
ナファモスタットメシル酸塩の晶析方法
【課題】ナファモスタットメシル酸塩が溶液中でゲル状の塊となることが抑制され、高純度のナファモスタットメシル酸塩を得ることのできるナファモスタットメシル酸塩の晶析方法を提供する。
【解決手段】体積比で水性溶媒:水=9:1〜1:9である水性溶媒と水との混合液に溶解したナファモスタットメシル酸塩に対して、−10〜45℃の温度範囲の下、アセトンを加える工程を有することを特徴とするナファモスタットメシル酸塩の晶析方法である。前記水性溶媒と水との混合液が、体積比で水性溶媒:水=4:6〜7:3の混合液であることが好ましい。
【解決手段】体積比で水性溶媒:水=9:1〜1:9である水性溶媒と水との混合液に溶解したナファモスタットメシル酸塩に対して、−10〜45℃の温度範囲の下、アセトンを加える工程を有することを特徴とするナファモスタットメシル酸塩の晶析方法である。前記水性溶媒と水との混合液が、体積比で水性溶媒:水=4:6〜7:3の混合液であることが好ましい。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ナファモスタットメシル酸塩の晶析方法に関し、詳しくは、ナファモスタットメシル酸塩が溶液中でゲル状の塊となることが抑制され、高純度のナファモスタットメシル酸塩を得ることのできるナファモスタットメシル酸塩の晶析方法に関する。
【背景技術】
【0002】
ナファモスタットメシル酸塩(NAF)の晶析は、一般に、ナファモスタットメシル酸塩の水溶液にアセトンを加える方法により行われる(特許文献1参照)。この方法では、ナファモスタットメシル酸塩水溶液を冷却するとナファモスタットメシル酸塩の析出とともに溶液がゲル状の塊となり、ナファモスタットメシル酸塩のろ過による分離が困難になることが知られている。そのため、ナファモスタットメシル酸塩水溶液を加温しながら晶析を行う必要があった。例えば、特許文献2では、25〜45℃でナファモスタットメシル酸塩水溶液の晶析を行うことが開示されている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2004−284982号公報
【特許文献2】特許第3796481号
【発明の概要】
【発明が解決しようとする課題】
【0004】
しかしながら、ナファモスタットメシル酸塩はエステル化合物であり、水溶液状態での安定性が悪く、加水分解して4−グアニジノ安息香酸や6−アミジノ−2−ナフトール等の分解物が生じることが知られている(薬学雑誌 105(5),512−516(1985))。このため、高純度のナファモスタットメシル酸塩を得るためには、なるべく低温でナファモスタットメシル酸塩溶液の晶析を行うことが望ましい。
【0005】
そこで本発明の目的は、ナファモスタットメシル酸塩が溶液中でゲル状の塊となることが抑制され、高純度のナファモスタットメシル酸塩を得ることのできるナファモスタットメシル酸塩の晶析方法を提供することにある。
【課題を解決するための手段】
【0006】
本発明者等は、上記課題を解決すべく鋭意検討した結果、水性溶媒と水との混合液にナファモスタットメシル酸塩を溶解して得られるナファモスタットメシル酸塩溶液は、冷却してもゲル状の塊にならず、該ナファモスタットメシル酸塩溶液にアセトンを加えることにより、高純度のナファモスタットメシル酸塩を得ることができることを見出し、本発明を完成するに至った。
【0007】
すなわち、本発明のナファモスタットメシル酸塩の晶析方法は、体積比で水性溶媒:水=9:1〜1:9である水性溶媒と水との混合液に溶解したナファモスタットメシル酸塩に対して、−10〜45℃の温度範囲の下、アセトンを加える工程を有することを特徴とするものである。
【0008】
本発明のナファモスタットメシル酸塩の晶析方法においては、前記水性溶媒と水との混合液が、体積比で水性溶媒:水=4:6〜7:3の混合液であることが好ましい。
【0009】
また、本発明のナファモスタットメシル酸塩の晶析方法においては、前記水性溶媒が、炭素原子数1〜3の低級アルコール、炭素原子数3または4のジアルキルケトン、R1−CN(式中、R1はメチル基またはエチル基である)で表されるニトリル、R2−C(=O)−NR3R4(式中、R2、R3およびR4はそれぞれ独立に水素原子、メチル基またはエチル基を表す。)で表されるアミド、R5−S(=O)−R6(式中、R5およびR6はそれぞれ独立にメチル基またはエチル基を表す。)で表されるジアルキルスルホキシド、ジオキサン、および、テトラヒドロフランからなる群から選ばれるものであることが好ましい。
【0010】
さらに、本発明のナファモスタットメシル酸塩の晶析方法においては、前記水性溶媒が、アセトン、メタノール、エタノール、イソプロピルアルコール、アセトニトリル、N,N−ジメチルホルムアミド、ジメチルスルホキシド、ジオキサン、および、テトラヒドロフランからなる群から選ばれるものであることが好ましい。
【0011】
本発明のナファモスタットメシル酸塩の結晶は、上記のナファモスタットメシル酸塩の晶析方法によって得られることを特徴とするものである。
【発明の効果】
【0012】
本発明により、ナファモスタットメシル酸塩が溶液中でゲル状の塊となることが抑制され、高純度のナファモスタットメシル酸塩を得ることのできるナファモスタットメシル酸塩の晶析方法を提供することができる。
【図面の簡単な説明】
【0013】
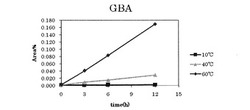
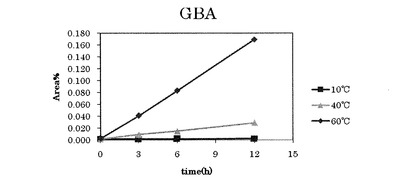
【図1】参考例1〜3におけるp−グアニジノ安息香酸の増加を表すグラフである。
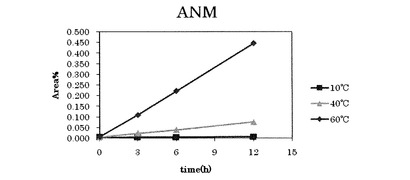
【図2】参考例1〜3における6−アミジノ−2−ナフトールの増加を表すグラフである。
【発明を実施するための形態】
【0014】
本発明のナファモスタットメシル酸塩の晶析方法は、水性溶媒と水との混合液に溶解したナファモスタットメシル酸塩に対して、−10〜45℃の温度範囲の下、アセトンを加える工程を有することを特徴とするものである。
ナファモスタットメシル酸塩は、下記式で表される化合物である4−[(アミノイミノメチル)アミノ]安息香酸6−(アミノイミノメチル)−2−ナフタレニル・2メタンスルホン酸の一般名である。

【0015】
ナファモスタットメシル酸塩を水性溶媒と水との混合液に溶解する方法は、公知のいずれの方法でもよく、例えば水性溶媒と水との混合液を35〜60℃に加温し、攪拌しながら溶解させる方法が好ましい。通常、ナファモスタットメシル酸塩を溶媒に溶解させるには、ある程度加温が必要であるが、あまり高温にするとナファモスタットメシル酸塩が分解する恐れがあるため、上記温度範囲で溶解させるのが好ましい。
【0016】
上記水性溶媒と水との混合液における水性溶媒とは、水との混和性を有する溶媒であればいずれでもよい。また、2種以上の水性触媒を用いてもよい。本発明において好ましい水性溶媒としては、炭素原子数1〜3の低級アルコール、炭素原子数3または4のジアルキルケトン、R1−CN(式中、R1はメチル基またはエチル基である)で表されるニトリル、R2−C(=O)−NR3R4(式中、R2、R3およびR4はそれぞれ独立に水素原子、メチル基またはエチル基を表す。)で表されるアミド、R5−S(=O)−R6(式中、R5およびR6はそれぞれ独立にメチル基またはエチル基を表す。)で表されるジアルキルスルホキシド、ジオキサン、および、テトラヒドロフランを挙げることができる。
【0017】
上記炭素原子数1〜3の低級アルコールとしては、例えば、メタノール、エタノール、イソプロピルアルコール等が挙げられる。
【0018】
上記炭素原子数3または4のジアルキルケトンとしては、アセトン、メチルエチルケトン等が挙げられる。
【0019】
R1−CN(式中、R1はメチル基またはエチル基である)で表されるニトリルとしては、アセトニトリル、プロピオニトリル等が挙げられる。
【0020】
R2−C(=O)−NR3R4(式中、R2、R3およびR4はそれぞれ独立に水素原子、メチル基またはエチル基を表す。)で表されるアミドとしては、N,N−ジメチルホルムアミド(DMF)、N,N−ジメチルアセトアミド、N,N−ジエチルホルムアミド、N,N−ジエチルアセトアミド等が挙げられる。
【0021】
R5−S(=O)−R6(式中、R5およびR6はそれぞれ独立にメチル基またはエチル基を表す。)で表されるジアルキルスルホキシドとしては、ジメチルスルホキシド(DMSO)、ジエチルスルホキシド等を挙げることができる。
【0022】
上記水性溶媒のうち、取り扱いの容易さなどから、アセトン、メタノール、エタノール、イソプロピルアルコール、アセトニトリル、N,N−ジメチルホルムアミド、ジメチルスルホキシド、ジオキサン、テトラヒドロフランが特に好ましい。
【0023】
上記水性溶媒と水との混合液における水性溶媒と水との比率は体積比で水性溶媒:水=1:9〜9:1であり、好ましくは、体積比で水性溶媒:水=4:6〜7:3である。
【0024】
本発明のナファモスタットメシル酸塩の晶析方法では、水性溶媒と水との混合液に溶解したナファモスタットメシル酸塩に対して、−10〜45℃の温度範囲の下、アセトンを加えるが、上記温度範囲は、好ましくは−5〜40℃、より好ましくは、−5〜20℃である。本発明のナファモスタットメシル酸塩の晶析方法は、低温であってもナファモスタットメシル酸塩溶液がゲル状の塊を生じることを抑制することができる。なるべく低温の方がナファモスタットメシル酸塩の分解をより防止できるので好ましいが、45℃近辺まで温度を上げてアセトンを加えても晶析は可能である。また、−10℃より低い温度については、コストの面で不利である。
加えるアセトンの量は任意であるが、ナファモスタットメシル酸塩溶液に対して、2〜7倍容量が好ましい。アセトンをあまり多く使用しても回収率はそれほど増加しないため、実用的ではない。
【0025】
上記方法により、析出したナファモスタットメシル酸塩は公知の分離方法により回収することができる。分離方法としては例えば、濾過などが挙げられる。
【0026】
本発明のナファモスタットメシル酸塩の結晶は、上記本発明のナファモスタットメシル酸塩の晶析方法によって得られることを特徴とするものである。
【実施例】
【0027】
次に、実施例によって本発明を更に詳細に説明するが、本発明は下記の実施例によって制限を受けるものではない。
【0028】
(実施例1)
[低温晶析操作]
粗ナファモスタットメシル酸塩(10.0g)をメタノール(32mL)と精製水(48mL)の混合液に加え、攪拌下で45℃に加温して溶解した。得られた溶液を3℃に冷却し、攪拌しながらアセトン(300mL)を注入し、晶析を行った。析出した結晶を濾取し乾燥することによりナファモスタットメシル酸塩(8.9g)を得た。回収率は89%であった。
【0029】
(実施例2)
[40℃晶析操作]
粗ナファモスタットメシル酸塩(10.0g)をメタノール(32mL)と精製水(48mL)の混合液に加え、攪拌下で45℃に加温して溶解した。得られた溶液を40℃に維持しながら、アセトン(300mL)を注入し、晶析を行った。析出した結晶を濾取し乾燥することによりナファモスタットメシル酸塩(8.8g)を得た。回収率は88%であった。
【0030】
(実施例3〜実施例10)
粗ナファモスタットメシル酸塩を35〜50℃で溶解させ、水性溶媒、および、低温晶析におけるアセトンを加える時の温度を下記表1に記載のものに変更した以外は、実施例1または実施例2と同様にしてナファモスタットメシル酸塩の低温晶析、40℃晶析を行った。結果を下記表1に示す。
【0031】
【表1】
【0032】
(実施例11)
粗ナファモスタットメシル酸塩(10.0g)をジメチルホルムアミド(32mL)と精製水(48mL)の混合液に加え、攪拌下で40℃に加温して溶解した。得られた溶液を−5℃に冷却し、攪拌しながらアセトン(300mL)を注入した。3時間同温度で晶析をし、析出した結晶を濾取し乾燥することによりナファモスタットメシル酸塩(8.3g)を得た。回収率は83%であった。
【0033】
(実施例12)
粗ナファモスタットメシル酸塩(10.0g)をエタノール(56mL)と精製水(24mL)の混合液に加え、攪拌下で60℃に加温して溶解した。得られた溶液を3℃に冷却し、攪拌しながらアセトン(300mL)を注入し、晶析を行った。析出した結晶を濾取し乾燥することによりナファモスタットメシル酸塩(9.7g)を得た。回収率は97%であった。
【0034】
(比較例1)
粗ナファモスタットメシル酸塩(10.0g)を精製水(80mL)に加え、攪拌下で40℃に加温して溶解した。得られた水溶液を冷却すると、15℃付近でゲル化してしまい、低温での晶析ができなかった。
【0035】
(比較例2)
粗ナファモスタットメシル酸塩(10.0g)を、非水溶性有機溶媒である酢酸エチル(32mL)と精製水(48mL)との混合液に加え、攪拌下で40℃に加温して溶解した。得られた水溶液を冷却すると、14℃付近でゲル化してしまい、低温での晶析ができなかった。
【0036】
(参考例1〜参考例3)
[ナファモスタットメシル酸塩の安定性]
ナファモスタットメシル酸塩(10.0g)をエタノール(80mL)と精製水(120mL)の混合液に加え、攪拌下で40℃に加温して溶解した。得られた溶液を10℃(参考例1)、40℃(参考例2)または60℃(参考例3)に維持しながら攪拌し、経時的にサンプリングを行いHPLCで解析した。HPLCは、日本薬局方のナファモスタットメシル酸塩の純度試験の試験条件に準じて行った。測定機器としてLC−2010AHT(株式会社島津製作所製)、カラムとしてInertsil−ODS3(5μm、4.6×250mm。ジーエルサイエンス株式会社製)を用い、移動相は0.1M酢酸(0.03M 1−ヘプタスルホン酸ナトリウム含有)/アセトニトリル(7:3)、流速は1.5mL/min、検出波長は260nmであった。
それぞれの温度について、所定の温度にしてから0時間後、3時間後、6時間後、12時間後のHPLCでの解析結果を下記表2〜表4および図1、図2に示す。なお、表中の数値は、各ピークのピーク面積(Area)の割合(%)を表す。図1および図2はそれぞれ、表2〜表4中のGBA(p−グアニジノ安息香酸)およびANM(6−アミジノ−2−ナフトール)に対応するピーク面積の数値をグラフにしたものである。
また、溶解12時間後にナファモスタットメシル酸塩溶液(15mL)にアセトン(55mL)を加えて析出した結晶を減圧乾燥後、HPLCで解析した。得られた結果を下記表5に示す。
【0037】
(参考例1)
[10℃で維持]
【表2】
※1:p−グアニジノ安息香酸
※2:6−アミジノ−2−ナフトール
※3:ナファモスタットメシル酸塩
【0038】
上記表2から明らかなように、ナファモスタットメシル酸塩溶液を10℃に維持した場合、ナファモスタットメシル酸塩の分解産物であるp−グアニジノ安息香酸および6−アミジノ−2−ナフトールは、時間毎の増加が見られず、ナファモスタットメシル酸塩の減少も見られなかった。
【0039】
(参考例2)
[40℃で維持]
【表3】
【0040】
上記表3から明らかなように、ナファモスタットメシル酸塩溶液を40℃に維持した場合、1時間あたり、p−グアニジノ安息香酸が0.002%増加し、6−アミジノ−2−ナフトールは0.006%増加した。一方、ナファモスタットメシル酸塩は1時間毎におよそ0.009%減少した。
【0041】
(参考例3)
[60℃で維持]
【表4】
【0042】
上記表4から明らかなように、ナファモスタットメシル酸塩溶液を60℃に維持した場合、1時間あたり、p−グアニジノ安息香酸が0.014%増加し、6−アミジノ−2−ナフトールは0.037%増加した。また、15minのピークが1時間毎に0.006%増加した。一方、ナファモスタットメシル酸塩は1時間毎におよそ0.059%減少した。
【0043】
[溶解12時間後にアセトン晶析によって得られた結晶]
【表5】
【0044】
上記表5から明らかなように、晶析を行うと高純度のナファモスタットメシル酸塩が得られるものの、60℃で維持した場合のように溶液中にナファモスタットメシル酸塩の分解に起因すると考えられる不純物が多く含まれると、晶析により得られるナファモスタットメシル酸塩中にその不純物が含まれてしまうことが確認できた。
【0045】
上記表2〜表4の結果から明らかなように、10℃ではナファモスタットメシル酸塩は12時間安定であったが、60℃でのp−グアニジノ安息香酸の増加量は、40℃の場合の7倍であり、60℃での6−アミジノ−2−ナフトールの増加量は、40℃の場合の6.2倍であった。一方、60℃でのナファモスタットメシル酸塩の減少量は、40℃の場合の6.6倍であった。これらの結果から、高温になるほどナファモスタットメシル酸塩の分解が促進されることが分かる。従って、より低温で晶析することが、分解を抑えつつ高純度のナファモスタットメシル酸塩を得る為には好ましいといえる。
【技術分野】
【0001】
本発明は、ナファモスタットメシル酸塩の晶析方法に関し、詳しくは、ナファモスタットメシル酸塩が溶液中でゲル状の塊となることが抑制され、高純度のナファモスタットメシル酸塩を得ることのできるナファモスタットメシル酸塩の晶析方法に関する。
【背景技術】
【0002】
ナファモスタットメシル酸塩(NAF)の晶析は、一般に、ナファモスタットメシル酸塩の水溶液にアセトンを加える方法により行われる(特許文献1参照)。この方法では、ナファモスタットメシル酸塩水溶液を冷却するとナファモスタットメシル酸塩の析出とともに溶液がゲル状の塊となり、ナファモスタットメシル酸塩のろ過による分離が困難になることが知られている。そのため、ナファモスタットメシル酸塩水溶液を加温しながら晶析を行う必要があった。例えば、特許文献2では、25〜45℃でナファモスタットメシル酸塩水溶液の晶析を行うことが開示されている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2004−284982号公報
【特許文献2】特許第3796481号
【発明の概要】
【発明が解決しようとする課題】
【0004】
しかしながら、ナファモスタットメシル酸塩はエステル化合物であり、水溶液状態での安定性が悪く、加水分解して4−グアニジノ安息香酸や6−アミジノ−2−ナフトール等の分解物が生じることが知られている(薬学雑誌 105(5),512−516(1985))。このため、高純度のナファモスタットメシル酸塩を得るためには、なるべく低温でナファモスタットメシル酸塩溶液の晶析を行うことが望ましい。
【0005】
そこで本発明の目的は、ナファモスタットメシル酸塩が溶液中でゲル状の塊となることが抑制され、高純度のナファモスタットメシル酸塩を得ることのできるナファモスタットメシル酸塩の晶析方法を提供することにある。
【課題を解決するための手段】
【0006】
本発明者等は、上記課題を解決すべく鋭意検討した結果、水性溶媒と水との混合液にナファモスタットメシル酸塩を溶解して得られるナファモスタットメシル酸塩溶液は、冷却してもゲル状の塊にならず、該ナファモスタットメシル酸塩溶液にアセトンを加えることにより、高純度のナファモスタットメシル酸塩を得ることができることを見出し、本発明を完成するに至った。
【0007】
すなわち、本発明のナファモスタットメシル酸塩の晶析方法は、体積比で水性溶媒:水=9:1〜1:9である水性溶媒と水との混合液に溶解したナファモスタットメシル酸塩に対して、−10〜45℃の温度範囲の下、アセトンを加える工程を有することを特徴とするものである。
【0008】
本発明のナファモスタットメシル酸塩の晶析方法においては、前記水性溶媒と水との混合液が、体積比で水性溶媒:水=4:6〜7:3の混合液であることが好ましい。
【0009】
また、本発明のナファモスタットメシル酸塩の晶析方法においては、前記水性溶媒が、炭素原子数1〜3の低級アルコール、炭素原子数3または4のジアルキルケトン、R1−CN(式中、R1はメチル基またはエチル基である)で表されるニトリル、R2−C(=O)−NR3R4(式中、R2、R3およびR4はそれぞれ独立に水素原子、メチル基またはエチル基を表す。)で表されるアミド、R5−S(=O)−R6(式中、R5およびR6はそれぞれ独立にメチル基またはエチル基を表す。)で表されるジアルキルスルホキシド、ジオキサン、および、テトラヒドロフランからなる群から選ばれるものであることが好ましい。
【0010】
さらに、本発明のナファモスタットメシル酸塩の晶析方法においては、前記水性溶媒が、アセトン、メタノール、エタノール、イソプロピルアルコール、アセトニトリル、N,N−ジメチルホルムアミド、ジメチルスルホキシド、ジオキサン、および、テトラヒドロフランからなる群から選ばれるものであることが好ましい。
【0011】
本発明のナファモスタットメシル酸塩の結晶は、上記のナファモスタットメシル酸塩の晶析方法によって得られることを特徴とするものである。
【発明の効果】
【0012】
本発明により、ナファモスタットメシル酸塩が溶液中でゲル状の塊となることが抑制され、高純度のナファモスタットメシル酸塩を得ることのできるナファモスタットメシル酸塩の晶析方法を提供することができる。
【図面の簡単な説明】
【0013】
【図1】参考例1〜3におけるp−グアニジノ安息香酸の増加を表すグラフである。
【図2】参考例1〜3における6−アミジノ−2−ナフトールの増加を表すグラフである。
【発明を実施するための形態】
【0014】
本発明のナファモスタットメシル酸塩の晶析方法は、水性溶媒と水との混合液に溶解したナファモスタットメシル酸塩に対して、−10〜45℃の温度範囲の下、アセトンを加える工程を有することを特徴とするものである。
ナファモスタットメシル酸塩は、下記式で表される化合物である4−[(アミノイミノメチル)アミノ]安息香酸6−(アミノイミノメチル)−2−ナフタレニル・2メタンスルホン酸の一般名である。
【0015】
ナファモスタットメシル酸塩を水性溶媒と水との混合液に溶解する方法は、公知のいずれの方法でもよく、例えば水性溶媒と水との混合液を35〜60℃に加温し、攪拌しながら溶解させる方法が好ましい。通常、ナファモスタットメシル酸塩を溶媒に溶解させるには、ある程度加温が必要であるが、あまり高温にするとナファモスタットメシル酸塩が分解する恐れがあるため、上記温度範囲で溶解させるのが好ましい。
【0016】
上記水性溶媒と水との混合液における水性溶媒とは、水との混和性を有する溶媒であればいずれでもよい。また、2種以上の水性触媒を用いてもよい。本発明において好ましい水性溶媒としては、炭素原子数1〜3の低級アルコール、炭素原子数3または4のジアルキルケトン、R1−CN(式中、R1はメチル基またはエチル基である)で表されるニトリル、R2−C(=O)−NR3R4(式中、R2、R3およびR4はそれぞれ独立に水素原子、メチル基またはエチル基を表す。)で表されるアミド、R5−S(=O)−R6(式中、R5およびR6はそれぞれ独立にメチル基またはエチル基を表す。)で表されるジアルキルスルホキシド、ジオキサン、および、テトラヒドロフランを挙げることができる。
【0017】
上記炭素原子数1〜3の低級アルコールとしては、例えば、メタノール、エタノール、イソプロピルアルコール等が挙げられる。
【0018】
上記炭素原子数3または4のジアルキルケトンとしては、アセトン、メチルエチルケトン等が挙げられる。
【0019】
R1−CN(式中、R1はメチル基またはエチル基である)で表されるニトリルとしては、アセトニトリル、プロピオニトリル等が挙げられる。
【0020】
R2−C(=O)−NR3R4(式中、R2、R3およびR4はそれぞれ独立に水素原子、メチル基またはエチル基を表す。)で表されるアミドとしては、N,N−ジメチルホルムアミド(DMF)、N,N−ジメチルアセトアミド、N,N−ジエチルホルムアミド、N,N−ジエチルアセトアミド等が挙げられる。
【0021】
R5−S(=O)−R6(式中、R5およびR6はそれぞれ独立にメチル基またはエチル基を表す。)で表されるジアルキルスルホキシドとしては、ジメチルスルホキシド(DMSO)、ジエチルスルホキシド等を挙げることができる。
【0022】
上記水性溶媒のうち、取り扱いの容易さなどから、アセトン、メタノール、エタノール、イソプロピルアルコール、アセトニトリル、N,N−ジメチルホルムアミド、ジメチルスルホキシド、ジオキサン、テトラヒドロフランが特に好ましい。
【0023】
上記水性溶媒と水との混合液における水性溶媒と水との比率は体積比で水性溶媒:水=1:9〜9:1であり、好ましくは、体積比で水性溶媒:水=4:6〜7:3である。
【0024】
本発明のナファモスタットメシル酸塩の晶析方法では、水性溶媒と水との混合液に溶解したナファモスタットメシル酸塩に対して、−10〜45℃の温度範囲の下、アセトンを加えるが、上記温度範囲は、好ましくは−5〜40℃、より好ましくは、−5〜20℃である。本発明のナファモスタットメシル酸塩の晶析方法は、低温であってもナファモスタットメシル酸塩溶液がゲル状の塊を生じることを抑制することができる。なるべく低温の方がナファモスタットメシル酸塩の分解をより防止できるので好ましいが、45℃近辺まで温度を上げてアセトンを加えても晶析は可能である。また、−10℃より低い温度については、コストの面で不利である。
加えるアセトンの量は任意であるが、ナファモスタットメシル酸塩溶液に対して、2〜7倍容量が好ましい。アセトンをあまり多く使用しても回収率はそれほど増加しないため、実用的ではない。
【0025】
上記方法により、析出したナファモスタットメシル酸塩は公知の分離方法により回収することができる。分離方法としては例えば、濾過などが挙げられる。
【0026】
本発明のナファモスタットメシル酸塩の結晶は、上記本発明のナファモスタットメシル酸塩の晶析方法によって得られることを特徴とするものである。
【実施例】
【0027】
次に、実施例によって本発明を更に詳細に説明するが、本発明は下記の実施例によって制限を受けるものではない。
【0028】
(実施例1)
[低温晶析操作]
粗ナファモスタットメシル酸塩(10.0g)をメタノール(32mL)と精製水(48mL)の混合液に加え、攪拌下で45℃に加温して溶解した。得られた溶液を3℃に冷却し、攪拌しながらアセトン(300mL)を注入し、晶析を行った。析出した結晶を濾取し乾燥することによりナファモスタットメシル酸塩(8.9g)を得た。回収率は89%であった。
【0029】
(実施例2)
[40℃晶析操作]
粗ナファモスタットメシル酸塩(10.0g)をメタノール(32mL)と精製水(48mL)の混合液に加え、攪拌下で45℃に加温して溶解した。得られた溶液を40℃に維持しながら、アセトン(300mL)を注入し、晶析を行った。析出した結晶を濾取し乾燥することによりナファモスタットメシル酸塩(8.8g)を得た。回収率は88%であった。
【0030】
(実施例3〜実施例10)
粗ナファモスタットメシル酸塩を35〜50℃で溶解させ、水性溶媒、および、低温晶析におけるアセトンを加える時の温度を下記表1に記載のものに変更した以外は、実施例1または実施例2と同様にしてナファモスタットメシル酸塩の低温晶析、40℃晶析を行った。結果を下記表1に示す。
【0031】
【表1】
【0032】
(実施例11)
粗ナファモスタットメシル酸塩(10.0g)をジメチルホルムアミド(32mL)と精製水(48mL)の混合液に加え、攪拌下で40℃に加温して溶解した。得られた溶液を−5℃に冷却し、攪拌しながらアセトン(300mL)を注入した。3時間同温度で晶析をし、析出した結晶を濾取し乾燥することによりナファモスタットメシル酸塩(8.3g)を得た。回収率は83%であった。
【0033】
(実施例12)
粗ナファモスタットメシル酸塩(10.0g)をエタノール(56mL)と精製水(24mL)の混合液に加え、攪拌下で60℃に加温して溶解した。得られた溶液を3℃に冷却し、攪拌しながらアセトン(300mL)を注入し、晶析を行った。析出した結晶を濾取し乾燥することによりナファモスタットメシル酸塩(9.7g)を得た。回収率は97%であった。
【0034】
(比較例1)
粗ナファモスタットメシル酸塩(10.0g)を精製水(80mL)に加え、攪拌下で40℃に加温して溶解した。得られた水溶液を冷却すると、15℃付近でゲル化してしまい、低温での晶析ができなかった。
【0035】
(比較例2)
粗ナファモスタットメシル酸塩(10.0g)を、非水溶性有機溶媒である酢酸エチル(32mL)と精製水(48mL)との混合液に加え、攪拌下で40℃に加温して溶解した。得られた水溶液を冷却すると、14℃付近でゲル化してしまい、低温での晶析ができなかった。
【0036】
(参考例1〜参考例3)
[ナファモスタットメシル酸塩の安定性]
ナファモスタットメシル酸塩(10.0g)をエタノール(80mL)と精製水(120mL)の混合液に加え、攪拌下で40℃に加温して溶解した。得られた溶液を10℃(参考例1)、40℃(参考例2)または60℃(参考例3)に維持しながら攪拌し、経時的にサンプリングを行いHPLCで解析した。HPLCは、日本薬局方のナファモスタットメシル酸塩の純度試験の試験条件に準じて行った。測定機器としてLC−2010AHT(株式会社島津製作所製)、カラムとしてInertsil−ODS3(5μm、4.6×250mm。ジーエルサイエンス株式会社製)を用い、移動相は0.1M酢酸(0.03M 1−ヘプタスルホン酸ナトリウム含有)/アセトニトリル(7:3)、流速は1.5mL/min、検出波長は260nmであった。
それぞれの温度について、所定の温度にしてから0時間後、3時間後、6時間後、12時間後のHPLCでの解析結果を下記表2〜表4および図1、図2に示す。なお、表中の数値は、各ピークのピーク面積(Area)の割合(%)を表す。図1および図2はそれぞれ、表2〜表4中のGBA(p−グアニジノ安息香酸)およびANM(6−アミジノ−2−ナフトール)に対応するピーク面積の数値をグラフにしたものである。
また、溶解12時間後にナファモスタットメシル酸塩溶液(15mL)にアセトン(55mL)を加えて析出した結晶を減圧乾燥後、HPLCで解析した。得られた結果を下記表5に示す。
【0037】
(参考例1)
[10℃で維持]
【表2】
※1:p−グアニジノ安息香酸
※2:6−アミジノ−2−ナフトール
※3:ナファモスタットメシル酸塩
【0038】
上記表2から明らかなように、ナファモスタットメシル酸塩溶液を10℃に維持した場合、ナファモスタットメシル酸塩の分解産物であるp−グアニジノ安息香酸および6−アミジノ−2−ナフトールは、時間毎の増加が見られず、ナファモスタットメシル酸塩の減少も見られなかった。
【0039】
(参考例2)
[40℃で維持]
【表3】
【0040】
上記表3から明らかなように、ナファモスタットメシル酸塩溶液を40℃に維持した場合、1時間あたり、p−グアニジノ安息香酸が0.002%増加し、6−アミジノ−2−ナフトールは0.006%増加した。一方、ナファモスタットメシル酸塩は1時間毎におよそ0.009%減少した。
【0041】
(参考例3)
[60℃で維持]
【表4】
【0042】
上記表4から明らかなように、ナファモスタットメシル酸塩溶液を60℃に維持した場合、1時間あたり、p−グアニジノ安息香酸が0.014%増加し、6−アミジノ−2−ナフトールは0.037%増加した。また、15minのピークが1時間毎に0.006%増加した。一方、ナファモスタットメシル酸塩は1時間毎におよそ0.059%減少した。
【0043】
[溶解12時間後にアセトン晶析によって得られた結晶]
【表5】
【0044】
上記表5から明らかなように、晶析を行うと高純度のナファモスタットメシル酸塩が得られるものの、60℃で維持した場合のように溶液中にナファモスタットメシル酸塩の分解に起因すると考えられる不純物が多く含まれると、晶析により得られるナファモスタットメシル酸塩中にその不純物が含まれてしまうことが確認できた。
【0045】
上記表2〜表4の結果から明らかなように、10℃ではナファモスタットメシル酸塩は12時間安定であったが、60℃でのp−グアニジノ安息香酸の増加量は、40℃の場合の7倍であり、60℃での6−アミジノ−2−ナフトールの増加量は、40℃の場合の6.2倍であった。一方、60℃でのナファモスタットメシル酸塩の減少量は、40℃の場合の6.6倍であった。これらの結果から、高温になるほどナファモスタットメシル酸塩の分解が促進されることが分かる。従って、より低温で晶析することが、分解を抑えつつ高純度のナファモスタットメシル酸塩を得る為には好ましいといえる。
【特許請求の範囲】
【請求項1】
体積比で水性溶媒:水=9:1〜1:9である水性溶媒と水との混合液に溶解したナファモスタットメシル酸塩に対して、−10〜45℃の温度範囲の下、アセトンを加える工程を有することを特徴とするナファモスタットメシル酸塩の晶析方法。
【請求項2】
前記水性溶媒と水との混合液が、体積比で水性溶媒:水=4:6〜7:3の混合液である請求項1記載のナファモスタットメシル酸塩の晶析方法。
【請求項3】
前記水性溶媒が、炭素原子数1〜3の低級アルコール、炭素原子数3または4のジアルキルケトン、R1−CN(式中、R1はメチル基またはエチル基である)で表されるニトリル、R2−C(=O)−NR3R4(式中、R2、R3およびR4はそれぞれ独立に水素原子、メチル基またはエチル基を表す。)で表されるアミド、R5−S(=O)−R6(式中、R5およびR6はそれぞれ独立にメチル基またはエチル基を表す。)で表されるジアルキルスルホキシド、ジオキサン、および、テトラヒドロフランからなる群から選ばれるものである請求項1または2記載のナファモスタットメシル酸塩の晶析方法。
【請求項4】
前記水性溶媒が、アセトン、メタノール、エタノール、イソプロピルアルコール、アセトニトリル、N,N−ジメチルホルムアミド、ジメチルスルホキシド、ジオキサン、および、テトラヒドロフランからなる群から選ばれるものである請求項1〜3のいずれか一項記載のナファモスタットメシル酸塩の晶析方法。
【請求項5】
請求項1〜4のいずれか一項記載のナファモスタットメシル酸塩の晶析方法によって得られることを特徴とするナファモスタットメシル酸塩の結晶。
【請求項1】
体積比で水性溶媒:水=9:1〜1:9である水性溶媒と水との混合液に溶解したナファモスタットメシル酸塩に対して、−10〜45℃の温度範囲の下、アセトンを加える工程を有することを特徴とするナファモスタットメシル酸塩の晶析方法。
【請求項2】
前記水性溶媒と水との混合液が、体積比で水性溶媒:水=4:6〜7:3の混合液である請求項1記載のナファモスタットメシル酸塩の晶析方法。
【請求項3】
前記水性溶媒が、炭素原子数1〜3の低級アルコール、炭素原子数3または4のジアルキルケトン、R1−CN(式中、R1はメチル基またはエチル基である)で表されるニトリル、R2−C(=O)−NR3R4(式中、R2、R3およびR4はそれぞれ独立に水素原子、メチル基またはエチル基を表す。)で表されるアミド、R5−S(=O)−R6(式中、R5およびR6はそれぞれ独立にメチル基またはエチル基を表す。)で表されるジアルキルスルホキシド、ジオキサン、および、テトラヒドロフランからなる群から選ばれるものである請求項1または2記載のナファモスタットメシル酸塩の晶析方法。
【請求項4】
前記水性溶媒が、アセトン、メタノール、エタノール、イソプロピルアルコール、アセトニトリル、N,N−ジメチルホルムアミド、ジメチルスルホキシド、ジオキサン、および、テトラヒドロフランからなる群から選ばれるものである請求項1〜3のいずれか一項記載のナファモスタットメシル酸塩の晶析方法。
【請求項5】
請求項1〜4のいずれか一項記載のナファモスタットメシル酸塩の晶析方法によって得られることを特徴とするナファモスタットメシル酸塩の結晶。
【図1】

【図2】
【図2】
【公開番号】特開2012−87099(P2012−87099A)
【公開日】平成24年5月10日(2012.5.10)
【国際特許分類】
【出願番号】特願2010−236239(P2010−236239)
【出願日】平成22年10月21日(2010.10.21)
【出願人】(508262342)桂化学株式会社 (1)
【Fターム(参考)】
【公開日】平成24年5月10日(2012.5.10)
【国際特許分類】
【出願日】平成22年10月21日(2010.10.21)
【出願人】(508262342)桂化学株式会社 (1)
【Fターム(参考)】
[ Back to top ]