
デオキシニバレノールの分解活性を有するタンパク質をコードする遺伝子
【課題】デオキシニバレノールを分解する活性を有するたんぱく質、及び該たんぱく質をコードする遺伝子の提供。
【解決手段】(A)特定な配列からなるアミノ酸配列、(B)特定な配列からなるアミノ酸配列(A)において1若しくは複数のアミノ酸が欠失、置換または付加されているアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列、又は(C)特定な配列からなるアミノ酸配列(A)と少なくとも80%以上の相同性を有するアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列であり、更に本発明は、(D)(A)とは別の特定な配列からなる塩基配列、又は(E)(D)の塩基配列において1若しくは複数の塩基が欠失、置換または付加されている塩基配列であって、デオキシニバレノール分解活性を有するタンパク質をコードする塩基配列である。
【解決手段】(A)特定な配列からなるアミノ酸配列、(B)特定な配列からなるアミノ酸配列(A)において1若しくは複数のアミノ酸が欠失、置換または付加されているアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列、又は(C)特定な配列からなるアミノ酸配列(A)と少なくとも80%以上の相同性を有するアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列であり、更に本発明は、(D)(A)とは別の特定な配列からなる塩基配列、又は(E)(D)の塩基配列において1若しくは複数の塩基が欠失、置換または付加されている塩基配列であって、デオキシニバレノール分解活性を有するタンパク質をコードする塩基配列である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、デオキシニバレノール分解活性を有するタンパク質、およびデオキシニバレノール分解活性を有するタンパク質をコードする新規遺伝子に関する。
【背景技術】
【0002】
かび毒は、ある種のかびが農作物に付着・増殖し、そこで産生する化学物質(天然毒素)のうち、人や家畜の健康に悪影響を及ぼすものの総称であり、現在300種類以上あるといわれている。代表的なかび毒として、アフラトキシン(ナッツ類、穀類)、デオキシニバレノール(穀類)、パツリン(りんご加工品)、オクラトキシンA(穀類、豆類)などがある。このうちわが国では平成20年8月現在、アフラトキシンB1(農作物を含む一般の食品)、デオキシニバレノール(小麦)、パツリン(リンゴ果汁)が食品衛生法に基づく規制の対象となっている。
【0003】
ムギ類を始めとする穀類に広く感染する赤かび病菌Fusarium graminearumが産生するデオキシニバレノール(以下DONと略称することがある。)、ニバレノール、T−2トキシンは、トリコテセン系マイコトキシンであり、ムギ生産地帯で大きな問題となっている。中でもデオキシニバレノールは、各国のムギ生産地帯で発生しているマイコトキシン汚染の主要因であり、摂取すると嘔吐、下痢などの急性中毒を引き起こすことから、世界的に本毒素の対策が喫緊の課題となっている。我が国でも平成14年5月に、厚生労働省においてコムギ粒の暫定基準濃度が1.1ppm以下と定められた。
【0004】
従来この対策としては、化学的手法により赤かび病菌を制御する方法が用いられてきた。また近年は植物体に残存する化学農薬の危険性が指摘されることから、微生物農薬がより安心で安全な植物病害防除剤として注目されている(例えば、特許文献1及び特許文献2を参照)。またバチルス・ズブチリスに属する菌株が産生するアイツリンAのフザリウムに対する増殖抑制効果を利用し、当該菌株の培養液で農産物を処理する方法が提案されている(特許文献3を参照。)。
【0005】
しかしデオキシニバレノールはトリコテセン骨格の構造を有し、121℃、20分の加圧・高温処理に対しても安定である。そのため、一旦赤かび病菌が発生した植物体においては、赤かび病を殺菌剤等で防除した後であっても、罹病した植物体の穂等に残存する。さらにデオキシニバレノールの特徴として、赤かび病が感染した植物体においては、発病したムギ粒等だけではなく、無病徴のムギ粒等にも蓄積される。このため、発病の有無にかかわらず、赤かび病菌汚染にされたムギ粒に蓄積されたデオキシニバレノールを低減する技術が求められている。
【0006】
そのためにデオキシニバレノールを直接除去する方法として、食品のレベルで物理的吸着により不活性化または排除する、家畜が食べてもそのまま排泄可能なカビ毒吸着剤(特許文献4を参照。)や、マイコトキシンを吸着除去する金属酸化物粒子複合体(特許文献5を参照。)が提案されている。
【0007】
一方で、食品となる以前に、直接穀物等に付着したデオキシニバレノールを除去することが望まれており、デオキシニバレノール分解微生物の探索が進められている。デオキシニバレノール分解微生物としては、現在のところ、土壌から分離されたグラム陰性菌Agrobacterium-RhizobiumグループのE3-39株(非特許文献1を参照。)、嫌気性細菌BBSH797(非特許文献2を参照。)、ムギ根圏土壌から分離されたDevosia属細菌に近縁のRS1株(特許文献6を参照。)、ムギ根圏土壌から分離されたNocardioides属に属するWSN05-2株、LS1株、SS1株およびSS2株(特許文献7を参照。)が報告されている。しかしながら、これまでにデオキシニバレノールの分解活性を有するタンパク質、およびデオキシニバレノール分解酵素をコードする遺伝子がデオキシニバレノール分解細菌株から同定された事例は皆無である。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2003−206212号公報
【特許文献2】特開2003−192515号公報
【特許文献3】特開平5−85911号公報
【特許文献4】特表2002−512011号公報
【特許文献5】特表2004−500214号公報
【特許文献6】特開2008−220179号公報
【特許文献7】特開2008−220184号公報
【非特許文献】
【0009】
【非特許文献1】Shima J. et. al (1997)Appl. Environ. Microbiol., 63: 3825-3830
【非特許文献2】FuchsE. et al (2002) Food Additives and Contaminants., 19: 379-386
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、デオキシニバレノールを分解する活性を有するタンパク質、及び該タンパク質をコードする遺伝子を提供することを課題とする。
【課題を解決するための手段】
【0011】
本発明者らは、霞ヶ浦湖水に生息する微生物の中から、デオキシニバレノールを効率よく分解することができる菌株を見出し、該菌株よりデオキシニバレノール分解酵素をコードする遺伝子のクローニングに成功し、本発明を完成するに至った。すなわち本発明は以下の通りである。
【0012】
<1> 下記(A)乃至(C)のいずれかのアミノ酸配列を含むデオキシニバレノール分解酵素タンパク質である。
(A)配列番号2に記載のアミノ酸配列。
(B)配列番号2に記載のアミノ酸配列において1若しくは複数のアミノ酸が欠失、置換または付加されているアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
(C)配列番号2に記載のアミノ酸配列と少なくとも80%以上の相同性を有するアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
<2> また本発明は、下記(A)乃至(C)のいずれかのアミノ酸配列をコードするデオキシニバレノール分解酵素遺伝子である。
(A)配列番号2に記載のアミノ酸配列。
(B)配列番号2に記載のアミノ酸配列において1若しくは複数のアミノ酸が欠失、置換または付加されているアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
(C)配列番号2に記載のアミノ酸配列と少なくとも80%以上の相同性を有するアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
<3> さらに本発明は、下記(D)乃至(F)のいずれかの塩基配列を含むデオキシニバレノール分解酵素遺伝子である。
(D)配列番号1で表される塩基配列。
(E)配列番号1に記載の塩基配列において1若しくは複数の塩基が欠失、置換または付加されている塩基配列であって、デオキシニバレノール分解活性を有するタンパク質をコードする塩基配列。
(F)配列番号1に記載の塩基配列とストリンジェントな条件下でハイブリダイズし、且つデオキシニバレノール分解活性を有するタンパク質をコードする塩基配列。
<4> さらに本発明は、前記の遺伝子を含む組み換えベクターである。
<5> さらに本発明は、前記の遺伝子を含む形質転換体である。
<6> さらに本発明は、前記の組換えベクターを用いる形質転換体である。
<7> さらに本発明は、前記の形質転換体を用いるデオキシニバレノールの分解方法である。
【発明の効果】
【0013】
本発明において、熱に安定で、難分解性であるデオキシニバレノールを分解するタンパク質、及び該タンパク質をコードする遺伝子を提供する。
【図面の簡単な説明】
【0014】
【図1】pKSM4511を保持するUT26株によるDON (20 mg/ml)の分解を示すDON分解活性アッセイHPLCサンプルのHPLCクロマトグラム図である。
【図2】pKSM4511の挿入DNA断片に存在するDON分解酵素遺伝子の同定を示す。
【図3】配列番号1(ORF6)の遺伝子を導入したUT26株によるDON(20 mg/ml)の分解を示すDON分解活性アッセイHPLCサンプルのHPLCクロマトグラム図である。
【図4】配列番号1の開始コドン付近の塩基配列である。
【図5】配列番号2のアミノ酸配列に存在するshort chain dehydrogenase/reductaseのモチーフを示した。
【発明を実施するための最良の形態】
【0015】
本発明は、Sphingobium sp. に近縁の新規微生物KSM1菌株に由来する配列番号1に示されるデオキシニバレノール分解酵素遺伝子、及び配列番号2に示されるデオキシニバレノール分解酵素に関するものである。以下本発明について詳説する。
【0016】
<本発明に係る遺伝子について>
本発明は、デオキシニバレノール分解酵素をコードする配列番号1に示される遺伝子である。配列番号1に示す塩基配列は、前記新規微生物KSM1株由来のデオキシニバレノール分解酵素遺伝子の開始コドンから終始コドンまでの、いわゆるオープンリーディングフレーム(以下ORFとする。)である。
【0017】
本発明に係るデオキシニバレノール分解酵素遺伝子は好ましくは、配列番号1に示す塩基配列からなるポリヌクレオチドからなるORFであるが、この部分に相当する部分が含まれる限り、いずれの遺伝子であってもよい。例えば、該ORFに5'および3'UTRを加えた遺伝子も本発明に含まれる。配列番号1に示す塩基配列からなるポリヌクレオチドは、該新規微生物KSM1株(受領番号FERMAP-21835)のゲノミックDNAから取得できる。
【0018】
本発明はまた、配列番号2に記載のデオキシニバレノールを分解するタンパク質と機能的に同等なタンパク質をコードする遺伝子を含む。ここで「デオキシニバレノールを分解するタンパク質と機能的に同等」とは、対象となるタンパク質が配列番号2に記載のタンパク質と同等の生物学的機能、即ちデオキシニバレノール分解代謝を触媒する機能を有することを指す。
【0019】
本発明の遺伝子には、例えば、配列番号2に記載のアミノ酸配列において1若しくは数個のアミノ酸が置換、欠失、付加および/または挿入されたアミノ酸配列からなるタンパク質をコードする変異体、誘導体、アレル、バリアントおよびホモログが含まれる。
【0020】
アミノ酸配列が改変されたタンパク質をコードする遺伝子を調製するための当業者によく知られた方法としては、例えば「Kramer, W.&Fritz,H.-J. Methods in Enzymology, 154: 350-367(1987)」に記載されている、部位特異的突然変異誘発(site-directedmutagenesis)法が挙げられる。また、塩基配列の変異によりコードするタンパク質のアミノ酸配列が変異することは、自然界においても生じ得る。このように天然型の本発明のデオキシニバレノール分解酵素(配列番号2)をコードする遺伝子においても、または該天然型デオキシニバレノール分解酵素に1もしくは数個のアミノ酸が置換、欠失もしくは付加したアミノ酸配列を有するタンパク質をコードする遺伝子であっても、天然型のアミノ酸配列と同等の機能を有するタンパク質をコードする限り、本発明の遺伝子に含まれる。
【0021】
また、たとえ塩基配列が変異した場合でも、それがタンパク質中のアミノ酸の変異を伴わない場合(縮重変異)もあり、このような縮重変異体も本発明の遺伝子に含まれる。対象となる遺伝子を構成するDNAの塩基の変異数は、アミノ酸レベルにおいて、100アミノ酸以内、好ましくは50アミノ酸以内、さらに好ましくは20アミノ酸以内、さらに好ましくは10アミノ酸以内である。
【0022】
上記遺伝子を獲得する方法は特に限定されるものではなく、一般的な方法が採用される。例えば、当該遺伝子を、それを有する生物のゲノムDNA、ゲノムDNAライブラリーなどから適切な制限酵素で切り出し、精製すればよい。即ち、本発明のデオキシニバレノール分解酵素遺伝子には、ゲノムDNAおよび化学合成DNAが含まれる。ゲノムDNAの調製は、当業者にとって常套手段を利用して行うことが可能である。ゲノムDNAは、例えば、対象生物からゲノムDNAを抽出し、ゲノミックライブラリー(ベクターとしては、プラスミド、ファージ、コスミド、BAC、PACなどが利用できる)を作成し、これを展開して、本発明のタンパク質をコードするDNA(配列番号1)を基に調製したプローブを用いてコロニーハイブリダイゼーションあるいはプラークハイブリダイゼーションを行うことにより調製することが可能である。
【0023】
また、本発明のデオキシニバレノール分解酵素をコードするDNA(配列番号1)に特異的なプライマーを作成し、これを利用したPCRを行うことによって調製することも可能である。
【0024】
なお、デオキシニバレノール分解活性を有する微生物(特許文献6、特許文献7、非特許文献1、非特許文献2)にも前記遺伝子を保有する可能性が考えられるが、前記KSM1株を用いれば、より確実に目的の遺伝子を単離することができる。
【0025】
次に本発明のデオキシニバレノール分解酵素遺伝子の獲得方法について説明する。具体的には、ゲノムの少なくとも一部がデータベース化されている微生物から本発明のデオキシニバレノール分解酵素遺伝子を獲得する場合には、上記ポリヌクレオチドの塩基配列(配列番号1)に基づいて相同性のある塩基配列をデータベース中から検索すればよい。例えば、汎用されている相同性検索アルゴリズムであるBLASTNによる塩基配列の相同性検索を好適に用いることができる。
【0026】
また、ゲノムがデータベース化されていない微生物の場合には、例えば、従来公知のDNAライブラリーを用いたハイブリダイゼーション法を用いることもできる。具体的には、適切なクローニングベクターを使用して対象となる微生物からゲノムライブラリーを調製する工程と、上記ポリヌクレオチドの少なくとも一部をプローブとして用いてハイブリダイゼーションを行い、ライブラリーから上記プローブにポジティブの断片を検出する工程とを含む方法を用いることができる。このように、本発明に係る遺伝子はプローブとしても有用である。プローブに用いる領域には、目的とする遺伝子に特異的な配列が含まれることが好ましい。また、プローブとして使用されるポリヌクレオチドの長さは、100bp以上が好ましい。
【0027】
より具体的には、配列番号2に記載のデオキシニバレノール分解酵素と機能的に同等なタンパク質をコードする遺伝子を調製するために、当業者によく知られた方法としては、「Southern,E.M. Journal of Molecular Biology, 98, 503(1975)」に記載のハイブリダイゼーション技術や、「Saiki, R.K. et al. Science, 230, 1350-1354(1985)、Saiki, R. K. et al. Science, 239,487-491(1988)」に記載のポリメラーゼ連鎖反応(PCR)技術を利用する方法が挙げられる。このようにハイブリダイズ技術やPCR技術により単離しうる本発明のデオキシニバレノール分解酵素と同等の機能を有するタンパク質をコードする遺伝子もまた本発明の遺伝子に含まれる。
【0028】
このような遺伝子を単離するためには、好ましくはストリンジェントな条件下でハイブリダイゼーション反応を行う。本発明においてストリンジェントなハイブリダイゼーション条件とは、0.2%SDS、0.2x SSC、65℃の条件、またはこれと同等以上のストリンジェンシーのハイブリダイゼーション/洗浄条件を指し、例えば、0.2% SDS、0.1x SSC、65℃の条件を用いれば、より相同性の高い遺伝子の効率的な単離を期待することができる。
【0029】
前記により単離された遺伝子は、それがコードするアミノ酸レベルにおいて、本発明のタンパク質のアミノ酸配列(配列番号2)と高い相同性を有すると考えられる。高い相同性とは、アミノ酸配列全体で、少なくとも70%以上、さらに好ましくは80%以上の配列の同一性を指す。配列の同一性は、FASTA検索(Pearson W.R. and D.J. Lipman (1988) Proc. Natl. Acad. Sci. USA. 85:2444-2448)やBLASTP検索により決定することができる。
【0030】
<本発明に係るタンパク質について>
本発明に係るタンパク質は、上記遺伝子によってコードされる配列番号2で表されるアミノ酸配列からなるタンパク質あるいは、配列番号2で表されるアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列からなり、かつ、デオキシニバレノール分解酵素の活性を有するタンパク質、もしくは、配列番号2で表されるアミノ酸配列からなるタンパク質とアミノ酸レベルで80%以上、さらに好ましくは90%以上95%以上の相同性を示し、かつ、デオキシニバレノール分解酵素の活性を有するタンパク質である。
【0031】
また、本発明に係るタンパク質は、タンパク質の精製や検出等を容易に行うために、公知のHAやFlag等の付加配列を末端に含ませてもよいし、融合タンパク質であってもよい。また、N-グリコシル化などの各種修飾を受けていてもよい。
【0032】
本発明による組換えベクターは、上記遺伝子が組み込まれたものである。上記ベクターは、公知の形質転換方法によって宿主に発現可能に導入されることにより、当該宿主において組み込まれた遺伝子あるいは遺伝子断片を発現させて本発明に係るタンパク質を得ることが出来るものである。なお、本発明によるタンパク質は下記のように組換え産生により得られたものでもよいし、微生物から単離、精製したものでもよく、その起源は特に限定されない。
【0033】
組み換えタンパク質を調製する場合には、通常、本発明のタンパク質をコードする遺伝子を適当な発現ベクターに挿入し、該ベクターを適当な宿主細胞に導入し、形質転換宿主細胞を培養して発現させたタンパク質を精製する。組み換えタンパク質は、精製を容易にするなどの目的で、他のタンパク質との融合タンパク質として発現させることも可能である。
【0034】
例えば、大腸菌を宿主としてマルトース結合タンパク質との融合タンパク質として調製する方法(米国New England BioLabs社発売のベクターpMALシリーズ)、グルタチオン-S-トランスフェラーゼ(GST)との融合タンパク質として調製する方法(AmershamPharmacia Biotech社発売のベクターpGEXシリーズ)、ヒスチジンタグを付加して調製する方法(Novagen社のpETシリーズ)などを利用することが可能である。宿主細胞としては、組み換えタンパク質の発現に適した細胞であれば特に制限はなく、上記の大腸菌の他、例えば、酵母等の真菌細胞、種々の動植物細胞などを用いることが可能である。
【0035】
宿主細胞へのベクターの導入には、当業者に公知の種々の方法を用いることが可能である。例えば、大腸菌への導入には、「Mandel, M.& Higa,A. Journal of Molecular Biology, 53, 158-162(1970)、Hanahan, D. Journal ofMolecular Biology, 166, 557-580(1983)」に記載のカルシウムイオンを利用した導入方法を用いることができる。宿主細胞内で発現させた組み換えタンパク質は、該宿主細胞またはその培養上清から、当業者に公知の方法により精製し、回収することが可能である。組み換えタンパク質を上記のマルトース結合タンパク質などとの融合タンパク質として発現させた場合には、容易にアフィニティー精製を行うことが可能である。
【0036】
得られた組換えタンパク質を用いれば、これに結合する抗体を調製することができる。例えば、ポリクローナル抗体は、精製した本発明のタンパク質若しくはその一部のペプチドをウサギなどの免疫動物に免疫し、一定期間の後に血液を採取し、血ぺいを除去することにより調製することが可能である。また、モノクローナル抗体は、上記タンパク質若しくはペプチドで免疫した動物の抗体産生細胞と骨腫瘍細胞とを融合させ、目的とする抗体を産生する単一クローンの細胞(ハイブリドーマ)を単離し、該細胞を増殖させ、細胞から抗体を得ることにより調製することができる。これにより得られた抗体は、本発明のタンパク質の精製や検出などに利用することが可能である。本発明には、本発明のタンパク質に結合する抗体が含まれる。
【0037】
<形質転換体について>
本発明の遺伝子を発現する形質転換体を作製する場合には、該遺伝子を適当なベクターに挿入して、これを対象の宿主細胞に導入する。
【0038】
本発明に係る形質転換体は、所望により組換えベクターに含まれた、上記遺伝子が導入されたものである。より具体的に言えば、本発明に係る形質転換体は、本発明によるデオキシニバレノール分解酵素をコードする遺伝子が導入されたものである。ここで、「遺伝子または組換えベクターが導入された」とは、公知の遺伝子工学的手法(遺伝子操作技術)により、宿主内に所望によりベクター内に組み込まれた遺伝子が、例えばプロモーターの作用により、発現可能に導入されることを意味する。
【0039】
本発明に係る形質転換体は、上記遺伝子を直接、あるいは上記遺伝子が組み込まれたベクターを宿主に導入することにより得られる。上記宿主は、特に限定されるものではなく、例えば、大腸菌などの原核生物、酵母などの真菌、あるいは、植物、昆虫細胞などを挙げることができ、中でも宿主細胞としては、原核生物が好ましい。
【0040】
本発明の遺伝子は、宿主細胞内で発現可能なプロモーター、例えば、大腸菌ではLacプロモーター、酵母では、GAPDHプロモーターやアルコールオキシダーゼプロモーター、植物細胞では、カリフラワーモザイクウイルス35Sプロモーター、昆虫細胞では、バキュロウイルスプロモーター等の下流域に連結して使用する。
【0041】
上記形質転換宿主細胞を含む形質転換体を作製する方法としては、具体的には、例えば、ポリエチレングリコール法、電気穿孔法(エレクトロポーレーション)、アグロバクテリウムを介する方法、パーティクルガン法など当業者に公知の種々の方法を用いることができる。
【0042】
前記形質転換植物を作出する手法について具体的には、「Datta,S.K. (1995) In Gene Transfer ToPlants(Potrykus I and Spangenberg Eds.) pp66-74」に記載のポリエチレングリコールによりプロトプラストへ遺伝子導入し植物体を再生させる方法、「Tokiet al (1992) Plant Physiol. 100, 1503-1507」に記載の電気パルスによりプロトプラストへ遺伝子導入し植物体を再生させる方法、「Christouet al. (1991) Bio/technology, 9: 957-962.」に記載のパーティクルガン法により細胞へ遺伝子を直接導入し植物体を再生させる方法、および「Hieiet al. (1994) Plant J. 6: 271-282.」に記載のアグロバクテリウムを介して遺伝子を導入し、植物体を再生させる方法などを挙げることができるが、特に限定されるものではない。また、上記形質転換方法は、宿主となる生物の種類に応じて適宜選択されることが好ましい。即ち、例えば宿主が酵母である場合は、塩化リチウム法が好ましい。
【0043】
本発明において使用することができるベクターとしては、従来から細菌、真菌などの形質転換に使用されているベクター、例えば、細菌用には、pUC、pBluescript、pET系、RK2-oriを有する広宿主域ベクターなど、酵母用には、pGYR、pFLD、pYES2等、植物用には、pBI121を挙げることができる。
【0044】
前記ベクターは、上記遺伝子またはその断片の他に、従来公知の遺伝子を発現させるための構成的プロモーター、例えば、GAPDHプロモーターやカリフラワーモザイクウイルス35Sプロモーター、または発現誘導性プロモーター、例えば、Lacプロモーターやアルコールオキシダーゼプロモーター、そして形質転換体の選抜を容易にする薬剤耐性遺伝子、例えば、アンピシリンあるいは、カナマイシン、ハイグロマイシン抵抗性遺伝子などを含んでいることが好ましい。
【実施例】
【0045】
以下、本発明を実施例によりさらに詳細に説明するが、本発明はこれら実施例に制限されるものではない。実施例で示した遺伝子組換え実験は、特に言及しない限り、「Molecular Cloning 2nd. Ed,Cold Spring Harbor Press ,1989」に記載の方法(以下単に常法という。)を用いて行った。
【0046】
<KSM1株ゲノミックライブラリーの構築>
(1)ゲノミックDNAの抽出
-80℃冷凍庫に保存したKSM1株のグリセロールストックを、R2A寒天培地(Merck社製)に植菌し、28℃で6日間培養した。寒天培地上に生育したKSM1株菌体を、表1に示すCDM培地に、100mg/mlの濃度にDON(Wako社)を添加した培地100 mlに植菌した。植菌後の培地を、28℃、暗所で振とう培養法により6日間培養した。培養後の培地を、9,200x gにて3分間遠心し、KSM1株菌体を集菌した。この菌体より、AquaPureGenomic DNA Isolation Kit (Bio-Rad社製)を用いてゲノミックDNAを抽出した。以上の操作により、約27 mgのKSM1株ゲノミックDNAを得た。
【0047】
【表1】

【0048】
(2)ゲノミックDNAのサイズ分画
前記により抽出したKSM1株ゲノミックDNAを含む溶液から、20-23 Kb程度のDNA断片のみを得る目的で、以下の手順により該KSM1株ゲノミックDNAのサイズ分画を行った。
(a) 前記KSM1株ゲノミックDNAを約5mg含む溶液10 mlを高速振とう機CM-1000(東京理化器械社製)を用いて8分間撹拌し、該DNA溶液中に含まれる20-23Kb程度のサイズのDNA断片の割合を高めた。
(b)前記攪拌後のDNA溶液を、低融点アガロースゲル(SeaPlaqueGTG Agarose、Lonza社製)を用いた電気泳動に供した。前記低融点アガロースゲルは、0.6 %(weight/volume)のものを用い、電気泳動は50Vで一時間行った。その際ゲルの両端のレーンにはλDNAをHindIII処理することによって作製したマーカー(以下λ/HindIIIマーカーという。)を供した。
(c)前記λ/HindIIIマーカーのレーン部分のアガロースゲルのみをメスを用いて切り取り、エチジウムブロマイドを含むTAE緩衝液(40mMTris-acetate, 1mM EDTA)中で20分間染色した。その後、UVイルミネーターで、切り取ったアガロースゲルにUVを照射しながら、λ/HindIIIマーカーのアガロースゲルのレーンの23 Kbのバンド部分にメスで印をつけた。
(d)前記により印をつけたλ/HindIIIマーカーのアガロースゲルのレーンと、前記KSM1株ゲノミックDNA試料を泳動したレーンとをUV非照射下で並べ、マーカーのレーンにつけた印を目安に23Kb前後のDNA試料を含むゲル画分を切り出した。切り出したゲルはエッペンドルフチューブに1本あたり約100 mlとなるように移した。
(e)b-agarase (Lonza社製)を用い、添付のプロトコールに従って切り出したアガロースゲルを溶解させた。
(f)前記アガロースゲル溶解液200 mlに、PCI (フェノール (pH8.0):クロロホルム:イソアミルアルコール=25:24:1)溶液を等量添加し攪拌した。本該アガロース溶解液・PCI混合溶液を21,600x g で5 分間遠心した。遠心後、水層を回収した。
(g)回収した該水層に2.5倍量のエタノールと0.1 倍量の3 M酢酸ナトリウム水溶液(pH 5.2)を添加し、撹拌したのちに氷上で10分間静置した。静置後、21,600x g で15 分間遠心し、DNAを沈殿させた。上澄を廃棄し、沈殿させた該DNAを含むエッペンドルフチューブに500mlの70 %エタノールを添加した。該チューブを数回静かに反転させた後、ただちに21,600x g で5 分間遠心した。遠心後、上澄を廃棄した。なお本操作を、以後単に「エタノール沈殿」という。
(h)蓋を開いた状態でエッペンドルフチューブを横に倒し、該チューブ内が乾燥するまで該チューブを静置した。なお本操作を、以後単に「風乾」という。
(i)風乾した後の沈殿物を20 mlのTE緩衝液(10 mMトリス塩酸、1 mMEDTA、pH8.0)に溶解させ、20-23 Kb程度の断片のKSM1株ゲノミックDNAを得た。
【0049】
(3)ゲノミックDNAの末端平滑化
前記20-23Kb程度の断片のKSM1株ゲノミックDNAの末端の平滑化を、DNABlunting Kit(タカラバイオ社製)を用い、その添付説明書に従って行った。本操作により末端平滑化済みゲノミックDNAを得た。ただしBlunting後のKSM1株ゲノミックDNAとコスミドベクターとのライゲーションに関しては、該DNABlunting Kitに付属の試薬は用いずに、下記の「ゲノミックDNAとコスミドベクターとのライゲーション」によりおこなった。
【0050】
(4)コスミドベクターの調製
KSM1株のゲノミックライブラリー作製にあたり、ベクターには「Ono,A. et al. (2007)Applied Microbiology and Biotechnology, 74 (2): 501-510」の記載に従い、以下の手順により調製した広宿主域のコスミドベクターであるpKS13Sを用いた。
(a)pKS13Sを保持する大腸菌DH5a株(タカラバイオ社製、以後単にDH5a株という。)の-80℃冷凍ストックより、テトラサイクリンを15mg/mlの濃度で含む10 mlのTerrific broth液体培地(培地組成:水1 L当たり、酵母エキス, 24 g; ペプトン, 12 g ; K2HPO4 , 9.4 g;KH2PO4, 2.2 gを入れ、全体で1,000mlとした。以下TB液体培地という。)にDH5aを一白金耳植菌し、37℃で一晩振とう培養を行った。
(b)振とう培養後の該培養液を、10,000 x g にて遠心して集菌した後、QuantumPrep Mini Prep Kit(Bio-Rad社製)を用い、該菌体よりpKS13SをTE緩衝液に抽出した。Quantum Prep Mini PrepKitの操作は添付説明書に従った。さらに抽出したpKS13Sを含むTE緩衝液をエタノール沈殿し、該沈殿物を滅菌済み超純水に溶解させ、pKS13Sの沈殿物溶解液を得た。
(c)前記pKS13SをScaIにより切断した。ScaI処理は、前記pKS13Sの沈殿物溶解液を5 mg、ScaIを30 unit含む100 mlの反応液中で、37℃で2時間行った。その後、該反応液をエタノール沈殿し、風乾後に31 mlの滅菌済み超純水に溶解させた。
(d)前記31 ml のpKS13S含有溶液に、BAP(タカラバイオ社製)に添付のアルカリフォスファターゼ反応用緩衝液を4 ml、BAPを5 ml添加して計40 mlとし、ピペッティングによってよく混合した後、65℃にて30分間静置した。30分間静置後に、60 mlのTE緩衝液を添加し、pKS13S含有溶液の総量を100 mlとした。
(e)前記pKS13S含有溶液にPCIを等量添加し攪拌し、その後該pKS13SとPCIの混合溶液を21,600x g で5 分間遠心し、遠心後、水層を回収した。本操作を3回反復した後に、回収した該水層をエタノール沈殿した。風乾後、20mlのTE緩衝液に溶解させ、コスミドベクターpKS13Sを得た。
【0051】
(5)ゲノミックDNAとコスミドベクターとのライゲーション
前記末端平滑化済みゲノミックDNAと、前記コスミドベクターpKS13Sを用い、以下の手順で該ゲノミックDNAと該コスミドベクターのライゲーションを行った。
(a)前記ゲノミックDNA約600 ngを含むTE緩衝液と前記コスミドベクター約2000 ngを含むTE緩衝液をエッペンドルフチューブ内で混合し、エタノール沈殿した。
(b)前記により得た沈殿物を風乾した後、該風乾物を滅菌済み超純水5 mlに溶解させた。
(c)前記により調製した該風乾済みDNA溶解液に、DNA Ligation Kit Ver.2.1 (タカラバイオ社製)のI液を10 ml、II液を5 ml加え、計20 mlの溶液とした。各溶液を混合後ただちに数回ピペッティングした。軽く遠心した後、26 ℃で一晩反応させ、KSM1株ゲノミックDNAとpKS13Sのライゲーション反応液を得た。
【0052】
(6)バクテリオファージへのin vitroパッケージング
本操作にはEPICENTRE社製のMaxPlaxLambda Packaging Extracts (-80 ℃保存)を用い、その操作は添付の説明書に従って行った。添加するDNA溶液には前記において調製したライゲーション反応液20mlのうちの8 mlを用いた。8ml の該ライゲーション反応液を65℃で15分間静置、さらに氷上で5分間静置した後に、添付説明書に記載の操作に用い、パッケージング反応液を得た。
【0053】
(7)E.coli XL-1 blue MR株への形質導入
E.coli XL-1 blue MR株(Stratagene社製)を0.2%マルトースおよび10 mM硫酸マグネシウムを含むLB培地(10 ml)において、37℃で約16時間振とう培養し、OD610=0.8〜0.9となったE.coli XL-1 blue MR株培養液を得た。該E.coli XL-1 blue MR株培養液の200 mlと、前記パッケージング反応液の40 mlをエッペンドルフチューブ内で混合した。この混合液を37℃で20分間静置培養した後、1mlのLB培地を添加し、37℃で1時間振とう培養した。その後、該培養液を室温にて500 x gで10分間遠心し、上澄を廃棄して菌体を集菌した。この菌体を100 mlのLB培地に懸濁し、形質転換体候補E.coli XL-1 blue MR株菌液を得た。
【0054】
(8)テトラサイクリン耐性形質転換体の選択と保存
15 mg/mlのテトラサイクリンを含むLB寒天培地に、前記形質転換体候補E.coli XL-1 blue MR株菌液を塗布し、37℃、暗所にて18時間培養し、E.coli XL-1 blue MR株のテトラサイクリン耐性形質転換体を生育させた。培養後、該E.coli XL-1 blue MR株テトラサイクリン耐性形質転換体を保存するため、該テトラサイクリン耐性形質転換体が生育したLB寒天培地上に、15%グリセロールを含むLB培地を2 ml添加し、該テトラサイクリン耐性形質転換体を該添加液に懸濁した。その後、該テトラサイクリン耐性形質転換体懸濁液を収集し、滅菌済みの1.5ml容スクリューチューブに菌体溶液を移し、該スクリューチューブを-80℃で保存した。
【0055】
前記「(7)E.coli XL-1 blue MR株への形質導入」及び「(8)テトラサイクリン耐性形質転換体の選択と保存」の操作を、1,800クローンのE.coli XL-1 blue MR株テトラサイクリン形質転換体が得られるまで繰り返し行った。
【0056】
<KSM1株ゲノミックライブラリーのSphingobium japonicum UT26株への導入>
(1)KSM1株ゲノムのコスミドライブラリーを構成する組換えコスミドの抽出
前記により-80℃冷凍ストックとして保存したE.coli XL-1 blue MR株テトラサイクリン耐性形質転換体を、テトラサイクリンを15mg/mlの濃度で含む10 mlのTB液体培地に一白金耳植菌し、37℃で一晩培養した。該E.coli XL-1 blue MR株テトラサイクリン耐性形質転換体の培養後、QuantumPrep Mini Prep Kit(Bio-Rad社製)により、組換えコスミドをTE緩衝液に抽出した。その後、該組換えコスミド含有TE緩衝液をエタノール沈殿した。沈殿物を風乾した後、該風乾済みDNAを滅菌済み超純水に溶解させた。以上の操作は1.800種の全組換えコスミドをカバーするよう行った。1,800種の組換えコスミドの全種を含むDNA溶液を、組換えコスミド1種あたりのモル数が概ね等しくなるように、かつ組換えコスミド濃度が約100ng/mlとなるように組換えコスミド溶液を調製し、KSM1株ゲノムのコスミドライブラリーを構成する組換えコスミドを調製した。
【0057】
(2)Sphingobium japonicum UT26株のコンピテントセルの作製
(a)「Nagata, Y. et al. (2007) Applied Microbiology andBiotechnology, 76:741-752」に記載のSphingobium japonicum UT26株 (以下UT26株という。)を、-80℃冷凍ストックより1/3LB寒天培地 (per liter:Bact tryptone, 3.33 g; Bact yeast extract 1.67 g; NaCl, 5 g; agar, 15 g)に植菌した。その後2日間、30 ℃で培養し、UT26菌体を生育させた。
(b)前記1/3LB寒天培地上に生育した該UT26菌体を回収し、氷冷した2 mlのMOPS-10%グリセロール溶液に移した。その後、該菌体を含むMOPS-10%グリセロール溶液を、4℃、5,800x gで2分間遠心し、UT26菌体を沈殿させた。
(c)前記遠心後の上澄を廃棄し、前記の沈殿させたUT26菌体に、氷冷したMOPS-10%グリセロール溶液を新たに1.5 ml加え、懸濁した。該懸濁液を4℃、7,400x gで2分間遠心した。本操作を3回繰り返した。
(d)前記遠心した上澄を残らず廃棄し、氷冷したMOPS-10%グリセロール溶液を50 ml加えて懸濁し、UT26菌体懸濁液を得た。該菌体懸濁液を形質転換に用いるUT26株のコンピテントセルとした。
【0058】
(3)UT26株への組換えコスミドの導入
前記UT26株のコンピテントセルに、前記KSM1株ゲノムのコスミドライブラリーを構成する組換えコスミド溶液(約100ng/ml)を5 ml添加し、組換えコスミド含有コンピテントセルを得た。その後、緩やかな数回のピペッティングによって該組換えコスミド含有コンピテントセルを撹拌し、1mm幅のエレクトロポレーション用キュベット(Bio-Rad社製)に移した。該組換えコスミド含有コンピテントセルを含むキュベットをGene Pulser(Bio-Rad社製)にセットし、電圧1.8 KV、抵抗値200 Ω、静電容量25 mFで通電した。通電後直ちにキュベット内の菌液を1mlの1/3 LB液体培地(per liter: Bact tryptone, 3.33 g; Bact yeast extract 1.67 g; NaCl, 5 g)に移し、30℃で2時間培養を行った。培養後、該培養液を5,800x gで2分間遠心し、上澄を100 ml程度を残して廃棄し、残した100 ml程度の菌液をピペッティングにより撹拌した。該菌液を、テトラサイクリンを15mg/mlの濃度で含む1/3LB寒天培地に塗布した。菌液を塗布後の寒天培地を、30℃で4日間培養し、テトラサイクリン耐性のUT26株形質転換体のコロニーを得た。本方法を以下エレクトロポレーション法という。
【0059】
<DON分解活性を有するUT26株組換え体の選択>
(1)UT26株形質転換体の植えつぎ
前記により寒天培地上に生育させたUT26株の形質転換体のコロニーを、テトラサイクリンを15 mg/ml含む1/3LB寒天培地に、約10mmの線を引くように植え継ぎ、3日間、30℃で培養を行った。その結果、それぞれのUT26株形質転換体が約10 mmの長さのコロニーを形成した。
【0060】
(2)DON分解活性アッセイ溶液の作製と培養
20 mg/mlのDONを含む1.5 mlのCDM培地をエッペンドルフチューブに添加し、これを100チューブ準備した。この該CDM培地を含むチューブ1本に、前記の約10mmの長さのコロニーとして生育したUT26組換え体を12クローン接種した。該UT26組換え体の接種にあたっては、約10 mmの長さのUT26組換え体コロニーのうち、3mm程度を採取して該CDM培地に接種し、懸濁した。このようにして得た懸濁液をDON分解活性アッセイ溶液とした。他のクローンについても同様に12クローンずつ1本のチューブに接種・懸濁し、100本のDON分解活性アッセイ溶液を作製した。各アッセイ溶液を撹拌したのち該アッセイ溶液を15ml容プラスチックチューブに移し、28 ℃で2日間振とう培養した。なお本項のDON分解活性アッセイ溶液の作製と培養方法を、以後単にDON分解活性アッセイ溶液培養法という。
【0061】
(3)DONの減少したDON分解活性アッセイ溶液の探索
振とう培養後の前記DON分解活性アッセイ溶液のチューブ100本の内容物の全量を、それぞれ1本ずつエッペンドルフチューブに移した。このDON分解活性アッセイ溶液が移されたエッペンドルフチューブを、常温、21,600x g にて2分間遠心し、該アッセイ溶液の上澄と菌体を分離した。上澄の液体培地を採取して口径0.20mmの滅菌フィルター(ADVANTEC社製)を用いてろ過し、ろ過後の該液体培地を高速液体クロマトグラフィー(HPLC)分析を下記HPLC分析条件で行った。なお本項のろ過後の液体培地を以後DON分解活性アッセイHPLCサンプルという。また、本項のDON分解活性アッセイ溶液の作製と培養方法を、以後単にDON分解活性アッセイ溶液探索法という。
【0062】
HPLC分析条件
分析装置:600 Controller, 717plus Autosampler, 2487 Dual λ Absorbance Detector
(Waters社)
カラム:Symmetry C18 5mm 3.9x150mm (Waters社)
溶媒:水/メタノール(85:15)
流速:1 ml/分
温度:室温
【0063】
DON濃度の減少の成否を100本の該DON分解活性アッセイHPLCサンプルについて検討することにより、DON分解活性が付与されたUT26形質転換体を含むDON分解活性アッセイ溶液を探索した。その結果、1本のDON分解活性アッセイ溶液「45番」において、他に新たなピークが出現することなくDONが減少していた。本結果から、該アッセイ溶液に接種された12クローンのうちの少なくとも1クローンにDONを減少させる活性があるものと考えられた。
【0064】
(4)DON分解活性を有するUT26形質転換体の確定
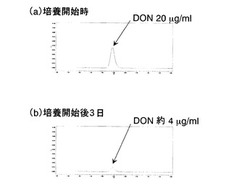
前記DON分解活性アッセイ溶液「45番」に含まれる12クローンのそれぞれを、個別に20mg/mlのDONを含む1 mlのCDM培地に接種してDON分解活性アッセイ溶液を作製し、DONの減少をHPLC分析により検討した。培養は前記DON分解活性アッセイ溶液培養法と同様に行った。HPLC分析は前記DON分解活性アッセイ溶液探索法と同様に行った。その結果、それらの12クローンのうちの1クローンである「クローン45-11」を接種した培養液から、図1に示す通りDON分解活性が検出された。本クローンの保持する組換えコスミドを「pKSM4511」と命名した。
【0065】
図1はDON分解活性アッセイ溶液から作製したDON分解活性アッセイHPLCサンプルのHPLCクロマトグラムを示し、図1において、(a)は培養開始時点のDON分解活性アッセイ溶液から作製したDON分解活性アッセイHPLCサンプルのHPLCクロマトグラムを、(b)は培養開始後3日の時点でのDON分解活性アッセイ溶液から作製したDON分解活性アッセイHPLCサンプルのHPLCクロマトグラムを示す。
【0066】
(5)クローン45-11のDON分解活性がpKSM4511に起因することの検証
前記クローン45-11は、図1に示す通り確かにDON分解活性を有していた。ここで、該クローンがDON分解活性を有していたことの原因として、a)クローン45-11が保持するpKSM4511の挿入DNA断片上にDON分解酵素遺伝子が存在する、b) pKSM4511を保持する宿主菌株であるUT26株のゲノムDNAにDON分解活性が生じるような突然変異が生じた、の2通りが考えられた。そこで、クローン45-11のDON分解活性がpKSM4511に起因するのか否かを検証することを目的として、クローン45-11から抽出したpKSM4511を再度UT26株に導入し直し、該再導入株にDON分解活性が付与されるか否かの検討を行った。
【0067】
クローン45-11からのpKSM4511の抽出は前記QuantumPrep Mini Prep Kitを用いて行い、TE緩衝液にpKSM4511を抽出した。その後、該pKSM4511含有TE緩衝液をPCI処理し、さらにエタノール沈殿してpKSM4511沈殿物を得た。該沈殿物を風乾したのち、滅菌超純水中に溶解させた。超純水に溶解させた該pKSM4511を、前記エレクトロポレーション法によってUT26株に導入し、pKSM4511含有UT26組換え体を得た。該UT26株組換え体を、前記DON分解活性アッセイ溶液培養法に従って、20mg/mlのDONを含むCDM培地に接種し、前記DON分解活性アッセイ溶液を調製した。該アッセイ溶液を30℃で3日間培養し、該アッセイ溶液を用いて前記DON分解活性アッセイHPLCサンプルを作製した。該DON分解活性アッセイHPLCサンプルを、前記HPLC分析条件で分析した結果、該UT26組換え体はクローン45-11と同様のDON分解活性を示した。以上の結果より、UT26にpKSM4511を導入することで、UT26にDON分解能が付与されることが明らかとなった。すなわち、クローン45-11のDON分解活性は、pKSM4511に起因することが示された。
【0068】
<pKSM4511上に存在するDON分解酵素遺伝子の同定>
(1)pKSM4511の大腸菌株への導入とpKSM4511の大腸菌株からの再抽出
pKSM4511を公知のヒートショック法によって大腸菌DH5a株に導入し、pKSM4511を保持する組換え大腸菌株を作製した。該組換え大腸菌株から前記QuantumPrep Mini Prep Kitを用いて再度pKSM4511を抽出し、以後、pKSM4511は全てこの大腸菌株から抽出したものを使用した。
【0069】
(2)pKSM4511のXhoI処理とDNA断片のクローニング
該pKSM4511をXhoI処理し、該XhoI反応液を0.8 %アガロースゲルを用いて電気泳動したところ、6本のバンド(それぞれ約6.9 Kb、 6.1 Kb、2.7 Kb、1.4Kb、400 bp、280 bp)が出現した。これらのバンドのうち、6.9 Kbおよび6.1 KbのDNA断片を、XhoI処理およびアルカリフォスファターゼ処理を行った広宿主域ベクターpNIT6012 (accession No. AB043476)に常法によってクローニングした。pNIT6012はカナマイシン耐性遺伝子由来の構成的発現プロモーターを備えており、プロモーター下流のマルチクローニングサイトに連結した遺伝子を大腸菌内およびUT26株内で構成的に発現させることが可能である。上記6.9Kbおよび6.1 KbのDNA断片をpNIT6012にクローニングして作製した組換えプラスミドをそれぞれp4511XX1およびp4511XX2と命名した。
【0070】
(3)pKSM4511のEcoRI処理
pKSM4511をEcoRI処理し、該EcoRI反応液を0.8 %アガロースゲルを用いて電気泳動したところ、6本のバンド(それぞれ約5.2 Kb, 4.7 Kb, 2.7Kb, 2.0 Kb, 1.6 Kb, 1.5 Kb)が出現した。これらのDNA断片の全てを、それぞれEcoRI処理およびアルカリフォスファターゼ処理を行ったpNIT6012に常法によってクローニングした。上記5.2 Kb、4.7 Kb、2.7Kb、2.0 Kb、1.6 Kb、および1.5 KbのDNA断片をpNIT6012にクローニングして作製した組換えプラスミドをそれぞれp4511EE1、p4511EE2、p4511EE3、p4511EE4、p4511EE5およびp4511EE6と命名した。
【0071】
(4)組換えプラスミドのUT26株への導入
(1)(2)で作製した上記計8種の組換えプラスミドをそれぞれ前記エレクトロポレーション法によってUT26株に導入し、UT26株組換え体を作出した。
【0072】
(5)UT26株組換え体のDON分解活性の確認
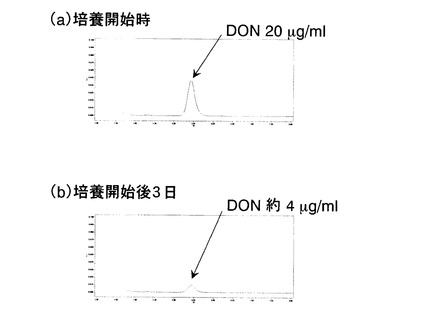
前記において作出した8種類のUT26株組換え体について、前記DON分解活性アッセイ溶液培養法、及び前記DON分解活性アッセイ溶液探索法により、DON分解活性の有無の検討を行った。その結果、p4511XX1を保持するUT26株組換え体でのみ、DON分解活性が検出された。図2にpKSM4511の挿入DNA断片に存在するDON分解酵素遺伝子の同定を示す。本結果から、DON分解酵素をコードする遺伝子は、その機能を発揮しうる単位として、p4511XX1の挿入DNA断片上、すなわちpKSM4511の2カ所のXhoI切断サイトに挟まれた約6.9Kbの領域に存在すると考えられた。
【0073】
(6)p4511XX1の挿入DNA断片およびその周辺領域のDNA塩基配列解析
図2に示す、pKSM4511挿入DNA断片上の2カ所のXhoI切断部位に挟まれた約6.9 Kbの領域、およびその周辺領域のDNA塩基配列解析を行った。塩基配列の決定にはBigDyeTerminator Ver.3.1 (ABI社製)とABI PRISM 3100 DNAシーケンサー (ABI社製)を用いた。p4511XX1の挿入DNA断片の末端シーケンス配列の決定にあたっては、鋳型DNAにp4511XX1を用い、プライマーにはpNIT6012のマルチクローニングサイト近傍のDNA塩基配列に基づいた下記配列番号3、及び配列番号4に示す2種のオリゴヌクレオチド(A)および(B)を用いた。その他のDNA領域はpKSM4511を鋳型DNAとしたプライマーウォーキング法により塩基配列を決定した。
【0074】
プライマー
(A)pNIT6012-MCS-CW
5’- gatttattcaacaaagccacgttgtgtctc-3’ (配列番号3)
(B)pNIT6012-MCS-CCW
5’-gattccgactcgtccaacatcaatacaacc -3’ (配列番号4)
【0075】
塩基配列を決定したp4511XX1挿入DNA断片を含む約6.5 Kbの解析の結果、図2に示すように、8個のオープンリーディングフレーム(ORF1-ORF8)が同定された(ただしORF1のみ遺伝子全長を含まない)。前記
【0076】
においてp4511XX1の挿入断片上にDON分解酵素遺伝子が存在すると考えられた。さらに図2に示す通り、p4511EE1を保持するUT26株形質転換体およびp4511EE5を保持するUT26株形質転換体にDON分解酵素活性が検出されなかったことから、これらのORFのうちORF6がDON分解活性に関与する遺伝子であると強く示唆された。
【0077】
p4511XX1を保持する組換えUT26株にDON分解活性が検出された理由として、ORF8がDON分解活性を有する酵素をコードする遺伝子であり、p4511XX1挿入DNA断片上のORF8部分配列の存在が該組換えUT26株にDON分解活性を付与したことが考えられた。そこで、ORF8がDON分解活性を有する酵素をコードする遺伝子であるか否かを検討する目的で、ORF8をUT26株に導入して作出したUT26形質転換体がDON分解活性を示すか否かの検証を行った。
【0078】
ORF8を常法によってpNIT6012にクローニングした組換えプラスミドp4511ORF8を構築し、該p4511ORF8をUT26株に導入してp4511ORF8含有組換えUT26株を作出した。該組換えUT26株のDON分解活性の有無の検討を行った結果、該組換えUT26株はDON分解活性を示さなかった(図2)。本結果から、ORF8がDON分解酵素遺伝子である可能性は否定された。また、本結果は、ORF6がDON分解活性をUT26株に付与する遺伝子であるとの示唆を支持するものであった。
【0079】
(7)p4511XX1挿入DNA断片上のORF6のPCR増幅
KSM1株ゲノミックDNAを鋳型DNAとして、下記配列番号5、及び配列番号6に示すオリゴヌクレオチド配列(C)(D)をプライマーとして用い、PCR法によってORF6を増幅した。配列番号7のオリゴヌクレオチド(C)にはXhoI切断部位(CTCGAG)を、配列番号8のオリゴヌクレオチド(D)にはSphI切断部位(GCATGC)を付加してある。PCR反応の酵素にはKODPlus Ver.2 (TOYOBO社製)を用い、以下のPCRプログラムで反応させた。反応液組成は添付説明書に従った。その結果、約800 bpのPCR増幅産物を得た。
【0080】
プライマー
(C)Xho-SD4511CR-F
5’- gccCTCGAGtagaaaaaggagaataaagtcatggc-3’ (配列番号5)
(D)Sph-4511CR-R
5’- gccGCATGCttatgccaacaccaaacagtcc-3’ (配列番号6)
制限酵素切断部位は大文字で示した。
【0081】
PCRプログラム
ステップ1 94℃ 2分 (1サイクル)
ステップ2 98℃ 10秒 → 51℃ 30秒 → 68℃ 50秒 (1サイクル)
ステップ3 98℃ 10秒 → 62℃ 30秒 → 68℃ 50秒 (29サイクル)
ステップ4 4℃(∞)
【0082】
(8)PCR増幅産物のpNITdSへのクローニング
前記ORF6がDON分解酵素遺伝子であるか否かを検討することを目的として、ORF6をベクターにクローニングしてUT26株に導入し、作出された組換えUT26株にDON分解能が付与されるか否かの検討を以下のように行った。
【0083】
ORF6の前記PCR増幅産物をXhoIおよびSphI処理によって切断し、該反応産物を0.8 %アガロースゲルを用いて電気泳動した。電気泳動後、該反応産物を含むゲル画分を切り出し、QIAEXII Gel Extraction Kit (QIAGEN社製)を用いて切り出したゲル画分からの該反応産物の抽出を行った。本操作により下記ライゲーション反応に用いる調製済みPCR反応産物を得た。
【0084】
一方、ORF6をクローニングするベクターにはpNITdS(SmaI切断部位に1塩基の置換があるためSmaIによる切断を受けないこと以外はpNIT6012と同一のベクター)を用いることとした。該ベクターをXhoIおよびSphI処理し、該制限酵素反応産物を0.8 %のアガロースゲルを用いて電気泳動した。電気泳動後、該反応産物を含むゲル画分を切り出し、該ゲル画分からの該反応産物の抽出を前記QIAEXII Gel Extraction Kitを用いて行った。以上の操作で下記のライゲーション反応に用いる調整済みpNITdSを得た。前記調製済みPCR反応産物と、前記調製済みpNITdSを混合し、TakaraDNA Ligation kit <Mighty Mix> (タカラバイオ社製)を用いてライゲーション反応を行った。本ライゲーション反応の操作は添付説明書の記載に従った。
【0085】
前記反応終了後、該ライゲーション反応産物を公知のヒートショック法により大腸菌DH5a株に導入し、テトラサイクリン耐性大腸菌形質転換体を得た。ORF6をクローニングした組換えpNITdSの挿入DNA断片の塩基配列は前記のプライマー(A)および(B)を用いて決定し、pKSM4511上のORF6の塩基配列がクローニングされていることを確認した。以上のの工程において作製した、ORF6をクローニングしたpNITdS組換えベクターをp4511CRと命名した。
【0086】
(9)ORF6がUT26株にDON分解活性を付与する遺伝子であることを証明
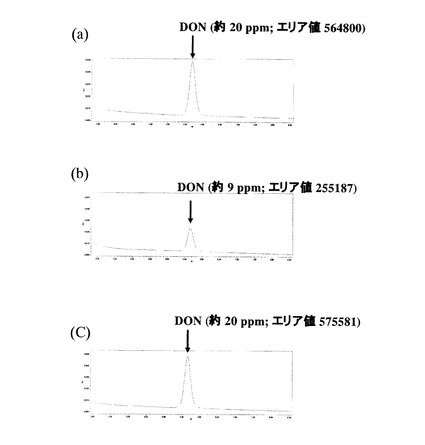
p4511CRをUT26株に導入した形質転換体を前記UT26株への組換えコスミドの導入における方法と同様の方法により導入し、前記DON分解活性アッセイ溶液培養法およびDON分解活性アッセイ溶液探索法により、DON分解活性の有無の検討を行った。ORF6を導入したUT26株組換え体と同じく20mg/mlのDONと反応させた。その結果、該UT26株組換え体はDON分解活性を示した。なおネガティブコントロールである野生型UT26株はDON分解活性を示さなかった。
【0087】
図3にp4511CRを導入したUT26株によるDON(20mg/ml)の分解について、DON分解活性アッセイ溶液から作製したDON分解活性アッセイHPLCサンプルのHPLCクロマトグラムを示す。(a)は培養開始時点のHPLCクロマトグラムを、(b)は培養開始後40時間の時点でのHPLCクロマトグラムを、(c)は野生型UT26株(ネガティブコントロール)の培養開始後40時間の時点でのHPLCクロマトグラムをそれぞれ示す。以上の結果から、p4511CRに含まれるORF6がUT26株にDON分解活性を付与する遺伝子であることが示された。
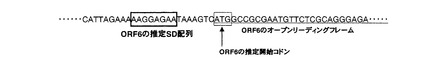
【0088】
<ORF6の配列解析>
ORF6は終止コドンを含めて全長786塩基対(配列番号1)で、推定261アミノ酸残基からなるタンパク質(配列番号2)をコードしていた。開始コドンの上流8bpから14 bpの領域には推定Shine Dalgano (SD)配列が認められた。図4に配列番号1の開始コドン付近の塩基配列を示す。図4において推定SD(ShineDalgano)配列を四角内に示した。本遺伝子の塩基配列をクエリとしてNCBIホームページ上でnucleotide Blast検索を行った。表2にBLASTサーチ結果を示す。サーチ結果はトップヒットのみを示したが、2009年11月9日の時点で有意な相同性を示す塩基配列は認められなかった。
【0089】
【表2】
【0090】
一方、配列番号2に示すアミノ酸配列をクエリとしてProtein Blast検索を行うと、30%以上の相同性を示す配列が多数検出された。ただし相同性が最も高かった配列で40 %の相同性にとどまった。ヒットした配列の殆どがshort chaindehydrogenase/reductaseとアノテーションされていた。最もE-valueの低い相同配列はAeromicrobium marinum DSM15272由来のshort chain dehydrogenaseであり、E-valueは4e-36、相同性は35 %であった。「short chaindehydrogenase/reductase (SDR)スーパーファミリー」は原核生物から菌類、高等動物、高等植物などの真核生物まで幅広く保存されている酵素群であり、N末端付近の補酵素結合モチーフ「Gly-X-X-X-Gly-X-Gly」と、shortchain dehydrogenase/reductase酵素活性に必須のモチーフ「Tyr-X-X-X-Lys」(Xは任意のアミノ酸)が保存されている(Kallberg,Y. et al.(2002)Eur.J.Biochem.269:4409-4417)。該スーパーファミリーに属する酵素の典型的な機能としては補酵素NAD(P)(H)依存的なアルコール脱水素反応またはカルボニル基還元反応の触媒である。該ORF6の推定アミノ酸配列においても、15番目から21番目のアミノ酸残基にGly-X-X-X-Gly-X-Glyのモチーフ(ORF6ではGly-Ala-Gly-Lys-Gly-Ile-Gly)が、また157番目から161番目のアミノ酸残基においてTyr-X-X-X-Lysのモチーフ(ORF6ではTyr-Gly-Ala-Ser-Lys)が存在することが確認された。
【0091】
図5に配列番号2のアミノ酸配列に比較的近縁なP450酵素群と配列番号2の系統樹を示す。以上の配列情報から、該ORF6はshort chain dehydrogenase/reductase(アルコール脱水素酵素またはカルボニル還元酵素)として、補酵素依存的にDON初発代謝を担う酵素をコードする遺伝子であると考えられた。
【産業上の利用可能性】
【0092】
本発明の遺伝子の利用により、デオキシニバレノール分解酵素の高発現微生物の作製とデオキシニバレノール汚染現場での利用、デオキシニバレノールの分解活性を備えデオキシニバレノールを蓄積させない新規組み換えムギの作製と栽培、ならびにデオキシニバレノール分解酵素製剤の製造など、デオキシニバレノール汚染問題の解決に向けた、新規の組み換え生物や酵素製剤の開発の可能性が喚起される。
【技術分野】
【0001】
本発明は、デオキシニバレノール分解活性を有するタンパク質、およびデオキシニバレノール分解活性を有するタンパク質をコードする新規遺伝子に関する。
【背景技術】
【0002】
かび毒は、ある種のかびが農作物に付着・増殖し、そこで産生する化学物質(天然毒素)のうち、人や家畜の健康に悪影響を及ぼすものの総称であり、現在300種類以上あるといわれている。代表的なかび毒として、アフラトキシン(ナッツ類、穀類)、デオキシニバレノール(穀類)、パツリン(りんご加工品)、オクラトキシンA(穀類、豆類)などがある。このうちわが国では平成20年8月現在、アフラトキシンB1(農作物を含む一般の食品)、デオキシニバレノール(小麦)、パツリン(リンゴ果汁)が食品衛生法に基づく規制の対象となっている。
【0003】
ムギ類を始めとする穀類に広く感染する赤かび病菌Fusarium graminearumが産生するデオキシニバレノール(以下DONと略称することがある。)、ニバレノール、T−2トキシンは、トリコテセン系マイコトキシンであり、ムギ生産地帯で大きな問題となっている。中でもデオキシニバレノールは、各国のムギ生産地帯で発生しているマイコトキシン汚染の主要因であり、摂取すると嘔吐、下痢などの急性中毒を引き起こすことから、世界的に本毒素の対策が喫緊の課題となっている。我が国でも平成14年5月に、厚生労働省においてコムギ粒の暫定基準濃度が1.1ppm以下と定められた。
【0004】
従来この対策としては、化学的手法により赤かび病菌を制御する方法が用いられてきた。また近年は植物体に残存する化学農薬の危険性が指摘されることから、微生物農薬がより安心で安全な植物病害防除剤として注目されている(例えば、特許文献1及び特許文献2を参照)。またバチルス・ズブチリスに属する菌株が産生するアイツリンAのフザリウムに対する増殖抑制効果を利用し、当該菌株の培養液で農産物を処理する方法が提案されている(特許文献3を参照。)。
【0005】
しかしデオキシニバレノールはトリコテセン骨格の構造を有し、121℃、20分の加圧・高温処理に対しても安定である。そのため、一旦赤かび病菌が発生した植物体においては、赤かび病を殺菌剤等で防除した後であっても、罹病した植物体の穂等に残存する。さらにデオキシニバレノールの特徴として、赤かび病が感染した植物体においては、発病したムギ粒等だけではなく、無病徴のムギ粒等にも蓄積される。このため、発病の有無にかかわらず、赤かび病菌汚染にされたムギ粒に蓄積されたデオキシニバレノールを低減する技術が求められている。
【0006】
そのためにデオキシニバレノールを直接除去する方法として、食品のレベルで物理的吸着により不活性化または排除する、家畜が食べてもそのまま排泄可能なカビ毒吸着剤(特許文献4を参照。)や、マイコトキシンを吸着除去する金属酸化物粒子複合体(特許文献5を参照。)が提案されている。
【0007】
一方で、食品となる以前に、直接穀物等に付着したデオキシニバレノールを除去することが望まれており、デオキシニバレノール分解微生物の探索が進められている。デオキシニバレノール分解微生物としては、現在のところ、土壌から分離されたグラム陰性菌Agrobacterium-RhizobiumグループのE3-39株(非特許文献1を参照。)、嫌気性細菌BBSH797(非特許文献2を参照。)、ムギ根圏土壌から分離されたDevosia属細菌に近縁のRS1株(特許文献6を参照。)、ムギ根圏土壌から分離されたNocardioides属に属するWSN05-2株、LS1株、SS1株およびSS2株(特許文献7を参照。)が報告されている。しかしながら、これまでにデオキシニバレノールの分解活性を有するタンパク質、およびデオキシニバレノール分解酵素をコードする遺伝子がデオキシニバレノール分解細菌株から同定された事例は皆無である。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2003−206212号公報
【特許文献2】特開2003−192515号公報
【特許文献3】特開平5−85911号公報
【特許文献4】特表2002−512011号公報
【特許文献5】特表2004−500214号公報
【特許文献6】特開2008−220179号公報
【特許文献7】特開2008−220184号公報
【非特許文献】
【0009】
【非特許文献1】Shima J. et. al (1997)Appl. Environ. Microbiol., 63: 3825-3830
【非特許文献2】FuchsE. et al (2002) Food Additives and Contaminants., 19: 379-386
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、デオキシニバレノールを分解する活性を有するタンパク質、及び該タンパク質をコードする遺伝子を提供することを課題とする。
【課題を解決するための手段】
【0011】
本発明者らは、霞ヶ浦湖水に生息する微生物の中から、デオキシニバレノールを効率よく分解することができる菌株を見出し、該菌株よりデオキシニバレノール分解酵素をコードする遺伝子のクローニングに成功し、本発明を完成するに至った。すなわち本発明は以下の通りである。
【0012】
<1> 下記(A)乃至(C)のいずれかのアミノ酸配列を含むデオキシニバレノール分解酵素タンパク質である。
(A)配列番号2に記載のアミノ酸配列。
(B)配列番号2に記載のアミノ酸配列において1若しくは複数のアミノ酸が欠失、置換または付加されているアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
(C)配列番号2に記載のアミノ酸配列と少なくとも80%以上の相同性を有するアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
<2> また本発明は、下記(A)乃至(C)のいずれかのアミノ酸配列をコードするデオキシニバレノール分解酵素遺伝子である。
(A)配列番号2に記載のアミノ酸配列。
(B)配列番号2に記載のアミノ酸配列において1若しくは複数のアミノ酸が欠失、置換または付加されているアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
(C)配列番号2に記載のアミノ酸配列と少なくとも80%以上の相同性を有するアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
<3> さらに本発明は、下記(D)乃至(F)のいずれかの塩基配列を含むデオキシニバレノール分解酵素遺伝子である。
(D)配列番号1で表される塩基配列。
(E)配列番号1に記載の塩基配列において1若しくは複数の塩基が欠失、置換または付加されている塩基配列であって、デオキシニバレノール分解活性を有するタンパク質をコードする塩基配列。
(F)配列番号1に記載の塩基配列とストリンジェントな条件下でハイブリダイズし、且つデオキシニバレノール分解活性を有するタンパク質をコードする塩基配列。
<4> さらに本発明は、前記の遺伝子を含む組み換えベクターである。
<5> さらに本発明は、前記の遺伝子を含む形質転換体である。
<6> さらに本発明は、前記の組換えベクターを用いる形質転換体である。
<7> さらに本発明は、前記の形質転換体を用いるデオキシニバレノールの分解方法である。
【発明の効果】
【0013】
本発明において、熱に安定で、難分解性であるデオキシニバレノールを分解するタンパク質、及び該タンパク質をコードする遺伝子を提供する。
【図面の簡単な説明】
【0014】
【図1】pKSM4511を保持するUT26株によるDON (20 mg/ml)の分解を示すDON分解活性アッセイHPLCサンプルのHPLCクロマトグラム図である。
【図2】pKSM4511の挿入DNA断片に存在するDON分解酵素遺伝子の同定を示す。
【図3】配列番号1(ORF6)の遺伝子を導入したUT26株によるDON(20 mg/ml)の分解を示すDON分解活性アッセイHPLCサンプルのHPLCクロマトグラム図である。
【図4】配列番号1の開始コドン付近の塩基配列である。
【図5】配列番号2のアミノ酸配列に存在するshort chain dehydrogenase/reductaseのモチーフを示した。
【発明を実施するための最良の形態】
【0015】
本発明は、Sphingobium sp. に近縁の新規微生物KSM1菌株に由来する配列番号1に示されるデオキシニバレノール分解酵素遺伝子、及び配列番号2に示されるデオキシニバレノール分解酵素に関するものである。以下本発明について詳説する。
【0016】
<本発明に係る遺伝子について>
本発明は、デオキシニバレノール分解酵素をコードする配列番号1に示される遺伝子である。配列番号1に示す塩基配列は、前記新規微生物KSM1株由来のデオキシニバレノール分解酵素遺伝子の開始コドンから終始コドンまでの、いわゆるオープンリーディングフレーム(以下ORFとする。)である。
【0017】
本発明に係るデオキシニバレノール分解酵素遺伝子は好ましくは、配列番号1に示す塩基配列からなるポリヌクレオチドからなるORFであるが、この部分に相当する部分が含まれる限り、いずれの遺伝子であってもよい。例えば、該ORFに5'および3'UTRを加えた遺伝子も本発明に含まれる。配列番号1に示す塩基配列からなるポリヌクレオチドは、該新規微生物KSM1株(受領番号FERMAP-21835)のゲノミックDNAから取得できる。
【0018】
本発明はまた、配列番号2に記載のデオキシニバレノールを分解するタンパク質と機能的に同等なタンパク質をコードする遺伝子を含む。ここで「デオキシニバレノールを分解するタンパク質と機能的に同等」とは、対象となるタンパク質が配列番号2に記載のタンパク質と同等の生物学的機能、即ちデオキシニバレノール分解代謝を触媒する機能を有することを指す。
【0019】
本発明の遺伝子には、例えば、配列番号2に記載のアミノ酸配列において1若しくは数個のアミノ酸が置換、欠失、付加および/または挿入されたアミノ酸配列からなるタンパク質をコードする変異体、誘導体、アレル、バリアントおよびホモログが含まれる。
【0020】
アミノ酸配列が改変されたタンパク質をコードする遺伝子を調製するための当業者によく知られた方法としては、例えば「Kramer, W.&Fritz,H.-J. Methods in Enzymology, 154: 350-367(1987)」に記載されている、部位特異的突然変異誘発(site-directedmutagenesis)法が挙げられる。また、塩基配列の変異によりコードするタンパク質のアミノ酸配列が変異することは、自然界においても生じ得る。このように天然型の本発明のデオキシニバレノール分解酵素(配列番号2)をコードする遺伝子においても、または該天然型デオキシニバレノール分解酵素に1もしくは数個のアミノ酸が置換、欠失もしくは付加したアミノ酸配列を有するタンパク質をコードする遺伝子であっても、天然型のアミノ酸配列と同等の機能を有するタンパク質をコードする限り、本発明の遺伝子に含まれる。
【0021】
また、たとえ塩基配列が変異した場合でも、それがタンパク質中のアミノ酸の変異を伴わない場合(縮重変異)もあり、このような縮重変異体も本発明の遺伝子に含まれる。対象となる遺伝子を構成するDNAの塩基の変異数は、アミノ酸レベルにおいて、100アミノ酸以内、好ましくは50アミノ酸以内、さらに好ましくは20アミノ酸以内、さらに好ましくは10アミノ酸以内である。
【0022】
上記遺伝子を獲得する方法は特に限定されるものではなく、一般的な方法が採用される。例えば、当該遺伝子を、それを有する生物のゲノムDNA、ゲノムDNAライブラリーなどから適切な制限酵素で切り出し、精製すればよい。即ち、本発明のデオキシニバレノール分解酵素遺伝子には、ゲノムDNAおよび化学合成DNAが含まれる。ゲノムDNAの調製は、当業者にとって常套手段を利用して行うことが可能である。ゲノムDNAは、例えば、対象生物からゲノムDNAを抽出し、ゲノミックライブラリー(ベクターとしては、プラスミド、ファージ、コスミド、BAC、PACなどが利用できる)を作成し、これを展開して、本発明のタンパク質をコードするDNA(配列番号1)を基に調製したプローブを用いてコロニーハイブリダイゼーションあるいはプラークハイブリダイゼーションを行うことにより調製することが可能である。
【0023】
また、本発明のデオキシニバレノール分解酵素をコードするDNA(配列番号1)に特異的なプライマーを作成し、これを利用したPCRを行うことによって調製することも可能である。
【0024】
なお、デオキシニバレノール分解活性を有する微生物(特許文献6、特許文献7、非特許文献1、非特許文献2)にも前記遺伝子を保有する可能性が考えられるが、前記KSM1株を用いれば、より確実に目的の遺伝子を単離することができる。
【0025】
次に本発明のデオキシニバレノール分解酵素遺伝子の獲得方法について説明する。具体的には、ゲノムの少なくとも一部がデータベース化されている微生物から本発明のデオキシニバレノール分解酵素遺伝子を獲得する場合には、上記ポリヌクレオチドの塩基配列(配列番号1)に基づいて相同性のある塩基配列をデータベース中から検索すればよい。例えば、汎用されている相同性検索アルゴリズムであるBLASTNによる塩基配列の相同性検索を好適に用いることができる。
【0026】
また、ゲノムがデータベース化されていない微生物の場合には、例えば、従来公知のDNAライブラリーを用いたハイブリダイゼーション法を用いることもできる。具体的には、適切なクローニングベクターを使用して対象となる微生物からゲノムライブラリーを調製する工程と、上記ポリヌクレオチドの少なくとも一部をプローブとして用いてハイブリダイゼーションを行い、ライブラリーから上記プローブにポジティブの断片を検出する工程とを含む方法を用いることができる。このように、本発明に係る遺伝子はプローブとしても有用である。プローブに用いる領域には、目的とする遺伝子に特異的な配列が含まれることが好ましい。また、プローブとして使用されるポリヌクレオチドの長さは、100bp以上が好ましい。
【0027】
より具体的には、配列番号2に記載のデオキシニバレノール分解酵素と機能的に同等なタンパク質をコードする遺伝子を調製するために、当業者によく知られた方法としては、「Southern,E.M. Journal of Molecular Biology, 98, 503(1975)」に記載のハイブリダイゼーション技術や、「Saiki, R.K. et al. Science, 230, 1350-1354(1985)、Saiki, R. K. et al. Science, 239,487-491(1988)」に記載のポリメラーゼ連鎖反応(PCR)技術を利用する方法が挙げられる。このようにハイブリダイズ技術やPCR技術により単離しうる本発明のデオキシニバレノール分解酵素と同等の機能を有するタンパク質をコードする遺伝子もまた本発明の遺伝子に含まれる。
【0028】
このような遺伝子を単離するためには、好ましくはストリンジェントな条件下でハイブリダイゼーション反応を行う。本発明においてストリンジェントなハイブリダイゼーション条件とは、0.2%SDS、0.2x SSC、65℃の条件、またはこれと同等以上のストリンジェンシーのハイブリダイゼーション/洗浄条件を指し、例えば、0.2% SDS、0.1x SSC、65℃の条件を用いれば、より相同性の高い遺伝子の効率的な単離を期待することができる。
【0029】
前記により単離された遺伝子は、それがコードするアミノ酸レベルにおいて、本発明のタンパク質のアミノ酸配列(配列番号2)と高い相同性を有すると考えられる。高い相同性とは、アミノ酸配列全体で、少なくとも70%以上、さらに好ましくは80%以上の配列の同一性を指す。配列の同一性は、FASTA検索(Pearson W.R. and D.J. Lipman (1988) Proc. Natl. Acad. Sci. USA. 85:2444-2448)やBLASTP検索により決定することができる。
【0030】
<本発明に係るタンパク質について>
本発明に係るタンパク質は、上記遺伝子によってコードされる配列番号2で表されるアミノ酸配列からなるタンパク質あるいは、配列番号2で表されるアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列からなり、かつ、デオキシニバレノール分解酵素の活性を有するタンパク質、もしくは、配列番号2で表されるアミノ酸配列からなるタンパク質とアミノ酸レベルで80%以上、さらに好ましくは90%以上95%以上の相同性を示し、かつ、デオキシニバレノール分解酵素の活性を有するタンパク質である。
【0031】
また、本発明に係るタンパク質は、タンパク質の精製や検出等を容易に行うために、公知のHAやFlag等の付加配列を末端に含ませてもよいし、融合タンパク質であってもよい。また、N-グリコシル化などの各種修飾を受けていてもよい。
【0032】
本発明による組換えベクターは、上記遺伝子が組み込まれたものである。上記ベクターは、公知の形質転換方法によって宿主に発現可能に導入されることにより、当該宿主において組み込まれた遺伝子あるいは遺伝子断片を発現させて本発明に係るタンパク質を得ることが出来るものである。なお、本発明によるタンパク質は下記のように組換え産生により得られたものでもよいし、微生物から単離、精製したものでもよく、その起源は特に限定されない。
【0033】
組み換えタンパク質を調製する場合には、通常、本発明のタンパク質をコードする遺伝子を適当な発現ベクターに挿入し、該ベクターを適当な宿主細胞に導入し、形質転換宿主細胞を培養して発現させたタンパク質を精製する。組み換えタンパク質は、精製を容易にするなどの目的で、他のタンパク質との融合タンパク質として発現させることも可能である。
【0034】
例えば、大腸菌を宿主としてマルトース結合タンパク質との融合タンパク質として調製する方法(米国New England BioLabs社発売のベクターpMALシリーズ)、グルタチオン-S-トランスフェラーゼ(GST)との融合タンパク質として調製する方法(AmershamPharmacia Biotech社発売のベクターpGEXシリーズ)、ヒスチジンタグを付加して調製する方法(Novagen社のpETシリーズ)などを利用することが可能である。宿主細胞としては、組み換えタンパク質の発現に適した細胞であれば特に制限はなく、上記の大腸菌の他、例えば、酵母等の真菌細胞、種々の動植物細胞などを用いることが可能である。
【0035】
宿主細胞へのベクターの導入には、当業者に公知の種々の方法を用いることが可能である。例えば、大腸菌への導入には、「Mandel, M.& Higa,A. Journal of Molecular Biology, 53, 158-162(1970)、Hanahan, D. Journal ofMolecular Biology, 166, 557-580(1983)」に記載のカルシウムイオンを利用した導入方法を用いることができる。宿主細胞内で発現させた組み換えタンパク質は、該宿主細胞またはその培養上清から、当業者に公知の方法により精製し、回収することが可能である。組み換えタンパク質を上記のマルトース結合タンパク質などとの融合タンパク質として発現させた場合には、容易にアフィニティー精製を行うことが可能である。
【0036】
得られた組換えタンパク質を用いれば、これに結合する抗体を調製することができる。例えば、ポリクローナル抗体は、精製した本発明のタンパク質若しくはその一部のペプチドをウサギなどの免疫動物に免疫し、一定期間の後に血液を採取し、血ぺいを除去することにより調製することが可能である。また、モノクローナル抗体は、上記タンパク質若しくはペプチドで免疫した動物の抗体産生細胞と骨腫瘍細胞とを融合させ、目的とする抗体を産生する単一クローンの細胞(ハイブリドーマ)を単離し、該細胞を増殖させ、細胞から抗体を得ることにより調製することができる。これにより得られた抗体は、本発明のタンパク質の精製や検出などに利用することが可能である。本発明には、本発明のタンパク質に結合する抗体が含まれる。
【0037】
<形質転換体について>
本発明の遺伝子を発現する形質転換体を作製する場合には、該遺伝子を適当なベクターに挿入して、これを対象の宿主細胞に導入する。
【0038】
本発明に係る形質転換体は、所望により組換えベクターに含まれた、上記遺伝子が導入されたものである。より具体的に言えば、本発明に係る形質転換体は、本発明によるデオキシニバレノール分解酵素をコードする遺伝子が導入されたものである。ここで、「遺伝子または組換えベクターが導入された」とは、公知の遺伝子工学的手法(遺伝子操作技術)により、宿主内に所望によりベクター内に組み込まれた遺伝子が、例えばプロモーターの作用により、発現可能に導入されることを意味する。
【0039】
本発明に係る形質転換体は、上記遺伝子を直接、あるいは上記遺伝子が組み込まれたベクターを宿主に導入することにより得られる。上記宿主は、特に限定されるものではなく、例えば、大腸菌などの原核生物、酵母などの真菌、あるいは、植物、昆虫細胞などを挙げることができ、中でも宿主細胞としては、原核生物が好ましい。
【0040】
本発明の遺伝子は、宿主細胞内で発現可能なプロモーター、例えば、大腸菌ではLacプロモーター、酵母では、GAPDHプロモーターやアルコールオキシダーゼプロモーター、植物細胞では、カリフラワーモザイクウイルス35Sプロモーター、昆虫細胞では、バキュロウイルスプロモーター等の下流域に連結して使用する。
【0041】
上記形質転換宿主細胞を含む形質転換体を作製する方法としては、具体的には、例えば、ポリエチレングリコール法、電気穿孔法(エレクトロポーレーション)、アグロバクテリウムを介する方法、パーティクルガン法など当業者に公知の種々の方法を用いることができる。
【0042】
前記形質転換植物を作出する手法について具体的には、「Datta,S.K. (1995) In Gene Transfer ToPlants(Potrykus I and Spangenberg Eds.) pp66-74」に記載のポリエチレングリコールによりプロトプラストへ遺伝子導入し植物体を再生させる方法、「Tokiet al (1992) Plant Physiol. 100, 1503-1507」に記載の電気パルスによりプロトプラストへ遺伝子導入し植物体を再生させる方法、「Christouet al. (1991) Bio/technology, 9: 957-962.」に記載のパーティクルガン法により細胞へ遺伝子を直接導入し植物体を再生させる方法、および「Hieiet al. (1994) Plant J. 6: 271-282.」に記載のアグロバクテリウムを介して遺伝子を導入し、植物体を再生させる方法などを挙げることができるが、特に限定されるものではない。また、上記形質転換方法は、宿主となる生物の種類に応じて適宜選択されることが好ましい。即ち、例えば宿主が酵母である場合は、塩化リチウム法が好ましい。
【0043】
本発明において使用することができるベクターとしては、従来から細菌、真菌などの形質転換に使用されているベクター、例えば、細菌用には、pUC、pBluescript、pET系、RK2-oriを有する広宿主域ベクターなど、酵母用には、pGYR、pFLD、pYES2等、植物用には、pBI121を挙げることができる。
【0044】
前記ベクターは、上記遺伝子またはその断片の他に、従来公知の遺伝子を発現させるための構成的プロモーター、例えば、GAPDHプロモーターやカリフラワーモザイクウイルス35Sプロモーター、または発現誘導性プロモーター、例えば、Lacプロモーターやアルコールオキシダーゼプロモーター、そして形質転換体の選抜を容易にする薬剤耐性遺伝子、例えば、アンピシリンあるいは、カナマイシン、ハイグロマイシン抵抗性遺伝子などを含んでいることが好ましい。
【実施例】
【0045】
以下、本発明を実施例によりさらに詳細に説明するが、本発明はこれら実施例に制限されるものではない。実施例で示した遺伝子組換え実験は、特に言及しない限り、「Molecular Cloning 2nd. Ed,Cold Spring Harbor Press ,1989」に記載の方法(以下単に常法という。)を用いて行った。
【0046】
<KSM1株ゲノミックライブラリーの構築>
(1)ゲノミックDNAの抽出
-80℃冷凍庫に保存したKSM1株のグリセロールストックを、R2A寒天培地(Merck社製)に植菌し、28℃で6日間培養した。寒天培地上に生育したKSM1株菌体を、表1に示すCDM培地に、100mg/mlの濃度にDON(Wako社)を添加した培地100 mlに植菌した。植菌後の培地を、28℃、暗所で振とう培養法により6日間培養した。培養後の培地を、9,200x gにて3分間遠心し、KSM1株菌体を集菌した。この菌体より、AquaPureGenomic DNA Isolation Kit (Bio-Rad社製)を用いてゲノミックDNAを抽出した。以上の操作により、約27 mgのKSM1株ゲノミックDNAを得た。
【0047】
【表1】
【0048】
(2)ゲノミックDNAのサイズ分画
前記により抽出したKSM1株ゲノミックDNAを含む溶液から、20-23 Kb程度のDNA断片のみを得る目的で、以下の手順により該KSM1株ゲノミックDNAのサイズ分画を行った。
(a) 前記KSM1株ゲノミックDNAを約5mg含む溶液10 mlを高速振とう機CM-1000(東京理化器械社製)を用いて8分間撹拌し、該DNA溶液中に含まれる20-23Kb程度のサイズのDNA断片の割合を高めた。
(b)前記攪拌後のDNA溶液を、低融点アガロースゲル(SeaPlaqueGTG Agarose、Lonza社製)を用いた電気泳動に供した。前記低融点アガロースゲルは、0.6 %(weight/volume)のものを用い、電気泳動は50Vで一時間行った。その際ゲルの両端のレーンにはλDNAをHindIII処理することによって作製したマーカー(以下λ/HindIIIマーカーという。)を供した。
(c)前記λ/HindIIIマーカーのレーン部分のアガロースゲルのみをメスを用いて切り取り、エチジウムブロマイドを含むTAE緩衝液(40mMTris-acetate, 1mM EDTA)中で20分間染色した。その後、UVイルミネーターで、切り取ったアガロースゲルにUVを照射しながら、λ/HindIIIマーカーのアガロースゲルのレーンの23 Kbのバンド部分にメスで印をつけた。
(d)前記により印をつけたλ/HindIIIマーカーのアガロースゲルのレーンと、前記KSM1株ゲノミックDNA試料を泳動したレーンとをUV非照射下で並べ、マーカーのレーンにつけた印を目安に23Kb前後のDNA試料を含むゲル画分を切り出した。切り出したゲルはエッペンドルフチューブに1本あたり約100 mlとなるように移した。
(e)b-agarase (Lonza社製)を用い、添付のプロトコールに従って切り出したアガロースゲルを溶解させた。
(f)前記アガロースゲル溶解液200 mlに、PCI (フェノール (pH8.0):クロロホルム:イソアミルアルコール=25:24:1)溶液を等量添加し攪拌した。本該アガロース溶解液・PCI混合溶液を21,600x g で5 分間遠心した。遠心後、水層を回収した。
(g)回収した該水層に2.5倍量のエタノールと0.1 倍量の3 M酢酸ナトリウム水溶液(pH 5.2)を添加し、撹拌したのちに氷上で10分間静置した。静置後、21,600x g で15 分間遠心し、DNAを沈殿させた。上澄を廃棄し、沈殿させた該DNAを含むエッペンドルフチューブに500mlの70 %エタノールを添加した。該チューブを数回静かに反転させた後、ただちに21,600x g で5 分間遠心した。遠心後、上澄を廃棄した。なお本操作を、以後単に「エタノール沈殿」という。
(h)蓋を開いた状態でエッペンドルフチューブを横に倒し、該チューブ内が乾燥するまで該チューブを静置した。なお本操作を、以後単に「風乾」という。
(i)風乾した後の沈殿物を20 mlのTE緩衝液(10 mMトリス塩酸、1 mMEDTA、pH8.0)に溶解させ、20-23 Kb程度の断片のKSM1株ゲノミックDNAを得た。
【0049】
(3)ゲノミックDNAの末端平滑化
前記20-23Kb程度の断片のKSM1株ゲノミックDNAの末端の平滑化を、DNABlunting Kit(タカラバイオ社製)を用い、その添付説明書に従って行った。本操作により末端平滑化済みゲノミックDNAを得た。ただしBlunting後のKSM1株ゲノミックDNAとコスミドベクターとのライゲーションに関しては、該DNABlunting Kitに付属の試薬は用いずに、下記の「ゲノミックDNAとコスミドベクターとのライゲーション」によりおこなった。
【0050】
(4)コスミドベクターの調製
KSM1株のゲノミックライブラリー作製にあたり、ベクターには「Ono,A. et al. (2007)Applied Microbiology and Biotechnology, 74 (2): 501-510」の記載に従い、以下の手順により調製した広宿主域のコスミドベクターであるpKS13Sを用いた。
(a)pKS13Sを保持する大腸菌DH5a株(タカラバイオ社製、以後単にDH5a株という。)の-80℃冷凍ストックより、テトラサイクリンを15mg/mlの濃度で含む10 mlのTerrific broth液体培地(培地組成:水1 L当たり、酵母エキス, 24 g; ペプトン, 12 g ; K2HPO4 , 9.4 g;KH2PO4, 2.2 gを入れ、全体で1,000mlとした。以下TB液体培地という。)にDH5aを一白金耳植菌し、37℃で一晩振とう培養を行った。
(b)振とう培養後の該培養液を、10,000 x g にて遠心して集菌した後、QuantumPrep Mini Prep Kit(Bio-Rad社製)を用い、該菌体よりpKS13SをTE緩衝液に抽出した。Quantum Prep Mini PrepKitの操作は添付説明書に従った。さらに抽出したpKS13Sを含むTE緩衝液をエタノール沈殿し、該沈殿物を滅菌済み超純水に溶解させ、pKS13Sの沈殿物溶解液を得た。
(c)前記pKS13SをScaIにより切断した。ScaI処理は、前記pKS13Sの沈殿物溶解液を5 mg、ScaIを30 unit含む100 mlの反応液中で、37℃で2時間行った。その後、該反応液をエタノール沈殿し、風乾後に31 mlの滅菌済み超純水に溶解させた。
(d)前記31 ml のpKS13S含有溶液に、BAP(タカラバイオ社製)に添付のアルカリフォスファターゼ反応用緩衝液を4 ml、BAPを5 ml添加して計40 mlとし、ピペッティングによってよく混合した後、65℃にて30分間静置した。30分間静置後に、60 mlのTE緩衝液を添加し、pKS13S含有溶液の総量を100 mlとした。
(e)前記pKS13S含有溶液にPCIを等量添加し攪拌し、その後該pKS13SとPCIの混合溶液を21,600x g で5 分間遠心し、遠心後、水層を回収した。本操作を3回反復した後に、回収した該水層をエタノール沈殿した。風乾後、20mlのTE緩衝液に溶解させ、コスミドベクターpKS13Sを得た。
【0051】
(5)ゲノミックDNAとコスミドベクターとのライゲーション
前記末端平滑化済みゲノミックDNAと、前記コスミドベクターpKS13Sを用い、以下の手順で該ゲノミックDNAと該コスミドベクターのライゲーションを行った。
(a)前記ゲノミックDNA約600 ngを含むTE緩衝液と前記コスミドベクター約2000 ngを含むTE緩衝液をエッペンドルフチューブ内で混合し、エタノール沈殿した。
(b)前記により得た沈殿物を風乾した後、該風乾物を滅菌済み超純水5 mlに溶解させた。
(c)前記により調製した該風乾済みDNA溶解液に、DNA Ligation Kit Ver.2.1 (タカラバイオ社製)のI液を10 ml、II液を5 ml加え、計20 mlの溶液とした。各溶液を混合後ただちに数回ピペッティングした。軽く遠心した後、26 ℃で一晩反応させ、KSM1株ゲノミックDNAとpKS13Sのライゲーション反応液を得た。
【0052】
(6)バクテリオファージへのin vitroパッケージング
本操作にはEPICENTRE社製のMaxPlaxLambda Packaging Extracts (-80 ℃保存)を用い、その操作は添付の説明書に従って行った。添加するDNA溶液には前記において調製したライゲーション反応液20mlのうちの8 mlを用いた。8ml の該ライゲーション反応液を65℃で15分間静置、さらに氷上で5分間静置した後に、添付説明書に記載の操作に用い、パッケージング反応液を得た。
【0053】
(7)E.coli XL-1 blue MR株への形質導入
E.coli XL-1 blue MR株(Stratagene社製)を0.2%マルトースおよび10 mM硫酸マグネシウムを含むLB培地(10 ml)において、37℃で約16時間振とう培養し、OD610=0.8〜0.9となったE.coli XL-1 blue MR株培養液を得た。該E.coli XL-1 blue MR株培養液の200 mlと、前記パッケージング反応液の40 mlをエッペンドルフチューブ内で混合した。この混合液を37℃で20分間静置培養した後、1mlのLB培地を添加し、37℃で1時間振とう培養した。その後、該培養液を室温にて500 x gで10分間遠心し、上澄を廃棄して菌体を集菌した。この菌体を100 mlのLB培地に懸濁し、形質転換体候補E.coli XL-1 blue MR株菌液を得た。
【0054】
(8)テトラサイクリン耐性形質転換体の選択と保存
15 mg/mlのテトラサイクリンを含むLB寒天培地に、前記形質転換体候補E.coli XL-1 blue MR株菌液を塗布し、37℃、暗所にて18時間培養し、E.coli XL-1 blue MR株のテトラサイクリン耐性形質転換体を生育させた。培養後、該E.coli XL-1 blue MR株テトラサイクリン耐性形質転換体を保存するため、該テトラサイクリン耐性形質転換体が生育したLB寒天培地上に、15%グリセロールを含むLB培地を2 ml添加し、該テトラサイクリン耐性形質転換体を該添加液に懸濁した。その後、該テトラサイクリン耐性形質転換体懸濁液を収集し、滅菌済みの1.5ml容スクリューチューブに菌体溶液を移し、該スクリューチューブを-80℃で保存した。
【0055】
前記「(7)E.coli XL-1 blue MR株への形質導入」及び「(8)テトラサイクリン耐性形質転換体の選択と保存」の操作を、1,800クローンのE.coli XL-1 blue MR株テトラサイクリン形質転換体が得られるまで繰り返し行った。
【0056】
<KSM1株ゲノミックライブラリーのSphingobium japonicum UT26株への導入>
(1)KSM1株ゲノムのコスミドライブラリーを構成する組換えコスミドの抽出
前記により-80℃冷凍ストックとして保存したE.coli XL-1 blue MR株テトラサイクリン耐性形質転換体を、テトラサイクリンを15mg/mlの濃度で含む10 mlのTB液体培地に一白金耳植菌し、37℃で一晩培養した。該E.coli XL-1 blue MR株テトラサイクリン耐性形質転換体の培養後、QuantumPrep Mini Prep Kit(Bio-Rad社製)により、組換えコスミドをTE緩衝液に抽出した。その後、該組換えコスミド含有TE緩衝液をエタノール沈殿した。沈殿物を風乾した後、該風乾済みDNAを滅菌済み超純水に溶解させた。以上の操作は1.800種の全組換えコスミドをカバーするよう行った。1,800種の組換えコスミドの全種を含むDNA溶液を、組換えコスミド1種あたりのモル数が概ね等しくなるように、かつ組換えコスミド濃度が約100ng/mlとなるように組換えコスミド溶液を調製し、KSM1株ゲノムのコスミドライブラリーを構成する組換えコスミドを調製した。
【0057】
(2)Sphingobium japonicum UT26株のコンピテントセルの作製
(a)「Nagata, Y. et al. (2007) Applied Microbiology andBiotechnology, 76:741-752」に記載のSphingobium japonicum UT26株 (以下UT26株という。)を、-80℃冷凍ストックより1/3LB寒天培地 (per liter:Bact tryptone, 3.33 g; Bact yeast extract 1.67 g; NaCl, 5 g; agar, 15 g)に植菌した。その後2日間、30 ℃で培養し、UT26菌体を生育させた。
(b)前記1/3LB寒天培地上に生育した該UT26菌体を回収し、氷冷した2 mlのMOPS-10%グリセロール溶液に移した。その後、該菌体を含むMOPS-10%グリセロール溶液を、4℃、5,800x gで2分間遠心し、UT26菌体を沈殿させた。
(c)前記遠心後の上澄を廃棄し、前記の沈殿させたUT26菌体に、氷冷したMOPS-10%グリセロール溶液を新たに1.5 ml加え、懸濁した。該懸濁液を4℃、7,400x gで2分間遠心した。本操作を3回繰り返した。
(d)前記遠心した上澄を残らず廃棄し、氷冷したMOPS-10%グリセロール溶液を50 ml加えて懸濁し、UT26菌体懸濁液を得た。該菌体懸濁液を形質転換に用いるUT26株のコンピテントセルとした。
【0058】
(3)UT26株への組換えコスミドの導入
前記UT26株のコンピテントセルに、前記KSM1株ゲノムのコスミドライブラリーを構成する組換えコスミド溶液(約100ng/ml)を5 ml添加し、組換えコスミド含有コンピテントセルを得た。その後、緩やかな数回のピペッティングによって該組換えコスミド含有コンピテントセルを撹拌し、1mm幅のエレクトロポレーション用キュベット(Bio-Rad社製)に移した。該組換えコスミド含有コンピテントセルを含むキュベットをGene Pulser(Bio-Rad社製)にセットし、電圧1.8 KV、抵抗値200 Ω、静電容量25 mFで通電した。通電後直ちにキュベット内の菌液を1mlの1/3 LB液体培地(per liter: Bact tryptone, 3.33 g; Bact yeast extract 1.67 g; NaCl, 5 g)に移し、30℃で2時間培養を行った。培養後、該培養液を5,800x gで2分間遠心し、上澄を100 ml程度を残して廃棄し、残した100 ml程度の菌液をピペッティングにより撹拌した。該菌液を、テトラサイクリンを15mg/mlの濃度で含む1/3LB寒天培地に塗布した。菌液を塗布後の寒天培地を、30℃で4日間培養し、テトラサイクリン耐性のUT26株形質転換体のコロニーを得た。本方法を以下エレクトロポレーション法という。
【0059】
<DON分解活性を有するUT26株組換え体の選択>
(1)UT26株形質転換体の植えつぎ
前記により寒天培地上に生育させたUT26株の形質転換体のコロニーを、テトラサイクリンを15 mg/ml含む1/3LB寒天培地に、約10mmの線を引くように植え継ぎ、3日間、30℃で培養を行った。その結果、それぞれのUT26株形質転換体が約10 mmの長さのコロニーを形成した。
【0060】
(2)DON分解活性アッセイ溶液の作製と培養
20 mg/mlのDONを含む1.5 mlのCDM培地をエッペンドルフチューブに添加し、これを100チューブ準備した。この該CDM培地を含むチューブ1本に、前記の約10mmの長さのコロニーとして生育したUT26組換え体を12クローン接種した。該UT26組換え体の接種にあたっては、約10 mmの長さのUT26組換え体コロニーのうち、3mm程度を採取して該CDM培地に接種し、懸濁した。このようにして得た懸濁液をDON分解活性アッセイ溶液とした。他のクローンについても同様に12クローンずつ1本のチューブに接種・懸濁し、100本のDON分解活性アッセイ溶液を作製した。各アッセイ溶液を撹拌したのち該アッセイ溶液を15ml容プラスチックチューブに移し、28 ℃で2日間振とう培養した。なお本項のDON分解活性アッセイ溶液の作製と培養方法を、以後単にDON分解活性アッセイ溶液培養法という。
【0061】
(3)DONの減少したDON分解活性アッセイ溶液の探索
振とう培養後の前記DON分解活性アッセイ溶液のチューブ100本の内容物の全量を、それぞれ1本ずつエッペンドルフチューブに移した。このDON分解活性アッセイ溶液が移されたエッペンドルフチューブを、常温、21,600x g にて2分間遠心し、該アッセイ溶液の上澄と菌体を分離した。上澄の液体培地を採取して口径0.20mmの滅菌フィルター(ADVANTEC社製)を用いてろ過し、ろ過後の該液体培地を高速液体クロマトグラフィー(HPLC)分析を下記HPLC分析条件で行った。なお本項のろ過後の液体培地を以後DON分解活性アッセイHPLCサンプルという。また、本項のDON分解活性アッセイ溶液の作製と培養方法を、以後単にDON分解活性アッセイ溶液探索法という。
【0062】
HPLC分析条件
分析装置:600 Controller, 717plus Autosampler, 2487 Dual λ Absorbance Detector
(Waters社)
カラム:Symmetry C18 5mm 3.9x150mm (Waters社)
溶媒:水/メタノール(85:15)
流速:1 ml/分
温度:室温
【0063】
DON濃度の減少の成否を100本の該DON分解活性アッセイHPLCサンプルについて検討することにより、DON分解活性が付与されたUT26形質転換体を含むDON分解活性アッセイ溶液を探索した。その結果、1本のDON分解活性アッセイ溶液「45番」において、他に新たなピークが出現することなくDONが減少していた。本結果から、該アッセイ溶液に接種された12クローンのうちの少なくとも1クローンにDONを減少させる活性があるものと考えられた。
【0064】
(4)DON分解活性を有するUT26形質転換体の確定
前記DON分解活性アッセイ溶液「45番」に含まれる12クローンのそれぞれを、個別に20mg/mlのDONを含む1 mlのCDM培地に接種してDON分解活性アッセイ溶液を作製し、DONの減少をHPLC分析により検討した。培養は前記DON分解活性アッセイ溶液培養法と同様に行った。HPLC分析は前記DON分解活性アッセイ溶液探索法と同様に行った。その結果、それらの12クローンのうちの1クローンである「クローン45-11」を接種した培養液から、図1に示す通りDON分解活性が検出された。本クローンの保持する組換えコスミドを「pKSM4511」と命名した。
【0065】
図1はDON分解活性アッセイ溶液から作製したDON分解活性アッセイHPLCサンプルのHPLCクロマトグラムを示し、図1において、(a)は培養開始時点のDON分解活性アッセイ溶液から作製したDON分解活性アッセイHPLCサンプルのHPLCクロマトグラムを、(b)は培養開始後3日の時点でのDON分解活性アッセイ溶液から作製したDON分解活性アッセイHPLCサンプルのHPLCクロマトグラムを示す。
【0066】
(5)クローン45-11のDON分解活性がpKSM4511に起因することの検証
前記クローン45-11は、図1に示す通り確かにDON分解活性を有していた。ここで、該クローンがDON分解活性を有していたことの原因として、a)クローン45-11が保持するpKSM4511の挿入DNA断片上にDON分解酵素遺伝子が存在する、b) pKSM4511を保持する宿主菌株であるUT26株のゲノムDNAにDON分解活性が生じるような突然変異が生じた、の2通りが考えられた。そこで、クローン45-11のDON分解活性がpKSM4511に起因するのか否かを検証することを目的として、クローン45-11から抽出したpKSM4511を再度UT26株に導入し直し、該再導入株にDON分解活性が付与されるか否かの検討を行った。
【0067】
クローン45-11からのpKSM4511の抽出は前記QuantumPrep Mini Prep Kitを用いて行い、TE緩衝液にpKSM4511を抽出した。その後、該pKSM4511含有TE緩衝液をPCI処理し、さらにエタノール沈殿してpKSM4511沈殿物を得た。該沈殿物を風乾したのち、滅菌超純水中に溶解させた。超純水に溶解させた該pKSM4511を、前記エレクトロポレーション法によってUT26株に導入し、pKSM4511含有UT26組換え体を得た。該UT26株組換え体を、前記DON分解活性アッセイ溶液培養法に従って、20mg/mlのDONを含むCDM培地に接種し、前記DON分解活性アッセイ溶液を調製した。該アッセイ溶液を30℃で3日間培養し、該アッセイ溶液を用いて前記DON分解活性アッセイHPLCサンプルを作製した。該DON分解活性アッセイHPLCサンプルを、前記HPLC分析条件で分析した結果、該UT26組換え体はクローン45-11と同様のDON分解活性を示した。以上の結果より、UT26にpKSM4511を導入することで、UT26にDON分解能が付与されることが明らかとなった。すなわち、クローン45-11のDON分解活性は、pKSM4511に起因することが示された。
【0068】
<pKSM4511上に存在するDON分解酵素遺伝子の同定>
(1)pKSM4511の大腸菌株への導入とpKSM4511の大腸菌株からの再抽出
pKSM4511を公知のヒートショック法によって大腸菌DH5a株に導入し、pKSM4511を保持する組換え大腸菌株を作製した。該組換え大腸菌株から前記QuantumPrep Mini Prep Kitを用いて再度pKSM4511を抽出し、以後、pKSM4511は全てこの大腸菌株から抽出したものを使用した。
【0069】
(2)pKSM4511のXhoI処理とDNA断片のクローニング
該pKSM4511をXhoI処理し、該XhoI反応液を0.8 %アガロースゲルを用いて電気泳動したところ、6本のバンド(それぞれ約6.9 Kb、 6.1 Kb、2.7 Kb、1.4Kb、400 bp、280 bp)が出現した。これらのバンドのうち、6.9 Kbおよび6.1 KbのDNA断片を、XhoI処理およびアルカリフォスファターゼ処理を行った広宿主域ベクターpNIT6012 (accession No. AB043476)に常法によってクローニングした。pNIT6012はカナマイシン耐性遺伝子由来の構成的発現プロモーターを備えており、プロモーター下流のマルチクローニングサイトに連結した遺伝子を大腸菌内およびUT26株内で構成的に発現させることが可能である。上記6.9Kbおよび6.1 KbのDNA断片をpNIT6012にクローニングして作製した組換えプラスミドをそれぞれp4511XX1およびp4511XX2と命名した。
【0070】
(3)pKSM4511のEcoRI処理
pKSM4511をEcoRI処理し、該EcoRI反応液を0.8 %アガロースゲルを用いて電気泳動したところ、6本のバンド(それぞれ約5.2 Kb, 4.7 Kb, 2.7Kb, 2.0 Kb, 1.6 Kb, 1.5 Kb)が出現した。これらのDNA断片の全てを、それぞれEcoRI処理およびアルカリフォスファターゼ処理を行ったpNIT6012に常法によってクローニングした。上記5.2 Kb、4.7 Kb、2.7Kb、2.0 Kb、1.6 Kb、および1.5 KbのDNA断片をpNIT6012にクローニングして作製した組換えプラスミドをそれぞれp4511EE1、p4511EE2、p4511EE3、p4511EE4、p4511EE5およびp4511EE6と命名した。
【0071】
(4)組換えプラスミドのUT26株への導入
(1)(2)で作製した上記計8種の組換えプラスミドをそれぞれ前記エレクトロポレーション法によってUT26株に導入し、UT26株組換え体を作出した。
【0072】
(5)UT26株組換え体のDON分解活性の確認
前記において作出した8種類のUT26株組換え体について、前記DON分解活性アッセイ溶液培養法、及び前記DON分解活性アッセイ溶液探索法により、DON分解活性の有無の検討を行った。その結果、p4511XX1を保持するUT26株組換え体でのみ、DON分解活性が検出された。図2にpKSM4511の挿入DNA断片に存在するDON分解酵素遺伝子の同定を示す。本結果から、DON分解酵素をコードする遺伝子は、その機能を発揮しうる単位として、p4511XX1の挿入DNA断片上、すなわちpKSM4511の2カ所のXhoI切断サイトに挟まれた約6.9Kbの領域に存在すると考えられた。
【0073】
(6)p4511XX1の挿入DNA断片およびその周辺領域のDNA塩基配列解析
図2に示す、pKSM4511挿入DNA断片上の2カ所のXhoI切断部位に挟まれた約6.9 Kbの領域、およびその周辺領域のDNA塩基配列解析を行った。塩基配列の決定にはBigDyeTerminator Ver.3.1 (ABI社製)とABI PRISM 3100 DNAシーケンサー (ABI社製)を用いた。p4511XX1の挿入DNA断片の末端シーケンス配列の決定にあたっては、鋳型DNAにp4511XX1を用い、プライマーにはpNIT6012のマルチクローニングサイト近傍のDNA塩基配列に基づいた下記配列番号3、及び配列番号4に示す2種のオリゴヌクレオチド(A)および(B)を用いた。その他のDNA領域はpKSM4511を鋳型DNAとしたプライマーウォーキング法により塩基配列を決定した。
【0074】
プライマー
(A)pNIT6012-MCS-CW
5’- gatttattcaacaaagccacgttgtgtctc-3’ (配列番号3)
(B)pNIT6012-MCS-CCW
5’-gattccgactcgtccaacatcaatacaacc -3’ (配列番号4)
【0075】
塩基配列を決定したp4511XX1挿入DNA断片を含む約6.5 Kbの解析の結果、図2に示すように、8個のオープンリーディングフレーム(ORF1-ORF8)が同定された(ただしORF1のみ遺伝子全長を含まない)。前記
【0076】
においてp4511XX1の挿入断片上にDON分解酵素遺伝子が存在すると考えられた。さらに図2に示す通り、p4511EE1を保持するUT26株形質転換体およびp4511EE5を保持するUT26株形質転換体にDON分解酵素活性が検出されなかったことから、これらのORFのうちORF6がDON分解活性に関与する遺伝子であると強く示唆された。
【0077】
p4511XX1を保持する組換えUT26株にDON分解活性が検出された理由として、ORF8がDON分解活性を有する酵素をコードする遺伝子であり、p4511XX1挿入DNA断片上のORF8部分配列の存在が該組換えUT26株にDON分解活性を付与したことが考えられた。そこで、ORF8がDON分解活性を有する酵素をコードする遺伝子であるか否かを検討する目的で、ORF8をUT26株に導入して作出したUT26形質転換体がDON分解活性を示すか否かの検証を行った。
【0078】
ORF8を常法によってpNIT6012にクローニングした組換えプラスミドp4511ORF8を構築し、該p4511ORF8をUT26株に導入してp4511ORF8含有組換えUT26株を作出した。該組換えUT26株のDON分解活性の有無の検討を行った結果、該組換えUT26株はDON分解活性を示さなかった(図2)。本結果から、ORF8がDON分解酵素遺伝子である可能性は否定された。また、本結果は、ORF6がDON分解活性をUT26株に付与する遺伝子であるとの示唆を支持するものであった。
【0079】
(7)p4511XX1挿入DNA断片上のORF6のPCR増幅
KSM1株ゲノミックDNAを鋳型DNAとして、下記配列番号5、及び配列番号6に示すオリゴヌクレオチド配列(C)(D)をプライマーとして用い、PCR法によってORF6を増幅した。配列番号7のオリゴヌクレオチド(C)にはXhoI切断部位(CTCGAG)を、配列番号8のオリゴヌクレオチド(D)にはSphI切断部位(GCATGC)を付加してある。PCR反応の酵素にはKODPlus Ver.2 (TOYOBO社製)を用い、以下のPCRプログラムで反応させた。反応液組成は添付説明書に従った。その結果、約800 bpのPCR増幅産物を得た。
【0080】
プライマー
(C)Xho-SD4511CR-F
5’- gccCTCGAGtagaaaaaggagaataaagtcatggc-3’ (配列番号5)
(D)Sph-4511CR-R
5’- gccGCATGCttatgccaacaccaaacagtcc-3’ (配列番号6)
制限酵素切断部位は大文字で示した。
【0081】
PCRプログラム
ステップ1 94℃ 2分 (1サイクル)
ステップ2 98℃ 10秒 → 51℃ 30秒 → 68℃ 50秒 (1サイクル)
ステップ3 98℃ 10秒 → 62℃ 30秒 → 68℃ 50秒 (29サイクル)
ステップ4 4℃(∞)
【0082】
(8)PCR増幅産物のpNITdSへのクローニング
前記ORF6がDON分解酵素遺伝子であるか否かを検討することを目的として、ORF6をベクターにクローニングしてUT26株に導入し、作出された組換えUT26株にDON分解能が付与されるか否かの検討を以下のように行った。
【0083】
ORF6の前記PCR増幅産物をXhoIおよびSphI処理によって切断し、該反応産物を0.8 %アガロースゲルを用いて電気泳動した。電気泳動後、該反応産物を含むゲル画分を切り出し、QIAEXII Gel Extraction Kit (QIAGEN社製)を用いて切り出したゲル画分からの該反応産物の抽出を行った。本操作により下記ライゲーション反応に用いる調製済みPCR反応産物を得た。
【0084】
一方、ORF6をクローニングするベクターにはpNITdS(SmaI切断部位に1塩基の置換があるためSmaIによる切断を受けないこと以外はpNIT6012と同一のベクター)を用いることとした。該ベクターをXhoIおよびSphI処理し、該制限酵素反応産物を0.8 %のアガロースゲルを用いて電気泳動した。電気泳動後、該反応産物を含むゲル画分を切り出し、該ゲル画分からの該反応産物の抽出を前記QIAEXII Gel Extraction Kitを用いて行った。以上の操作で下記のライゲーション反応に用いる調整済みpNITdSを得た。前記調製済みPCR反応産物と、前記調製済みpNITdSを混合し、TakaraDNA Ligation kit <Mighty Mix> (タカラバイオ社製)を用いてライゲーション反応を行った。本ライゲーション反応の操作は添付説明書の記載に従った。
【0085】
前記反応終了後、該ライゲーション反応産物を公知のヒートショック法により大腸菌DH5a株に導入し、テトラサイクリン耐性大腸菌形質転換体を得た。ORF6をクローニングした組換えpNITdSの挿入DNA断片の塩基配列は前記のプライマー(A)および(B)を用いて決定し、pKSM4511上のORF6の塩基配列がクローニングされていることを確認した。以上のの工程において作製した、ORF6をクローニングしたpNITdS組換えベクターをp4511CRと命名した。
【0086】
(9)ORF6がUT26株にDON分解活性を付与する遺伝子であることを証明
p4511CRをUT26株に導入した形質転換体を前記UT26株への組換えコスミドの導入における方法と同様の方法により導入し、前記DON分解活性アッセイ溶液培養法およびDON分解活性アッセイ溶液探索法により、DON分解活性の有無の検討を行った。ORF6を導入したUT26株組換え体と同じく20mg/mlのDONと反応させた。その結果、該UT26株組換え体はDON分解活性を示した。なおネガティブコントロールである野生型UT26株はDON分解活性を示さなかった。
【0087】
図3にp4511CRを導入したUT26株によるDON(20mg/ml)の分解について、DON分解活性アッセイ溶液から作製したDON分解活性アッセイHPLCサンプルのHPLCクロマトグラムを示す。(a)は培養開始時点のHPLCクロマトグラムを、(b)は培養開始後40時間の時点でのHPLCクロマトグラムを、(c)は野生型UT26株(ネガティブコントロール)の培養開始後40時間の時点でのHPLCクロマトグラムをそれぞれ示す。以上の結果から、p4511CRに含まれるORF6がUT26株にDON分解活性を付与する遺伝子であることが示された。
【0088】
<ORF6の配列解析>
ORF6は終止コドンを含めて全長786塩基対(配列番号1)で、推定261アミノ酸残基からなるタンパク質(配列番号2)をコードしていた。開始コドンの上流8bpから14 bpの領域には推定Shine Dalgano (SD)配列が認められた。図4に配列番号1の開始コドン付近の塩基配列を示す。図4において推定SD(ShineDalgano)配列を四角内に示した。本遺伝子の塩基配列をクエリとしてNCBIホームページ上でnucleotide Blast検索を行った。表2にBLASTサーチ結果を示す。サーチ結果はトップヒットのみを示したが、2009年11月9日の時点で有意な相同性を示す塩基配列は認められなかった。
【0089】
【表2】
【0090】
一方、配列番号2に示すアミノ酸配列をクエリとしてProtein Blast検索を行うと、30%以上の相同性を示す配列が多数検出された。ただし相同性が最も高かった配列で40 %の相同性にとどまった。ヒットした配列の殆どがshort chaindehydrogenase/reductaseとアノテーションされていた。最もE-valueの低い相同配列はAeromicrobium marinum DSM15272由来のshort chain dehydrogenaseであり、E-valueは4e-36、相同性は35 %であった。「short chaindehydrogenase/reductase (SDR)スーパーファミリー」は原核生物から菌類、高等動物、高等植物などの真核生物まで幅広く保存されている酵素群であり、N末端付近の補酵素結合モチーフ「Gly-X-X-X-Gly-X-Gly」と、shortchain dehydrogenase/reductase酵素活性に必須のモチーフ「Tyr-X-X-X-Lys」(Xは任意のアミノ酸)が保存されている(Kallberg,Y. et al.(2002)Eur.J.Biochem.269:4409-4417)。該スーパーファミリーに属する酵素の典型的な機能としては補酵素NAD(P)(H)依存的なアルコール脱水素反応またはカルボニル基還元反応の触媒である。該ORF6の推定アミノ酸配列においても、15番目から21番目のアミノ酸残基にGly-X-X-X-Gly-X-Glyのモチーフ(ORF6ではGly-Ala-Gly-Lys-Gly-Ile-Gly)が、また157番目から161番目のアミノ酸残基においてTyr-X-X-X-Lysのモチーフ(ORF6ではTyr-Gly-Ala-Ser-Lys)が存在することが確認された。
【0091】
図5に配列番号2のアミノ酸配列に比較的近縁なP450酵素群と配列番号2の系統樹を示す。以上の配列情報から、該ORF6はshort chain dehydrogenase/reductase(アルコール脱水素酵素またはカルボニル還元酵素)として、補酵素依存的にDON初発代謝を担う酵素をコードする遺伝子であると考えられた。
【産業上の利用可能性】
【0092】
本発明の遺伝子の利用により、デオキシニバレノール分解酵素の高発現微生物の作製とデオキシニバレノール汚染現場での利用、デオキシニバレノールの分解活性を備えデオキシニバレノールを蓄積させない新規組み換えムギの作製と栽培、ならびにデオキシニバレノール分解酵素製剤の製造など、デオキシニバレノール汚染問題の解決に向けた、新規の組み換え生物や酵素製剤の開発の可能性が喚起される。
【特許請求の範囲】
【請求項1】
下記(A)乃至(C)のいずれかのアミノ酸配列を含むデオキシニバレノール分解酵素タンパク質。
(A)配列番号2に記載のアミノ酸配列。
(B)配列番号2に記載のアミノ酸配列において1若しくは複数のアミノ酸が欠失、置換または付加されているアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
(C)配列番号2に記載のアミノ酸配列と少なくとも80%以上の相同性を有するアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
【請求項2】
下記(A)乃至(C)のいずれかのアミノ酸配列をコードするデオキシニバレノール分解酵素遺伝子。
(A)配列番号2に記載のアミノ酸配列。
(B)配列番号2に記載のアミノ酸配列において1若しくは複数のアミノ酸が欠失、置換または付加されているアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
(C)配列番号2に記載のアミノ酸配列と少なくとも80%以上の相同性を有するアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
【請求項3】
下記(D)乃至(F)のいずれかの塩基配列を含むデオキシニバレノール分解酵素遺伝子。
(D)配列番号1で表される塩基配列。
(E)配列番号1に記載の塩基配列において1若しくは複数の塩基が欠失、置換または付加されている塩基配列であって、デオキシニバレノール分解活性を有するタンパク質をコードする塩基配列。
(F)配列番号1に記載の塩基配列とストリンジェントな条件下でハイブリダイズし、且つデオキシニバレノール分解活性を有するタンパク質をコードする塩基配列。
【請求項4】
請求項2又は請求項3に記載の遺伝子を含む組み換えベクター。
【請求項5】
請求項2又は請求項3に記載の遺伝子を含む形質転換体。
【請求項6】
請求項4に記載の組み換えベクターを用いる形質転換体。
【請求項7】
請求項5又は請求項6に記載の形質転換体を用いるデオキシニバレノールの分解方法。
【請求項1】
下記(A)乃至(C)のいずれかのアミノ酸配列を含むデオキシニバレノール分解酵素タンパク質。
(A)配列番号2に記載のアミノ酸配列。
(B)配列番号2に記載のアミノ酸配列において1若しくは複数のアミノ酸が欠失、置換または付加されているアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
(C)配列番号2に記載のアミノ酸配列と少なくとも80%以上の相同性を有するアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
【請求項2】
下記(A)乃至(C)のいずれかのアミノ酸配列をコードするデオキシニバレノール分解酵素遺伝子。
(A)配列番号2に記載のアミノ酸配列。
(B)配列番号2に記載のアミノ酸配列において1若しくは複数のアミノ酸が欠失、置換または付加されているアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
(C)配列番号2に記載のアミノ酸配列と少なくとも80%以上の相同性を有するアミノ酸配列であって、デオキシニバレノール分解活性を有するアミノ酸配列。
【請求項3】
下記(D)乃至(F)のいずれかの塩基配列を含むデオキシニバレノール分解酵素遺伝子。
(D)配列番号1で表される塩基配列。
(E)配列番号1に記載の塩基配列において1若しくは複数の塩基が欠失、置換または付加されている塩基配列であって、デオキシニバレノール分解活性を有するタンパク質をコードする塩基配列。
(F)配列番号1に記載の塩基配列とストリンジェントな条件下でハイブリダイズし、且つデオキシニバレノール分解活性を有するタンパク質をコードする塩基配列。
【請求項4】
請求項2又は請求項3に記載の遺伝子を含む組み換えベクター。
【請求項5】
請求項2又は請求項3に記載の遺伝子を含む形質転換体。
【請求項6】
請求項4に記載の組み換えベクターを用いる形質転換体。
【請求項7】
請求項5又は請求項6に記載の形質転換体を用いるデオキシニバレノールの分解方法。
【図1】

【図2】
【図3】
【図4】
【図5】

【図2】
【図3】
【図4】
【図5】
【公開番号】特開2011−103863(P2011−103863A)
【公開日】平成23年6月2日(2011.6.2)
【国際特許分類】
【出願番号】特願2009−265762(P2009−265762)
【出願日】平成21年11月20日(2009.11.20)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成20年度、農林水産省プロジェクト研究、生産・流通・加工工程における体系的な危害要因の特性解明とリスク低減技術の開発(麦類のかび毒汚染防止・低減技術の開発)、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(501245414)独立行政法人農業環境技術研究所 (60)
【Fターム(参考)】
【公開日】平成23年6月2日(2011.6.2)
【国際特許分類】
【出願日】平成21年11月20日(2009.11.20)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成20年度、農林水産省プロジェクト研究、生産・流通・加工工程における体系的な危害要因の特性解明とリスク低減技術の開発(麦類のかび毒汚染防止・低減技術の開発)、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(501245414)独立行政法人農業環境技術研究所 (60)
【Fターム(参考)】
[ Back to top ]