
トランス−4−ヒドロキシ−L−プロリンの製造法
【課題】 工業的に有利にトランス−4−ヒドロキシ−L−プロリンを製造するための方法を提供する。
【解決手段】 2−ケトグルタル酸および2価鉄イオンの存在下、遊離のL−プロリンに作用して、トランス−4−ヒドロキシ−L−プロリンを生成する、L−プロリン4位水酸化酵素活性を有する蛋白質をコードする遺伝子を含むDNA断片をベクターに組み込んで得られる組換え体DNAを保有し、且つ、L−プロリンの生合成系の活性の強化された形質転換体、および該形質転換体形質転換体を培養し、該培養物、菌体または菌体処理物を酵素源として、2−ケトグルタル酸および2価鉄イオンの存在下、培養液または水性媒体中で、L−プロリンをトランス−4−ヒドロキシ−L−プロリンに変換させ、生成したトランス−4−ヒドロキシ−L−プロリンを該培養物または該水性媒体より採取することを特徴とするトランス−4−ヒドロキシ−L−プロリンの製造法を提供する。
【解決手段】 2−ケトグルタル酸および2価鉄イオンの存在下、遊離のL−プロリンに作用して、トランス−4−ヒドロキシ−L−プロリンを生成する、L−プロリン4位水酸化酵素活性を有する蛋白質をコードする遺伝子を含むDNA断片をベクターに組み込んで得られる組換え体DNAを保有し、且つ、L−プロリンの生合成系の活性の強化された形質転換体、および該形質転換体形質転換体を培養し、該培養物、菌体または菌体処理物を酵素源として、2−ケトグルタル酸および2価鉄イオンの存在下、培養液または水性媒体中で、L−プロリンをトランス−4−ヒドロキシ−L−プロリンに変換させ、生成したトランス−4−ヒドロキシ−L−プロリンを該培養物または該水性媒体より採取することを特徴とするトランス−4−ヒドロキシ−L−プロリンの製造法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、消炎剤として利用されているN−アセチルヒドロキシプロリン,カルバペネム系抗生物質等の医薬品の合成原料または食品添加物として有用なトランス−4−ヒドロキシ−L−プロリンを工業的に製造する方法、該方法に有用なL−プロリン4位水酸化酵素遺伝子を含有し、かつ、プロリン合成系の活性の強化された形質転換体に関する。
【背景技術】
【0002】
微生物を用いトランス−4−ヒドロキシ−L−プロリンを製造する方法としては、
1)エッシェリヒア属に属する微生物を用い、4−ヒドロキシ−2−オキソグルタル酸からトランス−4−ヒドロキシ−L−プロリンを製造する方法(特許文献1参照)、
2)細菌やかび類を用い直接発酵生産する方法(特許文献2、3および4参照) 、
3)ストレプトミセス属に属する微生物を用い、L−プロリンから製造する方法(非特許文献1、2、3および4参照)が知られている。
【0003】
従来のトランス−4−ヒドロキシ−L−プロリンの製造方法は、1)4−ヒドロキシ−2−オキソグルタル酸等のトランス−4−ヒドロキシ−L−プロリンを製造するための基質が高価で入手困難である、2)トランス−4−ヒドロキシ−L−プロリンの生産性が低い、3)トランス−4−ヒドロキシ−L−プロリンの製造に関与する酵素の活性が極めて微弱である、等の理由から、工業化は困難である。トランス−4−ヒドロキシ−L−プロリンの製造に関与する酵素については、L−プロリン4位水酸化酵素をダクチロスポランジウム.エスピー.RH1(Dactylosporangium sp.RH1 )より精製したとの報告(特許文献5参照)、および前述のストレプトマイセス属に属する微生物より精製したとの報告(非特許文献5参照)がある。また、2−ケトグルタル酸および2価鉄イオンの存在下において、遊離のL−プロリンをトランス−4−ヒドロキシ−L−プロリンに変換させる活性を有する、L−プロリン4位水酸化酵素をコードする遺伝子を前述のダクチロスポランジウム.エスピー.RH1(Dactylosporangiumsp.RH1 )よりクローニングし、L−プロリン4位水酸化酵素遺伝子を含むプラスミドpRH71を取得し、さらにトリプトファンタンデムプロモーター支配下でL−プロリン4位水酸化酵素遺伝子を発現させるプラスミドpTr2−4OHを構築し、大腸菌で活性を発現させたという報告(非特許文献6参照)がある。
【0004】
活性の高いL−プロリン4位水酸化酵素を用い、工業的に有利にトランス−4−ヒドロキシ−L−プロリンを製造する方法が求められている。
一方、プロリン生合成経路に関する酵素およびその遺伝子は大腸菌などですでに明らかになっている(非特許文献7および8参照)。また、プロリン生合成は例えば大腸菌などでは生合成経路の第一酵素であるガンマ−グルタミル キナーゼ(GK)が、生成物であるプロリンのフィードバック阻害を受けることにより調節されている事が知られている(非特許文献9参照)。
【0005】
更に、GKをコードする遺伝子proBに変異を生じさせることにより、プロリンのフィードバック阻害を著しく減少させたGKを生成させ、プロリン高生産株を造成できることが知られている(非特許文献10、11および12参照)。該proBに変異を起こさせた遺伝子としてproB74が知られており、proB74の変異点はproBのコーディングリージョンの5’末端から319番目のGがAに置換することにより、proB蛋白のN末端から107番目のアスパラギン酸がアスパラギンに変化していることが知られている(非特許文献13参照)。
プロリン分解に関する酵素系、それをコードする遺伝子、遺伝子系の制御についても、大腸菌、サルモネラ菌などで研究が進んでおりよく知られている(非特許文献14および15参照)。
【0006】
プロリンの生合成系の活性の強化された株を用いたトランス−4−ヒドロキシ−L−プロリンの製造法は知られていない。
【特許文献1】特開平3−266995号公報
【特許文献2】欧州特許出願公開第0547898号明細書
【特許文献3】特開平5−236980号公報
【0007】
【特許文献4】特開平6−245782号公報
【特許文献5】特開平7−322885
【非特許文献1】ジャーナル・オブ・バイオロジカル・ケミストリー(J. Biol. Chem.)、254巻、6684〜6690ページ(1979年)
【非特許文献2】バイオケミカル・アンド・バイオフィジカル・リサーチ・コミュニケーション(Biochem.Biophys. Res.Comm. )、120巻,45〜51ページ(1984年)
【0008】
【非特許文献3】テトラヘドロン・レターズ(Tetrahedron Letters ),34巻,7489〜7492ページ(1993年)
【非特許文献4】テトラヘドロン・レターズ(Tetrahedron Letters )、35巻,4649〜4652ページ(1994年)
【非特許文献5】バイオケミカル・ジャーナル(Biochem.J.)、313巻、185−191ページ、(1966年)
【非特許文献6】日本農芸化学会1996年度大会、講演要旨集257ページ
【0009】
【非特許文献7】ジャーナル・オブ・ジェネラル・マイクロバイオロジー(J.Gen.Microbiol.)、118巻、287−293ページ(1980年)
【非特許文献8】バイオキミカ・バイオフィジカ・アクタ(Biochim.Biophys.Acta)、104巻、397〜404ページ(1965年)
【非特許文献9】バイオキミカ・バイオフィジカ・アクタ(Biochim.Biophys.Acta)、192巻、462〜467ページ(1969年)
【0010】
【非特許文献10】モレキュラー・ジェネラル・ジェネティクス(Mol.Gen.Genet.)、182巻、82−86ページ(1981年)
【非特許文献11】ジャーナル・オブ・バクテリオロジー(J.Bacteriol.)、156巻、1249−1262ページ(1983年)
【非特許文献12】ジャーナル・オブ・バクテリオロジー(J.Bacteriol.)、164巻、1088−1093ページ(1985年)
【非特許文献13】ジーン(Gene)、64巻、199−205ページ(1988年)
【0011】
【非特許文献14】ジャーナル・オブ・バクテリオロジー(J.Bacteriol.)、146巻、895−901ページ(1981年)
【非特許文献15】ジャーナル・オブ・モレキュラー・バイオロジー (J.Mol.Biol.)、148巻、21−44ページ(1981年)
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明の課題は、L−プロリン4位水酸化酵素を用いて、効率的にトランス−4−ヒドロキシ−L−プロリンを製造する方法において、より工業的に有利にトランス−4−ヒドロキシ−L−プロリンを製造するために、L−プロリン4位水酸化酵素遺伝子を含有し、これを高効率で発現させる組換え体DNAを保持し、かつ、プロリン生合成系の活性の強化された形質転換体を提供し、該形質転換体を用いてトランス−4−ヒドロキシ−L−プロリンを工業的に安価に製造する方法を提供することにある。
【課題を解決するための手段】
【0013】
本発明は、2−ケトグルタル酸および2価鉄イオンの存在下、遊離のL−プロリンに作用して、トランス−4−ヒドロキシ−L−プロリンを生成する、L−プロリン4位水酸化酵素活性を有する蛋白質をコードする遺伝子を含むDNA断片をベクターに組み込んで得られる組換え体DNAを保有し、且つ、L−プロリンの生合成系の活性の強化された形質転換体、および該形質転換体を培養し、該培養物、菌体またはそれらの処理物を酵素源として、2−ケトグルタル酸および2価鉄イオンの存在下、水性媒体中で、L−プロリンをトランス−4−ヒドロキシ−L−プロリンに変換させ、生成したトランス−4−ヒドロキシ−L−プロリンを該水性媒体より採取することを特徴とするトランス−4−ヒドロキシ−L−プロリンの製造法に関する。
【発明の効果】
【0014】
本発明によれば、医薬品の合成原料または食品添加物として有用なトランス−4−ヒドロキシ−L−プロリンを工業的に製造する方法、該方法に有用なL−プロリン4位水酸化酵素遺伝子を含有し、かつ、プロリン合成系の活性の強化された形質転換体を提供することができる。
【発明を実施するための最良の形態】
【0015】
以下に本発明を詳細に説明する。
本発明にかかわるL−プロリン4位水酸化酵素は、2−ケトグルタル酸および2価鉄イオンの存在下において、遊離のL−プロリンを水酸化してトランス−4−ヒドロキシ−L−プロリンを生成する酵素である。
本発明のL−プロリン4位水酸化酵素をコードするDNAを含むプラスミッドとしては、例えばpRH71があげられる。pRH71を含む大腸菌であるEscherichia coli SOLR/pRH71 は、平成7年3月2日付けで工業技術院生命工学工業技術研究所、日本国茨城県つくば市東1丁目1番3号(郵便番号305)にFERM BP−5025として寄託されている。
【0016】
L−プロリン4位水酸化酵素遺伝子を宿主中で発現させるためには、まず、L−プロリン4位水酸化酵素遺伝子を含むDNA断片を、制限酵素類あるいはDNA分解酵素類で、L−プロリン4位水酸化酵素遺伝子を含む適当な長さのDNA断片とした後に、発現ベクター中プロモーターの下流に挿入し、次いで上記DNAを挿入した発現ベクターを、発現ベクターに適合した宿主中に導入する。宿主としては、目的とする遺伝子を発現できるものは全て用いることができる。例えば、エッシェリヒア属、セラチア属、コリネバクテリウム属、ブレビバクテリウム属、シュードモナス属、バチルス属、等に属する微生物菌株の他、酵母菌株や動物細胞宿主等をあげることができる。
【0017】
発現ベクターとしては、上記宿主において自立複製可能ないしは染色体中への組込みが可能で、L−プロリン4位水酸化酵素遺伝子を転写できる位置にプロモーターを含有しているものが用いられる。
大腸菌等の微生物を宿主として用いる場合は、L−プロリン4位水酸化酵素発現ベクターは微生物中で自立複製可能であると同時に、プロモーター、リボソーム結合配列、L−プロリン4位水酸化酵素遺伝子、転写終結配列、より構成されていることが好ましい。プロモーターを制御する遺伝子が含まれていてもよい。
【0018】
発現ベクターとしては、例えば、pBTrp2、pBTac1、pBTac2(いずれもベーリンガーマンハイム社より市販)、pKYP10( 特開昭58−110600)、pKYP200〔アグリカルチャラル・バイオロジカル・ケミストリー(Agric. Biol.Chem.), 48巻, 669〜675ページ(1984年) 〕、pLSA1〔アグリカルチャラル・バイオロジカル・ケミストリー(Agric. Biol.Chem.),53巻,277ページ(1989年)〕、pGEL1〔プロシーディング・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc. Natl.Acad.Sci., USA) ,82巻,4306ページ( 1985年)〕、pBluescript (STRATAGENE社)、pTrs30〔エシェリヒア・コリJM109/pTrS30 (FERM BP−5407)より調製〕およびpTrs32〔エシェリヒア・コリ JM109/pTrS32 (FERM BP−5408)より調製〕等を例示することができる。
【0019】
プロモーターとしては、大腸菌等の宿主中で発現できるものであればいかなるものでもよい。例えば、trpプロモーター(Ptrp)、lacプロモーター(Plac)、PL プロモーターPR プロモーターなどの、大腸菌やファージ等に由来するプロモーターをあげることができる。またPtrpを2つ直列させたプロモーター(Ptrpx2)、tacプロモーターのように人為的に設計改変されたプロモーター等も用いることができる。
【0020】
リボソーム結合配列としては、大腸菌等の宿主中で発現できるものであればいかなるものでもよいが、リボソーム結合配列と開始コドンとの間を適当な距離(例えば6〜18塩基)に調節したプラスミッドを用いることが好ましい。
L−プロリン4位水酸化酵素遺伝子はL−プロリン4位水酸化酵素をコードする遺伝子であればいずれも用いることができるが、該遺伝子のDNA配列を宿主微生物での発現に最適なコドンとなるように、塩基を置換して用いることにより高発現化が達成できる。大腸菌を宿主として、発現に最適なコドンとなるように塩基を置換したL−プロリン4位水酸化酵素遺伝子の具体例として、配列番号1で示される塩基配列等をあげることができる。
【0021】
本遺伝子の発現には転写終結配列は必ずしも必要ではないが、好適には構造遺伝子直下に転写終結配列を配置することが望ましい。
宿主としては、Escherichia coli XL1-Blue 、Escherichia coli XL2-Blue、Escherichia coli DH1、Escherichia coliMC1000 、 Escherichia coli KY3276、Escherichia coliW1485、Escherichia coli JM109、Escherichia coli HB101、Escherichia coli No.49、Escherichia coli W3110、Escherichia coliNY49 、Bacillus subtilis 、Bacillus amyloliquefacines、Brevibacterium immariophilum ATCC14068、Brevibacterium saccharolyticumATCC14066、Brevibacterium flavum ATCC14067 、Brevibacterium lactofermentumATCC13869 、Corynebacterium glutamicum ATCC13032、Corynebacterium acetoacidophilum ATCC13870、Microbacterium ammoniaphilumATCC15354等をあげることができる。
【0022】
酵母菌株を宿主として用いる場合には、発現ベクターとして、例えば、YEp13(ATCC37115 )、YEp24(ATCC37051 )、YCp50(ATCC37419 )等を例示することができる。 プロモーターとしては、酵母菌株の宿主中で発現できるものであればいかなるものでもよい。例えば、ヘキソースキナーゼ等の解糖系の遺伝子のプロモーター、gal1 プロモーター、gal 10プロモーター、ヒートショック蛋白質プロモーター、MFα1 プロモーター、CUP 1 プロモーター等のプロモーターをあげることができる。
【0023】
宿主としては、Saccharomyces cerevisae 、Schizosaccharomyces pombe 、Kluyveromyces lactis、Trichosporon pullulans、Schwanniomyces alluvius 等をあげることができる。
動物細胞を宿主として用いる場合には、発現ベクターとして、例えば、pcDNAI/Amp、pcDNAI、pcDM8(いずれもフナコシ社より市販)等を例示することができる。プロモーターとしては、動物細胞の宿主中で発現できるものであればいかなるものでもよい。例えば、ヒトCMVのIE(immediate early)遺伝子のプロモーター等のプロモーターをあげることができる。また、ヒトCMVのIE遺伝子のエンハンサーをプロモーターと共に用いてもよい。宿主としては、ナマルバ細胞、HBT5637(特開昭63−299)、COS細胞、CHO細胞等をあげることができる。
【0024】
動物細胞へのDNAの導入法としては、動物細胞にDNAを導入することができればいかなる方法も用いることができる。例えば、エレクトロポーレーション法〔Miyajiら:サイトテクノロジー(Cytotechnology), 3, 133(1990)〕、リン酸カルシウム法(特開平2-227075)、リポフェクション法〔フィリップ・エル・フェルグナー(PhilipL. Felgner)ら:プロシーディング・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス (Proc. Natl. Acad. Sci., USA), 84, 7413(1987)〕等を用いることができる。形質転換株の取得および培養は、特開平2-227075あるいは特開平2-257891に記載されている方法に準じて行なうことができる。
【0025】
宿主をプロリン生合成系の活性の強化されたものにするためには、1)宿主中のプロリン合成系遺伝子のコピー数を増加させる、2)プロリンによってフィードバック阻害を受けるプロリン合成系の酵素をコードする遺伝子に変異を生ぜしめ、プロリンによるフィードバック阻害が著しく減少した酵素をコードする遺伝子を宿主に導入する、3)プロリンの分解活性を欠失させる、または4)これらを組み合わせることにより達成することができる。
【0026】
プロリン合成系遺伝子またはプロリンによるフィードバック阻害が著しく減少したプロリン合成系酵素をコードする遺伝子は宿主の染色体上に存在しても良いし、またプラスミドなどの宿主中のベクター上に存在してもよい。
プロリンによるフィードバック阻害が著しく減少したプロリン合成系酵素をコードする遺伝子としては、proB74遺伝子、DHPrproB遺伝子[ジーン(gene)、39巻、109−112ページ、(1984年)]等、プロリンによるフィードバック阻害が著しく減少したプロリン合成系酵素をコードする遺伝子をあげることができる。
【0027】
また、プロリン合成系遺伝子またはプロリンによるフィードバック阻害が著しく減少したプロリン合成系酵素をコードする遺伝子をプロリン4位水酸化酵素遺伝子と共にプラスミドなどの宿主中のベクター上に存在させる場合は、両遺伝子が一つのプラスミド上に存在していてもよいし、別の共存可能なプラスミド上に存在していてもよい。
共存可能なプラスミドとしては、例えば、大腸菌においては、コリシンE1系のプラスミド(例、pBR322)とpACYC系のプラスミド、コリシンE1系のプラスミドとF因子系のプラスミド、コリシンE1系のプラスミドとR因子系のプラスミド等をあげることができる。
【0028】
プロリン生合成系遺伝子を宿主中で発現させるためには、前述のL−プロリン4位水酸化酵素遺伝子での発現と同様の方法を用いることができる。
宿主のプロリン分解活性を欠失させるには、変異剤で宿主を処理した後、例えば細菌などではPro−TTCプレート[アプライド・アンド・エンバイロメンタル・マイクロバイオロジー(Appl.Environ,Microbiol,)、33巻、434−444ページ、(1977年)]上で白色のコロニーを形成するものを選択することで取得できる。
【0029】
更に、大腸菌等ではput遺伝子の適当な位置にトランスポゾンが挿入されることでプロリンの資化性が失われることがわかっている[ジェネティクス(Genetics)、92巻、741−747ページ(1979年)]ので、トランスポゾンを挿入した後、薬剤耐性と前述したPro−TTCプレートにより、プロリン分解活性欠失株を取得することもできる。
【0030】
また、トランスポゾンによりプロリン分解活性が欠失した株より、P1トランスダクション[A Short Course In Bacterial Genetics, A Laboratory Manual andHandbook for Esherichia coli and Related Bacteria, J. H. Miller, Cold SpringHarbor Laboratory Press, 1922, Laboratory Manual, pp.263 ]により別の菌株にプロリン分解活性欠失という形質を移すこともできる。
【0031】
上記のようにしてプロリン生合成系の活性の強化された宿主を用いて得られた形質転換体の培養は、通常の培養方法に従って行うことができる。
大腸菌や酵母菌等の微生物を宿主として用いた形質転換体を培養する培地は、微生物が資化し得る炭素源、窒素源、無機塩類等を含有し、形質転換体の培養を効率的に行える培地であれば天然培地、合成培地のいずれでもよい。
【0032】
炭素源としては、それぞれの微生物が資化し得るものであればよく、グルコース、フラクトース、スクロース、これらを含有する糖蜜、デンプンあるいはデンプン加水分解物等の炭水化物、酢酸、プロピオン酸等の有機酸、エタノール、プロパノールなどのアルコール類が用いられる。
窒素源としては、アンモニア、塩化アンモニウム、硫酸アンモニウム、酢酸アンモニウム、りん酸アンモニウム、等の各種無機酸や有機酸のアンモニウム塩、その他含窒素化合物、並びに、ペプトン、肉エキス、酵母エキス、コーンスチープリカー、カゼイン加水分解物、大豆粕および大豆粕加水分解物、各種発酵菌体およびその消化物等が用いられる。
【0033】
無機物としては、りん酸第一カリウム、りん酸第二カリウム、りん酸マグネシウム、硫酸マグネシウム、塩化ナトリウム、硫酸第一鉄、硫酸マンガン、硫酸銅、炭酸カルシウム等が用いられる。
培養は、振盪培養または深部通気攪拌培養などの好気的条件下で行う。培養温度は15〜40℃がよく、培養時間は、通常16〜100時間である。培養中pHは、3. 0〜9. 0に保持する。pHの調整は、無機あるいは有機の酸、アルカリ溶液、尿素、炭酸カルシウム、アンモニアなどを用いて行う。
【0034】
また培養中必要に応じて、アンピシリンやテトラサイクリン等の抗生物質を培地に添加してもよい。
プロモーターとして誘導性のプロモーターを用いた発現ベクターで形質転換した微生物を培養するときには、必要に応じてインデューサーを培地に添加してもよい。例えば、lacプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはイソプロピル−β−D−チオガラクトピラノシド(IPTG)等を、trpプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはインドールアクリル酸(IAA)等を培地に添加してもよい。
【0035】
動物細胞を宿主として用いた形質転換体を培養する培地は、一般に使用されているRPMI1640培地、EagleのMEM培地またはこれら培地に牛胎児血清等を添加した培地等が用いられる。
培養は、5%CO2 存在下等の条件下で行う。培養温度は35〜37℃がよく、培養時間は、通常3〜7日間である。
【0036】
また培養中必要に応じて、カナマイシン、ペニシリン等の抗生物質を培地に添加してもよい。
このように培養して得た形質転換体中には、遺伝子源として用いた微生物菌株、例えばダクチロスポランジウム・エスピーRH1等およびpTr2−4OHを保有する組換え大腸菌と比較してL−プロリン4位水酸化酵素が著量生成蓄積しており、プロリン生合成活性も強く、トランス−4−ヒドロキシ−L−プロリン製造を、遺伝子源として用いた微生物菌株、例えばダクチロスポランジウム・エスピーRH1等およびpTr2−4OHを保有する組換え大腸菌を用いた場合と比較して、はるかに効率的に行うことができる。
【0037】
該形質転換体中にL−プロリン4位水酸化酵素が生成していることは、培養物、菌体またはそれらの処理物を、酵素反応に適した水性媒体中に、L−プロリン、二価鉄イオン、2−ケトグルタル酸とともに加え、また必要に応じて界面活性剤や有機溶剤を添加することにより、生成するトランス−4−ヒドロキシ−L−プロリンを検出することにより知ることができる。生成されたL−プロリン4位水酸化酵素の活性は、下記測定条件下、1分間に1nmolのトランス−4−ヒドロキシ−L−プロリンを生成する活性を1単位(U)として表示する。なお、ここでは、微生物菌体に加え、動物細胞を含めて菌体と呼ぶ。
【0038】
L−プロリン4位水酸化酵素活性の測定:
12mM L−プロリン、24mM 2−ケトグルタル酸、4mM 硫酸第一鉄および8mM L−アスコルビン酸を含有する240mMのMES〔2−(N−モルホリノ)エタンスルホン酸〕緩衝液(pH6. 5)に菌体、菌体処理物または酵素標品等を添加して合計250μlとし、35℃、10分間反応する。反応液を100℃、2分間加熱して反応を停止した後に、反応液中に生成したトランス−4−ヒドロキシ−L−プロリンを高速液体クロマトグラフィー(以下、HPLCと略記する)を用いて定量する。
【0039】
定量にはトランス−4−ヒドロキシ−L−プロリンを定量できる方法であればどのような方法を用いてもよいが、例えば、反応液中のトランス−4−ヒドロキシ−L−プロリンを配位子交換クロマトグラフィーカラム、例えば、株式会社住化分析センター製SUMICHIRAL OA5000 等を用いてHPLCで分離溶出後、7−クロロ−4−ニトロベンゾ−2−オキサ−1,3−ジアゾ−ル(以下、NBDと略記する)によってポストカラム誘導体化し検出する方法(ポストカラム誘導体化法) 、あるいは反応液中の目的化合物をあらかじめNBD誘導体化しておき、これをHPLCを用いた逆相クロマトグラフィーにかけてNBD誘導体化物を分離後検出する方法〔WilliamJ. Lindblad and Robert F. Diegelmann, アナリティカル・バイオケミストリー(Analytical Biochemistry)、138巻、390〜395ページ、1984年〕(プレカラム誘導体化法)等があげられる。NBD誘導体化物の検出は、いずれもその蛍光( 励起波長503nm、蛍光波長541nm)測定によって行われる。
【0040】
上記のようにして培養菌体中にL−プロリン4位水酸化酵素の生成が確認された形質転換体を、前述の形質転換体の培養条件と同様の条件で培養し、トランス−4−ヒドロキシ−L−プロリンを生成蓄積させ、該培養物よりトランス−4−ヒドロキシ−L−プロリンを採取することにより、トランス−4−ヒドロキシ−L−プロリンを製造することができる。
【0041】
該培養において、必要に応じて2−ケトグルタル酸および2価鉄イオンを培養液中に添加してもよい。
本発明により製造されたトランス−4−ヒドロキシ−L−プロリンは、前述のポストカラム誘導体化法やプレカラム誘導体化法によって、定量分析することができる。
【実施例1】
【0042】
L−プロリン4位水酸化酵素高発現プラスミッドの構築
配列番号2記載のDNAと配列番号3記載のDNAをアプライド・バイオシステムズ(Applied Biosystems)社製380A・DNA合成機を用いて合成した。 該合成DNAの3’末端25bpは互いに相補的な配列となるように設計されている。さらに、該合成DNAには、Dactylosporangium sp. RH1 由来のL−プロリン4位水酸化酵素蛋白のN末端部分をコードする塩基配列が含有されているが、該塩基配列には、Dactylosporangium sp. RH1 由来の塩基配列を、大腸菌での発現に最適なコドンとなるように部位特異的な塩基置換がほどこされている。
【0043】
該合成DNAをプライマーかつ鋳型としてPCRを行った。
PCRはPfuDNAポリメラーゼ(STRATAGENE社製)0.5U、Pfu DNAポリメラーゼ用×10緩衝液(STRATAGENE社製)2μl、DMSO 2μl、各2.5mMdNTP液1μl、配列番号2記載の合成DNAと配列番号3記載の合成DNA各2μMを含む反応液20μlを用い、96℃、5分間のインキュベーションの後、96℃−2分間、50℃−1分間、75℃−1分間のインキュベーション工程を35回繰り返す条件で行った。
【0044】
反応終了後、該反応液を、15%ポリアクリルアミドゲル(アトー株式会社製、パジェルNPU-15L )電気泳動にかけ、107bpの増幅断片の生成を確認した。
該107bpの増幅断片を日本エイドー株式会社製のダヴィンチくん(ペンタッチリカバリーNB−7000型)を用いて回収後、該107bpのDNA断片の両末端をHind IIIおよびSal I で切断した。
【0045】
該Hind III−Sal I 切断断片をBio, Inc. 製 MERmaid Kitを用いて回収した。回収液量は16μlとなった。
参考例1に記載の方法に従って作成したプラスミドpTr2−4OHDNAをBam HIおよびPvu IIで切断後、反応液をアガロースゲル電気泳動にかけ、2つの断片が生成していることを確認した。該断片の内、L−プロリン水酸化酵素の構造遺伝子を含む長い断片をバイオラッド社製のPrep-A-geneを用いて回収し、宝酒造社製のブランティングキットを用いて末端を平滑化した後、宝酒造社製のライゲーションキットを用いて連結環状化した。
【0046】
該連結環状化したプラスミドを用い、E. coli JM109 株を常法に従って形質転換し、該形質転換体を50μg/mlのアンピシリンを含むLB寒天培地に塗布後、37℃で1晩培養した。
生育してきた形質転換体のコロニーより常法に従ってプラスミドを抽出し、その構造を制限酵素消化により確認した。
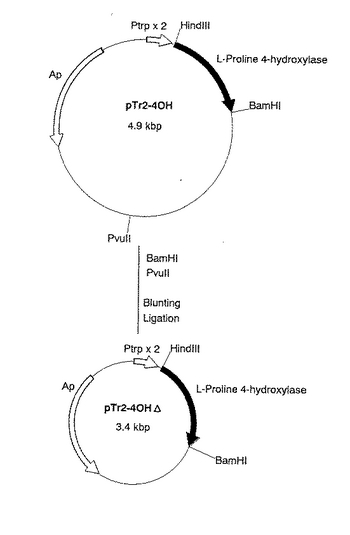
【0047】
以上の結果、pTr2−4OHの一部の配列を削除したプラスミドpTr2−4OHΔ(図1)を得た。
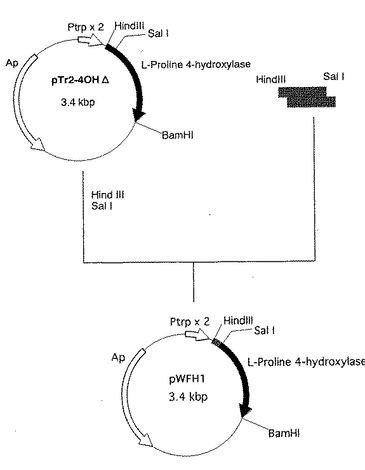
取得したプラスミドpTr2−4OHΔDNAをHind IIIおよびSal I で切断後、該切断部位に、HindIIIおよびSal I 処理した上記PCR増幅断片を、宝酒造社製のライゲーションキットを用いて、挿入した。
【0048】
得られたプラスミドを用い、E. coli XL1-Blue MRF' 株を常法に従って形質転換し、該形質転換体を50μg/mlのアンピシリンを含むLB寒天培地に塗布後、37℃で1晩培養した。 生育してきた形質転換体のコロニーより常法に従ってプラスミドを抽出し、プラスミドpWFH1(図2)を取得した。
該プラスミドの構造を制限酵素消化により確認した。PCR増幅断片挿入部分についてはアプライドバイオシステムズ社製の塩基配列決定キット(Taq DyeDeoxyTM Terminator Cycle Sequencing Kit)を用いて、塩基配列を決定した。該塩基配列を配列番号1に示す。
【0049】
該プラスミドは、構造遺伝子の5’末端からSal I 部位においてDactylosporangium sp.RH1 由来の塩基配列と一部異なる以外は、Dactylosporangium sp. RH1 由来のL−プロリン4位水酸化酵素とまったく同じアミノ酸配列をコードする構造遺伝子DNA断片が、Ptrp×2の転写方向と同方向に挿入された構造を有していた(図2)。
第1表に示したように、pWFH1を保有する形質転換体は、遺伝子源として用いたダクチロスポランジウムエスピーRH1株と比較して1400倍、pTr2−4OHを保有する形質転換体と比較して約5.4倍のL−プロリン水酸化酵素を生産していた。
【0050】
【表1】

【実施例2】
【0051】
プロリン分解系欠損株の造成
E. coli ATCC12435 のプロリン分解に関与する遺伝子putAを以下の方法で破壊し、プロリン分解系欠損株を造成した。
国立遺伝学研究所保存菌株であるE. coli ME8395 株[F- :pyrD34trp-45 his-68 thyA25 thi deoR33 galK35xyl-7 mtl-2 malA1 rpsL118 λR ( λ) -appA1 putA::Tn5 (Mu+) ]を35μg/mlのカナマイシンを含有するLB培地に植菌し、終夜培養した。
【0052】
該培養液100μlにP1ファージ液100μlを添加・混合し、5分間放置した。
該放置液を10mM CaCl2 を含む3mlのLB軟寒天培地と混合し、10mMCaCl2 を含むLB寒天培地上に重層後、37℃で7時間培養した。 培養後、得られた寒天培地表面のライゼートを10mMCaCl2 を含む2mlのLB培地中に回収した。 該回収液に0.5mlのクロロホルムを添加し、ボルテックスミキサーを用いて混合後、3000rpmで15分間遠心し、得られた上清をP1ファージライゼートとして用いた。
【0053】
該P1ファージライゼート液50μlと10mM CaCl2 を含むLB培地で培養したE. coli ATCC12435 培養液100μlとを混合し、37℃で20分間静置した。該静置液をエフ・トップ・サイトレイト(F-top-citrate:NaCl 8. 5g/ l、クエン酸2ナトリウム100mM、寒天0. 7%)3mlと混合し、35μg/mlのカナマイシンを含有するLB寒天培地上に塗布後、37℃で1日培養した。
【0054】
該培養により生育してきたカナマイシン耐性株を0.85%のNaClに懸濁した後、Pro−TTCプレート(K2 HPO4 7g/ l、KH2 PO4 3g/ l、MgSO4 0. 1g/ l、プロテオースペプトン2g/l、2,3,5-トリフェニルテトラゾリウムクロライド0. 025g/ l、L- Pro 2g/ l、寒天15g/ l、pH7. 2) に塗布し、37℃で1〜2日間培養した。
【0055】
該培養によりPro−TTCプレート上に白色のコロニーを形成した株を、プロリンを分解資化する能力の失われた株として選択し、プロリン分解活性欠損株E. coliWT1株を取得した。該菌株は平成8年8月7日付けで工業技術院生命工学工業技術研究所、日本国茨城県つくば市東1丁目1番3号(郵便番号305)にFERM BP−5618として寄託されている。
【実施例3】
【0056】
プロリン生合成系遺伝子proB74およびproA発現プラスミドの構築
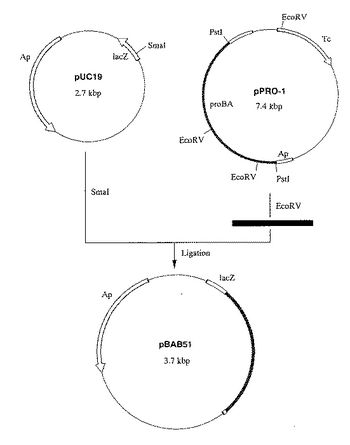
大腸菌由来のγ-glutamylkinaseをコードするproB遺伝子をプロリンによるフィードバック阻害に対して脱感作された変異型遺伝子に変換したproB74遺伝子および大腸菌由来のγ-glutamylphosphate reductase をコードするproA遺伝子の発現プラスミドpPF1を以下の方法により構築した。
大腸菌由来のproBおよびproA遺伝子を含むプラスミドpPRO−1[大腸菌K83株(FERM BP−2807)より取得]をEco RVで消化し、アガロースゲル電気泳動後、Prep-A-gene DNAPurification System (BIO-RAD社製)でproB遺伝子の一部を含む約1kbのDNA断片を取得した。
該DNA断片をpUC19(宝酒造社製)のSmaI 消化物と連結し、プラスミドpBAB51を得た(図3)。該プラスミド中のproB遺伝子を、以下の方法により、公知[A. M. Dandekarand S. L. Uratsu, J. Bacteriol. 170, 5943 (1988)] のプロリンによるフィードバック阻害に対して脱感作された変異型遺伝子proB74に変換した。
【0057】
配列番号4に示したオリゴヌクレオチドA1および配列番号5に示したオリゴヌクレオチドA2をアプライド・バイオシステムズ(Applied Biosystems)社製380A・DNA合成機を用いて合成した。オリゴヌクレオチドA1とM13プライマーM3(宝酒造社製、カタログNo.3831)を一組のプライマーとして、pBAB51を鋳型として用い、PCR法により変異型proB遺伝子であるproB74遺伝子の部分配列を増幅した。
【0058】
PCRは、pBAB51を0.1μg、オリゴヌクレオチドA1とM13プライマーM3を各2μM、Taq DNA ポリメラーゼ(宝酒造社製)を1U、dNTPミクスチャー(宝酒造社製、カタログNo.4030)を1.6μl、添付バッファー2μlを含む反応液20μlを用い、94℃−30秒間、52℃−30秒間、72℃−1分間のインキュベーション工程を30回繰り返し、最後に72℃−5分間インキュベーションすることにより行った。
【0059】
同様の方法で、オリゴヌクレオチドA2とM13プライマーRV(宝酒造社製、カタログNo.3830A)を一組のプライマーとして、pBAB51を鋳型として用い、PCR法により変異型proB遺伝子の部分配列を増幅した。
PCR法により増幅された該2種類のDNAをそれぞれアガロースゲル電気泳動にかけた後、Prep-A-gene DNA Purification System (BIO-RAD社製)を用いて精製した。
【0060】
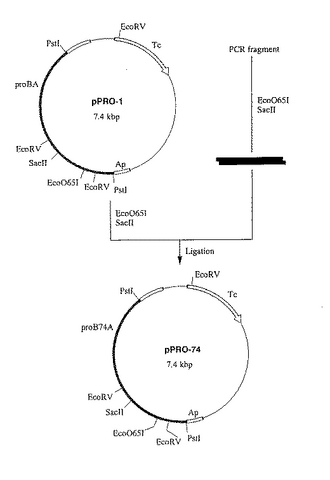
該2種の精製DNA断片を混合したものを鋳型とし、M13プライマーM3およびM13プライマーRVをプライマーとして用い、上記と同様の方法により、PCRおよび精製を行い、proB74遺伝子の塩基配列を含む約1kbのDNA断片を取得した。
該DNA断片をEco O65IおよびSac IIで消化し、EcoO65I−Sac II消化断片を取得した。
【0061】
該消化断片と、pPRO−1のEco O65I−Sac II消化DNA断片(約6.8kbp)とを連結し、pPRO−1のproB遺伝子を脱感作型proB74遺伝子に変換したプラスミドpPRO74を作製した(図4)。
配列番号6に示したオリゴヌクレオチドp1および配列番号7に示したオリゴヌクレオチドp2をアプライド・バイオシステムズ(Applied Biosystems)社製380A・DNA合成機を用いて合成した。
【0062】
該p1、p2をプライマーとして、プラスミドpPRO74を鋳型として用い、PCR法によりproB74およびproA遺伝子を増幅した。
PCRは、pPRO74を0.1μg、p1、p2を各2μM、タカラ・イーエックス・タック(TaKaRa Ex Taq :宝酒造社製、コードRR001Q)1U、dNTPミクスチャー(宝酒造社製、カタログNo.4030)を1.6μl含む反応液20μlを用い、94℃−1分間、42℃−2分間、72℃−3分間のインキュベーション工程を30回繰り返すことにより行った。
【0063】
該反応液をアガロースゲル電気泳動にかけ、proB74およびproAを含む2370bpの増幅断片を常法により抽出し、GENECLEANII KIT(BIO 101, INC. 製)を用いて該DNA断片を回収した。回収した2370bpのDNA断片をHind IIIおよびBamHIで切断後、エタノール沈殿法によりエタノール沈殿を得た。該エタノール沈殿を5μlのTEに溶解し、proB74およびproA断片として用いた。
【0064】
該proB74およびproA断片と、プラスミドpSTV29(宝酒造社製)のHind III−Bam HI消化DNA断片とを宝酒造社製のDNAライゲーションキットを用いて連結し、proB74およびproA断片の挿入されたプラスミドを構築した。該プラスミドをE.coli JM109 株に常法に従って形質転換後、30μg/mlのクロラムフェニコール、0.1mMIPTG、40μg/mlのX−Galを含むLB寒天培地に塗布し、37℃で一晩培養した。
【0065】
該培養により白色コロニーを形成した株より常法に従ってプラスミドを抽出し、該プラスミドの構造を制限酵素消化により確認した。 上記の方法により、脱感作型γ-glutamyl kinaseのN末端にβ−ガラクトシダーゼのαフラグメントのN末端8アミノ酸残基(lacZ.Nterm)が結合した融合蛋白をコードする遺伝子およびproA遺伝子がPlac支配下に存在するプラスミドpPF1を取得した(図5)。
【0066】
該融合蛋白の構造遺伝子(lacZ.Nterm-proB74 )の塩基配列とアミノ酸配列を配列番号8に示す。
【実施例4】
【0067】
プロリン生合成系遺伝子発現プラスミドを含む形質転換体によるトランス−4−ヒドロキシ−L−プロリンの生産
実施例3で構築したプラスミドpPF1を用いて、実施例2で取得したE. coli WT1 株を形質転換し、形質転換株E. coliWT1/pPF1を取得した。
実施例1で構築したプロリン4位水酸化酵素発現プラスミドpWFH1で、実施例2で取得したWT1株を形質転換しE. coli WT1/pWFH1 を取得した。
【0068】
該形質転換株E. coli WT1/pWFH1 より塩化カルシウム法にてコンピテントセルを作製し、実施例3で作製したプラスミドpPF1を導入し、両プラスミドを保有する形質転換株E.coli WT1/pWFH1/pPF1を30μg/ mlのクロラムフェニコールと50μg/ mlのアンピシリンを含むLB培地に生育してくるコロニーを選択することにより取得した。
WT1 、WT1/pPF1、WT1/pWFH1およびWT1/pWFH1/pPF1をそれぞれカナマイシン37.5μg/ ml、アンピシリン100μg/ ml、クロラムフェニコール30μg/ ml、そしてアンピシリン100μg/mlおよびクロラムフェニコール30μg/ mlを含むLB培地で37℃で16時間培養した。
【0069】
該培養液それぞれ100μlを、2%(W/ V)の炭酸カルシウムを含む10mlのMed7培地(グルコース2%、硫酸アンモニウム1%、K2 HPO40. 1%、NaCl 0. 2%、MgSO4 0. 05%、FeSO4 0. 0278%、CaCl20. 0015%、ペプトン0. 8%)をいれた太型試験管(φ20×200mm)に植菌し、30℃で24時間培養した。
【0070】
該培養上清中のL−プロリンおよびトランス−4−ヒドロキシ−L−プロリン含量を第2表に示した。
【0071】
【表2】
【0072】
プロリン生合成系遺伝子発現プラスミドpPF1保有菌株においてはL−プロリンの生成が著しく増加し、L−プロリン4位水酸化酵素発現プラスミドpWFH1およびプロリン生合成系遺伝子発現プラスミドpPF1の共存する形質転換体においてはL−プロリン4位水酸化酵素発現プラスミドを単独で保持する形質転換体に比べてトランス−4−ヒドロキシ−L−プロリンの生成量が顕著に増加することが確認できた。
【実施例5】
【0073】
L ―プロリン4位水酸化酵素とプロリン生合成系遺伝子proB74およびproA発現プラスミドの構築
Dactylosporangium sp. RH1 由来のL−プロリン4 位水酸化酵素遺伝子、プロリン生合成系遺伝子proB74およびproAの全ての遺伝子を単一のプラスミド上に保持し、これを発現させるような発現プラスミドpWFP1を以下の方法により構築した。
【0074】
実施例1で作成したpWFH1をテンプレートとし、PCR法にてL−プロリン4位水酸化酵素の構造遺伝子を増幅した。
PCRはpWFH1プラスミド0.1μg 、Pfu DNAポリメラーゼ(stratagene社製)0.5 U、Pfu DNAポリメラーゼ用×10緩衝液(STRATAGENE社製)2μl、DMSO 2μl、各2.5mMdNTP液1μl、配列番号9記載の合成DNAと配列番号10記載の合成DNA各2μMを含む反応液20μlを用い、96℃、5分間のインキュベーションの後、96℃−2分間、58℃−1分間、75℃−3分間のインキュベーション工程を30回繰り返す条件で行った。
【0075】
反応終了後、該反応液をアガロースゲル電気泳動にかけ、L−プロリン4 位水酸化酵素遺伝子を含む約800bpの増幅断片を常法により抽出し、GENECLEAN II KIT(BIO 101, INC. 製)を用いて該DNA断片を回収した。該回収DNA断片をHindIIIおよびEco RIで切断後、アガロースゲル電気泳動にかけ、GENECLEAN II KIT(BIO 101, INC. 製)を用いてL−プロリン4位水酸化酵素遺伝子を有する該Hind III−Eco RI切断DNA断片を回収した。
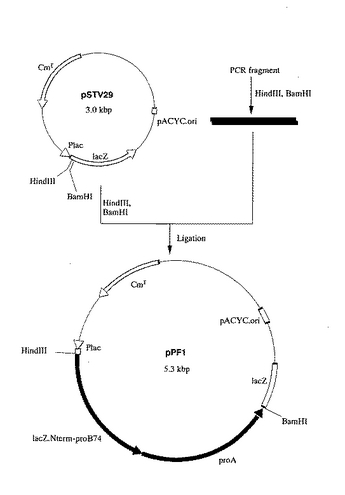
該L−プロリン4位水酸化酵素遺伝子断片と、プラスミドpBluescriptII KS(+)(stratagene社製)のHind III―EcoRI消化DNA断片とを宝酒造社製のDNAライゲーションキットを用いて連結し、L−プロリン4位水酸化酵素断片の挿入されたプラスミドpBII―4OHを構築した(図6)。
実施例3で作成したpPRO74をテンプレートとしPCR法にてproB74およびproA遺伝子を増幅した。
【0076】
PCRはpPRO74プラスミド0.1 μg 、タカラ・イーエックス・タック(TaKaRa Ex Taq :宝酒造社製、コードRR001Q)1U、タカラ・イーエックス・タック用×10緩衝液(宝酒造社製)2μl、各2.5mMdNTP液1.6μl、配列番号11記載の合成DNAと配列番号12記載の合成DNA各2μMを含む反応液20μlを用い、94℃−1分間、42℃−2分間、75℃−3分間のインキュベーション工程を30回繰り返す条件で行った。
【0077】
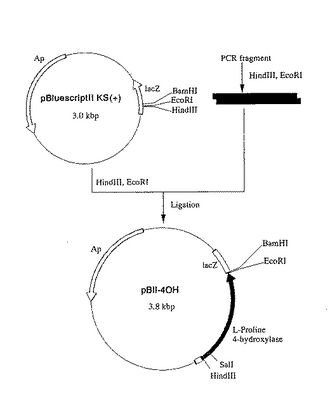
反応終了後、該反応液をアガロースゲル電気泳動にかけ、proB74およびproA遺伝子を含む約2.3kbpの増幅断片を常法により抽出し、GENECLEANII KIT(BIO 101, INC. 製)を用いて該DNA断片を回収した。回収したDNA断片をBam HIおよびEco RIで切断後、フェノール/クロロホルム処理、エタノール沈殿を行い、該DNA断片を回収した。このproB74およびproA遺伝子を含むDNA断片と、上記プラスミドpBII―4OHのHindIII―Eco RI消化DNA断片とを宝酒造社製のDNAライゲーションキットを用いて連結し、L−プロリン4位水酸化酵素遺伝子、proB74およびproAを含むプラスミドpBII―4OHBAを構築した(図7)。
【0078】
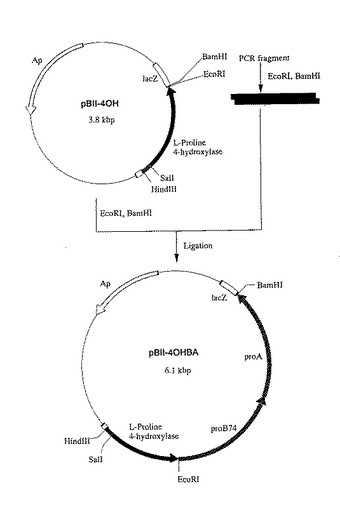
プラスミドpBII―4OHBAをHind IIIおよびBam HIで消化し、アガロースゲル電気泳動にかけ、L−プロリン4位水酸化酵素遺伝子、proB74およびproAを含む約3.16kbpのDNA断片を取得した。また、実施例1で作成したpWFH1をHindIIIおよびBam HIで消化し、アガロースゲル電気泳動にかけ、L−プロリン4位水酸化酵素遺伝子を含まず、複製開始点を含む方の約2.6kbpのDNA断片を取得した。取得した2つのDNA断片を宝酒造社製のDNAライゲーションキットを用いて連結し、トリプトファンタンデムプロモーター下でL−プロリン4位水酸化酵素、proB74タンパク、proAタンパクを発現させうるプラスミドpWFP1を構築した(図8)。
【実施例6】
【0079】
形質転換体E. coli WT1/pWFH1/pPF1およびE.coli WT1/pWFP1 を用いたトランス−4−ヒドロキシ−L−プロリンの生産
形質転換株WT1/pWFH1/pPF1株をアンピシリン100μg/mlおよびクロラムフェニコール30μg/mlを含む50ml Med4G培地[グルコース 2%、ポリペプトン(日本製薬社製) 1%、イーストエキストラクト(Difco社製) 0.5%、NaCl 1%、炭酸カルシウム 2%、pH7.0]に、形質転換株WT1/pWFP1株をアンピシリン100μg/ mlを含む50ml Med4G培地に、それぞれ植菌した後、30℃で16時間振とう培養した。
【0080】
該培養液を種培養液とし、2リッターのMed7培地を入れた5リッタージャーファーメンターにそれぞれ植菌し、WT1/pWFH1/pPF1株の培養においては、アンピシリン100μg/ mlおよびクロラムフェニコール30μg/ mlを、WT1/pWFP1株の培養においては、アンピシリン100μg/ mlを更に添加し、培養温度30℃、撹拌数400回転/分、通気量1リッター/培養液1リッター/分の条件で培養した。
【0081】
また、WT1/pWFH1/pPF1の培養においては、培養8時間目に、IPTGを0.2mMとなるように添加した。更に、両菌株とも、培養24時間目に、培養開始時と同種、同濃度の抗生物質を添加した。
培養中、グルコースをほぼ1%の濃度となるように適時培養液に添加し、NH4OHを用いてpH6. 5に下限コントロールした。また、培養5時間以降に、撹拌数を250〜700rpmに変化させることにより、培養液の溶存酸素濃度が培養開始時点の1/15となるようにコントロールした。
【0082】
99時間培養後、該培養液を遠心分離し、得られた上清中のトランス−4−ヒドロキシ−L−プロリン含量を定量した結果、WT1/pWFH1/pPF1株において156mM(20.5g/ l)、WT1/pWFP1 株において191mM(25.0g/ l)のトランス−4−ヒドロキシ−L−プロリンの生成蓄積が認められた。
【実施例7】
【0083】
L−プロリン4位水酸化酵素遺伝子およびプロリン生合成系遺伝子発現プラスミドを含む形質転換体によるトランスー4―ヒドロキシーL−プロリンの生産
実施例5で構築したプラスミドpWFP1を用いて、実施例2で取得したE. coliWT1株を形質転換し、形質転換株E. coli WT1/pWFP1 を取得した。
WT1/pWFP1 をアンピシリン100μg/mlを含むLB培地で37℃で16時間培養した。該培養液100μlを、2%(W/V)の炭酸カルシウムを含む10mlのMed7培地を入れた太型試験管(φ20×200mm)に植菌し、30℃で24時間培養した。 該培養上清中のL−プロリンは0.56g/Lであり、トランス−4−ヒドロキシ−L−プロリンは2.4g/Lであった。
【実施例8】
【0084】
形質転換体の菌体を用いたトランス−4−ヒドロキシ−L−プロリンの生産
形質転換体E. coli ATCC12435/pTr2-4OHを50μg/mlのアンピシリンを含む3mlLB培地に植菌し30℃、一晩振とう培養後、該培養液を遠心分離し、E.coli ATCC12435/pTr2-4OHの湿菌体を取得した。
形質転換体E. coli ATCC12435/pWFH1 をアンピシリン100μg/mlを含む100ml Med4培地〔ポリペプトン(日本製薬社製)1%、イーストエキストラクト(Difco社製)0.5%、NaCl1%〕に植菌し、30℃で16時間振とう培養した。
【0085】
該培養液を、2リッターのMed7培地(グルコース2%、硫酸アンモニウム1%、K2HPO4 0.1%、NaCl0.2%、MgSO4 0.05%、FeSO4 0.0278%、CaCl2 0.0015%、ペプトン0.4%)を入れた5リッタージャーファーメンターに植菌後、L−Proを200mM添加し、培養温度33℃、通気量1リッター/培養液1リッター/分という条件で48時間培養した。
【0086】
溶存酸素濃度が初期値の15分の1に達した時点で、撹拌数400回転/分を基準とし、700回転/分を上限として攪拌数を変化させることにより、溶存酸素濃度を初期値の15分の1となるようにコントロールした。
培養中、グルコースは無くならぬように、L−プロリンは約50mMとなるように適時添加し、NH4OHを用いて、pH6.5に下限コントロールした。
【0087】
該培養により得られた培養液を遠心分離し、E. coli ATCC12435/pTr2-4OHおよび E. coli ATCC12435/pWFH1 の湿菌体を取得した。
該両菌株の湿菌体は必要に応じて−20℃で凍結保存することが可能で、使用時に解凍し用いることができる。
反応液〔240mMのMES緩衝液(pH6. 5)中に、12mM L−プロリン、24mM 2−ケトグルタル酸、4mM 硫酸第一鉄および8mM L−アスコルビン酸を含有する〕250μlに湿菌体量として4%(w/v)となるように加え、35℃で10分間反応した。反応液を100℃、2分間加熱することにより反応を停止した。
【0088】
該反応停止液を遠心分離し、得られた上清中のトランス−4−ヒドロキシ−L−プロリン含量を定量した結果、E. coli ATCC12435/pTr2-4OH株において3mmol/l、E.coli ATCC12435/pWFH1 株において16mmol/lのトランス−4−ヒドロキシ−L−プロリンの生産が認められた。L−プロリン4位水酸化酵素に対する菌体活性はそれぞれ、7.5および40U/mg・wet cells であった。
【0089】
参考例1 L−プロリン4位水酸化酵素発現プラスミッドの構築
配列番号13記載のセンス鎖DNAプライマーと、配列番号14に記載のアンチセンス鎖DNAプライマーをアプライド・バイオシステムズ(Applied Biosystems)社製380A・DNA合成機を用いて合成した。該合成DNAをプライマーとして、pRH71[該プラスミドを含有するEscherichia coli SOLR/pRH71 (FERM BP−5025)より定法により取得]を鋳型としてPCRを行った。
【0090】
PCRは、pRH71を0. 1μg、センス鎖およびアンチセンス鎖DNAプライマー各2μM、Pfu DNAポリメラーゼ(STRATAGENE社製)0.125U/μl、DMSO10%、Tris・HCl(pH8. 2)20mM、KCl10mM、硫酸アンモニウム6mM、塩化マグネシウム2mM、TritonX−1000.1%、Bovine Serum Albumine 10ng/μlを含む反応液20μlを用い、96℃、5分間のインキュベーション後、96℃−2分間、58℃−1分間、75℃−1分間のインキュベーション工程を30回繰り返す条件で行った。
【0091】
反応終了後、該反応液をアガロースゲル電気泳動にかけ、L−プロリン4位水酸化酵素の構造遺伝子をコードする844bpの増幅断片が生成されていることを確認した。
該844bpの増幅断片をアガロースゲルより常法により抽出し、バイオラッド社製のPrep-A-gene を用いて断片を回収した。回収した844bpのDNA断片の両末端をHind IIIおよびBamHIで切断後、エタノール沈殿法により、エタノール沈殿を得た。該エタノール沈殿を5μlのTEに溶解した。
【0092】
pBR322を基本とし、Ptrpを2つ直列させたプロモーターPtrp×2を持つプラスミドpKYP200〔アグリカルチャラル・バイオロジカル・ケミストリー(Agric. Biol. Chem.), 48巻, 669〜675ページ(1984年) 〕と合成リンカーを組み合わせて作製されたATGベクターpTrS32〔エシェリヒア・コリJM109/pTrS32 (FERM BP−5408)より調製〕をHind IIIおよびBam HIで切断した。該切断部位に、HindIIIおよびBam HIで切断処理した上記のL−プロリン4位水酸化酵素構造遺伝子断片を、宝酒造社製のライゲーションキットを用いて、挿入した。
【0093】
得られたプラスミドを用い、E. coli XL1-Blue MRF' 株を常法に従って形質転換し、該形質転換体を50μg/mlのアンピシリンを含むLB寒天培地に塗布後、37℃で1晩培養した。
生育してきた形質転換体のコロニーより常法に従ってプラスミドを抽出し、その構造を制限酵素消化により確認した。構造遺伝子部分についてはアプライドバイオシステムズ社製の塩基配列決定キット(Taq DyeDeoxyTM Terminator Cycle Sequencing Kit)を用いて、塩基配列を決定し、配列番号15で示した塩基配列であることを確認した。
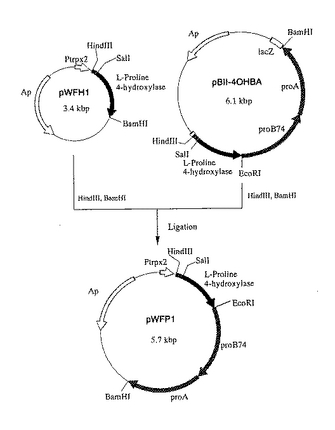
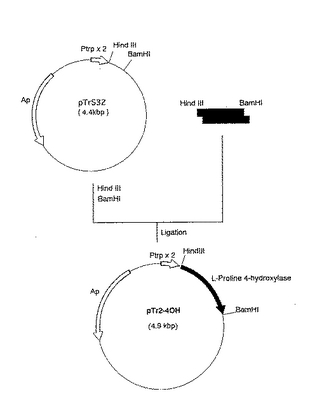
該方法により、Ptrp×2の転写方向と同方向にL−プロリン4位水酸化酵素の構造遺伝子をコードするDNA断片が挿入されたプラスミドpTr2−4OH(図9)を得た。
【0094】
参考例2 ダクチロスポランジウム・エスピーの菌体を用いたトランス−4−ヒドロキシ−L−プロリンの生産
SR3培地(グルコース1.0%、可溶性澱粉1.0%、酵母エキス0.5%、トリプトン0.5%、肉エキス0.3%およびリン酸マグネシウム0.05%を含み、6N NaOHでpH7.2に調整した培地)を試験管に10mlずつ分注し、120℃、20分間殺菌した。この培地に、HT寒天平板培地に生育したダクチロスポランジウム・エスピー(Dactylosporangium sp.)RH1 を一白金耳植菌し、28℃、2日間振盪培養し、種培養液として用いた
Df1培地(可溶性澱粉5%、ソイビーンミール1.5%、リン酸1カリウム0.05%、硫酸マグネシウム7水塩0.05%および炭酸カルシウム0.5%を含み、6N NaOHでpH7.0に調整した培地)を試験管に10mlずつ分注し、120℃、20分間殺菌した。この培地に、上記種培養液1mlを無菌的に接種し、28℃、2日間振盪培養した。
【0095】
得られた培養液を8000rpm、10分間、4℃で遠心分離し、菌体を取得した。
該菌体を80mMTES[N−トリス(ヒドロキシメチル)メチル−2−アミノエタンスルホン酸]緩衝液(pH7. 5)で洗浄後、遠心分離し、湿菌体を取得した。
該湿菌体150mgを、1. 5mlの反応液[4mM L−プロリン、8mM α−ケト−グルタル酸、4mM L−アスコルビン酸、2mM 硫酸第一鉄、を含有する80mMTES緩衝液(pH7.5)にナイミーン溶液(ナイミーンS−215(日本油脂株式会社製)4gをキシレン10mlに溶解)を1. 4%( v/v) 添加]に懸濁し、30℃、30分間反応を行った。
【0096】
反応終了後、該反応液を遠心分離し、得られた上清中のトランス−4−ヒドロキシ−L−プロリン含量を定量した結果、84μmol/lトランス−4−ヒドロキシ−L−プロリンの生産が認められた。該菌株における、L−プロリン4位水酸化酵素にたいする菌体活性は0.028U/mg・ wet cells であった。
【0097】
「配列表フリーテキスト」
配列番号1−配列番号15において塩基置換されたDNA
配列番号2−合成DNA
配列番号3−合成DNA
配列番号4−合成DNA
配列番号5−合成DNA
配列番号6−合成DNA
配列番号7−合成DNA
配列番号8−融合蛋白質の構造遺伝子
配列番号9−合成DNA
配列番号10−合成DNA
配列番号11−合成DNA
配列番号12−合成DNA
配列番号13−合成DNA
配列番号14−合成DNA
【図面の簡単な説明】
【0098】
【図1】図1は、プラスミドpTr2−4OHΔの造成工程を示す図である。 図中、黒塗の太線で示した部分が、L−プロリン4位水酸化酵素遺伝子を含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、Ptrp×2は大腸菌由来のトリプトファンオペロンのプロモーターを2つ直列させたプロモーター(タンデムトリプトファンプロモーター)を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0099】
【図2】図2は、プラスミドpWFH1の造成工程を示す図である。 図中、網掛けの太線で示した部分が、Hind IIIおよびSal I 処理したPCR増幅断片の挿入部分を示す。黒塗の太線で示した部分が、Dactylosporangiumsp. RH1 由来のL−プロリン4位水酸化酵素遺伝子を含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、Ptrp×2は大腸菌由来のトリプトファンオペロンのプロモーターを2つ直列させたプロモーター(タンデムトリプトファンプロモーター)を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0100】
【図3】図3は、プラスミドpBAB51の造成工程を示す図である。 図中、黒の網掛けで示した部分が、プロリン合成系酵素遺伝子proB74およびproAを含む部分である。ApはpBR322由来のアンピシリン耐性遺伝子を、lacZはβ- ガラクトシダーゼαフラグメント構造遺伝子を示す。TcはpBR322由来のテトラサイクリン耐性遺伝子を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0101】
【図4】図4は、プラスミドpPRO74の造成工程を示す図である。 図中、黒の網掛けで示した部分が、プロリン合成系酵素遺伝子proB74およびproAを含む部分である。黒塗りの部分はproB74とproBで唯一異なる塩基対を含むproB74遺伝子の一部分を示す。TcはpBR322由来のテトラサイクリン耐性遺伝子を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0102】
【図5】図5は、プラスミドpPF1の造成工程を示す図である。 図中、黒塗の太線で示した部分が、プロリン合成系酵素遺伝子proB74およびproAを含む部分を示す。CmrはTn9由来のクロラムフェニコール耐性遺伝子を含む部分を示す。pACYC.oriはpACYC184由来の複製開始基点を、Placはlacプロモーターを、lacZはβ- ガラクトシダーゼαフラグメント構造遺伝子を示す。lacZ.Nterm- proB74はβ- ガラクトシダーゼαフラグメント構造遺伝子のN末端部分とproB74遺伝子との融合した遺伝子を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0103】
【図6】図6は、プラスミドpBII−4OHの造成工程を示す図である。 図中、黒塗の太線で示した部分が、L−プロリン4位水酸化酵素遺伝子を含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、lacZはβ- ガラクトシダーゼαフラグメント構造遺伝子を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0104】
【図7】図7は、プラスミドpBII−4OHBAの造成工程を示す図である。 図中、黒塗の太線で示した部分が、L−プロリン4位水酸化酵素遺伝子を含む部分を示す。黒の網掛け部分はプロリン合成系酵素遺伝子proB74およびproAを含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、lacZはβ- ガラクトシダーゼαフラグメント構造遺伝子を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0105】
【図8】図8は、プラスミドpWFP1の造成工程を示す図である。 図中、黒塗の太線で示した部分が、L−プロリン4位水酸化酵素遺伝子を含む部分を示す。黒の網掛け部分はプロリン合成系酵素遺伝子proB74およびproAを含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、lacZはβ- ガラクトシダーゼαフラグメント構造遺伝子を示す。Ptrp×2は大腸菌由来のトリプトファンオペロンのプロモーターを2つ直列させたプロモーター(タンデムトリプトファンプロモーター)を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0106】
【図9】図9は、プラスミドpTr2−4OHの造成工程を示す図である。 図中、黒塗の太線で示した部分が、L−プロリン4位水酸化酵素遺伝子を含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、Ptrp×2は大腸菌由来のトリプトファンオペロンのプロモーターを2つ直列させたプロモーター(タンデムトリプトファンプロモーター)を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【技術分野】
【0001】
本発明は、消炎剤として利用されているN−アセチルヒドロキシプロリン,カルバペネム系抗生物質等の医薬品の合成原料または食品添加物として有用なトランス−4−ヒドロキシ−L−プロリンを工業的に製造する方法、該方法に有用なL−プロリン4位水酸化酵素遺伝子を含有し、かつ、プロリン合成系の活性の強化された形質転換体に関する。
【背景技術】
【0002】
微生物を用いトランス−4−ヒドロキシ−L−プロリンを製造する方法としては、
1)エッシェリヒア属に属する微生物を用い、4−ヒドロキシ−2−オキソグルタル酸からトランス−4−ヒドロキシ−L−プロリンを製造する方法(特許文献1参照)、
2)細菌やかび類を用い直接発酵生産する方法(特許文献2、3および4参照) 、
3)ストレプトミセス属に属する微生物を用い、L−プロリンから製造する方法(非特許文献1、2、3および4参照)が知られている。
【0003】
従来のトランス−4−ヒドロキシ−L−プロリンの製造方法は、1)4−ヒドロキシ−2−オキソグルタル酸等のトランス−4−ヒドロキシ−L−プロリンを製造するための基質が高価で入手困難である、2)トランス−4−ヒドロキシ−L−プロリンの生産性が低い、3)トランス−4−ヒドロキシ−L−プロリンの製造に関与する酵素の活性が極めて微弱である、等の理由から、工業化は困難である。トランス−4−ヒドロキシ−L−プロリンの製造に関与する酵素については、L−プロリン4位水酸化酵素をダクチロスポランジウム.エスピー.RH1(Dactylosporangium sp.RH1 )より精製したとの報告(特許文献5参照)、および前述のストレプトマイセス属に属する微生物より精製したとの報告(非特許文献5参照)がある。また、2−ケトグルタル酸および2価鉄イオンの存在下において、遊離のL−プロリンをトランス−4−ヒドロキシ−L−プロリンに変換させる活性を有する、L−プロリン4位水酸化酵素をコードする遺伝子を前述のダクチロスポランジウム.エスピー.RH1(Dactylosporangiumsp.RH1 )よりクローニングし、L−プロリン4位水酸化酵素遺伝子を含むプラスミドpRH71を取得し、さらにトリプトファンタンデムプロモーター支配下でL−プロリン4位水酸化酵素遺伝子を発現させるプラスミドpTr2−4OHを構築し、大腸菌で活性を発現させたという報告(非特許文献6参照)がある。
【0004】
活性の高いL−プロリン4位水酸化酵素を用い、工業的に有利にトランス−4−ヒドロキシ−L−プロリンを製造する方法が求められている。
一方、プロリン生合成経路に関する酵素およびその遺伝子は大腸菌などですでに明らかになっている(非特許文献7および8参照)。また、プロリン生合成は例えば大腸菌などでは生合成経路の第一酵素であるガンマ−グルタミル キナーゼ(GK)が、生成物であるプロリンのフィードバック阻害を受けることにより調節されている事が知られている(非特許文献9参照)。
【0005】
更に、GKをコードする遺伝子proBに変異を生じさせることにより、プロリンのフィードバック阻害を著しく減少させたGKを生成させ、プロリン高生産株を造成できることが知られている(非特許文献10、11および12参照)。該proBに変異を起こさせた遺伝子としてproB74が知られており、proB74の変異点はproBのコーディングリージョンの5’末端から319番目のGがAに置換することにより、proB蛋白のN末端から107番目のアスパラギン酸がアスパラギンに変化していることが知られている(非特許文献13参照)。
プロリン分解に関する酵素系、それをコードする遺伝子、遺伝子系の制御についても、大腸菌、サルモネラ菌などで研究が進んでおりよく知られている(非特許文献14および15参照)。
【0006】
プロリンの生合成系の活性の強化された株を用いたトランス−4−ヒドロキシ−L−プロリンの製造法は知られていない。
【特許文献1】特開平3−266995号公報
【特許文献2】欧州特許出願公開第0547898号明細書
【特許文献3】特開平5−236980号公報
【0007】
【特許文献4】特開平6−245782号公報
【特許文献5】特開平7−322885
【非特許文献1】ジャーナル・オブ・バイオロジカル・ケミストリー(J. Biol. Chem.)、254巻、6684〜6690ページ(1979年)
【非特許文献2】バイオケミカル・アンド・バイオフィジカル・リサーチ・コミュニケーション(Biochem.Biophys. Res.Comm. )、120巻,45〜51ページ(1984年)
【0008】
【非特許文献3】テトラヘドロン・レターズ(Tetrahedron Letters ),34巻,7489〜7492ページ(1993年)
【非特許文献4】テトラヘドロン・レターズ(Tetrahedron Letters )、35巻,4649〜4652ページ(1994年)
【非特許文献5】バイオケミカル・ジャーナル(Biochem.J.)、313巻、185−191ページ、(1966年)
【非特許文献6】日本農芸化学会1996年度大会、講演要旨集257ページ
【0009】
【非特許文献7】ジャーナル・オブ・ジェネラル・マイクロバイオロジー(J.Gen.Microbiol.)、118巻、287−293ページ(1980年)
【非特許文献8】バイオキミカ・バイオフィジカ・アクタ(Biochim.Biophys.Acta)、104巻、397〜404ページ(1965年)
【非特許文献9】バイオキミカ・バイオフィジカ・アクタ(Biochim.Biophys.Acta)、192巻、462〜467ページ(1969年)
【0010】
【非特許文献10】モレキュラー・ジェネラル・ジェネティクス(Mol.Gen.Genet.)、182巻、82−86ページ(1981年)
【非特許文献11】ジャーナル・オブ・バクテリオロジー(J.Bacteriol.)、156巻、1249−1262ページ(1983年)
【非特許文献12】ジャーナル・オブ・バクテリオロジー(J.Bacteriol.)、164巻、1088−1093ページ(1985年)
【非特許文献13】ジーン(Gene)、64巻、199−205ページ(1988年)
【0011】
【非特許文献14】ジャーナル・オブ・バクテリオロジー(J.Bacteriol.)、146巻、895−901ページ(1981年)
【非特許文献15】ジャーナル・オブ・モレキュラー・バイオロジー (J.Mol.Biol.)、148巻、21−44ページ(1981年)
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明の課題は、L−プロリン4位水酸化酵素を用いて、効率的にトランス−4−ヒドロキシ−L−プロリンを製造する方法において、より工業的に有利にトランス−4−ヒドロキシ−L−プロリンを製造するために、L−プロリン4位水酸化酵素遺伝子を含有し、これを高効率で発現させる組換え体DNAを保持し、かつ、プロリン生合成系の活性の強化された形質転換体を提供し、該形質転換体を用いてトランス−4−ヒドロキシ−L−プロリンを工業的に安価に製造する方法を提供することにある。
【課題を解決するための手段】
【0013】
本発明は、2−ケトグルタル酸および2価鉄イオンの存在下、遊離のL−プロリンに作用して、トランス−4−ヒドロキシ−L−プロリンを生成する、L−プロリン4位水酸化酵素活性を有する蛋白質をコードする遺伝子を含むDNA断片をベクターに組み込んで得られる組換え体DNAを保有し、且つ、L−プロリンの生合成系の活性の強化された形質転換体、および該形質転換体を培養し、該培養物、菌体またはそれらの処理物を酵素源として、2−ケトグルタル酸および2価鉄イオンの存在下、水性媒体中で、L−プロリンをトランス−4−ヒドロキシ−L−プロリンに変換させ、生成したトランス−4−ヒドロキシ−L−プロリンを該水性媒体より採取することを特徴とするトランス−4−ヒドロキシ−L−プロリンの製造法に関する。
【発明の効果】
【0014】
本発明によれば、医薬品の合成原料または食品添加物として有用なトランス−4−ヒドロキシ−L−プロリンを工業的に製造する方法、該方法に有用なL−プロリン4位水酸化酵素遺伝子を含有し、かつ、プロリン合成系の活性の強化された形質転換体を提供することができる。
【発明を実施するための最良の形態】
【0015】
以下に本発明を詳細に説明する。
本発明にかかわるL−プロリン4位水酸化酵素は、2−ケトグルタル酸および2価鉄イオンの存在下において、遊離のL−プロリンを水酸化してトランス−4−ヒドロキシ−L−プロリンを生成する酵素である。
本発明のL−プロリン4位水酸化酵素をコードするDNAを含むプラスミッドとしては、例えばpRH71があげられる。pRH71を含む大腸菌であるEscherichia coli SOLR/pRH71 は、平成7年3月2日付けで工業技術院生命工学工業技術研究所、日本国茨城県つくば市東1丁目1番3号(郵便番号305)にFERM BP−5025として寄託されている。
【0016】
L−プロリン4位水酸化酵素遺伝子を宿主中で発現させるためには、まず、L−プロリン4位水酸化酵素遺伝子を含むDNA断片を、制限酵素類あるいはDNA分解酵素類で、L−プロリン4位水酸化酵素遺伝子を含む適当な長さのDNA断片とした後に、発現ベクター中プロモーターの下流に挿入し、次いで上記DNAを挿入した発現ベクターを、発現ベクターに適合した宿主中に導入する。宿主としては、目的とする遺伝子を発現できるものは全て用いることができる。例えば、エッシェリヒア属、セラチア属、コリネバクテリウム属、ブレビバクテリウム属、シュードモナス属、バチルス属、等に属する微生物菌株の他、酵母菌株や動物細胞宿主等をあげることができる。
【0017】
発現ベクターとしては、上記宿主において自立複製可能ないしは染色体中への組込みが可能で、L−プロリン4位水酸化酵素遺伝子を転写できる位置にプロモーターを含有しているものが用いられる。
大腸菌等の微生物を宿主として用いる場合は、L−プロリン4位水酸化酵素発現ベクターは微生物中で自立複製可能であると同時に、プロモーター、リボソーム結合配列、L−プロリン4位水酸化酵素遺伝子、転写終結配列、より構成されていることが好ましい。プロモーターを制御する遺伝子が含まれていてもよい。
【0018】
発現ベクターとしては、例えば、pBTrp2、pBTac1、pBTac2(いずれもベーリンガーマンハイム社より市販)、pKYP10( 特開昭58−110600)、pKYP200〔アグリカルチャラル・バイオロジカル・ケミストリー(Agric. Biol.Chem.), 48巻, 669〜675ページ(1984年) 〕、pLSA1〔アグリカルチャラル・バイオロジカル・ケミストリー(Agric. Biol.Chem.),53巻,277ページ(1989年)〕、pGEL1〔プロシーディング・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc. Natl.Acad.Sci., USA) ,82巻,4306ページ( 1985年)〕、pBluescript (STRATAGENE社)、pTrs30〔エシェリヒア・コリJM109/pTrS30 (FERM BP−5407)より調製〕およびpTrs32〔エシェリヒア・コリ JM109/pTrS32 (FERM BP−5408)より調製〕等を例示することができる。
【0019】
プロモーターとしては、大腸菌等の宿主中で発現できるものであればいかなるものでもよい。例えば、trpプロモーター(Ptrp)、lacプロモーター(Plac)、PL プロモーターPR プロモーターなどの、大腸菌やファージ等に由来するプロモーターをあげることができる。またPtrpを2つ直列させたプロモーター(Ptrpx2)、tacプロモーターのように人為的に設計改変されたプロモーター等も用いることができる。
【0020】
リボソーム結合配列としては、大腸菌等の宿主中で発現できるものであればいかなるものでもよいが、リボソーム結合配列と開始コドンとの間を適当な距離(例えば6〜18塩基)に調節したプラスミッドを用いることが好ましい。
L−プロリン4位水酸化酵素遺伝子はL−プロリン4位水酸化酵素をコードする遺伝子であればいずれも用いることができるが、該遺伝子のDNA配列を宿主微生物での発現に最適なコドンとなるように、塩基を置換して用いることにより高発現化が達成できる。大腸菌を宿主として、発現に最適なコドンとなるように塩基を置換したL−プロリン4位水酸化酵素遺伝子の具体例として、配列番号1で示される塩基配列等をあげることができる。
【0021】
本遺伝子の発現には転写終結配列は必ずしも必要ではないが、好適には構造遺伝子直下に転写終結配列を配置することが望ましい。
宿主としては、Escherichia coli XL1-Blue 、Escherichia coli XL2-Blue、Escherichia coli DH1、Escherichia coliMC1000 、 Escherichia coli KY3276、Escherichia coliW1485、Escherichia coli JM109、Escherichia coli HB101、Escherichia coli No.49、Escherichia coli W3110、Escherichia coliNY49 、Bacillus subtilis 、Bacillus amyloliquefacines、Brevibacterium immariophilum ATCC14068、Brevibacterium saccharolyticumATCC14066、Brevibacterium flavum ATCC14067 、Brevibacterium lactofermentumATCC13869 、Corynebacterium glutamicum ATCC13032、Corynebacterium acetoacidophilum ATCC13870、Microbacterium ammoniaphilumATCC15354等をあげることができる。
【0022】
酵母菌株を宿主として用いる場合には、発現ベクターとして、例えば、YEp13(ATCC37115 )、YEp24(ATCC37051 )、YCp50(ATCC37419 )等を例示することができる。 プロモーターとしては、酵母菌株の宿主中で発現できるものであればいかなるものでもよい。例えば、ヘキソースキナーゼ等の解糖系の遺伝子のプロモーター、gal1 プロモーター、gal 10プロモーター、ヒートショック蛋白質プロモーター、MFα1 プロモーター、CUP 1 プロモーター等のプロモーターをあげることができる。
【0023】
宿主としては、Saccharomyces cerevisae 、Schizosaccharomyces pombe 、Kluyveromyces lactis、Trichosporon pullulans、Schwanniomyces alluvius 等をあげることができる。
動物細胞を宿主として用いる場合には、発現ベクターとして、例えば、pcDNAI/Amp、pcDNAI、pcDM8(いずれもフナコシ社より市販)等を例示することができる。プロモーターとしては、動物細胞の宿主中で発現できるものであればいかなるものでもよい。例えば、ヒトCMVのIE(immediate early)遺伝子のプロモーター等のプロモーターをあげることができる。また、ヒトCMVのIE遺伝子のエンハンサーをプロモーターと共に用いてもよい。宿主としては、ナマルバ細胞、HBT5637(特開昭63−299)、COS細胞、CHO細胞等をあげることができる。
【0024】
動物細胞へのDNAの導入法としては、動物細胞にDNAを導入することができればいかなる方法も用いることができる。例えば、エレクトロポーレーション法〔Miyajiら:サイトテクノロジー(Cytotechnology), 3, 133(1990)〕、リン酸カルシウム法(特開平2-227075)、リポフェクション法〔フィリップ・エル・フェルグナー(PhilipL. Felgner)ら:プロシーディング・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス (Proc. Natl. Acad. Sci., USA), 84, 7413(1987)〕等を用いることができる。形質転換株の取得および培養は、特開平2-227075あるいは特開平2-257891に記載されている方法に準じて行なうことができる。
【0025】
宿主をプロリン生合成系の活性の強化されたものにするためには、1)宿主中のプロリン合成系遺伝子のコピー数を増加させる、2)プロリンによってフィードバック阻害を受けるプロリン合成系の酵素をコードする遺伝子に変異を生ぜしめ、プロリンによるフィードバック阻害が著しく減少した酵素をコードする遺伝子を宿主に導入する、3)プロリンの分解活性を欠失させる、または4)これらを組み合わせることにより達成することができる。
【0026】
プロリン合成系遺伝子またはプロリンによるフィードバック阻害が著しく減少したプロリン合成系酵素をコードする遺伝子は宿主の染色体上に存在しても良いし、またプラスミドなどの宿主中のベクター上に存在してもよい。
プロリンによるフィードバック阻害が著しく減少したプロリン合成系酵素をコードする遺伝子としては、proB74遺伝子、DHPrproB遺伝子[ジーン(gene)、39巻、109−112ページ、(1984年)]等、プロリンによるフィードバック阻害が著しく減少したプロリン合成系酵素をコードする遺伝子をあげることができる。
【0027】
また、プロリン合成系遺伝子またはプロリンによるフィードバック阻害が著しく減少したプロリン合成系酵素をコードする遺伝子をプロリン4位水酸化酵素遺伝子と共にプラスミドなどの宿主中のベクター上に存在させる場合は、両遺伝子が一つのプラスミド上に存在していてもよいし、別の共存可能なプラスミド上に存在していてもよい。
共存可能なプラスミドとしては、例えば、大腸菌においては、コリシンE1系のプラスミド(例、pBR322)とpACYC系のプラスミド、コリシンE1系のプラスミドとF因子系のプラスミド、コリシンE1系のプラスミドとR因子系のプラスミド等をあげることができる。
【0028】
プロリン生合成系遺伝子を宿主中で発現させるためには、前述のL−プロリン4位水酸化酵素遺伝子での発現と同様の方法を用いることができる。
宿主のプロリン分解活性を欠失させるには、変異剤で宿主を処理した後、例えば細菌などではPro−TTCプレート[アプライド・アンド・エンバイロメンタル・マイクロバイオロジー(Appl.Environ,Microbiol,)、33巻、434−444ページ、(1977年)]上で白色のコロニーを形成するものを選択することで取得できる。
【0029】
更に、大腸菌等ではput遺伝子の適当な位置にトランスポゾンが挿入されることでプロリンの資化性が失われることがわかっている[ジェネティクス(Genetics)、92巻、741−747ページ(1979年)]ので、トランスポゾンを挿入した後、薬剤耐性と前述したPro−TTCプレートにより、プロリン分解活性欠失株を取得することもできる。
【0030】
また、トランスポゾンによりプロリン分解活性が欠失した株より、P1トランスダクション[A Short Course In Bacterial Genetics, A Laboratory Manual andHandbook for Esherichia coli and Related Bacteria, J. H. Miller, Cold SpringHarbor Laboratory Press, 1922, Laboratory Manual, pp.263 ]により別の菌株にプロリン分解活性欠失という形質を移すこともできる。
【0031】
上記のようにしてプロリン生合成系の活性の強化された宿主を用いて得られた形質転換体の培養は、通常の培養方法に従って行うことができる。
大腸菌や酵母菌等の微生物を宿主として用いた形質転換体を培養する培地は、微生物が資化し得る炭素源、窒素源、無機塩類等を含有し、形質転換体の培養を効率的に行える培地であれば天然培地、合成培地のいずれでもよい。
【0032】
炭素源としては、それぞれの微生物が資化し得るものであればよく、グルコース、フラクトース、スクロース、これらを含有する糖蜜、デンプンあるいはデンプン加水分解物等の炭水化物、酢酸、プロピオン酸等の有機酸、エタノール、プロパノールなどのアルコール類が用いられる。
窒素源としては、アンモニア、塩化アンモニウム、硫酸アンモニウム、酢酸アンモニウム、りん酸アンモニウム、等の各種無機酸や有機酸のアンモニウム塩、その他含窒素化合物、並びに、ペプトン、肉エキス、酵母エキス、コーンスチープリカー、カゼイン加水分解物、大豆粕および大豆粕加水分解物、各種発酵菌体およびその消化物等が用いられる。
【0033】
無機物としては、りん酸第一カリウム、りん酸第二カリウム、りん酸マグネシウム、硫酸マグネシウム、塩化ナトリウム、硫酸第一鉄、硫酸マンガン、硫酸銅、炭酸カルシウム等が用いられる。
培養は、振盪培養または深部通気攪拌培養などの好気的条件下で行う。培養温度は15〜40℃がよく、培養時間は、通常16〜100時間である。培養中pHは、3. 0〜9. 0に保持する。pHの調整は、無機あるいは有機の酸、アルカリ溶液、尿素、炭酸カルシウム、アンモニアなどを用いて行う。
【0034】
また培養中必要に応じて、アンピシリンやテトラサイクリン等の抗生物質を培地に添加してもよい。
プロモーターとして誘導性のプロモーターを用いた発現ベクターで形質転換した微生物を培養するときには、必要に応じてインデューサーを培地に添加してもよい。例えば、lacプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはイソプロピル−β−D−チオガラクトピラノシド(IPTG)等を、trpプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはインドールアクリル酸(IAA)等を培地に添加してもよい。
【0035】
動物細胞を宿主として用いた形質転換体を培養する培地は、一般に使用されているRPMI1640培地、EagleのMEM培地またはこれら培地に牛胎児血清等を添加した培地等が用いられる。
培養は、5%CO2 存在下等の条件下で行う。培養温度は35〜37℃がよく、培養時間は、通常3〜7日間である。
【0036】
また培養中必要に応じて、カナマイシン、ペニシリン等の抗生物質を培地に添加してもよい。
このように培養して得た形質転換体中には、遺伝子源として用いた微生物菌株、例えばダクチロスポランジウム・エスピーRH1等およびpTr2−4OHを保有する組換え大腸菌と比較してL−プロリン4位水酸化酵素が著量生成蓄積しており、プロリン生合成活性も強く、トランス−4−ヒドロキシ−L−プロリン製造を、遺伝子源として用いた微生物菌株、例えばダクチロスポランジウム・エスピーRH1等およびpTr2−4OHを保有する組換え大腸菌を用いた場合と比較して、はるかに効率的に行うことができる。
【0037】
該形質転換体中にL−プロリン4位水酸化酵素が生成していることは、培養物、菌体またはそれらの処理物を、酵素反応に適した水性媒体中に、L−プロリン、二価鉄イオン、2−ケトグルタル酸とともに加え、また必要に応じて界面活性剤や有機溶剤を添加することにより、生成するトランス−4−ヒドロキシ−L−プロリンを検出することにより知ることができる。生成されたL−プロリン4位水酸化酵素の活性は、下記測定条件下、1分間に1nmolのトランス−4−ヒドロキシ−L−プロリンを生成する活性を1単位(U)として表示する。なお、ここでは、微生物菌体に加え、動物細胞を含めて菌体と呼ぶ。
【0038】
L−プロリン4位水酸化酵素活性の測定:
12mM L−プロリン、24mM 2−ケトグルタル酸、4mM 硫酸第一鉄および8mM L−アスコルビン酸を含有する240mMのMES〔2−(N−モルホリノ)エタンスルホン酸〕緩衝液(pH6. 5)に菌体、菌体処理物または酵素標品等を添加して合計250μlとし、35℃、10分間反応する。反応液を100℃、2分間加熱して反応を停止した後に、反応液中に生成したトランス−4−ヒドロキシ−L−プロリンを高速液体クロマトグラフィー(以下、HPLCと略記する)を用いて定量する。
【0039】
定量にはトランス−4−ヒドロキシ−L−プロリンを定量できる方法であればどのような方法を用いてもよいが、例えば、反応液中のトランス−4−ヒドロキシ−L−プロリンを配位子交換クロマトグラフィーカラム、例えば、株式会社住化分析センター製SUMICHIRAL OA5000 等を用いてHPLCで分離溶出後、7−クロロ−4−ニトロベンゾ−2−オキサ−1,3−ジアゾ−ル(以下、NBDと略記する)によってポストカラム誘導体化し検出する方法(ポストカラム誘導体化法) 、あるいは反応液中の目的化合物をあらかじめNBD誘導体化しておき、これをHPLCを用いた逆相クロマトグラフィーにかけてNBD誘導体化物を分離後検出する方法〔WilliamJ. Lindblad and Robert F. Diegelmann, アナリティカル・バイオケミストリー(Analytical Biochemistry)、138巻、390〜395ページ、1984年〕(プレカラム誘導体化法)等があげられる。NBD誘導体化物の検出は、いずれもその蛍光( 励起波長503nm、蛍光波長541nm)測定によって行われる。
【0040】
上記のようにして培養菌体中にL−プロリン4位水酸化酵素の生成が確認された形質転換体を、前述の形質転換体の培養条件と同様の条件で培養し、トランス−4−ヒドロキシ−L−プロリンを生成蓄積させ、該培養物よりトランス−4−ヒドロキシ−L−プロリンを採取することにより、トランス−4−ヒドロキシ−L−プロリンを製造することができる。
【0041】
該培養において、必要に応じて2−ケトグルタル酸および2価鉄イオンを培養液中に添加してもよい。
本発明により製造されたトランス−4−ヒドロキシ−L−プロリンは、前述のポストカラム誘導体化法やプレカラム誘導体化法によって、定量分析することができる。
【実施例1】
【0042】
L−プロリン4位水酸化酵素高発現プラスミッドの構築
配列番号2記載のDNAと配列番号3記載のDNAをアプライド・バイオシステムズ(Applied Biosystems)社製380A・DNA合成機を用いて合成した。 該合成DNAの3’末端25bpは互いに相補的な配列となるように設計されている。さらに、該合成DNAには、Dactylosporangium sp. RH1 由来のL−プロリン4位水酸化酵素蛋白のN末端部分をコードする塩基配列が含有されているが、該塩基配列には、Dactylosporangium sp. RH1 由来の塩基配列を、大腸菌での発現に最適なコドンとなるように部位特異的な塩基置換がほどこされている。
【0043】
該合成DNAをプライマーかつ鋳型としてPCRを行った。
PCRはPfuDNAポリメラーゼ(STRATAGENE社製)0.5U、Pfu DNAポリメラーゼ用×10緩衝液(STRATAGENE社製)2μl、DMSO 2μl、各2.5mMdNTP液1μl、配列番号2記載の合成DNAと配列番号3記載の合成DNA各2μMを含む反応液20μlを用い、96℃、5分間のインキュベーションの後、96℃−2分間、50℃−1分間、75℃−1分間のインキュベーション工程を35回繰り返す条件で行った。
【0044】
反応終了後、該反応液を、15%ポリアクリルアミドゲル(アトー株式会社製、パジェルNPU-15L )電気泳動にかけ、107bpの増幅断片の生成を確認した。
該107bpの増幅断片を日本エイドー株式会社製のダヴィンチくん(ペンタッチリカバリーNB−7000型)を用いて回収後、該107bpのDNA断片の両末端をHind IIIおよびSal I で切断した。
【0045】
該Hind III−Sal I 切断断片をBio, Inc. 製 MERmaid Kitを用いて回収した。回収液量は16μlとなった。
参考例1に記載の方法に従って作成したプラスミドpTr2−4OHDNAをBam HIおよびPvu IIで切断後、反応液をアガロースゲル電気泳動にかけ、2つの断片が生成していることを確認した。該断片の内、L−プロリン水酸化酵素の構造遺伝子を含む長い断片をバイオラッド社製のPrep-A-geneを用いて回収し、宝酒造社製のブランティングキットを用いて末端を平滑化した後、宝酒造社製のライゲーションキットを用いて連結環状化した。
【0046】
該連結環状化したプラスミドを用い、E. coli JM109 株を常法に従って形質転換し、該形質転換体を50μg/mlのアンピシリンを含むLB寒天培地に塗布後、37℃で1晩培養した。
生育してきた形質転換体のコロニーより常法に従ってプラスミドを抽出し、その構造を制限酵素消化により確認した。
【0047】
以上の結果、pTr2−4OHの一部の配列を削除したプラスミドpTr2−4OHΔ(図1)を得た。
取得したプラスミドpTr2−4OHΔDNAをHind IIIおよびSal I で切断後、該切断部位に、HindIIIおよびSal I 処理した上記PCR増幅断片を、宝酒造社製のライゲーションキットを用いて、挿入した。
【0048】
得られたプラスミドを用い、E. coli XL1-Blue MRF' 株を常法に従って形質転換し、該形質転換体を50μg/mlのアンピシリンを含むLB寒天培地に塗布後、37℃で1晩培養した。 生育してきた形質転換体のコロニーより常法に従ってプラスミドを抽出し、プラスミドpWFH1(図2)を取得した。
該プラスミドの構造を制限酵素消化により確認した。PCR増幅断片挿入部分についてはアプライドバイオシステムズ社製の塩基配列決定キット(Taq DyeDeoxyTM Terminator Cycle Sequencing Kit)を用いて、塩基配列を決定した。該塩基配列を配列番号1に示す。
【0049】
該プラスミドは、構造遺伝子の5’末端からSal I 部位においてDactylosporangium sp.RH1 由来の塩基配列と一部異なる以外は、Dactylosporangium sp. RH1 由来のL−プロリン4位水酸化酵素とまったく同じアミノ酸配列をコードする構造遺伝子DNA断片が、Ptrp×2の転写方向と同方向に挿入された構造を有していた(図2)。
第1表に示したように、pWFH1を保有する形質転換体は、遺伝子源として用いたダクチロスポランジウムエスピーRH1株と比較して1400倍、pTr2−4OHを保有する形質転換体と比較して約5.4倍のL−プロリン水酸化酵素を生産していた。
【0050】
【表1】
【実施例2】
【0051】
プロリン分解系欠損株の造成
E. coli ATCC12435 のプロリン分解に関与する遺伝子putAを以下の方法で破壊し、プロリン分解系欠損株を造成した。
国立遺伝学研究所保存菌株であるE. coli ME8395 株[F- :pyrD34trp-45 his-68 thyA25 thi deoR33 galK35xyl-7 mtl-2 malA1 rpsL118 λR ( λ) -appA1 putA::Tn5 (Mu+) ]を35μg/mlのカナマイシンを含有するLB培地に植菌し、終夜培養した。
【0052】
該培養液100μlにP1ファージ液100μlを添加・混合し、5分間放置した。
該放置液を10mM CaCl2 を含む3mlのLB軟寒天培地と混合し、10mMCaCl2 を含むLB寒天培地上に重層後、37℃で7時間培養した。 培養後、得られた寒天培地表面のライゼートを10mMCaCl2 を含む2mlのLB培地中に回収した。 該回収液に0.5mlのクロロホルムを添加し、ボルテックスミキサーを用いて混合後、3000rpmで15分間遠心し、得られた上清をP1ファージライゼートとして用いた。
【0053】
該P1ファージライゼート液50μlと10mM CaCl2 を含むLB培地で培養したE. coli ATCC12435 培養液100μlとを混合し、37℃で20分間静置した。該静置液をエフ・トップ・サイトレイト(F-top-citrate:NaCl 8. 5g/ l、クエン酸2ナトリウム100mM、寒天0. 7%)3mlと混合し、35μg/mlのカナマイシンを含有するLB寒天培地上に塗布後、37℃で1日培養した。
【0054】
該培養により生育してきたカナマイシン耐性株を0.85%のNaClに懸濁した後、Pro−TTCプレート(K2 HPO4 7g/ l、KH2 PO4 3g/ l、MgSO4 0. 1g/ l、プロテオースペプトン2g/l、2,3,5-トリフェニルテトラゾリウムクロライド0. 025g/ l、L- Pro 2g/ l、寒天15g/ l、pH7. 2) に塗布し、37℃で1〜2日間培養した。
【0055】
該培養によりPro−TTCプレート上に白色のコロニーを形成した株を、プロリンを分解資化する能力の失われた株として選択し、プロリン分解活性欠損株E. coliWT1株を取得した。該菌株は平成8年8月7日付けで工業技術院生命工学工業技術研究所、日本国茨城県つくば市東1丁目1番3号(郵便番号305)にFERM BP−5618として寄託されている。
【実施例3】
【0056】
プロリン生合成系遺伝子proB74およびproA発現プラスミドの構築
大腸菌由来のγ-glutamylkinaseをコードするproB遺伝子をプロリンによるフィードバック阻害に対して脱感作された変異型遺伝子に変換したproB74遺伝子および大腸菌由来のγ-glutamylphosphate reductase をコードするproA遺伝子の発現プラスミドpPF1を以下の方法により構築した。
大腸菌由来のproBおよびproA遺伝子を含むプラスミドpPRO−1[大腸菌K83株(FERM BP−2807)より取得]をEco RVで消化し、アガロースゲル電気泳動後、Prep-A-gene DNAPurification System (BIO-RAD社製)でproB遺伝子の一部を含む約1kbのDNA断片を取得した。
該DNA断片をpUC19(宝酒造社製)のSmaI 消化物と連結し、プラスミドpBAB51を得た(図3)。該プラスミド中のproB遺伝子を、以下の方法により、公知[A. M. Dandekarand S. L. Uratsu, J. Bacteriol. 170, 5943 (1988)] のプロリンによるフィードバック阻害に対して脱感作された変異型遺伝子proB74に変換した。
【0057】
配列番号4に示したオリゴヌクレオチドA1および配列番号5に示したオリゴヌクレオチドA2をアプライド・バイオシステムズ(Applied Biosystems)社製380A・DNA合成機を用いて合成した。オリゴヌクレオチドA1とM13プライマーM3(宝酒造社製、カタログNo.3831)を一組のプライマーとして、pBAB51を鋳型として用い、PCR法により変異型proB遺伝子であるproB74遺伝子の部分配列を増幅した。
【0058】
PCRは、pBAB51を0.1μg、オリゴヌクレオチドA1とM13プライマーM3を各2μM、Taq DNA ポリメラーゼ(宝酒造社製)を1U、dNTPミクスチャー(宝酒造社製、カタログNo.4030)を1.6μl、添付バッファー2μlを含む反応液20μlを用い、94℃−30秒間、52℃−30秒間、72℃−1分間のインキュベーション工程を30回繰り返し、最後に72℃−5分間インキュベーションすることにより行った。
【0059】
同様の方法で、オリゴヌクレオチドA2とM13プライマーRV(宝酒造社製、カタログNo.3830A)を一組のプライマーとして、pBAB51を鋳型として用い、PCR法により変異型proB遺伝子の部分配列を増幅した。
PCR法により増幅された該2種類のDNAをそれぞれアガロースゲル電気泳動にかけた後、Prep-A-gene DNA Purification System (BIO-RAD社製)を用いて精製した。
【0060】
該2種の精製DNA断片を混合したものを鋳型とし、M13プライマーM3およびM13プライマーRVをプライマーとして用い、上記と同様の方法により、PCRおよび精製を行い、proB74遺伝子の塩基配列を含む約1kbのDNA断片を取得した。
該DNA断片をEco O65IおよびSac IIで消化し、EcoO65I−Sac II消化断片を取得した。
【0061】
該消化断片と、pPRO−1のEco O65I−Sac II消化DNA断片(約6.8kbp)とを連結し、pPRO−1のproB遺伝子を脱感作型proB74遺伝子に変換したプラスミドpPRO74を作製した(図4)。
配列番号6に示したオリゴヌクレオチドp1および配列番号7に示したオリゴヌクレオチドp2をアプライド・バイオシステムズ(Applied Biosystems)社製380A・DNA合成機を用いて合成した。
【0062】
該p1、p2をプライマーとして、プラスミドpPRO74を鋳型として用い、PCR法によりproB74およびproA遺伝子を増幅した。
PCRは、pPRO74を0.1μg、p1、p2を各2μM、タカラ・イーエックス・タック(TaKaRa Ex Taq :宝酒造社製、コードRR001Q)1U、dNTPミクスチャー(宝酒造社製、カタログNo.4030)を1.6μl含む反応液20μlを用い、94℃−1分間、42℃−2分間、72℃−3分間のインキュベーション工程を30回繰り返すことにより行った。
【0063】
該反応液をアガロースゲル電気泳動にかけ、proB74およびproAを含む2370bpの増幅断片を常法により抽出し、GENECLEANII KIT(BIO 101, INC. 製)を用いて該DNA断片を回収した。回収した2370bpのDNA断片をHind IIIおよびBamHIで切断後、エタノール沈殿法によりエタノール沈殿を得た。該エタノール沈殿を5μlのTEに溶解し、proB74およびproA断片として用いた。
【0064】
該proB74およびproA断片と、プラスミドpSTV29(宝酒造社製)のHind III−Bam HI消化DNA断片とを宝酒造社製のDNAライゲーションキットを用いて連結し、proB74およびproA断片の挿入されたプラスミドを構築した。該プラスミドをE.coli JM109 株に常法に従って形質転換後、30μg/mlのクロラムフェニコール、0.1mMIPTG、40μg/mlのX−Galを含むLB寒天培地に塗布し、37℃で一晩培養した。
【0065】
該培養により白色コロニーを形成した株より常法に従ってプラスミドを抽出し、該プラスミドの構造を制限酵素消化により確認した。 上記の方法により、脱感作型γ-glutamyl kinaseのN末端にβ−ガラクトシダーゼのαフラグメントのN末端8アミノ酸残基(lacZ.Nterm)が結合した融合蛋白をコードする遺伝子およびproA遺伝子がPlac支配下に存在するプラスミドpPF1を取得した(図5)。
【0066】
該融合蛋白の構造遺伝子(lacZ.Nterm-proB74 )の塩基配列とアミノ酸配列を配列番号8に示す。
【実施例4】
【0067】
プロリン生合成系遺伝子発現プラスミドを含む形質転換体によるトランス−4−ヒドロキシ−L−プロリンの生産
実施例3で構築したプラスミドpPF1を用いて、実施例2で取得したE. coli WT1 株を形質転換し、形質転換株E. coliWT1/pPF1を取得した。
実施例1で構築したプロリン4位水酸化酵素発現プラスミドpWFH1で、実施例2で取得したWT1株を形質転換しE. coli WT1/pWFH1 を取得した。
【0068】
該形質転換株E. coli WT1/pWFH1 より塩化カルシウム法にてコンピテントセルを作製し、実施例3で作製したプラスミドpPF1を導入し、両プラスミドを保有する形質転換株E.coli WT1/pWFH1/pPF1を30μg/ mlのクロラムフェニコールと50μg/ mlのアンピシリンを含むLB培地に生育してくるコロニーを選択することにより取得した。
WT1 、WT1/pPF1、WT1/pWFH1およびWT1/pWFH1/pPF1をそれぞれカナマイシン37.5μg/ ml、アンピシリン100μg/ ml、クロラムフェニコール30μg/ ml、そしてアンピシリン100μg/mlおよびクロラムフェニコール30μg/ mlを含むLB培地で37℃で16時間培養した。
【0069】
該培養液それぞれ100μlを、2%(W/ V)の炭酸カルシウムを含む10mlのMed7培地(グルコース2%、硫酸アンモニウム1%、K2 HPO40. 1%、NaCl 0. 2%、MgSO4 0. 05%、FeSO4 0. 0278%、CaCl20. 0015%、ペプトン0. 8%)をいれた太型試験管(φ20×200mm)に植菌し、30℃で24時間培養した。
【0070】
該培養上清中のL−プロリンおよびトランス−4−ヒドロキシ−L−プロリン含量を第2表に示した。
【0071】
【表2】
【0072】
プロリン生合成系遺伝子発現プラスミドpPF1保有菌株においてはL−プロリンの生成が著しく増加し、L−プロリン4位水酸化酵素発現プラスミドpWFH1およびプロリン生合成系遺伝子発現プラスミドpPF1の共存する形質転換体においてはL−プロリン4位水酸化酵素発現プラスミドを単独で保持する形質転換体に比べてトランス−4−ヒドロキシ−L−プロリンの生成量が顕著に増加することが確認できた。
【実施例5】
【0073】
L ―プロリン4位水酸化酵素とプロリン生合成系遺伝子proB74およびproA発現プラスミドの構築
Dactylosporangium sp. RH1 由来のL−プロリン4 位水酸化酵素遺伝子、プロリン生合成系遺伝子proB74およびproAの全ての遺伝子を単一のプラスミド上に保持し、これを発現させるような発現プラスミドpWFP1を以下の方法により構築した。
【0074】
実施例1で作成したpWFH1をテンプレートとし、PCR法にてL−プロリン4位水酸化酵素の構造遺伝子を増幅した。
PCRはpWFH1プラスミド0.1μg 、Pfu DNAポリメラーゼ(stratagene社製)0.5 U、Pfu DNAポリメラーゼ用×10緩衝液(STRATAGENE社製)2μl、DMSO 2μl、各2.5mMdNTP液1μl、配列番号9記載の合成DNAと配列番号10記載の合成DNA各2μMを含む反応液20μlを用い、96℃、5分間のインキュベーションの後、96℃−2分間、58℃−1分間、75℃−3分間のインキュベーション工程を30回繰り返す条件で行った。
【0075】
反応終了後、該反応液をアガロースゲル電気泳動にかけ、L−プロリン4 位水酸化酵素遺伝子を含む約800bpの増幅断片を常法により抽出し、GENECLEAN II KIT(BIO 101, INC. 製)を用いて該DNA断片を回収した。該回収DNA断片をHindIIIおよびEco RIで切断後、アガロースゲル電気泳動にかけ、GENECLEAN II KIT(BIO 101, INC. 製)を用いてL−プロリン4位水酸化酵素遺伝子を有する該Hind III−Eco RI切断DNA断片を回収した。
該L−プロリン4位水酸化酵素遺伝子断片と、プラスミドpBluescriptII KS(+)(stratagene社製)のHind III―EcoRI消化DNA断片とを宝酒造社製のDNAライゲーションキットを用いて連結し、L−プロリン4位水酸化酵素断片の挿入されたプラスミドpBII―4OHを構築した(図6)。
実施例3で作成したpPRO74をテンプレートとしPCR法にてproB74およびproA遺伝子を増幅した。
【0076】
PCRはpPRO74プラスミド0.1 μg 、タカラ・イーエックス・タック(TaKaRa Ex Taq :宝酒造社製、コードRR001Q)1U、タカラ・イーエックス・タック用×10緩衝液(宝酒造社製)2μl、各2.5mMdNTP液1.6μl、配列番号11記載の合成DNAと配列番号12記載の合成DNA各2μMを含む反応液20μlを用い、94℃−1分間、42℃−2分間、75℃−3分間のインキュベーション工程を30回繰り返す条件で行った。
【0077】
反応終了後、該反応液をアガロースゲル電気泳動にかけ、proB74およびproA遺伝子を含む約2.3kbpの増幅断片を常法により抽出し、GENECLEANII KIT(BIO 101, INC. 製)を用いて該DNA断片を回収した。回収したDNA断片をBam HIおよびEco RIで切断後、フェノール/クロロホルム処理、エタノール沈殿を行い、該DNA断片を回収した。このproB74およびproA遺伝子を含むDNA断片と、上記プラスミドpBII―4OHのHindIII―Eco RI消化DNA断片とを宝酒造社製のDNAライゲーションキットを用いて連結し、L−プロリン4位水酸化酵素遺伝子、proB74およびproAを含むプラスミドpBII―4OHBAを構築した(図7)。
【0078】
プラスミドpBII―4OHBAをHind IIIおよびBam HIで消化し、アガロースゲル電気泳動にかけ、L−プロリン4位水酸化酵素遺伝子、proB74およびproAを含む約3.16kbpのDNA断片を取得した。また、実施例1で作成したpWFH1をHindIIIおよびBam HIで消化し、アガロースゲル電気泳動にかけ、L−プロリン4位水酸化酵素遺伝子を含まず、複製開始点を含む方の約2.6kbpのDNA断片を取得した。取得した2つのDNA断片を宝酒造社製のDNAライゲーションキットを用いて連結し、トリプトファンタンデムプロモーター下でL−プロリン4位水酸化酵素、proB74タンパク、proAタンパクを発現させうるプラスミドpWFP1を構築した(図8)。
【実施例6】
【0079】
形質転換体E. coli WT1/pWFH1/pPF1およびE.coli WT1/pWFP1 を用いたトランス−4−ヒドロキシ−L−プロリンの生産
形質転換株WT1/pWFH1/pPF1株をアンピシリン100μg/mlおよびクロラムフェニコール30μg/mlを含む50ml Med4G培地[グルコース 2%、ポリペプトン(日本製薬社製) 1%、イーストエキストラクト(Difco社製) 0.5%、NaCl 1%、炭酸カルシウム 2%、pH7.0]に、形質転換株WT1/pWFP1株をアンピシリン100μg/ mlを含む50ml Med4G培地に、それぞれ植菌した後、30℃で16時間振とう培養した。
【0080】
該培養液を種培養液とし、2リッターのMed7培地を入れた5リッタージャーファーメンターにそれぞれ植菌し、WT1/pWFH1/pPF1株の培養においては、アンピシリン100μg/ mlおよびクロラムフェニコール30μg/ mlを、WT1/pWFP1株の培養においては、アンピシリン100μg/ mlを更に添加し、培養温度30℃、撹拌数400回転/分、通気量1リッター/培養液1リッター/分の条件で培養した。
【0081】
また、WT1/pWFH1/pPF1の培養においては、培養8時間目に、IPTGを0.2mMとなるように添加した。更に、両菌株とも、培養24時間目に、培養開始時と同種、同濃度の抗生物質を添加した。
培養中、グルコースをほぼ1%の濃度となるように適時培養液に添加し、NH4OHを用いてpH6. 5に下限コントロールした。また、培養5時間以降に、撹拌数を250〜700rpmに変化させることにより、培養液の溶存酸素濃度が培養開始時点の1/15となるようにコントロールした。
【0082】
99時間培養後、該培養液を遠心分離し、得られた上清中のトランス−4−ヒドロキシ−L−プロリン含量を定量した結果、WT1/pWFH1/pPF1株において156mM(20.5g/ l)、WT1/pWFP1 株において191mM(25.0g/ l)のトランス−4−ヒドロキシ−L−プロリンの生成蓄積が認められた。
【実施例7】
【0083】
L−プロリン4位水酸化酵素遺伝子およびプロリン生合成系遺伝子発現プラスミドを含む形質転換体によるトランスー4―ヒドロキシーL−プロリンの生産
実施例5で構築したプラスミドpWFP1を用いて、実施例2で取得したE. coliWT1株を形質転換し、形質転換株E. coli WT1/pWFP1 を取得した。
WT1/pWFP1 をアンピシリン100μg/mlを含むLB培地で37℃で16時間培養した。該培養液100μlを、2%(W/V)の炭酸カルシウムを含む10mlのMed7培地を入れた太型試験管(φ20×200mm)に植菌し、30℃で24時間培養した。 該培養上清中のL−プロリンは0.56g/Lであり、トランス−4−ヒドロキシ−L−プロリンは2.4g/Lであった。
【実施例8】
【0084】
形質転換体の菌体を用いたトランス−4−ヒドロキシ−L−プロリンの生産
形質転換体E. coli ATCC12435/pTr2-4OHを50μg/mlのアンピシリンを含む3mlLB培地に植菌し30℃、一晩振とう培養後、該培養液を遠心分離し、E.coli ATCC12435/pTr2-4OHの湿菌体を取得した。
形質転換体E. coli ATCC12435/pWFH1 をアンピシリン100μg/mlを含む100ml Med4培地〔ポリペプトン(日本製薬社製)1%、イーストエキストラクト(Difco社製)0.5%、NaCl1%〕に植菌し、30℃で16時間振とう培養した。
【0085】
該培養液を、2リッターのMed7培地(グルコース2%、硫酸アンモニウム1%、K2HPO4 0.1%、NaCl0.2%、MgSO4 0.05%、FeSO4 0.0278%、CaCl2 0.0015%、ペプトン0.4%)を入れた5リッタージャーファーメンターに植菌後、L−Proを200mM添加し、培養温度33℃、通気量1リッター/培養液1リッター/分という条件で48時間培養した。
【0086】
溶存酸素濃度が初期値の15分の1に達した時点で、撹拌数400回転/分を基準とし、700回転/分を上限として攪拌数を変化させることにより、溶存酸素濃度を初期値の15分の1となるようにコントロールした。
培養中、グルコースは無くならぬように、L−プロリンは約50mMとなるように適時添加し、NH4OHを用いて、pH6.5に下限コントロールした。
【0087】
該培養により得られた培養液を遠心分離し、E. coli ATCC12435/pTr2-4OHおよび E. coli ATCC12435/pWFH1 の湿菌体を取得した。
該両菌株の湿菌体は必要に応じて−20℃で凍結保存することが可能で、使用時に解凍し用いることができる。
反応液〔240mMのMES緩衝液(pH6. 5)中に、12mM L−プロリン、24mM 2−ケトグルタル酸、4mM 硫酸第一鉄および8mM L−アスコルビン酸を含有する〕250μlに湿菌体量として4%(w/v)となるように加え、35℃で10分間反応した。反応液を100℃、2分間加熱することにより反応を停止した。
【0088】
該反応停止液を遠心分離し、得られた上清中のトランス−4−ヒドロキシ−L−プロリン含量を定量した結果、E. coli ATCC12435/pTr2-4OH株において3mmol/l、E.coli ATCC12435/pWFH1 株において16mmol/lのトランス−4−ヒドロキシ−L−プロリンの生産が認められた。L−プロリン4位水酸化酵素に対する菌体活性はそれぞれ、7.5および40U/mg・wet cells であった。
【0089】
参考例1 L−プロリン4位水酸化酵素発現プラスミッドの構築
配列番号13記載のセンス鎖DNAプライマーと、配列番号14に記載のアンチセンス鎖DNAプライマーをアプライド・バイオシステムズ(Applied Biosystems)社製380A・DNA合成機を用いて合成した。該合成DNAをプライマーとして、pRH71[該プラスミドを含有するEscherichia coli SOLR/pRH71 (FERM BP−5025)より定法により取得]を鋳型としてPCRを行った。
【0090】
PCRは、pRH71を0. 1μg、センス鎖およびアンチセンス鎖DNAプライマー各2μM、Pfu DNAポリメラーゼ(STRATAGENE社製)0.125U/μl、DMSO10%、Tris・HCl(pH8. 2)20mM、KCl10mM、硫酸アンモニウム6mM、塩化マグネシウム2mM、TritonX−1000.1%、Bovine Serum Albumine 10ng/μlを含む反応液20μlを用い、96℃、5分間のインキュベーション後、96℃−2分間、58℃−1分間、75℃−1分間のインキュベーション工程を30回繰り返す条件で行った。
【0091】
反応終了後、該反応液をアガロースゲル電気泳動にかけ、L−プロリン4位水酸化酵素の構造遺伝子をコードする844bpの増幅断片が生成されていることを確認した。
該844bpの増幅断片をアガロースゲルより常法により抽出し、バイオラッド社製のPrep-A-gene を用いて断片を回収した。回収した844bpのDNA断片の両末端をHind IIIおよびBamHIで切断後、エタノール沈殿法により、エタノール沈殿を得た。該エタノール沈殿を5μlのTEに溶解した。
【0092】
pBR322を基本とし、Ptrpを2つ直列させたプロモーターPtrp×2を持つプラスミドpKYP200〔アグリカルチャラル・バイオロジカル・ケミストリー(Agric. Biol. Chem.), 48巻, 669〜675ページ(1984年) 〕と合成リンカーを組み合わせて作製されたATGベクターpTrS32〔エシェリヒア・コリJM109/pTrS32 (FERM BP−5408)より調製〕をHind IIIおよびBam HIで切断した。該切断部位に、HindIIIおよびBam HIで切断処理した上記のL−プロリン4位水酸化酵素構造遺伝子断片を、宝酒造社製のライゲーションキットを用いて、挿入した。
【0093】
得られたプラスミドを用い、E. coli XL1-Blue MRF' 株を常法に従って形質転換し、該形質転換体を50μg/mlのアンピシリンを含むLB寒天培地に塗布後、37℃で1晩培養した。
生育してきた形質転換体のコロニーより常法に従ってプラスミドを抽出し、その構造を制限酵素消化により確認した。構造遺伝子部分についてはアプライドバイオシステムズ社製の塩基配列決定キット(Taq DyeDeoxyTM Terminator Cycle Sequencing Kit)を用いて、塩基配列を決定し、配列番号15で示した塩基配列であることを確認した。
該方法により、Ptrp×2の転写方向と同方向にL−プロリン4位水酸化酵素の構造遺伝子をコードするDNA断片が挿入されたプラスミドpTr2−4OH(図9)を得た。
【0094】
参考例2 ダクチロスポランジウム・エスピーの菌体を用いたトランス−4−ヒドロキシ−L−プロリンの生産
SR3培地(グルコース1.0%、可溶性澱粉1.0%、酵母エキス0.5%、トリプトン0.5%、肉エキス0.3%およびリン酸マグネシウム0.05%を含み、6N NaOHでpH7.2に調整した培地)を試験管に10mlずつ分注し、120℃、20分間殺菌した。この培地に、HT寒天平板培地に生育したダクチロスポランジウム・エスピー(Dactylosporangium sp.)RH1 を一白金耳植菌し、28℃、2日間振盪培養し、種培養液として用いた
Df1培地(可溶性澱粉5%、ソイビーンミール1.5%、リン酸1カリウム0.05%、硫酸マグネシウム7水塩0.05%および炭酸カルシウム0.5%を含み、6N NaOHでpH7.0に調整した培地)を試験管に10mlずつ分注し、120℃、20分間殺菌した。この培地に、上記種培養液1mlを無菌的に接種し、28℃、2日間振盪培養した。
【0095】
得られた培養液を8000rpm、10分間、4℃で遠心分離し、菌体を取得した。
該菌体を80mMTES[N−トリス(ヒドロキシメチル)メチル−2−アミノエタンスルホン酸]緩衝液(pH7. 5)で洗浄後、遠心分離し、湿菌体を取得した。
該湿菌体150mgを、1. 5mlの反応液[4mM L−プロリン、8mM α−ケト−グルタル酸、4mM L−アスコルビン酸、2mM 硫酸第一鉄、を含有する80mMTES緩衝液(pH7.5)にナイミーン溶液(ナイミーンS−215(日本油脂株式会社製)4gをキシレン10mlに溶解)を1. 4%( v/v) 添加]に懸濁し、30℃、30分間反応を行った。
【0096】
反応終了後、該反応液を遠心分離し、得られた上清中のトランス−4−ヒドロキシ−L−プロリン含量を定量した結果、84μmol/lトランス−4−ヒドロキシ−L−プロリンの生産が認められた。該菌株における、L−プロリン4位水酸化酵素にたいする菌体活性は0.028U/mg・ wet cells であった。
【0097】
「配列表フリーテキスト」
配列番号1−配列番号15において塩基置換されたDNA
配列番号2−合成DNA
配列番号3−合成DNA
配列番号4−合成DNA
配列番号5−合成DNA
配列番号6−合成DNA
配列番号7−合成DNA
配列番号8−融合蛋白質の構造遺伝子
配列番号9−合成DNA
配列番号10−合成DNA
配列番号11−合成DNA
配列番号12−合成DNA
配列番号13−合成DNA
配列番号14−合成DNA
【図面の簡単な説明】
【0098】
【図1】図1は、プラスミドpTr2−4OHΔの造成工程を示す図である。 図中、黒塗の太線で示した部分が、L−プロリン4位水酸化酵素遺伝子を含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、Ptrp×2は大腸菌由来のトリプトファンオペロンのプロモーターを2つ直列させたプロモーター(タンデムトリプトファンプロモーター)を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0099】
【図2】図2は、プラスミドpWFH1の造成工程を示す図である。 図中、網掛けの太線で示した部分が、Hind IIIおよびSal I 処理したPCR増幅断片の挿入部分を示す。黒塗の太線で示した部分が、Dactylosporangiumsp. RH1 由来のL−プロリン4位水酸化酵素遺伝子を含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、Ptrp×2は大腸菌由来のトリプトファンオペロンのプロモーターを2つ直列させたプロモーター(タンデムトリプトファンプロモーター)を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0100】
【図3】図3は、プラスミドpBAB51の造成工程を示す図である。 図中、黒の網掛けで示した部分が、プロリン合成系酵素遺伝子proB74およびproAを含む部分である。ApはpBR322由来のアンピシリン耐性遺伝子を、lacZはβ- ガラクトシダーゼαフラグメント構造遺伝子を示す。TcはpBR322由来のテトラサイクリン耐性遺伝子を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0101】
【図4】図4は、プラスミドpPRO74の造成工程を示す図である。 図中、黒の網掛けで示した部分が、プロリン合成系酵素遺伝子proB74およびproAを含む部分である。黒塗りの部分はproB74とproBで唯一異なる塩基対を含むproB74遺伝子の一部分を示す。TcはpBR322由来のテトラサイクリン耐性遺伝子を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0102】
【図5】図5は、プラスミドpPF1の造成工程を示す図である。 図中、黒塗の太線で示した部分が、プロリン合成系酵素遺伝子proB74およびproAを含む部分を示す。CmrはTn9由来のクロラムフェニコール耐性遺伝子を含む部分を示す。pACYC.oriはpACYC184由来の複製開始基点を、Placはlacプロモーターを、lacZはβ- ガラクトシダーゼαフラグメント構造遺伝子を示す。lacZ.Nterm- proB74はβ- ガラクトシダーゼαフラグメント構造遺伝子のN末端部分とproB74遺伝子との融合した遺伝子を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0103】
【図6】図6は、プラスミドpBII−4OHの造成工程を示す図である。 図中、黒塗の太線で示した部分が、L−プロリン4位水酸化酵素遺伝子を含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、lacZはβ- ガラクトシダーゼαフラグメント構造遺伝子を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0104】
【図7】図7は、プラスミドpBII−4OHBAの造成工程を示す図である。 図中、黒塗の太線で示した部分が、L−プロリン4位水酸化酵素遺伝子を含む部分を示す。黒の網掛け部分はプロリン合成系酵素遺伝子proB74およびproAを含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、lacZはβ- ガラクトシダーゼαフラグメント構造遺伝子を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0105】
【図8】図8は、プラスミドpWFP1の造成工程を示す図である。 図中、黒塗の太線で示した部分が、L−プロリン4位水酸化酵素遺伝子を含む部分を示す。黒の網掛け部分はプロリン合成系酵素遺伝子proB74およびproAを含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、lacZはβ- ガラクトシダーゼαフラグメント構造遺伝子を示す。Ptrp×2は大腸菌由来のトリプトファンオペロンのプロモーターを2つ直列させたプロモーター(タンデムトリプトファンプロモーター)を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【0106】
【図9】図9は、プラスミドpTr2−4OHの造成工程を示す図である。 図中、黒塗の太線で示した部分が、L−プロリン4位水酸化酵素遺伝子を含む部分を示す。ApはpBR322由来のアンピシリン耐性遺伝子を、Ptrp×2は大腸菌由来のトリプトファンオペロンのプロモーターを2つ直列させたプロモーター(タンデムトリプトファンプロモーター)を示す。矢印は遺伝子の転写並びに翻訳の方向を示す。なお図中にはプラスミドの造成に関係する制限酵素部位のみを示してある。
【特許請求の範囲】
【請求項1】
配列番号15で示される塩基配列を有するDNA、または配列番号15で示される塩基配列において宿主微生物での発現に最適なコドンとなるように塩基の置換された塩基配列を有するDNA断片をベクターに組み込んで得られる組換え体DNAを保有し、且つ、L−プロリンの生合成系の活性の強化された形質転換体。
【請求項2】
配列番号15で示される塩基配列において宿主微生物での発現に最適なコドンとなるように塩基の置換された塩基配列を有するDNAが、配列番号1で示される塩基配列を有するDNAである、請求項1記載の形質転換体。
【請求項3】
L−プロリン生合成系の活性の強化が、宿主中のプロリン合成系遺伝子のコピー数を増加させる、プロリンによってフィードバック阻害を受けるプロリン合成系酵素遺伝子に変異を生ぜしめ、プロリンによるフィードバック阻害が著しく減少した酵素をコードする遺伝子を宿主に導入する、プロリンの分解活性を欠失させる、またはこれらを組み合わせることにより達成されることを特徴とする、請求項1記載の形質転換体。
【請求項4】
L−プロリン生合成系の活性の強化が、L−プロリンの生合成に係わる酵素をコードする遺伝子の導入により達成されることを特徴とする、請求項1記載の形質転換体。
【請求項5】
遺伝子が、大腸菌由来のproBまたはproA遺伝子である、請求項4記載の形質転換体。
【請求項6】
遺伝子が、L−プロリンによるフィードバック阻害の低減したプロリン生合成に係わる酵素をコードする遺伝子である、請求項4記載の形質転換体。
【請求項7】
遺伝子が、大腸菌由来のproB74遺伝子である、請求項6記載の形質転換体。
【請求項8】
請求項1〜7いずれか1項に記載の形質転換体を培地に培養して得られる培養物、菌体またはそれらの処理物を酵素源として、2−ケトグルタル酸および2価鉄イオンの存在下、水性媒体中で、L−プロリンをトランス−4−ヒドロキシ−L−プロリンに変換させ、生成したトランス−4−ヒドロキシ−L−プロリンを該水性媒体より採取することを特徴とするトランス−4−ヒドロキシ−L−プロリンの製造法。
【請求項9】
水性媒体が培養液であることを特徴とする、請求項8記載の製造法。
【請求項1】
配列番号15で示される塩基配列を有するDNA、または配列番号15で示される塩基配列において宿主微生物での発現に最適なコドンとなるように塩基の置換された塩基配列を有するDNA断片をベクターに組み込んで得られる組換え体DNAを保有し、且つ、L−プロリンの生合成系の活性の強化された形質転換体。
【請求項2】
配列番号15で示される塩基配列において宿主微生物での発現に最適なコドンとなるように塩基の置換された塩基配列を有するDNAが、配列番号1で示される塩基配列を有するDNAである、請求項1記載の形質転換体。
【請求項3】
L−プロリン生合成系の活性の強化が、宿主中のプロリン合成系遺伝子のコピー数を増加させる、プロリンによってフィードバック阻害を受けるプロリン合成系酵素遺伝子に変異を生ぜしめ、プロリンによるフィードバック阻害が著しく減少した酵素をコードする遺伝子を宿主に導入する、プロリンの分解活性を欠失させる、またはこれらを組み合わせることにより達成されることを特徴とする、請求項1記載の形質転換体。
【請求項4】
L−プロリン生合成系の活性の強化が、L−プロリンの生合成に係わる酵素をコードする遺伝子の導入により達成されることを特徴とする、請求項1記載の形質転換体。
【請求項5】
遺伝子が、大腸菌由来のproBまたはproA遺伝子である、請求項4記載の形質転換体。
【請求項6】
遺伝子が、L−プロリンによるフィードバック阻害の低減したプロリン生合成に係わる酵素をコードする遺伝子である、請求項4記載の形質転換体。
【請求項7】
遺伝子が、大腸菌由来のproB74遺伝子である、請求項6記載の形質転換体。
【請求項8】
請求項1〜7いずれか1項に記載の形質転換体を培地に培養して得られる培養物、菌体またはそれらの処理物を酵素源として、2−ケトグルタル酸および2価鉄イオンの存在下、水性媒体中で、L−プロリンをトランス−4−ヒドロキシ−L−プロリンに変換させ、生成したトランス−4−ヒドロキシ−L−プロリンを該水性媒体より採取することを特徴とするトランス−4−ヒドロキシ−L−プロリンの製造法。
【請求項9】
水性媒体が培養液であることを特徴とする、請求項8記載の製造法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2006−20641(P2006−20641A)
【公開日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願番号】特願2005−232987(P2005−232987)
【出願日】平成17年8月11日(2005.8.11)
【分割の表示】特願平8−232724の分割
【原出願日】平成8年9月3日(1996.9.3)
【出願人】(000001029)協和醗酵工業株式会社 (276)
【Fターム(参考)】
【公開日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願日】平成17年8月11日(2005.8.11)
【分割の表示】特願平8−232724の分割
【原出願日】平成8年9月3日(1996.9.3)
【出願人】(000001029)協和醗酵工業株式会社 (276)
【Fターム(参考)】
[ Back to top ]