
トランスフェクション剤としてのリポポリアミン及びその医薬的使用
【課題】in vitro及び/又はin vivo細胞トランスフェクション、特に核酸のベクター導入に有効に使用可能な新規化合物を提供する。
【解決手段】式(I)において、R1、R2及びR3は各々独立して水素原子又は−(CH2)q−NRR’基を表し、R4は一般式(II)の基を表し、式中、R6及びR7はC10〜C22脂肪族基などを表し、R5は置換された天然アミノ酸側鎖などを表す。

【解決手段】式(I)において、R1、R2及びR3は各々独立して水素原子又は−(CH2)q−NRR’基を表し、R4は一般式(II)の基を表し、式中、R6及びR7はC10〜C22脂肪族基などを表し、R5は置換された天然アミノ酸側鎖などを表す。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はリポポリアミンファミリーに属する新規化合物、該化合物を含有する医薬組成物、核酸のin vivo及び/又はin vitroトランスフェクションのためのその適用並びにその製造方法に関する。
【発明の開示】
【0002】
多数の遺伝病は1種以上の核酸の発現障害及び/又は異常発現即ち発現不足又は過剰発現に関連している。遺伝子治療はクローン化遺伝子のin vivo又はin vitro細胞発現によりこのような遺伝異常を治療することを主眼とする。
【0003】
今日、この種の遺伝情報の細胞内送達方法として数種の方法が提案されている。特に、化学的又は生化学的ベクターを利用するものが挙げられる。合成ベクターはトランスフェクトしようとするDNAを圧縮する機能と、その細胞固定並びにその細胞質膜及び場合により2つの核膜通過を助長する機能との2つの主機能をもつ。
【0004】
このトランスフェクション方法は、カチオン脂質の利用に基づく技術の開発により著しい進歩を遂げた。例えば、正電荷をもつカチオン脂質であるN−[1−(2,3−ジオレイルオキシ)プロピル]−N,N,N−トリメチルアンモニウムクロリド(DOTMA)は、負電荷をもつDNAとリポソーム又は小ベジクル形態で自然に相互作用し、細胞膜と融合することが可能な脂質−DNA複合体を形成し、こうしてDNAの細胞内送達を可能にすることが判明した。しかし、この分子はトランスフェクションの点では有効であるが、非生分解性であり、細胞に毒性であるという欠点がある。
【0005】
DOTMA以来、所謂「スペーサー」アームを介してアミノ基に結合した親油基というこの構造モデルで他のカチオン脂質も開発された。そのうちでは、親油基として2個の脂肪酸又は1個のコレステロール誘導体を含み、更に、場合によりアミノ基として第4級アンモニウム基を含むものを挙げることができる。このカチオン脂質の分類の代表例として、DOTAP、DOBT又はChOTBを特に挙げることができる。この他、DOSCやChOSC等のように、第4級アンモニウム基の代わりにコリン基の存在を特徴とする化合物もある。しかし、一般にこれらの化合物のトランスフェクション活性は依然としてかなり低い。
【0006】
別の分類のカチオン脂質であるリポポリアミンも報告されている。本発明の意味では、リポポリアミンなる用語は所謂スペーサー領域を介して親油領域と結合した少なくとも1個の親水ポリアミン領域を含む両親媒性分子を意味する。リポポリアミンの正電荷をもつポリアミン領域は負電荷をもつ核酸と可逆的に結合することができる。この相互作用は核酸を著しく圧縮する。親油領域は脂質膜から形成される核脂質粒子を覆うことにより、このイオン相互作用を外部媒質に対して非感受性にする。この種の化合物では、カチオン基は2個の第1級アンモニウム基と2個の第2級アンモニウム基との4個のアンモニウム基を含むL−5−カルボキシスペルミン基により置換することができる。この種の化合物としては特にDOGSとDPPESが挙げられる。これらのリポポリアミンは一次内分泌細胞のトランスフェクションに特に有効である。
【0007】
実際に、理想的な合成トランスフェクション剤は広い細胞範囲で高いトランスフェクションレベルをもち、使用用量で無毒性であるか又は非常に低毒性であり、更に処理細胞のレベルで副作用が全くないように生分解性でなければならない。
【0008】
本発明は詳細には、独自のポリアミンフラクションをもち、in vitro及び/又はin vivo細胞トランスフェクション、特に核酸のベクター導入に有効に使用可能な新規リポポリアミンを提案することを目的とする。
【0009】
本発明の第1の目的は一般式I:
【0010】
【化6】
[式中、
R1、R2及びR3は各々独立して水素原子又は−(CH2)q−NRR’基を表し、
qはR1、R2及びR3の各基で独立して1、2、3、4、5及び6であり、
R及びR’は各々独立して水素原子又は−(CH2)q’−NH2基を表し、q’はR及びR’の各基で独立して1、2、3、4、5及び6であり、
m、n及びpは各々独立して0〜6の整数を表し、但しnが2以上のとき、mは異なる値をとることができ、R3は一般式I中で異なる意味をもち、
R4は一般式II:
【0011】
【化7】
の基を表し、式中、
R6及びR7は各々独立して水素原子又は飽和もしくは不飽和C10〜C22脂肪族基を表し、2個の基の少なくとも一方は水素以外のものであり、
uは0〜10から選択される整数であり、但しuが2以上の整数であるとき、R5、X、Y及びrは各モチーフ[X−(CHR5)r−Y]中で異なる意味をもつことができ、
Xは酸素原子、硫黄原子又はモノアルキルもしくは非モノアルキルアミン基を表し、
Yはカルボニル基又はメチレン基を表し、
R5は水素原子又は場合により置換された天然アミノ酸側鎖を表し、
rは1〜10の整数を表し、但しrが1であるとき、R5は天然アミノ酸側鎖を表し、rが2以上であるとき、R5は水素原子を表す]により表されることを特徴とするD、L又はDL形リポポリアミン及びその塩を提供することである。
【0012】
天然アミノ酸側鎖とは、特に本発明の意味では例えばアルギニン側鎖のようにアミジニウムモチーフを含む鎖を意味する。上述のように、この鎖は飽和もしくは不飽和のC1〜C24直鎖、分枝鎖もしくは環状脂肪族基(例えばコレステリル基、アラキドニル基又はレチノイル基)又はモノもしくはポリ芳香族基(例えば置換又は非置換ベンジルオキシカルボニル、ベンジルエステル及びローダミニル誘導体)により置換されていてもよい。
【0013】
一般式(I)のこれらの新規生成物は医薬的に許容可能な非毒性塩の形態をとることができる。これらの非毒性塩は鉱酸(塩酸、硫酸、臭化水素酸、リン酸、硝酸)、有機酸(酢酸、プロピオン酸、コハク酸、マレイン酸、ヒドロキシマレイン酸、安息香酸、フマル酸、メタンスルホン酸又は蓚酸)、無機塩(水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化カルシウム)又は有機塩(トリエチルアミン、ピペリジン、ベンジルアミン等の第3級アミン)との塩を含む。
【0014】
本発明による化合物の例としては、特に下記亜一般式:
【0015】
【化8】
(式中、R4、R6及びR7は上記と同義である)の化合物を挙げることができる。上記式中、好ましくはR4はNR6R7であり、R6及びR7は亜式III〜XIIにおいて(CH2)17CH3、(CH2)11CH3、(CH2)13CH3又は(CH2)12CH3から選択される同一基を表す。
【0016】
特に有利な態様において、本発明の化合物は該化合物が結合した核酸の移送を指向させることが可能なターゲティング成分を更に含む。好ましくは、このターゲティング成分はR5置換基により表されるアミノ酸側鎖のレベルで一般式Iの化合物に組み込まれる。より好ましくは、ターゲティング成分は本発明による化合物に共有又は非共有的に結合される。
【0017】
このようなターゲティング成分としては、所定細胞型又は所望の所定組織(腫瘍細胞、肝細胞、造血細胞等)に核酸の移送を指向させることが可能な細胞外ターゲティング成分が挙げられる。例えば、標的細胞型の表面に存在する細胞レセプターのリガンドが挙げられ、例えば糖、葉酸、トランスフェリン、インスリン、アシアロオロソムコイドタンパク質又は細胞外レセプターにより認識される任意の生物活性分子等である。また、例えばトランスフェクトされるDNAを核の内部に優先的に蓄積する核局在シグナル配列(nls)のように、所定の優先的細胞区画(ミトコンドリア、核等)に向けて核酸移送を指向させることが可能な細胞内ターゲティングでもよい。
【0018】
より一般には、本発明の範囲で利用可能なターゲティング成分としては糖類、ペプチド、オリゴヌクレオチド、ステロイド又は脂質が挙げられる。好ましくは、糖類及び/又はペブチド(例えば抗体又は抗体フラグメント、細胞レセプターのリガンド又はそのフラグメント、レセプター又はレセプターフラグメント等)である。特に、成長因子レセプター、サイトカインレセプター、細胞レクチンレセプター又は接着タンパク質レセプター(例えばインテグリン)のリガンドが挙げられる。また、トランスフェリンのレセプター、脂質HDL及びLDLも挙げられる。ターゲティング成分は更に、アシアロ糖タンパク質レセプター等のレクチンをターゲティングすることが可能な糖や、イムノグロブリンのFcフラグメントのレセプターをターゲティングすることが可能な抗体Fabフラグメントでもよい。
【0019】
同様に、一般式Iの化合物の例えばアミノ酸側鎖R5にビオチン、ローダミン、葉酸型のマーカー物質を結合することも考えられる。このマーカー物質は更に、インテグリン型の接着タンパク質の一次及び/又は二次レセプターの認識エピトープ
Arg−Gly−Aspを含む直鎖又は環状ペプチド又はプソイドペプチド配列でもよい。
【0020】
本発明のリポポリアミンの例としては、後記実施例に詳細に記載する下記化合物を特に挙げることができる。
【0021】
【化9】
【0022】
本発明の特に代表的な化合物としては、下記一般式の化合物を特に挙げることができる。
【0023】
【化10】
【0024】
本発明の範囲内で、本発明によるリポポリアミンを誘導する非対称官能化ポリアミンを製造するための新規固相方法も開発された。
【0025】
従来、非対称官能化ポリアミンの製造は直鎖又は分枝鎖対称ポリアミンに選択的に修飾を導入する必要により制限されている。ポリアミンの第1アミノ基と第2アミノ基の化学的差異により、アルキル化、ミカエリス型付加又はアシル化等の反応を実施するためには高い選択性が必要である。更に、このような化学的差異により要求される選択性は、ポリアミンで行われるような複数の第1及び第2アミン群での選択的アシル化に適合しない。このような制約に対する従来の対応は、「ポリアミノ化」することが可能なアミノ基と適当に保護された非対称官能基をもつモノマーブロックから非対称官能化ポリアミンを構築する方法であった。従って、このアプローチには複数段階からなる面倒な合成ストラテジーが必要であり、特に官能基の直交保護が必要であった。
【0026】
本発明の範囲内で開発された方法は、詳細には上記型の合成に伴う従来の欠点を解消するを目的とする。この方法は、対称ポリアミンのアルキル化又は還元アルキル化により一次アミノ基のみで選択的に官能化されたポリアミンを容易且つ迅速に得られるという顕著な利点がある。
【0027】
本発明の方法の原理は固相合成法の利用に基づき、アルキル化剤とポリアミンの二分子反応を助長し、ポリアミンのポリアルキル化を回避する。より詳細には、本発明は固体支持体に共有結合したアルキル化剤と対称ポリアミンの二分子反応により予め得られた少なくとも1個の非対称ポリアミンフラクションに少なくとも1個の脂質フラクションを結合することを特徴とする方法に関する。このアプローチによると、アルキル化剤はエステル化又はアミド化によりポリマー支持体に共有結合している。対称ポリアミンは二分子反応により固相アルキル化剤と反応し、支持体に結合した単官能化非対称ポリアミンを生じる。生成物の遊離アミンは一般にBOC又はZ型の保護基により固相で保護されており、最終的に生成物は固相支持体から分離される。これらのポリアミノ酸を適宜脂質フラクションに結合すると、所望のトランスフェクション剤が得られる。この結合には、例えばBOP、Pybop、BopCl及びDCC等の慣用ペプチド結合剤を利用することができる。本方法は、場合によりトレーサーペプチド、糖類又は蛍光プローブを分子に導入して完全トランスフェクション剤を固体支持体に結合することも可能である。当然のことながら、遊離リポポリアミンにこの型のグラフトを実施できることが判明した。
【0028】
本方法の実施可能性は、複数の直鎖又は分枝鎖非対称官能化ポリアミノ酸の合成により立証された。
【0029】
本発明は更に、上記のようなリポポリアミンの直接又は医薬組成物としての任意の治療的適用にも関する。
【0030】
上述のように、一般式Iの化合物は核酸のin vitro及びin vivoトランスフェクションに特に有利であることが判明した。これらの化合物はDNAを有効に圧縮し、非常に低毒性であるという利点がある。
【0031】
本発明の組成物から最大限の効果を得るためには、核酸の負電荷に対する該当リポポリアミンの正電荷の比であるR比が最適となるように、一般式Iの化合物と核酸の夫々の比率を決定することが好ましい。この最適比は特にin vivo又はin vitroの使用方式や、トランスフェクトしようとする細胞型により異なり、場合に応じて最適化される。この最適化は当業者に自明である。
【0032】
本発明の医薬組成物において、ポリヌクレオチドはデオキシリボ核酸でもリボ核酸でもよい。天然又は人工起源のいずれの配列でもよく、特にゲノムDNA、cDNA、mRNA、tRNA、rRNA、ハイブリッド配列又は修飾もしくは非修飾オリゴヌクレオチドの合成もしくは半合成配列が挙げられる。これらの核酸はヒト、動物、植物、細菌、ウイルス等に由来するものであり得る。これらの核酸は当業者に公知の任意方法により得られ、特に、バンクスクリーニング、化学的合成又はバンクスクリーニングにより得られた配列の化学的もしくは酵素的修飾を含む混合方法により得られる。更に、プラスミドベクター等のベクターに組み込んでもよい。
【0033】
特にデオキシリボ核酸については、1本鎖でも2本鎖でもよいし、短いオリゴヌクレオチドでもより長い配列でもよい。これらのデオキシリボ核酸は治療遺伝子、転写又は複製調節配列、修飾又は非修飾アンチセンス配列、他の細胞成分との結合領域等を含み得る。
【0034】
本発明の意味では、治療遺伝子とは特に治療効果をもつタンパク質産物をコードする任意遺伝子を意味する。こうしてコードされるタンパク質産物はタンパク質、ペプチド等であり得る。このタンパク質産物はターゲット細胞に対して同種であり得る(即ちターゲット細胞が疾病をもたないときにターゲット細胞で正常に発現される産物)。この場合、タンパク質の発現は例えば細胞における不十分な発現や、不活性であるか又は修飾により弱活性となったタンパク質の発現を解消し、あるいは前記タンパク質を過剰発現させることができる。治療遺伝子は安定性の増加、活性改変等の性質をもつ細胞タンパク質の突然変異体をコードするものでもよい。タンパク質産物はターゲット細胞に対して異種でもよい。その場合には、発現されるタンパク質は例えば細胞に欠失している活性を補充又は付加し、疾病に対抗するこ又は免疫応答を刺激することができる。
【0035】
本発明の意味での治療物質としては、より特定的には酵素、血液誘導体、ホルモン、リンホカイン(インターロイキン、インターフェロン、TNF等(仏国特許出願第9203120号))、成長因子、神経伝達物質又はその前駆物質もしくは合成酵素、栄養因子(BDNF、CNTF、NGF、IGF、GMF、aFGF、bFGF、NT3、NT5、HARP/プレイオトロフィン等、ジストロフィン又はミニジストロフィン(仏国特許出願第9111947号))、膵臓線維症に関連するタンパク質CFTR、腫瘍抑制遺伝子(p53、Rb、Rap1A、DCC、k−rev等(仏国特許出願第9304745号))、凝血に関与する因子(VII、VIII、IX因子)をコードする遺伝子、DNAの修復に関与する遺伝子、自殺遺伝子(チミジンキナーゼ、シトシンデアミナーゼ)、ヘモグロビン又は他の輸送タンパク質の遺伝子、アポリポタンパク質A−I、A−II、A−IV、B、C−I、C−II、C−III、D、E、F、G、H、J及びアポ(a)から選択されるアポリポタンパク質型の脂質の代謝に関与するタンパク質に対応する遺伝子、代謝酵素(例えばリポタンパク質リパーゼ、肝性リパーゼ、レシチンコレステロールアシルトランスフェラーゼ、7−α−コレステロールヒドロキシラーゼ、ホスファチジン酸ホスファターゼ)、脂質輸送タンパク質(例えばコレステロールエステル輸送タンパク質やリン脂質輸送タンパク質)、HDL結合タンパク質又は例えばLDLレセプター、レムナントキロミクロンレセプター及びスカベンジャーレセプター等から選択されるレセプターを挙げることができる。
【0036】
治療核酸は更に、ターゲット細胞で発現されると遺伝子発現又は細胞mRNA転写を調節することが可能なアンチセンス遺伝子又は配列でもよい。このような配列は、ヨーロッパ特許第140308号に記載の技術により、例えば細胞mRNAの相補的RNAターゲット細胞に転写され、こうしてそのタンパク質翻訳を阻止することができる。治療遺伝子は更に、ターゲットRNAを選択的に破壊することが可能なリボソームをコードする配列も含む(ヨーロッパ特許第321201号)。
【0037】
上述のように、核酸は更に、ヒト又は動物で免疫応答を発生することが可能な抗原ペプチドをコードする1個以上の遺伝子を含んでいてもよい。従って、この特定態様によると、本発明はヒト又は動物で特に微生物、ウイルス又は癌に対するワクチン又は免疫治療を実現することができる。特に、エプスタイン・バールウイルス、HIVウイルス、B型肝炎ウイルス(ヨーロッパ特許第185573号)、偽狂犬病ウイルス、シンシチウム形成ウイルス、他のウイルスの特異的抗原ペプチド又は腫瘍特異的抗原ペプチド(ヨーロッパ特許第259212号)を挙げることができる。
【0038】
好ましくは、核酸は更に細胞又は所望臓器において治療遺伝子及び/又は抗原ペプチドをコードする遺伝子の発現を可能にする配列も含む。このような配列としては、これらの配列が感染細胞で機能できるときに該当遺伝子の発現に天然に関与する配列が挙げられる。異なる起源の配列でもよい(他のタンパク質の発現に関与、あるいは合成配列でもよい)。特に、真核又はウイルス遺伝子のプロモーター配列が挙げられる。例えば、感染させようとする細胞のゲノムに由来するプロモーター配列が挙げられる。また、ウイルスのゲノムに由来するプロモーター配列でもよい。この点では、例えばE1A、MLP、CMV、RSV等の遺伝子のプロモーターを挙げることができる。更に、活性化配列や調節配列等を付加してこれらの発現配列を修飾してもよい。誘導又は抑制プロモーターでもよい。
【0039】
更に、核酸は合成治療物質をターゲット細胞の分泌経路に導くシグナル配列も特に治療遺伝子の上流に含んでいてもよい。このシグナル配列は治療物質の天然シグナル配列でもよいし、他の任意の機能的シグナル配列又は人工シグナル配列でもよい。核酸は更に合成治療物質を細胞の特定区画に導くシグナル配列も含んでいてもよい。
【0040】
別の態様において、本発明は核酸と、本発明のリポポリアミンと、リポポリアミン/核酸複合体に結合してトランスフェクション能を改善することが可能なアジュバントを含む組成物に関する。本願出願人は実際に、リポポリアミン/核酸複合体に結合することが可能な所定のアジュバント(例えば脂質、ペプチド又はタンパク質)の存在下でリポポリアミンのトランスフェクション能を予想外に増加できることを立証した。
【0041】
この点で、本発明の組成物はアジュバントとして1種以上の中性脂質を含み得る。このような組成物は特にR比が小さいときに特に有利である。本願出願人は実際に、中性脂質を加えると核脂質粒子の形成を改善し、驚くべきことに細胞膜を撹乱して粒子の細胞侵入を助長できることを立証した。
【0042】
より好ましくは、本発明の範囲内で使用する中性脂質は2個の脂肪鎖をもつ脂質である。
【0043】
生理的条件下で両性イオン性又はイオン電荷をもたない天然又は合成脂質を使用すると特に有利である。脂質は特にジオレオイルホスファチジルエタノールアミン(DOPE)、オレオイルパルミトイルホスファチジルエタノールアミン(POPE)、ジステアロイルホスファチジルエタノールアミン、ジパルミトイルホスファチジルエタノールアミン、ジミリストイルホスファチジルエタノールアミン及びその1〜3倍N−メチル化誘導体、ホスファチジルグリセロール、ジアシルグリセロール、グリコシルジアシルグリセロール、セレブロシド(例えば特にガラクトセレブロシド)、スフィンゴ脂質(例えば特にスフィンゴミエリン)又はアシアロガングリオシド(例えば特にアシアロGM1及びGM2)から選択することができる。
【0044】
これらの種々の脂質は当業者に周知の慣用技術により合成するか又は臓器(例えば脳)もしく胎児から抽出することにより得られる。特に、天然脂質の抽出は有機溶媒を用いて実施することができる(Lehninger,Biochemistry参照)。
【0045】
ごく最近になって本願出願人は前記核酸の凝縮レベルに直接又は間接的に作用する化合物をアジュバントとして使用すると特に有利であることも立証した(WO96/25508)。
【0046】
リポポリアミンをベースとするトランスフェクション組成物中にこのような化合物が存在すると、この組成物の量を著しく低減でき、この組成物のトランスフェクション活性を損なわずに毒物学的面で有益な結果が得られる。それどころか、組成物のトランスフェクションレベルは向上するという利点がある。
【0047】
核酸の凝縮レベルに作用する化合物とは、核酸を直接又は間接的に圧縮する化合物として定義される。より詳細には、この化合物はトランスフェクトしようとする核酸のレベルに直接作用するか、又はこの核酸の凝縮に直接関与する付加化合物のレベルに作用することができる。核酸のレベルに直接作用するものが好ましい。
【0048】
好ましい態様によると、核酸の凝縮レベルに作用するこの物質は全体又は一部をペプチドモチーフ(KTPKKAKKP)及び/又は(ATPAKKAA)から構成され、モチーフ数は2〜10である。本発明による化合物の構造において、これらのモチーフは連続又は不連続的に反復し得る。従って、これらのモチーフは生化学的結合(例えば1個以上のアミノ酸)又は化学的結合により分離することができる。このような物質は全体又は一部がヒストン、ヌクレオリン、プロタミン及び/又はそれらの誘導体の1種から誘導されるものでもよい。
【0049】
好ましくは、本発明の組成物は核酸1当量当たりアジュバント0.01〜20当量(重量/重量)、より好ましくは0.5〜5当量を含む。
【0050】
本発明による組成物は局所、皮膚、経口、直腸、膣、非経口、鼻腔内、静脈内、筋肉内、皮下、眼内、経皮等の経路で投与するように調剤することができる。好ましくは、本発明の医薬組成物は特に所望臓器のレベルに直接注射するための注射用製剤又は局所投与(皮膚及び/又は粘膜)用として医薬的に許容可能なキャリヤーを含有する。このようなキャリヤーとしては、特に等張滅菌溶液又は場合に応じて滅菌水もしくは生理的血清を加えると注射可能な溶質を構成することが可能な乾燥組成物、特に凍結乾燥組成物が挙げられる。注射に使用する核酸の用量及び投与回数は種々のパラメーター、特に使用する投与方法、該当疾病、発現させようとする遺伝子、又は所望の治療期間に応じて選択できる。特に投与方法については、組織又は循環経路への直接注射や、培養細胞処理後の注射又は移植によるin vivo再移植が挙げられる。
【0051】
従って、本発明は上記条件下で上記疾病を治療することが可能な核酸と一般式Iの化合物のin vivo又はin vitro併用投与を特徴とする、疾病治療に特に有利な方法を提供する。より詳細には、この方法はタンパク質又は核酸産物の不足に起因する疾病に適用することができ、投与する核酸は前記タンパク質産物をコードするか又は前記核酸産物を含む。
【0052】
本発明は、in vivo又はin vitro細胞トランスフェクションのための本発明のリポポリアミンの全使用に及ぶ。
【0053】
以下、実施例及び図面により本発明をより詳細に説明するが、これらの説明は例示に過ぎず、本発明を制限するとみなすべきではない。
【0054】
略語及び記号
AcOEt:酢酸エチル
BOC:t−ブトキシカルボニル
BOP:ベンゾトリアゾール−1−イルオキシトリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート
DCC:ジシクロヘキシルカルボジイミド
DCU:ジシクロヘキシル尿素
DMAP:4−ジメチルアミノピリジン
DMF:ジメチルホルムアミド
DMSO:ジメチルスルホキシド
DODA:ジオクタデシルアミン
EP:石油エーテル
EtOH:エタノール
NEt3:トリエチルアミン
Rf:前端保持率
TFA:トリフルオロ酢酸
THF:テトラヒドロフラン
TMS:テトラメチルシラン
UV:紫外線
SPPS:固相ペプチド合成
HPLC:高圧液体クロマトグラフィー
Z:ベンジルオキシカルボニル
ClZ:p−クロロベンジルオキシカルボニル
A−化学合成用材料及び方法
1.材料
a)化合物
−出発ポリアミンは例えばスペルミジン、スペルミン、トリス(2−アミノエチル)アミン、フェニレンジアミン、ジアミノエタン(プロパン、ブタン、ペンタン、ヘキサン等)等の市販品でもよいし、分枝鎖ポリアミン製造用として市販されている例えばジアミノエタン(プロパン、ブタン、ペンタン、ヘキサン等)、アミン、スペルミジン、スペルミン等のアミンの完全なシアノエチル化により常法で合成してもよい。
−アルキル化剤はアルキル化方法に応じて次のように選択される。慣用アルキル化にはブロモ酢酸、ω−ハロゲノカルボン酸を使用し、還元アルキル化にはω−アルデヒドカルボン酸(例えばグリオキサル酸、コハク酸準アルデヒド等)又はケト酸(例えばアセト酢酸又はピルビン酸等)を使用する。
−使用するポリマーは固相ペプチド合成(メリフィールド合成)用に市販されている樹脂、例えば遊離酸基をもつ生成物を提供するO−クロロトリチルクロリド樹脂、HMP樹脂や、Rink型樹脂である。ポリアミノ酸は、固相で予備合成したブロモアルキル基をもつペプチド又はω−アルデヒド酸で直接合成することができる。
−Aldrich製品ジオクタデシルアミン、トリエチルアミン、トリフルオロ酢酸、BOP、DMAP、クロロギ酸ベンジルは市販品である。NaCl及びNaHCO3溶液は飽和溶液であり、KHSO4溶液は0.5Mである。
b)物理的測定
陽子NMRスペクトルはBruker 400スペクトロメーターで600MHzで記録した。
【0055】
質量スペクトルはAPI−MS/IIIで測定した。
c)クロマトグラフィー技術
HPLC分析はMerck−Hitachi装置にオート
サンプラーAS−2000A、インテリジェントポンプL−6200A及び紫外−可視検出器L−4000を取り付け、分析用分離には220nm、分取用分離には235nmの波長に調節して実施した。分析用分離カラムはPerkin−Elmer製BU−300 aquapore Butyl 7m,300A 300×4.6mmカラムを使用し、分取用分離カラムはBiorad製Biosil C18 HL 90−10 250×10mmカラムを使用した。移動相はH2O(0.1%TFA)及びアセトニトリル(0.1%TFA)である。分析用分離流速は1ml/min、分取用分離流速は4ml/minに調節する。
【0056】
薄層クロマトグラフィー(TLC)は厚さ0.2mmのMerckシリカゲルプレートで実施した。
【0057】
カラムクロマトグラフィーは粒度0.063〜0.200mmのシリカゲル60 Merckで実施した。
【0058】
カラムクロマトグラフィーの検出は紫外線(254nm)を用いるか、アミン又はアミドの検出にはニンヒドリンを用いて150℃に加熱しながらニンヒドリンのエタノール溶液(40mg/100ml EtOH)を噴霧(弱いスプレー)し、第1級アミンの検出にはフルオレサミンを用いて溶液(40mg/100mlアセトン)を噴霧し、あるいはヨードを用いてヨード粉末でプレートを覆うことにより実施した。
【0059】
カラムクロマトグラフィーは粒度0.063〜0.200mmのシリカゲル60 Merckで実施した。
d)固相合成SPPS法
固相合成は手動製造用SPPSペプチド合成用手動反応器で実施し、撹拌機はFlask Shaker モデルA5−6021である。SPPSでポリアミンの固相結合及びポリアミンの保護後にKaiser試験を行う[Kaiser,E.,Colescolt,D.L.,Bossiner,C.D.及びCook,P.I. Anal.Biochem.34(2),595(1970)]。実施例でSPPSに使用した樹脂はスイス、NOVABIOCHEM製クロロトリチルクロリド樹脂である。
2.一般操作方法
a)(N,N,N’,N’−テトラアミノプロピル)−1−4−ジアミノブタンの製造により例示する対称ポリアミンの合成
2リットル容三頚丸底フラスコに1−4−ジアミノブタン147gと脱イオン水1000mlを仕込む。溶液を磁気撹拌する。温度を38℃に維持しながら等圧フラスコによりアクリロニトリル443gを1時間かけて加える。次いで還流冷却器を取り付け、1時間かけて混合物を水浴で80℃まで昇温させる。フルオレサミン試験は陰性であり、過剰のアクリロニトリルを40℃で減圧蒸発させる。
【0060】
2相が得られる。下部の有機相を分離し、水300mlで洗浄し、1000ml容丸底フラスコに移す。水/メタノール(1:1v/v)混合物170mlを加える。一晩結晶化させる。翌日、結晶を細孔度3の500ml容ガラス濾過器で濾過する。
【0061】
ケーキをガラス濾過器でメタノール(2×170ml)とエーテル(2×150ml)で洗浄する。生成物を減圧デシケーター(26mm)で一晩連続乾燥する。こうして生成物461g(収率93%)が得られる。生成物をNMRとMSにより分析した処、分析値は整合していた。生成物を精製せずに水素化する。
【0062】
1リットル容イノックスオートクレーブに上記ポリニトリル30g(0.1モル)を仕込む。同時にビーカーでエタノール140ml(95%)とNaOH8g(0.2モル)の溶液を調製する。NaOHが可溶化したらこの溶液をオートクレーブに加える。オートクレーブに窒素を通し、炭素担持ラネーニッケル8mlを加える。オートクレーブを閉じる。初期水素化圧は52気圧であり、周囲温度で5時間かけて28.5気圧まで下げる。懸濁液を濾紙で濾過し、濾渣をエタノール(2×25ml)で洗浄し、濾液を減圧乾燥して濃縮する。油状物を水30mlと混合し、CH2Cl2 100mlで抽出する。有機相をMgSO4で乾燥し、濾過した後、減圧蒸発させる。黄色っぽい液体油状物(27g、収率85%)が得られる。
【0063】
生成物をTLC(単スポット)、NMR及びMSにより分析した処、分析値は整合していた。生成物を精製せずに使用する。
b)方法A:酸基のポリマー支持体固定
クロロトリチルクロリド樹脂(5g、1.2mmol Cl/g樹脂)をSPPS反応器に仕込み、CH2Cl2 50mlを加え、混合物を5分間撹拌する。ブロモ酢酸(1.05g、7mmol)、次いでDIEA(0.95ml,7.5mmol)を加える。反応器を周囲温度で2時間撹拌する。液体を濾過し、樹脂をCH2Cl2とiPrOH(10×50)及びMeOH(2×50ml)で洗浄する。最後に樹脂を窒素流下に乾燥する。
c)方法B:ポリアミンとブロモアセチル樹脂の反応
ポリアミン(10モル倍)をCH2Cl2 50mlに溶かし、方法Aにより得られた生成物を収容する反応器に加える。反応器を周囲温度で2時間撹拌する。溶媒を濾過し、樹脂をCH2Cl2とiPrOH(10×50ml)で洗浄すると、Kaiser試験は陽性である。
d)樹脂上のポリアミノ酸の保護
方法C:
ジ−tert−ブチルジカーボネート(48mmol)とDIEA(50mmol)をCH2Cl2(50ml)に溶かし、方法Bにより得られた生成物を収容する反応器に加える。反応器を一晩撹拌する。翌日、Kaiser試験は陰性である。溶媒を濾過し、樹脂をCH2Cl2とiPrOH(10×50ml)、MeOH(2×50ml)及びエーテル(2×50ml)で交互に洗浄する。樹脂を窒素流下に乾燥する。Kaiser試験は依然として陰性である。
方法D:
方法Bにより得られた樹脂(1.5g)を丸底フラスコに入れ、CH2Cl2(20ml)、次いでDIEA(20mmol)を加える。混合物を磁気撹拌し、クロロギ酸ベンジル(14mmol)を5分間かけて滴下する。pHはDIEAを加えて11に維持する。一晩後、樹脂をSPPS反応器に移し、濾過し、CH2Cl2とiPrOH(10×20ml)及びエーテル(2×20ml)で交互に洗浄する。樹脂を窒素流下に乾燥する。
e)方法E:樹脂からの保護ポリアミノ酸の分離
磁気棒を備える250ml容丸底フラスコに、方法C及びDにより得られた樹脂を加える。CH2Cl2 50mlとCF3CH2OH 25mlからなる溶液を加え、混合物を2時間撹拌する。溶液を濾過し、樹脂をCH2Cl2(2×10ml)で洗浄し、こうして得られた有機相を集めて減圧蒸発させる。次に、CHCl3/MeOH(9:1)を溶離剤としてSiO2上でフラッシュクロマトグラフィーにより生成物を精製する。生成物を含むフラクションをTLCにより同定する。(詳細については下記実施例参照。)
f)方法F:アミノ酸とジリピジルアミンの結合
Boc−アミノ酸(10mmol)とC12〜C22ジリピジルアミン(10mmol)を250ml容丸底フラスコに加える。CHCl3(100ml)を加え、完全に溶解するまで混合物を撹拌する。次いでTEA(30mmol)とBOP(33mmol)を加える。TEAを加えてpHを10に維持し、反応物を2時間撹拌する。反応の完了後(TLC)、クロロホルムを蒸発させ、固形分を酢酸エチル(300ml)にとる。有機相をKHSO4(4×100ml)、NaHCO3(4×100ml)及びNaCl(4×100ml)で洗浄する。有機相をMgSO4で乾燥し、濾過し、減圧蒸発させる。生成物をTLC、NMR及びMSにより分析し、精製せずに使用する。収率は約90%である。
g)保護ポリアミノ酸とジリピジルアミド酸の結合と、Boc及びZ保護基の分離
方法G:
方法Fにより得られた生成物(9mmol)を磁気棒を備える丸底フラスコに入れ、冷TFA(4℃)を加える(30ml)。溶液を1時間撹拌する。TFAを減圧蒸発させる。DMF(70ml)を加えて生成物を溶かす。TEA(30mmol)を加えた後、方法Eにより得られた保護ポリアミノ酸(9mmol)を加える。pHを10に調整し、BOP(33mmol)を加える。溶液を2時間撹拌後、TLCを行う。結合の完了後(TLC)、KHSO4溶液(700ml)を加え、生成物を酢酸エチル(3×100ml)で抽出する。有機相をKHSO4(3×50)、NaHCO4(3×50)及びNaCl(3×50ml)で洗浄し、MgSO4で乾燥し、濾過し、減圧蒸発させる。生成物をNMR、TLC及びMSにより分析し、予備精製せずに脱保護する。TFA(50ml)を生成物に加え、溶液を1.5時間撹拌し、TFAを蒸発させる。生成物がTFAで分離可能なZ又はClZ基をまだ含んでいる場合には、方法Hを直接実施する。最終生成物を準分取HPLCにより精製する(実施例参照)。
方法H:
方法Gにより得られた生成物がZ又はClZ基を含んでいる場合には、磁気棒を備える丸底フラスコに入れ、10ml MeOH/g生成物に溶かす。Pd/C(10%、1g/g生成物)とギ酸アンモニウム(1g/g生成物)を周囲温度で加える。HPLCにより水素化を行う。2時間後に反応を終了し、混合物を濾過し、濾渣をMeOH 10mlで洗浄する。軟蒸留水を加え、溶液を凝固させ、凍結乾燥する。最終生成物を分取HPLCにより精製する。
h)方法I:Boc保護基の脱保護
丸底フラスコ中のBoc基(1mmol)を含む生成物にトリフルオロ酢酸(50ml)を加える。溶液を1.5時間撹拌し、TFAを蒸発させる。アミンを完全に脱保護し、精製せずに結合に使用する。
B−生物学的試験材料及び方法
1.in vitro遺伝子導入に使用したプラスミド
プラスミドpCMV−LUCは、ベクタープラスミドpcDNA3(Invitrogen)から抽出したヒトサイトメガロウイルス(CMV)のプロモーターを含むMlu I−Hind IIIフラグメントの挿入により、プラスミドpGL2−Basic Vector(Promega)又はプラスミドpGL2−Control Vector(Promega)から誘導される構築物である。
2.トランスフェクションに使用した溶液の調製プロトコール
本発明に記載する生成物はエタノール又は水に20mMに溶かした後、最終エタノール濃度が10%未満となるように水で希釈する。
【0064】
生理的血清(0.15M NaCl)で希釈した核酸溶液を1/1(v/v)の比でリポフェクタント溶液に加える。ボルテックス均質化と周囲温度で15分間のインキュベーション後、DNA/リポフェクタント溶液を最終濃度9%(v/v)でウェルに分配し、無タンパク質(血清)成長培地で洗浄し、無血清又は非無血清培地で再培養した。
C−in vivoアッセイ材料
1.材料
a)実験モデル:
−雌成体(>8週)マウスC57/BL6。
−腫瘍フラグメントを脇腹の皮下に移植して動物間で継代することにより得られる3LL(ルイス肺癌)型腫瘍。
b)使用したプラスミド:
pXL2622:ルシフェラーゼをコードする遺伝子の上流にpCDNA3(Invitrogen)から抽出したサイトメガロウイルス(CMV)のプロモーターを挿入することにより、基本プラスミドpGL2(Promega)から誘導される。このプラスミドはPEG沈殿法(Ausubel)により得られ、 DNA約10μg/μlの濃度で4℃で10mM Tris,1mM EDTA,pH8中に保存する。
2.プロトコール
注射溶液:まずトランスフェクトしようとするDNAを緩衝液に可溶化した後、配列番号1のペプチド(KTPKKAKKP)2を加え、20分後に高濃度(20又は40mM)のカチオン脂質溶液を混合物に加える。全生成物の添加後、混合物はDNA(最終濃度0.5mg/ml)、ペプチド(0.75mg/ml)及びカチオン脂質以外に、150mM NaCl、5% D−グルコース及び5mM MES,pH6.2を含有している。溶液の調製から20〜30分後に注射を行う。
【実施例】
【0065】
実施例1:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlyN[(CH2)17CH3]2(6)の合成
a−{Boc−[3−(Boc−{4−[Boc−(3−Boc−アミノプロピル)アミノ]ブチル}アミノ)プロピル]アミノ}酢酸(3)の合成
方法Aにより得られた樹脂を方法B、C及びEによりスペルミンと反応させる。保護生成物をSiO2上でクロマトグラフィーにより精製する。収率は40%である。
TLC:Rf=0.32(CHCl3/MeOH,9:1)。
HPLC,Rt=4.22分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,d(ppm)):1.40(4s,36H:C(CH3)3); 1.46(mt,4H:ブチルの中心のCH2CH2); 1.64及び1.74(2mt,各2H:プロピルの中心のCH2); 2.96(t,J=7Hz,2H:CH2NCOO); 3.15(mt,8H:CH2NCH2); 3.23(t,J=7.5Hz,2H:CH2NCOO); 3.83(s,2H:OCONCH2COO)。
MH+:661。
b−{Z−[3−(Z−{4−[Z−(3−Z−アミノプロピル)アミノ]ブチル}アミノ)プロピル]アミノ}酢酸(4)の合成
方法Aにより得られた樹脂を方法B、C及びDによりスペルミンと反応させる。保護生成物をSiO2上でクロマトグラフィーにより精製する。収率は20%である。
TLC:Rf=0.85(CHCl3/MeOH,8:2)。
HPLC,Rt=6.92分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度413K,d(ppm)):1.49(mt,4H:ブチルの中心のCH2CH2); 1.74及び1.81(2mt,各2H:プロピルの中心のCH2); 3.07(q,J=7Hz,2H:CH2NCOOベンジル); 3.15−3.30(mt,8H:CH2NCH2); 3.33(t,J=7.5Hz,2H:NCH2COO); 3.70(s,2H:OCONCH2COO); 5.07,5.10,5.12及び5.13(4s,各2H:ArCH2OCON); 6.65(mf,1H:NHCO); 7.25−7.40(mt,20H:芳香族H)。
MH+:797。
c−Boc−Gly−ジオクタデシルアミド(5)
方法FによりBoc−Glyをジオクタデシルアミンに結合する(収率90%)。
TLC:Rf=0.9(CHCl3/MeOH,9:1)。
MH+:679。
1HNMRスペクトル(300MHz,CDCl3,d(ppm)):0.89(t,J=7Hz,6H:CH3); 1.29(mt,60H;脂肪鎖の中心のCH2); 1.49(s,9H:C(CH3)3); 1.55(mt,4H;各脂肪鎖の1CH2); 3.15及び3.33(2t,J=7.5Hz,各2H:脂肪鎖のNCH2); 3.95(d,J=5Hz,2H:OCONCH2CON); 5.57(mf,1H:CONH)。
d−H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlyN[(CH2)18]2(6)
生成物(3)と(5)又は(4)と(5)を方法Gにより結合する。生成物をBoc保護生成物は方法G、Z保護生成物は方法Hに記載したように脱保護する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=15.35分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
BYK20531HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,温度300K,d(ppm)):0.83(t,J=7Hz,6H:CH3); 1.23(mt,60H:脂肪鎖の中心のCH2); 1.43及び1.53(2mt,各2H:各脂肪鎖の1CH2); 1.63(m,4H:ブチルの中心のCH2CH2); 1.96(mt,4H:プロピルの中心のCH2); 2.93,3.00及び3.22(3mt,合計16H:NCH2); 3.83(s,2H:NCH2CON); 4.03(s,2H:CONCH2CON)。
MH+:821。
【0066】
実施例2:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2CON[(CH2)18]2(7)の合成
生成物(3)を方法Fによりジオクタデシルアミンと結合し、方法Gにより脱保護する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=15.2分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
1HNMRスペクトル(400MHz,CF3COOD 2/3とCD3COODd4 1/3の混合物中,d(ppm)):0.78(t,J=7Hz,6H:CH3); 1.20(mt,60H:脂肪鎖の中心のCH2); 1.52(mt,4H:各脂肪鎖の1CH2); 1.80(mt,4H:ブチルの中心のCH2CH2); 2.23及び2.32(2mt,各2H:プロピルの中心のCH2); 3.10−3.40(3mt,合計16H:NCH2); 4.15(s,2H:NCH2CON)。
MH+:764。
【0067】
実施例3:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COArgN[(CH2)18]2(9)の合成
a−Boc−Arg(Z2)ジオクタデシルアミド(8)
本生成物は、方法FによりBocArg(Z2)とジオクタデシルアミンを結合して合成する(収率91%)。
TLC:Rf=0.9(CHCl3/MeOH,9:1)。
MH+:1046。
b−H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COArgN[(CH2)18]2(9)
生成物(3)又は(4)を方法Gにより生成物(8)と結合し、方法G(Boc)及び/又はH(Z)により脱保護する。最終生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=13.83分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6,d(ppm))):0.90(t,J=7Hz,6H:CH3);1.28(mt,60H:脂肪鎖のCH2);1.40−1.80(mt,12H:CH2);1.93(m,4H:プロピルの中心のCH2);2.80−3.10(3mt,16H:NCH2及び脂肪鎖のNCH2);3.42(mt,2H:アミドのCH2N);3.77(mt,2H:NCH2CON);4.67(mt,1H:NCHCON);6.80−7.50(広幅mf,2H:NH2);7.78,7.92,8.80及び9.03(夫々mt及び3mf、夫々1H,2H,4H及び1H:CONH,NH及びNH2)。MH+:920。
【0068】
実施例4:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COArg(Z)2N[(CH2)17CH3]2(10)の合成
生成物(3)を方法Gにより生成物(8)と結合し、同一方法によりBoc基を分離する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=17.75分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO,d(ppm)):0.87(t,J=7Hz,6H:CH3);1.25(mt,60H:脂肪鎖の中心のCH2);1.40及び1.57(2mt,各2H:各脂肪鎖の1CH2);1.65(mt,8H:ブチルの中心のCH2CH2);1.95(mt,4H:プロピルの中心のCH2);2.85−3.05(mt,合計14H:NCH2);3.23(t,J=7.5Hz,2H:NCH2);3.75(s,2H:NCH2CON);3.85及び3.95(2mt,各1H:CH2NC);4.67(mt,1H:CONCHCON);5.07及び5.25(夫々限界AB及びs,J=13.5Hz,各2H:NCOOCH2Ar);7.25−7.45(mt,10H:芳香族H);7.95,8.85,9.90及び9.20(4mf:互換H)。
MH+:1188。
【0069】
実施例5:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(ローダミン)N[(CH2)17CH3]2(13)の合成
a−Boc−Lys(Z)ジオクタデシルアミド(11)
本生成物は、方法FによりBocLys(ClZ)をジオクタデシルアミンと結合することにより合成した(収率89%)。
TLC:Rf=0.92(CHCl3/MeOH,9:1)。
MH+:918。
b−BocHN(CH2)3NBoc(CH2)4NBoc(CH2)3NBocCH2COLys(ClZ)N[(CH2)17CH3]2(12)
(Bocを脱保護せずに)生成物(3)を方法Gにより生成物(11)に結合する。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度423K,d(ppm)):0.92(t,J=6.5Hz,6H:CH3);1.32(mt,60H:脂肪鎖の中心のCH2);1.44(2s,合計36H:C(CH3)3);1.50−1.80(mt,16H:各脂肪鎖の1CH2,ブチルの中心のCH2CH2,CH2CH2CH2及びプロピルの中心のCH2);3.00(q,J=6.5Hz,2H:OCONCH2);3.05(q,J=6.5Hz,2H:CH2NCOO);3.15−3.40(mt,14H:脂肪鎖のNCH2,CH2NCH2及びCH2NCH2CH2N);3.80(s,2H:OCONCH2CON);4.75(mt,1H:CONCHCON);5.15(s,2H:NCOOCH2Ar);5.97及び6.53(2mt,各1H:OCONH及びNHCOO);7.08(d,J=7.5Hz,1H:CONH);7.30−7.50(mt,4H:芳香族H)。
c−H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(ローダミン)N[(CH2)17CH3]2(13)
生成物(12)のリジン上のClZ基を方法Hにより分離し、こうして得られた生成物を減圧乾燥し、エーテルにとり、NaHCO3とNaClで濯ぐ。エーテルをMgSO4で乾燥し、減圧蒸発させる。
【0070】
脱保護生成物77mg(60μM)をMeOH 3mlに溶かし、DIEA(64μL)を加えた後、テトラメチルローダミンイソチオシアネート(30mg,68μM)を加え、溶液を17時間撹拌し、反応をTLCによりモニターする。翌日、溶液を減圧乾燥して濃縮する。次にTFA(4ml)を加え、1時間撹拌する。TFAを蒸発させ、粗生成物を準分取HPCLにより精製する(最終収率30%)。
TLC:Rf=0.05(MeOH)。
HPLC(準分取),Rt=61.55分(H2O/MeCN:3分[100/0]、3〜45分[0/100]、45〜140分[0/100])。
MH+:1335。
【0071】
実施例6:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(ビオチニル)N[(CH2)17CH3]2(14)の合成
生成物(12)を方法Hにより脱保護し(271mg,0.21mmol)、DMF(10ml)に溶かす。DIEA(0.11ml)を加えた後、ビオチン(56.4mg,0.23mmol)とBOP(102mg,0.23mmol;pHは10(DIEA)に維持)を加え、反応の終了をフルオレサミン試験により確認する。生成物を方法Fに記載したように回収し、精製せずにTFA(5ml)で1時間脱保護する。TFAを蒸発させ、生成物を準分取HPLCにより精製する(収率50%)。
HPLC,Rt=13.12分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
MH+:1118。
【0072】
実施例7:
{H2N(CH2)3}2N(CH2)4N{(CH2)3NH2}(CH2)3NHCH2COGlyN[(CH2)17CH3]2(16)の合成
a−{BocNH(CH2)3}2N(CH2)4N{(CH2)3NHBoc}(CH2)3NBocCH2COOH(15)
生成物(1)を方法Bによりポリマーに固定し、方法Cにより保護し、方法Eにより樹脂から分離する。生成物をSiO2上で精製する(収率35%)。
TLC:Rf=0.2(CHCl3/MeOH,8:2)。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,温度433K,d(ppm)):1.42(s,36H:C(CH3)3);1.56(mt,4H:ブチルの中心のCH2CH2);1.65−1.85(mt,8H:プロピルの中心のCH2);2.76(mt,12H:CH2N(CH2)2);3.06(t,J=6.5Hz,6H:OCONCH2);3.29(mt,2H:NCH2);3.86(s,2H:OCONCH2COO)。
MH+:775。
b−{H2N(CH2)3}2N(CH2)4N{(CH2)3NH2}(CH2)3NHCH2COGlyN[(CH2)17CH3]2(16)
生成物(15)を方法Gにより生成物(5)と結合する。生成物を方法Gにより脱保護し、準分取HPLCにより精製し、フラクションを分析HPLCにより分析し、凍結乾燥する(収率55%)。
HPLC(準分取),Rt=38.72分(H2O/MeCN:10分[100/0]、10〜45分[0/100]、45〜140分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度386K,d(ppm)):0.90(t,J=7Hz,6H:CH3);1.30(m,60H:脂肪鎖の中心のCH2);1.55(mt,4H:各脂肪鎖の1CH2);1.65(mt,4H:ブチルの中心のCH2CH2);1.97(mt,8H:プロピルの中心のCH2);2.80−3.05,3.06及び3.28(夫々mt及び2t,J=7.5Hz,18H,2H及び4H:NCH2);3.80(s,2H:NCH2CON);4.03(d,J=5.5Hz,2H:CONCH2CON);6.00−9.00(広幅mf:NH2及びNH);8.27(mt,1H:CONH)。
MH+:935。
【0073】
実施例8:
{H2N(CH2)3}2N(CH2)4N{(CH2)3NH2}(CH2)3NHCH2CON[(CH2)17CH3]2(17)の合成
(3)の代わりに生成物(15)を使用することにより生成物(7)と同様に合成する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析し、凍結乾燥する。
HPLC(準分取),Rt=38分(H2O/MeCN:10分[100/0]、10〜45分[0/100]、45〜140分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,d(ppm)):0.88(t,J=7Hz,6H:CH3);1.29(mt,60H:脂肪鎖の中心のCH2);1.52(mt,4H:各脂肪鎖の1CH2);1.68(mt,4H:ブチルの中心のCH2CH2);1.90−2,10(mt,8H:プロピルの中心のCH2);2.90,2.95−3.15,3.18及び3.15(夫々t,mt及び広幅2t,J=7.5Hz,合計24H:NCH2);4.02(s,2H:NCH2CON)。
MH+:878。
【0074】
実施例9:
{H2N(CH2)2}2N(CH2)2NHCH2COGlyN[(CH2)17CH3]2(19)の合成
a−{BocNH(CH2)2}2N(CH2)2NBocCH2COOH(18)
生成物(1)の代わりにトリス(アミノエチル)アミンを使用することにより生成物(15)と同様に合成する(収率29%)。
TLC:Rf=0.55(CHCl3/MeOH,8:2)。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度393K,d(ppm)):1.44(s,27H:C(CH3)3);2.58(t,J=6.5Hz,4H:CH2NCH2);2.66(t,J=7Hz,2H:NCH2);
3.04(q,J=6.5Hz,4H:OCONCH2);
3.28(t,J=7Hz,2H:OCONCH2);
3.76(s,2H:OCONCH2COO);6.06(mf,2H:CONH)。
MH+:505。
b−{H2N(CH2)2}2N(CH2)2NHCH2COGlyN[(CH2)17CH3]2(19)
(15)の代わりに(18)を使用することにより生成物(17)と同様に合成する(収率65%)。
HPLC,Rt=122分(H2O/MeCN:10分[100/0]、10〜45分[0/100]、45〜140分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,d(ppm)):0.87(t,J=7Hz,6H:CH3);1.15−1.35(mt,60H:脂肪鎖の中心のCH2);1.45及び1.55(2mt,各2H:各脂肪鎖の1CH2);2.64(t,J=5.5Hz,4H:CH2NCH2);2.75(t,J=6Hz,2H:NCH2);2.95(t,J=5.5Hz,4H:NCH2);3.08(t,J=6Hz,2H:NCH2);3.25(mt,4H:脂肪鎖のNCH2); 3.88(s,2H:NCH2CON);4.06(d,J=5Hz,2H:CONCH2CON);7.75(残留広幅mf:NH);8.68(残留t,J=5Hz:CONH)。
MH+:765。
【0075】
実施例10:
{H2N(CH2)2}2N(CH2)2NHCH2CON[(CH2)17CH3]2(20)の合成
(5)の代わりにジオクタデシルアミンを使用することにより生成物(19)と同様に合成する(収率73%)。
HPLC,Rt=100.1分(H2O/MeCN:10分[100/0]、10〜45分[0/100]、45〜140分[0/100])。
MH+:708。
【0076】
実施例11:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLysN[(CH2)17CH3]2(21)(RPR127888A)の合成
生成物(12)を方法H(Cl−Z)、次いで方法Iにより脱保護する。最終生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=11.76分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:892。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度393K,d(ppm)):0.91(t,J=7Hz,6H:脂肪鎖のCH3);1.31(mt,60H:脂肪鎖の中心の(CH2)15);1.35−1.75(mt,10H:各脂肪鎖の1CH2,リシルの中心の(CH2)3);1.75(mt,4H:ブチルの中心の(CH2)2);2.00(mt,4H:プロピルの中心のCH2);2.82,2.98,3.06及び3.10−3.50(夫々2t,mt及び2mf、J=7Hz,合計18H:リシルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.62(s,2H:NCH2CON);4.73(q,J=7Hz,1H:リシルのCONCHCON);8.18(d,J=7Hz,1H:リシルのCONH)。
【0077】
実施例12:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(Cl−Z)N[(CH2)17CH3]2(22)(RPR122759A)の合成
生成物(12)を方法Iにより脱保護する。最終生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=16.79分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1060。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度373K,d(ppm)):0.91(t,J=7Hz,6H:脂肪鎖のCH3);1.31(mt,60H:脂肪鎖の中心の(CH2)15);1.30−1.75(mt,10H:各脂肪鎖の1CH2,リシルの中心の(CH2)3);1.72(mt,4H:ブチルの中心の(CH2)2);1.95(mt,4H:プロピルのCH2);2.98,3.06及び2.90−3.50(夫々2mt及びmf,合計18H:リシルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.59(s,2H:NCH2CON);4.75(q,J=7Hz,1H:リシルのCONCHCON);5.16(s,2H:COOCH2Ar);6.85(mf,1H:OCONH);7.35−7.55(mt,5H:芳香族H);8.15(mf,1H:リシルのCONH)。
【0078】
実施例13:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(CHO)N[(CH2)17CH3]2(24)(RPR122760A)の合成
a−NHBoc(CH2)3NBoc(CH2)4NBoc(CH2)3NBocCH2COLysN[(CH2)17CH3]2(23)
生成物(12)を方法H(Cl−Z)により脱保護し(収率65%)、精製せずに使用する。
HPLC,Rt=20.82分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
b−NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(CHO)N[(CH2)17CH3]2(24)
生成物(23)を方法Gによりギ酸と結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=13.60分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:920。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度383K,d(ppm)):0.92(t,J=7Hz,6H:脂肪鎖のCH3);1.31(mt,60H:脂肪鎖の中心の(CH2)15);1.35−1.70(mt,10H:各脂肪鎖の1CH2,リシルの中心の(CH2)3);1.73(mt,4H:ブチルの中心の(CH2)2);1.98(mt,4H:プロピルのCH2);2.85−3.50(mt,18H:リシルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2); 3.62(s,2H:NCH2CON);4.75(mt,1H:リシルのCONCHCON);7.60(mf,1H:CONH);8.05(広幅s,1H:アルデヒドのCH);8.18(mf,1H:リシルのCONH)。
【0079】
実施例14:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys[コレステリル]N[(CH2)17CH3]2(25)(RPR128142A)の合成
(BOP試薬を使用せずに)生成物(23)を方法Gによりクロロギ酸コレステリルと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=21.66分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1304。
1HNMRスペクトル(600MHz,(CD3)2SO d6,d(ppm)):0.68及び0.98(2s,各3H:コレステリルの18位CH3及び19位CH3);0.86(mt,12H:脂肪鎖のCH3並びにコレステリルの26及び27位CH3);0.91(d,J=7Hz,3H:コレステリルの21位CH3);1.31(mt,60H:脂肪鎖の中心の(CH2)15);0.80−2.30(mt,42H:各脂肪鎖の1CH2,コレステリルの1、2、4、7、11、12、15、16、22、23及び24位のCH2,コレステリルの8、9、14、17、20及び25位のCH,リシルの中心の(CH2)3並びにプロピルのCH2);1.65(mt,4H:ブチルの中心の(CH2)2);2.88及び2.96(2mt,合計14H:リシルのNCH2,ブチルのNCH2,プロピルのNCH2);3.20−3.50(mt,4H:脂肪鎖のNCH2);3.64(s,2H:NCH2CON);4.23(mt,1H:コレステリルの3位CH);4.63(mt,1H:リシルのCONCHCON);5.30(mt,1H:コレステリルの6位CH);6.98(mt,1H:NHCOO);7.90(mt,1H:リシルのCONH);8.60−9.01(互換mf)。
【0080】
実施例15:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys[アラキドニル]N[(CH2)17CH3]2(26)(RPR130605)の合成
生成物(23)を方法Gにより窒素流下で遮光下にアラキドン酸と結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=20.67分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1177。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,温度393K,d(ppm)):0.90(t,J=7Hz,6H:脂肪鎖のCH3); 0.91(t,J=7Hz,3H:アラキドニルのCH3);
1.31(mt,60H:脂肪鎖の中心の(CH2)15);
1.35−1.75(mt,18H:各脂肪鎖の1CH2,アラキドニルの中心の(CH2)3,アラキドニルの中心のCH2及びリシルの中心の(CH2)3);1.75(mt,4H:ブチルの中心の(CH2)2);2.02(mt,4H:プロピルのCH2);2.10(mt,6H:COCH2及びアラキドニルの2個の=CCH2);2.80,2.97,3.06及び3.10−3.50(夫々mt,t,mt及び2mf,J=7Hz,合計24H:アラキドニルの=CCH2C=,リシルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.62(s,2H:NCH2CON);4.73(dd,J=8及び5Hz,1H:リシルのCONCHCON);5.38(mt,8H:アラキドニルのCH=CH)。
【0081】
実施例16:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGluN[(CH2)17CH3]2(28)(RPR126097A)の合成
a−Boc−Glu(O−Bz)−ジオクタデシルアミン(27)
本生成物は方法FによりBoc−Glu(OBz)とジオクタデシルアミンを結合して合成する(収率90%)。
TLC:Rf=0.88(CHCl3/MeOH,9:1)。
b−NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGluN[(CH2)17CH3]2(28)
生成物(3)又は(4)を方法G、次いで方法H(Cl−Z脱保護)により生成物(27)と結合する。最終生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=14.64分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:893。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度383K,d(ppm)):0.90(t,J=7Hz,6H:脂肪鎖のCH3);1.30(mt,60H:脂肪鎖の中心の(CH2)15);1.56(mf,4H:各脂肪鎖の1CH2);1.60−2.00(mt,2H:グルタリルの中心のCH2);1.73(mt,4H:ブチルの中心のCH2)2);1.98(mt,4H:プロピルのCH2);2.32(t,J=7Hz,2H:グルタリルのCOCH2);3.00,3.06及び3.45(夫々t及び2mt,J=7Hz,合計16H:ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.65(広幅s,2H:NCH2CON);4.85(mt,1H:グルタリルのCONCHCON);8.19(広幅s,1H:グルタリルのCONH)。
【0082】
実施例17:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(O−Bz)N[(CH2)17CH3]2(29)(RPR123027A)の合成
生成物(3)を方法Gにより生成物(27)と結合し、脱保護(Boc)する。最終生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=16.02分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:983。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度413K,d(ppm)):0.89(t,J=7Hz,6H:脂肪鎖のCH3);1.30(mt,60H:脂肪鎖の中心の(CH2)15);1.55(mf,4H:各脂肪鎖の1CH2);1.72(mt,4H:ブチルの中心の(CH2)2);1.75−2.00(mt,2H:グルタリルの中心のCH2);1.99(mt,4H:プロピルのCH2);2.47(t,J=7Hz,2H:グルタリルのCOCH2);2.95,3.05及び3.40(3mt,合計16H:ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.62(広幅s,2H:NCH2CON);4.85(mt,1H:グルタリルのCONCHCON);5.14(限界AB,J=12Hz,2H:ベンジルのCH2);7.35(mt,5H:ベンジルの芳香族H);8.23(mf,1H:グルタリルのCONH)。
【0083】
実施例18:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(ガラクトサミド)N[(CH2)17CH3]2(31)(RPR130596A)の合成
a−BocNH(CH2)3NBoc(CH2)4NBoc(CH2)3NBocCH2COGluN[(CH2)17CH3]2(30)
生成物(3)を生成物(27)に結合し、側鎖のOBz保護基を方法H(Cl−Z)により分離し、精製せずに使用する。
HPLC,Rt=22.84分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1293。
b−NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(ガラクトサミド)N[(CH2)17CH3]2(31)
生成物(30)を方法Gにより塩酸D(+)−ガラクトサミンと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=13.71分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1054。
【0084】
実施例19:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(ガラクトサミド)N[(CH2)17CH3]2(32)(RPR130595A)の合成
生成物(30)を方法Gにより塩酸D(+)−ガラクトサミンと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=12.27分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1054。
【0085】
実施例20:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(マンノサミド)N[(CH2)17CH3]2(33)(RPR130598A)の合成
生成物(30)を方法Gにより塩酸D(+)−マンノサミンと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=12.98分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1054。
【0086】
実施例21:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(N(CH3)2)N[(CH2)17CH3]2(34)(RPR131111A)の合成
生成物(30)を方法Gによりジメチルアミンと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=14.44分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:920。
1HNMRスペクトル(400MHz,(CD3)2SO d6,d(ppm)):0.89(t,J=7Hz,6H:脂肪鎖のCH3);1.25(mt,60H:脂肪鎖の中心の(CH2)15);1.43及び1.60(2mt,各2H:各脂肪鎖の1CH2);1.65(mt,4H:ブチルの中心の(CH2)2);1.65及び1.85−2.00(2mt,各1H:グルタリルの中心のCH2);1.95(mt,4H:プロピルのCH2);2.32(限界AB,2H:グルタリルのCOCH2);2.80及び2.92(2s,各3H:CON(CH3)2);2.85−3.05(mt,12H:ブチルのNCH2,プロピルのNCH2);3.00,3.22,3.45及び3.58(4mt,各1H:脂肪鎖のNCH2);3.78(AB,J=16Hz,2H:NCH2CON);4.75(mt,1H:グルタリルのCONCHCON);8.72(d,J=7.5Hz,1H:グルタリルのCONH);8.85及び8.90−9.15(互換mf)。
【0087】
実施例22:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlyN[(CH2)11CH3]2(35)(RPR122767A)の合成
ジオクタデシルアミンの代わりにジドデシルアミンを使用する以外は生成物(6)と同様に合成する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=9.54分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:653。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度403K,d(ppm)):0.93(t,J=7Hz,6H:脂肪鎖のCH3);1.33(mt,36H:脂肪鎖の中心の(CH2)9);1.58(mt,4H:各脂肪鎖の1CH2);1.75(mt,4H:ブチルの中心の(CH2)2);1.95及び2.00(2mt,各2H:プロピルの中心のCH2);2.98及び3.00(2mt,合計12H:ブチルのNCH2及びプロピルのNCH2);3.30(t,J=7Hz,4H:脂肪鎖のNCH2);3.58(s,2H:NCH2CON);4.05(s,2H:グルシルのCONCH2CON)。
【0088】
実施例23:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlyN[(CH2)12CH3]2(36)(RPR122774A)の合成
ジオクタデシルアミンの代わりにジトリドデシルアミンを使用する以外は生成物(6)と同様に合成する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=10.64分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:681。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度393K,d(ppm)):0.91(t,J=7Hz,6H:脂肪鎖のCH3);1.33(mt,40H:脂肪鎖の中心の(CH2)10);1.58(mt,4H:各脂肪鎖の1CH2);1.75(mt,4H:ブチルの中心の(CH2)2);2.00(mt,4H:プロピルの中心のCH2);2.98及び3.00(2t,J=7Hz,合計12H:ブチルのNCH2及びプロピルのNCH2);3.32(t,J=7Hz,4H:脂肪鎖のNCH2);3.65(s,2H:NCH2CON);4.06(d,J=4Hz,2H:グルシルのCONCH2CON);8.60(広幅s,1H:グルシルのCONH)。
【0089】
実施例24:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlyN[(CH2)13CH3]2(37)(RPR122766A)の合成
ジオクタデシルアミンの代わりにジテトラドデシルアミンを使用する以外は生成物(6)と同様に合成する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=9.92分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:709。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度393K,d(ppm)):0.90(t,J=7Hz,6H:脂肪鎖のCH3);1.31(mt,44H:脂肪鎖の中心の(CH2)11);1.58(mt,4H:各脂肪鎖の1CH2);1.76(mt,4H:ブチルの中心の(CH2)2);2.00(mt,4H:プロピルの中心のCH2);2.98及び3.08(夫々mt及びt,J=7Hz,合計12H:ブチルのNCH2及びプロピルのNCH2);3.30(t,J=7Hz,4H:脂肪鎖のNCH2);3.65(s,2H:NCH2CON);4.06(d,J=4Hz,2H:グルシルのCONCH2CON); 8.10(mf,1H:グルシルのCONH)。
【0090】
実施例25:
NH2(CH2)3NH(CH2)4N[(CH2)3NH2]CH2COGlyN[(CH2)17CH3]2(39)(RPR126096A)の合成
a−BocNH(CH2)3NBoc(CH2)4N[(CH2)3NHBoc]CH2CO2H(38)の合成
生成物(3)の合成中にSiO2上で精製時に副産物(38)を回収する。収率は8%である。
TLC:Rf=0.32(CHCl3/MeOH,9:1)。
MH+:561。
1HNMRスペクトル(400MHz,(CD3)2SO d6,d(ppm)):1.30−1.61(mt,4H:ブチルの中心の(CH2)2);1.40(s,27H:C(CH3)3);1.56(mt,4H:プロピルのCH2);2.68及び3.11(夫々広幅t及びt、J=7Hz,各4H:ブチルのNCH2及びプロピルのNCH2);2.90及び2.96(2q,J=7Hz,各2H:プロピルのBocNHCH2);3.18(s,2H:NCH2COO)。
b−NH2(CH2)3NH(CH2)4[N(CH2)3NH2]CH2COGlyN[(CH2)17CH3]2(39)
生成物(38)と(5)を方法Gにより結合する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。HPLC,Rt=13.60分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:821。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,d(ppm)):0.87(t,J=7Hz,6H:各脂肪鎖のCH3);1.28(mt,60H:脂肪鎖の中心の(CH2)15);1.46及び1.54(2mt,各2H:各脂肪鎖の1CH2);1.63(mt,4H:ブチルの中心の(CH2)2);1.91(mt,4H:プロピルのCH2);2.85−3.15(mt,12H:ブチルのNCH2及びプロピルのNCH2);3.24(mt,4H:脂肪鎖のNCH2);3.76(mf,2H:NCH2CON);4.05(広幅s,2H:グリシルのCONCH2CON)。
【0091】
実施例26:
NH2(CH2)3NH(CH2)4NH(CH2)3NH(CH2)3CON[(CH2)17CH3]2(41)(RPR122786A)の合成
a−BocNH(CH2)3NBoc(CH2)4NBoc(CH2)3NBoc(CH2)3CO2H(40)
溶液としてのNaCNBH3とコハク酸準アルデヒドの存在下にスペルミンでアルキル還元を実施することにより生成物(40)を合成する。
【0092】
200ml容丸底フラスコにスペルミン1.8g、メタノール60ml及びNaCNBH3 0.138gを仕込む。溶液を激しく磁気撹拌する。等圧フラスコによりコハク酸準アルデヒド5.5ml(15%)とメタノール30mlの溶液を100分間かけて注入する。撹拌を100分間維持する。アミンをBoc基により次のように保護する。媒質にTEA2.8mlを注いだ後、メタノール30mlに可溶化したジ−tert−ブチルジカーボネート8.8gを加える。撹拌下に一晩維持する。媒質を減圧濃縮し、生成物を酢酸エチルにとり、NaHCO33×50mlで抽出し、水相を集めてエーテル(3×100ml)で濯ぐ。KHSO4を加えて水相のpHを3まで下げ、生成物41の沈殿による濁りを確認し、混合物を酢酸エチル(3×100ml)で抽出する。有機相をMgSO4で乾燥し、減圧蒸発させる。生成物を精製せずに使用する。
b−NH2(CH2)3NH(CH2)4NH(CH2)3NH(CH2)3CON[(CH2)17CH3]2(41)
生成物(40)とジオクタデシルアミンを方法Gにより結合する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=15.04分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:792。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度383K,d(ppm)):0.85(t,J=7Hz,6H:脂肪鎖のCH3);1.22(mt,60H:脂肪鎖の中心の(CH2)15);1.48(mt,4H:各脂肪鎖の1CH2);1.72(mt,4H:ブチルの中心の(CH2)2);1.88(mt,2H:アミノペンタノイルの中心のCH2);1.99(mt,4H:プロピルのCH2);2.42(t,J=7Hz,2H:アミノペンタノイルのCOCH2);2.96,3.03及び3.22(3mt,合計18H:アミノペンタノイルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2)。
【0093】
実施例27:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2CONH(CH2)5CON[(CH2)17CH3]2(42)(RPR128506A)の合成
BocGlyの代わりにBoc6−アミノカプロン酸を使用する以外は生成物(6)と同様に合成する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=13.94分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:877。
1HNMRスペクトル(300MHz,(CD3)2SO d6,d(ppm)):0.87(t,J=7Hz,6H:脂肪鎖のCH3);1.28(mt,60H:脂肪鎖の中心の(CH2)15);1.48(mt,10H:各脂肪鎖の1CH2及びアミノヘキサノイルの中心の(CH2)3);1.65(mt,4H:ブチルの中心の(CH2)2);1.95(mt,4H:プロピルのCH2);2.27(t,J=7Hz,2H:アミノヘキサノイルのCOCH2);2.85−3.30(mt,18H:アミノヘキサノイルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.70(広幅s,2H:NCH2CON);7.90−9.10(互換mf)。
【0094】
実施例28:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(11−アミドウンデカニルヘプタ−O−アセチルラクトース)N[(CH2)17CH3]2(43)(RPR130765A)の合成
生成物(30)を方法Gにより11−アミノウンデカニルヘプタ−O−アセチルラクトースと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=15.91分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1680。
【0095】
実施例29:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COAsm(β−NAc(Ac)3)N[(CH2)17CH3]2(45)(RPR131283A)の合成
a−Fmoc−Asm−β−Glc−NAc(Ac)3−ジオクタデシルアミン(44)
本生成物は方法FによりFmoc−Asm−β−Glc−NAc(Ac)3−OHとジオクタデシルアミンを結合することにより合成する。
TLC:Rf=0.67(CHCl3/MeOH,9:1)。
HPLC,Rt=25.31分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
b−NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COAsm(β−NAc(Ac)3)N[(CH2)17CH3]2(45)
生成物(45)のFmoc基を分離する。
【0096】
生成物(45)0.7gにDMF20mlとジエチルアミン2mlを注ぐ。撹拌下に6時間維持した後、媒質を減圧濃縮する。得られた生成物を方法Gにより生成物(3)に結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=15.35分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1207。
【0097】
実施例30:
生成物(6)の大規模溶液合成
溶液としてのNaCNBH3とグリオキシル酸の存在下にスペルミンでアルキル還元を実施することにより生成物(6)を合成する。
【0098】
2l容丸底フラスコにスペルミン18.2g、メタノール500ml及びNaCNBH3 2gを仕込む。溶液を激しく磁気撹拌する。等圧フラスコによりメタノール300ml中のグリオキシル酸8.45gの溶液を100分間かけて注入する。撹拌を一晩維持する。アミンをBoc基により次のように保護する。媒質にTEA14mlを注いだ後、THF200mlに可溶化したジ−tert−ブチルジカーボネート100gを加える。撹拌下に一晩維持する。媒質を減圧濃縮し、生成物を酢酸エチル(250ml)にとり、KHSO4(6×100ml)、次いで飽和NaCl溶液(3×100)で濯ぎ、MgSO4で乾燥し、減圧蒸発させる。CHCl3/MeOH(9:1)を溶離剤としてシリカカラムで生成物を精製する。生成物を含むフラクションをTLCにより同定し、再び集めて減圧蒸発させると、生成物(6)10gが得られる(合計合成収率17%)。
【0099】
分析HPLC、質量スペクトル及びNMRの分析値は固相法により得られる生成物に一致する。
【0100】
実施例31:
(7)RPR120534A、(6)120535A及び(9)RPR120531Aによるトランスフェクション効率に及ぼす電荷比(アミン/リン酸)の効果
2cm2の指数増殖期の細胞[NIH 3T3、3LL又はウサギSMC]1×105個のサンプルを、5%CO2下で電荷比を変えてリポフェクタント/pCMV−LUC溶液により37℃で2時間処理し、各サンプルに核酸2μgを投与する。最終濃度8%のウシ胎児血清を添加後、CO2インキュベーターで40時間インキュベーション後にリポーター遺伝子の発現を調べる。
【0101】
ルシフェラーゼ活性はルシフェリン、補酵素A及びATPの存在下で10秒間の発光[RLU=相対光単位]により定量し、処理細胞2000個に対する割合として表す。得られた結果を図1、2及び3に示す。
【0102】
これらの図から明らかなように、脂質部分とポリアミンの間の「スペーサー」アームにグリシンが存在すると、低いナノモルカチオン脂質/μgDNA比で良好なトランスフェクション効率が得られる。
【0103】
実施例32:
(20)RPR120527A、(19)120528A、(17)RPR120526A及び(16)RPR120525Aによるトランスフェクション効率に及ぼす電荷比(アミン/リン酸)の効果
2cm2の指数増殖期のNIH 3T3細胞1×105個のサンプルを、5%CO2下で電荷比を変えてリポフェクタント/pCMV−LUC溶液により37℃で2時間処理し、各サンプルに核酸1μgを投与する。最終濃度8%のウシ胎児血清を添加後、CO2インキュベーターで40時間インキュベーション後にリポーター遺伝子の発現を調べる。
【0104】
ルシフェラーゼ活性は細胞の溶解後に得られる上清中で10秒間の発光[RLU=相対光単位]により定量し、タンパク質1mgに対する割合として表す。得られた結果を図4に示す。「スペーサー」アームにグリシン残基が存在すると有利であることは本実施例からも明白である。
【0105】
実施例33:
(6)120535、(41)RPR122786及び(42)RPR128506によるトランスフェクション効率に及ぼすスペーサー長の効果
2cm2の指数増殖期の細胞[NIH 3T3及びHeLa]1×105個のサンプルを、血清タンパク質の不在下で37℃、5%CO2下の湿潤雰囲気でカチオン脂質濃度を変えてカチオン脂質/pCMV−Luc混合物により2時間処理する。次いで細胞増殖培地に最終濃度8%のウシ胎児血清を加え、CO2インキュベーターで更に40時間インキュベーション後にトランス遺伝子の発現を測定する。
【0106】
スペーサーの構造特性は以下の通りである。
【0107】
【表1】
【0108】
下表Iは得られた結果を示し、試験材料の各々で得られた最大効率を表す。
【0109】
【表2】
【0110】
トランスフェクション効率は処理細胞2103個当たりのRLU/10秒として与える。括弧内はナノモル脂質/μgDNA比を示す。
【0111】
実験1及び2では異なるプラスミドを使用し、実験1及び2で夫々細胞1105個当たりDNA1μg及び0.5μgの用量とする。
【0112】
実施例34:
(6)RPR120535によるトランスフェクション効率に及ぼすスペーサー構造の効果
補助細胞系(3LL細胞)を導入した以外は上記実施例に記載したと同一の実験条件下で、Arg型、Lys型又はGlu型の基で「スペーサー」を置換することにより修飾したカチオン脂質(6)RPR120535を用いて得られたトランスフェクション効率を比較した。
【0113】
種々のスペーサーの構造は以下の通りである。
【0114】
【表3】
【0115】
トランスフェクション効率は処理細胞2103個当たりのRLU/10秒として与える。
実験1及び2では異なるプラスミドを使用し、実験1及び2で夫々細胞1105個当たりDNA0.5μg及び1μgの用量とした。結果を分析した処、該当細胞に応じて、好ましくは置換したアミノ酸鎖の存在は良好なトランスフェクション効率をもたらすことが判明した。
【表4】
【0116】
実施例34:
(9)RPR120531Aによるリポフェクタント/DNA混合物中のDOPEの存在の効果
実施例31で使用したと同一のプロトコールに従い、トランスフェクション混合物にDNAを添加する前にカチオン脂質(9)RPR120531Aに種々のモル比でDOPE(ジオレオイルホスファチジルエタノールアミン)を加えた。
細胞の溶解後に上清中のルシフェラーゼ活性を定量し、タンパク質1mgに対する割合を計算する(図5)。
リポフェクタント混合物中にDOPEが存在すると、(9)RPR120531Aの濃度が低い場合にトランスフェクション効率を改善することができる。
【0117】
実施例35:
(6)RPR120535A、(9)120531A及び(10)RPR121650Aによるトランスフェクション効率に及ぼす血清の効果
2cm2の指数増殖期の細胞[NIH 3T3又はHeLa]1×105個のサンプルを、血清の不在下で2時間又は培地中の血清の存在下でリポフェクタント/pCMV−LUC溶液(リポフェクタント3ナノモル/μgDNA)により処理する。本実施例では各サンプルに核酸2μgを投与する。細胞溶解液の上清中のルシフェラーゼの発現を調べ、RLU/10秒で表し、タンパク質1mgに対する割合を計算する。結果を分析した処、血清の存在はトランスフェクションにさほど影響しないことが判明した。
【0118】
実施例36:
(20)RPR120527A、(19)120528A、(17)RPR120526A及び(16)RPR120525AによるDNA/リポフェクタント混合物中の核酸濃度の効果
実施例31に記載した条件下で、ナノモルリポフェクタント/μgDNA比を最適化し、NIH 3T3細胞のサンプルにトランスフェクトする(実施例32参照、(20)RPR120527A及び(19)RPR120528Aでは比=6、(17)RPR120526A及び(16)RPR120525Aでは比=3)。各サンプルに加えるDNAの量は0.5〜2μgとする。結果を図6に示す。
指数増殖期の細胞1×105個にプラスミドDNA1μgをトランスフェクトするのが好ましい選択であると思われ、実際に、DNAの使用量が増加すると細胞に接触するカチオン脂質の濃度が増加し、場合によっては毒性の問題を生じる。DNA濃度が低いと、トランスフェクション効率との比例が得られなくなる。
【0119】
実施例37:
本発明によるリポポリアミンのin vivoトランスフェクション試験
腫瘍移植後7日のマウスをケタミン+キシラジンで麻酔し、上記のように調製した本発明によるリポポリアミンを含む溶液を注射する。注射から2日後に腫瘍組織を取り出し、計量後に細切し、溶解用緩衝液(Promega Cell Lysis Buffer E153 A)500μl中で粉砕する。遠心分離(20000gで10分間)後、10μlを分取し、試薬(Promega Luciferase Assay Substrate)50μlと混合後に得られた合計発光をルミノメーターLumat LB 9501(Berthold)で測定し、10秒間積分することによりルシフェラーゼ活性を評価する。得られた活性を腫瘍溶解液の合計上清中の推定RLU(相対光単位)又は注射DNAμg当たりのRLUとして表す。表IIIは得られた結果を示す。
【表5】
【図面の簡単な説明】
【0120】
【図1】種々のカチオン脂質によるNIH 3T3細胞(マウス胎児繊維芽細胞)の処理後のトランスフェクション効率の測定を表すグラフである。
【図2】種々のカチオン脂質によるウサギSMC細胞(ウサギ大動脈平滑筋細胞の一次培養物)の処理後のトランスフェクション効率の測定を表すグラフである。
【図3】種々のカチオン脂質による3LL細胞(ルイス肺癌)の処理後のトランスフェクション効率の測定を表すグラフである。
【図4】種々のカチオン脂質によるNIH 3T3細胞(マウス胎児繊維芽細胞)の処理後のトランスフェクション効率の測定を表すグラフである。
【図5】3LL細胞のトランスフェクション効率に及ぼすDOPE濃度の効果を表すグラフである。
【図6】ナノモルリポフェクタント/μgDNA比を一定にしてDNA量を変えた場合のNIH 3T3細胞のトランスフェクションを表すグラフである。
【技術分野】
【0001】
本発明はリポポリアミンファミリーに属する新規化合物、該化合物を含有する医薬組成物、核酸のin vivo及び/又はin vitroトランスフェクションのためのその適用並びにその製造方法に関する。
【発明の開示】
【0002】
多数の遺伝病は1種以上の核酸の発現障害及び/又は異常発現即ち発現不足又は過剰発現に関連している。遺伝子治療はクローン化遺伝子のin vivo又はin vitro細胞発現によりこのような遺伝異常を治療することを主眼とする。
【0003】
今日、この種の遺伝情報の細胞内送達方法として数種の方法が提案されている。特に、化学的又は生化学的ベクターを利用するものが挙げられる。合成ベクターはトランスフェクトしようとするDNAを圧縮する機能と、その細胞固定並びにその細胞質膜及び場合により2つの核膜通過を助長する機能との2つの主機能をもつ。
【0004】
このトランスフェクション方法は、カチオン脂質の利用に基づく技術の開発により著しい進歩を遂げた。例えば、正電荷をもつカチオン脂質であるN−[1−(2,3−ジオレイルオキシ)プロピル]−N,N,N−トリメチルアンモニウムクロリド(DOTMA)は、負電荷をもつDNAとリポソーム又は小ベジクル形態で自然に相互作用し、細胞膜と融合することが可能な脂質−DNA複合体を形成し、こうしてDNAの細胞内送達を可能にすることが判明した。しかし、この分子はトランスフェクションの点では有効であるが、非生分解性であり、細胞に毒性であるという欠点がある。
【0005】
DOTMA以来、所謂「スペーサー」アームを介してアミノ基に結合した親油基というこの構造モデルで他のカチオン脂質も開発された。そのうちでは、親油基として2個の脂肪酸又は1個のコレステロール誘導体を含み、更に、場合によりアミノ基として第4級アンモニウム基を含むものを挙げることができる。このカチオン脂質の分類の代表例として、DOTAP、DOBT又はChOTBを特に挙げることができる。この他、DOSCやChOSC等のように、第4級アンモニウム基の代わりにコリン基の存在を特徴とする化合物もある。しかし、一般にこれらの化合物のトランスフェクション活性は依然としてかなり低い。
【0006】
別の分類のカチオン脂質であるリポポリアミンも報告されている。本発明の意味では、リポポリアミンなる用語は所謂スペーサー領域を介して親油領域と結合した少なくとも1個の親水ポリアミン領域を含む両親媒性分子を意味する。リポポリアミンの正電荷をもつポリアミン領域は負電荷をもつ核酸と可逆的に結合することができる。この相互作用は核酸を著しく圧縮する。親油領域は脂質膜から形成される核脂質粒子を覆うことにより、このイオン相互作用を外部媒質に対して非感受性にする。この種の化合物では、カチオン基は2個の第1級アンモニウム基と2個の第2級アンモニウム基との4個のアンモニウム基を含むL−5−カルボキシスペルミン基により置換することができる。この種の化合物としては特にDOGSとDPPESが挙げられる。これらのリポポリアミンは一次内分泌細胞のトランスフェクションに特に有効である。
【0007】
実際に、理想的な合成トランスフェクション剤は広い細胞範囲で高いトランスフェクションレベルをもち、使用用量で無毒性であるか又は非常に低毒性であり、更に処理細胞のレベルで副作用が全くないように生分解性でなければならない。
【0008】
本発明は詳細には、独自のポリアミンフラクションをもち、in vitro及び/又はin vivo細胞トランスフェクション、特に核酸のベクター導入に有効に使用可能な新規リポポリアミンを提案することを目的とする。
【0009】
本発明の第1の目的は一般式I:
【0010】
【化6】
[式中、
R1、R2及びR3は各々独立して水素原子又は−(CH2)q−NRR’基を表し、
qはR1、R2及びR3の各基で独立して1、2、3、4、5及び6であり、
R及びR’は各々独立して水素原子又は−(CH2)q’−NH2基を表し、q’はR及びR’の各基で独立して1、2、3、4、5及び6であり、
m、n及びpは各々独立して0〜6の整数を表し、但しnが2以上のとき、mは異なる値をとることができ、R3は一般式I中で異なる意味をもち、
R4は一般式II:
【0011】
【化7】
の基を表し、式中、
R6及びR7は各々独立して水素原子又は飽和もしくは不飽和C10〜C22脂肪族基を表し、2個の基の少なくとも一方は水素以外のものであり、
uは0〜10から選択される整数であり、但しuが2以上の整数であるとき、R5、X、Y及びrは各モチーフ[X−(CHR5)r−Y]中で異なる意味をもつことができ、
Xは酸素原子、硫黄原子又はモノアルキルもしくは非モノアルキルアミン基を表し、
Yはカルボニル基又はメチレン基を表し、
R5は水素原子又は場合により置換された天然アミノ酸側鎖を表し、
rは1〜10の整数を表し、但しrが1であるとき、R5は天然アミノ酸側鎖を表し、rが2以上であるとき、R5は水素原子を表す]により表されることを特徴とするD、L又はDL形リポポリアミン及びその塩を提供することである。
【0012】
天然アミノ酸側鎖とは、特に本発明の意味では例えばアルギニン側鎖のようにアミジニウムモチーフを含む鎖を意味する。上述のように、この鎖は飽和もしくは不飽和のC1〜C24直鎖、分枝鎖もしくは環状脂肪族基(例えばコレステリル基、アラキドニル基又はレチノイル基)又はモノもしくはポリ芳香族基(例えば置換又は非置換ベンジルオキシカルボニル、ベンジルエステル及びローダミニル誘導体)により置換されていてもよい。
【0013】
一般式(I)のこれらの新規生成物は医薬的に許容可能な非毒性塩の形態をとることができる。これらの非毒性塩は鉱酸(塩酸、硫酸、臭化水素酸、リン酸、硝酸)、有機酸(酢酸、プロピオン酸、コハク酸、マレイン酸、ヒドロキシマレイン酸、安息香酸、フマル酸、メタンスルホン酸又は蓚酸)、無機塩(水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化カルシウム)又は有機塩(トリエチルアミン、ピペリジン、ベンジルアミン等の第3級アミン)との塩を含む。
【0014】
本発明による化合物の例としては、特に下記亜一般式:
【0015】
【化8】
(式中、R4、R6及びR7は上記と同義である)の化合物を挙げることができる。上記式中、好ましくはR4はNR6R7であり、R6及びR7は亜式III〜XIIにおいて(CH2)17CH3、(CH2)11CH3、(CH2)13CH3又は(CH2)12CH3から選択される同一基を表す。
【0016】
特に有利な態様において、本発明の化合物は該化合物が結合した核酸の移送を指向させることが可能なターゲティング成分を更に含む。好ましくは、このターゲティング成分はR5置換基により表されるアミノ酸側鎖のレベルで一般式Iの化合物に組み込まれる。より好ましくは、ターゲティング成分は本発明による化合物に共有又は非共有的に結合される。
【0017】
このようなターゲティング成分としては、所定細胞型又は所望の所定組織(腫瘍細胞、肝細胞、造血細胞等)に核酸の移送を指向させることが可能な細胞外ターゲティング成分が挙げられる。例えば、標的細胞型の表面に存在する細胞レセプターのリガンドが挙げられ、例えば糖、葉酸、トランスフェリン、インスリン、アシアロオロソムコイドタンパク質又は細胞外レセプターにより認識される任意の生物活性分子等である。また、例えばトランスフェクトされるDNAを核の内部に優先的に蓄積する核局在シグナル配列(nls)のように、所定の優先的細胞区画(ミトコンドリア、核等)に向けて核酸移送を指向させることが可能な細胞内ターゲティングでもよい。
【0018】
より一般には、本発明の範囲で利用可能なターゲティング成分としては糖類、ペプチド、オリゴヌクレオチド、ステロイド又は脂質が挙げられる。好ましくは、糖類及び/又はペブチド(例えば抗体又は抗体フラグメント、細胞レセプターのリガンド又はそのフラグメント、レセプター又はレセプターフラグメント等)である。特に、成長因子レセプター、サイトカインレセプター、細胞レクチンレセプター又は接着タンパク質レセプター(例えばインテグリン)のリガンドが挙げられる。また、トランスフェリンのレセプター、脂質HDL及びLDLも挙げられる。ターゲティング成分は更に、アシアロ糖タンパク質レセプター等のレクチンをターゲティングすることが可能な糖や、イムノグロブリンのFcフラグメントのレセプターをターゲティングすることが可能な抗体Fabフラグメントでもよい。
【0019】
同様に、一般式Iの化合物の例えばアミノ酸側鎖R5にビオチン、ローダミン、葉酸型のマーカー物質を結合することも考えられる。このマーカー物質は更に、インテグリン型の接着タンパク質の一次及び/又は二次レセプターの認識エピトープ
Arg−Gly−Aspを含む直鎖又は環状ペプチド又はプソイドペプチド配列でもよい。
【0020】
本発明のリポポリアミンの例としては、後記実施例に詳細に記載する下記化合物を特に挙げることができる。
【0021】
【化9】
【0022】
本発明の特に代表的な化合物としては、下記一般式の化合物を特に挙げることができる。
【0023】
【化10】
【0024】
本発明の範囲内で、本発明によるリポポリアミンを誘導する非対称官能化ポリアミンを製造するための新規固相方法も開発された。
【0025】
従来、非対称官能化ポリアミンの製造は直鎖又は分枝鎖対称ポリアミンに選択的に修飾を導入する必要により制限されている。ポリアミンの第1アミノ基と第2アミノ基の化学的差異により、アルキル化、ミカエリス型付加又はアシル化等の反応を実施するためには高い選択性が必要である。更に、このような化学的差異により要求される選択性は、ポリアミンで行われるような複数の第1及び第2アミン群での選択的アシル化に適合しない。このような制約に対する従来の対応は、「ポリアミノ化」することが可能なアミノ基と適当に保護された非対称官能基をもつモノマーブロックから非対称官能化ポリアミンを構築する方法であった。従って、このアプローチには複数段階からなる面倒な合成ストラテジーが必要であり、特に官能基の直交保護が必要であった。
【0026】
本発明の範囲内で開発された方法は、詳細には上記型の合成に伴う従来の欠点を解消するを目的とする。この方法は、対称ポリアミンのアルキル化又は還元アルキル化により一次アミノ基のみで選択的に官能化されたポリアミンを容易且つ迅速に得られるという顕著な利点がある。
【0027】
本発明の方法の原理は固相合成法の利用に基づき、アルキル化剤とポリアミンの二分子反応を助長し、ポリアミンのポリアルキル化を回避する。より詳細には、本発明は固体支持体に共有結合したアルキル化剤と対称ポリアミンの二分子反応により予め得られた少なくとも1個の非対称ポリアミンフラクションに少なくとも1個の脂質フラクションを結合することを特徴とする方法に関する。このアプローチによると、アルキル化剤はエステル化又はアミド化によりポリマー支持体に共有結合している。対称ポリアミンは二分子反応により固相アルキル化剤と反応し、支持体に結合した単官能化非対称ポリアミンを生じる。生成物の遊離アミンは一般にBOC又はZ型の保護基により固相で保護されており、最終的に生成物は固相支持体から分離される。これらのポリアミノ酸を適宜脂質フラクションに結合すると、所望のトランスフェクション剤が得られる。この結合には、例えばBOP、Pybop、BopCl及びDCC等の慣用ペプチド結合剤を利用することができる。本方法は、場合によりトレーサーペプチド、糖類又は蛍光プローブを分子に導入して完全トランスフェクション剤を固体支持体に結合することも可能である。当然のことながら、遊離リポポリアミンにこの型のグラフトを実施できることが判明した。
【0028】
本方法の実施可能性は、複数の直鎖又は分枝鎖非対称官能化ポリアミノ酸の合成により立証された。
【0029】
本発明は更に、上記のようなリポポリアミンの直接又は医薬組成物としての任意の治療的適用にも関する。
【0030】
上述のように、一般式Iの化合物は核酸のin vitro及びin vivoトランスフェクションに特に有利であることが判明した。これらの化合物はDNAを有効に圧縮し、非常に低毒性であるという利点がある。
【0031】
本発明の組成物から最大限の効果を得るためには、核酸の負電荷に対する該当リポポリアミンの正電荷の比であるR比が最適となるように、一般式Iの化合物と核酸の夫々の比率を決定することが好ましい。この最適比は特にin vivo又はin vitroの使用方式や、トランスフェクトしようとする細胞型により異なり、場合に応じて最適化される。この最適化は当業者に自明である。
【0032】
本発明の医薬組成物において、ポリヌクレオチドはデオキシリボ核酸でもリボ核酸でもよい。天然又は人工起源のいずれの配列でもよく、特にゲノムDNA、cDNA、mRNA、tRNA、rRNA、ハイブリッド配列又は修飾もしくは非修飾オリゴヌクレオチドの合成もしくは半合成配列が挙げられる。これらの核酸はヒト、動物、植物、細菌、ウイルス等に由来するものであり得る。これらの核酸は当業者に公知の任意方法により得られ、特に、バンクスクリーニング、化学的合成又はバンクスクリーニングにより得られた配列の化学的もしくは酵素的修飾を含む混合方法により得られる。更に、プラスミドベクター等のベクターに組み込んでもよい。
【0033】
特にデオキシリボ核酸については、1本鎖でも2本鎖でもよいし、短いオリゴヌクレオチドでもより長い配列でもよい。これらのデオキシリボ核酸は治療遺伝子、転写又は複製調節配列、修飾又は非修飾アンチセンス配列、他の細胞成分との結合領域等を含み得る。
【0034】
本発明の意味では、治療遺伝子とは特に治療効果をもつタンパク質産物をコードする任意遺伝子を意味する。こうしてコードされるタンパク質産物はタンパク質、ペプチド等であり得る。このタンパク質産物はターゲット細胞に対して同種であり得る(即ちターゲット細胞が疾病をもたないときにターゲット細胞で正常に発現される産物)。この場合、タンパク質の発現は例えば細胞における不十分な発現や、不活性であるか又は修飾により弱活性となったタンパク質の発現を解消し、あるいは前記タンパク質を過剰発現させることができる。治療遺伝子は安定性の増加、活性改変等の性質をもつ細胞タンパク質の突然変異体をコードするものでもよい。タンパク質産物はターゲット細胞に対して異種でもよい。その場合には、発現されるタンパク質は例えば細胞に欠失している活性を補充又は付加し、疾病に対抗するこ又は免疫応答を刺激することができる。
【0035】
本発明の意味での治療物質としては、より特定的には酵素、血液誘導体、ホルモン、リンホカイン(インターロイキン、インターフェロン、TNF等(仏国特許出願第9203120号))、成長因子、神経伝達物質又はその前駆物質もしくは合成酵素、栄養因子(BDNF、CNTF、NGF、IGF、GMF、aFGF、bFGF、NT3、NT5、HARP/プレイオトロフィン等、ジストロフィン又はミニジストロフィン(仏国特許出願第9111947号))、膵臓線維症に関連するタンパク質CFTR、腫瘍抑制遺伝子(p53、Rb、Rap1A、DCC、k−rev等(仏国特許出願第9304745号))、凝血に関与する因子(VII、VIII、IX因子)をコードする遺伝子、DNAの修復に関与する遺伝子、自殺遺伝子(チミジンキナーゼ、シトシンデアミナーゼ)、ヘモグロビン又は他の輸送タンパク質の遺伝子、アポリポタンパク質A−I、A−II、A−IV、B、C−I、C−II、C−III、D、E、F、G、H、J及びアポ(a)から選択されるアポリポタンパク質型の脂質の代謝に関与するタンパク質に対応する遺伝子、代謝酵素(例えばリポタンパク質リパーゼ、肝性リパーゼ、レシチンコレステロールアシルトランスフェラーゼ、7−α−コレステロールヒドロキシラーゼ、ホスファチジン酸ホスファターゼ)、脂質輸送タンパク質(例えばコレステロールエステル輸送タンパク質やリン脂質輸送タンパク質)、HDL結合タンパク質又は例えばLDLレセプター、レムナントキロミクロンレセプター及びスカベンジャーレセプター等から選択されるレセプターを挙げることができる。
【0036】
治療核酸は更に、ターゲット細胞で発現されると遺伝子発現又は細胞mRNA転写を調節することが可能なアンチセンス遺伝子又は配列でもよい。このような配列は、ヨーロッパ特許第140308号に記載の技術により、例えば細胞mRNAの相補的RNAターゲット細胞に転写され、こうしてそのタンパク質翻訳を阻止することができる。治療遺伝子は更に、ターゲットRNAを選択的に破壊することが可能なリボソームをコードする配列も含む(ヨーロッパ特許第321201号)。
【0037】
上述のように、核酸は更に、ヒト又は動物で免疫応答を発生することが可能な抗原ペプチドをコードする1個以上の遺伝子を含んでいてもよい。従って、この特定態様によると、本発明はヒト又は動物で特に微生物、ウイルス又は癌に対するワクチン又は免疫治療を実現することができる。特に、エプスタイン・バールウイルス、HIVウイルス、B型肝炎ウイルス(ヨーロッパ特許第185573号)、偽狂犬病ウイルス、シンシチウム形成ウイルス、他のウイルスの特異的抗原ペプチド又は腫瘍特異的抗原ペプチド(ヨーロッパ特許第259212号)を挙げることができる。
【0038】
好ましくは、核酸は更に細胞又は所望臓器において治療遺伝子及び/又は抗原ペプチドをコードする遺伝子の発現を可能にする配列も含む。このような配列としては、これらの配列が感染細胞で機能できるときに該当遺伝子の発現に天然に関与する配列が挙げられる。異なる起源の配列でもよい(他のタンパク質の発現に関与、あるいは合成配列でもよい)。特に、真核又はウイルス遺伝子のプロモーター配列が挙げられる。例えば、感染させようとする細胞のゲノムに由来するプロモーター配列が挙げられる。また、ウイルスのゲノムに由来するプロモーター配列でもよい。この点では、例えばE1A、MLP、CMV、RSV等の遺伝子のプロモーターを挙げることができる。更に、活性化配列や調節配列等を付加してこれらの発現配列を修飾してもよい。誘導又は抑制プロモーターでもよい。
【0039】
更に、核酸は合成治療物質をターゲット細胞の分泌経路に導くシグナル配列も特に治療遺伝子の上流に含んでいてもよい。このシグナル配列は治療物質の天然シグナル配列でもよいし、他の任意の機能的シグナル配列又は人工シグナル配列でもよい。核酸は更に合成治療物質を細胞の特定区画に導くシグナル配列も含んでいてもよい。
【0040】
別の態様において、本発明は核酸と、本発明のリポポリアミンと、リポポリアミン/核酸複合体に結合してトランスフェクション能を改善することが可能なアジュバントを含む組成物に関する。本願出願人は実際に、リポポリアミン/核酸複合体に結合することが可能な所定のアジュバント(例えば脂質、ペプチド又はタンパク質)の存在下でリポポリアミンのトランスフェクション能を予想外に増加できることを立証した。
【0041】
この点で、本発明の組成物はアジュバントとして1種以上の中性脂質を含み得る。このような組成物は特にR比が小さいときに特に有利である。本願出願人は実際に、中性脂質を加えると核脂質粒子の形成を改善し、驚くべきことに細胞膜を撹乱して粒子の細胞侵入を助長できることを立証した。
【0042】
より好ましくは、本発明の範囲内で使用する中性脂質は2個の脂肪鎖をもつ脂質である。
【0043】
生理的条件下で両性イオン性又はイオン電荷をもたない天然又は合成脂質を使用すると特に有利である。脂質は特にジオレオイルホスファチジルエタノールアミン(DOPE)、オレオイルパルミトイルホスファチジルエタノールアミン(POPE)、ジステアロイルホスファチジルエタノールアミン、ジパルミトイルホスファチジルエタノールアミン、ジミリストイルホスファチジルエタノールアミン及びその1〜3倍N−メチル化誘導体、ホスファチジルグリセロール、ジアシルグリセロール、グリコシルジアシルグリセロール、セレブロシド(例えば特にガラクトセレブロシド)、スフィンゴ脂質(例えば特にスフィンゴミエリン)又はアシアロガングリオシド(例えば特にアシアロGM1及びGM2)から選択することができる。
【0044】
これらの種々の脂質は当業者に周知の慣用技術により合成するか又は臓器(例えば脳)もしく胎児から抽出することにより得られる。特に、天然脂質の抽出は有機溶媒を用いて実施することができる(Lehninger,Biochemistry参照)。
【0045】
ごく最近になって本願出願人は前記核酸の凝縮レベルに直接又は間接的に作用する化合物をアジュバントとして使用すると特に有利であることも立証した(WO96/25508)。
【0046】
リポポリアミンをベースとするトランスフェクション組成物中にこのような化合物が存在すると、この組成物の量を著しく低減でき、この組成物のトランスフェクション活性を損なわずに毒物学的面で有益な結果が得られる。それどころか、組成物のトランスフェクションレベルは向上するという利点がある。
【0047】
核酸の凝縮レベルに作用する化合物とは、核酸を直接又は間接的に圧縮する化合物として定義される。より詳細には、この化合物はトランスフェクトしようとする核酸のレベルに直接作用するか、又はこの核酸の凝縮に直接関与する付加化合物のレベルに作用することができる。核酸のレベルに直接作用するものが好ましい。
【0048】
好ましい態様によると、核酸の凝縮レベルに作用するこの物質は全体又は一部をペプチドモチーフ(KTPKKAKKP)及び/又は(ATPAKKAA)から構成され、モチーフ数は2〜10である。本発明による化合物の構造において、これらのモチーフは連続又は不連続的に反復し得る。従って、これらのモチーフは生化学的結合(例えば1個以上のアミノ酸)又は化学的結合により分離することができる。このような物質は全体又は一部がヒストン、ヌクレオリン、プロタミン及び/又はそれらの誘導体の1種から誘導されるものでもよい。
【0049】
好ましくは、本発明の組成物は核酸1当量当たりアジュバント0.01〜20当量(重量/重量)、より好ましくは0.5〜5当量を含む。
【0050】
本発明による組成物は局所、皮膚、経口、直腸、膣、非経口、鼻腔内、静脈内、筋肉内、皮下、眼内、経皮等の経路で投与するように調剤することができる。好ましくは、本発明の医薬組成物は特に所望臓器のレベルに直接注射するための注射用製剤又は局所投与(皮膚及び/又は粘膜)用として医薬的に許容可能なキャリヤーを含有する。このようなキャリヤーとしては、特に等張滅菌溶液又は場合に応じて滅菌水もしくは生理的血清を加えると注射可能な溶質を構成することが可能な乾燥組成物、特に凍結乾燥組成物が挙げられる。注射に使用する核酸の用量及び投与回数は種々のパラメーター、特に使用する投与方法、該当疾病、発現させようとする遺伝子、又は所望の治療期間に応じて選択できる。特に投与方法については、組織又は循環経路への直接注射や、培養細胞処理後の注射又は移植によるin vivo再移植が挙げられる。
【0051】
従って、本発明は上記条件下で上記疾病を治療することが可能な核酸と一般式Iの化合物のin vivo又はin vitro併用投与を特徴とする、疾病治療に特に有利な方法を提供する。より詳細には、この方法はタンパク質又は核酸産物の不足に起因する疾病に適用することができ、投与する核酸は前記タンパク質産物をコードするか又は前記核酸産物を含む。
【0052】
本発明は、in vivo又はin vitro細胞トランスフェクションのための本発明のリポポリアミンの全使用に及ぶ。
【0053】
以下、実施例及び図面により本発明をより詳細に説明するが、これらの説明は例示に過ぎず、本発明を制限するとみなすべきではない。
【0054】
略語及び記号
AcOEt:酢酸エチル
BOC:t−ブトキシカルボニル
BOP:ベンゾトリアゾール−1−イルオキシトリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート
DCC:ジシクロヘキシルカルボジイミド
DCU:ジシクロヘキシル尿素
DMAP:4−ジメチルアミノピリジン
DMF:ジメチルホルムアミド
DMSO:ジメチルスルホキシド
DODA:ジオクタデシルアミン
EP:石油エーテル
EtOH:エタノール
NEt3:トリエチルアミン
Rf:前端保持率
TFA:トリフルオロ酢酸
THF:テトラヒドロフラン
TMS:テトラメチルシラン
UV:紫外線
SPPS:固相ペプチド合成
HPLC:高圧液体クロマトグラフィー
Z:ベンジルオキシカルボニル
ClZ:p−クロロベンジルオキシカルボニル
A−化学合成用材料及び方法
1.材料
a)化合物
−出発ポリアミンは例えばスペルミジン、スペルミン、トリス(2−アミノエチル)アミン、フェニレンジアミン、ジアミノエタン(プロパン、ブタン、ペンタン、ヘキサン等)等の市販品でもよいし、分枝鎖ポリアミン製造用として市販されている例えばジアミノエタン(プロパン、ブタン、ペンタン、ヘキサン等)、アミン、スペルミジン、スペルミン等のアミンの完全なシアノエチル化により常法で合成してもよい。
−アルキル化剤はアルキル化方法に応じて次のように選択される。慣用アルキル化にはブロモ酢酸、ω−ハロゲノカルボン酸を使用し、還元アルキル化にはω−アルデヒドカルボン酸(例えばグリオキサル酸、コハク酸準アルデヒド等)又はケト酸(例えばアセト酢酸又はピルビン酸等)を使用する。
−使用するポリマーは固相ペプチド合成(メリフィールド合成)用に市販されている樹脂、例えば遊離酸基をもつ生成物を提供するO−クロロトリチルクロリド樹脂、HMP樹脂や、Rink型樹脂である。ポリアミノ酸は、固相で予備合成したブロモアルキル基をもつペプチド又はω−アルデヒド酸で直接合成することができる。
−Aldrich製品ジオクタデシルアミン、トリエチルアミン、トリフルオロ酢酸、BOP、DMAP、クロロギ酸ベンジルは市販品である。NaCl及びNaHCO3溶液は飽和溶液であり、KHSO4溶液は0.5Mである。
b)物理的測定
陽子NMRスペクトルはBruker 400スペクトロメーターで600MHzで記録した。
【0055】
質量スペクトルはAPI−MS/IIIで測定した。
c)クロマトグラフィー技術
HPLC分析はMerck−Hitachi装置にオート
サンプラーAS−2000A、インテリジェントポンプL−6200A及び紫外−可視検出器L−4000を取り付け、分析用分離には220nm、分取用分離には235nmの波長に調節して実施した。分析用分離カラムはPerkin−Elmer製BU−300 aquapore Butyl 7m,300A 300×4.6mmカラムを使用し、分取用分離カラムはBiorad製Biosil C18 HL 90−10 250×10mmカラムを使用した。移動相はH2O(0.1%TFA)及びアセトニトリル(0.1%TFA)である。分析用分離流速は1ml/min、分取用分離流速は4ml/minに調節する。
【0056】
薄層クロマトグラフィー(TLC)は厚さ0.2mmのMerckシリカゲルプレートで実施した。
【0057】
カラムクロマトグラフィーは粒度0.063〜0.200mmのシリカゲル60 Merckで実施した。
【0058】
カラムクロマトグラフィーの検出は紫外線(254nm)を用いるか、アミン又はアミドの検出にはニンヒドリンを用いて150℃に加熱しながらニンヒドリンのエタノール溶液(40mg/100ml EtOH)を噴霧(弱いスプレー)し、第1級アミンの検出にはフルオレサミンを用いて溶液(40mg/100mlアセトン)を噴霧し、あるいはヨードを用いてヨード粉末でプレートを覆うことにより実施した。
【0059】
カラムクロマトグラフィーは粒度0.063〜0.200mmのシリカゲル60 Merckで実施した。
d)固相合成SPPS法
固相合成は手動製造用SPPSペプチド合成用手動反応器で実施し、撹拌機はFlask Shaker モデルA5−6021である。SPPSでポリアミンの固相結合及びポリアミンの保護後にKaiser試験を行う[Kaiser,E.,Colescolt,D.L.,Bossiner,C.D.及びCook,P.I. Anal.Biochem.34(2),595(1970)]。実施例でSPPSに使用した樹脂はスイス、NOVABIOCHEM製クロロトリチルクロリド樹脂である。
2.一般操作方法
a)(N,N,N’,N’−テトラアミノプロピル)−1−4−ジアミノブタンの製造により例示する対称ポリアミンの合成
2リットル容三頚丸底フラスコに1−4−ジアミノブタン147gと脱イオン水1000mlを仕込む。溶液を磁気撹拌する。温度を38℃に維持しながら等圧フラスコによりアクリロニトリル443gを1時間かけて加える。次いで還流冷却器を取り付け、1時間かけて混合物を水浴で80℃まで昇温させる。フルオレサミン試験は陰性であり、過剰のアクリロニトリルを40℃で減圧蒸発させる。
【0060】
2相が得られる。下部の有機相を分離し、水300mlで洗浄し、1000ml容丸底フラスコに移す。水/メタノール(1:1v/v)混合物170mlを加える。一晩結晶化させる。翌日、結晶を細孔度3の500ml容ガラス濾過器で濾過する。
【0061】
ケーキをガラス濾過器でメタノール(2×170ml)とエーテル(2×150ml)で洗浄する。生成物を減圧デシケーター(26mm)で一晩連続乾燥する。こうして生成物461g(収率93%)が得られる。生成物をNMRとMSにより分析した処、分析値は整合していた。生成物を精製せずに水素化する。
【0062】
1リットル容イノックスオートクレーブに上記ポリニトリル30g(0.1モル)を仕込む。同時にビーカーでエタノール140ml(95%)とNaOH8g(0.2モル)の溶液を調製する。NaOHが可溶化したらこの溶液をオートクレーブに加える。オートクレーブに窒素を通し、炭素担持ラネーニッケル8mlを加える。オートクレーブを閉じる。初期水素化圧は52気圧であり、周囲温度で5時間かけて28.5気圧まで下げる。懸濁液を濾紙で濾過し、濾渣をエタノール(2×25ml)で洗浄し、濾液を減圧乾燥して濃縮する。油状物を水30mlと混合し、CH2Cl2 100mlで抽出する。有機相をMgSO4で乾燥し、濾過した後、減圧蒸発させる。黄色っぽい液体油状物(27g、収率85%)が得られる。
【0063】
生成物をTLC(単スポット)、NMR及びMSにより分析した処、分析値は整合していた。生成物を精製せずに使用する。
b)方法A:酸基のポリマー支持体固定
クロロトリチルクロリド樹脂(5g、1.2mmol Cl/g樹脂)をSPPS反応器に仕込み、CH2Cl2 50mlを加え、混合物を5分間撹拌する。ブロモ酢酸(1.05g、7mmol)、次いでDIEA(0.95ml,7.5mmol)を加える。反応器を周囲温度で2時間撹拌する。液体を濾過し、樹脂をCH2Cl2とiPrOH(10×50)及びMeOH(2×50ml)で洗浄する。最後に樹脂を窒素流下に乾燥する。
c)方法B:ポリアミンとブロモアセチル樹脂の反応
ポリアミン(10モル倍)をCH2Cl2 50mlに溶かし、方法Aにより得られた生成物を収容する反応器に加える。反応器を周囲温度で2時間撹拌する。溶媒を濾過し、樹脂をCH2Cl2とiPrOH(10×50ml)で洗浄すると、Kaiser試験は陽性である。
d)樹脂上のポリアミノ酸の保護
方法C:
ジ−tert−ブチルジカーボネート(48mmol)とDIEA(50mmol)をCH2Cl2(50ml)に溶かし、方法Bにより得られた生成物を収容する反応器に加える。反応器を一晩撹拌する。翌日、Kaiser試験は陰性である。溶媒を濾過し、樹脂をCH2Cl2とiPrOH(10×50ml)、MeOH(2×50ml)及びエーテル(2×50ml)で交互に洗浄する。樹脂を窒素流下に乾燥する。Kaiser試験は依然として陰性である。
方法D:
方法Bにより得られた樹脂(1.5g)を丸底フラスコに入れ、CH2Cl2(20ml)、次いでDIEA(20mmol)を加える。混合物を磁気撹拌し、クロロギ酸ベンジル(14mmol)を5分間かけて滴下する。pHはDIEAを加えて11に維持する。一晩後、樹脂をSPPS反応器に移し、濾過し、CH2Cl2とiPrOH(10×20ml)及びエーテル(2×20ml)で交互に洗浄する。樹脂を窒素流下に乾燥する。
e)方法E:樹脂からの保護ポリアミノ酸の分離
磁気棒を備える250ml容丸底フラスコに、方法C及びDにより得られた樹脂を加える。CH2Cl2 50mlとCF3CH2OH 25mlからなる溶液を加え、混合物を2時間撹拌する。溶液を濾過し、樹脂をCH2Cl2(2×10ml)で洗浄し、こうして得られた有機相を集めて減圧蒸発させる。次に、CHCl3/MeOH(9:1)を溶離剤としてSiO2上でフラッシュクロマトグラフィーにより生成物を精製する。生成物を含むフラクションをTLCにより同定する。(詳細については下記実施例参照。)
f)方法F:アミノ酸とジリピジルアミンの結合
Boc−アミノ酸(10mmol)とC12〜C22ジリピジルアミン(10mmol)を250ml容丸底フラスコに加える。CHCl3(100ml)を加え、完全に溶解するまで混合物を撹拌する。次いでTEA(30mmol)とBOP(33mmol)を加える。TEAを加えてpHを10に維持し、反応物を2時間撹拌する。反応の完了後(TLC)、クロロホルムを蒸発させ、固形分を酢酸エチル(300ml)にとる。有機相をKHSO4(4×100ml)、NaHCO3(4×100ml)及びNaCl(4×100ml)で洗浄する。有機相をMgSO4で乾燥し、濾過し、減圧蒸発させる。生成物をTLC、NMR及びMSにより分析し、精製せずに使用する。収率は約90%である。
g)保護ポリアミノ酸とジリピジルアミド酸の結合と、Boc及びZ保護基の分離
方法G:
方法Fにより得られた生成物(9mmol)を磁気棒を備える丸底フラスコに入れ、冷TFA(4℃)を加える(30ml)。溶液を1時間撹拌する。TFAを減圧蒸発させる。DMF(70ml)を加えて生成物を溶かす。TEA(30mmol)を加えた後、方法Eにより得られた保護ポリアミノ酸(9mmol)を加える。pHを10に調整し、BOP(33mmol)を加える。溶液を2時間撹拌後、TLCを行う。結合の完了後(TLC)、KHSO4溶液(700ml)を加え、生成物を酢酸エチル(3×100ml)で抽出する。有機相をKHSO4(3×50)、NaHCO4(3×50)及びNaCl(3×50ml)で洗浄し、MgSO4で乾燥し、濾過し、減圧蒸発させる。生成物をNMR、TLC及びMSにより分析し、予備精製せずに脱保護する。TFA(50ml)を生成物に加え、溶液を1.5時間撹拌し、TFAを蒸発させる。生成物がTFAで分離可能なZ又はClZ基をまだ含んでいる場合には、方法Hを直接実施する。最終生成物を準分取HPLCにより精製する(実施例参照)。
方法H:
方法Gにより得られた生成物がZ又はClZ基を含んでいる場合には、磁気棒を備える丸底フラスコに入れ、10ml MeOH/g生成物に溶かす。Pd/C(10%、1g/g生成物)とギ酸アンモニウム(1g/g生成物)を周囲温度で加える。HPLCにより水素化を行う。2時間後に反応を終了し、混合物を濾過し、濾渣をMeOH 10mlで洗浄する。軟蒸留水を加え、溶液を凝固させ、凍結乾燥する。最終生成物を分取HPLCにより精製する。
h)方法I:Boc保護基の脱保護
丸底フラスコ中のBoc基(1mmol)を含む生成物にトリフルオロ酢酸(50ml)を加える。溶液を1.5時間撹拌し、TFAを蒸発させる。アミンを完全に脱保護し、精製せずに結合に使用する。
B−生物学的試験材料及び方法
1.in vitro遺伝子導入に使用したプラスミド
プラスミドpCMV−LUCは、ベクタープラスミドpcDNA3(Invitrogen)から抽出したヒトサイトメガロウイルス(CMV)のプロモーターを含むMlu I−Hind IIIフラグメントの挿入により、プラスミドpGL2−Basic Vector(Promega)又はプラスミドpGL2−Control Vector(Promega)から誘導される構築物である。
2.トランスフェクションに使用した溶液の調製プロトコール
本発明に記載する生成物はエタノール又は水に20mMに溶かした後、最終エタノール濃度が10%未満となるように水で希釈する。
【0064】
生理的血清(0.15M NaCl)で希釈した核酸溶液を1/1(v/v)の比でリポフェクタント溶液に加える。ボルテックス均質化と周囲温度で15分間のインキュベーション後、DNA/リポフェクタント溶液を最終濃度9%(v/v)でウェルに分配し、無タンパク質(血清)成長培地で洗浄し、無血清又は非無血清培地で再培養した。
C−in vivoアッセイ材料
1.材料
a)実験モデル:
−雌成体(>8週)マウスC57/BL6。
−腫瘍フラグメントを脇腹の皮下に移植して動物間で継代することにより得られる3LL(ルイス肺癌)型腫瘍。
b)使用したプラスミド:
pXL2622:ルシフェラーゼをコードする遺伝子の上流にpCDNA3(Invitrogen)から抽出したサイトメガロウイルス(CMV)のプロモーターを挿入することにより、基本プラスミドpGL2(Promega)から誘導される。このプラスミドはPEG沈殿法(Ausubel)により得られ、 DNA約10μg/μlの濃度で4℃で10mM Tris,1mM EDTA,pH8中に保存する。
2.プロトコール
注射溶液:まずトランスフェクトしようとするDNAを緩衝液に可溶化した後、配列番号1のペプチド(KTPKKAKKP)2を加え、20分後に高濃度(20又は40mM)のカチオン脂質溶液を混合物に加える。全生成物の添加後、混合物はDNA(最終濃度0.5mg/ml)、ペプチド(0.75mg/ml)及びカチオン脂質以外に、150mM NaCl、5% D−グルコース及び5mM MES,pH6.2を含有している。溶液の調製から20〜30分後に注射を行う。
【実施例】
【0065】
実施例1:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlyN[(CH2)17CH3]2(6)の合成
a−{Boc−[3−(Boc−{4−[Boc−(3−Boc−アミノプロピル)アミノ]ブチル}アミノ)プロピル]アミノ}酢酸(3)の合成
方法Aにより得られた樹脂を方法B、C及びEによりスペルミンと反応させる。保護生成物をSiO2上でクロマトグラフィーにより精製する。収率は40%である。
TLC:Rf=0.32(CHCl3/MeOH,9:1)。
HPLC,Rt=4.22分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,d(ppm)):1.40(4s,36H:C(CH3)3); 1.46(mt,4H:ブチルの中心のCH2CH2); 1.64及び1.74(2mt,各2H:プロピルの中心のCH2); 2.96(t,J=7Hz,2H:CH2NCOO); 3.15(mt,8H:CH2NCH2); 3.23(t,J=7.5Hz,2H:CH2NCOO); 3.83(s,2H:OCONCH2COO)。
MH+:661。
b−{Z−[3−(Z−{4−[Z−(3−Z−アミノプロピル)アミノ]ブチル}アミノ)プロピル]アミノ}酢酸(4)の合成
方法Aにより得られた樹脂を方法B、C及びDによりスペルミンと反応させる。保護生成物をSiO2上でクロマトグラフィーにより精製する。収率は20%である。
TLC:Rf=0.85(CHCl3/MeOH,8:2)。
HPLC,Rt=6.92分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度413K,d(ppm)):1.49(mt,4H:ブチルの中心のCH2CH2); 1.74及び1.81(2mt,各2H:プロピルの中心のCH2); 3.07(q,J=7Hz,2H:CH2NCOOベンジル); 3.15−3.30(mt,8H:CH2NCH2); 3.33(t,J=7.5Hz,2H:NCH2COO); 3.70(s,2H:OCONCH2COO); 5.07,5.10,5.12及び5.13(4s,各2H:ArCH2OCON); 6.65(mf,1H:NHCO); 7.25−7.40(mt,20H:芳香族H)。
MH+:797。
c−Boc−Gly−ジオクタデシルアミド(5)
方法FによりBoc−Glyをジオクタデシルアミンに結合する(収率90%)。
TLC:Rf=0.9(CHCl3/MeOH,9:1)。
MH+:679。
1HNMRスペクトル(300MHz,CDCl3,d(ppm)):0.89(t,J=7Hz,6H:CH3); 1.29(mt,60H;脂肪鎖の中心のCH2); 1.49(s,9H:C(CH3)3); 1.55(mt,4H;各脂肪鎖の1CH2); 3.15及び3.33(2t,J=7.5Hz,各2H:脂肪鎖のNCH2); 3.95(d,J=5Hz,2H:OCONCH2CON); 5.57(mf,1H:CONH)。
d−H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlyN[(CH2)18]2(6)
生成物(3)と(5)又は(4)と(5)を方法Gにより結合する。生成物をBoc保護生成物は方法G、Z保護生成物は方法Hに記載したように脱保護する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=15.35分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
BYK20531HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,温度300K,d(ppm)):0.83(t,J=7Hz,6H:CH3); 1.23(mt,60H:脂肪鎖の中心のCH2); 1.43及び1.53(2mt,各2H:各脂肪鎖の1CH2); 1.63(m,4H:ブチルの中心のCH2CH2); 1.96(mt,4H:プロピルの中心のCH2); 2.93,3.00及び3.22(3mt,合計16H:NCH2); 3.83(s,2H:NCH2CON); 4.03(s,2H:CONCH2CON)。
MH+:821。
【0066】
実施例2:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2CON[(CH2)18]2(7)の合成
生成物(3)を方法Fによりジオクタデシルアミンと結合し、方法Gにより脱保護する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=15.2分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
1HNMRスペクトル(400MHz,CF3COOD 2/3とCD3COODd4 1/3の混合物中,d(ppm)):0.78(t,J=7Hz,6H:CH3); 1.20(mt,60H:脂肪鎖の中心のCH2); 1.52(mt,4H:各脂肪鎖の1CH2); 1.80(mt,4H:ブチルの中心のCH2CH2); 2.23及び2.32(2mt,各2H:プロピルの中心のCH2); 3.10−3.40(3mt,合計16H:NCH2); 4.15(s,2H:NCH2CON)。
MH+:764。
【0067】
実施例3:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COArgN[(CH2)18]2(9)の合成
a−Boc−Arg(Z2)ジオクタデシルアミド(8)
本生成物は、方法FによりBocArg(Z2)とジオクタデシルアミンを結合して合成する(収率91%)。
TLC:Rf=0.9(CHCl3/MeOH,9:1)。
MH+:1046。
b−H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COArgN[(CH2)18]2(9)
生成物(3)又は(4)を方法Gにより生成物(8)と結合し、方法G(Boc)及び/又はH(Z)により脱保護する。最終生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=13.83分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6,d(ppm))):0.90(t,J=7Hz,6H:CH3);1.28(mt,60H:脂肪鎖のCH2);1.40−1.80(mt,12H:CH2);1.93(m,4H:プロピルの中心のCH2);2.80−3.10(3mt,16H:NCH2及び脂肪鎖のNCH2);3.42(mt,2H:アミドのCH2N);3.77(mt,2H:NCH2CON);4.67(mt,1H:NCHCON);6.80−7.50(広幅mf,2H:NH2);7.78,7.92,8.80及び9.03(夫々mt及び3mf、夫々1H,2H,4H及び1H:CONH,NH及びNH2)。MH+:920。
【0068】
実施例4:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COArg(Z)2N[(CH2)17CH3]2(10)の合成
生成物(3)を方法Gにより生成物(8)と結合し、同一方法によりBoc基を分離する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=17.75分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO,d(ppm)):0.87(t,J=7Hz,6H:CH3);1.25(mt,60H:脂肪鎖の中心のCH2);1.40及び1.57(2mt,各2H:各脂肪鎖の1CH2);1.65(mt,8H:ブチルの中心のCH2CH2);1.95(mt,4H:プロピルの中心のCH2);2.85−3.05(mt,合計14H:NCH2);3.23(t,J=7.5Hz,2H:NCH2);3.75(s,2H:NCH2CON);3.85及び3.95(2mt,各1H:CH2NC);4.67(mt,1H:CONCHCON);5.07及び5.25(夫々限界AB及びs,J=13.5Hz,各2H:NCOOCH2Ar);7.25−7.45(mt,10H:芳香族H);7.95,8.85,9.90及び9.20(4mf:互換H)。
MH+:1188。
【0069】
実施例5:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(ローダミン)N[(CH2)17CH3]2(13)の合成
a−Boc−Lys(Z)ジオクタデシルアミド(11)
本生成物は、方法FによりBocLys(ClZ)をジオクタデシルアミンと結合することにより合成した(収率89%)。
TLC:Rf=0.92(CHCl3/MeOH,9:1)。
MH+:918。
b−BocHN(CH2)3NBoc(CH2)4NBoc(CH2)3NBocCH2COLys(ClZ)N[(CH2)17CH3]2(12)
(Bocを脱保護せずに)生成物(3)を方法Gにより生成物(11)に結合する。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度423K,d(ppm)):0.92(t,J=6.5Hz,6H:CH3);1.32(mt,60H:脂肪鎖の中心のCH2);1.44(2s,合計36H:C(CH3)3);1.50−1.80(mt,16H:各脂肪鎖の1CH2,ブチルの中心のCH2CH2,CH2CH2CH2及びプロピルの中心のCH2);3.00(q,J=6.5Hz,2H:OCONCH2);3.05(q,J=6.5Hz,2H:CH2NCOO);3.15−3.40(mt,14H:脂肪鎖のNCH2,CH2NCH2及びCH2NCH2CH2N);3.80(s,2H:OCONCH2CON);4.75(mt,1H:CONCHCON);5.15(s,2H:NCOOCH2Ar);5.97及び6.53(2mt,各1H:OCONH及びNHCOO);7.08(d,J=7.5Hz,1H:CONH);7.30−7.50(mt,4H:芳香族H)。
c−H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(ローダミン)N[(CH2)17CH3]2(13)
生成物(12)のリジン上のClZ基を方法Hにより分離し、こうして得られた生成物を減圧乾燥し、エーテルにとり、NaHCO3とNaClで濯ぐ。エーテルをMgSO4で乾燥し、減圧蒸発させる。
【0070】
脱保護生成物77mg(60μM)をMeOH 3mlに溶かし、DIEA(64μL)を加えた後、テトラメチルローダミンイソチオシアネート(30mg,68μM)を加え、溶液を17時間撹拌し、反応をTLCによりモニターする。翌日、溶液を減圧乾燥して濃縮する。次にTFA(4ml)を加え、1時間撹拌する。TFAを蒸発させ、粗生成物を準分取HPCLにより精製する(最終収率30%)。
TLC:Rf=0.05(MeOH)。
HPLC(準分取),Rt=61.55分(H2O/MeCN:3分[100/0]、3〜45分[0/100]、45〜140分[0/100])。
MH+:1335。
【0071】
実施例6:
H2N(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(ビオチニル)N[(CH2)17CH3]2(14)の合成
生成物(12)を方法Hにより脱保護し(271mg,0.21mmol)、DMF(10ml)に溶かす。DIEA(0.11ml)を加えた後、ビオチン(56.4mg,0.23mmol)とBOP(102mg,0.23mmol;pHは10(DIEA)に維持)を加え、反応の終了をフルオレサミン試験により確認する。生成物を方法Fに記載したように回収し、精製せずにTFA(5ml)で1時間脱保護する。TFAを蒸発させ、生成物を準分取HPLCにより精製する(収率50%)。
HPLC,Rt=13.12分(H2O/MeCN:3分[40/60]、3〜20分[0/100]、35分[0/100])。
MH+:1118。
【0072】
実施例7:
{H2N(CH2)3}2N(CH2)4N{(CH2)3NH2}(CH2)3NHCH2COGlyN[(CH2)17CH3]2(16)の合成
a−{BocNH(CH2)3}2N(CH2)4N{(CH2)3NHBoc}(CH2)3NBocCH2COOH(15)
生成物(1)を方法Bによりポリマーに固定し、方法Cにより保護し、方法Eにより樹脂から分離する。生成物をSiO2上で精製する(収率35%)。
TLC:Rf=0.2(CHCl3/MeOH,8:2)。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,温度433K,d(ppm)):1.42(s,36H:C(CH3)3);1.56(mt,4H:ブチルの中心のCH2CH2);1.65−1.85(mt,8H:プロピルの中心のCH2);2.76(mt,12H:CH2N(CH2)2);3.06(t,J=6.5Hz,6H:OCONCH2);3.29(mt,2H:NCH2);3.86(s,2H:OCONCH2COO)。
MH+:775。
b−{H2N(CH2)3}2N(CH2)4N{(CH2)3NH2}(CH2)3NHCH2COGlyN[(CH2)17CH3]2(16)
生成物(15)を方法Gにより生成物(5)と結合する。生成物を方法Gにより脱保護し、準分取HPLCにより精製し、フラクションを分析HPLCにより分析し、凍結乾燥する(収率55%)。
HPLC(準分取),Rt=38.72分(H2O/MeCN:10分[100/0]、10〜45分[0/100]、45〜140分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度386K,d(ppm)):0.90(t,J=7Hz,6H:CH3);1.30(m,60H:脂肪鎖の中心のCH2);1.55(mt,4H:各脂肪鎖の1CH2);1.65(mt,4H:ブチルの中心のCH2CH2);1.97(mt,8H:プロピルの中心のCH2);2.80−3.05,3.06及び3.28(夫々mt及び2t,J=7.5Hz,18H,2H及び4H:NCH2);3.80(s,2H:NCH2CON);4.03(d,J=5.5Hz,2H:CONCH2CON);6.00−9.00(広幅mf:NH2及びNH);8.27(mt,1H:CONH)。
MH+:935。
【0073】
実施例8:
{H2N(CH2)3}2N(CH2)4N{(CH2)3NH2}(CH2)3NHCH2CON[(CH2)17CH3]2(17)の合成
(3)の代わりに生成物(15)を使用することにより生成物(7)と同様に合成する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析し、凍結乾燥する。
HPLC(準分取),Rt=38分(H2O/MeCN:10分[100/0]、10〜45分[0/100]、45〜140分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,d(ppm)):0.88(t,J=7Hz,6H:CH3);1.29(mt,60H:脂肪鎖の中心のCH2);1.52(mt,4H:各脂肪鎖の1CH2);1.68(mt,4H:ブチルの中心のCH2CH2);1.90−2,10(mt,8H:プロピルの中心のCH2);2.90,2.95−3.15,3.18及び3.15(夫々t,mt及び広幅2t,J=7.5Hz,合計24H:NCH2);4.02(s,2H:NCH2CON)。
MH+:878。
【0074】
実施例9:
{H2N(CH2)2}2N(CH2)2NHCH2COGlyN[(CH2)17CH3]2(19)の合成
a−{BocNH(CH2)2}2N(CH2)2NBocCH2COOH(18)
生成物(1)の代わりにトリス(アミノエチル)アミンを使用することにより生成物(15)と同様に合成する(収率29%)。
TLC:Rf=0.55(CHCl3/MeOH,8:2)。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度393K,d(ppm)):1.44(s,27H:C(CH3)3);2.58(t,J=6.5Hz,4H:CH2NCH2);2.66(t,J=7Hz,2H:NCH2);
3.04(q,J=6.5Hz,4H:OCONCH2);
3.28(t,J=7Hz,2H:OCONCH2);
3.76(s,2H:OCONCH2COO);6.06(mf,2H:CONH)。
MH+:505。
b−{H2N(CH2)2}2N(CH2)2NHCH2COGlyN[(CH2)17CH3]2(19)
(15)の代わりに(18)を使用することにより生成物(17)と同様に合成する(収率65%)。
HPLC,Rt=122分(H2O/MeCN:10分[100/0]、10〜45分[0/100]、45〜140分[0/100])。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,d(ppm)):0.87(t,J=7Hz,6H:CH3);1.15−1.35(mt,60H:脂肪鎖の中心のCH2);1.45及び1.55(2mt,各2H:各脂肪鎖の1CH2);2.64(t,J=5.5Hz,4H:CH2NCH2);2.75(t,J=6Hz,2H:NCH2);2.95(t,J=5.5Hz,4H:NCH2);3.08(t,J=6Hz,2H:NCH2);3.25(mt,4H:脂肪鎖のNCH2); 3.88(s,2H:NCH2CON);4.06(d,J=5Hz,2H:CONCH2CON);7.75(残留広幅mf:NH);8.68(残留t,J=5Hz:CONH)。
MH+:765。
【0075】
実施例10:
{H2N(CH2)2}2N(CH2)2NHCH2CON[(CH2)17CH3]2(20)の合成
(5)の代わりにジオクタデシルアミンを使用することにより生成物(19)と同様に合成する(収率73%)。
HPLC,Rt=100.1分(H2O/MeCN:10分[100/0]、10〜45分[0/100]、45〜140分[0/100])。
MH+:708。
【0076】
実施例11:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLysN[(CH2)17CH3]2(21)(RPR127888A)の合成
生成物(12)を方法H(Cl−Z)、次いで方法Iにより脱保護する。最終生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=11.76分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:892。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度393K,d(ppm)):0.91(t,J=7Hz,6H:脂肪鎖のCH3);1.31(mt,60H:脂肪鎖の中心の(CH2)15);1.35−1.75(mt,10H:各脂肪鎖の1CH2,リシルの中心の(CH2)3);1.75(mt,4H:ブチルの中心の(CH2)2);2.00(mt,4H:プロピルの中心のCH2);2.82,2.98,3.06及び3.10−3.50(夫々2t,mt及び2mf、J=7Hz,合計18H:リシルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.62(s,2H:NCH2CON);4.73(q,J=7Hz,1H:リシルのCONCHCON);8.18(d,J=7Hz,1H:リシルのCONH)。
【0077】
実施例12:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(Cl−Z)N[(CH2)17CH3]2(22)(RPR122759A)の合成
生成物(12)を方法Iにより脱保護する。最終生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=16.79分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1060。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度373K,d(ppm)):0.91(t,J=7Hz,6H:脂肪鎖のCH3);1.31(mt,60H:脂肪鎖の中心の(CH2)15);1.30−1.75(mt,10H:各脂肪鎖の1CH2,リシルの中心の(CH2)3);1.72(mt,4H:ブチルの中心の(CH2)2);1.95(mt,4H:プロピルのCH2);2.98,3.06及び2.90−3.50(夫々2mt及びmf,合計18H:リシルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.59(s,2H:NCH2CON);4.75(q,J=7Hz,1H:リシルのCONCHCON);5.16(s,2H:COOCH2Ar);6.85(mf,1H:OCONH);7.35−7.55(mt,5H:芳香族H);8.15(mf,1H:リシルのCONH)。
【0078】
実施例13:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(CHO)N[(CH2)17CH3]2(24)(RPR122760A)の合成
a−NHBoc(CH2)3NBoc(CH2)4NBoc(CH2)3NBocCH2COLysN[(CH2)17CH3]2(23)
生成物(12)を方法H(Cl−Z)により脱保護し(収率65%)、精製せずに使用する。
HPLC,Rt=20.82分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
b−NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys(CHO)N[(CH2)17CH3]2(24)
生成物(23)を方法Gによりギ酸と結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=13.60分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:920。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度383K,d(ppm)):0.92(t,J=7Hz,6H:脂肪鎖のCH3);1.31(mt,60H:脂肪鎖の中心の(CH2)15);1.35−1.70(mt,10H:各脂肪鎖の1CH2,リシルの中心の(CH2)3);1.73(mt,4H:ブチルの中心の(CH2)2);1.98(mt,4H:プロピルのCH2);2.85−3.50(mt,18H:リシルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2); 3.62(s,2H:NCH2CON);4.75(mt,1H:リシルのCONCHCON);7.60(mf,1H:CONH);8.05(広幅s,1H:アルデヒドのCH);8.18(mf,1H:リシルのCONH)。
【0079】
実施例14:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys[コレステリル]N[(CH2)17CH3]2(25)(RPR128142A)の合成
(BOP試薬を使用せずに)生成物(23)を方法Gによりクロロギ酸コレステリルと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=21.66分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1304。
1HNMRスペクトル(600MHz,(CD3)2SO d6,d(ppm)):0.68及び0.98(2s,各3H:コレステリルの18位CH3及び19位CH3);0.86(mt,12H:脂肪鎖のCH3並びにコレステリルの26及び27位CH3);0.91(d,J=7Hz,3H:コレステリルの21位CH3);1.31(mt,60H:脂肪鎖の中心の(CH2)15);0.80−2.30(mt,42H:各脂肪鎖の1CH2,コレステリルの1、2、4、7、11、12、15、16、22、23及び24位のCH2,コレステリルの8、9、14、17、20及び25位のCH,リシルの中心の(CH2)3並びにプロピルのCH2);1.65(mt,4H:ブチルの中心の(CH2)2);2.88及び2.96(2mt,合計14H:リシルのNCH2,ブチルのNCH2,プロピルのNCH2);3.20−3.50(mt,4H:脂肪鎖のNCH2);3.64(s,2H:NCH2CON);4.23(mt,1H:コレステリルの3位CH);4.63(mt,1H:リシルのCONCHCON);5.30(mt,1H:コレステリルの6位CH);6.98(mt,1H:NHCOO);7.90(mt,1H:リシルのCONH);8.60−9.01(互換mf)。
【0080】
実施例15:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COLys[アラキドニル]N[(CH2)17CH3]2(26)(RPR130605)の合成
生成物(23)を方法Gにより窒素流下で遮光下にアラキドン酸と結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=20.67分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1177。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,温度393K,d(ppm)):0.90(t,J=7Hz,6H:脂肪鎖のCH3); 0.91(t,J=7Hz,3H:アラキドニルのCH3);
1.31(mt,60H:脂肪鎖の中心の(CH2)15);
1.35−1.75(mt,18H:各脂肪鎖の1CH2,アラキドニルの中心の(CH2)3,アラキドニルの中心のCH2及びリシルの中心の(CH2)3);1.75(mt,4H:ブチルの中心の(CH2)2);2.02(mt,4H:プロピルのCH2);2.10(mt,6H:COCH2及びアラキドニルの2個の=CCH2);2.80,2.97,3.06及び3.10−3.50(夫々mt,t,mt及び2mf,J=7Hz,合計24H:アラキドニルの=CCH2C=,リシルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.62(s,2H:NCH2CON);4.73(dd,J=8及び5Hz,1H:リシルのCONCHCON);5.38(mt,8H:アラキドニルのCH=CH)。
【0081】
実施例16:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGluN[(CH2)17CH3]2(28)(RPR126097A)の合成
a−Boc−Glu(O−Bz)−ジオクタデシルアミン(27)
本生成物は方法FによりBoc−Glu(OBz)とジオクタデシルアミンを結合して合成する(収率90%)。
TLC:Rf=0.88(CHCl3/MeOH,9:1)。
b−NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGluN[(CH2)17CH3]2(28)
生成物(3)又は(4)を方法G、次いで方法H(Cl−Z脱保護)により生成物(27)と結合する。最終生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=14.64分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:893。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度383K,d(ppm)):0.90(t,J=7Hz,6H:脂肪鎖のCH3);1.30(mt,60H:脂肪鎖の中心の(CH2)15);1.56(mf,4H:各脂肪鎖の1CH2);1.60−2.00(mt,2H:グルタリルの中心のCH2);1.73(mt,4H:ブチルの中心のCH2)2);1.98(mt,4H:プロピルのCH2);2.32(t,J=7Hz,2H:グルタリルのCOCH2);3.00,3.06及び3.45(夫々t及び2mt,J=7Hz,合計16H:ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.65(広幅s,2H:NCH2CON);4.85(mt,1H:グルタリルのCONCHCON);8.19(広幅s,1H:グルタリルのCONH)。
【0082】
実施例17:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(O−Bz)N[(CH2)17CH3]2(29)(RPR123027A)の合成
生成物(3)を方法Gにより生成物(27)と結合し、脱保護(Boc)する。最終生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=16.02分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:983。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度413K,d(ppm)):0.89(t,J=7Hz,6H:脂肪鎖のCH3);1.30(mt,60H:脂肪鎖の中心の(CH2)15);1.55(mf,4H:各脂肪鎖の1CH2);1.72(mt,4H:ブチルの中心の(CH2)2);1.75−2.00(mt,2H:グルタリルの中心のCH2);1.99(mt,4H:プロピルのCH2);2.47(t,J=7Hz,2H:グルタリルのCOCH2);2.95,3.05及び3.40(3mt,合計16H:ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.62(広幅s,2H:NCH2CON);4.85(mt,1H:グルタリルのCONCHCON);5.14(限界AB,J=12Hz,2H:ベンジルのCH2);7.35(mt,5H:ベンジルの芳香族H);8.23(mf,1H:グルタリルのCONH)。
【0083】
実施例18:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(ガラクトサミド)N[(CH2)17CH3]2(31)(RPR130596A)の合成
a−BocNH(CH2)3NBoc(CH2)4NBoc(CH2)3NBocCH2COGluN[(CH2)17CH3]2(30)
生成物(3)を生成物(27)に結合し、側鎖のOBz保護基を方法H(Cl−Z)により分離し、精製せずに使用する。
HPLC,Rt=22.84分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1293。
b−NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(ガラクトサミド)N[(CH2)17CH3]2(31)
生成物(30)を方法Gにより塩酸D(+)−ガラクトサミンと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=13.71分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1054。
【0084】
実施例19:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(ガラクトサミド)N[(CH2)17CH3]2(32)(RPR130595A)の合成
生成物(30)を方法Gにより塩酸D(+)−ガラクトサミンと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=12.27分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1054。
【0085】
実施例20:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(マンノサミド)N[(CH2)17CH3]2(33)(RPR130598A)の合成
生成物(30)を方法Gにより塩酸D(+)−マンノサミンと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=12.98分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1054。
【0086】
実施例21:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(N(CH3)2)N[(CH2)17CH3]2(34)(RPR131111A)の合成
生成物(30)を方法Gによりジメチルアミンと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=14.44分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:920。
1HNMRスペクトル(400MHz,(CD3)2SO d6,d(ppm)):0.89(t,J=7Hz,6H:脂肪鎖のCH3);1.25(mt,60H:脂肪鎖の中心の(CH2)15);1.43及び1.60(2mt,各2H:各脂肪鎖の1CH2);1.65(mt,4H:ブチルの中心の(CH2)2);1.65及び1.85−2.00(2mt,各1H:グルタリルの中心のCH2);1.95(mt,4H:プロピルのCH2);2.32(限界AB,2H:グルタリルのCOCH2);2.80及び2.92(2s,各3H:CON(CH3)2);2.85−3.05(mt,12H:ブチルのNCH2,プロピルのNCH2);3.00,3.22,3.45及び3.58(4mt,各1H:脂肪鎖のNCH2);3.78(AB,J=16Hz,2H:NCH2CON);4.75(mt,1H:グルタリルのCONCHCON);8.72(d,J=7.5Hz,1H:グルタリルのCONH);8.85及び8.90−9.15(互換mf)。
【0087】
実施例22:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlyN[(CH2)11CH3]2(35)(RPR122767A)の合成
ジオクタデシルアミンの代わりにジドデシルアミンを使用する以外は生成物(6)と同様に合成する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=9.54分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:653。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度403K,d(ppm)):0.93(t,J=7Hz,6H:脂肪鎖のCH3);1.33(mt,36H:脂肪鎖の中心の(CH2)9);1.58(mt,4H:各脂肪鎖の1CH2);1.75(mt,4H:ブチルの中心の(CH2)2);1.95及び2.00(2mt,各2H:プロピルの中心のCH2);2.98及び3.00(2mt,合計12H:ブチルのNCH2及びプロピルのNCH2);3.30(t,J=7Hz,4H:脂肪鎖のNCH2);3.58(s,2H:NCH2CON);4.05(s,2H:グルシルのCONCH2CON)。
【0088】
実施例23:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlyN[(CH2)12CH3]2(36)(RPR122774A)の合成
ジオクタデシルアミンの代わりにジトリドデシルアミンを使用する以外は生成物(6)と同様に合成する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=10.64分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:681。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度393K,d(ppm)):0.91(t,J=7Hz,6H:脂肪鎖のCH3);1.33(mt,40H:脂肪鎖の中心の(CH2)10);1.58(mt,4H:各脂肪鎖の1CH2);1.75(mt,4H:ブチルの中心の(CH2)2);2.00(mt,4H:プロピルの中心のCH2);2.98及び3.00(2t,J=7Hz,合計12H:ブチルのNCH2及びプロピルのNCH2);3.32(t,J=7Hz,4H:脂肪鎖のNCH2);3.65(s,2H:NCH2CON);4.06(d,J=4Hz,2H:グルシルのCONCH2CON);8.60(広幅s,1H:グルシルのCONH)。
【0089】
実施例24:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlyN[(CH2)13CH3]2(37)(RPR122766A)の合成
ジオクタデシルアミンの代わりにジテトラドデシルアミンを使用する以外は生成物(6)と同様に合成する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=9.92分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:709。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度393K,d(ppm)):0.90(t,J=7Hz,6H:脂肪鎖のCH3);1.31(mt,44H:脂肪鎖の中心の(CH2)11);1.58(mt,4H:各脂肪鎖の1CH2);1.76(mt,4H:ブチルの中心の(CH2)2);2.00(mt,4H:プロピルの中心のCH2);2.98及び3.08(夫々mt及びt,J=7Hz,合計12H:ブチルのNCH2及びプロピルのNCH2);3.30(t,J=7Hz,4H:脂肪鎖のNCH2);3.65(s,2H:NCH2CON);4.06(d,J=4Hz,2H:グルシルのCONCH2CON); 8.10(mf,1H:グルシルのCONH)。
【0090】
実施例25:
NH2(CH2)3NH(CH2)4N[(CH2)3NH2]CH2COGlyN[(CH2)17CH3]2(39)(RPR126096A)の合成
a−BocNH(CH2)3NBoc(CH2)4N[(CH2)3NHBoc]CH2CO2H(38)の合成
生成物(3)の合成中にSiO2上で精製時に副産物(38)を回収する。収率は8%である。
TLC:Rf=0.32(CHCl3/MeOH,9:1)。
MH+:561。
1HNMRスペクトル(400MHz,(CD3)2SO d6,d(ppm)):1.30−1.61(mt,4H:ブチルの中心の(CH2)2);1.40(s,27H:C(CH3)3);1.56(mt,4H:プロピルのCH2);2.68及び3.11(夫々広幅t及びt、J=7Hz,各4H:ブチルのNCH2及びプロピルのNCH2);2.90及び2.96(2q,J=7Hz,各2H:プロピルのBocNHCH2);3.18(s,2H:NCH2COO)。
b−NH2(CH2)3NH(CH2)4[N(CH2)3NH2]CH2COGlyN[(CH2)17CH3]2(39)
生成物(38)と(5)を方法Gにより結合する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。HPLC,Rt=13.60分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:821。
1HNMRスペクトル(400MHz,(CD3)2SO d6にCD3COOD d4数滴を滴下,d(ppm)):0.87(t,J=7Hz,6H:各脂肪鎖のCH3);1.28(mt,60H:脂肪鎖の中心の(CH2)15);1.46及び1.54(2mt,各2H:各脂肪鎖の1CH2);1.63(mt,4H:ブチルの中心の(CH2)2);1.91(mt,4H:プロピルのCH2);2.85−3.15(mt,12H:ブチルのNCH2及びプロピルのNCH2);3.24(mt,4H:脂肪鎖のNCH2);3.76(mf,2H:NCH2CON);4.05(広幅s,2H:グリシルのCONCH2CON)。
【0091】
実施例26:
NH2(CH2)3NH(CH2)4NH(CH2)3NH(CH2)3CON[(CH2)17CH3]2(41)(RPR122786A)の合成
a−BocNH(CH2)3NBoc(CH2)4NBoc(CH2)3NBoc(CH2)3CO2H(40)
溶液としてのNaCNBH3とコハク酸準アルデヒドの存在下にスペルミンでアルキル還元を実施することにより生成物(40)を合成する。
【0092】
200ml容丸底フラスコにスペルミン1.8g、メタノール60ml及びNaCNBH3 0.138gを仕込む。溶液を激しく磁気撹拌する。等圧フラスコによりコハク酸準アルデヒド5.5ml(15%)とメタノール30mlの溶液を100分間かけて注入する。撹拌を100分間維持する。アミンをBoc基により次のように保護する。媒質にTEA2.8mlを注いだ後、メタノール30mlに可溶化したジ−tert−ブチルジカーボネート8.8gを加える。撹拌下に一晩維持する。媒質を減圧濃縮し、生成物を酢酸エチルにとり、NaHCO33×50mlで抽出し、水相を集めてエーテル(3×100ml)で濯ぐ。KHSO4を加えて水相のpHを3まで下げ、生成物41の沈殿による濁りを確認し、混合物を酢酸エチル(3×100ml)で抽出する。有機相をMgSO4で乾燥し、減圧蒸発させる。生成物を精製せずに使用する。
b−NH2(CH2)3NH(CH2)4NH(CH2)3NH(CH2)3CON[(CH2)17CH3]2(41)
生成物(40)とジオクタデシルアミンを方法Gにより結合する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=15.04分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:792。
1HNMRスペクトル(400MHz,(CD3)2SO d6,温度383K,d(ppm)):0.85(t,J=7Hz,6H:脂肪鎖のCH3);1.22(mt,60H:脂肪鎖の中心の(CH2)15);1.48(mt,4H:各脂肪鎖の1CH2);1.72(mt,4H:ブチルの中心の(CH2)2);1.88(mt,2H:アミノペンタノイルの中心のCH2);1.99(mt,4H:プロピルのCH2);2.42(t,J=7Hz,2H:アミノペンタノイルのCOCH2);2.96,3.03及び3.22(3mt,合計18H:アミノペンタノイルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2)。
【0093】
実施例27:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2CONH(CH2)5CON[(CH2)17CH3]2(42)(RPR128506A)の合成
BocGlyの代わりにBoc6−アミノカプロン酸を使用する以外は生成物(6)と同様に合成する。生成物を準分取HPLCにより精製し、フラクションをHPLCにより分析する。
HPLC,Rt=13.94分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:877。
1HNMRスペクトル(300MHz,(CD3)2SO d6,d(ppm)):0.87(t,J=7Hz,6H:脂肪鎖のCH3);1.28(mt,60H:脂肪鎖の中心の(CH2)15);1.48(mt,10H:各脂肪鎖の1CH2及びアミノヘキサノイルの中心の(CH2)3);1.65(mt,4H:ブチルの中心の(CH2)2);1.95(mt,4H:プロピルのCH2);2.27(t,J=7Hz,2H:アミノヘキサノイルのCOCH2);2.85−3.30(mt,18H:アミノヘキサノイルのNCH2,ブチルのNCH2,プロピルのNCH2及び脂肪鎖のNCH2);3.70(広幅s,2H:NCH2CON);7.90−9.10(互換mf)。
【0094】
実施例28:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COGlu(11−アミドウンデカニルヘプタ−O−アセチルラクトース)N[(CH2)17CH3]2(43)(RPR130765A)の合成
生成物(30)を方法Gにより11−アミノウンデカニルヘプタ−O−アセチルラクトースと結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=15.91分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1680。
【0095】
実施例29:
NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COAsm(β−NAc(Ac)3)N[(CH2)17CH3]2(45)(RPR131283A)の合成
a−Fmoc−Asm−β−Glc−NAc(Ac)3−ジオクタデシルアミン(44)
本生成物は方法FによりFmoc−Asm−β−Glc−NAc(Ac)3−OHとジオクタデシルアミンを結合することにより合成する。
TLC:Rf=0.67(CHCl3/MeOH,9:1)。
HPLC,Rt=25.31分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
b−NH2(CH2)3NH(CH2)4NH(CH2)3NHCH2COAsm(β−NAc(Ac)3)N[(CH2)17CH3]2(45)
生成物(45)のFmoc基を分離する。
【0096】
生成物(45)0.7gにDMF20mlとジエチルアミン2mlを注ぐ。撹拌下に6時間維持した後、媒質を減圧濃縮する。得られた生成物を方法Gにより生成物(3)に結合する。生成物を準分取HPLCにより精製し、フラクションを分析HPLCにより分析する。
HPLC,Rt=15.35分(H2O/MeCN:3分[60/40]、3〜20分[0/100]、35分[0/100])。
MH+:1207。
【0097】
実施例30:
生成物(6)の大規模溶液合成
溶液としてのNaCNBH3とグリオキシル酸の存在下にスペルミンでアルキル還元を実施することにより生成物(6)を合成する。
【0098】
2l容丸底フラスコにスペルミン18.2g、メタノール500ml及びNaCNBH3 2gを仕込む。溶液を激しく磁気撹拌する。等圧フラスコによりメタノール300ml中のグリオキシル酸8.45gの溶液を100分間かけて注入する。撹拌を一晩維持する。アミンをBoc基により次のように保護する。媒質にTEA14mlを注いだ後、THF200mlに可溶化したジ−tert−ブチルジカーボネート100gを加える。撹拌下に一晩維持する。媒質を減圧濃縮し、生成物を酢酸エチル(250ml)にとり、KHSO4(6×100ml)、次いで飽和NaCl溶液(3×100)で濯ぎ、MgSO4で乾燥し、減圧蒸発させる。CHCl3/MeOH(9:1)を溶離剤としてシリカカラムで生成物を精製する。生成物を含むフラクションをTLCにより同定し、再び集めて減圧蒸発させると、生成物(6)10gが得られる(合計合成収率17%)。
【0099】
分析HPLC、質量スペクトル及びNMRの分析値は固相法により得られる生成物に一致する。
【0100】
実施例31:
(7)RPR120534A、(6)120535A及び(9)RPR120531Aによるトランスフェクション効率に及ぼす電荷比(アミン/リン酸)の効果
2cm2の指数増殖期の細胞[NIH 3T3、3LL又はウサギSMC]1×105個のサンプルを、5%CO2下で電荷比を変えてリポフェクタント/pCMV−LUC溶液により37℃で2時間処理し、各サンプルに核酸2μgを投与する。最終濃度8%のウシ胎児血清を添加後、CO2インキュベーターで40時間インキュベーション後にリポーター遺伝子の発現を調べる。
【0101】
ルシフェラーゼ活性はルシフェリン、補酵素A及びATPの存在下で10秒間の発光[RLU=相対光単位]により定量し、処理細胞2000個に対する割合として表す。得られた結果を図1、2及び3に示す。
【0102】
これらの図から明らかなように、脂質部分とポリアミンの間の「スペーサー」アームにグリシンが存在すると、低いナノモルカチオン脂質/μgDNA比で良好なトランスフェクション効率が得られる。
【0103】
実施例32:
(20)RPR120527A、(19)120528A、(17)RPR120526A及び(16)RPR120525Aによるトランスフェクション効率に及ぼす電荷比(アミン/リン酸)の効果
2cm2の指数増殖期のNIH 3T3細胞1×105個のサンプルを、5%CO2下で電荷比を変えてリポフェクタント/pCMV−LUC溶液により37℃で2時間処理し、各サンプルに核酸1μgを投与する。最終濃度8%のウシ胎児血清を添加後、CO2インキュベーターで40時間インキュベーション後にリポーター遺伝子の発現を調べる。
【0104】
ルシフェラーゼ活性は細胞の溶解後に得られる上清中で10秒間の発光[RLU=相対光単位]により定量し、タンパク質1mgに対する割合として表す。得られた結果を図4に示す。「スペーサー」アームにグリシン残基が存在すると有利であることは本実施例からも明白である。
【0105】
実施例33:
(6)120535、(41)RPR122786及び(42)RPR128506によるトランスフェクション効率に及ぼすスペーサー長の効果
2cm2の指数増殖期の細胞[NIH 3T3及びHeLa]1×105個のサンプルを、血清タンパク質の不在下で37℃、5%CO2下の湿潤雰囲気でカチオン脂質濃度を変えてカチオン脂質/pCMV−Luc混合物により2時間処理する。次いで細胞増殖培地に最終濃度8%のウシ胎児血清を加え、CO2インキュベーターで更に40時間インキュベーション後にトランス遺伝子の発現を測定する。
【0106】
スペーサーの構造特性は以下の通りである。
【0107】
【表1】
【0108】
下表Iは得られた結果を示し、試験材料の各々で得られた最大効率を表す。
【0109】
【表2】
【0110】
トランスフェクション効率は処理細胞2103個当たりのRLU/10秒として与える。括弧内はナノモル脂質/μgDNA比を示す。
【0111】
実験1及び2では異なるプラスミドを使用し、実験1及び2で夫々細胞1105個当たりDNA1μg及び0.5μgの用量とする。
【0112】
実施例34:
(6)RPR120535によるトランスフェクション効率に及ぼすスペーサー構造の効果
補助細胞系(3LL細胞)を導入した以外は上記実施例に記載したと同一の実験条件下で、Arg型、Lys型又はGlu型の基で「スペーサー」を置換することにより修飾したカチオン脂質(6)RPR120535を用いて得られたトランスフェクション効率を比較した。
【0113】
種々のスペーサーの構造は以下の通りである。
【0114】
【表3】
【0115】
トランスフェクション効率は処理細胞2103個当たりのRLU/10秒として与える。
実験1及び2では異なるプラスミドを使用し、実験1及び2で夫々細胞1105個当たりDNA0.5μg及び1μgの用量とした。結果を分析した処、該当細胞に応じて、好ましくは置換したアミノ酸鎖の存在は良好なトランスフェクション効率をもたらすことが判明した。
【表4】
【0116】
実施例34:
(9)RPR120531Aによるリポフェクタント/DNA混合物中のDOPEの存在の効果
実施例31で使用したと同一のプロトコールに従い、トランスフェクション混合物にDNAを添加する前にカチオン脂質(9)RPR120531Aに種々のモル比でDOPE(ジオレオイルホスファチジルエタノールアミン)を加えた。
細胞の溶解後に上清中のルシフェラーゼ活性を定量し、タンパク質1mgに対する割合を計算する(図5)。
リポフェクタント混合物中にDOPEが存在すると、(9)RPR120531Aの濃度が低い場合にトランスフェクション効率を改善することができる。
【0117】
実施例35:
(6)RPR120535A、(9)120531A及び(10)RPR121650Aによるトランスフェクション効率に及ぼす血清の効果
2cm2の指数増殖期の細胞[NIH 3T3又はHeLa]1×105個のサンプルを、血清の不在下で2時間又は培地中の血清の存在下でリポフェクタント/pCMV−LUC溶液(リポフェクタント3ナノモル/μgDNA)により処理する。本実施例では各サンプルに核酸2μgを投与する。細胞溶解液の上清中のルシフェラーゼの発現を調べ、RLU/10秒で表し、タンパク質1mgに対する割合を計算する。結果を分析した処、血清の存在はトランスフェクションにさほど影響しないことが判明した。
【0118】
実施例36:
(20)RPR120527A、(19)120528A、(17)RPR120526A及び(16)RPR120525AによるDNA/リポフェクタント混合物中の核酸濃度の効果
実施例31に記載した条件下で、ナノモルリポフェクタント/μgDNA比を最適化し、NIH 3T3細胞のサンプルにトランスフェクトする(実施例32参照、(20)RPR120527A及び(19)RPR120528Aでは比=6、(17)RPR120526A及び(16)RPR120525Aでは比=3)。各サンプルに加えるDNAの量は0.5〜2μgとする。結果を図6に示す。
指数増殖期の細胞1×105個にプラスミドDNA1μgをトランスフェクトするのが好ましい選択であると思われ、実際に、DNAの使用量が増加すると細胞に接触するカチオン脂質の濃度が増加し、場合によっては毒性の問題を生じる。DNA濃度が低いと、トランスフェクション効率との比例が得られなくなる。
【0119】
実施例37:
本発明によるリポポリアミンのin vivoトランスフェクション試験
腫瘍移植後7日のマウスをケタミン+キシラジンで麻酔し、上記のように調製した本発明によるリポポリアミンを含む溶液を注射する。注射から2日後に腫瘍組織を取り出し、計量後に細切し、溶解用緩衝液(Promega Cell Lysis Buffer E153 A)500μl中で粉砕する。遠心分離(20000gで10分間)後、10μlを分取し、試薬(Promega Luciferase Assay Substrate)50μlと混合後に得られた合計発光をルミノメーターLumat LB 9501(Berthold)で測定し、10秒間積分することによりルシフェラーゼ活性を評価する。得られた活性を腫瘍溶解液の合計上清中の推定RLU(相対光単位)又は注射DNAμg当たりのRLUとして表す。表IIIは得られた結果を示す。
【表5】
【図面の簡単な説明】
【0120】
【図1】種々のカチオン脂質によるNIH 3T3細胞(マウス胎児繊維芽細胞)の処理後のトランスフェクション効率の測定を表すグラフである。
【図2】種々のカチオン脂質によるウサギSMC細胞(ウサギ大動脈平滑筋細胞の一次培養物)の処理後のトランスフェクション効率の測定を表すグラフである。
【図3】種々のカチオン脂質による3LL細胞(ルイス肺癌)の処理後のトランスフェクション効率の測定を表すグラフである。
【図4】種々のカチオン脂質によるNIH 3T3細胞(マウス胎児繊維芽細胞)の処理後のトランスフェクション効率の測定を表すグラフである。
【図5】3LL細胞のトランスフェクション効率に及ぼすDOPE濃度の効果を表すグラフである。
【図6】ナノモルリポフェクタント/μgDNA比を一定にしてDNA量を変えた場合のNIH 3T3細胞のトランスフェクションを表すグラフである。
【特許請求の範囲】
【請求項1】
一般式I:
【化1】
[式中、
R1、R2及びR3は各々独立して水素原子又は−(CH2)q−NRR’基を表し、
qはR1、R2及びR3の各基で独立して1、2、3、4、5及び6であり、
R及びR’は各々独立して水素原子又は−(CH2)q’−NH2基を表し、q’はR及びR’の各基で独立して1、2、3、4、5及び6であり、
m、n及びpは各々独立して0〜6の整数を表し、但しnが2以上のとき、mは異なる値をとることができ、R3は一般式I中で異なる意味をもち、
R4は一般式II:
【化2】
の基を表し、式中、
R6及びR7は各々独立して水素原子又は飽和もしくは不飽和C10〜C22脂肪族基を表し、2個の基の少なくとも一方は水素以外のものであり、
uは0〜10から選択される整数であり、但しuが2以上の整数であるとき、R5、X、Y及びrは各モチーフ[X−(CHR5)r−Y]中で異なる意味をもつことができ、
Xは酸素原子、硫黄原子又はモノアルキルもしくは非モノアルキルアミン基を表し、
Yはカルボニル基又はメチレン基を表し、
R5は水素原子又は場合により置換された天然アミノ酸側鎖を表し、
rは1〜10の整数を表し、但しrが1であるとき、R5は置換又は非置換天然アミノ酸側鎖を表し、rが2以上であるとき、R5は水素原子を表す]により表されることを特徴し、但し、N−デシル尿素ならびに下記式:
【化3】
に示す化合物を除く、D、L又はDL形リポポリアミン及びその塩。
【請求項2】
下記亜式:
【化4】
(式中、R4、R6及びR7は請求項1に定義した意味をもつ)の1個により表されることを特徴とする請求項1に記載のリポポリアミン。
【請求項3】
R4がNR6R7基であり、R6及びR7は亜式III〜XIにおいて(CH2)17CH3、(CH2)11CH3、(CH2)13CH3又は(CH2)12CH3から選択される同一基を表すことを特徴とする請求項2に記載のリポポリアミン。
【請求項4】
細胞外又は細胞内ターゲティング成分に結合していることを特徴とする請求項1から3のいずれか一項に記載のリポポリアミン。
【請求項5】
R5置換基により表されるアミノ酸側鎖のレベルにターゲティング成分を含むことを特徴とする請求項4に記載のリポポリアミン。
【請求項6】
前記ターゲティング成分が標的細胞型の表面に存在する細胞レセプターのリガンドであることを特徴とする請求項4又は5に記載のリポポリアミン。
【請求項7】
前記ターゲティング成分が糖類、ペプチド、脂質HDLもしくはLDL、ステロイド及び/又はオリゴヌクレオチドから選択されることを特徴とする請求項4から6のいずれか一項に記載のリポポリアミン。
【請求項8】
前記ターゲティング成分が核局在シグナル配列により表されることを特徴とする請求項4又は5に記載のリポポリアミン。
【請求項9】
R5により表されるアミノ酸側鎖のレベルにビオチン、ローダミン、葉酸又はエピトープArg−Gly−Aspを含む直鎖もしくは環状ペプチドもしくはプソイドペプチド配列型のマーカー物質を含むことを特徴とする請求項1から8のいずれか一項に記載のリポポリアミン。
【請求項10】
下記化合物から選択されることを特徴とする請求項1から9のいずれか一項に記載のリポポリアミン。
【化5】
【請求項1】
一般式I:
【化1】
[式中、
R1、R2及びR3は各々独立して水素原子又は−(CH2)q−NRR’基を表し、
qはR1、R2及びR3の各基で独立して1、2、3、4、5及び6であり、
R及びR’は各々独立して水素原子又は−(CH2)q’−NH2基を表し、q’はR及びR’の各基で独立して1、2、3、4、5及び6であり、
m、n及びpは各々独立して0〜6の整数を表し、但しnが2以上のとき、mは異なる値をとることができ、R3は一般式I中で異なる意味をもち、
R4は一般式II:
【化2】
の基を表し、式中、
R6及びR7は各々独立して水素原子又は飽和もしくは不飽和C10〜C22脂肪族基を表し、2個の基の少なくとも一方は水素以外のものであり、
uは0〜10から選択される整数であり、但しuが2以上の整数であるとき、R5、X、Y及びrは各モチーフ[X−(CHR5)r−Y]中で異なる意味をもつことができ、
Xは酸素原子、硫黄原子又はモノアルキルもしくは非モノアルキルアミン基を表し、
Yはカルボニル基又はメチレン基を表し、
R5は水素原子又は場合により置換された天然アミノ酸側鎖を表し、
rは1〜10の整数を表し、但しrが1であるとき、R5は置換又は非置換天然アミノ酸側鎖を表し、rが2以上であるとき、R5は水素原子を表す]により表されることを特徴し、但し、N−デシル尿素ならびに下記式:
【化3】
に示す化合物を除く、D、L又はDL形リポポリアミン及びその塩。
【請求項2】
下記亜式:
【化4】
(式中、R4、R6及びR7は請求項1に定義した意味をもつ)の1個により表されることを特徴とする請求項1に記載のリポポリアミン。
【請求項3】
R4がNR6R7基であり、R6及びR7は亜式III〜XIにおいて(CH2)17CH3、(CH2)11CH3、(CH2)13CH3又は(CH2)12CH3から選択される同一基を表すことを特徴とする請求項2に記載のリポポリアミン。
【請求項4】
細胞外又は細胞内ターゲティング成分に結合していることを特徴とする請求項1から3のいずれか一項に記載のリポポリアミン。
【請求項5】
R5置換基により表されるアミノ酸側鎖のレベルにターゲティング成分を含むことを特徴とする請求項4に記載のリポポリアミン。
【請求項6】
前記ターゲティング成分が標的細胞型の表面に存在する細胞レセプターのリガンドであることを特徴とする請求項4又は5に記載のリポポリアミン。
【請求項7】
前記ターゲティング成分が糖類、ペプチド、脂質HDLもしくはLDL、ステロイド及び/又はオリゴヌクレオチドから選択されることを特徴とする請求項4から6のいずれか一項に記載のリポポリアミン。
【請求項8】
前記ターゲティング成分が核局在シグナル配列により表されることを特徴とする請求項4又は5に記載のリポポリアミン。
【請求項9】
R5により表されるアミノ酸側鎖のレベルにビオチン、ローダミン、葉酸又はエピトープArg−Gly−Aspを含む直鎖もしくは環状ペプチドもしくはプソイドペプチド配列型のマーカー物質を含むことを特徴とする請求項1から8のいずれか一項に記載のリポポリアミン。
【請求項10】
下記化合物から選択されることを特徴とする請求項1から9のいずれか一項に記載のリポポリアミン。
【化5】
【図1】

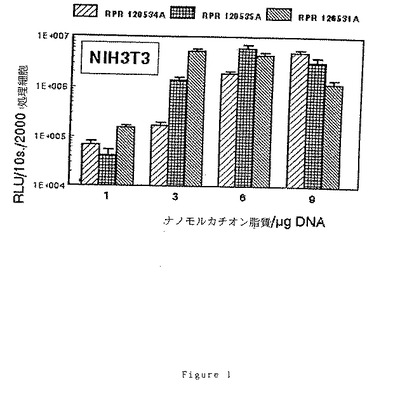
【図2】
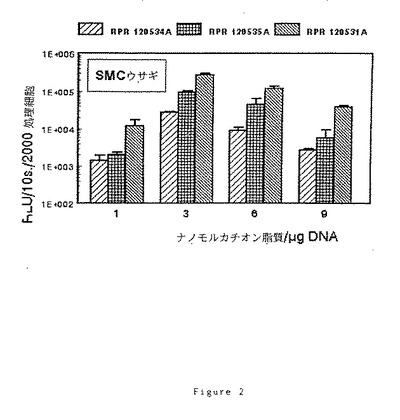
【図3】
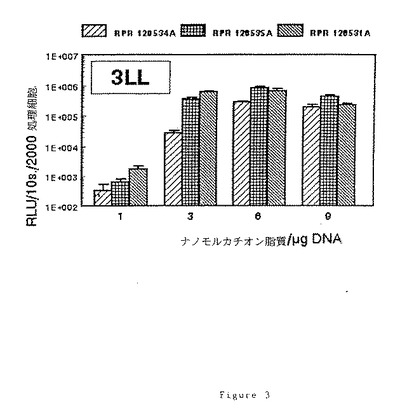
【図4】
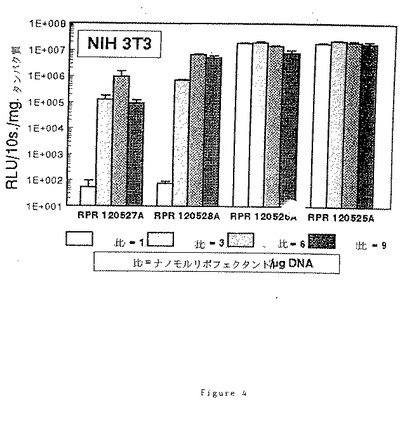
【図5】
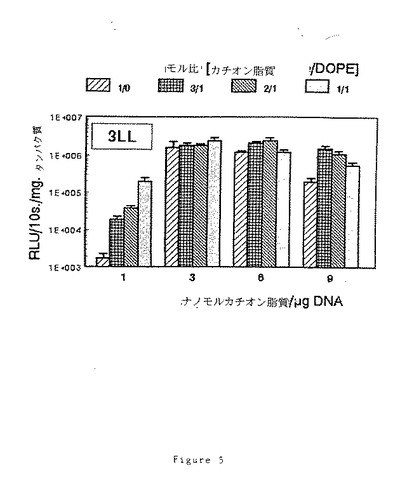
【図6】
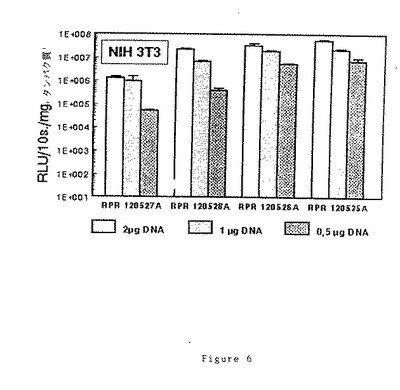
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2009−29790(P2009−29790A)
【公開日】平成21年2月12日(2009.2.12)
【国際特許分類】
【出願番号】特願2008−161421(P2008−161421)
【出願日】平成20年6月20日(2008.6.20)
【分割の表示】特願平9−518621の分割
【原出願日】平成8年11月8日(1996.11.8)
【出願人】(500152119)アバンテイス・フアルマ・エス・アー (65)
【Fターム(参考)】
【公開日】平成21年2月12日(2009.2.12)
【国際特許分類】
【出願日】平成20年6月20日(2008.6.20)
【分割の表示】特願平9−518621の分割
【原出願日】平成8年11月8日(1996.11.8)
【出願人】(500152119)アバンテイス・フアルマ・エス・アー (65)
【Fターム(参考)】
[ Back to top ]