
トランスジェニック植物細胞に由来する安定な免疫予防的および治療的組成物ならびにその産生方法
本発明は、一般的に免疫学の分野に関し、免疫保護組成物およびトランスジェニック植物細胞からそのような組成物を調製する方法を提供する。本発明はまた、タンパク質産生(例えば、酵素、毒素、細胞受容体、リガンド、シグナル伝達物質、サイトカイン、またはトランスジェニック植物細胞培養物において発現される他のタンパク質の組換えによる産生)の分野に関し、これらのタンパク質を含む組成物を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、一般的に免疫学の分野に関し、免疫保護組成物およびトランスジェニック植物細胞からそのような組成物を調製する方法を提供する。本発明はまた、タンパク質産生(例えば、酵素、毒素、細胞受容体、リガンド、シグナル伝達物質、サイトカイン、またはトランスジェニック植物細胞培養物において発現される他のタンパク質の組換えによる産生)の分野に関し、これらのタンパク質を含む組成物を提供する。
【0002】
関連出願の相互参照
本出願は、全ての図、表、アミノ酸配列、およびポリヌクレオチド配列を含む、その全内容物が参照により本明細書に組み入れられる、2003年5月5日に提出された米国特許仮出願第60/467,999号の恩典を主張する。
【背景技術】
【0003】
発明の背景
特定の病原体に対する全身免疫は、外来物質に反応した生得のまたはT-細胞/B-細胞性免疫系の活性化が原因で起こる。それらの物質はしばしば、特定の病原性微生物または特定の病原物質に対して保護するように設計されたワクチンの抗原となりうる。病原体に対する曝露はしばしば、病原性微生物に絶えず曝露され、チャレンジされる粘膜表面を通して起こる。
【0004】
粘膜および口腔免疫によって、呼吸器、消化管、および尿生殖器の粘膜表面によって分泌される、および全ての分泌腺からの分泌物においてsIgA(分泌IgA)抗体の産生が起こる。McGhee, J.R.ら、Annals N.Y. Acad. Sci. 409(1983)。これらのsIgA抗体は、粘膜表面上で病原体の定着を防止するように作用し(Williams, R.C.ら、Science 177、697(1972);McNabb, P.C.ら、Ann. Rev. Microbiol. 35、477(1981))、粘膜表面を通しての定着または浸潤を防止するための免疫防御メカニズムの重要な特徴である。sIgAの産生は、分泌腺もしくは組織の局所免疫によって、またはGALT(腸管関連リンパ様組織もしくはパイエル板)もしくはBALT(気管支関連リンパ様組織)のいずれかに対する抗原の提示によって刺激することができる。Cebra, J.J.ら、Cold Spring Harbor Symp. Quant. Biol. 41、210(1976);Bienenstock, J.M.、Adv. Exp. Med. Biol. 107、53(1978);Weisz-Carrington, P.ら、J. Immunol. 123、1705(1979);McCaughan, G.ら、Internal Rev. Physiol 28、131(1983)。膜性ミクロフォルド細胞、またはM細胞として知られる細胞は、GALTおよびBALTの表面を覆い、他の分泌性粘膜表面に会合する可能性がある。M細胞は、粘膜表面に隣接する内腔から抗原を採取して、そのような抗原を抗原提示細胞(樹状細胞およびマクロファージ)に移動させるように作用し、次に抗原提示細胞が抗原をTリンパ球に提示する(T-細胞依存的抗原の場合)。次に、B細胞が刺激を受けて、増殖して遊走し、最終的に、提示された抗原に対するIgAを産生する抗体分泌プラズマ細胞へと変化する。抗原がGALTおよびBALTの上に存在するM細胞によって取り込まれると、全身性の粘膜免疫が起こり、抗原に対するsIgAが体内の全ての分泌組織によって産生される。Cebraら、上記;Bienenstockら、上記;Weinz-Carringtonら、上記:McCaughanら、上記。したがって、口腔曝露による免疫保護は、全身性の粘膜免疫応答を刺激するための重要な経路であり、さらに、口腔および消化管における分泌性免疫応答の局所刺激が起こる。
【0005】
その上、粘膜免疫は、都合よく子孫に伝達されうる。新生児の免疫は、初乳および/または乳汁を通して受動的に獲得される可能性がある。これは、催乳性免疫と呼ばれており、生後間もない動物を保護する効率的な方法である。sIgAは、乳汁における主要な免疫グロブリンであり、粘膜免疫によって最も効率的に誘導される。
【0006】
腸管関連リンパ様組織のパイエル板の上に存在するM細胞は、多様な抗原材料および粒子を取り込むことができる(Sneller, M.C. and Strober, W.、J. Inf. Dis. 154、737(1986))。M細胞はラテックスおよびポリスチレン球体、木炭、マイクロカプセル、ならびに他の可溶性および微粒子物質を取り込むことができることから、輸送される材料の任意の特異的な接着型の特性とは無関係に、GALTに多様な材料を輸送することができる。したがって、植物由来免疫保護抗原として適当な大きさの安定かつ強力な粒子を産生するための組成物および手段があれば、動物病原体に対する植物産生粘膜ワクチンの開発を大きく促進するであろう。
【0007】
組換えDNA技術は、ワクチンを含む薬学および獣医学医薬品の安全性、品質、有効性、および費用において実質的な改善を提供した。植物によって産生される粘膜ワクチンは、Curtiss & Cardineauによって発明された。参照により本明細書に組み入れられる、米国特許第5,654,184号;第5,679,880号;および第5,686,079号を参照されたい。Arntzen、MasonおよびLamを含む他の研究者らは、免疫保護抗原を発現するトランスジェニック植物およびその産生法を記述した。例えば、参照により本明細書に組み入れられる、米国特許第5,484,717号;第5,914,123号;第6,034,298号;第6,136,320号;第6,194,560号;および第6,395,964号を参照されたい。
【0008】
既定の培地における細胞培養を用いて植物細胞を産生すれば、産生プロセスから病原体混入物を伝搬するリスクが本質的になくなり、増殖培地に動物起源の成分が存在する必要がなくなる。植物細胞は、ワクチンの製造において現在用いられている通常の増殖培地と比較して、一般的に安価であり、取り扱いおよび調製が容易である。
【0009】
植物系において産生された薬理学的または関連生物活性を有するワクチン抗原およびタンパク質は、通常の産生システムに対して多くの長所を提供する。植物由来のサブユニットタンパク質は、毒性型に復帰することができない(通常に、または組換えによって産生された生きたベクターのワクチンの特徴)。通常の産生法によって産生されるサブユニットタンパク質は、タンパク質の不安定性および生化学的抽出問題のために産生および精製が難しく、グリコシル化を必要とするサブユニットワクチン組成物は、原核細胞において産生すると、グリコシル化されないであろう。
【0010】
植物は、如何なる単一の通常のまたは哺乳類由来の組換えDNA系から誘導することが難しい独自の利益を提供する。従来、サブユニットワクチンまたは生物活性タンパク質は:1)産生できないほど低い収率のために組換えまたは通常の起源から精製することが難しい;2)タンパク質分解、pH、または精製の際に用いられる溶媒のために不安定である;3)それらが天然でないために有効性が低い、または精製プロセスが重要なエピトープを変性させる;および4)哺乳類系において産生された場合に、生物起源の外来材料によって妨害される(先に記述)。
【発明の開示】
【0011】
発明の概要
本発明は、免疫原または他のポリペプチドを発現するように遺伝的に形質転換された機械的または物理的に破壊された植物細胞が、ワクチン、工業、製薬、および薬理学調製物において有用な生物活性タンパク質および免疫保護粒子を産生するという予想外の知見に基づいている。さらに、これらのタンパク質は、製剤およびその後の加工機能において安定性および強健性を示す。
【0012】
本発明は、後期指数増殖期および増殖静止期において植物細胞培養物に蓄積する少なくとも一つの免疫保護抗原または機能的タンパク質を発現する形質転換植物細胞から調製された粒子を含む、安定かつ有効な組成物を作製する方法を提供する。抗原または機能的タンパク質は、植物細胞の細胞質細胞壁および膜領域に蓄積して、機械的もしくは物理的破壊、または何らかの他の手段によって粒子の形で放出されうる。さらに、抗原または機能的タンパク質は、植物細胞の細胞質細胞壁および膜に生物活性型で安定化され、請求の方法の際におよびその後も安定かつ活性である。さらなる態様において、抗原または機能的タンパク質を産生する方法には、下等植物、単子葉植物または双子葉植物、細胞および培養を用いることが含まれる。方法のさらなる態様は、AIV(トリインフルエンザウイルス)のHA(赤血球凝集素)タンパク質、1型糖タンパク質;トリNDV(ニューカッスル病ウイルス)のHN(赤血球凝集素/ノイラミニダーゼ)タンパク質、2型糖タンパク質(参照により本明細書に組み入れられる、米国特許第5,310,678号を参照されたい);伝染性ファブリキウス嚢病ウイルス(IBDV)の構造タンパク質、VP2;酵素ADPリボシルトランスフェラーゼ(大腸菌の熱不安定毒素のLT-Aサブユニット);二つのサブユニットで構成される大腸菌の細菌毒素LT、口蹄疫ウイルス(FMDV)のようなピコルナウイルス、ポリオウイルス、ヒトライノウイルス(HRV)、A型肝炎ウイルス(HAV)、免疫不全ウイルス(HIV)、ヒト乳頭腫ウイルス(HPV)、単純ヘルペスウイルス(HSV)、および呼吸合胞体ウイルス(RSV)が含まれるがこれらに限定されないヒトウイルス、が含まれるがこれらに限定されない、特定の病原性ウイルスの免疫保護粒子における免疫原性タンパク質の産生を提供する。本発明はまた、静止期において細胞質細胞壁および膜構造に蓄積する少なくとも一つの免疫保護抗原または生物活性タンパク質を有する、機械的破壊のような手段によって容易に抽出することができる、凍結保存、凍結乾燥または懸濁液においても安定である、および天然のタンパク質と類似の特徴を有する、植物細胞可溶性抽出物を含む生物活性組成物を提供する。さらに、これらのタンパク質は、カリフラワーモザイクウイルスのS35、カッサバ葉脈モザイクウイルス、アグロバクテリウム・ツメファシエンス(Agrobacterium tumerfacians)のモノピン/オクトピンプロモーターが含まれるがこれらに限定されない、いくつかの異なるタイプのプロモーター系によって発現されると、後期指数増殖期および静止期において沈着される。組換え型蛋白質および植物細胞材料を含むこれらの組成物は、一つまたはそれ以上の薬学的に許容されるアジュバント、希釈剤、担体、または賦形剤と会合させることができる。さらなる態様において、組成物には、下等植物、単子葉または双子葉植物由来粒子と共に、特定の植物細胞および培養物に由来する粒子が含まれる。請求の組成物のさらなる態様は、植物細胞において産生される酵素ADPリボシラーゼ;構造タンパク質VP2;1型糖タンパク質;および2型糖タンパク質を含む。トリNDVのHNタンパク質およびAIVのHAタンパク質を含む、特定の病原性ウイルスの特異的免疫原性タンパク質も同様に、本発明の態様である。
【0013】
配列の概要
図1aおよび1bに示すSEQ ID NO:1および2は、NDV株「Lasota」のHN遺伝子の植物最適化コード配列およびタンパク質配列である。
【0014】
図10に示すSEQ ID NO:3および4は、AIV A/turkey/Wisconsin/68(H5N9)のHA遺伝子のDNAおよびタンパク質配列である。
【0015】
SEQ ID NO:5は、pCP!H上でCsVMVプロモーターを末端に配置するために用いられるPCRプライマーである。
【0016】
SEQ ID NO:6は、pCP!H上でCsVMVプロモーターを末端に配置するために用いられるPCRプライマーである。
【0017】
SEQ ID NO:7はNco I部位を作製するために用いられる変異誘発プライマーである。
【0018】
SEQ ID NO:8は、5'領域と相補的なフォワードプライマーである。
【0019】
SEQ ID NO:9はXhoI I部位を作製するために用いられる変異誘発プライマーである。
【0020】
図14に示すSEQ ID NO:10は、伝染性ファブリキウス嚢病ウイルスのVP2遺伝子のDNA配列である。
【0021】
SEQ ID NO:11は、E/91 VP2(1425塩基)の変種をコードする植物最適化DNA配列である。E/91植物最適化VP2のコード領域は、塩基16位〜1383位(塩基1371個)を含む。フレームストップ6個が塩基1384〜1425位に認められる。
【0022】
SEQ ID NO:12は、E/91 VP2コード領域(SEQ ID NO:11)の植物最適化版によってコードされるE/91 VP2タンパク質の配列を含む。
【0023】
SEQ ID NO:13は、読み取り枠6個において翻訳終止(「終止」)コドンをコードするDNA配列である。配列は、形質転換の際のDNA組み込み後の偶然のオープンリーディングフレームの翻訳を終了させるために用いられ、これにはSac I BstE IIおよびBgl II制限酵素認識部位が含まれる(Tsukamoto K., Kojima, C., Komori, Y., Tanimura, N., Mase, M., and Yamaguchi, S.(1999)、「Protection of chickens against very virulent infectious bursal disease virus(IBDV)and Marek's disease virus(MDV)with a recombinant MDV expressing IBDV VP2」、Virol. 257:352〜362)。
【0024】
発明の詳細な説明
免疫原または免疫保護抗原は、動物が免疫原を有する病原体に対する将来の曝露に対して保護されるように、健康な動物において生得の液性および/または細胞性免疫応答を誘発する物質である。これらの病原体は典型的に、ウイルス、細菌、真菌、および原虫のような要因である。免疫原はまた、細胞壁成分およびウイルス外皮タンパク質を含む病原体の抗原性の一部であってもよい。
【0025】
生物活性タンパク質には、糖質(例えば、α-アミラーゼ[細菌α-アミラーゼ(例えば、枯草菌)、真菌α-アミラーゼ(例えば、アスペルギルス・ニガー、アルカリα-アミラーゼ];β-アミラーゼ;セルラーゼ;β-グルカナーゼ;エキソ-β-1,4-グルカナーゼ、エンド-β-1,4-グルカナーゼ;β-グルコシダーゼ;デキストラナーゼ;デキストリナーゼ;α-ガラクトシダーゼ(メリビアーゼ);グルコアミラーゼ;ヘミセルラーゼ/ペントサナーゼ/キシラナーゼ;インベルターゼ;ラクターゼ;ナリンギナーゼ;ペクチナーゼ;プルラナーゼ);プロテアーゼ(例えば、酸プロテナーゼ;アルカリプロテアーゼ;ブロメライン;ペプシン;アミノペプチダーゼ;エンドペプチダーゼ;サブチリシン);リパーゼ、およびエステラーゼ(例えば、ホスホリパーゼ;プレガストリックエステラーゼ;ホスファターゼ;アミノアシラーゼ;グルタミナーゼ;ライソザイム;ペニシリンアシラーゼ;イソメラーゼ);オキシレダクターゼ(例えば、アルコールデヒドロゲナーゼ;アミノ酸オキシダーゼ;カタラーゼ;クロロペルオキシダーゼ;ペルオキシダーゼ);リアーゼ(例えば、アセトラクテートデカルボキシラーゼ;アスパラギン酸β-デカルボキシラーゼ;ヒスチダーゼ);またはトランスフェラーゼ(例えば、シクロデキストリングリコシルトランスフェラーゼ)を含む、酵素、毒素、細胞受容体、リガンド、シグナル伝達物質、サイトカイン、またはトランスジェニック植物細胞培養において発現される他のタンパク質が含まれるがこれらに限定されない。これらの酵素(または毒素、細胞受容体、リガンド、シグナル伝達物質、または本発明の発現系において発現のために適したサイトカイン)をコードするポリヌクレオチドは、EMBL、SWISSPROT、またはNCBIデータベースのような市販のデータベースから得ることができる。典型的に、トランスジェニック植物細胞培養物において産生された生物活性タンパク質は、天然資源から単離された同じタンパク質と機能的または構造活性において同等である。
【0026】
生物活性タンパク質粒子は、本発明の方法によって調製される生物活性タンパク質を発現するトランスジェニック植物細胞に由来する、組換え型蛋白質、植物タンパク質、脂質、糖質、核酸またはその組み合わせからなる異種粒子または凝集物として定義される。特定の態様において、生物活性タンパク質粒子は、脂質小胞、膜断片、細胞壁断片、細胞下オルガネラもしくは断片、または典型的に形質転換植物細胞の後期指数増殖期および増殖静止期に由来する貯蔵タンパク質の一部となりうる、またはそれらに会合しうる。請求される粒子は非常に安定であり、非常に安定かつ生物学的に活性なコンフォメーションで組換え型蛋白質を維持する。他の態様において、粒子は、植物細胞に導入された組換え型遺伝子からのタンパク質を発現する後期指数増殖期または増殖静止期トランスジェニック細胞培養物を機械的または物理的に破壊することによって、緩衝液または培養上清において容易に懸濁することができる材料として定義される。
【0027】
免疫保護粒子は、免疫保護抗原を発現するように遺伝子操作されているトランスジェニック植物細胞に由来する、またはそこから得られる。請求の免疫保護粒子は、ヒトを含む動物に適切に投与されると、免疫原を有する病原体に対する将来の曝露に対して保護を提供する操作されたトランスジェニック植物細胞に由来する組換え型免疫保護抗原、タンパク質、脂質、糖質、核酸、またはその組み合わせからなる異種粒子または凝集物である。請求の粒子は、非常に安定であり、非常に安定かつ生物学的に活性なコンフォメーションで組換え型蛋白質を維持する。免疫保護粒子は、操作された細胞の機械的または物理的破壊の後に、細胞の破片を免疫保護粒子から分離することによって得られる。特定の態様において、粒子は、脂質小胞、膜断片、細胞壁断片、細胞下オルガネラもしくは断片、または形質転換植物細胞の後期指数増殖期および増殖静止期に由来する貯蔵タンパク質の一部となりうる、またはそれらに会合しうる。他の態様において、粒子は、植物細胞に導入された組換え型遺伝子からのタンパク質を発現する後期指数増殖期または増殖静止期トランスジェニック細胞培養物を機械的または物理的に破壊することによって、緩衝液または培養上清において容易に懸濁することができる材料として定義される。
【0028】
下等植物は、シダ、裸子植物、針葉樹、トクサ、ヒカゲノカズラ、ゼニゴケ、キンギョ藻、苔、紅藻類、褐藻類、配偶体、シダ植物の胞子体、および緑藻類を含む任意の非顕花植物として定義される。苔が特に好ましい。
【0029】
ワクチン接種およびワクチン接種することは、宿主免疫系が刺激されて、病原体のその後の曝露に対する宿主反応に関連したその後の望ましくない病態を予防または減弱させるように、免疫原調製物、免疫保護粒子、または病原体の免疫原性調製物、またはその非毒性型もしくはその一部を宿主に接種することによって、病原体に対する保護を提供する手段として定義される。
【0030】
ワクチンは、少なくとも一つの免疫保護抗原性物質を含む、ヒトを含む動物にワクチン接種するために用いられる組成物である。
【0031】
病原性微生物は、感染した動物において疾患を引き起こす、および/または生理的状態を誘導する/制御する、細菌、ウイルス、真菌、または原虫である。
【0032】
本明細書の目的に関して、アジュバントは、免疫原または抗原に対する免疫応答を加速、増加、適度にする、または増強する物質である。アジュバントは典型的に、液性および細胞性免疫応答の双方を増強するが、他方が存在しない場合に反応が増加することがアジュバントの定義として適格である。その上、アジュバントおよびその使用は、免疫学者に周知であり、典型的に、免疫原の用量が限られている場合、免疫原の免疫原性が低い場合、または投与経路が最適下である場合に、免疫応答を増強するために用いられる。このように、「アジュバント量」は、所定の免疫原または抗原に対する免疫応答を増強することができるアジュバントの量である。「アジュバント量」に等しい質量は変化して、免疫原の特徴、投与される免疫原の量、宿主種、投与経路、免疫原の投与プロトコールが含まれるがこれらに限定されない多様な要因に依存する。「アジュバント量」は、特定の一連の状況を考慮して、普通の実験によって容易に定量することができる。これは十分に当業者の範囲内であり、典型的に様々な量の投与される免疫原およびアジュバントに対して普通の用量反応決定を用いる。反応は、酵素結合イムノソルベントアッセイ、ラジオイムノアッセイ、赤血球凝集アッセイ等を用いて、免疫原に対する血清抗体力価または細胞性反応を決定することによって測定される。
【0033】
本発明はまた、免疫保護もしくは生物活性タンパク質もしくは粒子を含む薬学的および獣医学組成物、または一つもしくはそれ以上の薬学的に許容されるアジュバント、担体、希釈剤、および賦形剤と併用した本発明の組成物を提供する。そのような薬学的組成物はまた、ワクチンとも呼ばれ、薬学およびワクチンの技術分野において周知の方法で製剤化される。
【0034】
投与または投与することは、ヒトを含む動物の体内に物質を導入することとして定義され、これには経口、鼻腔内、点眼、直腸内、膣内、および非経口経路が含まれる。請求される組成物は、皮下(SQ)、筋肉内(IM)、静脈内(IV)、腹腔内(IP)、皮内(ID)、鼻腔内、点眼、口腔粘膜(IN)、または経口投与が含まれるがこれらに限定されない任意の投与経路によって、個々に、または他の治療物質と併用して投与してもよい。粘膜経路が特に好ましく、経口経路が最も好ましい。
【0035】
有効量は、ヒトまたは動物が、病原体によって開始される攻撃に有効に抵抗するため、および糖尿病に対する自己免疫抗原のような、動物の生理的要件に反応するために十分な、ヒトまたは動物における免疫応答を誘発するために必要な量である。そのようなヒトまたは動物に投与される用量は、特定の免疫保護粒子、または粒子の組み合わせ、ヒトまたは動物の状態、および選択された投与経路を含む関連する状況に照らして医師、獣医師、または訓練された化学者によって決定されるであろう。一般的に、有効量は約1 ng〜約0.5 mgであり、好ましくは約1 μg〜約50 μgである。家禽におけるニューカッスル病ウイルス(HN抗原)の場合、有効量は約0.5 μg〜約50 μg、好ましくはSQ経路によって約2.5 μg〜約5 μgである。IN/点眼粘膜経路の場合、家禽におけるHNの有効量は、約0.5 μg〜約50 μg、好ましくは約5 μg〜約25 μg、より好ましくは約10 μg〜約12 μgである。トリインフルエンザウイルス(HA抗原)の場合、有効量は、約0.5 μg〜約50 μg、好ましくは約1 μg〜約30 μg、より好ましくはIN/点眼経路によって約24 μg〜約26 μgであり、好ましくはSQ経路によって約1 μg〜約5 μgである。家禽における伝染性ファブリキウス嚢病(VP2抗原)の場合、有効量は0.5 μg〜約50 μg、好ましくは約5 μg〜約25 μg、より好ましくはSQ経路によって約5 μg〜約20 μgである。LT抗原の場合、経口有効量は約50 ng〜約250 ng、好ましくは約100 ng〜約200 ngである。LT抗原の場合、SQまたはIN/点眼有効量は約50 ng〜約100 μg、好ましくは約1 μg〜約25 μg、より好ましくは約2 μg〜約10 μgである。本明細書において示した用量範囲は、いずれにせよ本発明の範囲を制限すると解釈されず、熟練した医師の一般的指針として示される。
【0036】
本明細書において、トリは、典型的に翼に変化した前足、鱗状の脚、嘴を有し、硬い殻に包まれた卵を産む、鳥綱の任意の温血脊椎動物として定義される。本明細書の目的に関して、好ましいトリの群は、家畜用ニワトリ、七面鳥、ダチョウ、アヒル、ガチョウ、およびコーニッシュ種の闘鶏である。より好ましい群は、家畜用ニワトリおよび七面鳥である。本発明の目的に関して最も好ましいトリは、ブロイラーおよび産卵鳥(家禽)を含む家畜用ニワトリである。
【0037】
本発明の方法および組成物は、ヒトを含む動物、好ましくはトリ(家禽)、ウシ、ヒツジ、ヤギ、ブタ、ウマ、ネコ、イヌ、およびラマのような家畜用動物、最も好ましくはトリを免疫および保護することに向けられる。これらの動物種の特定のものは、多数の胃を有し、植物を分解するために特異的な消化酵素を有しえて、そうでなければ、他のタイプの経口ワクチンを容易に不活化する可能性がある。本発明を制限することを意味しないが、トランスジェニック植物細胞およびそれに由来する組成物を摂取することによって、扁桃を含む口腔粘膜部位で動物の免疫が起こりうる。
【0038】
本発明の目的に関して、膜配列という用語は、当業者がその用語を理解する意味であることを企図する。膜固定配列には、膜貫通タンパク質配列が含まれ、天然に存在する多くのタンパク質において認められる。そのような膜固定配列は大きさが多様であるが、常に、膜内の疎水性環境に都合がよい親油性または脂肪族側鎖を有する一連のアミノ酸を含む。RNAの翻訳および翻訳後プロセシングの際に、固定配列が組み入れられ、細胞膜に埋もれて、細胞膜成分にタンパク質を固定または緩く結合させるように機能し、細胞内外の水性環境にタンパク質の親水性部分を露出させて、それらと相互作用させる。
【0039】
本明細書における貯蔵封入体または貯蔵タンパク質は、窒素源として植物が利用するタンパク質であると定義され、これらのタンパク質は、植物のライフサイクルの非生産相(例えば静止期)のあいだに貯蔵され、細胞が活動的増殖へと誘導された場合に、エネルギーおよび窒素源として急速に利用される。
【0040】
本明細書において、トランスジェニック植物は、植物細胞培養、植物細胞株、植物組織培養、下等植物、単子葉植物、双子葉植物、または形質転換された植物細胞もしくはプロトプラストに由来するその子孫であると定義され、形質転換植物のゲノムは、実験技術によって導入され、同じ種の天然の非トランスジェニック植物細胞には通常存在しない外来DNAを含む。「トランスジェニック植物」および「形質転換植物」という用語は時に、そのDNAが外因性DNA分子を含む植物を定義するための同義語として当技術分野において用いられている。
【0041】
植物において免疫保護抗原を発現するための遺伝子カセットの構築は、Sambrookら(1989)およびAusubelら(1987)、「Current Protocols in Molerular Biology」、John Wiley and Sons、ニューヨーク、ニューヨーク州に開示される方法のような、周知の方法を利用して容易に行われる。本発明にはまた、それらが発現時に開示の作用を有することができるように、免疫保護抗原をコードする開示の配列と実質的な配列相同性を有するDNA配列が含まれる。本出願において用いられるように、「実質的な配列相同性」という用語は、ヌクレオチド配列(DNAまたはRNAの場合)またはアミノ酸配列(タンパク質またはポリペプチドの場合)が、もう一つのヌクレオチドまたはアミノ酸配列と実質的、機能的、または構造的同等性を示すことを示すために用いられる。実質的な配列相同性を有する配列間の任意の機能的または構造的な差は小さいであろう;すなわち、それらは本出願において示されるように機能する配列の能力に影響を及ぼさないであろう。本明細書に開示の配列と実質的な配列相同性を有する配列は通常、変異のような、開示の配列の変種であるが、合成配列であってもよい。
【0042】
ほとんどの場合、本明細書において特に開示された配列と95%相同性を有する配列は、同等物として機能して、多くの場合かなり低い相同性、例えば75%または80%が許容されるであろう。重要でないこれらの配列の部分を特定することは時間がかかるが、普通のことであり、十分に当業者の範囲内である。オリゴヌクレオチド配列を改変する例としての技術には、ポリヌクレオチド媒介、部位特異的変異誘発を用いることが含まれる。Zollerら(1984);Higuchiら(1988);Hoら(1989);Hortonら(1989);および「PCR Technology:Principles and Applications for DNA Amplifications」Erlich(編)(1989)を参照されたい。
【0043】
ほとんどの場合、組換え型DNAによるタンパク質産生のために用いられる哺乳類細胞、細菌細胞、または他の宿主ベクター系は、再生された培養環境に置いた場合に再利用することができるタンパク質貯蔵を確立しない。大腸菌に関して記述される封入体、またはバキュロウイルス、グラニュローシスウイルス、もしくはバシラス・スリンギエンシスの結晶タンパク質は、様々な生物学的目的のために宿主系によって貯蔵される。しかしながら、いずれも、再培養の際または休止期もしくは静止期から新たな増殖の活性化の際の窒素源として植物が利用することができる貯蔵区画にタンパク質を入れることは示されていない。
【0044】
NT-1増殖の静止期の後期段階で細胞における貯蔵区画または安定な部位にタンパク質を入れることは、インビトロで培養したトランスジェニック植物細胞におけるタンパク質発現の予想された特徴ではなかった。電子顕微鏡から、白色体に暗い中心部があること、そして細胞壁および膜に隣接する細胞質区画にタンパク質に対する免疫金標識結合が存在することが示されている(実施例16を参照されたい)。さらに、NT-1系が、発現されたタンパク質のタイプまたは用いられる転写プロモーター系によらず、安定な区画にタンパク質を貯蔵できることは、これまでに例のない知見である。首尾よく発現されているタンパク質には、いくつかの異なるクラスのタンパク質が含まれる:1)酵素ADPリボシルトランスフェラーゼ、大腸菌のLTエンテロトキシンのLTA区画;2)大腸菌に由来するLTAおよびLTBサブユニットの双方を含む十分に形成されたおよび機能的LTホロトキシン;3)伝染性ファブリキウス嚢病ウイルス(IBDV)の構造タンパク質;4)トリインフルエンザウイルス(AIV)の1型ウイルス糖タンパク質赤血球凝集素(HA);および5)ニューカッスル病ウイルスの2型ウイルス糖タンパク質。それぞれの場合において、発現されたタンパク質の生物活性は、それぞれの病原体に由来する天然のタンパク質ほど強力ではないものの強力であることが判明した。各タンパク質に関連した有効性は、外来タンパク質の発現のために用いられる単一のタイプの宿主細胞に関する予想外の特徴である。保存された外来タンパク質のもう一つの予想外の特徴は、それが安定であることであり、先に記述したように、タンパク質を容易に単離できることである(例えば、機械的破壊によって)。懸濁されたタンパク質またはタンパク質含有粒子は、シグナルまたは安定性を失うことなく凍結乾燥、凍結、乳化、ホモジナイズ、または微少溶液操作することができる。本発明のタンパク質および粒子を液体型で2〜7℃で長期保存すると、長い半減期を示す;如何なる安定化剤も加えなくても、単純な機械的攪拌によって産生された抽出物から、NDVのHNタンパク質に関して予測半減期1〜2年、および大腸菌のLTに関して13〜15ヶ月の調製物が得られた。本発明に従って産生されたタンパク質は、極めて強健であり、免疫応答を増強することができる様々なタイプの製剤にすることができる。
【0045】
請求の方法と一致する物理的または機械的細胞破壊技術には、超音波、微少溶液操作または他の剪断型の方法、高剪断ローター/ステーター法、フレンチプレスまたは他の加圧法、およびホモジナイゼーション法が含まれるがこれらに限定されない。初期の研究開発活動によって、回収された細胞からHNタンパク質を免疫保護粒子の形で抽出するためには高圧破壊エネルギーが必要であることが示された。小さい発酵器アッセイ容量(1〜10 ml)からHN免疫保護粒子を放出するためには、超音波破壊が利用されたが、採取された細胞容積が1 Lを超える場合には、HN免疫保護粒子を回収するために超音波破壊はそれほど有効でなく(>35%)、規模を拡大することができないことが示されている。
【0046】
Microfludics Inc.の固定開口部圧破壊装置を用いる力量滴定試験から、破壊圧はNT1-CHN-18細胞から回収されるHN免疫保護粒子の収量と比例することが示された。HN粒子の最高の回収は、18,000 psigである機器の最高圧の設定で得られた。最高圧であっても、総HNタンパク質の40%を超える量が廃棄された細胞破片分画に存在した。この後者の実験の圧力滴定曲線から、より高い溶解圧が、廃棄される細胞破片分画におけるHNタンパク質の量を有意に減少させる可能性があることが示唆される。
【0047】
Microfludics社の製品ラインは、一定の細胞破壊技術において「第二世代」であると見なされている。Microfludics社の機器は、水撃ポンプによって高い流速で空にされるリザーバーに接続された固定された0.1 mmの乱流(「Y」外形)開口部に、懸濁細胞を通過させることによって細胞破壊を行う。ラムが上に上がると逆止め弁が開き、次のサイクルのためにリザーバーを満たす。Microfludics Incは、およそ10 ml*/分〜10,000 L*/時間の規模で同等性を主張する。細胞の分解は、(1)開口部(内破)による加速、(2)細胞破壊を引き起こす開口部先端と噴出室とのあいだの圧力の差、および/または(3)噴出室標的への減速、の結果であると考えられている。細胞の超微細構造(すなわち、細胞壁)、細胞濃度、破壊エネルギー(psig)、および分解緩衝液の組成は、溶解効率に影響を及ぼす最も重要な変数であると見なされる。
【0048】
より初期の第一世代機器は、Aminco Inc.によって連続的フレンチプレスセルとして産生された。これらは、開口部の直径と水圧を手動で制御することを除いて、Microfludics社の機器と類似である。これらの後者の機器は、主に試料容積50 ml未満の研究開発活動に用いられる。第三世代の一定細胞破壊機器(DeBEE, Inc.およびConstant Systems, Inc.)には、より高い操作圧(60,000 psigまで)、圧力の変動を減少させるための二重の試料室、および真空下で操作される試料放出室のような改善が含まれた。これらの改善は、第一および第二世代の機器と比較して改善された分解効率を有すると報告された。
【0049】
請求の方法の清澄段階には、重力沈降、遠心、浮上分離、接線流を含む濾過、ならびに全ての形のカラムおよびHPLC法を含むクロマトグラフィー技術が含まれるがこれらに限定されない任意の分離技術が含まれる。好ましい方法は、数分間の約1000 g〜約5000 gの低速遠心である。
【0050】
本発明の構築物を調製する場合、適切な方向、および適当であれば適切な読み取り枠でDNA配列を提供するために、様々なDNA断片を操作してもよい。DNA断片を結合するためにアダプターもしくはリンカーを用いてもよく、または都合のよい制限部位、過剰なDNAの除去、制限部位の除去等を提供する他の手段を含んでもよい。
【0051】
様々な段階を行う場合、所望の宿主細胞にその後導入するために、プロモーター/対象遺伝子を含むベクターを増幅するためにクローニングを用いる。広範なクローニングベクターが市販されており、クローニングベクターには、大腸菌において機能的な複製系および形質転換細胞の選択を可能にするマーカーが含まれる。例としてのベクターには、pBR322、pUCシリーズ、pACYC184、Bluescriptシリーズ(Stratagene)等が含まれる。このように、配列を適当な制限部位でベクターに挿入してもよく、得られたプラスミドを用いて大腸菌宿主(例えば、大腸菌株HB101、JM101、およびDH5α)を形質転換してもよく、適当な栄養培地において大腸菌を増殖させて、細胞を回収して溶解し、プラスミドを回収してもよい。分析は、配列分析、制限分析、電気泳動等を含んでもよい。各操作後、最終構築物において用いられるDNA配列を制限して次の配列に結合させて、部分的構築物のそれぞれを同じまたは異なるプラスミドにおいてクローニングしてもよい。
【0052】
ベクターは市販されている、または植物細胞の形質転換のために容易に調製することができる。一般的に、プラスミドまたはウイルスベクターは、所定の宿主における異種DNA配列の維持および発現にとって必要な全てのDNA制御配列を含むべきである。そのような制御配列には一般的に、リーダー配列、翻訳開始シグナルコドンをコードするDNA配列、翻訳終止コドン、メッセンジャーRNAプロセシングを制御する3' UTRシグナルをコードするDNA配列が含まれる。任意の特定の種において発現を最適にする適当なエレメントの選択は、本開示の教示を利用して当業者の問題である。最後に、ベクターは望ましくは、ベクターを含む宿主細胞の同定を可能にする表現型特性を提供することができるマーカー遺伝子を有しなければならない。
【0053】
植物細胞に挿入された外来コード配列の活性は、インサートに隣接する内因性植物DNAの影響に依存する。一般的に、異種遺伝子の挿入は、如何なる形質転換技術を用いても無作為であるように思われる。しかしながら、植物細胞へのDNAの部位特異的組換えによって植物を産生する技術は現在存在する(国際公開公報第91/09957号を参照されたい)。プロモーターの制御下で所望の配列または配列の発現が得られる任意の方法または方法の組み合わせが許容される。
【0054】
本発明は、植物細胞を形質転換する任意の特定の方法に限定されない。植物細胞にDNAを導入する技術は、当業者に周知である。外来DNAを植物細胞に輸送する四つの基本的な方法が記述されている。化学法(Graham and van der Eb、Virology 54(02):536〜539、1973;Zatloukal、Wagner, Cotten, Phillips, Plank, Steinlein, Curiel, Birnstiel、Ann. N.Y. Acad. Sci.、660:136〜153、1992);マイクロインジェクション(Capecchi、Cell 22(2):479〜488、1980)、電気穿孔(Wong and Neumann、Biochim. Biophys. Res. Commun. 107(2):584〜587、1982;Fromm, Taylor, Walbot、Proc. Natl. Acad. Sci. USA、 82(17):5824〜5828、1985;米国特許第5,384,253号)、および遺伝子銃(Johnston and Tang、Methods Cell Biol., 43(A):353〜365、1994;Fynan, Webster, Fuller, Haynes, Santoro, Robinson、Proc. Natl. Acad. Sci. USA 90(24):11478〜11482、1993)を含む物理法;ウイルス法(Clapp、Clin. Perinatol.、20(1):155〜168、1993;Lu, Xiao, Clapp, Li, Broxmeyer、J. Exp. Med. 178(6):2089〜2096、1993;Eglitis and Anderson、Biotechniques 6(7):608〜614、1988;Eglitis, Kantoff, Kohn, Karson, Moen, Lothrop, Blaese, Anderson、Avd. Exp. Med. Biol.、241:19〜27、1988)および受容体媒介法(Curiel, Agarwal, Wagner, Cotten、Proc. Natl. Acad. Sci. USA、88(19):8850〜8854、1991;Curiel, Wagner, Cotten, Birnstiel, Agarwal, Li, Loechel, Hu、Hum. Gen. Ther. 3(2):147〜154(1992);Wagnerら、Proc. Natl. Acad. Sci. USA、89(13):6099〜6103、1992)。
【0055】
電気穿孔によって植物細胞にDNAを導入することは、当業者に周知である。ペクチン分解酵素のような植物細胞壁分解酵素は、レシピエント細胞を無処置細胞と比較して電気穿孔による形質転換に対する感受性を高めるために用いられる。電気穿孔による形質転換を行うために、懸濁培養細胞、胚形成カルス、未成熟胚、または他の構築組織のような脆弱な組織を直接用いてもよい。一般的に、ペクチン分解酵素または制御された方法での機械的損傷によって標的植物材料の細胞壁を部分的に分解する必要がある。そのような処置植物材料は、電気穿孔によって外来DNAを受け入れる準備が整っている。
【0056】
植物細胞に外来形質転換DNAを輸送するもう一つの方法は、微小弾丸衝突である。この方法において、微粒子を外来DNAによってコーティングして噴射力によって細胞に輸送する。そのような微粒子は典型的に、タングステン、金、プラチナ、および類似の金属製である。微小弾丸衝突の長所は、プロトプラストの単離(Cristouら、1988、Plant Physiol.、87:671〜674)もアグロバクテリウム感染に対する感受性も必要ない点である。加速によってトウモロコシ細胞にDNAを輸送する方法の例としての態様は、バイオリスティック粒子輸送系であり、これは懸濁培養トウモロコシ細胞で覆われたフィルター表面上にスクリーンを通してDNAをコーティングした粒子または細胞を噴射するために用いることができる。スクリーンは、分子が大きい凝集物でレシピエント細胞に輸送されないように粒子を分散させる。衝突の場合、好ましくは懸濁細胞をフィルターまたは固体培養培地において濃縮する。または未成熟胚または他の標的細胞を固体培養培地上で整列させてもよい。衝突させる細胞を、マクロ弾丸停止板の下で適当な距離で配置する。衝突形質転換において、最大数の安定な形質転換体が得られるように衝突前の培養条件および衝突パラメータを最適にしてもよい。衝突のための物理的および生物学的パラメータの両者がこの技術において重要である。物理的要因は、DNA/微小弾丸沈殿物の操作に伴う要因、または微小弾丸の飛行および速度に影響を及ぼす要因である。生物学的要因には、衝突前および直後の細胞の操作に伴う全ての段階、衝突に関連した外傷を軽減するために役立つ標的細胞の浸透圧調節、ならびに直線状DNAまたは無傷のスーパーコイルプラスミドのような形質転換DNAの特性が含まれる。
【0057】
アグロバクテリウムによる移入は、プロトプラストから無傷の植物を再生する必要がなく、DNAを植物全体の組織に導入することができることから、植物細胞に外来DNAを導入するために広く適用されている系である。植物細胞にDNAを導入するためにアグロバクテリウム媒介植物組み込みベクターを用いることは、当技術分野において周知である。例えば、Fraleyら、1985、Biotechnology、3:629;Rogersら、1987、Meth. in Enzymol.、153:253〜277に記述される方法を参照されたい。さらに、Ti-DNAの組み込みは、比較的正確なプロセスであり、再配列はほとんどない。移入されるDNAの領域は、境界配列によって定義され、介在DNAは通常、Spielmannら、1986、Mol. Gen. Genet.、205:34;Jorgensenら、1987、Mol. Gen. Genet.、207:471に記述されるように、植物ゲノムに挿入される。
【0058】
現代のアグロバクテリウム形質転換ベクターは、アグロバクテリウムのみならず大腸菌において複製することができ、簡便に操作することができる。その上、アグロバクテリウム媒介遺伝子移入に関するベクターの最近の技術の進歩によって、様々なタンパク質またはポリペプチドを発現することができるベクターの構築を促進するために、ベクターにおける遺伝子の整列および制限部位が改善された。挿入されたポリペプチドコード遺伝子を直接発現させるためのプロモーターおよびポリアデニル部位に隣接する簡便な多重リンカー領域は、本発明の目的にとって適している。さらに、アームを有するおよびアームを有しないTi遺伝子を含むアグロバクテリウムは、形質転換に用いることができる。
【0059】
植物プロトプラストの形質転換は、リン酸カルシウム沈殿、ポリエチレングリコール処置、電気穿孔、およびこれらの処置の組み合わせに基づく方法を用いて行うことができる(例えば、Potrykusら、1985、Mol. Gen. Genet.、199:183;Marcotteら、Nature、335:454、1988)。これらのシステムを異なる植物種に適用することは、プロトプラストから特定の種の再生能に依存する。
【0060】
植物細胞を形質転換、選択、および抗原の発現に関してチェックした後、場合によっては、稔性の植物全体を再生することが可能である。これは選択した植物種に大きく依存するであろう。多数の植物種を再生する方法は、文献に報告されており、当業者に周知である。本発明を実践する場合、一般的に長い再生段階を回避することによって、培養して急速に規模を拡大することができる植物細胞を形質転換することが好ましい。さらに、植物細胞培養を用いることによって、農地で産生する必要がなく、遺伝子の逸出および食物の混入の機会が大きく減少する。NT-1およびBY-2(An, G.、1985、Plant Physiol. 79、568〜570)のようなタバコ懸濁細胞培養物は、これらの株が培養での取り扱いが特に容易であること、容易に形質転換されること、安定な組み込み事象を生じること、および凍結保存できることから好ましい。
【0061】
タバコ懸濁細胞株NT-1は、本発明の実践にとって適している。NT-1細胞は当初、タバコ(Nicotiana tabacum L.cv. bright yellow 2)から作製された。NT-1細胞株は広く用いられ、容易に入手可能である;しかしながら、如何なるタバコ懸濁細胞株も本発明の実践にとって適合する。NT-1細胞株のが起源は不明であることは注目に値する。その上、細胞株は多様であるように思われ、培養条件に反応して変化する傾向がある。下記の実施例において用いるために適したNT-1細胞は、American Type Culture Collectionからアクセッション番号ATCC 74840として入手可能である。同様に、その全内容物が参照により本明細書に組み入れられる、米国特許第6,140,075号を参照されたい。
【0062】
実験室規模のシェーカーから何千リットルものバイオリアクター容器に至るまで、多くの植物細胞培養技術および系が記述されており、植物細胞培養の技術分野において周知である。例えば、Fischer, R.ら、1999 Biotechnol. Appl. Biochem. 30、109〜112およびDoran, P.、2000、Current Opionions in Biotechnology 11、199〜204を参照されたい。形質転換植物細胞を望ましい量まで培養した後、それらを回収して、軽く洗浄して、破壊するために適切な緩衝液に入れる。異なる多くの緩衝液が本発明に適合性である。一般的に、緩衝液は、膜を可溶化するために用いることができる強い洗浄剤を含まない、中性pHまたは中性pH付近の等張緩衝塩水溶液である。好ましい緩衝液には、ダルベッコリン酸緩衝生理食塩液および1 mM EDTAを含むPBSが含まれる。
【0063】
一つの態様において、細胞を超音波によって破壊することができる。洗浄細胞を約0.01 gm/ml〜約5.0 gm/mlの範囲、好ましくは約0.1 gm/ml〜約0.5 gm/mlの範囲(洗浄湿重量細胞/緩衝液の容量)の緩衝液に入れる。市販の多くの超音波装置は、本発明に適合性であり、超音波処置時間は約5〜約20秒、好ましくは約15〜約20秒である。得られた大きさは数ミクロンから数百ミクロンの範囲となる可能性があり、免疫保護タンパク質または他の生物活性タンパク質を露出してもよい。
【0064】
実施例1:ベクター
遺伝子の構築:NDV株「Lasota」のHN遺伝子(Genbankアクセッション番号AF077761号)、AIV株ATurkey/Wisconsin/68のHA遺伝子、IBDV株E19のVP2遺伝子(Genbankアクセッション番号X00493)、および大腸菌のLT遺伝子のコード配列を、コドンの使用、ならびに偽性mRNAプロセシングおよび不安定性またはゲノムDNAのメチル化を媒介しうる望ましくない配列モチーフの存在に関して分析した。Adang MJ, Brody MS, Cardineau G, Eagan N, Roush RT, Shewmaker CK, Jones A, Oakes JV, McBride KE(1993)、「The construction and expression of Bacillus thuringiensis cryIIIA gene in protoplasts and potato plants」、Plant Mol Biol 21:1131〜1145を参照されたい。植物最適化コード配列をトマトおよびジャガイモのコドンの使用を反映したハイブリッドコドン嗜好性によって設計した(Ausubel F.ら編(1994)、「Current Protocols in Molecular Biology」、第3巻、p. A.1C.3 Haq TA, Mason HS, Clements JD, Arntzen CJ(1995)、「Oral immunization with a recombinant bacterial antigen produced in transgenic plants.」、Science 268:714〜716)。HNの設計された配列を図1に示す。合成HN遺伝子は市販の供給元(Retrogen)によって構築され、pCR-Bluntにクローニングされた遺伝子の5'(p4187-4203-1)または3'(p42111-4235-1c-1)半分のいずれかを含む二つの異なるプラスミドとして受領した。
【0065】
プラスミドの構築:アグロバクテリウム媒介植物形質転換のためのバイナリベクターは、図2に示すベクターpBBV-PHAS-iaaHに基づいて構築され、これは、参照により本明細書に組み入れられる米国特許第5,879,903号、第5,637,489号、第5,276,268号および第5,273,894号に記述され、国際公開公報第97/48819号に記述される構成的カッサバ葉脈モザイクウイルス(CsVMV)プロモーターによって促進される、植物選択マーカーホスフィノトリシンアセチルトランスフェラーゼ(PAT)を用いる。本発明者らは、最初にpBBV-PHAS-iaaHのPacIによる消化によってiaaH遺伝子およびファセオリンプロモーター配列を欠失して、再ライゲーションしてpCVMV-PATを形成した。次に本発明者らは、一つのHindIII部位をクレノウ酵素で塞ぐことによってこれを欠失させ、再ライゲーションしてpCP!Hを形成した。本発明者らは、鋳型pCP!H上でプライマー

を用いて、PCRによってCsVMVプロモーターを末端に配置して、EcoRV-消化、T-テールpBluescriptKSにおいて産物をクローニングしてpKS-CVM7を作製した。pCP!Hのマップを図3に示す。本発明者らは三つのインサート断片:NcoI/PstIにおいてHN 5'半分、PstI/KpnIにおいてHN 3'半分、およびKpnI-EcoRIにおいて大豆vspB 3'エレメント(Haq 1995)によってベクターpKS-CVM7/NcoI-EcoRIをライゲーションすることによって、HN発現カセットpKS-CHNを構築した。次に、バイナリT-DNAベクターpCHNを、ベクターpCP!H/AscI-EcoRIとpKS-CHNのAscI-EcoRI断片とのライゲーションによって構築した。pCHNのマップを図4に示す。
【0066】
参照により本明細書に組み入れられる、米国特許第5,824,798号に記述される顆粒結合デンプンシンターゼ(GBSS)プロモーターを用いて他のベクターを作製した。これらの構築物は、中国のジャガイモ栽培品種「Dongnong」に関してGenbankアクセッション番号X83220の配列から設計したプライマーを用いて、ジャガイモ(Solanum tuberosum L.cv.「Desiree」)のゲノムDNAから増幅したプロモーター断片を用いて作製した。変異誘発プライマー「GSS-Nco」
を−1800 bpで5'領域と相補的なフォワードプライマー「GSS-1.8F」
と共に用いて翻訳開始コドンに重なり合うNco I部位を作製した;1825 bpのPCR産物をT-テールpBluescriptKSにおいてクローニングして、pKS-GBNを作製して、シークエンシングした。変異誘発プライマー「GSS-Xho」
をプライマー「GSS-1.8F」と共に用いて、転写開始部位のまさに3'でXhoI部位を作製した;1550 bp PCR産物をT-テールpBluescriptKSにおいてクローニングして、pKS-GBXを作製し、シークエンシングした。
【0067】
参照により本明細書に組み入れられる、米国特許第5,891,665号に記述されるTEV 5'UTR(非翻訳領域)を含むGBSSプロモーター発現カセットは、HindIII/XhoIによって消化したベクターpTH210を、pKS-GBXのHindIII/XhoI断片とライゲーションすることによって構築し、これによってCaMV 35Sプロモーターを811 bp GBSSプロモーターに置換して、pTH252Aを作製した。Haq TA, Mason HS, Clements JD, Arntzen CJ(1995)、「Oral immunization with a recombinant bacterial antigen produced in transgenic plants」、Science 268:714〜716を参照されたい。NcoI/PstIにおいてHN 5'半分およびPstI/KpnIにおいてHN 3'半分をライゲーションすることによって、HN遺伝子をpTH252A/NcoI-KpnIに挿入して、pHN252Aを作製した。NsiIおよびEcoRIによって消化したベクターpGLTB(図11に示す)と、pHN252A/NsiI-KpnIおよびpTH210/KpnI-EcoRIの断片とのライゲーションによって、バイナリT-DNAベクターpgHNを作製した。pgHNのマップを図5に示す。
【0068】
参照により本明細書に組み入れられる、米国特許第5,824,798号に記述されるGBSS 5' UTRを、そのイントロンと共に含むGBSSプロモーター発現カセットを、HindIII/NcoIによって消化したベクターpTH210(Haq1995)とpKS-GBNのHindIII/NcoI断片とのライゲーションによって構築し、これによって(カリフラワーモザイクウイルス)CaMV35Sプロモーター/TEV 5' UTRを1084 bp GBSSプロモーター/5' UTRに置換して、pTH251Aを作製する。バイナリT-DNAベクターpgHN151は、ベクターpCLT105(図12に示す)を断片pTH251A/HindIII-NcoIおよびpHN252A/NcoI-KpnIとライゲーションすることによって作製した。pgHN151のマップを図6に示す。
【0069】
そのイントロンおよびマメのファセオリン3'エレメント(参照により本明細書に組み入れられる、米国特許第5,270,200号、第6,184,437号、第6,320,101号に記述される)と共にGBSS 5'UTRを含むGBSSプロモーター発現カセットを構築した。第一に、pCP!Hを唯一のKpnI部位で消化して、T4 DNAポリメラーゼによって平滑末端にし、再度ライゲーションして、KpnI部位が除去されたpCP!HKを作製した。pCP!HKをNsiIによって消化した後、T4 DNAポリメラーゼによって平滑末端にした後、PacIによって消化した。得られたベクターを、SacIによって消化したpgHN151からの2848 bp断片とライゲーションした後、T4 DNAポリメラーゼによって平滑末端にして、PacIによって消化してpgHN153を作製した。pgHN153のマップを図7に示す。
【0070】
キメラ構成的プロモーター(4OCS△MAS、参照により本明細書に組み入れられる、米国特許第5,001,060号、第5,573,932号、および第5,290,924号)を用いて、HNに関するもう一つの発現ベクターを構築した。プラスミドpAGM149をEcoRVによって消化して、BamHIによって部分消化した。この断片をPmeI/PstIによって消化したpCHNと、pKS-CHNのBamHI/PstIによる消化によって得られた合成HN遺伝子の5'半分とにライゲーションした。得られたpMHNを図8に示す。
【0071】
AIV A/Turkey/Wisconsin/68(H5N9)のHA遺伝子を含むプラスミドは、David Suarez(SEPRL、アテンズ、ジョージア州)から得た(図10)。これをPCRによって、5'末端で制限部位NcoIおよび3'末端でKpnI部位を付加するように調製し、35Sプロモーター、TEV 5'-UTRおよびvspB 3'末端を有するベクターpIBT210.1(Haqら、1995)に挿入した。発現カセットを、HindIIIおよびEcoRI(部分的)による消化によってバイナリベクターpGPTV-Kan(Beckerら、Plant Mol Biol 1992;20:1195〜7)に転移させて、pIBT-HAOを作製した。pIBT-HAOからのHA遺伝子/vspB3'末端断片は、NcoIおよびEcoRI(部分的)による消化によって、pKS-CVM7に挿入して、pKS-CHAを作製した。CsVMVプロモーター、HA遺伝子、およびvspB3'末端を含むカセットを、AscIおよびEcoRI(部分的)による消化によってpKS-CHAから得て、これをpCP!Hにライゲーションして図9に示されるpCHAを作製した。
【0072】
大腸菌株H10407のLT-B遺伝子をコードする植物最適化配列は、当技術分野で既知である(Mason HS、Haq TA、Clements JD, Arntzen CJ, 1998、Vaccine 16:1336〜1343)。大腸菌株H10407のLT-A遺伝子をコードする植物最適化配列は、その全教示が参照により本明細書に組み入れられる、当初、米国特許仮出願第60/113,507号として提出された国際公開公報第00/37609号に記述された。国際公開公報第00/37609号は、実施例2におけるタバコNT-1細胞のアグロバクテリウム・ツメファシエンスによる植物細胞の形質転換のために用いられる三つのバイナリT-DNAベクター(pSLT102、pSLT105、pSLT107)の構築を記述している。得られた形質転換NT-1細胞株(SLT102、SLT105、およびSLT107)は、LT-Bおよび改変型LT-Aサブユニットからなる完全に構築されたLTおよびLT類似体を発現して蓄積した。図12は、Ala72をArg72に置換する改変LT-A遺伝子を含むpSLT107を図示する。SLT102およびSLT105発現産物は、それらがLT-A遺伝子において異なる変化(pSLT102ではSer63がLys63に、pSLT105ではArg192がGly192に)を含むことを除き、同一であった。これらの株は、核染色体DNAに安定に組み入れられたプラスミドのT-DNA領域の未確認数のコピーを含む。トランスジェニックNT1細胞は、ガングリオシド結合五量体に構築されるLT-Bサブユニットを、ガングリオシド依存的ELISAによって決定した場合に、総可溶性タンパク質の0.4%までのレベルで蓄積した。トランスジェニックNT1細胞はまた、改変LT-Aサブユニットも蓄積し、そのいくつかは、LT-A特異的抗体を用いるガングリオシド依存的ELISAによって決定した場合に、LT-B五量体を構築した。
【0073】
アグロバクテリウムによる植物細胞形質転換のバイナリベクターは、VP2および選択マーカー発現カセットを付加するために、AgeIリンカーによって唯一のBamHI部位で改変された基本バイナリベクター(pBBV)から構築した。VP2は、アグロバクテリウム・ツメファシエンス(Atu)ORF24(Genbankアクセッション番号X00493号)3' UTRと共に、RB7 MARエレメント(米国特許第5,773,689号、米国特許第5,773,695号、米国特許第6,239,328号、国際公開公報第94/07902号および国際公開公報第97/27207号)およびCsVMVプロモーターに隣接する。選択マーカーPATは、シロイヌナズナ(At)ユビキチン10プロモーター(Plant J. 1997、11(5):1017;Plant Mol. Biol. 1993、21(5):895;Genetics、1995、139(2):921)およびAtu ORF 1(米国特許第5,428,147号;Plant Molecular Biology 1983、2:335;Genbankアクセッション番号X00493号)3' UTRによって調節され、得られたプラスミドpDAB2423を図13に示す。
【0074】
伝染性ファブリキウス嚢病(IBD)ウイルスの非常に有毒な株Ehime 91(J Vet Med Sci. 1992、54(1):153;JVI 2002、76(11):5637)を用いて、株UK661(Genbankアクセッション番号NC_004178号)からのアミノ酸番号454〜456位と共に、報告されたVP2アミノ酸配列(Genbankアクセッション番号AB024076号)に基づいて、VP2植物最適化ヌクレオチド配列を産生した(VP2配列に関しては図14を参照されたい)。
【0075】
実施例2:トランスジェニックタバコの調製
形質転換の3〜4日前に、NT-1の1週間培養物を、NT-1培養物 2 mlをNT-1培地40 mlに加えることによって新鮮な培地で培養した。培養物を25±1℃の暗所で100 rpmで維持した。
【0076】
対象発現ベクターを含むアグロバクテリウム・ツメファシエンスを、50 mg/lスペクチノマイシンを含むLB培地のプレートにグリセロール保存液から画線培養した。細菌培養物を30℃の暗所で24〜48時間インキュベートした。1ウェル形成コロニーを選択して、50 mg/Lスペクチノマイシンを含むYM培地3 mlに移した。液体培養物を250 rpmのインキュベーター内で30℃の暗所で、OD600が0.5〜0.6となるまでインキュベートした。これには約24時間を要した。
(または、粉末型のYMを購入することができる(Gibco BRL;カタログ番号10090-011)。液体培養培地を作製する場合、11.1 gを水1 Lに加える。)
【0077】
形質転換の1日後、20 mMアセトシリンゴン1 μlをNT-1培養物1 mlに加えた、アセトシリンゴン保存液は形質転換日にエタノールにおいて作製した。NT-1細胞に損傷を与えて形質転換効率を増加させた。損傷を与える場合、懸濁培養物を孔径の広い5 ml滅菌ピペットの中を繰り返し(20回)上下させて損傷を与えた。懸濁液4 mlを10、60×15 mmペトリ皿のそれぞれに移した。プレート1枚を非形質転換対象として用いるために除いておいた。アグロバクテリウム懸濁液約50〜100 μlを残りの9枚のプレートのそれぞれに加えた。プレートをパラフィルムで覆って、暗所の100 rpmのシェーカーにおいて25±1℃で3日間インキュベートした。
【0078】
細胞を滅菌の50 ml遠心管に移して、最終容量をNTC培地(500 mg/Lカルベニシリンを含むNT-1培地、オートクレーヴ後加えた)によって45 mlとした。それらを混合した後、スィングバケットローターを備えた遠心機において1000 rpmで10分間遠心した。上清を除去して、得られた沈降物をNTC 45 mlに再懸濁させた。洗浄を繰り返した。懸濁液を遠心して、上清を捨て、沈降物をNTC 40 mlに再懸濁させた。5 mlの少量を、NTCB10培地(10 mg/lバイアラホスを添加した8 g/lアガー/アガーによって固化したNTC培地、オートクレーヴ後加えた)を含む各ペトリ皿(150×15 mm)に播種した。プレートをパラフィルムによって覆って、25±1℃の暗所で維持した。培養室に移す前に、プレートを層流のフード内で開いたままにして、過剰量の液体を蒸発させた。6〜8週間後、推定の形質転換体が出現した。それらを選択して新鮮なNTCB5(5 mg/lバイアラホスを添加した8 g/lアガー/アガーによって固化したNTC培地、オートクレーヴ後加えた)に移した。プレートをパラフィルムによって覆って、25±1℃の暗所で維持した。
【0079】
推定の形質転換体は、死んだ非形質転換細胞のバックグラウンドにおいて小さいカルス集団として出現した。これらのカルスをNTCB5培地に移して、数週間増殖させた。各推定の形質転換体の一部をELISA分析のために選択した。ELISAを少なくとも2回行った後、最高の抗原レベルを示す株を選択した。各選択株のそれぞれのカルス材料の量を、プレート培養および時に液体培養において増幅させた。
【0080】
実施例3:抗原の調製
蠕動性ポンプおよびシリコンチューブを用いて、細胞を発酵器の採取口から採取した。細胞を30 μmスペクトロメッシュを含む円錐形の濾過装置にポンプで加えて、真空によって細胞を濾過して湿細胞ケークを形成した。1 mMエチレンジアミン四酢酸(EDTA;カタログ番号E(884、Sigma Aldrich)を添加したダルベッコリン酸緩衝生理食塩液(カタログ番号21-031-CV、Mediatech, Inc)を含む冷溶解緩衝液に、細胞を濾過細胞1 gあたり緩衝液2 mlの比で懸濁させた。細胞のスラリーを処理するまで5℃で維持した。微少溶液操作の前に、細胞をSilverson L4RTミキサーを用いて6000 rpmで5〜10分間ホモジナイズすることができる。100 μm Z形相互作用チャンバー(H10Z)を備えたMicrofludicsモデル110 Lマイクロフルイダイザーに、冷溶解緩衝液約200 mlを加える。チャンバーの圧力を18,000 PSIに設定して、相互作用チャンバー、入り口、および出力ラインを氷で覆った。試料を流速100 ml/分でマイクロフルイダイザーの中を通過させて、溶解細胞懸濁液を氷中で回収した。処理した溶液を、4℃、2800×gで15分間遠心することによって細胞塊を除去した。放出されたHN、HA、LTまたはVP2タンパク質を含む上清を細胞破片の沈降物から分離して、-80℃で保存した。2800×gの沈降物を2倍量の新鮮な溶解緩衝液において再懸濁させて、5℃で16時間インキュベートして、細胞破片に会合したままであるHNタンパク質を抽出した。細胞破片を、スィングバケットローターにおいて2800×g、4℃で15分間沈降させた。上清をデカントして−80℃で保存した。
【0081】
冷蔵保存からの処理は、バルク材料を2800×g、4℃で15分間遠心することによって行い、上清を真空下で30 μmスペクトラメッシュによって濾過した。上清のバルク材料は、産物の標的分子量より小さい分子量カットオフメンブレンを用いてPall Centramate接線流システムを用いて濃縮することができる。入り口および保持ラインを産物容器に向けて、産物プールを10〜20倍濃縮する。濾液を産物の漏出に関して試験する。濃縮終了後、CentramateユニットにDPBS〜500 mlを一気に流し、この洗浄液を最終の濃縮プールに加えた。濃縮物を4℃または−80℃で保存する。
【0082】
超音波処理の場合、HN、HA、またはゼロ対照のいずれかを発現する全湿潤NT-1細胞を細胞培養物から直接回収して、スペクトラメッシュ30フィルターをブフナー漏斗に入れて、細胞および培地をわずかな真空を用いて濾紙に注ぐことによって過剰量の培地を濾過して除去した。細胞0.5 gを緩衝液(ダルベッコリン酸緩衝生理食塩液および1 mM EDTA)2 mlに加えて氷中で15〜20秒間超音波処理した。超音波処理は、出力制御8、デューティサイクル60で交換可能なマイクロチップを備えたBranson 450超音波装置を用いて様々な時間行った。超音波処理物を使用するまで氷中に入れた。細胞量が多い場合、超音波処理に必要な時間はそれに比例して増加するであろう(例えば、細胞250 gの場合、超音波処理は8〜10分間に増加されるであろう)。
【0083】
実施例4:抗原の抽出
非洗浄剤処置が形質転換NT-1細胞からELISAシグナルを放出して、生物活性を保持できるか否かを調べるために、洗浄剤を含まない処置、および様々なレベルの超音波または微少溶液操作の処置の比較を含む一連の処置を設定した。結果は、抽出緩衝液における20秒より長い超音波時間がNT-1細胞株を有するpCHNからのHNの赤血球凝集活性を完全に破壊するが、ELISAシグナルは破壊しないという点において顕著であった。対称的に、DPBSにおいて20秒のみの超音波処理を行うと、ELISAシグナルによって検出可能な抗原を放出したのみならず、可溶性タンパク質抽出物は優れた赤血球凝集活性を示した(表1を参照されたい)。
【0084】
強い洗浄剤または高濃度の洗浄剤を用いずに抽出された植物由来HNを、赤血球凝集阻害アッセイにおける抗原として用いて、天然のウイルスに対して産生されたポリクローナル抗体が植物由来HNによるRBCの凝集を認識して阻害できるか否かを決定した。結果は、天然の抗体が、天然のウイルスと同様に、植物由来HNの赤血球凝集エピトープを認識するであろうことを示している(表2)。表2からのデータはまた、対照NT-1細胞または非赤血球凝集タンパク質を発現するNT-1細胞が赤血球を凝集せず、NDV特異的血清によっても影響を受けないことを示している。本実験において、植物由来タンパク質の抽出物を4 HA(赤血球凝集)単位まで希釈した後、NDV特異的ポリクローナル抗血清によって処置した。4 HA単位は、血清の滴定のために用いられる標準的なウイルス量である。
【0085】
上記のデータは、強い洗浄剤を利用せずに、細胞破壊量を減少させる抽出法を用いることによって、HNまたはHAを発現する形質転換NT-1細胞株の赤血球凝集活性を保持する細胞外分画が産生されることを示している。非洗浄剤抽出NT-1細胞からのHNタンパク質が、ワクチン有効性に関連する可能性があるさらなる生物活性を有するか否かを調べるために、HN抽出物をニワトリ細胞受容体に対する結合能に関して調べた。免疫蛍光染色では、天然のウイルスまたはpCHN-18抽出物によって処置したニワトリ胚線維芽細胞(CEF)を区別できないことが示された。このように、植物由来HNは、標的細胞表面上の受容体に対するウイルス様結合能を保持する。
【0086】
表1および2のデータを、先に記述した赤血球凝集および免疫蛍光アッセイと複合すると、本発明のトランスジェニックNT-1細胞に由来するHNタンパク質が免疫および生物学的特徴の双方を保持することを示唆している。同様に、タンパク質および免疫保護粒子は、洗浄剤の非存在下で有効かつ天然型で植物細胞から放出されうる。先に提供した最も重要なデータは、天然のウイルスに対する抗血清が、HAI試験において植物由来HNを認識するであろう点である。赤血球凝集阻害(HAI)活性がバックグラウンドより少なくとも4倍高い力価を含むニワトリはほぼ常に、毒性ウイルスからのチャレンジに対して確実に保護される。
【0087】
HNの収量が他の機械的破壊手段によって増加しうるか否かを決定するために、細胞を上記の微少溶液操作に曝露した。様々な圧力を用いて、破壊の作用およびHNタンパク質の生物活性を調べた。表3は試験の結果を示す。データは、HNタンパク質の単位質量あたりの赤血球凝集活性の量が、この破壊法を用いた場合10倍より大きく変化しうるが、タンパク質濃度は約20%増加するに過ぎないことを示唆している。これらのデータから、超音波処理によってごく部分的に放出されたに過ぎないより大きい粒子径にHNタンパク質が組み入れられ、生物活性を保持するより小さい粒子径が存在しうることが示唆される。より均一な抽出物を生じる破壊法を用いると、さらなる活性ポリペプチドの回収することができる。
【0088】
実施例5:定量的ELISAs
定量的ELISA VP2
Nunc Maxisorp 96ウェルマイクロタイターELISAプレートを、0.01 Mホウ酸緩衝液において1:2000倍希釈したニワトリ抗IBDVポリクローナル抗血清(SPAFASロット番号G0148)によって100 μl/ウェルを用いてコーティングした。プレートを5℃で一晩インキュベートした。プレートを300 μl/ウェルPBS-T(0.05%Tween20を含む1×PBS、Sigmaカタログ番号P-1379)によって3回洗浄した。各ウェルをブロッキング緩衝液(5%(w/v)脱脂粉乳(Difcoカタログ番号232100)のPBS溶液)200 μlと共に37℃で1時間インキュベートした。ウェルをPBS-Tを用いて300 μl/ウェルで3回洗浄した。IBDV参照抗原(BEI不活化IBDV D-78株、ロット番号220903IBDV)を、ブロッキング緩衝液において最終濃度1000 ng/ml VP2となるように希釈した。試料をブロッキング緩衝液によって予め希釈した。希釈した参照抗原および実験抗原試料を、試料200 μlをB列に1試料あたり2ウェルずつ加え、残りのウェルにブロッキング緩衝液100 μlを加えることによってプレートに加えた。100 μl/ウェルを混合して移動させることによって連続2倍希釈を行って、参照または試料につき希釈液6個を作製した。プレートを37℃で1時間インキュベートした後、PBS-Tにおいて3回洗浄し、ブロッキング緩衝液において1:10,000倍希釈したR-63モノクローナル抗体腹水(IBDV VP2特異的、ロット番号190903R-63)100 μlをウェルに加えて、37℃で1時間インキュベートした。プレートをPBS-Tによって3回洗浄した。ブロッキング緩衝液において1:2000倍希釈したヤギ抗マウスIgGペルオキシダーゼ標識抗体結合体(KPLカタログ番号074-1806)を100 μl/ウェルで加えて、プレートを37℃で1時間インキュベートした。プレートをPBS-Tにおいて3回洗浄し、ABTS基質(KPLカタログ番号50-66-01)100 μlを各プレートに加えて、室温で約5分間インキュベートした。Tecan Sunriseプレートリーダーを用いて波長405 nmでの吸光度を測定した。Tecan Magellanソフトウェアを用いてデータを転送して表示した。直線回帰および定量分析は、マイクロソフトオフィスエクセル2003を用いて行った。
【0089】
Nunc Maxisorp 96ウェルマイクロタイターELISAプレートを、100 μl/ウェルを用いて0.01 Mホウ酸緩衝液において混合GM1ガングリオシド5 μg/ウェルによってコーティングした。プレートを室温で一晩インキュベートした。プレートをPBS-Tによって3回洗浄した(0.05%Tween 20を含む1×、Sigma、ロット番号120K0248)。次に、各ウェルを5%(w/v)脱脂粉乳のPBS-0.05%Tween20溶液を含むブロッキング緩衝液200 μlと共に37℃で1時間インキュベートした。ウェルをPBS-Tを用いて250 μl/ウェルによって3回洗浄した。LT参照抗原(またはLT-B参照抗原)を50 ng/mlに希釈した。試料をブロッキング緩衝液において予め希釈した。希釈した参照抗原および試料を、試料200 μlをA列に加え、残りのウェルにブロッキング緩衝液100 μlを加えることによってプレートに加えた。100 μl/ウェルを混合して移動させることによって連続2倍希釈を行った。プレートを37℃で1時間インキュベートした後、PBS-Tにおいて3回洗浄し、ブロッキング緩衝液において希釈したLT-AまたはLT-B特異的抗血清100 μlをウェルに加えて、37℃で1時間インキュベートした。プレートをPBS-Tによって3回洗浄して、ペルオキシダーゼ標識抗体結合体100 μlを加えて、37℃で1時間インキュベートした。プレートをPBS-Tにおいて3回洗浄し、TMB基質50 μlを各プレートに加えた。基質を添加した20分後にTMB停止溶液を加えた。Tecan Sunriseプレートリーダーを用いて波長450 nmでの吸光度を測定した。Tecan Magellanソフトウェアを用いてデータを転送して表示した。直線回帰および定量分析は、マイクロソフトオフィスエクセル2000バージョン9.0.3821 SR-1を用いて行った。
【0090】
定量的ELISA HN
HNに関する定量的ELISAを、アッセイを行う前日にプレートをコーティングすることによって行うことができる。捕獲抗体(ウサギ抗HNの50%グリセロール溶液、0.01 Mホウ酸緩衝液において希釈(1:500倍))50 μl/ウェルを平底96ウェルマイクロタイタープレートの各ウェルに加える。プレートの蓋をして、2℃〜7℃で一晩(12〜18時間)インキュベートする。コーティングしたELISAプレートを室温で平衡にした後(約20〜30分)、PBS-Tによって1回の洗浄あたり200〜300 μl/ウェルで3回洗浄する。3%スキムミルクブロッキング溶液200 μl/ウェルを加えることによって、非特異的反応を防止するためにプレート全体をブロックする。プレートを37℃±2℃で2時間(+10分)インキュベートした(プレートの蓋または同等物で覆う)。HN参照抗原(Ag)を1%スキムミルクブロッカーにおいて250 ng HA/mlの濃度になるように加える;実験抗原を1%ブロッカーによって希釈する。HN ELISAプレートをPBS-Tによって1回洗浄して、希釈したHN参照抗原100 μl/ウェルおよびHN試験試料を列Bに加える;1%ブロッカー50 μl/ウェルを残りの全てのウェルに加える;列Bから50 μl/ウェルを列Gまで移し、次に移す前にピペットによって4〜5回混合することによって試料をプレートの下段方向に連続希釈する。プレートの蓋をして、37℃±2℃で1時間インキュベート(+10分)する;ELISAプレートをPBS-Tによって3回洗浄する。NDV HN 4A腹水の50%グリセロール溶液(1:2000倍希釈)50を3%ブロッカーにおいて各ウェルに加えて、プレートの蓋をして37℃±2℃で1時間インキュベート(+10分)する。ELISAプレートをPBS-Tによって3回洗浄して、3%ブロッカーにおいて50%グリセロール(1:3000倍希釈)においてウサギ抗マウスIgG 50 μlを各ウェルに加える;プレートに蓋をして37℃±2℃で1時間(+10分)インキュベートする。ELISAプレートをPBS-Tによって3回洗浄して、ABTSペルオキシダーゼ基質溶液(RT(室温)で少なくとも30分間平衡にする)50 μlを各ウェルに加える。プレートの蓋をしてRTの暗所で15〜20分間インキュベートする。ウェルの吸光度(OD)を波長405 nm(492 nm参照フィルターによって)で読み取る。HN参照抗原の最初の希釈液は、0.7〜1.0 ODの範囲内となるはずであり、これをELISAの陽性対照とする。
【0091】
定量的ELISA HA
HAの定量的ELISAに関して、アッセイを行う前日にプレートをコーティングする。捕獲抗体(ヤギ抗Hav5の50%グリセロール溶液、0.01 Mホウ酸緩衝液において希釈(1:1000))50 μl/ウェルを平底96ウェルマイクロタイタープレートの各ウェルに加える。プレートの蓋をして、2℃〜7℃で一晩(12〜18時間)インキュベートする。コーティングしたELISAプレートを室温で平衡にして(約20〜30分)、PBS-Tによって200〜300 μl/ウェルで3回洗浄した。3%スキムミルクブロッキング溶液200 μl/ウェルを加えることによって非特異的反応を防止するためにプレート全体をブロックする。プレートを37℃±2℃で2時間(+10分)インキュベートする(プレートの蓋または同等物によって覆う)。AIV-HA(尿膜腔液)参照抗原を1%スキムミルクブロッカーにおいて1000 ng HA/mlの濃度で加える;実験抗原を1%ブロッカーにおいて希釈する。HA ELISAプレートをPBS-Tによって1回洗浄して、希釈したHA参照抗原およびHA試験試料100 μl/ウェルを列Bに加える;1%ブロッカー50 μl/ウェルを残りの全てのウェルに加える;50 μl/ウェルを列B〜列Gまで移動させて、各移動の前にピペットによって4〜5回混合することによって、試料をプレートの下段方向に連続希釈する。プレートに蓋をして37℃±2℃で1時間(+10分)インキュベートする;ELISAプレートをPBS-Tによって3回洗浄する。ニワトリ抗AIVポリクローナル抗血清の50%グリセロール溶液(1:2000倍)50 μlを3%ブロッカーにおいて各ウェルに加えて、プレートの蓋をして37℃±2℃で1時間(+10分)インキュベートする。ELISAプレートをPBS-Tによって3回洗浄した後、ヤギ抗ニワトリIgGの50%グリセロール(1:3000)3%ブロッカー溶液50 μlを各ウェルに加える;プレートに蓋をして37℃±2℃で1時間(+10分)インキュベートする。ELISAプレートをPBS-Tによって3回洗浄して、ABTSペルオキシダーゼ基質溶液(RTで少なくとも30分間平衡にする)50 μlを各ウェルに加える。プレートに蓋をしてRTの暗所で15〜20分間インキュベートする。ウェルの吸光度(OD)を波長405 nm(492 nm参照フィルターによる)で読み取った。HA参照抗原の初回希釈液は0.7〜1.0 ODの範囲内となるはずであり、これをELISAの陽性対照とする。
【0092】
定量的ELISA LTおよびLTB
Nunc Maxis 96ウェルマイクロタイターELISAプレートを100 μl/ウェルを用いて0.01 Mホウ酸緩衝液において混合GM1ガングリオシド5 μg/ウェルによってコーティングした。プレートを室温で一晩インキュベートした。プレートをPBS-Tによって3回洗浄した。各ウェルを、5%(w/v)脱脂粉乳のPBS-T溶液を含むブロッキング緩衝液200 μlと共に37℃で1時間インキュベートした。ウェルをPBS-Tを用いて250 μl/ウェルによって3回洗浄した。参照抗原および試料抗原をプレートに加える前にPBS-Tによって1:1に混合した。LT参照抗原およびLTB参照抗原を第一のウェルにおいて50 ng/mlに希釈した。試料を、試料200 μlをA列に加え、残りのウェルにブロッキング緩衝液100 μlを加えることによってプレートに加えた。100 μl/ウェルを混合して移動させることによって連続2倍希釈を行った。プレートを37℃で1時間インキュベートした後、PBS-Tにおいて3回洗浄し、ブロッキング緩衝液において希釈した抗血清100 μlをウェルに加えて、37℃で1時間インキュベートした。プレートをPBS-Tによって3回洗浄した後、抗体結合体100 μlを加えて、37℃で1時間インキュベートした。プレートをPBS-Tによって3回洗浄し、TMB基質50 μlを各プレートに加えて、基質を加えた20分後にTMB停止溶液を加えた。Tecan Sunriseプレートリーダーを用いて波長450 nmでの吸光度を測定した。Tecan Magellanソフトウェアを用いてデータを転送して表示した。直線回帰および定量分析は、マイクロソフトオフィスエクセル2000バージョン9.0.3821 SR-1を用いて行った。
【0093】
実施例6:血清ELISA
血清ELISA LT
血液を断頭(生後0〜7日齢のトリ)または翼の羽板もしくは頚静脈での静脈穿刺によって採取した。トリを断頭によって、または断頭前にCO2に1〜5分間曝露することによって安楽死させた。血液を動物施設から実験室に運び、2〜7℃で45分放置して、凝血を進行させて濃縮させた。血液試料を37℃の温浴に10分間移した後Beckman GPR遠心機において2〜7℃で2500 rpmで20分間遠心した。各試験管から血清を無菌的に採取して、0.5〜1.5 mlの少量にして凍結用チューブ(Nunc)に入れ、使用するまで−18℃で保存した。血清ELISAの場合、ガングリオシド吸着段階は、1.5 μg/ウェルまたは15 μg/mlを利用して2〜7℃で一晩インキュベートした。プレートをPBS-Tによって3回洗浄した後、3%スキムミルクPBSと共に37℃で1時間ブロックした。血清試料あたりの抗体の力価を測定するために、ガングリオシドを吸着させた後、 2.5 μg/ml LT-BまたはLTのブロッキング緩衝液溶液100 μlをウェルあたり加えて、37℃で1時間インキュベートした。プレートをPBS-Tによって3回洗浄した後、ブロッキング緩衝液において希釈した血清試料200 μlを列Aに加えて、ブロッキング緩衝液100 μlを残りの列に加えた。特に明記していなければ血清の開始希釈液は、ブロッキング緩衝液において1:10倍であった。血清試料を連続2倍希釈した後、プレートを37℃で1時間インキュベートした後PBS-Tにおいて3回洗浄した。ヤギ抗ニワトリ結合体をHRPによって標識して、これを加え、37℃で1時間インキュベートした。プレートを洗浄してABTS 100 μlを加えて、Tecan Sunriseプレートリーダーを用いて、陽性対照が405/492の2波長での吸光度0.7〜1.0を示すまでインキュベートした。Tecan Magellanソフトウェアを用いてデータを転送して表示した。直線回帰および定量分析は、マイクロソフトオフィスエクセル2000バージョン9.0.3821 SR-1を用いて行った。マイクロソフトエクセル2000バージョン9.0.3821 SR-1を用いて、各治療群の血清の幾何平均力価(GMT)を決定した。これらの計算に関して、バックグラウンドELISA力価<10に値1を与えた。処置したトリの最小二乗平均値の対照との差を、最小二乗分析を用いて決定した。処置は、非ワクチン接種非チャレンジ対照群と処置群とのあいだに有意差があれば有効であるとした。
【0094】
血清ELISA NDV-HN
0.01 Mホウ酸緩衝液によって希釈した(1:2000倍)ウサギα-NDVプール抗血清(50%グリセロール水溶液と1:2混合)(100 μl/ウェル)によってプレートをコーティングする。プレートを2〜7℃で一晩インキュベートする;蓋をして室温で約20〜30分間プレートを平衡にする。プレートをPBS-T(1×PBS+0.05%Tween20)によって300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって3回洗浄する。プレートを5%スキムミルクのPBS-T(ブロッキング緩衝液)溶液(200 μl/ウェル)によってブロックして、37℃で2時間インキュベートする。プレートをPBS-Tによって300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって1回洗浄する。NDV尿膜腔液をブロッキング緩衝液において1:200倍希釈する。希釈した抗原を100 μl/ウェルでプレートに加えて、プレートを37℃で1時間インキュベートする。プレートをPBS-Tによって300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって洗浄する。試験ニワトリ血清試料を希釈する(1:50倍)。陰性対照血清(1:50倍)(陰性対照27NOV00)を希釈する。陽性対照血清(1:10,000または1:20,000)(SPAFASニワトリα-NDV血清)を希釈する。血清試料は全てブロッキング緩衝液において希釈する。陰性対照血清100 μl/ウェルを縦列1の列B〜Gに加える;陽性対照血清200 μl/ウェルを縦列2〜3の列Aに加える;試験血清試料200 μgl/ウェルを列Aの適当なカラムに加える。これによって、プレートあたり試料4個を試料あたり希釈液8個で行うことができる。残りの全てのウェルにブロッキング緩衝液100 μl/ウェルを加える;陽性対照血清および試験血清試料をプレートの下段に向かって連続2倍希釈する。試料をプレートのA列からH列まで希釈して、残りの100 μl/ウェルを捨てる。プレートを37℃で1時間インキュベートして、PBS-T 300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって3回洗浄する。ヤギαニワトリIgG(H&L)-HRP(1:3000倍)をブロッキング緩衝液において希釈する。希釈した結合体100 μl/ウェルを各プレートに加える;結合体をプレートに加えた後、RTの暗所でABTS基質を平衡にする。プレートを37℃で1時間インキュベートする;プレートを300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によってPBS-Tによって3回洗浄する。予め加温したABTS基質100 μl/ウェルを各プレートに加える。プレート間を2〜3分間離す。陽性対照の最初の希釈液の吸光度が0.7〜1.0に達した場合に、Tecan Sunriseプレートリーダーを用いて、プレートを二波長405/490 nmで読み取る。
【0095】
血清ELISA AIV-HA
0.01 Mホウ酸緩衝液において希釈した(1:1000倍)ウサギα-HAプール抗血清によってプレートをコーティングして、蓋をして2〜7℃で一晩プレートをインキュベートした。室温で約20〜30分間プレートを平衡にして、PBS-T(PBS保存液+0.05%Tween20)によって300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって3回洗浄した。5%スキムミルクのPBS-T溶液(ブロッキング緩衝液)によってプレートをブロックして(200 μl/ウェル)、プレートを37℃で1時間インキュベートした。プレートをPBS-Tによって300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって1回洗浄する。不活化T/W/68 AIV尿膜腔液(1:100倍)をブロッキング緩衝液溶液において希釈して、希釈した抗原100 μl/ウェルをプレートに加えた;プレートを37℃で1時間インキュベートする。PBS-T 300 μl/ウェルによってTitertek M96プレートウォッシャーまたは同等物を用いてプレートを洗浄する。試験ニワトリ血清試料(1:50倍)を希釈する;陰性対照血清(1:50倍)を希釈する;陽性対照血清(1:25600倍)(USDA/SEPRLニワトリα-AIV(T/W/68抗血清))をブロッキング緩衝液において希釈する。陰性対照血清100 μl/ウェルを縦列1の列B〜Gに加える;陽性対照血清200 μl/ウェルを縦列2〜3のA列に加える;試験血清試料200 μl/ウェルを適当な縦列のA列に加える;ブロッキング緩衝液100 μl/ウェルを残りの全てのウェルに加える。陽性対照血清および試験血清試料をプレートの下段に向かって連続2倍希釈して、残った100 μl/ウェルを捨てて、プレートを37℃で1時間インキュベートする。PBS-T(300 μl/ウェル)によってTitertek M96プレートウォッシャーまたは同等物を用いてプレートを3回洗浄する。ヤギα-ニワトリIgG(H&L)HRP(1:3000倍)をブロッキング緩衝液において希釈して、希釈した結合体100 μl/ウェルを各プレートに加える。結合体をプレートに加えた後、ABTS基質をRTの暗所で平衡にする。プレートを37℃で1時間インキュベートして、PBS-T 300 μl/ウェルによってTitertek M96プレートウォッシャーまたは同等物を用いてプレートを3回洗浄する。平衡にしたABTS基質を各プレートに100 μl/ウェルで加える;プレート間に2〜3分の間隔を空ける。陽性対照の最初の希釈液の吸光度が0.7〜1.0に達した場合に、Tecan Sunriseプレートリーダーを用いて、プレートを二波長405/490 nmで読み取る。
【0096】
血清ELISA IBDV-VP2
血液および血清の採取は、LTに関する血清ELISAに関して先に記述した通りに行った。血清ELISAに関して、ニワトリ抗IBDVを1.0 μg/mlの0.1 Mホウ酸緩衝液において2〜7℃で一晩インキュベートしてプレートに吸着させた。プレートをPBS-Tによって3回洗浄した後、3%スキムミルクPBS-T溶液によって37℃で1時間ブロックした。ブロッキング緩衝液において希釈したニワトリ血清試料200 μlをA列に加えてブロッキング緩衝液100 μlを残りの列に加える。特に明記していない限り、血清の開始希釈液はブロッキング緩衝液において1:10倍であった。血清試料を連続2倍希釈した後、プレートを37℃で1時間インキュベートして、PBS-Tによって3回洗浄した。HRPによって標識したヤギ抗ニワトリ結合体を加えて、37℃で1時間インキュベートした。プレートを洗浄して、ABTS 100 μlを加えて、陽性対照の吸光度がTecan Sunriseプレートリーダーを用いて、プレートを二波長405/490 nmで読み取った場合に0.7〜1.0に達するまでインキュベートする。LTに関する血清ELISAに関して先に記述した通りに、データをTecan Magellanソフトウェアを用いてデータを転送して表示した。
【0097】
実施例7:赤血球の赤血球凝集および赤血球凝集阻害
赤血球凝集
ニワトリ赤血球のAlsevers溶液(CRBC)をColorado Serum(L# 8152)から得た。1%CRBC溶液を調製するために、5 mlを15 ml遠心管に移して、250×gで10分間遠心した。上清およびバフィーコートをRBC沈降物からピペットによって採取した;沈降物を1×DPBS(ダルベッコリン酸緩衝生理食塩液)(L# 003435E JRH)に再懸濁させることによって2回洗浄して、250×gで10分間遠心した。沈降物をDPBSにおいて1%(v/v)に再懸濁させた。懸濁液の濃度を確認するために、400 μlを脱イオン水1.6 mlに移して、激しく混合することによって細胞を溶解した。OD540は0.4〜0.5のあいだであった。1%溶液は使用するまで2〜7℃で保存した。赤血球凝集を試験するために、96ウェルU底ディッシュ(Falcon)にまず、Static Guard(商標)を噴霧して、ペーパータオルにしみこませた。ウイルス試料をDPBSにおいて予め1:2倍希釈して、DPBS 50 μlを96ウェルディッシュの各ウェルに加えた。希釈したウイルスを第一の列に加えて、ウイルス試料あたりの望ましい回数の希釈液を得るまで連続2倍希釈した。1%CRBC 50 μlを各ウェルに加えてプレートを600 rpmで20秒間混合した。プレートを湿ったペーパータオルの上に置いて、対照ウェルにおけるCRBC(DPBSおよびCRBSが1:1比)がプレートの底に沈降するまで、または2〜7℃で少なくとも1時間インキュベートした。終点は、100%凝集を提供するシリーズにおける最後のウェルの希釈であった。
【0098】
赤血球凝集阻害(HAI)
ウイルスをDPBSにおいて予め希釈して4〜8 HA単位/50 μl(本明細書に記述のウイルスの力価測定に基づく)を得た。DPBS 25 μl/ウェルを縦列1および3〜12に用いて異なるプレートを作製した;血清25 μlを縦列1および3のウェルに加えた。縦列3における血清を10ウェルを通して連続希釈した。予め力価を測定したウイルス(25 μl)を縦列3〜12の全てのウェルに加えて、600 rpmで20秒間混合した;プレートを室温で1時間±15分インキュベートした。1%CRBC 50μlをウェルに加えて、600 rpmで20秒間混合して、湿潤室においてAIVに関して2〜7℃で一晩、およびNDVに関して2〜7℃で1〜2時間インキュベートした。血清の力価は赤血球凝集を100%阻害する連続希釈液の最後のウェルである。
【0099】
実施例8:ウサギにおける抗原性
上記のように、非洗浄剤緩衝液において抽出された免疫保護粒子における植物由来タンパク質が動物種において抗体を産生するか否かを調べるために、HAおよびHNタンパク質の双方を調製して、ウサギに接種した。3ヶ月齢のニュージーランドホワイトウサギに、表4に示す投与スケジュールに従って植物由来HA-AIVまたはHV-NDVを接種した。初回接種の場合、抗原をフロイントの完全アジュバント(CFA)と混合して、追加接種の場合には全てフロイントの不完全アジュバントを用いた。双方のタンパク質によって誘導された抗体力価を表5に示す。結果は、2回の接種後、HAI抗体力価が双方のタンパク質によって誘導され、このことはNT-1細胞の後期増殖相から調製した本発明の植物由来免疫保護粒子が哺乳類において、天然のタンパク質を認識することができる抗体を誘導することを示している。データから、植物由来HNおよびHAが天然に由来するHNおよびHAタンパク質と共有する特徴を有することが示唆される。植物由来AIV-HA接種ウサギの力価は、NDV-HN植物由来タンパク質によって誘導された力価より高かった。このことは、AIV-HAタンパク質の単位あたりのAIV/HAタンパク質の全般的生物活性(赤血球凝集)がNDV-HNより低かったことから、重要であるかも知れない(表4、縦欄4)。
【0100】
実施例9:発現された抗原の有効性および生物活性:家禽およびインビトロ細胞障害性におけるチャレンジ
ニューカッスル病ウイルス(NDV)のチャレンジ試験
植物由来HNタンパク質の有効性を調べるために、2日齢および10日齢のトリを用いて二つの異なる試験においてHNタンパク質を接種した。これらの試験に用いられる試験#16の用量濃度を表6に示す。全てのワクチン接種物をシェーカーフラスコにおいて25℃で15〜20日増殖させたNT-1細胞の可溶性分画から調製した。双方の試験において用いられるアジュバントは、Corixa、Inc.のMPL-TDMであった。鼻腔内群には、MPL単独をアジュバントとして投与した。
【0101】
2日齢のSPFニワトリに様々な経路によって、NT-1由来生物活性(赤血球凝集陽性)NDV-HNタンパク質を表6に示す接種あたりのHNタンパク質の量で用いて接種した。この試験の血清およびチャレンジの結果を表7に示す。対照群は全て予想どおりに反応した。接種物においてNDV-HN抗原を投与しなかったトリは、100%死亡率を示したが、SQによって天然のNDV 20 μgを投与した対照のトリは100%生存した。植物由来HN抗原群を用いた実験処置において、#3群において75%保護(アジュバントと共にSQ接種)および#4群において80%保護(アジュバントと共にSQ接種)を認めた。INおよび経口経路によって接種した残りの治療群は100%死亡率を示した。しかしながら、6群ではトリ2羽に死亡の遅れを認め、これらのトリがワクチン接種に対して感作されている可能性があること(例えば、表10、列14を参照されたい)、およびこの経路によって投与する場合には有効性を増強するために異なる製剤を必要とすることを示している。その後の試験(#18)において、10日齢のSPFトリに表8のスケジュールに記述した用量を接種した。対照群1群(#3)、非ワクチン接種非チャレンジ処置を用いて、ケージおよび施設がニワトリの全般的な健康に有害な作用を及ぼさないことを示した。この試験における対照群も、予想どおりに反応した。双方の試験からのトリを同じ施設でチャレンジすることから、処置群#2を試験16(表7)および18(表9)の双方の陽性対照とした。その全てにNT1細胞に由来するHNをSQ接種した残りの群では、群#7において100%生存、群5および6のそれぞれにおいて80%生存を認め、そして4群において60%生存を認めた(表9を参照されたい)。
【0102】
トリインフルエンザ(AIV)のチャレンジ試験
異なる試験において、ブロイラーニワトリに、植物由来赤血球凝集タンパク質(HA)をワクチン接種した。トリインフルエンザウイルス(AIV)株A/turkey/Wisc/68(H5N9)の植物由来HAタンパク質遺伝子配列を、NT1 CHN-18に関して記述したベクター系を用いてNT1細胞に形質転換した。HA-AIVタンパク質のトランスジェニック植物細胞産生のためのNT1株の命名はCHA-13であった。ニワトリをふ化後3日齢で得て、トリ10羽を各治療群に関してケージに無作為に入れた。各治療群の用量を表11に示す。トリを試験の0、14、および28日目に3回投与して、血液試料を0日、21日、35、および45日に採取した。各血液試料からの血清をHAI力価に関して分析して、35日目にトリをジョージア州アテンズのSoutheast Poultry Research Laboratoryに輸送して、そこでトリに毒性AIV(ニワトリ/ペンシルバニア/1370/1983)をチャレンジした。表11に示したデータから、CHA-13 NT1株に由来するHAタンパク質30 μg用量によって、ワクチン調製物の2回投与後であってもHAI陽性力価への血清変換が得られたことが示される。チャレンジすると、ワクチン接種群は全て、AIVに対して保護を示した:50またはそれ以上のチャレンジスコアは疾患または臨床病態を示している。製剤によらず、全ての群は、チャレンジ時に天然のAIVに対して非常に類似の力価を示し、このことは植物由来タンパク質によって誘導される天然のウイルスに対するメモリー反応を示している(縦欄4、表11)。
【0103】
伝染性ファブリキウス嚢病ウイルス(IBDV)のチャレンジ試験
上記の試験は、本発明に従うトランスジェニック植物細胞に由来する二つのタイプの糖タンパク質が、それらが毒性チャレンジから標的種を保護できるという点において非常に有効であることを示している。さらなる試験において、IBDVからの非グリコシル化構造タンパク質VP2を、CHN-18およびCHA-13の場合と類似のベクターおよびプロモーター構築物によってNT-1細胞に形質転換した。本明細書に記述の得られたトランスフェクト細胞をCVP2-002と命名した。本試験において、SFGニワトリをふ化後7、21、および35日目に、NT-1対照細胞溶解物、IBDV(形質転換事象CVP2-002)からのVP2タンパク質を発現するトランスジェニックNT細胞からの細胞溶解物、および市販の不活化伝染性ファブリキウス嚢病ウイルス(IBDV)ワクチン(Lohman Animal Health)Vi Bursa K+Vをワクチン接種した。NT-1対照細胞を10 L発酵器において増殖させて、継代6代目のCVP2-002細胞をシェーカーフラスコにおいて増殖させた。細胞を植物10〜14日後に回収して、18,000 PSIで100 μm Z形相互作用チャンバーを備えたMicrofludics 110 L微小溶液装置の中に通過させることによって溶解した。得られた細胞溶解物を2000×gでの遠心によって透明にした。透明にした上清を凍結乾燥によって濃縮した。ワクチンをアジュバントと共に調製して、各ワクチンのVP2濃度をワクチン接種前にELISAによって決定した。表12は、ワクチン製剤、投与経路、各ワクチン接種日でのVP2濃度を記述する。血液試料をふ化後21、35、および42日目に採取して、血清ELISAにおいて抗体反応に関して、およびIBDV血清中和(SN)アッセイにおいて中和抗体力価に関して試験した。トリに、IBDVの50 EID50胚由来STC株を両側の点眼によってチャレンジした。トリをチャレンジ後10日目に安楽死させた。ファブリキウス嚢対体重(BBW)および脾臓対体重(SBW)比を各トリに関して決定した。各トリからのファブリキウス嚢組織をホルマリンによって固定して、嚢胞枯渇によって示されるIBDV関連病変に関して採点した。BBW比、SBW比、および嚢病変スコアを非チャレンジ対照トリと比較した。非チャレンジおよび対照のあいだでBBWに統計学的有意差がなければトリは、チャレンジから保護されたと採点された。表13は、各ワクチン群に関する血清学およびチャレンジ結果を要約し、これは、植物由来VP2抗原が通常の死菌IBDワクチンより実際に大きい(ELISAによって)血清反応を生じることを示している。さらに、BBWによって測定したチャレンジに対する保護は、植物由来VP2が通常の死菌IBDワクチンと共に保護することを示している(表13の列4〜列10を比較)。
【0104】
Y1副腎細胞における熱不安定性毒素の細胞障害性
マウスのY1副腎細胞をATCC(CCL-79、L#1353400)から購入した。細胞のバイアルを37℃で融解して、F-12K培地(Gibco L#1089716)において15%ドナーウマ血清(Quad-5 L#2212)、2.5%ウシ胎児血清(JRH L# 7N2326)、1%グルタマックス-1(Gibco L#1080323)からなる増殖培地10 mlを含む25 cm2T-フラスコ(Corning)に入れた。細胞を5%CO2において37℃でインキュベートした。細胞を、各継代時にこの増殖培地で、LTおよびCT細胞障害性アッセイのために維持した。アッセイするために、細胞を96ウェル細胞培養プレート(Nunc)に播種して80%コンフルエンスに達するまで増殖させた。LT毒素をF-12K増殖培地において1 μg/mlに希釈した。毒素を、プレートのA列に予め希釈した試料100 μlを加えることによって、96ウェルマイクロタイタープレートにおいて2倍連続希釈によってさらに希釈した。次に、A列の50 μlを次のウェルの増殖培地50 μlに移すことによって2倍連続希釈を行った。試料の各希釈液を、試料または細胞の利用率に応じてY1副腎細胞の1〜4ウェルに移した。LT毒素の終点力価は、50%細胞障害性(細胞死)を得るために必要なタンパク質の量(EC50)である(Guidryら、1997;Dontaら、1974)。用いた毒素は、NT-1トランスジェニック細胞株SLT105、SLT107およびSLT102においてそれぞれ産生された大腸菌の熱不安定毒素のG192、R72、およびK63単アミノ酸遺伝子置換変異体であった。LT毒素の三つの変異体型は、インビトロバイオアッセイおよびインビボで毒素であることが報告されており、G192、R72、K63は野生型LT毒素よりそれぞれ約10倍、100倍、および1000倍毒性が低い(Rappuoliら、1999、Immunology Today 20:293〜500)。植物において産生されたLT変異体濃度を、Y1副腎アッセイ(表14を参照されたい)において大腸菌からのLT野生型毒素と比較したところ、結果は、植物由来毒素によって、大腸菌に由来する同じ変異体に関して認められる類似のレベルの感受性が得られることを示している。さらに、LT毒素の定量はG1ガングリオシド捕獲ELISA法によって決定されることから、植物産生毒素は完全に構築されたホロトキシンを模倣する(Guidryら、1993、Inf. and Imm. 65:4943〜4950)。
【0105】
CHA-13およびCHN-18からの植物産生免疫保護粒子の粘膜輸送
粘膜表面に輸送される非複製植物由来抗原が、強力な免疫保護材料であるか否かを決定するために、接種のために均一な乳剤を作製するために微少溶液操作によって調製した製剤を用いて、眼および鼻腔粘膜表面に抗原を直接接種することによってトリの試験を行った。7日齢のブロイラーニワトリをケージに無作為に入れて(1群5羽)、2.6〜16.7 μg/トリの抗原を接種した(表15を参照されたい)。トリにワクチンの3用量を0、14、および21日目に投与して、トリは試験終了時42日齢であった。抗原にはCHN-18トランスジェニック植物細胞に由来するHN、CHA-13トランスジェニック植物細胞に由来するHA、および感染ニワトリ胚の尿膜腔液に由来する不活化トリインフルエンザウイルスが含まれた。抗原調製物は、先の実施例3に記述したように調製した。様々な製剤において異なるアジュバント5個を用いて、免疫応答を、赤血球凝集阻害および血清ELISAに関して血清学によって決定した(表15を参照されたい)。試験の結果を表16に示す。一つの製剤を除く三つの用量によって全て、CHN-18からの植物由来HNを接種したトリでは血清変換が起こった。製剤の一つによって、CHA-13からのHAを接種したトリでは血清変換が起こり、二つの製剤によって、不活化AIV抗原を接種したトリの血清変換が起こった。一つのアジュバントは全ての反応群に共通し、コレステロールと混合したQuil Aであった。結果から、植物由来の非複製抗原が、粘膜表面への接種によってトリにいて血清学的反応を提供したことが示される。
【0106】
実施例10:トランスジェニック細胞における発現タンパク質の産生および蓄積
10 Lバイオリアクターまたはシェーカーフラスコにおいて増殖させたトランスジェニック細胞培養物から発現されたrDNAを図15〜18に示す。実施例2において先に記述した培地にCHN-18、CHA-13、SLT102、またはCVP2-002トランスジェニック細胞を産生するNT-1トランスジェニック細胞培養物を、培養12日後に回収した(静止期)。次に、シェーカーフラスコからの接種物を、Pluronic L61消泡剤1 mlを含む増殖培地10 Lを含む10 L Bioflow 3000発酵器(New Brunswick)に無菌的に移した。細胞の産生は、100 rpmで攪拌しながら30%溶存酸素で2.5 L/分で通気しながら25℃で行う;細胞の産生は9〜15日間行う。15 ml遠心管に発酵培養物10 mlを加えて、2000×gで10分間遠心することによって、細胞沈降容積(PCV)を決定し、細胞容積を測定して、接種日から10日目までの細胞増殖、または静止期培養を追跡するパラメータとして評価した。データは、約3日間の遅滞の後、培養物の指数増殖期が3日〜7日までのあいだに起こり、その後培養物は、挿入された遺伝子および用いたプロモーター系によらず、分析した各トランスジェニック細胞株に関して静止期に達し始めた。CHN-18の場合、測定可能なHNタンパク質の量を毎日追跡した。1日目、新たな細胞増殖の前に、抽出することができるHN ELISAシグナルを認め、これは培養12日でシェーカーフラスコから採取した接種物に存在するHNの量を表す。しかしながら、HNは急速に分解し、細胞が静止期に達した培養約6日後まで検出されず、細胞が静止期を通過した後も細胞に蓄積され続ける。HN発現は異なる二つの測定によって追跡し、黒三角は定量的ELISAによって測定したHNタンパク質を表し、黒四角は赤血球凝集を表す。定量的ELISAは、HNタンパク質産生に対してより感受性が高く、単量体または重合化HNタンパク質の双方を測定するが、赤血球凝集は、赤血球を凝集することができる二量体または重合化タンパク質のみを測定し、より多くのタンパク質が蓄積して初めて、このように赤血球凝集活性を測定することができる(図15)。タンパク質の増殖後期産生の現象は、発現されるタンパク質(大腸菌のホロトキシンLT、トリインフルエンザウイルスの赤血球凝集素タンパク質(HA)、伝染性ファブリキウス嚢病ウイルスのVP2構造タンパク質、またはニューカッスル病ウイルスの赤血球凝集素-ノイラミニダーゼタンパク質(HN))によらず認められる(図16、17、および18を参照されたい)。
【0107】
CHA-13およびCVP2-002の産生および増殖曲線をそれぞれ、図16および17に示す。CHA-13に関して、増殖(PCV)は、接種後2日目に始まり、接種後10日目で静止期に入る。ショ糖は接種後2日まで消費され、デキストロースは接種後6日まで消費される。HAの蓄積は、接種後6日目に始まり(中期対数増殖期)14日まで増加する。細胞増殖は接種後2日目に始まり、接種後9または10日目で静止期に入る。ショ糖は接種後2日まで消費され、デキストロースは接種後5日まで消費される。VP2蓄積は接種後7日目に始まり(中期対数増殖期)、14日まで増加する。
【0108】
大腸菌熱不安定毒素(LT)に関するK63変異体を発現するSLT102トランスジェニックNT-1細胞株を図18に示す。LT毒素は5日および6日のあいだに蓄積し始め、沈降細胞容積はこの実験では示されないが、他のNT-1トランスジェニック細胞株の容積と類似である。
【0109】
実施例11:植物産生タンパク質の安定性
組換え型または天然起源から抽出したタンパク質はしばしば、プロテアーゼ、グリコシラーゼ、リパーゼ、またはタンパク質および細胞成分と共に精製された他の酵素のために不安定である。NT-1細胞から単離されたタンパク質および免疫保護粒子は、固有に安定であり、多くの異なるタイプの下流のプロセシング活性に対して強健である。図19において、CHN-18細胞を静止期の10 L発酵器から採取して、濾過し、遠心によって透明にして、実施例3に記載の方法に従って1回微少溶液化した。上清を0.2または0.45ミクロンのフィルターに通過させて、濾過または微少溶液化による操作の際に導入された可能性がある如何なる細菌物質も除去し、安定化剤をこれらの懸濁液に加えなかったが、安定性は、これらのトランスジェニック細胞に由来するタンパク質に対して固有である。次に、材料を2〜7℃、25℃で保存するか、または−80℃で凍結した;材料は全ての温度で安定であることが判明したが、最も興味深いことに25℃(外界温度)で維持すると、単離タンパク質は、安定であることが判明した(図19に示す)。月毎にシグナルの変動を認めたが、単離されたタンパク質の量は、数ヶ月後顕著な安定性を示し、これらのデータから計算することができる半減期は、予測半減期8ヶ月(0.45ミクロン試料)および0.2ミクロン濾過試料では1年より長いことを示している。
【0110】
実施例12:抗原の細胞下局在
共焦点レーザー走査顕微鏡(CLSM)
共焦点レーザー走査顕微鏡を形質転換MHN-41およびCHN-18においてHN抗原を局在するために行った。局在技法のために用いた抗体は、IgG精製ウサギ抗HNポリクローナル(HN ELISAにおける捕獲Ab)およびHN Mab 4A−腹水からの非精製型(HN ELISAにおける検出Ab)であった。画像は以下の技法を用いて培養植物細胞から得た。非形質転換対照細胞(NT-Ctrl.)を含む細胞を1000 gで5分間遠心して、3.7%ホルムアルデヒドにおいて15分間固定した。細胞をPBSによって各洗浄について5分間2回洗浄した。細胞を3000 gで2分間(毎回)遠心して、0.5%TritonX-100および1%ペクトリアーゼによって15分間処置した。H2Oによる洗浄を行って、H2Oをメタノール(−20℃)に置換した;異なるウェルを有するコーティングしたスライドガラスに、ピペットによって細胞を載せて、空気乾燥させた(フード内で20〜30分間)。PBSによって洗浄した後、3%BSA/PBSによって30分間ブロックした。一次抗体(1%BSA-PBS-T(0.05%Tween-20を含むPBSを37℃で1時間、またはRTで1.5〜2時間)インキュベートする)。PBS-Tによって3回洗浄する。Cy5/Cy2(1:100倍)によって標識した二次抗体をRTで1時間インキュベートして、スライドガラスをPBS-Tによって3回洗浄して封入した。
【0111】
NT対照細胞ではRb抗HNポリクローナルまたはHN Mab 4Aによって染色を認めなかった。双方のHN特異的抗体によって、静止期細胞の細胞質全体に明るい染色(核には認めない)をMHC-41細胞株全体に認めた。(図20および21を参照されたい)。
【0112】
電子顕微鏡
発現されたタンパク質が蓄積されている場所を確立するために、トランスジェニック植物細胞を培養10日後に回収して、以下のように薄切片および免疫金標識のために調製した。免疫金標識は、10日齢の感染ニワトリ卵胚から採取した尿膜腔液のニューカッスル病ウイルス調製物から精製したHNタンパク質によって免疫したウサギからの精製IgGを用いて行った。形態学的特徴を定義するために、細胞懸濁液を3%グルタルアルデヒドの0.1 Mリン酸緩衝溶液(pH 6.8)において3時間固定した。次に、それらをリン酸緩衝液において、緩衝液を4回交換して1時間洗浄した。細胞を2%四酸化オスミウムのリン酸緩衝溶液において1時間後固定した。濃度を増加させたエタノール(25%、50%、75%、95%、および100%、各段階15分)およびプロピレンオキサイドにおいて細胞を脱水した。細胞をプロピレンオキサイド/Epon 812混合物において一晩放置した後、Epon 812において抱埋し、60℃で2日間重合化した。切片をLKB Ultrotome IIIによって切断して、2%酢酸ウラニル水溶液およびクエン酸鉛によって染色して、Hitachi 7500透過型電子顕微鏡を80 kVで操作して調べた。
【0113】
免疫金染色に関して、グルタルアルデヒド固定細胞をリン酸緩衝液(0.02 Mグリシンを加える)洗浄後、エタノールの濃度を増加させて脱水した。次に、細胞をLR White樹脂に一晩浸潤させて、最終的に抱埋し、50℃で24時間重合化した。
【0114】
ニッケルグリッド上に封入した切片を、1%ウシ血清アルブミン溶液と共にpH 7.4のPBS緩衝液において20分間インキュベートして、非特異的部位をブロックした。次に、細胞を一次抗体(PBSにおいて1:150倍希釈)と共に室温で2時間インキュベートした。次に、PBS-BSAによって6回洗浄して(各3分)、ヤギ抗ウサギAB(PBSにおいて1:150倍希釈)に結合させた金コロイド(15 nm)と共に室温で2時間インキュベートした。細胞をPBSにおいて5分間の洗浄を4回行った後、水によって1分の洗浄を2回行った後、グリッドを酢酸ウラニルによって5分間染色した。EM画像によって、対照細胞と、HNタンパク質を発現するトランスジェニック細胞とのあいだに二つの主要な差が示され、最初の色素体/白色体は、トランスジェニック細胞において蓄積するが正常細胞には蓄積しない暗い顆粒を示し(図22)、第二に、免疫金染色顆粒は、トランスジェニック細胞の細胞壁近傍で蓄積することが認められうるが、対照細胞は認めることができない(図23)。典型的に、宿主細胞において発現される遺伝子産物は、細胞の指数増殖期に起こり、一般的に小胞体、ゴルジ体、および細胞における他のタンパク質合成小構造において染色されうる。図19および20に示す共焦点画像と共に、データは、電子顕微鏡によって、タンパク質が細胞膜および細胞壁において産生され、沈着されるが、核、葉緑体、ミトコンドリア、小胞体、およびゴルジ体には蓄積を認めないことを示している。電子顕微鏡から、後期静止相の細胞が肥大した液胞および圧縮された細胞質および核を有することが示される。共焦点画像は、タンパク質が、細胞全体の細胞質細胞壁および膜に対して圧縮されていることを示唆している。
【0115】
細胞におけるタンパク質産生および蓄積が、細胞がもはや活性な代謝状態ではない後期指数増殖期および静止期まで明白でないことは珍しい。増殖フラスコまたは発酵器における細胞の接種時に発現されたタンパク質シグナル(24時間)が急激に減少したこと(実施例15を参照されたい)は、窒素源としてタンパク質を用いること、そして細胞が活性な増殖を終了した場合には窒素源(タンパク質)の貯蔵が起こりうることを示している。この現象は、上記の実施例において記述される植物細胞培養において産生されたトランスジェニックタンパク質に対して独自の特徴である。細胞壁および膜近傍にタンパク質が存在することは、機械的破壊によって予想外に容易にタンパク質を単離できることを説明するために役立つ。各タンパク質のクラスが安定かつ有効な形で容易に単離されうることもまた、予想外である。如何なる単一のタンパク質もしばしば、組換え型DNA発現を調べるために選択された任意の外来宿主系において合成されうるが、多くのタンパク質、特に膜貫通結合糖タンパク質はしばしば、低レベルで産生されるが、一つの宿主系は同じ方法で二つの糖タンパク質を発現しない。本明細書に記述する細胞培養トランスジェニック系において、少なくとも五つのクラスのタンパク質(酵素、1型ウイルス糖タンパク質、2型ウイルス糖タンパク質、LT毒素、および構造的非グリコシル化タンパク質VP2)が、同じ宿主系において首尾よく類似のレベルで発現されている。さらに、タンパク質は、タンパク質のクラスによらず、後期静止期において蓄積し、転写カセットまたはプロモーター系は同じ物理的または機械的破壊法を用いて細胞から容易に除去することができる。これらのトランスジェニック細胞によって発現されるタンパク質のクラスによらず、各タンパク質は、生物学的に活性である安定な形での単離に成功している。
【0116】
実施例13:伝染性ファブリキウス嚢病植物最適化VP2抗原遺伝子
伝染性ファブリキウス嚢病(IBD)ウイルス(またはIBDV)のウイルス原因物質は、二つに分かれたRNAゲノムである(J. Virol. (1979)、32:593)。完全長のRNA1がポリタンパク質に翻訳され、これがペプチドVP2、VP3、およびVP4にプロセシングされる。ゲノムRNAのインシリコ(in silico)逆転写を行って、天然のRNAのタンパク質コード能に対応するDNA配列を得ることができる。天然のE/91 VP2タンパク質をコードするIBDVのEhime91(E/91)株の誘導DNA配列の1359塩基対(bp)は、Genbankアクセッション番号AB024076号として利用できる。この配列の分析によって、最適な植物遺伝子発現と共に、最適でないコドン組成にとって有害であると考えられるいくつかの配列モチーフが存在することが判明した(例えば、米国特許第5,380,831号を参照されたい)。単子葉植物と共に双子葉植物において組換え型VP2タンパク質の産生を改善するために、天然のE/91タンパク質のカルボキシ末端でイソロイシン、アラニン、およびバリン残基が1個ずつ付加されていることを除き、天然のE/91 VP2タンパク質と本質的に同一であるタンパク質(本明細書においてSEQ ID NO:12として開示される)をコードする「植物最適化」DNA配列(SEQ ID NO:11)を作製した。これらの付加アミノ酸のコドンは、J. Castonら(J Virol(2001)、75:10815)の報告に基づいて含めたが、彼らの報告はVP2カプシドアセンブリの最適なVP2プロセシング部位が、451位(Leu対Ile)を除いてE/91の配列と同一であるIBD株UK661(Genbankアクセッション番号NC_004178)VP2配列によってコードされる453位ではなくて最後のアミノ酸であるアミノ酸456位で起こることを示している。このように、アミノ酸454、455、および456位(Ile、Ala、Val)は、VP2遺伝子のアミノ酸456位を操作するためにUK661株に由来した。天然のE/91配列によってコードされるVP2タンパク質、および植物最適化コード領域によってコードされるVP2タンパク質は、99.3%同一であり、アミノ酸454、455、および456位が異なるに過ぎない。対照的に、天然のE/91 VP2コード領域の誘導DNAおよび植物最適化DNAは80.3%同一であるに過ぎない。
【0117】
外来遺伝子を植物の染色体に無作為に組み入れると、如何なる特定の組み込み部位も、組み込み部位に隣接する遺伝子制御エレメントおよびオープンリーディングフレームから新しい異常なタンパク質の偶発的な産生を行う部位となる可能性が存在する。これらの望ましくないそしておそらく有害なタンパク質の産生を消失させるために役立つように、可能性がある六個全ての読み取り枠において翻訳終止コドンをコードするさらなる塩基をVP2コード領域の下流に含めた(SEQ ID NO:13に開示する「万能ターミネーター」)。その後のクローニング段階を可能にするために、これらの制限酵素の認識部位を含む塩基をこの有用な配列に含める。
【0118】
実施例14:植物細胞における伝染性ファブリキウス嚢病植物最適化VP2抗原遺伝子の発現のための基本バイナリベクターpDAB2423の構築
IBD VP2遺伝子(SEQ ID NO:11)の植物最適化ヌクレオチド配列を含む双子葉植物発現ベクターを構築した。基本バイナリベクター(BBV)骨格(図24)を用いて、AgeIリンカーを付加することによって唯一のBamHI部位で改変を行った。新しいバイナリベクター(pDAB2407、図25)によって、T-DNA境界配列のあいだでVP2と選択マーカー発現カセットとのAge1/AgeIライゲーションを行うことができる(pDAB2423、図31)。
【0119】
発現カセットは、DAS5 P60C2からの合成VP2配列(図26、PICOSCRIPT、ハウストン、テキサス州)をBbsIおよびSacI制限酵素によって切除することによって構築した。CsVMVプロモーターおよびアグロバクテリウム・ツメファシエンス(Atu)ORF24 3' UTRをコードするpDAB2406(図27)をNcoIおよびSacIによって切断した。pDAB2406骨格およびVP2インサート断片を、pDAB2406のNcoIおよびSacI部位でライゲーションして、pDAB2415(図28)を得た。ライゲーションしたDNAを大腸菌DH5αコンピテント細胞(Invitrogen)に形質転換して、陽性クローンに関してスクリーニングを行った。HindIII×MluI酵素を用いて陽性クローンを制限分析によって同定し、インサート/ベクター接合部を超えたシークエンシングによって確認した。
【0120】
pDAB2415サブクローンを確認した後、プラスミドをNotIによって切断して、CsVMV/VP2/ORF24断片を単離した。RB7 MARエレメント(米国特許第5,773,689号、米国特許第5,773,695号、米国特許第6,239,328号、国際公開公報第94/07902号、および国際公開公報第97/27207号)およびシロイヌナズナ(At)ユビキチン10(Ubi 10)プロモーター(Plant J. 1997、11(5):1017;Plant Mol. Biol. 1993、21(5):895;Genetics 1995、139(2):921)によって調節される選択マーカー、PAT、およびAtu ORF1 3' UTR(米国特許第5,428,147号;Plant Molecular Biology 1983、2:335;Genbankアクセッション番号X00493)をコードするpDAB2418(図29)をNotIによって直線にした。pDAB2418骨格およびpDAB2415インサート断片を、ゲルにおいて単離して、精製し、NotI部位でライゲーションすると、pDAB2416(図30)が得られた。ライゲーションしたDNAを大腸菌に形質転換して、コロニーをBglIIおよびSacIによる制限酵素消化物によってスクリーニングした。陽性サブクローンのさらなる確認は、NotI接合部を超えるシークエンシングによって確認した。次に、pDAB2416プラスミドをAgeIによって切断して、ベクター骨格からMAR/CsVMV/VP2/ORF24/Ubi10/PAT/ORF1発現カセットを除去した。pDAB2407も同様に、AgeIによって切断して、バイナリベクターを直線にして、適当な断片をライゲーションしてpDAB2423を形成した。ライゲーションしたDNAを形質転換した後、コロニーをHindIIIおよびXhoI消化物を用いてスクリーニングした。採取したコロニー30個のうち、12個が陽性であった。陽性クローンを、NcoI、PmeI、およびPstI酵素を用いて制限消化物によってさらに分析した。最終確認のため、クローンをT-DNA境界配列のあいだで十分にシークエンシングした。
【0121】
(表1)植物由来HNの赤血球凝集活性に及ぼす抽出法の比較
1天然のNDVを2分間超音波処理した。抽出緩衝液−50 mMアスコルビン酸ナトリウム、1 mM EDTA、1 mM PMSF、および0.1%TritonX-100、pH 7.2;DPBS−ダルベッコリン酸緩衝生理食塩液;sonic−超音波処理;F/T−凍結融解;nd−本試験では実施していない
【0122】
(表2)植物由来HNおよび天然のウイルスの赤血球凝集阻害(HAI)活性の比較
1保存ウイルスは4HA単位であり、保存ウイルスの1:4倍希釈に等しい。これは抗体の力価を測定するために用いられるウイルスの濃度であり、ウイルスの4 HA単位を妨害する終点希釈抗体が、抗体調製物のHAI力価であると考えられる。
【0123】
(表3)微少溶液操作の際に様々な圧力を用いたCHN-18トランスジェニック細胞の細胞抽出物の赤血球凝集の比較
MF−微少溶液操作;PSI−ポンド/平方インチ;S/N−上清
【0124】
(表4)ウサギに接種するための用量レベル
1全てのNT-1試料を非凍結乾燥材料から提供した。BCA−ビシンコニン酸、Pierce Chemical BCAタンパク質アッセイキットの主成分;TSP−総可溶性タンパク質。
CHN-7およびCHN-18はNDVからのHNタンパク質を発現する二つの異なるトランスジェニック細胞株であり、CHA-13およびCHA-47はAIVのHAタンパク質を発現する二つの異なるトランスジェニック細胞株である。
【0125】
(表5)トランスジェニック植物細胞CHA-13およびCHN-18に由来するAIV-HAおよびNDV-HNタンパク質を摂取したウサギからの血清学の結果
S/N−上清;HAI−赤血球凝集阻害血清力価
【0126】
(表6)家禽の試験#16に関して接種あたりに用いられる用量レベル
1湿重量細胞あたり赤血球凝集素/ノイラミニダーゼ(HN)の発現に基づく用量;IN−鼻腔内;SQ−皮下;OG−経口針;OF−飼料混合物
【0127】
(表7)家禽の試験#16のCHN-18に関する血清学およびチャレンジ結果(NDVチャレンジ)
全てのトリに、NDVのTexas GB株102 EID50(卵感染用量)を接種した。トリには最後のワクチン接種後24日目にチャレンジした。太字のトリの数は、遅れて死亡を認めた。表9を参照されたい。HAI−赤血球凝集阻害血清力価;na−適用されない。
【0128】
(表8)試験#18に関する接種あたり用いられる抗原の用量レベル
【0129】
(表9)試験#18からのCHN-18の血清学およびチャレンジ結果(NDVチャレンジ)
トリには、NDVのTexas GB株104 EID50 を接種した1群および非チャレンジ対照である3群を除き、全てNDVのTexas GB株102 EID50(卵感染用量)を接種した。トリは最後のワクチン接種後31日目にチャレンジした。赤血球凝集阻害(HAI)力価は、3回の実験の平均力価として報告する。
【0130】
(表10)試験#16および#18に関するチャレンジ後の死亡日(NDVチャレンジ)
*5群のトリ1羽はこの日付で記録しなかった。
【0131】
(表11)CHA-13に関する血清学およびチャレンジ結果(AIVチャレンジ)
1赤血球凝集阻害(HIA)力価は2回の実験の50%終点として報告し、値1は、バックグラウンドを示す。
2チャレンジスコアは、結膜炎=1;抑うつ=2;運動失調=3;麻痺/斜頚=4;死亡/安楽死=5の複合評定によって決定した。
3CorixaアジュバントモノホスホリルリピッドA(MPL)トレハロースジコリノミコレート(TDM)
【0132】
(表12)家禽の試験の接種あたりに用いられる用量レベル
1SQ−皮下;OG−経口針投与;IB−嚢内;SQ OG−1回目の投与は皮下、2回目および3回目は経口針による投与;ドレークオイル乳剤−nCVP2-002と共に等量の5%ドレークオイル、1%Tween80、0.33%スパン80;CGI−精製セルキャップ由来ニワトリγインターフェロン:LAP−nCVP2-002と共に等量のレシチンアクリル酸ポリマー水中油型乳剤
【0133】
(表13)CVP2-002家禽試験の血清学およびチャレンジ結果(IBDVチャレンジ)
1血清中和(SN)力価の範囲は、処置あたりのトリの群に関して血清力価の最低値から最高値までである。
2血清ELISA力価の範囲は、処置あたりのトリの群に関して血清力価の最低値から最高値までである。
3処置群において各トリに関してファブリキウス嚢対体重(BBW)を測定して、各チャレンジ群を非チャレンジ群(#1)と比較し、p値を提供する統計学的有意差が得られる;0.05より大きいp値は保護的であると考えられる。
na−適用されない
【0134】
(表14)様々な熱不安定毒素調製物を用いたY1マウス副腎細胞細胞障害性
1実施例6に記載のようにガングリオシド定量捕獲ELISAを用いて各抗原をアッセイする。
2LT濃度は、LTA特異的抗体を用いてガングリオシドによって捕獲されたLTBに結合するLTAタンパク質の量を表す。
3LTB特異的抗体を用いて毒素のLTB定量に基づく濃度。
LT−熱不安定毒素;LTA−熱不安定毒素のAサブユニット;LTB−熱不安定毒素のBサブユニット。
【0135】
(表15)植物由来抗原および天然の抗原の粘膜輸送(鼻腔内および点眼);処置群あたり投与した用量
1試験に用いた抗原には、CHA-13およびCHN-18植物由来抗原と共に不活化トリインフルエンザウイルス(不活化AIV)が含まれ、日付は10 L発酵器から採取した日付を指し、バッチ番号を示す。
2処置に用いたアジュバントは全て、0.05 mlを眼に点眼することによっておよび各鼻孔に0.2 mlを滴下することによって点眼経路および鼻腔内INによって投与した。LAP−レシチンアクリル酸ポリマー;Quil Aは、樹皮(Quillia saponaria)からのサポニンである;LT−大腸菌からの熱不安定毒素;chol−コレステロール;oil−ドラケオール鉱油。
3投与した用量レベルは、ワクチンをアジュバントと共に混合する前に、バルク抗原に関する定量的ELISAから採取した抗原の希釈液または組み合わせを用いて決定した。
4陽性対照試料はSQ−皮下接種によって投与した。
【0136】
(表16)植物由来抗原および天然の抗原の粘膜輸送ワクチン輸送(鼻腔内および点眼);処置群あたり投与した用量
1,2情報に関しては表15を参照されたい。
3赤血球凝集阻害(HAI)血清力価は、いずれもニワトリ胚の尿膜腔液に由来する不活化AIVおよびNDV抗原を用いて決定した。双方の抗原を各処置の対照として用い、報告された血清力価は、その処置に関する抗原に対する特異的反応であった。
NR−反応なし。
【図面の簡単な説明】
【0137】
本特許のファイルは、カラーで印刷される少なくとも一つの図面を含む。カラーの図面を有する本特許または特許出願の出版物のコピーは、要請に応じて、必要な料金を支払えば特許商標庁から提供されるであろう。
【図1】(SEQ ID NO:1および2)NDV株「Lasota」のHN遺伝子の植物最適化コード配列およびタンパク質配列を示す。
【図2】構成的CsVMV(カッサバ葉脈モザイクウイルス)プロモーターによって促進され、MAS 3'(マンノピンシンターゼ)エレメントによって終止する植物選択マーカーPAT(ホスフィノトリシンアセチルトランスフェラーゼ)を含むpBBV-PHAS-iaaHのマップを示す。DNAの境界を定めるアグロバクテリウムのLBおよびRB(左右T-DNA境界配列)エレメントが植物ゲノムに組み入れられている。
【図3】免疫保護抗原を発現するための多様な植物発現ベクターの開始プラスミドとして用いられる「鋳型ベクター」であるpC!Hのマップを示す。
【図4】NDV HNタンパク質のpCHN発現ベクターのマップを示す。HN発現ベクターまたはカセットは、構成的CsVMVプロモーターによって促進され、大豆vspB 3'エレメントによって終止する。
【図5】NDV HNタンパク質のpgHN発現ベクターのマップを示す。HN発現カセットは、TEV 5' UTRと共に塊茎特異的GBSSプロモーターによって促進され、大豆vspB 3'エレメントによって終止する。
【図6】NDV HNタンパク質のpgHN151発現ベクターのマップを示す。HN発現カセットは、その本来の5' UTRおよびイントロンと共に塊茎特異的GBSSプロモーターによって促進され、大豆vspB 3'エレメントによって終止する。ベクターは、CsVMVプロモーターによって促進されMAS 3'エレメントによって終止する植物選択マーカーPATを含むpBBV-PHAS-iaaHに由来する。DNAの境界を示すLBおよびRB、すなわち左右T-DNA境界配列エレメントが植物ゲノムに組み入れられている。
【図7】NDV HNタンパク質のpgHN153発現ベクターのマップを示す。HN発現カセットは、その本来の 5' UTRおよびイントロンと共に塊茎特異的GBSSプロモーターによって促進され、マメファセオリン3'エレメントによって終止する。ベクターは、CsVMVプロモーターによって促進され、MAS 3'エレメントによって終止する植物選択マーカーPATを含むpBBV-PHAS-iaaHに由来する。DNAの境界を示すLBおよびRB、すなわち左右T-DNA境界配列エレメントが植物ゲノムに組み入れられている。
【図8】NDV HNタンパク質のpMHN発現ベクターのマップを示す。HN発現カセットは、構成的4OCS△MASプロモーター(P2方向)によって促進され、大豆vspB 3'エレメントによって終止する。ベクターは、CsVMVプロモーターによって促進され、MAS 3'エレメントによって終止する植物選択マーカーPATを含むpBBV-PHAS-iaaHに由来する。DNAの境界を示すLBおよびRB、すなわち左右T-DNA境界配列エレメントが植物ゲノムに組み入れられている。
【図9】AIV A/turkey/Wisconsin/68(H5N9)のHA遺伝子のpCHA発現ベクターのマップを示す。
【図10】(SEQ ID NO:3および4)AIV A/turkey/Wisconsin/68(H5N9)のHA遺伝子のDNAおよびタンパク質配列を示す。
【図11】pGLTB中間ベクターのマップを示す。
【図12】pCLT105中間ベクターのマップを示す。
【図13】pDAB2423を示す。VP2をコードするバイナリベクターを示す。
【図14】IBDV伝染性ファブリキウス嚢病(IBD)ウイルスのVP2遺伝子のDNA配列、非常に毒性の高い株Ehime 91を示す。
【図15】CHN-18、CHA-13、SLT102、およびCVP2-002に関する発現されたタンパク質の産生、増殖、および蓄積を示す。10 L発酵器におけるCHA-13の増殖の結果を示す。黒四角:接種後様々な時間での試料10 mlからの細胞沈降容積(PCV)を用いたCn-18 NT-1トランスジェニック細胞の増殖。黒三角:実施例7に記述する定量的ELISAアッセイを用いたHNタンパク質(10 L発酵器あたり)の蓄積。黒菱形は、実施例8に記述の赤血球凝集力価の蓄積であり、1日目に認められた赤血球凝集力価は、13日振とう培養物からの接種物である。
【図16】CHN-18、CHA-13、SLT102、およびCVP2-002に関する発現されたタンパク質の産生、増殖、および蓄積を示す。10 L発酵器におけるCHA-13の増殖の結果を示す。黒四角:接種後様々な時間での試料10 mlからの細胞沈降容積(PCV)を用いたCHA-13 NT-1トランスジェニック細胞の増殖である。白三角は、ショ糖濃度を示し、ショ糖は、炭素源として用いられ、これはデキストロース(白四角)に急速に変換されて、培養物への接種後48時間ではもはや検出できない。定量的ELISAによるHAタンパク質の蓄積は、CHA-13 NT-1の細胞抽出物からの黒三角によって示される。
【図17】CHN-18、CHA-13、SLT102、およびCVP2-002に関する発現されたタンパク質の産生、増殖、および蓄積を示す。10 L発酵器におけるCVP2-002の増殖の結果を示す。黒菱形は、接種後様々な時間での試料10 mlからの細胞沈降容積(PCV)を用いたCVP2-002 NT-1トランスジェニック細胞の増殖を示す。白三角は、ショ糖濃度を示し、ショ糖は、炭素源として用いられ、これはデキストロース(*)に急速に変換されて、培養物への接種後48時間ではもはや検出できない。定量的ELISAによるHAタンパク質の蓄積は、CVP2-002 NT-1の細胞抽出物からの黒三角によって示される。
【図18】CHN-18、CHA-13、SLT102、およびCVP2-002に関する発現されたタンパク質の産生、増殖、および蓄積を示す。シェーカーフラスコ培養におけるLTの蓄積、濃度は実施例7におけるLT定量的ELISAによって決定した。増殖曲線は本試験では決定しなかったが、SLT-102 NT-1細胞のPCVは、図15〜17におけるNT-1トランスジェニック細胞株に関して認められた場合と同じ増殖および産生を示す。
【図19】トランスジェニック細胞株CHN-18からのタンパク質の安定な産生。CHN-18 NT-1から調製した試料の定量的ELISAシグナルの安定性。CHN-18からの上清は、実施例3に記載の10 L発酵器から細胞を回収することによって単離した。細胞を破壊するために、1回微少溶液操作した後、試料を0.45ミクロンまたは0.2ミクロンのフィルターによって濾過して、25℃で保存した。
【図20】共焦点走査顕微鏡。MHN-41染色細胞を示す。緑色:Rb抗HNポリクローナル抗体に関して標識したCy2色素によって染色した細胞(左上)。青色:4A腹水に関して標識したCy5色素によって染色した細胞(右下)。赤色:ヨウ化プロピジウム染色核(右上)。水色/赤:緑、青、および赤色の画像のデジタル融合画像。細胞の核には、いずれの抗体による染色も認めない。細胞壁全体および細胞内細胞質の膜に沿って強く染色された領域のために、液胞は区別できない。
【図21】共焦点走査顕微鏡。対照NT-1細胞を示す。対照NT-1細胞の染色、左のパネルはRb抗HNポリクローナル抗体または4A腹水のいずれかによって染色した細胞である:右のパネルはヨウ化プロピジウム染色NT-1細胞である。
【図22】トランスジーンによって産生されたポリペプチドの局在を示す電子顕微鏡写真を示す。NT-1対照細胞、CHN-18トランスジェニック細胞、およびMHN-41トランスジェニック細胞の四酸化オスミウム固定細胞の電子顕微鏡写真。各フレームの倍率を示し、対照細胞は倍率16,000倍、CHN-18は50,000倍、およびMHN-41は26,000倍である。
【図23】トランスジーンによって産生されたポリペプチドの局在を示す電子顕微鏡写真を示す。NT-1対照細胞、CHN-18トランスジェニック細胞、およびMN-41トランスジェニック細胞の電子顕微鏡の免疫金染色。
【図24】バイナリ、中間および発現ベクターのマップを示す。基本バイナリベクター(BBV)のマップを示す。
【図25】バイナリ、中間および発現ベクターのマップを示す。中間pDAB2407のマップを示す。
【図26】バイナリ、中間および発現ベクターのマップを示す。PICOSCRIPT(ハウストン、テキサス州)によって提供されたBluescriptベクターにおける合成されたVP2を示す。
【図27】バイナリ、中間および発現ベクターのマップを示す。中間ベクターpDAB2406のマップを示す。
【図28】バイナリ、中間および発現ベクターのマップを示す。中間ベクターpDAB2415のマップを示す。
【図29】バイナリ、中間および発現ベクターのマップを示す。中間ベクターpDBA2418のマップを示す。
【図30】バイナリ、中間および発現ベクターのマップを示す。中間ベクターpDAB2416のマップを示す。
【図31】バイナリ、中間および発現ベクターのマップを示す。CsVMVプロモーターによって促進され、上流のRB7 MARエレメントと共にAtu ORF24 3' UTRによって終止するVP2を示す双子葉植物発現ベクターpDAB2423のマップを示す。選択マーカーPATは、At Ubi 10プロモーターおよびAtu ORF1 3' UTRによって調節される。
【技術分野】
【0001】
発明の分野
本発明は、一般的に免疫学の分野に関し、免疫保護組成物およびトランスジェニック植物細胞からそのような組成物を調製する方法を提供する。本発明はまた、タンパク質産生(例えば、酵素、毒素、細胞受容体、リガンド、シグナル伝達物質、サイトカイン、またはトランスジェニック植物細胞培養物において発現される他のタンパク質の組換えによる産生)の分野に関し、これらのタンパク質を含む組成物を提供する。
【0002】
関連出願の相互参照
本出願は、全ての図、表、アミノ酸配列、およびポリヌクレオチド配列を含む、その全内容物が参照により本明細書に組み入れられる、2003年5月5日に提出された米国特許仮出願第60/467,999号の恩典を主張する。
【背景技術】
【0003】
発明の背景
特定の病原体に対する全身免疫は、外来物質に反応した生得のまたはT-細胞/B-細胞性免疫系の活性化が原因で起こる。それらの物質はしばしば、特定の病原性微生物または特定の病原物質に対して保護するように設計されたワクチンの抗原となりうる。病原体に対する曝露はしばしば、病原性微生物に絶えず曝露され、チャレンジされる粘膜表面を通して起こる。
【0004】
粘膜および口腔免疫によって、呼吸器、消化管、および尿生殖器の粘膜表面によって分泌される、および全ての分泌腺からの分泌物においてsIgA(分泌IgA)抗体の産生が起こる。McGhee, J.R.ら、Annals N.Y. Acad. Sci. 409(1983)。これらのsIgA抗体は、粘膜表面上で病原体の定着を防止するように作用し(Williams, R.C.ら、Science 177、697(1972);McNabb, P.C.ら、Ann. Rev. Microbiol. 35、477(1981))、粘膜表面を通しての定着または浸潤を防止するための免疫防御メカニズムの重要な特徴である。sIgAの産生は、分泌腺もしくは組織の局所免疫によって、またはGALT(腸管関連リンパ様組織もしくはパイエル板)もしくはBALT(気管支関連リンパ様組織)のいずれかに対する抗原の提示によって刺激することができる。Cebra, J.J.ら、Cold Spring Harbor Symp. Quant. Biol. 41、210(1976);Bienenstock, J.M.、Adv. Exp. Med. Biol. 107、53(1978);Weisz-Carrington, P.ら、J. Immunol. 123、1705(1979);McCaughan, G.ら、Internal Rev. Physiol 28、131(1983)。膜性ミクロフォルド細胞、またはM細胞として知られる細胞は、GALTおよびBALTの表面を覆い、他の分泌性粘膜表面に会合する可能性がある。M細胞は、粘膜表面に隣接する内腔から抗原を採取して、そのような抗原を抗原提示細胞(樹状細胞およびマクロファージ)に移動させるように作用し、次に抗原提示細胞が抗原をTリンパ球に提示する(T-細胞依存的抗原の場合)。次に、B細胞が刺激を受けて、増殖して遊走し、最終的に、提示された抗原に対するIgAを産生する抗体分泌プラズマ細胞へと変化する。抗原がGALTおよびBALTの上に存在するM細胞によって取り込まれると、全身性の粘膜免疫が起こり、抗原に対するsIgAが体内の全ての分泌組織によって産生される。Cebraら、上記;Bienenstockら、上記;Weinz-Carringtonら、上記:McCaughanら、上記。したがって、口腔曝露による免疫保護は、全身性の粘膜免疫応答を刺激するための重要な経路であり、さらに、口腔および消化管における分泌性免疫応答の局所刺激が起こる。
【0005】
その上、粘膜免疫は、都合よく子孫に伝達されうる。新生児の免疫は、初乳および/または乳汁を通して受動的に獲得される可能性がある。これは、催乳性免疫と呼ばれており、生後間もない動物を保護する効率的な方法である。sIgAは、乳汁における主要な免疫グロブリンであり、粘膜免疫によって最も効率的に誘導される。
【0006】
腸管関連リンパ様組織のパイエル板の上に存在するM細胞は、多様な抗原材料および粒子を取り込むことができる(Sneller, M.C. and Strober, W.、J. Inf. Dis. 154、737(1986))。M細胞はラテックスおよびポリスチレン球体、木炭、マイクロカプセル、ならびに他の可溶性および微粒子物質を取り込むことができることから、輸送される材料の任意の特異的な接着型の特性とは無関係に、GALTに多様な材料を輸送することができる。したがって、植物由来免疫保護抗原として適当な大きさの安定かつ強力な粒子を産生するための組成物および手段があれば、動物病原体に対する植物産生粘膜ワクチンの開発を大きく促進するであろう。
【0007】
組換えDNA技術は、ワクチンを含む薬学および獣医学医薬品の安全性、品質、有効性、および費用において実質的な改善を提供した。植物によって産生される粘膜ワクチンは、Curtiss & Cardineauによって発明された。参照により本明細書に組み入れられる、米国特許第5,654,184号;第5,679,880号;および第5,686,079号を参照されたい。Arntzen、MasonおよびLamを含む他の研究者らは、免疫保護抗原を発現するトランスジェニック植物およびその産生法を記述した。例えば、参照により本明細書に組み入れられる、米国特許第5,484,717号;第5,914,123号;第6,034,298号;第6,136,320号;第6,194,560号;および第6,395,964号を参照されたい。
【0008】
既定の培地における細胞培養を用いて植物細胞を産生すれば、産生プロセスから病原体混入物を伝搬するリスクが本質的になくなり、増殖培地に動物起源の成分が存在する必要がなくなる。植物細胞は、ワクチンの製造において現在用いられている通常の増殖培地と比較して、一般的に安価であり、取り扱いおよび調製が容易である。
【0009】
植物系において産生された薬理学的または関連生物活性を有するワクチン抗原およびタンパク質は、通常の産生システムに対して多くの長所を提供する。植物由来のサブユニットタンパク質は、毒性型に復帰することができない(通常に、または組換えによって産生された生きたベクターのワクチンの特徴)。通常の産生法によって産生されるサブユニットタンパク質は、タンパク質の不安定性および生化学的抽出問題のために産生および精製が難しく、グリコシル化を必要とするサブユニットワクチン組成物は、原核細胞において産生すると、グリコシル化されないであろう。
【0010】
植物は、如何なる単一の通常のまたは哺乳類由来の組換えDNA系から誘導することが難しい独自の利益を提供する。従来、サブユニットワクチンまたは生物活性タンパク質は:1)産生できないほど低い収率のために組換えまたは通常の起源から精製することが難しい;2)タンパク質分解、pH、または精製の際に用いられる溶媒のために不安定である;3)それらが天然でないために有効性が低い、または精製プロセスが重要なエピトープを変性させる;および4)哺乳類系において産生された場合に、生物起源の外来材料によって妨害される(先に記述)。
【発明の開示】
【0011】
発明の概要
本発明は、免疫原または他のポリペプチドを発現するように遺伝的に形質転換された機械的または物理的に破壊された植物細胞が、ワクチン、工業、製薬、および薬理学調製物において有用な生物活性タンパク質および免疫保護粒子を産生するという予想外の知見に基づいている。さらに、これらのタンパク質は、製剤およびその後の加工機能において安定性および強健性を示す。
【0012】
本発明は、後期指数増殖期および増殖静止期において植物細胞培養物に蓄積する少なくとも一つの免疫保護抗原または機能的タンパク質を発現する形質転換植物細胞から調製された粒子を含む、安定かつ有効な組成物を作製する方法を提供する。抗原または機能的タンパク質は、植物細胞の細胞質細胞壁および膜領域に蓄積して、機械的もしくは物理的破壊、または何らかの他の手段によって粒子の形で放出されうる。さらに、抗原または機能的タンパク質は、植物細胞の細胞質細胞壁および膜に生物活性型で安定化され、請求の方法の際におよびその後も安定かつ活性である。さらなる態様において、抗原または機能的タンパク質を産生する方法には、下等植物、単子葉植物または双子葉植物、細胞および培養を用いることが含まれる。方法のさらなる態様は、AIV(トリインフルエンザウイルス)のHA(赤血球凝集素)タンパク質、1型糖タンパク質;トリNDV(ニューカッスル病ウイルス)のHN(赤血球凝集素/ノイラミニダーゼ)タンパク質、2型糖タンパク質(参照により本明細書に組み入れられる、米国特許第5,310,678号を参照されたい);伝染性ファブリキウス嚢病ウイルス(IBDV)の構造タンパク質、VP2;酵素ADPリボシルトランスフェラーゼ(大腸菌の熱不安定毒素のLT-Aサブユニット);二つのサブユニットで構成される大腸菌の細菌毒素LT、口蹄疫ウイルス(FMDV)のようなピコルナウイルス、ポリオウイルス、ヒトライノウイルス(HRV)、A型肝炎ウイルス(HAV)、免疫不全ウイルス(HIV)、ヒト乳頭腫ウイルス(HPV)、単純ヘルペスウイルス(HSV)、および呼吸合胞体ウイルス(RSV)が含まれるがこれらに限定されないヒトウイルス、が含まれるがこれらに限定されない、特定の病原性ウイルスの免疫保護粒子における免疫原性タンパク質の産生を提供する。本発明はまた、静止期において細胞質細胞壁および膜構造に蓄積する少なくとも一つの免疫保護抗原または生物活性タンパク質を有する、機械的破壊のような手段によって容易に抽出することができる、凍結保存、凍結乾燥または懸濁液においても安定である、および天然のタンパク質と類似の特徴を有する、植物細胞可溶性抽出物を含む生物活性組成物を提供する。さらに、これらのタンパク質は、カリフラワーモザイクウイルスのS35、カッサバ葉脈モザイクウイルス、アグロバクテリウム・ツメファシエンス(Agrobacterium tumerfacians)のモノピン/オクトピンプロモーターが含まれるがこれらに限定されない、いくつかの異なるタイプのプロモーター系によって発現されると、後期指数増殖期および静止期において沈着される。組換え型蛋白質および植物細胞材料を含むこれらの組成物は、一つまたはそれ以上の薬学的に許容されるアジュバント、希釈剤、担体、または賦形剤と会合させることができる。さらなる態様において、組成物には、下等植物、単子葉または双子葉植物由来粒子と共に、特定の植物細胞および培養物に由来する粒子が含まれる。請求の組成物のさらなる態様は、植物細胞において産生される酵素ADPリボシラーゼ;構造タンパク質VP2;1型糖タンパク質;および2型糖タンパク質を含む。トリNDVのHNタンパク質およびAIVのHAタンパク質を含む、特定の病原性ウイルスの特異的免疫原性タンパク質も同様に、本発明の態様である。
【0013】
配列の概要
図1aおよび1bに示すSEQ ID NO:1および2は、NDV株「Lasota」のHN遺伝子の植物最適化コード配列およびタンパク質配列である。
【0014】
図10に示すSEQ ID NO:3および4は、AIV A/turkey/Wisconsin/68(H5N9)のHA遺伝子のDNAおよびタンパク質配列である。
【0015】
SEQ ID NO:5は、pCP!H上でCsVMVプロモーターを末端に配置するために用いられるPCRプライマーである。
【0016】
SEQ ID NO:6は、pCP!H上でCsVMVプロモーターを末端に配置するために用いられるPCRプライマーである。
【0017】
SEQ ID NO:7はNco I部位を作製するために用いられる変異誘発プライマーである。
【0018】
SEQ ID NO:8は、5'領域と相補的なフォワードプライマーである。
【0019】
SEQ ID NO:9はXhoI I部位を作製するために用いられる変異誘発プライマーである。
【0020】
図14に示すSEQ ID NO:10は、伝染性ファブリキウス嚢病ウイルスのVP2遺伝子のDNA配列である。
【0021】
SEQ ID NO:11は、E/91 VP2(1425塩基)の変種をコードする植物最適化DNA配列である。E/91植物最適化VP2のコード領域は、塩基16位〜1383位(塩基1371個)を含む。フレームストップ6個が塩基1384〜1425位に認められる。
【0022】
SEQ ID NO:12は、E/91 VP2コード領域(SEQ ID NO:11)の植物最適化版によってコードされるE/91 VP2タンパク質の配列を含む。
【0023】
SEQ ID NO:13は、読み取り枠6個において翻訳終止(「終止」)コドンをコードするDNA配列である。配列は、形質転換の際のDNA組み込み後の偶然のオープンリーディングフレームの翻訳を終了させるために用いられ、これにはSac I BstE IIおよびBgl II制限酵素認識部位が含まれる(Tsukamoto K., Kojima, C., Komori, Y., Tanimura, N., Mase, M., and Yamaguchi, S.(1999)、「Protection of chickens against very virulent infectious bursal disease virus(IBDV)and Marek's disease virus(MDV)with a recombinant MDV expressing IBDV VP2」、Virol. 257:352〜362)。
【0024】
発明の詳細な説明
免疫原または免疫保護抗原は、動物が免疫原を有する病原体に対する将来の曝露に対して保護されるように、健康な動物において生得の液性および/または細胞性免疫応答を誘発する物質である。これらの病原体は典型的に、ウイルス、細菌、真菌、および原虫のような要因である。免疫原はまた、細胞壁成分およびウイルス外皮タンパク質を含む病原体の抗原性の一部であってもよい。
【0025】
生物活性タンパク質には、糖質(例えば、α-アミラーゼ[細菌α-アミラーゼ(例えば、枯草菌)、真菌α-アミラーゼ(例えば、アスペルギルス・ニガー、アルカリα-アミラーゼ];β-アミラーゼ;セルラーゼ;β-グルカナーゼ;エキソ-β-1,4-グルカナーゼ、エンド-β-1,4-グルカナーゼ;β-グルコシダーゼ;デキストラナーゼ;デキストリナーゼ;α-ガラクトシダーゼ(メリビアーゼ);グルコアミラーゼ;ヘミセルラーゼ/ペントサナーゼ/キシラナーゼ;インベルターゼ;ラクターゼ;ナリンギナーゼ;ペクチナーゼ;プルラナーゼ);プロテアーゼ(例えば、酸プロテナーゼ;アルカリプロテアーゼ;ブロメライン;ペプシン;アミノペプチダーゼ;エンドペプチダーゼ;サブチリシン);リパーゼ、およびエステラーゼ(例えば、ホスホリパーゼ;プレガストリックエステラーゼ;ホスファターゼ;アミノアシラーゼ;グルタミナーゼ;ライソザイム;ペニシリンアシラーゼ;イソメラーゼ);オキシレダクターゼ(例えば、アルコールデヒドロゲナーゼ;アミノ酸オキシダーゼ;カタラーゼ;クロロペルオキシダーゼ;ペルオキシダーゼ);リアーゼ(例えば、アセトラクテートデカルボキシラーゼ;アスパラギン酸β-デカルボキシラーゼ;ヒスチダーゼ);またはトランスフェラーゼ(例えば、シクロデキストリングリコシルトランスフェラーゼ)を含む、酵素、毒素、細胞受容体、リガンド、シグナル伝達物質、サイトカイン、またはトランスジェニック植物細胞培養において発現される他のタンパク質が含まれるがこれらに限定されない。これらの酵素(または毒素、細胞受容体、リガンド、シグナル伝達物質、または本発明の発現系において発現のために適したサイトカイン)をコードするポリヌクレオチドは、EMBL、SWISSPROT、またはNCBIデータベースのような市販のデータベースから得ることができる。典型的に、トランスジェニック植物細胞培養物において産生された生物活性タンパク質は、天然資源から単離された同じタンパク質と機能的または構造活性において同等である。
【0026】
生物活性タンパク質粒子は、本発明の方法によって調製される生物活性タンパク質を発現するトランスジェニック植物細胞に由来する、組換え型蛋白質、植物タンパク質、脂質、糖質、核酸またはその組み合わせからなる異種粒子または凝集物として定義される。特定の態様において、生物活性タンパク質粒子は、脂質小胞、膜断片、細胞壁断片、細胞下オルガネラもしくは断片、または典型的に形質転換植物細胞の後期指数増殖期および増殖静止期に由来する貯蔵タンパク質の一部となりうる、またはそれらに会合しうる。請求される粒子は非常に安定であり、非常に安定かつ生物学的に活性なコンフォメーションで組換え型蛋白質を維持する。他の態様において、粒子は、植物細胞に導入された組換え型遺伝子からのタンパク質を発現する後期指数増殖期または増殖静止期トランスジェニック細胞培養物を機械的または物理的に破壊することによって、緩衝液または培養上清において容易に懸濁することができる材料として定義される。
【0027】
免疫保護粒子は、免疫保護抗原を発現するように遺伝子操作されているトランスジェニック植物細胞に由来する、またはそこから得られる。請求の免疫保護粒子は、ヒトを含む動物に適切に投与されると、免疫原を有する病原体に対する将来の曝露に対して保護を提供する操作されたトランスジェニック植物細胞に由来する組換え型免疫保護抗原、タンパク質、脂質、糖質、核酸、またはその組み合わせからなる異種粒子または凝集物である。請求の粒子は、非常に安定であり、非常に安定かつ生物学的に活性なコンフォメーションで組換え型蛋白質を維持する。免疫保護粒子は、操作された細胞の機械的または物理的破壊の後に、細胞の破片を免疫保護粒子から分離することによって得られる。特定の態様において、粒子は、脂質小胞、膜断片、細胞壁断片、細胞下オルガネラもしくは断片、または形質転換植物細胞の後期指数増殖期および増殖静止期に由来する貯蔵タンパク質の一部となりうる、またはそれらに会合しうる。他の態様において、粒子は、植物細胞に導入された組換え型遺伝子からのタンパク質を発現する後期指数増殖期または増殖静止期トランスジェニック細胞培養物を機械的または物理的に破壊することによって、緩衝液または培養上清において容易に懸濁することができる材料として定義される。
【0028】
下等植物は、シダ、裸子植物、針葉樹、トクサ、ヒカゲノカズラ、ゼニゴケ、キンギョ藻、苔、紅藻類、褐藻類、配偶体、シダ植物の胞子体、および緑藻類を含む任意の非顕花植物として定義される。苔が特に好ましい。
【0029】
ワクチン接種およびワクチン接種することは、宿主免疫系が刺激されて、病原体のその後の曝露に対する宿主反応に関連したその後の望ましくない病態を予防または減弱させるように、免疫原調製物、免疫保護粒子、または病原体の免疫原性調製物、またはその非毒性型もしくはその一部を宿主に接種することによって、病原体に対する保護を提供する手段として定義される。
【0030】
ワクチンは、少なくとも一つの免疫保護抗原性物質を含む、ヒトを含む動物にワクチン接種するために用いられる組成物である。
【0031】
病原性微生物は、感染した動物において疾患を引き起こす、および/または生理的状態を誘導する/制御する、細菌、ウイルス、真菌、または原虫である。
【0032】
本明細書の目的に関して、アジュバントは、免疫原または抗原に対する免疫応答を加速、増加、適度にする、または増強する物質である。アジュバントは典型的に、液性および細胞性免疫応答の双方を増強するが、他方が存在しない場合に反応が増加することがアジュバントの定義として適格である。その上、アジュバントおよびその使用は、免疫学者に周知であり、典型的に、免疫原の用量が限られている場合、免疫原の免疫原性が低い場合、または投与経路が最適下である場合に、免疫応答を増強するために用いられる。このように、「アジュバント量」は、所定の免疫原または抗原に対する免疫応答を増強することができるアジュバントの量である。「アジュバント量」に等しい質量は変化して、免疫原の特徴、投与される免疫原の量、宿主種、投与経路、免疫原の投与プロトコールが含まれるがこれらに限定されない多様な要因に依存する。「アジュバント量」は、特定の一連の状況を考慮して、普通の実験によって容易に定量することができる。これは十分に当業者の範囲内であり、典型的に様々な量の投与される免疫原およびアジュバントに対して普通の用量反応決定を用いる。反応は、酵素結合イムノソルベントアッセイ、ラジオイムノアッセイ、赤血球凝集アッセイ等を用いて、免疫原に対する血清抗体力価または細胞性反応を決定することによって測定される。
【0033】
本発明はまた、免疫保護もしくは生物活性タンパク質もしくは粒子を含む薬学的および獣医学組成物、または一つもしくはそれ以上の薬学的に許容されるアジュバント、担体、希釈剤、および賦形剤と併用した本発明の組成物を提供する。そのような薬学的組成物はまた、ワクチンとも呼ばれ、薬学およびワクチンの技術分野において周知の方法で製剤化される。
【0034】
投与または投与することは、ヒトを含む動物の体内に物質を導入することとして定義され、これには経口、鼻腔内、点眼、直腸内、膣内、および非経口経路が含まれる。請求される組成物は、皮下(SQ)、筋肉内(IM)、静脈内(IV)、腹腔内(IP)、皮内(ID)、鼻腔内、点眼、口腔粘膜(IN)、または経口投与が含まれるがこれらに限定されない任意の投与経路によって、個々に、または他の治療物質と併用して投与してもよい。粘膜経路が特に好ましく、経口経路が最も好ましい。
【0035】
有効量は、ヒトまたは動物が、病原体によって開始される攻撃に有効に抵抗するため、および糖尿病に対する自己免疫抗原のような、動物の生理的要件に反応するために十分な、ヒトまたは動物における免疫応答を誘発するために必要な量である。そのようなヒトまたは動物に投与される用量は、特定の免疫保護粒子、または粒子の組み合わせ、ヒトまたは動物の状態、および選択された投与経路を含む関連する状況に照らして医師、獣医師、または訓練された化学者によって決定されるであろう。一般的に、有効量は約1 ng〜約0.5 mgであり、好ましくは約1 μg〜約50 μgである。家禽におけるニューカッスル病ウイルス(HN抗原)の場合、有効量は約0.5 μg〜約50 μg、好ましくはSQ経路によって約2.5 μg〜約5 μgである。IN/点眼粘膜経路の場合、家禽におけるHNの有効量は、約0.5 μg〜約50 μg、好ましくは約5 μg〜約25 μg、より好ましくは約10 μg〜約12 μgである。トリインフルエンザウイルス(HA抗原)の場合、有効量は、約0.5 μg〜約50 μg、好ましくは約1 μg〜約30 μg、より好ましくはIN/点眼経路によって約24 μg〜約26 μgであり、好ましくはSQ経路によって約1 μg〜約5 μgである。家禽における伝染性ファブリキウス嚢病(VP2抗原)の場合、有効量は0.5 μg〜約50 μg、好ましくは約5 μg〜約25 μg、より好ましくはSQ経路によって約5 μg〜約20 μgである。LT抗原の場合、経口有効量は約50 ng〜約250 ng、好ましくは約100 ng〜約200 ngである。LT抗原の場合、SQまたはIN/点眼有効量は約50 ng〜約100 μg、好ましくは約1 μg〜約25 μg、より好ましくは約2 μg〜約10 μgである。本明細書において示した用量範囲は、いずれにせよ本発明の範囲を制限すると解釈されず、熟練した医師の一般的指針として示される。
【0036】
本明細書において、トリは、典型的に翼に変化した前足、鱗状の脚、嘴を有し、硬い殻に包まれた卵を産む、鳥綱の任意の温血脊椎動物として定義される。本明細書の目的に関して、好ましいトリの群は、家畜用ニワトリ、七面鳥、ダチョウ、アヒル、ガチョウ、およびコーニッシュ種の闘鶏である。より好ましい群は、家畜用ニワトリおよび七面鳥である。本発明の目的に関して最も好ましいトリは、ブロイラーおよび産卵鳥(家禽)を含む家畜用ニワトリである。
【0037】
本発明の方法および組成物は、ヒトを含む動物、好ましくはトリ(家禽)、ウシ、ヒツジ、ヤギ、ブタ、ウマ、ネコ、イヌ、およびラマのような家畜用動物、最も好ましくはトリを免疫および保護することに向けられる。これらの動物種の特定のものは、多数の胃を有し、植物を分解するために特異的な消化酵素を有しえて、そうでなければ、他のタイプの経口ワクチンを容易に不活化する可能性がある。本発明を制限することを意味しないが、トランスジェニック植物細胞およびそれに由来する組成物を摂取することによって、扁桃を含む口腔粘膜部位で動物の免疫が起こりうる。
【0038】
本発明の目的に関して、膜配列という用語は、当業者がその用語を理解する意味であることを企図する。膜固定配列には、膜貫通タンパク質配列が含まれ、天然に存在する多くのタンパク質において認められる。そのような膜固定配列は大きさが多様であるが、常に、膜内の疎水性環境に都合がよい親油性または脂肪族側鎖を有する一連のアミノ酸を含む。RNAの翻訳および翻訳後プロセシングの際に、固定配列が組み入れられ、細胞膜に埋もれて、細胞膜成分にタンパク質を固定または緩く結合させるように機能し、細胞内外の水性環境にタンパク質の親水性部分を露出させて、それらと相互作用させる。
【0039】
本明細書における貯蔵封入体または貯蔵タンパク質は、窒素源として植物が利用するタンパク質であると定義され、これらのタンパク質は、植物のライフサイクルの非生産相(例えば静止期)のあいだに貯蔵され、細胞が活動的増殖へと誘導された場合に、エネルギーおよび窒素源として急速に利用される。
【0040】
本明細書において、トランスジェニック植物は、植物細胞培養、植物細胞株、植物組織培養、下等植物、単子葉植物、双子葉植物、または形質転換された植物細胞もしくはプロトプラストに由来するその子孫であると定義され、形質転換植物のゲノムは、実験技術によって導入され、同じ種の天然の非トランスジェニック植物細胞には通常存在しない外来DNAを含む。「トランスジェニック植物」および「形質転換植物」という用語は時に、そのDNAが外因性DNA分子を含む植物を定義するための同義語として当技術分野において用いられている。
【0041】
植物において免疫保護抗原を発現するための遺伝子カセットの構築は、Sambrookら(1989)およびAusubelら(1987)、「Current Protocols in Molerular Biology」、John Wiley and Sons、ニューヨーク、ニューヨーク州に開示される方法のような、周知の方法を利用して容易に行われる。本発明にはまた、それらが発現時に開示の作用を有することができるように、免疫保護抗原をコードする開示の配列と実質的な配列相同性を有するDNA配列が含まれる。本出願において用いられるように、「実質的な配列相同性」という用語は、ヌクレオチド配列(DNAまたはRNAの場合)またはアミノ酸配列(タンパク質またはポリペプチドの場合)が、もう一つのヌクレオチドまたはアミノ酸配列と実質的、機能的、または構造的同等性を示すことを示すために用いられる。実質的な配列相同性を有する配列間の任意の機能的または構造的な差は小さいであろう;すなわち、それらは本出願において示されるように機能する配列の能力に影響を及ぼさないであろう。本明細書に開示の配列と実質的な配列相同性を有する配列は通常、変異のような、開示の配列の変種であるが、合成配列であってもよい。
【0042】
ほとんどの場合、本明細書において特に開示された配列と95%相同性を有する配列は、同等物として機能して、多くの場合かなり低い相同性、例えば75%または80%が許容されるであろう。重要でないこれらの配列の部分を特定することは時間がかかるが、普通のことであり、十分に当業者の範囲内である。オリゴヌクレオチド配列を改変する例としての技術には、ポリヌクレオチド媒介、部位特異的変異誘発を用いることが含まれる。Zollerら(1984);Higuchiら(1988);Hoら(1989);Hortonら(1989);および「PCR Technology:Principles and Applications for DNA Amplifications」Erlich(編)(1989)を参照されたい。
【0043】
ほとんどの場合、組換え型DNAによるタンパク質産生のために用いられる哺乳類細胞、細菌細胞、または他の宿主ベクター系は、再生された培養環境に置いた場合に再利用することができるタンパク質貯蔵を確立しない。大腸菌に関して記述される封入体、またはバキュロウイルス、グラニュローシスウイルス、もしくはバシラス・スリンギエンシスの結晶タンパク質は、様々な生物学的目的のために宿主系によって貯蔵される。しかしながら、いずれも、再培養の際または休止期もしくは静止期から新たな増殖の活性化の際の窒素源として植物が利用することができる貯蔵区画にタンパク質を入れることは示されていない。
【0044】
NT-1増殖の静止期の後期段階で細胞における貯蔵区画または安定な部位にタンパク質を入れることは、インビトロで培養したトランスジェニック植物細胞におけるタンパク質発現の予想された特徴ではなかった。電子顕微鏡から、白色体に暗い中心部があること、そして細胞壁および膜に隣接する細胞質区画にタンパク質に対する免疫金標識結合が存在することが示されている(実施例16を参照されたい)。さらに、NT-1系が、発現されたタンパク質のタイプまたは用いられる転写プロモーター系によらず、安定な区画にタンパク質を貯蔵できることは、これまでに例のない知見である。首尾よく発現されているタンパク質には、いくつかの異なるクラスのタンパク質が含まれる:1)酵素ADPリボシルトランスフェラーゼ、大腸菌のLTエンテロトキシンのLTA区画;2)大腸菌に由来するLTAおよびLTBサブユニットの双方を含む十分に形成されたおよび機能的LTホロトキシン;3)伝染性ファブリキウス嚢病ウイルス(IBDV)の構造タンパク質;4)トリインフルエンザウイルス(AIV)の1型ウイルス糖タンパク質赤血球凝集素(HA);および5)ニューカッスル病ウイルスの2型ウイルス糖タンパク質。それぞれの場合において、発現されたタンパク質の生物活性は、それぞれの病原体に由来する天然のタンパク質ほど強力ではないものの強力であることが判明した。各タンパク質に関連した有効性は、外来タンパク質の発現のために用いられる単一のタイプの宿主細胞に関する予想外の特徴である。保存された外来タンパク質のもう一つの予想外の特徴は、それが安定であることであり、先に記述したように、タンパク質を容易に単離できることである(例えば、機械的破壊によって)。懸濁されたタンパク質またはタンパク質含有粒子は、シグナルまたは安定性を失うことなく凍結乾燥、凍結、乳化、ホモジナイズ、または微少溶液操作することができる。本発明のタンパク質および粒子を液体型で2〜7℃で長期保存すると、長い半減期を示す;如何なる安定化剤も加えなくても、単純な機械的攪拌によって産生された抽出物から、NDVのHNタンパク質に関して予測半減期1〜2年、および大腸菌のLTに関して13〜15ヶ月の調製物が得られた。本発明に従って産生されたタンパク質は、極めて強健であり、免疫応答を増強することができる様々なタイプの製剤にすることができる。
【0045】
請求の方法と一致する物理的または機械的細胞破壊技術には、超音波、微少溶液操作または他の剪断型の方法、高剪断ローター/ステーター法、フレンチプレスまたは他の加圧法、およびホモジナイゼーション法が含まれるがこれらに限定されない。初期の研究開発活動によって、回収された細胞からHNタンパク質を免疫保護粒子の形で抽出するためには高圧破壊エネルギーが必要であることが示された。小さい発酵器アッセイ容量(1〜10 ml)からHN免疫保護粒子を放出するためには、超音波破壊が利用されたが、採取された細胞容積が1 Lを超える場合には、HN免疫保護粒子を回収するために超音波破壊はそれほど有効でなく(>35%)、規模を拡大することができないことが示されている。
【0046】
Microfludics Inc.の固定開口部圧破壊装置を用いる力量滴定試験から、破壊圧はNT1-CHN-18細胞から回収されるHN免疫保護粒子の収量と比例することが示された。HN粒子の最高の回収は、18,000 psigである機器の最高圧の設定で得られた。最高圧であっても、総HNタンパク質の40%を超える量が廃棄された細胞破片分画に存在した。この後者の実験の圧力滴定曲線から、より高い溶解圧が、廃棄される細胞破片分画におけるHNタンパク質の量を有意に減少させる可能性があることが示唆される。
【0047】
Microfludics社の製品ラインは、一定の細胞破壊技術において「第二世代」であると見なされている。Microfludics社の機器は、水撃ポンプによって高い流速で空にされるリザーバーに接続された固定された0.1 mmの乱流(「Y」外形)開口部に、懸濁細胞を通過させることによって細胞破壊を行う。ラムが上に上がると逆止め弁が開き、次のサイクルのためにリザーバーを満たす。Microfludics Incは、およそ10 ml*/分〜10,000 L*/時間の規模で同等性を主張する。細胞の分解は、(1)開口部(内破)による加速、(2)細胞破壊を引き起こす開口部先端と噴出室とのあいだの圧力の差、および/または(3)噴出室標的への減速、の結果であると考えられている。細胞の超微細構造(すなわち、細胞壁)、細胞濃度、破壊エネルギー(psig)、および分解緩衝液の組成は、溶解効率に影響を及ぼす最も重要な変数であると見なされる。
【0048】
より初期の第一世代機器は、Aminco Inc.によって連続的フレンチプレスセルとして産生された。これらは、開口部の直径と水圧を手動で制御することを除いて、Microfludics社の機器と類似である。これらの後者の機器は、主に試料容積50 ml未満の研究開発活動に用いられる。第三世代の一定細胞破壊機器(DeBEE, Inc.およびConstant Systems, Inc.)には、より高い操作圧(60,000 psigまで)、圧力の変動を減少させるための二重の試料室、および真空下で操作される試料放出室のような改善が含まれた。これらの改善は、第一および第二世代の機器と比較して改善された分解効率を有すると報告された。
【0049】
請求の方法の清澄段階には、重力沈降、遠心、浮上分離、接線流を含む濾過、ならびに全ての形のカラムおよびHPLC法を含むクロマトグラフィー技術が含まれるがこれらに限定されない任意の分離技術が含まれる。好ましい方法は、数分間の約1000 g〜約5000 gの低速遠心である。
【0050】
本発明の構築物を調製する場合、適切な方向、および適当であれば適切な読み取り枠でDNA配列を提供するために、様々なDNA断片を操作してもよい。DNA断片を結合するためにアダプターもしくはリンカーを用いてもよく、または都合のよい制限部位、過剰なDNAの除去、制限部位の除去等を提供する他の手段を含んでもよい。
【0051】
様々な段階を行う場合、所望の宿主細胞にその後導入するために、プロモーター/対象遺伝子を含むベクターを増幅するためにクローニングを用いる。広範なクローニングベクターが市販されており、クローニングベクターには、大腸菌において機能的な複製系および形質転換細胞の選択を可能にするマーカーが含まれる。例としてのベクターには、pBR322、pUCシリーズ、pACYC184、Bluescriptシリーズ(Stratagene)等が含まれる。このように、配列を適当な制限部位でベクターに挿入してもよく、得られたプラスミドを用いて大腸菌宿主(例えば、大腸菌株HB101、JM101、およびDH5α)を形質転換してもよく、適当な栄養培地において大腸菌を増殖させて、細胞を回収して溶解し、プラスミドを回収してもよい。分析は、配列分析、制限分析、電気泳動等を含んでもよい。各操作後、最終構築物において用いられるDNA配列を制限して次の配列に結合させて、部分的構築物のそれぞれを同じまたは異なるプラスミドにおいてクローニングしてもよい。
【0052】
ベクターは市販されている、または植物細胞の形質転換のために容易に調製することができる。一般的に、プラスミドまたはウイルスベクターは、所定の宿主における異種DNA配列の維持および発現にとって必要な全てのDNA制御配列を含むべきである。そのような制御配列には一般的に、リーダー配列、翻訳開始シグナルコドンをコードするDNA配列、翻訳終止コドン、メッセンジャーRNAプロセシングを制御する3' UTRシグナルをコードするDNA配列が含まれる。任意の特定の種において発現を最適にする適当なエレメントの選択は、本開示の教示を利用して当業者の問題である。最後に、ベクターは望ましくは、ベクターを含む宿主細胞の同定を可能にする表現型特性を提供することができるマーカー遺伝子を有しなければならない。
【0053】
植物細胞に挿入された外来コード配列の活性は、インサートに隣接する内因性植物DNAの影響に依存する。一般的に、異種遺伝子の挿入は、如何なる形質転換技術を用いても無作為であるように思われる。しかしながら、植物細胞へのDNAの部位特異的組換えによって植物を産生する技術は現在存在する(国際公開公報第91/09957号を参照されたい)。プロモーターの制御下で所望の配列または配列の発現が得られる任意の方法または方法の組み合わせが許容される。
【0054】
本発明は、植物細胞を形質転換する任意の特定の方法に限定されない。植物細胞にDNAを導入する技術は、当業者に周知である。外来DNAを植物細胞に輸送する四つの基本的な方法が記述されている。化学法(Graham and van der Eb、Virology 54(02):536〜539、1973;Zatloukal、Wagner, Cotten, Phillips, Plank, Steinlein, Curiel, Birnstiel、Ann. N.Y. Acad. Sci.、660:136〜153、1992);マイクロインジェクション(Capecchi、Cell 22(2):479〜488、1980)、電気穿孔(Wong and Neumann、Biochim. Biophys. Res. Commun. 107(2):584〜587、1982;Fromm, Taylor, Walbot、Proc. Natl. Acad. Sci. USA、 82(17):5824〜5828、1985;米国特許第5,384,253号)、および遺伝子銃(Johnston and Tang、Methods Cell Biol., 43(A):353〜365、1994;Fynan, Webster, Fuller, Haynes, Santoro, Robinson、Proc. Natl. Acad. Sci. USA 90(24):11478〜11482、1993)を含む物理法;ウイルス法(Clapp、Clin. Perinatol.、20(1):155〜168、1993;Lu, Xiao, Clapp, Li, Broxmeyer、J. Exp. Med. 178(6):2089〜2096、1993;Eglitis and Anderson、Biotechniques 6(7):608〜614、1988;Eglitis, Kantoff, Kohn, Karson, Moen, Lothrop, Blaese, Anderson、Avd. Exp. Med. Biol.、241:19〜27、1988)および受容体媒介法(Curiel, Agarwal, Wagner, Cotten、Proc. Natl. Acad. Sci. USA、88(19):8850〜8854、1991;Curiel, Wagner, Cotten, Birnstiel, Agarwal, Li, Loechel, Hu、Hum. Gen. Ther. 3(2):147〜154(1992);Wagnerら、Proc. Natl. Acad. Sci. USA、89(13):6099〜6103、1992)。
【0055】
電気穿孔によって植物細胞にDNAを導入することは、当業者に周知である。ペクチン分解酵素のような植物細胞壁分解酵素は、レシピエント細胞を無処置細胞と比較して電気穿孔による形質転換に対する感受性を高めるために用いられる。電気穿孔による形質転換を行うために、懸濁培養細胞、胚形成カルス、未成熟胚、または他の構築組織のような脆弱な組織を直接用いてもよい。一般的に、ペクチン分解酵素または制御された方法での機械的損傷によって標的植物材料の細胞壁を部分的に分解する必要がある。そのような処置植物材料は、電気穿孔によって外来DNAを受け入れる準備が整っている。
【0056】
植物細胞に外来形質転換DNAを輸送するもう一つの方法は、微小弾丸衝突である。この方法において、微粒子を外来DNAによってコーティングして噴射力によって細胞に輸送する。そのような微粒子は典型的に、タングステン、金、プラチナ、および類似の金属製である。微小弾丸衝突の長所は、プロトプラストの単離(Cristouら、1988、Plant Physiol.、87:671〜674)もアグロバクテリウム感染に対する感受性も必要ない点である。加速によってトウモロコシ細胞にDNAを輸送する方法の例としての態様は、バイオリスティック粒子輸送系であり、これは懸濁培養トウモロコシ細胞で覆われたフィルター表面上にスクリーンを通してDNAをコーティングした粒子または細胞を噴射するために用いることができる。スクリーンは、分子が大きい凝集物でレシピエント細胞に輸送されないように粒子を分散させる。衝突の場合、好ましくは懸濁細胞をフィルターまたは固体培養培地において濃縮する。または未成熟胚または他の標的細胞を固体培養培地上で整列させてもよい。衝突させる細胞を、マクロ弾丸停止板の下で適当な距離で配置する。衝突形質転換において、最大数の安定な形質転換体が得られるように衝突前の培養条件および衝突パラメータを最適にしてもよい。衝突のための物理的および生物学的パラメータの両者がこの技術において重要である。物理的要因は、DNA/微小弾丸沈殿物の操作に伴う要因、または微小弾丸の飛行および速度に影響を及ぼす要因である。生物学的要因には、衝突前および直後の細胞の操作に伴う全ての段階、衝突に関連した外傷を軽減するために役立つ標的細胞の浸透圧調節、ならびに直線状DNAまたは無傷のスーパーコイルプラスミドのような形質転換DNAの特性が含まれる。
【0057】
アグロバクテリウムによる移入は、プロトプラストから無傷の植物を再生する必要がなく、DNAを植物全体の組織に導入することができることから、植物細胞に外来DNAを導入するために広く適用されている系である。植物細胞にDNAを導入するためにアグロバクテリウム媒介植物組み込みベクターを用いることは、当技術分野において周知である。例えば、Fraleyら、1985、Biotechnology、3:629;Rogersら、1987、Meth. in Enzymol.、153:253〜277に記述される方法を参照されたい。さらに、Ti-DNAの組み込みは、比較的正確なプロセスであり、再配列はほとんどない。移入されるDNAの領域は、境界配列によって定義され、介在DNAは通常、Spielmannら、1986、Mol. Gen. Genet.、205:34;Jorgensenら、1987、Mol. Gen. Genet.、207:471に記述されるように、植物ゲノムに挿入される。
【0058】
現代のアグロバクテリウム形質転換ベクターは、アグロバクテリウムのみならず大腸菌において複製することができ、簡便に操作することができる。その上、アグロバクテリウム媒介遺伝子移入に関するベクターの最近の技術の進歩によって、様々なタンパク質またはポリペプチドを発現することができるベクターの構築を促進するために、ベクターにおける遺伝子の整列および制限部位が改善された。挿入されたポリペプチドコード遺伝子を直接発現させるためのプロモーターおよびポリアデニル部位に隣接する簡便な多重リンカー領域は、本発明の目的にとって適している。さらに、アームを有するおよびアームを有しないTi遺伝子を含むアグロバクテリウムは、形質転換に用いることができる。
【0059】
植物プロトプラストの形質転換は、リン酸カルシウム沈殿、ポリエチレングリコール処置、電気穿孔、およびこれらの処置の組み合わせに基づく方法を用いて行うことができる(例えば、Potrykusら、1985、Mol. Gen. Genet.、199:183;Marcotteら、Nature、335:454、1988)。これらのシステムを異なる植物種に適用することは、プロトプラストから特定の種の再生能に依存する。
【0060】
植物細胞を形質転換、選択、および抗原の発現に関してチェックした後、場合によっては、稔性の植物全体を再生することが可能である。これは選択した植物種に大きく依存するであろう。多数の植物種を再生する方法は、文献に報告されており、当業者に周知である。本発明を実践する場合、一般的に長い再生段階を回避することによって、培養して急速に規模を拡大することができる植物細胞を形質転換することが好ましい。さらに、植物細胞培養を用いることによって、農地で産生する必要がなく、遺伝子の逸出および食物の混入の機会が大きく減少する。NT-1およびBY-2(An, G.、1985、Plant Physiol. 79、568〜570)のようなタバコ懸濁細胞培養物は、これらの株が培養での取り扱いが特に容易であること、容易に形質転換されること、安定な組み込み事象を生じること、および凍結保存できることから好ましい。
【0061】
タバコ懸濁細胞株NT-1は、本発明の実践にとって適している。NT-1細胞は当初、タバコ(Nicotiana tabacum L.cv. bright yellow 2)から作製された。NT-1細胞株は広く用いられ、容易に入手可能である;しかしながら、如何なるタバコ懸濁細胞株も本発明の実践にとって適合する。NT-1細胞株のが起源は不明であることは注目に値する。その上、細胞株は多様であるように思われ、培養条件に反応して変化する傾向がある。下記の実施例において用いるために適したNT-1細胞は、American Type Culture Collectionからアクセッション番号ATCC 74840として入手可能である。同様に、その全内容物が参照により本明細書に組み入れられる、米国特許第6,140,075号を参照されたい。
【0062】
実験室規模のシェーカーから何千リットルものバイオリアクター容器に至るまで、多くの植物細胞培養技術および系が記述されており、植物細胞培養の技術分野において周知である。例えば、Fischer, R.ら、1999 Biotechnol. Appl. Biochem. 30、109〜112およびDoran, P.、2000、Current Opionions in Biotechnology 11、199〜204を参照されたい。形質転換植物細胞を望ましい量まで培養した後、それらを回収して、軽く洗浄して、破壊するために適切な緩衝液に入れる。異なる多くの緩衝液が本発明に適合性である。一般的に、緩衝液は、膜を可溶化するために用いることができる強い洗浄剤を含まない、中性pHまたは中性pH付近の等張緩衝塩水溶液である。好ましい緩衝液には、ダルベッコリン酸緩衝生理食塩液および1 mM EDTAを含むPBSが含まれる。
【0063】
一つの態様において、細胞を超音波によって破壊することができる。洗浄細胞を約0.01 gm/ml〜約5.0 gm/mlの範囲、好ましくは約0.1 gm/ml〜約0.5 gm/mlの範囲(洗浄湿重量細胞/緩衝液の容量)の緩衝液に入れる。市販の多くの超音波装置は、本発明に適合性であり、超音波処置時間は約5〜約20秒、好ましくは約15〜約20秒である。得られた大きさは数ミクロンから数百ミクロンの範囲となる可能性があり、免疫保護タンパク質または他の生物活性タンパク質を露出してもよい。
【0064】
実施例1:ベクター
遺伝子の構築:NDV株「Lasota」のHN遺伝子(Genbankアクセッション番号AF077761号)、AIV株ATurkey/Wisconsin/68のHA遺伝子、IBDV株E19のVP2遺伝子(Genbankアクセッション番号X00493)、および大腸菌のLT遺伝子のコード配列を、コドンの使用、ならびに偽性mRNAプロセシングおよび不安定性またはゲノムDNAのメチル化を媒介しうる望ましくない配列モチーフの存在に関して分析した。Adang MJ, Brody MS, Cardineau G, Eagan N, Roush RT, Shewmaker CK, Jones A, Oakes JV, McBride KE(1993)、「The construction and expression of Bacillus thuringiensis cryIIIA gene in protoplasts and potato plants」、Plant Mol Biol 21:1131〜1145を参照されたい。植物最適化コード配列をトマトおよびジャガイモのコドンの使用を反映したハイブリッドコドン嗜好性によって設計した(Ausubel F.ら編(1994)、「Current Protocols in Molecular Biology」、第3巻、p. A.1C.3 Haq TA, Mason HS, Clements JD, Arntzen CJ(1995)、「Oral immunization with a recombinant bacterial antigen produced in transgenic plants.」、Science 268:714〜716)。HNの設計された配列を図1に示す。合成HN遺伝子は市販の供給元(Retrogen)によって構築され、pCR-Bluntにクローニングされた遺伝子の5'(p4187-4203-1)または3'(p42111-4235-1c-1)半分のいずれかを含む二つの異なるプラスミドとして受領した。
【0065】
プラスミドの構築:アグロバクテリウム媒介植物形質転換のためのバイナリベクターは、図2に示すベクターpBBV-PHAS-iaaHに基づいて構築され、これは、参照により本明細書に組み入れられる米国特許第5,879,903号、第5,637,489号、第5,276,268号および第5,273,894号に記述され、国際公開公報第97/48819号に記述される構成的カッサバ葉脈モザイクウイルス(CsVMV)プロモーターによって促進される、植物選択マーカーホスフィノトリシンアセチルトランスフェラーゼ(PAT)を用いる。本発明者らは、最初にpBBV-PHAS-iaaHのPacIによる消化によってiaaH遺伝子およびファセオリンプロモーター配列を欠失して、再ライゲーションしてpCVMV-PATを形成した。次に本発明者らは、一つのHindIII部位をクレノウ酵素で塞ぐことによってこれを欠失させ、再ライゲーションしてpCP!Hを形成した。本発明者らは、鋳型pCP!H上でプライマー
を用いて、PCRによってCsVMVプロモーターを末端に配置して、EcoRV-消化、T-テールpBluescriptKSにおいて産物をクローニングしてpKS-CVM7を作製した。pCP!Hのマップを図3に示す。本発明者らは三つのインサート断片:NcoI/PstIにおいてHN 5'半分、PstI/KpnIにおいてHN 3'半分、およびKpnI-EcoRIにおいて大豆vspB 3'エレメント(Haq 1995)によってベクターpKS-CVM7/NcoI-EcoRIをライゲーションすることによって、HN発現カセットpKS-CHNを構築した。次に、バイナリT-DNAベクターpCHNを、ベクターpCP!H/AscI-EcoRIとpKS-CHNのAscI-EcoRI断片とのライゲーションによって構築した。pCHNのマップを図4に示す。
【0066】
参照により本明細書に組み入れられる、米国特許第5,824,798号に記述される顆粒結合デンプンシンターゼ(GBSS)プロモーターを用いて他のベクターを作製した。これらの構築物は、中国のジャガイモ栽培品種「Dongnong」に関してGenbankアクセッション番号X83220の配列から設計したプライマーを用いて、ジャガイモ(Solanum tuberosum L.cv.「Desiree」)のゲノムDNAから増幅したプロモーター断片を用いて作製した。変異誘発プライマー「GSS-Nco」
を−1800 bpで5'領域と相補的なフォワードプライマー「GSS-1.8F」
と共に用いて翻訳開始コドンに重なり合うNco I部位を作製した;1825 bpのPCR産物をT-テールpBluescriptKSにおいてクローニングして、pKS-GBNを作製して、シークエンシングした。変異誘発プライマー「GSS-Xho」
をプライマー「GSS-1.8F」と共に用いて、転写開始部位のまさに3'でXhoI部位を作製した;1550 bp PCR産物をT-テールpBluescriptKSにおいてクローニングして、pKS-GBXを作製し、シークエンシングした。
【0067】
参照により本明細書に組み入れられる、米国特許第5,891,665号に記述されるTEV 5'UTR(非翻訳領域)を含むGBSSプロモーター発現カセットは、HindIII/XhoIによって消化したベクターpTH210を、pKS-GBXのHindIII/XhoI断片とライゲーションすることによって構築し、これによってCaMV 35Sプロモーターを811 bp GBSSプロモーターに置換して、pTH252Aを作製した。Haq TA, Mason HS, Clements JD, Arntzen CJ(1995)、「Oral immunization with a recombinant bacterial antigen produced in transgenic plants」、Science 268:714〜716を参照されたい。NcoI/PstIにおいてHN 5'半分およびPstI/KpnIにおいてHN 3'半分をライゲーションすることによって、HN遺伝子をpTH252A/NcoI-KpnIに挿入して、pHN252Aを作製した。NsiIおよびEcoRIによって消化したベクターpGLTB(図11に示す)と、pHN252A/NsiI-KpnIおよびpTH210/KpnI-EcoRIの断片とのライゲーションによって、バイナリT-DNAベクターpgHNを作製した。pgHNのマップを図5に示す。
【0068】
参照により本明細書に組み入れられる、米国特許第5,824,798号に記述されるGBSS 5' UTRを、そのイントロンと共に含むGBSSプロモーター発現カセットを、HindIII/NcoIによって消化したベクターpTH210(Haq1995)とpKS-GBNのHindIII/NcoI断片とのライゲーションによって構築し、これによって(カリフラワーモザイクウイルス)CaMV35Sプロモーター/TEV 5' UTRを1084 bp GBSSプロモーター/5' UTRに置換して、pTH251Aを作製する。バイナリT-DNAベクターpgHN151は、ベクターpCLT105(図12に示す)を断片pTH251A/HindIII-NcoIおよびpHN252A/NcoI-KpnIとライゲーションすることによって作製した。pgHN151のマップを図6に示す。
【0069】
そのイントロンおよびマメのファセオリン3'エレメント(参照により本明細書に組み入れられる、米国特許第5,270,200号、第6,184,437号、第6,320,101号に記述される)と共にGBSS 5'UTRを含むGBSSプロモーター発現カセットを構築した。第一に、pCP!Hを唯一のKpnI部位で消化して、T4 DNAポリメラーゼによって平滑末端にし、再度ライゲーションして、KpnI部位が除去されたpCP!HKを作製した。pCP!HKをNsiIによって消化した後、T4 DNAポリメラーゼによって平滑末端にした後、PacIによって消化した。得られたベクターを、SacIによって消化したpgHN151からの2848 bp断片とライゲーションした後、T4 DNAポリメラーゼによって平滑末端にして、PacIによって消化してpgHN153を作製した。pgHN153のマップを図7に示す。
【0070】
キメラ構成的プロモーター(4OCS△MAS、参照により本明細書に組み入れられる、米国特許第5,001,060号、第5,573,932号、および第5,290,924号)を用いて、HNに関するもう一つの発現ベクターを構築した。プラスミドpAGM149をEcoRVによって消化して、BamHIによって部分消化した。この断片をPmeI/PstIによって消化したpCHNと、pKS-CHNのBamHI/PstIによる消化によって得られた合成HN遺伝子の5'半分とにライゲーションした。得られたpMHNを図8に示す。
【0071】
AIV A/Turkey/Wisconsin/68(H5N9)のHA遺伝子を含むプラスミドは、David Suarez(SEPRL、アテンズ、ジョージア州)から得た(図10)。これをPCRによって、5'末端で制限部位NcoIおよび3'末端でKpnI部位を付加するように調製し、35Sプロモーター、TEV 5'-UTRおよびvspB 3'末端を有するベクターpIBT210.1(Haqら、1995)に挿入した。発現カセットを、HindIIIおよびEcoRI(部分的)による消化によってバイナリベクターpGPTV-Kan(Beckerら、Plant Mol Biol 1992;20:1195〜7)に転移させて、pIBT-HAOを作製した。pIBT-HAOからのHA遺伝子/vspB3'末端断片は、NcoIおよびEcoRI(部分的)による消化によって、pKS-CVM7に挿入して、pKS-CHAを作製した。CsVMVプロモーター、HA遺伝子、およびvspB3'末端を含むカセットを、AscIおよびEcoRI(部分的)による消化によってpKS-CHAから得て、これをpCP!Hにライゲーションして図9に示されるpCHAを作製した。
【0072】
大腸菌株H10407のLT-B遺伝子をコードする植物最適化配列は、当技術分野で既知である(Mason HS、Haq TA、Clements JD, Arntzen CJ, 1998、Vaccine 16:1336〜1343)。大腸菌株H10407のLT-A遺伝子をコードする植物最適化配列は、その全教示が参照により本明細書に組み入れられる、当初、米国特許仮出願第60/113,507号として提出された国際公開公報第00/37609号に記述された。国際公開公報第00/37609号は、実施例2におけるタバコNT-1細胞のアグロバクテリウム・ツメファシエンスによる植物細胞の形質転換のために用いられる三つのバイナリT-DNAベクター(pSLT102、pSLT105、pSLT107)の構築を記述している。得られた形質転換NT-1細胞株(SLT102、SLT105、およびSLT107)は、LT-Bおよび改変型LT-Aサブユニットからなる完全に構築されたLTおよびLT類似体を発現して蓄積した。図12は、Ala72をArg72に置換する改変LT-A遺伝子を含むpSLT107を図示する。SLT102およびSLT105発現産物は、それらがLT-A遺伝子において異なる変化(pSLT102ではSer63がLys63に、pSLT105ではArg192がGly192に)を含むことを除き、同一であった。これらの株は、核染色体DNAに安定に組み入れられたプラスミドのT-DNA領域の未確認数のコピーを含む。トランスジェニックNT1細胞は、ガングリオシド結合五量体に構築されるLT-Bサブユニットを、ガングリオシド依存的ELISAによって決定した場合に、総可溶性タンパク質の0.4%までのレベルで蓄積した。トランスジェニックNT1細胞はまた、改変LT-Aサブユニットも蓄積し、そのいくつかは、LT-A特異的抗体を用いるガングリオシド依存的ELISAによって決定した場合に、LT-B五量体を構築した。
【0073】
アグロバクテリウムによる植物細胞形質転換のバイナリベクターは、VP2および選択マーカー発現カセットを付加するために、AgeIリンカーによって唯一のBamHI部位で改変された基本バイナリベクター(pBBV)から構築した。VP2は、アグロバクテリウム・ツメファシエンス(Atu)ORF24(Genbankアクセッション番号X00493号)3' UTRと共に、RB7 MARエレメント(米国特許第5,773,689号、米国特許第5,773,695号、米国特許第6,239,328号、国際公開公報第94/07902号および国際公開公報第97/27207号)およびCsVMVプロモーターに隣接する。選択マーカーPATは、シロイヌナズナ(At)ユビキチン10プロモーター(Plant J. 1997、11(5):1017;Plant Mol. Biol. 1993、21(5):895;Genetics、1995、139(2):921)およびAtu ORF 1(米国特許第5,428,147号;Plant Molecular Biology 1983、2:335;Genbankアクセッション番号X00493号)3' UTRによって調節され、得られたプラスミドpDAB2423を図13に示す。
【0074】
伝染性ファブリキウス嚢病(IBD)ウイルスの非常に有毒な株Ehime 91(J Vet Med Sci. 1992、54(1):153;JVI 2002、76(11):5637)を用いて、株UK661(Genbankアクセッション番号NC_004178号)からのアミノ酸番号454〜456位と共に、報告されたVP2アミノ酸配列(Genbankアクセッション番号AB024076号)に基づいて、VP2植物最適化ヌクレオチド配列を産生した(VP2配列に関しては図14を参照されたい)。
【0075】
実施例2:トランスジェニックタバコの調製
形質転換の3〜4日前に、NT-1の1週間培養物を、NT-1培養物 2 mlをNT-1培地40 mlに加えることによって新鮮な培地で培養した。培養物を25±1℃の暗所で100 rpmで維持した。
【0076】
対象発現ベクターを含むアグロバクテリウム・ツメファシエンスを、50 mg/lスペクチノマイシンを含むLB培地のプレートにグリセロール保存液から画線培養した。細菌培養物を30℃の暗所で24〜48時間インキュベートした。1ウェル形成コロニーを選択して、50 mg/Lスペクチノマイシンを含むYM培地3 mlに移した。液体培養物を250 rpmのインキュベーター内で30℃の暗所で、OD600が0.5〜0.6となるまでインキュベートした。これには約24時間を要した。
(または、粉末型のYMを購入することができる(Gibco BRL;カタログ番号10090-011)。液体培養培地を作製する場合、11.1 gを水1 Lに加える。)
【0077】
形質転換の1日後、20 mMアセトシリンゴン1 μlをNT-1培養物1 mlに加えた、アセトシリンゴン保存液は形質転換日にエタノールにおいて作製した。NT-1細胞に損傷を与えて形質転換効率を増加させた。損傷を与える場合、懸濁培養物を孔径の広い5 ml滅菌ピペットの中を繰り返し(20回)上下させて損傷を与えた。懸濁液4 mlを10、60×15 mmペトリ皿のそれぞれに移した。プレート1枚を非形質転換対象として用いるために除いておいた。アグロバクテリウム懸濁液約50〜100 μlを残りの9枚のプレートのそれぞれに加えた。プレートをパラフィルムで覆って、暗所の100 rpmのシェーカーにおいて25±1℃で3日間インキュベートした。
【0078】
細胞を滅菌の50 ml遠心管に移して、最終容量をNTC培地(500 mg/Lカルベニシリンを含むNT-1培地、オートクレーヴ後加えた)によって45 mlとした。それらを混合した後、スィングバケットローターを備えた遠心機において1000 rpmで10分間遠心した。上清を除去して、得られた沈降物をNTC 45 mlに再懸濁させた。洗浄を繰り返した。懸濁液を遠心して、上清を捨て、沈降物をNTC 40 mlに再懸濁させた。5 mlの少量を、NTCB10培地(10 mg/lバイアラホスを添加した8 g/lアガー/アガーによって固化したNTC培地、オートクレーヴ後加えた)を含む各ペトリ皿(150×15 mm)に播種した。プレートをパラフィルムによって覆って、25±1℃の暗所で維持した。培養室に移す前に、プレートを層流のフード内で開いたままにして、過剰量の液体を蒸発させた。6〜8週間後、推定の形質転換体が出現した。それらを選択して新鮮なNTCB5(5 mg/lバイアラホスを添加した8 g/lアガー/アガーによって固化したNTC培地、オートクレーヴ後加えた)に移した。プレートをパラフィルムによって覆って、25±1℃の暗所で維持した。
【0079】
推定の形質転換体は、死んだ非形質転換細胞のバックグラウンドにおいて小さいカルス集団として出現した。これらのカルスをNTCB5培地に移して、数週間増殖させた。各推定の形質転換体の一部をELISA分析のために選択した。ELISAを少なくとも2回行った後、最高の抗原レベルを示す株を選択した。各選択株のそれぞれのカルス材料の量を、プレート培養および時に液体培養において増幅させた。
【0080】
実施例3:抗原の調製
蠕動性ポンプおよびシリコンチューブを用いて、細胞を発酵器の採取口から採取した。細胞を30 μmスペクトロメッシュを含む円錐形の濾過装置にポンプで加えて、真空によって細胞を濾過して湿細胞ケークを形成した。1 mMエチレンジアミン四酢酸(EDTA;カタログ番号E(884、Sigma Aldrich)を添加したダルベッコリン酸緩衝生理食塩液(カタログ番号21-031-CV、Mediatech, Inc)を含む冷溶解緩衝液に、細胞を濾過細胞1 gあたり緩衝液2 mlの比で懸濁させた。細胞のスラリーを処理するまで5℃で維持した。微少溶液操作の前に、細胞をSilverson L4RTミキサーを用いて6000 rpmで5〜10分間ホモジナイズすることができる。100 μm Z形相互作用チャンバー(H10Z)を備えたMicrofludicsモデル110 Lマイクロフルイダイザーに、冷溶解緩衝液約200 mlを加える。チャンバーの圧力を18,000 PSIに設定して、相互作用チャンバー、入り口、および出力ラインを氷で覆った。試料を流速100 ml/分でマイクロフルイダイザーの中を通過させて、溶解細胞懸濁液を氷中で回収した。処理した溶液を、4℃、2800×gで15分間遠心することによって細胞塊を除去した。放出されたHN、HA、LTまたはVP2タンパク質を含む上清を細胞破片の沈降物から分離して、-80℃で保存した。2800×gの沈降物を2倍量の新鮮な溶解緩衝液において再懸濁させて、5℃で16時間インキュベートして、細胞破片に会合したままであるHNタンパク質を抽出した。細胞破片を、スィングバケットローターにおいて2800×g、4℃で15分間沈降させた。上清をデカントして−80℃で保存した。
【0081】
冷蔵保存からの処理は、バルク材料を2800×g、4℃で15分間遠心することによって行い、上清を真空下で30 μmスペクトラメッシュによって濾過した。上清のバルク材料は、産物の標的分子量より小さい分子量カットオフメンブレンを用いてPall Centramate接線流システムを用いて濃縮することができる。入り口および保持ラインを産物容器に向けて、産物プールを10〜20倍濃縮する。濾液を産物の漏出に関して試験する。濃縮終了後、CentramateユニットにDPBS〜500 mlを一気に流し、この洗浄液を最終の濃縮プールに加えた。濃縮物を4℃または−80℃で保存する。
【0082】
超音波処理の場合、HN、HA、またはゼロ対照のいずれかを発現する全湿潤NT-1細胞を細胞培養物から直接回収して、スペクトラメッシュ30フィルターをブフナー漏斗に入れて、細胞および培地をわずかな真空を用いて濾紙に注ぐことによって過剰量の培地を濾過して除去した。細胞0.5 gを緩衝液(ダルベッコリン酸緩衝生理食塩液および1 mM EDTA)2 mlに加えて氷中で15〜20秒間超音波処理した。超音波処理は、出力制御8、デューティサイクル60で交換可能なマイクロチップを備えたBranson 450超音波装置を用いて様々な時間行った。超音波処理物を使用するまで氷中に入れた。細胞量が多い場合、超音波処理に必要な時間はそれに比例して増加するであろう(例えば、細胞250 gの場合、超音波処理は8〜10分間に増加されるであろう)。
【0083】
実施例4:抗原の抽出
非洗浄剤処置が形質転換NT-1細胞からELISAシグナルを放出して、生物活性を保持できるか否かを調べるために、洗浄剤を含まない処置、および様々なレベルの超音波または微少溶液操作の処置の比較を含む一連の処置を設定した。結果は、抽出緩衝液における20秒より長い超音波時間がNT-1細胞株を有するpCHNからのHNの赤血球凝集活性を完全に破壊するが、ELISAシグナルは破壊しないという点において顕著であった。対称的に、DPBSにおいて20秒のみの超音波処理を行うと、ELISAシグナルによって検出可能な抗原を放出したのみならず、可溶性タンパク質抽出物は優れた赤血球凝集活性を示した(表1を参照されたい)。
【0084】
強い洗浄剤または高濃度の洗浄剤を用いずに抽出された植物由来HNを、赤血球凝集阻害アッセイにおける抗原として用いて、天然のウイルスに対して産生されたポリクローナル抗体が植物由来HNによるRBCの凝集を認識して阻害できるか否かを決定した。結果は、天然の抗体が、天然のウイルスと同様に、植物由来HNの赤血球凝集エピトープを認識するであろうことを示している(表2)。表2からのデータはまた、対照NT-1細胞または非赤血球凝集タンパク質を発現するNT-1細胞が赤血球を凝集せず、NDV特異的血清によっても影響を受けないことを示している。本実験において、植物由来タンパク質の抽出物を4 HA(赤血球凝集)単位まで希釈した後、NDV特異的ポリクローナル抗血清によって処置した。4 HA単位は、血清の滴定のために用いられる標準的なウイルス量である。
【0085】
上記のデータは、強い洗浄剤を利用せずに、細胞破壊量を減少させる抽出法を用いることによって、HNまたはHAを発現する形質転換NT-1細胞株の赤血球凝集活性を保持する細胞外分画が産生されることを示している。非洗浄剤抽出NT-1細胞からのHNタンパク質が、ワクチン有効性に関連する可能性があるさらなる生物活性を有するか否かを調べるために、HN抽出物をニワトリ細胞受容体に対する結合能に関して調べた。免疫蛍光染色では、天然のウイルスまたはpCHN-18抽出物によって処置したニワトリ胚線維芽細胞(CEF)を区別できないことが示された。このように、植物由来HNは、標的細胞表面上の受容体に対するウイルス様結合能を保持する。
【0086】
表1および2のデータを、先に記述した赤血球凝集および免疫蛍光アッセイと複合すると、本発明のトランスジェニックNT-1細胞に由来するHNタンパク質が免疫および生物学的特徴の双方を保持することを示唆している。同様に、タンパク質および免疫保護粒子は、洗浄剤の非存在下で有効かつ天然型で植物細胞から放出されうる。先に提供した最も重要なデータは、天然のウイルスに対する抗血清が、HAI試験において植物由来HNを認識するであろう点である。赤血球凝集阻害(HAI)活性がバックグラウンドより少なくとも4倍高い力価を含むニワトリはほぼ常に、毒性ウイルスからのチャレンジに対して確実に保護される。
【0087】
HNの収量が他の機械的破壊手段によって増加しうるか否かを決定するために、細胞を上記の微少溶液操作に曝露した。様々な圧力を用いて、破壊の作用およびHNタンパク質の生物活性を調べた。表3は試験の結果を示す。データは、HNタンパク質の単位質量あたりの赤血球凝集活性の量が、この破壊法を用いた場合10倍より大きく変化しうるが、タンパク質濃度は約20%増加するに過ぎないことを示唆している。これらのデータから、超音波処理によってごく部分的に放出されたに過ぎないより大きい粒子径にHNタンパク質が組み入れられ、生物活性を保持するより小さい粒子径が存在しうることが示唆される。より均一な抽出物を生じる破壊法を用いると、さらなる活性ポリペプチドの回収することができる。
【0088】
実施例5:定量的ELISAs
定量的ELISA VP2
Nunc Maxisorp 96ウェルマイクロタイターELISAプレートを、0.01 Mホウ酸緩衝液において1:2000倍希釈したニワトリ抗IBDVポリクローナル抗血清(SPAFASロット番号G0148)によって100 μl/ウェルを用いてコーティングした。プレートを5℃で一晩インキュベートした。プレートを300 μl/ウェルPBS-T(0.05%Tween20を含む1×PBS、Sigmaカタログ番号P-1379)によって3回洗浄した。各ウェルをブロッキング緩衝液(5%(w/v)脱脂粉乳(Difcoカタログ番号232100)のPBS溶液)200 μlと共に37℃で1時間インキュベートした。ウェルをPBS-Tを用いて300 μl/ウェルで3回洗浄した。IBDV参照抗原(BEI不活化IBDV D-78株、ロット番号220903IBDV)を、ブロッキング緩衝液において最終濃度1000 ng/ml VP2となるように希釈した。試料をブロッキング緩衝液によって予め希釈した。希釈した参照抗原および実験抗原試料を、試料200 μlをB列に1試料あたり2ウェルずつ加え、残りのウェルにブロッキング緩衝液100 μlを加えることによってプレートに加えた。100 μl/ウェルを混合して移動させることによって連続2倍希釈を行って、参照または試料につき希釈液6個を作製した。プレートを37℃で1時間インキュベートした後、PBS-Tにおいて3回洗浄し、ブロッキング緩衝液において1:10,000倍希釈したR-63モノクローナル抗体腹水(IBDV VP2特異的、ロット番号190903R-63)100 μlをウェルに加えて、37℃で1時間インキュベートした。プレートをPBS-Tによって3回洗浄した。ブロッキング緩衝液において1:2000倍希釈したヤギ抗マウスIgGペルオキシダーゼ標識抗体結合体(KPLカタログ番号074-1806)を100 μl/ウェルで加えて、プレートを37℃で1時間インキュベートした。プレートをPBS-Tにおいて3回洗浄し、ABTS基質(KPLカタログ番号50-66-01)100 μlを各プレートに加えて、室温で約5分間インキュベートした。Tecan Sunriseプレートリーダーを用いて波長405 nmでの吸光度を測定した。Tecan Magellanソフトウェアを用いてデータを転送して表示した。直線回帰および定量分析は、マイクロソフトオフィスエクセル2003を用いて行った。
【0089】
Nunc Maxisorp 96ウェルマイクロタイターELISAプレートを、100 μl/ウェルを用いて0.01 Mホウ酸緩衝液において混合GM1ガングリオシド5 μg/ウェルによってコーティングした。プレートを室温で一晩インキュベートした。プレートをPBS-Tによって3回洗浄した(0.05%Tween 20を含む1×、Sigma、ロット番号120K0248)。次に、各ウェルを5%(w/v)脱脂粉乳のPBS-0.05%Tween20溶液を含むブロッキング緩衝液200 μlと共に37℃で1時間インキュベートした。ウェルをPBS-Tを用いて250 μl/ウェルによって3回洗浄した。LT参照抗原(またはLT-B参照抗原)を50 ng/mlに希釈した。試料をブロッキング緩衝液において予め希釈した。希釈した参照抗原および試料を、試料200 μlをA列に加え、残りのウェルにブロッキング緩衝液100 μlを加えることによってプレートに加えた。100 μl/ウェルを混合して移動させることによって連続2倍希釈を行った。プレートを37℃で1時間インキュベートした後、PBS-Tにおいて3回洗浄し、ブロッキング緩衝液において希釈したLT-AまたはLT-B特異的抗血清100 μlをウェルに加えて、37℃で1時間インキュベートした。プレートをPBS-Tによって3回洗浄して、ペルオキシダーゼ標識抗体結合体100 μlを加えて、37℃で1時間インキュベートした。プレートをPBS-Tにおいて3回洗浄し、TMB基質50 μlを各プレートに加えた。基質を添加した20分後にTMB停止溶液を加えた。Tecan Sunriseプレートリーダーを用いて波長450 nmでの吸光度を測定した。Tecan Magellanソフトウェアを用いてデータを転送して表示した。直線回帰および定量分析は、マイクロソフトオフィスエクセル2000バージョン9.0.3821 SR-1を用いて行った。
【0090】
定量的ELISA HN
HNに関する定量的ELISAを、アッセイを行う前日にプレートをコーティングすることによって行うことができる。捕獲抗体(ウサギ抗HNの50%グリセロール溶液、0.01 Mホウ酸緩衝液において希釈(1:500倍))50 μl/ウェルを平底96ウェルマイクロタイタープレートの各ウェルに加える。プレートの蓋をして、2℃〜7℃で一晩(12〜18時間)インキュベートする。コーティングしたELISAプレートを室温で平衡にした後(約20〜30分)、PBS-Tによって1回の洗浄あたり200〜300 μl/ウェルで3回洗浄する。3%スキムミルクブロッキング溶液200 μl/ウェルを加えることによって、非特異的反応を防止するためにプレート全体をブロックする。プレートを37℃±2℃で2時間(+10分)インキュベートした(プレートの蓋または同等物で覆う)。HN参照抗原(Ag)を1%スキムミルクブロッカーにおいて250 ng HA/mlの濃度になるように加える;実験抗原を1%ブロッカーによって希釈する。HN ELISAプレートをPBS-Tによって1回洗浄して、希釈したHN参照抗原100 μl/ウェルおよびHN試験試料を列Bに加える;1%ブロッカー50 μl/ウェルを残りの全てのウェルに加える;列Bから50 μl/ウェルを列Gまで移し、次に移す前にピペットによって4〜5回混合することによって試料をプレートの下段方向に連続希釈する。プレートの蓋をして、37℃±2℃で1時間インキュベート(+10分)する;ELISAプレートをPBS-Tによって3回洗浄する。NDV HN 4A腹水の50%グリセロール溶液(1:2000倍希釈)50を3%ブロッカーにおいて各ウェルに加えて、プレートの蓋をして37℃±2℃で1時間インキュベート(+10分)する。ELISAプレートをPBS-Tによって3回洗浄して、3%ブロッカーにおいて50%グリセロール(1:3000倍希釈)においてウサギ抗マウスIgG 50 μlを各ウェルに加える;プレートに蓋をして37℃±2℃で1時間(+10分)インキュベートする。ELISAプレートをPBS-Tによって3回洗浄して、ABTSペルオキシダーゼ基質溶液(RT(室温)で少なくとも30分間平衡にする)50 μlを各ウェルに加える。プレートの蓋をしてRTの暗所で15〜20分間インキュベートする。ウェルの吸光度(OD)を波長405 nm(492 nm参照フィルターによって)で読み取る。HN参照抗原の最初の希釈液は、0.7〜1.0 ODの範囲内となるはずであり、これをELISAの陽性対照とする。
【0091】
定量的ELISA HA
HAの定量的ELISAに関して、アッセイを行う前日にプレートをコーティングする。捕獲抗体(ヤギ抗Hav5の50%グリセロール溶液、0.01 Mホウ酸緩衝液において希釈(1:1000))50 μl/ウェルを平底96ウェルマイクロタイタープレートの各ウェルに加える。プレートの蓋をして、2℃〜7℃で一晩(12〜18時間)インキュベートする。コーティングしたELISAプレートを室温で平衡にして(約20〜30分)、PBS-Tによって200〜300 μl/ウェルで3回洗浄した。3%スキムミルクブロッキング溶液200 μl/ウェルを加えることによって非特異的反応を防止するためにプレート全体をブロックする。プレートを37℃±2℃で2時間(+10分)インキュベートする(プレートの蓋または同等物によって覆う)。AIV-HA(尿膜腔液)参照抗原を1%スキムミルクブロッカーにおいて1000 ng HA/mlの濃度で加える;実験抗原を1%ブロッカーにおいて希釈する。HA ELISAプレートをPBS-Tによって1回洗浄して、希釈したHA参照抗原およびHA試験試料100 μl/ウェルを列Bに加える;1%ブロッカー50 μl/ウェルを残りの全てのウェルに加える;50 μl/ウェルを列B〜列Gまで移動させて、各移動の前にピペットによって4〜5回混合することによって、試料をプレートの下段方向に連続希釈する。プレートに蓋をして37℃±2℃で1時間(+10分)インキュベートする;ELISAプレートをPBS-Tによって3回洗浄する。ニワトリ抗AIVポリクローナル抗血清の50%グリセロール溶液(1:2000倍)50 μlを3%ブロッカーにおいて各ウェルに加えて、プレートの蓋をして37℃±2℃で1時間(+10分)インキュベートする。ELISAプレートをPBS-Tによって3回洗浄した後、ヤギ抗ニワトリIgGの50%グリセロール(1:3000)3%ブロッカー溶液50 μlを各ウェルに加える;プレートに蓋をして37℃±2℃で1時間(+10分)インキュベートする。ELISAプレートをPBS-Tによって3回洗浄して、ABTSペルオキシダーゼ基質溶液(RTで少なくとも30分間平衡にする)50 μlを各ウェルに加える。プレートに蓋をしてRTの暗所で15〜20分間インキュベートする。ウェルの吸光度(OD)を波長405 nm(492 nm参照フィルターによる)で読み取った。HA参照抗原の初回希釈液は0.7〜1.0 ODの範囲内となるはずであり、これをELISAの陽性対照とする。
【0092】
定量的ELISA LTおよびLTB
Nunc Maxis 96ウェルマイクロタイターELISAプレートを100 μl/ウェルを用いて0.01 Mホウ酸緩衝液において混合GM1ガングリオシド5 μg/ウェルによってコーティングした。プレートを室温で一晩インキュベートした。プレートをPBS-Tによって3回洗浄した。各ウェルを、5%(w/v)脱脂粉乳のPBS-T溶液を含むブロッキング緩衝液200 μlと共に37℃で1時間インキュベートした。ウェルをPBS-Tを用いて250 μl/ウェルによって3回洗浄した。参照抗原および試料抗原をプレートに加える前にPBS-Tによって1:1に混合した。LT参照抗原およびLTB参照抗原を第一のウェルにおいて50 ng/mlに希釈した。試料を、試料200 μlをA列に加え、残りのウェルにブロッキング緩衝液100 μlを加えることによってプレートに加えた。100 μl/ウェルを混合して移動させることによって連続2倍希釈を行った。プレートを37℃で1時間インキュベートした後、PBS-Tにおいて3回洗浄し、ブロッキング緩衝液において希釈した抗血清100 μlをウェルに加えて、37℃で1時間インキュベートした。プレートをPBS-Tによって3回洗浄した後、抗体結合体100 μlを加えて、37℃で1時間インキュベートした。プレートをPBS-Tによって3回洗浄し、TMB基質50 μlを各プレートに加えて、基質を加えた20分後にTMB停止溶液を加えた。Tecan Sunriseプレートリーダーを用いて波長450 nmでの吸光度を測定した。Tecan Magellanソフトウェアを用いてデータを転送して表示した。直線回帰および定量分析は、マイクロソフトオフィスエクセル2000バージョン9.0.3821 SR-1を用いて行った。
【0093】
実施例6:血清ELISA
血清ELISA LT
血液を断頭(生後0〜7日齢のトリ)または翼の羽板もしくは頚静脈での静脈穿刺によって採取した。トリを断頭によって、または断頭前にCO2に1〜5分間曝露することによって安楽死させた。血液を動物施設から実験室に運び、2〜7℃で45分放置して、凝血を進行させて濃縮させた。血液試料を37℃の温浴に10分間移した後Beckman GPR遠心機において2〜7℃で2500 rpmで20分間遠心した。各試験管から血清を無菌的に採取して、0.5〜1.5 mlの少量にして凍結用チューブ(Nunc)に入れ、使用するまで−18℃で保存した。血清ELISAの場合、ガングリオシド吸着段階は、1.5 μg/ウェルまたは15 μg/mlを利用して2〜7℃で一晩インキュベートした。プレートをPBS-Tによって3回洗浄した後、3%スキムミルクPBSと共に37℃で1時間ブロックした。血清試料あたりの抗体の力価を測定するために、ガングリオシドを吸着させた後、 2.5 μg/ml LT-BまたはLTのブロッキング緩衝液溶液100 μlをウェルあたり加えて、37℃で1時間インキュベートした。プレートをPBS-Tによって3回洗浄した後、ブロッキング緩衝液において希釈した血清試料200 μlを列Aに加えて、ブロッキング緩衝液100 μlを残りの列に加えた。特に明記していなければ血清の開始希釈液は、ブロッキング緩衝液において1:10倍であった。血清試料を連続2倍希釈した後、プレートを37℃で1時間インキュベートした後PBS-Tにおいて3回洗浄した。ヤギ抗ニワトリ結合体をHRPによって標識して、これを加え、37℃で1時間インキュベートした。プレートを洗浄してABTS 100 μlを加えて、Tecan Sunriseプレートリーダーを用いて、陽性対照が405/492の2波長での吸光度0.7〜1.0を示すまでインキュベートした。Tecan Magellanソフトウェアを用いてデータを転送して表示した。直線回帰および定量分析は、マイクロソフトオフィスエクセル2000バージョン9.0.3821 SR-1を用いて行った。マイクロソフトエクセル2000バージョン9.0.3821 SR-1を用いて、各治療群の血清の幾何平均力価(GMT)を決定した。これらの計算に関して、バックグラウンドELISA力価<10に値1を与えた。処置したトリの最小二乗平均値の対照との差を、最小二乗分析を用いて決定した。処置は、非ワクチン接種非チャレンジ対照群と処置群とのあいだに有意差があれば有効であるとした。
【0094】
血清ELISA NDV-HN
0.01 Mホウ酸緩衝液によって希釈した(1:2000倍)ウサギα-NDVプール抗血清(50%グリセロール水溶液と1:2混合)(100 μl/ウェル)によってプレートをコーティングする。プレートを2〜7℃で一晩インキュベートする;蓋をして室温で約20〜30分間プレートを平衡にする。プレートをPBS-T(1×PBS+0.05%Tween20)によって300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって3回洗浄する。プレートを5%スキムミルクのPBS-T(ブロッキング緩衝液)溶液(200 μl/ウェル)によってブロックして、37℃で2時間インキュベートする。プレートをPBS-Tによって300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって1回洗浄する。NDV尿膜腔液をブロッキング緩衝液において1:200倍希釈する。希釈した抗原を100 μl/ウェルでプレートに加えて、プレートを37℃で1時間インキュベートする。プレートをPBS-Tによって300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって洗浄する。試験ニワトリ血清試料を希釈する(1:50倍)。陰性対照血清(1:50倍)(陰性対照27NOV00)を希釈する。陽性対照血清(1:10,000または1:20,000)(SPAFASニワトリα-NDV血清)を希釈する。血清試料は全てブロッキング緩衝液において希釈する。陰性対照血清100 μl/ウェルを縦列1の列B〜Gに加える;陽性対照血清200 μl/ウェルを縦列2〜3の列Aに加える;試験血清試料200 μgl/ウェルを列Aの適当なカラムに加える。これによって、プレートあたり試料4個を試料あたり希釈液8個で行うことができる。残りの全てのウェルにブロッキング緩衝液100 μl/ウェルを加える;陽性対照血清および試験血清試料をプレートの下段に向かって連続2倍希釈する。試料をプレートのA列からH列まで希釈して、残りの100 μl/ウェルを捨てる。プレートを37℃で1時間インキュベートして、PBS-T 300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって3回洗浄する。ヤギαニワトリIgG(H&L)-HRP(1:3000倍)をブロッキング緩衝液において希釈する。希釈した結合体100 μl/ウェルを各プレートに加える;結合体をプレートに加えた後、RTの暗所でABTS基質を平衡にする。プレートを37℃で1時間インキュベートする;プレートを300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によってPBS-Tによって3回洗浄する。予め加温したABTS基質100 μl/ウェルを各プレートに加える。プレート間を2〜3分間離す。陽性対照の最初の希釈液の吸光度が0.7〜1.0に達した場合に、Tecan Sunriseプレートリーダーを用いて、プレートを二波長405/490 nmで読み取る。
【0095】
血清ELISA AIV-HA
0.01 Mホウ酸緩衝液において希釈した(1:1000倍)ウサギα-HAプール抗血清によってプレートをコーティングして、蓋をして2〜7℃で一晩プレートをインキュベートした。室温で約20〜30分間プレートを平衡にして、PBS-T(PBS保存液+0.05%Tween20)によって300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって3回洗浄した。5%スキムミルクのPBS-T溶液(ブロッキング緩衝液)によってプレートをブロックして(200 μl/ウェル)、プレートを37℃で1時間インキュベートした。プレートをPBS-Tによって300 μl/ウェルでTitertek M96プレートウォッシャーまたは同等物によって1回洗浄する。不活化T/W/68 AIV尿膜腔液(1:100倍)をブロッキング緩衝液溶液において希釈して、希釈した抗原100 μl/ウェルをプレートに加えた;プレートを37℃で1時間インキュベートする。PBS-T 300 μl/ウェルによってTitertek M96プレートウォッシャーまたは同等物を用いてプレートを洗浄する。試験ニワトリ血清試料(1:50倍)を希釈する;陰性対照血清(1:50倍)を希釈する;陽性対照血清(1:25600倍)(USDA/SEPRLニワトリα-AIV(T/W/68抗血清))をブロッキング緩衝液において希釈する。陰性対照血清100 μl/ウェルを縦列1の列B〜Gに加える;陽性対照血清200 μl/ウェルを縦列2〜3のA列に加える;試験血清試料200 μl/ウェルを適当な縦列のA列に加える;ブロッキング緩衝液100 μl/ウェルを残りの全てのウェルに加える。陽性対照血清および試験血清試料をプレートの下段に向かって連続2倍希釈して、残った100 μl/ウェルを捨てて、プレートを37℃で1時間インキュベートする。PBS-T(300 μl/ウェル)によってTitertek M96プレートウォッシャーまたは同等物を用いてプレートを3回洗浄する。ヤギα-ニワトリIgG(H&L)HRP(1:3000倍)をブロッキング緩衝液において希釈して、希釈した結合体100 μl/ウェルを各プレートに加える。結合体をプレートに加えた後、ABTS基質をRTの暗所で平衡にする。プレートを37℃で1時間インキュベートして、PBS-T 300 μl/ウェルによってTitertek M96プレートウォッシャーまたは同等物を用いてプレートを3回洗浄する。平衡にしたABTS基質を各プレートに100 μl/ウェルで加える;プレート間に2〜3分の間隔を空ける。陽性対照の最初の希釈液の吸光度が0.7〜1.0に達した場合に、Tecan Sunriseプレートリーダーを用いて、プレートを二波長405/490 nmで読み取る。
【0096】
血清ELISA IBDV-VP2
血液および血清の採取は、LTに関する血清ELISAに関して先に記述した通りに行った。血清ELISAに関して、ニワトリ抗IBDVを1.0 μg/mlの0.1 Mホウ酸緩衝液において2〜7℃で一晩インキュベートしてプレートに吸着させた。プレートをPBS-Tによって3回洗浄した後、3%スキムミルクPBS-T溶液によって37℃で1時間ブロックした。ブロッキング緩衝液において希釈したニワトリ血清試料200 μlをA列に加えてブロッキング緩衝液100 μlを残りの列に加える。特に明記していない限り、血清の開始希釈液はブロッキング緩衝液において1:10倍であった。血清試料を連続2倍希釈した後、プレートを37℃で1時間インキュベートして、PBS-Tによって3回洗浄した。HRPによって標識したヤギ抗ニワトリ結合体を加えて、37℃で1時間インキュベートした。プレートを洗浄して、ABTS 100 μlを加えて、陽性対照の吸光度がTecan Sunriseプレートリーダーを用いて、プレートを二波長405/490 nmで読み取った場合に0.7〜1.0に達するまでインキュベートする。LTに関する血清ELISAに関して先に記述した通りに、データをTecan Magellanソフトウェアを用いてデータを転送して表示した。
【0097】
実施例7:赤血球の赤血球凝集および赤血球凝集阻害
赤血球凝集
ニワトリ赤血球のAlsevers溶液(CRBC)をColorado Serum(L# 8152)から得た。1%CRBC溶液を調製するために、5 mlを15 ml遠心管に移して、250×gで10分間遠心した。上清およびバフィーコートをRBC沈降物からピペットによって採取した;沈降物を1×DPBS(ダルベッコリン酸緩衝生理食塩液)(L# 003435E JRH)に再懸濁させることによって2回洗浄して、250×gで10分間遠心した。沈降物をDPBSにおいて1%(v/v)に再懸濁させた。懸濁液の濃度を確認するために、400 μlを脱イオン水1.6 mlに移して、激しく混合することによって細胞を溶解した。OD540は0.4〜0.5のあいだであった。1%溶液は使用するまで2〜7℃で保存した。赤血球凝集を試験するために、96ウェルU底ディッシュ(Falcon)にまず、Static Guard(商標)を噴霧して、ペーパータオルにしみこませた。ウイルス試料をDPBSにおいて予め1:2倍希釈して、DPBS 50 μlを96ウェルディッシュの各ウェルに加えた。希釈したウイルスを第一の列に加えて、ウイルス試料あたりの望ましい回数の希釈液を得るまで連続2倍希釈した。1%CRBC 50 μlを各ウェルに加えてプレートを600 rpmで20秒間混合した。プレートを湿ったペーパータオルの上に置いて、対照ウェルにおけるCRBC(DPBSおよびCRBSが1:1比)がプレートの底に沈降するまで、または2〜7℃で少なくとも1時間インキュベートした。終点は、100%凝集を提供するシリーズにおける最後のウェルの希釈であった。
【0098】
赤血球凝集阻害(HAI)
ウイルスをDPBSにおいて予め希釈して4〜8 HA単位/50 μl(本明細書に記述のウイルスの力価測定に基づく)を得た。DPBS 25 μl/ウェルを縦列1および3〜12に用いて異なるプレートを作製した;血清25 μlを縦列1および3のウェルに加えた。縦列3における血清を10ウェルを通して連続希釈した。予め力価を測定したウイルス(25 μl)を縦列3〜12の全てのウェルに加えて、600 rpmで20秒間混合した;プレートを室温で1時間±15分インキュベートした。1%CRBC 50μlをウェルに加えて、600 rpmで20秒間混合して、湿潤室においてAIVに関して2〜7℃で一晩、およびNDVに関して2〜7℃で1〜2時間インキュベートした。血清の力価は赤血球凝集を100%阻害する連続希釈液の最後のウェルである。
【0099】
実施例8:ウサギにおける抗原性
上記のように、非洗浄剤緩衝液において抽出された免疫保護粒子における植物由来タンパク質が動物種において抗体を産生するか否かを調べるために、HAおよびHNタンパク質の双方を調製して、ウサギに接種した。3ヶ月齢のニュージーランドホワイトウサギに、表4に示す投与スケジュールに従って植物由来HA-AIVまたはHV-NDVを接種した。初回接種の場合、抗原をフロイントの完全アジュバント(CFA)と混合して、追加接種の場合には全てフロイントの不完全アジュバントを用いた。双方のタンパク質によって誘導された抗体力価を表5に示す。結果は、2回の接種後、HAI抗体力価が双方のタンパク質によって誘導され、このことはNT-1細胞の後期増殖相から調製した本発明の植物由来免疫保護粒子が哺乳類において、天然のタンパク質を認識することができる抗体を誘導することを示している。データから、植物由来HNおよびHAが天然に由来するHNおよびHAタンパク質と共有する特徴を有することが示唆される。植物由来AIV-HA接種ウサギの力価は、NDV-HN植物由来タンパク質によって誘導された力価より高かった。このことは、AIV-HAタンパク質の単位あたりのAIV/HAタンパク質の全般的生物活性(赤血球凝集)がNDV-HNより低かったことから、重要であるかも知れない(表4、縦欄4)。
【0100】
実施例9:発現された抗原の有効性および生物活性:家禽およびインビトロ細胞障害性におけるチャレンジ
ニューカッスル病ウイルス(NDV)のチャレンジ試験
植物由来HNタンパク質の有効性を調べるために、2日齢および10日齢のトリを用いて二つの異なる試験においてHNタンパク質を接種した。これらの試験に用いられる試験#16の用量濃度を表6に示す。全てのワクチン接種物をシェーカーフラスコにおいて25℃で15〜20日増殖させたNT-1細胞の可溶性分画から調製した。双方の試験において用いられるアジュバントは、Corixa、Inc.のMPL-TDMであった。鼻腔内群には、MPL単独をアジュバントとして投与した。
【0101】
2日齢のSPFニワトリに様々な経路によって、NT-1由来生物活性(赤血球凝集陽性)NDV-HNタンパク質を表6に示す接種あたりのHNタンパク質の量で用いて接種した。この試験の血清およびチャレンジの結果を表7に示す。対照群は全て予想どおりに反応した。接種物においてNDV-HN抗原を投与しなかったトリは、100%死亡率を示したが、SQによって天然のNDV 20 μgを投与した対照のトリは100%生存した。植物由来HN抗原群を用いた実験処置において、#3群において75%保護(アジュバントと共にSQ接種)および#4群において80%保護(アジュバントと共にSQ接種)を認めた。INおよび経口経路によって接種した残りの治療群は100%死亡率を示した。しかしながら、6群ではトリ2羽に死亡の遅れを認め、これらのトリがワクチン接種に対して感作されている可能性があること(例えば、表10、列14を参照されたい)、およびこの経路によって投与する場合には有効性を増強するために異なる製剤を必要とすることを示している。その後の試験(#18)において、10日齢のSPFトリに表8のスケジュールに記述した用量を接種した。対照群1群(#3)、非ワクチン接種非チャレンジ処置を用いて、ケージおよび施設がニワトリの全般的な健康に有害な作用を及ぼさないことを示した。この試験における対照群も、予想どおりに反応した。双方の試験からのトリを同じ施設でチャレンジすることから、処置群#2を試験16(表7)および18(表9)の双方の陽性対照とした。その全てにNT1細胞に由来するHNをSQ接種した残りの群では、群#7において100%生存、群5および6のそれぞれにおいて80%生存を認め、そして4群において60%生存を認めた(表9を参照されたい)。
【0102】
トリインフルエンザ(AIV)のチャレンジ試験
異なる試験において、ブロイラーニワトリに、植物由来赤血球凝集タンパク質(HA)をワクチン接種した。トリインフルエンザウイルス(AIV)株A/turkey/Wisc/68(H5N9)の植物由来HAタンパク質遺伝子配列を、NT1 CHN-18に関して記述したベクター系を用いてNT1細胞に形質転換した。HA-AIVタンパク質のトランスジェニック植物細胞産生のためのNT1株の命名はCHA-13であった。ニワトリをふ化後3日齢で得て、トリ10羽を各治療群に関してケージに無作為に入れた。各治療群の用量を表11に示す。トリを試験の0、14、および28日目に3回投与して、血液試料を0日、21日、35、および45日に採取した。各血液試料からの血清をHAI力価に関して分析して、35日目にトリをジョージア州アテンズのSoutheast Poultry Research Laboratoryに輸送して、そこでトリに毒性AIV(ニワトリ/ペンシルバニア/1370/1983)をチャレンジした。表11に示したデータから、CHA-13 NT1株に由来するHAタンパク質30 μg用量によって、ワクチン調製物の2回投与後であってもHAI陽性力価への血清変換が得られたことが示される。チャレンジすると、ワクチン接種群は全て、AIVに対して保護を示した:50またはそれ以上のチャレンジスコアは疾患または臨床病態を示している。製剤によらず、全ての群は、チャレンジ時に天然のAIVに対して非常に類似の力価を示し、このことは植物由来タンパク質によって誘導される天然のウイルスに対するメモリー反応を示している(縦欄4、表11)。
【0103】
伝染性ファブリキウス嚢病ウイルス(IBDV)のチャレンジ試験
上記の試験は、本発明に従うトランスジェニック植物細胞に由来する二つのタイプの糖タンパク質が、それらが毒性チャレンジから標的種を保護できるという点において非常に有効であることを示している。さらなる試験において、IBDVからの非グリコシル化構造タンパク質VP2を、CHN-18およびCHA-13の場合と類似のベクターおよびプロモーター構築物によってNT-1細胞に形質転換した。本明細書に記述の得られたトランスフェクト細胞をCVP2-002と命名した。本試験において、SFGニワトリをふ化後7、21、および35日目に、NT-1対照細胞溶解物、IBDV(形質転換事象CVP2-002)からのVP2タンパク質を発現するトランスジェニックNT細胞からの細胞溶解物、および市販の不活化伝染性ファブリキウス嚢病ウイルス(IBDV)ワクチン(Lohman Animal Health)Vi Bursa K+Vをワクチン接種した。NT-1対照細胞を10 L発酵器において増殖させて、継代6代目のCVP2-002細胞をシェーカーフラスコにおいて増殖させた。細胞を植物10〜14日後に回収して、18,000 PSIで100 μm Z形相互作用チャンバーを備えたMicrofludics 110 L微小溶液装置の中に通過させることによって溶解した。得られた細胞溶解物を2000×gでの遠心によって透明にした。透明にした上清を凍結乾燥によって濃縮した。ワクチンをアジュバントと共に調製して、各ワクチンのVP2濃度をワクチン接種前にELISAによって決定した。表12は、ワクチン製剤、投与経路、各ワクチン接種日でのVP2濃度を記述する。血液試料をふ化後21、35、および42日目に採取して、血清ELISAにおいて抗体反応に関して、およびIBDV血清中和(SN)アッセイにおいて中和抗体力価に関して試験した。トリに、IBDVの50 EID50胚由来STC株を両側の点眼によってチャレンジした。トリをチャレンジ後10日目に安楽死させた。ファブリキウス嚢対体重(BBW)および脾臓対体重(SBW)比を各トリに関して決定した。各トリからのファブリキウス嚢組織をホルマリンによって固定して、嚢胞枯渇によって示されるIBDV関連病変に関して採点した。BBW比、SBW比、および嚢病変スコアを非チャレンジ対照トリと比較した。非チャレンジおよび対照のあいだでBBWに統計学的有意差がなければトリは、チャレンジから保護されたと採点された。表13は、各ワクチン群に関する血清学およびチャレンジ結果を要約し、これは、植物由来VP2抗原が通常の死菌IBDワクチンより実際に大きい(ELISAによって)血清反応を生じることを示している。さらに、BBWによって測定したチャレンジに対する保護は、植物由来VP2が通常の死菌IBDワクチンと共に保護することを示している(表13の列4〜列10を比較)。
【0104】
Y1副腎細胞における熱不安定性毒素の細胞障害性
マウスのY1副腎細胞をATCC(CCL-79、L#1353400)から購入した。細胞のバイアルを37℃で融解して、F-12K培地(Gibco L#1089716)において15%ドナーウマ血清(Quad-5 L#2212)、2.5%ウシ胎児血清(JRH L# 7N2326)、1%グルタマックス-1(Gibco L#1080323)からなる増殖培地10 mlを含む25 cm2T-フラスコ(Corning)に入れた。細胞を5%CO2において37℃でインキュベートした。細胞を、各継代時にこの増殖培地で、LTおよびCT細胞障害性アッセイのために維持した。アッセイするために、細胞を96ウェル細胞培養プレート(Nunc)に播種して80%コンフルエンスに達するまで増殖させた。LT毒素をF-12K増殖培地において1 μg/mlに希釈した。毒素を、プレートのA列に予め希釈した試料100 μlを加えることによって、96ウェルマイクロタイタープレートにおいて2倍連続希釈によってさらに希釈した。次に、A列の50 μlを次のウェルの増殖培地50 μlに移すことによって2倍連続希釈を行った。試料の各希釈液を、試料または細胞の利用率に応じてY1副腎細胞の1〜4ウェルに移した。LT毒素の終点力価は、50%細胞障害性(細胞死)を得るために必要なタンパク質の量(EC50)である(Guidryら、1997;Dontaら、1974)。用いた毒素は、NT-1トランスジェニック細胞株SLT105、SLT107およびSLT102においてそれぞれ産生された大腸菌の熱不安定毒素のG192、R72、およびK63単アミノ酸遺伝子置換変異体であった。LT毒素の三つの変異体型は、インビトロバイオアッセイおよびインビボで毒素であることが報告されており、G192、R72、K63は野生型LT毒素よりそれぞれ約10倍、100倍、および1000倍毒性が低い(Rappuoliら、1999、Immunology Today 20:293〜500)。植物において産生されたLT変異体濃度を、Y1副腎アッセイ(表14を参照されたい)において大腸菌からのLT野生型毒素と比較したところ、結果は、植物由来毒素によって、大腸菌に由来する同じ変異体に関して認められる類似のレベルの感受性が得られることを示している。さらに、LT毒素の定量はG1ガングリオシド捕獲ELISA法によって決定されることから、植物産生毒素は完全に構築されたホロトキシンを模倣する(Guidryら、1993、Inf. and Imm. 65:4943〜4950)。
【0105】
CHA-13およびCHN-18からの植物産生免疫保護粒子の粘膜輸送
粘膜表面に輸送される非複製植物由来抗原が、強力な免疫保護材料であるか否かを決定するために、接種のために均一な乳剤を作製するために微少溶液操作によって調製した製剤を用いて、眼および鼻腔粘膜表面に抗原を直接接種することによってトリの試験を行った。7日齢のブロイラーニワトリをケージに無作為に入れて(1群5羽)、2.6〜16.7 μg/トリの抗原を接種した(表15を参照されたい)。トリにワクチンの3用量を0、14、および21日目に投与して、トリは試験終了時42日齢であった。抗原にはCHN-18トランスジェニック植物細胞に由来するHN、CHA-13トランスジェニック植物細胞に由来するHA、および感染ニワトリ胚の尿膜腔液に由来する不活化トリインフルエンザウイルスが含まれた。抗原調製物は、先の実施例3に記述したように調製した。様々な製剤において異なるアジュバント5個を用いて、免疫応答を、赤血球凝集阻害および血清ELISAに関して血清学によって決定した(表15を参照されたい)。試験の結果を表16に示す。一つの製剤を除く三つの用量によって全て、CHN-18からの植物由来HNを接種したトリでは血清変換が起こった。製剤の一つによって、CHA-13からのHAを接種したトリでは血清変換が起こり、二つの製剤によって、不活化AIV抗原を接種したトリの血清変換が起こった。一つのアジュバントは全ての反応群に共通し、コレステロールと混合したQuil Aであった。結果から、植物由来の非複製抗原が、粘膜表面への接種によってトリにいて血清学的反応を提供したことが示される。
【0106】
実施例10:トランスジェニック細胞における発現タンパク質の産生および蓄積
10 Lバイオリアクターまたはシェーカーフラスコにおいて増殖させたトランスジェニック細胞培養物から発現されたrDNAを図15〜18に示す。実施例2において先に記述した培地にCHN-18、CHA-13、SLT102、またはCVP2-002トランスジェニック細胞を産生するNT-1トランスジェニック細胞培養物を、培養12日後に回収した(静止期)。次に、シェーカーフラスコからの接種物を、Pluronic L61消泡剤1 mlを含む増殖培地10 Lを含む10 L Bioflow 3000発酵器(New Brunswick)に無菌的に移した。細胞の産生は、100 rpmで攪拌しながら30%溶存酸素で2.5 L/分で通気しながら25℃で行う;細胞の産生は9〜15日間行う。15 ml遠心管に発酵培養物10 mlを加えて、2000×gで10分間遠心することによって、細胞沈降容積(PCV)を決定し、細胞容積を測定して、接種日から10日目までの細胞増殖、または静止期培養を追跡するパラメータとして評価した。データは、約3日間の遅滞の後、培養物の指数増殖期が3日〜7日までのあいだに起こり、その後培養物は、挿入された遺伝子および用いたプロモーター系によらず、分析した各トランスジェニック細胞株に関して静止期に達し始めた。CHN-18の場合、測定可能なHNタンパク質の量を毎日追跡した。1日目、新たな細胞増殖の前に、抽出することができるHN ELISAシグナルを認め、これは培養12日でシェーカーフラスコから採取した接種物に存在するHNの量を表す。しかしながら、HNは急速に分解し、細胞が静止期に達した培養約6日後まで検出されず、細胞が静止期を通過した後も細胞に蓄積され続ける。HN発現は異なる二つの測定によって追跡し、黒三角は定量的ELISAによって測定したHNタンパク質を表し、黒四角は赤血球凝集を表す。定量的ELISAは、HNタンパク質産生に対してより感受性が高く、単量体または重合化HNタンパク質の双方を測定するが、赤血球凝集は、赤血球を凝集することができる二量体または重合化タンパク質のみを測定し、より多くのタンパク質が蓄積して初めて、このように赤血球凝集活性を測定することができる(図15)。タンパク質の増殖後期産生の現象は、発現されるタンパク質(大腸菌のホロトキシンLT、トリインフルエンザウイルスの赤血球凝集素タンパク質(HA)、伝染性ファブリキウス嚢病ウイルスのVP2構造タンパク質、またはニューカッスル病ウイルスの赤血球凝集素-ノイラミニダーゼタンパク質(HN))によらず認められる(図16、17、および18を参照されたい)。
【0107】
CHA-13およびCVP2-002の産生および増殖曲線をそれぞれ、図16および17に示す。CHA-13に関して、増殖(PCV)は、接種後2日目に始まり、接種後10日目で静止期に入る。ショ糖は接種後2日まで消費され、デキストロースは接種後6日まで消費される。HAの蓄積は、接種後6日目に始まり(中期対数増殖期)14日まで増加する。細胞増殖は接種後2日目に始まり、接種後9または10日目で静止期に入る。ショ糖は接種後2日まで消費され、デキストロースは接種後5日まで消費される。VP2蓄積は接種後7日目に始まり(中期対数増殖期)、14日まで増加する。
【0108】
大腸菌熱不安定毒素(LT)に関するK63変異体を発現するSLT102トランスジェニックNT-1細胞株を図18に示す。LT毒素は5日および6日のあいだに蓄積し始め、沈降細胞容積はこの実験では示されないが、他のNT-1トランスジェニック細胞株の容積と類似である。
【0109】
実施例11:植物産生タンパク質の安定性
組換え型または天然起源から抽出したタンパク質はしばしば、プロテアーゼ、グリコシラーゼ、リパーゼ、またはタンパク質および細胞成分と共に精製された他の酵素のために不安定である。NT-1細胞から単離されたタンパク質および免疫保護粒子は、固有に安定であり、多くの異なるタイプの下流のプロセシング活性に対して強健である。図19において、CHN-18細胞を静止期の10 L発酵器から採取して、濾過し、遠心によって透明にして、実施例3に記載の方法に従って1回微少溶液化した。上清を0.2または0.45ミクロンのフィルターに通過させて、濾過または微少溶液化による操作の際に導入された可能性がある如何なる細菌物質も除去し、安定化剤をこれらの懸濁液に加えなかったが、安定性は、これらのトランスジェニック細胞に由来するタンパク質に対して固有である。次に、材料を2〜7℃、25℃で保存するか、または−80℃で凍結した;材料は全ての温度で安定であることが判明したが、最も興味深いことに25℃(外界温度)で維持すると、単離タンパク質は、安定であることが判明した(図19に示す)。月毎にシグナルの変動を認めたが、単離されたタンパク質の量は、数ヶ月後顕著な安定性を示し、これらのデータから計算することができる半減期は、予測半減期8ヶ月(0.45ミクロン試料)および0.2ミクロン濾過試料では1年より長いことを示している。
【0110】
実施例12:抗原の細胞下局在
共焦点レーザー走査顕微鏡(CLSM)
共焦点レーザー走査顕微鏡を形質転換MHN-41およびCHN-18においてHN抗原を局在するために行った。局在技法のために用いた抗体は、IgG精製ウサギ抗HNポリクローナル(HN ELISAにおける捕獲Ab)およびHN Mab 4A−腹水からの非精製型(HN ELISAにおける検出Ab)であった。画像は以下の技法を用いて培養植物細胞から得た。非形質転換対照細胞(NT-Ctrl.)を含む細胞を1000 gで5分間遠心して、3.7%ホルムアルデヒドにおいて15分間固定した。細胞をPBSによって各洗浄について5分間2回洗浄した。細胞を3000 gで2分間(毎回)遠心して、0.5%TritonX-100および1%ペクトリアーゼによって15分間処置した。H2Oによる洗浄を行って、H2Oをメタノール(−20℃)に置換した;異なるウェルを有するコーティングしたスライドガラスに、ピペットによって細胞を載せて、空気乾燥させた(フード内で20〜30分間)。PBSによって洗浄した後、3%BSA/PBSによって30分間ブロックした。一次抗体(1%BSA-PBS-T(0.05%Tween-20を含むPBSを37℃で1時間、またはRTで1.5〜2時間)インキュベートする)。PBS-Tによって3回洗浄する。Cy5/Cy2(1:100倍)によって標識した二次抗体をRTで1時間インキュベートして、スライドガラスをPBS-Tによって3回洗浄して封入した。
【0111】
NT対照細胞ではRb抗HNポリクローナルまたはHN Mab 4Aによって染色を認めなかった。双方のHN特異的抗体によって、静止期細胞の細胞質全体に明るい染色(核には認めない)をMHC-41細胞株全体に認めた。(図20および21を参照されたい)。
【0112】
電子顕微鏡
発現されたタンパク質が蓄積されている場所を確立するために、トランスジェニック植物細胞を培養10日後に回収して、以下のように薄切片および免疫金標識のために調製した。免疫金標識は、10日齢の感染ニワトリ卵胚から採取した尿膜腔液のニューカッスル病ウイルス調製物から精製したHNタンパク質によって免疫したウサギからの精製IgGを用いて行った。形態学的特徴を定義するために、細胞懸濁液を3%グルタルアルデヒドの0.1 Mリン酸緩衝溶液(pH 6.8)において3時間固定した。次に、それらをリン酸緩衝液において、緩衝液を4回交換して1時間洗浄した。細胞を2%四酸化オスミウムのリン酸緩衝溶液において1時間後固定した。濃度を増加させたエタノール(25%、50%、75%、95%、および100%、各段階15分)およびプロピレンオキサイドにおいて細胞を脱水した。細胞をプロピレンオキサイド/Epon 812混合物において一晩放置した後、Epon 812において抱埋し、60℃で2日間重合化した。切片をLKB Ultrotome IIIによって切断して、2%酢酸ウラニル水溶液およびクエン酸鉛によって染色して、Hitachi 7500透過型電子顕微鏡を80 kVで操作して調べた。
【0113】
免疫金染色に関して、グルタルアルデヒド固定細胞をリン酸緩衝液(0.02 Mグリシンを加える)洗浄後、エタノールの濃度を増加させて脱水した。次に、細胞をLR White樹脂に一晩浸潤させて、最終的に抱埋し、50℃で24時間重合化した。
【0114】
ニッケルグリッド上に封入した切片を、1%ウシ血清アルブミン溶液と共にpH 7.4のPBS緩衝液において20分間インキュベートして、非特異的部位をブロックした。次に、細胞を一次抗体(PBSにおいて1:150倍希釈)と共に室温で2時間インキュベートした。次に、PBS-BSAによって6回洗浄して(各3分)、ヤギ抗ウサギAB(PBSにおいて1:150倍希釈)に結合させた金コロイド(15 nm)と共に室温で2時間インキュベートした。細胞をPBSにおいて5分間の洗浄を4回行った後、水によって1分の洗浄を2回行った後、グリッドを酢酸ウラニルによって5分間染色した。EM画像によって、対照細胞と、HNタンパク質を発現するトランスジェニック細胞とのあいだに二つの主要な差が示され、最初の色素体/白色体は、トランスジェニック細胞において蓄積するが正常細胞には蓄積しない暗い顆粒を示し(図22)、第二に、免疫金染色顆粒は、トランスジェニック細胞の細胞壁近傍で蓄積することが認められうるが、対照細胞は認めることができない(図23)。典型的に、宿主細胞において発現される遺伝子産物は、細胞の指数増殖期に起こり、一般的に小胞体、ゴルジ体、および細胞における他のタンパク質合成小構造において染色されうる。図19および20に示す共焦点画像と共に、データは、電子顕微鏡によって、タンパク質が細胞膜および細胞壁において産生され、沈着されるが、核、葉緑体、ミトコンドリア、小胞体、およびゴルジ体には蓄積を認めないことを示している。電子顕微鏡から、後期静止相の細胞が肥大した液胞および圧縮された細胞質および核を有することが示される。共焦点画像は、タンパク質が、細胞全体の細胞質細胞壁および膜に対して圧縮されていることを示唆している。
【0115】
細胞におけるタンパク質産生および蓄積が、細胞がもはや活性な代謝状態ではない後期指数増殖期および静止期まで明白でないことは珍しい。増殖フラスコまたは発酵器における細胞の接種時に発現されたタンパク質シグナル(24時間)が急激に減少したこと(実施例15を参照されたい)は、窒素源としてタンパク質を用いること、そして細胞が活性な増殖を終了した場合には窒素源(タンパク質)の貯蔵が起こりうることを示している。この現象は、上記の実施例において記述される植物細胞培養において産生されたトランスジェニックタンパク質に対して独自の特徴である。細胞壁および膜近傍にタンパク質が存在することは、機械的破壊によって予想外に容易にタンパク質を単離できることを説明するために役立つ。各タンパク質のクラスが安定かつ有効な形で容易に単離されうることもまた、予想外である。如何なる単一のタンパク質もしばしば、組換え型DNA発現を調べるために選択された任意の外来宿主系において合成されうるが、多くのタンパク質、特に膜貫通結合糖タンパク質はしばしば、低レベルで産生されるが、一つの宿主系は同じ方法で二つの糖タンパク質を発現しない。本明細書に記述する細胞培養トランスジェニック系において、少なくとも五つのクラスのタンパク質(酵素、1型ウイルス糖タンパク質、2型ウイルス糖タンパク質、LT毒素、および構造的非グリコシル化タンパク質VP2)が、同じ宿主系において首尾よく類似のレベルで発現されている。さらに、タンパク質は、タンパク質のクラスによらず、後期静止期において蓄積し、転写カセットまたはプロモーター系は同じ物理的または機械的破壊法を用いて細胞から容易に除去することができる。これらのトランスジェニック細胞によって発現されるタンパク質のクラスによらず、各タンパク質は、生物学的に活性である安定な形での単離に成功している。
【0116】
実施例13:伝染性ファブリキウス嚢病植物最適化VP2抗原遺伝子
伝染性ファブリキウス嚢病(IBD)ウイルス(またはIBDV)のウイルス原因物質は、二つに分かれたRNAゲノムである(J. Virol. (1979)、32:593)。完全長のRNA1がポリタンパク質に翻訳され、これがペプチドVP2、VP3、およびVP4にプロセシングされる。ゲノムRNAのインシリコ(in silico)逆転写を行って、天然のRNAのタンパク質コード能に対応するDNA配列を得ることができる。天然のE/91 VP2タンパク質をコードするIBDVのEhime91(E/91)株の誘導DNA配列の1359塩基対(bp)は、Genbankアクセッション番号AB024076号として利用できる。この配列の分析によって、最適な植物遺伝子発現と共に、最適でないコドン組成にとって有害であると考えられるいくつかの配列モチーフが存在することが判明した(例えば、米国特許第5,380,831号を参照されたい)。単子葉植物と共に双子葉植物において組換え型VP2タンパク質の産生を改善するために、天然のE/91タンパク質のカルボキシ末端でイソロイシン、アラニン、およびバリン残基が1個ずつ付加されていることを除き、天然のE/91 VP2タンパク質と本質的に同一であるタンパク質(本明細書においてSEQ ID NO:12として開示される)をコードする「植物最適化」DNA配列(SEQ ID NO:11)を作製した。これらの付加アミノ酸のコドンは、J. Castonら(J Virol(2001)、75:10815)の報告に基づいて含めたが、彼らの報告はVP2カプシドアセンブリの最適なVP2プロセシング部位が、451位(Leu対Ile)を除いてE/91の配列と同一であるIBD株UK661(Genbankアクセッション番号NC_004178)VP2配列によってコードされる453位ではなくて最後のアミノ酸であるアミノ酸456位で起こることを示している。このように、アミノ酸454、455、および456位(Ile、Ala、Val)は、VP2遺伝子のアミノ酸456位を操作するためにUK661株に由来した。天然のE/91配列によってコードされるVP2タンパク質、および植物最適化コード領域によってコードされるVP2タンパク質は、99.3%同一であり、アミノ酸454、455、および456位が異なるに過ぎない。対照的に、天然のE/91 VP2コード領域の誘導DNAおよび植物最適化DNAは80.3%同一であるに過ぎない。
【0117】
外来遺伝子を植物の染色体に無作為に組み入れると、如何なる特定の組み込み部位も、組み込み部位に隣接する遺伝子制御エレメントおよびオープンリーディングフレームから新しい異常なタンパク質の偶発的な産生を行う部位となる可能性が存在する。これらの望ましくないそしておそらく有害なタンパク質の産生を消失させるために役立つように、可能性がある六個全ての読み取り枠において翻訳終止コドンをコードするさらなる塩基をVP2コード領域の下流に含めた(SEQ ID NO:13に開示する「万能ターミネーター」)。その後のクローニング段階を可能にするために、これらの制限酵素の認識部位を含む塩基をこの有用な配列に含める。
【0118】
実施例14:植物細胞における伝染性ファブリキウス嚢病植物最適化VP2抗原遺伝子の発現のための基本バイナリベクターpDAB2423の構築
IBD VP2遺伝子(SEQ ID NO:11)の植物最適化ヌクレオチド配列を含む双子葉植物発現ベクターを構築した。基本バイナリベクター(BBV)骨格(図24)を用いて、AgeIリンカーを付加することによって唯一のBamHI部位で改変を行った。新しいバイナリベクター(pDAB2407、図25)によって、T-DNA境界配列のあいだでVP2と選択マーカー発現カセットとのAge1/AgeIライゲーションを行うことができる(pDAB2423、図31)。
【0119】
発現カセットは、DAS5 P60C2からの合成VP2配列(図26、PICOSCRIPT、ハウストン、テキサス州)をBbsIおよびSacI制限酵素によって切除することによって構築した。CsVMVプロモーターおよびアグロバクテリウム・ツメファシエンス(Atu)ORF24 3' UTRをコードするpDAB2406(図27)をNcoIおよびSacIによって切断した。pDAB2406骨格およびVP2インサート断片を、pDAB2406のNcoIおよびSacI部位でライゲーションして、pDAB2415(図28)を得た。ライゲーションしたDNAを大腸菌DH5αコンピテント細胞(Invitrogen)に形質転換して、陽性クローンに関してスクリーニングを行った。HindIII×MluI酵素を用いて陽性クローンを制限分析によって同定し、インサート/ベクター接合部を超えたシークエンシングによって確認した。
【0120】
pDAB2415サブクローンを確認した後、プラスミドをNotIによって切断して、CsVMV/VP2/ORF24断片を単離した。RB7 MARエレメント(米国特許第5,773,689号、米国特許第5,773,695号、米国特許第6,239,328号、国際公開公報第94/07902号、および国際公開公報第97/27207号)およびシロイヌナズナ(At)ユビキチン10(Ubi 10)プロモーター(Plant J. 1997、11(5):1017;Plant Mol. Biol. 1993、21(5):895;Genetics 1995、139(2):921)によって調節される選択マーカー、PAT、およびAtu ORF1 3' UTR(米国特許第5,428,147号;Plant Molecular Biology 1983、2:335;Genbankアクセッション番号X00493)をコードするpDAB2418(図29)をNotIによって直線にした。pDAB2418骨格およびpDAB2415インサート断片を、ゲルにおいて単離して、精製し、NotI部位でライゲーションすると、pDAB2416(図30)が得られた。ライゲーションしたDNAを大腸菌に形質転換して、コロニーをBglIIおよびSacIによる制限酵素消化物によってスクリーニングした。陽性サブクローンのさらなる確認は、NotI接合部を超えるシークエンシングによって確認した。次に、pDAB2416プラスミドをAgeIによって切断して、ベクター骨格からMAR/CsVMV/VP2/ORF24/Ubi10/PAT/ORF1発現カセットを除去した。pDAB2407も同様に、AgeIによって切断して、バイナリベクターを直線にして、適当な断片をライゲーションしてpDAB2423を形成した。ライゲーションしたDNAを形質転換した後、コロニーをHindIIIおよびXhoI消化物を用いてスクリーニングした。採取したコロニー30個のうち、12個が陽性であった。陽性クローンを、NcoI、PmeI、およびPstI酵素を用いて制限消化物によってさらに分析した。最終確認のため、クローンをT-DNA境界配列のあいだで十分にシークエンシングした。
【0121】
(表1)植物由来HNの赤血球凝集活性に及ぼす抽出法の比較
1天然のNDVを2分間超音波処理した。抽出緩衝液−50 mMアスコルビン酸ナトリウム、1 mM EDTA、1 mM PMSF、および0.1%TritonX-100、pH 7.2;DPBS−ダルベッコリン酸緩衝生理食塩液;sonic−超音波処理;F/T−凍結融解;nd−本試験では実施していない
【0122】
(表2)植物由来HNおよび天然のウイルスの赤血球凝集阻害(HAI)活性の比較
1保存ウイルスは4HA単位であり、保存ウイルスの1:4倍希釈に等しい。これは抗体の力価を測定するために用いられるウイルスの濃度であり、ウイルスの4 HA単位を妨害する終点希釈抗体が、抗体調製物のHAI力価であると考えられる。
【0123】
(表3)微少溶液操作の際に様々な圧力を用いたCHN-18トランスジェニック細胞の細胞抽出物の赤血球凝集の比較
MF−微少溶液操作;PSI−ポンド/平方インチ;S/N−上清
【0124】
(表4)ウサギに接種するための用量レベル
1全てのNT-1試料を非凍結乾燥材料から提供した。BCA−ビシンコニン酸、Pierce Chemical BCAタンパク質アッセイキットの主成分;TSP−総可溶性タンパク質。
CHN-7およびCHN-18はNDVからのHNタンパク質を発現する二つの異なるトランスジェニック細胞株であり、CHA-13およびCHA-47はAIVのHAタンパク質を発現する二つの異なるトランスジェニック細胞株である。
【0125】
(表5)トランスジェニック植物細胞CHA-13およびCHN-18に由来するAIV-HAおよびNDV-HNタンパク質を摂取したウサギからの血清学の結果
S/N−上清;HAI−赤血球凝集阻害血清力価
【0126】
(表6)家禽の試験#16に関して接種あたりに用いられる用量レベル
1湿重量細胞あたり赤血球凝集素/ノイラミニダーゼ(HN)の発現に基づく用量;IN−鼻腔内;SQ−皮下;OG−経口針;OF−飼料混合物
【0127】
(表7)家禽の試験#16のCHN-18に関する血清学およびチャレンジ結果(NDVチャレンジ)
全てのトリに、NDVのTexas GB株102 EID50(卵感染用量)を接種した。トリには最後のワクチン接種後24日目にチャレンジした。太字のトリの数は、遅れて死亡を認めた。表9を参照されたい。HAI−赤血球凝集阻害血清力価;na−適用されない。
【0128】
(表8)試験#18に関する接種あたり用いられる抗原の用量レベル
【0129】
(表9)試験#18からのCHN-18の血清学およびチャレンジ結果(NDVチャレンジ)
トリには、NDVのTexas GB株104 EID50 を接種した1群および非チャレンジ対照である3群を除き、全てNDVのTexas GB株102 EID50(卵感染用量)を接種した。トリは最後のワクチン接種後31日目にチャレンジした。赤血球凝集阻害(HAI)力価は、3回の実験の平均力価として報告する。
【0130】
(表10)試験#16および#18に関するチャレンジ後の死亡日(NDVチャレンジ)
*5群のトリ1羽はこの日付で記録しなかった。
【0131】
(表11)CHA-13に関する血清学およびチャレンジ結果(AIVチャレンジ)
1赤血球凝集阻害(HIA)力価は2回の実験の50%終点として報告し、値1は、バックグラウンドを示す。
2チャレンジスコアは、結膜炎=1;抑うつ=2;運動失調=3;麻痺/斜頚=4;死亡/安楽死=5の複合評定によって決定した。
3CorixaアジュバントモノホスホリルリピッドA(MPL)トレハロースジコリノミコレート(TDM)
【0132】
(表12)家禽の試験の接種あたりに用いられる用量レベル
1SQ−皮下;OG−経口針投与;IB−嚢内;SQ OG−1回目の投与は皮下、2回目および3回目は経口針による投与;ドレークオイル乳剤−nCVP2-002と共に等量の5%ドレークオイル、1%Tween80、0.33%スパン80;CGI−精製セルキャップ由来ニワトリγインターフェロン:LAP−nCVP2-002と共に等量のレシチンアクリル酸ポリマー水中油型乳剤
【0133】
(表13)CVP2-002家禽試験の血清学およびチャレンジ結果(IBDVチャレンジ)
1血清中和(SN)力価の範囲は、処置あたりのトリの群に関して血清力価の最低値から最高値までである。
2血清ELISA力価の範囲は、処置あたりのトリの群に関して血清力価の最低値から最高値までである。
3処置群において各トリに関してファブリキウス嚢対体重(BBW)を測定して、各チャレンジ群を非チャレンジ群(#1)と比較し、p値を提供する統計学的有意差が得られる;0.05より大きいp値は保護的であると考えられる。
na−適用されない
【0134】
(表14)様々な熱不安定毒素調製物を用いたY1マウス副腎細胞細胞障害性
1実施例6に記載のようにガングリオシド定量捕獲ELISAを用いて各抗原をアッセイする。
2LT濃度は、LTA特異的抗体を用いてガングリオシドによって捕獲されたLTBに結合するLTAタンパク質の量を表す。
3LTB特異的抗体を用いて毒素のLTB定量に基づく濃度。
LT−熱不安定毒素;LTA−熱不安定毒素のAサブユニット;LTB−熱不安定毒素のBサブユニット。
【0135】
(表15)植物由来抗原および天然の抗原の粘膜輸送(鼻腔内および点眼);処置群あたり投与した用量
1試験に用いた抗原には、CHA-13およびCHN-18植物由来抗原と共に不活化トリインフルエンザウイルス(不活化AIV)が含まれ、日付は10 L発酵器から採取した日付を指し、バッチ番号を示す。
2処置に用いたアジュバントは全て、0.05 mlを眼に点眼することによっておよび各鼻孔に0.2 mlを滴下することによって点眼経路および鼻腔内INによって投与した。LAP−レシチンアクリル酸ポリマー;Quil Aは、樹皮(Quillia saponaria)からのサポニンである;LT−大腸菌からの熱不安定毒素;chol−コレステロール;oil−ドラケオール鉱油。
3投与した用量レベルは、ワクチンをアジュバントと共に混合する前に、バルク抗原に関する定量的ELISAから採取した抗原の希釈液または組み合わせを用いて決定した。
4陽性対照試料はSQ−皮下接種によって投与した。
【0136】
(表16)植物由来抗原および天然の抗原の粘膜輸送ワクチン輸送(鼻腔内および点眼);処置群あたり投与した用量
1,2情報に関しては表15を参照されたい。
3赤血球凝集阻害(HAI)血清力価は、いずれもニワトリ胚の尿膜腔液に由来する不活化AIVおよびNDV抗原を用いて決定した。双方の抗原を各処置の対照として用い、報告された血清力価は、その処置に関する抗原に対する特異的反応であった。
NR−反応なし。
【図面の簡単な説明】
【0137】
本特許のファイルは、カラーで印刷される少なくとも一つの図面を含む。カラーの図面を有する本特許または特許出願の出版物のコピーは、要請に応じて、必要な料金を支払えば特許商標庁から提供されるであろう。
【図1】(SEQ ID NO:1および2)NDV株「Lasota」のHN遺伝子の植物最適化コード配列およびタンパク質配列を示す。
【図2】構成的CsVMV(カッサバ葉脈モザイクウイルス)プロモーターによって促進され、MAS 3'(マンノピンシンターゼ)エレメントによって終止する植物選択マーカーPAT(ホスフィノトリシンアセチルトランスフェラーゼ)を含むpBBV-PHAS-iaaHのマップを示す。DNAの境界を定めるアグロバクテリウムのLBおよびRB(左右T-DNA境界配列)エレメントが植物ゲノムに組み入れられている。
【図3】免疫保護抗原を発現するための多様な植物発現ベクターの開始プラスミドとして用いられる「鋳型ベクター」であるpC!Hのマップを示す。
【図4】NDV HNタンパク質のpCHN発現ベクターのマップを示す。HN発現ベクターまたはカセットは、構成的CsVMVプロモーターによって促進され、大豆vspB 3'エレメントによって終止する。
【図5】NDV HNタンパク質のpgHN発現ベクターのマップを示す。HN発現カセットは、TEV 5' UTRと共に塊茎特異的GBSSプロモーターによって促進され、大豆vspB 3'エレメントによって終止する。
【図6】NDV HNタンパク質のpgHN151発現ベクターのマップを示す。HN発現カセットは、その本来の5' UTRおよびイントロンと共に塊茎特異的GBSSプロモーターによって促進され、大豆vspB 3'エレメントによって終止する。ベクターは、CsVMVプロモーターによって促進されMAS 3'エレメントによって終止する植物選択マーカーPATを含むpBBV-PHAS-iaaHに由来する。DNAの境界を示すLBおよびRB、すなわち左右T-DNA境界配列エレメントが植物ゲノムに組み入れられている。
【図7】NDV HNタンパク質のpgHN153発現ベクターのマップを示す。HN発現カセットは、その本来の 5' UTRおよびイントロンと共に塊茎特異的GBSSプロモーターによって促進され、マメファセオリン3'エレメントによって終止する。ベクターは、CsVMVプロモーターによって促進され、MAS 3'エレメントによって終止する植物選択マーカーPATを含むpBBV-PHAS-iaaHに由来する。DNAの境界を示すLBおよびRB、すなわち左右T-DNA境界配列エレメントが植物ゲノムに組み入れられている。
【図8】NDV HNタンパク質のpMHN発現ベクターのマップを示す。HN発現カセットは、構成的4OCS△MASプロモーター(P2方向)によって促進され、大豆vspB 3'エレメントによって終止する。ベクターは、CsVMVプロモーターによって促進され、MAS 3'エレメントによって終止する植物選択マーカーPATを含むpBBV-PHAS-iaaHに由来する。DNAの境界を示すLBおよびRB、すなわち左右T-DNA境界配列エレメントが植物ゲノムに組み入れられている。
【図9】AIV A/turkey/Wisconsin/68(H5N9)のHA遺伝子のpCHA発現ベクターのマップを示す。
【図10】(SEQ ID NO:3および4)AIV A/turkey/Wisconsin/68(H5N9)のHA遺伝子のDNAおよびタンパク質配列を示す。
【図11】pGLTB中間ベクターのマップを示す。
【図12】pCLT105中間ベクターのマップを示す。
【図13】pDAB2423を示す。VP2をコードするバイナリベクターを示す。
【図14】IBDV伝染性ファブリキウス嚢病(IBD)ウイルスのVP2遺伝子のDNA配列、非常に毒性の高い株Ehime 91を示す。
【図15】CHN-18、CHA-13、SLT102、およびCVP2-002に関する発現されたタンパク質の産生、増殖、および蓄積を示す。10 L発酵器におけるCHA-13の増殖の結果を示す。黒四角:接種後様々な時間での試料10 mlからの細胞沈降容積(PCV)を用いたCn-18 NT-1トランスジェニック細胞の増殖。黒三角:実施例7に記述する定量的ELISAアッセイを用いたHNタンパク質(10 L発酵器あたり)の蓄積。黒菱形は、実施例8に記述の赤血球凝集力価の蓄積であり、1日目に認められた赤血球凝集力価は、13日振とう培養物からの接種物である。
【図16】CHN-18、CHA-13、SLT102、およびCVP2-002に関する発現されたタンパク質の産生、増殖、および蓄積を示す。10 L発酵器におけるCHA-13の増殖の結果を示す。黒四角:接種後様々な時間での試料10 mlからの細胞沈降容積(PCV)を用いたCHA-13 NT-1トランスジェニック細胞の増殖である。白三角は、ショ糖濃度を示し、ショ糖は、炭素源として用いられ、これはデキストロース(白四角)に急速に変換されて、培養物への接種後48時間ではもはや検出できない。定量的ELISAによるHAタンパク質の蓄積は、CHA-13 NT-1の細胞抽出物からの黒三角によって示される。
【図17】CHN-18、CHA-13、SLT102、およびCVP2-002に関する発現されたタンパク質の産生、増殖、および蓄積を示す。10 L発酵器におけるCVP2-002の増殖の結果を示す。黒菱形は、接種後様々な時間での試料10 mlからの細胞沈降容積(PCV)を用いたCVP2-002 NT-1トランスジェニック細胞の増殖を示す。白三角は、ショ糖濃度を示し、ショ糖は、炭素源として用いられ、これはデキストロース(*)に急速に変換されて、培養物への接種後48時間ではもはや検出できない。定量的ELISAによるHAタンパク質の蓄積は、CVP2-002 NT-1の細胞抽出物からの黒三角によって示される。
【図18】CHN-18、CHA-13、SLT102、およびCVP2-002に関する発現されたタンパク質の産生、増殖、および蓄積を示す。シェーカーフラスコ培養におけるLTの蓄積、濃度は実施例7におけるLT定量的ELISAによって決定した。増殖曲線は本試験では決定しなかったが、SLT-102 NT-1細胞のPCVは、図15〜17におけるNT-1トランスジェニック細胞株に関して認められた場合と同じ増殖および産生を示す。
【図19】トランスジェニック細胞株CHN-18からのタンパク質の安定な産生。CHN-18 NT-1から調製した試料の定量的ELISAシグナルの安定性。CHN-18からの上清は、実施例3に記載の10 L発酵器から細胞を回収することによって単離した。細胞を破壊するために、1回微少溶液操作した後、試料を0.45ミクロンまたは0.2ミクロンのフィルターによって濾過して、25℃で保存した。
【図20】共焦点走査顕微鏡。MHN-41染色細胞を示す。緑色:Rb抗HNポリクローナル抗体に関して標識したCy2色素によって染色した細胞(左上)。青色:4A腹水に関して標識したCy5色素によって染色した細胞(右下)。赤色:ヨウ化プロピジウム染色核(右上)。水色/赤:緑、青、および赤色の画像のデジタル融合画像。細胞の核には、いずれの抗体による染色も認めない。細胞壁全体および細胞内細胞質の膜に沿って強く染色された領域のために、液胞は区別できない。
【図21】共焦点走査顕微鏡。対照NT-1細胞を示す。対照NT-1細胞の染色、左のパネルはRb抗HNポリクローナル抗体または4A腹水のいずれかによって染色した細胞である:右のパネルはヨウ化プロピジウム染色NT-1細胞である。
【図22】トランスジーンによって産生されたポリペプチドの局在を示す電子顕微鏡写真を示す。NT-1対照細胞、CHN-18トランスジェニック細胞、およびMHN-41トランスジェニック細胞の四酸化オスミウム固定細胞の電子顕微鏡写真。各フレームの倍率を示し、対照細胞は倍率16,000倍、CHN-18は50,000倍、およびMHN-41は26,000倍である。
【図23】トランスジーンによって産生されたポリペプチドの局在を示す電子顕微鏡写真を示す。NT-1対照細胞、CHN-18トランスジェニック細胞、およびMN-41トランスジェニック細胞の電子顕微鏡の免疫金染色。
【図24】バイナリ、中間および発現ベクターのマップを示す。基本バイナリベクター(BBV)のマップを示す。
【図25】バイナリ、中間および発現ベクターのマップを示す。中間pDAB2407のマップを示す。
【図26】バイナリ、中間および発現ベクターのマップを示す。PICOSCRIPT(ハウストン、テキサス州)によって提供されたBluescriptベクターにおける合成されたVP2を示す。
【図27】バイナリ、中間および発現ベクターのマップを示す。中間ベクターpDAB2406のマップを示す。
【図28】バイナリ、中間および発現ベクターのマップを示す。中間ベクターpDAB2415のマップを示す。
【図29】バイナリ、中間および発現ベクターのマップを示す。中間ベクターpDBA2418のマップを示す。
【図30】バイナリ、中間および発現ベクターのマップを示す。中間ベクターpDAB2416のマップを示す。
【図31】バイナリ、中間および発現ベクターのマップを示す。CsVMVプロモーターによって促進され、上流のRB7 MARエレメントと共にAtu ORF24 3' UTRによって終止するVP2を示す双子葉植物発現ベクターpDAB2423のマップを示す。選択マーカーPATは、At Ubi 10プロモーターおよびAtu ORF1 3' UTRによって調節される。
【特許請求の範囲】
【請求項1】
以下の段階を含む、免疫保護粒子または生物活性タンパク質粒子を作製する方法:
a)少なくとも一つの免疫保護抗原または少なくとも一つの生物活性タンパク質をコードするポリヌクレオチドによって植物細胞を形質転換する段階:
b)該形質転換植物細胞の増殖、および該植物細胞における該免疫保護抗原または該生物活性タンパク質の蓄積を可能にする条件で、該形質転換植物細胞を培養する段階;
c)該培養形質転換細胞を回収および洗浄する段階;
d)該洗浄形質転換細胞を溶解緩衝液に再懸濁させる段階;
e)該再懸濁細胞を、免疫保護粒子または生物活性タンパク質粒子が形成されるように物理的または機械的に破壊する段階;ならびに
f)該免疫保護粒子または該生物活性タンパク質粒子から細胞の破片を分離する段階。
【請求項2】
形質転換植物細胞が、下等植物細胞、単子葉植物細胞、および双子葉植物細胞からなる群より選択される、請求項1記載の方法。
【請求項3】
形質転換植物細胞がタバコ細胞株培養物である、請求項2記載の方法。
【請求項4】
免疫保護粒子または生物活性タンパク質粒子が、形質転換植物細胞の後期指数増殖期および増殖静止期に由来する、請求項1記載の方法。
【請求項5】
物理的または機械的破壊が、超音波処理、微少溶液操作もしくは他の剪断型方法、高剪断ローター/ステーター法、フレンチプレスもしくは他の加圧法、またはホモジナイゼーションによって行われる、請求項1記載の方法。
【請求項6】
物理的または機械的破壊が20秒間までの超音波処理によって行われる、請求項5記載の方法。
【請求項7】
方法の段階が、細胞を破壊する強い洗浄剤または他の化学物質の非存在下で行われる、請求項1記載の方法。
【請求項8】
免疫保護抗原がトリウイルスからのタンパク質である、請求項1記載の方法。
【請求項9】
免疫保護抗原が、ニューカッスル病ウイルス(NDV)の赤血球凝集素/ノイラミニダーゼタンパク質、トリインフルエンザウイルス(AIV)の赤血球凝集素タンパク質、および伝染性ファブリキウス嚢病ウイルスのVP2タンパク質からなる群より選択される、請求項1記載の方法。
【請求項10】
免疫保護抗原が、SEQ ID NO:2、SEQ ID NO:4、およびSEQ ID NO:12からなる群より選択される、請求項1記載の方法。
【請求項11】
植物細胞が、NT-1、BY-2、CHN-18、CHA-13、CVP2、およびMHN-41からなる群より選択される、請求項1記載の方法。
【請求項12】
ポリヌクレオチドがサブユニットワクチンをコードする、請求項1記載の方法。
【請求項13】
生物活性タンパク質が、酵素、毒素、細胞受容体、リガンド、シグナル伝達物質、サイトカインからなる群より選択される、請求項1記載の方法。
【請求項14】
細胞破片から分離されている免疫保護粒子または生物活性タンパク質粒子を含む上清を回収する段階をさらに含む、請求項1記載の方法。
【請求項15】
免疫保護粒子または生物活性タンパク質粒子から、可溶性の安定な生物活性タンパク質を単離する段階をさらに含む、請求項1記載の方法。
【請求項16】
上清から可溶性の安定な生物活性タンパク質または免疫保護タンパク質を単離する段階をさらに含む、請求項14記載の方法。
【請求項17】
請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、または16のいずれか一項記載の方法によって産生された単離免疫保護粒子または生物活性タンパク質粒子。
【請求項18】
一つまたはそれ以上の薬学的に許容されるアジュバント、希釈剤、担体、または賦形剤と混合して請求項17記載の免疫保護粒子または生物活性タンパク質粒子を含む組成物。
【請求項19】
免疫保護粒子が少なくとも一つの免疫保護抗原を含む、請求項18記載の組成物。
【請求項20】
免疫保護抗原が、ニューカッスル病ウイルス(NDV)の赤血球凝集素/ノイラミニダーゼタンパク質、トリインフルエンザウイルス(AIV)の赤血球凝集素タンパク質、および伝染性ファブリキウス嚢病ウイルスのVP2タンパク質からなる群より選択される、請求項19記載の組成物。
【請求項21】
免疫保護抗原が、SEQ ID NO:2、SEQ ID NO:4、およびSEQ ID NO:12からなる群より選択される、請求項20記載の組成物。
【請求項22】
ワクチン接種するため、免疫するため、免疫応答を刺激するため、特異的抗体の産生を刺激するため、または細胞性免疫応答を刺激するために十分な量の請求項18記載の組成物の用量を、動物またはヒトに投与する段階を含む、動物をワクチン接種または免疫する方法。
【請求項23】
組成物が、ニューカッスル病ウイルス(NDV)の赤血球凝集素/ノイラミニダーゼタンパク質、トリインフルエンザウイルス(AIV)の赤血球凝集素タンパク質、および伝染性ファブリキウス嚢病ウイルスのVP2タンパク質からなる群より選択される免疫保護抗原を提供する、請求項22記載の方法。
【請求項24】
組成物が筋肉内、静脈内、経口、鼻腔内、粘膜内、または皮下投与される、請求項22記載の方法。
【請求項1】
以下の段階を含む、免疫保護粒子または生物活性タンパク質粒子を作製する方法:
a)少なくとも一つの免疫保護抗原または少なくとも一つの生物活性タンパク質をコードするポリヌクレオチドによって植物細胞を形質転換する段階:
b)該形質転換植物細胞の増殖、および該植物細胞における該免疫保護抗原または該生物活性タンパク質の蓄積を可能にする条件で、該形質転換植物細胞を培養する段階;
c)該培養形質転換細胞を回収および洗浄する段階;
d)該洗浄形質転換細胞を溶解緩衝液に再懸濁させる段階;
e)該再懸濁細胞を、免疫保護粒子または生物活性タンパク質粒子が形成されるように物理的または機械的に破壊する段階;ならびに
f)該免疫保護粒子または該生物活性タンパク質粒子から細胞の破片を分離する段階。
【請求項2】
形質転換植物細胞が、下等植物細胞、単子葉植物細胞、および双子葉植物細胞からなる群より選択される、請求項1記載の方法。
【請求項3】
形質転換植物細胞がタバコ細胞株培養物である、請求項2記載の方法。
【請求項4】
免疫保護粒子または生物活性タンパク質粒子が、形質転換植物細胞の後期指数増殖期および増殖静止期に由来する、請求項1記載の方法。
【請求項5】
物理的または機械的破壊が、超音波処理、微少溶液操作もしくは他の剪断型方法、高剪断ローター/ステーター法、フレンチプレスもしくは他の加圧法、またはホモジナイゼーションによって行われる、請求項1記載の方法。
【請求項6】
物理的または機械的破壊が20秒間までの超音波処理によって行われる、請求項5記載の方法。
【請求項7】
方法の段階が、細胞を破壊する強い洗浄剤または他の化学物質の非存在下で行われる、請求項1記載の方法。
【請求項8】
免疫保護抗原がトリウイルスからのタンパク質である、請求項1記載の方法。
【請求項9】
免疫保護抗原が、ニューカッスル病ウイルス(NDV)の赤血球凝集素/ノイラミニダーゼタンパク質、トリインフルエンザウイルス(AIV)の赤血球凝集素タンパク質、および伝染性ファブリキウス嚢病ウイルスのVP2タンパク質からなる群より選択される、請求項1記載の方法。
【請求項10】
免疫保護抗原が、SEQ ID NO:2、SEQ ID NO:4、およびSEQ ID NO:12からなる群より選択される、請求項1記載の方法。
【請求項11】
植物細胞が、NT-1、BY-2、CHN-18、CHA-13、CVP2、およびMHN-41からなる群より選択される、請求項1記載の方法。
【請求項12】
ポリヌクレオチドがサブユニットワクチンをコードする、請求項1記載の方法。
【請求項13】
生物活性タンパク質が、酵素、毒素、細胞受容体、リガンド、シグナル伝達物質、サイトカインからなる群より選択される、請求項1記載の方法。
【請求項14】
細胞破片から分離されている免疫保護粒子または生物活性タンパク質粒子を含む上清を回収する段階をさらに含む、請求項1記載の方法。
【請求項15】
免疫保護粒子または生物活性タンパク質粒子から、可溶性の安定な生物活性タンパク質を単離する段階をさらに含む、請求項1記載の方法。
【請求項16】
上清から可溶性の安定な生物活性タンパク質または免疫保護タンパク質を単離する段階をさらに含む、請求項14記載の方法。
【請求項17】
請求項1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、または16のいずれか一項記載の方法によって産生された単離免疫保護粒子または生物活性タンパク質粒子。
【請求項18】
一つまたはそれ以上の薬学的に許容されるアジュバント、希釈剤、担体、または賦形剤と混合して請求項17記載の免疫保護粒子または生物活性タンパク質粒子を含む組成物。
【請求項19】
免疫保護粒子が少なくとも一つの免疫保護抗原を含む、請求項18記載の組成物。
【請求項20】
免疫保護抗原が、ニューカッスル病ウイルス(NDV)の赤血球凝集素/ノイラミニダーゼタンパク質、トリインフルエンザウイルス(AIV)の赤血球凝集素タンパク質、および伝染性ファブリキウス嚢病ウイルスのVP2タンパク質からなる群より選択される、請求項19記載の組成物。
【請求項21】
免疫保護抗原が、SEQ ID NO:2、SEQ ID NO:4、およびSEQ ID NO:12からなる群より選択される、請求項20記載の組成物。
【請求項22】
ワクチン接種するため、免疫するため、免疫応答を刺激するため、特異的抗体の産生を刺激するため、または細胞性免疫応答を刺激するために十分な量の請求項18記載の組成物の用量を、動物またはヒトに投与する段階を含む、動物をワクチン接種または免疫する方法。
【請求項23】
組成物が、ニューカッスル病ウイルス(NDV)の赤血球凝集素/ノイラミニダーゼタンパク質、トリインフルエンザウイルス(AIV)の赤血球凝集素タンパク質、および伝染性ファブリキウス嚢病ウイルスのVP2タンパク質からなる群より選択される免疫保護抗原を提供する、請求項22記載の方法。
【請求項24】
組成物が筋肉内、静脈内、経口、鼻腔内、粘膜内、または皮下投与される、請求項22記載の方法。
【図2】

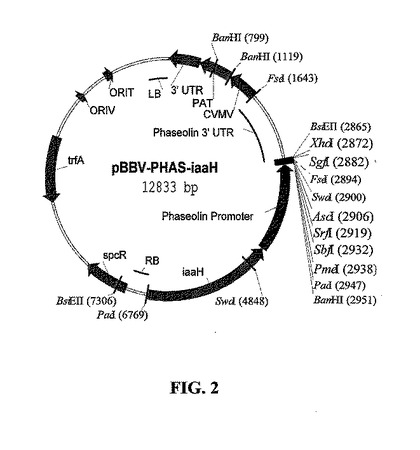
【図3】
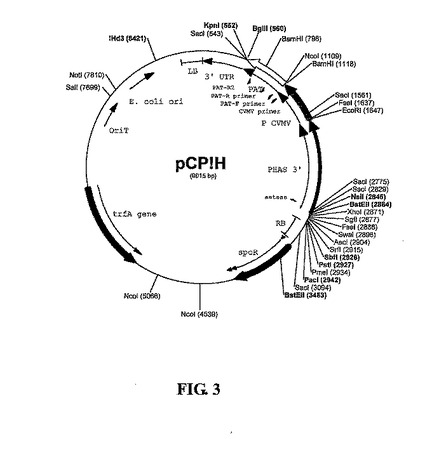
【図4】
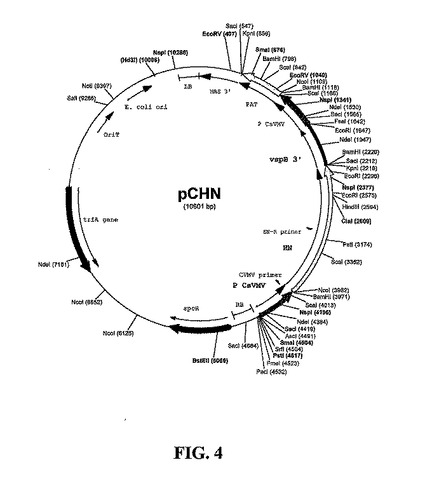
【図5】
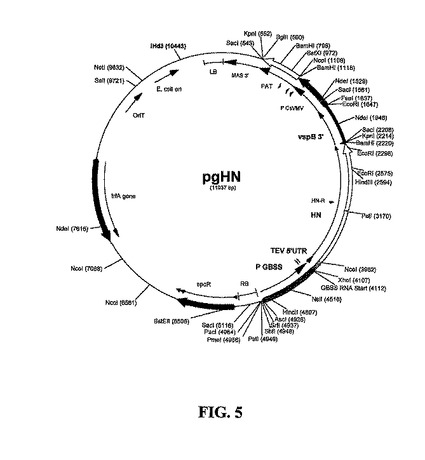
【図6】
【図7】
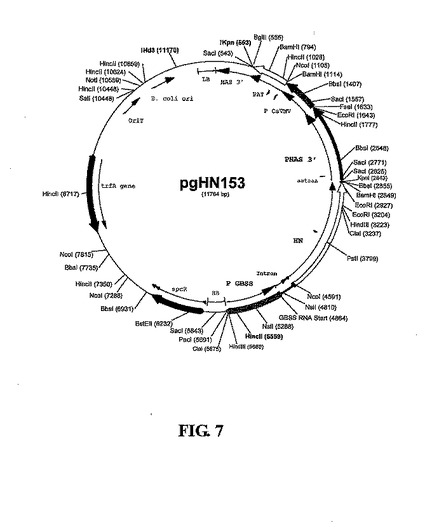
【図8】
【図9】
【図10】

【図11】
【図12】
【図13】
【図14】

【図15】
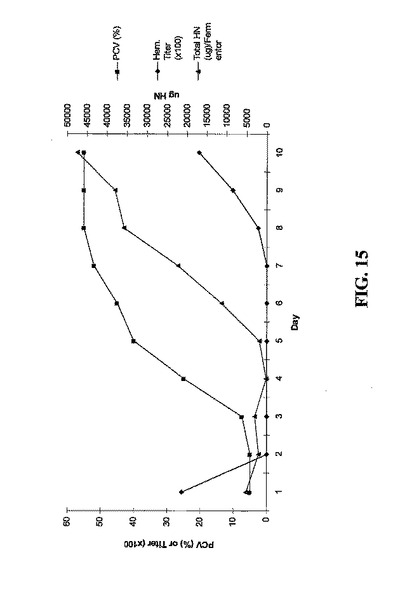
【図16】
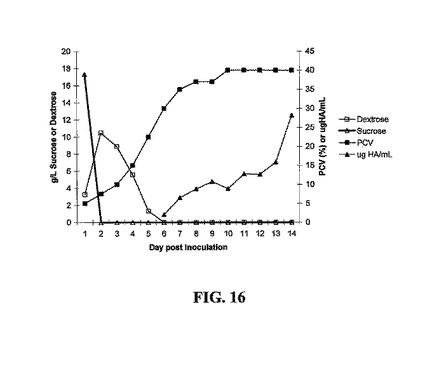
【図17】
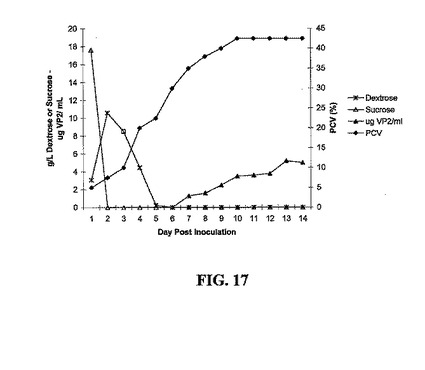
【図18】
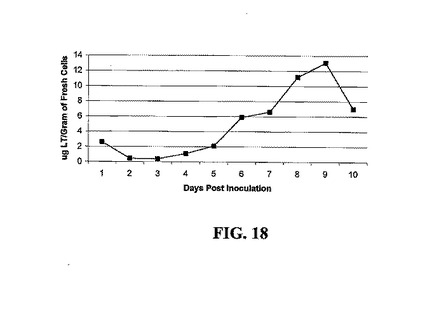
【図19】
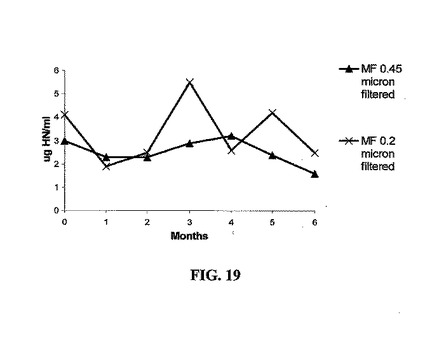
【図20】

【図21】

【図22】
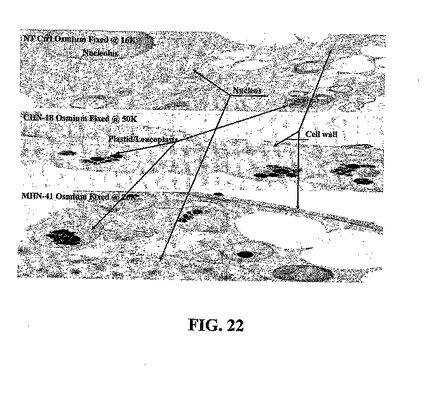
【図23】
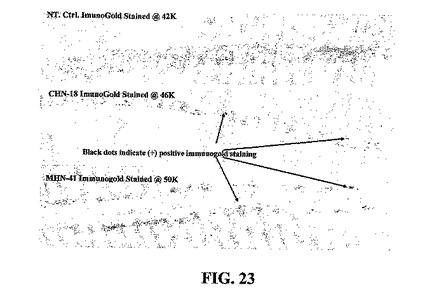
【図24】
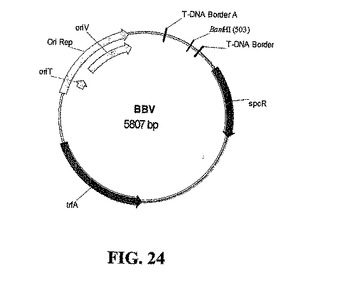
【図25】
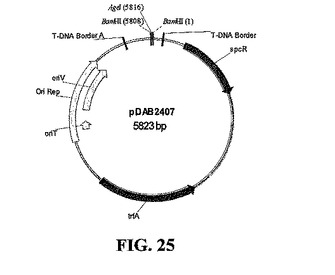
【図26】
【図27】
【図28】
【図29】
【図30】
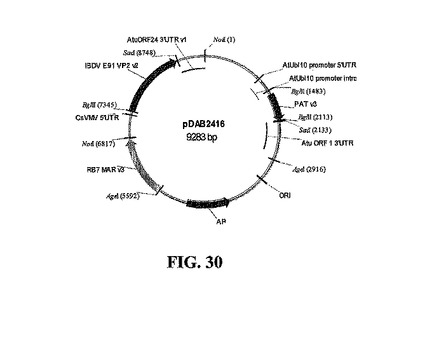
【図31】
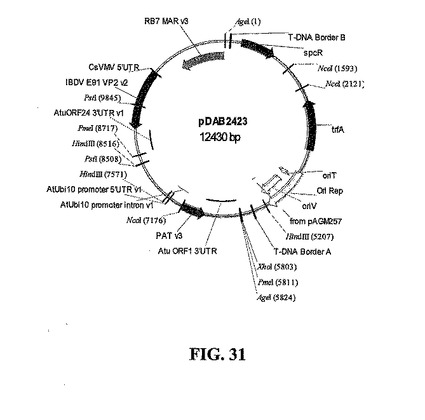
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【公表番号】特表2007−526215(P2007−526215A)
【公表日】平成19年9月13日(2007.9.13)
【国際特許分類】
【出願番号】特願2006−514295(P2006−514295)
【出願日】平成16年5月4日(2004.5.4)
【国際出願番号】PCT/US2004/013965
【国際公開番号】WO2004/098530
【国際公開日】平成16年11月18日(2004.11.18)
【出願人】(505412443)ダウ アグロサイエンス リミテッド ライアビリティー カンパニー (10)
【Fターム(参考)】
【公表日】平成19年9月13日(2007.9.13)
【国際特許分類】
【出願日】平成16年5月4日(2004.5.4)
【国際出願番号】PCT/US2004/013965
【国際公開番号】WO2004/098530
【国際公開日】平成16年11月18日(2004.11.18)
【出願人】(505412443)ダウ アグロサイエンス リミテッド ライアビリティー カンパニー (10)
【Fターム(参考)】
[ Back to top ]