
トナー及び二成分系現像剤
【課題】耐久性や低温定着性を達成しつつ、画像の面内のグロスの均一性を確保したトナーを提供すること。
【解決手段】結着樹脂およびワックスを少なくとも含有するトナーであって、結着樹脂は、ポリエステル樹脂を含有し、ワックスの含有量が、結着樹脂100質量部に対して、3.0質量部以上20.0質量部以下であり、トナーの深さ方向に対してのワックス偏在度合いが制御されていることを特徴とする、トナー。
【解決手段】結着樹脂およびワックスを少なくとも含有するトナーであって、結着樹脂は、ポリエステル樹脂を含有し、ワックスの含有量が、結着樹脂100質量部に対して、3.0質量部以上20.0質量部以下であり、トナーの深さ方向に対してのワックス偏在度合いが制御されていることを特徴とする、トナー。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、電子写真法および静電記録法に用いられるトナー及び二成分系現像剤に関する。
【背景技術】
【0002】
電子写真法において、静電荷像を現像する工程は摩擦帯電されたトナーを静電荷像とのクーロン力を利用して静電荷像上に付着させて画像形成する。トナーを用いて静電荷像を現像するための現像剤には、磁性体を樹脂中に分散した磁性トナーを用いる一成分系現像剤と、非磁性トナーと磁性キャリアを混合して用いる二成分系現像剤とに大別される。
特に、高画質を要求されるフルカラー複写機またはフルカラープリンタ等のフルカラー画像形成装置では、後者が好適に用いられている。
フルカラー画像形成装置は、近年POD用途やグラフィック用途に用いられている。このような分野では従来以上の高速・高画質が要求されている。高画質として要求されることの一つとして、画像の面内のグロス均一性がある。画像の面内のグロス均一性は、定着部材とトナーとの剥離性に相関がある。剥離性が良い場合、画像の面内のグロスは画像のどの部分でも同じ(均一)になる。
それに対し、剥離性が悪いトナーの場合、剥離が不十分だった部分は画像表面が剥離時にあらされ、画像の面内のグロスが低下してしまうことがある。このため、グロスが高い部分と低い部分が同じ画像の中に出てしまうことがある(不均一になる)。
このような課題を解決するために、定着部材にオイルを塗布する手法が提案されてきた。このような手法の場合、剥離性は良いが、塗布したオイルにより過度のグロスが出てしまいグロスのコントロールが困難である。
また、トナー中にワックスを多く添加することでも剥離性を改善できるが、耐久性の低下やトナーの耐熱保存性が低下する場合がある。
トナーの耐久性や耐熱保存性改善の目的で、トナー粒子の表面から0.3μmまでの深さ領域に存在するワックス比率を規定したトナーの提案がなされている。(特許文献1、特許文献2)
上記提案のトナーは、トナーの保存性能や、現像部材の汚染は低減されていたが、グロスの均一性の悪化が生じる場合があり、トナーの剥離性は不十分であった。
このようにいずれの提案においても、耐久性や低温定着性を達成しつつ、画像のグロスの均一性を確保したトナー及び二成分系現像剤は得られていない。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2004−246345号公報
【特許文献2】特開2006−258901号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明は、耐久性や低温定着性を達成しつつ、画像の面内のグロスの均一性を確保したトナー及び二成分系現像剤を提供する。
【課題を解決するための手段】
【0005】
本発明者らは鋭意検討を重ねた結果、低温定着性に優れたトナーにおいて、十分な耐久性、耐熱保存性を達成しつつ、画像の面内のグロスの均一性を改善させるためには、トナー中のワックス偏在状態をコントロールすることが必要であることを見出した。
すなわち、結着樹脂、ワックス、及び外添剤としての無機微粉体を少なくとも含有する
トナーであって、前記結着樹脂はポリエステル樹脂を含有し、前記ワックスの含有量が、前記結着樹脂100質量部に対して、3.0質量部以上20.0質量部以下であり、前記トナーは、ATR法を用い、ATR結晶としてGe、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPa、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPbとし、ATR法を用い、ATR結晶としてKRS5、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPc、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPdとしたときに、下記式(1)の関係を満たすことを特徴とするトナー。
1.05≦P1/P2≦2.00 ・・・ 式(1)
[前記式(1)において、前記最大吸収ピーク強度Paは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pbは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pcは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pdは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、P1=Pa/Pb、P2=Pc/Pdである。]
【発明の効果】
【0006】
本発明によれば、耐久性や低温定着性を達成しつつ、画像の面内のグロスの均一性を確保したトナー及び二成分系現像剤を提供できる。
【図面の簡単な説明】
【0007】
【図1】表面改質装置の模式図。
【図2】ATR結晶としてGeを用い測定したFT−IRスペクトルの一例。
【図3】交流インピーダンス測定法の構成を表す模式図。
【図4】インピーダンス測定で得られたCole−Coleプロットをフィッティングするために用いるフィッティング回路。
【図5】図4に示した回路のインピーダンスのCole−Coleプロットを表すグラフ図。
【発明を実施するための形態】
【0008】
以下、本発明を実施するための形態について説明する。
本発明で使用するトナーは、結着樹脂、ワックス、及び外添剤としての無機微粉体を少なくとも含有するトナーであって、かつ、該結着樹脂がポリエステル樹脂を含み、トナーの深さ方向に対してのワックス偏在度合いをコントロールしたトナーである。本発明のトナーは当該構成をとることで、耐久性能と画像の面内のグロス均一性(以下単にグロス均一性ともいう)を両立することができる。
まず、トナー表面からトナー中心部に向かうトナーの深さ方向において、トナー表面から約0.3μmの間にワックスを多量に存在させることでグロス均一性を改善することができる。これは、トナー表面近傍の、結着樹脂に対するワックスの存在比率(以下単にワックス比率ともいう)が多いために、定着部材とトナー層の剥離性が良好であり、結果、画像のどの部分でも同じように剥離することに起因する。
しかしながら、ワックスは分子量が結着樹脂に比べ小さく軟らかいために、トナー中にワックスが多く存在するとトナー表面に外添剤が埋め込まれやすくなる。その結果、トナ
ーとキャリア間の付着力が増加し、トナーがキャリアから像担持体へ現像されにくくなり、耐久時の画像濃度が低下してしまうことがある。
そこで、発明者らは鋭意検討し、トナー表面から約1.0μmの、結着樹脂に対するワックスの存在比率をコントロールすることで、上記耐久時の画像濃度の低下を抑制できることを見出した。
そのメカニズムは明確ではないが、発明者らは以下のように推察している。
外添剤の埋め込みは、トナー表層だけではなく、その下層のやわらかさも関与している。例えば、トナーの最表層のワックス比率が高かったとしても、その下層が固い樹脂の層で構成されていれば、外添剤はその機能を失うほどには埋め込まれない。この外添剤の埋め込みに関与するのがトナー表層から約1.0μmの範囲である。このため、この範囲に存在するワックス比率をコントロールすることで、耐久時の画像濃度の低下を抑制できる。このため、トナー表面からトナー中心部に向かうトナーの深さ方向において、トナー表面から約0.3μmの間の、結着樹脂に対するワックスの存在比率を、約1.0μmの間の、結着樹脂に対するワックスの存在比率より多くすることが重要である。
また、当該構成を採用することで、トナー表面に多く存在するワックスがトナー中のワックスの染み出しをさらに促進する。これは、トナー表面に存在するワックスが溶けることにより、トナー内部からトナー表面へのワックスの通り道が形成され、定着時に効果的にワックスが染み出すことができるためだと思われる。染み出したワックスは、離型性をより高められるため、グロス均一性が向上する。
一方、トナー表面にワックスを偏析させないトナーの場合、ワックスを多量に添加しなくてはならず、その結果、トナーの耐久性が低下する場合がある。
さらに、トナー表面よりもやや内側のワックス比率をトナー表面のワックス比率より少なくすることで、耐久性もさらに向上する。これは、トナー表層はワックス比率が多いため、外添剤が埋め込まれやすい。それに対し、トナー表面のやや内側はトナー表層に比べワックス比率が少ないために、外添剤が埋め込まれにくい。このため、ワックス比率が高いトナー表層からワックス比率が低いトナーやや内側にいくにしたがって、外添剤の埋め込みが抑制される。
上記理由から、トナー表面から約0.3μmの間の結着樹脂に対するワックスの存在比率[A]、及び、約1.0μmの間の結着樹脂に対するワックスの存在比率[B]を調整し、当該[A]の[B]に対する比([A]/[B])を制御することで、初めて耐久性とグロスの均一性を確保できる。
【0009】
具体的には、本発明のトナーは、ATR法を用い、ATR結晶としてGe、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPa、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPbとし、
ATR法を用い、ATR結晶としてKRS5、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPc、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPdとしたときに、下記式(1)の関係を満たしていることを特徴とする。
1.05≦P1/P2≦2.00 ・・・ 式(1)
[前記式(1)において、前記最大吸収ピーク強度Paは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pbは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pcは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pdは、1713cm−1以上1723cm−
1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、P1=Pa/Pb、P2=Pc/Pdである。]
【0010】
上記ATR(Attenuated Total Reflection)法とは、試料より高い屈折率を有する結晶(ATR結晶)に、試料を密着させ、臨界角以上の入射角で赤外光を結晶に入射させると、光は密着した試料と結晶の界面で全反射を繰り返し出射する。この時、赤外光は試料と結晶の界面で反射するのではなく、試料側にわずかににじみこんでから全反射する。このにじみこみ深さは、波長、入射角及びATR結晶の屈折率に依存する。
dp=λ/(2πn1)×[sin2θ―(n2/n1)2]−1/2
dp:にじみ込み深さ
n1:試料の屈折率(本発明では1.5としている)
n2:ATR結晶の屈折率(ATR結晶がGeの場合の屈折率;4.0、ATR結晶がKRS5の場合の屈折率;2.4)
θ:入射角
このため、ATR結晶の屈折率や入射角を変えることでにじみこみ深さの異なるFT−IRスペクトルを得ることができる。
例えば、ATR法において、ATR結晶にGe(n2=4.0)を用い、2000cm−1(λ=5μm)の光を、入射角45°の条件で測定した場合、上記式を用いると、にじみこみ深さdpは0.3μmになる。一方、ATR結晶にKRS5<臭沃化タリウム[臭化タリウムTlBr(42モル%)+ヨウ化タリウム Tll(58モル%)]の組成をもつタリウムハライドの混晶>(n2=2.4)を用い入射角45°の条件で測定した場合、にじみこみ深さは1.0μmとなる。
【0011】
本発明のトナーでは、結着樹脂はポリエステル樹脂を含む樹脂である。
このため、1713cm−1以上1723cm−1以下の範囲の吸収ピークは、主に結着樹脂由来の−CO―の伸縮振動に起因するピークである。
結着樹脂由来のピークとしては、上記以外にも芳香環のCHの面外変角振動等様々なピークが検出されるが、1500cm−1以下の範囲には、ピークが数多く存在し、結着樹脂のピークだけを分離することが困難であり、正確な数値を算出できない。このため、他のピークとの分離が容易な1713cm−1以上1723cm−1以下の範囲の吸収ピークを結着樹脂由来のピークとして用いる。
また、2843cm−1以上2853cm−1以下の範囲の吸収ピークは、主にワックス由来の−CH2−の伸縮振動(対称)に起因するピークである。
ワックスのピークとしては、上記以外にも1450cm−1以上1500cm−1以下にCH2の面内変角振動のピークが検出されるが、結着樹脂由来のピークとも重なり合ってしまい、ワックスのピークを分離することが困難である。このため、他のピークとの分離が容易な2843cm−1以上2853cm−1以下の範囲の吸収ピークをワックス由来のピークとして用いる。
本発明において、上記結着樹脂由来の最大吸収ピーク強度(Pb、Pd)及びワックス由来の最大吸収ピーク強度(Pa、Pc)が、結着樹脂及びワックスの存在量に相関することを見出した。そこで、ワックス由来の最大吸収ピーク強度を結着樹脂由来の最大吸収ピーク強度で割ることで、結着樹脂に対するワックスの存在比率を算出している。
ここで、Pa及びPcを求めるに当たり、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引く理由は、ベースラインの影響を排除し、真のピーク強度を算出するためである。3050cm−1と2600cm−1付近には吸収ピークがないため、この2点の平均値を算出することで、ベースライン強度を算出できる。
一方、Pb及びPdを求めるに当たり、1713cm−1以上1723cm−1以下の
範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引く理由も、ベースラインの影響を排除し、真のピーク強度を算出するためである。3050cm−1と2600cm−1付近には吸収ピークがないため、この2点の平均値を算出することで、ベースライン強度を算出できる。
【0012】
そして、トナー表面から約0.3μmの間の結着樹脂に対するワックスの存在比率(P1)は、ATR法を用い、ATR結晶としてGe(n2=4.0)、赤外光入射角として45°の条件で測定し得られた、上記Pa及びPbから算出される(P1=Pa/Pb)。ここでP1は、トナーの表面近傍の、結着樹脂に対するワックスの存在比率を表すことが必要である。さらなる、(すなわち、トナー表面から約0.3μmより小さい距離)トナー表面近傍の結着樹脂に対するワックスの存在比率を測定する場合、ATR結晶への赤外光(IR)の入射角を大きくすることが考えられる。しかし、入射角を大きくしていくにつれてIRスペクトルの強度が低下してくる。その結果、数値の信頼性が低下してしまう。このため、発明者らはIRのスペクトルの強度が確保できる、入射角45°の条件で測定を行い、トナー表面から約0.3μmの間の結着樹脂に対するワックスの存在比率をトナー表面近傍の結着樹脂に対するワックスの存在比率(P1)とした。
先に述べたとおり、P1の値をコントロールすることで、グロス均一性を改善することができる。具体的には、P1は0.20以上1.50以下であることが好ましく、より好ましくは、0.30以上1.20以下である。
P1は、ワックスの種類及び結着樹脂に対するワックスの添加量の調整、トナー中おけるワックスの分散状態又はワックスの偏在状態を制御することができる樹脂の添加、トナーの製造工程における熱風を用いたトナーの改質処理の実施により、上記範囲に制御することが可能である。これらの具体例は後述する。
【0013】
一方、トナー表面から約1.0μmの間の結着樹脂に対するワックスの存在比率(P2)は、ATR法を用い、ATR結晶としてKRS5(n2=2.4)、赤外光入射角として45°の条件で測定し得られた上記Pc及びPdから算出される(P2=Pc/Pd)。先に述べたとおり、P2をコントロールすることで、耐久時の画像濃度の低下を抑制することができる。具体的には、P2は0.10以上0.70以下であることが好ましく、より好ましくは、0.20以上0.60以下である。
P2は、ワックスの結着樹脂に対する添加量を調整することで上記範囲に制御することが可能である。
当該ワックスの添加量は、結着樹脂100質量部に対して、3.0質量部以上20.0質量部以下であり、好ましくは3.5質量部以上16.0質量部以下である。
【0014】
そして、耐久時の画像濃度の低下の抑制と画像の面内のグロス均一性の両立させるためには、P1/P2が1.05以上2.00以下であり、好ましくは1.25以上1.90以下である。
P1/P2が2.00より大きい場合、トナーの表面近傍に偏在するワックス比率が高すぎる。このようなトナーの場合、表面近傍に偏在するワックスが多すぎるために、外添剤がトナー表面に埋め込まれやすくなる。その結果、トナーとキャリア間の付着力が増加し、トナーがキャリアから像担持体へ現像されにくくなり、耐久時の画像濃度が低下する。一方、P1/P2が1.05未満のトナーは、トナーの内部に偏在するワックス比率が高く、耐久時の外添剤の埋め込みを抑制することが出来ないことに加えて、グロス均一性が顕著に低下する。
P1/P2は、ワックスの種類及び結着樹脂に対するワックスの添加量の調整、トナー中おけるワックスの分散状態又はワックスの偏在状態を制御することができる樹脂の添加、トナーの製造工程における熱風を用いたトナーの改質処理の実施により、上記範囲に制御することが可能である。
【0015】
本発明のトナーに用いられるワックスとしては、特に限定されず、公知の物を使用することができる。以下、好適に使用できる物を例示する。低分子量ポリエチレン、低分子量ポリプロピレン、アルキレン共重合体、マイクロクリスタリンワックス、パラフィンワックス、フィッシャー・トロプシュワックスの如き脂肪族炭化水素系ワックス;酸化ポリエチレンワックスの如き脂肪族炭化水素系ワックスの酸化物;脂肪族炭化水素系エステルワックスの如き脂肪酸エステルを主成分とするワックス;及び脱酸カルナバワックスの如き脂肪酸エステルを一部または全部を脱酸化したもの。ベヘニン酸モノグリセリドの如き脂肪酸と多価アルコールの部分エステル化物;植物性油脂を水素添加することによって得られるヒドロキシル基を有するメチルエステル化合物等。
なかでも、炭化水素系ワックス、特に脂肪族炭化水素系ワックスが高湿環境下における耐久時の画像濃度の低下を防止できることから、より好ましい。
本発明において、ワックスの最大吸熱ピークのピーク温度は、55℃以上140℃以下であることが、耐熱保存性及びホットオフセット性の改善のため、好ましい。
ワックスの最大吸熱ピークを55℃以上にすることで、ワックスが保管時に溶解することを防止でき、その結果、耐熱保存性を向上することができる。また、ワックスの最大吸熱ピークを140℃以下とすることで、定着時にワックスが溶融し、巻きつきを抑制することができる。
【0016】
次に、結着樹脂について説明する。本発明において、結着樹脂はトナーの構成要素を十分に結着するための樹脂である。後述するトナーのワックス偏在状態を制御するための樹脂は、本発明においては結着樹脂に該当しない。
本発明のトナーに用いられる結着樹脂は、低温定着性の観点から、シャープメルト性の高いポリエステル樹脂を含有する。結着樹脂に占めるポリエステル樹脂の割合は、50質量%以上であることが好ましく、70質量%以上であることがより好ましく、90質量%以上であることがさらに好ましく、100質量%であることが特に好ましい。本発明に使用される結着樹脂は、ポリエステル樹脂を含む以外に、低温定着性に影響を与えない範囲で、他の樹脂を含むこともできる。当該樹脂としては、例えば、熱可塑性結着樹脂などが挙げられる。具体的には、スチレン、パラクロロスチレン、α−メチルスチレン等のスチレン類の単独重合体又は共重合体(スチレン系樹脂);アクリル酸メチル、アクリル酸エチル、アクリル酸n−プロピル、アクリル酸n−ブチル、アクリル酸ラウリル、アクリル酸2−エチルヘキシル、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸n−プロピル、メタクリル酸ラウリル、メタクリル酸2−エチルヘキシル等のビニル基を有するエステル類の単独重合体又は共重合体(ビニル系樹脂);アクリロニトリル、メタクリロニトリル等のビニルニトリル類の単独重合体又は共重合体(ビニル系樹脂);ビニルメチルエーテル、ビニルイソブチルエーテル等のビニルエーテル類の単独重合体又は共重合体(ビニル系樹脂);ビニルメチルケトン、ビニルエチルケトン、ビニルイソプロペニルケトン等のビニルケトン類の単独重合体又は共重合体(ビニル系樹脂);エチレン、プロピレン、ブタジエン、イソプレン等のオレフィン類の単独重合体又は共重合体(オレフィン系樹脂);エポキシ樹脂、上述以外のポリエステル系樹脂、ポリウレタン樹脂、ポリアミド樹脂、セルロース樹脂、ポリエーテル樹脂等の非ビニル縮合系樹脂、及びこれらの非ビニル縮合系樹脂とビニル系モノマーとのグラフト重合体などが挙げられる。これらの樹脂は、1種のみを上記ポリエステル樹脂に含有させて用いてもよいし、2種以上を含有させて用いてもよい。
【0017】
本発明に用いられるポリエステル樹脂はアルコールモノマーとカルボン酸モノマーが縮重合したものが用いられる。アルコールモノマーとしては以下のものが挙げられる。
ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン、ポリオキシプロピレン(3.3)−2,2−ビス(4−ヒドロキシフェニル)プロパン、ポリオキシエチレン(2.0)−2,2−ビス(4−ヒドロキシフェニル)プロパン、ポリオキシプロピレン(2.0)−ポリオキシエチレン(2.0)−2,2−ビス(4−
ヒドロキシフェニル)プロパン、ポリオキシプロピレン(6)−2,2−ビス(4−ヒドロキシフェニル)プロパン等のビスフェノールAのアルキレンオキシド付加物、エチレングリコール、ジエチレングリコール、トリエチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、ネオペンチルグリコール、1,4−ブテンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,4−シクロヘキサンジメタノール、ジプロピレングリコール、ポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレングリコール、ビスフェノールA、水素添加ビスフェノールA、ソルビトール、1,2,3,6−ヘキサンテトロール、1,4−ソルビタン、ペンタエリスリトール、ジペンタエリスリトール、トリペンタエリスリトール、1,2,4−ブタントリオール、1,2,5−ペンタントリオール、グリセロール、2−メチルプロパントリオール、2−メチル−1,2,4−ブタントリオール、トリメチロールエタン、トリメチロールプロパン、1,3,5−トリヒドロキシメチルベンゼン。
一方、カルボン酸モノマーとしては、以下のものが挙げられる。
フタル酸、イソフタル酸及びテレフタル酸の如き芳香族ジカルボン酸類又はその無水物;コハク酸、アジピン酸、セバシン酸及びアゼライン酸の如きアルキルジカルボン酸類又はその無水物;炭素数6〜18のアルキル基又はアルケニル基で置換されたコハク酸もしくはその無水物;フマル酸、マレイン酸及びシトラコン酸の如き不飽和ジカルボン酸類又はその無水物。
また、その他のモノマーとしては、以下のものが挙げられる。
グリセリン、ソルビット、ソルビタン、さらには例えばノボラック型フェノール樹脂のオキシアルキレンエーテル等の多価アルコール類;トリメリット酸、ピロメリット酸、ベンゾフェノンテトラカルボン酸やその無水物等の多価カルボン酸類。
それらの中でも、特に、下記一般式(1)で表されるビスフェノール誘導体を2価アルコールモノマー成分とし、2価以上のカルボン酸又はその酸無水物、又はその低級アルキルエステルとからなるカルボン酸成分(例えば、フマル酸、マレイン酸、無水マレイン酸、フタル酸、テレフタル酸、トリメリット酸、ピロメリット酸等)を酸モノマー成分として、これらのポリエステルユニット成分で縮重合した樹脂が良好な帯電特性を有するので好ましい。
【0018】
【化1】

(式中、Rはエチレン基又はプロピレン基を示し、x及びyはそれぞれ1以上の整数であり、かつx+yの平均値は2〜10である。)
【0019】
本発明に用いられる結着樹脂として、低温定着性を達成するために、軟化点(Tm)が好ましくは60℃以上120℃以下、より好ましくは60℃以上100℃以下の樹脂を用いるとよい。
【0020】
本発明において、トナーのワックス偏在状態を制御するための樹脂(以下、ワックス偏在制御樹脂)を、結着樹脂とは別に添加することが好ましい。
ワックス偏在制御樹脂の添加量としては、結着樹脂100質量部に対して、1質量部以上20質量部以下であることが、トナー表面近傍の結着樹脂に対するワックスの存在比率を制御するために好ましい。
上記ワックス偏在制御樹脂としては、結着樹脂に極性が近い部位、ワックスに極性が近い部位の両方を有するものであればどのようなものでもかまわない。具体的には、スチレン系モノマー、並びに、窒素(N)含有ビニルモノマー、カルボキシル基含有モノマー、
水酸基含有モノマー、アクリル酸エステルモノマー及びメタアクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーを用いて合成された共重合体と、ポリオレフィンとがグラフト共重合した樹脂が好適に例示できる。
グラフト共重合させることにより、共重合体成分とポリオレフィン成分が一体となり、ワックス偏在状態をコントロールすることができる。グラフト共重合していない場合、ワックス偏在状態をコントロールしにくくなる。また、グラフト共重合していない樹脂に熱をかけ溶解させると共重合体成分とポリオレフィン成分が2相に分離したり、白濁を生じたりする場合がある。
スチレン系モノマーと窒素含有ビニルモノマー、カルボキシル基含有モノマー、水酸基含有モノマー、アクリル酸エステルモノマー及びメタクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーとを用いて合成された共重合体を合成するために用いることのできるモノマーとしては、次のようなものが挙げられる。
スチレン系モノマーとしては、スチレン、o−メチルスチレン、m−メチルスチレン、p−メチルスチレン、p−メトキシスチレン、p−フェニルスチレン、p−クロルスチレン、3,4−ジクロルスチレン、p−エチルスチレン、2,4−ジメチルスチレン、p−n−ブチルスチレン、p−tert−ブチルスチレン、p−n−ヘキシルスチレン、p−n−オクチルスチレン、p−n−ノニルスチレン、p−n−デシルスチレン、p−n−ドデシルスチレンの如きスチレン及びその誘導体が挙げられる。
窒素含有ビニル系モノマーとしては、メタクリル酸ジメチルアミノエチル、メタクリル酸ジエチルアミノエチルの如きアミノ基含有α−メチレン脂肪族モノカルボン酸エステル類;アクリロニトリル、メタクリロニトリル、アクリルアミドの如きアクリル酸もしくはメタクリル酸誘導体が挙げられる。
カルボキシル基含有モノマーとしては、マレイン酸、シトラコン酸、イタコン酸、アルケニルコハク酸、フマル酸、メサコン酸の如き不飽和二塩基酸;マレイン酸無水物、シトラコン酸無水物、イタコン酸無水物、アルケニルコハク酸無水物の如き不飽和二塩基酸無水物;マレイン酸メチルハーフエステル、マレイン酸エチルハーフエステル、マレイン酸ブチルハーフエステル、シトラコン酸メチルハーフエステル、シトラコン酸エチルハーフエステル、シトラコン酸ブチルハーフエステル、イタコン酸メチルハーフエステル、アルケニルコハク酸メチルハーフエステル、フマル酸メチルハーフエステル、メサコン酸メチルハーフエステルの如き不飽和二塩基酸のハーフエステル;ジメチルマレイン酸、ジメチルフマル酸の如き不飽和二塩基酸エステル;アクリル酸、メタクリル酸、クロトン酸、ケイヒ酸の如きα,β−不飽和酸;クロトン酸無水物、ケイヒ酸無水物の如きα,β−不飽和酸無水物、該α,β−不飽和酸と低級脂肪酸との無水物;アルケニルマロン酸、アルケニルグルタル酸、アルケニルアジピン酸、これらの酸無水物及びこれらのモノエステルが挙げられる。
水酸基含有モノマーとしては、2−ヒドロキシエチルアクリレート、2−ヒドロキシエチルメタクリレート、2−ヒドロキシプロピルメタクリレートなどのアクリル酸又はメタクリル酸エステル類、4−(1−ヒドロキシ−1−メチルブチル)スチレン、4−(1−ヒドロキシ−1−メチルヘキシル)スチレンが挙げられる。
アクリル酸エステルモノマーとしては、アクリル酸メチル、アクリル酸エチル、アクリル酸n−ブチル、アクリル酸イソブチル、アクリル酸プロピル、アクリル酸n−オクチル、アクリル酸ドデシル、アクリル酸2−エチルヘキシル、アクリル酸ステアリル、アクリル酸2−クロルエチル、アクリル酸フェニルの如きアクリル酸エステル類が挙げられる。
メタクリル酸エステルモノマーとしては、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸プロピル、メタクリル酸n−ブチル、メタクリル酸イソブチル、メタクリル酸n−オクチル、メタクリル酸ドデシル、メタクリル酸2−エチルヘキシル、メタクリル酸ステアリル、メタクリル酸フェニル、メタクリル酸ジメチルアミノエチル、メタクリル酸ジエチルアミノエチルの如きα−メチレン脂肪族モノカルボン酸エステル類が挙げられる。
その中でも特に、スチレン−アクリロニトリル−メチルメタクリレートの三元共重合体
が好ましい。
スチレン系モノマー、並びに、窒素含有ビニルモノマー、カルボキシル基含有モノマー、水酸基含有モノマー、アクリル酸エステルモノマー及びメタアクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーを用いて合成された共重合体は、ゲルパーミエーションクロマトグラフィー(GPC)により測定された分子量分布において、重量平均分子量(Mw)が5,000〜100,000であり、数平均分子量(Mn)が1,500〜15,000であり、重量平均分子量(Mw)と数平均分子量(Mn)との比(Mw/Mn)が2〜40であることが好ましい。
また、スチレン系モノマー、並びに、窒素含有ビニルモノマー、カルボキシル基含有モノマー、水酸基含有モノマー、アクリル酸エステルモノマー及びメタアクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーを用いて合成された共重合体は、該トナー中にトナーの質量を基準として0.1〜20質量%含有されていることが好ましい。
スチレン系モノマー、並びに、窒素含有ビニルモノマー、カルボキシル基含有モノマー、水酸基含有モノマー、アクリル酸エステルモノマー及びメタアクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーを用いて合成された共重合体とのグラフト重合に用いられる上記ポリオレフィンは、示差走査熱量測定(DSC)装置によって測定される昇温時の吸熱曲線において、最大吸熱ピークの極大値が70〜130℃にあることが離型性の観点から好ましい。なお、ポリオレフィンとしては、ポリエチレン、ポリプロピレン、エチレン/プロピレン共重合体、エチレン/1−ブテン共重合体、プロピレン/1−ヘキセン共重合体が好適に例示できる。また、本発明においては、ポリマー構造がポリオレフィンの構造を有していれば良く、モノマーが必ずしもオレフィン構造を有している必要はない。
また、スチレン系モノマー、並びに、窒素含有ビニルモノマー、カルボキシル基含有モノマー、水酸基含有モノマー、アクリル酸エステルモノマー及びメタアクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーを用いて合成された共重合体の含有量(W1)、及びポリオレフィンの含有量(W2)の質量比としては、0.01≦W2/W1≦1、を満足することが好ましい。
【0021】
本発明において、必要に応じてトナーは着色剤を含有することが可能であり、以下のものが挙げられる。なお、着色剤には、顔料を単独で使用してもかまわないが、染料と顔料とを併用してその鮮明度を向上させた方がフルカラー画像の画質の点からより好ましい。
黒色着色剤としては、カーボンブラック;磁性体;イエロー着色剤とマゼンタ着色剤及びシアン着色剤とを用いて黒色に調色したものが挙げられる。
マゼンタトナー用着色顔料としては、以下のものが挙げられる。C.I.ピグメントレッド1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、21、22、23、30、31、32、37、38、39、40、41、48:1、48:2、48:3、48:4、48:5、49、50、51、52、53、54、55、57:1、58、60、63、64、68、81:1、81:2、81:3、81:4、81:5、83、87、88、89、90、112、114、122、123、146、147、150、163、184、185、202、206、207、209、238、269;C.I.ピグメントバイオレット19;C.I.バットレッド1、2、10、13、15、23、29、35。
マゼンタトナー用染料としては、以下のものが挙げられる。C.I.ソルベントレッド1、3、8、23、24、25、27、30、49、81、82、83、84、100、109、121;C.I.ディスパースレッド9;C.I.ソルベントバイオレット8、13、14、21、27;C.I.ディスパーバイオレット1の如き油溶染料、C.I.ベーシックレッド1、2、9、12、13、14、15、17、18、22、23、24、27、29、32、34、35、36、37、38、39、40;C.I.ベーシックバイオレット1、3、7、10、14、15、21、25、26、27、28の如き塩基性染料。
シアントナー用着色顔料としては、以下のものが挙げられる。C.I.ピグメントブルー2、3、15:3、15:4、16、17;C.I.バットブルー6;C.I.アシッドブルー45、フタロシアニン骨格にフタルイミドメチル基を1〜5個置換した銅フタロシアニン顔料。シアン用着色染料としては、C.I.ソルベントブルー70がある。
イエロー用着色顔料としては、以下のものが挙げられる。C.I.ピグメントイエロー1、2、3、4、5、6、7、10、11、12、13、14、15、16、17、23、62、65、73、74、83、93、94、95、97、109、110、111、120、127、128、129、147、151、154、155、168、174、175、176、180、181、185;C.I.バットイエロー1、3、20。イエロー用着色染料としては、C.I.ソルベントイエロー162がある。
着色剤の使用量は、結着樹脂100質量部に対して、好ましくは0.1質量部以上30質量部以下であり、より好ましくは0.5質量部以上20質量部以下である。
【0022】
本発明のトナーは、結着樹脂およびワックスを少なくとも含有するトナー粒子に、流動性向上のため、外添剤として無機微粉体が添加されているトナーである。外添剤としては、シリカ、酸化チタン、酸化アルミニウムの如き無機微粉体が好ましい。無機微粉体は、シラン化合物、シリコーンオイル又はそれらの混合物の如き疎水化剤で疎水化されていることが好ましい。
外添剤としては、個数平均粒径が5nm以上300nm以下の外添剤を用いることができるが、耐ストレス性を向上させるためには、80nm以上300nm以下の外添剤を用いることがより好ましい。個数平均粒径が80nm以上300nm以下の外添剤は、耐久時等にトナー表面に埋め込まれたとしても、粒径が大きいために流動性の低下を低減することができる。このため、耐久時の画像濃度の低下を抑制できる。
外添剤は、トナー粒子100質量部に対して0.1質量部以上5.0質量部以下使用されることが好ましい。
トナー粒子と外添剤との混合は、ヘンシェルミキサーの如き公知の混合機を用いることができる。
【0023】
上記トナー粒子及びトナーの製造方法についても、特に限定されず、従来公知の製造方法を用いることができる。
ここでは、粉砕法を用いたトナー製造の手順について説明する。
原料混合工程では、トナー粒子を構成する材料として、結着樹脂及びワックス、並びに必要に応じて、ワックス偏在制御樹脂、着色剤及び荷電制御剤等の他の成分を所定量秤量して配合し、混合する。混合装置の一例としては、ダブルコン・ミキサー、V型ミキサー、ドラム型ミキサー、スーパーミキサー、ヘンシェルミキサー、ナウタミキサ、メカノハイブリッド(日本コークス工業社製)が挙げられる。
次に、混合した材料を溶融混練して、結着樹脂中にワックス等を分散させる。その溶融混練工程では、加圧ニーダー、バンバリィミキサーの如きバッチ式練り機や、連続式の練り機を用いることができる。連続生産できる優位性から、1軸又は2軸押出機が主流となっている。例えば、KTK型2軸押出機(神戸製鋼所社製)、TEM型2軸押出機(東芝機械社製)、PCM混練機(池貝鉄工製)、2軸押出機(ケイ・シー・ケイ社製)、コ・ニーダー(ブス社製)、ニーデックス(日本コークス工業社製)が挙げられる。
更に、溶融混練することによって得られる樹脂組成物は、2本ロール等で圧延され、冷却工程で水などによって冷却してもよい。
ついで、樹脂組成物の冷却物は、粉砕工程で所望の粒径にまで粉砕される。粉砕工程では、クラッシャー、ハンマーミル、フェザーミルの如き粉砕機で粗粉砕した後、更に、クリプトロンシステム(川崎重工業社製)、スーパーローター(日清エンジニアリング社製)、ターボ・ミル(ターボ工業製)やエアージェット方式による微粉砕機で微粉砕する。
その後、必要に応じて慣性分級方式のエルボージェット(日鉄鉱業社製)、遠心力分級方式のターボプレックス(ホソカワミクロン社製)、TSPセパレータ(ホソカワミクロ
ン社製)、ファカルティ(ホソカワミクロン社製)の如き分級機や篩分機を用いて分級し、トナー粒子を得る。
本発明においては、粉砕後、または、分級後に、熱風を用いてトナーの表面処理を行うことが、トナー表面近傍のワックス比率をコントロールし、耐久性能と画像の面内のグロス均一性を両立させるために、好ましい。
上記熱風を用いた表面処理としては、熱風でトナーの表面を溶融状態にすることができ、かつ、熱風で処理されたトナーを冷風で冷却できる方式を採用できる手段であればどのようなものでもかまわない。例えば、ハイブリタイゼーションシステム(奈良機械製作所製)、メカノフージョンシステム(ホソカワミクロン社製)、ファカルティ(ホソカワミクロン社製)、メテオレインボー MR Type(日本ニューマチック社製)などを用いることが可能である。
また、本発明においては、上記微粉砕物を得た後、熱風により表面処理を行い、続いて分級をすることにより得られたトナーであることが好ましい。若しくは、予め分級したものを、熱風により表面処理を行っても良い。
ここで、上記熱風を用いた表面処理の方法の概略を、図1を用いて説明するが、これに限定されるものではない。図1は本発明で用いた表面処理装置の一例を示した断面図である。具体的には、上記微粉砕物(ここでは、トナー粒子ともいう)を得た後、当該表面処理装置に供給する。そして、トナー粒子供給口(100)から供給されたトナー粒子(114)は、高圧エア供給ノズル(115)から噴射されるインジェクションエアにより加速され、その下方にある気流噴射部材(102)へ向かう。気流噴射部材(102)からは拡散エアが噴射され、この拡散エアによりトナー粒子が外側方向へ拡散する。この時、インジェクションエアの流量と拡散エアの流量とを調節することにより、トナーの拡散状態をコントロールすることができる。また、トナー粒子の融着防止を目的として、トナー粒子供給口(100)の外周、表面処理装置外周及び移送配管(116)の外周には冷却ジャケット(106)が設けられている。尚、該冷却ジャケットには冷却水(好ましくはエチレングリコール等の不凍液)を通水することが好ましい。一方、拡散エアにより拡散したトナー粒子は、熱風供給口(101)から供給された熱風により、トナー粒子の表面が処理される。この時、熱風の吐出温度は100℃以上、300℃以下であることが好ましく、150℃以上、250℃以下であることがより好ましい。熱風の温度が100℃未満の場合にはトナー粒子の表面を溶融状態にすることができない場合がある。また、300℃を超える場合には溶融状態が進みすぎる事で、ワックスを過度にトナー表面に偏析させる場合や、トナー粒子同士の合一に起因する、トナー粒子の粗大化や融着が生じる場合がある。
熱風により表面が処理されたトナー粒子は、装置上部外周に設けた冷風供給口(103)から供給される冷風により冷却される。この時、装置内の温度分布の制御、トナー粒子の表面状態をコントロールする目的で、装置の本体側面に設けた第二の冷風供給口(104)から冷風を導入することが好ましい。第二の冷風供給口(104)の出口はスリット形状、ルーバー形状、多孔板形状、メッシュ形状等を用いる事ができ、導入方向は中心方向へ水平、装置壁面に沿う方向が、目的に応じて選択可能である。
この時、上記冷風温度は−50℃以上、10℃以下であることが好ましく、−40℃以上、8℃以下であることがより好ましい。また、上記冷風は除湿された冷風であることが好ましい。具体的には、冷風の絶対水分量が5g/m3以下であることが好ましい。更に好ましくは、3g/m3以下である。
これらの冷風温度が−50℃未満の場合には装置内の温度が下がりすぎてしまい、本来の目的である熱による処理が十分に為されず、トナーの表面を溶融状態にすることができない場合がある。また、10℃を超える場合には、装置内における熱風ゾーンの制御が不十分になり、表面処理時にワックスを過度にトナー表面に偏析させることがある。
その後、冷却されたトナー粒子は、ブロワーで吸引され、移送配管(116)を通じて、サイクロン等で回収される。
【0024】
本発明のトナーは、トナー単独で構成される一成分系の現像剤として使用することも可能であるが、トナーと磁性キャリアを含む二成分系現像剤としても用いることができる。すなわち、本発明の二成分系現像剤は、本発明のトナーと磁性キャリアを含む二成分系現像剤である。ここで、磁性キャリアは、特に限定されず、公知の磁性キャリアが使用できる。鉄、銅、亜鉛、ニッケル、コバルト、マンガン、及び、クロム元素から選ばれる元素単独または複合のフェライトで構成されるキャリアが例示できる。また、磁性体が樹脂中に分散されている磁性体含有樹脂キャリアコアの表面に樹脂成分を含有する磁性体含有樹脂キャリアや、多孔質磁性体(多孔質フェライトを含む)に樹脂が含浸された樹脂含浸キャリアなども、上記磁性キャリアとして挙げられる。
しかしながら、二成分系現像剤が、交流インピーダンス測定により得られるインピーダンスZの周波数依存特性Z(ω)を、下記(2)式で表されるフィッティング関数により、フィッティングしたときのパラメータαが、1000V/cmの電界下において、0.70以上0.95以下であることを特徴とする磁性キャリア、及び、本発明のトナーを含有する二成分系現像剤である場合、トナーが本発明のトナーの様に、トナー表面近傍の結着樹脂に対するワックスの存在比率が高いトナーであっても、現像性の更なる向上が図れるため好ましい。
【0025】
【数1】
【0026】
そのメカニズムについて、発明者らは以下のように考えている。本発明のトナーは、トナー表面近傍の結着樹脂に対するワックスの存在比率が高いトナーである。このようなトナーを用いた場合、トナー表面近傍に存在するワックスの影響でトナーと磁性キャリアの付着力が高まり、トナーが磁性キャリアから像担時体へ現像されにくくなる場合がある。この現象を抑制するためには、トナーが現像された後の磁性キャリア表面に残ったカウンター電荷を、すばやく逃がす必要がある。これにより、トナーを引き戻す力が弱くなり現像性が向上する。例えば、磁性キャリアの抵抗を低くすることで、磁性キャリア同士を通じて、現像剤担持体へカウンター電荷を逃がす手法が従来から知られている。しかし、単に抵抗の低い磁性キャリアを用いた場合には、ハーフトーン部などトナーが薄層で現像されるような場合、抵抗の低い磁性キャリアにより潜像電位が乱されガサツキを生じてしまう場合がある。そこで、発明者らは鋭意検討の結果、現像性向上のためには、抵抗をさげカウンター電荷を逃がすのではなく、磁性キャリアの電気的特性を制御することで、現像性をさらに向上できることを見出した。
具体的には、磁性キャリアの交流インピーダンス測定により得られるインピーダンスZの周波数依存特性Z(ω)を、上記式(2)で書かれるフィッティング関数によりフィッティングしたときのパラメータαを、1000V/cmの電界下において0.70以上0.95以下にすることである。
上記パラメータαは、磁性キャリアの交流インピーダンス測定により、複素インピーダンスの周波数特性を測定し、下記式(3)式で表されるCole−Coleの式によるフィッティングから算出でき、磁性キャリアの持つ導電の時定数分布の度合いを表すことができる。該パラメータαは、0≦α≦1の範囲をとり、導電の時定数分布の広がりが大きい程、小さな値となる。
【0027】
【数2】
【0028】
本発明において、パラメータα、1000V/cmの電界下において、0.70以上0.95以下にすることで、一粒子の磁性キャリア内部に極端に電荷移動の時定数の小さい部分から極端に電荷移動の時定数の大きい部分が存在することになる。
このような磁性キャリアであれば、外部電界が印加された時に、磁性キャリア内部の電気伝導が局所的に抑制され大きな分極が形成される。分極した磁性キャリアは、磁性キャリアの外部電界を歪めるため、磁性キャリアに付着したトナーが受ける電界は強められることになる。その結果、磁性キャリアの抵抗を低めなくとも、トナーが磁性キャリアから飛翔しやすくなる。
このようにパラメータαを0.70以上0.95以下にコントロールすることで、磁性キャリアの抵抗を下げることなく、トナーにかかる電界を強め、現像性を高めることが可能になる。
パラメータαを上記範囲にコントロールするためには、磁性キャリアの内部構造を均一なものではなく不均一にすることが必要である。磁性キャリアの内部構造を不均一にすることで、一粒子の磁性キャリア内部に極端に電荷移動の時定数の小さい部分から極端に電荷移動の時定数の大きい部分が存在させることができる。具体的には、多孔質磁性コア粒子に樹脂を含有させた磁性キャリアを用いることが好ましい。当該多孔質磁性コア粒子の材質としては、フェライトが好ましい。そして、多孔質磁性コア粒子を構成している結晶粒子同士のつながりの状態に、一粒子のキャリア内部で十分に「ばらつき」を持たせることが特に好ましい。
そのためには、後述する多孔質磁性コア粒子の製造過程で、仮焼フェライト微粉砕品の体積基準の50%粒径(D50)を、0.5μm以上5.0μm以下、また、体積基準の90%粒径(D90)を、3.0μm以上10.0μm以下とすることが好ましい。
仮焼フェライト微粉砕品を上記の粒径にするために、例えば、ボールミルやビーズミルでは用いるボールやビーズの素材、粒径、運転時間を制御することが好ましい。具体的には、仮焼フェライト微粉砕品の粒径を小さくするためには、比重の重いボールを用いたり、粉砕時間を長くすればよい。また、仮焼フェライト微粉砕品の粒度分布を広くするためには、比重の重いボールやビーズを用い、粉砕時間を短くすることで得ることができる。また、粒径の異なる複数の仮焼フェライトを混合することでも分布の広い仮焼フェライトを得ることができる。このようにして、広い粒度分布の仮焼フェライト微粉砕品を用いることで、パラメータαが0.70以上0.95以下である磁性キャリアを得ることができる。
【0029】
以下、多孔質磁性コア粒子としてフェライトを用いた磁性キャリアの製造方法を例示する。ここで、フェライトとは次式で表される焼結体である。
(M12O)x(M2O)y(Fe2O3)z
(式中、M1は1価、M2は2価の金属であり、x+y+z=1.0とした時、x及びyは、それぞれ0≦(x,y)≦0.8であり、zは、0.2<z<1.0である。)
該式中において、M1及びM2としては、Li、Fe、Mn、Mg、Sr、Cu、Zn、Ni、Co、Caからなる群から選ばれる1種類以上の金属原子を用いることが好ましい。
具体的には、Li系フェライト(例えば、(Li2O)a(Fe2O3)b(0.0<
a<0.4,0.6≦b<1.0、a+b=1)、(Li2O)a(SrO)b(Fe2O3)c(0.0<a<0.4、0.0<b<0.2、0.4≦c<1.0、a+b+c=1));Mn系フェライト(例えば、(MnO)a(Fe2O3)b(0.0<a<0.5、0.5≦b<1.0、a+b=1));Mn−Mg系フェライト(例えば、(MnO)a(MgO)b(Fe2O3)c(0.0<a<0.5、0.0<b<0.5、0.5≦c<1.0、a+b+c=1));Mn−Mg−Sr系フェライト(例えば、(MnO)a(MgO)b(SrO)c(Fe2O3)d(0.0<a<0.5、0.0<b<0.5、0.0<c<0.5、0.5≦d<1.0、a+b+c+d=1);Cu−Zn系フェライト(例えば、(CuO)a(ZnO)b(Fe2O3)c(0.0<a<0.5、0.0<b<0.5、0.5≦c<1.0、a+b+c=1)が挙げられる。上記フェライトは微量の他の金属を含有していてもよい。
結晶の成長速度を容易にコントロールでき、多孔質磁性コア粒子の細孔径分布を好適にコントロールできる観点から、Mn元素を含有する、Mn系フェライト、Mn−Mg系フェライト、Mn−Mg−Sr系フェライトが好ましい。
【0030】
上記多孔質磁性コア粒子は、以下のような工程で製造することができる。
<工程1(秤量・混合工程)>
フェライトの原料を、秤量し、混合する。フェライトの原料としては、例えば以下のものが挙げられる。Li、Fe、Zn、Ni、Mn、Mg、Co、Cu、Ba、Sr、Y、Ca、Si、V、Bi、In、Ta、Zr、B、Mo、Na、Sn、Ti、Cr、Al、希土類金属の金属粒子、酸化物、水酸化物、シュウ酸塩、炭酸塩。
粉砕・混合する装置としては、以下のものが挙げられる。ボールミル、遊星ミル、ジオットミル、振動ミル。フェライトの原料に水を添加しスラリーとした状態で混合する、湿式のボールミルが混合性と多孔質構造の形成のためには好ましい。具体的には、ボールミル中に、秤量したフェライト原料及びボール、湿式の場合は更に水を入れ、0.1時間以上20.0時間以下、粉砕・混合する。
<工程2(仮焼成工程)>
粉砕・混合したフェライト原料を、スプレードライヤーを用いて、造粒・乾燥した後、大気中で焼成温度700℃以上1000℃以下にして、0.5時間以上5.0時間以下仮焼成し、原料をフェライトにする。温度1000℃を超えると焼結が進み、多孔質にするための粒径まで粉砕することができにくくなる場合がある。焼成する装置としては、バーナー式焼却炉、ロータリー式焼却炉、電気炉が挙げられる。
<工程3(粉砕工程)>
工程2で作製した仮焼フェライトを粉砕機で微粉砕する。粉砕機としては、クラッシャーやハンマーミル、ボールミル、ビーズミル、遊星ミル、ジオットミルが挙げられる。
上述のように、仮焼フェライト微粉砕品の体積基準の50%粒径(D50)は、0.5μm以上5.0μm以下、体積基準の90%粒径(D90)は3.0μm以上10.0μm以下とすることが好ましい。こうすることで、多孔質磁性コアの結晶粒子(グレイン)同士の繋がりの状態に、一粒子の磁性キャリア内部で十分に「ばらつき」を持たせることができ、パラメータαを1000V/cmの電界下において、0.70以上0.95以下にコントロールすることができる。
仮焼フェライト微粉砕品を上記の粒径にするために、例えば、ボールミルやビーズミルでは用いるボールやビーズの素材、粒径、運転時間を制御することが好ましい。具体的には、仮焼フェライト微粉砕品の粒径を小さくするためには、比重の重いボールを用いたり、粉砕時間を長くすればよい。また、仮焼フェライト微粉砕品の粒度分布を広くするためには、比重の重いボールやビーズを用い、粉砕時間を短くすることで得ることができる。また、粒径の異なる複数の仮焼フェライト微粉砕品を混合することでも分布の広い仮焼フェライト微粉砕品を得ることができる。
ボールやビーズの素材としては、所望の粒径・分布が得られば、特に限定されない。例えば、以下のものが挙げられる。ソーダガラス(比重2.5g/cm3)、ソーダレスガ
ラス(比重2.6g/cm3)、高比重ガラス(比重2.7g/cm3)等のガラスや、石英(比重2.2g/cm3)、チタニア(比重3.9g/cm3)、窒化ケイ素(比重3.2g/cm3)、アルミナ(比重3.6g/cm3)、ジルコニア(比重6.0g/cm3)、スチール(比重7.9g/cm3)、ステンレス(比重8.0g/cm3)。中でも、アルミナ、ジルコニア、ステンレスは、耐磨耗性に優れているために好ましい。
ボールやビーズの粒径は、所望の粒径・分布が得られれば、特に限定されない。例えば、ボールとしては、直径(φ)が5mm以上60mm以下のものが好適に用いられる。また、ビーズとしては直径(φ)が0.03mm以上5mm未満のものが好適に用いられる。また、ボールミルやビーズミルは、粉砕効率が高く仮焼フェライト微粉砕品の粒度分布コントロールが容易になるため、乾式粉砕よりも、水を添加しスラリーとした状態で粉砕を行う、湿式粉砕の方がより好ましい。
<工程4(造粒工程)>
得られた仮焼フェライト微粉砕品に対し、水、バインダーと、必要に応じて、孔調整剤を加える。工程3において、湿式で粉砕した場合は、フェライトスラリー中に含まれている水も考慮し、バインダーと必要に応じて孔調整剤を加えることが好ましい。多孔質の程度をコントロールするため、スラリーの固形分濃度を50質量%以上80質量%以下にして、造粒することが好ましい。得られたフェライトスラリーを、噴霧乾燥機を用い、温度100℃以上200℃以下の加温雰囲気下で、乾燥・造粒する。
噴霧乾燥機としては、スプレードライヤーがフェライトスラリーの粒径を所望に調整できるため、好適に使用できる。多孔質磁性コア粒子の粒径は、スプレードライヤーに用いられるディスクの回転数、噴霧量を適宜選択してコントロールできる。
<工程5(本焼成工程)>
次に、得られた造粒品を温度800℃以上1300℃以下で1時間以上24時間以下焼成する。焼成温度が1000℃以上1200℃以下であれば、上記磁性キャリアのパラメータαをコントロールしやすいことからより好ましい。
<工程6(選別工程)>
得られた焼成物を解砕した後に、必要に応じて、分級や篩で篩分して粗大粒子や微粒子を除去してもよい。当該選別工程を経て得られた多孔質磁性コア粒子の体積基準の50%粒径(D50)は、18.0μm以上68.0μm以下であることが、画像へのキャリア付着とガサツキの抑制のためより望ましい。
【0031】
次に、得られた多孔質磁性コア粒子に樹脂を充填し、磁性キャリアを得る。ここで、樹脂を充填した後にさらに樹脂で磁性キャリアの表面を被覆すれば、磁性キャリアとしての物理的強度を高めることが可能となるため好ましい。得られた磁性キャリアの体積基準の50%粒径(D50)は、20.0μm以上70.0μm以下であることが、ドラム上へのキャリア付着抑制と、画質の観点より好ましい。
上記多孔質磁性コア粒子に樹脂を充填する方法は、多孔質磁性コア粒子の奥の孔まで樹脂を充填する方法と、多孔質磁性コア粒子の表面の孔のみに樹脂を充填する方法の2つがある。具体的な充填方法は、特に限定されないが、樹脂と溶剤を混合した樹脂溶液を多孔質磁性コア粒子の孔へ充填させる方法が好ましい。
上記樹脂溶液における樹脂固形分の量は、好ましくは5質量%以上30質量%以下であり、より好ましくは6質量%以上20質量%以下である。30質量%より樹脂量の多い樹脂溶液を用いると粘度が高いため多孔質磁性コア粒子の孔に樹脂溶液を均一に充填しにくい。また、5質量%未満であると樹脂量が少なく、多孔質磁性コア粒子へ均一な充填ができず、摩擦帯電量の部分的なバラツキの要因となる場合がある。
上記多孔質磁性コア粒子の孔に充填する樹脂としては特に限定されず、熱可塑性樹脂、熱硬化性樹脂のどちらを用いてもかまわない。しかしながら、多孔質磁性コア粒子に対する親和性が高いものであることが好ましい。親和性が高い樹脂を用いた場合には、多孔質磁性コア粒子の孔への樹脂の充填時に、同時に多孔質磁性コア粒子表面も樹脂で覆うことが容易になる。
具体的には、シリコーン樹脂または変性シリコーン樹脂が、多孔質磁性コア粒子に対する親和性が高いため好ましい。
市販品として、以下のものが挙げられる。シリコーン樹脂では、信越化学社製のKR271、KR255、KR152、東レ・ダウコーニング社製のSR2400、SR2405、SR2410、SR2411。変性シリコーン樹脂では、信越化学社製のKR206(アルキッド変性)、KR5208(アクリル変性)、ES1001N(エポキシ変性)、KR305(ウレタン変性)、東レ・ダウコーニング社製のSR2115(エポキシ変性)、SR2110(アルキッド変性)。
多孔質磁性コア粒子内の孔に樹脂を充填させる方法としては、上記樹脂を溶剤に希釈し、これを多孔質磁性コア粒子内の孔に添加する方法が採用できる。ここで用いられる溶剤は、樹脂を溶解できるものであれば特に限定されない。有機溶剤に可溶な樹脂である場合は、有機溶剤として、トルエン、キシレン、セルソルブブチルアセテート、メチルエチルケトン、メチルイソブチルケトン、メタノールが挙げられる。また、水溶性の樹脂またはエマルジョンタイプの樹脂である場合には、溶剤として水を用いればよい。多孔質磁性コア粒子の孔に、樹脂を充填する方法としては、浸漬法、スプレー法、ハケ塗り法、及び流動床の如き塗布方法により多孔質磁性コア粒子を樹脂溶液に含浸させ、その後、溶剤を揮発させる方法が挙げられる。
【0032】
本発明の二成分系現像剤において、トナーと磁性キャリアの混合比率は、磁性キャリア100質量部に対してトナーを2質量部以上35質量部以下とすることが好ましく、4質量部以上25質量部以下とすることがより好ましい。トナーを上記範囲とすることで、高い画像濃度を達成し、かつ、トナーの飛散を低減することができる。
【0033】
以下、トナー及び磁性キャリアの各種物性の測定法について説明する。
<P1及びP2の算出方法>
FT−IRスペクトルは、ユニバーサルATR測定アクセサリー(Universal
ATR Sampling Accessory)を装着したフーリエ変換赤外分光分析装置(Spectrum One;PerkinElmer社製)を用い、ATR法で測定する。具体的な測定手順と、P1、P2及びP1をP2で除した[P1/P2]の算出方法は以下の通りである。
赤外光の入射角は45°に設定する。ATR結晶としては、GeのATR結晶(屈折率=4.0)、KRS5のATR結晶(屈折率=2.4)を用いる。その他の条件は以下の通りである。
Range
Start :4000cm−1
End:600cm−1(GeのATR結晶)
400cm−1(KRS5のATR結晶)
Duration
Scan number:16
Resolution:4.00cm−1
Advanced :CO2/H2O補正あり
[P1の算出方法]
(1)GeのATR結晶(屈折率=4.0)を装置に装着する。
(2)Scan typeをBackground、UnitsをEGYに設定し、バックグラウンドを測定する。
(3)Scan typeをSample、UnitsをAに設定する。
(4)トナーをATR結晶の上に、0.01g精秤する。
(5)圧力アームでサンプルを加圧する。(Force Gaugeは90)
(6)サンプルを測定する。
(7)得られたFT−IRスペクトルを、Automatic Correctionで
ベースライン補正をする。
(8)2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pa1)
(9)3050cm−1と2600cm−1の吸収ピーク強度の平均値を算出する。(Pa2)
(10)Pa1−Pa2=Paとする。当該Paを2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度と規定する。
(11)1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pb1)
(12)1763cm−1と1630cm−1の吸収ピーク強度の平均値を算出する(Pb2)
(13)Pb1−Pb2=Pbとする。当該Pbを1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度と規定する。
(14)Pa/Pb=P1とする。
図2にATR結晶としてGeを用い測定したFT−IRスペクトルの一例を示す。
[P2の算出方法]
(1)KRS5のATR結晶(屈折率=2.4)を装置に装着する。
(2)トナーをATR結晶の上に、0.01g精秤する。
(3)圧力アームでサンプルを加圧する。(Force Gaugeは90)
(4)サンプルを測定する。
(5)得られたFT−IRスペクトルを、Automatic Correctionでベースライン補正をする。
(6)2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pc1)
(7)3050cm−1と2600cm−1の吸収ピーク強度の平均値を算出する。(Pc2)
(8)Pc1−Pc2=Pcとする。当該Pcを2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度と規定する。
(9)1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pd1)
(10)1763cm−1と1630cm−1の吸収ピーク強度の平均値を算出する(Pd2)
(11)Pd1−Pd2=Pdとする。当該Pdを1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度と規定する。
(12)Pc/Pd=P2とする。
[P1/P2の算出方法]
上記のようにして求めたP1とP2を用い、P1/P2を算出する。
【0034】
<樹脂の軟化点の測定方法>
樹脂の軟化点の測定は、定荷重押し出し方式の細管式レオメータ「流動特性評価装置 フローテスターCFT−500D」(島津製作所社製)を用い、装置付属のマニュアルに従って行う。本装置では、測定試料の上部からピストンによって一定荷重を加えつつ、シリンダに充填した測定試料を昇温させて溶融し、シリンダ底部のダイから溶融された測定試料を押し出し、この際のピストン降下量と温度との関係を示す流動曲線を得ることができる。
本発明においては、「流動特性評価装置 フローテスターCFT−500D」に付属のマニュアルに記載の「1/2法における溶融温度」を軟化点とする。尚、1/2法における溶融温度とは、次のようにして算出されたものである。まず、流出が終了した時点におけるピストンの降下量Smaxと、流出が開始した時点におけるピストンの降下量Sminとの差の1/2を求める(これをXとする。X=(Smax−Smin)/2)。そし
て、流動曲線においてピストンの降下量がXとSminの和と、交わるときの流動曲線の温度が、1/2法における溶融温度である。
測定試料には、1.0gの樹脂を、25℃の環境下で、錠剤成型圧縮機(NT−100H、エヌピーエーシステム社製)を用いて10MPaで、60秒間圧縮成型し、直径8mmの円柱状としたものを用いる。一方、CFT−500Dの測定条件は、以下の通りである。
試験モード:昇温法
開始温度:40℃
到達温度:200℃
測定間隔:1.0℃
昇温速度:4.0℃/min
ピストン断面積:1.000cm2
試験荷重(ピストン荷重):10.0kgf(0.9807MPa)
予熱時間:300秒
ダイの穴の直径:1.0mm
ダイの長さ:1.0mm
【0035】
<トナーの平均円形度の算出方法>
トナーの平均円形度は、フロー式粒子像分析装置「FPIA−3000」(シスメックス社製)によって、校正作業時の測定及び解析条件で測定する。
フロー式粒子像分析装置「FPIA−3000」(シスメックス社製)の測定原理は、流れている粒子を静止画像として撮像し、画像解析を行うというものである。試料チャンバーへ加えられた試料は、試料吸引シリンジによって、フラットシースフローセルに送り込まれる。フラットシースフローに送り込まれた試料は、シース液に挟まれて扁平な流れを形成する。フラットシースフローセル内を通過する試料に対しては、1/60秒間隔でストロボ光が照射されており、流れている粒子を静止画像として撮影することが可能である。また、扁平な流れであるため、焦点の合った状態で撮像される。粒子像はCCDカメラで撮像され、撮像された画像は512×512画素の画像処理解像度(一画素あたり0.37×0.37μm)で画像処理され、各粒子像の輪郭抽出を行い、粒子像の投影面積Sや周囲長L等が計測される。
次に、上記面積Sと周囲長Lを用いて円相当径と円形度を求める。円相当径とは、粒子像の投影面積と同じ面積を持つ円の直径のことであり、円形度Cは、円相当径から求めた円の周囲長を粒子投影像の周囲長で割った値として定義され、次式で算出される。
円形度C=2×(π×S)1/2/L
粒子像が円形の時に円形度は1.000になり、粒子像外周の凹凸の程度が大きくなればなるほど円形度は小さい値になる。各粒子の円形度を算出後、円形度0.200乃至1.000の範囲を800分割し、得られた円形度の相加平均値を算出し、その値を平均円形度とする。
具体的な測定方法は、以下の通りである。まず、ガラス製の容器中に予め不純固形物などを除去したイオン交換水20mlを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を0.2ml加える。更に測定試料を0.02g加え、超音波分散器を用いて2分間分散処理を行い、測定用の分散液とする。その際、分散液の温度が10℃以上40℃以下となる様に適宜冷却する。超音波分散器としては、発振周波数50kHz、電気的出力150Wの卓上型の超音波洗浄器分散器(例えば「VS−150」(ヴェルヴォクリーア社製))を用い、水槽内には所定量のイオン交換水を入れ、この水槽中に前記コンタミノンNを2ml添加する。
測定には、標準対物レンズ(10倍)を搭載した前記フロー式粒子像分析装置を用い、シース液にはパーティクルシース「PSE−900A」(シスメックス社製)を使用する
。前記手順に従い調製した分散液を前記フロー式粒子像分析装置に導入し、HPF測定モードで、トータルカウントモードにて3000個のトナー粒子を計測する。そして、粒子解析時の2値化閾値を85%とし、解析粒子径を円相当径1.985μm以上、39.69μm未満に限定し、トナー粒子の平均円形度を求める。
測定にあたっては、測定開始前に標準ラテックス粒子(例えば、Duke Scientific社製の「RESEARCH AND TEST PARTICLES Latex Microsphere Suspensions 5200A」をイオン交換水で希釈)を用いて自動焦点調整を行う。その後、測定開始から2時間毎に焦点調整を実施することが好ましい。
なお、本願実施例では、シスメックス社による校正作業が行われた、シスメックス社が発行する校正証明書の発行を受けたフロー式粒子像分析装置を使用する。解析粒子径を円相当径1.985μm以上、39.69μm未満に限定した以外は、校正証明を受けた時の測定及び解析条件で測定を行う。
【0036】
<トナーの重量平均粒径(D4)の測定方法>
トナーの重量平均粒径(D4)は、以下のようにして算出する。測定装置としては、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer 3」(登録商標、ベックマン・コールター社製)を用いる。測定条件の設定及び測定データの解析は、付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いる。尚、測定は実効測定チャンネル数2万5千チャンネルで行う。
測定に使用する電解水溶液は、特級塩化ナトリウムをイオン交換水に溶解して濃度が1質量%となるようにしたもの、例えば、「ISOTON II」(ベックマン・コールター社製)が使用できる。
尚、測定、解析を行う前に、以下のように専用ソフトの設定を行う。
専用ソフトの「標準測定方法(SOM)を変更」画面において、コントロールモードの総カウント数を50000粒子に設定し、測定回数を1回、Kd値は「標準粒子10.0μm」(ベックマン・コールター社製)を用いて得られた値を設定する。「閾値/ノイズレベルの測定ボタン」を押すことで、閾値とノイズレベルを自動設定する。また、カレントを1600μAに、ゲインを2に、電解液をISOTON IIに設定し、「測定後のアパーチャーチューブのフラッシュ」にチェックを入れる。
専用ソフトの「パルスから粒径への変換設定」画面において、ビン間隔を対数粒径に、粒径ビンを256粒径ビンに、粒径範囲を2μmから60μmまでに設定する。
具体的な測定法は以下の通りである。
(1)Multisizer 3専用のガラス製250ml丸底ビーカーに前記電解水溶液200mlを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行う。そして、専用ソフトの「アパーチャーのフラッシュ」機能により、アパーチャーチューブ内の汚れと気泡を除去しておく。
(2)ガラス製の100ml平底ビーカーに前記電解水溶液30mlを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を0.3ml加える。
(3)発振周波数50kHzの発振器2個を、位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispersion System Tetora150」(日科機バイオス社製)を準備する。超音波分散器の水槽内に3.3lのイオン交換水を入れ、この水槽中にコンタミノンNを2ml添加する。(4)前記(2)のビーカーを前記超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させる。そして、ビーカー内の電解水溶液液面の共振状態が最大となるようにビーカーの高さ位置を調整する。
(5)前記(4)のビーカー内の電解水溶液に超音波を照射した状態で、トナー10mgを少量ずつ前記電解水溶液に添加し、分散させる。そして、さらに60秒間超音波分散処理を継続する。尚、超音波分散にあたっては、水槽の水温が10℃以上40℃以下となる様に適宜調節する。
(6)サンプルスタンド内に設置した前記(1)の丸底ビーカーに、ピペットを用いてトナーを分散した前記(5)の電解質水溶液を滴下し、測定濃度が5%となるように調整する。そして、測定粒子数が50000個になるまで測定を行う。
(7)測定データを装置付属の前記専用ソフトにて解析を行い、重量平均粒径(D4)を算出する。尚、専用ソフトでグラフ/体積%と設定したときの、「分析/体積統計値(算術平均)」画面の「平均径」が重量平均粒径(D4)である。
【0037】
<樹脂のピーク分子量(Mp)、数平均分子量(Mn)、重量平均分子量(Mw)の測定方法>
ピーク分子量(Mp)、数平均分子量(Mn)、重量平均分子量(Mw)は、ゲルパーミエーションクロマトグラフィー(GPC)により、以下のようにして測定する。
まず、室温で24時間かけて、試料(樹脂)をテトラヒドロフラン(THF)に溶解する。そして、得られた溶液を、ポア径が0.2μmの耐溶剤性メンブランフィルター「マエショリディスク」(東ソー社製)で濾過してサンプル溶液を得る。尚、サンプル溶液は、THFに可溶な成分の濃度が0.8質量%となるように調整する。このサンプル溶液を用いて、以下の条件で測定する。
装置 :HLC8120 GPC(検出器:RI)(東ソー社製)
カラム :Shodex KF−801、802、803、804、805、
806、807の7連(昭和電工社製)
溶離液 :テトラヒドロフラン(THF)
流速 :1.0ml/min
オーブン温度 :40.0℃
試料注入量 :0.10ml
試料の分子量の算出にあたっては、標準ポリスチレン樹脂(例えば、商品名「TSKスタンダード ポリスチレン F−850、F−450、F−288、F−128、F−80、F−40、F−20、F−10、F−4、F−2、F−1、A−5000、A−2500、A−1000、A−500」、東ソ−社製)を用いて作成した分子量校正曲線を使用する。
【0038】
<ワックスの最大吸熱ピークのピーク温度、樹脂のガラス転移温度Tg>
ワックスの最大吸熱ピークのピーク温度は、示差走査熱量分析装置「Q1000」(TA Instruments社製)を用いてASTM D3418−82に準じて測定する。装置検出部の温度補正はインジウムと亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いる。具体的には、ワックスを10mg精秤し、これをアルミニウム製のパンの中に入れ、リファレンスとして空のアルミニウム製のパンを用い、測定温度範囲30以上200℃以下の間で、昇温速度10℃/minで測定を行う。尚、測定においては、一度200℃まで昇温させ、続いて30℃まで降温し、その後に再度昇温を行う。この2度目の昇温過程での温度30以上200℃以下の範囲におけるDSC曲線の最大の吸熱ピークを、ワックスの最大吸熱ピークとする。
また、樹脂のガラス転移温度(Tg)は、ワックスの最大吸熱ピークのピーク温度測定と同様に、樹脂を10mg精秤し測定する。温度40℃以上100℃以下の範囲において比熱変化が得られる。このときの比熱変化前と比熱変化後のベースラインの中間点の線と示差熱曲線との交点を、樹脂のガラス転移温度Tgとする。
【0039】
<外添剤の個数平均粒径の測定方法>
外添剤の個数平均粒径は、外添剤を透過電子顕微鏡で観察し、100個の粒子の長径を
測定して個数平均粒子径を求める。トナー粒子上の粒子径を測定する場合は、走査電子顕微鏡で観察し、100個の粒子の長径を測定して個数平均粒径を求める。なお、測定時の倍率は40000倍とし、0.5nm以上の粒子を対象とする。
【0040】
<磁性キャリアの体積基準の50%粒径(D50)及び90%粒径(D90)>
粒度分布測定は、レーザー回折・散乱方式の粒度分布測定装置「マイクロトラックMT3300EX」(日機装社製)にて測定を行う。
磁性キャリアの体積基準の50%粒径(D50)及び90%粒径(D90)の測定には、乾式測定用の試料供給機「ワンショットドライ型サンプルコンディショナーTurbotrac」(日機装社製)を装着して行う。Turbotracの供給条件として、真空源として集塵機を用い、風量33リットル/sec、圧力17kPaとする。制御は、ソフトウエア上で自動的に行い、体積基準の累積値である50%粒径(D50)及び90%粒径(D90)を求める。制御及び解析は付属ソフト(バージョン10.3.3−202D)を用いて行う。
測定条件は下記の通りである。
SetZero時間 :10秒
測定時間 :10秒
測定回数 :1回
粒子屈折率 :1.81
粒子形状 :非球形
測定上限 :1408μm
測定下限 :0.243μm
測定環境 :23℃/50%RH
【0041】
<磁性キャリアのパラメータαの算出方法>
パラメータαの算出法について図面に従って詳細に説明する。磁性キャリアのパラメータαの値は、以下の手順で算出することができる。
(サンプルの秤量)
磁性キャリアを、電極面積が4.9cm2である円筒型電極(直径2.5cm)を有するサンプルホルダに、電極間に100Nの押し圧をかけたときに封入したサンプルの厚みが0.95mm以上1.05mm以下の範囲となるように封入し、封入された磁性キャリアを秤量した。
(測定回路の説明)
上記のサンプルホルダの電極間に図3に示すように配線を行い、サンプルホルダの電極間に100Nの押し圧をかけた状態において、サンプルホルダ内部に封入した磁性キャリアの交流インピーダンス測定を行った。尚、本測定では、電界下におけるαを求めるために、直流電圧を印加した状態における交流インピーダンス測定を行う。このため、図3に示すように、交流成分である正弦波電圧Vacに直流成分である直流電圧Voを重畳した交番バイアスをサンプルホルダの電極間に印加する。ここで、正弦波電圧Vacの振幅は実効値で1V、直流電圧Voは磁性キャリアにかかる電界が1000V/cmとなるようにした。詳細は、後述する。
更に、このときにab間に流れる応答電流の交流成分のみを取り出し、解析することで、直流電界下におけるインピーダンスを測定した。インピーダンス測定装置としては、Solartron社製1260型周波数応答解析装置(FRA)及び、同社製1296型誘電率測定インターフェイスを用いた。上記交番バイアスに用いられる、直流電圧Voは、波形発振器から出力した直流電圧信号を、Trek社製PZD2000型高電圧電源で増幅して得た。又、正弦波電圧Vacは1296型誘電率測定インターフェイスのSAMPLE−HI端子より出力される。更に図3に示すように、測定系にコンデンサーC1(66μF)及びツェナーダイオードD1(5V)を配置することで、正弦波電圧Vacに直流電圧Voを重畳することで、上記交番バイアスを得た。
又、応答電流は、図3中の抵抗器R2(10kΩ)、コンデンサーC2(33μF)、
及びツェナーダイオードD2(5V)を用いた分流回路を用いることで、直流成分と交流成分に分離することができる。このとき、コンデンサーC2側に流れる交流成分のみを1260型インピーダンスアナライザのINPUT−V1−LO端子および、1296型誘電率測定インターフェイスのSAMPLE−LO端子に入力し、応答電流波形の解析を行い、インピーダンスを測定した。尚、図3中の抵抗器R1(10kΩ)は保護抵抗であり、測定系に流れる最大電流量を制限する。
(複素インピーダンスの測定)
本実施例では、Solartron社製インピーダンス測定ソフトウエアSMaRTを用いて、インピーダンスの自動測定を行った。SMaRTでは、所定の周波数fの正弦波電圧と正弦波電圧に対する応答電流から、周波数fに対する複素インピーダンス(下記式)を測定することができる。
【0042】
【数3】
【0043】
インピーダンスの周波数特性を測定するために、上記正弦波電圧の周波数f(Hz)は、1Hzから1MHzまでの範囲で複数の周波数でインピーダンス測定を行った。
具体的には、1.0、1.6、2.5、4.0、6.3、1.0×101、1.6×101、2.5×101、4.0×101、6.3×101、1.0×102、1.6×102、2.5×102、4.0×102、6.3×102、1.0×103、1.6×103、2.5×103、4.0×103、6.3×103、1.0×104、1.6×104、2.5×104、4.0×104、6.3×104、1.0×105、1.6×105、2.5×105、4.0×105、6.3×105、1.0×106Hzで測定を行った。
正弦波電圧Vacの振幅は実効値で1Vとした。
上記のように、1Hzから1MHzの周波数範囲において複数の周波数で測定した複素インピーダンスZを複素平面状にプロットすることで、所謂Cole−Coleプロット(ナイキスト線図)を作成した。
【0044】
(等価回路によるフィッティング)
次に、交流インピーダンス測定で得られた複素インピーダンス測定データからパラメータαを求める方法について具体的に説明する。
作成したCole−Coleプロットは、Solartron社製解析ソフトウエアZView2のInstant Fit機能を使用し、図4に示した等価回路の複素インピ
ーダンスと対応させてフィッティングを行い、インピーダンス測定データのフィッティングパラメータとしてαを求めた。ここで、図4においてRs、Rは抵抗であり、CPEはConstant Phase Element(定相要素)と呼ばれる回路素子であり、CPEの複素インピーダンスZCPEの周波数特性は下記(4)式で表される。
【0045】
【数4】
ここで、ωはインピーダンス測定の角周波数、iは虚数単位である。又、αは0.00
から1.00までの実数のパラメータである。TはCPEの静電容量を反映したパラメータであり、α=1のときはコンデンサーの静電容量となり、F(ファラッド)の次元を持つ。
【0046】
図4のフィッティング回路全体のインピーダンスは、下式のように表され、最終的に下記式(5)式で表される。
Z(ω)=Rs+(1/R+1/ZCFE)−1
=Rs+(1/R+1/((iω)αT)−1)−1
【数5】
【0047】
尚、Rsはフィッティングの精度を向上させるためにフィッティング回路に導入した仮想的な抵抗であるため、負の値をとってもよい。
図5は図4の回路において、Rs=100Ω、R=1×105Ωとし、T=2×10−10Fα・Ωα−1、α=1.0、α=0.9、α=0.8、およびα=0.7におけるCole−Coleプロットを示している。Cole−Coleプロットの形状から解るように、上記(5)式におけるαはCole−Coleプロットの描く円弧の歪み対応したパラメータである。
【0048】
(電界下におけるα)
1000V/cmの電界下における磁性キャリアのαの値は以下のように求めた。
インピーダンスを測定する際に、サンプルに印加される平均の電界強度Esampleは、インピーダンス測定時に電極間のサンプルが分担する電圧の直流成分Vsampleと電極間距離Lを用いて、Vsample/Lで表される。Vsampleの値は、図3の回路図に示したa点における電位とb点における電位の差分により測定することができる。本測定では、Tktronix社製の高電圧プローブP6015Aを用いて、a点及びb点における電位を測定し、それらの電位の差分によりサンプルホルダの電極間の分担電圧Vsampleを測定した。又、Vsampleの値は、高電圧電源から出力される直流電圧Voの電圧を変化させることで調整した。このようにして、複数の電界強度Eにおいてインピーダンス測定を行い、各電界強度下におけるαを求め、グラフにプロットすることで、1000V/cmの電界下における磁性キャリアのαを算出した。
【実施例】
【0049】
以下、本発明の具体的実施例について説明するが、本発明はこれらの実施例に限定されるものではない。尚、以下の配合における部数は特に説明が無い場合は質量部である。
〔結着樹脂の製造例1〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 22.6質量部無水トリメリット酸 1.7質量部ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
75.6質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、200℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させ、その後、10mmHgに減圧し1時間反応させ、樹脂1を合成した。GPCで求めた樹脂1の分子量は、重量平均分子量(Mw)6200、数平均分子量(Mn)2500であり、ピーク分子量(Mp)2900、ガラス転移点は55℃、軟化点は93℃であった。
【0050】
〔結着樹脂の製造例2〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 17.2質量部ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
76.6質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、220℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させた。
さらに、無水トリメリット酸6.1質量部を加え、180℃に加熱し、2時間反応させ樹脂2を合成した。GPCで求めた樹脂2の分子量は、重量平均分子量(Mw)86000、数平均分子量(Mn)6000、ピーク分子量(Mp)12800、ガラス転移点は62℃、軟化点は132℃であった。
【0051】
〔結着樹脂の製造例3〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 19.6質量部無水トリメリット酸 5.1質量部ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
75.3質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、200℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させ、その後、10mmHgに減圧し1時間反応させ、樹脂3を合成した。GPCで求めた樹脂3の分子量は、重量平均分子量(Mw)32000、数平均分子量(Mn)5500であり、ピーク分子量(Mp)8700、ガラス転移点は58℃、軟化点は106℃であった。
【0052】
〔結着樹脂の製造例4〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 17.2質量部ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
76.6質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、220℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させた。
さらに、無水トリメリット酸17.2質量部を加え、180℃に加熱し、2時間反応させ樹脂4を合成した。GPCで求めた樹脂4の分子量は、重量平均分子量(Mw)125000、数平均分子量(Mn)6400、ピーク分子量(Mp)14800、ガラス転移点は61℃、軟化点は141℃であった。
【0053】
〔結着樹脂の製造例5〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 19.6質量部無水トリメリット酸 5.1質量部ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
75.3質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、200℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させ、その後、10mmHgに減圧し1時間反応させ、樹脂5を合成した。GPCで求めた樹脂5の分子量は、重量平均分子量(Mw)18000、数平均分子量(Mn)4800であり、ピーク分子量(Mp)8000、ガラス転移点は58℃、軟化点は99℃で
あった。
【0054】
〔結着樹脂の製造例6〕
スチレン 77.4質量部アクリル酸−n−ブチル 20.6質量部メタクリル酸 2.0質量部を反応容器に添加し、該混合液を110℃まで昇温した。窒素雰囲気下にラジカル重合開始剤であるtert−ブチルハイドロパーオキサイド1部をキシレン10部に溶解したものを該混合液に30分かけて滴下した。さらにその温度で該混合液を10時間保温してラジカル重合反応を終了させた。さらに該混合液を加熱しながら減圧し、脱溶剤することにより樹脂5を得た。GPCで求めた樹脂5の分子量は、重量平均分子量(Mw)36000、数平均分子量(Mn)8000であり、ピーク分子量(Mp)13000、ガラス転移点(Tg)58℃、軟化点 113℃であった。
【0055】
〔ワックス偏在制御樹脂の製造例1〕
反応容器中に下記材料を入れ、十分溶解させた。
キシレン 25.0質量部
低分子量ポリプロピレン 9.3質量部
(三洋化成工業(株)製 ビスコール660P:軟化点 145℃)
さらに
スチレン 68.5質量部
メチルメタクリレート 3.6質量部
アクリロニトリル 9.0質量部
ジ-t-ブチルパーオキシヘキサヒドロテレフタレート 2.7質量部
キシレン 15.0質量部
の混合溶液を180℃で、4時間かけて滴下後、さらに170℃で1時間保持した後、有機溶剤を留去した。得られた樹脂を冷延・固化後、粉砕して、ビニル系共重合体とポリプロピレンがグラフト結合したワックス偏在制御樹脂1を得た。
得られたワックス偏在制御樹脂1をビーカー中で180℃に加熱したところ、分離したり、白濁を生じることはなかった。
【0056】
〔ワックス偏在制御樹脂の製造例2〕
低分子量ポリプロピレン(軟化点145℃)の添加量を9.3質量部から、20.5質量部に変更した以外は、ワックス偏在制御樹脂の製造例1と同様に、ビニル系共重合体とポリプロピレンがグラフト結合したワックス偏在制御樹脂2を得た。
得られたワックス偏在制御樹脂2をビーカー中で180℃に加熱したところ、分離したり、白濁を生じることはなかった。
【0057】
〔ワックス偏在制御樹脂の製造例3〕
低分子量ポリプロピレン(軟化点145℃)の添加量を9.3質量部から、32.6質量部に変更した以外は、ワックス偏在制御樹脂の製造例1と同様に、ビニル系共重合体とポリプロピレンがグラフト結合したワックス偏在制御樹脂3を得た。
得られたワックス偏在制御樹脂3をビーカー中で180℃に加熱したところ、分離したり、白濁を生じることはなかった。
【0058】
〔ワックス偏在制御樹脂の製造例4〕
低分子量ポリプロピレン(軟化点145℃)の添加量を9.3質量部から、0.9質量部に変更した以外は、ワックス偏在制御樹脂の製造例1と同様に、ビニル系共重合体とポリプロピレンがグラフト結合したワックス偏在制御樹脂4を得た。
得られたワックス偏在制御樹脂4をビーカー中で180℃に加熱したところ、分離した
り、白濁を生じることはなかった。
【0059】
〔トナーの製造例1〕
樹脂1 50.0質量部樹脂4 50.0質量部ワックス偏在制御樹脂1 5.0質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク105℃) 3.5質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 3.5質量部C.I.ピグメントブルー15:3 5.0質量部
上記材料をヘンシェルミキサー(FM−75型、三井三池化工機(株)製)でよく混合した後、温度130℃に設定した二軸混練機(PCM−30型、池貝鉄工(株)製)にて混練した。得られた混練物を冷却し、ハンマーミルにて1mm以下に粗粉砕し、粗砕物を得た。得られた粗砕物を、高圧気体を用いた衝突式気流粉砕機を用いて微粉砕した。
次に、得られた微粉砕物を図1に示す表面改質装置により表面改質を行った。表面改質時の条件として、原料供給速度が2.0kg/hr、熱風流量が4.5m3/min、熱風の吐出温度が210℃、冷風温度が3℃、冷風流量が3.0m3/min、絶対水分量が3g/m3で表面改質を行った。次に、コアンダ効果を利用した風力分級機(エルボジェットラボEJ−L3、日鉄鉱業社製)で分級しで微粉及び粗粉を同時に分級除去、トナー粒子1を得た。
得られたトナー粒子1 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が200nmであり、ヘキサメチルジシラザンで処理されたゾルゲル法シリカ微粉体を2.0質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー1を得た。
【0060】
〔トナーの製造例2〕
樹脂1 50.0質量部樹脂2 50.0質量部ワックス偏在制御樹脂2 10.0質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク105℃) 16.0質量部C.I.ピグメントブルー15:3 5.0質量部
上記材料を用い、表面改質時に熱風の吐出温度を210℃から230℃に変更した以外は、トナー製造例1と同様にして、トナー粒子2を得た。
得られたトナー粒子2 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が150nmであり、ヘキサメチルジシラザンで処理されたヒュームド法シリカ微粉体を3.0質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー2を得た。
【0061】
〔トナーの製造例3〕
樹脂5 100.0質量部ワックス偏在制御樹脂1 5.0質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 3.5質量部C.I.ピグメントブルー15:3 5.0質量部
上記材料を用い、表面改質時に熱風の吐出温度を210℃から250℃に変更した以外は、トナー製造例1と同様にして、トナー粒子3を得た。
得られたトナー粒子3 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が300nmであり、ヘキサメチルジシラザンで処理されたゾルゲル法シリカ微粉体を1.0質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機
(株)製)で混合し、トナー3を得た。
【0062】
〔トナーの製造例4〕
樹脂5 100.0質量部ワックス偏在制御樹脂3 5.0質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 10.0質量部C.I.ピグメントブルー15:3 5.0質量部
上記材料を用い、表面改質時に熱風の吐出温度を210℃から190℃に変更した以外は、トナー製造例1と同様にして、トナー粒子4を得た。
得られたトナー粒子4 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が80nmであり、ヘキサメチルジシラザンで処理されたゾルゲル法シリカ微粉体を2.0質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー4を得た。
【0063】
〔トナーの製造例5〕
樹脂5 100.0質量部ワックス偏在制御樹脂4 3.0質量部精製ノルマルパラフィン(DSC最大吸熱ピーク75℃) 3.0質量部C.I.ピグメントブルー15:3 5.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にして、トナー5を得た。
【0064】
〔トナーの製造例6〕
樹脂5 100.0質量部ワックス偏在制御樹脂1 15.0質量部ベヘン酸ベヘニル(DSC最大吸熱ピーク74℃) 18.0質量部C.I.ピグメントブルー15:3 5.0質量部
混練する材料を上記材料とする以外は、トナー製造例4と同様にして、トナー6を得た。
【0065】
〔トナーの製造例7〕
ワックス偏在制御樹脂1 5.0質量部を0.0質量部に変更した以外は、トナー製造例1と同様にトナー7を得た。
【0066】
〔トナーの製造例8〕
樹脂3 100.0質量部精製ノルマルパラフィン(DSC最大吸熱ピーク75℃) 2.0質量部C.I.ピグメントブルー15:3 5.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にして、トナー粒子8を得た。得られたトナー粒子8 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が30nmであり、ヘキサメチルジシラザンで処理されたヒュームド法シリカ微粉体を0.5質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー8を得た。
【0067】
〔トナーの製造例9〕
樹脂6 100.0質量部ベヘン酸ベヘニル(DSC最大吸熱ピーク74℃) 23.0質量部C.I.ピグメントブルー15:3 5.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にして、トナー粒子9を得た。得られたトナー粒子9 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が30nmであり、ヘキサメチルジシラザンで処理されたヒュームド法シリカ微粉体を0.5質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー9を得た。
【0068】
〔トナーの製造例10〕
樹脂1 50.0質量部樹脂4 50.0質量部ワックス偏在制御樹脂2 20.0質量部ベヘン酸ベヘニル(DSC最大吸熱ピーク74℃) 15.0質量部C.I.ピグメントブルー15:3 5.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にして、トナー粒子10を得た。得られたトナー粒子10 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が30nmであり、ヘキサメチルジシラザンで処理されたヒュームド法シリカ微粉体を0.5質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー10を得た。
【0069】
〔トナーの製造例11〕
表面改質時の冷風温度を3℃から25℃に、絶対水分量を3g/m3から10g/m3に変更した以外は、トナー製造例1と同様にしてトナー11を得た。
【0070】
〔トナーの製造例12〕
樹脂1 50.0質量部樹脂4 50.0質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク105℃) 3.5質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 3.5質量部C.I.ピグメントブルー15:3 5.0質量部を用いトナー製造例11と同様にしてトナー12を得た。
【0071】
〔トナーの製造例13〕
表面改質処理を行わないこと以外は、トナー製造例1と同様にしてトナー13を得た。
【0072】
〔トナーの製造例14〕
(微粒子分散液1の調製)
水 68.4質量部
メタクリル酸エチレンオキサイド付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業製) 1.0質量部
スチレン 8.5質量部
メタクリル酸 8.0質量部
アクリル酸ブチル 11.0質量部
過硫酸アンモニウム 0.1質量部
以上を、撹拌棒及び温度計をセットした反応容器に仕込み、4000回転/分で1時間攪拌した。その後、80℃まで昇温し、4時間反応させた。さらに、1%過硫酸アンモニウム水溶液を3.0質量部加え、80℃で5時間熟成して微粒子分散液1を得た。
(水相1の調製)
水 80.0質量部
微粒子分散液1 8.0質量部
ドデシルジフェニルエーテルジスルホン酸ナトリウムの48.3%水溶液(エレミノールMON−7:三洋化成工業製) 3.5質量部
酢酸エチル 8.5質量部
以上を混合撹拌し、水相1を得た。
(プレポリマー1の調製)
冷却管、撹拌機及び窒素導入管の付いた反応容器中に、
樹脂1 41.0質量部
イソホロンジイソシアネート 8.9質量部
酢酸エチル 50.0質量部
以上を入れ100℃で5時間反応し、プレポリマー1を得た。
(ケチミン化合物1の調製)
撹拌棒及び温度計をセットした反応容器に、
イソホロンジアミン 17.5質量部
メチルエチルケトン 7.5質量部
以上を入れ、50℃で5時間反応を行い、ケチミン化合物1を得た。
(原料溶解液1の調製)
撹拌棒及び温度計をセットした容器に、
樹脂1 37.8質量部
C.I.ピグメントブルー15:3 10.0質量部
カルナバワックス 10.0質量部
酢酸エチル 94.7質量部
以上を入れ、80℃で5時間、攪拌しながら保持した。その後、2時間で30℃に冷却後、1時間攪拌し、原料溶解液1を得た。
(顔料・ワックス分散液1の調製)
原料溶解液1 50.0質量部を容器に移し、ビーズミル(ウルトラビスコミル:アイメックス社製)を用いて、送液速度1kg/hr、ディスク周速度6m/秒、0.5mmジルコニアビーズを70体積%充填、3パスの条件で分散を行った。
次いで、樹脂1の65%酢酸エチル溶液 50.0質量部を加え、上記条件のビーズミルで2パスし、顔料・ワックス分散液1を得た。顔料・ワックス分散液1の固形分濃度は50%であった。
(トナーの調製)
顔料・ワックス分散液1 74.2質量部
プレポリマー1 11.4質量部
ケチミン化合物1 0.3質量部
以上を容器に入れ、TKホモミキサー(特殊機化製)で5,000rpmで2分間混合した後、容器に水相1 120.0質量部を加え、TKホモミキサーで、回転数13,000rpmで25分間混合し乳化スラリー1を得た。
撹拌機及び温度計をセットした容器に、乳化スラリー1を投入し、30℃で8時間脱溶剤した後、45℃で7時間熟成を行い、分散スラリー1を得た。
分散スラリー1 100部を減圧濾過した後、イオン交換水100質量部を加え、TKホモミキサーで混合(回転数12,000rpmで10分間)した後濾過した。
さらに、5%水酸化ナトリウム水溶液100部を加え、TKホモミキサーで混合(回転数12,000rpmで10分間)した後、減圧濾過した。
次に、10%塩酸100質量部を加え、TKホモミキサーで混合(回転数12,000rpmで10分間)した後濾過した。
最後に、イオン交換水300質量部を加え、TKホモミキサーで混合(回転数12,000rpmで10分間)した後濾過する操作を2回行い、濾過ケーキ1を得た。
濾過ケーキ1を循風乾燥機にて40℃で48時間乾燥し、目開き75μmメッシュで篩い、トナー粒子14を得た。
得られたトナー粒子14 100.0質量部に、無機微粒子として、個数平均粒径が4
0nmであり、i−ブチルトリメトキシシランで処理されたの酸化チタン微粉体1.0質量部、個数平均粒径が20nmであり、ヘキサメチルジシラザンで処理されたのヒュームド法シリカ微粉体を0.5質量部の外添混合し、トナー14を得た。
【0073】
トナー1乃至14の物性を表1に示す。
【表1】
【0074】
(多孔質磁性コアの製造例1)
<工程1(秤量・混合工程)>
Fe2O3 59.7質量%
MnCO3 34.4質量%
Mg(OH)2 4.8質量%
SrCO3 1.1質量%
となるようにフェライト原材料を秤量した。その後、ジルコニア(φ10mm)のボールを用いた乾式ボールミルで2時間粉砕・混合した。
<工程2(仮焼成工程)>
粉砕・混合した後、バーナー式焼成炉を用い大気中で950℃で2時間焼成し、仮焼フェライトを作製した。
フェライトの組成は、下記の通り。
(MnO)a(MgO)b(SrO)c(Fe2O3)d
上記式において、a=0.39、b=0.11、c=0.01、d=0.49
<工程3(粉砕工程)>
クラッシャーで0.5mm程度に粉砕し仮焼フェライトを得た。その後、仮焼フェライトを半分に分けた。仮焼フェライトの半分をジルコニア(φ10mm)のボールを用い、仮焼フェライト100質量部に対し、水を30質量部加え、湿式ボールミルで2時間粉砕し、フェライトスラリー1−1を得た。フェライトスラリー1−1の半分を、さらにジルコニアのビーズ(φ1.0mm)を用いた湿式ビーズミルで3時間粉砕し、フェライトスラリー1−2を得た。フェライトスラリー1−1と1−2をジルコニアのビーズ(φ1.0mm)を用いた湿式ビーズミルで10分間混合し、フェライトスラリー1(仮焼フェライト微粉砕品1)を得た。得られた仮焼フェライト微粉砕品1の、体積基準の50%粒径(D50)は1.3μm、体積基準の90%粒径(D90)は2.3μmであった。
<工程4(造粒工程)>
得られた仮焼フェライト微粉砕品1に、バインダーとして上記仮焼フェライト100質量部に対してポリビニルアルコール2.0質量部を添加し、スプレードライヤー(製造元:大川原化工機)で、36μmの球状粒子に造粒した。
<工程5(本焼成工程)>
焼成雰囲気をコントロールするために、電気炉にて窒素雰囲気下(酸素濃度0.01体積%以下)で、1050℃で4時間焼成した。
<工程6(選別工程)>
凝集した粒子を解砕した後に、目開き250μmの篩で篩分して粗大粒子を除去し、多孔質磁性コア1を得た。
【0075】
<多孔質磁性コアの製造例2>
多孔質磁性コア製造例1のうち、工程3のフェライトスラリー1−2を得るための湿式ビーズミルの粉砕時間を3時間から2時間に変更した。得られた仮焼フェライト微粉砕品は、D50=1.4μm、D90=3.0μmであった。また、工程5の焼成温度を1050℃から1100℃に変更した。
上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア2を得た。
【0076】
<多孔質磁性コアの製造例3>
多孔質磁性コア製造例1のうち、工程3のフェライトスラリー1−2を得るための湿式ビーズミルの粉砕時間を3時間から1時間に変更した。得られた仮焼フェライト微粉砕品は、D50=1.7μm、D90=5.0μmであった。また、工程5の焼成温度を1050℃から1150℃に変更した。
上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア3を得た。
【0077】
<多孔質磁性コアの製造例4>
<工程1(秤量・混合工程)>
Fe2O3 62.4質量%
MnCO3 30.5質量%
Mg(OH)2 6.4質量%
SrCO3 0.7質量%
をジルコニア(φ10mm)のボールを用いた乾式ボールミルで2時間粉砕・混合した。<工程2(仮焼成工程)>
粉砕・混合した後、バーナー式焼成炉を用い大気中で950℃で2時間焼成し、仮焼フェライトを作製した。
<工程3(粉砕工程)>
クラッシャーで0.5mm程度に粉砕し仮焼フェライトを得た。仮焼フェライトをステンレス(φ10mm)のボールを用い、仮焼フェライト100質量部に対し、水を30質量部加え、湿式ボールミルで1時間粉砕し、フェライトスラリー4−1を得た。フェライトスラリー4−1を、さらにステンレスのビーズ(φ1.0mm)を用いた湿式ビーズミルで4時間粉砕し、フェライトスラリー4(仮焼フェライト微粉砕品4)を得た。得られた仮焼フェライト微粉砕品4の、体積基準の50%粒径(D50)は1.4μm、体積基準の90%粒径(D90)は1.8μmであった。
<工程4(造粒工程)>
得られた仮焼フェライト微粉砕品4に、バインダーとして上記仮焼フェライト100質量部に対して、ポリビニルアルコール2.0質量部を添加し、スプレードライヤー(製造元:大川原化工機)で、36μmの球状粒子に造粒した。
<工程5(本焼成工程)>
焼成雰囲気をコントロールするために、電気炉にて窒素雰囲気下(酸素濃度0.01体積%以下)で、1100℃で4時間焼成した。
<工程6(選別工程)>
凝集した粒子を解砕した後に、目開き250μmの篩で篩分して粗大粒子を除去し、多孔質磁性コア4を得た。
【0078】
<磁性キャリアの製造例1>
[樹脂液の調製]
(樹脂液1)
シリコーン樹脂溶液(SR2411 東レ・ダウコーニング) 20.0質量部
(20質量%トルエン溶液における粘度 1.1×10−6m2/sec)
γ−アミノプロピルトリエトキシシラン 0.5質量部
トルエン 79.5質量部
以上を、ボールミル(ソーダガラスボール φ10mm)を用いて1時間混合し、樹脂液1を得た。
(樹脂液2)
シリコーン樹脂溶液(SR2410 東レ・ダウコーニング) 20.0質量部
(20質量%トルエン溶液における粘度 2.9×10−6m2/sec)
γ−アミノプロピルトリエトキシシラン 0.3質量部
トルエン 79.7質量部
以上を、ボールミル(ソーダガラスボール φ10mm)を用いて1時間混合し、樹脂液2を得た。
[磁性キャリア1の製造]
工程例1(樹脂充填工程例1):
多孔質磁性コア1の100.0質量部を万能攪拌混合機(ダルトン社製)に入れ、減圧下、60℃に加熱しながら撹拌する。続いて、樹脂液1を多孔質磁性コア1に対し樹脂成分として15質量部になるように添加し4時間加熱を続け、溶剤を除去した。得られた試料をドラムミキサ(杉山重工業製)に移し、窒素雰囲気下に200℃で2時間熱処理して、開口70μmのメッシュで分級して、磁性キャリア1aを得た。
工程例2(樹脂コート工程例1):
磁性キャリア1aの100.0質量部をナウタミキサ(ホソカワミクロン社製)に投入し、さらに、樹脂液2を樹脂成分として1.0質量部になるようにナウタミキサに投入した。減圧下で70℃に加熱し、1.7S−1(100rpm)で混合し、2時間かけて溶媒除去及び塗布操作を行った。その後、得られた試料をドラムミキサ(杉山重工業製)に移し、窒素雰囲気下、温度200℃で2時間熱処理した後、開口70μmのメッシュで分級して磁性キャリア1を得た。
【0079】
<磁性キャリアの製造例2、3、及び4>
磁性キャリアの製造例1において、多孔質磁性コア1を多孔質磁性コア2に、樹脂液1を多孔質磁性コア2に対し樹脂成分として10質量部になるように添加した以外は、磁性キャリアの製造例1と同様にして、磁性キャリア2を得た。
磁性キャリアの製造例1において、多孔質磁性コア1を多孔質磁性コア3に、樹脂液1を多孔質磁性コア3に対し樹脂成分として8質量部になるように添加した以外は、磁性キャリアの製造例1と同様にして、磁性キャリア3を得た。
磁性キャリアの製造例1において、工程例2(樹脂コート工程例1)を行わなかった以外は、磁性キャリアの製造例1と同様にして、磁性キャリア4を得た。
【0080】
<磁性キャリアの製造例5>
体積基準の50%粒径(D50)が32μmのマグネタイト粒子100質量部に、シリコーン樹脂(信越化学社製:KR271)1質量部、γ―アミノプロピルトリエトキシシラン0.5質量部、トルエン98.5質量部の混合液を、添加し、さらに溶液減圧ニーダーで撹拌混合しながら70℃、5時間減圧乾燥を行い、溶剤を除去した。その後、140℃で2時間焼き付け処理して、篩振とう機(300MM−2型、筒井理化学機械:75μ
m開口)で篩い、磁性キャリア5を得た。
【0081】
磁性キャリア1乃至5の物性を表2に示す。
【表2】
【0082】
〔実施例1乃至8及び比較例1乃至8〕
次に、このように作製したトナーと磁性キャリアを表3の組み合わせで二成分系現像剤を作製した。二成分系現像剤は、磁性キャリア100質量部に対して、トナー9質量部の配合割合とし、V型混合機で5分間混合した。
【0083】
【表3】
【0084】
<二成分系現像剤の評価>
得られた二成分系現像剤の低温定着性、グロス均一性、画像濃度、画像濃度変動、及び現像性の各種評価を、温度23℃/湿度50%RH(以下「N/N」)と、温度30℃/湿度80%RH(以下「H/H」)の2つの環境で実施した。詳細な評価方法及び評価基準は後述する。上記各種評価用の画像形成装置としては、キヤノン製カラー複写機imageRUNNER iRC3580改造機を用いた。改造箇所は、現像ドラムに対する現像スリーブ周速を標準機に比して1.5倍としたこと、及び、プロセススピードが245mm/secとなるようしたことである。なお、上記二成分系現像剤は、画像形成装置のシアン用現像器に入れて評価を行った。評価結果を表4及び表5に示す。
【0085】
<低温定着性の評価>
紙は、カラー複写機・プリンタ用普通紙 CS−814(A4、81.4g/m2)(キヤノンマーケティングジャパン株式会社より販売)を用いた。
FFH画像(以下、ベタ部)のトナーの紙上への載り量が0.6mg/cm2となるように直流電圧VDCを調整し、未定着のFFH画像を得た。
その後、キヤノン製プロダクション向け複写機imagePRESS C1+の外部定着器(ベルト&ローラ定着器)をプロセススピードが300mm/secとなるように改造したものを定着性評価に用いた。
上記外部定着器における定着温度を100〜200℃の範囲で10℃おきに調整し、各温度で上記未定着のFFH画像を定着し定着画像を得た。得られた定着画像を4.9kPaの荷重をかけたレンズクリーニングワイパー(ダスパー 小津産業株式会社製)で5往復摺擦し、摺擦前後の画像濃度の濃度低下率が10%以下になる点を定着温度とし、下記の評価基準に従って評価した。
A:定着温度120℃以下 (非常に良好)
B:定着温度130℃ (良好)
C:定着温度140℃ (本発明において許容レベル)
D:定着温度150℃以上 (本発明において許容できないレベル)
【0086】
<グロス均一性の評価>
紙は、CLC用 厚口用紙 NS−700(A4、157g/m2)(キヤノンマーケティングジャパン株式会社より販売)を用いた。
低温定着性評価と同様に、ベタ部のトナーの紙上への載り量が0.6mg/cm2となるように直流電圧VDCを調整し、紙の四隅及び中央部に、4cm×4cmの正方形のベタ画像を印刷した未定着画像を得た。
その後、キヤノン製プロダクション向け複写機imagePRESS C1+の外部定着器をプロセススピード100mm/secとなるように改造したものを用い、グロス均一性評価を行った。具体的には、外部定着器における定着温度を180℃に調整し、未定着画像を定着した。当該定着画像の四隅及び中央部にあるベタ画像の各グロス値を測定し、その最大値と最小値の差を以下の基準で判断した。
A:1.0未満
B:1.0以上2.0未満(良い)
C:2.0以上3.0未満(本発明において許容レベル)
D:3.0以上(本発明において許容できないレベル)
【0087】
<画像濃度の評価>
現像スリーブには、周波数2.0kHz、Vpp1.3kVの交流電圧と直流電圧VDCを印加した。直流電圧VDCはベタ部のトナーの紙上への載り量が0.6mg/cm2となるように調整した。FFH画像を印刷し、画像濃度(反射濃度)を求めた(初期)。その後、画像比率1%で50000枚印刷し、50000枚印刷後に、再びFFH画像を印刷し、画像濃度(反射濃度)を測定した(50000枚印刷後)。紙は、CS−814(A4、81.4g/m2)(キヤノンマーケティングジャパン株式会社より販売)を用いた。画像濃度(反射濃度)は、分光濃度計500シリーズ(X−Rite社製)を用いて測定し、以下の基準で判断した。
A:1.50以上
B:1.40以上1.50未満(良い)
C:1.30以上1.40未満(本発明において許容レベル)
D:1.30未満(本発明において許容できないレベル)
【0088】
<画像濃度変動の評価>
現像スリーブには、周波数2.0kHz、Vpp1.3kVの交流電圧と直流電圧VDCを印加した。直流電圧VDCはベタ部のトナーの紙上への載り量が0.6mg/cm2となるように調整した。FFH画像を印刷し、画像濃度(反射濃度)を求めた。
画像濃度は、分光濃度計500シリーズ(X−Rite社製)を用いて測定した。
その後、現像器内の現像剤のトナー比率が9質量部で一定になるようにトナーを補給しながら、画像比率20%で100枚印刷し、初期と同様にFFH画像を印刷し、画像濃度(反射濃度)を測定した。
画像比率20%で100枚印刷する前後のFFH画像の画像濃度(反射濃度)の差を算出し、以下の基準で評価を行った。
A:0.10未満
B:0.10以上0.20未満(良い)
C:0.20以上0.30未満(本発明において許容レベル)
D:0.30以上(本発明において許容できないレベル)
【0089】
<現像性>
直流電圧VDCを300Vに固定し、周波数2.0kHzでVppを0.7kVから0.1kV刻みで2.0kVまで変えた交流電圧と直流電圧VDCを印加した。トナー載り量が0.50mg/cm2となるときのVppを求めた。
A:Vppが1.4kV未満 (非常に良好)
B:Vppが1.4kV以上1.6kV未満(良好)
C:Vppが1.6kV以上1.9kV未満(本発明において許容レベル)
D:Vppが1.9kV以上 (本発明において許容できないレベル)
【0090】
【表4】
【0091】
【表5】
【技術分野】
【0001】
本発明は、電子写真法および静電記録法に用いられるトナー及び二成分系現像剤に関する。
【背景技術】
【0002】
電子写真法において、静電荷像を現像する工程は摩擦帯電されたトナーを静電荷像とのクーロン力を利用して静電荷像上に付着させて画像形成する。トナーを用いて静電荷像を現像するための現像剤には、磁性体を樹脂中に分散した磁性トナーを用いる一成分系現像剤と、非磁性トナーと磁性キャリアを混合して用いる二成分系現像剤とに大別される。
特に、高画質を要求されるフルカラー複写機またはフルカラープリンタ等のフルカラー画像形成装置では、後者が好適に用いられている。
フルカラー画像形成装置は、近年POD用途やグラフィック用途に用いられている。このような分野では従来以上の高速・高画質が要求されている。高画質として要求されることの一つとして、画像の面内のグロス均一性がある。画像の面内のグロス均一性は、定着部材とトナーとの剥離性に相関がある。剥離性が良い場合、画像の面内のグロスは画像のどの部分でも同じ(均一)になる。
それに対し、剥離性が悪いトナーの場合、剥離が不十分だった部分は画像表面が剥離時にあらされ、画像の面内のグロスが低下してしまうことがある。このため、グロスが高い部分と低い部分が同じ画像の中に出てしまうことがある(不均一になる)。
このような課題を解決するために、定着部材にオイルを塗布する手法が提案されてきた。このような手法の場合、剥離性は良いが、塗布したオイルにより過度のグロスが出てしまいグロスのコントロールが困難である。
また、トナー中にワックスを多く添加することでも剥離性を改善できるが、耐久性の低下やトナーの耐熱保存性が低下する場合がある。
トナーの耐久性や耐熱保存性改善の目的で、トナー粒子の表面から0.3μmまでの深さ領域に存在するワックス比率を規定したトナーの提案がなされている。(特許文献1、特許文献2)
上記提案のトナーは、トナーの保存性能や、現像部材の汚染は低減されていたが、グロスの均一性の悪化が生じる場合があり、トナーの剥離性は不十分であった。
このようにいずれの提案においても、耐久性や低温定着性を達成しつつ、画像のグロスの均一性を確保したトナー及び二成分系現像剤は得られていない。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2004−246345号公報
【特許文献2】特開2006−258901号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明は、耐久性や低温定着性を達成しつつ、画像の面内のグロスの均一性を確保したトナー及び二成分系現像剤を提供する。
【課題を解決するための手段】
【0005】
本発明者らは鋭意検討を重ねた結果、低温定着性に優れたトナーにおいて、十分な耐久性、耐熱保存性を達成しつつ、画像の面内のグロスの均一性を改善させるためには、トナー中のワックス偏在状態をコントロールすることが必要であることを見出した。
すなわち、結着樹脂、ワックス、及び外添剤としての無機微粉体を少なくとも含有する
トナーであって、前記結着樹脂はポリエステル樹脂を含有し、前記ワックスの含有量が、前記結着樹脂100質量部に対して、3.0質量部以上20.0質量部以下であり、前記トナーは、ATR法を用い、ATR結晶としてGe、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPa、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPbとし、ATR法を用い、ATR結晶としてKRS5、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPc、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPdとしたときに、下記式(1)の関係を満たすことを特徴とするトナー。
1.05≦P1/P2≦2.00 ・・・ 式(1)
[前記式(1)において、前記最大吸収ピーク強度Paは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pbは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pcは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pdは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、P1=Pa/Pb、P2=Pc/Pdである。]
【発明の効果】
【0006】
本発明によれば、耐久性や低温定着性を達成しつつ、画像の面内のグロスの均一性を確保したトナー及び二成分系現像剤を提供できる。
【図面の簡単な説明】
【0007】
【図1】表面改質装置の模式図。
【図2】ATR結晶としてGeを用い測定したFT−IRスペクトルの一例。
【図3】交流インピーダンス測定法の構成を表す模式図。
【図4】インピーダンス測定で得られたCole−Coleプロットをフィッティングするために用いるフィッティング回路。
【図5】図4に示した回路のインピーダンスのCole−Coleプロットを表すグラフ図。
【発明を実施するための形態】
【0008】
以下、本発明を実施するための形態について説明する。
本発明で使用するトナーは、結着樹脂、ワックス、及び外添剤としての無機微粉体を少なくとも含有するトナーであって、かつ、該結着樹脂がポリエステル樹脂を含み、トナーの深さ方向に対してのワックス偏在度合いをコントロールしたトナーである。本発明のトナーは当該構成をとることで、耐久性能と画像の面内のグロス均一性(以下単にグロス均一性ともいう)を両立することができる。
まず、トナー表面からトナー中心部に向かうトナーの深さ方向において、トナー表面から約0.3μmの間にワックスを多量に存在させることでグロス均一性を改善することができる。これは、トナー表面近傍の、結着樹脂に対するワックスの存在比率(以下単にワックス比率ともいう)が多いために、定着部材とトナー層の剥離性が良好であり、結果、画像のどの部分でも同じように剥離することに起因する。
しかしながら、ワックスは分子量が結着樹脂に比べ小さく軟らかいために、トナー中にワックスが多く存在するとトナー表面に外添剤が埋め込まれやすくなる。その結果、トナ
ーとキャリア間の付着力が増加し、トナーがキャリアから像担持体へ現像されにくくなり、耐久時の画像濃度が低下してしまうことがある。
そこで、発明者らは鋭意検討し、トナー表面から約1.0μmの、結着樹脂に対するワックスの存在比率をコントロールすることで、上記耐久時の画像濃度の低下を抑制できることを見出した。
そのメカニズムは明確ではないが、発明者らは以下のように推察している。
外添剤の埋め込みは、トナー表層だけではなく、その下層のやわらかさも関与している。例えば、トナーの最表層のワックス比率が高かったとしても、その下層が固い樹脂の層で構成されていれば、外添剤はその機能を失うほどには埋め込まれない。この外添剤の埋め込みに関与するのがトナー表層から約1.0μmの範囲である。このため、この範囲に存在するワックス比率をコントロールすることで、耐久時の画像濃度の低下を抑制できる。このため、トナー表面からトナー中心部に向かうトナーの深さ方向において、トナー表面から約0.3μmの間の、結着樹脂に対するワックスの存在比率を、約1.0μmの間の、結着樹脂に対するワックスの存在比率より多くすることが重要である。
また、当該構成を採用することで、トナー表面に多く存在するワックスがトナー中のワックスの染み出しをさらに促進する。これは、トナー表面に存在するワックスが溶けることにより、トナー内部からトナー表面へのワックスの通り道が形成され、定着時に効果的にワックスが染み出すことができるためだと思われる。染み出したワックスは、離型性をより高められるため、グロス均一性が向上する。
一方、トナー表面にワックスを偏析させないトナーの場合、ワックスを多量に添加しなくてはならず、その結果、トナーの耐久性が低下する場合がある。
さらに、トナー表面よりもやや内側のワックス比率をトナー表面のワックス比率より少なくすることで、耐久性もさらに向上する。これは、トナー表層はワックス比率が多いため、外添剤が埋め込まれやすい。それに対し、トナー表面のやや内側はトナー表層に比べワックス比率が少ないために、外添剤が埋め込まれにくい。このため、ワックス比率が高いトナー表層からワックス比率が低いトナーやや内側にいくにしたがって、外添剤の埋め込みが抑制される。
上記理由から、トナー表面から約0.3μmの間の結着樹脂に対するワックスの存在比率[A]、及び、約1.0μmの間の結着樹脂に対するワックスの存在比率[B]を調整し、当該[A]の[B]に対する比([A]/[B])を制御することで、初めて耐久性とグロスの均一性を確保できる。
【0009】
具体的には、本発明のトナーは、ATR法を用い、ATR結晶としてGe、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPa、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPbとし、
ATR法を用い、ATR結晶としてKRS5、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPc、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPdとしたときに、下記式(1)の関係を満たしていることを特徴とする。
1.05≦P1/P2≦2.00 ・・・ 式(1)
[前記式(1)において、前記最大吸収ピーク強度Paは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pbは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pcは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pdは、1713cm−1以上1723cm−
1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、P1=Pa/Pb、P2=Pc/Pdである。]
【0010】
上記ATR(Attenuated Total Reflection)法とは、試料より高い屈折率を有する結晶(ATR結晶)に、試料を密着させ、臨界角以上の入射角で赤外光を結晶に入射させると、光は密着した試料と結晶の界面で全反射を繰り返し出射する。この時、赤外光は試料と結晶の界面で反射するのではなく、試料側にわずかににじみこんでから全反射する。このにじみこみ深さは、波長、入射角及びATR結晶の屈折率に依存する。
dp=λ/(2πn1)×[sin2θ―(n2/n1)2]−1/2
dp:にじみ込み深さ
n1:試料の屈折率(本発明では1.5としている)
n2:ATR結晶の屈折率(ATR結晶がGeの場合の屈折率;4.0、ATR結晶がKRS5の場合の屈折率;2.4)
θ:入射角
このため、ATR結晶の屈折率や入射角を変えることでにじみこみ深さの異なるFT−IRスペクトルを得ることができる。
例えば、ATR法において、ATR結晶にGe(n2=4.0)を用い、2000cm−1(λ=5μm)の光を、入射角45°の条件で測定した場合、上記式を用いると、にじみこみ深さdpは0.3μmになる。一方、ATR結晶にKRS5<臭沃化タリウム[臭化タリウムTlBr(42モル%)+ヨウ化タリウム Tll(58モル%)]の組成をもつタリウムハライドの混晶>(n2=2.4)を用い入射角45°の条件で測定した場合、にじみこみ深さは1.0μmとなる。
【0011】
本発明のトナーでは、結着樹脂はポリエステル樹脂を含む樹脂である。
このため、1713cm−1以上1723cm−1以下の範囲の吸収ピークは、主に結着樹脂由来の−CO―の伸縮振動に起因するピークである。
結着樹脂由来のピークとしては、上記以外にも芳香環のCHの面外変角振動等様々なピークが検出されるが、1500cm−1以下の範囲には、ピークが数多く存在し、結着樹脂のピークだけを分離することが困難であり、正確な数値を算出できない。このため、他のピークとの分離が容易な1713cm−1以上1723cm−1以下の範囲の吸収ピークを結着樹脂由来のピークとして用いる。
また、2843cm−1以上2853cm−1以下の範囲の吸収ピークは、主にワックス由来の−CH2−の伸縮振動(対称)に起因するピークである。
ワックスのピークとしては、上記以外にも1450cm−1以上1500cm−1以下にCH2の面内変角振動のピークが検出されるが、結着樹脂由来のピークとも重なり合ってしまい、ワックスのピークを分離することが困難である。このため、他のピークとの分離が容易な2843cm−1以上2853cm−1以下の範囲の吸収ピークをワックス由来のピークとして用いる。
本発明において、上記結着樹脂由来の最大吸収ピーク強度(Pb、Pd)及びワックス由来の最大吸収ピーク強度(Pa、Pc)が、結着樹脂及びワックスの存在量に相関することを見出した。そこで、ワックス由来の最大吸収ピーク強度を結着樹脂由来の最大吸収ピーク強度で割ることで、結着樹脂に対するワックスの存在比率を算出している。
ここで、Pa及びPcを求めるに当たり、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引く理由は、ベースラインの影響を排除し、真のピーク強度を算出するためである。3050cm−1と2600cm−1付近には吸収ピークがないため、この2点の平均値を算出することで、ベースライン強度を算出できる。
一方、Pb及びPdを求めるに当たり、1713cm−1以上1723cm−1以下の
範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引く理由も、ベースラインの影響を排除し、真のピーク強度を算出するためである。3050cm−1と2600cm−1付近には吸収ピークがないため、この2点の平均値を算出することで、ベースライン強度を算出できる。
【0012】
そして、トナー表面から約0.3μmの間の結着樹脂に対するワックスの存在比率(P1)は、ATR法を用い、ATR結晶としてGe(n2=4.0)、赤外光入射角として45°の条件で測定し得られた、上記Pa及びPbから算出される(P1=Pa/Pb)。ここでP1は、トナーの表面近傍の、結着樹脂に対するワックスの存在比率を表すことが必要である。さらなる、(すなわち、トナー表面から約0.3μmより小さい距離)トナー表面近傍の結着樹脂に対するワックスの存在比率を測定する場合、ATR結晶への赤外光(IR)の入射角を大きくすることが考えられる。しかし、入射角を大きくしていくにつれてIRスペクトルの強度が低下してくる。その結果、数値の信頼性が低下してしまう。このため、発明者らはIRのスペクトルの強度が確保できる、入射角45°の条件で測定を行い、トナー表面から約0.3μmの間の結着樹脂に対するワックスの存在比率をトナー表面近傍の結着樹脂に対するワックスの存在比率(P1)とした。
先に述べたとおり、P1の値をコントロールすることで、グロス均一性を改善することができる。具体的には、P1は0.20以上1.50以下であることが好ましく、より好ましくは、0.30以上1.20以下である。
P1は、ワックスの種類及び結着樹脂に対するワックスの添加量の調整、トナー中おけるワックスの分散状態又はワックスの偏在状態を制御することができる樹脂の添加、トナーの製造工程における熱風を用いたトナーの改質処理の実施により、上記範囲に制御することが可能である。これらの具体例は後述する。
【0013】
一方、トナー表面から約1.0μmの間の結着樹脂に対するワックスの存在比率(P2)は、ATR法を用い、ATR結晶としてKRS5(n2=2.4)、赤外光入射角として45°の条件で測定し得られた上記Pc及びPdから算出される(P2=Pc/Pd)。先に述べたとおり、P2をコントロールすることで、耐久時の画像濃度の低下を抑制することができる。具体的には、P2は0.10以上0.70以下であることが好ましく、より好ましくは、0.20以上0.60以下である。
P2は、ワックスの結着樹脂に対する添加量を調整することで上記範囲に制御することが可能である。
当該ワックスの添加量は、結着樹脂100質量部に対して、3.0質量部以上20.0質量部以下であり、好ましくは3.5質量部以上16.0質量部以下である。
【0014】
そして、耐久時の画像濃度の低下の抑制と画像の面内のグロス均一性の両立させるためには、P1/P2が1.05以上2.00以下であり、好ましくは1.25以上1.90以下である。
P1/P2が2.00より大きい場合、トナーの表面近傍に偏在するワックス比率が高すぎる。このようなトナーの場合、表面近傍に偏在するワックスが多すぎるために、外添剤がトナー表面に埋め込まれやすくなる。その結果、トナーとキャリア間の付着力が増加し、トナーがキャリアから像担持体へ現像されにくくなり、耐久時の画像濃度が低下する。一方、P1/P2が1.05未満のトナーは、トナーの内部に偏在するワックス比率が高く、耐久時の外添剤の埋め込みを抑制することが出来ないことに加えて、グロス均一性が顕著に低下する。
P1/P2は、ワックスの種類及び結着樹脂に対するワックスの添加量の調整、トナー中おけるワックスの分散状態又はワックスの偏在状態を制御することができる樹脂の添加、トナーの製造工程における熱風を用いたトナーの改質処理の実施により、上記範囲に制御することが可能である。
【0015】
本発明のトナーに用いられるワックスとしては、特に限定されず、公知の物を使用することができる。以下、好適に使用できる物を例示する。低分子量ポリエチレン、低分子量ポリプロピレン、アルキレン共重合体、マイクロクリスタリンワックス、パラフィンワックス、フィッシャー・トロプシュワックスの如き脂肪族炭化水素系ワックス;酸化ポリエチレンワックスの如き脂肪族炭化水素系ワックスの酸化物;脂肪族炭化水素系エステルワックスの如き脂肪酸エステルを主成分とするワックス;及び脱酸カルナバワックスの如き脂肪酸エステルを一部または全部を脱酸化したもの。ベヘニン酸モノグリセリドの如き脂肪酸と多価アルコールの部分エステル化物;植物性油脂を水素添加することによって得られるヒドロキシル基を有するメチルエステル化合物等。
なかでも、炭化水素系ワックス、特に脂肪族炭化水素系ワックスが高湿環境下における耐久時の画像濃度の低下を防止できることから、より好ましい。
本発明において、ワックスの最大吸熱ピークのピーク温度は、55℃以上140℃以下であることが、耐熱保存性及びホットオフセット性の改善のため、好ましい。
ワックスの最大吸熱ピークを55℃以上にすることで、ワックスが保管時に溶解することを防止でき、その結果、耐熱保存性を向上することができる。また、ワックスの最大吸熱ピークを140℃以下とすることで、定着時にワックスが溶融し、巻きつきを抑制することができる。
【0016】
次に、結着樹脂について説明する。本発明において、結着樹脂はトナーの構成要素を十分に結着するための樹脂である。後述するトナーのワックス偏在状態を制御するための樹脂は、本発明においては結着樹脂に該当しない。
本発明のトナーに用いられる結着樹脂は、低温定着性の観点から、シャープメルト性の高いポリエステル樹脂を含有する。結着樹脂に占めるポリエステル樹脂の割合は、50質量%以上であることが好ましく、70質量%以上であることがより好ましく、90質量%以上であることがさらに好ましく、100質量%であることが特に好ましい。本発明に使用される結着樹脂は、ポリエステル樹脂を含む以外に、低温定着性に影響を与えない範囲で、他の樹脂を含むこともできる。当該樹脂としては、例えば、熱可塑性結着樹脂などが挙げられる。具体的には、スチレン、パラクロロスチレン、α−メチルスチレン等のスチレン類の単独重合体又は共重合体(スチレン系樹脂);アクリル酸メチル、アクリル酸エチル、アクリル酸n−プロピル、アクリル酸n−ブチル、アクリル酸ラウリル、アクリル酸2−エチルヘキシル、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸n−プロピル、メタクリル酸ラウリル、メタクリル酸2−エチルヘキシル等のビニル基を有するエステル類の単独重合体又は共重合体(ビニル系樹脂);アクリロニトリル、メタクリロニトリル等のビニルニトリル類の単独重合体又は共重合体(ビニル系樹脂);ビニルメチルエーテル、ビニルイソブチルエーテル等のビニルエーテル類の単独重合体又は共重合体(ビニル系樹脂);ビニルメチルケトン、ビニルエチルケトン、ビニルイソプロペニルケトン等のビニルケトン類の単独重合体又は共重合体(ビニル系樹脂);エチレン、プロピレン、ブタジエン、イソプレン等のオレフィン類の単独重合体又は共重合体(オレフィン系樹脂);エポキシ樹脂、上述以外のポリエステル系樹脂、ポリウレタン樹脂、ポリアミド樹脂、セルロース樹脂、ポリエーテル樹脂等の非ビニル縮合系樹脂、及びこれらの非ビニル縮合系樹脂とビニル系モノマーとのグラフト重合体などが挙げられる。これらの樹脂は、1種のみを上記ポリエステル樹脂に含有させて用いてもよいし、2種以上を含有させて用いてもよい。
【0017】
本発明に用いられるポリエステル樹脂はアルコールモノマーとカルボン酸モノマーが縮重合したものが用いられる。アルコールモノマーとしては以下のものが挙げられる。
ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン、ポリオキシプロピレン(3.3)−2,2−ビス(4−ヒドロキシフェニル)プロパン、ポリオキシエチレン(2.0)−2,2−ビス(4−ヒドロキシフェニル)プロパン、ポリオキシプロピレン(2.0)−ポリオキシエチレン(2.0)−2,2−ビス(4−
ヒドロキシフェニル)プロパン、ポリオキシプロピレン(6)−2,2−ビス(4−ヒドロキシフェニル)プロパン等のビスフェノールAのアルキレンオキシド付加物、エチレングリコール、ジエチレングリコール、トリエチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,4−ブタンジオール、ネオペンチルグリコール、1,4−ブテンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,4−シクロヘキサンジメタノール、ジプロピレングリコール、ポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレングリコール、ビスフェノールA、水素添加ビスフェノールA、ソルビトール、1,2,3,6−ヘキサンテトロール、1,4−ソルビタン、ペンタエリスリトール、ジペンタエリスリトール、トリペンタエリスリトール、1,2,4−ブタントリオール、1,2,5−ペンタントリオール、グリセロール、2−メチルプロパントリオール、2−メチル−1,2,4−ブタントリオール、トリメチロールエタン、トリメチロールプロパン、1,3,5−トリヒドロキシメチルベンゼン。
一方、カルボン酸モノマーとしては、以下のものが挙げられる。
フタル酸、イソフタル酸及びテレフタル酸の如き芳香族ジカルボン酸類又はその無水物;コハク酸、アジピン酸、セバシン酸及びアゼライン酸の如きアルキルジカルボン酸類又はその無水物;炭素数6〜18のアルキル基又はアルケニル基で置換されたコハク酸もしくはその無水物;フマル酸、マレイン酸及びシトラコン酸の如き不飽和ジカルボン酸類又はその無水物。
また、その他のモノマーとしては、以下のものが挙げられる。
グリセリン、ソルビット、ソルビタン、さらには例えばノボラック型フェノール樹脂のオキシアルキレンエーテル等の多価アルコール類;トリメリット酸、ピロメリット酸、ベンゾフェノンテトラカルボン酸やその無水物等の多価カルボン酸類。
それらの中でも、特に、下記一般式(1)で表されるビスフェノール誘導体を2価アルコールモノマー成分とし、2価以上のカルボン酸又はその酸無水物、又はその低級アルキルエステルとからなるカルボン酸成分(例えば、フマル酸、マレイン酸、無水マレイン酸、フタル酸、テレフタル酸、トリメリット酸、ピロメリット酸等)を酸モノマー成分として、これらのポリエステルユニット成分で縮重合した樹脂が良好な帯電特性を有するので好ましい。
【0018】
【化1】
(式中、Rはエチレン基又はプロピレン基を示し、x及びyはそれぞれ1以上の整数であり、かつx+yの平均値は2〜10である。)
【0019】
本発明に用いられる結着樹脂として、低温定着性を達成するために、軟化点(Tm)が好ましくは60℃以上120℃以下、より好ましくは60℃以上100℃以下の樹脂を用いるとよい。
【0020】
本発明において、トナーのワックス偏在状態を制御するための樹脂(以下、ワックス偏在制御樹脂)を、結着樹脂とは別に添加することが好ましい。
ワックス偏在制御樹脂の添加量としては、結着樹脂100質量部に対して、1質量部以上20質量部以下であることが、トナー表面近傍の結着樹脂に対するワックスの存在比率を制御するために好ましい。
上記ワックス偏在制御樹脂としては、結着樹脂に極性が近い部位、ワックスに極性が近い部位の両方を有するものであればどのようなものでもかまわない。具体的には、スチレン系モノマー、並びに、窒素(N)含有ビニルモノマー、カルボキシル基含有モノマー、
水酸基含有モノマー、アクリル酸エステルモノマー及びメタアクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーを用いて合成された共重合体と、ポリオレフィンとがグラフト共重合した樹脂が好適に例示できる。
グラフト共重合させることにより、共重合体成分とポリオレフィン成分が一体となり、ワックス偏在状態をコントロールすることができる。グラフト共重合していない場合、ワックス偏在状態をコントロールしにくくなる。また、グラフト共重合していない樹脂に熱をかけ溶解させると共重合体成分とポリオレフィン成分が2相に分離したり、白濁を生じたりする場合がある。
スチレン系モノマーと窒素含有ビニルモノマー、カルボキシル基含有モノマー、水酸基含有モノマー、アクリル酸エステルモノマー及びメタクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーとを用いて合成された共重合体を合成するために用いることのできるモノマーとしては、次のようなものが挙げられる。
スチレン系モノマーとしては、スチレン、o−メチルスチレン、m−メチルスチレン、p−メチルスチレン、p−メトキシスチレン、p−フェニルスチレン、p−クロルスチレン、3,4−ジクロルスチレン、p−エチルスチレン、2,4−ジメチルスチレン、p−n−ブチルスチレン、p−tert−ブチルスチレン、p−n−ヘキシルスチレン、p−n−オクチルスチレン、p−n−ノニルスチレン、p−n−デシルスチレン、p−n−ドデシルスチレンの如きスチレン及びその誘導体が挙げられる。
窒素含有ビニル系モノマーとしては、メタクリル酸ジメチルアミノエチル、メタクリル酸ジエチルアミノエチルの如きアミノ基含有α−メチレン脂肪族モノカルボン酸エステル類;アクリロニトリル、メタクリロニトリル、アクリルアミドの如きアクリル酸もしくはメタクリル酸誘導体が挙げられる。
カルボキシル基含有モノマーとしては、マレイン酸、シトラコン酸、イタコン酸、アルケニルコハク酸、フマル酸、メサコン酸の如き不飽和二塩基酸;マレイン酸無水物、シトラコン酸無水物、イタコン酸無水物、アルケニルコハク酸無水物の如き不飽和二塩基酸無水物;マレイン酸メチルハーフエステル、マレイン酸エチルハーフエステル、マレイン酸ブチルハーフエステル、シトラコン酸メチルハーフエステル、シトラコン酸エチルハーフエステル、シトラコン酸ブチルハーフエステル、イタコン酸メチルハーフエステル、アルケニルコハク酸メチルハーフエステル、フマル酸メチルハーフエステル、メサコン酸メチルハーフエステルの如き不飽和二塩基酸のハーフエステル;ジメチルマレイン酸、ジメチルフマル酸の如き不飽和二塩基酸エステル;アクリル酸、メタクリル酸、クロトン酸、ケイヒ酸の如きα,β−不飽和酸;クロトン酸無水物、ケイヒ酸無水物の如きα,β−不飽和酸無水物、該α,β−不飽和酸と低級脂肪酸との無水物;アルケニルマロン酸、アルケニルグルタル酸、アルケニルアジピン酸、これらの酸無水物及びこれらのモノエステルが挙げられる。
水酸基含有モノマーとしては、2−ヒドロキシエチルアクリレート、2−ヒドロキシエチルメタクリレート、2−ヒドロキシプロピルメタクリレートなどのアクリル酸又はメタクリル酸エステル類、4−(1−ヒドロキシ−1−メチルブチル)スチレン、4−(1−ヒドロキシ−1−メチルヘキシル)スチレンが挙げられる。
アクリル酸エステルモノマーとしては、アクリル酸メチル、アクリル酸エチル、アクリル酸n−ブチル、アクリル酸イソブチル、アクリル酸プロピル、アクリル酸n−オクチル、アクリル酸ドデシル、アクリル酸2−エチルヘキシル、アクリル酸ステアリル、アクリル酸2−クロルエチル、アクリル酸フェニルの如きアクリル酸エステル類が挙げられる。
メタクリル酸エステルモノマーとしては、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸プロピル、メタクリル酸n−ブチル、メタクリル酸イソブチル、メタクリル酸n−オクチル、メタクリル酸ドデシル、メタクリル酸2−エチルヘキシル、メタクリル酸ステアリル、メタクリル酸フェニル、メタクリル酸ジメチルアミノエチル、メタクリル酸ジエチルアミノエチルの如きα−メチレン脂肪族モノカルボン酸エステル類が挙げられる。
その中でも特に、スチレン−アクリロニトリル−メチルメタクリレートの三元共重合体
が好ましい。
スチレン系モノマー、並びに、窒素含有ビニルモノマー、カルボキシル基含有モノマー、水酸基含有モノマー、アクリル酸エステルモノマー及びメタアクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーを用いて合成された共重合体は、ゲルパーミエーションクロマトグラフィー(GPC)により測定された分子量分布において、重量平均分子量(Mw)が5,000〜100,000であり、数平均分子量(Mn)が1,500〜15,000であり、重量平均分子量(Mw)と数平均分子量(Mn)との比(Mw/Mn)が2〜40であることが好ましい。
また、スチレン系モノマー、並びに、窒素含有ビニルモノマー、カルボキシル基含有モノマー、水酸基含有モノマー、アクリル酸エステルモノマー及びメタアクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーを用いて合成された共重合体は、該トナー中にトナーの質量を基準として0.1〜20質量%含有されていることが好ましい。
スチレン系モノマー、並びに、窒素含有ビニルモノマー、カルボキシル基含有モノマー、水酸基含有モノマー、アクリル酸エステルモノマー及びメタアクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーを用いて合成された共重合体とのグラフト重合に用いられる上記ポリオレフィンは、示差走査熱量測定(DSC)装置によって測定される昇温時の吸熱曲線において、最大吸熱ピークの極大値が70〜130℃にあることが離型性の観点から好ましい。なお、ポリオレフィンとしては、ポリエチレン、ポリプロピレン、エチレン/プロピレン共重合体、エチレン/1−ブテン共重合体、プロピレン/1−ヘキセン共重合体が好適に例示できる。また、本発明においては、ポリマー構造がポリオレフィンの構造を有していれば良く、モノマーが必ずしもオレフィン構造を有している必要はない。
また、スチレン系モノマー、並びに、窒素含有ビニルモノマー、カルボキシル基含有モノマー、水酸基含有モノマー、アクリル酸エステルモノマー及びメタアクリル酸エステルモノマーから選ばれる1種又は2種以上のモノマーを用いて合成された共重合体の含有量(W1)、及びポリオレフィンの含有量(W2)の質量比としては、0.01≦W2/W1≦1、を満足することが好ましい。
【0021】
本発明において、必要に応じてトナーは着色剤を含有することが可能であり、以下のものが挙げられる。なお、着色剤には、顔料を単独で使用してもかまわないが、染料と顔料とを併用してその鮮明度を向上させた方がフルカラー画像の画質の点からより好ましい。
黒色着色剤としては、カーボンブラック;磁性体;イエロー着色剤とマゼンタ着色剤及びシアン着色剤とを用いて黒色に調色したものが挙げられる。
マゼンタトナー用着色顔料としては、以下のものが挙げられる。C.I.ピグメントレッド1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、21、22、23、30、31、32、37、38、39、40、41、48:1、48:2、48:3、48:4、48:5、49、50、51、52、53、54、55、57:1、58、60、63、64、68、81:1、81:2、81:3、81:4、81:5、83、87、88、89、90、112、114、122、123、146、147、150、163、184、185、202、206、207、209、238、269;C.I.ピグメントバイオレット19;C.I.バットレッド1、2、10、13、15、23、29、35。
マゼンタトナー用染料としては、以下のものが挙げられる。C.I.ソルベントレッド1、3、8、23、24、25、27、30、49、81、82、83、84、100、109、121;C.I.ディスパースレッド9;C.I.ソルベントバイオレット8、13、14、21、27;C.I.ディスパーバイオレット1の如き油溶染料、C.I.ベーシックレッド1、2、9、12、13、14、15、17、18、22、23、24、27、29、32、34、35、36、37、38、39、40;C.I.ベーシックバイオレット1、3、7、10、14、15、21、25、26、27、28の如き塩基性染料。
シアントナー用着色顔料としては、以下のものが挙げられる。C.I.ピグメントブルー2、3、15:3、15:4、16、17;C.I.バットブルー6;C.I.アシッドブルー45、フタロシアニン骨格にフタルイミドメチル基を1〜5個置換した銅フタロシアニン顔料。シアン用着色染料としては、C.I.ソルベントブルー70がある。
イエロー用着色顔料としては、以下のものが挙げられる。C.I.ピグメントイエロー1、2、3、4、5、6、7、10、11、12、13、14、15、16、17、23、62、65、73、74、83、93、94、95、97、109、110、111、120、127、128、129、147、151、154、155、168、174、175、176、180、181、185;C.I.バットイエロー1、3、20。イエロー用着色染料としては、C.I.ソルベントイエロー162がある。
着色剤の使用量は、結着樹脂100質量部に対して、好ましくは0.1質量部以上30質量部以下であり、より好ましくは0.5質量部以上20質量部以下である。
【0022】
本発明のトナーは、結着樹脂およびワックスを少なくとも含有するトナー粒子に、流動性向上のため、外添剤として無機微粉体が添加されているトナーである。外添剤としては、シリカ、酸化チタン、酸化アルミニウムの如き無機微粉体が好ましい。無機微粉体は、シラン化合物、シリコーンオイル又はそれらの混合物の如き疎水化剤で疎水化されていることが好ましい。
外添剤としては、個数平均粒径が5nm以上300nm以下の外添剤を用いることができるが、耐ストレス性を向上させるためには、80nm以上300nm以下の外添剤を用いることがより好ましい。個数平均粒径が80nm以上300nm以下の外添剤は、耐久時等にトナー表面に埋め込まれたとしても、粒径が大きいために流動性の低下を低減することができる。このため、耐久時の画像濃度の低下を抑制できる。
外添剤は、トナー粒子100質量部に対して0.1質量部以上5.0質量部以下使用されることが好ましい。
トナー粒子と外添剤との混合は、ヘンシェルミキサーの如き公知の混合機を用いることができる。
【0023】
上記トナー粒子及びトナーの製造方法についても、特に限定されず、従来公知の製造方法を用いることができる。
ここでは、粉砕法を用いたトナー製造の手順について説明する。
原料混合工程では、トナー粒子を構成する材料として、結着樹脂及びワックス、並びに必要に応じて、ワックス偏在制御樹脂、着色剤及び荷電制御剤等の他の成分を所定量秤量して配合し、混合する。混合装置の一例としては、ダブルコン・ミキサー、V型ミキサー、ドラム型ミキサー、スーパーミキサー、ヘンシェルミキサー、ナウタミキサ、メカノハイブリッド(日本コークス工業社製)が挙げられる。
次に、混合した材料を溶融混練して、結着樹脂中にワックス等を分散させる。その溶融混練工程では、加圧ニーダー、バンバリィミキサーの如きバッチ式練り機や、連続式の練り機を用いることができる。連続生産できる優位性から、1軸又は2軸押出機が主流となっている。例えば、KTK型2軸押出機(神戸製鋼所社製)、TEM型2軸押出機(東芝機械社製)、PCM混練機(池貝鉄工製)、2軸押出機(ケイ・シー・ケイ社製)、コ・ニーダー(ブス社製)、ニーデックス(日本コークス工業社製)が挙げられる。
更に、溶融混練することによって得られる樹脂組成物は、2本ロール等で圧延され、冷却工程で水などによって冷却してもよい。
ついで、樹脂組成物の冷却物は、粉砕工程で所望の粒径にまで粉砕される。粉砕工程では、クラッシャー、ハンマーミル、フェザーミルの如き粉砕機で粗粉砕した後、更に、クリプトロンシステム(川崎重工業社製)、スーパーローター(日清エンジニアリング社製)、ターボ・ミル(ターボ工業製)やエアージェット方式による微粉砕機で微粉砕する。
その後、必要に応じて慣性分級方式のエルボージェット(日鉄鉱業社製)、遠心力分級方式のターボプレックス(ホソカワミクロン社製)、TSPセパレータ(ホソカワミクロ
ン社製)、ファカルティ(ホソカワミクロン社製)の如き分級機や篩分機を用いて分級し、トナー粒子を得る。
本発明においては、粉砕後、または、分級後に、熱風を用いてトナーの表面処理を行うことが、トナー表面近傍のワックス比率をコントロールし、耐久性能と画像の面内のグロス均一性を両立させるために、好ましい。
上記熱風を用いた表面処理としては、熱風でトナーの表面を溶融状態にすることができ、かつ、熱風で処理されたトナーを冷風で冷却できる方式を採用できる手段であればどのようなものでもかまわない。例えば、ハイブリタイゼーションシステム(奈良機械製作所製)、メカノフージョンシステム(ホソカワミクロン社製)、ファカルティ(ホソカワミクロン社製)、メテオレインボー MR Type(日本ニューマチック社製)などを用いることが可能である。
また、本発明においては、上記微粉砕物を得た後、熱風により表面処理を行い、続いて分級をすることにより得られたトナーであることが好ましい。若しくは、予め分級したものを、熱風により表面処理を行っても良い。
ここで、上記熱風を用いた表面処理の方法の概略を、図1を用いて説明するが、これに限定されるものではない。図1は本発明で用いた表面処理装置の一例を示した断面図である。具体的には、上記微粉砕物(ここでは、トナー粒子ともいう)を得た後、当該表面処理装置に供給する。そして、トナー粒子供給口(100)から供給されたトナー粒子(114)は、高圧エア供給ノズル(115)から噴射されるインジェクションエアにより加速され、その下方にある気流噴射部材(102)へ向かう。気流噴射部材(102)からは拡散エアが噴射され、この拡散エアによりトナー粒子が外側方向へ拡散する。この時、インジェクションエアの流量と拡散エアの流量とを調節することにより、トナーの拡散状態をコントロールすることができる。また、トナー粒子の融着防止を目的として、トナー粒子供給口(100)の外周、表面処理装置外周及び移送配管(116)の外周には冷却ジャケット(106)が設けられている。尚、該冷却ジャケットには冷却水(好ましくはエチレングリコール等の不凍液)を通水することが好ましい。一方、拡散エアにより拡散したトナー粒子は、熱風供給口(101)から供給された熱風により、トナー粒子の表面が処理される。この時、熱風の吐出温度は100℃以上、300℃以下であることが好ましく、150℃以上、250℃以下であることがより好ましい。熱風の温度が100℃未満の場合にはトナー粒子の表面を溶融状態にすることができない場合がある。また、300℃を超える場合には溶融状態が進みすぎる事で、ワックスを過度にトナー表面に偏析させる場合や、トナー粒子同士の合一に起因する、トナー粒子の粗大化や融着が生じる場合がある。
熱風により表面が処理されたトナー粒子は、装置上部外周に設けた冷風供給口(103)から供給される冷風により冷却される。この時、装置内の温度分布の制御、トナー粒子の表面状態をコントロールする目的で、装置の本体側面に設けた第二の冷風供給口(104)から冷風を導入することが好ましい。第二の冷風供給口(104)の出口はスリット形状、ルーバー形状、多孔板形状、メッシュ形状等を用いる事ができ、導入方向は中心方向へ水平、装置壁面に沿う方向が、目的に応じて選択可能である。
この時、上記冷風温度は−50℃以上、10℃以下であることが好ましく、−40℃以上、8℃以下であることがより好ましい。また、上記冷風は除湿された冷風であることが好ましい。具体的には、冷風の絶対水分量が5g/m3以下であることが好ましい。更に好ましくは、3g/m3以下である。
これらの冷風温度が−50℃未満の場合には装置内の温度が下がりすぎてしまい、本来の目的である熱による処理が十分に為されず、トナーの表面を溶融状態にすることができない場合がある。また、10℃を超える場合には、装置内における熱風ゾーンの制御が不十分になり、表面処理時にワックスを過度にトナー表面に偏析させることがある。
その後、冷却されたトナー粒子は、ブロワーで吸引され、移送配管(116)を通じて、サイクロン等で回収される。
【0024】
本発明のトナーは、トナー単独で構成される一成分系の現像剤として使用することも可能であるが、トナーと磁性キャリアを含む二成分系現像剤としても用いることができる。すなわち、本発明の二成分系現像剤は、本発明のトナーと磁性キャリアを含む二成分系現像剤である。ここで、磁性キャリアは、特に限定されず、公知の磁性キャリアが使用できる。鉄、銅、亜鉛、ニッケル、コバルト、マンガン、及び、クロム元素から選ばれる元素単独または複合のフェライトで構成されるキャリアが例示できる。また、磁性体が樹脂中に分散されている磁性体含有樹脂キャリアコアの表面に樹脂成分を含有する磁性体含有樹脂キャリアや、多孔質磁性体(多孔質フェライトを含む)に樹脂が含浸された樹脂含浸キャリアなども、上記磁性キャリアとして挙げられる。
しかしながら、二成分系現像剤が、交流インピーダンス測定により得られるインピーダンスZの周波数依存特性Z(ω)を、下記(2)式で表されるフィッティング関数により、フィッティングしたときのパラメータαが、1000V/cmの電界下において、0.70以上0.95以下であることを特徴とする磁性キャリア、及び、本発明のトナーを含有する二成分系現像剤である場合、トナーが本発明のトナーの様に、トナー表面近傍の結着樹脂に対するワックスの存在比率が高いトナーであっても、現像性の更なる向上が図れるため好ましい。
【0025】
【数1】
【0026】
そのメカニズムについて、発明者らは以下のように考えている。本発明のトナーは、トナー表面近傍の結着樹脂に対するワックスの存在比率が高いトナーである。このようなトナーを用いた場合、トナー表面近傍に存在するワックスの影響でトナーと磁性キャリアの付着力が高まり、トナーが磁性キャリアから像担時体へ現像されにくくなる場合がある。この現象を抑制するためには、トナーが現像された後の磁性キャリア表面に残ったカウンター電荷を、すばやく逃がす必要がある。これにより、トナーを引き戻す力が弱くなり現像性が向上する。例えば、磁性キャリアの抵抗を低くすることで、磁性キャリア同士を通じて、現像剤担持体へカウンター電荷を逃がす手法が従来から知られている。しかし、単に抵抗の低い磁性キャリアを用いた場合には、ハーフトーン部などトナーが薄層で現像されるような場合、抵抗の低い磁性キャリアにより潜像電位が乱されガサツキを生じてしまう場合がある。そこで、発明者らは鋭意検討の結果、現像性向上のためには、抵抗をさげカウンター電荷を逃がすのではなく、磁性キャリアの電気的特性を制御することで、現像性をさらに向上できることを見出した。
具体的には、磁性キャリアの交流インピーダンス測定により得られるインピーダンスZの周波数依存特性Z(ω)を、上記式(2)で書かれるフィッティング関数によりフィッティングしたときのパラメータαを、1000V/cmの電界下において0.70以上0.95以下にすることである。
上記パラメータαは、磁性キャリアの交流インピーダンス測定により、複素インピーダンスの周波数特性を測定し、下記式(3)式で表されるCole−Coleの式によるフィッティングから算出でき、磁性キャリアの持つ導電の時定数分布の度合いを表すことができる。該パラメータαは、0≦α≦1の範囲をとり、導電の時定数分布の広がりが大きい程、小さな値となる。
【0027】
【数2】
【0028】
本発明において、パラメータα、1000V/cmの電界下において、0.70以上0.95以下にすることで、一粒子の磁性キャリア内部に極端に電荷移動の時定数の小さい部分から極端に電荷移動の時定数の大きい部分が存在することになる。
このような磁性キャリアであれば、外部電界が印加された時に、磁性キャリア内部の電気伝導が局所的に抑制され大きな分極が形成される。分極した磁性キャリアは、磁性キャリアの外部電界を歪めるため、磁性キャリアに付着したトナーが受ける電界は強められることになる。その結果、磁性キャリアの抵抗を低めなくとも、トナーが磁性キャリアから飛翔しやすくなる。
このようにパラメータαを0.70以上0.95以下にコントロールすることで、磁性キャリアの抵抗を下げることなく、トナーにかかる電界を強め、現像性を高めることが可能になる。
パラメータαを上記範囲にコントロールするためには、磁性キャリアの内部構造を均一なものではなく不均一にすることが必要である。磁性キャリアの内部構造を不均一にすることで、一粒子の磁性キャリア内部に極端に電荷移動の時定数の小さい部分から極端に電荷移動の時定数の大きい部分が存在させることができる。具体的には、多孔質磁性コア粒子に樹脂を含有させた磁性キャリアを用いることが好ましい。当該多孔質磁性コア粒子の材質としては、フェライトが好ましい。そして、多孔質磁性コア粒子を構成している結晶粒子同士のつながりの状態に、一粒子のキャリア内部で十分に「ばらつき」を持たせることが特に好ましい。
そのためには、後述する多孔質磁性コア粒子の製造過程で、仮焼フェライト微粉砕品の体積基準の50%粒径(D50)を、0.5μm以上5.0μm以下、また、体積基準の90%粒径(D90)を、3.0μm以上10.0μm以下とすることが好ましい。
仮焼フェライト微粉砕品を上記の粒径にするために、例えば、ボールミルやビーズミルでは用いるボールやビーズの素材、粒径、運転時間を制御することが好ましい。具体的には、仮焼フェライト微粉砕品の粒径を小さくするためには、比重の重いボールを用いたり、粉砕時間を長くすればよい。また、仮焼フェライト微粉砕品の粒度分布を広くするためには、比重の重いボールやビーズを用い、粉砕時間を短くすることで得ることができる。また、粒径の異なる複数の仮焼フェライトを混合することでも分布の広い仮焼フェライトを得ることができる。このようにして、広い粒度分布の仮焼フェライト微粉砕品を用いることで、パラメータαが0.70以上0.95以下である磁性キャリアを得ることができる。
【0029】
以下、多孔質磁性コア粒子としてフェライトを用いた磁性キャリアの製造方法を例示する。ここで、フェライトとは次式で表される焼結体である。
(M12O)x(M2O)y(Fe2O3)z
(式中、M1は1価、M2は2価の金属であり、x+y+z=1.0とした時、x及びyは、それぞれ0≦(x,y)≦0.8であり、zは、0.2<z<1.0である。)
該式中において、M1及びM2としては、Li、Fe、Mn、Mg、Sr、Cu、Zn、Ni、Co、Caからなる群から選ばれる1種類以上の金属原子を用いることが好ましい。
具体的には、Li系フェライト(例えば、(Li2O)a(Fe2O3)b(0.0<
a<0.4,0.6≦b<1.0、a+b=1)、(Li2O)a(SrO)b(Fe2O3)c(0.0<a<0.4、0.0<b<0.2、0.4≦c<1.0、a+b+c=1));Mn系フェライト(例えば、(MnO)a(Fe2O3)b(0.0<a<0.5、0.5≦b<1.0、a+b=1));Mn−Mg系フェライト(例えば、(MnO)a(MgO)b(Fe2O3)c(0.0<a<0.5、0.0<b<0.5、0.5≦c<1.0、a+b+c=1));Mn−Mg−Sr系フェライト(例えば、(MnO)a(MgO)b(SrO)c(Fe2O3)d(0.0<a<0.5、0.0<b<0.5、0.0<c<0.5、0.5≦d<1.0、a+b+c+d=1);Cu−Zn系フェライト(例えば、(CuO)a(ZnO)b(Fe2O3)c(0.0<a<0.5、0.0<b<0.5、0.5≦c<1.0、a+b+c=1)が挙げられる。上記フェライトは微量の他の金属を含有していてもよい。
結晶の成長速度を容易にコントロールでき、多孔質磁性コア粒子の細孔径分布を好適にコントロールできる観点から、Mn元素を含有する、Mn系フェライト、Mn−Mg系フェライト、Mn−Mg−Sr系フェライトが好ましい。
【0030】
上記多孔質磁性コア粒子は、以下のような工程で製造することができる。
<工程1(秤量・混合工程)>
フェライトの原料を、秤量し、混合する。フェライトの原料としては、例えば以下のものが挙げられる。Li、Fe、Zn、Ni、Mn、Mg、Co、Cu、Ba、Sr、Y、Ca、Si、V、Bi、In、Ta、Zr、B、Mo、Na、Sn、Ti、Cr、Al、希土類金属の金属粒子、酸化物、水酸化物、シュウ酸塩、炭酸塩。
粉砕・混合する装置としては、以下のものが挙げられる。ボールミル、遊星ミル、ジオットミル、振動ミル。フェライトの原料に水を添加しスラリーとした状態で混合する、湿式のボールミルが混合性と多孔質構造の形成のためには好ましい。具体的には、ボールミル中に、秤量したフェライト原料及びボール、湿式の場合は更に水を入れ、0.1時間以上20.0時間以下、粉砕・混合する。
<工程2(仮焼成工程)>
粉砕・混合したフェライト原料を、スプレードライヤーを用いて、造粒・乾燥した後、大気中で焼成温度700℃以上1000℃以下にして、0.5時間以上5.0時間以下仮焼成し、原料をフェライトにする。温度1000℃を超えると焼結が進み、多孔質にするための粒径まで粉砕することができにくくなる場合がある。焼成する装置としては、バーナー式焼却炉、ロータリー式焼却炉、電気炉が挙げられる。
<工程3(粉砕工程)>
工程2で作製した仮焼フェライトを粉砕機で微粉砕する。粉砕機としては、クラッシャーやハンマーミル、ボールミル、ビーズミル、遊星ミル、ジオットミルが挙げられる。
上述のように、仮焼フェライト微粉砕品の体積基準の50%粒径(D50)は、0.5μm以上5.0μm以下、体積基準の90%粒径(D90)は3.0μm以上10.0μm以下とすることが好ましい。こうすることで、多孔質磁性コアの結晶粒子(グレイン)同士の繋がりの状態に、一粒子の磁性キャリア内部で十分に「ばらつき」を持たせることができ、パラメータαを1000V/cmの電界下において、0.70以上0.95以下にコントロールすることができる。
仮焼フェライト微粉砕品を上記の粒径にするために、例えば、ボールミルやビーズミルでは用いるボールやビーズの素材、粒径、運転時間を制御することが好ましい。具体的には、仮焼フェライト微粉砕品の粒径を小さくするためには、比重の重いボールを用いたり、粉砕時間を長くすればよい。また、仮焼フェライト微粉砕品の粒度分布を広くするためには、比重の重いボールやビーズを用い、粉砕時間を短くすることで得ることができる。また、粒径の異なる複数の仮焼フェライト微粉砕品を混合することでも分布の広い仮焼フェライト微粉砕品を得ることができる。
ボールやビーズの素材としては、所望の粒径・分布が得られば、特に限定されない。例えば、以下のものが挙げられる。ソーダガラス(比重2.5g/cm3)、ソーダレスガ
ラス(比重2.6g/cm3)、高比重ガラス(比重2.7g/cm3)等のガラスや、石英(比重2.2g/cm3)、チタニア(比重3.9g/cm3)、窒化ケイ素(比重3.2g/cm3)、アルミナ(比重3.6g/cm3)、ジルコニア(比重6.0g/cm3)、スチール(比重7.9g/cm3)、ステンレス(比重8.0g/cm3)。中でも、アルミナ、ジルコニア、ステンレスは、耐磨耗性に優れているために好ましい。
ボールやビーズの粒径は、所望の粒径・分布が得られれば、特に限定されない。例えば、ボールとしては、直径(φ)が5mm以上60mm以下のものが好適に用いられる。また、ビーズとしては直径(φ)が0.03mm以上5mm未満のものが好適に用いられる。また、ボールミルやビーズミルは、粉砕効率が高く仮焼フェライト微粉砕品の粒度分布コントロールが容易になるため、乾式粉砕よりも、水を添加しスラリーとした状態で粉砕を行う、湿式粉砕の方がより好ましい。
<工程4(造粒工程)>
得られた仮焼フェライト微粉砕品に対し、水、バインダーと、必要に応じて、孔調整剤を加える。工程3において、湿式で粉砕した場合は、フェライトスラリー中に含まれている水も考慮し、バインダーと必要に応じて孔調整剤を加えることが好ましい。多孔質の程度をコントロールするため、スラリーの固形分濃度を50質量%以上80質量%以下にして、造粒することが好ましい。得られたフェライトスラリーを、噴霧乾燥機を用い、温度100℃以上200℃以下の加温雰囲気下で、乾燥・造粒する。
噴霧乾燥機としては、スプレードライヤーがフェライトスラリーの粒径を所望に調整できるため、好適に使用できる。多孔質磁性コア粒子の粒径は、スプレードライヤーに用いられるディスクの回転数、噴霧量を適宜選択してコントロールできる。
<工程5(本焼成工程)>
次に、得られた造粒品を温度800℃以上1300℃以下で1時間以上24時間以下焼成する。焼成温度が1000℃以上1200℃以下であれば、上記磁性キャリアのパラメータαをコントロールしやすいことからより好ましい。
<工程6(選別工程)>
得られた焼成物を解砕した後に、必要に応じて、分級や篩で篩分して粗大粒子や微粒子を除去してもよい。当該選別工程を経て得られた多孔質磁性コア粒子の体積基準の50%粒径(D50)は、18.0μm以上68.0μm以下であることが、画像へのキャリア付着とガサツキの抑制のためより望ましい。
【0031】
次に、得られた多孔質磁性コア粒子に樹脂を充填し、磁性キャリアを得る。ここで、樹脂を充填した後にさらに樹脂で磁性キャリアの表面を被覆すれば、磁性キャリアとしての物理的強度を高めることが可能となるため好ましい。得られた磁性キャリアの体積基準の50%粒径(D50)は、20.0μm以上70.0μm以下であることが、ドラム上へのキャリア付着抑制と、画質の観点より好ましい。
上記多孔質磁性コア粒子に樹脂を充填する方法は、多孔質磁性コア粒子の奥の孔まで樹脂を充填する方法と、多孔質磁性コア粒子の表面の孔のみに樹脂を充填する方法の2つがある。具体的な充填方法は、特に限定されないが、樹脂と溶剤を混合した樹脂溶液を多孔質磁性コア粒子の孔へ充填させる方法が好ましい。
上記樹脂溶液における樹脂固形分の量は、好ましくは5質量%以上30質量%以下であり、より好ましくは6質量%以上20質量%以下である。30質量%より樹脂量の多い樹脂溶液を用いると粘度が高いため多孔質磁性コア粒子の孔に樹脂溶液を均一に充填しにくい。また、5質量%未満であると樹脂量が少なく、多孔質磁性コア粒子へ均一な充填ができず、摩擦帯電量の部分的なバラツキの要因となる場合がある。
上記多孔質磁性コア粒子の孔に充填する樹脂としては特に限定されず、熱可塑性樹脂、熱硬化性樹脂のどちらを用いてもかまわない。しかしながら、多孔質磁性コア粒子に対する親和性が高いものであることが好ましい。親和性が高い樹脂を用いた場合には、多孔質磁性コア粒子の孔への樹脂の充填時に、同時に多孔質磁性コア粒子表面も樹脂で覆うことが容易になる。
具体的には、シリコーン樹脂または変性シリコーン樹脂が、多孔質磁性コア粒子に対する親和性が高いため好ましい。
市販品として、以下のものが挙げられる。シリコーン樹脂では、信越化学社製のKR271、KR255、KR152、東レ・ダウコーニング社製のSR2400、SR2405、SR2410、SR2411。変性シリコーン樹脂では、信越化学社製のKR206(アルキッド変性)、KR5208(アクリル変性)、ES1001N(エポキシ変性)、KR305(ウレタン変性)、東レ・ダウコーニング社製のSR2115(エポキシ変性)、SR2110(アルキッド変性)。
多孔質磁性コア粒子内の孔に樹脂を充填させる方法としては、上記樹脂を溶剤に希釈し、これを多孔質磁性コア粒子内の孔に添加する方法が採用できる。ここで用いられる溶剤は、樹脂を溶解できるものであれば特に限定されない。有機溶剤に可溶な樹脂である場合は、有機溶剤として、トルエン、キシレン、セルソルブブチルアセテート、メチルエチルケトン、メチルイソブチルケトン、メタノールが挙げられる。また、水溶性の樹脂またはエマルジョンタイプの樹脂である場合には、溶剤として水を用いればよい。多孔質磁性コア粒子の孔に、樹脂を充填する方法としては、浸漬法、スプレー法、ハケ塗り法、及び流動床の如き塗布方法により多孔質磁性コア粒子を樹脂溶液に含浸させ、その後、溶剤を揮発させる方法が挙げられる。
【0032】
本発明の二成分系現像剤において、トナーと磁性キャリアの混合比率は、磁性キャリア100質量部に対してトナーを2質量部以上35質量部以下とすることが好ましく、4質量部以上25質量部以下とすることがより好ましい。トナーを上記範囲とすることで、高い画像濃度を達成し、かつ、トナーの飛散を低減することができる。
【0033】
以下、トナー及び磁性キャリアの各種物性の測定法について説明する。
<P1及びP2の算出方法>
FT−IRスペクトルは、ユニバーサルATR測定アクセサリー(Universal
ATR Sampling Accessory)を装着したフーリエ変換赤外分光分析装置(Spectrum One;PerkinElmer社製)を用い、ATR法で測定する。具体的な測定手順と、P1、P2及びP1をP2で除した[P1/P2]の算出方法は以下の通りである。
赤外光の入射角は45°に設定する。ATR結晶としては、GeのATR結晶(屈折率=4.0)、KRS5のATR結晶(屈折率=2.4)を用いる。その他の条件は以下の通りである。
Range
Start :4000cm−1
End:600cm−1(GeのATR結晶)
400cm−1(KRS5のATR結晶)
Duration
Scan number:16
Resolution:4.00cm−1
Advanced :CO2/H2O補正あり
[P1の算出方法]
(1)GeのATR結晶(屈折率=4.0)を装置に装着する。
(2)Scan typeをBackground、UnitsをEGYに設定し、バックグラウンドを測定する。
(3)Scan typeをSample、UnitsをAに設定する。
(4)トナーをATR結晶の上に、0.01g精秤する。
(5)圧力アームでサンプルを加圧する。(Force Gaugeは90)
(6)サンプルを測定する。
(7)得られたFT−IRスペクトルを、Automatic Correctionで
ベースライン補正をする。
(8)2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pa1)
(9)3050cm−1と2600cm−1の吸収ピーク強度の平均値を算出する。(Pa2)
(10)Pa1−Pa2=Paとする。当該Paを2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度と規定する。
(11)1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pb1)
(12)1763cm−1と1630cm−1の吸収ピーク強度の平均値を算出する(Pb2)
(13)Pb1−Pb2=Pbとする。当該Pbを1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度と規定する。
(14)Pa/Pb=P1とする。
図2にATR結晶としてGeを用い測定したFT−IRスペクトルの一例を示す。
[P2の算出方法]
(1)KRS5のATR結晶(屈折率=2.4)を装置に装着する。
(2)トナーをATR結晶の上に、0.01g精秤する。
(3)圧力アームでサンプルを加圧する。(Force Gaugeは90)
(4)サンプルを測定する。
(5)得られたFT−IRスペクトルを、Automatic Correctionでベースライン補正をする。
(6)2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pc1)
(7)3050cm−1と2600cm−1の吸収ピーク強度の平均値を算出する。(Pc2)
(8)Pc1−Pc2=Pcとする。当該Pcを2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度と規定する。
(9)1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pd1)
(10)1763cm−1と1630cm−1の吸収ピーク強度の平均値を算出する(Pd2)
(11)Pd1−Pd2=Pdとする。当該Pdを1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度と規定する。
(12)Pc/Pd=P2とする。
[P1/P2の算出方法]
上記のようにして求めたP1とP2を用い、P1/P2を算出する。
【0034】
<樹脂の軟化点の測定方法>
樹脂の軟化点の測定は、定荷重押し出し方式の細管式レオメータ「流動特性評価装置 フローテスターCFT−500D」(島津製作所社製)を用い、装置付属のマニュアルに従って行う。本装置では、測定試料の上部からピストンによって一定荷重を加えつつ、シリンダに充填した測定試料を昇温させて溶融し、シリンダ底部のダイから溶融された測定試料を押し出し、この際のピストン降下量と温度との関係を示す流動曲線を得ることができる。
本発明においては、「流動特性評価装置 フローテスターCFT−500D」に付属のマニュアルに記載の「1/2法における溶融温度」を軟化点とする。尚、1/2法における溶融温度とは、次のようにして算出されたものである。まず、流出が終了した時点におけるピストンの降下量Smaxと、流出が開始した時点におけるピストンの降下量Sminとの差の1/2を求める(これをXとする。X=(Smax−Smin)/2)。そし
て、流動曲線においてピストンの降下量がXとSminの和と、交わるときの流動曲線の温度が、1/2法における溶融温度である。
測定試料には、1.0gの樹脂を、25℃の環境下で、錠剤成型圧縮機(NT−100H、エヌピーエーシステム社製)を用いて10MPaで、60秒間圧縮成型し、直径8mmの円柱状としたものを用いる。一方、CFT−500Dの測定条件は、以下の通りである。
試験モード:昇温法
開始温度:40℃
到達温度:200℃
測定間隔:1.0℃
昇温速度:4.0℃/min
ピストン断面積:1.000cm2
試験荷重(ピストン荷重):10.0kgf(0.9807MPa)
予熱時間:300秒
ダイの穴の直径:1.0mm
ダイの長さ:1.0mm
【0035】
<トナーの平均円形度の算出方法>
トナーの平均円形度は、フロー式粒子像分析装置「FPIA−3000」(シスメックス社製)によって、校正作業時の測定及び解析条件で測定する。
フロー式粒子像分析装置「FPIA−3000」(シスメックス社製)の測定原理は、流れている粒子を静止画像として撮像し、画像解析を行うというものである。試料チャンバーへ加えられた試料は、試料吸引シリンジによって、フラットシースフローセルに送り込まれる。フラットシースフローに送り込まれた試料は、シース液に挟まれて扁平な流れを形成する。フラットシースフローセル内を通過する試料に対しては、1/60秒間隔でストロボ光が照射されており、流れている粒子を静止画像として撮影することが可能である。また、扁平な流れであるため、焦点の合った状態で撮像される。粒子像はCCDカメラで撮像され、撮像された画像は512×512画素の画像処理解像度(一画素あたり0.37×0.37μm)で画像処理され、各粒子像の輪郭抽出を行い、粒子像の投影面積Sや周囲長L等が計測される。
次に、上記面積Sと周囲長Lを用いて円相当径と円形度を求める。円相当径とは、粒子像の投影面積と同じ面積を持つ円の直径のことであり、円形度Cは、円相当径から求めた円の周囲長を粒子投影像の周囲長で割った値として定義され、次式で算出される。
円形度C=2×(π×S)1/2/L
粒子像が円形の時に円形度は1.000になり、粒子像外周の凹凸の程度が大きくなればなるほど円形度は小さい値になる。各粒子の円形度を算出後、円形度0.200乃至1.000の範囲を800分割し、得られた円形度の相加平均値を算出し、その値を平均円形度とする。
具体的な測定方法は、以下の通りである。まず、ガラス製の容器中に予め不純固形物などを除去したイオン交換水20mlを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を0.2ml加える。更に測定試料を0.02g加え、超音波分散器を用いて2分間分散処理を行い、測定用の分散液とする。その際、分散液の温度が10℃以上40℃以下となる様に適宜冷却する。超音波分散器としては、発振周波数50kHz、電気的出力150Wの卓上型の超音波洗浄器分散器(例えば「VS−150」(ヴェルヴォクリーア社製))を用い、水槽内には所定量のイオン交換水を入れ、この水槽中に前記コンタミノンNを2ml添加する。
測定には、標準対物レンズ(10倍)を搭載した前記フロー式粒子像分析装置を用い、シース液にはパーティクルシース「PSE−900A」(シスメックス社製)を使用する
。前記手順に従い調製した分散液を前記フロー式粒子像分析装置に導入し、HPF測定モードで、トータルカウントモードにて3000個のトナー粒子を計測する。そして、粒子解析時の2値化閾値を85%とし、解析粒子径を円相当径1.985μm以上、39.69μm未満に限定し、トナー粒子の平均円形度を求める。
測定にあたっては、測定開始前に標準ラテックス粒子(例えば、Duke Scientific社製の「RESEARCH AND TEST PARTICLES Latex Microsphere Suspensions 5200A」をイオン交換水で希釈)を用いて自動焦点調整を行う。その後、測定開始から2時間毎に焦点調整を実施することが好ましい。
なお、本願実施例では、シスメックス社による校正作業が行われた、シスメックス社が発行する校正証明書の発行を受けたフロー式粒子像分析装置を使用する。解析粒子径を円相当径1.985μm以上、39.69μm未満に限定した以外は、校正証明を受けた時の測定及び解析条件で測定を行う。
【0036】
<トナーの重量平均粒径(D4)の測定方法>
トナーの重量平均粒径(D4)は、以下のようにして算出する。測定装置としては、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer 3」(登録商標、ベックマン・コールター社製)を用いる。測定条件の設定及び測定データの解析は、付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いる。尚、測定は実効測定チャンネル数2万5千チャンネルで行う。
測定に使用する電解水溶液は、特級塩化ナトリウムをイオン交換水に溶解して濃度が1質量%となるようにしたもの、例えば、「ISOTON II」(ベックマン・コールター社製)が使用できる。
尚、測定、解析を行う前に、以下のように専用ソフトの設定を行う。
専用ソフトの「標準測定方法(SOM)を変更」画面において、コントロールモードの総カウント数を50000粒子に設定し、測定回数を1回、Kd値は「標準粒子10.0μm」(ベックマン・コールター社製)を用いて得られた値を設定する。「閾値/ノイズレベルの測定ボタン」を押すことで、閾値とノイズレベルを自動設定する。また、カレントを1600μAに、ゲインを2に、電解液をISOTON IIに設定し、「測定後のアパーチャーチューブのフラッシュ」にチェックを入れる。
専用ソフトの「パルスから粒径への変換設定」画面において、ビン間隔を対数粒径に、粒径ビンを256粒径ビンに、粒径範囲を2μmから60μmまでに設定する。
具体的な測定法は以下の通りである。
(1)Multisizer 3専用のガラス製250ml丸底ビーカーに前記電解水溶液200mlを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行う。そして、専用ソフトの「アパーチャーのフラッシュ」機能により、アパーチャーチューブ内の汚れと気泡を除去しておく。
(2)ガラス製の100ml平底ビーカーに前記電解水溶液30mlを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を0.3ml加える。
(3)発振周波数50kHzの発振器2個を、位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispersion System Tetora150」(日科機バイオス社製)を準備する。超音波分散器の水槽内に3.3lのイオン交換水を入れ、この水槽中にコンタミノンNを2ml添加する。(4)前記(2)のビーカーを前記超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させる。そして、ビーカー内の電解水溶液液面の共振状態が最大となるようにビーカーの高さ位置を調整する。
(5)前記(4)のビーカー内の電解水溶液に超音波を照射した状態で、トナー10mgを少量ずつ前記電解水溶液に添加し、分散させる。そして、さらに60秒間超音波分散処理を継続する。尚、超音波分散にあたっては、水槽の水温が10℃以上40℃以下となる様に適宜調節する。
(6)サンプルスタンド内に設置した前記(1)の丸底ビーカーに、ピペットを用いてトナーを分散した前記(5)の電解質水溶液を滴下し、測定濃度が5%となるように調整する。そして、測定粒子数が50000個になるまで測定を行う。
(7)測定データを装置付属の前記専用ソフトにて解析を行い、重量平均粒径(D4)を算出する。尚、専用ソフトでグラフ/体積%と設定したときの、「分析/体積統計値(算術平均)」画面の「平均径」が重量平均粒径(D4)である。
【0037】
<樹脂のピーク分子量(Mp)、数平均分子量(Mn)、重量平均分子量(Mw)の測定方法>
ピーク分子量(Mp)、数平均分子量(Mn)、重量平均分子量(Mw)は、ゲルパーミエーションクロマトグラフィー(GPC)により、以下のようにして測定する。
まず、室温で24時間かけて、試料(樹脂)をテトラヒドロフラン(THF)に溶解する。そして、得られた溶液を、ポア径が0.2μmの耐溶剤性メンブランフィルター「マエショリディスク」(東ソー社製)で濾過してサンプル溶液を得る。尚、サンプル溶液は、THFに可溶な成分の濃度が0.8質量%となるように調整する。このサンプル溶液を用いて、以下の条件で測定する。
装置 :HLC8120 GPC(検出器:RI)(東ソー社製)
カラム :Shodex KF−801、802、803、804、805、
806、807の7連(昭和電工社製)
溶離液 :テトラヒドロフラン(THF)
流速 :1.0ml/min
オーブン温度 :40.0℃
試料注入量 :0.10ml
試料の分子量の算出にあたっては、標準ポリスチレン樹脂(例えば、商品名「TSKスタンダード ポリスチレン F−850、F−450、F−288、F−128、F−80、F−40、F−20、F−10、F−4、F−2、F−1、A−5000、A−2500、A−1000、A−500」、東ソ−社製)を用いて作成した分子量校正曲線を使用する。
【0038】
<ワックスの最大吸熱ピークのピーク温度、樹脂のガラス転移温度Tg>
ワックスの最大吸熱ピークのピーク温度は、示差走査熱量分析装置「Q1000」(TA Instruments社製)を用いてASTM D3418−82に準じて測定する。装置検出部の温度補正はインジウムと亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いる。具体的には、ワックスを10mg精秤し、これをアルミニウム製のパンの中に入れ、リファレンスとして空のアルミニウム製のパンを用い、測定温度範囲30以上200℃以下の間で、昇温速度10℃/minで測定を行う。尚、測定においては、一度200℃まで昇温させ、続いて30℃まで降温し、その後に再度昇温を行う。この2度目の昇温過程での温度30以上200℃以下の範囲におけるDSC曲線の最大の吸熱ピークを、ワックスの最大吸熱ピークとする。
また、樹脂のガラス転移温度(Tg)は、ワックスの最大吸熱ピークのピーク温度測定と同様に、樹脂を10mg精秤し測定する。温度40℃以上100℃以下の範囲において比熱変化が得られる。このときの比熱変化前と比熱変化後のベースラインの中間点の線と示差熱曲線との交点を、樹脂のガラス転移温度Tgとする。
【0039】
<外添剤の個数平均粒径の測定方法>
外添剤の個数平均粒径は、外添剤を透過電子顕微鏡で観察し、100個の粒子の長径を
測定して個数平均粒子径を求める。トナー粒子上の粒子径を測定する場合は、走査電子顕微鏡で観察し、100個の粒子の長径を測定して個数平均粒径を求める。なお、測定時の倍率は40000倍とし、0.5nm以上の粒子を対象とする。
【0040】
<磁性キャリアの体積基準の50%粒径(D50)及び90%粒径(D90)>
粒度分布測定は、レーザー回折・散乱方式の粒度分布測定装置「マイクロトラックMT3300EX」(日機装社製)にて測定を行う。
磁性キャリアの体積基準の50%粒径(D50)及び90%粒径(D90)の測定には、乾式測定用の試料供給機「ワンショットドライ型サンプルコンディショナーTurbotrac」(日機装社製)を装着して行う。Turbotracの供給条件として、真空源として集塵機を用い、風量33リットル/sec、圧力17kPaとする。制御は、ソフトウエア上で自動的に行い、体積基準の累積値である50%粒径(D50)及び90%粒径(D90)を求める。制御及び解析は付属ソフト(バージョン10.3.3−202D)を用いて行う。
測定条件は下記の通りである。
SetZero時間 :10秒
測定時間 :10秒
測定回数 :1回
粒子屈折率 :1.81
粒子形状 :非球形
測定上限 :1408μm
測定下限 :0.243μm
測定環境 :23℃/50%RH
【0041】
<磁性キャリアのパラメータαの算出方法>
パラメータαの算出法について図面に従って詳細に説明する。磁性キャリアのパラメータαの値は、以下の手順で算出することができる。
(サンプルの秤量)
磁性キャリアを、電極面積が4.9cm2である円筒型電極(直径2.5cm)を有するサンプルホルダに、電極間に100Nの押し圧をかけたときに封入したサンプルの厚みが0.95mm以上1.05mm以下の範囲となるように封入し、封入された磁性キャリアを秤量した。
(測定回路の説明)
上記のサンプルホルダの電極間に図3に示すように配線を行い、サンプルホルダの電極間に100Nの押し圧をかけた状態において、サンプルホルダ内部に封入した磁性キャリアの交流インピーダンス測定を行った。尚、本測定では、電界下におけるαを求めるために、直流電圧を印加した状態における交流インピーダンス測定を行う。このため、図3に示すように、交流成分である正弦波電圧Vacに直流成分である直流電圧Voを重畳した交番バイアスをサンプルホルダの電極間に印加する。ここで、正弦波電圧Vacの振幅は実効値で1V、直流電圧Voは磁性キャリアにかかる電界が1000V/cmとなるようにした。詳細は、後述する。
更に、このときにab間に流れる応答電流の交流成分のみを取り出し、解析することで、直流電界下におけるインピーダンスを測定した。インピーダンス測定装置としては、Solartron社製1260型周波数応答解析装置(FRA)及び、同社製1296型誘電率測定インターフェイスを用いた。上記交番バイアスに用いられる、直流電圧Voは、波形発振器から出力した直流電圧信号を、Trek社製PZD2000型高電圧電源で増幅して得た。又、正弦波電圧Vacは1296型誘電率測定インターフェイスのSAMPLE−HI端子より出力される。更に図3に示すように、測定系にコンデンサーC1(66μF)及びツェナーダイオードD1(5V)を配置することで、正弦波電圧Vacに直流電圧Voを重畳することで、上記交番バイアスを得た。
又、応答電流は、図3中の抵抗器R2(10kΩ)、コンデンサーC2(33μF)、
及びツェナーダイオードD2(5V)を用いた分流回路を用いることで、直流成分と交流成分に分離することができる。このとき、コンデンサーC2側に流れる交流成分のみを1260型インピーダンスアナライザのINPUT−V1−LO端子および、1296型誘電率測定インターフェイスのSAMPLE−LO端子に入力し、応答電流波形の解析を行い、インピーダンスを測定した。尚、図3中の抵抗器R1(10kΩ)は保護抵抗であり、測定系に流れる最大電流量を制限する。
(複素インピーダンスの測定)
本実施例では、Solartron社製インピーダンス測定ソフトウエアSMaRTを用いて、インピーダンスの自動測定を行った。SMaRTでは、所定の周波数fの正弦波電圧と正弦波電圧に対する応答電流から、周波数fに対する複素インピーダンス(下記式)を測定することができる。
【0042】
【数3】
【0043】
インピーダンスの周波数特性を測定するために、上記正弦波電圧の周波数f(Hz)は、1Hzから1MHzまでの範囲で複数の周波数でインピーダンス測定を行った。
具体的には、1.0、1.6、2.5、4.0、6.3、1.0×101、1.6×101、2.5×101、4.0×101、6.3×101、1.0×102、1.6×102、2.5×102、4.0×102、6.3×102、1.0×103、1.6×103、2.5×103、4.0×103、6.3×103、1.0×104、1.6×104、2.5×104、4.0×104、6.3×104、1.0×105、1.6×105、2.5×105、4.0×105、6.3×105、1.0×106Hzで測定を行った。
正弦波電圧Vacの振幅は実効値で1Vとした。
上記のように、1Hzから1MHzの周波数範囲において複数の周波数で測定した複素インピーダンスZを複素平面状にプロットすることで、所謂Cole−Coleプロット(ナイキスト線図)を作成した。
【0044】
(等価回路によるフィッティング)
次に、交流インピーダンス測定で得られた複素インピーダンス測定データからパラメータαを求める方法について具体的に説明する。
作成したCole−Coleプロットは、Solartron社製解析ソフトウエアZView2のInstant Fit機能を使用し、図4に示した等価回路の複素インピ
ーダンスと対応させてフィッティングを行い、インピーダンス測定データのフィッティングパラメータとしてαを求めた。ここで、図4においてRs、Rは抵抗であり、CPEはConstant Phase Element(定相要素)と呼ばれる回路素子であり、CPEの複素インピーダンスZCPEの周波数特性は下記(4)式で表される。
【0045】
【数4】
ここで、ωはインピーダンス測定の角周波数、iは虚数単位である。又、αは0.00
から1.00までの実数のパラメータである。TはCPEの静電容量を反映したパラメータであり、α=1のときはコンデンサーの静電容量となり、F(ファラッド)の次元を持つ。
【0046】
図4のフィッティング回路全体のインピーダンスは、下式のように表され、最終的に下記式(5)式で表される。
Z(ω)=Rs+(1/R+1/ZCFE)−1
=Rs+(1/R+1/((iω)αT)−1)−1
【数5】
【0047】
尚、Rsはフィッティングの精度を向上させるためにフィッティング回路に導入した仮想的な抵抗であるため、負の値をとってもよい。
図5は図4の回路において、Rs=100Ω、R=1×105Ωとし、T=2×10−10Fα・Ωα−1、α=1.0、α=0.9、α=0.8、およびα=0.7におけるCole−Coleプロットを示している。Cole−Coleプロットの形状から解るように、上記(5)式におけるαはCole−Coleプロットの描く円弧の歪み対応したパラメータである。
【0048】
(電界下におけるα)
1000V/cmの電界下における磁性キャリアのαの値は以下のように求めた。
インピーダンスを測定する際に、サンプルに印加される平均の電界強度Esampleは、インピーダンス測定時に電極間のサンプルが分担する電圧の直流成分Vsampleと電極間距離Lを用いて、Vsample/Lで表される。Vsampleの値は、図3の回路図に示したa点における電位とb点における電位の差分により測定することができる。本測定では、Tktronix社製の高電圧プローブP6015Aを用いて、a点及びb点における電位を測定し、それらの電位の差分によりサンプルホルダの電極間の分担電圧Vsampleを測定した。又、Vsampleの値は、高電圧電源から出力される直流電圧Voの電圧を変化させることで調整した。このようにして、複数の電界強度Eにおいてインピーダンス測定を行い、各電界強度下におけるαを求め、グラフにプロットすることで、1000V/cmの電界下における磁性キャリアのαを算出した。
【実施例】
【0049】
以下、本発明の具体的実施例について説明するが、本発明はこれらの実施例に限定されるものではない。尚、以下の配合における部数は特に説明が無い場合は質量部である。
〔結着樹脂の製造例1〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 22.6質量部無水トリメリット酸 1.7質量部ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
75.6質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、200℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させ、その後、10mmHgに減圧し1時間反応させ、樹脂1を合成した。GPCで求めた樹脂1の分子量は、重量平均分子量(Mw)6200、数平均分子量(Mn)2500であり、ピーク分子量(Mp)2900、ガラス転移点は55℃、軟化点は93℃であった。
【0050】
〔結着樹脂の製造例2〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 17.2質量部ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
76.6質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、220℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させた。
さらに、無水トリメリット酸6.1質量部を加え、180℃に加熱し、2時間反応させ樹脂2を合成した。GPCで求めた樹脂2の分子量は、重量平均分子量(Mw)86000、数平均分子量(Mn)6000、ピーク分子量(Mp)12800、ガラス転移点は62℃、軟化点は132℃であった。
【0051】
〔結着樹脂の製造例3〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 19.6質量部無水トリメリット酸 5.1質量部ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
75.3質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、200℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させ、その後、10mmHgに減圧し1時間反応させ、樹脂3を合成した。GPCで求めた樹脂3の分子量は、重量平均分子量(Mw)32000、数平均分子量(Mn)5500であり、ピーク分子量(Mp)8700、ガラス転移点は58℃、軟化点は106℃であった。
【0052】
〔結着樹脂の製造例4〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 17.2質量部ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
76.6質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、220℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させた。
さらに、無水トリメリット酸17.2質量部を加え、180℃に加熱し、2時間反応させ樹脂4を合成した。GPCで求めた樹脂4の分子量は、重量平均分子量(Mw)125000、数平均分子量(Mn)6400、ピーク分子量(Mp)14800、ガラス転移点は61℃、軟化点は141℃であった。
【0053】
〔結着樹脂の製造例5〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 19.6質量部無水トリメリット酸 5.1質量部ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
75.3質量部チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、200℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させ、その後、10mmHgに減圧し1時間反応させ、樹脂5を合成した。GPCで求めた樹脂5の分子量は、重量平均分子量(Mw)18000、数平均分子量(Mn)4800であり、ピーク分子量(Mp)8000、ガラス転移点は58℃、軟化点は99℃で
あった。
【0054】
〔結着樹脂の製造例6〕
スチレン 77.4質量部アクリル酸−n−ブチル 20.6質量部メタクリル酸 2.0質量部を反応容器に添加し、該混合液を110℃まで昇温した。窒素雰囲気下にラジカル重合開始剤であるtert−ブチルハイドロパーオキサイド1部をキシレン10部に溶解したものを該混合液に30分かけて滴下した。さらにその温度で該混合液を10時間保温してラジカル重合反応を終了させた。さらに該混合液を加熱しながら減圧し、脱溶剤することにより樹脂5を得た。GPCで求めた樹脂5の分子量は、重量平均分子量(Mw)36000、数平均分子量(Mn)8000であり、ピーク分子量(Mp)13000、ガラス転移点(Tg)58℃、軟化点 113℃であった。
【0055】
〔ワックス偏在制御樹脂の製造例1〕
反応容器中に下記材料を入れ、十分溶解させた。
キシレン 25.0質量部
低分子量ポリプロピレン 9.3質量部
(三洋化成工業(株)製 ビスコール660P:軟化点 145℃)
さらに
スチレン 68.5質量部
メチルメタクリレート 3.6質量部
アクリロニトリル 9.0質量部
ジ-t-ブチルパーオキシヘキサヒドロテレフタレート 2.7質量部
キシレン 15.0質量部
の混合溶液を180℃で、4時間かけて滴下後、さらに170℃で1時間保持した後、有機溶剤を留去した。得られた樹脂を冷延・固化後、粉砕して、ビニル系共重合体とポリプロピレンがグラフト結合したワックス偏在制御樹脂1を得た。
得られたワックス偏在制御樹脂1をビーカー中で180℃に加熱したところ、分離したり、白濁を生じることはなかった。
【0056】
〔ワックス偏在制御樹脂の製造例2〕
低分子量ポリプロピレン(軟化点145℃)の添加量を9.3質量部から、20.5質量部に変更した以外は、ワックス偏在制御樹脂の製造例1と同様に、ビニル系共重合体とポリプロピレンがグラフト結合したワックス偏在制御樹脂2を得た。
得られたワックス偏在制御樹脂2をビーカー中で180℃に加熱したところ、分離したり、白濁を生じることはなかった。
【0057】
〔ワックス偏在制御樹脂の製造例3〕
低分子量ポリプロピレン(軟化点145℃)の添加量を9.3質量部から、32.6質量部に変更した以外は、ワックス偏在制御樹脂の製造例1と同様に、ビニル系共重合体とポリプロピレンがグラフト結合したワックス偏在制御樹脂3を得た。
得られたワックス偏在制御樹脂3をビーカー中で180℃に加熱したところ、分離したり、白濁を生じることはなかった。
【0058】
〔ワックス偏在制御樹脂の製造例4〕
低分子量ポリプロピレン(軟化点145℃)の添加量を9.3質量部から、0.9質量部に変更した以外は、ワックス偏在制御樹脂の製造例1と同様に、ビニル系共重合体とポリプロピレンがグラフト結合したワックス偏在制御樹脂4を得た。
得られたワックス偏在制御樹脂4をビーカー中で180℃に加熱したところ、分離した
り、白濁を生じることはなかった。
【0059】
〔トナーの製造例1〕
樹脂1 50.0質量部樹脂4 50.0質量部ワックス偏在制御樹脂1 5.0質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク105℃) 3.5質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 3.5質量部C.I.ピグメントブルー15:3 5.0質量部
上記材料をヘンシェルミキサー(FM−75型、三井三池化工機(株)製)でよく混合した後、温度130℃に設定した二軸混練機(PCM−30型、池貝鉄工(株)製)にて混練した。得られた混練物を冷却し、ハンマーミルにて1mm以下に粗粉砕し、粗砕物を得た。得られた粗砕物を、高圧気体を用いた衝突式気流粉砕機を用いて微粉砕した。
次に、得られた微粉砕物を図1に示す表面改質装置により表面改質を行った。表面改質時の条件として、原料供給速度が2.0kg/hr、熱風流量が4.5m3/min、熱風の吐出温度が210℃、冷風温度が3℃、冷風流量が3.0m3/min、絶対水分量が3g/m3で表面改質を行った。次に、コアンダ効果を利用した風力分級機(エルボジェットラボEJ−L3、日鉄鉱業社製)で分級しで微粉及び粗粉を同時に分級除去、トナー粒子1を得た。
得られたトナー粒子1 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が200nmであり、ヘキサメチルジシラザンで処理されたゾルゲル法シリカ微粉体を2.0質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー1を得た。
【0060】
〔トナーの製造例2〕
樹脂1 50.0質量部樹脂2 50.0質量部ワックス偏在制御樹脂2 10.0質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク105℃) 16.0質量部C.I.ピグメントブルー15:3 5.0質量部
上記材料を用い、表面改質時に熱風の吐出温度を210℃から230℃に変更した以外は、トナー製造例1と同様にして、トナー粒子2を得た。
得られたトナー粒子2 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が150nmであり、ヘキサメチルジシラザンで処理されたヒュームド法シリカ微粉体を3.0質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー2を得た。
【0061】
〔トナーの製造例3〕
樹脂5 100.0質量部ワックス偏在制御樹脂1 5.0質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 3.5質量部C.I.ピグメントブルー15:3 5.0質量部
上記材料を用い、表面改質時に熱風の吐出温度を210℃から250℃に変更した以外は、トナー製造例1と同様にして、トナー粒子3を得た。
得られたトナー粒子3 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が300nmであり、ヘキサメチルジシラザンで処理されたゾルゲル法シリカ微粉体を1.0質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機
(株)製)で混合し、トナー3を得た。
【0062】
〔トナーの製造例4〕
樹脂5 100.0質量部ワックス偏在制御樹脂3 5.0質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 10.0質量部C.I.ピグメントブルー15:3 5.0質量部
上記材料を用い、表面改質時に熱風の吐出温度を210℃から190℃に変更した以外は、トナー製造例1と同様にして、トナー粒子4を得た。
得られたトナー粒子4 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が80nmであり、ヘキサメチルジシラザンで処理されたゾルゲル法シリカ微粉体を2.0質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー4を得た。
【0063】
〔トナーの製造例5〕
樹脂5 100.0質量部ワックス偏在制御樹脂4 3.0質量部精製ノルマルパラフィン(DSC最大吸熱ピーク75℃) 3.0質量部C.I.ピグメントブルー15:3 5.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にして、トナー5を得た。
【0064】
〔トナーの製造例6〕
樹脂5 100.0質量部ワックス偏在制御樹脂1 15.0質量部ベヘン酸ベヘニル(DSC最大吸熱ピーク74℃) 18.0質量部C.I.ピグメントブルー15:3 5.0質量部
混練する材料を上記材料とする以外は、トナー製造例4と同様にして、トナー6を得た。
【0065】
〔トナーの製造例7〕
ワックス偏在制御樹脂1 5.0質量部を0.0質量部に変更した以外は、トナー製造例1と同様にトナー7を得た。
【0066】
〔トナーの製造例8〕
樹脂3 100.0質量部精製ノルマルパラフィン(DSC最大吸熱ピーク75℃) 2.0質量部C.I.ピグメントブルー15:3 5.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にして、トナー粒子8を得た。得られたトナー粒子8 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が30nmであり、ヘキサメチルジシラザンで処理されたヒュームド法シリカ微粉体を0.5質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー8を得た。
【0067】
〔トナーの製造例9〕
樹脂6 100.0質量部ベヘン酸ベヘニル(DSC最大吸熱ピーク74℃) 23.0質量部C.I.ピグメントブルー15:3 5.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にして、トナー粒子9を得た。得られたトナー粒子9 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が30nmであり、ヘキサメチルジシラザンで処理されたヒュームド法シリカ微粉体を0.5質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー9を得た。
【0068】
〔トナーの製造例10〕
樹脂1 50.0質量部樹脂4 50.0質量部ワックス偏在制御樹脂2 20.0質量部ベヘン酸ベヘニル(DSC最大吸熱ピーク74℃) 15.0質量部C.I.ピグメントブルー15:3 5.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にして、トナー粒子10を得た。得られたトナー粒子10 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が30nmであり、ヘキサメチルジシラザンで処理されたヒュームド法シリカ微粉体を0.5質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー10を得た。
【0069】
〔トナーの製造例11〕
表面改質時の冷風温度を3℃から25℃に、絶対水分量を3g/m3から10g/m3に変更した以外は、トナー製造例1と同様にしてトナー11を得た。
【0070】
〔トナーの製造例12〕
樹脂1 50.0質量部樹脂4 50.0質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク105℃) 3.5質量部フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 3.5質量部C.I.ピグメントブルー15:3 5.0質量部を用いトナー製造例11と同様にしてトナー12を得た。
【0071】
〔トナーの製造例13〕
表面改質処理を行わないこと以外は、トナー製造例1と同様にしてトナー13を得た。
【0072】
〔トナーの製造例14〕
(微粒子分散液1の調製)
水 68.4質量部
メタクリル酸エチレンオキサイド付加物硫酸エステルのナトリウム塩(エレミノールRS−30、三洋化成工業製) 1.0質量部
スチレン 8.5質量部
メタクリル酸 8.0質量部
アクリル酸ブチル 11.0質量部
過硫酸アンモニウム 0.1質量部
以上を、撹拌棒及び温度計をセットした反応容器に仕込み、4000回転/分で1時間攪拌した。その後、80℃まで昇温し、4時間反応させた。さらに、1%過硫酸アンモニウム水溶液を3.0質量部加え、80℃で5時間熟成して微粒子分散液1を得た。
(水相1の調製)
水 80.0質量部
微粒子分散液1 8.0質量部
ドデシルジフェニルエーテルジスルホン酸ナトリウムの48.3%水溶液(エレミノールMON−7:三洋化成工業製) 3.5質量部
酢酸エチル 8.5質量部
以上を混合撹拌し、水相1を得た。
(プレポリマー1の調製)
冷却管、撹拌機及び窒素導入管の付いた反応容器中に、
樹脂1 41.0質量部
イソホロンジイソシアネート 8.9質量部
酢酸エチル 50.0質量部
以上を入れ100℃で5時間反応し、プレポリマー1を得た。
(ケチミン化合物1の調製)
撹拌棒及び温度計をセットした反応容器に、
イソホロンジアミン 17.5質量部
メチルエチルケトン 7.5質量部
以上を入れ、50℃で5時間反応を行い、ケチミン化合物1を得た。
(原料溶解液1の調製)
撹拌棒及び温度計をセットした容器に、
樹脂1 37.8質量部
C.I.ピグメントブルー15:3 10.0質量部
カルナバワックス 10.0質量部
酢酸エチル 94.7質量部
以上を入れ、80℃で5時間、攪拌しながら保持した。その後、2時間で30℃に冷却後、1時間攪拌し、原料溶解液1を得た。
(顔料・ワックス分散液1の調製)
原料溶解液1 50.0質量部を容器に移し、ビーズミル(ウルトラビスコミル:アイメックス社製)を用いて、送液速度1kg/hr、ディスク周速度6m/秒、0.5mmジルコニアビーズを70体積%充填、3パスの条件で分散を行った。
次いで、樹脂1の65%酢酸エチル溶液 50.0質量部を加え、上記条件のビーズミルで2パスし、顔料・ワックス分散液1を得た。顔料・ワックス分散液1の固形分濃度は50%であった。
(トナーの調製)
顔料・ワックス分散液1 74.2質量部
プレポリマー1 11.4質量部
ケチミン化合物1 0.3質量部
以上を容器に入れ、TKホモミキサー(特殊機化製)で5,000rpmで2分間混合した後、容器に水相1 120.0質量部を加え、TKホモミキサーで、回転数13,000rpmで25分間混合し乳化スラリー1を得た。
撹拌機及び温度計をセットした容器に、乳化スラリー1を投入し、30℃で8時間脱溶剤した後、45℃で7時間熟成を行い、分散スラリー1を得た。
分散スラリー1 100部を減圧濾過した後、イオン交換水100質量部を加え、TKホモミキサーで混合(回転数12,000rpmで10分間)した後濾過した。
さらに、5%水酸化ナトリウム水溶液100部を加え、TKホモミキサーで混合(回転数12,000rpmで10分間)した後、減圧濾過した。
次に、10%塩酸100質量部を加え、TKホモミキサーで混合(回転数12,000rpmで10分間)した後濾過した。
最後に、イオン交換水300質量部を加え、TKホモミキサーで混合(回転数12,000rpmで10分間)した後濾過する操作を2回行い、濾過ケーキ1を得た。
濾過ケーキ1を循風乾燥機にて40℃で48時間乾燥し、目開き75μmメッシュで篩い、トナー粒子14を得た。
得られたトナー粒子14 100.0質量部に、無機微粒子として、個数平均粒径が4
0nmであり、i−ブチルトリメトキシシランで処理されたの酸化チタン微粉体1.0質量部、個数平均粒径が20nmであり、ヘキサメチルジシラザンで処理されたのヒュームド法シリカ微粉体を0.5質量部の外添混合し、トナー14を得た。
【0073】
トナー1乃至14の物性を表1に示す。
【表1】
【0074】
(多孔質磁性コアの製造例1)
<工程1(秤量・混合工程)>
Fe2O3 59.7質量%
MnCO3 34.4質量%
Mg(OH)2 4.8質量%
SrCO3 1.1質量%
となるようにフェライト原材料を秤量した。その後、ジルコニア(φ10mm)のボールを用いた乾式ボールミルで2時間粉砕・混合した。
<工程2(仮焼成工程)>
粉砕・混合した後、バーナー式焼成炉を用い大気中で950℃で2時間焼成し、仮焼フェライトを作製した。
フェライトの組成は、下記の通り。
(MnO)a(MgO)b(SrO)c(Fe2O3)d
上記式において、a=0.39、b=0.11、c=0.01、d=0.49
<工程3(粉砕工程)>
クラッシャーで0.5mm程度に粉砕し仮焼フェライトを得た。その後、仮焼フェライトを半分に分けた。仮焼フェライトの半分をジルコニア(φ10mm)のボールを用い、仮焼フェライト100質量部に対し、水を30質量部加え、湿式ボールミルで2時間粉砕し、フェライトスラリー1−1を得た。フェライトスラリー1−1の半分を、さらにジルコニアのビーズ(φ1.0mm)を用いた湿式ビーズミルで3時間粉砕し、フェライトスラリー1−2を得た。フェライトスラリー1−1と1−2をジルコニアのビーズ(φ1.0mm)を用いた湿式ビーズミルで10分間混合し、フェライトスラリー1(仮焼フェライト微粉砕品1)を得た。得られた仮焼フェライト微粉砕品1の、体積基準の50%粒径(D50)は1.3μm、体積基準の90%粒径(D90)は2.3μmであった。
<工程4(造粒工程)>
得られた仮焼フェライト微粉砕品1に、バインダーとして上記仮焼フェライト100質量部に対してポリビニルアルコール2.0質量部を添加し、スプレードライヤー(製造元:大川原化工機)で、36μmの球状粒子に造粒した。
<工程5(本焼成工程)>
焼成雰囲気をコントロールするために、電気炉にて窒素雰囲気下(酸素濃度0.01体積%以下)で、1050℃で4時間焼成した。
<工程6(選別工程)>
凝集した粒子を解砕した後に、目開き250μmの篩で篩分して粗大粒子を除去し、多孔質磁性コア1を得た。
【0075】
<多孔質磁性コアの製造例2>
多孔質磁性コア製造例1のうち、工程3のフェライトスラリー1−2を得るための湿式ビーズミルの粉砕時間を3時間から2時間に変更した。得られた仮焼フェライト微粉砕品は、D50=1.4μm、D90=3.0μmであった。また、工程5の焼成温度を1050℃から1100℃に変更した。
上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア2を得た。
【0076】
<多孔質磁性コアの製造例3>
多孔質磁性コア製造例1のうち、工程3のフェライトスラリー1−2を得るための湿式ビーズミルの粉砕時間を3時間から1時間に変更した。得られた仮焼フェライト微粉砕品は、D50=1.7μm、D90=5.0μmであった。また、工程5の焼成温度を1050℃から1150℃に変更した。
上記以外は、多孔質磁性コア製造例1と同様にして、多孔質磁性コア3を得た。
【0077】
<多孔質磁性コアの製造例4>
<工程1(秤量・混合工程)>
Fe2O3 62.4質量%
MnCO3 30.5質量%
Mg(OH)2 6.4質量%
SrCO3 0.7質量%
をジルコニア(φ10mm)のボールを用いた乾式ボールミルで2時間粉砕・混合した。<工程2(仮焼成工程)>
粉砕・混合した後、バーナー式焼成炉を用い大気中で950℃で2時間焼成し、仮焼フェライトを作製した。
<工程3(粉砕工程)>
クラッシャーで0.5mm程度に粉砕し仮焼フェライトを得た。仮焼フェライトをステンレス(φ10mm)のボールを用い、仮焼フェライト100質量部に対し、水を30質量部加え、湿式ボールミルで1時間粉砕し、フェライトスラリー4−1を得た。フェライトスラリー4−1を、さらにステンレスのビーズ(φ1.0mm)を用いた湿式ビーズミルで4時間粉砕し、フェライトスラリー4(仮焼フェライト微粉砕品4)を得た。得られた仮焼フェライト微粉砕品4の、体積基準の50%粒径(D50)は1.4μm、体積基準の90%粒径(D90)は1.8μmであった。
<工程4(造粒工程)>
得られた仮焼フェライト微粉砕品4に、バインダーとして上記仮焼フェライト100質量部に対して、ポリビニルアルコール2.0質量部を添加し、スプレードライヤー(製造元:大川原化工機)で、36μmの球状粒子に造粒した。
<工程5(本焼成工程)>
焼成雰囲気をコントロールするために、電気炉にて窒素雰囲気下(酸素濃度0.01体積%以下)で、1100℃で4時間焼成した。
<工程6(選別工程)>
凝集した粒子を解砕した後に、目開き250μmの篩で篩分して粗大粒子を除去し、多孔質磁性コア4を得た。
【0078】
<磁性キャリアの製造例1>
[樹脂液の調製]
(樹脂液1)
シリコーン樹脂溶液(SR2411 東レ・ダウコーニング) 20.0質量部
(20質量%トルエン溶液における粘度 1.1×10−6m2/sec)
γ−アミノプロピルトリエトキシシラン 0.5質量部
トルエン 79.5質量部
以上を、ボールミル(ソーダガラスボール φ10mm)を用いて1時間混合し、樹脂液1を得た。
(樹脂液2)
シリコーン樹脂溶液(SR2410 東レ・ダウコーニング) 20.0質量部
(20質量%トルエン溶液における粘度 2.9×10−6m2/sec)
γ−アミノプロピルトリエトキシシラン 0.3質量部
トルエン 79.7質量部
以上を、ボールミル(ソーダガラスボール φ10mm)を用いて1時間混合し、樹脂液2を得た。
[磁性キャリア1の製造]
工程例1(樹脂充填工程例1):
多孔質磁性コア1の100.0質量部を万能攪拌混合機(ダルトン社製)に入れ、減圧下、60℃に加熱しながら撹拌する。続いて、樹脂液1を多孔質磁性コア1に対し樹脂成分として15質量部になるように添加し4時間加熱を続け、溶剤を除去した。得られた試料をドラムミキサ(杉山重工業製)に移し、窒素雰囲気下に200℃で2時間熱処理して、開口70μmのメッシュで分級して、磁性キャリア1aを得た。
工程例2(樹脂コート工程例1):
磁性キャリア1aの100.0質量部をナウタミキサ(ホソカワミクロン社製)に投入し、さらに、樹脂液2を樹脂成分として1.0質量部になるようにナウタミキサに投入した。減圧下で70℃に加熱し、1.7S−1(100rpm)で混合し、2時間かけて溶媒除去及び塗布操作を行った。その後、得られた試料をドラムミキサ(杉山重工業製)に移し、窒素雰囲気下、温度200℃で2時間熱処理した後、開口70μmのメッシュで分級して磁性キャリア1を得た。
【0079】
<磁性キャリアの製造例2、3、及び4>
磁性キャリアの製造例1において、多孔質磁性コア1を多孔質磁性コア2に、樹脂液1を多孔質磁性コア2に対し樹脂成分として10質量部になるように添加した以外は、磁性キャリアの製造例1と同様にして、磁性キャリア2を得た。
磁性キャリアの製造例1において、多孔質磁性コア1を多孔質磁性コア3に、樹脂液1を多孔質磁性コア3に対し樹脂成分として8質量部になるように添加した以外は、磁性キャリアの製造例1と同様にして、磁性キャリア3を得た。
磁性キャリアの製造例1において、工程例2(樹脂コート工程例1)を行わなかった以外は、磁性キャリアの製造例1と同様にして、磁性キャリア4を得た。
【0080】
<磁性キャリアの製造例5>
体積基準の50%粒径(D50)が32μmのマグネタイト粒子100質量部に、シリコーン樹脂(信越化学社製:KR271)1質量部、γ―アミノプロピルトリエトキシシラン0.5質量部、トルエン98.5質量部の混合液を、添加し、さらに溶液減圧ニーダーで撹拌混合しながら70℃、5時間減圧乾燥を行い、溶剤を除去した。その後、140℃で2時間焼き付け処理して、篩振とう機(300MM−2型、筒井理化学機械:75μ
m開口)で篩い、磁性キャリア5を得た。
【0081】
磁性キャリア1乃至5の物性を表2に示す。
【表2】
【0082】
〔実施例1乃至8及び比較例1乃至8〕
次に、このように作製したトナーと磁性キャリアを表3の組み合わせで二成分系現像剤を作製した。二成分系現像剤は、磁性キャリア100質量部に対して、トナー9質量部の配合割合とし、V型混合機で5分間混合した。
【0083】
【表3】
【0084】
<二成分系現像剤の評価>
得られた二成分系現像剤の低温定着性、グロス均一性、画像濃度、画像濃度変動、及び現像性の各種評価を、温度23℃/湿度50%RH(以下「N/N」)と、温度30℃/湿度80%RH(以下「H/H」)の2つの環境で実施した。詳細な評価方法及び評価基準は後述する。上記各種評価用の画像形成装置としては、キヤノン製カラー複写機imageRUNNER iRC3580改造機を用いた。改造箇所は、現像ドラムに対する現像スリーブ周速を標準機に比して1.5倍としたこと、及び、プロセススピードが245mm/secとなるようしたことである。なお、上記二成分系現像剤は、画像形成装置のシアン用現像器に入れて評価を行った。評価結果を表4及び表5に示す。
【0085】
<低温定着性の評価>
紙は、カラー複写機・プリンタ用普通紙 CS−814(A4、81.4g/m2)(キヤノンマーケティングジャパン株式会社より販売)を用いた。
FFH画像(以下、ベタ部)のトナーの紙上への載り量が0.6mg/cm2となるように直流電圧VDCを調整し、未定着のFFH画像を得た。
その後、キヤノン製プロダクション向け複写機imagePRESS C1+の外部定着器(ベルト&ローラ定着器)をプロセススピードが300mm/secとなるように改造したものを定着性評価に用いた。
上記外部定着器における定着温度を100〜200℃の範囲で10℃おきに調整し、各温度で上記未定着のFFH画像を定着し定着画像を得た。得られた定着画像を4.9kPaの荷重をかけたレンズクリーニングワイパー(ダスパー 小津産業株式会社製)で5往復摺擦し、摺擦前後の画像濃度の濃度低下率が10%以下になる点を定着温度とし、下記の評価基準に従って評価した。
A:定着温度120℃以下 (非常に良好)
B:定着温度130℃ (良好)
C:定着温度140℃ (本発明において許容レベル)
D:定着温度150℃以上 (本発明において許容できないレベル)
【0086】
<グロス均一性の評価>
紙は、CLC用 厚口用紙 NS−700(A4、157g/m2)(キヤノンマーケティングジャパン株式会社より販売)を用いた。
低温定着性評価と同様に、ベタ部のトナーの紙上への載り量が0.6mg/cm2となるように直流電圧VDCを調整し、紙の四隅及び中央部に、4cm×4cmの正方形のベタ画像を印刷した未定着画像を得た。
その後、キヤノン製プロダクション向け複写機imagePRESS C1+の外部定着器をプロセススピード100mm/secとなるように改造したものを用い、グロス均一性評価を行った。具体的には、外部定着器における定着温度を180℃に調整し、未定着画像を定着した。当該定着画像の四隅及び中央部にあるベタ画像の各グロス値を測定し、その最大値と最小値の差を以下の基準で判断した。
A:1.0未満
B:1.0以上2.0未満(良い)
C:2.0以上3.0未満(本発明において許容レベル)
D:3.0以上(本発明において許容できないレベル)
【0087】
<画像濃度の評価>
現像スリーブには、周波数2.0kHz、Vpp1.3kVの交流電圧と直流電圧VDCを印加した。直流電圧VDCはベタ部のトナーの紙上への載り量が0.6mg/cm2となるように調整した。FFH画像を印刷し、画像濃度(反射濃度)を求めた(初期)。その後、画像比率1%で50000枚印刷し、50000枚印刷後に、再びFFH画像を印刷し、画像濃度(反射濃度)を測定した(50000枚印刷後)。紙は、CS−814(A4、81.4g/m2)(キヤノンマーケティングジャパン株式会社より販売)を用いた。画像濃度(反射濃度)は、分光濃度計500シリーズ(X−Rite社製)を用いて測定し、以下の基準で判断した。
A:1.50以上
B:1.40以上1.50未満(良い)
C:1.30以上1.40未満(本発明において許容レベル)
D:1.30未満(本発明において許容できないレベル)
【0088】
<画像濃度変動の評価>
現像スリーブには、周波数2.0kHz、Vpp1.3kVの交流電圧と直流電圧VDCを印加した。直流電圧VDCはベタ部のトナーの紙上への載り量が0.6mg/cm2となるように調整した。FFH画像を印刷し、画像濃度(反射濃度)を求めた。
画像濃度は、分光濃度計500シリーズ(X−Rite社製)を用いて測定した。
その後、現像器内の現像剤のトナー比率が9質量部で一定になるようにトナーを補給しながら、画像比率20%で100枚印刷し、初期と同様にFFH画像を印刷し、画像濃度(反射濃度)を測定した。
画像比率20%で100枚印刷する前後のFFH画像の画像濃度(反射濃度)の差を算出し、以下の基準で評価を行った。
A:0.10未満
B:0.10以上0.20未満(良い)
C:0.20以上0.30未満(本発明において許容レベル)
D:0.30以上(本発明において許容できないレベル)
【0089】
<現像性>
直流電圧VDCを300Vに固定し、周波数2.0kHzでVppを0.7kVから0.1kV刻みで2.0kVまで変えた交流電圧と直流電圧VDCを印加した。トナー載り量が0.50mg/cm2となるときのVppを求めた。
A:Vppが1.4kV未満 (非常に良好)
B:Vppが1.4kV以上1.6kV未満(良好)
C:Vppが1.6kV以上1.9kV未満(本発明において許容レベル)
D:Vppが1.9kV以上 (本発明において許容できないレベル)
【0090】
【表4】
【0091】
【表5】
【特許請求の範囲】
【請求項1】
結着樹脂、ワックス、及び外添剤としての無機微粉体を少なくとも含有するトナーであって、
前記結着樹脂はポリエステル樹脂を含有し、
前記ワックスの含有量が、前記結着樹脂100質量部に対して、3.0質量部以上20.0質量部以下であり、
前記トナーは、ATR法を用い、ATR結晶としてGe、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPa、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPbとし、
ATR法を用い、ATR結晶としてKRS5、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPc、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPdとしたときに、下記式(1)の関係を満たすことを特徴とするトナー。
1.05≦P1/P2≦2.00 ・・・ 式(1)
[前記式(1)において、前記最大吸収ピーク強度Paは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pbは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pcは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pdは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、P1=Pa/Pb、P2=Pc/Pdである。]
【請求項2】
前記ワックスは炭化水素系ワックスであることを特徴とする請求項1に記載のトナー。
【請求項3】
トナーと磁性キャリアを含む二成分系現像剤において、前記トナーは請求項1または2に記載のトナーであることを特徴とする二成分系現像剤。
【請求項4】
前記磁性キャリアは、交流インピーダンス測定により得られるインピーダンスZの周波数依存特性Z(ω)を、下記(2)式で表されるフィッティング関数により、フィッティングしたときのパラメータαが、1000V/cmの電界下において、0.70以上0.95以下であることを特徴とする請求項3に記載の二成分系現像剤。
【数1】
【請求項1】
結着樹脂、ワックス、及び外添剤としての無機微粉体を少なくとも含有するトナーであって、
前記結着樹脂はポリエステル樹脂を含有し、
前記ワックスの含有量が、前記結着樹脂100質量部に対して、3.0質量部以上20.0質量部以下であり、
前記トナーは、ATR法を用い、ATR結晶としてGe、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPa、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPbとし、
ATR法を用い、ATR結晶としてKRS5、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPc、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPdとしたときに、下記式(1)の関係を満たすことを特徴とするトナー。
1.05≦P1/P2≦2.00 ・・・ 式(1)
[前記式(1)において、前記最大吸収ピーク強度Paは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pbは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pcは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、前記最大吸収ピーク強度Pdは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、P1=Pa/Pb、P2=Pc/Pdである。]
【請求項2】
前記ワックスは炭化水素系ワックスであることを特徴とする請求項1に記載のトナー。
【請求項3】
トナーと磁性キャリアを含む二成分系現像剤において、前記トナーは請求項1または2に記載のトナーであることを特徴とする二成分系現像剤。
【請求項4】
前記磁性キャリアは、交流インピーダンス測定により得られるインピーダンスZの周波数依存特性Z(ω)を、下記(2)式で表されるフィッティング関数により、フィッティングしたときのパラメータαが、1000V/cmの電界下において、0.70以上0.95以下であることを特徴とする請求項3に記載の二成分系現像剤。
【数1】
【図1】

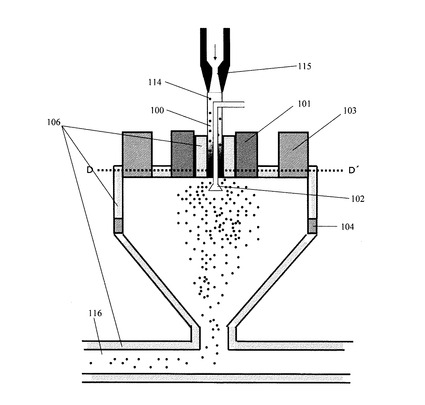
【図2】
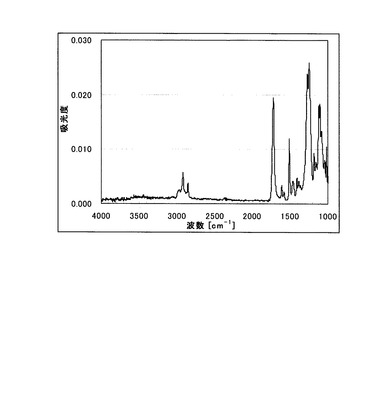
【図3】
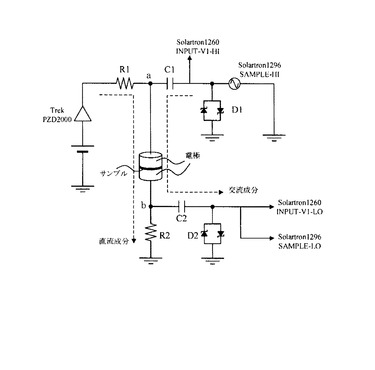
【図4】
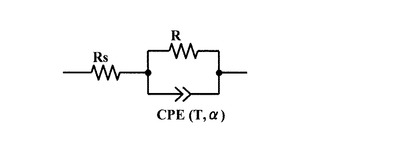
【図5】
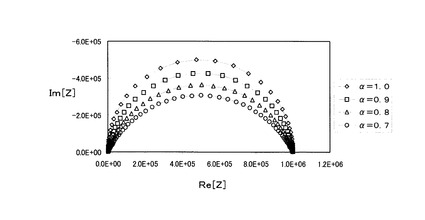
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2011−158758(P2011−158758A)
【公開日】平成23年8月18日(2011.8.18)
【国際特許分類】
【出願番号】特願2010−21123(P2010−21123)
【出願日】平成22年2月2日(2010.2.2)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
【公開日】平成23年8月18日(2011.8.18)
【国際特許分類】
【出願日】平成22年2月2日(2010.2.2)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
[ Back to top ]