
デキストラン生成酵素遺伝子、デキストラン生成酵素およびその製造方法、デキストランの製造方法
【課題】デキストリンデキストラナーゼの工業的生産が可能な程度に高いデキストラン合成能力を有する、新たなデキストリンデキストラナーゼ(デキストラン生成酵素)を提供する。
【解決手段】特定の塩基配列を有するデキストラン生成酵素遺伝子、この遺伝子を含有する組換えベクター、形質転換体およびそれを用いたデキストラン生成酵素の製造方法、デキストラン生成酵素、並びにデキストランの製造方法。この方法で製造されたデキストランおよび配糖体を含有する、食品、飼料、餌料、化粧料、または医薬品。
【解決手段】特定の塩基配列を有するデキストラン生成酵素遺伝子、この遺伝子を含有する組換えベクター、形質転換体およびそれを用いたデキストラン生成酵素の製造方法、デキストラン生成酵素、並びにデキストランの製造方法。この方法で製造されたデキストランおよび配糖体を含有する、食品、飼料、餌料、化粧料、または医薬品。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、デキストラン生成酵素遺伝子、この遺伝子を含有する組換えベクター、形質転換体およびそれを用いたデキストラン生成酵素の製造方法、デキストラン生成酵素、並びにデキストランの製造方法に関するものである。
【背景技術】
【0002】
デキストラン(α−1,6−グルカン)はロイコノストック属やストレプトコッカス属などに属する細菌を、蔗糖を炭素源として培養することにより、これらの細菌がデキストランスクラーゼを生産し、この酵素の作用により蔗糖から合成される。デキストランの分子構造はD−グルコースのみから成る高分子多糖類で、α−1,6−グルコシド結合を主体として、さらにα−1,2−,α−1,3−,α−1,4−グルコシド結合の分岐を有しているが、これら分岐の含有比はデキストランの起源により異なる。デキストランは、血液増量剤や代用血漿として用いられる他、優れたゲル濾過剤として医薬・生化学分野で広く利用されている。しかし、このデキストランスクラーゼを用いた蔗糖からのデキストランの合成は、蔗糖の構成糖のうちグルコースのみしか利用されないために、対糖収率が最高50%を超えることはない。
【0003】
一方、グルコノバクター属に属する細菌は、澱粉部分分解物を炭素源として培養することにより、デキストリンデキストラナーゼを生成し、この酵素の作用により澱粉部分分解物を基質としてデキストランの製造方法が報告されている(E.J.Hehre and D.M.Hamilton:Proc. Soc. Exp. Biol. and Med., 71,336-339 (1949)(非特許文献1) 、特許第3594650号公報(特許文献1))。この酵素の作用によれば、マルトースからのデキストランの合成はないものの、マルトトリオース以上のマルトオリゴ糖およびマルトデキストリンからデキストランを合成することができる。しかし本酵素を用いたデキストランの製造方法では、対糖収率が低く工業化が困難であった。
【0004】
この問題点の解決策として特開2001-258589号公報(特許文献2)には、デキストリンデキストラナーゼをα−グルコシダーゼと共存させることにより収率を向上させる方法が開示されている。
【非特許文献1】E.J.Hehre and D.M.Hamilton:Proc. Soc. Exp. Biol. and Med., 71,336-339 (1949)
【特許文献1】特許第3594650号公報
【特許文献2】特開2001-258589号公報
【発明の開示】
【発明が解決しようとする課題】
【0005】
上述のように、特許文献1に記載のグルコノバクター属に属する細菌由来のデキストリンデキストラナーゼは、生成量が少ないため工業用酵素として実用化されていない。また、特許文献2に記載のα−グルコシダーゼを併用する方法は、少ないデキストリンデキストラナーゼを効率よく使用する方法であって、該酵素の生産性向上の解決には至っていないという問題があった。
【0006】
デキストランの安全性は広く認められており、様々な食品用途にその利用が期待されている。しかし、非常に高価なためにほとんど利用されていない。デキストランを安価に製造し、その用途を広げるためには、デキストリンデキストラナーゼの安定供給が望まれている。
【0007】
そこで本発明の目的は、デキストリンデキストラナーゼの工業的生産が可能な程度に高いデキストラン合成能力を有する、新たなデキストリンデキストラナーゼ(デキストラン生成酵素)を提供することにある。
【課題を解決するための手段】
【0008】
そこで発明者は、バイオテクノロジー技術を用いて、これまで全く知られていないデキストリンデキストラナーゼの酵素遺伝子の取得に成功し、高発現させる方法や活性型酵素を製造する方法を発見した。このことにより、デキストリンデキストラナーゼの工業的生産方法を見出し、本発明を完成するに至った。
【0009】
本発明は以下の通りである。
[1]以下の(a)若しくは(b)のタンパク質をコードする、または以下の(c)若しくは(d)のDNAからなるデキストラン生成酵素遺伝子。
(a) 配列表に記載された配列番号2に示される1〜1284番のアミノ酸配列からなるタンパク質、
(b) アミノ酸配列(a)において1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列からなり、デキストラン生成活性を有するタンパク質、
(c) 配列表に記載された配列番号1に示される4728〜8579番の塩基配列からなるDNA、
(d) (c)に示すDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNA、
[2]以下の(e)若しくは(f)のタンパク質をコードするか、または以下の(g)若しくは(h)のDNAからなるデキストラン生成酵素遺伝子。
(e) 配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番のアミノ酸配列からなるタンパク質
(f) アミノ酸配列(e)において1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列からなり、デキストラン生成活性を有するタンパク質
(g) 配列表に記載された配列番号1に示される4728〜8189番、4728〜8021番、4728〜7814番、4728〜7634番、または4728〜7436番の塩基配列からなるDNA
(h) (g)に示すDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNA
[3](f)のタンパク質は、野生型のデキストラン生成酵素と同等またはそれより高いデキストラン生成活性を有する[2]に記載のデキストラン生成酵素遺伝子。
[4](h)のタンパク質は、野生型のデキストラン生成酵素またはそれより高いデキストラン生成活性を有する[2]に記載のデキストラン生成酵素遺伝子。
[5][1]〜[4]のいずれかに記載のデキストラン生成酵素遺伝子を含有する組換えベクター。
[6]ベクターが微生物で機能するベクターである[5]記載の組換えベクター。
[7]微生物で機能するベクターが、大腸菌、枯草菌、酵母またはカビで機能するベクターである[6]に記載の組換えベクター。
[8][5]〜[7]のいずれか一項に記載の組換えベクターを含む形質転換体。
[9]宿主が細菌、酵母またはカビである[8]に記載の形質転換体。
[10]宿主の細菌が大腸菌もしくは枯草菌であり、組換えベクターが[3または4記載の組換えベクターである[6]に記載の形質転換体。
[11]宿主の酵母がピチア属(Pichia属)、サッカロミセス属(Saccharomyces属)またはシゾサッカロミセス属(Schizosaccharomyces属)であり、組換えベクターが[5または6記載の組換えベクターである[8]に記載の形質転換体。
[12]宿主のカビが麹菌、クモノスカビであり、組換えベクターが[5または6記載の組換えベクターである[8]に記載の形質転換体。
[13][8]〜[12]のいずれか一項に記載の形質転換体を培養し、培養物からデキストラン生成酵素を採取することを特徴とするデキストラン生成酵素の製造方法。
[14][13]に記載のデキストラン生成酵素の製造方法にリフォールディング工程を含むことを特徴とするデキストラン生成酵素の製造方法。
[15][13]に記載のデキストラン生成酵素の製造方法に分子シャペロンと共発現することを特徴とするデキストラン生成酵素の製造方法。
[16]以下の(i)、(j)、(k)若しくは(l)のタンパク質からなるデキストラン生成酵素。
(i) 配列表に記載された配列番号2に示される1〜1284番のアミノ酸配列からなるタンパク質、
(j) アミノ酸配列(a)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質、
(k) 配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番のアミノ酸配列からなるタンパク質、
(l) アミノ酸配列(e)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質
[17](l)のタンパク質は、野生型のデキストラン生成酵素と同等またはそれより高いデキストラン生成活性を有する[16]に記載のデキストラン生成酵素。
[18][13]から[15]に記載の方法で製造されたデキストラン生成酵素または[16]または[17]に記載のデキストラン生成酵素を用いてデキストランを製造する方法。
[19][13]から[15]に記載の方法で製造されたデキストラン生成酵素または[16]または[17]に記載のデキストラン生成酵素を用いて配糖体を製造する方法。
[20][18]または[19]に記載の方法で製造されたデキストランおよび配糖体を含有する、食品、飼料、餌料、化粧料、または医薬品。
【発明の効果】
【0010】
本発明によれば、デキストリンデキストラナーゼの工業的生産が可能な程度に高いデキストラン合成能力を有する、新たなデキストリンデキストラナーゼ(デキストラン生成酵素)を提供することができる。
【発明を実施するための最良の形態】
【0011】
以下、デキストリンデキストラナーゼをDDaseと略すことがある。
【0012】
[デキストラン生成酵素遺伝子]
本発明によるデキストラン生成酵素遺伝子は、澱粉部分分解物の中から選ばれた基質に作用してデキストランを合成する反応を触媒するデキストラン生成酵素活性を有するタンパク質をコードするDNA配列であり、デキストラン生成反応を触媒するデキストラン生成酵素の部分アミノ酸配列情報をもとに、当該酵素の生産菌グルコノバクター・オキシダンス(Gluconobacter oxydans ATCC11894)から調製したゲノムDNAより当該酵素をコードする遺伝子を取得することに成功し、さらに細菌等の微生物での発現にも成功することによって決定されたものである。尚、グルコノバクター・オキシダンス(Gluconobacter oxydans)は、当初は、アセトバクター・カプスラタム(Acetobacter capsulatum)と分類されていたが、その後、グルコノバクター・オキシダンス(Gluconobacter oxydans)に分類が変更になった(Bergey's Manual of Determinating Bacteriology, Eighth Eddition, p251-253)。ATCC11894の菌株自体に変更はなく、ATCC11894のカタログにも、アセトバクター・カプスラタム(Acetobacter capsulatum)が、グルコノバクター・オキシダンス(Gluconobacter oxydans)に分類変更された旨、明記されている。
【0013】
[デキストラン生成酵素遺伝子のクローニング]
グルコノバクター(Gluconobacter)より得られるデキストラン生成酵素はデキストリンからデキスラトンを生成する反応を触媒する酵素である。このため、該酵素を生産する微生物の培地炭素源にデキストリンを用いると、該酵素は菌対外に分泌されデキストランを生成する。しかしながら、生成されたデキストランは、酵素と親和性が高く付着するため、酵素タンパク質とデキストランを分離するのが困難である。特定の蛋白質をコードする遺伝子を単離する場合、蛋白質の部分アミノ酸配列を決定し、その縮重コドンからなる混合オリゴヌクレオチドをプロープとして遺伝子ライブラリーから単離することが可能である。また、本発明において実施したようなPCRにより単離することも可能である。しかしながら、本酵素のように酵素タンパク質とデキストランとを分離することが困難な場合、該酵素のペプチドマップを明らかにすることは非常に困難である。すなわち、ペプチドマップを明らかにする際、変性剤による酵素の変性や、プロテアーゼにより酵素をペプチドへ分解することが必須であるが、該酵素の周りにデキストランが付着しているため、変性剤やプロテアーゼの作用をほとんど受けない。このため、蛋白質の部分アミノ酸配列を決定することは容易でない。
【0014】
そこで本発明者らは、まずデキストランをほとんど含まない酵素の培養条件を検討した。その結果、炭素源にマンニトールを用いることにより、デキストランをほとんど含まない酵素を得ることに成功した。これにより該酵素は、変性剤やプロテアーゼの作用を受けペプチドマップを明らかにすることが可能となり、いくつかの部分アミノ酸配列が決定された。さらにゲノムDNAを鋳型とするPCRによる部分断片の取得の後、その断片情報をもとにゲノムライブラリーのスクリーニングとアダプターPCRによって、初めて該酵素遺伝子のクローニングに成功した。
【0015】
[デキストラン生成酵素をコードする遺伝子を含むDNA断片]
本発明によるデキストラン生成酵素をコードしている遺伝子を含むDNA断片は、グルコノバクター(Gluconobacter)に属する菌体、より好ましくは、グルコノバクター・オキシダンス(Gluconobacter oxydans ATCC11894)より調製されるゲノムDNAのライブラリーより単離することができる。本発明によるデキストラン生成酵素遺伝子は、配列表に記載された配列番号2(デキストラン生成酵素遺伝子から推定されるアミノ酸配列およびそれをコードするDNAの塩基配列)のアミノ酸番号1〜1284で示されるアミノ酸配列、または該アミノ酸配列において1もしくは複数個のアミノ酸が置換、欠失、挿入もしくは付加されたアミノ酸配列を有し、かつ上記のデキストラン生成活性を有するタンパク質をコードするDNA配列である。
【0016】
本発明によるデキストラン生成酵素をコードするDNAの好ましい具体例としては、配列表に記載された配列番号1に示される4728〜8579番の塩基配列からなるDNA、およびこのDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNAが挙げられる。
【0017】
さらに本発明のデキストラン生成酵素遺伝子は、配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、若しくは1〜903番のアミノ酸配列からなるタンパク質、または該アミノ酸配列において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質をコードするDNA配列である。本発明者らの検討により、アミノ酸番号1〜1284で示されるアミノ酸配列の内、少なくともC末端側の904から1284番のアミノ酸配列を欠失するタンパク質は、アミノ酸番号1〜1284で示されるアミノ酸配列を有するタンパク質(野生型のデキストラン生成酵素)と同等またはそれより高いデキストラン生成活性を有することが見出された。
【0018】
さらに本発明によるデキストラン生成酵素をコードするDNAの好ましい具体例としては、配列表に記載された配列番号1に示される4728〜8189番、4728〜8021番、4728〜7814番、4728〜7634番、または4728〜7436番の塩基配列からなるDNA、およびこれらのDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNAを挙げることができる。
【0019】
本発明において、ストリンジェントな条件とは、特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいう。この条件を明確に数値化することは困難であるが、例えば、相同性の高いDNA同士、例えば80%以上、好ましくは90%以上の相同性を有するDNA同士がハイブリダイズし、それより低いDNA同士がハイブリダイズされない条件が挙げられる。具体的には、一般的なサザンハイブリダイゼーションの処理条件(ハイブリ溶液の塩濃度は、5XSSC,5Xdenhardt's液、0.02%SDS、0.5%スキムミルク溶液、100μg/mlサケ変性精子DNAからなる)よりも強い条件によりハイブリダイズすることを指し、この組成よりもハイブリ溶液の構成成分の濃度が高いか、ハイブリダイゼーション温度が55℃以上か、膜洗浄溶液が、0.1%SDS、0.5XSSCの組成より低い濃度の溶液であるか、膜洗浄温度が50℃以上の場合をいう。
【0020】
蛋白質のアミノ酸配列が与えられば、それをコードする塩基配列は、いわゆるコドン表を参照して決定することができる。よって配列番号2に示されるアミノ酸配列をコードする種々の塩基配列を適宜選択することが可能である。本発明の好ましい態様において、配列番号2のアミノ酸番号1〜1284、1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番に示されるアミノ酸配列をコードするDNA配列 とは、配列番号1に示される塩基配列の4728〜8579番、4728〜8189番、4728〜8021番、4728〜7814番、4728〜7634番、または4728〜7436番の配列を有するもの、およびその縮重関係にあるコドンが使用されている部分以外は同一の塩基配列を有し且つ上記のアミノ酸をコードする塩基配列、更には該配列において1もしくは複数(たとえば1もしくは数個)のコドンが置換、欠失、挿入もしくは付加された塩基配列をも包含するものである。配列番号1に示される塩基配列の4728〜8579番、4728〜8189番、4728〜8021番、4728〜7814番、4728〜7634番、または4728〜7436番までの配列を有するDNA断片は塩基配列が決定されていることから、そのDNA断片を取得する一つの手段は核酸合成の手法たとえば、DNA/RNAシンセサイザー(モデル392,アプライドバイオシステムズ)を用い、そのマニュアルに記載の方法に従って製造することができる。またこの配列は、前記したグルコノバクター(Gluconobacter)に属する菌体、より好ましくは、グルコノバクター・オキシダンス(Gluconobacter oxydans ATCC11894)から遺伝子工学的な手法を用いて得ることが出きる。例えば、Molecular Cloning: A Laboratory Manual (Sambrook、 Maniatis ら、 Cold Spring Harbour Laboratory Press(1989))などに記載の方法で好ましく行なうことができる。具体的な方法は、後記する実施例に詳細に説明されている。
【0021】
本明細書で言う「1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列」における「1から数個」の範囲は特には限定されないが、例えば、1から20個、好ましくは1から10個、より好ましくは1から7個、さらに好ましくは1から5個、特に好ましくは1から3個程度を意味する。
【0022】
[デキストラン生成酵素]
本発明によるデキストラン生成酵素は、澱粉部分分解物からデキストランを生成するものであり、より具体的には、マルトオリゴ糖および/またはマルトデキストリンよりデキストランを合成する。
【0023】
本発明の一つの態様によるデキストラン生成酵素のアミノ酸配列は、具体的には、配列表の配列番号2で示されるアミノ酸配列のアミノ酸番号1〜1284のアミノ酸配列、または該アミノ酸配列において1もしくは複数個のアミノ酸の挿入、置換、または欠失、若しくは両末端への付加がなされたものであって、且つ上記したデキストラン生成酵素活性を依然として保持する、改変された配列を包含するものとする。その改変配列におけるデキストラン生成酵素活性の保持とは、その活性を利用した実際の使用態様において、配列番号2に示される配列を全て有するポリペプチドと、同一の条件でほぼ同様の利用が可能な程度の活性が維持されていることをいうものとする。このような改変された配列は、配列番号2に示されている配列を参照すれば、当業者であれば格別の困難なしに選択し、製造可能であることは明らかである。
【0024】
さらに、本発明の別の態様によれば、本発明のもう一つの態様によるデキストラン生成酵素のアミノ酸配列は、配列表の配列番号2のアミノ酸番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番までに示されるアミノ酸配列において1もしくは複数個のアミノ酸の挿入、置換、または欠失、若しくは両末端への付加がなされたものであって、且つ上記したデキストラン生成酵素活性を依然として保持する改変された配列を包含するものとする。
【0025】
なお、後述のように本発明遺伝子を酵母で分泌発現させた場合、この酵素タンパク質は糖鎖が結合した形で得られ、糖鎖の切断によって糖鎖のない酵素タンパク質とすることができるが、これらもデキストラン生成酵素活性を保持している限り本発明の酵素に包含される。
【0026】
[デキストラン生成酵素をコードする遺伝子の発現/デキストラン生成酵素の製造]
1)発現ベクター
本発明によるデキストラン生成酵素をコードするDNA断片を、宿主細胞内で複製可能であるか、あるいは染色体に組み込まれかつ同遺伝子が発現可能な状態で含むDNA分子、特に発現ベクターの形態として宿主細胞の形質転換を行なえば、宿主細胞において本発明によるデキストラン生成酵素を産生させることができる。
【0027】
従って、本発明によれば、さらに本発明によるデキストラン生成酵素をコードする遺伝子を含んだDNA分子、特に発現ベクターが提供される。このDNA分子は、ベクター分子に本発明によるデキストラン生成酵素をコードするDNA断片を組み込むことによって得ることが出きる。本発明の好ましい態様によれば、このベクターはプラスミドである。
【0028】
本発明によるDNA分子の作成は、前掲のMolecular Cloning:A Laboratory Manualに記載の方法に準じて行なうことが出きる。
【0029】
本発明において利用されるベクターは、使用する宿主細胞の種類を勘案しながら、ウイルス、プラスミド、コスミドベクターなどから適宜選択できる。例えば、宿主細胞が枯草菌の場合はpUB系のプラスミド、大腸菌の場合はλファージ系のバクテリオファージ、pBR、pUC系のプラスミド、酵母の場合はYEp、YCp系、YIP系のベクター、あるいはpLeu4、pPPLeu4、pJPLeu系(特開平4−218382号公報に記載)などが挙げられる。
【0030】
このプラスミドは形質転換体の選択マーカーを含むのが好ましく、選択マーカーとしては薬剤耐性マーカー、栄養要求マーカー遺伝子を使用することができる。
【0031】
さらに、本発明による発現ベクターとしてのDNA分子は、デキストラン生成酵素遺伝子の発現に必要なDNA配列、例えばプロモーター、ターミネーター、リボゾーム結合部位、転写終結シグナルなどの転写調節信号、翻訳調節信号などを有しているのが好ましい。
【0032】
プロモーターとしては、枯草菌においてはズブチリシン、SPAC等のプロモーター、酵母ではアルコールデヒドロゲナーゼ(ADH)、酸性フォスファターゼ(PHO)、ガラクトース遺伝子(GAL)、グリセルアルデビド3リン酸脱水素酵素遺伝子(GAP)等のプロモーターが好ましく用いることができる。配列番号2に示しているアミノ酸配列のアミノ酸番号1番から1284番までの配列にはシグナルペプチド(アミノ酸番号1番〜25番)を含んでおり、後記する実施例で示すようにこの配列をそのまま、酵母由来のプロモーター、ターミネーターに挿入し酵母に導入して分泌発現をさせることが出きる。シグナルペプチドの使用は、培養上清からの精製も容易になるので好ましい。また、シグナルペプチドを枯草菌や酵母由来のもの(たとえばインベルターゼシグナル、酸性フォスファターゼシグナル、λ-ファクターシグナルなど)に置き換えることも好ましい。また、大腸菌においては、一般に慣用されるlacプロモーターやT7プロモーターの他、cspAプロモーター等を用い、分子シャペロンを同時に発現させる等の工夫をすればより効率的な発現が可能になることも考えられる。
【0033】
2)形質転換体/培養
本発明によれば、更に上記の発現ベクターを適当な宿主細胞に導入した形質転換細胞、および形質転換細胞を培養して培養物からデキストラン生成酵素を得るデキストラン生成酵素の製造法が提供される。
【0034】
形質転換細胞の培養は、使用宿主細胞に関して一般的な方法を用いることができ、通常1〜4日程度の培養により細胞内または細胞外の培養物中にデキストラン生成酵素が生成蓄積される。培養条件(培地、pH、温度等)に関しては、例えば、細菌では25〜37℃、酵母では25〜30℃、真核細胞では37℃程度が一般的であり、たとえば遺伝子発現実験マニュアル(講談社)等を参照することができる。
【0035】
宿主細胞としては、大腸菌、枯草菌等の細菌、カンディダ・ウチリス(Candida utilis)、サッカロミセス・セレビシエ(Saccaromyces cerevisiae)、ピチア・パストリス(Pichia Pastoris)等の酵母以外に、リゾープス・ニベウス(Rhizopus niveus)、リゾープス・デルマー(Rhizopus delemar)や高等真核生物(例えばCHO細胞など)を用いることができる。枯草菌としてはパチルス(Bacillus)属に属する微生物を用いることが好ましい。該属には蛋白質を菌体外へ分泌する株(たとえば、パチルス・スブチリス(Bacillus subtilis)など)が存在することが知られている。またプロテアーゼを殆ど分泌しない株も知られており、このような株を宿主として用いることも好ましい。本発明においては、宿主細胞として酵母、糸状菌または細菌が好ましいが、細菌がより好ましく、特に大腸菌やパチルス・スブチリス(Bacillus subtilis)が好ましい。後記する実施例に示すように、大腸菌を宿主としてこの遺伝子を発現させたところ、培地中に酵素活性が認められた。
【0036】
かくして調製された形質転換体の産生する組換えデキストラン生成酵素の単離・精製には、公知の分離、精製方法を適当に組み合わせて行なうことが出きる。これらの分離、精製方法としては例えば塩沈殿、溶媒沈殿のような溶解性の差を利用する方法、透析、限外濾過、ゲル濾過およびSDS−ポリアクリル電気泳動のような分子量の差を利用する方法、イオン交換クロマトグラフィーのような電荷の差を利用する方法、疎水クロマトグラフィー、逆相クロマトグラフィーのような疎水性の差を利用する方法、さらに等電点電気泳動のような等電点の差を利用する方法、この他アフィニティークロマトグラフィー等が挙げられる。具体的には、例えば実施例1に記載のようにデキストラン生成酵素を精製できるが、一般的な分離・精製法に関しては例えば蛋白質・酵素の基礎実験法(南江堂)等を参照することができる。
【0037】
[デキストランの製造]
本発明によれば、上記の組換えデキストラン生成酵素を用いたデキストランの製造方法が提供される。すなわち、本発明によるデキストランの製造法では、上述のような本発明によるデキストラン生成酵素を、澱粉部分加水分解物に作用させデキストランを製造する。
【0038】
この製造方法においては基質として、澱粉部分加水分解物の1種又は2種以上を用いる。澱粉部分加水分解物は主にマルトデキストリンであり、澱粉を常法により、酸または酵素により加水分解し、必要により分離精製することで得られる。澱粉の分解の程度は特に限度はない。また、澱粉部分加水分解物の一部であるマルトオリゴ糖等を澱粉部分加水分解物として用いることもできる。マルトオリゴ糖としては、例えば、マルトトリオース、マルトテトラオース、マルトペンタオース、マルトヘキサオース、マルトヘプタオースなどを挙げることができる。また、澱粉部分加水分解物としては、短鎖長アミロースを例示することもできる。マルトオリゴ糖は高純度の試薬レベルのものであっても、マルトオリゴ糖シラップのように純度の低いものであってもよい。反応液中の基質濃度(澱粉部分加水分解物濃度)は、特に制限はないが、例えば、0.1〜40%(w/w)の範囲であることができる。但し、より高濃度のデキストランを得るという観点からは、基質濃度も高い方が好ましい。
【0039】
この反応の際にはα−グルコシダーゼを併用することもでき、α−グルコシダーゼとしては、Bacillus属などの細菌、Aspergillus属、Mucor属、Penicillium属などの糸状菌、Candida属、Saccharomyces属、Schizosaccharomyces属などの酵母、イネ、ソバ、トウモロコシ、テンサイなどの植物や動物などに幅広く存在している。本発明に用いるα−グルコシダーゼとしては、これらの起源などは特に限定されるものではない。しかしながら、大量かつ安価で均質なα−グルコシダーゼを得るには、細菌、糸状菌や酵母などの微生物起源のα−グルコシダーゼを用いることが有利である。また、本発明においては、マルトテトラオース以上の重合度を有するマルトオリゴ糖およびマルトデキストリンは、デキストラン合成の基質として有効に利用されるために、α−グルコシダーゼは、これらの糖質に作用せず、マルトースおよび/またはマルトトリオースに主として作用するα−グルコシダーゼであることが望ましい。
【0040】
α−グルコシダーゼは、反応系にデキストリンデキストラナーゼの反応の当初から共存させても、反応の途中から共存させてもよい。反応系に存在し、デキストリンデキストラナーゼの反応を阻害するマルトースを効率よく分解し、グルコースとすることができるタイミング、あるいは反応効率の悪いマルトトリオースを分解できるタイミングであれば制限はない。尚、デキストリンデキストラナーゼは、グルコースでは阻害されない。
【0041】
反応系におけるデキストリンデキストラナーゼおよびα−グルコシダーゼの量は、基質である澱粉部分加水分解物の種類(分解の程度等)や濃度、さらには反応時間等を考慮して適宜決定することができる。また、デキストリンデキストラナーゼとα−グルコシダーゼの使用比率も、基質である澱粉部分加水分解物の種類や各酵素の起源(性能)に応じて適宜決定できる。但し、デキストラン合成量を考慮すると、α−グルコシダーゼの酵素活性量は、デキストリンデキストラナーゼの酵素活性量の50%以内であることが適当である。
【0042】
また、反応温度には特に制限はなく、使用するデキストリンデキストラナーゼおよびα−グルコシダーゼが安定に作用する温度域であればよい。但し、デキストランの合成効率という観点からは、デキストリンデキストラナーゼがより効率よく作用することが好ましい。尚、デキストリンデキストラナーゼはpH2.5〜6.5の範囲であれば、60℃以下の温度で活性を有することが知られている。反応時間は、反応温度や基質の濃度、使用する酵素の活性等を考慮して適宜決定できる。
【0043】
本発明の反応により、デキストランを含む水溶液が得られる。この水溶液から、エタノールなどを用いた有機溶媒による沈殿法、クロマト分画法や限外濾過膜による処理などを用いることによりデキストランを精製することができる。これらの方法は、単独またはいくつかの操作を組み合わせることにより、より効率的にデキストランを得ることができる。
【0044】
<デキストラン生成酵素の測定方法>
酵素活性測定法
40 mM マルトペンタオース 40μlに50mM 酢酸緩衝液(pH 4.5 ) 40 μlおよび40 μlの酵素溶液を加え、37℃にて反応を行った場合に、1分間当り1.0 μgのデキストランを合成する酵素量を1単位とする。なお、デキストランの定量は、以下に示すHPLC条件で測定することができる。
カラム:Aminex HPX-42A (バイオラド製)
カラム温度:75 ℃
流速: 0.5 ml/min
溶離液:精製水
【0045】
<デキストラン生成酵素>
本発明のデキストラン生成酵素は、以下の(i)、(j)、(k)若しくは(l)のタンパク質からなるデキストラン生成酵素である。各タンパク質については上記遺伝子についての説明で記載した通りである。
(i) 配列表に記載された配列番号2に示される1〜1284番のアミノ酸配列からなるタンパク質、
(j) アミノ酸配列(a)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質、
(k) 配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番のアミノ酸配列からなるタンパク質、
(l) アミノ酸配列(e)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質
【0046】
特に本発明の(k)のタンパク質からなるデキストラン生成酵素は、実施例にも示す通り、(i)のタンパク質からなるデキストラン生成酵素(野生型)より2倍〜3倍の範囲の高いデキストラン生成活性を有する。また、本発明の(l)のタンパク質からなるデキストラン生成酵素は、野生型のデキストラン生成酵素と同等またはそれより高いデキストラン生成活性を有するものであり、好ましくは、野生型より2倍以上の高いデキストラン生成活性を有するデキストラン生成酵素である。
【0047】
本発明のタンパク質の取得方法は特に制限されず、化学合成により合成したタンパク質でもよいし、遺伝子組み換え技術により作製した組み換えタンパク質でもよい。組み換えタンパク質を作製する場合には、先ず、本明細書の上記に記載した当該タンパク質をコードする遺伝子(DNA)を取得する。このDNAを適当な発現系に導入することにより、本発明のタンパク質を産生することができる。タンパク質の産生については前述のとおりである。
【0048】
本発明は、上記本発明の方法で製造されたデキストランおよび配糖体を含有する、食品、飼料、餌料、化粧料、または医薬品に関する。本発明において食品、飼料、餌料、化粧料、および医薬品には特に制限はない。
【実施例】
【0049】
以下に本発明の実施例を示すが、これは本発明を更に具体的に説明するためのものであり、本発明が以下の実施例の範囲のみに限定されるものではない。また、操作手順は特に記載しない限りMolecular Cloning: A Laboratory Manual (Sambrook、 Maniatis ら、Cold Spring Harbour Laboratory Press(1989))に記載の方法に従った。
【0050】
(実施例1)
DDaseの精製
DDaseの精製を鈴木らの方法1)に従って行った。まずグルコノバクター・オキシダンス(Gluconobacter oxydans ATCC11894)を100 mlの培地(0.5% yeast extract、2.5% mannitol、0.05% dextrin (HLD; 二村化学))により25℃で24時間振とう培養した。これを180 mlずつ分注した同培地5本に10 mlずつ植え継ぎ、25℃でさらに36時間振とう培養した。培養に500 ml容バッフル付き三角フラスコを用いた。
【0051】
培養液を3,800xg、4℃で20分間遠心分離し、培養上清を得た。これを30,000xg、4℃で40分間遠心分離し、沈殿を得た。この沈殿を20 mM 酢酸ナトリウム 緩衝液 (pH 4.5) 45 mlに溶解した。これをさらに3,600xg、4℃で10分間遠心分離し、1回目の遠心分離で除去されなかった菌体を除き、上清を得た。得られた上清を20 mM 酢酸ナトリウム 緩衝液 (pH 4.5) に対して透析した。この結果、電気泳動的に単一なDDaseを51 mg得た。
【0052】
(実施例2)
DDaseのN末端および内部部分アミノ酸配列の決定
アミノ酸配列の決定にプロテインシーケンサーProcise 491 (Applied Biosystems Inc.) を用いた。N末端アミノ酸配列をPVDF膜にエレクトロブロッティングにより転写した試料を用いて決定した。内部部分アミノ酸配列を本酵素のトリプシン消化ペプチドを用いて決定した。トリプシン消化ペプチドを以下に示す方法で調製した。96.8 μgのDDaseを減圧遠心濃縮器で乾固し、2 M 尿素 200μlに溶解した。これに1 mg/ml トリプシンを1μl加え、37 ℃で24時間保持した。この溶液に2-メルカプトエタノール 3.2μl加え窒素ガスを封入後、室温に16時間保持した。これに4-ビニルピロリドンを4.8μl加え、窒素ガスを封入し、室温に2時間置き、逆相HPLCの試料とした。HPLCを以下の条件で行った。カラムにCapcell Pak C18 UG120 (φ4.6 x 150 mm) を用いた。クロマトグラフィを流速1 ml/min、カラム温度50℃で行い、溶出を0.1% トリフルオロ酢酸中で0-50% アセトニトリル直線濃度勾配により行った。ペプチドを波長214 nmにより検出した。得られたペプチドのアミノ酸配列を決定した。
【0053】
その結果、N末端アミノ酸配列はADNSDEQFVA(配列番号3)であり、内部アミノ酸配列は、AEGLNAASQLASAMGEGNQA(配列番号4), XXXLNLGTDGQK(配列番号5), XATGINPGEVSSTTXDP(配列番号6), NVTPGQQAETYSPYFK(配列番号7), ASVYLK(配列番号8)の5つが得られた。
【0054】
(実施例3)
ゲノムDNAの調製
グルコノバクター・オキシダンス(Gluconobacter oxydans ATCC11894)を100 mlの培地 (0.5% yeast extract、2.5% mannitol) により30℃で48時間培養し、5,800xg、4℃で10分間遠心分離し菌体を回収した。これを20 mM EDTAを含む10 mM Tris-HCl 緩衝液 (pH 8.0)に懸濁し、リゾチームを4 mg加えて37℃で30分間保持した。これにプロテイナーゼ Kを2 mg加えて37℃に30分間保持し、10% SDSを1 ml加えてさらに1時間保持した。得られた溶液にフェノール抽出を3回、フェノール‐クロロホルム抽出を3回行うことによりタンパク質を除去し、核酸を抽出した。核酸をエタノール沈殿により回収し、15 mlのTE(1 mM EDTAを含む10 mM Tris-HCl 緩衝液 (pH 8.0))に溶解した。これに10 mg/ml RNase Aを300μl加え、37℃で30分間保持し、RNAを消化した。フェノール‐クロロホルム抽出を行い、エタノール沈殿によりDNAを回収した。これを1 mlのTEに溶解した。その結果、0.4 mgのゲノムDNAを得た。
【0055】
(実施例4)
DDase遺伝子のクローニング
N末端配列および内部配列より設計したオリゴヌクレオチド、5'-GCIGATAA T(A/T)(G/C)IGATGAGCAGTT(C/T)GT (sense) (配列番号9)および5'-TCICCIGGGTTIATICCIGT (A/T/C/G)GC (antisense) (配列番号10)を用いてPCRを行った。反応を50μlの系で行い、鋳型としてゲノムDNAを100 ng用い、プライマーの濃度を各々0.4μMとした。Taq ポリメラーゼを使用した。得られた増幅断片をpGEM T-Vector (Promega) にサブクローニングし、この塩基配列をジデオキシ法により解析した (pGEMDDとする)。ABI PRISM 310 Genetic Analyzer (Applied Biosystems Inc.) を使用した。
【0056】
限定ゲノムライブラリーを作成した。まずインサートの調製を行った。5μgのゲノムDNAをNcoIにより消化し、アガロースゲル電気泳動を行い、6,557-23,130 bpの消化断片をSephaglas BandPrep Kit (Amersham Pharmacia Biotech) を用いて回収した。次にベクターの調製を行った。pBluescript SK II (Stratagene)のマルチクローニングサイトにNcoIサイトを導入した。XhoIおよびEcoRI の突出末端部分をそれぞれ持ち、内部にNcoIサイトを持つ2本のオリゴヌクレオチド5'TCGAGCCATGGG(配列番号11)および5’AATTCCCATGGC(配列番号12)を96 ℃に1分間保持し、0.02 ℃/secの割合で37 ℃まで温度を下げアニールさせた。これをXhoIおよびEcoRIで消化したpBluescript SKIIに挿入した。これらの調製したインサートとベクターをT4 ligase(タカラバイオ)を用いて連結し、大腸菌DH5αをInoueらの方法2)に従って形質転換した。
【0057】
得られたコロニーをHybond-N+ (Amersham) に転写し、コロニーハイブリダイゼーションを行った。変性液(1.5 M NaCl、0.5 M NaOH)を浸み込ませたろ紙の上に転写後の膜を置き、室温に5分間保持後、中和液(1.5 M NaClを含む0.5 M Tris-HCl 緩衝液 (pH 7.5))で浸み込ませたろ紙の上で3分間保持した。得られた膜を2xSSC (0.3 M NaCl、0.3 M クエン酸ナトリウム)で洗浄し、風乾した。これにトランスイルミネーター上でUVを5分間照射し、ハイブリダイゼーションに用いた。プローブを以下のように調製した。まずオリゴヌクレオチド5'CCGATTACACGCTGCTTGGTGACGC (sense) (配列番号13)および5'TCATCGGTCGTCTGATTATTCAGTT (antisense) (配列番号14) を用いてPCRを行った。鋳型にpGEMDDを用い、Taq ポリメラーゼを用いた。次に、得られた増幅断片をSephaglas BandPrep Kitを用いて精製し、Alkphos Direct Labeling Reagents (Amersham Biocsiences) を用いて修飾した。ハイブリダイゼーションを以下のように行った。作製した膜をハイブリダイゼーション溶液 (0.5 M NaCl、0.4% blocking reagentを含むハイブリダイゼーション 緩衝液; blocking reagentおよびハイブリダイゼーション 緩衝液はいずれもAmersham Biocsiences社製) に浸漬し、55 ℃保持した (プレハイブリダイゼーション)。これに作製したプローブを加え、60℃で一晩保持した。得られた膜を第1洗浄緩衝液 (2 M 尿素、0.1% SDS、2 mM MgCl2および0.2% blocking reagentを含む50 mM リン酸ナトリウム 緩衝液 (pH 7.0)) に浸漬し、60 ℃で10分間保持することにより洗浄した。これを2回繰り返した。次に膜を第2洗浄緩衝液 (0.1 M NaClおよび2 mM MgCl2を含む50 mM Tris-HCl 緩衝液 (pH 10)) に浸漬し、室温で5分間振とうすることにより洗浄した。この操作を2回繰り返した。得られた膜にCDP-Star Detection Reagent (Amersham Bioscience) を均一に滴下し、室温に5分間保持した。これを用いてフィルムを感光させた。フィルムにはHyper film (Amersham Bioscience) を用いた。
【0058】
得られたポジティブクローンからプラスミドDNAを調製し、この塩基配列を決定した。このプラスミドDNAをpDD5'とする。得られた配列には本酵素の遺伝子の3'領域が含まれていなかったので、これをLA PCR in vitro Cloning Kit (タカラバイオ) を用いて取得した。まずゲノムDNA2.5μgをEcoRIにより消化し、エタノール沈殿後、Kitに添付のカセットを消化断片に連結した。これをPCRの鋳型として用いた。プライマーには得られた5'領域の配列から設計したオリゴヌクレオチド5'-CGTCAGGCTCTGGTCAACTC (sense) (配列番号15)およびカセットに相補的な配列を持つプライマーC1 (5'-GTACATATTGTCGTTAGAACGCGTAATACGACTCA) (配列番号16)を用いた。KOD DNA ポリメラーゼ (東洋紡) を用いた。この反応液1μlを鋳型としてnested PCRを行った。ここではプライマーとして5'-GGATCCAGGAAAATA TTGGTAGC (sense ) (配列番号17)およびカセットに相補的な配列を持つプライマーC2 (5'-CGTTAGAACGCGTAATACGACTCACTATAGGGAGA) (配列番号18)を用いた。得られたDNA断片をpBluescript SK IIにEcoRVサイトを介してサブクローニングし、塩基配列を決定した。この配列に基づき設計したプライマー5'-CGGTCGCCATGGTCCGCAATGAAGC (sense) (配列番号19)およびプライマーC1を用いてPCRを行った。鋳型にはゲノムDNA2.5μgをSalIにより消化し、同様にカセットを連結したものを用いた。同様にnested PCRを行った。ここではプライマーとして5'-TGGCAATGCCGGCGATACGAACGTT (sense) (配列番号20)およびプライマーC2を用いた。得られたDNA断片の塩基配列を決定した。これに基づきプライマー5'-GACGACGTCCGATGCCTATATCACG (sense) (配列番号21) を設計し、C1とともにPCRに用いた。ここでは鋳型としてゲノムDNA2.5μgをHindIIIにより消化したものに同様にカセットを連結したものを使用した。同様にnested PCRを行った。プライマー5'-CTGGACGCGATCGACAATCAAGGCA (sense) (配列番号22)およびプライマーC2を用いた。得られた増幅断片の塩基配列を決定した。以上の操作で得られた領域を1回のPCRにより増幅した。プライマーに5'-CGTCAGGCTCTGGTCAACTC (sense) (配列番号23)および5'-TCTGTAACGCCGCCAGTCTG (antisense) (配列番号24)を用いた。鋳型としてゲノムDNA100 ngを用い、KOD DNA ポリメラーゼを使用した。得られたDNA断片の配列を決定し、上記の3回のPCRにより明らかにした配列と同一であることを確認した。この配列を配列表1に示す。本配列中にN末端アミノ酸配列および全てのトリプシン消化ペプチドから得られた内部部分アミノ酸配列を確認することができたため、本遺伝子は目的遺伝子であると予想された。またこの増幅断片が挿入されたpBluescript SK IIをpDD3'とする。
【0059】
(実施例5)
発現プラスミドの構築
発現プラスミドを構築した。まずpDD5'およびpDD3'の2つのプラスミドDNAに分かれたDDaseのORFを一本につなげた。pDD5'およびpDD3'を鋳型としてPCRを行い、DDaseの5'末端側および3'末端側の領域をそれぞれ増幅した。5'末端側の増幅にはプライマー5'-AAAGTATGAAAGTGTAACTGTTGTG(S1 とする。sense) (配列番号25)および5'-GCTGGTCCG GTCGAATTCGACGACC (antisense) (配列番号26) を用い、3'末端側の増幅にはプライマー5'-CGTCAG GCTCTGGTCAACTC (sense) (配列番号27) および5'-GTTATCAGAAGGCATCTCGGTCACG (A1とする。antisense) (配列番号28)を用いた。得られた増幅断片をoverlap extension PCRによって連結した。プライマーにS1およびA1を用いた。得られたDNA断片をpBluescript SK IIにEcoRVサイトを介して挿入した。これをpDDaseSKとする。
【0060】
pET-23d (Novagene) を用いて発現プラスミドを構築した。このために3箇所のNcoIサイトを除き、ORFの5'および3'にそれぞれNcoIおよびXhoIサイトを導入した。5'-ACGCCATGGCTGACAACTCTGACGAGC (sense; DDNcoとする) (配列番号29)と5'-GCCCTCGCCCATAGCCGAGGCGAGT (antisense) (配列番号30)、5'-ACTCGCCTCGGCTATGGGC GAGGGC (sense) (配列番号31)と5'-ATTGCGGACCATAGCGACCGACGTG (antisense) (配列番号32)、5'-CACGTCG GTCGCTATGGTCCGCAAT (sense) (配列番号33)と5'-CACCGTGCCATGATCCCCGGCGGCG (anti- sense) (配列番号34) 、5'-CGCCGCCGGGGATCATGGCACGGTG (sense) (配列番号35) と5'-TCACTCGAGGACGA TGTTCATGCCCTGAA (antisense; DDXhoとする) (配列番号36)をそれぞれ用いてPCRを行った。鋳型にpDDaseSKを用いた。得られた増幅断片をそれぞれPCR Purification Kit (Qiagen) を用いて精製し、overlap extension PCRでの鋳型に用いた。ここではDDNcoおよびDDXhoをプライマーとした。得られた増幅断片およびpET-23dをそれぞれNcoIおよびXhoIで消化し、DNA Ligation Kit ver. 2 (タカラバイオ) を用いて連結した。これをpDDpETとする。
【0061】
次にpCold DNA I (タカラバイオ) を用いて発現プラスミドを構築した。pDDaseSKを鋳型としてPCRを行いORFの5'および3'にそれぞれSacIおよびXbaIサイトを導入した。プライマーに5'-AAAAAGAGCTCATGGCTGACAACTCTGACGAG (sense) (配列番号37)および5'-AA AAATCTAGATCAGGCGCCGACGATGTTCAT (antisense) (配列番号38)を用いた。この増幅断片およびpCold I DNAをそれぞれSacIおよびXbaIで消化し、上記の様に連結した。これをpDDColdとする。
【0062】
(実施例6)
DDaseの生産
まず大腸菌BL21 (DE3) をそれぞれpDDpETおよびpDDColdで形質転換した。次に分子シャペロンと共発現させるために各シャペロンプラスミドすなわちpG-KJE8、pGro7、pKJE7、pG-Tf2およびpTf16により形質転換した大腸菌BL21 (DE3) をそれぞれpDDpETおよびpDDColdで形質転換した。pG-KJE8、pGro7、pKJE7、pG-Tf2およびpTf16はそれぞれdnaK-dnaJ-grpEとgroES-groEL、groES-groEL、dnaK-dnaJ-grpE、groES-groEL-tigおよびtigのシャペロンチームを発現する。得られた形質転換体を50μg/ml アンピシリンを含む (分子シャペロンとの共発現では20μg/ml クロラムフェニコールも含む) LB培地2 mlで37 ℃で一晩培養し、同培地20 ml (ただしpG-KJE8との共発現では0.5 mg/ml L-アラビノースと5 ng/ml テトラサイクリン、pGro7、pKJE7およびpTf16では0.5 mg/ml L-アラビノース、pG-Tf2では5 ng/ml テトラサイクリンを含む) に植え継いだ。これを37℃でA600が0.5になるまで培養し、氷上で培養液を冷却した。これに0.1 M IPTGを200μl加え、タンパク質の生産を誘導した。誘導後の培養温度をpDDpETで形質転換したものでは28℃および15℃とし、pDDColdでは15℃として24時間培養した。これを5,800xg、4℃で2分間遠心分離し、菌体を回収した。得られた菌体を0.1 M 酢酸ナトリウム 緩衝液 (pH 4.5) 400μlに懸濁し、超音波により菌体を破砕した。これを13,000 xg、4℃で2分間遠心分離し、上清を得た。
【0063】
その結果、分子シャペロンの有無に関わらず生産された組換え酵素の大部分は封入体を形成し、不溶性画分に認められたが、無細胞抽出液にデキストラン合成活性が認められた(図1)。このことから単離した遺伝子がDDaseをコードすることを確認できた。DDaseのみを発現させた形質転換体の無細胞抽出液とマルトペンタオースとの反応18時間で、誘導後の培養を15℃で行ったものではタンパク質1 mgあたり1.79 mg、28℃では1.13 mgのデキストランが合成された。また、分子シャペロンと共発現させた場合にも図1に示されるようにデキストランの合成が確認された。
【0064】
なお、デキストランの合成量は以下のようにして求めた。14 mM マルトペンタオース、40 mM 酢酸緩衝液 (pH 4.5)、および酵素溶液からなる反応液50 μlを37℃に18時間保持した。このうち10 μlを0.5 M 酢酸緩衝液 (pH 6.0) 25 μlおよび74.3 μM Streptococcus mutans由来dextran glucosidase 15 μlと混合し、37℃に10分間保持した。その後、遊離したグルコースを定量した。すなわち、試料50 μlに2 M Tris-HCl buffer (pH 7.0) 100 μlおよびglucostat reagent (和光純薬工業) 20 μlを混合し、37℃に1時間保持した。このA505を測定し、0-500 μMの範囲で作成した標準曲線に基づきグルコース濃度を算出し、この値をもとに合成されたデキストラン量を求めた。
【0065】
(実施例7)
DDaseのリフォールディング
pDDpETで形質転換した大腸菌BL21 (DE3) を100μg/ml アンピシリンを含むLB培地15 mlで37 ℃で一晩培養し、同培地150 mlに植え継いだ。これを37℃でA600が0.5になるまで培養し、氷上で培養液を冷却した。これに0.1 M IPTGを1.5 ml加え、タンパク質の生産を誘導した。誘導後の培養を15℃で24時間行った。これを5,800xg、4℃で2分間遠心分離し、菌体を回収した。得られた菌体を0.5 M NaClを含む0.1 M 酢酸ナトリウム 緩衝液 (pH 4.5) 4 mlに懸濁し、超音波により菌体を破砕した。これを13,000 xg、4℃で5分間遠心分離し沈殿を得た。これを5 M 尿素、2% Triton X-100および5 mM EDTAを含む50 mM 酢酸ナトリウム 緩衝液 (pH 4.5) に懸濁し、室温に5分間保持した。これを同様に遠心分離し、沈殿を得た。これを50 mM 酢酸ナトリウム 緩衝液 (pH 4.5) 10 mlに懸濁した。このうち48μlに8 M グアニジン-HCl 150μlおよび4 M DTT 2μlを加え室温に1時間保持してタンパク質を変性させた。これより20μlをとり、0.05%各種界面活性剤および2 mM DL-シスチンを含む50 mM 酢酸ナトリウム 緩衝液 (pH 4.5) 1.4 mlと混合し、室温に1時間保持した。界面活性剤にTween 40、Tween 60、CTABおよびSB3-14を用いた。得られた溶液400μlと3% シクロアミロース 100μlを混合し、室温に24時間保持してこの酵素活性を測定した。対照としてDL-シスチンおよびシクロアミロースを加えないものもそれぞれ行った。
その結果を図2に示す。
【0066】
(実施例8)
発現プラスミドの構築
N末端およびC末端からそれぞれ欠失させた変異酵素を作製した。欠失させた領域を以下に示す。1-36 (N1)、1-83 (N2)、1-126 (N3)、1-179 (N4)、1-225 (N5)、1207-1284 (C1)、1155-1284 (C2)、1099-1284 (C3)、1030-1284 (C4)、970-1284 (C5)、904-1284 (C6)、853-1284 (C7)、786-1284 (C8)、734-1284 (C9)、663-1284 (C10)。これらの変異酵素の発現プラスミドを構築した。N末端部分を欠失させた変異酵素では以下に示すオリゴヌクレオチドをセンスプライマーとし、5'-AA AAATCTAGATCAGGCGCCGACGATGTTCAT (配列番号39)をアンチセンスプライマーとしてPCRを行った。使用したセンスプライマーの配列を示す。
N1, 5'-CCGGTGAGCTCCCAGCCACGACCAACGCGGTT(配列番号40); (SacIサイトを下線で示した)
N2, GCCAGGAGCTCCCCGGGGTCGTCGAA TTCGAC(配列番号41);
N3, CCAATGAGCTCACACCGGTGCCGGAGGATCAG(配列番号42);
N4, GCGAGGAGCTCGCGCCTA AGCAGCAATGGACA(配列番号43);
N5, AGACTGAGCTCCCGTATTTCAAATCCAACGAG(配列番号44)。
【0067】
C末端部分を欠失させた変異酵素では以下に示すオリゴヌクレオチドをアンチセンスプライマーとし、5'-AAAAAGAGCTCATGGCTGACAACTCTGACGAG(配列番号45)をセンスプライマーとしてPCRを行った。使用したアンチセンスプライマーの配列を示す。
C1, 5'-AGACCTCTAGAGTCGTTTGCGGTGATGTTC AT(配列番号46); (XbaIサイトを下線で示した)
C2, GTATATCTAGACGACTGGCTGGCGTTGATGGT(配列番号47);
C3, AACGTTCTAGAGCGGCCGGTATAG AGGAGGTC(配列番号48);
C4, GTCGTTCTAGAGGCCTGAATTTCCGTCAGAGG(配列番号49);
C5, TCGTTTCTAGAGTTCGT GTTGTCGATGGTTTC(配列番号50);
C6, GCGGTTCTAGACTGCCCGGTAATCGCAGGCAT(配列番号51);
C7, GGCGGTCTAG AGCCATTGTCATAGGACTGTTC(配列番号52);
C8, AGACCTCTAGAGTCCTGCGTCAGCGCATTTTC(配列番号53);
C9, AAAT GTCTAGACTGCGCATTCCAGAGCCCCGG(配列番号54);
C10, TAGTCTCTAGACCCAAGCCCGTTGCTACCAAT (配列番号55)。
【0068】
鋳型にpDDColdを用いた。得られた増幅断片およびpCold I DNAをそれぞれSacIおよびXbaIで消化し、上述した方法で連結した。作製した発現プラスミドの塩基配列を解析し、PCRが正確に行われたことを確認した。
【0069】
(実施例9)
N末端およびC末端を欠失させた変異酵素の生産とデキストラン生成
あらかじめpTf16で形質転換した大腸菌BL21 (DE3) を作製した発現プラスミドでさらに形質転換した。得られた形質転換体を50μg/ml アンピシリンおよび20μg/ml chloramphenicolを含むLB培地2 mlにより37℃で一晩培養した。これを0.5 mg/ml L-アラビノースを含む同培地20 mlに植え継ぎ、37℃でA600が0.5になるまで培養した。得られた培養液を氷上で冷却した。これに0.1 M IPTGを200μl加え、タンパク質の生産を誘導し、15℃で20時間培養した。これを5,800xg、4℃で2分間遠心分離し、菌体を回収した。得られた菌体を0.1 M 酢酸ナトリウム 緩衝液 (pH 4.5) 400μlに懸濁し、超音波により菌体を破砕した。これを13,000 xg、4℃で2分間遠心分離し、上清を得た。
【0070】
大腸菌により発現させた変異酵素は、野生型酵素と同様に全ての変異酵素は封入体を形成し、発現したタンパク質の大部分は不溶性画分に認められた。無細胞抽出液のデキストラン合成活性を測定したところ、N末端を欠失させた変異酵素はいずれも活性を示さなかったが、C末端を欠失させた変異酵素ではC1-C6が酵素活性を示し、C7-C10は活性を示さなかった。特にC2-C6は野生型酵素より高い活性を示した(図3)。
【0071】
(実施例10)
沢庵付け
大根25kgを常法に従い下漬し、サッカリン8gとともに中漬し、続いて、糠1.4kg、サッカリン8g、グリシン25g、グルタミン酸ソーダ170g、複合調味料58g,ソルビトール400g、食塩300g、焼酎(25度)280ml、および、実施例6もしくは9の方法で製造したデキストラン280gからなる床で仕上げ漬けした。
この製品は、製造2週間後においても粘度の低下がほとんどなく、糠は付着しており、艶、照りともに良好であった。
【0072】
(実施例11)
野沢菜
常法により下漬けされた野沢菜5kgを、醤油150ml、アミノ酸液150ml、水200ml、グルタミン酸ナトリウム30g、アミノ酸系粉末旨味料20g、コハク酸ナトリウム2g、50%乳酸5ml、食塩6gに、実施例6もしくは9の方法で製造したデキストラン10gの調味液に漬け、凍結貯蔵した。塩分控えめであるがドリップが少なく肉質の劣化が抑えられ、粘性を有したユニークな野沢菜であった。
【0073】
(実施例12)
シャンプー
実施例6もしくは9の方法で製造したデキストラン1.5w/w%水溶液としたもの80.0重量部に、エチルアルコール13.0重量部、グリセリン2.0重量部、香料0.3重量部、ポリオキシエチレン.ソルビタイ.モノラウレート1.5重量部を添加し、混合溶解してシャンプーを製造した。本品は、使用中の指の通りが極めて良く、かつ、洗髪後の毛髪の手触りがしなやかで、非常に使用感、爽快感に優れたシャンプーであった。
【産業上の利用可能性】
【0074】
本発明は、デキストラン製造および利用分野において有用である。
【図面の簡単な説明】
【0075】
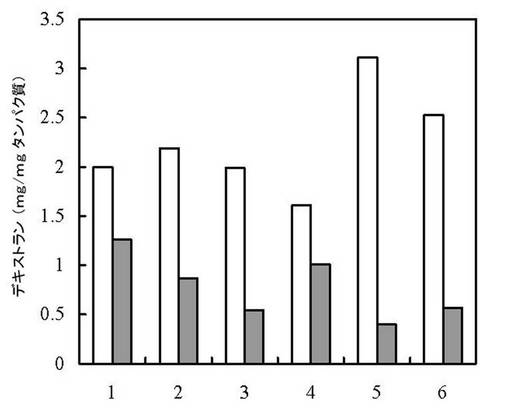
【図1】実施例6におけるデキストラン合成活性試験結果(発現ベクターとしてpET-23d を用い、シャペロン物質を用いた大腸菌でのDDaseの生成)。1:シャペロン物質無し; 2:pG-KJE8有り; 3:pGro7有り; 4:pKJE7有り; 5:pG-Tf2有り; 6:pTf16有り、白:15℃、灰色:28℃
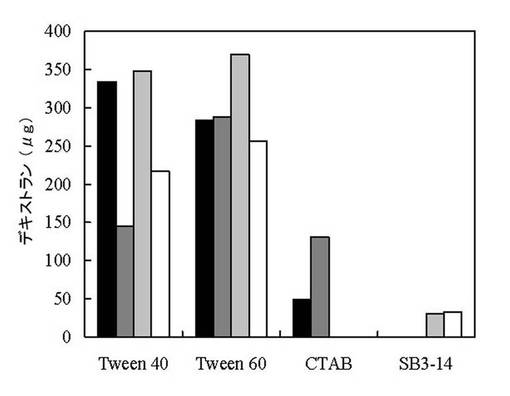
【図2】実施例7における酵素活性測定結果。黒は、DL-シスチン有り、シクロアミロース無しの結果、濃い灰色はDL-シスチン無し、シクロアミロース無しの結果、薄い灰色はDL-シスチン有り、シクロアミロース有りの結果、白はDL-シスチン無し、シクロアミロース有りの結果。
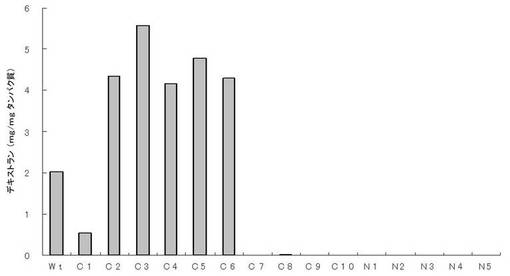
【図3】実施例9における変異酵素の酵素活性測定結果。
【技術分野】
【0001】
本発明は、デキストラン生成酵素遺伝子、この遺伝子を含有する組換えベクター、形質転換体およびそれを用いたデキストラン生成酵素の製造方法、デキストラン生成酵素、並びにデキストランの製造方法に関するものである。
【背景技術】
【0002】
デキストラン(α−1,6−グルカン)はロイコノストック属やストレプトコッカス属などに属する細菌を、蔗糖を炭素源として培養することにより、これらの細菌がデキストランスクラーゼを生産し、この酵素の作用により蔗糖から合成される。デキストランの分子構造はD−グルコースのみから成る高分子多糖類で、α−1,6−グルコシド結合を主体として、さらにα−1,2−,α−1,3−,α−1,4−グルコシド結合の分岐を有しているが、これら分岐の含有比はデキストランの起源により異なる。デキストランは、血液増量剤や代用血漿として用いられる他、優れたゲル濾過剤として医薬・生化学分野で広く利用されている。しかし、このデキストランスクラーゼを用いた蔗糖からのデキストランの合成は、蔗糖の構成糖のうちグルコースのみしか利用されないために、対糖収率が最高50%を超えることはない。
【0003】
一方、グルコノバクター属に属する細菌は、澱粉部分分解物を炭素源として培養することにより、デキストリンデキストラナーゼを生成し、この酵素の作用により澱粉部分分解物を基質としてデキストランの製造方法が報告されている(E.J.Hehre and D.M.Hamilton:Proc. Soc. Exp. Biol. and Med., 71,336-339 (1949)(非特許文献1) 、特許第3594650号公報(特許文献1))。この酵素の作用によれば、マルトースからのデキストランの合成はないものの、マルトトリオース以上のマルトオリゴ糖およびマルトデキストリンからデキストランを合成することができる。しかし本酵素を用いたデキストランの製造方法では、対糖収率が低く工業化が困難であった。
【0004】
この問題点の解決策として特開2001-258589号公報(特許文献2)には、デキストリンデキストラナーゼをα−グルコシダーゼと共存させることにより収率を向上させる方法が開示されている。
【非特許文献1】E.J.Hehre and D.M.Hamilton:Proc. Soc. Exp. Biol. and Med., 71,336-339 (1949)
【特許文献1】特許第3594650号公報
【特許文献2】特開2001-258589号公報
【発明の開示】
【発明が解決しようとする課題】
【0005】
上述のように、特許文献1に記載のグルコノバクター属に属する細菌由来のデキストリンデキストラナーゼは、生成量が少ないため工業用酵素として実用化されていない。また、特許文献2に記載のα−グルコシダーゼを併用する方法は、少ないデキストリンデキストラナーゼを効率よく使用する方法であって、該酵素の生産性向上の解決には至っていないという問題があった。
【0006】
デキストランの安全性は広く認められており、様々な食品用途にその利用が期待されている。しかし、非常に高価なためにほとんど利用されていない。デキストランを安価に製造し、その用途を広げるためには、デキストリンデキストラナーゼの安定供給が望まれている。
【0007】
そこで本発明の目的は、デキストリンデキストラナーゼの工業的生産が可能な程度に高いデキストラン合成能力を有する、新たなデキストリンデキストラナーゼ(デキストラン生成酵素)を提供することにある。
【課題を解決するための手段】
【0008】
そこで発明者は、バイオテクノロジー技術を用いて、これまで全く知られていないデキストリンデキストラナーゼの酵素遺伝子の取得に成功し、高発現させる方法や活性型酵素を製造する方法を発見した。このことにより、デキストリンデキストラナーゼの工業的生産方法を見出し、本発明を完成するに至った。
【0009】
本発明は以下の通りである。
[1]以下の(a)若しくは(b)のタンパク質をコードする、または以下の(c)若しくは(d)のDNAからなるデキストラン生成酵素遺伝子。
(a) 配列表に記載された配列番号2に示される1〜1284番のアミノ酸配列からなるタンパク質、
(b) アミノ酸配列(a)において1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列からなり、デキストラン生成活性を有するタンパク質、
(c) 配列表に記載された配列番号1に示される4728〜8579番の塩基配列からなるDNA、
(d) (c)に示すDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNA、
[2]以下の(e)若しくは(f)のタンパク質をコードするか、または以下の(g)若しくは(h)のDNAからなるデキストラン生成酵素遺伝子。
(e) 配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番のアミノ酸配列からなるタンパク質
(f) アミノ酸配列(e)において1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列からなり、デキストラン生成活性を有するタンパク質
(g) 配列表に記載された配列番号1に示される4728〜8189番、4728〜8021番、4728〜7814番、4728〜7634番、または4728〜7436番の塩基配列からなるDNA
(h) (g)に示すDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNA
[3](f)のタンパク質は、野生型のデキストラン生成酵素と同等またはそれより高いデキストラン生成活性を有する[2]に記載のデキストラン生成酵素遺伝子。
[4](h)のタンパク質は、野生型のデキストラン生成酵素またはそれより高いデキストラン生成活性を有する[2]に記載のデキストラン生成酵素遺伝子。
[5][1]〜[4]のいずれかに記載のデキストラン生成酵素遺伝子を含有する組換えベクター。
[6]ベクターが微生物で機能するベクターである[5]記載の組換えベクター。
[7]微生物で機能するベクターが、大腸菌、枯草菌、酵母またはカビで機能するベクターである[6]に記載の組換えベクター。
[8][5]〜[7]のいずれか一項に記載の組換えベクターを含む形質転換体。
[9]宿主が細菌、酵母またはカビである[8]に記載の形質転換体。
[10]宿主の細菌が大腸菌もしくは枯草菌であり、組換えベクターが[3または4記載の組換えベクターである[6]に記載の形質転換体。
[11]宿主の酵母がピチア属(Pichia属)、サッカロミセス属(Saccharomyces属)またはシゾサッカロミセス属(Schizosaccharomyces属)であり、組換えベクターが[5または6記載の組換えベクターである[8]に記載の形質転換体。
[12]宿主のカビが麹菌、クモノスカビであり、組換えベクターが[5または6記載の組換えベクターである[8]に記載の形質転換体。
[13][8]〜[12]のいずれか一項に記載の形質転換体を培養し、培養物からデキストラン生成酵素を採取することを特徴とするデキストラン生成酵素の製造方法。
[14][13]に記載のデキストラン生成酵素の製造方法にリフォールディング工程を含むことを特徴とするデキストラン生成酵素の製造方法。
[15][13]に記載のデキストラン生成酵素の製造方法に分子シャペロンと共発現することを特徴とするデキストラン生成酵素の製造方法。
[16]以下の(i)、(j)、(k)若しくは(l)のタンパク質からなるデキストラン生成酵素。
(i) 配列表に記載された配列番号2に示される1〜1284番のアミノ酸配列からなるタンパク質、
(j) アミノ酸配列(a)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質、
(k) 配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番のアミノ酸配列からなるタンパク質、
(l) アミノ酸配列(e)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質
[17](l)のタンパク質は、野生型のデキストラン生成酵素と同等またはそれより高いデキストラン生成活性を有する[16]に記載のデキストラン生成酵素。
[18][13]から[15]に記載の方法で製造されたデキストラン生成酵素または[16]または[17]に記載のデキストラン生成酵素を用いてデキストランを製造する方法。
[19][13]から[15]に記載の方法で製造されたデキストラン生成酵素または[16]または[17]に記載のデキストラン生成酵素を用いて配糖体を製造する方法。
[20][18]または[19]に記載の方法で製造されたデキストランおよび配糖体を含有する、食品、飼料、餌料、化粧料、または医薬品。
【発明の効果】
【0010】
本発明によれば、デキストリンデキストラナーゼの工業的生産が可能な程度に高いデキストラン合成能力を有する、新たなデキストリンデキストラナーゼ(デキストラン生成酵素)を提供することができる。
【発明を実施するための最良の形態】
【0011】
以下、デキストリンデキストラナーゼをDDaseと略すことがある。
【0012】
[デキストラン生成酵素遺伝子]
本発明によるデキストラン生成酵素遺伝子は、澱粉部分分解物の中から選ばれた基質に作用してデキストランを合成する反応を触媒するデキストラン生成酵素活性を有するタンパク質をコードするDNA配列であり、デキストラン生成反応を触媒するデキストラン生成酵素の部分アミノ酸配列情報をもとに、当該酵素の生産菌グルコノバクター・オキシダンス(Gluconobacter oxydans ATCC11894)から調製したゲノムDNAより当該酵素をコードする遺伝子を取得することに成功し、さらに細菌等の微生物での発現にも成功することによって決定されたものである。尚、グルコノバクター・オキシダンス(Gluconobacter oxydans)は、当初は、アセトバクター・カプスラタム(Acetobacter capsulatum)と分類されていたが、その後、グルコノバクター・オキシダンス(Gluconobacter oxydans)に分類が変更になった(Bergey's Manual of Determinating Bacteriology, Eighth Eddition, p251-253)。ATCC11894の菌株自体に変更はなく、ATCC11894のカタログにも、アセトバクター・カプスラタム(Acetobacter capsulatum)が、グルコノバクター・オキシダンス(Gluconobacter oxydans)に分類変更された旨、明記されている。
【0013】
[デキストラン生成酵素遺伝子のクローニング]
グルコノバクター(Gluconobacter)より得られるデキストラン生成酵素はデキストリンからデキスラトンを生成する反応を触媒する酵素である。このため、該酵素を生産する微生物の培地炭素源にデキストリンを用いると、該酵素は菌対外に分泌されデキストランを生成する。しかしながら、生成されたデキストランは、酵素と親和性が高く付着するため、酵素タンパク質とデキストランを分離するのが困難である。特定の蛋白質をコードする遺伝子を単離する場合、蛋白質の部分アミノ酸配列を決定し、その縮重コドンからなる混合オリゴヌクレオチドをプロープとして遺伝子ライブラリーから単離することが可能である。また、本発明において実施したようなPCRにより単離することも可能である。しかしながら、本酵素のように酵素タンパク質とデキストランとを分離することが困難な場合、該酵素のペプチドマップを明らかにすることは非常に困難である。すなわち、ペプチドマップを明らかにする際、変性剤による酵素の変性や、プロテアーゼにより酵素をペプチドへ分解することが必須であるが、該酵素の周りにデキストランが付着しているため、変性剤やプロテアーゼの作用をほとんど受けない。このため、蛋白質の部分アミノ酸配列を決定することは容易でない。
【0014】
そこで本発明者らは、まずデキストランをほとんど含まない酵素の培養条件を検討した。その結果、炭素源にマンニトールを用いることにより、デキストランをほとんど含まない酵素を得ることに成功した。これにより該酵素は、変性剤やプロテアーゼの作用を受けペプチドマップを明らかにすることが可能となり、いくつかの部分アミノ酸配列が決定された。さらにゲノムDNAを鋳型とするPCRによる部分断片の取得の後、その断片情報をもとにゲノムライブラリーのスクリーニングとアダプターPCRによって、初めて該酵素遺伝子のクローニングに成功した。
【0015】
[デキストラン生成酵素をコードする遺伝子を含むDNA断片]
本発明によるデキストラン生成酵素をコードしている遺伝子を含むDNA断片は、グルコノバクター(Gluconobacter)に属する菌体、より好ましくは、グルコノバクター・オキシダンス(Gluconobacter oxydans ATCC11894)より調製されるゲノムDNAのライブラリーより単離することができる。本発明によるデキストラン生成酵素遺伝子は、配列表に記載された配列番号2(デキストラン生成酵素遺伝子から推定されるアミノ酸配列およびそれをコードするDNAの塩基配列)のアミノ酸番号1〜1284で示されるアミノ酸配列、または該アミノ酸配列において1もしくは複数個のアミノ酸が置換、欠失、挿入もしくは付加されたアミノ酸配列を有し、かつ上記のデキストラン生成活性を有するタンパク質をコードするDNA配列である。
【0016】
本発明によるデキストラン生成酵素をコードするDNAの好ましい具体例としては、配列表に記載された配列番号1に示される4728〜8579番の塩基配列からなるDNA、およびこのDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNAが挙げられる。
【0017】
さらに本発明のデキストラン生成酵素遺伝子は、配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、若しくは1〜903番のアミノ酸配列からなるタンパク質、または該アミノ酸配列において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質をコードするDNA配列である。本発明者らの検討により、アミノ酸番号1〜1284で示されるアミノ酸配列の内、少なくともC末端側の904から1284番のアミノ酸配列を欠失するタンパク質は、アミノ酸番号1〜1284で示されるアミノ酸配列を有するタンパク質(野生型のデキストラン生成酵素)と同等またはそれより高いデキストラン生成活性を有することが見出された。
【0018】
さらに本発明によるデキストラン生成酵素をコードするDNAの好ましい具体例としては、配列表に記載された配列番号1に示される4728〜8189番、4728〜8021番、4728〜7814番、4728〜7634番、または4728〜7436番の塩基配列からなるDNA、およびこれらのDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNAを挙げることができる。
【0019】
本発明において、ストリンジェントな条件とは、特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいう。この条件を明確に数値化することは困難であるが、例えば、相同性の高いDNA同士、例えば80%以上、好ましくは90%以上の相同性を有するDNA同士がハイブリダイズし、それより低いDNA同士がハイブリダイズされない条件が挙げられる。具体的には、一般的なサザンハイブリダイゼーションの処理条件(ハイブリ溶液の塩濃度は、5XSSC,5Xdenhardt's液、0.02%SDS、0.5%スキムミルク溶液、100μg/mlサケ変性精子DNAからなる)よりも強い条件によりハイブリダイズすることを指し、この組成よりもハイブリ溶液の構成成分の濃度が高いか、ハイブリダイゼーション温度が55℃以上か、膜洗浄溶液が、0.1%SDS、0.5XSSCの組成より低い濃度の溶液であるか、膜洗浄温度が50℃以上の場合をいう。
【0020】
蛋白質のアミノ酸配列が与えられば、それをコードする塩基配列は、いわゆるコドン表を参照して決定することができる。よって配列番号2に示されるアミノ酸配列をコードする種々の塩基配列を適宜選択することが可能である。本発明の好ましい態様において、配列番号2のアミノ酸番号1〜1284、1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番に示されるアミノ酸配列をコードするDNA配列 とは、配列番号1に示される塩基配列の4728〜8579番、4728〜8189番、4728〜8021番、4728〜7814番、4728〜7634番、または4728〜7436番の配列を有するもの、およびその縮重関係にあるコドンが使用されている部分以外は同一の塩基配列を有し且つ上記のアミノ酸をコードする塩基配列、更には該配列において1もしくは複数(たとえば1もしくは数個)のコドンが置換、欠失、挿入もしくは付加された塩基配列をも包含するものである。配列番号1に示される塩基配列の4728〜8579番、4728〜8189番、4728〜8021番、4728〜7814番、4728〜7634番、または4728〜7436番までの配列を有するDNA断片は塩基配列が決定されていることから、そのDNA断片を取得する一つの手段は核酸合成の手法たとえば、DNA/RNAシンセサイザー(モデル392,アプライドバイオシステムズ)を用い、そのマニュアルに記載の方法に従って製造することができる。またこの配列は、前記したグルコノバクター(Gluconobacter)に属する菌体、より好ましくは、グルコノバクター・オキシダンス(Gluconobacter oxydans ATCC11894)から遺伝子工学的な手法を用いて得ることが出きる。例えば、Molecular Cloning: A Laboratory Manual (Sambrook、 Maniatis ら、 Cold Spring Harbour Laboratory Press(1989))などに記載の方法で好ましく行なうことができる。具体的な方法は、後記する実施例に詳細に説明されている。
【0021】
本明細書で言う「1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列」における「1から数個」の範囲は特には限定されないが、例えば、1から20個、好ましくは1から10個、より好ましくは1から7個、さらに好ましくは1から5個、特に好ましくは1から3個程度を意味する。
【0022】
[デキストラン生成酵素]
本発明によるデキストラン生成酵素は、澱粉部分分解物からデキストランを生成するものであり、より具体的には、マルトオリゴ糖および/またはマルトデキストリンよりデキストランを合成する。
【0023】
本発明の一つの態様によるデキストラン生成酵素のアミノ酸配列は、具体的には、配列表の配列番号2で示されるアミノ酸配列のアミノ酸番号1〜1284のアミノ酸配列、または該アミノ酸配列において1もしくは複数個のアミノ酸の挿入、置換、または欠失、若しくは両末端への付加がなされたものであって、且つ上記したデキストラン生成酵素活性を依然として保持する、改変された配列を包含するものとする。その改変配列におけるデキストラン生成酵素活性の保持とは、その活性を利用した実際の使用態様において、配列番号2に示される配列を全て有するポリペプチドと、同一の条件でほぼ同様の利用が可能な程度の活性が維持されていることをいうものとする。このような改変された配列は、配列番号2に示されている配列を参照すれば、当業者であれば格別の困難なしに選択し、製造可能であることは明らかである。
【0024】
さらに、本発明の別の態様によれば、本発明のもう一つの態様によるデキストラン生成酵素のアミノ酸配列は、配列表の配列番号2のアミノ酸番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番までに示されるアミノ酸配列において1もしくは複数個のアミノ酸の挿入、置換、または欠失、若しくは両末端への付加がなされたものであって、且つ上記したデキストラン生成酵素活性を依然として保持する改変された配列を包含するものとする。
【0025】
なお、後述のように本発明遺伝子を酵母で分泌発現させた場合、この酵素タンパク質は糖鎖が結合した形で得られ、糖鎖の切断によって糖鎖のない酵素タンパク質とすることができるが、これらもデキストラン生成酵素活性を保持している限り本発明の酵素に包含される。
【0026】
[デキストラン生成酵素をコードする遺伝子の発現/デキストラン生成酵素の製造]
1)発現ベクター
本発明によるデキストラン生成酵素をコードするDNA断片を、宿主細胞内で複製可能であるか、あるいは染色体に組み込まれかつ同遺伝子が発現可能な状態で含むDNA分子、特に発現ベクターの形態として宿主細胞の形質転換を行なえば、宿主細胞において本発明によるデキストラン生成酵素を産生させることができる。
【0027】
従って、本発明によれば、さらに本発明によるデキストラン生成酵素をコードする遺伝子を含んだDNA分子、特に発現ベクターが提供される。このDNA分子は、ベクター分子に本発明によるデキストラン生成酵素をコードするDNA断片を組み込むことによって得ることが出きる。本発明の好ましい態様によれば、このベクターはプラスミドである。
【0028】
本発明によるDNA分子の作成は、前掲のMolecular Cloning:A Laboratory Manualに記載の方法に準じて行なうことが出きる。
【0029】
本発明において利用されるベクターは、使用する宿主細胞の種類を勘案しながら、ウイルス、プラスミド、コスミドベクターなどから適宜選択できる。例えば、宿主細胞が枯草菌の場合はpUB系のプラスミド、大腸菌の場合はλファージ系のバクテリオファージ、pBR、pUC系のプラスミド、酵母の場合はYEp、YCp系、YIP系のベクター、あるいはpLeu4、pPPLeu4、pJPLeu系(特開平4−218382号公報に記載)などが挙げられる。
【0030】
このプラスミドは形質転換体の選択マーカーを含むのが好ましく、選択マーカーとしては薬剤耐性マーカー、栄養要求マーカー遺伝子を使用することができる。
【0031】
さらに、本発明による発現ベクターとしてのDNA分子は、デキストラン生成酵素遺伝子の発現に必要なDNA配列、例えばプロモーター、ターミネーター、リボゾーム結合部位、転写終結シグナルなどの転写調節信号、翻訳調節信号などを有しているのが好ましい。
【0032】
プロモーターとしては、枯草菌においてはズブチリシン、SPAC等のプロモーター、酵母ではアルコールデヒドロゲナーゼ(ADH)、酸性フォスファターゼ(PHO)、ガラクトース遺伝子(GAL)、グリセルアルデビド3リン酸脱水素酵素遺伝子(GAP)等のプロモーターが好ましく用いることができる。配列番号2に示しているアミノ酸配列のアミノ酸番号1番から1284番までの配列にはシグナルペプチド(アミノ酸番号1番〜25番)を含んでおり、後記する実施例で示すようにこの配列をそのまま、酵母由来のプロモーター、ターミネーターに挿入し酵母に導入して分泌発現をさせることが出きる。シグナルペプチドの使用は、培養上清からの精製も容易になるので好ましい。また、シグナルペプチドを枯草菌や酵母由来のもの(たとえばインベルターゼシグナル、酸性フォスファターゼシグナル、λ-ファクターシグナルなど)に置き換えることも好ましい。また、大腸菌においては、一般に慣用されるlacプロモーターやT7プロモーターの他、cspAプロモーター等を用い、分子シャペロンを同時に発現させる等の工夫をすればより効率的な発現が可能になることも考えられる。
【0033】
2)形質転換体/培養
本発明によれば、更に上記の発現ベクターを適当な宿主細胞に導入した形質転換細胞、および形質転換細胞を培養して培養物からデキストラン生成酵素を得るデキストラン生成酵素の製造法が提供される。
【0034】
形質転換細胞の培養は、使用宿主細胞に関して一般的な方法を用いることができ、通常1〜4日程度の培養により細胞内または細胞外の培養物中にデキストラン生成酵素が生成蓄積される。培養条件(培地、pH、温度等)に関しては、例えば、細菌では25〜37℃、酵母では25〜30℃、真核細胞では37℃程度が一般的であり、たとえば遺伝子発現実験マニュアル(講談社)等を参照することができる。
【0035】
宿主細胞としては、大腸菌、枯草菌等の細菌、カンディダ・ウチリス(Candida utilis)、サッカロミセス・セレビシエ(Saccaromyces cerevisiae)、ピチア・パストリス(Pichia Pastoris)等の酵母以外に、リゾープス・ニベウス(Rhizopus niveus)、リゾープス・デルマー(Rhizopus delemar)や高等真核生物(例えばCHO細胞など)を用いることができる。枯草菌としてはパチルス(Bacillus)属に属する微生物を用いることが好ましい。該属には蛋白質を菌体外へ分泌する株(たとえば、パチルス・スブチリス(Bacillus subtilis)など)が存在することが知られている。またプロテアーゼを殆ど分泌しない株も知られており、このような株を宿主として用いることも好ましい。本発明においては、宿主細胞として酵母、糸状菌または細菌が好ましいが、細菌がより好ましく、特に大腸菌やパチルス・スブチリス(Bacillus subtilis)が好ましい。後記する実施例に示すように、大腸菌を宿主としてこの遺伝子を発現させたところ、培地中に酵素活性が認められた。
【0036】
かくして調製された形質転換体の産生する組換えデキストラン生成酵素の単離・精製には、公知の分離、精製方法を適当に組み合わせて行なうことが出きる。これらの分離、精製方法としては例えば塩沈殿、溶媒沈殿のような溶解性の差を利用する方法、透析、限外濾過、ゲル濾過およびSDS−ポリアクリル電気泳動のような分子量の差を利用する方法、イオン交換クロマトグラフィーのような電荷の差を利用する方法、疎水クロマトグラフィー、逆相クロマトグラフィーのような疎水性の差を利用する方法、さらに等電点電気泳動のような等電点の差を利用する方法、この他アフィニティークロマトグラフィー等が挙げられる。具体的には、例えば実施例1に記載のようにデキストラン生成酵素を精製できるが、一般的な分離・精製法に関しては例えば蛋白質・酵素の基礎実験法(南江堂)等を参照することができる。
【0037】
[デキストランの製造]
本発明によれば、上記の組換えデキストラン生成酵素を用いたデキストランの製造方法が提供される。すなわち、本発明によるデキストランの製造法では、上述のような本発明によるデキストラン生成酵素を、澱粉部分加水分解物に作用させデキストランを製造する。
【0038】
この製造方法においては基質として、澱粉部分加水分解物の1種又は2種以上を用いる。澱粉部分加水分解物は主にマルトデキストリンであり、澱粉を常法により、酸または酵素により加水分解し、必要により分離精製することで得られる。澱粉の分解の程度は特に限度はない。また、澱粉部分加水分解物の一部であるマルトオリゴ糖等を澱粉部分加水分解物として用いることもできる。マルトオリゴ糖としては、例えば、マルトトリオース、マルトテトラオース、マルトペンタオース、マルトヘキサオース、マルトヘプタオースなどを挙げることができる。また、澱粉部分加水分解物としては、短鎖長アミロースを例示することもできる。マルトオリゴ糖は高純度の試薬レベルのものであっても、マルトオリゴ糖シラップのように純度の低いものであってもよい。反応液中の基質濃度(澱粉部分加水分解物濃度)は、特に制限はないが、例えば、0.1〜40%(w/w)の範囲であることができる。但し、より高濃度のデキストランを得るという観点からは、基質濃度も高い方が好ましい。
【0039】
この反応の際にはα−グルコシダーゼを併用することもでき、α−グルコシダーゼとしては、Bacillus属などの細菌、Aspergillus属、Mucor属、Penicillium属などの糸状菌、Candida属、Saccharomyces属、Schizosaccharomyces属などの酵母、イネ、ソバ、トウモロコシ、テンサイなどの植物や動物などに幅広く存在している。本発明に用いるα−グルコシダーゼとしては、これらの起源などは特に限定されるものではない。しかしながら、大量かつ安価で均質なα−グルコシダーゼを得るには、細菌、糸状菌や酵母などの微生物起源のα−グルコシダーゼを用いることが有利である。また、本発明においては、マルトテトラオース以上の重合度を有するマルトオリゴ糖およびマルトデキストリンは、デキストラン合成の基質として有効に利用されるために、α−グルコシダーゼは、これらの糖質に作用せず、マルトースおよび/またはマルトトリオースに主として作用するα−グルコシダーゼであることが望ましい。
【0040】
α−グルコシダーゼは、反応系にデキストリンデキストラナーゼの反応の当初から共存させても、反応の途中から共存させてもよい。反応系に存在し、デキストリンデキストラナーゼの反応を阻害するマルトースを効率よく分解し、グルコースとすることができるタイミング、あるいは反応効率の悪いマルトトリオースを分解できるタイミングであれば制限はない。尚、デキストリンデキストラナーゼは、グルコースでは阻害されない。
【0041】
反応系におけるデキストリンデキストラナーゼおよびα−グルコシダーゼの量は、基質である澱粉部分加水分解物の種類(分解の程度等)や濃度、さらには反応時間等を考慮して適宜決定することができる。また、デキストリンデキストラナーゼとα−グルコシダーゼの使用比率も、基質である澱粉部分加水分解物の種類や各酵素の起源(性能)に応じて適宜決定できる。但し、デキストラン合成量を考慮すると、α−グルコシダーゼの酵素活性量は、デキストリンデキストラナーゼの酵素活性量の50%以内であることが適当である。
【0042】
また、反応温度には特に制限はなく、使用するデキストリンデキストラナーゼおよびα−グルコシダーゼが安定に作用する温度域であればよい。但し、デキストランの合成効率という観点からは、デキストリンデキストラナーゼがより効率よく作用することが好ましい。尚、デキストリンデキストラナーゼはpH2.5〜6.5の範囲であれば、60℃以下の温度で活性を有することが知られている。反応時間は、反応温度や基質の濃度、使用する酵素の活性等を考慮して適宜決定できる。
【0043】
本発明の反応により、デキストランを含む水溶液が得られる。この水溶液から、エタノールなどを用いた有機溶媒による沈殿法、クロマト分画法や限外濾過膜による処理などを用いることによりデキストランを精製することができる。これらの方法は、単独またはいくつかの操作を組み合わせることにより、より効率的にデキストランを得ることができる。
【0044】
<デキストラン生成酵素の測定方法>
酵素活性測定法
40 mM マルトペンタオース 40μlに50mM 酢酸緩衝液(pH 4.5 ) 40 μlおよび40 μlの酵素溶液を加え、37℃にて反応を行った場合に、1分間当り1.0 μgのデキストランを合成する酵素量を1単位とする。なお、デキストランの定量は、以下に示すHPLC条件で測定することができる。
カラム:Aminex HPX-42A (バイオラド製)
カラム温度:75 ℃
流速: 0.5 ml/min
溶離液:精製水
【0045】
<デキストラン生成酵素>
本発明のデキストラン生成酵素は、以下の(i)、(j)、(k)若しくは(l)のタンパク質からなるデキストラン生成酵素である。各タンパク質については上記遺伝子についての説明で記載した通りである。
(i) 配列表に記載された配列番号2に示される1〜1284番のアミノ酸配列からなるタンパク質、
(j) アミノ酸配列(a)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質、
(k) 配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番のアミノ酸配列からなるタンパク質、
(l) アミノ酸配列(e)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質
【0046】
特に本発明の(k)のタンパク質からなるデキストラン生成酵素は、実施例にも示す通り、(i)のタンパク質からなるデキストラン生成酵素(野生型)より2倍〜3倍の範囲の高いデキストラン生成活性を有する。また、本発明の(l)のタンパク質からなるデキストラン生成酵素は、野生型のデキストラン生成酵素と同等またはそれより高いデキストラン生成活性を有するものであり、好ましくは、野生型より2倍以上の高いデキストラン生成活性を有するデキストラン生成酵素である。
【0047】
本発明のタンパク質の取得方法は特に制限されず、化学合成により合成したタンパク質でもよいし、遺伝子組み換え技術により作製した組み換えタンパク質でもよい。組み換えタンパク質を作製する場合には、先ず、本明細書の上記に記載した当該タンパク質をコードする遺伝子(DNA)を取得する。このDNAを適当な発現系に導入することにより、本発明のタンパク質を産生することができる。タンパク質の産生については前述のとおりである。
【0048】
本発明は、上記本発明の方法で製造されたデキストランおよび配糖体を含有する、食品、飼料、餌料、化粧料、または医薬品に関する。本発明において食品、飼料、餌料、化粧料、および医薬品には特に制限はない。
【実施例】
【0049】
以下に本発明の実施例を示すが、これは本発明を更に具体的に説明するためのものであり、本発明が以下の実施例の範囲のみに限定されるものではない。また、操作手順は特に記載しない限りMolecular Cloning: A Laboratory Manual (Sambrook、 Maniatis ら、Cold Spring Harbour Laboratory Press(1989))に記載の方法に従った。
【0050】
(実施例1)
DDaseの精製
DDaseの精製を鈴木らの方法1)に従って行った。まずグルコノバクター・オキシダンス(Gluconobacter oxydans ATCC11894)を100 mlの培地(0.5% yeast extract、2.5% mannitol、0.05% dextrin (HLD; 二村化学))により25℃で24時間振とう培養した。これを180 mlずつ分注した同培地5本に10 mlずつ植え継ぎ、25℃でさらに36時間振とう培養した。培養に500 ml容バッフル付き三角フラスコを用いた。
【0051】
培養液を3,800xg、4℃で20分間遠心分離し、培養上清を得た。これを30,000xg、4℃で40分間遠心分離し、沈殿を得た。この沈殿を20 mM 酢酸ナトリウム 緩衝液 (pH 4.5) 45 mlに溶解した。これをさらに3,600xg、4℃で10分間遠心分離し、1回目の遠心分離で除去されなかった菌体を除き、上清を得た。得られた上清を20 mM 酢酸ナトリウム 緩衝液 (pH 4.5) に対して透析した。この結果、電気泳動的に単一なDDaseを51 mg得た。
【0052】
(実施例2)
DDaseのN末端および内部部分アミノ酸配列の決定
アミノ酸配列の決定にプロテインシーケンサーProcise 491 (Applied Biosystems Inc.) を用いた。N末端アミノ酸配列をPVDF膜にエレクトロブロッティングにより転写した試料を用いて決定した。内部部分アミノ酸配列を本酵素のトリプシン消化ペプチドを用いて決定した。トリプシン消化ペプチドを以下に示す方法で調製した。96.8 μgのDDaseを減圧遠心濃縮器で乾固し、2 M 尿素 200μlに溶解した。これに1 mg/ml トリプシンを1μl加え、37 ℃で24時間保持した。この溶液に2-メルカプトエタノール 3.2μl加え窒素ガスを封入後、室温に16時間保持した。これに4-ビニルピロリドンを4.8μl加え、窒素ガスを封入し、室温に2時間置き、逆相HPLCの試料とした。HPLCを以下の条件で行った。カラムにCapcell Pak C18 UG120 (φ4.6 x 150 mm) を用いた。クロマトグラフィを流速1 ml/min、カラム温度50℃で行い、溶出を0.1% トリフルオロ酢酸中で0-50% アセトニトリル直線濃度勾配により行った。ペプチドを波長214 nmにより検出した。得られたペプチドのアミノ酸配列を決定した。
【0053】
その結果、N末端アミノ酸配列はADNSDEQFVA(配列番号3)であり、内部アミノ酸配列は、AEGLNAASQLASAMGEGNQA(配列番号4), XXXLNLGTDGQK(配列番号5), XATGINPGEVSSTTXDP(配列番号6), NVTPGQQAETYSPYFK(配列番号7), ASVYLK(配列番号8)の5つが得られた。
【0054】
(実施例3)
ゲノムDNAの調製
グルコノバクター・オキシダンス(Gluconobacter oxydans ATCC11894)を100 mlの培地 (0.5% yeast extract、2.5% mannitol) により30℃で48時間培養し、5,800xg、4℃で10分間遠心分離し菌体を回収した。これを20 mM EDTAを含む10 mM Tris-HCl 緩衝液 (pH 8.0)に懸濁し、リゾチームを4 mg加えて37℃で30分間保持した。これにプロテイナーゼ Kを2 mg加えて37℃に30分間保持し、10% SDSを1 ml加えてさらに1時間保持した。得られた溶液にフェノール抽出を3回、フェノール‐クロロホルム抽出を3回行うことによりタンパク質を除去し、核酸を抽出した。核酸をエタノール沈殿により回収し、15 mlのTE(1 mM EDTAを含む10 mM Tris-HCl 緩衝液 (pH 8.0))に溶解した。これに10 mg/ml RNase Aを300μl加え、37℃で30分間保持し、RNAを消化した。フェノール‐クロロホルム抽出を行い、エタノール沈殿によりDNAを回収した。これを1 mlのTEに溶解した。その結果、0.4 mgのゲノムDNAを得た。
【0055】
(実施例4)
DDase遺伝子のクローニング
N末端配列および内部配列より設計したオリゴヌクレオチド、5'-GCIGATAA T(A/T)(G/C)IGATGAGCAGTT(C/T)GT (sense) (配列番号9)および5'-TCICCIGGGTTIATICCIGT (A/T/C/G)GC (antisense) (配列番号10)を用いてPCRを行った。反応を50μlの系で行い、鋳型としてゲノムDNAを100 ng用い、プライマーの濃度を各々0.4μMとした。Taq ポリメラーゼを使用した。得られた増幅断片をpGEM T-Vector (Promega) にサブクローニングし、この塩基配列をジデオキシ法により解析した (pGEMDDとする)。ABI PRISM 310 Genetic Analyzer (Applied Biosystems Inc.) を使用した。
【0056】
限定ゲノムライブラリーを作成した。まずインサートの調製を行った。5μgのゲノムDNAをNcoIにより消化し、アガロースゲル電気泳動を行い、6,557-23,130 bpの消化断片をSephaglas BandPrep Kit (Amersham Pharmacia Biotech) を用いて回収した。次にベクターの調製を行った。pBluescript SK II (Stratagene)のマルチクローニングサイトにNcoIサイトを導入した。XhoIおよびEcoRI の突出末端部分をそれぞれ持ち、内部にNcoIサイトを持つ2本のオリゴヌクレオチド5'TCGAGCCATGGG(配列番号11)および5’AATTCCCATGGC(配列番号12)を96 ℃に1分間保持し、0.02 ℃/secの割合で37 ℃まで温度を下げアニールさせた。これをXhoIおよびEcoRIで消化したpBluescript SKIIに挿入した。これらの調製したインサートとベクターをT4 ligase(タカラバイオ)を用いて連結し、大腸菌DH5αをInoueらの方法2)に従って形質転換した。
【0057】
得られたコロニーをHybond-N+ (Amersham) に転写し、コロニーハイブリダイゼーションを行った。変性液(1.5 M NaCl、0.5 M NaOH)を浸み込ませたろ紙の上に転写後の膜を置き、室温に5分間保持後、中和液(1.5 M NaClを含む0.5 M Tris-HCl 緩衝液 (pH 7.5))で浸み込ませたろ紙の上で3分間保持した。得られた膜を2xSSC (0.3 M NaCl、0.3 M クエン酸ナトリウム)で洗浄し、風乾した。これにトランスイルミネーター上でUVを5分間照射し、ハイブリダイゼーションに用いた。プローブを以下のように調製した。まずオリゴヌクレオチド5'CCGATTACACGCTGCTTGGTGACGC (sense) (配列番号13)および5'TCATCGGTCGTCTGATTATTCAGTT (antisense) (配列番号14) を用いてPCRを行った。鋳型にpGEMDDを用い、Taq ポリメラーゼを用いた。次に、得られた増幅断片をSephaglas BandPrep Kitを用いて精製し、Alkphos Direct Labeling Reagents (Amersham Biocsiences) を用いて修飾した。ハイブリダイゼーションを以下のように行った。作製した膜をハイブリダイゼーション溶液 (0.5 M NaCl、0.4% blocking reagentを含むハイブリダイゼーション 緩衝液; blocking reagentおよびハイブリダイゼーション 緩衝液はいずれもAmersham Biocsiences社製) に浸漬し、55 ℃保持した (プレハイブリダイゼーション)。これに作製したプローブを加え、60℃で一晩保持した。得られた膜を第1洗浄緩衝液 (2 M 尿素、0.1% SDS、2 mM MgCl2および0.2% blocking reagentを含む50 mM リン酸ナトリウム 緩衝液 (pH 7.0)) に浸漬し、60 ℃で10分間保持することにより洗浄した。これを2回繰り返した。次に膜を第2洗浄緩衝液 (0.1 M NaClおよび2 mM MgCl2を含む50 mM Tris-HCl 緩衝液 (pH 10)) に浸漬し、室温で5分間振とうすることにより洗浄した。この操作を2回繰り返した。得られた膜にCDP-Star Detection Reagent (Amersham Bioscience) を均一に滴下し、室温に5分間保持した。これを用いてフィルムを感光させた。フィルムにはHyper film (Amersham Bioscience) を用いた。
【0058】
得られたポジティブクローンからプラスミドDNAを調製し、この塩基配列を決定した。このプラスミドDNAをpDD5'とする。得られた配列には本酵素の遺伝子の3'領域が含まれていなかったので、これをLA PCR in vitro Cloning Kit (タカラバイオ) を用いて取得した。まずゲノムDNA2.5μgをEcoRIにより消化し、エタノール沈殿後、Kitに添付のカセットを消化断片に連結した。これをPCRの鋳型として用いた。プライマーには得られた5'領域の配列から設計したオリゴヌクレオチド5'-CGTCAGGCTCTGGTCAACTC (sense) (配列番号15)およびカセットに相補的な配列を持つプライマーC1 (5'-GTACATATTGTCGTTAGAACGCGTAATACGACTCA) (配列番号16)を用いた。KOD DNA ポリメラーゼ (東洋紡) を用いた。この反応液1μlを鋳型としてnested PCRを行った。ここではプライマーとして5'-GGATCCAGGAAAATA TTGGTAGC (sense ) (配列番号17)およびカセットに相補的な配列を持つプライマーC2 (5'-CGTTAGAACGCGTAATACGACTCACTATAGGGAGA) (配列番号18)を用いた。得られたDNA断片をpBluescript SK IIにEcoRVサイトを介してサブクローニングし、塩基配列を決定した。この配列に基づき設計したプライマー5'-CGGTCGCCATGGTCCGCAATGAAGC (sense) (配列番号19)およびプライマーC1を用いてPCRを行った。鋳型にはゲノムDNA2.5μgをSalIにより消化し、同様にカセットを連結したものを用いた。同様にnested PCRを行った。ここではプライマーとして5'-TGGCAATGCCGGCGATACGAACGTT (sense) (配列番号20)およびプライマーC2を用いた。得られたDNA断片の塩基配列を決定した。これに基づきプライマー5'-GACGACGTCCGATGCCTATATCACG (sense) (配列番号21) を設計し、C1とともにPCRに用いた。ここでは鋳型としてゲノムDNA2.5μgをHindIIIにより消化したものに同様にカセットを連結したものを使用した。同様にnested PCRを行った。プライマー5'-CTGGACGCGATCGACAATCAAGGCA (sense) (配列番号22)およびプライマーC2を用いた。得られた増幅断片の塩基配列を決定した。以上の操作で得られた領域を1回のPCRにより増幅した。プライマーに5'-CGTCAGGCTCTGGTCAACTC (sense) (配列番号23)および5'-TCTGTAACGCCGCCAGTCTG (antisense) (配列番号24)を用いた。鋳型としてゲノムDNA100 ngを用い、KOD DNA ポリメラーゼを使用した。得られたDNA断片の配列を決定し、上記の3回のPCRにより明らかにした配列と同一であることを確認した。この配列を配列表1に示す。本配列中にN末端アミノ酸配列および全てのトリプシン消化ペプチドから得られた内部部分アミノ酸配列を確認することができたため、本遺伝子は目的遺伝子であると予想された。またこの増幅断片が挿入されたpBluescript SK IIをpDD3'とする。
【0059】
(実施例5)
発現プラスミドの構築
発現プラスミドを構築した。まずpDD5'およびpDD3'の2つのプラスミドDNAに分かれたDDaseのORFを一本につなげた。pDD5'およびpDD3'を鋳型としてPCRを行い、DDaseの5'末端側および3'末端側の領域をそれぞれ増幅した。5'末端側の増幅にはプライマー5'-AAAGTATGAAAGTGTAACTGTTGTG(S1 とする。sense) (配列番号25)および5'-GCTGGTCCG GTCGAATTCGACGACC (antisense) (配列番号26) を用い、3'末端側の増幅にはプライマー5'-CGTCAG GCTCTGGTCAACTC (sense) (配列番号27) および5'-GTTATCAGAAGGCATCTCGGTCACG (A1とする。antisense) (配列番号28)を用いた。得られた増幅断片をoverlap extension PCRによって連結した。プライマーにS1およびA1を用いた。得られたDNA断片をpBluescript SK IIにEcoRVサイトを介して挿入した。これをpDDaseSKとする。
【0060】
pET-23d (Novagene) を用いて発現プラスミドを構築した。このために3箇所のNcoIサイトを除き、ORFの5'および3'にそれぞれNcoIおよびXhoIサイトを導入した。5'-ACGCCATGGCTGACAACTCTGACGAGC (sense; DDNcoとする) (配列番号29)と5'-GCCCTCGCCCATAGCCGAGGCGAGT (antisense) (配列番号30)、5'-ACTCGCCTCGGCTATGGGC GAGGGC (sense) (配列番号31)と5'-ATTGCGGACCATAGCGACCGACGTG (antisense) (配列番号32)、5'-CACGTCG GTCGCTATGGTCCGCAAT (sense) (配列番号33)と5'-CACCGTGCCATGATCCCCGGCGGCG (anti- sense) (配列番号34) 、5'-CGCCGCCGGGGATCATGGCACGGTG (sense) (配列番号35) と5'-TCACTCGAGGACGA TGTTCATGCCCTGAA (antisense; DDXhoとする) (配列番号36)をそれぞれ用いてPCRを行った。鋳型にpDDaseSKを用いた。得られた増幅断片をそれぞれPCR Purification Kit (Qiagen) を用いて精製し、overlap extension PCRでの鋳型に用いた。ここではDDNcoおよびDDXhoをプライマーとした。得られた増幅断片およびpET-23dをそれぞれNcoIおよびXhoIで消化し、DNA Ligation Kit ver. 2 (タカラバイオ) を用いて連結した。これをpDDpETとする。
【0061】
次にpCold DNA I (タカラバイオ) を用いて発現プラスミドを構築した。pDDaseSKを鋳型としてPCRを行いORFの5'および3'にそれぞれSacIおよびXbaIサイトを導入した。プライマーに5'-AAAAAGAGCTCATGGCTGACAACTCTGACGAG (sense) (配列番号37)および5'-AA AAATCTAGATCAGGCGCCGACGATGTTCAT (antisense) (配列番号38)を用いた。この増幅断片およびpCold I DNAをそれぞれSacIおよびXbaIで消化し、上記の様に連結した。これをpDDColdとする。
【0062】
(実施例6)
DDaseの生産
まず大腸菌BL21 (DE3) をそれぞれpDDpETおよびpDDColdで形質転換した。次に分子シャペロンと共発現させるために各シャペロンプラスミドすなわちpG-KJE8、pGro7、pKJE7、pG-Tf2およびpTf16により形質転換した大腸菌BL21 (DE3) をそれぞれpDDpETおよびpDDColdで形質転換した。pG-KJE8、pGro7、pKJE7、pG-Tf2およびpTf16はそれぞれdnaK-dnaJ-grpEとgroES-groEL、groES-groEL、dnaK-dnaJ-grpE、groES-groEL-tigおよびtigのシャペロンチームを発現する。得られた形質転換体を50μg/ml アンピシリンを含む (分子シャペロンとの共発現では20μg/ml クロラムフェニコールも含む) LB培地2 mlで37 ℃で一晩培養し、同培地20 ml (ただしpG-KJE8との共発現では0.5 mg/ml L-アラビノースと5 ng/ml テトラサイクリン、pGro7、pKJE7およびpTf16では0.5 mg/ml L-アラビノース、pG-Tf2では5 ng/ml テトラサイクリンを含む) に植え継いだ。これを37℃でA600が0.5になるまで培養し、氷上で培養液を冷却した。これに0.1 M IPTGを200μl加え、タンパク質の生産を誘導した。誘導後の培養温度をpDDpETで形質転換したものでは28℃および15℃とし、pDDColdでは15℃として24時間培養した。これを5,800xg、4℃で2分間遠心分離し、菌体を回収した。得られた菌体を0.1 M 酢酸ナトリウム 緩衝液 (pH 4.5) 400μlに懸濁し、超音波により菌体を破砕した。これを13,000 xg、4℃で2分間遠心分離し、上清を得た。
【0063】
その結果、分子シャペロンの有無に関わらず生産された組換え酵素の大部分は封入体を形成し、不溶性画分に認められたが、無細胞抽出液にデキストラン合成活性が認められた(図1)。このことから単離した遺伝子がDDaseをコードすることを確認できた。DDaseのみを発現させた形質転換体の無細胞抽出液とマルトペンタオースとの反応18時間で、誘導後の培養を15℃で行ったものではタンパク質1 mgあたり1.79 mg、28℃では1.13 mgのデキストランが合成された。また、分子シャペロンと共発現させた場合にも図1に示されるようにデキストランの合成が確認された。
【0064】
なお、デキストランの合成量は以下のようにして求めた。14 mM マルトペンタオース、40 mM 酢酸緩衝液 (pH 4.5)、および酵素溶液からなる反応液50 μlを37℃に18時間保持した。このうち10 μlを0.5 M 酢酸緩衝液 (pH 6.0) 25 μlおよび74.3 μM Streptococcus mutans由来dextran glucosidase 15 μlと混合し、37℃に10分間保持した。その後、遊離したグルコースを定量した。すなわち、試料50 μlに2 M Tris-HCl buffer (pH 7.0) 100 μlおよびglucostat reagent (和光純薬工業) 20 μlを混合し、37℃に1時間保持した。このA505を測定し、0-500 μMの範囲で作成した標準曲線に基づきグルコース濃度を算出し、この値をもとに合成されたデキストラン量を求めた。
【0065】
(実施例7)
DDaseのリフォールディング
pDDpETで形質転換した大腸菌BL21 (DE3) を100μg/ml アンピシリンを含むLB培地15 mlで37 ℃で一晩培養し、同培地150 mlに植え継いだ。これを37℃でA600が0.5になるまで培養し、氷上で培養液を冷却した。これに0.1 M IPTGを1.5 ml加え、タンパク質の生産を誘導した。誘導後の培養を15℃で24時間行った。これを5,800xg、4℃で2分間遠心分離し、菌体を回収した。得られた菌体を0.5 M NaClを含む0.1 M 酢酸ナトリウム 緩衝液 (pH 4.5) 4 mlに懸濁し、超音波により菌体を破砕した。これを13,000 xg、4℃で5分間遠心分離し沈殿を得た。これを5 M 尿素、2% Triton X-100および5 mM EDTAを含む50 mM 酢酸ナトリウム 緩衝液 (pH 4.5) に懸濁し、室温に5分間保持した。これを同様に遠心分離し、沈殿を得た。これを50 mM 酢酸ナトリウム 緩衝液 (pH 4.5) 10 mlに懸濁した。このうち48μlに8 M グアニジン-HCl 150μlおよび4 M DTT 2μlを加え室温に1時間保持してタンパク質を変性させた。これより20μlをとり、0.05%各種界面活性剤および2 mM DL-シスチンを含む50 mM 酢酸ナトリウム 緩衝液 (pH 4.5) 1.4 mlと混合し、室温に1時間保持した。界面活性剤にTween 40、Tween 60、CTABおよびSB3-14を用いた。得られた溶液400μlと3% シクロアミロース 100μlを混合し、室温に24時間保持してこの酵素活性を測定した。対照としてDL-シスチンおよびシクロアミロースを加えないものもそれぞれ行った。
その結果を図2に示す。
【0066】
(実施例8)
発現プラスミドの構築
N末端およびC末端からそれぞれ欠失させた変異酵素を作製した。欠失させた領域を以下に示す。1-36 (N1)、1-83 (N2)、1-126 (N3)、1-179 (N4)、1-225 (N5)、1207-1284 (C1)、1155-1284 (C2)、1099-1284 (C3)、1030-1284 (C4)、970-1284 (C5)、904-1284 (C6)、853-1284 (C7)、786-1284 (C8)、734-1284 (C9)、663-1284 (C10)。これらの変異酵素の発現プラスミドを構築した。N末端部分を欠失させた変異酵素では以下に示すオリゴヌクレオチドをセンスプライマーとし、5'-AA AAATCTAGATCAGGCGCCGACGATGTTCAT (配列番号39)をアンチセンスプライマーとしてPCRを行った。使用したセンスプライマーの配列を示す。
N1, 5'-CCGGTGAGCTCCCAGCCACGACCAACGCGGTT(配列番号40); (SacIサイトを下線で示した)
N2, GCCAGGAGCTCCCCGGGGTCGTCGAA TTCGAC(配列番号41);
N3, CCAATGAGCTCACACCGGTGCCGGAGGATCAG(配列番号42);
N4, GCGAGGAGCTCGCGCCTA AGCAGCAATGGACA(配列番号43);
N5, AGACTGAGCTCCCGTATTTCAAATCCAACGAG(配列番号44)。
【0067】
C末端部分を欠失させた変異酵素では以下に示すオリゴヌクレオチドをアンチセンスプライマーとし、5'-AAAAAGAGCTCATGGCTGACAACTCTGACGAG(配列番号45)をセンスプライマーとしてPCRを行った。使用したアンチセンスプライマーの配列を示す。
C1, 5'-AGACCTCTAGAGTCGTTTGCGGTGATGTTC AT(配列番号46); (XbaIサイトを下線で示した)
C2, GTATATCTAGACGACTGGCTGGCGTTGATGGT(配列番号47);
C3, AACGTTCTAGAGCGGCCGGTATAG AGGAGGTC(配列番号48);
C4, GTCGTTCTAGAGGCCTGAATTTCCGTCAGAGG(配列番号49);
C5, TCGTTTCTAGAGTTCGT GTTGTCGATGGTTTC(配列番号50);
C6, GCGGTTCTAGACTGCCCGGTAATCGCAGGCAT(配列番号51);
C7, GGCGGTCTAG AGCCATTGTCATAGGACTGTTC(配列番号52);
C8, AGACCTCTAGAGTCCTGCGTCAGCGCATTTTC(配列番号53);
C9, AAAT GTCTAGACTGCGCATTCCAGAGCCCCGG(配列番号54);
C10, TAGTCTCTAGACCCAAGCCCGTTGCTACCAAT (配列番号55)。
【0068】
鋳型にpDDColdを用いた。得られた増幅断片およびpCold I DNAをそれぞれSacIおよびXbaIで消化し、上述した方法で連結した。作製した発現プラスミドの塩基配列を解析し、PCRが正確に行われたことを確認した。
【0069】
(実施例9)
N末端およびC末端を欠失させた変異酵素の生産とデキストラン生成
あらかじめpTf16で形質転換した大腸菌BL21 (DE3) を作製した発現プラスミドでさらに形質転換した。得られた形質転換体を50μg/ml アンピシリンおよび20μg/ml chloramphenicolを含むLB培地2 mlにより37℃で一晩培養した。これを0.5 mg/ml L-アラビノースを含む同培地20 mlに植え継ぎ、37℃でA600が0.5になるまで培養した。得られた培養液を氷上で冷却した。これに0.1 M IPTGを200μl加え、タンパク質の生産を誘導し、15℃で20時間培養した。これを5,800xg、4℃で2分間遠心分離し、菌体を回収した。得られた菌体を0.1 M 酢酸ナトリウム 緩衝液 (pH 4.5) 400μlに懸濁し、超音波により菌体を破砕した。これを13,000 xg、4℃で2分間遠心分離し、上清を得た。
【0070】
大腸菌により発現させた変異酵素は、野生型酵素と同様に全ての変異酵素は封入体を形成し、発現したタンパク質の大部分は不溶性画分に認められた。無細胞抽出液のデキストラン合成活性を測定したところ、N末端を欠失させた変異酵素はいずれも活性を示さなかったが、C末端を欠失させた変異酵素ではC1-C6が酵素活性を示し、C7-C10は活性を示さなかった。特にC2-C6は野生型酵素より高い活性を示した(図3)。
【0071】
(実施例10)
沢庵付け
大根25kgを常法に従い下漬し、サッカリン8gとともに中漬し、続いて、糠1.4kg、サッカリン8g、グリシン25g、グルタミン酸ソーダ170g、複合調味料58g,ソルビトール400g、食塩300g、焼酎(25度)280ml、および、実施例6もしくは9の方法で製造したデキストラン280gからなる床で仕上げ漬けした。
この製品は、製造2週間後においても粘度の低下がほとんどなく、糠は付着しており、艶、照りともに良好であった。
【0072】
(実施例11)
野沢菜
常法により下漬けされた野沢菜5kgを、醤油150ml、アミノ酸液150ml、水200ml、グルタミン酸ナトリウム30g、アミノ酸系粉末旨味料20g、コハク酸ナトリウム2g、50%乳酸5ml、食塩6gに、実施例6もしくは9の方法で製造したデキストラン10gの調味液に漬け、凍結貯蔵した。塩分控えめであるがドリップが少なく肉質の劣化が抑えられ、粘性を有したユニークな野沢菜であった。
【0073】
(実施例12)
シャンプー
実施例6もしくは9の方法で製造したデキストラン1.5w/w%水溶液としたもの80.0重量部に、エチルアルコール13.0重量部、グリセリン2.0重量部、香料0.3重量部、ポリオキシエチレン.ソルビタイ.モノラウレート1.5重量部を添加し、混合溶解してシャンプーを製造した。本品は、使用中の指の通りが極めて良く、かつ、洗髪後の毛髪の手触りがしなやかで、非常に使用感、爽快感に優れたシャンプーであった。
【産業上の利用可能性】
【0074】
本発明は、デキストラン製造および利用分野において有用である。
【図面の簡単な説明】
【0075】
【図1】実施例6におけるデキストラン合成活性試験結果(発現ベクターとしてpET-23d を用い、シャペロン物質を用いた大腸菌でのDDaseの生成)。1:シャペロン物質無し; 2:pG-KJE8有り; 3:pGro7有り; 4:pKJE7有り; 5:pG-Tf2有り; 6:pTf16有り、白:15℃、灰色:28℃
【図2】実施例7における酵素活性測定結果。黒は、DL-シスチン有り、シクロアミロース無しの結果、濃い灰色はDL-シスチン無し、シクロアミロース無しの結果、薄い灰色はDL-シスチン有り、シクロアミロース有りの結果、白はDL-シスチン無し、シクロアミロース有りの結果。
【図3】実施例9における変異酵素の酵素活性測定結果。
【特許請求の範囲】
【請求項1】
以下の(a)若しくは(b)のタンパク質をコードする、または以下の(c)若しくは(d)のDNAからなるデキストラン生成酵素遺伝子。
(a) 配列表に記載された配列番号2に示される1〜1284番のアミノ酸配列からなるタンパク質、
(b) アミノ酸配列(a)において1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列からなり、デキストラン生成活性を有するタンパク質、
(c) 配列表に記載された配列番号1に示される4728〜8579番の塩基配列からなるDNA、
(d) (c)に示すDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNA、
【請求項2】
以下の(e)若しくは(f)のタンパク質をコードするか、または以下の(g)若しくは(h)のDNAからなるデキストラン生成酵素遺伝子。
(e) 配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番のアミノ酸配列からなるタンパク質
(f) アミノ酸配列(e)において1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列からなり、デキストラン生成活性を有するタンパク質
(g) 配列表に記載された配列番号1に示される4728〜8189番、4728〜8021番、4728〜7814番、4728〜7634番、または4728〜7436番の塩基配列からなるDNA
(h) (g)に示すDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNA
【請求項3】
(f)のタンパク質は、野生型のデキストラン生成酵素と同等またはそれより高いデキストラン生成活性を有する請求項2に記載のデキストラン生成酵素遺伝子。
【請求項4】
(h)のタンパク質は、野生型のデキストラン生成酵素またはそれより高いデキストラン生成活性を有する請求項2に記載のデキストラン生成酵素遺伝子。
【請求項5】
請求項1〜4のいずれかに記載のデキストラン生成酵素遺伝子を含有する組換えベクター。
【請求項6】
ベクターが微生物で機能するベクターである請求項5記載の組換えベクター。
【請求項7】
微生物で機能するベクターが、大腸菌、枯草菌、酵母またはカビで機能するベクターである請求項6に記載の組換えベクター。
【請求項8】
請求項5〜7のいずれか一項に記載の組換えベクターを含む形質転換体。
【請求項9】
宿主が細菌、酵母またはカビである請求項8に記載の形質転換体。
【請求項10】
宿主の細菌が大腸菌もしくは枯草菌であり、組換えベクターが請求項3または4記載の組換えベクターである請求項6に記載の形質転換体。
【請求項11】
宿主の酵母がピチア属(Pichia属)、サッカロミセス属(Saccharomyces属)またはシゾサッカロミセス属(Schizosaccharomyces属)であり、組換えベクターが請求項5または6記載の組換えベクターである請求項8に記載の形質転換体。
【請求項12】
宿主のカビが麹菌、クモノスカビであり、組換えベクターが請求項5または6記載の組換えベクターである請求項8に記載の形質転換体。
【請求項13】
請求項8〜12のいずれか一項に記載の形質転換体を培養し、培養物からデキストラン生成酵素を採取することを特徴とするデキストラン生成酵素の製造方法。
【請求項14】
請求項13に記載のデキストラン生成酵素の製造方法にリフォールディング工程を含むことを特徴とするデキストラン生成酵素の製造方法。
【請求項15】
請求項13に記載のデキストラン生成酵素の製造方法に分子シャペロンと共発現することを特徴とするデキストラン生成酵素の製造方法。
【請求項16】
以下の(i)、(j)、(k)若しくは(l)のタンパク質からなるデキストラン生成酵素。
(i) 配列表に記載された配列番号2に示される1〜1284番のアミノ酸配列からなるタンパク質、
(j) アミノ酸配列(a)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質、
(k) 配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番のアミノ酸配列からなるタンパク質、
(l) アミノ酸配列(e)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質
【請求項17】
(l)のタンパク質は、野生型のデキストラン生成酵素と同等またはそれより高いデキストラン生成活性を有する請求項16に記載のデキストラン生成酵素。
【請求項18】
請求項13から15に記載の方法で製造されたデキストラン生成酵素または請求項16または17に記載のデキストラン生成酵素を用いてデキストランを製造する方法。
【請求項19】
請求項13から15に記載の方法で製造されたデキストラン生成酵素または請求項16または17に記載のデキストラン生成酵素を用いて配糖体を製造する方法。
【請求項20】
請求項18または19に記載の方法で製造されたデキストランおよび配糖体を含有する、食品、飼料、餌料、化粧料、または医薬品。
【請求項1】
以下の(a)若しくは(b)のタンパク質をコードする、または以下の(c)若しくは(d)のDNAからなるデキストラン生成酵素遺伝子。
(a) 配列表に記載された配列番号2に示される1〜1284番のアミノ酸配列からなるタンパク質、
(b) アミノ酸配列(a)において1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列からなり、デキストラン生成活性を有するタンパク質、
(c) 配列表に記載された配列番号1に示される4728〜8579番の塩基配列からなるDNA、
(d) (c)に示すDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNA、
【請求項2】
以下の(e)若しくは(f)のタンパク質をコードするか、または以下の(g)若しくは(h)のDNAからなるデキストラン生成酵素遺伝子。
(e) 配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番のアミノ酸配列からなるタンパク質
(f) アミノ酸配列(e)において1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列からなり、デキストラン生成活性を有するタンパク質
(g) 配列表に記載された配列番号1に示される4728〜8189番、4728〜8021番、4728〜7814番、4728〜7634番、または4728〜7436番の塩基配列からなるDNA
(h) (g)に示すDNAと相補的な塩基配列を有するDNAとストリンジェントな条件下でハイブリダイズし、かつデキストラン生成活性を有するタンパク質をコードするDNA
【請求項3】
(f)のタンパク質は、野生型のデキストラン生成酵素と同等またはそれより高いデキストラン生成活性を有する請求項2に記載のデキストラン生成酵素遺伝子。
【請求項4】
(h)のタンパク質は、野生型のデキストラン生成酵素またはそれより高いデキストラン生成活性を有する請求項2に記載のデキストラン生成酵素遺伝子。
【請求項5】
請求項1〜4のいずれかに記載のデキストラン生成酵素遺伝子を含有する組換えベクター。
【請求項6】
ベクターが微生物で機能するベクターである請求項5記載の組換えベクター。
【請求項7】
微生物で機能するベクターが、大腸菌、枯草菌、酵母またはカビで機能するベクターである請求項6に記載の組換えベクター。
【請求項8】
請求項5〜7のいずれか一項に記載の組換えベクターを含む形質転換体。
【請求項9】
宿主が細菌、酵母またはカビである請求項8に記載の形質転換体。
【請求項10】
宿主の細菌が大腸菌もしくは枯草菌であり、組換えベクターが請求項3または4記載の組換えベクターである請求項6に記載の形質転換体。
【請求項11】
宿主の酵母がピチア属(Pichia属)、サッカロミセス属(Saccharomyces属)またはシゾサッカロミセス属(Schizosaccharomyces属)であり、組換えベクターが請求項5または6記載の組換えベクターである請求項8に記載の形質転換体。
【請求項12】
宿主のカビが麹菌、クモノスカビであり、組換えベクターが請求項5または6記載の組換えベクターである請求項8に記載の形質転換体。
【請求項13】
請求項8〜12のいずれか一項に記載の形質転換体を培養し、培養物からデキストラン生成酵素を採取することを特徴とするデキストラン生成酵素の製造方法。
【請求項14】
請求項13に記載のデキストラン生成酵素の製造方法にリフォールディング工程を含むことを特徴とするデキストラン生成酵素の製造方法。
【請求項15】
請求項13に記載のデキストラン生成酵素の製造方法に分子シャペロンと共発現することを特徴とするデキストラン生成酵素の製造方法。
【請求項16】
以下の(i)、(j)、(k)若しくは(l)のタンパク質からなるデキストラン生成酵素。
(i) 配列表に記載された配列番号2に示される1〜1284番のアミノ酸配列からなるタンパク質、
(j) アミノ酸配列(a)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質、
(k) 配列表に記載された配列番号2に示される配列番号1〜1154番、1〜1098番、1〜1029番、1〜969番、または1〜903番のアミノ酸配列からなるタンパク質、
(l) アミノ酸配列(e)において1または複数のアミノ酸が欠失、置換または付加されたアミノ酸配列からなり、デキストラン生成活性を有するタンパク質
【請求項17】
(l)のタンパク質は、野生型のデキストラン生成酵素と同等またはそれより高いデキストラン生成活性を有する請求項16に記載のデキストラン生成酵素。
【請求項18】
請求項13から15に記載の方法で製造されたデキストラン生成酵素または請求項16または17に記載のデキストラン生成酵素を用いてデキストランを製造する方法。
【請求項19】
請求項13から15に記載の方法で製造されたデキストラン生成酵素または請求項16または17に記載のデキストラン生成酵素を用いて配糖体を製造する方法。
【請求項20】
請求項18または19に記載の方法で製造されたデキストランおよび配糖体を含有する、食品、飼料、餌料、化粧料、または医薬品。
【図1】


【図2】
【図3】
【図2】
【図3】
【公開番号】特開2007−181452(P2007−181452A)
【公開日】平成19年7月19日(2007.7.19)
【国際特許分類】
【出願番号】特願2006−322192(P2006−322192)
【出願日】平成18年11月29日(2006.11.29)
【出願人】(504173471)国立大学法人 北海道大学 (971)
【出願人】(000231453)日本食品化工株式会社 (68)
【Fターム(参考)】
【公開日】平成19年7月19日(2007.7.19)
【国際特許分類】
【出願日】平成18年11月29日(2006.11.29)
【出願人】(504173471)国立大学法人 北海道大学 (971)
【出願人】(000231453)日本食品化工株式会社 (68)
【Fターム(参考)】
[ Back to top ]