
ナノ粒子
周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物と、前記分子クラスター化合物に提供されるコア半導体材料とからなるナノ粒子であって、コア半導体材料は、周期表の第13族及び第15族からのイオンを組み込むナノ粒子。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は半導体ナノ粒子に関する。
【背景技術】
【0002】
多くの場合量子ドット又はナノ結晶と呼ばれる、例えば2〜50nmの範囲の寸法の粒子からなる化合物半導体の調製及び特徴づけに対して多くの関心が持たれてきた。これらの研究は、これらの物質のサイズ調整可能な電子的特性により主になされ、それは、光学的及び電子的装置などの多くの商業的応用において、又、多様な範囲に及ぶ他の応用において利用され得る。その範囲は、多くの新たな新進の応用があるが、生物学的ラベリング、太陽電池、触媒作用、生物学的イメージング、発光ダイオード、一般的な空間照明及びエレクトロルミネセンス・ディスプレイ、フォトルミネセンス・ディスプレイのように広範に亘る。
【0003】
最も多く研究されてきた半導体材料は、カルコゲニドII−VI(例えば第12族〜第16族)材料、例えばZnS、ZnSe、CdS、CdSe、CdTeである。最も顕著には、そのスペクトルの可視域上の光学調整能力のため、CdSeが非常に研究されている。いくつかの以前の例が文献において見られるが、より最近になって、再現可能な方法が、「ボトムアップ」技術によって開発され、それによって、粒子は原子毎に、「湿式」化学法を用いて調製される。
【0004】
共に個々の半導体ナノ粒子のサイズに関係する2つの根本的要因が、これらの粒子の独特の特性に関与している。第1は、大きい表面積対体積比である。粒子がより小さくなるにつれて、内部の原子に対する表面原子の数の比率が増加する。このため、材料全体の特性において表面特性が重要な役割を果たすことになる。第2の要因は、半導体ナノ粒子は、サイズと共に材料の電子的特性が変化すること、更に、粒子のサイズが減少するにつれて、量子閉じ込め効果のためバンドギャップが徐々により大きくなることである。この効果は、『箱の中の電子』の閉じ込めによって、対応するバルク半導体材料における連続バンドではなく、原子及び分子において観察されるのと類似の離散的エネルギー準位が生じている結果である。従って、半導体ナノ粒子においては、第1励起子遷移より大きなエネルギーを有する電磁放射(光子)の吸収によって生じた「電子及び正孔」は、物理パラメータのため、対応する巨視的結晶質材料においてより接近しているので、クーロンの相互作用は無視できない。これは狭バンド幅放出につながり、それは粒子サイズ及び組成に依存する。このように、量子ドットは対応する巨視的結晶質材料より高い運動エネルギーを有するので、第1の励起子遷移(バンドギャップ)はエネルギーにおいて粒径の減少と共に増加する。
【発明の開示】
【発明が解決しようとする課題】
【0005】
単一半導体材料と外側の有機不動態化層からなる単一コアナノ粒子は、欠陥で起こっている電子正孔再結合、及び非放射電子正孔再結合の原因になるナノ粒子表面上のダングリングボンドのため、量子効率が比較的低くなる可能性がある。
【0006】
欠陥及びダングリングボンドを除去する1つの方法は、バンドギャップがより広く、コア材料との格子不整合が小さい第2の無機材料をコア粒子の表面にエピタキシャルに成長させて、「コアシェル」粒子を生成することである。コアシェル粒子は、コアに閉じ込められたあらゆるキャリアを、非放射再結合の中心として働くおそれのある表面状態から離す。1つの例は、CdSeコアの表面に成長させてCdSe/ZnSコア/シェルナノ粒子を作成するZnSである。
【0007】
もう一つの方法は、量子ドット−量子井戸構造のような、「電子正孔」一対が単一のシェル層に完全に閉じ込められるコア/マルチシェル構造を調製することである。ここで、コアはバンドギャップの広い材料で出来ており、バンドギャップがより狭い材料の薄いシェルが続き、そして更なる広いバンドギャップ層によって覆われる。例えばわずかのHgSの単層を堆積させるためにコアナノ結晶の表面上でCdの代替にHgを使用して成長するCdS/HgS/CdSである。結果として生じる構造は、HgS層への光励起キャリアの閉じ込めを明らかに示した。
【0008】
如何なるコア、コアシェル又はコアマルチシェルナノ粒子においても、最終無機表面原子の周りの配位は、反応性の高い完全に配位されていない原子「ダングリングボンド」が粒子の表面上にあって不完全であり、これは粒子の凝集につながることがある。この問題は、保護用有機基で「裸の」表面原子を不動態化(キャッピング)することによって解決される。
【0009】
有機材料又はシース材料の最外側層(キャッピング剤)は、粒子凝集を抑制するのに役立ち、更にナノ粒子を、それらを囲んでいる化学環境から保護すると共に、他の無機、有機、又は生物学的材料との化学結合の手段を提供する。多くの場合、キャッピング剤は、ナノ粒子の作成が行われる溶媒であり、ルイス塩基化合物又は、不活性溶媒(例えば炭化水素)において薄められたルイス塩基化合物から成り、それによってナノ粒子の表面に対して供与型配位が可能である孤立電子対がある。
【0010】
高品質な半導体ナノ粒子の合成に関わる重要な問題は、粒子の均一性、サイズ分布、量子効率及び商業的応用におけるそれらの長期の化学的及び光安定性の使用である。初期のルートは従来のコロイド水性化学を利用したが、より最近の方法は、有機金属化合物を用いたナノ結晶子の動力学的に制御された沈殿を含んでいる。より最近の方法のほとんどは、Murray、Norris及びBawendiによって説明された当初の「核生成及び成長」方法(非特許文献1)に基づくが、当初用いられた有機金属前駆体以外のもの、例えば、酸化物(例えばCdO)、炭酸塩(例えばMCO3)、アセテート(例えばM(CH3CO2))及びアセチルアセトネート(例えばM[CH3COOCH=C(C−)CH3]2)(例えばM=Cd又はZn)を用いている。
【0011】
Murrayら(非特許文献1)は当初、金属アルキル(R2M)、M=Cd、Zn、Te、R=Me、Etの有機金属溶液、及びトリ−n−オクチルホスフィン(TOP)に溶解させたトリ−n−オクチルホスフィンスルフィド/セレニド(TOPS/Se)を用いた。これらの前駆体溶液を、調製されている材料に応じて温度範囲120〜400℃の熱いトリ−n−オクチルホスフィンオキシド(TOPO)に注入する。これによって、II−VI材料のTOPOコート/キャップ半導体ナノ粒子が製造される。粒子のサイズは、温度、キャッピング剤、用いられる前駆体の濃度及び合成が行なわれる延べ時間によって制御され、より高い温度、より高い前駆体濃度及び長期にわたる反応時間でより大きい粒子が得られる。この有機金属のルートは、よりよい単分散度及び高い粒子結晶化度を含む他の合成方法に勝る効果がある。上述の如く、この方法の多くのバリエーションが現在文献に見られ、単分散度及び量子収量の両方について高品質なコア及びコアシェルナノ粒子を日常的に生じさせる。
【0012】
単一源前駆体は、他の化合物半導体ナノ粒子と同様に、II−VIの半導体ナノ粒子材料の合成においても役立つということが分かった。ビス(ジアルキルジチオ/ジセレノカルバメート)カドミウム(II)/亜鉛(II)化合物、M(E2CNR2)2(M=Zn又はCd、E=S又はSe及びR=アルキル)は同様の『ワンポット』合成法において用いられてきて、それは、前駆体をトリ−n−オクチルホスフィン(TOP)に溶解させてから200℃を超える熱いトリ−n−オクチルホスフィンオキシド/トリ−n−オクチルホスフィン(TOPO/TOP)へ急速に注入することを含んでいた。
【0013】
根本的に、これらの製法は全て、高温での粒子の核生成に続いて低温での粒子成長という原理に依存している。更に言えば、2〜10nmの範囲のナノ粒子の単分散集合を有するため、ナノ粒子の核生成は、ナノ粒子の成長から適切に分離しなければならない。これは、一方又は両方の前駆体のより低温の溶液を、粒子の核生成を起こす、より高温の配位性溶媒(存在しない場合、他方の前駆体を含有する)へ急速に注入することによって達成される。注入時に突然それより冷たい溶液を添加することで、その後反応温度が低下し(添加する溶液の容量は通常全溶液の約1/3である)、更なる核生成が阻害される。粒子成長は、(用いる前駆体によって面触媒過程であるか又はオストワルド熟成を経て)その低下した温度で続き、このように核生成と成長が分離され、狭いナノ粒子のサイズ分布が得られる。この方法は、反応の全体にわたって合理的に均一温度を保ちながら1つの溶液を急速に他のものに添加することが出来る小規模合成でよく機能する。しかしながら、商業的応用において必要とされる、大容積の溶液同士を互いに急速に注入する必要がある、より大きな調製規模のとき、反応混合物中に著しい温度差が生じることがあり、これがその後、容認出来ない程の大きな粒子サイズ分布につながることがある。
【0014】
Cooney及び共同研究者ら(非特許文献2)は、II−VI分子クラスター[S4Cd10(SPh)16][Me3NH]4を用いて、CdSのII−VIナノ粒子を製造した。ここでヨウ素による表面キャッピングSPh−配位子の酸化も含まれた。この調製ルートでは、II−VIクラスターの大部分がイオンへ細分化され、これらが残りのII−VI([S4Cd10(SPh)16]4−)クラスターによって消費され、これらが後にCdSのII−VIナノ粒子へと成長する。
【0015】
Strouse及び共同研究者ら(非特許文献3)は、II−VIクラスターを用いた同様の合成手法を用いて、II−VIナノ粒子を成長させたが、化学剤ではなく熱分解(分散熱温度上昇)を採用して粒子成長を起こした。更に言えば、単一源前駆体[M10Se4(SPh)16][X]4、X=Li+又は(CH3)3NH+、M=Cd又はZnを熱分解し、それによって幾つかのクラスターの細分化を起こし、続いて、M及びSe自由イオンのスカベンジングから粒子成長させるか、又は、単に、クラスターが凝集して、最初は、より大きいクラスター、そして後に小さいナノ粒子、そして最終的に、より大きい粒子を調製した。
【0016】
Cooney(非特許文献2)の方法もStrouse(非特許文献3) の方法も、分子クラスターを用いてナノ粒子を成長させたが、クラスターからのイオンを用いて、クラスター凝集又はクラスターの細分化によって、より大きなナノ粒子を成長させた。いずれの場合においても、最初の分子クラスター上のより大きなナノ粒子を成長させるために必要とされるイオンを提供するために、別のナノ粒子前駆体組成物を用いなかった。更に、Cooney(非特許文献2)の方法もStrouse(非特許文献3)の方法も、最終的なナノ粒子において、最初の個々の分子クラスターの構造的整合性を保たなかった。更に又、これらの方法の両方とも、II―VIクラスターを用いてII―VIナノ粒子を形成することに制限されることが分かり、それは、これらの方法がより大きなナノ粒子を造るために分子クラスターの材料を用いることに制限されるという事実の必然的な結果である。従って、Cooney(非特許文献2)及び
Strouse(非特許文献3)の作業は、それらの方法論を用いて生成可能な材料の範囲に関して制限される。
【0017】
出願人の公開国際特許出願PCT/GB2005/001611号及びPCT/GB2006/004003号は、大量の高品質単分散の量子ドットを製造する方法を説明しており、それは以前の小規模の方法に関連する問題の多数を克服する。化学前駆体は、分子クラスター化合物の存在下で、又、分子クラスターの整合性が維持され、化学前駆体と反応する核生成中心を提供する明確に定義された既成のシード又はテンプレートの働きをするという条件下で提供され、産業的な応用に十分に大きな規模で高品質ナノ粒子を生成する。
【0018】
PCT/GB2005/001611及びPCT/GB2006/004003に説明される方法の重要な際立った特徴は、前駆体組成物のナノ粒子への転化がナノ粒子成長の全体にわたってその構造的整合性を保つ分子クラスター化合物の存在下で遂行されるということである。クラスター化合物の同一の分子は、ナノ粒子成長が開始されるシード又は核生成点の働きをする。このようにして、適当な明確に定義された核生成サイトが分子クラスターによるシステムですでに提供されるので、高温核形成工程はナノ粒子成長を開始するために必要ではない。クラスター化合物の分子は、ナノ粒子成長を導くテンプレートの働きをする。「分子クラスター」は、関連技術分野で広く理解されている用語であるが、明確にするため、ここでは、クラスター化合物の全ての分子が同じ相対分子式を所有するように十分に明確に定義された化学構造の3つ以上の金属原子及びそれらに関連した配位子のクラスターに関するものであると理解されるべきである。このように、1つのH2O分子が他のH2O分子と同一であるのと同様に、分子クラスターは互いに同一である。他の方法で使用される核生成サイトよりも非常に明確に定義されている核生成サイトを提供することにより、分子クラスター化合物を用いることで、本質的に単分散であるナノ粒子の集合が提供出来る。この方法の更なる有意な利点は、容易に規模を拡大することが出来るということである。適当な分子クラスター化合物を製造する方法は当該技術内で知られていて、その例はケンブリッジ結晶学データセンター(www.ccdc.ca.ac.uk)で見ることが出来る。
【0019】
現在まで広く研究されたナノ粒子材料の多数がカドミウムイオンを組み込むことは、前述の考察から理解されるだろう。しかしながら、カドミウムや、水銀及び鉛系材料のような他の重金属の使用に関連する多くの環境問題があり、そこで、カドミウムを含まないナノ粒子材料を開発することが必要である。特に、現在のカドミウム系材料と同等の単分散度及びサイズ調整可能な光ルミネセンススペクトルを示す、カドミウムを含まない量子ドットを製造することが望ましい。商業的には、このような材料を大規模に、高収量において、出来るだけ安価に製造することが要求される。
【0020】
本発明の目的は、ナノ粒子材料の製造のための先行技術の方法に内在する1つ又はそれ以上の問題に対処することである。更なる目的は、上記の商業的な要求の1つ又はそれ以上を満たす、カドミウムを含まない新たなナノ粒子材料を提供することである。
【課題を解決するための手段】
【0021】
本発明は、II―VI分子クラスター上に成長して、それによってII―VI分子クラスターを組み込むIII−Vナノ粒子に関する。本発明の第1の態様において、周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物と、前記分子クラスター化合物に提供されるコア半導体材料とからなるナノ粒子であって、コア半導体材料は、周期表の第13族及び第15族からのイオンを組み込むナノ粒子を提供する。
【0022】
本発明の第2の態様において、周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物と、前記分子クラスター化合物に提供されるコア半導体材料とを具えるナノ粒子の製造方法であって、コア半導体材料は周期表の第13族及び第15族からのイオンを組み込み、当該方法は、ナノ粒子コア材料のシーディング及び成長を可能にする条件の下で、周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物の存在下で、それぞれ周期表の第13族及び第15族からのイオンを含む第1及び第2のコア前駆体種を具えているナノ粒子コア前駆体組成物の、ナノ粒子コアの材料への転化を生じさせることを含む、ナノ粒子の製造方法を提供する。
【0023】
従って、本発明は、III−Vナノ粒子が大規模に比較的安価に製造することが出来る手段を提供する。
【0024】
分子クラスター化合物及びコア半導体材料が、前記分子クラスター化合物上の前記コア半導体材料の成長を可能にする互換性を持つ結晶相を有することが好ましい。最も好ましくは、分子クラスター化合物及びコア半導体材料は、同じ結晶相を有する。以下に説明する例において、硫化亜鉛クラスターを、リン化インジウムコア半導体ナノ粒子を成長させるために用いる。発明者は何らかの特定の理論によって制約されたくないが、ZnSクラスター/InPコアナノ粒子を得ることが可能だった理由は、ZnSクラスター及びInPコア材料が、両方とも六角形の結晶相を有し、従って、それらは互いに互換性を有し、ZnSクラスター上のInPの層の成長を容易にするという事実に起因するだろうと仮定される。
【0025】
1つの種類の材料のクラスターを組み込んでいるナノ粒子及びそのクラスター上に成長した異なる種類の材料のコアが低い量子収量を示すと予測されたが、驚くべきことに、本発明の第1の態様に係るナノ粒子が更なる準備後の処理の後、約20〜60%及び最高90%の範囲の比較的高い量子効率を示すことが観察された。更に、フッ化水素でナノ粒子の表面を洗浄すること及び/又はコア半導体材料上の半導体材料(例えばII―VI(VI)材料(例えばZnS))の一つ以上の更なる層を成長させることによって、量子収量を更に高めることが出来ることが断定された。
【0026】
好ましくは、分子クラスター化合物は、亜鉛イオンを組み込む。本発明の第1の態様のナノ粒子の好ましい実施形態において、分子クラスター化合物は、硫化物イオン、セレンイオン、及びテルル化物イオンからなる群から選択される第16族イオンを組み込む。
【0027】
コア半導体材料は、好ましくは、アルミニウムイオン、ガリウムイオン、及びインジウムイオンからなる群から選択される第13族イオンを組み込む。コア半導体材料は、窒化物イオン、ヒ化物イオン、及びアンチモンイオンからなる群から選択される第15族イオンを組み込んでもよい。
【0028】
好ましい実施形態において、本発明の第1の態様のナノ粒子は、前記ナノ粒子コア上に提供される第1の半導体材料を含む第1の層を具える。第1の半導体材料として、適切な半導体材料が選択されてもよく、例えば、IIA―VIB材料、IIB―VIB材料、II―V材料、III―V材料、III―IV材料、III―VI材料、IV―VI材料、又は遷移金属元素又はd―ブロック元素を組み込んでいる材料であるが、これに限定されない。好ましくは、第1の半導体材料は、周期表の第12族からのイオンを組み込む(例えば亜鉛イオン)。第1の半導体材料は、周期表の第16族からのイオンを組み込んでもよい(例えば硫化物イオン、セレンイオン又はテルル化物イオン)。
【0029】
ナノ粒子は、前記第1の層上に提供される第2の半導体材料を含む第2の層を具えてもよい。選択的に、半導体材料の更なる層又はシェルが、第2の層に提供されてもよい。第2及び以降の層は、適切な半導体材料から形成されてもよく、半導体材料は例えば、IIA―VIB材料、IIB―VIB材料、II―V材料、III―V材料、III―IV材料、III―VI材料、IV―VI材料、又は遷移金属元素又はd―ブロック元素を組み込んでいる材料であるが、これに限定されない。
【0030】
本発明の第1の態様に係るナノ粒子の製造の好ましい方法において、粒子成長を開始するために、ナノ粒子前駆体の存在下で溶媒(配位又は非配位)に、シーディングII―VI分子クラスターを入れる。シーディング分子クラスターは、反応溶液内に存在する他の前駆体から粒子成長を開始するためのテンプレートとして用いられる。シーディング剤として用いられる分子クラスターは、先に作成することが出来、又はシーディング剤としての働きをする前にその場で製造することも出来る。いくつかの前駆体は、分子クラスターとともに反応プロセスの初めに存在しても、存在しなくてもよいが、反応が進行して、温度が上昇するにつれて、更なる量の前駆体を溶液として滴状で、又は、固体として、周期的に反応に加えることが出来る。
【0031】
方法は、ナノ粒子前駆体組成物の所望のナノ粒子への転化に関係する。好適な前駆体には、成長しているナノ粒子に組み込まれる2つ以上のイオンを含む単一供給源前駆体、又は、成長しているナノ粒子において含まれる少なくとも1つのイオンをそれぞれ含む、2つ以上の別の前駆体を含むマルチ供給源前駆体が含まれる。ナノ粒子の最終的な所望の収率を形成するために必要な前駆体組成物の全量を、ナノ粒子の成長が始まる前に加えることが出来、或いは、前駆体組成物は、反応の全体にわたって段階的に加えることが出来る。
【0032】
前駆体のナノ粒子材料への転化は、任意の適当な溶媒において実行することが出来る。クラスター化合物の分子の整合性を維持することが重要であることが理解されるだろう。それゆえに、クラスター化合物及びナノ粒子前駆体が溶媒に導入されるときに、溶媒の温度は前記クラスター化合物の満足な溶解及び混合を確実にするために十分に高くなければならない(存在する化合物が完全に溶解することは、必要ではないが望ましい)が、クラスター化合物分子の整合性を崩壊させるほど高くてはならない。一旦クラスター化合物及び前駆体組成物が溶媒において十分によく溶解すると、このように形成される溶液の温度をある温度又は温度範囲まで上昇させ、該温度又は温度範囲は、ナノ粒子の成長が始まるのに十分に高いが、クラスター化合物分子の整合性に損傷を与えるほど高くない。温度を上昇させるにつれて、前駆体の更なる分量を反応物に、液滴の方法で、又は固体として加える。そして溶液の温度を、この温度又はこの温度範囲内で、所望の特性を具えたナノ粒子を形成するために必要な時間維持することが出来る。
【0033】
多様な適当な溶媒が利用出来る。使用する特定の溶媒は通常、反応種、即ちナノ粒子前駆体及び/又はクラスター化合物の性質、及び/又は形成されるナノ粒子のタイプに、少なくとも部分的に依存する。通常の媒体としては、ホスフィン(例えばTOP)、ホスフィンオキシド(例えばTOPO)、アミン(例えばHDA)、チオール(例えばオクタンチオール)のようなルイス塩基型配位性溶媒、又は、アルカン及びアルケンのような非配位性有機溶媒が挙げられる。非配位性溶媒が用いられる場合、以下の理由により、それは、キャッピング剤としての役割を果たす更なる配位剤の存在下で通常使用される。
【0034】
形成されているナノ粒子が量子ドットとして機能することを目的とする場合、完全には配位結合されなかった「ダングリングボンド」である表面原子をキャップして、非放射電子正孔再結合を最小にすると共に、量子効率を低下させ得る又はナノ粒子の凝集を形成し得る粒子凝集を抑制することが重要である。キャッピング剤又は不動態化剤としての役割を果たすことも出来る多くの異なる配位性溶媒が知られており、例えばTOP、TOPO、had又はミリスチン酸(テトラデカン酸)、長鎖アミン、官能化されたPEG鎖のような長鎖有機酸であるが、これらのキャッピング剤に限定されない。キャッピング剤としての役割を果たすことができない溶媒が選択される場合、ナノ粒子が成長する間、任意の望ましいキャッピング剤を反応混合物に加えることが出来る。上記のキャッピング剤は一般的にルイス塩基であり、ホスフィン型単歯状、多歯状配位子(トリオクチルホスフィン、トリフェニルホスフィン、t−ブチルホスフィン)、ホスフィンオキシド(トリオクチルホスフィンオキシド)、アルキルホスホン酸、アルキルアミン(ヘキサデシルアミン、オクチルアミン)、アリルアミン、ピリジン、オクタンチオール、長鎖脂肪酸、及びチオフェンが挙げられるが、ナノ粒子の周囲に保護シースを形成するオレイン酸及び有機ポリマーのような広範囲の他の試剤が利用出来る。
【0035】
量子ドットの最外側層(キャッピング剤)は、他の無機、有機又は生物学的材料に対する化学結合として用いることが出来る追加の官能基を処理する配位リガンドからなることもでき、それによって、官能基は量子ドット表面から離間する方に向き、その他利用可能な分子と結合/反応することが出来る。その分子は、1級、2級アミン、アルコール、カルボン酸、アジド、ヒドロキシル基等であるが、これに限定されない。量子ドットの最外側層(キャッピング剤)は、重合可能であると共に粒子の周囲にポリマーを形成するために用いることが出来る官能基を処理する配位リガンドからなることも出来る。
【0036】
最外側層(キャッピング剤)は、最も外側の無機層に直接結合された有機単位からなることもでき、又、粒子の表面に結合されずに、粒子の周囲にポリマーを形成するために、又は、更なる反応のために用いることが出来る官能基を有してもよい。
【0037】
本発明の第2の態様を構成する方法に関して、分子クラスター化合物が亜鉛イオンを組み込むことが好ましい。分子クラスター化合物は、好ましくは、硫化物イオン、セレンイオン及びテルル化物イオンからなる群から選択される第16族イオンを組み込む。
【0038】
第1及び第2のコア前駆体種は、前記コア前駆体組成物に含まれる別の実体でもよい。この場合、コア半導体材料の第13族イオンは、III―ホスフィン、III―(TMS)3、III―(アルキル)、III―(アリール)、III―(ミリスチン酸塩)3、III―(アセテート)3、III―(ミリスチン酸塩)(アセテート)2、III―(ミリスチン酸塩)2(アセテート)、及びIII―(III) アセチルアセトネートからなる群から選択される供給源に由来してもよい。コア半導体材料の第13族イオンは、有機金属化合物、配位化合物、無機塩及び/又は元素供給源に由来してもよい。更に、コア半導体材料の第15族イオンは、有機金属化合物、配位化合物、無機塩及び/又は元素供給源に由来してもよい。
【0039】
第1及び第2のコア前駆体種は、前記コア前駆体組成物に含まれる単体において組み合わせられてもよい。
【0040】
方法は、ナノ粒子コア材料上の第1の半導体材料から構成される第1の層を形成することを更に含んでもよい。好ましくは、第1の半導体材料の第1の層の形成は、第1の材料前駆体組成物の、前記第1の材料への転化を生じさせることを含む。第1の材料前駆体組成物が、第1の材料から構成される層に組み込まれる第3及び第4イオンを具えることが好ましい。第3及び第4イオンは、第1の材料前駆体組成物において含まれる別の実体でもよいか又は第1の材料前駆体組成物において含まれる単体において組み合わせられてもよい。
【0041】
第1の半導体材料の第1の層の形成の前にナノ粒子コア材料がフッ化水素に晒されることが好ましい。例において以下に更に詳細に述べるように、フッ化水素を含む溶液でナノ粒子コアの表面にエッチングすることによってナノ粒子の量子効率を上昇させることが出来る。
【0042】
本発明は、II―VI分子クラスターを用いて製造されるIII―Vナノ粒子に関係し、小さなナノ粒子の集合と比較して、クラスターは明確に定義された同一の分子実体であり、それは本質的に分子クラスターの特徴のない性質を欠いている。知られているIII―V分子クラスターは少数であるので、III―Vクラスターの代わりにII―VI分子クラスターが用いられる。III―V分子クラスターは、作成するのが困難で、通常、空気及び水分に影響を受けるのに対し、多数のII―VI分子クラスターが簡単な手順によって作成でき、それらの多くは空気及び水分に影響を受けない。III―V粒子は、多くのII―VI分子クラスターにシーディングされることができ、それゆえ、初めて、III―Vナノ粒子を成長させるためにIII―V分子クラスターを用いる必要又は利点がないことが、現在理解される。分子クラスターを用いることにより、III―V量子ドットを製造する従来の方法のように、高温核形成工程の必要がなく、それは大規模な合成が可能であることを意味する。
【0043】
更に、例えばInP及びGaP量子ドット及びそれらの合金のようなIII―Vナノ粒子材料の成長をシーディングするのに、[HNEt3]4[Zn10S4(SPh)16]のようなII―VI分子クラスターを用いることが可能である。II―VI分子クラスター、III及びVイオンの分子供給源、「分子フィードストック」の後に続く添加又はその場形成がなされて、それらは粒子成長を容易にするために消費される。これらの分子供給源は、オストワルトの熟成が生じることを抑制してナノ粒子サイズ範囲が分散することから抑制するために自由イオンの濃度を維持しながら、自由イオンの濃度を最低限に保つように、反応溶液と周期的に加えてもよい。
【0044】
ナノ粒子成長は、加熱(熱分解)によって、又はソルボサーマル法によって開始されてもよい。ソルボサーマルという用語は、ここでは、粒子成長を生じさせて維持するように行う反応溶液における加熱を指すように用いており、また、thermolsolvol、溶液熱分解及びリオサーマルとも呼ばれる方法も包含することを意図している。粒子の調製は、反応条件を変化させることによって達成されることもでき、例えば塩基又は酸の添加、圧力の上昇(即ち気圧より大きな圧力の使用)、マイクロ波放射等の電磁放射を加えること、又は当業者に知られている多くの他の方法の何れかによって達成される。
【0045】
III―V半導体量子ドットの調製のための条件
フィードストック
分子フィードストックは、ナノ粒子の成長のために必要な全ての元素が単一化合物前駆体の中に存在する単一源前駆体の形、又は成長したナノ粒子となるために必要な1又は複数の元素/イオンをそれぞれが含む前駆体の組み合わせであることが出来る。これらのフィードストックを反応の初めに加えるか、又は粒子成長の反応の全体にわたって周期的に加えてもよい。このフィードストックは、液体、溶液、固体、スラリー又は気体の形であることが出来る。
【0046】
シーディングクラスターのその場形成
シーディングテンプレートとして使用されるクラスターは、反応の前に予め調製して反応過程の初めに反応溶液に加えてもよく、又はナノ粒子の成長のために用いられる前駆体を加える前に、反応溶液においてその場で形成してもよい。
【0047】
作成される系の種類
半導体コア材料
本発明は、III−Vナノ粒子材料及びその合金の調製を目的としていて、量子ドット又はナノ結晶とも呼ばれる化合物半導体粒子を含む。これは、通常2〜100nmのサイズ範囲内で、以下を含むナノ粒子コア材料を含む。
周期表の第13族(III)からの第1元素と、周期表の第15族(V)からの第2元素と、又、三元、四元、及びドープ材料を含むIII−V材料。
【0048】
ナノ粒子コア材料の好ましい例は、BP、AlP、AlAs、AlSb;GaN、GaP、GaAs、GaSb;InN、InP、InAs、InSb、AlN、BNを含むが、これに制限されない。
【0049】
本発明の第1の態様に含まれるナノ粒子は、III及びVイオンを組み込んでいる二元相材料だけでなく、存在するIII及びVイオンに加えてそれぞれ1つか2つの更なる種類のイオンを組み込んでいる三元及び四元相ナノ粒子も含む。三元相ナノ粒子が3つの成分の材料から成り、四元相ナノ粒子が4つの成分の材料から成ることはいうまでもない。
【0050】
ドープされたナノ粒子は、1つ以上の主要な族又は希土類元素、多くの場合遷移金属又は希土類元素(例えばMn+又はCu2+であるがこれに限らない)からなるドーパントを更に組み込む上記の種類のナノ粒子を指す。
【0051】
外側の1つ又は複数の無機シェル
コアIII―Vナノ粒子上へ成長する任意のシェル又はそれに続く複数のシェルに用いられる材料はほとんどの場合、コア材料と類似の格子形材料だろう。即ち、コア上にエピタキシャルに成長することが出来るように、コア材料に近い格子整合を有するが、この適合性を有する材料に、必ずしも制限されるというわけではない。しかしながら、2つの材料(コア及びシェル)に互換性がない場合、半導体コアと半導体シェルとの間に、緩衝層をコアの外側に最初に成長させてもよい。コア上に成長する任意の緩衝層又はシェル層の材料は、以下を具える材料を含むことが出来る。
周期表の第2族からの第1元素と、周期表の第16族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むIIA−VIB(2−16)材料。ナノ粒子材料は、MgS、MgSe、MgTe、CaS、CaSe、CaTe、SrS、SrSe、SrTeを含むが、これに限定されない。
周期表の第12族からの第1元素と、周期表の第16族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むIIB−VIB(12−16)材料。ナノ粒子材料は、ZnS、ZnSe、ZnTe、CdS、CdSe、CdTe、HgS、HgSe、HgTeを含むが、これに限定されない。
周期表の第12族からの第1元素と、周期表の第15族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むII−V材料。ナノ粒子材料は、Zn3P2、Zn3As2、Cd3P2、Cd3As2、Cd3N2、Zn3N2を含むが、これに限定されない。
周期表の第13族からの第1元素と、周期表の第15族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むIII−V材料。ナノ粒子材料は、BP、AlP、AlAs、AlSb;GaN、GaP、GaAs、GaSb;InN、InP、InAs、InSb、AlN、BNを含むが、これに限定されない。
周期表の第13族からの第1元素と、周期表の第14族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むIII−IV材料。ナノ粒子材料は、B4C、Al4C3、Ga4Cを含むが、これに限定されない。
周期表の第13族からの第1元素と、周期表の第16族からの第2元素を組み込み、又、三元及び四元材料を含むIII−VI材料。ナノ粒子材料は、Al2S3、Al2Se3、Al2Te3、Ga2S3、Ga2Se3;In2S3、In2Se3、Ga2Te3、In2Te3を含むが、これに限定されない。
周期表の第14族からの第1元素と、周期表の第16族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むIV−VI材料。ナノ粒子材料は、PbS、PbSe、PbTe、Sb2Te3、SnS、SnSe、SnTeを含むが、これに限定されない。
周期表の遷移金属における任意の族からの第1元素と、周期表のd−ブロック元素の任意の族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むナノ粒子材料。ナノ粒子材料は、NiS、CrS、CuInS2、CuInSe2、CuGaS2、CuGaSe2を含むが、これに限定されない。
【0052】
ナノ粒子形状
ナノ粒子の形状は、球体に限定されず、任意の望ましい形状をとることができ、例えば桿体、球体、円板、四脚又は星形である。ナノ粒子の形状の制御は、反応粒子成長過程において、成長している粒子の特定の格子面と優先して結合し、その後特定の方向の粒子成長を阻害又は遅延させる化合物を加えることによって達成することが出来る。この効果を達成するために加えることが出来る化合物の非限定的な例は、ホスホン酸(n−テトラデシルホスホン酸、ヘキシルホスホン酸、1−デカンスルホン酸、12−ヒドロキシドデカン酸、n−オクタデシルホスホン酸)を含む。
【0053】
調製方法の概要
本発明は、配位性有機層のようなキャッピング剤によって粒子凝集及び周囲の化学環境から安定化されてよい実質的に純粋な単分散のナノ結晶粒子を提供することを目的としている。このようなナノ粒子を表す適当な化学式は、MEnLyであり、但し、M=III族元素、E=V族元素、L=配位子(例えば配位性有機層/キャッピング剤)、そして、n及びyが構成要素E及びLの適切な化学量論量を表す。
【0054】
半導体材料のナノ粒子を調製するための第1ステップは、分子クラスターをテンプレートとして用いて他の元素源前駆体からのナノ粒子の成長をシードすることである。ホスフィン、ホスフィンオキシド、アミン又は有機酸に限定されないが、このようなルイス塩基配位化合物、又はオレイン酸のようなキャッピング剤化合物を添加したアルカン(オクタデセンス)のような不活性溶媒である、キャッピング剤ともなり得る高沸点溶媒と、テンプレートとして用いられるべき少量のクラスターを混合することによってこれが達成される。反応温度が上昇するにつれて、M及びEの供給源前駆体が、液体であれば液滴で、又は多量の固体粉末として、周期的に加えられる。これに続いて、M供給源及びE供給源が、反応混合物に加えられる。M及びE前駆体供給源は、一方がMを含有し、他方がEを含有している2つの別々の前駆体の形であるか、又は単一分子内にM及びEの両方を含有する単一源前駆体の形であることが可能である。
【0055】
これに続き、成長したナノ粒子の形状を制御する能力を有する他の試剤を反応に添加してもしなくてもよい。これらの添加物は、成長しているナノ粒子の特定の面(格子面)と優先して結合し、それゆえ、粒子のその特定の方向に沿う粒子成長を阻害又は遅延させることが出来る化合物の形である。三元、四元又はドープ粒子を製造するために、他の元素源前駆体を反応に添加してもしなくてもよい。
【0056】
分子クラスターを配位化合物と混合し、それから、分子クラスターテンプレートの表面に粒子成長が始まるまで、反応混合物を一定の速度で加熱する。更なる量のM及びE前駆体を、適当な温度で反応混合物に加えてもよい。更なる前駆体の添加は、固体の前駆体又は前駆体を含む溶液を一定時間にわたって加えるバッチ添加、又は、溶液相前駆体の連続した滴加の形であってもよい。粒子の核生成及び成長を完全に分離するため、本発明では、反応の温度及び存在する前駆体の濃度によって制御される粒子サイズに関して高度な制御を示す。その場光学プローブ(in situ optical probe)によって、又は反応溶液の分取から、反応溶液のUV及び/又はPLスペクトルから証明されるように、所望の粒子サイズが得られると、温度は特定の度数だけ下降してもしなくてもよく、混合物を10分から72時間の期間にわたってアニールする。
【0057】
ME/NYコアシェル又はコアマルチシェル粒子を形成するための、形成されたナノ粒子の更なる継続的処理を行なってもよい。コアシェル粒子調製はナノ粒子隔離の前又は後に行い、これによって、ナノ粒子は反応から隔離されて新たな(きれいな)キャッピング剤(同じキャッピング剤化合物でも異なるキャッピング剤化合物でもよい)に再溶解し、これが結果としてより良質の量子ドットとなることが出来る。N源及びY源前駆体(N及びYは、MEコア量子ドット上に成長しているシェル又は層において必要とされる元素である)を反応混合物に加えるが、ME/NYコアシェル材料のコアシェル粒子を形成するために、一方がNを含有し、他方がYを含有している2つの別々の前駆体の形でも、又、単一分子内にN及びYを両方含有する単一源前駆体としてでもよい。
【0058】
所望のコアマルチシェル材料が形成されるまで、適当な元素前駆体でこの過程を繰り返してもよい。粒子の集合におけるナノ粒子サイズ及びサイズ分布は、成長時間、温度及び溶液中の反応物の濃度に依存し、温度が高くなると共に、大きいナノ粒子が製造される。
【0059】
シーディングに用いるクラスターのタイプ
本発明はII−VI分子クラスターの使用を含み、これによって用いられるクラスターは、集合における分子クラスターの匿名性を本質的に欠くナノ粒子と比較して、同一の分子的実体である。クラスターは、ナノ粒子の成長のための「胚型」テンプレートとして働き、これによって他の分子源前駆体はイオンを成長過程に貢献させるので、クラスターがその後粒子へと成長する。
【0060】
分子クラスターは、周期表の第12族からの第1のイオンを組み込み、周期表の第16族からの第2のイオンを組み込む。
【0061】
第12族イオンは、亜鉛、カドミウム及び水銀からなる群から選択されてもよい。
【0062】
第16族イオンは、硫化物、セレン化物及びテルル化物からなる群から選択されてもよい。
【0063】
用いられることが出来る分子クラスターの好ましい実施例は以下であるが、限定されない。
IIB−VIB
[{(PPh3)Hg}4(SPh)6]
(Ph4P)2[(SEt)5(Br)(HgBr)4]
(Ph4P)2[Hg4(SEt)5Br]
[Hg4Te12][N(CH2CH2Et)4]4
IIB−VIB
[RMEtBu]5但し、M=Zn、Cd、Hg;E=S、Se、Te;R=Me、Et、Ph
[X]4[E4M10(SR)16]但し、M=Zn、Cd、Hg;E=S、Se、Te、X=Me3NH+、Li+、Et3NH+
[Cd32S14(SPh)36]・L
[Hg10Se4(SePh)(PPh2nPr)4]
[Hg32Se14(SePh)36]
[Cd10Se4(SePh)12(PPr3)4]
[Cd32Sel4(SePh)36(PPh3)4]
[M4(SPh)12]+[X]2−但し、M=Zn、Cd、Hg;X=Me4N+、Li+
[Zn(SEt)Et]10
[MeMEiPr]但し、M=Zn、Cd、Hg;E=S、Se、Te
[RCdSR’]5但し、R=O(ClO3)、R’=PPh3、iPr
[Cd10E4(E’Ph)12(PR3)4]
但し、E=Te、Se、S、そして別に、E’=Te、Se、S
[Cd8Se(SePh)12Cl4]2−
[M4Te12]4−但し、M=Cd、Hg
[Ph12M18Cd10(PEt3)3]
但し、M=Te、Se
【0064】
無機コアのために用いられる前駆体
IIIイオン供給源
III―ホスフィン、III−(TMS)3、III―(アルキル)、III―(アリール)、III−(ミリスチン酸塩)3、III―(アセテート)3、III―(ミリスチン酸塩)(アセテート)2、III―(ミリスチン酸塩)2(アセテート)、III―(III)アセチルアセトネート
MR3但しM=Ga、In、Al、B;R=アルキル又はアリール基,例えばAlR3、GaR3、InR3(R=Me、Et、iPr)に限定されないが、このような有機金属化合物。
炭酸塩、M(CH3C)3但しM=B、Al、Ga、In:アセチルアセトネート(2,4−ペンタンジオネート)のようなβ−ジケトネート又はその誘導体、M[CH3COOCH=C(O−)CH3]2但しM=B、Al、Ga、Inのような配位化合物。
酸化物(例えばIn2O3、Ga2O3)又は、硝酸塩(例えばAl(NO3)3、In(NO3)3、Ga(NO3)3)のような無機塩。
B、Al、Ga、Inのような元素供給源。
【0065】
Vイオン供給源
NR3、PR3、AsR3、SbR3(R=Me、Et、tBu、iBu、Pri、Ph等);NHR2、PHR2、AsHR2、SbHR2(R=Me、Et、tBu、iBu、Pri、Ph等);NH2R、PH2R、AsH2R、SbH2R3(R=Me、Et,tBu、iBu、Pri、Ph等);PH3、AsH3;M(NMe)3、M=P、Sb、As;ジメチルドラジン(Me2NNH2);エチルアジド(Et−NNN);ヒドラジン(H2NNH2);Me3SiN3に限定されないが、このような有機金属化合物。
炭酸塩、MCO3 M=P、次炭酸ビスマス(BiO)2CO3;アセチルアセトネート(2,4−ペンタンジオネート)のようなβ−ジケトネート又はその誘導体、[CH3COCH=C(O−)CH3]3M、M=Biである;[CH3COOCH=C(O−)CH3]2M M=Bi;[CH3COOCH=C(O−)CH3]2Mに限定されないが、このような配位化合物。
酸化物P2O3、As2O3、Sb2O3、Sb2O4、Sb2O5、Bi2O3、又は硝酸塩Bi(NO3)3、Sn(NO3)4、Pb(NO3)2のような無機塩。
N、P、As、Sb、Biのような元素供給源。
【0066】
複合III−Vイオン供給源−ME単一源前駆体
III−V元素を具える化合物半導体ナノ粒子では、III及びVイオンが、単一源前駆体の形であることも可能であり、用いられる前駆体は、単一分子内にM及びEの両方を含む。
単一源前駆体は、有機金属化合物、無機塩又は配位化合物(MaEb)Lcであることが可能である。ここで、M及びEはナノ粒子内で必要とされる元素であり、Lはキャッピング配位子であり、a、b及びcは、M、E及びLの適切な化学量論量を表す数である。
III−Vコア半導体には、単一源前駆体は以下であることが可能であるが、それに限定されない。
GaNには、[(Me)2GaN(H)tBu]2[H2GaNH2]3、
Gapには、[Ph2GaP(SiMe3)3Ga(Ph)2Cl]、[Et2GaP(SiMe3)2]2、[Et2GaPEt2]3、[tBu2GaPH2]3、[Me2GaP(iPr)2]3[tBuGaPAr´]2、[tBu2GaP(H)C5H9]2、
GaAsには、Ga(AstBu2)3、[Et2GaAs(SiMe3)2]2、[tBu2GaAs(SiMe3)2]2、
GaSbには、[Et2GaSb(SiMe3)2]2、
InPには、[(Me3SiCH2)2InP(SiMe3)2]2、[R2InP(SiMe3)2]2、[Me2InPtBu2]2、
InSbには、[Me2InSbtBu2]3 [Et2InSb(SiMe3)2]3、[Me2InNEt2]2、[Et2AlAstBu2]2、
AlSbには、[tBu2AlSb(SiMe3)2]2、
GaAsには、[nBu2GaAstBu2]2、[Me2Ga2AstBu2]2、[Et2GaAstBu2]2
【0067】
任意の外側の無機シェル又はそれに続く複数のシェルのために用いられる前駆体(ME)
III―Vコア及びMEnLmシェル(但しM及びEは、シェル又は以降のシェル層の元素である)からなる化合物コア/シェル半導体ナノ粒子のために、元素Mの供給源は、反応に更に加えられてもよく、成長する粒子にEイオンの供給源を提供する能力を有する、任意のE含有種から成ることが出来る。
【0068】
M源
この前駆体は、有機金属化合物、無機塩、配位化合物又は元素供給源からなることが出来るが、これに限定されない。第1元素のためのII−VI、III―V、III―VI又はIV−Vの例は、以下を含むが、これに限定されない。
MR2、但しM=Mg、R=アルキル又はアリール基(MgtBu2);MR2、但しM=Zn、Cd、Te;R=アルキル又はアリール基(Me2Zn、Et2Zn、Me2Cd、Et2Cd);MR3、但しM=Ga、In、Al、B;R=アルキル又はアリール基[AlR3、GaR3、InR3(R=Me、Et、iPr)]に限定されないが、このような有機金属。
MCO3 M=Ca、Sr、Ba、[水酸化炭酸マグネシウム[(MgCO3)4Mg(OH)2];M(CO3)2 M=Zn、Cd、;MCO3 M=Pb:アセテート:M(CH3CO2)2 M=Mg、Ca、Sr、Ba;Zn、Cd、Hg;M(CH3C)3 M=B、Al、Ga、In:アセチルアセトネート(2,4−ペンタンジオネート)のようなβ−ジケトネート又はその誘導体、[CH3COOCH=C(O−)CH3]2 M=Mg、Ca、Sr、Ba、Zn、Cd、Hg;[CH3COOCH=C(O−)CH3]2 M=B、Al、Ga、In。オキザラートSrC2O4、CaC2O4、BaC2O4、SnC2O4に限定されないが、このような炭酸塩のような配位化合物。
酸化物SrO、ZnO、CdO、In2O3、Ga2O3、SnO2、PbO2;硝酸塩Mg(NO3)2、Ca(NO3)2、Sr(NO3)2、Ba(NO3)2、Cd(NO3)2、Zn(NO3)2、Hg(NO3)2、Al(NO3)3、In(NO3)3、Ga(NO3)3、Sn(NO3)4、Pb(NO3)2に限定されないが、このような無機塩。
Mg、Ca、Sr、Ba、Zn、Cd、Hg、B、Al、Ga、In、Sn、Pbに限定されないが、このような元素供給源。
【0069】
E源
反応に元素E源を更に加えてもよく、この元素E源は、成長している粒子にEイオン源を供給する能力を有する任意のE含有種から構成されることが出来る。
この前駆体は、有機金属化合物、無機塩、配位化合物又は元素供給源から構成されることが出来るが、これに限定されない。II−VI、III―V、III―VI又はIV−V半導体における元素Eの例は、以下を含むが、これに限定されない。
NR3、PR3、AsR3、SbR3(R=Me、Et、tBu、iBu、Pri、Ph等);NHR2、PHR2、AsHR2、SbHR2(R=Me、Et、tBu、iBu、Pri、Ph等);NH2R、PH2R、AsH2R、SbH2R3(R=Me、Et,tBu、iBu、Pri、Ph等);PH3、AsH3;M(NMe)3 M=P、Sb、As;ジメチルドラジン(Me2NNH2);エチルアジド(Et−NNN);ヒドラジン(H2NNH2);Me3SiN3に限定されないが、このような有機金属。
MR2(M=S、Se、Te;R=Me、Et、tBu、iBu等);HMR(M=S、Se、Te;R=Me、Et、tBu、iBu、iPr、Ph等);チオ尿素S=C(NH2)2;Se=C(NH2)2。
Sn(CH4)4、Sn(C4H9)、Sn(CH3)2(OOCH3)2。
炭酸塩、MCO3 M=P、次炭酸ビスマス(BiO)2CO3;M(CO3)2;アセテートM(CH3CO)2 M=S、Se、Te:M(CH3C)3 M=Sn、Pb:アセチルアセトネート(2,4−ペンタンジオネート)のようなβ−ジケトネート又はその誘導体、[CH3COOCH=C(O−)CH3]3M M=Bi;[CH3COOCH=C(O−)CH3]2M M=S、Se、Te:[CH3COOCH=C(O−)CH3]2M、M=Sn、Pb:チオ尿素、セレノ尿素(H2NC(=Se)NH2に限定されないが、このような配位化合物。
酸化物P2O3、As2O3、Sb2O3、Sb2O4、Sb2O5、Bi2O3、SO2、SeO2、TeO2、Sn2O、PbO、PbO2;硝酸塩Bi(NO3)3、Sn(NO3)4、Pb(NO3)2に限定されないが、このような無機塩。
元素供給源:Sn、Ge、N、P、As、Sb、Bi、S、Se、Te、Sn、Pb。
【0070】
複合III−Vイオン供給源−ME単一源前駆体
化学式MEの化合物半導体ナノ粒子シェルでは、元素M及びEが、単一源前駆体によって提供されることが可能であり、用いられる前駆体は、単一分子内にM及びEの両方を含む。この前駆体は、有機金属化合物、無機塩又は配位化合物、(MaEb)Lcであることが可能である。ここで、M及びEはナノ粒子内で必要とされる元素であり、Lはキャッピング配位子であり、a、b及びcは、適切な化学量論値である。
MがII族イオンであり、EがVI族イオンであるII―VI半導体シェルの非限定的な例は以下である。ビス(ジアルキルジチオ―カルバメート)M、(II)複合体又は、式M(S2CNR2)2の同族Se及びTe化合物 但しM=Zn、Cd、Hg;S=S、Se、O、Te及びR=アルキル基又はアリール基、CdS Cd[SSiMe3]2、Cd(SCNHNH2)2Cl2、Cd(SOCR)2・py、CdSe[Cd(SePh)2]2
III−V半導体シェルには、ME単一源前駆体の非限定的な例は以下である。
GaNには、[(Me)2GaN(H)tBu]2、[H2GaNH2]3、
Gapには、[Ph2GaP(SiMe3)3Ga(Ph)2Cl]、[Et2GaP(SiMe3)2]2、[Et2GaPEt2]3、[tBu2GaPH2]3[Me2GaP(iPr)2]3、[tBuGaPAr´]2、[tBu2GaP(H)C5H9]2、
GaAsには、Ga(AstBu2)3、[Et2GaAs(SiMe3)2]2、[tBu2GaAs(SiMe3)2]2、
GaSbには、[Et2GaSb(SiMe3)2]2、
InPには、[(Me3SiCH2)2InP(SiMe3)2]2、[R2InP(SiMe3)2]2、[Me2InPtBu2]2、
InSbには、[Me2InSbtBu2]3、[Et2InSb(SiMe3)2]3、[Me2InNEt2]2、[Et2AlAstBu2]2、
AlSbには、[tBu2AlSb(SiMe3)2]2、
GaAsには、[nBu2GaAstBu2]2、[Me2Ga2AstBu2]2、[Et2GaAstBu2]2
II―V半導体シェルの、ME単一源前駆体の非限定的な例は以下である。
Cd3P2には、[MeCdPtBu2]3、Cd[P(SiPh3)2]2、Zn3P2、Zn[P(SiPh3)2]2
IV―VI半導体シェルの、ME単一源前駆体の非限定的な例は以下である。
PbSには、鉛(II)ジチオカルバメート、
PbSeには、鉛(II)セレノカルバメート
【0071】
本発明は、これより、次の非限定的な例を参照し、次の図を参照して実証される。
【図面の簡単な説明】
【0072】
【図1】図1は、界面活性剤で覆われているナノ粒子の概略図であり、界面活性剤頭部基はナノクリスタルに対する強い親和性を有しており、界面活性剤の炭化水素鎖は、ナノクリスタルを溶媒に溶けやすくし、分散させるのに役立っている。
【図2】図2は、a)半導体InPコアからなる粒子、b)半導体InPコア及び半導体ZnSシェルからなる粒子、c)半導体InPコア、異なる半導体材料であるZnSeの緩衝層、及び外側の半導体ZnSシェルからなる粒子の概略図である。
【図3】図3は、分子シードとして[Zn10S4(SPh)16][X]4x=Li+又は(CH3)3NH+を用いたリン化インジウム量子ドット形成の概略図であり、酢酸カドミウム及びトリ―n―オクチルホスフィンセレニドの滴加、そして、カドミウム及びセレン元素源前駆体及び、キャッピング剤としてヘキサデシルアミンを用いた。
【図4】図4は、a)熱アニーリング前の反応混合物からの分離後、b)熱アニーリング後、のInP量子ドットのUV及びPL発光スペクトルを示す。
【図5】図5は、シーディングテンプレートとして用いられることが出来る分子クラスターを図示する。
【図6】図6は、反応混合物からの分離後のInP/ZnS量子ドットのUV及びPL発光スペクトルを示す。
【実施例】
【0073】
全ての合成及び操作は、乾燥無酸素アルゴン又は窒素雰囲気の下で、標準的なシュレンク及びグローブボックス技術を用いて実行した。全ての溶媒は分析用であって、使用前に適当な乾燥剤から精製される(THF、Et2O、トルエン、ヘキサン、ペンタンにはNa/K−ベンゾフェノン、メタノール及びエタノールにはマグネシウム、そしてアセトンには水素化カルシウム)。全ての化学物質は、分析用である。
【0074】
元素分析は、カルロエルバCHNS−O EA1108・元素分析計にて行った。UV−Vis吸収スペクトルは、Heλiosβサーモスペクトロニック上で測定した。光ルミネセンス(PL)スペクトルは、フルオロログ3(FL3-22)光分光器によって励起波長380nmで測定し、スペクトルは、2nmに設定された切れ込みで1秒の統合時間で得るか、又はOcean Optics 2000 USBプローブを用いてその場で測定した。粉末X線回折(PXRD)測定は、モノクロマトのCu−Kα放射線を使用してブルカーAXS D8回折計で行い、試料を平らに取り付け、10°から70°まで0.04°の刻み幅で2.5秒の計数率で走査した。測定は、20°〜60°にわたって、3°の角度で2θ値で、1秒の計測時間で0.04°ずつ視射角発生率検出器を用いて行なった。ナノ粒子の形態及びサイズ分布を観察するため、そして蛍光X線(EDAX)のエネルギー分散方式分析のために、フィリップスCM200透過電子顕微鏡を用いた。TEM及びEDAXのためのサンプルは、トルエンにおけるサンプルの希釈した懸濁液を一滴、銅グリッド(300メッシュ、Agar Scientific)に置くことによって調製した。過剰な溶媒は、室温で乾燥させた。
【0075】
通常の手順
II―VIクラスター、(例えば[HNEt3]4Zn10S4(SPh)16])及び少量の「フィードストック」前駆体即ち、例えばIn(ミリスチン酸塩)3のようなIII族前駆体及びP(TMS)3のようなV族元素前駆体をフィードストック前駆体として用いて、キャッピング剤を含む溶液に加え、それから温度を上昇させ、しばらくの間反応を撹拌した。その結果、III―V粒子形成のセットとなった。
【0076】
両フィードストック前駆体を更に滴加することによって、量子ドットの、PL発光最大の赤方偏移が起こり、それはその場PLプローブによって監視された。更なる前駆体をこの段階で加える場合、PL最大の赤方偏移はこれ以上起こらず、したがって、粒子はこれ以上成長しない。しかしながら、温度を上昇させると(5〜40℃)、PL最大は再度、赤方偏移を起こす。更に前駆体を反応溶液に加えると、再度PL最大は赤方偏移を起こす。従って、続いて反応温度を段々に上昇させるこの前駆体の添加のサイクルは、PL最大ピークが所望の発光状態になるまで繰り返すことが出来る。それから反応をより低い温度に冷却し、更なる期間アニールさせ、その期間の後、II―VIクラスターを組み込んでいるIII−Vナノ粒子を分離することが出来る。
【0077】
分子クラスターの選択
知られているIII―V分子クラスターは少数であるので、III―Vクラスターの代わりにII―VI分子クラスターが用いられる。III―V分子クラスターは、作成するのが困難で、通常、空気及び水分に影響を受けるのに対し、多数のII―VI分子クラスターが簡単な手順によって作成でき、II―VIクラスターは空気及び水分に影響を受けない。従って、III―V粒子は、II―VI分子クラスター上にシードされることができ、それゆえ、初めて、III―Vナノ粒子を成長させるためにIII―V分子クラスターを用いる理由又は利点がないことが、現在理解される。
【0078】
実施例1
InPナノ粒子の調製(赤発光)
60℃のセバシン酸ジ―n―ブチルエステル(200ml)及びミリスチン酸(10g)を、三つ口丸底フラスコに入れ、N2でパージし、その後ZnSクラスター[HNEt3]4Zn10S4(SPh)16](0.94g)を加えた。反応混合物を、それから30分間100℃に加熱し、その後[In2(Ac)3(MA)3](0.25M、12ml)を48ml/hrの速度で電子シリンジポンプを用いて15分間にわたって加えた。その後、同じ添加速度で(TMS)3P(0.25M、12ml)を加えた。添加が完了すると、反応の温度を180℃に上昇させ、更に、[In2(Ac)3(MA)3]及び(TMS)3Pを以下のように添加した。[In2(Ac)3(MA)3](16ml)と、続いて(TMS)3P(16ml)を加え、温度を200℃まで上昇させ、それから更に[In2(Ac)3(MA)3](10ml)を加えて、粒子を必要なサイズまで成長させ、したがって必要な赤い発光となる。それから1時間、温度を200℃で置き、そして160℃に下げ、それから、反応混合物を3日間アニールする。粒子は、アセトニトリルを用いて分離し、遠心分離して、収集した。粒子は5〜60%の量子効率を示した。
【0079】
実施例2
500〜700nmの発光を有するInP量子ドットの調製及びZnSシェルの成長
ジブチルエステル(100ml)及びミリスチン酸(10.0627g)を、三つ口フラスコに入れ、真空下で1時間、70℃でガスを除去した。この期間の後、窒素を導入し、90℃まで温度を上昇させた。ZnS分子クラスター[Et3NH4][Zn10S4(SPh)16](4.7076g)を加えて、混合物を45分間撹拌した。それから温度を100℃に上昇させ、その後、In(MA)3(1M、15ml)、続いて(TMS)3P(1M、15ml)を滴加した。温度を140℃に上昇させながら、反応混合物を攪拌した。140℃で、In(MA)3(1M、35ml)(5分間撹拌する)及び(TMS)3P(1M、35ml)を更に滴加した。それから温度を180℃までゆっくり上昇させ、In(MA)3(1M、55ml)、続いて、(TMS)3P(1M、40ml)を更に滴加した。上記の方法で前駆体を加えることによって、InPのナノ粒子は、発光最大が520nmから700nmまで徐々に増加する状態で成長することができ、それによって、所望の発光最大が得られたときに反応を止め、30分間その温度で撹拌することが出来る。この期間の後、温度を160℃まで下降させ、反応混合物を最長4日間アニールした(反応の温度よりも下の20〜40℃低い温度で)。この段階で紫外線ランプも、アニーリングを促進するために用いた。
【0080】
カニューレ技術によって、脱気した乾燥メタノール(約200ml)を添加してナノ粒子を分離した。沈殿物を沈殿させ、それから、メタノールを、フィルタスティックを用いてカニューレを介して除去した。脱気した乾燥クロロホルム(約10ml)を、固体を洗浄するために加えた。固体は真空下で1日乾燥させた。これにより、5.60gのInPコアナノ粒子を製造した。元素分析:最大PL=630nm、FWHM=70nm。
【0081】
後処理
上記において調製したInP量子ドットの量子収量を、薄いHF酸での洗浄によって増加させた。該ドットは、無水脱気クロロホルム(〜270ml)において溶解した。50mlの分量を取ってプラスチックフラスコに入れ、窒素で洗浄した。プラスチックシリンジを用いて、3mlの60%w/wHFを水に加え、脱気THF(17ml)に加えることで、HF溶液を調製した。HFを、InPドットに5時間にわたって滴加した。添加完了後、溶液を終夜撹拌した。塩化カルシウム水溶液で抽出し、エッチングされたInPドットを乾燥させることによって過剰HFを除去した。乾燥ドットは、将来使用するために、50mlのクロロホルムにおいて、再分散させた。最大567nm、FWHM60nm。この段階のコア材料の量子効率は25〜90%である。
【0082】
ZnSシェルの成長
HFエッチングしたInPコア粒子の20mlの分量を、三つ口フラスコにおいて乾燥させた。1.3gのミリスチン酸及び20mlのセバシン酸ジ―n―ブチルエステルを加えて、30分間脱気した。溶液を200℃まで加熱し、それから、1.2gの無水酢酸亜鉛を加え、そして、2mlの1M(TMS)2Sを(7.93ml/hrの速度で)滴加した。添加完了後に溶液を撹拌した。溶液を200℃で1時間保ち、それから室温に冷却した。粒子は、40mlの無水脱気メタノールを加えることによって分離し、遠心分離した。上澄み液体は処分し、残りの固体に、30mlの無水脱気ヘキサンを加えた。溶液を5時間沈殿させ、それから再度遠心分離した。上澄み液体を収集し、残りの固体を廃棄した。PL発光ピーク最大値=535nm、FWHM=65nm。この段階でナノ粒子コア/シェル材料の量子効率は35〜90%であった。
【0083】
実施例3
ミリスチン酸インジウム0.25Mの調製
In(Ac)3+1.5CH3(CH2)12COOH→In(Ac)1.5(OOC(CH2)12CH3)1.5
ミリスチン酸(70.798g、0.3100モル、1.5eqv)、酢酸インジウム(58.392g、0.200モル、1eqv)及びセバシン酸ジブチル(626.4ml、568.3g、1.864モル)を、1Lの丸底フラスコに入れ、真空下で二昼一夜、118℃まで加熱した。氷/塩/アセトンによって冷却し、プレトラップを設定し、これと、高真空溶媒トラップとで周期的に調べて、ミリスチン酸インジウムの合成の間、蓄積した凍結した酢酸を取り除いた。
【0084】
TMSP 0.25Mの調製
シュレンクチューブ及びシリンジを、終夜オーブン乾燥した。シュレンクチューブをそれから真空ラインへ動かして冷却した。セバシン酸ジブチルを真空下で脱気し、1時間にわたって穏やかに加熱し、その後92.74ml(0.294モル)を窒素下でシュレンクチューブに移動させた。TMSP(7.26ml、0.025モル)を窒素下で移動させ、セバシン酸ジブチルと混合した。
【0085】
グリーン発光を有するInP量子ドットの調製及びZnS/ZnOシェルの成長
ミリスチン酸(140.000g、0.444モル)及びセバシン酸ジブチル(2800ml)を、60℃〜90℃の温度で、高真空下で1時間脱気した。系をアルゴンでパージし、それから[Et3NH4][Zn10S4(SPh)16]クラスター(13.16g)をすぐに加えた。100℃の温度に到達するまで、反応混合物を15分間加熱し、この段階で、反応混合物を、それから30分間100℃で維持した。クラスターは、完全に溶解せず、白い懸濁液を形成した。
【0086】
ミリスチン酸インジウム(168ml、0.25M)の1回目の添加を1秒に3滴の速度で始め、そして温度は終始100℃に維持した。ミリスチン酸溶液の添加完了後、反応混合物を100℃で5分間撹拌した。それからTMSP(168ml、0.25M)を1秒に2滴の速度で加え、ここでも慎重に温度を監視した。添加が完了するとすぐに、反応混合物の温度を出来るだけ急速に180℃に上昇させ、絶対に超過しないよう注意した。180℃に達するとすぐに、ここでも1秒に3滴の速度で、ミリスチン酸インジウム(224ml、0.25M)を更に添加した。それから反応混合物を30分間180℃で保持した。それから、反応を160℃に冷却させ、温度を150〜160℃に維持しながら、3日間アニールした。
【0087】
アニール後、混合物を室温に冷却させ、その後500mlの分量を大きなビーカーに注入し、アセトニトリルによって沈殿させた。それから、凝集された溶液を遠心分離機チューブに移動させ、ペレットが形成されるまで遠心分離した。透明な上澄みは廃棄し、オレンジ沈殿物を最小量のクロロホルムに再溶解させ、終夜空気中に放置した。
【0088】
後処理
ミリスチン酸におけるプレエッチング
クロロホルムを回転蒸発器において除去し、固体はセバシン酸ジブチルエステル(600ml)に再溶解させた。それからミリスチン酸(80g)を加え、混合物を6時間140℃まで加熱した。混合物を冷却させ、上記と同様にアセトニトリルで沈殿させた。
【0089】
HFエッチング
クロロホルム(1750ml)にペレットを再溶解させ、この溶液をHFエッチングに用いた。500Wのタングステン球の前に溶液を置き、8mlのテトラヒドロフラン内の2mlのHF(48%)の溶液を撹拌しながらゆっくり加えた。溶液の光学発光スペクトルを光学手プローブによって監視し、それは強度が増すにつれて徐々に赤方偏移を始めた。記録している波長が青方偏移を始めた時点までHF溶液を加え、それから、HF/THF/CHCI3を空気で吹き飛ばした。結果として生じるペーストをアセトンで洗浄し、アセトニトリルで沈殿させ、それからヘキサンで洗浄して再度アセトニトリルに沈殿させた。それから沈殿物を最小量のセバシン酸ジブチルエステルに溶解させ、脱気してアルゴン下に置いた。
【0090】
ZnS/ZnOシェルの成長
上記のように形成されるInPナノ粒子を室温で1時間脱気した。カルボキシ―PEG―メチルエーテル(125.82g)も100℃で高真空下で1時間脱気し、それから室温に冷却させた。酢酸亜鉛(34.903g、0.1902モル)及び脱気InPナノ粒子を、冷却したエーテルに加え、それから5分間真空下に置いて、3回アルゴンでパージした。InPナノ粒子を含む溶液を180℃に加熱し、脱気オクタンチオール(8.3111g、0.0568モル)を120℃で急速に注入した。反応混合物を180℃で1時間保持し、そしてオクタノール(24.93g、30.04ml、0.1914モル)を1秒に1滴の速度で加えた。それから反応混合物を30分間加熱して180℃に戻し、その後室温に冷却してからヘキサン(350ml)を加えた。ゲル状の沈殿物から上部層は別の容器へ移し、別の容器へ移した溶液にアセトニトリル(700ml)を加えた。それから混合物を分液漏斗に加え、沈殿物が凝集し始めるまでアセトンを加えた。塊の周辺から溶液を排出し、遠心分離して全ての生成物を回収した。最後に塊及び遠心分離された沈殿物を脱気クロロホルムに溶解した。
【0091】
実施例4
グリーン発光を有するInP量子ドットの調製
例3において説明する手順に従って、ミリスチン酸インジウム及びTMSPを調製した。
【0092】
ミリスチン酸(70.000g、0.222モル)及びセバシン酸ジブチル(1400ml)を、60〜90℃の温度で1時間高真空下で脱気した。系をアルゴンでパージし、それから[Et3NH4][Zn10S4(SPh)16]クラスター(6.58g)をすぐに加えた。それから反応混合物を100℃の温度に到達するまで15分間加熱し、この段階で、反応混合物をそれから30分間100℃で維持した。クラスターは完全に溶解せず、白い懸濁液を形成した。
【0093】
ミリスチン酸インジウム(84ml、0.25M)の最初の添加を1秒に3滴の速度で始め、温度は終始100℃に維持した。ミリスチン酸溶液の添加完了後、反応混合物を5分間100℃で撹拌した。それからTMSP(84ml、0.25M)を1秒に2滴の速度で加え、ここでも慎重に温度を監視した。添加が完了するとすぐに、反応混合物の温度を出来るだけ急速に180℃に上昇させ、絶対に超過しないよう注意した。180℃に達するとすぐに、ここでも1秒に3滴の速度で、ミリスチン酸インジウム(112ml、0.25M)を更に添加した。それから反応混合物を30分間180℃で保持した。それから反応を160℃に冷却させ、温度を150〜160℃に維持しながら3日間アニールした。
【0094】
アニール後、混合物を室温に冷却させ、その後500mlの分量を大きなビーカーに注入し、アセトニトリルで沈殿させた。それから凝集した溶液を遠心分離機チューブに移動させ、ペレットが形成されるまで遠心分離した。透明な上澄みは廃棄し、オレンジ沈殿物を最小量のクロロホルムに再溶解させ、終夜空気中に放置した。
【0095】
後処理
ミリスチン酸におけるプレエッチング
クロロホルムを回転蒸発器において除去し、固体はセバシン酸ジブチルエステル(300ml)に再溶解させた。それからミリスチン酸(40g)を加え、混合物を6時間140℃まで加熱した。混合物を冷却させ、上記と同様にアセトニトリルで沈殿させた。
【0096】
HFエッチング
クロロホルム(900ml)にペレットを再溶解させ、この溶液をHFエッチングに用いた。500Wのタングステン球の前に溶液を置き、8mlのテトラヒドロフラン内の2mlのHF(48%)の溶液を撹拌しながらゆっくり加えた。溶液の光学発光スペクトルを光学手プローブによって監視し、それは強度が増すにつれて徐々に赤方偏移を始めた。記録している波長が青方偏移を始めた時点までHF溶液を加え、それから、HF/THF/CHCI3を空気で吹き飛ばした。結果として生じるペーストをアセトンで洗浄し、アセトニトリルで沈殿させ、それからヘキサンで洗浄して、再度アセトニトリルにおいて沈殿させた。それから沈殿物を最小量のセバシン酸ジブチルエステルに溶解させ、脱気してアルゴン下に置いた。
【0097】
実施例5
赤発光を有するInP量子ドットの調製
上記の手順に従って、ミリスチン酸インジウム及びTMSPを調製した。
【0098】
ミリスチン酸(120.000g、0.526モル)及びセバシン酸ジブチル(2400ml)を60〜90℃の温度で1時間高真空下で脱気した。系をアルゴンでパージし、それから[Et3NH4][Zn10S4(SPh)16]クラスター(11.28g)をすぐに加えた。それから反応混合物を温度が100℃に到達するまで15分間加熱し、この段階で、反応混合物をそれから30分間100℃で維持した。クラスターは完全に溶解せず、白い懸濁液を形成した。
【0099】
ミリスチン酸インジウム(144ml、0.25M)の最初の添加を1秒に3滴の速度で始め、温度は終始100℃に維持した。ミリスチン酸溶液の添加完了後、反応混合物を、5分間100℃で撹拌した。それからTMSP(144ml、0.25M)を1秒に2滴の速度で加え、ここでも慎重に温度を監視した。添加が完了するとすぐに、反応混合物の温度を出来るだけ急速に180℃に上昇させ、絶対に超過しないよう注意した(温度上昇には30分強しかかからなかった)。180℃に達するとすぐに、ここでも1秒に3滴の速度で、ミリスチン酸インジウム(192ml、0.25M)の2回目の添加を行った。再度反応を5分間180℃で保持し、その後TMSP(192ml、0.25M)の2回目の添加を1秒に2滴の速度で行った。添加が完了した直後に温度を200℃まで上昇させ、ここでも超過しないよう注意した。そしてミリスチン酸インジウム(120ml、0.25M)の最後の分量を滴加した。それから反応を200℃で60分間保持し、その後160℃に冷却した。それから、140〜160℃の間で温度を維持しながら反応混合物を3日間アニールした。
【0100】
アニール後、混合物を室温に冷却させ、その後500mlの分量を大きなビーカーに注入し、アセトニトリル(600ml)で沈殿させた。それから凝集した溶液を遠心分離機チューブに移動させ、4000rpmで5分間遠心分離した。透明な上澄みは廃棄し、赤い沈殿物をクロロホルムに再溶解させ、終夜空気中に放置した。
【0101】
後処理
ミリスチン酸におけるプレエッチング
クロロホルムを回転蒸発器において除去し、固体はセバシン酸ジブチルエステル(600ml)に再溶解させた。それからミリスチン酸(80g)を加え、混合物を6時間140℃まで加熱した。混合物を冷却させ、上記と同様にアセトニトリルで沈殿させた。
【0102】
HFエッチング
クロロホルム(1750ml)にペレットを再溶解させ、この溶液をHFエッチングに用いた。500Wのタングステン球の前に溶液を置き、8mlのテトラヒドロフラン内の2mlのHF(48%)の溶液を撹拌しながらゆっくり加えた。溶液の光学発光スペクトルを光学手プローブによって監視し、それは強度が増すにつれて徐々に赤方偏移を始めた。記録している波長が青方偏移を始めた時点までHF溶液を加え、それから、HF/THF/CHCI3を空気で吹き飛ばした。結果として生じるペーストをアセトンで洗浄し、アセトニトリルで沈殿させ、それからヘキサンで洗浄してもう一度アセトニトリルで沈殿させた。それから沈殿物を最小量のセバシン酸ジブチルエステルに溶解させ、脱気してアルゴン下に置いた。
【0103】
実施例6
赤発光を有するInP量子ドットの調製
ミリスチン酸(60.000g、0.263モル)及びセバシン酸ジブチル(1200ml)を60〜90℃の温度で1時間高真空下で脱気した。系をアルゴンでパージし、それから[Et3NH4][Zn10S4(SPh)16]クラスター(5.64g)をすぐに加えた。それから反応混合物を100℃の温度に到達するまで15分間加熱し、この段階で、反応混合物をそれから30分間100℃で維持した。クラスターは完全に溶解せず、白い懸濁液を形成した。
【0104】
ミリスチン酸インジウム(72ml、0.25M)の最初の添加を1秒に3滴の速度で始め、温度は終始100℃に維持した。ミリスチン酸溶液の添加完了後、反応混合物を5分間100℃で撹拌した。それからTMSP(72ml、0.25M)を1秒に2滴の速度で加え、ここでも慎重に温度を監視した。添加が完了するとすぐに、反応混合物の温度を出来るだけ急速に180℃に上昇させ、絶対に超過しないよう注意した(温度上昇には30分強しかかからなかった)。180℃に達するとすぐに、ここでも1秒に3滴の速度で、ミリスチン酸インジウム(96ml、0.25M)の2回目の添加を行った。再度反応を5分間180℃で保持し、その後TMSP(96ml、0.25M)の2回目の添加を1秒に2滴の速度で行った。添加が完了した直後に、温度を200℃まで上昇させ、ここでも超過しないよう注意した。それから反応を200℃で60分間保持し、その後160℃に冷却した。それから、反応混合物を140〜160℃の間で3日間アニールした。
【0105】
後処理
ミリスチン酸におけるプレエッチング
クロロホルムを回転蒸発器において除去し、固体はセバシン酸ジブチルエステル(300ml)に再溶解させた。それからミリスチン酸(40g)を加え、混合物を6時間140℃まで加熱した。混合物を冷却させ、上記と同様にアセトニトリルで沈殿させた。
【0106】
HFエッチング
クロロホルム(900ml)にペレットを再溶解させ、この溶液をHFエッチングに用いた。500Wのタングステン球の前に溶液を置き、8mlのテトラヒドロフラン内の2mlのHF(48%)の溶液を撹拌しながらゆっくり加えた。溶液の光学発光スペクトルを光学手プローブによって監視し、それは強度が増すにつれて徐々に赤方偏移を始めた。記録している波長が青方偏移を始めた時点までHF溶液を加え、それからHF/THF/CHCI3を空気で吹き飛ばした。結果として生じるペーストをアセトンで洗浄し、アセトニトリルで沈殿させ、それからヘキサンで洗浄してもう一度アセトニトリルで沈殿させた。それから沈殿物を最小量のセバシン酸ジブチルエステルに溶解させ、脱気してアルゴン下に置いた。
【0107】
実施例7
InMA3前駆体の調製

三つ口フラスコ(1リットル)において、In(Ac)3(100g)及びミリスチン酸(313g)を窒素下で2時間140℃まで加熱した。反応混合物は80℃に冷却し、反応フラスコを蒸留装置に取り付けた。反応混合物は5時間ゆるやかな真空下に置き、その後60℃で窒素下に終夜置いた。(反応のこの第1ステップにおいて60mLの酢酸を収集した)。1リットルの三角フラスコに分析用アセトン(400mL)を加えて静かに撹拌した。反応混合物を途切れなく撹拌しながらこのアセトンにゆっくり加えた。反応混合物を15分間撹拌し、それから放置した。底部に黄色の固体が沈殿し、過剰溶媒を別の容器へ移した。溶媒の変色が最小限で固体が白くなるまで、黄色の固体をアセトンで3回洗浄した。固体をブフナー漏斗及びフラスコ装置を用いて濾過し、それから真空下に終夜デシケータにおいて乾燥させた。最終生成物:InMA3乾燥粉末の質量=226.82g。
【0108】
InMA3(1モルの)前駆体溶液の調製
コンデンサ及び温度計を配備した250mLの三つ口フラスコにおいて、InMA3(100g)及びセバシン酸ジブチル(30mL)を100℃で1時間30分間完全に脱気した。溶液が無色になるまで反応温度を維持した。InMA3溶液は室温で凝固するので、用いるときには、およそ60℃の温度に保ち、しっかりと撹拌しなければならない。
【0109】
(TMSi)3P(1モルの)前駆体溶液の調製
250mLの三つ口フラスコにおいて、セバシン酸ジブチル(100mL)を100℃で2時間30分間脱気した。オーブン乾燥した大きなシュレンクチューブにおいて、セバシン酸ジブチル(71mL)、続いて(TMSi)3P(29mL)を窒素の強い流れの下で加えた。
【0110】
グリーン発光を有するInP量子ドットの調製及びZnS/ZnOシェルの成長
セバシン酸ジブチル(100mL)及びミリスチン酸(5.125g)を、コンデンサ及び温度計を配備した250mLの三つ口フラスコにおいて真空下で100℃で1時間脱気した。[Et3NH4]Zn10S4(SPh)16クラスター(0.47g)を窒素の強い流れの下でしっかりと撹拌しながら80℃で加えた。反応混合物を30分間しっかりと撹拌した。それから反応温度を100℃に上昇させた。
【0111】
InPナノ粒子のシーディングは、InMA3溶液(1M、3mL)をゆっくりと滴加することによって開始され、それは、それから5分間しっかりと撹拌した。5分間の後、(TMSi)3P(1M、3mL)をゆっくりと滴加した。それから反応温度を160℃に上昇させた。160℃でInMA3(1M、4mL)の2回目の滴加をゆっくり行った。反応混合物を5分間撹拌し、その後に(TMSi)3P(1M、4mL)の2回目の添加を行った。好ましくは、反応溶液はすべての添加の間しっかりと撹拌されるべきである。反応温度を190℃に上昇させ、1時間撹拌した。それから反応温度を160℃まで下降させ、そして反応混合物を3日間この温度でアニールした。
【0112】
光ルミネセンスを測定するために5nmの切れ込みを用いてもよい。3日間の後、反応混合物を室温に冷却した。無水アセトニトリル(100mL)の添加によってInPナノ粒子を窒素下で分離した。固体を遠心分離を用いて分離し、CHCl3(200mL)に再溶解した。
【0113】
後処理
HFエッチング
2mLの水性HF(58〜62%wt.溶液)とTHF(8mL分析用)とを組み合わせることによってHF溶液を調製した。250mLの三角フラスコで、HF(全体で1.4mL)を、上記のように調製されるクロロホルム中に分散しているInPナノ粒子に加えた。空気中でしっかりと撹拌しながら7〜8時間にわたってHF溶液を0.2mL滴加した。反応混合物に、サイズによってカットオフ波長を有するカットオフフィルタを通過した光の照射をした。光源として500Wのキセノン灯を用いた。
【0114】
エッチングの処理は、エッチングするナノ粒子の特定のバッチの規模に応じて、およそ10〜15時間後に終了した。CHCl3の蒸発によって、高発光性のHFエッチングされたInPナノ粒子を反応混合物から分離した。それからHFエッチングされたInPナノ粒子をアセトン(10〜15mL)で洗浄し、続いてアセトニトリル(100mL)を加えた。固体を遠心分離によって分離し、そしてアセトン(5mL)で2回目の洗浄を行い、その後セバシン酸ジブチル(バッチの規模によっておよそ20mL)に分散させた。効果的にエッチングされたInPナノ粒子は15〜30%のPL量子効率を有する強い発光を示した。
【0115】
この方法では、535nm(±5nm)及びFWHM<70nmで発光を有する約2グラムのInP量子ドットが生じた。
【0116】
ZnS/ZnOシェルの成長
セバシン酸ジブチル(15mL)及びミリスチン酸(5.2g)を、コンデンサに取り付けた温度計を具えた250mLの枝付き三つ口フラスコにおいて、1時間30分間100℃で脱気した。上記の様に調製されたInPナノ粒子(2.32g)を、別のフラスコにおいて1時間脱気した。セバシン酸ジブチル(15mL)及びミリスチン酸(5.2g)の反応混合物をおよそ60℃まで冷却し、そしてInPナノ粒子を加えた。反応混合物を更に45分間脱気した。
【0117】
窒素の強い流れの下で、しっかりと撹拌しながら、枝から一部に酢酸亜鉛(2.8gを)を加えた。反応混合物を15分間脱気し、脱気の間N2で数回洗浄した。反応温度を120℃に上昇させた。
【0118】
オクタンチオール(0.82mL)を120℃で速く加え、温度は最高220℃まで傾斜した。ZnSシェルを形成するために温度を1時間30分間220℃で保持した。それから反応溶液を180℃まで冷却した。
【0119】
180℃で1―オクタノール(2.2mL)をゆっくり滴加した。添加完了後、溶液を190℃で30分間撹拌し、ZnOシェルを形成した。
【0120】
反応混合物を室温に冷却し、終夜撹拌した。InP/ZnS/ZnOコア―シェル粒子を、無水アセトニトリル(80mL)を有するN2下で分離し、遠心分離によって収集した。粒子をトルエン(20mL)に分散させて、アセトニトリルで再沈殿させ、続いて遠心分離した。それから粒子をトルエン(20mL)に再分散させた。最終的な溶液を遠心分離し、不溶性物質を除去した。最終生成物:質量=1.62g、PL最大=521nm、FWHM=61nm、PLQY=64%。
【0121】
実施例8
赤発光を有するInP量子ドットの調製
セバシン酸ジブチル(100mL)及びミリスチン酸(5.125g)を、コンデンサ及び温度計を備えた250mLの三つ口フラスコにおいて真空下で、100℃で1時間脱気した。[Et3NH4]Zn10S4(SPh)16クラスター(0.47g)を窒素の強い流れの下で、しっかりと撹拌しながら80℃で加えた。反応混合物を、30分間しっかりと撹拌した。反応温度を100℃に上昇させた。
【0122】
InPナノ粒子のシーディングは、InMA3溶液(1M、3mL)をゆっくりと滴加することによって開始され、それは、それから5分間しっかりと撹拌した。5分間の後、(TMSi)3P(1M、3mL)をゆっくり滴加した。反応温度を160℃に上昇させた。160℃でInMA3(1M、4mL)の2回目の滴加をゆっくりと行った。反応混合物を5分間撹拌し、その後に(TMSi)3P(1M、4mL)の2回目の添加を行った。反応温度は180℃に上昇させ、InMA3溶液(1M、3mL)、続いて(TMSi)3P(1M、2.25mL)の3回目の添加を行った。好ましくは、反応溶液はすべての添加の間しっかりと撹拌されるべきである。反応温度を200℃に上昇させ、1時間撹拌した。それから反応温度を170℃まで下降させ、そして反応混合物を3日間この温度でアニールした。
【0123】
光ルミネセンスを測定するために5nmの切れ込みを用いてもよい。3日間の後、反応混合物を室温に冷却した。無水アセトニトリル(100mL)の添加によって、InPコアを窒素下で分離した。固体を遠心分離を用いて分離し、CHCl3(200mL)に再溶解した。
【0124】
後処理
HFエッチング
4mLの水性HF(58〜62%wt.溶液)とTHF(16mL分析用)とを組み合わせることによってHF溶液を調製した。250mLの三角フラスコで、HF(全体で5mL)を、クロロホルムにおいて分散しているInPナノ粒子に加えた。空気中でしっかりと撹拌しながら、5〜8時間にわたってHF溶液を0.2mL滴加した。反応混合物に、サイズによってカットオフ波長を有するカットオフフィルタを通過する光の照射をした。光源として500Wのキセノン灯を用いた。
【0125】
エッチングの処理は、エッチングするナノ粒子の特定のバッチの規模に応じて、およそ10〜15時間後に終了する。CHCl3の蒸発によって、高発光性のHFエッチングされたInPナノ粒子を反応混合物から分離した。それからHFエッチングされたInPナノ粒子をアセトン(10〜15mL)で洗浄し、続いてアセトニトリル(100mL)を加える。固体を遠心分離によって分離し、そしてアセトン(5mL)で2回目の洗浄を行い、セバシン酸ジブチル(バッチの規模によっておよそ20mL)に分散させた。効果的にエッチングされたInPナノ粒子は、15〜30%のPL量子効率を有する強い発光を示す。
【0126】
この方法では、610nm(±5nm)及びFWHM<90nmで発光を有する約2グラムのInP量子ドットが生じた。
【0127】
ZnS/ZnOシェルの成長
セバシン酸ジブチル(15mL)及びミリスチン酸(6.71g)を、コンデンサに取り付けた温度計を備えた250mLの枝付き三つ口フラスコにおいて1時間30分間100℃で脱気した。上記の様に調製されたInPナノ粒子(2.58g)を別のフラスコにおいて1時間脱気した。セバシン酸ジブチル(15mL)及びミリスチン酸(6.71g)の反応混合物をおよそ60℃まで冷却し、そしてInPナノ粒子を加えた。反応混合物を更に45分間脱気した。
【0128】
窒素の強い流れ下でしっかりと撹拌しながら、枝から一部に酢酸亜鉛(3.61g)を加えた。反応混合物を15分間脱気し、脱気の間N2で数回洗浄した。反応温度を120℃に上昇させた。
【0129】
オクタンチオール(1.1mL)を120℃で速く加え、温度は最高220℃まで傾斜した。ZnSシェルを形成するために温度を1時間30分間220℃で保持した。それから反応溶液を180℃まで冷却した。
【0130】
180℃で1―オクタノール(2.84mL)をゆっくり滴加した。添加完了後、溶液を190℃で30分間撹拌し、ZnOシェルを形成する。
【0131】
反応混合物を室温に冷却し、終夜撹拌した。InP/ZnS/ZnOコア―シェル粒子を、無水アセトニトリル(80mL)を有するN2下で分離し、遠心分離によって収集した。粒子をトルエン(30mL)に分散させてアセトニトリルで再沈殿させ、続いて遠心分離した。それから粒子をトルエン(50mL)に再分散させた。最終的な溶液を遠心分離し、不溶性物質を除去した。最終生成物:質量=3.2g、PL最大=605nm、FWHM=93nm、PLQY=61%。
【0132】
【非特許文献1】Murray, C. B.; Norris, D. J.; Bawendi, M. G. J. Am. Chem. Soc. 1993, 115, 8706。
【非特許文献2】LOver, T.; Bowmaker, G. A.; Seakins, J. M.; Cooney, R. P.; Henderson, W. J. Mater. Chem., 1997, 7(4), 647。
【非特許文献3】Cumberland, S. L.; Hanif, K. M.; Javier, A.; Khitov, K. A.; Strouse, G. F.; Woessner, S. M.; Yun, C. S. Chem. Mater. 2002, 14, 1576。
【技術分野】
【0001】
本発明は半導体ナノ粒子に関する。
【背景技術】
【0002】
多くの場合量子ドット又はナノ結晶と呼ばれる、例えば2〜50nmの範囲の寸法の粒子からなる化合物半導体の調製及び特徴づけに対して多くの関心が持たれてきた。これらの研究は、これらの物質のサイズ調整可能な電子的特性により主になされ、それは、光学的及び電子的装置などの多くの商業的応用において、又、多様な範囲に及ぶ他の応用において利用され得る。その範囲は、多くの新たな新進の応用があるが、生物学的ラベリング、太陽電池、触媒作用、生物学的イメージング、発光ダイオード、一般的な空間照明及びエレクトロルミネセンス・ディスプレイ、フォトルミネセンス・ディスプレイのように広範に亘る。
【0003】
最も多く研究されてきた半導体材料は、カルコゲニドII−VI(例えば第12族〜第16族)材料、例えばZnS、ZnSe、CdS、CdSe、CdTeである。最も顕著には、そのスペクトルの可視域上の光学調整能力のため、CdSeが非常に研究されている。いくつかの以前の例が文献において見られるが、より最近になって、再現可能な方法が、「ボトムアップ」技術によって開発され、それによって、粒子は原子毎に、「湿式」化学法を用いて調製される。
【0004】
共に個々の半導体ナノ粒子のサイズに関係する2つの根本的要因が、これらの粒子の独特の特性に関与している。第1は、大きい表面積対体積比である。粒子がより小さくなるにつれて、内部の原子に対する表面原子の数の比率が増加する。このため、材料全体の特性において表面特性が重要な役割を果たすことになる。第2の要因は、半導体ナノ粒子は、サイズと共に材料の電子的特性が変化すること、更に、粒子のサイズが減少するにつれて、量子閉じ込め効果のためバンドギャップが徐々により大きくなることである。この効果は、『箱の中の電子』の閉じ込めによって、対応するバルク半導体材料における連続バンドではなく、原子及び分子において観察されるのと類似の離散的エネルギー準位が生じている結果である。従って、半導体ナノ粒子においては、第1励起子遷移より大きなエネルギーを有する電磁放射(光子)の吸収によって生じた「電子及び正孔」は、物理パラメータのため、対応する巨視的結晶質材料においてより接近しているので、クーロンの相互作用は無視できない。これは狭バンド幅放出につながり、それは粒子サイズ及び組成に依存する。このように、量子ドットは対応する巨視的結晶質材料より高い運動エネルギーを有するので、第1の励起子遷移(バンドギャップ)はエネルギーにおいて粒径の減少と共に増加する。
【発明の開示】
【発明が解決しようとする課題】
【0005】
単一半導体材料と外側の有機不動態化層からなる単一コアナノ粒子は、欠陥で起こっている電子正孔再結合、及び非放射電子正孔再結合の原因になるナノ粒子表面上のダングリングボンドのため、量子効率が比較的低くなる可能性がある。
【0006】
欠陥及びダングリングボンドを除去する1つの方法は、バンドギャップがより広く、コア材料との格子不整合が小さい第2の無機材料をコア粒子の表面にエピタキシャルに成長させて、「コアシェル」粒子を生成することである。コアシェル粒子は、コアに閉じ込められたあらゆるキャリアを、非放射再結合の中心として働くおそれのある表面状態から離す。1つの例は、CdSeコアの表面に成長させてCdSe/ZnSコア/シェルナノ粒子を作成するZnSである。
【0007】
もう一つの方法は、量子ドット−量子井戸構造のような、「電子正孔」一対が単一のシェル層に完全に閉じ込められるコア/マルチシェル構造を調製することである。ここで、コアはバンドギャップの広い材料で出来ており、バンドギャップがより狭い材料の薄いシェルが続き、そして更なる広いバンドギャップ層によって覆われる。例えばわずかのHgSの単層を堆積させるためにコアナノ結晶の表面上でCdの代替にHgを使用して成長するCdS/HgS/CdSである。結果として生じる構造は、HgS層への光励起キャリアの閉じ込めを明らかに示した。
【0008】
如何なるコア、コアシェル又はコアマルチシェルナノ粒子においても、最終無機表面原子の周りの配位は、反応性の高い完全に配位されていない原子「ダングリングボンド」が粒子の表面上にあって不完全であり、これは粒子の凝集につながることがある。この問題は、保護用有機基で「裸の」表面原子を不動態化(キャッピング)することによって解決される。
【0009】
有機材料又はシース材料の最外側層(キャッピング剤)は、粒子凝集を抑制するのに役立ち、更にナノ粒子を、それらを囲んでいる化学環境から保護すると共に、他の無機、有機、又は生物学的材料との化学結合の手段を提供する。多くの場合、キャッピング剤は、ナノ粒子の作成が行われる溶媒であり、ルイス塩基化合物又は、不活性溶媒(例えば炭化水素)において薄められたルイス塩基化合物から成り、それによってナノ粒子の表面に対して供与型配位が可能である孤立電子対がある。
【0010】
高品質な半導体ナノ粒子の合成に関わる重要な問題は、粒子の均一性、サイズ分布、量子効率及び商業的応用におけるそれらの長期の化学的及び光安定性の使用である。初期のルートは従来のコロイド水性化学を利用したが、より最近の方法は、有機金属化合物を用いたナノ結晶子の動力学的に制御された沈殿を含んでいる。より最近の方法のほとんどは、Murray、Norris及びBawendiによって説明された当初の「核生成及び成長」方法(非特許文献1)に基づくが、当初用いられた有機金属前駆体以外のもの、例えば、酸化物(例えばCdO)、炭酸塩(例えばMCO3)、アセテート(例えばM(CH3CO2))及びアセチルアセトネート(例えばM[CH3COOCH=C(C−)CH3]2)(例えばM=Cd又はZn)を用いている。
【0011】
Murrayら(非特許文献1)は当初、金属アルキル(R2M)、M=Cd、Zn、Te、R=Me、Etの有機金属溶液、及びトリ−n−オクチルホスフィン(TOP)に溶解させたトリ−n−オクチルホスフィンスルフィド/セレニド(TOPS/Se)を用いた。これらの前駆体溶液を、調製されている材料に応じて温度範囲120〜400℃の熱いトリ−n−オクチルホスフィンオキシド(TOPO)に注入する。これによって、II−VI材料のTOPOコート/キャップ半導体ナノ粒子が製造される。粒子のサイズは、温度、キャッピング剤、用いられる前駆体の濃度及び合成が行なわれる延べ時間によって制御され、より高い温度、より高い前駆体濃度及び長期にわたる反応時間でより大きい粒子が得られる。この有機金属のルートは、よりよい単分散度及び高い粒子結晶化度を含む他の合成方法に勝る効果がある。上述の如く、この方法の多くのバリエーションが現在文献に見られ、単分散度及び量子収量の両方について高品質なコア及びコアシェルナノ粒子を日常的に生じさせる。
【0012】
単一源前駆体は、他の化合物半導体ナノ粒子と同様に、II−VIの半導体ナノ粒子材料の合成においても役立つということが分かった。ビス(ジアルキルジチオ/ジセレノカルバメート)カドミウム(II)/亜鉛(II)化合物、M(E2CNR2)2(M=Zn又はCd、E=S又はSe及びR=アルキル)は同様の『ワンポット』合成法において用いられてきて、それは、前駆体をトリ−n−オクチルホスフィン(TOP)に溶解させてから200℃を超える熱いトリ−n−オクチルホスフィンオキシド/トリ−n−オクチルホスフィン(TOPO/TOP)へ急速に注入することを含んでいた。
【0013】
根本的に、これらの製法は全て、高温での粒子の核生成に続いて低温での粒子成長という原理に依存している。更に言えば、2〜10nmの範囲のナノ粒子の単分散集合を有するため、ナノ粒子の核生成は、ナノ粒子の成長から適切に分離しなければならない。これは、一方又は両方の前駆体のより低温の溶液を、粒子の核生成を起こす、より高温の配位性溶媒(存在しない場合、他方の前駆体を含有する)へ急速に注入することによって達成される。注入時に突然それより冷たい溶液を添加することで、その後反応温度が低下し(添加する溶液の容量は通常全溶液の約1/3である)、更なる核生成が阻害される。粒子成長は、(用いる前駆体によって面触媒過程であるか又はオストワルド熟成を経て)その低下した温度で続き、このように核生成と成長が分離され、狭いナノ粒子のサイズ分布が得られる。この方法は、反応の全体にわたって合理的に均一温度を保ちながら1つの溶液を急速に他のものに添加することが出来る小規模合成でよく機能する。しかしながら、商業的応用において必要とされる、大容積の溶液同士を互いに急速に注入する必要がある、より大きな調製規模のとき、反応混合物中に著しい温度差が生じることがあり、これがその後、容認出来ない程の大きな粒子サイズ分布につながることがある。
【0014】
Cooney及び共同研究者ら(非特許文献2)は、II−VI分子クラスター[S4Cd10(SPh)16][Me3NH]4を用いて、CdSのII−VIナノ粒子を製造した。ここでヨウ素による表面キャッピングSPh−配位子の酸化も含まれた。この調製ルートでは、II−VIクラスターの大部分がイオンへ細分化され、これらが残りのII−VI([S4Cd10(SPh)16]4−)クラスターによって消費され、これらが後にCdSのII−VIナノ粒子へと成長する。
【0015】
Strouse及び共同研究者ら(非特許文献3)は、II−VIクラスターを用いた同様の合成手法を用いて、II−VIナノ粒子を成長させたが、化学剤ではなく熱分解(分散熱温度上昇)を採用して粒子成長を起こした。更に言えば、単一源前駆体[M10Se4(SPh)16][X]4、X=Li+又は(CH3)3NH+、M=Cd又はZnを熱分解し、それによって幾つかのクラスターの細分化を起こし、続いて、M及びSe自由イオンのスカベンジングから粒子成長させるか、又は、単に、クラスターが凝集して、最初は、より大きいクラスター、そして後に小さいナノ粒子、そして最終的に、より大きい粒子を調製した。
【0016】
Cooney(非特許文献2)の方法もStrouse(非特許文献3) の方法も、分子クラスターを用いてナノ粒子を成長させたが、クラスターからのイオンを用いて、クラスター凝集又はクラスターの細分化によって、より大きなナノ粒子を成長させた。いずれの場合においても、最初の分子クラスター上のより大きなナノ粒子を成長させるために必要とされるイオンを提供するために、別のナノ粒子前駆体組成物を用いなかった。更に、Cooney(非特許文献2)の方法もStrouse(非特許文献3)の方法も、最終的なナノ粒子において、最初の個々の分子クラスターの構造的整合性を保たなかった。更に又、これらの方法の両方とも、II―VIクラスターを用いてII―VIナノ粒子を形成することに制限されることが分かり、それは、これらの方法がより大きなナノ粒子を造るために分子クラスターの材料を用いることに制限されるという事実の必然的な結果である。従って、Cooney(非特許文献2)及び
Strouse(非特許文献3)の作業は、それらの方法論を用いて生成可能な材料の範囲に関して制限される。
【0017】
出願人の公開国際特許出願PCT/GB2005/001611号及びPCT/GB2006/004003号は、大量の高品質単分散の量子ドットを製造する方法を説明しており、それは以前の小規模の方法に関連する問題の多数を克服する。化学前駆体は、分子クラスター化合物の存在下で、又、分子クラスターの整合性が維持され、化学前駆体と反応する核生成中心を提供する明確に定義された既成のシード又はテンプレートの働きをするという条件下で提供され、産業的な応用に十分に大きな規模で高品質ナノ粒子を生成する。
【0018】
PCT/GB2005/001611及びPCT/GB2006/004003に説明される方法の重要な際立った特徴は、前駆体組成物のナノ粒子への転化がナノ粒子成長の全体にわたってその構造的整合性を保つ分子クラスター化合物の存在下で遂行されるということである。クラスター化合物の同一の分子は、ナノ粒子成長が開始されるシード又は核生成点の働きをする。このようにして、適当な明確に定義された核生成サイトが分子クラスターによるシステムですでに提供されるので、高温核形成工程はナノ粒子成長を開始するために必要ではない。クラスター化合物の分子は、ナノ粒子成長を導くテンプレートの働きをする。「分子クラスター」は、関連技術分野で広く理解されている用語であるが、明確にするため、ここでは、クラスター化合物の全ての分子が同じ相対分子式を所有するように十分に明確に定義された化学構造の3つ以上の金属原子及びそれらに関連した配位子のクラスターに関するものであると理解されるべきである。このように、1つのH2O分子が他のH2O分子と同一であるのと同様に、分子クラスターは互いに同一である。他の方法で使用される核生成サイトよりも非常に明確に定義されている核生成サイトを提供することにより、分子クラスター化合物を用いることで、本質的に単分散であるナノ粒子の集合が提供出来る。この方法の更なる有意な利点は、容易に規模を拡大することが出来るということである。適当な分子クラスター化合物を製造する方法は当該技術内で知られていて、その例はケンブリッジ結晶学データセンター(www.ccdc.ca.ac.uk)で見ることが出来る。
【0019】
現在まで広く研究されたナノ粒子材料の多数がカドミウムイオンを組み込むことは、前述の考察から理解されるだろう。しかしながら、カドミウムや、水銀及び鉛系材料のような他の重金属の使用に関連する多くの環境問題があり、そこで、カドミウムを含まないナノ粒子材料を開発することが必要である。特に、現在のカドミウム系材料と同等の単分散度及びサイズ調整可能な光ルミネセンススペクトルを示す、カドミウムを含まない量子ドットを製造することが望ましい。商業的には、このような材料を大規模に、高収量において、出来るだけ安価に製造することが要求される。
【0020】
本発明の目的は、ナノ粒子材料の製造のための先行技術の方法に内在する1つ又はそれ以上の問題に対処することである。更なる目的は、上記の商業的な要求の1つ又はそれ以上を満たす、カドミウムを含まない新たなナノ粒子材料を提供することである。
【課題を解決するための手段】
【0021】
本発明は、II―VI分子クラスター上に成長して、それによってII―VI分子クラスターを組み込むIII−Vナノ粒子に関する。本発明の第1の態様において、周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物と、前記分子クラスター化合物に提供されるコア半導体材料とからなるナノ粒子であって、コア半導体材料は、周期表の第13族及び第15族からのイオンを組み込むナノ粒子を提供する。
【0022】
本発明の第2の態様において、周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物と、前記分子クラスター化合物に提供されるコア半導体材料とを具えるナノ粒子の製造方法であって、コア半導体材料は周期表の第13族及び第15族からのイオンを組み込み、当該方法は、ナノ粒子コア材料のシーディング及び成長を可能にする条件の下で、周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物の存在下で、それぞれ周期表の第13族及び第15族からのイオンを含む第1及び第2のコア前駆体種を具えているナノ粒子コア前駆体組成物の、ナノ粒子コアの材料への転化を生じさせることを含む、ナノ粒子の製造方法を提供する。
【0023】
従って、本発明は、III−Vナノ粒子が大規模に比較的安価に製造することが出来る手段を提供する。
【0024】
分子クラスター化合物及びコア半導体材料が、前記分子クラスター化合物上の前記コア半導体材料の成長を可能にする互換性を持つ結晶相を有することが好ましい。最も好ましくは、分子クラスター化合物及びコア半導体材料は、同じ結晶相を有する。以下に説明する例において、硫化亜鉛クラスターを、リン化インジウムコア半導体ナノ粒子を成長させるために用いる。発明者は何らかの特定の理論によって制約されたくないが、ZnSクラスター/InPコアナノ粒子を得ることが可能だった理由は、ZnSクラスター及びInPコア材料が、両方とも六角形の結晶相を有し、従って、それらは互いに互換性を有し、ZnSクラスター上のInPの層の成長を容易にするという事実に起因するだろうと仮定される。
【0025】
1つの種類の材料のクラスターを組み込んでいるナノ粒子及びそのクラスター上に成長した異なる種類の材料のコアが低い量子収量を示すと予測されたが、驚くべきことに、本発明の第1の態様に係るナノ粒子が更なる準備後の処理の後、約20〜60%及び最高90%の範囲の比較的高い量子効率を示すことが観察された。更に、フッ化水素でナノ粒子の表面を洗浄すること及び/又はコア半導体材料上の半導体材料(例えばII―VI(VI)材料(例えばZnS))の一つ以上の更なる層を成長させることによって、量子収量を更に高めることが出来ることが断定された。
【0026】
好ましくは、分子クラスター化合物は、亜鉛イオンを組み込む。本発明の第1の態様のナノ粒子の好ましい実施形態において、分子クラスター化合物は、硫化物イオン、セレンイオン、及びテルル化物イオンからなる群から選択される第16族イオンを組み込む。
【0027】
コア半導体材料は、好ましくは、アルミニウムイオン、ガリウムイオン、及びインジウムイオンからなる群から選択される第13族イオンを組み込む。コア半導体材料は、窒化物イオン、ヒ化物イオン、及びアンチモンイオンからなる群から選択される第15族イオンを組み込んでもよい。
【0028】
好ましい実施形態において、本発明の第1の態様のナノ粒子は、前記ナノ粒子コア上に提供される第1の半導体材料を含む第1の層を具える。第1の半導体材料として、適切な半導体材料が選択されてもよく、例えば、IIA―VIB材料、IIB―VIB材料、II―V材料、III―V材料、III―IV材料、III―VI材料、IV―VI材料、又は遷移金属元素又はd―ブロック元素を組み込んでいる材料であるが、これに限定されない。好ましくは、第1の半導体材料は、周期表の第12族からのイオンを組み込む(例えば亜鉛イオン)。第1の半導体材料は、周期表の第16族からのイオンを組み込んでもよい(例えば硫化物イオン、セレンイオン又はテルル化物イオン)。
【0029】
ナノ粒子は、前記第1の層上に提供される第2の半導体材料を含む第2の層を具えてもよい。選択的に、半導体材料の更なる層又はシェルが、第2の層に提供されてもよい。第2及び以降の層は、適切な半導体材料から形成されてもよく、半導体材料は例えば、IIA―VIB材料、IIB―VIB材料、II―V材料、III―V材料、III―IV材料、III―VI材料、IV―VI材料、又は遷移金属元素又はd―ブロック元素を組み込んでいる材料であるが、これに限定されない。
【0030】
本発明の第1の態様に係るナノ粒子の製造の好ましい方法において、粒子成長を開始するために、ナノ粒子前駆体の存在下で溶媒(配位又は非配位)に、シーディングII―VI分子クラスターを入れる。シーディング分子クラスターは、反応溶液内に存在する他の前駆体から粒子成長を開始するためのテンプレートとして用いられる。シーディング剤として用いられる分子クラスターは、先に作成することが出来、又はシーディング剤としての働きをする前にその場で製造することも出来る。いくつかの前駆体は、分子クラスターとともに反応プロセスの初めに存在しても、存在しなくてもよいが、反応が進行して、温度が上昇するにつれて、更なる量の前駆体を溶液として滴状で、又は、固体として、周期的に反応に加えることが出来る。
【0031】
方法は、ナノ粒子前駆体組成物の所望のナノ粒子への転化に関係する。好適な前駆体には、成長しているナノ粒子に組み込まれる2つ以上のイオンを含む単一供給源前駆体、又は、成長しているナノ粒子において含まれる少なくとも1つのイオンをそれぞれ含む、2つ以上の別の前駆体を含むマルチ供給源前駆体が含まれる。ナノ粒子の最終的な所望の収率を形成するために必要な前駆体組成物の全量を、ナノ粒子の成長が始まる前に加えることが出来、或いは、前駆体組成物は、反応の全体にわたって段階的に加えることが出来る。
【0032】
前駆体のナノ粒子材料への転化は、任意の適当な溶媒において実行することが出来る。クラスター化合物の分子の整合性を維持することが重要であることが理解されるだろう。それゆえに、クラスター化合物及びナノ粒子前駆体が溶媒に導入されるときに、溶媒の温度は前記クラスター化合物の満足な溶解及び混合を確実にするために十分に高くなければならない(存在する化合物が完全に溶解することは、必要ではないが望ましい)が、クラスター化合物分子の整合性を崩壊させるほど高くてはならない。一旦クラスター化合物及び前駆体組成物が溶媒において十分によく溶解すると、このように形成される溶液の温度をある温度又は温度範囲まで上昇させ、該温度又は温度範囲は、ナノ粒子の成長が始まるのに十分に高いが、クラスター化合物分子の整合性に損傷を与えるほど高くない。温度を上昇させるにつれて、前駆体の更なる分量を反応物に、液滴の方法で、又は固体として加える。そして溶液の温度を、この温度又はこの温度範囲内で、所望の特性を具えたナノ粒子を形成するために必要な時間維持することが出来る。
【0033】
多様な適当な溶媒が利用出来る。使用する特定の溶媒は通常、反応種、即ちナノ粒子前駆体及び/又はクラスター化合物の性質、及び/又は形成されるナノ粒子のタイプに、少なくとも部分的に依存する。通常の媒体としては、ホスフィン(例えばTOP)、ホスフィンオキシド(例えばTOPO)、アミン(例えばHDA)、チオール(例えばオクタンチオール)のようなルイス塩基型配位性溶媒、又は、アルカン及びアルケンのような非配位性有機溶媒が挙げられる。非配位性溶媒が用いられる場合、以下の理由により、それは、キャッピング剤としての役割を果たす更なる配位剤の存在下で通常使用される。
【0034】
形成されているナノ粒子が量子ドットとして機能することを目的とする場合、完全には配位結合されなかった「ダングリングボンド」である表面原子をキャップして、非放射電子正孔再結合を最小にすると共に、量子効率を低下させ得る又はナノ粒子の凝集を形成し得る粒子凝集を抑制することが重要である。キャッピング剤又は不動態化剤としての役割を果たすことも出来る多くの異なる配位性溶媒が知られており、例えばTOP、TOPO、had又はミリスチン酸(テトラデカン酸)、長鎖アミン、官能化されたPEG鎖のような長鎖有機酸であるが、これらのキャッピング剤に限定されない。キャッピング剤としての役割を果たすことができない溶媒が選択される場合、ナノ粒子が成長する間、任意の望ましいキャッピング剤を反応混合物に加えることが出来る。上記のキャッピング剤は一般的にルイス塩基であり、ホスフィン型単歯状、多歯状配位子(トリオクチルホスフィン、トリフェニルホスフィン、t−ブチルホスフィン)、ホスフィンオキシド(トリオクチルホスフィンオキシド)、アルキルホスホン酸、アルキルアミン(ヘキサデシルアミン、オクチルアミン)、アリルアミン、ピリジン、オクタンチオール、長鎖脂肪酸、及びチオフェンが挙げられるが、ナノ粒子の周囲に保護シースを形成するオレイン酸及び有機ポリマーのような広範囲の他の試剤が利用出来る。
【0035】
量子ドットの最外側層(キャッピング剤)は、他の無機、有機又は生物学的材料に対する化学結合として用いることが出来る追加の官能基を処理する配位リガンドからなることもでき、それによって、官能基は量子ドット表面から離間する方に向き、その他利用可能な分子と結合/反応することが出来る。その分子は、1級、2級アミン、アルコール、カルボン酸、アジド、ヒドロキシル基等であるが、これに限定されない。量子ドットの最外側層(キャッピング剤)は、重合可能であると共に粒子の周囲にポリマーを形成するために用いることが出来る官能基を処理する配位リガンドからなることも出来る。
【0036】
最外側層(キャッピング剤)は、最も外側の無機層に直接結合された有機単位からなることもでき、又、粒子の表面に結合されずに、粒子の周囲にポリマーを形成するために、又は、更なる反応のために用いることが出来る官能基を有してもよい。
【0037】
本発明の第2の態様を構成する方法に関して、分子クラスター化合物が亜鉛イオンを組み込むことが好ましい。分子クラスター化合物は、好ましくは、硫化物イオン、セレンイオン及びテルル化物イオンからなる群から選択される第16族イオンを組み込む。
【0038】
第1及び第2のコア前駆体種は、前記コア前駆体組成物に含まれる別の実体でもよい。この場合、コア半導体材料の第13族イオンは、III―ホスフィン、III―(TMS)3、III―(アルキル)、III―(アリール)、III―(ミリスチン酸塩)3、III―(アセテート)3、III―(ミリスチン酸塩)(アセテート)2、III―(ミリスチン酸塩)2(アセテート)、及びIII―(III) アセチルアセトネートからなる群から選択される供給源に由来してもよい。コア半導体材料の第13族イオンは、有機金属化合物、配位化合物、無機塩及び/又は元素供給源に由来してもよい。更に、コア半導体材料の第15族イオンは、有機金属化合物、配位化合物、無機塩及び/又は元素供給源に由来してもよい。
【0039】
第1及び第2のコア前駆体種は、前記コア前駆体組成物に含まれる単体において組み合わせられてもよい。
【0040】
方法は、ナノ粒子コア材料上の第1の半導体材料から構成される第1の層を形成することを更に含んでもよい。好ましくは、第1の半導体材料の第1の層の形成は、第1の材料前駆体組成物の、前記第1の材料への転化を生じさせることを含む。第1の材料前駆体組成物が、第1の材料から構成される層に組み込まれる第3及び第4イオンを具えることが好ましい。第3及び第4イオンは、第1の材料前駆体組成物において含まれる別の実体でもよいか又は第1の材料前駆体組成物において含まれる単体において組み合わせられてもよい。
【0041】
第1の半導体材料の第1の層の形成の前にナノ粒子コア材料がフッ化水素に晒されることが好ましい。例において以下に更に詳細に述べるように、フッ化水素を含む溶液でナノ粒子コアの表面にエッチングすることによってナノ粒子の量子効率を上昇させることが出来る。
【0042】
本発明は、II―VI分子クラスターを用いて製造されるIII―Vナノ粒子に関係し、小さなナノ粒子の集合と比較して、クラスターは明確に定義された同一の分子実体であり、それは本質的に分子クラスターの特徴のない性質を欠いている。知られているIII―V分子クラスターは少数であるので、III―Vクラスターの代わりにII―VI分子クラスターが用いられる。III―V分子クラスターは、作成するのが困難で、通常、空気及び水分に影響を受けるのに対し、多数のII―VI分子クラスターが簡単な手順によって作成でき、それらの多くは空気及び水分に影響を受けない。III―V粒子は、多くのII―VI分子クラスターにシーディングされることができ、それゆえ、初めて、III―Vナノ粒子を成長させるためにIII―V分子クラスターを用いる必要又は利点がないことが、現在理解される。分子クラスターを用いることにより、III―V量子ドットを製造する従来の方法のように、高温核形成工程の必要がなく、それは大規模な合成が可能であることを意味する。
【0043】
更に、例えばInP及びGaP量子ドット及びそれらの合金のようなIII―Vナノ粒子材料の成長をシーディングするのに、[HNEt3]4[Zn10S4(SPh)16]のようなII―VI分子クラスターを用いることが可能である。II―VI分子クラスター、III及びVイオンの分子供給源、「分子フィードストック」の後に続く添加又はその場形成がなされて、それらは粒子成長を容易にするために消費される。これらの分子供給源は、オストワルトの熟成が生じることを抑制してナノ粒子サイズ範囲が分散することから抑制するために自由イオンの濃度を維持しながら、自由イオンの濃度を最低限に保つように、反応溶液と周期的に加えてもよい。
【0044】
ナノ粒子成長は、加熱(熱分解)によって、又はソルボサーマル法によって開始されてもよい。ソルボサーマルという用語は、ここでは、粒子成長を生じさせて維持するように行う反応溶液における加熱を指すように用いており、また、thermolsolvol、溶液熱分解及びリオサーマルとも呼ばれる方法も包含することを意図している。粒子の調製は、反応条件を変化させることによって達成されることもでき、例えば塩基又は酸の添加、圧力の上昇(即ち気圧より大きな圧力の使用)、マイクロ波放射等の電磁放射を加えること、又は当業者に知られている多くの他の方法の何れかによって達成される。
【0045】
III―V半導体量子ドットの調製のための条件
フィードストック
分子フィードストックは、ナノ粒子の成長のために必要な全ての元素が単一化合物前駆体の中に存在する単一源前駆体の形、又は成長したナノ粒子となるために必要な1又は複数の元素/イオンをそれぞれが含む前駆体の組み合わせであることが出来る。これらのフィードストックを反応の初めに加えるか、又は粒子成長の反応の全体にわたって周期的に加えてもよい。このフィードストックは、液体、溶液、固体、スラリー又は気体の形であることが出来る。
【0046】
シーディングクラスターのその場形成
シーディングテンプレートとして使用されるクラスターは、反応の前に予め調製して反応過程の初めに反応溶液に加えてもよく、又はナノ粒子の成長のために用いられる前駆体を加える前に、反応溶液においてその場で形成してもよい。
【0047】
作成される系の種類
半導体コア材料
本発明は、III−Vナノ粒子材料及びその合金の調製を目的としていて、量子ドット又はナノ結晶とも呼ばれる化合物半導体粒子を含む。これは、通常2〜100nmのサイズ範囲内で、以下を含むナノ粒子コア材料を含む。
周期表の第13族(III)からの第1元素と、周期表の第15族(V)からの第2元素と、又、三元、四元、及びドープ材料を含むIII−V材料。
【0048】
ナノ粒子コア材料の好ましい例は、BP、AlP、AlAs、AlSb;GaN、GaP、GaAs、GaSb;InN、InP、InAs、InSb、AlN、BNを含むが、これに制限されない。
【0049】
本発明の第1の態様に含まれるナノ粒子は、III及びVイオンを組み込んでいる二元相材料だけでなく、存在するIII及びVイオンに加えてそれぞれ1つか2つの更なる種類のイオンを組み込んでいる三元及び四元相ナノ粒子も含む。三元相ナノ粒子が3つの成分の材料から成り、四元相ナノ粒子が4つの成分の材料から成ることはいうまでもない。
【0050】
ドープされたナノ粒子は、1つ以上の主要な族又は希土類元素、多くの場合遷移金属又は希土類元素(例えばMn+又はCu2+であるがこれに限らない)からなるドーパントを更に組み込む上記の種類のナノ粒子を指す。
【0051】
外側の1つ又は複数の無機シェル
コアIII―Vナノ粒子上へ成長する任意のシェル又はそれに続く複数のシェルに用いられる材料はほとんどの場合、コア材料と類似の格子形材料だろう。即ち、コア上にエピタキシャルに成長することが出来るように、コア材料に近い格子整合を有するが、この適合性を有する材料に、必ずしも制限されるというわけではない。しかしながら、2つの材料(コア及びシェル)に互換性がない場合、半導体コアと半導体シェルとの間に、緩衝層をコアの外側に最初に成長させてもよい。コア上に成長する任意の緩衝層又はシェル層の材料は、以下を具える材料を含むことが出来る。
周期表の第2族からの第1元素と、周期表の第16族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むIIA−VIB(2−16)材料。ナノ粒子材料は、MgS、MgSe、MgTe、CaS、CaSe、CaTe、SrS、SrSe、SrTeを含むが、これに限定されない。
周期表の第12族からの第1元素と、周期表の第16族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むIIB−VIB(12−16)材料。ナノ粒子材料は、ZnS、ZnSe、ZnTe、CdS、CdSe、CdTe、HgS、HgSe、HgTeを含むが、これに限定されない。
周期表の第12族からの第1元素と、周期表の第15族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むII−V材料。ナノ粒子材料は、Zn3P2、Zn3As2、Cd3P2、Cd3As2、Cd3N2、Zn3N2を含むが、これに限定されない。
周期表の第13族からの第1元素と、周期表の第15族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むIII−V材料。ナノ粒子材料は、BP、AlP、AlAs、AlSb;GaN、GaP、GaAs、GaSb;InN、InP、InAs、InSb、AlN、BNを含むが、これに限定されない。
周期表の第13族からの第1元素と、周期表の第14族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むIII−IV材料。ナノ粒子材料は、B4C、Al4C3、Ga4Cを含むが、これに限定されない。
周期表の第13族からの第1元素と、周期表の第16族からの第2元素を組み込み、又、三元及び四元材料を含むIII−VI材料。ナノ粒子材料は、Al2S3、Al2Se3、Al2Te3、Ga2S3、Ga2Se3;In2S3、In2Se3、Ga2Te3、In2Te3を含むが、これに限定されない。
周期表の第14族からの第1元素と、周期表の第16族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むIV−VI材料。ナノ粒子材料は、PbS、PbSe、PbTe、Sb2Te3、SnS、SnSe、SnTeを含むが、これに限定されない。
周期表の遷移金属における任意の族からの第1元素と、周期表のd−ブロック元素の任意の族からの第2元素を組み込み、又、三元及び四元材料とドープ材料とを含むナノ粒子材料。ナノ粒子材料は、NiS、CrS、CuInS2、CuInSe2、CuGaS2、CuGaSe2を含むが、これに限定されない。
【0052】
ナノ粒子形状
ナノ粒子の形状は、球体に限定されず、任意の望ましい形状をとることができ、例えば桿体、球体、円板、四脚又は星形である。ナノ粒子の形状の制御は、反応粒子成長過程において、成長している粒子の特定の格子面と優先して結合し、その後特定の方向の粒子成長を阻害又は遅延させる化合物を加えることによって達成することが出来る。この効果を達成するために加えることが出来る化合物の非限定的な例は、ホスホン酸(n−テトラデシルホスホン酸、ヘキシルホスホン酸、1−デカンスルホン酸、12−ヒドロキシドデカン酸、n−オクタデシルホスホン酸)を含む。
【0053】
調製方法の概要
本発明は、配位性有機層のようなキャッピング剤によって粒子凝集及び周囲の化学環境から安定化されてよい実質的に純粋な単分散のナノ結晶粒子を提供することを目的としている。このようなナノ粒子を表す適当な化学式は、MEnLyであり、但し、M=III族元素、E=V族元素、L=配位子(例えば配位性有機層/キャッピング剤)、そして、n及びyが構成要素E及びLの適切な化学量論量を表す。
【0054】
半導体材料のナノ粒子を調製するための第1ステップは、分子クラスターをテンプレートとして用いて他の元素源前駆体からのナノ粒子の成長をシードすることである。ホスフィン、ホスフィンオキシド、アミン又は有機酸に限定されないが、このようなルイス塩基配位化合物、又はオレイン酸のようなキャッピング剤化合物を添加したアルカン(オクタデセンス)のような不活性溶媒である、キャッピング剤ともなり得る高沸点溶媒と、テンプレートとして用いられるべき少量のクラスターを混合することによってこれが達成される。反応温度が上昇するにつれて、M及びEの供給源前駆体が、液体であれば液滴で、又は多量の固体粉末として、周期的に加えられる。これに続いて、M供給源及びE供給源が、反応混合物に加えられる。M及びE前駆体供給源は、一方がMを含有し、他方がEを含有している2つの別々の前駆体の形であるか、又は単一分子内にM及びEの両方を含有する単一源前駆体の形であることが可能である。
【0055】
これに続き、成長したナノ粒子の形状を制御する能力を有する他の試剤を反応に添加してもしなくてもよい。これらの添加物は、成長しているナノ粒子の特定の面(格子面)と優先して結合し、それゆえ、粒子のその特定の方向に沿う粒子成長を阻害又は遅延させることが出来る化合物の形である。三元、四元又はドープ粒子を製造するために、他の元素源前駆体を反応に添加してもしなくてもよい。
【0056】
分子クラスターを配位化合物と混合し、それから、分子クラスターテンプレートの表面に粒子成長が始まるまで、反応混合物を一定の速度で加熱する。更なる量のM及びE前駆体を、適当な温度で反応混合物に加えてもよい。更なる前駆体の添加は、固体の前駆体又は前駆体を含む溶液を一定時間にわたって加えるバッチ添加、又は、溶液相前駆体の連続した滴加の形であってもよい。粒子の核生成及び成長を完全に分離するため、本発明では、反応の温度及び存在する前駆体の濃度によって制御される粒子サイズに関して高度な制御を示す。その場光学プローブ(in situ optical probe)によって、又は反応溶液の分取から、反応溶液のUV及び/又はPLスペクトルから証明されるように、所望の粒子サイズが得られると、温度は特定の度数だけ下降してもしなくてもよく、混合物を10分から72時間の期間にわたってアニールする。
【0057】
ME/NYコアシェル又はコアマルチシェル粒子を形成するための、形成されたナノ粒子の更なる継続的処理を行なってもよい。コアシェル粒子調製はナノ粒子隔離の前又は後に行い、これによって、ナノ粒子は反応から隔離されて新たな(きれいな)キャッピング剤(同じキャッピング剤化合物でも異なるキャッピング剤化合物でもよい)に再溶解し、これが結果としてより良質の量子ドットとなることが出来る。N源及びY源前駆体(N及びYは、MEコア量子ドット上に成長しているシェル又は層において必要とされる元素である)を反応混合物に加えるが、ME/NYコアシェル材料のコアシェル粒子を形成するために、一方がNを含有し、他方がYを含有している2つの別々の前駆体の形でも、又、単一分子内にN及びYを両方含有する単一源前駆体としてでもよい。
【0058】
所望のコアマルチシェル材料が形成されるまで、適当な元素前駆体でこの過程を繰り返してもよい。粒子の集合におけるナノ粒子サイズ及びサイズ分布は、成長時間、温度及び溶液中の反応物の濃度に依存し、温度が高くなると共に、大きいナノ粒子が製造される。
【0059】
シーディングに用いるクラスターのタイプ
本発明はII−VI分子クラスターの使用を含み、これによって用いられるクラスターは、集合における分子クラスターの匿名性を本質的に欠くナノ粒子と比較して、同一の分子的実体である。クラスターは、ナノ粒子の成長のための「胚型」テンプレートとして働き、これによって他の分子源前駆体はイオンを成長過程に貢献させるので、クラスターがその後粒子へと成長する。
【0060】
分子クラスターは、周期表の第12族からの第1のイオンを組み込み、周期表の第16族からの第2のイオンを組み込む。
【0061】
第12族イオンは、亜鉛、カドミウム及び水銀からなる群から選択されてもよい。
【0062】
第16族イオンは、硫化物、セレン化物及びテルル化物からなる群から選択されてもよい。
【0063】
用いられることが出来る分子クラスターの好ましい実施例は以下であるが、限定されない。
IIB−VIB
[{(PPh3)Hg}4(SPh)6]
(Ph4P)2[(SEt)5(Br)(HgBr)4]
(Ph4P)2[Hg4(SEt)5Br]
[Hg4Te12][N(CH2CH2Et)4]4
IIB−VIB
[RMEtBu]5但し、M=Zn、Cd、Hg;E=S、Se、Te;R=Me、Et、Ph
[X]4[E4M10(SR)16]但し、M=Zn、Cd、Hg;E=S、Se、Te、X=Me3NH+、Li+、Et3NH+
[Cd32S14(SPh)36]・L
[Hg10Se4(SePh)(PPh2nPr)4]
[Hg32Se14(SePh)36]
[Cd10Se4(SePh)12(PPr3)4]
[Cd32Sel4(SePh)36(PPh3)4]
[M4(SPh)12]+[X]2−但し、M=Zn、Cd、Hg;X=Me4N+、Li+
[Zn(SEt)Et]10
[MeMEiPr]但し、M=Zn、Cd、Hg;E=S、Se、Te
[RCdSR’]5但し、R=O(ClO3)、R’=PPh3、iPr
[Cd10E4(E’Ph)12(PR3)4]
但し、E=Te、Se、S、そして別に、E’=Te、Se、S
[Cd8Se(SePh)12Cl4]2−
[M4Te12]4−但し、M=Cd、Hg
[Ph12M18Cd10(PEt3)3]
但し、M=Te、Se
【0064】
無機コアのために用いられる前駆体
IIIイオン供給源
III―ホスフィン、III−(TMS)3、III―(アルキル)、III―(アリール)、III−(ミリスチン酸塩)3、III―(アセテート)3、III―(ミリスチン酸塩)(アセテート)2、III―(ミリスチン酸塩)2(アセテート)、III―(III)アセチルアセトネート
MR3但しM=Ga、In、Al、B;R=アルキル又はアリール基,例えばAlR3、GaR3、InR3(R=Me、Et、iPr)に限定されないが、このような有機金属化合物。
炭酸塩、M(CH3C)3但しM=B、Al、Ga、In:アセチルアセトネート(2,4−ペンタンジオネート)のようなβ−ジケトネート又はその誘導体、M[CH3COOCH=C(O−)CH3]2但しM=B、Al、Ga、Inのような配位化合物。
酸化物(例えばIn2O3、Ga2O3)又は、硝酸塩(例えばAl(NO3)3、In(NO3)3、Ga(NO3)3)のような無機塩。
B、Al、Ga、Inのような元素供給源。
【0065】
Vイオン供給源
NR3、PR3、AsR3、SbR3(R=Me、Et、tBu、iBu、Pri、Ph等);NHR2、PHR2、AsHR2、SbHR2(R=Me、Et、tBu、iBu、Pri、Ph等);NH2R、PH2R、AsH2R、SbH2R3(R=Me、Et,tBu、iBu、Pri、Ph等);PH3、AsH3;M(NMe)3、M=P、Sb、As;ジメチルドラジン(Me2NNH2);エチルアジド(Et−NNN);ヒドラジン(H2NNH2);Me3SiN3に限定されないが、このような有機金属化合物。
炭酸塩、MCO3 M=P、次炭酸ビスマス(BiO)2CO3;アセチルアセトネート(2,4−ペンタンジオネート)のようなβ−ジケトネート又はその誘導体、[CH3COCH=C(O−)CH3]3M、M=Biである;[CH3COOCH=C(O−)CH3]2M M=Bi;[CH3COOCH=C(O−)CH3]2Mに限定されないが、このような配位化合物。
酸化物P2O3、As2O3、Sb2O3、Sb2O4、Sb2O5、Bi2O3、又は硝酸塩Bi(NO3)3、Sn(NO3)4、Pb(NO3)2のような無機塩。
N、P、As、Sb、Biのような元素供給源。
【0066】
複合III−Vイオン供給源−ME単一源前駆体
III−V元素を具える化合物半導体ナノ粒子では、III及びVイオンが、単一源前駆体の形であることも可能であり、用いられる前駆体は、単一分子内にM及びEの両方を含む。
単一源前駆体は、有機金属化合物、無機塩又は配位化合物(MaEb)Lcであることが可能である。ここで、M及びEはナノ粒子内で必要とされる元素であり、Lはキャッピング配位子であり、a、b及びcは、M、E及びLの適切な化学量論量を表す数である。
III−Vコア半導体には、単一源前駆体は以下であることが可能であるが、それに限定されない。
GaNには、[(Me)2GaN(H)tBu]2[H2GaNH2]3、
Gapには、[Ph2GaP(SiMe3)3Ga(Ph)2Cl]、[Et2GaP(SiMe3)2]2、[Et2GaPEt2]3、[tBu2GaPH2]3、[Me2GaP(iPr)2]3[tBuGaPAr´]2、[tBu2GaP(H)C5H9]2、
GaAsには、Ga(AstBu2)3、[Et2GaAs(SiMe3)2]2、[tBu2GaAs(SiMe3)2]2、
GaSbには、[Et2GaSb(SiMe3)2]2、
InPには、[(Me3SiCH2)2InP(SiMe3)2]2、[R2InP(SiMe3)2]2、[Me2InPtBu2]2、
InSbには、[Me2InSbtBu2]3 [Et2InSb(SiMe3)2]3、[Me2InNEt2]2、[Et2AlAstBu2]2、
AlSbには、[tBu2AlSb(SiMe3)2]2、
GaAsには、[nBu2GaAstBu2]2、[Me2Ga2AstBu2]2、[Et2GaAstBu2]2
【0067】
任意の外側の無機シェル又はそれに続く複数のシェルのために用いられる前駆体(ME)
III―Vコア及びMEnLmシェル(但しM及びEは、シェル又は以降のシェル層の元素である)からなる化合物コア/シェル半導体ナノ粒子のために、元素Mの供給源は、反応に更に加えられてもよく、成長する粒子にEイオンの供給源を提供する能力を有する、任意のE含有種から成ることが出来る。
【0068】
M源
この前駆体は、有機金属化合物、無機塩、配位化合物又は元素供給源からなることが出来るが、これに限定されない。第1元素のためのII−VI、III―V、III―VI又はIV−Vの例は、以下を含むが、これに限定されない。
MR2、但しM=Mg、R=アルキル又はアリール基(MgtBu2);MR2、但しM=Zn、Cd、Te;R=アルキル又はアリール基(Me2Zn、Et2Zn、Me2Cd、Et2Cd);MR3、但しM=Ga、In、Al、B;R=アルキル又はアリール基[AlR3、GaR3、InR3(R=Me、Et、iPr)]に限定されないが、このような有機金属。
MCO3 M=Ca、Sr、Ba、[水酸化炭酸マグネシウム[(MgCO3)4Mg(OH)2];M(CO3)2 M=Zn、Cd、;MCO3 M=Pb:アセテート:M(CH3CO2)2 M=Mg、Ca、Sr、Ba;Zn、Cd、Hg;M(CH3C)3 M=B、Al、Ga、In:アセチルアセトネート(2,4−ペンタンジオネート)のようなβ−ジケトネート又はその誘導体、[CH3COOCH=C(O−)CH3]2 M=Mg、Ca、Sr、Ba、Zn、Cd、Hg;[CH3COOCH=C(O−)CH3]2 M=B、Al、Ga、In。オキザラートSrC2O4、CaC2O4、BaC2O4、SnC2O4に限定されないが、このような炭酸塩のような配位化合物。
酸化物SrO、ZnO、CdO、In2O3、Ga2O3、SnO2、PbO2;硝酸塩Mg(NO3)2、Ca(NO3)2、Sr(NO3)2、Ba(NO3)2、Cd(NO3)2、Zn(NO3)2、Hg(NO3)2、Al(NO3)3、In(NO3)3、Ga(NO3)3、Sn(NO3)4、Pb(NO3)2に限定されないが、このような無機塩。
Mg、Ca、Sr、Ba、Zn、Cd、Hg、B、Al、Ga、In、Sn、Pbに限定されないが、このような元素供給源。
【0069】
E源
反応に元素E源を更に加えてもよく、この元素E源は、成長している粒子にEイオン源を供給する能力を有する任意のE含有種から構成されることが出来る。
この前駆体は、有機金属化合物、無機塩、配位化合物又は元素供給源から構成されることが出来るが、これに限定されない。II−VI、III―V、III―VI又はIV−V半導体における元素Eの例は、以下を含むが、これに限定されない。
NR3、PR3、AsR3、SbR3(R=Me、Et、tBu、iBu、Pri、Ph等);NHR2、PHR2、AsHR2、SbHR2(R=Me、Et、tBu、iBu、Pri、Ph等);NH2R、PH2R、AsH2R、SbH2R3(R=Me、Et,tBu、iBu、Pri、Ph等);PH3、AsH3;M(NMe)3 M=P、Sb、As;ジメチルドラジン(Me2NNH2);エチルアジド(Et−NNN);ヒドラジン(H2NNH2);Me3SiN3に限定されないが、このような有機金属。
MR2(M=S、Se、Te;R=Me、Et、tBu、iBu等);HMR(M=S、Se、Te;R=Me、Et、tBu、iBu、iPr、Ph等);チオ尿素S=C(NH2)2;Se=C(NH2)2。
Sn(CH4)4、Sn(C4H9)、Sn(CH3)2(OOCH3)2。
炭酸塩、MCO3 M=P、次炭酸ビスマス(BiO)2CO3;M(CO3)2;アセテートM(CH3CO)2 M=S、Se、Te:M(CH3C)3 M=Sn、Pb:アセチルアセトネート(2,4−ペンタンジオネート)のようなβ−ジケトネート又はその誘導体、[CH3COOCH=C(O−)CH3]3M M=Bi;[CH3COOCH=C(O−)CH3]2M M=S、Se、Te:[CH3COOCH=C(O−)CH3]2M、M=Sn、Pb:チオ尿素、セレノ尿素(H2NC(=Se)NH2に限定されないが、このような配位化合物。
酸化物P2O3、As2O3、Sb2O3、Sb2O4、Sb2O5、Bi2O3、SO2、SeO2、TeO2、Sn2O、PbO、PbO2;硝酸塩Bi(NO3)3、Sn(NO3)4、Pb(NO3)2に限定されないが、このような無機塩。
元素供給源:Sn、Ge、N、P、As、Sb、Bi、S、Se、Te、Sn、Pb。
【0070】
複合III−Vイオン供給源−ME単一源前駆体
化学式MEの化合物半導体ナノ粒子シェルでは、元素M及びEが、単一源前駆体によって提供されることが可能であり、用いられる前駆体は、単一分子内にM及びEの両方を含む。この前駆体は、有機金属化合物、無機塩又は配位化合物、(MaEb)Lcであることが可能である。ここで、M及びEはナノ粒子内で必要とされる元素であり、Lはキャッピング配位子であり、a、b及びcは、適切な化学量論値である。
MがII族イオンであり、EがVI族イオンであるII―VI半導体シェルの非限定的な例は以下である。ビス(ジアルキルジチオ―カルバメート)M、(II)複合体又は、式M(S2CNR2)2の同族Se及びTe化合物 但しM=Zn、Cd、Hg;S=S、Se、O、Te及びR=アルキル基又はアリール基、CdS Cd[SSiMe3]2、Cd(SCNHNH2)2Cl2、Cd(SOCR)2・py、CdSe[Cd(SePh)2]2
III−V半導体シェルには、ME単一源前駆体の非限定的な例は以下である。
GaNには、[(Me)2GaN(H)tBu]2、[H2GaNH2]3、
Gapには、[Ph2GaP(SiMe3)3Ga(Ph)2Cl]、[Et2GaP(SiMe3)2]2、[Et2GaPEt2]3、[tBu2GaPH2]3[Me2GaP(iPr)2]3、[tBuGaPAr´]2、[tBu2GaP(H)C5H9]2、
GaAsには、Ga(AstBu2)3、[Et2GaAs(SiMe3)2]2、[tBu2GaAs(SiMe3)2]2、
GaSbには、[Et2GaSb(SiMe3)2]2、
InPには、[(Me3SiCH2)2InP(SiMe3)2]2、[R2InP(SiMe3)2]2、[Me2InPtBu2]2、
InSbには、[Me2InSbtBu2]3、[Et2InSb(SiMe3)2]3、[Me2InNEt2]2、[Et2AlAstBu2]2、
AlSbには、[tBu2AlSb(SiMe3)2]2、
GaAsには、[nBu2GaAstBu2]2、[Me2Ga2AstBu2]2、[Et2GaAstBu2]2
II―V半導体シェルの、ME単一源前駆体の非限定的な例は以下である。
Cd3P2には、[MeCdPtBu2]3、Cd[P(SiPh3)2]2、Zn3P2、Zn[P(SiPh3)2]2
IV―VI半導体シェルの、ME単一源前駆体の非限定的な例は以下である。
PbSには、鉛(II)ジチオカルバメート、
PbSeには、鉛(II)セレノカルバメート
【0071】
本発明は、これより、次の非限定的な例を参照し、次の図を参照して実証される。
【図面の簡単な説明】
【0072】
【図1】図1は、界面活性剤で覆われているナノ粒子の概略図であり、界面活性剤頭部基はナノクリスタルに対する強い親和性を有しており、界面活性剤の炭化水素鎖は、ナノクリスタルを溶媒に溶けやすくし、分散させるのに役立っている。
【図2】図2は、a)半導体InPコアからなる粒子、b)半導体InPコア及び半導体ZnSシェルからなる粒子、c)半導体InPコア、異なる半導体材料であるZnSeの緩衝層、及び外側の半導体ZnSシェルからなる粒子の概略図である。
【図3】図3は、分子シードとして[Zn10S4(SPh)16][X]4x=Li+又は(CH3)3NH+を用いたリン化インジウム量子ドット形成の概略図であり、酢酸カドミウム及びトリ―n―オクチルホスフィンセレニドの滴加、そして、カドミウム及びセレン元素源前駆体及び、キャッピング剤としてヘキサデシルアミンを用いた。
【図4】図4は、a)熱アニーリング前の反応混合物からの分離後、b)熱アニーリング後、のInP量子ドットのUV及びPL発光スペクトルを示す。
【図5】図5は、シーディングテンプレートとして用いられることが出来る分子クラスターを図示する。
【図6】図6は、反応混合物からの分離後のInP/ZnS量子ドットのUV及びPL発光スペクトルを示す。
【実施例】
【0073】
全ての合成及び操作は、乾燥無酸素アルゴン又は窒素雰囲気の下で、標準的なシュレンク及びグローブボックス技術を用いて実行した。全ての溶媒は分析用であって、使用前に適当な乾燥剤から精製される(THF、Et2O、トルエン、ヘキサン、ペンタンにはNa/K−ベンゾフェノン、メタノール及びエタノールにはマグネシウム、そしてアセトンには水素化カルシウム)。全ての化学物質は、分析用である。
【0074】
元素分析は、カルロエルバCHNS−O EA1108・元素分析計にて行った。UV−Vis吸収スペクトルは、Heλiosβサーモスペクトロニック上で測定した。光ルミネセンス(PL)スペクトルは、フルオロログ3(FL3-22)光分光器によって励起波長380nmで測定し、スペクトルは、2nmに設定された切れ込みで1秒の統合時間で得るか、又はOcean Optics 2000 USBプローブを用いてその場で測定した。粉末X線回折(PXRD)測定は、モノクロマトのCu−Kα放射線を使用してブルカーAXS D8回折計で行い、試料を平らに取り付け、10°から70°まで0.04°の刻み幅で2.5秒の計数率で走査した。測定は、20°〜60°にわたって、3°の角度で2θ値で、1秒の計測時間で0.04°ずつ視射角発生率検出器を用いて行なった。ナノ粒子の形態及びサイズ分布を観察するため、そして蛍光X線(EDAX)のエネルギー分散方式分析のために、フィリップスCM200透過電子顕微鏡を用いた。TEM及びEDAXのためのサンプルは、トルエンにおけるサンプルの希釈した懸濁液を一滴、銅グリッド(300メッシュ、Agar Scientific)に置くことによって調製した。過剰な溶媒は、室温で乾燥させた。
【0075】
通常の手順
II―VIクラスター、(例えば[HNEt3]4Zn10S4(SPh)16])及び少量の「フィードストック」前駆体即ち、例えばIn(ミリスチン酸塩)3のようなIII族前駆体及びP(TMS)3のようなV族元素前駆体をフィードストック前駆体として用いて、キャッピング剤を含む溶液に加え、それから温度を上昇させ、しばらくの間反応を撹拌した。その結果、III―V粒子形成のセットとなった。
【0076】
両フィードストック前駆体を更に滴加することによって、量子ドットの、PL発光最大の赤方偏移が起こり、それはその場PLプローブによって監視された。更なる前駆体をこの段階で加える場合、PL最大の赤方偏移はこれ以上起こらず、したがって、粒子はこれ以上成長しない。しかしながら、温度を上昇させると(5〜40℃)、PL最大は再度、赤方偏移を起こす。更に前駆体を反応溶液に加えると、再度PL最大は赤方偏移を起こす。従って、続いて反応温度を段々に上昇させるこの前駆体の添加のサイクルは、PL最大ピークが所望の発光状態になるまで繰り返すことが出来る。それから反応をより低い温度に冷却し、更なる期間アニールさせ、その期間の後、II―VIクラスターを組み込んでいるIII−Vナノ粒子を分離することが出来る。
【0077】
分子クラスターの選択
知られているIII―V分子クラスターは少数であるので、III―Vクラスターの代わりにII―VI分子クラスターが用いられる。III―V分子クラスターは、作成するのが困難で、通常、空気及び水分に影響を受けるのに対し、多数のII―VI分子クラスターが簡単な手順によって作成でき、II―VIクラスターは空気及び水分に影響を受けない。従って、III―V粒子は、II―VI分子クラスター上にシードされることができ、それゆえ、初めて、III―Vナノ粒子を成長させるためにIII―V分子クラスターを用いる理由又は利点がないことが、現在理解される。
【0078】
実施例1
InPナノ粒子の調製(赤発光)
60℃のセバシン酸ジ―n―ブチルエステル(200ml)及びミリスチン酸(10g)を、三つ口丸底フラスコに入れ、N2でパージし、その後ZnSクラスター[HNEt3]4Zn10S4(SPh)16](0.94g)を加えた。反応混合物を、それから30分間100℃に加熱し、その後[In2(Ac)3(MA)3](0.25M、12ml)を48ml/hrの速度で電子シリンジポンプを用いて15分間にわたって加えた。その後、同じ添加速度で(TMS)3P(0.25M、12ml)を加えた。添加が完了すると、反応の温度を180℃に上昇させ、更に、[In2(Ac)3(MA)3]及び(TMS)3Pを以下のように添加した。[In2(Ac)3(MA)3](16ml)と、続いて(TMS)3P(16ml)を加え、温度を200℃まで上昇させ、それから更に[In2(Ac)3(MA)3](10ml)を加えて、粒子を必要なサイズまで成長させ、したがって必要な赤い発光となる。それから1時間、温度を200℃で置き、そして160℃に下げ、それから、反応混合物を3日間アニールする。粒子は、アセトニトリルを用いて分離し、遠心分離して、収集した。粒子は5〜60%の量子効率を示した。
【0079】
実施例2
500〜700nmの発光を有するInP量子ドットの調製及びZnSシェルの成長
ジブチルエステル(100ml)及びミリスチン酸(10.0627g)を、三つ口フラスコに入れ、真空下で1時間、70℃でガスを除去した。この期間の後、窒素を導入し、90℃まで温度を上昇させた。ZnS分子クラスター[Et3NH4][Zn10S4(SPh)16](4.7076g)を加えて、混合物を45分間撹拌した。それから温度を100℃に上昇させ、その後、In(MA)3(1M、15ml)、続いて(TMS)3P(1M、15ml)を滴加した。温度を140℃に上昇させながら、反応混合物を攪拌した。140℃で、In(MA)3(1M、35ml)(5分間撹拌する)及び(TMS)3P(1M、35ml)を更に滴加した。それから温度を180℃までゆっくり上昇させ、In(MA)3(1M、55ml)、続いて、(TMS)3P(1M、40ml)を更に滴加した。上記の方法で前駆体を加えることによって、InPのナノ粒子は、発光最大が520nmから700nmまで徐々に増加する状態で成長することができ、それによって、所望の発光最大が得られたときに反応を止め、30分間その温度で撹拌することが出来る。この期間の後、温度を160℃まで下降させ、反応混合物を最長4日間アニールした(反応の温度よりも下の20〜40℃低い温度で)。この段階で紫外線ランプも、アニーリングを促進するために用いた。
【0080】
カニューレ技術によって、脱気した乾燥メタノール(約200ml)を添加してナノ粒子を分離した。沈殿物を沈殿させ、それから、メタノールを、フィルタスティックを用いてカニューレを介して除去した。脱気した乾燥クロロホルム(約10ml)を、固体を洗浄するために加えた。固体は真空下で1日乾燥させた。これにより、5.60gのInPコアナノ粒子を製造した。元素分析:最大PL=630nm、FWHM=70nm。
【0081】
後処理
上記において調製したInP量子ドットの量子収量を、薄いHF酸での洗浄によって増加させた。該ドットは、無水脱気クロロホルム(〜270ml)において溶解した。50mlの分量を取ってプラスチックフラスコに入れ、窒素で洗浄した。プラスチックシリンジを用いて、3mlの60%w/wHFを水に加え、脱気THF(17ml)に加えることで、HF溶液を調製した。HFを、InPドットに5時間にわたって滴加した。添加完了後、溶液を終夜撹拌した。塩化カルシウム水溶液で抽出し、エッチングされたInPドットを乾燥させることによって過剰HFを除去した。乾燥ドットは、将来使用するために、50mlのクロロホルムにおいて、再分散させた。最大567nm、FWHM60nm。この段階のコア材料の量子効率は25〜90%である。
【0082】
ZnSシェルの成長
HFエッチングしたInPコア粒子の20mlの分量を、三つ口フラスコにおいて乾燥させた。1.3gのミリスチン酸及び20mlのセバシン酸ジ―n―ブチルエステルを加えて、30分間脱気した。溶液を200℃まで加熱し、それから、1.2gの無水酢酸亜鉛を加え、そして、2mlの1M(TMS)2Sを(7.93ml/hrの速度で)滴加した。添加完了後に溶液を撹拌した。溶液を200℃で1時間保ち、それから室温に冷却した。粒子は、40mlの無水脱気メタノールを加えることによって分離し、遠心分離した。上澄み液体は処分し、残りの固体に、30mlの無水脱気ヘキサンを加えた。溶液を5時間沈殿させ、それから再度遠心分離した。上澄み液体を収集し、残りの固体を廃棄した。PL発光ピーク最大値=535nm、FWHM=65nm。この段階でナノ粒子コア/シェル材料の量子効率は35〜90%であった。
【0083】
実施例3
ミリスチン酸インジウム0.25Mの調製
In(Ac)3+1.5CH3(CH2)12COOH→In(Ac)1.5(OOC(CH2)12CH3)1.5
ミリスチン酸(70.798g、0.3100モル、1.5eqv)、酢酸インジウム(58.392g、0.200モル、1eqv)及びセバシン酸ジブチル(626.4ml、568.3g、1.864モル)を、1Lの丸底フラスコに入れ、真空下で二昼一夜、118℃まで加熱した。氷/塩/アセトンによって冷却し、プレトラップを設定し、これと、高真空溶媒トラップとで周期的に調べて、ミリスチン酸インジウムの合成の間、蓄積した凍結した酢酸を取り除いた。
【0084】
TMSP 0.25Mの調製
シュレンクチューブ及びシリンジを、終夜オーブン乾燥した。シュレンクチューブをそれから真空ラインへ動かして冷却した。セバシン酸ジブチルを真空下で脱気し、1時間にわたって穏やかに加熱し、その後92.74ml(0.294モル)を窒素下でシュレンクチューブに移動させた。TMSP(7.26ml、0.025モル)を窒素下で移動させ、セバシン酸ジブチルと混合した。
【0085】
グリーン発光を有するInP量子ドットの調製及びZnS/ZnOシェルの成長
ミリスチン酸(140.000g、0.444モル)及びセバシン酸ジブチル(2800ml)を、60℃〜90℃の温度で、高真空下で1時間脱気した。系をアルゴンでパージし、それから[Et3NH4][Zn10S4(SPh)16]クラスター(13.16g)をすぐに加えた。100℃の温度に到達するまで、反応混合物を15分間加熱し、この段階で、反応混合物を、それから30分間100℃で維持した。クラスターは、完全に溶解せず、白い懸濁液を形成した。
【0086】
ミリスチン酸インジウム(168ml、0.25M)の1回目の添加を1秒に3滴の速度で始め、そして温度は終始100℃に維持した。ミリスチン酸溶液の添加完了後、反応混合物を100℃で5分間撹拌した。それからTMSP(168ml、0.25M)を1秒に2滴の速度で加え、ここでも慎重に温度を監視した。添加が完了するとすぐに、反応混合物の温度を出来るだけ急速に180℃に上昇させ、絶対に超過しないよう注意した。180℃に達するとすぐに、ここでも1秒に3滴の速度で、ミリスチン酸インジウム(224ml、0.25M)を更に添加した。それから反応混合物を30分間180℃で保持した。それから、反応を160℃に冷却させ、温度を150〜160℃に維持しながら、3日間アニールした。
【0087】
アニール後、混合物を室温に冷却させ、その後500mlの分量を大きなビーカーに注入し、アセトニトリルによって沈殿させた。それから、凝集された溶液を遠心分離機チューブに移動させ、ペレットが形成されるまで遠心分離した。透明な上澄みは廃棄し、オレンジ沈殿物を最小量のクロロホルムに再溶解させ、終夜空気中に放置した。
【0088】
後処理
ミリスチン酸におけるプレエッチング
クロロホルムを回転蒸発器において除去し、固体はセバシン酸ジブチルエステル(600ml)に再溶解させた。それからミリスチン酸(80g)を加え、混合物を6時間140℃まで加熱した。混合物を冷却させ、上記と同様にアセトニトリルで沈殿させた。
【0089】
HFエッチング
クロロホルム(1750ml)にペレットを再溶解させ、この溶液をHFエッチングに用いた。500Wのタングステン球の前に溶液を置き、8mlのテトラヒドロフラン内の2mlのHF(48%)の溶液を撹拌しながらゆっくり加えた。溶液の光学発光スペクトルを光学手プローブによって監視し、それは強度が増すにつれて徐々に赤方偏移を始めた。記録している波長が青方偏移を始めた時点までHF溶液を加え、それから、HF/THF/CHCI3を空気で吹き飛ばした。結果として生じるペーストをアセトンで洗浄し、アセトニトリルで沈殿させ、それからヘキサンで洗浄して再度アセトニトリルに沈殿させた。それから沈殿物を最小量のセバシン酸ジブチルエステルに溶解させ、脱気してアルゴン下に置いた。
【0090】
ZnS/ZnOシェルの成長
上記のように形成されるInPナノ粒子を室温で1時間脱気した。カルボキシ―PEG―メチルエーテル(125.82g)も100℃で高真空下で1時間脱気し、それから室温に冷却させた。酢酸亜鉛(34.903g、0.1902モル)及び脱気InPナノ粒子を、冷却したエーテルに加え、それから5分間真空下に置いて、3回アルゴンでパージした。InPナノ粒子を含む溶液を180℃に加熱し、脱気オクタンチオール(8.3111g、0.0568モル)を120℃で急速に注入した。反応混合物を180℃で1時間保持し、そしてオクタノール(24.93g、30.04ml、0.1914モル)を1秒に1滴の速度で加えた。それから反応混合物を30分間加熱して180℃に戻し、その後室温に冷却してからヘキサン(350ml)を加えた。ゲル状の沈殿物から上部層は別の容器へ移し、別の容器へ移した溶液にアセトニトリル(700ml)を加えた。それから混合物を分液漏斗に加え、沈殿物が凝集し始めるまでアセトンを加えた。塊の周辺から溶液を排出し、遠心分離して全ての生成物を回収した。最後に塊及び遠心分離された沈殿物を脱気クロロホルムに溶解した。
【0091】
実施例4
グリーン発光を有するInP量子ドットの調製
例3において説明する手順に従って、ミリスチン酸インジウム及びTMSPを調製した。
【0092】
ミリスチン酸(70.000g、0.222モル)及びセバシン酸ジブチル(1400ml)を、60〜90℃の温度で1時間高真空下で脱気した。系をアルゴンでパージし、それから[Et3NH4][Zn10S4(SPh)16]クラスター(6.58g)をすぐに加えた。それから反応混合物を100℃の温度に到達するまで15分間加熱し、この段階で、反応混合物をそれから30分間100℃で維持した。クラスターは完全に溶解せず、白い懸濁液を形成した。
【0093】
ミリスチン酸インジウム(84ml、0.25M)の最初の添加を1秒に3滴の速度で始め、温度は終始100℃に維持した。ミリスチン酸溶液の添加完了後、反応混合物を5分間100℃で撹拌した。それからTMSP(84ml、0.25M)を1秒に2滴の速度で加え、ここでも慎重に温度を監視した。添加が完了するとすぐに、反応混合物の温度を出来るだけ急速に180℃に上昇させ、絶対に超過しないよう注意した。180℃に達するとすぐに、ここでも1秒に3滴の速度で、ミリスチン酸インジウム(112ml、0.25M)を更に添加した。それから反応混合物を30分間180℃で保持した。それから反応を160℃に冷却させ、温度を150〜160℃に維持しながら3日間アニールした。
【0094】
アニール後、混合物を室温に冷却させ、その後500mlの分量を大きなビーカーに注入し、アセトニトリルで沈殿させた。それから凝集した溶液を遠心分離機チューブに移動させ、ペレットが形成されるまで遠心分離した。透明な上澄みは廃棄し、オレンジ沈殿物を最小量のクロロホルムに再溶解させ、終夜空気中に放置した。
【0095】
後処理
ミリスチン酸におけるプレエッチング
クロロホルムを回転蒸発器において除去し、固体はセバシン酸ジブチルエステル(300ml)に再溶解させた。それからミリスチン酸(40g)を加え、混合物を6時間140℃まで加熱した。混合物を冷却させ、上記と同様にアセトニトリルで沈殿させた。
【0096】
HFエッチング
クロロホルム(900ml)にペレットを再溶解させ、この溶液をHFエッチングに用いた。500Wのタングステン球の前に溶液を置き、8mlのテトラヒドロフラン内の2mlのHF(48%)の溶液を撹拌しながらゆっくり加えた。溶液の光学発光スペクトルを光学手プローブによって監視し、それは強度が増すにつれて徐々に赤方偏移を始めた。記録している波長が青方偏移を始めた時点までHF溶液を加え、それから、HF/THF/CHCI3を空気で吹き飛ばした。結果として生じるペーストをアセトンで洗浄し、アセトニトリルで沈殿させ、それからヘキサンで洗浄して、再度アセトニトリルにおいて沈殿させた。それから沈殿物を最小量のセバシン酸ジブチルエステルに溶解させ、脱気してアルゴン下に置いた。
【0097】
実施例5
赤発光を有するInP量子ドットの調製
上記の手順に従って、ミリスチン酸インジウム及びTMSPを調製した。
【0098】
ミリスチン酸(120.000g、0.526モル)及びセバシン酸ジブチル(2400ml)を60〜90℃の温度で1時間高真空下で脱気した。系をアルゴンでパージし、それから[Et3NH4][Zn10S4(SPh)16]クラスター(11.28g)をすぐに加えた。それから反応混合物を温度が100℃に到達するまで15分間加熱し、この段階で、反応混合物をそれから30分間100℃で維持した。クラスターは完全に溶解せず、白い懸濁液を形成した。
【0099】
ミリスチン酸インジウム(144ml、0.25M)の最初の添加を1秒に3滴の速度で始め、温度は終始100℃に維持した。ミリスチン酸溶液の添加完了後、反応混合物を、5分間100℃で撹拌した。それからTMSP(144ml、0.25M)を1秒に2滴の速度で加え、ここでも慎重に温度を監視した。添加が完了するとすぐに、反応混合物の温度を出来るだけ急速に180℃に上昇させ、絶対に超過しないよう注意した(温度上昇には30分強しかかからなかった)。180℃に達するとすぐに、ここでも1秒に3滴の速度で、ミリスチン酸インジウム(192ml、0.25M)の2回目の添加を行った。再度反応を5分間180℃で保持し、その後TMSP(192ml、0.25M)の2回目の添加を1秒に2滴の速度で行った。添加が完了した直後に温度を200℃まで上昇させ、ここでも超過しないよう注意した。そしてミリスチン酸インジウム(120ml、0.25M)の最後の分量を滴加した。それから反応を200℃で60分間保持し、その後160℃に冷却した。それから、140〜160℃の間で温度を維持しながら反応混合物を3日間アニールした。
【0100】
アニール後、混合物を室温に冷却させ、その後500mlの分量を大きなビーカーに注入し、アセトニトリル(600ml)で沈殿させた。それから凝集した溶液を遠心分離機チューブに移動させ、4000rpmで5分間遠心分離した。透明な上澄みは廃棄し、赤い沈殿物をクロロホルムに再溶解させ、終夜空気中に放置した。
【0101】
後処理
ミリスチン酸におけるプレエッチング
クロロホルムを回転蒸発器において除去し、固体はセバシン酸ジブチルエステル(600ml)に再溶解させた。それからミリスチン酸(80g)を加え、混合物を6時間140℃まで加熱した。混合物を冷却させ、上記と同様にアセトニトリルで沈殿させた。
【0102】
HFエッチング
クロロホルム(1750ml)にペレットを再溶解させ、この溶液をHFエッチングに用いた。500Wのタングステン球の前に溶液を置き、8mlのテトラヒドロフラン内の2mlのHF(48%)の溶液を撹拌しながらゆっくり加えた。溶液の光学発光スペクトルを光学手プローブによって監視し、それは強度が増すにつれて徐々に赤方偏移を始めた。記録している波長が青方偏移を始めた時点までHF溶液を加え、それから、HF/THF/CHCI3を空気で吹き飛ばした。結果として生じるペーストをアセトンで洗浄し、アセトニトリルで沈殿させ、それからヘキサンで洗浄してもう一度アセトニトリルで沈殿させた。それから沈殿物を最小量のセバシン酸ジブチルエステルに溶解させ、脱気してアルゴン下に置いた。
【0103】
実施例6
赤発光を有するInP量子ドットの調製
ミリスチン酸(60.000g、0.263モル)及びセバシン酸ジブチル(1200ml)を60〜90℃の温度で1時間高真空下で脱気した。系をアルゴンでパージし、それから[Et3NH4][Zn10S4(SPh)16]クラスター(5.64g)をすぐに加えた。それから反応混合物を100℃の温度に到達するまで15分間加熱し、この段階で、反応混合物をそれから30分間100℃で維持した。クラスターは完全に溶解せず、白い懸濁液を形成した。
【0104】
ミリスチン酸インジウム(72ml、0.25M)の最初の添加を1秒に3滴の速度で始め、温度は終始100℃に維持した。ミリスチン酸溶液の添加完了後、反応混合物を5分間100℃で撹拌した。それからTMSP(72ml、0.25M)を1秒に2滴の速度で加え、ここでも慎重に温度を監視した。添加が完了するとすぐに、反応混合物の温度を出来るだけ急速に180℃に上昇させ、絶対に超過しないよう注意した(温度上昇には30分強しかかからなかった)。180℃に達するとすぐに、ここでも1秒に3滴の速度で、ミリスチン酸インジウム(96ml、0.25M)の2回目の添加を行った。再度反応を5分間180℃で保持し、その後TMSP(96ml、0.25M)の2回目の添加を1秒に2滴の速度で行った。添加が完了した直後に、温度を200℃まで上昇させ、ここでも超過しないよう注意した。それから反応を200℃で60分間保持し、その後160℃に冷却した。それから、反応混合物を140〜160℃の間で3日間アニールした。
【0105】
後処理
ミリスチン酸におけるプレエッチング
クロロホルムを回転蒸発器において除去し、固体はセバシン酸ジブチルエステル(300ml)に再溶解させた。それからミリスチン酸(40g)を加え、混合物を6時間140℃まで加熱した。混合物を冷却させ、上記と同様にアセトニトリルで沈殿させた。
【0106】
HFエッチング
クロロホルム(900ml)にペレットを再溶解させ、この溶液をHFエッチングに用いた。500Wのタングステン球の前に溶液を置き、8mlのテトラヒドロフラン内の2mlのHF(48%)の溶液を撹拌しながらゆっくり加えた。溶液の光学発光スペクトルを光学手プローブによって監視し、それは強度が増すにつれて徐々に赤方偏移を始めた。記録している波長が青方偏移を始めた時点までHF溶液を加え、それからHF/THF/CHCI3を空気で吹き飛ばした。結果として生じるペーストをアセトンで洗浄し、アセトニトリルで沈殿させ、それからヘキサンで洗浄してもう一度アセトニトリルで沈殿させた。それから沈殿物を最小量のセバシン酸ジブチルエステルに溶解させ、脱気してアルゴン下に置いた。
【0107】
実施例7
InMA3前駆体の調製
三つ口フラスコ(1リットル)において、In(Ac)3(100g)及びミリスチン酸(313g)を窒素下で2時間140℃まで加熱した。反応混合物は80℃に冷却し、反応フラスコを蒸留装置に取り付けた。反応混合物は5時間ゆるやかな真空下に置き、その後60℃で窒素下に終夜置いた。(反応のこの第1ステップにおいて60mLの酢酸を収集した)。1リットルの三角フラスコに分析用アセトン(400mL)を加えて静かに撹拌した。反応混合物を途切れなく撹拌しながらこのアセトンにゆっくり加えた。反応混合物を15分間撹拌し、それから放置した。底部に黄色の固体が沈殿し、過剰溶媒を別の容器へ移した。溶媒の変色が最小限で固体が白くなるまで、黄色の固体をアセトンで3回洗浄した。固体をブフナー漏斗及びフラスコ装置を用いて濾過し、それから真空下に終夜デシケータにおいて乾燥させた。最終生成物:InMA3乾燥粉末の質量=226.82g。
【0108】
InMA3(1モルの)前駆体溶液の調製
コンデンサ及び温度計を配備した250mLの三つ口フラスコにおいて、InMA3(100g)及びセバシン酸ジブチル(30mL)を100℃で1時間30分間完全に脱気した。溶液が無色になるまで反応温度を維持した。InMA3溶液は室温で凝固するので、用いるときには、およそ60℃の温度に保ち、しっかりと撹拌しなければならない。
【0109】
(TMSi)3P(1モルの)前駆体溶液の調製
250mLの三つ口フラスコにおいて、セバシン酸ジブチル(100mL)を100℃で2時間30分間脱気した。オーブン乾燥した大きなシュレンクチューブにおいて、セバシン酸ジブチル(71mL)、続いて(TMSi)3P(29mL)を窒素の強い流れの下で加えた。
【0110】
グリーン発光を有するInP量子ドットの調製及びZnS/ZnOシェルの成長
セバシン酸ジブチル(100mL)及びミリスチン酸(5.125g)を、コンデンサ及び温度計を配備した250mLの三つ口フラスコにおいて真空下で100℃で1時間脱気した。[Et3NH4]Zn10S4(SPh)16クラスター(0.47g)を窒素の強い流れの下でしっかりと撹拌しながら80℃で加えた。反応混合物を30分間しっかりと撹拌した。それから反応温度を100℃に上昇させた。
【0111】
InPナノ粒子のシーディングは、InMA3溶液(1M、3mL)をゆっくりと滴加することによって開始され、それは、それから5分間しっかりと撹拌した。5分間の後、(TMSi)3P(1M、3mL)をゆっくりと滴加した。それから反応温度を160℃に上昇させた。160℃でInMA3(1M、4mL)の2回目の滴加をゆっくり行った。反応混合物を5分間撹拌し、その後に(TMSi)3P(1M、4mL)の2回目の添加を行った。好ましくは、反応溶液はすべての添加の間しっかりと撹拌されるべきである。反応温度を190℃に上昇させ、1時間撹拌した。それから反応温度を160℃まで下降させ、そして反応混合物を3日間この温度でアニールした。
【0112】
光ルミネセンスを測定するために5nmの切れ込みを用いてもよい。3日間の後、反応混合物を室温に冷却した。無水アセトニトリル(100mL)の添加によってInPナノ粒子を窒素下で分離した。固体を遠心分離を用いて分離し、CHCl3(200mL)に再溶解した。
【0113】
後処理
HFエッチング
2mLの水性HF(58〜62%wt.溶液)とTHF(8mL分析用)とを組み合わせることによってHF溶液を調製した。250mLの三角フラスコで、HF(全体で1.4mL)を、上記のように調製されるクロロホルム中に分散しているInPナノ粒子に加えた。空気中でしっかりと撹拌しながら7〜8時間にわたってHF溶液を0.2mL滴加した。反応混合物に、サイズによってカットオフ波長を有するカットオフフィルタを通過した光の照射をした。光源として500Wのキセノン灯を用いた。
【0114】
エッチングの処理は、エッチングするナノ粒子の特定のバッチの規模に応じて、およそ10〜15時間後に終了した。CHCl3の蒸発によって、高発光性のHFエッチングされたInPナノ粒子を反応混合物から分離した。それからHFエッチングされたInPナノ粒子をアセトン(10〜15mL)で洗浄し、続いてアセトニトリル(100mL)を加えた。固体を遠心分離によって分離し、そしてアセトン(5mL)で2回目の洗浄を行い、その後セバシン酸ジブチル(バッチの規模によっておよそ20mL)に分散させた。効果的にエッチングされたInPナノ粒子は15〜30%のPL量子効率を有する強い発光を示した。
【0115】
この方法では、535nm(±5nm)及びFWHM<70nmで発光を有する約2グラムのInP量子ドットが生じた。
【0116】
ZnS/ZnOシェルの成長
セバシン酸ジブチル(15mL)及びミリスチン酸(5.2g)を、コンデンサに取り付けた温度計を具えた250mLの枝付き三つ口フラスコにおいて、1時間30分間100℃で脱気した。上記の様に調製されたInPナノ粒子(2.32g)を、別のフラスコにおいて1時間脱気した。セバシン酸ジブチル(15mL)及びミリスチン酸(5.2g)の反応混合物をおよそ60℃まで冷却し、そしてInPナノ粒子を加えた。反応混合物を更に45分間脱気した。
【0117】
窒素の強い流れの下で、しっかりと撹拌しながら、枝から一部に酢酸亜鉛(2.8gを)を加えた。反応混合物を15分間脱気し、脱気の間N2で数回洗浄した。反応温度を120℃に上昇させた。
【0118】
オクタンチオール(0.82mL)を120℃で速く加え、温度は最高220℃まで傾斜した。ZnSシェルを形成するために温度を1時間30分間220℃で保持した。それから反応溶液を180℃まで冷却した。
【0119】
180℃で1―オクタノール(2.2mL)をゆっくり滴加した。添加完了後、溶液を190℃で30分間撹拌し、ZnOシェルを形成した。
【0120】
反応混合物を室温に冷却し、終夜撹拌した。InP/ZnS/ZnOコア―シェル粒子を、無水アセトニトリル(80mL)を有するN2下で分離し、遠心分離によって収集した。粒子をトルエン(20mL)に分散させて、アセトニトリルで再沈殿させ、続いて遠心分離した。それから粒子をトルエン(20mL)に再分散させた。最終的な溶液を遠心分離し、不溶性物質を除去した。最終生成物:質量=1.62g、PL最大=521nm、FWHM=61nm、PLQY=64%。
【0121】
実施例8
赤発光を有するInP量子ドットの調製
セバシン酸ジブチル(100mL)及びミリスチン酸(5.125g)を、コンデンサ及び温度計を備えた250mLの三つ口フラスコにおいて真空下で、100℃で1時間脱気した。[Et3NH4]Zn10S4(SPh)16クラスター(0.47g)を窒素の強い流れの下で、しっかりと撹拌しながら80℃で加えた。反応混合物を、30分間しっかりと撹拌した。反応温度を100℃に上昇させた。
【0122】
InPナノ粒子のシーディングは、InMA3溶液(1M、3mL)をゆっくりと滴加することによって開始され、それは、それから5分間しっかりと撹拌した。5分間の後、(TMSi)3P(1M、3mL)をゆっくり滴加した。反応温度を160℃に上昇させた。160℃でInMA3(1M、4mL)の2回目の滴加をゆっくりと行った。反応混合物を5分間撹拌し、その後に(TMSi)3P(1M、4mL)の2回目の添加を行った。反応温度は180℃に上昇させ、InMA3溶液(1M、3mL)、続いて(TMSi)3P(1M、2.25mL)の3回目の添加を行った。好ましくは、反応溶液はすべての添加の間しっかりと撹拌されるべきである。反応温度を200℃に上昇させ、1時間撹拌した。それから反応温度を170℃まで下降させ、そして反応混合物を3日間この温度でアニールした。
【0123】
光ルミネセンスを測定するために5nmの切れ込みを用いてもよい。3日間の後、反応混合物を室温に冷却した。無水アセトニトリル(100mL)の添加によって、InPコアを窒素下で分離した。固体を遠心分離を用いて分離し、CHCl3(200mL)に再溶解した。
【0124】
後処理
HFエッチング
4mLの水性HF(58〜62%wt.溶液)とTHF(16mL分析用)とを組み合わせることによってHF溶液を調製した。250mLの三角フラスコで、HF(全体で5mL)を、クロロホルムにおいて分散しているInPナノ粒子に加えた。空気中でしっかりと撹拌しながら、5〜8時間にわたってHF溶液を0.2mL滴加した。反応混合物に、サイズによってカットオフ波長を有するカットオフフィルタを通過する光の照射をした。光源として500Wのキセノン灯を用いた。
【0125】
エッチングの処理は、エッチングするナノ粒子の特定のバッチの規模に応じて、およそ10〜15時間後に終了する。CHCl3の蒸発によって、高発光性のHFエッチングされたInPナノ粒子を反応混合物から分離した。それからHFエッチングされたInPナノ粒子をアセトン(10〜15mL)で洗浄し、続いてアセトニトリル(100mL)を加える。固体を遠心分離によって分離し、そしてアセトン(5mL)で2回目の洗浄を行い、セバシン酸ジブチル(バッチの規模によっておよそ20mL)に分散させた。効果的にエッチングされたInPナノ粒子は、15〜30%のPL量子効率を有する強い発光を示す。
【0126】
この方法では、610nm(±5nm)及びFWHM<90nmで発光を有する約2グラムのInP量子ドットが生じた。
【0127】
ZnS/ZnOシェルの成長
セバシン酸ジブチル(15mL)及びミリスチン酸(6.71g)を、コンデンサに取り付けた温度計を備えた250mLの枝付き三つ口フラスコにおいて1時間30分間100℃で脱気した。上記の様に調製されたInPナノ粒子(2.58g)を別のフラスコにおいて1時間脱気した。セバシン酸ジブチル(15mL)及びミリスチン酸(6.71g)の反応混合物をおよそ60℃まで冷却し、そしてInPナノ粒子を加えた。反応混合物を更に45分間脱気した。
【0128】
窒素の強い流れ下でしっかりと撹拌しながら、枝から一部に酢酸亜鉛(3.61g)を加えた。反応混合物を15分間脱気し、脱気の間N2で数回洗浄した。反応温度を120℃に上昇させた。
【0129】
オクタンチオール(1.1mL)を120℃で速く加え、温度は最高220℃まで傾斜した。ZnSシェルを形成するために温度を1時間30分間220℃で保持した。それから反応溶液を180℃まで冷却した。
【0130】
180℃で1―オクタノール(2.84mL)をゆっくり滴加した。添加完了後、溶液を190℃で30分間撹拌し、ZnOシェルを形成する。
【0131】
反応混合物を室温に冷却し、終夜撹拌した。InP/ZnS/ZnOコア―シェル粒子を、無水アセトニトリル(80mL)を有するN2下で分離し、遠心分離によって収集した。粒子をトルエン(30mL)に分散させてアセトニトリルで再沈殿させ、続いて遠心分離した。それから粒子をトルエン(50mL)に再分散させた。最終的な溶液を遠心分離し、不溶性物質を除去した。最終生成物:質量=3.2g、PL最大=605nm、FWHM=93nm、PLQY=61%。
【0132】
【非特許文献1】Murray, C. B.; Norris, D. J.; Bawendi, M. G. J. Am. Chem. Soc. 1993, 115, 8706。
【非特許文献2】LOver, T.; Bowmaker, G. A.; Seakins, J. M.; Cooney, R. P.; Henderson, W. J. Mater. Chem., 1997, 7(4), 647。
【非特許文献3】Cumberland, S. L.; Hanif, K. M.; Javier, A.; Khitov, K. A.; Strouse, G. F.; Woessner, S. M.; Yun, C. S. Chem. Mater. 2002, 14, 1576。
【特許請求の範囲】
【請求項1】
周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物と、前記分子クラスター化合物に提供されるコア半導体材料とからなるナノ粒子であって、コア半導体材料は、周期表の第13族及び第15族からのイオンを組み込むナノ粒子。
【請求項2】
分子クラスター化合物及びコア半導体材料は、前記分子クラスター化合物上の前記コア半導体材料の成長を可能にする互換性を持つ結晶相を有する請求項1に記載のナノ粒子。
【請求項3】
分子クラスター化合物及びコア半導体材料は、同じ結晶相を有する請求項1に記載のナノ粒子。
【請求項4】
分子クラスター化合物は、亜鉛イオンを組み込む上記請求項の何れかに記載のナノ粒子。
【請求項5】
分子クラスター化合物は、硫化物イオン、セレンイオン、及びテルル化物イオンからなる群から選択される第16族イオンを組み込む上記請求項の何れかに記載のナノ粒子。
【請求項6】
分子クラスター化合物は、[{(PPh3)Hg}4(SPh)6]、(Ph4P)2[(SEt)5(Br)(HgBr)4]、(Ph4P)2[Hg4(SEt)5Br]、及び[Hg4Te12][N(CH2CH2Et)4]4からなる群から選択される請求項1、2又は3に記載のナノ粒子。
【請求項7】
分子クラスター化合物は、[RMEtBu]5但しM=Zn,Cd,Hg、E=S,Se,Te、R=Me,Et,Ph、[X]4[E4M10(SR)16]但しM=Zn,Cd,Hg,E=S,Se,Te,X=Me3NH+,Li+,Et3NH+、[Cd32S14(SPh)36]・L、[Hg10Se4(SePh)(PPh2nPr)4]、[Hg32Se14(SePh)36]、[Cd10Se4(SePh)12(PPr3)4]、[Cd32Sel4(SePh)36(PPh3)4]、[M4(SPh)12]+[X]2−但しM=Zn,Cd,Hg,X=Me4N+,Li+、[Zn(SEt)Et]10、[MeMEiPr]但しM=Zn,Cd,Hg,E=S,Se,Te、[RCdSR’]5但しR=O(ClO3),R’=PPh3,iPr、[Cd10E4(E’Ph)12(PR3)4]但しE=Te,Se,Sそして別にE’=Te,Se,S、[Cd8Se(SePh)12Cl4]2−、[M4Te12]4−但しM=Cd,Hg、及び[Ph12M18Cd10(PEt3)3]但しM=Te,Seからなる群から選択される請求項1、2又は3に記載のナノ粒子。
【請求項8】
コア半導体材料は、アルミニウムイオン、ガリウムイオン、及びインジウムイオンからなる群から選択される第13族イオンを組み込む上記請求項の何れかに記載のナノ粒子。
【請求項9】
コア半導体材料の第13族イオンは、III―ホスフィン、III―(TMS)3、III―(アルキル)、III―(アリール)、III―(ミリスチン酸塩)3、III―(アセテート)3、III―(ミリスチン酸塩)(アセテート)2、III―(ミリスチン酸塩)2(アセテート)、及びIII―(III)アセチルアセトネートからなる群から選択される供給源に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項10】
コア半導体材料の第13族イオンは、有機金属化合物に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項11】
コア半導体材料の第13族イオンは、配位化合物に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項12】
コア半導体材料の第13族イオンは、無機塩に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項13】
コア半導体材料の第13族イオンは、元素供給源に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項14】
コア半導体材料は、窒化物イオン、ヒ化物イオン、及びアンチモンイオンからなる群から選択される第15族イオンを組み込む上記請求項の何れかに記載のナノ粒子。
【請求項15】
コア半導体材料の第15族イオンは、有機金属化合物に由来している請求項1乃至13の何れか1つに記載のナノ粒子。
【請求項16】
コア半導体材料の第15族イオンは、配位化合物に由来している請求項1乃至13の何れか1つに記載のナノ粒子。
【請求項17】
コア半導体材料の第15族イオンは、無機塩に由来している請求項1乃至13の何れか1つに記載のナノ粒子。
【請求項18】
コア半導体材料の第15族イオンは、元素供給源に由来している請求項1乃至13の何れか1つに記載のナノ粒子。
【請求項19】
コア半導体材料の第13族イオン及び第15族イオンは、[(Me)2GaN(H)tBu]2 [H2GaNH2]3 [Ph2GaP(SiMe3)3Ga(Ph)2Cl]、[Et2GaP(SiMe3)2]2、[Et2GaPEt2]3、[tBu2GaPH2]3、[Me2GaP(iPr)2]3、[tBuGaPAr´]2、[tBu2GaP(H)C5H9]2、Ga(AstBu2)3、[Et2GaAs(SiMe3)2]2、[tBu2GaAs(SiMe3)2]2、[Et2GaSb(SiMe3)2]2、[(Me3SiCH2)2InP(SiMe3)2]2、[R2InP(SiMe3)2]2、[Me2InPtBu2]2、[Me2InSbtBu2]3、[Et2InSb(SiMe3)2]3、[Me2InNEt2]2、[Et2AlAstBu2]2、[tBu2AlSb(SiMe3)2]2、[nBu2GaAstBu2]2、[Me2Ga2AstBu2]2、及び[Et2GaAstBu2]2からなる群から選択される単一供給源に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項20】
ナノ粒子は、約20〜60%の範囲の量子効率を示す上記請求項の何れかに記載のナノ粒子。
【請求項21】
ナノ粒子は、前記ナノ粒子コア上に提供される第1の半導体材料を含む第1の層を具える上記請求項の何れかに記載のナノ粒子。
【請求項22】
前記第1の半導体材料は、周期表の第12族からのイオンを組み込む請求項21に記載のナノ粒子。
【請求項23】
第1の半導体材料は、亜鉛イオンを組み込む請求項21に記載のナノ粒子。
【請求項24】
前記第1の半導体材料は、周期表の第16族からのイオンを組み込む請求項21、22又は23に記載のナノ粒子。
【請求項25】
第1の半導体材料は、硫化物イオン、セレンイオン、及びテルル化物イオンからなる群から選択される第16族イオンを組み込む請求項21、22又は23に記載のナノ粒子。
【請求項26】
ナノ粒子は、前記第1の層上に提供される第2の半導体材料を含む第2の層を具える上記請求項の何れかに記載のナノ粒子。
【請求項27】
周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物と、前記分子クラスター化合物上に提供されるコア半導体材料とを具えるナノ粒子を製造する方法であって、コア半導体材料は周期表の第13族及び第15族からのイオンを組み込み、当該方法は、ナノ粒子コア材料のシーディング及び成長を可能にする条件の下で、周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物の存在下で、それぞれ周期表の第13族及び第15族からのイオンを含む第1及び第2のコア前駆体種を具えているナノ粒子コア前駆体組成物の、ナノ粒子コアの材料への転化を生じさせることを含む、ナノ粒子の製造方法。
【請求項28】
分子クラスター化合物は、亜鉛イオンを組み込む請求項27に記載の方法。
【請求項29】
分子クラスター化合物は、硫化物イオン、セレンイオン、及びテルル化物イオンからなる群から選択される第16族イオンを組み込む請求項27又は28に記載の方法。
【請求項30】
分子クラスター化合物は、[{(PPh3)Hg}4(SPh)6]、(Ph4P)2[(SEt)5(Br)(HgBr)4]、(Ph4P)2[Hg4(SEt)5Br]、及び[Hg4Te12][N(CH2CH2Et)4]4からなる群から選択される請求項27に記載の方法。
【請求項31】
分子クラスター化合物は、[RMEtBu]5但しM=Zn,Cd,Hg、E=S,Se,Te、R=Me,Et,Ph、[X]4[E4M10(SR)16]但しM=Zn,Cd,Hg,E=S,Se,Te,X=Me3NH+,Li+,Et3NH+、[Cd32S14(SPh)36]・L、[Hg10Se4(SePh)(PPh2nPr)4]、[Hg32Se14(SePh)36]、[Cd10Se4(SePh)12(PPr3)4]、[Cd32Sel4(SePh)36(PPh3)4]、[M4(SPh)12]+[X]2−但しM=Zn,Cd,Hg,X=Me4N+,Li+、[Zn(SEt)Et]10、[MeMEiPr]但しM=Zn,Cd,Hg,E=S,Se,Te、[RCdSR’]5但しR=O(ClO3),R’=PPh3,iPr、[Cd10E4(E’Ph)12(PR3)4]但しE=Te,Se,Sそして別にE’=Te,Se,S、[Cd8Se(SePh)12Cl4]2−、[M4Te12]4−但しM=Cd,Hg、及び[Ph12M18Cd10(PEt3)3]但しM=Te,Seからなる群から選択される請求項27に記載の方法。
【請求項32】
コア半導体材料は、アルミニウムイオン、ガリウムイオン、及びインジウムイオンからなる群から選択される第13族イオンを組み込む請求項27乃至31の何れか1つに記載の方法。
【請求項33】
コア半導体材料は、窒化物イオン、ヒ化物イオン、及びアンチモンイオンからなる群から選択される第15族イオンを組み込む請求項27乃至32の何れか1つに記載の方法。
【請求項34】
前記第1及び第2のコア前駆体種は、前記コア前駆体組成物に含まれる別の実体である請求項27乃至33の何れか1つに記載の方法。
【請求項35】
コア半導体材料の第13族イオンは、III―ホスフィン、III―(TMS)3、III―(アルキル)、III―(アリール)、III―(ミリスチン酸塩)3、III―(アセテート)3、III―(ミリスチン酸塩)(アセテート)2、III―(ミリスチン酸塩)2(アセテート)、及びIII―(III) アセチルアセトネートからなる群から選択される供給源に由来している請求項34に記載の方法。
【請求項36】
コア半導体材料の第13族イオンは、有機金属化合物に由来している請求項34に記載の方法。
【請求項37】
コア半導体材料の第13族イオンは、配位化合物に由来している請求項34に記載の方法。
【請求項38】
コア半導体材料の第13族イオンは、無機塩に由来している請求項34に記載の方法。
【請求項39】
コア半導体材料の第13族イオンは、元素供給源に由来している請求項34に記載の方法。
【請求項40】
コア半導体材料の第15族イオンは、有機金属化合物に由来している請求項34乃至39の何れか1つに記載の方法。
【請求項41】
コア半導体材料の第15族イオンは、配位化合物に由来している請求項34乃至39の何れか1つに記載の方法。
【請求項42】
コア半導体材料の第15族イオンは、無機塩に由来している請求項34乃至39の何れか1つに記載の方法。
【請求項43】
コア半導体材料の第15族イオンは、元素供給源に由来している請求項34乃至39の何れか1つに記載の方法。
【請求項44】
前記第1及び第2のコア前駆体種は、前記コア前駆体組成物に含まれる単体において組み合わせられる請求項27乃至33の何れか1つに記載の方法。
【請求項45】
コア半導体材料の第13族イオン及び第15族イオンは、[(Me)2GaN(H)tBu]2 [H2GaNH2]3 [Ph2GaP(SiMe3)3Ga(Ph)2Cl]、[Et2GaP(SiMe3)2]2、[Et2GaPEt2]3、[tBu2GaPH2]3、[Me2GaP(iPr)2]3、[tBuGaPAr´]2、[tBu2GaP(H)C5H9]2、Ga(AstBu2)3、[Et2GaAs(SiMe3)2]2、[tBu2GaAs(SiMe3)2]2、[Et2GaSb(SiMe3)2]2、[(Me3SiCH2)2InP(SiMe3)2]2、[R2InP(SiMe3)2]2、[Me2InPtBu2]2、[Me2InSbtBu2]3、[Et2InSb(SiMe3)2]3、[Me2InNEt2]2、[Et2AlAstBu2]2、[tBu2AlSb(SiMe3)2]2、[nBu2GaAstBu2]2、[Me2Ga2AstBu2]2、及び[Et2GaAstBu2]2からなる群から選択される単一供給源に由来している請求項44に記載の方法。
【請求項46】
方法は、ナノ粒子コア材料上の第1の半導体材料から構成される第1の層を形成することを更に含む請求項27乃至45の何れか1つに記載の方法。
【請求項47】
第1の半導体材料の第1の層の形成は、第1の材料前駆体組成物の、前記第1の材料への転化を生じさせることを含む請求項46に記載の方法。
【請求項48】
第1の材料前駆体組成物が、第1の材料から構成される層に組み込まれる第3及び第4イオンを具える請求項47に記載の方法。
【請求項49】
第3及び第4イオンは、第1の材料前駆体組成物において含まれる別の実体である請求項48に記載の方法。
【請求項50】
第3及び第4イオンは、第1の材料前駆体組成物において含まれる単体において組み合わせられる請求項48に記載の方法。
【請求項51】
第1の半導体材料の第1の層の形成の前にナノ粒子コア材料がフッ化水素に晒される請求項46乃至50の何れか1つに記載の方法。
【請求項1】
周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物と、前記分子クラスター化合物に提供されるコア半導体材料とからなるナノ粒子であって、コア半導体材料は、周期表の第13族及び第15族からのイオンを組み込むナノ粒子。
【請求項2】
分子クラスター化合物及びコア半導体材料は、前記分子クラスター化合物上の前記コア半導体材料の成長を可能にする互換性を持つ結晶相を有する請求項1に記載のナノ粒子。
【請求項3】
分子クラスター化合物及びコア半導体材料は、同じ結晶相を有する請求項1に記載のナノ粒子。
【請求項4】
分子クラスター化合物は、亜鉛イオンを組み込む上記請求項の何れかに記載のナノ粒子。
【請求項5】
分子クラスター化合物は、硫化物イオン、セレンイオン、及びテルル化物イオンからなる群から選択される第16族イオンを組み込む上記請求項の何れかに記載のナノ粒子。
【請求項6】
分子クラスター化合物は、[{(PPh3)Hg}4(SPh)6]、(Ph4P)2[(SEt)5(Br)(HgBr)4]、(Ph4P)2[Hg4(SEt)5Br]、及び[Hg4Te12][N(CH2CH2Et)4]4からなる群から選択される請求項1、2又は3に記載のナノ粒子。
【請求項7】
分子クラスター化合物は、[RMEtBu]5但しM=Zn,Cd,Hg、E=S,Se,Te、R=Me,Et,Ph、[X]4[E4M10(SR)16]但しM=Zn,Cd,Hg,E=S,Se,Te,X=Me3NH+,Li+,Et3NH+、[Cd32S14(SPh)36]・L、[Hg10Se4(SePh)(PPh2nPr)4]、[Hg32Se14(SePh)36]、[Cd10Se4(SePh)12(PPr3)4]、[Cd32Sel4(SePh)36(PPh3)4]、[M4(SPh)12]+[X]2−但しM=Zn,Cd,Hg,X=Me4N+,Li+、[Zn(SEt)Et]10、[MeMEiPr]但しM=Zn,Cd,Hg,E=S,Se,Te、[RCdSR’]5但しR=O(ClO3),R’=PPh3,iPr、[Cd10E4(E’Ph)12(PR3)4]但しE=Te,Se,Sそして別にE’=Te,Se,S、[Cd8Se(SePh)12Cl4]2−、[M4Te12]4−但しM=Cd,Hg、及び[Ph12M18Cd10(PEt3)3]但しM=Te,Seからなる群から選択される請求項1、2又は3に記載のナノ粒子。
【請求項8】
コア半導体材料は、アルミニウムイオン、ガリウムイオン、及びインジウムイオンからなる群から選択される第13族イオンを組み込む上記請求項の何れかに記載のナノ粒子。
【請求項9】
コア半導体材料の第13族イオンは、III―ホスフィン、III―(TMS)3、III―(アルキル)、III―(アリール)、III―(ミリスチン酸塩)3、III―(アセテート)3、III―(ミリスチン酸塩)(アセテート)2、III―(ミリスチン酸塩)2(アセテート)、及びIII―(III)アセチルアセトネートからなる群から選択される供給源に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項10】
コア半導体材料の第13族イオンは、有機金属化合物に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項11】
コア半導体材料の第13族イオンは、配位化合物に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項12】
コア半導体材料の第13族イオンは、無機塩に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項13】
コア半導体材料の第13族イオンは、元素供給源に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項14】
コア半導体材料は、窒化物イオン、ヒ化物イオン、及びアンチモンイオンからなる群から選択される第15族イオンを組み込む上記請求項の何れかに記載のナノ粒子。
【請求項15】
コア半導体材料の第15族イオンは、有機金属化合物に由来している請求項1乃至13の何れか1つに記載のナノ粒子。
【請求項16】
コア半導体材料の第15族イオンは、配位化合物に由来している請求項1乃至13の何れか1つに記載のナノ粒子。
【請求項17】
コア半導体材料の第15族イオンは、無機塩に由来している請求項1乃至13の何れか1つに記載のナノ粒子。
【請求項18】
コア半導体材料の第15族イオンは、元素供給源に由来している請求項1乃至13の何れか1つに記載のナノ粒子。
【請求項19】
コア半導体材料の第13族イオン及び第15族イオンは、[(Me)2GaN(H)tBu]2 [H2GaNH2]3 [Ph2GaP(SiMe3)3Ga(Ph)2Cl]、[Et2GaP(SiMe3)2]2、[Et2GaPEt2]3、[tBu2GaPH2]3、[Me2GaP(iPr)2]3、[tBuGaPAr´]2、[tBu2GaP(H)C5H9]2、Ga(AstBu2)3、[Et2GaAs(SiMe3)2]2、[tBu2GaAs(SiMe3)2]2、[Et2GaSb(SiMe3)2]2、[(Me3SiCH2)2InP(SiMe3)2]2、[R2InP(SiMe3)2]2、[Me2InPtBu2]2、[Me2InSbtBu2]3、[Et2InSb(SiMe3)2]3、[Me2InNEt2]2、[Et2AlAstBu2]2、[tBu2AlSb(SiMe3)2]2、[nBu2GaAstBu2]2、[Me2Ga2AstBu2]2、及び[Et2GaAstBu2]2からなる群から選択される単一供給源に由来している請求項1乃至7の何れか1つに記載のナノ粒子。
【請求項20】
ナノ粒子は、約20〜60%の範囲の量子効率を示す上記請求項の何れかに記載のナノ粒子。
【請求項21】
ナノ粒子は、前記ナノ粒子コア上に提供される第1の半導体材料を含む第1の層を具える上記請求項の何れかに記載のナノ粒子。
【請求項22】
前記第1の半導体材料は、周期表の第12族からのイオンを組み込む請求項21に記載のナノ粒子。
【請求項23】
第1の半導体材料は、亜鉛イオンを組み込む請求項21に記載のナノ粒子。
【請求項24】
前記第1の半導体材料は、周期表の第16族からのイオンを組み込む請求項21、22又は23に記載のナノ粒子。
【請求項25】
第1の半導体材料は、硫化物イオン、セレンイオン、及びテルル化物イオンからなる群から選択される第16族イオンを組み込む請求項21、22又は23に記載のナノ粒子。
【請求項26】
ナノ粒子は、前記第1の層上に提供される第2の半導体材料を含む第2の層を具える上記請求項の何れかに記載のナノ粒子。
【請求項27】
周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物と、前記分子クラスター化合物上に提供されるコア半導体材料とを具えるナノ粒子を製造する方法であって、コア半導体材料は周期表の第13族及び第15族からのイオンを組み込み、当該方法は、ナノ粒子コア材料のシーディング及び成長を可能にする条件の下で、周期表の第12族及び第16族からのイオンを組み込んでいる分子クラスター化合物の存在下で、それぞれ周期表の第13族及び第15族からのイオンを含む第1及び第2のコア前駆体種を具えているナノ粒子コア前駆体組成物の、ナノ粒子コアの材料への転化を生じさせることを含む、ナノ粒子の製造方法。
【請求項28】
分子クラスター化合物は、亜鉛イオンを組み込む請求項27に記載の方法。
【請求項29】
分子クラスター化合物は、硫化物イオン、セレンイオン、及びテルル化物イオンからなる群から選択される第16族イオンを組み込む請求項27又は28に記載の方法。
【請求項30】
分子クラスター化合物は、[{(PPh3)Hg}4(SPh)6]、(Ph4P)2[(SEt)5(Br)(HgBr)4]、(Ph4P)2[Hg4(SEt)5Br]、及び[Hg4Te12][N(CH2CH2Et)4]4からなる群から選択される請求項27に記載の方法。
【請求項31】
分子クラスター化合物は、[RMEtBu]5但しM=Zn,Cd,Hg、E=S,Se,Te、R=Me,Et,Ph、[X]4[E4M10(SR)16]但しM=Zn,Cd,Hg,E=S,Se,Te,X=Me3NH+,Li+,Et3NH+、[Cd32S14(SPh)36]・L、[Hg10Se4(SePh)(PPh2nPr)4]、[Hg32Se14(SePh)36]、[Cd10Se4(SePh)12(PPr3)4]、[Cd32Sel4(SePh)36(PPh3)4]、[M4(SPh)12]+[X]2−但しM=Zn,Cd,Hg,X=Me4N+,Li+、[Zn(SEt)Et]10、[MeMEiPr]但しM=Zn,Cd,Hg,E=S,Se,Te、[RCdSR’]5但しR=O(ClO3),R’=PPh3,iPr、[Cd10E4(E’Ph)12(PR3)4]但しE=Te,Se,Sそして別にE’=Te,Se,S、[Cd8Se(SePh)12Cl4]2−、[M4Te12]4−但しM=Cd,Hg、及び[Ph12M18Cd10(PEt3)3]但しM=Te,Seからなる群から選択される請求項27に記載の方法。
【請求項32】
コア半導体材料は、アルミニウムイオン、ガリウムイオン、及びインジウムイオンからなる群から選択される第13族イオンを組み込む請求項27乃至31の何れか1つに記載の方法。
【請求項33】
コア半導体材料は、窒化物イオン、ヒ化物イオン、及びアンチモンイオンからなる群から選択される第15族イオンを組み込む請求項27乃至32の何れか1つに記載の方法。
【請求項34】
前記第1及び第2のコア前駆体種は、前記コア前駆体組成物に含まれる別の実体である請求項27乃至33の何れか1つに記載の方法。
【請求項35】
コア半導体材料の第13族イオンは、III―ホスフィン、III―(TMS)3、III―(アルキル)、III―(アリール)、III―(ミリスチン酸塩)3、III―(アセテート)3、III―(ミリスチン酸塩)(アセテート)2、III―(ミリスチン酸塩)2(アセテート)、及びIII―(III) アセチルアセトネートからなる群から選択される供給源に由来している請求項34に記載の方法。
【請求項36】
コア半導体材料の第13族イオンは、有機金属化合物に由来している請求項34に記載の方法。
【請求項37】
コア半導体材料の第13族イオンは、配位化合物に由来している請求項34に記載の方法。
【請求項38】
コア半導体材料の第13族イオンは、無機塩に由来している請求項34に記載の方法。
【請求項39】
コア半導体材料の第13族イオンは、元素供給源に由来している請求項34に記載の方法。
【請求項40】
コア半導体材料の第15族イオンは、有機金属化合物に由来している請求項34乃至39の何れか1つに記載の方法。
【請求項41】
コア半導体材料の第15族イオンは、配位化合物に由来している請求項34乃至39の何れか1つに記載の方法。
【請求項42】
コア半導体材料の第15族イオンは、無機塩に由来している請求項34乃至39の何れか1つに記載の方法。
【請求項43】
コア半導体材料の第15族イオンは、元素供給源に由来している請求項34乃至39の何れか1つに記載の方法。
【請求項44】
前記第1及び第2のコア前駆体種は、前記コア前駆体組成物に含まれる単体において組み合わせられる請求項27乃至33の何れか1つに記載の方法。
【請求項45】
コア半導体材料の第13族イオン及び第15族イオンは、[(Me)2GaN(H)tBu]2 [H2GaNH2]3 [Ph2GaP(SiMe3)3Ga(Ph)2Cl]、[Et2GaP(SiMe3)2]2、[Et2GaPEt2]3、[tBu2GaPH2]3、[Me2GaP(iPr)2]3、[tBuGaPAr´]2、[tBu2GaP(H)C5H9]2、Ga(AstBu2)3、[Et2GaAs(SiMe3)2]2、[tBu2GaAs(SiMe3)2]2、[Et2GaSb(SiMe3)2]2、[(Me3SiCH2)2InP(SiMe3)2]2、[R2InP(SiMe3)2]2、[Me2InPtBu2]2、[Me2InSbtBu2]3、[Et2InSb(SiMe3)2]3、[Me2InNEt2]2、[Et2AlAstBu2]2、[tBu2AlSb(SiMe3)2]2、[nBu2GaAstBu2]2、[Me2Ga2AstBu2]2、及び[Et2GaAstBu2]2からなる群から選択される単一供給源に由来している請求項44に記載の方法。
【請求項46】
方法は、ナノ粒子コア材料上の第1の半導体材料から構成される第1の層を形成することを更に含む請求項27乃至45の何れか1つに記載の方法。
【請求項47】
第1の半導体材料の第1の層の形成は、第1の材料前駆体組成物の、前記第1の材料への転化を生じさせることを含む請求項46に記載の方法。
【請求項48】
第1の材料前駆体組成物が、第1の材料から構成される層に組み込まれる第3及び第4イオンを具える請求項47に記載の方法。
【請求項49】
第3及び第4イオンは、第1の材料前駆体組成物において含まれる別の実体である請求項48に記載の方法。
【請求項50】
第3及び第4イオンは、第1の材料前駆体組成物において含まれる単体において組み合わせられる請求項48に記載の方法。
【請求項51】
第1の半導体材料の第1の層の形成の前にナノ粒子コア材料がフッ化水素に晒される請求項46乃至50の何れか1つに記載の方法。
【図1】

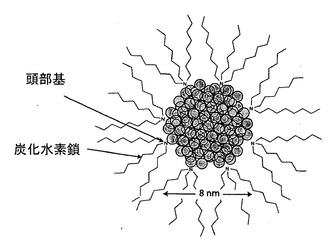
【図2】
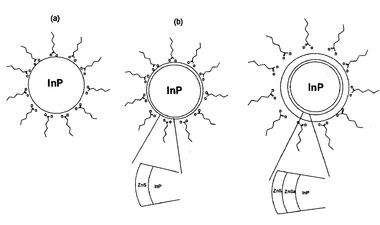
【図3】
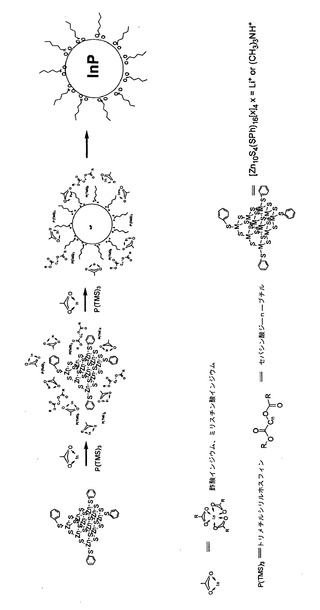
【図4】
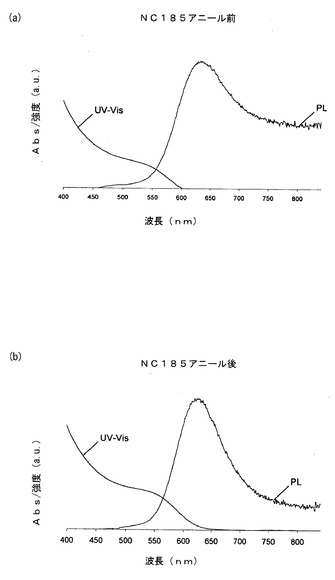
【図5】
【図6】
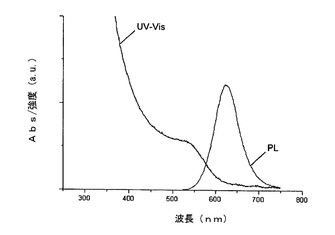
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2010−535262(P2010−535262A)
【公表日】平成22年11月18日(2010.11.18)
【国際特許分類】
【出願番号】特願2010−518730(P2010−518730)
【出願日】平成20年7月28日(2008.7.28)
【国際出願番号】PCT/GB2008/002560
【国際公開番号】WO2009/016354
【国際公開日】平成21年2月5日(2009.2.5)
【出願人】(509295262)ナノコ テクノロジーズ リミテッド (12)
【Fターム(参考)】
【公表日】平成22年11月18日(2010.11.18)
【国際特許分類】
【出願日】平成20年7月28日(2008.7.28)
【国際出願番号】PCT/GB2008/002560
【国際公開番号】WO2009/016354
【国際公開日】平成21年2月5日(2009.2.5)
【出願人】(509295262)ナノコ テクノロジーズ リミテッド (12)
【Fターム(参考)】
[ Back to top ]