
トランスフェクト細胞を含有するカプセル、それを調製する方法、並びに免疫及びワクチン接種するためのその使用
本発明は、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセル、これらのカプセルを調製する方法、対象の遺伝子によって分泌されたタンパク質の活性をin vivoで評価する方法であって、カプセルを多細胞生物に投与するステップと活性を検出するステップとを含む方法、このようなカプセルを含む医薬組成物、並びに対象のタンパク質を被検体に投与するためのその使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセル、これらのカプセルを調製する方法、及びこのカプセルを使用して、これらの細胞によって発現及び分泌されたタンパク質のin vivo活性を評価するための方法に関する。
【0002】
本発明はさらに、免疫及びワクチン接種用の抗原/免疫原及び/又はアジュバントの送達に使用することができる配合物、組成物、及び方法に関する。より詳細には、本発明は、より効率的及び効果的に免疫及びワクチン接種するための、対象の遺伝子を一過性にトランスフェクトした細胞を含有するカプセルに関する。
【背景技術】
【0003】
タンパク質機能のin vivo評価は、新たな治療法の開発、特に新たなタンパク質治療を開発するための、新たな可溶性タンパク質又は未知の機能を有する可溶性タンパク質の評価への重要な段階である。このようなin vivo評価又は試験には、薬物開発の特に初期に入手することが難しい相当量の精製タンパク質が必要とされる。この過程を短縮するためには、遺伝子をin vivoで直接発現させることが興味深い代替法である。
【0004】
プラスミドDNAのin vivoエレクトロポレーションは、コストが低く安全に利用できるので、遺伝子をin vivoで直接発現させる有望な方法である(Davis H.L.ら1999 Hum.Gen.Ther.4,151〜159)。骨格筋線維へのDNAのエレクトロポレーションは、適切な条件下で使用すると、DNAが細胞内に非常に効率的に取り込まれること、導入遺伝子が長期間発現されることなど多くの特定の利点があるが、エレクトロポレーションを筋肉に行う主要な利点は、血流中で生物活性タンパク質が産生及び分泌されるその潜在的可能性にあり、したがって離れた標的に作用を及ぼす、組換えタンパク質の局所的産生が可能となる(Feewell J.G.ら2001 Mol.Ther.3,574〜583;Kreissら1999 J.Gene Med.1,245〜250;Aihara Hら1998 Nat.Biotechnol.16,867〜870;Li S.ら2001 Gene Ther,8,400〜407)。
【0005】
遺伝子をin vivoで直接発現させる別の方法は、DNAの流体力学的送達である。この方法は、大量の裸のDNA水溶液の肝臓への急速静注を利用する。マウス及びラットに適用する場合、これは、肝臓での高レベルの遺伝子発現、並びに低レベルであるが腎臓、肺、心臓など他の器官での遺伝子発現を導入するための完全で効率的な手段である(Liu F.ら1999 Gene Ther 6(7),1258〜1266;Yang J.ら2001 Hepatology,33(4),848〜859;Maruyama H.ら2002 J.Gene.Med.4(3),333〜341;Hodges B.L.ら2003 Expert Opin.Biol.Ther,3(6),911〜918)。
【0006】
エレクトロポレーション及び流体力学的遺伝子送達法のどちらにおいても、導入遺伝子の分泌レベル又は産生タンパク質の転写後修飾に影響を与える可能性はほとんどない。
【0007】
in vivoでのタンパク質の機能を評価するのに適した第3の方法は、所望の遺伝子産物をin situで産生する、トランスジェニック細胞系のマイクロカプセル化細胞をマウス又はラットなどの実験動物に投与するものである。この手法では、細胞を宿主の免疫系から物理的に隔離するマトリックス内に細胞が封入される。栄養物、外部刺激、及び治療用タンパク質の通過が可能であるが、移植片拒絶の原因である免疫成分に不透過性であり、したがって移植片の生着を延長する選択透過膜が使用される(Lim F.ら1980 Science,210,908〜910)。このような方法は、遺伝子治療(Hortelano G.ら1996 Blood,87(12),5095〜5103;Visted T.ら2001 Neuro−Oncology 3,201〜210)、インスリン依存性糖尿病(Sun Y.ら1996 J.Clin.Invest 98,1417〜1422)、及び抗癌療法(Read T−A.ら1999 Int.J.Devl.Neuroscience,17(5〜6),653〜663)のために開発されてきた。導入遺伝子の発現及び翻訳後修飾のレベルの完全な制御は、カプセル化される細胞系の選択性、カプセル内の細胞密度、及び発現カセットに調節エレメントを含む可能性によって可能である。発現すべき治療用タンパク質の特性に応じいくつかの生体適合性ポリマーを使用して細胞を固定化することができる。アルギナート−ポリ−L−リシン−アルギナート(APA)カプセルが最も頻繁に使用されているが(Lim F.ら1980 Science,210,908〜910;Hortelano G.ら1996 Blood,87(12),5095〜5103;Visted T.ら2001 Neuro−Oncology 3,201〜210;Sun Y.ら1996 J.Clin.Invest 98,1417〜1422;Read T−A.ら1999 Int.J.Devi.Neuroscience,17(5〜6),653〜663;Strand B.L.ら2002 J.Microencapsulation 19(5),615〜630を参照のこと)、ポリ(メタクリル酸ヒドロキシエチル−co−メタクリル酸メチル)(HEMA−MMA)など他のポリマーも使用することができる(Lahooti S.ら2000 Biomaterials,21,987〜995)。カプセル化マトリックスは、完全に固体であるか、又は包含させた細胞を環境から隔離する膜のみと共に液体コアを含有することができる。
【0008】
Savelkoulら(Savelkoul Huub F.J.ら1994 Journal of Immunological Methods 170 185〜196頁)は、Il−4又はIL−5を産生する安定的にトランスフェクトされたサル細胞系CV1又はCHO−Kiの細胞の調製及びカプセル化(2.3及び2.4、186頁参照)、並びにマウスへの前記カプセル化細胞の腹腔内(i.p.)又は皮下(s.c.)注射を記載している。
【0009】
国際公開第93/00439号は、活性因子又は増強物質を産生する遺伝子改変した細胞系、特にラットNGF放出細胞系N8−21の細胞のカプセル化を開示している(実施例1)。
【0010】
上記の従来技術に記載のあらゆる例において、カプセル化細胞は、安定的にトランスフェクトされた細胞に由来する。
【0011】
安定的にトランスフェクトされた細胞の作製は、トランスフェクトする遺伝子を細胞のゲノム中に組み込むことを必要とするので、労力を要し時間がかかる。これは、トランスフェクト細胞を選択培地中で長時間培養した後、培養細胞を段階希釈して、安定的にトランスフェクトされ対象のタンパク質を十分な量で産生する細胞クローンを単離することによって達成される(Maniatisら参照)。
【0012】
安定的にトランスフェクトされた細胞は通常、対象のタンパク質が高産生されるかどうかで選択される。安定的にトランスフェクトされた細胞は、トランスフェクトした遺伝子を高度に長期間(数カ月まで)発現する利点がある。
【0013】
しかし、上記の方法は、複数のタンパク質のin vivo活性の評価など、高スループット及び/又は高スピードを必要とする用途には適していない。
【0014】
本発明によって取り組まれる課題は、高スループットに適しており短期間内(例えば、1又は2又は3週間)に実施することができる、タンパク質をin vivoで発現させる方法を提供すること、及び従来技術の方法の欠点を有していない、タンパク質のin vivo活性を評価するための手段を提供することである。
【0015】
上記の課題を、特許請求の範囲で定義される本発明によって解決する。
【0016】
脊椎動物の免疫系の活性化は、病原体及び悪性腫瘍から動物を防御するための重要な機構である。この免疫系は、体液性及び細胞性の部門を含めた多くの相互作用成分からなる。体液性免疫には、抗原に直接結合する抗体が関与する。体液性免疫のエフェクターとしての抗体分子は、Bリンパ球によって分泌される。細胞性免疫には、非自己抗原を産生する他の細胞を認識し死滅させる分化した細胞障害性Tリンパ球(CTL)が関与する。CTLは、MHC(主要組織適合遺伝子複合体)クラスI分子に結合した、標的細胞の表面上に出現する分解されたペプチド断片に応答する。細胞内で産生されたタンパク質は、細胞代謝の一部としてペプチドに連続的に分解されることが理解されている。こうした断片は、MHC分子に結合し、細胞表面に輸送される。したがって、細胞性免疫系は、体内のあらゆる細胞内で産生されたタンパク質のスペクトルを絶えずモニターしており、非自己抗原を産生するいかなる細胞をも排除するような態勢にある。
【0017】
ワクチン接種は、抗原/免疫原に対して応答するように動物をプライミングする過程である。この抗原は、精製タンパク質、死滅/弱毒病原体に含まれるタンパク質として、又は次いで宿主細胞内で抗原を発現する遺伝子として(遺伝子免疫)投与することができる。この過程には、T及びBリンパ球、他の種類のリンパ球様細胞、並びに抗原を処理し、この抗原を免疫系を活性化できる形で提示することができる分化した抗原提示細胞(APC)が関与する。
【0018】
ワクチンの有効性は、腫瘍又は病原体による、後の抗原チャレンジに対する防御の程度によって評価される。有効なワクチンは、最少回数の接種の後、疾患を標的とした介入に対する高力価で持続的な防御免疫を誘導することができる免疫原である。
【0019】
ワクチン開発に成功した手法の数は、病原菌の数とほとんど同じぐらいである。技術が発展するにつれ、防御抗原/免疫原の性質を分子レベルで定義することが可能となる。近年、無細胞ワクチンが、種々の病原体由来のサブユニット(すなわち多成分系ワクチン)で投与することができ、有害反応の数を減少させる潜在的可能性を有するので、ワクチン開発のための好まれる方法となっている。サブユニットワクチンは、病原微生物由来の限定された精製防御抗原で構成されている。驚くべき成功例がいくつかあるが、現在使用されているサブユニットワクチンは少数である。おそらく、サブユニットワクチンの使用を妨害する最も困難な理由は、抗原送達の問題である。最適な抗原送達のためには、抗原がその生物学的状況下で抗原提示細胞に送達される必要があり、すなわち抗原には、食細胞によって容易に認識され取り込まれるリスクがある。大部分の抗原は、寄生生物−宿主細胞間相互作用にとって重要な三次元構造を有しているが、こうした構造の多くは抗原精製の際に失われる。
【0020】
サブユニットワクチン製造に伴う問題の多くを回避するのに取られた1つの手法は、防御抗原をin vivoで発現するように遺伝子改変した、弱毒ワクチン及び弱毒細菌に基づく組換え生ワクチンビヒクル(すなわち、ワクシニアウイルス、アデノウイルス、サルモネラ、及びウシ型結核菌種カルメットゲラン菌すなわちBCGの組換え体)の開発である(Snapper,S.B.ら、Proc.Natl.Acad.Sci USA85:6987〜6991,1988;Jackett,P.S.ら、J.Clin.Micro.26:2313〜2318,1988;Lamb,J.R.ら、Rev.Infect.Dis.11:S443〜S447,1989;Shinnick,T.M.ら、Infect.Immun.56:446〜451,1988を参照のこと)。
【0021】
生細菌を使用したワクチン接種が研究されており、致死遺伝子をノックアウトするように突然変異が誘発された生細菌株を利用することが多い。生ワクチンには、抗原が先天性の免疫原の形で発現され;生送達システムが宿主内で複製及び持続し、したがって宿主の免疫系が再刺激され多回投与の必要性が回避され;生ベクター系は、抗原を精製する必要性をなくし、製造が安価であり;生ベクターは、複数の抗原を送達するように設計することができ、したがって個体にワクチン接種しなければならない回数が減少するという利点がある。しかし、この方法は、細菌が毒性に戻ることを可能にするさらなる突然変異が起こらないことを保障するような厳しい安全対策を必要とする。より確実な方法は、後に宿主がその抗体を産生できるタンパク質を発現する弱毒細菌を利用することである。細菌ベクターは、ワクチンの経口投与用に研究されることが多い。例えば、HIV、ライム病、及びエプスタイン−バーウイルスから防御するための経口投与用にサルモネラ菌ベースのワクチンが研究されている。
【0022】
また、バキュロウイルス、酵母、及び組織培養細胞が、ワクチンの製造において使用するために研究されてきた。各例は、バキュロウイルスを使用して、E型肝炎に対するワクチンで使用されるタンパク質を産生した米国特許第6287759号;酵母をベクターとして使用して、ワクチンで使用するためのヘリコバクターポリペプチドを産生した米国特許第6290962号;脊椎動物組織培養細胞を使用して、ワクチンで使用するための精製不活化デングウイルスを増殖させた米国特許第6254873号で示されている。こうした例のすべてにおいて、ベクターを使用して、後に精製されワクチンで使用されるはずの対象のタンパク質が産生された。
【0023】
遺伝子免疫は、特定のタンパク質をコードしている遺伝子を動物自身の細胞内で発現させることによりこのタンパク質に対する免疫応答を誘導する別の手法である。in vivoでの長期間の抗原提示によって生じた相当な抗原増幅及び免疫刺激は、抗原に対する確実な免疫(solid immunity)を誘導することができる。遺伝子免疫では、タンパク質精製及びアジュバントとの混合というどちらもワクチン開発に常に必要とされる困難な段階が省かれるため、特定のタンパク質に対する免疫応答を生じさせるためのワクチン接種プロトコールが簡素化される。遺伝子免疫は、タンパク質の単離を必要としないので、生化学的に精製すると高次構造エピトープ(conformational epitope)を失う可能性があるタンパク質にとって特に価値がある。また、遺伝子ワクチンは、干渉を誘発することなく又は有効性に影響を及ぼすことなく混合して送達することもでき(Tangら,1992;Barryら,1995)、複数の抗原に対するワクチン接種計画を簡素化することができる。
【0024】
ワクチンは、アジュバントを使用することによって増強されることが多い。ワクチンアジュバントは、ワクチン組成物中で任意の特定の抗原と共に得られる免疫応答を改善するのに有用である。アジュバントは、抗体及びエフェクターT細胞の量を増加させ、抗原の量及び注射の頻度を低減するのに使用される。抗原は、アジュバントを含まないワクチンとして投与されることもあるが、効果的なアジュバントなしでは、有用な免疫応答を刺激するのに十分な免疫原性を欠く抗原が多く存在する。アジュバントはまた、得られる免疫応答を増大させ、又は抗原投与量を低減することができるという点で、「自給自足」抗原の免疫応答を改善する。
【0025】
分子ワクチン接種のためのアジュバント及び送達システムに関する包括的な総説については、Sheikh NA,Al−Shamisi M,Morrow WJW;Delivery systems for molecular vaccination;Current Opinion in Molecular Therapeutics 2000,2:1(37−54)を参照されたい。
【発明の開示】
【発明が解決しようとする課題】
【0026】
ワクチン開発が困難であった病原体又は腫瘍に対するワクチンを開発するためには、及び/又は十分に利用されなかったことによる市販ワクチンの失敗を克服するためには、ワクチンの免疫性及び/又は副作用の低下を可能にする抗原/免疫原送達の新たな方法を開発しなければならない。
【0027】
抗原/免疫原を含む新たな医薬組成物及びワクチン送達法は、本出願に記載されており、適当な抗原及び/又はアジュバントをコードしている1種又は複数の遺伝子を一過性にトランスフェクトしたカプセル化細胞を送達した後、抗原/免疫原をin vivoで産生させるものである。この方法は、タンパク質ワクチンに伴う、抗原/免疫原の厄介な精製の必要性、又は遺伝子免疫に伴う、抗原/免疫原のin vivo産生が低いことによる低効率など、上記の他のワクチン及びワクチン接種法の欠点を克服する。
【課題を解決するための手段】
【0028】
本発明は、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルに関する。
【0029】
別の態様では、本発明は、細胞が動物細胞、好ましくは哺乳動物細胞である前記カプセルを提供する。
【0030】
さらなる態様では、前記カプセルは、対象の遺伝子が、タンパク質を分泌するためのシグナル配列に融合しているカプセルである。
【0031】
本発明のさらに別の態様では、カプセルは、発現カセット中に挿入された対象の遺伝子を含む。
【0032】
さらなる態様では、カプセルは、プラスミド中に挿入された対象の遺伝子を含む。
【0033】
本発明のさらに別の態様では、前記カプセルは、生体適合性ポリマー、アルギナート−ポリ−L−リシン−アルギナート(APA)を含む。
【0034】
さらなる態様では、カプセルは、分離サイズが90〜30kDa、好ましくは80〜60kDaである孔を有する生体適合性ポリマー膜を含む。
【0035】
本発明のさらなる態様では、カプセルは、平均直径が100〜1500μm、好ましくは250〜600μm、特に440〜530μmである。
【0036】
本発明の別の態様では、カプセルは、低剪断、微小重力条件下で維持された。
【0037】
本発明はさらに、カプセルを調製する方法を提供し、この方法は、細胞に対象の遺伝子を一過性にトランスフェクトするステップと、一過性にトランスフェクトされた細胞をカプセル化するステップとを含む。
【0038】
本発明はさらに、カプセルを調製する方法を提供し、この方法は、低剪断、微小重力条件下で維持するステップをさらに含む。
【0039】
本発明はさらに、対象の遺伝子によってコードされており、一過性にトランスフェクトしカプセル化した細胞によって発現及び分泌されたタンパク質のin vivo活性を評価する方法を提供し、この方法は、上記のカプセルを多細胞生物に投与するステップと、前記多細胞生物において前記タンパク質の活性を検出するステップとを含む。
【0040】
本発明はさらに、対象の遺伝子によってコードされており、一過性にトランスフェクトしカプセル化した細胞によって発現及び分泌されたタンパク質のin vivo活性を評価する方法を提供し、この方法は、上記のカプセルを、哺乳動物である多細胞生物に投与するステップを含む。
【0041】
本発明はさらに、対象の遺伝子によってコードされており、一過性にトランスフェクトしカプセル化した細胞によって発現及び分泌されたタンパク質のin vivo活性を評価する方法を提供し、この方法は、上記のカプセルを、マウス、ラット、イヌ、ヤギ、ヒツジ、ウシ、及びサルからなる群から選択される哺乳動物に投与するステップを含む。
【0042】
本発明はさらに、対象の遺伝子によってコードされており、一過性にトランスフェクトしカプセル化した細胞によって発現及び分泌されたタンパク質のin vivo活性を評価する方法を提供し、この方法は、i.p.注射によって実施される、上記のカプセルを多細胞生物に投与するステップを含む。
【0043】
本発明はさらに、動物におけるカプセル又は上記の方法の使用を提供し、この使用では、検出すべき活性が急速に生じ(例えば、注射後数時間又は1日若しくは複数日以内)、活性物質の投与後数日以内に完了する。
【0044】
本発明はさらに、動物におけるカプセル又は上記の方法の使用を提供し、この使用では、検出すべき活性が、急速に生じ(例えば、注射後数時間又は1日若しくは複数日以内)、活性物質の投与後数日以内に完了し、動物は、コンカナバリンA(ConA)誘発肝毒性を有するマウスである。
【0045】
本発明はさらに、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルを含む医薬組成物を提供する。
【0046】
本発明はさらに、細胞に、抗原/免疫原及び/又はアジュバントをコードしている遺伝子がトランスフェクトされている、上記の医薬組成物を提供する。
【0047】
本発明はさらに、抗原/免疫原が、細菌抗原、ウイルス抗原、真菌抗原、寄生虫抗原、又は腫瘍抗原である、上記の医薬組成物を提供する。
【0048】
本発明はさらに、カプセルが、(1種又は複数の)抗原及び/又はアジュバントをコードしている遺伝子をそれぞれトランスフェクトした少なくとも2種類の細胞を含み、この2種の遺伝子が同一でない、上記の医薬組成物を提供する。
【0049】
本発明はさらに、対象のタンパク質を被検体に投与するための、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルの使用を提供する。
【0050】
本発明はさらに、対象のタンパク質を被検体に投与するための、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルの使用を提供し、このタンパク質は、免疫及びワクチン接種用の抗原である。
【0051】
本発明はさらに、対象のタンパク質を被検体に投与するための、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルの使用を提供し、被検体が、ヒト;げっ歯類、イヌ、ブタ及びサルを含めた研究目的で飼育された動物並びにイヌ及びネコを含めた、家畜及び飼育動物からなる群から選択される。
【0052】
本発明はさらに、上記のカプセル又は医薬組成物を含むキット、及び前記カプセル又は組成物を被検体に投与するための手段を提供する。
【発明を実施するための最良の形態】
【0053】
本発明は、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルを提供する。
【0054】
用語「1種又は複数のカプセル」は、同義的に使用され、細胞を環境から隔離する生体適合性ポリマー膜のみと共に細胞を含有するカプセル化マトリックスを指す。
【0055】
用語「1種又は複数のカプセル」はさらに、生体適合性ポリマー膜内に封入又は包含された1種又は複数の細胞を指す。
【0056】
用語「一過性にトランスフェクトされた細胞」又は「対象の遺伝子を一過性にトランスフェクトした細胞」は、対象の遺伝子を当技術分野で既知の方法(Maniatisら参照)によって導入し、トランスフェクションの日から14日以内に、好ましくは10日以内に、対象の遺伝子によってコードされているタンパク質を発現する(1種又は複数の)細胞を指す。
【0057】
用語「一過性にトランスフェクトされた」又は「対象の遺伝子を一過性にトランスフェクトした」はさらに、当技術分野で既知の方法(Maniatisら参照)により、事前選択した細胞中に対象の遺伝子を導入し、ここで、前記細胞は対象の遺伝子を含有するが、この遺伝子は、前記細胞のゲノム中に組み込まれないこと、又は細胞内で複製することができるエピソームベクター上に位置することを指す。
【0058】
一過性にトランスフェクトされた細胞は、トランスフェクション後細胞を培養する際に選択培地によるどんな選択圧をもかけることなく得ることが好ましい。
【0059】
細胞培養で使用される選択培地は、当技術分野で周知である。長年にわたるトランスフェクション研究の間、いくつかの様々な薬剤選択マーカーが一般に使用されてきた。例えば、細菌のアミノグリコシドホスホトランスフェラーゼ遺伝子を含有する組換えベクターをトランスフェクトした細胞は、薬剤G−418の存在下で安定にトランスフェクトするかどうかで選択することができる(Southern,P.J.及びBerg,P.(1982)「SV40初期領域プロモーターの制御下にある細菌遺伝子を用いた哺乳動物細胞の抗生物質耐性への形質転換(Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter)」,J.Mol.Appl.Gen.1,327)。同様に、トランスフェクトされたベクターからのハイグロマイシンBホスホトランスフェラーゼ遺伝子の発現により、薬剤ハイグロマイシンBに対する耐性が得られる(Blochlinger,K.及びDiggelmann,H.(1984)「高等真核細胞を用いたDNA導入実験用の選択マーカーとしてのハイグロマイシンBホスホトランスフェラーゼ(Hygromycin B phosphotransferase as a selectable marker for DNA transfer experiments with higher eucaryotic cells)」,Mol.Cell.Biol.4,2929)。
【0060】
代替戦略は、所与の細胞系において欠陥がある必須の遺伝子を担うベクターを使用することである。例えば、ジヒドロ葉酸還元酵素(DHFR)遺伝子の発現に欠陥があるCHO細胞は、追加のヌクレオシドの存在下でしか生存しない。しかし、こうした細胞は、DHFR遺伝子を発現するDNAを安定にトランスフェクトすると、必要とされるヌクレオシドを合成する(Stark,G.R.及びWahl,G.M.(1984)Gene amplification.Ann.Rev.Biochem.53,447.)。DHFRをマーカーとして使用するさらなる利点は、用量を増やしながらメトトレキセートに細胞を暴露すると、DHFR及び関連するトランスフェクトしたDNAの遺伝子増幅が起こることである(Schimke,R.T.(1988)「培養細胞における遺伝子増幅(Gene amplification in cultured cells)」J.Biol.Chem.263,5989)。
【0061】
用語「安定にトランスフェクトされた」又は「対象の遺伝子を安定にトランスフェクトした」又は「安定なトランスフェクション」は、当技術分野で既知の方法(Maniatisら参照)により、事前選択した細胞中に対象の遺伝子を導入し、ここで、前記対象の遺伝子は、前記細胞のゲノム中に組み込まれないこと、又はエピソームベクター上に位置し細胞内で複製されることを指す。
【0062】
用語「対象の遺伝子」は、対象のタンパク質、すなわち、興味深い活性を有するタンパク質、又はその特性が興味深い、例えばそれを評価すべきタンパク質をコードしているゲノムDNA、cDNA、合成DNA、RNA、及び他のポリヌクレオチド、又はそのアナログを指す。
【0063】
驚くべきことに、本発明者らにより、対象の遺伝子を一過性にトランスフェクトしたカプセル化細胞は、多細胞生物に投与する(例えば、注射する)ことができ、対象の遺伝子によってコードされるタンパク質を、そのin vivo活性又は作用を検出できるような十分な量in vivoで産生することが判明した。
【0064】
さらに驚くべきことに、前記カプセル化細胞は、トランスフェクションの当日に、又はその翌日若しくは数日後にも多細胞生物に投与することができ、前記タンパク質のin vivo活性又は作用を検出することができる。
【0065】
したがって、本発明は、投与の開始から14日以内に、優先的には10日以内に、さらにより好ましくは5日以内に、対象の遺伝子によってコードされるタンパク質のin vivo活性を分析又は評価するための新規ツールを提供する。
【0066】
第1の態様では、本発明は、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有する新規カプセルを提供する。
【0067】
この細胞は、植物細胞又は動物細胞であってよい。この細胞は、動物細胞、特にハイブリドーマを含めた哺乳動物細胞であることが好ましい。特に好ましい細胞は、COS細胞、BHK細胞、VERO細胞、CHO細胞、rCHO−tPA細胞、rCHO−Hep B表面抗原細胞、HEK293細胞、rHEK293細胞である。本発明に従って培養することができるハイブリドーマの例には、例えば、DA4.4細胞、123A細胞、127A細胞、GAMMA細胞、及び67−9−B細胞が含まれる。本発明に従って培養することができる昆虫細胞の例には、例えば、鱗翅類細胞、Tn−368細胞、SF9細胞、rSF9細胞、及びHi−5細胞が含まれる(例えば、Ikonomou Lら2002,Nov〜Dec,18(6),1345〜1355を参照のこと)。本発明に従って培養することができる非哺乳動物細胞の例には、例えば、ブラウンブルヘッド細胞系が含まれる(例えば、Buck CDら1985,Feb,10(2),171〜84を参照のこと)。
【0068】
上記で定義した対象の遺伝子は発現され、細胞からタンパク質が分泌される。優先的には、このタンパク質は成熟タンパク質として分泌される。優先的には、このタンパク質は、発現する場合、タンパク質を分泌するためのシグナルペプチドを含む。検討中の遺伝子が、タンパク質のシグナルペプチドをコードしている配列を有していない場合、このような配列、例えば、シグナル配列、プレ配列及び/又はプロ配列をその遺伝子配列に融合して、対象の遺伝子をタンパク質として発現、輸送及び分泌しやすいようにすることができる。
【0069】
対象の遺伝子は、一般に、細胞の一過性トランスフェクションに適したベクターの一部である、プロモーター及び調節シグナルを含む発現カセット中に通常挿入される。
【0070】
適当なベクターは、プラスミド又はエピソームベクターである。
【0071】
細胞の一過性トランスフェクションに適したプラスミドの例は、pCEP4(Invitrogen、米国)、pEAKベクターファミリー(Edge Biosystems、米国)、又はpCDNA3.1(Invitrogen、米国)である。
【0072】
対象の遺伝子を担うベクターの細胞へのトランスフェクションは、任意のトランスフェクション法、特にポリアミン法、ポリエチレンイミン法(O.Boussifら1995 Proc.Natl.Acad.Sci.USA 92,7297〜7301頁)、リン酸カルシウム共沈法、又はリポフェクションによって実施することができる。ポリアミン法は、トランスフェクトした細胞の90%超が得られるので好ましい。ショットガン法又はエレクトロポレーションは、高い細胞死亡率を伴い、これにより死細胞のカプセル化が高い割合でもたらされるはずであり、したがってカプセルの生産性が低下するのでこの用途には好ましくない。
【0073】
生体適合性ポリマーは、安定に発現する細胞系の細胞をカプセル化するための、当技術分野で既知の生体適合性ポリマー、特にアルギナート塩、特にアルギナートナトリウム若しくはカリウム、及びアルギナート−ポリ−L−リシン−アルギナート(APA)、並びにポリ(メタクリル酸ヒドロキシエチル−co−メタクリル酸メチル)(HEMA−MMA)などの他のポリマーの中から選択することができる。好ましい生体適合性ポリマーはAPAである。
【0074】
このポリマーを工学処理して、選択透過膜、すなわち栄養物、酸素、外部刺激、及び分泌タンパク質に透過性があるが、移植片拒絶の原因である免疫成分に不透過性である膜が作製される。このような選択透過膜は通常、分離が90〜30、好ましくは90〜50又は80〜50kDa、さらにより好ましくは80〜60kDaである孔を有する。分離は、選択透過膜を通して拡散することができる分子のサイズを定義する。
【0075】
カプセルは通常、直径が100〜1500μm、好ましくは250〜600μm、特に440〜530μmである。
【0076】
本発明はまた、上記カプセルを調製する方法に関し、この方法は、細胞に対象の遺伝子を一過性にトランスフェクトするステップと、一過性にトランスフェクトされた細胞をカプセル化するステップとを含む。
【0077】
カプセル化の後、この細胞は、カプセル内で増殖し続け、対象の遺伝子によって分泌される成熟タンパク質を、カプセルを維持する培地中に拡散する。
【0078】
このカプセルは、成熟タンパク質のin vitro拡散を低減し、機械的な理由によるカプセルのどんな破壊をも避ける、低剪断、微小重力条件下で維持することが好ましい。
【0079】
本発明のカプセルを調製するすべてのステップは、短期間、通常1日又は2日以内に実施することができる。
【0080】
細胞を含有するカプセルを数カ月まで保存するために、−80℃で、又は液体窒素(−196℃)気相若しくは液相中で凍結させることができる。この凍結保存により、大量のカプセルを製造し、続いてその一定分量を様々な用途に(例えば、キット中に)使用することが可能となる。蘇生後、この細胞はカプセル内で増殖を再開し、対象のタンパク質を3〜10日間産生し続ける。
【0081】
さらなる態様では、本発明は、対象の遺伝子によってコードされており、一過性にトランスフェクトした細胞によって発現及び分泌されたタンパク質のin vivo活性を分析又は評価する方法に関し、この方法は、上記で定義したカプセルを多細胞生物に投与するステップと、前記タンパク質の活性を検出するステップとを含む。この活性は、局所活性又は全身活性であってよい。
【0082】
本発明の方法に従って産生することができるタンパク質は、例えば、絨毛性ゴナドトロピン、卵胞刺激ホルモン、ルトロピン絨毛性ゴナドトロピンホルモン、甲状腺刺激ホルモン、ヒト成長ホルモン、インターフェロン(例えば、インターフェロンβ−la、インターフェロンβ−lb)、インターフェロン受容体(例えば、インターフェロンγ受容体)、TNF受容体p55及びp75、TACI−Fc融合タンパク質、インターロイキン(例えば、インターロイキン1、インターロイキン2、インターロイキン3、インターロイキン4、インターロイキン5、インターロイキン6、インターロイキン8、インターロイキン10、インターロイキン11、インターロイキン12)、インターロイキン結合タンパク質(例えば、インターロイキン−18結合タンパク質)、増殖因子(例えば、エリスロポイエチン、顆粒球コロニー刺激因子、顆粒球マクロファージコロニー刺激因子、血小板由来増殖因子、酸性及び塩基性線維芽細胞増殖因子、角化細胞増殖因子、グリア細胞系由来神経栄養因子)、下垂体ペプチドホルモン、閉経期性腺刺激ホルモン、インスリン様成長因子(例えば、ソマトメジンC)、トロンボモジュリン、インスリン、第VIII因子、ソマトロピン、骨形成タンパク質2、ヒルジン、エポエチン、組換えLFA−3/IgG1融合タンパク質、グルコセレブロシダーゼ、並びにその突然変異タンパク質、断片、可溶形、機能性誘導体、及び融合タンパク質を含めた対象のどんなタンパク質とすることもできる。
【0083】
多細胞生物は、植物又は動物であってよい。特定の対象の動物は、マウス、ラット、イヌ、ヤギ、ヒツジ、ブタ、ウシ、サルなど薬学研究で一般に使用されている哺乳動物である。
【0084】
特に興味深い哺乳動物は、マウス又はラットなどのげっ歯類であり、マウス、例えばC57/BL6系マウスが好ましい。
【0085】
検出する活性は、例えば、体温、組織又は体液の色、組織又は体液(例えば、血液、血清、血漿、糞便、痰、滑液、脳脊髄液、又は尿)中の所与の物質の濃度など、任意のパラメータに関するものでよい。
【0086】
カプセルは、筋肉内(i.m.)、皮内、皮下(s.c.)又は腹腔内(i.p.)注射によって投与することができる。好ましい投与形態はi.p.注射である。別の好ましい形態はs.c.注射である。
【0087】
カプセルはin vivo条件下で安定であり、包含する細胞を分解又は放出しない。
【0088】
カプセルは、一過性発現が起こっている限り、すなわちカプセル投与(0日目)から通常3〜14日目の間は成熟タンパク質をin vivoで放出するが、その放出の大部分は通常、1日目〜4、5又は6日目に起こる。成熟タンパク質が大量に放出される期間は通常3〜5日目であるが、これは検出すべき局所及び/又は全身活性を誘導するのに通常十分である。慢性作用を検出するためなど、必要な場合は、カプセルの投与を2回以上それぞれ別の時点で実施して、例えば、大量の成熟タンパク質の放出をより長期間維持することができる。
【0089】
したがって、本発明のカプセルは、実験動物、例えばヒト疾患の動物モデルで使用することができる、薬学研究の有用なツールとなる。
【0090】
この動物又は動物モデルは、誘導された検出すべき局所及び/又は全身活性が急速に(すなわち、数時間又は1日若しくは複数日以内に)生じ、活性物質(対象のタンパク質)の投与後数日以内に完了するものが好ましい。
【0091】
このような動物モデルの一例は、以下で例示するが、コンカナバリンA(ConA)によって肝毒性が誘発されたマウスである。
【0092】
中毒性肝疾患は、ヒトの世界中の健康問題を代表し、薬理学的治療法がまだ発見されていない。例えば、肝硬変をもたらす活動性慢性肝炎は、活性化T細胞によって肝実質細胞が進行的に破壊される病状である。ConA誘発肝毒性は、マウスで説明されているT細胞依存性アポトーシス性及び壊死性肝損傷の3つの実験モデルのうちの1つである。活性化抗CD3モノクローナル抗体又はスーパー抗原SEBのいずれかで誘発したGal N(D−ガラクトサミン)感作マウスは、重篤なアポトーシス性肝損傷及び二次的な壊死性肝損傷を発症する。植物のT細胞分裂促進レクチンConAの非感作マウスへの注射によっても、壊死に先行する肝アポトーシスがもたらされる。ConAは、全身へのTNFα及びIFNγ、並びに他の種々のサイトカインの放出を誘発する。傷害の8時間後、トランスアミナーゼの放出は重篤な肝臓破壊を示す。
【0093】
TNFα及びIFNγはどちらも、肝障害を誘発する上で肝損傷の決定的に重要なメディエータである。例えば、TNFαは、ConA注射の後に産生される最初のサイトカインの1つであり、抗TNFα抗体は、疾患に対する防御を与える(Seinoら2001,Annals of surgery 234,681)。ConA処理動物の血中トランスアミナーゼレベルの低下から判断して、抗IFNγ抗血清はマウスを有意に防御するので、後で誘導されるIFNγも、肝損傷の決定的に重要なメディエータであると思われる(KuestersらGastroenteroloy 111,462)。ConA誘発肝毒性からの防御を与える処置を記載した様々な文献がある。rIL6の単回投与により、トランスアミナーゼの放出が完全に阻害されたが、同じ投与計画でTNF産生の阻害は40〜50%しか誘導されなかった(Mizuharaら1994,J.Exp.Med.179,1529〜1537)。
【0094】
数種の細胞型、CD4 T細胞、マクロファージ、及びナチュラルキラー細胞が肝障害に関与することが示されている(Kaneko J.Exp.Med.2000,191,105〜114)。抗CD4抗体は、T細胞の活性化及びその結果としての肝障害を遮断する(Tiegsら1992,J.Clin.Invest.90,196〜203)。CD8に対するモノクローナル抗体のマウスへの前処置は防御に失敗したが、マクロファージの欠失は肝炎の誘発を阻止した。
【0095】
興味深い動物モデルの別の例は、マウスLPS(リポ多糖)誘発TNFα放出モデルである。
【0096】
LPSは、脂質及び炭水化物をどちらも含有する大分子である。LPSは、グラム陰性菌の細胞壁の主成分である。LPSは、原型的なエンドトキシンとして働き、多くの細胞型において炎症誘発性サイトカインの分泌を促進する。LPSは、Toll受容体4を介して先天性免疫系の食細胞によって認識される(Triantafilou M.らTrends Immunol.2002 Jun;23(6):301〜4)。LPSは、マクロファージ又はミクログリアをin vitro又はin vivoで活性化するために広く使用されている(Tobias PSらImmunobiology.1993 Apr;187(3〜5):227〜32)。マウスのLPS誘発TNFα放出モデルはヒトの炎症状況に似ている。
【0097】
本発明はさらに、細胞に1種又は複数の遺伝子を一過性にトランスフェクトした上記のカプセルを含む医薬組成物を提供する。この医薬組成物は、製薬上許容される1種又は複数の担体、希釈剤、或いは賦形剤を追加的に場合によって含む。
【0098】
この遺伝子は、病原体由来の、1種又は複数の抗原/免疫原、或いは1種又は複数のその部分、或いは対象の1種又は複数のエピトープをコードすることができる。抗原/免疫原という用語は、宿主において免疫応答を誘発することが可能な任意のタンパク質又はその誘導体を指す。
【0099】
好ましい実施形態では、病原体は、任意のウイルス、クラミジア、マイコプラズマ、細菌、寄生生物、及び菌類からなる群から選択される。ウイルスには、ヘルペスウイルス、オルトミクソウイルス、ライノウイルス、ピコルナウイルス、アデノウイルス、パラミクソウイルス、コロナウイルス、ラブドウイルス、トガウイルス、フラビウイルス、ブンヤウイルス、風疹ウイルス、レオウイルス、ヘパドナウイルス、及びヒト免疫不全ウイルスを含めたレトロウイルスが含まれる。細菌には、マイコバクテリア、スピロヘータ、リケッチア、クラミジア、及びマイコプラズマが含まれる。菌類には、酵母及びカビが含まれる。寄生生物には、住血吸虫、リーシュマニア、及びマラリア原虫が含まれる。この一覧には、本明細書に記載の方法に従ってその防御免疫応答を生じさせることができるあらゆる潜在的な病原体が含まれていないことは理解されよう。
【0100】
特に、その遺伝子は、インフルエンザ血球凝集素、インフルエンザ核タンパク質、インフルエンザM2、破傷風毒素Cフラグメント、炭疽菌防御抗原、炭疽菌致死因子、炭疽菌発芽因子、狂犬病糖タンパク質、HBV表面抗原、HIV gp120、HIV gp160、マラリアCSP、マラリアSSP、マラリアMSP、マラリアpfg、ボツリヌス毒素A、及び結核菌HSPからなる群から選択される1種又は複数の抗原/免疫原をコードすることができる。
【0101】
別の実施形態では、遺伝子は、腫瘍由来の抗原/免疫原をコードすることができる。この腫瘍は、乳腺、卵巣、肺、脳、胃、腸管、膵臓、膀胱、前立腺、骨、又は造血性の細胞若しくは組織の腫瘍、或いは他の細胞又は組織の腫瘍とすることができる。特に、その遺伝子は、HER2/neu、ヒト癌胎児性抗原、及び前立腺特異的抗原からなる群から選択される1種又は複数の抗原/免疫原をコードすることができる。
【0102】
別の実施形態では、遺伝子は、抗体を生じるべく選択された任意の抗原/免疫原をコードすることができる。この抗体は、ポリクローナル抗体又はモノクローナル抗体とすることができる。この抗体は、特に、マウス抗体、キメラ抗体、ヒト化抗体、完全ヒト抗体、ドメイン抗体、又は短鎖抗体であってよい。この抗体は、例えば、研究、診断、治療、又は他の目的に有用であり得る。
【0103】
さらに別の実施形態では、遺伝子は、アジュバントをコードすることができる。このアジュバントは、共刺激分子、サイトカイン、又はケモカインとすることができる。好ましいアジュバントは、IL−2、IL−4、IL−6、IL−12、IL−18、GM−CSF、又はINFγである。
【0104】
別の実施形態では、医薬組成物は、(a)本明細書に記載の1種若しくは複数の抗原/免疫原及び/又はアジュバントをトランスフェクトした細胞を含有するか、或いは(b)本明細書に記載の1種若しくは複数の抗原/免疫原及び/又はアジュバントから様々に選択したものをトランスフェクトした細胞群を含有するカプセルを含む。カプセル内の細胞の選択は、ワクチン接種の必要性に適合させることができることは理解されよう。これは、抗原/免疫原及びアジュバントの適切な選択、発現ベクター及び発現カセットの適切な選択、対象の遺伝子を発現する細胞型若しくは細胞系の適切な選択、並びに/又は本明細書及び当技術分野で記載されているカプセル化材料の適切な選択によって達成することができる。
【0105】
本発明の別の実施形態は、本明細書に記載のカプセル又は医薬組成物を被検体に投与するステップを含む、免疫する方法である。
【0106】
本発明の別の実施形態は、対象のタンパク質を被検体に投与するための、本明細書に記載のカプセル又は医薬組成物の使用である。
【0107】
好ましい実施形態は、対象のタンパク質を被検体に投与するための、本明細書に記載のカプセル又は医薬組成物の使用であり、このタンパク質は、ワクチン接種用の抗原/免疫原である。
【0108】
好ましい実施形態は、対象のタンパク質を被検体に投与するための、本明細書に記載のカプセル又は医薬組成物の使用であり、このタンパク質は、抗体の標的として選択された抗原/免疫原である。特に、このカプセルは、被検体において、カプセル化細胞によって産生される事前選択した抗原に対する抗体応答を誘発させるのに使用される。
【0109】
本発明の別の実施形態は、本明細書に記載のカプセル又は医薬組成物を含むキット、及び前記カプセル又は組成物を被検体に適用するための手段である。
【0110】
この被検体は、動物とすることができ、有利には脊椎動物、例えば、哺乳動物、鳥類、爬虫類、両生類、又は魚類であり、より有利にはヒト、或いは飼育動物、家畜化動物、食物を生産する動物、飼料を生産する動物、家畜、狩猟動物、競争用動物、又は競技用動物、例えば、ウシ、イヌ、ネコ、ヤギ、ヒツジ、ブタ、若しくはウマ、又はさらには家禽、例えば、シチメンチョウ、カモ、若しくはニワトリである。別の実施形態では、この脊椎動物は、それだけには限らないが、げっ歯類(マウス、モルモット、及びラットを含む)、イヌ、ブタ、及びサルを含めた研究目的で飼育された動物である。本発明の最も好ましい実施形態では、脊椎動物はヒトである。
【0111】
本明細書で開示されるカプセル又は医薬組成物は優先的には、非経口経路で被検体に投与される。例えば、個体に、腹腔内、皮内、皮下、又は筋肉内の方法によって接種することができる。
【0112】
本発明の他の特徴及び利点は、限定的な特性ではなく例示的なものを有する以下の実施例から明らかとなろう。実施例は、好都合なことには、添付の図面を参照することによって読まれよう。
【実施例】
【0113】
(実施例1:IL−6をコードするDNAを一過性にトランスフェクトした細胞を含有するカプセルの調製、並びにマウスコンカナバリンAモデルにおけるIL−6血中レベル及びその活性の測定)
1.対象の遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製
1.1 細胞の培養及び維持
エプスタイン−バーウイルス核抗原を発現するヒト胎生腎293細胞系(HEK293−EBNA、Invitrogen、米国)の細胞を、スピナーフラスコ(Techne、英国)中で4mM L−グルタミン(Invitrogen)及び1ml/lフェノールレッド溶液(水中0.5%w/v、フェノールレッド:Sigma、米国)を補充したEx細胞VPRO無血清培地(維持培地、JRH、英国)中で懸濁状態で維持した。
【0114】
1.2 プラスミド調製
HEK293−EBNA細胞内でIL−6を発現させるためのプラスミドの構築
ヒトIL−6の完全コード配列(ORF)(図1、配列番号1)を含有するプラスミドをInvitrogen社から購入した(Invitrogen社クローンID CS0DI019YP05)。Gateway(商標)クローニング法(Invitrogen)を使用して、このORFを哺乳動物細胞発現ベクターpEAK12d(図2A及び2B)中にサブクローニングした。
【0115】
1.2.1 6HISタグ配列にインフレームで融合させたGateway適合性IL−6 ORFの作製
Gatewayクローニングプロセスの第1段階は、2ステップのPCR反応を含み、この反応では、5’末端でattBl組換え部位及びコザック配列に隣接し、3’末端で6ヒスチジン(6HIS)タグをインフレームでコードする配列、終止コドン及びattB2組換え部位に隣接するIL−6のORF(Gateway適合性cDNA)が作製される。第1のPCR反応液(最終量50μl中)は、25ngのCS0DI019YP05プラスミド、2μlのdNTP(5mM)、5μlの10×Pfxポリメラーゼ緩衝液、各0.5μlの遺伝子特異的プライマー(100μM)(EX1フォワード及びEX1リバース)、及び0.5μlのPlatinum Pfx DNAポリメラーゼ(Invitrogen)を含有する。PCR反応は、最初の変性ステップ95℃で2分間、続いて94℃で15秒間及び68℃で30秒間を12サイクル実施した。Wizard PCR prep DNA精製システム(Promega)を製造元の使用説明書に従って使用して、反応混合液からPCR産物を直接精製した。第2のPCR反応液(最終量50μl中)は、10μlの精製PCR産物、2μlのdNTP(5mM)、5μlの10×Pfxポリメラーゼ緩衝液、各0.5μlのGateway変換プライマー(100μM)(GCPフォワード及びGCPリバース)、及び0.5μlのPlatinum Pfx DNAポリメラーゼを含んでいた。第2のPCR反応の条件は、95℃で1分間;94℃で15秒間、45℃で30秒間、及び68℃で3.5分間を4サイクル;94℃で15秒間、55℃で30秒間、及び68℃で3.5分間を25サイクルであった。PCR産物を上記の通り精製した。
【0116】
1.2.2 GatewayエントリーベクターpDONR201及び発現ベクターpEAK12d中へのGateway適合性IL−6 ORFのサブクローニング
Gatewayクローニングプロセスの第2段階は、Gateway修飾PCR産物をGatewayエントリーベクターpDONR201(Invitrogen)中に以下の通りサブクローニングすることを含む。5μlの精製PCR産物を1.5μlのpDONR201ベクター(0.1μg/μl)、2μlのBP緩衝液、及び1.5μlのBPクロナーゼ酵素混合液(BP clonase enzyme mix)(Invitrogen)と共に室温で1時間インキュベートする。この反応をプロテイナーゼK(2μg)の添加によって停止させ、37℃でさらに10分間インキュベートした。この反応液の一定分量(2μl)を、Biorad Gene Pulserを使用したエレクトロポレーションにより大腸菌DH10B細胞に形質転換した。形質転換体をLBカナマイシンプレート上に蒔いた。Wizard Plus SV Miniprepsキット(Promega)を使用して、得られた1〜4個のコロニーからプラスミドミニプレップDNAを調製し、次いで1.5μlのプラスミド溶出物を、最終量10μl中に1.5μlのpEAK12dベクター(図2)(0.1μg/μl)、2μlのLR緩衝液、及び1.5μlのLRクロナーゼ(Invitrogen)を含有する組換え反応液に使用した。混合液を室温で1時間インキュベートし、プロテイナーゼK(2μg)の添加によって反応停止させ、37℃でさらに10分間インキュベートした。この反応液(1μl)の一定分量を使用して、エレクトロポレーションにより大腸菌DH10B細胞に形質転換した。
【0117】
pEAK12dプライマー(pEAK12d F及びpEAK12d R)をPCR用に使用した以外は上記の通りコロニーPCRを実施することにより、正しいインサートを含有するクローンを同定した。Qiaprep Turbo 9600ロボットシステム(Qiagen)を使用するか、又はWizard Plus SV miniprepsキット(Promega)を手動で使用して、正しいインサートを含有するクローンからプラスミドミニプレップDNAを単離し、pEAK12d F及びpEAK12d Rプライマーを使用して配列を確認した。
【0118】
配列を確認したクローンの培養物500mlから、プラスミドpEAK12d−1L6−6HIS(プラスミド番号11381、図3)のCsCI勾配精製maxi−prepDNAを調製し(Sambrookら,in Molecular Cloning,a Laboratory Manual,第2版,1989,Cold Spring Harbor Laboratory Press)、滅菌水で1μg/μlの濃度で再懸濁させ、−20℃で保存した。
表I
IL−6サブクローニング及びシークエンシング用プライマー
【0119】
【表1】

【0120】
1.3 細胞トランスフェクション
トランスフェクションの当日に、細胞を遠心分離し、スピナー容器(DasGip,D)中の播種培地である1%FBS及び4ml/l ITS−Xサプリメントを含有する250mlのDMEM/F12(1:1)培地(Invitrogen)で細胞1×106/mlの密度で再懸濁させた。ポリエチレンイミン(PEI)法を使用して(O.Boussifら1995 Proc.Natl.Acad.Sci.USA 92,7297〜7301頁)、細胞に2:1の比のPEI:DNAをトランスフェクトした。100mlの播種培地中で、500μgの相当するプラスミドDNAを1mgのPEI(Polysciences、米国)と混合し、室温で10分間インキュベートした。この混合物を細胞懸濁液に添加し、37℃で90分間インキュベートした。インキュベーション後、この細胞懸濁液を遠心分離し(200×g、10分間、4℃)、細胞ペレットを500mlの維持培地で再懸濁させた。細胞は、カプセル化するまで5%CO2、37℃の加湿雰囲気下でインキュベートした。
【0121】
1.4 細胞カプセル化
上記で得られたHEK293−EBNAトランスフェクト細胞及び野生型HEK293−EBNA細胞を、米国特許第6458296号に記載のエンカプスレータに類似するInotech researchエンカプスレータ(Inotech、スイス)を使用してアルギナート−ポリ−L−リシン−アルギナート(APA)カプセル内に封入した。細胞を遠心分離し(200×g、10分間、4℃)、2ml洗浄緩衝液で再懸濁させた(すべての薬品はInotech社、スイス)。この懸濁液に1.5%アルギナート溶液を細胞2.5×106/溶液mlの最終細胞濃度が得られるまでゆっくり添加した。このアルギナート−細胞懸濁液をカプセル化装置に接続したシリンジ(Braun Omnifit,Braun,D)で吸い取った。表1に示したパラメータ、及び表2に記載のプロトコールを使用してカプセル化を実施した。すべての緩衝液は、製造元の使用説明書に従い滅菌蒸留水中、無菌条件下で調製した。
【0122】
カプセル化の最終ステップの最後に、カプセルを100mlの維持培地で再懸濁させ、滅菌スピナー容器(Dasgip,D)に移した。カプセルをスピナー容器中で維持し、5%CO2、37℃の加湿雰囲気下で一晩、又は動物に注射するまでインキュベートした。
【0123】
カプセルは、顕微鏡で測定すると485±10μmの平均直径を有していた。
表2 カプセル化パラメータ
【0124】
【表2】
表3 カプセル化プロトコール
【0125】
【表3】
【0126】
1.5.低剪断、微小重力条件下でのカプセルの維持の改良
一方はスピナー容器中で、もう一方は、低剪断、微小重力(10−12g)条件下、Syntecon社/NASA(米国ヒューストン)によって製造された水平低速回転式容器(slow turning lateral vessel)STLV(商標)(米国特許第6730498号参照)を使用して、上記で得られたカプセルの維持について比較実験を実施した。この細胞は、空気中5%CO2、37℃の加湿インキュベータ内の維持培地中で維持した。試験されたカプセルは、野生型HEK293−EBNA細胞、又はh−IL−6のcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルであった。
【0127】
低剪断、微小重力下で、カプセルの目に見えた破壊が7日間起こらないことが示されたが、スピナー容器では、同じ期間無傷であったカプセルが、カプセルの破壊によって生じた浮遊している細胞によって目に見えて囲まれていった。h−IL−6のcDNAをトランスフェクトした細胞については、in vitroで放出されたh−IL−6は、スピナー容器よりも低剪断、微小重力下で実質的に少なかった。
【0128】
2 コンカナバリンAモデル(ConA)
材料及び方法:
2.1 動物
すべての試験でC57/BL6雄マウス(8週齢)を使用した。常時、実験群あたり10匹のマウスを使用した。マウスを12時間の明暗周期、標準条件下で維持し、放射線照射飼料及び水を任意に与えた。
【0129】
2.1 カプセル注射
カプセル懸濁液をインキュベータから取り出し、層流フード内で数分放置してカプセルを沈殿させた。上澄液を除去し、濃縮したカプセルをシリンジで注意深く吸い上げた。700μlのカプセルを0.7mmの針(参照番号53158.01、Polylabo、スイス)で各マウスにゆっくりi.p.注射した。
【0130】
2.2 コンカナバリンA注射
コンカナバリンA(ConA)はSigma社から購入した(参照番号C7275、Sigma,D)。カプセルの移植から72時間後、ConAを18mg/kgでi.v.注射した。ConA注射から1.30時間後と8時間後に血液試料を採取した。サイトカイン及びトランスアミナーゼレベルの測定は、以下に記載の通り実施した。
【0131】
2.4 読取り
2.4.1 血液試料採取
ConA注射から1.30時間後と8時間後に眼窩後の静脈洞から100μlの血液試料を採取した。屠殺するときに血液を心臓から採取した。
【0132】
2.4.2 血液試料中のサイトカイン及びトランスアミナーゼの検出
TH1/TH2 CBAアッセイ(参照番号551287、Beckton Dickinson、米国)を使用してサイトカインIL−2、IL−4、IL−5、TNFα、及びIFNγのレベルを測定した。COBAS装置(Hitachi、スイス)を使用して、アスパラギン酸アミノトランスフェラーゼ(ASAT)、アラニンアミノトランスフェラーゼ(ALAT)、UREAの各血中パラメータを測定した。
【0133】
2.4.3 血液及び細胞培養物上清中のh−IL6の検出
カプセル化及びトランスフェクトされたHEK細胞からのhIL−6のin vivoでの分泌を追跡するために、カプセル注射後12日間毎日、一日あたり5匹のマウスの血液を採取した。
並行してIn vitroで上清を10日目まで採取した。市販のELISAキット(R&D Duoset、参照番号DY206)により検出を血清中で行った。すべての試料を希釈緩衝液で2倍希釈した。
【0134】
2.5 結果:
2.5.1 トランスフェクトしなかったHEK293−EBNA細胞を含有するカプセルの注射
第1の実験では、トランスフェクトしなかったカプセル化HEK293−EBNA細胞を試験した。このようなカプセルは、正常な動物においてサイトカインレベル又は血中パラメータを変化させないことが以前に示されている。0日目及び2日目にこのカプセルを注射した。次いで、ConAを注射した。ConA注射から1.5時間後及び8時間後にトランスアミナーゼ及びTNF−αレベルを測定した。
【0135】
図1に結果を示すが、この図は、トランスフェクトしなかったHEK293−EBNA細胞を含むカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したTNFα、ASAT、及びALATレベルを示したヒストグラムを表す。
【0136】
その図に示す通り、トランスフェクトしなかったHEK293−EBNA細胞を含有するカプセルを注射したマウスにおいて、対照マウス(ConA注射のみを施したマウス)と比較して、NF−α又はトランスアミナーゼASAT及びALATレベルの有意な変化は認められなかった。
【0137】
2.5.2 hIL−6を一過性にトランスフェクトしたHEK細胞を含有するカプセルのIn vivo活性
hIL−6は、in vivoでConA誘発肝炎におけるトランスアミナーゼ及びTNF−αのレベルを低減することが分かっている(Mizuharaら1994,J.Exp.Med.,179,1529〜1537)。
【0138】
第1の実験では、hIL−6 HISをコードしているcDNAをトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを雄C57 Bl/6にi.p.又はs.c.注射した。カプセル注射の直後から毎日12日間連続で血中hIL−6レベルを測定した。
【0139】
図2Aに結果を示すが、この図は、hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをi.p.又はs.c.注射した後12日間連続してマウスの血液中で測定したhIL−6レベルのグラフを表す。
【0140】
この図で示される通り、血中hIL−6レベルは、i.p.カプセル注射後1日目から5日目の間でピーク値に達し、i.p.注射したマウスの血中hIL−6レベルの値は常にs.c.注射したマウスよりも実質的に大きかった。i.p.又はS.C.注射後8日目、血中hIL−6レベルは、基底値に近い値に達した。
【0141】
再現性:上記の通り別々にカプセル化され、様々な群に分けられた細胞の同一プール、又は上記の通り別々に調製した細胞の様々なプールのいずれかを用いてこの実験を数回反復すると、いずれの場合においても、血中hIL−6レベルは、カプセルのi.p.注射後1日目から5日目の間でピーク値に達し、血中hIL−6レベルの最大値、及び最初の5日間の積算値は有意差がない(20%未満)ことが示された。
【0142】
第2の実験では、hIL−6 HISをコードしているcDNAをトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを0日目及び3日目に雄C57 Bl/6にi.p.注射した。さらに、18mg/kg ConAをi.v.注射して肝炎を誘発させた。ConA注射から1.5時間後及び8時間後にTNF−α及びトランスアミナーゼレベルを測定した。
【0143】
図2B〜Dに結果を示すが、これら図は、hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したTNFα、ASAT、及びALATレベルを示すヒストグラムを表す。
【0144】
これらの図で示される通り、ConA注射の後、5%CO2、37℃の加湿雰囲気下でインキュベートしたカプセルを事前注射したConA処置マウスのTNF−α及びトランスアミナーゼレベルは、トランスフェクトしなかったカプセル化HEK細胞を注射したConA処置マウスと比較して有意に低かった。
【0145】
したがって、カプセルからin vivoで放出されたhIL−6は、ConA誘発肝炎においてトランスアミナーゼ及びTNF−αのレベルを低下させる既知の予測された作用を有する。
【0146】
2.5.3 HEK293−EBNA−hIL6−HISカプセルを様々な量注射した後の用量反応の確立
hIL−6 HISをコードしているcDNAを一過性にトランスフェクトした細胞を含有するカプセルに関する用量反応関係を確立するための実験を実施して、ConA POCモデルにおいてこの技術を使用できるかどうかを実証した。700、350及び100μlのカプセルの注射後1日目、2日目及び3日目に、全身のhIL−6タンパク質産生を測定した。
【0147】
図3に結果を示すが、この図は、hIL−6−6HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセル700、350、及び100μlのi.p.注射後3日間連続してマウスの血液中で測定したhIL−6レベルを示すヒストグラムを表す。図4A〜Cに結果を示すが、この図は、hIL−6−6HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをそれぞれ700、350、及び100μl注射したConA処置マウス、並びに対照マウスの血液試料中で測定したTNFα、ASAT、及びALATレベルを示すヒストグラムを表す。
【0148】
これらの図で示される通り、血液中のhIL−6タンパク質の産生、並びにTNFα及びトランスアミナーゼの下方制御に対する生物学的作用に関する用量効果を確立することができた。
【0149】
(実施例2:hepaCAM遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製、及びLPS誘発TNFα放出のマウスモデルにおけるその活性の測定)
1.hepaCAM遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製
1.1 細胞の培養及び維持:実施例1参照
【0150】
1.2 プラスミド調製
hepaCAM遺伝子(遺伝子の配列:配列番号7に関する図8A、及びタンパク質の配列:配列番号8に関する図8Bを参照のこと)を、国際公開第03/093316号に詳細に記載されている通り、発現ベクターpEAK12d中にクローニングした(Mei Chung MohらのJ Hepatol.2005 Jun;42(6):833〜41、Epub 2005 Apr7、及びJ Biol Chem.2005 Jul22;280(29):27366〜74、Epub 2005 May25も参照のこと)。
【0151】
簡単に言えば、先ず、2ステップPCRを使用してこの遺伝子をGateway(商標)クローニングシステム(Invitrogen)のpENTRベクター中にクローニングした。pEAK12dベクター中へのサブクローニングは、Gatewayクローニングマニュアルに従って実施した。この遺伝子をゲノム配列からの3つのエクソンのPCR増幅によってクローニングした。
【0152】
使用したプライマーは以下である。
【0153】
【化1】
【0154】
1.3 細胞トランスフェクション:実施例1参照
【0155】
1.4 細胞カプセル化:実施例1参照
【0156】
2.LPS誘発TNFα放出のマウスモデル
プロトコール
マウスにおけるLPS誘発TNFα放出のモデルは、国際公開第98/38179号に記載の通り設定した。
【0157】
一過性にトランスフェクトされたカプセル化HEK293−EBNA細胞の注射後3日目に、0.3mg/kg LPS(0111:B4、Sigma、スイス)をC3H/HeNマウス(それぞれ8匹のマウスからなる群)(Charles River、仏国)にi.p.又はs.c.注射した。90分後、血液試料を採取し、ELISAキットを使用して血漿TNFαを測定した。デキサメタゾン(0.1mg/kg、sc)をPBSに溶解させ、注射し、15分後LPS誘発を行った。
【0158】
結果
図8Cを参照のこと。
【0159】
トランスフェクトしなかった、又はhepaCAMを一過性にトランスフェクトしたカプセル化HEK293−EBNA細胞のいずれかをi.p.又はs.c.経路で注射したマウスにLPSを注射すると、それぞれTNFα放出の非阻害又は有意な阻害が認められた。LPS誘発サイトカイン放出を80%阻害したデキサメタゾンを陽性対照とした。したがって、HepaCAMは、LPS誘発TNFαレベルを有意に下方制御した。
【0160】
こうした結果から、hepaCAMがTNFα媒介の炎症疾患を治療するのに有用であろうことが示される。
【0161】
(実施例3:INSP114−SV2をコードしているDNAを一過性にトランスフェクトした細胞を含有するカプセルの調製、及びLPS誘発TNFα放出のマウスモデルにおけるその活性の測定)
1.INSP114−SV2遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製
1.5 細胞の培養及び維持:実施例1参照
【0162】
1.6 プラスミド調製
INSP114SV2遺伝子(遺伝子の配列:配列番号9に関する図9A、及びタンパク質の配列:配列番号10に関する図9Bを参照のこと)を国際公開第2004/085469号に詳細に記載されている通り、発現ベクターpEAK12d中にクローニングした。
【0163】
使用したプライマーは以下である。
【0164】
【表4】
【0165】
1.7 細胞トランスフェクション:実施例1参照
【0166】
1.8 細胞カプセル化:実施例1参照
【0167】
2.LPS誘発TNFα放出のマウスモデル
実施例2を参照のこと。
【0168】
結果
図9Bを参照のこと。
【0169】
トランスフェクトしなかった、又はINSP114SV2を一過性にトランスフェクトしたカプセル化HEK293−EBNA細胞のいずれかをi.p.経路で注射したマウスにLPSを注射すると、それぞれTNFα放出の非阻害又は有意な阻害が認められた。LPS誘発サイトカイン放出を80%阻害したデキサメタゾンを陽性対照とした。したがって、INSP114SV2は、LPS誘発TNFαレベルを有意に下方制御した。
【0170】
こうした結果から、INSP114SV2がTNFα媒介の炎症疾患を治療するのに有用であろうことが示される。
【0171】
(実施例4:EPO遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製、並びにマウスモデルにおける血中EPO濃度及びヘマトクリット値の測定)
1.EPO遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製
1.1 細胞の培養及び維持:(実施例1参照)
【0172】
1.2 プラスミド精製
EPO遺伝子(遺伝子の配列:配列番号5に関する図10A、及びタンパク質の配列:配列番号6に関する図10Bを参照のこと)を、IL−6遺伝子に関する実施例1に記載のものに類似するプロトコール及び以下のプライマーを使用して発現ベクターpEAK12d中にクローニングした。
【0173】
【表5】
【0174】
1.3 細胞トランスフェクション
1.3.1:EPO遺伝子を一過性にトランスフェクトした細胞の調製
トランスフェクションの当日に、HEK293−EBNA細胞を遠心分離し、スピナー容器(DasGip,D)中の播種培地である1%FBS及び4ml/l ITS−Xサプリメントを含有する250mlのDMEM/F12(1:1)培地(Invitrogen)で細胞1×106/mlの密度で再懸濁させた。ポリエチレンイミン(PEI)法を使用して(O.Boussifら1995 Proc.Natl.Acad.Sci.USA 92,7297〜7301頁)、細胞に2:1の比のPEI:DNAをトランスフェクトした。100mlの播種培地中で、500μgの相当するプラスミドDNAを1mgのPEI(Polysciences、米国)と混合し、室温で10分間インキュベートした。この混合物を細胞懸濁液に添加し、37℃で90分間インキュベートした。インキュベーション後、この細胞懸濁液を遠心分離し(200×g、10分間、4℃)、細胞ペレットを500mlの維持培地で再懸濁させた。細胞は、カプセル化するまで5%CO2、37℃の加湿雰囲気下でインキュベートした。
【0175】
1.3.2:EPO遺伝子をトランスフェクトした細胞の半安定プールの調製
この技術は、ピューロマイシン−N−アセチル−トランスフェラーゼ耐性遺伝子をその骨格中に含むpEAK12dに挿入されたEPO遺伝子のエピソームによる複製、及び細胞の半安定プールを選択するためのピューロマイシン選択圧の適用に基づく。
【0176】
EPO遺伝子の初回のトランスフェクション
HEK293−EBNA細胞をEx細胞VPRO無血清培地(細胞保存、維持培地、JRH)中で懸濁状態で維持した。トランスフェクションの当日に、細胞を計数し、遠心分離し(低速)、ペレットを所望の量のトランスフェクション培地、すなわち1%FCS(JRH)及び4ml/lインスリントランスフェリンセレニウム(Gibco)を補充したDMEM/F12(1:1)(FEME培地、Invitrogen)で再懸濁させて、生細胞1×106/mlの細胞濃度を得た。上記の1.2におけるPEAK12d中へのEPO遺伝子のクローニングによって得られたDNA保存液(1mg/ml保存液)をFEME培地に2mg/トランスフェクション1リットル量(2%eGFPレポーター遺伝子を同時トランスフェクト)で希釈した。次いで、トランスフェクション剤ポリエチレンイミン(PEI、4mg/リットル量、Polysciences)をcDNA溶液に添加し、ボルテックスでしっかり撹拌し(30秒間)、室温で10分間インキュベートした(「トランスフェクションミックス」の作製)。
【0177】
次いで、このトランスフェクションミックスをスピナー容器に添加し、CO2インキュベータ(5%CO2、37℃)内で90分間インキュベートした。90分後、PEI−EPO cDNA複合体のHEK293−EBNA細胞中への組込みが可能となり、細胞を遠心分離し、Ex細胞VPRO無血清新鮮培地に再懸濁させ、例えば、単細胞懸濁物として維持した。次いで、トランスフェクションから24〜48時間後、無血清培地中で増殖させた上記のEPO HEK293−EBNA細胞の試料を蛍光顕微鏡下で観察して、例えば、トランスフェクト細胞に蛍光をもたらしたレポーター遺伝子を視覚化した。蛍光が細胞の80%超で認められた場合に限り、この培養物をさらなる半安定圧選択のために維持した。
【0178】
ピューロマイシン2重圧選択
トランスフェクションから72時間後、EPOを発現するHEK293−EBNA細胞を懸濁培養により増殖させた培地を7.5μg/mlのピューロマイシンを含有するEx−細胞VPRO新鮮培地(Clontech)と交換し、このピューロマイシン選択圧を6日間維持した。この期間の間、乳酸濃度が1.3g/lを超えると(乳酸による増殖阻害)、選択圧培地を新鮮選択圧培地と交換した(依然ピューロマイシンを含有する)。蛍光顕微鏡及び依然増殖している(集団倍加)懸濁細胞によって視覚化すると、非組換え細胞は死滅し(細胞のわずか10%、PEIトランスフェクション効率が高いと85%を超える)、EPO組換え細胞は圧選択に耐えた。
【0179】
選択期間の最後に、培地を通常の増殖培地(ピューロマイシンなし)と交換し、細胞をタンパク質発現について2又は3週間モニターした。2回目のピューロマイシン選択圧(上記と同じ選択圧)を細胞懸濁物にさらに6日間施すと、例えば、すべての非組換え細胞が死滅することが確かめられた。
【0180】
細胞増殖をモニターし、例えば、カプセル化操作のために細胞を収集するのに十分な量の細胞を培養物中で維持しながら、この培養物の一部の凍結保存により少量の細胞保存液を置いておいた。
【0181】
得られた半安定細胞は、EPO遺伝子を約3カ月発現することができる。
【0182】
1.4 細胞カプセル化
1.3.1で得られた一過性にトランスフェクトした細胞、及び1.3.2で得られた半安定細胞を実施例1の1.4に記載の通りカプセル化した。
【0183】
2.マウスモデルにおける血中EPO濃度及びヘマトクリット値の測定
2.1 プロトコール
全ての試験で、C57/BL6雄マウス(8週齢)を使用した。常時、実験群あたり5匹のマウスを使用した。マウスを12時間の明暗周期、標準条件下で維持し、放射線照射飼料及び水を任意に与えた。
【0184】
カプセル懸濁液をインキュベータから取り出し、層流フード内で数分放置してカプセルを沈殿させた。上澄液を除去し、濃縮したカプセルをシリンジで注意深く吸い上げた。EPOを一過性にトランスフェクトした細胞、及びEPOをトランスフェクトした半安定細胞、又はトランスフェクトしなかった細胞を含有する700μlのカプセルを0.7mmの針(参照番号53158.01、Polylabo、スイス)で各マウスにゆっくりi.p.又はs.c.注射した。
【0185】
カプセル注射の後10日間連続して、R&D SystemsからのhEPO ELISAキット(Quantitin IVD Human EPO、カタログ番号DEP00)を使用して血中EPOレベルを追跡した。ヘマトクリット値を測定した。
【0186】
2.2 結果
図10C及び10Dから明らかであるが、一過性に又は安定にトランスフェクトしたカプセル化細胞を(i.p.又はs.c.経路)与えたマウスにおけるヘマトクリット値の増加(10D)に対応する血中EPO濃度の増加(10C)を実験の14日間測定した。トランスフェクトしなかったカプセル化細胞を与えた対照マウスではこのような効果は観察されなかった。意外なことに、また驚くべきことに、一過性にトランスフェクトしたカプセル化細胞を与えたマウスにおけるEPO濃度及びヘマトクリット値は、安定にトランスフェクトしたカプセル化細胞を与えたマウスよりも高いレベルに達した。
【0187】
(実施例5:mIL−18BP遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製、及びマウスモデルにおける血中mIL−18BPレベルの測定)
1.mIL−18BP遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製
1.1 細胞の培養及び維持:実施例1参照
【0188】
1.2 プラスミド精製
mIL−18BP遺伝子(遺伝子の配列:配列番号3に関する図11A、及びタンパク質の配列:配列番号4に関する図11B)を、Mallat Z.ら2002 Circ.Res.91,441〜448頁に詳細に記載されている通り、発現ベクターpEAK12d中にクローニングした。
【0189】
簡単に言えば、プラスミドpCEP4−mIL18BP−d 23を鋳型として使用して、増幅反応を行った。PCR産物をNotI+HinDIIIで消化し、次いでNucleospin(商標)スピンカラムキット(Macherey−Nagel)によって精製し、HinDIII+NotIで消化したpEAK8ベクター(Edge BioSystems,Gaithersburg、MD、米国)にライゲートした。
【0190】
1.3 細胞トランスフェクション:実施例1参照
【0191】
1.4 細胞カプセル化:実施例1参照
【0192】
2.マウスモデルにおける血中mIL−18BPレベルの測定
2.1 プロトコール
17匹のC57/BL6雄マウス(8週齡)の群を使用した。マウスを12時間の明暗周期、標準条件下で維持し、放射線照射飼料及び水を任意に与えた。
【0193】
カプセル懸濁液をインキュベータから取り出し、層流フード内で数分放置してカプセルを沈殿させた。上澄液を除去し、濃縮したカプセルをシリンジで注意深く吸い上げた。mIL−18BPを一過性にトランスフェクトした細胞を含有する700μlのカプセルを0.7mmの針(参照番号53158.01、Polylabo、スイス)で各マウスにゆっくりi.p.注射した。
【0194】
カプセル注射の後8日間連続して、m−IL18BP用に実験室で作製したELISAを使用して血中EPOレベルを追跡した。
【0195】
ELISAの手順は以下の通りである。
【0196】
Labsystemコンビプレート12EBというプレートを使用した。コーティングは、0.1mlの1×PBS中の5mg/ml抗マウスIL−18BPであった。抗原親和性により、ウサギ血清からポリクローナル抗体を精製した。O/N、4℃でインキュベートした後、1×PBS+0.05%Tween20を用いて洗浄し、1×PBS、0.2%BSA(BSA、Sigma、参照番号A−2153)を用いて37℃で1時間ブロッキングし、1×PBS+0.05%Tween20を用いて洗浄した。標準物は、mIL−18BP 300ng/ml〜0.1ng/mlであり、試料は1×PBS、0.1%BSA、0.05%Tween20中0.1ml、37℃で2時間である。1×PBS+0.05%Tween20を用いて洗浄を実施した。0.1mlのビオチニル化抗マウスIL18BP(Peprotech)0.3mg/mlを37℃で2時間添加した。1×PBS+0.05%Tween20を用いて洗浄を実施し、0.1mlのextravidin HRP(Sigma、参照番号E2886)1/5000を静かに添加し、室温で30分間放置した。0.05mlの20%H2SO4を用いて反応を停止させた。492nmで読み取った。
【0197】
2.2 結果
図11Cから明らかであるが、血中m−IL18BPレベルは、カプセルのi.p.注射後3日目から6日目の間でピーク値に達し、8日目のm−IL18BPレベルは依然非常に高いままであった。
【図面の簡単な説明】
【0198】
【図1】ヒトIL−6(hIL−6)の完全コード配列及びその翻訳配列(配列番号1及び2)を示す図である。
【図2A】哺乳動物細胞発現ベクターpEAK12dの制限酵素地図である。
【図2B】哺乳動物細胞発現ベクターpEAK12dの完全ヌクレオチド配列(配列番号11)を示す図である。
【図3】哺乳動物細胞発現ベクターpEAK12d−IL−6−6HISの制限酵素地図である。
【図4】トランスフェクトされなかったHEK293−EBNA細胞を含有するカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したTNFα、ASAT、及びALATレベルを示すヒストグラムである。
【図5A】hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをi.p.又はs.c.注射した後12日間連続してマウスの血液中で測定したhIL−6レベルのグラフである。
【図5B】hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したTNFαレベルを示すヒストグラムである。
【図5C】hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したASATレベルを示すヒストグラムである。
【図5D】hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したALATレベルを示すヒストグラムである。
【図6】hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセル700、350、及び100μlそれぞれのi.p.注射後3日間連続してマウスの血液中で測定したhIL−6レベルを示すヒストグラムである。
【図7A】hIL−6−6HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをそれぞれ700、350、及び100μl注射したConA処置マウス、並びに対照マウスの血液試料中で測定したTNFαレベルを示すヒストグラムである。
【図7B】hIL−6−6HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをそれぞれ700、350、及び100μl注射したConA処置マウス、並びに対照マウスの血液試料中で測定したASATレベルを示すヒストグラムである。
【図7C】hIL−6−6HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをそれぞれ700、350、及び100μl注射したConA処置マウス、並びに対照マウスの血液試料中で測定したALATレベルを示すヒストグラムである。
【図8A】hepaCAM遺伝子の配列(配列番号7)を示す図である。
【図8B】hepaCAM成熟タンパク質の配列(配列番号8)を示す図である。
【図8C】トランスフェクトしなかったカプセル化HEK293−EBNA細胞、及びhepaCAMをコードしている遺伝子を一過性にトランスフェクトした異なる量のカプセル化HEK293−EBNA細胞をi.p.注射した後の、LPS誘発TNFα放出のマウスモデルにおけるTNFαレベルを示すヒストグラムである。
【図8D】トランスフェクトしなかったカプセル化HEK293−EBNA細胞、及びhepaCAMをコードしている遺伝子を一過性にトランスフェクトした異なる量のカプセル化HEK293−EBNA細胞をs.c.注射した後の、LPS誘発TNFα放出のマウスモデルにおけるTNFαレベルを示すヒストグラムである。
【図9A】INSP114−SV2をコードしている遺伝子の配列(配列番号9)を示す図である。
【図9B】INSP114−SV2成熟タンパク質の配列(配列番号10)を示す図である。
【図9C】トランスフェクトしなかったカプセル化HEK293−EBNA細胞、及びINSP114−SV2遺伝子を一過性にトランスフェクトしたカプセル化HEK293−EBNA細胞をi.p.注射した後の、LPS誘発TNFα放出のマウスモデルにおけるTNFαレベルを示すヒストグラムである。
【図10A】ヒトエリトロポエチン(EPO)をコードしている遺伝子の配列(配列番号5)を示す図である。
【図10B】ヒトエリトロポエチン(EPO)成熟タンパク質の配列(配列番号6)を示す図である。
【図10C】EPOをコードしている遺伝子を一過性にトランスフェクトしたカプセル化HEK293−EBNA細胞、EPOをコードしている遺伝子を安定にトランスフェクトしたカプセル化HEK293−EBNA細胞、及び対照カプセル化HEK293−EBNA細胞(トランスフェクトしなかった)をi.p.又はs.c.注射してから0日目〜15日目にELISAによって測定したマウスの血中EPO濃度を示すグラフである。
【図10D】EPOをコードしている遺伝子を一過性にトランスフェクトしたHEK293−EBNA細胞、及びEPOをコードしている遺伝子を安定にトランスフェクトしたHEK293−EBNAをi.p.又はs.c.注射してから0日目〜10日目のマウスにおけるヘマトクリット値(ヘマトクリット値(Ht)又はパック細胞容積(PCV)は、赤血球が占める血液量の割合である)を示すグラフである。
【図11A】マウスIL−18結合タンパク質(m−IL18BP)の遺伝子の配列(配列番号3)を示す図である。
【図11B】マウスIL−18結合タンパク質(m−IL18BP)の成熟タンパク質の配列(配列番号4)を示す図である。
【図11C】m−IL18BPをコードしている遺伝子を一過性にトランスフェクトしたカプセル化HEK293−EBNA細胞のi.p.注射から0日目〜8日目にELISAによって測定したマウスの血中m−IL18BP濃度を示すヒストグラムである。
【技術分野】
【0001】
本発明は、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセル、これらのカプセルを調製する方法、及びこのカプセルを使用して、これらの細胞によって発現及び分泌されたタンパク質のin vivo活性を評価するための方法に関する。
【0002】
本発明はさらに、免疫及びワクチン接種用の抗原/免疫原及び/又はアジュバントの送達に使用することができる配合物、組成物、及び方法に関する。より詳細には、本発明は、より効率的及び効果的に免疫及びワクチン接種するための、対象の遺伝子を一過性にトランスフェクトした細胞を含有するカプセルに関する。
【背景技術】
【0003】
タンパク質機能のin vivo評価は、新たな治療法の開発、特に新たなタンパク質治療を開発するための、新たな可溶性タンパク質又は未知の機能を有する可溶性タンパク質の評価への重要な段階である。このようなin vivo評価又は試験には、薬物開発の特に初期に入手することが難しい相当量の精製タンパク質が必要とされる。この過程を短縮するためには、遺伝子をin vivoで直接発現させることが興味深い代替法である。
【0004】
プラスミドDNAのin vivoエレクトロポレーションは、コストが低く安全に利用できるので、遺伝子をin vivoで直接発現させる有望な方法である(Davis H.L.ら1999 Hum.Gen.Ther.4,151〜159)。骨格筋線維へのDNAのエレクトロポレーションは、適切な条件下で使用すると、DNAが細胞内に非常に効率的に取り込まれること、導入遺伝子が長期間発現されることなど多くの特定の利点があるが、エレクトロポレーションを筋肉に行う主要な利点は、血流中で生物活性タンパク質が産生及び分泌されるその潜在的可能性にあり、したがって離れた標的に作用を及ぼす、組換えタンパク質の局所的産生が可能となる(Feewell J.G.ら2001 Mol.Ther.3,574〜583;Kreissら1999 J.Gene Med.1,245〜250;Aihara Hら1998 Nat.Biotechnol.16,867〜870;Li S.ら2001 Gene Ther,8,400〜407)。
【0005】
遺伝子をin vivoで直接発現させる別の方法は、DNAの流体力学的送達である。この方法は、大量の裸のDNA水溶液の肝臓への急速静注を利用する。マウス及びラットに適用する場合、これは、肝臓での高レベルの遺伝子発現、並びに低レベルであるが腎臓、肺、心臓など他の器官での遺伝子発現を導入するための完全で効率的な手段である(Liu F.ら1999 Gene Ther 6(7),1258〜1266;Yang J.ら2001 Hepatology,33(4),848〜859;Maruyama H.ら2002 J.Gene.Med.4(3),333〜341;Hodges B.L.ら2003 Expert Opin.Biol.Ther,3(6),911〜918)。
【0006】
エレクトロポレーション及び流体力学的遺伝子送達法のどちらにおいても、導入遺伝子の分泌レベル又は産生タンパク質の転写後修飾に影響を与える可能性はほとんどない。
【0007】
in vivoでのタンパク質の機能を評価するのに適した第3の方法は、所望の遺伝子産物をin situで産生する、トランスジェニック細胞系のマイクロカプセル化細胞をマウス又はラットなどの実験動物に投与するものである。この手法では、細胞を宿主の免疫系から物理的に隔離するマトリックス内に細胞が封入される。栄養物、外部刺激、及び治療用タンパク質の通過が可能であるが、移植片拒絶の原因である免疫成分に不透過性であり、したがって移植片の生着を延長する選択透過膜が使用される(Lim F.ら1980 Science,210,908〜910)。このような方法は、遺伝子治療(Hortelano G.ら1996 Blood,87(12),5095〜5103;Visted T.ら2001 Neuro−Oncology 3,201〜210)、インスリン依存性糖尿病(Sun Y.ら1996 J.Clin.Invest 98,1417〜1422)、及び抗癌療法(Read T−A.ら1999 Int.J.Devl.Neuroscience,17(5〜6),653〜663)のために開発されてきた。導入遺伝子の発現及び翻訳後修飾のレベルの完全な制御は、カプセル化される細胞系の選択性、カプセル内の細胞密度、及び発現カセットに調節エレメントを含む可能性によって可能である。発現すべき治療用タンパク質の特性に応じいくつかの生体適合性ポリマーを使用して細胞を固定化することができる。アルギナート−ポリ−L−リシン−アルギナート(APA)カプセルが最も頻繁に使用されているが(Lim F.ら1980 Science,210,908〜910;Hortelano G.ら1996 Blood,87(12),5095〜5103;Visted T.ら2001 Neuro−Oncology 3,201〜210;Sun Y.ら1996 J.Clin.Invest 98,1417〜1422;Read T−A.ら1999 Int.J.Devi.Neuroscience,17(5〜6),653〜663;Strand B.L.ら2002 J.Microencapsulation 19(5),615〜630を参照のこと)、ポリ(メタクリル酸ヒドロキシエチル−co−メタクリル酸メチル)(HEMA−MMA)など他のポリマーも使用することができる(Lahooti S.ら2000 Biomaterials,21,987〜995)。カプセル化マトリックスは、完全に固体であるか、又は包含させた細胞を環境から隔離する膜のみと共に液体コアを含有することができる。
【0008】
Savelkoulら(Savelkoul Huub F.J.ら1994 Journal of Immunological Methods 170 185〜196頁)は、Il−4又はIL−5を産生する安定的にトランスフェクトされたサル細胞系CV1又はCHO−Kiの細胞の調製及びカプセル化(2.3及び2.4、186頁参照)、並びにマウスへの前記カプセル化細胞の腹腔内(i.p.)又は皮下(s.c.)注射を記載している。
【0009】
国際公開第93/00439号は、活性因子又は増強物質を産生する遺伝子改変した細胞系、特にラットNGF放出細胞系N8−21の細胞のカプセル化を開示している(実施例1)。
【0010】
上記の従来技術に記載のあらゆる例において、カプセル化細胞は、安定的にトランスフェクトされた細胞に由来する。
【0011】
安定的にトランスフェクトされた細胞の作製は、トランスフェクトする遺伝子を細胞のゲノム中に組み込むことを必要とするので、労力を要し時間がかかる。これは、トランスフェクト細胞を選択培地中で長時間培養した後、培養細胞を段階希釈して、安定的にトランスフェクトされ対象のタンパク質を十分な量で産生する細胞クローンを単離することによって達成される(Maniatisら参照)。
【0012】
安定的にトランスフェクトされた細胞は通常、対象のタンパク質が高産生されるかどうかで選択される。安定的にトランスフェクトされた細胞は、トランスフェクトした遺伝子を高度に長期間(数カ月まで)発現する利点がある。
【0013】
しかし、上記の方法は、複数のタンパク質のin vivo活性の評価など、高スループット及び/又は高スピードを必要とする用途には適していない。
【0014】
本発明によって取り組まれる課題は、高スループットに適しており短期間内(例えば、1又は2又は3週間)に実施することができる、タンパク質をin vivoで発現させる方法を提供すること、及び従来技術の方法の欠点を有していない、タンパク質のin vivo活性を評価するための手段を提供することである。
【0015】
上記の課題を、特許請求の範囲で定義される本発明によって解決する。
【0016】
脊椎動物の免疫系の活性化は、病原体及び悪性腫瘍から動物を防御するための重要な機構である。この免疫系は、体液性及び細胞性の部門を含めた多くの相互作用成分からなる。体液性免疫には、抗原に直接結合する抗体が関与する。体液性免疫のエフェクターとしての抗体分子は、Bリンパ球によって分泌される。細胞性免疫には、非自己抗原を産生する他の細胞を認識し死滅させる分化した細胞障害性Tリンパ球(CTL)が関与する。CTLは、MHC(主要組織適合遺伝子複合体)クラスI分子に結合した、標的細胞の表面上に出現する分解されたペプチド断片に応答する。細胞内で産生されたタンパク質は、細胞代謝の一部としてペプチドに連続的に分解されることが理解されている。こうした断片は、MHC分子に結合し、細胞表面に輸送される。したがって、細胞性免疫系は、体内のあらゆる細胞内で産生されたタンパク質のスペクトルを絶えずモニターしており、非自己抗原を産生するいかなる細胞をも排除するような態勢にある。
【0017】
ワクチン接種は、抗原/免疫原に対して応答するように動物をプライミングする過程である。この抗原は、精製タンパク質、死滅/弱毒病原体に含まれるタンパク質として、又は次いで宿主細胞内で抗原を発現する遺伝子として(遺伝子免疫)投与することができる。この過程には、T及びBリンパ球、他の種類のリンパ球様細胞、並びに抗原を処理し、この抗原を免疫系を活性化できる形で提示することができる分化した抗原提示細胞(APC)が関与する。
【0018】
ワクチンの有効性は、腫瘍又は病原体による、後の抗原チャレンジに対する防御の程度によって評価される。有効なワクチンは、最少回数の接種の後、疾患を標的とした介入に対する高力価で持続的な防御免疫を誘導することができる免疫原である。
【0019】
ワクチン開発に成功した手法の数は、病原菌の数とほとんど同じぐらいである。技術が発展するにつれ、防御抗原/免疫原の性質を分子レベルで定義することが可能となる。近年、無細胞ワクチンが、種々の病原体由来のサブユニット(すなわち多成分系ワクチン)で投与することができ、有害反応の数を減少させる潜在的可能性を有するので、ワクチン開発のための好まれる方法となっている。サブユニットワクチンは、病原微生物由来の限定された精製防御抗原で構成されている。驚くべき成功例がいくつかあるが、現在使用されているサブユニットワクチンは少数である。おそらく、サブユニットワクチンの使用を妨害する最も困難な理由は、抗原送達の問題である。最適な抗原送達のためには、抗原がその生物学的状況下で抗原提示細胞に送達される必要があり、すなわち抗原には、食細胞によって容易に認識され取り込まれるリスクがある。大部分の抗原は、寄生生物−宿主細胞間相互作用にとって重要な三次元構造を有しているが、こうした構造の多くは抗原精製の際に失われる。
【0020】
サブユニットワクチン製造に伴う問題の多くを回避するのに取られた1つの手法は、防御抗原をin vivoで発現するように遺伝子改変した、弱毒ワクチン及び弱毒細菌に基づく組換え生ワクチンビヒクル(すなわち、ワクシニアウイルス、アデノウイルス、サルモネラ、及びウシ型結核菌種カルメットゲラン菌すなわちBCGの組換え体)の開発である(Snapper,S.B.ら、Proc.Natl.Acad.Sci USA85:6987〜6991,1988;Jackett,P.S.ら、J.Clin.Micro.26:2313〜2318,1988;Lamb,J.R.ら、Rev.Infect.Dis.11:S443〜S447,1989;Shinnick,T.M.ら、Infect.Immun.56:446〜451,1988を参照のこと)。
【0021】
生細菌を使用したワクチン接種が研究されており、致死遺伝子をノックアウトするように突然変異が誘発された生細菌株を利用することが多い。生ワクチンには、抗原が先天性の免疫原の形で発現され;生送達システムが宿主内で複製及び持続し、したがって宿主の免疫系が再刺激され多回投与の必要性が回避され;生ベクター系は、抗原を精製する必要性をなくし、製造が安価であり;生ベクターは、複数の抗原を送達するように設計することができ、したがって個体にワクチン接種しなければならない回数が減少するという利点がある。しかし、この方法は、細菌が毒性に戻ることを可能にするさらなる突然変異が起こらないことを保障するような厳しい安全対策を必要とする。より確実な方法は、後に宿主がその抗体を産生できるタンパク質を発現する弱毒細菌を利用することである。細菌ベクターは、ワクチンの経口投与用に研究されることが多い。例えば、HIV、ライム病、及びエプスタイン−バーウイルスから防御するための経口投与用にサルモネラ菌ベースのワクチンが研究されている。
【0022】
また、バキュロウイルス、酵母、及び組織培養細胞が、ワクチンの製造において使用するために研究されてきた。各例は、バキュロウイルスを使用して、E型肝炎に対するワクチンで使用されるタンパク質を産生した米国特許第6287759号;酵母をベクターとして使用して、ワクチンで使用するためのヘリコバクターポリペプチドを産生した米国特許第6290962号;脊椎動物組織培養細胞を使用して、ワクチンで使用するための精製不活化デングウイルスを増殖させた米国特許第6254873号で示されている。こうした例のすべてにおいて、ベクターを使用して、後に精製されワクチンで使用されるはずの対象のタンパク質が産生された。
【0023】
遺伝子免疫は、特定のタンパク質をコードしている遺伝子を動物自身の細胞内で発現させることによりこのタンパク質に対する免疫応答を誘導する別の手法である。in vivoでの長期間の抗原提示によって生じた相当な抗原増幅及び免疫刺激は、抗原に対する確実な免疫(solid immunity)を誘導することができる。遺伝子免疫では、タンパク質精製及びアジュバントとの混合というどちらもワクチン開発に常に必要とされる困難な段階が省かれるため、特定のタンパク質に対する免疫応答を生じさせるためのワクチン接種プロトコールが簡素化される。遺伝子免疫は、タンパク質の単離を必要としないので、生化学的に精製すると高次構造エピトープ(conformational epitope)を失う可能性があるタンパク質にとって特に価値がある。また、遺伝子ワクチンは、干渉を誘発することなく又は有効性に影響を及ぼすことなく混合して送達することもでき(Tangら,1992;Barryら,1995)、複数の抗原に対するワクチン接種計画を簡素化することができる。
【0024】
ワクチンは、アジュバントを使用することによって増強されることが多い。ワクチンアジュバントは、ワクチン組成物中で任意の特定の抗原と共に得られる免疫応答を改善するのに有用である。アジュバントは、抗体及びエフェクターT細胞の量を増加させ、抗原の量及び注射の頻度を低減するのに使用される。抗原は、アジュバントを含まないワクチンとして投与されることもあるが、効果的なアジュバントなしでは、有用な免疫応答を刺激するのに十分な免疫原性を欠く抗原が多く存在する。アジュバントはまた、得られる免疫応答を増大させ、又は抗原投与量を低減することができるという点で、「自給自足」抗原の免疫応答を改善する。
【0025】
分子ワクチン接種のためのアジュバント及び送達システムに関する包括的な総説については、Sheikh NA,Al−Shamisi M,Morrow WJW;Delivery systems for molecular vaccination;Current Opinion in Molecular Therapeutics 2000,2:1(37−54)を参照されたい。
【発明の開示】
【発明が解決しようとする課題】
【0026】
ワクチン開発が困難であった病原体又は腫瘍に対するワクチンを開発するためには、及び/又は十分に利用されなかったことによる市販ワクチンの失敗を克服するためには、ワクチンの免疫性及び/又は副作用の低下を可能にする抗原/免疫原送達の新たな方法を開発しなければならない。
【0027】
抗原/免疫原を含む新たな医薬組成物及びワクチン送達法は、本出願に記載されており、適当な抗原及び/又はアジュバントをコードしている1種又は複数の遺伝子を一過性にトランスフェクトしたカプセル化細胞を送達した後、抗原/免疫原をin vivoで産生させるものである。この方法は、タンパク質ワクチンに伴う、抗原/免疫原の厄介な精製の必要性、又は遺伝子免疫に伴う、抗原/免疫原のin vivo産生が低いことによる低効率など、上記の他のワクチン及びワクチン接種法の欠点を克服する。
【課題を解決するための手段】
【0028】
本発明は、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルに関する。
【0029】
別の態様では、本発明は、細胞が動物細胞、好ましくは哺乳動物細胞である前記カプセルを提供する。
【0030】
さらなる態様では、前記カプセルは、対象の遺伝子が、タンパク質を分泌するためのシグナル配列に融合しているカプセルである。
【0031】
本発明のさらに別の態様では、カプセルは、発現カセット中に挿入された対象の遺伝子を含む。
【0032】
さらなる態様では、カプセルは、プラスミド中に挿入された対象の遺伝子を含む。
【0033】
本発明のさらに別の態様では、前記カプセルは、生体適合性ポリマー、アルギナート−ポリ−L−リシン−アルギナート(APA)を含む。
【0034】
さらなる態様では、カプセルは、分離サイズが90〜30kDa、好ましくは80〜60kDaである孔を有する生体適合性ポリマー膜を含む。
【0035】
本発明のさらなる態様では、カプセルは、平均直径が100〜1500μm、好ましくは250〜600μm、特に440〜530μmである。
【0036】
本発明の別の態様では、カプセルは、低剪断、微小重力条件下で維持された。
【0037】
本発明はさらに、カプセルを調製する方法を提供し、この方法は、細胞に対象の遺伝子を一過性にトランスフェクトするステップと、一過性にトランスフェクトされた細胞をカプセル化するステップとを含む。
【0038】
本発明はさらに、カプセルを調製する方法を提供し、この方法は、低剪断、微小重力条件下で維持するステップをさらに含む。
【0039】
本発明はさらに、対象の遺伝子によってコードされており、一過性にトランスフェクトしカプセル化した細胞によって発現及び分泌されたタンパク質のin vivo活性を評価する方法を提供し、この方法は、上記のカプセルを多細胞生物に投与するステップと、前記多細胞生物において前記タンパク質の活性を検出するステップとを含む。
【0040】
本発明はさらに、対象の遺伝子によってコードされており、一過性にトランスフェクトしカプセル化した細胞によって発現及び分泌されたタンパク質のin vivo活性を評価する方法を提供し、この方法は、上記のカプセルを、哺乳動物である多細胞生物に投与するステップを含む。
【0041】
本発明はさらに、対象の遺伝子によってコードされており、一過性にトランスフェクトしカプセル化した細胞によって発現及び分泌されたタンパク質のin vivo活性を評価する方法を提供し、この方法は、上記のカプセルを、マウス、ラット、イヌ、ヤギ、ヒツジ、ウシ、及びサルからなる群から選択される哺乳動物に投与するステップを含む。
【0042】
本発明はさらに、対象の遺伝子によってコードされており、一過性にトランスフェクトしカプセル化した細胞によって発現及び分泌されたタンパク質のin vivo活性を評価する方法を提供し、この方法は、i.p.注射によって実施される、上記のカプセルを多細胞生物に投与するステップを含む。
【0043】
本発明はさらに、動物におけるカプセル又は上記の方法の使用を提供し、この使用では、検出すべき活性が急速に生じ(例えば、注射後数時間又は1日若しくは複数日以内)、活性物質の投与後数日以内に完了する。
【0044】
本発明はさらに、動物におけるカプセル又は上記の方法の使用を提供し、この使用では、検出すべき活性が、急速に生じ(例えば、注射後数時間又は1日若しくは複数日以内)、活性物質の投与後数日以内に完了し、動物は、コンカナバリンA(ConA)誘発肝毒性を有するマウスである。
【0045】
本発明はさらに、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルを含む医薬組成物を提供する。
【0046】
本発明はさらに、細胞に、抗原/免疫原及び/又はアジュバントをコードしている遺伝子がトランスフェクトされている、上記の医薬組成物を提供する。
【0047】
本発明はさらに、抗原/免疫原が、細菌抗原、ウイルス抗原、真菌抗原、寄生虫抗原、又は腫瘍抗原である、上記の医薬組成物を提供する。
【0048】
本発明はさらに、カプセルが、(1種又は複数の)抗原及び/又はアジュバントをコードしている遺伝子をそれぞれトランスフェクトした少なくとも2種類の細胞を含み、この2種の遺伝子が同一でない、上記の医薬組成物を提供する。
【0049】
本発明はさらに、対象のタンパク質を被検体に投与するための、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルの使用を提供する。
【0050】
本発明はさらに、対象のタンパク質を被検体に投与するための、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルの使用を提供し、このタンパク質は、免疫及びワクチン接種用の抗原である。
【0051】
本発明はさらに、対象のタンパク質を被検体に投与するための、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルの使用を提供し、被検体が、ヒト;げっ歯類、イヌ、ブタ及びサルを含めた研究目的で飼育された動物並びにイヌ及びネコを含めた、家畜及び飼育動物からなる群から選択される。
【0052】
本発明はさらに、上記のカプセル又は医薬組成物を含むキット、及び前記カプセル又は組成物を被検体に投与するための手段を提供する。
【発明を実施するための最良の形態】
【0053】
本発明は、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセルを提供する。
【0054】
用語「1種又は複数のカプセル」は、同義的に使用され、細胞を環境から隔離する生体適合性ポリマー膜のみと共に細胞を含有するカプセル化マトリックスを指す。
【0055】
用語「1種又は複数のカプセル」はさらに、生体適合性ポリマー膜内に封入又は包含された1種又は複数の細胞を指す。
【0056】
用語「一過性にトランスフェクトされた細胞」又は「対象の遺伝子を一過性にトランスフェクトした細胞」は、対象の遺伝子を当技術分野で既知の方法(Maniatisら参照)によって導入し、トランスフェクションの日から14日以内に、好ましくは10日以内に、対象の遺伝子によってコードされているタンパク質を発現する(1種又は複数の)細胞を指す。
【0057】
用語「一過性にトランスフェクトされた」又は「対象の遺伝子を一過性にトランスフェクトした」はさらに、当技術分野で既知の方法(Maniatisら参照)により、事前選択した細胞中に対象の遺伝子を導入し、ここで、前記細胞は対象の遺伝子を含有するが、この遺伝子は、前記細胞のゲノム中に組み込まれないこと、又は細胞内で複製することができるエピソームベクター上に位置することを指す。
【0058】
一過性にトランスフェクトされた細胞は、トランスフェクション後細胞を培養する際に選択培地によるどんな選択圧をもかけることなく得ることが好ましい。
【0059】
細胞培養で使用される選択培地は、当技術分野で周知である。長年にわたるトランスフェクション研究の間、いくつかの様々な薬剤選択マーカーが一般に使用されてきた。例えば、細菌のアミノグリコシドホスホトランスフェラーゼ遺伝子を含有する組換えベクターをトランスフェクトした細胞は、薬剤G−418の存在下で安定にトランスフェクトするかどうかで選択することができる(Southern,P.J.及びBerg,P.(1982)「SV40初期領域プロモーターの制御下にある細菌遺伝子を用いた哺乳動物細胞の抗生物質耐性への形質転換(Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter)」,J.Mol.Appl.Gen.1,327)。同様に、トランスフェクトされたベクターからのハイグロマイシンBホスホトランスフェラーゼ遺伝子の発現により、薬剤ハイグロマイシンBに対する耐性が得られる(Blochlinger,K.及びDiggelmann,H.(1984)「高等真核細胞を用いたDNA導入実験用の選択マーカーとしてのハイグロマイシンBホスホトランスフェラーゼ(Hygromycin B phosphotransferase as a selectable marker for DNA transfer experiments with higher eucaryotic cells)」,Mol.Cell.Biol.4,2929)。
【0060】
代替戦略は、所与の細胞系において欠陥がある必須の遺伝子を担うベクターを使用することである。例えば、ジヒドロ葉酸還元酵素(DHFR)遺伝子の発現に欠陥があるCHO細胞は、追加のヌクレオシドの存在下でしか生存しない。しかし、こうした細胞は、DHFR遺伝子を発現するDNAを安定にトランスフェクトすると、必要とされるヌクレオシドを合成する(Stark,G.R.及びWahl,G.M.(1984)Gene amplification.Ann.Rev.Biochem.53,447.)。DHFRをマーカーとして使用するさらなる利点は、用量を増やしながらメトトレキセートに細胞を暴露すると、DHFR及び関連するトランスフェクトしたDNAの遺伝子増幅が起こることである(Schimke,R.T.(1988)「培養細胞における遺伝子増幅(Gene amplification in cultured cells)」J.Biol.Chem.263,5989)。
【0061】
用語「安定にトランスフェクトされた」又は「対象の遺伝子を安定にトランスフェクトした」又は「安定なトランスフェクション」は、当技術分野で既知の方法(Maniatisら参照)により、事前選択した細胞中に対象の遺伝子を導入し、ここで、前記対象の遺伝子は、前記細胞のゲノム中に組み込まれないこと、又はエピソームベクター上に位置し細胞内で複製されることを指す。
【0062】
用語「対象の遺伝子」は、対象のタンパク質、すなわち、興味深い活性を有するタンパク質、又はその特性が興味深い、例えばそれを評価すべきタンパク質をコードしているゲノムDNA、cDNA、合成DNA、RNA、及び他のポリヌクレオチド、又はそのアナログを指す。
【0063】
驚くべきことに、本発明者らにより、対象の遺伝子を一過性にトランスフェクトしたカプセル化細胞は、多細胞生物に投与する(例えば、注射する)ことができ、対象の遺伝子によってコードされるタンパク質を、そのin vivo活性又は作用を検出できるような十分な量in vivoで産生することが判明した。
【0064】
さらに驚くべきことに、前記カプセル化細胞は、トランスフェクションの当日に、又はその翌日若しくは数日後にも多細胞生物に投与することができ、前記タンパク質のin vivo活性又は作用を検出することができる。
【0065】
したがって、本発明は、投与の開始から14日以内に、優先的には10日以内に、さらにより好ましくは5日以内に、対象の遺伝子によってコードされるタンパク質のin vivo活性を分析又は評価するための新規ツールを提供する。
【0066】
第1の態様では、本発明は、対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有する新規カプセルを提供する。
【0067】
この細胞は、植物細胞又は動物細胞であってよい。この細胞は、動物細胞、特にハイブリドーマを含めた哺乳動物細胞であることが好ましい。特に好ましい細胞は、COS細胞、BHK細胞、VERO細胞、CHO細胞、rCHO−tPA細胞、rCHO−Hep B表面抗原細胞、HEK293細胞、rHEK293細胞である。本発明に従って培養することができるハイブリドーマの例には、例えば、DA4.4細胞、123A細胞、127A細胞、GAMMA細胞、及び67−9−B細胞が含まれる。本発明に従って培養することができる昆虫細胞の例には、例えば、鱗翅類細胞、Tn−368細胞、SF9細胞、rSF9細胞、及びHi−5細胞が含まれる(例えば、Ikonomou Lら2002,Nov〜Dec,18(6),1345〜1355を参照のこと)。本発明に従って培養することができる非哺乳動物細胞の例には、例えば、ブラウンブルヘッド細胞系が含まれる(例えば、Buck CDら1985,Feb,10(2),171〜84を参照のこと)。
【0068】
上記で定義した対象の遺伝子は発現され、細胞からタンパク質が分泌される。優先的には、このタンパク質は成熟タンパク質として分泌される。優先的には、このタンパク質は、発現する場合、タンパク質を分泌するためのシグナルペプチドを含む。検討中の遺伝子が、タンパク質のシグナルペプチドをコードしている配列を有していない場合、このような配列、例えば、シグナル配列、プレ配列及び/又はプロ配列をその遺伝子配列に融合して、対象の遺伝子をタンパク質として発現、輸送及び分泌しやすいようにすることができる。
【0069】
対象の遺伝子は、一般に、細胞の一過性トランスフェクションに適したベクターの一部である、プロモーター及び調節シグナルを含む発現カセット中に通常挿入される。
【0070】
適当なベクターは、プラスミド又はエピソームベクターである。
【0071】
細胞の一過性トランスフェクションに適したプラスミドの例は、pCEP4(Invitrogen、米国)、pEAKベクターファミリー(Edge Biosystems、米国)、又はpCDNA3.1(Invitrogen、米国)である。
【0072】
対象の遺伝子を担うベクターの細胞へのトランスフェクションは、任意のトランスフェクション法、特にポリアミン法、ポリエチレンイミン法(O.Boussifら1995 Proc.Natl.Acad.Sci.USA 92,7297〜7301頁)、リン酸カルシウム共沈法、又はリポフェクションによって実施することができる。ポリアミン法は、トランスフェクトした細胞の90%超が得られるので好ましい。ショットガン法又はエレクトロポレーションは、高い細胞死亡率を伴い、これにより死細胞のカプセル化が高い割合でもたらされるはずであり、したがってカプセルの生産性が低下するのでこの用途には好ましくない。
【0073】
生体適合性ポリマーは、安定に発現する細胞系の細胞をカプセル化するための、当技術分野で既知の生体適合性ポリマー、特にアルギナート塩、特にアルギナートナトリウム若しくはカリウム、及びアルギナート−ポリ−L−リシン−アルギナート(APA)、並びにポリ(メタクリル酸ヒドロキシエチル−co−メタクリル酸メチル)(HEMA−MMA)などの他のポリマーの中から選択することができる。好ましい生体適合性ポリマーはAPAである。
【0074】
このポリマーを工学処理して、選択透過膜、すなわち栄養物、酸素、外部刺激、及び分泌タンパク質に透過性があるが、移植片拒絶の原因である免疫成分に不透過性である膜が作製される。このような選択透過膜は通常、分離が90〜30、好ましくは90〜50又は80〜50kDa、さらにより好ましくは80〜60kDaである孔を有する。分離は、選択透過膜を通して拡散することができる分子のサイズを定義する。
【0075】
カプセルは通常、直径が100〜1500μm、好ましくは250〜600μm、特に440〜530μmである。
【0076】
本発明はまた、上記カプセルを調製する方法に関し、この方法は、細胞に対象の遺伝子を一過性にトランスフェクトするステップと、一過性にトランスフェクトされた細胞をカプセル化するステップとを含む。
【0077】
カプセル化の後、この細胞は、カプセル内で増殖し続け、対象の遺伝子によって分泌される成熟タンパク質を、カプセルを維持する培地中に拡散する。
【0078】
このカプセルは、成熟タンパク質のin vitro拡散を低減し、機械的な理由によるカプセルのどんな破壊をも避ける、低剪断、微小重力条件下で維持することが好ましい。
【0079】
本発明のカプセルを調製するすべてのステップは、短期間、通常1日又は2日以内に実施することができる。
【0080】
細胞を含有するカプセルを数カ月まで保存するために、−80℃で、又は液体窒素(−196℃)気相若しくは液相中で凍結させることができる。この凍結保存により、大量のカプセルを製造し、続いてその一定分量を様々な用途に(例えば、キット中に)使用することが可能となる。蘇生後、この細胞はカプセル内で増殖を再開し、対象のタンパク質を3〜10日間産生し続ける。
【0081】
さらなる態様では、本発明は、対象の遺伝子によってコードされており、一過性にトランスフェクトした細胞によって発現及び分泌されたタンパク質のin vivo活性を分析又は評価する方法に関し、この方法は、上記で定義したカプセルを多細胞生物に投与するステップと、前記タンパク質の活性を検出するステップとを含む。この活性は、局所活性又は全身活性であってよい。
【0082】
本発明の方法に従って産生することができるタンパク質は、例えば、絨毛性ゴナドトロピン、卵胞刺激ホルモン、ルトロピン絨毛性ゴナドトロピンホルモン、甲状腺刺激ホルモン、ヒト成長ホルモン、インターフェロン(例えば、インターフェロンβ−la、インターフェロンβ−lb)、インターフェロン受容体(例えば、インターフェロンγ受容体)、TNF受容体p55及びp75、TACI−Fc融合タンパク質、インターロイキン(例えば、インターロイキン1、インターロイキン2、インターロイキン3、インターロイキン4、インターロイキン5、インターロイキン6、インターロイキン8、インターロイキン10、インターロイキン11、インターロイキン12)、インターロイキン結合タンパク質(例えば、インターロイキン−18結合タンパク質)、増殖因子(例えば、エリスロポイエチン、顆粒球コロニー刺激因子、顆粒球マクロファージコロニー刺激因子、血小板由来増殖因子、酸性及び塩基性線維芽細胞増殖因子、角化細胞増殖因子、グリア細胞系由来神経栄養因子)、下垂体ペプチドホルモン、閉経期性腺刺激ホルモン、インスリン様成長因子(例えば、ソマトメジンC)、トロンボモジュリン、インスリン、第VIII因子、ソマトロピン、骨形成タンパク質2、ヒルジン、エポエチン、組換えLFA−3/IgG1融合タンパク質、グルコセレブロシダーゼ、並びにその突然変異タンパク質、断片、可溶形、機能性誘導体、及び融合タンパク質を含めた対象のどんなタンパク質とすることもできる。
【0083】
多細胞生物は、植物又は動物であってよい。特定の対象の動物は、マウス、ラット、イヌ、ヤギ、ヒツジ、ブタ、ウシ、サルなど薬学研究で一般に使用されている哺乳動物である。
【0084】
特に興味深い哺乳動物は、マウス又はラットなどのげっ歯類であり、マウス、例えばC57/BL6系マウスが好ましい。
【0085】
検出する活性は、例えば、体温、組織又は体液の色、組織又は体液(例えば、血液、血清、血漿、糞便、痰、滑液、脳脊髄液、又は尿)中の所与の物質の濃度など、任意のパラメータに関するものでよい。
【0086】
カプセルは、筋肉内(i.m.)、皮内、皮下(s.c.)又は腹腔内(i.p.)注射によって投与することができる。好ましい投与形態はi.p.注射である。別の好ましい形態はs.c.注射である。
【0087】
カプセルはin vivo条件下で安定であり、包含する細胞を分解又は放出しない。
【0088】
カプセルは、一過性発現が起こっている限り、すなわちカプセル投与(0日目)から通常3〜14日目の間は成熟タンパク質をin vivoで放出するが、その放出の大部分は通常、1日目〜4、5又は6日目に起こる。成熟タンパク質が大量に放出される期間は通常3〜5日目であるが、これは検出すべき局所及び/又は全身活性を誘導するのに通常十分である。慢性作用を検出するためなど、必要な場合は、カプセルの投与を2回以上それぞれ別の時点で実施して、例えば、大量の成熟タンパク質の放出をより長期間維持することができる。
【0089】
したがって、本発明のカプセルは、実験動物、例えばヒト疾患の動物モデルで使用することができる、薬学研究の有用なツールとなる。
【0090】
この動物又は動物モデルは、誘導された検出すべき局所及び/又は全身活性が急速に(すなわち、数時間又は1日若しくは複数日以内に)生じ、活性物質(対象のタンパク質)の投与後数日以内に完了するものが好ましい。
【0091】
このような動物モデルの一例は、以下で例示するが、コンカナバリンA(ConA)によって肝毒性が誘発されたマウスである。
【0092】
中毒性肝疾患は、ヒトの世界中の健康問題を代表し、薬理学的治療法がまだ発見されていない。例えば、肝硬変をもたらす活動性慢性肝炎は、活性化T細胞によって肝実質細胞が進行的に破壊される病状である。ConA誘発肝毒性は、マウスで説明されているT細胞依存性アポトーシス性及び壊死性肝損傷の3つの実験モデルのうちの1つである。活性化抗CD3モノクローナル抗体又はスーパー抗原SEBのいずれかで誘発したGal N(D−ガラクトサミン)感作マウスは、重篤なアポトーシス性肝損傷及び二次的な壊死性肝損傷を発症する。植物のT細胞分裂促進レクチンConAの非感作マウスへの注射によっても、壊死に先行する肝アポトーシスがもたらされる。ConAは、全身へのTNFα及びIFNγ、並びに他の種々のサイトカインの放出を誘発する。傷害の8時間後、トランスアミナーゼの放出は重篤な肝臓破壊を示す。
【0093】
TNFα及びIFNγはどちらも、肝障害を誘発する上で肝損傷の決定的に重要なメディエータである。例えば、TNFαは、ConA注射の後に産生される最初のサイトカインの1つであり、抗TNFα抗体は、疾患に対する防御を与える(Seinoら2001,Annals of surgery 234,681)。ConA処理動物の血中トランスアミナーゼレベルの低下から判断して、抗IFNγ抗血清はマウスを有意に防御するので、後で誘導されるIFNγも、肝損傷の決定的に重要なメディエータであると思われる(KuestersらGastroenteroloy 111,462)。ConA誘発肝毒性からの防御を与える処置を記載した様々な文献がある。rIL6の単回投与により、トランスアミナーゼの放出が完全に阻害されたが、同じ投与計画でTNF産生の阻害は40〜50%しか誘導されなかった(Mizuharaら1994,J.Exp.Med.179,1529〜1537)。
【0094】
数種の細胞型、CD4 T細胞、マクロファージ、及びナチュラルキラー細胞が肝障害に関与することが示されている(Kaneko J.Exp.Med.2000,191,105〜114)。抗CD4抗体は、T細胞の活性化及びその結果としての肝障害を遮断する(Tiegsら1992,J.Clin.Invest.90,196〜203)。CD8に対するモノクローナル抗体のマウスへの前処置は防御に失敗したが、マクロファージの欠失は肝炎の誘発を阻止した。
【0095】
興味深い動物モデルの別の例は、マウスLPS(リポ多糖)誘発TNFα放出モデルである。
【0096】
LPSは、脂質及び炭水化物をどちらも含有する大分子である。LPSは、グラム陰性菌の細胞壁の主成分である。LPSは、原型的なエンドトキシンとして働き、多くの細胞型において炎症誘発性サイトカインの分泌を促進する。LPSは、Toll受容体4を介して先天性免疫系の食細胞によって認識される(Triantafilou M.らTrends Immunol.2002 Jun;23(6):301〜4)。LPSは、マクロファージ又はミクログリアをin vitro又はin vivoで活性化するために広く使用されている(Tobias PSらImmunobiology.1993 Apr;187(3〜5):227〜32)。マウスのLPS誘発TNFα放出モデルはヒトの炎症状況に似ている。
【0097】
本発明はさらに、細胞に1種又は複数の遺伝子を一過性にトランスフェクトした上記のカプセルを含む医薬組成物を提供する。この医薬組成物は、製薬上許容される1種又は複数の担体、希釈剤、或いは賦形剤を追加的に場合によって含む。
【0098】
この遺伝子は、病原体由来の、1種又は複数の抗原/免疫原、或いは1種又は複数のその部分、或いは対象の1種又は複数のエピトープをコードすることができる。抗原/免疫原という用語は、宿主において免疫応答を誘発することが可能な任意のタンパク質又はその誘導体を指す。
【0099】
好ましい実施形態では、病原体は、任意のウイルス、クラミジア、マイコプラズマ、細菌、寄生生物、及び菌類からなる群から選択される。ウイルスには、ヘルペスウイルス、オルトミクソウイルス、ライノウイルス、ピコルナウイルス、アデノウイルス、パラミクソウイルス、コロナウイルス、ラブドウイルス、トガウイルス、フラビウイルス、ブンヤウイルス、風疹ウイルス、レオウイルス、ヘパドナウイルス、及びヒト免疫不全ウイルスを含めたレトロウイルスが含まれる。細菌には、マイコバクテリア、スピロヘータ、リケッチア、クラミジア、及びマイコプラズマが含まれる。菌類には、酵母及びカビが含まれる。寄生生物には、住血吸虫、リーシュマニア、及びマラリア原虫が含まれる。この一覧には、本明細書に記載の方法に従ってその防御免疫応答を生じさせることができるあらゆる潜在的な病原体が含まれていないことは理解されよう。
【0100】
特に、その遺伝子は、インフルエンザ血球凝集素、インフルエンザ核タンパク質、インフルエンザM2、破傷風毒素Cフラグメント、炭疽菌防御抗原、炭疽菌致死因子、炭疽菌発芽因子、狂犬病糖タンパク質、HBV表面抗原、HIV gp120、HIV gp160、マラリアCSP、マラリアSSP、マラリアMSP、マラリアpfg、ボツリヌス毒素A、及び結核菌HSPからなる群から選択される1種又は複数の抗原/免疫原をコードすることができる。
【0101】
別の実施形態では、遺伝子は、腫瘍由来の抗原/免疫原をコードすることができる。この腫瘍は、乳腺、卵巣、肺、脳、胃、腸管、膵臓、膀胱、前立腺、骨、又は造血性の細胞若しくは組織の腫瘍、或いは他の細胞又は組織の腫瘍とすることができる。特に、その遺伝子は、HER2/neu、ヒト癌胎児性抗原、及び前立腺特異的抗原からなる群から選択される1種又は複数の抗原/免疫原をコードすることができる。
【0102】
別の実施形態では、遺伝子は、抗体を生じるべく選択された任意の抗原/免疫原をコードすることができる。この抗体は、ポリクローナル抗体又はモノクローナル抗体とすることができる。この抗体は、特に、マウス抗体、キメラ抗体、ヒト化抗体、完全ヒト抗体、ドメイン抗体、又は短鎖抗体であってよい。この抗体は、例えば、研究、診断、治療、又は他の目的に有用であり得る。
【0103】
さらに別の実施形態では、遺伝子は、アジュバントをコードすることができる。このアジュバントは、共刺激分子、サイトカイン、又はケモカインとすることができる。好ましいアジュバントは、IL−2、IL−4、IL−6、IL−12、IL−18、GM−CSF、又はINFγである。
【0104】
別の実施形態では、医薬組成物は、(a)本明細書に記載の1種若しくは複数の抗原/免疫原及び/又はアジュバントをトランスフェクトした細胞を含有するか、或いは(b)本明細書に記載の1種若しくは複数の抗原/免疫原及び/又はアジュバントから様々に選択したものをトランスフェクトした細胞群を含有するカプセルを含む。カプセル内の細胞の選択は、ワクチン接種の必要性に適合させることができることは理解されよう。これは、抗原/免疫原及びアジュバントの適切な選択、発現ベクター及び発現カセットの適切な選択、対象の遺伝子を発現する細胞型若しくは細胞系の適切な選択、並びに/又は本明細書及び当技術分野で記載されているカプセル化材料の適切な選択によって達成することができる。
【0105】
本発明の別の実施形態は、本明細書に記載のカプセル又は医薬組成物を被検体に投与するステップを含む、免疫する方法である。
【0106】
本発明の別の実施形態は、対象のタンパク質を被検体に投与するための、本明細書に記載のカプセル又は医薬組成物の使用である。
【0107】
好ましい実施形態は、対象のタンパク質を被検体に投与するための、本明細書に記載のカプセル又は医薬組成物の使用であり、このタンパク質は、ワクチン接種用の抗原/免疫原である。
【0108】
好ましい実施形態は、対象のタンパク質を被検体に投与するための、本明細書に記載のカプセル又は医薬組成物の使用であり、このタンパク質は、抗体の標的として選択された抗原/免疫原である。特に、このカプセルは、被検体において、カプセル化細胞によって産生される事前選択した抗原に対する抗体応答を誘発させるのに使用される。
【0109】
本発明の別の実施形態は、本明細書に記載のカプセル又は医薬組成物を含むキット、及び前記カプセル又は組成物を被検体に適用するための手段である。
【0110】
この被検体は、動物とすることができ、有利には脊椎動物、例えば、哺乳動物、鳥類、爬虫類、両生類、又は魚類であり、より有利にはヒト、或いは飼育動物、家畜化動物、食物を生産する動物、飼料を生産する動物、家畜、狩猟動物、競争用動物、又は競技用動物、例えば、ウシ、イヌ、ネコ、ヤギ、ヒツジ、ブタ、若しくはウマ、又はさらには家禽、例えば、シチメンチョウ、カモ、若しくはニワトリである。別の実施形態では、この脊椎動物は、それだけには限らないが、げっ歯類(マウス、モルモット、及びラットを含む)、イヌ、ブタ、及びサルを含めた研究目的で飼育された動物である。本発明の最も好ましい実施形態では、脊椎動物はヒトである。
【0111】
本明細書で開示されるカプセル又は医薬組成物は優先的には、非経口経路で被検体に投与される。例えば、個体に、腹腔内、皮内、皮下、又は筋肉内の方法によって接種することができる。
【0112】
本発明の他の特徴及び利点は、限定的な特性ではなく例示的なものを有する以下の実施例から明らかとなろう。実施例は、好都合なことには、添付の図面を参照することによって読まれよう。
【実施例】
【0113】
(実施例1:IL−6をコードするDNAを一過性にトランスフェクトした細胞を含有するカプセルの調製、並びにマウスコンカナバリンAモデルにおけるIL−6血中レベル及びその活性の測定)
1.対象の遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製
1.1 細胞の培養及び維持
エプスタイン−バーウイルス核抗原を発現するヒト胎生腎293細胞系(HEK293−EBNA、Invitrogen、米国)の細胞を、スピナーフラスコ(Techne、英国)中で4mM L−グルタミン(Invitrogen)及び1ml/lフェノールレッド溶液(水中0.5%w/v、フェノールレッド:Sigma、米国)を補充したEx細胞VPRO無血清培地(維持培地、JRH、英国)中で懸濁状態で維持した。
【0114】
1.2 プラスミド調製
HEK293−EBNA細胞内でIL−6を発現させるためのプラスミドの構築
ヒトIL−6の完全コード配列(ORF)(図1、配列番号1)を含有するプラスミドをInvitrogen社から購入した(Invitrogen社クローンID CS0DI019YP05)。Gateway(商標)クローニング法(Invitrogen)を使用して、このORFを哺乳動物細胞発現ベクターpEAK12d(図2A及び2B)中にサブクローニングした。
【0115】
1.2.1 6HISタグ配列にインフレームで融合させたGateway適合性IL−6 ORFの作製
Gatewayクローニングプロセスの第1段階は、2ステップのPCR反応を含み、この反応では、5’末端でattBl組換え部位及びコザック配列に隣接し、3’末端で6ヒスチジン(6HIS)タグをインフレームでコードする配列、終止コドン及びattB2組換え部位に隣接するIL−6のORF(Gateway適合性cDNA)が作製される。第1のPCR反応液(最終量50μl中)は、25ngのCS0DI019YP05プラスミド、2μlのdNTP(5mM)、5μlの10×Pfxポリメラーゼ緩衝液、各0.5μlの遺伝子特異的プライマー(100μM)(EX1フォワード及びEX1リバース)、及び0.5μlのPlatinum Pfx DNAポリメラーゼ(Invitrogen)を含有する。PCR反応は、最初の変性ステップ95℃で2分間、続いて94℃で15秒間及び68℃で30秒間を12サイクル実施した。Wizard PCR prep DNA精製システム(Promega)を製造元の使用説明書に従って使用して、反応混合液からPCR産物を直接精製した。第2のPCR反応液(最終量50μl中)は、10μlの精製PCR産物、2μlのdNTP(5mM)、5μlの10×Pfxポリメラーゼ緩衝液、各0.5μlのGateway変換プライマー(100μM)(GCPフォワード及びGCPリバース)、及び0.5μlのPlatinum Pfx DNAポリメラーゼを含んでいた。第2のPCR反応の条件は、95℃で1分間;94℃で15秒間、45℃で30秒間、及び68℃で3.5分間を4サイクル;94℃で15秒間、55℃で30秒間、及び68℃で3.5分間を25サイクルであった。PCR産物を上記の通り精製した。
【0116】
1.2.2 GatewayエントリーベクターpDONR201及び発現ベクターpEAK12d中へのGateway適合性IL−6 ORFのサブクローニング
Gatewayクローニングプロセスの第2段階は、Gateway修飾PCR産物をGatewayエントリーベクターpDONR201(Invitrogen)中に以下の通りサブクローニングすることを含む。5μlの精製PCR産物を1.5μlのpDONR201ベクター(0.1μg/μl)、2μlのBP緩衝液、及び1.5μlのBPクロナーゼ酵素混合液(BP clonase enzyme mix)(Invitrogen)と共に室温で1時間インキュベートする。この反応をプロテイナーゼK(2μg)の添加によって停止させ、37℃でさらに10分間インキュベートした。この反応液の一定分量(2μl)を、Biorad Gene Pulserを使用したエレクトロポレーションにより大腸菌DH10B細胞に形質転換した。形質転換体をLBカナマイシンプレート上に蒔いた。Wizard Plus SV Miniprepsキット(Promega)を使用して、得られた1〜4個のコロニーからプラスミドミニプレップDNAを調製し、次いで1.5μlのプラスミド溶出物を、最終量10μl中に1.5μlのpEAK12dベクター(図2)(0.1μg/μl)、2μlのLR緩衝液、及び1.5μlのLRクロナーゼ(Invitrogen)を含有する組換え反応液に使用した。混合液を室温で1時間インキュベートし、プロテイナーゼK(2μg)の添加によって反応停止させ、37℃でさらに10分間インキュベートした。この反応液(1μl)の一定分量を使用して、エレクトロポレーションにより大腸菌DH10B細胞に形質転換した。
【0117】
pEAK12dプライマー(pEAK12d F及びpEAK12d R)をPCR用に使用した以外は上記の通りコロニーPCRを実施することにより、正しいインサートを含有するクローンを同定した。Qiaprep Turbo 9600ロボットシステム(Qiagen)を使用するか、又はWizard Plus SV miniprepsキット(Promega)を手動で使用して、正しいインサートを含有するクローンからプラスミドミニプレップDNAを単離し、pEAK12d F及びpEAK12d Rプライマーを使用して配列を確認した。
【0118】
配列を確認したクローンの培養物500mlから、プラスミドpEAK12d−1L6−6HIS(プラスミド番号11381、図3)のCsCI勾配精製maxi−prepDNAを調製し(Sambrookら,in Molecular Cloning,a Laboratory Manual,第2版,1989,Cold Spring Harbor Laboratory Press)、滅菌水で1μg/μlの濃度で再懸濁させ、−20℃で保存した。
表I
IL−6サブクローニング及びシークエンシング用プライマー
【0119】
【表1】
【0120】
1.3 細胞トランスフェクション
トランスフェクションの当日に、細胞を遠心分離し、スピナー容器(DasGip,D)中の播種培地である1%FBS及び4ml/l ITS−Xサプリメントを含有する250mlのDMEM/F12(1:1)培地(Invitrogen)で細胞1×106/mlの密度で再懸濁させた。ポリエチレンイミン(PEI)法を使用して(O.Boussifら1995 Proc.Natl.Acad.Sci.USA 92,7297〜7301頁)、細胞に2:1の比のPEI:DNAをトランスフェクトした。100mlの播種培地中で、500μgの相当するプラスミドDNAを1mgのPEI(Polysciences、米国)と混合し、室温で10分間インキュベートした。この混合物を細胞懸濁液に添加し、37℃で90分間インキュベートした。インキュベーション後、この細胞懸濁液を遠心分離し(200×g、10分間、4℃)、細胞ペレットを500mlの維持培地で再懸濁させた。細胞は、カプセル化するまで5%CO2、37℃の加湿雰囲気下でインキュベートした。
【0121】
1.4 細胞カプセル化
上記で得られたHEK293−EBNAトランスフェクト細胞及び野生型HEK293−EBNA細胞を、米国特許第6458296号に記載のエンカプスレータに類似するInotech researchエンカプスレータ(Inotech、スイス)を使用してアルギナート−ポリ−L−リシン−アルギナート(APA)カプセル内に封入した。細胞を遠心分離し(200×g、10分間、4℃)、2ml洗浄緩衝液で再懸濁させた(すべての薬品はInotech社、スイス)。この懸濁液に1.5%アルギナート溶液を細胞2.5×106/溶液mlの最終細胞濃度が得られるまでゆっくり添加した。このアルギナート−細胞懸濁液をカプセル化装置に接続したシリンジ(Braun Omnifit,Braun,D)で吸い取った。表1に示したパラメータ、及び表2に記載のプロトコールを使用してカプセル化を実施した。すべての緩衝液は、製造元の使用説明書に従い滅菌蒸留水中、無菌条件下で調製した。
【0122】
カプセル化の最終ステップの最後に、カプセルを100mlの維持培地で再懸濁させ、滅菌スピナー容器(Dasgip,D)に移した。カプセルをスピナー容器中で維持し、5%CO2、37℃の加湿雰囲気下で一晩、又は動物に注射するまでインキュベートした。
【0123】
カプセルは、顕微鏡で測定すると485±10μmの平均直径を有していた。
表2 カプセル化パラメータ
【0124】
【表2】
表3 カプセル化プロトコール
【0125】
【表3】
【0126】
1.5.低剪断、微小重力条件下でのカプセルの維持の改良
一方はスピナー容器中で、もう一方は、低剪断、微小重力(10−12g)条件下、Syntecon社/NASA(米国ヒューストン)によって製造された水平低速回転式容器(slow turning lateral vessel)STLV(商標)(米国特許第6730498号参照)を使用して、上記で得られたカプセルの維持について比較実験を実施した。この細胞は、空気中5%CO2、37℃の加湿インキュベータ内の維持培地中で維持した。試験されたカプセルは、野生型HEK293−EBNA細胞、又はh−IL−6のcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルであった。
【0127】
低剪断、微小重力下で、カプセルの目に見えた破壊が7日間起こらないことが示されたが、スピナー容器では、同じ期間無傷であったカプセルが、カプセルの破壊によって生じた浮遊している細胞によって目に見えて囲まれていった。h−IL−6のcDNAをトランスフェクトした細胞については、in vitroで放出されたh−IL−6は、スピナー容器よりも低剪断、微小重力下で実質的に少なかった。
【0128】
2 コンカナバリンAモデル(ConA)
材料及び方法:
2.1 動物
すべての試験でC57/BL6雄マウス(8週齢)を使用した。常時、実験群あたり10匹のマウスを使用した。マウスを12時間の明暗周期、標準条件下で維持し、放射線照射飼料及び水を任意に与えた。
【0129】
2.1 カプセル注射
カプセル懸濁液をインキュベータから取り出し、層流フード内で数分放置してカプセルを沈殿させた。上澄液を除去し、濃縮したカプセルをシリンジで注意深く吸い上げた。700μlのカプセルを0.7mmの針(参照番号53158.01、Polylabo、スイス)で各マウスにゆっくりi.p.注射した。
【0130】
2.2 コンカナバリンA注射
コンカナバリンA(ConA)はSigma社から購入した(参照番号C7275、Sigma,D)。カプセルの移植から72時間後、ConAを18mg/kgでi.v.注射した。ConA注射から1.30時間後と8時間後に血液試料を採取した。サイトカイン及びトランスアミナーゼレベルの測定は、以下に記載の通り実施した。
【0131】
2.4 読取り
2.4.1 血液試料採取
ConA注射から1.30時間後と8時間後に眼窩後の静脈洞から100μlの血液試料を採取した。屠殺するときに血液を心臓から採取した。
【0132】
2.4.2 血液試料中のサイトカイン及びトランスアミナーゼの検出
TH1/TH2 CBAアッセイ(参照番号551287、Beckton Dickinson、米国)を使用してサイトカインIL−2、IL−4、IL−5、TNFα、及びIFNγのレベルを測定した。COBAS装置(Hitachi、スイス)を使用して、アスパラギン酸アミノトランスフェラーゼ(ASAT)、アラニンアミノトランスフェラーゼ(ALAT)、UREAの各血中パラメータを測定した。
【0133】
2.4.3 血液及び細胞培養物上清中のh−IL6の検出
カプセル化及びトランスフェクトされたHEK細胞からのhIL−6のin vivoでの分泌を追跡するために、カプセル注射後12日間毎日、一日あたり5匹のマウスの血液を採取した。
並行してIn vitroで上清を10日目まで採取した。市販のELISAキット(R&D Duoset、参照番号DY206)により検出を血清中で行った。すべての試料を希釈緩衝液で2倍希釈した。
【0134】
2.5 結果:
2.5.1 トランスフェクトしなかったHEK293−EBNA細胞を含有するカプセルの注射
第1の実験では、トランスフェクトしなかったカプセル化HEK293−EBNA細胞を試験した。このようなカプセルは、正常な動物においてサイトカインレベル又は血中パラメータを変化させないことが以前に示されている。0日目及び2日目にこのカプセルを注射した。次いで、ConAを注射した。ConA注射から1.5時間後及び8時間後にトランスアミナーゼ及びTNF−αレベルを測定した。
【0135】
図1に結果を示すが、この図は、トランスフェクトしなかったHEK293−EBNA細胞を含むカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したTNFα、ASAT、及びALATレベルを示したヒストグラムを表す。
【0136】
その図に示す通り、トランスフェクトしなかったHEK293−EBNA細胞を含有するカプセルを注射したマウスにおいて、対照マウス(ConA注射のみを施したマウス)と比較して、NF−α又はトランスアミナーゼASAT及びALATレベルの有意な変化は認められなかった。
【0137】
2.5.2 hIL−6を一過性にトランスフェクトしたHEK細胞を含有するカプセルのIn vivo活性
hIL−6は、in vivoでConA誘発肝炎におけるトランスアミナーゼ及びTNF−αのレベルを低減することが分かっている(Mizuharaら1994,J.Exp.Med.,179,1529〜1537)。
【0138】
第1の実験では、hIL−6 HISをコードしているcDNAをトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを雄C57 Bl/6にi.p.又はs.c.注射した。カプセル注射の直後から毎日12日間連続で血中hIL−6レベルを測定した。
【0139】
図2Aに結果を示すが、この図は、hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをi.p.又はs.c.注射した後12日間連続してマウスの血液中で測定したhIL−6レベルのグラフを表す。
【0140】
この図で示される通り、血中hIL−6レベルは、i.p.カプセル注射後1日目から5日目の間でピーク値に達し、i.p.注射したマウスの血中hIL−6レベルの値は常にs.c.注射したマウスよりも実質的に大きかった。i.p.又はS.C.注射後8日目、血中hIL−6レベルは、基底値に近い値に達した。
【0141】
再現性:上記の通り別々にカプセル化され、様々な群に分けられた細胞の同一プール、又は上記の通り別々に調製した細胞の様々なプールのいずれかを用いてこの実験を数回反復すると、いずれの場合においても、血中hIL−6レベルは、カプセルのi.p.注射後1日目から5日目の間でピーク値に達し、血中hIL−6レベルの最大値、及び最初の5日間の積算値は有意差がない(20%未満)ことが示された。
【0142】
第2の実験では、hIL−6 HISをコードしているcDNAをトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを0日目及び3日目に雄C57 Bl/6にi.p.注射した。さらに、18mg/kg ConAをi.v.注射して肝炎を誘発させた。ConA注射から1.5時間後及び8時間後にTNF−α及びトランスアミナーゼレベルを測定した。
【0143】
図2B〜Dに結果を示すが、これら図は、hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したTNFα、ASAT、及びALATレベルを示すヒストグラムを表す。
【0144】
これらの図で示される通り、ConA注射の後、5%CO2、37℃の加湿雰囲気下でインキュベートしたカプセルを事前注射したConA処置マウスのTNF−α及びトランスアミナーゼレベルは、トランスフェクトしなかったカプセル化HEK細胞を注射したConA処置マウスと比較して有意に低かった。
【0145】
したがって、カプセルからin vivoで放出されたhIL−6は、ConA誘発肝炎においてトランスアミナーゼ及びTNF−αのレベルを低下させる既知の予測された作用を有する。
【0146】
2.5.3 HEK293−EBNA−hIL6−HISカプセルを様々な量注射した後の用量反応の確立
hIL−6 HISをコードしているcDNAを一過性にトランスフェクトした細胞を含有するカプセルに関する用量反応関係を確立するための実験を実施して、ConA POCモデルにおいてこの技術を使用できるかどうかを実証した。700、350及び100μlのカプセルの注射後1日目、2日目及び3日目に、全身のhIL−6タンパク質産生を測定した。
【0147】
図3に結果を示すが、この図は、hIL−6−6HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセル700、350、及び100μlのi.p.注射後3日間連続してマウスの血液中で測定したhIL−6レベルを示すヒストグラムを表す。図4A〜Cに結果を示すが、この図は、hIL−6−6HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをそれぞれ700、350、及び100μl注射したConA処置マウス、並びに対照マウスの血液試料中で測定したTNFα、ASAT、及びALATレベルを示すヒストグラムを表す。
【0148】
これらの図で示される通り、血液中のhIL−6タンパク質の産生、並びにTNFα及びトランスアミナーゼの下方制御に対する生物学的作用に関する用量効果を確立することができた。
【0149】
(実施例2:hepaCAM遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製、及びLPS誘発TNFα放出のマウスモデルにおけるその活性の測定)
1.hepaCAM遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製
1.1 細胞の培養及び維持:実施例1参照
【0150】
1.2 プラスミド調製
hepaCAM遺伝子(遺伝子の配列:配列番号7に関する図8A、及びタンパク質の配列:配列番号8に関する図8Bを参照のこと)を、国際公開第03/093316号に詳細に記載されている通り、発現ベクターpEAK12d中にクローニングした(Mei Chung MohらのJ Hepatol.2005 Jun;42(6):833〜41、Epub 2005 Apr7、及びJ Biol Chem.2005 Jul22;280(29):27366〜74、Epub 2005 May25も参照のこと)。
【0151】
簡単に言えば、先ず、2ステップPCRを使用してこの遺伝子をGateway(商標)クローニングシステム(Invitrogen)のpENTRベクター中にクローニングした。pEAK12dベクター中へのサブクローニングは、Gatewayクローニングマニュアルに従って実施した。この遺伝子をゲノム配列からの3つのエクソンのPCR増幅によってクローニングした。
【0152】
使用したプライマーは以下である。
【0153】
【化1】
【0154】
1.3 細胞トランスフェクション:実施例1参照
【0155】
1.4 細胞カプセル化:実施例1参照
【0156】
2.LPS誘発TNFα放出のマウスモデル
プロトコール
マウスにおけるLPS誘発TNFα放出のモデルは、国際公開第98/38179号に記載の通り設定した。
【0157】
一過性にトランスフェクトされたカプセル化HEK293−EBNA細胞の注射後3日目に、0.3mg/kg LPS(0111:B4、Sigma、スイス)をC3H/HeNマウス(それぞれ8匹のマウスからなる群)(Charles River、仏国)にi.p.又はs.c.注射した。90分後、血液試料を採取し、ELISAキットを使用して血漿TNFαを測定した。デキサメタゾン(0.1mg/kg、sc)をPBSに溶解させ、注射し、15分後LPS誘発を行った。
【0158】
結果
図8Cを参照のこと。
【0159】
トランスフェクトしなかった、又はhepaCAMを一過性にトランスフェクトしたカプセル化HEK293−EBNA細胞のいずれかをi.p.又はs.c.経路で注射したマウスにLPSを注射すると、それぞれTNFα放出の非阻害又は有意な阻害が認められた。LPS誘発サイトカイン放出を80%阻害したデキサメタゾンを陽性対照とした。したがって、HepaCAMは、LPS誘発TNFαレベルを有意に下方制御した。
【0160】
こうした結果から、hepaCAMがTNFα媒介の炎症疾患を治療するのに有用であろうことが示される。
【0161】
(実施例3:INSP114−SV2をコードしているDNAを一過性にトランスフェクトした細胞を含有するカプセルの調製、及びLPS誘発TNFα放出のマウスモデルにおけるその活性の測定)
1.INSP114−SV2遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製
1.5 細胞の培養及び維持:実施例1参照
【0162】
1.6 プラスミド調製
INSP114SV2遺伝子(遺伝子の配列:配列番号9に関する図9A、及びタンパク質の配列:配列番号10に関する図9Bを参照のこと)を国際公開第2004/085469号に詳細に記載されている通り、発現ベクターpEAK12d中にクローニングした。
【0163】
使用したプライマーは以下である。
【0164】
【表4】
【0165】
1.7 細胞トランスフェクション:実施例1参照
【0166】
1.8 細胞カプセル化:実施例1参照
【0167】
2.LPS誘発TNFα放出のマウスモデル
実施例2を参照のこと。
【0168】
結果
図9Bを参照のこと。
【0169】
トランスフェクトしなかった、又はINSP114SV2を一過性にトランスフェクトしたカプセル化HEK293−EBNA細胞のいずれかをi.p.経路で注射したマウスにLPSを注射すると、それぞれTNFα放出の非阻害又は有意な阻害が認められた。LPS誘発サイトカイン放出を80%阻害したデキサメタゾンを陽性対照とした。したがって、INSP114SV2は、LPS誘発TNFαレベルを有意に下方制御した。
【0170】
こうした結果から、INSP114SV2がTNFα媒介の炎症疾患を治療するのに有用であろうことが示される。
【0171】
(実施例4:EPO遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製、並びにマウスモデルにおける血中EPO濃度及びヘマトクリット値の測定)
1.EPO遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製
1.1 細胞の培養及び維持:(実施例1参照)
【0172】
1.2 プラスミド精製
EPO遺伝子(遺伝子の配列:配列番号5に関する図10A、及びタンパク質の配列:配列番号6に関する図10Bを参照のこと)を、IL−6遺伝子に関する実施例1に記載のものに類似するプロトコール及び以下のプライマーを使用して発現ベクターpEAK12d中にクローニングした。
【0173】
【表5】
【0174】
1.3 細胞トランスフェクション
1.3.1:EPO遺伝子を一過性にトランスフェクトした細胞の調製
トランスフェクションの当日に、HEK293−EBNA細胞を遠心分離し、スピナー容器(DasGip,D)中の播種培地である1%FBS及び4ml/l ITS−Xサプリメントを含有する250mlのDMEM/F12(1:1)培地(Invitrogen)で細胞1×106/mlの密度で再懸濁させた。ポリエチレンイミン(PEI)法を使用して(O.Boussifら1995 Proc.Natl.Acad.Sci.USA 92,7297〜7301頁)、細胞に2:1の比のPEI:DNAをトランスフェクトした。100mlの播種培地中で、500μgの相当するプラスミドDNAを1mgのPEI(Polysciences、米国)と混合し、室温で10分間インキュベートした。この混合物を細胞懸濁液に添加し、37℃で90分間インキュベートした。インキュベーション後、この細胞懸濁液を遠心分離し(200×g、10分間、4℃)、細胞ペレットを500mlの維持培地で再懸濁させた。細胞は、カプセル化するまで5%CO2、37℃の加湿雰囲気下でインキュベートした。
【0175】
1.3.2:EPO遺伝子をトランスフェクトした細胞の半安定プールの調製
この技術は、ピューロマイシン−N−アセチル−トランスフェラーゼ耐性遺伝子をその骨格中に含むpEAK12dに挿入されたEPO遺伝子のエピソームによる複製、及び細胞の半安定プールを選択するためのピューロマイシン選択圧の適用に基づく。
【0176】
EPO遺伝子の初回のトランスフェクション
HEK293−EBNA細胞をEx細胞VPRO無血清培地(細胞保存、維持培地、JRH)中で懸濁状態で維持した。トランスフェクションの当日に、細胞を計数し、遠心分離し(低速)、ペレットを所望の量のトランスフェクション培地、すなわち1%FCS(JRH)及び4ml/lインスリントランスフェリンセレニウム(Gibco)を補充したDMEM/F12(1:1)(FEME培地、Invitrogen)で再懸濁させて、生細胞1×106/mlの細胞濃度を得た。上記の1.2におけるPEAK12d中へのEPO遺伝子のクローニングによって得られたDNA保存液(1mg/ml保存液)をFEME培地に2mg/トランスフェクション1リットル量(2%eGFPレポーター遺伝子を同時トランスフェクト)で希釈した。次いで、トランスフェクション剤ポリエチレンイミン(PEI、4mg/リットル量、Polysciences)をcDNA溶液に添加し、ボルテックスでしっかり撹拌し(30秒間)、室温で10分間インキュベートした(「トランスフェクションミックス」の作製)。
【0177】
次いで、このトランスフェクションミックスをスピナー容器に添加し、CO2インキュベータ(5%CO2、37℃)内で90分間インキュベートした。90分後、PEI−EPO cDNA複合体のHEK293−EBNA細胞中への組込みが可能となり、細胞を遠心分離し、Ex細胞VPRO無血清新鮮培地に再懸濁させ、例えば、単細胞懸濁物として維持した。次いで、トランスフェクションから24〜48時間後、無血清培地中で増殖させた上記のEPO HEK293−EBNA細胞の試料を蛍光顕微鏡下で観察して、例えば、トランスフェクト細胞に蛍光をもたらしたレポーター遺伝子を視覚化した。蛍光が細胞の80%超で認められた場合に限り、この培養物をさらなる半安定圧選択のために維持した。
【0178】
ピューロマイシン2重圧選択
トランスフェクションから72時間後、EPOを発現するHEK293−EBNA細胞を懸濁培養により増殖させた培地を7.5μg/mlのピューロマイシンを含有するEx−細胞VPRO新鮮培地(Clontech)と交換し、このピューロマイシン選択圧を6日間維持した。この期間の間、乳酸濃度が1.3g/lを超えると(乳酸による増殖阻害)、選択圧培地を新鮮選択圧培地と交換した(依然ピューロマイシンを含有する)。蛍光顕微鏡及び依然増殖している(集団倍加)懸濁細胞によって視覚化すると、非組換え細胞は死滅し(細胞のわずか10%、PEIトランスフェクション効率が高いと85%を超える)、EPO組換え細胞は圧選択に耐えた。
【0179】
選択期間の最後に、培地を通常の増殖培地(ピューロマイシンなし)と交換し、細胞をタンパク質発現について2又は3週間モニターした。2回目のピューロマイシン選択圧(上記と同じ選択圧)を細胞懸濁物にさらに6日間施すと、例えば、すべての非組換え細胞が死滅することが確かめられた。
【0180】
細胞増殖をモニターし、例えば、カプセル化操作のために細胞を収集するのに十分な量の細胞を培養物中で維持しながら、この培養物の一部の凍結保存により少量の細胞保存液を置いておいた。
【0181】
得られた半安定細胞は、EPO遺伝子を約3カ月発現することができる。
【0182】
1.4 細胞カプセル化
1.3.1で得られた一過性にトランスフェクトした細胞、及び1.3.2で得られた半安定細胞を実施例1の1.4に記載の通りカプセル化した。
【0183】
2.マウスモデルにおける血中EPO濃度及びヘマトクリット値の測定
2.1 プロトコール
全ての試験で、C57/BL6雄マウス(8週齢)を使用した。常時、実験群あたり5匹のマウスを使用した。マウスを12時間の明暗周期、標準条件下で維持し、放射線照射飼料及び水を任意に与えた。
【0184】
カプセル懸濁液をインキュベータから取り出し、層流フード内で数分放置してカプセルを沈殿させた。上澄液を除去し、濃縮したカプセルをシリンジで注意深く吸い上げた。EPOを一過性にトランスフェクトした細胞、及びEPOをトランスフェクトした半安定細胞、又はトランスフェクトしなかった細胞を含有する700μlのカプセルを0.7mmの針(参照番号53158.01、Polylabo、スイス)で各マウスにゆっくりi.p.又はs.c.注射した。
【0185】
カプセル注射の後10日間連続して、R&D SystemsからのhEPO ELISAキット(Quantitin IVD Human EPO、カタログ番号DEP00)を使用して血中EPOレベルを追跡した。ヘマトクリット値を測定した。
【0186】
2.2 結果
図10C及び10Dから明らかであるが、一過性に又は安定にトランスフェクトしたカプセル化細胞を(i.p.又はs.c.経路)与えたマウスにおけるヘマトクリット値の増加(10D)に対応する血中EPO濃度の増加(10C)を実験の14日間測定した。トランスフェクトしなかったカプセル化細胞を与えた対照マウスではこのような効果は観察されなかった。意外なことに、また驚くべきことに、一過性にトランスフェクトしたカプセル化細胞を与えたマウスにおけるEPO濃度及びヘマトクリット値は、安定にトランスフェクトしたカプセル化細胞を与えたマウスよりも高いレベルに達した。
【0187】
(実施例5:mIL−18BP遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製、及びマウスモデルにおける血中mIL−18BPレベルの測定)
1.mIL−18BP遺伝子を一過性にトランスフェクトした細胞を含有するカプセルの調製
1.1 細胞の培養及び維持:実施例1参照
【0188】
1.2 プラスミド精製
mIL−18BP遺伝子(遺伝子の配列:配列番号3に関する図11A、及びタンパク質の配列:配列番号4に関する図11B)を、Mallat Z.ら2002 Circ.Res.91,441〜448頁に詳細に記載されている通り、発現ベクターpEAK12d中にクローニングした。
【0189】
簡単に言えば、プラスミドpCEP4−mIL18BP−d 23を鋳型として使用して、増幅反応を行った。PCR産物をNotI+HinDIIIで消化し、次いでNucleospin(商標)スピンカラムキット(Macherey−Nagel)によって精製し、HinDIII+NotIで消化したpEAK8ベクター(Edge BioSystems,Gaithersburg、MD、米国)にライゲートした。
【0190】
1.3 細胞トランスフェクション:実施例1参照
【0191】
1.4 細胞カプセル化:実施例1参照
【0192】
2.マウスモデルにおける血中mIL−18BPレベルの測定
2.1 プロトコール
17匹のC57/BL6雄マウス(8週齡)の群を使用した。マウスを12時間の明暗周期、標準条件下で維持し、放射線照射飼料及び水を任意に与えた。
【0193】
カプセル懸濁液をインキュベータから取り出し、層流フード内で数分放置してカプセルを沈殿させた。上澄液を除去し、濃縮したカプセルをシリンジで注意深く吸い上げた。mIL−18BPを一過性にトランスフェクトした細胞を含有する700μlのカプセルを0.7mmの針(参照番号53158.01、Polylabo、スイス)で各マウスにゆっくりi.p.注射した。
【0194】
カプセル注射の後8日間連続して、m−IL18BP用に実験室で作製したELISAを使用して血中EPOレベルを追跡した。
【0195】
ELISAの手順は以下の通りである。
【0196】
Labsystemコンビプレート12EBというプレートを使用した。コーティングは、0.1mlの1×PBS中の5mg/ml抗マウスIL−18BPであった。抗原親和性により、ウサギ血清からポリクローナル抗体を精製した。O/N、4℃でインキュベートした後、1×PBS+0.05%Tween20を用いて洗浄し、1×PBS、0.2%BSA(BSA、Sigma、参照番号A−2153)を用いて37℃で1時間ブロッキングし、1×PBS+0.05%Tween20を用いて洗浄した。標準物は、mIL−18BP 300ng/ml〜0.1ng/mlであり、試料は1×PBS、0.1%BSA、0.05%Tween20中0.1ml、37℃で2時間である。1×PBS+0.05%Tween20を用いて洗浄を実施した。0.1mlのビオチニル化抗マウスIL18BP(Peprotech)0.3mg/mlを37℃で2時間添加した。1×PBS+0.05%Tween20を用いて洗浄を実施し、0.1mlのextravidin HRP(Sigma、参照番号E2886)1/5000を静かに添加し、室温で30分間放置した。0.05mlの20%H2SO4を用いて反応を停止させた。492nmで読み取った。
【0197】
2.2 結果
図11Cから明らかであるが、血中m−IL18BPレベルは、カプセルのi.p.注射後3日目から6日目の間でピーク値に達し、8日目のm−IL18BPレベルは依然非常に高いままであった。
【図面の簡単な説明】
【0198】
【図1】ヒトIL−6(hIL−6)の完全コード配列及びその翻訳配列(配列番号1及び2)を示す図である。
【図2A】哺乳動物細胞発現ベクターpEAK12dの制限酵素地図である。
【図2B】哺乳動物細胞発現ベクターpEAK12dの完全ヌクレオチド配列(配列番号11)を示す図である。
【図3】哺乳動物細胞発現ベクターpEAK12d−IL−6−6HISの制限酵素地図である。
【図4】トランスフェクトされなかったHEK293−EBNA細胞を含有するカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したTNFα、ASAT、及びALATレベルを示すヒストグラムである。
【図5A】hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをi.p.又はs.c.注射した後12日間連続してマウスの血液中で測定したhIL−6レベルのグラフである。
【図5B】hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したTNFαレベルを示すヒストグラムである。
【図5C】hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したASATレベルを示すヒストグラムである。
【図5D】hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルを注射したConA処置マウス、及び対照マウスの血液試料中で測定したALATレベルを示すヒストグラムである。
【図6】hIL−6 HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセル700、350、及び100μlそれぞれのi.p.注射後3日間連続してマウスの血液中で測定したhIL−6レベルを示すヒストグラムである。
【図7A】hIL−6−6HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをそれぞれ700、350、及び100μl注射したConA処置マウス、並びに対照マウスの血液試料中で測定したTNFαレベルを示すヒストグラムである。
【図7B】hIL−6−6HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをそれぞれ700、350、及び100μl注射したConA処置マウス、並びに対照マウスの血液試料中で測定したASATレベルを示すヒストグラムである。
【図7C】hIL−6−6HISをコードしているcDNAを一過性にトランスフェクトしたHEK293−EBNA細胞を含有するカプセルをそれぞれ700、350、及び100μl注射したConA処置マウス、並びに対照マウスの血液試料中で測定したALATレベルを示すヒストグラムである。
【図8A】hepaCAM遺伝子の配列(配列番号7)を示す図である。
【図8B】hepaCAM成熟タンパク質の配列(配列番号8)を示す図である。
【図8C】トランスフェクトしなかったカプセル化HEK293−EBNA細胞、及びhepaCAMをコードしている遺伝子を一過性にトランスフェクトした異なる量のカプセル化HEK293−EBNA細胞をi.p.注射した後の、LPS誘発TNFα放出のマウスモデルにおけるTNFαレベルを示すヒストグラムである。
【図8D】トランスフェクトしなかったカプセル化HEK293−EBNA細胞、及びhepaCAMをコードしている遺伝子を一過性にトランスフェクトした異なる量のカプセル化HEK293−EBNA細胞をs.c.注射した後の、LPS誘発TNFα放出のマウスモデルにおけるTNFαレベルを示すヒストグラムである。
【図9A】INSP114−SV2をコードしている遺伝子の配列(配列番号9)を示す図である。
【図9B】INSP114−SV2成熟タンパク質の配列(配列番号10)を示す図である。
【図9C】トランスフェクトしなかったカプセル化HEK293−EBNA細胞、及びINSP114−SV2遺伝子を一過性にトランスフェクトしたカプセル化HEK293−EBNA細胞をi.p.注射した後の、LPS誘発TNFα放出のマウスモデルにおけるTNFαレベルを示すヒストグラムである。
【図10A】ヒトエリトロポエチン(EPO)をコードしている遺伝子の配列(配列番号5)を示す図である。
【図10B】ヒトエリトロポエチン(EPO)成熟タンパク質の配列(配列番号6)を示す図である。
【図10C】EPOをコードしている遺伝子を一過性にトランスフェクトしたカプセル化HEK293−EBNA細胞、EPOをコードしている遺伝子を安定にトランスフェクトしたカプセル化HEK293−EBNA細胞、及び対照カプセル化HEK293−EBNA細胞(トランスフェクトしなかった)をi.p.又はs.c.注射してから0日目〜15日目にELISAによって測定したマウスの血中EPO濃度を示すグラフである。
【図10D】EPOをコードしている遺伝子を一過性にトランスフェクトしたHEK293−EBNA細胞、及びEPOをコードしている遺伝子を安定にトランスフェクトしたHEK293−EBNAをi.p.又はs.c.注射してから0日目〜10日目のマウスにおけるヘマトクリット値(ヘマトクリット値(Ht)又はパック細胞容積(PCV)は、赤血球が占める血液量の割合である)を示すグラフである。
【図11A】マウスIL−18結合タンパク質(m−IL18BP)の遺伝子の配列(配列番号3)を示す図である。
【図11B】マウスIL−18結合タンパク質(m−IL18BP)の成熟タンパク質の配列(配列番号4)を示す図である。
【図11C】m−IL18BPをコードしている遺伝子を一過性にトランスフェクトしたカプセル化HEK293−EBNA細胞のi.p.注射から0日目〜8日目にELISAによって測定したマウスの血中m−IL18BP濃度を示すヒストグラムである。
【特許請求の範囲】
【請求項1】
対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセル。
【請求項2】
細胞が動物細胞、好ましくは哺乳動物細胞である、請求項1に記載のカプセル。
【請求項3】
対象の遺伝子が、タンパク質を分泌するためのシグナル配列に融合している、前記請求項のいずれかに記載のカプセル。
【請求項4】
対象の遺伝子が発現カセット中に挿入されている、前記請求項のいずれかに記載のカプセル。
【請求項5】
対象の遺伝子がプラスミド中に挿入されている、前記請求項のいずれかに記載のカプセル。
【請求項6】
生体適合性ポリマーがアルギナート−ポリ−L−リシン−アルギナート(APA)である、前記請求項のいずれかに記載のカプセル。
【請求項7】
生体適合性ポリマー膜が、分離サイズが90〜30kDa、好ましくは80〜60kDaである孔を有する、前記請求項のいずれかに記載のカプセル。
【請求項8】
平均直径が100〜1500μm、好ましくは250〜600μm、特に440〜530μmである、前記請求項のいずれかに記載のカプセル。
【請求項9】
低剪断、微小重力条件下で維持された、前記請求項のいずれかに記載のカプセル。
【請求項10】
カプセルを調製する方法であって、細胞に対象の遺伝子を一過性にトランスフェクトするステップと、一過性にトランスフェクトされた細胞をカプセル化するステップとを含む方法。
【請求項11】
低剪断、微小重力条件下で維持するステップをさらに含む、請求項10に記載の方法。
【請求項12】
対象の遺伝子によって発現及び分泌されたタンパク質のin vivo活性を評価する方法であって、請求項1から9までのいずれかに記載のカプセルを多細胞生物に投与するステップと、前記タンパク質の活性を検出するステップとを含む方法。
【請求項13】
多細胞生物が哺乳動物である、請求項12に記載の方法。
【請求項14】
哺乳動物が、マウス、ラット、イヌ、ヤギ、ヒツジ、ウシ、及びサルからなる群から選択される、請求項13に記載の方法。
【請求項15】
カプセルの投与が腹控内注射によって実施される、請求項14に記載の方法。
【請求項16】
検出すべきin vivo活性が前記タンパク質の投与後3〜14日、好ましくは3〜5日以内に完全に誘導される、動物における請求項12から15までのいずれかに記載の方法の使用。
【請求項17】
動物が、コンカナバリンA(ConA)誘発肝毒性を有するマウスである、請求項16に記載の使用。
【請求項18】
請求項1から9までのいずれかに記載のカプセルを含む医薬組成物。
【請求項19】
対象の遺伝子が、抗原/免疫原及び/又はアジュバントをコードしている、請求項18に記載の医薬組成物。
【請求項20】
抗原/免疫原が、細菌抗原、ウイルス抗原、真菌抗原、寄生虫抗原、又は腫瘍抗原である、請求項19に記載の医薬組成物。
【請求項21】
カプセルが、(1種又は複数の)抗原及び/又はアジュバントをコードしている遺伝子をそれぞれトランスフェクトした少なくとも2種類の細胞を含み、この2種の遺伝子が同一でない、請求項18から21までのいずれかに記載の医薬組成物。
【請求項22】
対象のタンパク質を被検体に投与するための、請求項1から9までのいずれかに記載のカプセル又は請求項18から21までのいずれかに記載の医薬組成物の使用。
【請求項23】
対象のタンパク質が免疫又はワクチン接種用の抗原である、請求項22に記載の使用。
【請求項24】
被検体が、ヒト;げっ歯類、イヌ、ブタ及びサルを含めた研究目的で飼育された動物並びにイヌ及びネコを含めた、家畜及び飼育動物からなる群から選択される、請求項22又は23のいずれかに記載の使用。
【請求項25】
請求項18から21までのいずれかに記載のカプセル又は医薬組成物を含むキット、及び前記組成物を適用するための又は前記組成物を被検体に適用するための手段。
【請求項26】
免疫又はワクチン接種用の医薬組成物を調製するための、請求項1から9までに記載のカプセルの使用。
【請求項1】
対象の遺伝子を一過性にトランスフェクトし、生体適合性ポリマー膜内に包含させた細胞を含有するカプセル。
【請求項2】
細胞が動物細胞、好ましくは哺乳動物細胞である、請求項1に記載のカプセル。
【請求項3】
対象の遺伝子が、タンパク質を分泌するためのシグナル配列に融合している、前記請求項のいずれかに記載のカプセル。
【請求項4】
対象の遺伝子が発現カセット中に挿入されている、前記請求項のいずれかに記載のカプセル。
【請求項5】
対象の遺伝子がプラスミド中に挿入されている、前記請求項のいずれかに記載のカプセル。
【請求項6】
生体適合性ポリマーがアルギナート−ポリ−L−リシン−アルギナート(APA)である、前記請求項のいずれかに記載のカプセル。
【請求項7】
生体適合性ポリマー膜が、分離サイズが90〜30kDa、好ましくは80〜60kDaである孔を有する、前記請求項のいずれかに記載のカプセル。
【請求項8】
平均直径が100〜1500μm、好ましくは250〜600μm、特に440〜530μmである、前記請求項のいずれかに記載のカプセル。
【請求項9】
低剪断、微小重力条件下で維持された、前記請求項のいずれかに記載のカプセル。
【請求項10】
カプセルを調製する方法であって、細胞に対象の遺伝子を一過性にトランスフェクトするステップと、一過性にトランスフェクトされた細胞をカプセル化するステップとを含む方法。
【請求項11】
低剪断、微小重力条件下で維持するステップをさらに含む、請求項10に記載の方法。
【請求項12】
対象の遺伝子によって発現及び分泌されたタンパク質のin vivo活性を評価する方法であって、請求項1から9までのいずれかに記載のカプセルを多細胞生物に投与するステップと、前記タンパク質の活性を検出するステップとを含む方法。
【請求項13】
多細胞生物が哺乳動物である、請求項12に記載の方法。
【請求項14】
哺乳動物が、マウス、ラット、イヌ、ヤギ、ヒツジ、ウシ、及びサルからなる群から選択される、請求項13に記載の方法。
【請求項15】
カプセルの投与が腹控内注射によって実施される、請求項14に記載の方法。
【請求項16】
検出すべきin vivo活性が前記タンパク質の投与後3〜14日、好ましくは3〜5日以内に完全に誘導される、動物における請求項12から15までのいずれかに記載の方法の使用。
【請求項17】
動物が、コンカナバリンA(ConA)誘発肝毒性を有するマウスである、請求項16に記載の使用。
【請求項18】
請求項1から9までのいずれかに記載のカプセルを含む医薬組成物。
【請求項19】
対象の遺伝子が、抗原/免疫原及び/又はアジュバントをコードしている、請求項18に記載の医薬組成物。
【請求項20】
抗原/免疫原が、細菌抗原、ウイルス抗原、真菌抗原、寄生虫抗原、又は腫瘍抗原である、請求項19に記載の医薬組成物。
【請求項21】
カプセルが、(1種又は複数の)抗原及び/又はアジュバントをコードしている遺伝子をそれぞれトランスフェクトした少なくとも2種類の細胞を含み、この2種の遺伝子が同一でない、請求項18から21までのいずれかに記載の医薬組成物。
【請求項22】
対象のタンパク質を被検体に投与するための、請求項1から9までのいずれかに記載のカプセル又は請求項18から21までのいずれかに記載の医薬組成物の使用。
【請求項23】
対象のタンパク質が免疫又はワクチン接種用の抗原である、請求項22に記載の使用。
【請求項24】
被検体が、ヒト;げっ歯類、イヌ、ブタ及びサルを含めた研究目的で飼育された動物並びにイヌ及びネコを含めた、家畜及び飼育動物からなる群から選択される、請求項22又は23のいずれかに記載の使用。
【請求項25】
請求項18から21までのいずれかに記載のカプセル又は医薬組成物を含むキット、及び前記組成物を適用するための又は前記組成物を被検体に適用するための手段。
【請求項26】
免疫又はワクチン接種用の医薬組成物を調製するための、請求項1から9までに記載のカプセルの使用。
【図1】


【図2A】
【図2B】

【図2B】

【図2B】

【図3】
【図4】
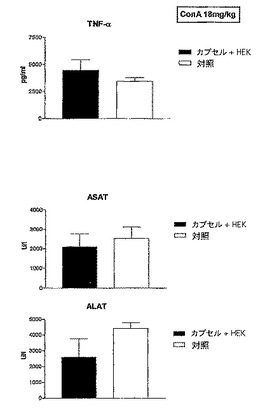
【図5A】
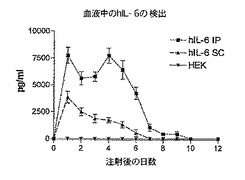
【図5B】
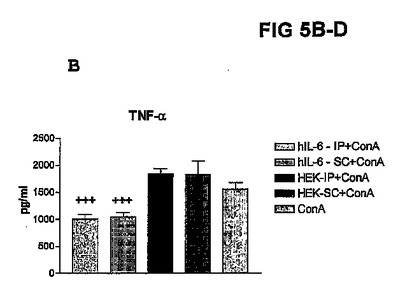
【図5C】
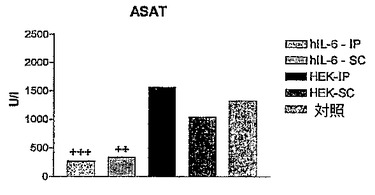
【図5D】
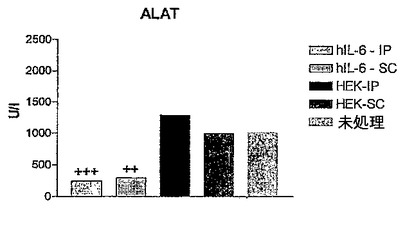
【図6】
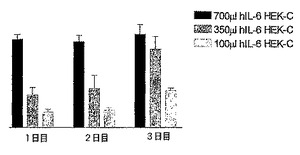
【図7】
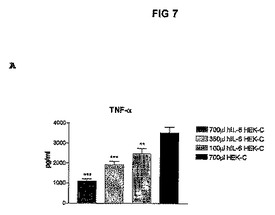
【図7】
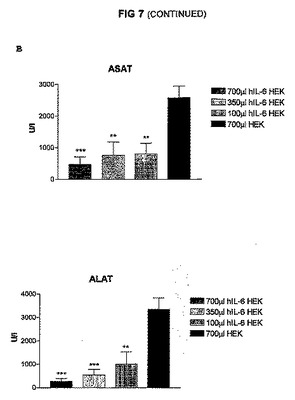
【図8A】

【図8B】

【図8C】
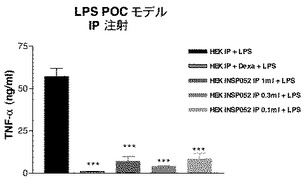
【図8D】
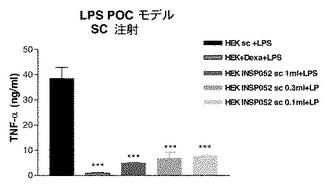
【図9A】

【図9B】

【図9C】
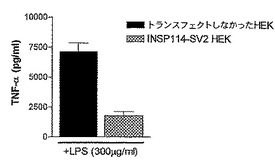
【図10A】

【図10B】

【図10C】
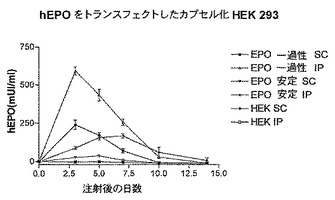
【図10D】
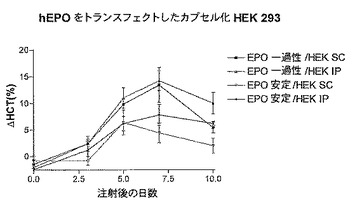
【図11A】

【図11B】

【図11C】
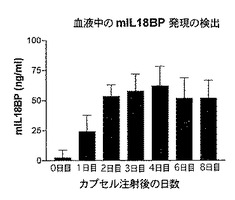
【図2A】
【図2B】
【図2B】
【図2B】
【図3】
【図4】
【図5A】
【図5B】
【図5C】
【図5D】
【図6】
【図7】
【図7】
【図8A】
【図8B】
【図8C】
【図8D】
【図9A】
【図9B】
【図9C】
【図10A】
【図10B】
【図10C】
【図10D】
【図11A】
【図11B】
【図11C】
【公表番号】特表2008−509127(P2008−509127A)
【公表日】平成20年3月27日(2008.3.27)
【国際特許分類】
【出願番号】特願2007−524420(P2007−524420)
【出願日】平成17年8月2日(2005.8.2)
【国際出願番号】PCT/IB2005/002294
【国際公開番号】WO2006/016238
【国際公開日】平成18年2月16日(2006.2.16)
【出願人】(507399427)ラボラトワ セローノ エス.エイ. (2)
【Fターム(参考)】
【公表日】平成20年3月27日(2008.3.27)
【国際特許分類】
【出願日】平成17年8月2日(2005.8.2)
【国際出願番号】PCT/IB2005/002294
【国際公開番号】WO2006/016238
【国際公開日】平成18年2月16日(2006.2.16)
【出願人】(507399427)ラボラトワ セローノ エス.エイ. (2)
【Fターム(参考)】
[ Back to top ]