
デュロキセチンアルコールを製造するための改良型生体触媒
本発明は、新規フェニルエタノールデヒドロゲナーゼ変異体、それらの製造方法;それらをコードする核酸配列、この配列を含有する発現カセット、ベクターおよび組換え微生物;光学活性の置換アルコールを生体触媒により合成する方法、およびこの変異体の使用に関するが、特に、この変異体によって触媒される生体触媒による合成ステップを含んでなる、デュロキセチンアルコールまたはデュロキセチンを製造する方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新しいタイプのフェニルエタノールデヒドロゲナーゼ変異体、それらの製造方法;それらをコードする核酸配列、そうした配列を含んでなる発現カセット、ベクターおよび組換え微生物;前記変異体を用いて光学活性の置換アルコールを生体触媒合成する方法;ならびに、特に、前記変異体によって触媒される生体触媒合成ステップを含んでなる、デュロキセチンアルコールもしくはデュロキセチンを調製する方法に関する。
【背景技術】
【0002】
デュロキセチンアルコール(3)は、デュロキセチン(4)の調製において重要な前駆体である(模式図1を参照されたい)が、このデュロキセチンはCymbalta(登録商標)という商品名で、特に抗うつ薬として販売されている。
【化1】

【0003】
模式図1:デュロキセチンアルコールを経由するデュロキセチンの調製
生成する中間体(TACA)(2)は、デヒドロゲナーゼを用いて調製することができる(WO2005/033094)。たとえば、Azoarcus菌種(新規名称Aromatoleum aromaticum)(Hoeffken et al., Biochemistry, vol. 45, No.1, 2006)由来のフェニルエタノールデヒドロゲナーゼEbN1は、Meerwein-Ponndorf還元と同様に、クロロケトン、3-クロロ-1-(チエニル-2-イル)-プロパン-1-オン(1)を、対応するクロロアルコールである(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オール(2)に還元する。このために、このデヒドロゲナーゼは、必要な還元当量をもたらす補酵素ニコチンアミドアデニンジヌクレオチド(NADH)を必要とする。この高価な補酵素は、二次的な「犠牲アルコール」(たとえば、2-プロパノールまたは2-ブタノール)の助けを借りることによって再生することが可能であり、その間に対応するケトン(たとえばアセトンまたは2-ブタノン)が生成される。比較的長鎖のアルコールがこの酵素には好ましいが、長鎖の方がやはり相当高価である。こうした理由から、2-ブタノールが犠牲アルコールとして使用される(模式図2を参照されたい)(WO2006/072465)。
【化2】
【0004】
模式図2:「犠牲アルコール」を用いた補酵素NADHの再生
野生型酵素EbN1およびその発現のために使用できる発現系は、WO2005/108590およびWO2006/094945に記載されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】国際公開第2005/033094号
【特許文献2】国際公開第2006/072465号
【特許文献3】国際公開第2005/108590号
【特許文献4】国際公開第2006/094945号
【非特許文献】
【0006】
【非特許文献1】Hoeffken et al., Biochemistry, vol. 45, No.1, 2006
【発明の概要】
【発明が解決しようとする課題】
【0007】
発明の簡単な説明
デュロキセチンの調製に使用することができる生体触媒の活性を高めることを本発明の目的とした。
【0008】
特に、TAC(1)からTACA(2)への酵素的還元を改善する生体触媒を提供することを目的とした。ここで達成されるべき改善は下記のとおりとすることができる:
- 反応速度を高めること
- 生成物の収率を高めること
- 生成物阻害の影響を受けにくくすること
- 補酵素再生の向上
- 上記を組み合わせること。
【課題を解決するための手段】
【0009】
驚くべきことに、この目的は、Azoarcus菌種に由来する上記フェニルアルコールデヒドロゲナーゼEbN1の特別な変異体を提供することによって達成された。
【0010】
具体的には、上記目的は、驚くべきことに、2つの異なる方法で達成された。第1の解決方法によれば、この生体触媒をコードする遺伝子の配列を、エラープローンポリメラーゼ連鎖反応(エラープローンPCR)によって偶然に変異させ、それによって多数のバリアントを生じ、そのバリアントから改善された変異体を選択することができた。これらの変異体を次に、さらに改良するために再度変異させた(人工分子進化法)。
【0011】
もう一つの解決方法は、選択された配列位置で、標的化された方法で飽和突然変異誘発を実施することであった。まず、デヒドロゲナーゼの結晶構造を端緒として(Hoeffken et al., Biochemistry, vol. 45, No.1, 2006)、適当な変異のための標的位置を、「合理的設計」によって決定した。次いで、こうした位置で飽和突然変異導入を実施した。
【図面の簡単な説明】
【0012】
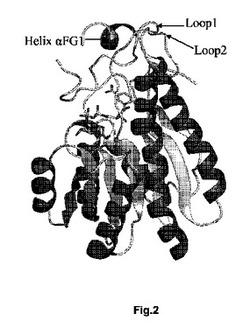
【図1】図1は、フェニルエタノールデヒドロゲナーゼEbN1の核酸コード配列(A)およびアミノ酸配列(B)を示す。
【図2】図2は、EbN1の1つのモノマーの、バンドモデルを示す。
【図3】図3は、さまざまな変異体のためのクローニングストラテジーを図示する。
【図4】図4は、いずれの場合も10 mM TA、TAAまたはTACAの存在下でフェニルエタノールデヒドロゲナーゼEbN1を阻害するための実験の結果を示すが、加えて、阻害物質なしでの対照バッチの結果も示す。実験はホールセルを用いて行った;いずれの場合も、25または50μlの細胞懸濁液をテストした。
【図5A】図5Aは、TACA存在下および非存在下で、Y151X型のさまざまな変異体による、2-ブタノールを用いた補酵素再生能力を示す。
【図5B】図5Bは、TACA存在下および非存在下で、T192X型のさまざまな変異体による、2-ブタノールを用いた補酵素再生能力を示す。
【図6A】図6Aは、補酵素再生を行わないTACテストにおけるT192X型のさまざまな変異体の活性を標準(LU11558)と比較して示す。
【図6B】図6Bは、補酵素再生を伴うTACテストにおけるT192X型のさまざまな変異体の活性を標準(LU11558)と比較して示す。
【図6C】図6Cは、TACAテストにおけるT192X型のさまざまな変異体の活性を標準(LU11558)と比較して示す。
【図7A】図7Aは、補酵素再生を行わないTACテストにおけるY151X型のさまざまな変異体の酵素活性を標準(LU11558)と比較して示す。
【図7B】図7Bは、補酵素再生を伴うTACテストにおけるY151X型のさまざまな変異体の酵素活性を標準(LU11558)と比較して示す。
【図7C】図7Cは、TACAテストにおけるY151X型のさまざまな変異体の活性を標準(LU11558)と比較して示す。
【図8A】図8Aは、TAC存在下よび非存在下での、Y151A-T192X 型変異体による、2-ブタノールを用いた補酵素の再生を示す。
【図8B】図8Bは、補酵素再生を伴うTACテストにおける、Y151A-T192X型変異体の活性を、対照(Y151A)と比較して示す。
【図9A】図9Aは、さまざまな反応混合物において変異体Y151Aを用いて達成されるTACAの収率を、いずれの場合も標準と比較し、異なるTAC濃度に応じて示す(400 mM)。
【図9B】図9Bは、さまざまな反応混合物において変異体Y151Aを用いて達成されるTACAの収率を、いずれの場合も標準と比較し、異なるTAC濃度に応じて示す(600 mM)。
【図10】図10は、野生型酵素EbN1における基質結合(TA)(合成A)または変異体Y151Aにおける基質結合(TA)(合成B)を、コンピューターアニメモデルとして示す。いずれの場合も下の図は、基質結合ポケットの拡大断面図を表す。
【図11】図11は、EbN1の活性中心の、コンピュータシミュレーション断面図を示す;両親媒性ヘリックス、ループ2、基質、および補酵素(NADH)の配置が強調されている。
【図12】図12は、部位特異的変異導入のためのクローニングストラテジーを示す。
【図13A】図13Aは、本発明のさまざまな点変異に関する活性テストの結果を示す。
【図13B】図13Bは、本発明のさまざまな点変異に関する活性テストの結果を示す。
【発明を実施するための形態】
【0013】
発明の詳細な説明
1. 一般的な用語の定義
「フェニルエタノールデヒドロゲナーゼ」(EC No.1.1.1)は、一般に、アセトフェノンからS-1-フェニルエタノールへの、NADH依存性の立体特異的還元を触媒する酵素である。「フェニルエタノールデヒドロゲナーゼ」または「フェニルエタノールデヒドロゲナーゼ活性を有する酵素」は、本発明に関しては特に、一般式Iのケトンから出発して、式IIの光学活性アルコールの酵素合成を触媒するものであって、具体的には3-クロロ-1-(チエニル-2-イル)-プロパン-1-オンと(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オールとの間の立体特異的平衡反応を触媒する。
【0014】
酵素反応の可逆性により、本発明は、両方向の、本明細書に記載される酵素反応に関する(すなわち還元当量の生成または消費を伴う)。
【0015】
「フェニルエタノールデヒドロゲナーゼ」の「機能的変異体」には、下記の、こうした酵素の「機能的同等物」が含まれる。
【0016】
「生体触媒プロセス」は、本発明の「フェニルエタノールデヒドロゲナーゼ」、または「フェニルエタノールデヒドロゲナーゼ活性」を有する酵素の、触媒活性の存在下で実施される任意のプロセス、すなわち、未精製の、もしくは精製された、溶解、分散もしくは固定化された酵素の存在下で行われるプロセス、またはそうした酵素活性を有する、もしくは発現する微生物ホールセルの存在下で行われるプロセスを表す。したがって、生体触媒プロセスは、酵素プロセスおよび微生物プロセスを含んでいる。
【0017】
「立体特異的」という用語は、本発明にしたがって調製される、少なくとも1つの不斉中心を有する化合物の、考えられるいくつかの立体異性体のうち1つが、たとえば少なくとも90%ee、特に少なくとも95%ee、または少なくとも98%ee、または少なくとも99%eeといった、高い「鏡像体過剰率」で、言い換えれば高い「鏡像体純度」で、本発明の酵素の作用によって生成されることを意味する。ee%値は、下記の式にしたがって計算される:
ee% = [XA-XB]/[ XA+XB]*100,
式中XAおよびXBはそれぞれ鏡像体AおよびBのモル分率である。
【0018】
さらに、本明細書では下記の略号を使用する。
【0019】
TAC = 3-クロロ-1-チオフェン-2-イル-プロパン-1-オン
TACA = 3-クロロ-1-チオフェン-2-イル-プロパン-1-オール
TA = 1-チオフェン-2-イル-プロペノン
TAA = 1-チオフェン-2-イル-プロパ-2-エン-1-オール
「低級アルコール」は特にモノオールであって、本明細書によれば低級アルキル基を含むものである。これは特にC1-C8-アルキル基、なかでもC1-C6-アルキル基であって、これらは分岐鎖または特に直鎖で、1から8個までの炭素原子、特に1、2、3、4、5または6個の炭素原子を有する。例としては、C1-C4-アルキル基、たとえばメチル、エチル、n-プロピル、イソプロピル、n-ブチル、2-ブチル、イソブチルもしくはtert-ブチル;さらに5個以上の炭素原子を有する基、たとえば、ペンチル、1-メチルブチル、2-メチルブチル、3-メチルブチル、2,2-ジメチルプロピル、1-エチルプロピル、1,1-ジメチルプロピル、1,2-ジメチルプロピル、1-メチルペンチル、2-メチルペンチル、3-メチルペンチル、4-メチルペンチル、n-ヘキシル、1,1-ジメチルブチル、1,2-ジメチルブチル、1,3-ジメチルブチル、2,2-ジメチルブチル、2,3-ジメチルブチル、および3,3-ジメチルブチルなどがある。
【0020】
「環」(Cyc)には、単環式もしくは多環式、飽和もしくは不飽和、炭素環もしくはヘテロ環、芳香族もしくは非芳香族であって、適宜、一置換もしくは多置換された環が含まれる。
【0021】
炭素環およびヘテロ環基Cycの例は、具体的には、単環式もしくは二環式、好ましくは単環式であって、4個以下、たとえば0、1もしくは2個の、O、NおよびSから選択される同一もしくは異なる環員ヘテロ原子を有する基である。
【0022】
このような炭素環もしくはヘテロ環は、特に3から12個まで、好ましくは4、5または6個の環員炭素原子を有する。例として挙げられるのは、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、それらの一価不飽和もしくは多価不飽和アナログ、たとえば、シクロブテニル、シクロペンテニル、シクロペンタジエニル、シクロヘキセニル、シクロヘプテニル、シクロヘキサジエニル、シクロヘプタジエニル、およびフェニル;ならびに5から7員環であって、O、NおよびSから選択される1から4個までのヘテロ原子を有する、飽和または一価もしくは多価不飽和ヘテロ環基である。具体的には、ピロリドン、テトラヒドロフラン、ピペリジン、モルホリン、ピロール、フラン、チオフェン、ピラゾール、イミダゾール、オキサゾール、チアゾール、ピリジン、ピラン、ピリミジン、ピリダジン、およびピラジンから導かれるヘテロ環基が挙げられる。
【0023】
前記炭素環もしくはヘテロ環のうち1つが、他のヘテロ環もしくは炭素環と縮合している二環式環基、たとえばクマロン、インドール、キノリンおよびナフタレンから誘導される基も挙げられる。
【0024】
Cyc基のさらに好ましい基はアリール基である。「アリール」は、単環式もしくは多環式、好ましくは単環式もしくは二環式の、適宜置換された芳香族基であって、具体的には、フェニル、もしくは任意の望ましい環上の位置で結合するナフチル、たとえば1-または2-ナフチルである。
【0025】
Cyc基は、任意の望ましい環上の位置で、好ましくは環員炭素原子を介して、結合することができる。
【0026】
適当なCyc基の例は、フェニル、ナフチル、2-チエニル、3-チエニル;2-フラニル、3-フラニル、2-ピリジル、3-ピリジルもしくは4-ピリジル;2-チアゾリル、4-チアゾリルもしくは5-チアゾリル;4-メチル-2-チエニル、3-エチル-2-チエニル、2-メチル-3-チエニル、4-プロピル-3-チエニル、5-n-ブチル-2-チエニル、4-メチル-3-チエニル、3-メチル-2-チエニル;3-クロロ-2-チエニル、4-ブロモ-3-チエニル、2-ヨード-3-チエニル、5-ヨード-3-チエニル、4-フルオロ-2-チエニル、2-ブロモ-3-チエニル、および4-クロロ-2-チエニルlである。
【0027】
Cyc基は、たとえば一置換もしくは多置換のように、1回または2回以上置換されていてもよい。好ましくは、置換基は環員炭素原子に結合する。適当な置換基の例は、ハロゲン、低級アルキル、低級アルケニル、低級アルコキシ、-OH、-SH、-NO2 またはNR2R3であって、このR2 およびR3は互いに無関係にH、メチルまたはエチルである。
【0028】
「ハロゲン」は、フッ素、塩素、臭素またはヨウ素であるが、特にフッ素または塩素である。
【0029】
「低級アルキル」は、好ましくは、2から8個まで、特に2から6個までの炭素原子を有する、直鎖もしくは分岐鎖アルキル基であって、たとえば、エチル、イソプロピルもしくはn-プロピル、n-ブチル、イソブチル、sec-ブチルもしくはtert-ブチル、n-ペンチルもしくは2-メチルブチル、n-ヘキシル、2-メチルペンチル、3-メチルペンチル、2-エチルブチルである。
【0030】
「低級アルコキシ」は、好ましくは、上記低級アルキル基に対応する酸素終端アナログである。
【0031】
「低級アルケニル」は、2から8個まで、特に2から6個までの炭素原子を有する前記アルキル基の、一価もしくは多価不飽和、好ましくは一価不飽和アナログであって、その二重結合は炭素鎖上の任意の望ましい位置にあるとすることができる。
【0032】
2. 本発明の好ましい実施形態
本発明は第1に、配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体を提供する。
【0033】
特に、本発明は、配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体に関するものであって、この変異体は、
(1)配列領域142〜153(ループ2とも呼ばれる)および
(2)配列領域190〜211(ヘリックスαFG1とも称される)
から選択される少なくとも1つの配列領域内に、少なくとも1つの変異を有する。
【0034】
特に、本発明は、
(3)配列領域93〜96(ループ1とも称される)
(4)配列領域241〜249(C末端)
(5)配列領域138〜141(結合ポケットの親水性領域、ループ2とも称される)
(6)Cys61および/またはCys83
から選択されるさらに他の配列領域中に、少なくとももう1つの変異を追加して有する、機能的なフェニルエタノールデヒドロゲナーゼ変異体に関する。
【0035】
さらに、本発明は、配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体に関するものであって、この変異体は、表1に記載の変異体から選択される。
【0036】
特に、以下の基の少なくとも1つが変異している変異体が挙げられるが:
T192、L197、M200、F201、L204、M246、L139、T140、T142、L146、I148、Y151、C61、C83、L186、
これらのアミノ酸はそれぞれ、他の任意の望ましい天然アミノ酸で置換されている。
【0037】
特に、本発明の変異体は、下記の変異のうち少なくとも1つを有する変異体から選択される:
a)単一変異:
Y151XA、ここでXAはA、R、N、E、Q、G、H、I、L、M、TまたはVである;
T192XB、ここでXBはA、E、G、I、P、S、W、VまたはLである;
b)多重変異:
Y151XA T192XB、ここでXA およびXB は上記の意味を持つ。
【0038】
本発明は、特に、下記の改変された部分配列のうち少なくとも1つによって特徴付けられる変異体を提供する:
(部分配列1) 142-TTYWX1KX2EAX3T-153(改変ループ2)および
(部分配列2) 190-ATX4EASAX5 SAX6X7DVX8PNMLQAI-211(改変ヘリックスαFG1)
これらの配列中、X1 〜X8は、互いに無関係に、任意の望ましいアミノ酸基であるが、基X1 〜X3 およびX4 〜X8の少なくとも1つは、配列番号2に記載の天然型酵素の天然アミノ酸基ではなく、具体的には
X1 はLである、またはI、V、A、M、FもしくはHで置換されている、
X2 はIである、またはL、V、A、M、FもしくはHで置換されている、
X3 はYである、またはA、R、N、E、Q、G、H、I、L、M、TもしくはVで置換されている;
あるいは、
X4 はTである、またはA、E、G、I、P、S、W、VもしくはLで置換されている、
X5 はLである、またはI、V、A、M、FもしくはHで置換されている、
X6 はMである、またはY、W、E、V、S、R、Q、K、I、H、G、F、EもしくはDで置換されている、
X7 はFである、またはG、K、T、Y、M、WもしくはRで置換されている、
X8 はLである、またはI、V、A、M、FもしくはHで置換されている。
【0039】
本発明はまた、特に、配列番号2のデヒドロゲナーゼの酵素活性の、すくなくとも約50%を依然として有する変異体、たとえば50から100%以上、たとえば100を超えて1000%までの活性を有する変異体など、に関するが、その活性はいずれの場合も、TACまたはTACAなどの標準物質を用いて標準的な条件下で測定される(下記、フェニルエタノールデヒドロゲナーゼ活性の測定に関する詳細を参照されたい)。
【0040】
特に、本発明はまた、配列番号2との配列同一性パーセントが少なくとも約70%、たとえば、70から99,9%まで、75から99.9%まで、80から99.9%まで、85から99.9%まで、90から99.9%まで、または95から99.9%までである変異体を提供する。
【0041】
特に本発明は、上記領域(1)〜(6)における少なくとも1つの変異に加えて、こうした領域の外側にあるアミノ酸基の25%までが、配列番号2と比較して、付加、欠失、挿入、置換、逆位もしくはそれらの組み合わせによって改変されている変異体も提供する。
【0042】
特に本発明は、補酵素NAD+またはNADHの存在下で3-クロロ-1-(チエニル-2-イル)-プロパン-1-オン(1)と(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オール(2)との間の立体特異的平衡反応を触媒する変異体を提供する。
【化3】
【0043】
本発明はさらに、本明細書に記載の変異体をコードする核酸配列を提供する。
【0044】
本発明はまた、少なくとも1つの調節核酸配列に機能的に連結された、本明細書に記載の少なくとも1つの核酸配列を含んでなる、発現カセットを提供する。
【0045】
本発明はさらに、本明細書に記載の少なくとも1つの発現カセットを含んでなるベクターを提供する。
【0046】
本発明はまた、少なくとも1つの、本明細書に記載の核酸配列、本明細書に記載の発現カセット、または本明細書に記載のベクターを含んでなる組換え微生物を与える。
【0047】
本発明はさらに、本明細書に記載のフェニルエタノールデヒドロゲナーゼ変異体を製造する方法を提供するが、その方法は、本明細書に記載の組換え微生物を培養すること、変異体をコードする核酸配列を発現させること、ならびに、必要に応じて、発現産物を単離することを含んでいる。
【0048】
さらに本発明は、式(II)の置換された光学活性アルコールを、微生物/酵素により合成する方法を与えるが、
【化4】
【0049】
式中、Cycは、単環式もしくは多環式、飽和もしくは不飽和、炭素環もしくはヘテロ環であって、適宜、一置換または多置換されていてもよく、
いずれの場合も、立体異性体として純粋な状態、または立体異性体の混合物であって、
その方法は、本明細書に記載のフェニルエタノールデヒドロゲナーゼ変異体の存在下で、適宜、特にNADHのような還元当量を添加して、生体触媒(微生物/酵素)により式(I)のケトンを還元することを含むものである。
【化5】
【0050】
特に、低級アルコール、たとえば具体的にはC1〜C6一価アルコールなどを犠牲アルコールとして使用して、還元当量再生条件下で反応を行う方法も与えられる。
【0051】
本発明の調製法を用いて、特に式(I)でCycがヘテロ環基、なかでもチエニル基である化合物を反応させる。
【0052】
特に、基本的に鏡像体として純粋な式(II)のアルコール、特に(S)-鏡像異性体を与える方法も提供される。
【0053】
特に、変異体が単離された状態で使用され、その結果、適宜、固体担体に固定化されている方法;または変異体が、適宜、固体担体に固定化された微生物細胞内で発現される方法も与えられる。適当な固体担体、たとえば、ビーズまたは膜などの高分子担体材料は、生体内変換および酵素リアクター技術の分野で当業者に知られている。
【0054】
また、特にデュロキセチンを調製する方法も与えられるが、その方法は
a)本明細書に記載の生体触媒による方法を用いた、3-クロロ-1-(チエニル-2-イル)-プロパン-1-オン(1)から(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オール(2)への微生物/酵素による還元;
【化6】
【0055】
b)メチルアミノ化によりデュロキセチンアルコール(3)を与える、アルコール(2)の化学的変換;
【化7】
【0056】
ならびに、最終的に
c)ナフチル基の挿入によるデュロキセチンアルコール(3)からデュロキセチン(4)へのの化学的変換
【化8】
【0057】
を含んでなる。
【0058】
さらに、本発明は、式(I)の置換ケトンを微生物/酵素により合成する方法を提供するが、
【化9】
【0059】
式中、Cycは、単環式もしくは多環式、飽和もしくは不飽和、炭素環もしくはヘテロ環、芳香族もしくは非芳香族の環であって、適宜、一置換または多置換されていてもよく、
その方法は、本明細書に記載のフェニルエタノールデヒドロゲナーゼ変異体の存在下で、適宜、特にNAD+のような、酸化当量を添加して、式(II)のアルコールを微生物/酵素により酸化することを含んでおり、
【化10】
【0060】
このアルコールは、いずれの場合も、立体異性体として純粋な状態、または立体異性体の混合物である。
【0061】
特に、この反応は、C1〜C6一価アルカノンを犠牲ケトンとして用いて、酸化当量再生条件下で行われる。
【0062】
使用する酵素変異体は、この場合、単離された形で使用され、たとえば適宜、固体担体に固定化されていてもよいが、固体担体に必要に応じて固定化された微生物細胞内で発現されていてもよい。
【0063】
最後に、本発明は、デュロキセチンアルコールおよび/またはデュロキセチンの調製において、本明細書に記載の酵素変異体を使用することに関する。
【0064】
3. 本発明のさらなる実施形態
3.1 タンパク質
本発明は、フェニルエタノールデヒドロゲナーゼ活性を有する、具体的に記載されたタンパク質および酵素に限定されることはなく、それと機能的に同等のものにまで及ぶ。
【0065】
具体的に記載された酵素の「機能的同等物」または類似物は、本発明との関連において、望ましい生物活性、たとえばフェニルエタノールデヒドロゲナーゼ活性を同様に有しているが記載された酵素とは異なるポリペプチドである。
【0066】
したがって、たとえば、「機能的同等物」は、本発明の「フェニルエタノールデヒドロゲナーゼ活性」用のテストにおいて、本明細書に記載のアミノ酸配列を有する酵素の活性を示す、酵素を意味すると考えられ、それは、少なくとも1%低い、または高い活性であるが、特に、少なくとも約5から10%、たとえば、少なくとも10%もしくは少なくとも20%、たとえば、少なくとも50%もしくは75%もしくは90%低い、または高い活性である。さらに、機能的同等物は好ましくは、pH 4〜11の間で安定であり、至適pHがpH 5〜10、たとえば特に、6.5〜9.5または7〜8または約7.5であって、かつ至適温度が15℃〜80℃または20℃〜70℃、たとえば、約30〜60℃または約35〜45℃、たとえば約40℃であれば有利である。
【0067】
本発明において、「フェニルエタノールデヒドロゲナーゼ活性」は、さまざまな既知のテストによって実証することができる。それらに限らず、実験の項に記載の標準条件下で、標準物質、たとえばTACもしくはTACAを使用するテスト(テスト1)、2)または3)の記述を参照されたい)、または4lリアクター内での生体内変換(イソプロパノールもしくは2-ブタノールを用いた補酵素再生を伴う、完全なTAC→TACA反応)を挙げることができる。
【0068】
本発明によれば、「機能的同等物」は、特に、前記アミノ酸配列の少なくとも1つの配列位置において、具体的に言及されたものとは異なるアミノ酸を有するにもかかわらず、前記生物活性の1つを有する、「変異体」を意味するとも考えられる。したがって、「機能的同等物」には、1つもしくは複数のアミノ酸の付加、置換、欠失および/または逆位によって得られる変異体が含まれるが、前記の改変は、それが本発明に一致する性質の特徴を有する変異体をもたらすならば、いかなる配列位置でも起こりうる。特に、変異体と未改変ポリペプチドとの間で反応性パターンが定性的に一致する、すなわち、たとえば同一基質が異なる速度で変換されるならば、機能的同等性はやはり存在する。適当なアミノ酸置換の例を下記の表にまとめる。
【0069】
元の基 置換例
Ala Ser
Arg Lys
Asn Gln; His
Asp Glu
Cys Ser
Gln Asn
Glu Asp
Gly Pro
His Asn ; Gln
Ile Leu; Val
Leu Ile; Val
Lys Arg ; Gln ; Glu
Met Leu ; Ile
Phe Met ; Leu ; Tyr
Ser Thr
Thr Ser
Trp Tyr
Tyr Trp ; Phe
Val Ile; Leu
記載されたポリペプチドの「前駆体」ならびにそうしたポリペプチドの「機能的誘導体」および「塩」も、上記の意味での「機能的同等物」である。
【0070】
ここで、「前駆体」は、望ましい生物活性の有無にかかわらず、ポリペプチドの天然もしくは合成の前駆体である。
【0071】
「塩」という表現は、本発明のタンパク質分子における、カルボキシル基の塩、およびアミノ基の酸付加塩の両方を意味するものと理解される。カルボキシル基の塩は、それ自体既知の方法で調製することができ、無機塩、たとえばナトリウム塩、カルシウム塩、アンモニウム塩、鉄塩および亜鉛塩、ならびに有機塩基、たとえば、トリエタノールアミン、アルギニン、リジン、ピペリジンなどのアミンとの塩もある。酸付加塩、たとえば、塩酸もしくは硫酸などの無機酸との塩、ならびに酢酸およびシュウ酸などの有機酸との塩も、同様に本発明により与えられる。
【0072】
本発明のポリペプチドの「機能的誘導体」は、同様に、既知の技術を用いて、機能的アミノ酸側鎖基またはそれらのNもしくはC末端で調製することができる。このような誘導体には、たとえば、カルボン酸基の脂肪族エステル、カルボン酸基のアミド(アンモニア、または一級もしくは二級アミンとの反応によって得られる);遊離アミノ基のN-アシル誘導体(アシル基との反応によって調製される);または遊離ヒドロキシ基のO-アシル誘導体(アシル基との反応によって調製される)が含まれる。
【0073】
「機能的同等物」には当然、他の生物から得られるポリペプチドおよび天然に存在するバリアントも含まれる。たとえば、配列比較によって、相同配列領域のエリアを決定し、本発明の具体的な規定に従って同等な酵素を決定することができる。
【0074】
「機能的同等物」には同様に、本発明のポリペプチドの断片、好ましくは個々のドメインもしくは配列モチーフも含まれており、それらはたとえば、望ましい生物学的機能を有するものである。
【0075】
さらに、「機能的同等物」は、前記ポリペプチド配列もしくはそれから誘導された機能的同等物の1つ、および少なくとも1つの他の機能的に異なる異種配列を、機能的にN末端もしくはC末端結合した融合タンパク質である(すなわち、融合タンパク質部分の相互の実質的な機能の減損はない)。このような異種配列の、限定的でない例としては、たとえば、シグナルペプチド、ヒスチジンアンカーまたは酵素がある。
【0076】
本発明によって含まれる「機能的同等物」は、具体的に記載されたタンパク質と相同である。これらは、具体的に記載されたアミノ酸配列の1つに対して、少なくとも60%、好ましくは少なくとも75%、特に少なくとも85%、たとえば、90、91、92、93、94、95、96、97、98または99%の相同性(言い換えれば、同一性)を有するが、それはPearson and Lipman, Proc. Natl. Acad, Sci. (USA) 85(8), 1988, 2444-2448によって、アルゴリズムに従って計算される。本発明の相同ペプチドの相同性すなわち同一性パーセントは、具体的には、本明細書に具体的に記載されるアミノ酸配列の1つの全長に基づく、アミノ酸基の同一性パーセントを意味する。
【0077】
同一性パーセントの値は、BLASTアラインメントのアルゴリズムblastp(タンパク質-タンパク質BLAST)に基づいて、または以下に与えられるClustalセッティングを用いて、確認することもできる。
【0078】
タンパク質に糖鎖付加(グリコシル化)の可能性がある場合、本発明の「機能的同等物」には、脱グリコシル化型またはグリコシル化型、ならびに糖鎖付加パターンの変更によって得られる修飾型の、上記のタイプのタンパク質が含まれる。
【0079】
本発明のタンパク質もしくはポリペプチドの相同体は、変異導入、たとえばタンパク質の点変異、延長もしくは短縮によって、作製することができる。
【0080】
本発明のタンパク質の相同体は、たとえばトランケート型変異体などの、変異体のコンビナトリアルライブラリーをスクリーニングすることによって同定することができる。たとえば合成オリゴヌクレオチド混合物の酵素的ライゲーションによって、核酸レベルでコンビナトリアル変異導入を行うことにより、多様なタンパク質バリアントのライブラリーを作製することができる。可能性のある相同体のライブラリーを縮重オリゴヌクレオチド配列から作製するために使用可能な、数多くの方法がある。縮重遺伝子配列の化学合成は、自動DNA合成装置で行い、その合成遺伝子を次に適当な発現ベクター内にライゲーションすることができる。遺伝子の縮重セットを使用することによって、可能性のあるタンパク質配列の望ましいセットをコードする全配列を1つの混合物として提供することができるようになる。縮重オリゴヌクレオチドの合成方法は当業者に知られている(たとえば、Narang, S.A. (1983) Tetrahedron 39:3; Itakura et al. (1984) Annu. Rev. Biochem. 53:323; Itakura et al., (1984) Science 198:1056; Ike et al. (1983) Nucleic Acids Res. 11:477)。
【0081】
点変異または短縮によって作成されたコンビナトリアルライブラリーの遺伝子産物をスクリーニングするための技法、ならびに特定の性質を有する遺伝子産物を目的としてcDNAライブラリーをスクリーニングするための技法は、先行技術でいくつか知られている。これらの技法は、本発明の相同体のコンビナトリアル変異導入によって作製された遺伝子ライブラリーの迅速スクリーニングに適合させることができる。大規模な遺伝子ライブラリーをスクリーニングするために最もよく使用される技法は、ハイスループット解析の根幹をなすものであって、その技法には、遺伝子ライブラリーを複製可能な発現ベクターにクローニングすること、その結果得られたベクターライブラリーを用いて適当な細胞を形質転換すること、ならびに、望ましい活性の検出により、遺伝子産物が検出された遺伝子をコードしているベクターを容易に単離できるような条件下で、コンビナトリアル遺伝子を発現させることが含まれる。リカーシブアンサンブル変異導入(recursive ensemble mutagenesis: REM)はライブラリー中の機能的変異体の頻度を高める技法であるが、この技法をスクリーニングテストと組み合わせて使用し、ホモログを同定することができる(Arkin and Yourvan (1992) PNAS 89:7811-7815; Delgrave et al. (1993) Protein Engineering 6(3):327-331)。
【0082】
3.2 核酸および構築物
3.2.1 核酸
本発明はまた、フェニルエタノールデヒドロゲナーゼ活性を有する酵素をコードする核酸配列を提供する。
【0083】
本発明はまた、本明細書に記載の具体的配列に対してある程度の同一性を示す核酸配列に関する。
【0084】
2つの核酸配列間の「同一性」は、個々の核酸の全長にわたるヌクレオチドの同一性、特に、Clustal法(Higgins DG, Sharp PM. Fast and sensitive multiple sequence alignments on a microcomputer. Comput Appl. Biosci. 1989 Apr; 5(2):151-1)を使用するInformax(USA)社製Vector NTI Suite 7.1ソフトウェアを用いて、下記のパラメーターを設定して比較することにより計算される同一性を意味すると理解される:
マルチプルアラインメントパラメーター:
ギャップ開始ペナルティ(Gap opening penalty) 10
ギャップ伸長ペナルティ(Gap extension penalty) 10
ギャップセパレーションペナルティ(Gap separation penalty range) 8
ギャップセパレーションペナルティ(Gap separation penalty) off
アラインメント遅延に関する%同一性(% identity for alignment delay) 40
残基特異的ギャップ(Residue specific gaps) off
親水性残基ギャップ(Hydrophilic residue gap) off
転移計量(Transition weighing) 0
ペアワイズアラインメントパラメーター:
FASTアルゴリズム on
K-タプルサイズ(K-tuple size) 1
ギャップペナルティ(Gap penalty) 3
ウィンドウサイズ(Window size) 5
Number of best diagonals 5
あるいはまた、同一性は、Chenna, Ramu, Sugawara, Hideaki, Koike,Tadashi, Lopez, Rodrigo, Gibson, Toby J, Higgins, Desmond G, Thompson, Julie D. Multiple sequence alignment with the Clustal series of programs. (2003) Nucleic Acids Res 31 (13):3497-500にしたがって、インターネットアドレスhttp://www.ebi.ac.uk/Tools/clustalw/index.html#によって、下記のパラメーターを用いて決定することもできる:
DNAギャップオープンペナルティ(DNA Gap Open Penalty) 15.0
DNAギャップエクステンションペナルティ(DNA Gap Extension Penalty) 6.66
DNAマトリックス(DNA Matrix) Identity
タンパク質ギャップオープンペナルティ(Protein Gap Open Penalty) 10.0
タンパク質ギャップエクステンションペナルティ(Protein Gap Extension Penalty)0.2
タンパク質マトリックス(Protein matrix) Gonnet
タンパク質/DNA ENDGAP(Protein/DNA ENDGAP) -1
タンパク質/DNA GAPDIST(Protein/DNA GAPDIST) 4
【0085】
本明細書に記載の核酸配列(一本鎖および二本鎖DNAおよびRNA配列、たとえばcDNAおよびmRNA)はすべて、それ自体公知の方法で、構成単位のヌクレオチドから化学合成によって、たとえば、二重らせんの構成単位である個々の重複する相補的核酸のフラグメント縮合によって、作製することができる。オリゴヌクレオチドの化学合成は、たとえば既知の方法で、ホスホアミダイト法(Voet, Voet, 2nd edition, Wiley Press New York, pages 896-897)にしたがって実施することができる。DNAポリメラーゼのクレノウ断片およびライゲーション反応により、合成オリゴヌクレオチドを付加しギャップを埋めること、ならびに一般的なクローニング法は、Sambrook et al. (1989), Molecular Cloning: A laboratory manual, Cold Spring Harbor Laboratory Pressに記載されている。
【0086】
本発明はまた、上記ポリペプチドおよびそれらの機能的同等物の1つをコードする核酸配列(一本鎖および二本鎖DNAおよびRNA配列、たとえばcDNAおよびmRNA)を提供するが、これらはたとえば人工ヌクレオチドアナログを用いて得られる。
【0087】
本発明は、本発明のポリペプチドもしくはタンパク質または生物学的活性を有するそのセグメントをコードする単離された核酸分子、ならびに、たとえば本発明のコード核酸の同定もしくは増幅のためにハイブリダイゼーションプローブもしくはプライマーとして使用することができる、核酸断片も提供する。
【0088】
さらに、本発明の核酸分子は、遺伝子のコード領域の3'-および/または5'-末端の非翻訳配列を含んでいてもよい。
【0089】
本発明にはさらに、具体的に記載されたヌクレオチド配列、またはそのセグメントに相補的な核酸分子が含まれる。
【0090】
本発明のヌクレオチド配列は、他の細胞型および生物において相同配列を同定および/またはクローニングするために使用することができる、プローブおよびプライマーの作製を可能にする。こうしたプローブもしくはプライマーは、通常、「ストリンジェントな」条件(下記参照)下で、本発明の核酸配列のセンス鎖の、または対応するアンチセンス鎖の、少なくとも約12、好ましくは少なくとも約25、たとえば約40、50または75個の連続したヌクレオチドとハイブリダイズするヌクレオチド配列領域を含有する。
【0091】
「単離された」核酸分子は、その核酸の天然起源の中に存在する他の核酸分子から分離されているが、その上、それが組換え技術によって作製されたならば、他の細胞物質もしくは培養培地を基本的に含んでおらず、化学合成されたならば化学的前駆体を含んでいないといえる。
【0092】
本発明の核酸は、分子生物学の標準的な技法、ならびに本発明により与えられる配列情報を用いて、単離することができる。たとえば、cDNAは、具体的に記載された完全な配列の1つもしくはそのセグメントをハイブリダイゼーションプローブとして使用し、標準的なハイブリダイゼーション法(たとえば、Sambrook, J., Fritsch, E.F. and Maniatis, T. Molecular Cloning: A Laboratory Manual. 2nd edition, Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989に記載)を用いることによって、適当なcDNAライブラリーから単離できる。さらに、開示されている配列の1つもしくはそのセグメントを含有する核酸分子を、この配列に基づいて作製されたオリゴヌクレオチドプライマーを用いてポリメラーゼ連鎖反応によって単離することができる。このようにして増幅された核酸を適当なベクターにクローニングして、DNA配列分析により特徴付けることができる。本発明のオリゴヌクレオチドは、たとえば自動DNA合成装置を用いて、標準的な合成法によっても調製することができる。
【0093】
本発明の核酸配列、またはその誘導体、前記配列のホモログもしくは一部は、たとえば、通常のハイブリダイゼーション法またはPCR技術を用いて、他の細菌から、たとえばゲノムライブラリーもしくはcDNAライブラリーを経て、単離することができる。これらのDNA配列は、標準条件下で本発明の配列とハイブリダイズする。
【0094】
「ハイブリダイゼーション」は、ポリヌクレオチドもしくはオリゴヌクレオチドが標準的な条件下で実質的に相補的な配列と結合する能力を意味すると理解されるが、この条件下では相補的でないパートナーとの間の非特異的結合は起こらない。このため、その配列は90-100%相補的であるといえる。互いに特異的に結合することができる相補配列の特性によって、相補配列は、たとえばノーザンブロットもしくはサザンブロット法において、またはPCRもしくはRT-PCRにおけるプライマー結合のために、有用となる。
【0095】
ハイブリダイゼーションのために、保存された領域の、短いオリゴヌクレオチドを使用することが有利である。しかしながら、本発明の核酸の長めの断片、または完全な配列をハイブリダイゼーションに使用することもできる。上記の標準的な条件は、使用する核酸(オリゴヌクレオチド、より長い断片、または完全な配列)に応じて、またはDNAかRNAか、どちらのタイプの核酸をハイブリダイゼーションに使用するのか、に応じて、さまざまである。たとえば、DNA:DNAハイブリッドの融解温度は、同じ長さのDNA:RNAハイブリッドの融解温度より約10℃低い。
【0096】
標準的な条件は、たとえば核酸に応じて、42℃から58℃までの、0.1から5 x SSC(1 X SSC = 0.15 M NaCl、15 mMクエン酸ナトリウム、pH 7.2)の濃度で、またはさらに50%ホルムアミド存在下の、緩衝水溶液の温度を意味すると理解されるべきであって、たとえば、5 x SSC、50%ホルムアミドで42℃である。DNA:DNAハイブリッドのハイブリダイゼーション条件は0.1 x SSCで約20℃から45℃までの温度であれば有利であるが、好ましくは約30℃から45℃までである。DNA:RNAハイブリッドについては、ハイブリダイゼーション条件は0.1 x SSCで約30℃から55℃までの温度であれば有利であるが、好ましくは約45℃から55℃までである。上記のハイブリダイゼーション温度は、ホルムアミドの非存在下で、長さが約100ヌクレオチドでG + C含量が50%である核酸に関する、融解温度の計算値の例である。DNAハイブリダイゼーションの実験条件は、遺伝学に関するテキスト、たとえば、Sambrook et al., “Molecular Cloning”, Cold Spring Harbor Laboratory, 1989に記載されており、たとえば、核酸の長さ、ハイブリッドの種類またはG + C含量の関数のような、当業者に知られている式を用いて、計算することができる。当業者は、ハイブリダイゼーションに関するさらに他の情報を、下記のテキストに見いだすことができる:Ausubel et al. (eds), 1985, Current Protocols in Molecular Biology, John Wiley & Sons, New York; Hames and Higgins (eds), 1985, Nucleic Acids Hybridization: A Practical Approach, IRL Press at Oxford University Press, Oxford; Brown (ed), 1991, Essential Molecular Biology: A Practical Approach, IRL Press at Oxford University Press, Oxford。
【0097】
「ハイブリダイゼーション」は特に、ストリンジェントな条件下で行うことができる。そのような条件は、たとえば、Sambrook, J., Fritsch, E.F., Maniatis, T., in: Molecular Cloning (A Laboratory Manual), 2nd edition, Cold Spring Harbor Laboratory Press, 1989, pages 9.31-9.57 or in Current Protocols in Molecular Biology, John Wiley & Sons, N.Y. (1989), 6.3.1-6.3.6に記載されている。
【0098】
「ストリンジェントな」ハイブリダイゼーション条件は、具体的には下記を意味すると理解される:50%ホルムアミド、5 x SSC(750 mM NaCl、75 mMクエン酸三ナトリウム)、50 mMリン酸ナトリウム(pH 7.6)、5x Denhardt溶液、10%デキストラン硫酸、および20 g/mlの変性、断片化処理したサケ精子DNAからなる溶液中で一晩42℃にてインキュベーションした後、65℃にて0.1 x SSC によるフィルターの洗浄ステップが続く。
【0099】
本発明はまた、具体的に記載された、もしくは推論できる核酸配列の、誘導体を提供する。
【0100】
上述のように、本発明に合致する他の核酸配列を、たとえば、配列番号1または3から誘導することが可能であり、そうした核酸配列は1つもしくは複数のヌクレオチドの付加、置換、挿入または欠失により前記とは異なるが、依然として望ましい特性を有するポリペプチドをコードする可能性がある。
【0101】
天然に存在するバリアント、たとえばスプライシングバリアントもしくはアレルバリアントなどのように、具体的に記載された配列と比べて、いわゆるサイレント変異を有する核酸配列、または特別な起源もしくは宿主生物のコドン使用頻度に応じて変更された核酸配列も、本発明にしたがって含めるものとする。
【0102】
同様に、保存的なヌクレオチド置換により得られる配列も与えられる(すなわち、当該アミノ酸は、同じ荷電、サイズ、極性、および/または溶解性を持つアミノ酸で置換される)。
【0103】
本発明はまた、配列多型により具体的に記載された核酸に由来する分子を提供する。これらの遺伝子多型は、自然変異により集団内の個体間に存在する可能性がある。こうした自然変異は通常、遺伝子のヌクレオチド配列において1から5%までの変異をもたらす。
【0104】
配列番号1または3の配列を有する本発明の核酸配列の誘導体は、たとえば、それに由来するアミノ酸レベルで、その全配列領域にわたって、少なくとも60%相同性を有する、好ましくは少なくとも80%相同性、特に好ましくは少なくとも90%相同性を有するアレルバリアントを意味すると理解されるべきである(アミノ酸レベルの相同性に関しては、ポリペプチドに関する上記の記述を参照すべきである)。その配列の一部の領域について、有利に、相同性は、より高くてもよい。
【0105】
さらに、誘導体は、当然のことながら、本発明の核酸のホモログ、具体的には、配列番号1または3のホモログ、たとえば、コードおよび非コードDNA配列の、真菌もしくは細菌ホモログ、短縮された配列、一本鎖DNAもしくはRNAも指している。
【0106】
さらに、誘導体は当然のことながら、たとえばプロモーターとの融合物を意味する。プロモーターは、上記ヌクレオチド配列の上流に連結されているが、そのプロモーターの機能および/または有効性を損なうことなしに、少なくとも1つのヌクレオチドの置換、少なくとも1つの挿入、逆位および/または欠失によりこれを改変することができる。その上、プロモーターの配列を改変することによってその有効性を高めることができるが、別種の生物に由来する、より有効なプロモーターによって完全に置き換えることもできる。
【0107】
3.2.2 機能的変異体の作製
さらに、本発明の酵素の機能的変異体を作製する方法が、当業者に知られている。
【0108】
使用する技術に応じて、当業者は、完全にランダムな変異、あるいはより標的化された変異を、遺伝子あるいは非コード核酸領域(これは、たとえば発現の調節のために重要である)内に挿入して、遺伝子ライブラリーを作製することができる。このために必要な分子生物学的方法は、当業者に知られており、たとえば、Sambrook and Russell, Molecular Cloning. 3rd edition, Cold Spring Harbor Laboratory Press 2001に記載されている。
【0109】
遺伝子を改変し、それによってその遺伝子によりコードされるタンパク質を改変する方法は、これまで長く当業者に周知されてきており、たとえば、以下のものがある:
- 部位特異的変異誘発、この方法では、遺伝子の1つもしくは複数のヌクレオチドを標的化された方法で置き換える(Trower MK (ed.) 1996; In vitro mutagenesis protocols. Humana Press, New Jersey);
- 飽和変異誘発、この方法では、遺伝子の任意の望ましい位置で、任意の望ましいアミノ酸に対するコドンを置換または付加することができる(Kegler-Ebo DM, Docktor CM, DiMaio D (1994) Nucleic Acids Res 22:1593; Barettino D, Feigenbutz M, Valcarel R, Stunnenberg HG (1994) Nucleic Acids Res 22:541; Barik S (1995) Mol Biotechnol 3:1);
- エラープローンポリメラーゼ連鎖反応(エラープローンPCR)、この方法では、不完全なDNAポリメラーゼによりヌクレオチド配列を変異させる(Eckert KA, Kunkel TA (1990) Nucleic Acids Res 18:3739);
- SeSaM法(配列飽和法)、この方法では、好ましい置換がポリメラーゼによって妨げられる(Schenk et al., Biospektrum, Vol. 3, 2006, 277-279);
- ミューテーター株への遺伝子の挿入、この場合、たとえば不完全なDNA修復メカニズムのために、ヌクレオチド配列の変異率の増加が生じる(Greener A, Callahan M, Jerpseth B (1996) An efficient random mutagenesis technique using an E.coli mutator strain. In: Trower MK (ed.) In vitro mutagenesis protocols. Humana Press, New Jersey);または
- DNAシャフリング、この方法では、近縁遺伝子のプールを作製して消化し、その断片をポリメラーゼ連鎖反応のテンプレートとして使用するが、鎖分離と再アニーリングを繰り返すことによって、最終的に全長のモザイク遺伝子が生成する(Stemmer WPC (1994) Nature 370:389; Stemmer WPC (1994) Proc Natl Acad Sci USA 91:10747)。
【0110】
いわゆる人工分子進化法(特に、Reetz MT and Jaeger K-E (1999), Topics Curr Chem 200:31; Zhao H, Moore JC, Volkov AA, Arnold FH (1999), Methods for optimizing industrial enzymes by directed evolution, in: Demain AL, Davies JE (ed.) Manual of industrial microbiology and biotechnology. American Society for Microbiologyに記載)を用いて、当業者は、標的化されたやり方で、しかも工業規模で機能的変異体を作製することができる。ここで、最初のステップにおいて、はじめに特定のタンパク質の遺伝子ライブラリーを作製するが、そのために上記の方法を使用することができる。その遺伝子ライブラリーを適当な方法で、たとえば、細菌によって、またはファージディスプレイシステムによって発現させる。
【0111】
望ましい特性におおむね合致する性質を有する機能的変異体を発現する、宿主生物の当該遺伝子に、さらに一連の変異を受けさせることができる。今ある機能的変異体が十分に望ましい性質を示すまで、変異および選択またはスクリーニングのステップを反復して繰り返すことができる。この反復法の結果として、限られた数の変異、たとえば1から5回の変異に段階的に取り組むことができ、問題の酵素活性への影響を評価して選択することができる。選択された変異体を次に、同様の方法で次の変異ステップに供することができる。結果として、検討すべき個々の変異体の数を、かなり少なくすることができる。
【0112】
配列番号2の酵素のエラープローン変異導入により得ることができた本発明の変異体の限定的でない例を、以下の表1にまとめる。
【表1】
【0113】
【0114】
【0115】
本発明の結果は、望ましい、改良された特性を有する酵素を標的化された方法でさらに作製するために必要とされる、当該酵素の構造および配列に関する重要な情報も与える。特に、いわゆる「ホットスポット」、すなわち標的変異を挿入することによって酵素の性質を改変するのに適している可能性のある配列セグメントを決定することができる。
【0116】
本発明の酵素の、このようなホットスポット領域の限定的でない例を、配列2に基づいて、以下にまとめる:
(1) 142〜153(ループ2)および
(2) 190〜211(ヘリックスαFG1)
(3) 93〜96(ループ1)
(4) 241〜249(C末端)
(5) 138〜141(結合ポケットの親水性領域)ならびに
(6) Cys61および/またはCys 83。
【0117】
同様に、おそらく酵素活性にほとんど影響を及ぼさないはずであって、潜在的な「サイレント変異」と呼ぶことができる、変異を実行することができる領域内の、アミノ酸配列位置に関する情報を引き出すことができる。こうした変異の位置は、配列番号2に関して下記の表2にまとめる。
【表2】
【0118】
3.2.3 構築物
さらに、本発明は、本発明のポリペプチドをコードする核酸配列を調節核酸配列の遺伝的制御下に含有する発現構築物;ならびにこれらの発現構築物の少なくとも1つを含有するベクターを提供する。
【0119】
本発明によれば、「発現ユニット」は、当然のことながら発現活性のある核酸を意味し、こうした核酸には本明細書で定義されたプロモーターが含まれるが、こうした核酸は、発現されるべき核酸もしくは遺伝子に機能的に連結された後、この核酸またはこの遺伝子の発現を調節し、したがって転写および翻訳を調節するものである。したがって、これに関連して、「調節核酸配列」という表現も用いられる。プロモーターに加えて、たとえばエンハンサーのような調節エレメントをさらに含んでいてもよい。
【0120】
本発明によれば、「発現カセット」または「発現構築物」は、当然のことながら、発現されるべき核酸または発現されるべき遺伝子に機能的に連結されている発現ユニットを意味する。したがって発現カセットは、発現ユニットとは違って、転写および翻訳を調節する核酸配列だけでなく、転写および翻訳の結果タンパク質として発現されるべき核酸配列も含んでいる。
【0121】
本発明の文脈において、「発現」または「過剰発現」という用語は、対応するDNAによってコードされる1つもしくは複数の酵素の、微生物における産生、またはその酵素の、微生物における細胞内活性の増加を言い表すものである。これを目的として、たとえば、遺伝子を生物に挿入すること、存在する遺伝子を別の遺伝子に置き換えること、1つもしくは複数の遺伝子のコピー数を増やすこと、強力なプロモーターを使用すること、または高活性を有する該当酵素をコードする遺伝子を使用することが可能であって、さらに、これらの手段を組み合わせることも選択できる。
【0122】
好ましくは、本発明のこうした構築物は、特定のコード配列の5’上流にプロモーターを、3’下流に転写終結配列を有し、必要に応じてさらに、通常の調節エレメントも含んでいるが、これらはいずれの場合も、機能しうるようにコード配列に連結されている。
【0123】
本発明によれば、「プロモーター」、「プロモーター活性を有する核酸」または「プロモーター配列」は、転写されるべき核酸に機能的に連結された状態で、この核酸の転写を調節する核酸を意味していると理解される。
【0124】
これに関連して、「機能的な」または「機能しうる」連結とは、たとえば、プロモーター活性を有する核酸の1つ、および転写されるべき核酸配列、ならびに必要に応じてさらに調節エレメント、たとえば核酸の転写を確実にする核酸配列、およびたとえばターミネーターなどが、核酸配列の転写時にそれぞれの調節エレメントがその機能を果たすことができるように、順に配列されていることを意味していると理解される。このために、化学的な意味での直接的な連結が必ずしも必要というわけではない。遺伝子制御配列、たとえばエンハンサー配列は、遠く離れた場所からも、あるいは他のDNA分子からでも、標的配列に対して機能を発揮することができる。転写されるべき核酸配列がプロモーター配列の後ろに(すなわち3’側に)ある配列配置が好ましく、2つの配列は共有結合で連結される。ここで、プロモーターと、遺伝子組み換えにより発現されるべき核酸配列との間隔は、200塩基対未満、または100塩基対未満、または50塩基対未満とすることができる。
【0125】
プロモーターおよびターミネーターに加えて、他の調節配列の例には、ターゲティング配列、エンハンサー、ポリアデニル化シグナル、選択可能なマーカー、増幅シグナル、複製開始点などがある。適当な調節配列は、たとえば、Goeddel, Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, CA (1990)に記載されている。
【0126】
本発明の核酸構築物には、特に、配列番号1または3の配列、またはその誘導体およびホモログ、ならびにそれらから得られる核酸配列であって、遺伝子発現を制御し、たとえば増加させるために有利となるように、1つもしくは複数の調節シグナルに、機能しうるように、もしくは機能的に連結されている前記核酸配列が含まれる。
【0127】
これらの調節配列のほかに、上記配列の天然の調節が、実際の構造遺伝子の上流に依然として残っている可能性があり、必要に応じて、天然の調節をスイッチオフして遺伝子の発現が増加するように、遺伝的に改変してもよかった。しかしながら、核酸構築物は、構造的によりシンプルにしてもよく、すなわち、追加の調節シグナルは、コード配列の上流に挿入されず、天然プロモーターは、その調節とともに、除去されなかった。その代わり、天然の調節配列は、もはや調節がなくなって遺伝子の発現が増加するように変異している。
【0128】
好ましい核酸構築物には、有利なことに、1つもしくは複数の前記「エンハンサー」配列がプロモーターに機能的に連結されて含まれており、この配列は核酸配列の発現増加を可能にする。追加の有利な配列、たとえば他の調節エレメントまたはターミネーターも、DNA配列の3’末端に挿入することができる。本発明の核酸は、構築物の1つもしくは複数のコピー中に存在することが考えられる。この構築物は、抗生物質耐性または栄養要求性を補完する遺伝子といったマーカーを、構築物の選択のために必要に応じて含んでいてもよい。
【0129】
適当な調節配列の例は、cos、tac、trp、tet、trp-tet、lpp、lac、lpp-lac、lacIq-、T7、T5、T3、gal、trc、ara、rhaP (rhaPBAD)SP6、lambda-PRなどのプロモーター、またはLambda-PLプロモーター中にあるが、これらはグラム陰性細菌において有利に使用される。さらに有利な調節配列が、たとえば、グラム陽性プロモーターamyおよびSPO2、酵母もしくは真菌プロモーターADC1、MFα、AC、P-60、CYC1、GAPDH、TEF、rp28、ADHの中にある。調節のために人工プロモーターを使用することも可能である。
【0130】
宿主生物での発現のために、宿主において遺伝子の最適な発現を可能にするベクター、たとえばプラスミドもしくはファージに核酸構築物を挿入するのは有利である。プラスミドおよびファージと同様にベクターは、当然のことながら、当業者に知られている任意の他のベクター、たとえば、SV40、CMV、バキュロウイルスおよびアデノウイルスなどのウイルス、トランスポゾン、ISエレメント、ファスミド(phasmid)、コスミド、および直鎖もしくは環状DNAを意味している。これらのベクターは、宿主生物において自律的に複製可能であり、または染色体性に複製することができる。これらのベクターは本発明の他の実施形態を構成する。
【0131】
適当なプラスミドはたとえば、大腸菌(E. coli)では、pLG338、pACYC184、pBR322、pUC18、pUC19、pKC30、pRep4、pHS1、pKK223-3、pDHE19.2、pHS2、pPLc236、pMBL24、pLG200、pUR290、pIN-III113-B1、λgt11もしくはpBdCI、ストレプトミセスでは、pIJ101、pIJ364、pIJ702もしくはpIJ361、バチルスでは、pUB110、pC194もしくはpBD214、コリネバクテリウムでは、pSA77もしくはpAJ667、真菌では、pALS1、pIL2もしくはpBB116、酵母では、2alphaM、pAG-1、YEp6、YEp13もしくはpEMBLYe23、または植物では、pLGV23、pGHlac+、pBIN19、pAK2004もしくはpDH51である。特定されたプラスミドは、可能性のあるプラスミドのうち小数選抜されたものである。さらに他のプラスミドが当業者によく知られており、たとえば、書籍Cloning Vectors (Eds. Pouwels P. H. et al. Elsevier, Amsterdam-New York-Oxford, 1985, ISBN 0 444 904018) に見いだすことができる。
【0132】
本発明の他の実施形態において、本発明の核酸構築物、または本発明の核酸を含有するベクターは、直鎖DNAの形で有利に微生物に導入することも、非相同もしくは相同組換えにより宿主生物のゲノムに組み込むことも有利に行うことができる。この直鎖DNAは、プラスミドのように直鎖状にしたベクターからなることもあるが、本発明の核酸構築物または核酸だけからなることもある。
【0133】
生物における異種遺伝子の最適な発現のために、その生物で使用される特異的な「コドン使用頻度」にしたがって核酸配列を改変することが有利である。「コドン使用頻度」は、当該生物に由来する他の既知遺伝子のコンピューター解析によって、容易に決定することができる。
【0134】
本発明の発現カセットは、適当なプロモーターを、適当なコード配列、および転写終結シグナルもしくはポリアデニル化シグナルに融合させることによって調製される。このために、一般的な組換えおよびクローニング技術が使用されるが、それはたとえば、T. Maniatis, E.F. Fritsch and J. Sambrook, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY (1989)、およびT.J. Silhavy, M.L. Berman and L.W. Enquist, Experiments with Gene Fusions, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY (1984)に、さらにAusubel, F.M. et al., Current Protocols in Molecular Biology, Greene Publishing Assoc. and Wiley Interscience (1987)に記載されている。
【0135】
適当な宿主生物における発現のために、組換え核酸構築物もしくは遺伝子構築物は、宿主特異的ベクターに挿入されることが有利であり、このベクターは宿主において遺伝子の最適な発現を可能にするものである。ベクターは当業者に周知であり、たとえば、“Cloning Vectors” (Pouwels P. H. et al., Ed., Elsevier, Amsterdam-New York-Oxford, 1985) に見いだすことができる。
【0136】
3.3 微生物
文脈に応じて、「微生物」という用語は、野生型の微生物もしくは遺伝的に改変された組換え微生物、またはその両者を意味すると理解することができる。
【0137】
本発明のベクターを用いることによって、たとえば少なくとも1つの本発明のベクターで形質転換された、本発明のポリペプチドを産生するために使用することができる組換え微生物を調製することができる。上記の本発明の組換え構築物を適当な宿主系に導入し発現させるのは好都合である。ここで、当業者に知られている一般的なクローニングおよびトランスフェクション法、たとえば、共沈殿、プロトプラスト融合、エレクトロポレーション、レトロウイルストランスフェクションなどを、特定の発現系で前記核酸を発現させるために使用することが好ましい。適当な系は、たとえば、Current Protocols in Molecular Biology, F. Ausubel et al., Ed., Wiley Interscience, New York 1997, or Sambrook et al. Molecular Cloning: A Laboratory Manual. 2nd edition., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989に記載されている。
【0138】
本発明の核酸もしくは核酸構築物に適した組換え宿主生物は、基本的にすべての原核または真核生物である。使用する宿主生物が細菌、真菌もしくは酵母などの微生物であれば好都合である。グラム陽性もしくはグラム陰性細菌、好ましくは、腸内細菌科(Enterobacteriaceae)、シュードモナス科(Pseudomonadaceae)、リゾビウム科(Rhizobiaceae)、ストレプトミセス科(Streptomycetaceae)またはノカルジア科(Nocardiaceae)に属する細菌、特に好ましくは、エシェリキア属(Escherichia)、シュードモナス属(Pseudomonas)、ストレプトミセス属(Streptomyces)、ノカルジア属(Nocardia)、バークホルデリア属(Burkholderia)、サルモネラ属(Salmonella)、アグロバクテリウム属(Agrobacterium)、クロストリジウム属(Clostridium)またはロドコッカス属(Rhodococcus)の細菌を使用すれば有利である。中でも特に好ましいのは、大腸菌Escherichia属およびEscherichia coli種である。その上、さらに有利な細菌を、αプロテオバクテリア、βプロテオバクテリアもしくはγプロテオバクテリア群に見いだすことができる。
【0139】
これに関連して、本発明の1つもしくは複数の宿主生物は、好ましくは、本発明に記載の上記定義にしたがってフェニルエタノールデヒドロゲナーゼ活性を有する酵素をコードする、少なくとも1つの核酸配列、核酸構築物またはベクターを含有する。
【0140】
本発明の方法で使用される生物は、宿主生物に応じて、当業者に知られている方法で増殖または培養する。一般に、微生物は液体培地中で、0℃から100℃まで、好ましくは10℃から60℃までの間の温度で、酸素ガスを供給して増殖させるが、この培地は炭素源を主として糖の形で、窒素源を、主に酵母エキスなどの有機窒素源または硫酸アンモニウムなどの塩の形で含有し、鉄、マンガンおよびマグネシウム塩などの微量元素、および必要に応じてビタミン類を含有する。ここで、栄養液のpHは、培養の間、一定値に維持すること、すなわち調節することができるが、調節されなくてもよい。培養は、バッチ式、半バッチ式、または連続式とすることができる。栄養物は、発酵培養開始時に最初に導入することができるが、半連続的または連続的に供給することもできる。
【0141】
3.4 本発明の酵素の組換え調製
本発明はさらに、本発明のポリペプチド、もしくは機能的、生物学的に活性のあるその断片を組換え調製する方法を提供するが、その方法はポリペプチド産生微生物を培養すること、必要に応じてポリペプチドの発現を誘導すること、ならびに培養物からポリペプチドを単離することを含んでいる。ポリペプチドは、必要ならば、このようにして工業規模で製造することもできる。
【0142】
本発明にしたがって調製された微生物は、連続培養することができるが、バッチ法で(バッチ培養)、または供給バッチ法(フィード法)もしくは反復供給バッチ法(反復フィード法)で不連続に培養することもできる。既知の培養法に関する概要は、Chmiel (Bioprozeβtechnik 1. Einfuehrung in die Bioverfahrenstechnik [Bioprocessing technology 1. Introduction to bioprocessing technology] (Gustav Fischer Verlag, Stuttgart, 1991))のテキスト、またはStorhas (Bioreaktoren und periphere Einrichtungen [Bioreactors and peripheral devices] (Vieweg Verlag, Braunschweig/Wiesbaden, 1994))のテキストに見いだすことができる。
【0143】
使用される培地は、個別の菌株の要求を適切に満足させなければならない。種々の微生物の培地に関する記述は、"Manual of Methods for General Bacteriology" from the American Society for Bacteriology (Washington D. C., USA, 1981) のハンドブックに見いだすことができる。
【0144】
本発明にしたがって使用することができるこれらの培地は、通常、1つもしくは複数の炭素源、窒素源、無機塩、ビタミン類および/または微量元素を含有する。
【0145】
好ましい炭素源は、糖類、たとえば、単糖、二糖もしくは多糖類である。非常に優れた炭素源は、たとえば、グルコース、フルクトース、マンノース、ガラクトース、リボース、ソルボース、リブロース、ラクトース、マルトース、スクロース、ラフィノース、デンプンまたはセルロースである。モラセスもしくは他の精糖副産物などの複雑な化合物を用いて、培地に糖類を加えることも可能である。異なる炭素源の混合物を添加することも有利であるかもしれない。他に考えられる炭素源は、油脂類、たとえば、大豆油、ヒマワリ油、ピーナッツ油およびココナツ油;脂肪酸、たとえば、パルミチン酸、ステアリン酸もしくはリノール酸;アルコール類、たとえばグリセロール、メタノールもしくはエタノール;ならびに有機酸、たとえば、酢酸もしくは乳酸である。
【0146】
窒素源は通常、有機もしくは無機窒素化合物、またはそうした化合物を含む材料である。窒素源の例には、アンモニアガス、またはアンモニウム塩、たとえば硫酸アンモニウム、塩化アンモニウム、リン酸アンモニウム、炭酸アンモニウムもしくは硝酸アンモニウム、硝酸塩、尿素、アミノ酸、または複雑な窒素源、たとえば、コーンスティープリカー、大豆粉、大豆タンパク質、酵母エキス、肉エキスなどがある。窒素源は、単独で、または混合物として使用することができる。
【0147】
培地中に含まれる可能性のある無機塩化合物としては、カルシウム、マグネシウム、ナトリウム、コバルト、モリブデン、カリウム、マンガン、亜鉛、銅および鉄の、塩化物塩、リン塩または硫酸塩がある。
【0148】
使用できるイオウ源は、無機イオウ含有化合物、たとえば、硫酸塩、亜硫酸塩、亜ジチオン酸塩、テトラチオン酸塩、チオ硫酸塩、硫化物であるが、有機イオウ化合物、たとえば、メルカプタン類およびチオール類もある。
【0149】
使用できるリン源は、リン酸、リン酸二水素カリウムもしくはリン酸水素二カリウム、または対応するナトリウム含有塩である。
【0150】
金属イオンを溶液中に維持するために、培地にキレート剤を添加することができる。特に適当なキレート剤には、ジヒドロキシフェノール類、たとえば、カテコールもしくはプロトカテク酸、またはクエン酸などの有機酸がある。
【0151】
本発明にしたがって使用される発酵培地は通常、他の成長因子、たとえば、ビタミン類もしくは成長促進物質を含有するが、これにはたとえば、ビオチン、リボフラビン、チアミン、葉酸、ニコチン酸、パントテン酸塩、およびピリドキシンがある。成長因子および塩類は複雑な培地成分、たとえば酵母エキス、モラセス、コーンスティープリカーなどに由来することも多い。さらに、適当な前駆体を培地に加えてもよい。培地化合物の正確な組成は、個別の実験に大きく左右され、具体的な場合ごとに個々に決定される。培地の最適化に関する情報は、"Applied Microbiol. Physiology, A Practical Approach" (ed. P.M. Rhodes, P.F. Stanbury, IRL Press (1997) p. 53-73, ISBN 0 19 963577 3) のテキストから入手することができる。増殖培地は、Standard 1 (Merck)またはBHI (Brain heart infusion, DIFCO)などのような供給業者から入手することもできる。
【0152】
すべての培地成分は、加熱(1.5バールで121℃にて20分)または無菌濾過によって、滅菌される。成分は合わせて滅菌してもよいが、必要ならば別々に滅菌することもできる。すべての培地成分が培養の開始時点で存在していてもよいが、適宜、連続的に、またはバッチ式に添加してもよい。
【0153】
培養温度は通常、15℃から45℃まで、好ましくは25℃から40℃までの間であって、実験の間、一定に保つことも、変化させることもできる。培地のpHは、5から8.5までの範囲、好ましくは7.0程度とすべきである。培養のためのpHは、培養中、塩基性化合物、たとえば、水酸化ナトリウム、水酸化カリウム、アンモニアもしくはアンモニア水、または酸性化合物、たとえば、リン酸もしくは硫酸の添加により制御することができる。発泡を抑えるために、消泡剤、たとえば、脂肪酸ポリグリコールエステルを使用することができる。プラスミドの安定性を維持するために、適当な選択物質、たとえば、抗生物質を培地に添加することができる。好気的条件を維持するために、酸素もしくは酸素含有気体混合物、たとえば外気が培養に導入される。培養温度は通常20℃から45℃までである。培養は、望ましい産物が最大限生成するまで続けられる。この目的は通常、10時間から160時間までの間に達成される。
【0154】
その後、発酵液はさらに処理される。必要な条件に応じて、たとえば、遠心分離、濾過、デカンテーション、もしくはこれらの方法を組み合わせた分離法によって、発酵液から生物体量を全部または一部除去することができるが、生物体量が前記液体中にすべて残されていてもよい。
【0155】
ポリペプチドが培地中に分泌されないならば、細胞を破壊し、その溶解物から、既知のタンパク質単離法により産物を得ることができる。細胞は適宜、超音波によって、高圧によって(たとえば、フレンチプレス圧力セル内で)、浸透圧溶解によって、界面活性剤、溶解酵素もしくは有機溶媒の作用によって、ホモジナイザーによって、または上記の方法のいくつかを組み合わせて、破壊することができる。
【0156】
ポリペプチドの精製は、既知のクロマトグラフィー法、たとえば、Q-Sepharoseクロマトグラフィーなどの分子篩クロマトグラフィー(ゲル濾過)、イオン交換クロマトグラフィーおよび疎水性クロマトグラフィーを用いて、ならびに他の常法、たとえば限外濾過、結晶化、塩析、透析およびネイティブゲル電気泳動によっても、達成することができる。適当な方法はたとえば、Cooper, T.G., Biochemische Arbeitsmethoden [Biochemical procedures], Verlag Walter de Gruyter, Berlin, New York or in Scopes, R., Protein Purification, Springer Verlag, New York, Heidelberg, Berlinに記載されている。
【0157】
組換えタンパク質を単離するために、そのcDNAをあるヌクレオチド配列だけ長く伸ばし、そうすることで、たとえばより簡単な精製に役立つ改変ポリペプチドもしくは融合タンパク質をコードする、ベクター系もしくはオリゴヌクレオチドを使用することは有利となるかもしれない。この種の適当な改変は、たとえば、アンカーとして機能するいわゆる「タグ」、たとえばヘキサヒスチジンアンカーとして知られる改変、または抗体によって抗原として認識されうるエピトープである(たとえば、Harlow, E. and Lane, D., 1988, Antibodies: A Laboratory Manual. Cold Spring Harbor (N.Y.) Pressに記載される)。こうしたアンカーは、タンパク質を、固体担体、たとえばポリマーマトリックスに固定するのに役立つ可能性があり、この担体はたとえば、クロマトグラフィーカラムの充填剤として、またはマイクロタイタープレートもしくは他の担体上で使用することができる。
【0158】
同時に、このようなアンカーは、タンパク質の認識のために使用することもできる。タンパク質を認識するために、通常のマーカー、たとえば、蛍光色素、基質との反応後に検出可能な生成物を形成する酵素マーカー、もしくは放射性マーカーを単独で使用することもできるが、タンパク質を誘導体化するためのアンカーと組み合わせて使用することもできる。
【0159】
本発明の変異体を発現させるために、WO2005/108590およびWO2006/094945の野生型酵素EbN1の発現、およびそのために使用することができる発現系に関する記述を参考にしてもよく、この記述は明確に引用される。
【0160】
3.5 酵素固定化
本発明の酵素は、遊離の状態で、または固定化された形で、本明細書に記載の方法に使用することができる。固定化酵素は、不活性担体に固定された酵素を意味すると理解される。適当な担体材料およびそれに固定化される酵素は、EP-A-1149849、EP-A-1 069 183およびDE A 100193773から、ならびにそれらに引用された元の文献から公知である。これに関連して、上記の明細書の記述を全体として参考とする。適当な担体材料には、たとえば、粘土、粘土鉱物、たとえばカオリナイト、珪藻土、パーライト、二酸化ケイ素、酸化アルミニウム、炭酸ナトリウム、炭酸カルシウム、セルロース粉末、アニオン交換体、合成ポリマー、たとえばポリスチレン、アクリル樹脂、フェノールホルムアルデヒド樹脂、ポリウレタンおよびポリオレフィン(ポリエチレンおよびポリプロピレンなど)がある。担体材料は、担体に結合した酵素を作製するために、通常、微粉化された粒子の形で使用されるが、多孔質の形状が好ましい。担体材料の粒径は、通常5 mm以下、特に2 mm以下である(粒度曲線)。同様に、デヒドロゲナーゼをホールセル触媒として使用する場合、遊離型もしくは固定化型を使用することができる。担体材料はたとえば、アルギン酸カルシウムおよびカラギーナンである。酵素は、細胞と同様に、グルタルアルデヒドを用いて直接架橋することができる(架橋してCLEAとなる)。対応する固定化法および他の固定化法が、たとえば、K. Drauz and H. Waldmann, Enzyme Catalysis in Organic Synthesis 2002, vol. III, 991-1032, Wiley-VCH, Weinheimの中でJ. Lalonde and A. Margolin “Immobilization of Enzymes” に記載されている。本発明の方法を実施するための、生体内変換およびバイオリアクターに関するさらに詳しい情報は、たとえば、Rehm et al (Ed) Biotechology, 2nd edition, vol. 3, chapter 17, VCH, Weinheimにも見いだすことができる。
【0161】
下記の限定的でない実施例を参照することによって、本発明をより詳細に説明することとする。
【0162】
(実施例)
【実施例1】
【0163】
飽和変異導入
1.1 分子モデリング
変異体は、酵素フェニルエタノールデヒドロゲナーゼEbN1の結晶構造(図2)を参考にして選択された。
【0164】
酵素の基質特異性は、2つのループ領域および1つのヘリックスによって決定される(図2のループ1および2、ならびにヘリックスαFG1)。ヘリックスαFG1はフレキシブルであって、基質結合後に活性中心を閉じる。ループ1のTyr93は基質結合ポケットを前側に向かって塞いでおり、それによって立体選択性に関与する。Tyr151はループ2に属し、結合ポケット内に向いている。Thr192はフレキシブルなヘリックスαFG1の一部であって、基質結合部位の方向に向いている。これらの2か所が選ばれたのは、基質の結合には影響を及ぼすが、触媒中心のアミノ酸および補酵素NAD、すなわち触媒メカニズムを妨害しないからである。
【0165】
1.2 飽和変異導入
初めに、Y151XおよびT192Xの位置で別々に飽和変異導入を行った(すなわちY151およびT192の位置を他のすべての19アミノ酸と置き換える、permutationとも呼ばれる)後、二重変異体(Y151A-T192X)を作製した。
【0166】
これは、いずれの場合も3回のポリメラーゼ連鎖反応で部位特異的変異導入を行うことによって実施された(図3のクローニングストラテジーを参照されたい)。ここで、DNAを増幅するために、以下のオリゴヌクレオチドを使用した:
ebn1H遺伝子断片を増幅するためのPCRを以下のように行った:100μlの反応混合物は、1μlテンプレート(約50 ngのベクターpDHE-ebn1H)、それぞれの場合について1μlオリゴヌクレオチド(20 ng)、2μl dNTPMix(Roche製、終濃度10 mM)、1μl Pfu-Ultra DNAポリメラーゼ(1 U/μl、Stratagene製)、10μl 10X Pfu-Ultraバッファー(Stratagene)、および80μl滅菌水を含有した。
【0167】
下記の温度プログラムをサーモサイクラー(Biometra)に設定した:95℃- 5分;30サイクル:95℃- 45秒、50℃- 45秒、72℃- 45秒;72℃- 10分; 10℃
1a) PCRオリゴヌクレオチド1および3 (T192Xについては6)
1b) PCRオリゴヌクレオチド2および4 (T192Xについては5)
2) PCR 1aおよびbから得られた産物をテンプレートとして、PCRオリゴヌクレオチド1および2(オーバーラップエクステンション)。
【0168】
上記から得られた増幅されたebn1H遺伝子を、GFXキット(GE Healthcare)を用いて1.2%アガロースゲルで精製した。
【0169】
増幅されたDNAを、制限酵素NdeIおよびHindIII(Fermentas)を用いて切断し、(同様にNdeI-HindIIIで切断された)ベクターpDHEの多重クローニング部位(MCS)にライゲートして、XL10ウルトラコンピテントセル(Stratagene)において形質転換した。こうした細胞の少量調製により、変異体のプラスミドDNAが得られた。ベクターpDHEは、DE 19848129もしくはWO2005/108590において、pDHE19.2ベクターとして記載されている。
【0170】
1.3 細胞の培養
初めに、ベクターpDHE-ebn1H-Y151XまたはpDHE-ebn1H-T192X(さらに後にpDHE-ebn1H-Y151A-T192X)を、菌株LU12037 (大腸菌派生株TG10 pAgro4 pHSG575 (TG10:大腸菌TG1に由来するRhaA- 派生株(Stratagene);pAgro4:Takeshita, S; M; M; Masahashi, W; T (1987) Gene 61, 63-74;pHSG575:T. Tomoyasu et al (2001), Mol. Microbiol. 40 (2))に形質転換したが、この菌株は、シャペロンGroEL/Sおよびlaclqリプレッサーを同時発現するものであって、Q-trayプレート上に蒔いて培養した。増殖したコロニーを、ピッキングロボット(Qpix)を用いて採取し、マイクロタイタープレート(MTP)内で抗生物質(100μMアンピシリン、20μMクロラムフェニコールおよび100μMスペクチノマイシン)を含有するCG前培養(Circular Growth, Gibco)に接種した。37℃にて200 rpmで5時間増殖させた後、細胞を、抗生物質(上記)および対応するインデューサー(ラムノース0.5 g/lおよびIPTG 0.1 mM)を含有するLB本培養に手作業で移した。16-18時間増殖後、細胞をテストに使用する。
【0171】
細胞を破壊するために、まずこれを遠心し、上清を取り除き、MTPに粘着フィルムを装着した。MTPを液体窒素中に約3秒間、完全に浸漬した後、再び実験台上において解凍した。室温での一時的解凍を伴う3回急速凍結で、もっともよくそろった結果が得られた。
【0172】
1.4 酵素阻害
TAC反応産物TACAまたは反応中に形成された二次的成分が、その反応を阻害することが判明している。基質は完全には変換されなかった。この反応は平衡反応であるにもかかわらず、たとえば4時間後に、細胞および/または基質が新たに添加されたが、それでそれ以上の反応は起こらなかった。さらに、結果として生じる2-ブタノンは、平衡をできる限り生成物側にシフトさせるために、蒸留により除去された。これらの方策にもかかわらず、完全な変換は達成されなかった。
【0173】
したがって、考えられるもっとも完全な変換を達成し、それによって容積時間当たりの高収率を達成するために、必ずしも活性は高くないが、より大量の生成物もしくは二次的成分に耐えられる変異体を見いだすことを目的とした。
【0174】
次のテストにおいて、TACAおよびTAもしくはTAAの両者を阻害剤としてテストした。このために、0.2 ml混合物(MTP)において、100 ml振盪フラスコでの培養から得られた細胞(LU11558; 過剰発現プラスミドとしてラムノース誘導性pDHE1650派生物を有する大腸菌TG10+ 菌株。シャペロンGroEL/Sおよびlaclqリプレッサーを同時発現する;野生型酵素EbN1が過剰発現される)50μl、1.75 mM NAD、および100 mM 2-ブタノールを80 mM TrisHClバッファーpH 8.0に添加した。いずれの場合も、10 mMのTA、TAAまたはTACAがそれに添加された。その後、光度計において340 nmで、NADHの生成を測定した。図4は、個々のVmax値(結果として生じた、時間当たりのNADH量)を示すことによって、フェニルエタノールデヒドロゲナーゼEbN1の阻害を説明するものである。対照(阻害剤なし)はブタノールを特徴とする。
【0175】
図4に見られるように、TACAはもっとも強い阻害を示す。TAはこの波長域に非常に強い吸収があるので、NADHの生成をここで検出することはできない。
【0176】
このため、他のアッセイでは阻害剤としてTACAを添加した。加えて、TACAは、以前行われた2-ブタノールおよびNADを用いた再生テストにも阻害剤として加えられた。適当なTACA濃度を決定するために、さまざまな濃度の細胞およびTACAをテストした。初めに、0から30 mMまでの一連の濃度、次に0から10 mMまでの一連の濃度を調製した。達成された結果(示さず)に基づいて、それぞれ25μlの細胞とともに10 mMのTACA濃度を他のテストで用いた。
【0177】
1.5 TACA添加および無添加での2-ブタノールテストの経過(補酵素の再生)
ここで、アミノ酸位置ごとのクローンで満たされた2つのマイクロタイタープレート(96ウェル)が選定された。マイクロプレートは完全に配列決定され、値は個々の変異に当てはめられた。
【0178】
細胞は上記のように培養した後、破壊し、最終的に100μlの水に再懸濁した。この細胞懸濁液25μlを新たなマイクロタイタープレートに入れ、水を足して容量100μlとした。次に基質溶液(終濃度:100 mM 2-ButOH、1.75 mM NAD、80 mM Tris-HCl pH 8.0、(10 mM TACA))を加え、NADHの生成を光度計で340 nmにて測定した。このテストから得られた結果を図5Aおよび5Bに示す。Vmax値を示す。
【0179】
図5Aおよび5Bから、変異体の大半がもはや補酵素を再生することができず、あるいは非常にゆっくりとしか再生できないことがわかる(ブタノールテスト、黒い棒グラフ)。しかしながら、ブタノールテストにTACAを添加することによって(淡色の棒グラフ)、これらの変異体は補酵素を再生することが可能であり、対照(野生型)よりずっと優れているといえる。おそらく、2-ブタノールが2-ブタノンになる代わりに、TACAがTACに酸化される。このような変異体は、野生型よりも、大量のTACAに耐えられる。
【0180】
抜け落ちている変異体(T192の位置についてN、D、Q、H、K、M、F、Y、ならびにY151の位置についてはC、F、S)が認められるが、これらは活性がない、または対照より劣っていた。次の変異体を、ラージスケールでテストするために、この実験から選択した:Y151A、E、G、H、ならびにT192A、G、L、I。
【0181】
1.6 陽性変異体の評価
このテストから出てくる陽性クローンを、次に、ラージスケールで調べた(100 ml振盪フラスコで培養)。ここでは、3つの異なるアッセイを行った:
【化11】
【0182】
それぞれのテストに関するテスト条件は:
テスト1)NADHを添加してTACからTACAへ還元
798.6μl 脱塩水
50μl 1M NaH2PO4 pH5
50μl NADH(100 mM原液(水溶液))
1.4μl TAC
100μl 振盪フラスコから得られた培養物の10倍濃縮粗抽出物
1000μl 最終容量
テスト2)完全な反応
730μl 脱塩水
50μl 1M NaH2PO4 pH5
20μl NAD 10 mM水溶液
100μl 100 mM TAC(1mlイソプロパノール中14μl)
100μl 振盪フラスコから得られた培養物の10倍濃縮粗抽出物
1000μl 最終容量
テスト3)TACAからTACへの酸化
798.6μl 脱塩水
50μl 1M NaH2PO4 pH5
50μl NAD(10 mM原液)
1.4μl TACA
100μl 振盪フラスコから得られた培養物の10倍濃縮粗抽出物
1000μl 最終容量
上記テストにおいて、テスト温度はいずれの場合も30℃とし、酵素濃度は0.1-10mg/mlとした。
【0183】
サンプルは濃塩酸で反応を止め、HPLCを用いて測定した。
【0184】
【0185】
【0186】
保持時間
TACA = 1.283分 (230 nm)
TAA = 0.910分 (230 nm)
TA = 1.168分 (260 nm)
TAC = 1.540分 (260 nm)
実験結果を次項にまとめる。
【0187】
1.6.1 変異体T192X
図6Aは、変異体T192LおよびT192Gが対照のLU11558よりも速やかにTACを減少させることを示す。しかしながら、完全な反応(図6B)を考えると野生型の方が活性が高く、それは他の変異体が同じように補酵素を再生することができないためである。
【0188】
図6Cでは、変異体T192Aが野生型より良好にTACAを酸化することができることが分かるが、生じるTAC濃度は非常に低いので、結果はテストの間ずっと変動する。
【0189】
1.6.2 変異体Y151A
図7Aにおいて、変異体Y151AおよびY151Hは補因子TACを、野生型(LU11558)より約4-5倍早く減少させることが分かる。しかしながら、完全な反応である、犠牲アルコール(ここでは2-プロパノール)を用いた補酵素再生を伴うTACからTACAへの還元を考えると、変異体Y151Aだけがなお活性がある(図7B)。全体を通しての活性は、対照の場合よりいくぶん低い。結合ポケットを大きくした結果として、「小さい」イソプロパノールは、反応器内で使用される2-ブタノールほど酸化反応をしない可能性がある。変異体Y151AおよびY151Hは、野生型より良好にTACAを酸化することができる(図7C)。
【0190】
変異体Y151Aを21 lスケールで培養し、その変異体を4 l反応器内で、2-ブタノールを再生剤として、かつ溶媒としても用いて、野生型と比較するために使用した。
【0191】
1.6.3 第二世代:変異体Y151A-T192X
変異体Y151Aが野生型より優れているので、この変異体を基に、第2の飽和変異導入をT192Xの位置で行った。
【0192】
2-ブタノールを用いた再生テスト(TACAの添加の有無による犠牲アルコールの再生を測定する、すなわちNADHの生成を光度計で測定する)の結果を図8Aに示す。
【0193】
変異体T192IおよびSは、対照(Y151A-T192T)より良好な結果となっている。したがってこれらをラージスケール(100 ml振盪フラスコでの培養)で検討した。さらに、変異体T192A、LおよびVも、対照とほぼ同じ活性を示すのでラージスケールで検討した。しかしながら、これら5つの変異体について完全な反応を考慮すると、対照がもっとも活性がある(図8Bを参照されたいが、ここで、犠牲アルコールとしてイソプロパノールを伴うTACAの生成はHPLCで測定される)ので、以後の実験では単一変異体Y151Aを使用した。
【0194】
1.7 4 l反応器
上記スクリーニングに由来する活性の高い変異体Y151Aは、21 lスケールで数回発酵させた。ラージスケールで対照LU11558と比較するために、一連の標準的な反応器バッチを実行した。
【0195】
1.7.1 バッチ
攪拌機および冷却器を取り付けた加熱可能な4 l反応器内で、初めに2 lの2-ブタノールを20 mM KH2PO4 バッファーpH 5.0に入れた。0.2 mM NAD (0.5 g) および400 mM TAC (275 g) もしくは 600 mM (420 g)を添加した。生体触媒(450 ml、7.0 g/l BTM)をホールセル(未処理の発酵産物)の形で添加することによって、反応をスタートさせた。発酵培地中の細胞を添加した時点で、pHは6に上昇した。二相反応混合物は、40℃にて減圧(100 mbar)下で攪拌した。ここで、2-ブタノール、2-ブタノンおよび水の混合物を、一段階で留去した。同時に、同量の、69%2-ブタノールおよび31%H2Oからなる溶液(2-ブタノンは別として蒸留物の組成に相当する)をフィードとして添加した。pH測定装置を用いてpHをチェックし、約5.5-6.0の間で一定に維持した。1時間ごとにサンプルを採取し、濃塩酸で反応を止め、HPLC(LJ31366)により分析した。8時間後、反応混合物を取り出した。
【0196】
1.7.2 野生型(LU11558)と変異体Y151A(LU14759)との比較
変異体がより大量の生成物/二次的成分に耐えられるかどうかをみるために、初めに、400 mM(70 g/l)TACとともに、次に600 mM(105 g/l)TACとともに、変異体について数回4 l反応器を運転した。
【0197】
図9Aは、変異体が平均で15-20%野生型より優れていることを示す。図に示すように、値は実験ごとに変動する(これは、個々の発酵にも左右される)が、違いは有意である。野生型の場合平均収率は67%±8%であるが、変異体の場合86%±7%であり、これらはいずれの場合も、別々の発酵である。TACA/(TAC+TA)比(淡色の棒グラフ)も、変異体の場合(4.8)、対照の場合(3.0)より有意にすぐれており、これは後に、メチルアミノ化において確実に明白となる。
【0198】
600 mM(図9B)による運転を考えると、ここでもまた、変異体は野生型より良好な結果をもたらす。しかしながら、TACA/(TAC+TA)比は、400 mM運転の場合より有意に悪化している。
【0199】
1.8 結果
合理的な計画によって、野生型より15-20%活性が高く、しかもデュロキセチンアルコールの中間体の調製中に、より大量のTACAおよび/またはTAに耐えられる変異体Y151A(LU14759)見いだすことができた。
【0200】
この結果は、一連の4 l反応器での反応においても確認され、それは小規模での製造プロセスを反映している。
【0201】
阻害剤TAを伴う酵素の結晶構造は解明されているので、酵素-基質複合体の信頼性のあるモデルを確立することができた。チロシン151のOH基が基質(この場合TA)のプロパノン側鎖のβ炭素原子に密接に接触しており、したがって弱いCH-O水素ブリッジを形成していることは、モデルから明白である(図10A)。チロシン151のアラニンへの変異の結果として、この相互作用は増大し、結合は弱くなる(図10B)。他の相互作用はすべて残存しており、結合ポケットのサイズが大きくなったにもかかわらず酵素の優れた選択性は変化しないことを意味している。その上、生成物のee値は> 99.5%である。
【実施例2】
【0202】
ランダム変異導入
2.1 ロボット設備のテスト開発
ランダム変異導入により作製される多数のサンプルを処理するために、(生成物を直接検出する)実験室におけるこれまでのHPLC分析の代わりに、ロボット生産ラインのための光度測定法を開発する必要があった。
【0203】
この目的のために、還元型補酵素NADHにおける還元は、340 nmでの吸光係数がεNAD<<εNADHであるので、この波長で測定することができる。最適なNADH濃度は0.02 mMであった。基質TACは1から2 mMの間で使用することができた。使用したバッファーは、TACからTACAへの還元が弱酸性の条件下で優先的に進行するので、50 mM NaH2PO4 pH 5.0とした。変異体を発現する細胞は、マイクロタイタープレート(MTP)内で直接、培養された。このために、ピッキングロボット(Qpix)を用いて寒天プレートからクローンを採取し、抗生物質(100μMアンピシリン、20μMクロラムフェニコールおよび100μMスペクチノマイシン)を入れたLB前培養培地に接種した。37℃にて200 rpmで24時間の増殖時間後、その細胞を、抗生物質およびインデューサー(ラムノース0.5 g/lおよびIPTG 0.1 mM)を含むLB本培養培地に手で移した。16-18時間増殖させた後、細胞をテストに使用した。
【0204】
予備的な実験から、培養で得られた細胞は、そうしないと活性が低すぎるので、アッセイ前に破壊しなければならないことが分かった。細胞を破壊するために、さまざまな方法が調べられたが、たとえば、細胞を4℃にて一晩保存すること、1-ブタノールおよび1.4-ブタンジオールの添加、および液体窒素による急速凍結などである。4℃保存、および液体窒素による急速凍結だけがうまくいったが、時間の節約のため窒素による処理が好ましい。この目的で、増殖させた細胞をまず遠心し、上清を除去し、MTPに粘着フィルムで封をした。MTPを約3秒間、液体窒素に完全に浸漬した後、再び下に置いて融解させた。もっとも一定した結果は、急速凍結と室温での一時的融解を4回繰り返した場合に達成された。
【0205】
2.2 ロボットテストの経過
細胞は上記のように培養した後破壊した。MTPにカバーを付けて、ロボット装置内の15℃のインキュベーターに入れた。Multidropにおいて、ウェルあたり100μlの水を、その後Packardにおいて細胞を再懸濁するために添加した。その後Multidropにおいて、基質溶液(終濃度:2 mM TAC、0.2 mM NADH、50 mM NaH2PO4 pH 5.0)を加え、NADHの減少を光度計で340nmにて測定した。
【0206】
このテストから得られた陽性クローンを次に、ラージスケールでさらによく検討した。このために、3つの異なるアッセイを行った。
【0207】
テストA 完全な反応:TACからTACAへの還元およびイソプロパノールによるNADH再生(50 mM NaH2PO4 pH 5.0、0.2 mM NAD、10 mM TAC、10%イソプロパノール)、測定HPLC LJ31366
【化12】
【0208】
テストB 光度計において、再生剤として2-ブタノールを用いたNADからNADHへの再生(80 mM TrisHCl pH 8.0、100 mM 2-ブタノール、1.75 mM NAD)
【化13】
【0209】
テストC 光度計において、NADHの添加によるTACからTACAへの還元(50 mM NaH2PO4 pH 5.0、0.2 mM NADH、1.4μl TAC pure (10 mM))
【化14】
【0210】
3つのアッセイから得られた結果の比較から、NADの再生(すなわちテストB)は、ロボットテストで始めに用いられたTACからTACAへの還元よりも、完全な反応の結果を有意によく反映することが明らかになった。
【0211】
予備的実験は同様に、2-ブタノールによる補酵素の再生(したがってNADHの生成)が、生体触媒を発現する細胞の場合にのみ起こることを示した。結果として、ロボットテストは、2-ブタノールによるNADH再生の検出に切り替えられた。しかしながら、これと並行してTACからTACAへの還元、したがってNADNの減少もやはり測定された。
【0212】
2.3 変更されたロボットテストの経過
細胞を培養した後破壊した。MTPにカバーを付けて、ロボット装置内の15℃のインキュベーターに入れた。Multidropにおいて、ウェルあたり100μlの水を、次に細胞を再懸濁するために添加した。これらから、細胞懸濁液20μl/ウェル(補酵素再生アッセイ用)もしくは70μl/ウェル(TAC還元アッセイ用)の2つの娘プレートが作製された。基質溶液(終濃度:還元:2 mM TAC、0.2 mM NADH、50 mM NaH2PO4 pH 5.0;再生: 100 mM 2-ブタノール、1.75 mM NAD、80 mM TrisHCl pH 8.0)を加え、NADHの生成を光度計で340nmにて測定した。
【0213】
2.4 ロボットテスト結果の評価
ロボットスクリーニングにおいて、NADHの生成/減少(還元/酸化)は光度計で測定した。このために、いずれの場合も、10個の測定値を10分間にわたって確認した。これらの値から増加を計算することによって、デヒドロゲナーゼの初発の活性を測定した。
【0214】
2.5 酵素反応の阻害
予備的な実験は、反応中に形成される生成物TACAまたは二次的成分が反応を阻害することを示した。基質は完全には変換されなかった。
【0215】
したがって、可能な限り完全な変換を達成することで容積時間当たりの高収率を達成するために、活性が高いだけでなく、より大量の生成物もしくは二次的成分に耐えられる変異体を見いだすことを目的とした。
【0216】
したがって、アッセイにTACAを添加した。このために、それまで2ブタノールおよびNADとともに行われた再生テストにさらに10 mMのTACAを添加した。
【0217】
生物量が限られるため、ロボットスクリーニングにおいて3つのテストをすべて並行して実施することはできなかった。したがって、TACからTACAへの還元反応は省略した。2-ブタノールによるNADHの再生反応は不変であった。TAC溶液は、TACA阻害のための基質溶液(100 mM 2-ブタノール、1.75 mM NAD、10 mM TACA、80 mM TrisHCl pH 8.0)に置き換えられた。ロボットテストの経過は、変化しないままであった。
【0218】
したがって、評価はTACA阻害の測定結果に適合した。
【0219】
2.6 変異体ライブラリーの調製:EbN1遺伝子のランダム変異導入
デヒドロゲナーゼをコードする配列内に変異を生じさせるために、エラープローンPCR反応を、MnCl2を添加して実施した。MnCl2とともに使用されたTaq-DNAポリメラーゼの特異性は低下したので、その結果として、MnCl2濃度が上昇すると、より多くの間違ったヌクレオチドが組み込まれ、したがってより多くの変異が生じる。
【0220】
PCRのために、開始領域および末端領域においても変異が起こるように、可能な限りもっとも短いDNA領域をカバーする、クローニング切断部位(NdeI - Hind III)を有する下記のオリゴヌクレオチドを選択した:
【0221】
バッチ
50μl PCRバッチ中:デヒドロゲナーゼ遺伝子を有する50 ngプラスミドDNA(pDHE-ebn1H)、それぞれの場合ごとに120 ngオリゴヌクレオチド、GCリッチ反応バッファー1x (Roche)、1/5容 GCリッチレゾリューション (Roche)、それぞれの場合ごとに0.2 mM dATP、dTTP、dCTP、dGTP、1 U Taq DNAポリメラーゼ。
【0222】
このバッチを95℃にて5分間加熱した(DNAの初回変性)後、85℃に冷却した。この温度で、MnCl2をさまざまな濃度(0.02 mM刻みで0-1 mM)で添加した。これは、MnCl2が完全に溶解するために必要であった。実際のPCRはその後、以下の温度プログラムにより開始された:4サイクル:95℃ 45秒、54℃ 45秒、72℃ 45秒;続いて26サイクル:95℃ 45秒、58℃ 45秒、72℃ 45秒;10℃で停止。
【0223】
PCR産物をアガロースゲル(Gfxキット)で精製した後、制限酵素NdeIおよびHindIII(いずれもNEB製)による切断を行った。ベクターpDHE(同様にNdeI/HindIIIで切断)にライゲートした後、形質転換を実施してXL10ウルトラコンピテントセル(Stratagene)に入れた。次に、クローンの一部(濃度当たり16クローン)を配列決定して、最適なMnCl2濃度を決定した。これに関連して、1-3塩基対までの交換を生じるMnCl2濃度を用いて、さらに実験を行った。このために、ライゲーションバッチをまずXL10ウルトラコンピテントセルに形質転換し、クローンを計数した後、すべてのクローンを寒天プレートからLB培地を用いて溶離した。細胞をさらにインキュベートすることなく、プラスミドDNAを単離し(Promegaキット)、このように単離されたDNAを生産菌株TG10に導入して形質転換したが、この菌株は、シャペロンpAgro pHSGを同時に発現するものであって、Q-trayプレート上に蒔かれた。この手順は、ライゲーション時の生産菌株TG10+(LU12037)の形質転換率が非常に低いために必要であり、できる限り多くの変異体が求められた。
【0224】
2.7 選択された変異体
上記の表1は、ロボットテストから選択されたクローンの概要を示す。これらのクローンは検証プレートから得られ、完全に配列決定された。これらをラージスケールで培養し、始めにエッペンドルフでテストした。しかしながら、変異体の大半の活性は、野生型と同程度であった。活性の高い変異体、たとえば、K114TおよびM200V F201Lは、21 lスケールで発酵培養し、0.5 l反応器でテストした。
【0225】
攪拌機および冷却器を取り付けた、加熱可能な0.5 l反応器内で、始めに250 mlの2-ブタノールを、20 mM KH2PO4 バッファーpH 5.0中に導入した。0.2 mM NAD(0.1 g)および100 mM TAC(35 g)を添加した。生体触媒(45 ml、5.5 g/l BTM)をホールセル(未処理発酵産物)の状態で添加することによって、反応を開始させた。発酵培地中の細胞を添加したとき、pHは6に上昇した。二相反応混合物は、減圧(110 mbar)下で40℃にて攪拌した。ここで、2-ブタノール、2-ブタノンおよび水を1段階で留去した。同時に、同量の、69%2-ブタノールおよび31%H2Oからなる溶液(2-ブタノンを除いて蒸留物の組成に相当する)をフィードとして添加した。pH測定装置を用いてpHをチェックし、約5.5-6.0の間で一定に維持した。1時間ごとにサンプルを採取し、濃塩酸で反応を止め、HPLC(LJ31366)により分析した。8時間後、反応混合物を取り出した。
【0226】
個別の段階のエラー解析は、もっとも大きなエラーがマイクロタイタープレートでの個別のクローンの増殖中に存在することを示す。マイクロタイタープレートでの増殖条件(温度、酸素導入など)は、発酵槽のように正確にコントロールできないので、これは驚くには当たらない。同一菌株の別々の発酵でさえ、約10%-15%ほど変動する。このロボットスクリーニングに関する全体としてのエラーは約35%である。すなわち、このスクリーニングにおいて、35%を超える増加を示す変異体だけが意味がある。
【実施例3】
【0227】
部位特異的変異導入および追加の飽和変異導入
追加して標的化された、選択された位置で、検討済みの単一変異(「部位特異的変異導入」)または飽和変異導入を実施した。
【0228】
3.1 変異のための位置の選択
酵素の基質結合ポケットは、ループ1、2およびヘリックスαFG1(図2)により形成される。大半の変異がこの領域から選択されたのは、これらのアミノ酸が基質結合および/または酵素活性に直接影響を及ぼすことが予想されるからである。
【0229】
ヘリックスは基質なしでは非常にフレキシブルであって(結晶状態では、電子密度は見られない)、基質結合時にのみ固定された状態となる。基質結合ポケットを含む活性中心は、疎水性領域と親水性領域に分けられる。疎水性部分は、おもに両親媒性ヘリックスαFG1によって形成されるが、このヘリックスは、基質結合後、基質結合ポケット上に蓋のように乗る。アミノ酸192から204までがこの領域にある。アミノ酸Thr192、Leu197、Met200およびLeu204の側鎖は結合ポケットの中に向いているが、アミノ酸Phe201はループの安定化に役立っている。OH基を有するトレオニン192は活性中心の疎水性領域と親水性領域の境界をなしている。Leu186はフレキシブルなヘリックスの開始部分にあり、活性中心が開いた状態および閉じた状態となるように、蝶番のように機能する。メチオニン246は、基質結合ポケットの末端にある。基質結合ポケットの反対側は、アミノ酸146から151までを含むループβEαFをなしている。ここでもまた、選択されたアミノ酸Leu146、Ile148およびTyr151の側鎖は結合ポケットの中に向いている。チロシン151の末端OH基は、活性中心の親水性部分の水素ブリッジネットワークの一部であるが、残りの残基は疎水性部分に属している。ほとんど親水性の活性中心の下側は、アミノ酸138-142のストランドを形成しているが、ここでLeu139、Thr140およびThr142は変異している。2つのシステイン62および83が変異のために選択されたのは、システインが通常、酸化の影響を受けやすく、それによって構造に悪影響を及ぼす可能性があるためである。
【0230】
図11は、活性中心からの断面図を示す。補酵素は紫色、基質(ここではTA)は緑色でマークし、変異したアミノ酸は強調してある。上部の領域において、両親媒性ヘリックスαFG1が見られ、下部領域にはloopβEαF(ループ2)が見られる。
【0231】
3.2 標的変異体の調製
始めに、DNA変異のそれぞれの位置について、望ましいDNA配列に対応する2つの相補的オリゴヌクレオチド(表2)を選択した。それに加えて、遺伝子全体に隣接するオリゴヌクレオチドをさらに2つ選択した(Mke123およびMke124、配列番号5および6)。部位特異的変異導入のクローニングストラテジーを図12に示す。
【0232】
その後、それぞれ遺伝子隣接オリゴヌクレオチドおよび望ましい変異を有するオリゴヌクレオチドを用いて、2つのPCR反応を行った。これによって、変異の代わりに、短い相補的領域を有する2つのPCR産物が与えられる。これら2つのPCR産物をテンプレートとして用いて、第2のPCRを、以前使用した遺伝子隣接オリゴヌクレオチドを再び使用して、行った。この反応から、望ましい変異を有する完全な遺伝子が与えられる。
【0233】
PCR産物を制限酵素NdeIおよびHindIIIで切断した後、pDHEベクターにライゲートした。コンピテントセルXL10 Gold (Stratagene)に入れて形質転換し、続いてプラスミドを単離した後、変異の成否を確認するためにそのプラスミドを配列決定した。活性を測定するために、このプラスミドを、シャペロンプラスミドpAgroおよびpHSGを有するTG10+コンピテントセル(LU12037)に入れて形質転換した。
【表3】
【0234】
【0235】
【0236】
【0237】
【0238】
3.3 標的変異体に対する活性テスト
図13Aは、NADHを添加したTACからTACAへの還元、ならびに再生を伴う全体としての反応(黒い棒グラフ)の両者を2.2項に記載のように検討した活性テストから得られた結果を示す。図13Bでは、再生を伴う全体反応のみ調べた。
【0239】
特に変異体L197Iは、野生型より3倍高い活性を示す。
【0240】
本明細書に引用された文献の開示について明確に参照する。
【技術分野】
【0001】
本発明は、新しいタイプのフェニルエタノールデヒドロゲナーゼ変異体、それらの製造方法;それらをコードする核酸配列、そうした配列を含んでなる発現カセット、ベクターおよび組換え微生物;前記変異体を用いて光学活性の置換アルコールを生体触媒合成する方法;ならびに、特に、前記変異体によって触媒される生体触媒合成ステップを含んでなる、デュロキセチンアルコールもしくはデュロキセチンを調製する方法に関する。
【背景技術】
【0002】
デュロキセチンアルコール(3)は、デュロキセチン(4)の調製において重要な前駆体である(模式図1を参照されたい)が、このデュロキセチンはCymbalta(登録商標)という商品名で、特に抗うつ薬として販売されている。
【化1】
【0003】
模式図1:デュロキセチンアルコールを経由するデュロキセチンの調製
生成する中間体(TACA)(2)は、デヒドロゲナーゼを用いて調製することができる(WO2005/033094)。たとえば、Azoarcus菌種(新規名称Aromatoleum aromaticum)(Hoeffken et al., Biochemistry, vol. 45, No.1, 2006)由来のフェニルエタノールデヒドロゲナーゼEbN1は、Meerwein-Ponndorf還元と同様に、クロロケトン、3-クロロ-1-(チエニル-2-イル)-プロパン-1-オン(1)を、対応するクロロアルコールである(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オール(2)に還元する。このために、このデヒドロゲナーゼは、必要な還元当量をもたらす補酵素ニコチンアミドアデニンジヌクレオチド(NADH)を必要とする。この高価な補酵素は、二次的な「犠牲アルコール」(たとえば、2-プロパノールまたは2-ブタノール)の助けを借りることによって再生することが可能であり、その間に対応するケトン(たとえばアセトンまたは2-ブタノン)が生成される。比較的長鎖のアルコールがこの酵素には好ましいが、長鎖の方がやはり相当高価である。こうした理由から、2-ブタノールが犠牲アルコールとして使用される(模式図2を参照されたい)(WO2006/072465)。
【化2】
【0004】
模式図2:「犠牲アルコール」を用いた補酵素NADHの再生
野生型酵素EbN1およびその発現のために使用できる発現系は、WO2005/108590およびWO2006/094945に記載されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】国際公開第2005/033094号
【特許文献2】国際公開第2006/072465号
【特許文献3】国際公開第2005/108590号
【特許文献4】国際公開第2006/094945号
【非特許文献】
【0006】
【非特許文献1】Hoeffken et al., Biochemistry, vol. 45, No.1, 2006
【発明の概要】
【発明が解決しようとする課題】
【0007】
発明の簡単な説明
デュロキセチンの調製に使用することができる生体触媒の活性を高めることを本発明の目的とした。
【0008】
特に、TAC(1)からTACA(2)への酵素的還元を改善する生体触媒を提供することを目的とした。ここで達成されるべき改善は下記のとおりとすることができる:
- 反応速度を高めること
- 生成物の収率を高めること
- 生成物阻害の影響を受けにくくすること
- 補酵素再生の向上
- 上記を組み合わせること。
【課題を解決するための手段】
【0009】
驚くべきことに、この目的は、Azoarcus菌種に由来する上記フェニルアルコールデヒドロゲナーゼEbN1の特別な変異体を提供することによって達成された。
【0010】
具体的には、上記目的は、驚くべきことに、2つの異なる方法で達成された。第1の解決方法によれば、この生体触媒をコードする遺伝子の配列を、エラープローンポリメラーゼ連鎖反応(エラープローンPCR)によって偶然に変異させ、それによって多数のバリアントを生じ、そのバリアントから改善された変異体を選択することができた。これらの変異体を次に、さらに改良するために再度変異させた(人工分子進化法)。
【0011】
もう一つの解決方法は、選択された配列位置で、標的化された方法で飽和突然変異誘発を実施することであった。まず、デヒドロゲナーゼの結晶構造を端緒として(Hoeffken et al., Biochemistry, vol. 45, No.1, 2006)、適当な変異のための標的位置を、「合理的設計」によって決定した。次いで、こうした位置で飽和突然変異導入を実施した。
【図面の簡単な説明】
【0012】
【図1】図1は、フェニルエタノールデヒドロゲナーゼEbN1の核酸コード配列(A)およびアミノ酸配列(B)を示す。
【図2】図2は、EbN1の1つのモノマーの、バンドモデルを示す。
【図3】図3は、さまざまな変異体のためのクローニングストラテジーを図示する。
【図4】図4は、いずれの場合も10 mM TA、TAAまたはTACAの存在下でフェニルエタノールデヒドロゲナーゼEbN1を阻害するための実験の結果を示すが、加えて、阻害物質なしでの対照バッチの結果も示す。実験はホールセルを用いて行った;いずれの場合も、25または50μlの細胞懸濁液をテストした。
【図5A】図5Aは、TACA存在下および非存在下で、Y151X型のさまざまな変異体による、2-ブタノールを用いた補酵素再生能力を示す。
【図5B】図5Bは、TACA存在下および非存在下で、T192X型のさまざまな変異体による、2-ブタノールを用いた補酵素再生能力を示す。
【図6A】図6Aは、補酵素再生を行わないTACテストにおけるT192X型のさまざまな変異体の活性を標準(LU11558)と比較して示す。
【図6B】図6Bは、補酵素再生を伴うTACテストにおけるT192X型のさまざまな変異体の活性を標準(LU11558)と比較して示す。
【図6C】図6Cは、TACAテストにおけるT192X型のさまざまな変異体の活性を標準(LU11558)と比較して示す。
【図7A】図7Aは、補酵素再生を行わないTACテストにおけるY151X型のさまざまな変異体の酵素活性を標準(LU11558)と比較して示す。
【図7B】図7Bは、補酵素再生を伴うTACテストにおけるY151X型のさまざまな変異体の酵素活性を標準(LU11558)と比較して示す。
【図7C】図7Cは、TACAテストにおけるY151X型のさまざまな変異体の活性を標準(LU11558)と比較して示す。
【図8A】図8Aは、TAC存在下よび非存在下での、Y151A-T192X 型変異体による、2-ブタノールを用いた補酵素の再生を示す。
【図8B】図8Bは、補酵素再生を伴うTACテストにおける、Y151A-T192X型変異体の活性を、対照(Y151A)と比較して示す。
【図9A】図9Aは、さまざまな反応混合物において変異体Y151Aを用いて達成されるTACAの収率を、いずれの場合も標準と比較し、異なるTAC濃度に応じて示す(400 mM)。
【図9B】図9Bは、さまざまな反応混合物において変異体Y151Aを用いて達成されるTACAの収率を、いずれの場合も標準と比較し、異なるTAC濃度に応じて示す(600 mM)。
【図10】図10は、野生型酵素EbN1における基質結合(TA)(合成A)または変異体Y151Aにおける基質結合(TA)(合成B)を、コンピューターアニメモデルとして示す。いずれの場合も下の図は、基質結合ポケットの拡大断面図を表す。
【図11】図11は、EbN1の活性中心の、コンピュータシミュレーション断面図を示す;両親媒性ヘリックス、ループ2、基質、および補酵素(NADH)の配置が強調されている。
【図12】図12は、部位特異的変異導入のためのクローニングストラテジーを示す。
【図13A】図13Aは、本発明のさまざまな点変異に関する活性テストの結果を示す。
【図13B】図13Bは、本発明のさまざまな点変異に関する活性テストの結果を示す。
【発明を実施するための形態】
【0013】
発明の詳細な説明
1. 一般的な用語の定義
「フェニルエタノールデヒドロゲナーゼ」(EC No.1.1.1)は、一般に、アセトフェノンからS-1-フェニルエタノールへの、NADH依存性の立体特異的還元を触媒する酵素である。「フェニルエタノールデヒドロゲナーゼ」または「フェニルエタノールデヒドロゲナーゼ活性を有する酵素」は、本発明に関しては特に、一般式Iのケトンから出発して、式IIの光学活性アルコールの酵素合成を触媒するものであって、具体的には3-クロロ-1-(チエニル-2-イル)-プロパン-1-オンと(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オールとの間の立体特異的平衡反応を触媒する。
【0014】
酵素反応の可逆性により、本発明は、両方向の、本明細書に記載される酵素反応に関する(すなわち還元当量の生成または消費を伴う)。
【0015】
「フェニルエタノールデヒドロゲナーゼ」の「機能的変異体」には、下記の、こうした酵素の「機能的同等物」が含まれる。
【0016】
「生体触媒プロセス」は、本発明の「フェニルエタノールデヒドロゲナーゼ」、または「フェニルエタノールデヒドロゲナーゼ活性」を有する酵素の、触媒活性の存在下で実施される任意のプロセス、すなわち、未精製の、もしくは精製された、溶解、分散もしくは固定化された酵素の存在下で行われるプロセス、またはそうした酵素活性を有する、もしくは発現する微生物ホールセルの存在下で行われるプロセスを表す。したがって、生体触媒プロセスは、酵素プロセスおよび微生物プロセスを含んでいる。
【0017】
「立体特異的」という用語は、本発明にしたがって調製される、少なくとも1つの不斉中心を有する化合物の、考えられるいくつかの立体異性体のうち1つが、たとえば少なくとも90%ee、特に少なくとも95%ee、または少なくとも98%ee、または少なくとも99%eeといった、高い「鏡像体過剰率」で、言い換えれば高い「鏡像体純度」で、本発明の酵素の作用によって生成されることを意味する。ee%値は、下記の式にしたがって計算される:
ee% = [XA-XB]/[ XA+XB]*100,
式中XAおよびXBはそれぞれ鏡像体AおよびBのモル分率である。
【0018】
さらに、本明細書では下記の略号を使用する。
【0019】
TAC = 3-クロロ-1-チオフェン-2-イル-プロパン-1-オン
TACA = 3-クロロ-1-チオフェン-2-イル-プロパン-1-オール
TA = 1-チオフェン-2-イル-プロペノン
TAA = 1-チオフェン-2-イル-プロパ-2-エン-1-オール
「低級アルコール」は特にモノオールであって、本明細書によれば低級アルキル基を含むものである。これは特にC1-C8-アルキル基、なかでもC1-C6-アルキル基であって、これらは分岐鎖または特に直鎖で、1から8個までの炭素原子、特に1、2、3、4、5または6個の炭素原子を有する。例としては、C1-C4-アルキル基、たとえばメチル、エチル、n-プロピル、イソプロピル、n-ブチル、2-ブチル、イソブチルもしくはtert-ブチル;さらに5個以上の炭素原子を有する基、たとえば、ペンチル、1-メチルブチル、2-メチルブチル、3-メチルブチル、2,2-ジメチルプロピル、1-エチルプロピル、1,1-ジメチルプロピル、1,2-ジメチルプロピル、1-メチルペンチル、2-メチルペンチル、3-メチルペンチル、4-メチルペンチル、n-ヘキシル、1,1-ジメチルブチル、1,2-ジメチルブチル、1,3-ジメチルブチル、2,2-ジメチルブチル、2,3-ジメチルブチル、および3,3-ジメチルブチルなどがある。
【0020】
「環」(Cyc)には、単環式もしくは多環式、飽和もしくは不飽和、炭素環もしくはヘテロ環、芳香族もしくは非芳香族であって、適宜、一置換もしくは多置換された環が含まれる。
【0021】
炭素環およびヘテロ環基Cycの例は、具体的には、単環式もしくは二環式、好ましくは単環式であって、4個以下、たとえば0、1もしくは2個の、O、NおよびSから選択される同一もしくは異なる環員ヘテロ原子を有する基である。
【0022】
このような炭素環もしくはヘテロ環は、特に3から12個まで、好ましくは4、5または6個の環員炭素原子を有する。例として挙げられるのは、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、それらの一価不飽和もしくは多価不飽和アナログ、たとえば、シクロブテニル、シクロペンテニル、シクロペンタジエニル、シクロヘキセニル、シクロヘプテニル、シクロヘキサジエニル、シクロヘプタジエニル、およびフェニル;ならびに5から7員環であって、O、NおよびSから選択される1から4個までのヘテロ原子を有する、飽和または一価もしくは多価不飽和ヘテロ環基である。具体的には、ピロリドン、テトラヒドロフラン、ピペリジン、モルホリン、ピロール、フラン、チオフェン、ピラゾール、イミダゾール、オキサゾール、チアゾール、ピリジン、ピラン、ピリミジン、ピリダジン、およびピラジンから導かれるヘテロ環基が挙げられる。
【0023】
前記炭素環もしくはヘテロ環のうち1つが、他のヘテロ環もしくは炭素環と縮合している二環式環基、たとえばクマロン、インドール、キノリンおよびナフタレンから誘導される基も挙げられる。
【0024】
Cyc基のさらに好ましい基はアリール基である。「アリール」は、単環式もしくは多環式、好ましくは単環式もしくは二環式の、適宜置換された芳香族基であって、具体的には、フェニル、もしくは任意の望ましい環上の位置で結合するナフチル、たとえば1-または2-ナフチルである。
【0025】
Cyc基は、任意の望ましい環上の位置で、好ましくは環員炭素原子を介して、結合することができる。
【0026】
適当なCyc基の例は、フェニル、ナフチル、2-チエニル、3-チエニル;2-フラニル、3-フラニル、2-ピリジル、3-ピリジルもしくは4-ピリジル;2-チアゾリル、4-チアゾリルもしくは5-チアゾリル;4-メチル-2-チエニル、3-エチル-2-チエニル、2-メチル-3-チエニル、4-プロピル-3-チエニル、5-n-ブチル-2-チエニル、4-メチル-3-チエニル、3-メチル-2-チエニル;3-クロロ-2-チエニル、4-ブロモ-3-チエニル、2-ヨード-3-チエニル、5-ヨード-3-チエニル、4-フルオロ-2-チエニル、2-ブロモ-3-チエニル、および4-クロロ-2-チエニルlである。
【0027】
Cyc基は、たとえば一置換もしくは多置換のように、1回または2回以上置換されていてもよい。好ましくは、置換基は環員炭素原子に結合する。適当な置換基の例は、ハロゲン、低級アルキル、低級アルケニル、低級アルコキシ、-OH、-SH、-NO2 またはNR2R3であって、このR2 およびR3は互いに無関係にH、メチルまたはエチルである。
【0028】
「ハロゲン」は、フッ素、塩素、臭素またはヨウ素であるが、特にフッ素または塩素である。
【0029】
「低級アルキル」は、好ましくは、2から8個まで、特に2から6個までの炭素原子を有する、直鎖もしくは分岐鎖アルキル基であって、たとえば、エチル、イソプロピルもしくはn-プロピル、n-ブチル、イソブチル、sec-ブチルもしくはtert-ブチル、n-ペンチルもしくは2-メチルブチル、n-ヘキシル、2-メチルペンチル、3-メチルペンチル、2-エチルブチルである。
【0030】
「低級アルコキシ」は、好ましくは、上記低級アルキル基に対応する酸素終端アナログである。
【0031】
「低級アルケニル」は、2から8個まで、特に2から6個までの炭素原子を有する前記アルキル基の、一価もしくは多価不飽和、好ましくは一価不飽和アナログであって、その二重結合は炭素鎖上の任意の望ましい位置にあるとすることができる。
【0032】
2. 本発明の好ましい実施形態
本発明は第1に、配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体を提供する。
【0033】
特に、本発明は、配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体に関するものであって、この変異体は、
(1)配列領域142〜153(ループ2とも呼ばれる)および
(2)配列領域190〜211(ヘリックスαFG1とも称される)
から選択される少なくとも1つの配列領域内に、少なくとも1つの変異を有する。
【0034】
特に、本発明は、
(3)配列領域93〜96(ループ1とも称される)
(4)配列領域241〜249(C末端)
(5)配列領域138〜141(結合ポケットの親水性領域、ループ2とも称される)
(6)Cys61および/またはCys83
から選択されるさらに他の配列領域中に、少なくとももう1つの変異を追加して有する、機能的なフェニルエタノールデヒドロゲナーゼ変異体に関する。
【0035】
さらに、本発明は、配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体に関するものであって、この変異体は、表1に記載の変異体から選択される。
【0036】
特に、以下の基の少なくとも1つが変異している変異体が挙げられるが:
T192、L197、M200、F201、L204、M246、L139、T140、T142、L146、I148、Y151、C61、C83、L186、
これらのアミノ酸はそれぞれ、他の任意の望ましい天然アミノ酸で置換されている。
【0037】
特に、本発明の変異体は、下記の変異のうち少なくとも1つを有する変異体から選択される:
a)単一変異:
Y151XA、ここでXAはA、R、N、E、Q、G、H、I、L、M、TまたはVである;
T192XB、ここでXBはA、E、G、I、P、S、W、VまたはLである;
b)多重変異:
Y151XA T192XB、ここでXA およびXB は上記の意味を持つ。
【0038】
本発明は、特に、下記の改変された部分配列のうち少なくとも1つによって特徴付けられる変異体を提供する:
(部分配列1) 142-TTYWX1KX2EAX3T-153(改変ループ2)および
(部分配列2) 190-ATX4EASAX5 SAX6X7DVX8PNMLQAI-211(改変ヘリックスαFG1)
これらの配列中、X1 〜X8は、互いに無関係に、任意の望ましいアミノ酸基であるが、基X1 〜X3 およびX4 〜X8の少なくとも1つは、配列番号2に記載の天然型酵素の天然アミノ酸基ではなく、具体的には
X1 はLである、またはI、V、A、M、FもしくはHで置換されている、
X2 はIである、またはL、V、A、M、FもしくはHで置換されている、
X3 はYである、またはA、R、N、E、Q、G、H、I、L、M、TもしくはVで置換されている;
あるいは、
X4 はTである、またはA、E、G、I、P、S、W、VもしくはLで置換されている、
X5 はLである、またはI、V、A、M、FもしくはHで置換されている、
X6 はMである、またはY、W、E、V、S、R、Q、K、I、H、G、F、EもしくはDで置換されている、
X7 はFである、またはG、K、T、Y、M、WもしくはRで置換されている、
X8 はLである、またはI、V、A、M、FもしくはHで置換されている。
【0039】
本発明はまた、特に、配列番号2のデヒドロゲナーゼの酵素活性の、すくなくとも約50%を依然として有する変異体、たとえば50から100%以上、たとえば100を超えて1000%までの活性を有する変異体など、に関するが、その活性はいずれの場合も、TACまたはTACAなどの標準物質を用いて標準的な条件下で測定される(下記、フェニルエタノールデヒドロゲナーゼ活性の測定に関する詳細を参照されたい)。
【0040】
特に、本発明はまた、配列番号2との配列同一性パーセントが少なくとも約70%、たとえば、70から99,9%まで、75から99.9%まで、80から99.9%まで、85から99.9%まで、90から99.9%まで、または95から99.9%までである変異体を提供する。
【0041】
特に本発明は、上記領域(1)〜(6)における少なくとも1つの変異に加えて、こうした領域の外側にあるアミノ酸基の25%までが、配列番号2と比較して、付加、欠失、挿入、置換、逆位もしくはそれらの組み合わせによって改変されている変異体も提供する。
【0042】
特に本発明は、補酵素NAD+またはNADHの存在下で3-クロロ-1-(チエニル-2-イル)-プロパン-1-オン(1)と(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オール(2)との間の立体特異的平衡反応を触媒する変異体を提供する。
【化3】
【0043】
本発明はさらに、本明細書に記載の変異体をコードする核酸配列を提供する。
【0044】
本発明はまた、少なくとも1つの調節核酸配列に機能的に連結された、本明細書に記載の少なくとも1つの核酸配列を含んでなる、発現カセットを提供する。
【0045】
本発明はさらに、本明細書に記載の少なくとも1つの発現カセットを含んでなるベクターを提供する。
【0046】
本発明はまた、少なくとも1つの、本明細書に記載の核酸配列、本明細書に記載の発現カセット、または本明細書に記載のベクターを含んでなる組換え微生物を与える。
【0047】
本発明はさらに、本明細書に記載のフェニルエタノールデヒドロゲナーゼ変異体を製造する方法を提供するが、その方法は、本明細書に記載の組換え微生物を培養すること、変異体をコードする核酸配列を発現させること、ならびに、必要に応じて、発現産物を単離することを含んでいる。
【0048】
さらに本発明は、式(II)の置換された光学活性アルコールを、微生物/酵素により合成する方法を与えるが、
【化4】
【0049】
式中、Cycは、単環式もしくは多環式、飽和もしくは不飽和、炭素環もしくはヘテロ環であって、適宜、一置換または多置換されていてもよく、
いずれの場合も、立体異性体として純粋な状態、または立体異性体の混合物であって、
その方法は、本明細書に記載のフェニルエタノールデヒドロゲナーゼ変異体の存在下で、適宜、特にNADHのような還元当量を添加して、生体触媒(微生物/酵素)により式(I)のケトンを還元することを含むものである。
【化5】
【0050】
特に、低級アルコール、たとえば具体的にはC1〜C6一価アルコールなどを犠牲アルコールとして使用して、還元当量再生条件下で反応を行う方法も与えられる。
【0051】
本発明の調製法を用いて、特に式(I)でCycがヘテロ環基、なかでもチエニル基である化合物を反応させる。
【0052】
特に、基本的に鏡像体として純粋な式(II)のアルコール、特に(S)-鏡像異性体を与える方法も提供される。
【0053】
特に、変異体が単離された状態で使用され、その結果、適宜、固体担体に固定化されている方法;または変異体が、適宜、固体担体に固定化された微生物細胞内で発現される方法も与えられる。適当な固体担体、たとえば、ビーズまたは膜などの高分子担体材料は、生体内変換および酵素リアクター技術の分野で当業者に知られている。
【0054】
また、特にデュロキセチンを調製する方法も与えられるが、その方法は
a)本明細書に記載の生体触媒による方法を用いた、3-クロロ-1-(チエニル-2-イル)-プロパン-1-オン(1)から(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オール(2)への微生物/酵素による還元;
【化6】
【0055】
b)メチルアミノ化によりデュロキセチンアルコール(3)を与える、アルコール(2)の化学的変換;
【化7】
【0056】
ならびに、最終的に
c)ナフチル基の挿入によるデュロキセチンアルコール(3)からデュロキセチン(4)へのの化学的変換
【化8】
【0057】
を含んでなる。
【0058】
さらに、本発明は、式(I)の置換ケトンを微生物/酵素により合成する方法を提供するが、
【化9】
【0059】
式中、Cycは、単環式もしくは多環式、飽和もしくは不飽和、炭素環もしくはヘテロ環、芳香族もしくは非芳香族の環であって、適宜、一置換または多置換されていてもよく、
その方法は、本明細書に記載のフェニルエタノールデヒドロゲナーゼ変異体の存在下で、適宜、特にNAD+のような、酸化当量を添加して、式(II)のアルコールを微生物/酵素により酸化することを含んでおり、
【化10】
【0060】
このアルコールは、いずれの場合も、立体異性体として純粋な状態、または立体異性体の混合物である。
【0061】
特に、この反応は、C1〜C6一価アルカノンを犠牲ケトンとして用いて、酸化当量再生条件下で行われる。
【0062】
使用する酵素変異体は、この場合、単離された形で使用され、たとえば適宜、固体担体に固定化されていてもよいが、固体担体に必要に応じて固定化された微生物細胞内で発現されていてもよい。
【0063】
最後に、本発明は、デュロキセチンアルコールおよび/またはデュロキセチンの調製において、本明細書に記載の酵素変異体を使用することに関する。
【0064】
3. 本発明のさらなる実施形態
3.1 タンパク質
本発明は、フェニルエタノールデヒドロゲナーゼ活性を有する、具体的に記載されたタンパク質および酵素に限定されることはなく、それと機能的に同等のものにまで及ぶ。
【0065】
具体的に記載された酵素の「機能的同等物」または類似物は、本発明との関連において、望ましい生物活性、たとえばフェニルエタノールデヒドロゲナーゼ活性を同様に有しているが記載された酵素とは異なるポリペプチドである。
【0066】
したがって、たとえば、「機能的同等物」は、本発明の「フェニルエタノールデヒドロゲナーゼ活性」用のテストにおいて、本明細書に記載のアミノ酸配列を有する酵素の活性を示す、酵素を意味すると考えられ、それは、少なくとも1%低い、または高い活性であるが、特に、少なくとも約5から10%、たとえば、少なくとも10%もしくは少なくとも20%、たとえば、少なくとも50%もしくは75%もしくは90%低い、または高い活性である。さらに、機能的同等物は好ましくは、pH 4〜11の間で安定であり、至適pHがpH 5〜10、たとえば特に、6.5〜9.5または7〜8または約7.5であって、かつ至適温度が15℃〜80℃または20℃〜70℃、たとえば、約30〜60℃または約35〜45℃、たとえば約40℃であれば有利である。
【0067】
本発明において、「フェニルエタノールデヒドロゲナーゼ活性」は、さまざまな既知のテストによって実証することができる。それらに限らず、実験の項に記載の標準条件下で、標準物質、たとえばTACもしくはTACAを使用するテスト(テスト1)、2)または3)の記述を参照されたい)、または4lリアクター内での生体内変換(イソプロパノールもしくは2-ブタノールを用いた補酵素再生を伴う、完全なTAC→TACA反応)を挙げることができる。
【0068】
本発明によれば、「機能的同等物」は、特に、前記アミノ酸配列の少なくとも1つの配列位置において、具体的に言及されたものとは異なるアミノ酸を有するにもかかわらず、前記生物活性の1つを有する、「変異体」を意味するとも考えられる。したがって、「機能的同等物」には、1つもしくは複数のアミノ酸の付加、置換、欠失および/または逆位によって得られる変異体が含まれるが、前記の改変は、それが本発明に一致する性質の特徴を有する変異体をもたらすならば、いかなる配列位置でも起こりうる。特に、変異体と未改変ポリペプチドとの間で反応性パターンが定性的に一致する、すなわち、たとえば同一基質が異なる速度で変換されるならば、機能的同等性はやはり存在する。適当なアミノ酸置換の例を下記の表にまとめる。
【0069】
元の基 置換例
Ala Ser
Arg Lys
Asn Gln; His
Asp Glu
Cys Ser
Gln Asn
Glu Asp
Gly Pro
His Asn ; Gln
Ile Leu; Val
Leu Ile; Val
Lys Arg ; Gln ; Glu
Met Leu ; Ile
Phe Met ; Leu ; Tyr
Ser Thr
Thr Ser
Trp Tyr
Tyr Trp ; Phe
Val Ile; Leu
記載されたポリペプチドの「前駆体」ならびにそうしたポリペプチドの「機能的誘導体」および「塩」も、上記の意味での「機能的同等物」である。
【0070】
ここで、「前駆体」は、望ましい生物活性の有無にかかわらず、ポリペプチドの天然もしくは合成の前駆体である。
【0071】
「塩」という表現は、本発明のタンパク質分子における、カルボキシル基の塩、およびアミノ基の酸付加塩の両方を意味するものと理解される。カルボキシル基の塩は、それ自体既知の方法で調製することができ、無機塩、たとえばナトリウム塩、カルシウム塩、アンモニウム塩、鉄塩および亜鉛塩、ならびに有機塩基、たとえば、トリエタノールアミン、アルギニン、リジン、ピペリジンなどのアミンとの塩もある。酸付加塩、たとえば、塩酸もしくは硫酸などの無機酸との塩、ならびに酢酸およびシュウ酸などの有機酸との塩も、同様に本発明により与えられる。
【0072】
本発明のポリペプチドの「機能的誘導体」は、同様に、既知の技術を用いて、機能的アミノ酸側鎖基またはそれらのNもしくはC末端で調製することができる。このような誘導体には、たとえば、カルボン酸基の脂肪族エステル、カルボン酸基のアミド(アンモニア、または一級もしくは二級アミンとの反応によって得られる);遊離アミノ基のN-アシル誘導体(アシル基との反応によって調製される);または遊離ヒドロキシ基のO-アシル誘導体(アシル基との反応によって調製される)が含まれる。
【0073】
「機能的同等物」には当然、他の生物から得られるポリペプチドおよび天然に存在するバリアントも含まれる。たとえば、配列比較によって、相同配列領域のエリアを決定し、本発明の具体的な規定に従って同等な酵素を決定することができる。
【0074】
「機能的同等物」には同様に、本発明のポリペプチドの断片、好ましくは個々のドメインもしくは配列モチーフも含まれており、それらはたとえば、望ましい生物学的機能を有するものである。
【0075】
さらに、「機能的同等物」は、前記ポリペプチド配列もしくはそれから誘導された機能的同等物の1つ、および少なくとも1つの他の機能的に異なる異種配列を、機能的にN末端もしくはC末端結合した融合タンパク質である(すなわち、融合タンパク質部分の相互の実質的な機能の減損はない)。このような異種配列の、限定的でない例としては、たとえば、シグナルペプチド、ヒスチジンアンカーまたは酵素がある。
【0076】
本発明によって含まれる「機能的同等物」は、具体的に記載されたタンパク質と相同である。これらは、具体的に記載されたアミノ酸配列の1つに対して、少なくとも60%、好ましくは少なくとも75%、特に少なくとも85%、たとえば、90、91、92、93、94、95、96、97、98または99%の相同性(言い換えれば、同一性)を有するが、それはPearson and Lipman, Proc. Natl. Acad, Sci. (USA) 85(8), 1988, 2444-2448によって、アルゴリズムに従って計算される。本発明の相同ペプチドの相同性すなわち同一性パーセントは、具体的には、本明細書に具体的に記載されるアミノ酸配列の1つの全長に基づく、アミノ酸基の同一性パーセントを意味する。
【0077】
同一性パーセントの値は、BLASTアラインメントのアルゴリズムblastp(タンパク質-タンパク質BLAST)に基づいて、または以下に与えられるClustalセッティングを用いて、確認することもできる。
【0078】
タンパク質に糖鎖付加(グリコシル化)の可能性がある場合、本発明の「機能的同等物」には、脱グリコシル化型またはグリコシル化型、ならびに糖鎖付加パターンの変更によって得られる修飾型の、上記のタイプのタンパク質が含まれる。
【0079】
本発明のタンパク質もしくはポリペプチドの相同体は、変異導入、たとえばタンパク質の点変異、延長もしくは短縮によって、作製することができる。
【0080】
本発明のタンパク質の相同体は、たとえばトランケート型変異体などの、変異体のコンビナトリアルライブラリーをスクリーニングすることによって同定することができる。たとえば合成オリゴヌクレオチド混合物の酵素的ライゲーションによって、核酸レベルでコンビナトリアル変異導入を行うことにより、多様なタンパク質バリアントのライブラリーを作製することができる。可能性のある相同体のライブラリーを縮重オリゴヌクレオチド配列から作製するために使用可能な、数多くの方法がある。縮重遺伝子配列の化学合成は、自動DNA合成装置で行い、その合成遺伝子を次に適当な発現ベクター内にライゲーションすることができる。遺伝子の縮重セットを使用することによって、可能性のあるタンパク質配列の望ましいセットをコードする全配列を1つの混合物として提供することができるようになる。縮重オリゴヌクレオチドの合成方法は当業者に知られている(たとえば、Narang, S.A. (1983) Tetrahedron 39:3; Itakura et al. (1984) Annu. Rev. Biochem. 53:323; Itakura et al., (1984) Science 198:1056; Ike et al. (1983) Nucleic Acids Res. 11:477)。
【0081】
点変異または短縮によって作成されたコンビナトリアルライブラリーの遺伝子産物をスクリーニングするための技法、ならびに特定の性質を有する遺伝子産物を目的としてcDNAライブラリーをスクリーニングするための技法は、先行技術でいくつか知られている。これらの技法は、本発明の相同体のコンビナトリアル変異導入によって作製された遺伝子ライブラリーの迅速スクリーニングに適合させることができる。大規模な遺伝子ライブラリーをスクリーニングするために最もよく使用される技法は、ハイスループット解析の根幹をなすものであって、その技法には、遺伝子ライブラリーを複製可能な発現ベクターにクローニングすること、その結果得られたベクターライブラリーを用いて適当な細胞を形質転換すること、ならびに、望ましい活性の検出により、遺伝子産物が検出された遺伝子をコードしているベクターを容易に単離できるような条件下で、コンビナトリアル遺伝子を発現させることが含まれる。リカーシブアンサンブル変異導入(recursive ensemble mutagenesis: REM)はライブラリー中の機能的変異体の頻度を高める技法であるが、この技法をスクリーニングテストと組み合わせて使用し、ホモログを同定することができる(Arkin and Yourvan (1992) PNAS 89:7811-7815; Delgrave et al. (1993) Protein Engineering 6(3):327-331)。
【0082】
3.2 核酸および構築物
3.2.1 核酸
本発明はまた、フェニルエタノールデヒドロゲナーゼ活性を有する酵素をコードする核酸配列を提供する。
【0083】
本発明はまた、本明細書に記載の具体的配列に対してある程度の同一性を示す核酸配列に関する。
【0084】
2つの核酸配列間の「同一性」は、個々の核酸の全長にわたるヌクレオチドの同一性、特に、Clustal法(Higgins DG, Sharp PM. Fast and sensitive multiple sequence alignments on a microcomputer. Comput Appl. Biosci. 1989 Apr; 5(2):151-1)を使用するInformax(USA)社製Vector NTI Suite 7.1ソフトウェアを用いて、下記のパラメーターを設定して比較することにより計算される同一性を意味すると理解される:
マルチプルアラインメントパラメーター:
ギャップ開始ペナルティ(Gap opening penalty) 10
ギャップ伸長ペナルティ(Gap extension penalty) 10
ギャップセパレーションペナルティ(Gap separation penalty range) 8
ギャップセパレーションペナルティ(Gap separation penalty) off
アラインメント遅延に関する%同一性(% identity for alignment delay) 40
残基特異的ギャップ(Residue specific gaps) off
親水性残基ギャップ(Hydrophilic residue gap) off
転移計量(Transition weighing) 0
ペアワイズアラインメントパラメーター:
FASTアルゴリズム on
K-タプルサイズ(K-tuple size) 1
ギャップペナルティ(Gap penalty) 3
ウィンドウサイズ(Window size) 5
Number of best diagonals 5
あるいはまた、同一性は、Chenna, Ramu, Sugawara, Hideaki, Koike,Tadashi, Lopez, Rodrigo, Gibson, Toby J, Higgins, Desmond G, Thompson, Julie D. Multiple sequence alignment with the Clustal series of programs. (2003) Nucleic Acids Res 31 (13):3497-500にしたがって、インターネットアドレスhttp://www.ebi.ac.uk/Tools/clustalw/index.html#によって、下記のパラメーターを用いて決定することもできる:
DNAギャップオープンペナルティ(DNA Gap Open Penalty) 15.0
DNAギャップエクステンションペナルティ(DNA Gap Extension Penalty) 6.66
DNAマトリックス(DNA Matrix) Identity
タンパク質ギャップオープンペナルティ(Protein Gap Open Penalty) 10.0
タンパク質ギャップエクステンションペナルティ(Protein Gap Extension Penalty)0.2
タンパク質マトリックス(Protein matrix) Gonnet
タンパク質/DNA ENDGAP(Protein/DNA ENDGAP) -1
タンパク質/DNA GAPDIST(Protein/DNA GAPDIST) 4
【0085】
本明細書に記載の核酸配列(一本鎖および二本鎖DNAおよびRNA配列、たとえばcDNAおよびmRNA)はすべて、それ自体公知の方法で、構成単位のヌクレオチドから化学合成によって、たとえば、二重らせんの構成単位である個々の重複する相補的核酸のフラグメント縮合によって、作製することができる。オリゴヌクレオチドの化学合成は、たとえば既知の方法で、ホスホアミダイト法(Voet, Voet, 2nd edition, Wiley Press New York, pages 896-897)にしたがって実施することができる。DNAポリメラーゼのクレノウ断片およびライゲーション反応により、合成オリゴヌクレオチドを付加しギャップを埋めること、ならびに一般的なクローニング法は、Sambrook et al. (1989), Molecular Cloning: A laboratory manual, Cold Spring Harbor Laboratory Pressに記載されている。
【0086】
本発明はまた、上記ポリペプチドおよびそれらの機能的同等物の1つをコードする核酸配列(一本鎖および二本鎖DNAおよびRNA配列、たとえばcDNAおよびmRNA)を提供するが、これらはたとえば人工ヌクレオチドアナログを用いて得られる。
【0087】
本発明は、本発明のポリペプチドもしくはタンパク質または生物学的活性を有するそのセグメントをコードする単離された核酸分子、ならびに、たとえば本発明のコード核酸の同定もしくは増幅のためにハイブリダイゼーションプローブもしくはプライマーとして使用することができる、核酸断片も提供する。
【0088】
さらに、本発明の核酸分子は、遺伝子のコード領域の3'-および/または5'-末端の非翻訳配列を含んでいてもよい。
【0089】
本発明にはさらに、具体的に記載されたヌクレオチド配列、またはそのセグメントに相補的な核酸分子が含まれる。
【0090】
本発明のヌクレオチド配列は、他の細胞型および生物において相同配列を同定および/またはクローニングするために使用することができる、プローブおよびプライマーの作製を可能にする。こうしたプローブもしくはプライマーは、通常、「ストリンジェントな」条件(下記参照)下で、本発明の核酸配列のセンス鎖の、または対応するアンチセンス鎖の、少なくとも約12、好ましくは少なくとも約25、たとえば約40、50または75個の連続したヌクレオチドとハイブリダイズするヌクレオチド配列領域を含有する。
【0091】
「単離された」核酸分子は、その核酸の天然起源の中に存在する他の核酸分子から分離されているが、その上、それが組換え技術によって作製されたならば、他の細胞物質もしくは培養培地を基本的に含んでおらず、化学合成されたならば化学的前駆体を含んでいないといえる。
【0092】
本発明の核酸は、分子生物学の標準的な技法、ならびに本発明により与えられる配列情報を用いて、単離することができる。たとえば、cDNAは、具体的に記載された完全な配列の1つもしくはそのセグメントをハイブリダイゼーションプローブとして使用し、標準的なハイブリダイゼーション法(たとえば、Sambrook, J., Fritsch, E.F. and Maniatis, T. Molecular Cloning: A Laboratory Manual. 2nd edition, Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989に記載)を用いることによって、適当なcDNAライブラリーから単離できる。さらに、開示されている配列の1つもしくはそのセグメントを含有する核酸分子を、この配列に基づいて作製されたオリゴヌクレオチドプライマーを用いてポリメラーゼ連鎖反応によって単離することができる。このようにして増幅された核酸を適当なベクターにクローニングして、DNA配列分析により特徴付けることができる。本発明のオリゴヌクレオチドは、たとえば自動DNA合成装置を用いて、標準的な合成法によっても調製することができる。
【0093】
本発明の核酸配列、またはその誘導体、前記配列のホモログもしくは一部は、たとえば、通常のハイブリダイゼーション法またはPCR技術を用いて、他の細菌から、たとえばゲノムライブラリーもしくはcDNAライブラリーを経て、単離することができる。これらのDNA配列は、標準条件下で本発明の配列とハイブリダイズする。
【0094】
「ハイブリダイゼーション」は、ポリヌクレオチドもしくはオリゴヌクレオチドが標準的な条件下で実質的に相補的な配列と結合する能力を意味すると理解されるが、この条件下では相補的でないパートナーとの間の非特異的結合は起こらない。このため、その配列は90-100%相補的であるといえる。互いに特異的に結合することができる相補配列の特性によって、相補配列は、たとえばノーザンブロットもしくはサザンブロット法において、またはPCRもしくはRT-PCRにおけるプライマー結合のために、有用となる。
【0095】
ハイブリダイゼーションのために、保存された領域の、短いオリゴヌクレオチドを使用することが有利である。しかしながら、本発明の核酸の長めの断片、または完全な配列をハイブリダイゼーションに使用することもできる。上記の標準的な条件は、使用する核酸(オリゴヌクレオチド、より長い断片、または完全な配列)に応じて、またはDNAかRNAか、どちらのタイプの核酸をハイブリダイゼーションに使用するのか、に応じて、さまざまである。たとえば、DNA:DNAハイブリッドの融解温度は、同じ長さのDNA:RNAハイブリッドの融解温度より約10℃低い。
【0096】
標準的な条件は、たとえば核酸に応じて、42℃から58℃までの、0.1から5 x SSC(1 X SSC = 0.15 M NaCl、15 mMクエン酸ナトリウム、pH 7.2)の濃度で、またはさらに50%ホルムアミド存在下の、緩衝水溶液の温度を意味すると理解されるべきであって、たとえば、5 x SSC、50%ホルムアミドで42℃である。DNA:DNAハイブリッドのハイブリダイゼーション条件は0.1 x SSCで約20℃から45℃までの温度であれば有利であるが、好ましくは約30℃から45℃までである。DNA:RNAハイブリッドについては、ハイブリダイゼーション条件は0.1 x SSCで約30℃から55℃までの温度であれば有利であるが、好ましくは約45℃から55℃までである。上記のハイブリダイゼーション温度は、ホルムアミドの非存在下で、長さが約100ヌクレオチドでG + C含量が50%である核酸に関する、融解温度の計算値の例である。DNAハイブリダイゼーションの実験条件は、遺伝学に関するテキスト、たとえば、Sambrook et al., “Molecular Cloning”, Cold Spring Harbor Laboratory, 1989に記載されており、たとえば、核酸の長さ、ハイブリッドの種類またはG + C含量の関数のような、当業者に知られている式を用いて、計算することができる。当業者は、ハイブリダイゼーションに関するさらに他の情報を、下記のテキストに見いだすことができる:Ausubel et al. (eds), 1985, Current Protocols in Molecular Biology, John Wiley & Sons, New York; Hames and Higgins (eds), 1985, Nucleic Acids Hybridization: A Practical Approach, IRL Press at Oxford University Press, Oxford; Brown (ed), 1991, Essential Molecular Biology: A Practical Approach, IRL Press at Oxford University Press, Oxford。
【0097】
「ハイブリダイゼーション」は特に、ストリンジェントな条件下で行うことができる。そのような条件は、たとえば、Sambrook, J., Fritsch, E.F., Maniatis, T., in: Molecular Cloning (A Laboratory Manual), 2nd edition, Cold Spring Harbor Laboratory Press, 1989, pages 9.31-9.57 or in Current Protocols in Molecular Biology, John Wiley & Sons, N.Y. (1989), 6.3.1-6.3.6に記載されている。
【0098】
「ストリンジェントな」ハイブリダイゼーション条件は、具体的には下記を意味すると理解される:50%ホルムアミド、5 x SSC(750 mM NaCl、75 mMクエン酸三ナトリウム)、50 mMリン酸ナトリウム(pH 7.6)、5x Denhardt溶液、10%デキストラン硫酸、および20 g/mlの変性、断片化処理したサケ精子DNAからなる溶液中で一晩42℃にてインキュベーションした後、65℃にて0.1 x SSC によるフィルターの洗浄ステップが続く。
【0099】
本発明はまた、具体的に記載された、もしくは推論できる核酸配列の、誘導体を提供する。
【0100】
上述のように、本発明に合致する他の核酸配列を、たとえば、配列番号1または3から誘導することが可能であり、そうした核酸配列は1つもしくは複数のヌクレオチドの付加、置換、挿入または欠失により前記とは異なるが、依然として望ましい特性を有するポリペプチドをコードする可能性がある。
【0101】
天然に存在するバリアント、たとえばスプライシングバリアントもしくはアレルバリアントなどのように、具体的に記載された配列と比べて、いわゆるサイレント変異を有する核酸配列、または特別な起源もしくは宿主生物のコドン使用頻度に応じて変更された核酸配列も、本発明にしたがって含めるものとする。
【0102】
同様に、保存的なヌクレオチド置換により得られる配列も与えられる(すなわち、当該アミノ酸は、同じ荷電、サイズ、極性、および/または溶解性を持つアミノ酸で置換される)。
【0103】
本発明はまた、配列多型により具体的に記載された核酸に由来する分子を提供する。これらの遺伝子多型は、自然変異により集団内の個体間に存在する可能性がある。こうした自然変異は通常、遺伝子のヌクレオチド配列において1から5%までの変異をもたらす。
【0104】
配列番号1または3の配列を有する本発明の核酸配列の誘導体は、たとえば、それに由来するアミノ酸レベルで、その全配列領域にわたって、少なくとも60%相同性を有する、好ましくは少なくとも80%相同性、特に好ましくは少なくとも90%相同性を有するアレルバリアントを意味すると理解されるべきである(アミノ酸レベルの相同性に関しては、ポリペプチドに関する上記の記述を参照すべきである)。その配列の一部の領域について、有利に、相同性は、より高くてもよい。
【0105】
さらに、誘導体は、当然のことながら、本発明の核酸のホモログ、具体的には、配列番号1または3のホモログ、たとえば、コードおよび非コードDNA配列の、真菌もしくは細菌ホモログ、短縮された配列、一本鎖DNAもしくはRNAも指している。
【0106】
さらに、誘導体は当然のことながら、たとえばプロモーターとの融合物を意味する。プロモーターは、上記ヌクレオチド配列の上流に連結されているが、そのプロモーターの機能および/または有効性を損なうことなしに、少なくとも1つのヌクレオチドの置換、少なくとも1つの挿入、逆位および/または欠失によりこれを改変することができる。その上、プロモーターの配列を改変することによってその有効性を高めることができるが、別種の生物に由来する、より有効なプロモーターによって完全に置き換えることもできる。
【0107】
3.2.2 機能的変異体の作製
さらに、本発明の酵素の機能的変異体を作製する方法が、当業者に知られている。
【0108】
使用する技術に応じて、当業者は、完全にランダムな変異、あるいはより標的化された変異を、遺伝子あるいは非コード核酸領域(これは、たとえば発現の調節のために重要である)内に挿入して、遺伝子ライブラリーを作製することができる。このために必要な分子生物学的方法は、当業者に知られており、たとえば、Sambrook and Russell, Molecular Cloning. 3rd edition, Cold Spring Harbor Laboratory Press 2001に記載されている。
【0109】
遺伝子を改変し、それによってその遺伝子によりコードされるタンパク質を改変する方法は、これまで長く当業者に周知されてきており、たとえば、以下のものがある:
- 部位特異的変異誘発、この方法では、遺伝子の1つもしくは複数のヌクレオチドを標的化された方法で置き換える(Trower MK (ed.) 1996; In vitro mutagenesis protocols. Humana Press, New Jersey);
- 飽和変異誘発、この方法では、遺伝子の任意の望ましい位置で、任意の望ましいアミノ酸に対するコドンを置換または付加することができる(Kegler-Ebo DM, Docktor CM, DiMaio D (1994) Nucleic Acids Res 22:1593; Barettino D, Feigenbutz M, Valcarel R, Stunnenberg HG (1994) Nucleic Acids Res 22:541; Barik S (1995) Mol Biotechnol 3:1);
- エラープローンポリメラーゼ連鎖反応(エラープローンPCR)、この方法では、不完全なDNAポリメラーゼによりヌクレオチド配列を変異させる(Eckert KA, Kunkel TA (1990) Nucleic Acids Res 18:3739);
- SeSaM法(配列飽和法)、この方法では、好ましい置換がポリメラーゼによって妨げられる(Schenk et al., Biospektrum, Vol. 3, 2006, 277-279);
- ミューテーター株への遺伝子の挿入、この場合、たとえば不完全なDNA修復メカニズムのために、ヌクレオチド配列の変異率の増加が生じる(Greener A, Callahan M, Jerpseth B (1996) An efficient random mutagenesis technique using an E.coli mutator strain. In: Trower MK (ed.) In vitro mutagenesis protocols. Humana Press, New Jersey);または
- DNAシャフリング、この方法では、近縁遺伝子のプールを作製して消化し、その断片をポリメラーゼ連鎖反応のテンプレートとして使用するが、鎖分離と再アニーリングを繰り返すことによって、最終的に全長のモザイク遺伝子が生成する(Stemmer WPC (1994) Nature 370:389; Stemmer WPC (1994) Proc Natl Acad Sci USA 91:10747)。
【0110】
いわゆる人工分子進化法(特に、Reetz MT and Jaeger K-E (1999), Topics Curr Chem 200:31; Zhao H, Moore JC, Volkov AA, Arnold FH (1999), Methods for optimizing industrial enzymes by directed evolution, in: Demain AL, Davies JE (ed.) Manual of industrial microbiology and biotechnology. American Society for Microbiologyに記載)を用いて、当業者は、標的化されたやり方で、しかも工業規模で機能的変異体を作製することができる。ここで、最初のステップにおいて、はじめに特定のタンパク質の遺伝子ライブラリーを作製するが、そのために上記の方法を使用することができる。その遺伝子ライブラリーを適当な方法で、たとえば、細菌によって、またはファージディスプレイシステムによって発現させる。
【0111】
望ましい特性におおむね合致する性質を有する機能的変異体を発現する、宿主生物の当該遺伝子に、さらに一連の変異を受けさせることができる。今ある機能的変異体が十分に望ましい性質を示すまで、変異および選択またはスクリーニングのステップを反復して繰り返すことができる。この反復法の結果として、限られた数の変異、たとえば1から5回の変異に段階的に取り組むことができ、問題の酵素活性への影響を評価して選択することができる。選択された変異体を次に、同様の方法で次の変異ステップに供することができる。結果として、検討すべき個々の変異体の数を、かなり少なくすることができる。
【0112】
配列番号2の酵素のエラープローン変異導入により得ることができた本発明の変異体の限定的でない例を、以下の表1にまとめる。
【表1】
【0113】
【0114】
【0115】
本発明の結果は、望ましい、改良された特性を有する酵素を標的化された方法でさらに作製するために必要とされる、当該酵素の構造および配列に関する重要な情報も与える。特に、いわゆる「ホットスポット」、すなわち標的変異を挿入することによって酵素の性質を改変するのに適している可能性のある配列セグメントを決定することができる。
【0116】
本発明の酵素の、このようなホットスポット領域の限定的でない例を、配列2に基づいて、以下にまとめる:
(1) 142〜153(ループ2)および
(2) 190〜211(ヘリックスαFG1)
(3) 93〜96(ループ1)
(4) 241〜249(C末端)
(5) 138〜141(結合ポケットの親水性領域)ならびに
(6) Cys61および/またはCys 83。
【0117】
同様に、おそらく酵素活性にほとんど影響を及ぼさないはずであって、潜在的な「サイレント変異」と呼ぶことができる、変異を実行することができる領域内の、アミノ酸配列位置に関する情報を引き出すことができる。こうした変異の位置は、配列番号2に関して下記の表2にまとめる。
【表2】
【0118】
3.2.3 構築物
さらに、本発明は、本発明のポリペプチドをコードする核酸配列を調節核酸配列の遺伝的制御下に含有する発現構築物;ならびにこれらの発現構築物の少なくとも1つを含有するベクターを提供する。
【0119】
本発明によれば、「発現ユニット」は、当然のことながら発現活性のある核酸を意味し、こうした核酸には本明細書で定義されたプロモーターが含まれるが、こうした核酸は、発現されるべき核酸もしくは遺伝子に機能的に連結された後、この核酸またはこの遺伝子の発現を調節し、したがって転写および翻訳を調節するものである。したがって、これに関連して、「調節核酸配列」という表現も用いられる。プロモーターに加えて、たとえばエンハンサーのような調節エレメントをさらに含んでいてもよい。
【0120】
本発明によれば、「発現カセット」または「発現構築物」は、当然のことながら、発現されるべき核酸または発現されるべき遺伝子に機能的に連結されている発現ユニットを意味する。したがって発現カセットは、発現ユニットとは違って、転写および翻訳を調節する核酸配列だけでなく、転写および翻訳の結果タンパク質として発現されるべき核酸配列も含んでいる。
【0121】
本発明の文脈において、「発現」または「過剰発現」という用語は、対応するDNAによってコードされる1つもしくは複数の酵素の、微生物における産生、またはその酵素の、微生物における細胞内活性の増加を言い表すものである。これを目的として、たとえば、遺伝子を生物に挿入すること、存在する遺伝子を別の遺伝子に置き換えること、1つもしくは複数の遺伝子のコピー数を増やすこと、強力なプロモーターを使用すること、または高活性を有する該当酵素をコードする遺伝子を使用することが可能であって、さらに、これらの手段を組み合わせることも選択できる。
【0122】
好ましくは、本発明のこうした構築物は、特定のコード配列の5’上流にプロモーターを、3’下流に転写終結配列を有し、必要に応じてさらに、通常の調節エレメントも含んでいるが、これらはいずれの場合も、機能しうるようにコード配列に連結されている。
【0123】
本発明によれば、「プロモーター」、「プロモーター活性を有する核酸」または「プロモーター配列」は、転写されるべき核酸に機能的に連結された状態で、この核酸の転写を調節する核酸を意味していると理解される。
【0124】
これに関連して、「機能的な」または「機能しうる」連結とは、たとえば、プロモーター活性を有する核酸の1つ、および転写されるべき核酸配列、ならびに必要に応じてさらに調節エレメント、たとえば核酸の転写を確実にする核酸配列、およびたとえばターミネーターなどが、核酸配列の転写時にそれぞれの調節エレメントがその機能を果たすことができるように、順に配列されていることを意味していると理解される。このために、化学的な意味での直接的な連結が必ずしも必要というわけではない。遺伝子制御配列、たとえばエンハンサー配列は、遠く離れた場所からも、あるいは他のDNA分子からでも、標的配列に対して機能を発揮することができる。転写されるべき核酸配列がプロモーター配列の後ろに(すなわち3’側に)ある配列配置が好ましく、2つの配列は共有結合で連結される。ここで、プロモーターと、遺伝子組み換えにより発現されるべき核酸配列との間隔は、200塩基対未満、または100塩基対未満、または50塩基対未満とすることができる。
【0125】
プロモーターおよびターミネーターに加えて、他の調節配列の例には、ターゲティング配列、エンハンサー、ポリアデニル化シグナル、選択可能なマーカー、増幅シグナル、複製開始点などがある。適当な調節配列は、たとえば、Goeddel, Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, CA (1990)に記載されている。
【0126】
本発明の核酸構築物には、特に、配列番号1または3の配列、またはその誘導体およびホモログ、ならびにそれらから得られる核酸配列であって、遺伝子発現を制御し、たとえば増加させるために有利となるように、1つもしくは複数の調節シグナルに、機能しうるように、もしくは機能的に連結されている前記核酸配列が含まれる。
【0127】
これらの調節配列のほかに、上記配列の天然の調節が、実際の構造遺伝子の上流に依然として残っている可能性があり、必要に応じて、天然の調節をスイッチオフして遺伝子の発現が増加するように、遺伝的に改変してもよかった。しかしながら、核酸構築物は、構造的によりシンプルにしてもよく、すなわち、追加の調節シグナルは、コード配列の上流に挿入されず、天然プロモーターは、その調節とともに、除去されなかった。その代わり、天然の調節配列は、もはや調節がなくなって遺伝子の発現が増加するように変異している。
【0128】
好ましい核酸構築物には、有利なことに、1つもしくは複数の前記「エンハンサー」配列がプロモーターに機能的に連結されて含まれており、この配列は核酸配列の発現増加を可能にする。追加の有利な配列、たとえば他の調節エレメントまたはターミネーターも、DNA配列の3’末端に挿入することができる。本発明の核酸は、構築物の1つもしくは複数のコピー中に存在することが考えられる。この構築物は、抗生物質耐性または栄養要求性を補完する遺伝子といったマーカーを、構築物の選択のために必要に応じて含んでいてもよい。
【0129】
適当な調節配列の例は、cos、tac、trp、tet、trp-tet、lpp、lac、lpp-lac、lacIq-、T7、T5、T3、gal、trc、ara、rhaP (rhaPBAD)SP6、lambda-PRなどのプロモーター、またはLambda-PLプロモーター中にあるが、これらはグラム陰性細菌において有利に使用される。さらに有利な調節配列が、たとえば、グラム陽性プロモーターamyおよびSPO2、酵母もしくは真菌プロモーターADC1、MFα、AC、P-60、CYC1、GAPDH、TEF、rp28、ADHの中にある。調節のために人工プロモーターを使用することも可能である。
【0130】
宿主生物での発現のために、宿主において遺伝子の最適な発現を可能にするベクター、たとえばプラスミドもしくはファージに核酸構築物を挿入するのは有利である。プラスミドおよびファージと同様にベクターは、当然のことながら、当業者に知られている任意の他のベクター、たとえば、SV40、CMV、バキュロウイルスおよびアデノウイルスなどのウイルス、トランスポゾン、ISエレメント、ファスミド(phasmid)、コスミド、および直鎖もしくは環状DNAを意味している。これらのベクターは、宿主生物において自律的に複製可能であり、または染色体性に複製することができる。これらのベクターは本発明の他の実施形態を構成する。
【0131】
適当なプラスミドはたとえば、大腸菌(E. coli)では、pLG338、pACYC184、pBR322、pUC18、pUC19、pKC30、pRep4、pHS1、pKK223-3、pDHE19.2、pHS2、pPLc236、pMBL24、pLG200、pUR290、pIN-III113-B1、λgt11もしくはpBdCI、ストレプトミセスでは、pIJ101、pIJ364、pIJ702もしくはpIJ361、バチルスでは、pUB110、pC194もしくはpBD214、コリネバクテリウムでは、pSA77もしくはpAJ667、真菌では、pALS1、pIL2もしくはpBB116、酵母では、2alphaM、pAG-1、YEp6、YEp13もしくはpEMBLYe23、または植物では、pLGV23、pGHlac+、pBIN19、pAK2004もしくはpDH51である。特定されたプラスミドは、可能性のあるプラスミドのうち小数選抜されたものである。さらに他のプラスミドが当業者によく知られており、たとえば、書籍Cloning Vectors (Eds. Pouwels P. H. et al. Elsevier, Amsterdam-New York-Oxford, 1985, ISBN 0 444 904018) に見いだすことができる。
【0132】
本発明の他の実施形態において、本発明の核酸構築物、または本発明の核酸を含有するベクターは、直鎖DNAの形で有利に微生物に導入することも、非相同もしくは相同組換えにより宿主生物のゲノムに組み込むことも有利に行うことができる。この直鎖DNAは、プラスミドのように直鎖状にしたベクターからなることもあるが、本発明の核酸構築物または核酸だけからなることもある。
【0133】
生物における異種遺伝子の最適な発現のために、その生物で使用される特異的な「コドン使用頻度」にしたがって核酸配列を改変することが有利である。「コドン使用頻度」は、当該生物に由来する他の既知遺伝子のコンピューター解析によって、容易に決定することができる。
【0134】
本発明の発現カセットは、適当なプロモーターを、適当なコード配列、および転写終結シグナルもしくはポリアデニル化シグナルに融合させることによって調製される。このために、一般的な組換えおよびクローニング技術が使用されるが、それはたとえば、T. Maniatis, E.F. Fritsch and J. Sambrook, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY (1989)、およびT.J. Silhavy, M.L. Berman and L.W. Enquist, Experiments with Gene Fusions, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY (1984)に、さらにAusubel, F.M. et al., Current Protocols in Molecular Biology, Greene Publishing Assoc. and Wiley Interscience (1987)に記載されている。
【0135】
適当な宿主生物における発現のために、組換え核酸構築物もしくは遺伝子構築物は、宿主特異的ベクターに挿入されることが有利であり、このベクターは宿主において遺伝子の最適な発現を可能にするものである。ベクターは当業者に周知であり、たとえば、“Cloning Vectors” (Pouwels P. H. et al., Ed., Elsevier, Amsterdam-New York-Oxford, 1985) に見いだすことができる。
【0136】
3.3 微生物
文脈に応じて、「微生物」という用語は、野生型の微生物もしくは遺伝的に改変された組換え微生物、またはその両者を意味すると理解することができる。
【0137】
本発明のベクターを用いることによって、たとえば少なくとも1つの本発明のベクターで形質転換された、本発明のポリペプチドを産生するために使用することができる組換え微生物を調製することができる。上記の本発明の組換え構築物を適当な宿主系に導入し発現させるのは好都合である。ここで、当業者に知られている一般的なクローニングおよびトランスフェクション法、たとえば、共沈殿、プロトプラスト融合、エレクトロポレーション、レトロウイルストランスフェクションなどを、特定の発現系で前記核酸を発現させるために使用することが好ましい。適当な系は、たとえば、Current Protocols in Molecular Biology, F. Ausubel et al., Ed., Wiley Interscience, New York 1997, or Sambrook et al. Molecular Cloning: A Laboratory Manual. 2nd edition., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989に記載されている。
【0138】
本発明の核酸もしくは核酸構築物に適した組換え宿主生物は、基本的にすべての原核または真核生物である。使用する宿主生物が細菌、真菌もしくは酵母などの微生物であれば好都合である。グラム陽性もしくはグラム陰性細菌、好ましくは、腸内細菌科(Enterobacteriaceae)、シュードモナス科(Pseudomonadaceae)、リゾビウム科(Rhizobiaceae)、ストレプトミセス科(Streptomycetaceae)またはノカルジア科(Nocardiaceae)に属する細菌、特に好ましくは、エシェリキア属(Escherichia)、シュードモナス属(Pseudomonas)、ストレプトミセス属(Streptomyces)、ノカルジア属(Nocardia)、バークホルデリア属(Burkholderia)、サルモネラ属(Salmonella)、アグロバクテリウム属(Agrobacterium)、クロストリジウム属(Clostridium)またはロドコッカス属(Rhodococcus)の細菌を使用すれば有利である。中でも特に好ましいのは、大腸菌Escherichia属およびEscherichia coli種である。その上、さらに有利な細菌を、αプロテオバクテリア、βプロテオバクテリアもしくはγプロテオバクテリア群に見いだすことができる。
【0139】
これに関連して、本発明の1つもしくは複数の宿主生物は、好ましくは、本発明に記載の上記定義にしたがってフェニルエタノールデヒドロゲナーゼ活性を有する酵素をコードする、少なくとも1つの核酸配列、核酸構築物またはベクターを含有する。
【0140】
本発明の方法で使用される生物は、宿主生物に応じて、当業者に知られている方法で増殖または培養する。一般に、微生物は液体培地中で、0℃から100℃まで、好ましくは10℃から60℃までの間の温度で、酸素ガスを供給して増殖させるが、この培地は炭素源を主として糖の形で、窒素源を、主に酵母エキスなどの有機窒素源または硫酸アンモニウムなどの塩の形で含有し、鉄、マンガンおよびマグネシウム塩などの微量元素、および必要に応じてビタミン類を含有する。ここで、栄養液のpHは、培養の間、一定値に維持すること、すなわち調節することができるが、調節されなくてもよい。培養は、バッチ式、半バッチ式、または連続式とすることができる。栄養物は、発酵培養開始時に最初に導入することができるが、半連続的または連続的に供給することもできる。
【0141】
3.4 本発明の酵素の組換え調製
本発明はさらに、本発明のポリペプチド、もしくは機能的、生物学的に活性のあるその断片を組換え調製する方法を提供するが、その方法はポリペプチド産生微生物を培養すること、必要に応じてポリペプチドの発現を誘導すること、ならびに培養物からポリペプチドを単離することを含んでいる。ポリペプチドは、必要ならば、このようにして工業規模で製造することもできる。
【0142】
本発明にしたがって調製された微生物は、連続培養することができるが、バッチ法で(バッチ培養)、または供給バッチ法(フィード法)もしくは反復供給バッチ法(反復フィード法)で不連続に培養することもできる。既知の培養法に関する概要は、Chmiel (Bioprozeβtechnik 1. Einfuehrung in die Bioverfahrenstechnik [Bioprocessing technology 1. Introduction to bioprocessing technology] (Gustav Fischer Verlag, Stuttgart, 1991))のテキスト、またはStorhas (Bioreaktoren und periphere Einrichtungen [Bioreactors and peripheral devices] (Vieweg Verlag, Braunschweig/Wiesbaden, 1994))のテキストに見いだすことができる。
【0143】
使用される培地は、個別の菌株の要求を適切に満足させなければならない。種々の微生物の培地に関する記述は、"Manual of Methods for General Bacteriology" from the American Society for Bacteriology (Washington D. C., USA, 1981) のハンドブックに見いだすことができる。
【0144】
本発明にしたがって使用することができるこれらの培地は、通常、1つもしくは複数の炭素源、窒素源、無機塩、ビタミン類および/または微量元素を含有する。
【0145】
好ましい炭素源は、糖類、たとえば、単糖、二糖もしくは多糖類である。非常に優れた炭素源は、たとえば、グルコース、フルクトース、マンノース、ガラクトース、リボース、ソルボース、リブロース、ラクトース、マルトース、スクロース、ラフィノース、デンプンまたはセルロースである。モラセスもしくは他の精糖副産物などの複雑な化合物を用いて、培地に糖類を加えることも可能である。異なる炭素源の混合物を添加することも有利であるかもしれない。他に考えられる炭素源は、油脂類、たとえば、大豆油、ヒマワリ油、ピーナッツ油およびココナツ油;脂肪酸、たとえば、パルミチン酸、ステアリン酸もしくはリノール酸;アルコール類、たとえばグリセロール、メタノールもしくはエタノール;ならびに有機酸、たとえば、酢酸もしくは乳酸である。
【0146】
窒素源は通常、有機もしくは無機窒素化合物、またはそうした化合物を含む材料である。窒素源の例には、アンモニアガス、またはアンモニウム塩、たとえば硫酸アンモニウム、塩化アンモニウム、リン酸アンモニウム、炭酸アンモニウムもしくは硝酸アンモニウム、硝酸塩、尿素、アミノ酸、または複雑な窒素源、たとえば、コーンスティープリカー、大豆粉、大豆タンパク質、酵母エキス、肉エキスなどがある。窒素源は、単独で、または混合物として使用することができる。
【0147】
培地中に含まれる可能性のある無機塩化合物としては、カルシウム、マグネシウム、ナトリウム、コバルト、モリブデン、カリウム、マンガン、亜鉛、銅および鉄の、塩化物塩、リン塩または硫酸塩がある。
【0148】
使用できるイオウ源は、無機イオウ含有化合物、たとえば、硫酸塩、亜硫酸塩、亜ジチオン酸塩、テトラチオン酸塩、チオ硫酸塩、硫化物であるが、有機イオウ化合物、たとえば、メルカプタン類およびチオール類もある。
【0149】
使用できるリン源は、リン酸、リン酸二水素カリウムもしくはリン酸水素二カリウム、または対応するナトリウム含有塩である。
【0150】
金属イオンを溶液中に維持するために、培地にキレート剤を添加することができる。特に適当なキレート剤には、ジヒドロキシフェノール類、たとえば、カテコールもしくはプロトカテク酸、またはクエン酸などの有機酸がある。
【0151】
本発明にしたがって使用される発酵培地は通常、他の成長因子、たとえば、ビタミン類もしくは成長促進物質を含有するが、これにはたとえば、ビオチン、リボフラビン、チアミン、葉酸、ニコチン酸、パントテン酸塩、およびピリドキシンがある。成長因子および塩類は複雑な培地成分、たとえば酵母エキス、モラセス、コーンスティープリカーなどに由来することも多い。さらに、適当な前駆体を培地に加えてもよい。培地化合物の正確な組成は、個別の実験に大きく左右され、具体的な場合ごとに個々に決定される。培地の最適化に関する情報は、"Applied Microbiol. Physiology, A Practical Approach" (ed. P.M. Rhodes, P.F. Stanbury, IRL Press (1997) p. 53-73, ISBN 0 19 963577 3) のテキストから入手することができる。増殖培地は、Standard 1 (Merck)またはBHI (Brain heart infusion, DIFCO)などのような供給業者から入手することもできる。
【0152】
すべての培地成分は、加熱(1.5バールで121℃にて20分)または無菌濾過によって、滅菌される。成分は合わせて滅菌してもよいが、必要ならば別々に滅菌することもできる。すべての培地成分が培養の開始時点で存在していてもよいが、適宜、連続的に、またはバッチ式に添加してもよい。
【0153】
培養温度は通常、15℃から45℃まで、好ましくは25℃から40℃までの間であって、実験の間、一定に保つことも、変化させることもできる。培地のpHは、5から8.5までの範囲、好ましくは7.0程度とすべきである。培養のためのpHは、培養中、塩基性化合物、たとえば、水酸化ナトリウム、水酸化カリウム、アンモニアもしくはアンモニア水、または酸性化合物、たとえば、リン酸もしくは硫酸の添加により制御することができる。発泡を抑えるために、消泡剤、たとえば、脂肪酸ポリグリコールエステルを使用することができる。プラスミドの安定性を維持するために、適当な選択物質、たとえば、抗生物質を培地に添加することができる。好気的条件を維持するために、酸素もしくは酸素含有気体混合物、たとえば外気が培養に導入される。培養温度は通常20℃から45℃までである。培養は、望ましい産物が最大限生成するまで続けられる。この目的は通常、10時間から160時間までの間に達成される。
【0154】
その後、発酵液はさらに処理される。必要な条件に応じて、たとえば、遠心分離、濾過、デカンテーション、もしくはこれらの方法を組み合わせた分離法によって、発酵液から生物体量を全部または一部除去することができるが、生物体量が前記液体中にすべて残されていてもよい。
【0155】
ポリペプチドが培地中に分泌されないならば、細胞を破壊し、その溶解物から、既知のタンパク質単離法により産物を得ることができる。細胞は適宜、超音波によって、高圧によって(たとえば、フレンチプレス圧力セル内で)、浸透圧溶解によって、界面活性剤、溶解酵素もしくは有機溶媒の作用によって、ホモジナイザーによって、または上記の方法のいくつかを組み合わせて、破壊することができる。
【0156】
ポリペプチドの精製は、既知のクロマトグラフィー法、たとえば、Q-Sepharoseクロマトグラフィーなどの分子篩クロマトグラフィー(ゲル濾過)、イオン交換クロマトグラフィーおよび疎水性クロマトグラフィーを用いて、ならびに他の常法、たとえば限外濾過、結晶化、塩析、透析およびネイティブゲル電気泳動によっても、達成することができる。適当な方法はたとえば、Cooper, T.G., Biochemische Arbeitsmethoden [Biochemical procedures], Verlag Walter de Gruyter, Berlin, New York or in Scopes, R., Protein Purification, Springer Verlag, New York, Heidelberg, Berlinに記載されている。
【0157】
組換えタンパク質を単離するために、そのcDNAをあるヌクレオチド配列だけ長く伸ばし、そうすることで、たとえばより簡単な精製に役立つ改変ポリペプチドもしくは融合タンパク質をコードする、ベクター系もしくはオリゴヌクレオチドを使用することは有利となるかもしれない。この種の適当な改変は、たとえば、アンカーとして機能するいわゆる「タグ」、たとえばヘキサヒスチジンアンカーとして知られる改変、または抗体によって抗原として認識されうるエピトープである(たとえば、Harlow, E. and Lane, D., 1988, Antibodies: A Laboratory Manual. Cold Spring Harbor (N.Y.) Pressに記載される)。こうしたアンカーは、タンパク質を、固体担体、たとえばポリマーマトリックスに固定するのに役立つ可能性があり、この担体はたとえば、クロマトグラフィーカラムの充填剤として、またはマイクロタイタープレートもしくは他の担体上で使用することができる。
【0158】
同時に、このようなアンカーは、タンパク質の認識のために使用することもできる。タンパク質を認識するために、通常のマーカー、たとえば、蛍光色素、基質との反応後に検出可能な生成物を形成する酵素マーカー、もしくは放射性マーカーを単独で使用することもできるが、タンパク質を誘導体化するためのアンカーと組み合わせて使用することもできる。
【0159】
本発明の変異体を発現させるために、WO2005/108590およびWO2006/094945の野生型酵素EbN1の発現、およびそのために使用することができる発現系に関する記述を参考にしてもよく、この記述は明確に引用される。
【0160】
3.5 酵素固定化
本発明の酵素は、遊離の状態で、または固定化された形で、本明細書に記載の方法に使用することができる。固定化酵素は、不活性担体に固定された酵素を意味すると理解される。適当な担体材料およびそれに固定化される酵素は、EP-A-1149849、EP-A-1 069 183およびDE A 100193773から、ならびにそれらに引用された元の文献から公知である。これに関連して、上記の明細書の記述を全体として参考とする。適当な担体材料には、たとえば、粘土、粘土鉱物、たとえばカオリナイト、珪藻土、パーライト、二酸化ケイ素、酸化アルミニウム、炭酸ナトリウム、炭酸カルシウム、セルロース粉末、アニオン交換体、合成ポリマー、たとえばポリスチレン、アクリル樹脂、フェノールホルムアルデヒド樹脂、ポリウレタンおよびポリオレフィン(ポリエチレンおよびポリプロピレンなど)がある。担体材料は、担体に結合した酵素を作製するために、通常、微粉化された粒子の形で使用されるが、多孔質の形状が好ましい。担体材料の粒径は、通常5 mm以下、特に2 mm以下である(粒度曲線)。同様に、デヒドロゲナーゼをホールセル触媒として使用する場合、遊離型もしくは固定化型を使用することができる。担体材料はたとえば、アルギン酸カルシウムおよびカラギーナンである。酵素は、細胞と同様に、グルタルアルデヒドを用いて直接架橋することができる(架橋してCLEAとなる)。対応する固定化法および他の固定化法が、たとえば、K. Drauz and H. Waldmann, Enzyme Catalysis in Organic Synthesis 2002, vol. III, 991-1032, Wiley-VCH, Weinheimの中でJ. Lalonde and A. Margolin “Immobilization of Enzymes” に記載されている。本発明の方法を実施するための、生体内変換およびバイオリアクターに関するさらに詳しい情報は、たとえば、Rehm et al (Ed) Biotechology, 2nd edition, vol. 3, chapter 17, VCH, Weinheimにも見いだすことができる。
【0161】
下記の限定的でない実施例を参照することによって、本発明をより詳細に説明することとする。
【0162】
(実施例)
【実施例1】
【0163】
飽和変異導入
1.1 分子モデリング
変異体は、酵素フェニルエタノールデヒドロゲナーゼEbN1の結晶構造(図2)を参考にして選択された。
【0164】
酵素の基質特異性は、2つのループ領域および1つのヘリックスによって決定される(図2のループ1および2、ならびにヘリックスαFG1)。ヘリックスαFG1はフレキシブルであって、基質結合後に活性中心を閉じる。ループ1のTyr93は基質結合ポケットを前側に向かって塞いでおり、それによって立体選択性に関与する。Tyr151はループ2に属し、結合ポケット内に向いている。Thr192はフレキシブルなヘリックスαFG1の一部であって、基質結合部位の方向に向いている。これらの2か所が選ばれたのは、基質の結合には影響を及ぼすが、触媒中心のアミノ酸および補酵素NAD、すなわち触媒メカニズムを妨害しないからである。
【0165】
1.2 飽和変異導入
初めに、Y151XおよびT192Xの位置で別々に飽和変異導入を行った(すなわちY151およびT192の位置を他のすべての19アミノ酸と置き換える、permutationとも呼ばれる)後、二重変異体(Y151A-T192X)を作製した。
【0166】
これは、いずれの場合も3回のポリメラーゼ連鎖反応で部位特異的変異導入を行うことによって実施された(図3のクローニングストラテジーを参照されたい)。ここで、DNAを増幅するために、以下のオリゴヌクレオチドを使用した:
ebn1H遺伝子断片を増幅するためのPCRを以下のように行った:100μlの反応混合物は、1μlテンプレート(約50 ngのベクターpDHE-ebn1H)、それぞれの場合について1μlオリゴヌクレオチド(20 ng)、2μl dNTPMix(Roche製、終濃度10 mM)、1μl Pfu-Ultra DNAポリメラーゼ(1 U/μl、Stratagene製)、10μl 10X Pfu-Ultraバッファー(Stratagene)、および80μl滅菌水を含有した。
【0167】
下記の温度プログラムをサーモサイクラー(Biometra)に設定した:95℃- 5分;30サイクル:95℃- 45秒、50℃- 45秒、72℃- 45秒;72℃- 10分; 10℃
1a) PCRオリゴヌクレオチド1および3 (T192Xについては6)
1b) PCRオリゴヌクレオチド2および4 (T192Xについては5)
2) PCR 1aおよびbから得られた産物をテンプレートとして、PCRオリゴヌクレオチド1および2(オーバーラップエクステンション)。
【0168】
上記から得られた増幅されたebn1H遺伝子を、GFXキット(GE Healthcare)を用いて1.2%アガロースゲルで精製した。
【0169】
増幅されたDNAを、制限酵素NdeIおよびHindIII(Fermentas)を用いて切断し、(同様にNdeI-HindIIIで切断された)ベクターpDHEの多重クローニング部位(MCS)にライゲートして、XL10ウルトラコンピテントセル(Stratagene)において形質転換した。こうした細胞の少量調製により、変異体のプラスミドDNAが得られた。ベクターpDHEは、DE 19848129もしくはWO2005/108590において、pDHE19.2ベクターとして記載されている。
【0170】
1.3 細胞の培養
初めに、ベクターpDHE-ebn1H-Y151XまたはpDHE-ebn1H-T192X(さらに後にpDHE-ebn1H-Y151A-T192X)を、菌株LU12037 (大腸菌派生株TG10 pAgro4 pHSG575 (TG10:大腸菌TG1に由来するRhaA- 派生株(Stratagene);pAgro4:Takeshita, S; M; M; Masahashi, W; T (1987) Gene 61, 63-74;pHSG575:T. Tomoyasu et al (2001), Mol. Microbiol. 40 (2))に形質転換したが、この菌株は、シャペロンGroEL/Sおよびlaclqリプレッサーを同時発現するものであって、Q-trayプレート上に蒔いて培養した。増殖したコロニーを、ピッキングロボット(Qpix)を用いて採取し、マイクロタイタープレート(MTP)内で抗生物質(100μMアンピシリン、20μMクロラムフェニコールおよび100μMスペクチノマイシン)を含有するCG前培養(Circular Growth, Gibco)に接種した。37℃にて200 rpmで5時間増殖させた後、細胞を、抗生物質(上記)および対応するインデューサー(ラムノース0.5 g/lおよびIPTG 0.1 mM)を含有するLB本培養に手作業で移した。16-18時間増殖後、細胞をテストに使用する。
【0171】
細胞を破壊するために、まずこれを遠心し、上清を取り除き、MTPに粘着フィルムを装着した。MTPを液体窒素中に約3秒間、完全に浸漬した後、再び実験台上において解凍した。室温での一時的解凍を伴う3回急速凍結で、もっともよくそろった結果が得られた。
【0172】
1.4 酵素阻害
TAC反応産物TACAまたは反応中に形成された二次的成分が、その反応を阻害することが判明している。基質は完全には変換されなかった。この反応は平衡反応であるにもかかわらず、たとえば4時間後に、細胞および/または基質が新たに添加されたが、それでそれ以上の反応は起こらなかった。さらに、結果として生じる2-ブタノンは、平衡をできる限り生成物側にシフトさせるために、蒸留により除去された。これらの方策にもかかわらず、完全な変換は達成されなかった。
【0173】
したがって、考えられるもっとも完全な変換を達成し、それによって容積時間当たりの高収率を達成するために、必ずしも活性は高くないが、より大量の生成物もしくは二次的成分に耐えられる変異体を見いだすことを目的とした。
【0174】
次のテストにおいて、TACAおよびTAもしくはTAAの両者を阻害剤としてテストした。このために、0.2 ml混合物(MTP)において、100 ml振盪フラスコでの培養から得られた細胞(LU11558; 過剰発現プラスミドとしてラムノース誘導性pDHE1650派生物を有する大腸菌TG10+ 菌株。シャペロンGroEL/Sおよびlaclqリプレッサーを同時発現する;野生型酵素EbN1が過剰発現される)50μl、1.75 mM NAD、および100 mM 2-ブタノールを80 mM TrisHClバッファーpH 8.0に添加した。いずれの場合も、10 mMのTA、TAAまたはTACAがそれに添加された。その後、光度計において340 nmで、NADHの生成を測定した。図4は、個々のVmax値(結果として生じた、時間当たりのNADH量)を示すことによって、フェニルエタノールデヒドロゲナーゼEbN1の阻害を説明するものである。対照(阻害剤なし)はブタノールを特徴とする。
【0175】
図4に見られるように、TACAはもっとも強い阻害を示す。TAはこの波長域に非常に強い吸収があるので、NADHの生成をここで検出することはできない。
【0176】
このため、他のアッセイでは阻害剤としてTACAを添加した。加えて、TACAは、以前行われた2-ブタノールおよびNADを用いた再生テストにも阻害剤として加えられた。適当なTACA濃度を決定するために、さまざまな濃度の細胞およびTACAをテストした。初めに、0から30 mMまでの一連の濃度、次に0から10 mMまでの一連の濃度を調製した。達成された結果(示さず)に基づいて、それぞれ25μlの細胞とともに10 mMのTACA濃度を他のテストで用いた。
【0177】
1.5 TACA添加および無添加での2-ブタノールテストの経過(補酵素の再生)
ここで、アミノ酸位置ごとのクローンで満たされた2つのマイクロタイタープレート(96ウェル)が選定された。マイクロプレートは完全に配列決定され、値は個々の変異に当てはめられた。
【0178】
細胞は上記のように培養した後、破壊し、最終的に100μlの水に再懸濁した。この細胞懸濁液25μlを新たなマイクロタイタープレートに入れ、水を足して容量100μlとした。次に基質溶液(終濃度:100 mM 2-ButOH、1.75 mM NAD、80 mM Tris-HCl pH 8.0、(10 mM TACA))を加え、NADHの生成を光度計で340 nmにて測定した。このテストから得られた結果を図5Aおよび5Bに示す。Vmax値を示す。
【0179】
図5Aおよび5Bから、変異体の大半がもはや補酵素を再生することができず、あるいは非常にゆっくりとしか再生できないことがわかる(ブタノールテスト、黒い棒グラフ)。しかしながら、ブタノールテストにTACAを添加することによって(淡色の棒グラフ)、これらの変異体は補酵素を再生することが可能であり、対照(野生型)よりずっと優れているといえる。おそらく、2-ブタノールが2-ブタノンになる代わりに、TACAがTACに酸化される。このような変異体は、野生型よりも、大量のTACAに耐えられる。
【0180】
抜け落ちている変異体(T192の位置についてN、D、Q、H、K、M、F、Y、ならびにY151の位置についてはC、F、S)が認められるが、これらは活性がない、または対照より劣っていた。次の変異体を、ラージスケールでテストするために、この実験から選択した:Y151A、E、G、H、ならびにT192A、G、L、I。
【0181】
1.6 陽性変異体の評価
このテストから出てくる陽性クローンを、次に、ラージスケールで調べた(100 ml振盪フラスコで培養)。ここでは、3つの異なるアッセイを行った:
【化11】
【0182】
それぞれのテストに関するテスト条件は:
テスト1)NADHを添加してTACからTACAへ還元
798.6μl 脱塩水
50μl 1M NaH2PO4 pH5
50μl NADH(100 mM原液(水溶液))
1.4μl TAC
100μl 振盪フラスコから得られた培養物の10倍濃縮粗抽出物
1000μl 最終容量
テスト2)完全な反応
730μl 脱塩水
50μl 1M NaH2PO4 pH5
20μl NAD 10 mM水溶液
100μl 100 mM TAC(1mlイソプロパノール中14μl)
100μl 振盪フラスコから得られた培養物の10倍濃縮粗抽出物
1000μl 最終容量
テスト3)TACAからTACへの酸化
798.6μl 脱塩水
50μl 1M NaH2PO4 pH5
50μl NAD(10 mM原液)
1.4μl TACA
100μl 振盪フラスコから得られた培養物の10倍濃縮粗抽出物
1000μl 最終容量
上記テストにおいて、テスト温度はいずれの場合も30℃とし、酵素濃度は0.1-10mg/mlとした。
【0183】
サンプルは濃塩酸で反応を止め、HPLCを用いて測定した。
【0184】
【0185】
【0186】
保持時間
TACA = 1.283分 (230 nm)
TAA = 0.910分 (230 nm)
TA = 1.168分 (260 nm)
TAC = 1.540分 (260 nm)
実験結果を次項にまとめる。
【0187】
1.6.1 変異体T192X
図6Aは、変異体T192LおよびT192Gが対照のLU11558よりも速やかにTACを減少させることを示す。しかしながら、完全な反応(図6B)を考えると野生型の方が活性が高く、それは他の変異体が同じように補酵素を再生することができないためである。
【0188】
図6Cでは、変異体T192Aが野生型より良好にTACAを酸化することができることが分かるが、生じるTAC濃度は非常に低いので、結果はテストの間ずっと変動する。
【0189】
1.6.2 変異体Y151A
図7Aにおいて、変異体Y151AおよびY151Hは補因子TACを、野生型(LU11558)より約4-5倍早く減少させることが分かる。しかしながら、完全な反応である、犠牲アルコール(ここでは2-プロパノール)を用いた補酵素再生を伴うTACからTACAへの還元を考えると、変異体Y151Aだけがなお活性がある(図7B)。全体を通しての活性は、対照の場合よりいくぶん低い。結合ポケットを大きくした結果として、「小さい」イソプロパノールは、反応器内で使用される2-ブタノールほど酸化反応をしない可能性がある。変異体Y151AおよびY151Hは、野生型より良好にTACAを酸化することができる(図7C)。
【0190】
変異体Y151Aを21 lスケールで培養し、その変異体を4 l反応器内で、2-ブタノールを再生剤として、かつ溶媒としても用いて、野生型と比較するために使用した。
【0191】
1.6.3 第二世代:変異体Y151A-T192X
変異体Y151Aが野生型より優れているので、この変異体を基に、第2の飽和変異導入をT192Xの位置で行った。
【0192】
2-ブタノールを用いた再生テスト(TACAの添加の有無による犠牲アルコールの再生を測定する、すなわちNADHの生成を光度計で測定する)の結果を図8Aに示す。
【0193】
変異体T192IおよびSは、対照(Y151A-T192T)より良好な結果となっている。したがってこれらをラージスケール(100 ml振盪フラスコでの培養)で検討した。さらに、変異体T192A、LおよびVも、対照とほぼ同じ活性を示すのでラージスケールで検討した。しかしながら、これら5つの変異体について完全な反応を考慮すると、対照がもっとも活性がある(図8Bを参照されたいが、ここで、犠牲アルコールとしてイソプロパノールを伴うTACAの生成はHPLCで測定される)ので、以後の実験では単一変異体Y151Aを使用した。
【0194】
1.7 4 l反応器
上記スクリーニングに由来する活性の高い変異体Y151Aは、21 lスケールで数回発酵させた。ラージスケールで対照LU11558と比較するために、一連の標準的な反応器バッチを実行した。
【0195】
1.7.1 バッチ
攪拌機および冷却器を取り付けた加熱可能な4 l反応器内で、初めに2 lの2-ブタノールを20 mM KH2PO4 バッファーpH 5.0に入れた。0.2 mM NAD (0.5 g) および400 mM TAC (275 g) もしくは 600 mM (420 g)を添加した。生体触媒(450 ml、7.0 g/l BTM)をホールセル(未処理の発酵産物)の形で添加することによって、反応をスタートさせた。発酵培地中の細胞を添加した時点で、pHは6に上昇した。二相反応混合物は、40℃にて減圧(100 mbar)下で攪拌した。ここで、2-ブタノール、2-ブタノンおよび水の混合物を、一段階で留去した。同時に、同量の、69%2-ブタノールおよび31%H2Oからなる溶液(2-ブタノンは別として蒸留物の組成に相当する)をフィードとして添加した。pH測定装置を用いてpHをチェックし、約5.5-6.0の間で一定に維持した。1時間ごとにサンプルを採取し、濃塩酸で反応を止め、HPLC(LJ31366)により分析した。8時間後、反応混合物を取り出した。
【0196】
1.7.2 野生型(LU11558)と変異体Y151A(LU14759)との比較
変異体がより大量の生成物/二次的成分に耐えられるかどうかをみるために、初めに、400 mM(70 g/l)TACとともに、次に600 mM(105 g/l)TACとともに、変異体について数回4 l反応器を運転した。
【0197】
図9Aは、変異体が平均で15-20%野生型より優れていることを示す。図に示すように、値は実験ごとに変動する(これは、個々の発酵にも左右される)が、違いは有意である。野生型の場合平均収率は67%±8%であるが、変異体の場合86%±7%であり、これらはいずれの場合も、別々の発酵である。TACA/(TAC+TA)比(淡色の棒グラフ)も、変異体の場合(4.8)、対照の場合(3.0)より有意にすぐれており、これは後に、メチルアミノ化において確実に明白となる。
【0198】
600 mM(図9B)による運転を考えると、ここでもまた、変異体は野生型より良好な結果をもたらす。しかしながら、TACA/(TAC+TA)比は、400 mM運転の場合より有意に悪化している。
【0199】
1.8 結果
合理的な計画によって、野生型より15-20%活性が高く、しかもデュロキセチンアルコールの中間体の調製中に、より大量のTACAおよび/またはTAに耐えられる変異体Y151A(LU14759)見いだすことができた。
【0200】
この結果は、一連の4 l反応器での反応においても確認され、それは小規模での製造プロセスを反映している。
【0201】
阻害剤TAを伴う酵素の結晶構造は解明されているので、酵素-基質複合体の信頼性のあるモデルを確立することができた。チロシン151のOH基が基質(この場合TA)のプロパノン側鎖のβ炭素原子に密接に接触しており、したがって弱いCH-O水素ブリッジを形成していることは、モデルから明白である(図10A)。チロシン151のアラニンへの変異の結果として、この相互作用は増大し、結合は弱くなる(図10B)。他の相互作用はすべて残存しており、結合ポケットのサイズが大きくなったにもかかわらず酵素の優れた選択性は変化しないことを意味している。その上、生成物のee値は> 99.5%である。
【実施例2】
【0202】
ランダム変異導入
2.1 ロボット設備のテスト開発
ランダム変異導入により作製される多数のサンプルを処理するために、(生成物を直接検出する)実験室におけるこれまでのHPLC分析の代わりに、ロボット生産ラインのための光度測定法を開発する必要があった。
【0203】
この目的のために、還元型補酵素NADHにおける還元は、340 nmでの吸光係数がεNAD<<εNADHであるので、この波長で測定することができる。最適なNADH濃度は0.02 mMであった。基質TACは1から2 mMの間で使用することができた。使用したバッファーは、TACからTACAへの還元が弱酸性の条件下で優先的に進行するので、50 mM NaH2PO4 pH 5.0とした。変異体を発現する細胞は、マイクロタイタープレート(MTP)内で直接、培養された。このために、ピッキングロボット(Qpix)を用いて寒天プレートからクローンを採取し、抗生物質(100μMアンピシリン、20μMクロラムフェニコールおよび100μMスペクチノマイシン)を入れたLB前培養培地に接種した。37℃にて200 rpmで24時間の増殖時間後、その細胞を、抗生物質およびインデューサー(ラムノース0.5 g/lおよびIPTG 0.1 mM)を含むLB本培養培地に手で移した。16-18時間増殖させた後、細胞をテストに使用した。
【0204】
予備的な実験から、培養で得られた細胞は、そうしないと活性が低すぎるので、アッセイ前に破壊しなければならないことが分かった。細胞を破壊するために、さまざまな方法が調べられたが、たとえば、細胞を4℃にて一晩保存すること、1-ブタノールおよび1.4-ブタンジオールの添加、および液体窒素による急速凍結などである。4℃保存、および液体窒素による急速凍結だけがうまくいったが、時間の節約のため窒素による処理が好ましい。この目的で、増殖させた細胞をまず遠心し、上清を除去し、MTPに粘着フィルムで封をした。MTPを約3秒間、液体窒素に完全に浸漬した後、再び下に置いて融解させた。もっとも一定した結果は、急速凍結と室温での一時的融解を4回繰り返した場合に達成された。
【0205】
2.2 ロボットテストの経過
細胞は上記のように培養した後破壊した。MTPにカバーを付けて、ロボット装置内の15℃のインキュベーターに入れた。Multidropにおいて、ウェルあたり100μlの水を、その後Packardにおいて細胞を再懸濁するために添加した。その後Multidropにおいて、基質溶液(終濃度:2 mM TAC、0.2 mM NADH、50 mM NaH2PO4 pH 5.0)を加え、NADHの減少を光度計で340nmにて測定した。
【0206】
このテストから得られた陽性クローンを次に、ラージスケールでさらによく検討した。このために、3つの異なるアッセイを行った。
【0207】
テストA 完全な反応:TACからTACAへの還元およびイソプロパノールによるNADH再生(50 mM NaH2PO4 pH 5.0、0.2 mM NAD、10 mM TAC、10%イソプロパノール)、測定HPLC LJ31366
【化12】
【0208】
テストB 光度計において、再生剤として2-ブタノールを用いたNADからNADHへの再生(80 mM TrisHCl pH 8.0、100 mM 2-ブタノール、1.75 mM NAD)
【化13】
【0209】
テストC 光度計において、NADHの添加によるTACからTACAへの還元(50 mM NaH2PO4 pH 5.0、0.2 mM NADH、1.4μl TAC pure (10 mM))
【化14】
【0210】
3つのアッセイから得られた結果の比較から、NADの再生(すなわちテストB)は、ロボットテストで始めに用いられたTACからTACAへの還元よりも、完全な反応の結果を有意によく反映することが明らかになった。
【0211】
予備的実験は同様に、2-ブタノールによる補酵素の再生(したがってNADHの生成)が、生体触媒を発現する細胞の場合にのみ起こることを示した。結果として、ロボットテストは、2-ブタノールによるNADH再生の検出に切り替えられた。しかしながら、これと並行してTACからTACAへの還元、したがってNADNの減少もやはり測定された。
【0212】
2.3 変更されたロボットテストの経過
細胞を培養した後破壊した。MTPにカバーを付けて、ロボット装置内の15℃のインキュベーターに入れた。Multidropにおいて、ウェルあたり100μlの水を、次に細胞を再懸濁するために添加した。これらから、細胞懸濁液20μl/ウェル(補酵素再生アッセイ用)もしくは70μl/ウェル(TAC還元アッセイ用)の2つの娘プレートが作製された。基質溶液(終濃度:還元:2 mM TAC、0.2 mM NADH、50 mM NaH2PO4 pH 5.0;再生: 100 mM 2-ブタノール、1.75 mM NAD、80 mM TrisHCl pH 8.0)を加え、NADHの生成を光度計で340nmにて測定した。
【0213】
2.4 ロボットテスト結果の評価
ロボットスクリーニングにおいて、NADHの生成/減少(還元/酸化)は光度計で測定した。このために、いずれの場合も、10個の測定値を10分間にわたって確認した。これらの値から増加を計算することによって、デヒドロゲナーゼの初発の活性を測定した。
【0214】
2.5 酵素反応の阻害
予備的な実験は、反応中に形成される生成物TACAまたは二次的成分が反応を阻害することを示した。基質は完全には変換されなかった。
【0215】
したがって、可能な限り完全な変換を達成することで容積時間当たりの高収率を達成するために、活性が高いだけでなく、より大量の生成物もしくは二次的成分に耐えられる変異体を見いだすことを目的とした。
【0216】
したがって、アッセイにTACAを添加した。このために、それまで2ブタノールおよびNADとともに行われた再生テストにさらに10 mMのTACAを添加した。
【0217】
生物量が限られるため、ロボットスクリーニングにおいて3つのテストをすべて並行して実施することはできなかった。したがって、TACからTACAへの還元反応は省略した。2-ブタノールによるNADHの再生反応は不変であった。TAC溶液は、TACA阻害のための基質溶液(100 mM 2-ブタノール、1.75 mM NAD、10 mM TACA、80 mM TrisHCl pH 8.0)に置き換えられた。ロボットテストの経過は、変化しないままであった。
【0218】
したがって、評価はTACA阻害の測定結果に適合した。
【0219】
2.6 変異体ライブラリーの調製:EbN1遺伝子のランダム変異導入
デヒドロゲナーゼをコードする配列内に変異を生じさせるために、エラープローンPCR反応を、MnCl2を添加して実施した。MnCl2とともに使用されたTaq-DNAポリメラーゼの特異性は低下したので、その結果として、MnCl2濃度が上昇すると、より多くの間違ったヌクレオチドが組み込まれ、したがってより多くの変異が生じる。
【0220】
PCRのために、開始領域および末端領域においても変異が起こるように、可能な限りもっとも短いDNA領域をカバーする、クローニング切断部位(NdeI - Hind III)を有する下記のオリゴヌクレオチドを選択した:
【0221】
バッチ
50μl PCRバッチ中:デヒドロゲナーゼ遺伝子を有する50 ngプラスミドDNA(pDHE-ebn1H)、それぞれの場合ごとに120 ngオリゴヌクレオチド、GCリッチ反応バッファー1x (Roche)、1/5容 GCリッチレゾリューション (Roche)、それぞれの場合ごとに0.2 mM dATP、dTTP、dCTP、dGTP、1 U Taq DNAポリメラーゼ。
【0222】
このバッチを95℃にて5分間加熱した(DNAの初回変性)後、85℃に冷却した。この温度で、MnCl2をさまざまな濃度(0.02 mM刻みで0-1 mM)で添加した。これは、MnCl2が完全に溶解するために必要であった。実際のPCRはその後、以下の温度プログラムにより開始された:4サイクル:95℃ 45秒、54℃ 45秒、72℃ 45秒;続いて26サイクル:95℃ 45秒、58℃ 45秒、72℃ 45秒;10℃で停止。
【0223】
PCR産物をアガロースゲル(Gfxキット)で精製した後、制限酵素NdeIおよびHindIII(いずれもNEB製)による切断を行った。ベクターpDHE(同様にNdeI/HindIIIで切断)にライゲートした後、形質転換を実施してXL10ウルトラコンピテントセル(Stratagene)に入れた。次に、クローンの一部(濃度当たり16クローン)を配列決定して、最適なMnCl2濃度を決定した。これに関連して、1-3塩基対までの交換を生じるMnCl2濃度を用いて、さらに実験を行った。このために、ライゲーションバッチをまずXL10ウルトラコンピテントセルに形質転換し、クローンを計数した後、すべてのクローンを寒天プレートからLB培地を用いて溶離した。細胞をさらにインキュベートすることなく、プラスミドDNAを単離し(Promegaキット)、このように単離されたDNAを生産菌株TG10に導入して形質転換したが、この菌株は、シャペロンpAgro pHSGを同時に発現するものであって、Q-trayプレート上に蒔かれた。この手順は、ライゲーション時の生産菌株TG10+(LU12037)の形質転換率が非常に低いために必要であり、できる限り多くの変異体が求められた。
【0224】
2.7 選択された変異体
上記の表1は、ロボットテストから選択されたクローンの概要を示す。これらのクローンは検証プレートから得られ、完全に配列決定された。これらをラージスケールで培養し、始めにエッペンドルフでテストした。しかしながら、変異体の大半の活性は、野生型と同程度であった。活性の高い変異体、たとえば、K114TおよびM200V F201Lは、21 lスケールで発酵培養し、0.5 l反応器でテストした。
【0225】
攪拌機および冷却器を取り付けた、加熱可能な0.5 l反応器内で、始めに250 mlの2-ブタノールを、20 mM KH2PO4 バッファーpH 5.0中に導入した。0.2 mM NAD(0.1 g)および100 mM TAC(35 g)を添加した。生体触媒(45 ml、5.5 g/l BTM)をホールセル(未処理発酵産物)の状態で添加することによって、反応を開始させた。発酵培地中の細胞を添加したとき、pHは6に上昇した。二相反応混合物は、減圧(110 mbar)下で40℃にて攪拌した。ここで、2-ブタノール、2-ブタノンおよび水を1段階で留去した。同時に、同量の、69%2-ブタノールおよび31%H2Oからなる溶液(2-ブタノンを除いて蒸留物の組成に相当する)をフィードとして添加した。pH測定装置を用いてpHをチェックし、約5.5-6.0の間で一定に維持した。1時間ごとにサンプルを採取し、濃塩酸で反応を止め、HPLC(LJ31366)により分析した。8時間後、反応混合物を取り出した。
【0226】
個別の段階のエラー解析は、もっとも大きなエラーがマイクロタイタープレートでの個別のクローンの増殖中に存在することを示す。マイクロタイタープレートでの増殖条件(温度、酸素導入など)は、発酵槽のように正確にコントロールできないので、これは驚くには当たらない。同一菌株の別々の発酵でさえ、約10%-15%ほど変動する。このロボットスクリーニングに関する全体としてのエラーは約35%である。すなわち、このスクリーニングにおいて、35%を超える増加を示す変異体だけが意味がある。
【実施例3】
【0227】
部位特異的変異導入および追加の飽和変異導入
追加して標的化された、選択された位置で、検討済みの単一変異(「部位特異的変異導入」)または飽和変異導入を実施した。
【0228】
3.1 変異のための位置の選択
酵素の基質結合ポケットは、ループ1、2およびヘリックスαFG1(図2)により形成される。大半の変異がこの領域から選択されたのは、これらのアミノ酸が基質結合および/または酵素活性に直接影響を及ぼすことが予想されるからである。
【0229】
ヘリックスは基質なしでは非常にフレキシブルであって(結晶状態では、電子密度は見られない)、基質結合時にのみ固定された状態となる。基質結合ポケットを含む活性中心は、疎水性領域と親水性領域に分けられる。疎水性部分は、おもに両親媒性ヘリックスαFG1によって形成されるが、このヘリックスは、基質結合後、基質結合ポケット上に蓋のように乗る。アミノ酸192から204までがこの領域にある。アミノ酸Thr192、Leu197、Met200およびLeu204の側鎖は結合ポケットの中に向いているが、アミノ酸Phe201はループの安定化に役立っている。OH基を有するトレオニン192は活性中心の疎水性領域と親水性領域の境界をなしている。Leu186はフレキシブルなヘリックスの開始部分にあり、活性中心が開いた状態および閉じた状態となるように、蝶番のように機能する。メチオニン246は、基質結合ポケットの末端にある。基質結合ポケットの反対側は、アミノ酸146から151までを含むループβEαFをなしている。ここでもまた、選択されたアミノ酸Leu146、Ile148およびTyr151の側鎖は結合ポケットの中に向いている。チロシン151の末端OH基は、活性中心の親水性部分の水素ブリッジネットワークの一部であるが、残りの残基は疎水性部分に属している。ほとんど親水性の活性中心の下側は、アミノ酸138-142のストランドを形成しているが、ここでLeu139、Thr140およびThr142は変異している。2つのシステイン62および83が変異のために選択されたのは、システインが通常、酸化の影響を受けやすく、それによって構造に悪影響を及ぼす可能性があるためである。
【0230】
図11は、活性中心からの断面図を示す。補酵素は紫色、基質(ここではTA)は緑色でマークし、変異したアミノ酸は強調してある。上部の領域において、両親媒性ヘリックスαFG1が見られ、下部領域にはloopβEαF(ループ2)が見られる。
【0231】
3.2 標的変異体の調製
始めに、DNA変異のそれぞれの位置について、望ましいDNA配列に対応する2つの相補的オリゴヌクレオチド(表2)を選択した。それに加えて、遺伝子全体に隣接するオリゴヌクレオチドをさらに2つ選択した(Mke123およびMke124、配列番号5および6)。部位特異的変異導入のクローニングストラテジーを図12に示す。
【0232】
その後、それぞれ遺伝子隣接オリゴヌクレオチドおよび望ましい変異を有するオリゴヌクレオチドを用いて、2つのPCR反応を行った。これによって、変異の代わりに、短い相補的領域を有する2つのPCR産物が与えられる。これら2つのPCR産物をテンプレートとして用いて、第2のPCRを、以前使用した遺伝子隣接オリゴヌクレオチドを再び使用して、行った。この反応から、望ましい変異を有する完全な遺伝子が与えられる。
【0233】
PCR産物を制限酵素NdeIおよびHindIIIで切断した後、pDHEベクターにライゲートした。コンピテントセルXL10 Gold (Stratagene)に入れて形質転換し、続いてプラスミドを単離した後、変異の成否を確認するためにそのプラスミドを配列決定した。活性を測定するために、このプラスミドを、シャペロンプラスミドpAgroおよびpHSGを有するTG10+コンピテントセル(LU12037)に入れて形質転換した。
【表3】
【0234】
【0235】
【0236】
【0237】
【0238】
3.3 標的変異体に対する活性テスト
図13Aは、NADHを添加したTACからTACAへの還元、ならびに再生を伴う全体としての反応(黒い棒グラフ)の両者を2.2項に記載のように検討した活性テストから得られた結果を示す。図13Bでは、再生を伴う全体反応のみ調べた。
【0239】
特に変異体L197Iは、野生型より3倍高い活性を示す。
【0240】
本明細書に引用された文献の開示について明確に参照する。
【特許請求の範囲】
【請求項1】
配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体。
【請求項2】
配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体であって、その変異体が明細書の表1に記載の変異体から選択される、前記変異体。
【請求項3】
配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体であって、その変異体が、
(1) 142〜153(ループ2)および
(2) 190〜211(ヘリックスαFG1)
から選択される少なくとも1つの配列領域内に、少なくとも1つの変異を有する、前記変異体。
【請求項4】
(3) 93〜96(ループ1)
(4) 241〜249(C末端)
(5) 138〜141(結合ポケットの親水性領域)および
(6) Cys61および/またはCys 83
から選択されるもう1つの配列領域内に、少なくとももう1つの変異を追加して有する、請求項2に記載の変異体。
【請求項5】
下記の基:T192、L197、M200、F201、L204、M246、L139、T140、T142、L146、I148、Y151、C61、C83、L186、のうち少なくとも1つが変異しており、そのそれぞれのアミノ酸が任意の望ましい他の天然アミノ酸で置き換えられている、請求項3または4に記載の変異体。
【請求項6】
下記の変異:
a)単一変異:
Y151XA、このXAはA、R、N、E、Q、G、H、I、L、M、TまたはVである;
T192XB、このXBはA、E、G、I、P、S、W、VまたはLである;
b)多重変異:
Y151XA T192XB、このXA およびXB は上記の意味を持つ;
のうち少なくとも1つを有する変異体から選択される、請求項1〜5のいずれか1つに記載の変異体。
【請求項7】
下記の改変された部分配列:
(1) 142-TTYWX1KX2EAX3T-153(改変ループ2)および
(2) 190-ATX4EASAX5 SAX6X7DVX8PNMLQAI-211(改変ヘリックスαFG1)
のうち少なくとも1つによって特徴付けられる、請求項1〜6のいずれか1つに記載の変異体であって、
これらの配列中、X1 〜X8は、互いに無関係に、任意の望ましいアミノ酸基であるが、基X1 〜X3 およびX4 〜X8の少なくとも1つは、配列番号2に記載の天然型酵素の天然アミノ酸基ではない、前記変異体。
【請求項8】
X1 はLである、またはI、V、A、M、FもしくはHで置換されている、
X2 はIである、またはL、V、A、M、FもしくはHで置換されている、
X3 はYである、またはA、R、N、E、Q、G、H、I、L、M、TもしくはVで置換されている;
あるいは、
X4 はTである、またはA、E、G、I、P、S、W、VもしくはLで置換されている、
X5 はLである、またはI、V、A、M、FもしくはHで置換されている、
X6 はMである、またはY、W、E、V、S、R、Q、K、I、H、G、F、EもしくはDで置換されている、
X7 はFである、またはG、K、T、Y、M、WもしくはRで置換されている、
X8 はLである、またはI、V、A、M、FもしくはHで置換されている、
請求項7に記載の変異体。
【請求項9】
配列番号2のデヒドロゲナーゼの酵素活性の、すくなくとも約50%を依然として有する、請求項1〜8のいずれか1つに記載の変異体。
【請求項10】
配列番号2に対して少なくとも約70%の配列同一性を有する、請求項1〜9のいずれか1つに記載の変異体。
【請求項11】
上記領域(1)〜(6)における少なくとも1つの変異に加えて、アミノ酸基の25%までが、配列番号2と比較して、付加、欠失、挿入、置換、逆位もしくはそれらの組み合わせにより改変されている、請求項1〜10のいずれか1つに記載の変異体。
【請求項12】
補酵素NAD+またはNADHの存在下で3-クロロ-1-(チエニル-2-イル)-プロパン-1-オン(1)と(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オール(2)との間の立体特異的平衡反応
【化1】
を触媒する、請求項1〜11のいずれか1つに記載の変異体。
【請求項13】
請求項1〜12のいずれか1つに記載の変異体をコードする核酸配列。
【請求項14】
少なくとも1つの調節核酸配列に機能的に連結された、請求項13に記載の少なくとも1つの核酸配列を含んでなる、発現カセット。
【請求項15】
請求項14に記載の少なくとも1つの発現カセットを含んでなるベクター。
【請求項16】
少なくとも1つの、請求項13に記載の核酸配列、請求項14に記載の発現カセット、または請求項15に記載のベクターを含んでなる、組換え微生物。
【請求項17】
請求項1〜12のいずれか1つに記載の変異体を製造する方法であって、請求項16に記載の組換え微生物を培養すること、その変異体をコードする核酸配列を発現させること、ならびに、必要に応じて発現産物を単離することを含んでなる、前記方法。
【請求項18】
式(II)の置換された光学活性アルコール
【化2】
を生体触媒により合成する方法であって、
式中、Cycは、単環式もしくは多環式、飽和もしくは不飽和、炭素環もしくはヘテロ環であって、適宜、一置換または多置換されていてもよく、
いずれの場合も、立体異性体として純粋な状態、または立体異性体の混合物であるが、
その方法が、請求項1〜12のいずれか1つに記載のフェニルエタノールデヒドロゲナーゼ変異体の存在下で、適宜、特にNADHのような還元当量を添加して、式(I)のケトン
【化3】
を微生物/酵素により還元することを含んでなる、前記方法。
【請求項19】
C1〜C6一価アルコールを犠牲アルコールとして使用して、還元当量再生条件下で反応が行われる、請求項18に記載の方法。
【請求項20】
Cycがヘテロ環基、特にチエニル基である、請求項18または19に記載の方法。
【請求項21】
基本的に鏡像体として純粋な式(II)のアルコール、特に(S)-鏡像異性体が与えられる、請求項18〜20のいずれか1つに記載の方法。
【請求項22】
変異体が単離された状態で使用され、その結果、変異体は、適宜、固体担体に固定化されている;または、適宜、固体担体に固定化された微生物細胞内で発現される、請求項18〜21のいずれか1つに記載の方法。
【請求項23】
a)請求項18〜22のいずれか1つに記載の方法を用いた、3-クロロ-1-(チエニル-2-イル)-プロパン-1-オン(1)から(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オール(2)への生体触媒による還元;
【化4】
b)メチルアミノ化によりデュロキセチンアルコール(3)を与える、アルコール(2)の化学的変換;
【化5】
ならびに、最終的に
c)ナフチル基の挿入によるデュロキセチンアルコール(3)からデュロキセチン(4)への化学的変換
【化6】
を含んでなる、デュロキセチンの調製方法。
【請求項24】
式(I)の置換ケトンを微生物/酵素により合成する方法であって、
【化7】
式中、Cycは、単環式もしくは多環式、飽和もしくは不飽和、炭素環もしくはヘテロ環であり、適宜、一置換または多置換されていてもよいが、
その方法が、請求項1〜12のいずれか1つに記載のフェニルエタノールデヒドロゲナーゼ変異体の存在下で、適宜、特にNAD+のような酸化当量を添加して、式(II)のアルコール
【化8】
であって、いずれの場合も、立体異性体として純粋な状態、または立体異性体の混合物であるもの、を微生物/酵素により酸化することを含んでなる、前記方法。
【請求項25】
犠牲ケトンとしてC1〜C6一価アルカノンを用いて、酸化当量再生条件下で反応が行われる、請求項24に記載の方法。
【請求項26】
変異体が単離された状態で使用され、その結果、変異体は、適宜、固体担体に固定化されている;または、適宜、固体担体に固定化された微生物細胞内で発現される、請求項24または25に記載の方法。
【請求項27】
デュロキセチンアルコールおよび/またはデュロキセチンの調製における、請求項1〜12のいずれか1つに記載の変異体の使用。
【請求項1】
配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体。
【請求項2】
配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体であって、その変異体が明細書の表1に記載の変異体から選択される、前記変異体。
【請求項3】
配列番号2に記載のアミノ酸配列を有するAzoarcus菌種のフェニルエタノールデヒドロゲナーゼEbN1に由来する、機能的なフェニルエタノールデヒドロゲナーゼ変異体であって、その変異体が、
(1) 142〜153(ループ2)および
(2) 190〜211(ヘリックスαFG1)
から選択される少なくとも1つの配列領域内に、少なくとも1つの変異を有する、前記変異体。
【請求項4】
(3) 93〜96(ループ1)
(4) 241〜249(C末端)
(5) 138〜141(結合ポケットの親水性領域)および
(6) Cys61および/またはCys 83
から選択されるもう1つの配列領域内に、少なくとももう1つの変異を追加して有する、請求項2に記載の変異体。
【請求項5】
下記の基:T192、L197、M200、F201、L204、M246、L139、T140、T142、L146、I148、Y151、C61、C83、L186、のうち少なくとも1つが変異しており、そのそれぞれのアミノ酸が任意の望ましい他の天然アミノ酸で置き換えられている、請求項3または4に記載の変異体。
【請求項6】
下記の変異:
a)単一変異:
Y151XA、このXAはA、R、N、E、Q、G、H、I、L、M、TまたはVである;
T192XB、このXBはA、E、G、I、P、S、W、VまたはLである;
b)多重変異:
Y151XA T192XB、このXA およびXB は上記の意味を持つ;
のうち少なくとも1つを有する変異体から選択される、請求項1〜5のいずれか1つに記載の変異体。
【請求項7】
下記の改変された部分配列:
(1) 142-TTYWX1KX2EAX3T-153(改変ループ2)および
(2) 190-ATX4EASAX5 SAX6X7DVX8PNMLQAI-211(改変ヘリックスαFG1)
のうち少なくとも1つによって特徴付けられる、請求項1〜6のいずれか1つに記載の変異体であって、
これらの配列中、X1 〜X8は、互いに無関係に、任意の望ましいアミノ酸基であるが、基X1 〜X3 およびX4 〜X8の少なくとも1つは、配列番号2に記載の天然型酵素の天然アミノ酸基ではない、前記変異体。
【請求項8】
X1 はLである、またはI、V、A、M、FもしくはHで置換されている、
X2 はIである、またはL、V、A、M、FもしくはHで置換されている、
X3 はYである、またはA、R、N、E、Q、G、H、I、L、M、TもしくはVで置換されている;
あるいは、
X4 はTである、またはA、E、G、I、P、S、W、VもしくはLで置換されている、
X5 はLである、またはI、V、A、M、FもしくはHで置換されている、
X6 はMである、またはY、W、E、V、S、R、Q、K、I、H、G、F、EもしくはDで置換されている、
X7 はFである、またはG、K、T、Y、M、WもしくはRで置換されている、
X8 はLである、またはI、V、A、M、FもしくはHで置換されている、
請求項7に記載の変異体。
【請求項9】
配列番号2のデヒドロゲナーゼの酵素活性の、すくなくとも約50%を依然として有する、請求項1〜8のいずれか1つに記載の変異体。
【請求項10】
配列番号2に対して少なくとも約70%の配列同一性を有する、請求項1〜9のいずれか1つに記載の変異体。
【請求項11】
上記領域(1)〜(6)における少なくとも1つの変異に加えて、アミノ酸基の25%までが、配列番号2と比較して、付加、欠失、挿入、置換、逆位もしくはそれらの組み合わせにより改変されている、請求項1〜10のいずれか1つに記載の変異体。
【請求項12】
補酵素NAD+またはNADHの存在下で3-クロロ-1-(チエニル-2-イル)-プロパン-1-オン(1)と(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オール(2)との間の立体特異的平衡反応
【化1】
を触媒する、請求項1〜11のいずれか1つに記載の変異体。
【請求項13】
請求項1〜12のいずれか1つに記載の変異体をコードする核酸配列。
【請求項14】
少なくとも1つの調節核酸配列に機能的に連結された、請求項13に記載の少なくとも1つの核酸配列を含んでなる、発現カセット。
【請求項15】
請求項14に記載の少なくとも1つの発現カセットを含んでなるベクター。
【請求項16】
少なくとも1つの、請求項13に記載の核酸配列、請求項14に記載の発現カセット、または請求項15に記載のベクターを含んでなる、組換え微生物。
【請求項17】
請求項1〜12のいずれか1つに記載の変異体を製造する方法であって、請求項16に記載の組換え微生物を培養すること、その変異体をコードする核酸配列を発現させること、ならびに、必要に応じて発現産物を単離することを含んでなる、前記方法。
【請求項18】
式(II)の置換された光学活性アルコール
【化2】
を生体触媒により合成する方法であって、
式中、Cycは、単環式もしくは多環式、飽和もしくは不飽和、炭素環もしくはヘテロ環であって、適宜、一置換または多置換されていてもよく、
いずれの場合も、立体異性体として純粋な状態、または立体異性体の混合物であるが、
その方法が、請求項1〜12のいずれか1つに記載のフェニルエタノールデヒドロゲナーゼ変異体の存在下で、適宜、特にNADHのような還元当量を添加して、式(I)のケトン
【化3】
を微生物/酵素により還元することを含んでなる、前記方法。
【請求項19】
C1〜C6一価アルコールを犠牲アルコールとして使用して、還元当量再生条件下で反応が行われる、請求項18に記載の方法。
【請求項20】
Cycがヘテロ環基、特にチエニル基である、請求項18または19に記載の方法。
【請求項21】
基本的に鏡像体として純粋な式(II)のアルコール、特に(S)-鏡像異性体が与えられる、請求項18〜20のいずれか1つに記載の方法。
【請求項22】
変異体が単離された状態で使用され、その結果、変異体は、適宜、固体担体に固定化されている;または、適宜、固体担体に固定化された微生物細胞内で発現される、請求項18〜21のいずれか1つに記載の方法。
【請求項23】
a)請求項18〜22のいずれか1つに記載の方法を用いた、3-クロロ-1-(チエニル-2-イル)-プロパン-1-オン(1)から(1S)-3-クロロ-1-(チエニル-2-イル)-プロパン-1-オール(2)への生体触媒による還元;
【化4】
b)メチルアミノ化によりデュロキセチンアルコール(3)を与える、アルコール(2)の化学的変換;
【化5】
ならびに、最終的に
c)ナフチル基の挿入によるデュロキセチンアルコール(3)からデュロキセチン(4)への化学的変換
【化6】
を含んでなる、デュロキセチンの調製方法。
【請求項24】
式(I)の置換ケトンを微生物/酵素により合成する方法であって、
【化7】
式中、Cycは、単環式もしくは多環式、飽和もしくは不飽和、炭素環もしくはヘテロ環であり、適宜、一置換または多置換されていてもよいが、
その方法が、請求項1〜12のいずれか1つに記載のフェニルエタノールデヒドロゲナーゼ変異体の存在下で、適宜、特にNAD+のような酸化当量を添加して、式(II)のアルコール
【化8】
であって、いずれの場合も、立体異性体として純粋な状態、または立体異性体の混合物であるもの、を微生物/酵素により酸化することを含んでなる、前記方法。
【請求項25】
犠牲ケトンとしてC1〜C6一価アルカノンを用いて、酸化当量再生条件下で反応が行われる、請求項24に記載の方法。
【請求項26】
変異体が単離された状態で使用され、その結果、変異体は、適宜、固体担体に固定化されている;または、適宜、固体担体に固定化された微生物細胞内で発現される、請求項24または25に記載の方法。
【請求項27】
デュロキセチンアルコールおよび/またはデュロキセチンの調製における、請求項1〜12のいずれか1つに記載の変異体の使用。
【図1】


【図2】
【図3】
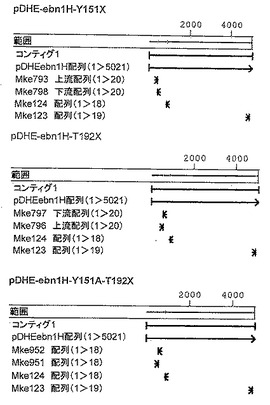
【図4】
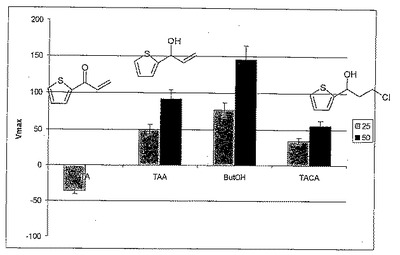
【図5A】
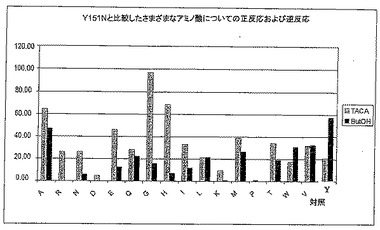
【図5B】
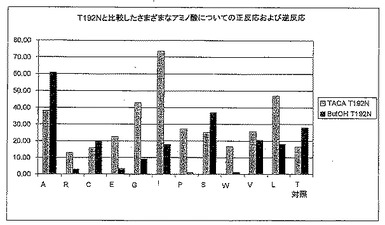
【図6A】
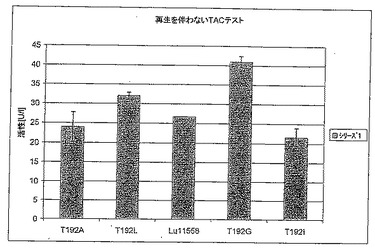
【図6B】
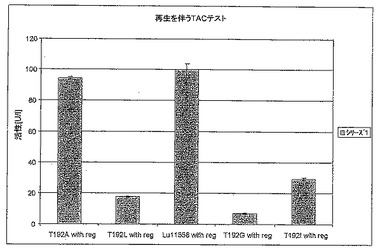
【図6C】
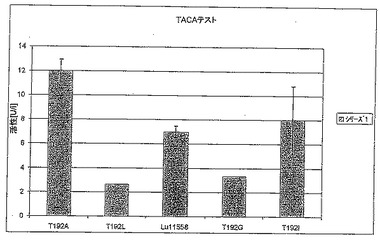
【図7A】
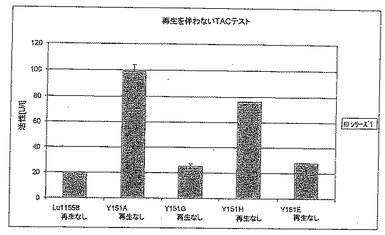
【図7B】
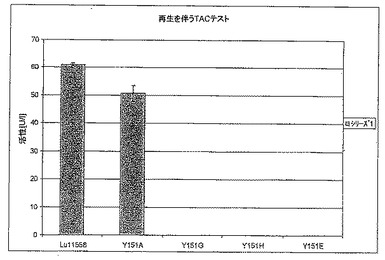
【図7C】
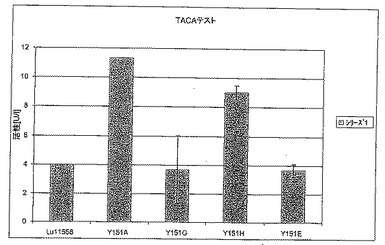
【図8A】
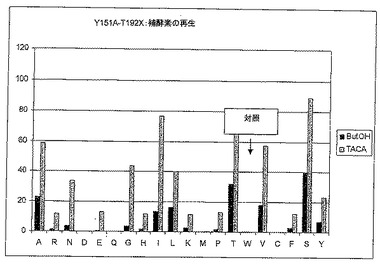
【図8B】
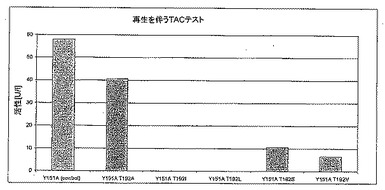
【図9A】
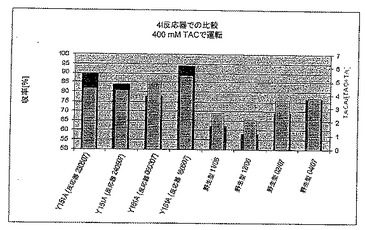
【図9B】
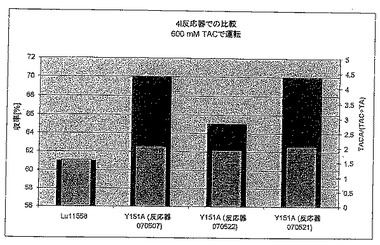
【図10】
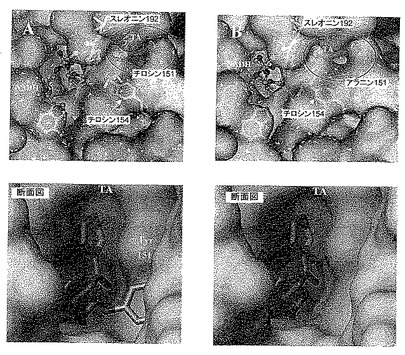
【図11】
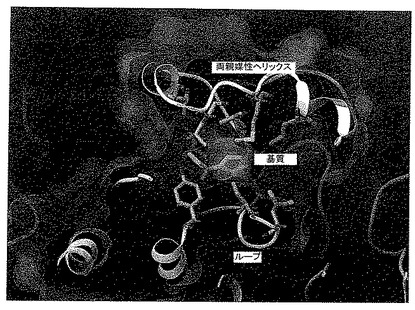
【図12】
【図13A】
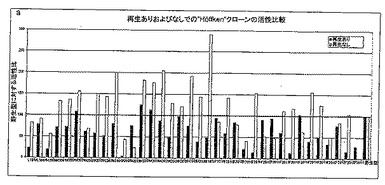
【図13B】
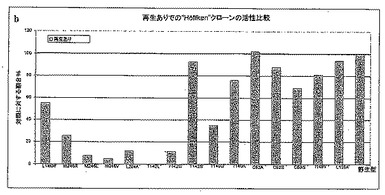
【図2】
【図3】
【図4】
【図5A】
【図5B】
【図6A】
【図6B】
【図6C】
【図7A】
【図7B】
【図7C】
【図8A】
【図8B】
【図9A】
【図9B】
【図10】
【図11】
【図12】
【図13A】
【図13B】
【公表番号】特表2012−511922(P2012−511922A)
【公表日】平成24年5月31日(2012.5.31)
【国際特許分類】
【出願番号】特願2011−541415(P2011−541415)
【出願日】平成21年12月16日(2009.12.16)
【国際出願番号】PCT/EP2009/067341
【国際公開番号】WO2010/079068
【国際公開日】平成22年7月15日(2010.7.15)
【出願人】(508020155)ビーエーエスエフ ソシエタス・ヨーロピア (2,842)
【氏名又は名称原語表記】BASF SE
【住所又は居所原語表記】D−67056 Ludwigshafen, Germany
【Fターム(参考)】
【公表日】平成24年5月31日(2012.5.31)
【国際特許分類】
【出願日】平成21年12月16日(2009.12.16)
【国際出願番号】PCT/EP2009/067341
【国際公開番号】WO2010/079068
【国際公開日】平成22年7月15日(2010.7.15)
【出願人】(508020155)ビーエーエスエフ ソシエタス・ヨーロピア (2,842)
【氏名又は名称原語表記】BASF SE
【住所又は居所原語表記】D−67056 Ludwigshafen, Germany
【Fターム(参考)】
[ Back to top ]