
データ解析方法及びそのシステム
【課題】 サンプル数が少なく、説明変数の数が膨大である場合でもオーバーフィットを克服してカテゴリー分類能力の高いデータ解析ができる方法を提供する。
【解決手段】 遺伝的アルゴリズムを用い、各サンプルにおける各カテゴリーの重み付け平均された帰属尤度に基づいて適応度を計算しつつ、多変量正規分布モデルの第1次最適解を複数個得て(ステップ21)、それら複数個の第1次最適解の重み付け平均最適解を算出して第2次最適解を求める(ステップ22)。
【解決手段】 遺伝的アルゴリズムを用い、各サンプルにおける各カテゴリーの重み付け平均された帰属尤度に基づいて適応度を計算しつつ、多変量正規分布モデルの第1次最適解を複数個得て(ステップ21)、それら複数個の第1次最適解の重み付け平均最適解を算出して第2次最適解を求める(ステップ22)。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、生体に関する情報を解析するデータ解析方法及びそのシステムに関する。
【背景技術】
【0002】
ヒトゲノムの解読以降、ゲノムに書かれた遺伝情報がどのように発現して機能しているのかを解明するポストゲノム時代に突入したと言われ、そのような動向を踏まえて遺伝子発現状態を測定する技術も年々進歩している。
遺伝子発現状態を効率的に測定する技術として次のものがあげられる。まず、トランスクリプトーム(mRNAの総体)を特定するものとして、基盤に複数種のDNAを担持し、それに相補的なmRNAを検出するDNAチップが知られている。代表的なDNAチップには、遺伝子チップやDNAマイクロアレイがある。
【0003】
また、プロテオーム(蛋白質の総体)を特定するものには、2次元電気泳動、抗体チップ、質量スペクトルを用いるものがある。さらにメタボローム(代謝中間体を含めた代謝産物の総体)を測定する手法にも質量分析などが適応されつつある。これらの測定技術は、生体の状態に関するパラメータを短時間に一挙に測定できることがそれまでの技術と比較して画期的であると言える。
【0004】
生体内の細胞の状態は遺伝子発現データによってよく記述されるため、従来の診断マーカーでは情報が不足している場面でも、精度のより高い診断が可能になるという期待も出てきている。特にカテゴリー分類された生体の状態を目的変数とし、DNAチップ等から得られた遺伝子発現データを説明変数としたカテゴリー分類解析法は患者に負担をかけることなく重大な疾病の早期発見、早期治療を可能にするものとして研究が行われている。
例えば、非特許文献1に示すように、P.Tanらはmahalanobis距離に基づく最尤帰属の方法を用いて癌細胞の複数カテゴリー分類を行なった。非特許文献1内では61細胞の9カテゴリー分類(以下、NCI90データと称する。データはhttp://genome-www.stanfoard.edu/suteeh/download/nci90)と、198細胞の14カテゴリー分類(以下、GCMデータと称する、データはhttp://www-genome.wimit.edu/mpr/publication/projects/Global Cancer Map/)とがそれぞれ13,32個の遺伝子の発現量で良好に説明されている。
【0005】
一方、単一のモデルだけでなく、複数個のモデルを組み合わせて安定した優秀な予測を得る方法がここ数年、情報処理の分野で活発に研究されている。bagging, boostingなどリサンプリングによって識別力の弱い学習モデルを複数個獲得し、それらの学習モデルを組み合わせて強い学習モデルにする方法論が盛んに研究されている。例えば、非特許文献2に示すように、Golubらは急性白血病の分類においてwv(weightedvoting)と呼ばれる方法を提案し、一つ一つの遺伝子の判定に重みを付けて票決することで優れた予測識別力が得られることを示している。
【0006】
【非特許文献1】C.H. Ooi & P.Tan;Genetic Algorithm applied to multi-class prediction for analysis of geneexpression data; Bioinformatics 19 (1) 37-44 (2003)
【非特許文献2】T.R. Golub. Science. 286, 531-537, 1999
【発明の開示】
【発明が解決しようとする課題】
【0007】
しかしながら、上記従来技術であれば下記のような課題がある。
(A)非特許文献1の方法に関する課題
遺伝子発現データの特徴はサンプル数Niが数十個程度と少なく、説明変数数Njが数百〜数万個のように膨大であることにある。手元のデータを綺麗に説明するモデルは簡単に得られるが、それらのほとんどは解析に用いられなかったデータへの応用力(以下、汎化力と呼ぶ)に乏しい。これはオーバーフィット(overfit)の問題と称されている。そこで手元のデータの一部をマスクし、予測実験(交差検証:crossvalidation, cv)を行って予測力の優れたモデルを選抜する方法が最近の傾向である。
しかし、遺伝子発現データほどNjが膨大な場合には、手元データのcv予測に優れているものの、真の汎化力に乏しい見せ掛けモデルである危険性が大きいことが分かってきた。これはある臨床グループによる研究が一見「完璧」でも実用場面に応用すると無効になることを意味する。このような事態を回避するためには、cvを超える新たな方法によってオーバーフィットを克服することが求められている。
前記非特許文献1ではP.Tanらは交差検証予測正解率だけで選抜すると独立テスト正解率が悪くなると報告しており、オーバーフィットを克服しているとは言えないものである。
【0008】
(B)非特許文献2の方法に関する課題
非特許文献2に示したリサンプリングの方法は複数カテゴリー分類において各カテゴリーあたりのサンプル数が多い場合には適しているが、少ない場合には不向きであると思われる。また、wv(weightedvoting)は一つ一つの遺伝子での判定というモデル自体が非常に単純であり、複数カテゴリー分類への適応性が明らかでない。
【0009】
本発明は上記課題に鑑みてなされたものであり、本発明の目的は、上記課題を解決できる、データ解析方法及びデータ解析システムを提供することにある。
具体的な目的の一例を示すと、以下の通りである。
(a)生体データの解析において、サンプル数が少なく、説明変数の数が膨大である場合でもオーバーフィットを克服してカテゴリー分類能力の高いデータ解析ができる方法及びそのシステムを提供する。
(b)生体データの解析において、集団学習の考え方を取り入れて比較的簡単な計算でカテゴリー分類能力の高いデータ解析ができる方法及びそのシステムを提供する。
なお、上記に記載した以外の発明の課題及びその解決手段は、後述する明細書内の記載において詳しく説明する。
【課題を解決するための手段】
【0010】
本発明は多面的に表現できるが、例えば、代表的なものを挙げると、次のように構成したものである。
第1発明のデータ解析方法は、コンピュータを用いて、カテゴリー分類された生体の状態を目的変数とし、遺伝子発現、生体関係物質の少なくとも一方に関するデータを説明変数とする相関モデルを決定する複数カテゴリー分類用のデータ解析方法であって、
カテゴリー分類された生体の状態と、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる訓練用サンプルの集合をコンピュータの記憶手段に記憶しておき、
遺伝的アルゴリズムに基づいて複数個の第1次最適解を得る第1次最適解取得処理と、
得られた複数個の第1次最適解の重み付け平均された第2次最適解を算出する重み付け平均最適解算出処理と、
を含んで前記相関モデル構築を行い、
前記第1次最適解取得処理が下記(a)〜(f)の各処理を含んで行われることを特徴とする。
(a)説明変数の候補を選択する説明変数候補選択処理、
(b)各個体が相関モデルの候補を表しており、それらの個体の集団である第1世代を生成する初期個体集団生成処理、
(c)各個体について、各サンプルの各カテゴリーへの帰属尤度を算出する帰属尤度算出処理、
(d)算出された帰属尤度に基づくサンプルの予測カテゴリーとサンプルが実際に帰属する実測カテゴリーとを比較し、その予測がどの程度、信頼性があるかという定量的な評価である適応度を算出する適応度算出処理、
(e)算出された適応度に基づいて、個体に関する突然変異、交叉、選抜などの遺伝的アルゴリズムに基づいて次の世代の個体集団を生成する次世代個体集団生成処理、
(f)少なくとも帰属尤度算出処理、適応度算出処理、次世代個体集団生成処理にわたる一連の処理を繰り返し行って、最も適応度の高い最適解を探索しつつ、所定終結条件を満足する複数個の第1次最適解を得る
【0011】
第2発明は、第1発明において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの帰属尤度を重み付け平均した重み付け平均帰属対数尤度で与えられることを特徴とする。
第3発明は、第1発明において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの中心へのマハラノビス距離を重み付け平均したもので与えられ、前記帰属尤度算出処理がその重み付け平均マハラノビス距離に基づいて重み付け平均化された帰属対数尤度を算出することを特徴とする。
【0012】
第4発明は、第1発明ないし第3発明のいずれか一つに記載の発明において、各第1次最適解の重み付け係数を同じ値に設定したことを特徴とする。
第5発明は、第1発明ないし第3発明のいずれか一つに記載の発明において、各第1次最適解の重み付け係数を各第1次最適解に対応するモデル尤度に基づいて決定することを特徴とする。
【0013】
第6発明は、第1発明ないし第5発明のいずれか一つに記載の発明において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返し(後述するRun)を複数回行う条件が含まれており、その各回で得られた最適解を第1次最適解取得処理における複数個の第1次最適解として設定することを特徴とする。
【0014】
第7発明は、第1発明ないし第6発明のいずれか一つに記載の発明において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返し(後述するRun)を少なくとも一回行う過程で、算出適応度が設定適応度を超えた最適解を複数個抽出することが含まれており、それらの最適解を第1次最適解取得処理における複数個の第1次最適解として設定することを特徴とする。
【0015】
第8発明は、第1発明ないし第7発明のいずれか一つに記載の発明において、前記適応度算出処理がAIC(Akaike'sInformation Criterion:赤池情報基準)に基づいて行われることを特徴とする。
第9発明は、第1発明ないし第7発明のいずれか一つに記載の発明において、前記適応度算出処理が交差検証成績に基づいて行われることを特徴とする。
【0016】
第10発明は、第1発明ないし第9発明のいずれか一つに記載の発明において、前記適応度算出処理が、前記帰属尤度算出処理によって算出された各サンプルの帰属尤度を全サンプルについて乗算することにより得られたモデル尤度に基づいて行われることを特徴とする。
【0017】
第11発明は、第1発明ないし第10発明のいずれか一つに記載の発明において、前記適応度算出処理が、前記帰属尤度算出処理によって算出された帰属尤度の中で各サンプルが一番大きな帰属尤度を有するカテゴリーに帰属すると予測し、その予測カテゴリーと実測カテゴリーとが一致する場合を識別正解、そうでない場合を識別不正解と判別する処理を含むことを特徴とする。
【0018】
第12発明は、第1発明ないし第11発明のいずれか一つに記載の発明において、前記相関モデルを多変量正規分布モデルとしたことを特徴とする。
第13発明は、第12発明において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をカテゴリーによらないものとして設定したことを特徴とする。
【0019】
第14発明は、第12発明において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をムーア・ペンローズの一般化逆行列を用いて求めることを特徴とする。
第15発明は、第1発明ないし第14発明のいずれか一つに記載の発明において、前記相関モデルを、多変量特徴空間の各サンプルを主成分分析によって得られた部分空間により複数カテゴリーに分類する相関モデルとしたことを特徴とする。
【0020】
第16発明は、第15発明において、前記相関モデルがSIMCAモデル(Soft Independent Modeling of Class Analogy)であることを特徴とする。
第17発明は、第1発明ないし第16発明のいずれか一つに記載の発明において、第1次最適解の数を3個以上〜50個以下とすることを特徴とする。
【0021】
第18発明データ解析方法は、第1発明ないし第17発明のいずれか一つに記載のデータ解析方法に基づいて構築された相関モデルに、予測用サンプルの選抜された説明変数を入力してカテゴリー分類された生体の状態を予測する出力を得ることを特徴とする。
【0022】
第19発明のデータ解析システムは、コンピュータを用いて、カテゴリー分類された生体の状態を目的変数とし、遺伝子発現、生体関係物質の少なくとも一方に関するデータを説明変数とする相関モデルを決定する複数カテゴリー分類用のデータ解析システムであって、
カテゴリー分類された生体の状態と、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる訓練用サンプルの集合を記憶する記憶手段と、
遺伝的アルゴリズムに基づいて複数個の第1次最適解を得る第1次最適解取得手段と、
得られた複数個の第1次最適解の重み付け平均された第2次最適解を算出する重み付け平均最適解算出手段と、
を備えて前記相関モデル構築を行い、
前記第1次最適解取得手段が下記(a)〜(f)の各手段を含んでいることを特徴とする。
(a)説明変数の候補を選択する説明変数候補選択手段、
(b)各個体が相関モデルの候補を表しており、それらの個体の集団である第1世代を生成する初期個体集団生成手段、
(c)各個体について、各サンプルの各カテゴリーへの帰属尤度を算出する帰属尤度算出手段、
(d)算出された帰属尤度に基づくサンプルの予測カテゴリーとサンプルが実際に帰属する実測カテゴリーとを比較し、その予測がどの程度、信頼性があるかという定量的な評価である適応度を算出する適応度算出手段、
(e)算出された適応度に基づいて、個体に関する突然変異、交叉、選抜などの遺伝的アルゴリズムに基づいて次の世代の個体集団を生成する次世代個体集団生成手段、
(f)最も適応度の高い最適解を探索しつつ、所定終結条件を満足する複数個の第1次最適解を得るために、少なくとも帰属尤度算出手段、適応度算出手段、次世代個体集団生成手段が行う一連の処理を複数回、繰り返して行なわせる繰返し処理手段
【0023】
第20発明は、第19発明において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの帰属尤度を重み付け平均した重み付け平均帰属対数尤度で与えられることを特徴とする。
第21発明は、第19発明において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの中心へのマハラノビス距離を重み付け平均したもので与えられ、前記帰属尤度算出手段がその重み付け平均マハラノビス距離に基づいて重み付け平均化された帰属対数尤度を算出することを特徴とする。
【0024】
第22発明は、第20発明ないし第21発明のいずれか一つに記載の発明において、各第1次最適解の重み付け係数を同じ値に設定したことを特徴とする。
第23発明は、第20発明ないし第21発明のいずれか一つに記載の発明において、各第1次最適解の重み付け係数を各第1次最適解に対応するモデル尤度に基づいて決定することを特徴とする。
【0025】
第24発明は、第19発明ないし第23発明のいずれか一つに記載の発明において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返し(後述するRun)を複数回行う条件が含まれており、その各回で得られた最適解を第1次最適解取得処理における複数個の第1次最適解として設定することを特徴とする。
【0026】
第25発明は、第19発明ないし第24発明のいずれか一つに記載の発明において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返し(後述するRun)を少なくとも一回行う過程で算出適応度が設定適応度を超えた最適解を複数個抽出することが含まれており、それらの最適解を第1次最適解取得処理における複数個の第1次最適解として設定することを特徴とする。
【0027】
第26発明は、第19発明ないし第25発明のいずれか一つに記載の発明において、前記適応度算出手段の行う適応度算出処理がAIC(Akaike'sInformation Criterion:赤池情報基準)に基づいて行われることを特徴とする。
第27発明は、第19発明ないし第25発明のいずれか一つに記載の発明において、前記適応度算出手段の行う適応度算出処理が交差検証成績に基づいて行われることを特徴とする。
【0028】
第28発明は、第19発明ないし第27発明のいずれか一つに記載の発明において、前記適応度算出手段の適応度算出処理が、前記帰属尤度算出手段によって算出された各サンプルの帰属尤度を全サンプルについて乗算することにより得られたモデル尤度に基づいて行われることを特徴とする。
第29発明は、第19発明ないし第28発明のいずれか一つに記載の発明において、前記適応度算出手段が、前記帰属尤度算出手段によって算出された帰属尤度の中で各サンプルが一番大きな帰属尤度を有するカテゴリーに帰属すると予測し、その予測カテゴリーと実測カテゴリーとが一致する場合を識別正解、そうでない場合を識別不正解と判別する判別手段を含むことを特徴とする。
【0029】
第30発明は、第19発明ないし第29発明のいずれか一つに記載の発明において、前記相関モデルを多変量正規分布モデルとしたことを特徴とする。
第31発明は、第30発明において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をカテゴリーによらないものとして設定したことを特徴とする。
【0030】
第32発明は、第30発明において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をムーア・ペンローズの一般化逆行列を用いて求めることを特徴とする。
第33発明は、第19発明ないし第32発明のいずれか一つに記載の発明において、前記相関モデルを、多変量特徴空間の各サンプルを主成分分析によって得られた部分空間により複数カテゴリーに分類する相関モデルとしたことを特徴とする。
【0031】
第34発明は、第33発明において、前記相関モデルがSIMCAモデル(Soft Independent Modeling of Class Analogy)であることを特徴とする。
第35発明は、第19発明ないし第34発明のいずれか一つに記載の発明において、第1次最適解の数を3個以上〜50個以下とすることを特徴とする。
【0032】
第36発明のデータ解析システムは、第19発明ないし第35発明のいずれか一つに記載のデータ解析システムに基づいて構築された相関モデルと、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる予測用サンプルを記憶する記憶手段とを備え、前記相関モデルに予測用サンプルの選抜された説明変数を入力してカテゴリー分類された生体の状態を予測する出力を得ることを特徴とする。
【0033】
以下、上記第1発明〜第36発明の各構成要素や変形例などについて説明する。
第1発明において「生体関係物質に関するデータ」の一例としては、「細胞内物質のデータ」が挙げられる。細胞内物質のデータとしては細胞内物質の量などが挙げられ、例えば、代謝中間体を含めた代謝産物全部であるメタボロームも含まれる。
なお、「細胞内物質のデータ」は「生体内に含まれる物質のデータ」を意味するものであり、血液、リンパ液、尿などの排泄物に含まれる物質の量も細胞の状態の反映であるという観点から、細胞内物質のデータの一種と解釈され、本発明では当然に対象となるべきものである。
これに対し、「生体関係物質」としては上記生体内に含まれる物質の他、生体が得る食物などに含まれる物質の量などが例示でき、それら物質に関するデータと生体の状態を解析する場合にも適用できるものである。
また、「生体」には人体の他、動物、植物、微生物などの各種生命体が含まれる。
【0034】
「遺伝子発現」の用語は、mRNA発現(トランスクリプトーム)やmRNAによる翻訳の結果として生じるタンパク質(プロテオーム)を含むものとして使用する。
遺伝子発現の量、生体関係物質の量は測定値に限定されるものではなく、加工計算された値、相対的な値、間接的に物質量を示す量でもよい。例えば、質量スペクトルでタンパク質の発現量を測定できることを応用して、カテゴリー分類された生体状態の状態と質量スペクトルとを直接関係づける相関モデルを構築することもできる。DNAチップで各スポットの測定量を説明変数として採用することもできるし、そのスポットの測定量の加工値を利用することもできる。
【0035】
本明細書において、「第1次最適解」とは、そのときの終結条件の範囲で改善がみられなくなるまで改良したことを意味しており、全ての説明変数の組み合わせの中で最適なものを見いだしたことを確認したことを意味するものではない。
第1発明において、記憶手段に記憶される「カテゴリー分類された生体の状態」には、カテゴリー分類された生体の状態を記憶する形態のみならず、その生体の状態を導出する元になるデータを入力し、各種の処理を行った結果として、記憶手段にカテゴリー分類された生体の状態を記憶させる形態も含まれる。
【0036】
第1発明において、複数個の第1次最適解の表現形態は重み付け平均最適解を算出でき、かつ第2次最適解の算出処理に用いることのできるデータ形式であれば特に限定されない。また、重み付け平均する方法は第2発明、第3発明のような表現形態のみに限定されず、なんらかの計算手法で実質的に複数の最適解のデータを数値的に重み付け平均する場合も含まれる。
【0037】
遺伝的アルゴリズムを採用して最適解を得る方法は、計算時間とのかねあいで予め設定した世代数の中で、予め設定した適応度よりも好ましい適応度になり、改善が見られない状態になったときが最適化されたと判断する方法が採用される。但し、第5発明に記載したように第1次最適解を各モデルのレベルに応じて重み付けする方法も採用できるので、多様性を確保するという点で、本発明における終結条件の設定は一般的な遺伝的アルゴリズムの場合に比べて広義に考えることができる。
【0038】
従って、第1発明における「所定終結条件」としては、設定世代数を超えた時、設定適応度以上のものを得た時、適応度の改善が見られなくなった時を適宜、組み合わせて設定することにより第1次最適解を得るようにする。さらに、後述する図5のように連続した計算処理によって複数個の第1次最適解を得る場合だけでなく、連続せず、試行錯誤的に一つ一つ別の計算フローによって個々の第1次最適解を取得し、その取得された第1次最適解を重み付け平均して第2次最適解を算出してもよい。
【0039】
また、複数個、第1次最適解を得る場合に、第7発明に記載したように第1次最適解の個数と、初期個体集団生成処理の再設定が行われる回数は同一である場合だけに限定されない。但し、多様性を確保するという観点においては、初期個体集団生成処理の再設定が行われる回毎に最も適応度の高い1個の最適解を得る第6発明の方法が好ましいと考えられる。
第1発明などに記載した「適応度」は少なくとも各サンプルの各カテゴリーへの帰属尤度を算出することを含んで算出される。
【0040】
各サンプルの帰属尤度としては、多変量正規分布モデルにおけるマハラノビス距離に基づく帰属尤度や、F検定などで求めることのできる帰属の信頼度などを用いることができる。また第11発明に記載したように分類識別が正解か不正解かを判断してそれを適応度の計算に反映させることもできる。
【0041】
第1次最適解におけるモデル選抜の評価基準としては例えば第8発明に記載されるように赤池情報基準(AIC)あるいはその応用をモデル選抜の基準にすることで良好なモデルが得られること確認した。
適応度の算出に赤池情報基準(AIC)を用いた場合には、モデル尤度とともに相関モデルの複雑さを表現するパラメータ数を用いる。このパラメータ数はモデルに用いる説明変数の数であったり、相関モデルを伝達する情報量などであっても良い。
第17発明に記載した集団学習に用いる第1次最適解の数は、集団としての多様性を確保する点から5個以上とし、計算時間が長くなりすぎないという点から20個以下とすることが好ましい。
【0042】
第19発明〜第36発明は、サーバー型コンピュータで構成される場合や、デスクトップ型コンピュータで構成される場合を問わずに適用できる。
サーバー型コンピュータで構成される場合は、第19発明のデータ解析システムをモデル構築処理サーバーで構成することもできる。
なお、第19発明〜第36発明はデータ解析装置又はデータ解析プログラムとしても認識できるものである。
【0043】
遺伝子発現を測定し、細胞を複数カテゴリーに分類する方法の実用場面を考えた場合、「解析に用いなかったデータを正しく予測すること」が問題となる。対策として集団学習によって予測力を向上させることが考えられるが、その集団学習の前提となることは各モデルの多様性を備えていることであり、このようなモデルを複数個得ることが重要になる。
【0044】
本発明者らは種々の検討の後に遺伝的アルゴリズム(GA)が単一の最適解を探索しつつも有限時間内には必ずしも最適解を得るものではなく、所定終結条件を満足する毎に異なった最適解を得る方法であることに注目した。通常は試行回数毎に異なった最適解を得ることはGAの不完全な側面と捉えられる。しかし本発明者らはその特徴を積極的に利用し、GAで得られた第1次最適解を組み合わせることで有効な集団学習が実現することを見出した。
【0045】
その一方法として次のような方法がある。まず、複数カテゴリー分類を行なうためのモデルとして多変量正規分布モデルを採用し、AICなどの何らかの最適指標(適応度)に基づいて多変量正規分布モデルの最適解をGA探索することで第1次最適解を得て、それらを1回(後述するRun)もしくは複数回実行して複数個の第1次最適解を得る。そして得られた複数個の第1次最適解から得られたマハラノビス距離又は帰属尤度の重み付け平均を算出して第2次最適解を得ることによって集団モデルとした。集団モデルにおいて、訓練用サンプル、予測用サンプルのカテゴリー帰属は各多変量正規分布モデルの場合と同じく尤度に基づいて算出される。
【0046】
複数カテゴリー分類を行なうためのモデルとしては、多変量正規分布モデルであることは必須ではない。例えば、SIMCAモデルも部分空間からのマハラノビス距離によって帰属尤度が決定され、その応用変形は直接的であることからSIMCAモデルの採用も充分期待されるところである。更にはマハラノビス距離という概念を有しない相関モデルであっても、帰属尤度の概念をもっているモデルであれば、本発明は適用可能となる。
【発明の効果】
【0047】
以下、各発明の効果などについて説明する。
なお、データ解析システムに係る発明の効果もデータ解析方法の効果に対応しているのでそれらの効果の記載は省略する。
第1発明であれば、遺伝的アルゴリズムに基づいて複数個の第1次最適解を得る第1次最適解取得処理と、得られた複数個の第1次最適解の重み付け平均された第2次最適解を算出する重み付け平均最適解算出処理とを含んで前記相関モデル構築を行うので、予め複数個得られた第1次最適解を集団学習したような形態となり、結果としてオーバーフィットを克服して複数カテゴリー分類の分析精度を高めた相関モデルを構築できる可能性を高めることができる。また、遺伝的アルゴリズムは初期個体集団から選択と交叉との組み合わせにより並設的に山登り探索を行うとともに突然変異によりランダムな変化を起こして局所安定解に陥ることを防止できる探索手法であるから、その性質上、集団学習を行う際に必要とされるモデルの多様性(あるいは相補性)を確保する場合に好ましいと言える。
【0048】
第2発明であれば、重み付け平均最適解を重み付け平均帰属対数尤度で与えることにより、カテゴリーの帰属尤度という概念を有する相関モデルであれば適用が可能になる。
第3発明であれば、重み付け平均最適解を統計的手法に広く用いられているマハラノビス距離を重み付け平均したもので与えることにより、各種モデルに広範囲に適用が可能になる。
【0049】
第4発明であれば、第1次最適解の重み付け係数を同じ値に設定することにより、各第1次最適解をその重要度において均等に扱うことができる。
第5発明であれば、各第1次最適解の重み付け係数を各第1次最適解に対応する相関モデルの尤度に基づいて決定することにより各相関モデルの確からしさを考慮して、確からしさの高いモデルは重み付けを大きく、確からしさの低いモデルは重み付けを小さくすることが可能になる。
【0050】
第6発明であれば、説明変数候補選択処理、初期個体集団生成処理の繰り返し回数を所定終結条件に基づいて複数回行って、それぞれの回における最適解を求めるので、複数回の初期個体集団の再設定処理が行われることになり、集団学習を行う際に必要とされるモデルの多様性(あるいは相補性)を確保しやすくなる。
【0051】
第7発明であれば、説明変数候補選択処理、初期個体集団生成処理の繰り返し回数がたとえ1回で得られた最適解であっても、適応度が設定適応度を超えた最適解であれば、それらの複数個の最適解を第1次最適解取得処理における複数個の第1次最適解として設定できるので、一定レベル以上で集団学習に必要な複数個の第1次最適解を少ない計算時間で得ることができる。
【0052】
第8発明であれば、適応度の算出においてAICを使用するので、相関モデルの尤度のみならず、相関モデルのパラメータ数も考慮でき、データの情報量に見合う最適な相関モデルを選定することが期待できる。
第9発明であれば、適応度の算出において交差検証成績を使用することにより、相関モデルを簡単な計算で最適化することができる。
【0053】
第10発明であれば、適応度を算出する場合に、各サンプルの帰属尤度を全サンプルについて乗算することにより得られたモデル尤度を使用することにより各帰属尤度の値を反映した値にすることができる。
第11発明であれば、適応度算出処理において、予測カテゴリーと実測カテゴリーと一致する場合を識別正解、そうでない場合を識別不正解と判別する処理を含んでいるので、適応度を自然に訓練用サンプルに適応したものとすることができる。
【0054】
第12発明であれば、多変量正規分布モデルは比較的計算が簡単で判別分析に使用する相関モデルとして研究が進んでいることから適用が行いやすい利点がある。
第13発明であれば、計算を単純化し、かつ共分散行列の推定精度を向上することができるとともに、特に共分散行列の次元を増加することが可能になるので、カテゴリー当たりのサンプルの数が少ない場合であっても多数の説明変数を採用してモデル構築が可能となる利点がある。
【0055】
第14発明であれば、多変量正規分布モデルの共分散逆行列をムーア・ペンローズの一般化逆行列を用いて求めるので、カテゴリー当たりのサンプル数が少ない場合にも多数の説明変数を採用してモデル構築が可能となる利点がある。
第18発明であれば、重み付け平均最適解を用いて集団学習的な手法を用いることによりオーバーフィットを克服し、分類能力の高いデータ解析ができる相関モデルを有しているので、予測用サンプルを入力した場合にカテゴリー分類された生体の状態を予測する精度の高い出力を得ることができる。
【発明を実施するための最良の形態】
【0056】
以下、上記各発明について数式、図面などを用いて、より具体的に説明する。
本実施形態では、本発明のデータ解析方法を複数カテゴリー分類のための遺伝子発現解析法に適用する。また、相関モデルとして後述する多変量正規分布モデルを採用するとともに、最適化処理として遺伝的アルゴリズムを採用する。
上記発明の内容を限定するものではないが、データ解析方法の一例のフローチャートを図1〜図6に示す。
【0057】
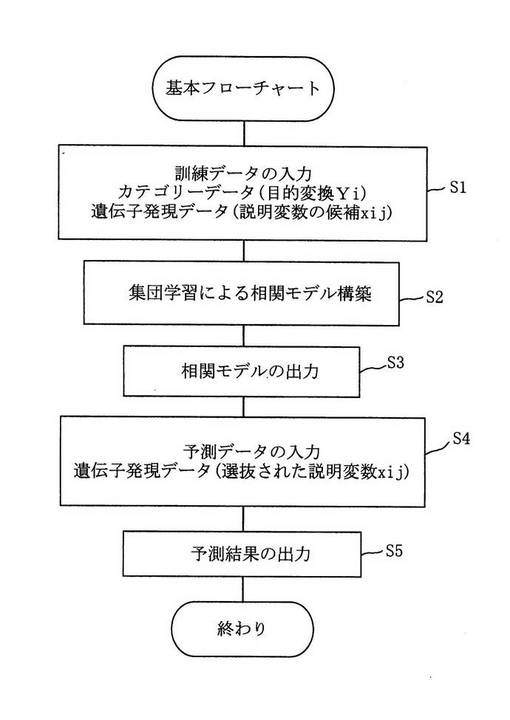
図1はデータ解析処理の全体を示すフローチャートであり、カテゴリーデータ(目的変数Yi)および遺伝子発現データ(説明変数の候補xij)からなる訓練データを入力した後(ステップ1)、集団学習による相関モデル(複数個の多変量正規分布モデル及びその重み)を構築し(ステップ2)、その多変量正規分布モデルを出力する(ステップ3)。
また、遺伝子発現データ(選抜された説明変数xij)を入力し(ステップ4)、前記多変量正規分布モデルに基づいて診断予測結果を出力する(ステップ5)。入出力データは必ずしも図1に厳密に従ったものである必要はなく、よく定義された加工方法による加工前あるいは加工後のものでもよい。
【0058】
また、モデル構築と診断予測は必ずしも時間的、空間的に連続した処理である必要はない。例えば、胃癌、肺癌、肝臓癌、……と診断された多数の患者の各遺伝子発現データを使用して図1のステップ1〜ステップ3の処理により、各癌診断の相関モデルの構築をデータ解析センターにおいて集中的に行い、そのデータ解析の成果として構築された癌診断用の相関モデルを世界各地の病院においてステップ4,ステップ5の処理を行って診断に利用する形態などが考えられる。
【0059】
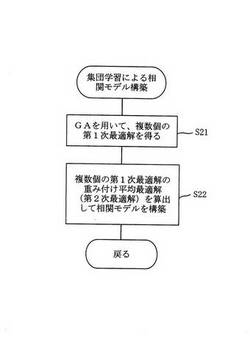
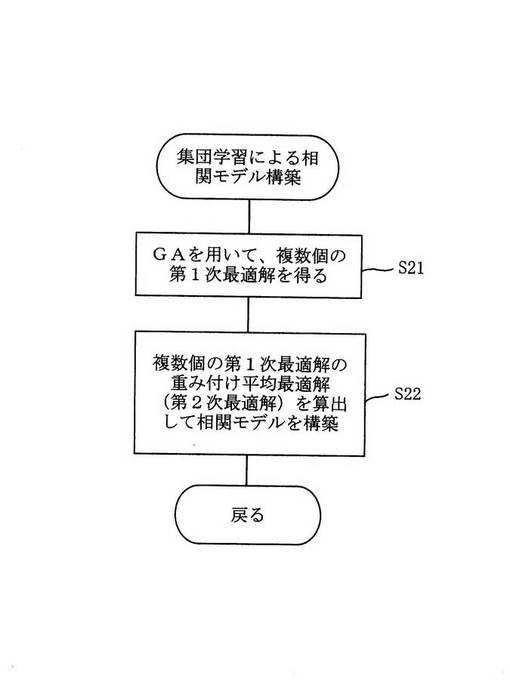
図2は図1のステップ2に示す集団学習による相関モデル構築処理のさらに詳しいフローチャートの一例である。
図2において遺伝的アルゴリズムを用い、各サンプルにおける各カテゴリーの重み付け平均された帰属尤度に基づいて適応度を計算しつつ、多変量正規分布モデルの第1次最適解を複数個得て(ステップ21)、それら複数個の第1次最適解の重み付け平均最適解を算出して第2次最適解を求める(ステップ22)。
【0060】
次に、図2のステップ21に示される多変量正規分布モデルの第1次最適解を得る処理について次の記号を用いて説明する。
iはサンプルのインデックス
jは説明変数(遺伝子発現量)のインデックス
kはカテゴリーのインデックス
Niはサンプルの個数
Njは説明変数の個数
Nkはカテゴリーの個数
Xiはサンプルiの説明変数ベクトル
xijはサンプルiの説明変数jの値
Xkはカテゴリーkの説明変数ベクトルの平均
[σk2] はカテゴリーkの共分散行列
Dikはサンプルiのカテゴリーkの中心へのマハラノビス距離
但し、上記Niはカテゴリーk毎に変化する値なので、そのカテゴリー毎に変化することを明示的に表示する場合は、以下に記述する数式及び添付するフローチャートにおいてNi(k)と表示する。
【0061】
まず、多変量正規分布モデル(多変量正規化ガウス確率分布密度関数モデルとも称される)では、各サンプルiの各カテゴリーkへの帰属尤度Likは次式で表される。
【0062】
【数1】

【0063】
で与えられる。ここで(xi−Xk)tは列ベクトル(xi−Xk)を転置した行ベクトルである。また、Dikはマハラノビス距離を表現している。
マハラノビス距離とは平均だけでなく、分散、共分散も考慮して定義された距離であり、サンプルiのカテゴリーkの中心へのマハラノビス距離の2乗Dik 2は、
【0064】
【数2】
【0065】
で示される。ここで、[σk2]はカテゴリーkの共分散行列であり、
【0066】
【数3】
【0067】
で与えられる。上記(3)式でΣ(i∈k)はカテゴリーkに属するサンプルiについての総和を意味している。なお、本実施形態では、上記(3)式の代わりに下記(4)式を用いる。
【0068】
【数4】
【0069】
(4)式を用いる理由は(A)計算の単純化、(B)共分散行列の推定精度向上、(C)共分散行列の次元を増加できる、などの利点があるからである。特に、(3)式で示す共分散行列[σk2] の次元はカテゴリーkに含まれる訓練サンプルの数より小さく、GCMデータでは7以下である。したがって8個を超える遺伝子を含むモデルは計算不能となる。[σ2]を用いることは前記P.Tanらの文献に記載されているが、本来カテゴリーごとに異なる[σk2] を同一視する弊害があるにも拘わらず、8遺伝子を超えるモデルを可能にするものであり、利点の方が大きいので(4)式を採用することにした。この(4)式を用いることは前記第13発明の一形態を示すものである。
【0070】
また、(4)式を用いる他に、[σk2]の逆行列[σk2] -1が計算不能の場合の別の対処方法として一般逆行列を用いることがある。行列Aの一般逆行列とは
AGA=A
を満たすGのことであり、A-1が計算不能の場合にはGは必ずしも一義的に決定されない。しかし、次の条件を追加したMoore-Penrose型(ムーア・ペンローズ)一般逆行列は一義的に決まり、特異値分解の方法によって求めることができる。
GAG=G, (GA)t=GA, (AG)t=AG
[σk2]のムーア・ペンローズ型一般逆行列[σk2] -1 MPを適用すると、カテゴリー当りのサンプル数が少ない場合にもカテゴリーごとに異なる[σk2]を用い、しかもある程度の数の遺伝子発現量を説明変数として利用することができる。このようにムーア・ペンローズ型一般逆行列を用いることは前記第14発明の一形態を示すものである。
【0071】
次に、相関モデルの適応度を算出する方法について説明する。
本実施形態では、前記適応度としてAIC(赤池情報基準)を最適化した多変量正規分布モデルを得る。
ここでAICとは、例えば、下記(5)式で計算される量であり、相関モデルの複雑さに依存する場合にその複雑さを表す第1項と、相関モデルの当てはまりの良さを表す第2項とのバランスを取った量である。
本実施形態ではAIC値が小さいほど優れたモデルとなるので、AIC値が小さくなるように多変量正規分布モデルを決定する。
【0072】
【数5】
【0073】
ここでkは両項のバランスを取るための調節パラメータであり、任意に設定できるものとする。k=0という特別なケースは最尤法に相当し、k=1は通常のAICの計算方法である。(5)式を使用する場合にk≠0の時は、モデルのパラメータ数Npも考慮して適応度を算出することになる。多変量正規分布モデルのパラメータ数Npを簡単に計算する場合は、説明変数の個数Njを採用することができる。
多変量正規分布モデルの尤度Lは例えば下記(6)式で定義することとした。
【0074】
【数6】
【0075】
但し、Liは訓練用サンプルiの尤度であり、Πiは訓練用サンプル集合の全サンプルi=1〜Niについての積を表す。この(6)式は前記第10発明の一形態を示したものである。また、帰属尤度Likは前記(1)式において説明したようにマハラビノス距離の2乗Dik 2によって算出できる値であり、サンプルiがカテゴリーkに帰属する信頼度である。
帰属尤度Likの算出方法としては、(1)式の他にF検定による下記(8)式の関係式を満たす量として算出することもできる。
【0076】
【数7】
【0077】
ここでF(Nf,P)は自由度Nf、危険率PのF検定の限界値、Nikはカテゴリーkに属するサンプルの個数である。このようなLikを求めることは、公知のF分布表あるいは公知のアルゴリズムを利用することによって可能である。あるサンプルiに対して最大のLikを与えるカテゴリーk=kmaxが最尤帰属カテゴリーであり、kmaxと実測カテゴリーが一致する場合を識別正解、そうでない場合を不正解と呼ぶ。
(6)式を用いた場合には識別正解と不正解との区別が鮮明ではない。そこで敢えて鮮明にするために、各iごとにLik (k=1〜Nk)を全て計算し、一番大きなカテゴリーに帰属するとして下記(9)式のようにLiを定義することもできる。ここでpenaltyは十分小さな量であり、例えば10-6である。この(9)式は前記第11発明の一形態を示したものである。
【0078】
【数8】
【0079】
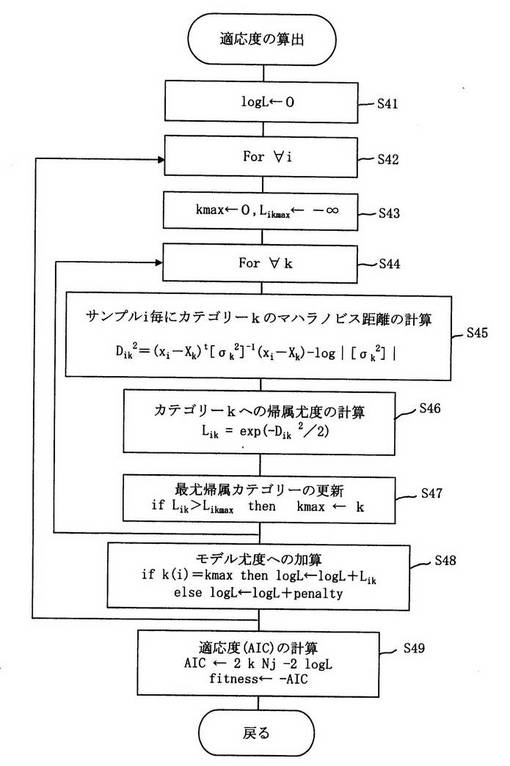
図3は適応度として上記赤池情報基準(-AIC)を用いた場合の、適応度の算出方法のフローチャートの一例である。
図3に示すようにサンプルi毎に各カテゴリーkの中心へのマハラノビス距離の2乗Dik 2を計算し(ステップ45)、(1)式に基づいて帰属の信頼度Likを計算し(ステップ46,ステップ47)、最尤帰属カテゴリーkmaxが実測カテゴリーk(i)と一致するかどうかの条件判定も行ないながらモデル尤度の計算を行ない(ステップ48)、適応度としてのAIC値を求める(ステップ49)。
図3に示すフローチャートは前記(9)式に準拠したものであり、前記(6)式に準拠する場合には最尤帰属の判定は不要であり、フローチャートは簡素なものになる。
【0080】
図4は図3のステップ45において記載したカテゴリーkの共分散逆行列[σk2] -1の算出フローチャートの一例である。
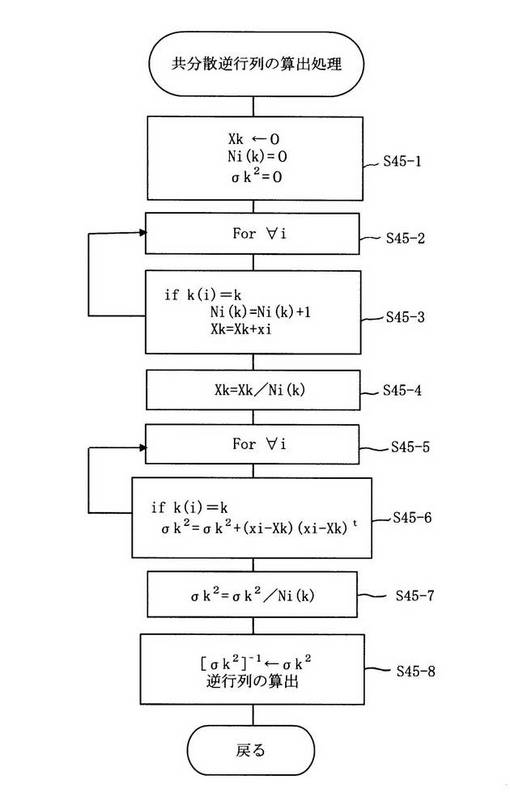
図4に示すようにカテゴリーkのサンプルの個数Ni(k)とカテゴリーkの説明変数ベクトルの平均Xkを算出し(ステップ45−2〜ステップ45−4)、算出されたNi(k)とXkに基づいてカテゴリーkの共分散行列σk2を算出し(ステップ45−5〜ステップ45−7)、カテゴリーkの共分散逆行列を算出する(ステップ45−8)。なお、本方法はカテゴリーkによらないものとして共分散逆行列を算出する方法やムーア・ペンローズの一般逆行列を用いて共分散逆行列を算出する方法に修正(ステップ45−7〜ステップ45−8)して実行する方法もある。
【0081】
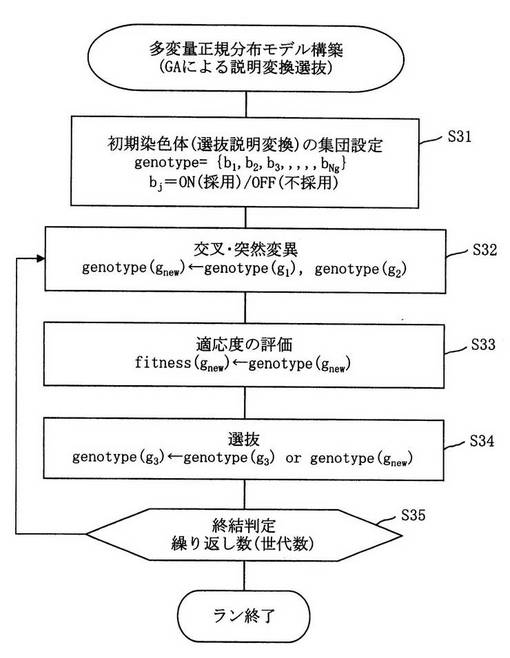
図5はモデル構築を遺伝的アルゴリズム(GA)で行う場合のフローチャートであり、このフローチャートを参照しつつ、遺伝的アルゴリズムについての予備的説明を行う。
遺伝的アルゴリズム(GA;Genetic Algorithm)は、進化論をヒントとした非線型最適化手法であり、ここ数年間、頻繁に用いられて定着してきた。遺伝的アルゴリズムを説明した文献としては、伊庭斉志;「遺伝的アルゴリズムの基礎」;オーム社;(1994)などがある。
GAでは最適化対象がビット列などで表現される。このような表現の形式を遺伝型(genotype)と呼び、ひとつひとつの遺伝型の具体的な文字列を染色体(chromosome)と呼ぶ。最適化の指標である適応度(fitness)は遺伝型より一義的に決定される。
【0082】
(処理1) 初期染色体プールの準備(図5のステップ31)
ランダムに染色体の集合を準備する。染色体の数はプールサイズと呼ばれる。
(処理2) 交叉、突然変異、選択による最適化(図5のステップ32〜ステップ34)
[2-1] 交叉(crossover)
集合より2つの染色体を選抜し、ビット列のランダムな交叉によって新しい染色体を創生する。交叉を1個所に限る一点交叉(onepoint crossover)と全ビット間で交叉を行なう一様交叉(unform crossover,UX)などがあり、一般的にはUX法が優れているとされている。
[2-2] 突然変異(mutation)
集合あるいは新しい染色体の任意のビットをランダムに選んで反転させる。
[2-3] 選択(selection)
各染色体の適応度を計算し、染色体プールを次世代のものに置き換える。適応度に比例した確率で染色体を選抜するルーレット方式や親世代と子世代との間で適応度の対決を行なうトーナメント方式などがある。これらいずれにおいても乱数が用いられることが多く、一度得られた最適染色体が失なわれることもある。このようなことが無いよう、プール内の最適染色体は必ず次世代に選抜されるとするエリート戦略なども用いられる。
【0083】
(処理3) 終結判定(図5のステップ35)
世代数がある回数に達した場合や、最適適応度に変化が見られなくなった場合に処理2は終了し、最適解は最適染色体の遺伝型およびその適応度として出力される。GAは基本的には乱数を用いた方法であるため、処理1〜処理3が何回か試みられ(Run)、その中の最適染色体が用いられることが多い。
【0084】
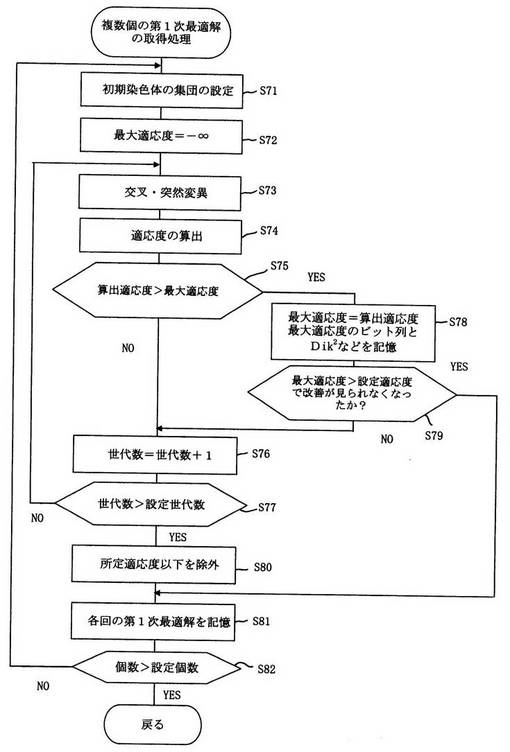
図6は図5に示した遺伝的アルゴリズムを用いた第1次最適解を複数個得るためのフローチャートの一例である。
図6において乱数を用いて初期染色体の集団をビット列で設定し(ステップ71)、予め最大適応度を−∞にするなどの初期設定を行い(ステップ72)、交叉・突然変異の処理を行い(ステップ73)、適応度(図6では値が大きいほどモデルが適合しているとする)を算出し(ステップ74)、算出された適応度が前の世代の最大適応度よりも大きいか否かを判別し(ステップ75)、算出された適応度が前の世代の最大適応度よりも大きくないと判別された場合は、世代を+1追加し(ステップ76)、終結判定基準として設定世代数よりも大きいか否かを判別し(ステップ77)、大きくないと判別された場合は、次の世代の交叉・突然変異の処理を実行する。
【0085】
一方、ステップ75において算出された適応度が前の世代の最大適応度よりも大きいと判別された場合は、算出適応度を最大適応度に設定するとともに、コンピュータの記憶装置に最大適応度のビット列とその時のマハラノビス距離の2乗Dik 2又は帰属尤度Likを記憶し(ステップ78)、終結判定基準として設定適応度よりもその最大適応度が大きくて、かつ算出適応度の改善傾向が見られなくなったか否かを判別し(ステップ79)、設定適応度よりもその最大適応度が大きくかつ改善傾向が見られなくなったと判別された場合は後述するステップ81へ移行し、それ以外の場合であると判別された場合は、ステップ76に移行する。
上記処理により算出された適応度が設定適応度を超え、かつ算出適応度の改善傾向が見られなくなった場合に計算のループを抜け出してその時のビット列とマハラノビス距離又は帰属尤度のデータを記憶装置に記憶することができる。
【0086】
また、ステップ77において設定世代数よりも大きいと判別された場合は、必要に応じて記憶された最大適応度が最低基準適応度よりも大きいか否かを判別して第1次最適解の候補として好ましくないものを除外し(ステップ80)、世代数による第1次最適解のデータと、ステップ79の条件で得られた第1次最適解のデータとを回(Run)ごとに整理して記憶装置に記憶する(ステップ81)。
【0087】
そしてステップ82において得られた第1次最適解の個数が、予め設定された設定個数よりも大きいか否かを判別して、設定個数得られていないと判別された場合は、新たなる回(Run)においてステップ71の初期染色体の集団設定から再び行い、第1次最適解の個数が設定個数得られたと判別された場合は一連の処理を終了する。
【0088】
次に、図2のステップ22に示した複数個の第1次最適解に基づいて重み付け平均最適解を算出する処理について説明する。
(1)式において示したように、帰属尤度Likはサンプルi毎に各カテゴリーkのマハラノビス距離を計算することにより求めることができる。このLikの算出は遺伝的アルゴリズムを用いた場合、回(Run)ごと(あるいは所定終結条件を満足した回数毎)に異なった最適解が得られることを利用する。これら複数の最適解を第1次最適解と言っていることは前述の通りである。そして、それらの第1次最適解の重み付け平均を求める。例えば、各回の第1次最適解のデータがマハラノビス距離で与えられる場合は、重み付け平均マハラノビス距離は、
【0089】
【数9】
【0090】
と表される。ここで、w(m)はm番目のモデルの重み付け係数、mは複数個得られるモデルの数、Dik 2(m)はmモデルのDik2の値を示す。この(10)式は前記第3発明の一形態を示したものである。
また、各回の第1次最適解のデータがマハラノビス距離でなく、直接的に帰属尤度Likで与えられる場合は、重み付け平均帰属対数尤度は、
【0091】
【数10】
【0092】
と表される。この(11)式は前記第2発明の一形態を示したものである。
なお、重み係数w(m)としては一定値を用いたり、各モデルの尤度を重み係数w(m)としたり、各モデルの尤度を重み係数w(m)を考慮した値とすることもできる。これらは前記第4発明、第5発明の一形態を示すものである。
【0093】
このように複数個得られた第1次最適解を用いて上記(10)式又は(11)式に基づいて第2次最適解を算出する(図2のステップ22)。この算出処理によってGAを用いた集団学習による相関モデルが構築されることになる。
【0094】
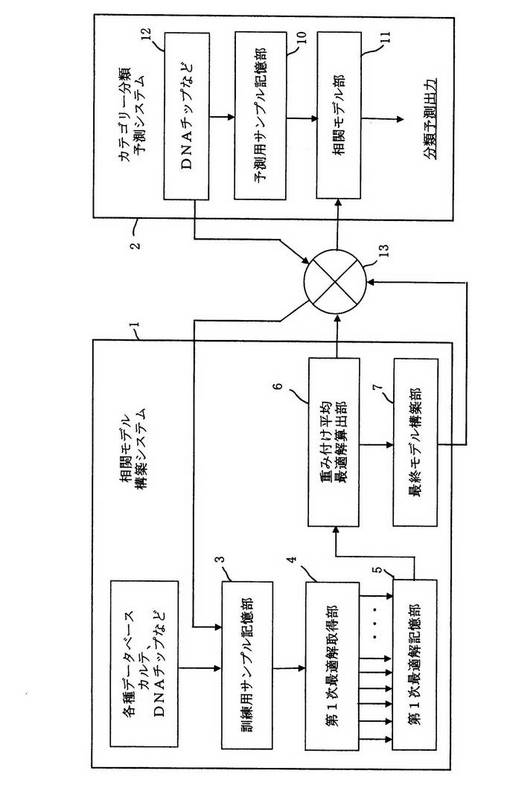
図7は本実施形態に係るデータ解析システムの一例を示す概略的なブロック図である。
本データ解析システムは、第19発明に記載したような相関モデル構築システム1と第36発明に記載したような予測サンプルを用いるカテゴリー分類予測システム2とに大別される。相関モデル構築システム1は、前記訓練用サンプルの記憶手段としての訓練用サンプル記憶部3と、前記第1次最適解取得手段としての第1次最適解取得部4と、複数の第1最適解を記憶する第1次最適解記憶部5と、前記第2次最適解を算出する重み付け平均最適解算出手段としての重み付け平均最適解算出部6とを備え、必要により最終モデル構築部7とを備えている。最終モデル構築部7は第2次最適解で得られる相関モデルをさらに改良、微調整する機能がある。
【0095】
訓練用サンプル記憶部3に入力される各データは大学、研究所が提供する生体関係情報を提供するウエブサイト、疾病関係データベースなどの各種データベースから得ることも可能である。また、DNAチップなどを用いて患者の遺伝子発現量のデータとカルテなどからの生体状態のカテゴリー分類データを取得して入力しても良い。
【0096】
カテゴリー分類予測システム2は、予測用サンプル記憶部10と、前記第2次最適解取得手段で構築された相関モデル(又は最終モデル構築部で構成された相関モデル)を予測用相関モデルとして設定した相関モデル部11とを備え、DNAチップ12などから取得した患者の遺伝子発現データに係る選抜した説明変数を入力することにより、どのカテゴリーに属するかの分類予測出力を得ることができるように構成してある。
【0097】
なお、図7では図示していないが、第1次最適解取得部4は、説明変数候補選択部、初期個体集団生成部、帰属尤度算出部、適応度算出部、次世代個体集団生成部、繰返し処理部を少なくとも含んで構成してある。
【0098】
本データ解析システムはスタンドアロン型のコンピュータでも、クライアントサーバー型のコンピュータでも構成できる。
スタンドアロン型のコンピュータで構成される場合は、相関モデル構築装置とカテゴリー分類予測装置として構成される場合が多い。本データ解析システムはネットワーク13上に配置された相関モデル構築サーバーと各病院等に設置されたカテゴリー分類予測コンピュータを連携させて構成することも可能である。この場合は、各病院等に設置されたカテゴリー分類予測コンピュータ側において、病院に設置されたDNAチップ12などを用いて患者の遺伝子発現量のデータとその病院のカルテから患者の生体状態のカテゴリー分類データを取得して、ネットワーク13を介して相関モデル構築サーバーへ訓練用サンプルの一部として送信することもできる。なお、本データ解析システムはネットワーク上の機能実現プログラムや記憶装置などで構成しても良い。
【実施例1】
【0099】
以下、具体的な実施例について説明する。
データは前記したGCMデータを用いた。それぞれ8サンプルよりなる18種類の癌細胞合計144個の訓練サンプル集合と、同じく18種類46個の独立テスト集合(予測用サンプル集合に相当)とより成る。各サンプルについて、約16,000遺伝子の発現データがあり、その中で訓練サンプル集合の分散の大きな上位1000個(重複を除き968個)を説明変数とした。
このようなデータでbagging等を用いると、カテゴリー当りのサンプル数が極端に少なくなり、有効なモデルが得られないと懸念される。
【0100】
識別モデルとしては多変量正規分布モデルを用い、最適化指標(適応度)としては交差検証予測不正解率(Pmis(cv))とAIC(赤池情報規準、AIC=2Nj-1ogL)について検討することとした。交差検証予測不正解率の方法はP.Tanらの方法に準じたものである。但し、P.TanらのGA/多変量正規分布モデルは交差検証予測不正解率(Pmis(cv))と独立テスト集合予測不正解率(Pmis(ind))との和を指標としてGAによって遺伝子を選抜している。しかし、この方法のようにモデル選抜に独立テスト予測不正解率を用いることは、モデルの予測精度を評価する上で問題と考えられるので交差検証予測不正解率(Pmis(cv))だけを用いて計算した。
一方、最適化指標にAICを用いる方法は前述のように本発明の重要な特徴の一つである。多変量正規分布モデルの共分散行列は(3)式及び(4)式において説明した議論により、P.Tanらの方法に準じてカテゴリーによらないものとした。即ち、σ2=Σkσk2で算出している。
【0101】
本実施例において多変量正規分布モデルの最適化のために使用したGAの設定は次のとおりである。
(1)genotype={b1,b2,b3,,,,,,bNg}
遺伝子発現量gjを説明変数に用いる場合にはbj=ON状態、
用いない場合にはbj=OFF状態とする。
(2)fitness=-AIC あるいは-Pmis(cv)
(3)プールサイズ=100
(4)初期染色体は、平均でmin_of(Ni, Ng, 300)/2個のビットがON状態となるように乱数を用いて準備する。ここでNiは訓練用サンプル数、Ngは説明変数候補の数、300は実装の都合上設定された定数である。
(5)交叉は、2つの染色体を乱数にて抽出し、各ビットをそれぞれ1/2の確率でいずれかの親染色体から引き継いだ子染色体を作成する。
【0102】
(6)突然変異は、ON状態ビット数が増加する変異と減少する変異とが同じ確率で発生するように乱数でビット反転を行なった。
(i)ON状態ビットの反転確率=1.1/ON状態ビット数
(ii)OFF状態ビットの反転確率=1.1/OFF状態ビット数
(7)選抜は、子染色体を作成する毎にトーナメント方式でプールを置換する。
(i)子染色体が新たな最適解となる場合は無条件で置換
(ii)トーナメント相手が最適解の場合には無置換
(iii)子染色体のfitness<トーナメント相手のfitnessの場合には0.25の確率で置換
(iv)子染色体のfitness>トーナメント相手のfitnessの場合には0.75の確率で置換
(8)終了は、交叉→突然変異→選抜の繰り返し回数の上限を最初100,000とし、最適解が見出される毎に1000追加されるものとした。
【0103】
上記GAの設定で、Pmis(cv)最適解、AIC最適解を最大世代数を9,000に設定して探索し、これを5回(5Run)繰り返し、それぞれ得られたモデルで独立テスト集合予測不正解数Nmis(ind)を検証した。これを表1のAICにおいてRun#の0,1,2,3,4で示した。またマハラノビス距離の5回の重み付け平均を用いた場合のNmis (ind)も同様に求め、これを表1のAICにおいてΣ5で示した。今回の実施では重み付け係数w(m)はモデルによらず一定とした。結果は表1に示すようにAIC最適解において重み付け平均を採用することによって不正解数Nmis(ind)=16となり、予測精度(Nmis(ind))の顕著な向上が確認された。
【0104】
比較のためPmis(cv)最適解でも同様の検討を行なった。表1のPmis(cv) 最適解を見て分かるようにAIC最適解よりPmis(cv) 最適解は劣るが、それでもPmis(cv) 最適解のΣ5はRun#の0,1,2,3,4に比べて改善されていることが分かる。
以上の訓練集合と独立テスト集合とをあわせて新たに訓練集合とし、AIC最適解をGAで探索したところ、誤分類率は訓練用サンプル集合で19/144、独立テスト集合で18/46であった。一方、両誤分類数+Njを最小とする解を探索したところ、誤分類率は訓練集合で11/144、独立テスト集合で10/46であった。これより、10/46〜18/46程度の誤分類率は多変量正規分布モデルと968遺伝子がもつ本質的な限界であると考えられる。なお、[σk2] を用いたモデリングでは少数遺伝子のフィット力に乏しいモデルしか得ることができなかった。
【0105】
【表1】
【実施例2】
【0106】
Golubらのデータ(AML/ALLデータ)に基いて加工されたデータにて解析を行なった。表2にその結果を示す。なお、1〜38が訓練用サンプル集合、39〜72が予測用サンプル集合である。表2において1〜18がそれぞれ複数個の第1次最適解としての多変量正規分布モデルの予測結果(○が正解、×が不正解)を示し、Σが集団学習による第2次最適解のモデルによる予測結果を示す。
表2に示すようにNmis(ind)=1の結果が得られた。各多変量正規分布モデルの予測はそれぞれ決して良好ではないが、集団判定によって飛躍的に改善している様子が分かる。
【0107】
【表2】
【図面の簡単な説明】
【0108】
【図1】図1はデータ解析処理の全体を示すフローチャートである。
【図2】図2は図1のステップ2に示す集団学習による相関モデル構築処理のさらに詳しいフローチャートの一例である。
【図3】図3は適応度を計算するフローチャートの一例である。
【図4】図4はカテゴリーkの共分散逆行列の算出フローチャートの一例である。
【図5】図5は遺伝的アルゴリズム(GA)を説明するためのフローチャートである。
【図6】図6は遺伝的アルゴリズムを用いた第1次最適解を複数個得るためのフローチャートの一例である。
【図7】図7はデータ解析システムの一例を示す概略的なブロック図である。
【符号の説明】
【0109】
3…訓練用サンプル記憶部、4…第1次最適解取得部、5…第1次最適解記憶部、6…重み付け平均最適解算出部、10…予測用サンプル記憶部、11…相関モデル部。
【技術分野】
【0001】
本発明は、生体に関する情報を解析するデータ解析方法及びそのシステムに関する。
【背景技術】
【0002】
ヒトゲノムの解読以降、ゲノムに書かれた遺伝情報がどのように発現して機能しているのかを解明するポストゲノム時代に突入したと言われ、そのような動向を踏まえて遺伝子発現状態を測定する技術も年々進歩している。
遺伝子発現状態を効率的に測定する技術として次のものがあげられる。まず、トランスクリプトーム(mRNAの総体)を特定するものとして、基盤に複数種のDNAを担持し、それに相補的なmRNAを検出するDNAチップが知られている。代表的なDNAチップには、遺伝子チップやDNAマイクロアレイがある。
【0003】
また、プロテオーム(蛋白質の総体)を特定するものには、2次元電気泳動、抗体チップ、質量スペクトルを用いるものがある。さらにメタボローム(代謝中間体を含めた代謝産物の総体)を測定する手法にも質量分析などが適応されつつある。これらの測定技術は、生体の状態に関するパラメータを短時間に一挙に測定できることがそれまでの技術と比較して画期的であると言える。
【0004】
生体内の細胞の状態は遺伝子発現データによってよく記述されるため、従来の診断マーカーでは情報が不足している場面でも、精度のより高い診断が可能になるという期待も出てきている。特にカテゴリー分類された生体の状態を目的変数とし、DNAチップ等から得られた遺伝子発現データを説明変数としたカテゴリー分類解析法は患者に負担をかけることなく重大な疾病の早期発見、早期治療を可能にするものとして研究が行われている。
例えば、非特許文献1に示すように、P.Tanらはmahalanobis距離に基づく最尤帰属の方法を用いて癌細胞の複数カテゴリー分類を行なった。非特許文献1内では61細胞の9カテゴリー分類(以下、NCI90データと称する。データはhttp://genome-www.stanfoard.edu/suteeh/download/nci90)と、198細胞の14カテゴリー分類(以下、GCMデータと称する、データはhttp://www-genome.wimit.edu/mpr/publication/projects/Global Cancer Map/)とがそれぞれ13,32個の遺伝子の発現量で良好に説明されている。
【0005】
一方、単一のモデルだけでなく、複数個のモデルを組み合わせて安定した優秀な予測を得る方法がここ数年、情報処理の分野で活発に研究されている。bagging, boostingなどリサンプリングによって識別力の弱い学習モデルを複数個獲得し、それらの学習モデルを組み合わせて強い学習モデルにする方法論が盛んに研究されている。例えば、非特許文献2に示すように、Golubらは急性白血病の分類においてwv(weightedvoting)と呼ばれる方法を提案し、一つ一つの遺伝子の判定に重みを付けて票決することで優れた予測識別力が得られることを示している。
【0006】
【非特許文献1】C.H. Ooi & P.Tan;Genetic Algorithm applied to multi-class prediction for analysis of geneexpression data; Bioinformatics 19 (1) 37-44 (2003)
【非特許文献2】T.R. Golub. Science. 286, 531-537, 1999
【発明の開示】
【発明が解決しようとする課題】
【0007】
しかしながら、上記従来技術であれば下記のような課題がある。
(A)非特許文献1の方法に関する課題
遺伝子発現データの特徴はサンプル数Niが数十個程度と少なく、説明変数数Njが数百〜数万個のように膨大であることにある。手元のデータを綺麗に説明するモデルは簡単に得られるが、それらのほとんどは解析に用いられなかったデータへの応用力(以下、汎化力と呼ぶ)に乏しい。これはオーバーフィット(overfit)の問題と称されている。そこで手元のデータの一部をマスクし、予測実験(交差検証:crossvalidation, cv)を行って予測力の優れたモデルを選抜する方法が最近の傾向である。
しかし、遺伝子発現データほどNjが膨大な場合には、手元データのcv予測に優れているものの、真の汎化力に乏しい見せ掛けモデルである危険性が大きいことが分かってきた。これはある臨床グループによる研究が一見「完璧」でも実用場面に応用すると無効になることを意味する。このような事態を回避するためには、cvを超える新たな方法によってオーバーフィットを克服することが求められている。
前記非特許文献1ではP.Tanらは交差検証予測正解率だけで選抜すると独立テスト正解率が悪くなると報告しており、オーバーフィットを克服しているとは言えないものである。
【0008】
(B)非特許文献2の方法に関する課題
非特許文献2に示したリサンプリングの方法は複数カテゴリー分類において各カテゴリーあたりのサンプル数が多い場合には適しているが、少ない場合には不向きであると思われる。また、wv(weightedvoting)は一つ一つの遺伝子での判定というモデル自体が非常に単純であり、複数カテゴリー分類への適応性が明らかでない。
【0009】
本発明は上記課題に鑑みてなされたものであり、本発明の目的は、上記課題を解決できる、データ解析方法及びデータ解析システムを提供することにある。
具体的な目的の一例を示すと、以下の通りである。
(a)生体データの解析において、サンプル数が少なく、説明変数の数が膨大である場合でもオーバーフィットを克服してカテゴリー分類能力の高いデータ解析ができる方法及びそのシステムを提供する。
(b)生体データの解析において、集団学習の考え方を取り入れて比較的簡単な計算でカテゴリー分類能力の高いデータ解析ができる方法及びそのシステムを提供する。
なお、上記に記載した以外の発明の課題及びその解決手段は、後述する明細書内の記載において詳しく説明する。
【課題を解決するための手段】
【0010】
本発明は多面的に表現できるが、例えば、代表的なものを挙げると、次のように構成したものである。
第1発明のデータ解析方法は、コンピュータを用いて、カテゴリー分類された生体の状態を目的変数とし、遺伝子発現、生体関係物質の少なくとも一方に関するデータを説明変数とする相関モデルを決定する複数カテゴリー分類用のデータ解析方法であって、
カテゴリー分類された生体の状態と、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる訓練用サンプルの集合をコンピュータの記憶手段に記憶しておき、
遺伝的アルゴリズムに基づいて複数個の第1次最適解を得る第1次最適解取得処理と、
得られた複数個の第1次最適解の重み付け平均された第2次最適解を算出する重み付け平均最適解算出処理と、
を含んで前記相関モデル構築を行い、
前記第1次最適解取得処理が下記(a)〜(f)の各処理を含んで行われることを特徴とする。
(a)説明変数の候補を選択する説明変数候補選択処理、
(b)各個体が相関モデルの候補を表しており、それらの個体の集団である第1世代を生成する初期個体集団生成処理、
(c)各個体について、各サンプルの各カテゴリーへの帰属尤度を算出する帰属尤度算出処理、
(d)算出された帰属尤度に基づくサンプルの予測カテゴリーとサンプルが実際に帰属する実測カテゴリーとを比較し、その予測がどの程度、信頼性があるかという定量的な評価である適応度を算出する適応度算出処理、
(e)算出された適応度に基づいて、個体に関する突然変異、交叉、選抜などの遺伝的アルゴリズムに基づいて次の世代の個体集団を生成する次世代個体集団生成処理、
(f)少なくとも帰属尤度算出処理、適応度算出処理、次世代個体集団生成処理にわたる一連の処理を繰り返し行って、最も適応度の高い最適解を探索しつつ、所定終結条件を満足する複数個の第1次最適解を得る
【0011】
第2発明は、第1発明において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの帰属尤度を重み付け平均した重み付け平均帰属対数尤度で与えられることを特徴とする。
第3発明は、第1発明において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの中心へのマハラノビス距離を重み付け平均したもので与えられ、前記帰属尤度算出処理がその重み付け平均マハラノビス距離に基づいて重み付け平均化された帰属対数尤度を算出することを特徴とする。
【0012】
第4発明は、第1発明ないし第3発明のいずれか一つに記載の発明において、各第1次最適解の重み付け係数を同じ値に設定したことを特徴とする。
第5発明は、第1発明ないし第3発明のいずれか一つに記載の発明において、各第1次最適解の重み付け係数を各第1次最適解に対応するモデル尤度に基づいて決定することを特徴とする。
【0013】
第6発明は、第1発明ないし第5発明のいずれか一つに記載の発明において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返し(後述するRun)を複数回行う条件が含まれており、その各回で得られた最適解を第1次最適解取得処理における複数個の第1次最適解として設定することを特徴とする。
【0014】
第7発明は、第1発明ないし第6発明のいずれか一つに記載の発明において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返し(後述するRun)を少なくとも一回行う過程で、算出適応度が設定適応度を超えた最適解を複数個抽出することが含まれており、それらの最適解を第1次最適解取得処理における複数個の第1次最適解として設定することを特徴とする。
【0015】
第8発明は、第1発明ないし第7発明のいずれか一つに記載の発明において、前記適応度算出処理がAIC(Akaike'sInformation Criterion:赤池情報基準)に基づいて行われることを特徴とする。
第9発明は、第1発明ないし第7発明のいずれか一つに記載の発明において、前記適応度算出処理が交差検証成績に基づいて行われることを特徴とする。
【0016】
第10発明は、第1発明ないし第9発明のいずれか一つに記載の発明において、前記適応度算出処理が、前記帰属尤度算出処理によって算出された各サンプルの帰属尤度を全サンプルについて乗算することにより得られたモデル尤度に基づいて行われることを特徴とする。
【0017】
第11発明は、第1発明ないし第10発明のいずれか一つに記載の発明において、前記適応度算出処理が、前記帰属尤度算出処理によって算出された帰属尤度の中で各サンプルが一番大きな帰属尤度を有するカテゴリーに帰属すると予測し、その予測カテゴリーと実測カテゴリーとが一致する場合を識別正解、そうでない場合を識別不正解と判別する処理を含むことを特徴とする。
【0018】
第12発明は、第1発明ないし第11発明のいずれか一つに記載の発明において、前記相関モデルを多変量正規分布モデルとしたことを特徴とする。
第13発明は、第12発明において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をカテゴリーによらないものとして設定したことを特徴とする。
【0019】
第14発明は、第12発明において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をムーア・ペンローズの一般化逆行列を用いて求めることを特徴とする。
第15発明は、第1発明ないし第14発明のいずれか一つに記載の発明において、前記相関モデルを、多変量特徴空間の各サンプルを主成分分析によって得られた部分空間により複数カテゴリーに分類する相関モデルとしたことを特徴とする。
【0020】
第16発明は、第15発明において、前記相関モデルがSIMCAモデル(Soft Independent Modeling of Class Analogy)であることを特徴とする。
第17発明は、第1発明ないし第16発明のいずれか一つに記載の発明において、第1次最適解の数を3個以上〜50個以下とすることを特徴とする。
【0021】
第18発明データ解析方法は、第1発明ないし第17発明のいずれか一つに記載のデータ解析方法に基づいて構築された相関モデルに、予測用サンプルの選抜された説明変数を入力してカテゴリー分類された生体の状態を予測する出力を得ることを特徴とする。
【0022】
第19発明のデータ解析システムは、コンピュータを用いて、カテゴリー分類された生体の状態を目的変数とし、遺伝子発現、生体関係物質の少なくとも一方に関するデータを説明変数とする相関モデルを決定する複数カテゴリー分類用のデータ解析システムであって、
カテゴリー分類された生体の状態と、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる訓練用サンプルの集合を記憶する記憶手段と、
遺伝的アルゴリズムに基づいて複数個の第1次最適解を得る第1次最適解取得手段と、
得られた複数個の第1次最適解の重み付け平均された第2次最適解を算出する重み付け平均最適解算出手段と、
を備えて前記相関モデル構築を行い、
前記第1次最適解取得手段が下記(a)〜(f)の各手段を含んでいることを特徴とする。
(a)説明変数の候補を選択する説明変数候補選択手段、
(b)各個体が相関モデルの候補を表しており、それらの個体の集団である第1世代を生成する初期個体集団生成手段、
(c)各個体について、各サンプルの各カテゴリーへの帰属尤度を算出する帰属尤度算出手段、
(d)算出された帰属尤度に基づくサンプルの予測カテゴリーとサンプルが実際に帰属する実測カテゴリーとを比較し、その予測がどの程度、信頼性があるかという定量的な評価である適応度を算出する適応度算出手段、
(e)算出された適応度に基づいて、個体に関する突然変異、交叉、選抜などの遺伝的アルゴリズムに基づいて次の世代の個体集団を生成する次世代個体集団生成手段、
(f)最も適応度の高い最適解を探索しつつ、所定終結条件を満足する複数個の第1次最適解を得るために、少なくとも帰属尤度算出手段、適応度算出手段、次世代個体集団生成手段が行う一連の処理を複数回、繰り返して行なわせる繰返し処理手段
【0023】
第20発明は、第19発明において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの帰属尤度を重み付け平均した重み付け平均帰属対数尤度で与えられることを特徴とする。
第21発明は、第19発明において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの中心へのマハラノビス距離を重み付け平均したもので与えられ、前記帰属尤度算出手段がその重み付け平均マハラノビス距離に基づいて重み付け平均化された帰属対数尤度を算出することを特徴とする。
【0024】
第22発明は、第20発明ないし第21発明のいずれか一つに記載の発明において、各第1次最適解の重み付け係数を同じ値に設定したことを特徴とする。
第23発明は、第20発明ないし第21発明のいずれか一つに記載の発明において、各第1次最適解の重み付け係数を各第1次最適解に対応するモデル尤度に基づいて決定することを特徴とする。
【0025】
第24発明は、第19発明ないし第23発明のいずれか一つに記載の発明において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返し(後述するRun)を複数回行う条件が含まれており、その各回で得られた最適解を第1次最適解取得処理における複数個の第1次最適解として設定することを特徴とする。
【0026】
第25発明は、第19発明ないし第24発明のいずれか一つに記載の発明において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返し(後述するRun)を少なくとも一回行う過程で算出適応度が設定適応度を超えた最適解を複数個抽出することが含まれており、それらの最適解を第1次最適解取得処理における複数個の第1次最適解として設定することを特徴とする。
【0027】
第26発明は、第19発明ないし第25発明のいずれか一つに記載の発明において、前記適応度算出手段の行う適応度算出処理がAIC(Akaike'sInformation Criterion:赤池情報基準)に基づいて行われることを特徴とする。
第27発明は、第19発明ないし第25発明のいずれか一つに記載の発明において、前記適応度算出手段の行う適応度算出処理が交差検証成績に基づいて行われることを特徴とする。
【0028】
第28発明は、第19発明ないし第27発明のいずれか一つに記載の発明において、前記適応度算出手段の適応度算出処理が、前記帰属尤度算出手段によって算出された各サンプルの帰属尤度を全サンプルについて乗算することにより得られたモデル尤度に基づいて行われることを特徴とする。
第29発明は、第19発明ないし第28発明のいずれか一つに記載の発明において、前記適応度算出手段が、前記帰属尤度算出手段によって算出された帰属尤度の中で各サンプルが一番大きな帰属尤度を有するカテゴリーに帰属すると予測し、その予測カテゴリーと実測カテゴリーとが一致する場合を識別正解、そうでない場合を識別不正解と判別する判別手段を含むことを特徴とする。
【0029】
第30発明は、第19発明ないし第29発明のいずれか一つに記載の発明において、前記相関モデルを多変量正規分布モデルとしたことを特徴とする。
第31発明は、第30発明において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をカテゴリーによらないものとして設定したことを特徴とする。
【0030】
第32発明は、第30発明において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をムーア・ペンローズの一般化逆行列を用いて求めることを特徴とする。
第33発明は、第19発明ないし第32発明のいずれか一つに記載の発明において、前記相関モデルを、多変量特徴空間の各サンプルを主成分分析によって得られた部分空間により複数カテゴリーに分類する相関モデルとしたことを特徴とする。
【0031】
第34発明は、第33発明において、前記相関モデルがSIMCAモデル(Soft Independent Modeling of Class Analogy)であることを特徴とする。
第35発明は、第19発明ないし第34発明のいずれか一つに記載の発明において、第1次最適解の数を3個以上〜50個以下とすることを特徴とする。
【0032】
第36発明のデータ解析システムは、第19発明ないし第35発明のいずれか一つに記載のデータ解析システムに基づいて構築された相関モデルと、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる予測用サンプルを記憶する記憶手段とを備え、前記相関モデルに予測用サンプルの選抜された説明変数を入力してカテゴリー分類された生体の状態を予測する出力を得ることを特徴とする。
【0033】
以下、上記第1発明〜第36発明の各構成要素や変形例などについて説明する。
第1発明において「生体関係物質に関するデータ」の一例としては、「細胞内物質のデータ」が挙げられる。細胞内物質のデータとしては細胞内物質の量などが挙げられ、例えば、代謝中間体を含めた代謝産物全部であるメタボロームも含まれる。
なお、「細胞内物質のデータ」は「生体内に含まれる物質のデータ」を意味するものであり、血液、リンパ液、尿などの排泄物に含まれる物質の量も細胞の状態の反映であるという観点から、細胞内物質のデータの一種と解釈され、本発明では当然に対象となるべきものである。
これに対し、「生体関係物質」としては上記生体内に含まれる物質の他、生体が得る食物などに含まれる物質の量などが例示でき、それら物質に関するデータと生体の状態を解析する場合にも適用できるものである。
また、「生体」には人体の他、動物、植物、微生物などの各種生命体が含まれる。
【0034】
「遺伝子発現」の用語は、mRNA発現(トランスクリプトーム)やmRNAによる翻訳の結果として生じるタンパク質(プロテオーム)を含むものとして使用する。
遺伝子発現の量、生体関係物質の量は測定値に限定されるものではなく、加工計算された値、相対的な値、間接的に物質量を示す量でもよい。例えば、質量スペクトルでタンパク質の発現量を測定できることを応用して、カテゴリー分類された生体状態の状態と質量スペクトルとを直接関係づける相関モデルを構築することもできる。DNAチップで各スポットの測定量を説明変数として採用することもできるし、そのスポットの測定量の加工値を利用することもできる。
【0035】
本明細書において、「第1次最適解」とは、そのときの終結条件の範囲で改善がみられなくなるまで改良したことを意味しており、全ての説明変数の組み合わせの中で最適なものを見いだしたことを確認したことを意味するものではない。
第1発明において、記憶手段に記憶される「カテゴリー分類された生体の状態」には、カテゴリー分類された生体の状態を記憶する形態のみならず、その生体の状態を導出する元になるデータを入力し、各種の処理を行った結果として、記憶手段にカテゴリー分類された生体の状態を記憶させる形態も含まれる。
【0036】
第1発明において、複数個の第1次最適解の表現形態は重み付け平均最適解を算出でき、かつ第2次最適解の算出処理に用いることのできるデータ形式であれば特に限定されない。また、重み付け平均する方法は第2発明、第3発明のような表現形態のみに限定されず、なんらかの計算手法で実質的に複数の最適解のデータを数値的に重み付け平均する場合も含まれる。
【0037】
遺伝的アルゴリズムを採用して最適解を得る方法は、計算時間とのかねあいで予め設定した世代数の中で、予め設定した適応度よりも好ましい適応度になり、改善が見られない状態になったときが最適化されたと判断する方法が採用される。但し、第5発明に記載したように第1次最適解を各モデルのレベルに応じて重み付けする方法も採用できるので、多様性を確保するという点で、本発明における終結条件の設定は一般的な遺伝的アルゴリズムの場合に比べて広義に考えることができる。
【0038】
従って、第1発明における「所定終結条件」としては、設定世代数を超えた時、設定適応度以上のものを得た時、適応度の改善が見られなくなった時を適宜、組み合わせて設定することにより第1次最適解を得るようにする。さらに、後述する図5のように連続した計算処理によって複数個の第1次最適解を得る場合だけでなく、連続せず、試行錯誤的に一つ一つ別の計算フローによって個々の第1次最適解を取得し、その取得された第1次最適解を重み付け平均して第2次最適解を算出してもよい。
【0039】
また、複数個、第1次最適解を得る場合に、第7発明に記載したように第1次最適解の個数と、初期個体集団生成処理の再設定が行われる回数は同一である場合だけに限定されない。但し、多様性を確保するという観点においては、初期個体集団生成処理の再設定が行われる回毎に最も適応度の高い1個の最適解を得る第6発明の方法が好ましいと考えられる。
第1発明などに記載した「適応度」は少なくとも各サンプルの各カテゴリーへの帰属尤度を算出することを含んで算出される。
【0040】
各サンプルの帰属尤度としては、多変量正規分布モデルにおけるマハラノビス距離に基づく帰属尤度や、F検定などで求めることのできる帰属の信頼度などを用いることができる。また第11発明に記載したように分類識別が正解か不正解かを判断してそれを適応度の計算に反映させることもできる。
【0041】
第1次最適解におけるモデル選抜の評価基準としては例えば第8発明に記載されるように赤池情報基準(AIC)あるいはその応用をモデル選抜の基準にすることで良好なモデルが得られること確認した。
適応度の算出に赤池情報基準(AIC)を用いた場合には、モデル尤度とともに相関モデルの複雑さを表現するパラメータ数を用いる。このパラメータ数はモデルに用いる説明変数の数であったり、相関モデルを伝達する情報量などであっても良い。
第17発明に記載した集団学習に用いる第1次最適解の数は、集団としての多様性を確保する点から5個以上とし、計算時間が長くなりすぎないという点から20個以下とすることが好ましい。
【0042】
第19発明〜第36発明は、サーバー型コンピュータで構成される場合や、デスクトップ型コンピュータで構成される場合を問わずに適用できる。
サーバー型コンピュータで構成される場合は、第19発明のデータ解析システムをモデル構築処理サーバーで構成することもできる。
なお、第19発明〜第36発明はデータ解析装置又はデータ解析プログラムとしても認識できるものである。
【0043】
遺伝子発現を測定し、細胞を複数カテゴリーに分類する方法の実用場面を考えた場合、「解析に用いなかったデータを正しく予測すること」が問題となる。対策として集団学習によって予測力を向上させることが考えられるが、その集団学習の前提となることは各モデルの多様性を備えていることであり、このようなモデルを複数個得ることが重要になる。
【0044】
本発明者らは種々の検討の後に遺伝的アルゴリズム(GA)が単一の最適解を探索しつつも有限時間内には必ずしも最適解を得るものではなく、所定終結条件を満足する毎に異なった最適解を得る方法であることに注目した。通常は試行回数毎に異なった最適解を得ることはGAの不完全な側面と捉えられる。しかし本発明者らはその特徴を積極的に利用し、GAで得られた第1次最適解を組み合わせることで有効な集団学習が実現することを見出した。
【0045】
その一方法として次のような方法がある。まず、複数カテゴリー分類を行なうためのモデルとして多変量正規分布モデルを採用し、AICなどの何らかの最適指標(適応度)に基づいて多変量正規分布モデルの最適解をGA探索することで第1次最適解を得て、それらを1回(後述するRun)もしくは複数回実行して複数個の第1次最適解を得る。そして得られた複数個の第1次最適解から得られたマハラノビス距離又は帰属尤度の重み付け平均を算出して第2次最適解を得ることによって集団モデルとした。集団モデルにおいて、訓練用サンプル、予測用サンプルのカテゴリー帰属は各多変量正規分布モデルの場合と同じく尤度に基づいて算出される。
【0046】
複数カテゴリー分類を行なうためのモデルとしては、多変量正規分布モデルであることは必須ではない。例えば、SIMCAモデルも部分空間からのマハラノビス距離によって帰属尤度が決定され、その応用変形は直接的であることからSIMCAモデルの採用も充分期待されるところである。更にはマハラノビス距離という概念を有しない相関モデルであっても、帰属尤度の概念をもっているモデルであれば、本発明は適用可能となる。
【発明の効果】
【0047】
以下、各発明の効果などについて説明する。
なお、データ解析システムに係る発明の効果もデータ解析方法の効果に対応しているのでそれらの効果の記載は省略する。
第1発明であれば、遺伝的アルゴリズムに基づいて複数個の第1次最適解を得る第1次最適解取得処理と、得られた複数個の第1次最適解の重み付け平均された第2次最適解を算出する重み付け平均最適解算出処理とを含んで前記相関モデル構築を行うので、予め複数個得られた第1次最適解を集団学習したような形態となり、結果としてオーバーフィットを克服して複数カテゴリー分類の分析精度を高めた相関モデルを構築できる可能性を高めることができる。また、遺伝的アルゴリズムは初期個体集団から選択と交叉との組み合わせにより並設的に山登り探索を行うとともに突然変異によりランダムな変化を起こして局所安定解に陥ることを防止できる探索手法であるから、その性質上、集団学習を行う際に必要とされるモデルの多様性(あるいは相補性)を確保する場合に好ましいと言える。
【0048】
第2発明であれば、重み付け平均最適解を重み付け平均帰属対数尤度で与えることにより、カテゴリーの帰属尤度という概念を有する相関モデルであれば適用が可能になる。
第3発明であれば、重み付け平均最適解を統計的手法に広く用いられているマハラノビス距離を重み付け平均したもので与えることにより、各種モデルに広範囲に適用が可能になる。
【0049】
第4発明であれば、第1次最適解の重み付け係数を同じ値に設定することにより、各第1次最適解をその重要度において均等に扱うことができる。
第5発明であれば、各第1次最適解の重み付け係数を各第1次最適解に対応する相関モデルの尤度に基づいて決定することにより各相関モデルの確からしさを考慮して、確からしさの高いモデルは重み付けを大きく、確からしさの低いモデルは重み付けを小さくすることが可能になる。
【0050】
第6発明であれば、説明変数候補選択処理、初期個体集団生成処理の繰り返し回数を所定終結条件に基づいて複数回行って、それぞれの回における最適解を求めるので、複数回の初期個体集団の再設定処理が行われることになり、集団学習を行う際に必要とされるモデルの多様性(あるいは相補性)を確保しやすくなる。
【0051】
第7発明であれば、説明変数候補選択処理、初期個体集団生成処理の繰り返し回数がたとえ1回で得られた最適解であっても、適応度が設定適応度を超えた最適解であれば、それらの複数個の最適解を第1次最適解取得処理における複数個の第1次最適解として設定できるので、一定レベル以上で集団学習に必要な複数個の第1次最適解を少ない計算時間で得ることができる。
【0052】
第8発明であれば、適応度の算出においてAICを使用するので、相関モデルの尤度のみならず、相関モデルのパラメータ数も考慮でき、データの情報量に見合う最適な相関モデルを選定することが期待できる。
第9発明であれば、適応度の算出において交差検証成績を使用することにより、相関モデルを簡単な計算で最適化することができる。
【0053】
第10発明であれば、適応度を算出する場合に、各サンプルの帰属尤度を全サンプルについて乗算することにより得られたモデル尤度を使用することにより各帰属尤度の値を反映した値にすることができる。
第11発明であれば、適応度算出処理において、予測カテゴリーと実測カテゴリーと一致する場合を識別正解、そうでない場合を識別不正解と判別する処理を含んでいるので、適応度を自然に訓練用サンプルに適応したものとすることができる。
【0054】
第12発明であれば、多変量正規分布モデルは比較的計算が簡単で判別分析に使用する相関モデルとして研究が進んでいることから適用が行いやすい利点がある。
第13発明であれば、計算を単純化し、かつ共分散行列の推定精度を向上することができるとともに、特に共分散行列の次元を増加することが可能になるので、カテゴリー当たりのサンプルの数が少ない場合であっても多数の説明変数を採用してモデル構築が可能となる利点がある。
【0055】
第14発明であれば、多変量正規分布モデルの共分散逆行列をムーア・ペンローズの一般化逆行列を用いて求めるので、カテゴリー当たりのサンプル数が少ない場合にも多数の説明変数を採用してモデル構築が可能となる利点がある。
第18発明であれば、重み付け平均最適解を用いて集団学習的な手法を用いることによりオーバーフィットを克服し、分類能力の高いデータ解析ができる相関モデルを有しているので、予測用サンプルを入力した場合にカテゴリー分類された生体の状態を予測する精度の高い出力を得ることができる。
【発明を実施するための最良の形態】
【0056】
以下、上記各発明について数式、図面などを用いて、より具体的に説明する。
本実施形態では、本発明のデータ解析方法を複数カテゴリー分類のための遺伝子発現解析法に適用する。また、相関モデルとして後述する多変量正規分布モデルを採用するとともに、最適化処理として遺伝的アルゴリズムを採用する。
上記発明の内容を限定するものではないが、データ解析方法の一例のフローチャートを図1〜図6に示す。
【0057】
図1はデータ解析処理の全体を示すフローチャートであり、カテゴリーデータ(目的変数Yi)および遺伝子発現データ(説明変数の候補xij)からなる訓練データを入力した後(ステップ1)、集団学習による相関モデル(複数個の多変量正規分布モデル及びその重み)を構築し(ステップ2)、その多変量正規分布モデルを出力する(ステップ3)。
また、遺伝子発現データ(選抜された説明変数xij)を入力し(ステップ4)、前記多変量正規分布モデルに基づいて診断予測結果を出力する(ステップ5)。入出力データは必ずしも図1に厳密に従ったものである必要はなく、よく定義された加工方法による加工前あるいは加工後のものでもよい。
【0058】
また、モデル構築と診断予測は必ずしも時間的、空間的に連続した処理である必要はない。例えば、胃癌、肺癌、肝臓癌、……と診断された多数の患者の各遺伝子発現データを使用して図1のステップ1〜ステップ3の処理により、各癌診断の相関モデルの構築をデータ解析センターにおいて集中的に行い、そのデータ解析の成果として構築された癌診断用の相関モデルを世界各地の病院においてステップ4,ステップ5の処理を行って診断に利用する形態などが考えられる。
【0059】
図2は図1のステップ2に示す集団学習による相関モデル構築処理のさらに詳しいフローチャートの一例である。
図2において遺伝的アルゴリズムを用い、各サンプルにおける各カテゴリーの重み付け平均された帰属尤度に基づいて適応度を計算しつつ、多変量正規分布モデルの第1次最適解を複数個得て(ステップ21)、それら複数個の第1次最適解の重み付け平均最適解を算出して第2次最適解を求める(ステップ22)。
【0060】
次に、図2のステップ21に示される多変量正規分布モデルの第1次最適解を得る処理について次の記号を用いて説明する。
iはサンプルのインデックス
jは説明変数(遺伝子発現量)のインデックス
kはカテゴリーのインデックス
Niはサンプルの個数
Njは説明変数の個数
Nkはカテゴリーの個数
Xiはサンプルiの説明変数ベクトル
xijはサンプルiの説明変数jの値
Xkはカテゴリーkの説明変数ベクトルの平均
[σk2] はカテゴリーkの共分散行列
Dikはサンプルiのカテゴリーkの中心へのマハラノビス距離
但し、上記Niはカテゴリーk毎に変化する値なので、そのカテゴリー毎に変化することを明示的に表示する場合は、以下に記述する数式及び添付するフローチャートにおいてNi(k)と表示する。
【0061】
まず、多変量正規分布モデル(多変量正規化ガウス確率分布密度関数モデルとも称される)では、各サンプルiの各カテゴリーkへの帰属尤度Likは次式で表される。
【0062】
【数1】
【0063】
で与えられる。ここで(xi−Xk)tは列ベクトル(xi−Xk)を転置した行ベクトルである。また、Dikはマハラノビス距離を表現している。
マハラノビス距離とは平均だけでなく、分散、共分散も考慮して定義された距離であり、サンプルiのカテゴリーkの中心へのマハラノビス距離の2乗Dik 2は、
【0064】
【数2】
【0065】
で示される。ここで、[σk2]はカテゴリーkの共分散行列であり、
【0066】
【数3】
【0067】
で与えられる。上記(3)式でΣ(i∈k)はカテゴリーkに属するサンプルiについての総和を意味している。なお、本実施形態では、上記(3)式の代わりに下記(4)式を用いる。
【0068】
【数4】
【0069】
(4)式を用いる理由は(A)計算の単純化、(B)共分散行列の推定精度向上、(C)共分散行列の次元を増加できる、などの利点があるからである。特に、(3)式で示す共分散行列[σk2] の次元はカテゴリーkに含まれる訓練サンプルの数より小さく、GCMデータでは7以下である。したがって8個を超える遺伝子を含むモデルは計算不能となる。[σ2]を用いることは前記P.Tanらの文献に記載されているが、本来カテゴリーごとに異なる[σk2] を同一視する弊害があるにも拘わらず、8遺伝子を超えるモデルを可能にするものであり、利点の方が大きいので(4)式を採用することにした。この(4)式を用いることは前記第13発明の一形態を示すものである。
【0070】
また、(4)式を用いる他に、[σk2]の逆行列[σk2] -1が計算不能の場合の別の対処方法として一般逆行列を用いることがある。行列Aの一般逆行列とは
AGA=A
を満たすGのことであり、A-1が計算不能の場合にはGは必ずしも一義的に決定されない。しかし、次の条件を追加したMoore-Penrose型(ムーア・ペンローズ)一般逆行列は一義的に決まり、特異値分解の方法によって求めることができる。
GAG=G, (GA)t=GA, (AG)t=AG
[σk2]のムーア・ペンローズ型一般逆行列[σk2] -1 MPを適用すると、カテゴリー当りのサンプル数が少ない場合にもカテゴリーごとに異なる[σk2]を用い、しかもある程度の数の遺伝子発現量を説明変数として利用することができる。このようにムーア・ペンローズ型一般逆行列を用いることは前記第14発明の一形態を示すものである。
【0071】
次に、相関モデルの適応度を算出する方法について説明する。
本実施形態では、前記適応度としてAIC(赤池情報基準)を最適化した多変量正規分布モデルを得る。
ここでAICとは、例えば、下記(5)式で計算される量であり、相関モデルの複雑さに依存する場合にその複雑さを表す第1項と、相関モデルの当てはまりの良さを表す第2項とのバランスを取った量である。
本実施形態ではAIC値が小さいほど優れたモデルとなるので、AIC値が小さくなるように多変量正規分布モデルを決定する。
【0072】
【数5】
【0073】
ここでkは両項のバランスを取るための調節パラメータであり、任意に設定できるものとする。k=0という特別なケースは最尤法に相当し、k=1は通常のAICの計算方法である。(5)式を使用する場合にk≠0の時は、モデルのパラメータ数Npも考慮して適応度を算出することになる。多変量正規分布モデルのパラメータ数Npを簡単に計算する場合は、説明変数の個数Njを採用することができる。
多変量正規分布モデルの尤度Lは例えば下記(6)式で定義することとした。
【0074】
【数6】
【0075】
但し、Liは訓練用サンプルiの尤度であり、Πiは訓練用サンプル集合の全サンプルi=1〜Niについての積を表す。この(6)式は前記第10発明の一形態を示したものである。また、帰属尤度Likは前記(1)式において説明したようにマハラビノス距離の2乗Dik 2によって算出できる値であり、サンプルiがカテゴリーkに帰属する信頼度である。
帰属尤度Likの算出方法としては、(1)式の他にF検定による下記(8)式の関係式を満たす量として算出することもできる。
【0076】
【数7】
【0077】
ここでF(Nf,P)は自由度Nf、危険率PのF検定の限界値、Nikはカテゴリーkに属するサンプルの個数である。このようなLikを求めることは、公知のF分布表あるいは公知のアルゴリズムを利用することによって可能である。あるサンプルiに対して最大のLikを与えるカテゴリーk=kmaxが最尤帰属カテゴリーであり、kmaxと実測カテゴリーが一致する場合を識別正解、そうでない場合を不正解と呼ぶ。
(6)式を用いた場合には識別正解と不正解との区別が鮮明ではない。そこで敢えて鮮明にするために、各iごとにLik (k=1〜Nk)を全て計算し、一番大きなカテゴリーに帰属するとして下記(9)式のようにLiを定義することもできる。ここでpenaltyは十分小さな量であり、例えば10-6である。この(9)式は前記第11発明の一形態を示したものである。
【0078】
【数8】
【0079】
図3は適応度として上記赤池情報基準(-AIC)を用いた場合の、適応度の算出方法のフローチャートの一例である。
図3に示すようにサンプルi毎に各カテゴリーkの中心へのマハラノビス距離の2乗Dik 2を計算し(ステップ45)、(1)式に基づいて帰属の信頼度Likを計算し(ステップ46,ステップ47)、最尤帰属カテゴリーkmaxが実測カテゴリーk(i)と一致するかどうかの条件判定も行ないながらモデル尤度の計算を行ない(ステップ48)、適応度としてのAIC値を求める(ステップ49)。
図3に示すフローチャートは前記(9)式に準拠したものであり、前記(6)式に準拠する場合には最尤帰属の判定は不要であり、フローチャートは簡素なものになる。
【0080】
図4は図3のステップ45において記載したカテゴリーkの共分散逆行列[σk2] -1の算出フローチャートの一例である。
図4に示すようにカテゴリーkのサンプルの個数Ni(k)とカテゴリーkの説明変数ベクトルの平均Xkを算出し(ステップ45−2〜ステップ45−4)、算出されたNi(k)とXkに基づいてカテゴリーkの共分散行列σk2を算出し(ステップ45−5〜ステップ45−7)、カテゴリーkの共分散逆行列を算出する(ステップ45−8)。なお、本方法はカテゴリーkによらないものとして共分散逆行列を算出する方法やムーア・ペンローズの一般逆行列を用いて共分散逆行列を算出する方法に修正(ステップ45−7〜ステップ45−8)して実行する方法もある。
【0081】
図5はモデル構築を遺伝的アルゴリズム(GA)で行う場合のフローチャートであり、このフローチャートを参照しつつ、遺伝的アルゴリズムについての予備的説明を行う。
遺伝的アルゴリズム(GA;Genetic Algorithm)は、進化論をヒントとした非線型最適化手法であり、ここ数年間、頻繁に用いられて定着してきた。遺伝的アルゴリズムを説明した文献としては、伊庭斉志;「遺伝的アルゴリズムの基礎」;オーム社;(1994)などがある。
GAでは最適化対象がビット列などで表現される。このような表現の形式を遺伝型(genotype)と呼び、ひとつひとつの遺伝型の具体的な文字列を染色体(chromosome)と呼ぶ。最適化の指標である適応度(fitness)は遺伝型より一義的に決定される。
【0082】
(処理1) 初期染色体プールの準備(図5のステップ31)
ランダムに染色体の集合を準備する。染色体の数はプールサイズと呼ばれる。
(処理2) 交叉、突然変異、選択による最適化(図5のステップ32〜ステップ34)
[2-1] 交叉(crossover)
集合より2つの染色体を選抜し、ビット列のランダムな交叉によって新しい染色体を創生する。交叉を1個所に限る一点交叉(onepoint crossover)と全ビット間で交叉を行なう一様交叉(unform crossover,UX)などがあり、一般的にはUX法が優れているとされている。
[2-2] 突然変異(mutation)
集合あるいは新しい染色体の任意のビットをランダムに選んで反転させる。
[2-3] 選択(selection)
各染色体の適応度を計算し、染色体プールを次世代のものに置き換える。適応度に比例した確率で染色体を選抜するルーレット方式や親世代と子世代との間で適応度の対決を行なうトーナメント方式などがある。これらいずれにおいても乱数が用いられることが多く、一度得られた最適染色体が失なわれることもある。このようなことが無いよう、プール内の最適染色体は必ず次世代に選抜されるとするエリート戦略なども用いられる。
【0083】
(処理3) 終結判定(図5のステップ35)
世代数がある回数に達した場合や、最適適応度に変化が見られなくなった場合に処理2は終了し、最適解は最適染色体の遺伝型およびその適応度として出力される。GAは基本的には乱数を用いた方法であるため、処理1〜処理3が何回か試みられ(Run)、その中の最適染色体が用いられることが多い。
【0084】
図6は図5に示した遺伝的アルゴリズムを用いた第1次最適解を複数個得るためのフローチャートの一例である。
図6において乱数を用いて初期染色体の集団をビット列で設定し(ステップ71)、予め最大適応度を−∞にするなどの初期設定を行い(ステップ72)、交叉・突然変異の処理を行い(ステップ73)、適応度(図6では値が大きいほどモデルが適合しているとする)を算出し(ステップ74)、算出された適応度が前の世代の最大適応度よりも大きいか否かを判別し(ステップ75)、算出された適応度が前の世代の最大適応度よりも大きくないと判別された場合は、世代を+1追加し(ステップ76)、終結判定基準として設定世代数よりも大きいか否かを判別し(ステップ77)、大きくないと判別された場合は、次の世代の交叉・突然変異の処理を実行する。
【0085】
一方、ステップ75において算出された適応度が前の世代の最大適応度よりも大きいと判別された場合は、算出適応度を最大適応度に設定するとともに、コンピュータの記憶装置に最大適応度のビット列とその時のマハラノビス距離の2乗Dik 2又は帰属尤度Likを記憶し(ステップ78)、終結判定基準として設定適応度よりもその最大適応度が大きくて、かつ算出適応度の改善傾向が見られなくなったか否かを判別し(ステップ79)、設定適応度よりもその最大適応度が大きくかつ改善傾向が見られなくなったと判別された場合は後述するステップ81へ移行し、それ以外の場合であると判別された場合は、ステップ76に移行する。
上記処理により算出された適応度が設定適応度を超え、かつ算出適応度の改善傾向が見られなくなった場合に計算のループを抜け出してその時のビット列とマハラノビス距離又は帰属尤度のデータを記憶装置に記憶することができる。
【0086】
また、ステップ77において設定世代数よりも大きいと判別された場合は、必要に応じて記憶された最大適応度が最低基準適応度よりも大きいか否かを判別して第1次最適解の候補として好ましくないものを除外し(ステップ80)、世代数による第1次最適解のデータと、ステップ79の条件で得られた第1次最適解のデータとを回(Run)ごとに整理して記憶装置に記憶する(ステップ81)。
【0087】
そしてステップ82において得られた第1次最適解の個数が、予め設定された設定個数よりも大きいか否かを判別して、設定個数得られていないと判別された場合は、新たなる回(Run)においてステップ71の初期染色体の集団設定から再び行い、第1次最適解の個数が設定個数得られたと判別された場合は一連の処理を終了する。
【0088】
次に、図2のステップ22に示した複数個の第1次最適解に基づいて重み付け平均最適解を算出する処理について説明する。
(1)式において示したように、帰属尤度Likはサンプルi毎に各カテゴリーkのマハラノビス距離を計算することにより求めることができる。このLikの算出は遺伝的アルゴリズムを用いた場合、回(Run)ごと(あるいは所定終結条件を満足した回数毎)に異なった最適解が得られることを利用する。これら複数の最適解を第1次最適解と言っていることは前述の通りである。そして、それらの第1次最適解の重み付け平均を求める。例えば、各回の第1次最適解のデータがマハラノビス距離で与えられる場合は、重み付け平均マハラノビス距離は、
【0089】
【数9】
【0090】
と表される。ここで、w(m)はm番目のモデルの重み付け係数、mは複数個得られるモデルの数、Dik 2(m)はmモデルのDik2の値を示す。この(10)式は前記第3発明の一形態を示したものである。
また、各回の第1次最適解のデータがマハラノビス距離でなく、直接的に帰属尤度Likで与えられる場合は、重み付け平均帰属対数尤度は、
【0091】
【数10】
【0092】
と表される。この(11)式は前記第2発明の一形態を示したものである。
なお、重み係数w(m)としては一定値を用いたり、各モデルの尤度を重み係数w(m)としたり、各モデルの尤度を重み係数w(m)を考慮した値とすることもできる。これらは前記第4発明、第5発明の一形態を示すものである。
【0093】
このように複数個得られた第1次最適解を用いて上記(10)式又は(11)式に基づいて第2次最適解を算出する(図2のステップ22)。この算出処理によってGAを用いた集団学習による相関モデルが構築されることになる。
【0094】
図7は本実施形態に係るデータ解析システムの一例を示す概略的なブロック図である。
本データ解析システムは、第19発明に記載したような相関モデル構築システム1と第36発明に記載したような予測サンプルを用いるカテゴリー分類予測システム2とに大別される。相関モデル構築システム1は、前記訓練用サンプルの記憶手段としての訓練用サンプル記憶部3と、前記第1次最適解取得手段としての第1次最適解取得部4と、複数の第1最適解を記憶する第1次最適解記憶部5と、前記第2次最適解を算出する重み付け平均最適解算出手段としての重み付け平均最適解算出部6とを備え、必要により最終モデル構築部7とを備えている。最終モデル構築部7は第2次最適解で得られる相関モデルをさらに改良、微調整する機能がある。
【0095】
訓練用サンプル記憶部3に入力される各データは大学、研究所が提供する生体関係情報を提供するウエブサイト、疾病関係データベースなどの各種データベースから得ることも可能である。また、DNAチップなどを用いて患者の遺伝子発現量のデータとカルテなどからの生体状態のカテゴリー分類データを取得して入力しても良い。
【0096】
カテゴリー分類予測システム2は、予測用サンプル記憶部10と、前記第2次最適解取得手段で構築された相関モデル(又は最終モデル構築部で構成された相関モデル)を予測用相関モデルとして設定した相関モデル部11とを備え、DNAチップ12などから取得した患者の遺伝子発現データに係る選抜した説明変数を入力することにより、どのカテゴリーに属するかの分類予測出力を得ることができるように構成してある。
【0097】
なお、図7では図示していないが、第1次最適解取得部4は、説明変数候補選択部、初期個体集団生成部、帰属尤度算出部、適応度算出部、次世代個体集団生成部、繰返し処理部を少なくとも含んで構成してある。
【0098】
本データ解析システムはスタンドアロン型のコンピュータでも、クライアントサーバー型のコンピュータでも構成できる。
スタンドアロン型のコンピュータで構成される場合は、相関モデル構築装置とカテゴリー分類予測装置として構成される場合が多い。本データ解析システムはネットワーク13上に配置された相関モデル構築サーバーと各病院等に設置されたカテゴリー分類予測コンピュータを連携させて構成することも可能である。この場合は、各病院等に設置されたカテゴリー分類予測コンピュータ側において、病院に設置されたDNAチップ12などを用いて患者の遺伝子発現量のデータとその病院のカルテから患者の生体状態のカテゴリー分類データを取得して、ネットワーク13を介して相関モデル構築サーバーへ訓練用サンプルの一部として送信することもできる。なお、本データ解析システムはネットワーク上の機能実現プログラムや記憶装置などで構成しても良い。
【実施例1】
【0099】
以下、具体的な実施例について説明する。
データは前記したGCMデータを用いた。それぞれ8サンプルよりなる18種類の癌細胞合計144個の訓練サンプル集合と、同じく18種類46個の独立テスト集合(予測用サンプル集合に相当)とより成る。各サンプルについて、約16,000遺伝子の発現データがあり、その中で訓練サンプル集合の分散の大きな上位1000個(重複を除き968個)を説明変数とした。
このようなデータでbagging等を用いると、カテゴリー当りのサンプル数が極端に少なくなり、有効なモデルが得られないと懸念される。
【0100】
識別モデルとしては多変量正規分布モデルを用い、最適化指標(適応度)としては交差検証予測不正解率(Pmis(cv))とAIC(赤池情報規準、AIC=2Nj-1ogL)について検討することとした。交差検証予測不正解率の方法はP.Tanらの方法に準じたものである。但し、P.TanらのGA/多変量正規分布モデルは交差検証予測不正解率(Pmis(cv))と独立テスト集合予測不正解率(Pmis(ind))との和を指標としてGAによって遺伝子を選抜している。しかし、この方法のようにモデル選抜に独立テスト予測不正解率を用いることは、モデルの予測精度を評価する上で問題と考えられるので交差検証予測不正解率(Pmis(cv))だけを用いて計算した。
一方、最適化指標にAICを用いる方法は前述のように本発明の重要な特徴の一つである。多変量正規分布モデルの共分散行列は(3)式及び(4)式において説明した議論により、P.Tanらの方法に準じてカテゴリーによらないものとした。即ち、σ2=Σkσk2で算出している。
【0101】
本実施例において多変量正規分布モデルの最適化のために使用したGAの設定は次のとおりである。
(1)genotype={b1,b2,b3,,,,,,bNg}
遺伝子発現量gjを説明変数に用いる場合にはbj=ON状態、
用いない場合にはbj=OFF状態とする。
(2)fitness=-AIC あるいは-Pmis(cv)
(3)プールサイズ=100
(4)初期染色体は、平均でmin_of(Ni, Ng, 300)/2個のビットがON状態となるように乱数を用いて準備する。ここでNiは訓練用サンプル数、Ngは説明変数候補の数、300は実装の都合上設定された定数である。
(5)交叉は、2つの染色体を乱数にて抽出し、各ビットをそれぞれ1/2の確率でいずれかの親染色体から引き継いだ子染色体を作成する。
【0102】
(6)突然変異は、ON状態ビット数が増加する変異と減少する変異とが同じ確率で発生するように乱数でビット反転を行なった。
(i)ON状態ビットの反転確率=1.1/ON状態ビット数
(ii)OFF状態ビットの反転確率=1.1/OFF状態ビット数
(7)選抜は、子染色体を作成する毎にトーナメント方式でプールを置換する。
(i)子染色体が新たな最適解となる場合は無条件で置換
(ii)トーナメント相手が最適解の場合には無置換
(iii)子染色体のfitness<トーナメント相手のfitnessの場合には0.25の確率で置換
(iv)子染色体のfitness>トーナメント相手のfitnessの場合には0.75の確率で置換
(8)終了は、交叉→突然変異→選抜の繰り返し回数の上限を最初100,000とし、最適解が見出される毎に1000追加されるものとした。
【0103】
上記GAの設定で、Pmis(cv)最適解、AIC最適解を最大世代数を9,000に設定して探索し、これを5回(5Run)繰り返し、それぞれ得られたモデルで独立テスト集合予測不正解数Nmis(ind)を検証した。これを表1のAICにおいてRun#の0,1,2,3,4で示した。またマハラノビス距離の5回の重み付け平均を用いた場合のNmis (ind)も同様に求め、これを表1のAICにおいてΣ5で示した。今回の実施では重み付け係数w(m)はモデルによらず一定とした。結果は表1に示すようにAIC最適解において重み付け平均を採用することによって不正解数Nmis(ind)=16となり、予測精度(Nmis(ind))の顕著な向上が確認された。
【0104】
比較のためPmis(cv)最適解でも同様の検討を行なった。表1のPmis(cv) 最適解を見て分かるようにAIC最適解よりPmis(cv) 最適解は劣るが、それでもPmis(cv) 最適解のΣ5はRun#の0,1,2,3,4に比べて改善されていることが分かる。
以上の訓練集合と独立テスト集合とをあわせて新たに訓練集合とし、AIC最適解をGAで探索したところ、誤分類率は訓練用サンプル集合で19/144、独立テスト集合で18/46であった。一方、両誤分類数+Njを最小とする解を探索したところ、誤分類率は訓練集合で11/144、独立テスト集合で10/46であった。これより、10/46〜18/46程度の誤分類率は多変量正規分布モデルと968遺伝子がもつ本質的な限界であると考えられる。なお、[σk2] を用いたモデリングでは少数遺伝子のフィット力に乏しいモデルしか得ることができなかった。
【0105】
【表1】
【実施例2】
【0106】
Golubらのデータ(AML/ALLデータ)に基いて加工されたデータにて解析を行なった。表2にその結果を示す。なお、1〜38が訓練用サンプル集合、39〜72が予測用サンプル集合である。表2において1〜18がそれぞれ複数個の第1次最適解としての多変量正規分布モデルの予測結果(○が正解、×が不正解)を示し、Σが集団学習による第2次最適解のモデルによる予測結果を示す。
表2に示すようにNmis(ind)=1の結果が得られた。各多変量正規分布モデルの予測はそれぞれ決して良好ではないが、集団判定によって飛躍的に改善している様子が分かる。
【0107】
【表2】
【図面の簡単な説明】
【0108】
【図1】図1はデータ解析処理の全体を示すフローチャートである。
【図2】図2は図1のステップ2に示す集団学習による相関モデル構築処理のさらに詳しいフローチャートの一例である。
【図3】図3は適応度を計算するフローチャートの一例である。
【図4】図4はカテゴリーkの共分散逆行列の算出フローチャートの一例である。
【図5】図5は遺伝的アルゴリズム(GA)を説明するためのフローチャートである。
【図6】図6は遺伝的アルゴリズムを用いた第1次最適解を複数個得るためのフローチャートの一例である。
【図7】図7はデータ解析システムの一例を示す概略的なブロック図である。
【符号の説明】
【0109】
3…訓練用サンプル記憶部、4…第1次最適解取得部、5…第1次最適解記憶部、6…重み付け平均最適解算出部、10…予測用サンプル記憶部、11…相関モデル部。
【特許請求の範囲】
【請求項1】
コンピュータを用いて、カテゴリー分類された生体の状態を目的変数とし、遺伝子発現、生体関係物質の少なくとも一方に関するデータを説明変数とする相関モデルを決定する複数カテゴリー分類用のデータ解析方法であって、
カテゴリー分類された生体の状態と、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる訓練用サンプルの集合をコンピュータの記憶手段に記憶しておき、
遺伝的アルゴリズムに基づいて複数個の第1次最適解を得る第1次最適解取得処理と、
得られた複数個の第1次最適解の重み付け平均された第2次最適解を算出する重み付け平均最適解算出処理と、
を含んで前記相関モデル構築を行い、
前記第1次最適解取得処理が下記(a)〜(f)の各処理を含んで行われることを特徴とするデータ解析方法。
(a)説明変数の候補を選択する説明変数候補選択処理、
(b)各個体が相関モデルの候補を表しており、それらの個体の集団である第1世代を生成する初期個体集団生成処理、
(c)各個体について、各サンプルの各カテゴリーへの帰属尤度を算出する帰属尤度算出処理、
(d)算出された帰属尤度に基づくサンプルの予測カテゴリーとサンプルが実際に帰属する実測カテゴリーとを比較し、その予測がどの程度、信頼性があるかという定量的な評価である適応度を算出する適応度算出処理、
(e)算出された適応度に基づいて、個体に関する突然変異、交叉、選抜などの遺伝的アルゴリズムに基づいて次の世代の個体集団を生成する次世代個体集団生成処理、
(f)少なくとも帰属尤度算出処理、適応度算出処理、次世代個体集団生成処理にわたる一連の処理を繰り返し行って、最も適応度の高い最適解を探索しつつ、所定終結条件を満足する複数個の第1次最適解を得る
【請求項2】
請求項1に記載のデータ解析方法において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの帰属尤度を重み付け平均した重み付け平均帰属対数尤度で与えられる、データ解析方法。
【請求項3】
請求項1に記載のデータ解析方法において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの中心へのマハラノビス距離を重み付け平均したもので与えられ、前記帰属尤度算出処理がその重み付け平均マハラノビス距離に基づいて重み付け平均化された帰属対数尤度を算出する、データ解析方法。
【請求項4】
請求項1ないし請求項3のいずれか一項に記載のデータ解析方法において、各第1次最適解の重み付け係数を同じ値に設定した、データ解析方法。
【請求項5】
請求項1ないし請求項3のいずれか一項に記載のデータ解析方法において、各第1次最適解の重み付け係数を各第1次最適解に対応するモデル尤度に基づいて決定する、データ解析方法。
【請求項6】
請求項1ないし請求項5のいずれか一項に記載のデータ解析方法において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返しを複数回行う条件が含まれており、その各回で得られた最適解を第1次最適解取得処理における複数個の第1次最適解として設定する、データ解析方法。
【請求項7】
請求項1ないし請求項6のいずれか一項に記載のデータ解析方法において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返しを少なくとも一回行う過程で、算出適応度が設定適応度を超えた最適解を複数個抽出することが含まれており、それらの最適解を第1次最適解取得処理における複数個の第1次最適解として設定する、データ解析方法。
【請求項8】
請求項1ないし請求項7のいずれか一項に記載のデータ解析方法において、前記適応度算出処理がAICに基づいて行われる、データ解析方法。
【請求項9】
請求項1ないし請求項7のいずれか一項に記載のデータ解析方法において、前記適応度算出処理が交差検証成績に基づいて行われる、データ解析方法。
【請求項10】
請求項1ないし請求項9のいずれか一項に記載のデータ解析方法において、前記適応度算出処理が、前記帰属尤度算出処理によって算出された各サンプルの帰属尤度を全サンプルについて乗算することにより得られたモデル尤度に基づいて行われる、データ解析方法。
【請求項11】
請求項1ないし請求項10のいずれか一項に記載のデータ解析方法において、前記適応度算出処理が、前記帰属尤度算出処理によって算出された帰属尤度の中で各サンプルが一番大きな帰属尤度を有するカテゴリーに帰属すると予測し、その予測カテゴリーと実測カテゴリーとが一致する場合を識別正解、そうでない場合を識別不正解と判別する処理を含む、データ解析方法。
【請求項12】
請求項1ないし請求項11のいずれか一項に記載のデータ解析方法において、前記相関モデルを多変量正規分布モデルとした、データ解析方法。
【請求項13】
請求項12に記載のデータ解析方法において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をカテゴリーによらないものとして設定した、データ解析方法。
【請求項14】
請求項12に記載のデータ解析方法において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をムーア・ペンローズの一般化逆行列を用いて求める、データ解析方法。
【請求項15】
請求項1ないし請求項14のいずれか一項に記載のデータ解析方法において、前記相関モデルを、多変量特徴空間の各サンプルを主成分分析によって得られた部分空間により複数カテゴリーに分類する相関モデルとした、データ解析方法。
【請求項16】
請求項15に記載のデータ解析方法において、前記相関モデルがSIMCAモデルである、データ解析方法。
【請求項17】
請求項1ないし請求項16のいずれか一項に記載のデータ解析方法において、第1次最適解の数を3個以上〜50個以下とする、データ解析方法。
【請求項18】
請求項1ないし請求項17のいずれか一項に記載のデータ解析方法に基づいて構築された相関モデルに、予測用サンプルの選抜された説明変数を入力してカテゴリー分類された生体の状態を予測する出力を得ることを特徴とする、データ解析方法。
【請求項19】
コンピュータを用いて、カテゴリー分類された生体の状態を目的変数とし、遺伝子発現、生体関係物質の少なくとも一方に関するデータを説明変数とする相関モデルを決定する複数カテゴリー分類用のデータ解析システムであって、
カテゴリー分類された生体の状態と、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる訓練用サンプルの集合を記憶する記憶手段と、
遺伝的アルゴリズムに基づいて複数個の第1次最適解を得る第1次最適解取得手段と、
得られた複数個の第1次最適解の重み付け平均された第2次最適解を算出する重み付け平均最適解算出手段と、
を備えて前記相関モデル構築を行い、
前記第1次最適解取得手段が下記(a)〜(f)の各手段を含んでいることを特徴とするデータ解析システム。
(a)説明変数の候補を選択する説明変数候補選択手段、
(b)各個体が相関モデルの候補を表しており、それらの個体の集団である第1世代を生成する初期個体集団生成手段、
(c)各個体について、各サンプルの各カテゴリーへの帰属尤度を算出する帰属尤度算出手段、
(d)算出された帰属尤度に基づくサンプルの予測カテゴリーとサンプルが実際に帰属する実測カテゴリーとを比較し、その予測がどの程度、信頼性があるかという定量的な評価である適応度を算出する適応度算出手段、
(e)算出された適応度に基づいて、個体に関する突然変異、交叉、選抜などの遺伝的アルゴリズムに基づいて次の世代の個体集団を生成する次世代個体集団生成手段、
(f)最も適応度の高い最適解を探索しつつ、所定終結条件を満足する複数個の第1次最適解を得るために、少なくとも帰属尤度算出手段、適応度算出手段、次世代個体集団生成手段が行う一連の処理を複数回、繰り返して行なわせる繰返し処理手段
【請求項20】
請求項19に記載のデータ解析システムにおいて、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの帰属尤度を重み付け平均した重み付け平均帰属対数尤度で与えられる、データ解析システム。
【請求項21】
請求項19に記載のデータ解析システムにおいて、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの中心へのマハラノビス距離を重み付け平均したもので与えられ、前記帰属尤度算出手段がその重み付け平均マハラノビス距離に基づいて重み付け平均化された帰属対数尤度を算出する、データ解析システム。
【請求項22】
請求項20ないし請求項21のいずれか一項に記載のデータ解析システムにおいて、各第1次最適解の重み付け係数を同じ値に設定した、データ解析システム。
【請求項23】
請求項20ないし請求項21のいずれか一項に記載のデータ解析システムにおいて、各第1次最適解の重み付け係数を各第1次最適解に対応するモデル尤度に基づいて決定する、データ解析システム。
【請求項24】
請求項19ないし請求項23のいずれか一項に記載のデータ解析システムにおいて、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返しを複数回行う条件が含まれており、その各回で得られた最適解を第1次最適解取得処理における複数個の第1次最適解として設定する、データ解析システム。
【請求項25】
請求項19ないし請求項24のいずれか一項に記載のデータ解析システムにおいて、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返しを少なくとも一回行う過程で算出適応度が設定適応度を超えた最適解を複数個抽出することが含まれており、それらの最適解を第1次最適解取得処理における複数個の第1次最適解として設定する、データ解析システム。
【請求項26】
請求項19ないし請求項25のいずれか一項に記載のデータ解析システムにおいて、前記適応度算出手段の行う適応度算出処理がAICに基づいて行われる、データ解析システム。
【請求項27】
請求項19ないし請求項25のいずれか一項に記載のデータ解析システムにおいて、前記適応度算出手段の行う適応度算出処理が交差検証成績に基づいて行われる、データ解析システム。
【請求項28】
請求項19ないし請求項27のいずれか一項に記載のデータ解析システムにおいて、前記適応度算出手段の適応度算出処理が、前記帰属尤度算出手段によって算出された各サンプルの帰属尤度を全サンプルについて乗算することにより得られたモデル尤度に基づいて行われる、データ解析システム。
【請求項29】
請求項19ないし請求項28のいずれか一項に記載のデータ解析システムにおいて、前記適応度算出手段が、前記帰属尤度算出手段によって算出された帰属尤度の中で各サンプルが一番大きな帰属尤度を有するカテゴリーに帰属すると予測し、その予測カテゴリーと実測カテゴリーとが一致する場合を識別正解、そうでない場合を識別不正解と判別する判別手段を含む、データ解析システム。
【請求項30】
請求項19ないし請求項29のいずれか一項に記載のデータ解析システムにおいて、前記相関モデルを多変量正規分布モデルとした、データ解析システム。
【請求項31】
請求項30に記載のデータ解析システムにおいて、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をカテゴリーによらないものとして設定した、データ解析システム。
【請求項32】
請求項30に記載のデータ解析システムにおいて、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をムーア・ペンローズの一般化逆行列を用いて求める、データ解析システム。
【請求項33】
請求項19ないし請求項32のいずれか一項に記載のデータ解析システムにおいて、前記相関モデルを、多変量特徴空間の各サンプルを主成分分析によって得られた部分空間により複数カテゴリーに分類する相関モデルとした、データ解析システム。
【請求項34】
請求項33に記載のデータ解析システムにおいて、前記相関モデルがSIMCAモデルである、データ解析システム。
【請求項35】
請求項19ないし請求項34のいずれか一項に記載のデータ解析システムにおいて、第1次最適解の数を3個以上〜50個以下とする、データ解析システム。
【請求項36】
請求項19ないし請求項35のいずれか一項に記載のデータ解析システムに基づいて構築された相関モデルと、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる予測用サンプルを記憶する記憶手段とを備え、前記相関モデルに予測用サンプルの選抜された説明変数を入力してカテゴリー分類された生体の状態を予測する出力を得ることを特徴とする、データ解析システム。
【請求項1】
コンピュータを用いて、カテゴリー分類された生体の状態を目的変数とし、遺伝子発現、生体関係物質の少なくとも一方に関するデータを説明変数とする相関モデルを決定する複数カテゴリー分類用のデータ解析方法であって、
カテゴリー分類された生体の状態と、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる訓練用サンプルの集合をコンピュータの記憶手段に記憶しておき、
遺伝的アルゴリズムに基づいて複数個の第1次最適解を得る第1次最適解取得処理と、
得られた複数個の第1次最適解の重み付け平均された第2次最適解を算出する重み付け平均最適解算出処理と、
を含んで前記相関モデル構築を行い、
前記第1次最適解取得処理が下記(a)〜(f)の各処理を含んで行われることを特徴とするデータ解析方法。
(a)説明変数の候補を選択する説明変数候補選択処理、
(b)各個体が相関モデルの候補を表しており、それらの個体の集団である第1世代を生成する初期個体集団生成処理、
(c)各個体について、各サンプルの各カテゴリーへの帰属尤度を算出する帰属尤度算出処理、
(d)算出された帰属尤度に基づくサンプルの予測カテゴリーとサンプルが実際に帰属する実測カテゴリーとを比較し、その予測がどの程度、信頼性があるかという定量的な評価である適応度を算出する適応度算出処理、
(e)算出された適応度に基づいて、個体に関する突然変異、交叉、選抜などの遺伝的アルゴリズムに基づいて次の世代の個体集団を生成する次世代個体集団生成処理、
(f)少なくとも帰属尤度算出処理、適応度算出処理、次世代個体集団生成処理にわたる一連の処理を繰り返し行って、最も適応度の高い最適解を探索しつつ、所定終結条件を満足する複数個の第1次最適解を得る
【請求項2】
請求項1に記載のデータ解析方法において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの帰属尤度を重み付け平均した重み付け平均帰属対数尤度で与えられる、データ解析方法。
【請求項3】
請求項1に記載のデータ解析方法において、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの中心へのマハラノビス距離を重み付け平均したもので与えられ、前記帰属尤度算出処理がその重み付け平均マハラノビス距離に基づいて重み付け平均化された帰属対数尤度を算出する、データ解析方法。
【請求項4】
請求項1ないし請求項3のいずれか一項に記載のデータ解析方法において、各第1次最適解の重み付け係数を同じ値に設定した、データ解析方法。
【請求項5】
請求項1ないし請求項3のいずれか一項に記載のデータ解析方法において、各第1次最適解の重み付け係数を各第1次最適解に対応するモデル尤度に基づいて決定する、データ解析方法。
【請求項6】
請求項1ないし請求項5のいずれか一項に記載のデータ解析方法において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返しを複数回行う条件が含まれており、その各回で得られた最適解を第1次最適解取得処理における複数個の第1次最適解として設定する、データ解析方法。
【請求項7】
請求項1ないし請求項6のいずれか一項に記載のデータ解析方法において、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返しを少なくとも一回行う過程で、算出適応度が設定適応度を超えた最適解を複数個抽出することが含まれており、それらの最適解を第1次最適解取得処理における複数個の第1次最適解として設定する、データ解析方法。
【請求項8】
請求項1ないし請求項7のいずれか一項に記載のデータ解析方法において、前記適応度算出処理がAICに基づいて行われる、データ解析方法。
【請求項9】
請求項1ないし請求項7のいずれか一項に記載のデータ解析方法において、前記適応度算出処理が交差検証成績に基づいて行われる、データ解析方法。
【請求項10】
請求項1ないし請求項9のいずれか一項に記載のデータ解析方法において、前記適応度算出処理が、前記帰属尤度算出処理によって算出された各サンプルの帰属尤度を全サンプルについて乗算することにより得られたモデル尤度に基づいて行われる、データ解析方法。
【請求項11】
請求項1ないし請求項10のいずれか一項に記載のデータ解析方法において、前記適応度算出処理が、前記帰属尤度算出処理によって算出された帰属尤度の中で各サンプルが一番大きな帰属尤度を有するカテゴリーに帰属すると予測し、その予測カテゴリーと実測カテゴリーとが一致する場合を識別正解、そうでない場合を識別不正解と判別する処理を含む、データ解析方法。
【請求項12】
請求項1ないし請求項11のいずれか一項に記載のデータ解析方法において、前記相関モデルを多変量正規分布モデルとした、データ解析方法。
【請求項13】
請求項12に記載のデータ解析方法において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をカテゴリーによらないものとして設定した、データ解析方法。
【請求項14】
請求項12に記載のデータ解析方法において、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をムーア・ペンローズの一般化逆行列を用いて求める、データ解析方法。
【請求項15】
請求項1ないし請求項14のいずれか一項に記載のデータ解析方法において、前記相関モデルを、多変量特徴空間の各サンプルを主成分分析によって得られた部分空間により複数カテゴリーに分類する相関モデルとした、データ解析方法。
【請求項16】
請求項15に記載のデータ解析方法において、前記相関モデルがSIMCAモデルである、データ解析方法。
【請求項17】
請求項1ないし請求項16のいずれか一項に記載のデータ解析方法において、第1次最適解の数を3個以上〜50個以下とする、データ解析方法。
【請求項18】
請求項1ないし請求項17のいずれか一項に記載のデータ解析方法に基づいて構築された相関モデルに、予測用サンプルの選抜された説明変数を入力してカテゴリー分類された生体の状態を予測する出力を得ることを特徴とする、データ解析方法。
【請求項19】
コンピュータを用いて、カテゴリー分類された生体の状態を目的変数とし、遺伝子発現、生体関係物質の少なくとも一方に関するデータを説明変数とする相関モデルを決定する複数カテゴリー分類用のデータ解析システムであって、
カテゴリー分類された生体の状態と、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる訓練用サンプルの集合を記憶する記憶手段と、
遺伝的アルゴリズムに基づいて複数個の第1次最適解を得る第1次最適解取得手段と、
得られた複数個の第1次最適解の重み付け平均された第2次最適解を算出する重み付け平均最適解算出手段と、
を備えて前記相関モデル構築を行い、
前記第1次最適解取得手段が下記(a)〜(f)の各手段を含んでいることを特徴とするデータ解析システム。
(a)説明変数の候補を選択する説明変数候補選択手段、
(b)各個体が相関モデルの候補を表しており、それらの個体の集団である第1世代を生成する初期個体集団生成手段、
(c)各個体について、各サンプルの各カテゴリーへの帰属尤度を算出する帰属尤度算出手段、
(d)算出された帰属尤度に基づくサンプルの予測カテゴリーとサンプルが実際に帰属する実測カテゴリーとを比較し、その予測がどの程度、信頼性があるかという定量的な評価である適応度を算出する適応度算出手段、
(e)算出された適応度に基づいて、個体に関する突然変異、交叉、選抜などの遺伝的アルゴリズムに基づいて次の世代の個体集団を生成する次世代個体集団生成手段、
(f)最も適応度の高い最適解を探索しつつ、所定終結条件を満足する複数個の第1次最適解を得るために、少なくとも帰属尤度算出手段、適応度算出手段、次世代個体集団生成手段が行う一連の処理を複数回、繰り返して行なわせる繰返し処理手段
【請求項20】
請求項19に記載のデータ解析システムにおいて、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの帰属尤度を重み付け平均した重み付け平均帰属対数尤度で与えられる、データ解析システム。
【請求項21】
請求項19に記載のデータ解析システムにおいて、前記重み付け平均最適解が、第1次最適解に係る各サンプルの各カテゴリーの中心へのマハラノビス距離を重み付け平均したもので与えられ、前記帰属尤度算出手段がその重み付け平均マハラノビス距離に基づいて重み付け平均化された帰属対数尤度を算出する、データ解析システム。
【請求項22】
請求項20ないし請求項21のいずれか一項に記載のデータ解析システムにおいて、各第1次最適解の重み付け係数を同じ値に設定した、データ解析システム。
【請求項23】
請求項20ないし請求項21のいずれか一項に記載のデータ解析システムにおいて、各第1次最適解の重み付け係数を各第1次最適解に対応するモデル尤度に基づいて決定する、データ解析システム。
【請求項24】
請求項19ないし請求項23のいずれか一項に記載のデータ解析システムにおいて、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返しを複数回行う条件が含まれており、その各回で得られた最適解を第1次最適解取得処理における複数個の第1次最適解として設定する、データ解析システム。
【請求項25】
請求項19ないし請求項24のいずれか一項に記載のデータ解析システムにおいて、前記(f)に記載した所定終結条件中に、説明変数候補選択処理、初期個体集団生成処理の繰り返しを少なくとも一回行う過程で算出適応度が設定適応度を超えた最適解を複数個抽出することが含まれており、それらの最適解を第1次最適解取得処理における複数個の第1次最適解として設定する、データ解析システム。
【請求項26】
請求項19ないし請求項25のいずれか一項に記載のデータ解析システムにおいて、前記適応度算出手段の行う適応度算出処理がAICに基づいて行われる、データ解析システム。
【請求項27】
請求項19ないし請求項25のいずれか一項に記載のデータ解析システムにおいて、前記適応度算出手段の行う適応度算出処理が交差検証成績に基づいて行われる、データ解析システム。
【請求項28】
請求項19ないし請求項27のいずれか一項に記載のデータ解析システムにおいて、前記適応度算出手段の適応度算出処理が、前記帰属尤度算出手段によって算出された各サンプルの帰属尤度を全サンプルについて乗算することにより得られたモデル尤度に基づいて行われる、データ解析システム。
【請求項29】
請求項19ないし請求項28のいずれか一項に記載のデータ解析システムにおいて、前記適応度算出手段が、前記帰属尤度算出手段によって算出された帰属尤度の中で各サンプルが一番大きな帰属尤度を有するカテゴリーに帰属すると予測し、その予測カテゴリーと実測カテゴリーとが一致する場合を識別正解、そうでない場合を識別不正解と判別する判別手段を含む、データ解析システム。
【請求項30】
請求項19ないし請求項29のいずれか一項に記載のデータ解析システムにおいて、前記相関モデルを多変量正規分布モデルとした、データ解析システム。
【請求項31】
請求項30に記載のデータ解析システムにおいて、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をカテゴリーによらないものとして設定した、データ解析システム。
【請求項32】
請求項30に記載のデータ解析システムにおいて、各サンプルの各カテゴリー中心へのマハラノビス距離を算出する場合に多変量正規分布モデルの共分散逆行列をムーア・ペンローズの一般化逆行列を用いて求める、データ解析システム。
【請求項33】
請求項19ないし請求項32のいずれか一項に記載のデータ解析システムにおいて、前記相関モデルを、多変量特徴空間の各サンプルを主成分分析によって得られた部分空間により複数カテゴリーに分類する相関モデルとした、データ解析システム。
【請求項34】
請求項33に記載のデータ解析システムにおいて、前記相関モデルがSIMCAモデルである、データ解析システム。
【請求項35】
請求項19ないし請求項34のいずれか一項に記載のデータ解析システムにおいて、第1次最適解の数を3個以上〜50個以下とする、データ解析システム。
【請求項36】
請求項19ないし請求項35のいずれか一項に記載のデータ解析システムに基づいて構築された相関モデルと、遺伝子発現、生体関係物質の少なくとも一方に関するデータからなる予測用サンプルを記憶する記憶手段とを備え、前記相関モデルに予測用サンプルの選抜された説明変数を入力してカテゴリー分類された生体の状態を予測する出力を得ることを特徴とする、データ解析システム。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2006−11724(P2006−11724A)
【公開日】平成18年1月12日(2006.1.12)
【国際特許分類】
【出願番号】特願2004−186573(P2004−186573)
【出願日】平成16年6月24日(2004.6.24)
【出願人】(000000354)石原産業株式会社 (289)
【公開日】平成18年1月12日(2006.1.12)
【国際特許分類】
【出願日】平成16年6月24日(2004.6.24)
【出願人】(000000354)石原産業株式会社 (289)
[ Back to top ]