
テトラ置換−5−アザスピロ[2.4]へプタン誘導体の製法およびその光学活性中間体
【課題】キノロンカルボン酸系抗菌薬の製造中間体として有用なテトラ置換−5−アザスピロ[2.4]ヘプタン誘導体の光学活性体の効率的な製法、およびその中間体の提供。
【解決手段】式(VII)

(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)で表わされる化合物を1)エステルの加水分解条件下にて処理し、2)カルボキシ基をアミノ基へ転位反応させ、3)還元条件下に処理し,4)脱保護反応に付すことをにより、(7S)−7−アミノ)−7−メチル−5−アザスピロ[2.4]ヘプタン等を得る。
【解決手段】式(VII)
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)で表わされる化合物を1)エステルの加水分解条件下にて処理し、2)カルボキシ基をアミノ基へ転位反応させ、3)還元条件下に処理し,4)脱保護反応に付すことをにより、(7S)−7−アミノ)−7−メチル−5−アザスピロ[2.4]ヘプタン等を得る。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、キノロンカルボン酸系抗菌薬の製造中間体として有用な、テトラ置換−5−アザスピロ[2.4]ヘプタン誘導体の光学活性体の効率的な製法、およびその光学活性中間体に関する。
【背景技術】
【0002】
7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン−5−イル基は、キノロンカルボン酸系抗菌薬の7位(ピリドベンズオキサジンカルボン酸誘導体では10位に相当)の置換基として優れており、この置換基を有するキノロン化合物は、抗菌活性、体内動態、および安全性に優れ、抗菌薬として有用である(特許文献1)。この7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン−5−イル基は、アミノ基が置換した7位の不斉に由来してこの置換基だけで一対の光学異性体関係となるキノロン化合物を生じさせるが、その一方が他方よりも強い抗菌活性を示し、その他の優れた生理活性をさらに発現させることが明らかとなっている(特許文献1)。
【0003】
この置換基を有するキノロンの製造には7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン化合物、しかも一方の光学活性体からなる化合物を利用するのが簡便である。この化合物の製造方法として下記に示す方法がある(特許文献1)。
【0004】
【化1】
【0005】
(式中、Bocは、第三級ブトキシカルボニル基を示し、R12は、炭素数1から6のアルキル基を意味する。)
【0006】
すなわち、上記化合物(A)の従来の製法は、アセト酢酸エステル化合物のケトン部分におけるストレッカー反応によってアミノ−シアノ化合物を得、この化合物のシアノ基をアミノメチル基へ変換した後にエステル部分(カルボン酸ユニット)と分子内閉環させてピロリドン誘導体に導くことを特徴としている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特願2005−146386号
【発明の概要】
【発明が解決しようとする課題】
【0008】
しかしながらこの方法では、例えば、ラネーニッケル等の金属触媒の存在下でシアノ基をアミノメチル基へ還元する際に、アミノ基が無保護のアミノ−シアノ化合物の場合、レトロストレッカー反応が進行する危惧があり、目的の成績体の収率低下や反応混合物の複雑化等の問題、さらに、精製の必要性等、工業的製法としてはさらに改良が必要であった。
また、光学活性な7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン−5−イル基を構築するための光学活性な7−アミノ−7−メチル−5−アザスピロ[2.4]へプタンの取得は、適宜な中間体でのキラルカラムを用いたHPLC分割法に拠っていた。しかしながら、HPLC分割法では量的な制限があり工業的に不利であることや、さらには不要の異性体が再利用できない等の問題があり、光学活性化合物の製法として十分に満足できる方法とは言い難かった。
【0009】
なお、光学活性な7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン−5−イル基を有するキノロン化合物では、異性体の一方が他方に比して優れた生理活性を示すことが判明したが(特許文献1)、この置換基の7位アミノ基の配位については詳細には明らかとなっておらず、(7R)−体と(7S)−体のどちらが優れた活性を示す異性体なのか具体的には明らかにされてはいなかった。
【0010】
したがって本発明の目的は、優れた生理活性を備えるキノロン化合物を提供できるキノロンカルボン酸系抗菌薬の7位(ピリドベンズオキサジンカルボン酸誘導体では10位に相当)置換基を構築するために有用で、特定の立体配置(7S)を有する、光学活性な(7S)−7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン誘導体の効率的な製造方法、およびそのための中間体化合物を提供することにある。
【課題を解決するための手段】
【0011】
本発明者は鋭意研究した結果、キラルビルディングブロックとして、(3S)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸エステル化合物を出発原料として用いることで、(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸エステル化合物を簡便、且つ、効率的に製造できることを見出した。
この(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸エステル化合物は結晶性に優れることから、7位のアミノ基の配位が(7S)であることがX線結晶構造解析によって容易に確定できた。この化合物の立体が確定できたことによって、この化合物を経由して調製される一連の化合物の配位も決定された。
【0012】
さらに本発明者は、スピロ環状構造の構築に際し、やはり(3S)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸エステル化合物を使用してこの化合物をエキソメチレン化してメチレン基の環付加反応を行うことで容易にスピロ環状構造が構築できることも見出した。
【0013】
さらに、これらの製造工程に基づき得られた(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸エステル誘導体(VII)から、(7S)−7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン誘導体を得る方法も見出した。
【0014】
すなわち、本発明は、式(I)
【0015】
【化2】
【0016】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物に、塩基存在下でメチル化剤を反応させて、式(II)
【0017】
【化3】
【0018】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物に、塩基存在下で式(III)
【0019】
【化4】
【0020】
(式中、R’は、水酸基の保護基を意味し、Xは、ハロゲン原子、または求核置換反応で用いられる脱離基を意味する。)
で表わされる化合物を反応させて、式(IV)
【0021】
【化5】
【0022】
(式中、R1、R、およびR’の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物の水酸基の保護基を脱保護して式(V)
【0023】
【化6】
【0024】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物の水酸基をハロゲン原子、または求核置換反応で用いられる脱離基に変換させて式(VI)
【0025】
【化7】
【0026】
(式中、Yは、ハロゲン原子、または求核置換反応で用いられる脱離基を意味し、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を塩基性条件下で処理することを特徴とする式(VII)
【0027】
【化8】
【0028】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物の製造方法に係るものである。
【0029】
また、本発明は、先の式(II)で表わされる化合物をエキソメチレン化して式(VIII)
【0030】
【化9】
【0031】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物のエキソメチレン基をシクロプロパン化反応に付することを特徴とする式(VII)
【0032】
【化10】
【0033】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物の製造方法に係るものである。
【0034】
また、本発明は、式(VII)
【0035】
【化11】
【0036】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物をエステルの加水分解条件下にて処理して式(IX)
【0037】
【化12】
【0038】
(式中、R1の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物のカルボキシ基をアミノ基へ転位反応させて式(X)
【0039】
【化13】
【0040】
(式中、R”は、水素原子、またはアミノ基の保護基を意味し、R1の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を直接に還元条件下、もしくはカルボニル基をチオカルボニル基へ変換後に還元条件下に処理して式(XI)
【0041】
【化14】
【0042】
(式中、R1およびR”の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を置換基R1の脱保護反応に付すことを特徴とする式(XII)
【0043】
【化15】
【0044】
(式中R”の定義は、先の定義と同一である。)
で表わされる化合物の製造方法に係るものである。
【0045】
さらに、本発明は、以下の化合物に係るものである。
式(II)
【0046】
【化16】
【0047】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0048】
式(IV)
【0049】
【化17】
【0050】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、またはカルボン酸とエステル結合を形成し得る不斉補助基を表わし、R’は、水酸基の保護基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0051】
式(V)
【0052】
【化18】
【0053】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0054】
式(VI)
【0055】
【化19】
【0056】
(式中、Yは、ハロゲン原子、または求核置換反応で用いられる脱離基を表わし、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0057】
式(VII)
【0058】
【化20】
【0059】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0060】
式(VIII)
【0061】
【化21】
【0062】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0063】
式(IX)
【0064】
【化22】
【0065】
(式中、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0066】
式(X)
【0067】
【化23】
【0068】
(式中、R”は、水素原子、またはアミノ基の保護基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0069】
式(XI)
【0070】
【化24】
【0071】
(式中、R”は、水素原子、またはアミノ基の保護基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0072】
式(XII)
【0073】
【化25】
【0074】
(式中、R”は、水素原子、またはアミノ基の保護基を意味する。)
で表わされる化合物。
【発明の効果】
【0075】
本発明の製造方法により、特定の立体配置(7S)が確定している、(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸エステル誘導体(VII)を合成中間体として用いることで、優れた抗菌活性、体内動態、および安全性を備えるキノロンカルボン酸系抗菌薬の7位(または、ピリドベンズオキサジンカルボン酸誘導体の10位に相当)置換基を構築するための製造原料として有用であり、光学活性な7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン誘導体の容易で効率的な製造が可能となった。
また、優れた抗菌活性、体内動態、および安全性を発現するのに重要である、特定の立体配置(7S)が確定した7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン誘導体の容易で効率的な製造およびそのための光学活性中間体化合物を提供することが可能となった。さらに、本発明者は、特願2005−146386号に記載され、本発明に係る7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン誘導体のエナンチオマーのうち、(7S)体のほうが(7R)体より高い抗菌活性を示すキノロンカルボン酸(ピリドベンズオキサジンカルボン酸)誘導体の側鎖であることを明らかにした。
【図面の簡単な説明】
【0076】
【図1】実施例5で得られた化合物のX線構造解析の結果を示す図である。
【発明を実施するための形態】
【0077】
本発明の製造方法の各工程について以下に詳細に説明する。
工程A:第一工程→第五工程[化合物(I)→化合物(VII)]
【0078】
【化26】
【0079】
(式中、R、R1、R2、XおよびYの定義は、先の定義と同一である。)
【0080】
工程1:
【化27】
【0081】
(式中、RおよびR1の定義は、先の定義と同一である。)
【0082】
本工程は、式(I)の化合物(以下、化合物(I)と表わす。また、他の番号の式の化合物も同様に表わす。)に、塩基存在下で、活性メチレン(もしくはメチン)のメチル化に通常使用される適宜なメチル化剤を反応させて、式(II)の化合物を得る工程である。なお化合物(I)は、ジャーナル オブ メディシナルケミストリー,第30巻,10号,1711頁(1987年)に記載された方法に従って得ることができる。
【0083】
式中、Rは、カルボン酸エステルを形成する基であればよい。例えば、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、−COORとして光学活性なエステルを形成する不斉補助基を挙げることができる。
Rで示されるアリール基としては、フェニル基またはナフチル基が挙げられ、これらは、ハロゲン原子、炭素数1から6のアルコキシ基、炭素数1から6のアルキル基、またはニトロ基を置換基として有していてもよい。
Rで示されるアラルキル基としては、特にベンジル構造を有する基がよい。具体的には、ベンジル基、パラニトロベンジル基、パラメトキシベンジル基、トリフェニルメチル基等を挙げることができる。
【0084】
Rで示される炭素数1から6のアルキル基としては、直鎖状または分技鎖状のいずれのアルキル基でもよく、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ノルマルブチル基、第三級ブチル基等を挙げることができる。アルキル基は、さらに1以上の基によって置換されていてもよく、例えば、ハロゲン原子、炭素数1から6のアルコキシ基等で置換されていてもよい。
【0085】
不斉補助基としては−COORとして光学活性なカルボン酸エステルを形成できる基であれば、特に制限はない。具体的には、(−)−メンチル基、(−)−8−フェニルメンチル基、(+)−ネオメンチル基、(S)−パントラクトン−2−イル基、(−)−ボルニル基、1−(R)−フェニルエチル基、1−(S)−フェニルエチル基等であり、いわゆる光学活性アルコール由来の置換基類を挙げることができる。この他には光学活性な鎖状や環状、ポリシクロ構造のアルキル基、光学活性なアラルキル基でもよい。なお、Rは、カルボキシ基の光学活性な保護基ということもできる。
【0086】
R1は、置換基を有していてもよいアラルキル基を意味する。具体的には、ベンジル基、パラニトロベンジル基、ジフェニルメチル基、およびトリフェニルメチル基等の分子内に不斉炭素を有しないアラルキル基、または、1−(R)−フェニルエチル基、1−(S)−フェニルエチル基、1−(R)−(1−ナフチル)エチル基、および1−(S)−(1−ナフチル)エチル基等の基のなかに不斉炭素を有する光学活性なアラルキル基を挙げることができる。好ましいのは、ベンジル基、および光学活性なアラルキル基であり、より具体的には、1−(R)−フェニルエチル基および1−(S)−フェニルエチル基である。
【0087】
RとR1の組み合わせに関して述べる。R1が、光学活性なアラルキル基(具体的には1−(R)−フェニルエチル基、1−(S)−フェニルエチル基等)である場合、Rとしては、炭素数1から6のアルキル基がよく、特に、第三級ブチル基を採用するのが反応成績体の単離精製操作等で簡便であり好ましい。一方、R1が、ベンジル基に代表される不斉炭素を有しないアラルキル基の場合、Rとしては、光学活性なカルボン酸エステルを形成し得る不斉補助基を採用するのが反応成績体の単離精製操作等が簡便となり好ましい。このような置換基Rの好ましい例としては、(−)−メンチル基、(−)−8−フェニルメンチル基、1−(R)−フェニルエチル基等汎用性の高い不斉補助基でよい。
【0088】
本工程は、塩基存在下にて、活性メチレン(もしくはメチン)のメチル化に使用される適宜なメチル化剤を反応させて実施するが、反応に用いることのできるメチル化剤としては、具体的には、メチルハライド、ジ硫酸ジメチル等を挙げることができ、さらに好ましくはヨウ化メチルである。
【0089】
本工程の反応に用いることのできる塩基としては有機または無機のいずれであってもよく、例えば、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、リチウム 2,2,6,6−テトラメチルピペリジン、n−ブチルリチウム、sec−ブチルリチウム、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類;トリエチルアミン、N,N−ジイソプロピルエチルアミン等のアルキルアミン類;4−メチルモルホリン、1,8−ジアザビシクロ[5,4,0]−7−ウンデセン、ピリジン等の飽和または不飽和の含窒素複素環化合物類;ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類等の有機塩基を用いることができる。また、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物類;水素化ナトリウム、水素化カリウム等のアルカリ金属の水素化物類;ナトリウムアミド等のアルカリアミド類;炭酸ナトリウム、炭酸カリウム等アルカリ金属の炭酸塩類;炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属の炭酸水素塩類等の無機塩基を用いることができる。これらの塩基のなかで特に好ましい塩基としては、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類、水素化ナトリウム、水素化カリウム等のアルカリ金属の水素化物類を挙げることができる。
【0090】
本工程の反応は溶媒中で実施すればよいが、反応溶媒としては、テトラヒドロフラン、1,4−ジオキサン、ジエチルエーテル、ジイソプロピルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒;N,N−ジメチルアセトアミド、N,N−ジメチルホルムアミド、N−メチル−2−ピロリドン等のアミド系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;その他、ジメチルスルホキシド等の反応を阻害しないものを用いることができる。また、塩基としてナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類を用いる際には、メタノール、エタノール、第三級ブタノール等のアルコール系溶媒を用いることもできる。また、これらを混合溶媒として使用することも可能である。特に好ましい溶媒としては、テトラヒドロフラン、1,4−ジオキサン、N,N−ジメチルホルムアミド等を挙げることができる。
【0091】
反応温度は塩基の種類や使用する溶媒によって異なるが、−78℃から溶媒の沸点の範囲で行えばよく、好ましくは、−78℃から室温の範囲、もしくは氷冷下から溶媒の沸点の範囲である。
【0092】
メチル化反応によって対掌体関係の異性体混合物が生成するが、対掌体の各々はシリカゲルカラムによって容易に分離できる。先の非特許文献に記載の通り、メチルエステルのメチル化体はシリカゲルカラムでの分離は困難であったが、驚くべきことに、第三級ブチルエステルの場合はメチル化化合物がシリカゲルカラムで容易に分離できることが判明した。なお、目的の配位の対掌体は、シリカゲルカラムによってヘキサンおよび酢酸エチルの混合溶媒を溶出溶媒として使用して分離したときに、後から溶出されるより高極性の対掌体の方である。
【0093】
工程2:
【化28】
【0094】
(式中、R、R1およびR’の定義は、先の定義と同一である。)
【0095】
本工程は、化合物(II)に対して式(III)
【0096】
【化29】
【0097】
(式中、R、R1およびXの定義は、先の定義と同一である。)
で表わされる化合物を反応させて化合物(IV)を得る工程である。
【0098】
R’は、水酸基の保護基であればよいが、通常使用される保護基であればよく、特に制限はない。この様な保護基の例としては、アセチル基、メトキシアセチル基、トリフルオロアセチル基、クロロアセチル基、ピバロイル基、ホルミル基、ベンゾイル基等のアシル基類;第三級ブチル基、ベンジル基、パラメトキシベンジル基、パラニトロベンジル基、トリフェニルメチル基等のアルキル基類またはアラルキル基類;メトキシメチル基、第三級ブトキシメチル基、テトラヒドロピラニル基、2,2,2−トリクロロエトキシメチル基等のエーテル類;トリメチルシリル基、イソプロピルジメチルシリル基、第三級ブチルジメチルシリル基、トリベンジルシリル基、第三級ブチルジフェニルシリル基等の置換シリル基類を挙げることができる。これらの保護基のなかで好ましい保護基としては、塩基存在下の反応系において脱離や分解しない保護基である。好ましくは、アラルキル基、エーテル基、または置換シリル基であり、具体的には置換基を有していてもよいベンジル基、そしてメトキシメチル基、テトラヒドロピラニル基、第三級ブチルジメチルシリル基、第三級ブチルジフェニルシリル基等を挙げることができる。
【0099】
また、Xは、ハロゲン原子またはその他の脱離基を意味するが、ハロゲン原子としては、塩素原子、臭素原子、およびヨウ素原子が好ましく、特に、ヨウ素原子が好ましい。また、Xが、脱離基を意味する場合、いわゆる求核置換反応の脱離基であればよい。例えば、置換スルホニルオキシ基を挙げることができ、具体的には、メタンスルスルホニルオキシ基、トリフルオロベンゼンスルホニル基、トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基等を挙げることができる。特に好ましいXとして、ヨウ素原子を挙げることができる。
【0100】
本工程の反応は塩基存在下に実施するが、反応で使用される塩基としては、有機または無機のいずれであってもよく、例えば、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、リチウム2,2,6,6−テトラメチルピペリジン、n−ブチルリチウム、sec−ブチルリチウム、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類;トリエチルアミン、N,N−ジイソプロピルエチルアミン等のアルキルアミン類;4−メチルモルホリン、1,8−ジアザビシクロ[5,4,0]−7−ウンデセン、ピリジン等の飽和または不飽和の含窒素複素環化合物類;ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類等の有機塩基を用いることができる。また、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物類;水素化ナトリウム、水素化カリウム等のアルカリ金属の水素化物類;ナトリウムアミド等のアルカリアミド類等の無機塩基を用いることができる。これらの塩基のなかで特に好ましい塩基としては、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類を挙げることができる。
【0101】
本工程の反応は溶媒中で実施すればよいが、反応溶媒としては、ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;テトラヒドロフラン、1,4−ジオキサン、ジエチルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒;N,N−ジメチルアセトアミド、N,N−ジメチルホルムアミド、N−メチル−2−ピロリドン等のアミド系溶媒;その他、液体アンモニア、ジメチルスルホキシド等を用いることができる。また、塩基としてナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類を用いる際には、メタノール、エタノール、第三級ブタノール等のアルコール系溶媒を用いることもできる。また、これらを混合溶媒として使用することも可能である。特に好ましい溶媒としては、テトラヒドロフラン、トルエン、N,N−ジメチルホルムアミド等を挙げることができる。
【0102】
反応温度は塩基の種類や使用する溶媒によって異なるが、−78℃から溶媒の沸点の範囲で行えばよく、好ましくは−78℃から室温の範囲である。
【0103】
工程3:
【化30】
【0104】
(式中、R、R1、およびR’の定義は先の定義と同じである。)
【0105】
本工程は、化合物(IV)から水酸基の保護基を除去して化合物(V)を得る工程である。
【0106】
本工程における保護基の除去反応は、通常使用される除去方法を用いることができる。この除去方法(試薬、溶媒、反応条件)は、使用した保護基に応じた脱保護方法であって、さらに、化合物(IV)および化合物(V)の3位エステル基部分(COOR)に影響を与えない方法であれば、いかなる方法も使用できる。
【0107】
ここで、化合物(III)の水酸基の保護基の特に好ましい例であるベンジル基、メトキシメチル基、テトラヒドロピラニル基、第三級ブチルジメチルシリル基、第三級ブチルジフェニルシリル基等を例に脱保護条件を具体的に説明する。
【0108】
ベンジル基等のアラルキル基は、接触還元またはバーチ還元にて脱保護することができる。
メトキシメチル基等のエーテル基は、塩酸、硫酸、硝酸、フッ化水素酸、臭化水素酸、ヨウ化水素酸、トルエンスルホン酸、メタンスルホン酸、トリフルオロ酢酸、またはトリクロロ酢酸等の無機酸あるいは有機酸から適宜選択した酸で処理すれば脱保護することができるが、この場合の酸としては、COORのエステル加水分解を避ける酸を選択する必要がある。
第三級ブチルジメチルシリル基等の置換シリル基は、酸またはフッ素アニオンにより脱保護される。この場合の酸としては、酢酸、塩酸、フッ化水素酸等から当該シリル基の性質に応じた酸を選択することできるが、COORのエステル加水分解を避ける酸を選択する必要がある。また、フッ素アニオンとしては、フッ化テトラブチルアンモニウム等使用すればよい。
【0109】
脱保護反応は、化合物(IV)および化合物(V)が溶解する適宜な溶媒中で行えばよい。反応温度としては、使用する酸や脱保護試薬の種類、濃度、あるいは溶媒等の条件にしたがって設定すればよいが、−30〜100℃までの範囲の適宜な温度を選択すればよい。
【0110】
工程4:
【化31】
【0111】
(式中、R、R1、およびYの定義は、先の定義と同じである。)
本工程は、化合物(V)の水酸基をハロゲン原子、もしくは求核置換反応で使用されるその他の脱離基に変換して化合物(VI)を得る工程である。
【0112】
Yは、ハロゲン原子またはその他の脱離基を意味するが、ハロゲン原子としては、臭素原子およびヨウ素原子が好ましく、特に、ヨウ素原子が好ましい。また、Yが、その他の脱離基を意味する場合、いわゆる求核置換反応の脱離基であればよい。例えば、置換スルホニルオキシ基を挙げることができ、具体的には、トリフルオロメタンスルホニル基、メタンスルスルホニルオキシ基、トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基等を挙げることができる。この中では、特に、ベンゼンスルホニル基が好ましい。すなわち、Yとしては、ヨウ素原子とベンゼンスルホニル基が特に好ましい。
【0113】
本工程である水酸基のハロゲン化工程に関して述べる。アルコール性水酸基のハロゲン化反応は、直接ハロゲン化する方法と、一旦他のハロゲン化アルキルまたは脱離基を経由してハロゲン化する方法があり、いずれも適用することができる。
【0114】
アルコール性水酸基の臭素化反応としては、例えば、臭化水素による方法を挙げることができる。例えば、48%臭化水素酸を用いる方法であり、この場合、エーテル生成の副反応を防ぐ目的から濃硫酸を適量加える。また、臭化水素酸を臭化リチウムまたは臭化カリウムと硫酸から反応系内で発生させる方法も挙げることができる。さらに、三臭化リン、アミドと無機の酸臭化物から得られるビルスマイヤー試薬、そしてトリフェニルホスフィンジブロミド等により変換する方法も挙げることができる。この場合、通常、溶媒としてN,N−ジメチルホルムアミドを用いて反応を実施するのが好ましい。
【0115】
アルコール性水酸基のヨウ素置換反応としては、例えば、ヨウ化水素による方法を挙げることができる。具体的には、ヨウ化水素酸、ヨウ化カリウム−リン酸、または、ヨウ化カリウム−フッ化水素ピリジン溶液によるヨウ素化法を挙げることができる。また、三ヨウ化リンおよび四ヨウ化二リンを用いる方法も挙げることができる。さらに、好ましい方法としては、水酸基をメタンスルホニルオキシ基、トルエンスルホニルオキシ基等に代表される、置換スルホニルオキシ基等の求核置換反応の脱離基へ一旦変換した後、テトラヒドロフラン、アセトン、もしくはN,N−ジメチルホルムアミド等の溶媒中、ヨウ化リチウム、ヨウ化ナトリウム、またはヨウ化カリウムと反応させる方法を挙げることができる。
【0116】
一方、本工程における置換スルホニルオキシ基に具体的に代表される求核置換反応の脱離基の導入反応は、トリエチルアミン、ピリジン、または4−ジメチルピリジン等の有機塩基存在下、置換スルホニルクロリド、または置換スルホン酸無水物を反応させて実施すればよい。
【0117】
反応に使用される溶媒としては反応を阻害しないものであれば制限はないが、ジクロロメタン、1,2−ジクロロエタン等の塩素系溶媒、その他、テトラヒドロフラン、トルエン等を用いればよい。また、塩基としてピリジン等を使用する場合はこれらを溶媒に兼ねさせてもよい。
【0118】
反応温度は、塩基の種類や使用する溶媒により異なるが、通常、−78から100℃の範囲で行えばよく、好ましくは、−30℃から室温の範囲である。
【0119】
工程5:
【化32】
【0120】
(式中、R、R1、およびYの定義は先の定義と同じである。)
【0121】
本工程は、化合物(VI)を塩基存在下で処理して閉環させてスピロ環状構造を構築して化合物(VII)を得る工程である。
【0122】
本工程は、Yが、ハロゲン原子もしくはその他の脱離基を有する化合物(VI)の分子内アルキル化反応により、化合物(VII)を得る工程といえるが、この分子内アルキル化反応は、塩基存在下に実施すればよい。反応に用いられる塩基としては、有機または無機のいずれであってもよく、例えば、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、リチウム2,2,6,6−テトラメチルピペリジン、n−ブチルリチウム、sec−ブチルリチウム、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類;トリエチルアミン、N,N−ジイソプロピルエチルアミン等のアルキルアミン類;4−メチルモルホリン、1,8−ジアザビシクロ[5,4,0]−7−ウンデセン、ピリジン等の飽和または不飽和の含窒素複素環化合物類;ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類等の有機塩基を用いることができる。また、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物類;水素化ナトリウム、水素化カリウム等のアルカリ金属の水素化物類;ナトリウムアミド等のアルカリアミド類等の無機塩基を用いることができる。これらの塩基のなかで特に好ましい塩基としては、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類を挙げることができる。
【0123】
本工程の反応は溶媒中で実施すればよいが、反応溶媒としては、ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;テトラヒドロフラン、1,4−ジオキサン、ジエチルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒;N,N−ジメチルアセトアミド、N,N−ジメチルホルムアミド、N−メチル−2−ピロリドン等のアミド系溶媒;その他、液体アンモニア、ジメチルスルホキシド等を用いることができる。また、塩基としてナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類を用いる際には、メタノール、エタノール、第三級ブタノール等のアルコール系溶媒を用いることもできる。また、これらを混合溶媒として使用することも可能である。特に好ましい溶媒としては、テトラヒドロフラン、トルエン、N,N−ジメチルホルムアミド等を挙げることができる。
【0124】
反応温度は塩基の種類や使用する溶媒によって異なるが、−78℃から溶媒の沸点の範囲で行えばよく、好ましくは−78℃から室温の範囲である。
【0125】
化合物(VII)は、上記の工程3で用いられる化合物(III)の代わりに次式(III’)
【0126】
【化33】
【0127】
(式中、Xの定義は先の定義と同じである。X’は、Xと同一であるか、これとは異なるハロゲン原子またはその他の脱離基から選ばれる基を意味する。)
【0128】
で表わされる化合物を使用すれば、工程3から5の三工程分の反応は、連続、且つ、最小の一工程で行うことができる。化合物(III’)は、化合物(III)R’O基部分がハロゲンまたは脱離基となった化合物である。これを用いる反応は、下式によって示される。
【0129】
【化34】
【0130】
(式中、R、R1、X、およびX’の定義は先の定義と同様である。)
【0131】
この反応は、塩基存在下にて実施されるが、その反応条件は、工程3または5で記載した条件から、適宜、選択して用いればよい。
【0132】
上記の反応に用いることのできる化合物(III’)としては、例えば、1,2−ジブロモエタン、1,2−ジヨードエタン、1−ブロモ−2−クロロエタン、2−クロロエチル メタンスルホネート、2−クロロエチル トルエンスルホネート、2−ヨードエチル メタンスルホネート、2−ヨードエチル ベンゼンスルホネート等を挙げることができる。
【0133】
工程B:化合物(VII)の別途合成法
【化35】
【0134】
工程6:
【化36】
【0135】
(式中、RおよびR1の定義は先の定義と同じである。)
【0136】
本工程は、化合物(II)にエキソメチレン基を導入して、化合物(VIII)を得る工程である。この工程および次工程によって化合物(VII)を得る別の工程が提供される。
【0137】
本工程は、アミドのα位(活性メチレン)にエキソメチレンを導入する工程であるが、このための反応の例として、塩基存在下にて、N,N−ジメチルメチレンアンモニウム ヨージド(エッシェンモーザー塩)を用いる方法、リン酸エステルを導入した後にホルムアルデヒドとのホーナー・エモンズ反応に付する方法、メチルメトキシマグネシウム カーボネートを用いる方法、およびヒドロキシメチル化後に脱水反応に付する方法等を挙げることができる。
【0138】
より具体的には、塩基存在下にてアミドのα位をホルミル化し、これにホルムアルデヒドを作用させて脱カルボニルメチレン化を行う方法である。
【0139】
上記の反応は塩基存在下に実施するが、反応に用いられる塩基としては、例えば、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、リチウム2,2,6,6−テトラメチルピペリジン、n−ブチルリチウム、sec−ブチルリチウム、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類;ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類;等の有機塩基を用いることができる。これらの塩基のなかで特に好ましい塩基としては、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類を挙げることができる。
【0140】
本工程の反応は溶媒中で実施すればよいが、反応溶媒としては、ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;テトラヒドロフラン、1,4−ジオキサン、ジエチルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒;N,N−ジメチルアセトアミド、N,N−ジメチルホルムアミド、N−メチル−2−ピロリドン等のアミド系溶媒等を用いることができる。また、塩基としてナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類を用いる際には、メタノール、エタノール、第三級ブタノール等のアルコール系溶媒を用いることもできる。また、これらを混合溶媒として使用することも可能である。特に好ましい溶媒としては、テトラヒドロフラン、トルエン、N,N−ジメチルホルムアミド等を挙げることができる。
【0141】
反応温度は塩基の種類や使用する溶媒によって異なるが、−78℃から溶媒の沸点の範囲で行えばよく、好ましくは−78℃から室温の範囲である。
【0142】
工程7:
【化37】
【0143】
(式中、RおよびR1の定義は先の定義と同じである。)
【0144】
本工程は、化合物(VIII)のエナミド部分の二重結合エキソメチレン部分にメチレン基を環化付加反応させてシクロプロパン環を構築し、化合物(VII)を得る工程である。
【0145】
本工程を達成する反応の例として、ジアゾメタン−遷移金属触媒、ジヨードメタン−ジエチル亜鉛(シモンズ・スミス試薬)等を用いるカルベン、カルベノイド反応、サルファー イリド(コーリー試薬)を用いたマイケル付加反応を経由する方法等を挙げることができる。
【0146】
ジアゾメタン−遷移金属触媒系によるカルベノイド反応は、具体的には、化合物(VIII)を溶媒に溶解して触媒を添加し、汎用の方法にてあらかじめ調製したジアゾメタンのジエチルエーテル溶液を加えて反応させればよい。
【0147】
この反応で用いることのできる触媒としては、例えば、パラジウム(II)アセテート、パラジウム(II)アセチルアセトネート等を挙げることができる。
【0148】
この反応の反応溶媒としてはノルマルへキサン等の炭化水素系;テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン、ジエチルエーテル等のエーテル系;ジクロロメタン、ジクロロエタン等のハロゲン系;等を用いることができる。また、これらを混合溶媒として使用することも可能である。
反応温度は溶媒によって異なるが、−20℃から0℃の範囲で行えばよい。
【0149】
シモンズ・スミス試薬等を用いるカルベン反応は、具体的には、化合物(VIII)を溶媒に溶解後、炭素原としてはジヨードメタン、クロロヨードメタン等のハロゲン化メチレンを用い、ジエチル亜鉛、亜鉛−銅、サマリウム−塩化水銀(II)、トリメチルアルミニウム等のトリアルキルアルミニウム等の金属試薬存在下にて反応させればよい。
【0150】
この場合、ハロゲン化メチレンとしてはジヨードメタンが好ましく、また、金属試薬で好ましいのは、ジエチル亜鉛、亜鉛−銅である。
【0151】
反応溶媒としては、ノルマルへキサン等の脂肪族炭化水素系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン、ジエチルエーテル等のエーテル系溶媒;ジクロロメタン、ジクロロエタン等のハロゲン系溶媒;等を挙げることができる。また、これらを混合溶媒として使用することもできる。
【0152】
また、本反応は、添加物としてトリフルオロ酢酸を加えて実施してもよい。
反応温度は塩基の種類や使用する溶媒によって異なるが、−78℃から溶媒の沸点の範囲で行えばよい。
【0153】
工程Aによれば光学活性体(II)から立体を保持したまま化合物(VII)へ誘導することができ、高い光学純度をもつ化合物(VII)の合成が可能である。さらに、工程Bは、前者の方法に比較して、非常に簡便、且つ、短工程で目的とする化合物(VII)を得ることができる。いずれの方法においても試薬および反応操作は、工業化等の大量合成に特に不都合なものはない。したがって工程のより短い工程Bが有利である。
さらに、化合物(VII)は結晶性に優れることからこの化合物においてX線結晶構造解析を実施し、メチル基の置換位置である7位の不斉炭素部分の絶対配置が(7S)であると判明した。このことからより優れた生理活性であるキノロンを与える配置が7Sであることが確定した。
【0154】
工程C:アミノ基の導入
【0155】
【化38】
【0156】
化合物(VII)に対してアミノ基を導入する工程について説明する。
【0157】
工程8:
【化39】
【0158】
(式中、RおよびR1の定義は先の定義と同じである。)
【0159】
本工程は、化合物(VII)のエステルを加水分解し、遊離のカルボン酸化合物である化合物(IX)を得る工程である。
【0160】
この工程で使用される加水分解反応は、エステルの加水分解に通常用いられる塩基性条件または酸性条件、あるいは加水素化分解条件で実施される反応であればいかなる方法でもよい。Rが、置換基を有していてもよいアリール基や置換基を有していてもよい炭素数1から6のアルキル基の場合は、塩基性条件下または酸性条件下の加水分解条件にて、Rが、置換基を有していてもよい炭素数7から9のアラルキル基の場合は加水素化分解条件にて実施するのが好ましい。
【0161】
酸性条件での加水分解反応は、酸として通常、塩酸または硫酸を用いるが、ルイス酸、例えば、三塩化ホウ素等も使用することができる。溶媒には、酢酸、ギ酸、およびこれらと水との混合溶媒が適している。この酸性条件での加水分解に適したエステルは、第三級ブチルエステルである。第三級ブチルエステルの加水分解では、酸としてトリフルオロ酢酸、パラトルエンスルホン酸も使用することができる。酸は溶媒兼用で使用して実施してもよいが、ジクロロメタン等を溶媒として用いることもできる。また、ルイス酸とチオールまたはスルフィドの併用による反応も用いることができる。
【0162】
塩基性条件、いわゆるアルカリ加水分解反応は、一般的には、適宜な濃度の水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化バリウム等の水溶液を用いて実施すればよい。反応溶媒としては、好ましくは、メタノール、エタノール、テトラヒドロフラン、エチレングリコール等のアルコール系溶媒、またはこれらと水の混合溶媒を用いればよい。反応温度は、−15から100℃が適当であり、好ましくは0から50℃の範囲である。また、当該加水分解は、アルカリ水溶液および少量の界面活性剤を添加して行ってもよい。このような界面活性剤の例としては、テトラエチルアンモニウムクロリド、テトラブチルアンモニウムブロミド、テトラベンジルアンモニウムブロミド等を挙げることができる。なお、この該加水分解は、反応終了後、塩酸、硫酸、酢酸等の鉱酸、もしくは酸性イオン交換樹脂を用いることで遊離のカルボン酸化合物を得ることができる。
【0163】
また、エステルがベンジルエステル、ベンズヒドリルエステル、1−(R)−フェニルエチルエステル等のベンジル構造を有するアラルキルエステルは、触媒存在下の接触加水素化分解反応により除去され、カルボン酸化合物を得ることができる。この接触加水素化分解反応は、触媒としてパラジウム炭素触媒系を用いるのが好ましく、反応基質をメタノール、エタノール、テトラヒドロフラン等の溶媒に溶解後、常圧(1気圧)から適宜に加圧した水素雰囲気下、室温から50℃の温度範囲にて実施される。また、液体アンモニア中、金属リチウムによる還元でもカルボン酸体が得られる。
【0164】
このようにして合成される化合物(IX)
【0165】
【化40】
【0166】
(式中、R1の定義は先の定義と同一である。)
は、遊離のカルボン酸でもよいが、カルボキシ基の塩に変換してもよい。カルボキシ基の塩としては、例えば、リチウム塩、ナトリウム塩、カリウム塩等のアルカリ金属塩、マグネシウム塩、カルシウム塩等のアルカリ土類金属塩、アンモニウム塩、また、トリエチルアミン塩等、無機塩類、有機塩類のいずれでもよい。また、これらのカルボキシ基の塩は、水和物として存在することもある。
【0167】
工程9:
【化41】
【0168】
(式中、R1の定義は先の定義と同一である。R”は、水素原子またはアミノ基の保護基を意味する。)
【0169】
本工程は、化合物(IX)のカルボキシ基を(置換)アミノ基へ転位変換させて、化合物(X)を得る工程である。
【0170】
本工程で実施されるカルボキシ基の(置換)アミノ基への転換反応は、幾つかの方法が知られているが、カルボン酸を酸アジドへ変換し、イソシアナートヘ熱転位させた後、イソシアナートを加水分解してアミンとするクルチウス転位反応を用いるのが好ましい。
【0171】
カルボン酸の酸アジドへの変換反応は、炭素数1から6のアルキルクロロホルメートを塩基存在下でカルボン酸化合物に加え、生成した活性エステルをナトリウムアジド水溶液と反応させて実施することができる。この反応で使用し得るアルキルクロロホルメートとして、メチルクロロホルメート、エチルクロロホルメート、またはブチルクロロホルメートを用いるのが好ましい。反応に用いる塩基としては、トリエチルアミン、ジイソプロピルエチルアミン等の有機塩基類、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基を挙げることができる。この反応は、例えば、アセトン、ジクロロメタン、テトラヒドロフラン、トルエン、またはそれらの混合溶媒中、−20℃から室温にて実施するのが好ましい。
【0172】
この酸アジドを、アジドあるいはイソシアナートと非反応性の溶媒中で、使用する溶媒の沸点において加熱することでイソシアナートへ転位させる。好ましい非反応性溶媒としては、トルエン、キシレン、ベンゼン、またはこれらの混合物を挙げることができる。
【0173】
上述のカルボン酸から酸アジドを経てイソシアナ−トを直接得る方法としては、適宜な溶媒中、塩基存在下にてジフェニルホスホリルアジド(DPAA)を用いる簡便な方法が好ましい例として挙げることができる。
【0174】
次いで、上述の適宜な方法により生成したイソシアナ−トを酸の存在下、−5℃から室温において、または還流下で加熱しながら加水分解することにより、または、イソシアナートをアルカリの存在下、室温から110℃において加水分解することにより置換アミン化合物を得ることができる。この酸性条件下の加水分解で用いるのみ好適な酸としては、適宜な濃度の塩酸、臭化水素酸、または硫酸水溶液等を挙げることができる。これらの酸による加水分解により、生成物は遊離アミンもしくはその酸付加塩として得られる。また、アルカリ条件下の加水分解において用いる好ましいアルカリは、適宜な濃度の水酸化ナトリウム、水酸化カリウム水溶液等を挙げることができる。
【0175】
また、イソシアナートをアルコール系溶媒中、もしくはトルエン、キシレン、ベンゼン等のイソシアナートと非反応性の溶媒中にアルコールを添加させて反応させることにより、アミノ基がアルコキシカルボニル基で保護されたカルバメート誘導体を直接に得ることができる。この反応は、塩基、例えば、トリエチルアミン、ナトリウムアルコキシド等の存在下でも実施することができる。上記のアルコールとしては、メタノール、エタノール、イソプロパノール、ブタノール、第三級ブタノール、ベンジルアルコール等を挙げることができ、使用するアルコールの種類により相当するメチルカルバメート、エチルカルバメート、イソプロピルカルバメート、ブチルカルバメート、第三級ブチルカルバメート、およびベンジルカルバメートを得ることができる。特に好ましいアルコールとしては、第三級ブタノールを挙げることができる。上記反応において用いられるアルコールの量は、計算上、カルボン酸に対して1倍モル以上必要であるが、通常は、大過剰を使用すればよい。
【0176】
なお、本工程のカルボン酸のアミンへの転換反応の具体例として、実施例においてもクルチウス転位反応を用いる方法を記載するが、この他の転位反応を用いることもできる。例えば、カルボン酸から酸アミド(カルバモイル誘導体)を経てアミンを得るホフマン転位反応を挙げることができる。
【0177】
本工程で得られる化合物(X)
【0178】
【化42】
【0179】
(式中、R1およびR”の定義は先の定義と同一である。)
【0180】
において、R”が水素原子である場合、化合物(X)は、遊離アミン、またはその酸付加塩として存在することができる。酸付加塩の例としては、無機酸塩および有機酸塩を挙げることができる。これらの具体例としては、塩酸塩、硫酸塩、硝酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩等の無機酸塩類、あるいは、メタンスルホン酸塩、ベンゼンスルホン酸、トルエンスルホン酸塩等のスルホン酸塩、酢酸塩、クエン酸塩、マレイン酸塩、フマル酸塩、乳酸塩、シュウ酸塩等のカルボン酸塩等の有機酸塩類を挙げることができる。また、これらの酸付加塩は、水和物としても存在することができる。
【0181】
R”が、アミノ基の保護基の場合、通常用いられ、保護と脱保護が容易であり、次工程以降の反応に影響しないか、もしくは保護基自身が反応しないものであれば、いかなる保護基でもよい。このようなアミノ基の保護基としては一般に使用されている、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、置換基を有していてもよいアシル基、置換基を有していてもよいアラルキル基、および置換シリル基から選ばれるものであればよい。置換基を有していてもよいアルコキシカルボニル基としては、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、2,2,2−トリクロロエトキシカルボニル基等を挙げることができる。置換基を有していてもよいアシル基としては、アセチル基、メトキシアセチル基、トリフルオロアセチル基、クロロアセチル基、ピバロイル基、ホルミル基、ベンゾイル基等を挙げることができる。置換基を有していてもよいアラルキルオキシカルボニル基としては、ベンジルオキシカルボニル基、パラメトキシベンジルオキシカルボニル基、パラニトロベンジルオキシカルボニル基等を挙げることができる。置換シリル基としては、トリメチルシリル基、イソプロピルジメチルシリル基、第三級ブチルジメチルシリル基、トリベンジルシリル基、第三級ブチルジメチルシリル基等を挙げることができる。R”としては、これらのうち、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、および置換基を有していてもよいアシル基が好ましく、より具体的には、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、ベンジルオキシカルボニル基、アセチル基、およびトリフルオアセチル基が好ましく、特に、第三級ブトキシカルボニル基が好ましい。
【0182】
なお、脱保護が必要な場合は、保護基であるR”の性質に応じた脱保護条件を適宜に選択して用いればよい。
【0183】
工程10:
【化43】
【0184】
(式中、R1とR”の定義は先の定義と同一である。)
【0185】
本工程は、化合物(X)を酸アミドの還元反応条件下にて処理して、ピロリジン化合物(XI)を得る工程である。
【0186】
本工程における還元反応は、通常、金属水素化物を用いて実施される。この金属水素化物としては、例えば、水素化リチウムアルミニウム、水素ホウ素ナトリウム−塩化第二スズ等の水素化ホウ素ナトリウムとルイス酸の組み合わせ、ジボラン、ボラン−テトラヒドロフラン錯体等を挙げることができる。この中では、特に、水素化リチウムアルミニウム、ボラン−テトラヒドロフラン錯体が好ましい。
【0187】
反応溶媒としては、非反応性の溶媒を用いればよく、具体的には、ジエチルエーテル、テトラヒドロフラン、ジイソプロピルエーテル、1,4−ジオキサンのエーテル系溶媒が好ましく、特に、テトラヒドロフランを用いるのが好ましい。
【0188】
反応温度は、0℃から使用する溶媒の沸点、いわゆる溶媒が還流する加熱下の範囲で実施されるが、室温から還流条件下にて実施するのが好ましい。
【0189】
上述のような酸アミドの金属水素化物を用いる還元反応において、化合物Xのアミノ基は遊離でも保護基を有していてもよいが、R”が、水素原子である遊離のアミノ基を有する化合物(X)を用いて実施するのが好ましい。R”がアミノ基の保護基である場合は、その保護基が選択した還元条件下にて安定である必要がある。
【0190】
Rが、アミノ基の保護基である場合、その保護基は、前述の工程9にて詳細に記載した保護基と同様であるが、生成物(XI)の単離、精製を容易にするためには、本工程にて、アミドの還元後に保護基を導入する方がよい。
【0191】
また、酸アミドの還元反応として、アミドのカルボニル基を適宜な試薬を用いてチオカルボニル基へ変換してチオアミドを得た後、接触還元等による脱硫反応によっても実施することができる。このような場合、アミドのカルボニル基をチオカルボニル基へ変換する方法としては、ローソン試薬が汎用されており、ベンゼンやトルエン中にて還流して実施される。次いで、脱硫反応は、メタノール、エタノール、テトラヒドロフラン等の溶媒中、水素雰囲気下、ラネーニッケル触媒を用いて実施するのが好ましい。この反応は、エタノール中等のアルコール系溶媒中、水素ガスなしでも実施できる。
【0192】
本工程により得られる化合物(XI)
【0193】
【化44】
【0194】
(式中、R1およびR”は先の定義と同じ定義である。)
【0195】
は、遊離のアミン体(二塩基性体)、またはその酸付加塩として存在することができる。酸付加塩の例としては、無機酸塩および有機酸塩を挙げることができる。これらの具体例としては、塩酸塩、硫酸塩、硝酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩等の無機酸塩類、あるいは、メタンスルホン酸塩、ベンゼンスルホン酸、トルエンスルホン酸塩等のスルホン酸塩、酢酸塩、クエン酸塩、マレイン酸塩、フマル酸塩、乳酸塩、シュウ酸塩等のカルボン酸塩等の有機酸塩類を挙げることができる。また、これらの酸付加塩は、水和物としても存在することができる。
【0196】
工程11:
【化45】
【0197】
(式中、R1およびR”は先の定義と同じ定義である。)
【0198】
本工程は、化合物(XI)の5位のアミノ基(R1)の脱保護反応により、化合物(XII)を得る工程である。
【0199】
本反応における脱保護反応は、保護基であるR”の性質に応じた脱保護条件を適宜に選択して用いればよい。この場合、7位のアミノ基が保護基R”を有する場合は、選択的にR1のみ脱保護されてもよく、また、R1およびR”の双方が同時に脱保護されてもよいが、好ましくは前者である。具体的には、R1が、ベンジル基、ベンズヒドリル基、1−(R)−フェニルエチル基、1−(S)−フェニルエチル基である場合、通常、メタノール、エタノール、イソプロパノール等、化合物(XI)を溶解し得る溶媒中、常圧から加圧の水素雰囲気下、パラジウム炭素系触媒を用いる接触加水素分解反応により実施される。反応温度は、室温から50℃の範囲で行えばよい。水素ガスの替わりに、アルコール系溶媒中、ギ酸アンモニウム等の水素ドナーを用いる方法もある。また、液体アンモニア中、金属ナトリウムを用いる方法(バーチ還元)もある。R1が、トリフェニルメチル基である場合は、上記の脱保護条件に加えて、アセトン中、塩酸を反応させる方法もあるが、この場合、7位のアミノの保護基が塩酸酸性条件下で安定である必要がある。
【0200】
本工程で得られる化合物(XII)
【0201】
【化46】
【0202】
(式中、R”は先の定義と同じ定義である。)
は、遊離のアミン体(二塩基性体)、またはその酸付加塩として存在することができる。酸付加塩の例としては、無機酸塩および有機酸塩を挙げることができる。これらの具体例としては、塩酸塩、硫酸塩、硝酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩等の無機酸塩類、あるいは、メタンスルホン酸塩、ベンゼンスルホン酸、トルエンスルホン酸塩等のスルホン酸塩、酢酸塩、クエン酸塩、マレイン酸塩、フマル酸塩、乳酸塩、シュウ酸塩等のカルボン酸塩等の有機酸塩類を挙げることができる。また、これらの酸付加塩は、水和物としても存在することができる。
【実施例】
【0203】
以下に実施例を挙げて本願発明をさらに具体的に説明するが、これらはいかなる意味においても本願発明を限定的に解釈させるものではない。
【0204】
[参考例1]
5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0205】
【化47】
【0206】
羽撹拌下、5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸(1165g,4.994mol)のジクロロメタン(10L)懸濁液にO−第三級ブチル−N,N’−ジイソプロピルウレア(3020g,15.00mol)を室温で加えた後、内温の上昇と還流の開始を認めた後、氷水浴にて冷却した。反応液を室温まで冷却した後、氷水浴を外して1時間、次いで40℃で加熱しながら3時間撹拌した。次いで、反応液を氷水浴で冷却して1時間撹拌した後、不溶物をろ去し、ろ液を減圧乾固して得られた残留物をシリカゲルカラムクロマトグラフィー(シリカゲル:4kg;溶離液,ヘキサン:酢酸エチル=3:1)にて精製し、標記化合物(3位異性体混合物)925.2g(64%)を淡黄色シロップとして得た。ピロリジンの3位に由来した各ジアステレオマーは容易に分取可能であったが、次工程がエピメリ化を伴う反応であることから分取せず使用した。下記には、別途分取した異性体の各々の1H−NMRスペクトルを示した。
【0207】
低極性異性体:
1H−NMR(400MHz,CDCl3)δ:1.45(9H,s),1.54(3H,d,J=7.08Hz),2.59−2.74(2H,m),2.95−3.03(1H,m),3.14(1H,dd,J=9.77,8.79Hz),3.49(1H,dd,J=9.77,6.35Hz),5.50(1H,q,J=7.1Hz),7.26−7.36(5H,m).
高極性異性体:
1H−NMR(400MHz,CDCl3)δ:1.36(9H,s),1.53(3H,d,J=7.32Hz),2.59−2.75(2H,m),3.02−3.11(1H,m),3.16(1H,dd,J=10.01,5.62Hz),3.51(1H,dd,J=10.01,8.54Hz),5.50(1H,q,J=7.1Hz),7.24−7.36(5H,m).
【0208】
[参考例2]
5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(別途合成法)
5−オキソ−1−[(R)−1−フェニルエチル]ピロリジン−3−カルボン酸(500mg,2.14mmol)のジクロロメタン(2.67mL)溶液に、氷冷下、オキサリルクロライド(224μL,2.57mmol)、およびN,N−ジメチルホルムアミド(1滴)を加え、氷冷下にて15分間、次いで室温にて15分間攪拌した。トルエンを加えて溶媒を減圧乾固する操作を3回繰り返し、朱色油状物質603mgを得た。別の容器に準備した第三級ブタノール(614μL,6.42mmol)のジクロロメタン(2.67mL)溶液に、窒素雰囲気下、氷冷しながら上記の朱色油状物質(603mg)のジクロロメタン(2.67mL)溶液を滴下した。反応液にトリエチルアミン(448μL,3.21mmol)を加えて、室温で30分間攪拌した。水(10mL)を加えて、ジクロロメタン(10mL×2)にて抽出した。有機層を飽和食塩水(25mL)にて洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧濃縮後、トルエンを加えて減圧乾固する操作を3回繰り返した。得られた残留物558mgをショートシリカゲルカラムクロマトグラフィー(酢酸エチル100%にて溶出)で精製し、淡黄色の標記化合物523mg(85%)をジアステレオマー混合物(ジアステレオマー比=2.2:1.0)として得た。
【0209】
1H−NMR(400MHz,CDCl3)δ:1.36(2.81/9H,s),1.45(6.19/9H,s),1.53(0.93/3Hd,J=3.7Hz),1.54(2.06/3H,d,J=3.7Hz),2.59−2.75(2H,m),2.95−3.18(2H,m),3.47−3.53(1H,m),5.50(1H,q,J=7.1Hz),7.25−7.37(5H,m).
【0210】
[実施例1]
(3S)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0211】
【化48】
【0212】
窒素ガス雰囲気下、5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(30.05g,0.104mol)のN,N’−ジメチルホルムアミド(210mL)溶液に、攪拌下、ヨードメタン26.0mL(59.28g,0.418mol)、次いで水素化ナトリウム(55%油性、11.35g,0.260mol)を室温にて加えた。内温が上昇して約50℃に達した時、氷水浴にて30℃まで冷却し、次いで外温17℃の水浴に切り替えて23時間撹拌した。反応液を冷クエン酸水溶液(10%クエン酸1Lと氷500gの混合水)に注ぎ、30分間撹拌した後、酢酸エチル(800mL,500mL)にて抽出した。有機層を合わせ、飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固した。残留物をフラッシュシリカゲルカラムクロマトグラフィーにて精製し(ヘキサン:酢酸エチル=5:1→4:1溶出部)、高極性異性体として標記化合物10.63g(33.7%)を白色固体として得た。また、低極性異性体として(3R)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル14.91g(47.3%)を得た。
【0213】
1H−NMR(400MHz,CDCl3)δ:1.34(12H,s),1.52(3H,d,J=7.10Hz),2.27(1H,d,J=17.0Hz),2.93(1H,d,J=17.0Hz),3.05(1H,d,J=10.1Hz),3.32(1H,d,J=10.1Hz),5.50(1H,q,J=7.1Hz),7.23−7.38(5H,m).
【0214】
[実施例2]
(3S)−4−[2−(第三級ブチルジメチルシリル)ヒドロキシエチル]−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0215】
【化49】
【0216】
(3S)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(30.0g,98.9mmol)、および第三級ブチル(2−ヨードエトキシ)ジメチルシラン(36.8g,129mmol)の無水テトラヒドロフラン(288mL)溶液へ、−4℃にてリチウムビス(トリメチルシリル)アミド(1.0Mテトラヒドロフラン溶液,129mL,129mmol)を滴下し、2℃にて3.5時間攪拌した。反応液に飽和塩化アンモニウム水溶液(300mL)を加え、酢酸エチル(300mL,200mL)にて抽出した。有機層を飽和食塩水(200mL)にて洗浄し、無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固して、標記化合物54.1gを得た。尚、本成績体は、精製せずに次の工程に使用した。
MS(ESI)m/z:363(M−Boc+H)+.
【0217】
[実施例3]
(3S)−4−(2−ヒドロキシエチル)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0218】
【化50】
【0219】
前述のシリル体粗生成物(54.1g,98.9mmol)をテトラヒドロフラン(450mL)に溶解し、氷冷下、フッ化テトラブチルアンモニウム、1.0mol/Lテトラヒドロフラン溶液(148mL,148mmol)を滴下した後、室温にて2時間攪拌した。反応液を濃縮後、酢酸エチル(200mL,100mL)にて抽出した。有機層を10%炭酸水素ナトリウム水溶液(200mL)、クエン酸水溶液(300mL)、および飽和食塩水(100mL)にて洗浄し、無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固した。残留物をシリカゲルカラムクロマトグラフィー(へキサン−酢酸エチル=6:1→4:1→1:1溶出部)にて精製し、標記化合物29.1g(83.9mmol,85%)を無色透明シロップ状物質として得た。
【0220】
1H−NMR(400MHz,CDCl3)δ:1.28(3H,s),1.40(9H,s),1.51−1.53(1H,m),1.53(3H,d,J=7.1Hz),1.78−1.94(2H,m),2.90−3.08(2H,m),3.67−3.75(1H,m),3.80−3.91(1H,m),4.85−4.89(1H,m),5.43−5.53(1H,m),7.27−7.37(5H,m).
MS(ESI)m/z:348(M+H)+.
【0221】
[実施例4]
(3S)−4−[2−(ベンゼンスルホニル)オキシエチル]−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0222】
【化51】
【0223】
(3S)−4−(2−ヒドロキシエチル)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(29.1g,83.9mmol)のジクロロメタン(280mL)溶液に、氷冷下、トリエチルアミン(15.2mL,109mmol)、塩化ベンゼンスルホニル(11.8mL,92.3mmol)、および4−ジメチルアミノピリジン(1.02g,8.39mmol)を加え、混合物を室温にて19時間攪拌した。反応液に飽和塩化アンモニウム水溶液(280mL)を加え、有機層を分離して溶媒を減圧下に留去し、残留物を酢酸エチル(280mL,180mL)に溶解後、先の飽和塩化アンモニウム水溶液にて再び洗浄した。有機層を1mol/L塩酸水溶液(250mL)、飽和重曹水(250mL)、飽和食塩水(200mL)にて洗浄後、無水硫酸ナトリウムで乾燥し、ろ過後、ろ液を減圧乾固し、標記のベンゼンスルホニル体粗生成物(43.7g)を得た。尚、本成績体は精製せずに次の工程に使用した。
MS(ESI)m/z:510(M+Na)+.
【0224】
[実施例5]
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸第三級ブチル
【0225】
【化52】
【0226】
前述のベンゼンスルホニル体粗生成物(43.7g,83.9mmol)の無水テトラヒドロフラン(470mL)溶液に、氷冷下、ナトリウムビス(トリメチルシリル)アミド、1.0mol/Lテトラヒドロフラン溶液(109mL,109mmol)を加え、混合物を室温にて1時間攪拌した。反応液に飽和塩化アンモニウム水溶液(300mL)を加え、酢酸エチル(300mL,200mL)にて抽出し、有機層を飽和食塩水(200mL)にて洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1→2:1にて溶出)で精製して、標記化合物24.6g(89%、2工程)を白色固体として得た。
【0227】
mp:55−57℃.
[α]D25.1=122.1゜(c=0.517,CHCl3).
1H−NMR(400MHz,CDCl3)δ:0.72−0.77(1H,m),0.85−0.90(1H,m),1.04−1.13(2H,m),1.18(3H,s),1.32(9H,s),1.54(3H,d,J=7.1Hz),3.08(1H,d,J=9.8Hz),3.53(1H,d,J=9.8Hz),5.52(1H,q,J=7.1Hz),7.26−7.34(5H,m).
元素分析;C20H27NO3として:
計算値:C,72.92;H,8.26;N,4.25.
実測値:C,72.64;H,8.27;N,4.06.
MS(FAB)m/z:330(M+H)+.
HRMS(FAB)m/z:330.2069(Calcd for C20H28NO3 330.2069).
IR(ATR)ν:3066,2976,2933,2879,1720,1676,1481,1454,1433,1365,1329,1286,1238,1203cm-1.
【0228】
本化合物について7位の配位を決定するべくX線構造解析を実施した。その詳細は以下のとおりである。
【0229】
【表1】
【0230】
データ収集後、直接法で初期位相を決定し、完全行列最小自乗法で位相精密化を行った。精密化の際、非水素原子は非等方性温度因子を適用し、水素原子は計算で位置を決定して座標を固定した。本化合物中には不斉炭素が2個存在するが、そのうち1個の不斉炭素の絶対配置は既知であった。この絶対配置をもとに他方の不斉炭素の絶対配置を決定した。得られた結果を図1に示す。すなわち、標題化合物の7位に関する配位が(S)であることが確定された。そして、この化合物を経由して調製される一連の化合物の配位も決定されることとなった。
【0231】
[実施例6]
(3S)−4−(2−ヨードエチル)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0232】
【化53】
【0233】
(3S)−4−(2−ヒドロキシエチル)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(0.37g,1.07mmol)のジクロロメタン(7.5mL)溶液に氷冷下にてトリエチルアミン(0.22mL,1.6mmol)、および塩化メタンスルホニル(99μL,1.28mmol)を加え、混合物を室温にて2時間半攪拌した。反応液に水(20mL)を加え、クロロホルム(20mL×3)にて抽出した。抽出液を飽和食塩水(20mL)にて洗浄後、無水硫酸ナトリウムで乾燥し、ろ過し、ろ液を減圧乾固した。得られた残留物をアセトン(20mL)に溶解し、ヨウ化ナトリウム(0.32g,2.13mmol)を加え、80℃で17時間半攪拌した。反応系を減圧濃縮し、残留物に水(20mL)を加え、酢酸エチル(20mL×3)にて抽出した。抽出液を10%チオ硫酸ナトリウム水溶液(20mL)、および飽和食塩水(20mL)にて洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固して標記化合物0.47g(96%)を淡黄色油状物として得た。
【0234】
1H−NMR(400MHz,CDCl3)δ:1.20(3H,s),1.43(9H,s),1.50(3H,d,J=7.1Hz),2.00−2.10(1H,m),2.16−2.27(1H,m),2.85(1H,dd,J=9.0,4.9Hz),2.93(1H,d,J=10.0Hz),3.24(1H,d,J=10.0Hz),3.55(2H,dd,J=8.1,6.3Hz),5.48(1H,q,J=7.1Hz),7.25−7.37(5H,m).
MS(ESI)m/z:458(M+H)+.
【0235】
[実施例7]
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸第三級ブチル
【0236】
【化54】
【0237】
(3S)−4−(2−ヨードエチル)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(118mg,0.26mmol)の無水テトラヒドロフラン(2.4mL)溶液に氷冷下、カリウムビス(トリメチルシリル)アミド(0.5Mトルエン溶液,0.62mL,0.31mmol)を加え、混合物を氷冷下にて10分間攪拌した。反応液に飽和塩化アンモニウム水(20mL)を加え、酢酸エチル(20mL×3)にて抽出した。抽出液を10%チオ硫酸ナトリウム水溶液(20mL)および飽和食塩水(20mL)にて洗浄した。有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン−酢酸エチル4:1)で精製して標記化合物91mg(100%)を無色固体として得た。本成績体の1H−NMRデータは、実施例5に記載のデータと一致した。
MS(ESI)m/z:330(M+H)+.
【0238】
[実施例8]
(3S)−3−メチル−4−メチレン−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0239】
【化55】
【0240】
(3S)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(200mg,0.66mmol)を無水テトラヒドロフラン(4mL)に溶解し、0℃にて1.0Mリチウムヘキサメチルジシラジドのテトラヒドロフラン溶液(1.45mL,1.45mmol)を加え、同温で30分撹拌した。次いで、ギ酸エチル(69μL,0.86mmol)を0℃で加えた。同温にて1時間20分撹拌した後、N,N−ジメチルホルムアミド(4mL)、およびパラホルムアルデヒド(120mg)を加え、室温で17時間撹拌した。本反応液に1M塩酸水溶液を0℃で加え反応を停止させた。酢酸エチルで3回抽出し、合わせた有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧濃縮して得られた残留物を中圧液体クロマトグラフィーにて精製し(ヘキサン:酢酸エチル=1:1にて溶出)、標記化合物127mg(61%)を無色固体として得た。
【0241】
1H−NMR(400MHz,CDCl3)δ:1.30(9H,s),1.43(3H,s),1.57(3H,d,J=7.1Hz),3.03(1H,d,J=10.0Hz),3.56(1H,d,J=10.0Hz),5.50(1H,s),5.63(1H,q,J=7.1Hz),6.12(1H,s),7.24−7.35(5H,m).
IR(ATR)ν:1724,1687,1427,1369,1315,1279,1230,1161,1132,700cm-1.
MS(ESI)m/z:316(M+1)+.
HRMS(EI)m/z:315.1842(Calcd for C19H25NO3 315.1834).
【0242】
[実施例9]
(7S)−7−メチル−4−オキソ−5−[(1R)−1−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸第三級ブチル
【0243】
【化56】
【0244】
トリメチルスルホニウムヨージド(401mg,1.82mmol)のジメチルスルホキシド(8mL)溶液に、水素化ナトリウム(60%油状、76.1mg,1.74mmol)、および(3S)−3−メチル−4−メチレン−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(500mg,1.59mmol)のジメチルスルホキシド(24mL)溶液を、順次加え、90℃にて30分間攪拌した。反応液に10%クエン酸水溶液(32mL)、および水(96mL)を室温にて加え、酢酸エチル(200mL×2)にて抽出し、有機層を水(100mL)、および飽和食塩水(100mL)にて洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1→2:1にて溶出)で精製して、標記化合物315mg(60.3%)を無色結晶として得た。本成績体の1H−NMRとIRの各スペクトルデータは、実施例5に記載したデータと一致した。
MS(FAB+)m/z:330(M+H)+.
HRMS(FAB+)m/z:330.2069(Calcd for C20H28NO3 330.2069).
【0245】
[実施例10]
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸
【0246】
【化57】
【0247】
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸第三級ブチル(24.5g,74.4mmol)のジクロロメタン(120mL)溶液に、氷冷下、トリフルオロ酢酸(120mL)を滴下し、2時間撹拌した。反応混合物を減圧乾固し、残留物にトルエン(20mL)を加えて減圧乾固した後、氷冷下、残留物を1mol/l水酸化ナトリウム水溶液(300mL)に溶解した。この水溶液を酢酸エチル(350mL)にて洗浄後、水層に氷冷下濃塩酸(25mL)を加えpH2〜3とし、クロロホルム(300mL×2)にて抽出した。有機層を水(200mL)および飽和食塩水(100mL)にて洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた残留物にトルエン(20mL)を加えて減圧乾固した後、残留物をクロロホルム(20mL)に懸濁し、ヘキサン(200mL)を加え、結晶化させた。析出した固体をヘキサン(100mL)にて洗浄後、減圧乾燥して標記化合物20.48g(定量的)を白色固体として得た。本成績体は精製せずに次工程に使用した。
【0248】
1H−NMR(400MHz,CDCl3)δ:0.78−0.83(1H,m),0.90−0.95(1H,m),1.08−1.18(2H,m),1.24(3H,s),1.55(3H,d,J=7.3Hz),3.11(1H,d,J=10.0Hz),3.55(1H,d,J=10.0Hz),5.52(1H,q,J=7.1Hz),7.28−7.32(5H,m).
MS(ESI)m/z:274(M+H)+.
【0249】
[実施例11]
(7S)−7−アミノ−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン
【0250】
【化58】
【0251】
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸(20.4g,74.4mmol)、およびジフェニルリン酸アジド(17.6mL,81.8mmol)のトルエン(200mL)溶液へ、トリエチルアミン(20.7mL,149mmol)を加え、125℃のオイルバスで1時間加熱攪拌した。反応液を減圧濃縮してイソシアネート体粗生成物を得た。
得られたイソシアネート体粗生成物を1,4−ジオキサン(180mL)に溶解し、水(90mL)、および濃塩酸(90mL)を加えた後、50℃のオイルバスで1時間加熱攪拌した。反応液に水(200mL)を加え、酢酸エチル(200mL)にて洗浄後、水層に氷冷下にて10mol/L水酸化ナトリウム水溶液(170mL)を加えpH9〜10とし、トルエン(200mL×2)にて抽出した。有機層を飽和食塩水(100mL)にて洗浄し、無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧濃縮して標記化合物15.8g(64.7mmol)を淡黄色油状物として得た。尚、本成績体は、精製せずに次の工程に使用した。
【0252】
1H−NMR(400MHz,CDCl3)δ:0.72−0.78(2H,m),0.99−1.10(2H,m),1.08(3H,s),1.53(3H,d,J=7.4Hz),2.82(1H,d,J=9.6Hz),3.27(1H,d,J=9.6Hz),5.56(1H,q,J=7.1Hz),7.14−7.37(5H,m).
【0253】
[実施例12]
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン
【0254】
【化59】
【0255】
前述の(7S)−7−アミノ−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン(15.8g,64.7mmol)をトルエン(82mL)に溶解し、内温が70℃を越えないよう氷冷しながら、ナトリウム水素化ビス(2−メトキシエトキシ)アルミニウム、65%(重量)トルエン溶液(77.6mL,259mmol)のトルエン(6mL)溶液を15分かけて滴下した後に、80℃のオイルバスで10分間加熱攪拌した。反応液を氷冷し、25%(重量)水酸化ナトリウム水溶液(158mL)を滴下して反応を停止させた後、トルエン(135mL)にて抽出した。有機層を飽和食塩水(100mL)にて洗浄後、これにジ−第三級ブチルジカーボネート(15.6g,71.2mmol)を加えた。反応液を室温にて3時間攪拌後、溶媒を減圧留去して得られた残留物をシリカゲルカラムクロマトグラフィー(へキサン:酢酸エチル=8:1→4:1→1:1にて溶出)にて精製し、標記化合物18.0g(73%)を無色透明シロップ状物質として得た。
【0256】
1H−NMR(400MHz,CDCl3)δ:0.37−0.49(2H,m),0.62−0.68(1H,m),0.77−0.82(1H,m),1.20(3H,s),1.32(3H,d,J=6.6Hz),1.44(9H,s),2.46(2H,dd,J=33.2,9.3Hz),2.68(1H,d,J=8.8Hz),3.27(1H,q,J=6.6Hz),3.31−3.34(1H,m),4.71(1H,s),7.19−7.34(5H,m).
MS(ESI)m/z:331(M+H)+.
【0257】
[実施例13]
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−アザスピロ[2.4]ヘプタン
【0258】
【化60】
【0259】
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン(18.0g,54.5mmol)のメタノール(180mL)溶液へ10%パラジウム炭素触媒(52.8%含水,9.00g)を加え、水素ガス雰囲気下、室温にて18時間攪拌後、さらに40℃のオイルバスで5.5時間撹拌した。触媒をろ去後、溶媒を減圧乾固し、標的化合物の粗生成物13.4g(定量的)を白色固体として得た。
【0260】
1H−NMR(400MHz,CDCl3)δ:0.38−0.43(1H,m),0.54−0.61(2H,m),0.74−0.80(1H,m),1.08(3H,s),1.44(9H,s),2.75(1H,d,J=7.6Hz),2.78(1H,d,J=7.1Hz),3.13(1H,d,J=11.5Hz),3.73−3.77(1H,m),4.45(1H,s).
MS(ESI)m/z:227(M+H)+.
【0261】
[実施例14]
7−[(7S)−7−アミノ−7−メチル−5−アザスピロ[2.4]ヘプタン−5−イル]−6−フルオロ−1−[(1R,2S)−2−フルオロシクロプロピル]−8−メトキシ−1,4−ジヒドロ−4−オキソキノリン−3−カルボン酸
【0262】
【化61】
【0263】
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−アザスピロ[2.4]ヘプタン(13.4g,54.5mmol)、6,7−ジフルオロ−1−[(1R,2S)−2−フルオロシクロプロピル]−8−メトキシ−1,4−ジヒドロ−4−オキソキノリン−3−カルボン酸・ジフルオロボラン錯体(17.9g,49.5mmol)、およびトリエチルアミン(8.97mL,64.4mmol)をジメチルスルホキシド(52mL)に溶解し、40℃のオイルバスで17時間加熱攪拌した。反応液を冷水(1000mL)に注ぎ、析出した固体をろ取した。この固体にエタノール:水=5:1混合溶液(180mL)、およびトリエチルアミン(15mL)を加えて1.5時間加熱還流した。反応混合物を減圧乾固して得られた残留物を酢酸エチル(150mL×2)に溶解し、10%クエン酸水溶液(200mL)、水(200mL)、飽和食塩水(100mL)にて洗浄した。有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた残留物をクロロホルム:メタノール=9:1混合溶液(100mL)に溶解し、シリカゲル(10g)を加え1時間撹拌した。シリカゲルをろ去し、クロロホルム:メタノール=9:1混合溶液(50mL×2)にて洗浄し、ろ液を合わせて濃縮乾固した。残留物を氷冷下にて濃塩酸(200mL)に溶解後、室温にて30分間撹拌し、反応液をクロロホルム(400mL×5)にて洗浄した。水層に氷冷下10mol/L水酸化ナトリウム水溶液を加えpH11.8とし、次いで塩酸にてpH7.4に調整後、クロロホルム(1000mL×3)にて抽出した。有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた残留物をエタノールから再結晶精製し、減圧乾燥して標記化合物18.5g(79%)を淡桃色粉末として得た。
本成績体の1H−NMRをはじめとする機器データは、特願2005−146386号に記載されている実施例9の化合物のデータと完全に一致した。すなわち、特願2005−146386号に記載されている7−アミノ−7−メチル−5−アザスピロ[2.4]ヘプタン−5−イル基を有するキノロン誘導体のうち、高活性な化合物である実施例9に記載されたキノロン誘導体の7位の立体は、(7S)と判明した。
【技術分野】
【0001】
本発明は、キノロンカルボン酸系抗菌薬の製造中間体として有用な、テトラ置換−5−アザスピロ[2.4]ヘプタン誘導体の光学活性体の効率的な製法、およびその光学活性中間体に関する。
【背景技術】
【0002】
7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン−5−イル基は、キノロンカルボン酸系抗菌薬の7位(ピリドベンズオキサジンカルボン酸誘導体では10位に相当)の置換基として優れており、この置換基を有するキノロン化合物は、抗菌活性、体内動態、および安全性に優れ、抗菌薬として有用である(特許文献1)。この7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン−5−イル基は、アミノ基が置換した7位の不斉に由来してこの置換基だけで一対の光学異性体関係となるキノロン化合物を生じさせるが、その一方が他方よりも強い抗菌活性を示し、その他の優れた生理活性をさらに発現させることが明らかとなっている(特許文献1)。
【0003】
この置換基を有するキノロンの製造には7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン化合物、しかも一方の光学活性体からなる化合物を利用するのが簡便である。この化合物の製造方法として下記に示す方法がある(特許文献1)。
【0004】
【化1】
【0005】
(式中、Bocは、第三級ブトキシカルボニル基を示し、R12は、炭素数1から6のアルキル基を意味する。)
【0006】
すなわち、上記化合物(A)の従来の製法は、アセト酢酸エステル化合物のケトン部分におけるストレッカー反応によってアミノ−シアノ化合物を得、この化合物のシアノ基をアミノメチル基へ変換した後にエステル部分(カルボン酸ユニット)と分子内閉環させてピロリドン誘導体に導くことを特徴としている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特願2005−146386号
【発明の概要】
【発明が解決しようとする課題】
【0008】
しかしながらこの方法では、例えば、ラネーニッケル等の金属触媒の存在下でシアノ基をアミノメチル基へ還元する際に、アミノ基が無保護のアミノ−シアノ化合物の場合、レトロストレッカー反応が進行する危惧があり、目的の成績体の収率低下や反応混合物の複雑化等の問題、さらに、精製の必要性等、工業的製法としてはさらに改良が必要であった。
また、光学活性な7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン−5−イル基を構築するための光学活性な7−アミノ−7−メチル−5−アザスピロ[2.4]へプタンの取得は、適宜な中間体でのキラルカラムを用いたHPLC分割法に拠っていた。しかしながら、HPLC分割法では量的な制限があり工業的に不利であることや、さらには不要の異性体が再利用できない等の問題があり、光学活性化合物の製法として十分に満足できる方法とは言い難かった。
【0009】
なお、光学活性な7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン−5−イル基を有するキノロン化合物では、異性体の一方が他方に比して優れた生理活性を示すことが判明したが(特許文献1)、この置換基の7位アミノ基の配位については詳細には明らかとなっておらず、(7R)−体と(7S)−体のどちらが優れた活性を示す異性体なのか具体的には明らかにされてはいなかった。
【0010】
したがって本発明の目的は、優れた生理活性を備えるキノロン化合物を提供できるキノロンカルボン酸系抗菌薬の7位(ピリドベンズオキサジンカルボン酸誘導体では10位に相当)置換基を構築するために有用で、特定の立体配置(7S)を有する、光学活性な(7S)−7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン誘導体の効率的な製造方法、およびそのための中間体化合物を提供することにある。
【課題を解決するための手段】
【0011】
本発明者は鋭意研究した結果、キラルビルディングブロックとして、(3S)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸エステル化合物を出発原料として用いることで、(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸エステル化合物を簡便、且つ、効率的に製造できることを見出した。
この(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸エステル化合物は結晶性に優れることから、7位のアミノ基の配位が(7S)であることがX線結晶構造解析によって容易に確定できた。この化合物の立体が確定できたことによって、この化合物を経由して調製される一連の化合物の配位も決定された。
【0012】
さらに本発明者は、スピロ環状構造の構築に際し、やはり(3S)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸エステル化合物を使用してこの化合物をエキソメチレン化してメチレン基の環付加反応を行うことで容易にスピロ環状構造が構築できることも見出した。
【0013】
さらに、これらの製造工程に基づき得られた(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸エステル誘導体(VII)から、(7S)−7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン誘導体を得る方法も見出した。
【0014】
すなわち、本発明は、式(I)
【0015】
【化2】
【0016】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物に、塩基存在下でメチル化剤を反応させて、式(II)
【0017】
【化3】
【0018】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物に、塩基存在下で式(III)
【0019】
【化4】
【0020】
(式中、R’は、水酸基の保護基を意味し、Xは、ハロゲン原子、または求核置換反応で用いられる脱離基を意味する。)
で表わされる化合物を反応させて、式(IV)
【0021】
【化5】
【0022】
(式中、R1、R、およびR’の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物の水酸基の保護基を脱保護して式(V)
【0023】
【化6】
【0024】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物の水酸基をハロゲン原子、または求核置換反応で用いられる脱離基に変換させて式(VI)
【0025】
【化7】
【0026】
(式中、Yは、ハロゲン原子、または求核置換反応で用いられる脱離基を意味し、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を塩基性条件下で処理することを特徴とする式(VII)
【0027】
【化8】
【0028】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物の製造方法に係るものである。
【0029】
また、本発明は、先の式(II)で表わされる化合物をエキソメチレン化して式(VIII)
【0030】
【化9】
【0031】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物のエキソメチレン基をシクロプロパン化反応に付することを特徴とする式(VII)
【0032】
【化10】
【0033】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物の製造方法に係るものである。
【0034】
また、本発明は、式(VII)
【0035】
【化11】
【0036】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物をエステルの加水分解条件下にて処理して式(IX)
【0037】
【化12】
【0038】
(式中、R1の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物のカルボキシ基をアミノ基へ転位反応させて式(X)
【0039】
【化13】
【0040】
(式中、R”は、水素原子、またはアミノ基の保護基を意味し、R1の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を直接に還元条件下、もしくはカルボニル基をチオカルボニル基へ変換後に還元条件下に処理して式(XI)
【0041】
【化14】
【0042】
(式中、R1およびR”の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を置換基R1の脱保護反応に付すことを特徴とする式(XII)
【0043】
【化15】
【0044】
(式中R”の定義は、先の定義と同一である。)
で表わされる化合物の製造方法に係るものである。
【0045】
さらに、本発明は、以下の化合物に係るものである。
式(II)
【0046】
【化16】
【0047】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0048】
式(IV)
【0049】
【化17】
【0050】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、またはカルボン酸とエステル結合を形成し得る不斉補助基を表わし、R’は、水酸基の保護基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0051】
式(V)
【0052】
【化18】
【0053】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0054】
式(VI)
【0055】
【化19】
【0056】
(式中、Yは、ハロゲン原子、または求核置換反応で用いられる脱離基を表わし、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0057】
式(VII)
【0058】
【化20】
【0059】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0060】
式(VIII)
【0061】
【化21】
【0062】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0063】
式(IX)
【0064】
【化22】
【0065】
(式中、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0066】
式(X)
【0067】
【化23】
【0068】
(式中、R”は、水素原子、またはアミノ基の保護基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0069】
式(XI)
【0070】
【化24】
【0071】
(式中、R”は、水素原子、またはアミノ基の保護基を表わし、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【0072】
式(XII)
【0073】
【化25】
【0074】
(式中、R”は、水素原子、またはアミノ基の保護基を意味する。)
で表わされる化合物。
【発明の効果】
【0075】
本発明の製造方法により、特定の立体配置(7S)が確定している、(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸エステル誘導体(VII)を合成中間体として用いることで、優れた抗菌活性、体内動態、および安全性を備えるキノロンカルボン酸系抗菌薬の7位(または、ピリドベンズオキサジンカルボン酸誘導体の10位に相当)置換基を構築するための製造原料として有用であり、光学活性な7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン誘導体の容易で効率的な製造が可能となった。
また、優れた抗菌活性、体内動態、および安全性を発現するのに重要である、特定の立体配置(7S)が確定した7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン誘導体の容易で効率的な製造およびそのための光学活性中間体化合物を提供することが可能となった。さらに、本発明者は、特願2005−146386号に記載され、本発明に係る7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン誘導体のエナンチオマーのうち、(7S)体のほうが(7R)体より高い抗菌活性を示すキノロンカルボン酸(ピリドベンズオキサジンカルボン酸)誘導体の側鎖であることを明らかにした。
【図面の簡単な説明】
【0076】
【図1】実施例5で得られた化合物のX線構造解析の結果を示す図である。
【発明を実施するための形態】
【0077】
本発明の製造方法の各工程について以下に詳細に説明する。
工程A:第一工程→第五工程[化合物(I)→化合物(VII)]
【0078】
【化26】
【0079】
(式中、R、R1、R2、XおよびYの定義は、先の定義と同一である。)
【0080】
工程1:
【化27】
【0081】
(式中、RおよびR1の定義は、先の定義と同一である。)
【0082】
本工程は、式(I)の化合物(以下、化合物(I)と表わす。また、他の番号の式の化合物も同様に表わす。)に、塩基存在下で、活性メチレン(もしくはメチン)のメチル化に通常使用される適宜なメチル化剤を反応させて、式(II)の化合物を得る工程である。なお化合物(I)は、ジャーナル オブ メディシナルケミストリー,第30巻,10号,1711頁(1987年)に記載された方法に従って得ることができる。
【0083】
式中、Rは、カルボン酸エステルを形成する基であればよい。例えば、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、−COORとして光学活性なエステルを形成する不斉補助基を挙げることができる。
Rで示されるアリール基としては、フェニル基またはナフチル基が挙げられ、これらは、ハロゲン原子、炭素数1から6のアルコキシ基、炭素数1から6のアルキル基、またはニトロ基を置換基として有していてもよい。
Rで示されるアラルキル基としては、特にベンジル構造を有する基がよい。具体的には、ベンジル基、パラニトロベンジル基、パラメトキシベンジル基、トリフェニルメチル基等を挙げることができる。
【0084】
Rで示される炭素数1から6のアルキル基としては、直鎖状または分技鎖状のいずれのアルキル基でもよく、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ノルマルブチル基、第三級ブチル基等を挙げることができる。アルキル基は、さらに1以上の基によって置換されていてもよく、例えば、ハロゲン原子、炭素数1から6のアルコキシ基等で置換されていてもよい。
【0085】
不斉補助基としては−COORとして光学活性なカルボン酸エステルを形成できる基であれば、特に制限はない。具体的には、(−)−メンチル基、(−)−8−フェニルメンチル基、(+)−ネオメンチル基、(S)−パントラクトン−2−イル基、(−)−ボルニル基、1−(R)−フェニルエチル基、1−(S)−フェニルエチル基等であり、いわゆる光学活性アルコール由来の置換基類を挙げることができる。この他には光学活性な鎖状や環状、ポリシクロ構造のアルキル基、光学活性なアラルキル基でもよい。なお、Rは、カルボキシ基の光学活性な保護基ということもできる。
【0086】
R1は、置換基を有していてもよいアラルキル基を意味する。具体的には、ベンジル基、パラニトロベンジル基、ジフェニルメチル基、およびトリフェニルメチル基等の分子内に不斉炭素を有しないアラルキル基、または、1−(R)−フェニルエチル基、1−(S)−フェニルエチル基、1−(R)−(1−ナフチル)エチル基、および1−(S)−(1−ナフチル)エチル基等の基のなかに不斉炭素を有する光学活性なアラルキル基を挙げることができる。好ましいのは、ベンジル基、および光学活性なアラルキル基であり、より具体的には、1−(R)−フェニルエチル基および1−(S)−フェニルエチル基である。
【0087】
RとR1の組み合わせに関して述べる。R1が、光学活性なアラルキル基(具体的には1−(R)−フェニルエチル基、1−(S)−フェニルエチル基等)である場合、Rとしては、炭素数1から6のアルキル基がよく、特に、第三級ブチル基を採用するのが反応成績体の単離精製操作等で簡便であり好ましい。一方、R1が、ベンジル基に代表される不斉炭素を有しないアラルキル基の場合、Rとしては、光学活性なカルボン酸エステルを形成し得る不斉補助基を採用するのが反応成績体の単離精製操作等が簡便となり好ましい。このような置換基Rの好ましい例としては、(−)−メンチル基、(−)−8−フェニルメンチル基、1−(R)−フェニルエチル基等汎用性の高い不斉補助基でよい。
【0088】
本工程は、塩基存在下にて、活性メチレン(もしくはメチン)のメチル化に使用される適宜なメチル化剤を反応させて実施するが、反応に用いることのできるメチル化剤としては、具体的には、メチルハライド、ジ硫酸ジメチル等を挙げることができ、さらに好ましくはヨウ化メチルである。
【0089】
本工程の反応に用いることのできる塩基としては有機または無機のいずれであってもよく、例えば、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、リチウム 2,2,6,6−テトラメチルピペリジン、n−ブチルリチウム、sec−ブチルリチウム、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類;トリエチルアミン、N,N−ジイソプロピルエチルアミン等のアルキルアミン類;4−メチルモルホリン、1,8−ジアザビシクロ[5,4,0]−7−ウンデセン、ピリジン等の飽和または不飽和の含窒素複素環化合物類;ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類等の有機塩基を用いることができる。また、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物類;水素化ナトリウム、水素化カリウム等のアルカリ金属の水素化物類;ナトリウムアミド等のアルカリアミド類;炭酸ナトリウム、炭酸カリウム等アルカリ金属の炭酸塩類;炭酸水素ナトリウム、炭酸水素カリウム等のアルカリ金属の炭酸水素塩類等の無機塩基を用いることができる。これらの塩基のなかで特に好ましい塩基としては、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類、水素化ナトリウム、水素化カリウム等のアルカリ金属の水素化物類を挙げることができる。
【0090】
本工程の反応は溶媒中で実施すればよいが、反応溶媒としては、テトラヒドロフラン、1,4−ジオキサン、ジエチルエーテル、ジイソプロピルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒;N,N−ジメチルアセトアミド、N,N−ジメチルホルムアミド、N−メチル−2−ピロリドン等のアミド系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;その他、ジメチルスルホキシド等の反応を阻害しないものを用いることができる。また、塩基としてナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類を用いる際には、メタノール、エタノール、第三級ブタノール等のアルコール系溶媒を用いることもできる。また、これらを混合溶媒として使用することも可能である。特に好ましい溶媒としては、テトラヒドロフラン、1,4−ジオキサン、N,N−ジメチルホルムアミド等を挙げることができる。
【0091】
反応温度は塩基の種類や使用する溶媒によって異なるが、−78℃から溶媒の沸点の範囲で行えばよく、好ましくは、−78℃から室温の範囲、もしくは氷冷下から溶媒の沸点の範囲である。
【0092】
メチル化反応によって対掌体関係の異性体混合物が生成するが、対掌体の各々はシリカゲルカラムによって容易に分離できる。先の非特許文献に記載の通り、メチルエステルのメチル化体はシリカゲルカラムでの分離は困難であったが、驚くべきことに、第三級ブチルエステルの場合はメチル化化合物がシリカゲルカラムで容易に分離できることが判明した。なお、目的の配位の対掌体は、シリカゲルカラムによってヘキサンおよび酢酸エチルの混合溶媒を溶出溶媒として使用して分離したときに、後から溶出されるより高極性の対掌体の方である。
【0093】
工程2:
【化28】
【0094】
(式中、R、R1およびR’の定義は、先の定義と同一である。)
【0095】
本工程は、化合物(II)に対して式(III)
【0096】
【化29】
【0097】
(式中、R、R1およびXの定義は、先の定義と同一である。)
で表わされる化合物を反応させて化合物(IV)を得る工程である。
【0098】
R’は、水酸基の保護基であればよいが、通常使用される保護基であればよく、特に制限はない。この様な保護基の例としては、アセチル基、メトキシアセチル基、トリフルオロアセチル基、クロロアセチル基、ピバロイル基、ホルミル基、ベンゾイル基等のアシル基類;第三級ブチル基、ベンジル基、パラメトキシベンジル基、パラニトロベンジル基、トリフェニルメチル基等のアルキル基類またはアラルキル基類;メトキシメチル基、第三級ブトキシメチル基、テトラヒドロピラニル基、2,2,2−トリクロロエトキシメチル基等のエーテル類;トリメチルシリル基、イソプロピルジメチルシリル基、第三級ブチルジメチルシリル基、トリベンジルシリル基、第三級ブチルジフェニルシリル基等の置換シリル基類を挙げることができる。これらの保護基のなかで好ましい保護基としては、塩基存在下の反応系において脱離や分解しない保護基である。好ましくは、アラルキル基、エーテル基、または置換シリル基であり、具体的には置換基を有していてもよいベンジル基、そしてメトキシメチル基、テトラヒドロピラニル基、第三級ブチルジメチルシリル基、第三級ブチルジフェニルシリル基等を挙げることができる。
【0099】
また、Xは、ハロゲン原子またはその他の脱離基を意味するが、ハロゲン原子としては、塩素原子、臭素原子、およびヨウ素原子が好ましく、特に、ヨウ素原子が好ましい。また、Xが、脱離基を意味する場合、いわゆる求核置換反応の脱離基であればよい。例えば、置換スルホニルオキシ基を挙げることができ、具体的には、メタンスルスルホニルオキシ基、トリフルオロベンゼンスルホニル基、トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基等を挙げることができる。特に好ましいXとして、ヨウ素原子を挙げることができる。
【0100】
本工程の反応は塩基存在下に実施するが、反応で使用される塩基としては、有機または無機のいずれであってもよく、例えば、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、リチウム2,2,6,6−テトラメチルピペリジン、n−ブチルリチウム、sec−ブチルリチウム、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類;トリエチルアミン、N,N−ジイソプロピルエチルアミン等のアルキルアミン類;4−メチルモルホリン、1,8−ジアザビシクロ[5,4,0]−7−ウンデセン、ピリジン等の飽和または不飽和の含窒素複素環化合物類;ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類等の有機塩基を用いることができる。また、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物類;水素化ナトリウム、水素化カリウム等のアルカリ金属の水素化物類;ナトリウムアミド等のアルカリアミド類等の無機塩基を用いることができる。これらの塩基のなかで特に好ましい塩基としては、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類を挙げることができる。
【0101】
本工程の反応は溶媒中で実施すればよいが、反応溶媒としては、ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;テトラヒドロフラン、1,4−ジオキサン、ジエチルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒;N,N−ジメチルアセトアミド、N,N−ジメチルホルムアミド、N−メチル−2−ピロリドン等のアミド系溶媒;その他、液体アンモニア、ジメチルスルホキシド等を用いることができる。また、塩基としてナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類を用いる際には、メタノール、エタノール、第三級ブタノール等のアルコール系溶媒を用いることもできる。また、これらを混合溶媒として使用することも可能である。特に好ましい溶媒としては、テトラヒドロフラン、トルエン、N,N−ジメチルホルムアミド等を挙げることができる。
【0102】
反応温度は塩基の種類や使用する溶媒によって異なるが、−78℃から溶媒の沸点の範囲で行えばよく、好ましくは−78℃から室温の範囲である。
【0103】
工程3:
【化30】
【0104】
(式中、R、R1、およびR’の定義は先の定義と同じである。)
【0105】
本工程は、化合物(IV)から水酸基の保護基を除去して化合物(V)を得る工程である。
【0106】
本工程における保護基の除去反応は、通常使用される除去方法を用いることができる。この除去方法(試薬、溶媒、反応条件)は、使用した保護基に応じた脱保護方法であって、さらに、化合物(IV)および化合物(V)の3位エステル基部分(COOR)に影響を与えない方法であれば、いかなる方法も使用できる。
【0107】
ここで、化合物(III)の水酸基の保護基の特に好ましい例であるベンジル基、メトキシメチル基、テトラヒドロピラニル基、第三級ブチルジメチルシリル基、第三級ブチルジフェニルシリル基等を例に脱保護条件を具体的に説明する。
【0108】
ベンジル基等のアラルキル基は、接触還元またはバーチ還元にて脱保護することができる。
メトキシメチル基等のエーテル基は、塩酸、硫酸、硝酸、フッ化水素酸、臭化水素酸、ヨウ化水素酸、トルエンスルホン酸、メタンスルホン酸、トリフルオロ酢酸、またはトリクロロ酢酸等の無機酸あるいは有機酸から適宜選択した酸で処理すれば脱保護することができるが、この場合の酸としては、COORのエステル加水分解を避ける酸を選択する必要がある。
第三級ブチルジメチルシリル基等の置換シリル基は、酸またはフッ素アニオンにより脱保護される。この場合の酸としては、酢酸、塩酸、フッ化水素酸等から当該シリル基の性質に応じた酸を選択することできるが、COORのエステル加水分解を避ける酸を選択する必要がある。また、フッ素アニオンとしては、フッ化テトラブチルアンモニウム等使用すればよい。
【0109】
脱保護反応は、化合物(IV)および化合物(V)が溶解する適宜な溶媒中で行えばよい。反応温度としては、使用する酸や脱保護試薬の種類、濃度、あるいは溶媒等の条件にしたがって設定すればよいが、−30〜100℃までの範囲の適宜な温度を選択すればよい。
【0110】
工程4:
【化31】
【0111】
(式中、R、R1、およびYの定義は、先の定義と同じである。)
本工程は、化合物(V)の水酸基をハロゲン原子、もしくは求核置換反応で使用されるその他の脱離基に変換して化合物(VI)を得る工程である。
【0112】
Yは、ハロゲン原子またはその他の脱離基を意味するが、ハロゲン原子としては、臭素原子およびヨウ素原子が好ましく、特に、ヨウ素原子が好ましい。また、Yが、その他の脱離基を意味する場合、いわゆる求核置換反応の脱離基であればよい。例えば、置換スルホニルオキシ基を挙げることができ、具体的には、トリフルオロメタンスルホニル基、メタンスルスルホニルオキシ基、トルエンスルホニルオキシ基、ベンゼンスルホニルオキシ基等を挙げることができる。この中では、特に、ベンゼンスルホニル基が好ましい。すなわち、Yとしては、ヨウ素原子とベンゼンスルホニル基が特に好ましい。
【0113】
本工程である水酸基のハロゲン化工程に関して述べる。アルコール性水酸基のハロゲン化反応は、直接ハロゲン化する方法と、一旦他のハロゲン化アルキルまたは脱離基を経由してハロゲン化する方法があり、いずれも適用することができる。
【0114】
アルコール性水酸基の臭素化反応としては、例えば、臭化水素による方法を挙げることができる。例えば、48%臭化水素酸を用いる方法であり、この場合、エーテル生成の副反応を防ぐ目的から濃硫酸を適量加える。また、臭化水素酸を臭化リチウムまたは臭化カリウムと硫酸から反応系内で発生させる方法も挙げることができる。さらに、三臭化リン、アミドと無機の酸臭化物から得られるビルスマイヤー試薬、そしてトリフェニルホスフィンジブロミド等により変換する方法も挙げることができる。この場合、通常、溶媒としてN,N−ジメチルホルムアミドを用いて反応を実施するのが好ましい。
【0115】
アルコール性水酸基のヨウ素置換反応としては、例えば、ヨウ化水素による方法を挙げることができる。具体的には、ヨウ化水素酸、ヨウ化カリウム−リン酸、または、ヨウ化カリウム−フッ化水素ピリジン溶液によるヨウ素化法を挙げることができる。また、三ヨウ化リンおよび四ヨウ化二リンを用いる方法も挙げることができる。さらに、好ましい方法としては、水酸基をメタンスルホニルオキシ基、トルエンスルホニルオキシ基等に代表される、置換スルホニルオキシ基等の求核置換反応の脱離基へ一旦変換した後、テトラヒドロフラン、アセトン、もしくはN,N−ジメチルホルムアミド等の溶媒中、ヨウ化リチウム、ヨウ化ナトリウム、またはヨウ化カリウムと反応させる方法を挙げることができる。
【0116】
一方、本工程における置換スルホニルオキシ基に具体的に代表される求核置換反応の脱離基の導入反応は、トリエチルアミン、ピリジン、または4−ジメチルピリジン等の有機塩基存在下、置換スルホニルクロリド、または置換スルホン酸無水物を反応させて実施すればよい。
【0117】
反応に使用される溶媒としては反応を阻害しないものであれば制限はないが、ジクロロメタン、1,2−ジクロロエタン等の塩素系溶媒、その他、テトラヒドロフラン、トルエン等を用いればよい。また、塩基としてピリジン等を使用する場合はこれらを溶媒に兼ねさせてもよい。
【0118】
反応温度は、塩基の種類や使用する溶媒により異なるが、通常、−78から100℃の範囲で行えばよく、好ましくは、−30℃から室温の範囲である。
【0119】
工程5:
【化32】
【0120】
(式中、R、R1、およびYの定義は先の定義と同じである。)
【0121】
本工程は、化合物(VI)を塩基存在下で処理して閉環させてスピロ環状構造を構築して化合物(VII)を得る工程である。
【0122】
本工程は、Yが、ハロゲン原子もしくはその他の脱離基を有する化合物(VI)の分子内アルキル化反応により、化合物(VII)を得る工程といえるが、この分子内アルキル化反応は、塩基存在下に実施すればよい。反応に用いられる塩基としては、有機または無機のいずれであってもよく、例えば、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、リチウム2,2,6,6−テトラメチルピペリジン、n−ブチルリチウム、sec−ブチルリチウム、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類;トリエチルアミン、N,N−ジイソプロピルエチルアミン等のアルキルアミン類;4−メチルモルホリン、1,8−ジアザビシクロ[5,4,0]−7−ウンデセン、ピリジン等の飽和または不飽和の含窒素複素環化合物類;ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類等の有機塩基を用いることができる。また、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物類;水素化ナトリウム、水素化カリウム等のアルカリ金属の水素化物類;ナトリウムアミド等のアルカリアミド類等の無機塩基を用いることができる。これらの塩基のなかで特に好ましい塩基としては、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類を挙げることができる。
【0123】
本工程の反応は溶媒中で実施すればよいが、反応溶媒としては、ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;テトラヒドロフラン、1,4−ジオキサン、ジエチルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒;N,N−ジメチルアセトアミド、N,N−ジメチルホルムアミド、N−メチル−2−ピロリドン等のアミド系溶媒;その他、液体アンモニア、ジメチルスルホキシド等を用いることができる。また、塩基としてナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類を用いる際には、メタノール、エタノール、第三級ブタノール等のアルコール系溶媒を用いることもできる。また、これらを混合溶媒として使用することも可能である。特に好ましい溶媒としては、テトラヒドロフラン、トルエン、N,N−ジメチルホルムアミド等を挙げることができる。
【0124】
反応温度は塩基の種類や使用する溶媒によって異なるが、−78℃から溶媒の沸点の範囲で行えばよく、好ましくは−78℃から室温の範囲である。
【0125】
化合物(VII)は、上記の工程3で用いられる化合物(III)の代わりに次式(III’)
【0126】
【化33】
【0127】
(式中、Xの定義は先の定義と同じである。X’は、Xと同一であるか、これとは異なるハロゲン原子またはその他の脱離基から選ばれる基を意味する。)
【0128】
で表わされる化合物を使用すれば、工程3から5の三工程分の反応は、連続、且つ、最小の一工程で行うことができる。化合物(III’)は、化合物(III)R’O基部分がハロゲンまたは脱離基となった化合物である。これを用いる反応は、下式によって示される。
【0129】
【化34】
【0130】
(式中、R、R1、X、およびX’の定義は先の定義と同様である。)
【0131】
この反応は、塩基存在下にて実施されるが、その反応条件は、工程3または5で記載した条件から、適宜、選択して用いればよい。
【0132】
上記の反応に用いることのできる化合物(III’)としては、例えば、1,2−ジブロモエタン、1,2−ジヨードエタン、1−ブロモ−2−クロロエタン、2−クロロエチル メタンスルホネート、2−クロロエチル トルエンスルホネート、2−ヨードエチル メタンスルホネート、2−ヨードエチル ベンゼンスルホネート等を挙げることができる。
【0133】
工程B:化合物(VII)の別途合成法
【化35】
【0134】
工程6:
【化36】
【0135】
(式中、RおよびR1の定義は先の定義と同じである。)
【0136】
本工程は、化合物(II)にエキソメチレン基を導入して、化合物(VIII)を得る工程である。この工程および次工程によって化合物(VII)を得る別の工程が提供される。
【0137】
本工程は、アミドのα位(活性メチレン)にエキソメチレンを導入する工程であるが、このための反応の例として、塩基存在下にて、N,N−ジメチルメチレンアンモニウム ヨージド(エッシェンモーザー塩)を用いる方法、リン酸エステルを導入した後にホルムアルデヒドとのホーナー・エモンズ反応に付する方法、メチルメトキシマグネシウム カーボネートを用いる方法、およびヒドロキシメチル化後に脱水反応に付する方法等を挙げることができる。
【0138】
より具体的には、塩基存在下にてアミドのα位をホルミル化し、これにホルムアルデヒドを作用させて脱カルボニルメチレン化を行う方法である。
【0139】
上記の反応は塩基存在下に実施するが、反応に用いられる塩基としては、例えば、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、リチウム2,2,6,6−テトラメチルピペリジン、n−ブチルリチウム、sec−ブチルリチウム、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類;ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類;等の有機塩基を用いることができる。これらの塩基のなかで特に好ましい塩基としては、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド、ナトリウムビストリメチルシリルアミド、カリウムビストリメチルシリルアミド等の有機金属化合物類を挙げることができる。
【0140】
本工程の反応は溶媒中で実施すればよいが、反応溶媒としては、ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;テトラヒドロフラン、1,4−ジオキサン、ジエチルエーテル、1,2−ジメトキシエタン等のエーテル系溶媒;N,N−ジメチルアセトアミド、N,N−ジメチルホルムアミド、N−メチル−2−ピロリドン等のアミド系溶媒等を用いることができる。また、塩基としてナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三級ブトキシド、カリウム第三級ブトキシド等のアルカリ金属のアルコキシ化物類を用いる際には、メタノール、エタノール、第三級ブタノール等のアルコール系溶媒を用いることもできる。また、これらを混合溶媒として使用することも可能である。特に好ましい溶媒としては、テトラヒドロフラン、トルエン、N,N−ジメチルホルムアミド等を挙げることができる。
【0141】
反応温度は塩基の種類や使用する溶媒によって異なるが、−78℃から溶媒の沸点の範囲で行えばよく、好ましくは−78℃から室温の範囲である。
【0142】
工程7:
【化37】
【0143】
(式中、RおよびR1の定義は先の定義と同じである。)
【0144】
本工程は、化合物(VIII)のエナミド部分の二重結合エキソメチレン部分にメチレン基を環化付加反応させてシクロプロパン環を構築し、化合物(VII)を得る工程である。
【0145】
本工程を達成する反応の例として、ジアゾメタン−遷移金属触媒、ジヨードメタン−ジエチル亜鉛(シモンズ・スミス試薬)等を用いるカルベン、カルベノイド反応、サルファー イリド(コーリー試薬)を用いたマイケル付加反応を経由する方法等を挙げることができる。
【0146】
ジアゾメタン−遷移金属触媒系によるカルベノイド反応は、具体的には、化合物(VIII)を溶媒に溶解して触媒を添加し、汎用の方法にてあらかじめ調製したジアゾメタンのジエチルエーテル溶液を加えて反応させればよい。
【0147】
この反応で用いることのできる触媒としては、例えば、パラジウム(II)アセテート、パラジウム(II)アセチルアセトネート等を挙げることができる。
【0148】
この反応の反応溶媒としてはノルマルへキサン等の炭化水素系;テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン、ジエチルエーテル等のエーテル系;ジクロロメタン、ジクロロエタン等のハロゲン系;等を用いることができる。また、これらを混合溶媒として使用することも可能である。
反応温度は溶媒によって異なるが、−20℃から0℃の範囲で行えばよい。
【0149】
シモンズ・スミス試薬等を用いるカルベン反応は、具体的には、化合物(VIII)を溶媒に溶解後、炭素原としてはジヨードメタン、クロロヨードメタン等のハロゲン化メチレンを用い、ジエチル亜鉛、亜鉛−銅、サマリウム−塩化水銀(II)、トリメチルアルミニウム等のトリアルキルアルミニウム等の金属試薬存在下にて反応させればよい。
【0150】
この場合、ハロゲン化メチレンとしてはジヨードメタンが好ましく、また、金属試薬で好ましいのは、ジエチル亜鉛、亜鉛−銅である。
【0151】
反応溶媒としては、ノルマルへキサン等の脂肪族炭化水素系溶媒;ベンゼン、トルエン、キシレン等の芳香族炭化水素系溶媒;テトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン、ジエチルエーテル等のエーテル系溶媒;ジクロロメタン、ジクロロエタン等のハロゲン系溶媒;等を挙げることができる。また、これらを混合溶媒として使用することもできる。
【0152】
また、本反応は、添加物としてトリフルオロ酢酸を加えて実施してもよい。
反応温度は塩基の種類や使用する溶媒によって異なるが、−78℃から溶媒の沸点の範囲で行えばよい。
【0153】
工程Aによれば光学活性体(II)から立体を保持したまま化合物(VII)へ誘導することができ、高い光学純度をもつ化合物(VII)の合成が可能である。さらに、工程Bは、前者の方法に比較して、非常に簡便、且つ、短工程で目的とする化合物(VII)を得ることができる。いずれの方法においても試薬および反応操作は、工業化等の大量合成に特に不都合なものはない。したがって工程のより短い工程Bが有利である。
さらに、化合物(VII)は結晶性に優れることからこの化合物においてX線結晶構造解析を実施し、メチル基の置換位置である7位の不斉炭素部分の絶対配置が(7S)であると判明した。このことからより優れた生理活性であるキノロンを与える配置が7Sであることが確定した。
【0154】
工程C:アミノ基の導入
【0155】
【化38】
【0156】
化合物(VII)に対してアミノ基を導入する工程について説明する。
【0157】
工程8:
【化39】
【0158】
(式中、RおよびR1の定義は先の定義と同じである。)
【0159】
本工程は、化合物(VII)のエステルを加水分解し、遊離のカルボン酸化合物である化合物(IX)を得る工程である。
【0160】
この工程で使用される加水分解反応は、エステルの加水分解に通常用いられる塩基性条件または酸性条件、あるいは加水素化分解条件で実施される反応であればいかなる方法でもよい。Rが、置換基を有していてもよいアリール基や置換基を有していてもよい炭素数1から6のアルキル基の場合は、塩基性条件下または酸性条件下の加水分解条件にて、Rが、置換基を有していてもよい炭素数7から9のアラルキル基の場合は加水素化分解条件にて実施するのが好ましい。
【0161】
酸性条件での加水分解反応は、酸として通常、塩酸または硫酸を用いるが、ルイス酸、例えば、三塩化ホウ素等も使用することができる。溶媒には、酢酸、ギ酸、およびこれらと水との混合溶媒が適している。この酸性条件での加水分解に適したエステルは、第三級ブチルエステルである。第三級ブチルエステルの加水分解では、酸としてトリフルオロ酢酸、パラトルエンスルホン酸も使用することができる。酸は溶媒兼用で使用して実施してもよいが、ジクロロメタン等を溶媒として用いることもできる。また、ルイス酸とチオールまたはスルフィドの併用による反応も用いることができる。
【0162】
塩基性条件、いわゆるアルカリ加水分解反応は、一般的には、適宜な濃度の水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化バリウム等の水溶液を用いて実施すればよい。反応溶媒としては、好ましくは、メタノール、エタノール、テトラヒドロフラン、エチレングリコール等のアルコール系溶媒、またはこれらと水の混合溶媒を用いればよい。反応温度は、−15から100℃が適当であり、好ましくは0から50℃の範囲である。また、当該加水分解は、アルカリ水溶液および少量の界面活性剤を添加して行ってもよい。このような界面活性剤の例としては、テトラエチルアンモニウムクロリド、テトラブチルアンモニウムブロミド、テトラベンジルアンモニウムブロミド等を挙げることができる。なお、この該加水分解は、反応終了後、塩酸、硫酸、酢酸等の鉱酸、もしくは酸性イオン交換樹脂を用いることで遊離のカルボン酸化合物を得ることができる。
【0163】
また、エステルがベンジルエステル、ベンズヒドリルエステル、1−(R)−フェニルエチルエステル等のベンジル構造を有するアラルキルエステルは、触媒存在下の接触加水素化分解反応により除去され、カルボン酸化合物を得ることができる。この接触加水素化分解反応は、触媒としてパラジウム炭素触媒系を用いるのが好ましく、反応基質をメタノール、エタノール、テトラヒドロフラン等の溶媒に溶解後、常圧(1気圧)から適宜に加圧した水素雰囲気下、室温から50℃の温度範囲にて実施される。また、液体アンモニア中、金属リチウムによる還元でもカルボン酸体が得られる。
【0164】
このようにして合成される化合物(IX)
【0165】
【化40】
【0166】
(式中、R1の定義は先の定義と同一である。)
は、遊離のカルボン酸でもよいが、カルボキシ基の塩に変換してもよい。カルボキシ基の塩としては、例えば、リチウム塩、ナトリウム塩、カリウム塩等のアルカリ金属塩、マグネシウム塩、カルシウム塩等のアルカリ土類金属塩、アンモニウム塩、また、トリエチルアミン塩等、無機塩類、有機塩類のいずれでもよい。また、これらのカルボキシ基の塩は、水和物として存在することもある。
【0167】
工程9:
【化41】
【0168】
(式中、R1の定義は先の定義と同一である。R”は、水素原子またはアミノ基の保護基を意味する。)
【0169】
本工程は、化合物(IX)のカルボキシ基を(置換)アミノ基へ転位変換させて、化合物(X)を得る工程である。
【0170】
本工程で実施されるカルボキシ基の(置換)アミノ基への転換反応は、幾つかの方法が知られているが、カルボン酸を酸アジドへ変換し、イソシアナートヘ熱転位させた後、イソシアナートを加水分解してアミンとするクルチウス転位反応を用いるのが好ましい。
【0171】
カルボン酸の酸アジドへの変換反応は、炭素数1から6のアルキルクロロホルメートを塩基存在下でカルボン酸化合物に加え、生成した活性エステルをナトリウムアジド水溶液と反応させて実施することができる。この反応で使用し得るアルキルクロロホルメートとして、メチルクロロホルメート、エチルクロロホルメート、またはブチルクロロホルメートを用いるのが好ましい。反応に用いる塩基としては、トリエチルアミン、ジイソプロピルエチルアミン等の有機塩基類、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基を挙げることができる。この反応は、例えば、アセトン、ジクロロメタン、テトラヒドロフラン、トルエン、またはそれらの混合溶媒中、−20℃から室温にて実施するのが好ましい。
【0172】
この酸アジドを、アジドあるいはイソシアナートと非反応性の溶媒中で、使用する溶媒の沸点において加熱することでイソシアナートへ転位させる。好ましい非反応性溶媒としては、トルエン、キシレン、ベンゼン、またはこれらの混合物を挙げることができる。
【0173】
上述のカルボン酸から酸アジドを経てイソシアナ−トを直接得る方法としては、適宜な溶媒中、塩基存在下にてジフェニルホスホリルアジド(DPAA)を用いる簡便な方法が好ましい例として挙げることができる。
【0174】
次いで、上述の適宜な方法により生成したイソシアナ−トを酸の存在下、−5℃から室温において、または還流下で加熱しながら加水分解することにより、または、イソシアナートをアルカリの存在下、室温から110℃において加水分解することにより置換アミン化合物を得ることができる。この酸性条件下の加水分解で用いるのみ好適な酸としては、適宜な濃度の塩酸、臭化水素酸、または硫酸水溶液等を挙げることができる。これらの酸による加水分解により、生成物は遊離アミンもしくはその酸付加塩として得られる。また、アルカリ条件下の加水分解において用いる好ましいアルカリは、適宜な濃度の水酸化ナトリウム、水酸化カリウム水溶液等を挙げることができる。
【0175】
また、イソシアナートをアルコール系溶媒中、もしくはトルエン、キシレン、ベンゼン等のイソシアナートと非反応性の溶媒中にアルコールを添加させて反応させることにより、アミノ基がアルコキシカルボニル基で保護されたカルバメート誘導体を直接に得ることができる。この反応は、塩基、例えば、トリエチルアミン、ナトリウムアルコキシド等の存在下でも実施することができる。上記のアルコールとしては、メタノール、エタノール、イソプロパノール、ブタノール、第三級ブタノール、ベンジルアルコール等を挙げることができ、使用するアルコールの種類により相当するメチルカルバメート、エチルカルバメート、イソプロピルカルバメート、ブチルカルバメート、第三級ブチルカルバメート、およびベンジルカルバメートを得ることができる。特に好ましいアルコールとしては、第三級ブタノールを挙げることができる。上記反応において用いられるアルコールの量は、計算上、カルボン酸に対して1倍モル以上必要であるが、通常は、大過剰を使用すればよい。
【0176】
なお、本工程のカルボン酸のアミンへの転換反応の具体例として、実施例においてもクルチウス転位反応を用いる方法を記載するが、この他の転位反応を用いることもできる。例えば、カルボン酸から酸アミド(カルバモイル誘導体)を経てアミンを得るホフマン転位反応を挙げることができる。
【0177】
本工程で得られる化合物(X)
【0178】
【化42】
【0179】
(式中、R1およびR”の定義は先の定義と同一である。)
【0180】
において、R”が水素原子である場合、化合物(X)は、遊離アミン、またはその酸付加塩として存在することができる。酸付加塩の例としては、無機酸塩および有機酸塩を挙げることができる。これらの具体例としては、塩酸塩、硫酸塩、硝酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩等の無機酸塩類、あるいは、メタンスルホン酸塩、ベンゼンスルホン酸、トルエンスルホン酸塩等のスルホン酸塩、酢酸塩、クエン酸塩、マレイン酸塩、フマル酸塩、乳酸塩、シュウ酸塩等のカルボン酸塩等の有機酸塩類を挙げることができる。また、これらの酸付加塩は、水和物としても存在することができる。
【0181】
R”が、アミノ基の保護基の場合、通常用いられ、保護と脱保護が容易であり、次工程以降の反応に影響しないか、もしくは保護基自身が反応しないものであれば、いかなる保護基でもよい。このようなアミノ基の保護基としては一般に使用されている、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、置換基を有していてもよいアシル基、置換基を有していてもよいアラルキル基、および置換シリル基から選ばれるものであればよい。置換基を有していてもよいアルコキシカルボニル基としては、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、2,2,2−トリクロロエトキシカルボニル基等を挙げることができる。置換基を有していてもよいアシル基としては、アセチル基、メトキシアセチル基、トリフルオロアセチル基、クロロアセチル基、ピバロイル基、ホルミル基、ベンゾイル基等を挙げることができる。置換基を有していてもよいアラルキルオキシカルボニル基としては、ベンジルオキシカルボニル基、パラメトキシベンジルオキシカルボニル基、パラニトロベンジルオキシカルボニル基等を挙げることができる。置換シリル基としては、トリメチルシリル基、イソプロピルジメチルシリル基、第三級ブチルジメチルシリル基、トリベンジルシリル基、第三級ブチルジメチルシリル基等を挙げることができる。R”としては、これらのうち、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、および置換基を有していてもよいアシル基が好ましく、より具体的には、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、ベンジルオキシカルボニル基、アセチル基、およびトリフルオアセチル基が好ましく、特に、第三級ブトキシカルボニル基が好ましい。
【0182】
なお、脱保護が必要な場合は、保護基であるR”の性質に応じた脱保護条件を適宜に選択して用いればよい。
【0183】
工程10:
【化43】
【0184】
(式中、R1とR”の定義は先の定義と同一である。)
【0185】
本工程は、化合物(X)を酸アミドの還元反応条件下にて処理して、ピロリジン化合物(XI)を得る工程である。
【0186】
本工程における還元反応は、通常、金属水素化物を用いて実施される。この金属水素化物としては、例えば、水素化リチウムアルミニウム、水素ホウ素ナトリウム−塩化第二スズ等の水素化ホウ素ナトリウムとルイス酸の組み合わせ、ジボラン、ボラン−テトラヒドロフラン錯体等を挙げることができる。この中では、特に、水素化リチウムアルミニウム、ボラン−テトラヒドロフラン錯体が好ましい。
【0187】
反応溶媒としては、非反応性の溶媒を用いればよく、具体的には、ジエチルエーテル、テトラヒドロフラン、ジイソプロピルエーテル、1,4−ジオキサンのエーテル系溶媒が好ましく、特に、テトラヒドロフランを用いるのが好ましい。
【0188】
反応温度は、0℃から使用する溶媒の沸点、いわゆる溶媒が還流する加熱下の範囲で実施されるが、室温から還流条件下にて実施するのが好ましい。
【0189】
上述のような酸アミドの金属水素化物を用いる還元反応において、化合物Xのアミノ基は遊離でも保護基を有していてもよいが、R”が、水素原子である遊離のアミノ基を有する化合物(X)を用いて実施するのが好ましい。R”がアミノ基の保護基である場合は、その保護基が選択した還元条件下にて安定である必要がある。
【0190】
Rが、アミノ基の保護基である場合、その保護基は、前述の工程9にて詳細に記載した保護基と同様であるが、生成物(XI)の単離、精製を容易にするためには、本工程にて、アミドの還元後に保護基を導入する方がよい。
【0191】
また、酸アミドの還元反応として、アミドのカルボニル基を適宜な試薬を用いてチオカルボニル基へ変換してチオアミドを得た後、接触還元等による脱硫反応によっても実施することができる。このような場合、アミドのカルボニル基をチオカルボニル基へ変換する方法としては、ローソン試薬が汎用されており、ベンゼンやトルエン中にて還流して実施される。次いで、脱硫反応は、メタノール、エタノール、テトラヒドロフラン等の溶媒中、水素雰囲気下、ラネーニッケル触媒を用いて実施するのが好ましい。この反応は、エタノール中等のアルコール系溶媒中、水素ガスなしでも実施できる。
【0192】
本工程により得られる化合物(XI)
【0193】
【化44】
【0194】
(式中、R1およびR”は先の定義と同じ定義である。)
【0195】
は、遊離のアミン体(二塩基性体)、またはその酸付加塩として存在することができる。酸付加塩の例としては、無機酸塩および有機酸塩を挙げることができる。これらの具体例としては、塩酸塩、硫酸塩、硝酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩等の無機酸塩類、あるいは、メタンスルホン酸塩、ベンゼンスルホン酸、トルエンスルホン酸塩等のスルホン酸塩、酢酸塩、クエン酸塩、マレイン酸塩、フマル酸塩、乳酸塩、シュウ酸塩等のカルボン酸塩等の有機酸塩類を挙げることができる。また、これらの酸付加塩は、水和物としても存在することができる。
【0196】
工程11:
【化45】
【0197】
(式中、R1およびR”は先の定義と同じ定義である。)
【0198】
本工程は、化合物(XI)の5位のアミノ基(R1)の脱保護反応により、化合物(XII)を得る工程である。
【0199】
本反応における脱保護反応は、保護基であるR”の性質に応じた脱保護条件を適宜に選択して用いればよい。この場合、7位のアミノ基が保護基R”を有する場合は、選択的にR1のみ脱保護されてもよく、また、R1およびR”の双方が同時に脱保護されてもよいが、好ましくは前者である。具体的には、R1が、ベンジル基、ベンズヒドリル基、1−(R)−フェニルエチル基、1−(S)−フェニルエチル基である場合、通常、メタノール、エタノール、イソプロパノール等、化合物(XI)を溶解し得る溶媒中、常圧から加圧の水素雰囲気下、パラジウム炭素系触媒を用いる接触加水素分解反応により実施される。反応温度は、室温から50℃の範囲で行えばよい。水素ガスの替わりに、アルコール系溶媒中、ギ酸アンモニウム等の水素ドナーを用いる方法もある。また、液体アンモニア中、金属ナトリウムを用いる方法(バーチ還元)もある。R1が、トリフェニルメチル基である場合は、上記の脱保護条件に加えて、アセトン中、塩酸を反応させる方法もあるが、この場合、7位のアミノの保護基が塩酸酸性条件下で安定である必要がある。
【0200】
本工程で得られる化合物(XII)
【0201】
【化46】
【0202】
(式中、R”は先の定義と同じ定義である。)
は、遊離のアミン体(二塩基性体)、またはその酸付加塩として存在することができる。酸付加塩の例としては、無機酸塩および有機酸塩を挙げることができる。これらの具体例としては、塩酸塩、硫酸塩、硝酸塩、臭化水素酸塩、ヨウ化水素酸塩、リン酸塩等の無機酸塩類、あるいは、メタンスルホン酸塩、ベンゼンスルホン酸、トルエンスルホン酸塩等のスルホン酸塩、酢酸塩、クエン酸塩、マレイン酸塩、フマル酸塩、乳酸塩、シュウ酸塩等のカルボン酸塩等の有機酸塩類を挙げることができる。また、これらの酸付加塩は、水和物としても存在することができる。
【実施例】
【0203】
以下に実施例を挙げて本願発明をさらに具体的に説明するが、これらはいかなる意味においても本願発明を限定的に解釈させるものではない。
【0204】
[参考例1]
5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0205】
【化47】
【0206】
羽撹拌下、5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸(1165g,4.994mol)のジクロロメタン(10L)懸濁液にO−第三級ブチル−N,N’−ジイソプロピルウレア(3020g,15.00mol)を室温で加えた後、内温の上昇と還流の開始を認めた後、氷水浴にて冷却した。反応液を室温まで冷却した後、氷水浴を外して1時間、次いで40℃で加熱しながら3時間撹拌した。次いで、反応液を氷水浴で冷却して1時間撹拌した後、不溶物をろ去し、ろ液を減圧乾固して得られた残留物をシリカゲルカラムクロマトグラフィー(シリカゲル:4kg;溶離液,ヘキサン:酢酸エチル=3:1)にて精製し、標記化合物(3位異性体混合物)925.2g(64%)を淡黄色シロップとして得た。ピロリジンの3位に由来した各ジアステレオマーは容易に分取可能であったが、次工程がエピメリ化を伴う反応であることから分取せず使用した。下記には、別途分取した異性体の各々の1H−NMRスペクトルを示した。
【0207】
低極性異性体:
1H−NMR(400MHz,CDCl3)δ:1.45(9H,s),1.54(3H,d,J=7.08Hz),2.59−2.74(2H,m),2.95−3.03(1H,m),3.14(1H,dd,J=9.77,8.79Hz),3.49(1H,dd,J=9.77,6.35Hz),5.50(1H,q,J=7.1Hz),7.26−7.36(5H,m).
高極性異性体:
1H−NMR(400MHz,CDCl3)δ:1.36(9H,s),1.53(3H,d,J=7.32Hz),2.59−2.75(2H,m),3.02−3.11(1H,m),3.16(1H,dd,J=10.01,5.62Hz),3.51(1H,dd,J=10.01,8.54Hz),5.50(1H,q,J=7.1Hz),7.24−7.36(5H,m).
【0208】
[参考例2]
5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(別途合成法)
5−オキソ−1−[(R)−1−フェニルエチル]ピロリジン−3−カルボン酸(500mg,2.14mmol)のジクロロメタン(2.67mL)溶液に、氷冷下、オキサリルクロライド(224μL,2.57mmol)、およびN,N−ジメチルホルムアミド(1滴)を加え、氷冷下にて15分間、次いで室温にて15分間攪拌した。トルエンを加えて溶媒を減圧乾固する操作を3回繰り返し、朱色油状物質603mgを得た。別の容器に準備した第三級ブタノール(614μL,6.42mmol)のジクロロメタン(2.67mL)溶液に、窒素雰囲気下、氷冷しながら上記の朱色油状物質(603mg)のジクロロメタン(2.67mL)溶液を滴下した。反応液にトリエチルアミン(448μL,3.21mmol)を加えて、室温で30分間攪拌した。水(10mL)を加えて、ジクロロメタン(10mL×2)にて抽出した。有機層を飽和食塩水(25mL)にて洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧濃縮後、トルエンを加えて減圧乾固する操作を3回繰り返した。得られた残留物558mgをショートシリカゲルカラムクロマトグラフィー(酢酸エチル100%にて溶出)で精製し、淡黄色の標記化合物523mg(85%)をジアステレオマー混合物(ジアステレオマー比=2.2:1.0)として得た。
【0209】
1H−NMR(400MHz,CDCl3)δ:1.36(2.81/9H,s),1.45(6.19/9H,s),1.53(0.93/3Hd,J=3.7Hz),1.54(2.06/3H,d,J=3.7Hz),2.59−2.75(2H,m),2.95−3.18(2H,m),3.47−3.53(1H,m),5.50(1H,q,J=7.1Hz),7.25−7.37(5H,m).
【0210】
[実施例1]
(3S)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0211】
【化48】
【0212】
窒素ガス雰囲気下、5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(30.05g,0.104mol)のN,N’−ジメチルホルムアミド(210mL)溶液に、攪拌下、ヨードメタン26.0mL(59.28g,0.418mol)、次いで水素化ナトリウム(55%油性、11.35g,0.260mol)を室温にて加えた。内温が上昇して約50℃に達した時、氷水浴にて30℃まで冷却し、次いで外温17℃の水浴に切り替えて23時間撹拌した。反応液を冷クエン酸水溶液(10%クエン酸1Lと氷500gの混合水)に注ぎ、30分間撹拌した後、酢酸エチル(800mL,500mL)にて抽出した。有機層を合わせ、飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固した。残留物をフラッシュシリカゲルカラムクロマトグラフィーにて精製し(ヘキサン:酢酸エチル=5:1→4:1溶出部)、高極性異性体として標記化合物10.63g(33.7%)を白色固体として得た。また、低極性異性体として(3R)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル14.91g(47.3%)を得た。
【0213】
1H−NMR(400MHz,CDCl3)δ:1.34(12H,s),1.52(3H,d,J=7.10Hz),2.27(1H,d,J=17.0Hz),2.93(1H,d,J=17.0Hz),3.05(1H,d,J=10.1Hz),3.32(1H,d,J=10.1Hz),5.50(1H,q,J=7.1Hz),7.23−7.38(5H,m).
【0214】
[実施例2]
(3S)−4−[2−(第三級ブチルジメチルシリル)ヒドロキシエチル]−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0215】
【化49】
【0216】
(3S)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(30.0g,98.9mmol)、および第三級ブチル(2−ヨードエトキシ)ジメチルシラン(36.8g,129mmol)の無水テトラヒドロフラン(288mL)溶液へ、−4℃にてリチウムビス(トリメチルシリル)アミド(1.0Mテトラヒドロフラン溶液,129mL,129mmol)を滴下し、2℃にて3.5時間攪拌した。反応液に飽和塩化アンモニウム水溶液(300mL)を加え、酢酸エチル(300mL,200mL)にて抽出した。有機層を飽和食塩水(200mL)にて洗浄し、無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固して、標記化合物54.1gを得た。尚、本成績体は、精製せずに次の工程に使用した。
MS(ESI)m/z:363(M−Boc+H)+.
【0217】
[実施例3]
(3S)−4−(2−ヒドロキシエチル)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0218】
【化50】
【0219】
前述のシリル体粗生成物(54.1g,98.9mmol)をテトラヒドロフラン(450mL)に溶解し、氷冷下、フッ化テトラブチルアンモニウム、1.0mol/Lテトラヒドロフラン溶液(148mL,148mmol)を滴下した後、室温にて2時間攪拌した。反応液を濃縮後、酢酸エチル(200mL,100mL)にて抽出した。有機層を10%炭酸水素ナトリウム水溶液(200mL)、クエン酸水溶液(300mL)、および飽和食塩水(100mL)にて洗浄し、無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固した。残留物をシリカゲルカラムクロマトグラフィー(へキサン−酢酸エチル=6:1→4:1→1:1溶出部)にて精製し、標記化合物29.1g(83.9mmol,85%)を無色透明シロップ状物質として得た。
【0220】
1H−NMR(400MHz,CDCl3)δ:1.28(3H,s),1.40(9H,s),1.51−1.53(1H,m),1.53(3H,d,J=7.1Hz),1.78−1.94(2H,m),2.90−3.08(2H,m),3.67−3.75(1H,m),3.80−3.91(1H,m),4.85−4.89(1H,m),5.43−5.53(1H,m),7.27−7.37(5H,m).
MS(ESI)m/z:348(M+H)+.
【0221】
[実施例4]
(3S)−4−[2−(ベンゼンスルホニル)オキシエチル]−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0222】
【化51】
【0223】
(3S)−4−(2−ヒドロキシエチル)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(29.1g,83.9mmol)のジクロロメタン(280mL)溶液に、氷冷下、トリエチルアミン(15.2mL,109mmol)、塩化ベンゼンスルホニル(11.8mL,92.3mmol)、および4−ジメチルアミノピリジン(1.02g,8.39mmol)を加え、混合物を室温にて19時間攪拌した。反応液に飽和塩化アンモニウム水溶液(280mL)を加え、有機層を分離して溶媒を減圧下に留去し、残留物を酢酸エチル(280mL,180mL)に溶解後、先の飽和塩化アンモニウム水溶液にて再び洗浄した。有機層を1mol/L塩酸水溶液(250mL)、飽和重曹水(250mL)、飽和食塩水(200mL)にて洗浄後、無水硫酸ナトリウムで乾燥し、ろ過後、ろ液を減圧乾固し、標記のベンゼンスルホニル体粗生成物(43.7g)を得た。尚、本成績体は精製せずに次の工程に使用した。
MS(ESI)m/z:510(M+Na)+.
【0224】
[実施例5]
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸第三級ブチル
【0225】
【化52】
【0226】
前述のベンゼンスルホニル体粗生成物(43.7g,83.9mmol)の無水テトラヒドロフラン(470mL)溶液に、氷冷下、ナトリウムビス(トリメチルシリル)アミド、1.0mol/Lテトラヒドロフラン溶液(109mL,109mmol)を加え、混合物を室温にて1時間攪拌した。反応液に飽和塩化アンモニウム水溶液(300mL)を加え、酢酸エチル(300mL,200mL)にて抽出し、有機層を飽和食塩水(200mL)にて洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1→2:1にて溶出)で精製して、標記化合物24.6g(89%、2工程)を白色固体として得た。
【0227】
mp:55−57℃.
[α]D25.1=122.1゜(c=0.517,CHCl3).
1H−NMR(400MHz,CDCl3)δ:0.72−0.77(1H,m),0.85−0.90(1H,m),1.04−1.13(2H,m),1.18(3H,s),1.32(9H,s),1.54(3H,d,J=7.1Hz),3.08(1H,d,J=9.8Hz),3.53(1H,d,J=9.8Hz),5.52(1H,q,J=7.1Hz),7.26−7.34(5H,m).
元素分析;C20H27NO3として:
計算値:C,72.92;H,8.26;N,4.25.
実測値:C,72.64;H,8.27;N,4.06.
MS(FAB)m/z:330(M+H)+.
HRMS(FAB)m/z:330.2069(Calcd for C20H28NO3 330.2069).
IR(ATR)ν:3066,2976,2933,2879,1720,1676,1481,1454,1433,1365,1329,1286,1238,1203cm-1.
【0228】
本化合物について7位の配位を決定するべくX線構造解析を実施した。その詳細は以下のとおりである。
【0229】
【表1】
【0230】
データ収集後、直接法で初期位相を決定し、完全行列最小自乗法で位相精密化を行った。精密化の際、非水素原子は非等方性温度因子を適用し、水素原子は計算で位置を決定して座標を固定した。本化合物中には不斉炭素が2個存在するが、そのうち1個の不斉炭素の絶対配置は既知であった。この絶対配置をもとに他方の不斉炭素の絶対配置を決定した。得られた結果を図1に示す。すなわち、標題化合物の7位に関する配位が(S)であることが確定された。そして、この化合物を経由して調製される一連の化合物の配位も決定されることとなった。
【0231】
[実施例6]
(3S)−4−(2−ヨードエチル)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0232】
【化53】
【0233】
(3S)−4−(2−ヒドロキシエチル)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(0.37g,1.07mmol)のジクロロメタン(7.5mL)溶液に氷冷下にてトリエチルアミン(0.22mL,1.6mmol)、および塩化メタンスルホニル(99μL,1.28mmol)を加え、混合物を室温にて2時間半攪拌した。反応液に水(20mL)を加え、クロロホルム(20mL×3)にて抽出した。抽出液を飽和食塩水(20mL)にて洗浄後、無水硫酸ナトリウムで乾燥し、ろ過し、ろ液を減圧乾固した。得られた残留物をアセトン(20mL)に溶解し、ヨウ化ナトリウム(0.32g,2.13mmol)を加え、80℃で17時間半攪拌した。反応系を減圧濃縮し、残留物に水(20mL)を加え、酢酸エチル(20mL×3)にて抽出した。抽出液を10%チオ硫酸ナトリウム水溶液(20mL)、および飽和食塩水(20mL)にて洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固して標記化合物0.47g(96%)を淡黄色油状物として得た。
【0234】
1H−NMR(400MHz,CDCl3)δ:1.20(3H,s),1.43(9H,s),1.50(3H,d,J=7.1Hz),2.00−2.10(1H,m),2.16−2.27(1H,m),2.85(1H,dd,J=9.0,4.9Hz),2.93(1H,d,J=10.0Hz),3.24(1H,d,J=10.0Hz),3.55(2H,dd,J=8.1,6.3Hz),5.48(1H,q,J=7.1Hz),7.25−7.37(5H,m).
MS(ESI)m/z:458(M+H)+.
【0235】
[実施例7]
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸第三級ブチル
【0236】
【化54】
【0237】
(3S)−4−(2−ヨードエチル)−3−メチル−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(118mg,0.26mmol)の無水テトラヒドロフラン(2.4mL)溶液に氷冷下、カリウムビス(トリメチルシリル)アミド(0.5Mトルエン溶液,0.62mL,0.31mmol)を加え、混合物を氷冷下にて10分間攪拌した。反応液に飽和塩化アンモニウム水(20mL)を加え、酢酸エチル(20mL×3)にて抽出した。抽出液を10%チオ硫酸ナトリウム水溶液(20mL)および飽和食塩水(20mL)にて洗浄した。有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン−酢酸エチル4:1)で精製して標記化合物91mg(100%)を無色固体として得た。本成績体の1H−NMRデータは、実施例5に記載のデータと一致した。
MS(ESI)m/z:330(M+H)+.
【0238】
[実施例8]
(3S)−3−メチル−4−メチレン−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル
【0239】
【化55】
【0240】
(3S)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(200mg,0.66mmol)を無水テトラヒドロフラン(4mL)に溶解し、0℃にて1.0Mリチウムヘキサメチルジシラジドのテトラヒドロフラン溶液(1.45mL,1.45mmol)を加え、同温で30分撹拌した。次いで、ギ酸エチル(69μL,0.86mmol)を0℃で加えた。同温にて1時間20分撹拌した後、N,N−ジメチルホルムアミド(4mL)、およびパラホルムアルデヒド(120mg)を加え、室温で17時間撹拌した。本反応液に1M塩酸水溶液を0℃で加え反応を停止させた。酢酸エチルで3回抽出し、合わせた有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧濃縮して得られた残留物を中圧液体クロマトグラフィーにて精製し(ヘキサン:酢酸エチル=1:1にて溶出)、標記化合物127mg(61%)を無色固体として得た。
【0241】
1H−NMR(400MHz,CDCl3)δ:1.30(9H,s),1.43(3H,s),1.57(3H,d,J=7.1Hz),3.03(1H,d,J=10.0Hz),3.56(1H,d,J=10.0Hz),5.50(1H,s),5.63(1H,q,J=7.1Hz),6.12(1H,s),7.24−7.35(5H,m).
IR(ATR)ν:1724,1687,1427,1369,1315,1279,1230,1161,1132,700cm-1.
MS(ESI)m/z:316(M+1)+.
HRMS(EI)m/z:315.1842(Calcd for C19H25NO3 315.1834).
【0242】
[実施例9]
(7S)−7−メチル−4−オキソ−5−[(1R)−1−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸第三級ブチル
【0243】
【化56】
【0244】
トリメチルスルホニウムヨージド(401mg,1.82mmol)のジメチルスルホキシド(8mL)溶液に、水素化ナトリウム(60%油状、76.1mg,1.74mmol)、および(3S)−3−メチル−4−メチレン−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル(500mg,1.59mmol)のジメチルスルホキシド(24mL)溶液を、順次加え、90℃にて30分間攪拌した。反応液に10%クエン酸水溶液(32mL)、および水(96mL)を室温にて加え、酢酸エチル(200mL×2)にて抽出し、有機層を水(100mL)、および飽和食塩水(100mL)にて洗浄した。有機層を無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧乾固した。得られた残留物をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=3:1→2:1にて溶出)で精製して、標記化合物315mg(60.3%)を無色結晶として得た。本成績体の1H−NMRとIRの各スペクトルデータは、実施例5に記載したデータと一致した。
MS(FAB+)m/z:330(M+H)+.
HRMS(FAB+)m/z:330.2069(Calcd for C20H28NO3 330.2069).
【0245】
[実施例10]
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸
【0246】
【化57】
【0247】
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸第三級ブチル(24.5g,74.4mmol)のジクロロメタン(120mL)溶液に、氷冷下、トリフルオロ酢酸(120mL)を滴下し、2時間撹拌した。反応混合物を減圧乾固し、残留物にトルエン(20mL)を加えて減圧乾固した後、氷冷下、残留物を1mol/l水酸化ナトリウム水溶液(300mL)に溶解した。この水溶液を酢酸エチル(350mL)にて洗浄後、水層に氷冷下濃塩酸(25mL)を加えpH2〜3とし、クロロホルム(300mL×2)にて抽出した。有機層を水(200mL)および飽和食塩水(100mL)にて洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた残留物にトルエン(20mL)を加えて減圧乾固した後、残留物をクロロホルム(20mL)に懸濁し、ヘキサン(200mL)を加え、結晶化させた。析出した固体をヘキサン(100mL)にて洗浄後、減圧乾燥して標記化合物20.48g(定量的)を白色固体として得た。本成績体は精製せずに次工程に使用した。
【0248】
1H−NMR(400MHz,CDCl3)δ:0.78−0.83(1H,m),0.90−0.95(1H,m),1.08−1.18(2H,m),1.24(3H,s),1.55(3H,d,J=7.3Hz),3.11(1H,d,J=10.0Hz),3.55(1H,d,J=10.0Hz),5.52(1H,q,J=7.1Hz),7.28−7.32(5H,m).
MS(ESI)m/z:274(M+H)+.
【0249】
[実施例11]
(7S)−7−アミノ−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン
【0250】
【化58】
【0251】
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸(20.4g,74.4mmol)、およびジフェニルリン酸アジド(17.6mL,81.8mmol)のトルエン(200mL)溶液へ、トリエチルアミン(20.7mL,149mmol)を加え、125℃のオイルバスで1時間加熱攪拌した。反応液を減圧濃縮してイソシアネート体粗生成物を得た。
得られたイソシアネート体粗生成物を1,4−ジオキサン(180mL)に溶解し、水(90mL)、および濃塩酸(90mL)を加えた後、50℃のオイルバスで1時間加熱攪拌した。反応液に水(200mL)を加え、酢酸エチル(200mL)にて洗浄後、水層に氷冷下にて10mol/L水酸化ナトリウム水溶液(170mL)を加えpH9〜10とし、トルエン(200mL×2)にて抽出した。有機層を飽和食塩水(100mL)にて洗浄し、無水硫酸ナトリウムで乾燥後、ろ過し、ろ液を減圧濃縮して標記化合物15.8g(64.7mmol)を淡黄色油状物として得た。尚、本成績体は、精製せずに次の工程に使用した。
【0252】
1H−NMR(400MHz,CDCl3)δ:0.72−0.78(2H,m),0.99−1.10(2H,m),1.08(3H,s),1.53(3H,d,J=7.4Hz),2.82(1H,d,J=9.6Hz),3.27(1H,d,J=9.6Hz),5.56(1H,q,J=7.1Hz),7.14−7.37(5H,m).
【0253】
[実施例12]
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン
【0254】
【化59】
【0255】
前述の(7S)−7−アミノ−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン(15.8g,64.7mmol)をトルエン(82mL)に溶解し、内温が70℃を越えないよう氷冷しながら、ナトリウム水素化ビス(2−メトキシエトキシ)アルミニウム、65%(重量)トルエン溶液(77.6mL,259mmol)のトルエン(6mL)溶液を15分かけて滴下した後に、80℃のオイルバスで10分間加熱攪拌した。反応液を氷冷し、25%(重量)水酸化ナトリウム水溶液(158mL)を滴下して反応を停止させた後、トルエン(135mL)にて抽出した。有機層を飽和食塩水(100mL)にて洗浄後、これにジ−第三級ブチルジカーボネート(15.6g,71.2mmol)を加えた。反応液を室温にて3時間攪拌後、溶媒を減圧留去して得られた残留物をシリカゲルカラムクロマトグラフィー(へキサン:酢酸エチル=8:1→4:1→1:1にて溶出)にて精製し、標記化合物18.0g(73%)を無色透明シロップ状物質として得た。
【0256】
1H−NMR(400MHz,CDCl3)δ:0.37−0.49(2H,m),0.62−0.68(1H,m),0.77−0.82(1H,m),1.20(3H,s),1.32(3H,d,J=6.6Hz),1.44(9H,s),2.46(2H,dd,J=33.2,9.3Hz),2.68(1H,d,J=8.8Hz),3.27(1H,q,J=6.6Hz),3.31−3.34(1H,m),4.71(1H,s),7.19−7.34(5H,m).
MS(ESI)m/z:331(M+H)+.
【0257】
[実施例13]
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−アザスピロ[2.4]ヘプタン
【0258】
【化60】
【0259】
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン(18.0g,54.5mmol)のメタノール(180mL)溶液へ10%パラジウム炭素触媒(52.8%含水,9.00g)を加え、水素ガス雰囲気下、室温にて18時間攪拌後、さらに40℃のオイルバスで5.5時間撹拌した。触媒をろ去後、溶媒を減圧乾固し、標的化合物の粗生成物13.4g(定量的)を白色固体として得た。
【0260】
1H−NMR(400MHz,CDCl3)δ:0.38−0.43(1H,m),0.54−0.61(2H,m),0.74−0.80(1H,m),1.08(3H,s),1.44(9H,s),2.75(1H,d,J=7.6Hz),2.78(1H,d,J=7.1Hz),3.13(1H,d,J=11.5Hz),3.73−3.77(1H,m),4.45(1H,s).
MS(ESI)m/z:227(M+H)+.
【0261】
[実施例14]
7−[(7S)−7−アミノ−7−メチル−5−アザスピロ[2.4]ヘプタン−5−イル]−6−フルオロ−1−[(1R,2S)−2−フルオロシクロプロピル]−8−メトキシ−1,4−ジヒドロ−4−オキソキノリン−3−カルボン酸
【0262】
【化61】
【0263】
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−アザスピロ[2.4]ヘプタン(13.4g,54.5mmol)、6,7−ジフルオロ−1−[(1R,2S)−2−フルオロシクロプロピル]−8−メトキシ−1,4−ジヒドロ−4−オキソキノリン−3−カルボン酸・ジフルオロボラン錯体(17.9g,49.5mmol)、およびトリエチルアミン(8.97mL,64.4mmol)をジメチルスルホキシド(52mL)に溶解し、40℃のオイルバスで17時間加熱攪拌した。反応液を冷水(1000mL)に注ぎ、析出した固体をろ取した。この固体にエタノール:水=5:1混合溶液(180mL)、およびトリエチルアミン(15mL)を加えて1.5時間加熱還流した。反応混合物を減圧乾固して得られた残留物を酢酸エチル(150mL×2)に溶解し、10%クエン酸水溶液(200mL)、水(200mL)、飽和食塩水(100mL)にて洗浄した。有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた残留物をクロロホルム:メタノール=9:1混合溶液(100mL)に溶解し、シリカゲル(10g)を加え1時間撹拌した。シリカゲルをろ去し、クロロホルム:メタノール=9:1混合溶液(50mL×2)にて洗浄し、ろ液を合わせて濃縮乾固した。残留物を氷冷下にて濃塩酸(200mL)に溶解後、室温にて30分間撹拌し、反応液をクロロホルム(400mL×5)にて洗浄した。水層に氷冷下10mol/L水酸化ナトリウム水溶液を加えpH11.8とし、次いで塩酸にてpH7.4に調整後、クロロホルム(1000mL×3)にて抽出した。有機層を無水硫酸ナトリウムで乾燥後、溶媒を減圧留去した。得られた残留物をエタノールから再結晶精製し、減圧乾燥して標記化合物18.5g(79%)を淡桃色粉末として得た。
本成績体の1H−NMRをはじめとする機器データは、特願2005−146386号に記載されている実施例9の化合物のデータと完全に一致した。すなわち、特願2005−146386号に記載されている7−アミノ−7−メチル−5−アザスピロ[2.4]ヘプタン−5−イル基を有するキノロン誘導体のうち、高活性な化合物である実施例9に記載されたキノロン誘導体の7位の立体は、(7S)と判明した。
【特許請求の範囲】
【請求項1】
式(VII)
【化1】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物をエステルの加水分解条件下にて処理して式(IX)
【化2】
(式中、R1の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物のカルボキシ基をアミノ基へ転位反応させて式(X)
【化3】
(式中、R”は、水素原子、またはアミノ基の保護基を意味し、R1の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を直接に還元条件下、もしくはカルボニル基をチオカルボニル基へ変換後に還元条件下に処理して式(XI)
【化4】
(式中、R1およびR”の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を置換基R1の脱保護反応に付すことを特徴とする式(XII)
【化5】
(式中、R”の定義は、先の定義と同一である。)
で表わされる化合物の製造方法。
【請求項2】
式(VII)の化合物が、式(I)
【化6】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物に、塩基存在下でメチル化剤を反応させて、式(II)
【化7】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物に、塩基存在下で式(III)
【化8】
(式中、R’は、水酸基の保護基を意味し、Xは、ハロゲン原子、または求核置換反応で用いられる脱離基を意味する。)
で表わされる化合物を反応させて、式(IV)
【化9】
(式中、R1、R、およびR’の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物の水酸基の保護基を脱保護して式(V)
【化10】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物の水酸基をハロゲン原子、または求核置換反応で用いられるハロゲン原子以外の脱離基に変換させて式(VI)
【化11】
(式中、Yは、ハロゲン原子、または求核置換反応で用いられる脱離基を意味し、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を塩基性条件下で処理することによって製造されたものである請求項1に記載の製造方法。
【請求項3】
式(VII)の化合物が、式(II)
【化12】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物をエキソメチレン化して式(VIII)
【化13】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物のエキソメチレン基をシクロプロパン化反応に付すことによって製造されたものである請求項1に記載の製造方法。
【請求項4】
置換基R1が、1−(R)−フェニルエチル基、または1−(S)−フェニルエチル基である請求項1から3のいずれか一項に記載の製造方法。
【請求項5】
置換基Rが、炭素数1から6のアルキル基である請求項1から4のいずれか一項に記載の製造方法。
【請求項6】
置換基Rが、第三級ブチル基である請求項5に記載の製造方法。
【請求項7】
置換基R”が、水素原子である請求項1から6のいずれか一項に記載の製造方法。
【請求項8】
置換基R”が、アミノ基の保護基である請求項1から6のいずれか一項に記載の製造方法。
【請求項9】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、置換基を有していてもよいアシル基、置換基を有していてもよいアルキル基、置換基を有していてもよいアラルキル基、および置換シリル基からなる群から選ばれる保護基である請求項8に記載の製造方法。
【請求項10】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、および置換基を有していてもよいアシル基からなる群から選ばれる保護基である請求項8に記載の製造方法。
【請求項11】
アミノ基の保護基が、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、ベンジルオキシカルボニル基、アセチル基、またはトリフルオロアセチル基である請求項8に記載の製造方法。
【請求項12】
請求項1から11のいずれか一項に記載の(7S)−7−(第三級ブトキシカルボニル)アミノ−7−メチル−5−アザスピロ[2.4]へプタンの製造方法。
【請求項13】
請求項1から11のいずれか一項に記載の(7S)−7−アミノ−7−メチル−5−アザスピロ[2.4]へプタンの製造方法。
【請求項14】
式(II)
【化14】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項15】
置換基Rが、炭素数1から6のアルキル基である請求項14に記載の化合物。
【請求項16】
置換基Rが、第三級ブチル基である請求項15に記載の化合物。
【請求項17】
置換基R1が、1−(R)−フェニルエチル基、または1−(S)−フェニルエチル基である請求項14から16のいずれか一項に記載の化合物。
【請求項18】
(3S)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル。
【請求項19】
式(IV)
【化15】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項20】
Rが、炭素数1から6のアルキル基である請求項19に記載の化合物。
【請求項21】
Rが、第三級ブチル基である請求項20に記載の化合物。
【請求項22】
置換基R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項19から21のいずれか一項に記載の化合物。
【請求項23】
式(V)
【化16】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項24】
Rが、炭素数1から6のアルキル基である請求項23に記載の化合物。
【請求項25】
Rが、第三級ブチル基である請求項24に記載の化合物。
【請求項26】
置換基R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項23から25のいずれか一項に記載の化合物。
【請求項27】
式(VI)
【化17】
(式中、Yは、ハロゲン原子、または求核置換反応で用いられる脱離基を意味し、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項28】
Yが、ハロゲン原子である請求項27に記載の化合物。
【請求項29】
Yが、臭素原子またはヨウ素原子である請求項28に記載の化合物。
【請求項30】
Yが、メタンスルホニルオキシ基、トリフルオロメタンスルホニルオキシ基、トルエンスルホニルオキシ基、またはベンゼンスルホニルオキシ基である請求項27に記載の化合物。
【請求項31】
Yが、ベンゼンスルホニルオキシ基である請求項27に記載の化合物。
【請求項32】
Rが、炭素数1から6のアルキル基である請求項27から31のいずれか一項に記載の化合物。
【請求項33】
Rが、第三級ブチル基である請求項27から31のいずれか一項に記載の化合物。
【請求項34】
置換基R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項27から33のいずれか一項に記載の化合物。
【請求項35】
式(VIII)
【化18】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項36】
Rが、炭素数1から6のアルキル基である請求項35に記載の化合物。
【請求項37】
Rが、第三級ブチル基である請求項36に記載の化合物。
【請求項38】
R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項35から37のいずれか一項に記載の化合物。
【請求項39】
(3S)−3−メチル−4−メチレン−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル。
【請求項40】
式(IX)
【化19】
(式中、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項41】
R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項40に記載の化合物。
【請求項42】
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸、その塩、またはそれらの水和物。
【請求項43】
式(X)
【化20】
(式中、R”は、水素原子、またはアミノ基の保護基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項44】
R”が、アミノ基の保護基である請求項43に記載の化合物。
【請求項45】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、置換基を有していてもよいアシル基、置換基を有していてもよいアラルキル基、および置換シリル基からなる群から選ばれる基である請求項44に記載の化合物。
【請求項46】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、および置換基を有していてもよいアシル基である請求項44に記載の化合物。
【請求項47】
アミノ基の保護基が、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、ベンジルオキシカルボニル基、アセチル基、およびトリフルオロアセチル基である請求項44に記載の化合物。
【請求項48】
R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項43から47のいずれか一項に記載の化合物。
【請求項49】
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]へプタン。
【請求項50】
R”が、水素原子である請求項43に記載の化合物。
【請求項51】
R1が、1−(R)−フェニルエチル基、または1−(S)−フェニルエチル基である請求項50に記載の化合物。
【請求項52】
(7S)−7−アミノ−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]へプタン、その塩、またはそれらの水和物。
【請求項53】
式(XI)
【化21】
(式中、R”は、水素原子、またはアミノ基の保護基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項54】
R”が、アミノ基の保護基である請求項53に記載の化合物。
【請求項55】
アミノ基の保護基が置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、置換基を有していてもよいアシル基、置換基を有していてもよいアラルキル基、および置換シリル基からなる群から選ばれる基である請求項54に記載の化合物。
【請求項56】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、および置換基を有していてもよいアシル基である請求項54に記載の化合物。
【請求項57】
アミノ基の保護基が、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、ベンジルオキシカルボニル基、アセチル基、およびトリフルオロアセチル基である請求項54に記載の化合物。
【請求項58】
R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項53から57のいずれか一項に記載の化合物。
【請求項59】
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]へプタン、その塩、またはそれらの水和物。
【請求項60】
R”が、水素原子である請求項53に記載の化合物。
【請求項61】
R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項60に記載の化合物。
【請求項62】
(7S)−7−アミノ−7−メチル−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]へプタン、その塩、またはそれらの水和物。
【請求項63】
式(XII)
【化22】
(式中、R”は、水素原子、またはアミノ基の保護基を意味する。)
で表わされる化合物。
【請求項64】
R”が、アミノ基の保護基である請求項63に記載の化合物。
【請求項65】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、置換基を有していてもよいアシル基、置換基を有していてもよいアラルキル基、および置換シリル基からなる群から選ばれる基である請求項64に記載の化合物。
【請求項66】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、および置換基を有していてもよいアシル基である請求項64に記載の化合物。
【請求項67】
アミノ基の保護基が、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、ベンジルオキシカルボニル基、アセチル基、およびトリフルオロアセチル基である請求項64に記載の化合物。
【請求項68】
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−アザスピロ[2.4]へプタン、その塩、またはそれらの水和物。
【請求項69】
R”が、水素原子である請求項63に記載の化合物。
【請求項70】
(7S)−7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン、その塩、またはそれらの水和物。
【請求項1】
式(VII)
【化1】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物をエステルの加水分解条件下にて処理して式(IX)
【化2】
(式中、R1の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物のカルボキシ基をアミノ基へ転位反応させて式(X)
【化3】
(式中、R”は、水素原子、またはアミノ基の保護基を意味し、R1の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を直接に還元条件下、もしくはカルボニル基をチオカルボニル基へ変換後に還元条件下に処理して式(XI)
【化4】
(式中、R1およびR”の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を置換基R1の脱保護反応に付すことを特徴とする式(XII)
【化5】
(式中、R”の定義は、先の定義と同一である。)
で表わされる化合物の製造方法。
【請求項2】
式(VII)の化合物が、式(I)
【化6】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物に、塩基存在下でメチル化剤を反応させて、式(II)
【化7】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物に、塩基存在下で式(III)
【化8】
(式中、R’は、水酸基の保護基を意味し、Xは、ハロゲン原子、または求核置換反応で用いられる脱離基を意味する。)
で表わされる化合物を反応させて、式(IV)
【化9】
(式中、R1、R、およびR’の定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物の水酸基の保護基を脱保護して式(V)
【化10】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物の水酸基をハロゲン原子、または求核置換反応で用いられるハロゲン原子以外の脱離基に変換させて式(VI)
【化11】
(式中、Yは、ハロゲン原子、または求核置換反応で用いられる脱離基を意味し、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物を塩基性条件下で処理することによって製造されたものである請求項1に記載の製造方法。
【請求項3】
式(VII)の化合物が、式(II)
【化12】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物をエキソメチレン化して式(VIII)
【化13】
(式中、R1およびRの定義は、先の定義と同一である。)
で表わされる化合物を得、この化合物のエキソメチレン基をシクロプロパン化反応に付すことによって製造されたものである請求項1に記載の製造方法。
【請求項4】
置換基R1が、1−(R)−フェニルエチル基、または1−(S)−フェニルエチル基である請求項1から3のいずれか一項に記載の製造方法。
【請求項5】
置換基Rが、炭素数1から6のアルキル基である請求項1から4のいずれか一項に記載の製造方法。
【請求項6】
置換基Rが、第三級ブチル基である請求項5に記載の製造方法。
【請求項7】
置換基R”が、水素原子である請求項1から6のいずれか一項に記載の製造方法。
【請求項8】
置換基R”が、アミノ基の保護基である請求項1から6のいずれか一項に記載の製造方法。
【請求項9】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、置換基を有していてもよいアシル基、置換基を有していてもよいアルキル基、置換基を有していてもよいアラルキル基、および置換シリル基からなる群から選ばれる保護基である請求項8に記載の製造方法。
【請求項10】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、および置換基を有していてもよいアシル基からなる群から選ばれる保護基である請求項8に記載の製造方法。
【請求項11】
アミノ基の保護基が、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、ベンジルオキシカルボニル基、アセチル基、またはトリフルオロアセチル基である請求項8に記載の製造方法。
【請求項12】
請求項1から11のいずれか一項に記載の(7S)−7−(第三級ブトキシカルボニル)アミノ−7−メチル−5−アザスピロ[2.4]へプタンの製造方法。
【請求項13】
請求項1から11のいずれか一項に記載の(7S)−7−アミノ−7−メチル−5−アザスピロ[2.4]へプタンの製造方法。
【請求項14】
式(II)
【化14】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項15】
置換基Rが、炭素数1から6のアルキル基である請求項14に記載の化合物。
【請求項16】
置換基Rが、第三級ブチル基である請求項15に記載の化合物。
【請求項17】
置換基R1が、1−(R)−フェニルエチル基、または1−(S)−フェニルエチル基である請求項14から16のいずれか一項に記載の化合物。
【請求項18】
(3S)−3−メチル−5−オキソ−1−[(1R)−1−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル。
【請求項19】
式(IV)
【化15】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項20】
Rが、炭素数1から6のアルキル基である請求項19に記載の化合物。
【請求項21】
Rが、第三級ブチル基である請求項20に記載の化合物。
【請求項22】
置換基R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項19から21のいずれか一項に記載の化合物。
【請求項23】
式(V)
【化16】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項24】
Rが、炭素数1から6のアルキル基である請求項23に記載の化合物。
【請求項25】
Rが、第三級ブチル基である請求項24に記載の化合物。
【請求項26】
置換基R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項23から25のいずれか一項に記載の化合物。
【請求項27】
式(VI)
【化17】
(式中、Yは、ハロゲン原子、または求核置換反応で用いられる脱離基を意味し、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項28】
Yが、ハロゲン原子である請求項27に記載の化合物。
【請求項29】
Yが、臭素原子またはヨウ素原子である請求項28に記載の化合物。
【請求項30】
Yが、メタンスルホニルオキシ基、トリフルオロメタンスルホニルオキシ基、トルエンスルホニルオキシ基、またはベンゼンスルホニルオキシ基である請求項27に記載の化合物。
【請求項31】
Yが、ベンゼンスルホニルオキシ基である請求項27に記載の化合物。
【請求項32】
Rが、炭素数1から6のアルキル基である請求項27から31のいずれか一項に記載の化合物。
【請求項33】
Rが、第三級ブチル基である請求項27から31のいずれか一項に記載の化合物。
【請求項34】
置換基R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項27から33のいずれか一項に記載の化合物。
【請求項35】
式(VIII)
【化18】
(式中、Rは、置換基を有していてもよいアリール基、置換基を有していてもよいアラルキル基、置換基を有していてもよい炭素数1から6のアルキル基、または、カルボン酸とエステル結合を形成し得る不斉補助基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項36】
Rが、炭素数1から6のアルキル基である請求項35に記載の化合物。
【請求項37】
Rが、第三級ブチル基である請求項36に記載の化合物。
【請求項38】
R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項35から37のいずれか一項に記載の化合物。
【請求項39】
(3S)−3−メチル−4−メチレン−5−オキソ−1−[(1R)−フェニルエチル]ピロリジン−3−カルボン酸第三級ブチル。
【請求項40】
式(IX)
【化19】
(式中、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項41】
R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項40に記載の化合物。
【請求項42】
(7S)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]ヘプタン−7−カルボン酸、その塩、またはそれらの水和物。
【請求項43】
式(X)
【化20】
(式中、R”は、水素原子、またはアミノ基の保護基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項44】
R”が、アミノ基の保護基である請求項43に記載の化合物。
【請求項45】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、置換基を有していてもよいアシル基、置換基を有していてもよいアラルキル基、および置換シリル基からなる群から選ばれる基である請求項44に記載の化合物。
【請求項46】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、および置換基を有していてもよいアシル基である請求項44に記載の化合物。
【請求項47】
アミノ基の保護基が、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、ベンジルオキシカルボニル基、アセチル基、およびトリフルオロアセチル基である請求項44に記載の化合物。
【請求項48】
R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項43から47のいずれか一項に記載の化合物。
【請求項49】
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]へプタン。
【請求項50】
R”が、水素原子である請求項43に記載の化合物。
【請求項51】
R1が、1−(R)−フェニルエチル基、または1−(S)−フェニルエチル基である請求項50に記載の化合物。
【請求項52】
(7S)−7−アミノ−7−メチル−4−オキソ−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]へプタン、その塩、またはそれらの水和物。
【請求項53】
式(XI)
【化21】
(式中、R”は、水素原子、またはアミノ基の保護基を意味し、R1は、置換基を有していてもよいアラルキル基を意味する。)
で表わされる化合物。
【請求項54】
R”が、アミノ基の保護基である請求項53に記載の化合物。
【請求項55】
アミノ基の保護基が置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、置換基を有していてもよいアシル基、置換基を有していてもよいアラルキル基、および置換シリル基からなる群から選ばれる基である請求項54に記載の化合物。
【請求項56】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、および置換基を有していてもよいアシル基である請求項54に記載の化合物。
【請求項57】
アミノ基の保護基が、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、ベンジルオキシカルボニル基、アセチル基、およびトリフルオロアセチル基である請求項54に記載の化合物。
【請求項58】
R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項53から57のいずれか一項に記載の化合物。
【請求項59】
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]へプタン、その塩、またはそれらの水和物。
【請求項60】
R”が、水素原子である請求項53に記載の化合物。
【請求項61】
R1が、1−(R)−フェニルエチル基または1−(S)−フェニルエチル基である請求項60に記載の化合物。
【請求項62】
(7S)−7−アミノ−7−メチル−5−[(1R)−フェニルエチル]−5−アザスピロ[2.4]へプタン、その塩、またはそれらの水和物。
【請求項63】
式(XII)
【化22】
(式中、R”は、水素原子、またはアミノ基の保護基を意味する。)
で表わされる化合物。
【請求項64】
R”が、アミノ基の保護基である請求項63に記載の化合物。
【請求項65】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、置換基を有していてもよいアシル基、置換基を有していてもよいアラルキル基、および置換シリル基からなる群から選ばれる基である請求項64に記載の化合物。
【請求項66】
アミノ基の保護基が、置換基を有していてもよいアルコキシカルボニル基、置換基を有していてもよいアラルキルオキシカルボニル基、および置換基を有していてもよいアシル基である請求項64に記載の化合物。
【請求項67】
アミノ基の保護基が、メトキシカルボニル基、エトキシカルボニル基、第三級ブトキシカルボニル基、ベンジルオキシカルボニル基、アセチル基、およびトリフルオロアセチル基である請求項64に記載の化合物。
【請求項68】
(7S)−7−(第三級ブトキシカルボニルアミノ)−7−メチル−5−アザスピロ[2.4]へプタン、その塩、またはそれらの水和物。
【請求項69】
R”が、水素原子である請求項63に記載の化合物。
【請求項70】
(7S)−7−アミノ−7−メチル−5−アザスピロ[2.4]へプタン、その塩、またはそれらの水和物。
【図1】

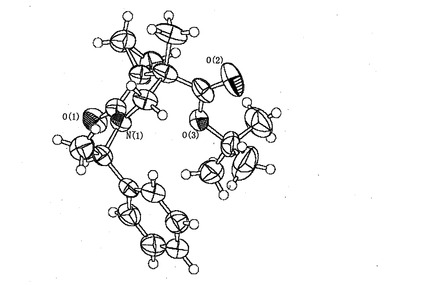
【公開番号】特開2012−254991(P2012−254991A)
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−158293(P2012−158293)
【出願日】平成24年7月17日(2012.7.17)
【分割の表示】特願2007−537660(P2007−537660)の分割
【原出願日】平成18年9月28日(2006.9.28)
【出願人】(307010166)第一三共株式会社 (196)
【Fターム(参考)】
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願日】平成24年7月17日(2012.7.17)
【分割の表示】特願2007−537660(P2007−537660)の分割
【原出願日】平成18年9月28日(2006.9.28)
【出願人】(307010166)第一三共株式会社 (196)
【Fターム(参考)】
[ Back to top ]