
ナノポアを用いたバイオポリマー決定方法、システム、及びキット
【課題】ナノポアを用いたバイオポリマーの計測方法は,非常に高速,安価分析であるが,バイオポリマーを構成するモノポリマ一つずつを判別する精度が低かった。
【解決手段】バイオポリマーのナノポアを介した両端にナノポアよりも大きな分子を結合し,外力によって前記バイオポリマーを往復運動させ,繰返し計測を行う。
【解決手段】バイオポリマーのナノポアを介した両端にナノポアよりも大きな分子を結合し,外力によって前記バイオポリマーを往復運動させ,繰返し計測を行う。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は,ナノポア構造を用いた,DNA,RNA,又はタンパク質等のバイオポリマーの高精度な計測,分析を行うための構造,方法に関する技術分野に属する。
【背景技術】
【0002】
病気の診断や創薬に対して,核酸(DNAやRNA)やタンパク質等のバイオポリマーを分析することは重要である。特にDNAは,生命の最も基本の構成体であるため,その分析,つまり塩基配列の決定は,上記目的に対して非常に重要となる。DNAの塩基配列を分析する主な方法としては,まず,化学もしくは酵素反応を利用して,配列決定したい鋳型DNAに対して,決まった末端塩基種をもつ様々な長さのDNA断片群を生成する。前記DNA断片群を4種の塩基種に対してそれぞれ生成する。次いで,ゲル電気泳動を用いて,これらのDNA断片群を分子量の順に分離する。前記DNA断片群を分離媒体に導入し電圧を印加する。DNA断片は全体としてマイナスの電荷をもつ高分子電解質であるため,マイナスからプラスの方向に電気泳動する。ゲル中では長いDNA断片ほど小さな易動度を持つので,1塩基長の違いしか持たないDNA断片同士をも分離することができる。分離後に,DNA断片の末端塩基種に応じた長さを計測することで,鋳型DNAにおける塩基種の位置が分かる。前記操作により鋳型DNAの塩基配列が決定される。上記方法において,DNA断片群の生成や電気泳動の作業は,非常に煩雑で分析時間も長く,ランニングコストも高価である。
【0003】
特許文献1および非特許文献1には,絶縁膜である脂質二重層の薄膜上にα−ヘモリジンを用いて形成した直径数ナノメートルの微細な開口,つまりナノポアを用いたバイオポリマーの分析方法が記載されている。2つの溶液槽の間にナノポアを設けた薄膜設置し,両溶液槽間で電圧勾配を設け,電流を計測する。バイオポリマーであるDNA分子を片方の溶液槽に設置すると,電圧勾配によりナノポアを通過し,別の溶液槽に移動する。DNA分子がナノポアを通過する際に,ナノポア内のイオンの流れを塞ぐため,電流の減少(封鎖電流)が生じる。この封鎖電流の大きさとその封鎖電流の継続時間を計測することにより,ナノポアを通過する個々のDNA分子の長さを検出可能となる。また,理論的にはDNA分子を構成する個々の塩基の種類の判別も可能となる。
【0004】
特許文献2および非特許文献2には,脂質二重層におけるα−ヘモリジンを用いたナノポアに代わり,絶縁膜であるシリコンナイトライド(Si3N4)膜に”ion−beam sculpting“と言う技術を用いてナノポアを作製し, 1本鎖DNA分子の封鎖電流の計測を行っている。
【0005】
非特許文献3では,ナノポアを通過するDNA分子の計測方法として,前述した封鎖電流とは異なる手段が提案されている。ナノポアの内表面に一対の金属電極を設け,この金属電極間をDNA鎖が通過する際のトンネル電流を計測する。この方法でも電極サイズなどが適切に制御されていれば,理論的には,ナノポアを通過するDNA分子を構成する個々の塩基の種類の判別も可能となる。
【0006】
特許文献3では,既知配列プローブのターゲット1本鎖DNAへのハイブリダイゼーションと,ナノポアを用いた前記既知配列プローブのハイブリダイゼーション位置の検出により,ターゲットDNAの塩基配列を決定する方法が示されている。既知配列のプローブを決定したいターゲット1本鎖DNA分子にハイブリダイゼーションさせ,前記ハイブリダイゼーションしたDNA分子をナノポアに通過させる。DNA分子がナノポアを通過する際にDNA分子の1本鎖部位と2本鎖部位で封鎖電流が異なるため,電流を計測することにより,既知配列プローブのハイブリダイゼーション位置を特定できる。複数種の異なる配列を持つ既知配列プローブで前記操作を行い,それぞれのターゲットDNA分子へのハイブリダイゼーション位置データを取得し,得られたデータをコンピュータアルゴリズム使って配列データに変換することで,ターゲットDNA分子の配列決定が可能となる。
【0007】
非特許文献4では,DNA分子がナノポア通過直後に印加電圧の正負を逆転することにより,同一DNA分子が再び同一のナノポアを通過する結果が示されている。
【0008】
非特許文献5では,ナノポア上でDNA分子の伸長反応を行い,封鎖電流計測により伸長反応の有無を確認している。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】米国特許第5,795,782
【特許文献2】米国特許第6,627,076
【特許文献3】米国特許公報第2007/0190542
【非特許文献】
【0010】
【非特許文献1】PNAS 1996, Vol.93, pp.13770-13773
【非特許文献2】NATURE 2001, Vol.412, pp.166-169
【非特許文献3】NANO LETTERS 2005, Vol.5, pp.421-424
【非特許文献4】nature nanotechnology 2007, Vol.2, pp.775-779
【非特許文献5】JACS 2008, Vol.130, pp.818-820
【発明の概要】
【発明が解決しようとする課題】
【0011】
電気泳動を用いたバイオポリマー,特にDNA分子の配列決定は分析時間も長く,ランニングコストも高価である。一方,特許文献1や非特許文献3に記載されたナノポアを用いたDNA分子の配列決定は,高速・安価な分析の可能性を持つが,それぞれ以下の問題がある。
【0012】
特許文献1では,DNA分子がナノポアを通過する時の各塩基種による封鎖電流の違いは僅かであるため,その判別をすることは困難であり,その精度は非常に低い。また,判別精度を向上するためには,ナノポア近傍の膜厚を1塩基分程度,つまりサブナノメートルにする必要があるが,現状の技術でその膜厚を達成するのは困難である。
【0013】
非特許文献3において,DNA分子がナノポアを通過する時のトンネル電流の大きさは,ナノポアを通過する塩基の種類の他に,その塩基のトンネル電流を計測する電極近傍での配向にも依存する。現状,通過する核酸の配向制御が困難であるため,塩基種分離の精度を上げるためには,繰返し計測が必要となる。
【0014】
一方,特許文献3で示される方法では,既知配列プローブのターゲットDNA分子へのハイブリダイゼーションした位置を検出するため,塩基1種類ずつを判別する必要は無い。しかしながら,1塩基レベルでの高精度なハイブリダイゼーション位置の検出が必要になる。また,同一ターゲットDNA分子に対して,複数種の異なる既知配列プローブを,それぞれ異なる場もしくは異なるタイミングでターゲットDNA分子にハイブリダイゼーションして,その位置を検出する必要がある。つまり,ターゲットDNA分子を増幅し,それらをそれぞれ異なる既知配列プローブが存在する異なる溶液に導入し,ターゲットDNA分子と既知配列プローブをハイブリダイゼーションさせて,それぞれ異なるナノポアもしくは異なるタイミングで同一ナノポアを用いて計測を行う必要がある。もしくは,ある既知配列プローブとハイブリダイゼーションしたターゲットDNA分子をナノポア計測した後に,抽出し,変性させて1本鎖に戻し,異なる既知配列プローブとハイブリさせて,再度ナノポア計測を行う事を繰返す必要がある。いずれの場合も,非常に煩雑な作業を要する。
【0015】
非特許文献4で示されるように,DNA分子がナノポアを通過した後に,逆電圧を印加することを繰返すことで,同一DNA分子を複数回同一ナノポアで計測できる。そのため,繰返し計測による計測精度の向上を図れる可能性はあるが,高精度なフィードバックによる電圧印加が必要になる。また,複数種のDNA分子が混在していた場合には,コンタミの恐れもある。さらに,DNA分子の方向もランダムとなる。
【0016】
非特許文献5では,ターゲットDNA分子の5’末端にpolyethylene glycol(PEG)−ビオチンを介したストレプトアビジンを結合させ,3’末端側には数塩基からなるDNAプライマーをハイブリダイゼーションさせ,ターゲットDNA分子の3’末端を2本鎖にすることにより,ナノポアを介したターゲットDNA分子の往復運動を行っている。前記往復運動を用いた繰返し計測により,高精度なナノポア計測ができる可能性がある。しかしながら,DNAプローブのターゲットDNA分子へのハイブリダイゼーションする位置をコントロールすることは難しく,ターゲットDNA分子の様々な位置にDNAプローブが結合し,往復運動ができないことがある。また,2本鎖DNAの直径は2nm程度であるため,ナノポア直径が2nm以上では,2本鎖部分がナノポアを通過してしまい,前記往復運動が困難となる。
【課題を解決するための手段】
【0017】
本発明は,核酸やタンパク質のようなバイオポリマー分子のナノポア計測において,計測精度向上のための繰返し計測を安定してできる方法,システム、及びキットを提供する。
【0018】
第一の溶液槽と第二の溶液槽を具備しており,両溶液槽間は薄膜で区切られており,薄膜にはナノメートルサイズの穴,つまりナノポアが設けられている。バイオポリマー分子の片端にナノポアの開口直径よりも大きなサイズの分子(第一ストッパ分子)を結合し,それを第一の溶液槽に導入する。外力を加えてバイオポリマーを駆動させてナノポアを通過させ,第二の溶液槽に移動させる。第一ストッパ分子により,ナノポアを通過するバイオポリマー分子は途中で移動を停止する。バイオポリマー分子の移動停止後に,第二の溶液槽に存在するナノポアの開口直径よりも大きな分子(第二ストッパ分子)をバイオポリマー分子のもう一方の片端に結合させる。次いで,外力を加えてバイオポリマー分子を,両溶液槽間を往復運動させながら計測を行い,バイオポリマー分子の同定を行う。
【0019】
第二ストッパ分子のバイオポリマー分子への結合は,バイオポリマー分子がナノポアを移動している間であっても良い。また,第二ストッパ分子をバイオポリマー分子に結合する際に,バイオポリマー分子を駆動させる外力を停止させても良いし,外力を与え続けても良い。第二ストッパ分子は,予め第二溶液槽に入れていても,バイオポリマー分子の片端が第二溶液槽に移動後に導入しても良い。両ストッパ分子は同じ分子であっても良いし,異なる分子であっても良い。バイオポリマーを駆動させる手段としては,バイオポリマー分子が電荷を持っているのであれば両溶液槽間に電圧勾配を生じさせても良いし,両溶液槽間でイオン組成を変えることによる電気化学的勾配を生じさせても良いし,溶液の流れを生じさて駆動させても良い。計測の手段としては,両溶液槽間を,ナノポアを介して流れる電流(封鎖電流)値であっても良いし,ナノポア内部に具備された電極間を流れる電流(トンネル電流)値であっても良い。また,バイオポリマー分子に蛍光体を標識し,ナノポア近傍で励起させ,その発光蛍光を検出しても良い。バイオポリマー分子の両端に両ストッパ分子が結合した状態で,バイオポリマー分子に,ある物質を結合させたり,分離させたりしても良い。例えば,非特許文献3のようにハイブリダイゼーションベースでのDNA分子配列決定の場合には,前記方法を用いて簡便かつ高精度に計測できる。DNA分子の両端にストッパ分子を結合させ,溶液槽に既知配列プローブを導入し,それをターゲットDNA分子にハイブリダイゼーションさせ,ターゲットDNA分子を,両溶液槽間を往復運動させてハイブリダイゼーションの位置を封鎖電流計測により検出する。次いで,変性操作により前記既知配列プローブをターゲットDNA分子から分離させ,別の既知配列プローブを用いて前記と同様の操作を繰返す。前記手段により,同一ターゲットDNA分子に対して異なる既知配列プローブを簡便に何回でもハイブリダイゼーションできる。また繰返し計測ができるため,ハイブリダイゼーションの位置を高精度に計測できる。
【0020】
本発明のバイオポリマー決定方法は,第1の溶液槽と,第2の溶液槽と,前記第1の溶液槽と前記第2の溶液槽とを区切りナノポアを備えた薄膜とを有する装置を用い,測定対象のバイオポリマーを構成するモノマーの並びを決定する方法であって,前記ナノポアよりも大きな第1の分子を一端に結合した前記バイオポリマーを前記第1の溶液槽に導入する工程と, 前記バイオポリマーを前記ナノポアを通して前記第1の溶液槽から前記第2の溶液槽に移動させる工程と,前記第2の溶液槽に,前記ナノポアよりも大きな第2の分子を導入し,前記バイオポリマーの他端に結合させる工程と,前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通して移動させる工程と,前記バイオポリマーの移動に伴い発生する信号の時間変化を測定し,前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出し,前記バイオポリマーを構成するモノマーの並びを決定することを特徴とする。
【0021】
本発明のバイオポリマー決定システムは,ナノポアを有する薄膜を備えた装置を用い,前記ナノポアの大きさよりも大きく,測定対象のバイオポリマーに結合させる分子を導入するシステムであって,前記装置は,前記薄膜によって区切られた第1及び第2の溶液槽と,前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通じて移動させる駆動手段と,前記分子が前記測定対象のバイオポリマーの一方に結合されたバイオポリマーを前記第1の溶液槽に導入する手段と,前記ナノポアを通過した前記バイオポリマーの他端に結合させる前記分子を前記第2の溶液槽に導入する手段と,前記駆動手段による前記バイオポリマーの移動に伴い発生する信号を検出する検出手段と,前記検出手段により検出した信号の時間変化を測定し,前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出する算出し,前記バイオポリマーを構成するモノマーの並びを決定する算出手段とを有することを特徴とする。
【0022】
本発明のバイオポリマー決定キットは,ナノポアを有する薄膜を備えた装置と,前記ナノポアの大きさよりも大きく,測定対象のバイオポリマーに結合させる分子とを有するキットであって,前記装置は,前記薄膜によって区切られた第1及び第2の溶液槽と,前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通じて移動させる駆動手段と,前記分子が前記測定対象のバイオポリマーの一方に結合されたバイオポリマーを前記第1の溶液槽に導入する手段と,前記ナノポアを通過した前記バイオポリマーの他端に結合させる前記分子を前記第2の溶液槽に導入する手段と,前記駆動手段による前記バイオポリマーの移動に伴い発生する信号を検出する検出手段と,前記検出手段により検出した信号の時間変化を測定し,前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出する算出し,前記バイオポリマーを構成するモノマーの並びを決定する算出手段とを有することを特徴とする。前記分子は,ストレプトアビジン,又は,DIG−抗DIG抗体結合により前記バイオポリマーに結合されるビーズであることを特徴とする。
【発明の効果】
【0023】
本発明により,バイオポリマー分子のナノポアを用いた安定した繰返し計測が可能になり,安価,高速,高精度な計測が可能となる。
【図面の簡単な説明】
【0024】
【図1】サンプル前処理時のサンプル溶液中の模式図
【図2】ベクターを利用したサンプル前処理法の模式図
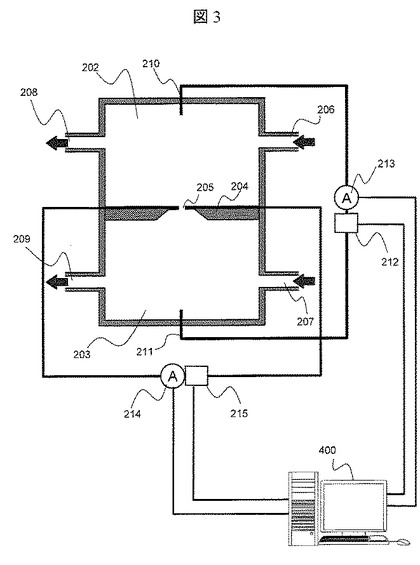
【図3】実施例1におけるナノポア装置の概略図
【図4】実施例1におけるナノポア近傍の拡大図
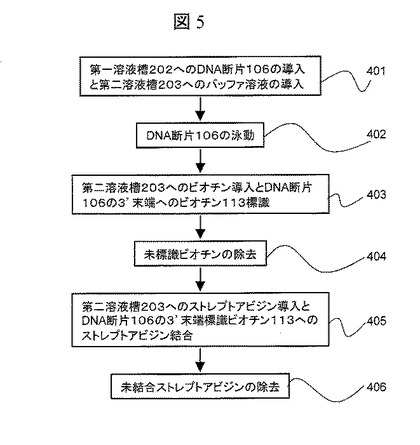
【図5】DNA断片の3’末端にストレプトアビジンを結合するフローチャート
【図6】DNA断片の3’末端にストレプトアビジンを結合する様子の模式図
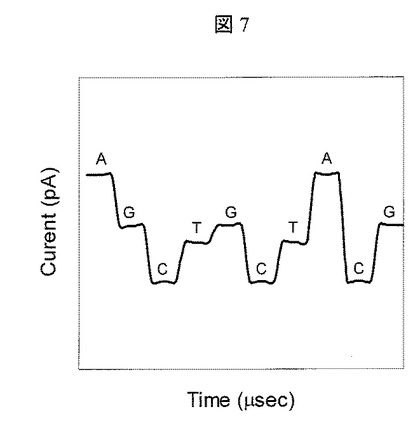
【図7】DNA分子ナノポア通過時のトンネル電流値の経時変化のグラフ
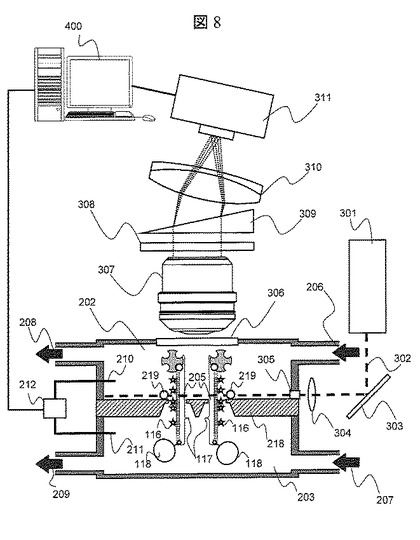
【図8】実施例2におけるナノポア装置の概略図
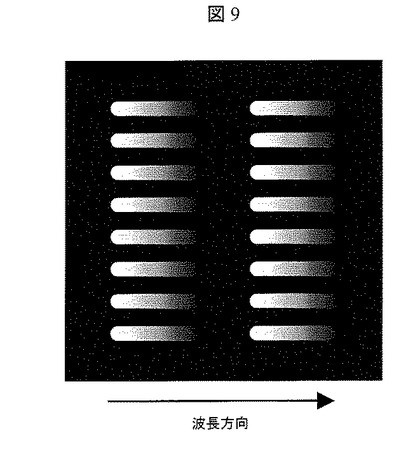
【図9】実施例2における各ナノポアからの発光のCCD上でイメージ図
【図10】DNA分子ナノポア通過時の各波長での信号強度の経時変化のグラフ
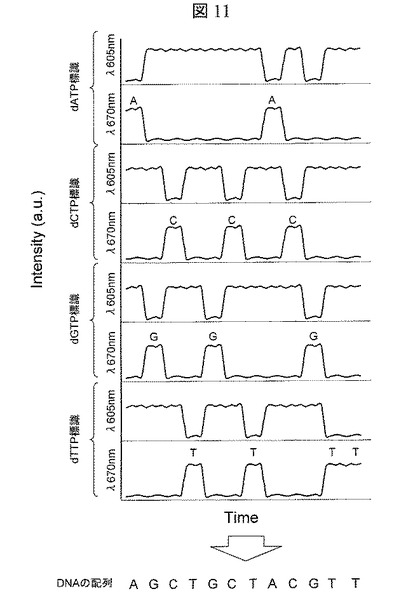
【図11】DNA分子ナノポア通過時の各塩基種における信号強度の経時変化のグラフ
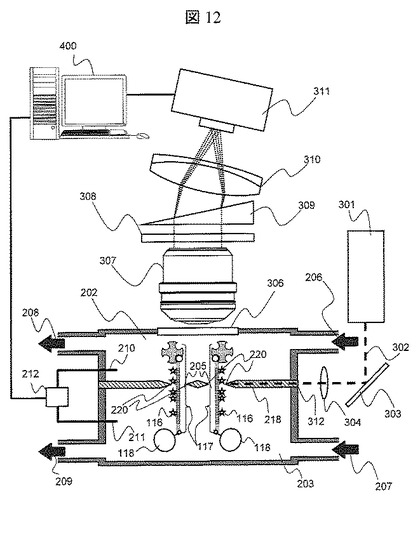
【図12】実施例3におけるナノポア装置の概略図
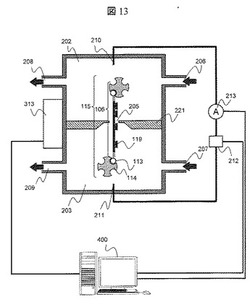
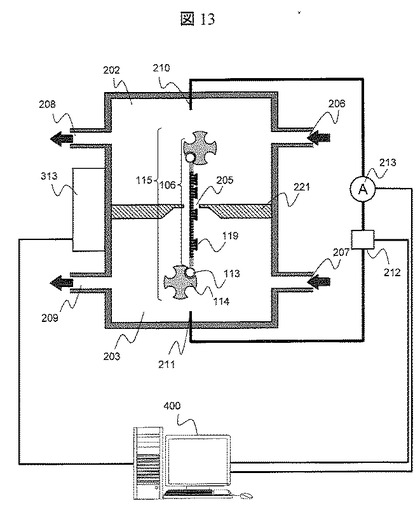
【図13】実施例4におけるナノポア装置の概略図
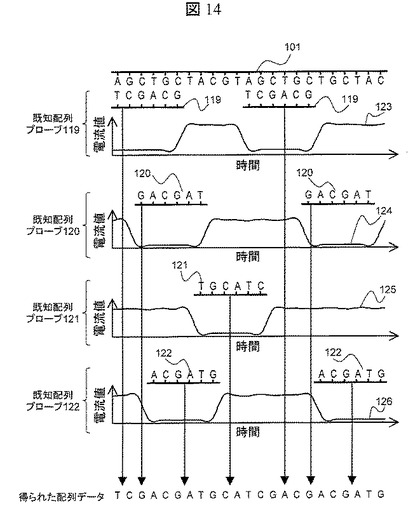
【図14】各既知配列プローブをハイブリダイゼーションさせたDNA分子のナノポア通過時の封鎖電流の経時変化グラフ
【発明を実施するための形態】
【0025】
以下,図面に従って本発明の実施の形態を説明する。
【実施例1】
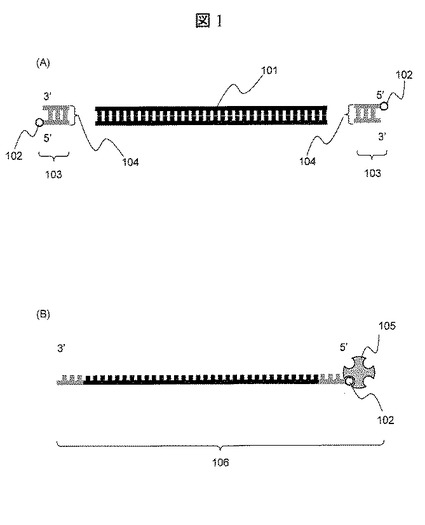
【0026】
本発明を用いたナノポアのトンネル電流計測によるDNA分子の塩基配列決定方法を説明する。図1(A)および(B)は,計測前のサンプル前処理時のサンプル溶液中の状態を模式的に示したものである。溶液中には,2本鎖になっているターゲットDNA分子101,5’末端にビオチン102が標識された2本鎖の合成プローブ103が混在している。2本鎖合成プローブ103のビオチンが標識されていない端面104は平滑末端処理されている。ターゲットDNA分子101の両末端に対して,S1 Nucleaseなどを用いて平滑末端処理を行う。ターゲットDNA分子101の平滑末端処理後に,リガーゼを用いて,ターゲットDNA分子101の両端に合成プローブ103をライゲーションさせる。前記ライゲーション反応終了後に,サンプルをアクリルアミドゲル電気泳動によりサイズ分離する。ターゲットDNA分子101の両端に合成プローブ103が結合したもののみを,アクリルアミドゲルより切出して蒸留水に溶出する。前記操作により,合成プローブ103同士がライゲーションしたもの,ターゲットDNA分子101同士がライゲーションしたものを除去できる。次いで,ストレプトアビジン105が入ったバッファ溶液を,両端に合成プローブが結合したターゲットDNA分子101が溶出された溶液に混入し,5’末端に標識されたビオチン102とストレプトアビジン105を結合させ,熱を加えてDNAを変性させ,図1(B)に示す5’末端にストレプトアビジンが結合したターゲットDNA分子101が含まれたDNA断片106を生成する。図2(A)および(B)は,DNA断片106の別の生成方法を模式的に示したものである。ターゲットDNA分子101をベクター107のマルチクローニングサイトの一部に導入する。次いで,ターゲットDNA分子101を挟みこむ状態で,ベクターのそれぞれ異なる制限酵素サイト108と109で制限酵素処理による切断を行う。そして、制限酵素サイト108の切断末端を有し,5’末端にビオチン102が標識された2本鎖の合成プローブ110と制限酵素サイト109の切断末端を有し,5’末端にビオチン102が標識された2本鎖の合成プローブ111を前記ベクターより切断処理した切断断片112に導入し,ライゲーション操作を行う。その後,5’末端に標識されたビオチン102とストレプトアビジン105を結合させ,熱を加えてDNAを変性させ,図1(B)に示す5’末端にストレプトアビジンが結合したターゲットDNA分子101が含まれたDNA断片106を生成する。
【0027】
図3は,本実施例で使用するナノポア装置の概略図である。ナノポア装置は,第一溶液槽202,第二溶液槽203,両溶液槽を分割するナノポア薄膜204で構成される。両溶液槽には,溶液を導入する導入口206,207および溶液を排出する排出口208,209がそれぞれ具備されている。ナノポア薄膜204を介して両溶液槽間で電圧勾配を設けるために,両溶液槽202,203には電極210,211が具備されており,前記電極210と211は極性可変の電圧源212および電流計213に接続されている。ナノポア薄膜204は,直径1nmのナノポア205が形成された絶縁体の薄膜で構成されている。ここでは,絶縁体薄膜の材料としてSi3N4を用いているが,他にSiO2やアスファルテンなどのプラスチック材料でもよい。さらに,Alなどの金属膜の表面に絶縁材量をコートした薄膜でも良い。ナノポア205の径としては,ここでは1nmとしているが,0.5nmから50nm程度あればよい。尚,ストッパ分子として用いたストレプトアビジンの大きさは5nm程度であり,ナノポアの大きさよりも十分大きい。ナノポアの径に対するストッパ分子の大きさとしては,DNA断片の進行を止めることが出来る大きさであればよいが,精度向上のためには,1.2〜50倍程度あることが望ましい。
【0028】
図4(A)は,ナノポア205近傍の拡大図である。また,図4(B)は図4(A)のa-a’での断面図である。ナノポア205内表面には一対の電極216と217が具備されており,前記電極216,217は電圧源215と電流計214に接続されている。
【0029】
以下,図5のフローチャートに沿ってDNA断片106の3’末端にストレプトアビジンを結合する方法を説明する。なお,図6に,DNA断片106の3’末端にストレプトアビジンを結合する様子を模式的に示す。
【0030】
まず、前記方法で得られたDNA断片106をバッファ溶液に混合し,導入口206より第一溶液槽202に,バッファ溶液のみを導入口207より第二溶液槽203に導入する(401)(図6(A))。電極210が陰極,電極211が陽極になるように電圧源212により電圧を印加し,DNA断片106を第一溶液槽202から第二溶液槽203に泳動させる(402)(図6(B))。前記電圧印加と同時に電流計213によってナノポア205を介して流れるイオンの流れを計測する。ストレプトアビジン105の大きさは5nm程度であるため直径1nmのナノポアは通過できず,DNA断片106は3’末端(ストレプトアビジン105が標識されていない末端)よりナノポア205に導入される。DNA断片106がナノポア205に導入されると電流値は減少する。先述したように,ストレプトアビジン105の大きさはナノポア205の直径よりも大きいため,ストレプトアビジン105がナノポア205を通過する直前で,DNA断片106の第二溶液槽203への移動が止まる。前記電流の減少を確認した後に,導入口207よりBiotin 3’End DNA Labeling Kitを第二溶液槽203に導入し,DNA断片106の3’末端にビオチン113を標識する(403)(図6(C))。
【0031】
ビオチン標識後に,導入口207よりバッファのみを導入し,未反応のビオチンを第二溶液槽203より除去する(404)。次いで,ストレプトアビジンを含んだ溶液を導入口207より導入し,前記DNA断片106の3’末端に標識されたビオチン113にストレプトアビジン114を結合させ,アレイ型DNA断片115を生成する(405)(図6(D))。導入口207よりバッファのみを導入し,未反応のストレプトアビジンを第二溶液槽203より除去する(406)。
【0032】
上記では,第一,第二ストッパ分子として同じ物質であるストレプトアビジンを使用したが,異なる物質を使用しても良い。例えば,第一,第二ストッパ分子に異なる物質を使用したアレイ型DNA断片115の別な生成方法として以下の方法がある。図2で示した方法において,合成プローブ110および111の切断末端とは異なる末端側の3’末端にDigoxigein(DIG)を標識しておく。それ以外は,前述した方法でDNA断片106を生成する。この時,DNA断片106の3’末端はDIG標識されている。次いで,前記3’末端がDIG標識されたDNA断片106を前述と同等の方法で,第一溶液槽202から第二溶液槽203へ泳動させる。DIG自身の大きさは,ナノポア205の直径1nmよりも十分小さいため,DNA断片106は3’末端よりナノポア205に導入される。ストレプトアビジン105によってDNA断片106の移動が停止した状態で,第二溶液槽203に直径が1nmよりも大きなビーズが標識された抗DIG抗体を導入し,DNA断片106の3’末端に標識されたDIGと結合させる。これにより,第一ストッパ分子としてストレプトアビジン,第二ストッパ分子としてDIG−抗DIG抗体結合を介したビーズから構成されるアレイ型DNA断片115を生成できる。ここでは,ビオチン・ストレプトアジビン以外としてDIG・抗DIG結合ビーズを用いた例を記載したが,他にもDNAの末端をチオール化して金粒子を結合させる方法や,DNAの末端をアミノ基修飾してカルボキシルキ修飾のビーズと脱水反応により結合させる方法等を用いてもよい。
【0033】
アレイ型DNA断片115生成後に,電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第二溶液槽203から第一溶液槽202に一定時間泳動させる。この泳動間に電極216,217によりトンネル電流を計測し,アレイ型DNA断片115を構成する塩基種を特定する。次いで,電極210を陰極に,電極211を陽極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第一溶液槽202から第二溶液槽203に一定時間泳動させる。この泳動間に電極216,217によりトンネル電流を計測する。前記両溶液槽間の泳動ならびにトンネル電流の計測を繰返す事により,同一ターゲットDNA分子のトンネル電流の計測が複数回でき,高精度な塩基種決定が可能となる。
【0034】
ここで,塩基種決定の方法を説明する。塩基にはATGCの4種類があるが,これらの塩基の種類ごとに固有の電流値が観測され,データ処理手段400に送られる。一例を図7に示す。予め,各塩基1種類のみが連なったポリマのナノポア通過時のトンネル電流を計測し,各塩基種に対応した電流値を求め,データ処理手段中のメモリに格納しておく。そして、データ処理手段400により,ターゲットDNAのトンネル電流計測時に得られた電流値と前記予め計測した各塩基種に対応した電流値を比較する事により,ターゲットDNAの塩基種決定を行う。
【0035】
前記往復運動計測における電圧極性の切替えは,ある一定時間で自動切替できるよう電圧源212を制御する。制御部は,データ処理手段中400に設けておけばよい。前記一定時間の設定は可変である。ストッパ分子がナノポアに近接するとナノポアを通過する電流の減少が計測できるため,前記電流の減少を引き金に電圧極性の切替えを行っても良い。
【0036】
本実施例では,1個のナノポアを使ったトンネル電流の計測を行っているが,多数のナノポアを使用して多数の異なるターゲットDNA分子のトンネル電流の同時計測により,スループットの向上も図れる。
【0037】
また、本実施例では説明用に、5’末端にストッパ分子が結合したDNAを第一溶液槽に導入してナノポアを通過させた後に3’末端にストッパ分子を結合させる例を記載しているが、同様の方法にて、3’末端にストッパ分子が結合したDNA断片を用い、ナノポアに導入した後5’末端にストッパ分子を結合させるのでもよい。
【実施例2】
【0038】
ナノポアの蛍光共鳴エネルギー移動(FRET)を利用した蛍光検出計測によるDNA分子の塩基配列決定方法を説明する。受容体としての蛍光体Cy5が標識されたターゲットDNA分子を以下の方法で生成した。実施例1の図2(B)を用いて説明したように,ターゲットDNA分子101が含まれ,5’末端にビオチンを介したストレプトアビジンが標識され,3’末端にDIGが標識されたDNA断片106を生成する。次いで,dCTP,dGTP,dTTP,Cy5が標識されたdATP,DNAポリメラーゼ,および前記DNA断片106の合成プローブ部分(110,111のどちらでも良い)と相補配列を持つプライマーを含む反応溶液と前記3’末端にDIGが標識されたDNA断片106を混合し,伸長反応を行う。前記伸長反応時には熱変性は行わない。同様の操作を,Cy5がdCTPのみに標識された反応溶液,dGTPのみに標識された反応溶液,dTTPのみに標識された反応溶液で,それぞれ異なる反応チューブで行い,ターゲットDNA断片101を含んだCy5蛍光体116が標識された2本鎖DNA断片117を生成する。
【0039】
図8は,本実施例で使用するナノポア装置の概略図である。ナノポア装置は,第一溶液槽202,第二溶液槽203,両溶液槽を分割するナノポア薄膜218で構成される。両溶液槽202,203には,溶液を導入する導入口206,207および溶液を排出する排出口208,209がそれぞれ具備されている。ナノポア薄膜218を介して両溶液槽間で電圧勾配を設けるために,両溶液槽202,203には電極210,211が具備されており,前記電極210と211は極性可変の電圧源212に接続されている。第一溶液槽202には,光透過性の照射窓305と検出窓306が具備されている。ナノポア薄膜218は,直径3nmのナノポア205が形成されたSi3N4薄膜で構成されている。ナノポア205は1μmの間隔で格子状に複数個形成されている。ナノポア205近傍には供与体として青色光で励起され605nmの発光蛍光するQdot(605)219が固定されている。
【0040】
レーザ光源(波長488nm)301より発振したレーザ光302は,ミラー303により角度を調整され,集光レンズ304により集光され,照射窓305を介してすべてのQdot219に照射される。ナノポア205近傍で発光した発光蛍光は,検出窓306を介して対物レンズ307によって集光され,フィルタ308により波長550nmから700nm以外の波長の光がカットされ,プリズム309により分光され,結像レンズ310によって図9に示すようにCCD311上に結像される。CCD311のデータは,データ処理手段400に格納される。図9にデータ処理手段400に格納されたCCD311上で結像されたイメージ図を示す。各スポットは各ナノポアからの輝点に対応しており,また,横軸は波長方向に対応している。
【0041】
前記方法で得られた2本鎖DNA断片117をバッファ溶液に混合し,導入口206より第一溶液槽202に,バッファ溶液のみを導入口207より第二溶液槽203に導入する。電極210が陰極,電極211が陽極になるように電圧源212により電圧を印加し,2本鎖DNA断片117を第一溶液槽202から第二溶液槽203に泳動させる。ストレプトアビジン105の大きさは5nm程度であるため直径3nmのナノポアは通過できず,2本鎖DNA断片117は,ストレプトアビジン105が標識されていない末端よりナノポア205に導入される。ストレプトアビジン105によって2本鎖DNA断片117の移動が停止した状態で,第二溶液槽203に直径が3nmよりも大きなビーズが標識された抗DIG抗体118を導入し,2本鎖DNA断片117の蛍光体116が標識されていない鎖の3’末端に標識されたDIGと結合させる。抗DIG抗体118標識後に,導入口207よりバッファのみを導入し,未反応の抗DIG抗体を第二溶液槽203より除去する。この間,電圧を印加し続ける。なお,各ナノポアに導入される2本鎖DNA断片117を構成するターゲットDNA分子は,同じものであってもよいし,異なるDNA分子であってもよい。検出された発光を統計的に処理することにより,それぞれの塩基種の特定ができる。同じDNAの分子の場合には,DNA分子の往復回数を減らして時間を短縮しつつ塩基種特定の精度が向上することができる。一方、異なる分子の場合には、同時に多くの分子を測定してスループットを向上することができる。
【0042】
2本鎖DNA断片117に抗DIG抗体118を標識した後に,レーザ光源301よりレーザ光302を発振し,Qdot219を励起する。次いで,電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,抗DIG抗体118標識2本鎖DNA断片117を第二溶液槽203から第一溶液槽202に一定時間泳動させる。この泳動間にQdot219近傍を2本鎖DNA断片117に標識されている蛍光体118が通過すると共鳴による励起エネルギーの移動が起こり,蛍光体118が発光するため,これら発光をCCD311で検出する。図10に,前記発光に対応するCCD311上でのスポットの内,波長605nmと波長670nmに対応するピクセルの強度の時間的変動を示す。波長605nmはQdot219の発光に対応し,波長670nmは蛍光体118の発光に対応する。検出された波長670nmに対応する信号強度の時間的変動から,蛍光体118の2本鎖DNA断片117の標識位置を算出できる。次いで,電極210を陰極に,電極211を陽極になるように電圧源212により電圧を印加し,2本鎖DNA断片117を第一溶液槽202から第二溶液槽203に一定時間泳動させる。この泳動間に前記と同様にCCD311によって蛍光検出する。前記泳動ならびに蛍光検出を繰返す事により,蛍光体118の2本鎖DNA断片117の標識位置を複数回計測でき,高精度な位置算出が可能となる。4種類の異なるdNTPに蛍光体が標識された2本鎖DNA断片117に対して前記と同様の泳動と蛍光検出の繰返し作業を行い,各波長に対して得られた信号強度の時間的変動を図11に示す。波長670nmに対応する信号強度の時間的変動により,各塩基種のターゲットDNA分子101上での位置が判別でき,各塩基種のデータを合わせることにより,ターゲットDNA分子101の塩基配列を決定する。
【0043】
前記往復運動計測における電圧極性の切替えは,ある一定時間で自動切替できるよう電圧源212を制御する。前記一定時間の設定は可変である。制御部は,データ処理手段中400に設けておけばよい。ストッパ分子がナノポアに近接するとナノポアを通過する電流の減少が計測できるため,前記電流の減少を引き金に電圧極性の切替えを行っても良い。
【0044】
本実施例では1種類の蛍光体のみを使用したが,4種のdNTPに異なる蛍光体を標識し,それらdNTPすべてが標識された2本鎖DNA断片117を生成し,それを前記と同様操作で蛍光検出することで,ターゲットDNA分子101の塩基配列を決定することも可能である。また,高い粘性のバッファ溶液の使用や泳動時の印加電圧を小さくすることにより,2本鎖DNA断片117の泳動速度を遅くする事ができ,高感度に蛍光検出することが可能となる。本実施例ではFRETの供与体としてQdotを使用したが蛍光体であっても良い。また,図8では2つのDNA断片を同時に計測する例を示しているが,1つであってもよいし,また3以上のDNA断片を同時に計測しても良い。
【実施例3】
【0045】
ナノポアの蛍光検出計測によるDNA分子の塩基配列決定方法を説明する。
図12は,本実施例で使用するナノポア装置の概略図である。レーザ光源301,フィルタ308,レーザ光のナノポア装置への照射位置およびナノポア薄膜の構成以外はすべて実施例2と同等である。ナノポア薄膜218を石英ガラスで作製し,表面を石英よりも低い屈折率の樹脂(例えばフロリナート)でコーティングする。この時,ナノポア近傍の鋭利な先端部220の樹脂を剥離しておく。レーザ光源(波長633nm)301より射出したレーザ光302は,集光レンズ304で集光され,ナノポア薄膜側面312に照射される。この時,レーザ光302はナノポア薄膜218内を全反射しながら伝播するが,先端部220は樹脂コーティングされていないため,両溶液槽内を満たすバッファ溶液に,近接場光として僅かに染み出す。レーザ光302はナノポア薄膜218内全体を全反射しながら伝播するため,すべての先端部220で近接場が生じる。ナノポア205近傍で発光した発光蛍光は,検出窓306を介して対物レンズ307によって集光され,フィルタ308により波長660nmから700nm以外の波長の光がカットされ,プリズム309により分光され,結像レンズ310によってCCD311上に結像される。CCD311のデータは,データ処理手段400に格納される。図12では2つのDNA断片を同時に計測する例を示しているが,1つであってもよいし,また3以上のDNA断片を同時に計測しても良い。
【0046】
実施例2と同等の方法で,2本鎖DNA断片117を生成および蛍光体116が標識されていない鎖の3’末端に標識されたDIGに直径が3nmよりも大きなビーズが標識された抗DIG抗体118を結合させる操作を行う。
【0047】
2本鎖DNA断片117に抗DIG抗体118を標識した後に,レーザ光源301よりレーザ光302を発振し,先端部220に近接場光を生成する。次いで,電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,抗DIG抗体118標識2本鎖DNA断片117を第二溶液槽203から第一溶液槽202に一定時間泳動させる。この泳動間に先端部220近傍の近接場光内を2本鎖DNA断片117に標識されている蛍光体118が通過すると蛍光体118が発光する。この発光をCCD311で検出し,検出された信号強度の時間的変動から,蛍光体118の2本鎖DNA断片117の標識位置を算出できる。次いで,電極210を陰極に,電極211を陽極になるように電圧源212により電圧を印加し,2本鎖DNA断片117を第一溶液槽202から第二溶液槽203に一定時間泳動させる。この泳動間に前記発光をCCD311で検出する。前記泳動ならびに蛍光検出を繰返す事により,蛍光体118の2本鎖DNA断片117の標識位置を複数回計測でき,高精度な位置算出が可能となる。4種類の異なるdNTPに蛍光体が標識された2本鎖DNA断片117に対して前記蛍光体標識位置検出の操作を行い,ターゲットDNA分子101の塩基配列を決定する。
【0048】
前記往復運動計測における電圧極性の切替えは,ある一定時間で自動切替できるよう電圧源212を制御する。前記一定時間の設定は可変である。制御部は,データ処理手段中400に設けておけばよい。ストッパ分子がナノポアに近接するとナノポアを通過する電流の減少が計測できるため,前記電流の減少を引き金に電圧極性の切替えを行っても良い。
【0049】
本実施例では1種類の蛍光体のみを使用したが,4種のdNTPに異なる蛍光体を標識し,それらdNTPすべてが標識された2本鎖DNA断片117を生成し,それを前記と同様操作で蛍光検出することで,ターゲットDNA分子101の塩基配列を決定することも可能である。また,高い粘性のバッファ溶液の使用や泳動時の印加電圧を小さくすることにより,2本鎖DNA断片117の泳動速度を遅くする事ができ,高感度に蛍光検出することが可能となる。
【実施例4】
【0050】
つづいて,ハイブリダイゼーションベースのDNA分子の塩基配列決定方法を説明する。実施例1と同等の方法で5’末端にストレプトアビジンが結合したターゲットDNA分子101が含まれたDNA断片106を生成する。
【0051】
図13に本実施例で使用するナノポア装置の概略図を示す。ナノポア装置は,第一溶液槽202,第二溶液槽203,両溶液槽を分割するナノポア薄膜221で構成される。両溶液槽には,溶液を導入する導入口206,207および溶液を排出する排出口208,209がそれぞれ具備されている。ナノポア薄膜221を介して両溶液槽間で電圧勾配を設けるために,両溶液槽には電極210,211が具備されており,前記電極210と211は極性可変の電圧源212におよび電流計213に接続されている。ナノポア薄膜221は,直径3nmのナノポア205が形成されたSi3N4薄膜で構成されている。温調ユニット313によって第一溶液槽202および第二溶液槽203内の溶液の温度を20℃から100℃まで調節できる。電圧源212の制御,温調ユニット313の制御,電流計213の電流値の取得,得られたデータの処理は,データ処理手段400によって行われる。
【0052】
前記方法で得られたDNA断片106をバッファ溶液に混合し導入口206より第一溶液槽202に,バッファ溶液のみを導入口207より第二溶液槽203に導入する。電極210が陰極,電極211が陽極になるように電圧源212により電圧を印加し,DNA断片106を第一溶液槽202から第二溶液槽203に泳動させる。前記電圧印加と同時に電流計213によって電流を計測する。ストレプトアビジン105の大きさは5nm程度であるため直径3nmのナノポアは通過できず,DNA断片106は3’末端(ストレプトアビジン105が標識されていない末端)よりナノポア205に導入される。DNA断片106がナノポア205に導入されると電流値は減少する。先述したように,ストレプトアビジン105の大きさはナノポア205の直径よりも大きいため,ストレプトアビジン105がナノポア205を通過する直前で,DNA断片106の第二溶液槽203への移動が止まる。前記電流の減少を確認した後に,導入口207よりBiotin 3’End DNA Labeling Kitを第二溶液槽203に導入し,DNA断片106の3’末端にビオチン113を標識する。ビオチン標識後に,導入口207よりバッファのみを導入し,未反応のビオチンを第二溶液槽203より除去する。ストレプトアビジンを含んだ溶液を導入口207より導入し,前記DNA断片106の3’末端に標識されたビオチン113にストレプトアビジン114を結合させ,アレイ型DNA断片115を生成する。導入口207よりバッファのみを導入し,未反応のストレプトアビジンを第二溶液槽203より除去する。
【0053】
アレイ型DNA断片115生成後に,導入口206および207より6塩基から構成される既知配列プローブ119を両溶液槽に導入し,アレイ型DNA断片115とハイブリダイゼーションさせる。ハイブリダイゼーション反応後に,導入口206および207よりバッファのみを導入し,未反応の既知配列プローブ119を両溶液槽203より除去する。電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第二溶液槽203から第一溶液槽202に一定時間泳動させ,次いで,電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第一溶液槽202から第二溶液槽203に一定時間泳動させる。前記電圧極性の切替えのタイミングは,ある一定時間で自動切替できるように電圧源212を制御する。前記一定時間の設定は可変である。ストッパ分子がナノポアに近接するとナノポアを通過する電流の減少が計測できるため,前記電流の減少を引き金に電圧極性の切替えを行っても良い。前記アレイ型DNA断片115の泳動間に電流計213より封鎖電流を計測する。計測された封鎖電流の時間的変動から,既知配列プローブ119のアレイ型DNA断片115へのハイブリダイゼーションした位置を算出できる。
【0054】
次いで,電圧印加を停止し,温調ユニット313を用いて第一溶液槽202,第二溶液槽203内の溶液の温度を95℃に一定時間加温し,熱変性により前記アレイ型DNA断片115から既知配列プローブ119を分離する。導入口206および207からバッファのみを導入し,既知配列プローブ119を両溶液槽より除去する。両溶液槽の温度を40℃に冷やし,導入口206および207から前記既知配列プローブ119とは異なる配列を持つ既知配列プローブを両溶液槽に導入し,アレイ型DNA断片115とハイブリダイゼーションさせる。ハイブリダイゼーション反応後に,導入口206および207よりバッファのみを導入し,未反応の前記既知配列プローブを両溶液槽より除去する。電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第二溶液槽203から第一溶液槽202に一定時間泳動させ,次いで,電極210を陰極に,電極211を陽極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第一溶液槽202から第二溶液槽203に一定時間泳動させる。この泳動間に電流計213より封鎖電流を計測する。
【0055】
既知配列プローブのアレイ型DNA断片へのハイブリダイゼーション,アレイ型DNA断片の泳動,封鎖電流計測,熱変性による既知配列プローブのアレイ型DNA断片から分離を,異なる配列の既知配列プローブで4n回(nは既知配列プローブの塩基長であり,本実施例では6となる)繰返し操作し,得られた各既知配列プローブのハイブリダイゼーション位置のデータをコンピュータアルゴリズムを使ってターゲットDNA分子101の塩基配列データに変換することが可能となる。具体的な方法を,図14を用いて説明する。ターゲットDNA101に対して,既知配列プローブ119をハイブリダイゼーションさせ,ナノポアを通過させて封鎖電流を計測すると,封鎖電流値の波形123が観測される。これを前述したような方法で繰返し計測を行い,既知配列プローブ119がターゲットDNA101どの位置にハイブリダイゼーションしたのかを予測する。前記操作を既知配列プローブ120,121,120で行い,得られた各既知配列プローブのターゲットDNA101へのハイブリダイゼーション位置を重ねる事で,ターゲットDNA101と相補的な配列を導き出すことができ,最終的にターゲットDNA101の配列データを得られる。
【0056】
本発明を利用することにより,ターゲットDNA分子の増幅無く,また,複数のナノポアを用いること無く,ハイブリダイゼーションベースのターゲットDNA分子塩基配列決定が可能となる。
【0057】
本実施例では,既知配列のプローブの長さは6塩基としている。既知配列プローブの長さが長いと,プローブ製作のコストの上昇やミスハイブリダイゼーションの増加が起こる。また,既知配列プローブの長さが短いと,計測分解能を上げないと,正確なプローブのハイブリダイゼーションの位置を計測できない。そのため,既知配列プローブの長さは3〜10塩基程度が望ましい。
【0058】
既知配列プローブのターゲットDNA分子へのハイブリダイゼーション位置を高精度に検出するために,封鎖電流計測の際に,電極210と211の極性を繰返し変化させて,複数回封鎖電流を計測しても良い。
【0059】
計測終了後にヌクレアーゼや酸などを用いてアレイ型DNA断片115を切断し,両溶液槽から前記アレイ型DNA断片115を除去する事により,ナノポア薄膜を再利用しても良い。
【0060】
既知配列プローブのハイブリダイゼーション位置の検出を,既知配列プローブの一部に蛍光体を標識し,実施例2や実施例3で示した蛍光検出を用いても良い。
【0061】
既知配列プローブのハイブリダイゼーション位置の検出を封鎖電流では無く,トンネル電流や蛍光検出することにより複数種のターゲット分子の同時計測が簡便に行え,スループットの向上が図ることができる。
【産業上の利用可能性】
【0062】
DNAシーケンサ
【符号の説明】
【0063】
101:ターゲットDNA分子,102:ビオチン,103:合成プローブ,104:端面,105:ストレプトアビジン,106:DNA断片,107:ベクター,108:制限酵素サイト,109:制限酵素サイト,110:合成プローブ,111:合成プローブ,112:切断断片,113:ビオチン,114:ストレプトアビジン,115:アレイ型DNA断片,116:蛍光体,117:2本鎖DNA断片,118:ビーズ標識抗DIG抗体,119:既知配列プローブ,120:既知配列プローブ,121:既知配列プローブ,122:既知配列プローブ,123:封鎖電流値の波形,124:封鎖電流値の波形,125:封鎖電流値の波形,126:封鎖電流値の波形,202:第一溶液槽,203:第二溶液槽,204:ナノポア薄膜,205:ナノポア,206:導入口,207:導入口,208:排出口,209:排出口,210:電極,211:電極,212:電圧源,213:電流計,214:電流計,215:電圧源,216:電極,217:電極,218:ナノポア薄膜,219:Qdot,220:先端部,221:ナノポア薄膜,301:レーザ光源,302:レーザ光,303:ミラー,304:集光レンズ,305:照射窓,306:検出窓,307:対物レンズ,308:フィルタ,309:プリズム,310:結像レンズ,311:CCD,312:ナノポア薄膜側面,313:温調ユニット,400:データ処理装置
【技術分野】
【0001】
本発明は,ナノポア構造を用いた,DNA,RNA,又はタンパク質等のバイオポリマーの高精度な計測,分析を行うための構造,方法に関する技術分野に属する。
【背景技術】
【0002】
病気の診断や創薬に対して,核酸(DNAやRNA)やタンパク質等のバイオポリマーを分析することは重要である。特にDNAは,生命の最も基本の構成体であるため,その分析,つまり塩基配列の決定は,上記目的に対して非常に重要となる。DNAの塩基配列を分析する主な方法としては,まず,化学もしくは酵素反応を利用して,配列決定したい鋳型DNAに対して,決まった末端塩基種をもつ様々な長さのDNA断片群を生成する。前記DNA断片群を4種の塩基種に対してそれぞれ生成する。次いで,ゲル電気泳動を用いて,これらのDNA断片群を分子量の順に分離する。前記DNA断片群を分離媒体に導入し電圧を印加する。DNA断片は全体としてマイナスの電荷をもつ高分子電解質であるため,マイナスからプラスの方向に電気泳動する。ゲル中では長いDNA断片ほど小さな易動度を持つので,1塩基長の違いしか持たないDNA断片同士をも分離することができる。分離後に,DNA断片の末端塩基種に応じた長さを計測することで,鋳型DNAにおける塩基種の位置が分かる。前記操作により鋳型DNAの塩基配列が決定される。上記方法において,DNA断片群の生成や電気泳動の作業は,非常に煩雑で分析時間も長く,ランニングコストも高価である。
【0003】
特許文献1および非特許文献1には,絶縁膜である脂質二重層の薄膜上にα−ヘモリジンを用いて形成した直径数ナノメートルの微細な開口,つまりナノポアを用いたバイオポリマーの分析方法が記載されている。2つの溶液槽の間にナノポアを設けた薄膜設置し,両溶液槽間で電圧勾配を設け,電流を計測する。バイオポリマーであるDNA分子を片方の溶液槽に設置すると,電圧勾配によりナノポアを通過し,別の溶液槽に移動する。DNA分子がナノポアを通過する際に,ナノポア内のイオンの流れを塞ぐため,電流の減少(封鎖電流)が生じる。この封鎖電流の大きさとその封鎖電流の継続時間を計測することにより,ナノポアを通過する個々のDNA分子の長さを検出可能となる。また,理論的にはDNA分子を構成する個々の塩基の種類の判別も可能となる。
【0004】
特許文献2および非特許文献2には,脂質二重層におけるα−ヘモリジンを用いたナノポアに代わり,絶縁膜であるシリコンナイトライド(Si3N4)膜に”ion−beam sculpting“と言う技術を用いてナノポアを作製し, 1本鎖DNA分子の封鎖電流の計測を行っている。
【0005】
非特許文献3では,ナノポアを通過するDNA分子の計測方法として,前述した封鎖電流とは異なる手段が提案されている。ナノポアの内表面に一対の金属電極を設け,この金属電極間をDNA鎖が通過する際のトンネル電流を計測する。この方法でも電極サイズなどが適切に制御されていれば,理論的には,ナノポアを通過するDNA分子を構成する個々の塩基の種類の判別も可能となる。
【0006】
特許文献3では,既知配列プローブのターゲット1本鎖DNAへのハイブリダイゼーションと,ナノポアを用いた前記既知配列プローブのハイブリダイゼーション位置の検出により,ターゲットDNAの塩基配列を決定する方法が示されている。既知配列のプローブを決定したいターゲット1本鎖DNA分子にハイブリダイゼーションさせ,前記ハイブリダイゼーションしたDNA分子をナノポアに通過させる。DNA分子がナノポアを通過する際にDNA分子の1本鎖部位と2本鎖部位で封鎖電流が異なるため,電流を計測することにより,既知配列プローブのハイブリダイゼーション位置を特定できる。複数種の異なる配列を持つ既知配列プローブで前記操作を行い,それぞれのターゲットDNA分子へのハイブリダイゼーション位置データを取得し,得られたデータをコンピュータアルゴリズム使って配列データに変換することで,ターゲットDNA分子の配列決定が可能となる。
【0007】
非特許文献4では,DNA分子がナノポア通過直後に印加電圧の正負を逆転することにより,同一DNA分子が再び同一のナノポアを通過する結果が示されている。
【0008】
非特許文献5では,ナノポア上でDNA分子の伸長反応を行い,封鎖電流計測により伸長反応の有無を確認している。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】米国特許第5,795,782
【特許文献2】米国特許第6,627,076
【特許文献3】米国特許公報第2007/0190542
【非特許文献】
【0010】
【非特許文献1】PNAS 1996, Vol.93, pp.13770-13773
【非特許文献2】NATURE 2001, Vol.412, pp.166-169
【非特許文献3】NANO LETTERS 2005, Vol.5, pp.421-424
【非特許文献4】nature nanotechnology 2007, Vol.2, pp.775-779
【非特許文献5】JACS 2008, Vol.130, pp.818-820
【発明の概要】
【発明が解決しようとする課題】
【0011】
電気泳動を用いたバイオポリマー,特にDNA分子の配列決定は分析時間も長く,ランニングコストも高価である。一方,特許文献1や非特許文献3に記載されたナノポアを用いたDNA分子の配列決定は,高速・安価な分析の可能性を持つが,それぞれ以下の問題がある。
【0012】
特許文献1では,DNA分子がナノポアを通過する時の各塩基種による封鎖電流の違いは僅かであるため,その判別をすることは困難であり,その精度は非常に低い。また,判別精度を向上するためには,ナノポア近傍の膜厚を1塩基分程度,つまりサブナノメートルにする必要があるが,現状の技術でその膜厚を達成するのは困難である。
【0013】
非特許文献3において,DNA分子がナノポアを通過する時のトンネル電流の大きさは,ナノポアを通過する塩基の種類の他に,その塩基のトンネル電流を計測する電極近傍での配向にも依存する。現状,通過する核酸の配向制御が困難であるため,塩基種分離の精度を上げるためには,繰返し計測が必要となる。
【0014】
一方,特許文献3で示される方法では,既知配列プローブのターゲットDNA分子へのハイブリダイゼーションした位置を検出するため,塩基1種類ずつを判別する必要は無い。しかしながら,1塩基レベルでの高精度なハイブリダイゼーション位置の検出が必要になる。また,同一ターゲットDNA分子に対して,複数種の異なる既知配列プローブを,それぞれ異なる場もしくは異なるタイミングでターゲットDNA分子にハイブリダイゼーションして,その位置を検出する必要がある。つまり,ターゲットDNA分子を増幅し,それらをそれぞれ異なる既知配列プローブが存在する異なる溶液に導入し,ターゲットDNA分子と既知配列プローブをハイブリダイゼーションさせて,それぞれ異なるナノポアもしくは異なるタイミングで同一ナノポアを用いて計測を行う必要がある。もしくは,ある既知配列プローブとハイブリダイゼーションしたターゲットDNA分子をナノポア計測した後に,抽出し,変性させて1本鎖に戻し,異なる既知配列プローブとハイブリさせて,再度ナノポア計測を行う事を繰返す必要がある。いずれの場合も,非常に煩雑な作業を要する。
【0015】
非特許文献4で示されるように,DNA分子がナノポアを通過した後に,逆電圧を印加することを繰返すことで,同一DNA分子を複数回同一ナノポアで計測できる。そのため,繰返し計測による計測精度の向上を図れる可能性はあるが,高精度なフィードバックによる電圧印加が必要になる。また,複数種のDNA分子が混在していた場合には,コンタミの恐れもある。さらに,DNA分子の方向もランダムとなる。
【0016】
非特許文献5では,ターゲットDNA分子の5’末端にpolyethylene glycol(PEG)−ビオチンを介したストレプトアビジンを結合させ,3’末端側には数塩基からなるDNAプライマーをハイブリダイゼーションさせ,ターゲットDNA分子の3’末端を2本鎖にすることにより,ナノポアを介したターゲットDNA分子の往復運動を行っている。前記往復運動を用いた繰返し計測により,高精度なナノポア計測ができる可能性がある。しかしながら,DNAプローブのターゲットDNA分子へのハイブリダイゼーションする位置をコントロールすることは難しく,ターゲットDNA分子の様々な位置にDNAプローブが結合し,往復運動ができないことがある。また,2本鎖DNAの直径は2nm程度であるため,ナノポア直径が2nm以上では,2本鎖部分がナノポアを通過してしまい,前記往復運動が困難となる。
【課題を解決するための手段】
【0017】
本発明は,核酸やタンパク質のようなバイオポリマー分子のナノポア計測において,計測精度向上のための繰返し計測を安定してできる方法,システム、及びキットを提供する。
【0018】
第一の溶液槽と第二の溶液槽を具備しており,両溶液槽間は薄膜で区切られており,薄膜にはナノメートルサイズの穴,つまりナノポアが設けられている。バイオポリマー分子の片端にナノポアの開口直径よりも大きなサイズの分子(第一ストッパ分子)を結合し,それを第一の溶液槽に導入する。外力を加えてバイオポリマーを駆動させてナノポアを通過させ,第二の溶液槽に移動させる。第一ストッパ分子により,ナノポアを通過するバイオポリマー分子は途中で移動を停止する。バイオポリマー分子の移動停止後に,第二の溶液槽に存在するナノポアの開口直径よりも大きな分子(第二ストッパ分子)をバイオポリマー分子のもう一方の片端に結合させる。次いで,外力を加えてバイオポリマー分子を,両溶液槽間を往復運動させながら計測を行い,バイオポリマー分子の同定を行う。
【0019】
第二ストッパ分子のバイオポリマー分子への結合は,バイオポリマー分子がナノポアを移動している間であっても良い。また,第二ストッパ分子をバイオポリマー分子に結合する際に,バイオポリマー分子を駆動させる外力を停止させても良いし,外力を与え続けても良い。第二ストッパ分子は,予め第二溶液槽に入れていても,バイオポリマー分子の片端が第二溶液槽に移動後に導入しても良い。両ストッパ分子は同じ分子であっても良いし,異なる分子であっても良い。バイオポリマーを駆動させる手段としては,バイオポリマー分子が電荷を持っているのであれば両溶液槽間に電圧勾配を生じさせても良いし,両溶液槽間でイオン組成を変えることによる電気化学的勾配を生じさせても良いし,溶液の流れを生じさて駆動させても良い。計測の手段としては,両溶液槽間を,ナノポアを介して流れる電流(封鎖電流)値であっても良いし,ナノポア内部に具備された電極間を流れる電流(トンネル電流)値であっても良い。また,バイオポリマー分子に蛍光体を標識し,ナノポア近傍で励起させ,その発光蛍光を検出しても良い。バイオポリマー分子の両端に両ストッパ分子が結合した状態で,バイオポリマー分子に,ある物質を結合させたり,分離させたりしても良い。例えば,非特許文献3のようにハイブリダイゼーションベースでのDNA分子配列決定の場合には,前記方法を用いて簡便かつ高精度に計測できる。DNA分子の両端にストッパ分子を結合させ,溶液槽に既知配列プローブを導入し,それをターゲットDNA分子にハイブリダイゼーションさせ,ターゲットDNA分子を,両溶液槽間を往復運動させてハイブリダイゼーションの位置を封鎖電流計測により検出する。次いで,変性操作により前記既知配列プローブをターゲットDNA分子から分離させ,別の既知配列プローブを用いて前記と同様の操作を繰返す。前記手段により,同一ターゲットDNA分子に対して異なる既知配列プローブを簡便に何回でもハイブリダイゼーションできる。また繰返し計測ができるため,ハイブリダイゼーションの位置を高精度に計測できる。
【0020】
本発明のバイオポリマー決定方法は,第1の溶液槽と,第2の溶液槽と,前記第1の溶液槽と前記第2の溶液槽とを区切りナノポアを備えた薄膜とを有する装置を用い,測定対象のバイオポリマーを構成するモノマーの並びを決定する方法であって,前記ナノポアよりも大きな第1の分子を一端に結合した前記バイオポリマーを前記第1の溶液槽に導入する工程と, 前記バイオポリマーを前記ナノポアを通して前記第1の溶液槽から前記第2の溶液槽に移動させる工程と,前記第2の溶液槽に,前記ナノポアよりも大きな第2の分子を導入し,前記バイオポリマーの他端に結合させる工程と,前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通して移動させる工程と,前記バイオポリマーの移動に伴い発生する信号の時間変化を測定し,前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出し,前記バイオポリマーを構成するモノマーの並びを決定することを特徴とする。
【0021】
本発明のバイオポリマー決定システムは,ナノポアを有する薄膜を備えた装置を用い,前記ナノポアの大きさよりも大きく,測定対象のバイオポリマーに結合させる分子を導入するシステムであって,前記装置は,前記薄膜によって区切られた第1及び第2の溶液槽と,前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通じて移動させる駆動手段と,前記分子が前記測定対象のバイオポリマーの一方に結合されたバイオポリマーを前記第1の溶液槽に導入する手段と,前記ナノポアを通過した前記バイオポリマーの他端に結合させる前記分子を前記第2の溶液槽に導入する手段と,前記駆動手段による前記バイオポリマーの移動に伴い発生する信号を検出する検出手段と,前記検出手段により検出した信号の時間変化を測定し,前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出する算出し,前記バイオポリマーを構成するモノマーの並びを決定する算出手段とを有することを特徴とする。
【0022】
本発明のバイオポリマー決定キットは,ナノポアを有する薄膜を備えた装置と,前記ナノポアの大きさよりも大きく,測定対象のバイオポリマーに結合させる分子とを有するキットであって,前記装置は,前記薄膜によって区切られた第1及び第2の溶液槽と,前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通じて移動させる駆動手段と,前記分子が前記測定対象のバイオポリマーの一方に結合されたバイオポリマーを前記第1の溶液槽に導入する手段と,前記ナノポアを通過した前記バイオポリマーの他端に結合させる前記分子を前記第2の溶液槽に導入する手段と,前記駆動手段による前記バイオポリマーの移動に伴い発生する信号を検出する検出手段と,前記検出手段により検出した信号の時間変化を測定し,前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出する算出し,前記バイオポリマーを構成するモノマーの並びを決定する算出手段とを有することを特徴とする。前記分子は,ストレプトアビジン,又は,DIG−抗DIG抗体結合により前記バイオポリマーに結合されるビーズであることを特徴とする。
【発明の効果】
【0023】
本発明により,バイオポリマー分子のナノポアを用いた安定した繰返し計測が可能になり,安価,高速,高精度な計測が可能となる。
【図面の簡単な説明】
【0024】
【図1】サンプル前処理時のサンプル溶液中の模式図
【図2】ベクターを利用したサンプル前処理法の模式図
【図3】実施例1におけるナノポア装置の概略図
【図4】実施例1におけるナノポア近傍の拡大図
【図5】DNA断片の3’末端にストレプトアビジンを結合するフローチャート
【図6】DNA断片の3’末端にストレプトアビジンを結合する様子の模式図
【図7】DNA分子ナノポア通過時のトンネル電流値の経時変化のグラフ
【図8】実施例2におけるナノポア装置の概略図
【図9】実施例2における各ナノポアからの発光のCCD上でイメージ図
【図10】DNA分子ナノポア通過時の各波長での信号強度の経時変化のグラフ
【図11】DNA分子ナノポア通過時の各塩基種における信号強度の経時変化のグラフ
【図12】実施例3におけるナノポア装置の概略図
【図13】実施例4におけるナノポア装置の概略図
【図14】各既知配列プローブをハイブリダイゼーションさせたDNA分子のナノポア通過時の封鎖電流の経時変化グラフ
【発明を実施するための形態】
【0025】
以下,図面に従って本発明の実施の形態を説明する。
【実施例1】
【0026】
本発明を用いたナノポアのトンネル電流計測によるDNA分子の塩基配列決定方法を説明する。図1(A)および(B)は,計測前のサンプル前処理時のサンプル溶液中の状態を模式的に示したものである。溶液中には,2本鎖になっているターゲットDNA分子101,5’末端にビオチン102が標識された2本鎖の合成プローブ103が混在している。2本鎖合成プローブ103のビオチンが標識されていない端面104は平滑末端処理されている。ターゲットDNA分子101の両末端に対して,S1 Nucleaseなどを用いて平滑末端処理を行う。ターゲットDNA分子101の平滑末端処理後に,リガーゼを用いて,ターゲットDNA分子101の両端に合成プローブ103をライゲーションさせる。前記ライゲーション反応終了後に,サンプルをアクリルアミドゲル電気泳動によりサイズ分離する。ターゲットDNA分子101の両端に合成プローブ103が結合したもののみを,アクリルアミドゲルより切出して蒸留水に溶出する。前記操作により,合成プローブ103同士がライゲーションしたもの,ターゲットDNA分子101同士がライゲーションしたものを除去できる。次いで,ストレプトアビジン105が入ったバッファ溶液を,両端に合成プローブが結合したターゲットDNA分子101が溶出された溶液に混入し,5’末端に標識されたビオチン102とストレプトアビジン105を結合させ,熱を加えてDNAを変性させ,図1(B)に示す5’末端にストレプトアビジンが結合したターゲットDNA分子101が含まれたDNA断片106を生成する。図2(A)および(B)は,DNA断片106の別の生成方法を模式的に示したものである。ターゲットDNA分子101をベクター107のマルチクローニングサイトの一部に導入する。次いで,ターゲットDNA分子101を挟みこむ状態で,ベクターのそれぞれ異なる制限酵素サイト108と109で制限酵素処理による切断を行う。そして、制限酵素サイト108の切断末端を有し,5’末端にビオチン102が標識された2本鎖の合成プローブ110と制限酵素サイト109の切断末端を有し,5’末端にビオチン102が標識された2本鎖の合成プローブ111を前記ベクターより切断処理した切断断片112に導入し,ライゲーション操作を行う。その後,5’末端に標識されたビオチン102とストレプトアビジン105を結合させ,熱を加えてDNAを変性させ,図1(B)に示す5’末端にストレプトアビジンが結合したターゲットDNA分子101が含まれたDNA断片106を生成する。
【0027】
図3は,本実施例で使用するナノポア装置の概略図である。ナノポア装置は,第一溶液槽202,第二溶液槽203,両溶液槽を分割するナノポア薄膜204で構成される。両溶液槽には,溶液を導入する導入口206,207および溶液を排出する排出口208,209がそれぞれ具備されている。ナノポア薄膜204を介して両溶液槽間で電圧勾配を設けるために,両溶液槽202,203には電極210,211が具備されており,前記電極210と211は極性可変の電圧源212および電流計213に接続されている。ナノポア薄膜204は,直径1nmのナノポア205が形成された絶縁体の薄膜で構成されている。ここでは,絶縁体薄膜の材料としてSi3N4を用いているが,他にSiO2やアスファルテンなどのプラスチック材料でもよい。さらに,Alなどの金属膜の表面に絶縁材量をコートした薄膜でも良い。ナノポア205の径としては,ここでは1nmとしているが,0.5nmから50nm程度あればよい。尚,ストッパ分子として用いたストレプトアビジンの大きさは5nm程度であり,ナノポアの大きさよりも十分大きい。ナノポアの径に対するストッパ分子の大きさとしては,DNA断片の進行を止めることが出来る大きさであればよいが,精度向上のためには,1.2〜50倍程度あることが望ましい。
【0028】
図4(A)は,ナノポア205近傍の拡大図である。また,図4(B)は図4(A)のa-a’での断面図である。ナノポア205内表面には一対の電極216と217が具備されており,前記電極216,217は電圧源215と電流計214に接続されている。
【0029】
以下,図5のフローチャートに沿ってDNA断片106の3’末端にストレプトアビジンを結合する方法を説明する。なお,図6に,DNA断片106の3’末端にストレプトアビジンを結合する様子を模式的に示す。
【0030】
まず、前記方法で得られたDNA断片106をバッファ溶液に混合し,導入口206より第一溶液槽202に,バッファ溶液のみを導入口207より第二溶液槽203に導入する(401)(図6(A))。電極210が陰極,電極211が陽極になるように電圧源212により電圧を印加し,DNA断片106を第一溶液槽202から第二溶液槽203に泳動させる(402)(図6(B))。前記電圧印加と同時に電流計213によってナノポア205を介して流れるイオンの流れを計測する。ストレプトアビジン105の大きさは5nm程度であるため直径1nmのナノポアは通過できず,DNA断片106は3’末端(ストレプトアビジン105が標識されていない末端)よりナノポア205に導入される。DNA断片106がナノポア205に導入されると電流値は減少する。先述したように,ストレプトアビジン105の大きさはナノポア205の直径よりも大きいため,ストレプトアビジン105がナノポア205を通過する直前で,DNA断片106の第二溶液槽203への移動が止まる。前記電流の減少を確認した後に,導入口207よりBiotin 3’End DNA Labeling Kitを第二溶液槽203に導入し,DNA断片106の3’末端にビオチン113を標識する(403)(図6(C))。
【0031】
ビオチン標識後に,導入口207よりバッファのみを導入し,未反応のビオチンを第二溶液槽203より除去する(404)。次いで,ストレプトアビジンを含んだ溶液を導入口207より導入し,前記DNA断片106の3’末端に標識されたビオチン113にストレプトアビジン114を結合させ,アレイ型DNA断片115を生成する(405)(図6(D))。導入口207よりバッファのみを導入し,未反応のストレプトアビジンを第二溶液槽203より除去する(406)。
【0032】
上記では,第一,第二ストッパ分子として同じ物質であるストレプトアビジンを使用したが,異なる物質を使用しても良い。例えば,第一,第二ストッパ分子に異なる物質を使用したアレイ型DNA断片115の別な生成方法として以下の方法がある。図2で示した方法において,合成プローブ110および111の切断末端とは異なる末端側の3’末端にDigoxigein(DIG)を標識しておく。それ以外は,前述した方法でDNA断片106を生成する。この時,DNA断片106の3’末端はDIG標識されている。次いで,前記3’末端がDIG標識されたDNA断片106を前述と同等の方法で,第一溶液槽202から第二溶液槽203へ泳動させる。DIG自身の大きさは,ナノポア205の直径1nmよりも十分小さいため,DNA断片106は3’末端よりナノポア205に導入される。ストレプトアビジン105によってDNA断片106の移動が停止した状態で,第二溶液槽203に直径が1nmよりも大きなビーズが標識された抗DIG抗体を導入し,DNA断片106の3’末端に標識されたDIGと結合させる。これにより,第一ストッパ分子としてストレプトアビジン,第二ストッパ分子としてDIG−抗DIG抗体結合を介したビーズから構成されるアレイ型DNA断片115を生成できる。ここでは,ビオチン・ストレプトアジビン以外としてDIG・抗DIG結合ビーズを用いた例を記載したが,他にもDNAの末端をチオール化して金粒子を結合させる方法や,DNAの末端をアミノ基修飾してカルボキシルキ修飾のビーズと脱水反応により結合させる方法等を用いてもよい。
【0033】
アレイ型DNA断片115生成後に,電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第二溶液槽203から第一溶液槽202に一定時間泳動させる。この泳動間に電極216,217によりトンネル電流を計測し,アレイ型DNA断片115を構成する塩基種を特定する。次いで,電極210を陰極に,電極211を陽極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第一溶液槽202から第二溶液槽203に一定時間泳動させる。この泳動間に電極216,217によりトンネル電流を計測する。前記両溶液槽間の泳動ならびにトンネル電流の計測を繰返す事により,同一ターゲットDNA分子のトンネル電流の計測が複数回でき,高精度な塩基種決定が可能となる。
【0034】
ここで,塩基種決定の方法を説明する。塩基にはATGCの4種類があるが,これらの塩基の種類ごとに固有の電流値が観測され,データ処理手段400に送られる。一例を図7に示す。予め,各塩基1種類のみが連なったポリマのナノポア通過時のトンネル電流を計測し,各塩基種に対応した電流値を求め,データ処理手段中のメモリに格納しておく。そして、データ処理手段400により,ターゲットDNAのトンネル電流計測時に得られた電流値と前記予め計測した各塩基種に対応した電流値を比較する事により,ターゲットDNAの塩基種決定を行う。
【0035】
前記往復運動計測における電圧極性の切替えは,ある一定時間で自動切替できるよう電圧源212を制御する。制御部は,データ処理手段中400に設けておけばよい。前記一定時間の設定は可変である。ストッパ分子がナノポアに近接するとナノポアを通過する電流の減少が計測できるため,前記電流の減少を引き金に電圧極性の切替えを行っても良い。
【0036】
本実施例では,1個のナノポアを使ったトンネル電流の計測を行っているが,多数のナノポアを使用して多数の異なるターゲットDNA分子のトンネル電流の同時計測により,スループットの向上も図れる。
【0037】
また、本実施例では説明用に、5’末端にストッパ分子が結合したDNAを第一溶液槽に導入してナノポアを通過させた後に3’末端にストッパ分子を結合させる例を記載しているが、同様の方法にて、3’末端にストッパ分子が結合したDNA断片を用い、ナノポアに導入した後5’末端にストッパ分子を結合させるのでもよい。
【実施例2】
【0038】
ナノポアの蛍光共鳴エネルギー移動(FRET)を利用した蛍光検出計測によるDNA分子の塩基配列決定方法を説明する。受容体としての蛍光体Cy5が標識されたターゲットDNA分子を以下の方法で生成した。実施例1の図2(B)を用いて説明したように,ターゲットDNA分子101が含まれ,5’末端にビオチンを介したストレプトアビジンが標識され,3’末端にDIGが標識されたDNA断片106を生成する。次いで,dCTP,dGTP,dTTP,Cy5が標識されたdATP,DNAポリメラーゼ,および前記DNA断片106の合成プローブ部分(110,111のどちらでも良い)と相補配列を持つプライマーを含む反応溶液と前記3’末端にDIGが標識されたDNA断片106を混合し,伸長反応を行う。前記伸長反応時には熱変性は行わない。同様の操作を,Cy5がdCTPのみに標識された反応溶液,dGTPのみに標識された反応溶液,dTTPのみに標識された反応溶液で,それぞれ異なる反応チューブで行い,ターゲットDNA断片101を含んだCy5蛍光体116が標識された2本鎖DNA断片117を生成する。
【0039】
図8は,本実施例で使用するナノポア装置の概略図である。ナノポア装置は,第一溶液槽202,第二溶液槽203,両溶液槽を分割するナノポア薄膜218で構成される。両溶液槽202,203には,溶液を導入する導入口206,207および溶液を排出する排出口208,209がそれぞれ具備されている。ナノポア薄膜218を介して両溶液槽間で電圧勾配を設けるために,両溶液槽202,203には電極210,211が具備されており,前記電極210と211は極性可変の電圧源212に接続されている。第一溶液槽202には,光透過性の照射窓305と検出窓306が具備されている。ナノポア薄膜218は,直径3nmのナノポア205が形成されたSi3N4薄膜で構成されている。ナノポア205は1μmの間隔で格子状に複数個形成されている。ナノポア205近傍には供与体として青色光で励起され605nmの発光蛍光するQdot(605)219が固定されている。
【0040】
レーザ光源(波長488nm)301より発振したレーザ光302は,ミラー303により角度を調整され,集光レンズ304により集光され,照射窓305を介してすべてのQdot219に照射される。ナノポア205近傍で発光した発光蛍光は,検出窓306を介して対物レンズ307によって集光され,フィルタ308により波長550nmから700nm以外の波長の光がカットされ,プリズム309により分光され,結像レンズ310によって図9に示すようにCCD311上に結像される。CCD311のデータは,データ処理手段400に格納される。図9にデータ処理手段400に格納されたCCD311上で結像されたイメージ図を示す。各スポットは各ナノポアからの輝点に対応しており,また,横軸は波長方向に対応している。
【0041】
前記方法で得られた2本鎖DNA断片117をバッファ溶液に混合し,導入口206より第一溶液槽202に,バッファ溶液のみを導入口207より第二溶液槽203に導入する。電極210が陰極,電極211が陽極になるように電圧源212により電圧を印加し,2本鎖DNA断片117を第一溶液槽202から第二溶液槽203に泳動させる。ストレプトアビジン105の大きさは5nm程度であるため直径3nmのナノポアは通過できず,2本鎖DNA断片117は,ストレプトアビジン105が標識されていない末端よりナノポア205に導入される。ストレプトアビジン105によって2本鎖DNA断片117の移動が停止した状態で,第二溶液槽203に直径が3nmよりも大きなビーズが標識された抗DIG抗体118を導入し,2本鎖DNA断片117の蛍光体116が標識されていない鎖の3’末端に標識されたDIGと結合させる。抗DIG抗体118標識後に,導入口207よりバッファのみを導入し,未反応の抗DIG抗体を第二溶液槽203より除去する。この間,電圧を印加し続ける。なお,各ナノポアに導入される2本鎖DNA断片117を構成するターゲットDNA分子は,同じものであってもよいし,異なるDNA分子であってもよい。検出された発光を統計的に処理することにより,それぞれの塩基種の特定ができる。同じDNAの分子の場合には,DNA分子の往復回数を減らして時間を短縮しつつ塩基種特定の精度が向上することができる。一方、異なる分子の場合には、同時に多くの分子を測定してスループットを向上することができる。
【0042】
2本鎖DNA断片117に抗DIG抗体118を標識した後に,レーザ光源301よりレーザ光302を発振し,Qdot219を励起する。次いで,電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,抗DIG抗体118標識2本鎖DNA断片117を第二溶液槽203から第一溶液槽202に一定時間泳動させる。この泳動間にQdot219近傍を2本鎖DNA断片117に標識されている蛍光体118が通過すると共鳴による励起エネルギーの移動が起こり,蛍光体118が発光するため,これら発光をCCD311で検出する。図10に,前記発光に対応するCCD311上でのスポットの内,波長605nmと波長670nmに対応するピクセルの強度の時間的変動を示す。波長605nmはQdot219の発光に対応し,波長670nmは蛍光体118の発光に対応する。検出された波長670nmに対応する信号強度の時間的変動から,蛍光体118の2本鎖DNA断片117の標識位置を算出できる。次いで,電極210を陰極に,電極211を陽極になるように電圧源212により電圧を印加し,2本鎖DNA断片117を第一溶液槽202から第二溶液槽203に一定時間泳動させる。この泳動間に前記と同様にCCD311によって蛍光検出する。前記泳動ならびに蛍光検出を繰返す事により,蛍光体118の2本鎖DNA断片117の標識位置を複数回計測でき,高精度な位置算出が可能となる。4種類の異なるdNTPに蛍光体が標識された2本鎖DNA断片117に対して前記と同様の泳動と蛍光検出の繰返し作業を行い,各波長に対して得られた信号強度の時間的変動を図11に示す。波長670nmに対応する信号強度の時間的変動により,各塩基種のターゲットDNA分子101上での位置が判別でき,各塩基種のデータを合わせることにより,ターゲットDNA分子101の塩基配列を決定する。
【0043】
前記往復運動計測における電圧極性の切替えは,ある一定時間で自動切替できるよう電圧源212を制御する。前記一定時間の設定は可変である。制御部は,データ処理手段中400に設けておけばよい。ストッパ分子がナノポアに近接するとナノポアを通過する電流の減少が計測できるため,前記電流の減少を引き金に電圧極性の切替えを行っても良い。
【0044】
本実施例では1種類の蛍光体のみを使用したが,4種のdNTPに異なる蛍光体を標識し,それらdNTPすべてが標識された2本鎖DNA断片117を生成し,それを前記と同様操作で蛍光検出することで,ターゲットDNA分子101の塩基配列を決定することも可能である。また,高い粘性のバッファ溶液の使用や泳動時の印加電圧を小さくすることにより,2本鎖DNA断片117の泳動速度を遅くする事ができ,高感度に蛍光検出することが可能となる。本実施例ではFRETの供与体としてQdotを使用したが蛍光体であっても良い。また,図8では2つのDNA断片を同時に計測する例を示しているが,1つであってもよいし,また3以上のDNA断片を同時に計測しても良い。
【実施例3】
【0045】
ナノポアの蛍光検出計測によるDNA分子の塩基配列決定方法を説明する。
図12は,本実施例で使用するナノポア装置の概略図である。レーザ光源301,フィルタ308,レーザ光のナノポア装置への照射位置およびナノポア薄膜の構成以外はすべて実施例2と同等である。ナノポア薄膜218を石英ガラスで作製し,表面を石英よりも低い屈折率の樹脂(例えばフロリナート)でコーティングする。この時,ナノポア近傍の鋭利な先端部220の樹脂を剥離しておく。レーザ光源(波長633nm)301より射出したレーザ光302は,集光レンズ304で集光され,ナノポア薄膜側面312に照射される。この時,レーザ光302はナノポア薄膜218内を全反射しながら伝播するが,先端部220は樹脂コーティングされていないため,両溶液槽内を満たすバッファ溶液に,近接場光として僅かに染み出す。レーザ光302はナノポア薄膜218内全体を全反射しながら伝播するため,すべての先端部220で近接場が生じる。ナノポア205近傍で発光した発光蛍光は,検出窓306を介して対物レンズ307によって集光され,フィルタ308により波長660nmから700nm以外の波長の光がカットされ,プリズム309により分光され,結像レンズ310によってCCD311上に結像される。CCD311のデータは,データ処理手段400に格納される。図12では2つのDNA断片を同時に計測する例を示しているが,1つであってもよいし,また3以上のDNA断片を同時に計測しても良い。
【0046】
実施例2と同等の方法で,2本鎖DNA断片117を生成および蛍光体116が標識されていない鎖の3’末端に標識されたDIGに直径が3nmよりも大きなビーズが標識された抗DIG抗体118を結合させる操作を行う。
【0047】
2本鎖DNA断片117に抗DIG抗体118を標識した後に,レーザ光源301よりレーザ光302を発振し,先端部220に近接場光を生成する。次いで,電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,抗DIG抗体118標識2本鎖DNA断片117を第二溶液槽203から第一溶液槽202に一定時間泳動させる。この泳動間に先端部220近傍の近接場光内を2本鎖DNA断片117に標識されている蛍光体118が通過すると蛍光体118が発光する。この発光をCCD311で検出し,検出された信号強度の時間的変動から,蛍光体118の2本鎖DNA断片117の標識位置を算出できる。次いで,電極210を陰極に,電極211を陽極になるように電圧源212により電圧を印加し,2本鎖DNA断片117を第一溶液槽202から第二溶液槽203に一定時間泳動させる。この泳動間に前記発光をCCD311で検出する。前記泳動ならびに蛍光検出を繰返す事により,蛍光体118の2本鎖DNA断片117の標識位置を複数回計測でき,高精度な位置算出が可能となる。4種類の異なるdNTPに蛍光体が標識された2本鎖DNA断片117に対して前記蛍光体標識位置検出の操作を行い,ターゲットDNA分子101の塩基配列を決定する。
【0048】
前記往復運動計測における電圧極性の切替えは,ある一定時間で自動切替できるよう電圧源212を制御する。前記一定時間の設定は可変である。制御部は,データ処理手段中400に設けておけばよい。ストッパ分子がナノポアに近接するとナノポアを通過する電流の減少が計測できるため,前記電流の減少を引き金に電圧極性の切替えを行っても良い。
【0049】
本実施例では1種類の蛍光体のみを使用したが,4種のdNTPに異なる蛍光体を標識し,それらdNTPすべてが標識された2本鎖DNA断片117を生成し,それを前記と同様操作で蛍光検出することで,ターゲットDNA分子101の塩基配列を決定することも可能である。また,高い粘性のバッファ溶液の使用や泳動時の印加電圧を小さくすることにより,2本鎖DNA断片117の泳動速度を遅くする事ができ,高感度に蛍光検出することが可能となる。
【実施例4】
【0050】
つづいて,ハイブリダイゼーションベースのDNA分子の塩基配列決定方法を説明する。実施例1と同等の方法で5’末端にストレプトアビジンが結合したターゲットDNA分子101が含まれたDNA断片106を生成する。
【0051】
図13に本実施例で使用するナノポア装置の概略図を示す。ナノポア装置は,第一溶液槽202,第二溶液槽203,両溶液槽を分割するナノポア薄膜221で構成される。両溶液槽には,溶液を導入する導入口206,207および溶液を排出する排出口208,209がそれぞれ具備されている。ナノポア薄膜221を介して両溶液槽間で電圧勾配を設けるために,両溶液槽には電極210,211が具備されており,前記電極210と211は極性可変の電圧源212におよび電流計213に接続されている。ナノポア薄膜221は,直径3nmのナノポア205が形成されたSi3N4薄膜で構成されている。温調ユニット313によって第一溶液槽202および第二溶液槽203内の溶液の温度を20℃から100℃まで調節できる。電圧源212の制御,温調ユニット313の制御,電流計213の電流値の取得,得られたデータの処理は,データ処理手段400によって行われる。
【0052】
前記方法で得られたDNA断片106をバッファ溶液に混合し導入口206より第一溶液槽202に,バッファ溶液のみを導入口207より第二溶液槽203に導入する。電極210が陰極,電極211が陽極になるように電圧源212により電圧を印加し,DNA断片106を第一溶液槽202から第二溶液槽203に泳動させる。前記電圧印加と同時に電流計213によって電流を計測する。ストレプトアビジン105の大きさは5nm程度であるため直径3nmのナノポアは通過できず,DNA断片106は3’末端(ストレプトアビジン105が標識されていない末端)よりナノポア205に導入される。DNA断片106がナノポア205に導入されると電流値は減少する。先述したように,ストレプトアビジン105の大きさはナノポア205の直径よりも大きいため,ストレプトアビジン105がナノポア205を通過する直前で,DNA断片106の第二溶液槽203への移動が止まる。前記電流の減少を確認した後に,導入口207よりBiotin 3’End DNA Labeling Kitを第二溶液槽203に導入し,DNA断片106の3’末端にビオチン113を標識する。ビオチン標識後に,導入口207よりバッファのみを導入し,未反応のビオチンを第二溶液槽203より除去する。ストレプトアビジンを含んだ溶液を導入口207より導入し,前記DNA断片106の3’末端に標識されたビオチン113にストレプトアビジン114を結合させ,アレイ型DNA断片115を生成する。導入口207よりバッファのみを導入し,未反応のストレプトアビジンを第二溶液槽203より除去する。
【0053】
アレイ型DNA断片115生成後に,導入口206および207より6塩基から構成される既知配列プローブ119を両溶液槽に導入し,アレイ型DNA断片115とハイブリダイゼーションさせる。ハイブリダイゼーション反応後に,導入口206および207よりバッファのみを導入し,未反応の既知配列プローブ119を両溶液槽203より除去する。電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第二溶液槽203から第一溶液槽202に一定時間泳動させ,次いで,電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第一溶液槽202から第二溶液槽203に一定時間泳動させる。前記電圧極性の切替えのタイミングは,ある一定時間で自動切替できるように電圧源212を制御する。前記一定時間の設定は可変である。ストッパ分子がナノポアに近接するとナノポアを通過する電流の減少が計測できるため,前記電流の減少を引き金に電圧極性の切替えを行っても良い。前記アレイ型DNA断片115の泳動間に電流計213より封鎖電流を計測する。計測された封鎖電流の時間的変動から,既知配列プローブ119のアレイ型DNA断片115へのハイブリダイゼーションした位置を算出できる。
【0054】
次いで,電圧印加を停止し,温調ユニット313を用いて第一溶液槽202,第二溶液槽203内の溶液の温度を95℃に一定時間加温し,熱変性により前記アレイ型DNA断片115から既知配列プローブ119を分離する。導入口206および207からバッファのみを導入し,既知配列プローブ119を両溶液槽より除去する。両溶液槽の温度を40℃に冷やし,導入口206および207から前記既知配列プローブ119とは異なる配列を持つ既知配列プローブを両溶液槽に導入し,アレイ型DNA断片115とハイブリダイゼーションさせる。ハイブリダイゼーション反応後に,導入口206および207よりバッファのみを導入し,未反応の前記既知配列プローブを両溶液槽より除去する。電極210を陽極に,電極211を陰極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第二溶液槽203から第一溶液槽202に一定時間泳動させ,次いで,電極210を陰極に,電極211を陽極になるように電圧源212により電圧を印加し,アレイ型DNA断片115を第一溶液槽202から第二溶液槽203に一定時間泳動させる。この泳動間に電流計213より封鎖電流を計測する。
【0055】
既知配列プローブのアレイ型DNA断片へのハイブリダイゼーション,アレイ型DNA断片の泳動,封鎖電流計測,熱変性による既知配列プローブのアレイ型DNA断片から分離を,異なる配列の既知配列プローブで4n回(nは既知配列プローブの塩基長であり,本実施例では6となる)繰返し操作し,得られた各既知配列プローブのハイブリダイゼーション位置のデータをコンピュータアルゴリズムを使ってターゲットDNA分子101の塩基配列データに変換することが可能となる。具体的な方法を,図14を用いて説明する。ターゲットDNA101に対して,既知配列プローブ119をハイブリダイゼーションさせ,ナノポアを通過させて封鎖電流を計測すると,封鎖電流値の波形123が観測される。これを前述したような方法で繰返し計測を行い,既知配列プローブ119がターゲットDNA101どの位置にハイブリダイゼーションしたのかを予測する。前記操作を既知配列プローブ120,121,120で行い,得られた各既知配列プローブのターゲットDNA101へのハイブリダイゼーション位置を重ねる事で,ターゲットDNA101と相補的な配列を導き出すことができ,最終的にターゲットDNA101の配列データを得られる。
【0056】
本発明を利用することにより,ターゲットDNA分子の増幅無く,また,複数のナノポアを用いること無く,ハイブリダイゼーションベースのターゲットDNA分子塩基配列決定が可能となる。
【0057】
本実施例では,既知配列のプローブの長さは6塩基としている。既知配列プローブの長さが長いと,プローブ製作のコストの上昇やミスハイブリダイゼーションの増加が起こる。また,既知配列プローブの長さが短いと,計測分解能を上げないと,正確なプローブのハイブリダイゼーションの位置を計測できない。そのため,既知配列プローブの長さは3〜10塩基程度が望ましい。
【0058】
既知配列プローブのターゲットDNA分子へのハイブリダイゼーション位置を高精度に検出するために,封鎖電流計測の際に,電極210と211の極性を繰返し変化させて,複数回封鎖電流を計測しても良い。
【0059】
計測終了後にヌクレアーゼや酸などを用いてアレイ型DNA断片115を切断し,両溶液槽から前記アレイ型DNA断片115を除去する事により,ナノポア薄膜を再利用しても良い。
【0060】
既知配列プローブのハイブリダイゼーション位置の検出を,既知配列プローブの一部に蛍光体を標識し,実施例2や実施例3で示した蛍光検出を用いても良い。
【0061】
既知配列プローブのハイブリダイゼーション位置の検出を封鎖電流では無く,トンネル電流や蛍光検出することにより複数種のターゲット分子の同時計測が簡便に行え,スループットの向上が図ることができる。
【産業上の利用可能性】
【0062】
DNAシーケンサ
【符号の説明】
【0063】
101:ターゲットDNA分子,102:ビオチン,103:合成プローブ,104:端面,105:ストレプトアビジン,106:DNA断片,107:ベクター,108:制限酵素サイト,109:制限酵素サイト,110:合成プローブ,111:合成プローブ,112:切断断片,113:ビオチン,114:ストレプトアビジン,115:アレイ型DNA断片,116:蛍光体,117:2本鎖DNA断片,118:ビーズ標識抗DIG抗体,119:既知配列プローブ,120:既知配列プローブ,121:既知配列プローブ,122:既知配列プローブ,123:封鎖電流値の波形,124:封鎖電流値の波形,125:封鎖電流値の波形,126:封鎖電流値の波形,202:第一溶液槽,203:第二溶液槽,204:ナノポア薄膜,205:ナノポア,206:導入口,207:導入口,208:排出口,209:排出口,210:電極,211:電極,212:電圧源,213:電流計,214:電流計,215:電圧源,216:電極,217:電極,218:ナノポア薄膜,219:Qdot,220:先端部,221:ナノポア薄膜,301:レーザ光源,302:レーザ光,303:ミラー,304:集光レンズ,305:照射窓,306:検出窓,307:対物レンズ,308:フィルタ,309:プリズム,310:結像レンズ,311:CCD,312:ナノポア薄膜側面,313:温調ユニット,400:データ処理装置
【特許請求の範囲】
【請求項1】
第1の溶液槽と、第2の溶液槽と、前記第1の溶液槽と前記第2の溶液槽とを区切りナノポアを備えた薄膜とを有する装置を用い、測定対象のバイオポリマーを構成するモノマーの並びを決定する方法であって、
前記ナノポアよりも大きな第1の分子を一端に結合した前記バイオポリマーを前記第1の溶液槽に導入する工程と、
前記バイオポリマーを前記ナノポアを通して前記第1の溶液槽から前記第2の溶液槽に移動させる工程と、
前記第2の溶液槽に、前記ナノポアよりも大きな第2の分子を導入し、前記バイオポリマーの他端に結合させる工程と、
前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通して移動させる工程と、
前記バイオポリマーの移動に伴い発生する信号の時間変化を測定し、前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出し、前記バイオポリマーを構成するモノマーの並びを決定することを特徴とするバイオポリマー決定方法。
【請求項2】
請求項1に記載のバイオポリマー決定方法において、前記バイオポリマーは核酸であり、核酸を構成する塩基配列を決定することを特徴とするバイオポリマー決定方法。
【請求項3】
請求項1に記載のバイオポリマー決定方法において、前記第1の溶液槽と前記第2の溶液槽との間の電流変化を、前記バイオポリマーの移動に伴い発生する信号として測定することを特徴とするバイオポリマー決定方法。
【請求項4】
請求項1に記載のバイオポリマー決定方法において、前記ナノポアを通過する前記バイオポリマーを挟むように設けられた電極に流れる電流変化を、前記バイオポリマーの移動に伴い発生する信号として測定することを特徴とするバイオポリマー決定方法。
【請求項5】
請求項1に記載のバイオポリマー決定方法において、前記バイオポリマーは特定の塩基を標識した核酸であって、前記バイオポリマーを移動させながら光を照射し、標識からの発光の時間変化を測定することを特徴とするバイオポリマー決定方法。
【請求項6】
請求項1に記載のバイオポリマー決定方法において、前記バイオポリマーは一本鎖核酸であって、前記第1,2の溶液槽のうち少なくとも一方に既知配列プローブを導入する工程を有し、前記1本鎖核酸の移動に伴い前記1本鎖核酸に結合した前記既知配列プローブの有無の信号の時間変化を測定することを特徴とするバイオポリマー決定方法。
【請求項7】
請求項1に記載のバイオポリマー決定方法において、前記第1の溶液槽と前記第2の溶液槽間で電気的な勾配をつけることにより、前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽間で移動させることを特徴とするバイオポリマー決定方法。
【請求項8】
請求項1に記載のバイオポリマー決定方法において、前記第1又は/及び第2の分子はストレプトアビジンであることを特徴とするバイオポリマー決定方法。
【請求項9】
請求項1に記載のバイオポリマー決定方法において、前記第1又は/及び第2の分子はDIG−抗DIG抗体結合により前記バイオポリマーに結合されるビーズであることを特徴とするバイオポリマー決定方法。
【請求項10】
ナノポアを有する薄膜を備えた装置を用い、前記ナノポアの大きさよりも大きく、測定対象のバイオポリマーに結合させる分子を導入するバイオポリマー決定システムであって、
前記装置は、前記薄膜によって区切られた第1及び第2の溶液槽と、
前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通じて移動させる駆動手段と、
前記分子が前記測定対象のバイオポリマーの一方に結合されたバイオポリマーを前記第1の溶液槽に導入する手段と、
前記ナノポアを通過した前記バイオポリマーの他端に結合させる前記分子を前記第2の溶液槽に導入する手段と、
前記駆動手段による前記バイオポリマーの移動に伴い発生する信号を検出する検出手段と、
前記検出手段により検出した信号の時間変化を測定し、前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出する算出し、前記バイオポリマーを構成するモノマーの並びを決定する算出手段とを有することを特徴とするバイオポリマー決定システム。
【請求項11】
請求項10に記載のバイオポリマー決定システムであって、前記検出手段は、前記第1の溶液槽と前記第2の溶液槽との間に設けられた電極であって、電流の時間変化を測定することを特徴とするバイオポリマー決定システム。
【請求項12】
請求項10に記載のバイオポリマー決定キットであって、前記バイオポリマーは特定のモノマーが蛍光標識され、前記装置は、前記ナノポアを通過する前記バイオポリマーに対して光を照射する光源を有し、前記前記検出手段は、光の照射による発光を検出することを特徴とするバイオポリマー決定システム。
【請求項13】
請求項10に記載のバイオポリマー決定システムであって、前記バイオポリマーは一本鎖核酸であって、既知プローブのセットを備え、前記装置は、前記第1,2の溶液槽のうち少なくとも一方に前記既知配列プローブを導入する手段を有し、前記検出手段は、前記1本鎖核酸の移動に伴い前記1本鎖核酸に結合した前記既知配列プローブの有無の信号の時間変化を測定することを特徴とするバイオポリマー決定システム。
【請求項14】
ナノポアを有する薄膜を備えた装置と、
前記ナノポアの大きさよりも大きく、測定対象のバイオポリマーに結合させる分子とを有するバイオポリマー決定キットであって、
前記装置は、
前記薄膜によって区切られた第1及び第2の溶液槽と、
前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通じて移動させる駆動手段と、
前記分子が前記測定対象のバイオポリマーの一方に結合されたバイオポリマーを前記第1の溶液槽に導入する手段と、
前記ナノポアを通過した前記バイオポリマーの他端に結合させる前記分子を前記第2の溶液槽に導入する手段と、
前記駆動手段による前記バイオポリマーの移動に伴い発生する信号を検出する検出手段と、
前記検出手段により検出した信号の時間変化を測定し、前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出する算出し、前記バイオポリマーを構成するモノマーの並びを決定する算出手段とを有することを特徴とするバイオポリマー決定キット。
【請求項15】
請求項14に記載のバイオポリマー決定キットであって、前記分子は、ストレプトアビジンであることを特徴とするバイオポリマー決定キット。
【請求項16】
請求項15に記載のバイオポリマー決定キットであって、前記分子は、DIG−抗DIG抗体結合により前記バイオポリマーに結合されるビーズであることを特徴とするバイオポリマー決定キット。
【請求項1】
第1の溶液槽と、第2の溶液槽と、前記第1の溶液槽と前記第2の溶液槽とを区切りナノポアを備えた薄膜とを有する装置を用い、測定対象のバイオポリマーを構成するモノマーの並びを決定する方法であって、
前記ナノポアよりも大きな第1の分子を一端に結合した前記バイオポリマーを前記第1の溶液槽に導入する工程と、
前記バイオポリマーを前記ナノポアを通して前記第1の溶液槽から前記第2の溶液槽に移動させる工程と、
前記第2の溶液槽に、前記ナノポアよりも大きな第2の分子を導入し、前記バイオポリマーの他端に結合させる工程と、
前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通して移動させる工程と、
前記バイオポリマーの移動に伴い発生する信号の時間変化を測定し、前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出し、前記バイオポリマーを構成するモノマーの並びを決定することを特徴とするバイオポリマー決定方法。
【請求項2】
請求項1に記載のバイオポリマー決定方法において、前記バイオポリマーは核酸であり、核酸を構成する塩基配列を決定することを特徴とするバイオポリマー決定方法。
【請求項3】
請求項1に記載のバイオポリマー決定方法において、前記第1の溶液槽と前記第2の溶液槽との間の電流変化を、前記バイオポリマーの移動に伴い発生する信号として測定することを特徴とするバイオポリマー決定方法。
【請求項4】
請求項1に記載のバイオポリマー決定方法において、前記ナノポアを通過する前記バイオポリマーを挟むように設けられた電極に流れる電流変化を、前記バイオポリマーの移動に伴い発生する信号として測定することを特徴とするバイオポリマー決定方法。
【請求項5】
請求項1に記載のバイオポリマー決定方法において、前記バイオポリマーは特定の塩基を標識した核酸であって、前記バイオポリマーを移動させながら光を照射し、標識からの発光の時間変化を測定することを特徴とするバイオポリマー決定方法。
【請求項6】
請求項1に記載のバイオポリマー決定方法において、前記バイオポリマーは一本鎖核酸であって、前記第1,2の溶液槽のうち少なくとも一方に既知配列プローブを導入する工程を有し、前記1本鎖核酸の移動に伴い前記1本鎖核酸に結合した前記既知配列プローブの有無の信号の時間変化を測定することを特徴とするバイオポリマー決定方法。
【請求項7】
請求項1に記載のバイオポリマー決定方法において、前記第1の溶液槽と前記第2の溶液槽間で電気的な勾配をつけることにより、前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽間で移動させることを特徴とするバイオポリマー決定方法。
【請求項8】
請求項1に記載のバイオポリマー決定方法において、前記第1又は/及び第2の分子はストレプトアビジンであることを特徴とするバイオポリマー決定方法。
【請求項9】
請求項1に記載のバイオポリマー決定方法において、前記第1又は/及び第2の分子はDIG−抗DIG抗体結合により前記バイオポリマーに結合されるビーズであることを特徴とするバイオポリマー決定方法。
【請求項10】
ナノポアを有する薄膜を備えた装置を用い、前記ナノポアの大きさよりも大きく、測定対象のバイオポリマーに結合させる分子を導入するバイオポリマー決定システムであって、
前記装置は、前記薄膜によって区切られた第1及び第2の溶液槽と、
前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通じて移動させる駆動手段と、
前記分子が前記測定対象のバイオポリマーの一方に結合されたバイオポリマーを前記第1の溶液槽に導入する手段と、
前記ナノポアを通過した前記バイオポリマーの他端に結合させる前記分子を前記第2の溶液槽に導入する手段と、
前記駆動手段による前記バイオポリマーの移動に伴い発生する信号を検出する検出手段と、
前記検出手段により検出した信号の時間変化を測定し、前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出する算出し、前記バイオポリマーを構成するモノマーの並びを決定する算出手段とを有することを特徴とするバイオポリマー決定システム。
【請求項11】
請求項10に記載のバイオポリマー決定システムであって、前記検出手段は、前記第1の溶液槽と前記第2の溶液槽との間に設けられた電極であって、電流の時間変化を測定することを特徴とするバイオポリマー決定システム。
【請求項12】
請求項10に記載のバイオポリマー決定キットであって、前記バイオポリマーは特定のモノマーが蛍光標識され、前記装置は、前記ナノポアを通過する前記バイオポリマーに対して光を照射する光源を有し、前記前記検出手段は、光の照射による発光を検出することを特徴とするバイオポリマー決定システム。
【請求項13】
請求項10に記載のバイオポリマー決定システムであって、前記バイオポリマーは一本鎖核酸であって、既知プローブのセットを備え、前記装置は、前記第1,2の溶液槽のうち少なくとも一方に前記既知配列プローブを導入する手段を有し、前記検出手段は、前記1本鎖核酸の移動に伴い前記1本鎖核酸に結合した前記既知配列プローブの有無の信号の時間変化を測定することを特徴とするバイオポリマー決定システム。
【請求項14】
ナノポアを有する薄膜を備えた装置と、
前記ナノポアの大きさよりも大きく、測定対象のバイオポリマーに結合させる分子とを有するバイオポリマー決定キットであって、
前記装置は、
前記薄膜によって区切られた第1及び第2の溶液槽と、
前記バイオポリマーを前記第1の溶液槽と前記第2の溶液槽との間で前記ナノポアを通じて移動させる駆動手段と、
前記分子が前記測定対象のバイオポリマーの一方に結合されたバイオポリマーを前記第1の溶液槽に導入する手段と、
前記ナノポアを通過した前記バイオポリマーの他端に結合させる前記分子を前記第2の溶液槽に導入する手段と、
前記駆動手段による前記バイオポリマーの移動に伴い発生する信号を検出する検出手段と、
前記検出手段により検出した信号の時間変化を測定し、前記信号をバイオポリマーを構成するモノマーの種類に応じたデータとして算出する算出し、前記バイオポリマーを構成するモノマーの並びを決定する算出手段とを有することを特徴とするバイオポリマー決定キット。
【請求項15】
請求項14に記載のバイオポリマー決定キットであって、前記分子は、ストレプトアビジンであることを特徴とするバイオポリマー決定キット。
【請求項16】
請求項15に記載のバイオポリマー決定キットであって、前記分子は、DIG−抗DIG抗体結合により前記バイオポリマーに結合されるビーズであることを特徴とするバイオポリマー決定キット。
【図1】


【図2】

【図3】
【図4】

【図5】
【図6】

【図7】
【図8】
【図9】
【図10】

【図11】
【図12】
【図13】
【図14】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2010−230614(P2010−230614A)
【公開日】平成22年10月14日(2010.10.14)
【国際特許分類】
【出願番号】特願2009−80867(P2009−80867)
【出願日】平成21年3月30日(2009.3.30)
【出願人】(501387839)株式会社日立ハイテクノロジーズ (4,325)
【Fターム(参考)】
【公開日】平成22年10月14日(2010.10.14)
【国際特許分類】
【出願日】平成21年3月30日(2009.3.30)
【出願人】(501387839)株式会社日立ハイテクノロジーズ (4,325)
【Fターム(参考)】
[ Back to top ]