
トランスグルタミナーゼによるIP3レセプターの修飾
【課題】トランスグルタミナーゼ(TG)によるIP3レセプター1(IP3R1)の生物活性の調節と疾患との相関に基づく該疾患の治療剤のスクリーニング法を提供する。
【解決手段】疾患関連細胞又は疾患モデル非ヒト動物において、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を指標にして該架橋によって引き起こされるオートファジーと該疾患との相関関係を測定し、さらに該疾患が該IP3R1のサブユニット間架橋に起因するときにTG2の阻害剤又は活性化剤を候補薬剤として該モデル非ヒト動物に投与して治療効果を調べることを含む、疾患の治療剤をスクリーニングする方法。
【解決手段】疾患関連細胞又は疾患モデル非ヒト動物において、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を指標にして該架橋によって引き起こされるオートファジーと該疾患との相関関係を測定し、さらに該疾患が該IP3R1のサブユニット間架橋に起因するときにTG2の阻害剤又は活性化剤を候補薬剤として該モデル非ヒト動物に投与して治療効果を調べることを含む、疾患の治療剤をスクリーニングする方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、トランスグルタミナーゼ2(「TG2」と称する)によるIP3レセプター1(「IP3R1」と称する)の修飾に関する。
【背景技術】
【0002】
すべての神経変性疾患の共通の特徴は、折り畳みの誤りと安定な代替コンフォメーションへの変換によって生じるタンパク質封入体の異常な沈着である(Selkoe, 2004)。前記封入体タンパク質の分析により、グルタミン(Gln)残基とリジン(Lys)残基との間のイソペプチド結合による共有結合性架橋の存在が明らかにされ、並びに、アルツハイマー(Alzheimer)病、ハンチントン(Huntington)病及びパーキンソン(Parkinson)病の患者の脳脊髄液に、切断されたイソペプチド部分が増加するという事実が明示された(Jeitner et al., 2009)。イソペプチド結合の形成はトランスグルタミナーゼ(TG)によって触媒され(Iismaa et al., 2009; LorandとGraham, 2003)、その活性、発現及び量は実際、上記疾患をもつ患者で増加する(Jeitner et al., 2009; LorandとGraham, 2003)。しかしながら、神経系でのTGの役割は完全には理解されていない。
【0003】
TGは、カルシウム(Ca2+)依存性酵素であり、アミド基転移や脱アミド化を含むGln残基の種々の翻訳後修飾を触媒する(Iismaa et al., 2009)。アミド基転移の結果、Nε-(γ-グルタミル)リジンイソペプチド結合を介してLys残基に架橋される。TGファミリーは、TG1〜TG7やXIII因子と称される8つの酵素/酵素前駆体(zymogen)からなる(Iismaa et al., 2009)。TGによるタンパク質の修飾は、種々の生理学的役割、例えばXIII因子による血液凝固、TGによる限局された表皮形成(Candi et al., 2005; ThacherとRice, 1985)、血小板中でのセロトニン結合(Dale et al., 2002; Walther et al., 2003)、及びAT1レセプターの調節(AbdAlla et al., 2004)に関与する。多くの疾患でのTGの病理学的役割もまた示されており、セリアック病(Hovhannisyan et al., 2008)から神経変性疾患(LorandとGraham, 2003)までの範囲である。TG2は脳内に広がっており、トランス(trans)型のレチノイン酸(RA)(神経モルフォゲン)で誘導可能である。TG2プロモーター領域はRAレセプター(RAR-RXR)の結合エレメントを含む(Nagy et al., 1996)。細胞死においてTG2は双面的であるが、これはTGが細胞に対して毒性的であったり保護的であったりしうるからである。トランスジェニックマウス又はノックアウトマウスを用いた最近の研究は一貫して、TG2が脳内の神経細胞死(Battaglia et al., 2007; Tucholski et al., 2006)や胸腺細胞の生存活性(Nanda et al., 2001)に関与しうることを示している。一方、TG2はまた、癌細胞では抗アポトーシス性であるが、これはTG2が癌の化学耐性や転移能と相関するからである(Verma et al., 2006)。Huntington病のR6/1モデルマウス(Mastroberardino et al., 2002)やR6/2モデルマウス(BaileyとJohnson, 2005)でのTG2の除去は、寿命の延長や運動機能の改善と関わりがあり、R6/2マウスでのTG2の過剰発現は一貫してカイニン酸刺激に対する細胞の脆弱性を実証している。しかしながら、TG2ノックアウトマウスはポリ-Gln凝集産生の低下を示していない(BaileyとJohnson, 2005)。したがって、タンパク質封入体自体のTG2依存性架橋が原因ではないと考えられており、その機序は未知である。
【0004】
脳の機能は、神経系のシグナル伝達によって調節されており、これには細胞内のCa2+ シグナル伝達が関与している(Berridge et al., 2000)。イノシトール1,4,5-トリホスフェートレセプター(IP3R)(これはER在留レセプターである。)は集合して、IP3に反応してERからCa2+を放出するテトラマーのチャネルを形成する(Foskett et al., 2007)。IP3は、種々の細胞表面レセプターの刺激によって産生される。IP3Rは構造的に3つのドメイン、すなわちIP3-結合コアと調節領域を含む大きな細胞質ドメイン、6つの膜貫通セグメントを含むチャネルドメイン、及び細胞質側の短いC末端尾部、に分けられる(Foskett et al., 2007)。IP3R1型(IP3R1)は専ら脳に存在し、長期増強 (LTP)と長期抑圧 (LTD)に関与する(Harnett et al., 2009; Nishiyama et al., 2000)。IP3R1を介するCa2+シグナル伝達及び恒常性は、細胞死や生存シグナル伝達に重要である(Berridge et al., 2000)。チトクロムc (Boehning et al., 2003)やBcl-2 (Rong et al., 2008)との相互作用は、ミトコンドリア依存性アポトーシスに関係している。最近の研究によって、IP3Rが、Bcl-2やBeclin 1との相互作用を通して(Vicencio et al., 2009)、オートファジーを調節していることが実証された(LevineとKroemer, 2008)。しかしながら、これらの調節因子がチャネル機能を制御して細胞運命を決定する正確な機序は依然としてほとんど知られていない。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Selkoe, D. J. (2004). Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol 6, 1054-1061
【非特許文献2】Jeitner, T. M., Pinto, J. T., Krasnikov, B. F., Horswill, M., and Cooper, A. J. (2009). Transglutaminases and neurodegeneration. J Neurochem 109 Suppl 1, 160-166
【非特許文献3】Iismaa, S. E., Mearns, B. M., Lorand, L., and Graham, R. M. (2009). Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev 89, 991-1023
【非特許文献4】Lorand, L., and Graham, R. M. (2003). Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 4, 140-156
【非特許文献5】Candi, E., Schmidt, R., and Melino, G. (2005). The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 6, 328-340
【非特許文献6】Thacher, S. M., and Rice, R. H. (1985). Keratinocyte-specific transglutaminase of cultured human epidermal cells: relation to cross-linked envelope formation and terminal differentiation. Cell 40, 685-695
【非特許文献7】Dale, G. L., Friese, P., Batar, P., Hamilton, S. F., Reed, G. L., Jackson, K. W., Clemetson, K. J., and Alberio, L. (2002). Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 415, 175-179
【非特許文献8】Walther, D. J., Peter, J. U., Winter, S., Holtje, M., Paulmann, N., Grohmann, M., Vowinckel, J., Alamo-Bethencourt, V., Wilhelm, C. S., Ahnert-Hilger, G., and Bader, M. (2003). Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 115, 851-862
【非特許文献9】AbdAlla, S., Lother, H., Langer, A., el Faramawy, Y., and Quitterer, U. (2004). Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell 119, 343-354
【非特許文献10】Hovhannisyan, Z., Weiss, A., Martin, A., Wiesner, M., Tollefsen, S., Yoshida, K., Ciszewski, C., Curran, S. A., Murray, J. A., David, C. S., et al. (2008). The role of HLA-DQ8 beta57 polymorphism in the anti-gluten T-cell response in coeliac disease. Nature 456, 534-538
【非特許文献11】Nagy, L., Saydak, M., Shipley, N., Lu, S., Basilion, J. P., Yan, Z. H., Syka, P., Chandraratna, R. A., Stein, J. P., Heyman, R. A., and Davies, P. J. (1996). Identification and characterization of a versatile retinoid response element (retinoic acid receptor response element-retinoid X receptor response element) in the mouse tissue transglutaminase gene promoter. J Biol Chem 271, 4355-4365
【非特許文献12】Battaglia, G., Farrace, M. G., Mastroberardino, P. G., Viti, I., Fimia, G. M., Van Beeumen, J., Devreese, B., Melino, G., Molinaro, G., Busceti, C. L., et al. (2007). Transglutaminase 2 ablation leads to defective function of mitochondrial respiratory complex I affecting neuronal vulnerability in experimental models of extrapyramidal disorders. J Neurochem 100, 36-49
【非特許文献13】Tucholski, J., Roth, K. A., and Johnson, G. V. (2006). Tissue transglutaminase overexpression in the brain potentiates calcium-induced hippocampal damage. J Neurochem 97, 582-594
【非特許文献14】Nanda, N., Iismaa, S. E., Owens, W. A., Husain, A., Mackay, F., and Graham, R. M. (2001). Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem 276, 20673-20678
【非特許文献15】Verma, A., Wang, H., Manavathi, B., Fok, J. Y., Mann, A. P., Kumar, R., and Mehta, K. (2006). Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res 66, 10525-10533
【非特許文献16】Mastroberardino, P. G., Iannicola, C., Nardacci, R., Bernassola, F., De Laurenzi, V., Melino, G., Moreno, S., Pavone, F., Oliverio, S., Fesus, L., and Piacentini, M. (2002). 'Tissue' transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ 9, 873-880
【非特許文献17】Bailey, C. D., and Johnson, G. V. (2005). Tissue transglutaminase contributes to disease progression in the R6/2 Huntington's disease mouse model via aggregate-independent mechanisms. J Neurochem 92, 83-92.
【非特許文献18】Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1, 11-21
【非特許文献19】Foskett, J. K., White, C., Cheung, K. H., and Mak, D. O. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87, 593-658
【非特許文献20】Harnett, M. T., Bernier, B. E., Ahn, K. C., and Morikawa, H. (2009). Burst-timing-dependent plasticity of NMDA receptor-mediated transmission in midbrain dopamine neurons. Neuron 62, 826-838
【非特許文献21】Nishiyama, M., Hong, K., Mikoshiba, K., Poo, M. M., and Kato, K. (2000). Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408, 584-588
【非特許文献22】Boehning, D., Patterson, R. L., Sedaghat, L., Glebova, N. O., Kurosaki, T., and Snyder, S. H. (2003). Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol 5, 1051-1061
【非特許文献23】Rong, Y. P., Aromolaran, A. S., Bultynck, G., Zhong, F., Li, X., McColl, K., Matsuyama, S., Herlitze, S., Roderick, H. L., Bootman, M. D., et al. (2008). Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Mol Cell 31, 255-265
【非特許文献24】Vicencio, J. M., Ortiz, C., Criollo, A., Jones, A. W., Kepp, O., Galluzzi, L., Joza, N., Vitale, I., Morselli, E., Tailler, M., et al. (2009). The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 16, 1006-1017
【非特許文献25】Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27-42
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、TG2によるIP3R1タンパク質のグルタミン残基の修飾又はサブユニット間架橋、それによって生じるIP3R1タンパク質のチャネル活性の抑制、或いはオートファジーによる細胞死の誘導、を利用した、TG2又はIP3R1活性の測定法、神経変性疾患や癌などの疾患の療法を提供することである。
【課題を解決するための手段】
【0007】
今回、本発明者らは、改良した電気泳動法を用いてIP3R1における共有結合性修飾の状態をモニターし、IP3R1がTG2の新規の基質であること、TG2は、テトラマーIP3R1内の互いに近傍にある2つのサブユニットのC末端領域と調節ドメインとの間のサブユニット間アミド基転移又はIP3R1のグルタミン残基の修飾を触媒して生物活性(すなわち、チャネル活性)を抑制又は阻害すること、TG2はまた、レチノイン酸(RA)依存的にIP3R1機能を損なうし、またオートファジーを調節すること、並びに、TG2によるIP3R1内の修飾が特定の疾患に関係するオートファジー細胞死を制御することを明らかにした。
【0008】
本発明は、要約すると、以下の特徴を包含する。
(1) 細胞においてTG2によってIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を形成してIP3R1生物活性を抑制し、それによってオートファジーを誘導して該細胞の細胞死を引き起こすことを含む、細胞増殖の抑制方法。
(2) 細胞が腫瘍細胞である、上記(1)に記載の方法。
【0009】
(3) 疾患関連細胞又は疾患モデル非ヒト動物において、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を指標にして該架橋によって引き起こされるオートファジーと該疾患との相関関係を測定し、さらに該疾患が該IP3R1のサブユニット間架橋に起因するときにTG2の阻害剤又は活性化剤を候補薬剤として該モデル非ヒト動物に投与して治療効果を調べることを含む、疾患の治療剤をスクリーニングする方法。
(4) 疾患が神経変性疾患又は癌である、上記(3)に記載の方法。
【0010】
(5) TG2及びIP3R1の生物活性をともに含む細胞を用いてそのIP3R1活性を、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋又は該架橋の脱アミド化を指標にして測定することを含む、細胞でのIP3R1活性の測定方法。
(6) 細胞が疾患関連細胞である、上記(5)に記載の方法。
【0011】
(7) TG2の活性を抑制することによるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の抑制によってオートファジーを抑制することを含む、神経変性疾患の治療方法。
(8) IP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の増強剤を有効成分として含む、癌の治療用組成物。
【0012】
(9) IP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の抑制剤を有効成分として含む、神経変性疾患の治療用組成物。
(10) TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を検出することを含む、IP3R1修飾の検出方法。
【0013】
(11) TG2によるIP3R1タンパク質のサブユニット間アミド基転移を介する該タンパク質のサブユニット間架橋を測定することを含む、IP3R1タンパク質チャネル活性のTG2阻害作用を測定する方法。
(12) TG2によるIP3R1タンパク質のサブユニット間架橋の脱アミド化を測定することを含む、TG2触媒活性の測定方法。
【0014】
(13) 測定が、SDS含有アガロースゲル電気泳動特異抗体との交差反応を用いて行うことを含む、上記(10)又は(11)に記載の方法。
(14) 配列番号1又は配列番号2のアミノ酸配列のそれぞれ2746位又は2707位のC末端側グルタミン残基(Q)を介してサブユニット間架橋されたテトラマーIP3R1タンパク質。
(15) TG2によってIP3R1タンパク質のC末端側グルタミン残基と一級アミン含有分子とを結合することを含む、IP3R1のグルタミン残基の修飾方法。
【発明の効果】
【0015】
本発明は、癌又は神経系において共に重要なタンパク質であるTG2とIP3R1とが相互作用して、これらのタンパク質が細胞内のIP3R1生物活性の調節や細胞死を伴うオートファジーに関与するという知見に基づいており、癌、神経変性疾患などの特定の疾患の治療のために本発明を利用することができるという利点が提供される。
【図面の簡単な説明】
【0016】
【図1−1】IP3R1のサブユニット間の架橋の内因性活性を示す。(A) 本発明者らが開発したSDS-アガロース電気泳動(AGE)による架橋の検出。IP3R1サブユニットを種々の濃度(0.01, 0.02, 0.05, 0.1, 0.2, 0.5, 1 mM(増加スロープで示す。))のビス[スルホスクシンイミジル]スベレート(BS)(上)又は1,8-ビス-マレイイミド-ジエチレングリコール(BM)(下)によって架橋した。(B) Triton X-100 (1)、Zwittergent 3-14 (2)、SDS (3)又は界面活性剤不含(4)で前処理したIP3R1を、0.5mM BS(上)又は0.1mM BM(下)を用いて架橋した。(C) Ca2+-依存性架橋の内因性活性。小脳P2/P3膜をバッファ又は細胞質画分と一緒にインキュベーションした。(D) アニオン交換カラムによる脳ライセートのクロマトグラム。左軸及び右軸はそれぞれ、吸光度(実線)及びNaCl勾配(破線)を示す。(E) 各溶出液、使用した細胞質画分 (Cyt)又は素通り画分(pass)を、小脳P2/P3膜と一緒にインキュベーションした。*印は、Ca2+-依存性断片である。(A〜C, E)のローマ数字は架橋したサブユニットを示し、これはThモノマー(335 kDa)及びダイマー(670 kDa)によって判断される。
【図1−2】(図1−1の続き)(F) ピーク画分(No. 14)又は精製TGを、シスタミン(2, 4, 10, 20, 40 mM)の存在又は非存在(第2レーン)下でP2/P3画分と一緒にインキュベーションした。(G) クマシーブリリアントブルー(CBB)染色によってIP3R1又はTG2 (*)が示された。精製IP3R1を精製TG2 (0, 1, 5, 20 μg/ml)と一緒にインキュベーションしAGEにかけた。(H) HeLa細胞中のサブユニット間架橋の内因性活性は、TG2 siRNAによって除去された。αTG2によるウエスタンブロット(WB)はTG2の細胞レベルを示す。アミドブラック(AB)染色を負荷対照のために使用した。右パネルはAGEの結果である。(I) DMSO又はRAで前処理したHeLa細胞の架橋活性。(J) RA又はDMSOで前処理した神経芽腫(SH-SY5Y)又は星状膠細胞腫(U87-MG)中のTG2の発現レベル。(K) SH-SY5Y又はU87-MG細胞中でのイオノマイシン(ION, 10μM)刺激によるIP3R1のサブユニット間架橋。
【図2−1】TG2によるIP3R1活性の阻害を示す。(A) 不活性化されたTG2の調製。煮沸したTG2 (中)又はNEM処理したTG2(下)は酵素活性を示さない。20個のスペクトルを重ねた。(B) TMR-cadの自己取り込みによるTG2活性の測定。(C) fura-2を用いたときのCa2+放出の代表的トレース。上の左と右のトレースは、対照(-TG)の、及びMg-ATPの添加によるCa2+負荷後のTG2-前処理(+TG) P2P3膜の、IP3-誘導Ca2+放出を示す。下のトレースは、TG2 (1〜4: 0, 5, 10, 20μg/ml TG), 10μg/ml煮沸TG2 (5)、或いは10μg/ml NEM-TG2 (6)の典型的な反応を表す。縦軸は、(340nmに対する510nmでの蛍光)対(380nmに対する510nmでの蛍光)の比を示す。(D) 棒グラフは、独立の3つの実験から得られたデータのまとめであり、ここで、膜はTG2 (0, 1, 2, 5, 10, 20, 50μg/ml)、10μg/ml煮沸TG2又は10μg/ml NEM-TG2で前処理された。IP3によるCa2+放出は、対照の%(平均+SD)として示されている。TG2のIC50値は、4パラメータ算定曲線関数を用いる非線形回帰分析によって計算された34nMであった。(E) BPは、TG2によって触媒される架橋を阻害した(BP: 1, 2, 5, 10, 20mM)。(F) TG2及びBPで前処理されたIP3R1を、アビジン-HRP又は4C11を用いて検出した。
【図2−2】(図2−1の続き)(G) 10mM BP又は10mM シスタミン(cystamine)(CA)の存在中で調製されたTG2処理膜からのCa2+放出。(H) アビジンによるIP3R1を介するCa2+放出の阻害。BPが取り込まれた膜 (BP+TG)又は取り込まれなかった膜(-TG)を10又は0μg/ml TG2で前処理した。Ca2+放出を0, 31, 63, 94, 125及び156μg/ml (スロープ)或いは156μg/ml (-TG)のアビジンの存在中で測定した。(I) 上のパネルは、RAによって分化された又は分化されなかったSH-SY5Y細胞でのIP3R1内のBPの顕著な取り込みを示す。下のパネルは、TG2及びIP3R1の量を示す。各レーンでは3μgタンパク質が負荷された。(J) fura-2比率の典型的トレースは、P2P3膜ベシクルでのIP3Rを介するIP3誘導Ca2+放出(左)、或いは、DMSO(対照)又はRAで前処理されたSH-SY5Y生細胞でのカバコール(carbachol; CCh)誘導Ca2+放出(右)を示す。(K) IP3誘導Ca2+放出は、0.1及び1μM IP3に応答するfura-2比率の変化(ΔR)によって表された。棒グラフは、3つの培養ディッシュから調製した3つのサンプルの平均+SDを示す。(L) SH-SY5Y生細胞でのカルバコール(CCh, 10μM)誘導Ca2+放出に対するRAの影響。*p<0.05, ***p<0.001 (two-tailed, n=3)
【図3−1】TG2がC末端付近でのIP3R1サブユニット間の架橋を触媒することを示す。BPが取り込まれた精製IP3R1を、(A)及び(B)で種々の濃度のカルパイン(Calp)或いは(B)で種々の濃度のリジル-エンドペプチダーゼ(Lep)によって消化した。得られた断片を、アビジン-HRP, 4C11, 10A6又は18A10を用いて検出した。矢印は、BPが取り込まれた(白)又はBPが取り込まれなかった(黒)主断片を示す。サンプルは、5%ゲル(A)又は7.5%ゲル(B)で泳動された。(C) IP3R1のC末端を認識する18A10抗体での前処理によってTG2によるIP3R1の架橋が阻害された。P2P3膜(1 mg/ml)は、氷上1時間抗体(なし, 18A10, 4C11)で、次いでTG2 (0, 2, 5, 10, 15, 20μg/ml)でインキュベーションした。
【図3−2】(図3−1の続き)(D) TG2による架橋後の免疫反応性。TG2 (0, 2, 5, 10, 20μg/ml)によって架橋されたIP3R1を4C11, 18A10及びpep6Abを用いて検出した。再現性は4回以上の別個の実験で確認された。TG2 (0, 5, 10, 20μg/ml)によって架橋された小脳IP3R1は、50μg/ml Calp (E)又は1μg/ml Lep (F)によって消化された。(G) 50μg/ml Calpでの消化後のTG2 (0, 5, 10, 20μg/ml)による架橋。(E〜G)の白矢印は、TG2によって著しく減少した際立ったバンドを示す。黒矢印は、TG2処理によって変化しなかったN末端断片を示す。
【図4−1】グルタミン2746とリジン-クラスター領域がTG2による架橋に必須であることを示す。(A) 脳サンプルからのTG2サブユニットの精製。アビジン-ビーズによって、CBBで染色される特異的基質(*)をほぼ精製することができたことを特記する。(B) WBによるIP3R1の検出。(C) IP3R1のグルタミン2746をTG2で修飾した。(A)に示したTG2サブユニットをIP3R1として同定した(配列番号1)。(C) IP3R1のイオンm/z 1935.99のC末端ペプチド(上)と2247.15のビオチン化ペプチド(下)をMS/MSでスキャニングした。bイオンとyイオンをMS/MSスペクトル上に記した。*印は修飾されたGln残基を示す。本発明者らは、MS分析によって帰属された63個のGln残基のうちGln2746のみが修飾されたことを見出した(配列番号1)。
【図4−2】(図4−1の続き)(D) サブユニット間架橋に重要なドメインの突然変異誘発。スキームはIP3R1の一次構造とCalpによる切断部位(C130)を示す。欠失変異体(d1〜d4)、点突然変異体(Q2746N)及び野生型(wt)を、スキーム中に描出した。低濃度ゲルによって、COS-7細胞で発現された欠失変異体のサブユニット間架橋が示された。黒と白の矢印はそれぞれ、架橋されたダイマーとその対応するサイズを示す。注目すべきことは、d3又はd4のいずれかがサブユニット間架橋をまったく示さないことである。(E) Q2746N突然変異体でのサブユニット間架橋の喪失。
【図5−1】TGが架橋の加水分解と脱アミド化を触媒することを示す。(A) 上のパネルはTMR-cadのピークを示す。中パネル及び下パネルは、TMR-cad、TG2及びNPQQPA(配列番号3)又はNPNQPA(配列番号4)を含む反応混合物のクロマトグラムである。白矢印は生成物を示す。(B) 上パネルは、精製した生成物を示す。中パネル又は下パネルは、該生成物、TG2及びCaCl2を含む反応混合物又は該生成物及びTG2を含む反応混合物のクロマトグラムを示す。(C) FITC及びTMR-cad (左)を結合したペプチドの典型的PDAデータ。二次元グラフは、ピークX (TMR-cad), Y (FITC-NPQQPA)及びZ (生成物)を示す上のクロマトグラムに相当する吸収スペクトルを示す。右パネルは精製された生成物を示す。
【図5−2】(図5−1の続き)(D) 左パネルに示したのは、(C)の生成物にTG2を添加した後の蛍光スペクトルの変化である。スペクトルは1分間隔で記録された。530 nmでの蛍光強度の経時的スキャニングを記録した(中及び右パネル)。(E)脱アミド化IP3R1テトラマーに対する特異抗体の特性化。スキームは、Q2746を含むGST融合タンパク質又は脱アミド化E2746を含むGST融合タンパク質を示す。ブロットしたQ2746又はE2746タンパク質(1, 3, 10 ng)は、αE2746、18A10及びαGST抗体を用いて検出された。αE2746と18A10はそれぞれE2746及びQ2746を特異的に検出することに注目されたい。(F) in vitroでのQ2746の酵素的脱アミド化。90分間TG2で前処理したP2P3画分中のIP3R1がαE2746、18A10及び4C11を用いて検出された。(G)脱アミド化IP3R1が精製IP3R1で検出された(レーン1:3μl,レーン2:10μl,レーン3:10倍希釈3μl,レーン2:10倍希釈10μl)。(H)脳サンプル中の脱アミド化IP3R1。ゲルは3匹の各マウスのサンプル(3mg/ml)の結果を示す(レーン1〜3)。
【図6−1】TG2及びIP3R1によるオートファジー調節を示す。(A) TG2 siRNAによるIP3R1修飾の喪失。HeLa細胞中の総IP3R1及び修飾IP3R1がそれぞれ4C11及びアビジン-HRPによって検出された。(B) siRNAによるTG2の除去によってLC3-IIの脂質化が減少したが、IP3R1をノックダウンすると該脂質化が高まった。HeLa細胞溶解物中の脂質化されたLC3 (LC3-II), TG2, IP3R1及びアクチンのレベルは、ウエスタンブロッティング(WB)によって評価された。棒グラフは、6つの独立の実験からの平均+SDのまとめである(*p<0.05, ***p<0.001, two-tailed)。ラパマイシン(1μM)又はスペルミジン(100μM)の存在下でのTG2のノックダウン(C)。p62(オートファジー基質)の細胞レベルは、WBによって評価された。
【図6−2】(図6−1の続き)ラパマイシン(1μM)又はスペルミジン(100μM)の存在下でのIP3R1 (D)のノックダウン。p62(オートファジー基質)の細胞レベルは、WBによって評価された。(E) TG2 siRNAによってLC3脂質化を損なうことに対するL-Gln(10mM)の影響。(F) Dan-cad (100μM)と2-APB (100μM)は、p62を蓄積することなくLC3の脂質化を強力に誘導した。
【図7−1】TG2及びIP3R1による空胞化と細胞死の制御を示す。(A) HeLa細胞での空胞化の形態図。この画像は、siRNAによるトランスフェクションの54時間後に得られた。(B) TG2又はIP3R1のノックダウンによる空胞化された細胞(中)又は空胞化されない細胞(右)の数の著しい変化。
【図7−2】(図7−1の続き)(C) 細胞死の評価。培養培地中のLDH活性はsiRNAによるトランスフェクションの48時間後に測定された。(*p<0.05, **p<0.01, ***p<0.001, ###p<0.001, two-tailed) (D) TG2及びIP3R1によって仲介される本発明者らの仮想的なオートファジーシグナル伝達モデル。正常な条件下(左パネル)で、オートファジーの誘導はIP3R1によって抑制される。活性化されたTG2がIP3R1を修飾し、オートファジーの抑制を解除する(右パネル)。その結果、オートファジーの空胞化が過剰になり細胞死が誘導される。
【図8】マウスIP3R1のアミノ酸配列(配列番号1)を示す。太字は、精密質量分析により検出されたペプチド配列を表す。また、下線は、TGにより修飾された部位を表す。
【図9】TG2によるIP3R1の酵素的調節の構造モデルを示す。(A) スキームはIP3R1のQ-ドナー(Q2746)及びK-ドナー(K-クラスター領域)を示す。C130ドメインが青で示され、IP3-結合コアを含むN180ドメインが灰色で示されている。膜貫通領域のヘリックスはN末端側からC末端側に番号付けられている(TM1-6)。(B) 絵は、細胞質側から描かれたテトラマーIP3R1内のQ-ドナー及びK-ドナーの配置を提示する。C末端のQ-ドナーは、近傍のサブユニットのK-ドナー領域のごく近くに位置する。本発明者らは、閉じた状態(B)から開いた状態(C)までのテトラマーIP3R1の立体配置の変化を提案する。ここで、Ca2+ (黄)は孔を通過する。TGによって触媒されるアミド基転移によって、閉じた立体配置をロックするための(D)に示される共有結合されたサブユニット間架橋(赤)が生じる。TGにより触媒されるBPの取り込みは、前記立体配置を留めることができない(E)。しかしながら、嵩高いアビジンとBPの化学量論的な結合は、立体障害によって、閉じたチャネル立体配置をロックする(F)。
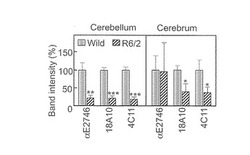
【図10】野性型マウス小脳および大脳のIP3R1をウエスタンブロッティングにより検出した結果である。縦軸は、野性型を100%としてバンドシグナル強度を計算したものである。R6/2の小脳ではE2746抗体、18A10抗体、4C11抗体、全てにおいて小脳IP3R1のシグナルが低下している。しかし、R6/2の大脳IP3R1では、E2746抗体でのIP3R1シグナルが他の抗体に比べて増強されていた。従って、これは特にR6/2の大脳に於いてIP3R1のQ2746がEに変換されていることを示唆する。
【発明を実施するための形態】
【0017】
本発明者らは、今回、IP3R1がTG2の新しい基質であり、TG2が酵素的にIP3R1の機能を損なうことを見出した。アミド基転移部位は、配列番号1(マウス)のQ2746又は配列番号2(ヒト)のQ2707であると決定され、この部位でTG2が脱アミド化も触媒する。TG2によるサブユニット間架橋には、隣接のサブユニットのK-クラスター領域が必要であり、このことは、TG2による共有結合によりサブユニットの立体配置を閉じた状態に固定することを示している。TG2によって触媒されるIP3R1の修飾は、レチノイン酸(RA)によって分化される神経芽腫で増強された。TG2はIP3R1と反対の方向にオートファジー事象と細胞死を制御したが、これは、TG2によるIP3R1の修飾がオートファジーの際の栄養飢餓からの細胞の生存を細胞死に変換する重要なシグナル伝達であることを支持している。
【0018】
<IP3R>
細胞膜上の受容体の活性化に伴いホスファチジルイノシトール4,5-ビスホスフェート(phosphatidylinositol 4,5-bisphosphate)が加水分解されると、細胞内セカンドメッセンジャーであるイノシトール1,4,5-三リン酸(inositol 1,4,5-trisphosphate (IP3))が生成する。IP3はIP3レセプター(IP3R)に結合することにより、細胞内カルシウム貯蔵オルガネラ(主に小胞体)からのCa2+放出を誘導する。このIP3/Ca2+シグナル経路において、IP3レセプターは、IP3のシグナルをCa2+のシグナルへ変換する役割を担っている。IP3レセプターは、テトラマー(4量体)構造の細胞内IP3誘導性Ca2+放出チャネル(IP3-gated Ca2+ release channel)である。哺乳動物では、3種の異なるIP3レセプターが存在する。IP3レセプター1型(IP3R1)は、中枢神経系、特に小脳において高発現している。ヒトIP3R1のアミノ酸配列及び塩基配列がGenBankに登録されており、そのAccession Numberは、例えば、NM_001099952.2(variant 1;配列番号2)、NM_002222.5(variant 2)及びNM_001168272.1(variant 3)である。マウスIP3R1は、2749アミノ酸(GenBank Accession No. NM_010585.3)からなる。IP3R1は、3つの機能的に異なる領域に分かれている。すなわち、N末端近傍にIP3結合ドメイン、C末端近傍に6回膜貫通領域を有するチャネル形成ドメイン、およびこれら2つの領域の間に制御領域が存在する。IP3結合ドメインの欠失突然変異体解析より、IP3レセプターのアミノ酸226〜578残基が特異的かつ高親和性のリガンド結合に必要な最小領域であることが示され、IP3結合コアと呼ばれている。
【0019】
IP3レセプターの活性化による細胞質Ca2+濃度の増加によって、多種多様な下流標的分子の活性が制御される。これら下流標的分子は、受精、発生、増殖、分泌、シナプス可塑性など多岐に渡る細胞応答において重要な役割を担っている(特開2007-284433)。
【0020】
<TG>
トランスグルタミナーゼ(TG)は、タンパク質分子間の架橋反応を触媒する酵素である。この酵素によって、その基質であるタンパク質中のグルタミン残基と別のタンパク質中のリシン残基又は一級アミンとの間で共有結合が形成されてタンパク質同士が結合される。
【0021】
ヒトTGには、8つのアイソザイム、例えば第8凝固因子、TG1〜TG7の存在が知られている。本発明に関わるTGは、特にTG2であり、この酵素は、細胞死や神経系疾患に関係するという報告がある(人見清隆,生化学,77巻6号,pp.552-558 (2005))。TGは、タンパク質の架橋化の他に、TGとタンパク質とから形成される複合体のグルタミン残基のグルタミン酸への脱アミド化も触媒することができる。
【0022】
TGは、カルシウムの存在によって活性化される酵素であり、アルツハイマー病、パーキンソン病、ハンチントン病等の神経疾患に関与することが知られるようになり、TG酵素の阻害剤がこれらの疾患の治療薬として有効であると考えられている(Hoffner G and Djian P, Front Biosci., 2005 Sep 1, 10:3078-3092; Duval E et al., Bioorg Med Chem Lett., 2005 Apr 1, 15(7):1885-1889)。タンパク質の異常架橋に関して、タンパク質のグルタミンのアミド基とリシンのアミノ基から脱アンモニアによりイソペプチド結合をつくる反応が架橋の主反応であり、この反応を引き起こすTGの阻害剤が、上記疾患等に効果的であろうとする作用機序が明らかにされた(Mastroberardino PG, et al., Cell Death Differ., 2002 Sep, 9(9):873-880)。その根拠をもとにしてTGの阻害剤を、アルツハイマー病、ハンチントン病、パーキンソン病、セリアック病、白内障、狂牛病、先天性葉状魚鱗症、先天性止血障害症、肝臓障害、自己免疫疾患、脳梗塞等の治療薬として開発する研究が増加している(WO03/033002号; WO2007/061074号; 特開2009-184988; 特開2007-169272; Lorand L, Proc Natl Acad Sci U S A., 1996 Dec 10, 93(25):14310-14313; Grierson AJ, et al., Neurosci Lett., 2001 Jan 26, 298(1):9-12; Watts RE, et al., J Med Chem., 2006 Dec 14, 49(25):7493-7501; Karpuj MV et al., Nat Med., 2002 Feb, 8(2):143-149; Thung GL et al., Chemistry & Biology 2008 Sept 22, 15:969-978; Tatsukawa H et al., Gastroenterology 2009. 136(5):1783-1795)。
【0023】
<詳細な説明>
本発明によれば、IP3R1タンパク質がTG2の基質であり、TG2が、細胞内で該タンパク質の架橋・修飾や該架橋タンパク質の脱アミド化を通じて細胞での細胞死やオートファジーに関与する。
【0024】
本発明は、配列番号1又は配列番号2のアミノ酸配列のそれぞれ2746位又は2707位に相当するC末端側グルタミン残基(Q)を介してサブユニット間架橋・修飾されたテトラマーIP3R1タンパク質を提供する。
【0025】
すなわち、IP3R1タンパク質がTG2の基質であることは、今回本発明者らが見出すまでは全く知られていなかった。上記のようにテトラマーIP3R1タンパク質の1つのサブユニットのアミノ酸配列のC末端に近い特定の1つのグルタミン残基がTG2酵素の触媒作用により別のサブユニットのK-クラスター領域(チャネル活性調節ドメイン)内のリシン残基との間で共有結合してサブユニット間架橋を形成する。
【0026】
本発明者らの知識によれば、これは、IP3R1がTG2の新規な基質であること、及びTG2が、テトラマーIP3R1の酵素的修飾によってIP3R1のチャネル活性を阻害することを示す最初の研究である。本発明者らは、C末端が近傍のIP3R1サブユニットに近い、及び、TG2が近接するサブユニットに該C末端を共有結合によって結合させ、チャネルを、閉じた立体配置に固定する、と結論付けた。
【0027】
これまでの報告では、C末端尾部がチャネル活性に重要であること(Nakade et al., 1991; Uchida et al., 2003)、並びに、C末端及びKクラスター領域の付近で相互作用する、チトクロムCやBcl-2などの、多くの調節因子がチャネル活性を調節すること(Boehning et al., 2003; Foskett et al., 2007; Rong et al., 2008)が実証されている。本発明者らは今回、これらの調節因子がC末端付近のサブユニット界面に結合して、他のオリゴマーチャネルに類似したサブユニット立体配置を制御することが可能であることを示す。IP3RはCa2+を必要とし、Ca2+は調節ドメインを含む細胞質ドメインを動かすことによってコンフォメーション変化を誘導し(Anyatonwu and Joseph, 2009; Hamada et al., 2002; Hamada et al., 2003)、チャネルと、K-クラスターの近傍に位置するCa2+-センサー用の重要な残基を開く(Foskett et al., 2007)。したがって、Ca2+依存性のコンフォメーション変化が、IP3結合からの刺激によってチャネルを開かせるサブユニット立体配置を自由にすることが提案される。おそらく、このことはCa2+が共アゴニストとしてチャネルのゲーティングに必須であることを示している。
【0028】
本発明はさらに、TG2及びIP3R1の生物活性をともに含む細胞中のIP3R1活性を、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋又は該架橋の脱アミド化を指標にして測定することを含む、細胞でのIP3R1活性の測定方法を提供する。
【0029】
TG2及びIP3R1の生物活性をともに含む細胞は、IP3R1が高発現する小脳や中枢神経系由来の細胞、とりわけ中枢神経系に関わる疾患関連細胞、例えばヒト神経芽細胞腫由来のSH-SY5Y細胞などであるが、これらに限定されないものとする。
【0030】
IP3R1タンパク質(具体的には、テトラマーIP3R1タンパク質)の架橋は、上記のテトラマーIP3R1の異なるサブユニット間の架橋であり、これによってIP3R1の生物活性、すなわちチャネル活性、が阻害又は抑制される。
【0031】
架橋の検出又は測定は、SDS含有アガロースゲル電気泳動及び特異抗体との反応によって行うことができる。後述の実施例では、電気泳動は、0.1%SDS及び1%アガロース含むゲルを用いて100Vで1〜2時間の泳動を行う。ゲルをメンブレンに転写し、特異抗体(例えば、4C11等のモノクローナル抗体)との交差反応を検出する。
【0032】
架橋が検出されるならば、IP3R1のチャネル活性が阻害又は抑制されることを示す。その相関性は、一方で、IP3R1のチャネル活性を測定し、互いの結果を対比することによって分かる。IP3R1のチャネル活性は、例えばZhouらの方法で測定することができる(Zhou et al., 2007)。
【0033】
上記架橋の脱アミド化は、IP3R1タンパク質のC末端側グルタミン残基(例えば配列番号1(マウス)のQ2746又は配列番号2(ヒト)のQ2707に相当する。)のグルタミン酸への変換を測定することによって検出又は測定できる。このために、IP3R1タンパク質の配列番号1又は配列番号2のアミノ酸配列等に基づいて設計された、上記C末端側グルタミン残基から変換されたグルタミン酸残基を含むペプチド(例えば、約7〜約15アミノ酸)と特異的に結合可能なポリクローナル抗体(例えばαE2746抗体)、或いはモノクローナル抗体を作製し、この抗体との交差反応を利用することができる。このようなモノクローナル抗体は、岩崎辰夫、単クローン抗体、ハイブリドーマとELISA(講談社、1983)などに記載の方法によって作製することができる。簡単に説明すると、上記ペプチドを免疫したマウスの脾臓細胞と哺乳動物ミエローマ細胞とをPEGの存在下で融合させて得られたハイブリドーマの中から、HAT培地で抗体産生ハイブリドーマを選択し、さらにグルタミン酸残基を含む領域と特異的に結合可能な抗体産生細胞を選別し、クローニング法か又はマウス腹腔内注射後の腹水から目的のモノクローナル抗体を得ることができる。ミエローマ細胞には、例えば、P3X63Ag8, NS-1, MPC-11, SP2, FO, S194, R210, P3U1などの細胞株が含まれる。
【0034】
上記架橋の検出又は測定はまた、IP3R1修飾の検出、或いはP3R1タンパク質のチャネル活性のTG2阻害作用を測定するために利用できる。
【0035】
すなわち、本発明はさらに、TG2によるIP3R1タンパク質のC末端側グルタミン残基の架橋を検出することを含む、IP3R1修飾の検出方法、或いは、TG2によるIP3R1タンパク質のサブユニット間アミド基転移を介する該タンパク質のサブユニット間架橋を測定することを含む、IP3R1タンパク質チャネル活性のTG2阻害作用を測定する方法をも提供する。
【0036】
さらに、上記の、TG2によるIP3R1タンパク質のサブユニット間架橋の脱アミド化を検出又は測定することによってTG2触媒活性を測定することも可能である。このとき、配列番号1又は配列番号2のアミノ酸配列のそれぞれ2746位又は2707位のC末端側グルタミン残基(Q)を介してサブユニット間架橋されたテトラマーIP3R1タンパク質を基質とすることができる。このように架橋されたテトラマーIP3R1タンパク質も本発明に包含される。
【0037】
従来、TG2の触媒活性は、例えばLorand, L et al., Anal. Biochem. 44:221-231, 1971などの方法で測定されているが、IP3R1タンパク質を基質とするTG2の活性測定については知られていなかった。本発明の方法で測定されたTG2活性と、従来の方法で測定されたTG2活性との相関をみることによって、本発明の測定方法の定量の妥当性が分かるはずである。
【0038】
TG2によるIP3R1の修飾(すなわち、テトラマーIP3R1のサブユニット間架橋)に関して、該修飾がレチノイン酸(RA)によって誘導されることが今回見出された。この知見は、IP3R1が、RAシグナル伝達によって神経機能を調節することができる可能性を示唆している。すなわち、TG2がIP3R1修飾によってレチノイドシグナル伝達を調節することができる新しい経路が存在する可能性を示唆している。一方、この関連で、RAシグナル伝達は神経の分化や変性に重要であることも知られているので(Maden, 2007)、TG2によるIP3R1の修飾と神経系(特に中枢神経系)の異常との関係が示唆される。
【0039】
本発明者らはさらに、TG2とIP3R1がともに、オートファジーの調節に関与しており、腫瘍細胞であるHeLa細胞でTG2の役割がIP3R1の役割と反対であること、すなわちTG2がIP3R1機能を損なうことを見出した。このことは、オートファジーがCa2+シグナル伝達(Hoyer-Hansen et al., 2007; Williams et al., 2008)及びIP3Rによって調節されるという従来の概念(Criollo et al., 2007; Vicencio et al., 2009)と矛盾しない。
【0040】
さらにまた、本発明者らは、TG2及びIP3R1が、リソソーム分解ではなくLC3の脂質化と空胞化を制御していることを見出し、IP3R1の除去によってLC3の脂質化と空胞化を増強したことを示すデータと合わせると、内因性のIP3R1がオートファジーを伴う空胞化を抑制するはずであり、これは従来の概念(Criollo et al., 2007; Vicencio et al., 2009)と矛盾しない。したがって、本発明は、細胞死に直結する、TG2及びIP3R1によるオートファジー調節に関する。すなわち、この調節は、TG2 siRNAなどのTG2阻害剤によって明らかにされるTG2によるオートファジー空胞化の抑制解除(disinhibition)、並びに、IP3R1によるオートファジー細胞死のTG2依存性制御である。本発明に係るこの考え方は、明らかな空胞化、LC3の脂質化及びTG2又はIP3R1のノックダウン時の細胞死、の細胞応答だけでなく、疾患状態にある神経変性の分子機構をも整然と解明する。すなわち、Huntington病及びAlzheimer病では、TGが活性化され(Iismaa et al., 2009)、及び、TG-産物であるγ-グルタミルアミンが増加する(Jeitner et al., 2009)が、本発明者らは、修飾IP3R1が、Huntington病モデルマウス(R6/2)(ポリグルタミン含有タンパク質が過剰発現される。)で専ら増大したことを確認した(図10)。TG2の活性化が酵素的にIP3R1を修飾し、そして、Ca2+シグナル伝達又はベクリン-1相互作用の制御不能によってオートファジーを伴う空胞化を抑制解除する(Vicencio et al., 2009)。正常の条件では、オートファジーは、リソソーム分解と結びつくことによって、折り畳みを誤ったタンパク質や栄養飢餓から細胞を保護する。しかしながら、TG2は、オートファジー空胞の過剰負荷を行い、したがって神経変性疾患での細胞死を誘導するはずである。重要なことは、本発明者らの知見が、TG2によるIP3R1の修飾が、細胞の保護から細胞死にオートファジーの固有の役割に切り替える重要な事象であるという考えに導くことである。オートファジーは、神経変性疾患でのタンパク質封入体のクリアランスに関与している(Rubinsztein et al., 2007)。
【0041】
一方、IP3R1によるオートファジー細胞死のTG2依存性制御を、癌細胞で利用するときには、癌細胞の細胞死に導くことが可能になる。このような癌細胞として、例えばヒト子宮頸癌細胞であるHeLa細胞などを例示することができるが、しかしTG2は各種の癌細胞で発現していることが周知であるから、そういうすべての癌細胞に適用可能であると考えられる。
【0042】
このように、疾患が神経変性疾患であれば、TG2によるIP3R1修飾を阻害する薬剤が治療に有効であるのに対して、疾患が癌(又は、悪性腫瘍)であれば、TG2によるIP3R1修飾を賦活化(又は活性化)する薬剤が治療に有効である。
【0043】
本発明者らの考えは、TG2によるIP3R1の修飾と疾患、特に神経変性疾患及び癌の治療法に光を与え、これはこれら疾患のための薬剤開発の新規の治療標的になりうることを示している。
【0044】
したがって、本発明はまた、細胞においてトランスグルタミナーゼ2(TG2)によってIP3レセプター1(IP3R1)タンパク質のC末端側グルタミン残基の修飾(すなわち、サブユニット間架橋)及び該修飾に伴って起こるIP3R1活性(すなわち、チャネル活性)の阻害を促進し、それによってオートファジーを誘導して該細胞の細胞死を引き起こすことを含む、細胞増殖の抑制方法を提供する。このような細胞の例は、腫瘍細胞である。
【0045】
本発明はさらに、TG2の活性を抑制することによるIP3R1タンパク質のC末端側グルタミン残基の修飾の抑制を介してオートファジーを抑制することを含む、神経変性疾患の治療方法を提供する。
【0046】
それゆえに、本発明はさらに、IP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の増強剤を有効成分として含む、癌の治療用組成物を提供する。
【0047】
癌の例は、すべての固形癌、血液癌などであり、固形癌には脳腫瘍、肺癌、大腸癌、肝臓癌、膵臓癌、胃癌、膀胱癌、前立腺癌、食道癌、皮膚癌、リンパ腫、骨癌などが非限定的に含まれる。また、血液癌には、白血病などが含まれる。
【0048】
前記架橋の増強剤は、TG2酵素の賦活剤や誘導剤であり、例えばレチノイン酸などを挙げることができる。
【0049】
またIP3R1タンパク質のグルタミン残基修飾の増強剤は、TGの基質となるアミノ供与体であり、例えばモノダンシルカダベリン、スペルミジン、スペルミンを挙げることができる。
【0050】
本発明はさらに、IP3R1タンパク質のC末端側グルタミン残基の架橋の抑制剤(又は阻害剤)を有効成分として含む、神経変性疾患の治療用組成物を提供する。
【0051】
神経変性疾患の例は、以下のものに限定されないが、アルツハイマー病、ハンチントン病、パーキンソン病などを含む。このような疾患では、脳内で蛋白質が異常な架橋反応が起こることによって不溶性の蛋白質または蛋白質凝集が生じることによって神経変性疾患が発症することが知られている。
【0052】
上記架橋・修飾の抑制剤は、TG2酵素の阻害剤であり、例えばTG2に対するsiRNA、miRNA、リボザイム、アンチセンスRNAなどの核酸、シスタミン、モノダンシルカダベリンなどの小分子(特許3012923号)や、新規のTG2阻害剤として報告された5-bromo-2-thienyl-(N-t-butyl N-benzyl)-aminoethyl keton等のβ-アミノケトン化合物を含む(Bioorg.Med.Chem.Lett, 20(3): 1141-4 (2010))ことができるし、或いは、IP3R1タンパク質のC末端側グルタミン残基の架橋の抑制剤であり、それは、例えば小分子、ペプチド、タンパク質、核酸、脂質、天然物、合成化合物などから選択可能であり、ここでタンパク質には例えばTG2やIP3R1架橋部位に対する抗体類、核酸にはIP3R1架橋部位に対する例えばsiRNA, miRNA,リボザイム、アンチセンスRNAなどがそれぞれ含まれる。
【0053】
本発明はさらに、疾患関連細胞又は疾患モデル非ヒト動物において、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋(又は、該架橋のレベル)を指標にして該架橋によって引き起こされるオートファジーと該疾患との相関関係を測定し、さらに該疾患が該IP3R1のサブユニット間架橋に起因するときにTG2の阻害剤又は活性化剤を候補薬剤として該モデル非ヒト動物に投与して治療効果を調べることを含む、疾患の治療剤をスクリーニングする方法を提供する。
【0054】
疾患は、例えば神経変性疾患又は癌である。
癌関連細胞は、上記例示のものを挙げることができる。
【0055】
疾患モデル非ヒト動物は、アルツハイマー病、ハンチントン病、パーキンソン病等の疾患のモデル動物(たとえばマウス、ラットなど)であり、これらの動物に候補薬剤を投与し、症状の軽減を観察することによって治療効果を確認することができる。モデル動物として、たとえばハンチントン病モデルマウス(J Neurol Sci, 231: 57-66 (Apr 15 2005))、アルツハイマー病モデルマウス(J. Clin. Invest., 116(3): 825-832 (2006))、パーキンソン病モデルマウス(PLoS Biol. 3(8): e303 (2005 August))等が知られている。腫瘍担持モデル非ヒト動物は、各種の腫瘍細胞株を動物の皮下に移植することによって作製することができる。
【0056】
TG2の阻害剤、IP3R1タンパク質のC末端側グルタミン残基の架橋の抑制剤、又はTG2の活性化剤の候補薬剤は、上記のような小分子、ペプチド、タンパク質、核酸、脂質、天然物、合成化合物などから選択可能であり、既存の又は新規の物質から選択されてよい。
【0057】
本発明はさらに、TG2によってIP3R1タンパク質のC末端側グルタミン残基と一級アミン含有分子とを結合することを含む、IP3R1のグルタミン残基の修飾方法を提供する。
【0058】
一級アミン含有分子は、モノアミン、ポリアミン、アミノ酸、リン脂質、ペプチド、タンパク質、核酸などを非限定的に含み、天然又は非天然由来のいずれの分子でもよい。後述の実施例では、テトラマー構造のIP3R1タンパク質のC末端側グルタミン残基が、IP3R1の別のサブユニットの調節ドメインのリジン残基と結合することを示すが、この事実から、他の一級アミン含有分子であってもIP3R1のC末端側グルタミン残基との結合がTG2によって触媒されることが明らかである。実際、IP3R1のC末端側グルタミン残基は、TG2の作用によって、ビオペンチルアミン(BP)、シスタミン(CA)、モノダンシルカダベリン(Dan-Cad)などの一級アミン含有分子と結合し、アミド基転移が生じた。
【実施例】
【0059】
以下に実施例を挙げて本発明をさらに説明するが、本発明はこれらの実施例に限定されないものとする。
<実験方法>
材料
N,N-ジメチル化カゼイン, すべてトランスのレチノイン酸(RA)及びカルバコール(CCh)をSigmaから購入した。Zwittergent 3-14, イオノマイシン及びカルパインをCalbiochemから得た。増強された化学ルミネッセンス(ECL)試薬及び西洋ワサビペルオキシダーゼ(HRP)-結合ストレプトアビジンをGE healthcareから購入した。5-(ビオチンアミド)ペンチルアミン(BP), 1,8-ビス-マレイイミド-ジエチレングリコール(BM)及びビス[スルホスクシンイミジル]スベレート(BS)をテルモ(Thermo)から購入した。IP3, fura-2及びfura-2 AMはDojindo (日本)から入手した。αTG2抗体(トランスグルタミナーゼII Ab-1, CUB 7402)をLabVisionから購入し、αLC3をMBLから購入した。精製TG2, チログロブリン及びリジル-エンドペプチダーゼ(Lep)を和光純薬から購入した。DMEM, ペニシリン-ストレプトマイシン混合溶液, HPLC用のアセトニトリル及びTriton X-100を半井テスクから購入した。牛胎児血清をJRH Biosciencesから入手した。SH-SY5Y及びU87-MG細胞系をAmerican Type Culture Collection (ATCC)から購入した。
【0060】
電気泳動によるIP3R1内のサブユニット間架橋の評価
マイクロウエーブ装置内で、0.1%SDS含有バッファにアガロースを溶解し、アガロースを加熱し手で回転し静かに混合した。1%アガロース溶液をアクリル板(Nihon Eido)上に注いだ。26レーンの櫛をアクリル板上に配置した。室温で30分後、装置を泳動バッファで充たし、櫛を注意深く取り出し、100Vで1〜2時間電気泳動を行った。泳動後すぐに、アガロースゲルを転写バッファ中に30分間置いた。分離した蛋白質をPVDF膜(Millipore)に転写した。分子マーカーは、牛チログロブリン(Th)を用いて作製した。5mg/ml Thは、4℃で一晩1mM BMを用いて架橋することによって前処理した。
【0061】
細胞培養
HeLa, SH-SY5Y, U87-MG及びCOS-7細胞を、10% (v/v)熱失活牛胎児血清、50U/mlペニシリン及び50μg/mlストレプトマイシンを補充したDulbecco改変Eagle培地(DMEM)中で培養した。すべての細胞を5% (v/v) CO2下で37℃に維持した。HeLa細胞及びSH-SY5Y細胞について20μMレチノイン酸(RA)を補充した分化培地を48時間おきに置換した。
【0062】
IP3R1のチャネル活性の測定
マウス小脳のP2/P3膜(3mg/ml蛋白質)を50mM HEPES-KOH、110mM KCl、10mM NaCl及び5mM KH2PO4を含有するバッファ(pH7.4)で3回洗浄してアミド転移反応用の酵素又は基質を除去した。小脳又はSH-SY5Y 細胞の10μlのP2/P3膜(2mg/ml蛋白質)を、石英キュベット中の、1mM 2-メルカプトエタノール、1μM CaCl2及び0.2μM Fura-2を含有する287μlのバッファに加えた。サンプルを、340及び380 nm励起光で発光し、510 nmで蛍光を測定した。P2/P3膜小胞中にCa2+を負荷するために、攪拌しながら3μlの100mM Mg-ATPをキュベットに添加した。15分後、IP3を添加した。fura-2のレシオ法測定を、30℃でCAF-110 (ジャスコ)又はF-2500分光光度計(日立)を用いて文献記載にしたがって行った(Zhou et al., 2007)。
【0063】
酵素によるアミド基転移
小脳膜を、50mM HEPES-NaOH、150mM NaCl、1mM EGTA及び2mM TCEPを含有するアミド基転移バッファ(pH7.5)で希釈して反応混合物を調製した。TG2によるアミド基転移を、小脳膜1mg/ml蛋白質、2mM CaCl2及び種々の濃度のTG2を含有する反応混合物を37℃で30分間インキュベーションすることによって行った。アミド基転移されたIP3Rは、説明がない場合にはすべて4C11を用いて検出された。
【0064】
蛋白質限定分解
カルパイン(Calbiochem)又はリジルエンドペプチダーゼ(Lep,和光純薬)による消化の前に、小脳膜又はTG2処理膜を、架橋バッファを用いて20,000×gでの遠心及び再懸濁によって3回洗浄した。種々の濃度のカルパイン又はLepと洗浄された膜とを37℃でインキュベーションすることによって消化を開始した。その混合物に1/15容量の100mM CaCl2を添加したのち、カルパインによる消化を開始した。
【0065】
TG2による組換えIP3R1のアミド基転移
3.5cmディッシュで培養されたCOS-7細胞を掻き取り器具で回収し、50mM HEPES-NaOH(pH7.5)、10mM KCl、2mM TCEP、1mM EDTA及びプロテアーゼ阻害剤(複数)を含むバッファ中でホモゲナイズした。800×gで回転して得た上清を、20,000×g、4℃で15分間遠心分離した。これによって得られた沈降物を前記バッファに懸濁し、Bradford法を用いてタンパク質濃度を決定した。それぞれのタンパク質濃度を調整し、5mM CaCl2及び1μg/ml TG2と一緒に30分間インキュベーションすることによって架橋を行った。
【0066】
ペプチド及びイソペプチドの分析
フォトダイオードアレイUV検出器を備えたHP1100システム(Agilent Technologies)を用いてHPLCを行った。得られた反応混合物を、10μl又は200μlループを介してC8カラム(Super-octyl TSK(登録商標)ゲル, 100mm×2.0mm(内径), 2μmレジン, 東ソー)に注入した。0.1% TFAを含有する10%から50%アセトニトリルの直線グラジエントを用いて200μl/分の流速で15分でペプチドを溶出した。2種類の発蛍光団を含むγ-分岐ペプチド(すなわち、FITC-Asn-Pro-Glu[γ-(TMR-cad)]-Gln-Pro-Ala(配列番号5))を酵素的に作製したが、このとき0.05mM TMR-cad、2.5mM CaCl2、25mM HEPES-NaOH(pH7.5)及び50μg/ml TG2を含有する溶液中で0.05mM FITC-Asn-Pro-Glu-Gln-Pro-Ala(配列番号6)をインキュベーションすることによって前記ペプチドを得た。TG2によるイソペプチドの加水分解を、F-2500装置中、10mM HEPES及び1mM CaCl2を含有する反応混合物を用いて30℃でFITC蛍光の変化に基づきモニターした。
【0067】
TG2によって脱アミド化されたマウスIP3R1の作製
小脳からのP2/P3画分(1mg/ml)を、TG2 (0, 1, 3, 10, 30, 100ng/ml)と一緒に、50mM HEPES-NaOH、150mM NaCl、1mM EGTA、3mM CaCl2及び2mM TCEPを含有するHEPESバッファ中、37℃、90分間インキュベーションした。
【0068】
HeLa細胞でのsiRNAによるノックダウン
siRNA(複数)とヒト陰性対照をB-bridgeから購入した。TG2 siRNAのヌクレオチド配列は、ggucaaugccgacguggua(配列番号7)であった。IP3R1ノックダウンを文献記載(Hattori et al., 2004)の方法で行った。リポフェクタミン2000(Invitrogen)を用いてsiRNAをHeLa細胞にトランスフェクションした。
【0069】
マウス小脳からのP2/P3画分の調製
小脳をddYマウス脳から取り出し、冷却したPBSで十分に洗浄し、10mM HEPES-NaOH, 0.32 Mスクロース, 1mM EDTA, 2mM TCEP及びプロテアーゼ阻害剤(0.2 mM PMSF, 10μMロイペプチン, 10μMペプスタチンA及び10μM E-64)から構成されるホモゲナイズバッファ(pH7.5)10容量中でホモゲナイズした。ホモゲネートを800×g、4℃で5分間遠心分離した。得られた上清を105,000×g、2〜4℃で60分間超遠心分離にかけた。沈降物を50mM HEPES-NaOH, 150mM NaCl, 1mM EDTA, 2mM TCEP及びプロテアーゼ阻害剤を含む再懸濁用バッファResB(pH7.5)に懸濁した。超遠心分離の上清をシトソル画分として使用した。この溶液のタンパク質濃度を見積もり、ResBにより3mg/mlに調整し、種々の実験に使用するまで液体窒素中ですばやく凍結した。
【0070】
IP3R1の化学的架橋
サブユニット間の化学的架橋を、25mM HEPES-NaOH, pH7.5, 75mM NaCl, 1mM TCEP, 0.5mM EDTA及び種々の濃度のBS又はBMを含む反応バッファ中でP2/P3画分(1.5mg/ml)をインキュベーションすることによって開始した。SDSサンプルバッファを添加して氷上30分間反応を停止し、得られたサンプルをAGE分析にかけた。P2/P3画分の膜を1 % (w/v)界面活性剤に可溶化し、20,000×g、4℃で15分間遠心分離した。500μM BS又は100μM BMを可溶化画分に添加し氷上で30分間インキュベーションした。
【0071】
アミド基転移活性の分画
ホモゲネートの超遠心分離の上清画分を、50mM NaCl含有10mM Trisバッファ(pH 7.5)で予め平衡化したアニオン交換カラム(TSKgel BioAssist Q, 50mm×4.6mm(内径), 東ソ)上にアプライした。架橋活性に富む画分を、50mMから750mM NaClの直線グラジエント、流速1ml/minを用いて溶出した。20〜30画分(1mlずつ)を回収し、架橋活性を測定した。
【0072】
TG2発現レベルの測定
SH-SY5Y細胞又はHeLa細胞を3.5cm又は6cmディッシュから掻き取り器具で回収し、SDSサンプルバッファ中で3分間煮沸し、SDS-PAGEにかけた。
【0073】
SH-SY5Y細胞由来のIP3R1中の架橋の評価
3.5cmディッシュから掻き取り器具で回収したSH-SY5Y細胞を55℃で15分間加熱し、5秒間音波処理を2回行った。IP3R1を4C11で検出した。
【0074】
TG2基質のアフィニティ精製
P2/P3画分(1 mg/mlタンパク質), 50mM HEPES-NaOH (pH 7.5), 150mM NaCl, 10mM 5-(ビオチンアミド)ペンチルアミン(BP), 1mM EGTA, 2mM CaCl2, 及び30ng/ml TGを含む反応溶液を37℃で30分間インキュベーションした。混合物を15,000rpm、4℃で30分間遠心分離し、ついで、50mM HEPES-NaOH (pH7.5)でペレットを洗浄した。1% CHAPSでタンパク質を可溶化し、アビジン-アガロースと混合した。1% CHAPS含有HEPESバッファで3回洗浄したのち、アビジン-アガロースに結合したビオチン取り込みタンパク質をSDS-PAGEにかけてCBB染色又はウエスタンブロッティングにかけた。CBB染色バンドを検出し、染色バンドを切り出し、ゲル内消化プロトコルによって消化し、MALDI TOF MS (ABI)を使用する質量分析によって分析した。
【0075】
プラスミド
C末端の130kDa断片をコードするcDNAを、哺乳動物発現ベクターpcDNA3.1zeo(+) (Invitrogen)中にサブクローニングした。PCR産物をNheI及びBamHIで消化し、pcDNA3.1zeo(+)中にサブクローニングした。突然変異体を構築するために、部位特異的突然変異誘発を、QuikChange II XL (Stratagene, La Jolla, CA)とプライマー(AATGTCAACCCAAACCAGCCGGCCTAG(配列番号8): Q2746N、ここで下線はコドン変更である。)を用いてC130を含むプラスミド上で行った。全ての変異体をDNA配列決定によって確認した。すべてのオリゴヌクレオチドの配列は要求に応じて入手可能である。
【0076】
NEM又は煮沸によるTG2の不活性化
文献記載に基づいてTG2を不活性化した(Folk, J. E. & Cole, P. W., J Biol Chem 241, 5518-25 (1966))。TG2を、20 mM HEPES-NaOH (pH7.5)中の5mM NEMと一緒に室温で30分間インキュベーションした。反応を、5 mM DTTと一緒に室温、30分間インキュベーションすることによって停止した。得られた混合物を、連続して2本のスピンカラム(BioRad)を通過させて遊離NEMを除去した。もう一つの不活性化TG2は20mM HEPES-NaOH (pH7.5)中で5分間煮沸することによって調製された。
【0077】
TG2活性の評価
TG2活性を、F-2500蛍光光度計(日立)中で測定した。このとき、N, N-ジメチルカゼインへのモノダンシル-カダベリンのTG2触媒取り込みの際の蛍光の増加速度をモニターした(Lorand, L. et al, Anal Biochem 44, 221-31 (1971))。TMR-cadのTG2中への自己取り込みは、37℃、30分間 1mM TMR-cad, 2mM CaCl2及び10μg/ml TG2をインキュベーションすることによって行った。蛍光強度をFluorImager595 (Amersham)を用いて検出した。
【0078】
2種類の発蛍光団を含む蛍光ペプチドの酵素的作製
N末端のαアミノ基にビオチン又はFITCが結合された合成ペプチドを林化成(大阪)から購入した。TG2によるペプチドアミド基転移のために、25mM HEPES-NaOH, pH7.5, 2.5mM CaCl2, 0.05mM TR-cad, 0.05mMペプチド、及び50μg/ml TG2若しくは50μg/ml 不活性化TG2のいずれかからなる反応混合物を37℃で10分間インキュベーションした。脱アミド化は、15/1容量のアミド基転移ペプチドを、1mM EGTA、20μg/ml TG2及び5mM CaCl2若しくはCaCl2不含のいずれか、を含む50mM HEPES-NaOH, pH7.5を含有する反応混合物中に加えて開始した。アミド基転移ペプチドの構造はナノESI-MS/MS分析(Aproscience Corp., 徳島)によって確認された。
【0079】
タンパク質アッセイ
P2/P3画分、シトソル画分又は精製酵素のタンパク質濃度をU-0080Dフォトダイオードアレイ分光光度計(日立)を用いて測定した。この装置はBradford法に従うものであり、また標準としてウシ血清アルブミンを用いた。
【0080】
脱アミド化IP3R1に対する特異抗体
マウスIP3R1中のQ2746の状態を解き明かすために、アミノ酸2738〜2749に対応する脱アミド化ペプチド(PPHMNVNPEQPA(配列番号9))を合成し、ペプチドのC末端に付加されたシステインを介してKLHに結合し、日本白ウサギに免疫した。生じたポリクローナル抗体を先ず、天然ペプチド(PPHMNVNPQQPA(配列番号10))を固定したアフィニティ樹脂にアプライし、素通り画分を、脱アミド化ペプチドを固定した樹脂にアプライした。脱アミド化ペプチドに対する特異抗体は100mMグリシン-HClバッファ(pH2.5)で溶出された。抗体の特異性をチェックするために、マウスIP3R1の組換えC末端タンパク質(これはGST融合蛋白質として発現された。)を調製した。PCR産物はGST融合ベクターpGEX-6Pに挿入された。
【0081】
IP3R1におけるアミド基転移の確認
SH-SY5Y細胞をトリプシンで処理し、均質に懸濁し、トリパンブルー染色を用いて計数したのち、10cmディッシュに0.3×106の密度でSH-SY5Y細胞を植え付け、20μM RA又はDMSOを含有する培養培地中でさらに6日間培養し、この間、2日おきに新鮮な培地が供給された。2 mM BPを培地に添加し、24時間後に細胞を掻き取り器具で回収した。コールドルーム内で1500rpm、5分間細胞を遠心分離し、さらに1500rpmで5分間回転することによってPBSで洗浄した。得られた細胞沈降物を液体窒素で急速に凍結し-80℃で保存した。開放チューブ内の凍結細胞に1%Triton X-100 (HE-T)含有HEPESバッファ0.5mlを加え、懸濁し、4℃で30分間回転した。MX-300遠心分離器(トミー)中15000rpm、4℃で15分間遠心分離した後の上清を、pep6-Ab (Nakade, S. et al, J Biol Chem 269, 6735-42 (1994))を結合したProtein A-Sepharose 4Bと混合し、4℃で2時間インキュベーションした。Sepharoseビーズを0.5 mlのHE-Tバッファで3回洗浄し、SDS-PAGEにかけた。免疫沈降したIP3R1を5%ゲルで分離し、PVDF膜に転写し、アビジン-HRP (1:2000希釈, GE healthcare)と一緒にインキュベーションした。ビオチン-アビジン複合体の量を、増強された化学ルミネッセンス(ECL)キット(GE healthcare)で検出した。
【0082】
SH-SY5Y細胞からのP2/P3画分の調製
10cmディッシュ中で培養した細胞を細胞掻き取り器具で回収し、遠心分離し、PBSで洗浄した。細胞を、200μlのホモゲナイズバッファ(pH7.5, 50mM HEPES-NaOH, 150mM NaCl, 1mM EDTA, 2mM TCEP, 及びプロテアーゼ阻害剤(0.2mM PMSF, 10μMロイペプチン, 10μMペプスタチンA, 及び10μM E-64)中に懸濁し、ハンディタイプの音波器UR-20P(トミー)を用いた音波処理によってホモゲナイズした。細胞溶解物を800×g、4℃で5分間遠心分離し、上清をTL100.4ローター(Beckman)中、100,000×g、4℃で60分間超遠心した。沈降物は、シトソル様培地又はHEPES-NaOHバッファ中に再懸濁された。
【0083】
SH-SY5Y細胞中の[Ca2+]iの測定
PBS中の3μM fura-2/AM (同仁)中で細胞を37℃1時間インキュベーションすることによって該細胞にfura-2を負荷した。細胞を回収し、トリパンブルー染色によって計数した。1500rpm、5分間の遠心分離のあと、HEPES緩衝生理食塩水(HBS)中に懸濁することにより細胞密度を調整し、濯ぎ、HBS中に細胞を再懸濁した。fura-2のレシオ法測定を、F-2500蛍光光度計(日立)を用いて30℃で行った。10μMカルバコール(carbachol; CCh)を添加することによって細胞を刺激した。
【0084】
<結果>
内因性酵素である組織トランスグルタミナーゼ(TG2)によるIP3R1のサブユニット間架橋
IP3Rはテトラマーとして機能するが、その巨大な質量(>1MDa)は、テトラマー構造を解析することを困難にさせている。したがって本発明者らは、IP3Rのテトラマー状態を明らかにするために新規のアガロースゲル電気泳動(AGE)法を開発した(図1)。化学架橋剤である1,8-ビス-マレイイミド-ジエチレングリコール(BM)及びビス[スルホスクシンイミジル]スベレート(BS)は、バンドのシフトにより判断されるように、IP3R1のサブユニットを架橋することができた(図1A)。Zwittergent 3-14 及びSDS(これらはともにIP3Rのサブユニット相互作用を破壊することが知られている。)は、バンドのシフトを阻害し(図1B)、開発したAGEがIP3Rサブユニット間の共有結合による架橋を評価するのに適することを確認した。
【0085】
次に、本発明者らは、AGEを生物学的サンプルに適用した。驚いたことに、本発明者らは、外因性架橋剤の非存在下でIP3R1がCa2+依存性の架橋を起こすことを見出した(図1C)。この知見は、脳シトソル内に内因性架橋剤が存在するはずであることを示した。本発明者らはアニオン交換クロマトグラフィーによる架橋活性を特徴づけることを試みた(図1D)。架橋活性は、クロマトグラム上約0.3M NaClと同等の画分中に富化されていた(図1E)。Ca2+依存性と溶出プロフィールにより、架橋がTGによって触媒される可能性が仮説された。そのあと、トランスグルタミナーゼ(TG)の阻害剤であるシスタミン(CA)を試験し、用量依存的な仕方でサブユニット間の架橋を明確に阻害することを観察した。精製した組織TG(TG2)は架橋を示した(図1F)。精製したIP3R1は精製TG2によって架橋されることが可能であり(図1G)、このことはIP3R1のサブユニット間の直接的な架橋を示している。この事実を確認するために、本発明者らは、siRNAによるTG2のノックダウンを使用した。TG2のノックダウンは、HeLa細胞ライセート中で内因性架橋活性を完全に抑制した(図1H)。HeLa細胞中のTG2はRAによって誘導されることが知られているため、RA処理した細胞中の架橋活性を調べた。RAによるTG2誘導により、明らかに架橋活性が増強された(図1I)。次に、本発明者らは、IP3R1が生細胞中でTG2によって架橋されうるかどうかを調べた。このとき本発明者らは神経芽腫SH-SY5Y細胞及びU87-MG細胞を使用した(図1J)。イオノマイシンで刺激されたSH-SY5Y細胞及びU87-MG細胞でIP3R1の架橋が観察された(図1K)。SH-SY5Y細胞中の架橋は、RA処理によるTG2レベルの誘導が原因していた。これらの結果は、TG2が、サブユニット間の架橋を触媒する内因性酵素であることを示している。
【0086】
TG2はIP3R1のチャネル活性を阻害する
IP3R1のチャネル活性に及ぼすTG2触媒による架橋の影響を評価するために、本発明者らは最初に、煮沸或いはN-エチルマレイイミド(NEM)処理による不活性TG2を作製した(図2A及び2B)。TG2がCa2+放出活性に影響するかどうかを決定するために、本発明者らは、マウス小脳からのP2/P3画分を用いてIP3により誘導されるCa2+放出活性を調べた。TG2による前処理は、ATP依存性Ca2+取り込みに影響することなくチャネル活性又は、Triton X-100によって見積もったCa2+の総プールを強く抑制し、この阻害は用量依存的であった(図2C)。煮沸TG2もNEM処理TG2もともに、チャネル活性を抑制し(図2C及び2D)、このことは、TG2による触媒活性がチャネル阻害に必須であることを示している。TG2依存性阻害はまったく再現可能であり、まとめたデータは、TG2のIC50値が34nMであることを示している(図2D)。
【0087】
次の疑問は、TG2によるIP3R1のアミド基転移がどのようにしてチャネル活性を阻害するかということであった。本発明者らは、TG2がIP3R1のグルタミン(Q)ドナー部位に取り込むことができるアミンドナーとして5-(ビオチンアミド)ファニルアミン(BP)を使用した。予想したとおり、BPはサブユニット間の架橋を阻害した(図2E)。精製IP3R1は、アビジン-HRPにより検出される強力なビオチンシグナルを示し(図2F)、このことは、BPがTG2によってIP3R1の中に直接取り込まれたことを示している。Ca2+放出アッセイによって、BP-取り込みがチャネル活性を完全に改善したことが示された(図2G)。
【0088】
TG2依存性阻害をさらに特徴づけるため、本発明者らは、IP3R1に取り込まれたビオチン部分に化学量論的に結合するアビジンのチャネル活性に対する影響を調べた。BPが取り込まれた膜にアビジンを添加することによってチャネル活性の用量依存的な阻害を見出した(図2H)。この知見は、Q-ドナー部位周辺の立体障害がTG2による阻害機序に重要であるという事実を強く支持している。生細胞でのTG2によるIP3Rのビオチン取り込みを確認するため、本発明者らはSH-SY5Y細胞を使用した。そして、本発明者らは、RAで分化されたSH-SY5Y生細胞中でのIP3R1の共有結合修飾を見出した(図2I)。RA処理は、IP3R1の発現レベルを検出可能に変化させなかったが、この事実はこれまでの報告(Mackrill et al., 1997)と一致した。本発明者らはまた、対照と比較して、RAで処理されたSH-SY5Y細胞から調製したIP3R1のチャネル活性の著しい減少を見出した(図2J及び2K)。さらに本発明者らは、CChによって誘導されるCa2+放出がRA処理SH-SY5Y細胞中で損なわれることを見出した(図2L)。合わせると、これらのデータはIP3R1がTG2の基質であること、並びにTG2がIP3R1を介してCa2+放出を阻害すること、と矛盾しない。数個の内因性アミンがTG2のアミンドナーであると報告されている(Dale et al., 2002; Iismaa et al., 2009; Walther et al., 2003)ので、TG2によるIP3R1の修飾の内因性アミンドナーを調べることには関心がある。
【0089】
C末端130kDaドメイン(C130)内のTG2によるサブユニット間架橋
Q-ドナーの正確な位置を決定するために、BPが取り込まれた精製IP3R1の限定消化を行った。アビジン-HRPを用いて検出された主バンドの見かけのサイズは130kDa及び85kDaであった(図3A)。4C11及び10A6の両方で検出された180kDa (N180)のサイズのN末端ドメイン(図3A)は、C末端の130kDaバンド(C130)又はC末端の85kDaバンド(C85)と比べてBPによってほとんど標識されていなかった(図3A)。これらのバンドはウエスタンブロッティングによってC末端断片として帰属された。さらに消化物は、矛盾なく、ビオチン標識バンドがC末端に相当し、4C11又は10A6によって検出された断片に相当しないことを示した(図4B)。次に本発明者らは、Q-ドナーがテトラマーIP3R1のサブユニット間架橋に関与しているかどうかを調べた。また、C末端結合抗体がTG2による架橋の形成を妨げるかどうかを調べた。IP3R1は、AGEによって判断されるように、TG2によって架橋された(図3C)が、18A10による前処理はTG2による架橋を阻害した(図3C)。この知見は、架橋がC末端の18A10エピトープ付近に位置していることを示している。さらに、本発明者らは、架橋されたIP3R1が、たとえそれが変性されたとしても18A10に対するその免疫反応性を喪失したことを見出した(図3D)。このことは、Q-ドナー部位が18A10認識にとって重要な残基であることを示した。TG2で前処理されたIP3R1の限定消化により、TG2によるC130及びC85の、しかしN180ではない、著しい減少が明示された(図4E)。変性後のリジルエンドペプチダーゼ(Lep)によるさらなる消化により、4C11でプローブされた断片に影響することなくC末端断片のTG2相関的減少が示された(図4F)。また、限定消化後の架橋の結果、C130の回収の顕著な減少が生じたが、一方、N180の量は、TG2によって変化しなかった(図4G)。これらの知見は、サブユニット間架橋がC130ドメイン内で起こるはずであるということを示している。
【0090】
Q2746はTG2の強力なQ-ドナーである
Q-ドナー部位を正確に決定するために、本発明者らは質量分析を行った。はじめに、本発明者らは、BPのin vitro取り込みによってTG2の内因性基質をプルダウンした。驚いたことに、300kDa付近にほぼ均質のバンドを見出した(図4A)。次に、本発明者らは、この300kDaタンパク質を同定するためにゲル内消化と質量分析を行った。断然最もよいスコアを示したデータがマウスIP3R1であったし、またその配列中のカバー率が2749個のアミノ酸の45%以上であり(図8)、このことから、IP3R1がP2P3膜中の小脳タンパク質のなかで好ましい基質であることが示された。BPの質量を計算し、C末端ペプチド(アミノ酸配列: IGLLGHLPPHMNVNPQQPA(配列番号9)が専らビオチン化されたことを見出した(図8)。この結果は、部分消化データ(図3)と矛盾しなかった。ウエスタンブロッティング分析により、IP3R1がビオチン化されたことを確認した(図4B)。本発明者らは、LC-MS及びそれに続くタンデム型質量分析(MS/MS)を用いて、ビオチン化ペプチドによる分画の際にTG2によって酵素的に修飾されるIP3R1の正確な残基を決定した。この分析によって、IP3R1中のグルタミン2746(Q2746)の1つのアミド基転移部位 (IGLLGHLPPHMNVNPQ*QPA(配列番号11); 図4C)が同定された。野生型ペプチドのMALDI MS/MSスペクトルと比較して、BP(311.1 Da)に対応した明瞭な質量シフトが修飾ペプチド中に検出された(図4C)。最近、TG2が、配列モチーフpQxP(又はT, S)l(p: 極性アミノ酸, x: 任意のアミノ酸, l: 脂肪族アミノ酸)(配列番号12)によって決定される基質特異性を有していることが報告された(Csosz et al., 2008)。NPQQPA配列(配列番号3)はこのモチーフと一致し、Q2746(しかしQ2747でない)がTG2の標的であることを確認した。
【0091】
次に本発明者らは、COS-7細胞中で過剰発現された組換えIP3R1タンパク質を用いてQ2746が架橋に必須であるかどうかを調べた。そのために本発明者らは、IP3R1(計算質量: 132.4kDa)のC末端ドメインを構築し、欠失変異体及びQ2746N変異体を作製した(図4D)。その結果、TG触媒反応のためのQ-ドナーがQ2746であることを確認した(図5D及び5E)。またアミノ酸1866〜1932に相当する領域もサブユニット架橋に必須であった(図5D)。この領域は2つのユニークなLys (K)-クラスター部位(NKKKD(配列番号13)及びRKKAKE(配列番号14))を有している。より短い断片(ES領域: アミノ酸2216〜2749)はテトラマーを形成するのに十分である(Sayers et al., 1997)ので、本発明者らは、このKクラスター領域(アミノ酸1866〜1932)がTG2によるサブユニット間架橋の間にK-ドナーとして作用するはずであると結論する。最近の論文では、このK-クラスター領域に相当する部位がユビキチン化可能であることが報告された(Sliter et al., 2008)。したがって、本発明者らのデータは、TG2による修飾もまたプロテアソーム分解過程と関連していることを示してしいる。
【0092】
TG2による架橋の切断と脱アミド化
本発明者らは、TG2がNPQQPA(配列番号3)と、アミンドナーであるTMR-cad (テトラメチルローダミン・カダベリン(tetramethylrhodamine cadaverine))との間の架橋を可逆的に切断するかどうかを調べた。NPQQPAペプチドはTMR-cadと反応して架橋生成物を生成した。この生成物は14分の保持時間のところで溶出したが、NPNQPA(配列番号4)ペプチドは溶出しなかった(図5A)。本発明者らは該生成物を精製し、その構造をナノMS/MS質量分析によって確認し、TG2による脱架橋反応にかけた(図5B)。前記生成物をTG2及びCaCl2とインキュベーションすることによってローダミンスペクトルをもつ生成物が生じたが、その保持時間はTMR-cadと一致した(図5B)。この反応はCa2+依存的であった。これらのデータから、TG2がCa2+依存的にNPQQPAペプチドとアミンドナーとの間のイソペプチド結合を切断可能であることが示された。
【0093】
リアルタイムでの切断をモニターするために、本発明者らは、蛍光共鳴エネルギー移動に基づく新しい蛍光プローブを作製した。すなわち、Q-ドナー及びアミンドナーとしてそれぞれFITC-結合NPQQPA (ピークY)及びTMR-cad (ピークX)を作製し使用した。TG2による酵素反応により、FITCスペクトルとローダミンスペクトル(ピークZ)の両方をもつ色原体が生じ、本発明者らは、得られた生成物を精製した(図5C)。この二蛍光含有ペプチドを用いて、リアルタイムで脱架橋反応を確認した。予想したとおり、TG2とCa2+の添加後にFITC蛍光の増強を観察し、そしてTG2による加水分解の間の良好な直線性を得た(図5D)。
【0094】
理論的見解は、イソペプチドの加水分解又はTG2による脱アミド化がGln位置でグルタミン酸(Glu: E)残基を生成するはずであることを示している(Iismaa et al., 2009)。TG2によってQ2746がGlu残基(E2746)に変換可能であるかどうかを試験するために、本発明者らはE2746含有ペプチドに対する特異抗体を作製した。アフィニティ精製によって、Q2746を認識しないE2746特異抗体(αE2746)を得ることを可能にした(図5E)。αE2746を用いて、本発明者らは、テトラマーIP3R1がTG2によって酵素的に脱アミド化されることを示す結果を得た(図5F; *)。小脳からのP2/P3画分(図5G)はαE2746によって検出されるシグナルを示すが、これはin vivoでIP3R1を修飾することができることを示している。本発明者らは、この抗体をHuntington舞踏病モデル(R6/2)(ポリグルタミン含有タンパク質が過剰発現される。)に使用した。本発明者らは、R6/2マウスで、野生型マウスと比べてαE2746シグナルが専ら増強されることを見出した(図5G)。脱アミド化によって生じたE2746の陰性電荷が、静電相互作用によってC末端と陽性電荷K-クラスター領域との間で誘引を引き起こす可能性がある。
【0095】
TG2及びIP3R1によるオートファジー調節
IP3R1はオートファジーに関与しているので(Vicencio et al., 2009)、本発明者らは次に、IP3R1によって調節されるオートファジー事象の調節におけるTG2の役割を調べた。本発明者らは、TG2 siRNAを用いてHeLa細胞での、TG2の除去と、それによる修飾IP3R1の欠損を確立した(図6A)。オートファジーマーカーである、相対量の脂質化LC3 (LC3-II) (Klionsky et al., 2008)は、LC3-I(LC3-IIの前駆体)と比較して、siRNAを使用するTG2の除去によって減少した(図6B)。これに対して、siRNAによるIP3R1の除去により、LC3−Iと比べてLC3-IIの著しい増加が生じた(図6B)。LC3-IIレベルに対するこれら逆作用は、今回の研究で証明されるように、TG2によるIP3R1の阻害と矛盾しない。
【0096】
TG2及びIP3R1の役割を証明するために、本発明者らはラパマイシン又はスペルミジンによって誘導されるオートファジーを調べた。4時間の1μMラパマイシン又は100μMスペルミジン処理により、対照siRNAでトランスフェクションされたHeLa細胞でLC3-IIの脂質化を誘導したが、TG2 siRNAはそれをブロックした(図6C)。IP3R1 siRNAは反対の作用を示したが、これは本発明者らの仮説の確認になった(図6D)。いずれの場合にも、p62 (オートファジー基質)はTG2又はIP3R1の除去の間に全く変化を示さなかった。これは、LC3-II by TG2又はIP3R1のノックダウンによるLC3-IIの定常状態レベルの変化が脂質化によるLC3-IのLC3-IIへの変換によるものであって分解過程の損害によるものではないことを示している。L-Glnは、LC3の脂質化を誘導しうる(図6E及び6F)。重要なことは、L-Glnによる脂質化がsiRNAを使用するTG2の除去によってブロックされたことであり(図6E)、このことは、TG2がL-Gln依存性のオートファジーシグナル伝達に関与していることを示している。本発明者らは、TG2の基質であるアミンドナーとしてのモノダンシル-カダベリン(Dan-cad)とIP3R1の阻害剤である2-APBの役割を調べ、LC3の脂質化を増強したことを見出した(図6F)。このとき、オートファジーの基質であるp62タンパク質の沈着はなかった。この知見は、TG2及びIP3R1が、LC3-IIの分解をブロックすることではなくLC3の脂質化を調節することを支持している。
【0097】
TG2及びIP3R1によるオートファジー調節を証明するために、本発明者らは、ノックダウン中のHeLa細胞の形態を調べた。対照siRNAを有するHeLa細胞は空胞化を示したが、TG2 siRNAでトランスフェクションされた細胞は空胞の損失を示した(図7A)。IP3R1 siRNAは、多数の空胞を生じさせた(図7A)。空胞化された細胞又は空胞化されなかった細胞の数の計数から(図7B)、図6に示されるように、空胞化に対するノックダウンの各影響は、オートファジーの間、LC3転換の影響と完全に一致する。
【0098】
本発明者らは、TG2及びIP3R1が細胞の保護又は死と相関するかどうかを調べた。IP3R1のノックダウンの場合、非接着細胞が顕微鏡下でかなり頻繁に観察された(データ示さず)。したがって、本発明者らは、LDHアッセイにより細胞死を評価した。LDH活性により、IP3R1のノックダウンとは逆に、TG2のノックダウンが細胞死を抑制することを示した(図7C)。このことから、明らかな空胞化とLC3の脂質化が、細胞の保護ではなく細胞死と相関することが示された。
【0099】
<考察>
本発明者らの知識によれば、これは、IP3R1がTG2の新規な基質であること、及びTG2が、テトラマーIP3R1の酵素的修飾によってIP3R1のチャネル活性を阻害することを示す最初の研究である。本発明者らは、C末端が近傍のIP3R1サブユニットに近い、及び、TG2が近接するサブユニットに該C末端を共有結合によって結合させ、チャネルを、閉じた立体配置に固定する、と結論付ける(図7)。
【0100】
これまでの報告では、C末端尾部がチャネル活性に重要であること(Nakade et al., 1991; Uchida et al., 2003)、並びに、C末端及びKクラスター領域の付近で相互作用する、チトクロムcやBcl-2などの、多くの調節因子がチャネル活性を調節すること(Boehning et al., 2003; Foskett et al., 2007; Rong et al., 2008)が実証されている。本発明者らは今回、これらの調節因子がC末端付近のサブユニット界面に結合して、他のオリゴマーチャネルに類似したサブユニット立体配置を制御することが可能であることを示す。ここで、他のオリゴマーチャネルには、例えばアセチルコリンレセプター、K-チャネル及びグルタミン酸チャネルが含まれ、それらについてサブユニットの立体配置はゲートの動作や調節に関与している(Changeux and Edelstein, 1998)。IP3R1のESドメイン(アミノ酸2216〜2749)は、テトラマー形成に十分である(Sayers et al., 1997)が、しかし、テトラマーESチャネルやテトラマーcasp-チャネル(アミノ酸1892〜2749)は常にそのチャネルを開いており、このことは、調節ドメインがチャネルドメインを閉じたままにしておく必要があることを示している(Nakayama et al., 2004)。このように、ESチャネル及びcaspチャネルは開いた状態で固有の立体配置を維持し、並びに、調節ドメインがサブユニットを閉じた立体配置に強制的にもたらすことができることが想起される。IP3RはCa2+を必要とし、Ca2+は調節ドメインを含む細胞質ドメインを動かすことによってコンフォメーション変化を誘導し(Anyatonwu and Joseph, 2009; Hamada et al., 2002; Hamada et al., 2003)、チャネルと、K-クラスターの近傍に位置するCa2+-センサー用の重要な残基を開く(Foskett et al., 2007)。したがって、Ca2+依存性のコンフォメーション変化が、IP3結合からの刺激によってチャネルを開かせるサブユニット立体配置を自由にすることが提案される。おそらく、このことはCa2+が共アドニストとしてチャネルのゲーティングに必須であることを示している。
【0101】
レチノイン酸(RA)は、SH-SY5Y及びHeLa細胞中でTG2によるIP3R1の修飾を誘導するが、このことは、RAが新規のRAシグナル伝達経路に関与していることを示している。脳では、RAはLTP、LTD (Chiang et al., 1998)及びシナプス可塑性(Aoto et al., 2008)をレチノイドレセプターを介して制御している。本発明者らの知見により、IP3R1が、RAシグナル伝達によって神経機能を調節することができる可能性が示唆された。その理由は、IP3R1によって調節されるIP3-Ca2+シグナル伝達がLTP及びLTDを含む種々の神経機能に関与しているからである(Harnett et al., 2009; Nishiyama et al., 2000)。TG2は、SH-SY5Y細胞(Tucholski et al., 2001)やHL60細胞(Antonyak et al., 2001)で、RA依存的分化を調節することが示されている。興味深いことに、 SH-SY5Y細胞内でのTG2の安定な発現がRAの不在下で神経表現型に自然発生的分化を誘導する(Tucholski et al., 2001)。本発明者らは、TG2がIP3R1修飾によってレチノイドシグナル伝達を調節することができる新しい経路を提案する。RAシグナル伝達は神経の分化や変性に重要である(Maden, 2007)。すなわち、今回の知見は、RAシグナル伝達の分子機構に新しい見識を提供することができる。
【0102】
本発明者らの研究により、TG2とIP3R1がともに、オートファジーの調節に関与しており、HeLa細胞でTG2の役割がIP3R1の役割と反対であることを実証しており、これはTG2がIP3R1機能を損なうという知見を支持している。このことは、オートファジーがCa2+シグナル伝達(Hoyer-Hansen et al., 2007; Williams et al., 2008)及びIP3R(Criollo et al., 2007; Vicencio et al., 2009)によって調節されるという従来の概念と矛盾しない。さらに、本発明者らの今回の研究は、siRNAによるTG2の除去によってラパマイシン又はL-Gln-依存性LC3脂質化がブロック(阻止)されること、並びに、TG2の基質である、Dan-cad及び、IP3R1のGln-ドナー含有C末端が、LC3-IIレベルを増加させることを明らかにした。更に又、細胞やマウス内のIP3R1を慢性的に欠損させると、 LC3-IIの減少が見られた。これらのデータは、TG2によって修飾されたIP3R1が新しいオートファジーシグナルとして機能するという代替的な考えを生じさせる。オートファジーは、内因性のオートファジーシグナルとしてのアミノ酸レベルに感受的であり、特に、アミノ酸トランスポーター(SLC1A5及びSLC7A5)を介するL-Gln取り込みと輸送がmTOR依存性のオートファジーシグナル伝達に重要である(Nicklin et al., 2009)。興味深いことに、このタイプの輸送はNγ-修飾Gln誘導体によってブロックされることが可能であった(Esslinger et al., 2005)。L-Glnによるオートファジー調節がTG2のノックダウンによって止められることを示す本発明者らのデータと合わせると、リソソーム分解後に修飾されたIP3R1から遊離するγ-グルタミルアミンがGlnの輸送及び/又は代謝を妨げてオートファジーを制御するはずであると提案される。poly-Glnタンパク質などのいくつかの病原性タンパク質は、Gln-ドナーであるTG2の強力な基質であり、また、γ-グルタミルアミンがHuntington病やAlzheimer病で増大する(Jeitner et al., 2009)。TG2によって修飾されるGlnがオートファジーシグナル伝達に関与することができるかどうかを決定することは興味あることである。本発明者らの結果は、アミン含有Dan-cad又は2-APBがLC3の脂質化を強く高めることを示しており、また、最近の文献は、TG2の内因性アミンドナーであるスペルミジンもオートファジーを誘導することを報告している(Eisenberg et al., 2009)ことから、TG2依存性のオートファジーシグナル伝達の考えが支持される。オートファジーの調節不能は、神経変性疾患に関係するタンパク質封入体の沈着に重要である(Rubinsztein et al., 2007)。本発明者らのデータは、TG2によるGln修飾に光を与え、これは上記の疾患のための薬剤開発の新規の治療標的になりうることを示している。
【0103】
さらにまた、本発明者らの研究により、TG2とIP3R1がともに、オートファジーの調節に関与しており、HeLa細胞でTG2の役割がIP3R1の役割と反対であることを実証しており、これはTG2がIP3R1機能を損なうという知見を支持している。本発明者らの結果は、p62含量から評価されるように、TG2及びIP3R1が、リソソーム分解ではなくLC3の脂質化と空胞化を制御していることを示した。IP3R1の除去によってLC3の脂質化と空胞化を増強したことを示す本発明者らのデータと合わせると、固有のIP3R1がオートファジーを伴う空胞化を抑制するはずであり(図7C)、従来の概念(Criollo et al., 2007; Vicencio et al., 2009)と矛盾しない。本発明者らは、細胞死に直結する、TG2及びIP3R1によるオートファジー調節を今回提案する(図7D)。この新しいモデルの態様は、TG2 siRNAによって明らかにされるTG2によるオートファジー空胞化の阻害解除(disinhibition)、並びに、IP3R1によるオートファジー細胞死のTG2依存性制御である。このモデルは、明らかな空胞化、LC3の脂質化及びTG2又はIP3R1のノックダウン時の細胞死、の細胞応答だけでなく、疾患状態下の神経変性の分子機構をも整然と解明する。Huntington病及びAlzheimer病では、TGが活性化され(Iismaa et al., 2009)、及び、TG-産物であるγ-グルタミルアミンが増加する(Jeitner et al., 2009)。本発明者らは、修飾IP3R1が、R6/2マウスで専ら増大したことを確認した(図10)。TG2の活性化が酵素的にIP3R1を修飾し、そして、Ca2+シグナル伝達又はベクリン-1相互作用の制御不能によってオートファジーを伴う空胞化を抑制解除する(Vicencio et al., 2009)。正常の条件では、オートファジーは、リソソーム分解と結びつくことによって、折り畳みを誤ったタンパク質や栄養飢餓から細胞を保護する。しかしながら、TG2は、オートファジー空胞の過剰負荷を行い、したがって神経変性疾患での細胞死を誘導するはずである。重要なことは、本発明者らの知見が、TG2によるIP3R1の修飾が、細胞の保護から細胞死にオートファジーの固有の役割に切り替える重要な事象であるという考えに導くことである(図7D)。このことは、Dictyostelium真核モデルでのランダムな突然変異誘発によって示されるオートファジーによる細胞死のシグナル伝達に対する分子的洞察を提供する(Lam et al., 2008)。オートファジーは、神経変性疾患でのタンパク質封入体のクリアランスに関与しているので(Rubinsztein et al., 2007)、本発明者らの考えは、TG2によるIP3R1の修飾に光を与え、これはこれら疾患のための薬剤開発の新規の治療標的になりうることを示している。
【0104】
まとめると、本発明者らは、IP3R1がTG2の新しい基質であり、TG2が酵素的にIP3R1の機能を損なうことを実証した。アミド基転移部位は、Q2746であると決定され、この部位でTG2もまた脱アミド化を触媒する。TG2によるサブユニット間架橋には、隣接のサブユニットのK-クラスター領域が必要であり、このことは、TG2による共有結合によりサブユニットの立体配置を閉じた状態に固定することを示している。TG2によって触媒されるIP3R1の修飾は、RAによって分化される神経芽腫で増強された。TG2はIP3R1と反対の方向にオートファジー事象と細胞死を制御したが、これは、TG2によるIP3R1の修飾がオートファジーの際の栄養飢餓からの細胞の生存を細胞死に変換する重要なシグナル伝達であることを支持している。
【0105】
参考文献
AbdAlla, S., Lother, H., Langer, A., el Faramawy, Y., and Quitterer, U. (2004). Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell 119, 343-354.
Antonyak, M. A., Singh, U. S., Lee, D. A., Boehm, J. E., Combs, C., Zgola, M. M., Page, R. L., and Cerione, R. A. (2001). Effects of tissue transglutaminase on retinoic acid-induced cellular differentiation and protection against apoptosis. J Biol Chem 276, 33582-33587.
Anyatonwu, G., and Joseph, S. K. (2009). Surface accessibility and conformational changes in the N-terminal domain of type I inositol trisphosphate receptors: studies using cysteine substitution mutagenesis. J Biol Chem 284, 8093-8102.
Aoto, J., Nam, C. I., Poon, M. M., Ting, P., and Chen, L. (2008). Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60, 308-320.
Bailey, C. D., and Johnson, G. V. (2005). Tissue transglutaminase contributes to disease progression in the R6/2 Huntington's disease mouse model via aggregate-independent mechanisms. J Neurochem 92, 83-92.
Battaglia, G., Farrace, M. G., Mastroberardino, P. G., Viti, I., Fimia, G. M., Van Beeumen, J., Devreese, B., Melino, G., Molinaro, G., Busceti, C. L., et al. (2007). Transglutaminase 2 ablation leads to defective function of mitochondrial respiratory complex I affecting neuronal vulnerability in experimental models of extrapyramidal disorders. J Neurochem 100, 36-49.
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1, 11-21.
Boehning, D., Patterson, R. L., Sedaghat, L., Glebova, N. O., Kurosaki, T., and Snyder, S. H. (2003). Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol 5, 1051-1061.
Candi, E., Schmidt, R., and Melino, G. (2005). The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 6, 328-340.
Changeux, J. P., and Edelstein, S. J. (1998). Allosteric receptors after 30 years. Neuron 21, 959-980.
Chiang, M. Y., Misner, D., Kempermann, G., Schikorski, T., Giguere, V., Sucov, H. M., Gage, F. H., Stevens, C. F., and Evans, R. M. (1998). An essential role for retinoid receptors RARbeta and RXRgamma in long-term potentiation and depression. Neuron 21, 1353-1361.
Criollo, A., Maiuri, M. C., Tasdemir, E., Vitale, I., Fiebig, A. A., Andrews, D., Molgo, J., Diaz, J., Lavandero, S., Harper, F., et al. (2007). Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 14, 1029-1039.
Csosz, E., Bagossi, P., Nagy, Z., Dosztanyi, Z., Simon, I., and Fesus, L. (2008). Substrate preference of transglutaminase 2 revealed by logistic regression analysis and intrinsic disorder examination. J Mol Biol 383, 390-402.
Dale, G. L., Friese, P., Batar, P., Hamilton, S. F., Reed, G. L., Jackson, K. W., Clemetson, K. J., and Alberio, L. (2002). Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 415, 175-179.
Eisenberg, T., Knauer, H., Schauer, A., Buttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., Ring, J., Schroeder, S., Magnes, C., Antonacci, L., et al. (2009). Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11, 1305-1314.
Esslinger, C. S., Cybulski, K. A., and Rhoderick, J. F. (2005). Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg Med Chem 13, 1111-1118.
Foskett, J. K., White, C., Cheung, K. H., and Mak, D. O. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87, 593-658.
Hamada, K., Miyata, T., Mayanagi, K., Hirota, J., and Mikoshiba, K. (2002). Two-state conformational changes in inositol 1,4,5-trisphosphate receptor regulated by calcium. J Biol Chem 277, 21115-21118.
Hamada, K., Terauchi, A., and Mikoshiba, K. (2003). Three-dimensional rearrangements within inositol 1,4,5-trisphosphate receptor by calcium. J Biol Chem 278, 52881-52889.
Harnett, M. T., Bernier, B. E., Ahn, K. C., and Morikawa, H. (2009). Burst-timing-dependent plasticity of NMDA receptor-mediated transmission in midbrain dopamine neurons. Neuron 62, 826-838.
Hattori, M., Suzuki, A. Z., Higo, T., Miyauchi, H., Michikawa, T., Nakamura, T., Inoue, T., and Mikoshiba, K. (2004). Distinct roles of inositol 1,4,5-trisphosphate receptor types 1 and 3 in Ca2+ signaling. J Biol Chem 279, 11967-11975.
Hovhannisyan, Z., Weiss, A., Martin, A., Wiesner, M., Tollefsen, S., Yoshida, K., Ciszewski, C., Curran, S. A., Murray, J. A., David, C. S., et al. (2008). The role of HLA-DQ8 beta57 polymorphism in the anti-gluten T-cell response in coeliac disease. Nature 456, 534-538.
Hoyer-Hansen, M., Bastholm, L., Szyniarowski, P., Campanella, M., Szabadkai, G., Farkas, T., Bianchi, K., Fehrenbacher, N., Elling, F., Rizzuto, R., et al. (2007). Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 25, 193-205.
Iismaa, S. E., Mearns, B. M., Lorand, L., and Graham, R. M. (2009). Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev 89, 991-1023.
Jeitner, T. M., Pinto, J. T., Krasnikov, B. F., Horswill, M., and Cooper, A. J. (2009). Transglutaminases and neurodegeneration. J Neurochem 109 Suppl 1, 160-166.
Klionsky, D. J., Abeliovich, H., Agostinis, P., Agrawal, D. K., Aliev, G., Askew, D. S., Baba, M., Baehrecke, E. H., Bahr, B. A., Ballabio, A., et al. (2008). Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151-175.
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27-42.
Lorand, L., and Graham, R. M. (2003). Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 4, 140-156.
Mackrill, J. J., Challiss, R. A., O'Connell D, A., Lai, F. A., and Nahorski, S. R. (1997). Differential expression and regulation of ryanodine receptor and myo-inositol 1,4,5-trisphosphate receptor Ca2+ release channels in mammalian tissues and cell lines. Biochem J 327 (Pt 1), 251-258.
Maden, M. (2007). Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci 8, 755-765.
Mastroberardino, P. G., Iannicola, C., Nardacci, R., Bernassola, F., De Laurenzi, V., Melino, G., Moreno, S., Pavone, F., Oliverio, S., Fesus, L., and Piacentini, M. (2002). 'Tissue' transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ 9, 873-880.
Nagy, L., Saydak, M., Shipley, N., Lu, S., Basilion, J. P., Yan, Z. H., Syka, P., Chandraratna, R. A., Stein, J. P., Heyman, R. A., and Davies, P. J. (1996). Identification and characterization of a versatile retinoid response element (retinoic acid receptor response element-retinoid X receptor response element) in the mouse tissue transglutaminase gene promoter. J Biol Chem 271, 4355-4365.
Nakade, S., Maeda, N., and Mikoshiba, K. (1991). Involvement of the C-terminus of the inositol 1,4,5-trisphosphate receptor in Ca2+ release analysed using region-specific monoclonal antibodies. Biochem J 277 (Pt 1), 125-131.
Nakayama, T., Hattori, M., Uchida, K., Nakamura, T., Tateishi, Y., Bannai, H., Iwai, M., Michikawa, T., Inoue, T., and Mikoshiba, K. (2004). The regulatory domain of the inositol 1,4,5-trisphosphate receptor is necessary to keep the channel domain closed: possible physiological significance of specific cleavage by caspase 3. Biochem J 377, 299-307.
Nanda, N., Iismaa, S. E., Owens, W. A., Husain, A., Mackay, F., and Graham, R. M. (2001). Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem 276, 20673-20678.
Nicklin, P., Bergman, P., Zhang, B., Triantafellow, E., Wang, H., Nyfeler, B., Yang, H., Hild, M., Kung, C., Wilson, C., et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521-534.
Nishiyama, M., Hong, K., Mikoshiba, K., Poo, M. M., and Kato, K. (2000). Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408, 584-588.
Rong, Y. P., Aromolaran, A. S., Bultynck, G., Zhong, F., Li, X., McColl, K., Matsuyama, S., Herlitze, S., Roderick, H. L., Bootman, M. D., et al. (2008). Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Mol Cell 31, 255-265.
Rubinsztein, D. C., Gestwicki, J. E., Murphy, L. O., and Klionsky, D. J. (2007). Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6, 304-312.
Sayers, L. G., Miyawaki, A., Muto, A., Takeshita, H., Yamamoto, A., Michikawa, T., Furuichi, T., and Mikoshiba, K. (1997). Intracellular targeting and homotetramer formation of a truncated inositol 1,4,5-trisphosphate receptor-green fluorescent protein chimera in Xenopus laevis oocytes: evidence for the involvement of the transmembrane spanning domain in endoplasmic reticulum targeting and homotetramer complex formation. Biochem J 323 (Pt 1), 273-280.
Selkoe, D. J. (2004). Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol 6, 1054-1061.
Sliter, D. A., Kubota, K., Kirkpatrick, D. S., Alzayady, K. J., Gygi, S. P., and Wojcikiewicz, R. J. (2008). Mass spectrometric analysis of type 1 inositol 1,4,5-trisphosphate receptor ubiquitination. J Biol Chem 283, 35319-35328.
Thacher, S. M., and Rice, R. H. (1985). Keratinocyte-specific transglutaminase of cultured human epidermal cells: relation to cross-linked envelope formation and terminal differentiation. Cell 40, 685-695.
Tucholski, J., Lesort, M., and Johnson, G. V. (2001). Tissue transglutaminase is essential for neurite outgrowth in human neuroblastoma SH-SY5Y cells. Neuroscience 102, 481-491.
Tucholski, J., Roth, K. A., and Johnson, G. V. (2006). Tissue transglutaminase overexpression in the brain potentiates calcium-induced hippocampal damage. J Neurochem 97, 582-594.
Uchida, K., Miyauchi, H., Furuichi, T., Michikawa, T., and Mikoshiba, K. (2003). Critical regions for activation gating of the inositol 1,4,5-trisphosphate receptor. J Biol Chem 278, 16551-16560.
Verma, A., Wang, H., Manavathi, B., Fok, J. Y., Mann, A. P., Kumar, R., and Mehta, K. (2006). Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res 66, 10525-10533.
Vicencio, J. M., Ortiz, C., Criollo, A., Jones, A. W., Kepp, O., Galluzzi, L., Joza, N., Vitale, I., Morselli, E., Tailler, M., et al. (2009). The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 16, 1006-1017.
Walther, D. J., Peter, J. U., Winter, S., Holtje, M., Paulmann, N., Grohmann, M., Vowinckel, J., Alamo-Bethencourt, V., Wilhelm, C. S., Ahnert-Hilger, G., and Bader, M. (2003). Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 115, 851-862.
Williams, A., Sarkar, S., Cuddon, P., Ttofi, E. K., Saiki, S., Siddiqi, F. H., Jahreiss, L., Fleming, A., Pask, D., Goldsmith, P., et al. (2008). Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 4, 295-305.
Zhou, H., Iwasaki, H., Nakamura, T., Nakamura, K., Maruyama, T., Hamano, S., Ozaki, S., Mizutani, A., and Mikoshiba, K. (2007). 2-Aminoethyl diphenylborinate analogues: selective inhibition for store-operated Ca2+ entry. Biochem Biophys Res Commun 352, 277-282.
【産業上の利用可能性】
【0106】
本発明により、神経変性疾患や癌などのTG2とIP3R1が関係する疾患の治療や治療剤の開発のために有用である。
【配列表フリーテキスト】
【0107】
配列番号12 1位:任意のアミノ酸残基
配列番号12 3位:任意のアミノ酸残基
配列番号12 4位:Pro, Thr又はSer残基
配列番号12 5位:脂肪族アミノ酸残基
【技術分野】
【0001】
本発明は、トランスグルタミナーゼ2(「TG2」と称する)によるIP3レセプター1(「IP3R1」と称する)の修飾に関する。
【背景技術】
【0002】
すべての神経変性疾患の共通の特徴は、折り畳みの誤りと安定な代替コンフォメーションへの変換によって生じるタンパク質封入体の異常な沈着である(Selkoe, 2004)。前記封入体タンパク質の分析により、グルタミン(Gln)残基とリジン(Lys)残基との間のイソペプチド結合による共有結合性架橋の存在が明らかにされ、並びに、アルツハイマー(Alzheimer)病、ハンチントン(Huntington)病及びパーキンソン(Parkinson)病の患者の脳脊髄液に、切断されたイソペプチド部分が増加するという事実が明示された(Jeitner et al., 2009)。イソペプチド結合の形成はトランスグルタミナーゼ(TG)によって触媒され(Iismaa et al., 2009; LorandとGraham, 2003)、その活性、発現及び量は実際、上記疾患をもつ患者で増加する(Jeitner et al., 2009; LorandとGraham, 2003)。しかしながら、神経系でのTGの役割は完全には理解されていない。
【0003】
TGは、カルシウム(Ca2+)依存性酵素であり、アミド基転移や脱アミド化を含むGln残基の種々の翻訳後修飾を触媒する(Iismaa et al., 2009)。アミド基転移の結果、Nε-(γ-グルタミル)リジンイソペプチド結合を介してLys残基に架橋される。TGファミリーは、TG1〜TG7やXIII因子と称される8つの酵素/酵素前駆体(zymogen)からなる(Iismaa et al., 2009)。TGによるタンパク質の修飾は、種々の生理学的役割、例えばXIII因子による血液凝固、TGによる限局された表皮形成(Candi et al., 2005; ThacherとRice, 1985)、血小板中でのセロトニン結合(Dale et al., 2002; Walther et al., 2003)、及びAT1レセプターの調節(AbdAlla et al., 2004)に関与する。多くの疾患でのTGの病理学的役割もまた示されており、セリアック病(Hovhannisyan et al., 2008)から神経変性疾患(LorandとGraham, 2003)までの範囲である。TG2は脳内に広がっており、トランス(trans)型のレチノイン酸(RA)(神経モルフォゲン)で誘導可能である。TG2プロモーター領域はRAレセプター(RAR-RXR)の結合エレメントを含む(Nagy et al., 1996)。細胞死においてTG2は双面的であるが、これはTGが細胞に対して毒性的であったり保護的であったりしうるからである。トランスジェニックマウス又はノックアウトマウスを用いた最近の研究は一貫して、TG2が脳内の神経細胞死(Battaglia et al., 2007; Tucholski et al., 2006)や胸腺細胞の生存活性(Nanda et al., 2001)に関与しうることを示している。一方、TG2はまた、癌細胞では抗アポトーシス性であるが、これはTG2が癌の化学耐性や転移能と相関するからである(Verma et al., 2006)。Huntington病のR6/1モデルマウス(Mastroberardino et al., 2002)やR6/2モデルマウス(BaileyとJohnson, 2005)でのTG2の除去は、寿命の延長や運動機能の改善と関わりがあり、R6/2マウスでのTG2の過剰発現は一貫してカイニン酸刺激に対する細胞の脆弱性を実証している。しかしながら、TG2ノックアウトマウスはポリ-Gln凝集産生の低下を示していない(BaileyとJohnson, 2005)。したがって、タンパク質封入体自体のTG2依存性架橋が原因ではないと考えられており、その機序は未知である。
【0004】
脳の機能は、神経系のシグナル伝達によって調節されており、これには細胞内のCa2+ シグナル伝達が関与している(Berridge et al., 2000)。イノシトール1,4,5-トリホスフェートレセプター(IP3R)(これはER在留レセプターである。)は集合して、IP3に反応してERからCa2+を放出するテトラマーのチャネルを形成する(Foskett et al., 2007)。IP3は、種々の細胞表面レセプターの刺激によって産生される。IP3Rは構造的に3つのドメイン、すなわちIP3-結合コアと調節領域を含む大きな細胞質ドメイン、6つの膜貫通セグメントを含むチャネルドメイン、及び細胞質側の短いC末端尾部、に分けられる(Foskett et al., 2007)。IP3R1型(IP3R1)は専ら脳に存在し、長期増強 (LTP)と長期抑圧 (LTD)に関与する(Harnett et al., 2009; Nishiyama et al., 2000)。IP3R1を介するCa2+シグナル伝達及び恒常性は、細胞死や生存シグナル伝達に重要である(Berridge et al., 2000)。チトクロムc (Boehning et al., 2003)やBcl-2 (Rong et al., 2008)との相互作用は、ミトコンドリア依存性アポトーシスに関係している。最近の研究によって、IP3Rが、Bcl-2やBeclin 1との相互作用を通して(Vicencio et al., 2009)、オートファジーを調節していることが実証された(LevineとKroemer, 2008)。しかしながら、これらの調節因子がチャネル機能を制御して細胞運命を決定する正確な機序は依然としてほとんど知られていない。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Selkoe, D. J. (2004). Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol 6, 1054-1061
【非特許文献2】Jeitner, T. M., Pinto, J. T., Krasnikov, B. F., Horswill, M., and Cooper, A. J. (2009). Transglutaminases and neurodegeneration. J Neurochem 109 Suppl 1, 160-166
【非特許文献3】Iismaa, S. E., Mearns, B. M., Lorand, L., and Graham, R. M. (2009). Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev 89, 991-1023
【非特許文献4】Lorand, L., and Graham, R. M. (2003). Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 4, 140-156
【非特許文献5】Candi, E., Schmidt, R., and Melino, G. (2005). The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 6, 328-340
【非特許文献6】Thacher, S. M., and Rice, R. H. (1985). Keratinocyte-specific transglutaminase of cultured human epidermal cells: relation to cross-linked envelope formation and terminal differentiation. Cell 40, 685-695
【非特許文献7】Dale, G. L., Friese, P., Batar, P., Hamilton, S. F., Reed, G. L., Jackson, K. W., Clemetson, K. J., and Alberio, L. (2002). Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 415, 175-179
【非特許文献8】Walther, D. J., Peter, J. U., Winter, S., Holtje, M., Paulmann, N., Grohmann, M., Vowinckel, J., Alamo-Bethencourt, V., Wilhelm, C. S., Ahnert-Hilger, G., and Bader, M. (2003). Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 115, 851-862
【非特許文献9】AbdAlla, S., Lother, H., Langer, A., el Faramawy, Y., and Quitterer, U. (2004). Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell 119, 343-354
【非特許文献10】Hovhannisyan, Z., Weiss, A., Martin, A., Wiesner, M., Tollefsen, S., Yoshida, K., Ciszewski, C., Curran, S. A., Murray, J. A., David, C. S., et al. (2008). The role of HLA-DQ8 beta57 polymorphism in the anti-gluten T-cell response in coeliac disease. Nature 456, 534-538
【非特許文献11】Nagy, L., Saydak, M., Shipley, N., Lu, S., Basilion, J. P., Yan, Z. H., Syka, P., Chandraratna, R. A., Stein, J. P., Heyman, R. A., and Davies, P. J. (1996). Identification and characterization of a versatile retinoid response element (retinoic acid receptor response element-retinoid X receptor response element) in the mouse tissue transglutaminase gene promoter. J Biol Chem 271, 4355-4365
【非特許文献12】Battaglia, G., Farrace, M. G., Mastroberardino, P. G., Viti, I., Fimia, G. M., Van Beeumen, J., Devreese, B., Melino, G., Molinaro, G., Busceti, C. L., et al. (2007). Transglutaminase 2 ablation leads to defective function of mitochondrial respiratory complex I affecting neuronal vulnerability in experimental models of extrapyramidal disorders. J Neurochem 100, 36-49
【非特許文献13】Tucholski, J., Roth, K. A., and Johnson, G. V. (2006). Tissue transglutaminase overexpression in the brain potentiates calcium-induced hippocampal damage. J Neurochem 97, 582-594
【非特許文献14】Nanda, N., Iismaa, S. E., Owens, W. A., Husain, A., Mackay, F., and Graham, R. M. (2001). Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem 276, 20673-20678
【非特許文献15】Verma, A., Wang, H., Manavathi, B., Fok, J. Y., Mann, A. P., Kumar, R., and Mehta, K. (2006). Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res 66, 10525-10533
【非特許文献16】Mastroberardino, P. G., Iannicola, C., Nardacci, R., Bernassola, F., De Laurenzi, V., Melino, G., Moreno, S., Pavone, F., Oliverio, S., Fesus, L., and Piacentini, M. (2002). 'Tissue' transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ 9, 873-880
【非特許文献17】Bailey, C. D., and Johnson, G. V. (2005). Tissue transglutaminase contributes to disease progression in the R6/2 Huntington's disease mouse model via aggregate-independent mechanisms. J Neurochem 92, 83-92.
【非特許文献18】Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1, 11-21
【非特許文献19】Foskett, J. K., White, C., Cheung, K. H., and Mak, D. O. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87, 593-658
【非特許文献20】Harnett, M. T., Bernier, B. E., Ahn, K. C., and Morikawa, H. (2009). Burst-timing-dependent plasticity of NMDA receptor-mediated transmission in midbrain dopamine neurons. Neuron 62, 826-838
【非特許文献21】Nishiyama, M., Hong, K., Mikoshiba, K., Poo, M. M., and Kato, K. (2000). Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408, 584-588
【非特許文献22】Boehning, D., Patterson, R. L., Sedaghat, L., Glebova, N. O., Kurosaki, T., and Snyder, S. H. (2003). Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol 5, 1051-1061
【非特許文献23】Rong, Y. P., Aromolaran, A. S., Bultynck, G., Zhong, F., Li, X., McColl, K., Matsuyama, S., Herlitze, S., Roderick, H. L., Bootman, M. D., et al. (2008). Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Mol Cell 31, 255-265
【非特許文献24】Vicencio, J. M., Ortiz, C., Criollo, A., Jones, A. W., Kepp, O., Galluzzi, L., Joza, N., Vitale, I., Morselli, E., Tailler, M., et al. (2009). The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 16, 1006-1017
【非特許文献25】Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27-42
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、TG2によるIP3R1タンパク質のグルタミン残基の修飾又はサブユニット間架橋、それによって生じるIP3R1タンパク質のチャネル活性の抑制、或いはオートファジーによる細胞死の誘導、を利用した、TG2又はIP3R1活性の測定法、神経変性疾患や癌などの疾患の療法を提供することである。
【課題を解決するための手段】
【0007】
今回、本発明者らは、改良した電気泳動法を用いてIP3R1における共有結合性修飾の状態をモニターし、IP3R1がTG2の新規の基質であること、TG2は、テトラマーIP3R1内の互いに近傍にある2つのサブユニットのC末端領域と調節ドメインとの間のサブユニット間アミド基転移又はIP3R1のグルタミン残基の修飾を触媒して生物活性(すなわち、チャネル活性)を抑制又は阻害すること、TG2はまた、レチノイン酸(RA)依存的にIP3R1機能を損なうし、またオートファジーを調節すること、並びに、TG2によるIP3R1内の修飾が特定の疾患に関係するオートファジー細胞死を制御することを明らかにした。
【0008】
本発明は、要約すると、以下の特徴を包含する。
(1) 細胞においてTG2によってIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を形成してIP3R1生物活性を抑制し、それによってオートファジーを誘導して該細胞の細胞死を引き起こすことを含む、細胞増殖の抑制方法。
(2) 細胞が腫瘍細胞である、上記(1)に記載の方法。
【0009】
(3) 疾患関連細胞又は疾患モデル非ヒト動物において、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を指標にして該架橋によって引き起こされるオートファジーと該疾患との相関関係を測定し、さらに該疾患が該IP3R1のサブユニット間架橋に起因するときにTG2の阻害剤又は活性化剤を候補薬剤として該モデル非ヒト動物に投与して治療効果を調べることを含む、疾患の治療剤をスクリーニングする方法。
(4) 疾患が神経変性疾患又は癌である、上記(3)に記載の方法。
【0010】
(5) TG2及びIP3R1の生物活性をともに含む細胞を用いてそのIP3R1活性を、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋又は該架橋の脱アミド化を指標にして測定することを含む、細胞でのIP3R1活性の測定方法。
(6) 細胞が疾患関連細胞である、上記(5)に記載の方法。
【0011】
(7) TG2の活性を抑制することによるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の抑制によってオートファジーを抑制することを含む、神経変性疾患の治療方法。
(8) IP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の増強剤を有効成分として含む、癌の治療用組成物。
【0012】
(9) IP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の抑制剤を有効成分として含む、神経変性疾患の治療用組成物。
(10) TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を検出することを含む、IP3R1修飾の検出方法。
【0013】
(11) TG2によるIP3R1タンパク質のサブユニット間アミド基転移を介する該タンパク質のサブユニット間架橋を測定することを含む、IP3R1タンパク質チャネル活性のTG2阻害作用を測定する方法。
(12) TG2によるIP3R1タンパク質のサブユニット間架橋の脱アミド化を測定することを含む、TG2触媒活性の測定方法。
【0014】
(13) 測定が、SDS含有アガロースゲル電気泳動特異抗体との交差反応を用いて行うことを含む、上記(10)又は(11)に記載の方法。
(14) 配列番号1又は配列番号2のアミノ酸配列のそれぞれ2746位又は2707位のC末端側グルタミン残基(Q)を介してサブユニット間架橋されたテトラマーIP3R1タンパク質。
(15) TG2によってIP3R1タンパク質のC末端側グルタミン残基と一級アミン含有分子とを結合することを含む、IP3R1のグルタミン残基の修飾方法。
【発明の効果】
【0015】
本発明は、癌又は神経系において共に重要なタンパク質であるTG2とIP3R1とが相互作用して、これらのタンパク質が細胞内のIP3R1生物活性の調節や細胞死を伴うオートファジーに関与するという知見に基づいており、癌、神経変性疾患などの特定の疾患の治療のために本発明を利用することができるという利点が提供される。
【図面の簡単な説明】
【0016】
【図1−1】IP3R1のサブユニット間の架橋の内因性活性を示す。(A) 本発明者らが開発したSDS-アガロース電気泳動(AGE)による架橋の検出。IP3R1サブユニットを種々の濃度(0.01, 0.02, 0.05, 0.1, 0.2, 0.5, 1 mM(増加スロープで示す。))のビス[スルホスクシンイミジル]スベレート(BS)(上)又は1,8-ビス-マレイイミド-ジエチレングリコール(BM)(下)によって架橋した。(B) Triton X-100 (1)、Zwittergent 3-14 (2)、SDS (3)又は界面活性剤不含(4)で前処理したIP3R1を、0.5mM BS(上)又は0.1mM BM(下)を用いて架橋した。(C) Ca2+-依存性架橋の内因性活性。小脳P2/P3膜をバッファ又は細胞質画分と一緒にインキュベーションした。(D) アニオン交換カラムによる脳ライセートのクロマトグラム。左軸及び右軸はそれぞれ、吸光度(実線)及びNaCl勾配(破線)を示す。(E) 各溶出液、使用した細胞質画分 (Cyt)又は素通り画分(pass)を、小脳P2/P3膜と一緒にインキュベーションした。*印は、Ca2+-依存性断片である。(A〜C, E)のローマ数字は架橋したサブユニットを示し、これはThモノマー(335 kDa)及びダイマー(670 kDa)によって判断される。
【図1−2】(図1−1の続き)(F) ピーク画分(No. 14)又は精製TGを、シスタミン(2, 4, 10, 20, 40 mM)の存在又は非存在(第2レーン)下でP2/P3画分と一緒にインキュベーションした。(G) クマシーブリリアントブルー(CBB)染色によってIP3R1又はTG2 (*)が示された。精製IP3R1を精製TG2 (0, 1, 5, 20 μg/ml)と一緒にインキュベーションしAGEにかけた。(H) HeLa細胞中のサブユニット間架橋の内因性活性は、TG2 siRNAによって除去された。αTG2によるウエスタンブロット(WB)はTG2の細胞レベルを示す。アミドブラック(AB)染色を負荷対照のために使用した。右パネルはAGEの結果である。(I) DMSO又はRAで前処理したHeLa細胞の架橋活性。(J) RA又はDMSOで前処理した神経芽腫(SH-SY5Y)又は星状膠細胞腫(U87-MG)中のTG2の発現レベル。(K) SH-SY5Y又はU87-MG細胞中でのイオノマイシン(ION, 10μM)刺激によるIP3R1のサブユニット間架橋。
【図2−1】TG2によるIP3R1活性の阻害を示す。(A) 不活性化されたTG2の調製。煮沸したTG2 (中)又はNEM処理したTG2(下)は酵素活性を示さない。20個のスペクトルを重ねた。(B) TMR-cadの自己取り込みによるTG2活性の測定。(C) fura-2を用いたときのCa2+放出の代表的トレース。上の左と右のトレースは、対照(-TG)の、及びMg-ATPの添加によるCa2+負荷後のTG2-前処理(+TG) P2P3膜の、IP3-誘導Ca2+放出を示す。下のトレースは、TG2 (1〜4: 0, 5, 10, 20μg/ml TG), 10μg/ml煮沸TG2 (5)、或いは10μg/ml NEM-TG2 (6)の典型的な反応を表す。縦軸は、(340nmに対する510nmでの蛍光)対(380nmに対する510nmでの蛍光)の比を示す。(D) 棒グラフは、独立の3つの実験から得られたデータのまとめであり、ここで、膜はTG2 (0, 1, 2, 5, 10, 20, 50μg/ml)、10μg/ml煮沸TG2又は10μg/ml NEM-TG2で前処理された。IP3によるCa2+放出は、対照の%(平均+SD)として示されている。TG2のIC50値は、4パラメータ算定曲線関数を用いる非線形回帰分析によって計算された34nMであった。(E) BPは、TG2によって触媒される架橋を阻害した(BP: 1, 2, 5, 10, 20mM)。(F) TG2及びBPで前処理されたIP3R1を、アビジン-HRP又は4C11を用いて検出した。
【図2−2】(図2−1の続き)(G) 10mM BP又は10mM シスタミン(cystamine)(CA)の存在中で調製されたTG2処理膜からのCa2+放出。(H) アビジンによるIP3R1を介するCa2+放出の阻害。BPが取り込まれた膜 (BP+TG)又は取り込まれなかった膜(-TG)を10又は0μg/ml TG2で前処理した。Ca2+放出を0, 31, 63, 94, 125及び156μg/ml (スロープ)或いは156μg/ml (-TG)のアビジンの存在中で測定した。(I) 上のパネルは、RAによって分化された又は分化されなかったSH-SY5Y細胞でのIP3R1内のBPの顕著な取り込みを示す。下のパネルは、TG2及びIP3R1の量を示す。各レーンでは3μgタンパク質が負荷された。(J) fura-2比率の典型的トレースは、P2P3膜ベシクルでのIP3Rを介するIP3誘導Ca2+放出(左)、或いは、DMSO(対照)又はRAで前処理されたSH-SY5Y生細胞でのカバコール(carbachol; CCh)誘導Ca2+放出(右)を示す。(K) IP3誘導Ca2+放出は、0.1及び1μM IP3に応答するfura-2比率の変化(ΔR)によって表された。棒グラフは、3つの培養ディッシュから調製した3つのサンプルの平均+SDを示す。(L) SH-SY5Y生細胞でのカルバコール(CCh, 10μM)誘導Ca2+放出に対するRAの影響。*p<0.05, ***p<0.001 (two-tailed, n=3)
【図3−1】TG2がC末端付近でのIP3R1サブユニット間の架橋を触媒することを示す。BPが取り込まれた精製IP3R1を、(A)及び(B)で種々の濃度のカルパイン(Calp)或いは(B)で種々の濃度のリジル-エンドペプチダーゼ(Lep)によって消化した。得られた断片を、アビジン-HRP, 4C11, 10A6又は18A10を用いて検出した。矢印は、BPが取り込まれた(白)又はBPが取り込まれなかった(黒)主断片を示す。サンプルは、5%ゲル(A)又は7.5%ゲル(B)で泳動された。(C) IP3R1のC末端を認識する18A10抗体での前処理によってTG2によるIP3R1の架橋が阻害された。P2P3膜(1 mg/ml)は、氷上1時間抗体(なし, 18A10, 4C11)で、次いでTG2 (0, 2, 5, 10, 15, 20μg/ml)でインキュベーションした。
【図3−2】(図3−1の続き)(D) TG2による架橋後の免疫反応性。TG2 (0, 2, 5, 10, 20μg/ml)によって架橋されたIP3R1を4C11, 18A10及びpep6Abを用いて検出した。再現性は4回以上の別個の実験で確認された。TG2 (0, 5, 10, 20μg/ml)によって架橋された小脳IP3R1は、50μg/ml Calp (E)又は1μg/ml Lep (F)によって消化された。(G) 50μg/ml Calpでの消化後のTG2 (0, 5, 10, 20μg/ml)による架橋。(E〜G)の白矢印は、TG2によって著しく減少した際立ったバンドを示す。黒矢印は、TG2処理によって変化しなかったN末端断片を示す。
【図4−1】グルタミン2746とリジン-クラスター領域がTG2による架橋に必須であることを示す。(A) 脳サンプルからのTG2サブユニットの精製。アビジン-ビーズによって、CBBで染色される特異的基質(*)をほぼ精製することができたことを特記する。(B) WBによるIP3R1の検出。(C) IP3R1のグルタミン2746をTG2で修飾した。(A)に示したTG2サブユニットをIP3R1として同定した(配列番号1)。(C) IP3R1のイオンm/z 1935.99のC末端ペプチド(上)と2247.15のビオチン化ペプチド(下)をMS/MSでスキャニングした。bイオンとyイオンをMS/MSスペクトル上に記した。*印は修飾されたGln残基を示す。本発明者らは、MS分析によって帰属された63個のGln残基のうちGln2746のみが修飾されたことを見出した(配列番号1)。
【図4−2】(図4−1の続き)(D) サブユニット間架橋に重要なドメインの突然変異誘発。スキームはIP3R1の一次構造とCalpによる切断部位(C130)を示す。欠失変異体(d1〜d4)、点突然変異体(Q2746N)及び野生型(wt)を、スキーム中に描出した。低濃度ゲルによって、COS-7細胞で発現された欠失変異体のサブユニット間架橋が示された。黒と白の矢印はそれぞれ、架橋されたダイマーとその対応するサイズを示す。注目すべきことは、d3又はd4のいずれかがサブユニット間架橋をまったく示さないことである。(E) Q2746N突然変異体でのサブユニット間架橋の喪失。
【図5−1】TGが架橋の加水分解と脱アミド化を触媒することを示す。(A) 上のパネルはTMR-cadのピークを示す。中パネル及び下パネルは、TMR-cad、TG2及びNPQQPA(配列番号3)又はNPNQPA(配列番号4)を含む反応混合物のクロマトグラムである。白矢印は生成物を示す。(B) 上パネルは、精製した生成物を示す。中パネル又は下パネルは、該生成物、TG2及びCaCl2を含む反応混合物又は該生成物及びTG2を含む反応混合物のクロマトグラムを示す。(C) FITC及びTMR-cad (左)を結合したペプチドの典型的PDAデータ。二次元グラフは、ピークX (TMR-cad), Y (FITC-NPQQPA)及びZ (生成物)を示す上のクロマトグラムに相当する吸収スペクトルを示す。右パネルは精製された生成物を示す。
【図5−2】(図5−1の続き)(D) 左パネルに示したのは、(C)の生成物にTG2を添加した後の蛍光スペクトルの変化である。スペクトルは1分間隔で記録された。530 nmでの蛍光強度の経時的スキャニングを記録した(中及び右パネル)。(E)脱アミド化IP3R1テトラマーに対する特異抗体の特性化。スキームは、Q2746を含むGST融合タンパク質又は脱アミド化E2746を含むGST融合タンパク質を示す。ブロットしたQ2746又はE2746タンパク質(1, 3, 10 ng)は、αE2746、18A10及びαGST抗体を用いて検出された。αE2746と18A10はそれぞれE2746及びQ2746を特異的に検出することに注目されたい。(F) in vitroでのQ2746の酵素的脱アミド化。90分間TG2で前処理したP2P3画分中のIP3R1がαE2746、18A10及び4C11を用いて検出された。(G)脱アミド化IP3R1が精製IP3R1で検出された(レーン1:3μl,レーン2:10μl,レーン3:10倍希釈3μl,レーン2:10倍希釈10μl)。(H)脳サンプル中の脱アミド化IP3R1。ゲルは3匹の各マウスのサンプル(3mg/ml)の結果を示す(レーン1〜3)。
【図6−1】TG2及びIP3R1によるオートファジー調節を示す。(A) TG2 siRNAによるIP3R1修飾の喪失。HeLa細胞中の総IP3R1及び修飾IP3R1がそれぞれ4C11及びアビジン-HRPによって検出された。(B) siRNAによるTG2の除去によってLC3-IIの脂質化が減少したが、IP3R1をノックダウンすると該脂質化が高まった。HeLa細胞溶解物中の脂質化されたLC3 (LC3-II), TG2, IP3R1及びアクチンのレベルは、ウエスタンブロッティング(WB)によって評価された。棒グラフは、6つの独立の実験からの平均+SDのまとめである(*p<0.05, ***p<0.001, two-tailed)。ラパマイシン(1μM)又はスペルミジン(100μM)の存在下でのTG2のノックダウン(C)。p62(オートファジー基質)の細胞レベルは、WBによって評価された。
【図6−2】(図6−1の続き)ラパマイシン(1μM)又はスペルミジン(100μM)の存在下でのIP3R1 (D)のノックダウン。p62(オートファジー基質)の細胞レベルは、WBによって評価された。(E) TG2 siRNAによってLC3脂質化を損なうことに対するL-Gln(10mM)の影響。(F) Dan-cad (100μM)と2-APB (100μM)は、p62を蓄積することなくLC3の脂質化を強力に誘導した。
【図7−1】TG2及びIP3R1による空胞化と細胞死の制御を示す。(A) HeLa細胞での空胞化の形態図。この画像は、siRNAによるトランスフェクションの54時間後に得られた。(B) TG2又はIP3R1のノックダウンによる空胞化された細胞(中)又は空胞化されない細胞(右)の数の著しい変化。
【図7−2】(図7−1の続き)(C) 細胞死の評価。培養培地中のLDH活性はsiRNAによるトランスフェクションの48時間後に測定された。(*p<0.05, **p<0.01, ***p<0.001, ###p<0.001, two-tailed) (D) TG2及びIP3R1によって仲介される本発明者らの仮想的なオートファジーシグナル伝達モデル。正常な条件下(左パネル)で、オートファジーの誘導はIP3R1によって抑制される。活性化されたTG2がIP3R1を修飾し、オートファジーの抑制を解除する(右パネル)。その結果、オートファジーの空胞化が過剰になり細胞死が誘導される。
【図8】マウスIP3R1のアミノ酸配列(配列番号1)を示す。太字は、精密質量分析により検出されたペプチド配列を表す。また、下線は、TGにより修飾された部位を表す。
【図9】TG2によるIP3R1の酵素的調節の構造モデルを示す。(A) スキームはIP3R1のQ-ドナー(Q2746)及びK-ドナー(K-クラスター領域)を示す。C130ドメインが青で示され、IP3-結合コアを含むN180ドメインが灰色で示されている。膜貫通領域のヘリックスはN末端側からC末端側に番号付けられている(TM1-6)。(B) 絵は、細胞質側から描かれたテトラマーIP3R1内のQ-ドナー及びK-ドナーの配置を提示する。C末端のQ-ドナーは、近傍のサブユニットのK-ドナー領域のごく近くに位置する。本発明者らは、閉じた状態(B)から開いた状態(C)までのテトラマーIP3R1の立体配置の変化を提案する。ここで、Ca2+ (黄)は孔を通過する。TGによって触媒されるアミド基転移によって、閉じた立体配置をロックするための(D)に示される共有結合されたサブユニット間架橋(赤)が生じる。TGにより触媒されるBPの取り込みは、前記立体配置を留めることができない(E)。しかしながら、嵩高いアビジンとBPの化学量論的な結合は、立体障害によって、閉じたチャネル立体配置をロックする(F)。
【図10】野性型マウス小脳および大脳のIP3R1をウエスタンブロッティングにより検出した結果である。縦軸は、野性型を100%としてバンドシグナル強度を計算したものである。R6/2の小脳ではE2746抗体、18A10抗体、4C11抗体、全てにおいて小脳IP3R1のシグナルが低下している。しかし、R6/2の大脳IP3R1では、E2746抗体でのIP3R1シグナルが他の抗体に比べて増強されていた。従って、これは特にR6/2の大脳に於いてIP3R1のQ2746がEに変換されていることを示唆する。
【発明を実施するための形態】
【0017】
本発明者らは、今回、IP3R1がTG2の新しい基質であり、TG2が酵素的にIP3R1の機能を損なうことを見出した。アミド基転移部位は、配列番号1(マウス)のQ2746又は配列番号2(ヒト)のQ2707であると決定され、この部位でTG2が脱アミド化も触媒する。TG2によるサブユニット間架橋には、隣接のサブユニットのK-クラスター領域が必要であり、このことは、TG2による共有結合によりサブユニットの立体配置を閉じた状態に固定することを示している。TG2によって触媒されるIP3R1の修飾は、レチノイン酸(RA)によって分化される神経芽腫で増強された。TG2はIP3R1と反対の方向にオートファジー事象と細胞死を制御したが、これは、TG2によるIP3R1の修飾がオートファジーの際の栄養飢餓からの細胞の生存を細胞死に変換する重要なシグナル伝達であることを支持している。
【0018】
<IP3R>
細胞膜上の受容体の活性化に伴いホスファチジルイノシトール4,5-ビスホスフェート(phosphatidylinositol 4,5-bisphosphate)が加水分解されると、細胞内セカンドメッセンジャーであるイノシトール1,4,5-三リン酸(inositol 1,4,5-trisphosphate (IP3))が生成する。IP3はIP3レセプター(IP3R)に結合することにより、細胞内カルシウム貯蔵オルガネラ(主に小胞体)からのCa2+放出を誘導する。このIP3/Ca2+シグナル経路において、IP3レセプターは、IP3のシグナルをCa2+のシグナルへ変換する役割を担っている。IP3レセプターは、テトラマー(4量体)構造の細胞内IP3誘導性Ca2+放出チャネル(IP3-gated Ca2+ release channel)である。哺乳動物では、3種の異なるIP3レセプターが存在する。IP3レセプター1型(IP3R1)は、中枢神経系、特に小脳において高発現している。ヒトIP3R1のアミノ酸配列及び塩基配列がGenBankに登録されており、そのAccession Numberは、例えば、NM_001099952.2(variant 1;配列番号2)、NM_002222.5(variant 2)及びNM_001168272.1(variant 3)である。マウスIP3R1は、2749アミノ酸(GenBank Accession No. NM_010585.3)からなる。IP3R1は、3つの機能的に異なる領域に分かれている。すなわち、N末端近傍にIP3結合ドメイン、C末端近傍に6回膜貫通領域を有するチャネル形成ドメイン、およびこれら2つの領域の間に制御領域が存在する。IP3結合ドメインの欠失突然変異体解析より、IP3レセプターのアミノ酸226〜578残基が特異的かつ高親和性のリガンド結合に必要な最小領域であることが示され、IP3結合コアと呼ばれている。
【0019】
IP3レセプターの活性化による細胞質Ca2+濃度の増加によって、多種多様な下流標的分子の活性が制御される。これら下流標的分子は、受精、発生、増殖、分泌、シナプス可塑性など多岐に渡る細胞応答において重要な役割を担っている(特開2007-284433)。
【0020】
<TG>
トランスグルタミナーゼ(TG)は、タンパク質分子間の架橋反応を触媒する酵素である。この酵素によって、その基質であるタンパク質中のグルタミン残基と別のタンパク質中のリシン残基又は一級アミンとの間で共有結合が形成されてタンパク質同士が結合される。
【0021】
ヒトTGには、8つのアイソザイム、例えば第8凝固因子、TG1〜TG7の存在が知られている。本発明に関わるTGは、特にTG2であり、この酵素は、細胞死や神経系疾患に関係するという報告がある(人見清隆,生化学,77巻6号,pp.552-558 (2005))。TGは、タンパク質の架橋化の他に、TGとタンパク質とから形成される複合体のグルタミン残基のグルタミン酸への脱アミド化も触媒することができる。
【0022】
TGは、カルシウムの存在によって活性化される酵素であり、アルツハイマー病、パーキンソン病、ハンチントン病等の神経疾患に関与することが知られるようになり、TG酵素の阻害剤がこれらの疾患の治療薬として有効であると考えられている(Hoffner G and Djian P, Front Biosci., 2005 Sep 1, 10:3078-3092; Duval E et al., Bioorg Med Chem Lett., 2005 Apr 1, 15(7):1885-1889)。タンパク質の異常架橋に関して、タンパク質のグルタミンのアミド基とリシンのアミノ基から脱アンモニアによりイソペプチド結合をつくる反応が架橋の主反応であり、この反応を引き起こすTGの阻害剤が、上記疾患等に効果的であろうとする作用機序が明らかにされた(Mastroberardino PG, et al., Cell Death Differ., 2002 Sep, 9(9):873-880)。その根拠をもとにしてTGの阻害剤を、アルツハイマー病、ハンチントン病、パーキンソン病、セリアック病、白内障、狂牛病、先天性葉状魚鱗症、先天性止血障害症、肝臓障害、自己免疫疾患、脳梗塞等の治療薬として開発する研究が増加している(WO03/033002号; WO2007/061074号; 特開2009-184988; 特開2007-169272; Lorand L, Proc Natl Acad Sci U S A., 1996 Dec 10, 93(25):14310-14313; Grierson AJ, et al., Neurosci Lett., 2001 Jan 26, 298(1):9-12; Watts RE, et al., J Med Chem., 2006 Dec 14, 49(25):7493-7501; Karpuj MV et al., Nat Med., 2002 Feb, 8(2):143-149; Thung GL et al., Chemistry & Biology 2008 Sept 22, 15:969-978; Tatsukawa H et al., Gastroenterology 2009. 136(5):1783-1795)。
【0023】
<詳細な説明>
本発明によれば、IP3R1タンパク質がTG2の基質であり、TG2が、細胞内で該タンパク質の架橋・修飾や該架橋タンパク質の脱アミド化を通じて細胞での細胞死やオートファジーに関与する。
【0024】
本発明は、配列番号1又は配列番号2のアミノ酸配列のそれぞれ2746位又は2707位に相当するC末端側グルタミン残基(Q)を介してサブユニット間架橋・修飾されたテトラマーIP3R1タンパク質を提供する。
【0025】
すなわち、IP3R1タンパク質がTG2の基質であることは、今回本発明者らが見出すまでは全く知られていなかった。上記のようにテトラマーIP3R1タンパク質の1つのサブユニットのアミノ酸配列のC末端に近い特定の1つのグルタミン残基がTG2酵素の触媒作用により別のサブユニットのK-クラスター領域(チャネル活性調節ドメイン)内のリシン残基との間で共有結合してサブユニット間架橋を形成する。
【0026】
本発明者らの知識によれば、これは、IP3R1がTG2の新規な基質であること、及びTG2が、テトラマーIP3R1の酵素的修飾によってIP3R1のチャネル活性を阻害することを示す最初の研究である。本発明者らは、C末端が近傍のIP3R1サブユニットに近い、及び、TG2が近接するサブユニットに該C末端を共有結合によって結合させ、チャネルを、閉じた立体配置に固定する、と結論付けた。
【0027】
これまでの報告では、C末端尾部がチャネル活性に重要であること(Nakade et al., 1991; Uchida et al., 2003)、並びに、C末端及びKクラスター領域の付近で相互作用する、チトクロムCやBcl-2などの、多くの調節因子がチャネル活性を調節すること(Boehning et al., 2003; Foskett et al., 2007; Rong et al., 2008)が実証されている。本発明者らは今回、これらの調節因子がC末端付近のサブユニット界面に結合して、他のオリゴマーチャネルに類似したサブユニット立体配置を制御することが可能であることを示す。IP3RはCa2+を必要とし、Ca2+は調節ドメインを含む細胞質ドメインを動かすことによってコンフォメーション変化を誘導し(Anyatonwu and Joseph, 2009; Hamada et al., 2002; Hamada et al., 2003)、チャネルと、K-クラスターの近傍に位置するCa2+-センサー用の重要な残基を開く(Foskett et al., 2007)。したがって、Ca2+依存性のコンフォメーション変化が、IP3結合からの刺激によってチャネルを開かせるサブユニット立体配置を自由にすることが提案される。おそらく、このことはCa2+が共アゴニストとしてチャネルのゲーティングに必須であることを示している。
【0028】
本発明はさらに、TG2及びIP3R1の生物活性をともに含む細胞中のIP3R1活性を、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋又は該架橋の脱アミド化を指標にして測定することを含む、細胞でのIP3R1活性の測定方法を提供する。
【0029】
TG2及びIP3R1の生物活性をともに含む細胞は、IP3R1が高発現する小脳や中枢神経系由来の細胞、とりわけ中枢神経系に関わる疾患関連細胞、例えばヒト神経芽細胞腫由来のSH-SY5Y細胞などであるが、これらに限定されないものとする。
【0030】
IP3R1タンパク質(具体的には、テトラマーIP3R1タンパク質)の架橋は、上記のテトラマーIP3R1の異なるサブユニット間の架橋であり、これによってIP3R1の生物活性、すなわちチャネル活性、が阻害又は抑制される。
【0031】
架橋の検出又は測定は、SDS含有アガロースゲル電気泳動及び特異抗体との反応によって行うことができる。後述の実施例では、電気泳動は、0.1%SDS及び1%アガロース含むゲルを用いて100Vで1〜2時間の泳動を行う。ゲルをメンブレンに転写し、特異抗体(例えば、4C11等のモノクローナル抗体)との交差反応を検出する。
【0032】
架橋が検出されるならば、IP3R1のチャネル活性が阻害又は抑制されることを示す。その相関性は、一方で、IP3R1のチャネル活性を測定し、互いの結果を対比することによって分かる。IP3R1のチャネル活性は、例えばZhouらの方法で測定することができる(Zhou et al., 2007)。
【0033】
上記架橋の脱アミド化は、IP3R1タンパク質のC末端側グルタミン残基(例えば配列番号1(マウス)のQ2746又は配列番号2(ヒト)のQ2707に相当する。)のグルタミン酸への変換を測定することによって検出又は測定できる。このために、IP3R1タンパク質の配列番号1又は配列番号2のアミノ酸配列等に基づいて設計された、上記C末端側グルタミン残基から変換されたグルタミン酸残基を含むペプチド(例えば、約7〜約15アミノ酸)と特異的に結合可能なポリクローナル抗体(例えばαE2746抗体)、或いはモノクローナル抗体を作製し、この抗体との交差反応を利用することができる。このようなモノクローナル抗体は、岩崎辰夫、単クローン抗体、ハイブリドーマとELISA(講談社、1983)などに記載の方法によって作製することができる。簡単に説明すると、上記ペプチドを免疫したマウスの脾臓細胞と哺乳動物ミエローマ細胞とをPEGの存在下で融合させて得られたハイブリドーマの中から、HAT培地で抗体産生ハイブリドーマを選択し、さらにグルタミン酸残基を含む領域と特異的に結合可能な抗体産生細胞を選別し、クローニング法か又はマウス腹腔内注射後の腹水から目的のモノクローナル抗体を得ることができる。ミエローマ細胞には、例えば、P3X63Ag8, NS-1, MPC-11, SP2, FO, S194, R210, P3U1などの細胞株が含まれる。
【0034】
上記架橋の検出又は測定はまた、IP3R1修飾の検出、或いはP3R1タンパク質のチャネル活性のTG2阻害作用を測定するために利用できる。
【0035】
すなわち、本発明はさらに、TG2によるIP3R1タンパク質のC末端側グルタミン残基の架橋を検出することを含む、IP3R1修飾の検出方法、或いは、TG2によるIP3R1タンパク質のサブユニット間アミド基転移を介する該タンパク質のサブユニット間架橋を測定することを含む、IP3R1タンパク質チャネル活性のTG2阻害作用を測定する方法をも提供する。
【0036】
さらに、上記の、TG2によるIP3R1タンパク質のサブユニット間架橋の脱アミド化を検出又は測定することによってTG2触媒活性を測定することも可能である。このとき、配列番号1又は配列番号2のアミノ酸配列のそれぞれ2746位又は2707位のC末端側グルタミン残基(Q)を介してサブユニット間架橋されたテトラマーIP3R1タンパク質を基質とすることができる。このように架橋されたテトラマーIP3R1タンパク質も本発明に包含される。
【0037】
従来、TG2の触媒活性は、例えばLorand, L et al., Anal. Biochem. 44:221-231, 1971などの方法で測定されているが、IP3R1タンパク質を基質とするTG2の活性測定については知られていなかった。本発明の方法で測定されたTG2活性と、従来の方法で測定されたTG2活性との相関をみることによって、本発明の測定方法の定量の妥当性が分かるはずである。
【0038】
TG2によるIP3R1の修飾(すなわち、テトラマーIP3R1のサブユニット間架橋)に関して、該修飾がレチノイン酸(RA)によって誘導されることが今回見出された。この知見は、IP3R1が、RAシグナル伝達によって神経機能を調節することができる可能性を示唆している。すなわち、TG2がIP3R1修飾によってレチノイドシグナル伝達を調節することができる新しい経路が存在する可能性を示唆している。一方、この関連で、RAシグナル伝達は神経の分化や変性に重要であることも知られているので(Maden, 2007)、TG2によるIP3R1の修飾と神経系(特に中枢神経系)の異常との関係が示唆される。
【0039】
本発明者らはさらに、TG2とIP3R1がともに、オートファジーの調節に関与しており、腫瘍細胞であるHeLa細胞でTG2の役割がIP3R1の役割と反対であること、すなわちTG2がIP3R1機能を損なうことを見出した。このことは、オートファジーがCa2+シグナル伝達(Hoyer-Hansen et al., 2007; Williams et al., 2008)及びIP3Rによって調節されるという従来の概念(Criollo et al., 2007; Vicencio et al., 2009)と矛盾しない。
【0040】
さらにまた、本発明者らは、TG2及びIP3R1が、リソソーム分解ではなくLC3の脂質化と空胞化を制御していることを見出し、IP3R1の除去によってLC3の脂質化と空胞化を増強したことを示すデータと合わせると、内因性のIP3R1がオートファジーを伴う空胞化を抑制するはずであり、これは従来の概念(Criollo et al., 2007; Vicencio et al., 2009)と矛盾しない。したがって、本発明は、細胞死に直結する、TG2及びIP3R1によるオートファジー調節に関する。すなわち、この調節は、TG2 siRNAなどのTG2阻害剤によって明らかにされるTG2によるオートファジー空胞化の抑制解除(disinhibition)、並びに、IP3R1によるオートファジー細胞死のTG2依存性制御である。本発明に係るこの考え方は、明らかな空胞化、LC3の脂質化及びTG2又はIP3R1のノックダウン時の細胞死、の細胞応答だけでなく、疾患状態にある神経変性の分子機構をも整然と解明する。すなわち、Huntington病及びAlzheimer病では、TGが活性化され(Iismaa et al., 2009)、及び、TG-産物であるγ-グルタミルアミンが増加する(Jeitner et al., 2009)が、本発明者らは、修飾IP3R1が、Huntington病モデルマウス(R6/2)(ポリグルタミン含有タンパク質が過剰発現される。)で専ら増大したことを確認した(図10)。TG2の活性化が酵素的にIP3R1を修飾し、そして、Ca2+シグナル伝達又はベクリン-1相互作用の制御不能によってオートファジーを伴う空胞化を抑制解除する(Vicencio et al., 2009)。正常の条件では、オートファジーは、リソソーム分解と結びつくことによって、折り畳みを誤ったタンパク質や栄養飢餓から細胞を保護する。しかしながら、TG2は、オートファジー空胞の過剰負荷を行い、したがって神経変性疾患での細胞死を誘導するはずである。重要なことは、本発明者らの知見が、TG2によるIP3R1の修飾が、細胞の保護から細胞死にオートファジーの固有の役割に切り替える重要な事象であるという考えに導くことである。オートファジーは、神経変性疾患でのタンパク質封入体のクリアランスに関与している(Rubinsztein et al., 2007)。
【0041】
一方、IP3R1によるオートファジー細胞死のTG2依存性制御を、癌細胞で利用するときには、癌細胞の細胞死に導くことが可能になる。このような癌細胞として、例えばヒト子宮頸癌細胞であるHeLa細胞などを例示することができるが、しかしTG2は各種の癌細胞で発現していることが周知であるから、そういうすべての癌細胞に適用可能であると考えられる。
【0042】
このように、疾患が神経変性疾患であれば、TG2によるIP3R1修飾を阻害する薬剤が治療に有効であるのに対して、疾患が癌(又は、悪性腫瘍)であれば、TG2によるIP3R1修飾を賦活化(又は活性化)する薬剤が治療に有効である。
【0043】
本発明者らの考えは、TG2によるIP3R1の修飾と疾患、特に神経変性疾患及び癌の治療法に光を与え、これはこれら疾患のための薬剤開発の新規の治療標的になりうることを示している。
【0044】
したがって、本発明はまた、細胞においてトランスグルタミナーゼ2(TG2)によってIP3レセプター1(IP3R1)タンパク質のC末端側グルタミン残基の修飾(すなわち、サブユニット間架橋)及び該修飾に伴って起こるIP3R1活性(すなわち、チャネル活性)の阻害を促進し、それによってオートファジーを誘導して該細胞の細胞死を引き起こすことを含む、細胞増殖の抑制方法を提供する。このような細胞の例は、腫瘍細胞である。
【0045】
本発明はさらに、TG2の活性を抑制することによるIP3R1タンパク質のC末端側グルタミン残基の修飾の抑制を介してオートファジーを抑制することを含む、神経変性疾患の治療方法を提供する。
【0046】
それゆえに、本発明はさらに、IP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の増強剤を有効成分として含む、癌の治療用組成物を提供する。
【0047】
癌の例は、すべての固形癌、血液癌などであり、固形癌には脳腫瘍、肺癌、大腸癌、肝臓癌、膵臓癌、胃癌、膀胱癌、前立腺癌、食道癌、皮膚癌、リンパ腫、骨癌などが非限定的に含まれる。また、血液癌には、白血病などが含まれる。
【0048】
前記架橋の増強剤は、TG2酵素の賦活剤や誘導剤であり、例えばレチノイン酸などを挙げることができる。
【0049】
またIP3R1タンパク質のグルタミン残基修飾の増強剤は、TGの基質となるアミノ供与体であり、例えばモノダンシルカダベリン、スペルミジン、スペルミンを挙げることができる。
【0050】
本発明はさらに、IP3R1タンパク質のC末端側グルタミン残基の架橋の抑制剤(又は阻害剤)を有効成分として含む、神経変性疾患の治療用組成物を提供する。
【0051】
神経変性疾患の例は、以下のものに限定されないが、アルツハイマー病、ハンチントン病、パーキンソン病などを含む。このような疾患では、脳内で蛋白質が異常な架橋反応が起こることによって不溶性の蛋白質または蛋白質凝集が生じることによって神経変性疾患が発症することが知られている。
【0052】
上記架橋・修飾の抑制剤は、TG2酵素の阻害剤であり、例えばTG2に対するsiRNA、miRNA、リボザイム、アンチセンスRNAなどの核酸、シスタミン、モノダンシルカダベリンなどの小分子(特許3012923号)や、新規のTG2阻害剤として報告された5-bromo-2-thienyl-(N-t-butyl N-benzyl)-aminoethyl keton等のβ-アミノケトン化合物を含む(Bioorg.Med.Chem.Lett, 20(3): 1141-4 (2010))ことができるし、或いは、IP3R1タンパク質のC末端側グルタミン残基の架橋の抑制剤であり、それは、例えば小分子、ペプチド、タンパク質、核酸、脂質、天然物、合成化合物などから選択可能であり、ここでタンパク質には例えばTG2やIP3R1架橋部位に対する抗体類、核酸にはIP3R1架橋部位に対する例えばsiRNA, miRNA,リボザイム、アンチセンスRNAなどがそれぞれ含まれる。
【0053】
本発明はさらに、疾患関連細胞又は疾患モデル非ヒト動物において、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋(又は、該架橋のレベル)を指標にして該架橋によって引き起こされるオートファジーと該疾患との相関関係を測定し、さらに該疾患が該IP3R1のサブユニット間架橋に起因するときにTG2の阻害剤又は活性化剤を候補薬剤として該モデル非ヒト動物に投与して治療効果を調べることを含む、疾患の治療剤をスクリーニングする方法を提供する。
【0054】
疾患は、例えば神経変性疾患又は癌である。
癌関連細胞は、上記例示のものを挙げることができる。
【0055】
疾患モデル非ヒト動物は、アルツハイマー病、ハンチントン病、パーキンソン病等の疾患のモデル動物(たとえばマウス、ラットなど)であり、これらの動物に候補薬剤を投与し、症状の軽減を観察することによって治療効果を確認することができる。モデル動物として、たとえばハンチントン病モデルマウス(J Neurol Sci, 231: 57-66 (Apr 15 2005))、アルツハイマー病モデルマウス(J. Clin. Invest., 116(3): 825-832 (2006))、パーキンソン病モデルマウス(PLoS Biol. 3(8): e303 (2005 August))等が知られている。腫瘍担持モデル非ヒト動物は、各種の腫瘍細胞株を動物の皮下に移植することによって作製することができる。
【0056】
TG2の阻害剤、IP3R1タンパク質のC末端側グルタミン残基の架橋の抑制剤、又はTG2の活性化剤の候補薬剤は、上記のような小分子、ペプチド、タンパク質、核酸、脂質、天然物、合成化合物などから選択可能であり、既存の又は新規の物質から選択されてよい。
【0057】
本発明はさらに、TG2によってIP3R1タンパク質のC末端側グルタミン残基と一級アミン含有分子とを結合することを含む、IP3R1のグルタミン残基の修飾方法を提供する。
【0058】
一級アミン含有分子は、モノアミン、ポリアミン、アミノ酸、リン脂質、ペプチド、タンパク質、核酸などを非限定的に含み、天然又は非天然由来のいずれの分子でもよい。後述の実施例では、テトラマー構造のIP3R1タンパク質のC末端側グルタミン残基が、IP3R1の別のサブユニットの調節ドメインのリジン残基と結合することを示すが、この事実から、他の一級アミン含有分子であってもIP3R1のC末端側グルタミン残基との結合がTG2によって触媒されることが明らかである。実際、IP3R1のC末端側グルタミン残基は、TG2の作用によって、ビオペンチルアミン(BP)、シスタミン(CA)、モノダンシルカダベリン(Dan-Cad)などの一級アミン含有分子と結合し、アミド基転移が生じた。
【実施例】
【0059】
以下に実施例を挙げて本発明をさらに説明するが、本発明はこれらの実施例に限定されないものとする。
<実験方法>
材料
N,N-ジメチル化カゼイン, すべてトランスのレチノイン酸(RA)及びカルバコール(CCh)をSigmaから購入した。Zwittergent 3-14, イオノマイシン及びカルパインをCalbiochemから得た。増強された化学ルミネッセンス(ECL)試薬及び西洋ワサビペルオキシダーゼ(HRP)-結合ストレプトアビジンをGE healthcareから購入した。5-(ビオチンアミド)ペンチルアミン(BP), 1,8-ビス-マレイイミド-ジエチレングリコール(BM)及びビス[スルホスクシンイミジル]スベレート(BS)をテルモ(Thermo)から購入した。IP3, fura-2及びfura-2 AMはDojindo (日本)から入手した。αTG2抗体(トランスグルタミナーゼII Ab-1, CUB 7402)をLabVisionから購入し、αLC3をMBLから購入した。精製TG2, チログロブリン及びリジル-エンドペプチダーゼ(Lep)を和光純薬から購入した。DMEM, ペニシリン-ストレプトマイシン混合溶液, HPLC用のアセトニトリル及びTriton X-100を半井テスクから購入した。牛胎児血清をJRH Biosciencesから入手した。SH-SY5Y及びU87-MG細胞系をAmerican Type Culture Collection (ATCC)から購入した。
【0060】
電気泳動によるIP3R1内のサブユニット間架橋の評価
マイクロウエーブ装置内で、0.1%SDS含有バッファにアガロースを溶解し、アガロースを加熱し手で回転し静かに混合した。1%アガロース溶液をアクリル板(Nihon Eido)上に注いだ。26レーンの櫛をアクリル板上に配置した。室温で30分後、装置を泳動バッファで充たし、櫛を注意深く取り出し、100Vで1〜2時間電気泳動を行った。泳動後すぐに、アガロースゲルを転写バッファ中に30分間置いた。分離した蛋白質をPVDF膜(Millipore)に転写した。分子マーカーは、牛チログロブリン(Th)を用いて作製した。5mg/ml Thは、4℃で一晩1mM BMを用いて架橋することによって前処理した。
【0061】
細胞培養
HeLa, SH-SY5Y, U87-MG及びCOS-7細胞を、10% (v/v)熱失活牛胎児血清、50U/mlペニシリン及び50μg/mlストレプトマイシンを補充したDulbecco改変Eagle培地(DMEM)中で培養した。すべての細胞を5% (v/v) CO2下で37℃に維持した。HeLa細胞及びSH-SY5Y細胞について20μMレチノイン酸(RA)を補充した分化培地を48時間おきに置換した。
【0062】
IP3R1のチャネル活性の測定
マウス小脳のP2/P3膜(3mg/ml蛋白質)を50mM HEPES-KOH、110mM KCl、10mM NaCl及び5mM KH2PO4を含有するバッファ(pH7.4)で3回洗浄してアミド転移反応用の酵素又は基質を除去した。小脳又はSH-SY5Y 細胞の10μlのP2/P3膜(2mg/ml蛋白質)を、石英キュベット中の、1mM 2-メルカプトエタノール、1μM CaCl2及び0.2μM Fura-2を含有する287μlのバッファに加えた。サンプルを、340及び380 nm励起光で発光し、510 nmで蛍光を測定した。P2/P3膜小胞中にCa2+を負荷するために、攪拌しながら3μlの100mM Mg-ATPをキュベットに添加した。15分後、IP3を添加した。fura-2のレシオ法測定を、30℃でCAF-110 (ジャスコ)又はF-2500分光光度計(日立)を用いて文献記載にしたがって行った(Zhou et al., 2007)。
【0063】
酵素によるアミド基転移
小脳膜を、50mM HEPES-NaOH、150mM NaCl、1mM EGTA及び2mM TCEPを含有するアミド基転移バッファ(pH7.5)で希釈して反応混合物を調製した。TG2によるアミド基転移を、小脳膜1mg/ml蛋白質、2mM CaCl2及び種々の濃度のTG2を含有する反応混合物を37℃で30分間インキュベーションすることによって行った。アミド基転移されたIP3Rは、説明がない場合にはすべて4C11を用いて検出された。
【0064】
蛋白質限定分解
カルパイン(Calbiochem)又はリジルエンドペプチダーゼ(Lep,和光純薬)による消化の前に、小脳膜又はTG2処理膜を、架橋バッファを用いて20,000×gでの遠心及び再懸濁によって3回洗浄した。種々の濃度のカルパイン又はLepと洗浄された膜とを37℃でインキュベーションすることによって消化を開始した。その混合物に1/15容量の100mM CaCl2を添加したのち、カルパインによる消化を開始した。
【0065】
TG2による組換えIP3R1のアミド基転移
3.5cmディッシュで培養されたCOS-7細胞を掻き取り器具で回収し、50mM HEPES-NaOH(pH7.5)、10mM KCl、2mM TCEP、1mM EDTA及びプロテアーゼ阻害剤(複数)を含むバッファ中でホモゲナイズした。800×gで回転して得た上清を、20,000×g、4℃で15分間遠心分離した。これによって得られた沈降物を前記バッファに懸濁し、Bradford法を用いてタンパク質濃度を決定した。それぞれのタンパク質濃度を調整し、5mM CaCl2及び1μg/ml TG2と一緒に30分間インキュベーションすることによって架橋を行った。
【0066】
ペプチド及びイソペプチドの分析
フォトダイオードアレイUV検出器を備えたHP1100システム(Agilent Technologies)を用いてHPLCを行った。得られた反応混合物を、10μl又は200μlループを介してC8カラム(Super-octyl TSK(登録商標)ゲル, 100mm×2.0mm(内径), 2μmレジン, 東ソー)に注入した。0.1% TFAを含有する10%から50%アセトニトリルの直線グラジエントを用いて200μl/分の流速で15分でペプチドを溶出した。2種類の発蛍光団を含むγ-分岐ペプチド(すなわち、FITC-Asn-Pro-Glu[γ-(TMR-cad)]-Gln-Pro-Ala(配列番号5))を酵素的に作製したが、このとき0.05mM TMR-cad、2.5mM CaCl2、25mM HEPES-NaOH(pH7.5)及び50μg/ml TG2を含有する溶液中で0.05mM FITC-Asn-Pro-Glu-Gln-Pro-Ala(配列番号6)をインキュベーションすることによって前記ペプチドを得た。TG2によるイソペプチドの加水分解を、F-2500装置中、10mM HEPES及び1mM CaCl2を含有する反応混合物を用いて30℃でFITC蛍光の変化に基づきモニターした。
【0067】
TG2によって脱アミド化されたマウスIP3R1の作製
小脳からのP2/P3画分(1mg/ml)を、TG2 (0, 1, 3, 10, 30, 100ng/ml)と一緒に、50mM HEPES-NaOH、150mM NaCl、1mM EGTA、3mM CaCl2及び2mM TCEPを含有するHEPESバッファ中、37℃、90分間インキュベーションした。
【0068】
HeLa細胞でのsiRNAによるノックダウン
siRNA(複数)とヒト陰性対照をB-bridgeから購入した。TG2 siRNAのヌクレオチド配列は、ggucaaugccgacguggua(配列番号7)であった。IP3R1ノックダウンを文献記載(Hattori et al., 2004)の方法で行った。リポフェクタミン2000(Invitrogen)を用いてsiRNAをHeLa細胞にトランスフェクションした。
【0069】
マウス小脳からのP2/P3画分の調製
小脳をddYマウス脳から取り出し、冷却したPBSで十分に洗浄し、10mM HEPES-NaOH, 0.32 Mスクロース, 1mM EDTA, 2mM TCEP及びプロテアーゼ阻害剤(0.2 mM PMSF, 10μMロイペプチン, 10μMペプスタチンA及び10μM E-64)から構成されるホモゲナイズバッファ(pH7.5)10容量中でホモゲナイズした。ホモゲネートを800×g、4℃で5分間遠心分離した。得られた上清を105,000×g、2〜4℃で60分間超遠心分離にかけた。沈降物を50mM HEPES-NaOH, 150mM NaCl, 1mM EDTA, 2mM TCEP及びプロテアーゼ阻害剤を含む再懸濁用バッファResB(pH7.5)に懸濁した。超遠心分離の上清をシトソル画分として使用した。この溶液のタンパク質濃度を見積もり、ResBにより3mg/mlに調整し、種々の実験に使用するまで液体窒素中ですばやく凍結した。
【0070】
IP3R1の化学的架橋
サブユニット間の化学的架橋を、25mM HEPES-NaOH, pH7.5, 75mM NaCl, 1mM TCEP, 0.5mM EDTA及び種々の濃度のBS又はBMを含む反応バッファ中でP2/P3画分(1.5mg/ml)をインキュベーションすることによって開始した。SDSサンプルバッファを添加して氷上30分間反応を停止し、得られたサンプルをAGE分析にかけた。P2/P3画分の膜を1 % (w/v)界面活性剤に可溶化し、20,000×g、4℃で15分間遠心分離した。500μM BS又は100μM BMを可溶化画分に添加し氷上で30分間インキュベーションした。
【0071】
アミド基転移活性の分画
ホモゲネートの超遠心分離の上清画分を、50mM NaCl含有10mM Trisバッファ(pH 7.5)で予め平衡化したアニオン交換カラム(TSKgel BioAssist Q, 50mm×4.6mm(内径), 東ソ)上にアプライした。架橋活性に富む画分を、50mMから750mM NaClの直線グラジエント、流速1ml/minを用いて溶出した。20〜30画分(1mlずつ)を回収し、架橋活性を測定した。
【0072】
TG2発現レベルの測定
SH-SY5Y細胞又はHeLa細胞を3.5cm又は6cmディッシュから掻き取り器具で回収し、SDSサンプルバッファ中で3分間煮沸し、SDS-PAGEにかけた。
【0073】
SH-SY5Y細胞由来のIP3R1中の架橋の評価
3.5cmディッシュから掻き取り器具で回収したSH-SY5Y細胞を55℃で15分間加熱し、5秒間音波処理を2回行った。IP3R1を4C11で検出した。
【0074】
TG2基質のアフィニティ精製
P2/P3画分(1 mg/mlタンパク質), 50mM HEPES-NaOH (pH 7.5), 150mM NaCl, 10mM 5-(ビオチンアミド)ペンチルアミン(BP), 1mM EGTA, 2mM CaCl2, 及び30ng/ml TGを含む反応溶液を37℃で30分間インキュベーションした。混合物を15,000rpm、4℃で30分間遠心分離し、ついで、50mM HEPES-NaOH (pH7.5)でペレットを洗浄した。1% CHAPSでタンパク質を可溶化し、アビジン-アガロースと混合した。1% CHAPS含有HEPESバッファで3回洗浄したのち、アビジン-アガロースに結合したビオチン取り込みタンパク質をSDS-PAGEにかけてCBB染色又はウエスタンブロッティングにかけた。CBB染色バンドを検出し、染色バンドを切り出し、ゲル内消化プロトコルによって消化し、MALDI TOF MS (ABI)を使用する質量分析によって分析した。
【0075】
プラスミド
C末端の130kDa断片をコードするcDNAを、哺乳動物発現ベクターpcDNA3.1zeo(+) (Invitrogen)中にサブクローニングした。PCR産物をNheI及びBamHIで消化し、pcDNA3.1zeo(+)中にサブクローニングした。突然変異体を構築するために、部位特異的突然変異誘発を、QuikChange II XL (Stratagene, La Jolla, CA)とプライマー(AATGTCAACCCAAACCAGCCGGCCTAG(配列番号8): Q2746N、ここで下線はコドン変更である。)を用いてC130を含むプラスミド上で行った。全ての変異体をDNA配列決定によって確認した。すべてのオリゴヌクレオチドの配列は要求に応じて入手可能である。
【0076】
NEM又は煮沸によるTG2の不活性化
文献記載に基づいてTG2を不活性化した(Folk, J. E. & Cole, P. W., J Biol Chem 241, 5518-25 (1966))。TG2を、20 mM HEPES-NaOH (pH7.5)中の5mM NEMと一緒に室温で30分間インキュベーションした。反応を、5 mM DTTと一緒に室温、30分間インキュベーションすることによって停止した。得られた混合物を、連続して2本のスピンカラム(BioRad)を通過させて遊離NEMを除去した。もう一つの不活性化TG2は20mM HEPES-NaOH (pH7.5)中で5分間煮沸することによって調製された。
【0077】
TG2活性の評価
TG2活性を、F-2500蛍光光度計(日立)中で測定した。このとき、N, N-ジメチルカゼインへのモノダンシル-カダベリンのTG2触媒取り込みの際の蛍光の増加速度をモニターした(Lorand, L. et al, Anal Biochem 44, 221-31 (1971))。TMR-cadのTG2中への自己取り込みは、37℃、30分間 1mM TMR-cad, 2mM CaCl2及び10μg/ml TG2をインキュベーションすることによって行った。蛍光強度をFluorImager595 (Amersham)を用いて検出した。
【0078】
2種類の発蛍光団を含む蛍光ペプチドの酵素的作製
N末端のαアミノ基にビオチン又はFITCが結合された合成ペプチドを林化成(大阪)から購入した。TG2によるペプチドアミド基転移のために、25mM HEPES-NaOH, pH7.5, 2.5mM CaCl2, 0.05mM TR-cad, 0.05mMペプチド、及び50μg/ml TG2若しくは50μg/ml 不活性化TG2のいずれかからなる反応混合物を37℃で10分間インキュベーションした。脱アミド化は、15/1容量のアミド基転移ペプチドを、1mM EGTA、20μg/ml TG2及び5mM CaCl2若しくはCaCl2不含のいずれか、を含む50mM HEPES-NaOH, pH7.5を含有する反応混合物中に加えて開始した。アミド基転移ペプチドの構造はナノESI-MS/MS分析(Aproscience Corp., 徳島)によって確認された。
【0079】
タンパク質アッセイ
P2/P3画分、シトソル画分又は精製酵素のタンパク質濃度をU-0080Dフォトダイオードアレイ分光光度計(日立)を用いて測定した。この装置はBradford法に従うものであり、また標準としてウシ血清アルブミンを用いた。
【0080】
脱アミド化IP3R1に対する特異抗体
マウスIP3R1中のQ2746の状態を解き明かすために、アミノ酸2738〜2749に対応する脱アミド化ペプチド(PPHMNVNPEQPA(配列番号9))を合成し、ペプチドのC末端に付加されたシステインを介してKLHに結合し、日本白ウサギに免疫した。生じたポリクローナル抗体を先ず、天然ペプチド(PPHMNVNPQQPA(配列番号10))を固定したアフィニティ樹脂にアプライし、素通り画分を、脱アミド化ペプチドを固定した樹脂にアプライした。脱アミド化ペプチドに対する特異抗体は100mMグリシン-HClバッファ(pH2.5)で溶出された。抗体の特異性をチェックするために、マウスIP3R1の組換えC末端タンパク質(これはGST融合蛋白質として発現された。)を調製した。PCR産物はGST融合ベクターpGEX-6Pに挿入された。
【0081】
IP3R1におけるアミド基転移の確認
SH-SY5Y細胞をトリプシンで処理し、均質に懸濁し、トリパンブルー染色を用いて計数したのち、10cmディッシュに0.3×106の密度でSH-SY5Y細胞を植え付け、20μM RA又はDMSOを含有する培養培地中でさらに6日間培養し、この間、2日おきに新鮮な培地が供給された。2 mM BPを培地に添加し、24時間後に細胞を掻き取り器具で回収した。コールドルーム内で1500rpm、5分間細胞を遠心分離し、さらに1500rpmで5分間回転することによってPBSで洗浄した。得られた細胞沈降物を液体窒素で急速に凍結し-80℃で保存した。開放チューブ内の凍結細胞に1%Triton X-100 (HE-T)含有HEPESバッファ0.5mlを加え、懸濁し、4℃で30分間回転した。MX-300遠心分離器(トミー)中15000rpm、4℃で15分間遠心分離した後の上清を、pep6-Ab (Nakade, S. et al, J Biol Chem 269, 6735-42 (1994))を結合したProtein A-Sepharose 4Bと混合し、4℃で2時間インキュベーションした。Sepharoseビーズを0.5 mlのHE-Tバッファで3回洗浄し、SDS-PAGEにかけた。免疫沈降したIP3R1を5%ゲルで分離し、PVDF膜に転写し、アビジン-HRP (1:2000希釈, GE healthcare)と一緒にインキュベーションした。ビオチン-アビジン複合体の量を、増強された化学ルミネッセンス(ECL)キット(GE healthcare)で検出した。
【0082】
SH-SY5Y細胞からのP2/P3画分の調製
10cmディッシュ中で培養した細胞を細胞掻き取り器具で回収し、遠心分離し、PBSで洗浄した。細胞を、200μlのホモゲナイズバッファ(pH7.5, 50mM HEPES-NaOH, 150mM NaCl, 1mM EDTA, 2mM TCEP, 及びプロテアーゼ阻害剤(0.2mM PMSF, 10μMロイペプチン, 10μMペプスタチンA, 及び10μM E-64)中に懸濁し、ハンディタイプの音波器UR-20P(トミー)を用いた音波処理によってホモゲナイズした。細胞溶解物を800×g、4℃で5分間遠心分離し、上清をTL100.4ローター(Beckman)中、100,000×g、4℃で60分間超遠心した。沈降物は、シトソル様培地又はHEPES-NaOHバッファ中に再懸濁された。
【0083】
SH-SY5Y細胞中の[Ca2+]iの測定
PBS中の3μM fura-2/AM (同仁)中で細胞を37℃1時間インキュベーションすることによって該細胞にfura-2を負荷した。細胞を回収し、トリパンブルー染色によって計数した。1500rpm、5分間の遠心分離のあと、HEPES緩衝生理食塩水(HBS)中に懸濁することにより細胞密度を調整し、濯ぎ、HBS中に細胞を再懸濁した。fura-2のレシオ法測定を、F-2500蛍光光度計(日立)を用いて30℃で行った。10μMカルバコール(carbachol; CCh)を添加することによって細胞を刺激した。
【0084】
<結果>
内因性酵素である組織トランスグルタミナーゼ(TG2)によるIP3R1のサブユニット間架橋
IP3Rはテトラマーとして機能するが、その巨大な質量(>1MDa)は、テトラマー構造を解析することを困難にさせている。したがって本発明者らは、IP3Rのテトラマー状態を明らかにするために新規のアガロースゲル電気泳動(AGE)法を開発した(図1)。化学架橋剤である1,8-ビス-マレイイミド-ジエチレングリコール(BM)及びビス[スルホスクシンイミジル]スベレート(BS)は、バンドのシフトにより判断されるように、IP3R1のサブユニットを架橋することができた(図1A)。Zwittergent 3-14 及びSDS(これらはともにIP3Rのサブユニット相互作用を破壊することが知られている。)は、バンドのシフトを阻害し(図1B)、開発したAGEがIP3Rサブユニット間の共有結合による架橋を評価するのに適することを確認した。
【0085】
次に、本発明者らは、AGEを生物学的サンプルに適用した。驚いたことに、本発明者らは、外因性架橋剤の非存在下でIP3R1がCa2+依存性の架橋を起こすことを見出した(図1C)。この知見は、脳シトソル内に内因性架橋剤が存在するはずであることを示した。本発明者らはアニオン交換クロマトグラフィーによる架橋活性を特徴づけることを試みた(図1D)。架橋活性は、クロマトグラム上約0.3M NaClと同等の画分中に富化されていた(図1E)。Ca2+依存性と溶出プロフィールにより、架橋がTGによって触媒される可能性が仮説された。そのあと、トランスグルタミナーゼ(TG)の阻害剤であるシスタミン(CA)を試験し、用量依存的な仕方でサブユニット間の架橋を明確に阻害することを観察した。精製した組織TG(TG2)は架橋を示した(図1F)。精製したIP3R1は精製TG2によって架橋されることが可能であり(図1G)、このことはIP3R1のサブユニット間の直接的な架橋を示している。この事実を確認するために、本発明者らは、siRNAによるTG2のノックダウンを使用した。TG2のノックダウンは、HeLa細胞ライセート中で内因性架橋活性を完全に抑制した(図1H)。HeLa細胞中のTG2はRAによって誘導されることが知られているため、RA処理した細胞中の架橋活性を調べた。RAによるTG2誘導により、明らかに架橋活性が増強された(図1I)。次に、本発明者らは、IP3R1が生細胞中でTG2によって架橋されうるかどうかを調べた。このとき本発明者らは神経芽腫SH-SY5Y細胞及びU87-MG細胞を使用した(図1J)。イオノマイシンで刺激されたSH-SY5Y細胞及びU87-MG細胞でIP3R1の架橋が観察された(図1K)。SH-SY5Y細胞中の架橋は、RA処理によるTG2レベルの誘導が原因していた。これらの結果は、TG2が、サブユニット間の架橋を触媒する内因性酵素であることを示している。
【0086】
TG2はIP3R1のチャネル活性を阻害する
IP3R1のチャネル活性に及ぼすTG2触媒による架橋の影響を評価するために、本発明者らは最初に、煮沸或いはN-エチルマレイイミド(NEM)処理による不活性TG2を作製した(図2A及び2B)。TG2がCa2+放出活性に影響するかどうかを決定するために、本発明者らは、マウス小脳からのP2/P3画分を用いてIP3により誘導されるCa2+放出活性を調べた。TG2による前処理は、ATP依存性Ca2+取り込みに影響することなくチャネル活性又は、Triton X-100によって見積もったCa2+の総プールを強く抑制し、この阻害は用量依存的であった(図2C)。煮沸TG2もNEM処理TG2もともに、チャネル活性を抑制し(図2C及び2D)、このことは、TG2による触媒活性がチャネル阻害に必須であることを示している。TG2依存性阻害はまったく再現可能であり、まとめたデータは、TG2のIC50値が34nMであることを示している(図2D)。
【0087】
次の疑問は、TG2によるIP3R1のアミド基転移がどのようにしてチャネル活性を阻害するかということであった。本発明者らは、TG2がIP3R1のグルタミン(Q)ドナー部位に取り込むことができるアミンドナーとして5-(ビオチンアミド)ファニルアミン(BP)を使用した。予想したとおり、BPはサブユニット間の架橋を阻害した(図2E)。精製IP3R1は、アビジン-HRPにより検出される強力なビオチンシグナルを示し(図2F)、このことは、BPがTG2によってIP3R1の中に直接取り込まれたことを示している。Ca2+放出アッセイによって、BP-取り込みがチャネル活性を完全に改善したことが示された(図2G)。
【0088】
TG2依存性阻害をさらに特徴づけるため、本発明者らは、IP3R1に取り込まれたビオチン部分に化学量論的に結合するアビジンのチャネル活性に対する影響を調べた。BPが取り込まれた膜にアビジンを添加することによってチャネル活性の用量依存的な阻害を見出した(図2H)。この知見は、Q-ドナー部位周辺の立体障害がTG2による阻害機序に重要であるという事実を強く支持している。生細胞でのTG2によるIP3Rのビオチン取り込みを確認するため、本発明者らはSH-SY5Y細胞を使用した。そして、本発明者らは、RAで分化されたSH-SY5Y生細胞中でのIP3R1の共有結合修飾を見出した(図2I)。RA処理は、IP3R1の発現レベルを検出可能に変化させなかったが、この事実はこれまでの報告(Mackrill et al., 1997)と一致した。本発明者らはまた、対照と比較して、RAで処理されたSH-SY5Y細胞から調製したIP3R1のチャネル活性の著しい減少を見出した(図2J及び2K)。さらに本発明者らは、CChによって誘導されるCa2+放出がRA処理SH-SY5Y細胞中で損なわれることを見出した(図2L)。合わせると、これらのデータはIP3R1がTG2の基質であること、並びにTG2がIP3R1を介してCa2+放出を阻害すること、と矛盾しない。数個の内因性アミンがTG2のアミンドナーであると報告されている(Dale et al., 2002; Iismaa et al., 2009; Walther et al., 2003)ので、TG2によるIP3R1の修飾の内因性アミンドナーを調べることには関心がある。
【0089】
C末端130kDaドメイン(C130)内のTG2によるサブユニット間架橋
Q-ドナーの正確な位置を決定するために、BPが取り込まれた精製IP3R1の限定消化を行った。アビジン-HRPを用いて検出された主バンドの見かけのサイズは130kDa及び85kDaであった(図3A)。4C11及び10A6の両方で検出された180kDa (N180)のサイズのN末端ドメイン(図3A)は、C末端の130kDaバンド(C130)又はC末端の85kDaバンド(C85)と比べてBPによってほとんど標識されていなかった(図3A)。これらのバンドはウエスタンブロッティングによってC末端断片として帰属された。さらに消化物は、矛盾なく、ビオチン標識バンドがC末端に相当し、4C11又は10A6によって検出された断片に相当しないことを示した(図4B)。次に本発明者らは、Q-ドナーがテトラマーIP3R1のサブユニット間架橋に関与しているかどうかを調べた。また、C末端結合抗体がTG2による架橋の形成を妨げるかどうかを調べた。IP3R1は、AGEによって判断されるように、TG2によって架橋された(図3C)が、18A10による前処理はTG2による架橋を阻害した(図3C)。この知見は、架橋がC末端の18A10エピトープ付近に位置していることを示している。さらに、本発明者らは、架橋されたIP3R1が、たとえそれが変性されたとしても18A10に対するその免疫反応性を喪失したことを見出した(図3D)。このことは、Q-ドナー部位が18A10認識にとって重要な残基であることを示した。TG2で前処理されたIP3R1の限定消化により、TG2によるC130及びC85の、しかしN180ではない、著しい減少が明示された(図4E)。変性後のリジルエンドペプチダーゼ(Lep)によるさらなる消化により、4C11でプローブされた断片に影響することなくC末端断片のTG2相関的減少が示された(図4F)。また、限定消化後の架橋の結果、C130の回収の顕著な減少が生じたが、一方、N180の量は、TG2によって変化しなかった(図4G)。これらの知見は、サブユニット間架橋がC130ドメイン内で起こるはずであるということを示している。
【0090】
Q2746はTG2の強力なQ-ドナーである
Q-ドナー部位を正確に決定するために、本発明者らは質量分析を行った。はじめに、本発明者らは、BPのin vitro取り込みによってTG2の内因性基質をプルダウンした。驚いたことに、300kDa付近にほぼ均質のバンドを見出した(図4A)。次に、本発明者らは、この300kDaタンパク質を同定するためにゲル内消化と質量分析を行った。断然最もよいスコアを示したデータがマウスIP3R1であったし、またその配列中のカバー率が2749個のアミノ酸の45%以上であり(図8)、このことから、IP3R1がP2P3膜中の小脳タンパク質のなかで好ましい基質であることが示された。BPの質量を計算し、C末端ペプチド(アミノ酸配列: IGLLGHLPPHMNVNPQQPA(配列番号9)が専らビオチン化されたことを見出した(図8)。この結果は、部分消化データ(図3)と矛盾しなかった。ウエスタンブロッティング分析により、IP3R1がビオチン化されたことを確認した(図4B)。本発明者らは、LC-MS及びそれに続くタンデム型質量分析(MS/MS)を用いて、ビオチン化ペプチドによる分画の際にTG2によって酵素的に修飾されるIP3R1の正確な残基を決定した。この分析によって、IP3R1中のグルタミン2746(Q2746)の1つのアミド基転移部位 (IGLLGHLPPHMNVNPQ*QPA(配列番号11); 図4C)が同定された。野生型ペプチドのMALDI MS/MSスペクトルと比較して、BP(311.1 Da)に対応した明瞭な質量シフトが修飾ペプチド中に検出された(図4C)。最近、TG2が、配列モチーフpQxP(又はT, S)l(p: 極性アミノ酸, x: 任意のアミノ酸, l: 脂肪族アミノ酸)(配列番号12)によって決定される基質特異性を有していることが報告された(Csosz et al., 2008)。NPQQPA配列(配列番号3)はこのモチーフと一致し、Q2746(しかしQ2747でない)がTG2の標的であることを確認した。
【0091】
次に本発明者らは、COS-7細胞中で過剰発現された組換えIP3R1タンパク質を用いてQ2746が架橋に必須であるかどうかを調べた。そのために本発明者らは、IP3R1(計算質量: 132.4kDa)のC末端ドメインを構築し、欠失変異体及びQ2746N変異体を作製した(図4D)。その結果、TG触媒反応のためのQ-ドナーがQ2746であることを確認した(図5D及び5E)。またアミノ酸1866〜1932に相当する領域もサブユニット架橋に必須であった(図5D)。この領域は2つのユニークなLys (K)-クラスター部位(NKKKD(配列番号13)及びRKKAKE(配列番号14))を有している。より短い断片(ES領域: アミノ酸2216〜2749)はテトラマーを形成するのに十分である(Sayers et al., 1997)ので、本発明者らは、このKクラスター領域(アミノ酸1866〜1932)がTG2によるサブユニット間架橋の間にK-ドナーとして作用するはずであると結論する。最近の論文では、このK-クラスター領域に相当する部位がユビキチン化可能であることが報告された(Sliter et al., 2008)。したがって、本発明者らのデータは、TG2による修飾もまたプロテアソーム分解過程と関連していることを示してしいる。
【0092】
TG2による架橋の切断と脱アミド化
本発明者らは、TG2がNPQQPA(配列番号3)と、アミンドナーであるTMR-cad (テトラメチルローダミン・カダベリン(tetramethylrhodamine cadaverine))との間の架橋を可逆的に切断するかどうかを調べた。NPQQPAペプチドはTMR-cadと反応して架橋生成物を生成した。この生成物は14分の保持時間のところで溶出したが、NPNQPA(配列番号4)ペプチドは溶出しなかった(図5A)。本発明者らは該生成物を精製し、その構造をナノMS/MS質量分析によって確認し、TG2による脱架橋反応にかけた(図5B)。前記生成物をTG2及びCaCl2とインキュベーションすることによってローダミンスペクトルをもつ生成物が生じたが、その保持時間はTMR-cadと一致した(図5B)。この反応はCa2+依存的であった。これらのデータから、TG2がCa2+依存的にNPQQPAペプチドとアミンドナーとの間のイソペプチド結合を切断可能であることが示された。
【0093】
リアルタイムでの切断をモニターするために、本発明者らは、蛍光共鳴エネルギー移動に基づく新しい蛍光プローブを作製した。すなわち、Q-ドナー及びアミンドナーとしてそれぞれFITC-結合NPQQPA (ピークY)及びTMR-cad (ピークX)を作製し使用した。TG2による酵素反応により、FITCスペクトルとローダミンスペクトル(ピークZ)の両方をもつ色原体が生じ、本発明者らは、得られた生成物を精製した(図5C)。この二蛍光含有ペプチドを用いて、リアルタイムで脱架橋反応を確認した。予想したとおり、TG2とCa2+の添加後にFITC蛍光の増強を観察し、そしてTG2による加水分解の間の良好な直線性を得た(図5D)。
【0094】
理論的見解は、イソペプチドの加水分解又はTG2による脱アミド化がGln位置でグルタミン酸(Glu: E)残基を生成するはずであることを示している(Iismaa et al., 2009)。TG2によってQ2746がGlu残基(E2746)に変換可能であるかどうかを試験するために、本発明者らはE2746含有ペプチドに対する特異抗体を作製した。アフィニティ精製によって、Q2746を認識しないE2746特異抗体(αE2746)を得ることを可能にした(図5E)。αE2746を用いて、本発明者らは、テトラマーIP3R1がTG2によって酵素的に脱アミド化されることを示す結果を得た(図5F; *)。小脳からのP2/P3画分(図5G)はαE2746によって検出されるシグナルを示すが、これはin vivoでIP3R1を修飾することができることを示している。本発明者らは、この抗体をHuntington舞踏病モデル(R6/2)(ポリグルタミン含有タンパク質が過剰発現される。)に使用した。本発明者らは、R6/2マウスで、野生型マウスと比べてαE2746シグナルが専ら増強されることを見出した(図5G)。脱アミド化によって生じたE2746の陰性電荷が、静電相互作用によってC末端と陽性電荷K-クラスター領域との間で誘引を引き起こす可能性がある。
【0095】
TG2及びIP3R1によるオートファジー調節
IP3R1はオートファジーに関与しているので(Vicencio et al., 2009)、本発明者らは次に、IP3R1によって調節されるオートファジー事象の調節におけるTG2の役割を調べた。本発明者らは、TG2 siRNAを用いてHeLa細胞での、TG2の除去と、それによる修飾IP3R1の欠損を確立した(図6A)。オートファジーマーカーである、相対量の脂質化LC3 (LC3-II) (Klionsky et al., 2008)は、LC3-I(LC3-IIの前駆体)と比較して、siRNAを使用するTG2の除去によって減少した(図6B)。これに対して、siRNAによるIP3R1の除去により、LC3−Iと比べてLC3-IIの著しい増加が生じた(図6B)。LC3-IIレベルに対するこれら逆作用は、今回の研究で証明されるように、TG2によるIP3R1の阻害と矛盾しない。
【0096】
TG2及びIP3R1の役割を証明するために、本発明者らはラパマイシン又はスペルミジンによって誘導されるオートファジーを調べた。4時間の1μMラパマイシン又は100μMスペルミジン処理により、対照siRNAでトランスフェクションされたHeLa細胞でLC3-IIの脂質化を誘導したが、TG2 siRNAはそれをブロックした(図6C)。IP3R1 siRNAは反対の作用を示したが、これは本発明者らの仮説の確認になった(図6D)。いずれの場合にも、p62 (オートファジー基質)はTG2又はIP3R1の除去の間に全く変化を示さなかった。これは、LC3-II by TG2又はIP3R1のノックダウンによるLC3-IIの定常状態レベルの変化が脂質化によるLC3-IのLC3-IIへの変換によるものであって分解過程の損害によるものではないことを示している。L-Glnは、LC3の脂質化を誘導しうる(図6E及び6F)。重要なことは、L-Glnによる脂質化がsiRNAを使用するTG2の除去によってブロックされたことであり(図6E)、このことは、TG2がL-Gln依存性のオートファジーシグナル伝達に関与していることを示している。本発明者らは、TG2の基質であるアミンドナーとしてのモノダンシル-カダベリン(Dan-cad)とIP3R1の阻害剤である2-APBの役割を調べ、LC3の脂質化を増強したことを見出した(図6F)。このとき、オートファジーの基質であるp62タンパク質の沈着はなかった。この知見は、TG2及びIP3R1が、LC3-IIの分解をブロックすることではなくLC3の脂質化を調節することを支持している。
【0097】
TG2及びIP3R1によるオートファジー調節を証明するために、本発明者らは、ノックダウン中のHeLa細胞の形態を調べた。対照siRNAを有するHeLa細胞は空胞化を示したが、TG2 siRNAでトランスフェクションされた細胞は空胞の損失を示した(図7A)。IP3R1 siRNAは、多数の空胞を生じさせた(図7A)。空胞化された細胞又は空胞化されなかった細胞の数の計数から(図7B)、図6に示されるように、空胞化に対するノックダウンの各影響は、オートファジーの間、LC3転換の影響と完全に一致する。
【0098】
本発明者らは、TG2及びIP3R1が細胞の保護又は死と相関するかどうかを調べた。IP3R1のノックダウンの場合、非接着細胞が顕微鏡下でかなり頻繁に観察された(データ示さず)。したがって、本発明者らは、LDHアッセイにより細胞死を評価した。LDH活性により、IP3R1のノックダウンとは逆に、TG2のノックダウンが細胞死を抑制することを示した(図7C)。このことから、明らかな空胞化とLC3の脂質化が、細胞の保護ではなく細胞死と相関することが示された。
【0099】
<考察>
本発明者らの知識によれば、これは、IP3R1がTG2の新規な基質であること、及びTG2が、テトラマーIP3R1の酵素的修飾によってIP3R1のチャネル活性を阻害することを示す最初の研究である。本発明者らは、C末端が近傍のIP3R1サブユニットに近い、及び、TG2が近接するサブユニットに該C末端を共有結合によって結合させ、チャネルを、閉じた立体配置に固定する、と結論付ける(図7)。
【0100】
これまでの報告では、C末端尾部がチャネル活性に重要であること(Nakade et al., 1991; Uchida et al., 2003)、並びに、C末端及びKクラスター領域の付近で相互作用する、チトクロムcやBcl-2などの、多くの調節因子がチャネル活性を調節すること(Boehning et al., 2003; Foskett et al., 2007; Rong et al., 2008)が実証されている。本発明者らは今回、これらの調節因子がC末端付近のサブユニット界面に結合して、他のオリゴマーチャネルに類似したサブユニット立体配置を制御することが可能であることを示す。ここで、他のオリゴマーチャネルには、例えばアセチルコリンレセプター、K-チャネル及びグルタミン酸チャネルが含まれ、それらについてサブユニットの立体配置はゲートの動作や調節に関与している(Changeux and Edelstein, 1998)。IP3R1のESドメイン(アミノ酸2216〜2749)は、テトラマー形成に十分である(Sayers et al., 1997)が、しかし、テトラマーESチャネルやテトラマーcasp-チャネル(アミノ酸1892〜2749)は常にそのチャネルを開いており、このことは、調節ドメインがチャネルドメインを閉じたままにしておく必要があることを示している(Nakayama et al., 2004)。このように、ESチャネル及びcaspチャネルは開いた状態で固有の立体配置を維持し、並びに、調節ドメインがサブユニットを閉じた立体配置に強制的にもたらすことができることが想起される。IP3RはCa2+を必要とし、Ca2+は調節ドメインを含む細胞質ドメインを動かすことによってコンフォメーション変化を誘導し(Anyatonwu and Joseph, 2009; Hamada et al., 2002; Hamada et al., 2003)、チャネルと、K-クラスターの近傍に位置するCa2+-センサー用の重要な残基を開く(Foskett et al., 2007)。したがって、Ca2+依存性のコンフォメーション変化が、IP3結合からの刺激によってチャネルを開かせるサブユニット立体配置を自由にすることが提案される。おそらく、このことはCa2+が共アドニストとしてチャネルのゲーティングに必須であることを示している。
【0101】
レチノイン酸(RA)は、SH-SY5Y及びHeLa細胞中でTG2によるIP3R1の修飾を誘導するが、このことは、RAが新規のRAシグナル伝達経路に関与していることを示している。脳では、RAはLTP、LTD (Chiang et al., 1998)及びシナプス可塑性(Aoto et al., 2008)をレチノイドレセプターを介して制御している。本発明者らの知見により、IP3R1が、RAシグナル伝達によって神経機能を調節することができる可能性が示唆された。その理由は、IP3R1によって調節されるIP3-Ca2+シグナル伝達がLTP及びLTDを含む種々の神経機能に関与しているからである(Harnett et al., 2009; Nishiyama et al., 2000)。TG2は、SH-SY5Y細胞(Tucholski et al., 2001)やHL60細胞(Antonyak et al., 2001)で、RA依存的分化を調節することが示されている。興味深いことに、 SH-SY5Y細胞内でのTG2の安定な発現がRAの不在下で神経表現型に自然発生的分化を誘導する(Tucholski et al., 2001)。本発明者らは、TG2がIP3R1修飾によってレチノイドシグナル伝達を調節することができる新しい経路を提案する。RAシグナル伝達は神経の分化や変性に重要である(Maden, 2007)。すなわち、今回の知見は、RAシグナル伝達の分子機構に新しい見識を提供することができる。
【0102】
本発明者らの研究により、TG2とIP3R1がともに、オートファジーの調節に関与しており、HeLa細胞でTG2の役割がIP3R1の役割と反対であることを実証しており、これはTG2がIP3R1機能を損なうという知見を支持している。このことは、オートファジーがCa2+シグナル伝達(Hoyer-Hansen et al., 2007; Williams et al., 2008)及びIP3R(Criollo et al., 2007; Vicencio et al., 2009)によって調節されるという従来の概念と矛盾しない。さらに、本発明者らの今回の研究は、siRNAによるTG2の除去によってラパマイシン又はL-Gln-依存性LC3脂質化がブロック(阻止)されること、並びに、TG2の基質である、Dan-cad及び、IP3R1のGln-ドナー含有C末端が、LC3-IIレベルを増加させることを明らかにした。更に又、細胞やマウス内のIP3R1を慢性的に欠損させると、 LC3-IIの減少が見られた。これらのデータは、TG2によって修飾されたIP3R1が新しいオートファジーシグナルとして機能するという代替的な考えを生じさせる。オートファジーは、内因性のオートファジーシグナルとしてのアミノ酸レベルに感受的であり、特に、アミノ酸トランスポーター(SLC1A5及びSLC7A5)を介するL-Gln取り込みと輸送がmTOR依存性のオートファジーシグナル伝達に重要である(Nicklin et al., 2009)。興味深いことに、このタイプの輸送はNγ-修飾Gln誘導体によってブロックされることが可能であった(Esslinger et al., 2005)。L-Glnによるオートファジー調節がTG2のノックダウンによって止められることを示す本発明者らのデータと合わせると、リソソーム分解後に修飾されたIP3R1から遊離するγ-グルタミルアミンがGlnの輸送及び/又は代謝を妨げてオートファジーを制御するはずであると提案される。poly-Glnタンパク質などのいくつかの病原性タンパク質は、Gln-ドナーであるTG2の強力な基質であり、また、γ-グルタミルアミンがHuntington病やAlzheimer病で増大する(Jeitner et al., 2009)。TG2によって修飾されるGlnがオートファジーシグナル伝達に関与することができるかどうかを決定することは興味あることである。本発明者らの結果は、アミン含有Dan-cad又は2-APBがLC3の脂質化を強く高めることを示しており、また、最近の文献は、TG2の内因性アミンドナーであるスペルミジンもオートファジーを誘導することを報告している(Eisenberg et al., 2009)ことから、TG2依存性のオートファジーシグナル伝達の考えが支持される。オートファジーの調節不能は、神経変性疾患に関係するタンパク質封入体の沈着に重要である(Rubinsztein et al., 2007)。本発明者らのデータは、TG2によるGln修飾に光を与え、これは上記の疾患のための薬剤開発の新規の治療標的になりうることを示している。
【0103】
さらにまた、本発明者らの研究により、TG2とIP3R1がともに、オートファジーの調節に関与しており、HeLa細胞でTG2の役割がIP3R1の役割と反対であることを実証しており、これはTG2がIP3R1機能を損なうという知見を支持している。本発明者らの結果は、p62含量から評価されるように、TG2及びIP3R1が、リソソーム分解ではなくLC3の脂質化と空胞化を制御していることを示した。IP3R1の除去によってLC3の脂質化と空胞化を増強したことを示す本発明者らのデータと合わせると、固有のIP3R1がオートファジーを伴う空胞化を抑制するはずであり(図7C)、従来の概念(Criollo et al., 2007; Vicencio et al., 2009)と矛盾しない。本発明者らは、細胞死に直結する、TG2及びIP3R1によるオートファジー調節を今回提案する(図7D)。この新しいモデルの態様は、TG2 siRNAによって明らかにされるTG2によるオートファジー空胞化の阻害解除(disinhibition)、並びに、IP3R1によるオートファジー細胞死のTG2依存性制御である。このモデルは、明らかな空胞化、LC3の脂質化及びTG2又はIP3R1のノックダウン時の細胞死、の細胞応答だけでなく、疾患状態下の神経変性の分子機構をも整然と解明する。Huntington病及びAlzheimer病では、TGが活性化され(Iismaa et al., 2009)、及び、TG-産物であるγ-グルタミルアミンが増加する(Jeitner et al., 2009)。本発明者らは、修飾IP3R1が、R6/2マウスで専ら増大したことを確認した(図10)。TG2の活性化が酵素的にIP3R1を修飾し、そして、Ca2+シグナル伝達又はベクリン-1相互作用の制御不能によってオートファジーを伴う空胞化を抑制解除する(Vicencio et al., 2009)。正常の条件では、オートファジーは、リソソーム分解と結びつくことによって、折り畳みを誤ったタンパク質や栄養飢餓から細胞を保護する。しかしながら、TG2は、オートファジー空胞の過剰負荷を行い、したがって神経変性疾患での細胞死を誘導するはずである。重要なことは、本発明者らの知見が、TG2によるIP3R1の修飾が、細胞の保護から細胞死にオートファジーの固有の役割に切り替える重要な事象であるという考えに導くことである(図7D)。このことは、Dictyostelium真核モデルでのランダムな突然変異誘発によって示されるオートファジーによる細胞死のシグナル伝達に対する分子的洞察を提供する(Lam et al., 2008)。オートファジーは、神経変性疾患でのタンパク質封入体のクリアランスに関与しているので(Rubinsztein et al., 2007)、本発明者らの考えは、TG2によるIP3R1の修飾に光を与え、これはこれら疾患のための薬剤開発の新規の治療標的になりうることを示している。
【0104】
まとめると、本発明者らは、IP3R1がTG2の新しい基質であり、TG2が酵素的にIP3R1の機能を損なうことを実証した。アミド基転移部位は、Q2746であると決定され、この部位でTG2もまた脱アミド化を触媒する。TG2によるサブユニット間架橋には、隣接のサブユニットのK-クラスター領域が必要であり、このことは、TG2による共有結合によりサブユニットの立体配置を閉じた状態に固定することを示している。TG2によって触媒されるIP3R1の修飾は、RAによって分化される神経芽腫で増強された。TG2はIP3R1と反対の方向にオートファジー事象と細胞死を制御したが、これは、TG2によるIP3R1の修飾がオートファジーの際の栄養飢餓からの細胞の生存を細胞死に変換する重要なシグナル伝達であることを支持している。
【0105】
参考文献
AbdAlla, S., Lother, H., Langer, A., el Faramawy, Y., and Quitterer, U. (2004). Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell 119, 343-354.
Antonyak, M. A., Singh, U. S., Lee, D. A., Boehm, J. E., Combs, C., Zgola, M. M., Page, R. L., and Cerione, R. A. (2001). Effects of tissue transglutaminase on retinoic acid-induced cellular differentiation and protection against apoptosis. J Biol Chem 276, 33582-33587.
Anyatonwu, G., and Joseph, S. K. (2009). Surface accessibility and conformational changes in the N-terminal domain of type I inositol trisphosphate receptors: studies using cysteine substitution mutagenesis. J Biol Chem 284, 8093-8102.
Aoto, J., Nam, C. I., Poon, M. M., Ting, P., and Chen, L. (2008). Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60, 308-320.
Bailey, C. D., and Johnson, G. V. (2005). Tissue transglutaminase contributes to disease progression in the R6/2 Huntington's disease mouse model via aggregate-independent mechanisms. J Neurochem 92, 83-92.
Battaglia, G., Farrace, M. G., Mastroberardino, P. G., Viti, I., Fimia, G. M., Van Beeumen, J., Devreese, B., Melino, G., Molinaro, G., Busceti, C. L., et al. (2007). Transglutaminase 2 ablation leads to defective function of mitochondrial respiratory complex I affecting neuronal vulnerability in experimental models of extrapyramidal disorders. J Neurochem 100, 36-49.
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1, 11-21.
Boehning, D., Patterson, R. L., Sedaghat, L., Glebova, N. O., Kurosaki, T., and Snyder, S. H. (2003). Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol 5, 1051-1061.
Candi, E., Schmidt, R., and Melino, G. (2005). The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 6, 328-340.
Changeux, J. P., and Edelstein, S. J. (1998). Allosteric receptors after 30 years. Neuron 21, 959-980.
Chiang, M. Y., Misner, D., Kempermann, G., Schikorski, T., Giguere, V., Sucov, H. M., Gage, F. H., Stevens, C. F., and Evans, R. M. (1998). An essential role for retinoid receptors RARbeta and RXRgamma in long-term potentiation and depression. Neuron 21, 1353-1361.
Criollo, A., Maiuri, M. C., Tasdemir, E., Vitale, I., Fiebig, A. A., Andrews, D., Molgo, J., Diaz, J., Lavandero, S., Harper, F., et al. (2007). Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 14, 1029-1039.
Csosz, E., Bagossi, P., Nagy, Z., Dosztanyi, Z., Simon, I., and Fesus, L. (2008). Substrate preference of transglutaminase 2 revealed by logistic regression analysis and intrinsic disorder examination. J Mol Biol 383, 390-402.
Dale, G. L., Friese, P., Batar, P., Hamilton, S. F., Reed, G. L., Jackson, K. W., Clemetson, K. J., and Alberio, L. (2002). Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 415, 175-179.
Eisenberg, T., Knauer, H., Schauer, A., Buttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., Ring, J., Schroeder, S., Magnes, C., Antonacci, L., et al. (2009). Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11, 1305-1314.
Esslinger, C. S., Cybulski, K. A., and Rhoderick, J. F. (2005). Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg Med Chem 13, 1111-1118.
Foskett, J. K., White, C., Cheung, K. H., and Mak, D. O. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87, 593-658.
Hamada, K., Miyata, T., Mayanagi, K., Hirota, J., and Mikoshiba, K. (2002). Two-state conformational changes in inositol 1,4,5-trisphosphate receptor regulated by calcium. J Biol Chem 277, 21115-21118.
Hamada, K., Terauchi, A., and Mikoshiba, K. (2003). Three-dimensional rearrangements within inositol 1,4,5-trisphosphate receptor by calcium. J Biol Chem 278, 52881-52889.
Harnett, M. T., Bernier, B. E., Ahn, K. C., and Morikawa, H. (2009). Burst-timing-dependent plasticity of NMDA receptor-mediated transmission in midbrain dopamine neurons. Neuron 62, 826-838.
Hattori, M., Suzuki, A. Z., Higo, T., Miyauchi, H., Michikawa, T., Nakamura, T., Inoue, T., and Mikoshiba, K. (2004). Distinct roles of inositol 1,4,5-trisphosphate receptor types 1 and 3 in Ca2+ signaling. J Biol Chem 279, 11967-11975.
Hovhannisyan, Z., Weiss, A., Martin, A., Wiesner, M., Tollefsen, S., Yoshida, K., Ciszewski, C., Curran, S. A., Murray, J. A., David, C. S., et al. (2008). The role of HLA-DQ8 beta57 polymorphism in the anti-gluten T-cell response in coeliac disease. Nature 456, 534-538.
Hoyer-Hansen, M., Bastholm, L., Szyniarowski, P., Campanella, M., Szabadkai, G., Farkas, T., Bianchi, K., Fehrenbacher, N., Elling, F., Rizzuto, R., et al. (2007). Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 25, 193-205.
Iismaa, S. E., Mearns, B. M., Lorand, L., and Graham, R. M. (2009). Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev 89, 991-1023.
Jeitner, T. M., Pinto, J. T., Krasnikov, B. F., Horswill, M., and Cooper, A. J. (2009). Transglutaminases and neurodegeneration. J Neurochem 109 Suppl 1, 160-166.
Klionsky, D. J., Abeliovich, H., Agostinis, P., Agrawal, D. K., Aliev, G., Askew, D. S., Baba, M., Baehrecke, E. H., Bahr, B. A., Ballabio, A., et al. (2008). Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151-175.
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27-42.
Lorand, L., and Graham, R. M. (2003). Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 4, 140-156.
Mackrill, J. J., Challiss, R. A., O'Connell D, A., Lai, F. A., and Nahorski, S. R. (1997). Differential expression and regulation of ryanodine receptor and myo-inositol 1,4,5-trisphosphate receptor Ca2+ release channels in mammalian tissues and cell lines. Biochem J 327 (Pt 1), 251-258.
Maden, M. (2007). Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci 8, 755-765.
Mastroberardino, P. G., Iannicola, C., Nardacci, R., Bernassola, F., De Laurenzi, V., Melino, G., Moreno, S., Pavone, F., Oliverio, S., Fesus, L., and Piacentini, M. (2002). 'Tissue' transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ 9, 873-880.
Nagy, L., Saydak, M., Shipley, N., Lu, S., Basilion, J. P., Yan, Z. H., Syka, P., Chandraratna, R. A., Stein, J. P., Heyman, R. A., and Davies, P. J. (1996). Identification and characterization of a versatile retinoid response element (retinoic acid receptor response element-retinoid X receptor response element) in the mouse tissue transglutaminase gene promoter. J Biol Chem 271, 4355-4365.
Nakade, S., Maeda, N., and Mikoshiba, K. (1991). Involvement of the C-terminus of the inositol 1,4,5-trisphosphate receptor in Ca2+ release analysed using region-specific monoclonal antibodies. Biochem J 277 (Pt 1), 125-131.
Nakayama, T., Hattori, M., Uchida, K., Nakamura, T., Tateishi, Y., Bannai, H., Iwai, M., Michikawa, T., Inoue, T., and Mikoshiba, K. (2004). The regulatory domain of the inositol 1,4,5-trisphosphate receptor is necessary to keep the channel domain closed: possible physiological significance of specific cleavage by caspase 3. Biochem J 377, 299-307.
Nanda, N., Iismaa, S. E., Owens, W. A., Husain, A., Mackay, F., and Graham, R. M. (2001). Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem 276, 20673-20678.
Nicklin, P., Bergman, P., Zhang, B., Triantafellow, E., Wang, H., Nyfeler, B., Yang, H., Hild, M., Kung, C., Wilson, C., et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521-534.
Nishiyama, M., Hong, K., Mikoshiba, K., Poo, M. M., and Kato, K. (2000). Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408, 584-588.
Rong, Y. P., Aromolaran, A. S., Bultynck, G., Zhong, F., Li, X., McColl, K., Matsuyama, S., Herlitze, S., Roderick, H. L., Bootman, M. D., et al. (2008). Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Mol Cell 31, 255-265.
Rubinsztein, D. C., Gestwicki, J. E., Murphy, L. O., and Klionsky, D. J. (2007). Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6, 304-312.
Sayers, L. G., Miyawaki, A., Muto, A., Takeshita, H., Yamamoto, A., Michikawa, T., Furuichi, T., and Mikoshiba, K. (1997). Intracellular targeting and homotetramer formation of a truncated inositol 1,4,5-trisphosphate receptor-green fluorescent protein chimera in Xenopus laevis oocytes: evidence for the involvement of the transmembrane spanning domain in endoplasmic reticulum targeting and homotetramer complex formation. Biochem J 323 (Pt 1), 273-280.
Selkoe, D. J. (2004). Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol 6, 1054-1061.
Sliter, D. A., Kubota, K., Kirkpatrick, D. S., Alzayady, K. J., Gygi, S. P., and Wojcikiewicz, R. J. (2008). Mass spectrometric analysis of type 1 inositol 1,4,5-trisphosphate receptor ubiquitination. J Biol Chem 283, 35319-35328.
Thacher, S. M., and Rice, R. H. (1985). Keratinocyte-specific transglutaminase of cultured human epidermal cells: relation to cross-linked envelope formation and terminal differentiation. Cell 40, 685-695.
Tucholski, J., Lesort, M., and Johnson, G. V. (2001). Tissue transglutaminase is essential for neurite outgrowth in human neuroblastoma SH-SY5Y cells. Neuroscience 102, 481-491.
Tucholski, J., Roth, K. A., and Johnson, G. V. (2006). Tissue transglutaminase overexpression in the brain potentiates calcium-induced hippocampal damage. J Neurochem 97, 582-594.
Uchida, K., Miyauchi, H., Furuichi, T., Michikawa, T., and Mikoshiba, K. (2003). Critical regions for activation gating of the inositol 1,4,5-trisphosphate receptor. J Biol Chem 278, 16551-16560.
Verma, A., Wang, H., Manavathi, B., Fok, J. Y., Mann, A. P., Kumar, R., and Mehta, K. (2006). Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res 66, 10525-10533.
Vicencio, J. M., Ortiz, C., Criollo, A., Jones, A. W., Kepp, O., Galluzzi, L., Joza, N., Vitale, I., Morselli, E., Tailler, M., et al. (2009). The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 16, 1006-1017.
Walther, D. J., Peter, J. U., Winter, S., Holtje, M., Paulmann, N., Grohmann, M., Vowinckel, J., Alamo-Bethencourt, V., Wilhelm, C. S., Ahnert-Hilger, G., and Bader, M. (2003). Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 115, 851-862.
Williams, A., Sarkar, S., Cuddon, P., Ttofi, E. K., Saiki, S., Siddiqi, F. H., Jahreiss, L., Fleming, A., Pask, D., Goldsmith, P., et al. (2008). Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 4, 295-305.
Zhou, H., Iwasaki, H., Nakamura, T., Nakamura, K., Maruyama, T., Hamano, S., Ozaki, S., Mizutani, A., and Mikoshiba, K. (2007). 2-Aminoethyl diphenylborinate analogues: selective inhibition for store-operated Ca2+ entry. Biochem Biophys Res Commun 352, 277-282.
【産業上の利用可能性】
【0106】
本発明により、神経変性疾患や癌などのTG2とIP3R1が関係する疾患の治療や治療剤の開発のために有用である。
【配列表フリーテキスト】
【0107】
配列番号12 1位:任意のアミノ酸残基
配列番号12 3位:任意のアミノ酸残基
配列番号12 4位:Pro, Thr又はSer残基
配列番号12 5位:脂肪族アミノ酸残基
【特許請求の範囲】
【請求項1】
細胞においてトランスグルタミナーゼ2(TG2)によってIP3レセプター1(IP3R1)タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を形成してIP3R1生物活性を抑制し、それによってオートファジーを誘導して該細胞の細胞死を引き起こすことを含む、細胞増殖の抑制方法。
【請求項2】
細胞が腫瘍細胞である、請求項1に記載の方法。
【請求項3】
疾患関連細胞又は疾患モデル非ヒト動物において、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を指標にして該架橋によって引き起こされるオートファジーと該疾患との相関関係を測定し、さらに該疾患が該IP3R1のサブユニット間架橋に起因するときにTG2の阻害剤又は活性化剤を候補薬剤として該モデル非ヒト動物に投与して治療効果を調べることを含む、疾患の治療剤をスクリーニングする方法。
【請求項4】
疾患が神経変性疾患又は癌である、請求項3に記載の方法。
【請求項5】
TG2及びIP3R1の生物活性をともに含む細胞を用いてそのIP3R1活性を、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋又は該架橋の脱アミド化を指標にして測定することを含む、細胞でのIP3R1活性の測定方法。
【請求項6】
細胞が疾患関連細胞である、請求項5に記載の方法。
【請求項7】
TG2の活性を抑制することによるIP3R11タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の抑制によってオートファジーを抑制することを含む、神経変性疾患の治療方法。
【請求項8】
IP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の増強剤を有効成分として含む、癌の治療用組成物。
【請求項9】
IP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の抑制剤を有効成分として含む、神経変性疾患の治療用組成物。
【請求項10】
TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を検出することを含む、IP3R1修飾の検出方法。
【請求項11】
TG2によるIP3R1タンパク質のサブユニット間アミド基転移を介する該タンパク質のサブユニット間架橋を測定することを含む、IP3R1タンパク質チャネル活性のTG2阻害作用を測定する方法。
【請求項12】
TG2によるIP3R1タンパク質のサブユニット間架橋の脱アミド化を測定することを含む、TG2触媒活性の測定方法。
【請求項13】
測定が、SDS含有アガロースゲル電気泳動及び特異抗体との交差反応を用いて行うことを含む、請求項10又は11に記載の方法。
【請求項14】
配列番号1又は配列番号2のアミノ酸配列のそれぞれ2746位又は2707位のC末端側グルタミン残基(Q)を介してサブユニット間架橋されたテトラマーIP3R1タンパク質。
【請求項15】
TG2によってIP3R1タンパク質のC末端側グルタミン残基と一級アミン含有分子とを結合することを含む、IP3R1のグルタミン残基の修飾方法。
【請求項1】
細胞においてトランスグルタミナーゼ2(TG2)によってIP3レセプター1(IP3R1)タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を形成してIP3R1生物活性を抑制し、それによってオートファジーを誘導して該細胞の細胞死を引き起こすことを含む、細胞増殖の抑制方法。
【請求項2】
細胞が腫瘍細胞である、請求項1に記載の方法。
【請求項3】
疾患関連細胞又は疾患モデル非ヒト動物において、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を指標にして該架橋によって引き起こされるオートファジーと該疾患との相関関係を測定し、さらに該疾患が該IP3R1のサブユニット間架橋に起因するときにTG2の阻害剤又は活性化剤を候補薬剤として該モデル非ヒト動物に投与して治療効果を調べることを含む、疾患の治療剤をスクリーニングする方法。
【請求項4】
疾患が神経変性疾患又は癌である、請求項3に記載の方法。
【請求項5】
TG2及びIP3R1の生物活性をともに含む細胞を用いてそのIP3R1活性を、TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋又は該架橋の脱アミド化を指標にして測定することを含む、細胞でのIP3R1活性の測定方法。
【請求項6】
細胞が疾患関連細胞である、請求項5に記載の方法。
【請求項7】
TG2の活性を抑制することによるIP3R11タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の抑制によってオートファジーを抑制することを含む、神経変性疾患の治療方法。
【請求項8】
IP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の増強剤を有効成分として含む、癌の治療用組成物。
【請求項9】
IP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋の抑制剤を有効成分として含む、神経変性疾患の治療用組成物。
【請求項10】
TG2によるIP3R1タンパク質のC末端側グルタミン残基を介するサブユニット間架橋を検出することを含む、IP3R1修飾の検出方法。
【請求項11】
TG2によるIP3R1タンパク質のサブユニット間アミド基転移を介する該タンパク質のサブユニット間架橋を測定することを含む、IP3R1タンパク質チャネル活性のTG2阻害作用を測定する方法。
【請求項12】
TG2によるIP3R1タンパク質のサブユニット間架橋の脱アミド化を測定することを含む、TG2触媒活性の測定方法。
【請求項13】
測定が、SDS含有アガロースゲル電気泳動及び特異抗体との交差反応を用いて行うことを含む、請求項10又は11に記載の方法。
【請求項14】
配列番号1又は配列番号2のアミノ酸配列のそれぞれ2746位又は2707位のC末端側グルタミン残基(Q)を介してサブユニット間架橋されたテトラマーIP3R1タンパク質。
【請求項15】
TG2によってIP3R1タンパク質のC末端側グルタミン残基と一級アミン含有分子とを結合することを含む、IP3R1のグルタミン残基の修飾方法。
【図1−1】


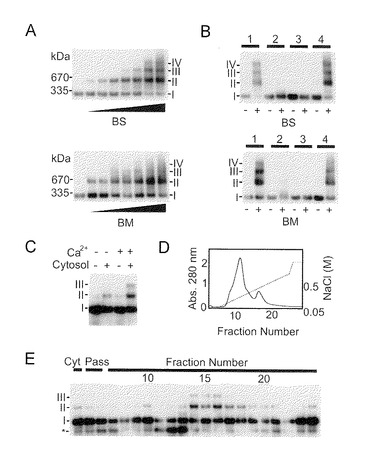
【図1−2】
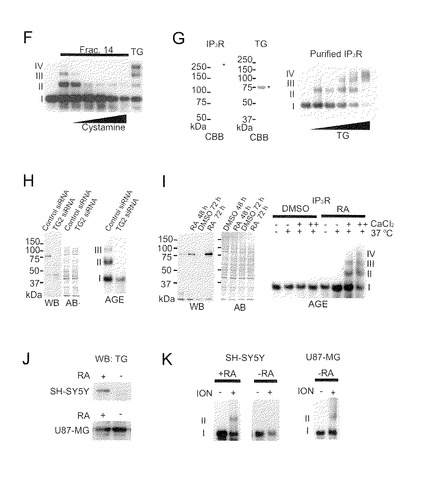
【図2−1】
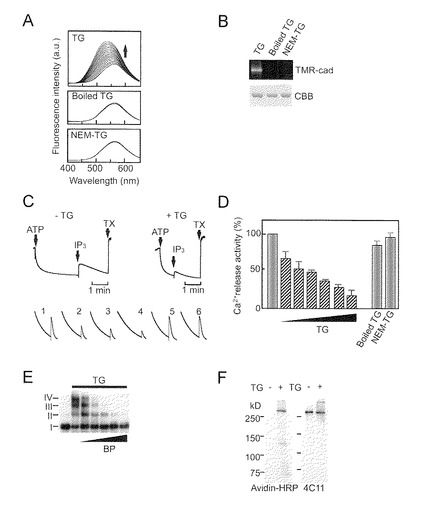
【図2−2】
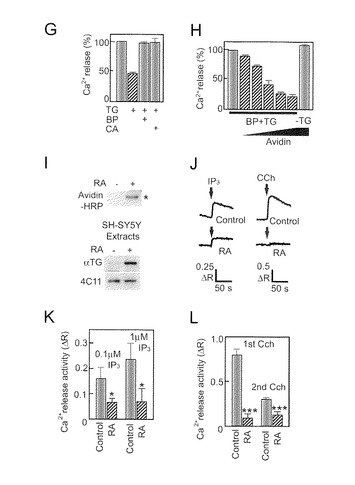
【図3−1】
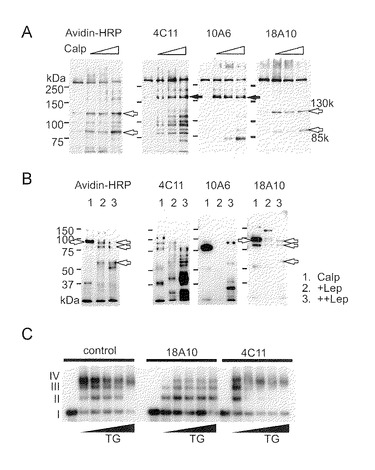
【図3−2】
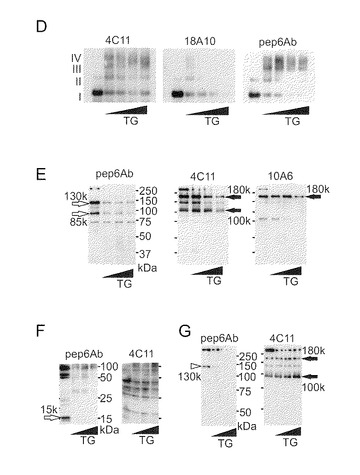
【図4−1】
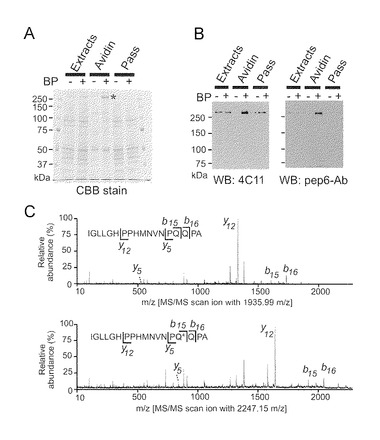
【図4−2】
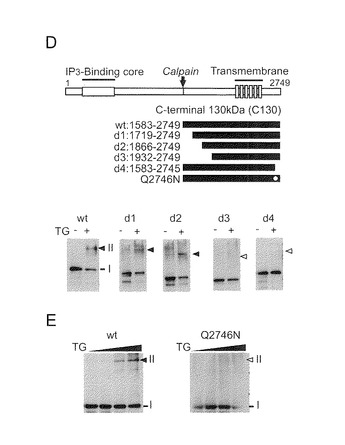
【図5−1】
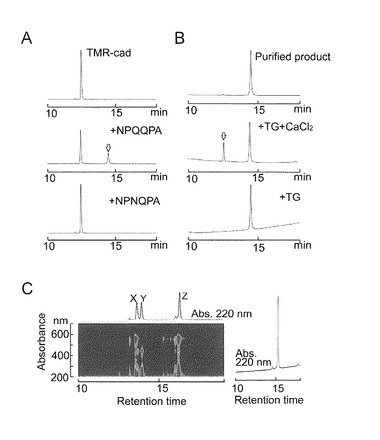
【図5−2】
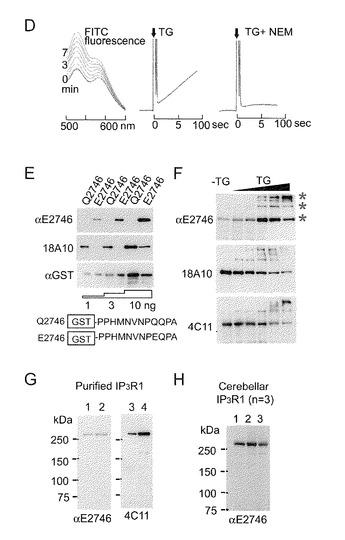
【図6−1】
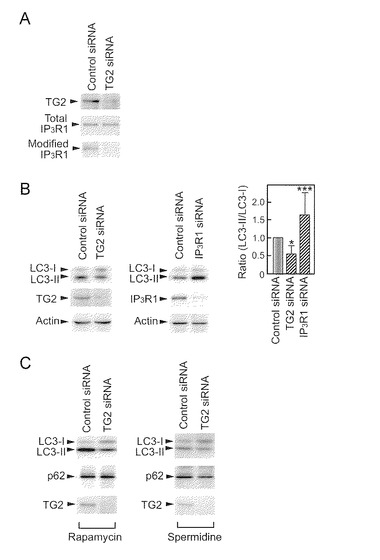
【図6−2】
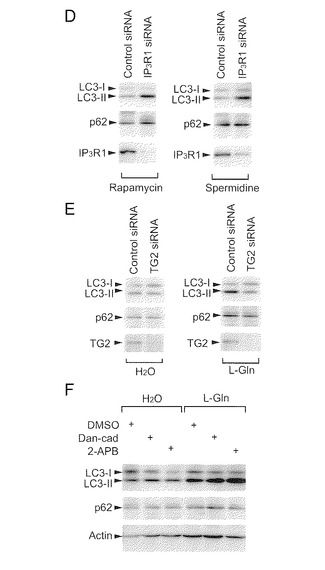
【図7−1】
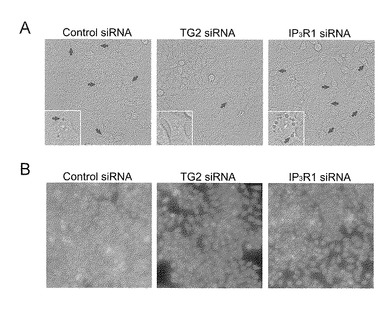
【図7−2】
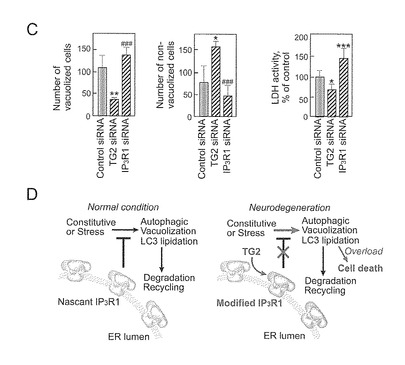
【図8】

【図9】
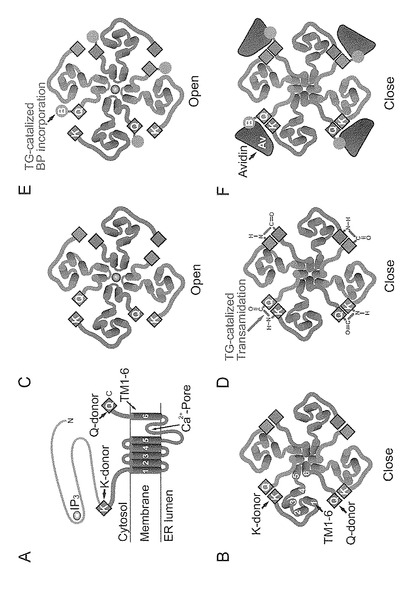
【図10】
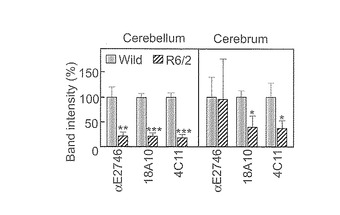
【図1−2】
【図2−1】
【図2−2】
【図3−1】
【図3−2】
【図4−1】
【図4−2】
【図5−1】
【図5−2】
【図6−1】
【図6−2】
【図7−1】
【図7−2】
【図8】
【図9】
【図10】
【公開番号】特開2011−219417(P2011−219417A)
【公開日】平成23年11月4日(2011.11.4)
【国際特許分類】
【出願番号】特願2010−90527(P2010−90527)
【出願日】平成22年4月9日(2010.4.9)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【Fターム(参考)】
【公開日】平成23年11月4日(2011.11.4)
【国際特許分類】
【出願日】平成22年4月9日(2010.4.9)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【Fターム(参考)】
[ Back to top ]