,753,656号;同第5,922,713号;同第5,874,433号;同第5,945,452号;同第6,274,581号;同第6,410,535号;同第6,441,038号;同第6,544,994号;および米国特許出願公開20020187958号;同第20020106645号;同第2002/0076782号;同第20030032616号(それぞれが参考として本明細書中に援用される)は、それぞれ非PKU処置のためにBH4組成物を投与する方法を記載する。これら特許のそれぞれが、当業者において公知のBH4組成物を投与する方法の一般的な教示を提供する参考として本明細書中に援用され、これらは本明細書に記載される処置に適合され得る。
【0076】
個々の要求は様々であるが、各成分の有効量の至適範囲の決定は、当該分野の技術内である。代表的なBH4の投薬量は、1日あたり体重1kgにつき約1〜約20mgを含み、これは通常、約5mg/日(1mg/kg×体重5kg)〜3000mg/日(30mg/kg×体重100kg)の量である。継続的な毎日投与が意図されるが、HPAに関して、Pheレベルの症状がある閾値レベルより下に減少された場合、BH4治療を中止することが望まれ得る。もちろん、Pheレベルがまた上昇する場合には、治療が再開され得る。適切な投薬量は、用量反応データと関連してPheの血中レベルを決定するための確立されたアッセイの使用を通して確かめられ得る。
【0077】
好ましい実施形態において、本発明の方法は、それを必要とする患者に1日用量約10mg/kg〜約20mg/kgのBH4を提供することが意図される。もちろん当業者は、投与により達成される効能に依存して、この用量の増加または減少を調整し得る。1日の用量は、単回用量で投与され得るか、あるいは代替的に都合のよい時間間隔で、複数回用量で投与され得る。例示的な実施形態において、1日用量は1mg/kg、2mg/kg、3mg/kg、4mg/kg、5mg/kg、6mg/kg、7mg/kg、8mg/kg、9mg/kg、10mg/kg、11mg/kg、12mg/kg、13mg/kg、14mg/kg、15mg/kg、16mg/kg、17mg/kg、18mg/kg、19mg/kg、20mg/kg、22mg/kg、24mg/kg、26mg/kg、28mg/kg、30mg/kg、32mg/kg、34mg/kg、36mg/kg、38mg/kg、40mg/kg、42mg/kg、44mg/kg、46mg/kg、48mg/kg、50mg/kgまたはそれ以上のmg/kgであり得る。
【0078】
本発明はさらに本発明の安定性処方物が、一酸化窒素合成酵素活性の増強から恩恵を受ける状態を患っている被験体および血管疾患、虚血性疾患、炎症性疾患、またはインスリン抵抗性を患っている患者の処置に使用され得る。この処置は、例えば、一酸化窒素合成酵素活性の欠損を軽減し得るか、または例えば、正常レベルを超えての一酸化窒素合成酵素活性の上昇を提供し得る。一酸化窒素合成酵素活性の欠損を患っている患者は葉酸前駆体、葉酸または葉酸誘導体を含む葉酸塩を使用する共処置による恩恵を受けることが示唆される。
【0079】
一酸化窒素は、血圧および血管緊張の調節において重要な生理学的役割を果たす血管内皮細胞により構成的に生産される。一酸化窒素の生物活性の欠損は、冠状動脈疾患、任意の動脈(冠状動脈、頚動脈、大脳動脈または末梢血管動脈を含む)のアテローム性硬化症、虚血再灌流傷害、高血圧、糖尿病、糖尿病性血管症、心臓血管疾患、末梢血管疾患を含む血管機能傷害、あるいは脳卒中のような虚血および/または炎症から派生する神経変性状態の病因と関連し、そのような病因としては、損傷した内皮、臓器および組織への不十分な酸素の流れ、上昇した全身血管抵抗(高血圧)、血管平滑筋の増殖、血管狭窄の進展(狭窄(narrowing))および炎症が挙げられることが示唆される。従って、任意のこれらの状態の処置は、本発明の方法に従って意図される。
【0080】
一酸化窒素合成酵素活性の増強はまた、参考として本明細書中に援用される米国特許番号第6,410,535号に記載されるように上昇したスーパーオキシドレベルの減少、増加したインスリン感受性およびインスリン抵抗性に関連した血管機能障害の減少をもたらすことがまた示唆される。従って、糖尿病(I型またはII型)、高インスリン血症またはインスリン抵抗性の処置は、本発明に従って意図される。インスリン抵抗性に関連する血管機能障害を有する疾患として、インスリン抵抗性により引き起こされるかまたはインスリン抵抗性により悪化される疾患、あるいはインスリン抵抗性により治癒が遅れる疾患(インスリン抵抗性に関する限りでは高血圧、高脂血症、動脈硬化、冠状血管狭窄性狭心症、労作性狭心症、脳血管狭窄性病変、脳血管不全、大脳血管攣縮、末梢循環障害、経皮経管冠状動脈形成(PTCA)または冠状動脈バイパス移植(CABG)に続く冠状動脈狭窄、肥満、インスリン依存性糖尿病、高インスリン血症、脂質代謝異常、冠状動脈硬化心疾患などのような)が挙げられる。これらの疾患を有する患者に投与される場合、BH4はNOSの機能を活性化し、NOの産生を増加させ、そして活性酸素種の産生を抑制することによりこれらの疾患を予防し得るか処置し得、血管内皮細胞の障害を改善することが意図される。
【0081】
本発明に従う組成物の適した用量は、受容者の年齢、健康状態および体重、もしあれば同時処置の種類、処置の頻度および望まれる作用の性質(すなわち、望まれる血漿Phe濃度の減少量)に依存することが理解される。投薬頻度はまた、Pheレベルに対する薬力学的作用にも依存する。この作用が単回用量により24時間持続する場合。しかしながら、最も好ましい投薬は、個々の被験体に合わせられ得、過度の実験なしに当業者により理解され決定可能である。これは代表的に標準的な用量の調整(例えば患者の体重が少ない場合、用量を減少させ)を包含する。
【0082】
BH4の投薬頻度は、薬剤の薬物動態学的パラメーターおよび投与経路に依存する。最適な薬学的処方物は、投与経路および望まれる投薬量に依存して当業者により決定される。例えば参考として本明細書中に援用されるRemington’s Pharmaceutical Sciences、第18版(1990、Mack Publ.Co,Easton PA 18042)PP1435 1712を参照のこと。そのような処方物は、物理的状態、安定性、投与した薬剤のインビボの放出速度およびインビボのクリアランス速度に影響を与え得る。投与経路に依存して、適した用量が体重、体表面積または臓器サイズに従って計算され得る。適した処置用量を決定するために必要な計算のさらなる修正が、過度の実験なしに、特に本明細書中に開示される投薬情報およびアッセイならびに動物またはヒトの臨床試験において観察される薬物動態学的データを鑑みて、当業者により慣習的に行われる。
【0083】
最終投薬レジメンは、担当医により、薬物の作用を変化させる因子(例えば薬物の比活性、患者の損傷の重症度および応答性、患者の年齢、状態、体重、性別および食事、任意の感染の重症度、投与の回数および他の臨床的要因)を考慮して決定される。研究が行われるにつれ、特定の疾患および状態のための適した投薬量レベルおよび処置の延長に関するさらなる情報が明らかになる。
【0084】
(V. 併用療法)
本発明のある方法は、本発明の安定性処方物と1つ以上の他の治療剤とを組み合わせた使用を包含する。
【0085】
そのような併用療法において、本発明の安定性処方物の投与は、第2の治療剤の投与と同時であり得るか、あるいは第2の治療剤の投与に先行するか後に続き得る(例えば、数分〜数時間におよぶ間隔で、両薬剤がオーバーラップする期間で治療効果を及ぼすことができる限り)。従って本発明は、第2の治療剤と使用するための本発明の安定性処方物を意図する。本発明はまた、本発明の安定なテトラヒドロビオプテリン処方物、前駆体処方物、誘導体処方物またはアナログ処方物を用いる投与のための医薬の調製において第2の治療剤の使用を意図する。
【0086】
テトラヒドロビオプテリン療法は、種々の形態のHPAを有する患者において治療結果をもたらすために食事性タンパク質の制限と組み合わせられ得る。例えば、望ましい治療結果(すなわち、血漿Phe濃度の減少および/または血漿Phe濃度の付随的な増加を生じることなくより多量のPhe/タンパク質摂取を許容する能力)を生じるために組み合わせた有効量でBH4組成物および低フェニルアラニン医療用タンパク質組成物を被験体に投与し得る。このプロセスは、BH4組成物および食事性タンパク質の治療組成物を同時に投与する工程を包含し得る。これは、単一の組成物または薬理学的タンパク質処方物を投与することにより達成され得、これらは食事性タンパク質の所要量をすべて含有し、またこのタンパク質処方物内にBH4を含有する。あるいは、食事性タンパク質(サプリメントまたは標準的なタンパク質の食事)はBH4の薬理学的処方物(錠剤、注射または飲料)とほぼ同時に摂取される。
【0087】
他の代替法において、BH4処置は、数分〜数時間におよぶ間隔で食事性タンパク質療法に先行し得るまたは後に続き得る。タンパク質およびBH4組成物が別々に投与される実施形態において、BH4が患者において有利な効果を依然として示すことができるように、有意な期間がそれぞれの送達の時間の間に終わらなかったことを一般的に保障する。そのような場合には、BH4を食事性タンパク質摂取の約2〜6時間(前または後)以内、最も好ましくは約1時間のみ時間を遅らせて投与することが意図される。ある実施形態において、BH4療法は、1日用量のBH4が無期限に患者に投与される継続的な治療であることが意図される。他の状況(例えばより軽度な形態のPKUおよびHPAのみを有する妊娠女性)において、BH4療法は、女性が妊娠している限りおよび/または母乳を与えているに限ってのみ、継続され得る。
【0088】
さらにBH4の送達および食事性タンパク質の調節のみをベースにした治療に加え、本発明の方法はまた、1つ以上のHPAの症状を特異的に標的とする第3の組成物との併用療法を意図する。例えば、HPAにより引き起こされるチロシンの欠乏は、神経伝達物質のドパミンおよびセロトニンの欠乏を生じることが公知である。従って、本発明の背景において、BH4および食事性タンパク質をベースにした方法は、食事中の減少したチロシンの量により生じる障害を治すために、L−ドパ、カルビドパおよび5−ヒドロキシトリプトファンの神経伝達物質の投与とさらに組み合わされ得ることが意図される。
【0089】
さらに、PAH(Christensenら.,Mol.Gent.And Metabol.76:313−318、2002;Christensenら.、Gene Therapy、7:1971−1978、2000)およびフェニルアラニンアンモニアリアーゼ(PAL Liuら.、Arts.Cells.Blood.Subs and Immob.Biotech.30(4)243−257、2002)の両方を用いる遺伝子治療は、当業者により意図されている。そのような遺伝子治療技術は、本発明の治療をベースに組み合わされたBH4/食事性タンパク質制限と組み合わせて使用され得る。さらなる併用療法において、フェニラーゼは、患者において低Phe濃度を打ち消すために注射可能な酵素として提供され得ることが意図される。フェニラーゼの投与がチロシンを生成しない(PAHの投与とは異なる)ので、そのような処置は、そのような患者に必須なアミノ酸であるチロシンを依然として生じる。そのためチロシンを用いる食事性の補充は、BH4療法と組み合わせてフェニラーゼを与えられている患者において望ましくあり得る。
【実施例】
【0090】
(VII.実施例)
以下の実施例は、本発明の好ましい実施形態を証明するために包含される。続く実施例に開示される技術は、本発明の実施において十分に機能することが本発明者らにより発見された技術を示し、従ってその実施のために好ましい様式を構成するとみなされ得ることは当業者により理解されるはずである。しかしながら、当業者は、本開示を鑑みて、多くの変更が開示される特定の実施形態においてなされ得、かつ本発明の精神および範囲から逸脱することなく、同様な結果または類似の結果を依然として得ることを理解すべきである。
【0091】
(実施例1)
(BH4の安定化した結晶化形態の調製)
その全体が参考として本明細書中に援用される国際公開番号第2005/065018号は、水和物または溶媒和物を含むBH4の多形を特徴付けるためのX線およびラーマンスペクトル研究ならびに多形が調製され得る例示的な結晶化条件を記載する。その全体が参考として本明細書中に援用される国際公開番号第2005/049000号は、BH4処置が適している種々の患者集団を記載し、BH4を用いてそのような患者を処置するための方法を記載する。その全体が参考として本明細書中に援用される国際公開番号第2005/049614号は、BH4を合成する方法を記載する。
【0092】
本明細書の全体にわたり引用される参考文献は、例示的な手順の詳細または本明細書の記載に補充的な他の詳細を提供するという範囲で、すべて具体的に参考として本明細書中に援用される。
【0093】
(実施例2)
(テトラヒドロビオプテリンの安定性錠剤処方物)
3つの錠剤処方物を、以下詳細に記載するように表Iに示す成分を混合することにより調製した。
【0094】
【表1】
表Iの各処方物に対して、まず4kgの6R−L−エリスロ−5,6,7,8−テトラヒドロビオプテリンジヒドロクロライド(SAPROPTERIN塩酸塩、Daiichi Suntory Pharma Co.、Ltd.、Japanから入手可能な)をブレンダーに充填し、BH4を10分間、1分あたり25回転(RPM)で混合することにより12キログラムのバッチを調製した。次いで6.91kgのD−マンニトール(PEARLITOL、Roquette America、Inc.、Keokuk、Iowaから入手可能)をブレンダーに加え、この混合物をさらに10分間、25RPMで混合した。次いで260グラムの無水二塩基リン酸カルシウム(Mallinckrodt Baker、Inc.、Phillipsburg、New Jerseyから入手可能な)ならびに(a)処方物Iにおいては、436グラムのヒドロキシプロピルセルロースおよび104グラムをブレンダーに加え、(b)処方物IIにおいては、540グラムのヒドロキシプロピルセルロースをブレンダーに加え;(c)処方物IIIにおいては、540グラムのポリビニルピロリドン(KOLLIDON CL、BASF Corporation、Florham Park、New Jerseyから入手可能)をブレンダーに加え、この混合物をさらに10分間、25RPMで混合した。200グラムのアスコルビン酸および120グラムのリボフラビンをブレンダーに加え、この混合物を3分間、25RPMで混合した。フマル酸ステアリルナトリウム滑沢剤(PRUV、Penwest Pharmaceuticals Co.、Danbury、Connecticutから入手可能)を25メッシュのステンレス鋼ふるいに通してバッグ中に濾取し、次いでブレンダーを、ふるいをかけた9kgのフマル酸ステアリルナトリウムを用いて充填し、生じた混合物を5分間、25RPMで混和した。
【0095】
次いで混和した各処方物の混合物をブレンダーから取り出し、150mg錠剤、300mg錠剤および600mg錠剤の調製のために、各処方物について3つのサンプルを集めた。各処方物について、この錠剤サンプル(150mg、300mgおよび600mg)を錠剤プレス(Jenn−Chiang Mahinery Co.、Ltd.、Taiwan、R.O.C.から入手可能な)に置き、ここでこの錠剤プレスのパラメーターを、4.5〜5.5ミリメートルの範囲の厚さおよび7KPの標的硬度を有する錠剤を提供するために設定した。
【0096】
次いで生じた錠剤を処方物の安定性を決定するために分析した。処方物の安定性を、異なる間隔で目視検査により時間にわたる外観の変化、米国薬局方推奨no.701を利用する処方物の崩壊、そして処方物の成分をアッセイすることにより化学的変化を研究した。安定性試験の結果を以下の表IIにまとめる。
【0097】
【表2】
安定性試験は、錠剤処方物IIIが、BH4の他の処方物より安定であることを示す。各薬学的な調製物は、BH4の送達に有用な処方物である。処方物IIIは、処方物Iおよび処方物IIより良好な安定性を示した。従って、好ましい実施形態の1つにおいて、安定化した錠剤処方物は最適な崩壊剤(例えば、クロスポビドンまたはヒドロキシプロピルセルロースよりもポリビニルピロリドンに類似の崩壊剤)を含有する。好ましい処方物は、処方物IIIである。他の適した錠剤処方物は、少なくとも0.01重量%または少なくとも0.05重量%または少なくとも0.1重量%の濃度で少なくともアスコルビン酸を含有する。
【0098】
(実施例3)
100mgテトラヒドロビオプテリンを含む300mg錠剤は、望ましい初期量のpolymorph Bを使用し、以下の乾燥製錠プロセスを使用して以下の表IIIに示される相対量の他の成分と混合して調製した。他の望ましい量のテトラヒドロビオプテリンを含有する錠剤は、同様な方法で調製され得る。
【0099】
6R−L−エリスロ−5,6,7,8−テトラヒドロビオプテリンおよびD−マンニトールを20メッシュふるい(約850μMより大きいサイズの粒子を濾別するように設計した)を用いて手でふるいにかけ、ブレンダーに置いた。この混合物を10分間、21RPMで混和した。次に、無水二塩基リン酸カルシウムおよびクロスポビドンを、20メッシュふるいを用いて手でふるいにかけ、そしてBH4およびD−マンニトールとともに10分間、21RPMで混和した。アスコルビン酸およびリボフラビンを、20メッシュふるいを用いて手でふるいにかけた後ブレンダーに加え、生じた混合物を10分間、21RPMで混和した。次に、フマル酸ステアリルナトリウムを、40メッシュふるいを用いて手でふるいにかけた後ブレンダーに加え、5分間、21RPMで混和した。次いでこの混和した混合物をバッグ内に注ぎ、錠剤に圧縮する前に均一性について試験した。
【0100】
【表3】
錠剤を、ホイルブリスターパックか1ボトルあたり45錠の量でHDPEボトルのどちらかに充填した。各タイプの充填した錠剤を、2つのバッチに分けた。1つのバッチを室温、25±2℃および60±5%相対湿度で貯蔵した。もう一方のバッチを加速試験条件下(40±2℃および75±5%相対湿度)で貯蔵した。規則的な間隔で、錠剤を貯蔵庫から取り出し、活性な薬学的成分(6R−L−エリスロ−5,6,7,8−テトラヒドロビオプテリン)の保持を試験した。300mg錠剤に関して例示的な結果を、以下の表IV、表V、表VIおよび表VIIに示す。室温または加速試験条件下で6ヶ月後貯蔵後、4つの各バッチは、HPLCアッセイにより少なくとも元の量の99%の6R−L−エリスロ−5,6,7,8−テトラヒドロビオプテリンの保持(1%未満の乾燥における損失)および3分以下の迅速な崩壊を示した。
【0101】
【表4】
【0102】
【表5】
【0103】
【表6】
【0104】
【表7】
【図面の簡単な説明】
【0105】
【図1】図1は、(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶形態Bについての特徴的なX線粉末回折パターンを示す。
【技術分野】
【0001】
(背景)
(分野)
本発明は、一般にヒトを処置するためのテトラヒドロビオプテリンあるいはそれらの前駆体、誘導体またはアナログの安定性錠剤処方物に関する。
【背景技術】
【0002】
(関連技術の背景)
テトラヒドロビオプテリン(時々BH4と呼ばれる)は、天然に存在するプテリンファミリーの生体アミンであり、フェニルアラニンヒドロキシラーゼ(PAH)、チロシンヒドロキシラーゼ、トリプトファンヒドロキシラーゼおよび一酸化窒素合成酵素を含む多くの異なる酵素に対する補因子である。プテリンは、還元型および酸化型で生理学的体液および組織中に存在するが、5,6,7,8,テトラヒドロビオプテリンのみが生物学的に活性である。これはキラル分子であり、この補因子の6Rエナンチオマーが、生物学的に活性なエナンチオマーであることが公知である。BH4に関する合成および疾患の詳細な概説は、非特許文献1を参照のこと。
【0003】
PAH酵素の欠失または変異したPAH酵素、あるいはPAHの補因子であるBH4の欠損によるPAHの活性の欠損は、アミノ酸のフェニルアラニン(Phe)の過剰として現れ、これは極めて軽度の形態の場合には高フェニルアラニン血症(HPA)、中程度または重篤な形態の場合にはフェニルケトン尿症(PKU)として公知である。PAHの欠損はまた、神経伝達物質の合成のための前駆体であるアミノ酸のチロシンの欠損を生じる。チロシンヒドロキシラーゼ活性またはトリプトファンヒドロキシラーゼ活性の欠損は、神経伝達物質の産生の欠損として現れ得る。
【0004】
フェニルケトン尿症におけるBH4の欠損の役割の解明にもかかわらず、BH4を用いる処置は提案されていない。なぜなら、フェニルアラニンを制限した食事療法が青年または成人において1年あたり6,000ドルであるのに対して、BH4を用いる処置は青年または成人において1年あたり30,000ドルと極めて高価なためである(非特許文献2)。BH4に関する別の重要な問題としては、本化合物は不安定であり、容易に室温で好気的酸化を受け(非特許文献3;特許文献1)、室温で8時間未満の貯蔵寿命を有する(非特許文献4)ことである。
【0005】
市場において入手可能な他のテトラヒドロビオプテリン製品は、特別にパッケージ化されるかまたは凍結して保存される必要がある。例えばSchirck’s Laboratoryにより販売される錠剤のラベリングは、この錠剤が凍結して保存されるべきであることを指定し、この製品が室温でたった2ヶ月の貯蔵寿命しか有しないことを提示する。BIOPTEN(テトラヒドロビオプテリンの顆粒)は、室温での安定性を維持するために、高価で、密封したホイルパッケージ化(hermetically−sealed foil packaging)を必要とする。そのようなBH4組成物の不安定性は、商業的に望ましくなく、不適当な貯蔵による有意な分解は患者の治療を妨げ得る。
【0006】
薬剤物質の多形相(polymorphic form)は、吸湿性、粒子の形状、密度、流動性および成形性を含む異なる物理的特性および機械的特性を示し得、このことは今度は、薬剤物質の加工および/また製剤の製造に影響を与え得る。薬学的加工における多形性の影響はまた、処方および製造プロセスに依存している。薬剤物質の多形相は、乾燥、製粉、微粉末化、湿式造粒、噴霧乾燥および圧縮のような一連の製造プロセスに曝露されると相変化を起こし得る。湿気および気温のような環境条件への曝露はまた、多形変化を誘導し得る。変化の程度は、一般的に多形の相対的な安定性、相変化の動的な障壁および加えた圧力に依存する。FDA Center for Drug Evaluation and Research(CDER)Draft Guidance for Industry ANDAs:Pharmaceutical Solid Polymorphism Chemistry, Manufacturing, and Controls Information,2004年12月を参照のこと。
【特許文献1】米国特許第4,701,455号明細書
【非特許文献1】Blauら、「Disorders of tetrahydrobiopterin and related biogenic amines.」:Scriver CR、Beaudet AL、Sly WS、Valle D、Childs B、Vogelstein B編。The Metabolic and Molecular Bases of Inherited Disease 第8版 New York:McGraw−Hill、2001年:1275〜1776
【非特許文献2】Hanley、N.Engl.J.Med、2003年 348(17):1723
【非特許文献3】Davisら、1988年、Eur.J.Biochem. Vol 173、345〜351
【非特許文献4】BerneggarおよびBlau、2002年、Mol.Genet.Metabol.77:304〜313
【発明の開示】
【発明が解決しようとする課題】
【0007】
従って、テトラヒドロビオプテリンの安定な固体処方物、およびそのような安定な処方物を製造するためのプロセスに対する必要性が残っている。本発明は、そのような必要性に取り込むことに関する。
【課題を解決するための手段】
【0008】
(発明の要旨)
本発明は、テトラヒドロビオプテリンの安定な固体処方物(特に安定な錠剤)、そのような処方物を生産するためのプロセスおよびそのような処方物を使用する処置方法に関する。
【0009】
本発明は、テトラヒドロビオプテリン、あるいはその前駆体、誘導体またはアナログの安定な固体処方物を提供し、これは延長した期間のための安定性を維持する。本発明の組成物は、室温で8時間より長く安定なBH4の安定な結晶形態および薬学的に受容可能なキャリア、希釈剤または賦形剤を含有し得る。本発明の例示的な安定な錠剤は、乾燥錠剤化プロセスを使用して調製され、室温で少なくとも6〜9ヶ月の貯蔵寿命を有することが示されている。
【0010】
本発明の別の局面は、安定な固体処方物を調製するための乾燥処方プロセスを提供し、このプロセスは、水を添加せずに、テトラヒドロビオプテリン、あるいはその前駆体、誘導体またはアナログと別の薬学的キャリア、希釈剤または賦形剤とを混合する工程を包含する。
【0011】
例示的な実施形態において、この活性な薬学的成分および賦形剤は乾燥混和され、圧縮される。この錠剤は、湿度が、約65%(±5%)以下に維持される湿度制御された部屋において加工される。一度加工されると、この錠剤は、プラスチックバッグの外側2層の間に満たされた乾燥ピロー(desiccant pillow)を有するプラスチックで三重に裏打ちされた耐水性容器に貯蔵される。従って、本発明は、初期量の(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形(好ましくはpolymorph B)と1つ以上の薬学的に受容可能な賦形剤とを混合する工程、およびこの混合物から錠剤を形成する工程を包含する乾燥処方方法を含み、ここでこの工程は、液体の水を加える工程を包含しない。例示的な粒子サイズとしては、例えば約0.2μm〜約500μm、約1μm〜約250μmまたは約2μm〜約200μm、あるいは約500μmより大きい、約600μmより大きい、約700μmより大きい、または約850μmより小さい粒子サイズが挙げられる。
【0012】
例示的な実施形態において、この錠剤は、以下に「polymorph B」として記載される(6R)−5,6,7,8−テトラヒドロビオプテリンの安定な結晶型を使用して最初に製造され、そして少なくとも約95%の活性薬学的成分(API)が、室温で3ヶ月後、6ヶ月後、または9ヶ月後、あるいは好ましくは12ヶ月以上(例えば、15ヶ月、18ヶ月、21ヶ月、2年、2.5年、3年またはそれ以上)後に保持される。好ましくは、この錠剤は、そのような期間、室温で貯蔵後、少なくとも約90%、91%、92%、93%、94%、95%、96%、97%、98%、99%または99.5%のAPIを保持している。この錠剤はまた、好ましくは、そのような期間後、2%以下、1.5%以下、1%以下、0.9%以下、0.8%以下、0.7%以下、0.6%以下の乾燥による損失を示す。例示的な錠剤は、テトラヒドロプテリンの活性薬学的成分の初期量が、約25mg、約50mg、約75mg、約100mg、約125mg、約150mg、約175mg、約200mg、約250mg、約300mg、約350mg、約400mg、約450mg、約500mg、約550mg、約600mg、約650mg、約700mg、約750mg、約800mg、約850mg、約900mgまたはそれ以上の用量で製造され得る。好ましい錠剤はまた、投与時にて迅速な崩壊(例えば、3分以下)を示し、投与の容易さを改善する。
【0013】
従って、本発明は、初期量のpolymorph Bとの名称の(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形および薬学的に受容可能な賦形剤を含有する安定性錠剤処方物を提供し、ここで室温および約60%の湿度で6ヶ月後、この安定性錠剤処方物は、(6R)−L−エリスロ−テトラヒドロビオプテリンの初期量の少なくとも約95%を保持し、ここでこの結晶多形は、塩酸塩として、d値(A)で表される以下の特性ピーク:8.7(vs)、5.63(m)、4.76(m)、4.40(m)、4.00(s)、3.23(s)、3.11(vs)、好ましくは8.7(vs)、6.9(w)、5.90(vw)、5.63(m)、5.07(m)、4.76(m)、4.40(m)、4.15(w)、4.00(s)、3.95(m)、3.52(m)、3.44(w)、3.32(m)、3.23 (s)、3.17(w)、3.11(vs)、3.06(w)、2.99(w)、2.96 (w)、2.94(m)、2.87(w)、2.84(s)、2.82(m)、2.69 (w)、2.59(w)および2.44(w)、を有するX線粉末回折パターンを示す。好ましくは、この錠剤は、(6R)−L−エリスロ−テトラヒドロビオプテリンの初期量の少なくとも約90%、91%、92%、93%、94%、95%、96%、97%、98%、99%または99.5%を保持する。
【0014】
安定な固体処方物は、好ましくは処方物の安定性または他の特性を改善する以下の付加的な成分:結合剤、崩壊剤、酸性抗酸化剤または滑沢剤、あるいはそれらの組み合わせ、を1つ以上含有する。1つの例示的な好ましい組成物は、無水二塩基リン酸カルシウム、クロスポビドン、アスコルビン酸およびフマル酸ステアリルを、必要に応じてマンニトールおよびリボフラビンとともに含有する。安定な固体処方物は、必要に応じて処置される状態に適した他の治療薬、例えば、葉酸塩(葉酸前駆体、葉酸または葉酸誘導体を含む);および/またはビタミン(ビタミンCおよび/またはビタミンB12のような);および/または神経伝達物質前駆体(L−ドパまたはカルビドパのような);および/または5−ヒドロキシトリプトファン;および/またはアルギニンを含有し得る。テトラヒドロビオプテリン(または前駆体または誘導体またはアナログ)および葉酸塩を含有する組成物、および必要に応じてさらにアルギニンを含有する組成物が、特に意図される。
【0015】
さらに本発明は、経口投与のための他の安定な固体処方物、例えば、同様に安定な特性を有するカプセル、丸剤またはトローチを意図する。
【0016】
さらに本発明の別の局面は、そのような安定な固体処方物を使用する処置方法を提供する。本発明は、そのような本発明の処方物が、代謝障害、特にアミノ酸代謝に関わる代謝障害における介入に有用であることを意図する。さらに詳しくは、この安定性処方物は、上昇したフェニルアラニンレベルまたは減少したチロシンレベルを示す被験体(例えば、高フェニルアラニン血症、軽度のフェニルケトン尿症または古典的に重篤なフェニルケトン尿症を患っている被験体);および一酸化窒素合成酵素活性の増強の恩恵を受ける状態(血管疾患、虚血性疾患、炎症性疾患、糖尿病またはインスリン抵抗性を含む)を患っている被験体の処置のために使用され得る。各処置に必要とされる総用量は、複数回用量または単回用量で投与され得る。この安定性処方物は、毎日またはある他の間隔(例えば、1日おき毎またはさらには1週毎に)で投与され得る。
【0017】
この安定性処方物は、単独で使用され得るかまたは、処置される障害(内在する疾患または臨床症状を含む)に適した他の治療との組み合わせで使用され得る。例えば、HPAに対して、本発明の安定性処方物は、タンパク質を制限した食事(例えば、被験体は、1日のタンパク質が約600mg以下または約300mg以下に制限される)との組み合わせで、必要に応じて、チロシン、バリン、イソロイシンおよびロイシンのような補充アミノ酸とともに投与され得る。この安定性処方物はまた、葉酸塩、アルギニン、ビタミンまたは神経伝達物質前駆体との組み合わせで投与され得る。別の例として、血管疾患、糖尿病またはインスリン抵抗性に対して、本発明の安定性処方物は、抗高血圧薬、抗血小板薬、コレステロール降下薬、インスリンまたは経口血糖降下薬のような他の治療薬との組み合わせで投与され得る。
【0018】
本発明の他の特徴および利点は、以下の詳細な説明により明らかになる。しかしながら、詳細な説明および特定の実施例は、本発明の好ましい実施形態を示すが、例証のみで与えられることが理解されるべきである。なぜなら本発明の精神および範囲内における種々の変更および改変は、この詳細な説明より当業者に明らかになるからである。
【発明を実施するための最良の形態】
【0019】
(好ましい実施形態の説明)
本発明は、活性成分の安定な結晶多形を維持する安定性処方物を提供する。以下にpolymorph Bとして記載される、室温で空気中の酸素および通常の湿度に対して安定である(6R)−5,6,7,8−テトラヒドロビオプテリンジヒドロクロライドの無水多形が同定された。しかしながら、相対湿度のパーセントが80%に近づくと、polymorph Bはさらに多くの水を吸収するようであり、その結晶形態を失い、酸化を起こしやすくなる。
【0020】
乾燥処方プロセスを使用することにより、この多形の安定な結晶構造は、最終生成物において維持される。対照的に、テトラヒドロビオプテリン組成物を調製するための他のプロセスは、本発明の生成物に比較して、安定性を欠いた生成物を生じる。
【0021】
本発明の安定性錠剤処方物は、乾燥処方プロセスにおいてpolymorph Bを使用して作られ、通常の室温および通常の湿度、ならびに加速試験条件下で、少なくとも6ヶ月間または9ヶ月間、最初の(6R)−5,6,7,8−テトラヒドロビオプテリンのうち、99%以上を保持することが示された。加速試験条件下(すなわち、高温および高湿度)で観察された安定性は、この錠剤処方物が通常の室温および通常の湿度で6ヶ月間または9ヶ月間よりはるかに長く安定であることを示す。
【0022】
本明細書中で用いられる場合、「貯蔵寿命」は、薬学的処方物が、特定の貯蔵条件下(例えば、通常の湿度で室温)で貯蔵される場合、薬学的処方物中の活性薬学的成分(API)が、最小の分解(例えば、約5%以下の分解)を有する間の貯蔵期間を意味する。
【0023】
本発明の安定性処方物の貯蔵寿命は以下のように測定され得る。試験される処方物は、1つ以上の異なるバッチに分けられ、代表的な貯蔵条件下(例えば、4℃(冷蔵庫)、または25℃(室温))で貯蔵され得る。薬学的処方物におけるAPIの分解もまた、薬剤物質の分解速度を増加させることを意図した過度の貯蔵条件下で、加速試験を使用して検出され得る。例えば、1つのバッチは、「負荷を加えられる(stressed)」(45℃の温度および75%湿度を維持するチャンバーに置かれる)であり得る。次いで処方物の各バッチのサンプルは、処方物において依然として存在するAPIの量を、異なる時間点(例えば、時間0、2週、1ヶ月、3ヶ月、6ヶ月、9ヶ月、1年、1.5年、2年、2.5年、3年またはより長い)で分析される。処方物中のAPIの分析は、高速液体クロマトグラフィー、結晶または粉末のX線回折、赤外線またはラーマンスペクトルの研究、顕微鏡、示差走査熱量測定、熱性重量分析、高温ステージ顕微鏡(hot−stage microscopy)および固体核磁気共鳴を含む種々の検出方法により実施され得る。ある特定の多形形態の維持は、例えば、粉末または結晶のX線回折研究または最初に多形を分析するために使用される任意の同じ手法を実施することにより決定され得る。
【0024】
(I.テトラヒドロビオプテリン、前駆体、誘導体およびアナログの合成)
テトラヒドロビオプテリン、前駆体、誘導体およびアナログを合成するための種々の方法が当該分野において公知である。米国特許第5,698,408号;同第2,601,215号;同第3505329号;同第4,540,783号;同第4,550,109号;同第4,587,340号;同第4,595,752号;同第4,649,197号;同第4,665,182号;同第4,701,455号;同第4,713,454号;同第4,937,342号;同第5,037,981号;同第5,198,547号;同第5,350,851号;同第5,401,844号;同第5,698,408号、カナダ出願CA2420374、欧州出願EP079 574、EP191 335およびサントリーの日本特許公開JP4−082888、JP59−021685およびJP9−157270およびSugimotoおよびMatsuura、Bull.Chem.Soc.Japan、48(12):3767〜3768(1975)、SugimotoおよびMatsuura、Bull.Chem.Soc.Japan、52(1):181〜183(1979)、Matsuuraら.、Chem.Lett.(日本)、735〜738(1984)、Matsuuraら.、Heterocycles、Vol.23、No.12、3115〜3120、1985およびWhiteleyら.、Anal Biochem.137(2):394〜6(1984)(それぞれは本明細書中に参考として援用される)はそれぞれ本発明の組成物として使用され得るジヒドロビオプテリン、BH4およびそれらの誘導体の作製方法を記載する。
【0025】
その全体が参考として本明細書中に援用される国際公開第2005049614号、米国特許第4,540,783号、特許第59−021685号、Schircksら.、Helv.Chim.Acta、60:211(1977)、Sugimotoら.、Bull.Chem.Soc.Jp、52(1):181(1979)、Sugimotoら.、Bull.Chem.Soc.Jp、48(12):3767(1975)、Visontiniら.、Helv.Chim.Acta、52:1225(1969)およびMatsuuraら.、Chem.Lett.、p735(1984)はBH4を合成する方法を記載する。
【0026】
明細書中に記載される組成物および方法における使用のためのアナログの非限定的な例としては、プテリジン、プテリン、ネオプテリン、ビオプテリン、7,8−ジヒドロビオプテリン、6−メチルテトラヒドロプテリンならびに他の6−置換したテトラヒドロプテリンおよび他の6−置換したテトラヒドロプテリン類、セピアプテリン、6,7−ジメチルテトラヒドロプテリン、6−メチルビオプテリンおよび他の6−置換したビオプテリン類、ならびに当該分野において記載される他のアナログが挙げられる。本明細書中に記載される組成物および方法の使用のための誘導体の非限定的な例としては、米国特許第4,758,571号;同第4,774,244号;同第6,162,806号;同第5,902,810号;同第2,955,110号;同第2,541,717号;同第2,603,643号;および同第4,371,514号に記載される誘導体が挙げられ、それらの開示が本明細書中に援用される。
【0027】
当該分野において公知の任意のそのような方法または他の方法が、本発明の安定性処方物および治療方法における使用のためにBH4あるいは前駆体、誘導体またはアナログの生成のために使用され得る。
【0028】
(II.6R−テトラヒドロビオプテリン塩酸塩の結晶多形)
BH4、および特にBH4のジヒドロクロライドは、結晶多形性を示すことが見出されている。BH4の構造は以下:
【0029】
【化1】
に示される。
【0030】
BH4のこの(6R)の形態は、生物学的に活性型として公知であるが、BH4は、周囲温度に不安定であることが公知である。
【0031】
BH4は、取り扱いが困難である。それゆえ、吸湿特性および酸化に対する感受性による物質の分解を防止するために窒素下で封入されたアンプル内で、BH4のジヒドロクロライド(Schircks Laboratories、Jona、Switzerland)として生産され提供された。米国特許第4,649,197号は、(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドおよび6(S)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドをそのジアステレオマーに分離することは、6(R,S)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの乏しい結晶性のため困難であることを開示する。欧州特許番号第0 079 574号は、テトラヒドロビオプテリンの調製を記載し、ここで固体のテトラヒドロビオプテリンジヒドロクロライドは、中間体として得られる。S.Matsuuraらは、Chemistry Letters 1984年、735〜738頁およびHeterocycles、Vol.23、No.12、1985年、3115〜3120頁に、無色針状結晶の形態の結晶性固体として、6(R)−テトラヒドロビオプテリンジヒドロクロライドを記載し、これはJ.Biochem.98、1341〜1348(1985)に開示されるX線解析により特徴付けられる。旋光度6.81°は結晶生成物であることが見出され、これはEP−A2−0 191 335の実施例6における白色結晶の形態の結晶性固体について報告される旋光度6.51°に極めて類似している。
【0032】
(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの開発中に得られた結果は、この化合物が、多形形態および溶媒和物を含む異なる結晶形態で存在し得ることを示した。1つのBH4の結晶多形がより安定であり、周囲条件下で分解に安定であることが見出された。
【0033】
(多形形態B)
最も安定であることが見出された結晶多形は、本明細書中で「形態B(form B)」あるいは「polymorph B」と呼ばれる。
【0034】
Polymorph Bは、約20℃より上で最も高い熱力学的安定性を有するわずかに吸湿性の無水物である。さらに、形態Bは、その熱安定性、標的とした条件による調製の可能性、その適切な形態および粒子サイズのため、容易に加工され取り扱われ得る。融点は260℃付近(ΔHf>140 J/g)であるが、融解前および融解中の分解のため、明確な融点は検出され得ない。これらの顕著な特性は、特に薬学的適用に適した多形形態Bを与え、これはしばしば上昇した温度で調製される。Polymorph Bは、0.2μm〜500μmの範囲であり得る粒子サイズを有する微細粉末として得られ得る。
【0035】
形態Bは、d値(A):8.7(vs)、6.9(w)、5.90(vw)、5.63(m)、5.07(m)、4.76(m)、4.40(m)、4.15(w)、4.00(s)、3.95(m)、3.52(m)、3.44(w)、3.32(m)、3.23(s)、3.17(w)、3.11(vs)、3.06(w)、2.99(w)、2.96(w)、2.94(m)、2.87(w)、2.84(s)、2.82(m)、2.69(w)、2.59(w)、2.44(w)で表されるX線粉末回折パターンを示す。図1は、(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの形態Bにより示される、特徴的なX線回折パターンのグラフである。本明細書中に使用される、以下の括弧内の略語は:(vs)=極めて強い強度;(s)=強い強度;(m)=中程度の強度;(w)=弱い強度;および(vw)=極めて弱い強度、を意味する。
【0036】
形態Bは、極めて大量(例えば、100キロスケール)に調製され得、延長した期間にわたり貯蔵され得る。
【0037】
結晶形態Bを含むすべての結晶形態(多形、水和物および溶媒和物)は、最も安定なpolymorph Bの調製のために使用され得る。Polymorph Bは、適した極性の非水溶性溶媒中において、非結晶性または他の形態の懸濁液の相平衡により得られ得る。
【0038】
BH4の他の形態は、BH4の他の形態を室温で溶媒中に分散し、多形形態Bを生成するのに十分な時間、この懸濁液を周囲温度で攪拌し、その後結晶形態Bを単離し、そして単離した形態Bから溶媒を除去することにより、形態Bに変換され得る。本明細書中に使用される周囲温度は、0℃〜60℃、好ましくは15℃〜40℃の範囲の温度を意味する。適用される温度は、処理中および攪拌中に段階的または継続的に温度を低下させることにより変更され得る。他の形態の形態Bへの変換のために適した溶媒は限定されないが、メタノール、エタノール、イソプロパノール、他のC3−アルコールおよびC4−アルコール、酢酸、アセトニトリル、テトラヒドロフラン、メチル(methy)−t−ブチルエーテル、1,4−ジオキサン、酢酸エチル、酢酸イソプロピル、他のC3−C6−酢酸塩、メチルエチルケトンおよび他のメチル−C3−C5アルキル−ケトンが挙げられる。完全な相平衡に至るまでの時間は、30時間までおよび好ましくは20時間以下までであり得る。
【0039】
Polymorph Bは、約5%まで水を含有する溶媒混合物(具体的にはエタノール、酢酸および水の混合物からなる)からの結晶化により得られ得る。(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドのPolymorph Bは、必要に応じて上昇した温度で、好ましくは形態Bより低いエネルギーの固体形態または(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの形態Bをエタノール、酢酸および水を含有する溶媒混合物中に溶解し、この溶液にシードを添加し、得られた懸濁液を冷却し、形成された結晶を単離することにより調製され得る。溶解は、室温または70℃までで、好ましくは50℃までで実施され得る。溶解のために最終溶媒混合物が使用され得るか、または出発物質が最初に水中に溶解され得、そして他の溶媒が両方または交互に溶媒に加えられ得るかである。この溶媒混合物の組成物は、水:酢酸:テトラヒドロフランを容量比1:3:2〜1:9:4および好ましくは1:5:4で含有し得る。この溶液は、好ましくは攪拌される。冷却は、−40℃〜0℃まで、好ましくは10℃〜30℃までの温度の低下を意味する。適したシードは、別のバッチ由来の多形形態Bあるいは同様なまたは同一の形態を有する結晶である。単離後、この結晶形態Bは、アセトンまたはテトラヒドロフランのような非溶媒を用いて洗浄され得、常法で乾燥され得る。
【0040】
Polymorph Bはまた、メタノール、エタノールおよび酢酸のような非溶媒の添加を通して水溶液からの結晶化により得られ得る。この結晶化手順および単離手順は、溶液を冷却することなしに室温で有利に実施され得る。そのためこのプロセスは、産業規模での実施に極めて適している。
【0041】
本明細書に記載される組成物および方法の1つの実施形態において、(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの多形形態Bを含む組成物は、周囲温度で水中において形態B以外の固体形態または(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの形態Bを溶解し、懸濁液を形成するのに十分な量の非溶媒の添加し、必要に応じてこの懸濁液をある時間攪拌し、そしてその後形成された結晶を単離することにより調製される。この組成物は、以下に記載されるように、さらに薬学的組成物に改変される。
【0042】
水溶液中における(6R)−L−エリスロ−テトラヒドロビオプテリンジヒドロクロライドの濃度は、この溶液を参照すると10〜80重量%、より好ましくは20〜60重量%であり得る。好ましい非溶媒(すなわち、BH4の懸濁液を調製するのに有用な溶媒)は、メタノール、エタノールおよび酢酸である。この非溶媒は、水溶液に加えられ得る。さらに好ましくは、この水溶液は非溶媒に加えられる。懸濁液形成後の攪拌時間は30時間まで、好ましくは20時間以下までであり得る。濾過および乾燥による単離は上に記載されるように公知の様式で実施される。
【0043】
多形形態Bは極めて安定な結晶形態であり、容易に濾過、乾燥され、薬学的処方物に望ましい粒子サイズに粉砕される。これらのすぐれた特性は、特に薬学的適用に適した多形形態Bを与える。
【0044】
(III 安定な薬学的処方物)
薬学的処方物は、初めに薬学的に受容可能なキャリアとともにテトラヒドロビオプテリンあるいはその前駆体または誘導体またはアナログの安定な結晶形態を含有する。本発明の安定性処方物は、好ましくは1つ以上の以下のさらなる成分:結合剤、崩壊剤、酸性抗酸化剤または滑沢剤あるいはそれらの組み合わせを含有し、これらは処方物の安定性または他の特性を改善する。好ましくは、安定性錠剤処方物は、結合剤および崩壊剤を必要に応じて酸性抗酸化剤とともに含有し、そして必要に応じてさらに滑沢剤を含有する。
【0045】
処方物を調製するために使用される(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形の初期量は、例えば、処方物の約30wt%〜約40wt%の範囲または約32wt%〜約35wt%の範囲あるいは約33wt%であり得る。
【0046】
結合剤は、錠剤処方物を維持するのを助ける。ある場合には、無水の結合剤が無水状態のpolymorph Bを貯蔵するために使用される。ある場合には、結合剤が乾燥剤として作用し得る。例示的な結合剤としては、無水二塩基リン酸カルシウムおよびその一水和物が挙げられる。
【0047】
本発明の安定性錠剤処方物における結合剤の例示的な濃度は、約1wt%〜約5wt%の間である。特に意図した濃度は、約1.5wt%〜3wt%の間である。また少なくとも約1.6wt%、1.7wt%、1.8wt%、1.9wt%、2.0wt%、2.1wt%、2.2wt%、2.3wt%、2.4wt%、2.5wt%、2.6wt%、2.7wt%、2.8wt%、2.9wt%および3.0wt%の結合剤の濃度が意図されるかまたは約3.1wt%、3.2wt%、3.3wt%、3.4wt%、3.5wt%、3.6wt%、3.7wt%、3.8wt%、3.9wt%、4.0wt%、4.1wt%、4.2wt%、4.3wt%、4.4wt%、4.5wt%、4.6wt%、4.7wt%、4.8wt%、4.9wt%および5.0wt%までの濃度が意図される。本発明の安定性錠剤処方物中の結合剤のテトラヒドロビオプテリンに対する重量比は、例えば約1:10から約1:20の範囲である。また、約1:10.25、1:10.5、1:10.75、1:11、1:11.25、1:11.5、1:11.75、1:12、1:12.25、1:12.5、1:12.75、1:13、1:13.25、1:13.5、1:13.75、1:14、1:14.25、1:14.5、1:14.75、1:15、1:15.25、1:15.5、1:15.75、1:16、1:16.25、1:16.5、1:16.75、1:17、1:17.25、1:17.5、1:17.75、1:18、1:18.25、1:18.5、1:18.75、1:19、1:19.25、1:19.5および1:19.75の重量比が意図される。
【0048】
崩壊剤は、水を吸収し膨張することにより固体処方物の迅速な崩壊を助ける。例示的な崩壊剤としては、ポリビニルピロリドン(PVP、例えばPOVIDONEという名で販売されている)、ポビドンの架橋した形態(CPVP、例えばCROSPOVIDONEという名で販売されている)、カルボキシメチルセルロースナトリウムの架橋した形態(NaCMC、例えばAC−DI−SOLという名で販売されている)、他の改変したセルロースおよび改変したデンプンが挙げられる。CPVPを用いて処方された錠剤は、PVPを用いて処方された錠剤より極めて迅速な崩壊を示した。
【0049】
本発明の安定性錠剤処方物中の崩壊剤の例示的濃度は、約1wt%〜約20wt%の間である。特に意図した濃度は、約3wt%と約10wt%との間である。また少なくとも約1.1wt%、1.2wt%、1.3wt%、1.4wt%、1.5wt%、1.6wt%、1.7wt%、1.8wt%、1.9wt%、2.0wt%、2.1wt%、2.2wt%、2.3wt%、2.4wt%、2.5wt%、2.6wt%、2.7wt%、2.8wt%、2.9wt%および3.0wt%の崩壊剤の濃度あるいは約4.0wt%、4.1wt%、4.2wt%、4.3wt%、4.4wt%、4.5wt%、4.6wt%、4.7wt%、4.8wt%、4.9wt%、5.0wt%、5.2wt%、5.4wt%、5.6wt%、5.7wt%、5.8wt%、6.0wt%、6.25wt%、6.5wt%、6.75wt%、8.0wt%、8.25wt%、8.5wt%、8.75wt%、9.0wt%、9.25wt%、9.5wt%、9.75wt%および10.0wt%までの崩壊剤の濃度が意図される。本発明の安定性錠剤処方物中の崩壊剤対テトラヒドロビオプテリンの重量比は、例えば約1:5〜約1:10の範囲である。また、約1:5.25、1:5.5、1:5.75、1:6.0、1:6.25、1:6.5、1:6.75、1:7.0、1:7.25、1:7.5、1:7.75、1:8.0、1:8.25、1:8.5、1:8.75、1:9.0、1:9.25、1:9.5および1:9.75の重量比が意図される。
【0050】
抗酸化剤が含まれ得、特に溶解後のテトラヒドロビオプテリン生成物を安定化させるのを助け得る。APIの低pH水溶液は、高pH溶液より安定である。例示的な酸性抗酸化剤としては、アスコルビン酸、アスコルビン酸パルミテートおよびアスコルビン酸ステアレートのようなアスコルビン酸の脂肪酸エステルおよびアスコルビン酸ナトリウム、アスコルビン酸カルシウムまたはアスコルビン酸カリウムのようなアスコルビン酸の塩が挙げられる。非酸性抗酸化剤もまた、安定性錠剤処方物に使用され得る。非酸性抗酸化剤の非限定的な例として、β−カロテン、α−トコフェロールが挙げられる。酸性添加物は、錠剤処方物の安定性を増強するために加えられ得、これらとしてはクエン酸またはリンゴ酸が挙げられる。
【0051】
本発明の安定性錠剤処方物中の抗酸化剤の例示的濃度は、約1wt%と約3wt%との間である。特に意図した濃度は、少なくとも約1.1wt%、1.2wt%、1.3wt%、1.4wt%、1.5wt%、1.6wt%、1.7wt%、1.8wt%、1.9wt%および2.0wt%であるかまたは約2.1wt%、2.2wt%、2.3wt%、2.4wt%、2.5wt%、2.6wt%、2.7wt%、2.8wt%、2.9wt%および3.0wt%までの濃度である。本発明の安定性錠剤処方物中の抗酸化剤対テトラヒドロビオプテリンの重量比は、例えば約1:5〜1:30の範囲である。また約1:5.5、1:6、1:6.5、1:7、1:7.5、1:8、1:8.5、1:9、1:9.5、1:10、1:10.5、1:11、1:11.5、1:12、1:12.5、1:13、1:13.5、1:14、1:14.5、1:15、1:15.5、1:16、1:16.5、1:17、1:17.5、1:18、1:18.5、1:19、1:19.5、1:20、1:20.5、1:21、1:21.5、1:22、1:22.5、1:23、1:23.5、1:24、1:24.5、1:25、1:25.5、1:26、1:26.5、1:27、1:27.5、1:28、1:28.5、1:29および1:29.5の重量比が意図される。
【0052】
Schirk’s Laboratoryの錠剤において、アスコルビン酸は、BH4に対して1:1の比で存在する。対照的に、本発明の安定性処方物中のアスコルビン酸の濃度ははるかに低く、例えばアスコルビン酸対BH4が1:20の重量比(mg/mg)である。従って本発明はまた、1:1未満(例えば、1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14、1:15、1:16、1:17、1:18または1:19であり、好ましくは1:10未満の比)のBH4に対する比でアスコルビン酸を含有する処方物を意図する。
【0053】
滑沢剤は固体処方物の安定性、硬度および均一性を改善する。例示的な滑沢剤としては、フマル酸ステアリルおよびステアリン酸マグネシウムが挙げられる。
【0054】
本発明の安定性錠剤処方物中の滑沢剤の例示的な濃度は、約0.1wt%と約2wt%との間である。特に意図した濃度は、約0.5wt%と1wt%との間である。また、少なくとも約0.1wt%、0.2wt%、0.3wt%、0.4wt%、0.5wt%、0.6wt%、0.7wt%、0.8wt%、0.9wt%および1.0wt%の滑沢剤の濃度、あるいは約1.1wt%、1.2wt%、1.3wt%、1.4wt%、1.5wt%、1.6wt%、1.7wt%、1.8wt%、1.9wt%および2.0wt%までの滑沢剤の濃度が意図される。本発明の安定性錠剤処方物中の滑沢剤対テトラヒドロビオプテリンの重量比は、例えば、約1:25〜1:65の範囲である。また、約1:26、1:27、1:28、1:29、1:30、1:31、1:32、1:33、1:34、1:35、1:36、1:37、1:38、1:39、1:40、1:41、1:42、1:43、1:44、1:45、1:46、1:47、1:48、1:49、1:50、1:51、1:52、1:53、1:54、1:55、1:56、1:57、1:58、1:59、1:60、1:61、1:62、1:63、1:64および1:65の重量比が意図される。
【0055】
安定な固体処方物は、必要に応じて処置される状態に適した他の治療薬、例えば、葉酸前駆体、葉酸または葉酸誘導体を含む葉酸塩;および/またはアルギニン;および/またはビタミンCおよび/またはビタミンB2(リボフラビン)および/またはビタミンB12のようなビタミン;および/またはL−ドパまたはカルビドパのような神経伝達物質前駆体;および/または5−ヒドロキシトリプトファンを含有し得る。
【0056】
葉酸前駆体、葉酸または葉酸誘導体を含む例示的な葉酸塩は、米国特許第6,011,040号および同第6,544,994号に開示され、これら両方は参考として本明細書中に援用され、葉酸(プテリルモノグルタミン酸塩)、ジヒドロ葉酸、テトラヒドロ葉酸、5−メチルテトラヒドロ葉酸、5,10−メチレンテトラヒドロ葉酸、5,10−メテニルテトラヒドロ葉酸、5,10−ホルムイミノテトラヒドロ葉酸、5−ホルミルテトラヒドロ葉酸(ロイコボリン)、10−ホルミルテトラヒドロ葉酸、10−メチルテトラヒドロ葉酸、1つ以上のホリルポリグルタミン酸塩、ジヒドロ葉酸塩またはテトラヒドロ葉酸塩を与えるために葉酸のプテリン部分のピラジン環またはホリルポリグルタミン酸塩のピラジン環が減少された化合物、あるいは種々の酸化レベルでN−5またはN−10部分が1つの炭素単位を保有する前述のすべての化合物の誘導体、あるいはそれらの薬学的に適合性の塩、あるいはそれらの2つ以上の組み合わせが挙げられる。例示的なテトラヒドロ葉酸塩としては、5−ホルミル−(6S)−テトラヒドロ葉酸、5−メチル−(6S)−テトラヒドロ葉酸、5,10−メチレン−(6R)−テトラヒドロ葉酸、5,10−メテニル−(6R)−テトラヒドロ葉酸、10−ホルミル−(6R)−テトラヒドロ葉酸、5−ホルムイミノ−(6S)−テトラヒドロ葉酸または(6S)−テトラヒドロ葉酸およびそれらの薬学的に受容可能な塩が挙げられる。例示的な塩としては、ナトリウム塩、カリウム塩、カルシウム塩またはアンモニウム塩が挙げられる。
【0057】
例示的なBH4対葉酸塩対アルギニンの相対重量比は、約1:10:10〜約10:1:1であり得る。
【0058】
必要に応じて本発明の安定性処方物はまた、マンニトール、ヒドロキシルプロピルセルロース、微結晶性セルロースまたはスクロース、トレハロース、メレジトース(melezitose)、プランテオース(planteose)およびラフィノースのような他の非還元糖も含み得る。還元糖は、BH4と反応し得る。
【0059】
経口投与の固体処方物を製造するための薬学的に受容可能な成分は、例えば、成分を混合し、必要に応じてそれらを微細に分割し、次いでカプセル(例えば硬ゼラチンまたは軟ゼラチンからなるカプセル)を充填するか、錠剤、丸剤またはトローチを圧縮することにより組み込まれ得る。コーティングは、丸剤を形成するために圧縮後適用され得る。
【0060】
薬学的に受容可能な成分は、種々のタイプの処方において周知であり、例えば天然ポリマーまたは合成ポリマーのような結合剤、賦形剤、滑沢剤、界面活性剤、甘味剤および香料、コーティング剤、保存料、色素、増粘剤、アジュバント、抗菌剤、抗酸化剤および種々の処方タイプのためのキャリアであり得る。本明細書に記載される組成物に有用な結合剤の非限定的な例としては、例えば、トラガカントゴム(gum tragacanth)、アカシア、デンプン、ゼラチン、ならびにジカルボン酸、アルキレングリコール、ポリアルキレングリコールおよび/または脂肪性のヒドロキシルカルボン酸のホモポリエステルまたはコポリエステル;ジカルボン酸、アルキレンジアミンおよび/または脂肪性のアミノカルボン酸のホモポリアミドまたはコポリアミド;関連したポリエステルポリアミドコポリマー、ポリ無水物、ポリオルトエステル、ポリホスファゼンおよびポリカーボネートのような生物学的に分解可能なポリマーが挙げられる。生物学的に分解可能なポリマーは線状であるか、分枝されるかまたは架橋され得る。具体的な例は、ポリグリコール酸、ポリ乳酸およびポリd,l−ラクチド/グリコリドである。ポリマーの他の例はポリオキサアルキレン(ポリオキサエチレン、ポリオキサプロピレンおよびそれらの混合ポリマー、ポリアクリルアミドおよびヒドロキシルアルキル化ポリアクリルアミド、ポリマレイン酸およびそれらのポリエステルまたはポリアミド、ポリアクリル酸およびそれらのポリエステルまたはポリアミド、ポリビニルアルコールおよびそれらのポリエステルまたはポリエーテル、ポリビニルイミダゾール、ポリビニルピロリドンおよびキトサンのような天然ポリマーのような水溶解性ポリマーである。
【0061】
本明細書中に記載される組成物に有用な賦形剤の非限定的な例としては、リン酸二カルシウムのようなリン酸塩が挙げられる。本明細書中に記載される組成物に有用な滑沢剤の非限定的な例としては、天然油または合成油、脂質、ろう、ステアリン酸マグネシウムのような脂肪酸塩が挙げられる。
【0062】
本明細書中に記載される組成物に有用な界面活性剤は、陰イオン性、陰イオン性、両性または中性であり得る。本明細書中に記載される組成物に有用な界面活性剤の非限定的な例としては、レシチン、リン脂質、硫酸オクチル、硫酸デシル、硫酸ドデシル、硫酸テトラデシル、硫酸ヘキサデシルおよび硫酸オクタデシル、オレイン酸ナトリウムまたはカプリン酸ナトリウム、1−オクタノイルアミノエタン−2−スルホン酸、1−デカノイルアミノエタン−2−スルホン酸、1−ドデカノイルアミノエタン−2−スルホン酸、1−テトラデカノイルアミノエタン−2−スルホン酸、1−ヘキサデカノイルアミノエタン−2−スルホン酸および1−オクタデカノイルアミノエタン−2−スルホン酸のような1−アシルアミノエタン−2−スルホン酸ならびにタウロコール酸およびタウロデオキシコール酸、胆汁酸および、コール酸、デオキシコール酸およびグリココール酸ナトリウムのようなそれらの塩、カプリン酸ナトリウムまたはラウリン酸ナトリウム、オレイン酸ナトリウム、ラウリル硫酸ナトリウム、セチル硫酸ナトリウム、硫酸化ヒマシ油およびジオクチルスルホコハク酸ナトリウム、コカミドプロピルベタインおよびラウリルベタイン、脂肪アルコール、コレステロール、モノステアリン酸グリセロールまたはジステアリン酸グリセロール、モノオレイン酸グリセロールまたはジオレイン酸グリセロールおよびモノパルミチン酸グリセロールまたはジパルミチン酸グリセロールおよびステアリン酸ポリオキシエチレンが挙げられる。
【0063】
本明細書中に記載される組成物に有用な甘味剤の非限定的な例としては、スクロース、フルクトース、ラクトースまたはアスパルテームが挙げられる。本明細書中に記載される組成物に有用な香料の非限定的な例としては、ペパーミント、ウインターグリーンオイルまたはチェリーの香料またはオレンジの香料のような果実の香料が挙げられる。本明細書中に記載される組成物に有用なコーティング剤の非限定的な例としては、ゼラチン、ろう、シェラック、糖または他の生物学的に分解可能なポリマーが挙げられる。本明細書中に記載される組成物に有用な保存料の非限定的な例としては、メチルパラベンまたはプロピルパラベン、ソルビン酸、クロロブタノール、フェノールおよびチメロサールが挙げられる。
【0064】
本明細書に記載される多形はまた、発泡性錠剤または粉末として処方され得、水性環境において崩壊し飲用溶液を提供する。徐放性処方物はまた、消化管中の体液と接触して活性物質の制御放出を達成するために、本明細書に記載される多形から調製され得、実質的に一定かつ有効な血漿中の活性物質レベルを提供する。結晶形態は本目的のために、生物学的に分解可能なポリマーのポリマーマトリックス、水溶性ポリマーのポリマーマトリックスまたは両方の混合物のポリマーマトリックスおよび必要に応じて適した界面活性剤に埋め込まれ得る。この文脈において埋め込みは、ポリマーのマトリックス中に微粒子を組み込むことを意味し得る。制御放出処方物はまた、公知の分散技術またはエマルジョンコーティング技術を介して分散した微粒子または乳化した微小な液滴(micro−droplet)のカプセル化により得られる。
【0065】
本明細書に記載される組成物に使用されるBH4は、好ましくは、ジヒドロクロライドとして処方されるが、BH4の他の塩の形態は望ましい生物学的活性を有し、結果としてBH4の他の塩の形態が使用され得ることが意図される。具体的には、無機酸または有機酸のBH4の塩が好ましい。代替のBH4塩の形態の非限定的な例としては、酢酸、クエン酸、シュウ酸、酒石酸、フマル酸およびマンデル酸のBH4の塩が挙げられる。
【0066】
薬学的に受容可能な塩基性付加塩は、アルカリ金属およびアルカリ土類金属または有機アミンのような金属またはアミンを用いて形成され得る。化合物の薬学的に受容可能な塩はまた、薬学的に受容可能な陽イオンを用いて調製され得る。適した薬学的に受容可能な陽イオンは、当業者において公知であり、それらとしてはアルカリ金属、アルカリ土類金属、アンモニウムおよび4級アンモニウムの陽イオンが挙げられる。炭酸塩または炭酸水素塩もまた可能である。陽イオンとして使用される金属の例は、ナトリウム、カリウム、マグネシウム、アンモニウム、カルシウムまたは第二鉄などである。適したアミンの例としては、イソプロピルアミン、トリメチルアミン、ヒスチジン、N,N’ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、ジシクロヘキシルアミン、エチレンジアミン、Nメチルグルカミンおよびプロカインが挙げられる。
【0067】
薬学的に受容可能な酸性付加塩としては、無機酸塩または有機酸塩が挙げられる。適した酸性塩としては、塩酸塩、酢酸塩、クエン酸塩、サリチル酸塩、硝酸塩、リン酸塩が挙げられる。他の適した薬学的に受容可能な塩は当業者において周知であり、例えば、酢酸、クエン酸、シュウ酸、酒石酸またはマンデル酸、塩酸、臭化水素酸、硫酸またはリン酸;有機カルボン酸、有機スルホン酸、有機スルホ酸(sulfo acid)または有機リン酸(phospho acid)またはN置換したスルファミン酸(例えば酢酸、プロピオン酸、グリコール酸、コハク酸、マレイン酸、ヒドロキシルマレイン酸、メチルマレイン酸、フマル酸、リンゴ酸、酒石酸、乳酸、シュウ酸、 グルコン酸、グルカル酸、グルクロン酸、クエン酸、安息香酸、ケイ皮酸、マンデル酸、サリチル酸、4アミノサリチル酸、2フェノキシ安息香酸、2アセトキシ安息香酸、パモ酸(embonic acid)、ニコチン酸またはイソニコチン酸)との塩;および天然においてタンパク質の合成に関与する20のαアミノ酸のようなアミノ酸(例えばグルタミン酸またはアスパラギン酸)との塩およびまたフェニル酢酸、メタンスルホン酸、エタンスルホン酸、2ヒドロキシエタンスルホン酸、エタン1,2ジスルホン酸、ベンゼンスルホン酸、4メチルベンゼンスルホン酸、ナフタレン2スルホン酸、ナフタレン1,5ジスルホン酸、2または3ホスホグリセリン酸、グルコース6リン酸、Nシクロヘキシルスルファミン酸(シクラメートの形成を伴う)、あるいはアスコルビン酸のような他の酸性有機化合物との塩が挙げられる。
【0068】
本発明の安定性処方物は、商業的に望まれる場合、例えば乾燥カプセルまたは乾燥ポーチを用いて提供されるHDPEボトル中;あるいはホイルの上にホイルを重ねた(foil−on−foil)ブリスター包装中、またはシースルーポリマーフィルムを含むブリスター包装中に錠剤または丸剤あるいはカプセルとして提供され得る。
【0069】
句「薬学的に受容可能または薬理学的に受容可能」は、動物またはヒトに投与した場合、副反応、アレルギー反応または他の有害反応を生じない分子の実体および組成を言及する。本明細書中に使用される場合、「薬学的に受容可能なキャリア」は任意かつすべての溶媒、分散媒、コーティング、抗菌剤および抗真菌剤、等張性物質および吸収遅延物質などを含む。薬学的に活性な物質のためにそのような溶媒や薬剤を使用することは、当業者において周知である。任意の従来の媒体または薬剤が治療組成物と配合禁忌である場合を除いて、治療組成物において溶媒や薬剤を使用することが意図される。補充的な活性成分はまた、組成物に組み込まれ得る。
【0070】
本明細書中に使用される場合、「薬学的に受容可能なキャリア」は任意のかつすべての溶媒、分散媒、コーティング、抗菌剤および抗真菌剤、等張性物質および吸収遅延物質などを含む。薬学的に活性な物質のためにそのような溶媒や薬剤を使用することは、当業者において周知である。任意の従来のよい媒体または薬剤が治療組成物と配合禁忌である場合を除いて、治療組成物において溶媒や薬剤を使用することが意図される。補充的な活性成分はまた、組成物に組み込まれ得る。
【0071】
本発明の薬学的組成物および処置方法は、ヒト医薬品および獣医医薬品の分野において有用であり得ると認識される。従って、処置される被験体は哺乳動物、好ましくはヒトまたは他の動物であり得る。獣医学的目的のために、被験体として、例えばウシ、ヒツジ、ブタ、ウマおよびヤギを含む家畜、イヌおよびネコのようなコンパニオン動物、外来動物および/または動物園の動物、マウス、ラット、ウサギ、モルモットおよびハムスターを含む実験動物;およびニワトリ、シチメンショウ、アヒルおよびガチョウのような家禽が挙げられる。
【0072】
(IV.安定性処方物を使用する処置方法)
本発明の安定性処方物は、例えば減少したフェニルアラニンヒドロキシラーゼ活性、チロシンヒドロキシラーゼ活性またはトリプトファンヒドロキシラーゼ活性により引き起こされ得る上昇したフェニルアラニンレベル、あるいは減少したチロシンレベルまたはトリプトファンレベルと関連する状態の処置に使用され得る。上昇したフェニルアラニンレベルと関連する状態としては具体的に、本明細書に記載されるように軽度および古典的両方のフェニルケトン尿症、高フェニルアラニン血症が挙げられ、例示的な患者集団として、本明細書に記載される患者のサブグループおよび正常より高いフェニルアラニンレベルを示す任意の他の患者が挙げられる。減少したチロシンレベルまたはトリプトファンレベルと関連する状態としては、神経伝達物質の欠損、パーキンソン病のような神経性障害および精神医学的障害、ジストニー、脊髄小脳の変性、痛み、疲労、うつ病、他の情動障害および統合失調症が挙げられる。
【0073】
安定性処方物はまたBH4欠損(例えば限定されないが、ドパ反応性失調症(DRD)、セピアプテリンレダクターゼ(SR)欠乏症またはジヒドロプテリジンレダクターゼ(DHPR)欠乏症を含むBH4合成のための経路を欠損していることによる)を患っている患者を処置するために使用され得る。
【0074】
本発明の安定性処方物を用いる処置に適した被験体として、治療なしで上昇した血漿Phe濃度(例えば、1800mM/Lより高い、または1600mMより高い、1400mMより高い、1200mMより高い、1000mMより高い、800mMより高い、または600mMより高い、420mMより高い、300mMより高い、200μMより高い、または180mMより高い)を有する被験体が挙げられる。一般的に、軽度のPKUは600μM/Lまでの血漿Phe濃度として、中程度のPKUは600μM/L〜約1200μM/Lの間の血漿Phe濃度として、および古典的または重篤なPKUは1200μM/Lより高い血漿Phe濃度として分類される。好ましくは、安定性処方物のみを用いる処置またはタンパク質を制限した食事とともに安定性処方物を用いる処置は、被験体の血漿フェニルアラニン濃度を600mM未満、または500mM未満、または360mM±15mM未満、または200mM未満、または100mM未満に減少させる。他の適した被験体として、減少したフェニルアラニンヒドロキシラーゼ(PAH)活性を有すると診断された被験体が挙げられる。減少したPAH活性は、PAH酵素の変異(例えばPAHの触媒ドメインの変異またはF39L、L48S、I65T、R68S、A104D、S110C、D129G、E178G、V190A、P211T、R241C、R261Q、A300S、L308F、A313T、K320N、A373T、V388M E390G、A395P、P407SおよびY414Cからなる群より選択される1つ以上の変異);あるいは妊娠した女性、妊娠する可能性のある子供を出産する(child−bearing)年齢の女性、あるいは0歳と3歳との間、または0〜2歳、0〜1.5歳または0〜1歳の幼児である被験体;または単回用量BH4負荷試験または4用量負荷試験または7日負荷試験のような複数回用量負荷試験に対して24時間以内に応答しないと診断された被験体に起因し得る。例示的な患者集団および例示的なBH4負荷試験は、国際公開第2005/049000号に開示され、その全体が参考として援用される。
【0075】
米国特許第4,752,573号;同第4,758,571号;同第4,774,244号;同第4,920,122号;同第5,753,656号;同第5,922,713号;同第5,874,433号;同第5,945,452号;同第6,274,581号;同第6,410,535号;同第6,441,038号;同第6,544,994号;および米国特許出願公開20020187958号;同第20020106645号;同第2002/0076782号;同第20030032616号(それぞれが参考として本明細書中に援用される)は、それぞれ非PKU処置のためにBH4組成物を投与する方法を記載する。これら特許のそれぞれが、当業者において公知のBH4組成物を投与する方法の一般的な教示を提供する参考として本明細書中に援用され、これらは本明細書に記載される処置に適合され得る。
【0076】
個々の要求は様々であるが、各成分の有効量の至適範囲の決定は、当該分野の技術内である。代表的なBH4の投薬量は、1日あたり体重1kgにつき約1〜約20mgを含み、これは通常、約5mg/日(1mg/kg×体重5kg)〜3000mg/日(30mg/kg×体重100kg)の量である。継続的な毎日投与が意図されるが、HPAに関して、Pheレベルの症状がある閾値レベルより下に減少された場合、BH4治療を中止することが望まれ得る。もちろん、Pheレベルがまた上昇する場合には、治療が再開され得る。適切な投薬量は、用量反応データと関連してPheの血中レベルを決定するための確立されたアッセイの使用を通して確かめられ得る。
【0077】
好ましい実施形態において、本発明の方法は、それを必要とする患者に1日用量約10mg/kg〜約20mg/kgのBH4を提供することが意図される。もちろん当業者は、投与により達成される効能に依存して、この用量の増加または減少を調整し得る。1日の用量は、単回用量で投与され得るか、あるいは代替的に都合のよい時間間隔で、複数回用量で投与され得る。例示的な実施形態において、1日用量は1mg/kg、2mg/kg、3mg/kg、4mg/kg、5mg/kg、6mg/kg、7mg/kg、8mg/kg、9mg/kg、10mg/kg、11mg/kg、12mg/kg、13mg/kg、14mg/kg、15mg/kg、16mg/kg、17mg/kg、18mg/kg、19mg/kg、20mg/kg、22mg/kg、24mg/kg、26mg/kg、28mg/kg、30mg/kg、32mg/kg、34mg/kg、36mg/kg、38mg/kg、40mg/kg、42mg/kg、44mg/kg、46mg/kg、48mg/kg、50mg/kgまたはそれ以上のmg/kgであり得る。
【0078】
本発明はさらに本発明の安定性処方物が、一酸化窒素合成酵素活性の増強から恩恵を受ける状態を患っている被験体および血管疾患、虚血性疾患、炎症性疾患、またはインスリン抵抗性を患っている患者の処置に使用され得る。この処置は、例えば、一酸化窒素合成酵素活性の欠損を軽減し得るか、または例えば、正常レベルを超えての一酸化窒素合成酵素活性の上昇を提供し得る。一酸化窒素合成酵素活性の欠損を患っている患者は葉酸前駆体、葉酸または葉酸誘導体を含む葉酸塩を使用する共処置による恩恵を受けることが示唆される。
【0079】
一酸化窒素は、血圧および血管緊張の調節において重要な生理学的役割を果たす血管内皮細胞により構成的に生産される。一酸化窒素の生物活性の欠損は、冠状動脈疾患、任意の動脈(冠状動脈、頚動脈、大脳動脈または末梢血管動脈を含む)のアテローム性硬化症、虚血再灌流傷害、高血圧、糖尿病、糖尿病性血管症、心臓血管疾患、末梢血管疾患を含む血管機能傷害、あるいは脳卒中のような虚血および/または炎症から派生する神経変性状態の病因と関連し、そのような病因としては、損傷した内皮、臓器および組織への不十分な酸素の流れ、上昇した全身血管抵抗(高血圧)、血管平滑筋の増殖、血管狭窄の進展(狭窄(narrowing))および炎症が挙げられることが示唆される。従って、任意のこれらの状態の処置は、本発明の方法に従って意図される。
【0080】
一酸化窒素合成酵素活性の増強はまた、参考として本明細書中に援用される米国特許番号第6,410,535号に記載されるように上昇したスーパーオキシドレベルの減少、増加したインスリン感受性およびインスリン抵抗性に関連した血管機能障害の減少をもたらすことがまた示唆される。従って、糖尿病(I型またはII型)、高インスリン血症またはインスリン抵抗性の処置は、本発明に従って意図される。インスリン抵抗性に関連する血管機能障害を有する疾患として、インスリン抵抗性により引き起こされるかまたはインスリン抵抗性により悪化される疾患、あるいはインスリン抵抗性により治癒が遅れる疾患(インスリン抵抗性に関する限りでは高血圧、高脂血症、動脈硬化、冠状血管狭窄性狭心症、労作性狭心症、脳血管狭窄性病変、脳血管不全、大脳血管攣縮、末梢循環障害、経皮経管冠状動脈形成(PTCA)または冠状動脈バイパス移植(CABG)に続く冠状動脈狭窄、肥満、インスリン依存性糖尿病、高インスリン血症、脂質代謝異常、冠状動脈硬化心疾患などのような)が挙げられる。これらの疾患を有する患者に投与される場合、BH4はNOSの機能を活性化し、NOの産生を増加させ、そして活性酸素種の産生を抑制することによりこれらの疾患を予防し得るか処置し得、血管内皮細胞の障害を改善することが意図される。
【0081】
本発明に従う組成物の適した用量は、受容者の年齢、健康状態および体重、もしあれば同時処置の種類、処置の頻度および望まれる作用の性質(すなわち、望まれる血漿Phe濃度の減少量)に依存することが理解される。投薬頻度はまた、Pheレベルに対する薬力学的作用にも依存する。この作用が単回用量により24時間持続する場合。しかしながら、最も好ましい投薬は、個々の被験体に合わせられ得、過度の実験なしに当業者により理解され決定可能である。これは代表的に標準的な用量の調整(例えば患者の体重が少ない場合、用量を減少させ)を包含する。
【0082】
BH4の投薬頻度は、薬剤の薬物動態学的パラメーターおよび投与経路に依存する。最適な薬学的処方物は、投与経路および望まれる投薬量に依存して当業者により決定される。例えば参考として本明細書中に援用されるRemington’s Pharmaceutical Sciences、第18版(1990、Mack Publ.Co,Easton PA 18042)PP1435 1712を参照のこと。そのような処方物は、物理的状態、安定性、投与した薬剤のインビボの放出速度およびインビボのクリアランス速度に影響を与え得る。投与経路に依存して、適した用量が体重、体表面積または臓器サイズに従って計算され得る。適した処置用量を決定するために必要な計算のさらなる修正が、過度の実験なしに、特に本明細書中に開示される投薬情報およびアッセイならびに動物またはヒトの臨床試験において観察される薬物動態学的データを鑑みて、当業者により慣習的に行われる。
【0083】
最終投薬レジメンは、担当医により、薬物の作用を変化させる因子(例えば薬物の比活性、患者の損傷の重症度および応答性、患者の年齢、状態、体重、性別および食事、任意の感染の重症度、投与の回数および他の臨床的要因)を考慮して決定される。研究が行われるにつれ、特定の疾患および状態のための適した投薬量レベルおよび処置の延長に関するさらなる情報が明らかになる。
【0084】
(V. 併用療法)
本発明のある方法は、本発明の安定性処方物と1つ以上の他の治療剤とを組み合わせた使用を包含する。
【0085】
そのような併用療法において、本発明の安定性処方物の投与は、第2の治療剤の投与と同時であり得るか、あるいは第2の治療剤の投与に先行するか後に続き得る(例えば、数分〜数時間におよぶ間隔で、両薬剤がオーバーラップする期間で治療効果を及ぼすことができる限り)。従って本発明は、第2の治療剤と使用するための本発明の安定性処方物を意図する。本発明はまた、本発明の安定なテトラヒドロビオプテリン処方物、前駆体処方物、誘導体処方物またはアナログ処方物を用いる投与のための医薬の調製において第2の治療剤の使用を意図する。
【0086】
テトラヒドロビオプテリン療法は、種々の形態のHPAを有する患者において治療結果をもたらすために食事性タンパク質の制限と組み合わせられ得る。例えば、望ましい治療結果(すなわち、血漿Phe濃度の減少および/または血漿Phe濃度の付随的な増加を生じることなくより多量のPhe/タンパク質摂取を許容する能力)を生じるために組み合わせた有効量でBH4組成物および低フェニルアラニン医療用タンパク質組成物を被験体に投与し得る。このプロセスは、BH4組成物および食事性タンパク質の治療組成物を同時に投与する工程を包含し得る。これは、単一の組成物または薬理学的タンパク質処方物を投与することにより達成され得、これらは食事性タンパク質の所要量をすべて含有し、またこのタンパク質処方物内にBH4を含有する。あるいは、食事性タンパク質(サプリメントまたは標準的なタンパク質の食事)はBH4の薬理学的処方物(錠剤、注射または飲料)とほぼ同時に摂取される。
【0087】
他の代替法において、BH4処置は、数分〜数時間におよぶ間隔で食事性タンパク質療法に先行し得るまたは後に続き得る。タンパク質およびBH4組成物が別々に投与される実施形態において、BH4が患者において有利な効果を依然として示すことができるように、有意な期間がそれぞれの送達の時間の間に終わらなかったことを一般的に保障する。そのような場合には、BH4を食事性タンパク質摂取の約2〜6時間(前または後)以内、最も好ましくは約1時間のみ時間を遅らせて投与することが意図される。ある実施形態において、BH4療法は、1日用量のBH4が無期限に患者に投与される継続的な治療であることが意図される。他の状況(例えばより軽度な形態のPKUおよびHPAのみを有する妊娠女性)において、BH4療法は、女性が妊娠している限りおよび/または母乳を与えているに限ってのみ、継続され得る。
【0088】
さらにBH4の送達および食事性タンパク質の調節のみをベースにした治療に加え、本発明の方法はまた、1つ以上のHPAの症状を特異的に標的とする第3の組成物との併用療法を意図する。例えば、HPAにより引き起こされるチロシンの欠乏は、神経伝達物質のドパミンおよびセロトニンの欠乏を生じることが公知である。従って、本発明の背景において、BH4および食事性タンパク質をベースにした方法は、食事中の減少したチロシンの量により生じる障害を治すために、L−ドパ、カルビドパおよび5−ヒドロキシトリプトファンの神経伝達物質の投与とさらに組み合わされ得ることが意図される。
【0089】
さらに、PAH(Christensenら.,Mol.Gent.And Metabol.76:313−318、2002;Christensenら.、Gene Therapy、7:1971−1978、2000)およびフェニルアラニンアンモニアリアーゼ(PAL Liuら.、Arts.Cells.Blood.Subs and Immob.Biotech.30(4)243−257、2002)の両方を用いる遺伝子治療は、当業者により意図されている。そのような遺伝子治療技術は、本発明の治療をベースに組み合わされたBH4/食事性タンパク質制限と組み合わせて使用され得る。さらなる併用療法において、フェニラーゼは、患者において低Phe濃度を打ち消すために注射可能な酵素として提供され得ることが意図される。フェニラーゼの投与がチロシンを生成しない(PAHの投与とは異なる)ので、そのような処置は、そのような患者に必須なアミノ酸であるチロシンを依然として生じる。そのためチロシンを用いる食事性の補充は、BH4療法と組み合わせてフェニラーゼを与えられている患者において望ましくあり得る。
【実施例】
【0090】
(VII.実施例)
以下の実施例は、本発明の好ましい実施形態を証明するために包含される。続く実施例に開示される技術は、本発明の実施において十分に機能することが本発明者らにより発見された技術を示し、従ってその実施のために好ましい様式を構成するとみなされ得ることは当業者により理解されるはずである。しかしながら、当業者は、本開示を鑑みて、多くの変更が開示される特定の実施形態においてなされ得、かつ本発明の精神および範囲から逸脱することなく、同様な結果または類似の結果を依然として得ることを理解すべきである。
【0091】
(実施例1)
(BH4の安定化した結晶化形態の調製)
その全体が参考として本明細書中に援用される国際公開番号第2005/065018号は、水和物または溶媒和物を含むBH4の多形を特徴付けるためのX線およびラーマンスペクトル研究ならびに多形が調製され得る例示的な結晶化条件を記載する。その全体が参考として本明細書中に援用される国際公開番号第2005/049000号は、BH4処置が適している種々の患者集団を記載し、BH4を用いてそのような患者を処置するための方法を記載する。その全体が参考として本明細書中に援用される国際公開番号第2005/049614号は、BH4を合成する方法を記載する。
【0092】
本明細書の全体にわたり引用される参考文献は、例示的な手順の詳細または本明細書の記載に補充的な他の詳細を提供するという範囲で、すべて具体的に参考として本明細書中に援用される。
【0093】
(実施例2)
(テトラヒドロビオプテリンの安定性錠剤処方物)
3つの錠剤処方物を、以下詳細に記載するように表Iに示す成分を混合することにより調製した。
【0094】
【表1】
表Iの各処方物に対して、まず4kgの6R−L−エリスロ−5,6,7,8−テトラヒドロビオプテリンジヒドロクロライド(SAPROPTERIN塩酸塩、Daiichi Suntory Pharma Co.、Ltd.、Japanから入手可能な)をブレンダーに充填し、BH4を10分間、1分あたり25回転(RPM)で混合することにより12キログラムのバッチを調製した。次いで6.91kgのD−マンニトール(PEARLITOL、Roquette America、Inc.、Keokuk、Iowaから入手可能)をブレンダーに加え、この混合物をさらに10分間、25RPMで混合した。次いで260グラムの無水二塩基リン酸カルシウム(Mallinckrodt Baker、Inc.、Phillipsburg、New Jerseyから入手可能な)ならびに(a)処方物Iにおいては、436グラムのヒドロキシプロピルセルロースおよび104グラムをブレンダーに加え、(b)処方物IIにおいては、540グラムのヒドロキシプロピルセルロースをブレンダーに加え;(c)処方物IIIにおいては、540グラムのポリビニルピロリドン(KOLLIDON CL、BASF Corporation、Florham Park、New Jerseyから入手可能)をブレンダーに加え、この混合物をさらに10分間、25RPMで混合した。200グラムのアスコルビン酸および120グラムのリボフラビンをブレンダーに加え、この混合物を3分間、25RPMで混合した。フマル酸ステアリルナトリウム滑沢剤(PRUV、Penwest Pharmaceuticals Co.、Danbury、Connecticutから入手可能)を25メッシュのステンレス鋼ふるいに通してバッグ中に濾取し、次いでブレンダーを、ふるいをかけた9kgのフマル酸ステアリルナトリウムを用いて充填し、生じた混合物を5分間、25RPMで混和した。
【0095】
次いで混和した各処方物の混合物をブレンダーから取り出し、150mg錠剤、300mg錠剤および600mg錠剤の調製のために、各処方物について3つのサンプルを集めた。各処方物について、この錠剤サンプル(150mg、300mgおよび600mg)を錠剤プレス(Jenn−Chiang Mahinery Co.、Ltd.、Taiwan、R.O.C.から入手可能な)に置き、ここでこの錠剤プレスのパラメーターを、4.5〜5.5ミリメートルの範囲の厚さおよび7KPの標的硬度を有する錠剤を提供するために設定した。
【0096】
次いで生じた錠剤を処方物の安定性を決定するために分析した。処方物の安定性を、異なる間隔で目視検査により時間にわたる外観の変化、米国薬局方推奨no.701を利用する処方物の崩壊、そして処方物の成分をアッセイすることにより化学的変化を研究した。安定性試験の結果を以下の表IIにまとめる。
【0097】
【表2】
安定性試験は、錠剤処方物IIIが、BH4の他の処方物より安定であることを示す。各薬学的な調製物は、BH4の送達に有用な処方物である。処方物IIIは、処方物Iおよび処方物IIより良好な安定性を示した。従って、好ましい実施形態の1つにおいて、安定化した錠剤処方物は最適な崩壊剤(例えば、クロスポビドンまたはヒドロキシプロピルセルロースよりもポリビニルピロリドンに類似の崩壊剤)を含有する。好ましい処方物は、処方物IIIである。他の適した錠剤処方物は、少なくとも0.01重量%または少なくとも0.05重量%または少なくとも0.1重量%の濃度で少なくともアスコルビン酸を含有する。
【0098】
(実施例3)
100mgテトラヒドロビオプテリンを含む300mg錠剤は、望ましい初期量のpolymorph Bを使用し、以下の乾燥製錠プロセスを使用して以下の表IIIに示される相対量の他の成分と混合して調製した。他の望ましい量のテトラヒドロビオプテリンを含有する錠剤は、同様な方法で調製され得る。
【0099】
6R−L−エリスロ−5,6,7,8−テトラヒドロビオプテリンおよびD−マンニトールを20メッシュふるい(約850μMより大きいサイズの粒子を濾別するように設計した)を用いて手でふるいにかけ、ブレンダーに置いた。この混合物を10分間、21RPMで混和した。次に、無水二塩基リン酸カルシウムおよびクロスポビドンを、20メッシュふるいを用いて手でふるいにかけ、そしてBH4およびD−マンニトールとともに10分間、21RPMで混和した。アスコルビン酸およびリボフラビンを、20メッシュふるいを用いて手でふるいにかけた後ブレンダーに加え、生じた混合物を10分間、21RPMで混和した。次に、フマル酸ステアリルナトリウムを、40メッシュふるいを用いて手でふるいにかけた後ブレンダーに加え、5分間、21RPMで混和した。次いでこの混和した混合物をバッグ内に注ぎ、錠剤に圧縮する前に均一性について試験した。
【0100】
【表3】
錠剤を、ホイルブリスターパックか1ボトルあたり45錠の量でHDPEボトルのどちらかに充填した。各タイプの充填した錠剤を、2つのバッチに分けた。1つのバッチを室温、25±2℃および60±5%相対湿度で貯蔵した。もう一方のバッチを加速試験条件下(40±2℃および75±5%相対湿度)で貯蔵した。規則的な間隔で、錠剤を貯蔵庫から取り出し、活性な薬学的成分(6R−L−エリスロ−5,6,7,8−テトラヒドロビオプテリン)の保持を試験した。300mg錠剤に関して例示的な結果を、以下の表IV、表V、表VIおよび表VIIに示す。室温または加速試験条件下で6ヶ月後貯蔵後、4つの各バッチは、HPLCアッセイにより少なくとも元の量の99%の6R−L−エリスロ−5,6,7,8−テトラヒドロビオプテリンの保持(1%未満の乾燥における損失)および3分以下の迅速な崩壊を示した。
【0101】
【表4】
【0102】
【表5】
【0103】
【表6】
【0104】
【表7】
【図面の簡単な説明】
【0105】
【図1】図1は、(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶形態Bについての特徴的なX線粉末回折パターンを示す。
【特許請求の範囲】
【請求項1】
polymorph Bと称される(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形の初期量および薬学的に受容可能な賦形剤を含有する安定性錠剤処方物であって、この安定性錠剤処方物が、室温および約60%の湿度で6ヶ月後、(6R)−L−エリスロ−テトラヒドロビオプテリンの該初期量の少なくとも約95%
を保持し、
ここで該結晶多形が塩酸塩として、d値(A)で表される以下の特性ピーク:8.7(vs)、5.63(m)、4.76(m)、4.40(m)、4.00(s)、3.23(s)、3.11(vs)、好ましくは8.7(vs)、6.9(w)、5.90(vw)、5.63(m)、5.07(m)、4.76(m)、4.40(m)、4.15(w)、4.00(s)、3.95(m)、3.52(m)、3.44(w)、3.32(m)、3.23(s)、3.17(w)、3.11(vs)、3.06(w)、2.99(w)、2.96(w)、2.94(m)、2.87(w)、2.84(s)、2.82(m)、2.69(w)、2.59(w)および2.44(w)、を有するX線粉末回折パターンを示す、安定性錠剤処方物。
【請求項2】
前記安定性錠剤処方物が、室温および約60%の湿度で9ヶ月後、(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量の少なくとも約95%を保持する、請求項1に記載の安定性錠剤処方物。
【請求項3】
前記安定性錠剤処方物が、(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量の少なくとも約98%を保持する、請求項1または2に記載の安定性錠剤処方物。
【請求項4】
(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量が、前記処方物の約30重量%〜約40重量%の範囲である、請求項1に記載の安定性錠剤処方物。
【請求項5】
(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量が、前記処方物の約32重量%〜約35重量%の範囲である、請求項1に記載の安定性錠剤処方物。
【請求項6】
(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量が、前記処方物の約33重量%の範囲である、請求項1に記載の安定性錠剤処方物。
【請求項7】
各錠剤中の(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量が、約100mgである、請求項1〜5のいずれかに記載の安定性錠剤処方物。
【請求項8】
各錠剤中の(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量が、約200mgである、請求項1〜5のいずれかに記載の安定性錠剤処方物。
【請求項9】
さらに結合剤を含有する、請求項1〜8のいずれかに記載の安定性錠剤処方物。
【請求項10】
前記結合剤が、無水二塩基リン酸カルシウムである、請求項9に記載の安定性錠剤処方物。
【請求項11】
前記結合剤が、約1重量%〜約5重量%の範囲である、請求項9〜10のいずれかに記載の安定性錠剤処方物。
【請求項12】
前記結合剤が、約1.5重量%〜約3重量%の範囲である、請求項9〜10のいずれかに記載の安定性錠剤処方物。
【請求項13】
テトラヒドロビオプテリンに対する結合剤の重量比が、約1:10〜約1:20の範囲である、請求項9〜12のいずれかに記載の安定性錠剤処方物。
【請求項14】
テトラヒドロビオプテリンに対する結合剤の重量比が、約1:15である、請求項13に記載の安定性錠剤処方物。
【請求項15】
さらに崩壊剤を含有する、請求項1〜14のいずれかに記載の安定性錠剤処方物。
【請求項16】
前記崩壊剤が、クロスポビドンである、請求項15に記載の安定性錠剤処方物。
【請求項17】
前記崩壊剤が、約3重量%〜約10重量%の範囲である、請求項15〜16のいずれかに記載の安定性錠剤処方物。
【請求項18】
前記崩壊剤が、約3重量%〜約5重量%の範囲である、請求項17に記載の安定性錠剤処方物。
【請求項19】
テトラヒドロビオプテリンに対する崩壊剤の重量比が、約1:5〜約1:10の範囲である、請求項1〜18のいずれかに記載の安定性錠剤処方物。
【請求項20】
テトラヒドロビオプテリンに対する崩壊剤の重量比が、約1:7.5である、請求項19に記載の安定性錠剤処方物。
【請求項21】
さらに酸性抗酸化剤を含有する、請求項1〜20のいずれかに記載の安定性錠剤処方物。
【請求項22】
前記酸性抗酸化剤が、アスコルビン酸である、請求項21に記載の安定性錠剤処方物。
【請求項23】
前記酸性抗酸化剤が、約1重量%〜約3重量%の範囲である、請求項21〜22のいずれかに記載の安定性錠剤処方物。
【請求項24】
テトラヒドロビオプテリンに対する酸性抗酸化剤の重量比が、約1:5〜約1:30の範囲である、請求項1〜23のいずれかに記載の安定性錠剤処方物。
【請求項25】
テトラヒドロビオプテリンに対する酸性抗酸化剤の重量比が、約1:20である、請求項24に記載の安定性錠剤処方物。
【請求項26】
さらに滑沢剤を含有する、請求項1〜25のいずれかに記載の安定性錠剤処方物。
【請求項27】
前記滑沢剤が、フマル酸ステアリルである、請求項26に記載の安定性錠剤処方物。
【請求項28】
前記滑沢剤が、約0.1重量%〜約2重量%の範囲である、請求項25〜26のいずれかに記載の安定性錠剤処方物。
【請求項29】
前記滑沢剤が、約0.5重量%〜約1重量%の範囲である、請求項28に記載の安定性錠剤処方物。
【請求項30】
テトラヒドロビオプテリンに対する滑沢剤の重量比が、約1:25〜約1:65の範囲である、請求項1〜29のいずれかに記載の安定性錠剤処方物。
【請求項31】
テトラヒドロビオプテリンに対する滑沢剤の重量比が、約1:45である、請求項30に記載の安定性錠剤処方物。
【請求項32】
さらにビタミンB2(リボフラビン)を含有する、請求項1〜31のいずれかに記載の安定性錠剤処方物。
【請求項33】
さらにビタミンB12を含有する、請求項1〜32のいずれかに記載の安定性錠剤処方物。
【請求項34】
さらに葉酸塩を含有する、請求項1〜33のいずれかに記載の安定性錠剤処方物。
【請求項35】
さらにアルギニンを含有する、請求項1〜34のいずれかに記載の安定性錠剤処方物。
【請求項36】
前記葉酸塩が、葉酸(プテロイルモノグルタミン酸塩)、ジヒドロ葉酸、テトラヒドロ葉酸、5−メチルテトラヒドロ葉酸、5,10−メチレンテトラヒドロ葉酸、5,10−メテニルテトラヒドロ葉酸、5,10−ホルムイミノテトラヒドロ葉酸、5−ホルミルテトラヒドロ葉酸(ロイコボリン)、10−ホルミルテトラヒドロ葉酸、10−メチルテトラヒドロ葉酸、ホリルポリグルタミン酸塩、ジヒドロ葉酸塩、テトラヒドロ葉酸塩、5−ホルミル−(6S)−テトラヒドロ葉酸、5−メチル−(6S)−テトラヒドロ葉酸、5,10−メチレン−(6R)−テトラヒドロ葉酸、5,10−メテニル−(6R)−テトラヒドロ葉酸、10−ホルミル−(6R)−テトラヒドロ葉酸、5−ホルムイミノ−(6S)−テトラヒドロ葉酸または(6S)−テトラヒドロ葉酸、あるいはそれらの薬学的に受容可能な塩である、請求項34に記載の安定性錠剤処方物。
【請求項37】
(6R)−L−エリスロ−テトラヒドロビオプテリンの安定性錠剤処方物であって、該処方物の約32重量%〜約35重量%の範囲で(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形の初期量、約3重量%〜約5重量%の範囲でクロスポビドン、約1.5重量%〜約3重量%の範囲で無水二塩基リン酸カルシウムおよび約0.5重量%〜約1重量%の範囲でフマル酸ステアリルを含有し、
ここで該安定性錠剤処方物は、室温で少なくとも6ヶ月の貯蔵寿命を有し、そして
ここで該結晶多形は、塩酸塩として、d値(A)で表される以下の特性ピーク:8.7(vs)、5.63(m)、4.76(m)、4.40(m)、4.00(s)、3.23(s)、3.11(vs)、好ましくは8.7(vs)、6.9(w)、5.90(vw)、5.63(m)、5.07(m)、4.76(m)、4.40(m)、4.15(w)、4.00(s)、3.95(m)、3.52(m)、3.44(w)、3.32(m)、3.23(s)、3.17(w)、3.11(vs)、3.06(w)、2.99(w)、2.96(w)、2.94(m)、2.87(w)、2.84(s)、2.82(m)、2.69(w)、2.59(w)および2.44(w)、を有するX線粉末回折パターンを示す、(6R)−L−エリスロ−テトラヒドロビオプテリンの安定性錠剤処方物。
【請求項38】
室温で少なくとも9ヶ月の貯蔵寿命を有する、請求項37に記載の安定性錠剤処方物。
【請求項39】
前記(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形の初期量が約33重量%であり、クロスポビドンが約4.5重量%であり、無水二塩基リン酸カルシウムが約2重量%であり、そしてフマル酸ステアリルが約0.75重量%である、請求項37または請求項38に記載の安定性錠剤処方物。
【請求項40】
前記(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形の初期量および1つ以上の薬学的に受容可能な賦形剤を混合する工程、ならびにこの混合物から錠剤を形成する工程を含み、ここで該工程が液体の水を加える工程を包含しない、請求項1〜39のいずれかに記載の安定性錠剤処方物を製造する方法。
【請求項41】
前記1つ以上の薬学的に受容可能な賦形剤が、1つの結合剤を含む、請求項40に記載の方法。
【請求項42】
前記結合剤が、無水二塩基リン酸カルシウムである、請求項41に記載の方法。
【請求項43】
前記1つ以上の薬学的に受容可能な賦形剤が、1つの崩壊剤を含む、請求項1〜42のいずれかに記載の方法。
【請求項44】
前記崩壊剤が、クロスポビドンである、請求項43に記載の方法。
【請求項45】
前記1つ以上の薬学的に受容可能な賦形剤が、1つの酸性抗酸化剤を含む、請求項1〜44のいずれかに記載の方法。
【請求項46】
前記酸性抗酸化剤が、アスコルビン酸である、請求項45に記載の方法。
【請求項47】
前記1つ以上の薬学的に受容可能な賦形剤が、1つの滑沢剤を含む、請求項1〜46のいずれか一項に記載の方法。
【請求項48】
前記滑沢剤が、ステアリン酸ホルミルである、請求項47に記載の方法。
【請求項1】
polymorph Bと称される(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形の初期量および薬学的に受容可能な賦形剤を含有する安定性錠剤処方物であって、この安定性錠剤処方物が、室温および約60%の湿度で6ヶ月後、(6R)−L−エリスロ−テトラヒドロビオプテリンの該初期量の少なくとも約95%
を保持し、
ここで該結晶多形が塩酸塩として、d値(A)で表される以下の特性ピーク:8.7(vs)、5.63(m)、4.76(m)、4.40(m)、4.00(s)、3.23(s)、3.11(vs)、好ましくは8.7(vs)、6.9(w)、5.90(vw)、5.63(m)、5.07(m)、4.76(m)、4.40(m)、4.15(w)、4.00(s)、3.95(m)、3.52(m)、3.44(w)、3.32(m)、3.23(s)、3.17(w)、3.11(vs)、3.06(w)、2.99(w)、2.96(w)、2.94(m)、2.87(w)、2.84(s)、2.82(m)、2.69(w)、2.59(w)および2.44(w)、を有するX線粉末回折パターンを示す、安定性錠剤処方物。
【請求項2】
前記安定性錠剤処方物が、室温および約60%の湿度で9ヶ月後、(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量の少なくとも約95%を保持する、請求項1に記載の安定性錠剤処方物。
【請求項3】
前記安定性錠剤処方物が、(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量の少なくとも約98%を保持する、請求項1または2に記載の安定性錠剤処方物。
【請求項4】
(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量が、前記処方物の約30重量%〜約40重量%の範囲である、請求項1に記載の安定性錠剤処方物。
【請求項5】
(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量が、前記処方物の約32重量%〜約35重量%の範囲である、請求項1に記載の安定性錠剤処方物。
【請求項6】
(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量が、前記処方物の約33重量%の範囲である、請求項1に記載の安定性錠剤処方物。
【請求項7】
各錠剤中の(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量が、約100mgである、請求項1〜5のいずれかに記載の安定性錠剤処方物。
【請求項8】
各錠剤中の(6R)−L−エリスロ−テトラヒドロビオプテリンの前記初期量が、約200mgである、請求項1〜5のいずれかに記載の安定性錠剤処方物。
【請求項9】
さらに結合剤を含有する、請求項1〜8のいずれかに記載の安定性錠剤処方物。
【請求項10】
前記結合剤が、無水二塩基リン酸カルシウムである、請求項9に記載の安定性錠剤処方物。
【請求項11】
前記結合剤が、約1重量%〜約5重量%の範囲である、請求項9〜10のいずれかに記載の安定性錠剤処方物。
【請求項12】
前記結合剤が、約1.5重量%〜約3重量%の範囲である、請求項9〜10のいずれかに記載の安定性錠剤処方物。
【請求項13】
テトラヒドロビオプテリンに対する結合剤の重量比が、約1:10〜約1:20の範囲である、請求項9〜12のいずれかに記載の安定性錠剤処方物。
【請求項14】
テトラヒドロビオプテリンに対する結合剤の重量比が、約1:15である、請求項13に記載の安定性錠剤処方物。
【請求項15】
さらに崩壊剤を含有する、請求項1〜14のいずれかに記載の安定性錠剤処方物。
【請求項16】
前記崩壊剤が、クロスポビドンである、請求項15に記載の安定性錠剤処方物。
【請求項17】
前記崩壊剤が、約3重量%〜約10重量%の範囲である、請求項15〜16のいずれかに記載の安定性錠剤処方物。
【請求項18】
前記崩壊剤が、約3重量%〜約5重量%の範囲である、請求項17に記載の安定性錠剤処方物。
【請求項19】
テトラヒドロビオプテリンに対する崩壊剤の重量比が、約1:5〜約1:10の範囲である、請求項1〜18のいずれかに記載の安定性錠剤処方物。
【請求項20】
テトラヒドロビオプテリンに対する崩壊剤の重量比が、約1:7.5である、請求項19に記載の安定性錠剤処方物。
【請求項21】
さらに酸性抗酸化剤を含有する、請求項1〜20のいずれかに記載の安定性錠剤処方物。
【請求項22】
前記酸性抗酸化剤が、アスコルビン酸である、請求項21に記載の安定性錠剤処方物。
【請求項23】
前記酸性抗酸化剤が、約1重量%〜約3重量%の範囲である、請求項21〜22のいずれかに記載の安定性錠剤処方物。
【請求項24】
テトラヒドロビオプテリンに対する酸性抗酸化剤の重量比が、約1:5〜約1:30の範囲である、請求項1〜23のいずれかに記載の安定性錠剤処方物。
【請求項25】
テトラヒドロビオプテリンに対する酸性抗酸化剤の重量比が、約1:20である、請求項24に記載の安定性錠剤処方物。
【請求項26】
さらに滑沢剤を含有する、請求項1〜25のいずれかに記載の安定性錠剤処方物。
【請求項27】
前記滑沢剤が、フマル酸ステアリルである、請求項26に記載の安定性錠剤処方物。
【請求項28】
前記滑沢剤が、約0.1重量%〜約2重量%の範囲である、請求項25〜26のいずれかに記載の安定性錠剤処方物。
【請求項29】
前記滑沢剤が、約0.5重量%〜約1重量%の範囲である、請求項28に記載の安定性錠剤処方物。
【請求項30】
テトラヒドロビオプテリンに対する滑沢剤の重量比が、約1:25〜約1:65の範囲である、請求項1〜29のいずれかに記載の安定性錠剤処方物。
【請求項31】
テトラヒドロビオプテリンに対する滑沢剤の重量比が、約1:45である、請求項30に記載の安定性錠剤処方物。
【請求項32】
さらにビタミンB2(リボフラビン)を含有する、請求項1〜31のいずれかに記載の安定性錠剤処方物。
【請求項33】
さらにビタミンB12を含有する、請求項1〜32のいずれかに記載の安定性錠剤処方物。
【請求項34】
さらに葉酸塩を含有する、請求項1〜33のいずれかに記載の安定性錠剤処方物。
【請求項35】
さらにアルギニンを含有する、請求項1〜34のいずれかに記載の安定性錠剤処方物。
【請求項36】
前記葉酸塩が、葉酸(プテロイルモノグルタミン酸塩)、ジヒドロ葉酸、テトラヒドロ葉酸、5−メチルテトラヒドロ葉酸、5,10−メチレンテトラヒドロ葉酸、5,10−メテニルテトラヒドロ葉酸、5,10−ホルムイミノテトラヒドロ葉酸、5−ホルミルテトラヒドロ葉酸(ロイコボリン)、10−ホルミルテトラヒドロ葉酸、10−メチルテトラヒドロ葉酸、ホリルポリグルタミン酸塩、ジヒドロ葉酸塩、テトラヒドロ葉酸塩、5−ホルミル−(6S)−テトラヒドロ葉酸、5−メチル−(6S)−テトラヒドロ葉酸、5,10−メチレン−(6R)−テトラヒドロ葉酸、5,10−メテニル−(6R)−テトラヒドロ葉酸、10−ホルミル−(6R)−テトラヒドロ葉酸、5−ホルムイミノ−(6S)−テトラヒドロ葉酸または(6S)−テトラヒドロ葉酸、あるいはそれらの薬学的に受容可能な塩である、請求項34に記載の安定性錠剤処方物。
【請求項37】
(6R)−L−エリスロ−テトラヒドロビオプテリンの安定性錠剤処方物であって、該処方物の約32重量%〜約35重量%の範囲で(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形の初期量、約3重量%〜約5重量%の範囲でクロスポビドン、約1.5重量%〜約3重量%の範囲で無水二塩基リン酸カルシウムおよび約0.5重量%〜約1重量%の範囲でフマル酸ステアリルを含有し、
ここで該安定性錠剤処方物は、室温で少なくとも6ヶ月の貯蔵寿命を有し、そして
ここで該結晶多形は、塩酸塩として、d値(A)で表される以下の特性ピーク:8.7(vs)、5.63(m)、4.76(m)、4.40(m)、4.00(s)、3.23(s)、3.11(vs)、好ましくは8.7(vs)、6.9(w)、5.90(vw)、5.63(m)、5.07(m)、4.76(m)、4.40(m)、4.15(w)、4.00(s)、3.95(m)、3.52(m)、3.44(w)、3.32(m)、3.23(s)、3.17(w)、3.11(vs)、3.06(w)、2.99(w)、2.96(w)、2.94(m)、2.87(w)、2.84(s)、2.82(m)、2.69(w)、2.59(w)および2.44(w)、を有するX線粉末回折パターンを示す、(6R)−L−エリスロ−テトラヒドロビオプテリンの安定性錠剤処方物。
【請求項38】
室温で少なくとも9ヶ月の貯蔵寿命を有する、請求項37に記載の安定性錠剤処方物。
【請求項39】
前記(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形の初期量が約33重量%であり、クロスポビドンが約4.5重量%であり、無水二塩基リン酸カルシウムが約2重量%であり、そしてフマル酸ステアリルが約0.75重量%である、請求項37または請求項38に記載の安定性錠剤処方物。
【請求項40】
前記(6R)−L−エリスロ−テトラヒドロビオプテリンの結晶多形の初期量および1つ以上の薬学的に受容可能な賦形剤を混合する工程、ならびにこの混合物から錠剤を形成する工程を含み、ここで該工程が液体の水を加える工程を包含しない、請求項1〜39のいずれかに記載の安定性錠剤処方物を製造する方法。
【請求項41】
前記1つ以上の薬学的に受容可能な賦形剤が、1つの結合剤を含む、請求項40に記載の方法。
【請求項42】
前記結合剤が、無水二塩基リン酸カルシウムである、請求項41に記載の方法。
【請求項43】
前記1つ以上の薬学的に受容可能な賦形剤が、1つの崩壊剤を含む、請求項1〜42のいずれかに記載の方法。
【請求項44】
前記崩壊剤が、クロスポビドンである、請求項43に記載の方法。
【請求項45】
前記1つ以上の薬学的に受容可能な賦形剤が、1つの酸性抗酸化剤を含む、請求項1〜44のいずれかに記載の方法。
【請求項46】
前記酸性抗酸化剤が、アスコルビン酸である、請求項45に記載の方法。
【請求項47】
前記1つ以上の薬学的に受容可能な賦形剤が、1つの滑沢剤を含む、請求項1〜46のいずれか一項に記載の方法。
【請求項48】
前記滑沢剤が、ステアリン酸ホルミルである、請求項47に記載の方法。
【図1】

【公表番号】特表2008−520574(P2008−520574A)
【公表日】平成20年6月19日(2008.6.19)
【国際特許分類】
【出願番号】特願2007−541419(P2007−541419)
【出願日】平成17年11月16日(2005.11.16)
【国際出願番号】PCT/US2005/041252
【国際公開番号】WO2006/055511
【国際公開日】平成18年5月26日(2006.5.26)
【出願人】(506136483)バイオマリン ファーマシューティカル インコーポレイテッド (18)
【Fターム(参考)】
【公表日】平成20年6月19日(2008.6.19)
【国際特許分類】
【出願日】平成17年11月16日(2005.11.16)
【国際出願番号】PCT/US2005/041252
【国際公開番号】WO2006/055511
【国際公開日】平成18年5月26日(2006.5.26)
【出願人】(506136483)バイオマリン ファーマシューティカル インコーポレイテッド (18)
【Fターム(参考)】
[ Back to top ]