
トリアゾロ[4,5−D]ピリミジン化合物の新規な結晶形及び非晶形
【課題】本発明は、式(I)の化学化合物の新規形態を提供する。
【解決手段】本発明は、式(I)の化合物の諸形態、特に結晶形及び非晶形、より特定すると、4つの結晶形と1つの非晶形に関する。さらに本発明は、そのような諸形態の製造法、結晶形及び/又は非晶形の化合物を含んでなる医薬組成物、及びそのような諸形態の治療使用に関する。
【解決手段】本発明は、式(I)の化合物の諸形態、特に結晶形及び非晶形、より特定すると、4つの結晶形と1つの非晶形に関する。さらに本発明は、そのような諸形態の製造法、結晶形及び/又は非晶形の化合物を含んでなる医薬組成物、及びそのような諸形態の治療使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、化学化合物の諸形態、特に結晶形及び非晶形、より特定すると、4つの結晶形と1つの非晶形に関する。さらに本発明は、そのような諸形態の製造法、結晶形及び/又は非晶形の化合物を含んでなる医薬組成物、及びそのような諸形態の治療使用に関する。
【背景技術】
【0002】
医薬組成物の製剤では、簡便に処理されて加工され得る形態で薬物が存在することが重要である。このことは、商業的に実行可能な製造法を得る視点からだけでなく、活性化合物を含んでなる医薬製剤の後続の製造という点からも重要である。有効成分の化学的安定性、固体状態の安定性、及び貯蔵寿命も非常に重要な要因である。医薬物質と、それを含有する組成物は、活性成分の物理化学的特性(例、その化学組成、密度、吸湿性、及び溶解性)において有意な変化を示すことなく、かなりの時間にわたり効果的に保存されることが可能であるべきである。さらに、可能な限り純粋である形態で薬物を提供することができることも重要である。非晶性の材料は、この点で重大な問題を提起する場合がある。例えば、そのような材料は、処理することや製剤化することが結晶性の材料よりも通常は困難であり、信頼し得ない溶解性を提供し、しばしば不安定で化学的に不純であることが見出される。当業者が理解されるように、薬物が安定な結晶形で容易に入手され得るならば、上記の諸問題は解決される可能性がある。このように、商業的に実行可能で製剤的に許容される医薬組成物の製造では、どこでも可能であれば、実質的に結晶性で、安定な形態で薬物を提供することが望ましい。しかしながら、注目すべきことは、この目標が必ずしも達成可能でないことである。実際、典型的には、ある化合物の結晶化の挙動がどのようであるかを分子構造だけから予測することは可能ではなく、このことは、通常、経験的にしか決定し得ないのである。
【0003】
血小板の付着及び凝集は、動脈血栓症における初発事象である。内皮下表面への血小板付着のプロセスは損傷した血管壁の修復に重要な役割を担う可能性があるが、これにより開始される血小板凝集は、生きた血管床へ急性の血栓閉塞を沈殿させる可能性があり、これが心筋梗塞や不安定狭心症のような、重い病態を伴う事象をもたらす。これら病態を予防するか又は緩和させるために使用される血栓溶解や血管形成のような介入も、血小板仲介性の閉塞若しくは再閉塞により弱められる。
【発明の概要】
【発明が解決しようとする課題】
【0004】
アデノシン5’−二リン酸(ADP)は、血栓症の重要なメディエーターとして作用することが見出されてきた。ADP誘導性の血小板凝集は、血小板の膜上に位置するP2T受容体サブタイプにより仲介される。P2T受容体(P2YADP又はP2TACとしても知られる)は、血小板の凝集/活性化を仲介することに主に関与し、まだクローン化されていないGタンパク質共役型受容体である。この受容体の薬理学的特性は、例えば、Humphries et al., Br. J. Pharmacology (1994), 113, 1057-1063 と Fagura et al., Br. J. Pharmacology (1998) 124, 157-164 による参考文献に記載されている。最近、この受容体での拮抗薬が他の抗血栓剤を越える重要な改良点を提供することが示された(J. Med. Chem. (1999) 42, 213 を参照のこと)。国際特許出願WO9905143は、P2T(P2YADP又はP2TAC)拮抗薬としての活性を有するトリアゾロ[4,5−d]ピリミジン化合物の系列を包括的に開示する。(以下に図示されるような)式(I)の化合物は、国際特許出願WO9905143の包括的な範囲に含まれるものの、そこで特別に開示されているわけではない。この化合物は、P2T(P2YADP又はP2TAC)拮抗薬として高い効力を示す。それはまた、驚くほど高い代謝安定性及びバイオアベイラビリティを有する。
【課題を解決するための手段】
【0005】
従って、本発明は、式(I):
【0006】
【化1】

【0007】
の実質的に結晶形の化合物に関する。
式(I)の化合物は、慣用的に、{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオールと命名される。
【0008】
式(I)の化合物は、以下、多型I、多型II、多型III、及び多型IVと呼ばれる、4つの異なる実質的に結晶性の形態で存在し得る。多型とは、化合物の特定の結晶形である。
【0009】
多型の諸形態が、互いに関して、そして非結晶状態に関して異なる物理特性を有することは、特に化合物が工業スケールで製造されるか又は使用されるときに、化合物の化学的及び製剤的処理に対して顕著に影響を及ぼす可能性がある。
【0010】
本発明の1つの側面では、式(I)の化合物の好ましい結晶形は、多型I、多型II、多型III、及び/又は多型IVの形態で存在する。
本発明の他の側面では、式(I)の化合物の好ましい結晶形が多型Iである。
【0011】
本発明の別の側面では、式(I)の化合物の好ましい結晶形が多型IIである。
本発明のさらなる側面では、式(I)の化合物の好ましい結晶形が多型IIIである。
【0012】
本発明の追加の側面では、式(I)の化合物の好ましい結晶形が多型IVである。
本発明のさらなる側面では、式(I)の化合物が実質的に非晶形で存在する。非晶形では、結晶形において(例えば、多型において)通常存在する三次元の長距離数(long range order)が存在せず、非晶形における分子の互いに対する位置は本質的に無秩序である(B. C. Hancock と G. Zografi, J. Pharm. Sci. (1997) 86, 1 を参照のこと)。式(I)の化合物の非晶形をフォームαと呼ぶ。
【0013】
我々は、式(I)の化合物を結晶形と非晶形で単離した。これらの形態は、実質的に、又は本質的に水分がないまま(「無水」形)で存在し得る。故に、本発明の1つの側面では、式(I)の化合物の無水形が結晶形又は非晶形で提供される。「実質的に純粋で本質的に無水形である」という用語の使用は、結晶格子構造の内側又は結晶格子構造の外側に、水を含む何らかの溶媒の存在を排除するものではない。無水形は、化合物1分子につき0.4未満の水分子を有する(40%未満水和している)。好ましくは、無水形は、化合物1分子につき0.1未満の水分子を含有する。
【0014】
多型I、II、III、及びIVは、その融解開始点(onset of melting)、粉末X線回折パターン、及び/又は単結晶X線データを参照して区別され得る。
多型Iは、実質的に純粋で、本質的に無水形である場合、146〜152℃の範囲、例えば約151℃にある融解開始点を有する。
【0015】
多型IIは、実質的に純粋で、本質的に無水形である場合、136〜139℃の範囲、例えば約137.5℃にある融解開始点を有する。
多型IIIは、実質的に純粋で、本質的に無水形である場合、127〜132℃の範囲、例えば約132℃にある融解開始点を有する。
【0016】
多型IVは、実質的に純粋で、本質的に無水形である場合、典型的には約139℃である融解開始点を有する。
フォームαは、典型的には、融解に先立ってガラス転移を受けた後で、上記多型の1つ、例えば多型IIへ結晶化する。
【0017】
融点は、パーキン・エルマーDSC7装置を使用する、示差走査熱量測定(DSC)を用いて決定した。融解開始点は、ベースラインからの有意な変化が起こる点として定義され、パーキン・エルマーのPyrisソフトウェアにより測定した。理解されるように、他のタイプの装置によるか、又は本明細書の記載とは異なる条件を使用することによって、別の融点読取り値が与えられる可能性がある。従って、引用される数値を絶対値として解釈してはならない。当業者は、正確な融点の値が化合物の純度、サンプル重量、加熱速度、及び粒子サイズにより影響され得ることを理解されよう。
【0018】
多型Iは、実質的に純粋で、本質的に無水形である場合、高強度の特定ピークを5.3°(±0.1°)、20.1°(±0.1°)、20.7°(±0.1°)、21.0°(±0.1°)、及び21.3°(±0.1°)2θに含有するX線粉末回折パターンを有する。より好ましくは、実質的に純粋で本質的に無水の多型Iは、特定ピークを5.3°(±0.1°)、8.0°(±0.1°)、9.6°(±0.1°)、13.9°(±0.1°)、15.3°(±0.1°)、20.1°(±0.1°)、20.7°(±0.1°)、21.0°(±0.1°)、21.3°(±0.1°)、26.2°(±0.1°)、及び27.5°(±0.1°)2θに含有するX線粉末回折パターンを有する。
【0019】
多型IIは、実質的に純粋で、本質的に無水形である場合、高強度の特定ピークを5.5°(±0.1°)、13.5°(±0.1°)、18.3°(±0.1°)、22.7°(±0.1°)、及び24.3°(±0.1°)2θに含有するX線粉末回折パターンを有する。より好ましくは、実質的に純粋で本質的に無水の多型IIは、特定ピークを5.5°(±0.1°)、6.8°(±0.1°)、10.6°(±0.1°)、13.5°(±0.1°)、14.9°(±0.1°)、18.3°(±0.1°)、19.2°(±0.1°)、22.7°(±0.1°)、24.3°(±0.1°)、及び27.1°(±0.1°)2θに含有するX線粉末回折パターンを有する。
【0020】
多型IIIは、実質的に純粋で、本質的に無水形である場合、高強度の特定ピークを14.0°(±0.1°)、17.4°(±0.1°)、18.4°(±0.1°)、21.4°(±0.1°)、及び24.1°(±0.1°)2θに含有するX線粉末回折パターンを有する。より好ましくは、実質的に純粋で本質的に無水の多型IIIは、特定ピークを5.6°(±0.1°)、12.5°(±0.1°)、14.0°(±0.1°)、17.4°(±0.1°)、18.4°(±0.1°)、21.4°(±0.1°)、22.2°(±0.1°)、22.9°(±0.1°)、24.1°(±0.1°)、及び24.5°(±0.1°)2θに含有するX線粉末回折パターンを有する。
【0021】
多型IVは、実質的に純粋で、本質的に無水形である場合、高強度の特定ピークを4.9°(±0.1°)、9.2°(±0.1°)、11.6°(±0.1°)、15.6°(±0.1°)、及び16.4°(±0.1°)2θに含有するX線粉末回折パターンを有する。より好ましくは、実質的に純粋で本質的に無水の多型IVは、特定ピークを4.9°(±0.1°)、6.0°(±0.1°)、9.2°(±0.1°)、11.6°(±0.1°)、12.8°(±0.1°)、15.6°(±0.1°)、16.4°(±0.1°)、17.2°(±0.1°)、及び18.1°(±0.1°)2θに含有するX線粉末回折パターンを有する。
【0022】
フォームαは、実質的に純粋で、本質的に無水形である場合、明瞭なピークを含有しないX線粉末回折パターンを有する。
多型II、多型III、多型IV、及びフォームαのX線回折データは、ジーメンスD5000装置を使用して入手した。多型IのX線回折データは、フィリップスのX’Pert MPD機器を使用して入手した。装置及び/又は条件が異なれば、やや異なるデータが作成され得ることが理解されよう。従って、引用される数値を絶対値として解釈してはならない。
【0023】
本発明の別の側面では、溶媒和形、例えば水和形(「水和物」)が形成され得る。故に、本発明のこの側面では、結晶形の式(I)の化合物の水和物が提供される。水和物は、化合物1分子につき0.8以上の水分子を有する(80%以上水和している)。半水和物は、化合物1分子につき0.4〜0.8の水分子を有する(40〜80%水和している)。
【0024】
本発明のさらなる特徴では、式(I)の化合物の結晶形及び/又は非晶形の混合物が提供される。好ましくは、混合物は、多型I、多型II、多型III、多型IV、及び/又はフォームαの混合物である。より好ましくは、本発明は、多型IIと多型IIIの任意の混合物を提供する。
【0025】
本発明のさらなる特徴では、式(I)の化合物の好適な溶媒からの結晶化による、式(I)の化合物の結晶形の製造法が提供される。好ましくは、溶媒は、エタノール、酢酸エチル、イソプロパノール、イソオクタン、アセトニトリル、水、又はこれらの混合物の群から選択される。より好ましくは、溶媒は、エタノール、酢酸エチル、イソプロパノール、イソオクタン、水、又はこれらの混合物の群から選択される。好適には、溶媒は、メタノール及び水の混合物、エタノール、酢酸エチル、エタノール及び水の混合物、イソプロパノール及び水の混合物、酢酸エチル及びイソオクタンの混合物、及びアセトニトリルの群から選択される。
【0026】
式(I)の化合物は、WO9905143の記載に類似した方法により製造され得る。
結晶化を始めるには、式(I)の化合物の結晶を用いた種入れ(seeding)が必要とされ得る。選択される多型を得るには、求められる多型を用いた種入れが必要となり得る。式(I)の化合物の適正な溶媒系からの結晶化は、例えば、冷却、溶媒蒸発、及び/又は抗溶媒(anti−solvent:式(I)の化合物がほとんど溶けない溶媒;好適な抗溶媒の例には、ヘプタン又はイソオクタンが含まれる)の追加により過飽和にさせることによって達成され得る。結晶化の温度及び時間は、溶液中の化合物の濃度、使用する溶媒系、及び、採用する結晶化の方法に依存して変化する。
【0027】
結晶形の式(I)の化合物は、当業者によく知られた技術、例えば、デカント(decanting)、濾過、又は遠心分離の技術を使用して、上記の反応混合物から単離され得る。同様に、結晶形の式(I)の化合物は、周知の方法に従って、乾燥され得る。
【0028】
非結晶性の材料、化学不純物のような不純物をさらに減らすためか、又はある多型を別の多型へ、又は水和物若しくは無水形へと結晶形を変換するために、同じか又は異なる溶媒系を使用して、場合によっては再結晶の工程が実施され得る。さらに、非結晶性の材料を減らすには、この固形物を高湿度に曝露する工程といった、条件付けの工程を実施してよい。
【0029】
好ましくは、結晶化は、反応溶液から直接行なわれる。他のやり方では、結晶化は後続の溶液から実施される。
本発明のさらなる特徴では、多型Iを製造する方法が提供され、前記方法は、ゆっくりと結晶成長する多型I由来の多型Iのわずかな種結晶(seed crystals)を多型IIの融成物から入手する工程、及び、これを使用して式(I)の化合物と、メタノール/水のような好適な混合溶媒系を含んでなる反応混合物に種入れする工程を含む。
【0030】
本発明のさらなる特徴では、多型IIを製造する方法が提供され、前記方法は、酢酸エチルのような好適な溶媒中での結晶化を含む。
本発明のさらなる特徴では、多型IIIを製造する方法が提供され、前記方法は、アルコール、例えばエタノール若しくはイソプロピルアルコール(IPA)のような好適な溶媒中での結晶化、特に、多型IIIの結晶を用いた種入れ、又は、IPAのような好適な溶媒中での式(I)の化合物のスラリー化を含む。
【0031】
本発明のさらなる特徴では、多型IVを製造する方法が提供され、前記方法は、アセトニトリルのような好適な溶媒からの結晶化、特に、多型IVの結晶を用いた種入れ、又は、アセトニトリルのような好適な溶媒中での式(I)の化合物をスラリー化させる時間を含む。
【0032】
本発明のさらなる特徴は、実質的に多型IIがない多型IIIを製造する方法を提供し、前記方法は、例えば、C1-6脂肪族アルコール/水溶媒系(好ましくは、IPA/水)において、5〜65℃の温度で1〜10日間、式(I)の化合物をスラリー化させる工程を含む。
【0033】
本発明のさらなる特徴では、実質的に非晶形である式(I)の化合物の製造法が提供され、前記方法は、好適な溶媒系、例えばエタノール/水を使用して、式(I)の化合物の溶液を凍結乾燥させるか、又はスプレー乾燥させる工程を含む。
【0034】
「実質的にない(substantially free)」という用語は、他の多型が10%未満であること、好ましくは5%未満であることを意味する。
本発明のさらなる側面では、上記方法のいずれでも入手可能な化合物が提供される。
【0035】
結晶形及び/又は非晶形の式(I)の化合物は、P2T(P2YADP又はP2TAC)受容体拮抗薬として作用する。従って、結晶形及び/又は非晶形の式(I)の化合物は、組合せ療法を含む療法に有用である。特に、結晶形の式(I)の化合物は、冠状動脈、脳血管、又は末梢血管の疾患を有する患者の動脈血栓合併症の治療若しくは予防における使用に適用される。動脈血栓合併症には、不安定狭心症、血栓若しくは塞栓性の卒中のようなアテローム性動脈硬化症の一次動脈血栓合併症、一過性虚血発作、末梢血管障害、血栓溶解を伴うか又は伴わない心筋梗塞、冠血管形成術(PTCA)を含む血管形成術、動脈内膜切除術、ステント置換、冠血管と他の血管の移植手術といった、アテローム動脈硬化症における介入による動脈合併症、偶発的若しくは外科的外傷や皮膚及び筋肉のフラップを含む再建手術後の組織サルベージ(tissue salvage)のような外科的若しくは機械的な損傷の血栓合併症、播種性血管内凝固のような拡散性の血栓/血小板消費因子を伴う病態、血栓性血小板減少性紫斑病、溶血性尿毒症候群、敗血症の血栓合併症、成人呼吸窮迫症候群、抗リン脂質症候群、ヘパリン誘導性の血小板減少症及び子癇前症/子癇症、又は深在性静脈血栓症のような静脈血栓症、静脈閉塞症、血小板血症を含む骨髄増殖疾患のような血液学的病態、鎌状赤血球病;[又は、心肺バイパス及び体外での膜式酸素加(微小血栓塞栓症の予防)のような、機械的に誘導される in vivo での血小板活性化、血液製品(例、血小板濃縮物)の保存中の使用のような、機械的に誘導されるin vitroでの血小板活性化、又は腎透析及びプラズマフェレーシス中のシャント閉塞の予防では]血管炎、動脈炎、糸球体腎炎、炎症性腸疾患、及び臓器移植拒絶のような血管損傷/炎症に続発する血栓症、偏頭痛のような病態、レイノー現象、アテローム性プラーク形成/進行、狭窄/再狭窄のような血管壁の根底にある炎症性疾患プロセスに血小板が貢献し得る病態、及び、血小板と血小板由来因子が免疫学的疾患プロセスに関連していると思われる喘息のような他の炎症病態が含まれる。さらなる適応症には、CNS障害の治療と腫瘍の増殖及び拡散の予防が含まれる。
【0036】
本発明のさらなる側面によれば、ヒト若しくは動物の身体の治療による処置の方法に使用される、結晶形及び/又は非晶形の式(I)の化合物が提供される。
本発明の追加の特徴によれば、医薬品としての使用のための、結晶形及び/又は非晶形の式(I)の化合物が提供される。好ましくは、結晶形及び/又は非晶形の式(I)の化合物は、ヒトのような温血動物においてP2T(P2YADP又はP2TAC)受容体に拮抗する医薬品として使用される。より好ましくは、結晶形及び/又は非晶形の式(I)の化合物は、冠動脈、脳血管、又は末梢血管の疾患を有する、ヒトのような温血動物の患者の動脈血栓合併症を治療するか又は予防するための医薬品として使用される。
【0037】
本発明によれば、結晶形及び/又は非晶形である式(I)の化合物の、P2T(P2YADP又はP2TAC)受容体の拮抗薬として使用される医薬品の製造における使用がさらに提供される。特に、結晶形及び/又は非晶形である式(I)の化合物の、冠動脈、脳血管、又は末梢血管の疾患を有する患者の動脈血栓合併症の治療若しくは予防に使用される医薬品の製造における使用がさらに提供される。
【0038】
本発明はまた、冠動脈、脳血管、又は末梢血管の疾患を有する患者における動脈血栓合併症の治療若しくは予防の方法を提供し、前記方法は、そのような障害に罹患しているか又はそれに罹りやすいヒトへ、結晶形及び/又は非晶形である式(I)の化合物の治療有効量を投与することを含む。
【0039】
結晶形及び/又は非晶形の式(I)の化合物は、溶液剤、懸濁液剤、HFAエアゾール剤、及び乾燥粉末製剤の形態で局所的に、例えば肺及び/又は気道へ;又は、錠剤、丸剤、カプセル剤、シロップ剤、散剤、又は顆粒剤の形態で全身的に、例えば経口投与により;又は、無菌の非経口溶液剤若しくは懸濁液剤の形態での非経口投与により、皮下投与により、又は坐剤の形態での直腸投与により、又は経皮的に、投与され得る。
【0040】
結晶形及び/又は非晶形の式(I)の化合物は、それ自身でか、又は、結晶形及び/又は非晶形の式(I)の化合物を製剤的に許容される希釈剤、アジュバント、及び/又は担体と組合せて含んでなる医薬組成物として投与され得る。故に、本発明のさらなる特徴として、結晶形及び/又は非晶形の式(I)の化合物を製剤的に許容される希釈剤、アジュバント、及び/又は担体と一緒に含んでなる医薬組成物が提供される。特に好ましいのは、有害なアレルギー反応のような有害反応を引き起こし得る材料を含有しない組成物である。
【0041】
結晶形及び/又は非晶形である式(I)の化合物の乾燥粉末製剤及び加圧HFAエアゾール剤は、経口若しくは経鼻吸入により投与され得る。吸入のために、結晶形及び/又は非晶形の式(I)の化合物は、望ましくは、粉砕される。結晶形及び/又は非晶形の式(I)の化合物はまた、乾燥粉末吸入器の手段により投与され得る。吸入器は単一若しくは複数用量の吸入器であり得て、呼気作動性の乾燥粉末吸入器であり得る。
【0042】
1つの可能性は、結晶形及び/又は非晶形の式(I)の粉砕化合物を、担体物質、例えば、単糖、二糖、若しくは多糖、糖アルコール、又は別のポリオールと混合することである。好適な担体には、糖類とデンプンが含まれる。他のやり方では、結晶形及び/又は非晶形の式(I)の粉砕化合物が別の物質により被覆され得る。粉末混合物はまた、結晶形及び/又は非晶形である式(I)の活性化合物の所望量をそれぞれ含有する、硬ゼラチンカプセル剤へ調剤され得る。
【0043】
もう1つの可能性は、粉砕された粉末を、吸入法の間に壊れる球体へ加工することである。この球状化した粉末が、多用量吸入器、例えば、Turbuhaler(登録商標)として知られるもの(患者により吸入される所望量の投薬単位が目盛られている)の薬物レザバーへ充填され得る。この系を用いて、式(I)の活性化合物が、担体物質を伴うか又は伴わずに患者へ投与される。結晶形及び/又は非晶形の式(I)の化合物を含んでなる医薬組成物は、好便にも、経口投与用の錠剤、丸剤、カプセル剤、シロップ剤、散剤、又は顆粒剤;非経口投与用の無菌の非経口若しくは皮下溶液剤、懸濁液剤、又は直腸投与用の坐剤であり得る。
【0044】
経口投与では、結晶形及び/又は非晶形の式(I)の化合物が、アジュバント若しくは担体(例えば、乳糖、サッカロース、ソルビトール、マンニトール、ジャガイモデンプン、トウモロコシデンプン、又はアミロペクチンのようなデンプン、セルロース誘導体)、ゼラチン若しくはポリビニルピロリドンのような結合剤、及び、ステアリン酸マグネシウム、ステアリン酸カルシウム、ポリエチレングリコール、ワックス、パラフィン、等のような潤滑剤とともに混合されてから、錠剤へ圧縮され得る。被覆錠剤が必要とされる場合、上記のように製造された芯が、例えば、アラビアゴム、ゼラチン、タルク、二酸化チタン、等を含有し得る濃縮糖溶液で被覆され得る。他のやり方では、錠剤を、易揮発性の有機溶媒若しくは水性溶媒のいずれかに溶けた好適なポリマーで被覆してもよい。
【0045】
軟ゼラチンカプセル剤の製造では、結晶形及び/又は非晶形の式(I)の化合物が、例えば、植物油又はポリエチレングリコールと混合され得る。硬ゼラチンカプセル剤は、上記の錠剤用の賦形剤、例えば、乳糖、サッカロース、ソルビトール、マンニトール、デンプン、セルロース誘導体、又はゼラチンのいずれか一方を使用して、本化合物の顆粒を含有し得る。また、薬物の液体若しくは半固体製剤を硬ゼラチンカプセル剤へ充填してもよい。
【0046】
経口適用の液体調製物は、シロップ剤若しくは懸濁液剤、例えば、結晶形及び/又は非晶形の式(I)の化合物を含有する溶液剤の形態であり得て、残りは、糖とエタノール、水、グリセロール、及びプロピレングリコールの混合物である。場合により、そのような液体調製物は、着色剤、芳香剤、サッカリン、及び、増粘剤としてのカルボキシメチルセルロース、又は当業者に知られた他の賦形剤を含有し得る。
【0047】
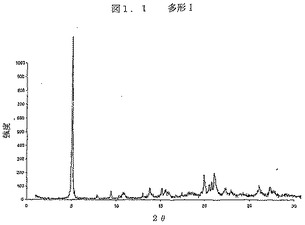
図1.1は、フィリップスのX’Pert MPD機器を、θ−θ配置で、1°〜40°2θの走査範囲にわたり、0.02°2θ増分につき2〜5秒の露出で使用して得られた、多型IのX線回折パターンである。X線は、40kVと50mAで作動する銅ロングファイン(long-fine)集束管により発生させた。X線の波長は1.5406A(オングストローム)であった。
【0048】
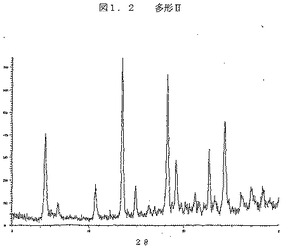
図1.2は、ジーメンスのD5000機器を、θ−θ配置で、2°〜30°2θの走査範囲にわたり、0.02°2θ増分につき4秒の露出で使用して得られた、多型IIのX線回折パターンである。X線は、45kVと40mAで作動する銅ロングファイン(long-fine)集束管により発生させた。X線の波長は1.5406A(オングストローム)であった。約10mgの化合物を置いたゼロ・バックグラウンドを使用してデータを収集した。このホルダーは、非回折面に沿って切削されてから、光学的に平面の仕上げにまで研磨された、単結晶のシリコンから製造された。この表面へのX線の入射は、ブラッグ消光により無効とされた。
【0049】
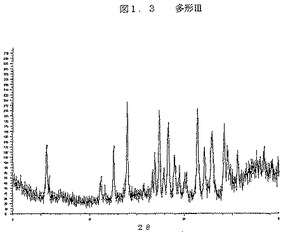
図1.3は、ジーメンスのD5000機器を上記のように使用して得られた、多型IIIのX線回折パターンである。
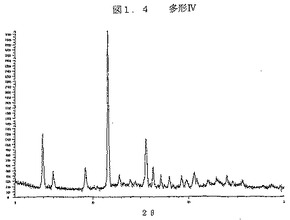
図1.4は、ジーメンスのD5000機器を上記のように使用して得られた、多型IVのX線回折パターンである。
【0050】
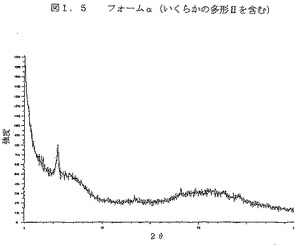
図1.5は、ジーメンスのD5000機器を上記のように使用して得られた、フォームαのX線回折パターンである。
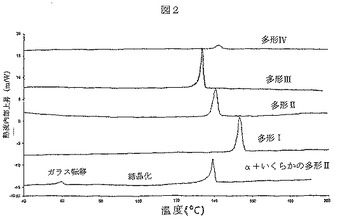
図2は、パーキン・エルマーDSC7装置を使用して得られた、多型I、II、III及びIVとフォームαのDSCグラフを示す。パンのタイプは、穿孔した蓋の付いたアルミニウムであった。サンプル重量は1〜3mgであった。この方法は、窒素ガス流(30ml/分)の下で実施し、試験した温度範囲は30℃〜325℃であり、10℃/分の一定速度で温度を増加させた。
【0051】
30ミクロンを超えるサイズの粒子と非単一性の縦横比を有するサンプルの分析は、ピークの相対強度に影響を及ぼす可能性があることを理解しておくべきである。当業者はまた、サンプルが回折計中にある正確な高さと回折計のゼロ較正により反射の位置が影響を受けることも理解されよう。サンプル表面の平面性もわずかに効果を及ぼす可能性がある。従って、提示される回折パターンデータを絶対値として解釈してはならない。
【図面の簡単な説明】
【0052】
【図1.1】図1.1は、フィリップスのX’Pert MPD機器を、θ−θ配置で、1°〜40°2θの走査範囲にわたり、0.02°2θ増分につき2〜5秒の露出で使用して得られた、多型IのX線回折パターンである。X線は、40kVと50mAで作動する銅ロングファイン(long-fine)集束管により発生させた。X線の波長は1.5406A(オングストローム)であった。
【図1.2】図1.2は、ジーメンスのD5000機器を、θ−θ配置で、2°〜30°2θの走査範囲にわたり、0.02°2θ増分につき4秒の露出で使用して得られた、多型IIのX線回折パターンである。X線は、45kVと40mAで作動する銅ロングファイン(long-fine)集束管により発生させた。X線の波長は1.5406A(オングストローム)であった。約10mgの化合物を置いたゼロ・バックグラウンドを使用してデータを収集した。このホルダーは、非回折面に沿って切削されてから、光学的に平面の仕上げにまで研磨された、単結晶のシリコンから製造された。この表面へのX線の入射は、ブラッグ消光により無効とされた。
【図1.3】図1.3は、ジーメンスのD5000機器を上記のように使用して得られた、多型IIIのX線回折パターンである。
【図1.4】図1.4は、ジーメンスのD5000機器を上記のように使用して得られた、多型IVのX線回折パターンである。
【図1.5】図1.5は、ジーメンスのD5000機器を上記のように使用して得られた、フォームαのX線回折パターンである。
【図2】図2は、パーキン・エルマーDSC7装置を使用して得られた、多型I、II、III及びIVとフォームαのDSCグラフを示す。パンのタイプは、穿孔した蓋の付いたアルミニウムであった。サンプル重量は1〜3mgであった。この方法は、窒素ガス流(30ml/分)の下で実施し、試験した温度範囲は30℃〜325℃であり、10℃/分の一定速度で温度を増加させた。
【実施例】
【0053】
本発明は、以下の非限定的な実施例により説明され得る。
【実施例1】
【0054】
多型Iの形態の{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
第1部
多型IIの形態である式(I)の化合物(2mg)を、以下のように、DSC中で加熱し、冷却した:35〜143〜35〜148〜35〜148〜35℃。このアニーリング法は、DSCにより示されるような純粋な多型Iの結晶化をもたらした。
第2部
式(I)の化合物、メタノール5ml/g、及び、水7.3ml/gと少量の多型Iの種を含んでなる溶液を、30℃で結晶化させた。XRPDとDSCにより、実質的に純粋な多型Iが形成されたことを確認した。
【実施例2】
【0055】
多型IIの形態の{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
45mgの式(I)の化合物へクロロホルム(150μl)を加え、この混合物を、蒸気浴上で溶けるまで温めた。生じた溶液を一晩放置して結晶化させ、窒素流の下で乾燥させた。XRPDとDSCにより、実質的に純粋な多型IIが形成されたことを確認した。
【実施例3】
【0056】
多型IIIの形態の{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
10mgの式(I)の化合物へエタノール(200μl)を加え、この混合物を、蒸気浴上で溶けるまで温めた。生じた溶液を一晩放置して結晶化させた。XRPDとDSCにより、多型II及びIIIの混合物が形成されたことを確認した。この材料を使用して、より大きいスケールの調製物へ種入れした:191mgの多型IIを1mlの50%イソプロパノール水溶液中でスラリー化させた。このスラリーへ、15mgの混合多型II/IIIの種を加えた。2日後には、XRPDにより示されるように、多型IIIへの完全な変換が起きていた。
【実施例4】
【0057】
多型IVの形態の{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
10mgの式(I)の化合物へアセトニトリル(0.12ml)を加え、この混合物を、蒸気浴上で溶けるまで温めた。この温溶液を、熱水の水冷器においてゆっくりと冷やした。生じた結晶を窒素下で乾燥させた。XRPDは、これが別個の多型であることを示した。
【実施例5】
【0058】
主にフォームαの形態の{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
式(I)の化合物(218mg)をエタノールの50%水溶液(24ml)に溶かした。この溶液へ、さらに14.5mlの水を1滴ずつ加えた。次いで、生じた飽和溶液を、以下の条件下でVirtis機器を使用して凍結乾燥させた(真空:2170mT、運転時間:20.2時間、濃縮温度:−52℃、周囲温度:20.3℃)。
【0059】
[参考実施例1]
{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
トリフルオロ酢酸(15ml)及び水(15ml)中の{3aR−[3aα,4α,6α(1R*,2S*),6aα]}−2−[6−({7−[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル}テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−イル)オキシ]エタノール(方法A,0.59g)の溶液を室温で30分撹拌した。この反応混合物を、水(150ml)中の重炭酸ナトリウム(21g)の溶液へ慎重に加え、30分撹拌した。この混合物を酢酸エチルで抽出し、これを乾燥させて蒸発させた。残渣を精製(SiO2,溶出液として酢酸エチル)して、表題化合物(0.44g)を得た。
MS(APCI)523(M+H+,100%);
【0060】
【化2】
【0061】
出発材料の製造
出発材料は、市販されているか、又は既知の材料から標準法により容易に製造される。例えば、以下の反応は、上記の反応で使用されるいくつかの出発材料の製法の例示であって、その限定ではない。
【0062】
方法A
{3aR−[3aα,4α,6α(1R*,2S*),6aα]}−2−[6−({7−[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル}テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−イル)オキシ]エタノール
THF(1ml)中の{3aR−[3aα,4α,6α(1R*,2S*),6aα]}−{[6−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル)−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−イル)オキシ}酢酸、メチルエステル(方法B,0.76g)の氷冷溶液へ、DIBAL−H(登録商標)(ヘキサン中の1.0M溶液、5.15ml)を加え、この溶液をこの温度で2時間撹拌した。この反応混合物を真空で濃縮し、残渣を酢酸エチル(75ml)に溶かした。酒石酸ナトリウムカリウムの飽和水溶液(75ml)を加え、この混合物を16時間激しく撹拌した。有機物を採取し、水相を酢酸エチル(2x50ml)で再抽出した。合わせた有機物を乾燥させ、濃縮し、残渣を精製(SiO2,溶出液としてイソヘキサン:酢酸エチル 1:1)して、表題化合物(0.63g)を得た。MS(APCI)563(M+H+,100%)。
【0063】
方法B
{3aR−[3aα,4α,6α(1R*,2S*),6aα]}−{[6−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル)−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−イル)オキシ}酢酸、メチルエステル
ジクロロメタン(25ml)中の[3aR−(3aα,4α,6α,6aα)]−({6−[7−ブロモ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール}オキシ)酢酸、メチルエステル(方法D,0.80g)及び(1R−trans)−2−(3,4−ジフルオロフェニル)シクロプロパンアミン、[R−(R*,R*)]−2,3−ジヒドロキシブタンジオエート(1:1)(方法C,0.61g)の混合物へN,N−ジイソプロピルエチルアミン(0.85ml)を加えた。生じた溶液を室温で16時間撹拌してから、真空で濃縮した。精製(SiO2,溶出液としてイソヘキサン:酢酸エチル 3:1)により、表題化合物(0.77g)を無色の泡状物として得た。MS(APCI)591(M+H+,100%)。
【0064】
方法C
(1R−trans)−2−(3,4−ジフルオロフェニル)シクロプロパンアミン、[R−(R*,R*)]−2,3−ジヒドロキシブタンジオエート(1:1)
表題化合物は、WO9905143に記載の方法に従って製造され得る。
【0065】
方法D
[3aR−(3aα,4α,6α,6aα)]−({6−[7−ブロモ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール}オキシ)酢酸、メチルエステル
ブロモホルム(30ml)中の[3aR−(3aα,4α,6α,6aα)]−({6−[7−アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール}オキシ)酢酸、メチルエステル(方法E,1.1g)及び亜硝酸イソアミル(2.4ml)を80℃で30分加熱した。冷やした反応混合物を精製(SiO2,溶出液として酢酸エチル:イソヘキサン 1:4)して、表題化合物(0.44g)を得た。MS(APCI)502/4(M+H+),504(100%)。
【0066】
方法E
[3aR−(3aα,4α,6α,6aα)]−({6−[7−アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール}オキシ)酢酸、メチルエステル
THF(25ml)中の[3aR−(3aα,4α,6α,6aα)]−6−[7−アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール(方法F,0.50g)の溶液へ0℃でブチルリチウム(2.5N/ヘキサンの0.62ml)を加えた。20分後、この懸濁液をTHF(10ml)中のトリフルオロメタンスルホニルオキシ酢酸メチルエステル(0.34g)(Biton, Tetrahedron, 1995, 51, 10513 の方法に従って製造)の溶液で処理した。生じた溶液を室温まで温めてから濃縮し、精製(SiO2,溶出液として酢酸エチル:イソヘキサン 4:6)して、表題化合物(0.25g)を得た。MS(APCI)439(M+H+,100%)。
【0067】
方法F
[3aR−(3aα,4α,6α,6aα)]−6−[7−アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール
0.88アンモニア(5ml)を含有する、THF(200ml)中の[3aR−(3aα,4α,6α,6aα)]−6−[7−クロロ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール(方法G,13.2g)を2時間撹拌してから濃縮乾固させ、水と酢酸エチルの間で残渣を分画した。有機物を乾燥させてから濃縮し、表題化合物(12.5g)を得た。MS(APCI)367(M+H+,100%)。
【0068】
方法G
[3aR−(3aα,4α,6α,6aα)]−6−[7−クロロ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール
アセトニトリル(100ml)中の[3aR−(3aα,4α,6α,6aα)]−6−{[5−アミノ−6−クロロ−2−(プロピルチオ)ピリミジン−4−イル]アミノ}−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール(方法H,2.0g)の溶液へ亜硝酸イソアミル(1.1ml)を加え、この溶液を70℃で1時間加熱した。冷やした反応混合物を濃縮して精製(SiO2,溶出液として酢酸エチル:イソヘキサン 1:3)し、表題化合物(1.9g)を得た。MS(APCI)386(M+H+,100%)。
【0069】
方法H
[3aR−(3aα,4α,6α,6aα)]−6−{[5−アミノ−6−クロロ−2−(プロピルチオ)ピリミジン−4−イル]アミノ}−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール
酢酸(100ml)中の[3aR−(3aα,4α,6α,6aα)]−6−{[6−クロロ−5−ニトロ−2−(プロピルチオ)ピリミジン−4−イル]アミノ}テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール(方法I,2.7g)の撹拌溶液へ鉄粉(3.0g)を加えた。この反応混合物を室温で2時間撹拌し、半量まで濃縮し、酢酸エチルで希釈し、水で洗浄した。有機相を乾燥させ、濃縮して表題化合物(2.0g)を得た。MS(APCI)375(M+H+,100%)。
【0070】
方法I
[3aR−(3aα,4α,6α,6aα)]−6−{[6−クロロ−5−ニトロ−2−(プロピルチオ)ピリミジン−4−イル]アミノ}テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール
THF(600ml)中の[3aR−(3aα,4α,6α,6aα)]−6−アミノテトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール塩酸塩(方法J,10.0g)及びN,N−ジイソプロピルエチルアミン(35ml)の溶液を1時間撹拌した。この混合物を濾過し、この溶液をTHF(1000ml)中の4,6−ジクロロ−5−ニトロ−2−(プロピルチオ)ピリミジン(WO9703084,25.6g)の溶液へ1時間にわたり加え、さらに2時間撹拌した。溶媒量を真空で減らし、酢酸エチル(1000ml)を加えた。この混合物を水で洗浄し、有機層を乾燥させ、蒸発させて、精製(SiO2,溶出液としてイソヘキサン−酢酸エチル)し、表題化合物(14.2g)を得た。MS(APCI)405(M+H+,100%)。
【0071】
方法J
[3aR−(3aα,4α,6α,6aα)]−6−アミノテトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール塩酸塩
6M HCl(100ml)/メタノール(500ml)中の[1R−(1α,2β,3β,4α)]−2,3,4−トリヒドロキシシクロペンテニルイミド二炭酸、ビス(1,1−ジメチルエチル)エステル(方法K,17.4g)を18時間撹拌した。この混合物を蒸発させてから、トルエン(4x200ml)とともに共沸させ、無色の粉末(8.7g)を得た。この固形物を、2,2−ジメトキシプロパン(25ml)及び濃HCl(0.2ml)を含有するアセトン(250ml)に懸濁してから、還流下で2時間加熱した。この混合物を冷やし、蒸発させ、トルエン(3x200ml)とともに共沸させた。残渣を20%水性酢酸に溶かし、2時間撹拌した。この混合物を蒸発させ、トルエン(4x200ml)とともに共沸させて、表題化合物(10.1g)を得た。MS(APCI)174(M+H+,100%)。
【0072】
方法K
[1R−(1α,2β,3β,4α)]−2,3,4−トリヒドロキシシクロペンテニルイミド二炭酸、ビス(1,1−ジメチルエチル)エステル
THF(500ml)/水(50ml)中の(1R−cis)−ビス(1,1−ジメチルエチル)−4−ヒドロキシ−2−シクロペンテニルイミド二炭酸(方法L,17.1g)の溶液へN−メチルモルホリン−N−オキシド(9.4g)に次いで、四酸化オスミウム(10ml,t−ブタノール中の2.5%溶液)を加えた。この混合物を室温で4日間撹拌してから、亜硫酸水素ナトリウム(6.0g)で処理した。この懸濁液を珪藻土に通して濾過し、生成物を精製(SiO2,溶出液として酢酸エチル:イソヘキサン 1:1)し、表題化合物(19.1g)を得た。
【0073】
【化3】
【0074】
方法L
(1R−cis)−ビス(1,1−ジメチルエチル)−4−ヒドロキシ−2−シクロペンテニルイミド二炭酸
THF(30ml)中のエーテル洗浄済み水素化ナトリウム(オイル中の60%分散液;0.31g)の懸濁液へ、イミド二炭酸ビス−(1,1−ジメチルエチル)エステル(1.84g)を加えた。この混合物を40℃で1時間撹拌した。次いで、この混合物へ、周囲温度で(1S−cis)−4−アセトキシ−2−シクロペンテン−1−オール(0.5g)とテトラキス(トリフェニルホスフィン)パラジウム(0)(0.18g)を加えた。この反応混合物を24時間撹拌してから精製(SiO2,溶出液として酢酸エチル:ヘキサン 1:9)し、表題化合物(0.90g)を無色の固形物として得た。
【0075】
【化4】
【0076】
[実施例2]
以下に、ヒトでの治療又は予防に使用される、結晶形及び/又は非晶形の式(I)の化合物(以下、化合物X)を含有する、代表的な医薬剤形を示す:
(a)錠剤I mg/錠
化合物X 100
ラクトース(Ph.Eur) 182.75
クロスカルメロースナトリウム 12.0
トウモロコシデンプンペースト(5% w/vペースト)2.25
ステアリン酸マグネシウム 3.0
(b)錠剤II mg/錠
化合物X 50
ラクトース(Ph.Eur) 223.75
クロスカルメロースナトリウム 6.0
トウモロコシデンプン 15.0
ポリビニルピロリドン(5% w/vペースト) 2.25
ステアリン酸マグネシウム 3.0
(c)錠剤III mg/錠
化合物X 1.0
ラクトース(Ph.Eur) 93.25
クロスカルメロースナトリウム 4.0
トウモロコシデンプンペースト(5%w/vペースト) 0.75
ステアリン酸マグネシウム 1.0
(d)カプセル剤 mg/カプセル
化合物X 10
ラクトース(Ph.Eur) 488.5
ステアリン酸マグネシウム 1.5
(e)注射剤I (50mg/ml)
化合物X 5.0% w/v
1N 水酸化ナトリウム溶液 15.0% w/v
0.1N 塩酸 (pH7.6へ調整)
ポリエチレングリコール 400 4.5% w/v
注射水 100%へ
(f)注射剤II (10mg/ml)
化合物X 1.0% w/v
リン酸ナトリウム BP 3.6% w/v
0.1N 水酸化ナトリウム溶液 15.0% v/v
注射水 100%へ
(g)注射剤III (1mg/ml,pH6へ緩衝化)
化合物X 0.1% w/v
リン酸ナトリウム BP 2.26% w/v
クエン酸 0.38% w/v
ポリエチレングリコール 400 3.5% w/v
注射水 100%へ
註:
上記の製剤は、製剤技術分野でよく知られている従来法により得られる。錠剤(a)〜(c)は、従来手段により、例えば酢酸セルロースフタル酸塩のコーティング剤を施して腸溶外皮化してもよい。
【0077】
NMRスペクトルは、Varian Unity Inova300若しくは400分光計で測定した;NMRデータは、主要な診断用プロトンについてデルタ値の形式で引用され、他に明記しなければ、過重水素ジメチルスルホキシド(DMSO−d6)を溶媒として使用して、内部標準としてのテトラメチルシラン(TMS)に対する百万分率(ppm)で示される;例えば、それはプロトンNMRスペクトル中でロータマーの存在を示し、主要ロータマーの化学シフトだけが引用される;カップリング定数(J)は、Hzで示される。
【0078】
質量スペクトル(MS)は、以下のように測定した:EIスペクトルは、VG70−250S若しくはFinnigan Mat Incos−XL分光計で入手し、FABスペクトルは、VG70−250SEQ分光計で入手し、ESI及びAPCIスペクトルは、Finnigan Mat SSQ7000若しくはMicromass Platform分光計で入手した。
【0079】
調製用HPLC分離は、全般に、BDSC−18逆相シリカを充填したNovapak(登録商標)、Bondapak(登録商標)、又はHypersil(登録商標)カラムを使用して実施した。
【0080】
フラッシュクロマトグラフィー(実施例では、(SiO2)として示される)は、Fisher Matrixシリカ、35〜70μmを使用して行なった。
略号
THF テトラヒドロフラン
XRPD X線粉末回折
DSC 示差走査熱量測定
【技術分野】
【0001】
本発明は、化学化合物の諸形態、特に結晶形及び非晶形、より特定すると、4つの結晶形と1つの非晶形に関する。さらに本発明は、そのような諸形態の製造法、結晶形及び/又は非晶形の化合物を含んでなる医薬組成物、及びそのような諸形態の治療使用に関する。
【背景技術】
【0002】
医薬組成物の製剤では、簡便に処理されて加工され得る形態で薬物が存在することが重要である。このことは、商業的に実行可能な製造法を得る視点からだけでなく、活性化合物を含んでなる医薬製剤の後続の製造という点からも重要である。有効成分の化学的安定性、固体状態の安定性、及び貯蔵寿命も非常に重要な要因である。医薬物質と、それを含有する組成物は、活性成分の物理化学的特性(例、その化学組成、密度、吸湿性、及び溶解性)において有意な変化を示すことなく、かなりの時間にわたり効果的に保存されることが可能であるべきである。さらに、可能な限り純粋である形態で薬物を提供することができることも重要である。非晶性の材料は、この点で重大な問題を提起する場合がある。例えば、そのような材料は、処理することや製剤化することが結晶性の材料よりも通常は困難であり、信頼し得ない溶解性を提供し、しばしば不安定で化学的に不純であることが見出される。当業者が理解されるように、薬物が安定な結晶形で容易に入手され得るならば、上記の諸問題は解決される可能性がある。このように、商業的に実行可能で製剤的に許容される医薬組成物の製造では、どこでも可能であれば、実質的に結晶性で、安定な形態で薬物を提供することが望ましい。しかしながら、注目すべきことは、この目標が必ずしも達成可能でないことである。実際、典型的には、ある化合物の結晶化の挙動がどのようであるかを分子構造だけから予測することは可能ではなく、このことは、通常、経験的にしか決定し得ないのである。
【0003】
血小板の付着及び凝集は、動脈血栓症における初発事象である。内皮下表面への血小板付着のプロセスは損傷した血管壁の修復に重要な役割を担う可能性があるが、これにより開始される血小板凝集は、生きた血管床へ急性の血栓閉塞を沈殿させる可能性があり、これが心筋梗塞や不安定狭心症のような、重い病態を伴う事象をもたらす。これら病態を予防するか又は緩和させるために使用される血栓溶解や血管形成のような介入も、血小板仲介性の閉塞若しくは再閉塞により弱められる。
【発明の概要】
【発明が解決しようとする課題】
【0004】
アデノシン5’−二リン酸(ADP)は、血栓症の重要なメディエーターとして作用することが見出されてきた。ADP誘導性の血小板凝集は、血小板の膜上に位置するP2T受容体サブタイプにより仲介される。P2T受容体(P2YADP又はP2TACとしても知られる)は、血小板の凝集/活性化を仲介することに主に関与し、まだクローン化されていないGタンパク質共役型受容体である。この受容体の薬理学的特性は、例えば、Humphries et al., Br. J. Pharmacology (1994), 113, 1057-1063 と Fagura et al., Br. J. Pharmacology (1998) 124, 157-164 による参考文献に記載されている。最近、この受容体での拮抗薬が他の抗血栓剤を越える重要な改良点を提供することが示された(J. Med. Chem. (1999) 42, 213 を参照のこと)。国際特許出願WO9905143は、P2T(P2YADP又はP2TAC)拮抗薬としての活性を有するトリアゾロ[4,5−d]ピリミジン化合物の系列を包括的に開示する。(以下に図示されるような)式(I)の化合物は、国際特許出願WO9905143の包括的な範囲に含まれるものの、そこで特別に開示されているわけではない。この化合物は、P2T(P2YADP又はP2TAC)拮抗薬として高い効力を示す。それはまた、驚くほど高い代謝安定性及びバイオアベイラビリティを有する。
【課題を解決するための手段】
【0005】
従って、本発明は、式(I):
【0006】
【化1】
【0007】
の実質的に結晶形の化合物に関する。
式(I)の化合物は、慣用的に、{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオールと命名される。
【0008】
式(I)の化合物は、以下、多型I、多型II、多型III、及び多型IVと呼ばれる、4つの異なる実質的に結晶性の形態で存在し得る。多型とは、化合物の特定の結晶形である。
【0009】
多型の諸形態が、互いに関して、そして非結晶状態に関して異なる物理特性を有することは、特に化合物が工業スケールで製造されるか又は使用されるときに、化合物の化学的及び製剤的処理に対して顕著に影響を及ぼす可能性がある。
【0010】
本発明の1つの側面では、式(I)の化合物の好ましい結晶形は、多型I、多型II、多型III、及び/又は多型IVの形態で存在する。
本発明の他の側面では、式(I)の化合物の好ましい結晶形が多型Iである。
【0011】
本発明の別の側面では、式(I)の化合物の好ましい結晶形が多型IIである。
本発明のさらなる側面では、式(I)の化合物の好ましい結晶形が多型IIIである。
【0012】
本発明の追加の側面では、式(I)の化合物の好ましい結晶形が多型IVである。
本発明のさらなる側面では、式(I)の化合物が実質的に非晶形で存在する。非晶形では、結晶形において(例えば、多型において)通常存在する三次元の長距離数(long range order)が存在せず、非晶形における分子の互いに対する位置は本質的に無秩序である(B. C. Hancock と G. Zografi, J. Pharm. Sci. (1997) 86, 1 を参照のこと)。式(I)の化合物の非晶形をフォームαと呼ぶ。
【0013】
我々は、式(I)の化合物を結晶形と非晶形で単離した。これらの形態は、実質的に、又は本質的に水分がないまま(「無水」形)で存在し得る。故に、本発明の1つの側面では、式(I)の化合物の無水形が結晶形又は非晶形で提供される。「実質的に純粋で本質的に無水形である」という用語の使用は、結晶格子構造の内側又は結晶格子構造の外側に、水を含む何らかの溶媒の存在を排除するものではない。無水形は、化合物1分子につき0.4未満の水分子を有する(40%未満水和している)。好ましくは、無水形は、化合物1分子につき0.1未満の水分子を含有する。
【0014】
多型I、II、III、及びIVは、その融解開始点(onset of melting)、粉末X線回折パターン、及び/又は単結晶X線データを参照して区別され得る。
多型Iは、実質的に純粋で、本質的に無水形である場合、146〜152℃の範囲、例えば約151℃にある融解開始点を有する。
【0015】
多型IIは、実質的に純粋で、本質的に無水形である場合、136〜139℃の範囲、例えば約137.5℃にある融解開始点を有する。
多型IIIは、実質的に純粋で、本質的に無水形である場合、127〜132℃の範囲、例えば約132℃にある融解開始点を有する。
【0016】
多型IVは、実質的に純粋で、本質的に無水形である場合、典型的には約139℃である融解開始点を有する。
フォームαは、典型的には、融解に先立ってガラス転移を受けた後で、上記多型の1つ、例えば多型IIへ結晶化する。
【0017】
融点は、パーキン・エルマーDSC7装置を使用する、示差走査熱量測定(DSC)を用いて決定した。融解開始点は、ベースラインからの有意な変化が起こる点として定義され、パーキン・エルマーのPyrisソフトウェアにより測定した。理解されるように、他のタイプの装置によるか、又は本明細書の記載とは異なる条件を使用することによって、別の融点読取り値が与えられる可能性がある。従って、引用される数値を絶対値として解釈してはならない。当業者は、正確な融点の値が化合物の純度、サンプル重量、加熱速度、及び粒子サイズにより影響され得ることを理解されよう。
【0018】
多型Iは、実質的に純粋で、本質的に無水形である場合、高強度の特定ピークを5.3°(±0.1°)、20.1°(±0.1°)、20.7°(±0.1°)、21.0°(±0.1°)、及び21.3°(±0.1°)2θに含有するX線粉末回折パターンを有する。より好ましくは、実質的に純粋で本質的に無水の多型Iは、特定ピークを5.3°(±0.1°)、8.0°(±0.1°)、9.6°(±0.1°)、13.9°(±0.1°)、15.3°(±0.1°)、20.1°(±0.1°)、20.7°(±0.1°)、21.0°(±0.1°)、21.3°(±0.1°)、26.2°(±0.1°)、及び27.5°(±0.1°)2θに含有するX線粉末回折パターンを有する。
【0019】
多型IIは、実質的に純粋で、本質的に無水形である場合、高強度の特定ピークを5.5°(±0.1°)、13.5°(±0.1°)、18.3°(±0.1°)、22.7°(±0.1°)、及び24.3°(±0.1°)2θに含有するX線粉末回折パターンを有する。より好ましくは、実質的に純粋で本質的に無水の多型IIは、特定ピークを5.5°(±0.1°)、6.8°(±0.1°)、10.6°(±0.1°)、13.5°(±0.1°)、14.9°(±0.1°)、18.3°(±0.1°)、19.2°(±0.1°)、22.7°(±0.1°)、24.3°(±0.1°)、及び27.1°(±0.1°)2θに含有するX線粉末回折パターンを有する。
【0020】
多型IIIは、実質的に純粋で、本質的に無水形である場合、高強度の特定ピークを14.0°(±0.1°)、17.4°(±0.1°)、18.4°(±0.1°)、21.4°(±0.1°)、及び24.1°(±0.1°)2θに含有するX線粉末回折パターンを有する。より好ましくは、実質的に純粋で本質的に無水の多型IIIは、特定ピークを5.6°(±0.1°)、12.5°(±0.1°)、14.0°(±0.1°)、17.4°(±0.1°)、18.4°(±0.1°)、21.4°(±0.1°)、22.2°(±0.1°)、22.9°(±0.1°)、24.1°(±0.1°)、及び24.5°(±0.1°)2θに含有するX線粉末回折パターンを有する。
【0021】
多型IVは、実質的に純粋で、本質的に無水形である場合、高強度の特定ピークを4.9°(±0.1°)、9.2°(±0.1°)、11.6°(±0.1°)、15.6°(±0.1°)、及び16.4°(±0.1°)2θに含有するX線粉末回折パターンを有する。より好ましくは、実質的に純粋で本質的に無水の多型IVは、特定ピークを4.9°(±0.1°)、6.0°(±0.1°)、9.2°(±0.1°)、11.6°(±0.1°)、12.8°(±0.1°)、15.6°(±0.1°)、16.4°(±0.1°)、17.2°(±0.1°)、及び18.1°(±0.1°)2θに含有するX線粉末回折パターンを有する。
【0022】
フォームαは、実質的に純粋で、本質的に無水形である場合、明瞭なピークを含有しないX線粉末回折パターンを有する。
多型II、多型III、多型IV、及びフォームαのX線回折データは、ジーメンスD5000装置を使用して入手した。多型IのX線回折データは、フィリップスのX’Pert MPD機器を使用して入手した。装置及び/又は条件が異なれば、やや異なるデータが作成され得ることが理解されよう。従って、引用される数値を絶対値として解釈してはならない。
【0023】
本発明の別の側面では、溶媒和形、例えば水和形(「水和物」)が形成され得る。故に、本発明のこの側面では、結晶形の式(I)の化合物の水和物が提供される。水和物は、化合物1分子につき0.8以上の水分子を有する(80%以上水和している)。半水和物は、化合物1分子につき0.4〜0.8の水分子を有する(40〜80%水和している)。
【0024】
本発明のさらなる特徴では、式(I)の化合物の結晶形及び/又は非晶形の混合物が提供される。好ましくは、混合物は、多型I、多型II、多型III、多型IV、及び/又はフォームαの混合物である。より好ましくは、本発明は、多型IIと多型IIIの任意の混合物を提供する。
【0025】
本発明のさらなる特徴では、式(I)の化合物の好適な溶媒からの結晶化による、式(I)の化合物の結晶形の製造法が提供される。好ましくは、溶媒は、エタノール、酢酸エチル、イソプロパノール、イソオクタン、アセトニトリル、水、又はこれらの混合物の群から選択される。より好ましくは、溶媒は、エタノール、酢酸エチル、イソプロパノール、イソオクタン、水、又はこれらの混合物の群から選択される。好適には、溶媒は、メタノール及び水の混合物、エタノール、酢酸エチル、エタノール及び水の混合物、イソプロパノール及び水の混合物、酢酸エチル及びイソオクタンの混合物、及びアセトニトリルの群から選択される。
【0026】
式(I)の化合物は、WO9905143の記載に類似した方法により製造され得る。
結晶化を始めるには、式(I)の化合物の結晶を用いた種入れ(seeding)が必要とされ得る。選択される多型を得るには、求められる多型を用いた種入れが必要となり得る。式(I)の化合物の適正な溶媒系からの結晶化は、例えば、冷却、溶媒蒸発、及び/又は抗溶媒(anti−solvent:式(I)の化合物がほとんど溶けない溶媒;好適な抗溶媒の例には、ヘプタン又はイソオクタンが含まれる)の追加により過飽和にさせることによって達成され得る。結晶化の温度及び時間は、溶液中の化合物の濃度、使用する溶媒系、及び、採用する結晶化の方法に依存して変化する。
【0027】
結晶形の式(I)の化合物は、当業者によく知られた技術、例えば、デカント(decanting)、濾過、又は遠心分離の技術を使用して、上記の反応混合物から単離され得る。同様に、結晶形の式(I)の化合物は、周知の方法に従って、乾燥され得る。
【0028】
非結晶性の材料、化学不純物のような不純物をさらに減らすためか、又はある多型を別の多型へ、又は水和物若しくは無水形へと結晶形を変換するために、同じか又は異なる溶媒系を使用して、場合によっては再結晶の工程が実施され得る。さらに、非結晶性の材料を減らすには、この固形物を高湿度に曝露する工程といった、条件付けの工程を実施してよい。
【0029】
好ましくは、結晶化は、反応溶液から直接行なわれる。他のやり方では、結晶化は後続の溶液から実施される。
本発明のさらなる特徴では、多型Iを製造する方法が提供され、前記方法は、ゆっくりと結晶成長する多型I由来の多型Iのわずかな種結晶(seed crystals)を多型IIの融成物から入手する工程、及び、これを使用して式(I)の化合物と、メタノール/水のような好適な混合溶媒系を含んでなる反応混合物に種入れする工程を含む。
【0030】
本発明のさらなる特徴では、多型IIを製造する方法が提供され、前記方法は、酢酸エチルのような好適な溶媒中での結晶化を含む。
本発明のさらなる特徴では、多型IIIを製造する方法が提供され、前記方法は、アルコール、例えばエタノール若しくはイソプロピルアルコール(IPA)のような好適な溶媒中での結晶化、特に、多型IIIの結晶を用いた種入れ、又は、IPAのような好適な溶媒中での式(I)の化合物のスラリー化を含む。
【0031】
本発明のさらなる特徴では、多型IVを製造する方法が提供され、前記方法は、アセトニトリルのような好適な溶媒からの結晶化、特に、多型IVの結晶を用いた種入れ、又は、アセトニトリルのような好適な溶媒中での式(I)の化合物をスラリー化させる時間を含む。
【0032】
本発明のさらなる特徴は、実質的に多型IIがない多型IIIを製造する方法を提供し、前記方法は、例えば、C1-6脂肪族アルコール/水溶媒系(好ましくは、IPA/水)において、5〜65℃の温度で1〜10日間、式(I)の化合物をスラリー化させる工程を含む。
【0033】
本発明のさらなる特徴では、実質的に非晶形である式(I)の化合物の製造法が提供され、前記方法は、好適な溶媒系、例えばエタノール/水を使用して、式(I)の化合物の溶液を凍結乾燥させるか、又はスプレー乾燥させる工程を含む。
【0034】
「実質的にない(substantially free)」という用語は、他の多型が10%未満であること、好ましくは5%未満であることを意味する。
本発明のさらなる側面では、上記方法のいずれでも入手可能な化合物が提供される。
【0035】
結晶形及び/又は非晶形の式(I)の化合物は、P2T(P2YADP又はP2TAC)受容体拮抗薬として作用する。従って、結晶形及び/又は非晶形の式(I)の化合物は、組合せ療法を含む療法に有用である。特に、結晶形の式(I)の化合物は、冠状動脈、脳血管、又は末梢血管の疾患を有する患者の動脈血栓合併症の治療若しくは予防における使用に適用される。動脈血栓合併症には、不安定狭心症、血栓若しくは塞栓性の卒中のようなアテローム性動脈硬化症の一次動脈血栓合併症、一過性虚血発作、末梢血管障害、血栓溶解を伴うか又は伴わない心筋梗塞、冠血管形成術(PTCA)を含む血管形成術、動脈内膜切除術、ステント置換、冠血管と他の血管の移植手術といった、アテローム動脈硬化症における介入による動脈合併症、偶発的若しくは外科的外傷や皮膚及び筋肉のフラップを含む再建手術後の組織サルベージ(tissue salvage)のような外科的若しくは機械的な損傷の血栓合併症、播種性血管内凝固のような拡散性の血栓/血小板消費因子を伴う病態、血栓性血小板減少性紫斑病、溶血性尿毒症候群、敗血症の血栓合併症、成人呼吸窮迫症候群、抗リン脂質症候群、ヘパリン誘導性の血小板減少症及び子癇前症/子癇症、又は深在性静脈血栓症のような静脈血栓症、静脈閉塞症、血小板血症を含む骨髄増殖疾患のような血液学的病態、鎌状赤血球病;[又は、心肺バイパス及び体外での膜式酸素加(微小血栓塞栓症の予防)のような、機械的に誘導される in vivo での血小板活性化、血液製品(例、血小板濃縮物)の保存中の使用のような、機械的に誘導されるin vitroでの血小板活性化、又は腎透析及びプラズマフェレーシス中のシャント閉塞の予防では]血管炎、動脈炎、糸球体腎炎、炎症性腸疾患、及び臓器移植拒絶のような血管損傷/炎症に続発する血栓症、偏頭痛のような病態、レイノー現象、アテローム性プラーク形成/進行、狭窄/再狭窄のような血管壁の根底にある炎症性疾患プロセスに血小板が貢献し得る病態、及び、血小板と血小板由来因子が免疫学的疾患プロセスに関連していると思われる喘息のような他の炎症病態が含まれる。さらなる適応症には、CNS障害の治療と腫瘍の増殖及び拡散の予防が含まれる。
【0036】
本発明のさらなる側面によれば、ヒト若しくは動物の身体の治療による処置の方法に使用される、結晶形及び/又は非晶形の式(I)の化合物が提供される。
本発明の追加の特徴によれば、医薬品としての使用のための、結晶形及び/又は非晶形の式(I)の化合物が提供される。好ましくは、結晶形及び/又は非晶形の式(I)の化合物は、ヒトのような温血動物においてP2T(P2YADP又はP2TAC)受容体に拮抗する医薬品として使用される。より好ましくは、結晶形及び/又は非晶形の式(I)の化合物は、冠動脈、脳血管、又は末梢血管の疾患を有する、ヒトのような温血動物の患者の動脈血栓合併症を治療するか又は予防するための医薬品として使用される。
【0037】
本発明によれば、結晶形及び/又は非晶形である式(I)の化合物の、P2T(P2YADP又はP2TAC)受容体の拮抗薬として使用される医薬品の製造における使用がさらに提供される。特に、結晶形及び/又は非晶形である式(I)の化合物の、冠動脈、脳血管、又は末梢血管の疾患を有する患者の動脈血栓合併症の治療若しくは予防に使用される医薬品の製造における使用がさらに提供される。
【0038】
本発明はまた、冠動脈、脳血管、又は末梢血管の疾患を有する患者における動脈血栓合併症の治療若しくは予防の方法を提供し、前記方法は、そのような障害に罹患しているか又はそれに罹りやすいヒトへ、結晶形及び/又は非晶形である式(I)の化合物の治療有効量を投与することを含む。
【0039】
結晶形及び/又は非晶形の式(I)の化合物は、溶液剤、懸濁液剤、HFAエアゾール剤、及び乾燥粉末製剤の形態で局所的に、例えば肺及び/又は気道へ;又は、錠剤、丸剤、カプセル剤、シロップ剤、散剤、又は顆粒剤の形態で全身的に、例えば経口投与により;又は、無菌の非経口溶液剤若しくは懸濁液剤の形態での非経口投与により、皮下投与により、又は坐剤の形態での直腸投与により、又は経皮的に、投与され得る。
【0040】
結晶形及び/又は非晶形の式(I)の化合物は、それ自身でか、又は、結晶形及び/又は非晶形の式(I)の化合物を製剤的に許容される希釈剤、アジュバント、及び/又は担体と組合せて含んでなる医薬組成物として投与され得る。故に、本発明のさらなる特徴として、結晶形及び/又は非晶形の式(I)の化合物を製剤的に許容される希釈剤、アジュバント、及び/又は担体と一緒に含んでなる医薬組成物が提供される。特に好ましいのは、有害なアレルギー反応のような有害反応を引き起こし得る材料を含有しない組成物である。
【0041】
結晶形及び/又は非晶形である式(I)の化合物の乾燥粉末製剤及び加圧HFAエアゾール剤は、経口若しくは経鼻吸入により投与され得る。吸入のために、結晶形及び/又は非晶形の式(I)の化合物は、望ましくは、粉砕される。結晶形及び/又は非晶形の式(I)の化合物はまた、乾燥粉末吸入器の手段により投与され得る。吸入器は単一若しくは複数用量の吸入器であり得て、呼気作動性の乾燥粉末吸入器であり得る。
【0042】
1つの可能性は、結晶形及び/又は非晶形の式(I)の粉砕化合物を、担体物質、例えば、単糖、二糖、若しくは多糖、糖アルコール、又は別のポリオールと混合することである。好適な担体には、糖類とデンプンが含まれる。他のやり方では、結晶形及び/又は非晶形の式(I)の粉砕化合物が別の物質により被覆され得る。粉末混合物はまた、結晶形及び/又は非晶形である式(I)の活性化合物の所望量をそれぞれ含有する、硬ゼラチンカプセル剤へ調剤され得る。
【0043】
もう1つの可能性は、粉砕された粉末を、吸入法の間に壊れる球体へ加工することである。この球状化した粉末が、多用量吸入器、例えば、Turbuhaler(登録商標)として知られるもの(患者により吸入される所望量の投薬単位が目盛られている)の薬物レザバーへ充填され得る。この系を用いて、式(I)の活性化合物が、担体物質を伴うか又は伴わずに患者へ投与される。結晶形及び/又は非晶形の式(I)の化合物を含んでなる医薬組成物は、好便にも、経口投与用の錠剤、丸剤、カプセル剤、シロップ剤、散剤、又は顆粒剤;非経口投与用の無菌の非経口若しくは皮下溶液剤、懸濁液剤、又は直腸投与用の坐剤であり得る。
【0044】
経口投与では、結晶形及び/又は非晶形の式(I)の化合物が、アジュバント若しくは担体(例えば、乳糖、サッカロース、ソルビトール、マンニトール、ジャガイモデンプン、トウモロコシデンプン、又はアミロペクチンのようなデンプン、セルロース誘導体)、ゼラチン若しくはポリビニルピロリドンのような結合剤、及び、ステアリン酸マグネシウム、ステアリン酸カルシウム、ポリエチレングリコール、ワックス、パラフィン、等のような潤滑剤とともに混合されてから、錠剤へ圧縮され得る。被覆錠剤が必要とされる場合、上記のように製造された芯が、例えば、アラビアゴム、ゼラチン、タルク、二酸化チタン、等を含有し得る濃縮糖溶液で被覆され得る。他のやり方では、錠剤を、易揮発性の有機溶媒若しくは水性溶媒のいずれかに溶けた好適なポリマーで被覆してもよい。
【0045】
軟ゼラチンカプセル剤の製造では、結晶形及び/又は非晶形の式(I)の化合物が、例えば、植物油又はポリエチレングリコールと混合され得る。硬ゼラチンカプセル剤は、上記の錠剤用の賦形剤、例えば、乳糖、サッカロース、ソルビトール、マンニトール、デンプン、セルロース誘導体、又はゼラチンのいずれか一方を使用して、本化合物の顆粒を含有し得る。また、薬物の液体若しくは半固体製剤を硬ゼラチンカプセル剤へ充填してもよい。
【0046】
経口適用の液体調製物は、シロップ剤若しくは懸濁液剤、例えば、結晶形及び/又は非晶形の式(I)の化合物を含有する溶液剤の形態であり得て、残りは、糖とエタノール、水、グリセロール、及びプロピレングリコールの混合物である。場合により、そのような液体調製物は、着色剤、芳香剤、サッカリン、及び、増粘剤としてのカルボキシメチルセルロース、又は当業者に知られた他の賦形剤を含有し得る。
【0047】
図1.1は、フィリップスのX’Pert MPD機器を、θ−θ配置で、1°〜40°2θの走査範囲にわたり、0.02°2θ増分につき2〜5秒の露出で使用して得られた、多型IのX線回折パターンである。X線は、40kVと50mAで作動する銅ロングファイン(long-fine)集束管により発生させた。X線の波長は1.5406A(オングストローム)であった。
【0048】
図1.2は、ジーメンスのD5000機器を、θ−θ配置で、2°〜30°2θの走査範囲にわたり、0.02°2θ増分につき4秒の露出で使用して得られた、多型IIのX線回折パターンである。X線は、45kVと40mAで作動する銅ロングファイン(long-fine)集束管により発生させた。X線の波長は1.5406A(オングストローム)であった。約10mgの化合物を置いたゼロ・バックグラウンドを使用してデータを収集した。このホルダーは、非回折面に沿って切削されてから、光学的に平面の仕上げにまで研磨された、単結晶のシリコンから製造された。この表面へのX線の入射は、ブラッグ消光により無効とされた。
【0049】
図1.3は、ジーメンスのD5000機器を上記のように使用して得られた、多型IIIのX線回折パターンである。
図1.4は、ジーメンスのD5000機器を上記のように使用して得られた、多型IVのX線回折パターンである。
【0050】
図1.5は、ジーメンスのD5000機器を上記のように使用して得られた、フォームαのX線回折パターンである。
図2は、パーキン・エルマーDSC7装置を使用して得られた、多型I、II、III及びIVとフォームαのDSCグラフを示す。パンのタイプは、穿孔した蓋の付いたアルミニウムであった。サンプル重量は1〜3mgであった。この方法は、窒素ガス流(30ml/分)の下で実施し、試験した温度範囲は30℃〜325℃であり、10℃/分の一定速度で温度を増加させた。
【0051】
30ミクロンを超えるサイズの粒子と非単一性の縦横比を有するサンプルの分析は、ピークの相対強度に影響を及ぼす可能性があることを理解しておくべきである。当業者はまた、サンプルが回折計中にある正確な高さと回折計のゼロ較正により反射の位置が影響を受けることも理解されよう。サンプル表面の平面性もわずかに効果を及ぼす可能性がある。従って、提示される回折パターンデータを絶対値として解釈してはならない。
【図面の簡単な説明】
【0052】
【図1.1】図1.1は、フィリップスのX’Pert MPD機器を、θ−θ配置で、1°〜40°2θの走査範囲にわたり、0.02°2θ増分につき2〜5秒の露出で使用して得られた、多型IのX線回折パターンである。X線は、40kVと50mAで作動する銅ロングファイン(long-fine)集束管により発生させた。X線の波長は1.5406A(オングストローム)であった。
【図1.2】図1.2は、ジーメンスのD5000機器を、θ−θ配置で、2°〜30°2θの走査範囲にわたり、0.02°2θ増分につき4秒の露出で使用して得られた、多型IIのX線回折パターンである。X線は、45kVと40mAで作動する銅ロングファイン(long-fine)集束管により発生させた。X線の波長は1.5406A(オングストローム)であった。約10mgの化合物を置いたゼロ・バックグラウンドを使用してデータを収集した。このホルダーは、非回折面に沿って切削されてから、光学的に平面の仕上げにまで研磨された、単結晶のシリコンから製造された。この表面へのX線の入射は、ブラッグ消光により無効とされた。
【図1.3】図1.3は、ジーメンスのD5000機器を上記のように使用して得られた、多型IIIのX線回折パターンである。
【図1.4】図1.4は、ジーメンスのD5000機器を上記のように使用して得られた、多型IVのX線回折パターンである。
【図1.5】図1.5は、ジーメンスのD5000機器を上記のように使用して得られた、フォームαのX線回折パターンである。
【図2】図2は、パーキン・エルマーDSC7装置を使用して得られた、多型I、II、III及びIVとフォームαのDSCグラフを示す。パンのタイプは、穿孔した蓋の付いたアルミニウムであった。サンプル重量は1〜3mgであった。この方法は、窒素ガス流(30ml/分)の下で実施し、試験した温度範囲は30℃〜325℃であり、10℃/分の一定速度で温度を増加させた。
【実施例】
【0053】
本発明は、以下の非限定的な実施例により説明され得る。
【実施例1】
【0054】
多型Iの形態の{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
第1部
多型IIの形態である式(I)の化合物(2mg)を、以下のように、DSC中で加熱し、冷却した:35〜143〜35〜148〜35〜148〜35℃。このアニーリング法は、DSCにより示されるような純粋な多型Iの結晶化をもたらした。
第2部
式(I)の化合物、メタノール5ml/g、及び、水7.3ml/gと少量の多型Iの種を含んでなる溶液を、30℃で結晶化させた。XRPDとDSCにより、実質的に純粋な多型Iが形成されたことを確認した。
【実施例2】
【0055】
多型IIの形態の{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
45mgの式(I)の化合物へクロロホルム(150μl)を加え、この混合物を、蒸気浴上で溶けるまで温めた。生じた溶液を一晩放置して結晶化させ、窒素流の下で乾燥させた。XRPDとDSCにより、実質的に純粋な多型IIが形成されたことを確認した。
【実施例3】
【0056】
多型IIIの形態の{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
10mgの式(I)の化合物へエタノール(200μl)を加え、この混合物を、蒸気浴上で溶けるまで温めた。生じた溶液を一晩放置して結晶化させた。XRPDとDSCにより、多型II及びIIIの混合物が形成されたことを確認した。この材料を使用して、より大きいスケールの調製物へ種入れした:191mgの多型IIを1mlの50%イソプロパノール水溶液中でスラリー化させた。このスラリーへ、15mgの混合多型II/IIIの種を加えた。2日後には、XRPDにより示されるように、多型IIIへの完全な変換が起きていた。
【実施例4】
【0057】
多型IVの形態の{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
10mgの式(I)の化合物へアセトニトリル(0.12ml)を加え、この混合物を、蒸気浴上で溶けるまで温めた。この温溶液を、熱水の水冷器においてゆっくりと冷やした。生じた結晶を窒素下で乾燥させた。XRPDは、これが別個の多型であることを示した。
【実施例5】
【0058】
主にフォームαの形態の{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
式(I)の化合物(218mg)をエタノールの50%水溶液(24ml)に溶かした。この溶液へ、さらに14.5mlの水を1滴ずつ加えた。次いで、生じた飽和溶液を、以下の条件下でVirtis機器を使用して凍結乾燥させた(真空:2170mT、運転時間:20.2時間、濃縮温度:−52℃、周囲温度:20.3℃)。
【0059】
[参考実施例1]
{1S−[1α,2α,3β(1S*,2R*),5β]}−3−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ}−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル)−5−(2−ヒドロキシエトキシ)シクロペンタン−1,2−ジオール
トリフルオロ酢酸(15ml)及び水(15ml)中の{3aR−[3aα,4α,6α(1R*,2S*),6aα]}−2−[6−({7−[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル}テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−イル)オキシ]エタノール(方法A,0.59g)の溶液を室温で30分撹拌した。この反応混合物を、水(150ml)中の重炭酸ナトリウム(21g)の溶液へ慎重に加え、30分撹拌した。この混合物を酢酸エチルで抽出し、これを乾燥させて蒸発させた。残渣を精製(SiO2,溶出液として酢酸エチル)して、表題化合物(0.44g)を得た。
MS(APCI)523(M+H+,100%);
【0060】
【化2】
【0061】
出発材料の製造
出発材料は、市販されているか、又は既知の材料から標準法により容易に製造される。例えば、以下の反応は、上記の反応で使用されるいくつかの出発材料の製法の例示であって、その限定ではない。
【0062】
方法A
{3aR−[3aα,4α,6α(1R*,2S*),6aα]}−2−[6−({7−[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]ピリミジン−3−イル}テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−イル)オキシ]エタノール
THF(1ml)中の{3aR−[3aα,4α,6α(1R*,2S*),6aα]}−{[6−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル)−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−イル)オキシ}酢酸、メチルエステル(方法B,0.76g)の氷冷溶液へ、DIBAL−H(登録商標)(ヘキサン中の1.0M溶液、5.15ml)を加え、この溶液をこの温度で2時間撹拌した。この反応混合物を真空で濃縮し、残渣を酢酸エチル(75ml)に溶かした。酒石酸ナトリウムカリウムの飽和水溶液(75ml)を加え、この混合物を16時間激しく撹拌した。有機物を採取し、水相を酢酸エチル(2x50ml)で再抽出した。合わせた有機物を乾燥させ、濃縮し、残渣を精製(SiO2,溶出液としてイソヘキサン:酢酸エチル 1:1)して、表題化合物(0.63g)を得た。MS(APCI)563(M+H+,100%)。
【0063】
方法B
{3aR−[3aα,4α,6α(1R*,2S*),6aα]}−{[6−(7−{[2−(3,4−ジフルオロフェニル)シクロプロピル]アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル)−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−イル)オキシ}酢酸、メチルエステル
ジクロロメタン(25ml)中の[3aR−(3aα,4α,6α,6aα)]−({6−[7−ブロモ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール}オキシ)酢酸、メチルエステル(方法D,0.80g)及び(1R−trans)−2−(3,4−ジフルオロフェニル)シクロプロパンアミン、[R−(R*,R*)]−2,3−ジヒドロキシブタンジオエート(1:1)(方法C,0.61g)の混合物へN,N−ジイソプロピルエチルアミン(0.85ml)を加えた。生じた溶液を室温で16時間撹拌してから、真空で濃縮した。精製(SiO2,溶出液としてイソヘキサン:酢酸エチル 3:1)により、表題化合物(0.77g)を無色の泡状物として得た。MS(APCI)591(M+H+,100%)。
【0064】
方法C
(1R−trans)−2−(3,4−ジフルオロフェニル)シクロプロパンアミン、[R−(R*,R*)]−2,3−ジヒドロキシブタンジオエート(1:1)
表題化合物は、WO9905143に記載の方法に従って製造され得る。
【0065】
方法D
[3aR−(3aα,4α,6α,6aα)]−({6−[7−ブロモ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール}オキシ)酢酸、メチルエステル
ブロモホルム(30ml)中の[3aR−(3aα,4α,6α,6aα)]−({6−[7−アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール}オキシ)酢酸、メチルエステル(方法E,1.1g)及び亜硝酸イソアミル(2.4ml)を80℃で30分加熱した。冷やした反応混合物を精製(SiO2,溶出液として酢酸エチル:イソヘキサン 1:4)して、表題化合物(0.44g)を得た。MS(APCI)502/4(M+H+),504(100%)。
【0066】
方法E
[3aR−(3aα,4α,6α,6aα)]−({6−[7−アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール}オキシ)酢酸、メチルエステル
THF(25ml)中の[3aR−(3aα,4α,6α,6aα)]−6−[7−アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール(方法F,0.50g)の溶液へ0℃でブチルリチウム(2.5N/ヘキサンの0.62ml)を加えた。20分後、この懸濁液をTHF(10ml)中のトリフルオロメタンスルホニルオキシ酢酸メチルエステル(0.34g)(Biton, Tetrahedron, 1995, 51, 10513 の方法に従って製造)の溶液で処理した。生じた溶液を室温まで温めてから濃縮し、精製(SiO2,溶出液として酢酸エチル:イソヘキサン 4:6)して、表題化合物(0.25g)を得た。MS(APCI)439(M+H+,100%)。
【0067】
方法F
[3aR−(3aα,4α,6α,6aα)]−6−[7−アミノ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール
0.88アンモニア(5ml)を含有する、THF(200ml)中の[3aR−(3aα,4α,6α,6aα)]−6−[7−クロロ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール(方法G,13.2g)を2時間撹拌してから濃縮乾固させ、水と酢酸エチルの間で残渣を分画した。有機物を乾燥させてから濃縮し、表題化合物(12.5g)を得た。MS(APCI)367(M+H+,100%)。
【0068】
方法G
[3aR−(3aα,4α,6α,6aα)]−6−[7−クロロ−5−(プロピルチオ)−3H−1,2,3−トリアゾロ[4,5−d]−ピリミジン−3−イル]−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール
アセトニトリル(100ml)中の[3aR−(3aα,4α,6α,6aα)]−6−{[5−アミノ−6−クロロ−2−(プロピルチオ)ピリミジン−4−イル]アミノ}−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール(方法H,2.0g)の溶液へ亜硝酸イソアミル(1.1ml)を加え、この溶液を70℃で1時間加熱した。冷やした反応混合物を濃縮して精製(SiO2,溶出液として酢酸エチル:イソヘキサン 1:3)し、表題化合物(1.9g)を得た。MS(APCI)386(M+H+,100%)。
【0069】
方法H
[3aR−(3aα,4α,6α,6aα)]−6−{[5−アミノ−6−クロロ−2−(プロピルチオ)ピリミジン−4−イル]アミノ}−テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール
酢酸(100ml)中の[3aR−(3aα,4α,6α,6aα)]−6−{[6−クロロ−5−ニトロ−2−(プロピルチオ)ピリミジン−4−イル]アミノ}テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール(方法I,2.7g)の撹拌溶液へ鉄粉(3.0g)を加えた。この反応混合物を室温で2時間撹拌し、半量まで濃縮し、酢酸エチルで希釈し、水で洗浄した。有機相を乾燥させ、濃縮して表題化合物(2.0g)を得た。MS(APCI)375(M+H+,100%)。
【0070】
方法I
[3aR−(3aα,4α,6α,6aα)]−6−{[6−クロロ−5−ニトロ−2−(プロピルチオ)ピリミジン−4−イル]アミノ}テトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール
THF(600ml)中の[3aR−(3aα,4α,6α,6aα)]−6−アミノテトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール塩酸塩(方法J,10.0g)及びN,N−ジイソプロピルエチルアミン(35ml)の溶液を1時間撹拌した。この混合物を濾過し、この溶液をTHF(1000ml)中の4,6−ジクロロ−5−ニトロ−2−(プロピルチオ)ピリミジン(WO9703084,25.6g)の溶液へ1時間にわたり加え、さらに2時間撹拌した。溶媒量を真空で減らし、酢酸エチル(1000ml)を加えた。この混合物を水で洗浄し、有機層を乾燥させ、蒸発させて、精製(SiO2,溶出液としてイソヘキサン−酢酸エチル)し、表題化合物(14.2g)を得た。MS(APCI)405(M+H+,100%)。
【0071】
方法J
[3aR−(3aα,4α,6α,6aα)]−6−アミノテトラヒドロ−2,2−ジメチル−4H−シクロペンタ−1,3−ジオキソル−4−オール塩酸塩
6M HCl(100ml)/メタノール(500ml)中の[1R−(1α,2β,3β,4α)]−2,3,4−トリヒドロキシシクロペンテニルイミド二炭酸、ビス(1,1−ジメチルエチル)エステル(方法K,17.4g)を18時間撹拌した。この混合物を蒸発させてから、トルエン(4x200ml)とともに共沸させ、無色の粉末(8.7g)を得た。この固形物を、2,2−ジメトキシプロパン(25ml)及び濃HCl(0.2ml)を含有するアセトン(250ml)に懸濁してから、還流下で2時間加熱した。この混合物を冷やし、蒸発させ、トルエン(3x200ml)とともに共沸させた。残渣を20%水性酢酸に溶かし、2時間撹拌した。この混合物を蒸発させ、トルエン(4x200ml)とともに共沸させて、表題化合物(10.1g)を得た。MS(APCI)174(M+H+,100%)。
【0072】
方法K
[1R−(1α,2β,3β,4α)]−2,3,4−トリヒドロキシシクロペンテニルイミド二炭酸、ビス(1,1−ジメチルエチル)エステル
THF(500ml)/水(50ml)中の(1R−cis)−ビス(1,1−ジメチルエチル)−4−ヒドロキシ−2−シクロペンテニルイミド二炭酸(方法L,17.1g)の溶液へN−メチルモルホリン−N−オキシド(9.4g)に次いで、四酸化オスミウム(10ml,t−ブタノール中の2.5%溶液)を加えた。この混合物を室温で4日間撹拌してから、亜硫酸水素ナトリウム(6.0g)で処理した。この懸濁液を珪藻土に通して濾過し、生成物を精製(SiO2,溶出液として酢酸エチル:イソヘキサン 1:1)し、表題化合物(19.1g)を得た。
【0073】
【化3】
【0074】
方法L
(1R−cis)−ビス(1,1−ジメチルエチル)−4−ヒドロキシ−2−シクロペンテニルイミド二炭酸
THF(30ml)中のエーテル洗浄済み水素化ナトリウム(オイル中の60%分散液;0.31g)の懸濁液へ、イミド二炭酸ビス−(1,1−ジメチルエチル)エステル(1.84g)を加えた。この混合物を40℃で1時間撹拌した。次いで、この混合物へ、周囲温度で(1S−cis)−4−アセトキシ−2−シクロペンテン−1−オール(0.5g)とテトラキス(トリフェニルホスフィン)パラジウム(0)(0.18g)を加えた。この反応混合物を24時間撹拌してから精製(SiO2,溶出液として酢酸エチル:ヘキサン 1:9)し、表題化合物(0.90g)を無色の固形物として得た。
【0075】
【化4】
【0076】
[実施例2]
以下に、ヒトでの治療又は予防に使用される、結晶形及び/又は非晶形の式(I)の化合物(以下、化合物X)を含有する、代表的な医薬剤形を示す:
(a)錠剤I mg/錠
化合物X 100
ラクトース(Ph.Eur) 182.75
クロスカルメロースナトリウム 12.0
トウモロコシデンプンペースト(5% w/vペースト)2.25
ステアリン酸マグネシウム 3.0
(b)錠剤II mg/錠
化合物X 50
ラクトース(Ph.Eur) 223.75
クロスカルメロースナトリウム 6.0
トウモロコシデンプン 15.0
ポリビニルピロリドン(5% w/vペースト) 2.25
ステアリン酸マグネシウム 3.0
(c)錠剤III mg/錠
化合物X 1.0
ラクトース(Ph.Eur) 93.25
クロスカルメロースナトリウム 4.0
トウモロコシデンプンペースト(5%w/vペースト) 0.75
ステアリン酸マグネシウム 1.0
(d)カプセル剤 mg/カプセル
化合物X 10
ラクトース(Ph.Eur) 488.5
ステアリン酸マグネシウム 1.5
(e)注射剤I (50mg/ml)
化合物X 5.0% w/v
1N 水酸化ナトリウム溶液 15.0% w/v
0.1N 塩酸 (pH7.6へ調整)
ポリエチレングリコール 400 4.5% w/v
注射水 100%へ
(f)注射剤II (10mg/ml)
化合物X 1.0% w/v
リン酸ナトリウム BP 3.6% w/v
0.1N 水酸化ナトリウム溶液 15.0% v/v
注射水 100%へ
(g)注射剤III (1mg/ml,pH6へ緩衝化)
化合物X 0.1% w/v
リン酸ナトリウム BP 2.26% w/v
クエン酸 0.38% w/v
ポリエチレングリコール 400 3.5% w/v
注射水 100%へ
註:
上記の製剤は、製剤技術分野でよく知られている従来法により得られる。錠剤(a)〜(c)は、従来手段により、例えば酢酸セルロースフタル酸塩のコーティング剤を施して腸溶外皮化してもよい。
【0077】
NMRスペクトルは、Varian Unity Inova300若しくは400分光計で測定した;NMRデータは、主要な診断用プロトンについてデルタ値の形式で引用され、他に明記しなければ、過重水素ジメチルスルホキシド(DMSO−d6)を溶媒として使用して、内部標準としてのテトラメチルシラン(TMS)に対する百万分率(ppm)で示される;例えば、それはプロトンNMRスペクトル中でロータマーの存在を示し、主要ロータマーの化学シフトだけが引用される;カップリング定数(J)は、Hzで示される。
【0078】
質量スペクトル(MS)は、以下のように測定した:EIスペクトルは、VG70−250S若しくはFinnigan Mat Incos−XL分光計で入手し、FABスペクトルは、VG70−250SEQ分光計で入手し、ESI及びAPCIスペクトルは、Finnigan Mat SSQ7000若しくはMicromass Platform分光計で入手した。
【0079】
調製用HPLC分離は、全般に、BDSC−18逆相シリカを充填したNovapak(登録商標)、Bondapak(登録商標)、又はHypersil(登録商標)カラムを使用して実施した。
【0080】
フラッシュクロマトグラフィー(実施例では、(SiO2)として示される)は、Fisher Matrixシリカ、35〜70μmを使用して行なった。
略号
THF テトラヒドロフラン
XRPD X線粉末回折
DSC 示差走査熱量測定
【特許請求の範囲】
【請求項1】
式(I):
【化1】
の実質的に結晶形の化合物。
【請求項2】
実質的に無水形で存在する、請求項1に記載の式(I)の化合物。
【請求項3】
高強度の特定ピークを5.3°(±0.1°)、20.1°(±0.1°)、20.7°(±0.1°)、21.0°(±0.1°)、及び21.3°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1に記載の式(I)の化合物。
【請求項4】
特定ピークを5.3°(±0.1°)、8.0°(±0.1°)、9.6°(±0.1°)、13.9°(±0.1°)、15.3°(±0.1°)、20.1°(±0.1°)、20.7°(±0.1°)、21.0°(±0.1°)、21.3°(±0.1°)、26.2°(±0.1°)、及び27.5°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1〜3に記載の式(I)の化合物。
【請求項5】
146〜152℃の範囲にある融解開始点を有する示差走査熱量測定曲線により特性決定される、請求項1、3、又は4のいずれか1項に記載の式(I)の化合物。
【請求項6】
高強度の特定ピークを5.5°(±0.1°)、13.5°(±0.1°)、18.3°(±0.1°)、22.7°(±0.1°)、及び24.3°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1に記載の式(I)の化合物。
【請求項7】
特定ピークを5.5°(±0.1°)、6.8°(±0.1°)、10.6°(±0.1°)、13.5°(±0.1°)、14.9°(±0.1°)、18.3°(±0.1°)、19.2°(±0.1°)、22.7°(±0.1°)、24.3°(±0.1°)、及び27.1°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1又は6に記載の式(I)の化合物。
【請求項8】
136〜139℃の範囲にある融解開始点を有する示差走査熱量測定曲線により特性決定される、請求項1〜6、又は7のいずれか1項に記載の式(I)の化合物。
【請求項9】
高強度の特定ピークを14.0°(±0.1°)、17.4°(±0.1°)、18.4°(±0.1°)、21.4°(±0.1°)、及び24.1°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1に記載の式(I)の化合物。
【請求項10】
特定ピークを5.6°(±0.1°)、12.5°(±0.1°)、14.0°(±0.1°)、17.4°(±0.1°)、18.4°(±0.1°)、21.4°(±0.1°)、22.2°(±0.1°)、22.9°(±0.1°)、24.1°(±0.1°)、及び24.5°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1又は9に記載の式(I)の化合物。
【請求項11】
127〜132℃の範囲にある融解開始点を有する示差走査熱量測定曲線により特性決定される、請求項1、9、又は10のいずれか1項に記載の式(I)の化合物。
【請求項12】
高強度の特定ピークを4.9°(±0.1°)、9.2°(±0.1°)、11.6°(±0.1°)、15.6°(±0.1°)、及び16.4°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1に記載の式(I)の化合物。
【請求項13】
特定ピークを4.9°(±0.1°)、6.0°(±0.1°)、9.2°(±0.1°)、11.6°(±0.1°)、12.8°(±0.1°)、15.6°(±0.1°)、16.4°(±0.1°)、17.2°(±0.1°)、及び18.1°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1又は12に記載の式(I)の化合物。
【請求項14】
約139℃に融解開始点を有する示差走査熱量測定曲線により特性決定される、請求項1、12、又は13のいずれか1項に記載の式(I)の化合物。
【請求項15】
実質的に非晶形の式(I)の化合物。
【請求項16】
水和物の形態である、式(I)の化合物。
【請求項17】
請求項6〜8のいずれか1項に記載の式(I)の化合物と請求項9〜11のいずれか1項に記載の式(I)の化合物との混合物。
【請求項18】
式(I)の化合物が、低級アルキルアセテート、低級アルキルアルコール、脂肪族及び芳香族の炭化水素、ジアルキルエーテル、ジアルキルケトン、アセトニトリル、水、又はこれらの混合物の群から選択される溶媒から結晶化される、請求項1に記載の化合物の製造法。
【請求項19】
溶媒が、エタノール、酢酸エチル、イソプロパノール、イソオクタン、アセトニトリル、水、又はこれらの混合物の群から選択される、請求項18に記載の方法。
【請求項20】
溶媒が、メタノール及び水の混合物、エタノール、酢酸エチル、エタノール及び水の混合物、イソプロパノール及び水の混合物、酢酸エチル及びイソオクタンの混合物、及びアセトニトリルの群から選択される、請求項19に記載の方法。
【請求項21】
溶媒がメタノール及び水の混合物である請求項18〜20のいずれか1項による、請求項3〜5のいずれか1項に記載の化合物の製造法。
【請求項22】
(結晶)種を使用する、請求項3〜5のいずれか1項に記載の化合物の製造法。
【請求項23】
請求項6〜8のいずれか1項に記載の化合物を融かすことによって種が製造される、請求項22に記載の方法。
【請求項24】
溶媒が酢酸エチルである請求項18〜20のいずれか1項による、請求項6〜8のいずれか1項に記載の化合物の製造法。
【請求項25】
溶媒がアルコールである請求項18〜20のいずれか1項による、請求項9〜11のいずれか1項に記載の化合物の製造法。
【請求項26】
式(I)の化合物をIPA/水溶媒系において5〜65℃の温度でスラリー化させる工程を含む、請求項9〜11のいずれか1項に記載の化合物の製造法。
【請求項27】
溶媒がアセトニトリルである請求項18〜20のいずれか1項による方法を含む、請求項12〜14のいずれか1項に記載の化合物の製造法。
【請求項28】
医薬品としての使用のための、請求項1〜17のいずれか1項に記載の化合物。
【請求項29】
請求項1〜17のいずれか1項に記載の化合物を製剤的に許容されるアジュバント、希釈剤、又は担体と混合して含んでなる医薬組成物。
【請求項30】
冠動脈、脳血管、又は末梢血管の疾患を有する患者における動脈血栓合併症の予防に使用される、請求項1〜17のいずれか1項に記載の化合物を含んでなる医薬組成物。
【請求項31】
請求項1〜17のいずれか1項に記載の化合物の、冠動脈、脳血管、又は末梢血管の疾患を有する患者における動脈血栓合併症の予防に使用される医薬品の製造における使用。
【請求項32】
冠動脈、脳血管、又は末梢血管の疾患を有する患者における動脈血栓合併症の治療若しくは予防の方法であって、そのような障害に罹患しているか又はそれに罹りやすいヒトへ、請求項1〜17のいずれか1項に記載の化合物の治療有効量を投与することを含む、前記方法。
【請求項1】
式(I):
【化1】
の実質的に結晶形の化合物。
【請求項2】
実質的に無水形で存在する、請求項1に記載の式(I)の化合物。
【請求項3】
高強度の特定ピークを5.3°(±0.1°)、20.1°(±0.1°)、20.7°(±0.1°)、21.0°(±0.1°)、及び21.3°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1に記載の式(I)の化合物。
【請求項4】
特定ピークを5.3°(±0.1°)、8.0°(±0.1°)、9.6°(±0.1°)、13.9°(±0.1°)、15.3°(±0.1°)、20.1°(±0.1°)、20.7°(±0.1°)、21.0°(±0.1°)、21.3°(±0.1°)、26.2°(±0.1°)、及び27.5°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1〜3に記載の式(I)の化合物。
【請求項5】
146〜152℃の範囲にある融解開始点を有する示差走査熱量測定曲線により特性決定される、請求項1、3、又は4のいずれか1項に記載の式(I)の化合物。
【請求項6】
高強度の特定ピークを5.5°(±0.1°)、13.5°(±0.1°)、18.3°(±0.1°)、22.7°(±0.1°)、及び24.3°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1に記載の式(I)の化合物。
【請求項7】
特定ピークを5.5°(±0.1°)、6.8°(±0.1°)、10.6°(±0.1°)、13.5°(±0.1°)、14.9°(±0.1°)、18.3°(±0.1°)、19.2°(±0.1°)、22.7°(±0.1°)、24.3°(±0.1°)、及び27.1°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1又は6に記載の式(I)の化合物。
【請求項8】
136〜139℃の範囲にある融解開始点を有する示差走査熱量測定曲線により特性決定される、請求項1〜6、又は7のいずれか1項に記載の式(I)の化合物。
【請求項9】
高強度の特定ピークを14.0°(±0.1°)、17.4°(±0.1°)、18.4°(±0.1°)、21.4°(±0.1°)、及び24.1°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1に記載の式(I)の化合物。
【請求項10】
特定ピークを5.6°(±0.1°)、12.5°(±0.1°)、14.0°(±0.1°)、17.4°(±0.1°)、18.4°(±0.1°)、21.4°(±0.1°)、22.2°(±0.1°)、22.9°(±0.1°)、24.1°(±0.1°)、及び24.5°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1又は9に記載の式(I)の化合物。
【請求項11】
127〜132℃の範囲にある融解開始点を有する示差走査熱量測定曲線により特性決定される、請求項1、9、又は10のいずれか1項に記載の式(I)の化合物。
【請求項12】
高強度の特定ピークを4.9°(±0.1°)、9.2°(±0.1°)、11.6°(±0.1°)、15.6°(±0.1°)、及び16.4°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1に記載の式(I)の化合物。
【請求項13】
特定ピークを4.9°(±0.1°)、6.0°(±0.1°)、9.2°(±0.1°)、11.6°(±0.1°)、12.8°(±0.1°)、15.6°(±0.1°)、16.4°(±0.1°)、17.2°(±0.1°)、及び18.1°(±0.1°)2θに含有するX線粉末回折パターンにより特性決定される、請求項1又は12に記載の式(I)の化合物。
【請求項14】
約139℃に融解開始点を有する示差走査熱量測定曲線により特性決定される、請求項1、12、又は13のいずれか1項に記載の式(I)の化合物。
【請求項15】
実質的に非晶形の式(I)の化合物。
【請求項16】
水和物の形態である、式(I)の化合物。
【請求項17】
請求項6〜8のいずれか1項に記載の式(I)の化合物と請求項9〜11のいずれか1項に記載の式(I)の化合物との混合物。
【請求項18】
式(I)の化合物が、低級アルキルアセテート、低級アルキルアルコール、脂肪族及び芳香族の炭化水素、ジアルキルエーテル、ジアルキルケトン、アセトニトリル、水、又はこれらの混合物の群から選択される溶媒から結晶化される、請求項1に記載の化合物の製造法。
【請求項19】
溶媒が、エタノール、酢酸エチル、イソプロパノール、イソオクタン、アセトニトリル、水、又はこれらの混合物の群から選択される、請求項18に記載の方法。
【請求項20】
溶媒が、メタノール及び水の混合物、エタノール、酢酸エチル、エタノール及び水の混合物、イソプロパノール及び水の混合物、酢酸エチル及びイソオクタンの混合物、及びアセトニトリルの群から選択される、請求項19に記載の方法。
【請求項21】
溶媒がメタノール及び水の混合物である請求項18〜20のいずれか1項による、請求項3〜5のいずれか1項に記載の化合物の製造法。
【請求項22】
(結晶)種を使用する、請求項3〜5のいずれか1項に記載の化合物の製造法。
【請求項23】
請求項6〜8のいずれか1項に記載の化合物を融かすことによって種が製造される、請求項22に記載の方法。
【請求項24】
溶媒が酢酸エチルである請求項18〜20のいずれか1項による、請求項6〜8のいずれか1項に記載の化合物の製造法。
【請求項25】
溶媒がアルコールである請求項18〜20のいずれか1項による、請求項9〜11のいずれか1項に記載の化合物の製造法。
【請求項26】
式(I)の化合物をIPA/水溶媒系において5〜65℃の温度でスラリー化させる工程を含む、請求項9〜11のいずれか1項に記載の化合物の製造法。
【請求項27】
溶媒がアセトニトリルである請求項18〜20のいずれか1項による方法を含む、請求項12〜14のいずれか1項に記載の化合物の製造法。
【請求項28】
医薬品としての使用のための、請求項1〜17のいずれか1項に記載の化合物。
【請求項29】
請求項1〜17のいずれか1項に記載の化合物を製剤的に許容されるアジュバント、希釈剤、又は担体と混合して含んでなる医薬組成物。
【請求項30】
冠動脈、脳血管、又は末梢血管の疾患を有する患者における動脈血栓合併症の予防に使用される、請求項1〜17のいずれか1項に記載の化合物を含んでなる医薬組成物。
【請求項31】
請求項1〜17のいずれか1項に記載の化合物の、冠動脈、脳血管、又は末梢血管の疾患を有する患者における動脈血栓合併症の予防に使用される医薬品の製造における使用。
【請求項32】
冠動脈、脳血管、又は末梢血管の疾患を有する患者における動脈血栓合併症の治療若しくは予防の方法であって、そのような障害に罹患しているか又はそれに罹りやすいヒトへ、請求項1〜17のいずれか1項に記載の化合物の治療有効量を投与することを含む、前記方法。
【図1.1】

【図1.2】
【図1.3】
【図1.4】
【図1.5】
【図2】
【図1.2】
【図1.3】
【図1.4】
【図1.5】
【図2】
【公開番号】特開2012−149093(P2012−149093A)
【公開日】平成24年8月9日(2012.8.9)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−104575(P2012−104575)
【出願日】平成24年5月1日(2012.5.1)
【分割の表示】特願2002−500875(P2002−500875)の分割
【原出願日】平成13年5月31日(2001.5.31)
【出願人】(300022641)アストラゼネカ アクチボラグ (581)
【Fターム(参考)】
【公開日】平成24年8月9日(2012.8.9)
【国際特許分類】
【出願番号】特願2012−104575(P2012−104575)
【出願日】平成24年5月1日(2012.5.1)
【分割の表示】特願2002−500875(P2002−500875)の分割
【原出願日】平成13年5月31日(2001.5.31)
【出願人】(300022641)アストラゼネカ アクチボラグ (581)
【Fターム(参考)】
[ Back to top ]