
ドーパミンニューロンの増強産生に関する方法と材料
【課題】Frizzledレセプターのいずれのリガンドも使用せず、細胞におけるドーパミン作動性ニューロン表現型の誘導、又は促進する方法、その方法で得られたドーパミン作動性ニューロン等を提供する。
【解決手段】神経幹細胞、神経系前駆若しくは神経前駆細胞、又は他の幹細胞においてGSK-3βを阻害することによって、該細胞における神経の運命の誘導、及び特定の神経表現型の誘導と増強、及び特に中脳ドーパミン作用性ニューロンの表現型誘導とその増強を行う。
【解決手段】神経幹細胞、神経系前駆若しくは神経前駆細胞、又は他の幹細胞においてGSK-3βを阻害することによって、該細胞における神経の運命の誘導、及び特定の神経表現型の誘導と増強、及び特に中脳ドーパミン作用性ニューロンの表現型誘導とその増強を行う。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、神経幹細胞、神経系前駆若しくは神経前駆体細胞、又は他の幹細胞における神経の運命誘導に関する。また本発明は、特定の神経表現型の誘導とその増強、特に中脳ドーパミン作動性ニューロンの表現型の誘導とその増強に関する。
【0002】
本発明の局面と実施形態は、GSK-3βの阻害調節に関連する。これは、GSK-3βの阻害剤である物質、又は、例えばβ-カテニンに作用したり、若しくはそれを模倣することによって、例えばβ-カテニンを安定化したり、その分解を阻害したり、若しくはそのリン酸化を阻害することによって、又はGSK-3βの基質に作用することによって、GSK-3β活性に影響する物質の利用に関連してもよい。本明細書で「GSK-3βの阻害」又は「GSK-3β阻害」というときは、これらのアプローチのいずれかを意味する。Frizzledレセプターに対するWnt及びいかなるリガンドの利用も考慮しない。「GSK-3β阻害剤」とは、その文脈が別の内容を意味しない限り、GSK-3βに直接に作用してこれを阻害する物質の利用をいう。
【0003】
いくつかの実施形態では、GSK-3β阻害はアップレギュレート、又はダウンレギュレートであり得る。すなわち、阻害は、誘導されても、あるいは縮小されてもよい。本明細書では、主として阻害の誘導について述べているが、GSK-3β阻害の調節がダウンレギュレーション、すなわち、阻害の縮小による類似の実施形態も本発明によって提供される。
【背景技術】
【0004】
パーキンソン病(PD)は非常によく知られた神経変性疾患であり、その病因は中脳のドーパミン作動性(DA)ニューロン(以下、神経細胞又は単に神経ということもある)の選択的、かつ進行性の欠損によって特徴付けられる。神経表現型の誘導の増強は、パーキンソン病や他の重篤な衰弱性神経変性疾患の治療を可能にする潜在性を有している。
【0005】
従来、好成績な結果をもたらすものとしてヒト胎児の中脳組織がパーキンソン病患者に移植されていた。しかし、本発明を利用した特定の細胞補充療法の開発によれば、移植の取り組みに先立つ実行上の、あるいは倫理上の困難を克服できる。特に、本発明によれば、胚組織又は胚性細胞の利用のいずれの必要性も減じ、あるいは排除できると同時に、移植用の細胞調製の開発を可能にする。幹細胞は、一般に廃棄される組織である臍帯から得ることができる。その他の選択肢は、成体幹細胞を例えば、骨髄、血液、皮膚、目、嗅球または嗅覚器上皮から得ることである。
【0006】
以前(WO00/66713 and Wagner ら, 1999)に、本発明者の研究室は、腹側中脳のタイプIアストロサイト/初期グリア細胞から得ることのできる一又は二以上の因子の存在下においてドーパミン作動性ニューロン表現型の誘導がNurr1を発現している細胞内で増強されることを示した。
【0007】
さらなる研究により、Nurr1を発現している細胞の神経表現型の誘導を増強する上でWnt因子が有効であることが判明した。これらの結果は、WO2004/029229で発表している。
【0008】
特に、WO2004/029229で開示したように、DA ニューロンの出現時まで背側中脳よりもVMで高レベルで発現されるあらゆるWntが、神経幹、前駆細胞若しくは前駆体細胞、又は他の幹細胞、又は神経細胞において、増殖、自己複製、ドーパミン作動性の誘導、生存、分化、及び/又は成熟化を増強することによりドーパミン作動性ニューロンの発生を誘導、又は促進する上で有用であることが判明した。
【0009】
すなわち、Wnt-1は、ドーパミン作動性前駆体の増殖とドーパミン作動性前駆体、及び/又は幹細胞のドーパミン作動性ニューロンへの成熟化を促進し、
Wnt-7aは、ドーパミン作動性前駆体の増殖を促進し、またドーパミン作動性ニューロンへの分化を可能にし、
Wnt-3aは、ドーパミン作動性前駆体、及び/又は幹細胞の増殖、及び/又は自己複製を促進し、
Wnt-2は、Nurr1+前駆体によって細胞周期からの離脱とドーパミン作動性ニューロン表現型の獲得を促進し、そして
Wnt-5aは、神経幹細胞、前駆細胞若しくは前駆体細胞でのドーパミン作動性表現型の誘導で、また神経細胞でのドーパミン作動性の誘導若しくは分化を増強することにおいて、最も有効であることが判明した。
【0010】
Wnt-1は、ドーパミン作動性前駆体、及び/又は幹細胞の分化と成熟化においてWnt-3aとWnt-5aよりも有効である。
【0011】
特定の神経表現型の誘導には、遺伝学的なシグナルと後成的なシグナルの双方の統合が必要である。発生中の中脳におけるドーパミン作動性ニューロンの誘導は、オーファン核内レセプターNurr1を必要とする (Zetterstroemら, 1997; Saucedo-Cardenasら, 1998; Castilloら, 1998)が、神経幹細胞においてはNurr1の発現がドーパミン作動性表現型を誘導するのに十分とは言えない(Wagner ら, 1999)。Nurr1と発生中の腹側中脳タイプIアストロサイト/初期グリア細胞から得られる未知の可溶性シグナルとの組み合わせにより、神経幹細胞での中脳ドーパミン作動性表現型の誘導が充足される (Wagnerら, 1999)。Wnt-5aは、このようなシグナルの一部であり、またWntファミリータンパク質の一員である。Wntファミリーは、Wnt-1, -2, -3a, -5a 及び -7aを含み、発生上で調節され、分化上では中脳ドーパミン作動性ニューロンの発生を制御する。部分精製されたWnt-1, -2, -5a 及び -7aは、二つの異なる機構でE14.5中脳DAニューロンの数を増加したが、Wnt-3aは増加しなかった。Wnt-1と-7aは、主にNurr1前駆体の増殖を増加し、またそれらのドーパミン作動性ニューロンへの分化を可能にした。Wnt-2は、Nurr1+前駆体によるドーパミン作動性ニューロンの表現型の獲得と細胞周期からの離脱を示した。Wnt-5aは、主として神経のDA表現型を獲得したNurr1前駆体の割合を増加した。これらの発見と一致して、Wnt-5aはNurr1発現ニューロン、又は皮質E13.5前駆体においてドーパミン作動性ニューロンを誘導する点で中脳アストロサイト/初期グリア細胞と同程度に有効であった。さらに、Frizzled 8のシステインリッチドメインは、Nurr1発現ニューロン前駆体培養にてドーパミン作動性表現型をもつ細胞の増加におけるVM T1A-、Wnt-1若しくはWnt-5aの介在する効果と基底の効果を効率的に妨げた。また、神経幹細胞での内因性のWnt又はFGF-8の効果は、Nurr1+中脳神経球に及んだ。このようにデータは、Wntが部分的に異なる機構で、Nurr1発現前駆体/幹細胞においてDA表現型を持つ神経の発生を独立に調節していることを示している。
【0012】
これらの発見は、Wntリガンドを腹側中脳神経発生期間における増殖、自己複製、分化、及び運命決定のキーとなるレギュレーターとして位置づけている。さらに、Wntは、幹細胞の発生をドーパミン作動性ニューロンに導くのに利用され、それによって、パーキンソン病に対する細胞補充療法でのそれらの治療の潜在性が活用されるかもしれない。
【0013】
胚性幹細胞、神経幹細胞、及び多分化能幹細胞は、神経、アストロサイト、およびオリゴデンドロサイトを含む神経細胞系統へ分化する能力を有している。さらに、幹細胞は、脳移植用の原材料として利用され、増幅され、分離され得る(Snyder, E. Y. ら Cell 68, 33-51 (1992); Rosenthal, A. Neuron 20, 169-172 (1998); Bain ら, 1995; Gage, F.H., ら Ann. Rev. Neurosci. 18, 159-192 (1995); Okabe ら, 1996; Weiss, S. ら Trends Neurosci. 19, 387-393 (1996); Snyder, E. Y. ら Clin. Neurosci. 3, 310-316 (1996); Martinez-Serrano, A. ら Trends Neurosci. 20, 530-538 (1997); McKay, R. Science 276, 66-71 (1997); Deacon ら, 1998; Studer, L. ら Nature Neurosci. 1, 290-295 (1998); Bjorklund and Lindvall 2000; Brustle ら, 1999; Lee ら, 2000; Shuldiner ら, 2000 and 2001; Reubinoff ら, 2000 and 2001; Tropepe ら, 2001; Zhang ら, 2001; Price and Williams 2001; Arenas 2002; Bjorklund ら, 2002; Rossi and Cattaneo, 2002; Gottlieb ら, 2002)。
【0014】
ほとんどの神経変性疾患は、神経細胞数に影響を及ぼす。さらに、損傷の多くは、特異的な神経化学表現型を生じる。例えば、ヒトパーキンソン病で欠損する主要な細胞型は、中脳ドーパミン作動性ニューロンである。神経組織の移植による特定の神経細胞数の機能的な補充は、神経変性疾患を治療する上で魅力的な治療戦略を提示する(Rosenthal, A. Neuron 20, 169-172 (1998))。他の選択肢は、再生を促進し、幹細胞、前駆細胞、前駆体細胞の発生及び/又は追加を修復し、若しくは導くのに必要なシグナルの直接的な注入、あるいはそれらの機能を調節する薬物の投与であろう。
【0015】
幹/前駆、又は前駆体細胞は、増幅可能で、かつ特定の神経表現型となる指示を受け得るので、移植療法にとっては理想的な材料である。一般に一人の患者を治療するために、数人のドナー由来の組織が必要であったが、一つの幹細胞が多くの患者を治療するために利用できるので、これらの細胞によってヒト胚性組織の移植のための使用を取り巻く倫理上、及び実施上の問題を回避できるであろう。
【0016】
しかし、Wntは可溶性に乏しい。これは、セットアップされた臨床現場でそれらの利用を制限し、また治療学上の応用を困難にしている。
【特許文献1】WO00/66713
【特許文献2】WO2004/029229
【非特許文献1】Wagner ら, 1999 Nat Biotechnol.;17 (7):653-659. (ref 22)
【非特許文献2】Zetterstrom ら, Science 1997 Apr 11;276(5310):248-50
【非特許文献3】Saucedo-Cardenas ら, Proc Natl Acad Sci U S A 1998 Mar 31;95(7):4013-8
【非特許文献4】Castillo ら, Mol Cell Neurosci 1998 May;11(1-2):36-46
【非特許文献5】Wagner ら, 1999 Nat Biotechnol.;17 (7):653-659. (ref 22)
【非特許文献6】Snyder, E. Y. ら Cell 68, 33-51 (1992)
【非特許文献7】Rosenthal, A. Neuron 20, 169-172 (1998)
【非特許文献8】Bain ら,1995, Dev. Biol., 168, 342-357
【非特許文献9】Gage, F.H., ら Ann. Rev. Neurosci. 18, 159-192 (1995)
【非特許文献10】Okabe ら, 1996, Mech. Dev., 59, 89-102
【非特許文献11】Weiss, S. ら Trends Neurosci. 19, 387-393 (1996)
【非特許文献12】Snyder, E. Y. ら Clin. Neurosci. 3, 310-316 (1996)
【非特許文献13】Martinez-Serrano, A. ら Trends Neurosci. 20, 530-538 (1997)
【非特許文献14】McKay, R. Science 276, 66-71 (1997)
【非特許文献15】Deacon ら, 1998, Exp. Neurol., 149, 28-41
【非特許文献16】Studer, L. ら Nature Neurosci. 1, 290-295 (1998)
【非特許文献17】Bjorklund A, Lindvall O. Nat Neurosci. 2000 Jun;3(6):537-44
【非特許文献18】Brustle ら, 1999, Science 285, 754-756
【非特許文献19】Lee ら, 2000. Nat Biotechnol.;18 (6):675-679. (ref 31)
【非特許文献20】Schuldiner ら, 2000, PNAS. USA 97, 11307-11312
【非特許文献21】Schuldiner ら, 2001; Brain Res. 913, 201-5
【非特許文献22】Reubinoff ら, 2000, Nat Biotech., 18, 399-404
【非特許文献23】Reubinoff ら, 2001, Nat Biotech., 19, 1134-1140
【非特許文献24】Tropepe.ら, 2001, Neuron 30,65-78
【非特許文献25】Zhang ら, 2001, Nat Biotech., 19, 1129-1133
【非特許文献26】Price J. & Williams BP. Current Opinion in Neurobiology 11, 564-567 (2001)
【非特許文献27】Arenas 2002. Brain Res Bull.; 57 (6):795-808. (ref 28)
【非特許文献28】Bjorklund ら, 2002, Proc Natl Acad Sci USA. Feb 19, 99(4): 2344-9
【非特許文献29】Rossi F. & Cattaneo E. Nat Rev Neurosci. 2002 May;3(5):401-9
【非特許文献30】Gottlieb DI. Annu Rev Neurosci. 2002;25:381-407
【発明の開示】
【0017】
本発明は、GSK-3βの阻害、例えば、GSK-3β自身を標的とすることにより、若しくはβ-カテニン活性を模倣若しくは増強することにより、細胞におけるドーパミン作動性ニューロン表現型の誘導、又は促進することに関する。本発明は、実用的な治療法の手段とWntリガンドに代わる代替法として、Wntシグナル伝達を形質導入するシグナル伝達経路の活性化を提供する。WntリガンドとFrizzledレセプターのいずれのリガンドも本発明では使用しない。
【0018】
GSK-3βは、多機能性セリン/スレオニンキナーゼであり、β-カテニンの分解に不可欠である。参照:Doble and Woodgett J Cell Sci. 2003 Apr 1;116(Pt 7):1175-86. Human accession number: NM_002093。
【0019】
本明細書中で開示したように、本発明によれば、細胞内でGSK-3βを阻害することにより、培養物内であろうが脳内であろうが、またその阻害が直接的であろうが間接的であろうが、ドーパミン作動性ニューロン、前駆体細胞、前駆細胞、若しくは幹細胞の増殖及び/又は自己複製の誘導又は促進することが可能となる。及び/又は、ドーパミン作動性ニューロンの産出量を高めることによりドーパミン作動性ニューロン、前駆体、前駆細胞、若しくは幹細胞の生存、分化、成熟化の促進を可能にし、及び/又は、in vitro、in vivoで幹細胞、前駆、前駆体細胞、若しくは神経細胞における神経のドーパミン作動性運命の誘導が可能となる。
【0020】
本発明は、GSK-3βの阻害により神経分化を増加することを提供する。そして、さらにNurr1を発現している前駆体のTH+ニューロンへの変換を介してドーパミン作動性ニューロン集団内でのドーパミン作動性ニューロン数を増加させる方法を提供する。
【0021】
GSK-3βは、直接的に又は間接的に阻害されてよい。阻害は、例えば上流又は下流のシグナル因子に作用することによって、例えば、dishevelled、アキシン、FRAT/GBP、若しくはカゼインキナーゼ1の分解や機能欠損、又はβ-カテニンの過剰発現若しくは安定化若しくは分解の阻害、若しくはβ-カテニンリン酸化の阻害によって、間接的になされてもよい。本明細書でGSK3の阻害とは、文中特に示した場合を除き、これらのアプローチのいずれかを意味し、実際、Frizzledレセプターのリガンド、例えばWntによる場合を除いたいずれのアプローチも意味する。
【0022】
GSK-3β阻害剤のインディルビン(indirubin)ファミリーは、当初、慢性骨髄性白血病を含む様々な慢性病に対して使用される伝統的な漢方薬から分離された(19)。ごく最近になって、幹細胞治療においてGSK-3β阻害剤としてのインディルビンファミリーの潜在的な応用法が報告された(15)。DA前駆体の分化における化学的なGSK-3β阻害剤の新しい性質について記載した我々の結果は、PD用細胞補充療法の開発にとって重要な意味を持つ。現在の治療アプローチは、ヒト胎児中脳DA前駆体をPD患者の新線条体へ移植することに焦点が当てられている(27)。これらのアプローチの成功は、移植されるDA神経数に決定的に依存し、また1患者あたり5人以上の胎児を必要とする(27、28)。VM前駆体移植に対するさらに可能性のあるアプローチは、DAニューロンに前分化したヒト幹細胞の移植を含んでいる。しかしながら、ヒト幹細胞のDAニューロンへの分化(29、30)は、マウス幹細胞に関して報告されたそれよりも困難であることが判明し(22、31-33)、幹細胞調製で与えねばならない新規の分化シグナルが必要であることが強調された。我々の結果は、GSK-3β阻害が前駆体のDA分化を効率的に促進することを示し、またそのような阻害はPD患者へ移植する前の細胞調製に利用し得ることを示唆している。
【0023】
β-カテニンシグナル伝達は、一般に中枢神経系の発生において神経系前駆細胞の増殖に関与している(2、34、35)。実際、神経幹/前駆細胞において活性なプロモーターの制御下でβ-カテニンを恒常的に発現させると、完全な神経管の増殖を生じる(34,35)。これは、前駆細胞増殖におけるβ-カテニンの役割を支持している。しかしながら、これらの実験は、神経分化期間の後期発生段階におけるβ-カテニンの可能性のある役割に関しては直接的に見ていない。
【0024】
我々は、以前にWnt-1とWnt-5aがVM前駆体培養物内でVM前駆体の増殖とDA分化をそれぞれ促進することを示した。さらに、我々は、in vivo でDA前駆体が分化する場所であるVMドメインにおいて、E10.5胚でNurr1/β−カテニンの共局在とTCF/LEF転写活性を見出した(7)。これらの結果は、Wnt/β-カテニンシグナル伝達がDA前駆体において増殖と分化の両方を調節できることを示唆している。
【0025】
本明細書では、GSK-3β阻害とβ-カテニン安定化が、前駆体のDAニューロンへの分化と、総神経細胞集団のうちのDAニューロンの割合の両方を増加することを報告する。
【0026】
我々の結果は、皮膚(38−40)などの他の器官の分化と同様に、感覚神経の分化(37)や樹上突起形成(10)におけるWnt/β-カテニンシグナルについての役割を支持している最近の発表と一致している。このように、我々のデータは細胞分化におけるβ-カテニンの一般的な役割についての証拠を提供し、またGSK-3β阻害は神経及びグリアの前駆体の両方を含む細胞調製品においてDA分化を促進することから細胞補充療法に非常によく適しているかもしれないことを示している。
【0027】
GSK-3βの阻害剤は、当該技術分野で利用可能である。以前から知られているGSK阻害剤はリチウム(マグネシウム競合剤)と亜鉛である。GSK-3βは、系統発生学的にはCDKと非常に密接に関連している。研究により、GSK-3βとある程度のCDKに対して作用することが判明している少なくとも4つのATP競合キナーゼ阻害剤クラスが存在することが明らかとなった。すなわち、ヒメナルジシン類(hymenaldisines)、インディルビン類(indirubins)、アロイシン類(aloisines)、そしてパウロン類(paullones)である。最近、マレイミド(maleimides)、チアゾール誘導体、チエニル及びメチル・ハロメチル・ケトン、並びに酸化アミノピリミジンを含む、より強力で特異的なGSK3阻害剤が同定された。参考文献は以下のものを含む:Knockaert ら J Biol Chem. 2002 Jul 12;277(28):25493-501; Hoessel ら (1999) Nat Cell Biol. ; 1 (1): 60-67; Leclerc ら (2001) J Biol Chem; 276 (1): 251-260; Conde ら J. Med. Chem. 46, 4631-4633 (2003); Cline ら Diabetes 51, 2903-2910 (2003); Ring ら Diabetes 52, 588-595 (2003); Bhat ら J. Biol. Chem. 278, 45937-45945 (2003); Kuo et al J. Med. Chem. 46, 4021-4031 (2003)。
【0028】
当該技術分野には、GSK-3β阻害の調節に利用できる物質を同定するための様々な技術がある。例えば、供試化合物の、GSK3の基質(β-カテニン、グリコーゲンシンターゼ、Tau、elf2B、CREB、c-jun)のリン酸化を阻害するが、他の酵素(サイクリン依存性キナーゼ、プロテインキナーゼA、B、C、SAPキナーゼ、JNK、MAPK、MEK、カゼインキナーゼ、Rhoファミリー、PDK1等)の基質のリン酸化は阻害しない能力を評価する。
【0029】
GSK-3β阻害に有用であるとして同定された化合物(例えば、GSK-3β阻害剤)は、それらの活性を増強する目的で、当業者に周知のいずれかの方法で改変することができる。
【0030】
好ましい実施形態において、阻害剤はヒメナルジシン、アロイシン、マレイミド、チアゾール、チエニル及びメチルハロメチルケトン、酸化アミノピリミジン、インディルビン(例えば、インディルビン−3−モノオキシム(indirubin−3−monoxime:I3M))、又はパウロン(例えば、ケンパウロン(kenpaullone:KP))のようなGSK-3βに対して特異的なものである。特に好ましい実施形態において、阻害剤はケンパウロンである。参考文献:Leclerc ら (2001) J Biol Chem; 276 (1): 251-260; Hoessel ら (1999) Nat Cell Biol. ; 1 (1): 60-67; Polychronopoulos ら J Med Chem. 2004 Feb 12;47(4):935-46; Conde ら J. Med. Chem. 46, 4631-4633 (2003); Cline ら Diabetes 51, 2903-2910 (2003); Ring ら Diabetes 52, 588-595 (2003); Bhat ら J. Biol. Chem. 278, 45937-45945 (2003); Kuo ら J. Med. Chem. 46, 4021-4031 (2003)。
【0031】
本発明のいかなる局面や実施形態も、神経細胞、すなわち神経に応用することができ、また使用し得る。本開示において「神経の細胞」とは、ニューロン(神経細胞)であってよい。
【0032】
ドーパミン作動性ニューロンが豊富な細胞調製品は、パーキンソン病や他の疾患における細胞補充療法に利用してもよく、また、ドーパミン作動性ニューロンにおけるシグナル伝達事象とin vitroでのドーパミン作動性ニューロンへの薬物の効果を例えば、ハイスループット・スクリーニングで研究するのに利用してもよい。
【0033】
本発明の局面と実施形態は、特許請求の範囲に示されている。
【発明を実施するための最良の形態】
【0034】
一つの形態として、本発明は幹細胞、神経幹細胞、又は神経系前駆若しくは前駆体細胞においてドーパミン作動性ニューロンの運命を誘導する方法、又は神経細胞でのドーパミン作動性誘導若しくは分化を増強する方法、又はドーパミン作動性前駆体若しくは前駆細胞若しくはNurr1を発現している幹細胞を増幅する方法を提供する。この方法は、GSK-3β阻害を調節すること(例えば、GSK-3βの阻害剤で、β-カテニンを過剰発現若しくは供給することで、β-カテニンのリン酸化を阻害することで、又はβ-カテニンの分解を阻害することで、又はGSK-3β阻害の他の調節で細胞を処理することによって)、それによりドーパミン作動性ニューロンを生じさせることである。本方法は予めNurr1サブファミリーの核内レセプターを細胞内の基底レベルよりも高く発現することを任意に含む。
【0035】
GSK-3β阻害(又は、論述したように間接的にGSK-3βを阻害すること)を調節すること、例えば、誘導することは、in vivo, ex vivo, in vitro若しくは培養物内で行えばよい。in vitro, ex vivo, 若しくは培養物内で処理することが好ましいかもしれない。
【0036】
本発明の方法において、GSK-3β阻害に作用する物質で処理することは、物質と細胞を接触させることによってもよい。このような物質(例えば、GSK-3β阻害剤)での処理は、幹、前駆若しくは前駆体細胞を含む培養物、又はin vivo におけるそのような細胞にその物質を与えることによってでもよい。もし、GSK-3β阻害剤の処理に代って、GSK-3βの阻害を間接的に行うのならば、間接的阻害を生じる処理(例えば、β-カテニンの安定化、β-カテニンの過剰発現を生じさせること等)が、in vivoで同様に実行されてもよい。または可能であれば、in vitro、ex vivo若しくは培養物内で実行されてもよい。
【0037】
GSK-3β阻害で作用する物質の供給に加えて、幹、神経幹、前駆若しくは前駆体細胞、又は神経細胞をin vitro又はin vivoでタイプIアストロサイト/グリア細胞と共培養してもよいし、それらの細胞若しくはそれらの細胞から得られた因子と接触させてもよい。
【0038】
共培養された若しくはホスト細胞は、別の幹、神経幹、前駆若しくは前駆体細胞、又は神経細胞であってもよい。
【0039】
Nurr1(Lawら, 1992; Xingら, 1997; Castillo, 1997; GenBank nos. S53744, U72345, U86783)は、チロイドホルモン/レチノイン酸核内レセプタースーパーファミリーの転写因子の一つである。WO00/66713とWagnerら(1999)によって以前に報告されたように、神経幹細胞若しくは神経系前駆細胞での基底レベルを超えるNurr1の発現は、神経運命方向に分化する細胞数を増加する。神経運命の誘導は、in vitro又はin vivoで実行されてもよい。移植前若しくはその後に幹細胞又は神経幹、前駆若しくは前駆体細胞を神経運命に分化誘導する能力は、WO00/66713とWagnerら(1999)によって以前に報告されたように、グリア由来の信号によっても増強できる。
【0040】
Nurr1はNR4Aサブファミリーの一員である。本発明の方法はNurr1を使用することに限定されないが、Nurr1が好ましいかもしれない。また、方法はNR4Aサブファミリーのいずれかの核内レセプターを細胞内で基底レベルを超えて発現することを含み得る。NR4Aサブファミリーのレセプターは、Nurr1/NR4A2、Nor1/NR4A3、そしてNGFI-B/NR4A1を含む。したがって、本発明の方法では、NR4Aサブファミリーの核内レセプター(例えば、Nurr1、Nor1、又はNGFI-B)が、細胞内で基底レベルを超えて発現されてもよい。NR4Aサブファミリーメンバーのアクセッション・ナンバーは、例えば、以下の通りである。
【0041】
NGF-IB タンパク質: NP775181 NP775180 NP002126;
NGFI-B ヌクレオチド: NM_173158 NM_173157 NM_002135;
Nor-1 タンパク質: NP775292 NP775291 NP775290 NP008912S71930 Q92570;
Nor-1 ヌクレオチド: NM_005413 NM_173200 NM_173199 NM_173198 NM_006981。
【0042】
グリコタンパク質のWntファミリーのメンバーは、可溶性に乏しく(Bradley and Brown, 1990 and 1995)、また中脳の発生において発現している(Parr ら, 1993)。Wntは、中脳−後脳発生(McMahon and Bradley, 1990; Thomas and Capecchi, 1990)、神経パターニング(Kiecker and Niehrs, 2001; Nordstrom ら, 2002; Houart ら, 2002)、前駆体増殖(Taipale and Beachy, 2001; Chenn and Walsh, 2002; Megason and McMahon, 2002)、そして、神経系(Dorsky ら, 1998; Baker ら, 1999; Wilson ら, 2001; Garcia-Castro ら, 2002; Muroyama ら, 2002)を含む複数の組織での運命決定(Kispert ら, 1998; Ross ら, 2000; Hartmann and Tabin, 2001; Marvin ら, 2001; Schneider and Mercola, 2001; Tzahor and Lassar, 2001; Pandur ら, 2002)を調節している。
【0043】
本明細書中で用いられる時、「Wntポリペプチド」、「Wntグリコタンパク質」、また「Wntリガンド」とは、細胞間相互作用を調節する分泌タンパク質のWingless-intファミリーのメンバーをいう。Wntは、DrosophilaやCaenorhabditis elegansからXenopus、ゼブラフィッシュ、そして哺乳動物に至るまで高度に保存されている。哺乳動物で現在知られている19のWntタンパク質は、二つの細胞表面レセプター型に結合する。すなわち、現在10個のレセプターによって構成される7回膜貫通型ドメインFrizzled レセプターファミリー、並びに低密度リポタンパク質レセプター関連タンパク質5と6(LRP-5、-6)及びKremen1とKremen2のレセプターである。Wntによって運ばれたシグナルは、3つの既知シグナル伝達経路を介して細胞内に導入される。(1)いわゆるカノニカルシグナル伝達経路:当該経路ではGSK-3βが抑制されることにより、β−カテニンがリン酸化されず、分解を受けない。その後、β−カテニンは核内移行してTCFと複合体を形成し、Wnt標的遺伝子の転写を活性化する。(2)Jnkを介した平面極性及びコンバージェンス伸展(convergence-extension)経路、そして(3)イノシトール1,4,5三リン酸(IP3)/カルシウム経路:当該経路ではカルシニューリンが活性化T細胞の核内因子(NF-AT)を脱リン酸化して、それを活性化する(Saneyoshi ら Nature. 2002 May 16;417(6886):295-9)。レビューに関しては、いずれかのウェブブラウザで閲覧可能なWntホームページ(www.stanford.edu/~rnusse/wntwindow.html)をご覧いただきたい。Wntシグナル伝達に関連する他の共レセプターは、チロシンキナーゼレセプターRor1とRor2(Oishi I ら Genes Cells. 2003 Jul;8(7):645-54)、触媒的に不活性化する受容体型チロシンキナーゼをコードするderailed/RYKレセプターファミリー(Yoshikawa ら, Nature. 2003 Apr 10;422(6932):583-8)を含んでいる。
【0044】
「幹細胞」とは、自己再生可能ないずれの細胞タイプも意味する。もしそれが胚性幹(ES)細胞である場合には、一個体における全細胞を生じさせることができ、また、もしそれが多分化能若しくは神経幹細胞である場合には、神経、アストロサイト、オリゴデンドロサイトを含む神経系における全細胞を生じさせることができる。幹細胞は、以下の一又は二以上のマーカーを発現していてもよい。すなわち、記載のように(Tropepeら Neuron. 2001 Apr;30(1):65-78; Xu ら Nat Biotechnol. 2001 Oct;19(10):971-4)、Oct-4、Sox1-3、Nanog(Chambers ら, 2003, Cell, May 30;113(5):643-55.)、ステージ特異的胚性抗原(SSEA-1, -3, -4)、そして腫瘍拒絶抗原TRA-1-60及び-1-81等である。神経幹細胞は、一又は二以上の以下のマーカーを発現していてもよい。すなわち、ネスチン、p75ニューロトロフィンレセプター、Notch1、SSEA-1(Capela and Temple Neuron. 2002 Aug 29;35(5):865-75)等である。
【0045】
「神経系前駆細胞」とは、多分化表現型、及び/又は幹細胞と比べるとより限定された分化能を持った神経幹細胞の嬢細胞、子孫細胞を意味する。前駆体細胞は、発生期間に神経と直接的な系統関連にあるものだけでなく、限定的な環境的条件下で分化転換若しくは再分化を誘導され得る、又は神経表現型を獲得する他のいずれの細胞をも意味する。
【0046】
幹細胞、神経幹細胞、又は神経系前駆細胞若しくは前駆体細胞は、骨髄、皮膚、目、鼻粘膜上皮、若しくは臍帯、若しくは神経系部位(例えば、小脳、脳室帯、副脳室帯、線条体、中脳、後脳、大脳皮質、又は海馬状隆起由来のもの)を含む。それらは、脊椎動物の組織体から得てもよいし、それ由来のものであってもよい。ここでいう脊椎動物とは、例えば、ヒト、又はヒト以外の哺乳動物であるウサギ、モルモット、ラット、マウス若しくは他のげっ歯類動物、ネコ、イヌ、ブタ、ヒツジ、ヤギ、ウシ、ウマ、若しくは霊長類動物のような哺乳動物、又はニワトリのような鳥でよい。
【0047】
本発明の好ましい実施形態では、成体幹/前駆/前駆体細胞がin vitro、ex vivo、又はin vivoで使用される。これは、同意成人(例えば、細胞を得ることに対して)と、適切な倫理委員会による承認を必要とする。ヒト胚/胎児を細胞源として使用する場合、ヒト胚は、通常使用されずに破壊されるか、いつまでも保管されるもの、特に妊娠困難な夫婦のIVF(体外受精)治療目的のために作成されたヒト胚である。通常IVFでは、移植及び究極的には妊娠に使用する数よりも多くのヒト胚の作成を伴う。このような予備胚は、一般には破壊される。破壊される以外にない胚は、関係者、特に直接関連する卵子提供者、及び/又は精子提供者の適切な同意をもって、パーキンソン病のような重度の神経変性疾患患者の利益に対して倫理上肯定的な方法で使用することができる。本発明自体は、その開発のいずれのステージにおいてもヒト胚の使用に関連しない。既に述べたように、本発明は恐ろしい病気に有益な治療法の開発を可能にすると同時に、ヒト胚に直接由来する材料を使用するに際して生じ得る必要性を最小限にしている。
【0048】
いくつかの好ましい実施形態において、幹若しくは前駆若しくは前駆体細胞は、GSK-3β阻害剤、若しくは論述したようなGSK-3βを阻害する他の手段等のGSK-3β阻害に作用する物質と接触される他、本発明のいずれかの局面に従って処理、及び/又は使用され、適切な同意を得た同意成人若しくは子供、例えば、本発明に従って製造され、及び/又は内在性のドーパミン作動性ニューロンの発生、若しくは機能を促進、若しくは誘導するためにGSK-3β阻害、GSK-3β阻害剤、若しくはGSK-3βを間接的に阻害する物質、及び/又は一又は二以上のタイプIアストロサイト/初期グリア細胞由来の因子で処理された神経を移植で戻すことによって治療されることになる疾患を持つ患者から得られる。
【0049】
幹若しくは前駆若しくは前駆体細胞が誘導される神経運命は、未分化表現型又は原始的な神経表現型を示してもよい。細胞は、一個体のどの細胞タイプをも生じ得る全能性細胞、又は複数の異なる神経表現型を生じ得る多分化能細胞、又は通常の発生期間では、より限られた表現型を生じ得るが、in vitroで適当な環境因子に晒された場合、その他の細胞を生じ得る前駆体若しくは前駆細胞であってもよい。細胞は、特定の神経運命と関連するマーカー(例えば、チロシン水酸化酵素)を欠いていてもよい。
【0050】
VM前駆体がGSK-3β阻害の調節、特に例えばGSK-3β阻害剤によるGSK-3β阻害の誘導を受け、あるいはその他GSK-3βの阻害を受ける本発明の神経運命を誘導する方法において、増加した割合の細胞が神経運命を取るように誘導され得る。ドーパミン作動性の誘導や分化が神経細胞で増強され得る。好ましい実施形態では、腹側中脳においてTH+/TuJ1+細胞の割合で3〜5倍増加する。
【0051】
培養条件は、異なる密度で、異なる培地で、異なる基質で、又は他の細胞集団の存在下で細胞を培養することを含むが、これに制限されることはない。
【0052】
「細胞内で基底レベルを超えるNurr1の発現」とは、in vivoで非病的条件下の(未改変の)細胞で発現しているレベルよりも多くのNurr1を発現することを意味する。同様に、細胞内での基底レベルを超えるNurr1サブファミリーの核内レセプターの発現とは、in vivoで非病的条件下の(未改変の)細胞で発現しているレベルよりも多くの核内レセプターを発現することを意味する。基底レベルを超える発現は、転写の、翻訳の、翻訳後の、薬理学的な、人工のアップレギュレーション及び過剰発現を含む。基底レベルを超える核内レセプターの発現を、本明細書ではNurr1に関して記載する。この開示は、またNurr1サブファミリーの他のメンバーに対しても当てはめることができ、またそのサブファミリーの他の核内レセプターである、例えば、Nor1やNGFI-Bなどを用いて本発明の方法に使用することができる。このように、Nurr1が例示されてはいるが、本発明の方法はNurr1に限定されず、Nurr1サブファミリーのあらゆる核内レセプターに及ぶ。
【0053】
基底レベルを超えるNurr1の発現は、当業者に周知のいずれの方法によって達成されてもよい。一例として、基底レベルを超える発現は、本来のゲノムNurr1の調節を調節することによって誘導されてもよい。これは、Nurr1 mRNA若しくはタンパク質の分解を阻害すること若しくは妨げることにより、又はNurr1の転写及び/又は翻訳を増加すること、例えば、Nurr1の転写をアップレギュレートする線維芽細胞増殖因子8(FGF8)(Rosenthal, A., (1998) Cell, 93(5),755-766)と細胞とを接触させることにより、及び/又は異種調節配列をNurr1の本来の調節領域の内部若しくはその近傍に導入することにより、及び/又はNurr1の本来の調節領域を、例えば相同的組み換えによって上記のような異種調節配列と置き換えることにより、及び/又はNurr1の転写、翻訳、若しくは機能を負に調節する、妨げる若しくはダウンレギュレートするような分子、例えば、Nurr2(Ohkura, ら, (1999) Biochim Biophys Acta 14444: 69-79)を破壊、若しくはダウンレギュレートすることにより、及び/又はNurr1 mRNA若しくはタンパク質を細胞内に直接マイクロインジェクションすることにより、行われてもよい。
【0054】
転写は、幹、神経幹、前駆体、前駆細胞、若しくは神経細胞に対して、例えば、細胞を転写活性化因子と接触させることにより、あるいはその活性化因子をコードした核酸で細胞を形質転換することによって前記活性化因子のレベルを増大することにより、増加されてもよい。代替方法として、転写はNurr1転写阻害物質に対するアンチセンス核酸で細胞を形質転換することにより増加させてもよい。
【0055】
したがって、幹、神経幹、前駆体、前駆細胞、若しくは神経細胞で神経運命を誘導、又は誘導を増強する本発明の方法は、FGF8やFGF20(Ohmachi ら, 2000)と細胞を接触させることを含んでいてもよい。
【0056】
内在性のNurr1の転写、及び/又は翻訳を増加させる代替法若しくは追加法として、基底レベルを超えるNurr1の発現が、幹、神経幹、前駆体、前駆細胞、若しくは神経細胞内への一又は二以上のNurr1の追加のコピーを導入することによって生じるものであってもよい。
【0057】
したがって、別の局面において、本発明は、幹細胞、神経幹細胞、神経系前駆、前駆体、若しくは神経細胞において神経運命を誘導する、及び/又はドーパミン作動性の発生の誘導を増強する方法、又は神経細胞においてドーパミン作動性の誘導若しくは分化を増強する方法を提供し、該方法は、GSK-3β阻害を調節する物質、例えば、GSK-3β阻害剤、若しくはGSK-3βを阻害する手段と細胞を接触させることに加えて、Nurr1で細胞を形質転換することを含んでいる。
【0058】
幹、神経幹、前駆体、前駆細胞、若しくは神経細胞への形質転換は、in vitro又はex vivoで行うことができる。細胞が誘導される神経運命は、本明細書中で論じられるタイプであればよい。例えば、原始神経表現型を示してもよいし、特定の神経運命に関連するマーカーを欠いていてもよい。さらに本発明は、Nurr1で形質転換され、またGSK-3β阻害と接触した幹細胞、神経幹細胞、若しくは前駆若しくは前駆体細胞を提供する。
【0059】
形質転換されたNurr1は、追加のゲノムベクターに含まれていてもよく、あるいはゲノム内に、好ましくは安定的に組み込まれてもよい。以下でより詳細に述べるように、該Nurr1は、幹細胞、又は神経幹細胞、前駆体若しくは前駆細胞、又は神経細胞内で基底レベルを超える発現を駆動するプロモーターに機能可能なように連結されてもよい。
【0060】
「機能可能なように連結」とは、プロモーターから開始される転写について適当な位置と方向性で、同じ核酸分子の一部として連結していることを意味する。
【0061】
細胞内に遺伝子を導入する方法は、当業者には周知である。ベクターは、Nurr1がベクター上に留まっていようがいまいが、ゲノム内に取り込まれようが、幹、若しくは神経幹、前駆体若しくは前駆細胞、又は神経細胞中にNurr1を導入するために使用されてよい。適切な調節配列を有し、プロモーター配列、ターミネーター断片、ポリA配列、エンハンサー配列を含んだ適当なベクターが選択、又は構築され得る。ベクターは、マーカー遺伝子、及び適当な他の配列を含んでいてもよい。調節配列は、幹、若しくは細胞幹、前駆体若しくは前駆細胞、又は神経細胞内でNurr1の発現、及び/又はGSK-3βの阻害(直接的であろうが、間接的であろうが)を駆動することができる。例えば、ベクターは追加のゲノム発現ベクターでもよいし、又は調節配列がNurr1と共にゲノム内に組み込まれてもよい。ベクターは、プラスミド又はウィルス性のものであってもよい。
【0062】
Nurr1をユーザの制御下に置くために、Nurr1は外部から誘導可能な遺伝子プロモーターの制御下に置かれてもよい。プロモーターに対して使用される用語「誘導可能」は、当業者には十分理解されている。要するに、誘導可能なプロモーターの制御下で発現が「スイッチ オン」され、又は作動刺激に応答して発現が増加されることである。刺激の性質は、プロモーター間で多様である。いくつかの誘導可能なプロモーターは、適切な刺激のない状態では発現(又は非発現)をほとんど生じさせないか、又は検出不能なレベルしか生じさせない。刺激のない状態での発現のレベルがいかなるものであっても、いずれの誘導可能なプロモーターも適当な刺激の存在下で発現が増加され得る。誘導可能なプロモーターの一例としては、遺伝子発現がテトラサイクリン類似物によって調節されるテトラサイクリン オン/オフ系(Gossenら, 1995)がある。
【0063】
さらなる詳細に関しては、例えば、Molecular Cloning: a Laboratory Manual: 3rd edition, Sambrook and Russell, 2001, Cold Spring Harbor Laboratory Pressをご覧いただきたい。核酸操作、例えば、核酸構築の調製、突然変異誘発、配列決定、細胞内へのDNAの導入、そして遺伝子発現、及びタンパク質の解析に関する数多くの既知の技術とプロトコルが、Current Protocols in Molecular Biology, Ausubelら編, John Wiley & Sons, 1992 、又はその後の版に詳細に記述されている。
【0064】
当業者に周知の抗生物質耐性若しくは感受性遺伝子のようなマーカー遺伝子が、目的の核酸を含むクローンを同定するために使用されてもよい。クローンは、同定されるか、又は例えばサザンブロットハイブリダイゼーションによる結合実験によってさらに調べられてもよい。
【0065】
Nurr1を含む核酸は、ホストの幹、神経幹、前駆、前駆体、若しくは神経細胞のゲノム内に組み込まれてもよい。組み込みは、標準的な技術に従ってゲノムとの組み換えを促進する形質転換された核酸配列を含むことで促進されてもよい。組み込まれる核酸は、幹細胞、または神経幹、前駆若しくは前駆体細胞、又は神経細胞でNurr1遺伝子の発現を駆動できる調節配列を含んでいてもよい。核酸は、ゲノム上の一部位であって、幹、若しくは神経幹、前駆体若しくは前駆細胞、又は神経細胞内でNurr1コーディング配列がその発現を駆動、及び/又は制御できる調節エレメントの制御下となるように、その組み込みを導く配列を含んでいてもよい。本明細書中で述べるように、組み込まれる核酸は、幹細胞、又は神経幹、前駆体若しくは前駆細胞、又は神経細胞内でNurr1を形質転換するために使用されるベクター由来であってもよい。
【0066】
Nurr1を含む核酸の導入は、その核酸が直鎖状であろうが、枝分かれ状であろうが、環状であろうが、一般に「形質転換」ということができるが、限定されることはない。該導入には、いずれの利用可能な技術を使用してもよい。適切な技術としては、リン酸カルシウム−トランスフェクション法、DEAE-デキストラン法、PEI、エレクトロポレーション法、マイクロインジェクションのような機械的技術、直接的DNAの取り込み、レセプターを介したDNA転移、レトロウィルス若しくは他のウィルスを用いた形質導入、そしてリポソーム若しくは脂質仲介型トランスフェクション等が挙げられる。選択された遺伝子構築物を細胞内に導入するに際し、当業者に周知の考慮すべきことがある。当業者には明白ではあるが、幹細胞、又は神経幹、前駆体若しくは前駆細胞、又は神経細胞にNurr1を導入するための特定の形質転換法の選択は本発明の本質ではなく、それに限定されるものでもない。
【0067】
幹、又は神経幹、前駆体若しくは前駆細胞、又は神経細胞のNurr1によるin vivoトランスフェクションに関する適切なベクターと技術は、当業者に周知である。適切なベクターには、アデノウィルス、アデノ随伴ウィルス、パポーバウィルス、ワクシニアウィルス、ヘルペスウィルス、レンチウィルス、そしてレトロウィルスが含まれる。無能化ウィルスベクターは、感染ウィルス粒子の生産に必要な遺伝子を発現するヘルパー細胞株内で製造されてもよい。適当なヘルパー細胞株は、当業者には周知である。例として、Fallaux, F.J., ら, (1996) Hum Gene Ther 7(2), 215-222; Willenbrink, W., ら, (1994) J Virol 68(12), 8413-8417; Cosset, F.L., ら, (1993) Virology 193(1), 385-395; Highkin, M.K., ら, (1991) Poult Sci 70(4), 970-981; Dougherty, J.P., ら, (1989) J Virol 63(7), 3209-3212; Salmons, B., ら, (1989) Biochem Biophys Res Commun 159(3), 1191-1198; Sorge, J., ら, (1984) Mol Cell Biol 4(9), 1730-1737; Wang, S., ら, (1997) Gene Ther 4(11), 1132-1141; Moore, K.W., ら, (1990) Science 248(4960), 1230-1234; Reiss, C.S., ら, (1987) J Immunol 139(3), 711-714をご覧いただきたい。ヘルパー細胞株は、通常、ウィルスゲノムをパッケージする機構によって認識される配列を失っている。該細胞株は、核酸を含まないウィルス粒子を生産する。完全なパッケージングシグナルを含むウィルスベクターは、ヘルパー細胞において送達される遺伝子(例えば、Nurr1)や他の配列と共に感染性ウィルス粒子内にパッケージされる。これを幹、又は神経幹、前駆体若しくは前駆細胞、又は神経細胞への遺伝子送達用に利用してもよい。
【0068】
内在性のNurr1の転写、及び/又は翻訳を増加させる代替法若しくは追加として、基底レベルを超えるNurr1の発現は、マイクロインジェクション、又は例えば細胞浸透性ペプチド、TAT、トランスポータン(transportan)、アンテナペディア−ペネトラチンペプチド(Antennapedia penetratin peptides)(Lindsay 2002)を含む、他の担体ベースの、若しくはタンパク質送達のシステムによって、一又は二以上の追加のNurr1タンパク質コピーを幹、神経幹、前駆体、前駆細胞、又は神経細胞内に導入して生じるものであってもよい。
【0069】
さらなる局面において、本発明は、幹細胞、神経幹細胞若しくは前駆若しくは前駆体細胞、若しくは神経細胞において特定の神経運命を誘導する方法であって、該幹細胞若しくは前駆細胞若しくは神経細胞が基底レベルを超えるNurr1を発現する方法を提供する。該方法は、細胞を、例えばGSK-3βの阻害剤のようなGSK-3βの阻害を調節する物質と、又はβ-カテニンの過剰発現、若しくは安定化、若しくは分解の阻害、又はβ-カテニンのリン酸化の阻害を引き起こす物質と、又は記述したような他のGSK-3β阻害誘導物質と、そして場合によっては、タイプIアストロサイト/グリア細胞によって供給される、若しくはそれらから得られる一又は二以上の任意因子と共に、接触させることを含んでいる。因子は、共培養することで、又は幹、前駆、若しくは前駆体細胞、又は神経細胞とタイプIアストロサイト/グリア細胞とを接触させることによって供給されてもよい。この方法は、in vitro、又はin vivoで行ってよい。基底レベルを超えるNurr1を発現する幹細胞、又は神経幹、前駆体、若しくは前駆細胞、又は神経細胞は、Nurr1での細胞の形質転換によって生産されてもよい。
【0070】
因子は、不死化したアストロサイト/グリア細胞によって供給されても、又はそれらから得られてもよい。因子は、グリア細胞株、例えば、アストロサイト、若しくは放射状グリア、若しくは未成熟グリア中脳細胞株によって供給されても、又はそれらから得られてもよい。細胞株は、同種の細胞集団を提供する。
【0071】
本発明の重要な局面は、細胞内での基底レベルを超えるNurr1の発現と、腹側中脳のタイプIアストロサイト/初期グリア細胞によって供給される若しくはそれらから得られる一又は二以上の因子との接触を含むプロセスによって、ドーパミン作動性ニューロンがin vitroにおいて幹細胞、又は前駆若しくは前駆体細胞から発生し得る一方で、ドーパミン作動性の運命の誘導がGSK-3β阻害物質との接触で増強、又は促進されるという発見に基づいている。
【0072】
本発明は、多数のドーパミン作動性ニューロンの発生を可能とする。これらのドーパミン作動性ニューロンは、パーキンソン病で退化したり、又は損傷を受けた若しくは失われた細胞と置換するための原材料として使用することができる。
【0073】
基底レベルを超えるNurr1を発現している細胞は、GSK-3βを阻害するための手段と接触させる際に有糸分裂し得る。
【0074】
本発明の方法において、細胞は付加的に、塩基性繊維芽細胞増殖因子(bFGF)、上皮細胞増殖因子(EGF)、そしてレチノイドX受容体(RXR)の活性化剤、例えば、合成レチノイド類似物SR11237(Gendimenico, G. J., ら, (1994) J Invest Dermatol 102(5), 676-80)、9−シス−レチノール 、又はドコサヘキサエン酸 (DHA)、又は LG849(Mata de Urquiza ら, 2000)、から選択された一又は二以上の物質と接触させてもよい。以下で実験的に立証されるように、本発明に従って細胞を一又は二以上のこれらの物質で処理することは、ドーパミン作動性の運命をとる幹、前駆、若しくは前駆体細胞の割合を増やすため、又は神経細胞におけるドーパミン作動性の誘導若しくは分化を促進するために使用することができる。本発明に従ってドーパミン作動性の運命を誘導する、又は神経細胞でドーパミン作動性の誘導や分化を増強する方法は、成長因子のFGFファミリーの一員、例えば、FGF4、FGF8 又はFGF20と細胞を接触させることを含んでいてもよい。
【0075】
有利な点は、細胞を二以上の上記物質と接触させてもよいことである。本発明者は、図らずもbFGF 又はEGFの有益な効果が飽和量でも相加的であることを発見した。この発見は、これらの物質が異なる機構を介して作用できることを示唆している。
【0076】
ドーパミン作動性表現型を誘導する方法は、幹細胞、神経幹細胞、又は神経前駆若しくは前駆体細胞、又は神経細胞を、GSK-3β阻害を調節するか又はGSK-3βを阻害するための手段(直接的なGSK-3β阻害剤であろうが、間接的にGSK-3を阻害する手段、例えば、β-カテニンの過剰発現、若しくは安定化、若しくは分解の阻害、又はβ-カテニンのリン酸化の阻害であろうが)と、そして任意に、腹側中脳のタイプIアストロサイト/グリア細胞によって供給される、若しくはそれらから得られる一又は二以上のさらなる因子とを接触させる前に、例えば、該細胞を腹側中脳タイプIアストロサイト/グリア細胞若しくはそれらから得られる因子と接触する前、又は共培養する前に、bFGF 及び/又はEGF で前処理することを含んでいてもよい。
【0077】
この任意の前処理ステップは、以前にWO00/66713とWagnerらによる1999年の論文で報告されたさらに二つの予期せぬ発見から生まれた。その発見とは、(i)基底レベルを超えるNurr1を発現及び高い増殖を示す神経幹細胞株が腹側中脳のタイプIアストロサイト/グリア細胞と共培養されるとドーパミン作動性の運命への誘導が促進されることが立証されたこと、そして(ii)無血清培地(SFM)内でのbFGF 及び/又はEGF処理後、SFMのみに移した後でも、基底レベルを超えるNurr1を発現しているほとんどの幹細胞株が高い増殖のベースラインを維持していたこと、である。
【0078】
ドーパミン作動性表現型の誘導方法は、幹細胞、又は神経幹、若しくは前駆、若しくは前駆体細胞を成長因子のFGFファミリーの一員、例えば、FGF2、FGF4、 FGF8、又はFGF20で前処理することを含んでいてもよい。
【0079】
細胞は、付加的に一又は二以上のWntリガンド、例えば、WO2004/029229に記載されているようなもので処理されてもよい。
【0080】
神経運命を幹、神経幹若しくは前駆若しくは前駆体細胞で誘導する、又は神経細胞でドーパミン作動性の誘導や分化を増強する本発明に係る方法は、神経運命用のマーカーを検出することを含んでいてもよい。β-チューブリンIII(TuJ1)は神経運命のマーカーの一つである(Menezes, J. R., ら, (1994) J Neurosci 14(9), 5399-5416)。他の神経マーカーとしては、神経フィラメントとMAP2がある。もし、特定の神経表現型が誘導されるのならば、マーカーはその表現型に特異的なものにすべきである。ドーパミン作動性の運命については、チロシン水酸化酵素(TH)、ドーパミントランスポーター(DAT)、そしてドーパミンレセプターの発現が、例えば、免疫反応性やin situ ハイブリダイゼーションによって検出されればよい。チロシン水酸化酵素は、DA細胞用の主要なマーカーである。ドーパミンと代謝産物の量、及び/又は放出は、例えば、高圧液体クロマトグラフィー(HPLC)(Cooper, J. R., ら, The Biochemical Basis of Neuropharmacology, 7th Edition, (1996) Oxford University Press)によって検出されればよい。(TH/ドーパミン/DATの存在下で)ドーパミンβ水酸化酵素、及びGABA又はGADの欠如も、またドーパミン作動性の運命を表す。付加的なマーカーとしては2型アルデヒド脱水素酵素(ADH-2)、GIRK2、 Lmx1b、そしてPtx3がある。
【0081】
マーカーの検出は、当業者に周知のいずれの方法に従って行われてもよい。検出方法は、マーカーをコードしている核酸配列に結合することができる特異的結合メンバーを使用することができ、この特異的結合メンバーは、その配列とハイブリダイズできる核酸プローブ又は核酸配列若しくはそれによってコードされたポリペプチドに対して特異性をもつ免疫グロブリン/抗体ドメインを含んでおり、配列やポリペプチドへの特異的結合メンバーの結合を検出できるように標識されていてもよい。「特異的結合メンバー」は、マーカーに対して特別な特異性を有しており、通常条件では、他の種に優先してそのマーカーに結合する。別の方法として、マーカーが特異的なmRNAである場合、特異的なオリゴヌクレオチドプライマーに対する結合、及び例えばポリメラーゼ連鎖反応(PCR)で増幅することによって検出してもよい。
【0082】
核酸プローブとプライマーは、ストリンジェントな条件下でマーカーとハイブリダイズしてもよい。適切な条件としては、例えば、約80−90%の同一性をもつマーカー配列の検出に関してであれば、ハイブリダイゼーションが0.25M Na2HPO4(pH7.2),6.5% SDS,10%デキストラン硫酸溶液中にて42℃で一晩処理された後、0.1×SSC,0.1%SDS溶液中にて、55℃で最終洗浄することが挙げられる。90%以上の同一性があるマーカー配列の検出に関して、ハイブリダイゼーションの適切な条件は、0.25M Na2HPO4(pH7.2),6.5%SDS,10%デキストラン硫酸溶液中にて65℃で一晩処理され、0.1×SSC,0.1%SDS溶液中にて、60℃で最終洗浄することが挙げられる。
【0083】
別の局面において、本発明は、本明細書中に開示したいずれか一の方法に従って製造された神経を提供する。神経は原始的な神経表現型を有していてもよい。該神経は、複数の異なる神経表現型を生じることができてもよい。神経は、特定の神経表現型を有していてもよい。その表現型は、基底レベルを超えるNurr1を発現している幹、神経幹、前駆、前駆体、若しくは神経細胞と接触する因子を供給した、又はそれらの因子が得られたアストロサイト/グリア細胞のタイプによって、及び/又は幹、神経幹、前駆、前駆体、若しくは神経細胞が共培養、又は接触したアストロサイト/グリア細胞のタイプによって影響される。好ましい実施形態においては、神経がドーパミン作動性表現型を有する。
【0084】
神経は、神経内で分子の発現を駆動できるプロモーターに機能可能なように連結された神経保護若しくは神経修復の性質をもつ分子をコードする核酸を含んでいてもよい。プロモーターは、例えば、TetONキメラプロモーターといった誘導可能なプロモーターであることができ、それにより、いかなる損傷性の過剰発現も防ぐことができる。プロモーターは特定の神経表現型と関連するもの、例えば、THプロモーターやNurr1プロモーターであってもよい。
【0085】
コードされた分子は、その発現により神経がその環境から独立することになるようなものでよく、すなわち、その生存が、例えば、それが移植される神経環境下における一又は二以上の因子又は状態の存在に依存しないようにすることができる。例として、神経は、神経内で分子の発現を駆動できるプロモーターに機能可能なように連結された、以下に記載する一又は二以上の神経保護、若しくは神経修復性分子をコードする核酸を含んでいてもよい。
【0086】
追加若しくは代替法として、コードされた分子の発現が、その神経周辺の細胞環境の神経保護、若しくは神経修復に機能してもよい。この方法において、神経は、神経保護と神経修復の性質をもった分子を送達するようにする細胞遺伝子併用療法において使用されてもよい。
【0087】
神経保護と神経修復の性質をもつ分子の例として、以下のものが挙げられる。
【0088】
(i)神経変性を補償し、それを阻害することのできる神経栄養因子。一例は、ドーパミン作動性ニューロンからの発芽を促進し、チロシン水酸化酵素発現を増加する有効性の高い神経生存因子であるグリア由来神経栄養成長因子(GDNF)(Tomacら,(1995) Nature, 373, 335-339; Arenasら, (1995) Neuron, 15,1465-1473)である。GDNF軸策伸張増強により、GDNFは、それらが局所する環境に神経を分布させるために神経の能力を増大してもよい。GDNFファミリーのその他の神経栄養分子には、ニュールツリン(Neurturin)、ペルセフィン(Persephin)、そしてアルテミン(Artemin)が含まれる。ニューロトロフィンファミリーの神経栄養因子には、神経成長因子(NGF)、脳由来神経栄養因子(BDNF)、そしてニューロトロフィン−3、−4/5、及び−6がある。神経栄養活性をもつ他の因子には、例えば、FGF2、4、8、及び20といったFGFファミリーのメンバー、Wnt-1、-2、-5a、-3a、及び7aを含むWntファミリーのメンバー、BMP2、4、5、及び7、nodal、アクチビン、並びにGDFを含むBMPファミリーのメンバー、TGFα/βファミリーのメンバー等がある。
【0089】
(ii)抗アポトーシス性分子: 細胞死において中心的な役割を果たすBcl2。Bcl2の過剰発現は本来的に生じる細胞死と虚血(Martinouら, (1994) Neuron, 1017-1030)から神経を保護する。その他の神経に対する抗アポトーシス性分子には、BclX-Lがある。
【0090】
(iii)神経が分布し、その環境での連絡の形成する上で神経を援助する、例えばエフリンのような、軸索の再生、及び/又は伸張及び/又はガイドをする分子: エフリンは、チロシンキナーゼレセプターを活性化できる膜結合型リガンドのクラスであることが明らかとなっている。エフリンは、神経発生に関わっている(Irvingら, (1996) Dev. Biol., 173, 26-38; Krullら, (1997) Curr. Biol. 7, 571-580; Frisenら, (1998) Neuron, 20, 235-243; Gaoら, (1996) PNAS, 93, 11161-11166; Torresら, (1998) Neuron, 21, 1453-1463; Winslowら, (1995) Neuron, 14, 973-981; Yueら, (1999) J Neurosci 19(6), 2090-2101)。軸索伸張に関連するその他の分子のファミリーに、ネトリン、セマフォリン、そしてスリット(Nakamotoら Curr Biol. 2004 Feb 3;14(3):R121-3)などがある。
【0091】
(iv)転写因子、例えば、ホメオボックス ドメイン タンパク質Ptx3(Smidt, M. P.ら, (1997) Proc Natl Acad Sci USA, 94(24), 13305-13310)、Lmx1b、 Pax2、 Pax5、Pax8、若しくはengrailed 1若しくは2(Wurst and Bally-Cuif, 2001; Rhinn and Brand, 2001)、又はベーシック へリックス−ループ−へリックス ファミリーの神経原性遺伝子。
【0092】
本発明に従った、又は該発明で使用する神経は、一又は二以上の他の細胞タイプ、例えば幹、神経幹、前駆体若しくは前駆細胞、または神経細胞を、実質上含まない。神経は、抗体と磁気ビーズ、又は蛍光標識細胞分取装置(FACS)による細胞外エピトープの認識に基づくものを含めて、当業者に周知のいずれかの技術を用いて神経幹若しく前駆細胞から分離されてもよい。例として、幹、神経幹、前駆体若しくは前駆細胞では見られるが、神経では見られない分子の細胞外領域に対する抗体を使用してもよい。このような分子には、Notch 1、CD133、SSEA1、prominin1/2、RPTPb/phosphocan、TIS21、及びグリア細胞株由来神経栄養因子レセプターGFRα、又はNCAMが含まれる。抗体と結合する幹細胞は、補体曝露によって溶解されても、又は、例えば、磁気選別によって(Johanssonら, (1999) Cell, 96, 25-34)分離されてもよい。もしも、目的のレシピエントに対して異種の抗体が使用されたのであれば、そのときは、いずれも、例えば、そのような細胞選別方法を免れた幹、神経幹若しくは前駆若しくは前駆細胞も、異種抗体で標識され、そしてレシピエントの免疫機構の一次標的となる。別の方法として、希望する表現型を獲得する細胞は、細胞外エピトープに対する抗体、又は細胞タイプ特異的プロモーターの制御下にある蛍光タンパク質を含む導入遺伝子の発現によっても、分離することができる。例として、ドーパミン作動性ニューロンは、TH、DAT、Ptx3、Nurr1、又はドーパミン作動性ニューロンにより特異的に利用される他のプロモーターの制御下で発現した蛍光タンパク質で分離できる。
【0093】
本発明の方法は、希望する表現型をもった神経幹、前駆若しくは前駆体細胞、又は他の幹細胞、または神経細胞を富化するために、付加的なネガティブ又はポジティブ選択法を含んでいてもよい。
【0094】
ネガティブ選択は、DAニューロンを集めるために使用されてもよい。非DAニューロン用の選択的な神経毒が使用されてもよい。例えば、5−7−ジヒドロキシトリプタミン(5-7-dihydroxytryptamine)(セロトニン作動性ニューロン除去用)、又はサポニン若しくは毒素に結合した、若しくは補体追加後に一緒にされる抗体、例えば、GABA、又はセロトニン トランスポーターに対する抗体(GABA作動性、又はセロトニン作動性ニューロン除去用)が挙げられる。本発明の方法は、神経幹、前駆若しくは前駆細胞、又は他の幹、若しくは神経細胞を、好ましくはin vitroで、例えば、細胞を含むin vitro 培養物にネガティブ選択剤を添加することにより、又はネガティブ選択剤の存在下で細胞を培養することにより、ネガティブ選択剤にて処理すること、又は接触させることを付加的に含んでいてもよい。ネガティブ選択物質は、希望する細胞タイプ以外の細胞タイプに対して選択する。例えば、本発明が、ドーパミン作動ニューロン表現型を促進すること、増強すること、又は誘導することに関連する場合、ネガティブ選択剤は、DAニューロン以外の細胞、並びにDAニューロンを生じる細胞、例えば、幹細胞、及び神経幹、前駆体及び前駆細胞のような細胞に対して選択されてもよい。したがって、ネガティブ選択剤は、非DA表現型をもつ、例えば、非DAニューロンといった、分化した細胞に逆らって、選択してもよい。ネガティブ選択剤は、希望する細胞タイプ以外の細胞の増殖を減少、又は阻害、及び/又は殺してもよい。ネガティブ選択剤は、DAニューロン以外の細胞ニューロンの母数を減少する選択的神経毒素であってもよい。例えば、ネガティブ選択剤は、5−7−ジヒドロキシトリプタミン(セロトニン作動性ニューロンを減少するため)であってもよい。ネガティブ選択物質は、非DAニューロンに対する抗体、若しくは抗体断片であってもよい。該抗体や該抗体断片(例えば、scFvやFab)は、サポニン又は毒素に結合される。例えば、抗体は、GABAトランスポーター(GABA作動性ニューロンを減少するため)に特異的なものであってもよい。
【0095】
本発明の方法において、神経幹、前駆若しくは前駆細胞、又は他の幹若しくは神経細胞は、抗酸化剤(例えば、アスコルビン酸)、低酸素分割、及び/又は低酸素誘導因子(例えば、HIF又はエリスロポエチン)存在下で育ててもよい。
【0096】
本発明はさらに多様な局面と実施形態で、例えばパーキンソン症候群やパーキンソン病をもつ個体の治療において、物質、又は、もしその物質が自然界でペプチド(ペプチド、ポリペプチド、又はタンパク質)であるのならばそれをコードする核酸を個体に投与して、内在性の、若しくは外来的に供給される幹、前駆若しくは前駆体細胞、又は神経細胞のいずれかに作用することにより脳内でドーパミン作動性ニューロンの発生を誘導させ、促進させ、又は増強させ、及び/又は、欠損を阻害し、若しくは妨げ、又は生存、若しくは形態的分化、若しくは成熟化、又はドーパミン作動性ニューロンの神経突起生成、若しくはシナプス形成、又は機能的アウトプットを促進することを含む治療方法における、GSK-3βを阻害する物質、例えばGSK-3β阻害剤の使用を提供する。物質は、例えば、薬学上許容できる賦型剤や担体を含むどんな適当な組成物で投与されてもよい。また、神経変性疾患、パーキンソン症候群、又はパーキンソン病の治療用の薬剤製造に使用されてもよい。物質、又はそれをコードしている核酸は、中枢神経系、及び/又は脳に投与されるか、又はそれらを標的としてもよい。
【0097】
本発明は、様々な局面において、本明細書中で開示されたいずれか一つの方法に従って製造された神経だけでなく、そのような神経、幹、前駆、若しくは前駆細胞、及び/又はGSK−3βの阻害調節をする、例えばGSK−3β阻害剤のような物質を含む医薬組成物、薬剤、薬、又はその他の組成物、そのような神経、幹、前駆、若しくは前駆体細胞、又は神経細胞及び/又は物質若しくは組成物の医療方法における使用、例えば、パーキンソン病や他の(例えば神経変性の)疾患の治療(予防療法を含む)のための、そのような神経、幹、前駆若しくは前駆体細胞又は神経細胞及び/又は物質若しくは組成物の患者に対する投与を含む方法、例えば、パーキンソン病その他(例えば、神経変性疾患)の治療のための投与用組成物の製造でのそのような神経又は細胞及び/又は物質の使用、並びに、そのような神経又は細胞及び/又は物質を、薬剤上許容できる賦形剤、媒体、又は担体と、また、任意で一又は二以上の他の成分、例えば、神経保護分子、神経修復分子、レチノイド、成長因子、アストロサイト/グリア細胞、抗アポトーシス因子、又は、幹、前駆若しくは前駆体細胞又は神経細胞で、又はホスト脳内で遺伝子発現を調節する因子と、を混ぜ合わせることを含む医薬組成物の製造方法、に及ぶ。そのような任意の成分は、その環境から神経を独立にすることができる。例えば、その生存が、その環境における一又は二以上の因子又は条件の存在に依存しないようにすることができる。例として、医薬組成物を製造する方法は、腹側中脳の発生において見られる一又は二以上の因子と神経を混ぜることを含んでいてもよい。神経は、GDNF及び/又はニューツリン(NTN)と混ぜてもよい。医薬組成物を製造する方法は、腹側中脳の発生において見られる一又は二以上の因子と共に神経内でBcLXLを発現させることを含んでいてもよい。
【0098】
本発明は、本発明に従って生産された神経、幹、前駆若しくは前駆体細胞、又は神経細胞、及び/又は、GSK-3β阻害で作用する物質、例えば、GSK-3β阻害剤、及び一又は二以上の付加的な成分を含む組成物を提供する。本発明に係り、かつ本発明に従って使用される医薬組成物は、神経若しくは細胞に加えて、薬剤上許容できる賦形剤、担体、バッファー、保存料、安定化剤、抗酸化剤、または、当業者に周知の他の材料を含んでいてもよい。そのような材料は当然無毒であるべきで、かつ神経活性を妨害すべきではない。キャリーや他の材料の正確な性質は投与の経路に依存する。組成物は、一又は二以上の神経保護分子、神経修復分子、レチノイド、成長因子、アストロサイト/グリア細胞、又は、幹、神経幹、前駆体若しくは前駆細胞、又は神経細胞において遺伝子発現を調節する因子を含んでいてもよい。そのような物質は、上述したように、神経をその環境から独立させ得る。
【0099】
液体医薬組成物は、通常水、石油、動物性、若しくは植物性油、鉱物油、または合成油などの液体の担体を含む。生理的食塩水、組織若しくは細胞培養培地、細胞外基質タンパク質、ヘパラン、コンドロイチン硫酸、デキストロース若しくは他の糖質液、又はエチレングリコール、プロピレングリコール、ポリエチレングリコール等のグリコールが含まれていてもよい。
【0100】
組成物は、発熱物質フリーで、適切なpH、等張性、及び安定性を有する非経口許容水溶液の形状であってもよい。当業者であれば、例えば、塩化ナトリウム、リンゲル輸液、又は乳酸リンゲル輸液等の等張媒体を用いて適切な溶液を調製できる。組成物は、人工脳脊髄液を用いて調製してもよい。
【0101】
本発明は、本発明に従って製造された神経、及び/又は、GSK-3β阻害物質(例えば、GSK-3β阻害剤)の、医学療法、特に神経細胞における変性、損傷、欠損、疾患に関連した病状の治療における使用に及ぶ。さらに、本発明は、特定の表現型の神経、及び/又はGSK-3β阻害を調節する、又は与える物質(例えば、GSK-3β阻害剤)の、その表現型の神経の再生、損傷、又は欠損に関連する状態、病気、又は疾患の治療における使用を提供し得る。より特に、本発明はヒトパーキンソン病の治療におけるドーパミン作動性ニューロン、及び/又はGSK-3β阻害物質の使用を提供する。本発明は、特に神経変性病(例えば、パーキンソン病)の治療用の材料と方法に関連するとはいえ、それに限定はされない。例として、本発明は、脊髄、及び/又は大脳皮質の変性、若しくは損傷の治療、又はNurr1+細胞を含む神経系の他の領域の治療に及ぶ。
【0102】
投与した細胞が二以上の異なる神経表現型を生じることのできる幹、前駆、若しくは前駆体である治療方法では、神経、細胞、及び/又はWntリガンド又は組成物は、希望する特定の神経運命への細胞分化を指令するアストロサイト/グリア細胞を含む領域に導入されてもよい。細胞、及び/又はWntリガンド又は組成物は、例えば、腹側中脳にインジェクションされてもよい。そこで細胞はタイプIアストロサイト/グリア細胞と相互作用し、ドーパミン作動性表現型をとるように誘導されてもよい。別途、又は加えて、移植される組成物は、上述のように、その発生を特定の神経運命に向かわせる一又は二以上の因子と、例えば、タイプIアストロサイト/グリア細胞と組み合わせた神経、又は細胞を含んでいてもよい。
【0103】
細胞は、当業者において知られる技術(例えば、Lindvall, O., (1998) Mov. Disord. 13, Suppl. 1:83-7、Freed, C.R.ら, (1997) Cell Transplant, 6, 201-202; Kordowerら, (1995) New England Journal of Medicine, 332, 1118-1124; Freed, C.R.,(1992) New England Journal of Medicine, 327, 1549-1555)によって患者に移植されてもよい。また、Freedら J Neurol. 2003 Oct;250 Suppl 3:III44-6; Dunnettら (2001) Nat Rev Neurosci. 2001, 2(5):365-9; Lindvall and Hagell (2000) Prog Brain Res. 2000, 127:299-320; Mendez ら J Neurosurg. 2000 May;92(5):863-9; Hagellら. Mov Disord. 2000 Mar;15(2):224-9; Hauserら. Arch Neurol. 1999 Feb;56(2):179-87; Lindvall Mov Disord. 1998;13 Suppl 1:83-7. Review.; Olanowら. Trends Neurosci. 1996 Mar;19(3):102-9. Review; Remyら. Ann Neurol. 1995 Oct;38(4):580-8; Kordowerら. N Engl J Med. 1995 Apr 27;332(17):1118-24; Lindvallら. Ann Neurol. 1994 Feb;35(2):172-80; Hofferら. Exp Neurol. 1992 Dec;118(3):243-52; Lindvallら. Arch Neurol. 1989 Jun;46(6):615-31をご覧いただきたい。
【0104】
本発明に従った組成物の投与は、好ましくは、個体に対して有益性を示すのに十分な「予防上有効な量」又は、「治療上有効な量」(場合によっては、予防法は治療法とみなしてもよいが、)で行う。投与される実際の量、及び、割合、投与の時間経過は、何を治療するかの性質とひどさによる。治療処方、例えば、容量の決定は、一般の開業医や他の医師の責任の範囲にある。
【0105】
組成物質は、単独で、又は他の治療との組み合わせで、治療状態によって同時か、あるいは連続して投与してもよい。
【0106】
本明細書で提供される方法は、原材料としてin vivoで、若しくはin vitroで、若しくは細胞株で、初代細胞を使用して実施してもよい。in vitroで増やした細胞の有利な点は、実質上、製造される神経の数に限界がないことである。
【0107】
移植された細胞の免疫学的拒絶に関連して起こり得る不都合を改善する目的で、幹、若しくは前駆、若しくは前駆体細胞を、患者から分離し、希望の表現型に誘導してもよい。その後、細胞を患者に移植してもよい。有利な点として、分離された幹、若しくは前駆、若しくは前駆体細胞は、多数の免疫適合性神経細胞が製造されるように、細胞株の確立に使用されてもよい。さらには、個々の患者のために適切な細胞選択が可能な免疫学的適合性の範囲をカバーする細胞バンクを任意で設立する。ある個体由来の幹、神経幹、前駆体若しくは前駆細胞、又は神経細胞は、該細胞若しくはその子孫が第二の個体に導入される時に、拒絶を改善するために変更されてもよい。例として、ドナー細胞の一又は二以上のMHCアレルを、例えば、相同的組換えによってレシピエントのそれと置換してもよい。免疫抑制処理が患者に対して施されてもよい。
【0108】
もし、不死化癌遺伝子を有する細胞株由来の細胞が患者への移植に使用されたならば、癌遺伝子は、その細胞を患者に移植する前にCRE-loxPシステムを用いて除くことができる(Westerman, K. A. ら Proc. Natl. Acad Sci. USA 93, 8971 (1996))。患者の体温では不活性な不死化癌遺伝子を用いてもよい。
【0109】
本発明で使用される幹、神経幹、前駆体、前駆、若しくは神経細胞として、胚性幹細胞、胎児幹細胞、成人骨髄若しくは造血幹細胞、メサンギオブラスト(mesangioblast)及びH6(Flaxら Nature Biotech 16 (1998))等のヒト細胞株がある。さらなる例は、Gageら. Ann. Rev. Neurosci. 18, 159-192 (1995) and Gottlieb 2002に列挙されている。
【0110】
本考察は、神経幹細胞、又は神経前駆若しくは前駆体細胞に関して行われたが、本明細書中で提供される方法は、他の幹、前駆若しくは前駆体細胞で、神経運命の誘導に応用されてもよい。そのような細胞例には、非神経系に関連する幹細胞が含まれる。本方法は、間質若しくは造血幹細胞、及び/又は、表皮由来の増殖性細胞に応用されてもよい。造血細胞は血液、又は骨髄生検から回収されてもよい。間質細胞は、骨髄生検から回収されてもよい。上皮細胞は、皮膚生検により、若しくは掻き取り(例えば口腔粘膜からの)により回収されてもよい。神経表現型は、in vivoでは、それらの幹、前駆、若しくは前駆体細胞の生理学的運命でないので、この誘導プロセスは、分化転換、又は脱分化と神経再分化といってもよい。そのような細胞を神経運命に誘導する方法は、非神経表現型と関連する遺伝子、例えば、それらの細胞の分化を非神経運命に向かうことを抑制する、及び/又は反対にする遺伝子に対するアンチセンスレギュレーターの使用を含んでいてもよい。
【0111】
本発明の方法は、神経運命になることが決まっていない幹細胞に応用されてもよい。本方法は、異なる分化機構に関連する二以上の嬢幹細胞を生じ得る幹細胞に応用されてもよい。そのような幹細胞の例として、胚性幹細胞、造血幹細胞、骨髄間質細胞。表皮由来の増殖細胞、及び神経幹細胞が挙げられる。
【0112】
上記で論述したように、GSK-3β阻害、及び/又は腹側中脳タイプIアストロサイト、又はグリア細胞から得られた一又は二以上の因子と組み合わせて、基底レベルを超えるNurr1の発現を必要とするプロセスによりドーパミン作動性ニューロンが、幹、前駆、若しくは前駆体細胞から生じ得るということを、本開示は実証している。
【0113】
さらなる多様な局面において、本発明は、神経幹、又は前駆若しくは前駆体細胞でドーパミン作動性運命の誘導を増強する、あるいは、例えばGSK-3β阻害剤のようにGSK-3βの阻害を調節する手段として、又は例えばβ−カテニンの過剰発現若しくは安定化若しくは分解阻害、若しくはβ−カテニンのリン酸化の阻害を生じさせることでGSK-3βを間接的に阻害する手段として機能することによって、基底レベルを超えるNurr1を発現している神経細胞においてドーパミン作動性の誘導、若しくは分化を増強する1以上の因子をスクリーニングするアッセイ、及び方法の供給、またそれによって同定された1以上の因子と関係している。
【0114】
本発明は、直接的又は間接的に、GSK−3βを阻害できる、あるいはその他、GSK−3βの阻害を調節でき、かつ単独で又は組み合わせて、基底レベルを超えるNurr1を発現している幹、神経幹、又は前駆若しくは前駆体細胞、又は神経細胞におけるドーパミン作動性の運命を増強、増加、又は誘導を強化できる1以上の因子をスクリーニングする方法を提供する。本発明のさらなる局面は、基底レベルを超えるNurr1を発現する幹、神経幹、又は前駆若しくは前駆体細胞、又は神経細胞の、そのような幹、前駆若しくは前駆体細胞、又は神経細胞等においてドーパミン作動性の運命の誘導を増強するGSK-3βを阻害する、またはその阻害を調節する手段をスクリーニングし、又は探索し、及び/又は、入手し/同定することにおける使用を提供する。
【0115】
試験物質は、まずGSK-3βの阻害を調節する能力について、例えば、GSK-3を阻害すること、すなわち、GSK-3βの直接の阻害剤であるか、又はβ−カテニンの過剰発現若しくは安定化若しくは分解阻害、若しくはβ−カテニンのリン酸化の阻害を起こし得るかどうかという能力について、試験することができる。ひとたび能力を有することが同定されたら、本明細書で開示したように、その物質を、基底レベルを超えるNurr1を発現している幹、神経幹、または前駆若しくは前駆体細胞、又は神経細胞において、ドーパミン作動性の運命の増強、増加、又は誘導の強化の能力について試験することができる。
【0116】
スクリーニング方法の一部として、試験物質を、まずGSK-3β、若しくはβ−カテニン、グリコーゲンシンターゼ、tau、elf2B、 CREB、又はc-junなどの基質との結合、又は相互作用について試験することができる。
【0117】
結合又は相互作用は、当業者に知られる多数の技術のいずれかによって定性又は定量的に測定することができる。試験物質と幹、若しくは前駆、若しくは前駆体細胞間の相互作用は、検出可能な標識でどちらか一方を標識し、固体支持物上に固定化しておいてもよい他方と接触させることにより、例えば、固体支持物に結合した抗体を用いることにより、又はBiacore systemを始めとしてそれ自体が知られている他のテクノロジーを介することによって、調べることができる。
【0118】
スクリーニング方法は、1種以上の試験物質の存在下で、幹、神経幹、又は前駆若しくは前駆細胞、又は神経細胞を培養すること、及びドーパミン作動性表現型への分化について、例えば、本明細書中で考察したように、ドーパミン作動性表現型のマーカーを検出することによって、細胞を分析することを含んでいてもよい。チロシン水酸化酵素(TH)は、ドーパミン作動性表現型のマーカーの一つである。他のドーパミン作動性表現型マーカーには、ドーパミントランスポーター(DAT)、ドーパミンレセプター、アミノ酸脱炭酸酵素(AADC)、核内レセプターNurr1、ホメオボックスPtx3、及びLmx1bの発現などがある。神経フィラメントやβ−チューブリンIIIのような神経マーカーも、先のマーカーの一つに加えて細胞により発現されるべきである。ドーパミン作動性ニューロンを検出する他の方法として、ドーパミンの放出、及び例えば、GFPのルシフェラーゼと結合したTHプロモーターなどのレポーターシステムの検出がある。
【0119】
ドーパミン作動性ニューロンの検出のためのこれらのマーカーと方法は、本発明のいずれの局面と実施形態においても、ドーパミン作動性の運命誘導がうまくいったことを決定するのに有用であることに注意すべきである。
【0120】
本発明に従ってスクリーニングされる物質はいずれも、天然、又は合成の化合物であってもよい。
【0121】
活性の尺度としてのGSK-3βによるリン酸化は、当業界で利用できる多様な技術のいずれを用いて測定してもよい。
【0122】
本発明のさらなる局面においては、以下のものを含むアッセイ法が提供される。
【0123】
(a)GSK-3βの存在下で、GSK−3βに対するリン酸化部位、例えばグリコーゲンシンターゼではSer652とSer 648、β―カテニンではSer37 とThr41、elF2BではSer535、CREBではThr129、c-JUNではThr239を含む基質と試験物質とをGSK-3βが前記基質を普通にリン酸化する条件下で接触ようにすること、そして
(b)前記基質のリン酸化を測定すること。そのような方法により、GSK-3β阻害で機能する物質を同定できる。
【0124】
リン酸化は、例えば基質をビーズやプレート上などに固定することによって、そして部位がリン酸化されているときと部位がリン酸化されていないときとで異なる親和性をもつ関連するリン酸化部位に結合する抗体、又は他の結合分子を用いてリン酸化を検出することによって測定してもよい。このような抗体は、いずれの標準的な技術的手段によって得られてもよい。例えば、リン酸化ペプチドの使用が挙げられる。基質のリン酸化形態と非リン酸化形態とを識別する結合分子の結合については、当業者が利用できるいずれの技術を用いて評価してもよい。また、それは蛍光等の適当な標識の有無を測定することと関連していてもよい。リン酸化は、例えばシンチレーターを含浸したビーズやプレート上などに基質を固定することにより、標準シンチレーションプロキシメトリーアッセイ等で、放射性リン酸の取り込みを測定することで決定されるリン酸化により測定されてもよい。基質へのリン酸の取り込みは、トリクロロ酢酸のような酸での沈殿とニトロセルロースフィルター紙のような適当な材料上への沈殿物の回収に続いて放射標識したリン酸の取り込みの測定により測定されてもよい。基質のSDS−PAGE分離を使用した後に放射標識の検出を行ってもよい。
【0125】
GSK-3β阻害のスクリーニングに関する他のアプローチには、以下の一又は二以上のいずれかの考察が含まれる。
【0126】
(i)野生型β−カテニン、又は例えば、蛍光標識若しくは酵素のような標識若しくはレポーターでタグ付けされたβ−カテニンの安定化、又は核内移行。
【0127】
(ii)既知のβ−カテニン標的遺伝子の発現調節についての解析。そのような遺伝子のいくつかは、その調節領域にTCF/LEFファミリーのメンバーのためのコンセンサスな結合部位を含んでいる。これらには、c-myc、Cyclin D、Tcf-1、PPARdelta、c-jun、fra-1、uPAR、matrix metalloproteinase MMP-7、Axin-2、Nr-CAM、ITF-2、Gastrin、CD44、EphB/ephrin-B、BMP4、claudin-1、Survivin、VEGF、FGF18、FoxN1、matrix metalloproteinase-26、Frizzled 7、Follistatin、Id2、siamois、twin、Xnr3、fibronectin、myogenic bHLH、engrailed-2、connexin43、connexin 30、retinoic acid receptor gamma、dharma/bozozok、Cdx4、nacre、Stra6、Wrch-1、TNF family 41BB ligand、ephrinB1、Stra6、autotaxin、及びISLR、Twist、Stromelysin、Brachyury、Proglucagon、Osteocalcin、cyclooxygenase-2、Irx3、Six3、neurogenin 1、WISP-1、WISP-2、IGF-II、Proliferin-2、Proliferin-3、Emp、IGF-I、VEGF-C、MDR1、COX-2、IL-6、Periostin、Cdx1、betaTrCP、sFRP-2、Pitx2、Eda (TNF-related)、E-cadherin、Keratin、movo1、mBTEB2、FGF4、ret、Ubx、wingless、Dpp、Engrailed-1、Dfrizzled2、shavenbaby、stripe、Nemo、が含まれるが、これらに限定はされない(例えば、いずれかのウェブブラウザで閲覧可能な伝達経路の標的を記載したスタンフォードウェブサイトhttp://www.stanford.edu/~rnusse/pathways/targets.html updated in June 2004を参照いただきたい)。これらの遺伝子の調節は、β−カテニンタンパク質のレベルを生化学的、及び/又は免疫学的な方法で測定すること、又はハイブリダイゼーション、又は/及びPCRによってNurr1 mRNAレベルを測定すること、又は組換えプロモーター/レポーター遺伝子発現システムを用いてNurr1遺伝子発現のレベルを測定することを含む広く多様な技術によってモニターされてもよい。この後者のアプローチは、Nurr1の内在性の発現を調節するために必要とされるシス調節エレメントにレポーター遺伝子を機能可能なように連結したようなものからなるDNA構築物を薬物スクリーニングのために使用される細胞内部で発現することに関連している。
【0128】
(iii)β−カテニンレスポンスエレメントを含む、又はレポーター遺伝子若しくは複数のレポーター遺伝子に連結された構築物の調節。これによって、発現レベルの測定に代わり、又はそれと共同でβ−カテニンの特異的な転写促進の活性をモニタリングすることが可能となる。これによって、特定の処理がGSK-3β阻害を生じるか、又はドーパミン作動性表現型の獲得に影響するかを測定するための、より速く、かつ高度な処理の方法が可能となる。使用のできるレポーター構築物は、生物発光性タンパク質、または酵素活性、ルシフェラーゼ・クロラムフェニコール・アセチルトランスフェラーゼ、β−ガラクトシダーゼ、β−ラクタマーゼ、若しくは他の抗生物質耐性遺伝子、ニューロトロフィン、又はサイトカイン等を含んでいてもよい。
【0129】
(iv)遺伝子の発見。β−カテニン活性は、おそらくTH遺伝子をより強くアップレギュレートする。β−カテニン活性化は、おそらくドーパミン作動性表現型の特定化に関与する下流の遺伝子のいくつかの発現レベルを変化させる。そのような多数の遺伝子は以前に十分に同定され、若しくは特徴付けられたかもしれないが、いくつかについては、未だ同定されていないかもしれない。さらに、たとえ同定されていたとしても、ドーパミン作動性表現型を特定化するそれらの機能は、従来十分には理解されていなかったかもしれない。これらの遺伝子は、ドーパミン作動性ニューロンの欠損を補償するための治療のための未だ知られていない標的となるかもしれない。標的遺伝子の同定は、細胞(幹、前駆、前駆体、Nurr1を発現している、若しくは神経の細胞など)において、β−カテニンを恒常的に、又は誘導可能なプロモーターの下で過剰発現すること、又は内在性の、若しくは人工のβ−カテニン遺伝子の発現を誘導することによって調べられてもよい。刺激細胞と非刺激細胞とで、遺伝子発現、既知タンパク質の翻訳後修飾、イオンチャネル機能、又は特定分子の細胞内局在について評価することにより、解析は可能である。技術には、サブトラクティブ・ハイブリダイゼーション法、ディファレンシャル・ディスプレー法、SAGE、生化学的分画、電気生理学、細胞化学的技術が含まれていてもよい。Wnt分野で使用される技術には、Xenopus(アフリカツメガエル)での二次軸形成、体節筋形成、腎臓上皮誘導、軸索リモデリング、ニューロトロフィン3の発現、心筋細胞の凝集、細胞形質転換アッセイ、そして造血幹細胞の増幅が含まれる(引用に関しては、いずれかのウェブブラウザで閲覧可能なアッセイを記載しているスタンフォードウェブサイトhttp://www.stanford.edu/~rnusse/assays/assays.html updated November 2000をご覧いただきたい)。
【0130】
スクリーニング方法は、細胞間の表現型の違いを解析するいずれかの既知の方法を使用してもよく、DNA、mRNA、cDNA、又はポリペプチドレベルであってもよい。ディフェレンシャル・スクリーニングと遺伝子スクリーニングが、そのような二つの技術に該当する。本明細書で述べたスクリーニング方法のいずれかによって同定された物質は、本明細書で述べた他のスクリーニング方法で試験物質として使用されてもよい。
【0131】
スクリーニング方法は、例えば、マウスcDNA発現アレイといった核発現アレイを使用してもよい。このアプローチでは、異なる核酸分子のアレイがフィルター、水晶、又は他の表面上に、例えば核酸とフィルターのクロスリンクによって配置されている。試験溶液や抽出液を得て、その中で核酸を例えば蛍光によって標識する。その後、試験溶液や抽出液は、フィルター、又はジーンチップにアプライされる。フィルター、又はジーンチップ上の試験核酸のハイブリダイゼーションが測定され、コントロール溶液で行われたハイブリダイゼーションと比較される。試験サンプルとコントロールサンプルで得られたハイブリダイゼーション間の違いが、異なる核酸の量を示している。核酸アレイにおけるさらなる情報に関しては、いずれかのウェブブラウザで閲覧可能なClontech社のウェブサイト(例えば、www.clontech.com)、又はAffymetrix社のウェブサイト(例えば、www.affymetrix.com)をご覧いただきたい。
【0132】
本明細書でのスクリーニング方法は、基底レベルを超えて発現したNurr1に関して記載しているが、本開示は、例えば、Nor-1とNGFI-B といったNurr1サブファミリーの全核内レセプターにも及んでいる。このように、Nurr1は例示として記載されているいるに過ぎず、これに限定されるものではない。本発明のいずれの方法においても、Nurr1、又は、例えばNor-1やNGFI-Bといった他のいずれのレセプターをも含むNurr1サブファミリーの核内レセプターが、細胞内で基底レベルを超えて発現されて構わない。
【0133】
スクリーニング方法は、例えば、増殖、及び/又は自己複製、及び/又は分化、及び/又は生存を増強するような、及び/又はドーパミン作動性の運命の獲得若しくは誘導を促進するような、及び/又は幹、神経幹、前駆体、前駆若しくは神経細胞におけるドーパミン作動性ニューロンの分化を誘導するような、及び/又はGSK-3βを阻害する物質の存在下で基底レベルを超えるNurr1を発現している神経細胞でのドーパミン作動性誘導と分化を増強するようなNurr1の標的遺伝子、及び/又は因子若しくは複数因子を同定するために、幹、若しくは前駆、若しくは前駆体、若しくは神経細胞と、GSK-3βの阻害に作用する物質の存在下で基底レベルを超えるNurr1を発現する幹、若しくは前駆、若しくは前駆体、若しくは神経細胞とを比較することを含んでいてもよい。ひとたび標的遺伝子、及び/又は因子が同定されたならば、それらは、さらなる方法で分離され、及び/又は精製され、及び/又はクローン化され、そして使用されてもよい。
【0134】
スクリーニング方法は、混合物から物質、若しくは複数物質を精製、及び/又は分離することを含んでいてもよい。その方法は、GSK-3βの阻害の存在下で基底レベルを超えるNur1を発現している幹細胞、神経幹細胞、又は神経前駆若しくは前駆体細胞、又は神経細胞と相互作用する一又は二以上の混合物フラクションの能力を測定することを含んでいてもよい。能力とは、例えば、それらの細胞に対する結合力、及び/又は増殖、及び/又は自己複製を促進する能力、及び/又は幹、神経幹、前駆体、前駆、若しくは神経細胞などで、ドーパミン作動性の表現型や運命の誘導、獲得、分化、又は発生を増強する能力が該当する。精製、及び/又は分離は、当業者に知られるいずれの方法を用いてもよい。
【0135】
スクリーニング方法は、試験物質をコードする核酸を機能可能なように連結した誘導可能なプロモーターを使用してもよい。そのような構築物は、ホスト細胞内に取り込まれ、プロモーターの発現許容条件下と非許容条件下で、その細胞の一又は二以上の性質が測定され、また比較される。測定される性質は、基底レベルを超えるNurr1を発言している幹、神経幹、前駆体、前駆、若しくは神経細胞でドーパミン作動性表現型を誘導するホスト細胞の能力であり得る。許容条件と非許容条件間でのホスト細胞の能力の違いは、その試験物質が、単独、あるいは組み合わせのどちらかで、幹、神経幹、又は前駆若しくは前駆体細胞での増殖、及び/又は自己複製、及び/又はドーパミン作動性の運命の誘導、ドーパミン作動性分化、生存若しくは発生を増強するため又はGSK-3βの阻害の存在下で基底レベルを超えるNurr1を発現する神経細胞でのドーパミン作動性誘導、又は分化を増強できることを示している。
【0136】
本発明のスクリーニング方法の正確な形態は、日常的な技能と知識で当業者によって変更されても構わない。
【0137】
本発明で提供されたいずれか一つの方法で同定された因子若しくは複数因子は分離され、及び/又は精製され、及び/又はさらに調べられてもよい。それは、製造されても構わない。
【0138】
さらなる多様な局面において、本発明はさらに本明細書で開示された方法のいずれか一つで同定された因子、そのような因子を含む医薬組成物、薬剤、薬、又は他の組成物(その組成物は、基底レベルを超えるNurr1を発現している幹、神経幹、又は前駆若しくは前駆細胞、又は神経を含んでいてもよい)、基底レベルを超えるNurr1を発現している幹、神経幹、又は前駆若しくは前駆体細胞由来のドーパミン作動性ニューロンの誘導、及び/又は形態的分化、若しくは成熟化、及び/又は生存、及び/又は神経突起生成、及び/又はシナプシス生成、及び/又は機能的アウトプットを増強するための前記因子の使用、そのような因子、又は組成物の医療方法での使用、そのような因子、又は組成物を患者へ投与することからなる、例えば、ドーパミン作動性ニューロンの変性、損傷、欠損、又は障害若しくは影響に関連する病状の治療(予防治療を含んでいてもよい)のための、例えば、パーキンソン病や他の神経変性疾患の治療のための方法、例えば、パーキンソン病や他の病気(例えば、神経変性疾患)の治療のための投与用の組成物、若しくは薬剤の製造におけるそのような因子の使用、そしてそのような因子を薬剤上許容できる賦形剤、媒体、若しくは担体、及び任意で他の成分と混合することからなる医薬組成物を作る方法を提供する。
【0139】
関連する局面で、本発明は幹、神経幹、前駆体若しくは前駆細胞でドーパミン作動性の運命を誘導するために、あるいは基底レベルを超えるNurr1を発現している神経細胞でドーパミン作動性の誘導、又は分化を増強するために、GSK-3β阻害で直接的に、又は間接的に作用する物質の能力を調節する物質のスクリーニングの方法を提供する。
【0140】
従って、本発明の方法は基底レベルを超えるNurr1を発現している幹、神経幹、前駆体、前駆若しくは神経細胞で増殖、自己複製、ドーパミン作動性発生、分化、成熟化、及び/又は、ドーパミン作動性運命の獲得、を誘導するために、GSK-3β阻害剤の能力を調節する物質をスクリーニングすることができる。
【0141】
そのような方法は、一又は二以上の以下の(i)〜(iii)を含んでいてもよい。
【0142】
(i)GSK-3βの阻害の存在下で基底レベルを超えるNurr1を発現する幹、神経幹、前駆、前駆体、若しくは神経細胞を提供すること。ここでGSK-3βの阻害の存在とは、例えばGSK-3β阻害剤、又はβ−カテニンの過剰発現若しくは安定化若しくは分解の阻害、若しくはβ−カテニンのリン酸化の阻害が可能な薬剤、又はGSK-3β若しくはGSK-3βの基質と結合すること、若しくは相互作用すること、GSK-3βの基質リン酸化、β−カテニンの核局在、β−カテニン標的遺伝子、若しくはレポーター遺伝子に連結したβ−カテニンレスポンスエレメントの発現の調節、及び一、又は二以上の試験物質が該当する。
【0143】
(ii)ドーパミン作動性の運命又は表現型を取った、及び/又は阻害剤に応答した細胞の割合を解析すること。
【0144】
(iii)、試験物質を欠如した状態で比較可能な反応培地と条件下で、ドーパミン作動性の運命を取った細胞と、ドーパミン作動性の運命又は表現型を取った、及び/又は阻害剤に応答した細胞の総数の割合を比較すること。処理済の、及び未処理の細胞間でのドーパミン作動性ニューロンの割合の違いは、関連する試験物質の調節効果を示している。
【0145】
そのようなスクリーニングの方法は、以下の(i)〜(iii)を含んでいてもよい。
【0146】
(i)基底レベルを超えるNurr1を発現する幹、神経幹、前駆体、若しくは前駆細胞、又は神経細胞を一又は二以上の試験物質の存在下でGSK-3βの阻害剤(直接的阻害剤であろうが、間接的阻害剤であろうが)に接触させること。
【0147】
(ii)ドーパミン作動性の運命又は表現型を取った、及び/又は阻害剤に応答した幹、神経幹、前駆体、若しくは前駆細胞、又は神経細胞の割合を解析すること。
【0148】
(iii)試験物質が欠如した、又は存在する比較可能な反応条件下で、ドーパミン作動性の運命又は表現型を取った、及び/又は阻害剤に応答した幹、神経幹、前駆体、若しくは前駆細胞、又は神経細胞の割合と、ドーパミン作動性の運命又は表現型を取った、及び/又はWntに応答した幹、前駆体、前駆、若しくは神経細胞の数とを比較すること。
【0149】
このようなスクリーニング方法は、比較可能なヒト以外の同一の動物にてin vivoで細胞に対して、又はin vitroで、又は培養物で実施されてもよい。
【0150】
GSK-3β阻害を調節する物質の同定に続いて、その物質をさらに調査してもよい。該物質は、製造されても、及び/又は調製、すなわち、薬剤、医学組成物、又は薬等の組成物の製造、又は処方で使用されてもよい。その調節活性に関してテストされた物質はいずれも、天然の、又は合成された化合物であってもよい。
【0151】
例えば直接的なGSK-3β阻害剤の活性増強といったGSK-3β阻害を増強するモジュレーターを、例えば阻害剤と共に提供することができ、これら二つの物質を、例えば、医薬組成物、及び多様な局面における本発明の他の実施形態で一緒に使用してもよい。
【0152】
既に開示したように、GSK-3β阻害を調節する手段を本発明に従って使用して、ドーパミン作動性表現型を復活させることができる。さらなる実施形態において、GSK-3β阻害の調節は、GSK-3阻害療法に関与する遺伝子の転写活性を調節する物質を投与することによって、又は、GSK-3β阻害を調節する遺伝子を遺伝子療法を介して投与することによって達成してもよい。In vitroで本発明者は、GSK3阻害を与える手段としてのβーカテニン投与が、神経細胞からドーパミン作動性ニューロンの発生増加を生じることを示した。脳の神経細胞内にGSK3阻害を調節する遺伝子を導入することにより、患者の脳内で、ドーパミン作動性細胞の数を増やすことができる。別の方法として、GSK3阻害を調節できる物質でPD患者を治療することができる。このような物質は、上述したタイプのドラッグディスカバリーアッセイ法を介して同定することができる。
【0153】
本発明は、診断上の適用についても提供する。GSK3阻害はドーパミン作動性表現型の獲得を促進し、また、その表現型の欠損はパーキンソン病に関連しているので、Dishevelled、アキシン、FRAT/GBP、カゼインキナーゼ1、β−カテニンのレベル、及び/又は機能の変化を含むGSK-3β阻害の異常は、パーキンソン病の病因の基礎となるかもしれない。GSK-3β阻害に関与する遺伝子、mRNA、若しくはタンパク質の構造的な、又は機能的な異常が、遺伝子、mRNA、若しくはタンパク質の異常なレベル、及び/又は機能を引き起こすのかもしれない。また、PDの臨床上の発症の前後でこれらの欠陥、及び/又は異常が表面化するかもしれない。したがって、DNA、mRNA、若しくはタンパク質レベルで、そのような異常性についてスクリーニングすることは、PDの前駆症状の検出を提供するかもしれないし、あるいは診断の確認を提供するかもしれない。これらの技術は、脳生検、脳脊髄液、血液、唾液、尿、若しくは他の体液、又は組織に適用できる可能性がある。
【0154】
以下、添付の図面を参照して、本発明の局面と実施形態を例示する。別の局面と実施形態は、当業者には自明であろう。本明細書に記載した全ての文献は、参照によって本明細書中に組み込まれるものとする。
【0155】
タイプIアストロサイト又はグリア細胞の存在下でNurr1を発現している幹、神経幹、前駆体、又は前駆細胞でのドーパミン作動性ニューロンの誘導とドーパミン作動性前駆体の増殖、及び/又は自己複製を実証している、そしてFGF(例えば、FGF8)やレチノイドのような付加的因子と前記細胞との接触の際に得られる付加的な結果を明らかにしたW000/66713で詳述される実験的結果は、参照によって特別に本明細書に組み込まれる。
【0156】
ドーパミン作動性ニューロンの分化におけるWntの利用に関するWO2004/029229で詳述された実験結果もまた、参照によって特別に本明細書に組み込まれる。
【実施例】
【0157】
<GSK-3β阻害は、VM前駆体培養物でDAニューロン数を増加する>
ラットVMにおける前駆体細胞は、胎生期11日目から16日目(E11-E16)でDA表現型を獲得する(16)。神経上皮で増殖している前駆体が、腹側に移動し始めると、それらはNurr1の発現を開始する(17)。それによって、サイクリン依存性キナーゼ(CDK)阻害剤p57に関連する機構により細胞周期から離脱し、またDA表現型を獲得する(18)。本発明者は以前に、WntがVM前駆体でDA分化を促進することを示した(7)。細胞内カノニカルWntシグナル伝達因子がDAニューロン発生を調節できるかどうかを調べる目的で、本発明者はラットVM E14.5前駆体培養物に対して、β−カテニン分解に不可欠な酵素であるGSK-3βに対する二種のATP競合型阻害剤の用量を増加していく処理を行った。前駆体培養物へのインディルビンー3−モノオキシム(Indirubin-3-monoxime:I3M)(19、20)、又はケンパウロン(KP)(13)の添加は、I3Mについては3〜5μM(5倍増)での、またKPについては15μM(3倍増)での最大効率をもって用量依存的様式でTHポジティブ細胞の数を増加した(図1Aと1C)。これらの濃度において、KPはGSK-3β選択的阻害剤であるが、I3MはGSK-3βとCDKの両方を阻害する点において、これらの化合物は異なっている(20、13)。興味深いことに、本発明者は、またKP又はI3Mでの黒質、SN4741(21)由来のDA前駆体細胞系統の処理が、GSK-3βタンパク質レベルのダウンレギュレーションを誘導することも見出した(図1B)。
【0158】
<GSK-3β阻害はVM前駆体の分化を増加する>
本発明者は、次にGSK-3βの阻害がVM前駆体の一般的な神経とグリアの分化に影響するかどうかを見た。6の免疫細胞化学法からわかるように(図2Aと2B)、本発明者は、I3MとKPのいずれもがアストロサイト(グリア繊維性アストロサイトタンパク質(GFAP)ポジティブ細胞/ヘキスト33258)(図2A)、未成熟神経(β−チューブリン タイプIII(TuJ1)ポジティブ細胞/ヘキスト33258)(図2B)の総数を増加することを見出した。重要なのは、GSK-3β阻害が、成熟神経(微小管結合タンパク質2(MAP2)ポジティブ細胞/ヘキスト33258)の数の増加(図2C)、及びVMにおけるTH+/TuJ1+細胞の割合の3から5倍の増加によって示されるように(図2D及び図2B)、DAニューロン分化における非常に著しい増加をも誘導したことである。興味深いことに、本発明者は、以前にNurr1+幹/前駆体細胞のDAニューロンへの分化がグリア由来のシグナルに依存することを示唆した(22,23)。このように、グリア細胞の分化の増加は、DAニューロンの分化の増強にも貢献し得る可能性がある。
【0159】
<GSK-3βはVM前駆体培養物において細胞の生存又は増殖を調節しない>
GSK-3β阻害が増殖を促進しているかどうかを測定する目的で、本発明者は培養物における細胞総数と増殖マーカーであるBedUを取り込んだ細胞の数を調べた。ヘキスト33258染色からわかるように、GSK-3β阻害剤での処理は3日後の培養物内の細胞総数を変化させなかった(図3A)。さらに、VM前駆体が固定前に6時間 BedUパルスで曝露された場合、KPはBedUの取り込みに有意に影響しなかった(図3B)。しかし、I3MがCDKを阻害した以前の報告(20,13)に一致して、I3M処理はBrdU+細胞の数を減少させることがわかった(図3B)。このように、本発明者の結果は、GSK-3β阻害によるTH+ニューロンの増加は、増殖を通して達成されないことを示唆している。本発明者は、次にGSK-3β阻害剤が生存を通してDA細胞の数を増加できるかどうかを調べた。GSK-3β/CDK阻害剤であるI3Mは活性型カスパーゼ3免疫反応性細胞の数を減少させたのに対し、選択的GSK-3β阻害剤であるKPは効果がなかった(図3C−D)。より重要なことは、TH+細胞の約5パーセントは活性型カスパーゼ3にポジティブに染色したが、二つのGSK-3β阻害剤7のいずれもCaspase+/TH+細胞の数を減少しなかった事実(データは示さず)が判明したことである。これはGSK-3β阻害がアポトーシスの減少を通してTH+ニューロンの数の増加を誘導しないことを示唆している。DA分化におけるGSK-3β阻害の効果を確認する目的で、本発明者は未成熟DAニューロンの性質を示すMN9D細胞(24)における選択的GSK-3β阻害剤KPの効果を調べた。VM前駆体細胞のように、KPでの処理は、細胞総数(ヘキスト33258で分析した)又はBrdUの取り込みのレベルを変えなかった(図3E)。しかし、KP処理は、VM前駆体培養物で見られた(図2C)ようなMN9D細胞における形態的分化と神経突起伸展を増加した(図3F)。Nurr1過剰発現後のMN9D細胞の形態学的分化における比較可能な効果の報告(18)は、これらの細胞とVM前駆体とがNurr1とGSK-3β阻害剤に対する応答において分化能を共有していることを示唆している。合わせて、本発明者の結果が前駆体細胞分化におけるGSK-3βの機能を示唆し、またこのGSK-3β阻害がVM前駆体におい
てDA分化を促進するかどうかを調べることを促した。
【0160】
<GSK-3βの阻害は、Nurr1+前駆体のDAニューロンへの転換を通してDA細胞数を増加する>
本発明者は、まずGSK-3β阻害が分化したVM、DAニューロンに特有な遺伝子の発現を促進するかどうかを見た。リアルタイムRT-PCR解析は、KP処理が癌原遺伝子c-retのmRNAレベルの増加を生じさせることを示した(図4A)。c-retは、VM前駆体培養物のWnt5aでの処理(7)や、あるいはWnt1で処理されたカテコールアミン作動性PC12細胞において(25)アップレギュレートされたことも報告されている。この結果は、GSK-3β阻害がDA前駆体の分化を増加する上で働くかもしれないことを示唆している。それゆえ本発明者はKPで処理したNurr1+/ TH-前駆体のNurr1+/ TH+ DAニューロンへの分化を調べ、この8特異的GSK-3β阻害剤がTH/Nurr1両ポジティブ細胞の数を2.5倍に増加することを見出した(図4B−C)。このように、本発明者の結果は、GSK-3β阻害がTH+ニューロンの数の増加を誘導する機構はVM前駆体の増殖や生存を促進することによるものではなく、Nurr1+ VM前駆体の分化を促進することによるものであることを示した。
【0161】
<β−カテニンの安定化はVM前駆体のDAニューロンへの分化を増加する>
本発明者は以前にWntがVM培養物においてDA分化を促進すること、及びin vivoでのE10.5 VM前駆体においてNurr1/β−カテニンの共局在とTCF/LEF転写活性があることを示した(7)。しかし、最近ではGSK-3βがβ―カテニン非依存的経路において微小管の安定化を通して軸索リモデリングを仲介することが記述されている(26)。したがって、DA分化の機構をさらに研究する目的で、本発明者はKP又はI3Mで処理したVM前駆体培養物においてβ―カテニンタンパク質のレベルを解析した。免疫ブロッティングは、βーカテニンタンパク質のレベルの増加を示した。これは、阻害剤での処理によりβーカテニンの安定化が増加したことを示している(図5A)。さらに両阻害剤は、THタンパク質のレベルを増加した(図5A)。これらの結果は、β−カテニンがDA前駆体におけるGSK-3β阻害剤とWntの効果のいくつかを仲介できることを示唆している。初期DA分化におけるβ−カテニン安定化の役割の重要性を調べる目的で、本発明者はβーカテニン発現ベクターでVM前駆体をトランスフェクトした。VM前駆体細胞におけるβ−カテニンの過剰発現は、空のコントロールベクターと比べてTHポジティブ細胞数の2倍増を誘導した(図5BとC)。9。
【0162】
<方法>
(リアルタイムPCRと遺伝子発現の定量化)
総RNAは、in vitroで3日間、GSK-3β阻害剤で処理したE14.5VM前駆体培養物(6cm2ディッシュで2.5×106細胞)から分離した。逆転写反応、リアルタイムRT−PCR、またc-retプライマー配列の説明に関しては、Castelo-Branco らによる2003年の文献(7)をご覧いただきたい。古典的18SRNA量の内部標準はAmbion社(Austin, USA)から、またPCRプライマーはDNA Technology A/S社(Aarhus, Denmark)から購入した。以下のPCRプログラムは、ABI PRISM 5700 Detection System(PE AppliedBiosystems社, Foster City, CA, USA)におけるSYBR Green検出用(94℃2分、94℃30秒と59℃30秒と72℃15秒を35−40サイクル、そして80℃5秒)を用いた。結果の統計分析は、両側ウィルコクソン符号順位検定によって行った。全検定の有意性は、p<0.05のレベルで仮定した(* p<0,05; ** 0.01<p<0.001; *** p<0.001)
(前駆体とMN9Dの培養及び処理)
時期交尾されたSprague-Dawley系ラットから得られたE14.5VM(動物実験のための倫理上の承認は、Stockholms Norra Djurfoersoeks Etiska Naemndにより許可された)を解剖し、機械的に分離し、そして5μg/mlインシュリン、100μg/mlトランスフェリン、100μMプトリッシン、20nMプロゲステロン、30nMセレン、6mg/mlグルコース、そして1mg/mlBSA(全てSigma社から購入)を含むF12とMEM(Invitrogen社)の1:1混合からなる無血清N2培地が入ったポリDリジン被覆プレート(Falcon社)上に1cm2あたりの最終密度が1×105細胞となるようにプレーティングした。培養液は、固定前に、5%CO2で37℃インキュベータ内にて3日、in vitroで保持した。MN9DとSN4741細胞は、Choiらによる1991年の文献とSonらによる1999年の文献に記載されたように育てた。MN9D培養物はin vitroで4日後に固定した。I3MとKP(Alexis Biochemicals社, Goeteborg, Sweden)はDMSOで希釈し、プレーティング時に培地に添加した。DMSO単体では、TH+12ニューロンの数を増加せず、またMN9Dの形態学的分化に影響しなかった(データは示さず)。10μMのBrdUは固定の6時間前に培地に加えた。
【0163】
(構築とトランスフェクション)
ヒトβーカテニンcDNA(Dr. Steven Byersより贈与)を、CMVエンハンサー下にニワトリβ−アクチンプロモーターを有するpCAIP2ベクター中にサブクローンした。空のpCAIP2ベクターをコントロールとして使用した。初代細胞をリポフェクタミン2000(Invitrogen社)を用いて、製品使用書に従ってトランスフェクトし、in vitroで2日後、免疫細胞化学法によって評価した。
【0164】
(免疫細胞化学法)
細胞を氷冷した4%パラホルムアルデヒドで20分間固定し、PBSで洗浄した。免疫細胞化学法の詳細なプロトコルは、Castelo-Branco らによる2003年の文献(7)をご覧いただきたい。以下の一次、及び二次抗体、すなわちマウス抗TH(1:1000希釈 - Immunostar社)、ウサギ抗TH(1:250 - Pelfreeze社)、マウス抗βIIIチューブリン(1:1000 - Promega社)、ウサギ抗Nurr1(1:1000 - Santa Cruzs社)、マウス抗MAP2(1:200 - Sigma社)、ウサギ抗GFAP(1:400 - Dako社)、マウス抗活性型カスパーゼIII(1:100 - Cell Signaling社)、マウス抗BrdU(1:100 - Dako社)、またラット抗BrdU(1:100 - Abcam社)、ビオチン化 1:500、シアニン-2、若しくはローダミン結合されたウマ抗マウスIgG 1:200又はヤギ抗ウサギIgG 1:200(Vector and Jackson Laboratories社から)を使用した。染色手順の最後で培養物をヘキスト33258試薬と共に10分間インキュベートした。BrdU免疫細胞化学法は、2NのHClでの30分間のインユベーションを含んでいる。PBS中の染色された細胞を室温においてZeiss Axioplan 100M microscope (LD Achrostigmat 20x, 0,3 PH1 . 0-2 and LD Achroplan 40x, 0,60 Korr PH2 . 0-2)で画像を捉え、Hamamatsu camera C4742-95(QEDイメージング ソフトウェアを用いて)で撮影した。
【0165】
(免疫ブロッティング)
細胞を20mMのTris(pH7.5)、140mMのNaCl、10%のグリセロール、1%のIGEPAL、1mMのβ-グリセロール フォスファターゼ、1mMのNa3VO4、そして完全なプロテアーゼ阻害剤(Roche社)を含む変性RIPAバッファ中で15分間溶解した。プレートから細胞を掻き取り、細胞溶解物を微量遠心管内で5分間遠心した。上清をBCA kit(PIERCE社)を用いてタンパク質測定用のよごれていないチューブに移し、Laemmliバッファー中で保存した。等量のタンパク質をポリアクリルアミドゲル電気泳動法(10%ポリアクリルアミド)で解析した。タンパク質は、ポリビニリデン ジフルオリド メンブレンに転写した。0.1% Tweenと3% BSAを含んだPBSでブロッキングした後、メンブレンは以下の一次抗体と共に4℃で一晩インキュベートした(マウス抗β-カテニン 1:500,BD社;ウサギ抗GSK-3β 1:1000,Cell Signaling社;ウサギ抗TH 1:500, PelFreeze社; そして マウス抗β−アクチン 1:500, Abcam社)。洗浄後、メンブレンを二次抗マウス抗体(1:10000,Pharmacia Amersham社)共役アルカリフォスファターゼと共に、室温で1時間インキュベートし、続いて化学発光を増強するために製品説明書に従って発光させた。
【0166】
(統計解析)
免疫細胞化学データの定量は、13から5の独立した実験条件あたり3つのウェルにおいて10回の非重複野由来の平均値±標準誤差を表している。RT−PCR実験に関しては、5回の独立実験を解析し、またGSK-3β阻害剤用量反応に関しては、2セットの実験を解析した。統計解析は図のレジェンドに記載したように、p<0.05 (* p<0,05; ** 0,01<p<0,001; *** p<0,001)のレベルで仮定された全検定に関する有意性をもって、Prism 4 (GraphPad software, San Diego, USA)で行われた。統計検定はサンプル集団の分布に従って選択した。
【0167】
<参照文献>
Airaksinen MS, Saarma M. (2002) Nat Rev Neurosci. 3, 383-394
Akerud ら, J. Neurosci. 21, 8108-18 (2001).
Arenas 2002. Brain Res Bull.; 57 (6):795-808. (ref 28)
Baek SH ら, (2003) Proc Natl Acad Sci U S A. 100, 3245-3250
Bain ら, 2003. Biochem J.; 371 (Pt 1): 199-204. (ref 13)
Bain ら,1995, Dev. Biol., 168, 342-357
Baker ら, Genes Dev 1999 Dec 1;13(23):3149-59
Bamji ら, 2003. Neuron; 40 (4):719-731 (ref 11)
Barberi ら, 2003 Nat Biotechnol. 21 (10):1200-1207 (ref 33)
Bennett ら, 2002. J Biol Chem.; 277 (34):30998-31004 (ref 14)
Bjorklund A, Lindvall O. Nat Neurosci. 2000 Jun;3(6):537-44.
Bjorklund ら, 2002, Proc Natl Acad Sci USA. Feb 19, 99(4): 2344-9
Bradley RS, Brown AM. EMBO J 9(5):1569-75 (1990).
Bradley RS, Brown AM. Mol Cell Biol 15(8):4616-22 (1995).
Brustle ら, 1999, Science 285, 754-756.
Capela and Temple, 2002, Neuron 35, 865-875.
Castelo-Branco ら, 2003. Proc Natl Acad Sci U S A. 100 (22): 12747-12752. (ref 7)
Castillo (1997) Genomics, 41, 250-257
Castillo ら, Mol Cell Neurosci 1998 May;11(1-2):36-46
Castro ら, 2001. J Biol Chem.; 276 (46):43277-43284 (ref 18)
Chambers ら, 2003, Cell May 30;113(5):643-55. Chenn ら.. 2002. Science; 297 (5580):365-369. (ref 34)
Choi ら, 1991 Brain Res. ;552 (1):67-76. (ref 24)
Ciani ら, 2004. J Cell Biol.;164 (2):243-253 (ref 26)
Cohen ら, 2001. Nat Rev Mol Cell Biol.; 2 (10):769-776. (ref 12)
Conacci-Sorrell ら, (2002) Genes Dev. 16, 2058-2072.
Danielian ら, 1996. Nature; 383 (6598):332-334. (ref 3)
Dann CE, ら, (2001) Nature 412, 86-90.
Deacon ら, 1998, Exp. Neurol., 149, 28-41.
Dorsky ら, 1998. Nature; 396 (6709): 370-373. (ref 1)
Dunnett ら, 2001. Nat Rev Neurosci.; 2 (5):365-369. (ref 27)
Foster ら, Int J Dev Neurosci 1988;6(4):367-86
Garcia-Castro ら, Science. 2002 Aug 2;297(5582):848-51
Gossen, ら, (1995) Science, 268, 1766-1769
Gottlieb DI. Annu Rev Neurosci. 2002;25:381-407.
Granholm AC, Reyland M, Albeck D, Sanders L, Gerhardt G, Hoernig G, Shen L,
Westphal H, Hoffer B. (2000) J Neurosci. 20, 3182-3190.
Hall AC, Lucas FR, Salinas PC. (2000) Cell 100, 525-535.
Hall ら, 2003. Glia.;43 (1):47-51. (ref 23)
Hartmann C, Tabin CJ. Cell 2001 Feb 9;104(3):341-51
Hoessel ら, 1999. Nat Cell Biol. ; 1 (1): 60-67. (ref 19)
Hornstein ら, 2004. J Neurosci Res.; 76 (2):169-173. (ref 30)
Houart ら, Neuron. 2002 Jul 18;35(2):255.
Hsieh ら, Proc Natl Acad Sci U S A. 96(7):3546-51 (1999).
Huelsken ら, 2001. Cell.; 105 (4): 533-545 (ref 38)
Huelsken ら, 2002. J Cell Sci.; 115 (Pt 21):3977-3978. (ref 9)
Kiecker ら, Development 2001 Nov;128(21):4189-201
Kim ら, 2002. Nature. 418 (6893):50-56 (ref 32)
Kioussi C., ら, (2002) Cell 111, 673-685, 38
Kispert ら, Development 1998 Nov;125(21):4225-34
Krylova O, ら, (2002) Neuron 35, 1043-1056.
Law, ら, (1992) Mol Endocrinol 6, 2129-2135
Leclerc ら, 2001. J Biol Chem; 276 (1): 251-260. (ref 20)
Lee ら, 2000. Nat Biotechnol.;18 (6):675-679. (ref 31)
Lee ら, 2004. Science. 303 (5660):1020-1023 (ref 37)
Lindsay 2002; Cur. Op. Pharmacol., 2: 587-594.
Martinez Arias ら, 1999, Curr. Op. Genetics and dev. 9, 447-454.
Marvin ら, Genes Dev 2001 Feb 1;15(3):316-27.
Mata de Urquiza ら, Science 2000 Dec 15; 290: 2140-2144.
McMahon ら, 1990. Cell; 62 (6):1073-1085. (ref 4)
Megason ら, 2002. Development; 129 (9): 2087-2098. (ref 2)
Merrill ら, 2001. Genes Dev.; 15 (13): 1688-1705. (ref 39)
Muroyama ら, Genes Dev. 16(5):548-53 (2002).
Nordstrom ら, Nat Neurosci 2002 Jun;5(6):525-32
Nunes I., ら, Proc Natl Acad Sci U S A. 100, 4245-4250 (2003)
Ohmachi ら, 2000; Biochem. Biophys. Res. Comm.277, 355-360.
Oishi I, ら, 2003, Genes Cells. 2003 Jul;8(7):645-54.
Okabe ら, 1996, Mech. Dev., 59, 89-102.
Pandur ら, Nature. 2002 Aug 8;418(6898):636-41.
Paratcha G., Ledda F., Ibanez C.F. (2003) Cell 113, in press.
Parr ら, Development 119(1):247-61(1993).
Patapoutian A, Reichardt LF. (2000) Curr Opin Neurobiol. 10, 392-399.
PCT/IB03/04598
Perrone-Capano ら, 2000. Neuroscience and Biobehavioral Reviews, 24, 119-124 (ref 16)
Pinson ら, 2000. Nature; 407 (6803):535-538. (ref 6)
Price J. & Williams BP. Current Opinion in Neurobiology 11, 564-567 (2001).
Reubinoff ら, 2000, Nat Biotech., 18, 399-404.
Reubinoff ら, 2001, Nat Biotech., 19, 1134-1140.
Reya T, ら, (2003) Nature 423, 409-414.
Rhinn M, Brand M. Curr Opin Neurobiol 2001 Feb;11(1):34-42
Ross ら, Science 2000 Aug 11;289(5481):950-3
Rossi F. & Cattaneo E. Nat Rev Neurosci. 2002 May;3(5):401-9.
Saneyoshi ら, Nature 417(6886):295-9 (2002).
Sato ら, 2004. Nat Med; 10 (1): 55-63. (ref 15)
Saucedo-Cardenas ら, Proc Natl Acad Sci U S A 1998 Mar 31;95(7):4013-8
Schaar ら, 1993 Exp Neurol.;124 (2):368-371. (ref 36)
Schneider ら, Genes Dev 2001 Feb 1;15(3):304-15
Schuldiner ら, 2000, PNAS. USA 97, 11307-11312.
Schuldiner ら, 2001; Brain Res. 913, 201-5.
Shimizu ら, Cell Growth Differ 1997 Dec;8(12):1349-58
Shtutman ら, Proc Natl Acad Sci U S A 96(10):5522-7 (1999).
Smidt ら, Proc Natl Acad Sci U S A. 94(24):13305-10 (1997).
Smidt ら, Nat Neurosci. 3, 337-341 (2000).
Son ら, 1999. J Neurosci; 19 (1): 10-20. (ref 21)
Song ら, Nature 2002 May 2;417(6884):39-44
Storch ら, 2001. Exp Neurol.; 170 (2):317-325. (ref 20)
Taipale J, Beachy PA. Nature. 2001 May 17;411(6835):349-54.
Tetsu ら, Nature 398(6726):422-6 (1999).
Thomas ら, 1990. Nature; 346 (6287):847-850. (ref 5)
Tropepe.ら, 2001, Neuron 30,65-78.
Trupp M ら, (1996). Nature 381, 785-789
Tumbar ら, 2004. Science; 303 (5656):359-363 (ref 40)
Tzahor E, Lassar AB. Genes Dev 2001 Feb 1;15(3):255-60
Van Den Munckhof P.ら, 2003 Development 130, 2535-2542
Wagner ら, 1999 Nat Biotechnol.;17 (7):653-659. (ref 22)
Wallen ら,1999. Exp Cell Res; 253 (2): 737-746. (ref 17)
Willert ら, 2003. Nature; 423 (6938):448-452 (ref 8)
Wilson ら, Nature 2001 May 17;411(6835):325-30
Wurst W, Bally-Cuif L. Nat Rev Neurosci 2001 Feb;2(2):99-108
Xing, ら, (1997) Mol Brain Res 47, 251-261
Ye ら, Cell 93(5):755-66 (1998).
Yoshikawa S., ら, Nature. 2003 Apr 10;422(6932):583-8.
Yu ら, 2003. Nat Neurosci.; 6 (11):1169-1177. (ref 10)
Zechner ら, 2003. Dev Biol; 258 (2):406-418. (ref 35)
Zetterstrom ら, Science 1997 Apr 11;276(5310):248-50
Zhang ら, 2001, Nat Biotech., 19, 1129-1133.
Zheng ら, 1996. Oncogene.; 12 (3): 555-562. (ref 25)
【図面の簡単な説明】
【0168】
【図1】KP及びI3MはVM前駆体培養物内でTH+ニューロンの数を増加する。
【0169】
図1A:E14.5 VM前駆体での用量反応実験は、15μMのKP及び3〜5μMのI3MがTH+細胞数を増加する上で最適用量であることを示している。
【0170】
図1B:免疫ブロッッティングからわかるように、15μMのKP及び5μMのI3Mで24時間処理したSN4741細胞は、GSK-3βタンパク質レベルが低下する。ローディングコントロール:β−アクチン。免疫染色法は、15μMのKP及び5μMのI3Mでの処理後三日で、TH免疫反応性細胞数の増加を示した。統計解析は、Fisher多重比較検定(Fisher’s post -hoc test)での一元配置分散分析を用いて行われた。
【図2】KP及びI3Mでの処理は、神経とグリアの双方への、そして特にDAニューロンへのVM前駆体の分化を増大する。抗GFAP抗体(A)、抗Tuj1抗体及び抗TH抗体(B)、そして抗MAP2抗体(C)での免疫染色法は、15μMのKP及び5μMのI3Mでの処理後三日で、アストロサイト、未熟及び成熟神経の数の増加、並びにTH+ DAニューロンの割合の著しい増加を示した(BとD)。統計分析は、両側独立t検定を用いて行われた。
【図3】GSK-3β阻害は、VM前駆体、又はMN9D細胞の生存と増殖を調節しないが、それらの形態的な分化を誘導する。
【0171】
図3Aは、ヘキスト33258染色からわかるように、15μMのKP及び5μMのI3Mでの処理は培養3日後のVM細胞の総数に変化を起こさなかったことを示している。
【0172】
図3Bは、BrdUの取込みからわかるように、増殖は15μMのKPでは影響を受けなかったが、5μMのI3Mで有意に減少したことを示している。
【0173】
図3Cは、活性型カスパーゼ3の免疫染色は、VM前駆体培養物において15μMのKPでは変化しなかったが、5μMのI3Mで減少したことを示している。
【0174】
図3Dは、15μMのKPで4日間処理したMN9D細胞が、細胞総数(ヘキスト33258)、又はそれらの増殖(BrdUの取込み)を変えなかったことを示している。α−活性型カスパーゼ3での免疫染色は、5μMのI3Mによる処理後3日でポジティブな細胞数の減少を示したが、KPによる処理では示さなかった。4日間の15μMのKPによる処理は、MN9D細胞の形態的な分化を誘導した。統計解析は、両側独立t検定(A)、クラスカル−ウォリス検定(Kruskal-Wallis test)(B)、ボンフェローニ多重比較検定(Boneferroni’s multiple comparison)での一元配置分散分析(C)を用いて行われた。
【図4】KPはc-ret mRNAをアップレギュレートし、またNurr1+前駆体のTH+ニューロンへの転換を増加する。
【0175】
図4Aは、リアルタイムRT-PCR解析によりc-ret mRNAレベルが15μMのKPによる処理でアップレギュレートされることが明らかとなったことを示している。
【0176】
図4Bは、15μMのKPがVM Nurr1+前駆体のTH+ニューロンへの転換を増加したことを示している。TH/Nurr1二重免疫染色は、15μMのKP処理後三日で免疫反応性の細胞数の増加を示している。統計解析は、両側ウィルコクソン符号順位検定(two-tailed Wilcoxon’s signed rank test)(A)及び両側独立t検定(B)を用いて行われた。
【図5】GSK-3β阻害剤は、β−カテニンを安定化し、またβ−カテニンの過剰発現は、TH+ニューロンの数を増加する。
【0177】
図5Aは、免疫染色からわかるように、15μMのKP又は5μMのI3Mで24時間VM前駆体を処理した結果、β−カテニンとTHタンパク質の両レベルが増加したことを示している。ローディング・コントロール:βアクチン。
【0178】
図5Bは、βーカテニンでのトランスフェクション後2日で、空のコントロールベクターでのトランスフェクションと比べて、TH−免疫染色のポジティブ細胞の数が2倍に増加したことを示している。統計解析は、両側独立t検定(B)を用いて行われた。
【技術分野】
【0001】
本発明は、神経幹細胞、神経系前駆若しくは神経前駆体細胞、又は他の幹細胞における神経の運命誘導に関する。また本発明は、特定の神経表現型の誘導とその増強、特に中脳ドーパミン作動性ニューロンの表現型の誘導とその増強に関する。
【0002】
本発明の局面と実施形態は、GSK-3βの阻害調節に関連する。これは、GSK-3βの阻害剤である物質、又は、例えばβ-カテニンに作用したり、若しくはそれを模倣することによって、例えばβ-カテニンを安定化したり、その分解を阻害したり、若しくはそのリン酸化を阻害することによって、又はGSK-3βの基質に作用することによって、GSK-3β活性に影響する物質の利用に関連してもよい。本明細書で「GSK-3βの阻害」又は「GSK-3β阻害」というときは、これらのアプローチのいずれかを意味する。Frizzledレセプターに対するWnt及びいかなるリガンドの利用も考慮しない。「GSK-3β阻害剤」とは、その文脈が別の内容を意味しない限り、GSK-3βに直接に作用してこれを阻害する物質の利用をいう。
【0003】
いくつかの実施形態では、GSK-3β阻害はアップレギュレート、又はダウンレギュレートであり得る。すなわち、阻害は、誘導されても、あるいは縮小されてもよい。本明細書では、主として阻害の誘導について述べているが、GSK-3β阻害の調節がダウンレギュレーション、すなわち、阻害の縮小による類似の実施形態も本発明によって提供される。
【背景技術】
【0004】
パーキンソン病(PD)は非常によく知られた神経変性疾患であり、その病因は中脳のドーパミン作動性(DA)ニューロン(以下、神経細胞又は単に神経ということもある)の選択的、かつ進行性の欠損によって特徴付けられる。神経表現型の誘導の増強は、パーキンソン病や他の重篤な衰弱性神経変性疾患の治療を可能にする潜在性を有している。
【0005】
従来、好成績な結果をもたらすものとしてヒト胎児の中脳組織がパーキンソン病患者に移植されていた。しかし、本発明を利用した特定の細胞補充療法の開発によれば、移植の取り組みに先立つ実行上の、あるいは倫理上の困難を克服できる。特に、本発明によれば、胚組織又は胚性細胞の利用のいずれの必要性も減じ、あるいは排除できると同時に、移植用の細胞調製の開発を可能にする。幹細胞は、一般に廃棄される組織である臍帯から得ることができる。その他の選択肢は、成体幹細胞を例えば、骨髄、血液、皮膚、目、嗅球または嗅覚器上皮から得ることである。
【0006】
以前(WO00/66713 and Wagner ら, 1999)に、本発明者の研究室は、腹側中脳のタイプIアストロサイト/初期グリア細胞から得ることのできる一又は二以上の因子の存在下においてドーパミン作動性ニューロン表現型の誘導がNurr1を発現している細胞内で増強されることを示した。
【0007】
さらなる研究により、Nurr1を発現している細胞の神経表現型の誘導を増強する上でWnt因子が有効であることが判明した。これらの結果は、WO2004/029229で発表している。
【0008】
特に、WO2004/029229で開示したように、DA ニューロンの出現時まで背側中脳よりもVMで高レベルで発現されるあらゆるWntが、神経幹、前駆細胞若しくは前駆体細胞、又は他の幹細胞、又は神経細胞において、増殖、自己複製、ドーパミン作動性の誘導、生存、分化、及び/又は成熟化を増強することによりドーパミン作動性ニューロンの発生を誘導、又は促進する上で有用であることが判明した。
【0009】
すなわち、Wnt-1は、ドーパミン作動性前駆体の増殖とドーパミン作動性前駆体、及び/又は幹細胞のドーパミン作動性ニューロンへの成熟化を促進し、
Wnt-7aは、ドーパミン作動性前駆体の増殖を促進し、またドーパミン作動性ニューロンへの分化を可能にし、
Wnt-3aは、ドーパミン作動性前駆体、及び/又は幹細胞の増殖、及び/又は自己複製を促進し、
Wnt-2は、Nurr1+前駆体によって細胞周期からの離脱とドーパミン作動性ニューロン表現型の獲得を促進し、そして
Wnt-5aは、神経幹細胞、前駆細胞若しくは前駆体細胞でのドーパミン作動性表現型の誘導で、また神経細胞でのドーパミン作動性の誘導若しくは分化を増強することにおいて、最も有効であることが判明した。
【0010】
Wnt-1は、ドーパミン作動性前駆体、及び/又は幹細胞の分化と成熟化においてWnt-3aとWnt-5aよりも有効である。
【0011】
特定の神経表現型の誘導には、遺伝学的なシグナルと後成的なシグナルの双方の統合が必要である。発生中の中脳におけるドーパミン作動性ニューロンの誘導は、オーファン核内レセプターNurr1を必要とする (Zetterstroemら, 1997; Saucedo-Cardenasら, 1998; Castilloら, 1998)が、神経幹細胞においてはNurr1の発現がドーパミン作動性表現型を誘導するのに十分とは言えない(Wagner ら, 1999)。Nurr1と発生中の腹側中脳タイプIアストロサイト/初期グリア細胞から得られる未知の可溶性シグナルとの組み合わせにより、神経幹細胞での中脳ドーパミン作動性表現型の誘導が充足される (Wagnerら, 1999)。Wnt-5aは、このようなシグナルの一部であり、またWntファミリータンパク質の一員である。Wntファミリーは、Wnt-1, -2, -3a, -5a 及び -7aを含み、発生上で調節され、分化上では中脳ドーパミン作動性ニューロンの発生を制御する。部分精製されたWnt-1, -2, -5a 及び -7aは、二つの異なる機構でE14.5中脳DAニューロンの数を増加したが、Wnt-3aは増加しなかった。Wnt-1と-7aは、主にNurr1前駆体の増殖を増加し、またそれらのドーパミン作動性ニューロンへの分化を可能にした。Wnt-2は、Nurr1+前駆体によるドーパミン作動性ニューロンの表現型の獲得と細胞周期からの離脱を示した。Wnt-5aは、主として神経のDA表現型を獲得したNurr1前駆体の割合を増加した。これらの発見と一致して、Wnt-5aはNurr1発現ニューロン、又は皮質E13.5前駆体においてドーパミン作動性ニューロンを誘導する点で中脳アストロサイト/初期グリア細胞と同程度に有効であった。さらに、Frizzled 8のシステインリッチドメインは、Nurr1発現ニューロン前駆体培養にてドーパミン作動性表現型をもつ細胞の増加におけるVM T1A-、Wnt-1若しくはWnt-5aの介在する効果と基底の効果を効率的に妨げた。また、神経幹細胞での内因性のWnt又はFGF-8の効果は、Nurr1+中脳神経球に及んだ。このようにデータは、Wntが部分的に異なる機構で、Nurr1発現前駆体/幹細胞においてDA表現型を持つ神経の発生を独立に調節していることを示している。
【0012】
これらの発見は、Wntリガンドを腹側中脳神経発生期間における増殖、自己複製、分化、及び運命決定のキーとなるレギュレーターとして位置づけている。さらに、Wntは、幹細胞の発生をドーパミン作動性ニューロンに導くのに利用され、それによって、パーキンソン病に対する細胞補充療法でのそれらの治療の潜在性が活用されるかもしれない。
【0013】
胚性幹細胞、神経幹細胞、及び多分化能幹細胞は、神経、アストロサイト、およびオリゴデンドロサイトを含む神経細胞系統へ分化する能力を有している。さらに、幹細胞は、脳移植用の原材料として利用され、増幅され、分離され得る(Snyder, E. Y. ら Cell 68, 33-51 (1992); Rosenthal, A. Neuron 20, 169-172 (1998); Bain ら, 1995; Gage, F.H., ら Ann. Rev. Neurosci. 18, 159-192 (1995); Okabe ら, 1996; Weiss, S. ら Trends Neurosci. 19, 387-393 (1996); Snyder, E. Y. ら Clin. Neurosci. 3, 310-316 (1996); Martinez-Serrano, A. ら Trends Neurosci. 20, 530-538 (1997); McKay, R. Science 276, 66-71 (1997); Deacon ら, 1998; Studer, L. ら Nature Neurosci. 1, 290-295 (1998); Bjorklund and Lindvall 2000; Brustle ら, 1999; Lee ら, 2000; Shuldiner ら, 2000 and 2001; Reubinoff ら, 2000 and 2001; Tropepe ら, 2001; Zhang ら, 2001; Price and Williams 2001; Arenas 2002; Bjorklund ら, 2002; Rossi and Cattaneo, 2002; Gottlieb ら, 2002)。
【0014】
ほとんどの神経変性疾患は、神経細胞数に影響を及ぼす。さらに、損傷の多くは、特異的な神経化学表現型を生じる。例えば、ヒトパーキンソン病で欠損する主要な細胞型は、中脳ドーパミン作動性ニューロンである。神経組織の移植による特定の神経細胞数の機能的な補充は、神経変性疾患を治療する上で魅力的な治療戦略を提示する(Rosenthal, A. Neuron 20, 169-172 (1998))。他の選択肢は、再生を促進し、幹細胞、前駆細胞、前駆体細胞の発生及び/又は追加を修復し、若しくは導くのに必要なシグナルの直接的な注入、あるいはそれらの機能を調節する薬物の投与であろう。
【0015】
幹/前駆、又は前駆体細胞は、増幅可能で、かつ特定の神経表現型となる指示を受け得るので、移植療法にとっては理想的な材料である。一般に一人の患者を治療するために、数人のドナー由来の組織が必要であったが、一つの幹細胞が多くの患者を治療するために利用できるので、これらの細胞によってヒト胚性組織の移植のための使用を取り巻く倫理上、及び実施上の問題を回避できるであろう。
【0016】
しかし、Wntは可溶性に乏しい。これは、セットアップされた臨床現場でそれらの利用を制限し、また治療学上の応用を困難にしている。
【特許文献1】WO00/66713
【特許文献2】WO2004/029229
【非特許文献1】Wagner ら, 1999 Nat Biotechnol.;17 (7):653-659. (ref 22)
【非特許文献2】Zetterstrom ら, Science 1997 Apr 11;276(5310):248-50
【非特許文献3】Saucedo-Cardenas ら, Proc Natl Acad Sci U S A 1998 Mar 31;95(7):4013-8
【非特許文献4】Castillo ら, Mol Cell Neurosci 1998 May;11(1-2):36-46
【非特許文献5】Wagner ら, 1999 Nat Biotechnol.;17 (7):653-659. (ref 22)
【非特許文献6】Snyder, E. Y. ら Cell 68, 33-51 (1992)
【非特許文献7】Rosenthal, A. Neuron 20, 169-172 (1998)
【非特許文献8】Bain ら,1995, Dev. Biol., 168, 342-357
【非特許文献9】Gage, F.H., ら Ann. Rev. Neurosci. 18, 159-192 (1995)
【非特許文献10】Okabe ら, 1996, Mech. Dev., 59, 89-102
【非特許文献11】Weiss, S. ら Trends Neurosci. 19, 387-393 (1996)
【非特許文献12】Snyder, E. Y. ら Clin. Neurosci. 3, 310-316 (1996)
【非特許文献13】Martinez-Serrano, A. ら Trends Neurosci. 20, 530-538 (1997)
【非特許文献14】McKay, R. Science 276, 66-71 (1997)
【非特許文献15】Deacon ら, 1998, Exp. Neurol., 149, 28-41
【非特許文献16】Studer, L. ら Nature Neurosci. 1, 290-295 (1998)
【非特許文献17】Bjorklund A, Lindvall O. Nat Neurosci. 2000 Jun;3(6):537-44
【非特許文献18】Brustle ら, 1999, Science 285, 754-756
【非特許文献19】Lee ら, 2000. Nat Biotechnol.;18 (6):675-679. (ref 31)
【非特許文献20】Schuldiner ら, 2000, PNAS. USA 97, 11307-11312
【非特許文献21】Schuldiner ら, 2001; Brain Res. 913, 201-5
【非特許文献22】Reubinoff ら, 2000, Nat Biotech., 18, 399-404
【非特許文献23】Reubinoff ら, 2001, Nat Biotech., 19, 1134-1140
【非特許文献24】Tropepe.ら, 2001, Neuron 30,65-78
【非特許文献25】Zhang ら, 2001, Nat Biotech., 19, 1129-1133
【非特許文献26】Price J. & Williams BP. Current Opinion in Neurobiology 11, 564-567 (2001)
【非特許文献27】Arenas 2002. Brain Res Bull.; 57 (6):795-808. (ref 28)
【非特許文献28】Bjorklund ら, 2002, Proc Natl Acad Sci USA. Feb 19, 99(4): 2344-9
【非特許文献29】Rossi F. & Cattaneo E. Nat Rev Neurosci. 2002 May;3(5):401-9
【非特許文献30】Gottlieb DI. Annu Rev Neurosci. 2002;25:381-407
【発明の開示】
【0017】
本発明は、GSK-3βの阻害、例えば、GSK-3β自身を標的とすることにより、若しくはβ-カテニン活性を模倣若しくは増強することにより、細胞におけるドーパミン作動性ニューロン表現型の誘導、又は促進することに関する。本発明は、実用的な治療法の手段とWntリガンドに代わる代替法として、Wntシグナル伝達を形質導入するシグナル伝達経路の活性化を提供する。WntリガンドとFrizzledレセプターのいずれのリガンドも本発明では使用しない。
【0018】
GSK-3βは、多機能性セリン/スレオニンキナーゼであり、β-カテニンの分解に不可欠である。参照:Doble and Woodgett J Cell Sci. 2003 Apr 1;116(Pt 7):1175-86. Human accession number: NM_002093。
【0019】
本明細書中で開示したように、本発明によれば、細胞内でGSK-3βを阻害することにより、培養物内であろうが脳内であろうが、またその阻害が直接的であろうが間接的であろうが、ドーパミン作動性ニューロン、前駆体細胞、前駆細胞、若しくは幹細胞の増殖及び/又は自己複製の誘導又は促進することが可能となる。及び/又は、ドーパミン作動性ニューロンの産出量を高めることによりドーパミン作動性ニューロン、前駆体、前駆細胞、若しくは幹細胞の生存、分化、成熟化の促進を可能にし、及び/又は、in vitro、in vivoで幹細胞、前駆、前駆体細胞、若しくは神経細胞における神経のドーパミン作動性運命の誘導が可能となる。
【0020】
本発明は、GSK-3βの阻害により神経分化を増加することを提供する。そして、さらにNurr1を発現している前駆体のTH+ニューロンへの変換を介してドーパミン作動性ニューロン集団内でのドーパミン作動性ニューロン数を増加させる方法を提供する。
【0021】
GSK-3βは、直接的に又は間接的に阻害されてよい。阻害は、例えば上流又は下流のシグナル因子に作用することによって、例えば、dishevelled、アキシン、FRAT/GBP、若しくはカゼインキナーゼ1の分解や機能欠損、又はβ-カテニンの過剰発現若しくは安定化若しくは分解の阻害、若しくはβ-カテニンリン酸化の阻害によって、間接的になされてもよい。本明細書でGSK3の阻害とは、文中特に示した場合を除き、これらのアプローチのいずれかを意味し、実際、Frizzledレセプターのリガンド、例えばWntによる場合を除いたいずれのアプローチも意味する。
【0022】
GSK-3β阻害剤のインディルビン(indirubin)ファミリーは、当初、慢性骨髄性白血病を含む様々な慢性病に対して使用される伝統的な漢方薬から分離された(19)。ごく最近になって、幹細胞治療においてGSK-3β阻害剤としてのインディルビンファミリーの潜在的な応用法が報告された(15)。DA前駆体の分化における化学的なGSK-3β阻害剤の新しい性質について記載した我々の結果は、PD用細胞補充療法の開発にとって重要な意味を持つ。現在の治療アプローチは、ヒト胎児中脳DA前駆体をPD患者の新線条体へ移植することに焦点が当てられている(27)。これらのアプローチの成功は、移植されるDA神経数に決定的に依存し、また1患者あたり5人以上の胎児を必要とする(27、28)。VM前駆体移植に対するさらに可能性のあるアプローチは、DAニューロンに前分化したヒト幹細胞の移植を含んでいる。しかしながら、ヒト幹細胞のDAニューロンへの分化(29、30)は、マウス幹細胞に関して報告されたそれよりも困難であることが判明し(22、31-33)、幹細胞調製で与えねばならない新規の分化シグナルが必要であることが強調された。我々の結果は、GSK-3β阻害が前駆体のDA分化を効率的に促進することを示し、またそのような阻害はPD患者へ移植する前の細胞調製に利用し得ることを示唆している。
【0023】
β-カテニンシグナル伝達は、一般に中枢神経系の発生において神経系前駆細胞の増殖に関与している(2、34、35)。実際、神経幹/前駆細胞において活性なプロモーターの制御下でβ-カテニンを恒常的に発現させると、完全な神経管の増殖を生じる(34,35)。これは、前駆細胞増殖におけるβ-カテニンの役割を支持している。しかしながら、これらの実験は、神経分化期間の後期発生段階におけるβ-カテニンの可能性のある役割に関しては直接的に見ていない。
【0024】
我々は、以前にWnt-1とWnt-5aがVM前駆体培養物内でVM前駆体の増殖とDA分化をそれぞれ促進することを示した。さらに、我々は、in vivo でDA前駆体が分化する場所であるVMドメインにおいて、E10.5胚でNurr1/β−カテニンの共局在とTCF/LEF転写活性を見出した(7)。これらの結果は、Wnt/β-カテニンシグナル伝達がDA前駆体において増殖と分化の両方を調節できることを示唆している。
【0025】
本明細書では、GSK-3β阻害とβ-カテニン安定化が、前駆体のDAニューロンへの分化と、総神経細胞集団のうちのDAニューロンの割合の両方を増加することを報告する。
【0026】
我々の結果は、皮膚(38−40)などの他の器官の分化と同様に、感覚神経の分化(37)や樹上突起形成(10)におけるWnt/β-カテニンシグナルについての役割を支持している最近の発表と一致している。このように、我々のデータは細胞分化におけるβ-カテニンの一般的な役割についての証拠を提供し、またGSK-3β阻害は神経及びグリアの前駆体の両方を含む細胞調製品においてDA分化を促進することから細胞補充療法に非常によく適しているかもしれないことを示している。
【0027】
GSK-3βの阻害剤は、当該技術分野で利用可能である。以前から知られているGSK阻害剤はリチウム(マグネシウム競合剤)と亜鉛である。GSK-3βは、系統発生学的にはCDKと非常に密接に関連している。研究により、GSK-3βとある程度のCDKに対して作用することが判明している少なくとも4つのATP競合キナーゼ阻害剤クラスが存在することが明らかとなった。すなわち、ヒメナルジシン類(hymenaldisines)、インディルビン類(indirubins)、アロイシン類(aloisines)、そしてパウロン類(paullones)である。最近、マレイミド(maleimides)、チアゾール誘導体、チエニル及びメチル・ハロメチル・ケトン、並びに酸化アミノピリミジンを含む、より強力で特異的なGSK3阻害剤が同定された。参考文献は以下のものを含む:Knockaert ら J Biol Chem. 2002 Jul 12;277(28):25493-501; Hoessel ら (1999) Nat Cell Biol. ; 1 (1): 60-67; Leclerc ら (2001) J Biol Chem; 276 (1): 251-260; Conde ら J. Med. Chem. 46, 4631-4633 (2003); Cline ら Diabetes 51, 2903-2910 (2003); Ring ら Diabetes 52, 588-595 (2003); Bhat ら J. Biol. Chem. 278, 45937-45945 (2003); Kuo et al J. Med. Chem. 46, 4021-4031 (2003)。
【0028】
当該技術分野には、GSK-3β阻害の調節に利用できる物質を同定するための様々な技術がある。例えば、供試化合物の、GSK3の基質(β-カテニン、グリコーゲンシンターゼ、Tau、elf2B、CREB、c-jun)のリン酸化を阻害するが、他の酵素(サイクリン依存性キナーゼ、プロテインキナーゼA、B、C、SAPキナーゼ、JNK、MAPK、MEK、カゼインキナーゼ、Rhoファミリー、PDK1等)の基質のリン酸化は阻害しない能力を評価する。
【0029】
GSK-3β阻害に有用であるとして同定された化合物(例えば、GSK-3β阻害剤)は、それらの活性を増強する目的で、当業者に周知のいずれかの方法で改変することができる。
【0030】
好ましい実施形態において、阻害剤はヒメナルジシン、アロイシン、マレイミド、チアゾール、チエニル及びメチルハロメチルケトン、酸化アミノピリミジン、インディルビン(例えば、インディルビン−3−モノオキシム(indirubin−3−monoxime:I3M))、又はパウロン(例えば、ケンパウロン(kenpaullone:KP))のようなGSK-3βに対して特異的なものである。特に好ましい実施形態において、阻害剤はケンパウロンである。参考文献:Leclerc ら (2001) J Biol Chem; 276 (1): 251-260; Hoessel ら (1999) Nat Cell Biol. ; 1 (1): 60-67; Polychronopoulos ら J Med Chem. 2004 Feb 12;47(4):935-46; Conde ら J. Med. Chem. 46, 4631-4633 (2003); Cline ら Diabetes 51, 2903-2910 (2003); Ring ら Diabetes 52, 588-595 (2003); Bhat ら J. Biol. Chem. 278, 45937-45945 (2003); Kuo ら J. Med. Chem. 46, 4021-4031 (2003)。
【0031】
本発明のいかなる局面や実施形態も、神経細胞、すなわち神経に応用することができ、また使用し得る。本開示において「神経の細胞」とは、ニューロン(神経細胞)であってよい。
【0032】
ドーパミン作動性ニューロンが豊富な細胞調製品は、パーキンソン病や他の疾患における細胞補充療法に利用してもよく、また、ドーパミン作動性ニューロンにおけるシグナル伝達事象とin vitroでのドーパミン作動性ニューロンへの薬物の効果を例えば、ハイスループット・スクリーニングで研究するのに利用してもよい。
【0033】
本発明の局面と実施形態は、特許請求の範囲に示されている。
【発明を実施するための最良の形態】
【0034】
一つの形態として、本発明は幹細胞、神経幹細胞、又は神経系前駆若しくは前駆体細胞においてドーパミン作動性ニューロンの運命を誘導する方法、又は神経細胞でのドーパミン作動性誘導若しくは分化を増強する方法、又はドーパミン作動性前駆体若しくは前駆細胞若しくはNurr1を発現している幹細胞を増幅する方法を提供する。この方法は、GSK-3β阻害を調節すること(例えば、GSK-3βの阻害剤で、β-カテニンを過剰発現若しくは供給することで、β-カテニンのリン酸化を阻害することで、又はβ-カテニンの分解を阻害することで、又はGSK-3β阻害の他の調節で細胞を処理することによって)、それによりドーパミン作動性ニューロンを生じさせることである。本方法は予めNurr1サブファミリーの核内レセプターを細胞内の基底レベルよりも高く発現することを任意に含む。
【0035】
GSK-3β阻害(又は、論述したように間接的にGSK-3βを阻害すること)を調節すること、例えば、誘導することは、in vivo, ex vivo, in vitro若しくは培養物内で行えばよい。in vitro, ex vivo, 若しくは培養物内で処理することが好ましいかもしれない。
【0036】
本発明の方法において、GSK-3β阻害に作用する物質で処理することは、物質と細胞を接触させることによってもよい。このような物質(例えば、GSK-3β阻害剤)での処理は、幹、前駆若しくは前駆体細胞を含む培養物、又はin vivo におけるそのような細胞にその物質を与えることによってでもよい。もし、GSK-3β阻害剤の処理に代って、GSK-3βの阻害を間接的に行うのならば、間接的阻害を生じる処理(例えば、β-カテニンの安定化、β-カテニンの過剰発現を生じさせること等)が、in vivoで同様に実行されてもよい。または可能であれば、in vitro、ex vivo若しくは培養物内で実行されてもよい。
【0037】
GSK-3β阻害で作用する物質の供給に加えて、幹、神経幹、前駆若しくは前駆体細胞、又は神経細胞をin vitro又はin vivoでタイプIアストロサイト/グリア細胞と共培養してもよいし、それらの細胞若しくはそれらの細胞から得られた因子と接触させてもよい。
【0038】
共培養された若しくはホスト細胞は、別の幹、神経幹、前駆若しくは前駆体細胞、又は神経細胞であってもよい。
【0039】
Nurr1(Lawら, 1992; Xingら, 1997; Castillo, 1997; GenBank nos. S53744, U72345, U86783)は、チロイドホルモン/レチノイン酸核内レセプタースーパーファミリーの転写因子の一つである。WO00/66713とWagnerら(1999)によって以前に報告されたように、神経幹細胞若しくは神経系前駆細胞での基底レベルを超えるNurr1の発現は、神経運命方向に分化する細胞数を増加する。神経運命の誘導は、in vitro又はin vivoで実行されてもよい。移植前若しくはその後に幹細胞又は神経幹、前駆若しくは前駆体細胞を神経運命に分化誘導する能力は、WO00/66713とWagnerら(1999)によって以前に報告されたように、グリア由来の信号によっても増強できる。
【0040】
Nurr1はNR4Aサブファミリーの一員である。本発明の方法はNurr1を使用することに限定されないが、Nurr1が好ましいかもしれない。また、方法はNR4Aサブファミリーのいずれかの核内レセプターを細胞内で基底レベルを超えて発現することを含み得る。NR4Aサブファミリーのレセプターは、Nurr1/NR4A2、Nor1/NR4A3、そしてNGFI-B/NR4A1を含む。したがって、本発明の方法では、NR4Aサブファミリーの核内レセプター(例えば、Nurr1、Nor1、又はNGFI-B)が、細胞内で基底レベルを超えて発現されてもよい。NR4Aサブファミリーメンバーのアクセッション・ナンバーは、例えば、以下の通りである。
【0041】
NGF-IB タンパク質: NP775181 NP775180 NP002126;
NGFI-B ヌクレオチド: NM_173158 NM_173157 NM_002135;
Nor-1 タンパク質: NP775292 NP775291 NP775290 NP008912S71930 Q92570;
Nor-1 ヌクレオチド: NM_005413 NM_173200 NM_173199 NM_173198 NM_006981。
【0042】
グリコタンパク質のWntファミリーのメンバーは、可溶性に乏しく(Bradley and Brown, 1990 and 1995)、また中脳の発生において発現している(Parr ら, 1993)。Wntは、中脳−後脳発生(McMahon and Bradley, 1990; Thomas and Capecchi, 1990)、神経パターニング(Kiecker and Niehrs, 2001; Nordstrom ら, 2002; Houart ら, 2002)、前駆体増殖(Taipale and Beachy, 2001; Chenn and Walsh, 2002; Megason and McMahon, 2002)、そして、神経系(Dorsky ら, 1998; Baker ら, 1999; Wilson ら, 2001; Garcia-Castro ら, 2002; Muroyama ら, 2002)を含む複数の組織での運命決定(Kispert ら, 1998; Ross ら, 2000; Hartmann and Tabin, 2001; Marvin ら, 2001; Schneider and Mercola, 2001; Tzahor and Lassar, 2001; Pandur ら, 2002)を調節している。
【0043】
本明細書中で用いられる時、「Wntポリペプチド」、「Wntグリコタンパク質」、また「Wntリガンド」とは、細胞間相互作用を調節する分泌タンパク質のWingless-intファミリーのメンバーをいう。Wntは、DrosophilaやCaenorhabditis elegansからXenopus、ゼブラフィッシュ、そして哺乳動物に至るまで高度に保存されている。哺乳動物で現在知られている19のWntタンパク質は、二つの細胞表面レセプター型に結合する。すなわち、現在10個のレセプターによって構成される7回膜貫通型ドメインFrizzled レセプターファミリー、並びに低密度リポタンパク質レセプター関連タンパク質5と6(LRP-5、-6)及びKremen1とKremen2のレセプターである。Wntによって運ばれたシグナルは、3つの既知シグナル伝達経路を介して細胞内に導入される。(1)いわゆるカノニカルシグナル伝達経路:当該経路ではGSK-3βが抑制されることにより、β−カテニンがリン酸化されず、分解を受けない。その後、β−カテニンは核内移行してTCFと複合体を形成し、Wnt標的遺伝子の転写を活性化する。(2)Jnkを介した平面極性及びコンバージェンス伸展(convergence-extension)経路、そして(3)イノシトール1,4,5三リン酸(IP3)/カルシウム経路:当該経路ではカルシニューリンが活性化T細胞の核内因子(NF-AT)を脱リン酸化して、それを活性化する(Saneyoshi ら Nature. 2002 May 16;417(6886):295-9)。レビューに関しては、いずれかのウェブブラウザで閲覧可能なWntホームページ(www.stanford.edu/~rnusse/wntwindow.html)をご覧いただきたい。Wntシグナル伝達に関連する他の共レセプターは、チロシンキナーゼレセプターRor1とRor2(Oishi I ら Genes Cells. 2003 Jul;8(7):645-54)、触媒的に不活性化する受容体型チロシンキナーゼをコードするderailed/RYKレセプターファミリー(Yoshikawa ら, Nature. 2003 Apr 10;422(6932):583-8)を含んでいる。
【0044】
「幹細胞」とは、自己再生可能ないずれの細胞タイプも意味する。もしそれが胚性幹(ES)細胞である場合には、一個体における全細胞を生じさせることができ、また、もしそれが多分化能若しくは神経幹細胞である場合には、神経、アストロサイト、オリゴデンドロサイトを含む神経系における全細胞を生じさせることができる。幹細胞は、以下の一又は二以上のマーカーを発現していてもよい。すなわち、記載のように(Tropepeら Neuron. 2001 Apr;30(1):65-78; Xu ら Nat Biotechnol. 2001 Oct;19(10):971-4)、Oct-4、Sox1-3、Nanog(Chambers ら, 2003, Cell, May 30;113(5):643-55.)、ステージ特異的胚性抗原(SSEA-1, -3, -4)、そして腫瘍拒絶抗原TRA-1-60及び-1-81等である。神経幹細胞は、一又は二以上の以下のマーカーを発現していてもよい。すなわち、ネスチン、p75ニューロトロフィンレセプター、Notch1、SSEA-1(Capela and Temple Neuron. 2002 Aug 29;35(5):865-75)等である。
【0045】
「神経系前駆細胞」とは、多分化表現型、及び/又は幹細胞と比べるとより限定された分化能を持った神経幹細胞の嬢細胞、子孫細胞を意味する。前駆体細胞は、発生期間に神経と直接的な系統関連にあるものだけでなく、限定的な環境的条件下で分化転換若しくは再分化を誘導され得る、又は神経表現型を獲得する他のいずれの細胞をも意味する。
【0046】
幹細胞、神経幹細胞、又は神経系前駆細胞若しくは前駆体細胞は、骨髄、皮膚、目、鼻粘膜上皮、若しくは臍帯、若しくは神経系部位(例えば、小脳、脳室帯、副脳室帯、線条体、中脳、後脳、大脳皮質、又は海馬状隆起由来のもの)を含む。それらは、脊椎動物の組織体から得てもよいし、それ由来のものであってもよい。ここでいう脊椎動物とは、例えば、ヒト、又はヒト以外の哺乳動物であるウサギ、モルモット、ラット、マウス若しくは他のげっ歯類動物、ネコ、イヌ、ブタ、ヒツジ、ヤギ、ウシ、ウマ、若しくは霊長類動物のような哺乳動物、又はニワトリのような鳥でよい。
【0047】
本発明の好ましい実施形態では、成体幹/前駆/前駆体細胞がin vitro、ex vivo、又はin vivoで使用される。これは、同意成人(例えば、細胞を得ることに対して)と、適切な倫理委員会による承認を必要とする。ヒト胚/胎児を細胞源として使用する場合、ヒト胚は、通常使用されずに破壊されるか、いつまでも保管されるもの、特に妊娠困難な夫婦のIVF(体外受精)治療目的のために作成されたヒト胚である。通常IVFでは、移植及び究極的には妊娠に使用する数よりも多くのヒト胚の作成を伴う。このような予備胚は、一般には破壊される。破壊される以外にない胚は、関係者、特に直接関連する卵子提供者、及び/又は精子提供者の適切な同意をもって、パーキンソン病のような重度の神経変性疾患患者の利益に対して倫理上肯定的な方法で使用することができる。本発明自体は、その開発のいずれのステージにおいてもヒト胚の使用に関連しない。既に述べたように、本発明は恐ろしい病気に有益な治療法の開発を可能にすると同時に、ヒト胚に直接由来する材料を使用するに際して生じ得る必要性を最小限にしている。
【0048】
いくつかの好ましい実施形態において、幹若しくは前駆若しくは前駆体細胞は、GSK-3β阻害剤、若しくは論述したようなGSK-3βを阻害する他の手段等のGSK-3β阻害に作用する物質と接触される他、本発明のいずれかの局面に従って処理、及び/又は使用され、適切な同意を得た同意成人若しくは子供、例えば、本発明に従って製造され、及び/又は内在性のドーパミン作動性ニューロンの発生、若しくは機能を促進、若しくは誘導するためにGSK-3β阻害、GSK-3β阻害剤、若しくはGSK-3βを間接的に阻害する物質、及び/又は一又は二以上のタイプIアストロサイト/初期グリア細胞由来の因子で処理された神経を移植で戻すことによって治療されることになる疾患を持つ患者から得られる。
【0049】
幹若しくは前駆若しくは前駆体細胞が誘導される神経運命は、未分化表現型又は原始的な神経表現型を示してもよい。細胞は、一個体のどの細胞タイプをも生じ得る全能性細胞、又は複数の異なる神経表現型を生じ得る多分化能細胞、又は通常の発生期間では、より限られた表現型を生じ得るが、in vitroで適当な環境因子に晒された場合、その他の細胞を生じ得る前駆体若しくは前駆細胞であってもよい。細胞は、特定の神経運命と関連するマーカー(例えば、チロシン水酸化酵素)を欠いていてもよい。
【0050】
VM前駆体がGSK-3β阻害の調節、特に例えばGSK-3β阻害剤によるGSK-3β阻害の誘導を受け、あるいはその他GSK-3βの阻害を受ける本発明の神経運命を誘導する方法において、増加した割合の細胞が神経運命を取るように誘導され得る。ドーパミン作動性の誘導や分化が神経細胞で増強され得る。好ましい実施形態では、腹側中脳においてTH+/TuJ1+細胞の割合で3〜5倍増加する。
【0051】
培養条件は、異なる密度で、異なる培地で、異なる基質で、又は他の細胞集団の存在下で細胞を培養することを含むが、これに制限されることはない。
【0052】
「細胞内で基底レベルを超えるNurr1の発現」とは、in vivoで非病的条件下の(未改変の)細胞で発現しているレベルよりも多くのNurr1を発現することを意味する。同様に、細胞内での基底レベルを超えるNurr1サブファミリーの核内レセプターの発現とは、in vivoで非病的条件下の(未改変の)細胞で発現しているレベルよりも多くの核内レセプターを発現することを意味する。基底レベルを超える発現は、転写の、翻訳の、翻訳後の、薬理学的な、人工のアップレギュレーション及び過剰発現を含む。基底レベルを超える核内レセプターの発現を、本明細書ではNurr1に関して記載する。この開示は、またNurr1サブファミリーの他のメンバーに対しても当てはめることができ、またそのサブファミリーの他の核内レセプターである、例えば、Nor1やNGFI-Bなどを用いて本発明の方法に使用することができる。このように、Nurr1が例示されてはいるが、本発明の方法はNurr1に限定されず、Nurr1サブファミリーのあらゆる核内レセプターに及ぶ。
【0053】
基底レベルを超えるNurr1の発現は、当業者に周知のいずれの方法によって達成されてもよい。一例として、基底レベルを超える発現は、本来のゲノムNurr1の調節を調節することによって誘導されてもよい。これは、Nurr1 mRNA若しくはタンパク質の分解を阻害すること若しくは妨げることにより、又はNurr1の転写及び/又は翻訳を増加すること、例えば、Nurr1の転写をアップレギュレートする線維芽細胞増殖因子8(FGF8)(Rosenthal, A., (1998) Cell, 93(5),755-766)と細胞とを接触させることにより、及び/又は異種調節配列をNurr1の本来の調節領域の内部若しくはその近傍に導入することにより、及び/又はNurr1の本来の調節領域を、例えば相同的組み換えによって上記のような異種調節配列と置き換えることにより、及び/又はNurr1の転写、翻訳、若しくは機能を負に調節する、妨げる若しくはダウンレギュレートするような分子、例えば、Nurr2(Ohkura, ら, (1999) Biochim Biophys Acta 14444: 69-79)を破壊、若しくはダウンレギュレートすることにより、及び/又はNurr1 mRNA若しくはタンパク質を細胞内に直接マイクロインジェクションすることにより、行われてもよい。
【0054】
転写は、幹、神経幹、前駆体、前駆細胞、若しくは神経細胞に対して、例えば、細胞を転写活性化因子と接触させることにより、あるいはその活性化因子をコードした核酸で細胞を形質転換することによって前記活性化因子のレベルを増大することにより、増加されてもよい。代替方法として、転写はNurr1転写阻害物質に対するアンチセンス核酸で細胞を形質転換することにより増加させてもよい。
【0055】
したがって、幹、神経幹、前駆体、前駆細胞、若しくは神経細胞で神経運命を誘導、又は誘導を増強する本発明の方法は、FGF8やFGF20(Ohmachi ら, 2000)と細胞を接触させることを含んでいてもよい。
【0056】
内在性のNurr1の転写、及び/又は翻訳を増加させる代替法若しくは追加法として、基底レベルを超えるNurr1の発現が、幹、神経幹、前駆体、前駆細胞、若しくは神経細胞内への一又は二以上のNurr1の追加のコピーを導入することによって生じるものであってもよい。
【0057】
したがって、別の局面において、本発明は、幹細胞、神経幹細胞、神経系前駆、前駆体、若しくは神経細胞において神経運命を誘導する、及び/又はドーパミン作動性の発生の誘導を増強する方法、又は神経細胞においてドーパミン作動性の誘導若しくは分化を増強する方法を提供し、該方法は、GSK-3β阻害を調節する物質、例えば、GSK-3β阻害剤、若しくはGSK-3βを阻害する手段と細胞を接触させることに加えて、Nurr1で細胞を形質転換することを含んでいる。
【0058】
幹、神経幹、前駆体、前駆細胞、若しくは神経細胞への形質転換は、in vitro又はex vivoで行うことができる。細胞が誘導される神経運命は、本明細書中で論じられるタイプであればよい。例えば、原始神経表現型を示してもよいし、特定の神経運命に関連するマーカーを欠いていてもよい。さらに本発明は、Nurr1で形質転換され、またGSK-3β阻害と接触した幹細胞、神経幹細胞、若しくは前駆若しくは前駆体細胞を提供する。
【0059】
形質転換されたNurr1は、追加のゲノムベクターに含まれていてもよく、あるいはゲノム内に、好ましくは安定的に組み込まれてもよい。以下でより詳細に述べるように、該Nurr1は、幹細胞、又は神経幹細胞、前駆体若しくは前駆細胞、又は神経細胞内で基底レベルを超える発現を駆動するプロモーターに機能可能なように連結されてもよい。
【0060】
「機能可能なように連結」とは、プロモーターから開始される転写について適当な位置と方向性で、同じ核酸分子の一部として連結していることを意味する。
【0061】
細胞内に遺伝子を導入する方法は、当業者には周知である。ベクターは、Nurr1がベクター上に留まっていようがいまいが、ゲノム内に取り込まれようが、幹、若しくは神経幹、前駆体若しくは前駆細胞、又は神経細胞中にNurr1を導入するために使用されてよい。適切な調節配列を有し、プロモーター配列、ターミネーター断片、ポリA配列、エンハンサー配列を含んだ適当なベクターが選択、又は構築され得る。ベクターは、マーカー遺伝子、及び適当な他の配列を含んでいてもよい。調節配列は、幹、若しくは細胞幹、前駆体若しくは前駆細胞、又は神経細胞内でNurr1の発現、及び/又はGSK-3βの阻害(直接的であろうが、間接的であろうが)を駆動することができる。例えば、ベクターは追加のゲノム発現ベクターでもよいし、又は調節配列がNurr1と共にゲノム内に組み込まれてもよい。ベクターは、プラスミド又はウィルス性のものであってもよい。
【0062】
Nurr1をユーザの制御下に置くために、Nurr1は外部から誘導可能な遺伝子プロモーターの制御下に置かれてもよい。プロモーターに対して使用される用語「誘導可能」は、当業者には十分理解されている。要するに、誘導可能なプロモーターの制御下で発現が「スイッチ オン」され、又は作動刺激に応答して発現が増加されることである。刺激の性質は、プロモーター間で多様である。いくつかの誘導可能なプロモーターは、適切な刺激のない状態では発現(又は非発現)をほとんど生じさせないか、又は検出不能なレベルしか生じさせない。刺激のない状態での発現のレベルがいかなるものであっても、いずれの誘導可能なプロモーターも適当な刺激の存在下で発現が増加され得る。誘導可能なプロモーターの一例としては、遺伝子発現がテトラサイクリン類似物によって調節されるテトラサイクリン オン/オフ系(Gossenら, 1995)がある。
【0063】
さらなる詳細に関しては、例えば、Molecular Cloning: a Laboratory Manual: 3rd edition, Sambrook and Russell, 2001, Cold Spring Harbor Laboratory Pressをご覧いただきたい。核酸操作、例えば、核酸構築の調製、突然変異誘発、配列決定、細胞内へのDNAの導入、そして遺伝子発現、及びタンパク質の解析に関する数多くの既知の技術とプロトコルが、Current Protocols in Molecular Biology, Ausubelら編, John Wiley & Sons, 1992 、又はその後の版に詳細に記述されている。
【0064】
当業者に周知の抗生物質耐性若しくは感受性遺伝子のようなマーカー遺伝子が、目的の核酸を含むクローンを同定するために使用されてもよい。クローンは、同定されるか、又は例えばサザンブロットハイブリダイゼーションによる結合実験によってさらに調べられてもよい。
【0065】
Nurr1を含む核酸は、ホストの幹、神経幹、前駆、前駆体、若しくは神経細胞のゲノム内に組み込まれてもよい。組み込みは、標準的な技術に従ってゲノムとの組み換えを促進する形質転換された核酸配列を含むことで促進されてもよい。組み込まれる核酸は、幹細胞、または神経幹、前駆若しくは前駆体細胞、又は神経細胞でNurr1遺伝子の発現を駆動できる調節配列を含んでいてもよい。核酸は、ゲノム上の一部位であって、幹、若しくは神経幹、前駆体若しくは前駆細胞、又は神経細胞内でNurr1コーディング配列がその発現を駆動、及び/又は制御できる調節エレメントの制御下となるように、その組み込みを導く配列を含んでいてもよい。本明細書中で述べるように、組み込まれる核酸は、幹細胞、又は神経幹、前駆体若しくは前駆細胞、又は神経細胞内でNurr1を形質転換するために使用されるベクター由来であってもよい。
【0066】
Nurr1を含む核酸の導入は、その核酸が直鎖状であろうが、枝分かれ状であろうが、環状であろうが、一般に「形質転換」ということができるが、限定されることはない。該導入には、いずれの利用可能な技術を使用してもよい。適切な技術としては、リン酸カルシウム−トランスフェクション法、DEAE-デキストラン法、PEI、エレクトロポレーション法、マイクロインジェクションのような機械的技術、直接的DNAの取り込み、レセプターを介したDNA転移、レトロウィルス若しくは他のウィルスを用いた形質導入、そしてリポソーム若しくは脂質仲介型トランスフェクション等が挙げられる。選択された遺伝子構築物を細胞内に導入するに際し、当業者に周知の考慮すべきことがある。当業者には明白ではあるが、幹細胞、又は神経幹、前駆体若しくは前駆細胞、又は神経細胞にNurr1を導入するための特定の形質転換法の選択は本発明の本質ではなく、それに限定されるものでもない。
【0067】
幹、又は神経幹、前駆体若しくは前駆細胞、又は神経細胞のNurr1によるin vivoトランスフェクションに関する適切なベクターと技術は、当業者に周知である。適切なベクターには、アデノウィルス、アデノ随伴ウィルス、パポーバウィルス、ワクシニアウィルス、ヘルペスウィルス、レンチウィルス、そしてレトロウィルスが含まれる。無能化ウィルスベクターは、感染ウィルス粒子の生産に必要な遺伝子を発現するヘルパー細胞株内で製造されてもよい。適当なヘルパー細胞株は、当業者には周知である。例として、Fallaux, F.J., ら, (1996) Hum Gene Ther 7(2), 215-222; Willenbrink, W., ら, (1994) J Virol 68(12), 8413-8417; Cosset, F.L., ら, (1993) Virology 193(1), 385-395; Highkin, M.K., ら, (1991) Poult Sci 70(4), 970-981; Dougherty, J.P., ら, (1989) J Virol 63(7), 3209-3212; Salmons, B., ら, (1989) Biochem Biophys Res Commun 159(3), 1191-1198; Sorge, J., ら, (1984) Mol Cell Biol 4(9), 1730-1737; Wang, S., ら, (1997) Gene Ther 4(11), 1132-1141; Moore, K.W., ら, (1990) Science 248(4960), 1230-1234; Reiss, C.S., ら, (1987) J Immunol 139(3), 711-714をご覧いただきたい。ヘルパー細胞株は、通常、ウィルスゲノムをパッケージする機構によって認識される配列を失っている。該細胞株は、核酸を含まないウィルス粒子を生産する。完全なパッケージングシグナルを含むウィルスベクターは、ヘルパー細胞において送達される遺伝子(例えば、Nurr1)や他の配列と共に感染性ウィルス粒子内にパッケージされる。これを幹、又は神経幹、前駆体若しくは前駆細胞、又は神経細胞への遺伝子送達用に利用してもよい。
【0068】
内在性のNurr1の転写、及び/又は翻訳を増加させる代替法若しくは追加として、基底レベルを超えるNurr1の発現は、マイクロインジェクション、又は例えば細胞浸透性ペプチド、TAT、トランスポータン(transportan)、アンテナペディア−ペネトラチンペプチド(Antennapedia penetratin peptides)(Lindsay 2002)を含む、他の担体ベースの、若しくはタンパク質送達のシステムによって、一又は二以上の追加のNurr1タンパク質コピーを幹、神経幹、前駆体、前駆細胞、又は神経細胞内に導入して生じるものであってもよい。
【0069】
さらなる局面において、本発明は、幹細胞、神経幹細胞若しくは前駆若しくは前駆体細胞、若しくは神経細胞において特定の神経運命を誘導する方法であって、該幹細胞若しくは前駆細胞若しくは神経細胞が基底レベルを超えるNurr1を発現する方法を提供する。該方法は、細胞を、例えばGSK-3βの阻害剤のようなGSK-3βの阻害を調節する物質と、又はβ-カテニンの過剰発現、若しくは安定化、若しくは分解の阻害、又はβ-カテニンのリン酸化の阻害を引き起こす物質と、又は記述したような他のGSK-3β阻害誘導物質と、そして場合によっては、タイプIアストロサイト/グリア細胞によって供給される、若しくはそれらから得られる一又は二以上の任意因子と共に、接触させることを含んでいる。因子は、共培養することで、又は幹、前駆、若しくは前駆体細胞、又は神経細胞とタイプIアストロサイト/グリア細胞とを接触させることによって供給されてもよい。この方法は、in vitro、又はin vivoで行ってよい。基底レベルを超えるNurr1を発現する幹細胞、又は神経幹、前駆体、若しくは前駆細胞、又は神経細胞は、Nurr1での細胞の形質転換によって生産されてもよい。
【0070】
因子は、不死化したアストロサイト/グリア細胞によって供給されても、又はそれらから得られてもよい。因子は、グリア細胞株、例えば、アストロサイト、若しくは放射状グリア、若しくは未成熟グリア中脳細胞株によって供給されても、又はそれらから得られてもよい。細胞株は、同種の細胞集団を提供する。
【0071】
本発明の重要な局面は、細胞内での基底レベルを超えるNurr1の発現と、腹側中脳のタイプIアストロサイト/初期グリア細胞によって供給される若しくはそれらから得られる一又は二以上の因子との接触を含むプロセスによって、ドーパミン作動性ニューロンがin vitroにおいて幹細胞、又は前駆若しくは前駆体細胞から発生し得る一方で、ドーパミン作動性の運命の誘導がGSK-3β阻害物質との接触で増強、又は促進されるという発見に基づいている。
【0072】
本発明は、多数のドーパミン作動性ニューロンの発生を可能とする。これらのドーパミン作動性ニューロンは、パーキンソン病で退化したり、又は損傷を受けた若しくは失われた細胞と置換するための原材料として使用することができる。
【0073】
基底レベルを超えるNurr1を発現している細胞は、GSK-3βを阻害するための手段と接触させる際に有糸分裂し得る。
【0074】
本発明の方法において、細胞は付加的に、塩基性繊維芽細胞増殖因子(bFGF)、上皮細胞増殖因子(EGF)、そしてレチノイドX受容体(RXR)の活性化剤、例えば、合成レチノイド類似物SR11237(Gendimenico, G. J., ら, (1994) J Invest Dermatol 102(5), 676-80)、9−シス−レチノール 、又はドコサヘキサエン酸 (DHA)、又は LG849(Mata de Urquiza ら, 2000)、から選択された一又は二以上の物質と接触させてもよい。以下で実験的に立証されるように、本発明に従って細胞を一又は二以上のこれらの物質で処理することは、ドーパミン作動性の運命をとる幹、前駆、若しくは前駆体細胞の割合を増やすため、又は神経細胞におけるドーパミン作動性の誘導若しくは分化を促進するために使用することができる。本発明に従ってドーパミン作動性の運命を誘導する、又は神経細胞でドーパミン作動性の誘導や分化を増強する方法は、成長因子のFGFファミリーの一員、例えば、FGF4、FGF8 又はFGF20と細胞を接触させることを含んでいてもよい。
【0075】
有利な点は、細胞を二以上の上記物質と接触させてもよいことである。本発明者は、図らずもbFGF 又はEGFの有益な効果が飽和量でも相加的であることを発見した。この発見は、これらの物質が異なる機構を介して作用できることを示唆している。
【0076】
ドーパミン作動性表現型を誘導する方法は、幹細胞、神経幹細胞、又は神経前駆若しくは前駆体細胞、又は神経細胞を、GSK-3β阻害を調節するか又はGSK-3βを阻害するための手段(直接的なGSK-3β阻害剤であろうが、間接的にGSK-3を阻害する手段、例えば、β-カテニンの過剰発現、若しくは安定化、若しくは分解の阻害、又はβ-カテニンのリン酸化の阻害であろうが)と、そして任意に、腹側中脳のタイプIアストロサイト/グリア細胞によって供給される、若しくはそれらから得られる一又は二以上のさらなる因子とを接触させる前に、例えば、該細胞を腹側中脳タイプIアストロサイト/グリア細胞若しくはそれらから得られる因子と接触する前、又は共培養する前に、bFGF 及び/又はEGF で前処理することを含んでいてもよい。
【0077】
この任意の前処理ステップは、以前にWO00/66713とWagnerらによる1999年の論文で報告されたさらに二つの予期せぬ発見から生まれた。その発見とは、(i)基底レベルを超えるNurr1を発現及び高い増殖を示す神経幹細胞株が腹側中脳のタイプIアストロサイト/グリア細胞と共培養されるとドーパミン作動性の運命への誘導が促進されることが立証されたこと、そして(ii)無血清培地(SFM)内でのbFGF 及び/又はEGF処理後、SFMのみに移した後でも、基底レベルを超えるNurr1を発現しているほとんどの幹細胞株が高い増殖のベースラインを維持していたこと、である。
【0078】
ドーパミン作動性表現型の誘導方法は、幹細胞、又は神経幹、若しくは前駆、若しくは前駆体細胞を成長因子のFGFファミリーの一員、例えば、FGF2、FGF4、 FGF8、又はFGF20で前処理することを含んでいてもよい。
【0079】
細胞は、付加的に一又は二以上のWntリガンド、例えば、WO2004/029229に記載されているようなもので処理されてもよい。
【0080】
神経運命を幹、神経幹若しくは前駆若しくは前駆体細胞で誘導する、又は神経細胞でドーパミン作動性の誘導や分化を増強する本発明に係る方法は、神経運命用のマーカーを検出することを含んでいてもよい。β-チューブリンIII(TuJ1)は神経運命のマーカーの一つである(Menezes, J. R., ら, (1994) J Neurosci 14(9), 5399-5416)。他の神経マーカーとしては、神経フィラメントとMAP2がある。もし、特定の神経表現型が誘導されるのならば、マーカーはその表現型に特異的なものにすべきである。ドーパミン作動性の運命については、チロシン水酸化酵素(TH)、ドーパミントランスポーター(DAT)、そしてドーパミンレセプターの発現が、例えば、免疫反応性やin situ ハイブリダイゼーションによって検出されればよい。チロシン水酸化酵素は、DA細胞用の主要なマーカーである。ドーパミンと代謝産物の量、及び/又は放出は、例えば、高圧液体クロマトグラフィー(HPLC)(Cooper, J. R., ら, The Biochemical Basis of Neuropharmacology, 7th Edition, (1996) Oxford University Press)によって検出されればよい。(TH/ドーパミン/DATの存在下で)ドーパミンβ水酸化酵素、及びGABA又はGADの欠如も、またドーパミン作動性の運命を表す。付加的なマーカーとしては2型アルデヒド脱水素酵素(ADH-2)、GIRK2、 Lmx1b、そしてPtx3がある。
【0081】
マーカーの検出は、当業者に周知のいずれの方法に従って行われてもよい。検出方法は、マーカーをコードしている核酸配列に結合することができる特異的結合メンバーを使用することができ、この特異的結合メンバーは、その配列とハイブリダイズできる核酸プローブ又は核酸配列若しくはそれによってコードされたポリペプチドに対して特異性をもつ免疫グロブリン/抗体ドメインを含んでおり、配列やポリペプチドへの特異的結合メンバーの結合を検出できるように標識されていてもよい。「特異的結合メンバー」は、マーカーに対して特別な特異性を有しており、通常条件では、他の種に優先してそのマーカーに結合する。別の方法として、マーカーが特異的なmRNAである場合、特異的なオリゴヌクレオチドプライマーに対する結合、及び例えばポリメラーゼ連鎖反応(PCR)で増幅することによって検出してもよい。
【0082】
核酸プローブとプライマーは、ストリンジェントな条件下でマーカーとハイブリダイズしてもよい。適切な条件としては、例えば、約80−90%の同一性をもつマーカー配列の検出に関してであれば、ハイブリダイゼーションが0.25M Na2HPO4(pH7.2),6.5% SDS,10%デキストラン硫酸溶液中にて42℃で一晩処理された後、0.1×SSC,0.1%SDS溶液中にて、55℃で最終洗浄することが挙げられる。90%以上の同一性があるマーカー配列の検出に関して、ハイブリダイゼーションの適切な条件は、0.25M Na2HPO4(pH7.2),6.5%SDS,10%デキストラン硫酸溶液中にて65℃で一晩処理され、0.1×SSC,0.1%SDS溶液中にて、60℃で最終洗浄することが挙げられる。
【0083】
別の局面において、本発明は、本明細書中に開示したいずれか一の方法に従って製造された神経を提供する。神経は原始的な神経表現型を有していてもよい。該神経は、複数の異なる神経表現型を生じることができてもよい。神経は、特定の神経表現型を有していてもよい。その表現型は、基底レベルを超えるNurr1を発現している幹、神経幹、前駆、前駆体、若しくは神経細胞と接触する因子を供給した、又はそれらの因子が得られたアストロサイト/グリア細胞のタイプによって、及び/又は幹、神経幹、前駆、前駆体、若しくは神経細胞が共培養、又は接触したアストロサイト/グリア細胞のタイプによって影響される。好ましい実施形態においては、神経がドーパミン作動性表現型を有する。
【0084】
神経は、神経内で分子の発現を駆動できるプロモーターに機能可能なように連結された神経保護若しくは神経修復の性質をもつ分子をコードする核酸を含んでいてもよい。プロモーターは、例えば、TetONキメラプロモーターといった誘導可能なプロモーターであることができ、それにより、いかなる損傷性の過剰発現も防ぐことができる。プロモーターは特定の神経表現型と関連するもの、例えば、THプロモーターやNurr1プロモーターであってもよい。
【0085】
コードされた分子は、その発現により神経がその環境から独立することになるようなものでよく、すなわち、その生存が、例えば、それが移植される神経環境下における一又は二以上の因子又は状態の存在に依存しないようにすることができる。例として、神経は、神経内で分子の発現を駆動できるプロモーターに機能可能なように連結された、以下に記載する一又は二以上の神経保護、若しくは神経修復性分子をコードする核酸を含んでいてもよい。
【0086】
追加若しくは代替法として、コードされた分子の発現が、その神経周辺の細胞環境の神経保護、若しくは神経修復に機能してもよい。この方法において、神経は、神経保護と神経修復の性質をもった分子を送達するようにする細胞遺伝子併用療法において使用されてもよい。
【0087】
神経保護と神経修復の性質をもつ分子の例として、以下のものが挙げられる。
【0088】
(i)神経変性を補償し、それを阻害することのできる神経栄養因子。一例は、ドーパミン作動性ニューロンからの発芽を促進し、チロシン水酸化酵素発現を増加する有効性の高い神経生存因子であるグリア由来神経栄養成長因子(GDNF)(Tomacら,(1995) Nature, 373, 335-339; Arenasら, (1995) Neuron, 15,1465-1473)である。GDNF軸策伸張増強により、GDNFは、それらが局所する環境に神経を分布させるために神経の能力を増大してもよい。GDNFファミリーのその他の神経栄養分子には、ニュールツリン(Neurturin)、ペルセフィン(Persephin)、そしてアルテミン(Artemin)が含まれる。ニューロトロフィンファミリーの神経栄養因子には、神経成長因子(NGF)、脳由来神経栄養因子(BDNF)、そしてニューロトロフィン−3、−4/5、及び−6がある。神経栄養活性をもつ他の因子には、例えば、FGF2、4、8、及び20といったFGFファミリーのメンバー、Wnt-1、-2、-5a、-3a、及び7aを含むWntファミリーのメンバー、BMP2、4、5、及び7、nodal、アクチビン、並びにGDFを含むBMPファミリーのメンバー、TGFα/βファミリーのメンバー等がある。
【0089】
(ii)抗アポトーシス性分子: 細胞死において中心的な役割を果たすBcl2。Bcl2の過剰発現は本来的に生じる細胞死と虚血(Martinouら, (1994) Neuron, 1017-1030)から神経を保護する。その他の神経に対する抗アポトーシス性分子には、BclX-Lがある。
【0090】
(iii)神経が分布し、その環境での連絡の形成する上で神経を援助する、例えばエフリンのような、軸索の再生、及び/又は伸張及び/又はガイドをする分子: エフリンは、チロシンキナーゼレセプターを活性化できる膜結合型リガンドのクラスであることが明らかとなっている。エフリンは、神経発生に関わっている(Irvingら, (1996) Dev. Biol., 173, 26-38; Krullら, (1997) Curr. Biol. 7, 571-580; Frisenら, (1998) Neuron, 20, 235-243; Gaoら, (1996) PNAS, 93, 11161-11166; Torresら, (1998) Neuron, 21, 1453-1463; Winslowら, (1995) Neuron, 14, 973-981; Yueら, (1999) J Neurosci 19(6), 2090-2101)。軸索伸張に関連するその他の分子のファミリーに、ネトリン、セマフォリン、そしてスリット(Nakamotoら Curr Biol. 2004 Feb 3;14(3):R121-3)などがある。
【0091】
(iv)転写因子、例えば、ホメオボックス ドメイン タンパク質Ptx3(Smidt, M. P.ら, (1997) Proc Natl Acad Sci USA, 94(24), 13305-13310)、Lmx1b、 Pax2、 Pax5、Pax8、若しくはengrailed 1若しくは2(Wurst and Bally-Cuif, 2001; Rhinn and Brand, 2001)、又はベーシック へリックス−ループ−へリックス ファミリーの神経原性遺伝子。
【0092】
本発明に従った、又は該発明で使用する神経は、一又は二以上の他の細胞タイプ、例えば幹、神経幹、前駆体若しくは前駆細胞、または神経細胞を、実質上含まない。神経は、抗体と磁気ビーズ、又は蛍光標識細胞分取装置(FACS)による細胞外エピトープの認識に基づくものを含めて、当業者に周知のいずれかの技術を用いて神経幹若しく前駆細胞から分離されてもよい。例として、幹、神経幹、前駆体若しくは前駆細胞では見られるが、神経では見られない分子の細胞外領域に対する抗体を使用してもよい。このような分子には、Notch 1、CD133、SSEA1、prominin1/2、RPTPb/phosphocan、TIS21、及びグリア細胞株由来神経栄養因子レセプターGFRα、又はNCAMが含まれる。抗体と結合する幹細胞は、補体曝露によって溶解されても、又は、例えば、磁気選別によって(Johanssonら, (1999) Cell, 96, 25-34)分離されてもよい。もしも、目的のレシピエントに対して異種の抗体が使用されたのであれば、そのときは、いずれも、例えば、そのような細胞選別方法を免れた幹、神経幹若しくは前駆若しくは前駆細胞も、異種抗体で標識され、そしてレシピエントの免疫機構の一次標的となる。別の方法として、希望する表現型を獲得する細胞は、細胞外エピトープに対する抗体、又は細胞タイプ特異的プロモーターの制御下にある蛍光タンパク質を含む導入遺伝子の発現によっても、分離することができる。例として、ドーパミン作動性ニューロンは、TH、DAT、Ptx3、Nurr1、又はドーパミン作動性ニューロンにより特異的に利用される他のプロモーターの制御下で発現した蛍光タンパク質で分離できる。
【0093】
本発明の方法は、希望する表現型をもった神経幹、前駆若しくは前駆体細胞、又は他の幹細胞、または神経細胞を富化するために、付加的なネガティブ又はポジティブ選択法を含んでいてもよい。
【0094】
ネガティブ選択は、DAニューロンを集めるために使用されてもよい。非DAニューロン用の選択的な神経毒が使用されてもよい。例えば、5−7−ジヒドロキシトリプタミン(5-7-dihydroxytryptamine)(セロトニン作動性ニューロン除去用)、又はサポニン若しくは毒素に結合した、若しくは補体追加後に一緒にされる抗体、例えば、GABA、又はセロトニン トランスポーターに対する抗体(GABA作動性、又はセロトニン作動性ニューロン除去用)が挙げられる。本発明の方法は、神経幹、前駆若しくは前駆細胞、又は他の幹、若しくは神経細胞を、好ましくはin vitroで、例えば、細胞を含むin vitro 培養物にネガティブ選択剤を添加することにより、又はネガティブ選択剤の存在下で細胞を培養することにより、ネガティブ選択剤にて処理すること、又は接触させることを付加的に含んでいてもよい。ネガティブ選択物質は、希望する細胞タイプ以外の細胞タイプに対して選択する。例えば、本発明が、ドーパミン作動ニューロン表現型を促進すること、増強すること、又は誘導することに関連する場合、ネガティブ選択剤は、DAニューロン以外の細胞、並びにDAニューロンを生じる細胞、例えば、幹細胞、及び神経幹、前駆体及び前駆細胞のような細胞に対して選択されてもよい。したがって、ネガティブ選択剤は、非DA表現型をもつ、例えば、非DAニューロンといった、分化した細胞に逆らって、選択してもよい。ネガティブ選択剤は、希望する細胞タイプ以外の細胞の増殖を減少、又は阻害、及び/又は殺してもよい。ネガティブ選択剤は、DAニューロン以外の細胞ニューロンの母数を減少する選択的神経毒素であってもよい。例えば、ネガティブ選択剤は、5−7−ジヒドロキシトリプタミン(セロトニン作動性ニューロンを減少するため)であってもよい。ネガティブ選択物質は、非DAニューロンに対する抗体、若しくは抗体断片であってもよい。該抗体や該抗体断片(例えば、scFvやFab)は、サポニン又は毒素に結合される。例えば、抗体は、GABAトランスポーター(GABA作動性ニューロンを減少するため)に特異的なものであってもよい。
【0095】
本発明の方法において、神経幹、前駆若しくは前駆細胞、又は他の幹若しくは神経細胞は、抗酸化剤(例えば、アスコルビン酸)、低酸素分割、及び/又は低酸素誘導因子(例えば、HIF又はエリスロポエチン)存在下で育ててもよい。
【0096】
本発明はさらに多様な局面と実施形態で、例えばパーキンソン症候群やパーキンソン病をもつ個体の治療において、物質、又は、もしその物質が自然界でペプチド(ペプチド、ポリペプチド、又はタンパク質)であるのならばそれをコードする核酸を個体に投与して、内在性の、若しくは外来的に供給される幹、前駆若しくは前駆体細胞、又は神経細胞のいずれかに作用することにより脳内でドーパミン作動性ニューロンの発生を誘導させ、促進させ、又は増強させ、及び/又は、欠損を阻害し、若しくは妨げ、又は生存、若しくは形態的分化、若しくは成熟化、又はドーパミン作動性ニューロンの神経突起生成、若しくはシナプス形成、又は機能的アウトプットを促進することを含む治療方法における、GSK-3βを阻害する物質、例えばGSK-3β阻害剤の使用を提供する。物質は、例えば、薬学上許容できる賦型剤や担体を含むどんな適当な組成物で投与されてもよい。また、神経変性疾患、パーキンソン症候群、又はパーキンソン病の治療用の薬剤製造に使用されてもよい。物質、又はそれをコードしている核酸は、中枢神経系、及び/又は脳に投与されるか、又はそれらを標的としてもよい。
【0097】
本発明は、様々な局面において、本明細書中で開示されたいずれか一つの方法に従って製造された神経だけでなく、そのような神経、幹、前駆、若しくは前駆細胞、及び/又はGSK−3βの阻害調節をする、例えばGSK−3β阻害剤のような物質を含む医薬組成物、薬剤、薬、又はその他の組成物、そのような神経、幹、前駆、若しくは前駆体細胞、又は神経細胞及び/又は物質若しくは組成物の医療方法における使用、例えば、パーキンソン病や他の(例えば神経変性の)疾患の治療(予防療法を含む)のための、そのような神経、幹、前駆若しくは前駆体細胞又は神経細胞及び/又は物質若しくは組成物の患者に対する投与を含む方法、例えば、パーキンソン病その他(例えば、神経変性疾患)の治療のための投与用組成物の製造でのそのような神経又は細胞及び/又は物質の使用、並びに、そのような神経又は細胞及び/又は物質を、薬剤上許容できる賦形剤、媒体、又は担体と、また、任意で一又は二以上の他の成分、例えば、神経保護分子、神経修復分子、レチノイド、成長因子、アストロサイト/グリア細胞、抗アポトーシス因子、又は、幹、前駆若しくは前駆体細胞又は神経細胞で、又はホスト脳内で遺伝子発現を調節する因子と、を混ぜ合わせることを含む医薬組成物の製造方法、に及ぶ。そのような任意の成分は、その環境から神経を独立にすることができる。例えば、その生存が、その環境における一又は二以上の因子又は条件の存在に依存しないようにすることができる。例として、医薬組成物を製造する方法は、腹側中脳の発生において見られる一又は二以上の因子と神経を混ぜることを含んでいてもよい。神経は、GDNF及び/又はニューツリン(NTN)と混ぜてもよい。医薬組成物を製造する方法は、腹側中脳の発生において見られる一又は二以上の因子と共に神経内でBcLXLを発現させることを含んでいてもよい。
【0098】
本発明は、本発明に従って生産された神経、幹、前駆若しくは前駆体細胞、又は神経細胞、及び/又は、GSK-3β阻害で作用する物質、例えば、GSK-3β阻害剤、及び一又は二以上の付加的な成分を含む組成物を提供する。本発明に係り、かつ本発明に従って使用される医薬組成物は、神経若しくは細胞に加えて、薬剤上許容できる賦形剤、担体、バッファー、保存料、安定化剤、抗酸化剤、または、当業者に周知の他の材料を含んでいてもよい。そのような材料は当然無毒であるべきで、かつ神経活性を妨害すべきではない。キャリーや他の材料の正確な性質は投与の経路に依存する。組成物は、一又は二以上の神経保護分子、神経修復分子、レチノイド、成長因子、アストロサイト/グリア細胞、又は、幹、神経幹、前駆体若しくは前駆細胞、又は神経細胞において遺伝子発現を調節する因子を含んでいてもよい。そのような物質は、上述したように、神経をその環境から独立させ得る。
【0099】
液体医薬組成物は、通常水、石油、動物性、若しくは植物性油、鉱物油、または合成油などの液体の担体を含む。生理的食塩水、組織若しくは細胞培養培地、細胞外基質タンパク質、ヘパラン、コンドロイチン硫酸、デキストロース若しくは他の糖質液、又はエチレングリコール、プロピレングリコール、ポリエチレングリコール等のグリコールが含まれていてもよい。
【0100】
組成物は、発熱物質フリーで、適切なpH、等張性、及び安定性を有する非経口許容水溶液の形状であってもよい。当業者であれば、例えば、塩化ナトリウム、リンゲル輸液、又は乳酸リンゲル輸液等の等張媒体を用いて適切な溶液を調製できる。組成物は、人工脳脊髄液を用いて調製してもよい。
【0101】
本発明は、本発明に従って製造された神経、及び/又は、GSK-3β阻害物質(例えば、GSK-3β阻害剤)の、医学療法、特に神経細胞における変性、損傷、欠損、疾患に関連した病状の治療における使用に及ぶ。さらに、本発明は、特定の表現型の神経、及び/又はGSK-3β阻害を調節する、又は与える物質(例えば、GSK-3β阻害剤)の、その表現型の神経の再生、損傷、又は欠損に関連する状態、病気、又は疾患の治療における使用を提供し得る。より特に、本発明はヒトパーキンソン病の治療におけるドーパミン作動性ニューロン、及び/又はGSK-3β阻害物質の使用を提供する。本発明は、特に神経変性病(例えば、パーキンソン病)の治療用の材料と方法に関連するとはいえ、それに限定はされない。例として、本発明は、脊髄、及び/又は大脳皮質の変性、若しくは損傷の治療、又はNurr1+細胞を含む神経系の他の領域の治療に及ぶ。
【0102】
投与した細胞が二以上の異なる神経表現型を生じることのできる幹、前駆、若しくは前駆体である治療方法では、神経、細胞、及び/又はWntリガンド又は組成物は、希望する特定の神経運命への細胞分化を指令するアストロサイト/グリア細胞を含む領域に導入されてもよい。細胞、及び/又はWntリガンド又は組成物は、例えば、腹側中脳にインジェクションされてもよい。そこで細胞はタイプIアストロサイト/グリア細胞と相互作用し、ドーパミン作動性表現型をとるように誘導されてもよい。別途、又は加えて、移植される組成物は、上述のように、その発生を特定の神経運命に向かわせる一又は二以上の因子と、例えば、タイプIアストロサイト/グリア細胞と組み合わせた神経、又は細胞を含んでいてもよい。
【0103】
細胞は、当業者において知られる技術(例えば、Lindvall, O., (1998) Mov. Disord. 13, Suppl. 1:83-7、Freed, C.R.ら, (1997) Cell Transplant, 6, 201-202; Kordowerら, (1995) New England Journal of Medicine, 332, 1118-1124; Freed, C.R.,(1992) New England Journal of Medicine, 327, 1549-1555)によって患者に移植されてもよい。また、Freedら J Neurol. 2003 Oct;250 Suppl 3:III44-6; Dunnettら (2001) Nat Rev Neurosci. 2001, 2(5):365-9; Lindvall and Hagell (2000) Prog Brain Res. 2000, 127:299-320; Mendez ら J Neurosurg. 2000 May;92(5):863-9; Hagellら. Mov Disord. 2000 Mar;15(2):224-9; Hauserら. Arch Neurol. 1999 Feb;56(2):179-87; Lindvall Mov Disord. 1998;13 Suppl 1:83-7. Review.; Olanowら. Trends Neurosci. 1996 Mar;19(3):102-9. Review; Remyら. Ann Neurol. 1995 Oct;38(4):580-8; Kordowerら. N Engl J Med. 1995 Apr 27;332(17):1118-24; Lindvallら. Ann Neurol. 1994 Feb;35(2):172-80; Hofferら. Exp Neurol. 1992 Dec;118(3):243-52; Lindvallら. Arch Neurol. 1989 Jun;46(6):615-31をご覧いただきたい。
【0104】
本発明に従った組成物の投与は、好ましくは、個体に対して有益性を示すのに十分な「予防上有効な量」又は、「治療上有効な量」(場合によっては、予防法は治療法とみなしてもよいが、)で行う。投与される実際の量、及び、割合、投与の時間経過は、何を治療するかの性質とひどさによる。治療処方、例えば、容量の決定は、一般の開業医や他の医師の責任の範囲にある。
【0105】
組成物質は、単独で、又は他の治療との組み合わせで、治療状態によって同時か、あるいは連続して投与してもよい。
【0106】
本明細書で提供される方法は、原材料としてin vivoで、若しくはin vitroで、若しくは細胞株で、初代細胞を使用して実施してもよい。in vitroで増やした細胞の有利な点は、実質上、製造される神経の数に限界がないことである。
【0107】
移植された細胞の免疫学的拒絶に関連して起こり得る不都合を改善する目的で、幹、若しくは前駆、若しくは前駆体細胞を、患者から分離し、希望の表現型に誘導してもよい。その後、細胞を患者に移植してもよい。有利な点として、分離された幹、若しくは前駆、若しくは前駆体細胞は、多数の免疫適合性神経細胞が製造されるように、細胞株の確立に使用されてもよい。さらには、個々の患者のために適切な細胞選択が可能な免疫学的適合性の範囲をカバーする細胞バンクを任意で設立する。ある個体由来の幹、神経幹、前駆体若しくは前駆細胞、又は神経細胞は、該細胞若しくはその子孫が第二の個体に導入される時に、拒絶を改善するために変更されてもよい。例として、ドナー細胞の一又は二以上のMHCアレルを、例えば、相同的組換えによってレシピエントのそれと置換してもよい。免疫抑制処理が患者に対して施されてもよい。
【0108】
もし、不死化癌遺伝子を有する細胞株由来の細胞が患者への移植に使用されたならば、癌遺伝子は、その細胞を患者に移植する前にCRE-loxPシステムを用いて除くことができる(Westerman, K. A. ら Proc. Natl. Acad Sci. USA 93, 8971 (1996))。患者の体温では不活性な不死化癌遺伝子を用いてもよい。
【0109】
本発明で使用される幹、神経幹、前駆体、前駆、若しくは神経細胞として、胚性幹細胞、胎児幹細胞、成人骨髄若しくは造血幹細胞、メサンギオブラスト(mesangioblast)及びH6(Flaxら Nature Biotech 16 (1998))等のヒト細胞株がある。さらなる例は、Gageら. Ann. Rev. Neurosci. 18, 159-192 (1995) and Gottlieb 2002に列挙されている。
【0110】
本考察は、神経幹細胞、又は神経前駆若しくは前駆体細胞に関して行われたが、本明細書中で提供される方法は、他の幹、前駆若しくは前駆体細胞で、神経運命の誘導に応用されてもよい。そのような細胞例には、非神経系に関連する幹細胞が含まれる。本方法は、間質若しくは造血幹細胞、及び/又は、表皮由来の増殖性細胞に応用されてもよい。造血細胞は血液、又は骨髄生検から回収されてもよい。間質細胞は、骨髄生検から回収されてもよい。上皮細胞は、皮膚生検により、若しくは掻き取り(例えば口腔粘膜からの)により回収されてもよい。神経表現型は、in vivoでは、それらの幹、前駆、若しくは前駆体細胞の生理学的運命でないので、この誘導プロセスは、分化転換、又は脱分化と神経再分化といってもよい。そのような細胞を神経運命に誘導する方法は、非神経表現型と関連する遺伝子、例えば、それらの細胞の分化を非神経運命に向かうことを抑制する、及び/又は反対にする遺伝子に対するアンチセンスレギュレーターの使用を含んでいてもよい。
【0111】
本発明の方法は、神経運命になることが決まっていない幹細胞に応用されてもよい。本方法は、異なる分化機構に関連する二以上の嬢幹細胞を生じ得る幹細胞に応用されてもよい。そのような幹細胞の例として、胚性幹細胞、造血幹細胞、骨髄間質細胞。表皮由来の増殖細胞、及び神経幹細胞が挙げられる。
【0112】
上記で論述したように、GSK-3β阻害、及び/又は腹側中脳タイプIアストロサイト、又はグリア細胞から得られた一又は二以上の因子と組み合わせて、基底レベルを超えるNurr1の発現を必要とするプロセスによりドーパミン作動性ニューロンが、幹、前駆、若しくは前駆体細胞から生じ得るということを、本開示は実証している。
【0113】
さらなる多様な局面において、本発明は、神経幹、又は前駆若しくは前駆体細胞でドーパミン作動性運命の誘導を増強する、あるいは、例えばGSK-3β阻害剤のようにGSK-3βの阻害を調節する手段として、又は例えばβ−カテニンの過剰発現若しくは安定化若しくは分解阻害、若しくはβ−カテニンのリン酸化の阻害を生じさせることでGSK-3βを間接的に阻害する手段として機能することによって、基底レベルを超えるNurr1を発現している神経細胞においてドーパミン作動性の誘導、若しくは分化を増強する1以上の因子をスクリーニングするアッセイ、及び方法の供給、またそれによって同定された1以上の因子と関係している。
【0114】
本発明は、直接的又は間接的に、GSK−3βを阻害できる、あるいはその他、GSK−3βの阻害を調節でき、かつ単独で又は組み合わせて、基底レベルを超えるNurr1を発現している幹、神経幹、又は前駆若しくは前駆体細胞、又は神経細胞におけるドーパミン作動性の運命を増強、増加、又は誘導を強化できる1以上の因子をスクリーニングする方法を提供する。本発明のさらなる局面は、基底レベルを超えるNurr1を発現する幹、神経幹、又は前駆若しくは前駆体細胞、又は神経細胞の、そのような幹、前駆若しくは前駆体細胞、又は神経細胞等においてドーパミン作動性の運命の誘導を増強するGSK-3βを阻害する、またはその阻害を調節する手段をスクリーニングし、又は探索し、及び/又は、入手し/同定することにおける使用を提供する。
【0115】
試験物質は、まずGSK-3βの阻害を調節する能力について、例えば、GSK-3を阻害すること、すなわち、GSK-3βの直接の阻害剤であるか、又はβ−カテニンの過剰発現若しくは安定化若しくは分解阻害、若しくはβ−カテニンのリン酸化の阻害を起こし得るかどうかという能力について、試験することができる。ひとたび能力を有することが同定されたら、本明細書で開示したように、その物質を、基底レベルを超えるNurr1を発現している幹、神経幹、または前駆若しくは前駆体細胞、又は神経細胞において、ドーパミン作動性の運命の増強、増加、又は誘導の強化の能力について試験することができる。
【0116】
スクリーニング方法の一部として、試験物質を、まずGSK-3β、若しくはβ−カテニン、グリコーゲンシンターゼ、tau、elf2B、 CREB、又はc-junなどの基質との結合、又は相互作用について試験することができる。
【0117】
結合又は相互作用は、当業者に知られる多数の技術のいずれかによって定性又は定量的に測定することができる。試験物質と幹、若しくは前駆、若しくは前駆体細胞間の相互作用は、検出可能な標識でどちらか一方を標識し、固体支持物上に固定化しておいてもよい他方と接触させることにより、例えば、固体支持物に結合した抗体を用いることにより、又はBiacore systemを始めとしてそれ自体が知られている他のテクノロジーを介することによって、調べることができる。
【0118】
スクリーニング方法は、1種以上の試験物質の存在下で、幹、神経幹、又は前駆若しくは前駆細胞、又は神経細胞を培養すること、及びドーパミン作動性表現型への分化について、例えば、本明細書中で考察したように、ドーパミン作動性表現型のマーカーを検出することによって、細胞を分析することを含んでいてもよい。チロシン水酸化酵素(TH)は、ドーパミン作動性表現型のマーカーの一つである。他のドーパミン作動性表現型マーカーには、ドーパミントランスポーター(DAT)、ドーパミンレセプター、アミノ酸脱炭酸酵素(AADC)、核内レセプターNurr1、ホメオボックスPtx3、及びLmx1bの発現などがある。神経フィラメントやβ−チューブリンIIIのような神経マーカーも、先のマーカーの一つに加えて細胞により発現されるべきである。ドーパミン作動性ニューロンを検出する他の方法として、ドーパミンの放出、及び例えば、GFPのルシフェラーゼと結合したTHプロモーターなどのレポーターシステムの検出がある。
【0119】
ドーパミン作動性ニューロンの検出のためのこれらのマーカーと方法は、本発明のいずれの局面と実施形態においても、ドーパミン作動性の運命誘導がうまくいったことを決定するのに有用であることに注意すべきである。
【0120】
本発明に従ってスクリーニングされる物質はいずれも、天然、又は合成の化合物であってもよい。
【0121】
活性の尺度としてのGSK-3βによるリン酸化は、当業界で利用できる多様な技術のいずれを用いて測定してもよい。
【0122】
本発明のさらなる局面においては、以下のものを含むアッセイ法が提供される。
【0123】
(a)GSK-3βの存在下で、GSK−3βに対するリン酸化部位、例えばグリコーゲンシンターゼではSer652とSer 648、β―カテニンではSer37 とThr41、elF2BではSer535、CREBではThr129、c-JUNではThr239を含む基質と試験物質とをGSK-3βが前記基質を普通にリン酸化する条件下で接触ようにすること、そして
(b)前記基質のリン酸化を測定すること。そのような方法により、GSK-3β阻害で機能する物質を同定できる。
【0124】
リン酸化は、例えば基質をビーズやプレート上などに固定することによって、そして部位がリン酸化されているときと部位がリン酸化されていないときとで異なる親和性をもつ関連するリン酸化部位に結合する抗体、又は他の結合分子を用いてリン酸化を検出することによって測定してもよい。このような抗体は、いずれの標準的な技術的手段によって得られてもよい。例えば、リン酸化ペプチドの使用が挙げられる。基質のリン酸化形態と非リン酸化形態とを識別する結合分子の結合については、当業者が利用できるいずれの技術を用いて評価してもよい。また、それは蛍光等の適当な標識の有無を測定することと関連していてもよい。リン酸化は、例えばシンチレーターを含浸したビーズやプレート上などに基質を固定することにより、標準シンチレーションプロキシメトリーアッセイ等で、放射性リン酸の取り込みを測定することで決定されるリン酸化により測定されてもよい。基質へのリン酸の取り込みは、トリクロロ酢酸のような酸での沈殿とニトロセルロースフィルター紙のような適当な材料上への沈殿物の回収に続いて放射標識したリン酸の取り込みの測定により測定されてもよい。基質のSDS−PAGE分離を使用した後に放射標識の検出を行ってもよい。
【0125】
GSK-3β阻害のスクリーニングに関する他のアプローチには、以下の一又は二以上のいずれかの考察が含まれる。
【0126】
(i)野生型β−カテニン、又は例えば、蛍光標識若しくは酵素のような標識若しくはレポーターでタグ付けされたβ−カテニンの安定化、又は核内移行。
【0127】
(ii)既知のβ−カテニン標的遺伝子の発現調節についての解析。そのような遺伝子のいくつかは、その調節領域にTCF/LEFファミリーのメンバーのためのコンセンサスな結合部位を含んでいる。これらには、c-myc、Cyclin D、Tcf-1、PPARdelta、c-jun、fra-1、uPAR、matrix metalloproteinase MMP-7、Axin-2、Nr-CAM、ITF-2、Gastrin、CD44、EphB/ephrin-B、BMP4、claudin-1、Survivin、VEGF、FGF18、FoxN1、matrix metalloproteinase-26、Frizzled 7、Follistatin、Id2、siamois、twin、Xnr3、fibronectin、myogenic bHLH、engrailed-2、connexin43、connexin 30、retinoic acid receptor gamma、dharma/bozozok、Cdx4、nacre、Stra6、Wrch-1、TNF family 41BB ligand、ephrinB1、Stra6、autotaxin、及びISLR、Twist、Stromelysin、Brachyury、Proglucagon、Osteocalcin、cyclooxygenase-2、Irx3、Six3、neurogenin 1、WISP-1、WISP-2、IGF-II、Proliferin-2、Proliferin-3、Emp、IGF-I、VEGF-C、MDR1、COX-2、IL-6、Periostin、Cdx1、betaTrCP、sFRP-2、Pitx2、Eda (TNF-related)、E-cadherin、Keratin、movo1、mBTEB2、FGF4、ret、Ubx、wingless、Dpp、Engrailed-1、Dfrizzled2、shavenbaby、stripe、Nemo、が含まれるが、これらに限定はされない(例えば、いずれかのウェブブラウザで閲覧可能な伝達経路の標的を記載したスタンフォードウェブサイトhttp://www.stanford.edu/~rnusse/pathways/targets.html updated in June 2004を参照いただきたい)。これらの遺伝子の調節は、β−カテニンタンパク質のレベルを生化学的、及び/又は免疫学的な方法で測定すること、又はハイブリダイゼーション、又は/及びPCRによってNurr1 mRNAレベルを測定すること、又は組換えプロモーター/レポーター遺伝子発現システムを用いてNurr1遺伝子発現のレベルを測定することを含む広く多様な技術によってモニターされてもよい。この後者のアプローチは、Nurr1の内在性の発現を調節するために必要とされるシス調節エレメントにレポーター遺伝子を機能可能なように連結したようなものからなるDNA構築物を薬物スクリーニングのために使用される細胞内部で発現することに関連している。
【0128】
(iii)β−カテニンレスポンスエレメントを含む、又はレポーター遺伝子若しくは複数のレポーター遺伝子に連結された構築物の調節。これによって、発現レベルの測定に代わり、又はそれと共同でβ−カテニンの特異的な転写促進の活性をモニタリングすることが可能となる。これによって、特定の処理がGSK-3β阻害を生じるか、又はドーパミン作動性表現型の獲得に影響するかを測定するための、より速く、かつ高度な処理の方法が可能となる。使用のできるレポーター構築物は、生物発光性タンパク質、または酵素活性、ルシフェラーゼ・クロラムフェニコール・アセチルトランスフェラーゼ、β−ガラクトシダーゼ、β−ラクタマーゼ、若しくは他の抗生物質耐性遺伝子、ニューロトロフィン、又はサイトカイン等を含んでいてもよい。
【0129】
(iv)遺伝子の発見。β−カテニン活性は、おそらくTH遺伝子をより強くアップレギュレートする。β−カテニン活性化は、おそらくドーパミン作動性表現型の特定化に関与する下流の遺伝子のいくつかの発現レベルを変化させる。そのような多数の遺伝子は以前に十分に同定され、若しくは特徴付けられたかもしれないが、いくつかについては、未だ同定されていないかもしれない。さらに、たとえ同定されていたとしても、ドーパミン作動性表現型を特定化するそれらの機能は、従来十分には理解されていなかったかもしれない。これらの遺伝子は、ドーパミン作動性ニューロンの欠損を補償するための治療のための未だ知られていない標的となるかもしれない。標的遺伝子の同定は、細胞(幹、前駆、前駆体、Nurr1を発現している、若しくは神経の細胞など)において、β−カテニンを恒常的に、又は誘導可能なプロモーターの下で過剰発現すること、又は内在性の、若しくは人工のβ−カテニン遺伝子の発現を誘導することによって調べられてもよい。刺激細胞と非刺激細胞とで、遺伝子発現、既知タンパク質の翻訳後修飾、イオンチャネル機能、又は特定分子の細胞内局在について評価することにより、解析は可能である。技術には、サブトラクティブ・ハイブリダイゼーション法、ディファレンシャル・ディスプレー法、SAGE、生化学的分画、電気生理学、細胞化学的技術が含まれていてもよい。Wnt分野で使用される技術には、Xenopus(アフリカツメガエル)での二次軸形成、体節筋形成、腎臓上皮誘導、軸索リモデリング、ニューロトロフィン3の発現、心筋細胞の凝集、細胞形質転換アッセイ、そして造血幹細胞の増幅が含まれる(引用に関しては、いずれかのウェブブラウザで閲覧可能なアッセイを記載しているスタンフォードウェブサイトhttp://www.stanford.edu/~rnusse/assays/assays.html updated November 2000をご覧いただきたい)。
【0130】
スクリーニング方法は、細胞間の表現型の違いを解析するいずれかの既知の方法を使用してもよく、DNA、mRNA、cDNA、又はポリペプチドレベルであってもよい。ディフェレンシャル・スクリーニングと遺伝子スクリーニングが、そのような二つの技術に該当する。本明細書で述べたスクリーニング方法のいずれかによって同定された物質は、本明細書で述べた他のスクリーニング方法で試験物質として使用されてもよい。
【0131】
スクリーニング方法は、例えば、マウスcDNA発現アレイといった核発現アレイを使用してもよい。このアプローチでは、異なる核酸分子のアレイがフィルター、水晶、又は他の表面上に、例えば核酸とフィルターのクロスリンクによって配置されている。試験溶液や抽出液を得て、その中で核酸を例えば蛍光によって標識する。その後、試験溶液や抽出液は、フィルター、又はジーンチップにアプライされる。フィルター、又はジーンチップ上の試験核酸のハイブリダイゼーションが測定され、コントロール溶液で行われたハイブリダイゼーションと比較される。試験サンプルとコントロールサンプルで得られたハイブリダイゼーション間の違いが、異なる核酸の量を示している。核酸アレイにおけるさらなる情報に関しては、いずれかのウェブブラウザで閲覧可能なClontech社のウェブサイト(例えば、www.clontech.com)、又はAffymetrix社のウェブサイト(例えば、www.affymetrix.com)をご覧いただきたい。
【0132】
本明細書でのスクリーニング方法は、基底レベルを超えて発現したNurr1に関して記載しているが、本開示は、例えば、Nor-1とNGFI-B といったNurr1サブファミリーの全核内レセプターにも及んでいる。このように、Nurr1は例示として記載されているいるに過ぎず、これに限定されるものではない。本発明のいずれの方法においても、Nurr1、又は、例えばNor-1やNGFI-Bといった他のいずれのレセプターをも含むNurr1サブファミリーの核内レセプターが、細胞内で基底レベルを超えて発現されて構わない。
【0133】
スクリーニング方法は、例えば、増殖、及び/又は自己複製、及び/又は分化、及び/又は生存を増強するような、及び/又はドーパミン作動性の運命の獲得若しくは誘導を促進するような、及び/又は幹、神経幹、前駆体、前駆若しくは神経細胞におけるドーパミン作動性ニューロンの分化を誘導するような、及び/又はGSK-3βを阻害する物質の存在下で基底レベルを超えるNurr1を発現している神経細胞でのドーパミン作動性誘導と分化を増強するようなNurr1の標的遺伝子、及び/又は因子若しくは複数因子を同定するために、幹、若しくは前駆、若しくは前駆体、若しくは神経細胞と、GSK-3βの阻害に作用する物質の存在下で基底レベルを超えるNurr1を発現する幹、若しくは前駆、若しくは前駆体、若しくは神経細胞とを比較することを含んでいてもよい。ひとたび標的遺伝子、及び/又は因子が同定されたならば、それらは、さらなる方法で分離され、及び/又は精製され、及び/又はクローン化され、そして使用されてもよい。
【0134】
スクリーニング方法は、混合物から物質、若しくは複数物質を精製、及び/又は分離することを含んでいてもよい。その方法は、GSK-3βの阻害の存在下で基底レベルを超えるNur1を発現している幹細胞、神経幹細胞、又は神経前駆若しくは前駆体細胞、又は神経細胞と相互作用する一又は二以上の混合物フラクションの能力を測定することを含んでいてもよい。能力とは、例えば、それらの細胞に対する結合力、及び/又は増殖、及び/又は自己複製を促進する能力、及び/又は幹、神経幹、前駆体、前駆、若しくは神経細胞などで、ドーパミン作動性の表現型や運命の誘導、獲得、分化、又は発生を増強する能力が該当する。精製、及び/又は分離は、当業者に知られるいずれの方法を用いてもよい。
【0135】
スクリーニング方法は、試験物質をコードする核酸を機能可能なように連結した誘導可能なプロモーターを使用してもよい。そのような構築物は、ホスト細胞内に取り込まれ、プロモーターの発現許容条件下と非許容条件下で、その細胞の一又は二以上の性質が測定され、また比較される。測定される性質は、基底レベルを超えるNurr1を発言している幹、神経幹、前駆体、前駆、若しくは神経細胞でドーパミン作動性表現型を誘導するホスト細胞の能力であり得る。許容条件と非許容条件間でのホスト細胞の能力の違いは、その試験物質が、単独、あるいは組み合わせのどちらかで、幹、神経幹、又は前駆若しくは前駆体細胞での増殖、及び/又は自己複製、及び/又はドーパミン作動性の運命の誘導、ドーパミン作動性分化、生存若しくは発生を増強するため又はGSK-3βの阻害の存在下で基底レベルを超えるNurr1を発現する神経細胞でのドーパミン作動性誘導、又は分化を増強できることを示している。
【0136】
本発明のスクリーニング方法の正確な形態は、日常的な技能と知識で当業者によって変更されても構わない。
【0137】
本発明で提供されたいずれか一つの方法で同定された因子若しくは複数因子は分離され、及び/又は精製され、及び/又はさらに調べられてもよい。それは、製造されても構わない。
【0138】
さらなる多様な局面において、本発明はさらに本明細書で開示された方法のいずれか一つで同定された因子、そのような因子を含む医薬組成物、薬剤、薬、又は他の組成物(その組成物は、基底レベルを超えるNurr1を発現している幹、神経幹、又は前駆若しくは前駆細胞、又は神経を含んでいてもよい)、基底レベルを超えるNurr1を発現している幹、神経幹、又は前駆若しくは前駆体細胞由来のドーパミン作動性ニューロンの誘導、及び/又は形態的分化、若しくは成熟化、及び/又は生存、及び/又は神経突起生成、及び/又はシナプシス生成、及び/又は機能的アウトプットを増強するための前記因子の使用、そのような因子、又は組成物の医療方法での使用、そのような因子、又は組成物を患者へ投与することからなる、例えば、ドーパミン作動性ニューロンの変性、損傷、欠損、又は障害若しくは影響に関連する病状の治療(予防治療を含んでいてもよい)のための、例えば、パーキンソン病や他の神経変性疾患の治療のための方法、例えば、パーキンソン病や他の病気(例えば、神経変性疾患)の治療のための投与用の組成物、若しくは薬剤の製造におけるそのような因子の使用、そしてそのような因子を薬剤上許容できる賦形剤、媒体、若しくは担体、及び任意で他の成分と混合することからなる医薬組成物を作る方法を提供する。
【0139】
関連する局面で、本発明は幹、神経幹、前駆体若しくは前駆細胞でドーパミン作動性の運命を誘導するために、あるいは基底レベルを超えるNurr1を発現している神経細胞でドーパミン作動性の誘導、又は分化を増強するために、GSK-3β阻害で直接的に、又は間接的に作用する物質の能力を調節する物質のスクリーニングの方法を提供する。
【0140】
従って、本発明の方法は基底レベルを超えるNurr1を発現している幹、神経幹、前駆体、前駆若しくは神経細胞で増殖、自己複製、ドーパミン作動性発生、分化、成熟化、及び/又は、ドーパミン作動性運命の獲得、を誘導するために、GSK-3β阻害剤の能力を調節する物質をスクリーニングすることができる。
【0141】
そのような方法は、一又は二以上の以下の(i)〜(iii)を含んでいてもよい。
【0142】
(i)GSK-3βの阻害の存在下で基底レベルを超えるNurr1を発現する幹、神経幹、前駆、前駆体、若しくは神経細胞を提供すること。ここでGSK-3βの阻害の存在とは、例えばGSK-3β阻害剤、又はβ−カテニンの過剰発現若しくは安定化若しくは分解の阻害、若しくはβ−カテニンのリン酸化の阻害が可能な薬剤、又はGSK-3β若しくはGSK-3βの基質と結合すること、若しくは相互作用すること、GSK-3βの基質リン酸化、β−カテニンの核局在、β−カテニン標的遺伝子、若しくはレポーター遺伝子に連結したβ−カテニンレスポンスエレメントの発現の調節、及び一、又は二以上の試験物質が該当する。
【0143】
(ii)ドーパミン作動性の運命又は表現型を取った、及び/又は阻害剤に応答した細胞の割合を解析すること。
【0144】
(iii)、試験物質を欠如した状態で比較可能な反応培地と条件下で、ドーパミン作動性の運命を取った細胞と、ドーパミン作動性の運命又は表現型を取った、及び/又は阻害剤に応答した細胞の総数の割合を比較すること。処理済の、及び未処理の細胞間でのドーパミン作動性ニューロンの割合の違いは、関連する試験物質の調節効果を示している。
【0145】
そのようなスクリーニングの方法は、以下の(i)〜(iii)を含んでいてもよい。
【0146】
(i)基底レベルを超えるNurr1を発現する幹、神経幹、前駆体、若しくは前駆細胞、又は神経細胞を一又は二以上の試験物質の存在下でGSK-3βの阻害剤(直接的阻害剤であろうが、間接的阻害剤であろうが)に接触させること。
【0147】
(ii)ドーパミン作動性の運命又は表現型を取った、及び/又は阻害剤に応答した幹、神経幹、前駆体、若しくは前駆細胞、又は神経細胞の割合を解析すること。
【0148】
(iii)試験物質が欠如した、又は存在する比較可能な反応条件下で、ドーパミン作動性の運命又は表現型を取った、及び/又は阻害剤に応答した幹、神経幹、前駆体、若しくは前駆細胞、又は神経細胞の割合と、ドーパミン作動性の運命又は表現型を取った、及び/又はWntに応答した幹、前駆体、前駆、若しくは神経細胞の数とを比較すること。
【0149】
このようなスクリーニング方法は、比較可能なヒト以外の同一の動物にてin vivoで細胞に対して、又はin vitroで、又は培養物で実施されてもよい。
【0150】
GSK-3β阻害を調節する物質の同定に続いて、その物質をさらに調査してもよい。該物質は、製造されても、及び/又は調製、すなわち、薬剤、医学組成物、又は薬等の組成物の製造、又は処方で使用されてもよい。その調節活性に関してテストされた物質はいずれも、天然の、又は合成された化合物であってもよい。
【0151】
例えば直接的なGSK-3β阻害剤の活性増強といったGSK-3β阻害を増強するモジュレーターを、例えば阻害剤と共に提供することができ、これら二つの物質を、例えば、医薬組成物、及び多様な局面における本発明の他の実施形態で一緒に使用してもよい。
【0152】
既に開示したように、GSK-3β阻害を調節する手段を本発明に従って使用して、ドーパミン作動性表現型を復活させることができる。さらなる実施形態において、GSK-3β阻害の調節は、GSK-3阻害療法に関与する遺伝子の転写活性を調節する物質を投与することによって、又は、GSK-3β阻害を調節する遺伝子を遺伝子療法を介して投与することによって達成してもよい。In vitroで本発明者は、GSK3阻害を与える手段としてのβーカテニン投与が、神経細胞からドーパミン作動性ニューロンの発生増加を生じることを示した。脳の神経細胞内にGSK3阻害を調節する遺伝子を導入することにより、患者の脳内で、ドーパミン作動性細胞の数を増やすことができる。別の方法として、GSK3阻害を調節できる物質でPD患者を治療することができる。このような物質は、上述したタイプのドラッグディスカバリーアッセイ法を介して同定することができる。
【0153】
本発明は、診断上の適用についても提供する。GSK3阻害はドーパミン作動性表現型の獲得を促進し、また、その表現型の欠損はパーキンソン病に関連しているので、Dishevelled、アキシン、FRAT/GBP、カゼインキナーゼ1、β−カテニンのレベル、及び/又は機能の変化を含むGSK-3β阻害の異常は、パーキンソン病の病因の基礎となるかもしれない。GSK-3β阻害に関与する遺伝子、mRNA、若しくはタンパク質の構造的な、又は機能的な異常が、遺伝子、mRNA、若しくはタンパク質の異常なレベル、及び/又は機能を引き起こすのかもしれない。また、PDの臨床上の発症の前後でこれらの欠陥、及び/又は異常が表面化するかもしれない。したがって、DNA、mRNA、若しくはタンパク質レベルで、そのような異常性についてスクリーニングすることは、PDの前駆症状の検出を提供するかもしれないし、あるいは診断の確認を提供するかもしれない。これらの技術は、脳生検、脳脊髄液、血液、唾液、尿、若しくは他の体液、又は組織に適用できる可能性がある。
【0154】
以下、添付の図面を参照して、本発明の局面と実施形態を例示する。別の局面と実施形態は、当業者には自明であろう。本明細書に記載した全ての文献は、参照によって本明細書中に組み込まれるものとする。
【0155】
タイプIアストロサイト又はグリア細胞の存在下でNurr1を発現している幹、神経幹、前駆体、又は前駆細胞でのドーパミン作動性ニューロンの誘導とドーパミン作動性前駆体の増殖、及び/又は自己複製を実証している、そしてFGF(例えば、FGF8)やレチノイドのような付加的因子と前記細胞との接触の際に得られる付加的な結果を明らかにしたW000/66713で詳述される実験的結果は、参照によって特別に本明細書に組み込まれる。
【0156】
ドーパミン作動性ニューロンの分化におけるWntの利用に関するWO2004/029229で詳述された実験結果もまた、参照によって特別に本明細書に組み込まれる。
【実施例】
【0157】
<GSK-3β阻害は、VM前駆体培養物でDAニューロン数を増加する>
ラットVMにおける前駆体細胞は、胎生期11日目から16日目(E11-E16)でDA表現型を獲得する(16)。神経上皮で増殖している前駆体が、腹側に移動し始めると、それらはNurr1の発現を開始する(17)。それによって、サイクリン依存性キナーゼ(CDK)阻害剤p57に関連する機構により細胞周期から離脱し、またDA表現型を獲得する(18)。本発明者は以前に、WntがVM前駆体でDA分化を促進することを示した(7)。細胞内カノニカルWntシグナル伝達因子がDAニューロン発生を調節できるかどうかを調べる目的で、本発明者はラットVM E14.5前駆体培養物に対して、β−カテニン分解に不可欠な酵素であるGSK-3βに対する二種のATP競合型阻害剤の用量を増加していく処理を行った。前駆体培養物へのインディルビンー3−モノオキシム(Indirubin-3-monoxime:I3M)(19、20)、又はケンパウロン(KP)(13)の添加は、I3Mについては3〜5μM(5倍増)での、またKPについては15μM(3倍増)での最大効率をもって用量依存的様式でTHポジティブ細胞の数を増加した(図1Aと1C)。これらの濃度において、KPはGSK-3β選択的阻害剤であるが、I3MはGSK-3βとCDKの両方を阻害する点において、これらの化合物は異なっている(20、13)。興味深いことに、本発明者は、またKP又はI3Mでの黒質、SN4741(21)由来のDA前駆体細胞系統の処理が、GSK-3βタンパク質レベルのダウンレギュレーションを誘導することも見出した(図1B)。
【0158】
<GSK-3β阻害はVM前駆体の分化を増加する>
本発明者は、次にGSK-3βの阻害がVM前駆体の一般的な神経とグリアの分化に影響するかどうかを見た。6の免疫細胞化学法からわかるように(図2Aと2B)、本発明者は、I3MとKPのいずれもがアストロサイト(グリア繊維性アストロサイトタンパク質(GFAP)ポジティブ細胞/ヘキスト33258)(図2A)、未成熟神経(β−チューブリン タイプIII(TuJ1)ポジティブ細胞/ヘキスト33258)(図2B)の総数を増加することを見出した。重要なのは、GSK-3β阻害が、成熟神経(微小管結合タンパク質2(MAP2)ポジティブ細胞/ヘキスト33258)の数の増加(図2C)、及びVMにおけるTH+/TuJ1+細胞の割合の3から5倍の増加によって示されるように(図2D及び図2B)、DAニューロン分化における非常に著しい増加をも誘導したことである。興味深いことに、本発明者は、以前にNurr1+幹/前駆体細胞のDAニューロンへの分化がグリア由来のシグナルに依存することを示唆した(22,23)。このように、グリア細胞の分化の増加は、DAニューロンの分化の増強にも貢献し得る可能性がある。
【0159】
<GSK-3βはVM前駆体培養物において細胞の生存又は増殖を調節しない>
GSK-3β阻害が増殖を促進しているかどうかを測定する目的で、本発明者は培養物における細胞総数と増殖マーカーであるBedUを取り込んだ細胞の数を調べた。ヘキスト33258染色からわかるように、GSK-3β阻害剤での処理は3日後の培養物内の細胞総数を変化させなかった(図3A)。さらに、VM前駆体が固定前に6時間 BedUパルスで曝露された場合、KPはBedUの取り込みに有意に影響しなかった(図3B)。しかし、I3MがCDKを阻害した以前の報告(20,13)に一致して、I3M処理はBrdU+細胞の数を減少させることがわかった(図3B)。このように、本発明者の結果は、GSK-3β阻害によるTH+ニューロンの増加は、増殖を通して達成されないことを示唆している。本発明者は、次にGSK-3β阻害剤が生存を通してDA細胞の数を増加できるかどうかを調べた。GSK-3β/CDK阻害剤であるI3Mは活性型カスパーゼ3免疫反応性細胞の数を減少させたのに対し、選択的GSK-3β阻害剤であるKPは効果がなかった(図3C−D)。より重要なことは、TH+細胞の約5パーセントは活性型カスパーゼ3にポジティブに染色したが、二つのGSK-3β阻害剤7のいずれもCaspase+/TH+細胞の数を減少しなかった事実(データは示さず)が判明したことである。これはGSK-3β阻害がアポトーシスの減少を通してTH+ニューロンの数の増加を誘導しないことを示唆している。DA分化におけるGSK-3β阻害の効果を確認する目的で、本発明者は未成熟DAニューロンの性質を示すMN9D細胞(24)における選択的GSK-3β阻害剤KPの効果を調べた。VM前駆体細胞のように、KPでの処理は、細胞総数(ヘキスト33258で分析した)又はBrdUの取り込みのレベルを変えなかった(図3E)。しかし、KP処理は、VM前駆体培養物で見られた(図2C)ようなMN9D細胞における形態的分化と神経突起伸展を増加した(図3F)。Nurr1過剰発現後のMN9D細胞の形態学的分化における比較可能な効果の報告(18)は、これらの細胞とVM前駆体とがNurr1とGSK-3β阻害剤に対する応答において分化能を共有していることを示唆している。合わせて、本発明者の結果が前駆体細胞分化におけるGSK-3βの機能を示唆し、またこのGSK-3β阻害がVM前駆体におい
てDA分化を促進するかどうかを調べることを促した。
【0160】
<GSK-3βの阻害は、Nurr1+前駆体のDAニューロンへの転換を通してDA細胞数を増加する>
本発明者は、まずGSK-3β阻害が分化したVM、DAニューロンに特有な遺伝子の発現を促進するかどうかを見た。リアルタイムRT-PCR解析は、KP処理が癌原遺伝子c-retのmRNAレベルの増加を生じさせることを示した(図4A)。c-retは、VM前駆体培養物のWnt5aでの処理(7)や、あるいはWnt1で処理されたカテコールアミン作動性PC12細胞において(25)アップレギュレートされたことも報告されている。この結果は、GSK-3β阻害がDA前駆体の分化を増加する上で働くかもしれないことを示唆している。それゆえ本発明者はKPで処理したNurr1+/ TH-前駆体のNurr1+/ TH+ DAニューロンへの分化を調べ、この8特異的GSK-3β阻害剤がTH/Nurr1両ポジティブ細胞の数を2.5倍に増加することを見出した(図4B−C)。このように、本発明者の結果は、GSK-3β阻害がTH+ニューロンの数の増加を誘導する機構はVM前駆体の増殖や生存を促進することによるものではなく、Nurr1+ VM前駆体の分化を促進することによるものであることを示した。
【0161】
<β−カテニンの安定化はVM前駆体のDAニューロンへの分化を増加する>
本発明者は以前にWntがVM培養物においてDA分化を促進すること、及びin vivoでのE10.5 VM前駆体においてNurr1/β−カテニンの共局在とTCF/LEF転写活性があることを示した(7)。しかし、最近ではGSK-3βがβ―カテニン非依存的経路において微小管の安定化を通して軸索リモデリングを仲介することが記述されている(26)。したがって、DA分化の機構をさらに研究する目的で、本発明者はKP又はI3Mで処理したVM前駆体培養物においてβ―カテニンタンパク質のレベルを解析した。免疫ブロッティングは、βーカテニンタンパク質のレベルの増加を示した。これは、阻害剤での処理によりβーカテニンの安定化が増加したことを示している(図5A)。さらに両阻害剤は、THタンパク質のレベルを増加した(図5A)。これらの結果は、β−カテニンがDA前駆体におけるGSK-3β阻害剤とWntの効果のいくつかを仲介できることを示唆している。初期DA分化におけるβ−カテニン安定化の役割の重要性を調べる目的で、本発明者はβーカテニン発現ベクターでVM前駆体をトランスフェクトした。VM前駆体細胞におけるβ−カテニンの過剰発現は、空のコントロールベクターと比べてTHポジティブ細胞数の2倍増を誘導した(図5BとC)。9。
【0162】
<方法>
(リアルタイムPCRと遺伝子発現の定量化)
総RNAは、in vitroで3日間、GSK-3β阻害剤で処理したE14.5VM前駆体培養物(6cm2ディッシュで2.5×106細胞)から分離した。逆転写反応、リアルタイムRT−PCR、またc-retプライマー配列の説明に関しては、Castelo-Branco らによる2003年の文献(7)をご覧いただきたい。古典的18SRNA量の内部標準はAmbion社(Austin, USA)から、またPCRプライマーはDNA Technology A/S社(Aarhus, Denmark)から購入した。以下のPCRプログラムは、ABI PRISM 5700 Detection System(PE AppliedBiosystems社, Foster City, CA, USA)におけるSYBR Green検出用(94℃2分、94℃30秒と59℃30秒と72℃15秒を35−40サイクル、そして80℃5秒)を用いた。結果の統計分析は、両側ウィルコクソン符号順位検定によって行った。全検定の有意性は、p<0.05のレベルで仮定した(* p<0,05; ** 0.01<p<0.001; *** p<0.001)
(前駆体とMN9Dの培養及び処理)
時期交尾されたSprague-Dawley系ラットから得られたE14.5VM(動物実験のための倫理上の承認は、Stockholms Norra Djurfoersoeks Etiska Naemndにより許可された)を解剖し、機械的に分離し、そして5μg/mlインシュリン、100μg/mlトランスフェリン、100μMプトリッシン、20nMプロゲステロン、30nMセレン、6mg/mlグルコース、そして1mg/mlBSA(全てSigma社から購入)を含むF12とMEM(Invitrogen社)の1:1混合からなる無血清N2培地が入ったポリDリジン被覆プレート(Falcon社)上に1cm2あたりの最終密度が1×105細胞となるようにプレーティングした。培養液は、固定前に、5%CO2で37℃インキュベータ内にて3日、in vitroで保持した。MN9DとSN4741細胞は、Choiらによる1991年の文献とSonらによる1999年の文献に記載されたように育てた。MN9D培養物はin vitroで4日後に固定した。I3MとKP(Alexis Biochemicals社, Goeteborg, Sweden)はDMSOで希釈し、プレーティング時に培地に添加した。DMSO単体では、TH+12ニューロンの数を増加せず、またMN9Dの形態学的分化に影響しなかった(データは示さず)。10μMのBrdUは固定の6時間前に培地に加えた。
【0163】
(構築とトランスフェクション)
ヒトβーカテニンcDNA(Dr. Steven Byersより贈与)を、CMVエンハンサー下にニワトリβ−アクチンプロモーターを有するpCAIP2ベクター中にサブクローンした。空のpCAIP2ベクターをコントロールとして使用した。初代細胞をリポフェクタミン2000(Invitrogen社)を用いて、製品使用書に従ってトランスフェクトし、in vitroで2日後、免疫細胞化学法によって評価した。
【0164】
(免疫細胞化学法)
細胞を氷冷した4%パラホルムアルデヒドで20分間固定し、PBSで洗浄した。免疫細胞化学法の詳細なプロトコルは、Castelo-Branco らによる2003年の文献(7)をご覧いただきたい。以下の一次、及び二次抗体、すなわちマウス抗TH(1:1000希釈 - Immunostar社)、ウサギ抗TH(1:250 - Pelfreeze社)、マウス抗βIIIチューブリン(1:1000 - Promega社)、ウサギ抗Nurr1(1:1000 - Santa Cruzs社)、マウス抗MAP2(1:200 - Sigma社)、ウサギ抗GFAP(1:400 - Dako社)、マウス抗活性型カスパーゼIII(1:100 - Cell Signaling社)、マウス抗BrdU(1:100 - Dako社)、またラット抗BrdU(1:100 - Abcam社)、ビオチン化 1:500、シアニン-2、若しくはローダミン結合されたウマ抗マウスIgG 1:200又はヤギ抗ウサギIgG 1:200(Vector and Jackson Laboratories社から)を使用した。染色手順の最後で培養物をヘキスト33258試薬と共に10分間インキュベートした。BrdU免疫細胞化学法は、2NのHClでの30分間のインユベーションを含んでいる。PBS中の染色された細胞を室温においてZeiss Axioplan 100M microscope (LD Achrostigmat 20x, 0,3 PH1 . 0-2 and LD Achroplan 40x, 0,60 Korr PH2 . 0-2)で画像を捉え、Hamamatsu camera C4742-95(QEDイメージング ソフトウェアを用いて)で撮影した。
【0165】
(免疫ブロッティング)
細胞を20mMのTris(pH7.5)、140mMのNaCl、10%のグリセロール、1%のIGEPAL、1mMのβ-グリセロール フォスファターゼ、1mMのNa3VO4、そして完全なプロテアーゼ阻害剤(Roche社)を含む変性RIPAバッファ中で15分間溶解した。プレートから細胞を掻き取り、細胞溶解物を微量遠心管内で5分間遠心した。上清をBCA kit(PIERCE社)を用いてタンパク質測定用のよごれていないチューブに移し、Laemmliバッファー中で保存した。等量のタンパク質をポリアクリルアミドゲル電気泳動法(10%ポリアクリルアミド)で解析した。タンパク質は、ポリビニリデン ジフルオリド メンブレンに転写した。0.1% Tweenと3% BSAを含んだPBSでブロッキングした後、メンブレンは以下の一次抗体と共に4℃で一晩インキュベートした(マウス抗β-カテニン 1:500,BD社;ウサギ抗GSK-3β 1:1000,Cell Signaling社;ウサギ抗TH 1:500, PelFreeze社; そして マウス抗β−アクチン 1:500, Abcam社)。洗浄後、メンブレンを二次抗マウス抗体(1:10000,Pharmacia Amersham社)共役アルカリフォスファターゼと共に、室温で1時間インキュベートし、続いて化学発光を増強するために製品説明書に従って発光させた。
【0166】
(統計解析)
免疫細胞化学データの定量は、13から5の独立した実験条件あたり3つのウェルにおいて10回の非重複野由来の平均値±標準誤差を表している。RT−PCR実験に関しては、5回の独立実験を解析し、またGSK-3β阻害剤用量反応に関しては、2セットの実験を解析した。統計解析は図のレジェンドに記載したように、p<0.05 (* p<0,05; ** 0,01<p<0,001; *** p<0,001)のレベルで仮定された全検定に関する有意性をもって、Prism 4 (GraphPad software, San Diego, USA)で行われた。統計検定はサンプル集団の分布に従って選択した。
【0167】
<参照文献>
Airaksinen MS, Saarma M. (2002) Nat Rev Neurosci. 3, 383-394
Akerud ら, J. Neurosci. 21, 8108-18 (2001).
Arenas 2002. Brain Res Bull.; 57 (6):795-808. (ref 28)
Baek SH ら, (2003) Proc Natl Acad Sci U S A. 100, 3245-3250
Bain ら, 2003. Biochem J.; 371 (Pt 1): 199-204. (ref 13)
Bain ら,1995, Dev. Biol., 168, 342-357
Baker ら, Genes Dev 1999 Dec 1;13(23):3149-59
Bamji ら, 2003. Neuron; 40 (4):719-731 (ref 11)
Barberi ら, 2003 Nat Biotechnol. 21 (10):1200-1207 (ref 33)
Bennett ら, 2002. J Biol Chem.; 277 (34):30998-31004 (ref 14)
Bjorklund A, Lindvall O. Nat Neurosci. 2000 Jun;3(6):537-44.
Bjorklund ら, 2002, Proc Natl Acad Sci USA. Feb 19, 99(4): 2344-9
Bradley RS, Brown AM. EMBO J 9(5):1569-75 (1990).
Bradley RS, Brown AM. Mol Cell Biol 15(8):4616-22 (1995).
Brustle ら, 1999, Science 285, 754-756.
Capela and Temple, 2002, Neuron 35, 865-875.
Castelo-Branco ら, 2003. Proc Natl Acad Sci U S A. 100 (22): 12747-12752. (ref 7)
Castillo (1997) Genomics, 41, 250-257
Castillo ら, Mol Cell Neurosci 1998 May;11(1-2):36-46
Castro ら, 2001. J Biol Chem.; 276 (46):43277-43284 (ref 18)
Chambers ら, 2003, Cell May 30;113(5):643-55. Chenn ら.. 2002. Science; 297 (5580):365-369. (ref 34)
Choi ら, 1991 Brain Res. ;552 (1):67-76. (ref 24)
Ciani ら, 2004. J Cell Biol.;164 (2):243-253 (ref 26)
Cohen ら, 2001. Nat Rev Mol Cell Biol.; 2 (10):769-776. (ref 12)
Conacci-Sorrell ら, (2002) Genes Dev. 16, 2058-2072.
Danielian ら, 1996. Nature; 383 (6598):332-334. (ref 3)
Dann CE, ら, (2001) Nature 412, 86-90.
Deacon ら, 1998, Exp. Neurol., 149, 28-41.
Dorsky ら, 1998. Nature; 396 (6709): 370-373. (ref 1)
Dunnett ら, 2001. Nat Rev Neurosci.; 2 (5):365-369. (ref 27)
Foster ら, Int J Dev Neurosci 1988;6(4):367-86
Garcia-Castro ら, Science. 2002 Aug 2;297(5582):848-51
Gossen, ら, (1995) Science, 268, 1766-1769
Gottlieb DI. Annu Rev Neurosci. 2002;25:381-407.
Granholm AC, Reyland M, Albeck D, Sanders L, Gerhardt G, Hoernig G, Shen L,
Westphal H, Hoffer B. (2000) J Neurosci. 20, 3182-3190.
Hall AC, Lucas FR, Salinas PC. (2000) Cell 100, 525-535.
Hall ら, 2003. Glia.;43 (1):47-51. (ref 23)
Hartmann C, Tabin CJ. Cell 2001 Feb 9;104(3):341-51
Hoessel ら, 1999. Nat Cell Biol. ; 1 (1): 60-67. (ref 19)
Hornstein ら, 2004. J Neurosci Res.; 76 (2):169-173. (ref 30)
Houart ら, Neuron. 2002 Jul 18;35(2):255.
Hsieh ら, Proc Natl Acad Sci U S A. 96(7):3546-51 (1999).
Huelsken ら, 2001. Cell.; 105 (4): 533-545 (ref 38)
Huelsken ら, 2002. J Cell Sci.; 115 (Pt 21):3977-3978. (ref 9)
Kiecker ら, Development 2001 Nov;128(21):4189-201
Kim ら, 2002. Nature. 418 (6893):50-56 (ref 32)
Kioussi C., ら, (2002) Cell 111, 673-685, 38
Kispert ら, Development 1998 Nov;125(21):4225-34
Krylova O, ら, (2002) Neuron 35, 1043-1056.
Law, ら, (1992) Mol Endocrinol 6, 2129-2135
Leclerc ら, 2001. J Biol Chem; 276 (1): 251-260. (ref 20)
Lee ら, 2000. Nat Biotechnol.;18 (6):675-679. (ref 31)
Lee ら, 2004. Science. 303 (5660):1020-1023 (ref 37)
Lindsay 2002; Cur. Op. Pharmacol., 2: 587-594.
Martinez Arias ら, 1999, Curr. Op. Genetics and dev. 9, 447-454.
Marvin ら, Genes Dev 2001 Feb 1;15(3):316-27.
Mata de Urquiza ら, Science 2000 Dec 15; 290: 2140-2144.
McMahon ら, 1990. Cell; 62 (6):1073-1085. (ref 4)
Megason ら, 2002. Development; 129 (9): 2087-2098. (ref 2)
Merrill ら, 2001. Genes Dev.; 15 (13): 1688-1705. (ref 39)
Muroyama ら, Genes Dev. 16(5):548-53 (2002).
Nordstrom ら, Nat Neurosci 2002 Jun;5(6):525-32
Nunes I., ら, Proc Natl Acad Sci U S A. 100, 4245-4250 (2003)
Ohmachi ら, 2000; Biochem. Biophys. Res. Comm.277, 355-360.
Oishi I, ら, 2003, Genes Cells. 2003 Jul;8(7):645-54.
Okabe ら, 1996, Mech. Dev., 59, 89-102.
Pandur ら, Nature. 2002 Aug 8;418(6898):636-41.
Paratcha G., Ledda F., Ibanez C.F. (2003) Cell 113, in press.
Parr ら, Development 119(1):247-61(1993).
Patapoutian A, Reichardt LF. (2000) Curr Opin Neurobiol. 10, 392-399.
PCT/IB03/04598
Perrone-Capano ら, 2000. Neuroscience and Biobehavioral Reviews, 24, 119-124 (ref 16)
Pinson ら, 2000. Nature; 407 (6803):535-538. (ref 6)
Price J. & Williams BP. Current Opinion in Neurobiology 11, 564-567 (2001).
Reubinoff ら, 2000, Nat Biotech., 18, 399-404.
Reubinoff ら, 2001, Nat Biotech., 19, 1134-1140.
Reya T, ら, (2003) Nature 423, 409-414.
Rhinn M, Brand M. Curr Opin Neurobiol 2001 Feb;11(1):34-42
Ross ら, Science 2000 Aug 11;289(5481):950-3
Rossi F. & Cattaneo E. Nat Rev Neurosci. 2002 May;3(5):401-9.
Saneyoshi ら, Nature 417(6886):295-9 (2002).
Sato ら, 2004. Nat Med; 10 (1): 55-63. (ref 15)
Saucedo-Cardenas ら, Proc Natl Acad Sci U S A 1998 Mar 31;95(7):4013-8
Schaar ら, 1993 Exp Neurol.;124 (2):368-371. (ref 36)
Schneider ら, Genes Dev 2001 Feb 1;15(3):304-15
Schuldiner ら, 2000, PNAS. USA 97, 11307-11312.
Schuldiner ら, 2001; Brain Res. 913, 201-5.
Shimizu ら, Cell Growth Differ 1997 Dec;8(12):1349-58
Shtutman ら, Proc Natl Acad Sci U S A 96(10):5522-7 (1999).
Smidt ら, Proc Natl Acad Sci U S A. 94(24):13305-10 (1997).
Smidt ら, Nat Neurosci. 3, 337-341 (2000).
Son ら, 1999. J Neurosci; 19 (1): 10-20. (ref 21)
Song ら, Nature 2002 May 2;417(6884):39-44
Storch ら, 2001. Exp Neurol.; 170 (2):317-325. (ref 20)
Taipale J, Beachy PA. Nature. 2001 May 17;411(6835):349-54.
Tetsu ら, Nature 398(6726):422-6 (1999).
Thomas ら, 1990. Nature; 346 (6287):847-850. (ref 5)
Tropepe.ら, 2001, Neuron 30,65-78.
Trupp M ら, (1996). Nature 381, 785-789
Tumbar ら, 2004. Science; 303 (5656):359-363 (ref 40)
Tzahor E, Lassar AB. Genes Dev 2001 Feb 1;15(3):255-60
Van Den Munckhof P.ら, 2003 Development 130, 2535-2542
Wagner ら, 1999 Nat Biotechnol.;17 (7):653-659. (ref 22)
Wallen ら,1999. Exp Cell Res; 253 (2): 737-746. (ref 17)
Willert ら, 2003. Nature; 423 (6938):448-452 (ref 8)
Wilson ら, Nature 2001 May 17;411(6835):325-30
Wurst W, Bally-Cuif L. Nat Rev Neurosci 2001 Feb;2(2):99-108
Xing, ら, (1997) Mol Brain Res 47, 251-261
Ye ら, Cell 93(5):755-66 (1998).
Yoshikawa S., ら, Nature. 2003 Apr 10;422(6932):583-8.
Yu ら, 2003. Nat Neurosci.; 6 (11):1169-1177. (ref 10)
Zechner ら, 2003. Dev Biol; 258 (2):406-418. (ref 35)
Zetterstrom ら, Science 1997 Apr 11;276(5310):248-50
Zhang ら, 2001, Nat Biotech., 19, 1129-1133.
Zheng ら, 1996. Oncogene.; 12 (3): 555-562. (ref 25)
【図面の簡単な説明】
【0168】
【図1】KP及びI3MはVM前駆体培養物内でTH+ニューロンの数を増加する。
【0169】
図1A:E14.5 VM前駆体での用量反応実験は、15μMのKP及び3〜5μMのI3MがTH+細胞数を増加する上で最適用量であることを示している。
【0170】
図1B:免疫ブロッッティングからわかるように、15μMのKP及び5μMのI3Mで24時間処理したSN4741細胞は、GSK-3βタンパク質レベルが低下する。ローディングコントロール:β−アクチン。免疫染色法は、15μMのKP及び5μMのI3Mでの処理後三日で、TH免疫反応性細胞数の増加を示した。統計解析は、Fisher多重比較検定(Fisher’s post -hoc test)での一元配置分散分析を用いて行われた。
【図2】KP及びI3Mでの処理は、神経とグリアの双方への、そして特にDAニューロンへのVM前駆体の分化を増大する。抗GFAP抗体(A)、抗Tuj1抗体及び抗TH抗体(B)、そして抗MAP2抗体(C)での免疫染色法は、15μMのKP及び5μMのI3Mでの処理後三日で、アストロサイト、未熟及び成熟神経の数の増加、並びにTH+ DAニューロンの割合の著しい増加を示した(BとD)。統計分析は、両側独立t検定を用いて行われた。
【図3】GSK-3β阻害は、VM前駆体、又はMN9D細胞の生存と増殖を調節しないが、それらの形態的な分化を誘導する。
【0171】
図3Aは、ヘキスト33258染色からわかるように、15μMのKP及び5μMのI3Mでの処理は培養3日後のVM細胞の総数に変化を起こさなかったことを示している。
【0172】
図3Bは、BrdUの取込みからわかるように、増殖は15μMのKPでは影響を受けなかったが、5μMのI3Mで有意に減少したことを示している。
【0173】
図3Cは、活性型カスパーゼ3の免疫染色は、VM前駆体培養物において15μMのKPでは変化しなかったが、5μMのI3Mで減少したことを示している。
【0174】
図3Dは、15μMのKPで4日間処理したMN9D細胞が、細胞総数(ヘキスト33258)、又はそれらの増殖(BrdUの取込み)を変えなかったことを示している。α−活性型カスパーゼ3での免疫染色は、5μMのI3Mによる処理後3日でポジティブな細胞数の減少を示したが、KPによる処理では示さなかった。4日間の15μMのKPによる処理は、MN9D細胞の形態的な分化を誘導した。統計解析は、両側独立t検定(A)、クラスカル−ウォリス検定(Kruskal-Wallis test)(B)、ボンフェローニ多重比較検定(Boneferroni’s multiple comparison)での一元配置分散分析(C)を用いて行われた。
【図4】KPはc-ret mRNAをアップレギュレートし、またNurr1+前駆体のTH+ニューロンへの転換を増加する。
【0175】
図4Aは、リアルタイムRT-PCR解析によりc-ret mRNAレベルが15μMのKPによる処理でアップレギュレートされることが明らかとなったことを示している。
【0176】
図4Bは、15μMのKPがVM Nurr1+前駆体のTH+ニューロンへの転換を増加したことを示している。TH/Nurr1二重免疫染色は、15μMのKP処理後三日で免疫反応性の細胞数の増加を示している。統計解析は、両側ウィルコクソン符号順位検定(two-tailed Wilcoxon’s signed rank test)(A)及び両側独立t検定(B)を用いて行われた。
【図5】GSK-3β阻害剤は、β−カテニンを安定化し、またβ−カテニンの過剰発現は、TH+ニューロンの数を増加する。
【0177】
図5Aは、免疫染色からわかるように、15μMのKP又は5μMのI3Mで24時間VM前駆体を処理した結果、β−カテニンとTHタンパク質の両レベルが増加したことを示している。ローディング・コントロール:βアクチン。
【0178】
図5Bは、βーカテニンでのトランスフェクション後2日で、空のコントロールベクターでのトランスフェクションと比べて、TH−免疫染色のポジティブ細胞の数が2倍に増加したことを示している。統計解析は、両側独立t検定(B)を用いて行われた。
【特許請求の範囲】
【請求項1】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞において増殖、自己複製、ドーパミン作動性の誘導、生存、分化、及び/又は成熟化を増強することによってドーパミン作動性ニューロンの発生を誘導又は促進する方法であって、
細胞内で基底レベルを超えるNurr1サブファミリーの核内レセプターを発現させることと、
細胞内でFrizzledレセプターに対するリガンド以外の手段によってGSK-3β阻害を調節することにより増殖、自己複製、生存、及び/又はドーパミン作動性の誘導、分化、生存、又は神経のドーパミン作動性表現型の獲得を生じ、又は増強することを含んでなる方法。
【請求項2】
核内レセプターがNurr1である請求項1に記載の方法。
【請求項3】
核内レセプターがNor1又はNGFI-Bである請求項1に記載の方法。
【請求項4】
Nurr1 DNAで細胞を形質転換すること、又はNurr1 RNAを細胞内に導入することによって基底レベルを超えるNurr1を発現させることを含む請求項1から3のいずれか一に記載の方法。
【請求項5】
細胞にNurr1タンパク質を導入することによって基底レベルを超えるNurr1を発現させることを含む請求項1から3のいずれか一に記載の方法。
【請求項6】
細胞内にNurr1タンパク質を保つことによって基底レベルを超えるNurr1を発現させることを含む請求項1から3のいずれか一に記載の方法。
【請求項7】
GSK-3β阻害は、ヒメナルジシン(hymenaldisine)類、アロイシン(aloisine)類、マレイミド類、チアゾール類、チエニル及びメチルハロメチルケトン類、酸化アミノピリミジン類、インディルビン類並びにパウロン(paullone)類からなるグループから選択されるGSK-3β阻害剤によるものである請求項1から6のいずれか一に記載の方法。
【請求項8】
阻害剤は、酸化アミノピリミジン、又はパウロンである請求項7に記載の方法。
【請求項9】
阻害剤は、ケンパウロン(kenpaullone)である請求項8に記載の方法。
【請求項10】
GSK-3β阻害は、β−カテニンの安定化によるものである請求項1から6のいずれか一に記載の方法。
【請求項11】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞は、GSK-3β阻害で処理される際に、有糸分裂、及び/又は自己複製の可能な細胞である請求項1から10のいずれか一に記載の方法。
【請求項12】
前記神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を付加的に成長因子FGFファミリーの一員と接触させることを特徴とする請求項1から11のいずれか一に記載の方法。
【請求項13】
前記神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞をレチノイド若しくはレチノイド誘導体、レチノイドXレセプター(RXR)の活性化剤、レチノイド酸レセプター(RAR)の抑制剤、9−シス−レチナール、DHA、SR11237、又はLG849と接触させることを特徴とする請求項1から12のいずれか一に記載の方法。
【請求項14】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を、細胞をGSK-3β阻害の調節で処理する前に、又はそれと同時に、bFGF、及び/又はEGF、及び/又はFGF-8、及び/又はLIF、及び/又はShhで処理することを特徴とする請求項1から13のいずれか一に記載の方法。
【請求項15】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を抗酸化剤、アスコルビン酸、低酸素分圧、又は低酸素誘導因子の存在下で生長させることを特徴とする請求項1から14のいずれか一に記載の方法。
【請求項16】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を腹側中脳アストロサイト、又は初期グリア細胞の存在下で生長、及び/又は分化させることを特徴とする請求項1から15のいずれか一に記載の方法。
【請求項17】
GSK-3β阻害の調節は、細胞を含むin vitro培養物に対してなされることを特徴とする請求項1から16のいずれか一に記載の方法。
【請求項18】
GSK-3β阻害の調節が神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞でのタンパク質の発現によって、又はGSK-3β阻害の調節をする物質をコードする核酸を用いた形質転換によって生じることを特徴とする請求項17に記載の方法。
【請求項19】
GSK-3β阻害の調節が神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞と共培養される細胞からの発現によって生じ、該共培養細胞が、タイプIアストロサイト若しくは初期グリア細胞以外の細胞である、又はGSK-3β阻害をする物質をコードする核酸で形質転換されたホスト細胞である、又は導入されたGSK-3βを含む細胞であることを特徴とする請求項17に記載の方法。
【請求項20】
共培養された、タイプIアストロサイト若しくは初期グリア細胞以外の細胞又はホスト細胞は、別の幹、神経幹、前駆若しくは前駆体細胞、又は神経細胞である請求項19に記載の方法。
【請求項21】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞は、GSK-3β阻害の調節をする物質をコードする核酸から発現するように作られたものである請求項17に記載の方法。
【請求項22】
GSK-3β阻害の調節をする物質が細胞内に導入されることを特徴とする請求項17に記載の方法。
【請求項23】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を、任意に腹側中脳の初期グリア細胞、又はタイプIアストロサイトと共培養することをさらに含む請求項1から22のいずれか一に記載の方法。
【請求項24】
タイプIアストロサイトは、不死化された細胞株、又は腹側中脳以外の領域のアストロサイト株である請求項23に記載の方法。
【請求項25】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を、非ドーパミン作動性ニューロンに抗して選択するネガティブ選択剤と接触させることを付加的に含む請求項1から24のいずれか一に記載の方法。
【請求項26】
一又は二以上の付加的な成分を含む組成物にニューロンを処方することをさらに含むことを特徴とする請求項1から25のいずれか一に記載の方法。
【請求項27】
組成物は、薬剤上許容できる賦形剤を含むことを特徴とする請求項26に記載の方法。
【請求項28】
組成物を個体に投与することをさらに含む請求項27に記載の方法。
【請求項29】
ニューロンを個体の脳内に移植することを特徴とする請求項28に記載の方法。
【請求項30】
細胞を個体においてin situで処理してNurr1発現を増加することを特徴とする請求項1から16のいずれか一に記載の方法。
【請求項31】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を、Frizzeledレセプターに対するリガンド以外のGSK-3β阻害を調節する物質で、個体においてin situで処理することを特徴とする請求項1から16のいずれか一に記載の方法。
【請求項32】
GSK-3β阻害を調節する物質をコードする核酸を細胞内に導入することを特徴とする請求項31に記載の方法。
【請求項33】
GSK-3β阻害を調節する物質を細胞内に導入することを特徴とする請求項31に記載の方法。
【請求項34】
細胞を個体においてin situで処理して、基底レベルを超えるNurr1発現を増加することを特徴とする請求項31に記載の方法。
【請求項35】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞は、個体に内在性のものである請求項30から34のいずれか一に記載の方法。
【請求項36】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞は、個体内に移植によって外来的に供給されるものである請求項30から34のいずれか一に記載の方法。
【請求項37】
個体がパーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患を有することを特徴とする請求項28から36のいずれか一に記載の方法。
【請求項38】
前記方法に従って製造されたニューロンの個体治療用の薬剤製造における使用をさらに含む請求項1から27のいずれか一に記載の方法。
【請求項39】
薬剤は、個体脳内への移植用である請求項38に記載の方法。
【請求項40】
個体がパーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患を有することを特徴とする請求項39に記載の方法。
【請求項41】
請求項1から25のいずれか一に記載の方法に従って製造されたドーパミン作動性ニューロン。
【請求項42】
神経変性疾患の治療用に使用される薬剤をスクリーニングする方法における請求項41に記載のドーパミン作動性ニューロンの使用。
【請求項43】
単独、若しくは組み合わせで、基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞において、増殖、自己複製、生存、及び/又はドーパミン作動性の発生、誘導、分化、若しくは成熟を増強する1以上の因子を得る方法であって、
(a)基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を、一以上の試験物質の存在下及び非存在下においてGSK-3β阻害のレギュレーターで処理すること、そして
(b)細胞の増殖、自己複製、生存、及び/又はドーパミン作動性の発生、誘導、分化、若しくは成熟を測定し、そして増殖、自己複製、生存、及び/又はドーパミン作動性の発生、誘導、分化、若しくは成熟の程度を、一以上の試験物質の存在下及び非存在下で比較することによって前記1以上の因子を得ることを含んでなる方法。
【請求項44】
細胞を含むin vitro培養物において該細胞をGSK-3β阻害のレギュレーターで処理することを特徴とする請求項43に記載の方法。
【請求項45】
GSK-3β阻害のレギュレーターをコードする核酸の細胞内導入によって、細胞を該レギュレーターで処理することを特徴とする請求項43に記載の方法。
【請求項46】
GSK-3β阻害のレギュレーターの細胞内導入によって細胞を該レギュレーターで処理することを特徴とする請求項43に記載の方法。
【請求項47】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を腹側中脳の初期グリア細胞又はタイプIアストロサイトと共培養することをさらに含む請求項43から46のいずれか一に記載の方法。
【請求項48】
基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞において増殖、自己複製、生存、及び/又はドーパミン作動性の発生、誘導、分化、又は成熟を増強できる1以上の因子を単離された、及び又は精製された形態で提供することを特徴とする請求項43から47のいずれか一に記載の方法。
【請求項49】
基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞において増殖、自己複製、生存、及び/又はドーパミン作動性の発生、誘導、分化、若しくは成熟を増強できる1以上の因子を、一以上の付加的な成分を含む組成物に処方することを特徴とする請求項43から48のいずれか一に記載の方法。
【請求項50】
組成物が基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を含むことを特徴とする請求項49に記載の方法。
【請求項51】
組成物は、薬剤上許容できる賦形剤を含むことを特徴とする請求項49又は50に記載の方法。
【請求項52】
組成物を個体に投与することをさらに含む請求項51に記載の方法。
【請求項53】
組成物を個体の脳内に移植することを特徴とする請求項52に記載の方法。
【請求項54】
個体がパーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患を有することを特徴とする請求項53に記載の方法。
【請求項55】
パーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患治療用の薬剤製造における、Frizzeledレセプターのリガンド以外でGSK-3βの阻害を調節する物質の使用。
【請求項56】
パーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患治療用の薬剤製造における、Frizzeledレセプターのリガンド以外のGSK-3β阻害のレギュレーターで処理した神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞であって、基底レベルを超えるNurr1サブファミリーの核内レセプターを発現している該細胞の使用。
【請求項57】
GSK-3β阻害は、ヒメナルジシン(hymenaldisine)類、アロイシン(aloisine)類、マレイミド類、チアゾール類、チエニル及びメチルハロメチルケトン類、酸化アミノピリミジン類、すなわちインディルビン類並びにパウロン(paullone)類からなるグループから選択されるGSK-3β阻害剤によって提供される請求項55又は56に記載の使用。
【請求項58】
阻害剤は、酸化ピリミジン又はパウロンである請求項57に記載の使用。
【請求項59】
阻害剤は、ケンパウロン(kenpaullone)である請求項58に記載の使用。
【請求項60】
GSK-3β阻害は、β−カテニンの安定化によることを特徴とする請求項55又は56に記載の使用。
【請求項61】
ドーパミン作動性ニューロンの発生を誘導、又は促進する物質を得る方法であって、細胞内で基底レベルを超えるNurr1サブファミリーの核内レセプターを発現する神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞、又はドーパミン作動性ニューロンにおいてGSK-3βの阻害を調節する試験物質の能力を測定することを含んでなる方法。
【請求項62】
細胞内でGSK-3β阻害を与える試験物質の能力を測定することを含む請求項61に記載の方法。
【請求項63】
細胞内でGSK-3βを阻害する試験物質の能力を測定することを含む請求項61又は62に記載の方法。
【請求項64】
細胞内でβ−カテニンを安定化する物質の能力を測定することを含む請求項61又は62に記載の方法。
【請求項65】
ドーパミン作動性ニューロンの発生を誘導又は促進する作用物質を得る方法であって、
(i)試験物質のGSK-3βの阻害を調節する能力を測定し、それによってGSK-3β阻害を調節する物質を同定すること、
(ii)(i)で同定されたGSK-3β阻害を調節する物質と共に、細胞内で基底レベルを超えるNurr1サブファミリーの核内レセプターを発現する神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を提供することによって、GSK-3β阻害を調節する物質のドーパミン作動性ニューロンの発生を誘導又は促進する能力を測定し、それによって前記作用物質を得ることを含んでなる方法。
【請求項66】
GSK-3β阻害を調節する物質がGSK-3βを阻害することを特徴とする請求項65に記載の方法。
【請求項67】
GSK-3β阻害を調節する物質がβーカテニンを安定化することを特徴とする請求項65に記載の方法。
【請求項68】
一以上の付加的な成分を含む組成物中に前記作用物質を処方することをさらに含む請求項61から67のいずれか一に記載の方法。
【請求項69】
組成物が基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を含むことを特徴とする請求項68に記載の方法。
【請求項70】
組成物が薬剤上許容できる賦形剤を含むことを特徴とする請求項68又は69に記載の方法。
【請求項71】
組成物を個体に投与することをさらに含む請求項70に記載の方法。
【請求項72】
組成物を個体の脳内に移植することを特徴とする請求項71に記載の方法。
【請求項73】
個体がパーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患を有することを特徴とする請求項72に記載の方法。
【請求項74】
前駆症状性パーキンソン病の検出、又はパーキンソン病の診断若しくは診断の確認の方法であって、患者又は患者から得たサンプルにおいてGSK-3β阻害の異常の存在を決定することを含んでなる方法。
【請求項75】
GSK-3β阻害の異常の存在を患者から得たニューロンで決定することを特徴とする請求項74に記載の方法。
【請求項76】
異常をGSK-3β活性で測定することを特徴とする請求項74又は75に記載の方法。
【請求項77】
異常をβ−カテニンの安定化で測定することを特徴とする請求項74又は75に記載の方法。
【請求項1】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞において増殖、自己複製、ドーパミン作動性の誘導、生存、分化、及び/又は成熟化を増強することによってドーパミン作動性ニューロンの発生を誘導又は促進する方法であって、
細胞内で基底レベルを超えるNurr1サブファミリーの核内レセプターを発現させることと、
細胞内でFrizzledレセプターに対するリガンド以外の手段によってGSK-3β阻害を調節することにより増殖、自己複製、生存、及び/又はドーパミン作動性の誘導、分化、生存、又は神経のドーパミン作動性表現型の獲得を生じ、又は増強することを含んでなる方法。
【請求項2】
核内レセプターがNurr1である請求項1に記載の方法。
【請求項3】
核内レセプターがNor1又はNGFI-Bである請求項1に記載の方法。
【請求項4】
Nurr1 DNAで細胞を形質転換すること、又はNurr1 RNAを細胞内に導入することによって基底レベルを超えるNurr1を発現させることを含む請求項1から3のいずれか一に記載の方法。
【請求項5】
細胞にNurr1タンパク質を導入することによって基底レベルを超えるNurr1を発現させることを含む請求項1から3のいずれか一に記載の方法。
【請求項6】
細胞内にNurr1タンパク質を保つことによって基底レベルを超えるNurr1を発現させることを含む請求項1から3のいずれか一に記載の方法。
【請求項7】
GSK-3β阻害は、ヒメナルジシン(hymenaldisine)類、アロイシン(aloisine)類、マレイミド類、チアゾール類、チエニル及びメチルハロメチルケトン類、酸化アミノピリミジン類、インディルビン類並びにパウロン(paullone)類からなるグループから選択されるGSK-3β阻害剤によるものである請求項1から6のいずれか一に記載の方法。
【請求項8】
阻害剤は、酸化アミノピリミジン、又はパウロンである請求項7に記載の方法。
【請求項9】
阻害剤は、ケンパウロン(kenpaullone)である請求項8に記載の方法。
【請求項10】
GSK-3β阻害は、β−カテニンの安定化によるものである請求項1から6のいずれか一に記載の方法。
【請求項11】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞は、GSK-3β阻害で処理される際に、有糸分裂、及び/又は自己複製の可能な細胞である請求項1から10のいずれか一に記載の方法。
【請求項12】
前記神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を付加的に成長因子FGFファミリーの一員と接触させることを特徴とする請求項1から11のいずれか一に記載の方法。
【請求項13】
前記神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞をレチノイド若しくはレチノイド誘導体、レチノイドXレセプター(RXR)の活性化剤、レチノイド酸レセプター(RAR)の抑制剤、9−シス−レチナール、DHA、SR11237、又はLG849と接触させることを特徴とする請求項1から12のいずれか一に記載の方法。
【請求項14】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を、細胞をGSK-3β阻害の調節で処理する前に、又はそれと同時に、bFGF、及び/又はEGF、及び/又はFGF-8、及び/又はLIF、及び/又はShhで処理することを特徴とする請求項1から13のいずれか一に記載の方法。
【請求項15】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を抗酸化剤、アスコルビン酸、低酸素分圧、又は低酸素誘導因子の存在下で生長させることを特徴とする請求項1から14のいずれか一に記載の方法。
【請求項16】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を腹側中脳アストロサイト、又は初期グリア細胞の存在下で生長、及び/又は分化させることを特徴とする請求項1から15のいずれか一に記載の方法。
【請求項17】
GSK-3β阻害の調節は、細胞を含むin vitro培養物に対してなされることを特徴とする請求項1から16のいずれか一に記載の方法。
【請求項18】
GSK-3β阻害の調節が神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞でのタンパク質の発現によって、又はGSK-3β阻害の調節をする物質をコードする核酸を用いた形質転換によって生じることを特徴とする請求項17に記載の方法。
【請求項19】
GSK-3β阻害の調節が神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞と共培養される細胞からの発現によって生じ、該共培養細胞が、タイプIアストロサイト若しくは初期グリア細胞以外の細胞である、又はGSK-3β阻害をする物質をコードする核酸で形質転換されたホスト細胞である、又は導入されたGSK-3βを含む細胞であることを特徴とする請求項17に記載の方法。
【請求項20】
共培養された、タイプIアストロサイト若しくは初期グリア細胞以外の細胞又はホスト細胞は、別の幹、神経幹、前駆若しくは前駆体細胞、又は神経細胞である請求項19に記載の方法。
【請求項21】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞は、GSK-3β阻害の調節をする物質をコードする核酸から発現するように作られたものである請求項17に記載の方法。
【請求項22】
GSK-3β阻害の調節をする物質が細胞内に導入されることを特徴とする請求項17に記載の方法。
【請求項23】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を、任意に腹側中脳の初期グリア細胞、又はタイプIアストロサイトと共培養することをさらに含む請求項1から22のいずれか一に記載の方法。
【請求項24】
タイプIアストロサイトは、不死化された細胞株、又は腹側中脳以外の領域のアストロサイト株である請求項23に記載の方法。
【請求項25】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を、非ドーパミン作動性ニューロンに抗して選択するネガティブ選択剤と接触させることを付加的に含む請求項1から24のいずれか一に記載の方法。
【請求項26】
一又は二以上の付加的な成分を含む組成物にニューロンを処方することをさらに含むことを特徴とする請求項1から25のいずれか一に記載の方法。
【請求項27】
組成物は、薬剤上許容できる賦形剤を含むことを特徴とする請求項26に記載の方法。
【請求項28】
組成物を個体に投与することをさらに含む請求項27に記載の方法。
【請求項29】
ニューロンを個体の脳内に移植することを特徴とする請求項28に記載の方法。
【請求項30】
細胞を個体においてin situで処理してNurr1発現を増加することを特徴とする請求項1から16のいずれか一に記載の方法。
【請求項31】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を、Frizzeledレセプターに対するリガンド以外のGSK-3β阻害を調節する物質で、個体においてin situで処理することを特徴とする請求項1から16のいずれか一に記載の方法。
【請求項32】
GSK-3β阻害を調節する物質をコードする核酸を細胞内に導入することを特徴とする請求項31に記載の方法。
【請求項33】
GSK-3β阻害を調節する物質を細胞内に導入することを特徴とする請求項31に記載の方法。
【請求項34】
細胞を個体においてin situで処理して、基底レベルを超えるNurr1発現を増加することを特徴とする請求項31に記載の方法。
【請求項35】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞は、個体に内在性のものである請求項30から34のいずれか一に記載の方法。
【請求項36】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞は、個体内に移植によって外来的に供給されるものである請求項30から34のいずれか一に記載の方法。
【請求項37】
個体がパーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患を有することを特徴とする請求項28から36のいずれか一に記載の方法。
【請求項38】
前記方法に従って製造されたニューロンの個体治療用の薬剤製造における使用をさらに含む請求項1から27のいずれか一に記載の方法。
【請求項39】
薬剤は、個体脳内への移植用である請求項38に記載の方法。
【請求項40】
個体がパーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患を有することを特徴とする請求項39に記載の方法。
【請求項41】
請求項1から25のいずれか一に記載の方法に従って製造されたドーパミン作動性ニューロン。
【請求項42】
神経変性疾患の治療用に使用される薬剤をスクリーニングする方法における請求項41に記載のドーパミン作動性ニューロンの使用。
【請求項43】
単独、若しくは組み合わせで、基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞において、増殖、自己複製、生存、及び/又はドーパミン作動性の発生、誘導、分化、若しくは成熟を増強する1以上の因子を得る方法であって、
(a)基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を、一以上の試験物質の存在下及び非存在下においてGSK-3β阻害のレギュレーターで処理すること、そして
(b)細胞の増殖、自己複製、生存、及び/又はドーパミン作動性の発生、誘導、分化、若しくは成熟を測定し、そして増殖、自己複製、生存、及び/又はドーパミン作動性の発生、誘導、分化、若しくは成熟の程度を、一以上の試験物質の存在下及び非存在下で比較することによって前記1以上の因子を得ることを含んでなる方法。
【請求項44】
細胞を含むin vitro培養物において該細胞をGSK-3β阻害のレギュレーターで処理することを特徴とする請求項43に記載の方法。
【請求項45】
GSK-3β阻害のレギュレーターをコードする核酸の細胞内導入によって、細胞を該レギュレーターで処理することを特徴とする請求項43に記載の方法。
【請求項46】
GSK-3β阻害のレギュレーターの細胞内導入によって細胞を該レギュレーターで処理することを特徴とする請求項43に記載の方法。
【請求項47】
神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を腹側中脳の初期グリア細胞又はタイプIアストロサイトと共培養することをさらに含む請求項43から46のいずれか一に記載の方法。
【請求項48】
基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞において増殖、自己複製、生存、及び/又はドーパミン作動性の発生、誘導、分化、又は成熟を増強できる1以上の因子を単離された、及び又は精製された形態で提供することを特徴とする請求項43から47のいずれか一に記載の方法。
【請求項49】
基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞において増殖、自己複製、生存、及び/又はドーパミン作動性の発生、誘導、分化、若しくは成熟を増強できる1以上の因子を、一以上の付加的な成分を含む組成物に処方することを特徴とする請求項43から48のいずれか一に記載の方法。
【請求項50】
組成物が基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を含むことを特徴とする請求項49に記載の方法。
【請求項51】
組成物は、薬剤上許容できる賦形剤を含むことを特徴とする請求項49又は50に記載の方法。
【請求項52】
組成物を個体に投与することをさらに含む請求項51に記載の方法。
【請求項53】
組成物を個体の脳内に移植することを特徴とする請求項52に記載の方法。
【請求項54】
個体がパーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患を有することを特徴とする請求項53に記載の方法。
【請求項55】
パーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患治療用の薬剤製造における、Frizzeledレセプターのリガンド以外でGSK-3βの阻害を調節する物質の使用。
【請求項56】
パーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患治療用の薬剤製造における、Frizzeledレセプターのリガンド以外のGSK-3β阻害のレギュレーターで処理した神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞であって、基底レベルを超えるNurr1サブファミリーの核内レセプターを発現している該細胞の使用。
【請求項57】
GSK-3β阻害は、ヒメナルジシン(hymenaldisine)類、アロイシン(aloisine)類、マレイミド類、チアゾール類、チエニル及びメチルハロメチルケトン類、酸化アミノピリミジン類、すなわちインディルビン類並びにパウロン(paullone)類からなるグループから選択されるGSK-3β阻害剤によって提供される請求項55又は56に記載の使用。
【請求項58】
阻害剤は、酸化ピリミジン又はパウロンである請求項57に記載の使用。
【請求項59】
阻害剤は、ケンパウロン(kenpaullone)である請求項58に記載の使用。
【請求項60】
GSK-3β阻害は、β−カテニンの安定化によることを特徴とする請求項55又は56に記載の使用。
【請求項61】
ドーパミン作動性ニューロンの発生を誘導、又は促進する物質を得る方法であって、細胞内で基底レベルを超えるNurr1サブファミリーの核内レセプターを発現する神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞、又はドーパミン作動性ニューロンにおいてGSK-3βの阻害を調節する試験物質の能力を測定することを含んでなる方法。
【請求項62】
細胞内でGSK-3β阻害を与える試験物質の能力を測定することを含む請求項61に記載の方法。
【請求項63】
細胞内でGSK-3βを阻害する試験物質の能力を測定することを含む請求項61又は62に記載の方法。
【請求項64】
細胞内でβ−カテニンを安定化する物質の能力を測定することを含む請求項61又は62に記載の方法。
【請求項65】
ドーパミン作動性ニューロンの発生を誘導又は促進する作用物質を得る方法であって、
(i)試験物質のGSK-3βの阻害を調節する能力を測定し、それによってGSK-3β阻害を調節する物質を同定すること、
(ii)(i)で同定されたGSK-3β阻害を調節する物質と共に、細胞内で基底レベルを超えるNurr1サブファミリーの核内レセプターを発現する神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を提供することによって、GSK-3β阻害を調節する物質のドーパミン作動性ニューロンの発生を誘導又は促進する能力を測定し、それによって前記作用物質を得ることを含んでなる方法。
【請求項66】
GSK-3β阻害を調節する物質がGSK-3βを阻害することを特徴とする請求項65に記載の方法。
【請求項67】
GSK-3β阻害を調節する物質がβーカテニンを安定化することを特徴とする請求項65に記載の方法。
【請求項68】
一以上の付加的な成分を含む組成物中に前記作用物質を処方することをさらに含む請求項61から67のいずれか一に記載の方法。
【請求項69】
組成物が基底レベルを超えるNurr1を発現している神経幹、前駆若しくは前駆体細胞、又は他の幹若しくは神経細胞を含むことを特徴とする請求項68に記載の方法。
【請求項70】
組成物が薬剤上許容できる賦形剤を含むことを特徴とする請求項68又は69に記載の方法。
【請求項71】
組成物を個体に投与することをさらに含む請求項70に記載の方法。
【請求項72】
組成物を個体の脳内に移植することを特徴とする請求項71に記載の方法。
【請求項73】
個体がパーキンソン病、パーキンソン症候群、神経欠損、又は神経変性疾患を有することを特徴とする請求項72に記載の方法。
【請求項74】
前駆症状性パーキンソン病の検出、又はパーキンソン病の診断若しくは診断の確認の方法であって、患者又は患者から得たサンプルにおいてGSK-3β阻害の異常の存在を決定することを含んでなる方法。
【請求項75】
GSK-3β阻害の異常の存在を患者から得たニューロンで決定することを特徴とする請求項74に記載の方法。
【請求項76】
異常をGSK-3β活性で測定することを特徴とする請求項74又は75に記載の方法。
【請求項77】
異常をβ−カテニンの安定化で測定することを特徴とする請求項74又は75に記載の方法。
【図1】


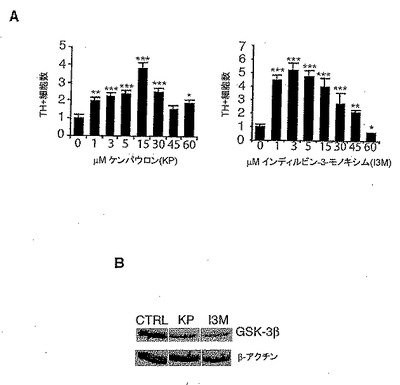
【図2】
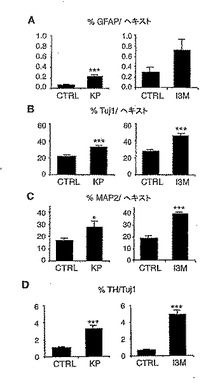
【図3】
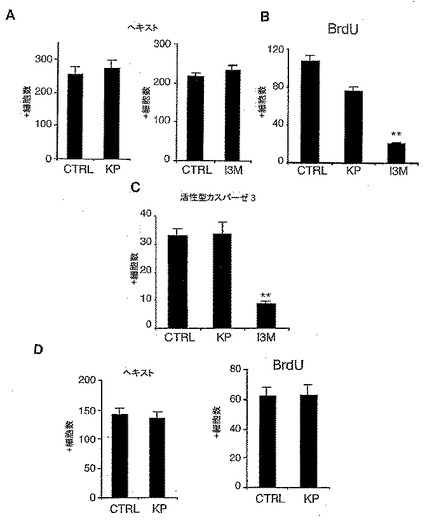
【図4】
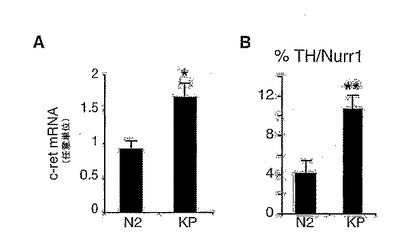
【図5】
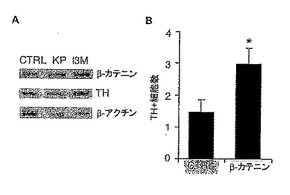
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2008−511304(P2008−511304A)
【公表日】平成20年4月17日(2008.4.17)
【国際特許分類】
【出願番号】特願2007−529034(P2007−529034)
【出願日】平成17年9月2日(2005.9.2)
【国際出願番号】PCT/IB2005/002803
【国際公開番号】WO2006/024947
【国際公開日】平成18年3月9日(2006.3.9)
【出願人】(505107077)ニューロ セラピューティクス エービー (4)
【Fターム(参考)】
【公表日】平成20年4月17日(2008.4.17)
【国際特許分類】
【出願日】平成17年9月2日(2005.9.2)
【国際出願番号】PCT/IB2005/002803
【国際公開番号】WO2006/024947
【国際公開日】平成18年3月9日(2006.3.9)
【出願人】(505107077)ニューロ セラピューティクス エービー (4)
【Fターム(参考)】
[ Back to top ]